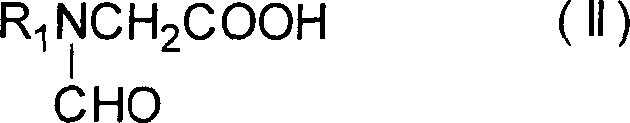-
Titel der
Erfindung
-
Verfahren zum Herstellen von 1-substituierten
Pyrrol-3-carbonsäure-Derivaten
-
Hintergrund der Erfindung
-
Erfindungsgebiet
-
Die vorliegende Erfindung betrifft
ein Verfahren zum Herstellen von 1-substituierten Pyrrol-3-carbonsäure-Derivaten, die als
Medizin oder als Rohmaterialien oder Zwischenprodukte für die Synthese
von Medizin nützlich
sind.
-
Derivate der 1-substituierten Pyrrol-3-carbonsäure sind
wichtige Verbindungen, die als Medizin für die Behandlung von Hyperlipidämie, Medizin
zur Verbesserung einer Störung
der peripheren Zirkulation oder als Rohmaterialien oder Zwischenprodukte
für die
Synthese von Medizin verwendet werden. Es waren jedoch keine industriellen
Verfahren zum Herstellen von Derivaten von 1-substituierten Pyrrol-3-carbonsäuren bekannt, in
denen die 2-, 4- und 5-Positionen alle Wasserstoffatome sind und
die eine genügende
Wirtschaftlichkeit und operative Einfachheit haben.
-
Genauer ist ein Verfahren der Induzierung
einer elektrophilen Substitutionsreaktion an der 3-Position eines
Pyrrol-Ringes, beispielsweise als Verfahren zum Herstellen von Pyrrol-3-carbonsäure-Derivaten
beschrieben worden (z.B. M. Kakushima et al., J. Org. Chem., Bd.
48, 3214 (1983); C. Cativiela et al., Org. Prep. Proced. Int., Bd.
18, 283 (1986); B. L. Bray et al., J. Org. Chem., Bd. 55, 6317 (1990);
etc.). Aufgrund der Eigenschaften des Pyrrols tritt die elektrophile
Substitutionssreaktion im Pyrrol-Ring nicht notwendigerweise ausschließlich an
der 3-Position auf. Es kann Fälle
geben, wo Nebenprodukte wie Pyrrol-2-carbonsäure-Derivate abgetrennt werden
müssen.
Zusätzlich
wird bei diesen Verfahren zuerst eine spezifische Gruppe, wie beispielsweise
eine Arylsulfonyl-Gruppe oder eine Triisopropylsilyl-Gruppe an die
1-Position eingeführt
und dann wird die elektrophile Substitutionsreaktion an der 3-Position
induziert. Aus diesem Grund muß die
Gruppe, die zuerst an der 1-Position eingefügt worden ist, vor dem Einfügen der
Zielgruppe zuerst entfernt werden, um eine Verbindung mit einer
Alkyl-Gruppe oder ähnlichem
an der 1-Position zu substituieren. Dies macht komplizierte, zeitaufwendige
Reaktionsschritte notwendig und verursacht dadurch Nachteile dieser
Verfahren.
-
Als alternative Verfahren zum direkten
Synthetisieren von Pyrrol-3-carbonsäure-Derivaten ist von einem
Verfahren zum Umsetzen eines Rohmaterials wie beispielsweise γ-Ketoester
in einer Cyclisierungsreaktion (z. B. japanische Patentveröffentlichung
Nr. 104658/1994) und einen Verfahren zum Umsetzen von p-Toluolsulfonylisocyanid
mit einem Acrylsäureester
in Gegenwart von Natriumhydrid (z. B. A. M, van Lausen et al., Tetrahedron
Lett., Bd. 52, 5337 (1972)) berichtet worden.
-
Da die Pyrrol-3-carbonsäure-Derivate,
die auf diese Weise hergestellt werden jedoch ein Wasserstoffatom
an der 1-Position haben, wird ein zusätzlicher Schritt zum Einführen einer
Zielgruppe benötigt,
um eine Verbindung mit einer Alkyl-Gruppe oder ähnlichem zu bekommen, die an
der 1-Position substituiert wird. Dies benötigt auch zusätzliche
Schritte. Außerdem
sind die zuletzt genannten Verfahren ökonomisch nicht zu bevorzugen,
da sie teure Reagenzien notwendig machen und die Zielverbindung
nur in einer geringen Ausbeute produzieren.
-
Zusätzlich zu diesen Verfahren
ist ein Verfahren zur Synthese von Pyrrol-3-carbonsäureestern
mit einer Substitutionsgruppe an der 1-Position vorgeschlagen worden.
Dieses Verfahren umfaßt
das Umsetzen eines Esters der Propiolsäure, ein Säureanhydrid und ein Aminosäure-Derivat
(z. B. Albert Padwa et al., J. Org. Chem., Bd. 47, 786 (1982), Piero
Dalla Croce et al., Heterocycles, Bd. 27, 2825 (1988)). Dieses Verfahren
erzeugt jedoch nur Verbindungen mit Substitutionsgruppen, die an
der 2-Position und/oder der 5-Position eingefügt sind.
-
D. h., daß keine industriell nützlichen
Verfahren zum Herstellen von 1-substituierten Pyrrol-3-carbonsäure-Derivaten mit Wasserstoffatomen
sowohl an der 2-, 4- und 5-Position bekannt waren, und deswegen
ist die Bedeutung der Derivate der 1-substituierten Pyrrol-3-carbonsäure, in
der an der 2-, 4- und 5-Position Wasserstoffatome sind, bisher nicht
untersucht worden.
-
Im Hinblick auf diese Situation haben
die Erfinder der vorliegenden Erfindung ausgedehnte Studien zur
Entwicklung eines Verfahrens zum Herstellen von 1-substituierten
Pyrrol-3-carbonsäure-Derivaten
und zur Synthese von Medikamenten unter Verwendung von Derivaten
von 1-substituierter Pyrrol-3-carbonsäure, in der
die 2-, 4- und 5-Positionen Wasserstoffatome sind, als synthetisches
Material oder als Zwischenprodukt studiert worden. Als Ergebnis
haben die Erfinder gefunden, daß 1-substituierte
Pyrrol-3-carbonsäure-Derivate leicht in
einer kurzen Zeit und in einer hohen Ausbeute hergestellt werden
können,
wenn N-substituiertes N-Formylglycin als Ausgangsmaterial verwendet
wird. Dieser Befund hat zur Vollendung der vorliegenden Erfindung
geführt.
-
Zusammenfassung der Erfindung
-
Demgemäß ist es ein Ziel der vorliegenden
Erfindung, ein Verfahren zum Herstellen eines 1-substituierten Pyrrol-3-carbonsäure-Derivats
der folgenden Formel (I)
zur Verfügung zu stellen (wobei R
1 und R
2 unabhängig voneinander
eine Alkyl-, Aralkyl- oder Aryl-Gruppe darstellen), umfassend das
Umsetzen einer Verbindung der folgenden Formel (II)
(wobei R
1 wie
oben definiert ist), einer Verbindung der folgenden Formel (III)
HC≡C-COOR2 (III)(wobei
R
2 wie oben definiert ist) und eines Säureanhydrids
umfaßt.
-
Weitere Ziele, Eigenschaften und
Vorteile der Erfindung werden aus der folgenden Beschreibung leicht
offensichtlich.
-
Detaillierte Beschreibung
der Erfindung und bevorzugte Ausführungsformen
-
Als bevorzugte Beispiele von R1 in Formel (I) des 1-substituierten Pyrrol-3-Carbonsäure-Derivats,
das durch das erfindungsgemäße Verfahren
erhalten wird, werden lineare oder verzweigte Alkyl-Gruppen, beispielsweise
eine Methyl-Gruppe, Ethyl-Gruppe, n-Propyl-Gruppe und Isopropyl-Gruppe,
Aralkyl-Gruppen,
wie beispielsweise eine Benzyl-Gruppe und 3,4-Dimethoxyphenylmethyl-Gruppe
und Aryl-Gruppen, wie beispielsweise eine Phenyl-Gruppe, 2,6-Dimethylphenyl-Gruppe
und eine Naphthyl-Gruppe angegeben. Bevorzugte Beispiele für R2 schließen
lineare oder verzweigte Alkyl-Gruppen, wie beispielsweise eine Methyl-Gruppe, Ethyl-Gruppe,
n-Propyl-Gruppe
und Isopropyl-Gruppe, Aralkyl-Gruppen, wie beispielsweise die Benzyl-Gruppe
und Aryl-Gruppen, wie beispielsweise die Phenyl-Gruppe ein.
