JP4397990B2 - 3−アルキルフラバノノール誘導体の精製法 - Google Patents
3−アルキルフラバノノール誘導体の精製法 Download PDFInfo
- Publication number
- JP4397990B2 JP4397990B2 JP00039899A JP39899A JP4397990B2 JP 4397990 B2 JP4397990 B2 JP 4397990B2 JP 00039899 A JP00039899 A JP 00039899A JP 39899 A JP39899 A JP 39899A JP 4397990 B2 JP4397990 B2 JP 4397990B2
- Authority
- JP
- Japan
- Prior art keywords
- derivative
- alkylflavanone
- alkylflavanonol
- crystallization
- impurity
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Landscapes
- Pyrane Compounds (AREA)
Description
【発明の属する技術分野】
本発明は、養毛・育毛剤として有用な次の一般式(1)
【0002】
【化3】
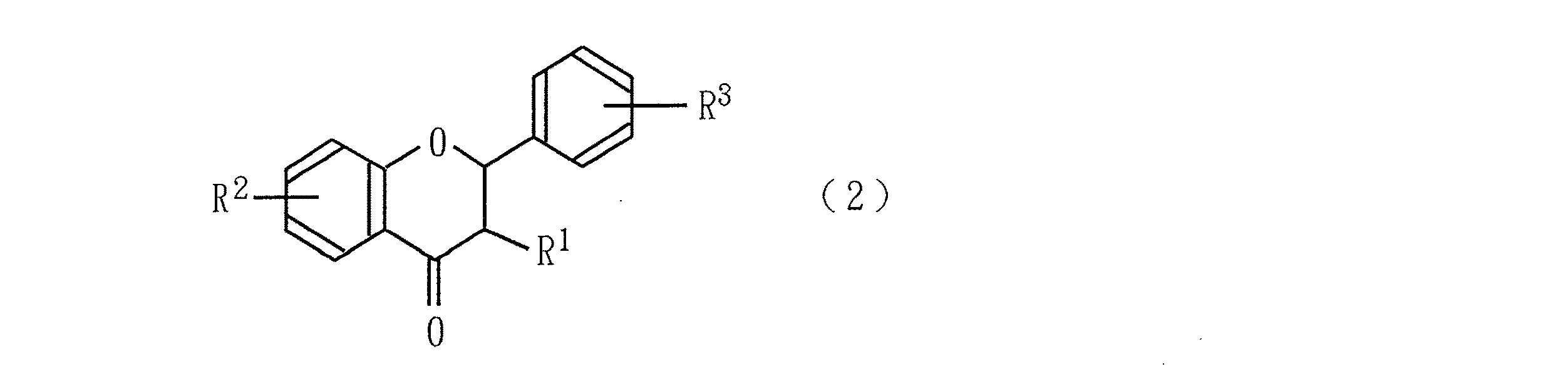
〔式中、R1 は炭素数1〜12のアルキル基を示し、R2 及びR3 は、ハロゲン原子若しくは炭素数1〜12のアルコキシル基が置換していてもよい炭素数1〜12のアルキル基、炭素数1〜12のアルコキシル基が置換していてもよい炭素数1〜12のアルコキシル基、水素原子、水酸基、シアノ基又はハロゲン原子を示す。〕
で表わされる3−アルキルフラバノノール誘導体の精製法に関する。
【0004】
【従来の技術】
養毛・育毛剤として有用な化合物である上記一般式(1)で表わされる3−アルキルフラバノノール誘導体の製造方法としては、特開平8−157464号公報に記載の如く、次の反応式に従って反応を行う方法が挙げられる。
【0005】
【化4】
〔式中、R1 、R2 及びR3 は前記と同じものを示す〕
【0007】
すなわち、o−ヒドロキシアシルベンゼン類(4)とベンズアルデヒド類(3)を反応させて、3−アルキルフラバノン類(2)を得、次いでこれを過酸化水素で酸化し、カラムクロマトグラフィー精製の後、晶析精製を行うことにより、目的とする3−アルキルフラバノノール誘導体(1)を得る方法が開示されている。
【0008】
【発明が解決しようとする課題】
しかし、上記製造方法では、精製法としてカラムクロマトグラフィーを用いており、工業的に大量に精製・製造するには不向きであるという問題点があった。また、原料である3−アルキルフラバノン誘導体(2)の溶解度が目的物である3−アルキルフラバノノール誘導体(1)よりも低いため、工業的に通常用いられる精製方法である晶析では、酸化反応後に不純物として残存する原料の3−アルキルフラバノン誘導体(2)を完全に除くことが困難であり、十分に精製できないという問題点を有していた。
従って、本発明の目的は、このような問題がなく、3−アルキルフラバノノール誘導体(1)を工業的に有利に精製・製造できる方法を提供することにある。
【0009】
【課題を解決するための手段】
本発明は、次の一般式(2)
【0010】
【化5】
〔式中、R1 は炭素数1〜12のアルキル基を示し、R2 及びR3 は、ハロゲン原子若しくは炭素数1〜12のアルコキシル基が置換していてもよい炭素数1〜12のアルキル基、炭素数1〜12のアルコキシル基が置換していてもよい炭素数1〜12のアルコキシル基、水素原子、水酸基、シアノ基又はハロゲン原子を示す。〕
で表わされる3−アルキルフラバノン誘導体を不純物として含む、次の一般式(1)
【0012】
【化6】
〔式中、R1 、R2 及びR3 は前記と同じものを示す〕
で表わされる3−アルキルフラバノノール誘導体を、塩基共存下で晶析することを特徴とする3−アルキルフラバノノール誘導体の高純度精製法を提供するものである。
【0014】
【発明の実施の形態】
本発明において、3−アルキルフラバノン誘導体(2)及び3−アルキルフラバノノール誘導体(1)は、それぞれ一般式(2)及び(1)で表わされるものであるが、式中、R1 は炭素数1〜5の直鎖又は分岐鎖のアルキル基が好ましく、特にメチル基が好ましい。R2 及びR3 は水素原子、炭素数1〜5の直鎖又は分岐鎖のアルキル基が好ましく、特に水素原子又はメチル基が好ましい。
【0015】
3−アルキルフラバノン誘導体(2)及び3−アルキルフラバノノール誘導体(1)として、最も好ましいものは、式中のR1 及びR3 がメチル基であり、R2 が水素原子である、3,4′−ジメチルフラバノン及び3,4′−ジメチル−3−ヒドロキシフラバノンである。
【0016】
なお、本発明において原料となる3−アルキルフラバノン誘導体(2)には、trans体とcis体の立体異性体が存在し、また、trans体とcis体のそれぞれについて光学異性体が存在するが、本発明の製造法では、これらの異性体のいずれを用いても良く、これらの混合物を用いてもよい。
【0017】
本発明の晶析に用いる、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)中の、3−アルキルフラバノン誘導体(2)及び3−アルキルフラバノノール誘導体(1)の含有量は、好ましくは3−アルキルフラバノン誘導体(2)が0.1〜50%、3−アルキルフラバノノール誘導体(1)99.9〜50%程度含有するものを用いる。なお、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)には、3−アルキルフラバノン誘導体(2)及び3−アルキルフラバノノール誘導体(1)の他、3−アルキルフラバノン誘導体(2)の酸化で生成するようなその他の不純物を含んでいてもよい。その他の不純物としては、例えば安息香酸、ケイヒ酸、サリチル酸、カテコールの各誘導体等が挙げられる。
【0018】
本発明の晶析に用いる、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)は、後述するような3−アルキルフラバノン誘導体(2)の酸化粗生成物をそのまま用いてもよいし、水洗等の予備的精製を行ったものでもよい。また、本発明の晶析に用いる、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)は、乾燥したものでもよく、また、水等の溶媒を含んだものでもよい。ただし、水を含有する場合には、水の含有量が35%以下程度であることが望ましい。
【0019】
本精製法において、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)の製造方法は前記したように、特開平8−157464号公報又はJ. Med. Chem.(ジャーナル オブ メディシナルケミストリー)33巻1948頁(1990年)に記載の方法で、3−アルキルフラバノン誘導体(2)を中間体として得た後、これを酸化する方法が挙げられる。すなわち、前記反応式に従ってo−ヒドロキシアシルベンゼン類(4)とベンズアルデヒド類(3)を有機酸とアミンの存在下で縮合するか、又は無機塩基の存在下で縮合して、3−アルキルフラバノン誘導体(2)を塩基存在下、有機過酸化物又は過酸化水素で酸化することにより、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)を得る方法が挙げられる。
【0020】
上記酸化反応終了後の後処理方法は、通常の過酸化物を用いた反応の後処理方法に準じて行う。すなわち、残存する有機過酸化物を水洗除去、もしくは、必要に応じて亜硫酸水素ナトリウム水溶液等で分解した後、本発明の晶析精製に供する。
【0021】
本発明の精製法は、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)を、塩基存在下で晶析することに特徴がある。
【0022】
本発明の晶析に用いる塩基としては、不純物である3−アルキルフラバノン誘導体(2)の3位の水素を引き抜くことができ、3−アルキルフラバノン誘導体(2)をより溶媒への溶解性のよいα−アルキルカルコン誘導体にできる塩基であればよく、中でも、水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物;ナトリウムメトキシド、ナトリウムエトキシド等の金属アルコキシドが好ましく、特に水酸化カリウムが最も好ましい。なお、水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物は、フレーク、ペレット等の固形物の形態で用いても、また、水溶液の形態(水酸化ナトリウム、水酸化カリウムの場合は20〜50%程度の水溶液が市販されている)で用いてもよい。
【0023】
塩基の使用量は、用いる3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)の純度、溶媒の種類・量等により変化するが、一般に3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)に対して0.03〜1.0重量倍、好ましくは0.05〜0.5重量倍、より好ましくは0.05〜0.3重量倍程度用いる。
【0024】
本発明の晶析に用いる溶媒としては、不純物である3−アルキルフラバノン誘導体(2)の3位の水素を引き抜くことにより生成するα−アルキルカルコン誘導体を溶解できる溶媒であればよく、メタノール、エタノール、n−プロパノール、イソプロピルアルコール、n−ブタノール、イソブチルアルコール、sec−ブチルアルコール、t−ブチルアルコール等の炭素数4以下のアルコール系溶媒、又は、これら炭素数4以下のアルコール系溶媒と水との混合溶媒がより好ましく、その中でも3−アルキルフラバノン誘導体(2)の3位の水素を引き抜くことにより生成するα−アルキルカルコン誘導体の溶解度がよいことから、イソプロピルアルコールと水の混合溶媒が最も好ましい。
【0025】
溶媒の使用量は、用いる3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)の純度、溶媒の種類、塩基の種類・量等によって変化するが、一般に、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)に対して1〜20重量倍、好ましくは1〜10重量倍、より好ましくは2〜7重量倍程度である。
【0026】
本発明の晶析では、溶媒に水を含有してもしなくてもよいが、水を含有した場合の方が、目的物である3−アルキルフラバノノール誘導体(1)の回収率がよい傾向があるので、好ましくは水を含有させた溶媒を用いる。水を含有させる場合の水の使用量は、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)に対して0.01〜1重量倍、好ましくは0.05〜0.5重量倍、より好ましくは0.05〜0.3重量倍程度である。なお、晶析に用いる3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)に水分が含有されている場合には、晶析溶媒に水を添加しなくてもよい。
【0027】
晶析の際に、塩基は最初から3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)の溶液に加えておいてもよいが、塩基を高温で3−アルキルフラバノノール誘導体(1)に作用させると、trans−cis異性化等の副反応が進行することがあるので、好ましくは、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)の溶液に、塩基水溶液を添加することで結晶を生成させて、晶析を行う。
【0028】
晶析温度は、用いる3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)の純度、溶媒の種類、塩基の種類・量、塩基水溶液の添加方法等によって変化するが、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)の溶液に、塩基水溶液を添加する場合は、好ましくは0〜60℃、より好ましくは30〜50℃で塩基水溶液を添加し、その後好ましくは−40〜30℃、より好ましくは−30〜30℃程度まで冷却して晶析を行う。また、3−アルキルフラバノン誘導体(2)を不純物として含む3−アルキルフラバノノール誘導体(1)の溶液に、塩基を最初から加えておく場合は、好ましくは10〜50℃で加温後、好ましくは−40〜30℃程度まで冷却して晶析を行う。
【0029】
上記操作により析出した結晶をろ過することにより、3−アルキルフラバノノール誘導体(1)を純度よく得ることができるが、更に残存する不純物や晶析で用いた塩基を除くために、水、アルコール系溶媒、水−アルコール系溶媒の混合溶媒等及びこれらの2種以上で1又は2回以上洗浄してもよい。ここで用いるアルコール系溶媒として好ましいものとしては、メタノール、エタノール、n−プロパノール、イソプロピルアルコール、n−ブタノール、イソブチルアルコール、sec−ブチルアルコール、t−ブチルアルコール等の炭素数4以下のアルコール系溶媒が挙げられ、このうちイソプロピルアルコールがより好ましい。
【0030】
このようにして得られた結晶を、必要に応じ、常圧又は減圧下で乾燥することにより、3−アルキルフラバノノール誘導体(1)を高純度で得ることができる。
【0031】
本発明方法で精製した3−アルキルフラバノノール誘導体(1)は養毛・育毛料として有用であり、高純度であるため、皮膚外用剤として、医薬品、医薬部外品、化粧品等に有利に用いることができる。
【0032】
【実施例】
参考例 トランス−3,4′−ジメチル−3−ヒドロキシフラバノンの製造:窒素雰囲気下、フラスコに2′−ヒドロキシプロピオフェノン1682g(11.2mol)、4−メチルベンズアルデヒド1413g(11.76mol)、イソプロピルアルコール3364g、ピペリジン954g(11.2mol)を仕込み、ここに酢酸673g(11.2mol)を約10分間かけて滴下した。滴下終了後、更に80〜90℃で24時間攪拌した。次に、得られた反応混合物を70〜75℃まで冷却してトランス−3,4′−ジメチルフラバノンの種晶0.25gを添加し、30〜40℃まで冷却してイオン交換水1400gを加え、更に0〜10℃まで冷却した。析出してきた結晶をろ取し、得られた結晶を50%イソプロピルアルコール水溶液及び水で洗浄し、減圧乾燥することにより、トランス−3,4′−ジメチルフラバノン2506g(収率88.7%)を得た。
次いで、SUS316製反応槽に、上記で得られたトランス−3,4′−ジメチルフラバノン126.16g(0.5mol)及びイソプロピルアルコール189.24gを仕込み、次いで50%水酸化カリウム水溶液44.88g(KOH:0.4mol)を添加した。次にこの混合物に、35〜50℃で攪拌しながら70%t−ブチルヒドロパーオキシド水溶液128.74g(1.0mol)を約4時間かけて滴下した。40〜50℃で1時間攪拌した後、50%水酸化カリウム水溶液8.42g(KOH:0.15mol;初期添加分と合わせてKOH:0.55mol)を約1時間かけて滴下した。滴下終了後、更に40〜50℃で8時間攪拌した。次いで、この反応混合物を約15℃まで冷却した後、水252.3gを加え、更に約5℃まで冷却した。析出してきた結晶をろ取し、得られた結晶を水で洗浄した後、減圧乾燥することにより、シス−及びトランス−3,4′−ジメチルフラバノンを不純物として含むトランス−3,4′−ジメチル−3−ヒドロキシフラバノン粗製物86.3g(収率64.3%;HPLC純度82.0%、シス−及びトランス−3,4−ジメチルフラバノンを17.4%含有)を得た。
【0033】
比較例1 トランス−3,4′−ジメチル−3−ヒドロキシフラバノンの晶析精製:
フラスコに、シス−及びトランス−3,4′−ジメチルフラバノンを不純物として含むトランス−3,4′−ジメチル−3−ヒドロキシフラバノン粗製物10g(HPLC純度92.9%、シス−及びトランス−3,4′−ジメチルフラバノンを4.4%含有)及びイソプロピルアルコール50gを仕込み、加熱溶解させた後、約20℃まで冷却した。析出してきた結晶をろ取し、得られた結晶をイソプロピルアルコール10gで洗浄後、減圧乾燥した(回収率80%)。得られた結晶をHPLC分析したところ、トランス−3,4′−ジメチル−3−ヒドロキシフラバノンを96.3%、シス−及びトランス−3,4′−ジメチルフラバノンを3.1%含有していた。
【0034】
比較例2〜9
晶析/洗浄溶媒のイソプロピルアルコールを、表1に示す溶媒に変更した以外は、比較例1と同様の方法で晶析を行った。
【0035】
【表1】
実施例1 トランス−3,4′−ジメチル−3−ヒドロキシフラバノンの晶析精製:
フラスコに、シス−及びトランス−3,4′−ジメチルフラバノンを不純物として含むトランス−3,4′−ジメチル−3−ヒドロキシフラバノン粗製物85.7g(HPLC純度82.0%、シス−及びトランス−3,4′−ジメチルフラバノンを17.4%含有)及びイソプロピルアルコール257.1gを仕込み、約55℃付近で溶解させた後、約40℃まで冷却し、50%水酸化カリウム水溶液34.28gを滴下した。滴下終了後、この混合物を約5℃まで冷却し、析出してきた結晶をろ取し、得られた結晶を88%イソプロピルアルコール水溶液128.6gで洗浄した(この晶析法を1法とする)。ついで、脱塩基のために、水257.1g、イソプロピルアルコール51.4gと水205.7g、及び水257.1gで順次洗浄した(この脱塩基洗浄法をa法とする)後、減圧乾燥することにより、精製シランス−3,4′−ジメチル−3−ヒドロキシフラバノン62.0g(回収率72.3%;HPLC純度100%)を得た。
【0037】
実施例2 トランス−3,4′−ジメチル−3−ヒドロキシフラバノンの晶析精製:
フラスコに、シス−及びトランス−3,4′−ジメチルフラバノンを不純物として含むトランス−3,4′−ジメチル−3−ヒドロキシフラバノン粗製物85.7g(HPLC純度91.8%、シス−及びトランス−3,4′−ジメチルフラバノンを7.3%含有)、イソプロピルアルコール265.7g、及び50%水酸化カリウム水溶液34.28gを仕込み、加熱溶解後、約20℃まで冷却した。次に、析出してきた結晶をろ取し、得られた結晶を88%イソプロピルアルコール水溶液128.6gで洗浄した(この晶析法を2法とする)。次いで、脱塩基のために、20%イソプロピルアルコール257.1gで2回洗浄した(この脱塩基洗浄法をb法とする)後、減圧乾燥することにより、精製トランス−3,4′−ジメチル−3−ヒドロキシフラバノン69.4g(回収率81.0%;HPLC純度99.7%)を得た。
【0038】
実施例3〜18
表2に示す条件以外は、実施例1又は2と同様の方法で晶析を行った。なお、晶析条件及び塩基洗浄条件は、実施例1又は2中の晶析法1法又は2法、及び、洗浄法a法又はb法により行った。
【0039】
【表2】
【表3】
【発明の効果】
本発明によれば、養毛・育毛剤として有用な化合物である3−アルキルフラバノノール誘導体(1)を高純度で、しかも工業的に有利に精製することができる。
Claims (1)
- 次の一般式(2)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP00039899A JP4397990B2 (ja) | 1999-01-05 | 1999-01-05 | 3−アルキルフラバノノール誘導体の精製法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP00039899A JP4397990B2 (ja) | 1999-01-05 | 1999-01-05 | 3−アルキルフラバノノール誘導体の精製法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2000198779A JP2000198779A (ja) | 2000-07-18 |
| JP4397990B2 true JP4397990B2 (ja) | 2010-01-13 |
Family
ID=11472712
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP00039899A Expired - Fee Related JP4397990B2 (ja) | 1999-01-05 | 1999-01-05 | 3−アルキルフラバノノール誘導体の精製法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4397990B2 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9341618B2 (en) | 2007-02-22 | 2016-05-17 | Kao Corporation | Method for evaluating or screening hair growth-regulating agent |
| EP2742930B1 (en) * | 2011-08-09 | 2018-10-10 | Kao Corporation | Cosmetic emulsion for eyebrows or eyelashes |
| EP3733155B1 (en) | 2017-12-27 | 2022-11-23 | Kao Corporation | Method for manufacturing composition that includes poorly water-soluble aromatic compound |
-
1999
- 1999-01-05 JP JP00039899A patent/JP4397990B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2000198779A (ja) | 2000-07-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2004527577A (ja) | 4−フェニル酪酸の合成 | |
| JP3913329B2 (ja) | (±)−クロマンカルボン酸の光学分割法 | |
| EP1951657B1 (en) | Process for the manufacture of iohexol | |
| JP4397990B2 (ja) | 3−アルキルフラバノノール誘導体の精製法 | |
| JP3777408B2 (ja) | カルボン酸誘導体の製造法 | |
| KR910003709B1 (ko) | 부틸 3'-(1h-테트라졸-5-일)옥사닐레이트의 제조법 | |
| CN109265385B (zh) | 一种手性催化剂的合成工艺 | |
| JPH05286889A (ja) | アリール酢酸及びそれらのアルカリ金属塩の製造方法 | |
| EP0093511A1 (en) | Method for producing and optically active 2,2-dimethylcyclopropanecarboxylic acid | |
| JPH029576B2 (ja) | ||
| JP3640319B2 (ja) | ベンズアミド誘導体の製造方法 | |
| CN111072450A (zh) | 一种烯丙醇类衍生物的合成方法 | |
| EA011763B1 (ru) | Способы получения венлафаксина и формы i венлафаксина гидрохлорида | |
| JP4093608B2 (ja) | 光学活性2−フェノキシプロピオン酸の製造方法 | |
| JP3823385B2 (ja) | 2,4,5−トリフルオロ−3−ヨ−ド安息香酸およびそのエステル類の製造方法 | |
| JP3726315B2 (ja) | ケトン酸エステルの精製法 | |
| JPS59186942A (ja) | 粗製1,4−ジヒドロキシ−2−ナフトエ酸又はその塩の精製法 | |
| JP2005194243A (ja) | メントール誘導体およびその製造方法 | |
| JP2571939B2 (ja) | シクロペンテノン誘導体及びその製造法 | |
| JP3777407B2 (ja) | カルボン酸誘導体の製造法 | |
| JPS61282347A (ja) | 1,2−ジニトロフエノ−ルアルコキシアルキルエ−テルの製法 | |
| JP2003137835A (ja) | (r)−3−ヒドロキシ−3−(2−フェニルエチル)ヘキサン酸の製造方法 | |
| JP2576598B2 (ja) | 光学活性1−メチル−3−フェニルプロピルアミンの製法 | |
| JP3234838B2 (ja) | 2,4,5−トリフルオロ−3−ヒドロキシ安息香酸の製造方法 | |
| JP3378273B2 (ja) | d−フタル酸水素−2−exo−ノルボルニルの製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050715 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20050715 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090623 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090810 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20091020 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20091022 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121030 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131030 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |