WO2013005735A1 - α-オレフィン重合方法 - Google Patents
α-オレフィン重合方法 Download PDFInfo
- Publication number
- WO2013005735A1 WO2013005735A1 PCT/JP2012/066960 JP2012066960W WO2013005735A1 WO 2013005735 A1 WO2013005735 A1 WO 2013005735A1 JP 2012066960 W JP2012066960 W JP 2012066960W WO 2013005735 A1 WO2013005735 A1 WO 2013005735A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- component
- olefin
- catalyst
- compound
- contact
- Prior art date
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F10/04—Monomers containing three or four carbon atoms
- C08F10/06—Propene
Definitions
- the present invention relates to a method for producing an ⁇ -olefin polymer, particularly a propylene polymer, which is a general-purpose resin.
- an ⁇ -olefin polymer having high stereoregularity can be obtained with long and long catalytic efficiency.
- the ⁇ -olefin polymer obtained by the method of the present invention has an advantage that the presence ratio of fine powder is very small, and therefore such ⁇ -olefin polymer is excellent in production stability and moldability. .
- a method for producing crystalline polyolefin by polymerizing an ⁇ -olefin such as propylene and 1-butene in the presence of a stereoregular catalyst has been proposed in many prior arts and is known.
- these polymerization methods (a) a highly active titanium solid catalyst component containing magnesium, titanium, halogen and an electron donor as essential components, (b) an organoaluminum compound catalyst component, and (C) an electron donor catalyst component.
- a number of prior art documents have proposed a method in which a highly stereoregular polymer can be obtained with a high catalytic activity by polymerizing an ⁇ -olefin in the presence of a catalyst to be formed.
- Patent Document 1 Japanese Patent No. 2637076 uses a solid titanium catalyst component formed by contacting a magnesium compound, a titanium compound, and a polyvalent carboxylic acid compound as an electron donor, and an organoaluminum compound. Propylene is prepolymerized in the pre-stage polymerization step, and then the prepolymerized solid catalyst component, the organoaluminum compound, and the organosilicon compound catalyst component as the electron donor are used, and propylene and other ⁇ -olefin are used in the post-stage polymerization step. A method for producing a propylene block copolymer by copolymerizing the above is disclosed.
- Example 1 of Patent Document 1 includes a solid catalyst component obtained by prepolymerizing propylene in a prepolymerization step (preliminary polymerization step), triethylaluminum (an organoaluminum compound), n-propyltriethoxysilane (an electron donor), Is added to a propylene-substituted autoclave to polymerize propylene, and then an ethylene-propylene mixed gas is added to the system to obtain an ethylene-propylene copolymer.
- Patent Document 1 does not disclose any preliminary contact between the prepolymerization catalyst, the organoaluminum compound, and the electron donor before the polymerization, and the contact order of these compounds.
- Patent Document 2 Japanese Patent No. 2740503 discloses a titanium solid catalyst component obtained by reacting a magnesium compound with a tetravalent titanium compound and reacting it with a polyvalent carboxylic acid ester, and an organometallic catalyst. It is disclosed that an ⁇ -olefin is prepolymerized using a component and an organosilicon compound, and then the ⁇ -olefin is polymerized using a prepolymerized solid catalyst component.
- Example 2 of Patent Document 2 propylene was prepolymerized using a titanium catalyst component containing titanium, magnesium and diisobutyl phthalate, triethylaluminum and diphenyldimethoxysilane, and the resulting prepolymerized catalyst, triethylaluminum and It is disclosed that diphenyldimethoxysilane is added to an autoclave containing propylene to polymerize propylene.
- Patent Document 2 does not disclose any preliminary contact between the prepolymerization catalyst, the organoaluminum compound, and the electron donor before polymerization, and the contact order of these compounds.
- Patent Document 3 Japanese Patent No. 3984304 is obtained by contacting a solid titanium catalyst component containing magnesium, titanium, halogen and an electron donor, an organometallic compound catalyst component, and an organosilicon compound. It is disclosed that polypropylene is produced using an olefin polymerization catalyst formed from a contact product, a 1,3-diether compound, and an organometallic compound catalyst component.
- propylene was prepolymerized using a solid catalyst component formed from magnesium chloride, titanium tetrachloride, and diisobutyl phthalate, triethylaluminum, and dicyclopentyldimethoxysilane, and then the resulting prepolymerization was performed.
- Patent Document 3 does not disclose any preliminary contact between the prepolymerization catalyst, the organoaluminum compound, and the electron donor before polymerization, and the contact order of these compounds.
- Patent Document 4 Japanese Patent Publication No. 6-23406
- propylene is prepolymerized using a titanium complex containing a halogenated magnesium and a halogenated titanium and an organoaluminum compound, and then the resulting solid catalyst is obtained.
- a method for producing a propylene polymer using a component, an organoaluminum compound catalyst component, and an organosilicon compound is disclosed.
- Example 1 of Patent Document 4 discloses that propylene is polymerized by adding a prepolymerization catalyst, tert-butylmethyldimethoxysilane, and triethylaluminum to an autoclave.
- Patent Document 4 does not disclose any preliminary contact between the prepolymerization catalyst, the organoaluminum compound and the electron donor before polymerization, and the contact order of these compounds.
- propylene is prepolymerized in advance when using a solid titanium catalyst component containing magnesium, titanium, halogen and an electron donor compound.
- a prepolymerized catalyst for the main polymerization the prior art in which the prepolymerized catalyst, the organoaluminum compound and the electron donor compound are preliminarily brought into contact with each other, or the order of contact between them is examined. There is no literature.
- the present invention polymerizes ⁇ -olefin in the presence of a catalyst formed from a solid catalyst component containing magnesium, titanium, halogen and an electron donor compound as essential components, an organoaluminum compound, and an electron donor compound.
- a prepolymerized catalyst is obtained by prepolymerizing an ⁇ -olefin using the solid catalyst component in the presence of a small amount of an organoaluminum compound, and then the obtained prepolymerized catalyst, organoaluminum compound and electron donation
- the contact product obtained by previously contacting the body compound is previously contacted before polymerizing the ⁇ -olefin, and then the ⁇ -olefin is polymerized.
- aspects of the present invention are as follows: 1. (A) a solid catalyst component containing magnesium, titanium, halogen and an electron donor compound as essential components; A process for producing an ⁇ -olefin polymer using a catalyst comprising (B) an organoaluminum compound; and (C) an electron donor compound, (1) pre-polymerizing ⁇ -olefin in the presence of component (A) and component (B); (2) bringing the obtained prepolymerized catalyst into contact with a contact product obtained by bringing the components (B) and (C) into contact in advance; (3) The method as described above, wherein the prepolymerization catalyst brought into contact with the contact product is added to a polymerization reaction vessel charged with ⁇ -olefin to polymerize the ⁇ -olefin.
- the present invention provides (A) a solid catalyst component containing magnesium, titanium, halogen and an electron donor compound as essential components; (B) an organoaluminum compound; and (C) a catalyst containing an electron donor compound.
- a method for producing an olefin polymer wherein (1) ⁇ -olefin is prepolymerized in the presence of component (A) and component (B); (2) the prepolymerized catalyst obtained, and (B And (3) the prepolymerized catalyst brought into contact with the contact product is added to the polymerization reaction vessel charged with ⁇ -olefin. The ⁇ -olefin is polymerized.
- the ⁇ -olefin to be polymerized in the present invention is ethylene and an ⁇ -olefin having 3 to 20 carbon atoms, such as propylene, 1-butene, 4-methyl-1-pentene, 1-hexene, 1-octene, 1-octene, A wide variety of common ⁇ -olefins such as decene, 1-dodecene, 1-tetradecene, and the like can be selected. In particular, the present invention exhibits a significant effect in the polymerization of propylene.
- the solid catalyst component which is the component (A) used in the present invention contains magnesium, titanium, halogen and an electron donor compound as essential components.
- this solid catalyst component many prior art documents have proposed the manufacturing method. Specifically, this solid catalyst component can be obtained by bringing a magnesium compound, a titanium compound and an electron donor compound into contact with each other.
- a method of depositing a solid titanium complex by reacting a liquid magnesium compound with a liquid titanium compound in the presence or absence of an electron donor compound; (3) a solid magnesium compound and liquid titanium; A method of reacting a compound and an electron donor compound; (4) a titanium compound is further reacted with the product obtained in (2) or (3) above; (5) A method of further reacting an electron donor compound and a titanium compound with those obtained in (1), (2) and (3) above; (6)
- Magnesium compound or magnesium compound and electron donor compound The complex compound is pulverized in the presence or absence of an electron donor
- the solid catalyst component that is the component (A) used in the present invention can be obtained by various methods such as a method of treating with a halogen compound or an aromatic hydrocarbon. Kill.
- a tetravalent titanium compound represented by (R is a hydrocarbon group, X is a halogen, and 0 ⁇ g ⁇ 4) is preferable. More specifically, titanium tetrahalides such as TiCl 4 , TiBr 4 , TiI 4 ; Ti (OCH 3 ) Cl 3 , Ti (OC 2 H 5 ) Cl 3 , Ti (O n —C 4 H 9 ) Cl 3 , Ti (OC 2 H 5 ) Br 3 , Ti (OisoC 4 H 9 ) Br 3 and other trihalogenated alkoxytitanium; Ti (OCH 3 ) 2 Cl 2 , Ti (OC 2 H 5 ) 2 Cl 2 , Ti Dihalogenated alkoxytitanium such as (O n —C 4 H 9 ) 2 Cl 2 , Ti (OC 2 H 5 ) 2 Br 2 ; Ti (OCH 3 ) 3 Cl, Ti (OC 2 H 5 ) 3 Cl, Ti ( O n -C 4 H 9) 3
- the magnesium compound used for the preparation of the solid catalyst component (A) used in the present invention is a magnesium compound having a magnesium-carbon bond or a magnesium-hydrogen bond, such as dimethylmagnesium, diethylmagnesium, dipropylmagnesium, dibutylmagnesium, diamylmagnesium. , Dihexyl magnesium, didecyl magnesium, ethyl magnesium chloride, propyl magnesium chloride, butyl magnesium chloride, hexyl magnesium chloride, amyl magnesium chloride, butyl ethoxy magnesium, ethyl butyl magnesium, butyl magnesium hydride and the like.
- magnesium compounds can be used, for example, in the form of a complex compound with organic aluminum or the like, and may be in a liquid state or a solid state.
- Further suitable magnesium compounds include magnesium halides such as magnesium chloride, magnesium bromide, magnesium iodide, magnesium fluoride; methoxy magnesium chloride, ethoxy magnesium chloride, isopropoxy magnesium chloride, butoxy magnesium chloride, octoxy magnesium chloride, and the like.
- Alkoxymagnesium halides Alkoxymagnesium halides; aryloxymagnesium halides such as phenoxymagnesium chloride and methylphenoxymagnesium chloride; alkoxymagnesiums such as ethoxymagnesium, isopropoxymagnesium, butoxymagnesium, n-octoxymagnesium and 2-ethylhexoxymagnesium; Allyloxymagnesium such as sigmamagnesium and dimethylphenoxymagnesium; Lau Magnesium phosphate, such as carboxylic acid salts of magnesium such as magnesium stearate and the like.
- the electron donor compound used for the preparation of the solid catalyst component (A) of the present invention is generally referred to as “internal electron donor”.
- electron donor compounds include alcohols, phenols, ketones, aldehydes, carboxylic acids, esters of organic or inorganic acids, ethers, acid amides, oxygenated electron donors such as acid anhydrides, ammonia, amines, Nitrogen-containing electron donors such as nitrile and isocyanate can be mentioned.
- it has 1 to 18 carbon atoms such as methanol, ethanol, propanol, pentanol, hexanol, octanol, 2-ethylhexanol, dodecanol, octadecyl alcohol, benzyl alcohol, phenylethyl alcohol, cumyl alcohol, isopropylbenzyl alcohol, and the like.
- Alcohols having 6 to 25 carbon atoms which may have an alkyl group such as phenol, cresol, xylenol, ethylphenol, propylphenol, cumylphenol, nonylphenol, naphthol; acetone, methyl ethyl ketone, methyl isobutyl ketone, C3-C15 ketones such as acetophenone and benzophenone; acetaldehyde, propionaldehyde, octylaldehyde, benzaldehyde Aldehydes having 2 to 15 carbon atoms, such as tolualdehyde, naphthaldehyde; methyl formate, ethyl acetate, vinyl acetate, propyl acetate, octyl acetate, cyclohexyl acetate, ethyl propionate, methyl butyrate, ethyl valerate, ethyl stearate
- Acid amides acid anhydrides such as benzoic anhydride and phthalic anhydride; amines such as methylamine, ethylamine, diethylamine, tributylamine, piperidine, tribenzylamine, aniline, pyridine, picoline, tetramethylethylenediamine; acetonitrile, And nitriles such as benzonitrile and tolunitrile; These electron donor compounds can be used in combination of two or more.
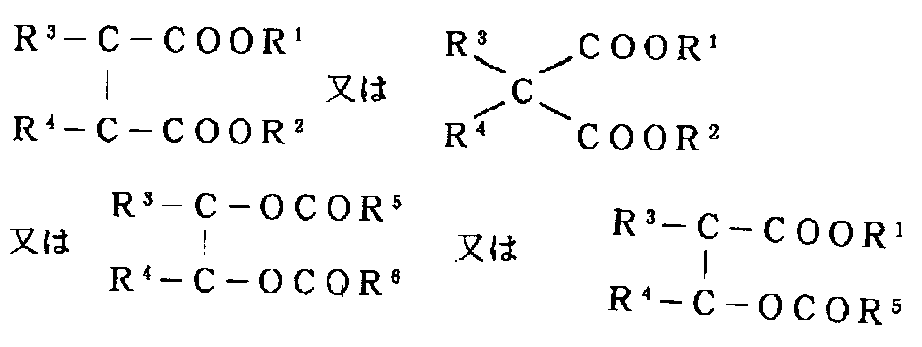
- the electron donor compound that is desirably contained in the solid catalyst component is an ester, and more preferred is a general formula:
- R 1 is a substituted or unsubstituted hydrocarbon group
- R 2 , R 5 and R 6 are hydrogen or a substituted or unsubstituted hydrocarbon group
- R 3 and R 4 are hydrogen or a substituted or unsubstituted carbon group.
- R 3 and R 4 may be linked to each other.
- substituted hydrocarbon groups of R 1 ⁇ R 6 Including a heteroatom such as N, O, S, etc., and having a group such as C—O—C, COOR, COOH, OH, SO 3 H, —C—N—C—, NH 2 And those having a skeleton represented by:
- dicarboxylic acid diesters (polycarboxylic acid diesters) in which at least one of R 1 and R 2 is an alkyl group having 2 or more carbon atoms are particularly preferable.
- preferable polyvalent carboxylic acid diesters include diethyl succinate, dibutyl succinate, diethyl methyl succinate, diethyl diisopropyl succinate, diisobutyl ⁇ -methyl glutarate, dibutyl methyl malonate, diethyl malonate, diethyl ethyl malonate.
- polyvalent hydroxy compound esters can be used as the electron donor compound.
- preferable ones include 1,2-diacetoxybenzene, 1-methyl-2,3-diacetoxybenzene, and 2,3-diacetoxy.
- Naphthalene, ethylene glycol dipivalate, butanediol pivalate, etc. can be mentioned.
- hydroxy-substituted carboxylic acid esters that can be preferably used as an electron donor compound include benzoylethyl salicylate, acetylisobutyl salicylate, and acetylmethyl salicylate.
- esters preferred are those having the skeleton of the general formula described above, and more preferred are esters of phthalic acid, maleic acid, substituted malonic acid and the like with alcohols having 2 or more carbon atoms. Particularly preferred is a diester of phthalic acid and an alcohol having 2 or more carbon atoms.
- RCOOR ′ Another electron donor compound that can be supported on the solid catalyst component is RCOOR ′ (R and R ′ are hydrocarbyl groups which may have a substituent, and at least one of them is branched (alicyclic). And a ring-containing chain group).
- R and R ′ are hydrocarbyl groups which may have a substituent, and at least one of them is branched (alicyclic). And a ring-containing chain group).
- R and R ′ are a group as described above, the other may be the above group, or another group such as a linear or cyclic group.
- esters such as dimethylacetic acid, trimethylacetic acid, ⁇ -methylbutyric acid, ⁇ -methylbutyric acid, methacrylic acid, and benzoylacetic acid, and various monocarboxylic acids such as isopropanol, isobutyl alcohol, and tert-butyl alcohol. Esters can be exemplified.
- a carbonate ester can be selected as the electron donor compound.
- Specific examples include diethyl carbonate, ethylene carbonate, diisopropyl carbonate, phenylethyl carbonate, diphenyl carbonate, and the like.
- halogen atom constituting the solid catalyst component examples include fluorine, chlorine, bromine, iodine or a mixture thereof, and chlorine is particularly preferable.
- organoaluminum compound (B) used in the method of the present invention examples include trialkylaluminum such as triethylaluminum and tributylaluminum, trialkenylaluminum such as triisoprenylaluminum, diethylaluminum ethoxide, and dibutylaluminum butoxide. dialkylaluminum alkoxides, ethylaluminum sesquichloride ethoxide such, in addition to alkylaluminum sesqui alkoxides such as butyl sesquichloride butoxide, R 1 2.5 Al (OR 2 ) 0.
- Dialkylaluminum halides such as modified alkylaluminum, diethylaluminum chloride, dibutylaluminum chloride, diethylaluminum bromide
- Partially halogens such as alkylaluminum sesquichloride, ethylaluminum sesquichloride, alkylaluminum sesquihalogenides such as ethylaluminum sesquibromide, alkylaluminum dichloride, propylaluminum dichloride, butylaluminum dibromide, etc.
- Partially hydrogenated alkylaluminum such as alkylaluminum alkylated, diethylaluminum hydride, dialkylaluminum hydride such as dibutylaluminum hydride, alkylaluminum dihydride such as ethylaluminum dihydride, propylaluminum dihydride, ethylaluminum ethoxychloride , Butylaluminum butoxycyclyl, Ethylaluminum et It can be selected from partially alkoxylated and halogenated alkylaluminums such as xybromide.
- the electron donor compound which is the component (C) used in the method of the present invention is generally referred to as “external electron donor”.
- an organosilicon compound is preferably used as such an electron donor compound.
- Preferred organosilicon compounds include, for example, trimethylmethoxysilane, trimethylethoxysilane, dimethyldimethoxysilane, dimethyldiethoxysilane, diisopropyldimethoxysilane, t-butylmethyldimethoxysilane, t-butylmethyldiethoxysilane, and t-amylmethyldiethoxy.
- Silane diphenyldimethoxysilane, phenylmethyldimethoxysilane, diphenyldiethoxysilane, bis o-tolyldimethoxysilane, bism-tolyldimethoxysilane, bisp-tolyldimethoxysilane, bisp-tolyldiethoxysilane, bisethylphenyldimethoxysilane , Dicyclopentyldimethoxysilane, dicyclohexyldimethoxysilane, cyclohexylmethyldimethoxysilane, cyclohexylmethyldiethoxysilane, Tiltrimethoxysilane, ethyltriethoxysilane, vinyltrimethoxysilane, methyltrimethoxysilane, n-propyltriethoxysilane, decyltrimethoxysilane, decyltriethoxysilane, pheny
- the method of the present invention comprises the following sequence: (1) prepolymerizing an ⁇ -olefin in the presence of component (A) and component (B); (2) bringing the obtained prepolymerized catalyst into contact with a contact product obtained by bringing the components (B) and (C) into contact in advance; (3) It is characterized in that it is carried out by polymerizing ⁇ -olefin using a prepolymerization catalyst brought into contact with the contact product.
- ⁇ -olefin is prepolymerized in the presence of the solid catalyst component (A) and the organoaluminum compound (B).
- the amount of the component (B) used is small compared with the amount of the component (B) used in the main polymerization, and is usually 0.00 by the component (B) with respect to 1 mol of Ti atom in the component (A). 5 to 50 mol, preferably 1.0 to 20 mol. If the amount of the component (B) is excessively increased in the prepolymerization stage, the storage stability of the resulting prepolymerization catalyst may be lowered.
- the ⁇ -olefin is prepolymerized using the component (A).
- the ⁇ -olefin prepolymerization is a step of forming a chain of ⁇ -olefin, which becomes a foothold for the subsequent main polymerization of ⁇ -olefin, in the solid catalyst component.
- the prepolymerization can be carried out by a known method such as a batch method or a continuous method, but a batch method is preferred in that a high-concentration solid catalyst can be used and the amount of ⁇ -olefin used for the prepolymerization is controlled.
- the prepolymerization can be carried out in the absence of a solvent or in an inert medium, but prepolymerization in an inert hydrocarbon medium is preferred.
- examples of such an inert hydrocarbon solvent include hydrocarbons such as butane, pentane, hexane, heptane, octane, benzene, toluene, xylene, and ethylbenzene.
- the prepolymerization is usually performed at 40 ° C or lower, preferably 30 ° C or lower, more preferably 20 ° C or lower.
- the amount of ⁇ -olefin to be prepolymerized is 0.5 to 300 g, preferably 1 to 100 g, more preferably 2 to 30 g, per 1 g of the solid catalyst.
- Examples of the ⁇ -olefin used in the prepolymerization include ethylene and ⁇ -olefins having 3 to 20 carbon atoms such as propylene, 1-butene, 4-methyl-1-pentene, 1-hexene, 1-octene and 1-decene. , 1-dodecene, 1-tetradecene and the like, but it is preferable to use propylene alone or propylene and ethylene and / or a small amount of ⁇ -olefin.
- hydrogen can be used as a molecular weight regulator as needed at the time of prepolymerization.
- the prepolymerized catalyst obtained in the above step (1) is brought into contact with a contact product obtained by bringing the component (B) and the electron donor compound (C) into contact in advance.
- the component ratio is 0.005 to 2.0 mol, preferably 0.01 mol to 1.0 mol, relative to 1 mol of the component (B).
- it is performed at 50 ° C. or lower, preferably 40 ° C. or lower, more preferably 30 ° C. or lower.
- Contacting can be carried out in an inert solvent.
- this contact method is not specifically limited, For example, each component can be contacted using piping, a metal container, a metal container with a stirrer, etc.
- preliminary contact time Time for preliminarily contacting the prepolymerized catalyst with the contact product obtained by previously contacting the component (B) and the component (C) outside the polymerization system (referred to herein as “preliminary contact time”).
- the preliminary contact time is preferably 10 minutes or less, and may be an extremely short time such as 1 minute or less, 0.5 minutes or less, or 0.25 minutes or less.
- the contact product is continuously supplied to the polymerization reactor. When it does, it can carry out in the pipe
- the amount of the component (B) and the component (C) to be brought into contact with the prepolymerization catalyst is 30 to 3000 mol as the component (B) and 0.03 to 0.03 as the component (C) with respect to 1 mol of Ti atoms contained in the prepolymerization catalyst.
- the amount is preferably 3000 mol, but may vary depending on the types of the component (B) and the component (C) used.
- the inert solvent that may be used when the component (B) is contacted with the component (C) and the prepolymerized catalyst is preliminarily contacted with the contact product is, for example, butane, pentane, hexane, heptane, octane. , Hydrocarbon compounds such as benzene, toluene and xylene ethylbenzene.
- the contact of each component is usually performed at 50 ° C. or lower, preferably 40 ° C. or lower, more preferably 30 ° C. or lower.
- the pre-catalyzed catalyst thus obtained is added to the polymerization reaction system charged with ⁇ -olefin, and the main polymerization of ⁇ -olefin is carried out.
- the polymerization may be batch-wise, semi-continuous, or continuous, but is preferably continuous for industrial use.
- the polymerization temperature is from room temperature to 150 ° C, preferably from 40 ° C to 100 ° C.
- the pressure is generally from normal pressure to 10 MPa, preferably from 0.5 to 60 MPa.
- the polymerization time is usually 10 hours or less, preferably 10 minutes to 5 hours.
- ⁇ -olefin used in the polymerization examples include ethylene and ⁇ -olefins having 3 to 20 carbon atoms such as propylene, 1-butene, 4-methyl-1-pentene, 1-hexene, 1-octene, 1-octene, Decene, 1-dodecene, 1-tetradecene and the like can be mentioned.
- the present invention includes a propylene / ⁇ -olefin random copolymer or a propylene / ⁇ -olefin block copolymer. It is particularly suitable for production.
- ⁇ -olefins other than propylene used for copolymerization with propylene examples include ethylene and butene-1.
- hydrogen can be used as a molecular weight regulator.
- the yield of the polymer can be improved (that is, the catalytic activity can be improved) by the method of the present invention.
- the second aspect of the present invention is (A) a solid catalyst component containing magnesium, titanium, halogen and an electron donor compound as essential components; (B) an organoaluminum compound; and (C) a catalyst comprising an electron donor compound.
- A Preliminarily polymerizing ⁇ -olefin in the presence of component (A) and component (B); (2) resulting prepolymerization The catalyst is brought into contact with the component (B); (3) The prepolymerized catalyst brought into contact with the component (B) is added to the polymerization reaction vessel charged with the component (C) and the ⁇ -olefin, and the ⁇ -olefin is added.
- the method is characterized by polymerizing.
- the second aspect of the present invention is different from the first aspect in that the preliminary polymerization catalyst and only the component (B) are first pre-contacted in the step (2), and further obtained in the step (2).
- the prepolymerization catalyst preliminarily contacted only with the component (B) is added to a polymerization reaction vessel charged with the component (C) and the ⁇ -olefin to polymerize the ⁇ -olefin. That is, the prepolymerized catalyst obtained in step (1) is contacted with only component (B) in step (2).
- the prepolymerization catalyst and the component (B) are outside the polymerization system (for example, in a pipe connected to a reactor, or in a metal container, Preliminary contact is made (in a metal vessel with agitation or an adder provided in the polymerization reaction vessel).
- the time for preliminarily contacting the prepolymerization catalyst and the component (B) outside the polymerization system preferably does not exceed 20 minutes.
- the preliminary contact time is preferably 10 minutes or less, and may be an extremely short time such as 1 minute or less, 0.5 minutes or less, or 0.25 minutes or less.
- a small amount of the component (B) (preferably 0.5% of the component (B) with respect to 1 mol of Ti atom in the component (A).
- the amount of the component (B) to be brought into contact with the prepolymerization catalyst is preferably 30 to 3000 mol with respect to 1 mol of Ti atom in the prepolymerization catalyst, but the component (B) used and the component (C) used in the next step are preferably used.
- the preliminary polymerization catalyst and the component (B) are preliminarily contacted in advance outside the polymerization system for a specific preliminary contact time. It can be changed as appropriate.
- the inert solvent that may be used when the prepolymerization catalyst and the component (B) are preliminarily contacted with each other include hydrocarbon compounds such as butane, pentane, hexane, heptane, octane, benzene, toluene, xylene, and ethylbenzene. Can be mentioned.
- the amount of ⁇ -olefin to be prepolymerized is 0.5 to 300 g, preferably 1 to 100 g, more preferably 2 to 30 g, per 1 g of the solid catalyst.
- component (C) 0.005 to 2.0 mol relative to 1 mol of component (B) contacted in step (2)
- ⁇ -olefin a polymerization reaction system in which the pre-contact catalyst thus obtained was charged with component (C) (0.005 to 2.0 mol relative to 1 mol of component (B) contacted in step (2)) and ⁇ -olefin.
- the main polymerization of ⁇ -olefin is carried out.
- the yield of the polymer can be improved (that is, the catalytic activity can be improved) by the method of the present invention.
- the yield of ⁇ -olefin polymer can be improved and the catalytic activity can be maintained for a long time.
- the ⁇ -olefin polymer obtained by the method of the present invention has a small proportion of fine powder, and thus can reduce troubles such as blockage of the production line due to fine powder agglomeration, so that it can be stably produced and has improved processing characteristics. Are better.
- FIG. 1 is a diagram showing the steps of the polymerization method according to the first embodiment of the present invention.
- FIG. 2 is a diagram showing the steps of the polymerization method according to the second aspect of the present invention.
- FIG. 3 is a diagram illustrating steps of a polymerization method of a comparative example.
- the solid catalyst component containing magnesium, titanium, halogen, and an electron donor compound as essential components which is the component (A) used in the present invention, can be appropriately prepared following conventional techniques known to those skilled in the art. it can.
- ⁇ -olefin is prepolymerized on the prepared solid catalyst component.
- An inert solvent eg, pentane, hexane, heptane, octane, etc.
- an inert gas eg, nitrogen, argon, etc.
- an inert solvent solution of an organoaluminum compound for example, triethylaluminum, triisobutylaluminum, etc.
- an organoaluminum compound for example, triethylaluminum, triisobutylaluminum, etc.
- ⁇ -olefin ethylene, propylene, 1-butene, etc.
- ⁇ -olefin ethylene, propylene, 1-butene, etc.
- the obtained prepolymerized catalyst solid phase
- the washed prepolymerized catalyst can be dissolved in an inert solvent to obtain a slurry-like prepolymerized catalyst.
- the organoaluminum compound as the component (B) and the electron donor compound as the component (C) are brought into contact with each other, and then the prepolymerized catalyst is brought into contact with the contact product obtained by the contact.
- ⁇ -olefin for example, a metal adder
- any container may be used as long as it has a function capable of pre-contacting these substances without difficulty and supplying the pre-contact catalyst to the polymerization reaction container.
- preliminary contact container any container may be used as long as it has a function capable of pre-contacting these substances without difficulty and supplying the pre-contact catalyst to the polymerization reaction container.
- An organoaluminum compound for example, triethylaluminum, triisobutylaluminum, etc.
- an electron donor compound for example, diisopropyldimethoxysilane, dicyclopentyldimethoxysilane, cyclohexylmethyldimethoxysilane, etc.
- the prepolymerized catalyst slurry obtained above is added here, and these materials are pre-contacted for 20 minutes or less, preferably 10 minutes or less, more preferably 1 minute or less, and most preferably 0.5 minutes or less. .
- the contacting temperature is usually 50 ° C. or lower, preferably 40 ° C., more preferably 30 ° C. or lower.
- ⁇ -olefin ethylene, propylene, butene, etc.
- An ⁇ -olefin (ethylene, propylene, butene, etc.) to be polymerized is added to a polymerization reaction vessel substituted with an inert gas (preferably nitrogen, argon, etc.), and heated to room temperature or higher, preferably 30 ° C. or higher.
- the pre-contact container containing the pre-polymerization catalyst pre-contacted above is pressurized, and the pre-contact catalyst is press-fitted into the polymerization reaction container.
- the temperature of the polymerization reaction vessel is raised (it varies depending on the type of ⁇ -olefin, but is generally from room temperature to 150 ° C., preferably from 40 ° C.
- the polymerization method of the second aspect of the present invention is essentially the same as the polymerization method of the first aspect.
- the solid catalyst component containing magnesium, titanium, halogen, and an electron donor compound as essential components which is the component (A) used in the present invention, can be appropriately prepared following conventional techniques known to those skilled in the art. it can.
- ⁇ -olefin is prepolymerized on the prepared solid catalyst component.
- An inert solvent eg, pentane, hexane, heptane, octane, etc.
- an inert gas eg, nitrogen, argon, etc.
- an inert solvent solution of an organoaluminum compound for example, triethylaluminum, triisobutylaluminum, etc.
- an organoaluminum compound for example, triethylaluminum, triisobutylaluminum, etc.
- ⁇ -olefin ethylene, propylene, 1-butene, etc.
- ⁇ -olefin ethylene, propylene, 1-butene, etc.
- the obtained prepolymerized catalyst solid phase
- the washed prepolymerized catalyst can be dissolved in an inert solvent to obtain a slurry-like prepolymerized catalyst.
- the organoaluminum compound as component (B) is brought into contact with the prepolymerization catalyst.
- a container preliminary contact container
- a polymerization reaction container autoclave or the like
- An organoaluminum compound eg, triethylaluminum, triisobutylaluminum, etc.
- the prepolymerized catalyst slurry obtained above is added here, and these materials are pre-contacted for 20 minutes or less, preferably 10 minutes or less, more preferably 1 minute or less, and most preferably 0.5 minutes or less.
- ⁇ -olefin ethylene, propylene, butene, etc.
- an inert gas preferably nitrogen, argon, etc.
- an electron donor compound for example, diisopropyldimethoxysilane, Cyclopentyldimethoxysilane, cyclohexylmethyldimethoxysilane, etc.
- the pre-contact container containing the pre-polymerization catalyst pre-contacted above is pressurized, and the pre-contact catalyst is press-fitted into the polymerization reaction container.
- the ⁇ -olefin is polymerized by raising the temperature of the polymerization reaction vessel (which varies depending on the type of ⁇ -olefin, but is generally from room temperature to 150 ° C., preferably from 40 ° C. to 100 ° C.). Unreacted ⁇ -olefin is purged from the polymerization reaction vessel to obtain the target ⁇ -olefin polymer.
- hydrogen or the like can be appropriately added to the polymerization reaction vessel.
- an ⁇ -olefin polymer having high stereoregularity and high polymerization yield can be obtained.
- the obtained ⁇ -olefin polymer has a small proportion of fine powder, and is therefore particularly excellent in processing characteristics.
- Example 1 Preparation of solid catalyst component A solid catalyst component was prepared according to the preparation method described in Examples of JP-A-9-25316. Specifically: 56.8 g of anhydrous magnesium chloride was completely dissolved at 120 ° C. in 100 g of absolute ethanol, 500 ml of petroleum oil CP15N manufactured by Idemitsu Kosan Co., Ltd. and 500 ml of silicone oil KF96 manufactured by Shin-Etsu Silicone Co., Ltd. under a nitrogen atmosphere. This mixture was stirred for 3 minutes at 120 ° C. and 3000 rpm using a TK homomixer manufactured by Tokushu Kika Kogyo Co., Ltd.
- the solid portion was collected by hot filtration. Thereafter, 500 ml of titanium tetrachloride was added to the reaction product and stirred, followed by reaction at 120 ° C. for 1 hour. After completion of the reaction, the solid part was again collected by hot filtration and washed 7 times with 1 liter of hexane at 60 ° C. and 3 times with 1 liter of hexane at room temperature.
- the inside of the adder was pressurized to 4 MPa with nitrogen, and the pre-contact catalyst was pressed into the autoclave.
- the autoclave was heated to 70 ° C., and propylene was polymerized for 60 minutes. After the polymerization, unreacted propylene was purged to obtain polypropylene.
- the obtained polypropylene was vacuum-dried at 60 ° C. for 16 hours, and MFR, XI, and polymerization activity were analyzed according to the above method.
- Examples 2 to 5 The same procedure as in Example 1 was conducted except that the preliminary contact time was changed to 0.5, 3, 10, and 20 minutes, respectively.
- Example 6 The prepolymerization catalyst was prepared in the same manner as in Examples 1 (1) and (2). Next, 1.0 mmol of diisopropyldimethoxysilane was added to the autoclave while feeding a small amount of nitrogen into the autoclave with an internal volume of 3 L, which was purged with nitrogen, and an adder was attached. After replacing the interior of the autoclave with propylene gas, 2400 mL of hydrogen and 18.5 mol of propylene were added at 25 ° C., and the mixture was stirred and heated to 30 ° C. 4.9 mmol of triethylaluminum was placed in a supplementer in a state where a small amount of nitrogen was fed.
- Example 7 to 9 The same procedure as in Example 1 was conducted except that the preliminary contact time was 0.25, 0.5 minutes, 3 minutes, and the polymerization time was 180 minutes, respectively.
- Comparative Example 2 The same procedure as in Comparative Example 1 was performed except that the polymerization time was 180 minutes.
- the catalytic activity is remarkably improved.
- the results of Example 2 (first aspect) and Example 6 (second aspect) having the same preliminary contact time (0.5 minutes) are compared with reference to Comparative Example 1, the activity increase rate Were 35.5% and 33.8%, respectively.
- Example 8 is a method according to the first aspect in which the preliminary contact time is 0.5 minutes and the polymerization time is 180 minutes, but the activity increase rate based on Comparative Example 2 is 39.3%. there were.
- the method of the present invention high catalytic activity can be maintained even if the polymerization time is extended.
- the ⁇ -olefin polymer obtained by the method of the present invention has a small proportion of fine powder.
- Example 1 and Comparative Example 1 cannot be directly compared because of different pre-contact times, but the ⁇ -olefin polymer obtained in Example 1 has a small proportion of fine powder present. I understand.
- Example 7 and Comparative Example 2 are compared, it can be seen that the ⁇ -olefin polymer obtained in Example 7 has a small proportion of fine powder. That is, the ⁇ -olefin polymer obtained by the method of the present invention can be stably produced because troubles such as blockage of the production line due to fine powder aggregation can be reduced, and it is particularly excellent in processing characteristics.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
Abstract
Description
1.(A)マグネシウム、チタン、ハロゲンおよび電子供与体化合物を必須成分として含有する固体触媒成分;
(B)有機アルミニウム化合物;および
(C)電子供与体化合物
を含む触媒を用いてα-オレフィン重合体を製造する方法であって、
(1)(A)成分と(B)成分との存在下に、α-オレフィンを予備重合させ;
(2)得られた予備重合触媒と、(B)成分と(C)成分とを予め接触させて得た接触物とを接触させ;
(3)該接触物と接触させた予備重合触媒を、α-オレフィンを装填した重合反応容器中に添加して、α-オレフィンを重合する
ことを特徴とする、前記方法。
2.工程(2)の接触時間が、20分間を超えない、上記1に記載の方法。
3.(C)成分が有機ケイ素化合物である、上記1または2に記載の方法。
4.(A)マグネシウム、チタン、ハロゲンおよび電子供与体化合物を必須成分として含有する固体触媒成分;
(B)有機アルミニウム化合物;および
(C)電子供与体化合物
を含む触媒を用いてα-オレフィン重合体を製造する方法であって、
(1)(A)成分と(B)成分との存在下に、α-オレフィンを予備重合させ;
(2)得られた予備重合触媒を(B)成分に接触させ;
(3)(B)成分に接触させた予備重合触媒を、(C)成分とα-オレフィンとを装填した重合反応容器中に添加して、α-オレフィンを重合する
ことを特徴とする、前記方法。
5.工程(2)の接触時間が、20分間を超えない、上記4に記載の方法。
6.(C)成分が有機ケイ素化合物である、上記4または5に記載の方法。

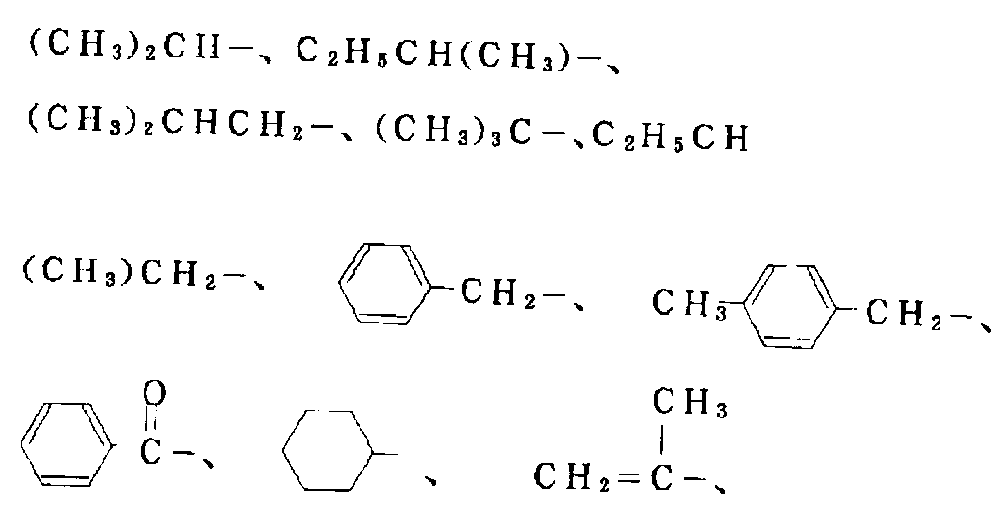
(2)得られた予備重合触媒と、(B)成分と(C)成分とを予め接触させて得た接触物とを接触させ;
(3)該接触物と接触させた予備重合触媒を用いて、α-オレフィンを重合する
で行われることに特徴がある。
[MFR]
JIS K 7210に準じ、温度230℃、荷重21.18Nの条件下で測定した。
[XI(キシレン不溶成分量)]
300mLフラスコに重合体試料2.5gおよび250mLのオルトキシレンを入れ、攪拌しながら沸騰温度で30分間溶解した。続いて、溶液を100℃に放冷した後、フラスコを25℃の恒温水槽に入れ、25℃になってから1時間経過後、ろ過を行い、回収したろ液のオルトキシレンを蒸発させ、残った残渣の重量を仕込みの重合体試料の重量で除した値を100倍し、XIを求めた。
[重合活性]
島津製作所株式会社製AA660を用い、原子吸光法により、生成したポリマーサンプル中のマグネシウム含有量を測定し、元の触媒に含まれるマグネシウム含有量から触媒1gあたりのポリマー重合量として求めた。
[微粉量測定]
重合ポリマーについて、Ro-Tap篩振とう機を使用して篩分けを行い、目開き125μの金属製メッシュを通過した微粉量を重量により求めた。
(1)固体触媒成分の調製
特開平9-25316の実施例に記載の調製法に従い、固体触媒成分を調製した。具体的には以下の通りである:
無水塩化マグネシウム56.8gを、無水エタノール100g、出光興産(株)製のワセリンオイルCP15N500mlおよび信越シリコーン(株)製のシリコーン油KF96 500ml中、窒素雰囲気下、120℃で完全に溶解させた。この混合物を、特殊機化工業(株)製のTKホモミキサーを用いて120℃、3000回転/分で3分間撹拌した。撹拌を保持しながら、2リットルの無水ヘプタン中に0℃を越えないような移送した。得られた白色固体は無水ヘプタンで十分に洗浄し室温下で真空乾燥した。得られたMgCl2 ・2.5C2 H5 OHの球状固体30gを無水ヘプタン200ml中に懸濁させた。0℃で撹拌しながら、四塩化チタン500mlを1時間かけて滴下した。次に、加熱を始めて40℃になったところで、フタル酸ジイソブチル4.96gを加えて、100℃まで約1時間で昇温させた。100℃で2時間反応させた後、熱時ろ過にて固体部分を採取した。その後、この反応物に四塩化チタン500mlを加え撹拌させた後、120℃で1時間反応させた。反応終了後、再度、熱時ろ過にて固体部分を採取し、60℃のヘキサン1リットルで7回、室温のヘキサン1リットルで3回洗浄した。TiCl4[C6H4(COOiC4H9)2]の調製四塩化チタン19gを含むヘキサン1リットルの溶液に、フタル酸ジイソブチル:C6H4(COOiC4H9)227.8gを、温度0℃を維持しながら約30分で滴下した。滴下終了後、40℃に昇温し30分間反応させた。反応終了後、固体部分を採取しヘキサン500mlで5回洗浄し目的物である固体触媒成分を得た。該固体触媒成分を分析したところ、チタン含有量およびマグネシウム含有量は、それぞれ2.3wt%および17.7wt%であった。
(2)予備重合触媒の調製
窒素置換した300mLの三口フラスコに少量の窒素をフィードしながら、精製ヘキサン100mLを加え冷却し、三口フラスコ内の温度を10℃以下にした。続いて、有機アルミニウム化合物としてトリエチルアルミニウムのヘキサン溶液をトリエチルアルミニウムが2.3mmol加わるように三口フラスコに添加し、さらに上記方法で調製した固体触媒成分500mgを三口フラスコ内へ加えた。その後、10℃で撹拌しながらプロピレンガス5gを三口フラスコ内に供給し、さらに90分間静置してプロピレンを完全に反応させた。続いて、少量の窒素をフィードしながら、三口フラスコ内の液相部分のみをシリンジで除去し、残った固相である予備重合触媒にさらに精製ヘキサン50mLを加え、液相のみを取り除くことで洗浄し、この洗浄操作を2回行った。洗浄後の予備重合触媒に再度少量の精製ヘキサンを加え、スラリー状態(3g/L)にして20℃で保存した。
(3)プロピレンの重合
窒素置換した内容積3Lのオートクレーブに少量の窒素をフィードしながら、追添器を取り付けた。オートクレーブ内をプロピレンガスで置換した後、25℃で水素2400mLとプロピレン18.5molとを加え撹拌し、30℃に昇温した。少量の窒素をフィードした状態の追添器に、トリエチルアルミニウム4.9mmolと、電子供与体化合物としてジイソプロピルジメトキシシラン1.0mmolとを入れた。上記で調製した予備重合触媒スラリー2.5ml(予備重合触媒として7.5mg)を追添器に入れ、0.25分間予備接触させた。続いて、追添器内を窒素で4MPaに加圧して、予備接触触媒をオートクレーブ内に圧入した。オートクレーブを70℃に昇温し、60分間プロピレンを重合した。重合終了後、未反応プロピレンをパージし、ポリプロピレンを得た。得られたポリプロピレンを60℃で16時間真空乾燥し、上記の方法に従い、MFR、XI、重合活性の分析を行った。
予備接触時間をそれぞれ0.5、3、10、20分と変えた以外は実施例1と同様に行った。
予備重合触媒の調製は、実施例1(1)~(2)と同様に行った。次いで、窒素置換した内容積3Lのオートクレーブに少量の窒素フィードしながら、オートクレーブ内にジイソプロピルジメトキシシラン1.0mmolを入れ、追添器を取り付けた。オートクレーブ内をプロピレンガスで置換した後、25℃で水素2400mLとプロピレン18.5molとを加え、撹拌し30℃に昇温した。少量の窒素をフィードした状態の追添器に、トリエチルアルミニウム4.9mmolを入れた。上記で調製した予備重合触媒スラリー2.5ml(予備重合触媒として7.5mg)を追添器に入れ、0.5分間予備接触させた。続いて、追添器内を窒素で4MPaに加圧し、予備接触触媒をオートクレーブ内に圧入した。オートクレーブを70℃に昇温し、60分間プロピレンを重合した。重合終了後、未反応プロピレンをパージし、ポリプロピレンを得た。得られたポリプロピレンを60℃で16時間真空乾燥し、上記の方法に従って、MFR、XI、重合活性の分析を行った。
予備重合触媒の調製は実施例1(1)~(2)と同様に行った。窒素置換した内容積3Lのオートクレーブに少量の窒素をフィードしながら、オートクレーブ内にトリエチルアルミニウム4.9mmolを入れ追添器を取り付けた。オートクレーブ内をプロピレンガスで置換した後、25℃で水素2400mLとプロピレン18.5molとを加え撹拌し30℃に昇温した。少量の窒素をフィードした状態の追添器に、ジイソプロピルジメトキシシラン1.0mmolを入れ、続いて上記で調製した予備重合触媒スラリー2.5ml(予備重合触媒として7.5mg)を追添器に入れ、0.5分間予備接触させた。追添器内を窒素で4MPaに加圧し、予備接触触媒をオートクレーブ内に圧入した。オートクレーブを70℃に昇温し、60分間プロピレンを重合した。重合終了後、未反応プロピレンをパージし、ポリプロピレンを得た。得られたポリプロピレンを60℃で16時間真空乾燥し、上記の方法に従い、MFR、XI、重合活性の分析を行った。
予備接触時間をそれぞれ、0.25、0.5分、3分、重合時間を180分とした以外は、実施例1と同様に行った。
重合時間を180分とした以外は比較例1と同様に行った。

Claims (6)
- (A)マグネシウム、チタン、ハロゲンおよび電子供与体化合物を必須成分として含有する固体触媒成分;
(B)有機アルミニウム化合物;および
(C)電子供与体化合物
を含む触媒を用いてα-オレフィン重合体を製造する方法であって、
(1)(A)成分と(B)成分との存在下に、α-オレフィンを予備重合させ;
(2)得られた予備重合触媒と、(B)成分と(C)成分とを予め接触させて得た接触物とを接触させ;
(3)該接触物と接触させた予備重合触媒を、α-オレフィンを装填した重合反応容器中に添加して、α-オレフィンを重合する
ことを特徴とする、前記方法。 - 工程(2)の接触時間が、20分間を超えない、請求項1に記載の方法。
- (C)成分が有機ケイ素化合物である、請求項1または2に記載の方法。
- (A)マグネシウム、チタン、ハロゲンおよび電子供与体化合物を必須成分として含有する固体触媒成分;
(B)有機アルミニウム化合物;および
(C)電子供与体化合物
を含む触媒を用いてα-オレフィン重合体を製造する方法であって、
(1)(A)成分と(B)成分との存在下に、α-オレフィンを予備重合させ;
(2)得られた予備重合触媒を(B)成分に接触させ;
(3)(B)成分に接触させた予備重合触媒を、(C)成分とα-オレフィンとを装填した重合反応容器中に添加して、α-オレフィンを重合する
ことを特徴とする、前記方法。 - 工程(2)の接触時間が、20分間を超えない、請求項4に記載の方法。
- (C)成分が有機ケイ素化合物である、請求項4または5に記載の方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/130,900 US9353198B2 (en) | 2011-07-06 | 2012-07-03 | Method for polymerizing alpha-olefin |
| CN201280031919.6A CN103635494B (zh) | 2011-07-06 | 2012-07-03 | α‑烯烃聚合方法 |
| KR1020147002863A KR101878532B1 (ko) | 2011-07-06 | 2012-07-03 | α-올레핀 중합 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-150001 | 2011-07-06 | ||
| JP2011150001A JP5918486B2 (ja) | 2011-07-06 | 2011-07-06 | α−オレフィン重合方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013005735A1 true WO2013005735A1 (ja) | 2013-01-10 |
Family
ID=47437083
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/066960 WO2013005735A1 (ja) | 2011-07-06 | 2012-07-03 | α-オレフィン重合方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9353198B2 (ja) |
| JP (1) | JP5918486B2 (ja) |
| KR (1) | KR101878532B1 (ja) |
| CN (1) | CN103635494B (ja) |
| WO (1) | WO2013005735A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8894719B2 (en) | 2002-12-20 | 2014-11-25 | Ossur Hf | Suspension liner system with seal |
| US8956422B2 (en) | 2011-08-22 | 2015-02-17 | Ossur Hf | Suspension liner with seal component |
| US9072611B2 (en) | 2009-01-21 | 2015-07-07 | Ossur Americas, Inc. | Sealing sheath for prosthetic liner and related methods |
| US9603726B2 (en) | 2002-12-20 | 2017-03-28 | Ossur Hf | Adjustable seal system, seal component and method for using the same |
| US10159585B2 (en) | 2016-04-25 | 2018-12-25 | Ossur Iceland Ehf | Prosthetic liner |
| US10322016B2 (en) | 2002-12-20 | 2019-06-18 | Ossur Iceland Ehf | Adjustable seal system, seal component and method for using the same |
| US10420657B2 (en) | 2015-10-15 | 2019-09-24 | Ossur Iceland Ehf | Adjustable seal system |
| US10945865B2 (en) | 2017-11-01 | 2021-03-16 | Ossur Iceland Ehf | Prosthetic socket sealing system |
| US11510793B2 (en) | 2017-11-28 | 2022-11-29 | Ossur Iceland Ehf | Adjustable seal system, seal component and method for using the same |
| US11523917B2 (en) | 2002-12-20 | 2022-12-13 | Ossur Hf | Suspension liner system with seal |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6598284B2 (ja) * | 2014-03-26 | 2019-10-30 | サンアロマー株式会社 | α−オレフィン類の重合方法 |
| JP6598283B2 (ja) * | 2014-11-10 | 2019-10-30 | サンアロマー株式会社 | α−オレフィン類の重合方法 |
| JP6598285B2 (ja) * | 2014-11-11 | 2019-10-30 | サンアロマー株式会社 | α−オレフィン類の重合方法 |
| KR102467598B1 (ko) * | 2017-11-28 | 2022-11-15 | 롯데케미칼 주식회사 | 올레핀 중합용 촉매 조성물, 이의 제조방법 및 이를 이용한 폴리올레핀의 제조방법 |
| CN110240668A (zh) * | 2019-06-24 | 2019-09-17 | 天津科技大学 | Ziegler-Natta催化剂的内给电子体、催化剂组分、制备方法及其应用 |
| JP7023322B2 (ja) * | 2020-05-27 | 2022-02-21 | 東邦チタニウム株式会社 | オレフィン類重合用触媒の製造方法 |
| EP4159773A4 (en) * | 2020-05-27 | 2024-06-19 | Toho Titanium Co., Ltd. | METHOD FOR PRODUCING A CATALYST FOR OLEFIN POLYMERIZATION, CATALYST FOR OLEFIN POLYMERIZATION AND METHOD FOR PRODUCING AN OLEFIN POLYMER |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10182740A (ja) * | 1987-02-02 | 1998-07-07 | Cosden Technol Inc | オレフインの重合方法 |
| JP2001055413A (ja) * | 1999-08-18 | 2001-02-27 | Japan Polychem Corp | α−オレフィン重合用触媒および重合方法 |
| JP2002500697A (ja) * | 1998-03-23 | 2002-01-08 | モンテル テクノロジー カンパニー ビーブイ | オレフィン重合用の予備重合された触媒成分 |
| JP2006176565A (ja) * | 2004-12-21 | 2006-07-06 | Sumitomo Chemical Co Ltd | 予備重合済付加重合用触媒成分、付加重合用触媒および付加重合体の製造方法 |
| JP2006316160A (ja) * | 2005-05-12 | 2006-11-24 | Japan Polypropylene Corp | オレフィン重合用触媒の保存方法 |
| JP2006528271A (ja) * | 2003-05-29 | 2006-12-14 | バセル ポリオレフィン イタリア エス.アール.エル. | 触媒成分の製造方法およびそれから得られる成分 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2591602B1 (fr) * | 1985-12-18 | 1988-02-26 | Atochem | Procede de traitement de catalyseurs spheriques de polymerisation des olefines. application du catalyseur obtenu a la polymerisation des olefines. |
| JP2637076B2 (ja) | 1986-07-21 | 1997-08-06 | 三井石油化学工業 株式会社 | プロピレンブロツク共重合体の製法 |
| US4767735A (en) | 1987-02-02 | 1988-08-30 | Cosden Technology, Inc. | Catalyst pretreatment process |
| US5122583A (en) * | 1987-02-02 | 1992-06-16 | Fina Technology, Inc. | Efficiency of a pre-polymerized catalyst |
| FR2629461B1 (fr) * | 1988-03-31 | 1993-05-07 | Bp Chimie Sa | Catalyseur de (co)polymerisation du propylene, supporte sur des particules spheriques de chlorure de magnesium et enrobe par du polypropylene, et procedes de preparation |
| IT1251679B (it) * | 1991-10-09 | 1995-05-19 | Enichem Polimeri | Catalizzatori per la polimerizzazione delle olefine |
| JPH0623406A (ja) | 1992-07-08 | 1994-02-01 | Kawasaki Steel Corp | 鋼片の連続熱間圧延方法 |
| JP3984304B2 (ja) | 1993-08-18 | 2007-10-03 | 三井化学株式会社 | オレフィン重合用触媒、これを用いるプロピレン系重合体の製造方法 |
| FI952175A (fi) * | 1995-05-05 | 1996-11-06 | Borealis As | Menetelmä ja katalysaattorikomponentti olefiinien homo- tai kopolymeroimiseksi |
| JP2740503B2 (ja) | 1996-08-28 | 1998-04-15 | 三井化学株式会社 | α−オレフインの重合方法 |
| DE69812710T2 (de) * | 1997-12-23 | 2003-10-23 | Borealis Technology Oy, Porvoo | Löslicher magnesiumhalogenidkomplex, herstellung und verwendung |
| KR100387734B1 (ko) * | 2000-06-17 | 2003-06-18 | 삼성종합화학주식회사 | 올레핀 중합용 촉매 및 중합방법 |
| US20090069171A1 (en) | 2005-05-12 | 2009-03-12 | Japan Polypropylene Corporation | Catalysts for olefin polymerization, process for production of the catalysts, and method for preservation thereof |
| EP1801157B1 (en) * | 2005-12-22 | 2010-09-22 | Borealis Technology Oy | Polypropylene composition comprising a propylene copolymer component |
| ATE522552T1 (de) * | 2007-12-28 | 2011-09-15 | Basell Poliolefine Srl | Katalysatorkomponenten zur olefinpolymerisation |
| JP2010168545A (ja) * | 2008-12-25 | 2010-08-05 | Sumitomo Chemical Co Ltd | α−オレフィン重合用触媒およびα−オレフィン重合体の製造方法 |
-
2011
- 2011-07-06 JP JP2011150001A patent/JP5918486B2/ja active Active
-
2012
- 2012-07-03 CN CN201280031919.6A patent/CN103635494B/zh not_active Expired - Fee Related
- 2012-07-03 WO PCT/JP2012/066960 patent/WO2013005735A1/ja active Application Filing
- 2012-07-03 US US14/130,900 patent/US9353198B2/en not_active Expired - Fee Related
- 2012-07-03 KR KR1020147002863A patent/KR101878532B1/ko active IP Right Grant
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10182740A (ja) * | 1987-02-02 | 1998-07-07 | Cosden Technol Inc | オレフインの重合方法 |
| JP2002500697A (ja) * | 1998-03-23 | 2002-01-08 | モンテル テクノロジー カンパニー ビーブイ | オレフィン重合用の予備重合された触媒成分 |
| JP2001055413A (ja) * | 1999-08-18 | 2001-02-27 | Japan Polychem Corp | α−オレフィン重合用触媒および重合方法 |
| JP2006528271A (ja) * | 2003-05-29 | 2006-12-14 | バセル ポリオレフィン イタリア エス.アール.エル. | 触媒成分の製造方法およびそれから得られる成分 |
| JP2006176565A (ja) * | 2004-12-21 | 2006-07-06 | Sumitomo Chemical Co Ltd | 予備重合済付加重合用触媒成分、付加重合用触媒および付加重合体の製造方法 |
| JP2006316160A (ja) * | 2005-05-12 | 2006-11-24 | Japan Polypropylene Corp | オレフィン重合用触媒の保存方法 |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8894719B2 (en) | 2002-12-20 | 2014-11-25 | Ossur Hf | Suspension liner system with seal |
| US10322016B2 (en) | 2002-12-20 | 2019-06-18 | Ossur Iceland Ehf | Adjustable seal system, seal component and method for using the same |
| US11523917B2 (en) | 2002-12-20 | 2022-12-13 | Ossur Hf | Suspension liner system with seal |
| US9056022B2 (en) | 2002-12-20 | 2015-06-16 | Ossur Hf | Suspension liner system with seal |
| US9060885B2 (en) | 2002-12-20 | 2015-06-23 | Ossur Hf | Suspension liner system with seal |
| US9066821B2 (en) | 2002-12-20 | 2015-06-30 | Ossur Hf | Suspension liner system with seal |
| US8911506B2 (en) | 2002-12-20 | 2014-12-16 | Ossur Hf | Suspension liner system with seal |
| US10898352B2 (en) | 2002-12-20 | 2021-01-26 | Ossur Hf | Suspension liner system with seal |
| US9603726B2 (en) | 2002-12-20 | 2017-03-28 | Ossur Hf | Adjustable seal system, seal component and method for using the same |
| US10828179B2 (en) | 2002-12-20 | 2020-11-10 | Ossur Iceland Ehf | Adjustable seal system, seal component and method for using the same |
| US9295567B2 (en) | 2002-12-20 | 2016-03-29 | Ossur Hf | Suspension liner system with seal |
| US9707106B2 (en) | 2002-12-20 | 2017-07-18 | Ossur Hf | Adjustable seal system, seal component and method for using the same |
| US9877851B2 (en) | 2002-12-20 | 2018-01-30 | Ossur Hf | Adjustable seal system, seal component and method for using the same |
| US10342682B2 (en) | 2002-12-20 | 2019-07-09 | Ossur Hf | Suspension liner system with seal |
| US9072611B2 (en) | 2009-01-21 | 2015-07-07 | Ossur Americas, Inc. | Sealing sheath for prosthetic liner and related methods |
| US9168157B2 (en) | 2009-01-21 | 2015-10-27 | Ossur Americas, Inc. | Sealing sheath for prosthetic liner and related methods |
| US10213325B2 (en) | 2011-08-22 | 2019-02-26 | Ossur Hf | Suspension liner with seal component |
| US10660768B2 (en) | 2011-08-22 | 2020-05-26 | Ossur Hf | Suspension liner with seal component |
| US9566175B2 (en) | 2011-08-22 | 2017-02-14 | Ossur Hf | Suspension liner with seal component |
| US11399968B2 (en) | 2011-08-22 | 2022-08-02 | Ossur Hf | Suspension liner with seal component |
| US8956422B2 (en) | 2011-08-22 | 2015-02-17 | Ossur Hf | Suspension liner with seal component |
| US10420657B2 (en) | 2015-10-15 | 2019-09-24 | Ossur Iceland Ehf | Adjustable seal system |
| US11844709B2 (en) | 2015-10-15 | 2023-12-19 | Ossur Iceland Ehf | Adjustable seal system |
| US10159585B2 (en) | 2016-04-25 | 2018-12-25 | Ossur Iceland Ehf | Prosthetic liner |
| US11123203B2 (en) | 2016-04-25 | 2021-09-21 | Ossur Iceland Ehf | Prosthetic liner |
| US10945865B2 (en) | 2017-11-01 | 2021-03-16 | Ossur Iceland Ehf | Prosthetic socket sealing system |
| US11510793B2 (en) | 2017-11-28 | 2022-11-29 | Ossur Iceland Ehf | Adjustable seal system, seal component and method for using the same |
Also Published As
| Publication number | Publication date |
|---|---|
| KR101878532B1 (ko) | 2018-07-13 |
| CN103635494B (zh) | 2016-12-14 |
| KR20140083968A (ko) | 2014-07-04 |
| JP5918486B2 (ja) | 2016-05-18 |
| US9353198B2 (en) | 2016-05-31 |
| JP2013014725A (ja) | 2013-01-24 |
| US20140148562A1 (en) | 2014-05-29 |
| CN103635494A (zh) | 2014-03-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5918486B2 (ja) | α−オレフィン重合方法 | |
| US5844046A (en) | Process for the preparation of olefin polymer | |
| JP2010043267A (ja) | オレフィン重合用の成分と触媒 | |
| US7220696B2 (en) | Solid titanium catalyst component for olefin polymerization, catalyst for olefin polymerization, and process for olefin polymerization | |
| JP2011508037A (ja) | オレフィン類の重合用触媒成分 | |
| JPH04218508A (ja) | α−オレフィン系重合体の製造方法 | |
| JP3688078B2 (ja) | オレフィン重合用触媒、予備重合触媒、オレフィンの重合方法 | |
| JP2732478B2 (ja) | オレフィンの重合方法 | |
| JPH04218507A (ja) | オレフィン重合用固体状チタン触媒成分、オレフィン重合用触媒およびオレフィンの重合方法 | |
| JP2004519530A (ja) | エチレンおよびα−オレフィン類を重合するための触媒担体の製造方法、得られる担体並びに対応する触媒 | |
| EP1505084A1 (en) | Solid catalyst component for olefin polymerization, catalyst for olefin polymerization and method for producing olefin polymer | |
| JPH0496911A (ja) | オレフィン重合用固体状チタン触媒成分、オレフイン重合用触媒およびオレフィンの重合方法 | |
| JP6598285B2 (ja) | α−オレフィン類の重合方法 | |
| JP2008163188A (ja) | プロピレンランダム共重合体粒子及びその製造方法 | |
| JP2004002742A (ja) | オレフィン重合用固体状チタン触媒成分、オレフィン重合用触媒およびオレフィンの重合方法 | |
| JP4233969B2 (ja) | オレフィン重合用固体状チタン触媒成分、オレフィン重合用触媒およびオレフィンの重合方法 | |
| JPH04218509A (ja) | 予備重合触媒、オレフィン重合用触媒およびオレフィンの重合方法 | |
| JP5734005B2 (ja) | α−オレフィン系重合体の製造方法 | |
| JP3195383B2 (ja) | 炭素数が3〜20のα−オレフィン重合用固体状触媒成分、これを含む重合用触媒および炭素数が3〜20のα−オレフィンの重合方法 | |
| JPH06279531A (ja) | オレフィン重合用固体状チタン触媒成分、オレフィン重合用触媒およびこれを用いるオレフィンの重合方法 | |
| JPH03294308A (ja) | 低結晶性または非晶性α―オレフィン共重合体の製造方法 | |
| JPH06279523A (ja) | オレフィン重合用固体状チタン触媒成分、オレフィン重合用触媒およびこれを用いるオレフィンの重合方法 | |
| JPH06279527A (ja) | オレフィン重合用触媒およびこれを用いるオレフィンの重合方法 | |
| CN114106223A (zh) | 一种用于烯烃聚合的催化剂体系和烯烃聚合方法 | |
| JP2941015B2 (ja) | オレフィン重合用固体状チタン触媒成分、オレフイン重合用触媒およびオレフィンの重合方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12807769 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14130900 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20147002863 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12807769 Country of ref document: EP Kind code of ref document: A1 |