-
Das erfindungsgemäße Verfahren umfaßt das Umsetzen
einer Verbindung der Formel (II) mit einer Verbindung der Formel
(III) und einem Säureanhydrid
entsprechend dem folgenden Reaktionsschema
wobei
R
1 und R
2 die gleiche
Bedeutung haben wie oben definiert.
-
Als Beispiele des Säureanhydrids
sind Essigsäureanhydrid,
Propionsäureanhydrid
und ähnliche
anzugeben, wobei Essigsäureanhydrid
besonders bevorzugt ist.
-
Die Verbindung (II), die das Ausgangsmaterial
ist, kann durch das Umsetzen einer Verbindung der Formel (IV) mit
einem Essigsäureanhydrid
und Ameisensäure
gemäß dem folgenden
Reaktionsschema leicht hergestellt werden.
wobei
R
1 die gleiche Bedeutung hat, wie zuvor
definiert.
-
N-substituiertes Glycin, wie beispielsweise
Sarcosin und N-Phenylglycin werden als spezifische Beispiele der
Verbindung (IV), die in dieser Reaktion verwendet wird, angegeben.
-
Die Verbindung (II) kann auch durch
ein Verfahren synthetisiert werden, das durch eine Publikation offenbart
wurde, oder durch ein Verfahren, das dem bekannten Verfahren ähnlich ist.
-
Die Verbindung (III), Propiolsäureester,
ist entweder selbst eine bekannte Verbindung oder eine Verbindung,
die nach einem in dem Fachgebiet bekannten Verfahren hergestellt
werden kann.
-
Die Cyclisierungsreaktion der Verbindung
(II) und der Verbindung (III) kann durch das Zugeben von mehr als
einem Mol des Säureanhydrids,
wie beispielsweise Essigsäureanhydrid,
und ungefähr
1 bis 5 Molen der Verbindung (III) je 1 mol der Verbindung (II),
und durch das Erwärmen
des Gemischs auf eine Temperatur von 80°C der Rückflußtemperaturen durchgeführt werden,
während
für ungefähr 4 bis
24 Stunden gerührt
wird. Diese Reaktion kann entweder in Gegenwart eines Säureanhydrids
(bevorzugt in Essigsäureanhydrid)
allein oder zusammen mit einem zusätzlichen Lösungsmittel durchgeführt werden,
das nicht in die Umsetzung eingeschlossen ist, wie beispielsweise
Toluol. Der erste Fall, in dem die Reaktion in einem Säureanhydrid
durchgeführt
wird, ist zum Erreichen einer hohen Ausbeute im allgemeinen mehr
bevorzugt. Es ist nicht notwendig zu sagen, daß Salze dieser Reaktionsteilnehmer
anstelle der Verbindungen selbst verwendet werden können.
-
Um die abgezielten 1-substituierten
Pyrrol-3-carbonsäure-Derivate (I) aus
dem Reaktionsgemisch zu erhalten, können konventionelle Reinigungsverfahren,
wie beispielsweise Destillierung, Rekristallisierung, und Säulenchromatographie,
entweder einzeln oder in Kombination von zwei oder mehr, verwendet
werden.
-
Das resultierende 1-substituierte
Pyrrol-3-carbonsäure-Derivat (I) ist als
Medikament oder als Rohmaterial oder als synthetisches Zwischenprodukt
für Medikamente
nützlich.
wenn es als Rohmaterial für
die Synthese von Medikamenten verwendet wird, wird die Verbindung
der Formel (I) durch das Entfernen der Gruppe R
2 beispielsweise
gemäß der folgenden
Reaktion in die Verbindung (I')
umgewandelt, bevor sie verschiedenen weiteren Reaktionen unterworfen
wird.
worin R
1 und
R
2 die gleichen Bedeutung haben wie zuvor
definiert.
-
Die oben gezeigte Umsetzung zum Umwandeln
der Verbindung der Formel (I) in die Verbindung der Formel (I') kann durch ein
bekanntes Verfahren, z. B. durch Behandeln der Verbindung der Formel
(I) mit einer Base wie beispielsweise Natriumhydroxid durchgeführt werden,
und durch das Umsetzen mit einer Säure wie beispielsweise Salzsäure.
-
Weitere Eigenschaften der Erfindung
werden im Verlaufe der folgenden Beschreibung der beispielhaften
Ausführungsformen
deutlich, die für
die Darstellung der Erfindung angegeben werden und nicht beabsichtigen,
diese zu begrenzen.
-
Beispiele
-
Referenzbeispiel 1
-
Synthese von N-Formylsarcosin:
-
243,5 g (2,733 mol) Sarcosin und
1500 g (32,58 mol) Ameisensäure
wurden in ein Reaktionsgefäß überführt, und
887,4 g (8,691 mol) Essigsäureanhydrid
wurden über
30 Minuten tröpfchenweise
zum Gemisch hinzugegeben, während
gekühlt
und gerührt
wurde. Das resultierende Gemisch wurde für 3 Stunden bei Raumtemperatur
gerührt.
-
Das resultierende leicht gelbe Reaktionsgemisch
wurde unter Vakuum konzentriert, 50 ml Wasser wurden zu dem Rest
hinzugegeben, und das erhaltene Gemisch wurde unter Vakuum verdampft.
Dieser Arbeitsschritt wurde wiederholt, und dann wurde 50 ml Toluol
zu dem Rest hinzugegeben, und das Gemisch wurde unter Vakuum verdampft.
Nach dem Wiederholen dieses Arbeitsschrittes wurden Saatkristalle
des N-Formylsarcosins hinzugegeben, um den Rest zu kristallisieren.
Nach dem vollständigen
Entfernen des Lösungsmittels
mit Hilfe einer Pumpe, wurden 1200 ml Ethylacetat hinzugegeben,
und das Gemisch wurde über
einem warmen Wasserbad bei 70°C
gerührt,
bis die Kristalle vollständig
gelöst
waren. Es wurden erneut Saatkristalle hinzugegeben, und das Gemisch
wurde über
Nacht gerührt.
Nach dem Rühren
für eine
Stunde bei 0°C
wurden die präzipitierten
Kristalle durch Filtrieren eingesammelt, um 278,6 g der Titelverbindung
zu erhalten (Ausbeute 87,1%).
Eigenschaften: farblose pulverartige
Kristalle
Schmelzpunkt: 85,0–87,0°C
-
Referenzbeispiel 2
-
Synthese von N-Benzyl-N-formylglycin:
-
19,33 g (100 mmol) N-Benzylglycinethylester
und 109,6 g (32,38 mol) Ameisensäure
wurden in ein Reaktionsgefäß überführt. Dann
wurden 32,46 g (316 mmol) Essigsäureanhydrid über 15 Minuten
tröpfchenweise zum
Gemisch hinzugegeben, während
gekühlt
und gerührt
wurde. Das resultierende Gemisch wurde für 1 Stunde bei 0°C und 2 Stunden
bei Raumtemperatur gerührt.
-
Das Reaktionsgemisch wurde unter
Vakuum konzentriert und 500 ml Ethylacetat wurden zu dem Rest hinzugegeben.
Die organische Schicht wurde mit einer halbgesättigten wäßrigen Kaliumcarbonat-Lösung, Wasser,
einer 15%igen wäßrigen Zitronensäure-Lösung, Wasser,
und gesättigter
Lauge in dieser Reihenfolge gewaschen, über wasserfreiem Natriumsulfat
getrocknet und unter Vakuum konzentriert, um 21,70 g einer hellgelben öligen Substanz
zu erhalten.
-
98 ml einer 2N wäßrigen Natriumhydroxid-Lösung wurden
zu der resultierenden öligen
Substanz hinzugegeben, und das Gemisch wurde für 30 Minuten bei 50°C und für 2 Stunden
bei Raumtemperatur gerührt.
-
Das Reaktionsgemisch wurde mit Ethylether
gewaschen, und 6N wäßrige Salzsäurelösung wurde
zu der Wasserschicht unter Rühren
hinzugegeben, um die Lösung
anzusäuern.
Nach der Zugabe von Natriumchlorid, wurde das Gemisch mit Ethylacetat
extrahiert (3-mal). Der Extrakt wurde mit gesättigter Lauge gewaschen, über wasserfreien
Natriumsulfat getrocknet, und unter Vakuum konzentriert. Die resultierenden
Rohkristalle wurden aus Chloroformisopropylether rekristallisiert,
um 17,27 g der Titelverbindung (Ausbeute 89,4%) zu erhalten.
Eigenschaften:
farblose prismenartige Kristalle
Schmelzpunkt: 124,0–127,0°C.
-
Referenzbeispiel 3
-
Synthese von N-Formyl-N-phenylglycin
-
7,56 g (50 mmol) an N-Phenylglycin
und 54,87 g (1,19 mol) Ameisensäure
wurden in Reaktionsgefäß übertragen
und 16,23 g (159 mmol) Essigsäureanhydrid
wurde tröpfchenweise über ungefähr 10 Minuten
unter Kühlen
und Rühren
hinzugegeben. Das Reaktionsgemisch wurde für eine Stunde bei 0°C und für 2 Stunden bei
Raumtemperatur gerührt.
Das erhaltene Reaktionsgemisch wurde unter Vakuum konzentriert,
um rohe Kristalle zu erhalten. Die Kristalle wurden aus Ethanolisopropylether
rekristallisiert, um 7,47 g der Titelverbindung (Ausbeute 83,4 59
zu erhalten.
Eigenschaften: hellgelbe pulvrige Kristalle
Schmelzpunkt:
169°C (zersetzt)
-
Beispiel 1
-
Synthese von Ethyl-1-methyl-3-pyrrolcarboxylat
(1):
-
Ein Gemisch aus 117,1 g (1 mol) N-Formylsarcosin
hergestellt im Referenzbeispiel 1, 98,10 g (1 mol) Ethylpropiolat,
und 638 ml Essigsäureanhydrid
wurden unter Verwendung eines Magnetrührers über einem Ölbad bei 130°C für 22 Stunden
gerührt.
Das Reaktionsgemisch wurde unter Vakuum konzentriert. Zu dem Rest wurden
100 ml Toluol hinzugegeben und das Gemisch wurde unter reduziertem
Druck verdampft. Dieses Verfahren wurde wiederholt, um eine braune ölige Substanz
zu erhalten. Diese ölige
Substanz wurde unter Vakuum destilliert, um 109,19 g (Ausbeute 71,3%)
eines farblosen oder leicht gelben Öls der Titelverbindung als Fraktion
mit einem Siedepunkt von 103–104°C bei 4 mmHg
zu erhalten.
IR (Film cm–1): 1701, 1544, 1250,
1218, 1113, 1026, 965, 763.
NMR (gemessen in CDCl3 unter
Verwendung von TMS als internem Standard/400 MHz/δppm):
1,32 (3H, t, J = 7,1 Hz), 3,66 (3H, s), 4,26 (2H, q, J = 7,1 Hz),
6,54 (1H, m), 6,57 (1H, m), 7,23 (1H, t, J = 1,9 Hz).
-
Beispiel 2
-
Synthese von Ethyl-1-methyl-3-pyrrolcarboxylat
(2):
-
Das gleiche Verfahren wie in Beispiel
1 wurde unter Verwendung von 15,64 g (0,134 mol) N-Formylsarcosin,
49,00 g (0,499 mol) Ethylpropiolat und 107 ml Essigsäureanhydrid
durchgeführt,
um 18,08 g (Ausbeute 88,4%) der Titelverbindung zu erhalten.
-
Beispiel 3
-
Synthese von Methyl-1-methyl-3-pyrrolcarboxylat:
-
Ein Gemisch aus 9,52 g (0,0813 mol)
N-Formylsarcosin, hergestellt im Referenzbeispiel 1, 25,56 g (0,304
mol) Methylpropiolat und 65 ml Essigsäureanhydrid wurde unter Verwendung
eines Magnetrührers über einem Ölbad bei
130°C für 24 Stunden
gerührt.
Das Reaktionsgemisch wurde unter Vakuum auf konzentriert. 30 ml
Toluol wurde zu dem Rest hinzugegeben und das Gemisch wurde unter
reduziertem Druck verdampft. Dieses Verfahren wurde wiederholt,
um eine braune ölige
Substanz zu erhalten. Diese ölige
Substanz wurde unter Vakuum destilliert, um 9,01 g (Ausbeute 79,6%)
eines farblosen oder leicht gelben Öls der Titelverbindung als
Fraktion mit einem Siedepunkt von 93 bis 96°C bei 4 mmHg zu erhalten.
IR
(Film cm–1):
1705, 1543, 1442, 1250, 1222, 1117, 764.
NMR (gemessen in CDCl3 unter Verwendung von TMS als internem Standard/400
MHz/δppm): 3,65 (3H, s), 3,78 (3H, s), 6,51– 6,58 (2H,
m), 7,22 (1H, s).
-
Beispiel 4
-
Synthese von Ethyl-1-benzyl-3-pyrrolcarboxylat:
-
Ein Gemisch aus 1,93 g (10 mmol)
im Referenzbeispiel 2 erhaltenen N-Benzyl-N-formylglycin, 3,65 g (37,2
mmol) Ethylpropiolat und 10 ml Essigsäureanhydrid wurde unter Verwendung
eines Magnetrührers über einem Ölbad bei
130°C für 20 Stunden
gerührt.
Das Reaktionsgemisch wurde unter Vakuum auf konzentriert. 15 ml
Toluol wurde zu dem Rest hinzugegeben und das Gemisch wurde unter
reduziertem Druck verdampft. Dieses Verfahren wurde wiederholt,
um eine braune ölige
Substanz zu erhalten. Diese ölige
Substanz wurde unter Verwendung von Kieselgel-Säulenchromatographie gereinigt
(Nr. 9385 Kieselgel, hergestellt von Merck Co., eluiert mit Ethylacetat
: Hexan = 1 : 3 V/V) um 2,156 g (Ausbeute 94,0 o) eines hellgelben Öls der Titelverbindung
zu erhalten.
IR (Film cm–1): 29801, 1702, 1541,
1508, 1455, 1373, 1221, 1112, 1027, 968, 763, 711.
NMR (gemessen
in CDCl3 unter Verwendung von TMS als internem
Standard/400 MHz/δppm): 1,33 (3H, t, J = 7,1 Hz), 4,26 (2H,
q, J = 7,1 Hz), 5,06 (2H, s), 6,59–6,64 (2H, m), 7,27–7,39 (4H,
m).
-
Beispiel 5
-
Synthese von Ethyl-1-phenyl-3-pyrrolcarboxylat:
-
Ein Gemisch aus 2,69 g (15 mmol)
N-Formyl-N-phenylglycin, hergestellt in Referenzbeispiel 3, 5,47
g (55,8 mmol) Ethylpropiolat und 15 ml Essigsäureanhydrid wurden unter Verwendung
eines Magnetrührers über einem Ölbad bei
130°C für 22 Stunden
gerührt.
Das Reaktionsgemisch wurde unter Vakuum auf konzentriert. 22 ml
Toluol wurden zu dem Rest hinzugegeben und das Gemisch wurde unter
reduziertem Druck verdampft. Dieses Verfahren wurde wiederholt,
um eine braune ölige
Substanz zu erhalten. Diese ölige
Substanz wurde unter Verwendung von Kieselgel-Säulenchromatographie (Nr. 93853
Kieselgel, hergestellt von Merck Co., eluiert mit Ethylacetat :
Hexan = 1 : 5 V/V) gereinigt, um 2,894 g (Ausbeute 89,6 g) eines
hellgelben Öls der
Titelverbindung zu erhalten.
IR (Film cm–1):
1709, 1600, 1544, 1509, 1260, 1224, 1138, 757, 692.
NMR (gemessen
in CDCl3 unter Verwendung von TMS als internem
Standard/400 MHz/δppm): 1,36 (3H, t, J = 7,1 Hz), 4,31 (2H,
q, J = 7,1 Hz), 6,76 (1H, br.s), 7,01 (1H, br.s), 7,31 (1H, t, J
= 7,2 Hz), 7,34–7,50
(4H, m), 7,68 (1H, s).
-
Referenzbeispiel 4
-
7,66 g (50 mmol) Ethyl-1-methyl-3-pyrrolcarboxylat,
hergestellt in Beispiel 1 und 37,5 ml (75 mmol) 2N wäßrige Natriumhydroxid-Lösung wurden
in ein Reaktionsgefäß geladen
und für
zwei Stunden im Rückfluß gehalten.
Das Reaktionsgemisch wurde auf 0°C
abgekühlt
und ungefähr
7 ml 6N Salzsäure-Lösung wurde
hinzugegeben während
gerührt
wurde. Nach der Zugabe von 15 g Natriumchlorid wurde das Gemisch
für eine Stunde über einem
Eis-Aceton-Bad gerührt,
um präzipitierte
Kristalle einzusammeln. Die Kristalle wurden mit kaltem Wasser gewaschen
und unter reduziertem Druck getrocknet, um 5,77 g (Ausbeute 92,2%)
farbloser oder leicht gelber pulvriger Kristalle der 1-Methyl-3-pyrrolcarbonsäure zu erhalten.
-
Da die in dem Verfahren der vorliegenden
Erfindung verwendete Reaktion keine Substituierungsreaktion an einem
Pyrrol-Ring ist, umfaßt
das Verfahren nicht die Nebenproduktion von Pyrrol-2-carbonsäure-Derivaten.
Deswegen besteht keine Notwendigkeit für ein Verfahren mit einem komplizierten
Schritt zur Trennung der Nebenprodukte. Ein zusätzlicher Vorteil in dem Verfahren
der vorliegenden Erfindung liegt in der Möglichkeit ein Pyrrol-3-carbonsäure-Derivat
mit einer substituierten Alkyl-Gruppe oder ähnlichen, die an der 1-Position
eingeführt
sind, direkt herzustellen.
-
Außerdem sichern die nicht-vorhandene
Verwendung jeglicher gefährlichen
Reagenzien und eine geringere Anzahl an Reaktionsschritten einfachere
Arbeitsschritte und eine drastisch verkürzte Verfahrensdauer. Zusätzlich machen
die Verwendung leicht erhältlicher
und günstiger
Rohmaterialien und die hohe Produktionsausbeute das Verfahren zur Herstellung
von 1-substituierten Pyrrol-3-carbonsäure-Derivaten industriell lebensfähig.
-
Außerdem werden die 1-substituierten
Pyrrol-3-carbonsäure-Derivate der vorliegenden
Erfindung es einfach machen, Medikamente unter Verwendung dieser
Derivate als Rohmaterialien zu entwickeln.
-
Offensichtlich sind zahlreiche Modifizierungen
und Variierungen der vorliegenden Erfindung im Hinblick auf die
obige Lehre möglich.
Es ist deswegen zu verstehen, daß die Erfindung anders als
spezifisch hier beschrieben im Schutzbereich der anhängenden
Ansprüche
durchgeführt
werden kann.
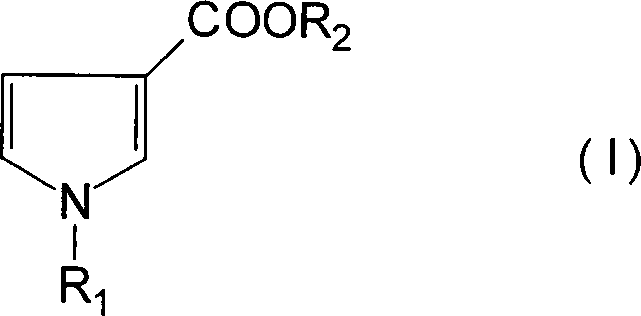 (wobei R1 wie oben definiert ist), einer Verbindung der folgenden Formel (III)
(wobei R1 wie oben definiert ist), einer Verbindung der folgenden Formel (III)



 wobei R1 wie oben definiert ist; einer Verbindung der folgenden Formel (III):
wobei R1 wie oben definiert ist; einer Verbindung der folgenden Formel (III):