JP5009109B2 - Hydrocarbon partial oxidation catalyst and method and apparatus for producing hydrogen-containing gas using the same - Google Patents
Hydrocarbon partial oxidation catalyst and method and apparatus for producing hydrogen-containing gas using the same Download PDFInfo
- Publication number
- JP5009109B2 JP5009109B2 JP2007237561A JP2007237561A JP5009109B2 JP 5009109 B2 JP5009109 B2 JP 5009109B2 JP 2007237561 A JP2007237561 A JP 2007237561A JP 2007237561 A JP2007237561 A JP 2007237561A JP 5009109 B2 JP5009109 B2 JP 5009109B2
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- partial oxidation
- reaction
- regeneration
- gas
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images


Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/584—Recycling of catalysts
Landscapes
- Hydrogen, Water And Hydrids (AREA)
- Carbon And Carbon Compounds (AREA)
- Catalysts (AREA)
Description
本発明は、炭化水素の部分酸化反応に係る触媒、例えばメタン、エタン、プロパンもしくはそれらのガスを主成分とする混合ガス、又は天然ガスから部分酸化反応により水素を含有する混合ガスを製造する触媒と、それを用いて水素を含有する混合ガスを製造する方法と装置に関するものである。 The present invention relates to a catalyst for a partial oxidation reaction of hydrocarbons, for example, a catalyst for producing a mixed gas containing methane, ethane, propane, or a gas thereof as a main component, or a mixed gas containing hydrogen from natural gas by a partial oxidation reaction. And a method and an apparatus for producing a mixed gas containing hydrogen using the same.
メタンガスや天然ガスを水素と一酸化炭素に転換する反応は、化学製品原料の合成ガスを得る方法として有用であるだけでなく、クリーンエネルギー源である水素の製造方法としても重要である。 The reaction of converting methane gas or natural gas into hydrogen and carbon monoxide is not only useful as a method for obtaining synthesis gas as a raw material for chemical products, but also important as a method for producing hydrogen as a clean energy source.
一方、燃料電池は発電の際に水しか排出せず、振動や騒音がないことから、エネルギー問題、環境問題の改善に大きく貢献すると期待されているが、燃料である水素の供給に問題があった。特に、小規模ビルや家庭用、船舶用等の比較的小型で分散配置された燃料電池については、その場で燃料を供給できる小型の燃料供給装置が求められている。 Fuel cells, on the other hand, emit only water during power generation and are free of vibrations and noise, so they are expected to make a significant contribution to improving energy and environmental issues. However, there are problems with the supply of hydrogen as a fuel. It was. In particular, for fuel cells that are relatively small and dispersedly arranged for small buildings, homes, ships, etc., there is a demand for small fuel supply devices that can supply fuel on the spot.
天然ガス等の化石燃料からの水素製造は、従来から、主として水蒸気改質法によって大規模に行われているが、通常800℃付近の高温で運転され、また水蒸気改質自体が吸熱反応であるため大量のエネルギー投入を必要とし、さらに副生物である二酸化炭素を大量に大気に放出するなどの問題点があった。このような大規模な水素製造装置を分散配置された小型燃料電池に接続することは不可能であり、また大規模に製造された水素をボンベで供給するにしても、運搬費用が高価となり、小型燃料電池の普及を妨げている。 Hydrogen production from fossil fuels such as natural gas has been conventionally carried out on a large scale mainly by the steam reforming method, but it is usually operated at a high temperature around 800 ° C., and the steam reforming itself is an endothermic reaction. For this reason, a large amount of energy was required, and a large amount of carbon dioxide as a by-product was released to the atmosphere. It is impossible to connect such a large-scale hydrogen production apparatus to small fuel cells that are dispersedly arranged, and even if hydrogen produced on a large scale is supplied by a cylinder, the transportation cost becomes expensive, This hinders the spread of small fuel cells.
水蒸気改質法以外に炭化水素から合成ガスや水素を製造する方法として、部分酸化法がある。飽和炭化水素の部分酸化から水素を製造する反応は次式のようになる。
CxH2(x+1)+(x/2)O2→xCO+(x+1)H2
部分酸化反応は発熱反応であるため外部からの大量のエネルギー投入は必要ないが、反応温度が高温になりやすく、高温に耐えうる反応容器材料に制約があり装置寿命も短くなる。そのため、比較的低い温度で反応を進める触媒が求められている。
In addition to the steam reforming method, there is a partial oxidation method as a method for producing synthesis gas or hydrogen from hydrocarbons. The reaction for producing hydrogen from the partial oxidation of saturated hydrocarbons is as follows:
C x H 2 (x + 1 ) + (x / 2) O 2 → xCO + (x + 1) H 2
Since the partial oxidation reaction is an exothermic reaction, it is not necessary to input a large amount of energy from the outside. However, the reaction temperature tends to be high, the reaction vessel material that can withstand the high temperature is limited, and the life of the apparatus is shortened. Therefore, there is a demand for a catalyst that proceeds the reaction at a relatively low temperature.
メタンと酸素から合成ガスや水素を製造するための部分酸化触媒としては、Ru又はRhをジルコニア又は安定化ジルコニアに担持させたもの(特許文献1参照。)や、Irを酸化チタンに担持させたもの(非特許文献1参照。)等が報告されているが、これらの触媒活性金属であるRu、Rh及びIrはいずれも高価な希少貴金属であり、実用的にはより安価な部分酸化触媒が求められていた。 As a partial oxidation catalyst for producing synthesis gas and hydrogen from methane and oxygen, Ru or Rh supported on zirconia or stabilized zirconia (see Patent Document 1), or Ir supported on titanium oxide. However, these catalytically active metals, Ru, Rh, and Ir are all expensive rare noble metals, and practically cheaper partial oxidation catalysts are available. It was sought after.
また、触媒による炭化水素の部分酸化反応では、炭素析出が起こりやすい。これらの提案の部分酸化触媒はメタンと酸素を原料ガスとして連続的に供給しながら触媒の活性の続く限り反応を継続させるのが原則であるので、触媒表面に炭素が析出すれば触媒が不活性化され短寿命となる問題点があった。 In the partial oxidation reaction of hydrocarbons by a catalyst, carbon deposition is likely to occur. In these proposed partial oxidation catalysts, in principle, the reaction is continued as long as the activity of the catalyst continues while continuously supplying methane and oxygen as raw material gases. Therefore, if carbon is deposited on the catalyst surface, the catalyst is inactive. There has been a problem of shortening the service life.
触媒活性金属として貴金属より安価なCoをアルミナに担持したCo/Al2O3部分酸化触媒も提案されている(非特許文献2参照。)。しかし、アルミナを担体とするCo担持触媒では、燃料ガスの転化率や生成物の水素選択率について、実用に堪えうるものは未だ得られていない。 There has also been proposed a Co / Al 2 O 3 partial oxidation catalyst in which Co, which is cheaper than a noble metal as a catalytically active metal, is supported on alumina (see Non-Patent Document 2). However, a Co-supported catalyst using alumina as a carrier has not yet been able to be practically used for the conversion rate of fuel gas and the hydrogen selectivity of the product.
本発明の対象とする部分酸化反応ではないが、メタンのCO2改質反応に対してCeO2−Al2O3担体にNiを担持した触媒を使用した報告がある(非特許文献3参照。)。そこでは、Ni担持量を5重量%に固定した上でCeO2が1〜5重量%のときが最適であると結論しており、しかも、その触媒が部分酸化反応に対しても有効であるかどうかは不明であった。 Although it is not the partial oxidation reaction which is the subject of the present invention, there is a report using a catalyst in which Ni is supported on a CeO 2 -Al 2 O 3 support for the CO 2 reforming reaction of methane (see Non-Patent Document 3). ). There, it is concluded that it is optimal when the amount of CeO 2 is 1 to 5% by weight with the Ni loading fixed at 5% by weight, and the catalyst is also effective for partial oxidation reaction. Whether it was unknown.
また従来の部分酸化法においては、メタンとともに純酸素を供給する必要があった。このためには部分酸化反応装置に大規模な酸素製造装置からガスラインを接続するか、酸素ボンベを運搬して接続する必要があり、システムが大型化かつ高コスト化してしまうという問題点があった。 In the conventional partial oxidation method, pure oxygen must be supplied together with methane. For this purpose, it is necessary to connect a gas line from a large-scale oxygen production apparatus to the partial oxidation reaction apparatus, or to transport and connect an oxygen cylinder, which increases the size and cost of the system. It was.
メタンの部分酸化反応に必要な酸素を、純酸素からではなく、触媒自身から供給する考え方もある。酸化物を触媒とし、その格子酸素を利用するものである。水素の貯蔵材料として大塚らによって研究された四酸化三鉄(Fe3O4)も、メタン分解による水素生成も行っていることから、このような触媒の例として考えることができる(非特許文献4、5参照。)。 There is a concept of supplying oxygen necessary for the partial oxidation reaction of methane not from pure oxygen but from the catalyst itself. An oxide is used as a catalyst, and lattice oxygen is used. Triiron tetroxide (Fe 3 O 4 ), which was studied by Otsuka et al. As a hydrogen storage material, can also be considered as an example of such a catalyst because it also produces hydrogen by methane decomposition (non-patent literature). 4 and 5).
また、触媒としてペロブスカイト酸化物を用い、触媒自身の酸素を使ってメタンを部分酸化し合成ガスを得るという研究結果が最近報告された(非特許文献6参照。)が、反応に900℃という高温を要するなど、実用的なシステムを設計するにはコスト面で問題となる課題が多い。 In addition, research results have recently been reported in which a perovskite oxide is used as a catalyst and methane is partially oxidized using the catalyst's own oxygen to obtain a synthesis gas (see Non-Patent Document 6). In order to design a practical system, there are many problems that are problematic in terms of cost.
本発明者らは、上記のような問題点を改良した部分酸化触媒として、酸化第二鉄を触媒活性成分とする新規触媒を開発し公表した。(非特許文献7)前発明によれば、担体としてはイットリアを含むことが必要であり、また貴金属であるロジウムの添加により高性能な部分酸化触媒が得られた。ロジウム添加量はわずかであり、活性主成分は安価な鉄の酸化物であるから、前発明の触媒は決して高価なものではない。しかし、より安価かつ希少性のない原料から触媒を製造することができれば、工業的にさらに有用であることは当然である。
メタンガスなどの炭化水素を原料として触媒自身の酸素により部分酸化を行うことができ、かつ再生することのできる触媒を見つけることができれば、従来の水蒸気改質法に替わる省エネルギーかつコンパクトで迅速起動可能な水素や合成ガスの製造につながる。このような水素製造は、クリーンな分散型電源である小型燃料電池への水素供給に最適であり、社会へのエネルギーの安定供給及び環境の改善に資する。 If hydrocarbons such as methane gas can be used as raw materials to perform partial oxidation with the catalyst's own oxygen and a catalyst that can be regenerated can be found, energy-saving, compact, and quick start-up can replace the conventional steam reforming method. This leads to the production of hydrogen and synthesis gas. Such hydrogen production is optimal for supplying hydrogen to small fuel cells, which are clean distributed power sources, and contributes to the stable supply of energy to society and the improvement of the environment.
本発明の第1の目的は、炭化水素の部分酸化反応による水素含有ガス製造に関して、部分酸化反応工程では酸素供給を必要とせず、安価で、炭素析出を抑えることができ、かつ再生可能な触媒を提供することである。
本発明の第2の目的は、その触媒を製造する方法を提供することである。
本発明の第3の目的は、その触媒を使用して水素含有ガスを製造する方法を提供することである。
本発明の第4の目的は、その触媒を使用して水素含有ガスを製造する装置を提供することである。
The first object of the present invention relates to the production of a hydrogen-containing gas by a partial oxidation reaction of hydrocarbons, which does not require oxygen supply in the partial oxidation reaction step, is inexpensive, can suppress carbon deposition, and can be regenerated. Is to provide.
A second object of the present invention is to provide a method for producing the catalyst.
A third object of the present invention is to provide a method for producing a hydrogen-containing gas using the catalyst.
The fourth object of the present invention is to provide an apparatus for producing a hydrogen-containing gas using the catalyst.
本発明者らは、鋭意検討を重ねた結果、ニッケルとクロムの各酸化物と、いわゆる担体としての役割を果たす酸化物から成る触媒を使用すれば、触媒自身の酸素を用いてメタンなど炭化水素の部分酸化反応が進行し、生成物中の水素選択率が高くなること、及び炭素析出を抑えることができることを見出し、さらにこの触媒は再生が容易であり、純酸素に限らず、空気等でも再生できることを確認し、本発明を完成するに至った。 As a result of intensive studies, the present inventors have found that if a catalyst comprising nickel and chromium oxides and an oxide that plays a role as a carrier is used, hydrocarbons such as methane can be obtained using the oxygen of the catalyst itself. It has been found that the partial oxidation reaction proceeds, the hydrogen selectivity in the product is increased, and carbon deposition can be suppressed. Further, this catalyst is easy to regenerate and is not limited to pure oxygen, but also air or the like. After confirming that it can be reproduced, the present invention has been completed.
本発明の部分酸化触媒は、外部から酸素を供給する必要なしに、メタンなどの炭化水素の部分酸化反応に対して良好な活性を示すことを主眼として開発されたものであり、炭化水素を部分酸化して水素と一酸化炭素を含有する混合ガスを生成させるものであって、ニッケルとクロムの各酸化物を含有することを特徴とする。メタンなどの炭化水素ガスと触媒自身を構成する酸化物の格子酸素が反応して、水素及び一酸化炭素を主成分とする合成ガスを生成する部分酸化反応を進行させることが可能であり、さらに反応で消費された格子酸素は、反応後の触媒を酸素、空気等で酸化することにより再生させることができるため、触媒の繰り返し使用が可能である。 The partial oxidation catalyst of the present invention was developed mainly for showing good activity for partial oxidation reaction of hydrocarbons such as methane without the need to supply oxygen from the outside. Oxidized to produce a mixed gas containing hydrogen and carbon monoxide, characterized by containing nickel and chromium oxides. It is possible for hydrocarbon gas such as methane and the lattice oxygen of the oxide constituting the catalyst itself to react to proceed a partial oxidation reaction that produces synthesis gas mainly composed of hydrogen and carbon monoxide, Lattice oxygen consumed in the reaction can be regenerated by oxidizing the catalyst after the reaction with oxygen, air, etc., so that the catalyst can be used repeatedly.
本発明の触媒を形成するための担体としては、通常の触媒に用いられる種々の酸化物を用いることができるが、その化学組成や製造方法によって、優れた部分酸化触媒が得られる場合と充分機能しない場合がある。担体としてはマグネシア(MgO)やシリカ(SiO2)、イットリア(Y2O3)が好ましく、アルミナ(Al2O3)やセリア(CeO2)を使用した場合には充分な触媒活性が得られないことが多い。 As the carrier for forming the catalyst of the present invention, various oxides used in ordinary catalysts can be used, but depending on the chemical composition and production method, an excellent partial oxidation catalyst can be obtained and function sufficiently. May not. As the support, magnesia (MgO), silica (SiO 2 ), and yttria (Y 2 O 3 ) are preferable. When alumina (Al 2 O 3 ) or ceria (CeO 2 ) is used, sufficient catalytic activity is obtained. Often not.
また、触媒の製造方法にも様々な手法がある。第1の製造方法は、触媒成分となる金属元素としてのニッケル及びクロム、並びに担体となる金属元素としてのマグネシウム、シリコン又はイットリウムの各金属元素を含む塩を溶解させた溶液を調製する工程と、前記溶液の溶媒を除去する乾燥工程と、乾燥工程後に酸化性雰囲気中で焼成する焼成工程とを含む方法である。この場合、マグネシウム、シリコン又はイットリウムは触媒成分とともに水溶液から酸化物になるが、本発明ではそのような形態のものも担体と称している。このように、材料の水溶液を予め混合してから溶媒を蒸発させた後、焼成して得た触媒では、部分酸化反応の好ましくない副反応である炭素析出が抑制される。 There are various methods for producing the catalyst. The first production method includes a step of preparing a solution in which a salt containing nickel and chromium as metal elements as catalyst components and magnesium, silicon or yttrium metal elements as metal elements as carriers is dissolved; The method includes a drying step of removing the solvent of the solution and a baking step of baking in an oxidizing atmosphere after the drying step. In this case, magnesium, silicon or yttrium is converted into an oxide from the aqueous solution together with the catalyst component. In the present invention, such a form is also called a carrier. Thus, in the catalyst obtained by previously mixing the aqueous solution of the material and evaporating the solvent and then calcining, carbon deposition, which is an undesirable side reaction of the partial oxidation reaction, is suppressed.
第2の製造方法は、ニッケルとクロムの各金属元素を含む塩を溶解させた溶液を、担体である酸化マグネシウム、シリカ又は酸化イットリウムに加える工程と、その後、溶液の溶媒を除去する乾燥工程と、乾燥工程後に酸化性雰囲気中で焼成する焼成工程とを含む方法である。 The second production method includes a step of adding a solution in which a salt containing nickel and chromium metal elements is dissolved to magnesium oxide, silica, or yttrium oxide as a carrier, and then a drying step of removing the solvent of the solution. And a baking step of baking in an oxidizing atmosphere after the drying step.
本発明の水素含有ガス製造方法は、炭化水素を含む原料ガスを加熱下で本発明の部分酸化触媒に接触させ、酸化剤としての酸素含有ガスを供給することなく、部分酸化触媒中の金属酸化物を構成する格子酸素により炭化水素を部分酸化して水素と一酸化炭素を含有する混合ガスを生成させる部分酸化工程と、部分酸化工程を経た部分酸化触媒を加熱下で酸素含有ガスと接触させて部分酸化触媒を再生する再生工程とを含んでいる。 The method for producing a hydrogen-containing gas of the present invention comprises contacting a raw material gas containing hydrocarbon with the partial oxidation catalyst of the present invention under heating, and oxidizing the metal in the partial oxidation catalyst without supplying an oxygen-containing gas as an oxidant. A partial oxidation step in which hydrocarbons are partially oxidized by lattice oxygen constituting the product to generate a mixed gas containing hydrogen and carbon monoxide, and a partial oxidation catalyst that has undergone the partial oxidation step is brought into contact with an oxygen-containing gas under heating. And a regeneration step for regenerating the partial oxidation catalyst.
再生工程で使用する酸素含有ガスは、純酸素ガスでもよいが、実施例に示されているように、酸素と不活性なガスとの混合ガスでもよく、水蒸気や空気でもよい。コストの面からは空気を使用するのが最も好ましい。 The oxygen-containing gas used in the regeneration process may be pure oxygen gas, but as shown in the examples, it may be a mixed gas of oxygen and an inert gas, or may be water vapor or air. From the viewpoint of cost, it is most preferable to use air.
本発明の水素含有ガス製造装置は、本発明の部分酸化触媒が保持された反応管と、その触媒を加熱する加熱炉と、メタンなどの炭化水素を含む原料ガスを反応管に送り触媒と接触させる原料ガス供給流路と、触媒再生に用いる酸素含有ガスを反応管に送り触媒と接触させる再生ガス供給流路とを備え、反応管中で、酸化剤としての酸素含有ガスの存在しない状態下で触媒中の金属酸化物を構成する格子酸素により原料ガスの炭化水素を部分酸化して水素と一酸化炭素を含有する混合ガスを生成させ、原料ガスが存在しない状態下で再生ガス供給流路からの酸素含有ガスにより部分酸化触媒を再生する。 The hydrogen-containing gas production apparatus of the present invention comprises a reaction tube holding the partial oxidation catalyst of the present invention, a heating furnace for heating the catalyst, and a raw material gas containing hydrocarbons such as methane is sent to the reaction tube and brought into contact with the catalyst. And a regeneration gas supply passage for sending an oxygen-containing gas used for catalyst regeneration to the reaction tube and bringing it into contact with the catalyst, in a state where no oxygen-containing gas as an oxidant is present in the reaction tube. In the catalyst, the hydrocarbon of the raw material gas is partially oxidized by lattice oxygen constituting the metal oxide in the catalyst to generate a mixed gas containing hydrogen and carbon monoxide, and the regeneration gas supply flow path in the absence of the raw material gas The partial oxidation catalyst is regenerated with the oxygen-containing gas from
反応管は部分酸化触媒が移動できないように固定された固定床反応管である場合には、反応管を2系列持てば、一方の部分酸化反応中に他方の触媒を再生することも可能であり、これにより連続的な水素含有ガス製造装置を実現できる。すなわち、その場合は、反応管と加熱炉の組が2組備えられ、原料ガス供給流路と再生ガス供給流路は切替え弁を介して両反応管に接続されており、加熱炉と切替え弁の制御により、一方の反応管での部分酸化反応中に他方の反応管での触媒を再生するようにするとともに、その操作を交互に切り替えることができるようになっている。 If the reaction tube is a fixed bed reaction tube that is fixed so that the partial oxidation catalyst cannot move, it is possible to regenerate the other catalyst during one partial oxidation reaction if there are two series of reaction tubes. Thus, a continuous hydrogen-containing gas production apparatus can be realized. That is, in that case, two sets of reaction tubes and heating furnaces are provided, and the source gas supply flow path and the regeneration gas supply flow path are connected to both reaction tubes via switching valves, and the heating furnace and switching valve are By controlling this, the catalyst in the other reaction tube is regenerated during the partial oxidation reaction in one reaction tube, and the operation can be switched alternately.
反応管は触媒が移動可能な状態で保持された移動床反応管である場合には、反応管が部分酸化反応を行わせる反応部と、反応部とは異なる場所で触媒再生を行わせる再生部とを備えており、反応部と再生部の間で触媒を搬送する搬送路が設けられており、原料ガス供給流路は反応部に接続され、再生ガス供給流路は再生部に接続されているようにすれば、触媒を移動させながら部分酸化反応と触媒再生を連続的に続けることができる。 When the reaction tube is a moving bed reaction tube held in a state where the catalyst is movable, the reaction unit in which the reaction tube performs the partial oxidation reaction and the regeneration unit in which the catalyst regeneration is performed at a location different from the reaction unit And a transport path for transporting the catalyst between the reaction section and the regeneration section is provided, the source gas supply flow path is connected to the reaction section, and the regeneration gas supply flow path is connected to the regeneration section. As a result, the partial oxidation reaction and the catalyst regeneration can be continuously continued while the catalyst is moved.
原料となる炭化水素としては、実施例ではメタンのみを取りあげているが、メタンに限るものではない。エタンやプロパンなどの飽和炭化水素であってもよい。しかし、炭化水素の炭素数が多くなると炭素の析出量が増す傾向があるので、メタンが最も好ましい。また、不飽和炭化水素でもよいが、やはり炭素の析出量が増す傾向があるので好ましくない。 In the examples, only methane is taken up as a hydrocarbon as a raw material, but it is not limited to methane. It may be a saturated hydrocarbon such as ethane or propane. However, methane is most preferable because the carbon deposition amount tends to increase as the number of carbon atoms in the hydrocarbon increases. Moreover, although unsaturated hydrocarbon may be used, it is not preferable because the carbon deposition amount tends to increase.
本発明の部分酸化触媒を用いることにより、部分酸化のために酸素供給ガスラインや酸素ボンベを接続する必要なしに、部分酸化法により水素と一酸化炭素を含む混合ガスを製造することができる。得られた混合ガスから水素を分離したり、水素と一酸化炭素の混合ガスを合成ガスとして有機化合物合成の原料に供したりすることができる。特に、小型燃料電池に好適な水素製造装置を実現できる。 By using the partial oxidation catalyst of the present invention, a mixed gas containing hydrogen and carbon monoxide can be produced by a partial oxidation method without the need to connect an oxygen supply gas line or an oxygen cylinder for partial oxidation. Hydrogen can be separated from the obtained mixed gas, or a mixed gas of hydrogen and carbon monoxide can be used as a synthesis gas for a raw material for organic compound synthesis. In particular, a hydrogen production apparatus suitable for a small fuel cell can be realized.
本発明に記載の触媒組成は、他に一般的に触媒に求められる特性、例えば機械的強度の向上などを得るための成分を、本発明の触媒組成に混合して触媒調製することを排除するものではない。また、本発明に記載の触媒組成は、触媒の製造工程で不可避的に混入する微量不純物成分を排除するものでもない。 The catalyst composition described in the present invention excludes the preparation of a catalyst by mixing other components for obtaining characteristics generally required for a catalyst, such as improvement in mechanical strength, with the catalyst composition of the present invention. It is not a thing. Further, the catalyst composition described in the present invention does not exclude trace impurity components inevitably mixed in the catalyst production process.
(1)MgO担体の調製
硝酸マグネシウム(Mg(NO3)2・6H2O)を空気流通下、600℃まで昇温した後5時間保持し、MgOを調製した。
(1) Preparation of MgO support Magnesium nitrate (Mg (NO 3 ) 2 · 6H 2 O) was heated to 600 ° C. under air flow and held for 5 hours to prepare MgO.
(2)Y2O3担体の調製
しゅう酸イットリウム(Wako, Y2(C2O4)3・4H2O)を空気流通下、室温から5℃/分の昇温速度で600℃まで昇温し、600℃で5時間保持しY2O3を調製した。
(2) Preparation of Y 2 O 3 carrier Yttrium oxalate (Wako, Y 2 (C 2 O 4 ) 3 · 4H 2 O) was raised from room temperature to 600 ° C. at a heating rate of 5 ° C./min. Warm and hold at 600 ° C. for 5 hours to prepare Y 2 O 3 .
(3)その他の担体の調製
CeO2もY2O3担体と同様の調製法で硝酸セリウム(III)六水和物(Wako, Ce(NO3)3・6H2O)から調製した。
他の担体(Al2O3(Merck)、La2O3(ナカライテスク)、SiO2(FUJI SILYSIA))はそのまま使用した。
(3) Preparation of Other Carriers CeO 2 was also prepared from cerium (III) nitrate hexahydrate (Wako, Ce (NO 3 ) 3 · 6H 2 O) by the same preparation method as the Y 2 O 3 carrier.
Other carriers (Al 2 O 3 (Merck), La 2 O 3 (Nacalai Tesque), SiO 2 (FUJI SILYSIA)) were used as they were.
(4)NiO(16)−Cr2O3(4)/MgO触媒の調製
所定量のNi(NO3)2・6H2O及びCr(NO3)3・9H2Oを溶解させた水溶液を、担体(MgO)1gに対して、Niとして16m mol、Crとして4m molとなるように加え、一夜放置後、蒸発乾固により乾燥させた。その後、空気流通下、室温から600℃まで昇温し、600℃で5時間保持し触媒を調製した。いわゆる含浸担持法であり、以下この方法で担持した場合、「(担持された物質)/(担体)」と表記する。
なお、触媒表示中のNiO(16)のような括弧内の数字は、担体1gあたりに担持された金属元素の量をm mol単位で表したものである。
(4) Preparation of NiO (16) -Cr 2 O 3 (4) / MgO catalyst An aqueous solution in which a predetermined amount of Ni (NO 3 ) 2 .6H 2 O and Cr (NO 3 ) 3 · 9H 2 O is dissolved To 1 g of carrier (MgO), Ni was added in an amount of 16 mmol and Cr as 4 mmol, and after standing overnight, it was dried by evaporation to dryness. Thereafter, the temperature was raised from room temperature to 600 ° C. under air circulation, and the catalyst was prepared by holding at 600 ° C. for 5 hours. This is a so-called impregnation supporting method, and hereinafter referred to as “(supported substance) / (support)” when supported by this method.
The numbers in parentheses such as NiO (16) in the catalyst display represent the amount of metal element supported per gram of support in millimoles.
(5)NiO(16)−Cr2O3(4)−MgO触媒の調製
所定量のNi(NO3)2・6H2O、Cr(NO3)3・9H2O及びMg(NO3)2・6H2Oを水に溶解させ、蒸発乾固により乾燥させた。その後、空気流通下、室温から600℃まで昇温し、600℃で5時間保持し触媒を調製した。以下この方法で調製した場合、「(担持された物質)−(担体)」と表記する。括弧内の数字の標記は、含浸担持法による触媒調製の場合と同様である。
また、焼成温度を800℃としたこと以外は上記と同様の方法で調製した触媒も作製した。
なお、比較例等、他の組成の触媒もそれぞれの成分の水溶性塩を用いて同様の方法で調製した。
(5) Preparation of NiO (16) -Cr 2 O 3 (4) -MgO catalyst Predetermined amounts of Ni (NO 3 ) 2 .6H 2 O, Cr (NO 3 ) 3 · 9H 2 O and Mg (NO 3 ) the 2 · 6H 2 O was dissolved in water and dried by evaporation to dryness. Thereafter, the temperature was raised from room temperature to 600 ° C. under air circulation, and the catalyst was prepared by holding at 600 ° C. for 5 hours. Hereinafter, when prepared by this method, it is expressed as “(supported substance)-(support)”. The numbers in parentheses are the same as in the catalyst preparation by the impregnation support method.
A catalyst prepared by the same method as described above was also prepared except that the calcination temperature was 800 ° C.
In addition, the catalyst of other compositions, such as a comparative example, was prepared by the same method using the water-soluble salt of each component.
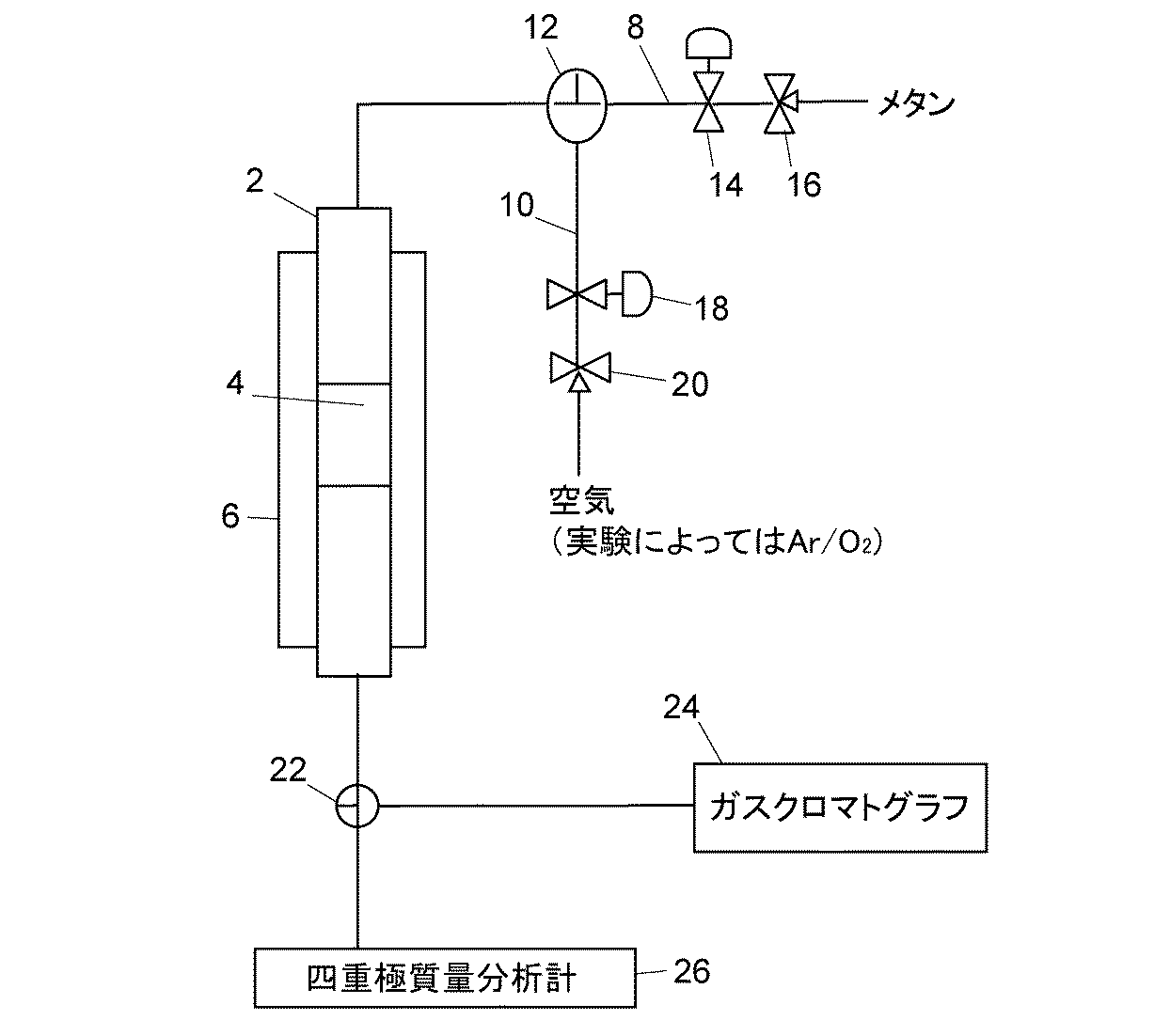
(5)評価のための実験装置
評価のための実験装置として、図1に示される反応装置を使用した。ただし、この反応装置はあくまで評価のための実験装置であり、実際にこの触媒を使用して水素含有ガスを製造する装置はこの反応装置に限定されるものではなく、各部の配置や規模は目的に応じて適宜変更することができる。
(5) Experimental apparatus for evaluation The reaction apparatus shown in FIG. 1 was used as an experimental apparatus for evaluation. However, this reactor is only an experimental device for evaluation, and an apparatus for actually producing a hydrogen-containing gas using this catalyst is not limited to this reactor. It can be changed as appropriate according to the situation.
図1の反応装置において、反応管2は石英ガラス管であり、例えばその内径が10mm、長さが250mmであり、内部には部分酸化触媒層4が充填されている。触媒層4は両側から石英製グラスウールで挟み込まれて反応管2内に固定されている。反応管2を加熱するために電気炉6が設けられており、反応管2が電気炉6中に収納されるように電気炉6に対して反応管2が位置決めされている。触媒層4に対して石英ガラス製熱電対保護管(図示略)が設置され、その中に熱電対(図示略)が通されて触媒層4と接触している。触媒層4の温度はその熱電対により検出され、その検出された温度が設定温度になるように、温度コントローラ(図示略)により電気炉6への通電が制御される。
In the reaction apparatus of FIG. 1, the
反応管2の一端には部分酸化反応のための原料ガスとしてメタンを供給する原料ガス供給流路8と、触媒再生時に酸素含有ガスとして酸素とアルゴンの混合ガス、水蒸気又は空気を供給する再生ガス供給流路10が、三方切替弁12により切り替えてガスを供給することができるように接続されている。いずれの流路8,10もそれぞれのガスを一定流量で供給するための質量流量制御器14,18を備えている。それぞれの質量流量制御器14,18の上流には開閉弁16,20が配置されている。
At one end of the
反応管2の他端は三方バルブ22を介してガスクロマトグラフ24及び質量分析計26に接続されている。質量分析計26として四重極質量分析計を用いているが他の形式の質量分析計でもよい。三方バルブ22の切替えにより、反応管2からの反応ガスをガスクロマトグラフ24又は質量分析計26に導いて適宜分析する。しかしながら、このような分析装置の配置は実験データ収集のための配置であり、実用化システムに不可欠なものではない。
The other end of the
なお、分析装置の使用条件の制約から、実験に用いるガスを希釈するときは、希釈ガスとしてアルゴンを用いた。これは、質量分析計で一酸化炭素を分析しようとすると、一酸化炭素と同じ質量数をもつ窒素は希釈ガスとしては使用できないからである。そして、アルゴンは不活性ガスであるため、触媒反応にはなんら影響しないことを確認済みである。 In addition, when diluting the gas used for the experiment, argon was used as the diluting gas because of restrictions on the use conditions of the analyzer. This is because, when attempting to analyze carbon monoxide with a mass spectrometer, nitrogen having the same mass number as carbon monoxide cannot be used as a diluent gas. And since argon is an inert gas, it has been confirmed that it has no influence on the catalytic reaction.
(6)本発明の部分酸化触媒を用いたメタンからの水素製造反応
製造した触媒は、メタンの部分酸化反応によって評価した。なお、分析装置の感度・分解能の制約から、メタンは不活性ガスであるアルゴンで希釈して導入したが、アルゴンは触媒活性の評価にはなんら影響するものではない。
(6) Hydrogen production reaction from methane using the partial oxidation catalyst of the present invention The produced catalyst was evaluated by a partial oxidation reaction of methane. Note that methane was diluted and introduced with argon, an inert gas, due to restrictions on the sensitivity and resolution of the analyzer, but argon has no effect on the evaluation of catalyst activity.
(7)本発明の触媒の再生反応
メタンの部分酸化による水素製造反応をある時間実施した後の触媒は、アルゴンで希釈した酸素ガス、又は空気により酸化し、再びメタンの部分酸化反応に供し、その性能によって完全に再生されたことを評価した。また、後で説明する図2に示すように、触媒のX線回折の結果によっても本発明の触媒は酸化によって再生することが確認された。
(7) Regeneration reaction of catalyst of the present invention The catalyst after carrying out hydrogen production reaction by partial oxidation of methane for a certain period of time is oxidized with oxygen gas diluted with argon or air, and again subjected to partial oxidation reaction of methane, It was evaluated that it was completely regenerated by its performance. Further, as shown in FIG. 2 to be described later, it was confirmed that the catalyst of the present invention was regenerated by oxidation also from the result of X-ray diffraction of the catalyst.
実際の部分酸化実験は、調製した触媒の0.5gを反応管に充填し、アルゴン:メタン=4:1の混合ガスを流量25ml/分、流速SV=3,000ml/g−cat・h(SVは空間速度)で流す環境下で、触媒温度を毎分10度の速度で700℃まで昇温した後、700℃に所定時間保持して行った。部分酸化と触媒再生を交互に繰り返して実験を行う場合には、上記の部分酸化反応を所定時間実施した後、700℃においてガスをアルゴン:酸素=4:1の混合ガスに切り替えて、25ml/分、空間速度SV=3,000ml/g−cat・hで部分酸化と同じ時間だけ反応させ、触媒を再生した。 In an actual partial oxidation experiment, 0.5 g of the prepared catalyst was charged into a reaction tube, a mixed gas of argon: methane = 4: 1 was supplied at a flow rate of 25 ml / min, and a flow rate SV = 3,000 ml / g-cat · h ( The temperature of the catalyst was raised to 700 ° C. at a rate of 10 degrees per minute in an environment where SV is flowing at a space velocity), and then held at 700 ° C. for a predetermined time. When the experiment is performed by alternately repeating partial oxidation and catalyst regeneration, the partial oxidation reaction is performed for a predetermined time, and then the gas is switched to a mixed gas of argon: oxygen = 4: 1 at 700 ° C. The catalyst was regenerated by reacting at a space velocity of SV = 3,000 ml / g-cat · h for the same time as the partial oxidation.
以上の反応条件はあくまで評価のための実験条件であり、実際に工業的に製造する際には目的に合せて適宜変更する。
以下、本発明を実施例によりさらに詳述する。
The above reaction conditions are experimental conditions for evaluation, and are appropriately changed according to the purpose when actually producing industrially.
Hereinafter, the present invention will be described in more detail with reference to examples.
焼成温度600℃で調製したNiO(16)−Cr2O3(4)−MgO触媒(MgO1gあたり16m molのNiOと4m molのCr2O3が含有されるよう、Mg、Ni、Crを総て溶解させた水溶液から調製した触媒)を用い、メタンの部分酸化反応−触媒再生の繰り返し実験を実施した。反応及び再生時間は10分ずつであり、3回繰り返した。反応温度と再生温度は1回目を600℃で、2回目を700℃で、3回目を800℃で行った。結果を表1に示す。600℃での反応では原料であるメタンの転化率が低いが、700℃以上の温度では90%以上の転化率が得られた。また600℃と700℃での反応では、CO選択率が高い一方、CO2選択率が低いことから、合成ガス(H2+CO)を生成する部分酸化反応が進行していることがわかる。800℃ではさらに高いメタン転化率を示すが、完全酸化の割合が高くなること、炭素析出がやや増加することなどから、本発明の触媒は700℃で充分にその機能を発揮するといえる。反応温度が低い方が、加熱のための投入エネルギーが少なくてすみ、工業的に有利であることは言うまでもない。 NiO (16) -Cr 2 O 3 (4) -MgO catalyst prepared at a calcination temperature of 600 ° C. (Mg, Ni, and Cr are added so that 16 mmol of NiO and 4 mmol of Cr 2 O 3 are contained per 1 g of MgO. Using a catalyst prepared from an aqueous solution dissolved in this manner, repeated experiments of methane partial oxidation reaction-catalyst regeneration were carried out. The reaction and regeneration time was 10 minutes each and repeated 3 times. The reaction temperature and regeneration temperature were 600 ° C for the first time, 700 ° C for the second time, and 800 ° C for the third time. The results are shown in Table 1. In the reaction at 600 ° C., the conversion rate of methane as a raw material was low, but at a temperature of 700 ° C. or higher, a conversion rate of 90% or higher was obtained. Further, in the reactions at 600 ° C. and 700 ° C., the CO selectivity is high, but the CO 2 selectivity is low, so that it is understood that the partial oxidation reaction for generating synthesis gas (H 2 + CO) proceeds. Although a higher methane conversion rate is exhibited at 800 ° C., it can be said that the catalyst of the present invention sufficiently performs its function at 700 ° C. because the rate of complete oxidation is increased and carbon deposition is slightly increased. It goes without saying that a lower reaction temperature requires less energy for heating and is industrially advantageous.
実施例1の触媒を用いて、700℃での部分酸化反応と700℃での再生反応を10回繰り返す実験を行った。表2に示す結果のように、10回の反応―再生を繰り返してもメタン転化率は90%前後を維持しており、本実施例の触媒が長寿命であることが示された。水素選択率は最初の1〜2回目は若干低めであるが、以降90%前後で推移した。 Using the catalyst of Example 1, an experiment was conducted in which a partial oxidation reaction at 700 ° C. and a regeneration reaction at 700 ° C. were repeated 10 times. As shown in Table 2, the methane conversion ratio was maintained at around 90% even after 10 reaction-regeneration cycles, indicating that the catalyst of this example had a long life. The hydrogen selectivity was slightly lower in the first and second rounds, but remained around 90% thereafter.
焼成温度700℃で調製したNiO(16)−Cr2O3(4)−MgO触媒についても、700℃での部分酸化反応と700℃での再生反応を10回繰り返す実験を行った。この実験の結果も表2に示す。10回の繰り返し実験を通してメタン転化率は90%以上、水素選択率、CO選択率とも安定して高い値で推移しており、活性、寿命ともに優れた触媒であることを示した。 For the NiO (16) -Cr 2 O 3 (4) -MgO catalyst prepared at a calcination temperature of 700 ° C., an experiment was conducted in which a partial oxidation reaction at 700 ° C. and a regeneration reaction at 700 ° C. were repeated 10 times. The results of this experiment are also shown in Table 2. Through 10 repeated experiments, the methane conversion was 90% or more, and the hydrogen selectivity and CO selectivity were both stable and high, indicating that the catalyst was excellent in both activity and life.
焼成温度800℃で調製したNiO(16)−Cr2O3(4)−MgO触媒についても、700℃での部分酸化反応と700℃での再生反応を10回繰り返す実験を行った。この実験の結果も表2に示す。実施例1の場合に比べ、メタン転化率はやや低下したが、水素選択率は初回が87.6%、2回目以降は90%を越えて推移しており、安定性に優れた触媒であることを示した。 For the NiO (16) -Cr 2 O 3 (4) -MgO catalyst prepared at a calcination temperature of 800 ° C., an experiment was performed in which the partial oxidation reaction at 700 ° C. and the regeneration reaction at 700 ° C. were repeated 10 times. The results of this experiment are also shown in Table 2. Compared to the case of Example 1, the methane conversion rate was slightly decreased, but the hydrogen selectivity was 87.6% for the first time and exceeded 90% for the second time and thereafter, and the catalyst was excellent in stability. Showed that.
Cr2O3の含有量を8m molと2倍に増量したほかは実施例1と同様の触媒、すなわちNiO(16)−Cr2O3(8)−MgO触媒((MgO1gあたり16m molのNiOと8m molのCr2O3が含有されるよう、Mg、Ni、Crを総て溶解させた水溶液から調製した触媒)について、メタンの部分酸化反応−触媒再生の繰り返し実験を実施した。反応及び再生時間は10分ずつであり、5回繰り返した。反応温度と再生温度は1回目を600℃で、2回目を650℃で、3回目を700℃で、4回目を750℃で、5回目を800℃で行った。結果を表3に示す。触媒性能は実施例1のものと殆ど変わらず、この程度のCr2O3の増量は触媒性能に影響を与えないことがわかる。 The same catalyst as in Example 1 except that the content of Cr 2 O 3 was doubled to 8 mmol, ie, NiO (16) -Cr 2 O 3 (8) -MgO catalyst ((16 mmol of NiO per 1 g of MgO) And a catalyst prepared from an aqueous solution in which Mg, Ni and Cr are completely dissolved so as to contain 8 mmol of Cr 2 O 3 , repeated experiments of methane partial oxidation reaction-catalyst regeneration were conducted. The regeneration time was 10 minutes each and was repeated 5. The reaction temperature and regeneration temperature were 600 ° C. for the first time, 650 ° C. for the second time, 700 ° C. for the third time, and 750 ° C. for the fourth time. The results were shown in Table 3. The results are shown in Table 3. The catalyst performance is almost the same as that of Example 1, and it can be seen that this increase in Cr 2 O 3 does not affect the catalyst performance.
NiOの含有量を32m molと2倍に増量したほかは実施例4と同様の触媒、すなわちNiO(32)−Cr2O3(8)−MgO触媒((MgO1gあたり32m molのNiOと8m molのCr2O3が含有されるよう、Mg、Ni、Crを総て溶解させた水溶液から調製した触媒)について、メタンの部分酸化反応−触媒再生の繰り返し実験を実施した。実施例4と同じく、反応及び再生時間は10分ずつであり、5回繰り返した。反応温度と再生温度も実施例4と同じく、1回目を600℃で、2回目を650℃で、3回目を700℃で、4回目を750℃で、5回目を800℃で行った。結果を表3に示す。
メタン転化率は実施例4と同様に高いものの、CO2とH2Oの発生が多くなることから、NiOの含有量を多くすると燃焼反応が起こりやすくなるものと考えられる。
The same catalyst as in Example 4 except that the NiO content was doubled to 32 mmol, that is, NiO (32) -Cr 2 O 3 (8) -MgO catalyst ((32 mmol of NiO per 8 g of MgO and 8 mmol) In the same manner as in Example 4, a methane partial oxidation reaction-catalyst regeneration repeated experiment was conducted on a catalyst prepared from an aqueous solution in which Mg, Ni, and Cr were all dissolved so that Cr 2 O 3 was contained. The reaction and regeneration time were 10 minutes each and repeated 5 times, the reaction temperature and regeneration temperature were also 600 ° C. for the first time, 650 ° C. for the second time, and 700 ° C. for the third time as in Example 4. The fourth time was 750 ° C. and the fifth time was 800 ° C. The results are shown in Table 3.
Although the methane conversion rate is high as in Example 4, the generation of CO 2 and H 2 O increases, so it is considered that the combustion reaction is likely to occur when the NiO content is increased.
MgO担体にNiとCrを含む水溶液を含浸担持させて調製したNiO(16)−Cr2O3(4)/MgO触媒について、メタンの部分酸化反応−触媒再生反応を各10分ずつ、3回繰り返す実験を実施した。反応温度は1回目を600℃で、2回目を700℃で、3回目を800℃で行った。結果は、実施例1と併せて表1に記載した。メタン転化率、水素選択性とも実施例1と同等の性能を示し、本実施例の調製方法でも優れた触媒が得られた。しかしながら、実施例1と比較してやや炭素析出量が多く、またCO選択率が低く完全酸化の比率が高い傾向が見られたことから、実施例1の方がより優れた触媒であるといえる。 For NiO (16) -Cr 2 O 3 (4) / MgO catalyst prepared by impregnating and supporting an aqueous solution containing Ni and Cr on an MgO support, the partial oxidation reaction of methane-catalyst regeneration reaction is performed 3 times, 10 minutes each. Repeated experiments were performed. The reaction temperature was 600 ° C for the first time, 700 ° C for the second time, and 800 ° C for the third time. The results are shown in Table 1 together with Example 1. Both the methane conversion rate and the hydrogen selectivity showed the same performance as in Example 1, and an excellent catalyst was obtained by the preparation method of this example. However, it can be said that Example 1 is a more excellent catalyst because the amount of deposited carbon is slightly larger than that of Example 1, and the CO selectivity is low and the ratio of complete oxidation is high.
担体としてシリカ(SiO2)を使用したほかは、実施例1と同様の方法で調製したNiO(16)−Cr2O3(4)−SiO2触媒(SiO21gあたり16m molのNiOと4m molのCr2O3が含有されるよう、Si、Ni、Crを総て溶解させた水溶液から調製した触媒)を用い、メタンの部分酸化反応−触媒再生の繰り返し実験を実施した。反応及び再生時間は10分ずつであり、3回繰り返した。反応温度は1回目を600℃で、2回目を700℃で、3回目を800℃で行った。結果を表4に示す。600℃での反応では原料であるメタンの転化率が低いが、700℃以上の温度では90%以上の転化率が得られた。800℃ではさらに高いメタン転化率を示すが、完全酸化の割合が高くなることなどから、本発明の触媒は実施例1と同様に700℃で充分にその機能を発揮するといえる。炭素析出はわずかしか認められず、優れた性能を示す。 A NiO (16) -Cr 2 O 3 (4) -SiO 2 catalyst prepared in the same manner as in Example 1 except that silica (SiO 2 ) was used as a support (16 mmol of NiO per 4 g of SiO 2 and 4 m Using a catalyst prepared from an aqueous solution in which all of Si, Ni, and Cr were dissolved so as to contain mol Cr 2 O 3 , repeated experiments of methane partial oxidation reaction-catalyst regeneration were performed. The reaction and regeneration time was 10 minutes each and repeated 3 times. The reaction temperature was 600 ° C for the first time, 700 ° C for the second time, and 800 ° C for the third time. The results are shown in Table 4. In the reaction at 600 ° C., the conversion rate of methane as a raw material was low, but at a temperature of 700 ° C. or higher, a conversion rate of 90% or higher was obtained. Although a higher methane conversion is exhibited at 800 ° C., it can be said that the catalyst of the present invention exhibits its function sufficiently at 700 ° C. as in Example 1 because the rate of complete oxidation becomes high. Only a small amount of carbon deposition is observed, indicating excellent performance.
担体としてイットリア(Y2O3)を使用したほかは、実施例1と同様の方法で調製したNiO(16)−Cr2O3(4)−Y2O3触媒(Y2O31gあたり16m molのNiOと4m molのCr2O3が含有されるよう、Y、Ni、Crを総て溶解させた水溶液から調製した触媒)を用い、メタンの部分酸化反応−触媒再生の繰り返し実験を実施した。反応及び再生時間は10分ずつであり、3回繰り返した。反応温度は1回目を600℃で、2回目を700℃で、3回目を800℃で行った。結果を表4に示す。600℃での反応では原料であるメタンの転化率が低いが、700℃では83.4%、800℃では93.3%と良好な転化率が得られた。特に700℃での反応においては、CO選択率が高く、かつ炭素析出が抑制されており、実施例1と同様に700℃で充分にその機能を発揮する触媒となっている。 A NiO (16) -Cr 2 O 3 (4) -Y 2 O 3 catalyst (per 1 g of Y 2 O 3 ) prepared in the same manner as in Example 1 except that yttria (Y 2 O 3 ) was used as the carrier. Using a catalyst prepared from an aqueous solution in which all of Y, Ni, and Cr are dissolved so that 16 mmol of NiO and 4 mmol of Cr 2 O 3 are contained, repeated experiments of partial oxidation reaction of methane-catalyst regeneration Carried out. The reaction and regeneration time was 10 minutes each and repeated 3 times. The reaction temperature was 600 ° C for the first time, 700 ° C for the second time, and 800 ° C for the third time. The results are shown in Table 4. In the reaction at 600 ° C., the conversion rate of methane as a raw material was low, but a good conversion rate of 83.4% at 700 ° C. and 93.3% at 800 ° C. was obtained. In particular, in the reaction at 700 ° C., the CO selectivity is high and carbon deposition is suppressed, and it is a catalyst that fully exhibits its function at 700 ° C. as in Example 1.
[比較例1]
Cr2O3を含有しないほかは実施例1と同様の触媒、すなわちNiO(16)−MgO触媒(MgO1gあたり16m molのNiOが含有されるよう、MgとNiを溶解させた水溶液から調製した触媒)について、メタンの部分酸化反応−触媒再生の繰り返し実験を実施した。反応及び再生時間は10分ずつであり、4回繰り返した。反応温度は1回目を500℃で、2回目を600℃で、3回目を700℃で、4回目を800℃で行った。結果を表4に示す。700℃以上でメタン転化率は上昇してくるが、炭素析出が多く実用には適さない。
[Comparative Example 1]
A catalyst similar to Example 1 except that it does not contain Cr 2 O 3 , that is, a NiO (16) -MgO catalyst (a catalyst prepared from an aqueous solution in which Mg and Ni are dissolved so as to contain 16 mmol of NiO per 1 g of MgO) ), Repeated experiments of partial oxidation reaction of methane-catalyst regeneration were conducted. The reaction and regeneration time was 10 minutes each and was repeated 4 times. The reaction temperature was 500 ° C for the first time, 600 ° C for the second time, 700 ° C for the third time, and 800 ° C for the fourth time. The results are shown in Table 4. Although the methane conversion increases at 700 ° C. or higher, carbon deposition is large and not suitable for practical use.
この触媒を用いて、700℃におけるメタンの部分酸化反応−触媒再生を各10分間ずつ10回繰り返す実験を行った結果を表5に示す。700℃での反応−再生を繰り返しても炭素析出量は一貫して多く、部分酸化反応触媒として不適である。 Table 5 shows the results of experiments in which partial oxidation reaction of methane at 700 ° C. and catalyst regeneration were repeated 10 times for 10 minutes each using this catalyst. Even if the reaction-regeneration at 700 ° C. is repeated, the amount of carbon deposition is consistently large and is not suitable as a partial oxidation reaction catalyst.
[比較例2]
担体としてセリア(CeO2)を使用したほかは実施例1と同様の触媒、すなわちNiO(16)−Cr2O3(4)−CeO2触媒(CeO21gあたり16m molのNiOと4m molのCr2O3が含有されるよう、Ce、Ni、Crを総て溶解させた水溶液から調製した触媒)について、メタンの部分酸化反応−触媒再生の繰り返し実験を実施した。反応及び再生時間は10分ずつであり、3回繰り返した。反応温度は1回目を700℃で、2回目を600℃で、3回目を800℃で行った。結果は表4に示すが、メタン転化率は700℃以上で高くなるものの、水素やCOの選択率が低い一方でCO2選択率が高いことから完全酸化反応が進行しており、MgOやSiO2を担体に使用した本発明の触媒のような部分酸化反応触媒とはなっていないことは明らかである。
[Comparative Example 2]
The same catalyst as in Example 1 except that ceria (CeO 2 ) was used as a support, ie, NiO (16) -Cr 2 O 3 (4) -CeO 2 catalyst (16 mmol NiO and 4 mmol mol per gram CeO 2 A catalyst prepared from an aqueous solution in which all of Ce, Ni, and Cr were dissolved so as to contain Cr 2 O 3 was subjected to repeated experiments of partial oxidation reaction of methane-catalyst regeneration. The reaction and regeneration time was 10 minutes each and repeated 3 times. The reaction temperature was 700 ° C for the first time, 600 ° C for the second time, and 800 ° C for the third time. The results are shown in Table 4. Although the methane conversion rate increases at 700 ° C. or higher, the complete oxidation reaction proceeds because the CO 2 selectivity is high while the selectivity of hydrogen and CO is low, and MgO and SiO It is clear that the catalyst is not a partial oxidation reaction catalyst such as the catalyst of the present invention using 2 as a support.
[比較例3]
担体としてセリア(CeO2)を使用したほかは実施例6と同様の触媒、すなわちCeO2担体にNiとCrを含む水溶液を含浸担持させて調製したNiO(16)−Cr2O3(4)/CeO2触媒について、メタンの部分酸化反応−触媒再生反応を各10分ずつ、温度500℃、600℃、700℃、800℃で実施した。メタン転化率は700℃以上で高くなるものの、CO2とH2Oの発生が多いことから燃焼反応が進行していることが明らかであり、部分酸化触媒としての活性は低い。
[Comparative Example 3]
NiO (16) -Cr 2 O 3 (4) prepared by impregnating and supporting an aqueous solution containing Ni and Cr on a CeO 2 carrier, except that ceria (CeO 2 ) was used as the carrier. For the / CeO 2 catalyst, a partial oxidation reaction of methane-catalyst regeneration reaction was carried out for 10 minutes each at temperatures of 500 ° C., 600 ° C., 700 ° C., and 800 ° C. Although the methane conversion rate increases at 700 ° C. or higher, it is clear that the combustion reaction proceeds because of the large amount of CO 2 and H 2 O, and the activity as a partial oxidation catalyst is low.
[比較例4]
担体として酸化ランタン(La2O3)を使用したほかは実施例1と同様の触媒、すなわちNiO(16)−Cr2O3(4)−La2O3触媒(La2O31gあたり16m molのNiOと4m molのCr2O3が含有されるよう、La、Ni、Crを総て溶解させた水溶液から調製した触媒)について、比較例2と同様の条件でメタンの部分酸化反応−触媒再生の3回繰り返し実験を実施した。この結果も表4に示すが、水素選択率が低い一方でCO2選択率が高く、メタンの部分酸化反応触媒としては不適である。
[Comparative Example 4]
The same catalyst as in Example 1 except that lanthanum oxide (La 2 O 3 ) was used as the carrier, ie, NiO (16) -Cr 2 O 3 (4) -La 2 O 3 catalyst (16 m per gram of La 2 O 3 (catalyst prepared from an aqueous solution in which all of La, Ni, and Cr are dissolved so as to contain 4 mol of NiO and 4 mmol of Cr 2 O 3 ), a partial oxidation reaction of methane under the same conditions as in Comparative Example 2 Experiments were repeated three times for catalyst regeneration. This result is also shown in Table 4, but the hydrogen selectivity is low but the CO 2 selectivity is high, which is not suitable as a catalyst for partial oxidation reaction of methane.
[比較例5]
担体としてアルミナ(Al2O3)を使用したほかは実施例6と同様の触媒、すなわちAl2O3担体にNiとCrを含む水溶液を含浸担持させて調製したNiO(16)−Cr2O3(4)/Al2O3触媒について、メタンの部分酸化反応−触媒再生反応を各10分ずつ、温度500℃、600℃、700℃、800℃で実施した。この結果も表4に示すが、メタン転化率は700℃以上で高くなるものの、CO選択率が低く、部分酸化反応活性に乏しいことがわかる。
[Comparative Example 5]
Similar catalyst except that an alumina (Al 2 O 3) as carrier as in Example 6, i.e. Al 2 O 3 carrier was prepared by impregnation with an aqueous solution containing Ni and Cr NiO (16) -Cr 2 O 3 For the (4) / Al 2 O 3 catalyst, a partial oxidation reaction of methane-catalyst regeneration reaction was carried out for 10 minutes each at temperatures of 500 ° C., 600 ° C., 700 ° C., and 800 ° C. Although this result is also shown in Table 4, it can be seen that the methane conversion increases at 700 ° C. or higher, but the CO selectivity is low and the partial oxidation reaction activity is poor.
[比較例6]
Cr2O3の替わりにCaOを使用したほかは実施例1と同様の触媒、すなわちNiO(16)−CaO(4)−MgO触媒(MgO1gあたり16m molのNiOと4m molのCaOが含有されるよう、Mg、Ni、Caを総て溶解させた水溶液から調製した触媒)について、メタンの部分酸化反応−触媒再生反応を各10分ずつ、温度500℃、600℃、700℃、800℃で実施した結果を表6に示す。700℃以上で部分酸化反応が進行するものの、この温度領域では炭素析出量が多く、実用触媒としては不適である。
[Comparative Example 6]
A catalyst similar to that of Example 1 except that CaO was used instead of Cr 2 O 3 , that is, a NiO (16) -CaO (4) -MgO catalyst (containing 16 mmol of NiO and 4 mmol of CaO per 1 g of MgO). For example, a catalyst prepared from an aqueous solution in which Mg, Ni, and Ca are completely dissolved) was subjected to partial oxidation reaction of methane-catalyst regeneration reaction at temperatures of 500 ° C, 600 ° C, 700 ° C, and 800 ° C for 10 minutes each. Table 6 shows the results. Although the partial oxidation reaction proceeds at 700 ° C. or higher, the carbon deposition amount is large in this temperature range, which is not suitable as a practical catalyst.
[比較例7]
Cr2O3の替わりにFe2O3を使用したほかは実施例1と同様の触媒、すなわちNiO(16)−Fe2O3(4)−MgO触媒(MgO1gあたり16m molのNiOと4m molのFe2O3が含有されるよう、Mg、Ni、Feを総て溶解させた水溶液から調製した触媒)について、比較例6と同様の実験を行った結果を表6に示す。どの温度でもCO2発生が多いことから、完全酸化反応が優勢となっていることが明らかであり、部分酸化反応触媒とはならない。
[Comparative Example 7]
The same catalyst as in Example 1 except that Fe 2 O 3 was used instead of Cr 2 O 3 , that is, NiO (16) -Fe 2 O 3 (4) -MgO catalyst (16 mmol of NiO and 4 mmol of MgO per 1 g of MgO). Table 6 shows the results of experiments similar to those of Comparative Example 6 for a catalyst prepared from an aqueous solution in which Mg, Ni, and Fe are all dissolved so that Fe 2 O 3 is contained. Since there is much CO 2 generation at any temperature, it is clear that the complete oxidation reaction is dominant and it does not become a partial oxidation reaction catalyst.
[比較例8]
Cr2O3の替わりにAl2O3を使用したほかは実施例1と同様の触媒、すなわちNiO(16)−Al2O3(4)−MgO触媒(MgO1gあたり16m molのNiOと4m molのAl2O3が含有されるよう、Mg、Ni、Alを総て溶解させた水溶液から調製した触媒)について、比較例6と同様の実験を行った結果を表6に示す。反応温度を800℃まで上げれば高いメタン転化率が得られ部分酸化反応が進行するが、炭素析出量が多いため実用的ではない。
[Comparative Example 8]
The same catalyst as in Example 1 except that Al 2 O 3 was used instead of Cr 2 O 3 , that is, NiO (16) -Al 2 O 3 (4) -MgO catalyst (16 mmol of NiO and 4 mmol of MgO per 1 g of MgO). Table 6 shows the results of experiments similar to those of Comparative Example 6 for a catalyst prepared from an aqueous solution in which Mg, Ni, and Al are all dissolved so that Al 2 O 3 is contained. If the reaction temperature is increased to 800 ° C., a high methane conversion rate is obtained and a partial oxidation reaction proceeds. However, since the amount of carbon deposition is large, it is not practical.
[比較例9]
Cr2O3の替わりにNd2O3を使用したほかは実施例1と同様の触媒、すなわちNiO(16)−Nd2O3(4)−MgO触媒(MgO1gあたり16m molのNiOと4m molのNd2O3が含有されるよう、Mg、Ni、Ndを総て溶解させた水溶液から調製した触媒)について、比較例6と同様の実験を行った結果を表6に示す。比較例8と同様、反応温度を800℃まで上げれば高いメタン転化率が得られ部分酸化反応が進行するが、炭素析出量が多いため実用的ではない。
[Comparative Example 9]
The same catalyst as in Example 1 except that Nd 2 O 3 was used instead of Cr 2 O 3 , that is, NiO (16) -Nd 2 O 3 (4) -MgO catalyst (16 mmol of NiO and 4 mmol of MgO per 1 g of MgO). Table 6 shows the results of experiments similar to those of Comparative Example 6 for a catalyst prepared from an aqueous solution in which Mg, Ni, and Nd were dissolved so that Nd 2 O 3 was contained. As in Comparative Example 8, if the reaction temperature is increased to 800 ° C., a high methane conversion rate is obtained and the partial oxidation reaction proceeds. However, since the amount of carbon deposition is large, it is not practical.
[比較例10]
Cr2O3の替わりにCo3O4を使用したほかは実施例1と同様の触媒、すなわちNiO(16)−Co3O4(4)−MgO触媒(MgO1gあたり16m molのNiOと4m molのCo3O4が含有されるよう、Mg、Ni、Coを総て溶解させた水溶液から調製した触媒)について、比較例6と同様の実験を行った結果を表6に示す。比較例8と同様、反応温度を800℃まで上げれば高いメタン転化率が得られ部分酸化反応が進行するが、炭素析出量が多いため実用的ではない。
[Comparative Example 10]
The same catalyst as in Example 1 except that Co 3 O 4 was used instead of Cr 2 O 3 , that is, NiO (16) -Co 3 O 4 (4) -MgO catalyst (16 mmol of NiO and 4 mmol of MgO per 1 g of MgO). Table 6 shows the results of experiments similar to those of Comparative Example 6 for a catalyst prepared from an aqueous solution in which Mg, Ni, and Co are all dissolved so that Co 3 O 4 is contained. As in Comparative Example 8, if the reaction temperature is increased to 800 ° C., a high methane conversion rate is obtained and the partial oxidation reaction proceeds. However, since the amount of carbon deposition is large, it is not practical.
(X線回折による触媒再生の確認)
本発明の触媒の格子酸素が部分酸化反応に関与し、再生可能であることを触媒のX線回折測定により確かめた。そのX線回折測定の結果を図2に示す。触媒としては実施例1の触媒を用いた。図中、(A)と(B)では縦軸のスケールが10倍異なる。
(Confirmation of catalyst regeneration by X-ray diffraction)
It was confirmed by X-ray diffraction measurement of the catalyst that the lattice oxygen of the catalyst of the present invention was involved in the partial oxidation reaction and could be regenerated. The result of the X-ray diffraction measurement is shown in FIG. The catalyst of Example 1 was used as the catalyst. In the figure, (A) and (B) are different in scale of the
a)は調製後の触媒のX線回折パターンである。図中○の記号で示されているNiMgO2のピークが認められ、Niは担体のMgOと反応して複合酸化物として存在していることがわかる。縦軸スケールを拡大すると、図中□の記号で示されているMgCr2O4も存在することが確かめられる。 a) is an X-ray diffraction pattern of the catalyst after preparation. A NiMgO 2 peak indicated by a symbol “◯” in the figure is recognized, and it can be seen that Ni is present as a composite oxide by reacting with MgO of the carrier. When the vertical scale is enlarged, it can be confirmed that MgCr 2 O 4 indicated by the symbol □ in the figure also exists.
この触媒上でメタンの部分酸化反応を行った後のX線回折パターンが(A)のb)、c)及び(B)のb)である。○の記号で示されているNiMgO2、□の記号で示されているMgCr2O4に加えて、△で示されている金属Niのピークが出現している。すなわち、触媒を構成するNiの複合酸化物の格子酸素がメタンの部分酸化反応に消費され、Niが還元されたことが明らかである。 The X-ray diffraction patterns after the partial oxidation reaction of methane on this catalyst are (A) b), c) and (B) b). NiMgO 2 shown by a symbol of ○, in addition to MgCr 2 O 4, which is indicated by the symbol of □, the peak of the metal Ni indicated by △ are emerging. That is, it is clear that the lattice oxygen of the Ni complex oxide constituting the catalyst was consumed in the partial oxidation reaction of methane, and Ni was reduced.
メタン部分酸化反応−触媒再生反応を10回繰り返した後の触媒のX線回折パターンを(A)のd)に示す。○で示されているNiMgO2のピークのみが見られ、調製直後の(A)のa)と同等の状態に戻っていることが確かめられた。
なお(B)のc)のパターンは、各ピークの同定のために合成した、Niを含まないCr2O3−MgOのX線回折パターンである。
The X-ray diffraction pattern of the catalyst after repeating the methane partial oxidation reaction-
Note that the pattern c) in (B) is an X-ray diffraction pattern of Cr 2 O 3 —MgO not containing Ni synthesized for identification of each peak.
(空気による触媒再生)
これまでに提示した実験は、部分酸化反応に供した後の触媒の再生にアルゴンガスで希釈した酸素を用いていた。しかし、再酸化による触媒再生を空気により行うことができれば、装置に酸素供給設備を設置する必要がなくなり、システムの小型化及び低コスト化が実現され、実用化に当たって大きな利点があることは明らかである。そこで、実施例1の触媒を用いて、空気による触媒再生を行った。結果を表7に示す。再生ガスに空気を用いた場合、分析を質量分析装置で行った実験装置の制約から炭素析出量、メタン転化率、CO選択率及びCO2選択率の測定はできなかったが、水素選択率、格子酸素転化率及び他の生成物量を見る限り、アルゴンガスで希釈した酸素による触媒再生と同等の結果が得られており、空気による触媒再生になんら問題のないことが明らかである。
(Catalyst regeneration by air)
The experiments presented so far have used oxygen diluted with argon gas for regeneration of the catalyst after being subjected to a partial oxidation reaction. However, if the catalyst regeneration by reoxidation can be performed with air, it is not necessary to install an oxygen supply facility in the apparatus, and the system can be reduced in size and cost, and it is clear that there are significant advantages in practical use. is there. Thus, catalyst regeneration using air was performed using the catalyst of Example 1. The results are shown in Table 7. When air was used as the regeneration gas, the amount of carbon deposition, methane conversion, CO selectivity, and CO 2 selectivity could not be measured due to the limitations of the experimental apparatus in which the analysis was performed with a mass spectrometer, but the hydrogen selectivity, As far as the lattice oxygen conversion rate and the amount of other products are observed, it is clear that the catalyst regeneration by oxygen diluted with argon gas is the same as the catalyst regeneration by air.
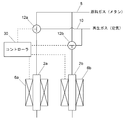
(水素含有ガス製造装置の実施例1)
図3に部分酸化触媒が移動できないように固定された固定床反応管を用いた水素含有ガス製造装置の実施例を概略的に示す。
第1の反応管2aに加熱炉6aが配置され、第2の反応管2bに加熱炉6bが配置されている。反応管2a,2bや加熱炉6a,6bは基本的には図1の評価用の装置のものと同じである。反応管2a,2b内には本発明の部分酸化触媒層が充填されている。
(Example 1 of a hydrogen-containing gas production apparatus)
FIG. 3 schematically shows an embodiment of a hydrogen-containing gas production apparatus using a fixed bed reaction tube fixed so that the partial oxidation catalyst cannot move.
A
一方の反応管2aの一端には原料ガスとしてメタンその他の炭化水素を供給する原料ガス供給流路8と、触媒再生時に酸素含有ガスとして空気を供給する再生ガス供給流路10が、三方切替弁12aにより切り替えてガスを供給することができるように接続されている。他方の反応管2bの一端にはその原料ガス供給流路8と再生ガス供給流路10が三方切替弁12bにより切り替えてガスを供給することができるように接続されている。原料ガス供給流路8と再生ガス供給流路10にはそれぞれ図1に示されているように開閉弁と質量流量制御器が設けられている。
A three-way switching valve is provided at one end of one
三方切替弁12aと12bはコントローラ30により同時に切り換えられ、反応管2aに原料ガスが供給されるときは反応管2bには再生ガスが供給され、逆に反応管2bに原料ガスが供給されるときは反応管2aには再生ガスが供給されるように制御される。
The three-
加熱炉6a,6bの温度もコントローラ30により制御され、それぞれの反応管2a,2bでの部分酸化反応又は触媒再生用の設定温度になるように調節される。部分酸化反応と触媒再生で反応管2a,2bの設定温度を変えないときは、コントローラ30により反応管2a,2bの温度が一定になるように制御される。
The temperatures of the
このようにして、加熱炉6a,6bと切替え弁12a,12bの制御により、一方の反応管での部分酸化反応中に他方の反応管での触媒を再生するようにするとともに、その操作を交互に切り替えることができる。
In this way, by controlling the
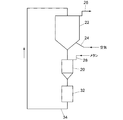
(水素含有ガス製造装置の実施例2)
図4に部分酸化触媒が移動可能な状態で保持された移動床反応管を用いた水素含有ガス製造装置の実施例を概略的に示す。
(Example 2 of a hydrogen-containing gas production apparatus)
FIG. 4 schematically shows an embodiment of a hydrogen-containing gas production apparatus using a moving bed reaction tube in which a partial oxidation catalyst is held in a movable state.
反応管は部分酸化反応を行わせる反応部20と、反応部20とは異なる場所で触媒再生を行わせる再生部22とを備えている。反応部20と再生部22はそれぞれ加熱炉を備えてそれぞれの設定温度になるように調節されている。反応部20と再生部22の設定温度は同じであってもよく、異なっていてもよい。
The reaction tube includes a
反応部20の上部に再生部22が配置されている。再生部22の下部には触媒再生のための酸素含有ガスとして空気を供給する再生ガス供給流路24が接続され、上部にはガス排出口26が設けられている。再生部22は再生された触媒を貯留することができるとともに、底部に開口をもち、再生された触媒をその開口から反応部20へ単位時間あたり一定量ずつ落下させる。
A
反応部20の上部には原料ガスとしてメタンその他の炭化水素を供給する原料ガス供給流路28が接続され、反応部20内で流動状態にある触媒と接触して部分酸化反応が行われる。反応部20の底部には開口が設けられ、反応部20の下部には触媒受け部32が設けられ、反応部20の底部の開口から落下した使用済みの触媒が触媒受け部32で受け止められる。
A raw material
触媒受け部32で受け止められた触媒を再生部22に搬送するために、搬送路34が設けられている。搬送路34は触媒を流路内のベルト又はスクリューにより押し上げて搬送するリフターになっている。
A
反応部20で部分酸化反応をした反応ガスは、搬送路34の流路を通って再生部22に送られ、再生部22のガス排出口26から使用済みの再生ガスとともに取り出される。
The reaction gas that has undergone the partial oxidation reaction in the
この水素含有ガス製造装置では、部分酸化反応と触媒再生が異なる場所で並行して実行され、触媒は反応部20と再生部22の間を循環するので、連続的に稼動させることができる。
In this hydrogen-containing gas production apparatus, the partial oxidation reaction and the catalyst regeneration are executed in parallel at different locations, and the catalyst circulates between the
本発明の部分酸化触媒並びにそれを用いた製造方法及び装置は、水素や合成ガスを製造するための原料となる水素含有混合ガスを製造するのに利用することができる。特に、本発明の触媒では、部分酸化反応においては触媒自身を構成する酸化物の格子酸素を利用し、反応後の触媒再生には空気中の酸素を利用できることから、極めてコンパクトかつ安価な水素含有混合ガス製造装置を構築することができる。かかる水素含有混合ガス製造装置は、例えば分散型電源としての燃料電池への燃料供給装置として好適である。触媒反応管を2系列組み込んで一方を反応に供している間に他方を再生するようにしたり、触媒反応管を移動床にして反応部と再生部の間で触媒を循環させるようにしたりすることにより、連続運転が可能となる。また、例えば日中に水素含有混合ガスを使用し、夜間は休止するような利用法が望まれるシステムでは、触媒を1系列のみ持ち、運転中に部分酸化反応を行い、夜間休止中に触媒再生を行うというコンパクトな装置構成が可能である。 The partial oxidation catalyst of the present invention and the production method and apparatus using the catalyst can be used to produce a hydrogen-containing mixed gas that is a raw material for producing hydrogen or synthesis gas. In particular, in the catalyst of the present invention, since the lattice oxygen of the oxide constituting the catalyst itself is used in the partial oxidation reaction, and oxygen in the air can be used for catalyst regeneration after the reaction, it is extremely compact and inexpensive. A mixed gas production apparatus can be constructed. Such a hydrogen-containing mixed gas production apparatus is suitable, for example, as a fuel supply apparatus for a fuel cell as a distributed power source. Incorporating two series of catalyst reaction tubes so that one is regenerated while the other is being used for the reaction, or using the catalyst reaction tube as a moving bed to circulate the catalyst between the reaction section and the regeneration section Thus, continuous operation becomes possible. For example, in a system that uses a hydrogen-containing mixed gas during the day and wants to stop during the night, only one series of catalyst is used, a partial oxidation reaction is performed during operation, and the catalyst is regenerated during the night out. A compact device configuration is possible.
2,2a,2b 反応管
4 部分酸化触媒層
6,6a,6b 電気炉
8 原料ガス供給流路
10 再生ガス供給流路
12,12a,12b 三方切替弁
20 反応部
22 再生部
30 コントローラ
32 触媒受け部
34 搬送路
2, 2a,
Claims (12)
少なくとも触媒成分と担体を含み、触媒成分はニッケルの酸化物とクロムの酸化物のみを含むことを特徴とする部分酸化触媒。 In a partial oxidation catalyst that partially oxidizes hydrocarbons to produce a mixed gas containing hydrogen and carbon monoxide,
A partial oxidation catalyst comprising at least a catalyst component and a support, wherein the catalyst component contains only nickel oxide and chromium oxide.
前記溶液の溶媒を除去する乾燥工程と、
前記乾燥工程後に酸化性雰囲気中で焼成する焼成工程と、
を含んで請求項1に記載の部分酸化触媒を製造する触媒製造方法。 A step of preparing only nickel and chromium, and magnesium as the metal element serving as a carrier, a mixed solution prepared by dissolving a salt containing the metal elements of silicon or yttrium as a metal element as a catalyst component,
A drying step of removing the solvent of the solution;
A firing step of firing in an oxidizing atmosphere after the drying step;
The catalyst manufacturing method which manufactures the partial oxidation catalyst of Claim 1 containing this.
その後、前記溶液の溶媒を除去する乾燥工程と、
前記乾燥工程後に酸化性雰囲気中で焼成する焼成工程と、
を含んで請求項1に記載の部分酸化触媒を製造する触媒製造方法。 Adding a solution in which a salt containing only nickel and chromium metal elements is dissolved to magnesium oxide (MgO), silica (SiO 2 ) or yttrium oxide (Y 2 O 3 ) as a carrier;
Thereafter, a drying step of removing the solvent of the solution;
A firing step of firing in an oxidizing atmosphere after the drying step;
The catalyst manufacturing method which manufactures the partial oxidation catalyst of Claim 1 containing this.
前記部分酸化工程を経た前記部分酸化触媒を加熱下で酸素含有ガスと接触させて前記部分酸化触媒を再生する再生工程と、
を含む水素含有ガス製造方法。 A metal in the partial oxidation catalyst is brought into contact with the partial oxidation catalyst according to any one of claims 1 to 4 under heating without supplying an oxygen-containing gas as an oxidant. A partial oxidation step of partially oxidizing the hydrocarbon with lattice oxygen constituting the oxide to generate a mixed gas containing hydrogen and carbon monoxide;
A regeneration step of regenerating the partial oxidation catalyst by contacting the partial oxidation catalyst that has undergone the partial oxidation step with an oxygen-containing gas under heating;
A method for producing a hydrogen-containing gas comprising:
前記触媒を加熱する加熱炉と、
炭化水素を含む原料ガスを前記反応管に送り前記触媒と接触させる原料ガス供給流路と、
触媒再生に用いる酸素含有ガスを前記反応管に送り前記部分酸化触媒と接触させる再生ガス供給流路と、を備え、
前記反応管中で、酸化剤としての酸素含有ガスの存在しない状態下で前記部分酸化触媒中の金属酸化物を構成する格子酸素により前記原料ガス中の炭化水素を部分酸化して水素と一酸化炭素を含有する混合ガスを生成させ、前記原料ガスが存在しない状態下で前記再生ガス供給流路からの酸素含有ガスにより前記部分酸化触媒を再生する水素含有ガス製造装置。 A reaction tube in which the partial oxidation catalyst according to any one of claims 1 to 4 is held,
A heating furnace for heating the catalyst;
A raw material gas supply channel for sending a raw material gas containing hydrocarbons to the reaction tube and contacting the catalyst;
A regeneration gas supply flow path for sending an oxygen-containing gas used for catalyst regeneration to the reaction tube and bringing it into contact with the partial oxidation catalyst,
In the reaction tube, in the absence of an oxygen-containing gas as an oxidant, the hydrocarbon in the raw material gas is partially oxidized by lattice oxygen constituting the metal oxide in the partial oxidation catalyst, and is oxidized with hydrogen. An apparatus for producing a hydrogen-containing gas, which generates a mixed gas containing carbon and regenerates the partial oxidation catalyst with an oxygen-containing gas from the regeneration gas supply flow path in a state where the source gas does not exist.
前記反応管と加熱炉の組が2組備えられ、
前記原料ガス供給流路と再生ガス供給流路は切替え弁を介して両反応管に接続されており、
前記加熱炉と切替え弁の制御により、一方の反応管での部分酸化反応中に他方の反応管での触媒を再生するようにするとともに、その操作を交互に切り替えることができるようになっている請求項10に記載の水素含有ガス製造装置。 The reaction tube is a fixed bed reaction tube fixed so that the partial oxidation catalyst cannot move,
Two sets of the reaction tube and the heating furnace are provided,
The source gas supply channel and the regeneration gas supply channel are connected to both reaction tubes via a switching valve,
By controlling the heating furnace and the switching valve, the catalyst in the other reaction tube is regenerated during the partial oxidation reaction in one reaction tube, and the operation can be switched alternately. The hydrogen-containing gas production apparatus according to claim 10.
前記反応部と再生部の間で前記触媒を搬送する搬送路が設けられており、
原料ガス供給流路は前記反応部に接続され、再生ガス供給流路は前記再生部に接続されている請求項10に記載の水素含有ガス製造装置。 The reaction tube is a moving bed reaction tube held in a state where the catalyst is movable, and the reaction tube performs a partial oxidation reaction and a regeneration for performing catalyst regeneration in a place different from the reaction unit. Department and
A transport path for transporting the catalyst between the reaction unit and the regeneration unit is provided;
The hydrogen-containing gas production apparatus according to claim 10, wherein the source gas supply flow path is connected to the reaction section, and the regeneration gas supply flow path is connected to the regeneration section.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007237561A JP5009109B2 (en) | 2007-09-13 | 2007-09-13 | Hydrocarbon partial oxidation catalyst and method and apparatus for producing hydrogen-containing gas using the same |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007237561A JP5009109B2 (en) | 2007-09-13 | 2007-09-13 | Hydrocarbon partial oxidation catalyst and method and apparatus for producing hydrogen-containing gas using the same |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009066520A JP2009066520A (en) | 2009-04-02 |
| JP5009109B2 true JP5009109B2 (en) | 2012-08-22 |
Family
ID=40603349
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007237561A Expired - Fee Related JP5009109B2 (en) | 2007-09-13 | 2007-09-13 | Hydrocarbon partial oxidation catalyst and method and apparatus for producing hydrogen-containing gas using the same |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5009109B2 (en) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5324265B2 (en) * | 2009-03-11 | 2013-10-23 | 関西電力株式会社 | Hydrocarbon partial oxidation catalyst and method and apparatus for producing hydrogen-containing gas using the same |
| JP5630987B2 (en) * | 2009-11-17 | 2014-11-26 | 中外炉工業株式会社 | Carburizing gas supply device |
| JP5784548B2 (en) * | 2012-06-12 | 2015-09-24 | フロンティア・ラボ株式会社 | Catalytic reaction simulation equipment |
| JP6131370B1 (en) | 2016-06-10 | 2017-05-17 | 千代田化工建設株式会社 | Syngas production catalyst carrier and production method thereof, synthesis gas production catalyst and production method thereof, and synthesis gas production method |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8714661D0 (en) * | 1987-06-23 | 1987-07-29 | British Petroleum Co Plc | Catalysts |
| FR2684567B1 (en) * | 1991-12-09 | 1994-11-04 | Atochem | MASS CATALYSTS BASED ON CHROME AND NICKEL OXIDES AND THEIR APPLICATION TO THE FLUORINATION OF HALOGENATED HYDROCARBONS. |
| GB2311790A (en) * | 1996-04-04 | 1997-10-08 | British Gas Plc | Production of synthesis gas from hydrocarbonaceous feedstock |
| FR2790750B1 (en) * | 1999-03-10 | 2001-04-20 | Air Liquide | PROCESS AND DEVICE FOR PRODUCING HYDROGEN BY THERMOCATALYTIC DECOMPOSITION OF HYDROCARBONS |
| JP3976498B2 (en) * | 2000-12-13 | 2007-09-19 | 独立行政法人科学技術振興機構 | Syngas production catalyst and synthesis gas production method |
-
2007
- 2007-09-13 JP JP2007237561A patent/JP5009109B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009066520A (en) | 2009-04-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Zhang et al. | Low temperature catalytic oxidation of propane over cobalt-cerium spinel oxides catalysts | |
| Silvester et al. | NiO supported on Al2O3 and ZrO2 oxygen carriers for chemical looping steam methane reforming | |
| Srisiriwat et al. | Oxidative steam reforming of ethanol over Ni/Al2O3 catalysts promoted by CeO2, ZrO2 and CeO2–ZrO2 | |
| Choudhary et al. | Energy‐efficient syngas production through catalytic oxy‐methane reforming reactions | |
| Zhu et al. | Catalytic partial oxidation of methane to synthesis gas over Ni–CeO2 | |
| Luengnaruemitchai et al. | Activity of different zeolite-supported Ni catalysts for methane reforming with carbon dioxide | |
| Cimino et al. | Insights into the cyclic CO2 capture and catalytic methanation over highly performing Li-Ru/Al2O3 dual function materials | |
| Białobok et al. | Ethanol combustion over strontium-and cerium-doped LaCoO3 catalysts | |
| JP5972678B2 (en) | Synthesis gas production catalyst and synthesis gas production method | |
| US6846475B1 (en) | Hydrogen refinement apparatus | |
| Pérez-Hernández et al. | Effect of Cu loading on CeO2 for hydrogen production by oxidative steam reforming of methanol | |
| Jin et al. | Engineering metal-oxide interface by depositing ZrO2 overcoating on Ni/Al2O3 for dry reforming of methane | |
| Zhou et al. | A novel catalyst with plate-type anodic alumina supports, Ni/NiAl2O4/γ-Al2O3/alloy, for steam reforming of methane | |
| KR101145122B1 (en) | Catalytic reaction between methanol and a peroxide | |
| KR20110074196A (en) | For reforming of methane and carbon dioxide, preparation of cobalt based catalysts and production method of synthesis gas | |
| KR100612956B1 (en) | High Performance Water Gas Shift Catalysts and A Method of Preparing The Same | |
| JP5009109B2 (en) | Hydrocarbon partial oxidation catalyst and method and apparatus for producing hydrogen-containing gas using the same | |
| US20060093550A1 (en) | High temperature stable non-noble metal catalyst, process for production of syngas using said catalyst | |
| WO2021116064A1 (en) | A method to capture and utilize co2 and an installation for capturing and utilizing co2 | |
| JP4859701B2 (en) | Hydrogen-containing gas production equipment | |
| Caro | Catalytic Membrane Reactors–Lab Curiosity or Key Enabling Technology? | |
| KR101245484B1 (en) | Water gas shift catalysts and method for producing syngas by Water gas shift reaction using the same | |
| JP5324265B2 (en) | Hydrocarbon partial oxidation catalyst and method and apparatus for producing hydrogen-containing gas using the same | |
| WO2014182020A1 (en) | Monolith catalyst for carbon dioxide reforming reaction, production method for same, and production method for synthesis gas using same | |
| JP4465478B2 (en) | Catalyst for hydrogen production |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100907 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20100907 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120118 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120131 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120330 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120529 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120530 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150608 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |