RU2259372C2 - Производные азаиндола - Google Patents
Производные азаиндола Download PDFInfo
- Publication number
- RU2259372C2 RU2259372C2 RU2002125494/04A RU2002125494A RU2259372C2 RU 2259372 C2 RU2259372 C2 RU 2259372C2 RU 2002125494/04 A RU2002125494/04 A RU 2002125494/04A RU 2002125494 A RU2002125494 A RU 2002125494A RU 2259372 C2 RU2259372 C2 RU 2259372C2
- Authority
- RU
- Russia
- Prior art keywords
- group
- compound
- methyl
- alkyl
- piperazine
- Prior art date
Links
- 0 C*c(c(I)c(c(N)c1P)P)c1N Chemical compound C*c(c(I)c(c(N)c1P)P)c1N 0.000 description 9
- PFYPDUUXDADWKC-UHFFFAOYSA-N CC(C)c1ccccn1 Chemical compound CC(C)c1ccccn1 PFYPDUUXDADWKC-UHFFFAOYSA-N 0.000 description 1
- FJBZRCOBIJJDSS-UHFFFAOYSA-N COc1nccc2c1[nH]cc2C(C(O)=O)=O Chemical compound COc1nccc2c1[nH]cc2C(C(O)=O)=O FJBZRCOBIJJDSS-UHFFFAOYSA-N 0.000 description 1
- LWDNSHAZYNFKHL-UHFFFAOYSA-N OC(C(c1c[nH]c2ccncc12)=O)=O Chemical compound OC(C(c1c[nH]c2ccncc12)=O)=O LWDNSHAZYNFKHL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Virology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Изобретение относится к новым производным азаиндола формулы I или его фармацевтически приемлемым солям:

где

выбирают из группы, состоящей из:

R1, R2, R3, R4 каждый независимо выбран из группы, состоящей из Н, С1-С6алкила, С2-С6алкенила, галогена, CN, фенила, нитро, OC(О)R15, C(О)R15, C(О)OR16, OR19, SR20 и NR21R22;
R15 независимо выбран из группы, включающей Н, C1-С6алкил и С2-С6алкенил;
R16, R19 и R20 каждый независимо выбран из группы, включающей Н, C1-С6алкил, С1-6алкил, замещенный от одного до трех атомов галогена;
R21 и R22 каждый независимо выбран из группы, включающей Н, ОН, C1-С6алкил;
R5 представляет собой (О)m, где m имеет значение 0 или 1;
n имеет значение 1 или 2;
R6 выбирают из группы, включающей Н, C1-C6алкил, C(О)R24, C(О)OR25;
при условии, что атомы углерода, которые содержат углерод-углеродную двойную связь указанного С3-С6алкенила не являются точкой присоединения к азоту, к которому присоединен R6;
R24 выбирают из группы, состоящей из Н, C1-C6алкила;
R25 представляет собой C1-С6алкил;
R7, R8, R9, R10, R11, R12, R13 и R14 каждый независимо выбран из группы, включающей Н, C1-С6алкил;
Ar выбирают из группы, включающей

Соединение I ингибирует вирус, вызываемый ВИЧ-1, что позволяет предположить возможность его использования в медицине. 21 з.п. ф-лы, 5 табл.
Description
Область техники
Настоящее изобретение относится к соединениям, обладающим лекарственными и биоактивными свойствами, их фармацевтическим композициям, а также способам их применения. В частности, изобретение касается производных азаиндолпиперазиндиамида, которые обладают уникальной антивирусной активностью. В особенности, настоящее изобретение касается соединений, пригодных для лечения ВИЧ-инфекции и СПИДа.
Уровень техники
ВИЧ-1 (вирус-1 человеческого иммунодефицита) остается главной медицинской проблемой, поскольку по всемирной оценке инфицированы 33.6 миллиона человек. Число случаев ВИЧ-инфекции и СПИДа (приобретенный синдром иммунодефицита) быстро повышается. В 1999 г. было сообщено о новых инфекциях у 5.6 миллионов человек и о 2.6 миллионах умерших от СПИДа. В настоящее время доступные лекарственные препараты для лечения ВИЧ-инфекции включают шесть нуклеозидных ингибиторов обратимости транскриптазы (RT) (зидовудин, диданозин, ставудин, ламивудин, залтитабин и абасавир) (zidovudine, didanosine, stavudine, lamivudine, zalcitabine and abacavir), три ненуклеозидных ингибитора обратимости транскриптазы (невирапин, делавирдин и эфавиренз) (nevirapine, delavirdine and efavirenz), а также пять пептидомиметических ингибиторов протеазы (саквинавир, индинавир, ритонавир, нельфинавир и ампренавир) (saquinavir, indinavir, ritonavir, nelfinavir and amprenavir). Каждый из указанных лекарственных препаратов способен только кратковременно ограничить размножение вирусов, если их используют как таковые. Однако, когда их используют в сочетании (комбинации), эти лекарственные препараты оказывают сильное воздействие на вирусы и прогрессирование болезни. Фактически, было подтверждено документально, что существенные сокращения в показателях смертности среди пациентов, заболевших СПИДом, являются следствием широкого применения комбинационной терапии. Однако, несмотря на эти впечатляющие результаты, от 30 до 50% пациентов в конечном счете не вылечиваются при применении комбинационной терапии лекарственных препаратов. Неэффективность действия лекарственных препаратов, несоответствие, ограниченное действие внутри некоторых типов клеток (например, большинство нуклеозидных аналогов не могут быть профосфорилированы в отдельных клетках) может служить оценкой неполного подавления восприимчивых вирусов. Кроме того, высокая скорость размножения и быстрое преобразование ВИЧ-1 вместе с частым включением мутаций, приводит к появлению устойчивости к лекарственным препаратам и лечение становится невозможным, когда присутствует сверхоптимальная концентрация лекарственных препаратов (Larder и Kemp; Gulick; Kuritzkes; Morris-Jones et al.; Schinazi et al.; Vacca и Condra; Flexner; Berkhout и Ren et al.; (Ref.6-14)). Таким образом, имеется необходимость создания новых анти-ВИЧ агентов, демонстрирующих отличные свойства устойчивости и благоприятную фармакокинетику, также как и безопасность, чтобы обеспечить альтернативное лечение.
В настоящее время в качестве запатентованных лекарственных препаратов против ВИЧ-1 доминируют либо нуклеозидные ингибиторы обратимости транскриптазы, либо ингибиторы пептидомиметической протеазы. Недавно полученные ненуклеозидные ингибиторы обратимости транскриптазы (NNRTIs) играют все более и более важную роль в терапии ВИЧ-инфекций (Pedersen & Pedersen, Ref.15). В литературе описаны по крайней мере 30 различных классов NNRTI (De Clercq, Ref.16) и несколько NNRTIs прошли клинические испытания. Дипиридодиазепинон (невиралин), бензоксазинон (эфавиренз) и производные бис(гетероарил) пиперазина (делавирдин) одобрены для клинического применения. Однако главным недостатком в развитии и применении NNRTIs является их склонность к быстрому появлению штаммов, резистентных к лекарственным препаратам как в культуре клеточной ткани, так и у подвергнутых лечению индивидуумов, в особенности тех, которых прошли монотерапию. Как следствие, возникает значительный интерес для выявления большого количества NNRTIs со сниженной способностью к развитию резистентности (Pedersen & Pedersen, Ref.15).
Имеются сообщения о нескоторых производных индола, включая индол-3-сульфоны, пиперазининдолы, пиразининдолы и производные 5Н-индоло[3,2-b][1,5]бензотиазепина, как об ингибиторах обратимости ВИЧ-1 транскриптазы (Greenlee et al., Ref.1; Williams et al., Ref.2; Romero et al., Ref.3; Font et al., Ref.17; Romero et al., Ref.18; Young et al., Ref.19; Genin et al., Ref.20; Silvestri et al., Ref.21). Индольные 2-карбоксамиды также были описаны как ингибиторы клеточной адгезии и ВИЧ-инфекции (Boschelli et al., US 5,424,329, Ref.4). И, наконец, были описаны как ингибиторы ВИЧ-1 протеазы природные продукты на основе 3-замещенного индола (семикохлиодинол А и В, дидеметиластерихинон и изокохлиодинол (Semicochliodinol А и В, didemethylasterriquinone и isocochliodinol) (Fredenhagen et al., Ref.22).
Ранее были описаны близкие по структуре производные азаиндоламида (Kato et al., Ref.23; Levacher et al., Ref.24; Mantovanini et al., Ref.5(a); Cassidy et al., Ref.5(b); Scherlock et al., Ref.5(с)). Однако эти структуры представляют собой скорее азаиндолмоноамиды, чем несимметричные производные азаиндолпиперазиндиамида и, кроме того, отсутствуют упоминания о применении этих соединений для лечения антивирусных инфекций, в частности ВИЧ-инфекции. Ничто в перечисленных ссылках не может быть определено, как раскрытие или предположение относительно новых соединений по настоящему изобретению и их применения для ингибирования ВИЧ-инфекции.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение включает в себя соединения формулы I или их фармацевтически приемлемые соли, которые являются эффективными антивирусными агентами, в частности действуют как ВИЧ ингибиторы.

где:

выбирают из группы, состоящей из
R1, R2, R3, R4 каждый независимо выбран из группы, состоящей из Н, C1-C6алкила,

С2-С6алкенила, галогена, -CN, фенила, нитро, OC(O)R15, C(O)R15, C(O)OR16, OR19, SR20 и NR21R22;
R15 независимо выбран из группы, включающей Н, C1-С6алкил и С2-С6алкенил;
R16, R19 и R20 каждый независимо выбран из группы, включающей Н, C1-С6алкил, C1-C6алкил, замещенный от одного до трех атомами галогена;
R21 и R22 каждый независимо выбран из группы, включающей Н, ОН, C1-С6алкил;
R5 представляет собой (O)m, где m имеет значение 0 или 1;
n имеет значение 1 или 2;
R6 выбирают из группы, включающей Н, С1-С6алкил, С3-С6алкенил, C(O)R24, C(O)OR25; при условии, что атомы углерода, которые содержат углерод-углеродную двойную связь указанного С3-С6алкенила, не являются точкой присоединения к азоту, к которому присоединен R6;
R24 выбирают из группы, состоящей из Н, C1-C6алкила;
R25 представляет собой C1-C6алкил;
R7, R8, R9, R10, R11, R12, R13 и R14 каждый независимо выбран из группы, включающей Н, C1-C6алкил;
Ar выбирают из группы, включающей

A1, А2, А3, А4, A5, B1, B2, В3, В4, C1, C2, С3 каждый независимо выбран из группы, включающей Н, галоген, C1-C6алкил, -CN, нитро, N3.
Предпочтительными являются соединения формулы I или их фармацевтически приемлемые соли, в которых R2-R4 независимо представляет собой Н, -ОСН3, -ОСН2CF3, -OiPr, -OnPr, CN, NO2, C1-С6алкил, NHOH, NH2, SR20 или N(СН3)2.
Также предпочтительны соединения формулы I, в которых один или два R7-R14 независимо представляют собой метил, а другие заместители представляют собой водород.
Также предпочтительны соединения формулы I, в которых один из A1-A5, В1-B4, C1-С3 представляют собой либо водород, галоген, или амино и остальные заместители представляют собой водород.
Также предпочтительны соединения формулы, приведенной ниже

где
R2 представляет собой Н, F, Cl, Br, OMe, CN или ОН;
R4 представляет собой C1-C6алкил, С2-С6алкенил, Cl, OMe, CN, ОН, Ph или -С(O)СН3;
n имеет значение 2;
R8, R9, R10, R11, R12, R13 и R14 каждый независимо является Н или СН3 при условии, что вплоть до двух этих заместителей могут быть метилом;
R1 представляет собой водород;
R5 является незамещенным;
R6 представляет собой водород или метил.
К наиболее предпочтительным относятся соединения или их фармацевтически приемлемые соли следующей формулы
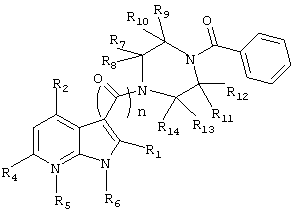
где
R2 представляет собой Н, -ОСН3, -OCH2CF3, -OPr, галоген, CN, NO2 или NHOH;
R4 представляет собой Н, -галоген, -CN или гидрокси;
Один или два члена R7-R14 представляют собой метил и оставшиеся члены представляют собой водород;
n имеет значение 2;
R1 представляет собой водород;
R5 представляет собой (O)m, где m представляют собой О;
R6 представляет собой водород, метил, или аллил.
К другим наиболее предпочтительным относятся соединения формулы

где
R2 выбирают из группы, включающей Н, F, Cl, Br, OMe, CN и ОН;
R4 выбирают из группы, включающей Н, C1-C6алкил, С2-С6алкенил, Cl, OMe, CN, ОН, фенил и -С(O)СН3;
n имеет значение 2;
R8, R9, R10, R11, R12, R13 и R14 каждый независимо является Н или СН3 при условии, что 0-2 членов группы R8, R9, R10, R11, R12, R13 и R14 могут быть СН3, а остальные члены группы R8, R9, R10, R11, R12, R13 и R14 являются Н;
R6 представляет собой Н или СН3.
К другим наиболее предпочтительным аспектам изобретения относятся соединения формулы, приведенной ниже

где
R4 выбирают из группы, включающей Н, C1-C6алкил, С2-С6алкенил, Cl, ОМе, CN, ОН, фенил и -С(O)СН3;
n имеет значение 2;
R8, R9, R10, R11, R12, R13 и R14 каждый независимо является Н или СН3 при условии, что 0-2 членов группы R8, R9, R10, R11, R12, R13 и R14 могут быть СН3, а остальные члены группы R8, R9, R10, R11, R12, R13 и R14 являются Н;
R6 представляет собой Н или СН3.
Поскольку соединения в соответствии с настоящим изобретением могут иметь асимметричные центры и соответственно существовать в виде смесей диастереоизомеров и энантиомеров, настоящее изобретение включает индивидуаьные формы диастереоизомеров и энантиомеров соединений формулы I.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Препаративные процессы и анти-ВИЧ-1 активность новых аналогов азаиндолпиперазиндиамида формулы I приведены ниже. Далее приведены также определения различных терминов.
Термин «С1-C6алкил», как его используют в описании и в формуле изобретения (если контекст не указывает на другое), означает линейные или разветвленные цепи алкильных групп, такие как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, амил, гексил и тому подобное. Аналогично "C1-6алкенил" или "C1-6алкинил" включает линейные или разветвленные цепочечные группы.
"Галоген" имеет отношение к хлору, брому, йоду или фтору.
Физиологически приемлемые соли и пролекарства соединений, описанные здесь, входят в границы настоящего изобретения. Термин ″фармацевтически приемлемые соли″, как его используют в описании и в формуле изобретения, как предполагается, включает нетоксичные основные аддитивные соли. Пригодные соли включают те, которые получают из органических и неорганическх кислот, таких как без ограничения соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота, метансульфоновая кислота, уксусная кислота, винная кислота, молочная кислота, сульфиновая кислота, лимонная кислота, малеиновая кислота, фумаровая кислота, сорбиновая кислота, аконитовая кислота, салициловая кислота, фталевая кислота и тому подобное. Термин ″фармацевтически приемлемая соль″, как его здесь используют, как предполагается, включает соли кислотных групп, таких как карбоксилат, с такими притивоионами, как аммоний, соли щелочных металлов, в частности натрия или калия, соли щелочноземельных металлов, в частности кальция или магния, и соли с пригодными органическими основаниями, такие как низшие алкиламины (метиламин, этиламин, циклогексиламин и тому подобное), или замещенные низшие алкиламины (например, гидроксилзамещенные алкиламины, такие как диэтаноламин, триэтаноламин или трис(гидроксиметил)-аминометан), или с основаниями, такими как пиперидин или морфолин.
В способе настоящего изобретения термин "антивирусное эффективное количество" означает общее количество каждого активного компонента способа, которое достаточно, чтобы показать заметное преимущество при лечении больных, то есть достигнуть исцеления острого состояния, характеризуемое ингибированием ВИЧ-инфекции. Когда термин применяют для индивидуальных активных инградиентов, вводимых по единому, он относится к одному ингредиенту. Когда термин применяют к комбинации, он имеет отношение к объединенному количеству активных ингредиентов, как результат терапевтического эффекта, при введении в комбинации, последовательно или одновременно. Термины "обрабатывать, лечение, обработка", как их используют в настоящем изобретении и в формуле изобретения, означает предотвращение или улучшение состояния при заболеваниях, связанных с ВИЧ-инфекцией.
Настоящее изобретение также направлено на комбинации (или сочетании) соединений с одним или более представителями, пригодными для лечения СПИДа. Например, соединения настоящего изобретения могут эффективно вводиться либо в периоды предварительного выявления и/или последующего выявления, в комбинации с эффективным количеством антивирусных препаратов против СПИДа, иммуномодуляторов, противоинфекционных препаратов или вакцин, таких как приведены в таблице 1.
| Таблица 1 АНТИВИРУСНЫЕ ПРЕПАРАТЫ |
||
| Лекарственный препарат, Название |
Производитель | Показания |
| 097 | Hoechst/Bayer | ВИЧ-инфекция, СПИД, ARC (ингибитор RT) |
| Ампренивир 141 W94 GW141 |
Glaxo Wellcome | ВИЧ-инфекция, СПИД, ARC (ингибитор протеазы) |
| Абакавир (1592U89) GW1592 |
Glaxo Wellcome | ВИЧ-инфекция, СПИД, ARC (ингибитор RT) |
| Ацематан | Carrington Labs (Irving, TX) | ARC |
| Ацикловир | Burroughs Wellcome | ВИЧ-инфекция, СПИД, ARC, в комбинации с AZT |
| AD-439 | Tanox Biosystems | ВИЧ-инфекция, СПИД, ARC |
| AD-519 | Tanox Biosystems | ВИЧ-инфекция, СПИД, ARC |
| Адефовир дипивоксил | Gilead Sciences | ВИЧ-инфекция |
| AL-721 | Ethigen (Los Angeles, CA) | ARC, PGL ВИЧ-инфекция положительная, СПИД |
| Альфа интерферон | Glaxo Wellcome | Саркома Капози, ВИЧ-инфекция в комбинации с w/ретровирусом |
| Анзалицин LM.427 | Adria Laboratories (Dublin, ОН) Erbamont (Stamford, CT) | ARC |
| Антитело, которое нейтрализует рН лабильный альфа абберантный интерферон | Advanced Biotherapy Concepts (Rockville, MD) | СПИД, ARC |
| AR177 | Aronex Pharm | ВИЧ-инфекция, СПИД, ARC |
| бета-фтор-ddA | Nat'l Cancer Institute | Связанные со СПИДом болезни |
| BMS-232623 (CGP-73547) | Bristol-Myers Squibb/ Novartis | ВИЧ-инфекция, СПИД, ARC (ингибитор протеазы) |
| BMS-234475 (CGP-61755) | Bristol-Myers Squibb/Novartis | ВИЧ-инфекция, СПИД, ARC (ингибитор протеазы) |
| Сl-1012 | Wamer-Lambert | ВИЧ-1 инфекция |
| Цидофовир | Gilead Science | CMV ретинит герпес, вирусная папилома |
| Сульфат курдлана | AJI Pharma USA | ВИЧ-инфекция |
| Цитомегаловирус иммуннглобулин | Medlmmune | CMV ретиниты |
| Цитовенодный Ганцикловир | Syntex | Угроза зрению CMV |
| перефирийные CMV ретиниты | ||
| Делавиридин | Pharmacia-Upjohn | ВИЧ-инфекция, СПИД, ARC (RT нгибитор) |
| Сульфат декстрана | Ueno Fine Chem. Inc. Ltd. (Osaka, Japan) | СПИД, ARC, ВИЧ-инфекция Положительная асимптоматика |
| ddC Дидезоксицитрон | Hoffinan-La Roche | ВИЧ-инфекция, СПИД, ARC |
| ddl Дидезоксицитрон | Bristol-Myers Squibb | ВИЧ-инфекция, СПИД, ARC; комбинация с AZT/d4T |
| DMP-450 | AVID(Camden,NJ) | ВИЧ-инфекция, СПИД, ARC (ингибитор протеазы) |
| Эфавиренц (DMP 266) (-)6-хлор-4-(S)-циклопропилэтинил-4(S)-трифтор-метил-1,4-дигидро-2Н-3,1-бензоксазин-2-он, СТОКРИН | DuPont Merck | ВИЧ-инфекция, СПИД. ARC (ненуклеозидный RT ингибитор) |
| EL10 | Elan Corp, PLC (Gainesville, GA) | ВИЧ-инфекция |
| Фамцикловир | Smith Kline | герпес опоясывающий лишай, герпес симплексный |
| FTC | Emory University | ВИЧ-инфекция, СПИД, ARC (ингибитор транскриптазы) |
| GS840 | Hoechst Marion Roussel | ВИЧ-инфекция, СПИД, ARC (ингибитор транскриптазы) |
| HBY097 | Gilead | ВИЧ-инфекция, СПИД, ARC (ненуклеозидные Ингибиторы транскриптазы) |
| Гиперицин | VIMRx Pharm. | ВИЧ-инфекция, СПИД, ARC |
| Рекомбинаторный интерферон бета человеческий | Triton Biosciences (Almeda, CA) | СПИД, Саркома Капози, ARC |
| Интерферон альфа-n3 | hiterferon Sciences | ARC, СПИД |
| Индинавир | Merck | ВИЧ-инфекция, СПИД, ARC, бессимптомная ВИЧ-инфекция положительная, также в комбинации с AZT/ddl/ddC |
| ISIS 2922 | ISIS Pharmaceuticals | CMV ретиниты |
| KNI-272 | Nat'l Cancer Institute diseases | болезни, ассоциированные с ВИЧ-инфекцией |
| Ламивудин, 3ТС | Glaxo Wellcome | ВИЧ-инфекция, СПИД. ARC (ингибитор транскриптазы); |
| также с AZT | ||
| Лабукавир | Bristol-Myers Squibb | CMV инфекция |
| Нелфинавир | Agouron Pharmaceuticals | ВИЧ-инфекция, СПИД, ARC (ингибитор протеазы) |
| Невирапин | Boeheringer bigleheim | ВИЧ-инфекция, СПИД, ARC (RT ингибитор) |
| Новапрен | Novaferon Labs, Inc. (Akron, OH) | ВИЧ-ингибитор инфекции |
| Пептид Т октановая последовательность | Peninsula Labs (Behnont, CA) | СПИД |
| Фосфоноформиат | Astra Pharm. Products, Inc. | CMV ретиниты, ВИЧ-инфекция, другие CMV инфекции |
| PNU-140690 | Pharmacia Upjohn | ВИЧ-инфекция, СПИД, ARC (ингибитор протеазы) |
| Пробукол | Vyrex Sheffield Med. Tech (Houston, TX) | ВИЧ-инфекция, СПИД |
| Ривонавир | Abbott | ВИЧ-инфекция, |
| СПИД, ARC (ингибитор протеазы) | ||
| Саквинавир | Hoffinann-LaRoche | ВИЧ-инфекция, СПИД, ARC (ингибитор протеазы) |
| Ставудин d4T Дидегидродезокситимидин | Bristol-Myers Squibb | ВИЧ-инфекция, СПИД, ARC |
| Валахщкловир | Glaxo Wellcome | Генитальные HSV & CMV инфекции |
| Виразол Рибавирин | Viratek/ICN (Costa Mesa, CA) | Бессимптомная ВИЧ-инфекция положительная, LAS, ARC |
| VX-478 | Vertex | ВИЧ-инфекция, СПИД, ARC |
| Залцитабин | Hofflnann-LaRoche | ВИЧ-инфекция, СПИД. ARC, с AZT |
| Зидовудин; AZT | Glaxo Wellcome | ВИЧ-инфекция, СПИД. ARC, Саркома Капози, в комбинации с др. терапиями |
| Таблица 2 ИММУНОМОДУЛЯТОРЫ |
||
| Лекарст. препарат, название | Производитель | Показания |
| AS-101 | Wyeth-Ayerst | СПИД |
| Бропиримин | Pharmacia Upjohn | Развитый СПИД |
| Ацеман | Carrington Labs, Inc. (Irving, TX) | СПИД, ARC |
| CL246.738 | American Cyanamid Lederle Labs | СПИД, Саркома Капози |
| EL10 | Elan Corp, PLC (Gainesville, GA) | ВИЧ-инфекция |
| FP-21399 | Fuki Immunol Pharm | Блоки ВИЧ-инфекций, объединенные с CD4+клетки |
| Гамма Интерферон | Genentech | ARC, в комбинации с w/TNF (фактор некроза опухоли) |
| Гранулоцитный Макрофаговая колония стимулирующего фактора | Genetics Institute Sandoz | СПИД |
| Гранулоцитный Макрофаговая колония стимулирующего фактора | Hoechst-Roussel Immunex | СПИД |
| Гранулоцитный Макрофаговая колония стимулирующего фактора | Schermg-Plough | СПИД, ВИЧ-инфекция комбинация с w/AZT |
| Hiv Core Particle Иммуностимулянт | Rorer | Серопозитивная ВИЧ-инфекция |
| IL-2 Интерлейкин-2 | Cetus | СПИД, в комбинации w/AZT |
| IL-2 Интерлейкин-2 | Hoffman-LaRoche Immunex | СПИД, ARC, ВИЧ-инфекция в комбинации w/AZT |
| IL-2 Интерлейкин-2 (альдеслукин) | Chiron | СПИД, увеличение CD4 в клеточном отсчете |
| Иммуноглобулин внутривенный (человека) | Cutter Biological (Berkeley, CA) | Педиатрия СПИДа, в комбинации w/AZT |
| IMREG-1 | Imreg (New Orleans, LA) | СПИД, Саркома Капози, ARC, PGL |
| IMREG-2 | Imreg | СПИД, Саркома Капози, ARC, PGL |
| (New Orleans, LA) | ||
| Имутиол диэтил-дитокарбамат | Merieux Institute | СПИД, ARC |
| Альфа-2 Интерферон | Schering Plough | Саркома Капози w/AZT, СПИД |
| Метионин Энкефалин | TNI Pharmaceutical (Chicago, IL) | СПИД, ARC |
| МТР-РЕ Мирамил-Трипетит | Ciba-Geigy Corp. | Саркома Капози |
| Гранулоционная колония фактора стимул. |
Amgen | СПИД, в комбинация w/AZT |
| Ремун | Immune Response Corp. | Иммунотерапия |
| rCD4 Рекомбинационный растворимый чел. CD4 | Genentech | СПИД, ARC |
| rCD4-lgG гибриды | СПИД, ARC | |
| Рекомбинационный растворимый чел. CD4 | ||
| Интерферон Альфа 2а | Hoffman-La Roche | Саркома Калози СПИД, ARC в комбинации w/AZT |
| SK&F106528 Растворимый Т4 | Smith Kline | ВИЧ-инфекция |
| Тимопентин | Immunobiology Research Institute (Annandale, NJ) | ВИЧ-инфекция |
| Фактор некроза опухоли; TNF | Genentech | ARC, в комбинации с w/гамма интерфероном |
| Таблица 3 ПРОТИВОИНФЕКЦИОННЫЕ |
||
| Лекарственный препарат название | Производитель | Показания |
| Клидамицин с Примахином | Pharmacia Upjohn | PCP |
| Флуконазол | Pfizer | Криптококковый менингит, кандидиоз |
| Pastille Нистагин Pastille | Squibb Corp. | Предотвращение орального кандидамикоза |
| Орнидол Эфлорнитин | Merrell Dow | PCP |
| Пентамидин Изетионат (ТМ & IV) | LyphoMed (Rosemont, IL) | PCP лечение |
| Триметоприн | Антибактериальный | |
| Триметоприн/ сульфа | Антибактериальный | |
| Пиритрексим | Burroughs Wellcome | РСР-лечение |
| Пентамидин изетион для ингаляции | Fisons Corporation | PCP-профилактика |
| Спирамицин | Rhone-Poulenc | Криптоспоридийная Диарея |
| менингит | ||
| Триметотриксат | Warner-Lambert | PCP |
| Даунорубицин | NeXstar, Sequus | Саркома капози |
| Рекомбинационный Человеческий эритропоэтин | Ortho Phaim. Corp | Серьезная ассоц. анемия с AZT терапией |
| Рекомбинационный Человеческий гормон | Serono | СПИД-связан. Изнурение, кахексия |
| Ацетат Мегестрола | Bristol-Myers Squibb | Лечение ассоц. анорексии, W/СПИД |
| Тестостерон | Alza, Smith Kline | СПИД-изнурение |
| Общее энтеральное питание | Norwich Eaton Pharmaceuticals | Диарея и малабсорбция, связанные СПИД |
Кроме того, соединения по настоящему изобретению могут быть применены в комбинациях, которые включают, более чем три анти-ВИЧ лекарственных препарата. Были исследованы комбинации четырех или даже пяти ВИЧ лекарственных препаратов и, как ожидается, соединения настоящего изобретения будут полезным компонентом таких комбинаций.
Кроме того, соединения по настоящему изобретению могут быть применены в комбинации с другим классом веществ для лечения СПИДа, которые называются ВИЧ входящими ингибиторами. Примеры таких ВИЧ входящих ингибиторов рассмотрены в DRUGS OF THE FUTURE 1999. 24(12), pp.1355-1362; CELL, Vol.9, pp.243-246, Oct.29, 1999; and DRUG DISCOVERY TODAY, Vol.5, №5, May 2000, pp.183-194.
Подразумевается, что набор комбинаций соединений по настоящему изобретению с противовирусными препаратами против СПИДа, иммуномодуляторами, противоинфекционными препаратами, ВИЧ входящими ингибиторами или вакцинами, не ограничивается списком в вышеупомянутой таблице, и он может включать в принципе любую комбинацию с любой фармацевтической структурой, полезной для лечения СПИДа.
Предпочтительными являются сочетания для одновременного или альтернативного лечения, содержащие соединения по настоящему изобретению и ингибиторы ВИЧ-протеазы и/или ненуклеозидный ингибитор - обратимости ВИЧ транскриптазы. Необязательным четвертым компонентом в комбинации является нуклеозидный ингибитор обратимости ВИЧ транскриптазы, такой как AZT, ЗТС, ddC или ddl. Предпочтительным ингибитором ВИЧ-протеазы является индинавир, который представляет собой сульфатную соль N-(2(R)-гидрокси-1-(S)-инданил)-2(R)-фенилметил-4-(S)-гидрокси-5-(1-(4-(3-пиридилметил)-2(S)-N'-(трет-бутилкарбоксамидо)-пиперазинил))-пентанамида этанолата, который синтезируют согласно US 5413999. Индинавир обычно вводят в виде дозы 800 мг три раза в день. Другие предпочтительные ингибиторы протеазы представляют собой нелфинавир и ритонавир. Другой предпочтительный ингибитор ВИЧ-протеазы представляет собой саквинавир, который вводят в дозе 600 или 1200 мг в день. И наконец, новый ингибитор протеазы, BMS-232632, который на текущий момент проходит клинические испытания, может стать предпочтительным ингибитором. Предпочтительные ненуклеозидные ингибиторы ВИЧ-обратимости транскриптазы включают эфавиренц. Получение ddC, ddl и AZT также описано в ЕРО 0484071. Указанные комбинации могут оказывать неожиданное воздействие на сокращение распространения и степени инфецирования ВИЧ-инфекцией. Предпочтительные сочетания включают следующие сочетания (1) индинавир с эфавиренцом и, необязательно, AZT, и/или 3ТС, и/или ddl и/или ddC; (2) индинавир и любой из AZT, и/или ddl, и/или ddC, и/или 3ТС, в особенности индинавир и AZT и 3ТС; (3) ставудин и ЗТС и/или зидовудин; (4) зидовудин и ламивудин, и 141W94 и 1592U89; (5) зидовудин и ламивудин.
В таких сочетаниях соединение по настоящему изобретению и другие активные агенты могут быть введены отдельно или все вместе. В дополнение к сказанному, введение одного элемента может быть осуществлено до, во время или последовательно с введением других агентов (а).
Родственные азаиндолы, такие как 4-азаиндол, 5-азаиндол, 6-азаиндол или 7-азаиндол, получают с помощью методик, описанных в литературе (Mahadevan et al., Ref.25(a)) или Hands et. al. Ref.25(b) и они являются коммерчески доступными (7-азаиндол из Aldrich Co.). В указанной ссылке, а также в аналогичных ссылках приведены некоторые примеры замещенных азаиндолов. Специалист в данной области может определить общую методику, которая распространяется на азаиндолы, имеющие различные заместители в исходных продуктах. Азаиндолы могут быть также получены через пути, описанные на схемах 1 и 2.

На схеме 1 синтез индола по Бартоли (Dobson et al., Ref.25(С)) распространен на получение замещенных азаиндолов. Нитропиридин 22 взаимодействует с избытком винилмагнийбромида при -78°С. После нагревания до -20°С получают желаемый азаиндол 1. Как правило, эти температурные интервалы являются оптимальными, но в конкретных примерах могут варьироваться обычно не более, чем на 20°С, однако случается и более, чтобы оптимизировать выход. Винилмагнийбромид является коммерческим продуктом, который получают в виде раствора в тетрагидрофуране или в некоторых случаях более оптимально его можно готовить в свежем виде из винилбромида и магния, используя известные методики. Винилмагнийхлорид также может быть применен в других примерах.

На схеме 2 ацетилен конденсируют с галогенпиридином 23 при использовании Pd(0) катализатора, чтобы получить 24. Последующая обработка основанием приводит к циклизации 24, что дает желаемый азаиндол 1 (Sakamoto et al., Ref.26). Пригодные основания для второй стадии включают метилат натрия, алкоксидные основания натрия, лития или калия.
Общие методики получения азаиндолпиперазиндиамида 5 формулы I описаны на схеме 3 и схеме 4.

Азаиндол 1, реагирует с MeMgl (метилмагнийиодид) и ZnCl2 (хлорид цинка), после чего добавляют ClCOCOOMe (метилхлороксоацетат), что дает глиоксилметиловый эфир 2 азаиндола (Shadrina et al., Ref.27). Альтернативно соединение 2 может быть получено путем взаимодействия азаиндола 1 с избытком ClCOCOOMe в присутствии AICI3 (хлорида алюминия) (Sycheva et al., Ref.28). Гидролиз метилового эфира 2 дает калийную соль 3, которую подвергают конденсации с монобензоилированными производными пиперазина 4 в присутствии DEPBT (3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3Н)-оном) и N,N-диизопропилэтиламином, обычно известных как основание Ханига, чтобы получить азаиндолпиперазиндиамид 5 (Li et al., Ref.29). Монобензоилированные производные пиперазина 4 могут быть получены согласно хорошо разработанной методике, такой как описана у Desai et al., Ref.30(a), Adamczyk et al., Ref.30(b), Rossen et al., Ref.30(c) и Wang et al., 30(д) и 30(е).

Альтернативная методика получения 5 включает обработку азаиндола 1, полученного как описано в литературе или из коммерческих источников с MeMgl и ZnCl2, с последующим добавлением ClCOCOCl (оксалилхлорида) либо в ТГФ (тетрагидрофуране), либо в этиловом эфире, что дает смесь желаемого продукта, глиоксилхлорида 6 и ацилхлорида 1, схема 4. Полученную смесь глиоксилхлорида 6 и ацилхлорида 7 затем конденсируют с монобензоилированными производными пиперазина 4 в основных условиях, что дает продукт 5 в виде смеси двух соединений (n=1 и 2).
Общие пути дальнейшей фунционализации азаиндольных колец приведены на схеме 5. Должно быть понятно, что символ RX означает, что он представляет собой общее определение остальных заместителей из R4-R2, которые находятся на азаиндольном кольце. Как показано на схеме 5, азаиндол может быть окислен до соответствующего N-оксидного производного 8 при использовании mCPBA (метахлорпербензойной кислотой) в ацетоне или ДМФА (диметилформамиде) (уравнение (ур.) 1, Harada et al., Ref.31 и Antonini et al., Ref.32). N-оксид 8 может быть превращен в различные замещенные производные азаиндола, используя хорошо известные реагенты, такие как оксихлорид фосфора (POCl3) (ур.2, Schneller et al., Ref.33(a)) или трибромид фосфора (ур.2, Wozniak et al., Ref.33(b)), реагенты Гриньяра RMgX (R=алкил, Х=Cl, Br или I) (ур.4, Shiotani et al., Ref.34), триметилсилилциамид (TMSCN) (yp.5, Minakata et al., Ref.35), Ac2O (yp.6, Klemm et al., Ref.36), тиол через тиолат натрия или другие тиолаты (ур.7, Shiotani et al., Ref.37), спирт через алкоксиды металла, как в ссылке 37 или (ур.8, Hayashida et al., Ref.38) и амин (ур.9, используя аммиак или амин в присутствии TsCl в хлорформе/воде как у Miura et al., Ref.39; или в аналогичных условиях, но 10% водным раствором NaOH, также описанным в Solekhova et al., Ref.40). В таких условиях (соответственно) атом хлора или брома, нитрильная группа, алкильная группа, гидроксильная группа, тиольная группа, апкоксигруппа и аминогруппа могут быть включены в пиридиновое кольцо. Аналогично тетраметиламмоний фторид (Me4NF) превращает N-оксиды 8 во фторазаиндолы (ур.3). Дальнейшие стандартные модификации ОН группы также хорошо обеспечивают алкоксифункцию (ур.6).


Нитрирование N-оксидов азаиндола приводит к включению нитрогруппы азаиндольное кольцо, как показано на схеме 6 (ур.10, Antonini et al., Ref.32). Нитрогруппа может быть затем замещена с помощью различных нуклефильных агентов, таких как OR, NR1R2 или SR, по хорошо известной химической схеме (ур.11, Regnouf De Vains et al., Ref.41(a), Miura et al., Ref.41(b), Profft et al., Ref.41(с)). Полученные N-оксиды 16 быстро восстанавливают до соответствующих азаиндолов 17, используя трихлорид фосфора (PCl3) (ур.12, Antonini et al., Ref.32 и Nesi et al., Ref.42) или другие восстанавливающие агенты. Аналогично нитрозамещенный N-оксид 15 может быть восстановлен до азаиндола 18, используя трихлорид фосфора (ур.13). Нитрогруппа соединения 18 может быть восстановлена до любого гидроксиламина (NHOH) (ур.14, Walser et al., Ref.43(a) и Barker et al., Ref.43(b)) или амино (NH2) группы (ур.15, Nesi et al., Ref.42 и Ayyangar et al., Ref.44) путем тщательного подбора различных условий восстановления.


Алкилирование атома азота в позиции 1 производных азаиндола может быть достигнуто, используя NaH в качестве основания, ДМФА как растворителя и алоилгалоида или сульфоната в качестве алкилирующего агента, согласно процессу, описанному в литературе (Mahadevan et al., Ref.45) (ур.16, схема 7).

Галоиды могут быть превращены в разнообразные функциональности, такие как нитрил (ур.17), аминогруппа (ур.18) и/или алкоксигруппа (ур.19) (Схема 8), используя хорошо разработанные методики. Примеры указанных типов превращений, раскрытых в равенстве, приведенном в ур.17, раскрыты в Sakamoto et al. (Ref.46(а)), где цианид меди используют для образования нитрила из галоида, у Halley et al. (Ref.46(b)), который получает нитрилы через цианид меди I в ДМФА, Yamaguchi et al. (Ref.46 (c)), Funhoff et al. (Ref.46(д)), а также используют CuCN в NMP, Shiotani et al. (Ref.37). Обычно реакция CuCN для замещения галогена требует нагревания. Температуры, такие как 145°С в течение 18 часов, как было найдено, являются предпочтительными, но эти условия могут варьироваться. Температура может быть повышена или понижена вплоть до 100°С и время реакции может варьироваться от столь небольшого как 30 минут, до столь длинного как 80 часов в зависимости от температуры реакции и субстрата. Как альтернатива уравнению 17, Klimesova et al. используют в качестве исходного первичный амид (который может быть получен из карбоновой кислоты, как широко описано) и оксихлоридфосфор для образования нитрила (Ref.47) и Katputzky et al. (Ref.48). Как показано в уравнении 18, галоиды могут быть замещены аминами или аммиаком. Некоторые примерные условия описаны в Shiotani et. al. ссылка 37 и в Katputzky et.al. ссылка 48. Например, нагревание галоида 9 в избытке первичного или вторичного амина как растворителя при температуре кипения (или между 20°С и 200°С) приводит к замещению галогена, что обеспечивает амины 27. В случае аммиака или летучих аминов может быть использован автоклав, как описано в Katputzky et al. ссылка 48, чтобы осуществлять реакцию без потери летучего амина при нагревании. Реакции могут быть отслежены с помощью ТСХ или жидкостной хроматографии, при этом температуру реакции поднимают пока осуществляется взаимодействие. Поскольку амин дорог, можно применять диоксаны или пиридины в качестве сорастворителей. Альтернативная методика применяет модифицированный метод палладиевого катализа по Hartwig (Yale) или Buchwald (MIT), чтобы вызвать замещения.в мягких условиях. Как показано в ур.19 на схеме 8, алкоксиды могут быть использованы для замещения галогенов в 9, чтобы обеспечить эфиры 26. Обычно указанное превращение лучшие осуществлять при добавлении натрия к раствору родственного спирта, чтобы образовать алканоат. В качестве альтернативы можно использовать сильное основание, такое как NaH, или NaN(SiMe3)2. Соответствующие литиевые или калийные основания, а также металлы также могут быть использованы. Обычно используют избыток основания относительно галогена, который должен быть замещен. Кроме того, обычно используют от двух до двадцати эквивалентов алконоата при десяти предпочтительных. Реакцию осуществляют при кипении с обратным холодильником или при температуре от 30°С до 200°С. Как правило, приблизительно 80°С являются оптимальными. Реакция протекает в течение от четырех до восьми часов до ее полного завершения, обычно от 12 до 48 часов. Как описано выше в ур.18, реакцию можно отслеживать. Обычные условия для замещения с помощью метилата натрия в метаноле раскрыты в Shiotani et al. ссылка 37, в общей методике, используемой для получения продуктов примеров 5а, 5с и 6 ссылки.
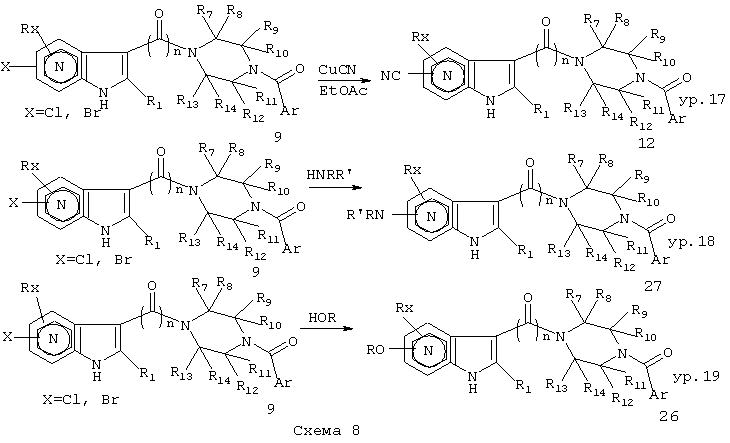
Нитрильная группа может быть превращена в карбоновую кислоту 28 (ур.20) при использовании водного раствора гидроксида натрия в этаноле по Miletin et al., Ref.49(а); или, используя КОН в водном растворе этанола по Shiotani et al., Ref.49(b); или, используя 6N HCl как в El Hadri et al., Ref.49(с)). Нитрильная группа может быть превращена в сложный эфир 29 (ур.21), используя метоксид натрия в метаноле по Heirtzler et al., Ref.50(а); или, используя HCl в метаноле по Norrby et al., Ref.50(b)). Нитрильная группа может быть превращена в амид 30 (ур.22), используя серную кислоту по Sitsun'Van et al., Ref.51(а); или, используя уксусную кислоту, трет-бутанол, серную кислоту и ацетонитрил по Reich et al., 51(b); или, используя MeOS(O)2F no Salfetnikova et al., 51(с)).

На схема 10 метильная группа на пиридиновом кольце может быть также окислена до карбоновой кислоты 28 при использовании К2Cr2O7 в 98% серной кислоте как в ур.23, Oki et al., Ref.52(а); или, используя триоксид хрома в конц. серной кислоте по Garelli et al., Ref.52(b); или, используя диоксид селена в пиридине по Koyama et al., Ref.52(с)). Карбоновая кислота может быть превращена в сложный эфир 29 при использовании HCl в 10% метаноле как в ур.24, Yasuda et al., Ref.53(а); или, используя тионилхлорида, а затем алкилалкоксид натрия по Levine et al., 53(b); или, используя спирт и РуВОР в NMM, DMAP и ДМФА по Hoemann, 53(с)).)). Карбоновая кислота может быть превращена в амид 30 при использовании водного раствора КОН с последующим оксалилхлоридом в бензоле, а затем триэтиламином в дихлорметане по (ур.25, Norman et al., Ref.54(а); или с помощью нагревания амина с кислотой по Jursic et al., 54(b); или путем присоединения амина к кислоте с помощью N,N-карбонилдиимидазола Strekowski et al., 54(с); или используя оксалилхлорид в диэтилэтиловом эфире и амин по Shi et al., 54(д)).

Альтернативная стратегия для синтеза соединения, содержащего различные заместители Аг, показана на схеме 11. Бензамидная часть диамида 5 может быть селективно гидролизована, что используют для получения промежуточного 31. Конденсация амина 31 с другими карбоновами кислотами в присутствии DEBPT и основания при использовании условий, описанных выше, для ранее описанных конденсаций обеспечивает другие новые диамиды 5.


Получение соединения 35, представленное на схеме 12, осуществляют из коммерчески доступного 32, как описано у dark, G. J.. ссылка 56. Методика Бартоли, описанная на схеме 1, использована для приготовления 4-метокси-6-азаиндола 36. Восстановление бромидов при использовании трансферного гидрирования обеспечивает желаемый 4-метоксииндол 37. Соединение 36 может быть превращено в разделяемую смесь монобромидов через селективный обмен бромида лития, используя трет-Buli при низких температурах, лежащих между от -100 до -78°, с последующим захолаживанием с помощью хлорида аммония.
Альтернативная методика, описанная на схеме 3 для ацилирования хлорметилоксалата в 3-положении, была применена к 37, что обеспечивает промежуточное 38. Затем следует осуществление методики на схеме 3, что обеспечивает соединение 39. Поскольку методика, описанная на схеме 12, является предпочтительным путем, по которому получают соединение 39 и другие соединения формулы I, был разработан альтернативный путь, приведенный на схеме 13 для получения таких соединений. Пиррол 40 получают по методике, описанной у Anderson, Н. J., ссылка 57; гидролиз эфира 40 осуществляют, используя стандартные условия, такие как гидроксид калия в этаноле при температуре окружающей среды в течение ~2 часов или пока не завершится образование 2-пирролкарбоксальдегид-4-оксоацетата калия. Раствор указанной карбоксилатной соли, гидрохлорида N-бензоилпиперазина, 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3Н)-она и триэтиламина в ДМФА перемешивают в течение приблизительно одного дня или пока не завершится реакция, что обеспечивает после выделения и крсталлизации амид 41. Амид/альдегид 41 перемешивают в виде взвеси в EtOH короткое время от 1 до 60 мин, охлаждают до 0°С (или до температуры между -15 и 20°), а затем перемешивают с гидрохлоридом глицинметилового эфира, триэтиламином (или альтернативно основанием Ханига, 2,6-лутидином или без основания) и цианборгидридом натрия, чтобы получить амин 42. Указанное превращение может быть выполнено, используя альдегид 41, гидрохлорид глицин метилового эфира и триацетоксиборгидрид натрия либо в дихлорметане, тетрагидрофуране или C1-C4 спирте как растворителе. Альтернативно свободное основание глицинметилового эфира может быть замещено по другой методике и дегидратирующий агент, такой как молекулярные сита, может быть использован в реакции перед добавлением боргидрида как восстанавливающего агента. Альтернативно указанное превращение может быть выполнено при осуществлении вначале защиты пиррольного азота бензоилом (из бензоилхлорида и третичного амина) или бензильного остатка (бензилбромида, NaH или DBU в ТГФ). Защитные группы могут быть удалены при желании, используя соответственно гидролиз водным раствором основания или гидрирование соответственно. Метиловый эфир 42 гидролизуют, используя карбонат калия в метаноле, чтобы получить после подкисления с HCl соответствующую карбоновую кислоту. Кислоту помещают в безводную метансульфоновую кислоту, содержащую пентоксид фосфора, который заранее готовят за период от 15 до 40 минут и нагревают приблизительно до 110° (обычно между 90 и 150°) в течение короткого времени приблизительно 15 минут, но обычно менее чем час и затем выливают на лед. Ацилирование или бензоилирование продукта при использовании, например, модифицированных условий реакции Шоттен-Баумана (дихлорметан, карбонат калия и бензоилхлорид) обеспечивает кетон 43. Реакция с диметоксипропаном и безводной п-толуолсульфокислотой приводит к промежуточному енолу эфира, который в реакции с хлоранилом обеспечивает соединение 39. Енол эфира может быть альтернативно приготовлен, используя триметилортоацетат и сульфоновую кислоту как катализатор. Азаиндолы, такие как 39, могут быть переведены в нитрилы, которые являются разнообразными промежуточными продуктами, путем окисления до N-оксида, а затем взаимодействием с DEPC и TEA или оксихлоридом фосфора, а затем CuCN в ДМФА. Детали реакций превращения 41 в 43-45 при использовании этих условий на аналогичном субстрате описаны в ссылке 58, Suzuki, H.; Iwata, С.; Sakurai, К.; Tokumoto, К.; Takahashi, H.; Hanada, M.; Yokoyama, Y.; Murakami, Y., Tetpahedron, 1997, 53(5), 1593-1606. Необходимо отметить, что на схемах 12 и 13 4b может быть замещен любым субстратом, представленным формулой 4 на схеме 4. Также должно быть отмечено, что индолы 37, 39, 44 и 45 могут быть переработаны, используя подходящие химические реакции, которые описаны на схемах 5-11 и которые описывают общую методику функционализации азаиндолов.

DEPBT=3-(диэтоксифосфорилокси)- 1.2.3-бензотриазин-4(3Н)-он
DMP=2,2-диметоксипропан
DEPC=диэтилцианофосфонат
Схема 13
Должно быть отменчено, что 2-хлор-5-фтор-3-нитропиридин может быть приготовлен с помощью методики в примере 5В в ссылке 59 Marfat et al. Химические процессы на схемах 1 и 3 обеспечивают производное, которое соответствует общей формуле 5 и имеет 6-азакольцо и R2=F и R4=Cl. В частности, реакцией 2-хлор-5-фтор-3-нитропиридина с 3 эквивалентами винилмагнийбромида, используя обычные условия, описанные здесь, обеспечивают 4-фтор-7-хлор-6-азаиндол с высокий выходом. Добавление указанного соединения к раствору трихлорида алюминия в дихлорметане с последующим перемешиванием при температуре окружающей среды в течение 30 минут с хлорметил или хлорэтилоксалатом обеспечивает эфир. Гидролиз с помощью КОН в стандартных условиях обеспечивает соль кислоты, которая реагирует с пиперазинами 4 (например, 1-бензоилпиперазином) в присутствии DEPBT в стандартных условиях, описанных здесь, что обеспечивает соединение 5, описанное выше. Соединение, содержащее бензоилпиперазин представляет собой N-(бензоил)-N'-[(4-фтор-7-хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазин и представляет собой соединение 5av. 7-хлорная часть в 5av может быть использована в методиках указанного изобретения, что обеспечивает желаемые производные, в котором R4 замещают согласно формуле изобретения. Например, экспонирование 5av с метилатом натрия в кипящем метаноле обеспечивает соединение 5aу, в котором 6-азаиндольное кольцо содержит 4-фтор- и 7-метоксизаместитель. Альтернативно 4-фтор-7-хлор-6-азаиндол может быть введен в реакцию с метилатом натрия, а затем проведен через последовательности, как описано выше, чтобы получить N-(бензоил)-N'-[(4-фтор-7-метокси-6-азаиндол-3-ил)-оксоацетил]-пиперазин, 5ау. 4-Фтор-7-хлор-6-азаиндол может также быть введен в реакцию с CuCN/ДМФА, как описано в ур.17, чтобы получить 7-цианопромежуточное содержание, которое может быть гидролизовано до кислоты, как описано в ур.21 схема 9, используя HCl в СН3ОН при комнатной температуре в течение 12 часов, с последующим нагреванием с обратным холодильником для завершения реакции. Кислота может быть превращена в мягких условиях в метиловый эфир добавлением диазометана в этиловом эфире к перемешиваемому раствору кислоты в диазометане при температуре окружающей среды или при более низкой температуре. Эти условия являются стандартными для использования диазометана, который приемлемо получают в виде раствора в диэтиловом эфире er Diazald®, в соответствии с инструкциями к набору от Aldrich Chemical Co. Метиловый эфир может быть проведен через ацилирование, используя оксалилхлорида, как показано в схема 4, а затем сконденсирован с пиперазином (бензоилпиперазином, например), чтобы получить соответствующий 4-фтор-7-карбометокси-6-азаиндол, который при добавлении к раствору метиламина в воде обеспечивает 5az, а именно N-(бензоил)-N'-[(4-фтор-7-(N-метилкарбоксамидо)-6-азаиндол-3-ил)-оксоацетил]-пиперазин. Такие же химические последовательности описанные выше для 4-фтор-7-хлориндола, могут быть выполнены, используя 7-хлор-4-азаиндол и (R)-3-метил-N-бензоилпиперазин 4а, что обеспечивает 5abc, которое представляет собой (R)-N-(бензоил)-3-метил-N'-[(7-метокси-4-азаиндол-3-ил)-оксоацетил]-пиперазин или 5abd, который представляет собой (R)-N-(бензоил)-3-метил-N'-[(7-(N-метикарбоксамидо)-4-азаиндол-3-ил)-оксоацетил]-пиперазин. Исходный 7-хлор-4-азаиндол представляет собой соединение 11 и его приготовление описано, например, в экпериментальной части.
Должно быть понятно, что в дополнение к соединениям 5a-5abd соединения 8, 11-30, 39, 44 и 45 являются соединениями формулы I, входят в область изобретения.
Детальное описание многих методик получения аналогов пиперазина для указанного изобретения и условия для проведения их реакций, описаны в РСТ WO 00/76521, опубликованного December 21, 2000.
В общих методиках замещения азаиндольного кольца, описанных выше, каждый способ может быть применен несколько раз и допустимы комбинации процессов для получения азаиндолов, содержащих различные заместители. Применение указанных процессов обеспечивает другие соединения формулы I.
Антивирусная активность
Антивирусная активность соединений была определена на HeLa CD4 CCR5 клетках, инфицированных монофекций, вызываемой ВИЧ-1 репортерным вирусом в присутствии соединения при концентрациях ≤10 мкМ. Вирусная инфекция оценивалась в течение 3 дней после заражения путем определения экспрессии люциферазы из интегрированной вирусной ДНК вируса в инфицированных клетках (Chen et al., Ref.55). Процент ингибирования для каждого соединения высчитывается путем определения уровня экспрессии люциферазы в клетках, инфицированных в присутствии каждого соединения как процент указанных клеток, исследуемых к клеткам, инфецированным в отсутствии соединения и вычитанием таким образом определенной величины из 100. Соединения, которые демонстрируют антивирусную активность без заметной токсичности при концентрации ≤10 мкМ, представлены в Таблице 4.
Таблица 4

| Соединение # | n | R7-14 | Средний % ингибирования при или ≤10 мкМ |
| 5а | 2 | R7-13=H, R14=(R)-Me | >99% |
| 5b | 2 | R7-8=R10-14=H, R9=Et | 90% |
| 5c | 1 | R7-8=R10-14-H, R9=Et | 80% |
| 5d | 2 | R7-14=H | 98% |
| 5е | 2 | R7-8=R10-14=H, R9=Me | 80% |
| 5f | 2 | R7·13=H, R14=(S)-Me | 80% |
| 5g | 2 | R7·13=H, R14=Et | 70% |
| 5h | 2 | R7-12=H, R13=R14=Me | 80% |
| 5i | 2 | R7-8=R10-13=H, R9=R14=Me | 89% |

| Соединение # | R | R14 | Средний % ингибирования при или ≤10 мкМ |
| 5j | Н | Н | 90% |
| 5k | Н | (R)-Me | >99% |

| Соединение # | R | R14 | Средний % ингибирования при или ≤10 мкМ |
| 5l | Н | (R)-Ме | >99% |

| Соединение # | R | R14 | Средний % ингибирования при или ≤10 мкМ |
| 5n | Н | (R)-Ме | 93% |

| Соединение # | Средний % ингибирования при или ≤10 мкМ |
| 5m | 60% |

| Соединение # | R2 | Средний % ингибирования при или ≤10 мкМ |
| 8а | H | 90% |
| 15а | NO2 | 70% |
| 16а | Оме | >99% |
| 16d | OEt | 88% |
| 16е | SPr | 50% |

| Соед. # | R2 | R4 | R14 | Средний % ингибирования при или ≤10 мкМ |
| 9а | Cl | H | (R)-Me | >99% |
| 9b | Н | Cl | (R)-Me | >99% |
| 10а | NO2 | F | (R)-Me | >99% |
| 11а | H (когда R4=Me), Me (когда R4=H) | Me (когда R2=H), H (когда R2=Me) | (R)-Me | 99% |
| 11b | H (когда R4=Ph), Ph (когда R4=H) | Ph (когда R2=H), H (когда R2=Ph) | (R)-Me | 85% |
| 11с | H (когда R4=винил), Винил (когда R4=H) | Винил (когда R2=H), H (когда R2=винил) | (R)-Me | 48% |
| 12a | H | CN | (R)-Me | >99% |
| 14a | H | ОН | (R)-Me | >99% |
| 17a | OMe | H | (H)-Me | >99% |
| 17d | OMe | H | (S)-Me | 98% |
| 17e | OMe | H | Me | 94% |
| 17b | ОСН2CF3 | H | (R)-Me | 99% |
| 17C | 0-i-P | H | (R)-Me | >99% |
| 18a | NO2 | H | (R)-Me | 80% |
| 19a | NHOH | H | (R)-Me | 98% |
| 20a | NH2 | H | (R)-Me | 95% |
| 17f | H | PrS | (R)-Me | >99% |

| Соединение # | Средний % ингибирования при или ≤10 мкМ |
| 13а | >99% |

| Соединение # | R | Средний % ингибирования при или ≤10 |
| 21а | Me | 70% |
| 21b | -СН2-СН=СН2 | 95% |

| Соед. # | R | R14 | Средний % ингибирования при или ≤10 мкМ |
| 5р | Н | Н | 40% |
| 5r | Н | (R)-Me | >99% |
| 5s | Н | (S)-Me | 56% |
| 5q | Н | Me | 97% |
| 5t | Cl | Н | >99% |
| 5u | Cl | (R)-Me | 99% |
| 5v | Ome | (R)-Me | >99% |
| 27c | Nme2 | (R)-Me | 63% |

| Соединение # | Средний % ингибирования при или ≤10 мкМ |
| 8b | 91% |

| Соединение # | R4 | R | Средний % ингибирования при или ≤10 мкМ |
| 5w | Н | Н | 98% |
| 5х | Me | Н | 99% |
| 5У | Cl | Н | >99% |
| 5z | ОМе | Me | 97% |
Экперементальные методики
Биология
«μM» означает микромоль; мкМ
«мл» или «мл» означает миллилитр;
«μl», означает микролитр; мкл
«mg» означает миллиграмм;
«nM» означает наномоль данных, представленных в виде средних значений
«а» имеет отношение к проценту ингибирования, при этом представленные данные означают значение по крайней мере двух экспериментов с дублированием значений для каждого эксперемента.
Материалы и экпериментальные методы используют, чтобы получить результаты переданные в Таблице 4, как описано ниже.
В Таблице 4 и далее применяются следующие определения:
Клетки:
• Получение вируса - клеточная линия эмбриональной почки человека, 293, размноженная в Dulbecco's Modified Eagle среде (Life Technologies, Gaithersburg, MD), содержащей 10% эмбриональной бычьей сыворотки (FBS, Sigma, St. Louis, МО).
• Вирусная инфекция - эпителиальная клеточная линия человека, HeLa, экспрессирующая ВИЧ-1 рецепторы CD4 и CCD5, размноженная в Dulbecco's Modified Eagle среде (Life Technologies, Gaithersburg, MD), содержащей 10% эмбриональной бычьей сыворотки (FBS, Sigma, St. Louis, МО) с добавлением 0.2 мг/мл генетицина (Life Technologies, Gaithersburg, MD) и 0.4 мг/мл зеоцина (Invitrogen, Carlsbad, CA).
Вирус - однократный инфекционный вирус-репортер, произведенный сотрансфекцией клеточной линии 293 эмбриональной почки человека с ВИЧ-1 покрытым ДНК вектором экспрессии и провирусной кДНК, содержащей покрытую делеционную мутацию и ген-репортер люциферазы, встроенный вместо ВИЧ-1 nef последовательности (Chen и др., ссылка 55). Трансфекции осуществляют с помощью lipofectAMINE PLUS реагента, как описано производителями (Life Technologies, Gaithersburg, MD).
Эксперименты
1. Соединение добавляют к HeLa CD4 CCR5 клеткам, помещенным на 96-ячеечную подложку при плотности клеток 5×104 клеток на ячейку в 100 мкл Dulbecco's Modified Eagle среды, содержащей 10% эмбриональной бычьей сыворотки в концентрации <20 мкМ.
2. Затем к клеткам на подложке добавляют 100 мкл однократного инфекционного вируса-репортера в Dulbecco's Modified Eagle среде и соединение при приблизительной численности инфекции (MOI) 0.01, обеспечивая конечный объем 200 мкл на ячейку и конечную концентрацию соединения <10 мкМ.
3. Образцы собирают через 72 часа после инфицирования.
4. Вирусную инфекцию исследуют, измеряя экспрессию люциферазы из вирусной ДНК в инфицированных клетках с использованием измерительного оборудования для гена-репортера люциферазы (Roche Molecular Biochemicals, Indianapolis, IN). Супернатанты инфицированных клеток удаляют и добавляют 50 мкл Dulbecco's Modified Eagle среды (без красного фенола) и 50 мкл измерительного реагента для люциферазы, приготовленного как описано производителем (Roche Molecular Biochemicals, Indianapolis, IN) на ячейку. Затем определяют активность люциферазы путем измерения люминесцентности, используя Wallac microbeta сцинтилляционный счетчик.
5. Рассчитывают процентное ингибирование для каждого соединения с помощью определения уровня экспрессии люциферазы в клетках, инфицированных в присутствии каждого соединения, к процентному соотношению уровня экспрессии люциферазы, измеренному для клеток, инфицированных при отсутствии соединения, и вычитают определенную величину из 100.
Способ экстраполяции % ингибирования при 10 мкМ
Данные в Таблице 4 получают, используя обычные процедуры, указанные выше, или используя следующие методы. Данные не представлены для всех соединений, так как данные для всех соединений представлены альтернативным способом в Таблице 5. Процентное ингибирование для каждого соединения рассчитывают определением уровня экспрессии люциферазы в клетках, инфицированных в присутствии каждого соединения, к процентному соотношению уровня экспрессии люциферазы, измеренному для клеток, инфицированных при отсутствии соединения и вычитая определенную величину из 100. Для соединений, тестируемых при концентрации менее чем 10 мкМ, процентное ингибирование при 10 мкМ определяют экстраполяцией, используя программу XLfit, устанавливающую кривую, Microsoft Excel программного обеспечения. Кривые получают по 10 точкам данных (% ингибирования, определенный при 10 концентрациях соединения), используя четыре параметра базовой модели (XLfit модель 205: у=А+((В-А)/(1+((С/х)D))), где А=минимум у, В=максимум у, С=logEC50, D=фактор наклона и х и у - известные значения данных. Экстраполяции осуществляют с найденными параметрами А и В.
Биологические данные, определенные как EC50
Таблица 5 представляет данные для соединений, сгруппированные по их ЕС50, которая обеспечивает дополнительный способ для сравнения антивирусной активности соединений по изобретению. Эти величины рассчитывают следующим способом. Эффективная концентрация пятидесятипроцентного ингибирования (ЕС50) рассчитывают с помощью Microsoft Excel XLfit, устанавливающую кривую, программного обеспечения. Для каждого соединения кривые получают из процентного ингибирования, рассчитанного при 10 различных концентрациях, используя четыре параметра базовой модели (модель 205).
| Таблица 5. Данные биологических испытаний, выраженные через ЕС50 |
||
| Соединения* с EC50s |
Соединения с EC50s>1 мкМ, но <5 мкМ | Соединения c EC50<1 мкМ |
| >0.4 нМ: 5 ac. | 5h, 11b, 18a, | 5а, 5b, 5c, 5d, |
| >0.5 мкМ: | 5е, 5f, 5g, 5i, 5j, | |
| 5м, 5р, 5s, | 5k, 5l, 5n, 5q, | |
| 5ab, 5ad, 5ae, | 5r, 5t, 5u, 5v, | |
| 16b, 16с, 16h, | 5w, 5x, 5y, 5z, 5ai, 5ak, | |
| 17f, 17g, 17h, | 8a, 8b, 9a, 9b, 10a, | |
| >5 мкМ: 5 af, | 11а, 12а, 13а, | |
| 5аr, 5ah, 8e, | 15а, 16а, 16d, | |
| 11c, 16e, 17g, | 17а, 17b, 17с, | |
| 17d, 17e, 19а, | ||
| 20а, 21а, 21b, | ||
| 27с, 39 | ||
*Некоторые из приведенных соединений тестируют при более низкой концентрации, чем их EC50, но они показывают некоторую способность вызывать ингибирование и таким образом они должны быть оценены для определения точной EC50 при более высокой концентрации.
Была предпринята попытка исключить соединения, которые не проявляют потенциала к ингибированию (к ним относятся те, которые не показывают некоторые возможности для ингибирования (те, которое могут иметь EC50>100 мкМ).
Химия
Все данные по жидкостной хроматографии (LC) получают на Shimadzu LC-10AS жидкостном хроматографе, используя SPD-10AV УФ-Vis детектор с масс-спектрометрическими данными (МС), определенными, используя Micromass Platform для LC в виде электроспрея.
LC/MS Метод (например, идентификация соединения)
Колонка A: YMC ODS-A S7 3.0×50 мм колонка
Колонка В: PHX-LUNA C18 4.6×30 мм колонка
Градиент: 100% Растворитель А/0% Растворитель В to 0%
Растворитель А/100% Растворитель В
Градиент по времени: 2 минуты
Время выдержки 1 минута
Скорость потока: 5 мл/мин
Длина волны детектора: 220 нм
Растворитель А: 10% СН3ОН/90% Н2О/0.1% трифторуксусной кислоты
Растворитель В: 10% H2O/90% СН3ОН/0.1% трифторуксусной кислоты
Соединения, очищенные препаративной HPLC, разбавляют в метаноле (1.2 мл) и очищают, используя следующую методику на Shimadzu LC-10A автоматизированной препаративной ВЭЖХ системе.
Методика препаративной ВЭЖХ (например, очистка соединения)
Методика очистки: Начальный градиент (30% В, 70% А) до конечного градиента (100% В, 0% А) в течение более 20 минут, выдержка 3 минуты (100% В, 0% А)
Растворитель А: 10% СН3ОН/90% H2O/0.1 % трифторуксусной кислоты.
Растворитель В: 10% H2O/90% СН3ОН/0.1 % трифторуксусной кислоты.
Колонка: YMC C18 S5 20х100 мм колонка
Длина волны детектора: 220 нм
Типичные методики и характеристики отдельных примеров
Типичная методика получения соединения по схеме 1
1) Получение Азаиндола 1

Получение азаиндола. Методика А: Получение 7-хлор-6-азаиндола 1е: 2-хлор-3-нитропиридин 22е (5.0 г) растворяют в сухом ТГФ (200 мл). Затем раствор охлаждают до -78°С, добавляют избыток винилмагнийбромида (1.0 М в ТГФ, 100 мл). Затем реакцию оставляют при -20°С на восемь часов перед захолаживанием 20%-ным NH4Cl (150 мл). Водную фазу экстрагируют EtOAc (3×150 мл). Объединенные органические слои сушат над MgSO4. После фильтрации и концентрации сырой продукт очищают на хроматографической колонке с силикагелем, что дает 1.5 г 7-хлор-6-азаиндола 1е с 31% выходом.
Ниже приведены характеристики соединений структурной формулы 1:

Соединение 1e, R=Cl, 7-хлор-6-азаиндол: 1Н ЯМР (500 мГц, CD3OD) δ 7.84 (д, 1Н, J=7.95 Гц), 7.76 (м, 2Н), 6.61 (д, 1Н, J=5.45 Гц). MS м/е: (М+Н)+ вычислено для C7H6ClN2: 153.02; найдено 152.93; HPLC (высокоэффективная жидкостная хроматография - ВЭЖХ) время задержки: 0.51 минут (колонка А).
Соединение 1f, R=ОМе, 7-Метокси-6-азаиндол: MS м/е: (М+Н)+ вычислено для C8H9SN2O: 149.07; найдено 149.00. ВЭЖХ время задержки: 0.42 минут (колонка А).
Характеристики соединений 1, имеющих следующую структуру, полученные с помощью методики, описанной выше:

Соединение 1g, R2=H, R4=Me, 7-Метил-4-азаиндол: MS м/е: (М+Н)+ вычислено для C8H9N2: 133.08; найдено 133.01. ВЭЖХ время задержки: 0.34 минут (колонка А).
Соединение 1ak, R2=Cl, R4=Me, 5-Хлор-7-метил-4-азаиндол: MS м/е: (М+Н)+ вычислено для C8H8ClN2: 167.04; найдено 166.99. ВЭЖХ время задержки: 1.22 минут (колонка В).

Получение азаиндола, Способ А: Получение 7-Бензилокси-4-азаиндол 1j: К раствору бензилового спирта (16.6 г) в 200 мл ДМФА медленно добавляют NaH (4.8 г). Смесь перемешивают при комнатной температуре в течение 2 часов, что дает бензоксид натрия, который переносят в раствор гидрохлорида 4-хлор-3-нитропиридина 22j (20 г) в ДМФА (100 мл). Полученную смесь выдерживают при перемешивании в течение 10 часов перед захолаживанием водой. Затем удаляют ДМФА под вакуумом, сырой продукт суспендируют в воде и экстрагируют EtOAc (3×250 мл). Органическую фазу сушат над MgSO4 и концентрируют с образованием остатка, который очищают путем перекристаллизации, что дает 6.1 г 4-бензокси-3-нитропиридина 22j.
Характеристика соединения 22j:
4-Бензилокси-3-нитропиридин: MS м/е: (М+Н)+ вычислено для С12Н11N2О3: 231.08; найдено 231.06. ВЭЖХ время задержки: 1.46 минут (колонка А).
Получение соединения 1j, 7-бензокси-4-азаиндол: Общая процедура и условия описаны для реакции по Бартоли, которую используют, чтобы получить соединение 1е, которое приедено ниже.
Характеристика соединения 1j:
Соединение 1j, 7-бензилокси-4-азаиндол: 1Н ЯМР (500 мгц, CDCl3) δ 8.64 (уширенный, 1Н), 8.34 (д, 1H, J=5.35 Гц), 7.40 (м, 6Н), 6.72 (д, 1Н, J=3.25 Гц), 6.67 (д. 1Н, J=5.45 Гц), 5.35 (с.2Н); 13С ЯМР (125 мГц, CDCl3) δ 151.1, 147.9, 145.2, 135.8, 128.8, 128.6, 127.9, 126.3, 119.6, 103.9, 99.6, 70.2. MS м/е: (М+Н)+ вычислено для С14Н13N2O: 225.10; найдено 225.03. ВЭЖХ время задержки: 1.11 минут (колонка А).
Получение азаиндола, типичный пример способа В: Получение 7-хлор-4-азаиндола 1i:

Избыток SnCl2 (25 г) осторожно добавляют в раствор гидрохлорида 4-хлор-3-нитропиридина (5 г) в концентрированной HCl и реакционную массу перемешивают в течение 12 часов. Концентрирование смеси под давлением приводит к смеси, которую затем нейтрализуют 2N NaOH до рН 6-7. Водную фазу экстрагируют EtOAc (5×100 мл). Органические слои затем объединяют, сушат над безводным растворм MgSO4 и концентрируют в вакууме, что дает сырой продукт (2.2 г), являющийся 4-хлор-3-нитропиридином, который достаточно чист для непосредственного применения в дальнейшей реакции.
7 г Сырого продукта из предыдущей стадии растворяют в 200 мл ТФК (трифторуксусной кислотой). Затем в перемешиваемый раствор осторожно добавляют 10.7 г NBS. После 8 часов растворитель удаляют в вакууме. Остаток растворяют в 2N NaOH (200 мл) и слой водного раствора экстрагируют EtOAc (3×200 мл). Объединенные органические слои сушат над MgSO4 и концентрируют, что дает сырой продукт, который затем очищают через перекристаллизацию в гексане, что дает 5 г 3-амино-2-бром-4-хлорлиридина.
Характеристика 3-амино-2-бром-4-хлорпиридина: MS м/е: (М+Н)+ вычислено для C5H5BrClN2: 206.93; найдено 206.86. ВЭЖХ время задержки: 1.32 минут (колонка В).
К раствору 3-амино-2-бром-4-хлорпиридина в 250 мл этилового эфира добавляют 8.4 г трифторуксусного ангидрида при 0°С. Через 10 минут добавляют 5.3 г Na2CO3 и реакционную смесь перемешивают при комнатной температуре в течение 10 часов, затем реакционную массу захолаживают водой (100 мл). Водную фазу экстрагируют EtOAc (3×150 мл). Объединенные органические слои сушат над MgSO4 и концентрируют с образованием остатка, который очищают на хроматографической колонке с силикагелем, что дает 3.7 г соединения 23i.
Характеристика соединения 23i:
2-Бром-4-хлор-3-трифторацетаминопиридин: MS м/е: (М+Н)+ вычислено для C7H4BrClF3N2O: 302.90; найдено 302.91. ВЭЖХ время задержки: 1.48 минут (колонка В).
Смесь соединения 23i (0.9 г), триметилсилилацетилена (0.49 г), Pd Cl2(PPh3)2 (0.1 г) и Cul (0.05 г) в Et3N (1.5 мл) нагревают до 100°С в герметизированной колбе в течение 10 часов. Затем растворитель удаляют под вакуумом. Остаток распределяют между водой (10 мл) и EtOAc (10 мл). Водную фазу экстрагируют EtOAc (2×10 мл). Объединенные органические слои сушат над MgSO4 и концентрируют под вакуумом, что дает сырой продукт 24i, который используют в дальнейшей реакции без очистки.
Характеристика соединения 24i:
Соединение 24i, 4-Хлор-3-трифторацетамидо-2-(триметилсилилэтинил)пиридин: MS м/е: (М+H)+ вычислено для С7Н4BrClF3N2О: 321.04; найдено 320.99. ВЭЖХ время задержки: 1.79 минут (колонка В).
Смесь соединения 24i (0.28 г) и этилата натрия (0.30 мл) в 20 мл этанола нагревают при кипении с обратным холодильником в течение 10 часов в атмосфере азота. Затем растворитель удаляют под вакуумом, остаток очищают, используя автоматическую систему Shimadzu для препаративной ВЭЖХ, что дает соединение 1i (0.1 г).
Характеристика соединения 1i:
Соединение 1i, 7-Хлор-4-азаиндол: 1H ЯМР (500 мГц, CD3OD) δ 8.50 (д, 1Н, J=6.20 Гц), 8.10 (д, 1H, J=3.20 Гц), 7.71 (д, 1Н, J=6.30 Гц), 6.91 (д, 1Н, J=3.25 Гц). MS м/е: (М+Н)+ вычислено для C7H6ClN2: 153.02; найдено 152.90. ВЭЖХ время задержки: 0.45 минут (колонка А).
1) Получение 3-глиоксилметилового эфира азаиндола 2

Ацилирование азаиндола, способ А: Получение метил (7-азаиндол-3-ил)-оксоацетата 2а: К раствору 7-азаиндола 1а (20.0 г, 0.169 моль) в сухом CH2Cl2 (1000 мл) добавляют 62.1 мл MeMgl (3.0M в Et2O, 0.186 моль) при комнатной температуре. Полученную смесь перемешивают также при комнатной температуре в течение 1 часа перед добавлением ZnCl2 (27.7 г, 0.203 моль). Через час по каплям вспрыскивают в раствор метилхлороксоацетат (24.9 г, 0.203 моль). Затем реакционную массу перемешивают в течение 8 часов, захолаживают метанолом, после чего все растворители упаривают, остаток распределяют между этилацетатом (500 мл) и Н2О (300 мл). Водную фазу нейтрализуют насыщенным Na2СО3 до рН 6-6.5 и экстрагируют EtOAc (3×500 мл). Затем органические слои объединяют, промывают 0.1 N HCl (3×200 мл), сушат над безводньм MgSO4 и концентрируют в вакууме, что дает сырой продукт 2а (14.3 г, 41.5%), который достаточно чист для дальнейшей реакции.

Ацилирование азаиндола, способ В: Получение метил(5-азаиндол-3-ил)-оксоацетата 2b: 5-Азаиндол 1b (0.5 г, 4.2 ммоль) добавляют к суспензии AlCl3 (2.8 г, 21.0 ммоль) в CH2Cl2 (100 мл). Перемешивание продолжают при комнатной температуре в течение 1 часа, затем добавляют по каплям метилхлороксоацетат (2.5 г, 21.0 ммоль). Реакционную массу перемешивают в течение 8 часов. Затем осторожно добавляют 20 мл СН3ОН для охлаждения реакционной массы и растворители удаляют под вакуумом. Остаток твердого вещества очищают с помощью колоночной хроматографии на силикагеле (EtOAc/СН3ОН=10:1), что дает 0.6 г (70%) ацилированного продукта 2b.
Характеристика соединений 2:
Соединение 2а, Метил (7-азаиндол-3-ил)-оксоацетат: 1Н ЯМР (300 мГц, DMSO-d6) δ 8.60 (с, 1Н), 8.47 (д, 1Н, J=7.86 Гц), 8.40 (д, 1Н, J=4.71 Гц), 7.34 (дд, 1Н, J=7.86, 4.77 Гц). 3.99 (с, 3Н); 13С ЯМР (75 мГц, DMSO-d6) δ 178.7, 163.3. 149.0, 145.1, 138.8, 129.7, 119.0, 118.0, 111.2, 52.7. MS м/е: (М+Н)+ вычислено для С10H9N2O3: 205.06; найдено 205.04. ВЭЖХ время задержки: 0.94 минут (колонка А).

Соединение 2b, Метил(5-азаиндол-3-ил)-оксоацетат: 1Н ЯМР (500 мГц, CD3OD) δ 9.61 (с, 1Н), 9.02 (с, 1Н), 8.59 (д, 1H, J=6.63 Гц), 8.15 (д, 1Н, J=6.60 Гц), 4.00 (с, 3Н); 13С ЯМР (125 мГц, CD3OD) δ 178.9, 163.0, 145.6, 144.2, 138.3, 135.0, 124.7, 116.3, 112.1, 53.8. MS м/е: (М+H)+ вычислено для С10H9N2О3: 205.06; найдено 205.04. ВЭЖХ время задержки: 0.32 минут (колонка А).

Соединение 2с, Метил (6-азаиндол-3-ил)-оксоацетат: MS м/е: (М+Н)+ вычислено для С10H9N2О3: 205.06; найдено 205.14. ВЭЖХ время задержки: 0.61 минут (колонка А).

Соединение 2d, Метил (4-азаиндол-3-ил)-оксоацетат: MS м/е: (М+Н)+ вычислено для С10Н9N2О3: 205.06; найдено 204.99. ВЭЖХ время задержки: 0.34 минут (колонка А).

Соединение 2е, Метил (7-хлор-6-азаиндол-3-ил)-оксоацетат; 1Н ЯМР (500 мГц, DMSO-d6) δ 8.66 (с, 1Н), 8.17 (д, 1Н, J=5.35 Гц), 8.05 (д, 1Н, J=5.30 Гц), 3.91 (с, 3Н); 13С ЯМР (125 мГц, DMSO-d6) δ 178.4, 162.7, 141.3, 140.9, 134.6, 133.0, 130.1, 115.4, 113.0, 52.8. MS м/е: (М+Н)+ вычислено для С10Н8ClN2О3: 239.02; найдено 238.97. ВЭЖХ время задержки: 1.18 минут (колонка А).

Соединение 2f, Метил (7-метокси-6-азаиндол-3-ил)-оксоацетат: MS м/е: (M+H)+ вычислено для C11H11N2O4: 235.07; найдено 234.95. ВЭЖХ время задержки: 0.95 минут (колонка А).

Соединение 2h, Метил (7-хлор-4-азаиндол-3-ил)-оксоацетат. MS м/е: (М+Н)+ вычислено для С10Н8ClN2О3: 239.02; найдено 238.97. ВЭЖХ время задержки: 0.60 минут (колонка А).

Соединение 2i, Метил (7-гидроксил-4-азаиндол-3-ил)-оксоацетат: MS м/е: (М+Н)+ вычислено для C10H9N2O4: 221.06; найдено 220.96. ВЭЖХ время задержки: 0.76 минут (колонка А).

Соединение 2ak, Метил (5-хлор-7-метил-4-азаиндол-3-ил)-оксоацетат: MS м/е: (М+Н)+ вычислено для С11Н10ClN2О3: 253.04; найдено 252.97. ВЭЖХ время задержки: 1.48 минут (колонка В).

Получение соединения 2j, метил (7-метоксил-1-метил-4-азаиндол-3-ил)-оксоацетат: К раствору соединения 2i (27 мг) в 10 мл сухого ДМФА добавляют 4.4 мг NaH. Через 1 час добавляют 26 мг Mel и смесь перемешивают при комнатной температуре в течение 10 часов. Затем удаляют под вакуумом ДМФА, что дает сырой продукт 2j, который используют в дальнейшей реакции без очистки.
Характеристика соединений 2j:
Соединение 2j, Метил (7-метокси-1-метил-4-азаиндол-3-ил)-оксоацетат: MS м/е: (М+Н)+ вычислено для C12H13N2O4 249.09; найдено 249.33. ВЭЖХ время задержки: 0.91 минут (колонка А).
2) Получение азаиндол 3-глиоксилата калия 3

Получение (7-Азаиндол-3-ил)-оксоацетата калия 3а: Соединение 2а (43 г, 0.21 моль) и К2СО3 (56.9 г, 0.41 моль) растворяют в СН3ОН (200 мл) и Н2О (200 мл). Через 8 часов продукт 3а выпадает из раствора в осадок. Фильтрование дает 43 г соединения 3а в виде белого твердого вещества с 90.4% выходом.
Характеристика соединений 3:
Соединение 3а, (7-Азаиндол-3-ил)-оксоацетат калия: 1Н ЯМР (300 мГц, DMSO-d6) δ 8.42 (д, 1H, J=7.86 Гц), 8.26 (д, 1Н, J=4.71 Гц), 8.14 (с, 1Н), 7.18 (дд, 1Н, J=7.86, 4.71Hz); 13C ЯМР (75 мГц, DMSO-d6) δ 169.4; 148.9, 143.6, 135.1, 129.3, 118.2, 117.5, 112.9. MS м/е: (М+Н)+ соответствующей кислоты соединения 3а (3а-К+Н) вычислено для С9Н7Н2O3: 191.05; найдено 190.97. ВЭЖХ время задержки: 0.48 минут (колонка А).

Соединение 3b, (5-Азаиндол-3-ил)-оксоацетат калия: MS м/е: (M+H)+ соответствующая кислота соединения 3b (3b-К+Н) вычислено для C9H7N2O3: 191.05; найдено 191.02. ВЭЖХ время задержки: 0.13 минут (колонка А).

Соединение 3с, (6-Азаиндол-3-ил)-оксоацетат калия: MS м/е: (М+Н)+ соответствующая кислота соединения 3с (3с-К+Н) вычислено для C9H7N2O3: 191.05; найдено: 190.99. (колонка А). ВЭЖХ время задержки: 0.23 минут

Соединение 3d, (4-Азаиндол-3-ил)-оксоацетат калийя: MS м/е: (М+Н)+ соответствующая кислота соединения 3d (3d-K+H) вычислено для С9Н7N2O3: 191.05; найдено 190.87. ВЭЖХ время задержки: 0.19 минут (колонка А).

Соединение 3е, (7-Хлор-6-азаиндол-3-ил)-оксоацетат калия: MS м/е: (М+Н)+ соответствующая кислота соединения 3е (3е-К+Н)+ вычислено для С9Н6ClN2O3: 225.01; найдено 224.99. ВЭЖХ время задержки: 0.93 минут (колонка А).

Соединение 3f, (7-Метокси-6-азаиндол-3-ил)-оксоацетат калия: MS м/е; (М+Н)+ соответствующая кислота соединения 3f (3f-K+H)+ вычислено для С10Н9N2O4: 221.06; найдено 220.97. ВЭЖХ время задержки: 0.45 минут (колонка А).

Соединение 3h, (7-Хлор-4-азаиндол-3-ил)-оксоацетат калия: MS м/е: (М+Н)+ соответствующая кислота соединения 3h (3h-K+H)* вычислено для С9Н6ClN2O3: 225.01; найдено 225.27. ВЭЖХ время задержки: 0.33 минут (колонка А).

Соединение 3j, (7-Метоксил-1-метил-4-азаиндол-3-ил)-оксоацетат калия: MS м/е: (М+Н)+ соответствующая кислота соединения 3j (3j-K+H)+ вычислено для С11H11N2O4 235.07; найдено 235.01. ВЭЖХ время задержки: 0.36 минут (колонка А).

Соединение 3ak, (5-Хлор-7-метил-4-азаиндол-3-ил)-оксоацетат калия: MS м/е: (М+Н)+ соответствующая кислота соединения 3ak (3аb-К+Н)+ вычислено для C10H8ClN2O3: 239.02; найдено 238.94. ВЭЖХ время задержки: 1.24 минут (колонка В).
1) Получение азаиндолпиперазиндиамида 5
Типичная процедура получения соединения по схеме 3

Получение (R)-Н-(бензоил)-3-метил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазина 5а: 7-Азаиндол 3-глиоксилат калия 3а (25.4 г, 0.111 моль), (R)-3-метил-N-бензоилпиперазин 4а (22.7 г, 0.111 моль), 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3Н)-он (DEPBT) (33.3 г, 0.111 моль) и основание Ханига (28.6 г, 0.222 моль) смешивают в 500 мл ДМФА. Полученную смесь перемешивают при комнатной температуре в течение 8 часов.
ДМФА удаляют путем упаривания при пониженном давлении и остаток распределяют между этилацетатом (2000 мл) и 5% водным раствором Na2СО3 (2×400 мл). Водный слой экстрагируют этилацетатом (3×300 мл). Органическую фазу объединяют и сушат над безводным MgSO4. Концентрация в вакууме дает сырой продукт, который очищают с помощью колоночной хроматографии на силикагеле с EtOAc/СН3ОН (50:1), что дает 33 г продукта 5а с 81% выходом.
Типичная процедура получения соединения по схеме 4

Получение N-(бензоил)-2-этил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазина 5b и N-(бензоил)-2-этил-N'-[(7-азаиндол-3-ил)-карбонил]-пиперазина 5с: К раствору 7-азаиндола 1а (1.0 г, 8.5 ммоль) в сухом диэтиловом эфире (20 мл) добавляют 3.1 мл MeMgl (3.0M в Et2O, 9.3 ммоль) при комнатной температуре. Полученную смесь также перемешивают при комнатной температуре в течение 1 часа, а затем добавляют ZnCl2 (1М в этиловом эфире, 10.2 мл, 10.2 ммоль). Через час осторожно вспрыскивают раствор оксалилхлорида (10.7 г, 85 ммоль). Затем реакционную массу перемешивают в течение 8 часов, растворитель и избыток оксаилхлорида удаляют под вакуумом, что дает остаток, содержащий смесь 6а и 7а.
Затем остаток растворяют в сухом СН3СН (8 мл), после чего последовательно добавляют в раствор монобензоилированный пиперазин 4b (0.25 г, 1.15 ммоль) и пиридин (1 г, 12.7 ммоль). Через час растворители удаляют и остаток очищают, используя Shimadzu автоматизированную систему для препаративной ВЭЖХ, что дает соединение 5b (20 мг, 0.6%) и соединение 5с (16 мг, 0.5%).
Характеристика соединений 5, имеющих следующую общую структуру:

Соединение 5а, n=2, R7-13=H, R14=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (300 мГц, CD3OD) δ 8.57 (д, 1Н, J=5.97 Гц), 8.38 (д, 1H, J=4.20 Гц),8.27 (м, 1H), 7.47 (с, 5Н), 7.35 (т, 1H, J=5.13 Гц), 4.75-2.87 (м, 7Н), 1.31 (уширенный, 3Н); 13С ЯМР (75 мГц, CD3OD) δ 185.6, 172.0, 166.3, 148.9, 144.6, 137.0, 134.8, 130.2, 129.9, 128.4, 126.6, 118.6, 118.0, 112.2, 61.3, 50.3. 45.1, 35.5, 14.9, 13.7. MS м/е: (М+H)+ вычислено для C21H21N4O3: 377.16; найдено 377.18. ВЭЖХ время задержки: 1.21 минут (колонка А).
Соединение 5ai, n=2, R7·13=H, R14=Me, N-(бензоил)-3-метил-N'-[(7-азаиндол-3-ил)-оксоацетпл]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H21N4O3: 377.16; найдено 377.0 5.
Соединение 5b, n=2, R7·8=R10·14=H, R9=Et, N-(бензоил)-2-этил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.63 (с, 1Н), 8.40 (с, 1H), 8.25 (м, 1H), 7.42 (м, 6Н), 4.70-2.90 (м, 7Н), 1.80-0.60 (м, 5Н); 13С ЯМР (125 мГц, CD3OD) δ 186.8, 174.2, 168.3, 149.6, 145.4, 138.8, 136.9, 132.6, 131.3, 130.0, 128.0. 120.2, 117.7, 114.1, 58.4, 52.2, 47.5. 44.8, 23.0, 10.9, 10.7. MS м/е: (М+Н)+ вычислено для С22Н23Н4O3: 391.18; найдено 391.22. ВЭЖХ время задержки: 1.35 минут (колонка А).
Соединение 5с, n=1, R7·8=R10·14=H, R9=Et, N-(бензоил)-2-этил-N'-[(7-азаиндол-3-ил)-карбонил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.33(м, 2Н), 7.87 (с, 1H), 7.47 (м, 5Н), 7.33 (м, 1H), 4.74-2.90 (м, 7Н), 1.78-0.75 (м, 5Н); 13С ЯМР (125 мГц, CD3OD) δ 168.0, 164.2, 162.8, 147.0, 142.8, 136.9, 133.1, 132.8, 131.3, 130.4, 130.0, 128.6, 118.4, 110.3, 57.0, 53.4, 46.7. 24.0, 10.7. MS м/е: (M+H)+ вычислено для C21H23N4O2: 363.18; найдено 363.22. ВЭЖХ время задержки: 1.14 минут (колонка А).
Соединение 5d, n=2, R7·12=H, N-(бензоил)-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, CD3OD) δ 8.62 (с, 1H), 8.44 (с, 1H), 8.26 (с, 1H), 7.46 (с, 5Н), 7.29 (м, 1H), 3.97-3.31 (м, 8Н). MS м/е: (М+Н)+ вычислено для C20H19N4O3: 363.15; найдено 363.24. ВЭЖХ время задержки: 1.18 минут (колонка А).
Соединение 5е, n=2, R7·8=R10·14=Н, R9=Me, N-(бензоил)-2-метил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.64 (с, 1H), 8.51 (с, 1H), 8.28 (м, 1H), 7.42 (м, 6Н), 4.48-2.90 (м, 7Н), 1.26 (м, 3Н); 13С ЯМР (125 мГц, CD3OD) δ 185.3, 171.4, 166.8, 164.0, 147.9, 143.6, 137.3, 135.3, 131.2. 129.8, 128.4, 126.2, 118.6, 112.4, 49.4, 45.9, 45.6, 45.1, 40.8, 40.4, 14.1. MS м/е: (М+Н)+ вычислено для C21H21N4O3: 377.16; найдено 377.21, ВЭЖХ время задержки: 1.26 минут (колонка А).
Соединение 5f, n=2, R7·13=Н, R14=(S)-Me, (S)-N-(бензоил)-3-метил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.64 (с, 1Н), 8.39 (с.1Н), 8.26 (м, 1H), 7.44 (м, 6Н), 4.71-3.79 (м, 7Н), 1.26 (м, 3Н); 13С ЯМР (125 мГц, CD3OD) δ 185.5, 171.9, 166.0, 158.4, 147.6, 143.5, 137.2, 134.8, 131.3, 129.8, 128.3, 126.6, 118.6, 112.4, 50.3, 45.1, 41.2, 40.3, 14.9, 13.7. MS м/е: (М+H)+ вычислено для C21H21N4O3: 377.16; найдено 377.21. ВЭЖХ время задержки: 1.25 минут (колонка А).
Соединение 5g, n=2, R7·13=Н, R14=Et, N-(бензоил)-3-этил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, CD3OD) δ 8.65 (уширенный, 1Н), 8.40 (с, 1H), 8.27 (м. 1Н), 7.46 (м, 6Н), 4.73-3.00 (м, 7Н), 1.80-0.58 (м, 5Н); 13С ЯМР (125 мГц, CD3OD) δ 187.1, 173.0, 168.0, 149.2, 145.0, 138.8, 136.4, 133.0, 131.4, 129.9, 128.2, 120.2, 114.1, 57.5, 46.0, 43.0, 37.5, 23.0, 10.7. MS м/е: (М+Н)+ вычислено для С22H23N4O3: 391.18; найдено 391.20. ВЭЖХ время задержки: 1.33 минут (колонка А).
Соединение 5h, n=2, R7·12=Н, R13=R14=Me, N-(бензоил)-3,3-диметил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С22Н23Н4O3: 391.18; найдено 390.98. ВЭЖХ время задержки: 1.22 минут (колонка А).
Соединение 5i, n=2, R7·8=R10·13=Н, R9=R14=Me, транс-N-(бензоил)-2,5-диметил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, CD3OD) δ 8.58 (м, 1H), 8.37 (д, 1H, J=15.7 Гц), 8.25 (м, 1H), 7.77 (м, 1H), 7.46 (м, 5H), 5.09-3.16 (м, 6Н), 1.30 (м, 6Н). I MS м/е: (М+Н)+ вычислено для С22Н23N4O3: 391.18; найдено 391.11. ВЭЖХ время задержки: 1.22 минут;(колонка А).
Соединение 5ab, n=2, R7·9=R10·13=Н, R14=i-Pr, N-(бензоил)-3-изопропил-N'-[(7-азаиндол-3-ил)-оксоацетил]'пиперазин: MS м/е: (М+Н)+ вычислено для C23H25N4O3: 405.19; найдено 405.22. ВЭЖХ время задержки: 1.52 минут (колонка А).
Соединение 5ас, n=2, R7·8=R10·14=Н, R9=i-Pr, N-(бензоил)-2-изопропил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C23H25N4O3: 405.19; найдено 405.25. ВЭЖХ время задержки: 1.53 минут (колонка А).
Соединение 5ad, n=1, R7·8=R10·14=H, R9=i-Pr, N-(бензоил)-2-изопропил-N'-[(7-азаиндол-3-ил)-карбонил]-пиперазин: MS м/е: (М+Н)+ вычислено для C22H25N4O2: 377.20; найдено 377.23. ВЭЖХ время задержки: 1.34 минут (колонка А).
Соединение 5ае, n=2, R7·8=R10·14=H, R9=пентил, транс-N-(бензоил)-2-пентил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С25Н29Н4O3: 433.22; найдено 433.42. ВЭЖХ время задержки: 1.74 минут (колонка А).
Характеристика соединения 5, имеющего следующую общую структуру:

Когда 
Соединение 5j, R14=H, N-(пиридин-2-ил)-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.65-7.30 (м, 8Н), 4.00-3.33 (м, 8Н). MS м/е: (М+Н)+ вычислено для С19Н18N5О3: 364.14; найдено 364.08. ВЭЖХ время задержки: 0.97 минут (колонка А).
Соединение 5k, R14=(R)-Me, (R)-N-(пиридин-2-ил)-3-метил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (300 мГц, CD3OD) δ 8.67-7.38 (м, 8Н), 4.76-3.00 (м, 7Н), 1.35 (м, 3Н); 13С ЯМР (75 мГц, CD3OD) δ 186.0, 168.9, 166.6, 152.9, 148.5, 144.0, 138.7, 137.8, 131.8, 125.6, 124.0, 119.0, 112.9, 51.3, 50.9, 50.7, 46.7, 46.2, 45.7, 42.6, 42.0, 41.8, 40.8, 36.6, 35.7, 15.5, 14.2. MS м/е: (М+H)+ вычислено для С20Н20N5О3: 378.16; найдено 378.14. ВЭЖХ время задержки: 1.02 минут (колонка А).
Когда 
Соединение 5l, R14=(R)-Me, (R)-N-(5-бромфуран-2-ил)-3-метил-N'-[(7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, CD3OD) δ 8.59 (д, 1H, J=9.4 гц), 8.37 (с, 1Н), 8.26 (м, 1Н), 7.34 (д, 1Н, J=10.1 Hz), 7.06 (с, 1H), 6.59 (с, 1Н), 4.56-3.16 (м, 7Н), 1.30 (м, 3Н); 13С ЯМР (125 мГц, CD3OD) δ 187.2, 167.8, 161.0, 150.1, 149.8, 145.8, 138.7, 132.1, 127.0, 120.5, 120.2, 119.8, 114.8, 113.9, 51.8, 47.0, 42.0, 37.0, 16.6, 15.4. MS м/е: (MtH)+ вычислено для C19H18BrN4O4: 445.05; найдено 445.18. ВЭЖХ время задержки: 1.35 минут (колонка А).

Характеристика соединения 5m
Соединение 5m, (R)-N-(бензоил)-3-метил-N'-[(5-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, CD3OD) δ 9.62 (уширенный, 1Н), 8.72 (м, 1H), 8.61 (д, 1Н, J=4.5 Гц), 8.16 (д, 1H, J=5.8 Гц), 7.51 (уширенный, 6Н), 4.90-3.10 (м, 7Н), 1.35 (уширенный, 3Н). MS м/е: (M+H)+ вычислено для C21H21N4O3 377.16, найдено 377.15. ВЭЖХ время задержки: 0.89 минут (колонка А).
Характеристика соединений 5, имеющих следующую общую структуру:

Соединение 5р, Х=Н, Y=Н, N-(бензоил)-N'-[(6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С20Н19N4O3 363.15, найдено 363.09. ВЭЖХ время задержки: 0.96 минут (колонка А).
Соединение 5q, Х=Н, Y=Me, N-(бензоил)-3-метил-N'-[(6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С21H21N4О3 377,16, найдено 377.11. ВЭЖХ время задержки: 0.99 минут (колонка А).
Соединение 5g, Х=Н, Y=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С21H21N4О3 377.16, найдено 377.10. ВЭЖХ время задержки: 0.99 минут (колонка А).
Соединение 5s, X=H, Y=(S)-Me, (S)-N-(бензоил)-3-метил-N'-[(6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H21N4O3 377.16, найдено 377.10. ВЭЖХ время задержки: 1.00 минут (колонка А).
Соединение 5t, Х=Cl, Y=H, N-(Бензоил)-N'-[(7-Хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C20H18ClN4O3 397.11, найдено 397.26. ВЭЖХ время задержки: 1.60 минут (колонка В).
Соединение 5u, Х=Cl, Y=(R)-Me, (R)-N-(Бензоил)-3-метил-N'-[(7-Хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H20ClN4O3 411.12, найдено 411.16. ВЭЖХ время задержки: 1.43 минут (колонка А).
Соединение 5v, Х=ОМе, Y=(R)-Me, (R)-N-(Бензоил)-3-метил-N'-[(7-метокси-6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H20ClN4O3 407.17, найдено 407.13. ВЭЖХ время задержки: 1.31 минут (колонка А).
Характеристика соединений 5, имеющих следующую общую структуру:

Соединение 5w, Х=H, Y=(R)-Me, Z=H, (R)-N-(Бензоил)-3-метил-N'-[(4-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H21N4O3 377.16, найдено 377.14. ВЭЖХ время задержки: 0.96 минут (колонка А).
Соединение 5х, Х=СН3, Y=(R)-Me, Z=H, (R)-N-(Бензоил)-3-метил-N'-[(7-метил-4-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С21Н21N4O3 391.18, найдено 391.15. ВЭЖХ время задержки: 1.15 минут (колонка А).
Соединение 5у, Х=Cl, Y=(R)-Me, Z=H, (R)-N-(Бензоил)-3-метил-N'-[(7-Хлор-4-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С21С20ClN4О3 411.12, найдено 411.04. ВЭЖХ время задержки: 1.10 минут (колонка А).
Соединение 5z, Х=ОМе, Y=(R)-Me, Z=Me, (R)-N-(Бензоил)-3-метил-N'-[(7-метокси-1-метил-4-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C23H25N4O4: 421.19, найдено 421.05. ВЭЖХ время задержки: 1.06 минут (колонка А).

Соединение 5ak, (R)-N-(Бензоил)-3-метил-N'-[(5-хлор-7-метил-4-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С22Н22ClN4O3 425.24, найдено 425.04. ВЭЖХ время задержки: 1.72 минут (колонка В).
Типичная процедура для получения соединений по схемам 5, 6 и 7
1) N-оксид образованием (уравнение 1, Схема 5)

Получение (R)-N-(бензоил)-3-метил-N'-[(7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазина 8а: 10 г 7-азаиндолпиперазиндиамида 5a (26.6 ммоль) растворяют в 250 мл ацетона. Затем добавляют в раствор 9.17 г mCPBA (53.1 ммоль). Продукт 8а выпадает в осадок из раствора в виде белого твердого вещества через 8 часов, после чего его собирают фильтрованием. После высушивания в вакууме получают 9.5 г соединения 8а с 91% выходом. Дальнейшая очистка не требуется.
Характеристика соединений 8, имеющих следующую общую структуру:

Соединение 8а, R=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (300 мГц, DMSO-d6) δ 8.30 (д, 1Н, J=12.2 Гц), 8.26 (д, 1Н, J=10.1 Гц), 8.00 (д, 1H, J=7.41 Гц), 7.41 (с, 5Н), 7.29 (м, 1Н), 4.57-2.80 (м, 7Н), 1.19 (уширенный, 3Н); 13С ЯМР (75 мГц, DMSO-d6) δ 186.2, 170.0, 165.0, 139.5, 136.9, 136.7, 135.5, 133.5, 129.7, 128.5, 126.9, 121.6, 119.9, 113.6, 49.4, 44.3, 15.9, 14.8. MS м/в: (М+Н)+ вычислено для C21H21N4O4: 393.16; найдено 393.16. ВЭЖХ время задержки: 1.05 минут (колонка А).
Соединение 8е, R=Н, N-(бензоил)-N'-[(7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C20H19N4O4: 379.14; найдено 379.02. ВЭЖХ время задержки: 1.15 минут (колонка А).
Соединение 8с, R=(S)-Me, (S)-N-(бензоил)-3-метил-N'-[(7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин. MS м/е: (М+Н)+ вычислено для C21H21N4O4: 393.16; найдено 393.05.
Соединение 8d, R=Me, N-(бензоил)-3-метил-N'-[(7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H21N4O4: 393.16; найдено 393.05.
Характеристика соединения 8b

Соединение 8b, (R)-N-(бензоил)-3-метил-N'-[(6-оксид-6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (M+H)+ вычислено для C21H21N4O4: 393.16; найдено 393.08. ВЭЖХ время задержки: 1.06 минут (колонка А).
Хлорирование (уравнение 2, Схема 5)

Получение (R)-N-(бензоил)-3-метил-N'-[(4-хлор-7-азаиндол-3-ил)-оксоацетил]-пиперазина 9а: 55 мг N-оксида 7-азаиндолпиперазиндиамида (0.14 ммоль) 8а растворяют в 5 мл POCl3. Реакционную смесь нагревают до 60°С в течение 4 часов. После охлаждения смесь выливают на охлажденный льдом насыщенный раствор NaHCO3 и водную фазу экстрагируют EtOAc (3×50 мл). Объединенные органические слои сушат над MgSO4 и концентрируют в вакууме. Сырой продукт очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает соединение 9а (15 мг, 26%).
Характеристика соединения 9а:
Соединение 9а, (R)-N-(бензоил)-3-метил-N'-[(4-хлор-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, DMSO-d6) 5 13.27 (уширенный, 1Н), 8.46 (м, 2Н), 7.43 (м, 6Н), 5.00-2.80 (м, 7Н), 1.23 (уширенный, 3Н). MS м/е: (М+Н)+ вычислено для С21Н20ClH4O3: 411.12; найдено: 411.09. ВЭЖХ время задержки: 1.32 минут (колонка А).
Нитрование N-оксида (уравнение 10, Схема 6)

Получение (R)-N-(бензоил)-3-метил-N'-[(4-нитро-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазина 15а: N-оксид 8а (10.8 г, 27.6 ммоль) растворяют в 200 мл трифторуксусной кислоты и 20 мл дымящейся азотной кислоты. Реакционную смесь перемешивают в течение 8 часов и захолаживают метанолом, затем фильтруют, фильтрат концентрируют в вакууме, что дает сырой продукт 15а в виде коричневого твердого вещества, которое переносят на следующую стадию без дальнейшей очистки. Небольшое количество сырого продукта очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает 3 мг вещества 15а.
Характеристика соединений 15, имеющих следующую общую структуру:

Соединение 15а, R=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-нитро-7-оксид-7-азаиндол-3-ип)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H20N5O6: 438.14; найдено 438.07. ВЭЖХ время задержки: 1.18 минут (колонка А).
Соединение 15b, R=(S)-Me, (S)-N-(бензоил)-3-метил-N'-[(4-нитро-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H20N5O6: 438.14; найдено 438.02. ВЭЖХ время задержки: 1.18 минут (колонка А).
Соединение 15с, R=Me, N-(бензоил)-3-метил-N'-[(4-нитро-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H20NsO6: 438.14; найдено 438.02. ВЭЖХ время задержки: 1.18 минут (колонка А).
4) Фторирование (уравнение 5, Схема 3)

Получение (R)-N-(бензоил)-3-метил-N'-[(4-нитро-6-фтор-7-азаиндол-3-ил)-оксоацетил]-пиперазина 10a: 20 мг сырого продукта N-оксида 4-нитро-7-азаиндол пиперазиндиамида 15а и избыток Me4NF (300 мг) растворяют в 5 мл DMSO-d6. Реакционную смесь нагревают до 100°С в течение 8 часов. После охлаждения DMSO-d6 удаляют с помощью продувания азотом. Остаток распределяют между этилацетатом (10 мл) и 2N NaOH раствором (10 мл). Водную фазу экстрагируют EtOAc (2×10 мл). Органические слои объединяют и концентрируют в вакууме, что дает остаток, который в дальнейшем очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает соединение 10a (8.3 мг).
Характеристика соединения 10a:
Соединение 10a: (R)-N-(бензоил)-3-метил-N'-[(4-нитро-6-фтор-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (300 мГц, ацетон-d6) δ 8.44 (д, 1Н, J=8.24 Гц), 7.47 (с, 6Н), 4.80-3.00 (м, 7Н), 1.29 (уширенный, 3Н). MS м/е: (М+Н)+ вычислено для C21H19FN5O5: 440.14; найдено 440.14. ВЭЖХ время задержки: 1.40 минут (колонка В).
5) Алкилирование и арилирование (уравнение 4, Схема 5)

Получение (R)-N-(бензоил)-3-метил-N'-[(4 или 6)-метил-7-азаиндол-3-ил)-оксоацетил]-пиперазина 11а: Избыток MeMgl (3М в ТГФ, 0.21 мл, 0.63 ммоль) добавляют в раствор N-оксид 7-азаиндолпиперазиндиамида 8а (25 мг, 0.064 ммоль). Реакционную смесь перемешивают при комнатной температуре и затем захолаживают метанолом. Растворители удаляют в вакууме, остаток разбавляют метанолом и очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает соединение 11а (6.7 мг, 27%).
Характеристика соединений 11, имеющих следующую общую структуру:

Соединение 11а: R=Me, (R)-N-(бензоил)-3-метил-N'-[(4 или 6)-метил-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С22Н23Н4O3: 391.18; найдено 391.17. ВЭЖХ время задержки: 1.35 минут (колонка В).
Соединение 11b: R=Ph, (R)-N-(бензоил)-3-метил-N'-[(4 или 6)-фенил-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C27H2SN4O3: 453.19; найдено 454.20. ВЭЖХ время задержки: 1.46 минут (колонка В).
Соединение 11с, R=СН=СН2, (R)-N-(бензоил)-3-метил-N'-[(4 или 6)-винил-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (M+Na)+ вычислено для C23H22N4NaO3: 425.16; найдено 425.23. ВЭЖХ время задержки: 1.12 минут (колонка А).
6) Нитрильное замещение и хлорирование (уравнение 5, Схема 5).

Получение (R)-N-(бензоил)-3-метил-N'-[(6-хлор-7-азаиндол-3-ил)-оксоацетил]-пиперазина 9b и (R)-N-(бензоил)-3-метил-N'-[(6-циано-7-азаиндол-3-ил)-оксоацетил]-пиперазина 12a: N-оксид 8а (0.20 г, 0.51 ммоль) суспендируют в 20 мл сухого ТГФ, к которому добавляют TMSCN (0.3 г, 3.0 ммоль) и BzCl (0.28 г, 2.0 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов и затем нагревают при кипении с обратным холодильником в течение 5 часов. После охлаждения смесь наливают в 100 мл насыщенного раствора NaHCO3 и водную фазу экстрагируют EtOAc (3×50 мл). Органическую фазу объединяют и концентрируют в вакууме с образованием остатка, который разбавляют метанолом и очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает соединение 12а (42 мг, 20%) и соединение 9b (23 мг, 11%).
Характеристика соединений 9b и 12а:
Соединение 9b, (R)-N-(бензоил)-3-метил-N'-[(6-хлор-7-азаиндол-З-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, DMSO-d6) δ 8.39 (м, 2Н), 7.42 (м, 6Н), 5.00-2.80 (м, 7Н), 1.19 (уширенный, 3Н); 13С ЯМР (125 мГц, DMSO-d6) δ 185.8, 170.0, 165.1, 147.9, 145.1, 137.4, 135.4, 132.2, 129.5, 128.3, 126.8, 118.6. 116.1, 111.8, 49.3, 47.2, 44.2, 15.6, 14.5. MS м/е: (М+H)+ вычислено для C21H20ClN4O3: 411.12; найдено 411.09. ВЭЖХ время задержки: 1.43 минут (колонка А).
Соединение 12а, (R)-N-(бензоил)-3-метил-N'-[(6-циано-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, DMSO-d6) δ 8.67 (м, 2Н), 7.86 (с, 1Н), 7.42 (м, 5Н), 4.80-2.80 (м, 7Н), 1.22 (уширенный, 3Н); 13С ЯМР (125 мГц, DMSO-d6) δ 185.7, 170.0, 164.8, 148.5, 140.9, 135.3, 130.3, 129.5, 128.3, 126.8, 126.2, 123.0, 120.4, 118.0, 111.8, 49.4, 47.3, 44.2, 15.6, 14.5. MS м/е: (М+H)+ вычислено для С22H20N5O3: 402.16; найдено 402.13. ВЭЖХ время задержки: 1.29 минут (колонка А).
7) Гидроксилирование (уравнение 6. Схема 5)

Получение (R)-N-(бензоил)-3-метил-N'-[(1-ацетил-6-ацетокси-7-азаиндол-3-ил)-оксоацетил]-пиперазина 13а: 20 мг N-оксида 7-азаиндолпиперазиндиамида 8а растворяют в 5 мл уксусного ангидрида (Ac2O). Реакционную смесь нагревают при кипении с обратным холодильником в течение 8 часов. После охлаждения растворители удаляют в вакууме, что дает продукт 13а, который достаточно чист для дальнейшей реакции.
Характеристика соединения 13а:
Соединение 13а, (R)-N-(бензоил)-3-метил-N'-[(1-ацетил-6-ацетокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (300 мГц, ацетон-d6) δ 8.67 (м, 2Н), 7.47 (с, 5Н), 7.27 (д, 1H, J=8.34 Гц), 4.90-2.80 (м, 7Н), 2.09 (с, 6Н), 1.30 (уширенный, 3Н); 13С ЯМР (75 мГц, ацетон-d6) δ 187.0, 170.8, 169.0, 168.6, 164.9, 155.3, 136.5, 134.7, 134.2, 133.2, 130.0, 129.8, 127.5, 118.9, 115.4, 113.8, 50.3, 45.4, 41.3, 36.3, 25.5, 20.5, 16.0, 14.8. MS м/е: (M+Na)+ вычислено для С25Н24N4O6Na: 499.16; найдено 499.15. ВЭЖХ время задержки: 1.46 минут (колонка В).
Получение (R)-N-(бензоил)-3-метил-N'-[(6-гидроксил-7-азаиндол-3-ил)-оксоацетил]-пиперазина 14а: Сырой продукт 13а и избыток К2СО3 (100 мг) смешивают с СН3ОН и Н2О (1:1). Реакционную смесь перемешивают в течение 8 часов. СН3ОН удаляют в вакууме, водную фазу экстрагируют EtOAc (3×10 мл) и органические слои объединяют и концентрируют. Полученный продукт очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает 1 мг соединения 14а (5% из соединение 8а).
Характеристика соединения 14а:
Соединение 14а, (R)-N-(бензоил)-3-метил-N'-[(6-гидроксил-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (M+H)+ вычислено для C21H21N4O4: 393.16; найдено 393.12. ВЭЖХ время задержки: 1.13 минут (колонка А).
8) Образование тиола (уравнение 7, Схема 5)

Получение (R)-N-(бензоил)-3-метил-N'-[(6-пропилтио-7-азаиндол-3-ил)-оксоацетил]-пиперазина 17f. К 100 мг раствора соединения 9а в 10 мл CHCl3 добавляют TsCl (63 мг) и раствор перемешивают в течение 5 минут. Затем добавляют 2 мл пропилтиола и реакционную смесь еще перемешивают в течение 8 часов. После концентрации сырой продукт очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает 1,4 мг соединения 17f.
Характеристика соединения 17f:
Соединение 17f, (R)-N-(бензоил)-3-метил-N'-[(6-пропилтио-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С24Н27Н4O3S: 451.18; найдено 451.09. ВЭЖХ время задержки: 1.45 минут (колонка А).
9) Замещение нитрогруппы (уравнение 11, Схема 6)

Получение (R)-N-(бензоил)-3-метил-N'-[(4-метокси-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазина 16а: 100 мг сырого соединения 15а из предыдущей стадии растворяют в 6 мл 0.5М MeONa в СН3ОН. Реакционную смесь нагревают при кипении с обратным холодильником в течение 8 часов и растворитель удаляют под вакуумом, что дает смесь, содержащую продукт 16а и другие неорганические соли. Указанную смесь используют на следующей стадии без дальнейшей очистки. Небольшую порцию смеси сырых продуктов очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает 5 мг соединения 16а.
Характеристика соединения 16, имеющего следующую общую структуру:

Соединение 16а, Х=ОМе, R=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-метокси-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (M+H)+ вычислено для C22H23N4O5 423.17, найдено 423.04. ВЭЖХ время задержки: 0.97 минут (колонка А).
Соединение 16f, Х=ОМе, R=(S)-Me, (S)-N-(бензоил)-3-метил-N'-[(4-метокси-7-оксид-7~азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C22H23N4O5 423.17, найдено 423.02.
Соединение 16g, X=OMe, R=Me, N-(бензоил)-3-метил-N'-[(4-метокси-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)* вычислено для С22Н23N4O5 423.17, найдено 423.03.
Соединение 16b, Х=ОСН2CF31, R=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-(2,2,2-трифторэтокси)-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.44 (уширенный, 1Н), 8.30 (м, 1Н), 7.50 (уширенный, 5Н), 7.14 (уширенный, 1Н), 4.90-3.10 (м, 9Н), 1.30 (м, 3Н). MS м/е: (М+Н)+ вычислено для С23Н22F3N4О5: 491.15; найдено 491.16. ВЭЖХ время задержки: 1.17 минут (колонка А).
Соединение 16с, Х=ОСН(СН3)2, R=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-(1-метилэтокси)-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, CD3OD) δ 8.48 (с, 1Н), 8.24 (м, 1H), 7.46 (м, 5Н), 7.13 (с, 1Н), 5.03-3.00 (м, 8Н), 1.49-1.15 (м, 9Н). MS м/е: (М+Н)+ вычислено для C24H27N4O5: 451.20; найдено 451.21. ВЭЖХ время задержки: 1.14 минут (колонка А).
Соединение 16d, Х=ОСН2СН3, R=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4~этокси-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С23Н25N4O5: 437.18; найдено 437.13. ВЭЖХ время задержки: 1.08 минут (колонка А).
Соединение 16е Х=SCH2CH2CH3, R=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-пропилтио-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, CD3OD) δ 8.24 (м, 2Н), 7.45 (м, 5Н), 7.25 (с, 1Н), 4.90-3.00 (м, 9Н), 1.81 (уширенный, 2Н), 1.30 (м, 6Н). MS м/е: (М+Н)+ вычислено для C24H27N4O4S: 467.18; найдено 467.14. ВЭЖХ время задержки: 1.30 минут (колонка А).
Соединение 16h, Х=NHMe, R=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-метиламино-7-оксид-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C22H24N5O4: 422.18; найдено 422,09. ВЭЖХ время задержки: 1.19 минут (колонка А).
10) Восстановление N-оксида (уравнение 12, Схема 6)

Получение (R)-N-(бензоил)-3-метил-N'-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазина 17а: 48 мг сырого продукта 16а суспендируют в 30 мл этилацетата при комнатной температуре. Добавляют 1 мл PCl3 и реакционную смесь перемешивают в течение 8 часов. Затем ее осторожно выливают на лед, захолаживая раствор 2N NaOH. После выделения органического слоя водную фазу экстрагируют EtOAc (6×80 мл). Органические слои объединяют и концентрируют в вакууме, что дает остаток, который очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает 38 мг соединения 17а.
Характеристика соединений 17, имеющих следующую общую структуру:

Соединение 17а, R=Ome, Х=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (300 мГц, CD3OD) δ 8.24 (д, 1Н, J=5.7 Гц), 8.21 (м, 1Н), 7.47 (с, 5Н), 6.90 (д, 1Н, J=5.7 Гц), 4.71-3.13 (м, 10Н), 1.26 (уширенный, 3Н); 13С ЯМР (75 мГц, CD3OD) δ 185.3, 172.0, 167.2, 161.2, 150.7, 146.6, 135.5, 134.8, 129.9, 128.3, 126.7, 112.8, 106.9, 100.6, 54.9, 50.2, 48.1, 45.1, 14.5, 13.8. MS м/е: (М+Н)+ вычислено для C22H23N4O4: 407.17; найдено 407.19. ВЭЖХ время задержки: 1.00 минут (колонка А).
Соединение 17d, R=Ome, X=(S)-Me, (S)-N-(бензоил)-3-метил-N'-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C22H23N4O4: 407.17; найдено 407.03.
Соединение 17е, R=Ome, Х=Me, N-(бензоил)-3-метил-N'-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C22H23N4O4: 407.17; найдено 407.03.
Соединение 17b, R=ОСН2CF3, X=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-(2,2,2-трифторэтокси)-7-азаиндол-3-ил)-оксоацетил)-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.33 (с, 1Н), 8.19 (м, 1H), 7.45 (м, 5Н), 7.05 (с, 1Н), 4.90-3.00 (м, 9Н), 1.29 (уширенный, 3Н); 13С ЯМР (125 мГц, CD3OD) δ 185.7, 174.0, 168.3, 162.0, 151.0, 146.1, 138.5, 136.4, 131.4, 130.0, 128.2, 114.8, 109.5, 103.6, 67.2, 66.9, 52.0, 47.0, 16.4, 15.3. MS м/е: (М+Н)+ вычислено для С23Н22F3Н4O4: 475.16; найдено 475.23. ВЭЖХ время задержки: 1.22 минут (колонка А).
Соединение 17с, R=ОСН(СН3)2, Х=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-(1-метилэтокси)-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1Н ЯМР (500 мГц, CD3OD) δ 8.42 (с, 1H), 8.24 (м, 1H), 7.47 (м, 5Н), 7.21 (с, 1H), 5.20-3.00 (м, 8Н), 1.51 (уширенный, 6Н), 1.22 (уширенный, 3Н); 13С ЯМР (125 мГц, CD3OD) δ 185.4, 173.6, 167.9, 166.1, 145.3, 141.4, 138.2, 136.4, 131.5, 129.7, 128.2, 113.9, 111.4, 104.0, 75.5, 54.4, 53.7, 51.8, 46.9, 22.1, 16.4, 15.3. MS м/е: (M+H)+ вычислено для С24Н27N4O4: 435.20; найдено 435.20. ВЭЖХ время задержки: 1.15 минут (колонка А).
Соединение 17м, R=ОСН2СН3, Х=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-этокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C23H25N4O4: 421.19; найдено 421.13. ВЭЖХ время задержки: 1.13 минут (колонка А).
Соединение 17g, R=SCH2CH2CH3, Х=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-пропилтио-7-азаиндол-3-ил)-оксоацетил)-пиперазин: MS м/е: (М+Н)+ вычислено для C24H27N4O4S: 451.18; найдено 451.13. ВЭЖХ время задержки: 1.50 минут (колонка А).
Соединение 17h, R=NHMe, X=(R)-Me, (R)-N-(бензоил)-3-метил-N'-[(4-метиламино-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (M+H)+ вычислено для С22Н24N5О3: 406.19; найдено 406.03. ВЭЖХ время задержки: 1.19 минут (колонка А).
Характеристика соединения 18а

Соединение 18а, (R)-N-(бензоил)-3-метил-N'-[(4-нитро-7-азаиндол-3-ил)-оксоацетил]-пиперазина: 1Н ЯМР (300 мГц, CD3OD) δ 8.58 (с, 1Н), 8.53 (м, 1Н), 7.64 (с, 1Н), 7.47 (с, 5Н), 4.90-3.00 (м, 7Н), 1.30 (уширенный, 3Н); 13С ЯМР (75 мГц, CD3OD) δ 184.1, 172.1, 165.6, 151.9, 149.6, 145.5, 139.4, 134.8, 129.7, 128.4, 126.7, 111.6, 111.2, 107.4, 53.7, 48.4, 45.9, 15.0, 13.7. MS м/е: (М+Н)+ вычислено для C21H20N5O5: 422.15; найдено 422.09. ВЭЖХ время задержки: 1.49 минут (колонка В).
11) Восстановление нитрогруппы до гидроксиамина (уравнение 14, Схема 6)

Получение (R)-N-(бензоил)-3-метил-N'-[(4-гидроксиламино-7-азаиндол-3-ил)-оксоацетил]-пиперазина 19а: 10 мг Pd (10% активированного углерода) добавляют к раствору соединения 18а (48 мг, 0.11 ммоль) в метаноле (10 мл) в атмосфере водорода. Реакционную смесь перемешивают в течение 8 часов при комнатной температуре. После фильтрования фильтрат концентрируют в вакууме с образованием остатка, который очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает соединение 19а (7.9 мг, 17%).
Характеристика соединения 19а:
Соединение 19а, (R)-N-(бензоил)-3-метил-N'-[(4-гидроксиламино-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е.: (М+Н)+ вычислено для C21H22N5O4: 408.17; найдено 408.21. ВЭЖХ время задержки: 1.03 минут (колонка А).
12) Восстановление нитрогруппы до аминогруппы (уравнение 15, Схема 6)

Получение (R)-N-(бензоил)-3-метил-N'-[(4-амино-7-азаиндол-3-ил)-оксоацетил]-пиперазина 20а: 114 мг Na2S·2H2O (1 ммоль) добавляют к раствору соединения 18а (20 мг, 0.048 ммоль) в СН3ОН (5 мл) и Н2О (5 мл). Реакционную смесь нагревают при кипении с обратным холодильником в течение 8 часов. После охлаждения реакционную смесь концентрируют в вакууме с образованием остатка, который очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает 4 мг соединения 20а (21.3%).
Характеристика соединения 20а:
Соединение 20а, (R)-N-(бензоил)-3-метил-N'-[(4-амино-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.16 (м, 1Н), 8.01(д, 1Н, J=8.1 Гц), 7.47 (м, 5Н). 6.66 (с, 1Н), 4.90-3.00 (м,7Н), 1.30 (уширенный, 3Н). MS м/е: (M+H)+ вычислено для С21Н22Н5O3: 392.17; найдено 392.14. ВЭЖХ время задержки: 0.96 минут (колонка А).
13) Алкилирование атома азота в положении 1 (уравнение 16, Схема 7)

Получение (R)-N-(бензоил)-3-метил-N'-[(1-метил-7-азаиндол-3-ил)-оксоацетил]-пиперазина 21 a: NaH (2 мг, 60% чистый, 0.05 ммоль) добавляют к раствору соединения 5а (10 мг, 0.027 ммоль) в ДМФА. Через 30 минут вспрыскивают Mel (5 мг, 0.035 ммоль) в смесь с помощью шприца. Реакционную смесь перемешивают в течение 8 часов при комнатной температуре и захолаживают метанолом. Смесь распределяют между этилацетатом (2 мл) и Н2О (2 мл). Водную фазу экстрагируют EtOAc (3×2 мл). Органические слои объединяют, сушат над безводным MgSO4 и концентрируют в вакууме, что дает сырой продукт, который очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает соединение 21а (2.5 мг, 24%). Характеристика соединений 21, имеющих следующую общую структуру:

Соединение 21а, R=Me, (R)-N-(бензоил)-3-метил-N'-[(1-метил-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) δ 8.56 (уширенный, 1Н), 8.42 (с, 1H), 8.30 (м, 1Н), 7.47 (м, 6Н), 4.90-3.00 (м,7Н), 3.96 (с, 3Н), 1.28 (уширенный, 3Н). MS м/е: (M+Na)+ вычислено для C22H22N4O3Na: 413.16; найдено 413.15. ВЭЖХ время задержки: 1.47 минут (колонка В).
Соединение 21b, R=СН2-СН=СН2, (R)-N-(бензоил)-3-метил-N'-[(1-аллил-7-азаиндол-3-ил)-оксоацетил]-пиперазин: 1H ЯМР (500 мГц, CD3OD) 5 8.37 (м, 3Н), 7.44 (м, 6Н), 6.08 (м, 1H), 5.22-3.06 (м, 11H), 1.27 (м, 3Н); 13С ЯМР (75 мГц, CD3OD) δ 184.2, 184.1, 170.8, 165.0, 146.7, 143.5, 137.9, 133.8, 131.4, 129.2, 128.8, 127.3, 125.6, 117.9, 117.4, 116.3, 110.3, 50.4, 49.7, 49.1, 45.7, 44.0, 41.0, 39.6, 34.8, 14.0, 12.8,. MS м/е: (М+Н)+ вычислено для С24Н25Н4O3: 417.19; найдено 417.11. ВЭЖХ время задержки: 1.43 минут (колонка А).
14) Реакции замещения галогена (уравнение 18, Схема 8)

Получение (R)-N-(бензоил)-3-метил-N'-[(7-диметиламино-6-азаиндол-3-ил)-оксоацетил]-пиперазина 27c: Смесь соединения 5u (50 мг) и 4 мл диметиламина (40% в воде) нагревают до 150°С в герметизированной колбе в течение 18 часов. Затем растворители удаляют под вакуумом и остаток очищают, используя Shimadzu автоматизированную систему для препаративной ВЭЖХ, что дает 10 мг соединения 27с.
Характеристика соединения 27с:
Соединение 27с, (R)-N-(бензоил)-3-метил-N'-[(7-диметиламино-6-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C23H26N5O3 420.20, найдено 420.16. ВЭЖХ время задержки: 1.13 минут (колонка А).
15) Модификация бензоильного остатка (уравнение 26, Схема 11)

Гидролиз бензоиламида; получение (R)-2-метил-N-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазина 31а: Соединение 17а (0.9 г) и КОН (2.0 г) перемешивают в растворе EtOH (15 мл) и воды (15 мл). Реакционную массу нагревают при кипении с обратным холодильником в течение 48 часов. Растворители удаляют под вакуумом и полученный остаток очищают с помощью колоночной хроматографии на силикагеле (EtOAc/Et3N=от 100:1 до 3:1), что дает 0.6 г соединения 31 а.
Характеристика соединения 31а:
Соединение 31 а, (R)-2-метил-N-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для С15Н19N4О3 303.15, найдено 303.09. ВЭЖХ время задержки: 0.29 минут (колонка А).

Образование диамида: Получение (R)-N-(4-азидо-2,3,5,6-тетрафторбензоил)-3-метил-N'-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазина 5n: Амин 31а (0.15 г), 4-азидо-2,3,5,6-тетрафторбензойную кислоту (0.12 г), 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3Н)-он (DEPBT) (0.15 г) и основание Ханига (0.5 мл) смешивают в 5 мл ДМФА. Затем смесь перемешивают при комнатной температуре в течение 8 часов. После этого растворители удаляют под вакуумом и остаток очищают, используя Shimadzu автоматизированную систему для препаративной ВЭЖХ, что дает 10 мг соединения 5n.
Характеристика соединения 5n:
Соединение 5n, (R)-N-(4-азидо-2,3,5,6-тетрафторбензоил)-3-метил-N'-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C22H18F4N7O4 520.14, найдено 520.05. ВЭЖХ время задержки: 1.42 минут (колонка А).
Соединение 5af, Ar=4,5-дибромфенил, (R)-N-(3,5-дибромбензил)-3-метил-N'-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C22H21Br2N4O4 562.99, найдено 562.99. ВЭЖХ время задержки: 1.54 минут (колонка А).
Соединение 5ag, Ar=4-[3-(трифторметил)-3Н-диазирин-3-ил]фенил, (R)-N-[4-(3-(трифторметил)-3Н-диазирин-3-ил)бензил]-3-метил-N'-[(4-метокси-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)* вычислено для С24Н22F3N6O4 515.17, найдено 515.02. ВЭЖХ время задержки: 1.55 минут (колонка А).
Новое уравнение:

Получение (R)-N-(бензоил)-3-метил-N'-[(4-гидроксил-7-азаиндол-3-ил)-оксоацетил]-пиперазина 5ah: Сырой продукт соединения 17а (100 мг) и избыток TMSI (0.25 мл) перемешивают в CHCl3. Затем реакционную смесь перемешивают в течение 6 дней. Растворитель удаляют в вакууме, сырой продукт очищают, используя автоматизированную систему Shimadzu для препаративной ВЭЖХ, что дает соединение 4.4 мг структуры 5ah.
Характеристика соединения 5ah:
Соединение 5ah, (R)-N-(бензоил)-3-метил-N'-[(4-гидроксил-7-азаиндол-3-ил)-оксоацетил]-пиперазин: MS м/е: (М+Н)+ вычислено для C21H21N4O4: 393.16; найдено 393.11. ВЭЖХ время задержки: 1.46 минут (колонка В).
Альтернативные методики, пригодные для синтеза соединения 39
Получение 5,7-дибром-4-метокси-7-азаиндола 36: Винилмагнийбромид (0.85 М в ТГФ, 97.7 мл, 83.0 ммоль) добавляют в течение более 30 минут к перемешиваемому раствору 2,6-дибром-3-метокси-5-нитропиридина (7.4 г, 23.7 ммоль) в ТГФ (160 мл) при -75°С. Раствор перемешивают один час при -75°С и в течение ночи при -20°С, повторно охлаждают до -75°С и захолаживают насыщенным водным раствором NH4Cl (-100 мл). Реакционной смеси дают нагреться до комнатной температуры, промывают рассолом (-100 мл) и экстрагируют Et2O (150 мл) и СН2Cl2 (2×100 мл). Объединенную органику сушат (MgSO4), фильтруют и концентрируют. Остаток очищают с помощью флэш-колоночной хроматографии (SiO2, 3:1 гексаны/EtOAc) с выходом 5,7-дибром-4-метокси-7-азаиндола 36 (1.10 г, 3.60 ммоль, 15%) в виде бледно-желтого твердого вещества.
Характеристика 36: 1Н ЯМР (500 мГц, CDCl3) 8.73 (уширенный с, 1Н), 7.41 (дд, J=3.1, 2.8 Гц, 1Н), 6.69 (д, J=3.1, 2.2 Гц, 1Н), 4.13 (с, 3Н); 13С ЯМР (125 мГц, CDCl3) 146.6, 133.7, 128.8, 127.5, 120.2, 115.6, 101.9, 60.7. MS м/е (М+Н)+ вычислено для C8H7Br2N2O: 304.88; найдено 304.88. ВЭЖХ время задержки: 1.31 минут (колонка А).
Получение 4-метокси-7-азаиндол 37: Раствор 5,7-дибром-4-метокси-7-азаиндола 36 (680 мг, 2.22 ммоль), 5% Pd/C (350 мг, 0.17 ммоль) и гидразина (2.5 мл, 80 ммоль) в EtOH нагревают при кипении с обратным холодильником в течение 1 часа. Реакционной смеси дают остыть до комнатной температуры, фильтруют через целит и фильтрат концентрируют. Водный раствор NH4OH (11% в Н2О, 45 мл) добавляют к остатку и раствор экстрагируют CH2Cl2 (3×30 мл). Объединенную органику сушат (MgSO4), фильтруют и концентрируют с выходом 4-метокси-7-азаиндола 37 (290 мг, 1.95 ммоль, 88%) в виде оранжевого твердого вещества.
Характеристика 37: 1Н ЯМР (500 мГц, CDCl3) 8.61 (уширенный с, 1Н), 8.52 (с, 1H), 7.88 (с, 1Н), 7.30 (д, J=2.9 Гц, 1Н), 6.69 (д, J=2.9 Гц, 1Н), 4.03 (с, 3Н). MS м/е (М+Н)+ вычислено для C8H9N2O: 149.06; найдено 148.99. ВЭЖХ время задержки: 0.61 минут (колонка А).
Получение 38: Треххлористый алюминий (67 мг, 0.50 ммоль) добавляют к раствору 4-метокси-6-азаиндола (15 мг, 0.10 ммоль) в СН2Cl2 (2 мл) и перемешивают при комнатной температуре в течение 30 мин. Добавляют метилхлорацетат (0.020 мл, 0.21 ммоль) и реакционную смесь перемешивают в течение ночи. Затем реакционную массу захолаживают СН3ОН (0.20 мл), перемешивают 5 часов, и после чего фильтруют (промывая CH2Cl2). Затем фильтрат промывают насыщенным водным раствором NH4OAc (2×10 мл) и Н2О (10 мл) и концентрируют с получением 38 (5 мг) в виде желтого твердого вещества.
Характеристика 38: 1H ЯМР (500 мГц, CDCl3) 8.65 (с, 1Н), 8.36 (с, 1Н), 8.02 (с, 1Н), 4.03 (с, 3Н), 3.96 (с, 3Н). MS м/е (М+H)+ вычислено для С11Н10N2O4: 235.06; найдено 234.96. ВЭЖХ время задержки: 0.63 минут (колонка А).
Получение N-бензоил-N'-[(2-карбоксальдегидпиррол-4-ил)-оксоацетил]-пиперазина 41: Раствор этил 4-оксоацетил-2-пирролкарбоксальдегида 40 (17.0 г, 87.1 ммоль) перемешивают два часа в 25 мл КОН (3.56 М в Н2О, 88.8 ммоль) и EtOH (400 мл). Образовавшийся белый осадок собирают с помощью фильтрования, промывают EtOH (-30 мл) и Et2O (~30 мл) и сушат под высоким вакуумом с получением 15.9 г 2-пирролкарбоксальдегида-4-оксоацетата калия в виде белого твердого вещества, которое используют без дальнейшей очистки. Раствор 2-пирролкарбоксальдегида-4-оксоацетата (3.96 г, 19.3 ммоль), N-бензоилпиперазин гидрохлорида (4.54 г, 19.7 ммоль), 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3Н)-она (5.88 г, 19.7 ммоль) и триэтиламина (3.2 мл, 23 ммоль) перемешивают в течение дня в ДМФА (50 мл). Реакционную смесь фильтруют в Н2O (300 мл), экстрагируют CH2Cl2 (3×200 мл) и объединенную органику концентрируют на роторном испарителе, чтобы удалить CH2Cl2. Затем сырой продукт (все еще в ДМФА) разбавляют Н2О (200 мл) и дают ему кристаллизоваться в течение 48 часов. После этого твердое вещество собирают с помощью фильтрования и сушат под высоким вакуумом (P2O5) с получением N-бензоил-N'-[(2-карбоксальдегидапиррол-4-ил)-оксоацетил]-пиперазина 41 (3.3 г, 9.7 ммоль, 45% более двух стадий) в виде светло-желтого твердого вещества. Дальнейшая очистка не требуется.
Характеристика 41: 1H ЯМР (500 мГц, CDCl3) 9.79 (с, 1Н), 9.63 (с, 1Н), 7.82 (с, 1H), 7.51-7.34 (м, 6Н), 4.05-3.35 (м, 8Н). MS м/е (М+H)+ вычислено для C18H18N3O4: 340.12; найдено 340.11. ВЭЖХ время задержки: 1.04 минут (колонка А).
Получение 42: N-бензоил-N'-[(2-карбоксальдегидпиррол-4-ил)-оксоацетил]-пиперазин 41 (3.3 г, 9.7 ммоль) перемешивают в виде суспензии в EtOH (100 мл) в течение 15 мин, охлаждают до 0°С и после чего он вступает в реакцию с гидрохлоридом глицинметилового эфира (3.66 г, 29.2 ммоль), триэтиламином (1.50 мл, 11 ммоль) и цианборгидридом натрия (672 мг, 10.7 ммоль). Реакционной смеси дают нагреться до комнатной температуры, перемешивают 24 часа и выливают на лед (~400 мл). Раствор экстрагируют EtOAc (3×300 мл) и объединенную органику промывают рассолом (300 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Остаток очищают с помощью препаративной тонкослойной хроматографии (SiO2, 9:1 EtOAc/СН3ОН, Rf=0.2) с выходом 42 (2.4 г, 5.8 ммоль, 60%) в виде белого твердого вещества.
Характеристика 42: 1Н ЯМР (500 мГц, CDCl3) 9.33 (с, 1Н), 7.49 (с, 1H), 7.58-7.32 (м, 5Н), 6.50 (с.1Н), 3.90-3.35 (м, 8Н), 3.81 (с, 2Н), 3.74 (с, 3Н), 3.40 (с.2Н). MS м/е (М+Н)+ вычислено для C21H25N4O5: 413.17; найдено 413.17. ВЭЖХ время задержки: 0.84 минут (колонка А).
Получение 43: Метиловый эфир 42 (485 мг, 1.17 ммоль) и К2СО3 (325 мг, 2.35 ммоль) в СН3ОН (6 мл) и H2O (6 мл) перемешивают при комнатной температуре в течение трех часов. Реакционную смесь затем захолаживают концентрированной HCl (0.40 мл) и концентрируют под высоким вакуумом. Часть твердого осадка (200 мг, 0.37 ммоль) добавляют к перемешиваемому раствору Р2O5 (400 мг, 1.4 ммоль) в метансульфокислоте (4.0 г, 42 ммоль) (которая уже была перемешана вместе с этиловым эфиром при 110°С в течение 45 мин) при 110°С и перемешивают в течение 15 мин. Реакционную смесь выливают на дробленный лед (~20 г), перемешивают 1 час, подщелачивают с К2СО3 (5.0 г, 38 ммоль), разбавляют CH2Cl2 (20 мл), бензоилхлоридом (1.0 мл, 8.5 ммоль) и перемешивают 1 час. Реакционную смесь экстрагируют СН2Cl2 (3×20 мл) и объединенную органику сушат (Na2SO4) и концентрируют при пониженном давлении. Остаток очищают с помощью препаративной тонкослойной хроматографии (SiO2, EtOAc, Rf=0.5) с выходом 43 (101 мг, 0.21 ммоль, 57%) в виде грязно-белого твердого вещества.
Характеристика 43: MS м/е (М+Н)+ вычислено для C27H24N4O5: 485.17; найдено 485.07. ВЭЖХ время задержки: 1.15 минут (колонка А).
Получение 39. R=ОМе, N-(бензоил)-N'-[(4-метокси-6-азаиндол-3-ил)-оксоацетил]-пиперазин:
В колбе объемом 1 л, оборудованной ловушкой Дина-Старка, гидрат n-толуолсульфокислоты (55 мг, 0.29 ммоль) и бензол (5 мл) нагревают при кипении с обратным холодильником в течение 1 часа. Раствор охлаждают до комнатной температуры и его подвергают взаимодействию с 2,2-диметоксипропаном (0.10 мл, 0.81 ммоль) и 43 (46 мг, 0.095 ммоль). Реакционную смесь перемешивают 1 час, разбавляют CH2Cl2 (2 мл), перемешивают 30 минут, затем окисляют с помощью тетрахлорбензохинона (150 мг, 0.61 ммоль) и перемешивают в течение ночи. Реакционную смесь выливают в 5% водный раствор NaOH (20 мл) и экстрагируют CH2 Cl2 (3×25 мл). Объединенную органику сушат (Na2SO4) и концентрируют при пониженном давлении. Остаток подвергают препаративной тонкослойной хроматографии (Et2O), основную часть вещества экстрагируют и повторно подвергают препаративной тонкослойной хроматографии (SiO2, 9:1 EtOAc/СН3ОН, Rf=0.15) с получением 39 (3 мг, 0.008 ммоль, 6%) в виде белого твердого вещества.

Соединение 39, R=ОМе, N-(бензоил)-3-метил-N'-[(4-метокси-6-азаиндол-3-ил)-оксоацетил]-пиперазин:
Характеристика 39: 1Н ЯМР (500 мГц, CD3OD) 8.49 (с, 1Н), 8.35 (с, 1Н), 7.98 (с, 1Н), 7.53-7.38 (м, 5Н), 4.02 (с, 3Н), 3.97-3.42 (м, 8Н). MS м/е (М+H)+ вычислено для C21H23N4O5: 393.15; найдено 393.13. ВЭЖХ время задержки: 0.85 минут (колонка А).

Получение 5av N-(бензоил)-N'-[(4-фтор-7-хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазина и 5av' (R)-N-(бензоил)-3-метил-N'-[(4-фтор-7-хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазина
Должно быть отмечено, что 2-хлор-5-фтор-3-нитропиридин может быть приготовлен по способу, описанному в примере 5В, из ссылки 59 Marfat et al. Схема, представленная ниже, раскрывает некоторые подробности, которые улучшают выход указанного синтеза. Химический процесс по Барроли на Схеме 1 используют, чтобы получить азаиндол 1zz с приведенными ниже подробностями.

Соединение zz1' (1.2 г, 0,01 моль) растворяют в 2.7 мл серной кислоты при комнатной температуре. Заранее сешанные дымящую азотную кислоту (1 мл) и серную кислоту добавляют по каплям при 5-10°С к раствору соединения zz1'. Реакционную смесь нагревают до 85°С в течение одного часа, затем охлаждают до комнатной температуры и выливают на лед (20 г). Желтое твердое вещество zz2' собирают фильтрованием, промывают водой и сушат на воздухе с получением 1.01 г соединения zz2'.
Соединение zz2' (500 мг, 3.16 ммоль) растворяют в оксихлориде фосфора (1.7 мл, 18.9 ммоль) и ДМФА (катализатор) при комнатной температуре. Реакцию нагревают до 110°С в течение 5 часов. Избыток POCl3 удаляют в вакууме. Остаток хроматографируют на силикагеле (CHCl3, 100%), что дает 176 мг продукта zz3'.
Соединение zz3' (140 мг, 0.79 ммоль) растворяют в ТГФ (5 мл) и охлаждают до -78°С под N2. Добавляют по каплям бромид винилмагния (1.0М в этиловом эфире, 1.2 мл). После упомянутого добавления реакционную смесь выдерживают при -20°С в течение около 15 часов. Реакционную массу затем захолаживают насыщенным раствором NH4Cl, экстрагируют EtOAc. Объединенные органические слои промывают рассолом, сушат над MgSO4, концентрируют и хроматографируют, что дает около 130 мг соединения 1zz.
Химические реакции на схеме 3 обеспечивают производные, которые соответствуют общей формуле 5 и имеют 6-азакольцо и R2=F и R4=Cl. В особенности, реакция 2-хлор-5-фтор-3-нитропиридина с 3 эквивалентами винилмагнийбромида при использовании типичных условий, описанных здесь, обеспечивает получение 4-фтор-7-хлор-6-азаиндола с высоким выходом. Прибавление этого соединения к раствору треххлористого алюминия в дихлорметане при перемешивании при температуре окружающей среды в течение последующих 30 минут, а затем с хлорметил или хлорэтилоксалатом обеспечивают получение эфира. Гидролиз с КОН как стандартном процессе обеспечивает получение соли кислоты, которая реагирует с пиперазинами 4 (например, 1-бензоилпиперазин) в присутствии DEPBT в стандартных условиях, описанных здесь, что обеспечивает получение соединения 5, описанного выше. Соединение с бензоилпиперазиновым остатком представляет собой N-(бензоил)-N'-[(4-фтор-7-хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазин, т.е. соединение, обозначенное 5av.
Соединение с (R)-метилбензоилпиперазиновым остатком представляет собой 5av' (R)-N-(бензоил)-3-метил-N'-[(4-фтор-7-хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазин, т.е. соединение, обозначенное 5av'
Характеристики 5av N-(бензоил)-N'-[(4-фтор-7-хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазин и 5av' (R)-N-(бензоил)-3-метил-N'-[(4-фтор-7-хлор-6-азаиндол-3-ил)-оксоацетил]-пиперазин

1Н ЯМР (500 мГц, CD3OD): 8.40 (с, 1H), 8.04 (с, 1Н), 7.46 (bc, 5Н), 3.80-3.50 (м, 8Н). LC/MS: (ES+) м/е (М+Н)+=415, RT=1.247.

1Н ЯМР (500 мГц, CD3OD): 8.42 (с, 1/2Н), 8.37 (с, 1/2Н), 8.03 (с, 1Н), 7.71~7.45 (м, 5Н), 4.72-3.05 (м, 7Н), 1.45~1.28 (м, 3Н). LC/MS: (ES+) м/е (М+Н)+=429, RT=1.297. LC/MS колонка: YMC ODS-A C18 S7 3.0×50 мм. Начало %В=0, окончание %В=100, Градиент время=2 мин, Скорость потока=5 мл/мин. Длина волны=220 нм. Растворитель А=10% СН3ОН - 90% H2O - 0.1% ТФК. Растворитель В=90% СН3ОН - 10% Н2O - 0.1% ТФК.
Аналогично могут быть получены соединения 5ау, 5az, 5abc и 5abd

5ау N-(бензоил)-N'-[(4-фтор-7-метокси-6-азаиндол-3-ил)-оксоацетил]-пиперазин

5az N-(бензоил)-N'-[(4-фтор-7-(N-метилкарбоксамидо)-6-азаиндол-3-ил)-оксоацетил]-пиперазин.

5abc (R)-N-(бензоил)-3-метил-N'-[(7-метокси-4-азаиндол-3-ил)-оксоацетил]-пиперазин

5abd (R)-N-(бензоил)-3-метил-N'-[(7-(N-метилкарбоксамидо)-4-азаиндол-3-ил)-оксоацетил]-пиперазин.
Соединения по настоящему изобретению могут быть введены орально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или путем ингаляции), с помощью спрея или ректально в виде состава дозированной формы, содержащей подходящие фармацевтически-приемлемые носители, адъюванты и наполнители.
Так, в соответствии с настоящим изобретением также обеспечивается способ лечения и фармацевтическая композиция для лечения вирусных инфекций, таких как ВИЧ инфекция и СПИД. Лечение включает введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей фармацевтический носитель и терапевтически эффективное количество соединения по настоящему изобретению.
Фармацевтическая композиция может быть в форме орально вводимых суспензий или таблеток: назальных спреев, стерильно приготовленных инъекций, например, в виде стерильных водных или масляных суспензий для инъекций или в виде суппозиторий.
В случае орального введения в виде суспензии указанные композиции готовят согласно известной из уровня техники технологии изготовления фармацевтических составов и могут содержать микрокристаллическую целлюлозу в качестве объемного наполнителя, алгиновую кислоту или алгинат натрия в качестве суспендирующего агента, метилцеллюлозу в качестве связующего компонента и подсластители/отдушки, известные из уровня техники. В качестве быстрорастворимых таблеток указанные соединения могут содержать микрокристаллическую целлюлозу, дикальций фосфат, крахмал, стеарат магния и лактозу и/или другие эксипиенты, связующие, разрыхлители, расширители, разбавители и смазки, известные из уровня техники.
Растворы и суспензии для инъекций могут быть составлены согласно известному уровню техники при использовании нетоксичных, парентерально приемлемых разбавителей или растворителей, таких как маннитол, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлорида натрия или подходящих диспергирующих, смачивающих и суспендирующих агентов, таких как стерильные масла, включая синтетические моно- или диглицериды, и жирные кислоты, включая олеиновую кислоту.
Соединения по настоящему изобретению могут быть введены человеку орально в дозе, находящейся в интервале от 1 до 100 мг/кг веса человека, разделенной на части. Одна предпочтительная доза находится в интервале от 1 до 10 мг/кг веса тела при оральном приеме по частям. Другая предпочтительная доза находится в интервале от 1 до 20 мг/кг веса тела при приеме орально по частям. Должно быть понятно, однако, что уровень конкретной дозы и частота ее введения любому пациенту может варьироваться в зависимости от различных факторов, включая активность конкретного применяемого соединения, метаболической устойчивости и времени действия активного соединения, возраста больного, его общего здоровья, пола, диеты, пути и времени введения, степени экскреции, комбинации лекарств, серьезности заболевания и проводимой общей терапии.
Сокращения или альтернативные названия
| ТФК | Трифторуксусная кислота |
| ДМФА | N,N-Диметилфopмaмид |
| ТГФ | Тетрагидрофуран |
| CH3OH | Метанол |
| Ether | Диэтилэтиловый эфир |
| DMSO | Диметилсульфоксид |
| EtOAc | Этилацетат |
| Ac | Ацетил |
| Bz | Бензоил |
| Me | Метил |
| Et | Этил |
| Pr | Пропил |
| Py | Пиридин |
| Hunig's Base | N,N-Диизопропилэтиламин |
| DEPBT | 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3Н)-он |
| DEPC | диэтилцианофосфат |
| DMP | 2,2-диметоксипропан |
| mCPBA | мета-хлорпербензойная кислота |
| Азаиндол | 1H-пирролопиридин |
| 4-азаиндол | 1Н-пирроло[3,2-b)]пиридин |
| 5-азаиндол | 1Н-пирроло[3,2-с]пиридин |
| 6-азаиндол | 1Н-пирроло[2,3-с]пиридин |
| 7-азаиндол | 1Н-Пирроло[2,3-b]пиридин |







Claims (22)
1. Производное азаиндола формулы I или его фармацевтически
приемлемая соль,

где

выбирают из группы, состоящей из

R1, R2, R3, R4 каждый независимо выбран из группы, состоящей из Н, C1-С6алкила, С2-С6алкенила, галогена, -CN, фенила, нитро, OC(O)R15, C(O)R15, C(O)OR16, OR19, SR20 и NR21R22;
R15 независимо выбран из группы, включающей Н, C1-С6алкил и С2-С6алкенил;
R16, R19 и R20 каждый независимо выбран из группы, включающей Н, C1-С6алкил, С1-6алкил, замещенный от одного до трех атомами галогена;
R21 и R22 каждый независимо выбран из группы, включающей Н, ОН, C1-С6алкил;
R5 представляет собой (O)m, где m имеет значение 0 или 1;
n имеет значение 1 или 2;
R6 выбирают из группы, включающей Н, C1-C6алкил, С3-C6алкенил, C(O)R24, C(O)OR25; при условии, что атомы углерода, которые содержат углерод-углеродную двойную связь указанного С3-С6алкенила не являются точкой присоединения к азоту, к которому присоединен R6;
R24 выбирают из группы, состоящей из Н, C1-C6алкила;
R25 представляет собой C1-C6алкил;
R7, R8, R9, R10, R11, R12, R13 и R14 каждый независимо выбран из группы, включающей Н, C1-С6алкил;
Ar выбирают из группы, включающей

A1, А2, А3, А4, A5, В1, В2, В3, В4, C1, C2, С3 каждый независимо выбран из группы, включающей Н, галоген, C1-С6алкил, -CN, нитро, N3.
2. Соединение по п.1 или его фрамацевтически приемлемая соль, выбранное из группы, включающей соединения 5а, 5b, 5c, 5d, 5e, 5f, 5g, 5h, 5i, и 5ai как определено ниже


3. Соединение по п.1 или его фармацевтически приемлемая соль, выбранное из группы, включающей соединения 5j, 5k и 5l как определено ниже


4. Соединение по п.1 или его фармацевтически приемлемая соль, имеющее формулу 5m, представленную ниже

5. Соединение по п.1 или его фармацевтически приемлемая соль, выбранное из группы, включающей соединения 8а, 15а, 16а, 16d и 16е, определенные ниже


6. Соединение по п.1 или его фармацевтически приемлемая соль, выбранное из группы, включающей соединения 9а, 9b, 10а, 11а, 11b, 11с, 12а, 14а, 17a-17f, 18a, 19а и 20а, определенные ниже


7. Соединение по п.6 или его фармацевтически приемлемая соль, в котором R2 представляет собой -ОМе, R4 представляет собой водород и R14 представляет собой (R)-метил.
8. Соединение по п.1 или его фармацевтически приемлемая соль, выбранное из группы, включающей соединения 13а, 21а, и 21b, определенные ниже


9. Соединение по п.1 или его фармацевтически приемлемая соль, в котором R2, R3 и R4 каждый независимо выбран из группы, включающей Н, -ОСН3, -ОСН2CF3, -OiPr, -OnPr, галоген, CN, NO2, C1-С6алкил, NHOH, NH2, Ph, SR20 и N(СН3)2.
10. Соединение по п.9 или его фармацевтически приемлемая соль, в котором n имеет значение 2; R1 выбирают из группы, включающей Н, C1-С6алкил и СН2СН=СН2;
и R5 представляет собой (O)m, где m имеет значение 0.
11. Соединение по п.10 или его фармацевтически приемлемая соль, в котором R7, R8, R9, R10, R11, R12, R13 и R14 каждый независимо является Н или СН3, в котором при условии, что один или два члена группы R7-R14 представляют собой СН3 и оставшиеся члены группы R7-R14 являются Н.
12. Соединение по п.11 или его фармацевтически приемлемая соль, в котором один из членов группы A1, А2, А3, А4, А5, B1, В2, В3, В4, С1, С2, С3, выбирают из группы, включающей водород, галоген и амино и оставшиеся члены группы A1, А2, А3, А4, А5, B1, В2, В3, В4, С1, С2, С3, представляют собой водород.
13. Соединение по п.1 или его фармацевтически приемлемая соль, формулы

где R2 выбирают из группы, включающей Н, -ОСН3, -ОСН2CF3, -OPr, галоген, CN, NO2 и NHOH;
R4 выбирают из группы, включающей Н, -галоген, -CN и гидрокси; и
R14 представляет собой СН3 или Н.
14. Соединение по п.1, в котором R4 выбирают из группы, включающей ОН, CN, галоген, -ОСОСН3 и C1-С6алкил.
15. Соединение по п.1 или его фармацевтически приемлемая соль, формулы

где R2 выбирают из группы, включающей Н, F, Cl, Br, OMe, CN или ОН;
R4 выбирают из группы, включающей Н, C1-С6алкил, С2-С6алкенил, -Cl, OMe, CN, ОН, Ph фенил или -С(O)СН3;
n имеет значение 2;
R8, R9, R10, R11, R12, R13 и R14 каждый независимо является Н или СН3, при условии, что 0-2 членов группы R8, R9, R10, R11, R12, R13 и R14 могут быть метилом и остальные члены группы R8, R9, R10, R11, R12, R13 и R14 являются Н;
и
R6 представляет собой водород или метил.
16. Соединение по п.1 или его фармацевтически приемлемая соль, выбранное из группы, включающей соединения 5р, 5r, 5s, 5q, 5t, 5u, 5v и 27с, определенные ниже


17. Соединение по п.1 или его фармацевтически приемлемая соль формулы

где R4 выбирают из группы, включающей H, С1-С6алкил, С2-С6алкенил, Cl, OMe, CN, ОН, фенил и -С(O)СН3;
n представляет собой 2;
R8, R9, R10, R11, R12, R13 и R14 каждый является независимо H или СН3, при условии, что 0-2 членов группы R8, R9, R10, R11, R12, R13 и R14, могут быть СН3, а остальные члены группы R8, R9, R10, R11, R12, R13 и R14 являются H; и
R6 представляет собой Н или СН3.
18. Соединение по п.1 или его фармацевтически приемлемая соль, выбранное из группы, включающей соединения 5w, 5x, 5y, 5z и 5ak, определенные ниже


19. Соединение по п.15, в котором R4, R7, R8, R9, R10, R11, R12, R13 и R14 являются Н; и R2 является - ОМе.
20. Соединение по п.15, в котором R2, R4, R7, R8, R9, R10, R11, R12, R13 и R14 являются Н.
21. Соединение по п.1 или его фармацевтически приемлемая соль формулы

где R2 представляет собой Н, F, Cl, Br, OMe, CN или ОН;
R4 выбирают из группы, включающей Н, C1-C6алкил, С1-С6алкенил, Cl, OMe, CN, ОН, фенил и -С(O)СН3;
n имеет значение 2;
R8, R9, R10, R11, R12, R13 и R14 каждый независимо является Н или СН3, при условии, что вплоть до двух других заместителей могут быть метилом;
R1 представляет собой водород;
R5 является незамещенным; и
R6 представляет собой водород или метил.
22. Соединение по п.1 или его фармацевтически приемлемая соль формулы

где R2 представляет собой Н, -ОСН3, -ОСН2CF3, -OPr, галоген, CN, NO2 или NHOH;
R4 представляет собой Н, -галоген, -CN или гидрокси;
один или два члена R7-R14 представляют собой метил и оставшиеся члены представляет собой водород;
n имеет значение 2;
R1 представляет собой водород;
R5 представляет собой (O)m, где m представляет собой 0; и
R6 представляет собой водород, метил или аллил.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18400400P | 2000-02-22 | 2000-02-22 | |
| US60/184,004 | 2000-02-22 | ||
| US60/184004 | 2000-02-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2002125494A RU2002125494A (ru) | 2004-03-20 |
| RU2259372C2 true RU2259372C2 (ru) | 2005-08-27 |
Family
ID=22675206
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2002125494/04A RU2259372C2 (ru) | 2000-02-22 | 2001-01-19 | Производные азаиндола |
Country Status (32)
| Country | Link |
|---|---|
| US (1) | US20020061892A1 (ru) |
| EP (1) | EP1257276B1 (ru) |
| JP (1) | JP3868814B2 (ru) |
| KR (1) | KR100496574B1 (ru) |
| CN (1) | CN1404392A (ru) |
| AR (1) | AR029469A1 (ru) |
| AT (1) | ATE314072T1 (ru) |
| AU (2) | AU2001232895B2 (ru) |
| BR (1) | BRPI0108485B1 (ru) |
| CA (1) | CA2400700C (ru) |
| CO (1) | CO5271657A1 (ru) |
| CY (1) | CY1107460T1 (ru) |
| DE (1) | DE60116260T2 (ru) |
| DK (1) | DK1257276T3 (ru) |
| EG (1) | EG24669A (ru) |
| ES (1) | ES2254367T3 (ru) |
| HK (1) | HK1052132A1 (ru) |
| HU (1) | HU229992B1 (ru) |
| IL (1) | IL150734A0 (ru) |
| MX (1) | MXPA02008113A (ru) |
| MY (1) | MY128458A (ru) |
| NO (1) | NO323564B1 (ru) |
| NZ (1) | NZ520317A (ru) |
| PE (1) | PE20020786A1 (ru) |
| PL (1) | PL357434A1 (ru) |
| PT (1) | PT1257276E (ru) |
| RU (1) | RU2259372C2 (ru) |
| TR (1) | TR200201961T2 (ru) |
| TW (1) | TWI293629B (ru) |
| UY (1) | UY26593A1 (ru) |
| WO (1) | WO2001062255A1 (ru) |
| ZA (1) | ZA200206073B (ru) |
Families Citing this family (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ527193A (en) * | 2001-02-02 | 2004-05-28 | Bristol Myers Squibb Co | Composition and antiviral activity of substituted azaindoleoxoacetic piperazine derivatives |
| US20040110785A1 (en) * | 2001-02-02 | 2004-06-10 | Tao Wang | Composition and antiviral activity of substituted azaindoleoxoacetic piperazine derivatives |
| US20030207910A1 (en) * | 2001-02-02 | 2003-11-06 | Tao Wang | Composition and antiviral activity of substituted azaindoleoxoacetic piperazine derivatives |
| US6825201B2 (en) * | 2001-04-25 | 2004-11-30 | Bristol-Myers Squibb Company | Indole, azaindole and related heterocyclic amidopiperazine derivatives |
| US20030236277A1 (en) | 2002-02-14 | 2003-12-25 | Kadow John F. | Indole, azaindole and related heterocyclic pyrrolidine derivatives |
| EP1476163A4 (en) * | 2002-02-23 | 2009-05-27 | Bristol Myers Squibb Co | METHOD FOR TREATING HIV INFECTION BY PREVENTING INTERACTION OF CD4 AND GP120 |
| US20040063744A1 (en) * | 2002-05-28 | 2004-04-01 | Tao Wang | Indole, azaindole and related heterocyclic 4-alkenyl piperidine amides |
| US20050075364A1 (en) * | 2003-07-01 | 2005-04-07 | Kap-Sun Yeung | Indole, azaindole and related heterocyclic N-substituted piperazine derivatives |
| CN1953985B (zh) * | 2004-03-15 | 2010-06-09 | 布里斯托尔-迈尔斯斯奎布公司 | 哌嗪和取代的哌啶抗病毒药物的前药 |
| US7745625B2 (en) | 2004-03-15 | 2010-06-29 | Bristol-Myers Squibb Company | Prodrugs of piperazine and substituted piperidine antiviral agents |
| US20050252408A1 (en) * | 2004-05-17 | 2005-11-17 | Richardson H W | Particulate wood preservative and method for producing same |
| CA2569887A1 (en) | 2004-06-09 | 2005-12-22 | Glaxo Group Limited | Pyrrolopyridine derivatives |
| US7498342B2 (en) | 2004-06-17 | 2009-03-03 | Plexxikon, Inc. | Compounds modulating c-kit activity |
| US20060100209A1 (en) * | 2004-11-09 | 2006-05-11 | Chong-Hui Gu | Formulations of 1-(4-benzoyl-piperazin-1-yl)-2-[4-methoxy-7-(3-methyl-[1,2,4]triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-3-yl]-ethane-1,2-dione |
| US20060100432A1 (en) * | 2004-11-09 | 2006-05-11 | Matiskella John D | Crystalline materials of 1-(4-benzoyl-piperazin-1-yl)-2-[4-methoxy-7-(3-methyl-[1,2,4]triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-3-yl]-ethane-1,2-dione |
| US7183284B2 (en) * | 2004-12-29 | 2007-02-27 | Bristol-Myers Squibb Company | Aminium salts of 1,2,3-triazoles as prodrugs of drugs including antiviral agents |
| US7601715B2 (en) | 2005-06-22 | 2009-10-13 | Bristol-Myers Squibb Company | Process for preparing triazole substituted azaindoleoxoacetic piperazine derivatives and novel salt forms produced therein |
| JP2009514790A (ja) | 2005-07-22 | 2009-04-09 | プロジェニクス・ファーマスーティカルズ・インコーポレイテッド | Hiv−1−感染患者におけるウイルス負荷を減少させる方法 |
| US7598380B2 (en) | 2005-08-03 | 2009-10-06 | Bristol-Myers Squibb Company | Method of preparation of azaindole derivatives |
| US7396830B2 (en) * | 2005-10-04 | 2008-07-08 | Bristol-Myers Squibb Company | Piperazine amidines as antiviral agents |
| US7851476B2 (en) * | 2005-12-14 | 2010-12-14 | Bristol-Myers Squibb Company | Crystalline forms of 1-benzoyl-4-[2-[4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-YL)-1-[(phosphonooxy)methyl]-1H-pyrrolo[2,3-C]pyridin-3-YL]-1,2-dioxoethyl]-piperazine |
| US7807671B2 (en) | 2006-04-25 | 2010-10-05 | Bristol-Myers Squibb Company | Diketo-piperazine and piperidine derivatives as antiviral agents |
| WO2008063888A2 (en) | 2006-11-22 | 2008-05-29 | Plexxikon, Inc. | Compounds modulating c-fms and/or c-kit activity and uses therefor |
| CA2690243A1 (en) * | 2007-06-18 | 2008-12-24 | Sanofi-Aventis | Pyrrole derivatives as p2y12 antagonists |
| NZ582772A (en) | 2007-07-17 | 2012-06-29 | Plexxikon Inc | Compounds and methods for kinase modulation, and indications therefor |
| WO2009011912A1 (en) * | 2007-07-18 | 2009-01-22 | Bristol-Myers Squibb Company | A composition for treating hiv comprising virus-like particles |
| PT2240482E (pt) | 2008-01-31 | 2013-09-06 | Sanofi Sa | Azaindole-3-carboxamidas cíclicas, sua preparação e sua utilização como fármacos |
| DK2323633T3 (da) * | 2008-09-04 | 2012-07-09 | Bristol Myers Squibb Co | Stabil, farmaceutisk sammensætning til optimeret indgivelse af en hiv-bindingshæmmer |
| EA022924B1 (ru) | 2009-04-03 | 2016-03-31 | Ф.Хоффманн-Ля Рош Аг | ТВЁРДАЯ ФОРМА {3-[5-(4-ХЛОРФЕНИЛ)-1Н-ПИРРОЛО[2,3-b]ПИРИДИН-3-КАРБОНИЛ]-2,4-ДИФТОРФЕНИЛ}АМИДА ПРОПАН-1-СУЛЬФОНОВОЙ КИСЛОТЫ И ЕЁ ПРИМЕНЕНИЕ |
| CA2780190C (en) | 2009-11-06 | 2020-05-05 | Plexxikon, Inc. | Compounds and methods for kinase modulation, and indications therefor |
| ES2585396T3 (es) | 2010-12-02 | 2016-10-05 | VIIV Healthcare UK (No.5) Limited | Alquilamidas como inhibidores de la unión del VIH |
| BR112013020041B1 (pt) | 2011-02-07 | 2021-11-23 | Plexxikon, Inc | Compostos e composições para a modulação de quinases e uso dos mesmos |
| TWI558702B (zh) | 2011-02-21 | 2016-11-21 | 普雷辛肯公司 | 醫藥活性物質的固態形式 |
| WO2012142080A1 (en) | 2011-04-12 | 2012-10-18 | Bristol-Myers Squibb Company | Thioamide, amidoxime and amidrazone derivatives as hiv attachment inhibitors |
| ES2616268T3 (es) | 2011-08-29 | 2017-06-12 | VIIV Healthcare UK (No.5) Limited | Derivados condensados de diamina bicíclica como inhibidores de la unión de VIH |
| WO2013033059A1 (en) | 2011-08-29 | 2013-03-07 | Bristol-Myers Squibb Company | Spiro bicyclic diamine derivatives as hiv attachment inhibitors |
| US9193725B2 (en) | 2012-03-14 | 2015-11-24 | Bristol-Meyers Squibb Company | Cyclic hydrazine derivatives as HIV attachment inhibitors |
| US9150570B2 (en) | 2012-05-31 | 2015-10-06 | Plexxikon Inc. | Synthesis of heterocyclic compounds |
| CN102746295B (zh) * | 2012-06-20 | 2014-10-15 | 上海阿达玛斯试剂有限公司 | 4位取代-7-氮杂吲哚的制备方法 |
| WO2014025854A1 (en) | 2012-08-09 | 2014-02-13 | Bristol-Myers Squibb Company | Piperidine amide derivatives as hiv attachment inhibitors |
| ES2616432T3 (es) | 2012-08-09 | 2017-06-13 | VIIV Healthcare UK (No.5) Limited | Derivados de alquenos tricíclicos como inhibidores de la unión del VIH |
| CN109528749B (zh) * | 2017-09-22 | 2023-04-28 | 上海交通大学医学院附属瑞金医院 | 长链非编码rna-h19在制备治疗垂体瘤药物中的用途 |
| WO2022076975A1 (en) | 2020-10-05 | 2022-04-14 | Enliven Therapeutics, Inc. | 5- and 6-azaindole compounds for inhibition of bcr-abl tyrosine kinases |
| CN115368320B (zh) * | 2020-11-24 | 2024-05-31 | 中国人民解放军海军军医大学 | 一类取代苯甲酰哌嗪类化合物及其应用 |
-
2001
- 2001-01-18 US US09/765,189 patent/US20020061892A1/en not_active Abandoned
- 2001-01-19 AU AU2001232895A patent/AU2001232895B2/en not_active Ceased
- 2001-01-19 PT PT01904970T patent/PT1257276E/pt unknown
- 2001-01-19 AT AT01904970T patent/ATE314072T1/de active
- 2001-01-19 CN CN01805504A patent/CN1404392A/zh active Pending
- 2001-01-19 AU AU3289501A patent/AU3289501A/xx active Pending
- 2001-01-19 BR BRPI0108485A patent/BRPI0108485B1/pt not_active IP Right Cessation
- 2001-01-19 MX MXPA02008113A patent/MXPA02008113A/es active IP Right Grant
- 2001-01-19 KR KR10-2002-7010888A patent/KR100496574B1/ko not_active IP Right Cessation
- 2001-01-19 WO PCT/US2001/002009 patent/WO2001062255A1/en active IP Right Grant
- 2001-01-19 NZ NZ520317A patent/NZ520317A/en not_active IP Right Cessation
- 2001-01-19 CA CA2400700A patent/CA2400700C/en not_active Expired - Fee Related
- 2001-01-19 DK DK01904970T patent/DK1257276T3/da active
- 2001-01-19 HU HU0301508A patent/HU229992B1/hu not_active IP Right Cessation
- 2001-01-19 PL PL01357434A patent/PL357434A1/xx not_active Application Discontinuation
- 2001-01-19 RU RU2002125494/04A patent/RU2259372C2/ru not_active IP Right Cessation
- 2001-01-19 TR TR2002/01961T patent/TR200201961T2/xx unknown
- 2001-01-19 ES ES01904970T patent/ES2254367T3/es not_active Expired - Lifetime
- 2001-01-19 JP JP2001561320A patent/JP3868814B2/ja not_active Expired - Fee Related
- 2001-01-19 IL IL15073401A patent/IL150734A0/xx unknown
- 2001-01-19 DE DE60116260T patent/DE60116260T2/de not_active Expired - Lifetime
- 2001-01-19 EP EP01904970A patent/EP1257276B1/en not_active Expired - Lifetime
- 2001-02-06 MY MYPI20010516A patent/MY128458A/en unknown
- 2001-02-07 TW TW090102639A patent/TWI293629B/zh not_active IP Right Cessation
- 2001-02-19 AR ARP010100742A patent/AR029469A1/es active IP Right Grant
- 2001-02-19 EG EG20010152A patent/EG24669A/xx active
- 2001-02-20 CO CO01013156A patent/CO5271657A1/es not_active Application Discontinuation
- 2001-02-21 UY UY26593A patent/UY26593A1/es not_active IP Right Cessation
- 2001-02-22 PE PE2001000190A patent/PE20020786A1/es not_active Application Discontinuation
-
2002
- 2002-07-30 ZA ZA200206073A patent/ZA200206073B/en unknown
- 2002-08-21 NO NO20023981A patent/NO323564B1/no not_active IP Right Cessation
-
2003
- 2003-04-23 HK HK03102905A patent/HK1052132A1/xx not_active IP Right Cessation
-
2006
- 2006-03-27 CY CY20061100420T patent/CY1107460T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2259372C2 (ru) | Производные азаиндола | |
| US6476034B2 (en) | Antiviral azaindole derivatives | |
| EP1467970B1 (en) | Hydroxynaphthyridinone carboxamides useful as hiv integrase inhibitors | |
| AU2001232895A1 (en) | Antiviral azaindole derivatives | |
| EP2646439B1 (en) | Alkyl amides as hiv attachment inhibitors | |
| EP1326611B1 (en) | Aza- and polyaza-naphthalenyl-carboxamides useful as hiv integrase inhibitors | |
| AU2014340110B2 (en) | Inhibitors of human immunodeficiency virus replication | |
| EP1943221B1 (en) | Piperazine amidines as antiviral agents | |
| US7601715B2 (en) | Process for preparing triazole substituted azaindoleoxoacetic piperazine derivatives and novel salt forms produced therein | |
| US7902204B2 (en) | Diazaindole-dicarbonyl-piperazinyl antiviral agents | |
| KR20010072471A (ko) | 항바이러스 인돌옥소아세틸 피페라진 유도체 | |
| US9505752B2 (en) | Piperidine amide derivatives as HIV attachment inhibitors | |
| ES2389478T3 (es) | Derivados de dicetopiperidina como inhibidores de la fijación del VIH | |
| HUE027396T2 (en) | Antiviral Activity and Composition of Substituted Axaindoloxoacetic Acid Piperazine Derivatives | |
| EP2013170B1 (en) | 4-squarylpiperazine derivatives as antiviral agents | |
| WO2013016441A1 (en) | Hiv integrase inhibitory oxoisoindoline sulfonamides | |
| EP1539714B1 (en) | 8-hydroxy-1-oxo-tetrahydropyrrolopyrazine compounds useful as hiv integrase inhibitors | |
| EP2696937B1 (en) | Thioamide, amidoxime and amidrazone derivatives as hiv attachment inhibitors | |
| US7183284B2 (en) | Aminium salts of 1,2,3-triazoles as prodrugs of drugs including antiviral agents | |
| EP1430058A1 (en) | Sodium salt of an hiv integrase inhibitor | |
| AU2002332521A1 (en) | Sodium salt of an HIV integrase inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PC41 | Official registration of the transfer of exclusive right |
Effective date: 20181031 |
|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20190120 |