CN1313861A - 2-(嘌呤-9-基)-四氢呋喃-3,4-二醇衍生物 - Google Patents
2-(嘌呤-9-基)-四氢呋喃-3,4-二醇衍生物 Download PDFInfo
- Publication number
- CN1313861A CN1313861A CN99809892A CN99809892A CN1313861A CN 1313861 A CN1313861 A CN 1313861A CN 99809892 A CN99809892 A CN 99809892A CN 99809892 A CN99809892 A CN 99809892A CN 1313861 A CN1313861 A CN 1313861A
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- alkyl
- represent
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/06—Heterocyclic radicals
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
按照本发明提供了:式(Ⅰ)的新化合物(式中R1、R2和R3如同在本说明书中所定义)、制备它们的方法、包含它们的制剂、以及它们在治疗炎症的疗法方面的应用。
Description
本发明涉及新的化合物、用于制备它们的方法、包含它们的药用制剂以及它们在治疗方面的运用。
炎症是对组织损伤或微生物侵入的主要反应,其特征在于在所述组织中白细胞粘附到内皮、血细胞渗出和活化来表示炎症。白细胞活化可导致有毒氧形式(例如过氧化物阴离子)的产生,以及颗粒产物(例如过氧化物酶和蛋白酶)的释放。循环的白细胞包括中性白细胞、嗜酸性粒细胞、嗜碱性粒细胞、单核细胞和淋巴细胞。炎症的各种形式包含不同类型的浸润白细胞,其特定分布型由所述组织中粘连分子、细胞因子和趋化因子表达的分布型来调节。
白细胞的主要功能是保护宿主使其不受诸如细菌和寄生虫之类的生物的侵入。一旦组织受损伤或感染,就发生一系列引起来自循环的白细胞被局部募集到受影响组织的事件。控制白细胞的募集,则便于对外来细胞或死细胞的有序破坏和吞噬作用,继之以组织修复和炎性浸润物的消退。然而,在慢性炎性状态下,募集常常是不适当的,炎症的消退被不适当地控制且炎性反应引起组织的破坏。
有来自体外研究和体内研究两者的证据暗示:在腺苷A2a受体上有活性的化合物将具有抗炎作用。Cronstein(1994)已综述了这领域。对分离的中性白细胞的研究表明了A2受体介导的对过氧化物产生的抑制、脱粒、聚集和粘附(Cronstein等,1983和1985;Burkey和Webster,1993;Richter,1992;Skubitz等,1988)。当使用对A2a受体的选择性超过对A2b受体的选择性的药剂(例如CGS21680)时,抑制的分布型看来与在A2a受体亚型上的作用一致(Dianzani等,1994)。腺苷激动剂也可以减量调节其它类别的白细胞(Elliot和Leonard,1989;Peachell等,1989)。对完整的动物的研究显示了通过腺苷和A2受体的活化而介导的甲氧蝶呤的抗炎效应(Asako等,1993;Cronstein等,1993和1994)。腺苷本身以及提高腺苷循环水平的化合物在体内也显示出抗炎效应(Green等,1991;Rosengren等,1995)。另外,在人体中,升高了的循环腺苷水平(作为腺苷脱氨酶缺乏症的结果)导致免疫抑制(Hirschorn,1993)。
在国际专利申请WO94/17090、WO96/02553、WO96/02543(Glaxo Group)中,描述了可用于治疗炎性疾病的某些取代的4’-羧酰胺基和4’-硫代酰胺基的腺苷衍生物。在AU 8771946(日本Hoechst)中,描述了可用于治疗痴呆的取代的4’-羧酰胺基腺苷衍生物。在EP-A-423776和EP-A-423777(Searle)中,描述了可用于治疗胃肠能动性紊乱的取代的4’-羟甲基腺苷衍生物。在BE-768925(Takeda)中,描述了可用作血小板凝聚反应抑制剂的取代的4’-羟甲基腺苷衍生物。在US 4663313、EP 139358和US 4767747(Warner Lambert)、US 4985409(Nippon Zoki)和US5043325(Whitby Research)中,描述了可用作抗高血压药或具有其它心血管活性的4’-羟甲基腺苷衍生物及其4’-酯。在US 5106837(ScrippsResearch Institute)中,描述了可用于治疗自身免疫病的4-羟甲基腺苷衍生物。在US 4704381(Boehringer Mannheim)中,描述了可用作抗过敏性剂的4’-羟甲基腺苷衍生物。在DT-A-2621470(Pharma-Waldhof)中,一般地描述了可用于治疗心脏病和循环障碍的某些4’-四唑基烷基腺苷衍生物。在US 5219840、GB 2203149和GB 2199036(Sandoz)、WO94/02497(US Dept.Health)、US 4968697和EP 277917(Ciba Geigy)、US 5424297(弗吉尼亚大学)和EP 232813(Warner Lambert)中,描述了在治疗心血管疾病方面有效的其它4’-羧酰胺基腺苷衍生物。在DT 2317770、DT2213180、US 4167565、US 3864483和US 3966917(Abbott Labs)、DT2034785(Boehringer Mannheim)、JP58174322和JP58167599(TanabeSeiyaku)、WO92/05177和US 5364862(Rhone Poulenc Rorer)、EP 66918(Procter和Gamble)、WO86/00310(Nelson)、EP 222330、US 4962194、WO88/03147和WO88/03148(Warner Lambert)及US 5219839、WO95/18817和WO93/14102(Lab UPSA)中,描述了在嘌呤环上的2位没有取代的其它4’-羧酰胺基腺苷衍生物。在WO95/11904(佛罗里达大学)中,描述了在嘌呤环上的2位没有取代的4’-羟甲基腺苷衍生物。在WO94/18215(Gensia)中,描述了可用作腺苷激酶抑制剂的4’-取代的腺苷衍生物。在EP 161128和EP 181129(Warner Lambert)及US 3983104(Schering)中,描述了其它的4’-卤代甲基、甲基、硫代烷基甲基或烷氧基甲基腺苷衍生物。其它的4’-羧酰胺基腺苷衍生物描述于US 7577528(NIH)、WO91/13082(Whitby Research)和WO95/02604(US Dept.Health)中。
在Baker等(1974)Tetrahedron 30,2939-2942中描述了某些含四唑的脱氧核苷酸,发现所述的脱氧核苷酸缺乏抗感染的活性。在Mester和Mester(1972)Pathologie-Biologie,20(Suppl)11-14中,描述了作为血小板凝聚反应抑制剂而显示活性的其它含四唑的腺苷衍生物。在Schmidt等(1974)Liebigs.Ann.Chem.1856-1863中,描述了某种含腈核糖衍生物。
其它的出版物包括:WO 98/16539(Novo Nordisk A/S),描述了用于治疗心肌缺血和大脑局部缺血以及癫痫的腺苷衍生物;WO 98/01426(Rone-Poulenc Rorer Pharmaceuticals Inc.),涉及具有抗高血压、保护心脏、抗局部缺血和抗分解脂肪特性的腺苷衍生物;以及WO 98/01459(Novo Nordisk A/S),描述了N,9-二取代的、在4’位置被未取代的噁唑基或异噁唑基取代的腺嘌呤衍生物及运用这样的化合物在人类中治疗涉及细胞因子的疾病。在本申请的最早优先权日之后,公布了WO 98/28319(Glaxo Group Limited);且它描述了4’-取代的四唑2-(嘌呤-9-基)-四氢呋喃-3,4-二醇衍生物。
目前我们已发现了一组具有广泛抗炎特性的新化合物,所述新化合物抑制白细胞的募集及白细胞的活化,并且所述新化合物是腺苷2a受体的激动剂。因此,在炎症部位牵涉白细胞的疾病中,在提供保护而不受白细胞诱导的组织损害方面,所述化合物具有潜在的治疗好处。在炎症的治疗方面,本发明的化合物也可以是皮质类固醇的较安全的替换物,皮质类固醇的运用可能由于其副作用分布型而受限制。
更具体地讲,本发明的化合物可显示出超过已知的A2a选择性激动剂的、改善的分布型,因为它们在人的A3受体上一般缺乏显著的激动剂活性。由于在白细胞(例如嗜酸性粒细胞)和其它炎性细胞(例如肥大细胞)上也发现了A3受体,且这些受体的活化可能具有促炎效应(Kohno等,1996;Van Schaick等1996);因而可认为这种分布型具有好处。甚至认为在哮喘病患者中,可能通过腺苷A3受体,介导腺苷的支气管收缩药效应(Kohno等,1996)。
这样,按照本发明,我们提供式(Ⅰ)的化合物,以及其盐和溶剂化物; 在式(Ⅰ)中,R1和R2独立地代表选自下面的基团:(ⅰ)C3-8环烷基-;(ⅱ)氢;(ⅲ)(芳基)2CHCH2-;(ⅳ)C3-8环烷基C1-6烷基-;(ⅴ)C1-8烷基-;(ⅵ)芳基C1-6烷基-;(ⅶ)R4R5N-C1-6烷基-;(ⅷ)C1-6烷基-CH(CH2OH)-;(ⅸ)芳基C1-5烷基-CH(CH2OH)-;(ⅹ)芳基C1-5烷基-C(CH2OH)2-;(ⅹⅰ)独立地用一个或更多个(例如1、2或3个)-(CH2)pR6基团取代的C3-8环烷基;(ⅹⅱ)H2NC(=NH)NHC1-6烷基-;(ⅹⅲ)下式的基团,
在式(Ⅰ)中,R1和R2独立地代表选自下面的基团:(ⅰ)C3-8环烷基-;(ⅱ)氢;(ⅲ)(芳基)2CHCH2-;(ⅳ)C3-8环烷基C1-6烷基-;(ⅴ)C1-8烷基-;(ⅵ)芳基C1-6烷基-;(ⅶ)R4R5N-C1-6烷基-;(ⅷ)C1-6烷基-CH(CH2OH)-;(ⅸ)芳基C1-5烷基-CH(CH2OH)-;(ⅹ)芳基C1-5烷基-C(CH2OH)2-;(ⅹⅰ)独立地用一个或更多个(例如1、2或3个)-(CH2)pR6基团取代的C3-8环烷基;(ⅹⅱ)H2NC(=NH)NHC1-6烷基-;(ⅹⅲ)下式的基团, 或基团中一个毗连X的亚甲基碳原子被甲基取代的这样的基团;或者,如果存在两个毗连X的亚甲基碳原子,则为这两个毗连X的亚甲基碳原子都被甲基取代的这样的基团;(ⅹⅳ)-C1-6烷基-OH;(ⅹⅴ)-C1-8卤烷基;(ⅹⅵ)下式的基团
或基团中一个毗连X的亚甲基碳原子被甲基取代的这样的基团;或者,如果存在两个毗连X的亚甲基碳原子,则为这两个毗连X的亚甲基碳原子都被甲基取代的这样的基团;(ⅹⅳ)-C1-6烷基-OH;(ⅹⅴ)-C1-8卤烷基;(ⅹⅵ)下式的基团 (ⅹⅶ)芳基;以及(ⅹⅷ)-(CH2)fSO2NHg(C1-4烷基-)2-g或-(CH2)fSO2NHg(芳基C1-4烷基-)2-g;R3代表甲基、乙基、-CH=CH2、正丙基、-CH2CH=CH2、-CH=CHCH3、异丙基、异丙烯基、环丙基、环丙烯基、环丙基甲基、环丙烯基甲基、环丁基、环丁烯基、-(CH2)q卤、-(CH2)hY(CH2)iH、-(CH2)hCOOCH3、-(CH2)hOCOCH3.-(CH2)hCON(CH2)mH((CH2)nH)、-(CH2)hCO(CH2)oH或-CH2C((CH2)uH)=NO(CH2)vH;Y代表O、S或N(CH2)j;如果a+b是在3至5的范围内,则a和b独立地代表0至4的整数;如果c+d+e是在2至3的范围内,则c、d和e独立地代表0至3的整数;f代表2或3,并且g代表0至2的整数;p代表0或1;q代表2或3;h代表2或3;i代表0至2的整数,以致h+i在2至4的范围内;j代表0至2的整数,以致h+i+j在2至4的范围内;m和n独立地代表0至2的整数,以致m+n在0至2的范围内;o代表0至2的整数,以致h+o在2至3的范围内;u和v独立地代表0或1,以致u+v在0至1的范围内;R4和R5独立地代表氢、C1-6烷基、芳基、芳基C1-6烷基-或NR4R5一起可代表吡咯烷基、哌啶基、吗啉基、氮杂环丁烷基、氮杂基、哌嗪基或N-C1-6烷基哌嗪基。R6代表OH、NH2、NHCOCH3或卤;R7代表氢、C1-6烷基、C1-6烷基芳基或-COC1-6烷基;X代表NR7、O、S、SO或SO2;前提是:如果R3代表甲基、乙基或异丙基,那么R1和/或R2必须独立地代表:(a)-(CH2)fSO2NHg(C1-4烷基-)2-g或-(CH2)fSO2NHg(芳基C1-4烷基-)2-g,这里f为2或3,并且g是0至2的整数;(b)用一个或更多个的-(CH2)pNHCOCH3基团独立地取代的C3-8环烷基;(c)下式的基团
(ⅹⅶ)芳基;以及(ⅹⅷ)-(CH2)fSO2NHg(C1-4烷基-)2-g或-(CH2)fSO2NHg(芳基C1-4烷基-)2-g;R3代表甲基、乙基、-CH=CH2、正丙基、-CH2CH=CH2、-CH=CHCH3、异丙基、异丙烯基、环丙基、环丙烯基、环丙基甲基、环丙烯基甲基、环丁基、环丁烯基、-(CH2)q卤、-(CH2)hY(CH2)iH、-(CH2)hCOOCH3、-(CH2)hOCOCH3.-(CH2)hCON(CH2)mH((CH2)nH)、-(CH2)hCO(CH2)oH或-CH2C((CH2)uH)=NO(CH2)vH;Y代表O、S或N(CH2)j;如果a+b是在3至5的范围内,则a和b独立地代表0至4的整数;如果c+d+e是在2至3的范围内,则c、d和e独立地代表0至3的整数;f代表2或3,并且g代表0至2的整数;p代表0或1;q代表2或3;h代表2或3;i代表0至2的整数,以致h+i在2至4的范围内;j代表0至2的整数,以致h+i+j在2至4的范围内;m和n独立地代表0至2的整数,以致m+n在0至2的范围内;o代表0至2的整数,以致h+o在2至3的范围内;u和v独立地代表0或1,以致u+v在0至1的范围内;R4和R5独立地代表氢、C1-6烷基、芳基、芳基C1-6烷基-或NR4R5一起可代表吡咯烷基、哌啶基、吗啉基、氮杂环丁烷基、氮杂基、哌嗪基或N-C1-6烷基哌嗪基。R6代表OH、NH2、NHCOCH3或卤;R7代表氢、C1-6烷基、C1-6烷基芳基或-COC1-6烷基;X代表NR7、O、S、SO或SO2;前提是:如果R3代表甲基、乙基或异丙基,那么R1和/或R2必须独立地代表:(a)-(CH2)fSO2NHg(C1-4烷基-)2-g或-(CH2)fSO2NHg(芳基C1-4烷基-)2-g,这里f为2或3,并且g是0至2的整数;(b)用一个或更多个的-(CH2)pNHCOCH3基团独立地取代的C3-8环烷基;(c)下式的基团
或基团中一个毗连X的亚甲基碳原子被甲基取代的这样的基团;或者,如果存在两个毗连X的亚甲基碳原子,则为这两个毗连X的亚甲基碳原子都被甲基取代的这样的基团;(d)下式的基团。
提及C1-6烷基的部分包括提及含有1至6个碳原子的脂族烃基的部分,所述脂族烃基可以是直链,或可以是分支的;并且可以是饱和的或不饱和的,尽管脂族烃基最好能是饱和的。可以类似地解释提及C1-4烷基、C1-5烷基、C2-4烷基和C1-8烷基的部分。
提及芳基的部分包括提及单环的和二环的碳环芳环(例如苯基、萘基)、以及包含1至3个选自N、O和S的杂原子的杂环芳环(例如吡啶基、嘧啶基、苯硫基、咪唑基、喹啉基、呋喃基、吡咯基、噁唑基)的部分,可以任选地取代所有这些芳环,例如,用C1-6烷基、卤素、羟基、硝基、C1-6烷氧基、氰基、氨基、SO2NH2或-CH2OH取代。
R1和R2的C3-8环烷基的实例包括单环烷基(例如环戊基、环己基)和二环烷基(例如,诸如外降冰片-2-基之类的降冰片基(norbornyl))。
R1和R2的(芳基)2CHCH2-的实例包括(苯基)2CHCH2-或者基团中一个或两个苯基部分被例如卤素或C1-4烷基取代的这样的基团。
R1和R2的C3-8环烷基C1-6烷基-的实例包括乙基环己基。
R1和R2的C1-8烷基的实例包括-(CH2)2C(Me)3、-CH(Et)2和CH2=C(Me)CH2CH2-。
R1和R2的芳基C1-6烷基-的实例包括:-(CH2)2苯基、-CH2苯基或基团中苯基被卤素(例如碘)、氨基、甲氧基、羟基、-CH2OH或SO2NH2(一次或更多次)取代的任一个基团;以及,任选地用氨基取代的-(CH2)2吡啶基(例如-(CH2)2吡啶-2-基);(CH2)2咪唑基或基团中咪唑基是用C1-6烷基(尤其是甲基)N-取代的这种基团。
R1和R2的R4R5N-C1-6烷基-的实例包括乙基-哌啶-1-基、乙基-吡咯烷-1-基、乙基-吗啉-1-基、-(CH2)2NH(吡啶-2-基)和-(CH2)2NH2。
R1和R2的C1-6烷基-CH(CH2OH)-的实例包括Me2CHCH(CH2OH)-。
R1和R2的芳基C1-5烷基-CH(CH2OH)-的实例包括苯基CH2CH(CH2OH)-,尤其是
R1和R2的芳基C1-5烷基-C(CH2OH)2-的实例包括苯基CH2C(CH2OH)2-。
R1和R2的、用一个或更多个-(CH2)pR6基团(例如1个、2个或3个这样的基团)独立地取代的C3-8环烷基的实例包括2-羟基-环戊基和4-氨基环己基(尤其是反式-4-氨基-环己基)。
R1和R2的H2NC(=NH)NHC1-6烷基的实例包括H2NC(=NH)NH(CH2)2-。
R1和R2的下式基团 的实例包括吡咯烷-3-基、哌啶-3-基、哌啶-4-基、或者其中环上氮被C1-6烷基(例如甲基)或苄基取代的衍生物、四氢-1,1-二氧化噻吩-3-基、四氢吡喃-4-基、四氢噻喃-4-基和1,1-二氧代-六氢-1.λ.6-噻喃-4-基(1,1-dioxo-hexahydro-l.lamda.6-thiopyran-4-yl)。
的实例包括吡咯烷-3-基、哌啶-3-基、哌啶-4-基、或者其中环上氮被C1-6烷基(例如甲基)或苄基取代的衍生物、四氢-1,1-二氧化噻吩-3-基、四氢吡喃-4-基、四氢噻喃-4-基和1,1-二氧代-六氢-1.λ.6-噻喃-4-基(1,1-dioxo-hexahydro-l.lamda.6-thiopyran-4-yl)。
R1和R2的-C1-6烷基-OH基团的实例包括-CH2CH2OH。
R1和R2的C1-8卤烷基的实例包括-CH2CH2Cl和(CH3)2ClC(CH2)3-。
R1和R2的下式基团的实例包括2-氧代吡咯烷-4-基、2-氧代吡咯烷-5-基或者其中环上氮被C1-6烷基(例如甲基)或苄基取代的衍生物。
R1和R2的芳基的实例包括任选地用卤素(例如氟,特别是4-氟)取代的苯基。
R7的C1-6烷基的实例包括甲基,而R7的C1-6烷基芳基包括苄基。R7的COC1-6烷基的实例包括-COCH3。
R3的C1-5烷基的实例包括正丙基和烯丙基。R3的C3-4环烷基的一个实例包括环丁基。R3的-(CH2)hO(CH2)iH的一个实例包括-(CH2)2OMe。用卤或羟基取代C2-4烷基的实例包括-(CH2)2Cl、-(CH2)2OH和-(CH2)3OH。
我们优选R1和R2两者并不都代表氢。
我们优选R1代表(芳基)2CHCH2-、C1-8烷基、氢或芳基C1-6烷基-。
我们特别优选R1代表(苯基)2CHCH2-、-CH(Et)2、氢或苯基乙基-的R1,尤其是(苯基)2CHCH2-。
我们优选R2代表R4R5N-C1-6烷基-、芳基C1-6烷基-、芳基C1-5烷基CH(CH2OH)-、芳基C1-6烷基或C1-6烷基-CH(CH2OH)-。
我们特别优选R2代表(CH2)2(哌啶-1-基)、2-(1-甲基-1H-咪唑-4-基)乙基、1S-羟甲基-2-苯基乙基、苯基乙基或1S-羟甲基-2-甲基丙基,尤其是-(CH2)2(哌啶-1-基)。
我们优选R3代表C1-3烷基(包括正丙基和2-丙烯基)、环丁基、环丙基甲基、-(CH2)2OCOCH3、-(CH2)2-3OH或-(CH2)2卤。更优选R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3或-(CH2)2-3OH。
我们特别优选R3代表-(CH2)2OCOCH3、-(CH2)2OH或-(CH2)3OH,尤其是R3代表-(CH2)2OCOCH3或-(CH2)2OH,最特别优选-(CH2)2OH。
我们优选R4和R5独立地代表氢、C1-6烷基或芳基,或者NR4R5一起代表吡咯烷基、哌啶基、吗啉基、氮杂环丁烷基、氮杂基、哌嗪基或N-甲基哌嗪基;
我们优选X代表NR7、O、S或SO2,特别优选X代表NR7或SO2,尤其优选X代表NR7。
我们优选a和b两者都代表2,或者a代表1且b代表2。
我们优选R7代表氢。
我们优选p代表0。我们优选q代表2。我们优选h代表2。我们优选i代表0或1,尤其是0。我们优选j代表1。我们优选m和n代表0或1。我们优选o代表1。我们优选u和v代表0。
我们优选R6代表OH或NH2,尤其是NH2。
我们优选c代表0,并且或者d代表2及e代表0,或者d代表1及e代表1。
式(Ⅰ)的表示表明了在四氢呋喃环周围位置的、的确如此的立体化学。如果侧链包含手性中心,则本发明延伸到对映体的混合物(包括外消旋混合物)和非对映异构体,以及单独的对映体。一般地,优选以纯化的单一对映体的形式使用式(Ⅰ)的化合物。
我们也提供用于制备式(Ⅰ)的化合物的方法,所述方法包括:(a)将式(Ⅱ)的对应化合物或其被保护的衍生物与式R2NH2的化合物或其被保护的衍生物反应,在式(Ⅱ)中,L代表一离去基团;(b)通过还原式(Ⅲ)的化合物或其被保护的衍生物,制备式(Ⅰ)中R1代
表氢的式(Ⅰ)的化合物 (c)将被保护的式(Ⅰ)化合物去保护;
(c)将被保护的式(Ⅰ)化合物去保护;
并且当希望或必需时,将式(Ⅰ)化合物或其盐转变为它
的另一种盐。
在过程(a)中,L代表一诸如卤之类的离去基团,所述卤例如氯或氟。一般地,将于存在一溶剂例如DMSO的情况下,通过加热试剂到温度50℃-150℃,进行过程(a)的该反应。对于该反应也优选存在一有机碱,例如一种三取代的有机胺(例如二异丙基乙胺)。在这些条件下,我们特别优选Hal代表氟(尤其是当R1代表氢时),因为该反应具有快速高效进行的趋势。
在过程(b)中,可以通过催化氢化来进行该还原反应,例如在标准条件下于钯/碳(Pd/C)上。
在过程(c)中,可于T W Greene“Protecting Groups in OrganicSynthesis”(J Wiley和Sons,1991)中找到保护基的实例和去除它们的方法。合适羟基的保护基包括通过水解可被去掉的烷基(例如甲基)、醛缩醇(例如丙酮化合物)和酰基(例如乙酰基或苯甲酰基),以及通过催化氢解可被去掉的芳烷基(例如苄基)。合适的胺的保护基包括在适当的情况下可以通过水解或氢解而被去掉的磺酰基(例如甲苯磺酰基)、酰基(例如苄氧羰基或叔丁氧羰基)和芳烷基(例如苄基)。
合适的式(Ⅰ)化合物的盐包括生理上可接受的盐,诸如衍生自有机酸或无机酸的酸加成盐,例如盐酸盐、氢溴化物、1-羟基-2-萘甲酸盐、甲磺酸盐、硫酸盐、磷酸盐、乙酸盐、苯甲酸盐、柠檬酸盐、琥珀酸盐、乳酸盐、酒石酸盐、延胡索酸盐和马来酸盐;如果适当,诸如碱金属盐之类的无机碱盐,例如钠盐。其它的式(Ⅰ)化合物的盐包括也许不是生理上可接受的、但可用于制备式(Ⅰ)化合物和其生理上可接受的盐的盐。这样的盐的实例包括三氟乙酸盐和甲酸盐。
式(Ⅰ)化合物的合适溶剂化物的实例包括水合物。
通过用一适当的酸处理式(Ⅰ)的游离碱,可以获得式(Ⅰ)化合物的酸加成盐。
通过将Ⅳ的化合物或其被保护的衍生物与式R1NH2的化合物反应,可制备式(Ⅱ)的化合物或其被保护的衍生物 L1和L2独立地代表诸如卤之类的离去基团,例如氯或氟。将优选在存在诸如有机胺碱之类的碱(例如二异丙基乙胺)的情况下,用诸如醇(例如异丙醇)之类的溶剂化物于高温(例如回流)进行这反应。
L1和L2独立地代表诸如卤之类的离去基团,例如氯或氟。将优选在存在诸如有机胺碱之类的碱(例如二异丙基乙胺)的情况下,用诸如醇(例如异丙醇)之类的溶剂化物于高温(例如回流)进行这反应。
通过将式(ⅢA)的化合物或其被保护的衍生物与式R2NH2的化合物在常规条件下反应,可制备式(Ⅲ)的化合物或其被保护的衍生物 在式(ⅢA)中,L代表一诸如卤之类的离去基团,例如氯或氟。
在式(ⅢA)中,L代表一诸如卤之类的离去基团,例如氯或氟。
通过将式(Ⅳ)的化合物或其被保护的衍生物与叠氮化物例如叠氮化钠在常规条件下反应,可制备式(ⅢA)的化合物或其被保护的衍生物。
通过将式中L代表一离去基团的式(Ⅴ)的化合物或其被保护的衍生物与2,6,二卤嘌呤例如2,6,二氯嘌呤反应,可制备式(Ⅳ)的化合物或其被保护的衍生物
我们优选使用式中核糖2-羟基和3-羟基被保护的式(Ⅴ)的化合物,例如用乙酰基保护。离去基团L可代表OH,但将优选代表C1-6烷氧基(例如甲氧基或乙氧基)、酯部分(例如乙酰氧基或苯甲酰氧基)或卤。优选的基团L是乙酰氧基。在存在一种路易斯酸(例如TMSOTf)和DUB的情况下,通过在一惰性溶剂例如乙腈中使反应物混合并温暖至比如说70-80℃,可进行该反应。
通过在水中用三氟乙酸处理式(Ⅵ)的化合物,可以从式中alk代表C1-6烷基例如甲基的式(Ⅵ)化合物制备式(Ⅴ)的化合物;继之以再保护,例如,通过在吡啶中与乙酸酐的反应。
可以从相应的1’-醇或1’-酯,例如乙酸酯,制备式(Ⅴ)中L代表卤的式(Ⅴ)化合物。通过用无水HCl或HBr处理,一般将发生反应。
通过用碘化三甲基硅烷处理,可直接制备1’-碘化物;而通过用DAST处理,可以制备1’-氟化物。一种惰性溶剂例如乙醚、二氯甲烷、四氢呋喃或CCl4,一般将是合适的。
按照方案1可制备式(Ⅵ)的化合物:
方案1
对于本领域技术熟练人员,适合于阶段1-6的一般条件该是已知的。也应意识到,在方案1中表明的试剂和条件是条件和可替换试剂的实例,并且对于本领域技术熟练人员,适合于完成同样化学转化的条件可能是已知的。例如,在阶段1中可用一种可替换的醇例如C1-6烷基醇,来产生式(Ⅶ)和(Ⅵ)化合物中的不同的C1-6烷氧基离去基团。也可以修改阶段1;因为用高氯酸(HClO4)和2,2二甲氧基丙烷,或者用乙酰氯(它具有保持高收率和避免使用高氯酸盐的优点),可代替使用HCl。在阶段3中可用可选择的反应条件,这样可利用乙酸乙酯、亚硫酰氯和气态氨(这具有避免氯化溶剂和避免合成讨厌的新戊酸铵杂质的优点)。在所述反应条件下用POCl3、TEA、DMF和乙酸乙酯同样可完成阶段4(这避免使用危险的DMAP)。从上述用于制备式(Ⅴ)化合物方法类推,可制备式(Ⅶ)化合物中需要除OMe之外的离去基团的式(Ⅶ)化合物。在阶段1中可用替代的基团来保护核糖上的2’羟基和3’羟基。我们也发现在甲苯中用叠氮三甲基硅烷和氧化二丁锡可合乎需要地完成阶段5。
在阶段6以后,可用常规的技术纯化不纯的产物,且尤其是在氮气压下运用快速层析条件。我们已发现,符合要求的条件包括将在最小体积的二氯甲烷中的该不纯产物装到一Keiselgel 60(Merck 9385)柱上,并用一具有在环己烷中的乙酸乙酯(10-40%)的梯度溶剂系统洗脱。
通过将式(Ⅴ)的化合物或其被保护的衍生物与式中L代表一诸如卤之类的离去基团例如氯或氟的式(Ⅷ)化合物反应,也可制备式(Ⅱ)的化合物及其被保护的衍生物;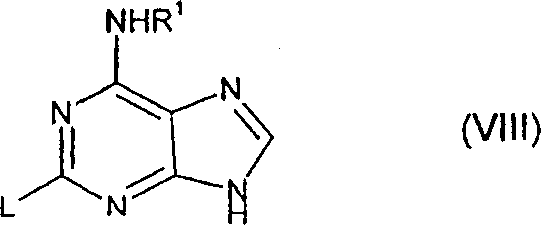 任选地,继之以脱去保护,或继之以脱去保护及再保护反应。
任选地,继之以脱去保护,或继之以脱去保护及再保护反应。

我们优选使用以被保护形式存在的式(Ⅴ)化合物。我们尤其优选以酯基的形式,至少保护核糖2位上的羟基,例如用乙酰基或苯甲酰基保护;因为这在偶联反应中有导致更大的立体选择性的趋势。我们优选用乙酰基保护2位和3位的羟基。先前描述了合适的离去基团L,优选的离去基团为乙酰氧基。
当L代表氟时(且极特别地,当R1代表氢时),尤其优选这方法;因为该反应一般快速而效率高,并且该反应有产生高结晶度产物的趋势。
在常规条件下,如果希望,可以将这反应的产物去保护;例如,通过在温和的碱性条件下(例如在存在碳酸钾的情况下)用一种醇(例如异丙醇)处理。
于一惰性溶剂例如乙腈中,在存在路易斯酸(例如TMSOTf)和任选地存在一种甲硅烷基化剂(例如BSA)的情况下,可以进行式(Ⅴ)化合物(以被保护形式)与式(Ⅷ)化合物的反应;继之以例如用水处理。当存在一种甲硅烷基化剂时,如果L代表卤,一般可省去所述的路易斯酸。
某些式(Ⅷ)的化合物是已知的。在常规条件下,通过式(Ⅸ)化合物与R1NH2的反应,可制备其它的式(Ⅷ)化合物 在式(Ⅸ)中,L1和L2独立地代表诸如卤(例如氯或氟)之类的离去基团。
在式(Ⅸ)中,L1和L2独立地代表诸如卤(例如氯或氟)之类的离去基团。
式R1NH2、R2NH2和Ⅸ的化合物或者是已知的,或者可用本身已知的常规方法制备这样的化合物。
作为本发明的又一方面,我们也提供新的方法,可用该方法无附带条件地提供式(Ⅰ)的化合物。
因而,我们提供用于无附带条件地制备式(Ⅰ)化合物的方法,所述方法包括:
(d)将式(Ⅹ)的对应化合物与式中L为一离去基团的式(Ⅺ)化合物反应
R3-L(Ⅺ);或者(e)将式(Ⅻ)的对应化合物与式(Ⅴ)的化合物或其被保护的衍生物反应
一般将在存在一种温和碱例如K2CO3和一种惰性有机溶剂例如DMF的情况下,通过使所述两种试剂混合而进行过程(d)。典型的离去基团L包括卤(例如溴)。
一般将于一惰性溶剂例如乙腈中,在存在路易斯酸(例如TMSOTf)和任选地存在一种甲硅烷基化剂(例如BSA)的情况下,进行过程(e);继之以例如用水处理。我们优选L代表乙酰氧基,并且优选以乙酰酯形式保护的所述两羟基。然后,一去保护步骤(用温和碱,例如K2CO3)将是必需的,以产生所述式(Ⅰ)的化合物。
用与用于制备式(Ⅰ)化合物的上述方法类似的方法,可制备式(Ⅹ)的化合物。如果由式(Ⅱ)、式(Ⅲ)、式(ⅢA)和/或式中R3被氢取代的式(Ⅳ)化合物的类似物制备式(Ⅹ)的化合物,则最好在四唑的N2位置保护这样的化合物。一种合适的保护基团是在有碱(例如K2CO3)的情况下,通过用苄基卤(例如苄基溴)处理未保护的所述四唑而可以被加入的苄基。在方案2中表示出了用于制备式(Ⅹ)化合物的一种说明性方法。
方案2 式(Ⅺ)化合物是已知的,或者用已知的方法可制备式(Ⅺ)化合物。可以制备式(Ⅻ)的化合物,例如遵循方案3:
式(Ⅺ)化合物是已知的,或者用已知的方法可制备式(Ⅺ)化合物。可以制备式(Ⅻ)的化合物,例如遵循方案3:
方案3
过程(d)和(e)特别适合于制备化合物(2R,3R,4S,5R)-2-[6-氨基-2-(1S-羟甲基-2-苯基-乙氨基)-嘌呤-9-基]-5-(2-乙基-2H-四唑-5-基)-四氢-呋喃-3,4-二醇、以及其盐和溶剂化物,尤其是马来酸盐。
我们优选过程(e)。
例如,根据式(Ⅰ)化合物对用化学吸引剂(例如N-甲酰甲硫氨酰-亮氨酰-苯丙氨酸(fMLP))刺激的中性白细胞产生超氧化物(O2 -)的抑制能力;可以证明式(Ⅰ)化合物抑制白细胞功能的潜力。因此,在发炎部位牵涉白细胞的疾病方面,式(Ⅰ)的化合物在提供保护而避免白细胞诱导的组织损害方面具有潜在的治疗益处。
本发明化合物具有潜在有益的抗炎效应的病情实例包括:呼吸道疾病,例如成人呼吸窘迫综合征(ARDS)、支气管炎(包括慢性支气管炎)、囊纤维化、哮喘(包括变应原诱发的哮喘反应)、慢性阻塞性肺病(COPD)、肺气肿、鼻炎和脓毒症性休克。其它有关的病情包括胃肠道疾病,例如肠的炎症包括炎性肠病(例如局限性回肠炎或溃疡性结肠炎)、幽门螺杆菌诱发的胃炎和由于暴露于辐射或暴露于变应原而继发的肠的炎症;和非类固醇性消炎药诱发的胃病。此外,可用本发明的化合物治疗诸如银屑病、过敏性皮炎和过敏性反应之类的皮肤病和具有炎性成分的中枢神经系统疾病,所述中枢神经系统疾病例如早老性痴呆和多发性硬化。
本发明化合物在病例中具有潜在有益的抗炎效应的其它病情实例包括心脏病,例如外周血管疾病、局部缺血后的再灌注损伤和特发性嗜酸性粒细胞过多综合征。
抑制淋巴细胞功能的本发明化合物可用作免疫抑制剂,因而在诸如类风湿性关节炎和糖尿病之类的自身免疫疾病的治疗方面有用。
在抑制转移方面,本发明的化合物同样可以是有用的。
主要关心的疾病包括哮喘和COPD。
本领域技术熟练人员将意识到本文提及治疗时不仅延伸至已确定病症的预防,而且延伸至其治疗。
如同上面提到的,在人类医学或兽医学方面,式(Ⅰ)的化合物是有用的,尤其是作为消炎药。
因而作为本发明的再一个方面,提供用于人类医学或兽医学、尤其是用于治疗对白细胞诱发的组织损害敏感的炎症患者的式(Ⅰ)化合物或其生理上可接受的盐或其溶剂化物。
按照本发明的另一个方面,提供式(Ⅰ)化合物或者其生理上可接受的盐或溶剂化物在制造用于治疗对白细胞诱发的组织损害敏感的炎症患者的药物方面的应用。
在又一方面或可选择的一个方面,提供治疗对白细胞诱发的组织损害敏感的炎症的人类或动物受治疗者的方法;该方法包括将有效量的式(Ⅰ)化合物或者其生理上可接受的盐或溶剂化物给予所述人类或动物受治疗者。
可以将根据本发明的化合物配制以任何方便的方式给予,因此本发明在本发明范围内也包括供抗炎治疗之用的药用组合物,所述药用组合物包括式(Ⅰ)的化合物或者其生理上可接受的盐或溶剂化物;如果需要,则加之一种或更多种生理上可接受的稀释剂或载体。
也提供用于制备这样的药用制剂的方法,包括混合的所述成分。
例如,可将根据本发明的化合物配制用于口服给药、口腔含化给药、非肠道给药、局部给药或直肠给药,优选非肠道给药或局部给药(例如用气雾剂)。
用于口服给药的片剂和胶囊可包含常规的赋形剂:诸如粘合剂之类,例如糖浆、阿拉伯胶、明胶、山梨糖醇、西黄蓍胶、淀粉胶浆、纤维素或聚乙烯吡咯酮;填充物,例如乳糖、微晶纤维素、糖、玉米淀粉、磷酸钙或山梨糖醇;润滑剂,例如硬脂酸镁、硬脂酸、滑石粉、聚乙二醇或硅石;崩解剂,例如马铃薯淀粉、交联羧甲纤维素钠或淀粉乙醇酸钠;或润湿剂,例如十二烷基硫酸钠。可以按照本领域众所周知的方法将所述片剂包衣。口服液体制剂可以为如下的形式:例如水性悬浮液或油性悬浮液、溶液、乳液、糖浆或酏剂;或者可以呈现为在使用前该干产品用水或其它合适的载体重建的干制品。这样的液体制剂可包含常规的添加剂,诸如悬浮剂,例如山梨糖醇糖浆、甲基纤维素、葡萄糖/糖浆、明胶、羟甲基纤维素、羧甲基纤维素、硬脂酸铝凝胶或氢化食用脂;乳化剂,例如卵磷脂、脱水山梨糖醇单油酸酯或阿拉伯胶);非水性载体(它们可以包括食用油),例如杏仁油、分馏的椰子油、油性的酯、丙二醇或乙醇;或防腐剂,例如对羟基苯甲酸甲酯、或对羟基苯甲酸丙酯、或山梨酸。在适当的时候,所述制剂也可包含缓冲盐、调味剂、着色剂和/或香化剂(例如甘露醇)。
对于口腔含化给药,所述组合物可以采用按常规方法配制的片剂或锭剂的形式。
也可以将所述化合物配制为栓剂,例如,包含诸如可可脂或其它甘油酯之类的常规栓剂基质。
根据本发明的化合物也可以配制用于通过大剂量注射或持续输注的非肠道给药,并且可以单位剂量的形式存在,例如,作为安瓿、管形瓶、小体积输注液或预先填充的注射器,或者,可以存在于添加了防腐剂的多剂量容器中。所述组合物也可以采用像溶液、悬浮液或在水性载体或者非水载体中的乳液这样的形式,并且可以包含配制剂(formulatoryagents),例如抗氧化剂、缓冲剂、抗微生物剂和/或张力调节剂。或者,所述活性成分可以以在使用前用合适的载体(例如无菌且无热原的水)重建的粉剂形式存在。通过将无菌粉剂无菌地装入独立的无菌容器中,或者通过将无菌溶液无菌地装到各个容器中并冻干,可制备干的固体物。
本文所用的所谓局部给药,我们包括通过吹入给药和吸入给药。用于局部给药的各种各样制剂类型的实例包括软膏、乳膏、洗剂、粉剂、阴道栓剂、喷雾剂、气雾剂、胶囊或者供吸入器或吹入器里使用的药筒、用于喷雾的溶液或滴剂(例如眼或鼻的滴剂)。
例如可以用一水性或油性基质,加上合适的增稠剂和/或胶凝剂和/或溶剂,配制软膏和乳膏。因而举例来说,这样的基质可包括水和/或油,或者诸如聚乙二醇之类的溶液;上面所述的油例如液体石蜡或植物油,该植物油例如花生油或蓖麻油。可以使用的增稠剂包括软石蜡、硬脂酸铝、鲸蜡硬脂醇、聚乙二醇、微晶蜡和蜂蜡。
可以用一水性或油性基质配制洗剂;洗剂一般也将包括一种或更多种的乳化剂、稳定剂、分散剂、悬浮剂或增稠剂。
借助于任何合适的粉剂基质,例如滑石粉、乳糖或淀粉,可以形成用于外部敷用的粉剂。用也包括一种或更多种分散剂、稳定剂或悬浮剂的水性或非水基质,可以配制滴剂。
例如,可以将喷雾组合物配制为水性溶液或悬浮液、或者利用一种合适的推进剂配制为由加压包装传递的气雾剂的;所述推进剂,例如为二氯二氟甲烷、三氯一氟甲烷、二氯四氟乙烷、1,1,1,2,3,3,3-七氟丙烷、1,1,1,2-四氟乙烷、二氧化碳或其它合适的气体。
可以用水性或非水性载体,并添加,例如增稠剂、缓冲盐或者调节pH的酸或碱、等渗性调节剂或抗氧化剂的药剂,配制鼻内喷雾剂。
可以配制供吸入器或吹入器里使用的、包含本发明化合物与一种合适粉剂基质(例如乳糖或淀粉)的粉末混合物的、例如明胶的胶囊和药筒、或例如层压铝箔的泡罩片。
可以用水性载体,添加例如酸或碱、缓冲盐、等渗调节剂或抗微生物剂的药剂配制用于通过喷雾吸入的溶液。通过将其过滤除菌或在一高压灭菌器中加热灭菌;或者它们可以作为非无菌制品存在。
根据本发明的药用组合物也可以与其它治疗剂一起联用,所述其它治疗剂例如:消炎药,例如皮质类固醇(例如丙酸氟替卡松、二丙酸倍氯米松、糠酸莫米松、曲安奈德或布地奈德)或者NSAID(例如色甘酸钠))或β-肾上腺素能药(例如沙美特罗、沙丁胺醇、福莫特罗、非诺特罗或特布他林以及其盐)或抗感染剂(例如抗生素、抗病毒药)。
因而,在再一方面,本发明提供包括式(Ⅰ)化合物或其生理上可接受的盐或其溶剂化物和另一种治疗活性剂的组合,所述另一种治疗活性剂例如一种诸如皮质类固醇或NSAID之类的消炎药。
可以方便第提供上述组合以用于所述形式的药用制剂中,因此包括如上定义的组合和其生理上可接受的一种稀释剂或载体的这样药用制剂代表本发明的再一方面。
可以以单独的药用制剂或联合的药用制剂中,或相继地或同时地给予这样的组合中的各个组分。本领域技术熟练人员将容易认识到已知治疗剂的适当剂量。
可以例如以0.01-500毫克/千克体重的量,方便地给予本发明的化合物;优选0.01-100毫克/千克体重,每日一至四次。当然,准确的剂量将取决于患者年龄和病症以及所选择的特定给药途径。
本发明的化合物的优点为:本发明化合物可能更具效力,显示出较大的选择性,具有的副作用较小,且作用的持续时间较长,它们通过优选的途径其生物利用度更大,如果通过吸入给予,则显示较小的系统活性,或者,它们比已知的类似化合物具有更多的、其它的所希望的特性。
尤其是,本发明化合物具有下面的优点,即:与迄今已知的化合物相比,本发明化合物可以对腺苷2a受体亚型显示出超过其它腺苷受体亚型(特别是A1和A3受体亚型)的、更大的选择性。
作为本发明的再一方面,我们提供某些作为新而有用的中间体的化合物。
按照下列筛选,可以在体外和体内测定本发明化合物的生物学活性:
(1)对腺苷2a受体亚型、腺苷1受体亚型和腺苷3受体亚型的激动剂活性。
按照基于Castanon和Spevak,1994之方法的一种方法,用相关的人腺苷受体基因转染的所述中国仓鼠卵巢(CHO)细胞,测定化合物对其它人腺苷受体的激动剂选择性。也用启动被分泌的胎盘碱性磷酸酶(SPAP)的基因的环AMP效应元件(Wood,1995)转染所述CHO细胞。根据通过在SPAP水平的变化而反映出的试验化合物对基础cAMP水平(A2a)或对forskolin提高的cAMP(A1和A3)的影响,确定试验化合物的效力。然后,以对于非选择性激动剂N-乙基甲酰胺腺苷(N-ethyl carboxamide adenosine)(NECA)之数据的比率,确定化合物的EC50值。
(2)在致敏的豚鼠中,抗原诱发的肺嗜酸性粒细胞的累积。
用美吡拉敏(1mg/kg,腹膜内)对卵清蛋白致敏的豚鼠进行给药,以保护豚鼠不患过敏性支气管痉挛。然后,在通过吸入途径供给本发明的化合物(呼吸所述化合物的气雾剂30分钟)后,立即进行卵清蛋白攻击(呼吸产生自50μg/ml卵清蛋白溶液的气雾剂30分钟)。在攻击24小时后,杀死所述豚鼠并灌洗肺。然后,获得对于支气管肺泡灌洗液的、总的和鉴别的白细胞记数,并测定出试验化合物产生嗜酸性粒细胞累积减少50%的剂量(Sanjar等,1992)。
参考文献:
Asako H,Wolf,RE,Granger,DN(1993),Gastroenterology 104,第31-37页;
Burkey TH,Webster,RO,(1993),Biochem.Biophys Acta 1175,第312-318页;
Castanon MJ,Spevak W,(1994),Biochem.Biophys Res.Commun.198,第626-631页;
Cronstein BN,Kramer SB,Weissmann G,Hirschhom R,(1983),Trans.Assoc.Am.Physicians 96,第384-91页;
Cronstein BN,Kramer SB,Rosenstein ED,Weissmann G,Hirschhom R,(1985),Ann N.Y.Acad.Sci.451,第291-301页;
Cronstein BN,Naime D,Ostad E,(1993),J Clin.Invest.92,第2675-82页;
Cronstein BN,Naime D,Ostad E,(1994),Adv.Exp.Med.Biol.,370,第411-6页;
Cronstein BN,(1994),J.Appl.Physiol 76,5-13页;
Dianzani C,Brunelleschi S,Viano I,Fantozzi R,(1994),Eur.J.Pharmacol 263,第223-226页;
Elliot KRF,Leonard EJ,(1989),FEBS Letters 254,第94-98页;
Green PG,Basbaum AI,Helms C,Levme JD,(1991),Proc Natl.AcadSci.88,第4162-4165页;
Hirschom R,(1993),Pediatr.Res 33,第S35-41页;
Kohno Y;Xiao-duo J;Mawhorter SD;Koshiba M;Jacobson KA.(1996).Blood 88第3569-3574页;
Peachell PT,Lichtenstein LM,Schleimer RP,(1989),BiochemPharmacol38,第1717-1725页;
Richter J,(1992),J.Leukocyte Biol.51,第270-275页;
Rosengren S,Bong GW,Firestein GS,(1995),J.Immunol.154,第5444-5451页;
Sanjar S,McCabe PJ,Fattah D,Humbles AA,Pole SM,(1992),Am.Rev.Repair.Dis.145,A40;
Skubitz KM,Wickman NW,Hammerschmidt DE,(1988),Blood 72第29-33页;
Van Schaick EA;Jacobson KA;Kim HO;Ijzerman AP;Danhof M.(1996)Eur J Pharmacol 308,第311-314页;
Wood KV.(1995)Curr Opinion BiotechnologY 6,第50-58页。
用下列的实施例阐明本发明:
实施例一般的实验细节
在用柱层析法纯化产物时,‘快速硅石’指的是用于层析的0.040-0.063mm目的硅胶(例如Merck Art 9385),用最高可达5p.s.i.施加的氮气压加速柱的洗脱。在用薄层层析(TLC)时,薄层层析指的是用5×10cm硅胶60 F254薄板(例如Merck Art 5719)的硅胶薄层层析。
在用制备性HPLC纯化产物时,如果不另外说明,则于一C18反相柱(1”Dynamax)上进行产物的纯化,用在水(包含0.1%三氟乙酸)中的乙腈(包含0.1%三氟乙酸)梯度洗脱,并且以化合物三氟乙酸盐的形式分离化合物。标准自动制备性HPLC柱,条件和洗脱液
用一Supelco ABZ+5μm 100mm×22mm内径(i.d.)的柱进行自动制备性高效液相层析(autoprep.HPLC),用一由ⅰ)溶于水的0.1%甲酸和ⅱ)在乙腈中的0.05%甲酸组成的溶剂混合物,以每分钟4ml的流速洗脱,以所述溶剂混合物中ⅱ)的百分率形式表示该洗脱液。如果不另外说明,则在20分钟期间以5-95%的梯度形式使用所述的洗脱液。LC/MS系统
所用的液相层析质谱(LC/MS)系统:
LC/MS系统A-A SupelcoABZ+,用溶剂洗脱3.3cm×4.6mm内径(i.d.)的柱,所述溶剂为:A-0.1%v/v甲酸+0.077%w/v乙酸铵水溶液和B-95∶5的乙腈∶水+0.05%v/v甲酸。运用下述梯度草案:100%A达0.7分钟;A+B混合物,梯度分布为在3.5分钟期间0-100%B;使保持在100%B达3.5分钟;在0.3分钟期间返回到0%B。使用正和负电喷雾电离。
LC/MS系统B-A Supelco ABZ+,用溶剂洗脱5cm×2.1mm内径(i.d.)的柱,所述溶剂为:A-0.1%v/v甲酸+0.077%w/v乙酸铵水溶液和B-95∶5的乙腈∶水+0.05%v/v甲酸。运用下述梯度草案:在3.5分钟期间0-100%B;使B保持在100%达1.50分钟;在0.50分钟期间返回到0%B。使用正和负电喷雾电离。
LC/MS系统C-A Supelco ABZ+,用溶剂洗脱内径(i.d.)3.3cm×4.6mm的柱,所述溶剂为:A-0.1%v/v甲酸+10mmol乙酸铵水溶液和B-95∶5的乙腈∶水+0.05%v/v甲酸。运用下述梯度草案:100%A达0.7分钟;A+B混合物,梯度分布为在3.7分钟期间0-100%B;使保持在100%B达0.9分钟;在0.2分钟期间返回到0%B。使用正和负电喷雾电离。新方法的实施例中间体A:2-溴次黄嘌呤
通过用溴来氧化巯基并由氢溴(hydrobromine)原位置换,由2-硫代黄嘌呤**制备这化合物。对于有关氧化和置换的参考文献,参见Beaman,A.G.;Gerster,J.F.;Robins,R K,J.Org.Chem,1962,27,986.1.
**Elion,G.B,;Lange,H.L.,Hitchings,G.H.,J.Am.Chem.Soc.,1956,78,217.中间体B:2-[(1S)-1-苄基-2-羟乙基]氨基-1,9-二氢-6H-嘌呤-6-酮
把在一个100ml圆底烧瓶中的10.0克(46.5mmol)中间体A与14.1克(93.0mmol)L-苯丙氨醇(phenylalaninol)于30ml 2-甲氧基乙醇中的混合物加热到回流过夜(>12小时)。冷却该混合物到周围温度,导致固体沉淀。通过加入150ml水,产生另外的沉淀。在搅拌这悬浮液1小时后,将其过滤,并用50ml水洗涤滤饼;在真空下干燥该滤饼,产出7.40克(56%)作为黄色固体的标题化合物。根据1HNMR,该产物为两种互变异构体的混合物。让合并的滤液与洗涤液在周围温度静置两天,过滤固体生成物并将其干燥,产生1.12克(8.4%)作为白色固体的产物。总的收率为64%。TCL(硅胶,在CH2Cl2中的50%甲醇;用254nm紫外光显示):Rf为0.9;2-溴次黄嘌呤的Rf为0.6。MS(ES-):m/z 284(M-1)-,1HNMR(主要的一种互变异构体,300Mhz)δ2.76-2.98(m,2H),3.49(m,2H),4.09(br s,1H),5.04(2,1H),6.36(d,J=8.1Hz,1H),7.19-7.38(m,5H),7.66(s,1H),10.5(s,1H),12.5(s,1H)。中间体C:乙酸(2S)-2-[(9-乙酰基-6-氧代-6,9-二氢-1H-嘌呤-2-基)氢基]-3-苯基丙酯
在周围温度下,向在一个25ml圆底烧瓶中的、悬浮于3.5ml DMF里的500毫克(1.75mmol)中间体B的悬浮液中连续加入0.66ml(7.02mmol)乙酸酐、5毫克(催化量)N,N-二甲基吡啶和0.98(7.02mmol)三乙胺。在周围温度下搅拌这混合物过夜。在用15ml水猝灭这悬浮液并搅拌它2小时后,过滤该悬浮液;并用10ml水洗涤该滤饼;在真空下于70-100℃干燥该滤饼,产出470毫克(73%)作为灰白色粉末的标题化合物。TCL(硅胶,在CH2Cl2中的10%甲醇;用254nm紫外光显示):Rf为0.45。MS(ES-):m/z 368(M-1)-,326(M-1-Ac)-,1H NMR(300Mhz)δ1.96(s,3H),2.80(s,3H),2.90(m,2H),4.16(m,2H),4.37(m,1H),6.70(br s,1H),7.16-7.31(m,5H),8.16(s,1H),10.8(s,1H)。中间体D:乙酸(2S)-2-[(6-氯-9H-嘌呤-2-基)氨基]-3-苯基丙酯
在周围温度下,向在一个100ml圆底烧瓶里的16.7ml(179mmol)三氯氧化磷中加入2.27ml(17.9mmol)N,N-二甲基苯胺。搅拌这混合物10分钟,然后,在15分钟期间,以两个等份加入4.40克(11.9mmol)6-羟基嘌呤中间体C。在回流状态加热这混合物15分钟。在该混合物冷却到周围温度后,在搅拌下,将该混合物慢慢加入到550ml冰水中。通过添加固体NaOAc将水性混合物中和至pH3.5,并用CH2Cl2萃取它(3X)。用含水的NaHCO3洗涤合并的有机层(2X),并经无水Na2SO4干燥该有机层,于真空下浓缩。在硅胶上将棕色油生成物层析,用溶于CH2Cl2中的5-10%甲醇洗脱,产出3.08克(75%)作为棕色固体的标题化合物。TCL(硅胶,在CH2Cl2中的10%甲醇;用254nm紫外光显示):Rf0.50。MS(ES-):m/z 344(M-1)-,346(M-1,同位素)-。1H NMR(300 MHz)d2.15(s,3H),2.98(m,2H),4.08-4.35(m,2H),4.49(m,1H),7.26-7.53(m,5H),7.64(br s,1H),8.25(s,1H),13.1(s,1H)。中间体E:(2S)-2-[(6-氨基-9H-嘌呤-2-基)氨基]-3-苯基-1-丙醇
将288毫克(0.834mmol)中间体D和25ml溶于甲醇中的2MNH3装入帕尔加压反应器里的一200ml的玻璃套管中。密封该反应器,并将其在90-100℃加热16小时。在冷却到周围温度后,在真空下蒸发该溶剂和过量的试剂。尽管通过TLC表明:反应不完全;仍在硅胶上将反应产物油层析。用溶于CH2Cl2中的10-15%甲醇洗脱,产出48毫克(20%)作为固体的标题化合物。进一步洗脱,产出156毫克(62%)作为脱乙酰基形式的、回收的原材料。TCL(硅胶,在CH2Cl2中的10%甲醇;用254nm紫外光显示):Rf0.22。1H NMR(300 MHz)d 2.61-2.80(m,2H),3.30-3.45(m,2H),3.95(m,1H),4.70(s,1H),5.65(d,J=8.0Hz),6.41(s,2H),6.99-7.26(m,5H),7.52(s,1H),12.1(s,1H)。中间体F:(3aS,4S,6R,6aR)-甲氧基-2,2-二甲基-四氢-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4-羧酸
向配备了一个添加漏斗、热电偶探针和氮气入口的1升的三颈圆底烧瓶中加入D-核糖(50克)和丙酮(400ml)。将这混合物冷却至-5℃,然后加入2,2-二甲氧基丙烷(100ml),继之以添加高氯酸(20ml)。让这反应混合物温暖至室温,然后短暂地搅拌一会儿;加入甲醇(70ml),并将这反应混合物搅拌过夜。把该反应溶液冷却至大约5℃,并逐滴加入大约95ml 30%的碳酸钠。让这反应混合物温暖,然后过滤。用乙酸乙酯(50ml)洗涤所产生的滤饼;在真空中于大约200毫巴浓缩这滤液,直到剩下的残留体积为250ml为止;用乙酸乙酯(200ml)稀释,并再次浓缩到170ml的残留体积。加入乙酸乙酯(200ml)和水(200ml),并混合两相,然后离这两相。用乙酸乙酯(200ml)洗涤该水相两次,并将两层分开。浓缩合并的有机萃取液至200ml的残留体积,并用乙酸乙酯(200ml)再次稀释,以提供6R-甲氧基-2,2-二甲基-四氢-(3aR,6aR)-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4R-基)-甲醇的乙酸乙酯溶液。
往一个2升的三颈圆底烧瓶中加入6R-甲氧基-2,2-二甲基-四氢-(3aR,6aR)-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4R-基)-甲醇的乙酸乙酯溶液、6%碳酸氢钠(158ml)、溴化钾(2.3克)和TEMPO(0.167克)。将这反应混合物冷却至-7℃。同时,将碳酸氢钠(6.8克)溶解到10-13%的次氯酸钠(400.5ml)中。在大约40分钟的期间,保持温度在15℃以下,逐滴加入这漂白溶液。将这反应混合物搅拌大约2小时,并添加10%的亚硫酸钠水溶液(47ml)。将这反应混合物搅拌15分钟,分离两相,用4M HCl将该水相调节到pH2,并用乙酸乙酯(225ml)萃取两次。在真空下浓缩该乙酸乙酯萃取液,提供白色残余物,将其用环己烷(90ml)研磨。过滤所述固体,并在真空下于45℃将其干燥,以提供作为白色固体的、熔点为126-129℃的标题产物(33.6克)(关于D-核糖的收率为46%)。中间体G:(3aS,4S,6R,6aR)-6-甲氧基-2,2-二甲基-四氢-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4-羧酸酰胺
向500ml的三颈圆底烧瓶中加入中间体F(20克)和乙酸乙酯(160ml),继之以添加亚硫酰氯(9.4ml)。在50℃将这反应溶液温暖2小时。以这样一速率加入气态氨(16克),使温度保持在40-60℃之间。加入水(120ml)。分离两层,并用乙酸乙酯(80ml)洗涤该水层两次。在真空下把合并的所述有机洗涤液浓缩到干。用环己烷(40ml)研磨该残余物,并过滤所述固体。用环己烷(40ml)洗涤该滤饼,并在真空下于45℃干燥所述固体,以提供作为淡褐色固体的、熔点为134-136℃的标题产物(16.7克)(收率83.9%)。TLC(95/5氯仿/甲醇/每50ml~5滴TFA/用磷钼酸喷雾)rf=0.49。中间体H:(3aS,4S,6R,6aR)-6-甲氧基-2,2-二甲基-四氢-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4-甲腈
向22升的三颈圆底烧瓶中加入中间体G(643克)、乙酸乙酯(7.72升)、N,N-二甲基甲酰胺(1.26升)和三乙胺(2.15升)。将这反应溶液冷却到大约0℃,然后,以这样一速率加入三氯氧化磷(1.38升),以致保持温度在25℃之下。搅拌该反应一个半小时。逐滴加入碳酸氢钾水溶液(20%,6.5升),且保持温度在20℃或低于20℃。分离两层,并用乙酸乙酯(3.5升)反萃取该水层。用20%碳酸氢钾(3.5升)洗涤合并的所述有机层两次,并浓缩它到大约1升的残留体积。将活性炭(15克)加入到稀的油中,并通过celite(80克)过滤。用乙酸乙酯(100ml)洗涤该滤饼。在真空中浓缩所述滤液,以提供作为红橙色油的标题产物(519克)(收率88%)。TLC(1∶1乙酸乙酯/环己烷,用磷钼酸试剂展开)rf=0.73。中间体I:5-(6R-甲氧基-2,2-二甲基-四氢-(3aR,6aR)-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4R-基)-1H-四唑
往一个3升的三颈圆底烧瓶中加入中间体H(200克)、甲苯(2升)、叠氮三甲基硅烷(332ml)和氧化二丁锡(24.9克)。将该反应混合物加热到60℃达15小时。在真空中浓缩该反应混合物到大约300ml的残留体积。加入甲苯(1升),并把这溶液再次浓缩至大约470ml的残留体积。加入甲苯(400ml)和水(19.8ml),并在室温下搅拌这混合物大约2小时。浓缩该混合物以提供大约250ml的残留物。随着加温,把该残余物溶解于甲苯(800ml)中,然后,让它冷却到室温,并搅拌它超过3天。过滤出固体,并用甲苯(250ml)洗涤该固体两次。在真空下干燥这产物,以提供作为白色固体的的标题产物(135克)(收率55%),熔点130℃。中间体J:2-乙基-5-(6R-甲氧基-2,2-二甲基-四氢-(3aR,6aR)-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4R-基)-2H-四唑
往一个1升的三颈圆底烧瓶中加入中间体I(31.8克)、碳酸钾(12.7克)和丙酮(238ml)。通过注射器将乙基碘(14.1ml)加入,并在42℃将该反应混合物温暖2.5至3小时。让该反应混合物冷至室温,然后加入环己烷(238ml)。过滤所产生的沉淀,并用环己烷(65ml)洗涤该滤饼三次。浓缩该滤液到195ml的残留体积,然后用环己烷(238ml)再次稀释。在0-5℃将这环己烷溶液冷却3天,过滤所产生的结晶固体(N1烷基化产物),并用环己烷(65ml)洗涤三次。在真空中浓缩合并的滤液,以提供作为一种油的中间体阶段的标题产物。在60℃将该油溶解于环己烷(200ml)中,并让这溶液冷却到室温,过滤该溶液。过滤所产生的结晶固体,并用环己烷(65ml)洗涤三次。浓缩合并的滤液,以提供作为黄色油的的标题产物;TLC(1∶1乙酸乙酯/环己烷,用磷钼酸试剂显现)rf=0.68。中间体K:rel-乙酸4R,5-二乙酰氧基-2R-(2-乙基-2H-四唑-5-基)-四氢-呋喃-3R-基酯
往一个圆底烧瓶中加入中间体J(5.0克),将溶于甲醇(50ml)中的乙酰氯(0.73克)的溶液加到该烧瓶里,并在300毫巴压力下将这反应溶液加热至回流。在8至9小时的期间,蒸馏法反应物;且在这段时间内,分批添加甲醇(135ml)以补充所述反应的体积。让该反应混合物冷却至室温,并加入吡啶(15ml)。在真空中浓缩该混合物,并用吡啶再次稀释。将乙酸乙酯(25ml)和乙酸酐(6.6克)加到该吡啶溶液中,并在室温下将所产生的混合物搅拌过夜。将该反应混合物冷却到5-10℃,并于20分钟期间,在保持温度低于10℃的同时,逐滴加入大约2M的硫酸(约45ml)。将两层分离,用大约0.7M的硫酸(约25ml)洗涤该有机层。用饱和碳酸氢钠与盐水洗涤该有机层,然后,在真空中浓缩,以提供一种浅黄色油,将其溶解于50ml乙酸乙酯中。加入乙酸酐(3.04克)和浓硫酸(0.65克),并将该反应混合物温暖到50℃达大约3.5小时。用饱和的碳酸氢钠溶液(25ml)猝灭该反应;在真空中浓缩中有机层,以提供作为黄色油的的标题产物(5.1克)(收率82%);TLC(1∶1乙酸乙酯/环己烷,用磷钼酸试剂显示)rf=0.44。中间体L:乙酸(2R,3S,4S,5R)-4-(乙酰氧基)-2-(6-氨基-2-[(1S)-1-苄基-2-羟乙基]氨基-9H-嘌呤-9-基)-5-(2-乙基-2H-1,2,3,4-四唑-5-基)四氢-3-呋喃酯
在周围温度下,向在10ml圆底烧瓶里的、于2.5ml乙腈中的65毫克(0.19mmol)中间体K和45毫克(0.16mmol)中间体E之混合物中相继加入88ml(0.36mmol)N,O-双(三甲代甲硅烷基)乙酰胺和34ml(0.19mmol)三氟甲磺酸三甲代甲硅烷基酯。将这黄色悬浮液加热到回流,且该悬浮液变成为暗黄色溶液。在于回流状态加热该混合物5小时后,冷却该混合物至周围温度,用2ml 10%的KHCO3进行猝灭,并用CH2Cl2(2×8ml)萃取。用10%的盐水(3ml)洗涤合并的所述有机层,并在真空下蒸发。在硅胶上层析黄色泡沫状生成物,用溶于CH2Cl2中的5%甲醇洗脱,产出70毫克(78%)作为固体的标题化合物。尽管纯度低,但对于下一步骤仍用该材料,而不进行进一步的纯化。TLC(硅胶,溶于CH2Cl2中的10%甲醇,用254nm紫外光显示):Rf0.54。实施例A:(2R,3R,4S,5R)-2-[6-氨基-2-(1S-羟甲基-2-苯基-乙氨基)-嘌呤-9-基]-5-(2-乙基-2H-四唑-5-基)-四氢-呋喃-3,4-二醇
在周围温度下,将70毫克(0.12 mmo1)中间体L与20毫克(0.15 mmol)无水K2CO3在5ml甲醇中的混合物搅拌2.5小时。将这混合物蒸发至几乎干燥,用2ml水稀释,并用EtOAc(3×5ml)萃取。用Na2SO4干燥合并的所述有机层,并在真空下蒸发。在硅胶上层析粗制的反应产物,用溶于CH2Cl2中的10%甲醇洗脱,产出24.5毫克(41%)作为固体的标题化合物。TLC(硅胶,溶于CH2Cl2中的10%甲醇,用254nm紫外光显示):Rf0.35。新的实施例中间体中间体1:2-苄基-5-(6-甲氧基-2,2-二甲基-四氢-(3aR,6aR)-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4R-基)-2H-四唑
在氮气下,往搅拌的、溶于二甲基甲酰胺(50ml)的中间体10(10g,41.3mM)溶液中加入碳酸钾(5.7g,41.3mM),继之以加入苄基溴(6ml,49.6mM)。在室温搅拌这混合物18小时。添加水(100ml),并用乙酸乙酯(2×100ml)萃取该混合物。合并这有机相,用水、盐水洗涤,并用硫酸镁干燥。通过用20%乙酸乙酯/环己烷洗脱的快速硅胶柱层析,纯化在减压下蒸发之后所获得的残余物;产生作为蜡状固体的该标题化合物(2.98克)。
TLC SiO2(在环己烷中的20%的乙酸乙酯)Rf=0.45中间体2:乙酸4R,5-二乙酰氧基-2R-(2-苄基-2H-四唑-5-基)-四氢-呋喃-3R-基酯
在室温,将TFA/水(40ml/4ml)的混合物加到中间体1(2.98g,8.9mM)中;并搅拌1小时。在减压下蒸发这混合物,并与甲苯一起共沸(3×20ml)。将这残余物溶解到二氯甲烷中(100ml),并加入二甲氨基吡啶(催化量)和三乙胺(40ml,356mM)。将这混合物冷却至0℃,并在15分钟期间逐滴加入乙酸酐(17ml,166mM)。让这混合物温暖至室温,并搅拌16小时。在减压下蒸发该混合物,并通过用50%乙酸乙酯/环己烷洗脱的快速硅胶柱层析,纯化该混合物;产生作为一种油的该标题化合物(2.44克)。
LC/MS系统A Rt=3.39分钟,m/z=279(MH+)。中间体3:乙酸4R-乙酰氧基-5R-(2-苄基-2H-四唑-5-基)-2R-(2,6-二氯-嘌呤-9-基)-四氢-呋喃-3R-基酯
在氮气下,向溶于乙腈(18ml)里的中间体2(2.43g,6mM)的搅拌的溶液中,加入1,8-重氮二环[5,4,0]十一碳-7烯(1.35ml,9mM),继之以加入2,6-二氯嘌呤(1.5克)。将这混合物冷却至0℃,并在15分钟期间逐滴加入三氟甲磺酸三甲代甲硅酯(1.87ml,10.2mM);让其温暖至20℃并搅拌38小时。用饱和的碳酸氢钠水溶液(35ml)猝灭这反应混合物,并用乙酸乙酯萃取(3×50ml)。合并所述的有机物,并用水(50ml)洗涤,用硫酸镁干燥,以及在减压下蒸发。通过用30%乙酸乙酯/环己烷洗脱的快速硅胶柱层析,纯化所获得的该残余物;产生作为一种油的该标题化合物(2.36克)。LC/MS系统B Rt=3.43分钟,m/z=535(MH+)。中间体4:乙酸4R-乙酰氧基-5R-(2-苄基-2H-四唑-5-基)-2R-[2-氯-6-(2,2-二苯基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3R-基酯
在氮气下,向溶于异丙醇(40ml)的中间体3(2.3g,4.3mM)的搅拌的溶液中,加入二异丙基乙胺(1.12ml,6.5mM),继之以加入二苯基乙胺(1.02克,5.2mM);将所产生的混合物加热至50℃18小时。在减压下蒸发该反应混合物,并通过用50%乙酸乙酯/环己烷洗脱的快速硅胶柱层析,纯化所获得该残余物;产生作为一种灰白色固体的该标题化合物(2.9克)。LC/MS系统B Rt=3.68分钟,m/z=694(MH+)。中间体5:(2R,3S,4R,5R)-2-(2-苄基-2H-四唑-5-基)-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇
在氮气下,将溶于二甲亚砜(1ml)中的中间体4(2.9g,4.2mM)和2-哌啶子基乙胺(3ml,20.9mM)的溶液加热至90℃72小时。让这混合物冷却,并通过用20%甲醇、79%氯仿和1%氨洗脱的快速硅胶柱层析,纯化该混合物;产生作为一种油的该标题化合物(1.6克)。LC/MS系统ARt=3.86分钟,m/z=702(MH+)。中间体6:(2R,3S,4R,5R)-2-(2H-四唑-5-基)-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇
在氮气下,向在10%披钯碳(1.6克)中添加溶于乙醇(50ml)的中间体5(1.67g,2.38mM)的溶液,继之以加入甲酸铵(0.72克,11.9mM)。加热这混合物至50℃4小时;通过一块Harborlite垫过滤。在减压下蒸发这滤液,产生作为一种浅黄色固体的该标题化合物(1.45克)。LC/MS系统A Rt=3.66分钟,m/z=612(MH+)。中间体7:(3aS,4S,6R,6aR)-甲氧基-2,2-二甲基-四氢-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4-羧酸
