FIELD OF THE INVENTION
This invention provides compounds having drug and bio-affecting properties, their pharmaceutical compositions and method of use. In particular, the invention is concerned with new heterocyclic amidopiperazine derivatives that possess unique antiviral activity. More particularly, the present invention relates to compounds useful for the treatment of HIV and AIDS. [0002]
BACKGROUND ART
HIV-1 (human immunodeficiency virus-1) infection remains a major medical problem, with an estimated 33.6 million people infected worldwide. The number of cases of HIV and AIDS (acquired immunodeficiency syndrome) has risen rapidly. In 1999, 5.6 million new infections were reported, and 2.6 million people died from AIDS. Currently available drugs for the treatment of HIV include six nucleoside reverse transcriptase (RT) inhibitors (zidovudine, didanosine, stavudine, lamivudine, zalcitabine and abacavir), three non-nucleoside reverse transcriptase inhibitors (nevirapine, delavirdine and efavirenz), and six peptidomimetic protease inhibitors (saquinavir, indinavir, ritonavir, nelfinavir, amprenavir and lopinavir). Each of these drugs can only transiently restrain viral replication if used alone. However, when used in combination, these drugs have a profound effect on viremia and disease progression. In fact, significant reductions in death rates among AIDS patients have been recently documented as a consequence of the widespread application of combination therapy. However, despite these impressive results, 30 to 50% of patients ultimately fail combination drug therapies. Insufficient drug potency, non-compliance, restricted tissue penetration and drug-specific limitations within certain cell types (e.g. most nucleoside analogs cannot be phosphorylated in resting cells) may account for the incomplete suppression of sensitive viruses. Furthermore, the high replication rate and rapid turnover of HIV-1 combined with the frequent incorporation of mutations, leads to the appearance of drug-resistant variants and treatment failures when sub-optimal drug concentrations are present (Larder and Kemp; Gulick; Kuritzkes; Morris-Jones et al; Schinazi et al; Vacca and Condra; Flexner; Berkhout and Ren et al; (Ref. 6-14)). Therefore, novel anti-HIV agents exhibiting distinct resistance patterns, and favorable pharmacokinetic as well as safety profiles are needed to provide more treatment options. [0003]
Currently marketed HIV-1 drugs are dominated by either nucleoside reverse transcriptase inhibitors or peptidomimetic protease inhibitors. Non-nucleoside reverse transcriptase inhibitors (NNRTIs) have recently gained an increasingly important role in the therapy of HIV infections (Pedersen & Pedersen, Ref 15). At least 30 different classes of NNRTI have been described in the literature (De Clercq, Ref. 16) and several NNRTIs have been evaluated in clinical trials. Dipyridodiazepinone (nevirapine), benzoxazinone (efavirenz) and bis(heteroaryl) piperazine derivatives (delavirdine) have been approved for clinical use. However, the major drawback to the development and application of NNRTIs is the propensity for rapid emergence of drug resistant strains, both in tissue cell culture and in treated individuals, particularly those subject to monotherapy. As a consequence, there is considerable interest in the identification of NNRTIs less prone to the development of resistance (Pedersen & Pedersen, Ref 15). A recent overview of non-nucleoside reverse transcriptase inhibitors: perspectives on novel therapeutic compounds and strategies for the treatment of HIV infection. has appeared (Buckheit , reference 99). A review covering both NRTI and NNRTIs has appeared (De clercq, reference 100). An overview of the current state of the HIV drugs has been published (De clercq, reference 101) [0004]
Several indole derivatives including indole-3-sulfones, piperazino indoles, pyrazino indoles, and 5H-indolo[3,2-b][1,5]benzothiazepine derivatives have been reported as HIV-1 reverse transciptase inhibitors (Greenlee et al, Ref. 1; Williams et al, Ref. 2; Romero et al, Ref. 3; Font et al, Ref. 17; Romero et al, Ref. 18; Young et al, Ref. 19; Genin et al, Ref. 20; Silvestri et al, Ref. 21). Indole 2-carboxamides have also been described as inhibitors of cell adhesion and HIV infection (Boschelli et al, U.S. Pat. No. 5,424,329, Ref. 4). 3-substituted indole natural products (Semicochliodinol A and B, didemethylasterriquinone and isocochliodinol) were disclosed as inhibitors of HIV-1 protease (Fredenhagen et al, Ref. 22). [0005]
Structurally related aza-indole amide derivatives have been disclosed previously (Kato et al, Ref. 23; Levacher et al, Ref. 24; Dompe Spa, WO-09504742, Ref. 5(a); SmithKline Beecham PLC, WO-09611929, Ref. 5(b); Schering Corp., US-05023265, Ref. 5(c)). However, these structures differ from those claimed herein in that they are aza-indole mono-amide rather than unsymmetrical aza-indole piperazine diamide derivatives, and there is no mention of the use of these compounds for treating viral infections, particularly HIV. Indole and azaindole piperazine containing derivatives have been disclosed in three different PCT patent applications (Reference 93-95) None of these applications discloses pyrrolidine compounds such as described in this invention. [0006]
Nothing in these references can be construed to disclose or suggest the novel compounds of this invention and their use to inhibit HIV infection. [0007]
REFERENCES CITED
Patent Documents [0008]
1. Greenlee, W. J.; Srinivasan, P. C. Indole reverse transcriptase inhibitors. U.S. Pat. No. 5,124,327. [0009]
2. Williams, T. M.; Ciccarone, T. M.; Saari, W. S.; Wai, J. S.; Greenlee, W. J.; Balani, S. K.; Goldman, M. E.; Theohrides, A. D. Indoles as inhibitors of HIV reverse transcriptase. European Patent 530907. [0010]
3. Romero, D. L.; Thomas, R. C.; Preparation of substituted indoles as anti-AIDS pharmaceuticals. PCT WO 93/01181. [0011]
4. Boschelli, D. H.; Connor, D. T.; Unangst, P. C. Indole-2-carboxamides as inhibitors of cell adhesion. U.S. Pat. No. 5,424,329. [0012]
5. (a) Mantovanini, M.; Melillo, G.; Daffonchio, L. Tropyl 7-azaindol-3-ylcarboxyamides as antitussive agents. PCT WO 95/04742 (Dompe Spa). (b) Cassidy, F.; Hughes, I.; Rahman, S.; Hunter, D. J. Bisheteroaryl-carbonyl and carboxamide derivatives with 5HT 2C/2B antagonists activity. PCT WO 96/11929. (c) Scherlock, M. H.; Tom, W. C. Substituted 1H-pyrrolopyridine-3-carboxamides. U.S. Pat. No. 5,023,265. [0013]
Other Publications [0014]
6. Larder, B. A.; Kemp, S. D. Multiple mutations in the HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). [0015] Science, 1989, 246,1155-1158.
7. Gulick, R. M. Current antiretroviral therapy: An overview. [0016] Quality of Life Research, 1997, 6, 471-474.
8. Kuritzkes, D. R. HIV resistance to current therapies. [0017] Antiviral Therapy, 1997, 2 (Supplement 3), 61-67.
9. Morris-Jones, S.; Moyle, G.; Easterbrook, P. J. Antiretroviral therapies in HIV-1 infection. [0018] Expert Opinion on Investigational Drugs, 1997, 6(8),1049-1061.
10. Schinazi, R. F.; Larder, B. A.; Mellors, J. W. Mutations in retroviral genes associated with drug resistance. [0019] International Antiviral News, 1997, 5,129-142.
11. Vacca, J. P.; Condra, J. H. Clinically effective HIV-1 protease inhibitors. [0020] Drug Discovery Today, 1997, 2, 261-272.
12. Flexner, D. HIV-protease inhibitors. [0021] Drug Therapy, 1998, 338, 1281-1292.
13. Berkhout, B. HIV-1 evolution under pressure of protease inhibitors: Climbing the stairs of viral fitness. [0022] J. Biomed. Sci., 1999, 6, 298-305.
14. Ren, S.; Lien, E. J. Development of HIV protease inhibitors: A survey. [0023] Prog. Drug Res., 1998, 51, 1-31.
15. Pedersen, O. S.; Pedersen, E. B. Non-nucleoside reverse transcriptase inhibitors: the NNRTI boom. [0024] Antiviral Chem. Chemother. 1999, 10, 285-314.
16. (a) De Clercq, E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. [0025] Antiviral Research, 1998, 38, 153-179. (b) De Clercq, E. Perspectives of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV infection. IL. Farmaco, 1999, 54, 26-45.
17. Font, M.; Monge, A.; Cuartero, A.; Elorriaga, A.; Martinez-Irujo, J. J.; Alberdi, E.; Santiago, E.; Prieto, I.; Lasarte, J. J.; Sarobe, P. and Borras, F. Indoles and pyrazino[4,5-b]indoles as nonnucleoside analog inhibitors of HIV-1 reverse transcriptase. [0026] Eur. J. Med. Chem., 1995, 30, 963-971.
18. Romero, D. L.; Morge, R. A.; Genin, M. J.; Biles, C.; Busso, M,; Resnick, L.; Althaus, I. W.; Reusser, F.; Thomas, R. C and Tarpley, W. G. Bis(heteroaryl)piperazine (BHAP) reverse transcriptase inhibitors: structure-activity relationships of novel substituted indole analogues and the identification of 1-[(5-methanesulfonamido-1H-indol-2-yl)-carbonyl]-4-[3-[1-methylethyl)amino]-pyridinyl]piperazine momomethansulfonate (U-90152S), a second generation clinical candidate. [0027] J. Med. Chem., 1993, 36, 1505-1508.
19. Young, S. D.; Amblard, M. C.; Britcher, S. F.; Grey, V. E.; Tran, L. O.; Lumma, W. C.; Huff, J. R.; Schleif, W. A.; Emini, E. E.; O'Brien, J. A.; Pettibone, D. J. 2-Heterocyclic indole-3-sulfones as inhibitors of HIV-reverse transcriptase. [0028] Bioorg. Med. Chem. Lett., 1995, 5, 491-496.
20. Genin, M. J.; Poel, T. J.; Yagi, Y.; Biles, C.; Althaus, I.; Keiser, B. J.; Kopta, L. A.; Friis, J. M.; Reusser, F.; Adams, W. J.; Olmsted, R. A.; Voorman, R. L.; Thomas, R. C. and Romero, D. L. Synthesis and bioactivity of novel bis(heteroaryl)piperazine (BHAP) reverse transcriptase inhibitors: structure-activity relationships and increased metabolic stability of novel substituted pyridine analogs. [0029] J. Med. Chem., 1996, 39, 5267-5275.
21. Silvestri, R.; Artico, M.; Bruno, B.; Massa, S.; Novellino, E.; Greco, G.; Marongiu, M. E.; Pani, A.; De Montis, A and La Colla, P. Synthesis and biological evaluation of 5H-indolo[3,2-b][1,5]benzothiazepine derivatives, designed as conformationally constrained analogues of the human immunodeficiency virus type 1 reverse transcriptase inhibitor L-737,126. [0030] Antiviral Chem. Chemother. 1998, 9, 139-148.
22. Fredenhagen, A.; Petersen, F.; Tintelnot-Blomley, M.; Rosel, J.; Mett, H and Hug, P. J. Semicochliodinol A and B: Inhibitors of HIV-1 protease and EGF-R protein Tyrosine Kinase related to Asterriquinones produced by the fungus [0031] Chrysosporium nerdarium. Antibiotics, 1997, 50, 395-401.
23. Kato, M.; Ito, K.; Nishino, S.; Yamakuni, H.; Takasugi, H. New 5-HT[0032] 3 (Serotonin-3) receptor antagonists. IV. Synthesis and structure-activity relationships of azabicycloalkaneacetamide derivatives. Chem. Pharm. Bull., 1995, 43, 1351-1357.
24. Levacher, V.; Benoit, R.; Duflos, J; Dupas, G.; Bourguignon, J.; Queguiner, G. Broadening the scope of NADH models by using chiral and non chiral pyrrolo [2,3-b] pyridine derivatives. [0033] Tetrahedron, 1991, 47, 429-440.
25. Shadrina, L. P.; Dormidontov, Yu.P.; Ponomarev, V, G.; Lapkin, I. I. Reactions of organomagnesium derivatives of 7-aza- and benzoindoles with diethyl oxalate and the reactivity of ethoxalylindoles. [0034] Khim. Geterotsikl. Soedin., 1987, 1206-1209.
26. Sycheva, T. V.; Rubtsov, N. M.; Sheinker, Yu. N.; Yakhontov, L. N. Some reactions of 5-cyano-6-chloro-7-azaindoles and lactam-lactim tautomerism in 5-cyano-6-hydroxy-7-azaindolines. [0035] Khim. Geterotsikl. Soedin., 1987, 100-106.
27. (a) Desai, M.; Watthey, J. W. H.; Zuckerman, M. A convenient preparation of 1-aroylpiperazines. [0036] Org. Prep. Proced. Int., 1976, 8, 85-86. (b) Adamczyk, M.; Fino, J. R. Synthesis of procainamide metabolites. N-acetyl desethylprocainamide and desethylprocainamide. Org. Prep. Proced. Int. 1996, 28, 470-474. (c) Rossen, K.; Weissman, S. A.; Sager, J.; Reamer, R. A.; Askin, D.; Volante, R. P.; Reider, P. J. Asymmetric Hydrogenation of tetrahydropyrazines: Synthesis of (S)-piperazine 2-tert-butylcarboxamide, an intermediate in the preparation of the HIV protease inhibitor Indinavir. Tetrahedron Lett., 1995, 36, 6419-6422. (d) Wang, T.; Zhang, Z.; Meanwell, N. A. Benzoylation of Dianions: Preparation of mono-Benzoylated Symmetric Secondary Diamines. J. Org. Chem., 1999, 64, 7661-7662.
28. Li, H.; Jiang, X.; Ye, Y. -H.; Fan, C.; Romoff, T.; Goodman, M. 3-(Diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT): A new coupling reagent with remarkable resistance to racemization. [0037] Organic Lett., 1999, 1, 91-93.
29. Harada, N.; Kawaguchi, T.; Inoue, I.; Ohashi, M.; Oda, K.; Hashiyama, T.; Tsujihara, K. Synthesis and antitumor activity of quaternary salts of 2-(2′-oxoalkoxy)-9-hydroxyellipticines. [0038] Chem. Pharm. Bull., 1997, 45, 134-137.
30. Schneller, S. W.; Luo, J. -K. Synthesis of 4-amino-1H-pyrrolo[2,3-b]pyridine (1,7-Dideazaadenine) and 1H-pyrrolo[2,3-b]pyridin-4-ol (1,7-Dideazahypoxanthine). [0039] J. Org. Chem., 1980, 45, 4045-4048.
31. Shiotani, S.; Tanigochi, K. Furopyridines. XXII [1]. Elaboration of the C-substitutents alpha to the heteronitrogen atom of furo[2,3-b]-, -[3.2-b]-, -[2.3-c]- and -[3,2-c]pyridine. [0040] J. Het. Chem., 1997, 34, 901-907.
32. Minakata, S.; Komatsu, M.; Ohshiro, Y. Regioselective functionalization of 1H-pyrrolo[2,3-b]pyridine via its N-oxide. [0041] Synthesis, 1992, 661-663.
33. Klemm, L. H.; Hartling, R. Chemistry of thienopyridines. XXIV. Two transformations of thieno[2,3-b]pyridine 7-oxide (1). [0042] J. Het. Chem., 1976, 13, 1197-1200.
34. Antonini, I.; Claudi, F.; Cristalli, G.; Franchetti, P.; Crifantini, M.; Martelli, S. Synthesis of 4-amino-1-□-D-ribofuranosyl-1H-pyrrolo[2,3-b]pyridine (1-Deazatubercidin) as a potential antitumor agent. [0043] J. Med. Chem., 1982, 25, 1258-1261.
35. (a) Regnouf De Vains, J. B.; Papet, A. L.; Marsura, A. New symmetric and unsymmetric polyfunctionalized 2,2′-bipyridines. [0044] J. Het. Chem., 1994, 31, 1069-1077. (b) Miura, Y.; Yoshida, M.; Hamana, M. Synthesis of 2,3-fused quinolines from 3-substituted quinoline 1-oxides. Part II, Heterocycles, 1993, 36, 1005-1016. (c) Profft, V. E.; Rolle, W. Uber 4-merkaptoverbindungendes 2-methylpyridins. J. Prakt. Chem., 1960, 283 (11), 22-34.
36. Nesi, R.; Giomi, D.; Turchi, S.; Tedeschi, P., Ponticelli, F. A new one step synthetic approach to the isoxazolo[4,5-b]pyridine system. [0045] Synth. Comm., 1992, 22, 2349-2355.
37. (a) Walser, A.; Zenchoff, G.; Fryer, R. I. Quinazolines and 1,4-benzodiazepines. 75. 7-Hydroxyaminobenzodiazepines and derivatives. [0046] J. Med. Chem., 1976, 19, 1378-1381. (b) Barker, G.; Ellis, G. P. Benzopyrone. Part I. 6-Amino- and 6-hydroxy-2-subtituted chromones. J. Chem. Soc., 1970, 2230-2233.
38. Ayyangar, N. R.; Lahoti, R J.; Daniel, T. An alternate synthesis of 3,4-diaminobenzophenone and mebendazole. [0047] Org. Prep. Proced. Int., 1991, 23, 627-631.
39. Mahadevan, I.; Rasmussen, M. Ambident heterocyclic reactivity: The alkylation of pyrrolopyridines (azaindoles, diazaindenes). Tetrahedron, 1993, 49, 7337-7352. [0048]
40. Chen, B. K.; Saksela, K.; Andino, R.; Baltimore, D. Distinct modes of human immunodeficiency type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. [0049] J. Virol., 1994, 68, 654-660.
41. Bodanszky, M.; Bodanszky, A. “[0050] The Practice of Peptide Synthesis” 2nd Ed., Springer-Veriag: Berlin Heidelberg, Germany, 1994.
42. Albericio, F. et al. [0051] J. Org. Chem. 1998, 63, 9678.
43. Knorr, R. et al. [0052] Tetrahedron Lett. 1989, 30, 1927.
44. (a) Jaszay Z. M. et al. [0053] Synth. Commun., 1998 28, 2761 and references cited therein; (b) Bernasconi, S. et al. Synthesis, 1980, 385.
45. (a) Jaszay Z. M. et al. [0054] Synthesis, 1989, 745 and references cited therein; (b) Nicolaou, K. C. et al. Angew. Chem. Int. Ed. 1999, 38, 1669.
46. Ooi, T. et al. [0055] Synlett. 1999, 729.
47. Ford, R. E. et al. [0056] J. Med. Chem. 1986, 29, 538.
48. (a) Yeung, K. -S. et al. Bristol-Myers Squibb Unpublished Results. (b) Wang, W. et al. [0057] Tetrahedron Lett. 1999, 40, 2501.
49. Brook, M. A. et al. [0058] Synthesis, 1983, 201.
50. Yamazaki, N. et al. [0059] Tetrahedron Lett. 1972, 5047.
51. Barry A. Bunin “The Combinatorial Index” 1998 Academic Press, San Diego/London pages 78-82. [0060]
52. Richard C. Larock Comprehensive Organic Transormations 2nd Ed. 1999, John Wiley and Sons New York. [0061]
53. M. D. Mullican et.al. [0062] J.Med. Chem. 1991, 34, 2186-2194.
54. Protective groups in organic synthesis 3rd ed./Theodora W. Greene and Peter G. M. Wuts. New York: Wiley, 1999. [0063]
55. Katritzky, Alan R. Lagowski, Jeanne M. The principles of heterocyclic ChemistryNew York: Academic Press, 1968 [0064]
56. Paquette, Leo A. Principles of modern heterocyclic chemistry New York: Benjamin. [0065]
57. Katritzky, Alan R.; Rees, Charles W.; Comprehensive heterocyclic chemistry: the structure, reactions, synthesis, and uses of heterocyclic compounds 1st ed.Oxford (Oxfordshire); New York: Pergamon Press, 1984. 8 v. [0066]
58. Katritzky, Alan RHandbook of heterocyclic 1st edOxford (Oxfordshire); New York: Pergamon Press, 1985. [0067]
59. Davies, David I Aromatic Heterocyclic Oxford; New York: Oxford University Press, 1991. [0068]
60. Ellis, G. P. Synthesis of fused Chichester [Sussex]; New York: Wiley, c1987-c1992. Chemistry of heterocyclic compounds; v. 47. [0069]
61. Joule, J. A Mills, K., Smith, G. F. Heterocyclic Chemistry, 3rd ed London; New York Chapman & Hall, 1995. [0070]
62. Katritzky, Alan R., Rees, Charles W., Scriven, Eric F. V. Comprehensive heterocyclic chemistry II: a review of the literature 1982-1995. [0071]
63. The structure, reactions, synthesis, and uses of heterocyclic compounds 1st ed. Oxford; New York: Pergamon, 1996. 11 v. in 12: ill.; 28 cm. [0072]
64. Eicher, Theophil, Hauptmann, Siegfried. The chemistry of heterocycles: structure, reactions, syntheses, and applications Stuttgart; New York: G. Thieme, 1995. [0073]
65. Grimmett, M. R. Imidazole and benzimidazole Synthesis London; San Diego: Academic Press, 1997. [0074]
66. Advances in heterocyclic chemistry. Published in New York by Academic Press, starting in 1963-present. [0075]
67. Gilchrist, T. L. (Thomas Lonsdale) Heterocyclic chemistry 3rd ed. Harlow, Essex: Longman, 1997. 414 p.: ill.; 24 cm. [0076]
68. Farina, Vittorio; Roth, Gregory P. Recent advances in the Stille reaction; [0077] Adv. Met.-Org. Chem. 1996, 5, 1-53.
69. Farina, Vittorio; Krishnamurthy, Venkat; Scott, William J. The Stille reaction; Org. React. (N. Y.) (1997), 50, 1-652. [0078]
70. Stille, J. K. [0079] Angew. Chem. Int. Ed. Engl. 1986, 25, 508-524.
71. Norio Miyaura and Akiro Suzuki [0080] Chem Rev. 1995, 95, 2457.
72. Home, D. A. [0081] Heterocycles 1994, 39, 139.
73. Kamitori, Y. et.al. [0082] Heterocycles, 1994, 37(1), 153.
74. Shawali, J. [0083] Heterocyclic Chem. 1976, 13, 989.
75. a) Kende, A. S.et al. [0084] Org. Photochem. Synth. 1972, 1, 92. b) Hankes, L. V.; Biochem. Prep. 1966, 11, 63. c) Synth. Meth. 22, 837.
76. Hulton et. al. [0085] Synth. Comm. 1979, 9, 789.
77. Pattanayak, B. K. et.al. [0086] Indian J. Chem. 1978, 16, 1030.
78. [0087] Chemische Berichte 1902, 35, 1545.
79. [0088] Chemische Berichte Ibid 1911, 44, 493.
80. Moubarak, I., Vessiere, R. [0089] Synthesis 1980, Vol. 1, 52-53.
81. [0090] Ind J. Chem. 1973, 11, 1260.
82. Roomi et.al. [0091] Can J. Chem. 1970, 48, 1689.
83. Sorrel, T. N. [0092] J. Org. Chem. 1994, 59, 1589.
84. Nitz, T. J. et. al. [0093] J. Org. Chem. 1994, 59, 5828-5832.
85. Bowden, K. et.al. [0094] J. Chem. Soc. 1946, 953.
86. Nitz, T. J. et. al. [0095] J. Org. Chem. 1994, 59, 5828-5832.
87. Scholkopf et. al. [0096] Angew. Int. Ed. Engl. 1971, 10(5), 333.
88. (a) Behun, J. D.; Levine, R. [0097] J. Org. Chem. 1961, 26, 3379. (b) Rossen, K.; Weissman, S. A.; Sager, J.; Reamer, R. A.; Askin, D.; Volante, R. P.; Reider, P. J. Asymmetric Hydrogenation of tetrahydropyrazines: Synthesis of (S)-piperazine 2-tert-butylcarboxamide, an intermediate in the preparation of the HIV protease inhibitor Indinavir. Tetrahedron Lett., 1995, 36, 6419-6422. (c) Jenneskens, L. W.; Mahy, J.; den Berg, E. M. M. de B. -v.; Van der Hoef, I.; Lugtenburg, J. Recl. Trav. Chim. Pays-Bas 1995, 114, 97.
89. Wang, T.; Zhang, Z.; Meanwell, N. A. Benzoylation of Dianions: Preparation of mono-Benzoylated Symmetric Secondary Diamines. [0098] J. Org. Chem., 1999, 64, 7661-7662.
90. (a) Adamczyk, M.; Fino, J. R. Synthesis of procainamide metabolites. N-acetyl desethylprocainamide and desethylprocainamide. [0099] Org. Prep. Proced. Int. 1996, 28, 470-474. (b) Wang, T.; Zhang, Z.; Meanwell, N. A. Regioselective mono-Benzoylation of Unsymmetrical Piperazines. J. Org. Chem., in press.
91. Masuzawa, K.; Kitagawa, M.; Uchida, H. [0100] Bull Chem. Soc. Jpn. 1967, 40, 244-245.
92. Furber, M.; Cooper, M. E.; Donald, D. K. [0101] Tetrahedron Lett. 1993, 34, 1351-1354.
93. Blair, Wade S.; Deshpande, Milind; Fang, Haiquan; Lin, Pin-fang; Spicer, Timothy P.; Wallace, Owen B.; Wang, Hui; Wang, Tao; Zhang, Zhongxing; Yeung, Kap-sun. Preparation of antiviral indoleoxoacetyl piperazine derivatives. PCT Int. Appl. (2000), 165 pp. WO 0076521 A1 [0102]
94. Wang, Tao; Wallace, Owen B.; Zhang, Zhongxing; Meanwell, Nicholas A.; Bender, John A. Preparation of antiviral azaindole derivatives. PCT Int. Appl. (2001), WO 0162255 A1 [0103]
95. Wallace, Owen B.; Wang, Tao; Yeung, Kap-Sun; Pearce, Bradley C.; Meanwell, Nicholas A.; Qiu, Zhilei; Fang, Haiquan; Xue, Qiufen May; Yin, Zhiwei. Composition and antiviral activity of substituted indoleoxoacetic piperazine derivatives. PCT Int. Appl. (2002), WO 0204440 A1 [0104]
96. J. L. Marco, S. T. Ingate, and P. M. Chinchon Tetrahedron 1999, 55, 7625-7644. [0105]
97. C. Thomas, F. Orecher, and P.Gmeiner Synthesis 1998, 1491. [0106]
98. M. P. Pavia, S. J. Lobbestael, C. P. Taylor, F. M. Hershenson, and D. W. Miskell [0107]
99. Buckheit, Robert W., Jr. Expert Opinion on Investigational Drugs 2001, 10(8), 1423-1442. [0108]
100. Balzarini, J.; De Clercq, E.. Antiretroviral Therapy 2001, 31-62. [0109]
101. E. De clercq Journal of Clinical Virology, 2001, 22, 73-89. [0110]
102. Merour, Jean-Yves; Joseph, Benoit. Curr. Org. Chem. (2001), 5(5), 471-506. [0111]
SUMMARY OF THE INVENTION
The present invention comprises compounds of Formula I, their pharmaceutical formulations, and their use in patients suffering from or susceptible to a virus such as HIV. The compounds of Formula I, which include nontoxic pharmaceutically acceptable salts and/or hydrates thereof, have the formula and meaning as described below. Each embodiment of a particular aspect of the invention depends from the preceding embodiment unless otherwise stated. [0112]
SUMMARY DESCRIPTION OF THE INVENTION
The present invention comprises compounds of Formula I, or pharmaceutically acceptable salts thereof, which are effective antiviral agents, particularly as inhibitors of HIV. [0113]
A first embodiment of a first aspect of the invention are compounds of Formula I, including pharmaceutically acceptable salts thereof,
[0114]
wherein: [0115]
Q is selected from the group consisting of:
[0117]
R[0118] 1, R2, R3, R4, and R5, are independently selected from the group consisting of hydrogen, halogen, cyano, nitro, COOR8, XR57, and B;
m is 1 or 2; [0119]
R[0120] 7 is (CH2)nR44 wherein n is 0-6;
R[0121] 6 is O or does not exist;
——represents a carbon-carbon bond or does not exist; [0122]
A is selected from the group consisting of C[0123] 1-6alkoxy, phenyl and D; wherein D is selected from the group consisting of pyridinyl, pyrimidinyl, pyrazinyl, triazinyl, furanyl, thienyl, pyrrolyl, imidazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, quinolinyl, isoquinolinyl, benzofuranyl, benzothienyl, benzoimidazolyl and benzothiazolyl; wherein said phenyl and D are independently optionally substituted with one or two of the same or different amino, halogen or trifluoromethyl;
B is selected from the group consisting of (C[0125] 1-6)alkyl, (C3-6)cycloalkyl, C(O)NR40R41, phenyl and heteroaryl; wherein said (C1-6)alkyl, phenyl and heteroaryl are independently optionally substituted with one to three same or different halogens or from one to three same or different substituents selected from F; F is selected from the group consisting of (C1-6)alkyl, phenyl, hydroxy, (C1-6)alkoxy, halogen, benzyl, —NR42C(O)—(C1-6)alkyl, —NR42R43, COOR54 and —CONR42; wherein said (C1-6)alkyl is optionally substituted with one to three same or different halogen;
R[0126] 8 is selected from the group consisting of hydrogen and (C1-6)alkyl;
R[0127] 9 is selected from the group consisting of hydrogen and methyl;
X is selected from the group consisting of NR[0128] 9, O and S;
R[0129] 40 and R41 are independently selected from the group consisting of hydrogen, (C1-6)alkyl, (C1-6)alkoxy, phenyl and heteroaryl; wherein said phenyl and heteroaryl are independently optionally substituted with one to three same or different halogen, methyl, or CF3 groups;
R[0130] 42 and R43 are independently selected from the group consisting of hydrogen and (C1-6)alkyl;
R[0131] 44 is selected from the group consisting of H, (C1-6)alkyl, CO(C1-6)alkyl, C(O)-phenyl and —CONRaRb;
R[0132] a and Rb are each independently H, (C1-6)alkyl or phenyl;
R[0133] 54 is selected from the group consisting of hydrogen and (C1-6)alkyl;
R[0134] 57 is (C1-6)alkyl; and
heteroaryl is selected from the group consisting of pyridinyl, pyrazinyl, pyridazinyl, pyrimidinyl, furanyl, thienyl, benzothienyl, thiazolyl, isothiazolyl, oxazolyl, benzooxazolyl, isoxazolyl, imidazolyl, benzoimidazolyl, 1H-imidazo[4,5-b]pyridin-2-yl, 1H-imidazo[4,5-c]pyridin-2-yl, oxadiazolyl, thiadiazolyl, pyrazolyl, tetrazolyl, tetrazinyl, triazinyl and triazolyl. [0135]
A more preferred embodiment of a first aspect of the invention are compounds of Formula I, including pharmaceutically acceptable salts thereof,
[0136]
wherein: [0137]
A is selected from the group consisting of phenyl and D; wherein D is selected from the group consisting of pyridinyl, furanyl and thienyl; wherein phenyl and D are independently optionally substituted with one or two of the same or different amino or halogen; [0139]
W is selected from the group consisting of
[0140]
R[0141] 1 is hydrogen; and
Q is a member selected from groups (A) and (B) consisting of
[0142]
provided R
[0143] 2 and R
3 are each independently hydrogen, methoxy or halogen; and R
4 and R
5 are selected from the group consisting of hydrogen, halogen, cyano, COOR
8, C(O)NHCH
3, C(O)NHheteroaryl, and heteroaryl; and
provided R[0144] 2 is hydrogen, methoxy or halogen;
R[0145] 3 and R4 are selected from the group consisting of hydrogen, halogen, methoxy, cyano, COOR8, C(O)NHCH3, C(O)NHheteroaryl and heteroaryl; and R6 does not exist;
and ——represents a carbon-carbon bond in (A) and (B). [0146]
Another embodiment of the present invention is a method for treating mammals infected with a virus, wherein said virus is HIV, comprising administering to said mammal an antiviral effective amount of a compound of Formula I, and one or more pharmaceutically acceptable carriers, excipients or diluents; optionally the compound of Formula I can be administered in combination with an antiviral effective amount of an AIDS treatment agent selected from the group consisting of: (a) an AIDS antiviral agent; (b) an anti-infective agent; (c) an immunomodulator; and (d) HIV entry inhibitors. [0147]
Another embodiment of the present invention is a pharmaceutical composition comprising an antiviral effective amount of a compound of Formula I and one or more pharmaceutically acceptable carriers, excipients, diluents and optionally in combination with an antiviral effective amount of an AIDS treatment agent selected from the group consisting of: (a) an AIDS antiviral agent; (b) an anti-infective agent; (c) an immunomodulator; and (d) HIV entry inhibitors. [0148]
DETAILED DESCRIPTION OF THE INVENTION
Since the compounds of the present invention, may possess asymmetric centers and therefore occur as mixtures of diastereomers and enantiomers, the present invention includes the individual diastereoisomeric and enantiomeric forms of the compounds of Formula I in addition to the mixtures thereof. [0149]
Definitions
The term “C[0150] 1-6 alkyl” as used herein and in the claims (unless specified otherwise) mean straight or branched chain alkyl groups such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl, amyl, hexyl and the like.
“Halogen” refers to chlorine, bromine, iodine or fluorine. [0151]
An “aryl” group refers to an all carbon monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups having a completely conjugated pi-electron system. Examples, without limitation, of aryl groups are phenyl, napthalenyl and anthracenyl. The aryl group may be substituted or unsubstituted. When substituted the substituted group(s) is preferably one or more selected from alkyl, cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, heteroaryloxy, heteroalicycloxy, thiohydroxy, thioaryloxy, thioheteroaryloxy, thioheteroalicycloxy, cyano, halogen, nitro, carbonyl, O-carbamyl, N-carbamyl, C-amido, N-amido, C-carboxy, O-carboxy, sulfinyl, sulfonyl, sulfonamido, trihalomethyl, ureido, amino and —NR[0152] xRy, wherein Rx and Ry are independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl, C-carboxy, sulfonyl, trihalomethyl, and, combined, a five- or six-member heteroalicyclic ring.
As used herein, a “heteroaryl” group refers to a monocyclic or fused ring (i.e., rings which share an adjacent pair of atoms) group having in the ring(s) one or more atoms selected from the group consisting of nitrogen, oxygen and sulfur and, in addition, having a completely conjugated pi-electron system. It should be noted that the term heteroaryl is intended to encompass an N-oxide of the parent heteroaryl if such an N-oxide is chemically feasible as is known in the art. Examples, without limitation, of heteroaryl groups are furyl, thienyl, benzothienyl, thiazolyl, imidazolyl, oxazolyl, oxadiazolyl, thiadiazolyl, benzothiazolyl, triazolyl, tetrazolyl, isoxazolyl, isothiazolyl, pyrrolyl, pyranyl, tetrahydropyranyl, pyrazolyl, pyridyl, pyrimidinyl, quinolinyl, isoquinolinyl, purinyl, carbazolyl, benzoxazolyl, benzimidazolyl, indolyl, isoindolyl, pyrazinyl. diazinyl, pyrazine, triazinyltriazine, tetrazinyl, and tetrazolyl. When substituted the substituted group(s) is preferably one or more selected from alkyl, cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, heteroaryloxy, heteroalicycloxy, thiohydroxy, thioaryloxy, thioheteroaryloxy, thioheteroalicycloxy, cyano, halogen, nitro, carbonyl, O-carbamyl, N-carbamyl, C-amido, N-amido, C-carboxy, O-carboxy, sulfinyl, sulfonyl, sulfonamido, trihalomethyl, ureido, amino, and —NR[0153] xRy, wherein Rx and Ry are as defined above.
As used herein, a “heteroalicyclic” group refers to a monocyclic or fused ring group having in the ring(s) one or more atoms selected from the group consisting of nitrogen, oxygen and sulfur. The rings may also have one or more double bonds. However, the rings do not have a completely conjugated pi-electron system. Examples, without limitation, of heteroalicyclic groups are azetidinyl, piperidyl, piperazinyl, imidazolinyl, thiazolidinyl, 3-pyrrolidin-1-yl, morpholinyl, thiomorpholinyl and tetrahydropyranyl. When substituted the substituted group(s) is preferably one or more selected from alkyl, cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, heteroaryloxy, heteroalicycloxy, thiohydroxy, thioalkoxy, thioaryloxy, thioheteroaryloxy, thioheteroalicycloxy, cyano, halogen, nitro, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, C-thioamido, N-amido, C-carboxy, O-carboxy, sulfinyl, sulfonyl, sulfonamido, trihalomethanesulfonamido, trihalomethanesulfonyl, silyl, guanyl, guanidino, ureido, phosphonyl, amino and —NR[0154] xRy, wherein Rx and Ry are as defined above.
An “alkyl” group refers to a saturated aliphatic hydrocarbon including straight chain and branched chain groups. Preferably, the alkyl group has 1 to 20 carbon atoms (whenever a numerical range; e.g., “1-20”, is stated herein, it means that the group, in this case the alkyl group may contain 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc. up to and including 20 carbon atoms). More preferably, it is a medium size alkyl having 1 to 10 carbon atoms. Most preferably, it is a lower alkyl having 1 to 4 carbon atoms. The alkyl group may be substituted or unsubstituted. When substituted, the substituent group(s) is preferably one or more individually selected from trihaloalkyl, cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, heteroaryloxy, heteroalicycloxy, thiohydroxy, thioalkoxy, thioaryloxy, thioheteroaryloxy, thioheteroalicycloxy, cyano, halo, nitro, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, C-thioamido, N-amido, C-carboxy, O-carboxy, sulfinyl, sulfonyl, sulfonamido, trihalomethanesulfonamido, trihalomethanesulfonyl, and combined, a five- or six-member heteroalicyclic ring. [0155]
A “cycloalkyl” group refers to an all-carbon monocyclic or fused ring (i.e., rings which share and adjacent pair of carbon atoms) group wherein one or more rings does not have a completely conjugated pi-electron system. Examples, without limitation, of cycloalkyl groups are cyclopropane, cyclobutane, cyclopentane, cyclopentene, cyclohexane, cyclohexadiene, cycloheptane, cycloheptatriene and adamantane. A cycloalkyl group may be substituted or unsubstituted. When substituted, the substituent group(s) is preferably one or more individually selected from alkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, heteroaryloxy, heteroalicycloxy, thiohydroxy, thioalkoxy, thioaryloxy, thioheteroarylloxy, thioheteroalicycloxy, cyano, halo, nitro, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, C-thioamido, N-amido, C-carboxy, O-carboxy, sulfinyl, sulfonyl, sulfonamido, trihalo-methanesulfonamido, trihalomethanesulfonyl, silyl, guanyl, guanidino, ureido, phosphonyl, amino and —NR[0156] xRy with Rx and Ry as defined above.
An “alkenyl” group refers to an alkyl group, as defined herein, consisting of at least two carbon atoms and at least one carbon-carbon double bond. [0157]
An “alkynyl” group refers to an alkyl group, as defined herein, consisting of at least two carbon atoms and at least one carbon-carbon triple bond. [0158]
A “hydroxy” group refers to an —OH group. [0159]
An “alkoxy” group refers to both an —O-alkyl and an —O-cycloalkyl group as defined herein. [0160]
An “aryloxy” group refers to both an —O-aryl and an —O-heteroaryl group, as defined herein. [0161]
A “heteroaryloxy” group refers to a heteroaryl-O— group with heteroaryl as defined herein. [0162]
A “heteroalicycloxy” group refers to a heteroalicyclic-O— group with heteroalicyclic as defined herein. [0163]
A “thiohydroxy” group refers to an —SH group. [0164]
A “thioalkoxy” group refers to both an S-alkyl and an —S-cycloalkyl group, as defined herein. [0165]
A “thioaryloxy” group refers to both an —S-aryl and an —S-heteroaryl group, as defined herein. [0166]
A “thioheteroaryloxy” group refers to a heteroaryl-S— group with heteroaryl as defined herein. [0167]
A “thioheteroalicycloxy” group refers to a heteroalicyclic-S— group with heteroalicyclic as defined herein. [0168]
A “carbonyl” group refers to a —C(═O)—R″ group, where R″ is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl (bonded through a ring carbon) and heteroalicyclic (bonded through a ring carbon), as each is defined herein. [0169]
An “aldehyde” group refers to a carbonyl group where R″ is hydrogen. [0170]
A “thiocarbonyl” group refers to a —C(═S)—R″ group, with R″ as defined herein. [0171]
A “Keto” group refers to a —CC(═O)C— group wherein the carbon on either or both sides of the C═O may be alkyl, cycloalkyl, aryl or a carbon of a heteroaryl or heteroaliacyclic group. [0172]
A “trihalomethanecarbonyl” group refers to a Z[0173] 3CC(═O)— group with said Z being a halogen.
A “C-carboxy” group refers to a —C(═O)O—R″ groups, with R″ as defined herein. [0174]
An “O-carboxy” group refers to a R″C(—O)O-group, with R″ as defined herein. [0175]
A “carboxylic acid” group refers to a C-carboxy group in which R″ is hydrogen. [0176]
A “trihalomethyl” group refers to a —CZ[0177] 3, group wherein Z is a halogen group as defined herein.
A “trihalomethanesulfonyl” group refers to an Z[0178] 3CS(═O)2— groups with Z as defined above.
A “trihalomethanesulfonamido” group refers to a Z[0179] 3CS(═O)2NRx— group with Z and RX as defined herein.
A “sulfinyl” group refers to a —S(═O)—R″ group, with R″ as defined herein and, in addition, as a bond only; i.e., —S(O)—. [0180]
A “sulfonyl” group refers to a —S(═O)[0181] 2R″ group with R″ as defined herein and, in addition as a bond only; i.e., —S(O)2—.
A “S-sulfonamido” group refers to a —S(═O)[0182] 2NRXRY, with RX and RY as defined herein.
A “N-Sulfonamido” group refers to a R″S(═O)[0183] 2NRX— group with Rx as defined herein.
A “O-carbamyl” group refers to a —OC(═O)NR[0184] xRy as defined herein.
A “N-carbamyl” group refers to a R[0185] xOC(═O)NRy group, with Rx and Ry as defined herein.
A “O-thiocarbamyl” group refers to a —OC(═S)NR[0186] xRy group with Rx and Ry as defined herein.
A “N-thiocarbamyl” group refers to a R[0187] xOC(═S)NRy— group with Rx and Ry as defined herein.
An “amino” group refers to an —NH[0188] 2 group.
A “C-amido” group refers to a —C(═O)NR[0189] xRy group with Rx and Ry as defined herein.
A “C-thioamido” group refers to a —C(═S)NR[0190] xRy group, with Rx and Ry as defined herein.
A “N-amido” group refers to a R[0191] xC(═O)NRy— group, with Rx and Ry as defined herein.
An “ureido” group refers to a —NR[0192] xC(═O)NRyRy2 group with Rx and Ry as defined herein and Ry2 defined the same as Rx and Ry.
A “guanidino” group refers to a —R[0193] xNC(═N)NRyRy2 group, with Rx, Ry and Ry2 as defined herein.
A “guanyl” group refers to a R[0194] xRyNC(═N)— group, with Rx and RY as defined herein.
A “cyano” group refers to a —CN group. [0195]
A “silyl” group refers to a —Si(R″)[0196] 3, with R″ as defined herein.
A “phosphonyl” group refers to a P(═O)(OR[0197] x)2 with Rx as defined herein.
A “hydrazino” group refers to a —NR[0198] xNRyRy2 group with Rx, Ry and Ry2 as defined herein.
Any two adjacent R groups may combine to form an additional aryl, cycloalkyl, heteroaryl or heterocyclic ring fused to the ring initially bearing those R groups. [0199]
It is known in the art that nitogen atoms in heteroaryl systems can be “participating in a heteroaryl ring double bond”, and this refers to the form of double bonds in the two tautomeric structures which comprise five-member ring heteroaryl groups. This dictates whether nitrogens can be substituted as well understood by chemists in the art. The disclosure and claims of the present invention are based on the known general principles of chemical bonding. It is understood that the claims do not encompass structures known to be unstable or not able to exist based on the literature. [0200]
Physiologically acceptable salts and prodrugs of compounds disclosed herein are within the scope of this invention. The term “pharmaceutically acceptable salt” as used herein and in the claims is intended to include nontoxic base addition salts. Suitable salts include those derived from organic and inorganic acids such as, without limitation, hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, methanesulfonic acid, acetic acid, tartaric acid, lactic acid, sulfinic acid, citric acid, maleic acid, fumaric acid, sorbic acid, aconitic acid, salicylic acid, phthalic acid, and the like. The term “pharmaceutically acceptable salt” as used herein is also intended to include salts of acidic groups, such as a carboxylate, with such counterions as ammonium, alkali metal salts, particularly sodium or potassium, alkaline earth metal salts, particularly calcium or magnesium, and salts with suitable organic bases such as lower alkylamines (methylamine, ethylamine, cyclohexylamine, and the like) or with substituted lower alkylamines (e.g. hydroxyl-substituted alkylamines such as diethanolamine, triethanolamine or tris(hydroxymethyl)-aminomethane), or with bases such as piperidine or morpholine. [0201]
In the method of the present invention, the term “antiviral effective amount” means the total amount of each active component of the method that is sufficient to show a meaningful patient benefit, i.e., healing of acute conditions characterized by inhibition of the HIV infection. When applied to an individual active ingredient, administered alone, the term refers to that ingredient alone. When applied to a combination, the term refers to combined amounts of the active ingredients that result in the therapeutic effect, whether administered in combination, serially or simultaneously. The terms “treat, treating, treatment” as used herein and in the claims means preventing or ameliorating diseases associated with HIV infection. [0202]
The present invention is also directed to combinations of the compounds with one or more agents useful in the treatment of AIDS. For example, the compounds of this invention may be effectively administered, whether at periods of pre-exposure and/or post-exposure, in combination with effective amounts of the AIDS antivirals, immunomodulators, antiinfectives, or vaccines, such as those in the following table.
[0203] | |
| |
| Drug Name | Manufacturer | Indication |
| |
| |
| 097 | Hoechst/Bayer | HIV infection, |
| | | AIDS, ARC |
| | | (non-nucleoside |
| | | reverse trans- |
| | | criptase (RT) |
| | | inhibitor) |
| Amprenivir | Glaxo Wellcome | HIV infection, |
| 141 W94 | | AIDS, ARC |
| GW 141 | | (protease inhibitor) |
| Abacavir (1592U89) | Glaxo Wellcome | HIV infection, |
| GW 1592 | | AIDS, ARC |
| | | (RT inhibitor) |
| Acemannan | Carrington Labs | ARC |
| | (Irving, TX) |
| Acyclovir | Burroughs Wellcome | HIV infection, AIDS, |
| | | ARC, in combination |
| | | with AZT |
| AD-439 | Tanox Biosystems | HIV infection, AIDS, |
| | | ARC |
| AD-519 | Tanox Biosystems | HIV infection, AIDS, |
| | | ARC |
| Adefovir dipivoxil | Gilead Sciences | HIV infection |
| AL-721 | Ethigen | ARC, PGL |
| | (Los Angeles, CA) | HIV positive, AIDS |
| Alpha Interferon | Glaxo Wellcome | Kaposi's sarcoma, |
| | | HIV in combination |
| | | w/Retrovir |
| Ansamycin | Adria Laboratories | ARC |
| LM 427 | (Dublin, OH) |
| | Erbamont |
| | (Stamford, CT) |
| Antibody which | Advanced Biotherapy | AIDS, ARC |
| Neutralizes pH | Concepts |
| Labile alpha aberrant | (Rockville, MD) |
| Interferon |
| AR177 | Aronex Pharm | HIV infection, AIDS, |
| | | ARC |
| Beta-fluoro-ddA | Nat'l Cancer Institute | AIDS-associated |
| | | diseases |
| BMS-232623 | Bristol-Myers Squibb/ | HIV infection, |
| (CGP-73547) | Novartis | AIDS, ARC |
| | | (protease inhibitor) |
| BMS-234475 | Bristol-Myers Squibb/ | HIV infection, |
| (CGP-61755) | Novartis | AIDS, ARC |
| | | (protease inhibitor) |
| CI-1012 | Warner-Lambert | HIV-1 infection |
| Cidofovir | Gilead Science | CMV retinitis, |
| | | herpes, papillomavirus |
| Curdlan sulfate | AJI Pharma USA | HIV infection |
| Cytomegalovirus | MedImmune | CMV retinitis |
| Immune globin |
| Cytovene | Syntex | Sight threatening |
| Ganciclovir | | CMV |
| | | peripheral CMV |
| | | retinitis |
| Delaviridine | Pharmacia-Upjohn | HIV infection, |
| | | AIDS, ARC |
| | | (RT inhibitor) |
| Dextran Sulfate | Ueno Fine Chem. | AIDS, ARC, HIV |
| | Ind. Ltd. (Osaka, | positive |
| | Japan) | asymptomatic |
| ddC | Hoffman-La Roche | HIV infection, AIDS, |
| Dideoxycytidine | | ARC |
| ddI | Bristol-Myers Squibb | HIV infection, AIDS, |
| Dideoxyinosine | | ARC; combination |
| | | with AZT/d4T |
| DMP-450 | AVID | HIV infection, |
| | (Camden, NJ) | AIDS, ARC |
| | | (protease inhibitor) |
| Efavirenz | DuPont Merck | HIV infection, |
| (DMP 266) | | AIDS, ARC |
| (-)6-Chloro-4-(S)- | | (non-nucleoside RT |
| cyclopropylethynyl- | | inhibitor) |
| 4(S)-trifluoro- |
| methyl-1,4-dihydro- |
| 2H-3,1-benzoxazin- |
| 2-one, STOCRINE |
| EL10 | Elan Corp, PLC | HIV infection |
| | (Gainesville, GA) |
| Famciclovir | Smith Kline | herpes zoster, |
| | | herpes simplex |
| FTC | Emory University | HIV infection, |
| | | AIDS, ARC |
| | | (reverse transcriptase |
| | | inhibitor) |
| GS 840 | Gilead | HIV infection, |
| | | AIDS, ARC |
| | | (reverse transcriptase |
| | | inhibitor) |
| HBY097 | Hoechst Marion | HIV infection, |
| | Roussel | AIDS, ARC |
| | | (non-nucleoside |
| | | reverse transcriptase |
| | | inhibitor) |
| Hypericin | VIMRx Pharm. | HIV infection, AIDS, |
| | | ARC |
| Recombinant Human | Triton Biosciences | AIDS, Kaposi's |
| Interferon Beta | (Almeda, CA) | sarcoma, ARC |
| Interferon alfa-n3 | Interferon Sciences | ARC, AIDS |
| Indinavir | Merck | HIV infection, AIDS, |
| | | ARC, asymptomatic |
| | | HIV positive, also in |
| | | combination with |
| | | AZT/ddI/ddC |
| ISIS 2922 | ISIS Pharmaceuticals | CMV retinitis |
| KNI-272 | Nat'l Cancer Institute | HIV-assoc. diseases |
| Lamivudine, 3TC | Glaxo Wellcome | HIV infection, |
| | | AIDS, ARC |
| | | (reverse |
| | | transcriptase |
| | | inhibitor); also |
| | | with AZT |
| Lobucavir | Bristol-Myers Squibb | CMV infection |
| Nelfinavir | Agouron | HIV infection, |
| | Pharmaceuticals | AIDS, ARC |
| | | (protease inhibitor) |
| Nevirapine | Boeheringer | HIV infection, |
| | Ingleheim | AIDS, ARC |
| | | (RT inhibitor) |
| Novapren | Novaferon Labs, Inc. | HIV inhibitor |
| | (Akron, OH) |
| Peptide T | Peninsula Labs | AIDS |
| Octapeptide | (Belmont, CA) |
| Sequence |
| Trisodium | Astra Pharm. | CMV retinitis, HIV |
| Phosphonoformate | Products, Inc. | infection, other CMV |
| | | infections |
| PNU-140690 | Pharmacia Upjohn | HIV infection, |
| | | AIDS, ARC |
| | | (protease inhibitor) |
| Probucol | Vyrex | HIV infection, AIDS |
| RBC-CD4 | Sheffield Med. | HIV infection, |
| | Tech (Houston, TX) | AIDS, ARC |
| Ritonavir | Abbott | HIV infection, |
| | | AIDS, ARC |
| | | (protease inhibitor) |
| Saquinavir | Hoffmann- | HIV infection, |
| | LaRoche | AIDS, ARC |
| | | (protease inbitor) |
| Stavudine; d4T | Bristol-Myers Squibb | HIV infection, AIDS, |
| Didehydrodeoxy- | | ARC |
| thymidine |
| Valaciclovir | Glaxo Wellcome | Genital HSV & CMV |
| | | infections |
| Virazole | Viratek/ICN | asymptomatic HIV |
| Ribavirin | (Costa Mesa, CA) | positive, LAS, ARC |
| VX-478 | Vertex | HIV infection, AIDS, |
| | | ARC |
| Zalcitabine | Hoffmann-LaRoche | HIV infection, AIDS, |
| | | ARC, with AZT |
| Zidovudine; AZT | Glaxo Wellcome | HIV infection, AIDS, |
| | | ARC, Kaposi's |
| | | sarcoma, in |
| | | combination with |
| | | other therapies |
| AS-101 | Wyeth-Ayerst | AIDS |
| Bropirimine | Pharmacia Upjohn | Advanced AIDS |
| Acemannan | Carrington Labs, Inc. | AIDS, ARC |
| | (Irving, TX) |
| CL246,738 | American Cyanamid | AIDS, Kaposi's |
| | Lederle Labs | sarcoma |
| EL10 | Elan Corp, PLC | HIV infection |
| | (Gainesville, GA) |
| FP-21399 | Fuki ImmunoPharm | Blocks HIV fusion |
| | | with CD4+ cells |
| Gamma Interferon | Genentech | ARC, in combination |
| | | w/TNF (tumor |
| | | necrosis factor) |
| Granulocyte | Genetics Institute | AIDS |
| Macrophage Colony | Sandoz |
| Stimulating Factor |
| Granulocyte | Hoechst-Roussel | AIDS |
| Macrophage Colony | Immunex |
| Stimulating Factor |
| Granulocyte | Schering-Plough | AIDS, |
| Macrophage Colony | | combination |
| Stimulating Factor | | w/AZT |
| HIV Core Particle | Rorer | Seropositive HIV |
| Immunostimulant |
| IL-2 | Cetus | AIDS, in combination |
| Interleukin-2 | | w/AZT |
| IL-2 | Hoffman-LaRoche | AIDS, ARC, HIV, in |
| Interleukin-2 | Immunex | combination w/AZT |
| IL-2 | Chiron | AIDS, increase in |
| Interleukin-2 | | CD4 cell counts |
| (aldeslukin) |
| Immune Globulin | Cutter Biological | Pediatric AIDS, in |
| Intravenous | (Berkeley, CA) | combination w/AZT |
| (human) |
| IMREG-1 | Imreg | AIDS, Kaposi's |
| | (New Orleans, LA) | sarcoma, ARC, PGL |
| IMREG-2 | Imreg | AIDS, Kaposi's |
| | (New Orleans, LA) | sarcoma, ARC, PGL |
| Imuthiol Diethyl | Merieux Institute | AIDS, ARC |
| Dithio Carbamate |
| Alpha-2 | Schering Plough | Kaposi's sarcoma |
| Interferon | | w/AZT, AIDS |
| Methionine- | TNI Pharmaceutical | AIDS, ARC |
| Enkephalin | (Chicago, IL) |
| MTP-PE | Ciba-Geigy Corp. | Kaposi's sarcoma |
| Muramyl-Tripeptide |
| Granulocyte | Amgen | AIDS, in combination |
| Colony Stimulating | | w/AZT |
| Factor |
| Remune | Immune Response | Immunotherapeutic |
| | Corp. |
| rCD4 | Genentech | AIDS, ARC |
| Recombinant |
| Soluble Human CD4 |
| rCD4-IgG | | AIDS, ARC |
| hybrids |
| Recombinant | Biogen | AIDS, ARC |
| Soluble Human CD4 |
| Interferon | Hoffman-La Roche | Kaposi's sarcoma |
| Alfa 2a | | AIDS, ARC, |
| | | in combination w/AZT |
| SK&F106528 | Smith Kline | HIV infection |
| Soluble T4 |
| Thymopentin | Immunobiology | HIV infection |
| | Research Institute |
| | (Annandale, NJ) |
| Tumor Necrosis | Genentech | ARC, in combination |
| Factor; TNF | | w/gamma Interferon |
| Clindamycin with | Pharmacia Upjohn | PCP |
| Primaquine |
| Fluconazole | Pfizer | Cryptococcal |
| | | meningitis, |
| | | candidiasis |
| Pastille | Squibb Corp. | Prevention of |
| Nystatin Pastille | | oral candidiasis |
| Ornidyl | Merrell Dow | PCP |
| Eflornithine |
| Pentamidine | LyphoMed | PCP treatment |
| Isethionate (IM & IV) | (Rosemont, IL) |
| Trimethoprim | | Antibacterial |
| Trimethoprim/sulfa | | Antibacterial |
| Piritrexim | Burroughs Wellcome | PCP treatment |
| Pentamidine | Fisons Corporation | PCP prophylaxis |
| Isethionate for |
| Inhalation |
| Spiramycin | Rhone-Poulene | Cryptosporidial |
| | diarrhea |
| Intraconazole- | Janssen-Pharm. | Histoplasmosis; |
| R51211 | | cryptococcal |
| | | meningitis |
| Trimetrexate | Wamer-Lambert | PCP |
| Daunorubicin | NeXstar, Sequus | Kaposi's sarcoma |
| Recombinant Human | Ortho Pharm. Corp. | Severe anemia |
| Erythropoietin | | assoc. with AZT |
| | | therapy |
| Recombinant Human | Serono | AIDS-related |
| Growth Hormone | | wasting, cachexia |
| Megestrol Acetate | Bristol-Myers Squibb | Treatment of |
| | | anorexia assoc. |
| | | W/AIDS |
| Testosterone | Alza, Smith Kline | AIDS-related wasting |
| Total Enteral | Norwich Eaton | Diarrhea and |
| Nutrition | Pharmaceuticals | malabsorption |
| | | related to AIDS |
| |
Additionally, the compounds of the invention herein may be used in combination with another class of agents for treating AIDS which are called HIV entry inhibitors. Examples of such HIV entry inhibitors are discussed in DRUGS OF THE FUTURE 1999, 24(12), pp. 1355-1362; CELL, Vol. 9, pp. 243-246, Oct. 29, 1999; and DRUG DISCOVERY TODAY, Vol. 5, No. 5, May 2000, pp. 183-194. [0204]
It will be understood that the scope of combinations of the compounds of this invention with AIDS antivirals, immunomodulators, anti-infectives, HIV entry inhibitors or vaccines is not limited to the list in the above Table, but includes in principle any combination with any pharmaceutical composition useful for the treatment of AIDS. [0205]
Preferred combinations are simultaneous or alternating treatments of with a compound of the present invention and an inhibitor of HIV protease and/or a non-nucleoside inhibitor of HIV reverse transcriptase. An optional fourth component in the combination is a nucleoside inhibitor of HIV reverse transcriptase, such as AZT, 3TC, ddC or ddI. A preferred inhibitor of HIV protease is indinavir, which is the sulfate salt of N-(2(R)-hydroxy-1-(S)-indanyl)-2(R)-phenylmethyl-4-(S)-hydroxy-5-(1-(4-(3-pyridyl-methyl)-2(S)-N′-(t-butylcarboxamido)-piperazinyl))-pentaneamide ethanolate, and is synthesized according to U.S. Pat. No. 5,413,999. Indinavir is generally administered at a dosage of 800 mg three times a day. Other preferred protease inhibitors are nelfinavir and ritonavir. Another preferred inhibitor of HIV protease is saquinavir which is administered in a dosage of 600 or 1200 mg tid. Preferred non-nucleoside inhibitors of HIV reverse transcriptase include efavirenz. The preparation of ddC, ddI and AZT are also described in EPO 0,484,071. These combinations may have unexpected effects on limiting the spread and degree of infection of HIV. Preferred combinations include those with the following (1) indinavir with efavirenz, and, optionally, AZT and/or 3TC and/or ddI and/or ddC; (2) indinavir, and any of AZT and/or ddI and/or ddC and/or 3TC, in particular, indinavir and AZT and 3TC; (3) stavudine and 3TC and/or zidovudine; (4) zidovudine and lamivudine and 141W94 and 1592U89; (5) zidovudine and lamivudine. [0206]
In such combinations the compound of the present invention and other active agents may be administered separately or in conjunction. In addition, the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s). [0207]
Abbreviations
The following abbreviations, most of which are conventional abbreviations well known to those skilled in the art, are used throughout the description of the invention and the examples. Some of the abbreviations used are as follows:
[0208] | |
| |
| h = | hour(s) |
| rt = | room temperature |
| mol = | mole(s) |
| mmol = | millimole(s) |
| g = | gram(s) |
| mg = | milligram(s) |
| mL = | milliliter(s) |
| TFA = | Trifluoroacetic Acid |
| DCE = | 1,2-Dichloroethane |
| CH2Cl2 = | Dichloromethane |
| TPAP = | tetrapropylammonium perruthenate |
| THF = | Tetrahydofuran |
| DEPBT = | 3-(Diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)- |
| | one |
| DMAP = | 4-dimethylaminopyridine |
| P-EDC = | Polymer supported 1-(3-dimethylaminopropyl)-3- |
| | ethylcarbodiimide |
| EDC = | 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide |
| DMF = | N,N-dimethylformamide |
| Hunig's Base = | N,N-Diisopropylethylamine |
| mCPBA = | meta-Chloroperbenzoic Acid |
| azaindole = | 1H-Pyrrolo-pyridine |
| 4-azaindole = | 1H-pyrrolo[3,2-b]pyridine |
| 5-azaindole = | 1H-Pyrrolo[3,2-c]pyridine |
| 6-azaindole = | 1H-pyrrolo[2,3-c]pyridine |
| 7-azaindole = | 1H-Pyrrolo[2,3-b]pyridine |
| PMB = | 4-Methoxybenzyl |
| DDQ = | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| OTf = | Trifluoromethanesulfonoxy |
| NMM = | 4-Methylmorpholine |
| PIP-COPh = | 1-Benzoylpiperazine |
| NaHMDS = | Sodium hexamethyldisilazide |
| EDAC = | 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide |
| TMS = | Trimethylsilyl |
| DCM = | Dichloromethane |
| DCE = | Dichloroethane |
| MeOH = | Methanol |
| THF = | Tetrahrdrofuran |
| EtOAc = | Ethyl Acetate |
| LDA = | Lithium diisopropylamide |
| TMP-Li = | 2,2,6,6-tetramethylpiperidinyl lithium |
| DME = | Dimethoxyethane |
| DIBALH = | Diisobutylaluminum hydride |
| HOBT = | 1-hydroxybenzotriazole |
| CBZ = | Benzyloxycarbonyl |
| PCC = | Pyridinium chlorochromate |
| |
Chemistry [0209]
The present invention comprises compounds of Formula I, their pharmaceutical formulations, and their use in patients suffering from or susceptible to HIV infection. The compounds of Formula I include pharmaceutically acceptable salts thereof. [0210]
The synthesis procedures and anti-HIV-1 activities of indoleoxoacetic pyrrolidine containing analogs are below. Procedures for making Z are described herein or in many cases references of Blair, Wang, Wallace, references 93-95 respectively. [0211]
Additional general procedures to construct substituted azaindole Q and Z of Formula I and intermediates useful for their synthesis are described in the following Schemes 1-16.
[0212]
Step A in Scheme 1 depicts the synthesis of an aza indole intermediate, 2, via the well known Bartoli reaction in which vinyl magnesium bromide reacts with an aryl or heteroaryl nitro group, such as in 1, to form a five-membered nitrogen containing ring as shown. Some references for the above transformation include: Bartoli et al. a) [0213] Tetrahedron Lett. 1989, 30, 2129. b) J. Chem. Soc. Perkin Trans. 1 1991, 2757. c) J. Chem. Soc. Perkin Trans. II 1991, 657. d) Synthesis (1999), 1594. In the preferred procedure, a solution of vinyl Magnesium bromide in THF (typically 1.0M but from 0.25 to 3.0M) is added dropwise to a solution of the nitro pyridine in THF at −78° under an inert atmosphere of either nitrogen or Argon. After addition is completed, the reaction temperature is allowed to warm to −20° and then is stirred for approximately 12 h before quenching with 20% aq ammonium chloride solution. The reaction is extracted with ethyl acetate and then worked up in a typical manner using a drying agent such as anhydrous magnesium sulfate or sodium sulfate. Products are generally purified using chromatography over Silica gel. Best results are generally achieved using freshly prepared vinyl Magnesium bromide. In some cases, vinyl Magnesium chloride may be substituted for vinyl Magnesium bromide.
Substituted azaindoles may be prepared by methods described in the literature or may be available from commercial sources. Thus there are many methods for carrying out step A in the literature and the specific examples are too numerous to even list. A review on the synthesis of 7-azaindoles has been published (Merour et. al. reference 102). Alternative syntheses of aza indoles and general methods for carrying out step A include, but are not limited to, those described in the following references (a-k below): a) Prokopov, A. A.; Yakhontov, L. N. [0214] Khim.-Farm. Zh. 1994, 28(7), 30-51; b) Lablache-Combier, A. Heteroaromatics. Photoinduced Electron Transfer 1988, Pt. C, 134-312; c) Saify, Zafar Said. Pak. J. Pharmacol. 1986, 2(2), 43-6; d) Bisagni, E. Jerusalem Symp. Quantum Chem. Biochem. 1972, 4, 439-45; e) Yakhontov, L. N. Usp. Khim. 1968, 37(7), 1258-87; f) Willette, R. E. Advan. Heterocycl. Chem. 1968, 9, 27-105; g) Mahadevan, I.; Rasmussen, M. Tetrahedron 1993, 49(33), 7337-52; h) Mahadevan, I.; Rasmussen, M. J. Heterocycl. Chem. 1992, 29(2), 359-67; i) Spivey, A. C.; Fekner, T.; Spey, S. E.; Adams, H. J. Org. Chem. 1999, 64(26), 9430-9443; j) Spivey, A. C.; Fekner, T.; Adams, H. Tetrahedron Lett. 1998, 39(48), 8919-8922; k) Advances in Heterocyclic Chemistry (Academic press) 1991, Vol. 52, pg 235-236 and references therein.
Step B. Intermediate 3 can be prepared by reaction of aza-indole, intermediate 2, with an excess of ClCOCOOMe in the presence of AlCl[0215] 3 (aluminum chloride) (Sycheva et al, Ref. 26, Sycheva, T. V.; Rubtsov, N. M.; Sheinker, Yu. N.; Yakhontov, L. N. Some reactions of 5-cyano-6-chloro-7-azaindoles and lactam-lactim tautomerism in 5-cyano-6-hydroxy-7-azaindolines. Khim. Geterotsikl. Soedin., 1987, 100-106). Typically an inert solvent such as CH2Cl2 is used but others such as THF, Et2O, DCE, dioxane, benzene, or toluene may find applicability either alone or in mixtures. Other oxalate esters such as ethyl or benzyl mono esters of oxalic acid could also suffice for either method shown above. More lipophilic esters ease isolation during aqueous extractions. Phenolic or substituted phenolic (such as pentafluorophenol) esters enable direct coupling of the HW(C═O)A group, such as a piperazine, in Step D without activation. Lewis acid catalysts, such as tin tetrachloride, titanium IV chloride, and aluminum chloride are employed in Step B with aluminum chloride being most preferred. Alternatively, the azaindole is treated with a Grignard reagent such as MeMgI (methyl magnesium iodide), methyl magnesium bromide or ethyl magnesium bromide and a zinc halide, such as ZnCl2 (zinc chloride) or zinc bromide, followed by the addition of an oxalyl chloride mono ester, such as ClCOCOOMe (methyl chlorooxoacetate) or another ester as above, to afford the aza-indole glyoxyl ester (Shadrina et al, Ref. 25). Oxalic acid esters such as methyl oxalate, ethyl oxalate or as above are used. Aprotic solvents such as CH2Cl2, Et2O, benzene, toluene, DCE, or the like may be used alone or in combination for this sequence. In addition to the oxalyl chloride mono esters, oxalyl chloride itself may be reacted with the azaindole and then further reacted with an appropriate amine, such as a piperazine derivative (See Scheme 52, for example).
Step C. Hydrolysis of the methyl ester, (intermediate 3, Scheme 1) affords a potassium salt of intermediate 4, which is coupled with mono-benzoylated piperazine derivatives as shown in Step D of Scheme 1. Some typical conditions employ methanolic or ethanolic sodium hydroxide followed by careful acidification with aqueous hydrochloric acid of varying molarity but 1M HCl is preferred. The acidification is not utilized in many cases as described above for the preferred conditions. Lithium hydroxide or potassium hydroxide could also be employed and varying amounts of water could be added to the alcohols. Propanols or butanols could also be used as solvents. Elevated temperatures up to the boiling points of the solvents may be utilized if ambient temperatures do not suffice. Alternatively, the hydrolysis may be carried out in a non polar solvent such as CH[0216] 2Cl2 or THF in the presence of Triton B. Temperatures of −78° C. to the boiling point of the solvent may be employed but −10° C. is preferred. Other conditions for ester hydrolysis are listed in reference 41 and both this reference and many of the conditions for ester hydrolysis are well known to chemists of average skill in the art.
Alternative Procedures for Step B and C: [0217]
Imidazolium Chloroaluminate: [0218]
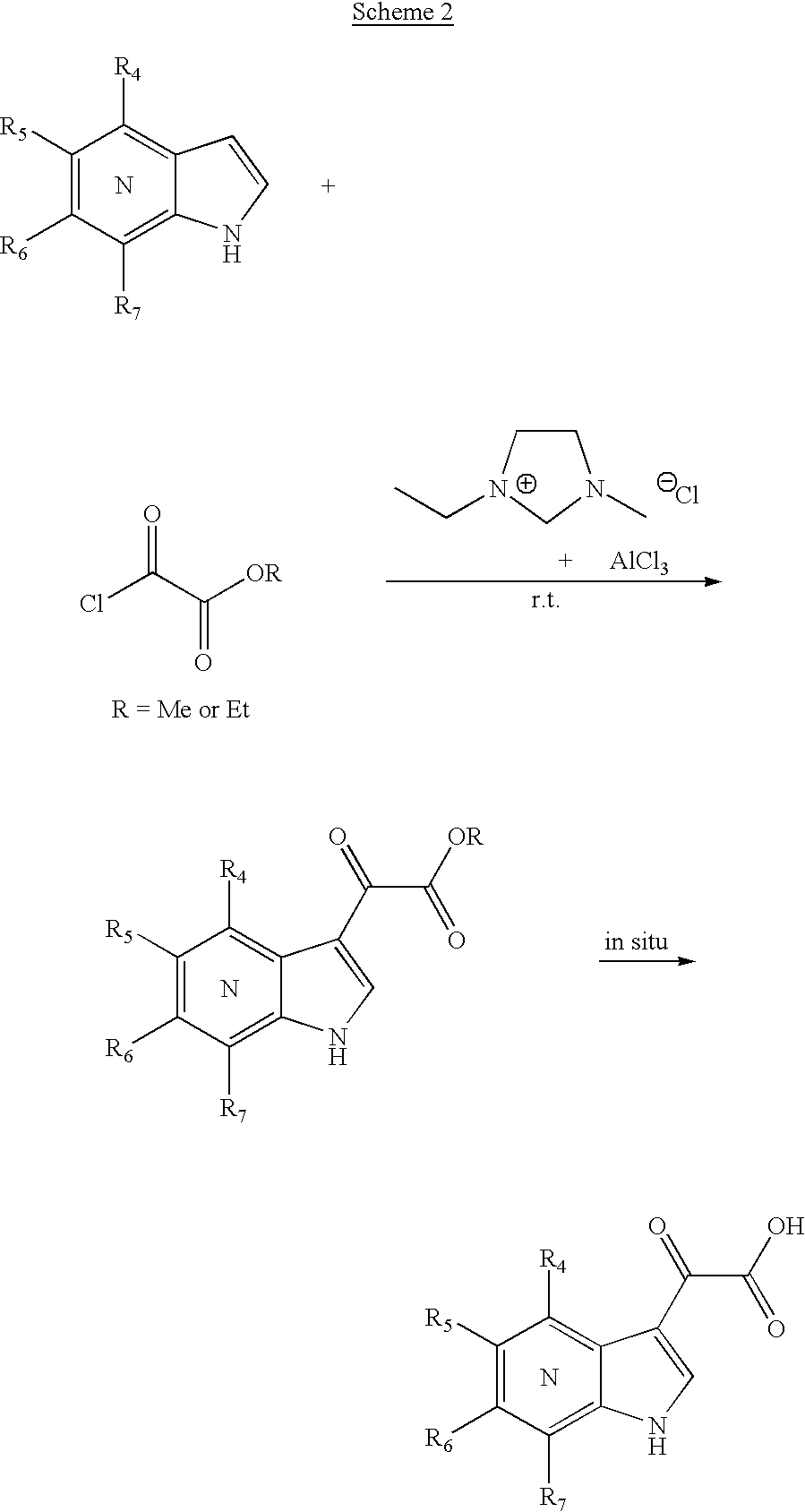
We found that ionic liquid 1-alkyl-3-alkylimidazolium chloroaluminate is generally useful in promoting the Friedel-Crafts type acylation of indoles and azaindoles. The ionic liquid is generated by mixing 1-alkyl-3-alkylimidazolium chloride with aluminium chloride at room temperature with vigorous stirring. 1:2 or 1:3 molar ratio of 1-alkyl-3-alkylimidazolium chloride to aluminium chloride is preferred. One particular useful imidazolium chloroaluminate for the acylation of azaindole with methyl or ethyl chlorooxoacetate is the 1-ethyl-3-methylimidazolium chloroaluminate. The reaction is typically performed at ambient temperature and the azaindoleglyoxyl ester can be isolated. More conveniently, we found that the glyoxyl ester can be hydrolyzed in situ at ambient temperature on prolonged reaction time (typically overnight) to give the corresponding glyoxyl acid for amide formation (Scheme 2).
[0219]
A representative experimental procedure is as follows: 1-ethyl-3-methylimidazolium chloride (2 equiv.; purchased from TCI; weighted under a stream of nitrogen) was stirred in an oven-dried round bottom flask at r.t. under a nitrogen atmosphere, and added aluminium chloride (6 equiv.; anhydrous powder packaged under argon in ampules purchased from Aldrich preferred; weighted under a stream of nitrogen). The mixture was vigorously stirred to form a liquid, which was then added azaindole (1 equiv.) and stirred until a homogenous mixture resulted. The reaction mixture was added dropwise ethyl or methyl chlorooxoacetate (2 equiv.) and then stirred at r.t. for 16 h. After which time, the mixture was cooled in an ice-water bath and the reaction quenched by carefully adding excess water. The precipitates were filtered, washed with water and dried under high vacuum to give the azaindoleglyoxyl acid. For some examples, 3 equivalents of 1-ethyl-3-methylimidazolium chloride and chlorooxoacetate may be required. [0220]
Related references: (1) Welton, T. [0221] Chem Rev. 1999, 99, 2071; (2) Surette, J. K. D.; Green, L.; Singer, R. D. Chem. Commun. 1996, 2753; (3) Saleh, R. Y. WO 0015594.
Step D. The acid intermediate, 4, from step C of Scheme 1 is coupled with an amine A(C═O)WH preferably in the presence of DEPBT (3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one) and N,N-diisopropylethylamine, commonly known as Hunig's base, to provide azaindole piperazine diamides. DEPBT was prepared according to the procedure of Ref. 28, Li, H.; Jiang, X.; Ye, Y. -H.; Fan, C.; Romoff, T.; Goodman, M. [0222] Organic Lett., 1999, 1, 91-93. Typically an inert solvent such as DMF or THF is used but other aprotic solvents could be used. The group W as referred to herein is described below.
The amide bond construction reaction could be carried out using the preferred conditions described above, the EDC conditions described below, other coupling conditions described in this application, or alternatively by applying the conditions or coupling reagents for amide bond construction described later in this application for construction of substituents R[0223] 1-R4. Some specific nonlimiting examples are given in this application.
It should be noted that in many cases reactions are depicted for only one position of an intermediate, such as the R
[0224] 5 position, for example. It is to be understood that such reactions could be used at other positions, such as R
2-R
4, of the various intermediates. Reaction conditions and methods given in the specific examples are broadly applicable to compounds with other substitution and other tranformations in this application. Schemes 1 and 2 describe general reaction schemes for taking appropriately substituted Q (indoles and azaindoles) and converting them to compounds of Formula I. While these schemes are very general, other permutations such as carrying a precursor or precursors to substituents R
2 through R
5 through the reaction scheme and then converting it to a compound of Formula I in the last step are also contemplated methods of this invention. Nonlimiting examples of such strategies follow in subsequent schemes.
The amide coupling with amine H—W—C(O)A is shown in Scheme 3, step a3. The group W as referred to herein is either
[0225]
One preferred method for carrying out this reaction is the use of the peptide coupling reagent 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT) and an amine H—W—C(O)A in DMF solvent containing a tertiary amine such as N,N-diisopropylethylamine. Commonly used amide bond coupling conditions, e.g. EDC with HOBT or DMAP, are also employed in some examples. Typical stoichiometries are given in the specific examples but these ratios may be modified. [0226]
The amide bond construction reactions depicted in step a3 or step a5 of Schemes 3 and 4 respectively could be carried out using the specialized conditions described herein or alternatively by applying the conditions or coupling reagents for amide bond construction described in Wallace, reference 95. Some specific nonlimiting examples are given in this application. [0227]
Additional procedures for synthesizing, modifying and attaching groups: WC(O)—A are contained in references 93-95 or described below except that the piperazine intermediates are replaced by the pyrrolidines described herein.
[0228]
Scheme 4a provides a more specific example of the transformations previously described in Scheme 1. Intermediates 6-10 are prepared by the methodologies as described for intermediates 1a-5a in Scheme 1. Scheme 5 is another embodiment of the transformations described in Schemes 1 and 4a. Conversion of the phenol to the chloride (Step S, Scheme 5) may be accomplished according to the procedures described in Reimann, E.; Wichmann, P.; Hoefner, G.; Sci. Pharm. 1996, 64(3), 637-646; and Katritzky, A. R.; Rachwal, S.; Smith, T. P.; Steel, P. J.;
[0229] J. Heterocycl. Chem. 1995, 32(3), 979-984. Step T of Scheme 5 can be carried out as described for Step A of Scheme 3. The bromo intermediate can then be converted into alkoxy, chloro, or fluoro intermediates as shown in Step U of Scheme 5. Scheme 2A describes the preferred method for preparing intermediate 6c or other closely related compounds containing a 4 methoxy group in the 6-azaindole system. When step U is the conversion of the bromide into alkoxy derivatives, the conversion may be carried out by reacting the bromide with an excess of sodium methoxide in methanol with cuprous salts, such as copper I bromide, copper I iodide, and copper I cyanide. The temperature may be carried out at temperatures of between ambient and 175° but most likely will be around 115° C. or 100° C. The reaction may be run in a pressure vessel or sealed tube to prevent escape of volatiles such as methanol. The preferred conditions utilize 3 UUq of sodium methoxide in methanol, CuBr as the reaction catalyst (0.2 to 3 equivalents with the preferred being 1 eq or less), and a reaction temperature of 115° C. The reaction is carried out in a sealed tube or sealed reaction vessel. The conversion of the bromide into alkoxy derivatives may also be carried out according to procedures described in Palucki, M.; Wolfe, J. P.; Buchwald, S. L.;
J. Am. Chem. Soc. 1997, 119(14), 3395-3396; Yamato, T.; Komine, M.; Nagano, Y.;
Org. Prep. Proc. Int. 1997, 29(3), 300-303; Rychnovsky, S. D.; Hwang, K.;
J. Org. Chem. 1994, 59(18), 5414-5418. Conversion of the bromide to the fluoro derivative (Step U, Scheme 2A) may be accomplished according to Antipin, I. S.; Vigalok, A. I.; Konovalov, A. I.;
Zh. Org. Khim. 1991, 27(7), 1577-1577; and Uchibori, Y.; Umeno, M.; Seto, H.; Qian, Z.; Yoshioka, H.;
Synlett. 1992, 4, 345-346. Conversion of the bromide to the chloro derivative (Step U, Scheme 2A) may be accomplished according to procedures described in Gilbert, E. J.; Van Vranken, D. L.;
J. Am. Chem. Soc. 1996, 118(23), 5500-5501; Mongin, F.; Mongin, O.; Trecourt, F.; Godard, A.; Queguiner, G.;
Tetrahedron Lett. 1996, 37(37), 6695-6698; and O'Connor, K. J.; Burrows, C. J.;
J. Org. Chem. 1991, 56(3), 1344-1346. Steps V, W and X of Scheme 2A are carried out according to the procedures previously described for Steps B, C, and D of Scheme 1, respectively. The steps of Scheme 5 may be carried out in a different order as shown in Scheme 6 and Scheme 7.
Scheme 8 shows the synthesis of 4-azaindole derivatives 1b-5b, 5-azaindole derivatives 1c-5c, and 7-azaindole derivatives 1d-5d. The methods used to synthesize 1b-5b, 1c-5c, and 1d-5d are the same methods described for the synthesis of 1a-5a as described in Scheme 3. It is understood, for the purposes of Scheme 8, that 1b is used to synthesize 2b-5b, 1c provides 2c-5c and 1d provides 2d-5d. [0230]
The compounds where there is a single carbonyl between the azaindole and group W can be prepared by the method of Kelarev, V. I.; Gasanov, S. Sh.; Karakhanov, R. A.; Polivin, Yu. N.; Kuatbekova, K. P.; Panina, M. E.;
[0231] Zh. Org. Khim 1992, 28(12), 2561-2568. In this method azaindoles are reacted with trichloroacetyl chloride in pyridine and then subsequently with KOH in methanol to provide the 3-carbomethoxy azaindoles shown in Scheme 4 which can then be hydrolyzed to the acid and carried through the coupling sequence with HW(C═O)A to provide the compounds of Formula I wherein a single carbonyl links the azaindole moiety and group W.
An alternative method for carrying out the sequence outlined in steps B-D (shown in Scheme 8) involves treating an azaindole, such as 11, obtained by procedures described in the literature or from commercial sources, with MeMgI and ZnCl
[0232] 2, followed by the addition of ClCOCOCl (oxalyl chloride) in either THF or Et
2O to afford a mixture of a glyoxyl chloride azaindole, 12a, and an acyl chloride azaindole, 12b. The resulting mixture of glyoxyl chloride azaindole and acyl chloride azaindole is then coupled with mono-benzoylated piperazine derivatives under basic conditions to afford the products of step D as a mixture of compounds, 13a and 13b, where either one or two carbonyl groups link the azaindole and group W. Separation via chromatographic methods which are well known in the art provides the pure 13a and 13b. This sequence is summarized in Scheme 10, below.
Scheme 11 depicts a general method for modifying the substituent A. Coupling of H—W—C(O)OtBu using the conditions described previously for W in Scheme 1, Step D provides Boc protected intermediate, 15. Intermediate 15 is then deprotected by treatment with an acid such as TFA, hydrochloric acid or formic acid using standard solvents or additives such as CH[0233] 2Cl2, dioxane, or anisole and temperatures between −78° C. and 100° C. Other acids such as aq hydrochloric or perchloric may also be used for deprotection. Alternatively other nitrogen protecting groups on W such as Cbz or TROC, may be utilized and could be removed via hydrogenation or treatment with zinc respectively. A stable silyl protecting group such as phenyl dimethylsilyl could also be employed as a nitrogen protecting group on W and can be removed with fluoride sources such as tetrabutylammonium fluoride. Finally, the free amine is coupled to acid A—C(O)OH using standard amine-acid coupling conditions such as those used to attach group W or as shown below for amide formation on positions R1-R4 to provide compound 16.
Scheme 12 a preferred method for preparing H—W—C(O)—A. Specific details are contained in the experimental section. Additional examples of the preparation of 3-amino and 3-aminomethyl pyrrolidines are described in Patane et al PCT Patent Application WO 98/57640. Scheme 13 depicts a specific route to compounds of the invention Q in which Q is an indole with a dicarbonyl at the 3 position and W is an aminomethyl pyrollidine attached to Q via the primary amine. A specific procedure where A is phenyl is contained in the experimental. Q could also be alternative indoles and azaindoles and A other substituents as needed to prepare compounds of the invention. Scheme 14 describes a similar sequence except that the amino methyl pyrrolidine, W is attached to the dicarbonyl via the secondary ring nitrogen and A is —OtBu. As shown, acidic removal of the tertbutoxycarbonyl group provides a free primary amine or amine hydrochloride which may be reacted with acyl chlorides or chloroformates to give additional compounds of the invention with various A groups. The example where A is phenyl is described in detail in the experimental section. Scheme 15 describes similar chemistry but shows how the 7-position of the indole may be funtionalized by an aldehyde, carboxylic acid, or methyl carboxamide. This sequence is also described in the experimental section. A chemist skilled in the art can recognize how this chemistry could be utilized on other examples where Q, W, and A are modified.
[0234]
As shown below in Scheme 16, step a13, suitable substituted indoles, such as the bromoindole intermediate, 10, may undergo metal mediated couplings with aryl groups, heterocycles, or vinyl stannanes to provide compounds within Formula I wherein R
[0235] 5 is aryl, heteroaryl, or heteroalicyclic for example. The bromoindole intermediates, 10 (or indole triflates or iodides) may undergo Stille-type coupling with heteroarylstannanes as shown in Scheme 17, step a14. Conditions for this reaction are well known in the art and references 72-74 as well as reference 91 provide numerous conditions in addition to the specific examples provided in Scheme 17 and in the specific embodiments. It can be well recognized that an indole stannane could also couple to a heterocyclic or aryl halide or triflate to construct compounds of Formula I. Suzuki coupling (reference 71) between the bromo intermediate, 10, and a suitable boronate could also be employed and some specific examples are contained in this application. Other Suzuki conditions, partners, and leaving groups have utility. Suzuki couplings between chloro intermediates are also feasible. If standard conditions fail new specialized catalysts and conditions can be employed. Procedures describing catalysts which are useful for coupling boronates with aryl and heteroaryl chlorides are known in the art (reference 100 a-g). The boronate could also be formed on the indole and then subjected to Suzuki coupling conditions. The same coupling methodologies may be used in the case where Q contains azaindoles rather than indoles.
Chemistry [0236]
All Liquid Chromatography (LC) data were recorded on a Shimadzu LC-10AS liquid chromatograph using a SPD-10AV UV-Vis detector with Mass Spectrometry (MS) data determined using a Micromass Platform for LC in electrospray mode. [0237]
LC/MS Method (i.e., Compound Identification) [0238]
Note: column A is used unless otherwise indicated in the preparation of intermediates or examples.
[0239] | |
| |
| Column A: | YMC ODS-A S7 3.0 × 50 mm column |
| Column B: | PHX-LUNA C18 4.6 × 30 mm column |
| Column C: | XTERRA ms C18 4.6 × 30 mm column |
| Column D: | YMC ODS-A C18 4.6 × 30 mm column |
| Column E: | YMC ODS-A C18 4.6 × 33 mm column |
| Column F: | YMC C18 S5 4.6 × 50 mm column |
| Column G: | XTERRA C18 S7 3.0 × 50 mm column |
| Gradient: | 100% Solvent A/0% Solvent B to 0% Solvent A/ |
| | 100% Solvent |
| Rt in min. | 2 minutes |
| Gradient time: |
| Hold time | 1 minute |
| Flow rate: | 5 mL/min |
| Detector Wavelength: | 220 nm |
| Solvent A: | 10% MeOH/90% H2O/0.1% Trifluoroacetic Acid |
| Solvent B: | 10% H2O/90% MeOH/0.1% Trifluoroacetic Acid |
| |
Compounds purified by preparative HPLC were diluted in MeOH (1.2 mL) and purified using the following methods on a Shimadzu LC-10A automated preparative HPLC system or on a Shimadzu LC-8A automated preparative HPLC system with detector (SPD-10AV UV-VIS) wavelength and solvent systems (A and B) the same as above. [0240]
Preparative HPLC Method (i.e., Compound Purification) [0241]
Purification Method: Initial gradient (40% B, 60% A) ramp to final gradient (100% B, 0% A) over 20 minutes, hold for 3 minutes (100% B, 0% A)
[0242] | |
| |
| Solvent A: | 10% MeOH/90% H2O/0.1% Trifluoroacetic Acid |
| Solvent B: | 10% H2O/90% MeOH/0.1% Trifluoroacetic Acid |
| Column: | YMC C18 S5 20 × 100 mm column |
| Detector Wavelength: | 220 nm |
| |
Intermediate 1 [0243]
Methanesulfonic acid 1-benzyl-pyrrolidin-3-ylmethyl ester
[0244]
This tranformation was carried out via the method in J. L. Marco et.al. reference 96. Methanesulfonyl chloride (0.10 mol, 7.8 mL) was added slowly to a solution of racemic 1-benzyl-pyrolidin-3-ol (0.085 mol, 15 g) in 150 mL of dichloromethane which was stirring under a nitogen atmosphere at a temperature of −20° C. The reaction was stirred for an additional 1.5 h after addition was completed. The reaction was poured into a separatory funnel containing additional dichloromethane and washed with five thirty mL portions of saturated aqueous sodium bicarbonate. The organic layer was washed with one portion of water and then one portion of saturated aq NaCl. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to provide 22.75 g of crude mesylate which was used directly following characterization by proton NMR and LC/MS. [0245]
Intermediate 2 [0246]
1-Benzyl-pyrrolidine-3-carbonitrile
[0247]
This tranformation was carried out using the method described in C. Thomas et. al. reference 97. The mesylate of racemic 1-benzyl-pyrrolidin-3-ol (0.039 mol, 10 g), prepared as described above, sodium cyanide (0.24 mol, 15 g) and tetrabutylammonium cyanide (10 g, 0.037 mol) in 75 mL of DMSO was stirred at 80-85° C. for 16 h. The reaction mixture was partitioned between diethyl ether and sat. aq sodium bicarbonate. The aqueous layer was extracted twice with ether and the combined organic layer was washed successively with sodium bicarbonate, water, and sat aq. NaCl. The organic layer was dried over anhydrous magnesium sulfate, concentrated, and purified by flash chromatography over silica gel using a gradient of 20 to 30% ethyl acetate in hexane to afford 5.5 g of the desired product. [0248]
Intermediate 3 [0249]
C-(1-Benzyl-pyrrolidin-3-yl)-methylamine
[0250]
This reaction was carried out according to the procedure in M. R. Pavia et.al. reference 98. The nitrite (0.03 mol, 5.5 g) was dissolved in THF and cooled to 0° C. Lithium aluminum hydride (0.03 mol, 1.14 g) was added into the solution in one portion. After the addition was finished, the colling bath was removed and the reaction was stirred at ambient temperature for 18 h. The reaction was filtered and the filtrate was aconcentrated in vacuo to provide the crude product which was used directly in the next reaction. [0251]
Intermediate 4 [0252]
(1-Benzyl-pyrrolidin-3-ylmethyl)-carbamic acid tert-butyl ester
[0253]
A mixture of crude amine (0.028 mol, 5.3 g), triethylamine (0.034 mol, 4.7 mL), and and ditertbutyl dicarbonate (0.034 mol, 7.4 g) in dichloromethane was stirred at ambient temperature for 3 h. The reaction was diluted with dichloromethane, and then washed with water and then sat aq NaCl. The organic extract was dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. Purification via flash chromatography over silica gel provided 4.2 g of the desired carbamate. 1H NMR (300 MHz, CDCl3): 7.32˜7.30(m, 5H), 3.59˜3.58(m, 2H), 3.17˜3.02(m, 2H), 2.73˜2.28(m, 5H), 2.04˜1.90(m, 1H), 1.60˜1.48(m,1H), 1.45(s,9H). [0254]
Intermediate 5 [0255]
Pyrrolidin-3-ylmethyl-carbamic acid tert-butyl ester
[0256]
The benzyl amine (4.2 g) and 2.1 g of 10% Pd/C in methanol was shaken under 50PSI of hydrogen on a Parr apparatus for 20 h. The reaction mixture was filtered through celite and concentrated in vacuo to give a residue which was used directly without further purification. 1H NMR (300 MHz, CDCl3): 3.15˜1.82(m, 8H), 1.55˜1.45(m, 1H), 1.43(s,9H). [0257]
Intermediate 6 [0258]
(1-Benzoyl-pyrrolidin-3-ylmethyl)-carbamic acid tert-butyl ester
[0259]
The amine (14.65 mmol, 2.93 g) was dissolved in dichloromethane and treated with DMAP (14.65 mmol, 1.79 g), EDC (14.65 mmol, 2.80 g), and then benzoic acid (13.18 mmol, 0.9 eq). The reaction was stirred for 16 h at ambient temperature and then diluted with dichloromethane. The organic layer was washed with water and sat aq. NaCl and dried over anhydrous sodium sulfate. The solution was filtered and concentrated in vacuo to provide a residue which was purified by by flash chromatography over silica gel to provide 3.06 g of the desired benzamide. 1H NMR (300 MHz, CDCl3): 7.55˜7.34(m, 5H), 3.80˜3.10(m, 6H), 2.55˜1.58(m, 3), 1.40(s,9H). [0260]
Intermediate 7 [0261]
(3-Aminomethyl-pyrrolidin-1-yl)-phenyl-methanone
[0262]
The carbamate was stirred in 20 mL of 4N HCl in dioxane for 20 h. The reaction mixture was concentrated on a rotary evaporator and dried under vacuum to provide the hydrochloride salt which was used directly without further purification.[0263]