CN100391963C - 制备c-芳基葡糖苷sglt2抑制剂的方法 - Google Patents
制备c-芳基葡糖苷sglt2抑制剂的方法 Download PDFInfo
- Publication number
- CN100391963C CN100391963C CNB2003801100401A CN200380110040A CN100391963C CN 100391963 C CN100391963 C CN 100391963C CN B2003801100401 A CNB2003801100401 A CN B2003801100401A CN 200380110040 A CN200380110040 A CN 200380110040A CN 100391963 C CN100391963 C CN 100391963C
- Authority
- CN
- China
- Prior art keywords
- compound
- group
- bromine
- formula
- ether
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon, or a metal, e.g. chelates, vitamin B12
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/20—Carbocyclic rings
- C07H15/207—Cyclohexane rings not substituted by nitrogen atoms, e.g. kasugamycins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H5/00—Compounds containing saccharide radicals in which the hetero bonds to oxygen have been replaced by the same number of hetero bonds to halogen, nitrogen, sulfur, selenium, or tellurium
- C07H5/04—Compounds containing saccharide radicals in which the hetero bonds to oxygen have been replaced by the same number of hetero bonds to halogen, nitrogen, sulfur, selenium, or tellurium to nitrogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H5/00—Compounds containing saccharide radicals in which the hetero bonds to oxygen have been replaced by the same number of hetero bonds to halogen, nitrogen, sulfur, selenium, or tellurium
- C07H5/04—Compounds containing saccharide radicals in which the hetero bonds to oxygen have been replaced by the same number of hetero bonds to halogen, nitrogen, sulfur, selenium, or tellurium to nitrogen
- C07H5/06—Aminosugars
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Emergency Medicine (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Endocrinology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pyrane Compounds (AREA)
Abstract
制备C-芳基葡糖苷SGLT2抑制剂及其中间体的方法,所述抑制剂用于治疗糖尿病及相关疾病。所述C-芳基葡糖苷可与氨基酸络合物形成试剂络合。
Description
发明的工业适用领域
本发明涉及C-芳基葡糖苷化合物,所述化合物为肠和肾中发现的钠依赖性葡萄糖转运蛋白(SGLT2)抑制剂,更具体地说涉及制备这种化合物例如1-C-(取代二苯基甲烷-3-基)-β-D-吡喃葡萄糖的方法,通过独特方法保证使用一锅法(one-pot)制备终产物而很少有中间体化合物必须分离的方式制备其有用中间体。本发明还涉及与氨基酸络合物形成剂形成的C-芳基葡糖苷化合物的结晶络合物。
发明背景
全世界约1亿人患有II型糖尿病[非胰岛素依赖型糖尿病(NIDDM)],其特征在于因过量肝葡萄糖生成和外周胰岛素抗性所致高血糖,高血糖的根本原因尚未完全清楚。据认为高血糖为发生糖尿病并发症的主要危险因素,很可能与出现在晚期NIDDM的胰岛素分泌减少直接有关。NIDDM患者的正常血浆葡萄糖预示胰岛素作用提高,因而抵消糖尿病并发症的发生。预期钠依赖性葡萄糖转运蛋白(SGLT2)抑制剂在肾中通过促进葡萄糖排泄能帮助血浆葡萄糖水平恢复正常,同时可能有助体重恢复正常。
高血糖为II型糖尿病的特点;持续控制糖尿病患者的血浆葡萄糖水平可抵消出现在晚期疾病的糖尿病并发症发生和β细胞缺乏。血浆葡萄糖通常在肾小球过滤,在近肾小管处主动重吸收。SGLT2看来是在此部位引起葡萄糖再摄取的主要转运蛋白。SGLT特异性抑制剂根皮苷或密切相关的类似物在糖尿病啮齿动物和狗中抑制该再摄取过程,通过促进葡萄糖排泄导致血浆葡萄糖水平正常化而无低血糖的副作用。据报道用SGLT2抑制剂长期(6个月)治疗Zucker糖尿病大鼠可提高胰岛素对糖血的反应,提高胰岛素的敏感性,延缓这些动物肾病和神经病的发作,未发现肾脏病变和血浆电解质失调。预期通过促进尿中葡萄糖排泄,糖尿病患者SGLT2的选择性抑制可使血浆葡萄糖水平恢复正常,因而提高胰岛素敏感性,延缓糖尿病并发症发生。
在肾脏中,90%的葡萄糖再摄取发生在肾皮质近小管前S1区段上皮细胞中,而SGLT2可能为引起该再摄取的主要转运蛋白。SGLT2为含14跨膜片段的672个氨基酸蛋白,其主要在肾近小管前S1区段中表达。SGLT2的底物特异性、钠依赖性和局部化与先前所述人肾皮质近小管中特征为高容量、低亲和性、钠依赖性葡萄糖转运蛋白的性质相一致。另外,杂交缺失研究提示,在近小管S1区段SGLT2为主要的Na+/葡萄糖协同转运蛋白,因为实际上所有在大鼠肾皮质mRNA中编码的钠依赖性葡萄糖转运活性被大鼠SGLT2特异性的反义寡核苷酸抑制。
SGLT2为某些类型的家族性葡糖尿的备选基因,所述家族性葡糖尿属于一种遗传异常,其中肾葡萄糖重吸收受到不同程度的损害。迄今为止,这些已研究过的综合征没有一种在染色体16上的SGLT2基因座绘制成图。但是涉及高度同源性啮齿类动物SGLT的研究强烈提示SGLT2为主要的肾钠依赖性葡萄糖转运蛋白,还提示已经绘图的葡糖尿基因座编码SGLT2调节基因。预计通过促进糖尿病患者的葡萄糖排泄来抑制SGLT2能降低血浆葡萄糖水平。
发现C-芳基葡糖苷这类SGLT2抑制剂与口服活性抗糖尿病药物那样作用。具体地说,发现这些C-芳基葡糖苷SGLT2抑制剂可用于治疗或延缓糖尿病的发作或进展,尤其是I型和II型糖尿病,包括糖尿病并发症例如视网膜病、神经病、肾病和延迟性伤口愈合,以及相关疾病如胰岛素抗性和葡萄糖体内平衡受损(IGH)、高血糖症、高胰岛素血症、高血脂肪酸或甘油水平、肥胖、高脂血症包括高甘油三酯血症、X综合征、高血压、动脉粥样硬化及相关疾病,以及用于提高高密度脂质水平。在Johannsson,J.Clin.Endrocrinol.Metab.,82,727-34(1997)中对统称为“X综合征”(又称代谢综合征)的病症和疾病有详细介绍。
这种C-芳基葡糖苷SGLT2抑制剂可单独使用或与现有治疗药物联合使用,所述药物包括磺酰脲、噻唑烷二酮、二甲双胍和胰岛素,以避免与使用这些其它药物通常相关的潜在的副作用。有关C-芳基葡糖苷及其衍生物的更详细内容可见于PCT国际申请WO 01/27128-A1、美国专利第6,414,126号、美国专利申请第10/151,436号和美国专利申请第10/117,914号,其所有公开内容通过引用结合到本文。
需要提供缩短步骤或一锅法操作、或任选多容器反应,其在制备终产物期间使产生的中间体最少以提高收率和纯度的制备C-芳基葡糖苷SGLT2抑制剂的方法。进一步需要这种方法采用立体有择性操作以便制备基本上对映体纯产物。这种方法可用于制备化合物包括但不限于1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖、1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-基)-β-D-吡喃葡萄糖、1-C-(6-氯-4′-乙氧基二苯基甲烷-3-基)-β-D-吡喃葡萄糖。还需要形成所述合成化合物的结晶络合物的方法。
发明概述
本发明涉及新的制备C-芳基葡糖苷的立体有择方法,在一个选项中,所述方法可按一锅法操作进行,所用步骤少于涉及一步或多步分离中间体反应产物的其它方法。本发明还包括在这些反应过程形成的某些中间体。在另一个实施方案中,本发明还包括制备C-芳基葡糖苷的结晶络合物。
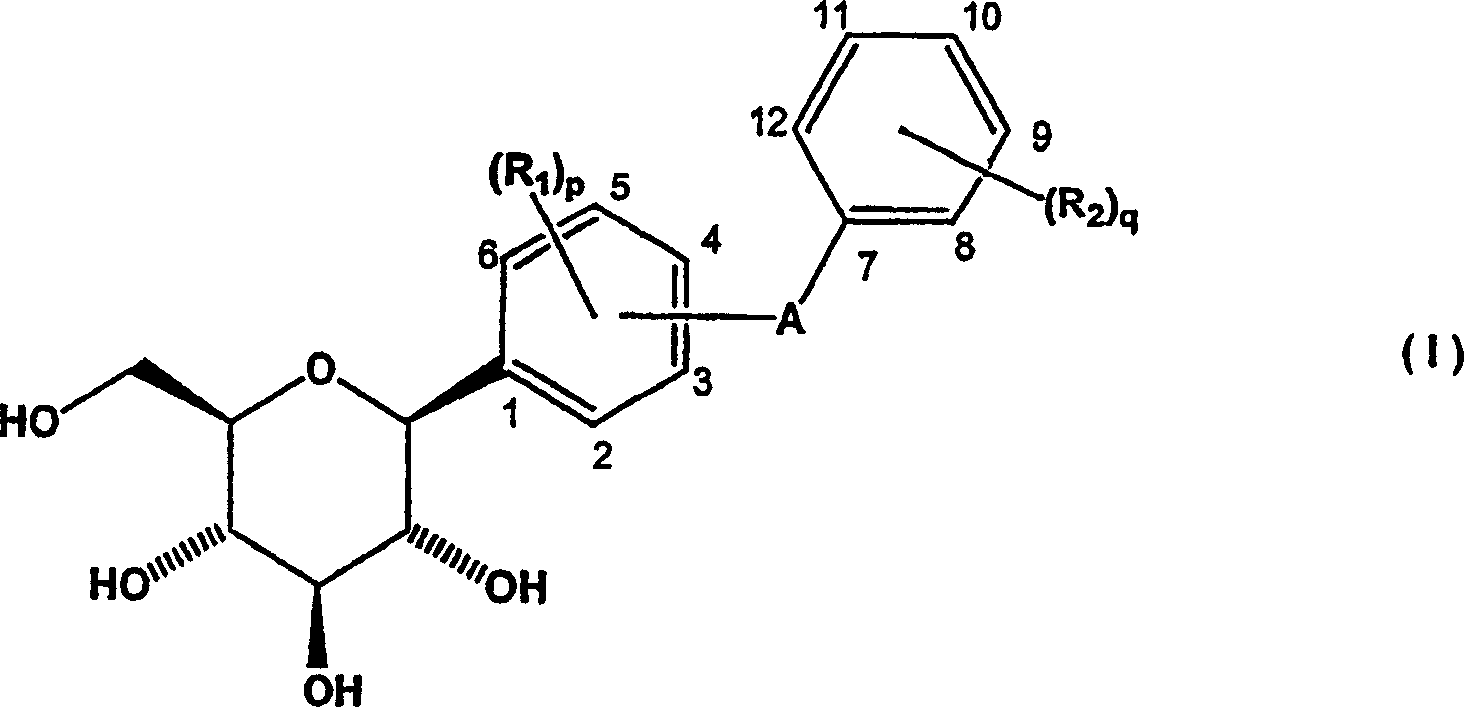
在本发明的一方面,提供一种制备式(I)化合物及其药学上可接受的络合物的方法

其中:
R1选自氢、羟基、溴、氯、氟、烷基、烷氧基、烷硫基和芳硫基,其中p为1至4,且条件是当溴、氯和氟存在时,只能在3-、4-和5-位中至少一个位置上存在;
R2选自氢、羟基、氯、氟、烷基、烷氧基和烷硫基,其中q为1至5;和
A选自共价键、O、S、NH和(CH2)n,其中n为1至3,且
条件是
当A在4-位时,R1不为溴;
当R1中的一个为溴;和
如果A在3-或6-位,则溴在5-位,
如果A在2-或5-位,则溴在3-位,和
当溴在3-位,且A在2-或5-位时,则在4-和6-位的R1基团相同且不为溴、氯或氟,和
当溴在5-位,且A在3-或6-位时,则在2-和4-位的R1基团相同且不为溴、氯或氟;所述方法包括:
a)用酸不稳定保护基团保护的内酯形成式(IV)化合物
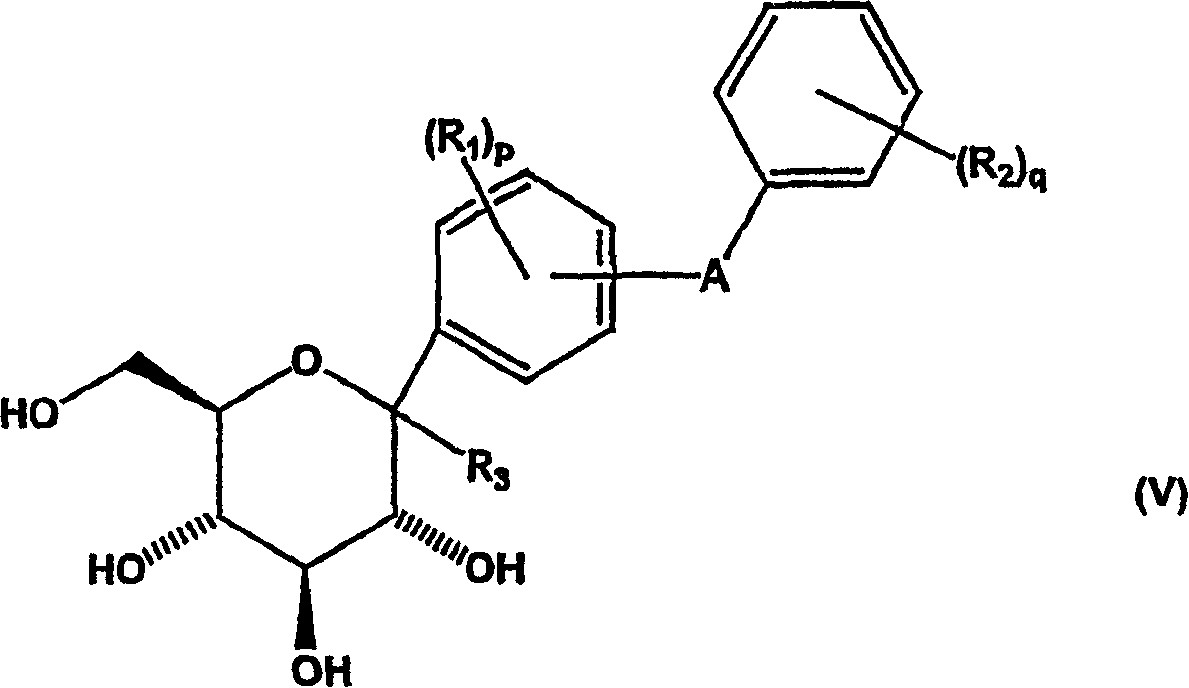
b)使式(IV)化合物苷化,同时除去酸不稳定保护基团以生成式(V)化合物;

c)使式(V)化合物与酰化剂反应形成式(VI)化合物;
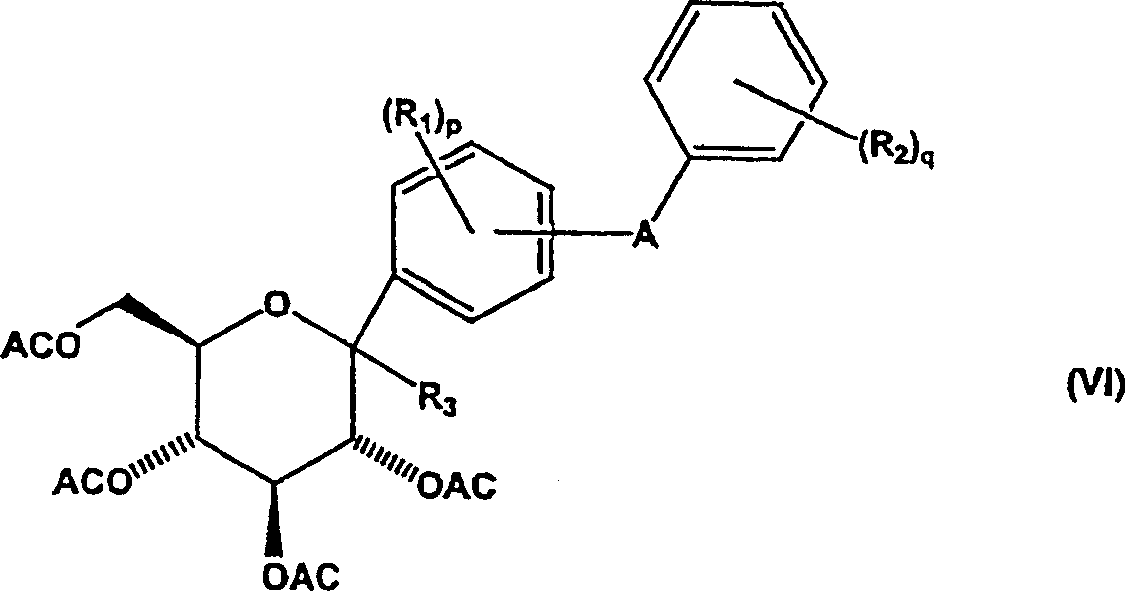
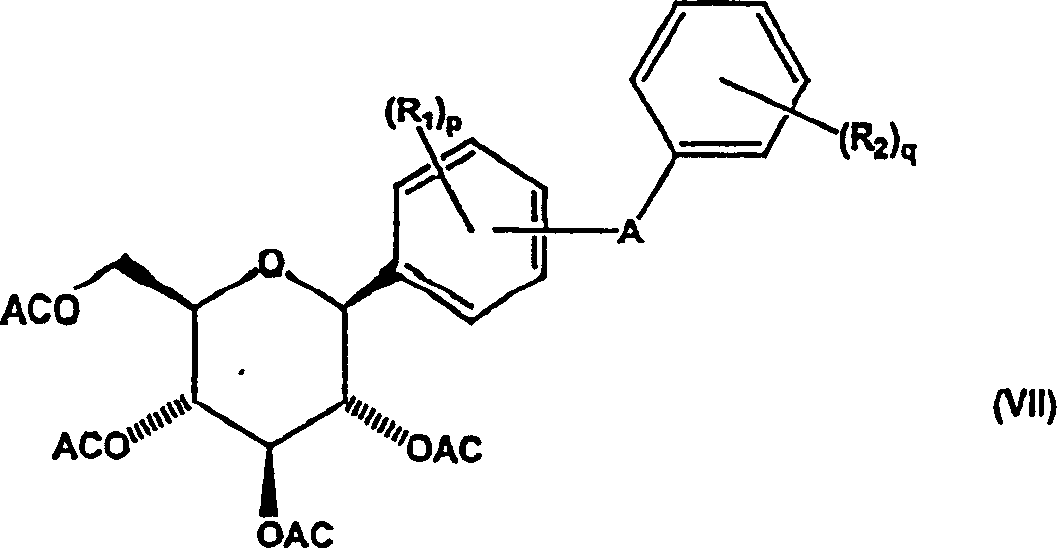
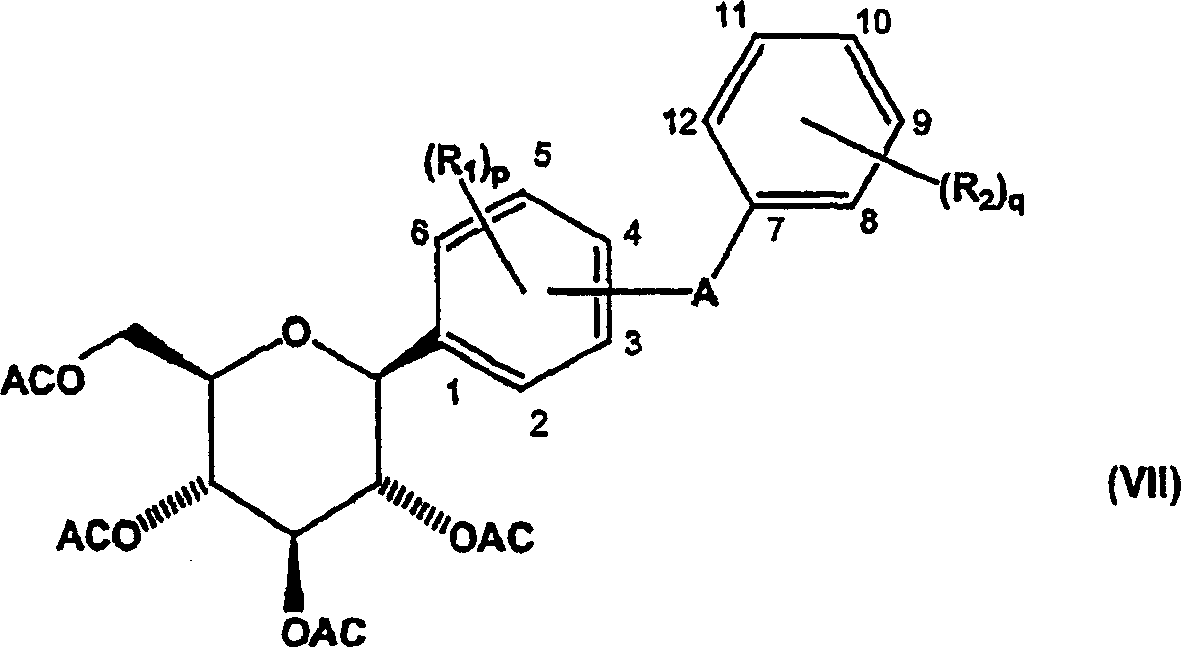
d)将式(VI)化合物还原以得到式(VII)化合物;和
e)除去式(VII)化合物的酰基保护基团以得到式(I)化合物;
其中R1、R2、A、p和q定义同上,且AC为酰基保护基团。
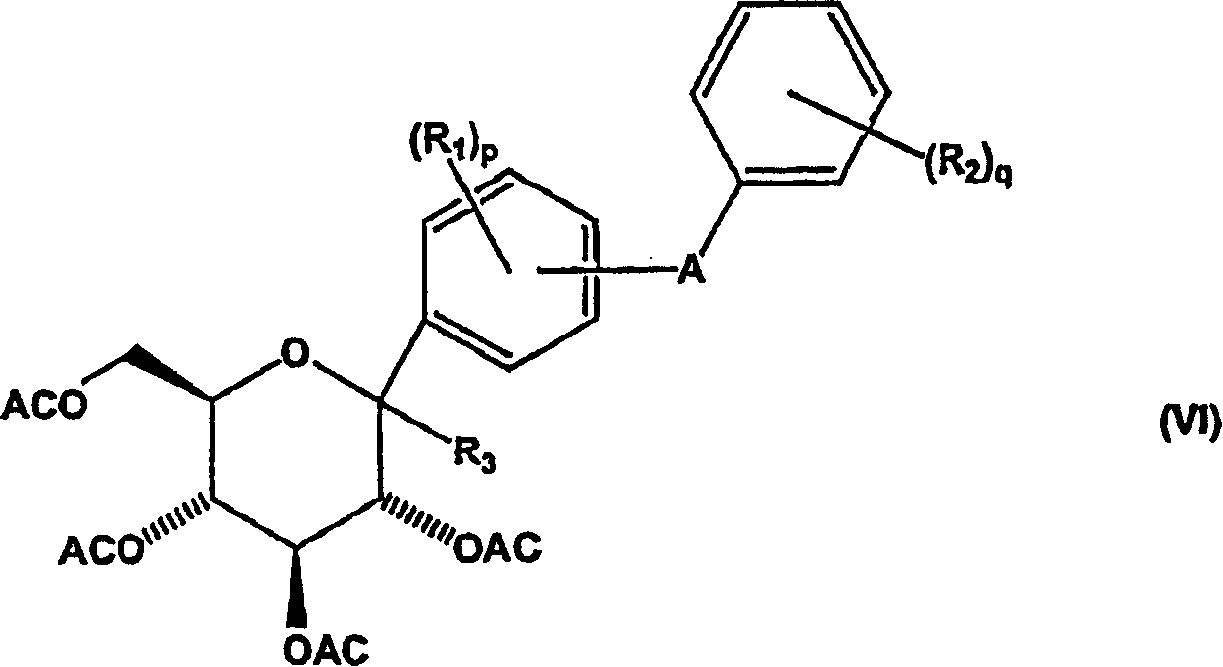
在再一个实施方案中,本发明包括一种制备C-芳基葡糖苷化合物的方法,该方法包括使式(VI)化合物与还原剂反应;

其中
R1选自氢、羟基、溴、氯、氟、烷基、烷氧基、烷硫基和芳硫基,其中p为1至4的整数,且条件是当溴、氯和氟存在时,只能在3-、4-和5-位中至少一个位置上存在;
R2选自氢、羟基、氯、氟、烷基、烷氧基和烷硫基,其中q为1至5的整数;和
A选自共价键、O、S、NH和(CH2)n,其中n为1至3的整数,且
条件是当A在4-位时,R1不为溴;
条件是当R1中的一个为溴;和
如果A在3-或6-位,则溴在5-位,
如果A在2-或5-位,则溴在3-位,和
当溴在3-位,且A在2-或5-位时,则在4-和6-位的R1基团相同,且不为溴、氯或氟,和
当溴在5-位,且A在3-或6-位时,则在2-和4-位的R1基团相同,且不为溴、氯或氟;
R3选自烷氧基、烯氧基、烷硫基和烯硫基;且
AC为酰基保护基团;
形成式(VII)化合物

在本发明的一个优选的实施方案中,提供一种制备式(LA)化合物及其药学上可接受的络合物的方法

其中:
R1选自氢、烷基和氯;和
R2选自氢、烷基、烷氧基和烷硫基。
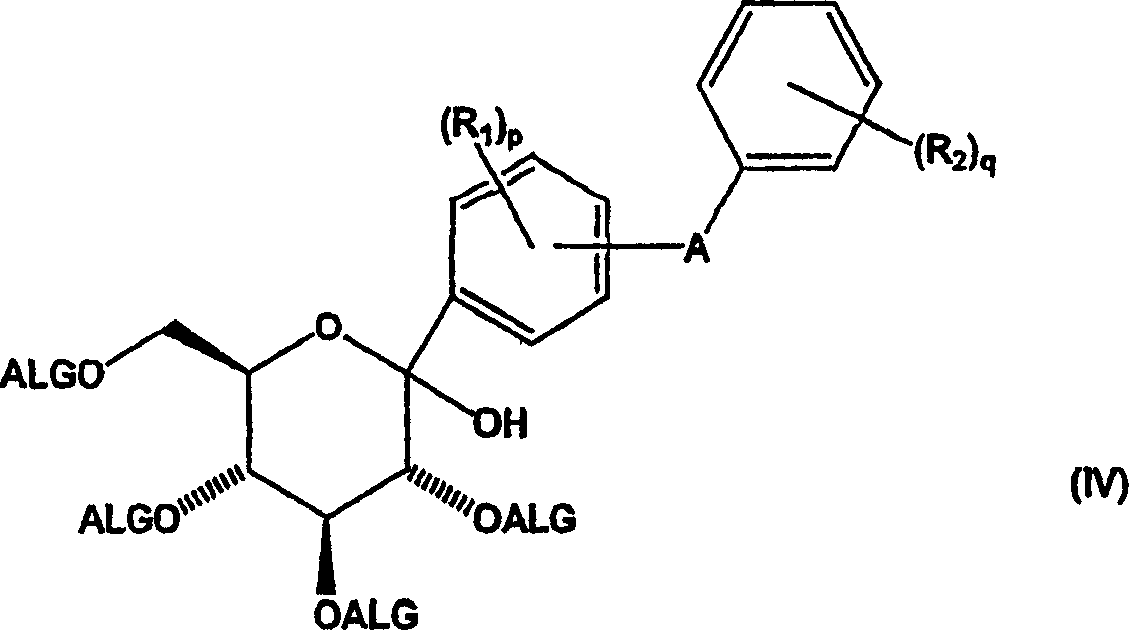
在本发明的另一方面,提供新的中间体化合物及其制备方法,用于制备式(I)化合物。本发明新的中间体化合物包括:
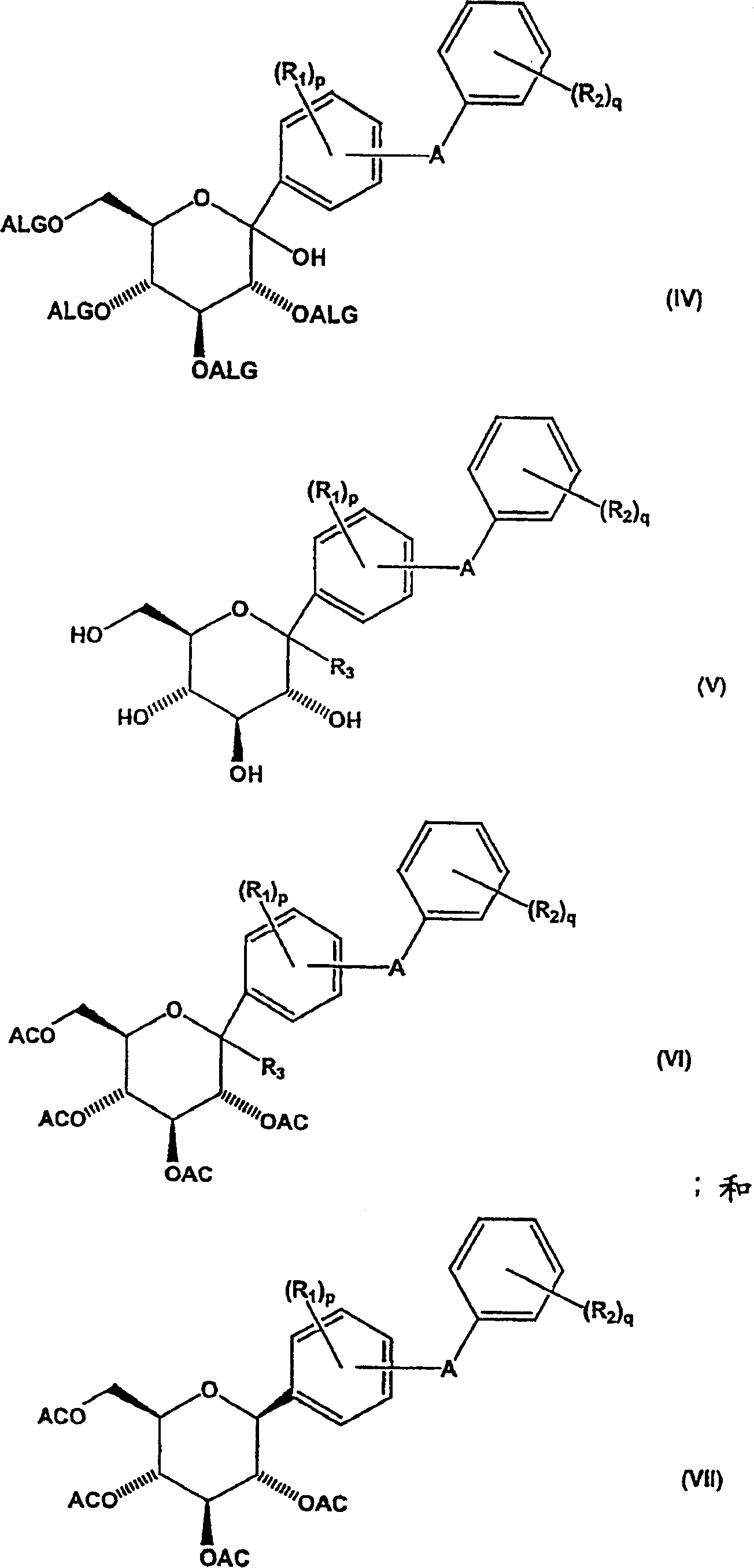
其中:
R1、R2、A、p和q定义同上;
R3选自烷氧基、烯氧基、烷硫基和烯硫基;
AC为酰基保护基团如CH3CO-;和
ALG为酸不稳定保护基团。
发明详述
本发明涉及制备C-芳基葡糖苷及其结晶络合物的方法,涉及制备中间体化合物的方法和涉及新的中间体化合物。本发明方法可按涉及一步或多步分离中间体的多容器反应进行;或任选按缩短步骤的反应进行,该反应所需步骤比多容器反应少或按其它常规方法制备这种化合物。因此本发明的某些方法可以低成本、低劳动力、高纯度和高收率制备C-芳基葡糖苷及其络合物和中间体。
现已发现在本发明方法的相应步骤中使用酸不稳定保护基团,尤其是成本合理的含甲硅烷基酸不稳定保护基团和酰基保护基团保护羟基,可显著提高合成本发明化合物的收率、立体有择性和经济性。在本发明的反应过程中,在取决于保护基团性质的酸性或碱性条件下,通过本领域已知的标准方法可完成羟基脱保护。关于使羟基脱保护的合适方法的概要见Greene,T.W.和Wuts,P.G.M.,Protecting Groups inOrganic Synthesis,第3版,John Wiley&Son,Inc.(1999),其通过引用结合到本文。
下列缩写为本发明说明书中使用的各种术语的定义。这些定义适用于在本说明书全文中(除非它们在特定的情况下另有限定)单独或作为较大基团的一部分使用的术语。
在本文中使用以下缩写:
Ph=苯基
t-Bu=叔丁基
Me=甲基
Et=乙基
TMS=三甲基甲硅烷基
THF=四氢呋喃
Tol=甲苯
BF3·Et2O=三氟化硼-乙醚合物
CH3CN=乙腈
EtOAc=乙酸乙酯
MeOH=甲醇
MSOH=甲磺酸
EtOH=乙醇
Et3SiH=三乙基甲硅烷
i-PrOH=异丙醇
Ac2O=乙酸酐
AcOH=乙酸
Et3N=三乙胺
DIPEA=i-Pr2NEt=N,N′-二异丙基乙胺
DMAP=4-二甲基氨基吡啶
n-BuLi=正丁基锂
NaOH=氢氧化钠
TLC=薄层层析
HPLC=高效液相色谱
GC=气相色谱
AP=面积百分率
KF=卡尔·费歇尔(Karl Fisher)
LOD=干燥失重
本文中使用的术语“葡糖苷”可与术语“苷”“吡喃葡萄糖”或“吡喃葡糖苷”等同使用并可互换,表示由羟基化合物与作为糖组分的葡萄糖结合衍生的缩醛分子。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“低级烷基”包括含1至8个碳的直链和支链烃,在本文中单独或作为另一基团的一部分使用的术语“烷基”和“烷”包括在直链中含1至20个、优选1至10个、更优选含有1至8个碳的直链和支链烃,例如甲基、乙基、丙基异丙基、丁基、叔丁基、异丁基、戊基、己基、异己基、庚基、4,4-二甲基戊基、辛基、2,2,4-三甲基戊基、壬基、癸基、十一烷基、十二烷基,其各种支链异构体等,以及包括1至4个取代基如卤素例如F、Br、Cl或I的这些基团或CF3、烷基、烷氧基、芳基、芳氧基、芳基(芳基)或二芳基、芳基烷基、芳基烷氧基、烯基、炔基、环烷基、环烯基、环烷基烷基、环烷基烷氧基、任选取代的氨基、羟基、羟基烷基、酰基、烷酰基、杂芳基、杂芳基氧基、杂环烷基、芳基杂芳基、芳基烷氧基羰基、杂芳基烷基、杂芳基烷氧基、芳氧基烷基、芳氧基芳基、烷基酰胺基、烷酰基氨基、芳基羰基氨基、硝基、氰基、巯基、卤代烷基、三卤代烷基和/或烷硫基。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“环烷基”包括含1至3个环的饱和或部分饱和(含1或2个双键)环状烃基,包括单环烷基、双环烷基和三环烷基,含形成环的总数为3至20个碳,优选3至10个碳形成环,该环可稠合到1或2个描述为芳基的芳环上,其包括环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环癸基、环十二烷基、环己烯基等,这些基团中任一基团可任选被1至4个以下的取代基取代:如卤素、烷基、烷氧基、羟基、芳基、芳氧基、芳基烷基、环烷基、烷基酰胺基、烷酰基氨基、氧代、酰基、芳基羰基氨基、氨基、硝基、氰基、巯基和/或烷硫基和/或任何烷基取代基。
在本文中单独或作为另一基团一部分使用的术语“环烯基”指含3至12个碳的环状烃、优选含5至10个碳和1或2个双键。示例性环烯基包括环戊烯基、环己烯基、环庚烯基、环辛烯基、环己二烯基和环庚二烯基,所述环烯基可任选被取代,如环烷基定义。
在本文中单独或作为另一基团一部分使用的术语“烷酰基”指与羰基连接的烷基。
除非另有说明,否则在本文中本身或作为另一基团一部分使用的术语“低级烯基”指2至8个碳的直链或支链基团,在本文中本身或作为另一基团一部分使用的术语“烯基”指2至20个碳的直链或支链基团,在直链中优选2至12个碳,更优选2至8个碳,其在直链中包括1至6个双键,例如乙烯基、2-丙烯基、3-丁烯基、2-丁烯基、4-戊烯基、3-戊烯基、2-己烯基、3-己烯基、2-庚烯基、3-庚烯基、4-庚烯基、3-辛烯基、3-壬烯基、4-癸烯基、3-十一碳烯基、4-十二碳烯基、4,8,12-十四碳三烯基等,且其可任选被1至4个以下的取代基取代:即卤素、卤代烷基、烷基、烷氧基、烯基、炔基、芳基、芳基烷基、环烷基、氨基、羟基、杂芳基、杂环烷基、烷酰基氨基、烷基酰胺基、芳基羰基氨基、硝基、氰基、巯基、烷硫基和/或本文中描述的任何烷基取代基。
除非另有说明,否则在本文中本身或作为另一基团一部分使用的术语“低级炔基”指2至8个碳的直链或支链基团,和在本文中本身或作为另一基团一部分使用的术语“炔基”指2至30个碳的直链或支链基团,在直链中优选2至12个碳,更优选2至8个碳,例如2-丙炔基、3-丁炔基、2-丁炔基、4-戊炔基、3-戊炔基、2-己炔基、3-己炔基、2-庚炔基、3-庚炔基、4-庚炔基、3-辛炔基、3-壬炔基、4-癸炔基、3-十一碳炔基、4-十二碳炔基等,其可任选被1至4个以下的取代基取代:即卤素、卤代烷基、烷基、烷氧基、烯基、炔基、芳基、芳基烷基、环烷基、氨基、杂芳基、杂环烷基、羟基、烷酰基氨基、烷基酰胺基、芳基羰基氨基、硝基、氰基、巯基和/或烷硫基和/或本文中描述的任何烷基取代基。
单独或作为另一基团一部分使用的术语“芳基烷基”、“芳基烯基,”和“芳基炔基”指具有芳基取代基的上述烷基、烯基和炔基。
当定义同上的烷基在两个不同碳原子上具有与其它基团连接的单键时,称其为“亚烷基”,可任选被取代,如以上“烷基”定义。
当定义同上的烯基和定义同上的炔基在两个不同碳原子上分别具有用于连接的单键时,其分别称为“亚烯基”和“亚炔基”,且可任选被取代,如以上“烯基”和“炔基”定义。
本文中定义的合适的亚烷基、亚烯基或亚炔基(CH2)s或(CH2)r(其中r为1至8,优选1至5,且s为1至5,优选1至3,包括亚烷基、亚烯基或亚炔基)可任选包括1、2或3个取代基,所述取代基包括烷基、烯基、卤素、氰基、羟基、烷氧基、氨基、硫代烷基、酮基、C3-C6环烷基、烷基羰基氨基或烷基羰基氧基。
在本文中单独或作为另一基团一部分使用的术语“卤素”或“卤代”指氯、溴、氟和碘。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“芳基”指在环部分含6至10个碳的单环和双环芳基(如苯基或包括1-萘基和2-萘基的萘基),可任选包括1至3个稠合到碳环或杂环的其它环(如芳基、环烷基、杂芳基或杂环烷基环,例如通过可用碳原子可任选被1、2或3个选自以下的基团取代:氢、卤素、卤代烷基、烷基、烷氧基、卤代烷氧基、烯基、三氟甲基、三氟甲氧基、炔基、环烷基-烷基、杂环烷基、杂环烷基烷基、芳基、杂芳基、芳基烷基、芳氧基、芳氧基烷基、芳基烷氧基、烷氧基羰基、芳基羰基、芳基烯基、氨基羰基芳基、芳硫基、芳基亚磺酰基、芳基偶氮基、杂芳基烷基、杂芳基烯基、杂芳基杂芳基、杂芳基氧基、羟基、硝基、氰基、氨基、其中所述氨基包括1或2个取代基(为烷基、芳基或定义中提及的任何其它芳基化合物)的取代氨基、巯基、烷硫基、芳硫基、杂芳基硫基、芳硫基烷基、烷氧基芳硫基、烷基羰基、芳基羰基、烷基氨基羰基、芳基氨基羰基、烷氧基羰基、氨基羰基、烷基羰基氧基、芳基羰基氧基、烷基羰基氨基、芳基羰基氨基、芳基亚磺酰基、芳基亚磺酰基烷基、芳基磺酰基氨基和芳基磺酰氨基羰基和/或本文中描述的任何烷基取代基。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“低级烷氧基”、“烷氧基”、“芳氧基”或“芳烷氧基”包括任何与氧原子连接的上述烷基、芳烷基或芳基。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“取代氨基”指被一个或两个取代基取代的氨基,所述取代基可以相同或不同,例如为烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、杂环烷基、杂环烷基烷基、环烷基、环烷基烷基、卤代烷基、羟基烷基、烷氧基烷基和硫代烷基。这些取代基可进一步被羧酸和/或上述任何烷基取代基取代。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“低级烷硫基”、“烷硫基”、“芳硫基”或“芳烷硫基”包括任何与硫原子连接的上述烷基、芳烷基或芳基。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“低级烷基氨基”、“烷基氨基”、“芳基氨基”或“芳基烷基氨基”包括任何与氮原子连接的上述烷基、芳基或芳基烷基。
除非另有说明,否则在本文中本身或作为另一基团一部分使用的术语“酰基”如本文定义指与羰基(C=O)连接的有机基团;酰基的实例包括与羰基连接的任何烷基取代基,例如烷酰基、烯酰基、芳酰基、芳烷酰基、杂芳酰基、环烷酰基、杂环烷酰基等。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“杂环烷基”指5元、6元或7元饱和或部分饱和环,所述环包括通过碳原子或杂原子连接1至2个杂原子如氮、氧和/或硫,如果可能任选通过连接(CH2)r(其中r为1、2或3)连接。以上基团可包括1至4个取代基例如烷基、卤素、氧代基和/或本文中描述的任何烷基取代基。另外,任何杂环烷基环可稠合到环烷基、芳基、杂芳基或杂环烷基环上。
除非另有说明,否则在本文中单独或作为另一基团一部分使用的术语“杂芳基”指5元或6元芳环,其包括1、2、3或4个例如氮、氧或硫的杂原子;包括稠合到芳基、环烷基、杂芳基或杂环烷基环(例如苯并噻吩基或吲哚基)上的这些环;和包括可能的N-氧化物。杂芳基可任选包括1至4个取代基例如上述任何烷基取代基。
在本文中单独或作为另一基团一部分使用的术语“杂环烷基烷基”指通过C原子或杂原子与(CH2)r链连接的定义同上的杂环烷基。
在本文中单独或作为另一基团一部分使用的术语“杂芳基烷基”或“杂芳基烯基”指通过C原子或杂原子连接到定义同上的-(CH2)r-链、亚烷基或亚烯基的定义同上的杂芳基。
本文中使用的术语“5元、6元或7元碳环或杂环”指定义同上的环烷基或环烯基,或定义同上的杂芳基或杂环芳基,例如噻二唑、四唑、咪唑或噁唑。
本文中使用的术语“多卤代烷基”指定义同上的“烷基”,其包括2至9个、优选2至5个卤素取代基,如F或Cl、优选F,如CF3CH2、CF3或CF3CF2CH2。
本文中使用的术语“多卤代烷氧基”指定义同上的“烷氧基”或“烷氧基”基团包括2至9个、优选2至5个卤素取代基,例如F或Cl、优选F,例如CF3CH2O、CF3O或CF3CF2CH2O。
在一个具体方面,本发明涉及一种新的制备式(I)化合物的方法,
其中:
R1选自氢、羟基、溴、氯、氟、烷基、烷氧基、烷硫基和芳硫基,其中p为1至4,且条件是当溴、氯和氟存在时,只能在3-、4-和5-位中至少一个位置上存在;
R2选自氢、羟基、氯、氟、烷基、烷氧基和烷硫基,其中q为1至5;和
A选自共价键、O、S、NH和(CH2)n,其中n为1至3,且
条件是当A在4-位时,R1不为溴;
条件是当R1中的一个为溴;和
如果A在3-或6-位,则溴在5-位,
如果A在2-或5-位,则溴在3-位,和
当溴在3-位,且A在2-或5-位时,则在4-和6-位的R1基团相同,且不为溴、氯或氟,和
当溴在5-位,且A在3-或6-位时,则在2-和4-位的R1基团相同,且不为溴、氯或氟。
在一个优选的实施方案中,本发明的新方法非常适用于制备优选的式(IA)所包括化合物及其药学上可接受的络合物,
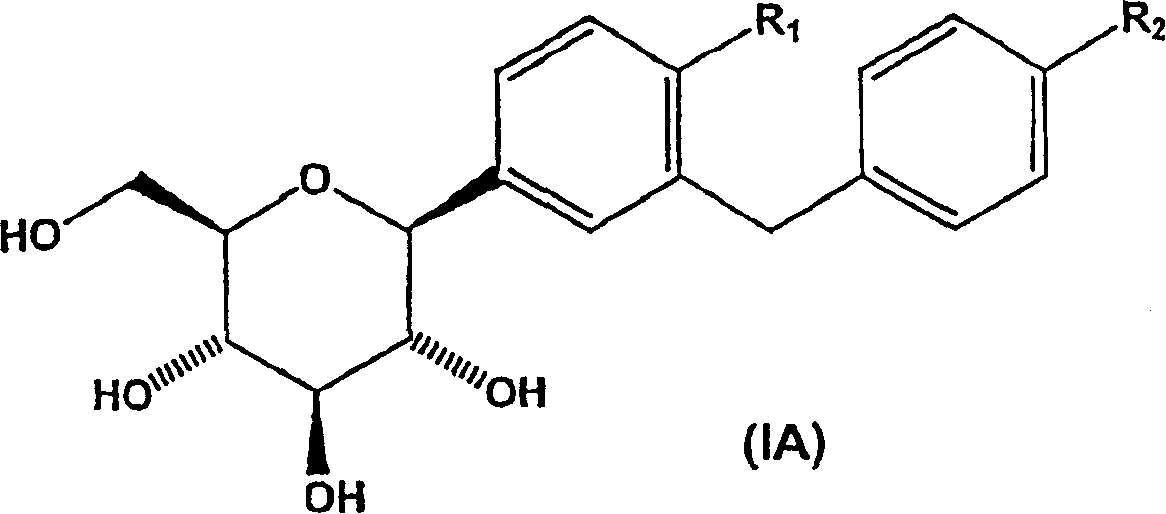
其中:
R1选自氢、烷基、氯;和
R2选自氢、烷基、烷氧基、烷硫基。
在一个特别优选的实施方案中,本发明的新方法尤其非常适用于制备更优选的上述式(IA)化合物及其药学上可接受的络合物,其中:
1)R1为氢,R2为乙基;
2)R1为氯,R2为乙氧基;和
3)R1为甲基,R2为甲硫基。
式(I)化合物具有温血动物包括人的肠和肾中钠依赖性葡萄糖转运蛋白抑制剂活性,因此可用于治疗糖尿病和糖尿病微血管和大血管并发症例如视网膜病、神经病、肾病和伤口愈合。另外,还发现式(I)化合物治疗或延缓糖尿病发作或发展特别有效,尤其是I型和II型糖尿病包括糖尿病并发症例如视网膜病、神经病、肾病、延迟性伤口愈合以及相关疾病例如胰岛素抗性与葡萄糖体内平衡损害(IGH)、高血糖、高胰岛素血症、高血脂肪酸或甘油水平、肥胖、高脂血症包括高甘油三酯血症、X综合征、高血压、动脉粥样硬化及相关疾病,以及对提高高密度脂质水平特别有效。统称为“X综合征”(又称为代谢综合征)的病症和疾病详见Johannsson,J.Clin.Endrocrinol.Metab.,82,727-34(1997),该文献通过引用结合到本文。
本发明还涉及新的中间体化合物,所述中间体可用于制备式(I)化合物。本发明新的中间体化合物包括:
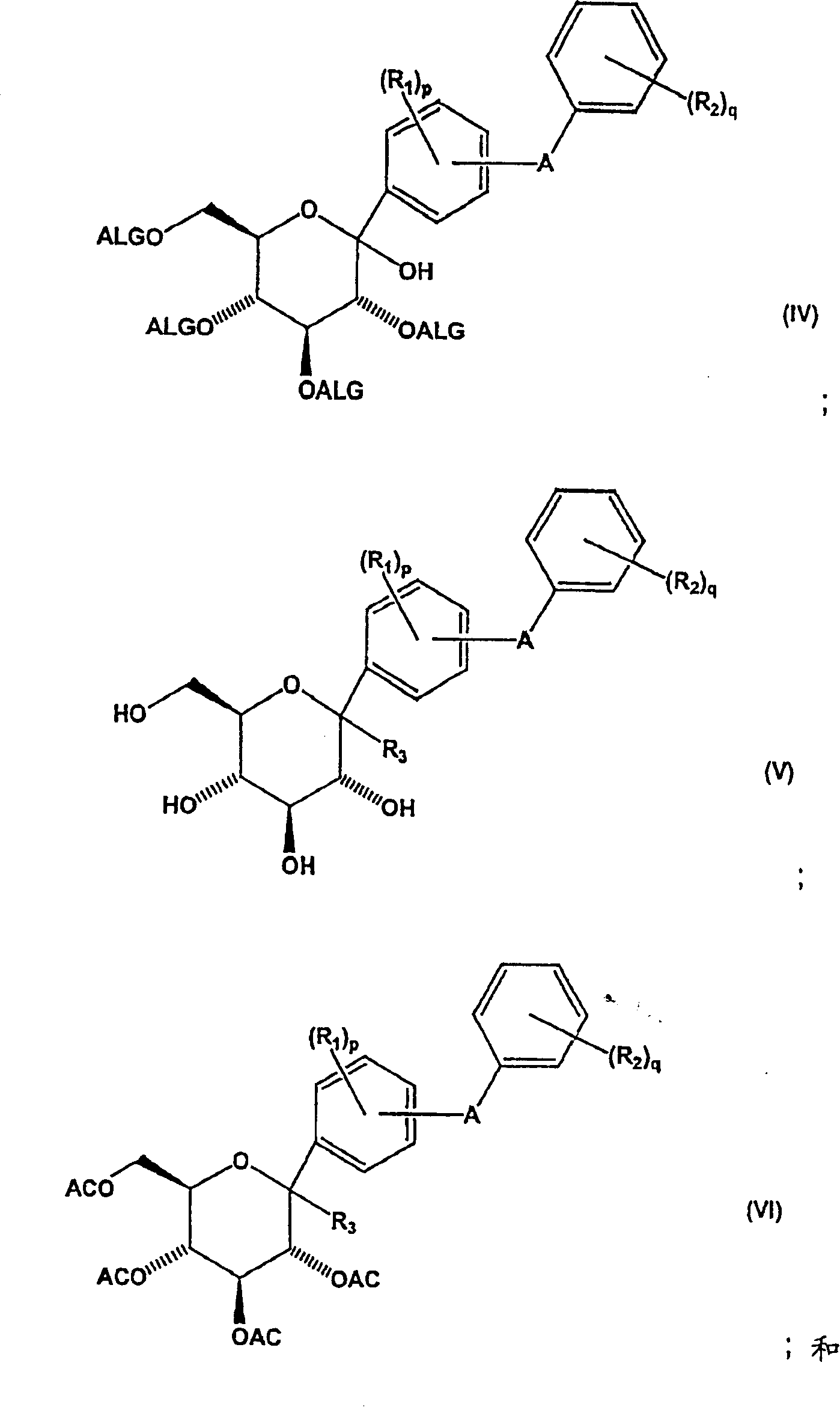
其中:
R1、R2、A、p和q定义同上述式(I)化合物;
R3选自烷氧基、烯氧基、烷硫基和烯硫基;
AC为酰基保护基团,例如CH3CO-;且
ALG为酸不稳定保护基团。
优选用于保护羟基的酸不稳定保护基团选自例如甲氧基甲基醚、甲硫基甲基醚、2-甲氧基乙氧基甲基醚、二(2-氯乙氧基)甲基醚、四氢吡喃醚、四氢噻喃醚、4-甲氧基四氢吡喃醚、4-甲氧基四氢噻喃醚、四氢呋喃醚、四氢噻吩醚、1-乙氧基乙基醚、1-甲基-1-甲氧基乙基醚、2-(苯基氧硒基(selenyl))乙基醚、叔丁基醚、烯丙基醚、三苯基甲基醚、α-萘基二苯基甲基醚、对甲氧基苯基二苯基甲基醚、三烷基甲硅烷基醚例如三甲基甲硅烷基醚和三乙基甲硅烷基醚、异丙基二甲基甲硅烷基醚、叔丁基二甲基甲硅烷基醚和叔丁基二苯基甲硅烷基醚。
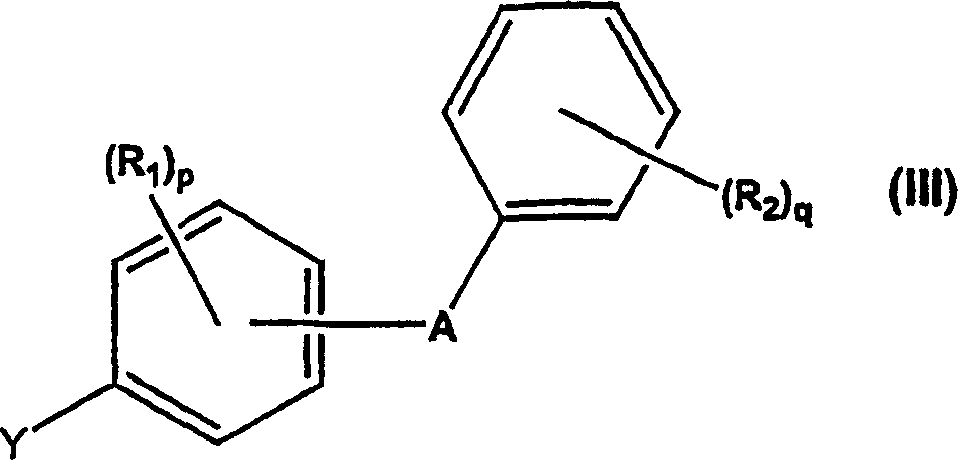
在本发明的一个具体方面,制备式(I)化合物的方法通常通过使具有羟基被酸不稳定保护基团(ALG)保护的葡糖酸-1,5-内酯与式(III)化合物反应进行
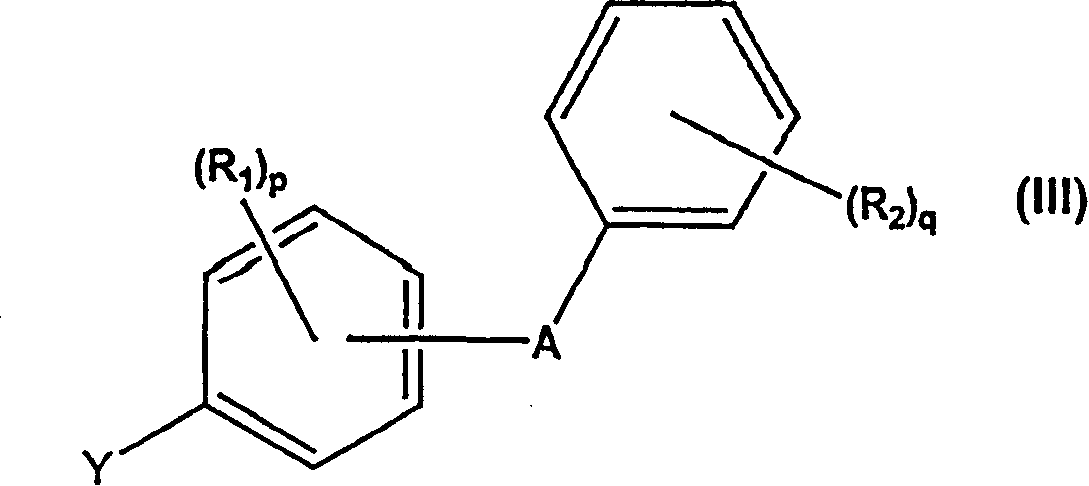
其中Y为金属,优选选自碱金属和碱土金属,得到新的式(IV)中间体化合物。然后用苷化试剂(例如甲磺酸)在亲核化合物例如醇存在下处理式(IV)中间体化合物,从而除去ALG保护基团,用定义同上的R3基团在异头碳-位取代羟基,得到新的式(V)苷中间体化合物。
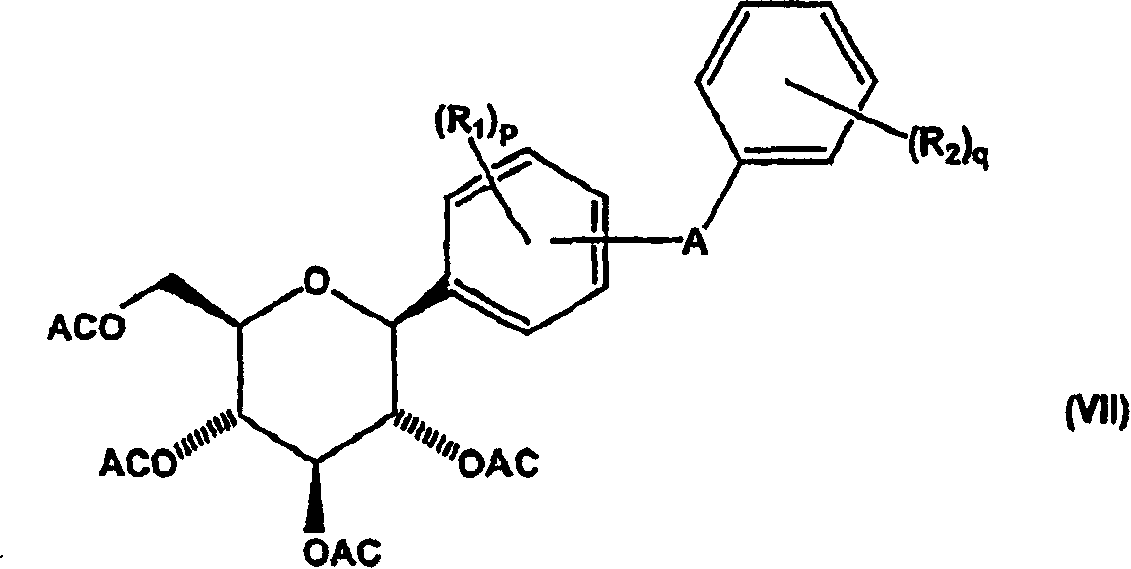
然后使式(V)中间体化合物与酰化剂反应,得到新的酰基保护的式(VI)中间体化合物。然后在活化剂(例如路易斯(Lewis)酸)存在下,用还原剂(例如甲硅烷)还原式(VI)中间体化合物,得到新的式VII中间体化合物。申请人发现本发明方法中的还原反应对需要的如式(VII)所示中间体化合物的β-芳基异构体具有非常高的立体有择性,其中与异头碳键合的二苯基取代基取向为平伏位置,相对于在吡喃糖环上的相邻酰基取代基为反式。
然后使式(VII)中间体化合物与酰基保护基团除去试剂反应使羟基脱保护,得到需要的最终式(I)β-D-吡喃葡萄糖化合物。
可任选用选自美国专利申请10/117,914中公开的氨基酸类络合物形成试剂例如L-苯丙氨酸进一步处理最终式(I)化合物,得到相应的式(I)化合物的结晶络合物,该专利申请通过引用结合到本文。
流程1

按照本发明的一个方面,可按流程1所示优选在溶剂例如甲苯存在下,如在低温下(例如-78℃)通过使式(II)化合物与式(III)化合物偶合制备新的式(IV)中间体化合物。在加入式(II)内酯化合物前,可将式(III)化合物活化以便在例如低温(例如-78℃)下、在溶剂例如THF中、在例如甲苯存在下与例如烷基-(碱金属)化合物例如n-BuLi和t-BuLi或烷基-(碱土金属)化合物偶合。
可通过用酸不稳定保护基团供给试剂(ALG-R)处理D-葡糖酸-1,5-内酯,通过酸不稳定保护基团(ALG)为羟基提供保护,从而制备式(II)化合物。
优选的酸不稳定保护基团可选自甲氧基甲基醚、甲硫基甲基醚、2-甲氧基乙氧基甲基醚、二(2-氯乙氧基)甲基醚、四氢吡喃醚、四氢噻喃醚、4-甲氧基四氢吡喃醚、4-甲氧基四氢噻喃醚、四氢呋喃醚、四氢噻吩醚、1-乙氧基乙基醚、1-甲基-1-甲氧基乙基醚、2-(苯基氧硒基)乙基醚、叔丁基醚、烯丙基醚、三苯基甲基醚、α-萘基二苯基甲基醚、对-甲氧基苯基二苯基甲基醚、三烷基甲硅烷基醚例如三甲基甲硅烷基醚和三乙基甲硅烷基醚,例如异丙基二甲基甲硅烷基醚、叔丁基二甲基甲硅烷基醚、叔丁基二苯基甲硅烷基醚及其组合。
更优选的酸不稳定保护基团选自甲氧基甲基醚、2-甲氧基乙氧基甲基醚、四氢吡喃醚、三甲基甲硅烷基醚、异丙基二甲基甲硅烷基醚、叔丁基二甲基甲硅烷基醚、叔丁基二苯基甲硅烷基醚及其组合。
在本发明中,合适的酸不稳定基团供给试剂可包括任何能够提供上述定义的用于保护羟基的相应ALG保护基团的试剂。这种试剂可包括但不限于氯化三甲基甲硅烷、三甲基甲硅烷基三氟甲磺酸、甲氧基甲基氯化物、苄氧基甲基氯化物、氯化三乙基甲硅烷、二氢呋喃、四氢吡喃等。
在本发明的再一方面,可按流程1所示例如在选自硫醇例如烷硫醇或烯硫醇的亲核化合物和例如醇例如甲醇、丁醇、乙醇、正丙醇和异丙醇存在下,通过用苷化试剂处理式(IV)中间体化合物制备新的式(V)中间体化合物,所述苷化试剂选自无机酸如盐酸、硫酸、硝酸等;有机酸如甲酸、乙酸、三氟乙酸、甲磺酸等;以及路易斯酸如三氟化硼乙醚合物、三氟甲磺酸钪(III)、异丙醇钛(IV)、氯化锡(IV)、溴化锌(II)和氯化锌(II)。通过除去酸不稳定保护基团(ALG),该苷化试剂能够促使羟基脱保护。
在本发明的再一方面,可按流程1所示通过用酰化剂处理式(V)中间体化合物制备新的式(VI)中间体化合物,所述酰化剂选自与式(V)化合物的羟基反应的酰基衍生物、酰卤如乙酰氯等;酸酐例如乙酸酐、丙酸酐等。优选酰化反应在以下条件下进行:例如在用于提高酰化反应反应活性的碱的存在下,例如三乙胺、三甲胺、N-N′-二异丙基乙胺(DIPEA)、吡啶和4-二甲氨基吡啶(DMAP);和反应溶剂例如甲苯中。
在本发明的再一方面,可按流程1所示,例如优选在活化基团包括路易斯酸例如BF3·Et2O和反应溶剂例如CH3CN、CH3CN/甲苯混合物或CH3CN/二氯甲烷混合物存在下,在环境温度(例如15℃)下通过用还原剂处理式(VI)化合物制备新的式(VII)中间体化合物,所述还原剂选自甲硅烷包括烷基甲硅烷,例如优选三烷基甲硅烷例如Et3SiH。
在本发明的再一方面,可按流程1所示,通过使式(VII)化合物在碱性条件下与选自碱例如NaOH的AC保护基团脱去试剂反应,促使羟基脱保护制备式(I)化合物。优选该反应在选自醇例如乙醇的溶剂存在下进行。
按照本发明,可按以下反应流程及其描述方法制备式(I)化合物。可理解有典型或优选的方法条件(即反应温度、时间、反应物的摩尔比、溶剂、压力等)给出时,除非另外说明,否则也可使用其它的方法条件。这些反应的示例性试剂和方法在下文和操作实施例中出现。
以下反应流程中的保护和脱保护可按本邻域通常已知的方法(见例如Greene,T.W.和Wuts,P.G.M.,Protecting Groups in OrganicSynthesis,第3版,1999[Wiley]),用本文所述的合适试剂进行。
流程2
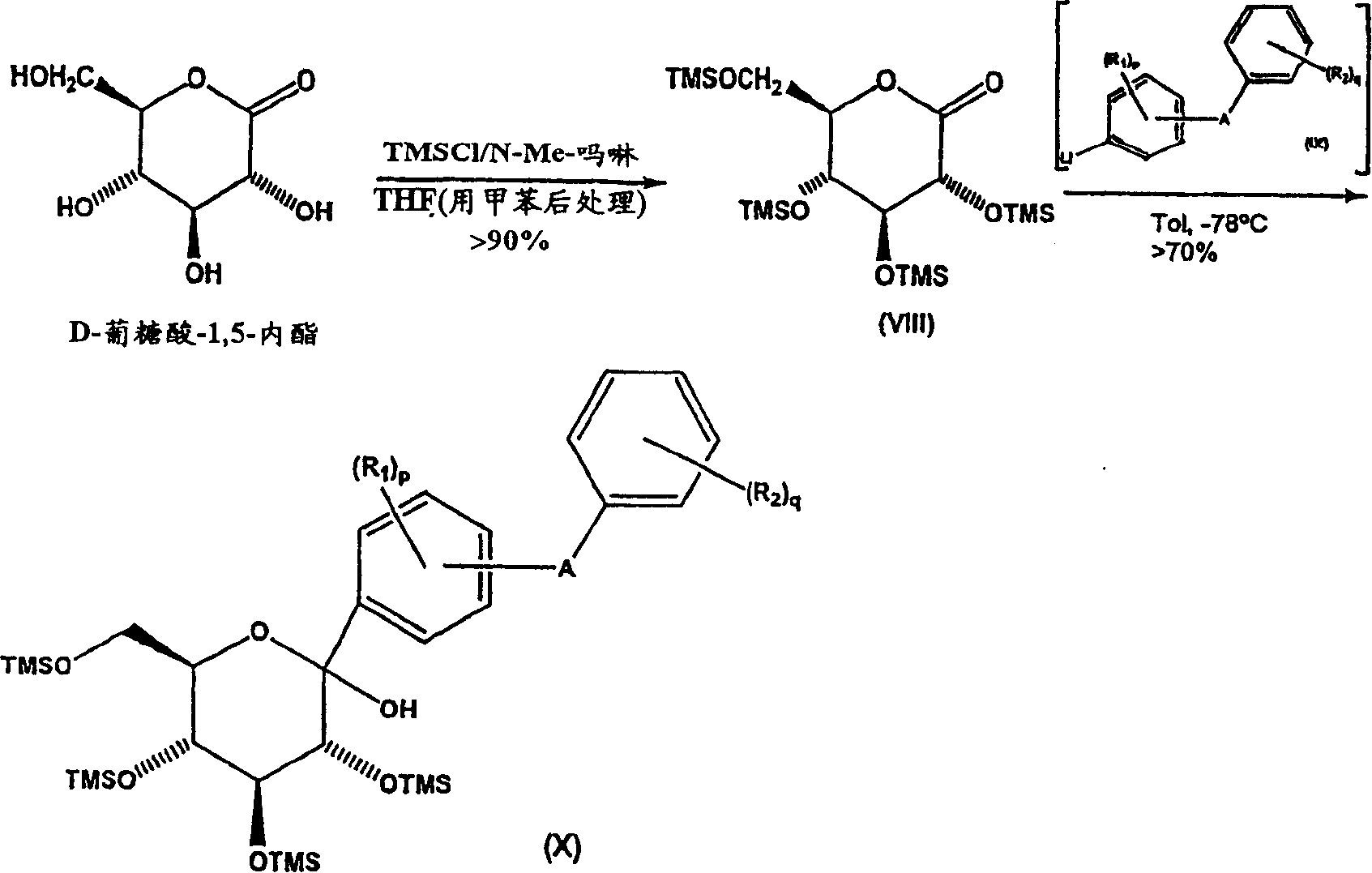
按照本发明,提供如流程2所示的一种制备式(VIII)中间体2,3,4,6-四-O-(取代甲硅烷基)-D-葡糖酸-1,5-内酯的方法,通过使D-葡糖酸-1,5-内酯和酸不稳定基团供给试剂(ALG-R)反应,优选含甲硅烷基的酸不稳定基团供给试剂,更优选卤化烷基甲硅烷化合物,最优选卤化三烷基甲硅烷化合物(例如氯化三甲基甲硅烷(TMSCl)),在碱例如N-Me-吗啉和非质子溶剂例如四氢呋喃存在下生成式(VIII)中间体化合物2,3,4,6-四-O-(取代甲硅烷基)-D-葡糖酸-1,5-内酯。该反应步骤最好为D-葡糖酸-1,5-内酯的那些欲保留(即不反应)羟基提供作为保护基团的酸不稳定保护基团(ALG),在随后的反应过程中制备终产物。
在反应溶剂例如甲苯存在下,在例如约-78℃下使得到的式(VIII)中间体化合物与式(IX)锂化阴离子偶合得到式(X)中间体化合物。
流程3

如流程3所示,在金属供体通常为碱金属或碱土金属供体例如锂供体,例如优选选自烷基-(碱金属)化合物包括烷基锂化合物,优选正丁基锂、仲丁基锂或叔丁基锂,和溶剂例如四氢呋喃∶甲苯或四氢呋喃∶庚烷(1∶4)存在下,通过其中X选自溴和碘的式(XIII)化合物原料的反应制备式(IX)锂化阴离子。
流程4

然后通过在异头碳-位用选自以下的取代基置换羟基使所得式(X)中间体化合物进行苷化:烷硫基、烯硫基、烯氧基和烷氧基,优选甲氧基。在本发明的一个优选的实施方案中,按照流程4,用酸优选甲磺酸处理式(X)化合物,在醇优选甲醇存在下除去含甲硅烷基的酸不稳定保护基团三甲基甲硅烷基(TMS),将异头碳羟基转化为甲氧基,得到式(XI)中间体化合物。
流程5

然后,通过选自以下的酰化剂使式(XI)中间体化合物的活性羟基酰化:酰基衍生物、酰卤例如乙酰氯等;酸酐例如乙酸酐、丙酸酐等。在本发明的一个优选的实施方案中,使式(XI)中间体化合物在N-N′-二异丙基乙胺(DIPEA)和4-二甲基氨基吡啶(DMAP)和溶剂例如甲苯存在下与乙酸酐反应,得到流程5所示式(XII)中间体化合物。
流程6
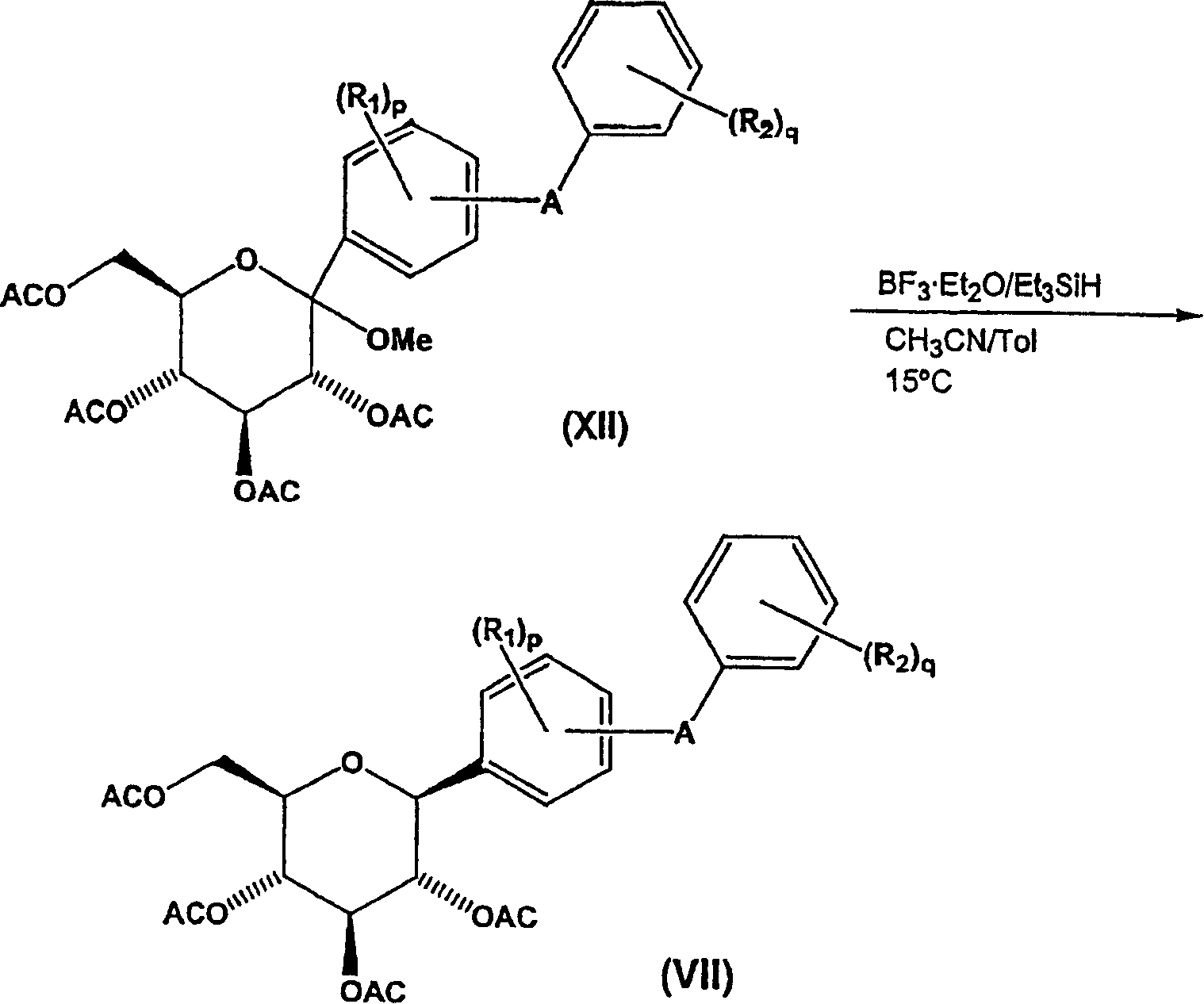
然后,如流程6所示,通过用还原剂例如甲硅烷,例如优选烷基甲硅烷,更优选三乙基甲硅烷处理化合物以除去式(XII)中间体化合物的甲氧基,得到式(VII)中间体化合物。优选该还原反应在活化基团例如BF3·Et2O和溶剂例如CH3CN、CH3CN/甲苯或CH3CN/二氯甲烷存在下,在约15℃下进行。如流程6所示,转化式(XII)化合物,生成式(VII)代表化合物的立体有择性β-芳基形式。
流程7
然后,如流程7所示,用选自碱,优选氢氧化钠的酰基保护基团脱去试剂处理式(VII)β-芳基中间体化合物以恢复羟基,得到需要的终产物式(I)化合物。优选在溶剂例如乙醇存在下进行酰基除去反应。
可任选用络合物形成试剂进一步处理需要的式(I)终产物,所述络合物形成试剂选自美国专利申请序号10/117,914中公开的氨基酸类例如L-苯丙氨酸,得到相应的式(I)化合物结晶络合物,该专利申请通过引用结合到本文。优选在溶剂例如水和乙醇的混合物的存在下进行该络合物形成反应。
适用于本发明最后脱保护步骤方法的碱包括但不限于碱金属硼氢化物氢化物例如硼氢化钠、氢化锂铝等;碱金属氢氧化物例如氢氧化钠、氢氧化钾等;醇盐例如甲醇钠、乙醇钠、叔丁醇钾等;碳酸钠和碳酸钾;和胺包括叔胺例如4-甲基吗啉、三乙胺、N-N′-二异丙基乙胺(DIPEA)等。优选的碱包括氢氧化钠和氢氧化钾;和芳胺例如咪唑、吡啶等。适用于本发明葡糖酸内酯甲硅烷基化步骤方法的碱包括叔胺例如4-甲基吗啉、三乙胺、N-N′-二异丙基乙胺(DIPEA)等以及芳胺例如咪唑、吡啶等。优选的碱包括叔胺,特别优选4-甲基吗啉。
适用于本发明芳基锂偶合和苷还原方法的非质子反应溶剂包括但不限于醚例如二氧六环、四氢呋喃等;芳烃例如苯、甲苯、二甲苯等;酯例如乙酸乙酯等;卤代烃例如氯仿、二氯甲烷等;腈例如乙腈等;酰胺例如N,N-二甲基甲酰胺、N,N-二甲基乙酰胺等以及亚砜例如二甲基亚砜等。优选的用于偶合反应的非质子溶剂为甲苯和四氢呋喃,这些溶剂的4∶1混合物为尤其优选的介质。而优选的用于苷还原步骤的非质子溶剂为CH3CN。
最重要的是发现与其它保护基团例如苄基相比,上述酸不稳定保护基团,尤其是那些含甲硅烷基的基团能使反应过程按一锅法操作进行,而形成较少的必须分离的中间体化合物。优选酸不稳定保护基团与D-葡糖酸-1,5-内酯的羟基反应,且优先保持与此部位键合直至需要除去该保护基团。
酸不稳定保护基团,尤其是本发明的三甲基甲硅烷基由于低成本和使用容易获得原料的结果非常易于大规模合成。此外,在本发明方法中使用该酸不稳定保护基团提供较为安全的合成方法,使不利的反应条件减至最小,而与使用苄基保护基团的现有技术方法相比,促进提高的立体有择性(35∶1)和产物纯度。
酸不稳定保护基团供给试剂的量通常大于化学计算量,并需要足够保护所有D-葡糖酸-1,5-内酯的目标羟基(合成期间不反应的羟基)。优选摩尔比大于4.0,更优选约6.0为必要的。优选保护基团反应在不超过10℃的温度下进行。
在本发明的一个优选的实施方案中,通过先将D-葡糖酸-1,5-内酯和非质子溶剂混合,然后加入碱,优选基于D-葡糖酸-1,5-内酯的量计算为约5至8摩尔当量,制备如流程2所示式(VIII)中间体化合物。然后加入酸不稳定基团供给试剂,优选加入6摩尔当量,得到式(VIII)中间体化合物,然后使其与式(IX)的锂化阴离子反应。
形成式(VIII)中间体化合物通常在约0℃至50℃,优选约35℃下进行直至反应完成,通常需约5小时。然后将反应混合物冷却,用溶剂稀释,用缓冲剂例如磷酸二氢钠中和,洗涤和蒸馏,得到式(VIII)中间体化合物。
然后,使式(VIII)中间体化合物与式(IX)锂化阴离子反应,得到式(X)中间体化合物,式(IX)锂化阴离子通过以下反应得到:使如流程3所示式(XIII)化合物的非质子溶剂溶液在低温通常-70℃以下,在惰性气氛下与烷基锂化合物优选正烷基锂化合物反应,更优选与略微大于化学计算量的正丁基锂反应。
使式(X)中间体化合物在亲核化合物例如低级醇例如优选甲醇、丁醇、乙醇、正丙醇、异丙醇等存在下与苷化试剂例如硫酸、盐酸、甲磺酸、对-甲苯磺酸及三氟甲磺酸例如优选甲磺酸反应。该反应在约40℃下进行。然后例如在30℃以下用猝灭剂例如碳酸氢钠猝灭反应溶液,得到流程4所示式(XI)中间体化合物。
通过过量的酰基供体例如乙酸酐将式(XI)中间体化合物转化为酰化衍生物。酰基供体的量应该足够提供使羟基最充分转化,通常为基于式(XI)化合物量计算为约4至6当量。例如反应在过量N-N′-二异丙基乙胺(DIPEA)和催化剂例如4-二甲基氨基吡啶(DMAP)存在下进行。反应温度通常维持在约35℃以下直至反应完成。然后例如按流程5所示,通过加入酸例如磷酸将反应pH降为约3或更低来猝灭反应。
在该方法的这一步,用还原剂处理式(XII)化合物,除去吡喃葡糖苷环上异头碳位的甲氧基。申请人确定最好用基于甲硅烷基的还原剂,优选三烷基甲硅烷,更优选三乙基甲硅烷,在酸的存在下将式(XII)吡喃葡糖苷化合物还原为式(VII)吡喃葡萄糖化合物,具有优异立体有择性约98%,收率通常为约80-90%。优选该还原反应按流程6所示在三氟化硼-乙醚合物(BF3·Et2O)和乙腈(CH3CN)存在下进行。与相应的苄基保护的中间体最明显不同地是,用乙酰基保护基团能使还原步骤不使用高度受阻甲硅烷、在非低温(crygenic)条件下进行,并具有优异立体有择性。
然后,用前述碱例如氢氧化钠或氢氧化锂,在溶剂例如低级醇(例如乙醇)存在下处理式(VII)中间体化合物,得到式(I)终产物。可任选使所得的式(I)化合物在合适的溶剂例如低级醇和水中,与选自氨基酸例如L-苯丙氨酸的络合物形成试剂反应,得到相应的结晶络合物例如L-苯丙氨酸络合物如式(I)化合物的L-苯丙氨酸络合物。
在本发明的再一方面,可使用上述方法制备式(I)C-芳基葡糖苷化合物,包括1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖、1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-基)-β-D-吡喃葡萄糖、1-C-(6-氯-4′-乙氧基二苯基甲烷-3-基)-β-D-吡喃葡萄糖及其络合物。
在本发明的一个更优选的实施方案中,用与上述步骤基本上相同的步骤制备1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖。具体地说,通过使式(XIII)的3-溴-4′-甲基二苯基甲烷与正烷基锂反应形成与式(VIII)2,3,4,6-四-O-(取代甲硅烷基)-D-葡糖酸-1,5-内酯反应的、其中R1为氢,R2为4′-乙基和A为-CH2-的式(IX)锂化阴离子。
在本发明的一个更优选的实施方案中,用与上述步骤基本上相同的步骤制备1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-基)-β-D-吡喃葡萄糖。具体地说,通过使式(XIII)的3-溴-4-甲基-4′-(甲硫基)二苯基甲烷与正烷基锂反应形成与式(VIII)2,3,4,6-四-O-(取代甲硅烷基)-D-葡糖酸-1,5-内酯反应的、其中R1为4-甲基,R2为4′-(甲硫基)和A为-CH2-的式(IX)锂化阴离子。
在本发明的一个更优选的实施方案中,用与上述步骤相同的步骤制备1-C-(6-氯-4′-乙氧基二苯基甲烷-3-基)-β-D-吡喃葡萄糖。具体地说,通过使式(XIII)3-溴-4-氯-4′-乙氧基二苯基甲烷与正烷基锂反应形成与式(VIII)2,3,4,6-四-O-(取代甲硅烷基)-D-葡糖酸-1,5-内酯反应的、其中R1为4-氯,R2为4′-乙氧基,A为-CH2-的式(IX)锂化阴离子。
在本领域已知式(XIII)化合物的原料,并可用本领域普通技术人员已知的标准方法容易地制备。
提供以下实施例仅为举例说明本发明,无意对本发明进行限定。例如最佳反应条件可随具体使用的反应物或溶剂而改变,但这种反应条件可由本领域的普通技术人员通过常规优化方法确定。
实施例1
制备2,3,4,6-四-O-三甲基甲硅烷基-1-C-(6-甲基-4′-(甲硫基)二苯基
甲烷-3-基)-α-D-吡喃葡萄糖

在1 L单颈圆底烧瓶中,将芳基溴化合物(1)(20.7g,67.4mmol,1.1当量)溶于四氢呋喃(THF)(61mL)和庚烷(245mL),冷却至-78℃,产生沉淀。在20分钟内向该多相反应混合物中滴加入2.3M n-BuLi(29.3mL,67.4mmol),产生微红色。30分钟后,在-78℃下将反应混合物移入盛有三甲基甲硅烷基内酯化合物(2)(29.5g,63.2mmol,1当量)和庚烷(306mL)的2-L单颈烧瓶中,得到无任何沉淀的混浊混合物。将反应混合物从冷浴中移开,用1%AcOH(290mL)猝灭,然后移至分液漏斗中。加入200ML乙酸乙酯(EtOAc),分离各层。有机层用水(1×200mL)和盐水(2×200mL)洗涤。水层用EtOAc反萃取。经TLC分析(约750mLEtOAc)已检测不到TMS保护的化合物(3)。合并的有机层经MgSO4干燥,过滤,浓缩,得到48g黄色泡沫状油,将其搅拌,在高真空下干燥约0.5小时。所得TMS保护的化合物(3)用于下一个实施例。
实施例2
制备甲基-1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-基)-α-D-吡喃葡
萄糖
将实施例1的TMS保护的化合物(3)(48g)溶于MeOH(196mL),然后加入甲磺酸(200μL)。将所得溶液升温至40℃,保持约20分钟。然后将溶液冷却至室温,浓缩。将残余物溶于EtOAc(200mL),用饱和NaHCO3水溶液(2×100mL)和盐水(2×100mL)洗涤。合并的水层用EtOAc(2×100mL)反萃取,合并的有机层经Na2SO4干燥,过滤,浓缩。使残余物在高真空下干燥过夜,然后在60℃下溶于甲苯(约75mL)。将所得混合物滴加到盛有450mL庚烷的圆底烧瓶中,产生白色沉淀。在室温下搅拌该混合物约3小时,然后过滤,得到27g白色固体化合物(4)。经HPLC分析,该白色固体的纯度为87%。滤液进行HPLC分析,证明没有化合物(4)。
实施例3
制备1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-基)-β-D-吡喃葡萄糖
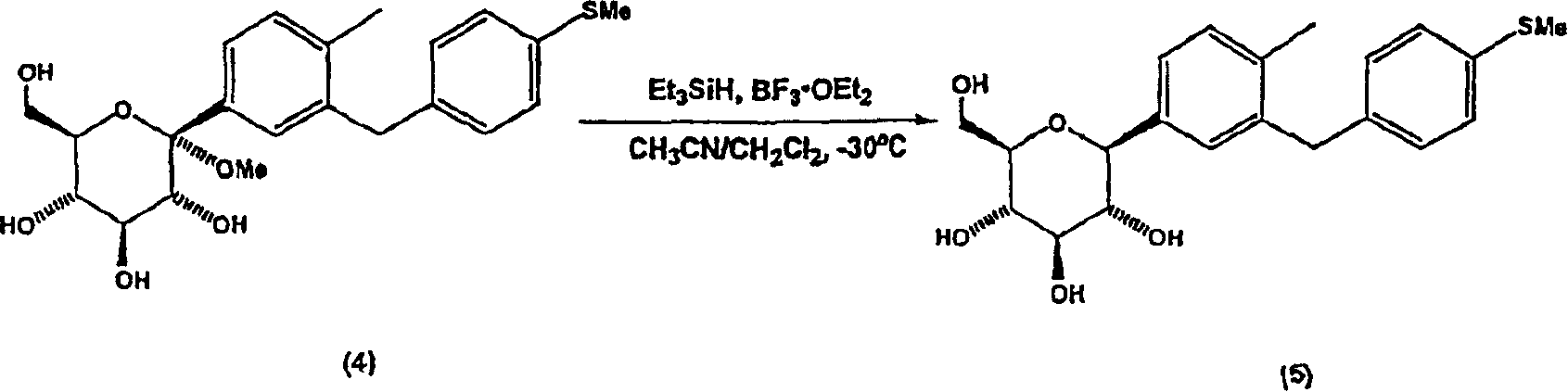
将实施例2的化合物(4)一分为二制备两份反应混合物。在第一个1-L圆底烧瓶中,将化合物(4)(13g,30.9mmol,1当量)溶于CH2Cl2(86mL)和CH3CN(223mL)中,冷却至-20℃,使原料沉淀。边搅拌,边加入Et3SiH(9.9mL,61.8mmol,2当量),随后加入BF3·OEt2(5.9mL,46.4mmol,1.5当量)。在第二个1-L圆底烧瓶中,通过将11g化合物(4)溶于CH2Cl2(73mL)、CH3CN(190mL)、Et3SiH(8.4mL)和BF3·OEt2(5.0mL),制备单独的反应混合物。使反应混合物静置约30分钟,产生橙色均相溶液。用饱和NaHCO3水溶液(约200mL)猝灭各反应混合物,然后升温至室温。合并反应混合物,减压除去有机溶剂。加入500MLEtOAc,在分液漏斗中分离各层。有机层用饱和NaHCO3水溶液(2×300mL)和盐水(2×200mL)洗涤。合并的水层用EtOAc反萃取直至通过TLC分析的洗涤液观测不到痕量所需产物(约600mL EtOAc)。合并的有机层经MgSO4干燥,过滤,浓缩,得到23g淡黄色泡沫状化合物(5)。通过将其转化为四乙酸酯结晶衍生物(6),然后按下述实施例4和5方法水解来纯化无定形化合物(5)。
实施例4
制备2,3,4,6-四-O-乙酰基-1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-
基)-β-D-吡喃葡萄糖
将实施例3的化合物(5)(8.3g,21.3mmol,1当量)溶于CH2Cl2(53mL)和吡啶(17mL,210mmol,9.9当量)中。将乙酸酐(20mL,212mmol,10当量)加入反应混合物,然后加入4-二甲基氨基吡啶(DMAP)(130mg,1.1mmol,0.05当量)。搅拌反应混合物约50分钟。加入水(200mL)和CH2Cl2(200mL),在分液漏斗中分离各层。有机层用1N HCl(3×200mL)和盐水(2×100mL)洗涤。合并的水层用CH2Cl2反萃取直至TLC分析证实萃取液中没有需要的产物存在。合并的有机层经MgSO4干燥,过滤,浓缩,得到11g淡黄色固体。用EtOAc/己烷经以下方法重结晶纯化所得产物:在50℃下将该固体溶于EtOAc(48mL),然后向混合物中加入己烷(119mL),将混合物缓慢冷却至室温,然后冷却至4℃过夜。重结晶后,得到7.7g白色固体化合物(6)(经HPLC检测纯度100%)。
实施例5
制备1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-基)-β-D-吡喃葡萄糖
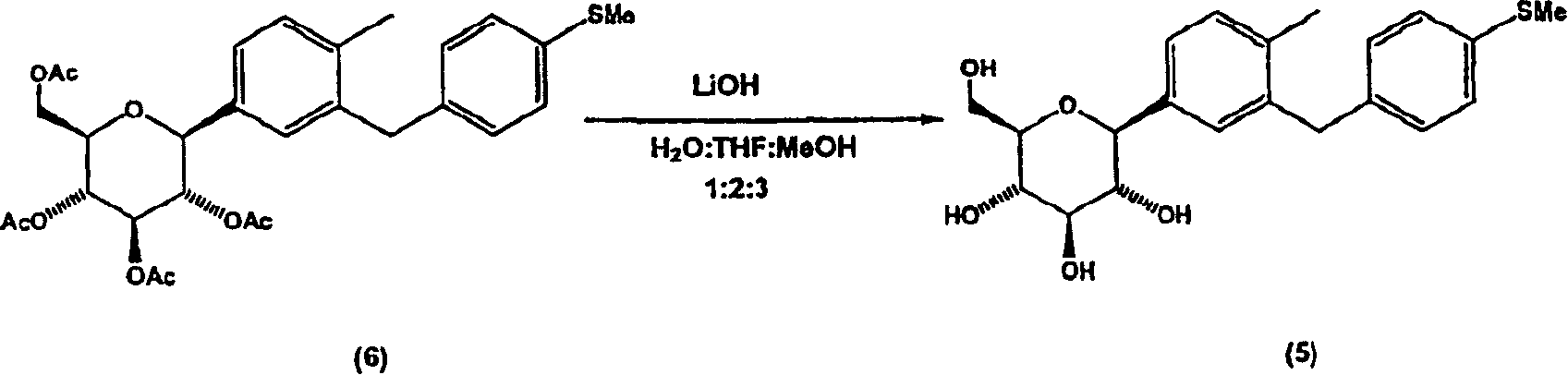
在室温下,将实施例4的化合物(6)(22.3g,39.9mmol,1当量)溶于THF/MeOH/H2O,搅拌。向该反应混合物中加入LiOH·H2O(1.65g,39.3mmol,1当量),得到浅黄色溶液。将反应混合物在室温下保持约5.2小时,然后减压浓缩。将残余物溶于EtOAc(300mL),然后加入饱和NaHCO3水溶液(200mL)。分离各层,有机层用NaHCO3(1×200mL)和盐水(1×200mL)洗涤。水层用EtOAc(3×300mL)反萃取。合并的有机层经MgSO4干燥,过滤,浓缩,得到15.5g 1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-基)-β-D-吡喃葡萄糖(5),为白色玻璃状固体。
实施例6
制备3-溴-2-甲基-4′-(甲硫基)二苯基甲烷
在一反应容器中使100.0G邻-甲苯甲酸悬浮于115mL二氯甲烷中形成浆状物。向浆状物中加入2.6g铁粉,随后在40分钟内加入143.1g溴,同时反应温度保持在约30℃。
稍微搅拌下,将所得反应混合物(151.0g)加入450mL乙醇中,随后加热至约70℃至80℃。然后将反应混合物冷却至环境温度。通过抽滤滤出3-溴-2-甲基-苯甲酸结晶,用乙醇水溶液洗涤。
向另一容器中加入770mL二氯甲烷。稍微搅拌下,向该反应容器中加入100.0 3-溴-2甲基-苯甲酸。在环境温度下向该反应容器中加入70.3g草酰氯和0.3mL二甲基甲酰胺,搅拌24小时,生成5-溴-2甲基-苯甲酰氯,然后将其溶于500mL二氯甲烷。向反应容器中加入57.8g茴香硫醚,冷却至10℃,随后先加入第一份12.4g氯化铝,搅拌15分钟。然后依次加入四份氯化铝,每次反应温度达到10℃。然后将反应混合物倾倒在300.0g冰和435mL 2N盐酸上,搅拌1小时。然后分离水层和有机层,有机层用210mL 2N盐酸洗涤,再用每份160mL的碳酸氢钠溶液洗涤两次。
然后除去二氯甲烷和水,随后加入270mL乙醇。将所得溶液冷却至0℃,得到3-溴-2-甲基-4′-(甲硫基)二苯基甲烷结晶。
然后按与实施例12中所述基本相同的方法处理上述制备的产物(5),得到1-C-(6-甲基-4′-(甲硫基)二苯基甲烷-3-基)-β-D-吡喃葡萄糖和L-苯丙氨酸络合物。
实施例7
制备2,3,4,6-四-O-(三甲基甲硅烷基)-D-葡糖酸-1,5-内酯
将700.0g D-葡糖酸-1,5-内酯、7L四氢呋喃和3185.0g 4-甲基吗啉加入60L反应容器中,保持温度在5℃以下。向容器中加入2590g氯化三甲基甲硅烷,将内容物在35℃下保持5小时。然后将该容器冷却至0℃。向容器中加入10.5L甲苯和14L水。分离水相和有机相,有机相用磷酸二氢盐一水合物水溶液洗涤除去过量的碱,将溶液pH调至7-8。
用水洗涤后,在50℃下减压(23英寸(in.)Hg柱)蒸馏有机相直至有机相的含水量不大于0.05%,得到浓度为约1.0g/10mL的标题化合物。
实施例8
制备3-溴-4′-乙基二苯基甲烷
A.3-溴-4′-乙基二苯基甲醇(benzylhydrol)
在Ar气下,向干燥Mg屑(4.4g,0.178mol)加入100mL无水Et2O,随后在1小时内加入对-溴乙苯(22g,0.119mol)的20mL Et2O溶液,搅拌过夜。(反应开始前加入0.5ml 1,2-二溴乙烷)。搅拌过夜后,缓慢加入间-溴苯甲醛(11g,0.06mol)的20mL Et2O溶液。用HPLC在约4至6小时期间监控所得明亮溶液以确定反应完成时间。反应物用饱和NH4Cl水溶液猝灭后,用EtOAc萃取3次。合并的有机层用盐水洗涤,经Na2SO4干燥,用旋转蒸发仪浓缩。将所得黄色油经硅胶层析,用5%EtOAc/己烷洗脱非极性杂质,用7-9%EtOAc/己烷洗脱浅黄色油状12.4g(71%)3-溴-4′-乙基二苯基甲醇。
B.3-溴-4′-乙基二苯基甲烷
在-30℃下搅拌A部分的3-溴-4′-乙基二苯基甲醇(12.4g,0.0426mol)的120mL MeCN溶液,向其中加入BF3Et2O(6.04g,0.0426mol),随后加入Et3SiH(9.9g,0.852mol)。在-30℃下搅拌1小时后,将该深色反应物缓慢升温至-5℃。经TLC确认反应完成后,加入饱和K2CO3水溶液猝灭反应。加入100mL H2O后,混合物用Et2O萃取3次。合并的有机层用盐水洗涤,经Na2SO4干燥。用旋转蒸发仪浓缩后得到浅黄色油状3-溴-4′-乙基二苯基甲烷(11.17g,95%),该产物使用时无需进一步纯化。
实施例9
制备甲基-1-C-(4′-乙基二苯基甲烷-3-基)-α-D-吡喃葡糖苷
在惰性氮气氛下,将895.0g 3-溴-4′-乙基二苯基甲烷(得自AustinChemicals of Chicago,lllinosis)、1.6L四氢呋喃和6.5L甲苯加入到20L反应容器中,冷却至-80℃。在30分钟内向反应容器中加入1.4L正丁基锂,同时保持反应温度约-80℃。搅拌该反应混合物,同时保持惰性气氛直至反应完成,得到3-锂-4′-乙基二苯基甲烷。
在惰性气氛、-80℃下,将反应混合物与实施例7制备的2,3,4,6-四-O-(三甲基甲硅烷基)-D-葡糖酸-1,5-内酯溶液混和,搅拌30分钟。
在30分钟内,将上述制备的锂化中间体化合物与443.0g(4.61mol)甲磺酸和7.2L甲醇溶液混和,同时保持反应容器的温度低于-60℃,得到甲基-1-C-(4′-乙基二苯基甲烷-3-基)-α-D-吡喃葡糖苷,收率76.9%。
实施例10
制备甲基-2,3,4,6-四-O-乙酰基-1-C-(4′-乙基二苯基甲烷-3-基)-α-
D-吡喃葡萄糖
在惰性氮气氛下,边搅拌,边将按照实施例9制备的550mL 60.2mg/mL的甲基-1-C-(4′-乙基二苯基甲烷-3-基)-α-D-吡喃葡糖苷的甲苯溶液和0.1g 4-(二甲基氨基)吡啶加入反应容器中,随后加入84mLN,N′-二异丙基乙胺(DIPEA)和40.9mL乙酸酐,同时保持温度不高于约35℃。搅拌反应混合物约4至7小时,得到甲基-2,3,4,6-四-O-乙酰基-1-C-(4′-乙基二苯基-甲烷-3-基)-α-D-吡喃葡糖苷。
然后用330ml 17%磷酸猝灭反应混合物直至下层水相pH不超过3。弃去含过量酸的下层水相。
用325mL水洗涤上层富有机相。常压浓缩有机相,得到甲基-2,3,4,6-四-O-乙酰基-1-C-(4′-乙基二苯基甲烷-3-基)-α-D-吡喃葡糖苷。
随后在不高于15℃的温度下,将232mL乙腈的1.5mL水溶液(1当量)加入该反应容器中。随后在20分钟内将40mL三乙基硅烷和21.1mL三氟化硼乙醚合物加入反应容器中,同时保持反应温度不高于25℃,搅拌反应物4-7小时,得到2,3,4,6-四-O-乙酰基-1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖,收率86%。
实施例11
制备1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖
在20℃下,将按实施例10制备的2.0g 2,3,4,6-四-O-乙酰基-1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖加入20mL乙醇中,随后加入0.83g氢氧化锂一水合物。搅拌混合物过夜,随后加入10mL水。加入2mL 6N盐酸使pH调为5.5。
将所得溶液蒸发至5 mL体积(大部分为乙醇),得到1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖。
实施例12
制备1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖L-苯丙氨酸
络合物
将按实施例11制备的1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖溶液与0.63g L-苯丙氨酸和45mL水混和。将混合物加热至83℃,然后搅拌下冷却至60℃。每下降2℃加入L-苯丙氨酸络合物晶种直至原先澄清溶液开始变为具有明显固体存在的稀浆状物。
在40℃至42℃下,搅拌所得浆状物4小时,然后在环境温度下搅拌过夜。然后过滤浆状物,洗涤,在40℃下干燥,得到1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖-L-苯丙氨酸络合物。
实施例13
制备1-C-(6-氯-4′-乙氧基二苯基甲烷-3-基)-β-D-吡喃葡萄糖
除用5-溴-2-氯-4′-乙氧基二苯基甲烷代替3-溴-4′-乙基二苯基甲烷外,用与实施例9-12所述方法基本相同的方法得到1-C-(6-氯-4′-乙氧基二苯基甲烷-3-基-β-D-吡喃葡萄糖及其相应的L-苯丙氨酸络合物。
或者,可按实施例14-21中所述方法制备1-C-(6-氯-4′-乙氧基二苯基甲烷-3-基)-β-D-吡喃葡萄糖。
实施例14
制备2-氯-5-溴苯甲酰氯
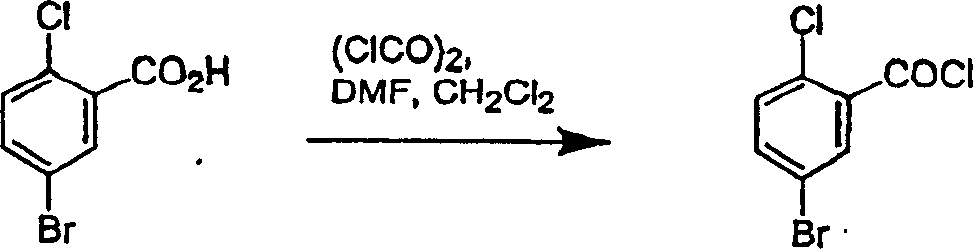
在20℃下,将8.00kg 2-氯-5-溴苯甲酸悬浮于80.00 L二氯甲烷(KF法测二氯甲烷H2O<0.1%)中,加入0.02L DMF。缓慢加入5.18Kg草酰氯,使内部温度保持在25℃以下。加入时,反应略微放热;产生HCl和CO2气体。使反应在20-25℃过夜;得到澄清溶液。将混合物真空浓缩至油状残余物并在40℃、真空下脱气。得到2-氯-5-溴苯甲酰氯:8.63kg(33,98mol,收率100,0%)
实施例15
制备2-氯-5-溴-4′-乙氧基二苯酮
将65.32L二氯甲烷加入反应器中,然后加入7.44kg氯化铝。将反应混合物冷却至0℃,缓慢加入6.81kg苯乙醚(phenetol),同时保持温度在0-5℃。完成加入后,将混合物冷却至0℃,搅拌15-20分钟,然后继续进行处理。
在另一个反应器中,用25.49L二氯甲烷稀释14.16kg 2-氯-5-溴苯甲酰氯。取样品进行HPLC分析。将该溶液加入到上述制备的混合物中,加入的速度要使温度保持在0-5℃。得到红色溶液。在0-5℃下搅拌混合物,取样品进行HPLC分析,每45分钟用MeOH猝灭。如果甲酯在1AP(面积%纯度)以下,可认为反应完全。通过在剧烈搅拌和冷却下将其加入至32.66L 2M HCl和16.33kg冰的混合物中,猝灭反应混合物。保持温度在25℃以下。在20-25℃下,搅拌该混合物20分钟,然后分离各相。有机相略微混浊。有机相用26.13L 2M HCl洗涤,合并的水相用13.06L二氯甲烷萃取。上一步合并的有机层用39.19L饱和碳酸氢钠溶液洗涤两次,每次搅拌至少30分钟。然后用13.06L二氯甲烷萃取合并的有机层。最后用19.6L盐水洗涤合并的有机层。真空浓缩有机相至干,在50-60℃下用32.66L乙醇溶解残余物。缓慢加入13.06L水,此时开始结晶。
在20-25℃下搅拌该浆状物1小时,然后再加入6.53L水,继续搅拌1小时。收集沉淀,滤饼用总量23.52L的水/乙醇(2∶1,预冷至-5℃)分次洗涤。干燥产物至最终含水量小于0.1%,得到17.31kg(91.3%)的标题二苯酮。产物为对-位和邻-位异构体的混合物(93∶7比率)。在后续反应步骤中除去不需要的邻-位异构体。
实施例16
制备2-氯-5-溴-4′-乙氧基二苯基甲烷

将10.81Kg 2-氯-5-溴-4′-乙氧基二苯酮溶于109.72L乙腈,将溶液冷却至10℃。加入9.99Kg三乙基硅烷,取样品进行HPLC分析。缓慢加入12.18Kg三氟化硼乙醚合物,反应温度保持在20℃以下。(通常无须加热,加入物料后温度可升至20-25℃)在此温度下搅拌反应混合物,不时冷却,每小时取样品进行HPLC分析直至反应完全。在反应期间形成沉淀。如果残存原料量小于0.1AP(4-6小时后),可认为反应完全。向该反应混合物中加入47.01L MTBE,混合物用47L饱和碳酸氢钠溶液洗涤两次。合并的水相用15.67L MTBE萃取;合并的有机相用15.67L盐水洗涤。真空浓缩有机相至干。然后将半固体残余物加热溶于21.55L乙醇。向溶液中放入晶种以便结晶,同时冷却至20-25℃过夜。在该步骤结束时,在0℃下搅拌浆状物1小时。收集沉淀,滤饼用3.92L预冷乙醇(-5-0℃)洗涤两次。在40℃、真空下将产物干燥至恒重(最终含水量:KF<0.08%),得量:7.80kg标题化合物(23.97mol,75,3%)。
实施例17
制备甲基-1-C-(2-氯-4′-乙氧基二苯基甲烷3-基)-α-D-吡喃葡萄糖
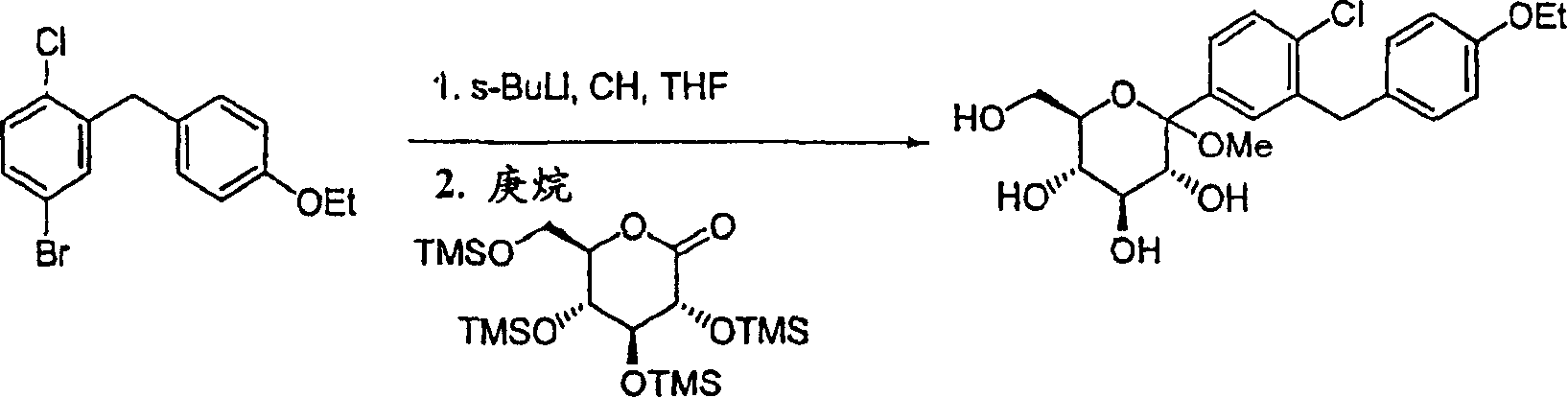
将11.97kg 2-氯-5-溴-4′-乙氧基-二苯基甲烷溶液加入盛有120LTHF的第一反应器中。取样品进行HPLC分析,然后将溶液冷却至-78℃。在第二反应器中,将17.16kg 2,3,4,6-四-O-三甲基甲硅烷基-D-葡糖酸内酯溶于87L庚烷,将溶液冷却至-78℃。向第一反应器中加入23.15kg s-BuLi(12%的环己烷溶液),以反应温度不超过-68℃的加入速度为宜。加入结束后,在-78℃下搅拌混合物1小时,然后取样品进行HPLC分析。在取样期间注意吸潮。(此时如果确定原料的含量小于3AP,反应可继续进行下步;否则,可能需要再加入s-BuLi)通过冷却管(-78℃)将第一反应器中的内容物加入到冷却的第二反应器(-78℃)中,以温度不超过-68℃的加入速度为宜。加入结束后,在-78℃下将第二反应器中的混合物搅拌30分钟,然后取样品进行HPLC分析。每小时重复取样一次直至反应完全。如果在连续两次抽样中未发现AP改变,可认为反应完全。将混合物升温至-40℃,然后非常小心地加入100L水。剧烈搅拌混合物10分钟,然后分离各相,水相用35L MTBE萃取。合并的有机相用35L盐水洗涤。然后在40℃、真空下,将有机相浓缩至油状残余物并小心脱气以除去所有挥发物。将油状残余物溶于100L甲醇,缓慢加入1.72kg甲磺酸。(在该阶段,反应温度可升至30℃)。将反应混合物在20-25℃下搅拌约12小时,然后加热至40℃保持3小时,取样品进行HPLC分析。在HPLC色谱图中,出现两个峰,其比率为95∶5至90∶10。如此鉴别的主要化合物与产物对应。加入2.49Kg三乙胺,将混合物真空浓缩至油状残余物。将残余物溶于150L乙酸乙酯,溶液用50L水洗涤两次。将有机相在真空中浓缩至油状残余物,小心脱气以除去所有挥发物。将该油状残余物溶于37L甲苯,将该甲苯溶液缓慢加入300L庚烷中。产物沉淀出;将悬浮液搅拌3分钟。收集沉淀,滤饼用极少量庚烷洗涤。然后使其在循环空气中干燥至恒重。得量:12.63kg标题化合物(28.78mol;78.3%)。
实施例18
制备甲基-2,3,4,6-四-O-乙酰基-1-C-(2-氯-4′-乙氧基二苯基甲烷-
3-基)-α-D-吡喃葡萄糖

将6.0Kg甲基-1-C-(2-氯-4′-乙氧基二苯基甲烷-3-基)-α-D-吡喃葡糖苷溶于30L THF,加入13.04kg DIPEA和0.06kg DMAP。取样进行HPLC分析。将混合物冷却至0-5℃,以温度不超过+5℃的速度加入9.14kg乙酸酐。加入结束后,在0-5℃下搅拌混合物1.5小时,然后取样进行HPLC分析。在0-5℃下再继续搅拌1.5小时,然后取样进行HPLC分析。加入30L预冷(5℃)MTBE,混合物用30L冰-水洗涤,然后在5℃下搅拌约30分钟。分离各层,水层用12L MTBE萃取。合并的有机相依次用12L 10%磷酸水溶液、12L饱和碳酸氢钠溶液2次和8L盐水洗涤。在40℃下真空蒸发溶剂,小心使油状残余物脱气,得到标题化合物:得量7.52kg(12.39mol,90.6%)。
实施例19
制备2,3,4,6-四-O-乙酰基-1-C-(2-氯-4′-乙氧基二苯基甲烷-3-基)-
β-D-吡喃葡萄糖

将5.01Kg甲基-2,3,4,6-四-O-乙酰基-1-C-(2-氯-4′-乙氧基二苯基甲烷-3-基)-β-D-吡喃葡糖苷(小心脱气,无残留MTBE)溶于25L乙腈。(通常K.F法测得溶液的含水量应为0.02-0.07%;更高含水量可能需要校正)将混合物冷却至10℃,加入0.15L水。加入3.04Kg三乙基硅烷,然后取样进行HPLC分析。以内部温度不超过15℃的速度加入2.34Kg三氟化硼乙醚合物。(加入结束后,温度通常升至约25℃。可能需要不时加热或冷却,反应时间通常为约18-20小时)继续搅拌直至至少90%的原料转化。(注意:可能需要加入额外的TES和BF3·Et2O)将混合物冷却至15℃,加入25L MTBE和14.7L饱和碳酸氢钠溶液。搅拌混合物20分钟,分离各相,有机相用另外的14.7L饱和碳酸氢钠溶液洗涤。合并的水相用6L MTBE萃取,合并的有机相用9L盐水洗涤。浓缩有机相至固体残余物。然后在50-60℃下将残余物溶于40L乙醇。加入0.5Kg活性碳,在回流下搅拌混合物10分钟。使该热溶液过滤,滤饼用4.4L热乙醇洗涤。使溶液在3小时内冷却至室温,然后冷却至0℃,搅拌1小时。收集沉淀,滤饼用8.8L冷(0-5℃)乙醇洗涤。在40℃、真空下使产物干燥至恒重。得量:3.10Kg标题化合物(5.36mmol,65.0%)。
实施例20
制备1-C-(2-氯4′-乙氧基二苯基甲烷-3-基)-β-D-吡喃葡萄糖

在20℃下,搅拌例如按实施例19制备的四乙酰化的β-C-葡糖苷(27.2g,49mmol)的480mL THF/MeOH/H2O(2∶3∶1)溶液,向其中加入LiOH一水合物(2.3g,57mmol)。搅拌过夜后,用旋转蒸发仪除去挥发物。将残余物溶于EtOAc(300mL),然后用盐水(150mL)洗涤1×,用含10mL 5%KHSO4水溶液的盐水(50mL)洗涤1×,最后用盐水(50mL)洗涤,经Na2SO4干燥。用旋转蒸发仪除去挥发物,将得到的油用极少量的CH2Cl2重蒸发,得到20.4g需要的标题C-芳基葡糖苷玻璃状灰白色固体。
实施例21
A.缩短步骤制备2,3,4,6-四-O-乙酰基-1-C-(4′-乙氧基二苯基甲烷- 3-基)-β-D-吡喃葡萄糖

将35Kg D-葡糖酸-1,5-内酯加入配备热电偶、机械搅拌器和加料漏斗的镍基合金反应器中。然后加入344.8Kg THF(无水)。随后将146.5Kg 4-甲基吗啉(约8当量)加入相同反应混合物中,将浆状物冷却至5℃。加入110.9Kg氯化三甲基甲硅烷(6当量),搅拌浆状物15分钟,之后在0.5小时内将反应混合物升温至30-35℃。5小时后反应完成(GC确定)。将反应混合物冷却至0-5℃,然后加入454.1kg甲苯。然后用700kg水猝灭反应。搅拌反应混合物10-15分钟,使各相分离。然后除去底部水层。向有机溶液中加入13.0Kg NaH2PO4的260Kg水溶液。搅拌反应混合物10分钟,然后再次分离各相。除去底部水层,向其中加入273kg水。搅拌混合物10分钟,再次分离所得水相。在40-60℃下,减压(通常23-25英寸Hg柱)蒸馏有机溶液,同时加入甲苯直至甲硅烷基化产物的甲苯溶液中KF法测得水<0.05%。可按需要,通过加入无水甲苯或蒸馏将甲硅烷基化产物的浓度调至~0.1g/mL。按GC分析法基于产物标准曲线(GC法)测得或通过减压除去溶剂至干得出:第一步的2,3,4,6-四-O-三甲基甲硅烷基-D-葡糖酸内酯收率为91-98%。
向另一配备热电偶、机械搅拌器和加料漏斗的反应器中加入44.8kg 5-溴-4′-乙基二苯基甲烷。然后加入78.8Kg无水THF,随后加入281kg甲苯。将反应器冷却至<-70℃。加入约48.9kg n-BuLi(2.5M的己烷溶液)。在<-70℃、N2下搅拌溶液直至经HPLC分析确定锂化完成。然后将该源自5-溴-4′-乙基二苯基甲烷的锂化阴离子溶液加入到冷的以上制备的2,3,4,6-四-O-三甲基甲硅烷基-D-葡糖酸内酯溶液中,加入速度以使温度保持<-70℃为宜。在<-70℃下搅拌混合物至少30分钟,取样进行HPLC分析,证实反应完成。加入22.2Kg CH3SO3H(1.4当量)的334kg MeOH溶液,同时保持温度<-60℃。通过以下方法取样品证实甲基苷化完成:取50μL反应混合物并用10mL CH3CN将其猝灭以进行HPLC分析。当确认反应完成后,加入碳酸氢钠水溶液(11KgNaHCO3的220kg水溶液)将其猝灭。用220kg水洗涤含产物的富有机层,分离各相,然后合并水相。合并的水层用223Kg乙酸乙酯萃取(根据需要操作该步骤)。合并富含产物的有机层,在35-60℃下减压(通常约25英寸Hg柱)蒸馏溶剂直至溶液用KF法测得H2O<0.07%,经GC分析测定,相对于甲苯EtOAC的量<1%。
用该方法进行偶合,得到产物的收率为72-89M%;通过基于产物标准曲线的HPLC分析测得该浓度。
向甲基苷溶液中加入0.11kg DMAP、64.33kg二异丙基乙胺和45.56Kg乙酸酐。在≤35℃下搅拌溶液直至HPLC分析确认反应完成。当中间体乙酰化产物的面积百分率(AP)以四乙酰化产物面积计≤2%时,可判断反应完成。该步骤通常的反应时间为4-7小时。
然后将H3PO4溶液(48.44Kg的528Kg水溶液)加入反应器中。得到的低部水层的pH≤3。(如果pH>3,可再加入H3PO4直至达到需要的pH)搅拌反应物10分钟,然后使各相分离。分离底层液相,有机相用245.54kg水洗涤,之后分离水洗层。常压下浓缩有机溶液至甲基-1-C-(4′-乙基二苯基甲烷-3-基)-α-D-吡喃葡糖苷(乙酰化中间体)的体积为4-6L/kg。
然后向盛有乙酰化中间体的反应器中加入174.8kg乙腈和1.6kg水(相对乙酰化中间体为1mol当量)。然后将混合物冷却至≤15℃,加入30.3kg Et3SiH。然后在至少20分钟内加入24.5kg BF3-Et2O(2.1当量),同时保持温度<15℃。搅拌反应混合物直至反应完成,需约4-7小时,通过HPLC分析判断。然后将反应混合物冷却至≤20℃。需要的2,2-二甲氧基丙烷的量基于测定残留Et3SiH量的GC分析和测定残留H2O量的KF法。
搅拌反应混合物直至通过GC分析确定Et3SiH完全消失,通常<2小时。在环境温度下加入NaHCO3水溶液(12.02kg NaHCO3的120.17kg水溶液)直至水相pH≥6。然后搅拌混合物至少10分钟,使各相澄清,此时除去底层水相。将20%NaCl(24.03kg NaCl的120.17kg水溶液)加入有机层中,分离水相。然后常压蒸馏富含产物的有机相直至除去大部分乙腈。
加入223.49Kg甲苯,继续蒸馏直至釜(pot)温达到至少112℃和甲基-1-C-(4′-乙基二苯基甲烷-3-基)-α-D-吡喃葡糖苷浓度达到4-6L/kg。同时保持温度>70℃,加入240Kg庚烷。然后在至少1小时内将溶液冷却至约60℃,将浆状物在60±10℃保持至少1小时。
然后,将浆状物在至少1小时内冷却至20±10℃。然后在Robatel离心机中过滤,滤饼用至少2倍滤饼体积的庚烷(57.7Kg)洗涤。湿滤饼在≤60℃、真空下干燥至LOD≤0.2%,得到≥40Kg标题化合物(86%,实验室HPLC:99 AP)。
B.制备1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖L-苯丙氨 酸络合物
1∶1∶1 L-苯丙氨酸:H2O
将68.70Kg水加入配备机械搅拌器和氮气入口的反应器中。将64.40Kg 2,3,4,6-四-O-乙酰基-1-C-(4′-乙基二苯基-甲烷-3-基)-β-D-吡喃葡萄糖加入同一反应器中。加入62.44Kg EtOH(SDA-3A 190标准),在室温(20℃)、氮气氛下,尽可能轻地搅拌该悬浮液。
在室温下加入0.1N NaOH水溶液(57.02kg)(中等搅拌),缓慢加热至40-50℃,之后搅拌混合物1-2小时直至过程控制HPLC测得溶液中去乙酰化产物的AP>97%(不包括溶剂前沿和系统峰)。
将溶液冷却至20℃,然后在室温下加入154.56kg去离子水;然后将溶液温度调至18-25℃。搅拌反应物1小时(混浊溶液)。然后加入浓HCl(37%,约123.44Kg)将pH调至6.3。加入20.20Kg L-苯丙氨酸,随后加入141.68kg水。将浆状物加热至75℃,然后使该澄清溶液通过过滤器。加入51.52Kg热(75℃)去离子水冲洗滤瓶,然后将洗涤液加入反应混合物,使溶剂在溶液中的组成调至~12%(体积)EtOH。将浆状物在75℃加热,然后将澄清溶液冷却至约57℃。加入322Gm标题化合物晶种。将浆状物在1小时内冷却至40℃,将釜温保持在40℃4小时。将浆状物在2小时内冷却至18-25℃,在此温度下搅拌12-16小时,之后将其通过Robatel离心机滤器过滤。滤饼用冷(<10℃)322kg水洗涤以除去副产物NaCl和NaOAc。继续用水洗涤直至洗涤液的电导率小于0.001Ω-1。滤饼用290kg EtOAc洗涤以除去剩余的产物。湿滤饼在18-25℃、真空下干燥至少4小时,然后在40℃下干燥至少12小时。当等份试样的KF法读数为含水2.8-3.6%时,停止干燥。分离1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖L-苯丙氨酸络合物,为白色固体(54-58Kg,80-88%)。
上述讨论公开和描述的内容仅为本发明的示例性实施方案。本领域技术人员将容易地从这种讨论及其附图和权利要求书中认识到,其中可进行各种改变、修改和变化,而没有偏离如权利要求书所限定的本发明的宗旨和范围。
Claims (34)
1.一种制备式(I)化合物的方法
其中:
R1选自氢、羟基、溴、氯、氟、C1-8烷基、C1-8烷氧基、C1-8烷硫基和C6-10芳硫基,其中p为1至4,且条件是当溴、氯和氟存在时,只能在3-、4-和5-位中至少一个位置上存在;
R2选自氢、羟基、氯、氟、C1-8烷基、C1-8烷氧基和C1-8烷硫基,其中q为1至5;和
A选自共价键、O、S、NH和(CH2)n,其中n为1至3,且
条件是当A在4-位时,R1不为溴;
条件是当R1中的一个为溴;和
如果A在3-或6-位,则溴在5-位,
如果A在2-或5-位,则溴在3-位,和
当溴在3-位,且A在2-或5-位时,则在4-和6-位的R1基团相同且不为溴、氯或氟,和
当溴在5-位,且A在3-或6-位时,则在2-和4-位的R1基团相同且不为溴、氯或氟,所述方法包括:
a)用酸不稳定保护基团保护的内酯形成式(IV)化合物
其中ALG为酸不稳定保护基团;
b)使式(IV)化合物苷化,同时除去所述酸不稳定保护基团以形成式(V)化合物;

c)使式(V)化合物与酰化剂反应形成式(VI)化合物;
d)将式(VI)化合物还原以得到式(VII)化合物;和
e)除去式(VII)化合物中的酰基保护基团以得到式(I)化合物;
式IV、V和VI中R1、R2、A、p和q的定义同式I化合物的定义,R3选自C1-8烷氧基、C2-8烯氧基、C1-8烷硫基和C2-8烯硫基,且AC为酰基保护基团。
2.权利要求1的方法,其中p为1至2。
3.权利要求1的方法,其中q为1至2。
4.权利要求1的方法,其中R1选自氢、C1-8烷基、氯和氟。
5.权利要求4的方法,其中p为1,且R1选自氢、氯和甲基。
6.权利要求5的方法,其中R1在4-位。
7.权利要求1的方法,其中R2选自氢、C1-8烷基、C1-8烷氧基和C1-8烷硫基。
8.权利要求7的方法,其中q为1,且R2选自乙基、乙氧基和甲硫基。
9.权利要求8的方法,其中R2在4-位。
10.权利要求1的方法,其中A为位于3-位的(CH2)。
11.权利要求1的方法,其中所述式(I)化合物为式(IA)化合物:

其中:
R1选自氢、C1-8烷基和氯;和
R2选自氢、C1-8烷基、C1-8烷氧基和C1-8烷硫基。
12.权利要求11的方法,其中R1为氢,且R2为乙基。
13.权利要求11的方法,其中R1为氯,且R2为乙氧基。
14.权利要求11的方法,其中R1为甲基,且R2为甲硫基。
15.权利要求1的方法,所述方法还包括使式(I)化合物与至少一种氨基酸络合物形成试剂反应以生成式(I)化合物药学上可接受的络合物。
16.权利要求15的方法,其中所述氨基酸为L-苯丙氨酸。
17.一种制备式VII的C-芳基葡糖苷化合物的方法,
所述方法包括使式(VI)化合物

其中:
R1选自氢、羟基、溴、氯、氟、C1-8烷基、C1-8烷氧基、C1-8烷硫基和C6-10芳硫基,其中p为1至4的整数,且条件是当溴、氯和氟存在时,只能在3-、4-和5-位中至少一个位置上存在;
R2选自氢、羟基、氯、氟、C1-8烷基、C1-8烷氧基和C1-8烷硫基,其中q为1至5的整数;
A选自共价键、O、S、NH和(CH2)n,其中n为1至3的整数,条件是
(i)当A在4-位时,R1不为溴;
(ii)当R1中的一个为溴;和
如果A在3-或6-位,则溴在5-位,
如果A在2-或5-位,则溴在3-位,和
当溴在3-位,且A在2-或5-位时,则在4-和6-位的R1基团相同且不为溴、氯或氟,和
当溴在5-位,且A在3-或6-位时,则在2-和4-位的R1基团相同且不为溴、氯或氟;
R3选自C1-8烷氧基、C2-8烯氧基、C1-8烷硫基和C2-8烯硫基;
且
AC为酰基保护基团;
与还原剂反应以形成式(VII)化合物

其中R1、R2、A、p和q同式(VI)化合物的定义。
18.权利要求17的方法,其中所述还原剂选自甲硅烷。
19.权利要求18的方法,其中所述还原剂为烷基甲硅烷。
20.权利要求17的方法,其中所述式(VI)化合物与所述还原剂间的反应在活化基团的存在下进行。
21.权利要求20的方法,其中所述活化基团选自路易斯酸。
22.权利要求17的方法,所述方法还包括使式(V)化合物与酰化剂反应形成式(VI)化合物,
其中R1、R2、R3、p、q和A定义同权利要求18。
23.权利要求22的方法,其中所述酰化剂选自酰基衍生物、酰卤、乙酰氯、酸酐、乙酸酐、丙酸酐及其组合。
24.权利要求22的方法,所述方法还包括使式(IV)化合物与苷化试剂反应形成式(V)化合物,
其中R1、R2、A、p和q定义同权利要求17,且ALG为酸不稳定保护基团。
25.权利要求24的方法,其中所述酸不稳定保护基团选自甲氧基甲基醚、甲硫基甲基醚、2-甲氧基乙氧基甲基醚、二(2-氯乙氧基)甲基醚、四氢吡喃醚、四氢噻喃醚、4-甲氧基四氢吡喃醚、4-甲氧基四氢噻喃醚、四氢呋喃醚、四氢噻吩醚、1-乙氧基乙基醚、1-甲基-1-甲氧基乙基醚、2-(苯基氧硒基)乙基醚、叔丁基醚、烯丙基醚、三苯基甲基醚、α-萘基二苯基甲基醚、对-甲氧基苯基二苯基甲基醚、三烷基甲硅烷基醚、三甲基甲硅烷基醚、三乙基甲硅烷基醚、异丙基二甲基甲硅烷基醚、叔丁基二甲基甲硅烷基醚、叔丁基二苯基甲硅烷基醚及其组合。
26.权利要求25的方法,其中所述酸不稳定保护基团选自甲氧基甲基醚、2-甲氧基乙氧基甲基醚、四氢吡喃醚、三甲基甲硅烷基醚、异丙基二甲基甲硅烷基醚、叔丁基二甲基甲硅烷基醚、叔丁基二苯基甲硅烷基醚及其组合。
27.权利要求24的方法,所述方法还包括使式(II)化合物

其中ALG的定义同权利要求24,与下式(III)化合物反应形成式(IV)化合物
其中R1、R2、A、p和q的定义同权利要求24,且Y为金属。
28.权利要求27的方法,其中Y选自碱金属和碱土金属。
29.权利要求27的方法,所述方法还包括使D-葡糖酸-1,5-内酯与酸不稳定保护基团供给试剂反应形成式(II)化合物。
30.权利要求29的方法,其中所述酸不稳定保护基团供给试剂选自氯化三甲基甲硅烷、三甲基甲硅烷基三氟甲磺酸、甲氧基甲基氯化物、二氢呋喃、苄氧基甲基氯化物、氯化三乙基甲硅烷和四氢吡喃。
31.一种制备式(VII)中间体化合物的方法

其中:
R1选自氢、羟基、溴、氯、氟、C1-8烷基、C1-8烷氧基、C1-8烷硫基和C6-10芳硫基,其中p为1至4,且条件是当溴、氯和氟存在时,只能在3-、4-和5-位中至少一个位置上存在;
R2选自氢、羟基、氯、氟、C1-8烷基、C1-8烷氧基和C1-8烷硫基,其中q为1至5;
R3选自C1-8烷氧基、C2-8烯氧基、C1-8烷硫基和C2-8烯硫基;和
A选自共价键、O、S、NH和(CH2)n,其中n为1至3,且
条件是当A在4-位时,R1不为溴;
条件是当R1中的一个为溴;和
如果A在3-或6-位,则溴在5-位,
如果A在2-或5-位,则溴在3-位,和
当溴在3-位,且A在2-或5-位时,则在4-和6-位的R1基团相同,且不为溴、氯或氟,和
当溴在5-位,且A在3-或6-位时,则在2-和4-位的R1基团相同,且不为溴、氯或氟;且
AC为酰基保护基团,所述方法包括使下式(VI)化合物反应形成式(VII)化合物,

其中R1、R2、R3、A、AC、p和q的定义同式(VII)化合物的定义。
32.一种式(VII)化合物
其中:
R1选自氢、羟基、溴、氯、氟、C1-8烷基、C1-8烷硫基和C6-10芳硫基,其中p为1至4,且条件是当溴、氯和氟存在时,只能在3-、4-和5-位中至少一个位置上存在;
R2选自氢、羟基、氯、氟、C1-8烷基、C1-8烷氧基和C1-8烷硫基,其中q为1至5;
A选自共价键、S、NH和(CH2)n,其中n为1至3,且
条件是当A在4-位时,R1不为溴;
条件是当R1中的一个为溴;和
如果A在3-或6-位,则溴在5-位,
如果A在2-或5-位,则溴在3-位,和
当溴在3-位,且A在2-或5-位时,则在4-和6-位的R1基团相同,且不为溴、氯或氟,和
当溴在5-位,且A在3-或6-位时,则在2-和4-位的R1基团相同,且不为溴、氯或氟;且
AC为酰基保护基团。
33.权利要求1的方法,其中所制备的化合物为1-C-(4′-乙基二苯基甲烷-3-基)-β-D-吡喃葡萄糖-L-苯丙氨酸络合物。
34.权利要求1的方法,其中所制备的化合物为式(I)化合物:

与氨基酸的络合物,所述氨基酸选自L-苯丙氨酸或L-脯氨酸。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US43784703P | 2003-01-03 | 2003-01-03 | |
| US60/437,847 | 2003-01-03 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2008100884952A Division CN101260130A (zh) | 2003-01-03 | 2003-12-23 | 制备c-芳基葡糖苷sglt2抑制剂的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1756759A CN1756759A (zh) | 2006-04-05 |
| CN100391963C true CN100391963C (zh) | 2008-06-04 |
Family
ID=32713238
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2003801100401A Expired - Fee Related CN100391963C (zh) | 2003-01-03 | 2003-12-23 | 制备c-芳基葡糖苷sglt2抑制剂的方法 |
| CNA2008100884952A Pending CN101260130A (zh) | 2003-01-03 | 2003-12-23 | 制备c-芳基葡糖苷sglt2抑制剂的方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2008100884952A Pending CN101260130A (zh) | 2003-01-03 | 2003-12-23 | 制备c-芳基葡糖苷sglt2抑制剂的方法 |
Country Status (12)
| Country | Link |
|---|---|
| US (4) | US7375213B2 (zh) |
| EP (1) | EP1581543A4 (zh) |
| JP (1) | JP2006516257A (zh) |
| KR (1) | KR20050090437A (zh) |
| CN (2) | CN100391963C (zh) |
| AU (1) | AU2003299966A1 (zh) |
| BR (1) | BR0317929A (zh) |
| CA (1) | CA2512389A1 (zh) |
| MX (1) | MXPA05007052A (zh) |
| PL (1) | PL378324A1 (zh) |
| TW (2) | TW200801030A (zh) |
| WO (1) | WO2004063209A2 (zh) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104109179A (zh) * | 2013-04-16 | 2014-10-22 | 杭州华东医药集团生物工程研究所有限公司 | 一类c-芳基葡萄糖苷衍生物、制备方法及其用途 |
| CN104478957A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 一种含腈基苯和双o-葡萄糖苷衍生物、其制备方法和用途 |
| CN104478968A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含噻唑基的o-半乳糖苷衍生物、其制备方法和用途 |
| CN104478965A (zh) * | 2015-01-15 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 一类烷氧基苯基s-葡萄糖苷衍生物、其制备方法和用途 |
| CN104478964A (zh) * | 2015-01-15 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含丙烯腈结构的苯o-葡萄糖苷衍生物、其制备方法和用途 |
| CN104478959A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含腈基联苯双葡萄糖苷结构化合物、其制备方法和用途 |
| CN104478963A (zh) * | 2015-01-15 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 一种含腈基苯s-葡萄糖苷结构的化合物及其用途 |
| CN104478966A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含苯胺噻唑基的o-半乳糖苷衍生物、其制备方法和用途 |
| CN104497070A (zh) * | 2015-01-15 | 2015-04-08 | 佛山市赛维斯医药科技有限公司 | 一种含三氟甲基苯s-葡萄糖苷结构的化合物和用途 |
| CN104530149A (zh) * | 2015-01-14 | 2015-04-22 | 佛山市赛维斯医药科技有限公司 | 卤代苯基双o-葡萄糖苷衍生物、其制备方法和用途 |
| CN105218329A (zh) * | 2015-10-15 | 2016-01-06 | 上海应用技术学院 | 一种列净类似物中间体及其制备方法 |
Families Citing this family (152)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100391963C (zh) * | 2003-01-03 | 2008-06-04 | 布里斯托尔-迈尔斯斯奎布公司 | 制备c-芳基葡糖苷sglt2抑制剂的方法 |
| PL1651658T5 (pl) | 2003-08-01 | 2020-11-30 | Mitsubishi Tanabe Pharma Corporation | Nowe związki o działaniu inhibitującym transporter zależny od sodu |
| US8785403B2 (en) | 2003-08-01 | 2014-07-22 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| WO2005038013A1 (en) | 2003-10-07 | 2005-04-28 | Isis Pharmaceuticals, Inc. | Artisense oligonucleotides optimized for kidney targeting |
| US20050191653A1 (en) * | 2003-11-03 | 2005-09-01 | Freier Susan M. | Modulation of SGLT2 expression |
| ES2387881T3 (es) * | 2004-03-16 | 2012-10-03 | Boehringer Ingelheim International Gmbh | Derivados de benceno sustituidos por glucopiranosilo, medicamentos que contienen estos compuestos, su uso y procedimiento para su preparación |
| EP1773800A1 (de) * | 2004-07-27 | 2007-04-18 | Boehringer Ingelheim International GmbH | D-glucopyranosyl-phenyl-substituierte cyclen, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| AR051446A1 (es) * | 2004-09-23 | 2007-01-17 | Bristol Myers Squibb Co | Glucosidos de c-arilo como inhibidores selectivos de transportadores de glucosa (sglt2) |
| EP1828216B1 (en) | 2004-12-16 | 2008-09-10 | Boehringer Ingelheim International GmbH | Glucopyranosyl-substituted benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| TW200637869A (en) * | 2005-01-28 | 2006-11-01 | Chugai Pharmaceutical Co Ltd | The spiroketal derivatives and the use as therapeutical agent for diabetes of the same |
| WO2006089872A1 (en) * | 2005-02-23 | 2006-08-31 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted ( (hetero)arylethynyl-benzyd-benzene derivatives and use thereof as sodium-dependent glucose cotransporter 2 (sglt2) inhibitors |
| CA2605245A1 (en) * | 2005-04-15 | 2006-10-19 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted (heteroaryloxy-benzyl)-benzene derivatives as sglt inhibitors |
| UA91546C2 (uk) | 2005-05-03 | 2010-08-10 | Бьорінгер Інгельхайм Інтернаціональ Гмбх | КРИСТАЛІЧНА ФОРМА 1-ХЛОР-4-(β-D-ГЛЮКОПІРАНОЗ-1-ИЛ)-2-[4-((S)-ТЕТРАГІДРОФУРАН-3-ІЛОКСИ)-БЕНЗИЛ]-БЕНЗОЛУ, СПОСІБ ЇЇ ОДЕРЖАННЯ ТА ЇЇ ЗАСТОСУВАННЯ ПРИ ПРИГОТУВАННІ ЛІКАРСЬКИХ ЗАСОБІВ |
| US7723309B2 (en) | 2005-05-03 | 2010-05-25 | Boehringer Ingelheim International Gmbh | Crystalline forms of 1-chloro-4-(β-D-glucopyranos-1-yl)-2-[4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-benzene, a method for its preparation and the use thereof for preparing medicaments |
| US7772191B2 (en) | 2005-05-10 | 2010-08-10 | Boehringer Ingelheim International Gmbh | Processes for preparing of glucopyranosyl-substituted benzyl-benzene derivatives and intermediates therein |
| TW200726755A (en) * | 2005-07-07 | 2007-07-16 | Astellas Pharma Inc | A crystalline choline salt of an azulene derivative |
| AR054871A1 (es) | 2005-07-27 | 2007-07-25 | Boehringer Ingelheim Int | Derivados de (hetero)cicloalquiletinil-bencil)-benceno sustituidos con glucopiranosilo, medicamentos que contienen dichos compuestos, sus uso y proceso para su fabricacion |
| WO2007025943A2 (en) * | 2005-08-30 | 2007-03-08 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| ATE491700T1 (de) | 2005-09-08 | 2011-01-15 | Boehringer Ingelheim Int | KRISTALLINE FORMEN VON 1-CHLORO-4-(ß-D- |
| AR056195A1 (es) * | 2005-09-15 | 2007-09-26 | Boehringer Ingelheim Int | Procedimientos para preparar derivados de (etinil-bencil)-benceno sustituidos de glucopiranosilo y compuestos intermedios de los mismos |
| EA200801773A1 (ru) | 2006-02-15 | 2009-02-27 | Бёрингер Ингельхайм Интернациональ Гмбх | Глюкопиранозилзамещенные производные бензонитрила, фармацевтические композиции, содержащие такие соединения, их применение и способ их получения |
| PE20080697A1 (es) | 2006-05-03 | 2008-08-05 | Boehringer Ingelheim Int | Derivados de benzonitrilo sustituidos con glucopiranosilo, composiciones farmaceuticas que contienen compuestos de este tipo, su uso y procedimiento para su fabricacion |
| US8673871B2 (en) | 2006-05-05 | 2014-03-18 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression ApoB |
| WO2007140191A2 (en) * | 2006-05-23 | 2007-12-06 | Theracos, Inc. | Glucose transport inhibitors and methods of use |
| US7919598B2 (en) * | 2006-06-28 | 2011-04-05 | Bristol-Myers Squibb Company | Crystal structures of SGLT2 inhibitors and processes for preparing same |
| AU2013200322B2 (en) * | 2006-06-28 | 2014-09-11 | Astrazeneca Ab | Crystalline solvates and complexes of (1s) -1, 5-anhydro-1-c-(3-((phenyl)methyl)phenyl)-d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetes |
| AU2014268177B2 (en) * | 2006-06-28 | 2016-05-19 | Astrazeneca Ab | Crystalline solvates and complexes of (1s) -1, 5-anhydro-1-c-(3-((phenyl)methyl)phenyl)-d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetes |
| JP5384343B2 (ja) * | 2006-08-15 | 2014-01-08 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | グルコピラノシル−置換シクロプロピルベンゼン誘導体、そのような化合物を含む医薬組成物、sglt阻害剤としてのそれらの使用及びそれらの製造方法 |
| TWI499414B (zh) | 2006-09-29 | 2015-09-11 | Lexicon Pharmaceuticals Inc | 鈉與葡萄糖第2型共同運輸體(co-transporter 2)的抑制物與其應用方法 |
| JP2010507629A (ja) * | 2006-10-27 | 2010-03-11 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 4−(β−D−グルコピラノス−1−イル)−1−メチル−2−[4−((S)−テトラヒドロフラン−3−イルオキシ)−ベンジル]−ベンゼンの結晶形、その製造方法及び医薬品を製造するための使用 |
| UY30730A1 (es) | 2006-12-04 | 2008-07-03 | Mitsubishi Tanabe Pharma Corp | Forma cristalina del hemihidrato de 1-(b (beta)-d-glucopiranosil) -4-metil-3-[5-(4-fluorofenil) -2-tienilmetil]benceno |
| WO2008075736A1 (ja) | 2006-12-21 | 2008-06-26 | Astellas Pharma Inc. | C-グリコシド誘導体の製造方法及びその合成中間体 |
| US7795228B2 (en) * | 2006-12-28 | 2010-09-14 | Theracos, Inc. | Spiroheterocyclic glycosides and methods of use |
| US7846945B2 (en) * | 2007-03-08 | 2010-12-07 | Lexicon Pharmaceuticals, Inc. | Piperdine-based inhibitors of sodium glucose co-transporter 2 and methods of their use |
| TW200904454A (en) | 2007-03-22 | 2009-02-01 | Bristol Myers Squibb Co | Methods for treating obesity employing an SGLT2 inhibitor and compositions thereof |
| TW200904405A (en) * | 2007-03-22 | 2009-02-01 | Bristol Myers Squibb Co | Pharmaceutical formulations containing an SGLT2 inhibitor |
| CN103254119B (zh) * | 2007-07-10 | 2016-07-06 | 莱西肯医药有限公司 | 钠-葡萄糖协同转运蛋白2的抑制剂及其用法 |
| BRPI0813840A2 (pt) * | 2007-07-26 | 2017-06-06 | Lexicon Pharmaceuticals Inc | métodos e compostos úteis para a preparação de inibidores de cotransportador 2 de glicose de sódio |
| CL2008002427A1 (es) | 2007-08-16 | 2009-09-11 | Boehringer Ingelheim Int | Composicion farmaceutica que comprende 1-cloro-4-(b-d-glucopiranos-1-il)-2-[4-((s)-tetrahidrofurano-3-iloxi)bencil]-benceno combinado con 1-[(4-metilquinazolin-2-il)metil]-3-metil-7-(2-butin-1-il)-8-(3-(r)-aminopiperidin-1-il)xantina; y su uso para tratar diabetes mellitus tipo 2. |
| KR101107425B1 (ko) | 2007-08-23 | 2012-01-19 | 테라코스, 인코포레이티드 | 벤질벤젠 유도체 및 사용 방법 |
| ME03072B (me) * | 2007-09-10 | 2019-01-20 | Janssen Pharmaceutica Nv | Postupak za dobijanje jedinjenja која su korisna као inhibiтori sgl т |
| CA2706018C (en) * | 2007-11-30 | 2015-11-24 | Boehringer Ingelheim International Gmbh | 1, 5-dihydro-pyrazolo [3,4-d]pyrimidin-4-one derivatives and their use as pde9a modulators for the treatment of cns disorders |
| UA101004C2 (en) | 2007-12-13 | 2013-02-25 | Теракос, Инк. | Derivatives of benzylphenylcyclohexane and use thereof |
| CN104387354A (zh) * | 2007-12-27 | 2015-03-04 | 阿斯利康公司 | Sglt2 抑制剂的晶体结构及其制备方法 |
| CN103319445B (zh) * | 2007-12-27 | 2016-01-20 | 阿斯利康公司 | Sglt2抑制剂的晶体结构及其制备方法 |
| CL2008003653A1 (es) | 2008-01-17 | 2010-03-05 | Mitsubishi Tanabe Pharma Corp | Uso de un inhibidor de sglt derivado de glucopiranosilo y un inhibidor de dppiv seleccionado para tratar la diabetes; y composicion farmaceutica. |
| EP2236137B1 (en) * | 2008-01-31 | 2015-06-24 | Astellas Pharma Inc. | Pharmaceutical composition for treatment of fatty liver diseases |
| WO2009117367A1 (en) * | 2008-03-18 | 2009-09-24 | Bristol-Myers Squibb Company | Method for treating cancers having high glucose requirements employing an sglt2 inhibitor and compositions thereof |
| UA105362C2 (en) * | 2008-04-02 | 2014-05-12 | Бьорингер Ингельхайм Интернациональ Гмбх | 1-heterocyclyl-1, 5-dihydro-pyrazolo [3, 4-d] pyrimidin-4-one derivatives and their use as pde9a modulators |
| RU2509773C2 (ru) | 2008-07-15 | 2014-03-20 | Теракос, Инк. | Дейтерированные бензилбензольные производные и способы применения |
| US8283454B2 (en) * | 2008-08-22 | 2012-10-09 | Theracos, Inc. | Processes for the preparation of SGLT2 inhibitors |
| KR101446454B1 (ko) * | 2008-08-28 | 2014-10-06 | 화이자 인코포레이티드 | 다이옥사-바이사이클로[3.2.1]옥테인-2,3,4-트라이올 유도체 |
| BRPI0918527A2 (pt) | 2008-09-08 | 2015-12-01 | Boehring Ingelheim Internat Gmbh | compostos para o tratamento de disturbios do cns |
| US9056850B2 (en) * | 2008-10-17 | 2015-06-16 | Janssen Pharmaceutica N.V. | Process for the preparation of compounds useful as inhibitors of SGLT |
| US20110046076A1 (en) | 2009-02-13 | 2011-02-24 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition, methods for treating and uses thereof |
| EP4327867A3 (en) * | 2009-02-13 | 2024-05-29 | Boehringer Ingelheim International GmbH | Pharmaceutical composition comprising glucopyranosyl diphenylmethane derivatives, pharmaceutical dosage form thereof, process for their preparation and uses thereof for improved glycemic control in a patient |
| NZ594567A (en) | 2009-03-31 | 2013-12-20 | Boehringer Ingelheim Int | 1-heterocyclyl-1, 5-dihydro-pyrazolo [3, 4-d] pyrimidin-4-one derivatives and their use as pde9a modulators |
| US20110009347A1 (en) * | 2009-07-08 | 2011-01-13 | Yin Liang | Combination therapy for the treatment of diabetes |
| SI2451797T1 (sl) | 2009-07-10 | 2013-07-31 | Janssen Pharmaceutica, N.V. | Postopek kristalizacije 1-(beta-d-glukopiranosil)-4-metil-3-(5-(4-fluorofenil)-2-tienilmetil) benzena |
| TW201118099A (en) * | 2009-08-12 | 2011-06-01 | Boehringer Ingelheim Int | New compounds for the treatment of CNS disorders |
| EA022032B1 (ru) * | 2009-09-30 | 2015-10-30 | Бёрингер Ингельхайм Интернациональ Гмбх | Способ получения глюкопиранозилзамещенных производных бензилбензола |
| MX2012002942A (es) * | 2009-09-30 | 2012-04-11 | Boehringer Ingelheim Int | Metodo para la preparacion de una forma cristalina de 1-cloro-4-(beta-d-glucopiranos-1-il)-2-(4-((s)-teteahidrofuran-3- iloxi)bencil)-benceno. |
| UY32919A (es) * | 2009-10-02 | 2011-04-29 | Boehringer Ingelheim Int | Composición farmacéutica, forma de dosificación farmacéutica, procedimiento para su preparación, mé todos para su tratamiento y sus usos |
| EP2488515B1 (en) | 2009-10-14 | 2017-01-04 | Janssen Pharmaceutica NV | Process for the preparation of compounds useful as inhibitors of sglt2 |
| JP5696156B2 (ja) | 2009-11-02 | 2015-04-08 | ファイザー・インク | ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール誘導体 |
| EA022365B1 (ru) | 2010-05-11 | 2015-12-30 | Янссен Фармацевтика Нв | Фармацевтические композиции, содержащие производные 1-(бета-d-глюкопиранозил)-2-тиенилметилбензола как ингибиторы нзпг |
| WO2011153712A1 (en) | 2010-06-12 | 2011-12-15 | Theracos, Inc. | Crystalline form of benzylbenzene sglt2 inhibitor |
| WO2012003811A1 (en) * | 2010-07-09 | 2012-01-12 | Zhejiang Beta Pharma Inc. | C-aryl glucoside sglt2 inhibitors and method |
| ES2626006T3 (es) | 2010-08-12 | 2017-07-21 | Boehringer Ingelheim International Gmbh | Derivados de 6-cicloalquil-1,5-dihidro-pirazolo[3,4-d]pirimidin-4-ona y su uso como inhibidores de PDE9A |
| WO2012025857A1 (en) | 2010-08-23 | 2012-03-01 | Hetero Research Foundation | Cycloalkyl methoxybenzyl phenyl pyran derivatives as sodium dependent glucose co transporter (sglt2) inhibitors |
| US8809345B2 (en) | 2011-02-15 | 2014-08-19 | Boehringer Ingelheim International Gmbh | 6-cycloalkyl-pyrazolopyrimidinones for the treatment of CNS disorders |
| CN103965176B (zh) | 2011-02-18 | 2016-03-16 | 凯惠药业(上海)有限公司 | 一种芳基糖苷类化合物及其制备方法和应用 |
| CN102167715B (zh) * | 2011-03-07 | 2013-04-24 | 上海惠斯生物科技有限公司 | 一种钠-葡萄糖协同运转蛋白-2原料药的共晶制备方法 |
| UY33937A (es) | 2011-03-07 | 2012-09-28 | Boehringer Ingelheim Int | Composiciones farmacéuticas que contienen inhibidores de dpp-4 y/o sglt-2 y metformina |
| CN102675378A (zh) * | 2011-03-09 | 2012-09-19 | 天津药物研究院 | 一类含环丙烷结构的c-葡萄糖苷衍生物、其制备方法和用途 |
| RS55056B1 (sr) | 2011-04-13 | 2016-12-30 | Janssen Pharmaceutica Nv | Proces za pripremu jedinjenja koja su korisna kao inhibitori sglt2 |
| TWI542596B (zh) | 2011-05-09 | 2016-07-21 | 健生藥品公司 | (2s,3r,4r,5s,6r)-2-(3-((5-(4-氟苯基)噻吩-2-基)甲基)-4-甲基苯基)-6-(羥甲基)四氫-2h-哌喃-3,4,5-三醇之l-脯胺酸及檸檬酸共晶體 |
| ES2448398T3 (es) | 2011-06-03 | 2014-03-13 | Ratiopharm Gmbh | Composición farmacéutica que comprende dapagliflozina y ciclodextrina |
| EP2714049A1 (en) | 2011-06-03 | 2014-04-09 | Ratiopharm GmbH | Pharmaceutical composition comprising dapagliflozin and cyclodextrin |
| WO2013000275A1 (zh) | 2011-06-25 | 2013-01-03 | 山东轩竹医药科技有限公司 | C-糖苷衍生物 |
| CN102408459B (zh) * | 2011-09-29 | 2014-07-23 | 天津药物研究院 | 一类含异头位烷基的苯基c-葡萄糖苷衍生物、其制备方法和用途 |
| BR112014010574A2 (pt) * | 2011-10-31 | 2017-05-02 | Scinopharm Taiwan Ltd | formas cristalinas e não cristalinas de inibidores sglt2 |
| US8952139B2 (en) | 2011-11-07 | 2015-02-10 | Scinopharm Taiwan, Ltd | Process for the preparation of β-C-aryl glucosides |
| TWI488861B (zh) * | 2011-11-07 | 2015-06-21 | 台灣神隆股份有限公司 | β-C-芳基葡萄糖苷之製備方法 |
| US9555001B2 (en) | 2012-03-07 | 2017-01-31 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition and uses thereof |
| US9192617B2 (en) | 2012-03-20 | 2015-11-24 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition, methods for treating and uses thereof |
| US9193751B2 (en) | 2012-04-10 | 2015-11-24 | Theracos, Inc. | Process for the preparation of benzylbenzene SGLT2 inhibitors |
| WO2013152476A1 (en) * | 2012-04-10 | 2013-10-17 | Theracos, Inc. | Process for the preparation of benzylbenzene sglt2 inhibitors |
| US9145434B2 (en) * | 2012-07-26 | 2015-09-29 | Boehringer Ingelheim International Gmbh | Crystalline complex of 1-cyano-2-(4-cyclopropyl-benzyl)-4-(ss-d-glucopyranos-1-yl)-benzene, methods for its preparation and the use thereof for preparing medicaments |
| CN103910769B (zh) | 2012-12-31 | 2018-10-02 | 上海璎黎药业有限公司 | 葡萄糖衍生物和脯氨酸的复合物、晶体、制备方法及应用 |
| ES2969245T3 (es) | 2013-03-14 | 2024-05-17 | Msd Int Gmbh | Formas cristalinas y métodos para preparar inhibidores de SGLT2 |
| HUE064190T2 (hu) | 2013-04-04 | 2024-03-28 | Boehringer Ingelheim Vetmedica Gmbh | Lófélék anyagcserezavarainak kezelése |
| US20140303097A1 (en) | 2013-04-05 | 2014-10-09 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition, methods for treating and uses thereof |
| ES2702174T3 (es) | 2013-04-05 | 2019-02-27 | Boehringer Ingelheim Int | Usos terapéuticos de empagliflozina |
| US11813275B2 (en) | 2013-04-05 | 2023-11-14 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition, methods for treating and uses thereof |
| MX2021004308A (es) | 2013-04-18 | 2022-10-26 | Boehringer Ingelheim Int | Empagliflozina para usarse en el tratamiento de micro y macroalbuminuria. |
| CN104250272B (zh) * | 2013-06-27 | 2018-10-09 | 上海方楠生物科技有限公司 | 一种利用微反应器制备列净类药物中间体的方法 |
| JP6574417B2 (ja) * | 2013-07-22 | 2019-09-11 | サンド・アクチエンゲゼルシヤフト | 非晶質ダパグリフロジンを含有する製剤 |
| WO2015040571A1 (en) | 2013-09-23 | 2015-03-26 | Ranbaxy Laboratories Limited | Process for the preparation of dapagliflozin |
| EP3049397A1 (en) | 2013-09-27 | 2016-08-03 | Sun Pharmaceutical Industries Ltd | Process for the purification of dapagliflozin |
| CN105611920B (zh) | 2013-10-12 | 2021-07-16 | 泰拉科斯萨普有限责任公司 | 羟基-二苯甲烷衍生物的制备 |
| US20160280619A1 (en) | 2013-10-31 | 2016-09-29 | Sun Pharmaceutical Industries Limited | Process for the preparation of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene |
| CA2930034C (en) | 2013-12-17 | 2022-08-16 | Boehringer Ingelheim Vetmedica Gmbh | Treatment of metabolic disorders in feline animals |
| WO2015101916A1 (en) | 2013-12-30 | 2015-07-09 | Mylan Laboratories Ltd. | Process for the preparation of empagliflozin |
| CN104761522B (zh) * | 2014-01-03 | 2017-02-15 | 山东轩竹医药科技有限公司 | 光学纯的苄基‑4‑氯苯基的c‑糖苷衍生物 |
| US9315438B2 (en) | 2014-01-03 | 2016-04-19 | Xuanzhu Pharma Co., Ltd | Optically pure benzyl-4-chlorophenyl-C-glucoside derivative |
| ES2712860T3 (es) * | 2014-01-23 | 2019-05-16 | Boehringer Ingelheim Vetmedica Gmbh | Tratamiento de trastornos metabólicos en animales caninos |
| US20170029398A1 (en) * | 2014-03-06 | 2017-02-02 | Srinivasan Thirumalai Rajan | Process for the preparation of (1s)-1,5-anhydro-1-c-{4-chloro-3-4[(4-ethoxyphenyl)methyl]phenyl]-glucitol and its solvate thereof |
| KR102662473B1 (ko) | 2014-04-01 | 2024-05-03 | 베링거잉겔하임베트메디카게엠베하 | 말과 동물에서 대사 장애의 치료 |
| CN105001213B (zh) | 2014-04-14 | 2020-08-28 | 上海迪诺医药科技有限公司 | C-芳基糖苷衍生物、其药物组合物、制备方法及应用 |
| EP3229814A4 (en) | 2014-06-23 | 2018-07-11 | Sun Pharmaceutical Industries Ltd | Co-crystal of dapagliflozin with citric acid |
| WO2016041470A1 (en) * | 2014-09-15 | 2016-03-24 | National Institute Of Biological Sciences, Beijing | Sglt-2 inhibitors |
| BR112017003570A2 (pt) | 2014-09-25 | 2017-12-05 | Boehringer Ingelheim Vetmedica Gmbh | tratamento de combinação de inibidores de sglt2 e agonistas de dopamina para prevenir distúrbios metabólicos em animais equinos |
| CN104478839A (zh) * | 2014-11-24 | 2015-04-01 | 苏州乔纳森新材料科技有限公司 | 一种达格列净的合成方法 |
| CN104496952B (zh) * | 2014-11-28 | 2017-04-19 | 深圳翰宇药业股份有限公司 | 一种达格列净的合成方法 |
| EP3226874A1 (en) * | 2014-12-03 | 2017-10-11 | Sun Pharmaceutical Industries Ltd | Processes for the preparation of ertugliflozin |
| CN105753827A (zh) * | 2014-12-17 | 2016-07-13 | 中美华世通生物医药科技(武汉)有限公司 | 制备化合物的方法 |
| CN104447907A (zh) * | 2015-01-14 | 2015-03-25 | 佛山市赛维斯医药科技有限公司 | 含硝基联苯双葡萄糖苷结构化合物、其制备方法和用途 |
| CN104447905A (zh) * | 2015-01-14 | 2015-03-25 | 佛山市赛维斯医药科技有限公司 | 一种含硝基苯和双o-葡萄糖苷衍生物、其制备方法和用途 |
| CN104478969A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含烷氧苯基噻唑基的o-半乳糖苷衍生物、其制备方法和用途 |
| CN104478962A (zh) * | 2015-01-15 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 一类卤代苯基s-葡萄糖苷衍生物、其制备方法和用途 |
| US10508128B2 (en) | 2015-02-09 | 2019-12-17 | Indoco Remedies Limited | Process for the preparation of SGLT inhibitor compounds |
| WO2016147197A1 (en) * | 2015-03-17 | 2016-09-22 | Harman Finochem Limited | A novel process for preparing (2s,3r,4r,5s,6r)-2-[4-chloro-3-(4-ethoxybenzyl)pheny 1] -6-(hy droxy methyl)tetrahydro-2h-py ran-3,4,5-triol and its amorphous form |
| WO2016161995A1 (en) | 2015-04-08 | 2016-10-13 | Zentiva, K.S. | Solid forms of amorphous dapagliflozin |
| WO2016178148A1 (en) * | 2015-05-05 | 2016-11-10 | Glenmark Pharmaceuticals Limited | Process for preparation of dapagliflozin |
| CN107995862B8 (zh) | 2015-08-27 | 2021-12-03 | 勃林格殷格翰维特梅迪卡有限公司 | 包含sglt-2抑制剂之液态药物组合物 |
| WO2017042683A1 (en) * | 2015-09-07 | 2017-03-16 | Dr. Reddy's Laboratories Limited | Isolated intermediate of dapagliflozin, process for the preparation of isolated intermediate of dapagliflozin, process for the preparation of dapagliflozin |
| US20170071970A1 (en) | 2015-09-15 | 2017-03-16 | Janssen Pharmaceutica Nv | Co-therapy comprising canagliflozin and phentermine for the treatment of obesity and obesity related disorders |
| EP3349762B1 (en) | 2015-09-15 | 2021-08-25 | Laurus Labs Limited | Co-crystals of sglt2 inhibitors, process for their preparation and pharmaceutical compositions thereof |
| WO2017060924A1 (en) * | 2015-10-09 | 2017-04-13 | Harman Finochem Limited | A novel pipecolic acid co-crystal of canagliflozin and process for the preparation thereof |
| WO2017099496A1 (ko) * | 2015-12-11 | 2017-06-15 | 동아에스티 주식회사 | 다파글리플로진의 신규 용매화물 및 이의 제조방법 |
| WO2017141202A1 (en) | 2016-02-17 | 2017-08-24 | Lupin Limited | Complex of sglt2 inhibitor and process for preparation thereof |
| WO2017221211A1 (en) * | 2016-06-24 | 2017-12-28 | Biocon Limited | Process for the preparation of dapagliflozin and its solvate thereof |
| WO2018142422A1 (en) * | 2017-02-02 | 2018-08-09 | Indoco Remedies Limited | Process for the preparation of dapagliflozin |
| US11020412B2 (en) | 2017-03-16 | 2021-06-01 | Inventia Healthcare Limited | Pharmaceutical composition comprising dapagliflozin |
| CN106938998B (zh) * | 2017-04-07 | 2018-07-03 | 四川智强医药科技开发有限公司 | 卡格列净有关物质的合成方法 |
| US11198703B2 (en) * | 2017-05-09 | 2021-12-14 | Piramal Enterprises Limited | Process for the preparation of SGLT2 inhibitors and intermediates thereof |
| CN107488156B (zh) * | 2017-09-04 | 2020-05-26 | 上海现代制药股份有限公司 | 一种无定型葡萄糖醇的合成方法 |
| CN108530408A (zh) * | 2018-04-13 | 2018-09-14 | 海门慧聚药业有限公司 | 制备达格列净的方法 |
| CN108997452A (zh) * | 2018-07-06 | 2018-12-14 | 陕西中医药大学 | 一种乙酰化二苯乙烯苷的合成方法 |
| WO2020151623A1 (zh) * | 2019-01-24 | 2020-07-30 | 北京盈科瑞创新药物研究有限公司 | 化合物、其制备方法及其作为药物中间体的应用 |
| CN111471040B (zh) * | 2019-01-24 | 2023-06-02 | 北京盈科瑞创新药物研究有限公司 | 一种糖苷类衍生物的合成方法及其中间体和应用 |
| CN110105459A (zh) * | 2019-05-27 | 2019-08-09 | 云南绿戎生物产业开发股份有限公司 | 一种三七多糖的提取方法 |
| CN110305118B (zh) * | 2019-06-20 | 2024-04-02 | 四川科伦药物研究院有限公司 | 一种适合工业生产恩格列净的合成方法 |
| EP4106732A1 (en) | 2020-02-21 | 2022-12-28 | Zaklady Farmaceutyczne Polpharma S.A. | Pharmaceutical composition comprising dapagliflozin |
| EP4114365A1 (en) | 2020-03-05 | 2023-01-11 | KRKA, d.d., Novo mesto | Pharmaceutical composition comprising sglt2 inhibitor |
| CN115867538A (zh) | 2020-06-05 | 2023-03-28 | 新梅斯托克公司 | 高纯的无定形达格列净的制备 |
| WO2021260617A1 (en) * | 2020-06-25 | 2021-12-30 | Hikal Limited | An improved process for preparation of dapagliflozin propanediol monohydrate |
| CN112159436A (zh) * | 2020-07-07 | 2021-01-01 | 杭州杜易科技有限公司 | 一种多羟基的糖类化合物的制备方法 |
| CN112500267A (zh) | 2020-12-04 | 2021-03-16 | 江苏慧聚药业有限公司 | 4-溴-2-(4’-乙氧基-苄基)-1-氯苯的制备 |
| CN113248464B (zh) * | 2021-05-31 | 2021-10-26 | 北京惠之衡生物科技有限公司 | 一种c-糖苷类衍生物的合成方法 |
| CN113461659B (zh) * | 2021-07-19 | 2022-11-11 | 上海应用技术大学 | 一种c-螺环列净类似物中间体及其制备方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5977328A (en) * | 1997-03-12 | 1999-11-02 | Krepinsky; Jiri J. | Polyvalent carbohydrate molecules |
| US6069238A (en) | 1998-09-30 | 2000-05-30 | Eli Lilly And Company | Spirocyclic C-glycosides |
| PH12000002657B1 (en) | 1999-10-12 | 2006-02-21 | Bristol Myers Squibb Co | C-aryl glucoside SGLT2 inhibitors |
| US6515117B2 (en) * | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| CA2444481A1 (en) | 2001-04-11 | 2002-10-24 | Bristol-Myers Squibb Company | Amino acid complexes of c-aryl glucosides for treatment of diabetes and method |
| CN100391963C (zh) * | 2003-01-03 | 2008-06-04 | 布里斯托尔-迈尔斯斯奎布公司 | 制备c-芳基葡糖苷sglt2抑制剂的方法 |
-
2003
- 2003-12-23 CN CNB2003801100401A patent/CN100391963C/zh not_active Expired - Fee Related
- 2003-12-23 BR BRPI0317929-0A patent/BR0317929A/pt not_active IP Right Cessation
- 2003-12-23 MX MXPA05007052A patent/MXPA05007052A/es unknown
- 2003-12-23 WO PCT/US2003/041373 patent/WO2004063209A2/en active Application Filing
- 2003-12-23 PL PL378324A patent/PL378324A1/pl unknown
- 2003-12-23 US US10/745,075 patent/US7375213B2/en active Active
- 2003-12-23 CN CNA2008100884952A patent/CN101260130A/zh active Pending
- 2003-12-23 AU AU2003299966A patent/AU2003299966A1/en not_active Abandoned
- 2003-12-23 CA CA002512389A patent/CA2512389A1/en not_active Abandoned
- 2003-12-23 EP EP03800231A patent/EP1581543A4/en not_active Withdrawn
- 2003-12-23 JP JP2004566603A patent/JP2006516257A/ja not_active Withdrawn
- 2003-12-23 KR KR1020057012462A patent/KR20050090437A/ko not_active Application Discontinuation
- 2003-12-30 TW TW096116680A patent/TW200801030A/zh unknown
- 2003-12-30 TW TW092137505A patent/TW200424213A/zh unknown
-
2007
- 2007-06-19 US US11/764,839 patent/US7932379B2/en active Active
-
2011
- 2011-04-25 US US13/093,430 patent/US20110201795A1/en not_active Abandoned
-
2014
- 2014-05-01 US US14/267,341 patent/US20140243517A1/en not_active Abandoned
Non-Patent Citations (4)
| Title |
|---|
| AgoTfa/SnCl4:a powerful new promoter combination in thearyl c-glycosidation of driverse range of sugar acetates andaromatic substrates. Takeshi Kuribayashi et al.Tetrahedron letters,Vol.39 . 1998 |
| AgoTfa/SnCl4:a powerful new promoter combination in thearyl c-glycosidation of driverse range of sugar acetates andaromatic substrates. Takeshi Kuribayashi et al.Tetrahedron letters,Vol.39 . 1998 * |
| c-glyocosylated diphenylmethanes andbenzophenones:thestille coupling reaction of c-glycosylatedaryl tins withbenzylbromides and acid chlorides. Takeshi Kuribayashi et al.J. Carboxydrate chemistry,Vol.18 No.4. 1999 |
| c-glyocosylated diphenylmethanes andbenzophenones:thestille coupling reaction of c-glycosylatedaryl tins withbenzylbromides and acid chlorides. Takeshi Kuribayashi et al.J. Carboxydrate chemistry,Vol.18 No.4. 1999 * |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104109179A (zh) * | 2013-04-16 | 2014-10-22 | 杭州华东医药集团生物工程研究所有限公司 | 一类c-芳基葡萄糖苷衍生物、制备方法及其用途 |
| CN104478957A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 一种含腈基苯和双o-葡萄糖苷衍生物、其制备方法和用途 |
| CN104478968A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含噻唑基的o-半乳糖苷衍生物、其制备方法和用途 |
| CN104478959A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含腈基联苯双葡萄糖苷结构化合物、其制备方法和用途 |
| CN104478966A (zh) * | 2015-01-14 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含苯胺噻唑基的o-半乳糖苷衍生物、其制备方法和用途 |
| CN104530149A (zh) * | 2015-01-14 | 2015-04-22 | 佛山市赛维斯医药科技有限公司 | 卤代苯基双o-葡萄糖苷衍生物、其制备方法和用途 |
| CN104478965A (zh) * | 2015-01-15 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 一类烷氧基苯基s-葡萄糖苷衍生物、其制备方法和用途 |
| CN104478964A (zh) * | 2015-01-15 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 含丙烯腈结构的苯o-葡萄糖苷衍生物、其制备方法和用途 |
| CN104478963A (zh) * | 2015-01-15 | 2015-04-01 | 佛山市赛维斯医药科技有限公司 | 一种含腈基苯s-葡萄糖苷结构的化合物及其用途 |
| CN104497070A (zh) * | 2015-01-15 | 2015-04-08 | 佛山市赛维斯医药科技有限公司 | 一种含三氟甲基苯s-葡萄糖苷结构的化合物和用途 |
| CN105218329A (zh) * | 2015-10-15 | 2016-01-06 | 上海应用技术学院 | 一种列净类似物中间体及其制备方法 |
| CN105218329B (zh) * | 2015-10-15 | 2017-05-03 | 上海应用技术学院 | 一种列净类似物中间体及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20140243517A1 (en) | 2014-08-28 |
| US20070238866A1 (en) | 2007-10-11 |
| JP2006516257A (ja) | 2006-06-29 |
| CA2512389A1 (en) | 2004-07-29 |
| US20040138439A1 (en) | 2004-07-15 |
| CN1756759A (zh) | 2006-04-05 |
| TW200801030A (en) | 2008-01-01 |
| EP1581543A4 (en) | 2008-03-19 |
| WO2004063209A2 (en) | 2004-07-29 |
| KR20050090437A (ko) | 2005-09-13 |
| MXPA05007052A (es) | 2005-08-18 |
| EP1581543A2 (en) | 2005-10-05 |
| AU2003299966A1 (en) | 2004-08-10 |
| BR0317929A (pt) | 2006-04-11 |
| US7375213B2 (en) | 2008-05-20 |
| WO2004063209A3 (en) | 2005-05-12 |
| TW200424213A (en) | 2004-11-16 |
| CN101260130A (zh) | 2008-09-10 |
| US7932379B2 (en) | 2011-04-26 |
| US20110201795A1 (en) | 2011-08-18 |
| PL378324A1 (pl) | 2006-03-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100391963C (zh) | 制备c-芳基葡糖苷sglt2抑制剂的方法 | |
| US10889574B2 (en) | Method for producing diphenylmethane derivative | |
| CN103965176B (zh) | 一种芳基糖苷类化合物及其制备方法和应用 | |
| SA99191247B1 (ar) | أميدات عطرية aromatic amides حديثة وطريقة لتحضيرها ولاستخدامها كعقاقير | |
| US20080167463A1 (en) | Process for Preparation of Gemcitabine Hydrochloride | |
| CN109988161A (zh) | 一种适合工业化生产恩格列净的制备方法 | |
| EP3148979A1 (en) | Process for preparation of canagliflozin | |
| WO2018142422A1 (en) | Process for the preparation of dapagliflozin | |
| EP3256482B1 (en) | Process for the preparation of sglt inhibitor compounds | |
| US20190048034A1 (en) | Method for producing alpha-halo-tetraacyl-glucose | |
| Su et al. | Facile synthesis of 2-deoxy-2-substituted D-arabinofuranose derivatives | |
| CN102234260A (zh) | 含二氟亚甲基基团的c-芳基葡糖苷衍生物 | |
| Graziani et al. | A mild and efficient approach for the regioselective silyl-mediated protection–deprotection of C-4 hydroxyl group on carbohydrates | |
| CN109071551A (zh) | 一种三氟甲基取代的吡喃衍生物的制备方法 | |
| CN102351860B (zh) | 一种帕尼培南及其中间体的合成方法 | |
| Ferrières et al. | Sulfur atom configuration of sulfinyl galactofuranosides determines different reactivities in glycosylation reactions | |
| Gómez et al. | Synthesis of 2, 3: 4, 6-di-O-isopropylidene-d-allopyranose from d-glucose | |
| CA2725842C (en) | Process for the glycosidation of colchicine and thiocolchicine | |
| CN105330706B (zh) | 卡格列净中间体的制备方法 | |
| CN105985296B (zh) | 一种可以工业化的lesinurad中间体1-萘基三唑硫酮的精制工艺 | |
| JPS60163892A (ja) | 光学活性な重水素化糖の製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1087716 Country of ref document: HK |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20080604 Termination date: 20100125 |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1087716 Country of ref document: HK |