向配备了一添加漏斗、热电偶探针和氮气入口的1升的三颈圆底烧瓶中加入D-核糖(50克)和丙酮(400ml)。将这混合物冷却至-5℃,然后加入2,2-二甲氧基丙烷(100ml),继之以添加高氯酸(20ml)。让这反应混合物温暖至室温,然后短暂地搅拌一会儿,加入甲醇(70ml),并将这反应混合物搅拌过夜。把该反应溶液冷却至大约5℃,并逐滴加入大约95ml30%的碳酸钠。让这反应混合物温暖,然后过滤;用乙酸乙酯(50ml)洗涤所产生的滤饼,在真空中于大约200毫巴浓缩这滤液,直到剩下的残留体积为250ml为止;用乙酸乙酯(200ml)稀释,并再次浓缩到170ml的残留体积。加入乙酸乙酯(200ml)和水(200ml),并混合两相,分离这两相。用乙酸乙酯(200ml)洗涤该水相两次,并将两层分开。浓缩合并的有机萃取液至200ml的残留体积,并用乙酸乙酯(200ml)再次稀释,以提供6R-甲氧基-2,2-二甲基-四氢-(3aR,6aR)-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4R-基)-甲醇的乙酸乙酯溶液。
往一个2升的三颈圆底烧瓶中加入6R-甲氧基-2,2-二甲基-四氢-(3aR,6aR)-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4R-基)-甲醇的乙酸乙酯溶液、6%碳酸氢钠(158ml)、溴化钾(2.3克)和TEMPO(0.167克)。将这反应混合物冷却至-7℃。同时,将碳酸氢钠(6.8克)溶解到10-13%的次氯酸钠(400.5ml)中。在大约40分钟的期间,保持温度在15℃以下,逐滴加入这漂白溶液。将这反应混合物搅拌大约2小时,并添加10%的亚硫酸钠水溶液(47ml)。将这反应混合物搅拌15分钟,分离两相,用4M HCl将该水相调节到pH2,并用乙酸乙酯(225ml)萃取两次。在真空下浓缩该乙酸乙酯萃取液,以提供白色残余物,将其用环己烷(90ml)研磨。过滤出所述固体,并在真空下于45℃将其干燥,以提供作为白色固体的、熔点为126-129℃的标题产物(33.6克)(46%为关于D-核糖的收率)。中间体8:(3aS,4S,6R,6aR)-6-甲氧基-2,2-二甲基-四氢-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4-羧酸酰胺
向500ml的三颈圆底烧瓶中加入中间体1(20克)和乙酸乙酯(160ml),继之以添加亚硫酰氯(9.4ml)。在50℃将这反应溶液温暖2小时。以这样一速率加入气态氨(16克),使温度保持在40-60℃之间。加入水(120ml)。分离两层,并用乙酸乙酯(80ml)洗涤该水层两次。在真空下把合并的所述有机洗涤液浓缩到干。用环己烷(40ml)研磨该残余物,并过滤所述固体。用环己烷(40ml)洗涤该滤饼,并在真空下于45℃干燥所述固体,以提供作为淡褐色固体的、熔点为134-136℃的标题产物(16.7克)(收率83.9%)。TLC(95/5氯仿/甲醇/每50ml~5滴TFA/磷钼酸喷雾)rf=0.49。中间体9:(3aS,4S,6R,6aR)-6-甲氧基-2,2-二甲基-四氢-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4-甲腈
向22升的三颈圆底烧瓶中加入中间体2(643克)、乙酸乙酯(7.72升)、N,N-二甲基甲酰胺(1.26升)和三乙胺(2.15升)。将这反应溶液冷却到大约0℃,然后,以这样一速率加入三氯氧化磷(1.38升),以致保持温度在25℃之下。搅拌该反应一个半小时。逐滴加入碳酸氢钾水溶液(20%,6.5升),且保持温度在20℃或低于20℃。分离两层,并用乙酸乙酯(3.5升)反萃取该水层。用20%碳酸氢钾(3.5升)洗涤合并的所述有机层两次,并浓缩它到大约1升的残留体积。将活性炭(15克)加入到稀的油中,并通过celite(80克)过滤混合物。用乙酸乙酯(100ml)洗涤该滤饼。在真空中浓缩所述滤液,以提供作为红橙色油的标题产物(519克)(收率88%)。TLC(1∶1乙酸乙酯/环己烷,用磷钼酸试剂展开)rf=0.73。中间体10:5-(6R-甲氧基-2,2-二甲基-四氢-(3aR,6aR)-呋喃并[3,4-d][1,3]间二氧杂环戊烯-4R-基)-1H-四唑
往一个3升的三颈圆底烧瓶中加入中间体3(200克)、甲苯(2升)、叠氮三甲基硅烷(332ml)和氧化二丁锡(24.9克)。将该反应混合物加热到60℃达15小时。在真空中浓缩该反应混合物到大约300ml的残留体积。加入甲苯(1升),并把这溶液再次浓缩至大约470ml的残留体积。加入甲苯(400ml)和水(19.8ml),并在室温下搅拌这混合物大约2小时。浓缩该混合物以提供大约250ml的残留物。在加温下,把该残余物溶解于甲苯(800ml)中,然后,让它冷却到室温,并搅拌它超过3天。过滤出固体,并用甲苯(250ml)洗涤该固体两次。在真空下干燥这产物,以提供作为白色固体的的标题产物(135克)(收率55%),熔点130℃。
实施例实施例1:2R,3R,4S,5R)-2-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-5-[2-(3-羟基-丙基)-2H-四唑-5-基]-四氢-呋喃-3,4-二醇双(三氟乙酸盐)
向溶于二甲基甲酰胺(1ml)的中间体6(0.06g,0.098mM)的溶液中添加碳酸钾(0.023g,0.167mM),继之以加入在一个密封了的管形瓶(例如Reacti-vialTM)中的3-溴丙醇(0.013ml,0.147mM),搅拌这反应混合物达18小时。过滤该混合物,并在减压下将滤液蒸发,且用制备性HPLC纯化;(用一Capital柱ODS2-IK5,内径(i.d.)15mm×20mm,以包含0.1%三氟乙酸的、5%至95%乙腈之30分钟梯度的方式),在冻干后产生作为白色固体的该标题化合物(0.022克)。LC/MS系统A Rt=3.59分钟,m/z=670(MH+)。实施例2:2R,3R,4S,5R)-2-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-5-(2-丙基-2H-四唑-5-基)-四氢-呋喃-3,4-二醇双(三氟乙酸盐)
用1-溴丙醇(0.013ml,0.147mM)以一种类似于实施例1的方法制备实施例2。在冻干后,产生作为白色固体的该标题化合物(0.026克)。LC/MS系统A Rt=3.76分钟,m/z=654(MH+)。实施例3:乙酸2-(5-{5R-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-3S,4R-二羟基-四氢-呋喃-2R-基}-四唑-2-基)-乙酯双(三氟乙酸盐)
用乙酸2-溴乙酯(0.017ml,0.147mM)以一种类似于实施例1的方法制备实施例3。在冻干后,产生作为白色固体的该标题化合物(0.029克)。LC/MS系统A Rt=3.68分钟,m/z=698(MH+)。实施例4:(2R,3S,4R,5R)-2-(2-环丙基甲基-2H-四唑-5-基)-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇双(三氟乙酸盐)
用(溴甲基)环丙烷(0.0165ml,0.147mM)以一种类似于实施例1的方法制备实施例4。在冻干后,产生作为白色固体的该标题化合物(0.023克)。LC/MS系统A Rt=3.74分钟,m/z=666(MH+)。实施例5:(2R,3R,4S,5R)-2-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-5-[2-(2-羟基-乙基)-2H-四唑-5-基]-四氢-呋喃-3,4-二醇双(三氟乙酸盐)
在氮气下,向在甲醇(1ml)中的实施例3(0.01克)添加甲醇钠溶液(0.005ml),在室温搅拌这混合物18小时。在减压下将该反应混合物蒸发,并用制备性HPLC纯化该反应混合物;(用一Capital柱ODS2-IK5,内径(i.d.)15mm×20mm,以5%至95%乙腈的30分钟梯度的方式)产生作为一种胶的该标题化合物(0.006克)。LC/MS系统C Rt=2.54分钟,m/z=656(MH+)。实施例6:(2R,3S,4R,5R)-2-[2-(2-氯-乙基)-2H-四唑-5-基]-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇双(三氟乙酸酯)
用1-溴-2-氯乙烷(0.012ml,0.147mM)以一种类似于实施例1的方法制备实施例6,在冻干后,产生作为白色固体的该标题化合物(0.004克)。LC/MS系统A=3.79分钟,m/z=674(MH+)。实施例7:(2R,3S,4R,5R)-2-(2-环丁基-2H-四唑-5-基)-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇双(三氟乙酸酯)
用溴环丁烷(0.014ml,0.147mM)以一种类似于实施例1的方法制备实施例7,在冻干后,产生作为白色固体的该标题化合物(0.008克)。LC/MS系统C=2.76分钟,m/z=666(MH+)。实施例8:(2R,3S,4R,5R)-2-(2-烯丙基-2H-四唑-5-基)-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇双(三氟乙酸酯)
在0℃用烯丙基溴(0.014ml,0.147mM)以一种类似于实施例1的方法制备实施例8,然后在0℃搅拌这混合物3小时,产生作为透明胶的该标题化合物(0.004克)。LC/MS系统C Rt=2.68分钟,m/z=652(MH+)。生物学数据
(A)对受体亚型的激动剂活性
在筛选(1)中,试验了各实施例的所述化合物(对受体亚型的激动剂活性),并且所得到的结果如下:
| 实施例号 | A2a | A1 | A3 |
| 1 | 22.64 | 434.8 | >93 |
| 2 | 30.95 | 755.4 | >93 |
| 3 | 16.59 | 310.5 | >93 |
| 4 | 37.24 | 1318.09 | >93 |
| 5 | 10.54 | 159.5 | >94 |
| 6 | 24.05 | 411.9 | >97 |
| 7 | 22.98 | 597.82 | >95 |
| 8 | 26.38 | >6131 | >165 |
数据为最小值,因为在试验后发现制剂包含大约20%的无活性杂质。
在所述表中表示出的值是作为与NECA的数据之比率的EC50值。
缩写TMS 三甲代甲硅烷基TFA 三氟乙酸DMF N,N-二甲基甲酰胺NECA N-乙基甲酰胺腺苷DAMP 4-二甲氨基吡啶TEMPO 2,2,6,6-四甲基-1-哌啶氧基,自由基TMSOTf 三氟甲磺酸三甲基甲硅烷基酯DUB 1,8-重氮二环[5,4,0]十一碳-7烯BSA 双三甲基甲硅烷基乙酰胺DCM 二氯甲烷DAST 二乙氨基三氟化硫Ph 苯基CDI 羰二咪唑NSAID 非类固醇性消炎药Bn 苄基
Claims (27)
1.式(Ⅰ)的化合物,以及其盐和溶剂化物;在式(Ⅰ)中,R1和R2独立地代表选自下面的基团:(ⅰ)C3-8环烷基-;(ⅱ)氢;(ⅲ)(芳基)2CHCH2-;(ⅳ)C3-8环烷基C1-6烷基-;(ⅴ)C1-8烷基-;(ⅵ)芳基C1-6烷基-;(ⅶ)R4R5N-C1-6烷基-;(ⅷ)C1-6烷基-CH(CH2OH)-;(ⅸ)芳基C1-5烷基-CH(CH2OH)-;(ⅹ)芳基C1-5烷基-C(CH2OH)2-;(ⅹⅰ)独立地用一个或更多个(例如1、2或3个)-(CH2)pR6基团取代的
C3-8环烷基;(ⅹⅱ)H2NC(=NH)NHC1-6烷基-;(ⅹⅲ)下式的基团,或基团中一个毗连X的亚甲基碳原子被甲基取代的这样的基团;或者,如果存在两个毗连X的亚甲基碳原子,则为这两个毗连X的亚甲基碳原子都被甲基取代的这样的基团;(ⅹⅳ)-C1-6烷基-OH;(ⅹⅴ)-C1-8卤烷基;(ⅹⅵ)下式的基团(ⅹⅶ)芳基;以及(ⅹⅷ)-(CH2)fSO2NHg(C1-4烷基-)2-g或-(CH2)fSO2NHg(芳基C1-4烷基-)2-g;R3代表甲基、乙基、-CH=CH2、正丙基、-CH2CH=CH2、-CH=CHCH3、异丙基、异丙烯基、环丙基、环丙烯基、环丙基甲基、环丙烯基甲基、环丁基、环丁烯基、-(CH2)q卤、-(CH2)hY(CH2)iH、-(CH2)hCOOCH3、-(CH2)hOCOCH3.-(CH2)hCON(CH2)mH((CH2)nH)、-(CH2)hCO(CH2)oH或-CH2C((CH2)uH)=NO(CH2)vH;Y代表O、S或N(CH2)j;如果a+b是在3至5的范围内,则a和b独立地代表0至4的整数;如果c+d+e是在2至3的范围内,则c、d和e独立地代表0至3的整数;f代表2或3,且g代表0至2的整数;p代表0或1;q代表2或3;h代表2或3;i代表0至2的整数,以致h+i在2至4的范围内;j代表0至2的整数,以致h+i+j在2至4的范围内;m和n独立地代表0至2的整数,以致m+n在0至2的范围内;o代表0至2的整数,以致h+o在2至3的范围内;u和v独立地代表0或1,以致u+v在0至1的范围内;R4和R5独立地代表氢、C1-6烷基、芳基、芳基C1-6烷基-或NR4R5一起可代表吡咯烷基、哌啶基、吗啉基、氮杂环丁烷基、氮杂基、哌嗪基或N-C1-6烷基哌嗪基。R6代表OH、NH2、NHCOCH3或卤;R7代表氢、C1-6烷基、C1-6烷基芳基或-COC1-6烷基;X代表NR7、O、S、SO或SO2;前提是:如果R3代表甲基、乙基或异丙基,那么R1和/或R2必须独立地代表:(a)-(CH2)fSO2NHg(C1-4烷基-)2-g或-(CH2)fSO2NHg(芳基C1-4烷基-)2-g,这里f为2或3,并且g是0至2的整数;(b)用一个或更多个的-(CH2)pNHCOCH3基团独立地取代的C3-8环烷
基;(c)下式的基团
或基团中一个毗连X的亚甲基碳原子被甲基取代的这样的基团;或者,如果存在两个毗连X的亚甲基碳原子,则为这两个毗连X的亚甲基碳原子都被甲基取代的这样的基团;
(d)下式的基团
2.按照权利要求1的式(Ⅰ)化合物,其中,R1和R2两者并不都代表氢。
3.按照权利要求1或2的化合物,其中,R1代表(芳基)2CHCH2-、C1-8烷基、氢或芳基C1-6烷基-。
4.按照权利要求1至3中的任何一项的化合物,其中,R1代表(苯基)2CHCH2-。
5.按照权利要求1至4中的任何一项的化合物,其中,R2代表R4R5N-C1-6烷基-、芳基C1-6烷基-、芳基C1-5烷基CH(CH2OH)-、芳基C1-6烷基或C1-6烷基-CH(CH2OH)-。
6.按照权利要求1至5中的任何一项的化合物,其中,R2代表-(CH2)2(哌啶-1-基)。
7.按照权利要求1至6中的任何一项的化合物,其中,R3代表C1-3烷基、环丁基、环丙基甲基、-(CH2)2OCOCH3、-(CH2)2-3OH或-(CH2)2卤。
8.按照权利要求1至7中的任何一项的化合物,其中,R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
9.按照权利要求1至8中的任何一项的化合物,其中,R4和R5独立地代表氢、C1-6烷基或芳基,或者NR4R5一起可代表吡咯烷基、哌啶基、吗啉基、氮杂环丁烷基、氮杂基、哌嗪基或N-甲基哌嗪基
10.按照权利要求1至9中的任何一项的化合物,其中,R6代表OH或NH2。
11.按照权利要求1至10中的任何一项的化合物,其中,X代表NR7、O、S、或SO2。
12.式(Ⅰ)的化合物,所述式(Ⅰ)的化合物为:
2R,3R,4S,5R)-2-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-5-[2-(3-羟基-丙基)-2H-四唑-5-基]-四氢-呋喃-3,4-二醇;
2R,3R,4S,5R)-2-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-5-(2-丙基-2H-四唑-5-基)-四氢-呋喃-3,4-二醇;
乙酸2-(5-{5R-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-3S,4R-二羟基-四氢-呋喃-2R-基}-四唑-2-基)-乙酯;
(2R,3S,4R,5R)-2-(2-环丙基甲基-2H-四唑-5-基)-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇;
(2R,3R,4S,5R)-2-[6-(2,2-二苯基-乙氨基)]-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-5-[2-(2-羟基-乙基)-2H-四唑-5-基]-四氢-呋喃-3,4-二醇;
(2R,3S,4R,5R)-2-[2-(2-氯-乙基)-2H-四唑-5-基]-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇;
(2R,3S,4R,5R)-2-(2-环丁基-2H-四唑-5-基)-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇;
(2R,3S,4R,5R)-2-(2-烯丙基-2H-四唑-5-基)-5-[6-(2,2-二苯基-乙氨基)-2-(2-哌啶-1-基-乙氨基)-嘌呤-9-基]-四氢-呋喃-3,4-二醇;或者其任何一种的盐或溶剂化物。
13.药用组合物,所述药用组合物包括与一种或更多种药学上可接受的稀释剂或载体相混合的、按照权利要求1至12中的任何一项定义的式(Ⅰ)化合物或者其药学上可接受的盐或溶剂化物。
14.用作药物的、按照权利要求1至12中的任何一项定义的式(Ⅰ)化合物或者其药学上可接受的盐或溶剂化物。
15.按照权利要求1至12中的任何一项定义的式(Ⅰ)化合物或者其药学上可接受的盐或溶剂化物在适合于治疗炎症的药物制造方面的应用。
16.治疗或预防炎症例如哮喘的方法,所述方法包括给予患者有效量的、按照权利要求1至12中的任何一项定义的式(Ⅰ)化合物或者其药学上可接受的盐或溶剂化物。
17.制备按照权利要求1至12中的任何一项定义的式(Ⅰ)化合物的方法,所述方法包括:
(a)将式中L代表一离去基团的式(Ⅱ)的对应化合物或其被保护的衍生物与式R2NH2的化合物或其被保护的衍生物反应, 其中,R1、R2和R3是如同按照权利要求1至12中的任何一项所定义的R1、R2和R3;或
其中,R1、R2和R3是如同按照权利要求1至12中的任何一项所定义的R1、R2和R3;或
(b)通过还原式(Ⅲ)的化合物或其被保护的衍生物,制备式(Ⅰ)中R1代表氢的式(Ⅰ)的化合物
其中,R2和R3是如同权利要求1至12中的任何一项所定义的R2和R3;或
(c)将被保护的式(Ⅰ)化合物脱去保护;
并且当希望或必需时,将式(Ⅰ)化合物或其盐转变为它
的另一种盐。
18.无附带条件地制备按照权利要求1至12中的任何一项所定义式(Ⅰ)化合物的方法,所述方法包括:
(a)将式(Ⅹ)的对应化合物 与式(Ⅺ)化合物反应
与式(Ⅺ)化合物反应
R3-L(Ⅺ)其中,L为一离去基团且R1、R2和R3是如同权利要求1至12中的任何一项所定义的R1、R2和R3;或者
(b)将式(Ⅻ)的对应化合物与式(Ⅴ)的化合物或其被保护的衍生物反应, 其中,R1和R2是如同权利要求1至12中的任何一项所定义的R1和R2。
其中,R1和R2是如同权利要求1至12中的任何一项所定义的R1和R2。
19.按照权利要求18的方法,所述方法是用于制备化合物(2R,3R,4S,5R)-2-[6-氨基-2-(1S-羟甲基-2-苯基-乙氨基)-嘌呤9-基]-5-(2-乙基-2H-四唑-5-基)-四氢-呋喃-3,4-二醇和其盐及溶剂化物的方法。
20.按照权利要求19的方法,所述方法包括将下式的对应化合物 与下式的化合物或其被保护的衍生物反应,其中L是一离去基团
与下式的化合物或其被保护的衍生物反应,其中L是一离去基团
21.式(Ⅱ)的化合物或其被保护的衍生物,
其中,L代表一离去基团,且R1是如同权利要求1至12中的任何一项所定义的R1;R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
22.式(Ⅲ)的化合物, 其中,R2是如同权利要求1至12中的任何一项所定义的R2且R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
其中,R2是如同权利要求1至12中的任何一项所定义的R2且R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
23.式(ⅢA)的化合物或其被保护的衍生物 其中,L代表一离去基团,且R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
其中,L代表一离去基团,且R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
24.式(Ⅳ)的化合物或其被保护的衍生物;其中,L1和L2独立地代表离去基团,且R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
25.式(Ⅴ)的化合物或其被保护的衍生物;其中,L代表一离去基团,且R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
26.式(Ⅵ)的化合物;其中,alk代表C1-6烷基例如甲基,且R3代表正丙基、2-丙烯基、环丁基、环丙基甲基、-(CH2)2OCOCH3、或-(CH2)2-3OH。
27.式(Ⅹ)的化合物或其被保护的衍生物; 其中,R1和R2是如同权利要求1至12中的任何一项所定义的R1和R2。
其中,R1和R2是如同权利要求1至12中的任何一项所定义的R1和R2。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9813538.7A GB9813538D0 (en) | 1998-06-23 | 1998-06-23 | Chemical compounds |
| GB9813538.7 | 1998-06-23 | ||
| GB9909482.3 | 1999-04-23 | ||
| GBGB9909482.3A GB9909482D0 (en) | 1999-04-23 | 1999-04-23 | 2-(Purin-9-yl)-tetahydrofuran-3,4-diol derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1313861A true CN1313861A (zh) | 2001-09-19 |
Family
ID=26313917
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN99809892A Pending CN1313861A (zh) | 1998-06-23 | 1999-06-23 | 2-(嘌呤-9-基)-四氢呋喃-3,4-二醇衍生物 |
Country Status (27)
| Country | Link |
|---|---|
| US (1) | US6495528B1 (zh) |
| EP (1) | EP1090022B1 (zh) |
| JP (1) | JP3933870B2 (zh) |
| KR (1) | KR20010071591A (zh) |
| CN (1) | CN1313861A (zh) |
| AP (1) | AP2000002016A0 (zh) |
| AT (1) | ATE246701T1 (zh) |
| AU (1) | AU750462B2 (zh) |
| BR (1) | BR9911482A (zh) |
| CA (1) | CA2335809A1 (zh) |
| CZ (1) | CZ20004868A3 (zh) |
| DE (1) | DE69910213T2 (zh) |
| EA (1) | EA200001224A1 (zh) |
| EE (1) | EE200000768A (zh) |
| ES (1) | ES2204139T3 (zh) |
| HR (1) | HRP20000894A2 (zh) |
| HU (1) | HUP0103025A3 (zh) |
| ID (1) | ID29609A (zh) |
| IL (1) | IL140396A0 (zh) |
| IS (1) | IS5779A (zh) |
| NO (1) | NO20006548L (zh) |
| NZ (1) | NZ509030A (zh) |
| PL (1) | PL345062A1 (zh) |
| SK (1) | SK19542000A3 (zh) |
| TR (1) | TR200100410T2 (zh) |
| WO (1) | WO1999067265A1 (zh) |
| YU (1) | YU81100A (zh) |
Families Citing this family (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6514949B1 (en) | 1994-07-11 | 2003-02-04 | University Of Virginia Patent Foundation | Method compositions for treating the inflammatory response |
| YU44900A (sh) | 1998-01-31 | 2003-01-31 | Glaxo Group Limited | Derivati 2-(purin-9-il)tetrahidrofuran-3,4-diola |
| CO4990969A1 (es) * | 1998-02-14 | 2000-12-26 | Glaxo Group Ltd | Derivados de 2-(purin-9-il)-tetrahidrofuran-3,4-diol |
| US6232297B1 (en) | 1999-02-01 | 2001-05-15 | University Of Virginia Patent Foundation | Methods and compositions for treating inflammatory response |
| US7427606B2 (en) | 1999-02-01 | 2008-09-23 | University Of Virginia Patent Foundation | Method to reduce inflammatory response in transplanted tissue |
| US7378400B2 (en) | 1999-02-01 | 2008-05-27 | University Of Virginia Patent Foundation | Method to reduce an inflammatory response from arthritis |
| ATE284712T1 (de) * | 2000-04-26 | 2005-01-15 | Eisai Co Ltd | Medizinische zusammensetzungen zur förderung der aktivierung der eingeweide |
| GB0022695D0 (en) | 2000-09-15 | 2000-11-01 | Pfizer Ltd | Purine Derivatives |
| GB0104555D0 (en) * | 2001-02-23 | 2001-04-11 | Glaxo Group Ltd | New Therapeutic method |
| NZ532062A (en) | 2001-10-01 | 2006-09-29 | Univ Virginia | 2-propynyl adenosine analogues having A2 adenosine recepter agonist activity and compositions thereof to treat inflammatory responses |
| GB0206655D0 (en) * | 2002-03-21 | 2002-05-01 | Glaxo Group Ltd | Novel process |
| TW200519106A (en) | 2003-05-02 | 2005-06-16 | Novartis Ag | Organic compounds |
| GB0401334D0 (en) | 2004-01-21 | 2004-02-25 | Novartis Ag | Organic compounds |
| TWI346109B (en) * | 2004-04-30 | 2011-08-01 | Otsuka Pharma Co Ltd | 4-amino-5-cyanopyrimidine derivatives |
| GB0411056D0 (en) | 2004-05-18 | 2004-06-23 | Novartis Ag | Organic compounds |
| PE20060272A1 (es) | 2004-05-24 | 2006-05-22 | Glaxo Group Ltd | (2r,3r,4s,5r,2'r,3'r,4's,5's)-2,2'-{trans-1,4-ciclohexanodiilbis-[imino(2-{[2-(1-metil-1h-imidazol-4-il)etil]amino}-9h-purin-6,9-diil)]}bis[5-(2-etil-2h-tetrazol-5-il)tetrahidro-3,4-furanodiol] como agonista a2a |
| WO2006028618A1 (en) | 2004-08-02 | 2006-03-16 | University Of Virginia Patent Foundation | 2-polycyclic propynyl adenosine analogs with modified 5'-ribose groups having a2a agonist activity |
| US7442687B2 (en) | 2004-08-02 | 2008-10-28 | The University Of Virginia Patent Foundation | 2-polycyclic propynyl adenosine analogs having A2A agonist activity |
| WO2006015357A2 (en) | 2004-08-02 | 2006-02-09 | University Of Virginia Patent Foundation | 2-propynyl adenosine analogs with modified 5'-ribose groups having a2a agonist activity |
| GT200500281A (es) | 2004-10-22 | 2006-04-24 | Novartis Ag | Compuestos organicos. |
| GB0424284D0 (en) | 2004-11-02 | 2004-12-01 | Novartis Ag | Organic compounds |
| GB0426164D0 (en) | 2004-11-29 | 2004-12-29 | Novartis Ag | Organic compounds |
| GB0500785D0 (en) * | 2005-01-14 | 2005-02-23 | Novartis Ag | Organic compounds |
| GB0505219D0 (en) * | 2005-03-14 | 2005-04-20 | Novartis Ag | Organic compounds |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| GB0514809D0 (en) * | 2005-07-19 | 2005-08-24 | Glaxo Group Ltd | Compounds |
| AR058104A1 (es) | 2005-10-21 | 2008-01-23 | Novartis Ag | Compuestos organicos |
| GB0523845D0 (en) * | 2005-11-23 | 2006-01-04 | Glaxo Group Ltd | Novel salts |
| GB0601951D0 (en) | 2006-01-31 | 2006-03-15 | Novartis Ag | Organic compounds |
| PT2322525E (pt) | 2006-04-21 | 2013-12-26 | Novartis Ag | Derivados de purina para utilização como agonistas do recetor de adenosina a2a |
| GB0607950D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| EP1889846A1 (en) * | 2006-07-13 | 2008-02-20 | Novartis AG | Purine derivatives as A2a agonists |
| EP1903044A1 (en) | 2006-09-14 | 2008-03-26 | Novartis AG | Adenosine Derivatives as A2A Receptor Agonists |
| JP2010504933A (ja) | 2006-09-29 | 2010-02-18 | ノバルティス アーゲー | Pi3k脂質キナーゼ阻害剤としてのピラゾロピリミジン |
| EP2089393A1 (en) | 2006-10-30 | 2009-08-19 | Novartis AG | Heterocyclic compounds as antiinflammatory agents |
| PL2231642T3 (pl) | 2008-01-11 | 2014-04-30 | Novartis Ag | Pirymidyny jako inhibitory kinazy |
| US20110281917A1 (en) | 2009-01-29 | 2011-11-17 | Darrin Stuart | Substituted Benzimidazoles for the Treatment of Astrocytomas |
| US8389526B2 (en) | 2009-08-07 | 2013-03-05 | Novartis Ag | 3-heteroarylmethyl-imidazo[1,2-b]pyridazin-6-yl derivatives |
| AU2010283806A1 (en) | 2009-08-12 | 2012-03-01 | Novartis Ag | Heterocyclic hydrazone compounds and their uses to treat cancer and inflammation |
| SG178454A1 (en) | 2009-08-17 | 2012-03-29 | Intellikine Inc | Heterocyclic compounds and uses thereof |
| JP5775871B2 (ja) | 2009-08-20 | 2015-09-09 | ノバルティス アーゲー | ヘテロ環式オキシム化合物 |
| UY33597A (es) | 2010-09-09 | 2012-04-30 | Irm Llc | Compuestos y composiciones como inhibidores de la trk |
| WO2012034095A1 (en) | 2010-09-09 | 2012-03-15 | Irm Llc | Compounds and compositions as trk inhibitors |
| WO2012107500A1 (en) | 2011-02-10 | 2012-08-16 | Novartis Ag | [1, 2, 4] triazolo [4, 3 -b] pyridazine compounds as inhibitors of the c-met tyrosine kinase |
| WO2012116237A2 (en) | 2011-02-23 | 2012-08-30 | Intellikine, Llc | Heterocyclic compounds and uses thereof |
| WO2012116217A1 (en) | 2011-02-25 | 2012-08-30 | Irm Llc | Compounds and compositions as trk inhibitors |
| JP5957526B2 (ja) | 2011-09-15 | 2016-07-27 | ノバルティス アーゲー | チロシンキナーゼとしての6−置換3−(キノリン−6−イルチオ)−[1,2,4]トリアゾロ[4,3−a]ピラジン |
| WO2013078440A2 (en) | 2011-11-23 | 2013-05-30 | Intellikine, Llc | Enhanced treatment regimens using mtor inhibitors |
| CN110507654A (zh) | 2012-04-03 | 2019-11-29 | 诺华有限公司 | 有酪氨酸激酶抑制剂的组合产品和其应用 |
| JP2016512835A (ja) | 2013-03-15 | 2016-05-09 | インテリカイン, エルエルシー | キナーゼ阻害剤の組み合わせ及びそれらの使用 |
| TW201605450A (zh) | 2013-12-03 | 2016-02-16 | 諾華公司 | Mdm2抑制劑與BRAF抑制劑之組合及其用途 |
| WO2016011658A1 (en) | 2014-07-25 | 2016-01-28 | Novartis Ag | Combination therapy |
| US10195208B2 (en) | 2014-07-31 | 2019-02-05 | Novartis Ag | Combination therapy |
| CN106632572B (zh) * | 2016-12-16 | 2018-08-14 | 中国科学院成都生物研究所 | 一种黄芪甲苷衍生物及其制备方法和应用 |
| TW202140550A (zh) | 2020-01-29 | 2021-11-01 | 瑞士商諾華公司 | 使用抗tslp抗體治療炎性或阻塞性氣道疾病之方法 |
Family Cites Families (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE768925A (fr) | 1970-06-30 | 1971-11-03 | Takeda Chemical Industries Ltd | Derives d'adenosine et procede de preparation |
| US4224438A (en) | 1970-07-14 | 1980-09-23 | Boehringer Mannheim Gmbh | Adenosine-5'-carboxylic acid amides |
| CA1019727A (en) | 1971-03-18 | 1977-10-25 | Abbott Laboratories | Adenosine-5'-carboxylic acid amides |
| BE789773A (fr) | 1971-10-08 | 1973-04-06 | Schering Ag | Adenosines n6 -substituees et leur procede de |
| US3864483A (en) | 1972-03-22 | 1975-02-04 | Abbott Lab | Adenosine-5{40 -carboxylic acid amides |
| CA1082695A (en) | 1972-04-10 | 1980-07-29 | Francis E. Fischer | Process for preparing adenosine-5'-carboxamides |
| US3966917A (en) | 1974-07-30 | 1976-06-29 | Abbott Laboratories | Platelet aggregation inhibitors |
| DE2621470A1 (de) | 1976-05-14 | 1977-12-01 | Pharma Waldhof Gmbh & Co | Nucleosidcarbonsaeurenitrile und ihre derivate, und verfahren zu ihrer herstellung |
| US4167565A (en) | 1976-11-08 | 1979-09-11 | Abbott Laboratories | Adenosine-5'-carboxamides and method of use |
| AU8379182A (en) | 1981-06-04 | 1982-12-09 | Procter & Gamble Company, The | Composition of salicylates and purine derivatives |
| JPS58167599A (ja) | 1982-03-29 | 1983-10-03 | Tanabe Seiyaku Co Ltd | アデノシン誘導体及びその製法 |
| JPS58174322A (ja) | 1982-04-07 | 1983-10-13 | Tanabe Seiyaku Co Ltd | 線溶促進剤 |
| CA1239397A (en) | 1983-08-01 | 1988-07-19 | James A. Bristol | N.sup.6-substituted adenosines |
| DE3406533A1 (de) | 1984-02-23 | 1985-08-29 | Boehringer Mannheim Gmbh, 6800 Mannheim | Verwendung von adenosin-derivaten als antiallergica und arzneimittel, die diese enthalten |
| US4496643A (en) | 1984-03-23 | 1985-01-29 | Eastman Kodak Company | Two-component dry electrostatic developer composition containing onium charge control agent |
| JPH0655756B2 (ja) | 1984-04-18 | 1994-07-27 | ネルソン・リサ−チ・アンド・デベロツプメント・カンパニ− | 心臓血管拡張薬としてのn−6置換アデノシン誘導体 |
| US5310731A (en) | 1984-06-28 | 1994-05-10 | Whitby Research, Inc. | N-6 substituted-5'-(N-substitutedcarboxamido)adenosines as cardiac vasodilators and antihypertensive agents |
| US4663313A (en) | 1984-10-26 | 1987-05-05 | Warner-Lambert Company | N6 -tricyclic adenosines for treating hypertension |
| AU575438B2 (en) | 1984-10-26 | 1988-07-28 | Warner-Lambert Company | N6 - substituted deoxyribose analogues of adenosines |
| US4738954A (en) | 1985-11-06 | 1988-04-19 | Warner-Lambert Company | Novel N6 -substituted-5'-oxidized adenosine analogs |
| US4755594A (en) | 1986-01-31 | 1988-07-05 | Warner-Lambert Company | N6 -substituted adenosines |
| US5106837A (en) | 1988-03-16 | 1992-04-21 | The Scripps Research Institute | Adenosine derivatives with therapeutic activity |
| JPH0696534B2 (ja) | 1986-04-25 | 1994-11-30 | ヘキストジヤパン株式会社 | 抗痴呆剤 |
| US4767747A (en) | 1986-08-28 | 1988-08-30 | Warner-Lambert Company | Method for treating congestive heart failure with N6 -acenaphthyl adenosine |
| AU8274187A (en) | 1986-10-31 | 1988-05-25 | Warner-Lambert Company | Heteroaromatic derivatives of adenosine |
| WO1988003147A1 (en) | 1986-10-31 | 1988-05-05 | Warner-Lambert Company | Selected n6-substituted adenosines having selective a2 binding activity |
| HU198950B (en) | 1986-12-15 | 1989-12-28 | Sandoz Ag | Process for producing new furanuronic acid derivatives and pharmaceutical compositions comprising such compounds |
| US4968697A (en) | 1987-02-04 | 1990-11-06 | Ciba-Geigy Corporation | 2-substituted adenosine 5'-carboxamides as antihypertensive agents |
| FI880405A (fi) | 1987-02-04 | 1988-08-05 | Ciba Geigy Ag | Adenosin-5'-karboxamidderivat. |
| US4962194A (en) | 1987-04-02 | 1990-10-09 | Warner-Lambert Company | Method of preparing 51,N6-disubstituted adenosines from inosines |
| US5219840A (en) | 1987-04-06 | 1993-06-15 | Sandoz Ltd. | Antihypertensive 9-(2,N6 -disubstituted adenyl) ribofuranuronic acid derivatives |
| LU87181A1 (fr) | 1987-04-06 | 1988-11-17 | Sandoz Sa | Nouveaux derives de l'acide furannuronique,leur preparation et leur utilisation comme medicaments |
| JPH0725785B2 (ja) | 1989-01-11 | 1995-03-22 | 日本臓器製薬株式会社 | アデノシン誘導体及び該化合物を有効成分として含有する医薬組成物 |
| KR910007655A (ko) | 1989-10-03 | 1991-05-30 | 엠. 피. 잭슨 | 치료용 뉴클레오시드 |
| CA2028002A1 (en) | 1989-10-19 | 1991-04-20 | Daniel P. Becker | Method of treating gastrointestinal motility disorders |
| US5055569A (en) | 1989-10-19 | 1991-10-08 | G. D. Searle & Co. | N-(6)-substituted adenosine compounds |
| US5140015A (en) | 1990-02-20 | 1992-08-18 | Whitby Research, Inc. | 2-aralkoxy and 2-alkoxy adenosine derivatives as coronary vasodilators and antihypertensive agents |
| WO1992005177A1 (en) | 1990-09-25 | 1992-04-02 | Rhone-Poulenc Rorer International (Holdings) Inc. | Compounds having antihypertensive and anti-ischemic properties |
| FR2685918B1 (fr) | 1992-01-08 | 1995-06-23 | Union Pharma Scient Appl | Nouveaux derives de l'adenosine, leurs procedes de preparation, compositions pharmaceutiques les contenant. |
| FR2687678B1 (fr) | 1992-01-31 | 1995-03-31 | Union Pharma Scient Appl | Nouveaux derives de l'adenosine, leurs procedes de preparation, compositions pharmaceutiques les contenant. |
| US5424297A (en) | 1992-04-27 | 1995-06-13 | University Of Virginia Alumni Patents Foundation | Adenosine dextran conjugates |
| WO1994002497A1 (en) | 1992-07-15 | 1994-02-03 | The United States Of America, Represented By The Secretary, Department Of Health And Human Services | Sulfo-derivatives of adenosine |
| GB9301000D0 (en) * | 1993-01-20 | 1993-03-10 | Glaxo Group Ltd | Chemical compounds |
| WO1994018215A1 (en) | 1993-02-03 | 1994-08-18 | Gensia, Inc. | Adenosine kinase inhibitors comprising lyxofuranosyl derivatives |
| US5773423A (en) | 1993-07-13 | 1998-06-30 | The United States Of America As Represented By The Department Of Health And Human Services | A3 adenosine receptor agonists |
| US5446046A (en) | 1993-10-28 | 1995-08-29 | University Of Florida Research Foundation | A1 adenosine receptor agonists and antagonists as diuretics |
| WO1995018817A1 (fr) | 1994-01-07 | 1995-07-13 | Laboratoires Upsa | Nouveaux derives de l'adenosine, leurs procedes de preparation, compositions pharmaceutiques les contenant |
| GB9414193D0 (en) | 1994-07-14 | 1994-08-31 | Glaxo Group Ltd | Compounds |
| GB9414208D0 (en) | 1994-07-14 | 1994-08-31 | Glaxo Group Ltd | Compounds |
| AU3255097A (en) | 1996-07-05 | 1998-02-02 | Novo Nordisk A/S | Novel (n)-alkoxyadenine derivatives acting as cytokine inhibitors |
| UA51716C2 (uk) | 1996-07-08 | 2002-12-16 | Авентіс Фармасьютікалз Продактс Інк. | Сполуки, що мають гіпотензивну, кардіопротекторну, анти-ішемічну та антиліполітичну властивості, фармацевтична композиція та способи лікування |
| AU4377397A (en) | 1996-10-14 | 1998-05-11 | Novo Nordisk A/S | Novel therapeutically active adenosine derivatives |
| TW528755B (en) * | 1996-12-24 | 2003-04-21 | Glaxo Group Ltd | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| ZA9810766B (en) | 1997-11-28 | 1999-05-25 | Mochida Pharm Co Ltd | Novel compounds having cgmp-pde inhibitory activity |
| YU44900A (sh) * | 1998-01-31 | 2003-01-31 | Glaxo Group Limited | Derivati 2-(purin-9-il)tetrahidrofuran-3,4-diola |
| CO4990969A1 (es) * | 1998-02-14 | 2000-12-26 | Glaxo Group Ltd | Derivados de 2-(purin-9-il)-tetrahidrofuran-3,4-diol |
-
1999
- 1999-06-23 US US09/720,390 patent/US6495528B1/en not_active Expired - Fee Related
- 1999-06-23 YU YU81100A patent/YU81100A/sh unknown
- 1999-06-23 AP APAP/P/2000/002016A patent/AP2000002016A0/en unknown
- 1999-06-23 CN CN99809892A patent/CN1313861A/zh active Pending
- 1999-06-23 KR KR1020007014720A patent/KR20010071591A/ko not_active Application Discontinuation
- 1999-06-23 CA CA002335809A patent/CA2335809A1/en not_active Abandoned
- 1999-06-23 ES ES99931113T patent/ES2204139T3/es not_active Expired - Lifetime
- 1999-06-23 DE DE69910213T patent/DE69910213T2/de not_active Expired - Lifetime
- 1999-06-23 NZ NZ509030A patent/NZ509030A/en not_active Application Discontinuation
- 1999-06-23 IL IL14039699A patent/IL140396A0/xx unknown
- 1999-06-23 JP JP2000555916A patent/JP3933870B2/ja not_active Expired - Fee Related
- 1999-06-23 EA EA200001224A patent/EA200001224A1/ru unknown
- 1999-06-23 TR TR2001/00410T patent/TR200100410T2/xx unknown
- 1999-06-23 AU AU47744/99A patent/AU750462B2/en not_active Ceased
- 1999-06-23 ID IDW20010189D patent/ID29609A/id unknown
- 1999-06-23 EE EEP200000768A patent/EE200000768A/xx unknown
- 1999-06-23 PL PL99345062A patent/PL345062A1/xx unknown
- 1999-06-23 WO PCT/EP1999/004269 patent/WO1999067265A1/en not_active Application Discontinuation
- 1999-06-23 AT AT99931113T patent/ATE246701T1/de not_active IP Right Cessation
- 1999-06-23 BR BR9911482-8A patent/BR9911482A/pt not_active IP Right Cessation
- 1999-06-23 CZ CZ20004868A patent/CZ20004868A3/cs unknown
- 1999-06-23 EP EP99931113A patent/EP1090022B1/en not_active Expired - Lifetime
- 1999-06-23 HU HU0103025A patent/HUP0103025A3/hu unknown
- 1999-06-23 SK SK1954-2000A patent/SK19542000A3/sk unknown
-
2000
- 2000-12-19 IS IS5779A patent/IS5779A/is unknown
- 2000-12-21 HR HR20000894A patent/HRP20000894A2/hr not_active Application Discontinuation
- 2000-12-21 NO NO20006548A patent/NO20006548L/no not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| EE200000768A (et) | 2002-04-15 |
| HUP0103025A3 (en) | 2002-04-29 |
| IS5779A (is) | 2000-12-19 |
| BR9911482A (pt) | 2002-01-22 |
| DE69910213D1 (de) | 2003-09-11 |
| AU4774499A (en) | 2000-01-10 |
| SK19542000A3 (sk) | 2001-09-11 |
| AU750462B2 (en) | 2002-07-18 |
| WO1999067265A1 (en) | 1999-12-29 |
| US6495528B1 (en) | 2002-12-17 |
| JP2002518512A (ja) | 2002-06-25 |
| HUP0103025A2 (hu) | 2001-12-28 |
| DE69910213T2 (de) | 2004-07-01 |
| NO20006548D0 (no) | 2000-12-21 |
| JP3933870B2 (ja) | 2007-06-20 |
| NO20006548L (no) | 2001-02-22 |
| EA200001224A1 (ru) | 2001-08-27 |
| CA2335809A1 (en) | 1999-12-29 |
| IL140396A0 (en) | 2002-02-10 |
| HRP20000894A2 (en) | 2001-12-31 |
| ID29609A (id) | 2001-09-06 |
| EP1090022B1 (en) | 2003-08-06 |
| EP1090022A1 (en) | 2001-04-11 |
| AP2000002016A0 (en) | 2000-12-31 |
| NZ509030A (en) | 2003-07-25 |
| YU81100A (sh) | 2003-10-31 |
| CZ20004868A3 (cs) | 2001-08-15 |
| KR20010071591A (ko) | 2001-07-28 |
| ES2204139T3 (es) | 2004-04-16 |
| PL345062A1 (en) | 2001-11-19 |
| ATE246701T1 (de) | 2003-08-15 |
| TR200100410T2 (tr) | 2001-06-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1313861A (zh) | 2-(嘌呤-9-基)-四氢呋喃-3,4-二醇衍生物 | |
| CN1313860A (zh) | 2-(嘌呤-9-基)-四氢呋喃-3,4-二醇衍生物 | |
| CN1246124A (zh) | 2-(嘌呤-9-基)-四氢呋喃-3,4-二醇衍生物 | |
| US6534486B1 (en) | 2-(purin-9-yl)-Tetrahydrofuran-3,4-diol derivatives | |
| CN1289338A (zh) | 2-(嘌呤-9-基)-四氢呋喃-3,4-二醇衍生物 | |
| US5905082A (en) | Crystalline oxathiolane derivatives | |
| US5424295A (en) | 9-β-D-arabinofuranasyl-2-amino-6-methaoxy-9H-purine | |
| CN1262556C (zh) | 特定的二核苷酸和它们作为粘膜纤毛清除和纤毛颤动频率调节剂的应用 | |
| KR20010082512A (ko) | 2-(푸린-9-일)-테트라히드로푸란-3,4-디올 유도체 | |
| JP2003176296A (ja) | 単環式ヌクレオシド、その類似体および使用 | |
| HRP20020676A2 (en) | Purine derivatives | |
| CN1516590A (zh) | 4′-取代的核苷 | |
| CN1478483A (zh) | 用于治疗乙型肝炎的β-L-2'-脱氧-核苷 | |
| CN1296011A (zh) | 嘌呤l-核苷、其类似物及其用途 | |
| US5492897A (en) | Method for treating T-cell lymphoblastic leukemia with ara-G nucleoside derivatives | |
| JPH0656877A (ja) | 抗ウイルス活性および抗ガン活性を有する2’−デオキシ−2’,2’−ジフルオロ(2,6,8−置換)−プリンヌクレオシド類およびその中間体 | |
| AU641533C (en) | Heterocyclic compounds | |
| MXPA00012926A (en) | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives | |
| MXPA00012895A (en) | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |