KR100639274B1 - 신규 아미디노벤질아민 유도체 및 트롬빈 억제제로서의 용도 - Google Patents
신규 아미디노벤질아민 유도체 및 트롬빈 억제제로서의 용도 Download PDFInfo
- Publication number
- KR100639274B1 KR100639274B1 KR1020017008826A KR20017008826A KR100639274B1 KR 100639274 B1 KR100639274 B1 KR 100639274B1 KR 1020017008826 A KR1020017008826 A KR 1020017008826A KR 20017008826 A KR20017008826 A KR 20017008826A KR 100639274 B1 KR100639274 B1 KR 100639274B1
- Authority
- KR
- South Korea
- Prior art keywords
- formula
- compound
- alkyl
- mmol
- group
- Prior art date
Links
- 0 CCC=C[C@](C)(*)C=C(*O)C=CCN Chemical compound CCC=C[C@](C)(*)C=C(*O)C=CCN 0.000 description 5
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biochemistry (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
본 발명은 특히 트롬빈의 억제가 요구되는 질환(예, 혈전증)의 치료에서 트립신 유사 프로테아제, 예를 들면 트롬빈의 경쟁적 억제제로서 또는 이의 전구약물로서, 또는 항응고제로서 유용한 하기 화학식 I의 화합물을 제공한다.
<화학식 I>

(여기서, R1, R2, Y, R3 및 R4는 명세서에 기재한 것과 같음)
트롬빈 억제제
Description
본 발명은 신규한 제약학적으로 유용한 화합물, 특히 트립신 유사 세린 프로테아제, 특히 트롬빈의 경쟁적 억제제 또는 그의 전구약물인 화합물, 이의 의약으로서의 용도, 이를 함유하는 제약 조성물 및 이를 제조하기 위한 합성 경로에 관한 것이다.
혈액 응고는 지혈 (즉, 손상된 혈관으로부터 혈액 손실의 방지) 및 혈전증 (즉, 혈관에서 혈괴를 형성하여 때때로 혈관 폐쇄를 일으킴) 모두에 관련된 중요한 과정이다.
응고는 복잡한 일련의 효소 반응의 결과이다. 이 연속된 반응에서 최종 단계 중의 하나는 전구 효소인 프로트롬빈의 활성 효소인 트롬빈으로의 전환이다.
트롬빈은 응고에 중요한 역할을 하는 것으로 알려져 있다. 이것은 혈소판을 활성화시켜서 혈소판 응집을 일으키고, 피브리노겐을 피브린 모노머(이것은 자발적으로 피브린 폴리머로 중합됨)로 전환시키고, 인자 XIII를 활성화시키는데, 활성화된 인자 XIII는 상기 폴리머를 가교결합시켜 불용성 피브린을 형성한다. 추가로, 트롬빈은 인자 V 및 인자 VIII을 활성화시켜서 프로트롬빈으로부터 트롬빈의 "양성 되먹임 (positive feedback)" 생산을 일으킨다.
효과적인 트롬빈 억제제는 혈소판의 응집 및 피브린의 형성 및 가교결합을 억제함으로써 항혈전 활성을 나타내는 것으로 생각된다. 게다가, 항혈전 활성은 양성 되먹임 기전의 효과적인 억제에 의해 증진되는 것으로 생각된다.
추가로, 트롬빈 억제제의 전구약물의 투여는 (a) 이 억제제 투여 후 일부 약물속도론적 성질, 및 (b) 상기 억제제와 관련된 일부 부작용의 이환율에서 개선을 일으킬 수 있을 것으로 알려져 있다.
저분자량 트롬빈 억제제의 초기 개발은 문헌[Claesson in Blood Coagul. Fibrinol. (1994) 5, 411]에 기술되어 있다.
블롬바크(Blombaeck) 등은 피브리노겐 Aα사슬에 대한 절단 부위 주위의 아미노산 서열 기재의 트롬빈 억제제를 보고했다 (J. Clin. Lab. Invest. 24, suppl. 107, 59, (1969)). 저자들은 논의된 아미노산 서열 중에서, 트리펩티드 서열 Phe-Val-Arg (P9-P2-P1, 이후 명세서에서 P3-P2-P1 서열로 지칭함)이 가장 효과적인 억제제라고 제안했다.
P1 위치에 α,ω-아미노알킬 구아니딘을 가진 디펩티딜 유도체 기재의 트롬빈 억제제는 미국 특허 제 4,346,078 호 및 국제 특허 출원 공개 제 93/11152 호에 공지되어 있다. 이와 유사한, 구조적으로 관련있는 디펩티딜 유도체도 보고되었다. 예를 들면, 국제 특허 출원 공개 제 94/29336 호는 예를 들면, P1 위치에 아미노메틸 벤즈아미딘류, 시클릭 아미노알킬 아미딘류 및 시클릭 아미노알킬 구아니딘류를 갖는 화합물을 개시하고 있다 (국제 특허 출원 공개 제 97/23499 호는 이 화합물 중의 일부의 전구약물을 개시하고 있다). 유럽 특허 출원 제 0 648 780 호는 예를 들면, P1 위치에 시클릭 아미노알킬 구아니딘류를 갖는 화합물을 개시하고 있다.
펩티딜 유도체를 기재로 하고, 또한 P1 위치에 시클릭 아미노알킬 구아니딘류(예, 3- 또는 4-아미노메틸-1-아미디노-피페리딘)를 갖는 트롬빈 억제제가 유럽 특허 출원 제 0 468 231 호, 제 0 559 046 호 및 제 0 641 779 호에 공지되어 있다.
P1 위치에 아르기닌 알데히드를 갖는 트리펩티딜 유도체 기재의 트롬빈 억제제는 유럽 특허 출원 제 0 185 390 호에 처음으로 개시되었다.
보다 최근에, P3 위치에서 개질된 아르기닌 알데히드 기재의 펩티딜 유도체가 보고되었다. 예를 들면, 국제 특허 출원 공개 제 93/18060 호는 P3 위치에 히드록시산을, 유럽 특허 출원 제 0 526 877 호는 데스-아미노산을, 유럽 특허 출원 제 0 542 525 호는 O-메틸 만델산을 갖는 것을 개시하고 있다.
P1 위치에 존재하는 친전자성 케톤 기재의 세린 프로테아제 (예, 트롬빈) 억제제도 공지되어 있다. 예를 들면, 유럽 특허 출원 제 0 195 212 호는 P1 위치에 펩티딜 α-케토 에스테르류 및 아미드류를, 유럽 특허 출원 제 0 362 002 호는 플루오로알킬아미드 케톤류를, 유럽 특허 출원 제 0 364 344 호는 α,β,δ-트리케토 화합물을, 유럽 특허 출원 제 0 530 167 호는 아르기닌의 α-알콕시 케톤 유도체를 갖는 것을 개시하고 있다.
기타 구조적으로 상이한, 아르기닌의 C-말단 보론산 유도체 및 이의 이소티 오우로늄 유사체 기재의 트립신 유사 세린 트로테아제 억제제가 유럽 특허 출원 제 0 293 881 호에 공지되어 있다.
보다 최근에, 펩티딜 유도체 기재의 트롬빈 억제제가 유럽 특허 출원 제 0 669 317 호 및 국제 특허 출원 공개 제 95/35309 호, 제 95/23609 호, 제 96/25426 호, 제 97/02284 호, 제 97/46577 호, 제 96/32110 호, 제 96/31504 호, 제 96/03374 호, 제 98/06740 호 및 제 97/49404 호에 개시되었다.
그러나, 트롬빈과 같은 트립신 유사 세린 프로테아제의 효과적인 억제제에 대한 필요성이 잔존해 있다. 또한, 경구적으로 생체이용률을 가지고, 다른 세린 프로테아제, 특히 지혈에 관련된 것보다 트롬빈을 선택적으로 억제하는 화합물에 대한 필요성이 존재한다. 트롬빈에 대하여 경쟁적인 억제 활성을 나타내는 화합물은 특히 항응고제로서 유용하고, 따라서 혈전증 및 관련 질환의 치료에 유용할 것으로 생각된다.
본 발명은 하기 화학식 I의 화합물 또는 그의 제약학적으로 허용되는 염 (이하, 명세서에서 "본 발명의 화합물"로 지칭함)을 제공한다:

(상기 식에서,
R1은 N(R5)R6 또는 S(O)mR7 치환체이고,
R2 및 R3는 독립적으로 할로, C1-4 알킬 또는 C1-4 알콕시 (후자의 두 개의 기는 임의로 할로에 의해 치환됨)로부터 선택되는 임의적 치환체이고,
Y는 임의로 C1-4 알킬, 메틸렌, =O 또는 히드록시에 의해 치환되는 C1-3 알킬렌이고,
R4는 H, OH, OR8a, C(O)OR8b 또는 R8c이고,
R5는 C1-6 알킬(임의로 할로에 의해 치환됨)이거나, 또는 R6, 및 R5
및 R6이 부착된 질소 원자와 함께, 임의로 산소 원자를 포함하고 및(또는) 임의로 =O기로 치환되는 3 내지 7원 질소 함유 고리이고,
R6은 C1-6 알킬 (임의로 할로에 의해 치환됨), 또는 C(O)R9이거나, 또는 R5, 및 R5 및 R6이 부착된 질소 원자와 함께, 임의로 산소 원자를 포함하고 및(또는) 임의로 =O기로 치환되는 3 내지 7원 질소 함유 고리이거나, 또는
N(R5)R6기는 하기 화학식 Ia의 구조적 단편이고,

R6a는 할로, C1-4 알킬 및 C1-4 알콕시 (후자의 두 개의 기는 임의로 할로에 의해 치환됨)로부터 선택되는 하나 이상의 임의적 치환체이고,
X는 CH 또는 N이고,
m은 0, 1 또는 2이고,
R7은 H, NH2 또는 C1-6 알킬이고,
R8a 및 R8b는 독립적으로 C1-10 알킬, C1-3 알킬페닐 또는 C
6-10 아릴이거나, 또는 R8a는 C(R10a)(R10b)OC(O)R11, C(R10a)(R10b
)N(H)C(O)OR12 또는 C(R10a)(R10b)OC(O)N(H)R12이고,
R8c는 C(R10a)(R10b)OC(O)R11, C(R10a)(R10b
)N(H)C(O)OR12 또는 C(R10a)(R10b)OC(O) N(H)R12이고,
R10a 및 R10b는 독립적으로 각 경우에서, H 또는 C1-4 알킬이고,
R11은 각 경우에서, C6-10 아릴, OR12 또는 C1-7 알킬 (후자의 기는 임의로 OH, CO2H 및 C6-10 아릴로부터 선택되는 치환체에 의해 치환됨)이고,
R12는 각 경우에서, C6-10 아릴 또는 C1-6 알킬 (후자의 기는 임의로 OH, CO2H 및 C6-10 아릴로부터 선택되는 치환체에 의해 치환됨)이고,
R9는 C1-8 알킬, Het1, C6-10 아릴, 또는 C6-10 아릴에 의해 치환되는 C1-4 알킬이고,
Het1은 산소, 질소 및(또는) 황으로부터 선택되는 하나 이상의 헤테로원자를 함유하고, 완전히 포화되거나 또는 부분적으로 포화되거나 또는 방향족일 수 있고 및(또는) 임의로 모노시클릭, 비시클릭 및(또는) 벤조 융합일 수 있는 4 내지 12원 헤테로시클릭 고리이고,
상기 각 아릴/페닐기 및 Het1기는 임의로 하나 이상의 할로, C1-4알킬 및(또는) C1-4 알콕시기 (후자의 두 개의 기는 임의로 하나 이상의 할로기에 의해 치환됨)에 의해 치환되고,
단, (a) m이 1 또는 2일 때, R7은 H가 아니고,
(b) m이 0일 때, R7은 NH2가 아니다.)
제약학적으로 허용되는 염은 무기산 (예, 수소 할로겐화물) 및 유기산 (예, 아세트산, 메탄술폰산 또는 트리플루오로아세트산) 부가염을 포함한다.
본 발명의 화합물은 호변이성 현상을 나타낼 수 있다. 모든 호변이성질체 형태 및 이들의 혼합물은 본 발명의 범위내에 포함된다. 언급할 수 있는 구체적인 호변이성질체 형태는 화학식 I의 화합물에서 아미딘 관능기에서의 이중 결합의 위치, 및 R4가 H가 아닐 때 치환체 R4의 위치와 관련된 것을 포함한다.
화학식 I의 화합물은 또한 2 이상의 비대칭 탄소 원자를 함유하고, 따라서 광학 및(또는) 부분입체이성 현상을 나타낼 수 있다. 모든 부분입체이성질체는 통상적인 기술, 예를 들면 크로마토그래피 또는 분별 결정을 사용하여 분리할 수 있다. 다양한 입체이성질체는 통상적인, 예를 들면 분별 결정 또는 HPLC 기술을 사용하여 상기 화합물의 라세미 또는 기타 혼합물의 분리에 의해 단리할 수 있다. 다른 식으로는, 목적하는 광학 이성질체는 라세미화 또는 에피머화를 일으키지 않을 조건 하에서 적당한 광학적으로 활성인 출발 물질을 반응시킴으로써, 또는 예를 들면 호모키랄산으로 유도체화한 후 통상적인 수단(예, HPLC, 실리카 상에서 크로마토그래피)에 의해 부분입체이성질체 유도체를 분리함으로써 제조할 수 있다. 모든 입체이성질체는 본 발명의 범위내에 포함된다.
본원 명세서에서 사용될 때, "아릴"이라는 용어는 페닐, 나프틸 등을 포함한다.
R2, R3, R5, R6, R6a, R7, R8a
, R8b, R9, R10a, R10b, R11 및 R12
일 수 있고, Y 및 아릴/페닐 및 Het1기를 치환할 수 있는 알킬기; R2, R3, R6a일 수 있고, 아릴/페닐 및 Het1기를 치환할 수 있는 알콕시기; R8a, R8b, R9, R11
및 R12일 수 있는 알킬페닐 또는 알킬아릴기의 알킬 부분; 및 Y일 수 있는 알킬렌기는, 탄소 원자수가 충분할 때 직쇄형 또는 분지쇄형일 수 있고, 및(또는) 포화 또는 불포화일 수 있고, 및(또는) 시클릭, 아시클릭 또는 부분 시클릭/아시클릭일 수 있고, 및(또는) 임의로 O 원자에 의해 단속될 수 있다. 당업자는 R2, R3, R5, R6, R
6a, R7, R8a, R8b, R9, R10a, R10b, R11 및 R12일 수 있고, Y 및 아릴/페닐 및 Het1기를 치환할 수 있는 알킬기가 시클릭이고 산소에 의해 단속되면, 테트라히드로푸라닐 또는 (적당한 경우) 테트라히드로피라닐과 같은 산소 함유 헤테로시클일 수 있다는 것을 인식할 것이다.
R2, R3 및 R6a일 수 있고, R2, R3, R5, R6, R6a 및 아릴/페닐 및 Het1기를 치환할 수 있는 할로기는 플루오로, 클로로, 브로모 및 요오도를 포함한다.
약어는 본원 명세서의 말미에 열거한다.
R5 및 R6이 그들이 부착된 질소 원자와 함께, 임의로 산소 원자를 포함하고 및(또는) =O기에 의해 치환되는 3 내지 7원 질소 함유(예, 피롤리딘) 고리일 때, 상기 고리는 바람직하게는 질소 원자에 대해 α인 탄소 원자에서 치환된다. 명확하게 하기 위해, R5 및 R6이 부착된 질소 원자는 고리에 존재해야 하는 질소 원자이다.
언급할 수 있는 본 발명의 화합물은
R2 및 R3가 독립적으로 할로 또는 C1-4 알킬(임의로 할로에 의해 치환됨)로부터 선택되는 임의적 치환체이고,
R5가 C1-6 알킬이거나, 또는 R6, 및 R5 및 R6이 부착된 질소 원자와 함께 임의로 =O기에 의해 치환되는 3 내지 7원 질소 함유 고리이고,
R6이 C1-6알킬 또는 C(O)R9이거나, 또는 R5, 및 R5 및 R6이 부착된 질소 원자와 함께 임의로 =O기에 의해 치환되는 3 내지 7원 질소 함유 고리이고,
R4가 OR8a 또는 C(O)OR8b일 때, R8a 및 R8b는 독립적으로 각 경우에서, C1-10알킬, C1-3 알킬페닐 또는 C6-10 아릴 (후자의 두 개의 기는 임의로 하나 이상의 할로, C1-4 알킬 및(또는) C1-4 알콕시기에 의해 치환됨)이고,
R9가 C1-6 알킬이고,
그 밖에 모든 다른 치환체는 상기 정의한 것인 화합물을 포함한다.
언급할 수 있는 추가의 본 발명의 화합물은 R4가 R8c가 아닌 것을 포함한다.
바람직한 본 발명의 화합물은
R2가, 존재하는 경우, 직쇄형 또는 분지쇄형 C1-4 알킬 또는 C1-4 알콕시 (양자 모두 임의로 할로에 의해 치환됨) 또는 할로(예, 클로로)이고,
R3이 존재하지 않거나, 또는 존재하는 경우, 직쇄형 또는 분지쇄형 C1-4 알킬 또는 할로이고,
R5가 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬이거나, 또는 R6, 및 R5
및 R6이 부착된 질소 원자와 함께 임의로 =O기에 의해 치환되는 4 내지 6원 질소 함유 고리이고,
R6이 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬 또는 C(O)-C1-6 알킬이거나, 또는 R5, 및 R5 및 R6이 부착된 질소 원자와 함께 임의로 =O기에 의해 치환되는 4 내지 6원 질소 함유 고리이고,
R7이 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬이고,
Y가 CH2 또는 (CH2)2인 것을 포함한다.
R4가 OR8a일 때, 바람직한 본 발명의 화합물은 R8a가 직쇄형 또는 분지쇄형 C1-6 알킬, C4-5 시클릭 알킬 (후자의 두 개의 기는 임의로 산소에 의해 단속될 수 있음), 또는 페닐 또는 C1-2 알킬페닐 (예, 벤질) (후자의 두 개의 기는 임의로 상기에 서와 같이 치환됨)이거나, 또는 R8a가 CH2OC(O)R11 (여기서, R11은 페닐, 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬 (후자의 기는 임의로 OH, CO2H 및 페닐로부터 선택되는 치환체에 의해 치환됨)임), 또는 OR12 (여기서, R12는 페닐, 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬 (후자의 기는 임의로 OH, CO2H 및 페닐로부터 선택되는 치환체에 의해 치환됨)임)인 것을 포함한다.
R4가 C(O)OR8b일 때, 바람직한 본 발명의 화합물은 R8b가 직쇄형 또는 분지쇄형 C1-2 알킬페닐 또는 페닐 (후자의 두 개의 기는 임의로 상기와 같이 치환됨)인 것을 포함한다.
바람직한 본 발명의 화합물은 R1이 페닐 고리가 또한 부착되어 있는 -CH(OH)-기에 대해 3-위치에서 페닐 고리에 부착된 것을 포함한다. 임의적 치환체 R2는 바람직하게는 페닐 고리가 또한 부착되어 있는 -CH(OH)-기에 대해 5-위치에서 페닐 고리에 부착된다.
N(R5)R6기가 화학식 Ia의 구조적 단편일 때, 이 단편은 바람직하게는 비치환된다.
더욱 바람직한 본 발명의 화합물은
R1이 N(R5)R6이고,
R3이 존재하지 않거나, 또는 존재하는 경우, 바람직하게는 페닐 고리가 또한 부착되어 있는 -CH2-기에 대해 2-위치에서 메틸 또는 클로로이고,
R8a가 직쇄형 또는 분지쇄형 C1-4 알킬 (임의로 산소에 의해 단속됨), 또는 산소에 의해 단속된 C4-5 시클릭 알킬이고,
R5가 C1-4알킬이거나, 또는 R6, 및 R5 및 R6이 부착된 질소 원자와 함께, 임의로 =O기에 의해 치환된 5 또는 6원 질소 함유 고리이고,
R6이 C1-4 알킬 또는 C(O)-C1-6 알킬(예, C(O)-C1-4 알킬)이거나, 또는 R5, 및 R5 및 R6이 부착된 질소 원자와 함께, 임의로 =O기에 의해 치환되는 5 또는 6원 질소 함유 고리인 것을 포함한다.
단편  이 S-배열인 화학식 I의 화합물이 바람직하다.
이 S-배열인 화학식 I의 화합물이 바람직하다.
단편 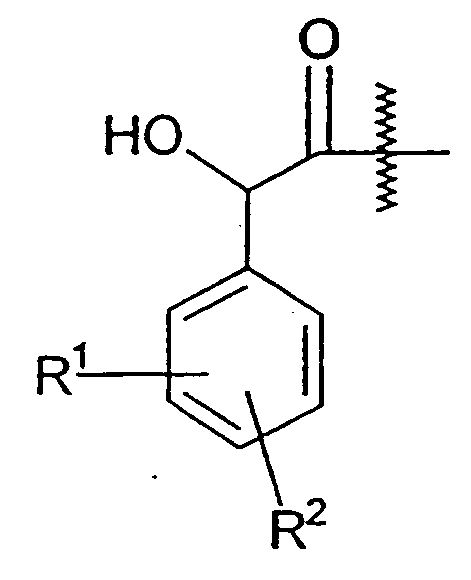 이 R-배열인 화학식 I의 화합물이 바람직하다.
이 R-배열인 화학식 I의 화합물이 바람직하다.
상기 두 개의 단편에서 결합 상의 파도모양의 선은 단편의 결합 위치를 나타낸다.
바람직한 화학식 I의 화합물은 이후 명세서에서 기술하는 실시예의 화합물을 포함한다.
제조
본 발명은 또한
(i) 하기 화학식 II의 화합물을, 예를 들면 커플링제 (예, EDC, DCC, HBTU, HATU, TBTU, PyBOP 또는 DMF 중의 옥살릴 클로라이드), 적당한 염기 (예, 피리딘, 2,4,6-트리메틸피리딘, 2,4,6-콜리딘, DMAP, TEA 또는 DIPEA) 및 적합한 유기 용매 (예, 디클로로메탄, 아세토니트릴 또는 DMF)의 존재 하에서, 하기 화학식 III의 화합물과 커플링하거나, 또는
(ii) 하기 화학식 IV의 화합물을, 예를 들면 커플링제 (예, DMF 중의 옥살릴 클로라이드, EDC, DCC, HBTU, HATU, PyBOP 또는 TBTU), 적당한 염기 (예, 피리딘, 2,4,6-트리메틸피리딘, DMAP, TEA, 2,4,6-콜리딘 또는 DIPEA) 및 적합한 유기 용매 (예, 디클로로메탄, 아세토니트릴 또는 DMF)의 존재 하에서, 하기 화학식 V의 화합물과 커플링하거나, 또는
(iii) R4가 OH 또는 OR8a인 화학식 I의 화합물의 경우에는, 하기 화학식 VI의 화합물을, 예를 들면 40 내지 60 ℃에서 적합한 염기(예, TEA) 및 적당한 유기 용매 (예, THF, CH3CN, DMF 또는 DMSO)의 존재 하에서, 임의로 하기 화학식 VI의 화합 물을, 예를 들면 0 ℃에서 저급 알킬(예, C1-6 알킬) 알콜(예, 에탄올)의 존재 하에서 기체 HCl로 전처리하여 하기 화학식 VIII의 화합물(필요하다면, 이 화합물은 단리할 수 있음)을 형성함으로써, 하기 화학식 VII의 화합물과 반응시키거나, 또는
(iv) R4가 OH 또는 OR8a인 화학식 I의 화합물의 경우에는, R4 대신 보호기 C(O)ORb1 (여기서, Rb1은 2-트리메틸실릴에틸, C1-6 알킬 또는 알킬페닐(예, 벤질)과 같은 기임)이 존재하는 화학식 I의 화합물에 대응하는 화합물을, 예를 들면 화학식 I의 화합물의 제조의 경우에는 상기한 것과 유사한 반응 조건(단계 (iii)) 하에서, 상기 정의한 화학식 VII의 화합물과 반응시키거나 (당업자는 이러한 반응에서 이중보호된 아미딘 (즉, C(O)ORb1 및 ORa기로 보호된) 유도체를 어떤 경우에서는, 목적한다면 단리할 수 있고, 이어서 통상적인 기술을 사용하여 C(O)ORb1기를 제거할 수 있다는 것을 알 것이다), 또는
(v) R4가 C(O)OR8b인 화학식 I의 화합물의 경우에는, R4가 H인 화학식 I의 화합물을, 예를 들면 0 ℃에서 적합한 염기 (예, NaOH) 및 적당한 유기 용매 (예, THF) 및(또는) 물의 존재 하에서, 하기 화학식 IX의 화합물과 반응시키거나, 또는
(vi) R4가 OR8a인 화학식 I의 화합물의 경우에는, R4가 OH인 대응 화학식 I의 화합물을, 예를 들면 0 ℃ 내지 환류 온도에서 임의로 적당한 용매 (예, DCM, THF, MeCN 또는 DMF) 및 적합한 염기 (예, Et3N 또는 피리딘)의 존재 하에서, 하기 화학 식 IXA의 화합물과 반응시키거나, 또는
(vii) R4가 R8c (여기서, R8c는 C(R10a)(R10b)OC(O)R
11 또는 C(R10a)(R10b) OC(O)N(H)R12임)인 화학식 I의 화합물의 경우에는, 하기 화학식 IXB의 대응 화합물을, 예를 들면 상기한 조건(공정 단계 (vi)) 하에서, 하기 화학식 IXC의 화합물과 반응시키거나, 또는
(viii) R4가 R8c인 화학식 I의 화합물의 경우에는, R4가 H인 대응 화학식 I의 화합물을, 예를 들면 상기한 조건(공정 단계 (vi)) 하에서, 하기 화학식 IXD의 화합물과 반응시키거나, 또는
(ix) R1이 S(O) 또는 S(O)2기를 포함하는 화학식 I의 화합물의 경우에는, R1이 S기를 포함하는 대응 화학식 I의 화합물을, 적당한 양의 적합한 산화제 (예, mCPBA 또는 포타슘 퍼옥시모노술페이트) 및 적당한 유기 용매 (예, CH2Cl2, 메탄올, 물 또는 이들의 혼합물 (예, 메탄올/물))의 존재 하에서 산화시키는
것을 포함하는 화학식 I의 화합물의 제조 방법을 제공한다.

(여기서, R1 및 R2는 상기 정의한 것임)

(여기서, Y, R3 및 R4는 상기 정의한 것임)

(여기서, R1, R2 및 Y는 상기 정의한 것임)

(여기서, R3 및 R4는 상기 정의한 것임)
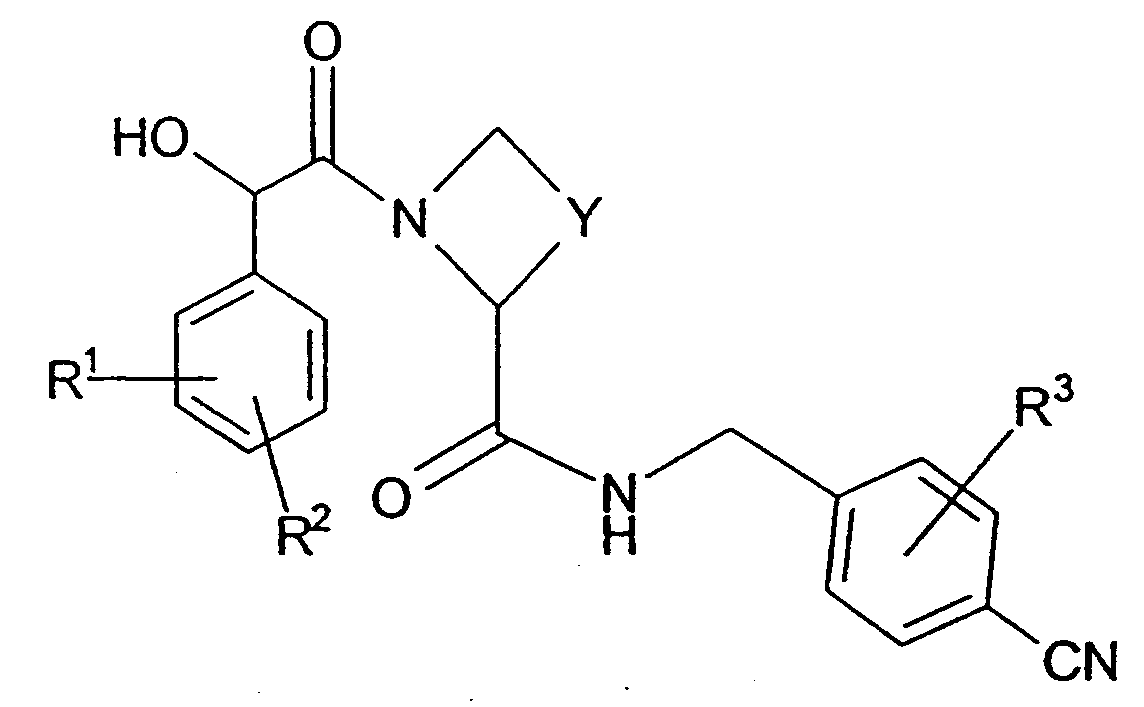
(여기서, R1,R2,Y 및 R3는 상기 정의한 것임)
(여기서, Ra는 H 또는 R8a이고, R8a는 상기 정의한 것임)

(여기서, Rc는 에틸과 같은 저급(예, C1-6) 알킬이고, R1, R2, Y 및 R3는 상기 정의한 것임)
(여기서, L1은 할로 또는 p-니트로페녹시와 같은 적합한 이탈기이고, R8b는 상기 정의한 것임)
(여기서, R8a 및 L1은 상기 정의한 것임)

(여기서, R1, R2, Y, R3, R10a 및 R10b는 상기에서 정의한 것임)
(여기서, R13은 R11 또는 N(H)R12이고, L1, R11 및 R12는 상기에서 정의한 것임)
(여기서, R14는 OC(O)R11, NHC(O)OR12 또는 OC(0)N(H)R12이고, L1, R10a, R10b, R11 및 R12는 상기에서 정의한 것임)
화학식 II의 화합물은 공지 및(또는) 표준 기술을 사용하여 얻을 수 있다. 예를 들면, 화학식 II의 화합물은 하기 화학식 X의 알데히드를
(a) 예를 들면, 실온 또는 승온된 온도 (예, 100 ℃ 미만)에서 적합한 유기 용매 (예, 클로로포름 또는 메틸렌 클로라이드) 및 필요한 경우, 적합한 염기 (예, TEA) 및(또는) 적합한 촉매 시스템 (예, 벤질암모늄 클로라이드 또는 요오도화 주석)의 존재 하에서, 하기 화학식 XI의 화합물과 반응시킨 후, 당업자에게 잘 알려진 조건 (예, 후술하는 조건) 하에서 가수분해하거나, 또는
(b) 예를 들면, NaHSO3 및 물의 존재 하에서 NaCN 또는 KCN과 반응시킨 후, 가수분해하거나, 또는
(c) 예를 들면, 승온(예, 실온 초과 100 ℃ 미만)에서 적합한 유기 용매 (예, 클로로포름) 및 필요한 경우, 적합한 촉매 시스템 (예, 벤질암모늄 클로라이드)의 존재 하에서 클로로포름과 반응시킨 후, 가수분해하거나, 또는
(d) 하기 화학식 XII의 화합물과 반응시킨 후, 당업자에게 잘 알려진 조건 하에서 산화적 절단(예, 오존분해 또는 오스뮴 또는 루테늄 촉매에 의함)시키거나, 또는
(e) 당업자에게 잘 알려진 조건 하에서 트리스(메틸티오)메탄과 반응시킨 후, 예를 들면 HgO 및 HBF4의 존재 하에서 가수분해함으로써 제조할 수 있다.

(여기서, R1 및 R2는 상기에서 정의한 것임)
(여기서, R″는 H 또는 (CH3)3Si임)
(여기서, M은 Mg 또는 Li임)
화학식 II의 화합물의 거울상이성질체 형태(즉, CO2H기의 경우에는 α-위치에 있는 탄소 원자에 대해서 상이한 배열의 치환체를 갖는 화합물)는 거울상이성질체특이성 유도체화 단계에 의해 분리할 수 있다. 이것은 예를 들면, 효소 공정에 의해 달성할 수 있다. 이러한 효소 공정은 예를 들면, 실온 내지 환류 온도에서 (예, 45 내지 55 ℃) 적합한 효소 (예, 리파제 PS Amano), 적당한 에스테르 (예, 비닐 아세테이트) 및 적합한 용매 (예, 메틸 tert-부틸 에테르)의 존재 하에서 α-OH기의 에스테르 교환 반응을 포함한다. 이어서, 유도체화된 이성질체는 통상적인 분리 기술(예, 크로마토그래피)에 의해 미반응 이성질체로부터 분리할 수 있다.
이러한 유도체화 단계에서 화학식 II의 화합물에 첨가되는 기는 임의의 추가의 반응 전 또는 화학식 I의 화합물의 합성에서 임의의 후기 단계에서 제거할 수 있다. 부가의 기는 통상적인 기술 (예, α-OH기의 에스테르에 대해서는, 당업자에게 공지된 조건 (예, 실온 내지 환류 온도에서 적합한 염기 (예, NaOH) 및 적당한 용매 (예, MeOH, 물 또는 이들의 혼합물) 존재) 하에서의 가수분해)을 사용하여 제거할 수 있다.
화학식 III의 화합물은 하기 화학식 XIII의 화합물을, 예를 들면 화학식 I의 화합물의 합성의 경우에는 상기한 조건 (예를 들면, 공정 단계 (i) 및 (ii) 참조) 하에서 상기에서 정의한 화학식 V의 화합물과 반응시킴으로써 제조할 수 있다.

(여기서, Y는 상기에서 정의한 것임)
화학식 IV의 화합물은 공지 기술을 사용하여 쉽게 얻을 수 있다. 예를 들 면, 화학식 IV의 화합물은 상기에서 정의한 화학식 II의 화합물을, 예를 들면 화학식 I의 화합물의 합성의 경우에는 상기한 조건 (예를 들면, 공정 단계 (i) 및 (ii) 참조) 하에서 상기에서 정의한 화학식 XIII의 화합물과 반응시킴으로써 제조할 수 있다.
화학식 V의 화합물은 문헌에 공지되어 있고, 및(또는) 공지 기술을 사용하여 제조할 수 있다. 예를 들면, 화학식 V의 화합물은 당업자에게 잘 알려진 조건 하에서 하기 화학식 XIV의 화합물을 환원시킴으로써 제조할 수 있다.

(여기서, R3 및 R4는 상기에서 정의한 것임)
화학식 VI의 화합물은 펩티드 커플링 기술에 따라, 예를 들면 화학식 I의 화합물의 경우에는 상기한 방법 (예를 들면, 공정 단계 (i) 및 (ii) 참조)과 유사한 방식으로 제조할 수 있다. 목적한다면, 화학식 VIII의 화합물도 이러한 방식으로 제조할 수 있다.
화학식 IXB의 화합물은 R4가 H인 대응 화학식 I의 화합물을, 예를 들면 당업자에게 공지된 조건 하에서 과량의 하기 화학식 XIVA의 화합물과 반응시킴으로써 제조할 수 있다:
(여기서, R10a 및 R10b는 상기에서 정의한 것임)
화학식 X의 화합물은 상업적으로 얻을 수 있고, 문헌에 잘 알려져 있거나 또는 공지 및(또는) 표준 기술을 사용하여 얻을 수 있다.
예를 들면, 화학식 X의 화합물은 적합한 환원제 (예, DIBAL-H)의 존재 하에서, 하기 화학식 XV의 화합물을 환원시킴으로써 제조할 수 있다:

(여기서, R1 및 R2는 상기에서 정의한 것임)
다른 식으로는, 화학식 X의 화합물은 적합한 산화제 (예, 피리디늄 클로로크로메이트 또는 DMSO 및 옥살릴 클로라이드의 혼합물)의 존재 하에서 하기 화학식 XVI의 화합물을 산화시킴으로써 제조할 수 있다:

(여기서, R1 및 R2는 상기에서 정의한 것임)
R1이 S(O) 또는 S(O)2기를 포함하는 화학식 II, IV, VI, VIII, X, XV 및 XVI의 화합물은 예를 들면 상기한 바와 같이, R1이 S기를 포함하는 대응 화학식 II, IV, VI, VIII, X, XV 및 XVI의 화합물을 산화시킴으로써 제조할 수 있다.
화학식 VII, IX, IXA, IXC, IXD, XI, XII, XIII, XIV, XIVA, XV 및 XVI의 화합물 및 이들의 유도체는 상업적으로 얻을 수 있고, 문헌에 알려져 있거나, 또는 상기한 것과 유사한 방법에 의하거나 또는 적당한 시약 및 반응 조건 (예, 후술함)을 사용하여 쉽게 얻을 수 있는 출발 물질로부터 표준 기술에 따라 통상적인 합성 방법에 의해 얻을 수 있다.
화학식 I, II, III, IV, V, VI, VII, VIII, IX, IXA, IXB, IXC, IXD, X, XIII, XIV, XV 및 XVI의 화합물에서 방향족 및(또는) 비방향족, 카르보시클릭 및 헤테로시클릭 고리상의 치환체는 당업자에게 잘 알려진 기술을 사용하여 도입되고 및(또는) 상호전환될 수 있다. 예를 들면, 니트로는 아미노로 환원될 수 있고, 아미노는 알킬화되거나 아실화되어 알킬- 및(또는) 아실아미노를 생성할 수 있고, 아미노는 피롤로(오산화인과 같은 촉매의 존재 하에서 2,5-디메톡시테트라히드로푸란 과의 축합에 의함)로 전환될 수 있고, 아미노는 (디아조화를 통해) 할로로 또는 (예, 1,4- 또는 1,5-디할로알킬 화합물 또는 β- 또는 γ-할로에스테르와의 반응을 통해) 질소 함유 고리 (임의로 =O기로 치환됨)로 전환될 수 있고, 요오도는 질소 함유 헤테로시클류(예를 들면, Buchwald 조건 하에서 이미다졸 또는 피페리딘의 처리에 의한 이미다졸릴 및 피페리디닐)로 전환될 수 있고, 질소 히드록시는 알킬화되어 알콕시를 생성할 수 있고, 알콕시는 가수분해되어 히드록시로 될 수 있고, 알켄은 수소화되어 알칸으로 될 수 있고, 할로는 수소화되어 H 등으로 될 수 있다. 이것과 관련하여, 화학식 XV의 화합물 (여기서, R1은 -N(CH3)2이고, R2
는 클로로 또는 메틸임)은 상업적으로 얻을 수 있는 요오도-클로로 또는 요오도-메틸 이중치환된 벤조산 메틸 에스테르로부터 예를 들면, 문헌[Wolfe 등, Tetrahedron Lett. 38, 6367 (1997)]에 기술된 Pd 촉매 아민화, 이어서 생성된 아닐린의 환원적 아민화 (예를 들면, HCHO 및 Na(CN)BH3와 같은 환원제 또는 Pt(IV) 옥사이드 및 수소의 혼합물을 사용하여) 또는 알킬화 (예를 들면, MeI 및 적당한 염기를 사용하여)를 사용하여 얻을 수 있다. R1이 -S(O)mCH3 (여기서, m은 상기에서 정의한 것임)이고, R2가 클로로 또는 메틸인 화학식 XV의 화합물은 문헌[타르벨 등, "Organic Synthesis", Coll. 제 III권, 809-811쪽 (1955)]에 기술된 바와 같이 상기 생성된 아닐린으로부터(또는 대응 벤조산으로부터) 디아조화 후, 디아조늄염을 에틸 크산탄 칼륨으로 처리하고, 이어서 이 중간체를 가수분해하여 대응 티오페놀을 생성함 으로써 얻을 수 있다. 이어서, 생성된 티오페놀을 알킬화 (예를 들면, 에탄올 중의 적합한 염기의 존재 하에서 적당한 알킬 요오다이드를 사용하여)한 후, (필요한 경우) 산화시켜(예를 들면, CH2Cl2 중의 mCPBA 또는 메탄올/물 중의 퍼옥시모노술폰산 칼륨을 사용하여) 술폰 또는 술폭사이드를 형성할 수 있다.
화학식 I의 화합물은 통상적인 기술을 사용하여 반응 혼합물로부터 단리할 수 있다.
당업자는 상기 공정에서 중간체 화합물의 관능기가 보호기에 의해 보호될 필요가 있을 수 있음을 알 것이다.
보호하기에 바람직한 관능기는 히드록시, 아미노, 알데히드, 2-히드록시카르복실산 및 카르복실산을 포함한다. 히드록시에 대한 적합한 보호기는 트리알킬실릴 또는 다아릴알킬실릴기(예, t-부틸디메틸실릴, t-부틸디페닐실릴 또는 트리메틸실릴) 및 테트라히드로피라닐을 포함한다. 카르복실산에 대한 적합한 보호기는 C1-6 알킬 또는 벤질 에스테르류를 포함한다. 아미노 및 아미디노에 대한 적합한 보호기는 t-부틸옥시카르보닐, 벤질옥시카르보닐 또는 2-트리메틸실릴에톡시카르보닐 (Teoc)를 포함한다. 아미디노 질소는 또한 히드록시 또는 알콕시기에 의해 보호될 수 있고, 단일- 또는 이중보호될 수 있다. 알데히드류는 예를 들면, 에틸렌 글리콜과 반응시킴으로써 아세탈로서 보호될 수 있다. 2-히드록시 카르복실산은 예를 들면, 아세톤과 축합시킴으로써 보호될 수 있다.
관능기의 보호 및 탈보호는 커플링 전 또는 후, 또는 상기 반응식에서 임의 의 다른 반응의 전 또는 후에 일어날 수 있다.
보호기는 당업자에게 잘 알려지고, 후술하는 기술에 따라 제거할 수 있다.
당업자는 다른 식으로, 어떤 경우에서는 더 편리한 방식으로 화학식 I의 화합물을 얻기 위해서, 상기 개별적인 공정 단계가 상이한 순서로 수행될 수 있고, 및(또는) 개별적인 반응이 전체 경로에서 상이한 단계에서 수행될 수 있다 (즉, 치환체는 특정 반응과 관련하여 상기한 것과 상이한 중간체에 첨가될 수 있고, 및(또는) 상기한 것과 상이한 중간체 상에서 화학적 변형이 수행될 수 있다)는 것을 알 것이다. 이것은 보호기에 대한 필요성을 부인하거나 또는 필요하게 한다.
예를 들면, 이것은 R4가 H가 아닌 화학식 I의 화합물의 합성에 있어서 특히 진실이다. 이 경우에서, OH, OR8a, C(O)OR8b 및(또는) R8c기는 상기한 공정 단계 (예를 들면, 공정 단계 (iii) 내지 (viii) 참조)를 사용하여 전체 합성에서 더 빠른 단계에서 도입될 수 있다. 추가로, 화학식 II 및 IV의 화합물의 만델산 OH기는 상기 커플링 단계 전에 보호될 필요가 있을 수 있다.
따라서, 관여된 화학의 순서 및 유형은 합성을 달성하기 위한 순서뿐만 아니라 보호기의 필요성 및 유형을 규정할 것이다.
보호기의 용도는 문헌 ["Protective Groups in Organic Chemistry", J W F McOmie 편저, Plenum Press (1973)] 및 문헌 ["Protective Groups in Organic Synthesis", 제 2판, T W Greene & P G M Wutz, Wiley-Interscience (1991)]에 완전하게 기술되어 있다.
화학식 I의 화합물의 보호된 유도체는 표준 탈보호 기술 (예, 수소화)을 사용하여 화학식 I의 화합물로 화학적으로 전환될 수 있다. 또한, 당업자는 화학식 I의 어떤 화합물이 화학식 I의 다른 화합물의 "보호된 유도체"로서 언급될 수 있다는 것을 알 것이다.
의약적 및 제약적 용도
본 발명의 화합물은 그 자체로서 약물학적 활성을 가질 수 있다. 이러한 활성을 가질 수 있는 본 발명의 화합물은 R4가 H인 것을 포함하나, 이에 제한되는 것은 아니다.
그러나, 화학식 I의 다른 화합물 (R4가 H가 아닌 것을 포함함)은 이러한 활성을 가지지 않을 수 있으나, 비경구적 또는 경구적으로 투여되고, 그 후에 체내에서 대사되어 약물학적으로 활성인 화합물 (R4가 H인 대응 화합물을 포함하나, 이에 제한되는 것은 아님)을 형성할 수 있다. 그러므로, 이러한 화합물 (약간의 약물학적 활성을 가질 수 있으나, 대사되어 형성되는 "활성" 화합물보다 훨씬 더 낮은 활성을 갖는 화합물도 포함함)은 활성 화합물의 "전구약물"로 기술할 수 있다.
따라서, 본 발명의 화합물은 이들이 약물학적 활성을 가지고 있고, 및(또는) 경구 또는 비경구적 투여 후 체내에서 대사되어 약물학적 활성을 가지는 화합물을 형성하기 때문에 유용하다. 따라서, 본 발명의 화합물은 제약 물질로 사용된다.
따라서, 본 발명은 추가로 제약 물질로서 사용하기 위한 본 발명의 화합물을 제공한다.
특히, 본 발명의 화합물은 예를 들면, 후술하는 시험에서 설명하는 바와 같이, 그 자체로서 강력한 트롬빈 억제제이고, 및(또는) (예를 들어, 전구약물의 경우) 투여 후 대사되어 강력한 트롬빈 억제제를 형성한다.
"트롬빈 억제제의 전구약물"은 경구 또는 비경구 투여 후 소정의 시간내에 (예, 약 1 시간) 실험적으로 검출가능한 양으로 트롬빈 억제제를 형성하는 화합물을 포함한다.
따라서, 본 발명의 화합물은 트롬빈의 억제가 요구되는 질환에 유용할 것으로 기대된다.
따라서, 본 발명의 화합물은 인간을 포함한 동물의 혈액 및 조직에서 혈전증 및 응고항진의 치료 및(또는) 예방에 사용된다.
응고항진이 혈전 색전 질병을 일으킬 수 있다는 것은 공지되어 있다. 언급할 수 있는 응고항진 및 혈전 색전 질병과 관련된 질환은 인자 V 변이 (인자 V Leiden)와 같은 선천성 또는 후천성 활성화 C 단백질 내성, 및 항혈전 III, 단백질 C, 단백질 S 또는 헤파린 보조인자 II의 선천성 또는 후천성 결핍을 포함한다. 기타 응고항진 및 혈전 색전 질병과 관련있는 것으로 알려진 질환은 순환하는 항인지질 항체 (루푸스 항응고제), 호모시스테인혈증, 헤파린 유도 혈소판감소증 및 섬유소용해 결손을 포함한다. 따라서, 본 발명의 화합물은 상기 질환의 치료적 및(또는) 예방적 처치 모두에서 사용된다.
본 발명의 화합물은 추가로 예를 들면, 알쯔하이머 질병과 같은 신경퇴행성 질병에서 응고항진의 징후 없이 트롬빈이 바람직하지 못할 정도로 과량인 질환의 치료에 사용된다.
언급할 수 있는 구체적인 질병 질환은 정맥 혈전증 및 폐 색전증, 동맥 혈전증 (예, 심근경색, 불안정성 협심증, 혈전증 기초 뇌졸중 및 말초 동맥 혈전증에서 볼 수 있음) 및 일반적으로 동맥 세동 동안 심방으로부터 또는 전층 심근 경색 후 좌심실로부터의 전신성 색전증의 치료적 및(또는) 예방적 처치를 포함한다.
게다가, 본 발명의 화합물은 혈전용해, 경피적 경관 혈관성형술 (PTA) 및 관상동맥 회로 수술후 재교합 (즉, 혈전증)의 예방, 미세수술 및 일반적인 혈관 수술 후 재혈전증의 방지에 유용할 것으로 기대된다.
추가의 적용은 세균, 다중 외상, 중독 또는 임의의 다른 기전에 의해 유발된 파종성 혈관내 응고의 치료적 및(또는) 예방적 처치; 혈액이 체내에서 혈관 이식물, 혈관 스텐트, 혈관 카테터, 기계학적 및 생물학적 인공 판막 또는 임의의 다른 의학적 장치와 같은 외부 표면과 접촉시 항응고적 처치; 및 심장-폐 기계를 사용하는 심혈관 수술 동안이나 또는 혈액투석시와 같이 혈액이 체외에서 의학적 장치와 접촉시 항응고적 처치를 포함한다.
응고 과정에 미치는 영향 이외에도, 트롬빈은 다수의 세포 (예, 호중구, 섬유아세포, 상피 세포 및 평활근 세포)를 활성화시키는 것으로 알려져 있다. 따라서, 본 발명의 화합물은 또한 특발성 및 성인 호흡 곤란증, 방사선 또는 화학요법 치료 후 폐 섬유증, 패혈성 쇽, 패혈증, 염증 반응 (부종을 포함하나, 이에 제한되는 것은 아님), 관상 동맥 질병과 같은 급성 또는 만성 동맥경화증, 뇌 동맥 질환, 말초 동맥 질환, 재관류 손상 및 경피적 경관 혈관성형술 (PTA) 후 재협착의 치료적 및(또는) 예방적 처치에 유용할 수 있다.
트립신 및(또는) 트롬빈을 억제하는 본 발명의 화합물은 또한 췌장염의 치료에 유용할 수 있다.
본 발명은 추가로 본 발명의 화합물 또는 이의 제약학적으로 허용되는 염의 치료적 유효량을 상기 질환에 걸려있거나 또는 걸리기 쉬운 환자에게 투여하는 것을 포함하는, 트롬빈의 억제가 요구되는 질환의 치료 방법을 제공한다.
본 발명의 화합물은 일반적으로 경구, 정맥내, 피하, 볼, 직장, 피부, 코, 기관, 기관지, 임의의 다른 비경구 경로에 의해 또는 흡입을 통해 제약학적으로 허용되는 제형에 유리 염기로서 또는 제약학적으로 허용되는 비독성 유기 또는 무기산 부가염으로서의 활성 화합물을 포함하는 제약 물질의 형태로 투여될 것이다. 질병, 치료해야 할 환자 및 투여 경로에 따라, 상기 조성물은 다양한 투여량으로 투여될 수 있다.
본 발명의 화합물은 또한 상이한 작용 기전을 가지는 임의의 항혈전제, 예를 들면 항혈소판제인 아세틸살리실산, 티클로피딘, 클로피도그렐, 트롬복산 수용체 및(또는) 합성효소 억제제, 피브리노겐 수용체 길항제, 프로스타시클린 유사체 및 포스포디에스테라제 억제제 및 ADP-수용체 (P2T) 길항제와 혼합될 수 있고, 및(또는) 이들과 함께 투여될 수 있다.
본 발명의 화합물은 추가로 혈전성 질병, 특히 심근 경색의 치료에서 조직 플라스미노겐 활성화제 (천연, 재조합 또는 개질된), 스트렙토키나제, 우로키나제, 프로우로키나제, 아니소일화 플라스미노겐-스트렙토키나제 활성화제 착물 (APSAC), 동물 침 샘 플라스미노겐 활성화제 등과 같은 혈전 용해제와 혼합될 수 있고, 및(또는) 이들과 함께 투여될 수 있다.
따라서, 본 발명은 추가로 제약학적으로 허용되는 보조제, 희석제 또는 담체와 혼합하여 본 발명의 화합물을 포함하는 제약 조성물을 제공한다.
인간의 치료에서 본 발명의 화합물의 적합한 일일 투여량은 경구 투여에서 는 체중(kg) 당 약 0.001 내지 100 mg이고, 비경구적 투여에서는 체중(kg) 당 약 0.001 내지 50 mg이다.
본 발명의 화합물은 종래 기술분야에서 알려진 화합물에 비해 더 효과적이고, 독성이 적고, 작용 시간이 더 길고, 작용 범위가 더 넓고, 더 강력하고, 부작용이 더 적고, 보다 쉽게 흡수되고 또는 기타 유용한 약물학적, 물리적 또는 화학적 성질을 가질 수 있거나, 또는 이러한 화합물로 대사될 수 있다는 장점이 있다.
생물학적 시험
시험 A
트롬빈 응혈 시간 (TT)의 측정
억제제 용액 25 ㎕를 혈장 25 ㎕와 3 분 동안 배양하였다. 이어서, pH 7.4의 완충 용액 중의 인간 트롬빈 (T 6769; Sigma Chem. Co 또는 Hematologic Technologies; 4.0 NIH units/ml) 25 ㎕를 첨가하고, 자동 장치 (KC 10; Amelung)에서 응혈 시간을 측정하였다.
트롬빈 응혈 시간 (TT)은 억제제가 존재할 때의 TT (TTi)에 대한 억제제가 없을 때의 TT (TTo)의 비율 뿐만 아니라 절대값(초)으로도 표현된다. 전자의 비율(범위 1-0)을 억제제의 농도(로그 변환함)에 대해 플롯하고, 하기 수학식에 따라 S자형 용량-반응 곡선으로 맞추었다:
y = a/[1+(x/IC50)s]
상기 식에서, a = 최대 범위, 즉 1; s = 용량-반응 곡선의 기울기; 및 IC50 = 응혈 시간이 2 배가 되는 억제제의 농도. 계산은 식을 0에서 시작하여 1에서 끝나도록 설정하여 소프트웨어 프로그램 GraFit Version 3 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, 영국 런던)을 사용하여 PC 상에서 수행하였다.
시험 B
색소생성 로보틱(Robotic) 검정에 의한 트롬빈 억제의 측정
트롬빈 억제제 역가를 96 개의 구멍을 가진 절반부 부피 미소적정 판 (Costar, 미국 매사츄세츠 캠브리지; 캐탈로그 번호 3690)을 사용하여 플라토 3300(Plato 3300) 로보틱 미소판 프로세서 (Rosys AG, CH-8634 Hombrechtikon, 스위스)에서 색소생성 기질법으로 측정하였다. DMSO 72 ㎕ 중의 시험 물질의 저장 용액 (0.1-1 mmol/l)을 DMSO로 1:3 (24 + 48 ㎕)으로 연속적으로 희석하여 10 개의 상이한 농도를 얻고, 이를 검정에서 샘플로 하여 분석하였다. 시험 샘플 2 ㎕를 검정 완충액 124 ㎕로 희석하고, 검정 완충액 중의 색소생성 기질 용액 (S-2366, Chromogenix, Molndal, 스웨덴) 12 ㎕ 및 최종적으로 검정 완충액 중의 α-트롬빈 용액 (인간 α-트롬빈, Sigma Chemical Co. 또는 Hematologic Technologies) 12 ㎕를 첨가하고, 상기 샘플을 혼합하였다. 최종 검정 농도는 시험 물질 0.00068-13.3 μmol/l, S-2366 0.30 mmol/l, α-트롬빈 0.020 NIHU/ml였다. 37 ℃에서 40 분 동안 배양하는 동안의 직선형의 흡광도 증가를 억제제가 없는 대조군과 비교한, 시험 샘플에 있어서의 억제 백분율의 계산에 사용하였다. 농도의 로그값 대 억제 % 곡선으로부터 트롬빈 활성을 50 % 억제하는 억제제 농도에 대응하는 IC50 로보틱 값을 계산하였다.
시험 C
인간 트롬빈에 대한 억제 상수 K
i
의 측정
37 ℃에서 코바스 바이오 원심분리식 분석기(Roche, Basel, 스위스) 상에서 수행하는 색소생성 기질법을 사용하여 Ki를 측정하였다. 인간 α-트롬빈을 다양한 농도의 시험 화합물과 배양한 후 잔류 효소 활성을 3 개의 상이한 기질 농도에서 405 nm에서의 광학 흡광도에서의 변화로서 측정하였다.
시험 화합물 용액 (일반적으로 BSA 10 g/l 함유 완충액 또는 식염수에 용해시킴) 100 ㎕를 BSA (10 g/l)를 함유하는 검정 완충액 (0.05 mol/l Tris-HCl pH 7.4, NaCl로 조절한 이온강도 0.15) 중의 인간 α-트롬빈 (Sigma Chemical Co) 200 ㎕와 혼합하고, 코바스 바이오에서 샘플로 하여 분석하였다. 샘플 60 ㎕를 물 20 ㎕와 함께 검정 완충액 중의 기질 S-2238 (Chromogenix AB, Molndal, 스웨덴) 320 ㎕에 첨가하고, 흡광도 변화 (△A/min)를 측정하였다. S-2238의 최종 농도는 16, 24 및 50 μmol/l였고, 트롬빈의 최종 농도는 0.125 NIHU/ml였다.
정상상태 반응 속도를 사용하여 딕손 플롯(Dixson plots), 즉 억제제 농도 대 1/(△A/min)의 그래프를 작성하였다. 가역성이고 경쟁적인 억제제의 경우에는, 상기 상이한 기질 농도의 데이타 점들은 전형적으로 x = -Ki에서 x축과 만나는 직선을 형성하는 것을 알 수 있다.
시험 D
활성화 부분 트롬보플라스틴 시간 (APTT)의 측정
APTT를 스타고(Stago)에 의해 제조된 시약 PTT 자동화 5를 사용하여 혼주(混注)한 구연산염 첨가 정상 인간 혈장에서 측정하였다. 억제제를 혈장에 첨가하고 (혈장 90 ㎕의 경우에는 억제제 용액 10 ㎕), 3분 동안 APTT 시약과 배양한 후, 염화칼슘 용액 (0.025 M) 100 ㎕를 첨가하고, 시약 제조자의 지시에 따라 응고 분석기 KC 10(Amelung)을 사용하여 APTT를 측정하였다.
응혈 시간은 억제제가 존재할 때의 APTT (APTTi)에 대한 억제제가 없을 때의 APTT (APTTo)의 비율 뿐만 아니라 절대값(초)으로도 표현된다. 전자의 비율(범위 1-0)을 억제제의 농도(로그 변환함)에 대해 플롯하고, 하기 수학식에 따라 S자형 용량-반응 곡선으로 맞추었다:
y = a/[1+(x/IC50)s]
상기 식에서, a = 최대 범위, 즉 1; s = 용량-반응 곡선의 기울기; 및 IC50 = 응혈 시간이 2 배가 되는 억제제의 농도. 계산은 식을 0에서 시작하여 1에서 끝나도록 설정하여 소프트웨어 프로그램 GraFit Version 3 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, 영국 런던)을 사용하여 PC 상에서 수행하였다.
IC50APTT는 활성화 부분 트롬보플라스틴 시간을 2 배로 하는 인간 혈장에서의 억제제의 농도로 정의된다.
시험 E
생체 밖에서 트롬빈 시간의 측정
에탄올:솔루톨(상표):물 (5:5:90)에 용해시킨 화학식 I의 화합물을 경구 또는 비경구적으로 투여한 후 트롬빈의 억제를 실험 전 하루 또는 이틀 전에 경동맥으로부터 혈액을 채취하기 위한 카테터를 장착시킨 의식이 있는 랫트에서 조사하였다. 실험 당일 1 부의 구연산 나트륨 용액 (0.13 mol/l) 및 9 부의 혈액을 함유하는 플라스틱 관에 화합물을 투여한 후 정해진 시간에 혈액 샘플을 회수하였다. 상기 관을 원심분리하여 혈소판이 적은 혈장을 얻었다. 이 혈장을 트롬빈 시간 또는 후술하는 에카린 응혈 시간 (ECT)의 측정에 사용하였다.
구연산염 첨가 랫트 혈장 100 ㎕를 식염수 용액 (0.9 %) 100 ㎕로 희석하고, 완충 용액 (pH 7.4) 100 ㎕ 중의 인간 트롬빈 (T 6769, Sigma Chem Co, USA 또는 Hematologic Technologies) 또는 에카린 (Pentapharm)을 첨가하여 혈장 응고를 개 시하였다. 자동화 장치 (KC 10, Amelung, 독일)에서 응혈 시간을 측정하였다.
화학식 I의 "전구약물" 화합물을 투여한 경우, 랫트 혈장에서 화학식 I의 적당한 활성 트롬빈 억제제 (예, 유리 아미딘 화합물)의 농도는 혼주한 구연산염 첨가 랫트 혈장에서의 트롬빈 시간 또는 에카린 응혈 시간을 식염수 중에 용해시킨 대응 "활성" 트롬빈 억제제의 공지 농도와 연관시키는 표준 곡선을 사용하여 측정하였다.
랫트에서 평가한 활성 트롬빈 억제제의 혈장 농도에 기초하여(트롬빈 시간 또는 ECT 연장이 상기 화합물에 기인한다고 가정함), 대응 화학식 I의 전구약물 화합물을 경구 및(또는) 비경구적으로 투여한 후의 곡선 하 면적 (AUCpd)을 사다리꼴 공식 및 데이타를 무한대로 외삽하는 것을 이용하여 계산하였다.
전구 약물을 경구 또는 비경구적으로 투여한 후의 활성 트롬빈 억제제의 생체이용률을 하기 수학식과 같이 계산하였다:
[(AUCpd/투여량)/(AUCactive, parenteral/투여량)] x 100
상기 식에서, AUCactive, parenteral은 대응 활성 트롬빈 억제제를 상기한 바와 같이 의식있는 랫트에 비경구적으로 투여한 후 얻은 AUC이다.
시험 F
생체 밖에서 뇨에서 트롬빈 시간의 측정
에탄올:솔루톨(상표):물 (5:5:90)에 용해시킨 본 발명의 "전구약물" 화합물을 경구 또는 비경구적으로 투여한 후 뇨로 배출된 "활성" 트롬빈 억제제의 양을 생체 밖에서 뇨에서 트롬빈 시간을 측정함으로써 평가하였다 (트롬빈 시간 연장이 전술한 화합물에 의해 일어난다고 가정함).
의식있는 랫트를 대사 케이지에 놓고, 본 발명의 화합물을 경구 투여한 후 24 시간 동안 뇨 및 배변을 따로 혼주하였다. 후술하는 바와 같이 혼주한 뇨 상에서 트롬빈 시간을 측정하였다.
혼주한 구연산염 첨가 정상 인간 혈장 100 ㎕를 농축한 랫트 뇨 또는 이의 식염수 희석액과 1 분 동안 배양하였다. 이어서, 완충 용액 (pH 7.4) 100 ㎕ 중의 인간 트롬빈 (T 6769, Sigma Chem Company)을 투여하여 혈장 응고를 개시하였다. 자동화 장치 (KC 10, Amelung)에서 응혈 시간을 측정하였다.
랫트 뇨에서 활성 트롬빈 억제제의 농도는 혼주한 구연산염 첨가 정상 인간 혈장에서의 트롬빈 시간을 농축 랫트 뇨 (또는 이의 식염수 희석액)에 용해시킨 전술한 활성 트롬빈 억제제의 공지 농도와 연관시키는 표준 곡선을 사용하여 측정하였다. 24 시간에 걸친 랫트의 총 뇨 생산량에 뇨에서의 전술한 활성 화합물의 평가된 평균 농도를 곱함으로써, 뇨에 배설된 활성 억제제의 양(AMOUNTpd)을 계산할 수 있었다.
전구 약물을 경구 또는 비경구적으로 투여한 후 활성 트롬빈 억제제의 생체이용률은 하기 수학식과 같이 계산하였다:
[(AMOUNTpd/투여량)/(AMOUNTactive,parenteral/투여량)] x 100
상기 식에서, AMOUNTactive,parenteral은 상기한 바와 같이 의식있는 랫트에 대응 활성 트롬빈 억제제를 비경구적으로 투여한 후 뇨에 배설된 양이다.
시험 G
시험관내 전구약물 화합물의 대사적 활성화
화학식 I의 전구약물 화합물을 37 ℃에서 인간 또는 랫트 간 균질액으로부터 제조한 간 과립체 또는 10 000 g(원심분리 속도를 지칭함) 상청액 분획 (즉, s9 분획)과 배양하였다. 배양액 중의 총 단백질 농도는 0.05 mol/l TRIS 완충액(pH 7.4)에 용해된 1 또는 3 mg/ml이었고, 보조인자인 NADH (2.5 mmol/l) 및 NADPH (0.8 mmol/l)도 존재했다. 배양액의 총 부피는 1.2 ml였다. 초기 전구약물 농도는 5 또는 10 μmol/l였다. 배양을 시작한 지 60분을 초과한 후 규칙적인 간격으로 배양물로부터 샘플을 혼주하였다. 배양물로부터 채취한 샘플 25 ㎕를 동일 부피의 인간 또는 랫트 혈장 및 적당한 양의 트롬빈과 혼합하고, 응고계량기 (KC 10; Amelung) 상에서 응혈 시간(즉, 트롬빈 시간)을 측정하였다. 형성된 "활성" 트롬빈 억제제의 양은 혼주한 구연산염 첨가 인간 또는 랫트 혈장에서의 트롬빈 시간을 대응 "활성 트롬빈 억제제"의 공지 농도와 연관시키는 표준 곡선을 사용하여 평가하였다.
"활성" 트롬빈 억제제의 양은 다른 식으로 또는 상기 방법에 부가하여 LC-MS를 사용하여 측정하였다.
본 발명은 하기 실시예에 의해 설명된다. 다르게 명시되지 않았다면, Pro 및 Aze 아미노산은 S-이성질체로 정의된다. 다르게 명시되지 않았다면 실시예들은 부분입체이성질체로 얻었다.
실시예 1
Ph(3-N(Me)
2
)-(R)- 또는 -(S)CH(OH)-C(O)-Aze-Pab x HOAc
(i) Ph(3-N(Me)
2
)-CHO
CH2Cl2 중의 1.9g(12.6mmol)의 Ph(3-N(Me)2)-CH2OH 및 8.8g(100mmol)의 MnO2의 혼합물을 실온에서 2.5일 동안 교반하였다. 혼합물을 세리트 (Celite
(Celite )을 통해 여과시키고 여과액을 증발시켰다. 조 생성물을 7:3의 이소-프로필에테르:트리메틸펜탄을 용리액으로 사용하여 실리카 겔 상에서 플래시 크로마토그래피하였다. 수득량 0.93g(50%).
)을 통해 여과시키고 여과액을 증발시켰다. 조 생성물을 7:3의 이소-프로필에테르:트리메틸펜탄을 용리액으로 사용하여 실리카 겔 상에서 플래시 크로마토그래피하였다. 수득량 0.93g(50%).

(ii) Ph(3-N(Me)
2
)-(R,S)CH(OSiMe
3
)CN
15mL의 CH2Cl2 중의 0.9g(6.0mmol)의 상기 단계(i)로부터의 Ph(3-N(Me)2-CHO 및 0.08mL(6.0mmol)의 Et3N의 혼합물에 0.75mL(6.0mmol)의 TMS-CN을 적가하였다. 반응 혼합물을 실온에서 24시간 동안 교반하였다. 추가의 0.08mL(6.1mmol)의 Et3N 및 0.75mL(6.0mmol)의 TMS-CN을 첨가하였으며 추가로 24시간 동안 교반하였다. 반응 혼합물을 증발시켜서 부표제 화합물 1.35g(90%)를 생성하였다.

(iii) Ph(3-(N(Me)
2
)-(R,S)CH(OH)-C(O)OH
1.35g(5.43mmol)의 상기 단계 (ii)로부터의 Ph(3-N(Me)2)-(R,S)CH(OSiMe3)CN
및 20mL의 진한 HCl의 혼합물을 실온에서 10분 동안 교반한 후, 90℃ 내지 100℃에서 (오일조에서) 3시간 동안 교반하였다. 반응 혼합물을 증발시키고, H2O를 첨가하였다. 산성 수층을 Et2O로 세척하고 10-15g의 양이온 교환 수지 IR-120 (이것을 2M NaOH 중에 현탁하여서 사전 준비된 양이온 교환제)에 놓은 후, 슬러리를 컬럼에 부었다. 이어서 양이온 교환제를 2 ×50mL의 2M HCl, 2 ×50mL의 H2O 로 세척한 후, pH가 중성이 될 때까지 H2O로 세척하고 생성물을 1M NH4OH 수용액으로 용리하였다. 합쳐진 수층을 증발시키고 냉동 건조시켜서 0.78g(74%)의 부표제 화합물을 생성하였다.

(iv) Ph(3-N(Me)
2
)-(R)- 또는 -(S)CH(OH)-C(O)OH x HCl
Ph(3-N(Me)2)-(R,S)CH(OH)-C(O)OH(상기 단계 (iii))의 거울상이성질체를 키랄셀TM OD를 고정상으로, 80:20:1의 n-헵탄:2-프로판올:포름산을 이동상으로 사용한 예비 HPLC에 의해 분리하였다. 마지막에 용리한 거울상이성질체를 증발시키고 냉동 건조시킨 후, 물에 재용해시켰으며, 3 당량의 1 M HCl을 첨가하였다. 용액을 냉동 건조시켜서 -63.7°의 [α]D
20(c=1.0, MeOH)을 가진 히드로클로라이드 염을 생성하였다. 분석 키랄 HPLC로 측정된 과량의 거울상은 97%이었다.
(v) Ph(3-N(Me)
2
)-(R)- 또는 -(S)CH(OH)-C(O)-Aze-Pab(Z)
0℃에서 1.03mL(6.15mmol)의 DIPEA을 10mL의 DMF 중의 0.36g(1.54mmol)의 분리되고 단리된 상기 단계 (iv)의 생성물인 Ph(3-N(Me)2)-(R)-또는 -(S)CH(OH)-C(O)OH x HCl, 0.743g(1.69mmol)의 H-Aze-Pab(Z) x HCl(국제 특허 출원 WO 제97/02284호 참조) 및 0.543g(1.69mmol)의 TBTU의 혼합물에 첨가하였다. 반응 혼합물을 실온에서 4일 동안 교반하였으며, H2O(400mL)에 부었고, NaHCO3 수용액을 첨가하여 pH을 10으로 조정하였다. 수층을 EtOAc로 추출한 후, 유기층을 NaHCO3 수용액, H2O 및 NaCl 수용액으로 세척하였고, Na2SO4 상에서 건조시키고 증발시켰다. CH2Cl2:MeOH(95:5)를 용리액으로 사용하여 실리카 겔 상에서 플래시 크로마토그래피하여서 조 생성물을 정제하였다. 생성물을 예비 HPLC에 의해 추가로 정제하여 203mg(24%)의 부표제 화합물을 얻었다.

(vi) Ph(3-N(Me)
2
)-(R)- 또는 -(S)CH(OH)-C(O)-Aze-Pab x HOAc
7mL의 EtOH 중의 112mg(0.206mmol)의 상기 단계 (v)로부터의 Ph(3-N(Me)2)-(R)- 또는 -(S)CH(OH)-C(O)-Aze-Pab(Z), 0.41mL의 HOAc 및 Pd/C 10%을 대기압 및 실온에서 3시간 동안 수소화하였다. 반응 혼합물을 세리트 을 통해 여과시키고, 여과액을 증발시키고, 냉동 건조시키기를 2회 반복하여서 90mg(93%)의 백색 결정을 생성하였다.
을 통해 여과시키고, 여과액을 증발시키고, 냉동 건조시키기를 2회 반복하여서 90mg(93%)의 백색 결정을 생성하였다.

실시예 2
Ph(3-N(Me)
2
)-(R)- 또는 -(S)CH(OH)-CO-Aze-Pab(OMe)
(i) 4-(아미노, 메톡시이미노메틸)벤질 아지드
10.5g(125mmol)의 O-메틸히드록시아민 히드로클로라이드, 56mL의 트리에틸아민 및 200mL의 메탄올의 혼합물을 디에틸 에테르 중의 22.5g(110mmol) WO 제94/29336호에 기술된 방법에 따라 제조된 4-에틸이미다벤질 아지드 히드로클로라이드에 첨가하였다. 반응 혼합물을 3 내지 4일 동안 실온에서 교반하였다. 대부분의 메탄올을 진공에서 증발시키고 에틸 아세테이트로 교체하였다. 유기층을 H2O, HOAc 수용액(1.5%, pH 4), NaHCO3 수용액으로 세척시키고 Na2SO4 상에서 건조시켰 다. 얻어진 용액을 에틸 아세테이트로 500mL가 되도록 희석시키고, 수득량을 평가하기 위해서 희석된 용액 중 25mL를 농축시켰다. 총 수득량은 약 20g이었다.

(ii) H-Pab(OMe)
200mg의 산화 백금을 200mL의 에탄올 중의 10g (0.049mol)의 상기 단계 (i)로부터의 4-(아미노,메톡시이미노메틸)벤질 아지드의 용액에 첨가하였다. 혼합물을 대기압하에서 8 시간 동안 수소화시키고, 세리트 을 통해 여과시키고 농축시켰다. 조 생성물은 다음 단계에서 직접 사용하였다.
을 통해 여과시키고 농축시켰다. 조 생성물은 다음 단계에서 직접 사용하였다.

(iii)Boc-Aze-Pab(OMe)
100mL의 DMF 중의 9.7g(48mmol) Boc-Aze-OH(국제 특허 출원 WO 제97/02284호 참조), 9.4g(52mmol)의 상기 단계 (ii)로부터의 H-Pab(OMe) 및 18.5g(58mmol)의 TBTU의 얼음으로 냉각된 용액에 17.5mL(105mmol)의 DIPEA를 첨가하고, 혼합물을 밤새 실온에서 교반하였다. 생성된 혼합물을 50mL의 물 위에 붓고, pH을 약 9로 조정하고, 혼합물을 EtOAc로 3번 추출하였다. 합쳐진 유기층을 NaHCO3 수용액, 물 및 염수로 세척하고, Na2SO4 상에서 건조시키고 농축시켰다. 조 생성물을 에틸아세테이트를 용리액으로 사용하는 실리카 겔 상의 플래시 크로마토그래피로 정제하였 다. 수득량은 11.9g(69%)이었다.

(iv)Aze-Pab(OMe) x 2HCl
250mL의 EtOAc 중의 9.4g(26mmol)의 상기 단계 (iii)으로부터의 Boc-Aze-Pab(OMe)의 용액을 HCl(g)으로 포화시켰다. 125mL의 무수 EtOH을 생성된 에멀젼에 첨가하고 혼합물을 10분 동안 초음파 분해하였다. 용액이 탁해질 때까지 EtOAc를 첨가하였고, 그 후 재빨리 부표제 화합물을 결정화하였다. 수득량 6.7g(77%).

(v)Ph(3-N(Me)
2
)-(R)- 또는 -(S)CH(OH)-C(O)-Aze-Pab(OMe)
3mL의 DMF 중의 118mg(0.51mmol)의 Ph(3-N(Me)2)-(R)- 또는 -(S)CH(OH)-C(O)OH x HCl(상기 실시예 1(iv) 참조) 및 214mg(0.56mmol)의 HATU의 혼합물을 0℃에서 1.5 시간 동안 교반시켰다. 189mg(0.56mmol)의 상기 단계 (iv)로부터의 H-Aze-Pab(OMe) x 2 HCl, 0.3mL( 2.25mmol)의 2,4,6-트리메틸피리딘 및 3mL의 DMF을 0℃에서 따로 혼합한 후 첫번째 혼합물에 적가하였다. 반응 혼합물을 0℃에서 3 시간 동안 교반하고, 냉장고에 3일 동안 놓고, 증발시켰다. 조 생성물을 예비 HPLC로 정제하여서 140mg(62%)의 표제 화합물을 얻었다.

실시예 3
Ph(3-SMe)-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab x TFA
(i) Ph(3-(SMe)-(R,S)CH(OTMS)CN
0℃, 질소 하에서 450mL의 CH2Cl2 중의 19.8g(130mmol)의 Ph(3-SMe)-CHO 및 2.1g(6.50mmol)의 ZnI2의 용액에 14.2g(143mmol)의 트리메틸실릴 시아나이드를 적가하였다. 25℃에서 밤새 교반한 후, 오렌지색 혼합물을 450mL의 H20로 켄칭(quench)시켰다. 유기층을 분리하고 300mL의 포화 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과시키고 진공에서 농축하여서, 오렌지색 오일로서 32.0g(98% 조 생성물)의 부표제 화합물을 얻었으며, 이 생성물은 정제하지 않고 사용하였다.

(ii)Ph(3-SMe)-(R,S)CH(OH)C(O)OH
250mL의 진한 HCl 중의 32.0g(130mmol)의 상기 단계 (i)로부터의 Ph(3-SMe)- (R,S)CH(OTMS)CN의 용액을 2.5 시간 동안 환류시켰다. 혼합물을 450mL의 6N NaOH을 사용하여 염기성화시키고, 3 ×300mL의 Et2O로 세척하여서 유기 불순물을 제거하였다. 수성 층을 150mL의 6N HCl을 사용하여 산성화시키고, 4 ×500mL의 EtOAc로 추출하였다. 합쳐진 추출물을 Na2SO4 상에서 건조시키고, 여과시키고 진공에서 농축시켜서 오렌지색 오일로서 22.6g(90% 조 생성물 수율)의 부표제 화합물을 생성하였으며, 이 생성물을 방치하자 황갈색 고체로 결정화하였다.
(iii)Ph(3-SMe)-(R) 또는 -(S)CH(OH)C(O)OH (a) 및 Ph(3-SMe)-(S) 또는 -(R)CH(OAc)C(O)OH (b)
2.0g(10.1mmol)의 Ph(3-SMe)-(R,S)CH(OH)C(O)OH(상기 단계 (ii) 참조), 1.0g의 리파아제 PS 아마노(Amano), 5.0mL의 비닐 아세테이트 및 5.0mL의 MTBE의 혼합물을 45℃에서 24시간 동안 가열시켰다. 혼합물을 여과시키고, 여과 케이크를 100mL의 EtOAc로 세척하였다. 여과액을 진공에서 농축시키고 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액의 혼합물로 용리하여 크로마토그래피하여 630mg(32%)의 황색 오일로 부표제 화합물(a)를 얻었고, 850mg(35%)의 황갈색 고체로 부표제 화합물 (b)를 얻었다.
부표제 화합물 (a):

부표제 화합물 (b):

(iv)Boc-Aze-Pab x HCOOH
3.0g(50mmol)의 암모늄 포르메이트 및 1.0g(5%)의 Pd/C을 50mL의 MeOH 중의 4.7g(10mmol)의 Boc-Aze-Pab(Z)(국제 특허 출원 WO 제94/29336호 참조)의 용액에 첨가하였다. 1.0g(22mmol)의 포름산을 첨가하고 혼합물을 30분 동안 교반하였다. 반응 혼합물을 하이플로(Hyflo)를 통해 여과시키고 용액을 농축시켰다. 조 생성물을 50mL의 CH2Cl2 중에 현탁시키고, 여과시키고 추가의 CH2Cl2로 세척시켰다. 고체 물질을 건조시키고, 추가 정제하지 않고 다음 단계에 사용하였다.
(v)Boc-Aze-Pab(Teoc)
100mL의 THF 중의 3.7g(10mmol)의 Boc-Aze-Pab x HCOOH (상기 단계 (iv) 참조)의 용액에 3.5g(12.3mmol)의 Teoc-p-니트로페닐 카르보네이트를 첨가한 후, 20mL의 물 중의 1.8g(13mmol)의 K2CO3의 용액을 2분에 걸쳐 첨가하였다. 생성된 용 액을 3일 동안 교반하고, 농축하고, 잔여물을 150mL의 EtOAc 및 50mL의 0.5M NaOH 수용액에 용해시켰다. 유기층을 2 ×50mL의 염수로 세척하고, Na2SO4 상에서 건조시키고 농축시켰다. 조 생성물을 실리카 겔 상에서 4:1의 메틸렌 클로라이드:아세톤으로 용리하여 플래시 크로마토그래피로 정제하였다. 수득량 4.6g(96%).

(vi)H-Aze-Pab(Teoc) x HCl
150mL 메틸렌 클로라이드 중의 4.6g(9.6mmol)의 Boc-Aze-Pab(Teoc)(상기 단계 (v) 참조)의 용액을 건조 HCl로 포화시켰다. 용액을 마개로 막은 플라스크 안에서 실온에서 10분 동안 놓아둔 후, 농축시켰다. 수득량 4.2g(97%).
(vii)Ph(3-SMe)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
8.0mL의 DMF 중의 300mg(1.51mmol)의 Ph(3-SMe)-(R) 또는 -(S)CH(OH)C(O)OH(상기 단계 (iii)(a)참조), 627mg(1.66mmol)의 H-Aze-Pab(Teoc)(상기 단계 (vi) 참조), 632mg(1.66mmol)의 TBTU 및 391mg(3.03mmol)의 DIPEA의 혼합물을 0℃에서 교반한 후, 25℃에서 밤새 교반하였다. 반응을 50mL의 H20로 켄칭시키고, 3×50mL의 EtOAc로 추출하였다. 합쳐진 추출물을 Na2SO4 상에서 건조시키고, 여과시키고 진공 에서 농축시켰다. 잔여물을 실리카 겔 상에서 9:1 CH2Cl2:MeOH로 용리하여 크로마토그래피하여서 150mg(18%)의 백색 고체의 부표제 화합물을 생성하였다.

(viii)Ph(3-SMe)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x TFA
2mL의 CH2Cl2 중의 80mg(0.19mmol)의 Ph(3-SMe)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 단계 (vii) 참조) 및 2.0mL의 TFA의 혼합물을 0℃에서 3시간 동안 교반하였다. 용액을 진공에서 농축시키고, 잔여물을 물 중에 용해시키고 냉동 건조시켜서 90mg(87%)의 표제 화합물을 생성하였다.

실시예 4
Ph(3-SO
2
Me)-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab x TFA
(i) Ph(3-SO 2 Me)-(R) 또는 -(S)CH(OH)C(O)OH
(i) Ph(3-SO 2 Me)-(R) 또는 -(S)CH(OH)C(O)OH
40mL의 MeOH 및 25mL의 H20 중의 890mg(4.49mmol)의 Ph(3-SMe)-(R) 또는 -(S)CH(OH)C(O)OH(상기 실시예 3(iii)(a)참조) 및 8.3g(13.5mmol)의 옥손 (Oxone
(Oxone )의 혼합물을 0℃에서 교반시키고, 25℃에서 밤새 교반하였다. 고체를 여과시키고 200mL의 EtOAc로 세척시켰다. 여과액을 진공에서 농축시키고, 50mL의 H2O로 희석시킨 후, 4×60mL의 EtOAc로 추출하였다. 합쳐진 유기 추출물을 Na2SO4
상에서 건조시키고, 여과시키고 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 크로마토그래피하여, 150mg (15%)의 백색 고체로 부표제 화합물을 얻었다.
)의 혼합물을 0℃에서 교반시키고, 25℃에서 밤새 교반하였다. 고체를 여과시키고 200mL의 EtOAc로 세척시켰다. 여과액을 진공에서 농축시키고, 50mL의 H2O로 희석시킨 후, 4×60mL의 EtOAc로 추출하였다. 합쳐진 유기 추출물을 Na2SO4
상에서 건조시키고, 여과시키고 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 크로마토그래피하여, 150mg (15%)의 백색 고체로 부표제 화합물을 얻었다.

(ii) Ph(3-SO
2
Me)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
10mL의 DMF 중의 400mg(1.74mmol)의 Ph(3-SO2Me)-(R) 또는 -(S)CH(OH) C(O)OH(상기 단계 (i) 참조), 720mg(1.91mmol)의 H-Aze-Pab(Teoc)(상기 실시예 3(vi) 참조), 995mg(1.91mmol)의 PyBOP 및 463mg(3.83mmol)의 2,4,6-콜리딘의 혼합물을 0℃에서 교반한 후, 25℃에서 밤새 교반하였다. 혼합물을 50mL의 H20로 켄칭시키고, 3 ×50mL의 EtOAc로 추출하였다. 합쳐진 추출물을 Na2SO4상에서 건조시키고, 여과시킨 후 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 15:1의 CHCl3:MeOH로 용리하여 크로마토그래피하여서 570mg(57%)의 백색 고체로 부표제 화 합물을 얻었다.

(iii) Ph(3-SO
2
Me)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x TFA
0.5mL의 메틸렌 클로라이드 중의 65mg(0.11mmol)의 Ph(3-SO2Me)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 단계 (ii) 참조)의 냉각된 용액에 3mL의 TFA를 첨가하고, 용액을 100 분 동안 교반하였다. 생성된 용액을 농축시키고, 물을 첨가하고, 수용액을 냉동 건조시켜서 60mg(96%)의 표제 화합물을 생성하였다.

실시예 5
Ph(3-Cl, 5-NMeAc)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x TFA
(i) Ph(3-Cl,5-NO
2
)-(R,S)CH(OTMS)CN
1.0 L의 CH2Cl2 중의 24.1g(0.13mol)의 3-클로로-5-니트로벤즈알데하이드의 용액에 2.1g(6.5mmol)의 ZnCl2을 첨가하였다. 생성된 현탁액을 0℃로 냉각시키고, 13.9g(0.14mol)의 트리메틸실릴 시아나이드를 5분에 걸쳐 첨가하였다. 용액을 0℃에서 3 시간 동안 교반하고, 25℃로 가온하고 18시간 동안 교반하였다. 반응물을 H20로 희석시키고, 유기물을 분리하고 Na2SO4 상에서 건조시키고 여과시킨 후 진공에서 농축시켜서 36.8g(99%)의 오일로 부표제 화합물을 얻었다.

(ii)Ph(3-Cl, 5-NO
2
)-(R,S)CH(OH)C(O)OH
600mL의 진한 HCl 중의 59.0g(0.209mol)의 Ph(3-Cl, 5-NO2)(R,S)CH(OTMS)CN(상기 단계 (i) 참조)의 용액을 3시간 동안 가열하여 환류시켰다. 용액을 냉각시키고 진공에서 500mL로 농축시켰다. 산성 용액을 Et2O로 4회 추출하고, 유기물을 염수로 2회 세척하고, Na2SO4 상에서 건조시키고 여과시킨 후 진공에서 농축시켜서 48.4g(93%)의 고체로 부표제 화합물을 얻었으며, 이 생성물을 추가 정제 없이 사용하였다.
(iii) Ph(3-
Cl, 5-NO
2
)-(R) 또는 -(S)CH(OH)C(O)OH (a) 및 Ph(3-Cl, 5-NO
2
)-(S) 또는 -(R)CH(OAc)C(O)OH (b)
300mL의 비닐 아세테이트 및 300mL의 MTBE 중의 17.1g(73.84mmol)의 Ph(3- Cl, 5-NO2)-(R,S)CH(OH)C(O)OH(상기 단계 (ii)참조) 및 8.5g의 리파아제 PS 아마노의 혼합물을 55℃에서 24시간 교반하였다. 반응물을 세리트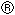 을 통해 여과시키고, 여과 케이크를 Et2O로 세척하였다. 여과액을 진공에서 농축시킨 후, 180:20:1의 CHCl3:CH3CN:TFA로 용리하여 실리카 겔 상에서 플래시 크로마토그래피하여서 7.1g (42%)의 부표제 화합물(a)를 고체로, 10.7g(52%)의 부표제 화합물 (b)를 고체로 얻었다.
을 통해 여과시키고, 여과 케이크를 Et2O로 세척하였다. 여과액을 진공에서 농축시킨 후, 180:20:1의 CHCl3:CH3CN:TFA로 용리하여 실리카 겔 상에서 플래시 크로마토그래피하여서 7.1g (42%)의 부표제 화합물(a)를 고체로, 10.7g(52%)의 부표제 화합물 (b)를 고체로 얻었다.
부표제 화합물 (a):

부표제 화합물 (b):

(iv)Ph(3-Cl,5-NH
2
)-(R) 또는 -(S)CH(OH)C(O)OH
200mL의 EtOH 중의 3.9g(16.8mmol)의 Ph(3-Cl, 5-NO2)-(R) 또는 -(S)CH(OH)
C(O)OH(상기 단계 (iii)(a) 참조) 및 0.4g의 산화 백금(IV)의 혼합물을 40℃의 수소 대기하에서 4시간 동안 교반하였다. 혼합물을 세리트 패드를 통해 여과시키 고 여과 케이크를 EtOH로 세척하였다. 여과액을 진공에서 농축시켜서 3.5g(약 100%)의 부표제 화합물을 부스러지는 포말로 얻었으며, 이 생성물을 추가의 정제 없이 사용하였다.
패드를 통해 여과시키 고 여과 케이크를 EtOH로 세척하였다. 여과액을 진공에서 농축시켜서 3.5g(약 100%)의 부표제 화합물을 부스러지는 포말로 얻었으며, 이 생성물을 추가의 정제 없이 사용하였다.

(v) Ph(3-Cl,5-NHMe)-(R) 또는 -(S)CH(OH)C(O)OH
방법 A:
400mL의 EtOH 중의 3.5g(16.8mmol)의 Ph(3-Cl,5-NH2)-(R) 또는 -(S)CH(OH)C
(O)OH(상기 단계 (iv) 참조) 및 1.8mL(23.9mmol)의 포름알데하이드(H2O 중의 37 중량%)의 혼합물을 25℃에서 18시간 동안 교반하였다. 용액을 진공에서 농축하여 부스러지는 포말을 얻고, 이것을 400mL 중의 0.35g의 산화 백금(IV)과 합쳐서, 수소 분위기하에서 48시간 동안 교반하였다. 혼합물을 세리트 패드를 통해 여과시키고 여과 케이크를 EtOH로 세척하였다. 유기물을 진공에서 농축시키고 실리카 겔 상에서 14:5:1의 CHCl3:MeOH:NH3로 용리하여 플래시 크로마토그래피하여서 1.0g (28%)의 부표제 화합물의 암모늄염을 부스러지는 포말로 얻었다. 부표제 화합물의 해당 암모늄염을 3:1 CH3CN:MeOH로 엠벌리트
패드를 통해 여과시키고 여과 케이크를 EtOH로 세척하였다. 유기물을 진공에서 농축시키고 실리카 겔 상에서 14:5:1의 CHCl3:MeOH:NH3로 용리하여 플래시 크로마토그래피하여서 1.0g (28%)의 부표제 화합물의 암모늄염을 부스러지는 포말로 얻었다. 부표제 화합물의 해당 암모늄염을 3:1 CH3CN:MeOH로 엠벌리트 (Amberlite
(Amberlite ) CG-50 패드를 통해 플러쉬하여 얻었다.
) CG-50 패드를 통해 플러쉬하여 얻었다.
방법 B:
500mL의 CH3CN 및 100mL의 MeOH 중의 8.67g(43.0mmol)의 Ph(3-Cl,5-NH2)-(R) 또는 -(S)CH(OH)C(O)OH(상기 단계 (iv)참조) 및 6.10g(43.0mmol)의 메틸 요오다이드의 혼합물을 50℃로 24시간 동안 가열하였다. 용액을 진공에서 농축시키고 14:5:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 실리카 겔 상에서 플래시 크로마토그래피하여서 2.9g(31%)의 부표제 화합물의 암모늄염을 고체로 얻었다. 부표제 화합물의 해당 암모늄염을 3:1 CH3CN:MeOH로 엠벌리트 CG-50 패드를 통해 플러쉬하여 얻었다.
CG-50 패드를 통해 플러쉬하여 얻었다.

(vi)Ph(3-Cl,5-NMeAc)-(R) 또는 -(S)CH(OH)C(O)OH
100mL의 MeOH 중의 1.0g(4.64mmol)의 Ph(3-Cl,5-NHMe)-(R) 또는 -(S)CH(OH)
C(O)OH(상기 단계 (v)참조)의 용액을 각각 40.47g(4.64mmol)씩 무수 아세트산으로 72 시간에 걸쳐 4번 처리하였다. 용액을 2N NaOH로 염기성화하고, 3시간 동안 교반하고 2N HCl로 중화한 후 진공에서 농축시켰다. 6:3:1의 CHCl3:MeOH:NH3로 용리하여 실리카 겔 상에서 2번 플래시 크로마토그래피하여서 0.83g(69%)의 부표제 화합물의 암모늄염을 부스러지는 포말로 얻었다. 부표제 화합물의 해당 암모늄염을 3:1 CH3CN:MeOH로 엠벌리트 CG-50 패드를 통해 플러쉬하여 얻었다.
CG-50 패드를 통해 플러쉬하여 얻었다.

(vii) Ph(3-Cl,5-NMeAc)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
15mL의 DMF 중의 0.34g(1.32mmol)의 Ph(3-Cl,5-NMeAc)-(R) 또는 -(S)CH(OH)
C(O)OH (상기 단계 (vi) 참조) 및 0.52g(1.39mmol)의 H-Aze-Pab(Teoc)(상기 실시예 3(vi) 참조)의 혼합물에 0℃에서 0.35g(2.90mmol)의 콜리딘 및 0.75g(1.45mmol)의 PyBOP를 첨가하였다. 용액을 0℃에서 2시간 동안 교반하고, 25℃로 가온하고, 2시간 동안 교반한 후 진공에서 농축시켰다. 실리카 겔 상에서 95:5의 CHCl3:EtOH로 용리하여 2번 플래시 크로마토그래피하여서 0.36g,(44%)의 부표제 화합물을 부스러지는 포말로 얻었다.

(viii) Ph(3-Cl,5-NMeAc)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x TFA
5.0mL의 TFA 중의 73mg(0.12mmol)의 Ph(3-Cl,5-NMeAc)-(R) 또는 -(S)CH(OH)
C(O)-Aze-Pab(Teoc)(상기 단계 (vii) 참조)의 용액을 실온에서 80분 동안 교반한후, 생성된 용액을 증발시켜서 건조시켰다. 남은 고체를 물에 용해시키고, 용액을 냉동 건조시켜서 70mg(98%)의 표제 화합물을 포말로 얻었다.

실시예 6
Ph(3-Cl, 5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x 2TFA
(i) 3-클로로-5-N,N-디메틸아미노벤질 알콜
750mL의 EtOH 중의 12.5g(66.6mmol)의 3-클로로-5-니트로벤질 알콜의 용액에 1.25g의 산화 백금(IV)을 첨가하였다. 생성된 현탁액을 수소로 3시간 동안 세정하였다. 97mL(1.3mmol)의 포름알데히드 용액(H2O 중의 37 중량%)을 첨가하고, 혼합물을 수소 분위기 하에서 8 시간 동안 교반하였다. 용액을 세리트 패드를 통해 여과시키고 진공에서 농축시켜서 조 생성물을 생성하였다. 실리카 겔 상에서 7:3의 Hex:EtOAc로 용리하여 플래시 크로마토그래피하여서 8.2g(66%)의 부표제 화합물을 오일로 얻었다.
패드를 통해 여과시키고 진공에서 농축시켜서 조 생성물을 생성하였다. 실리카 겔 상에서 7:3의 Hex:EtOAc로 용리하여 플래시 크로마토그래피하여서 8.2g(66%)의 부표제 화합물을 오일로 얻었다.

(ii) 3-클로로-5-N,N-디메틸아미노벤즈알데하이드
100mL의 CH2Cl2 중의 7.58g(97.0mmol)의 DMSO의 용액에 6.16g(48.5mmol)의 옥살릴 클로라이드를 -78℃에서 10분에 걸쳐 첨가하였다. -78℃에서 추가 15분 후, 100mL의 CH2Cl2 중의 8.18g(44.1mmol)의 3-클로로-5-N,N-디메틸아미노벤질 알콜(상기 단계 (i)을 참조)의 용액을 15분에 걸쳐 첨가하였다. 생성된 용액을 -78℃에서 1시간 동안 교반한 후, 28.5g(220.5mmol)의 DIPEA을 첨가하였다. 용액을 25℃로 가온하고 18 시간 동안 교반한 후, 진공에서 농축하여 조 생성물을 생성하였다. 실리카 겔 상에서 5:1의 Hex:EtOAc로 용리하여 플래시 크로마토그래피하여서 7.50g(93%)의 부표제 화합물을 황색 고체로 얻었다.

(iii) Ph(3-Cl,5-NMe
2
)-(R,S)CH(OTMS)CN
300mL의 CH2Cl2 중의 7.5g(40.8mmol)의 3-클로로-5-N,N-디메틸아미노벤즈알데하이드(상기 단계 (ii)참조)의 용액에 0.65g(2.04mmol)의 ZnI2을 첨가하였다. 생성된 현탁액을 0℃로 냉각시키고 4.5g(44.9mmol)의 트리메틸실릴 시아나이드를 5분에 걸쳐 첨가하였다. 용액을 0℃에서 1시간 동안 교반한 후, 25℃로 가온하고 2시간 동안 교반하였다. 생성된 혼합물을 H20로 희석시키고, 유기물을 분리하고, Na2SO4 상에서 건조시키고 여과시킨 후 진공에서 농축시켜서 11.7g(110%)의 부표제 화합물 을 오일로 얻었다.

(iv) Ph(3-Cl, 5-NMe
2
)-(R,S)CH(OH)C(O)OH
11.7g(41.4mmol)의 Ph(3-Cl, 5-NMe2)-(R,S)CH(OTMS)CN(상기 단계 (iii)참조)을 300mL의 진한 HCl에 용해시키고 1.5 시간 동안 가열하여 환류하였다. 용액을 냉각시키고, 진공에서 농축시켰다. 잔여물을 H2O에 용해시키고, NaHCO3로 중화시키고 진공에서 농축시켰다. 유기물과 염의 혼합물을 MeOH 중에서 슬러리화시키고 여과시킨 후 농축시켜서 조 생성물을 얻었다. 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:진한 NH4OH 수용액으로 용리하여 플래시 크로마토그래피시켜서 9.0g(95%)의 부표제 화합물의 암모늄염을 고체로 얻었다.
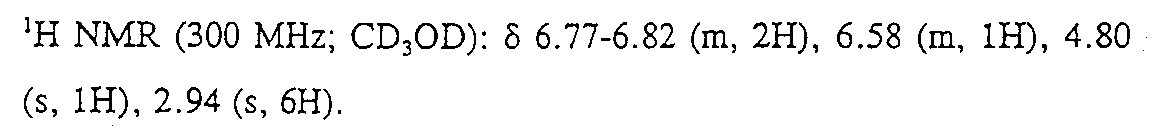
(v) Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)OH (a) 및 Ph(3-Cl,5-NMe
2
)-(S) 또는 -(R)CH(OAc)C(O)OH (b)
10mL의 비닐 아세테이트 및 10mL의 MTBE 중의 1.0g의 Ph(3-Cl,5-NMe2)-(R,S)CH(OH)C(O)OH(상기 단계 (iv) 참조) 및 0.5g의 리파아제 PS 아마노의 혼합물을 45℃에서 48 시간 동안 교반하였다. 반응물을 세리트 패드를 통해 여과시키 고 여과 케이크를 MeOH로 세척하였다. 여과액을 진공에서 농축시키고 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하여서 0.40g(40%)의 부표제 화합물 (a)를 부수러지는 포말로, 0.45g(38%)의 부표제 화합물 (b)를 부수러지는 포말로 얻었다. 부표제 화합물 (a)는 CH2Cl2 및 MeOH로부터 결정화하여서 추가 정제할 수 있다.
패드를 통해 여과시키 고 여과 케이크를 MeOH로 세척하였다. 여과액을 진공에서 농축시키고 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하여서 0.40g(40%)의 부표제 화합물 (a)를 부수러지는 포말로, 0.45g(38%)의 부표제 화합물 (b)를 부수러지는 포말로 얻었다. 부표제 화합물 (a)는 CH2Cl2 및 MeOH로부터 결정화하여서 추가 정제할 수 있다.
부표제 화합물 (a):

부표제 화합물 (b):

(vi) Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
15mL의 DMF 중의 0.11g(0.48mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH) C(O)OH(상기 단계 (v)(a) 참조) 및 0.20g(0.53mmol)의 H-Aze-Pab(Teoc)(상기 실시예 3(vi) 참조)의 혼합물에 0℃에서 0.12g(0.96mmol)의 DIPEA 및 0.17g( 0.53mmol)의 TBTU를 첨가하였다. 용액을 0℃에서 2시간 동안 교반하고, 25℃로 가온하고 18 시간 교반한 후 진공에서 농축시켰다. 실리카 겔 상에서 100:0에서 95:5의 CH2Cl2:MeOH의 농도 구배로 용리하여 플래시 크로마토그래피하여서 0.25g의 부표제 화합물을 얻은 후, 이것을 실리카 겔 상에서 30:1의 EtOAc:MeOH로 용리하여 또 다시 플래시 크로마토그래피하여서 0.22g(78%)의 부스러지는 포말로 부표제 화합물을 얻었다.

(vii) Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x 2TFA
84mg(0.14mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 단계 (vi) 참조)의 냉각된 용액에 4mL의 TFA를 첨가하고, 생성된 용액을 0℃에서 2시간 동안 교반하였다. 용액을 농축시켜서 잔여물을 생성하고 이것을 물에 용해시킨 후 냉동 건조하였다. 78mg(81%)의 백색 분말로 표제 화합물을 얻었다.

실시예 7
Ph(3-NMeAc)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x HAOAc
(i)Ph(3-NO
2
)-(R) 또는 -(S)CH(OH)C(O)OH (a) 및 Ph(3-NO
2
)-(S) 또는 -(R)CH(OAc)C(O)OH (b)
150mL의 비닐 아세테이트 및 375mL의 MTBE 중의 25g(126mmol)의 Ph(3-NO2)-(R,S)CH(OH)C(O)OH, 12.5g의 리파아제 PS 아마노를 45℃에서 24시간 동안 가열하였다. 반응물을 여과시키고 여과 케이크를 500mL의 EtOAc로 세척하였다. 여과액을 진공에서 농축시키고 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액의 혼합물로 용리하여 크로마토그래피하여서 9.0g(36%)의 황색 오일로 부표제 화합물 (a) 및 6.5g(21%)의 황갈색 고체로 부표제 화합물 (b)를 얻었다.
부표제 화합물 (a):

부표제 화합물 (b):

(ii)Ph(3-NH
2
)-(R) 또는 -(S)CH(OH)C(O)OH
200mL의 MeOH 중의 8.0g(40.6mmol)의 Ph(3-NO2)-(R) 또는 -(S)CH(OH)C(O)OH( 상기 단계 (i)(a) 참조) 및 800mg의 탄소상의 10% 팔라듐의 혼합물을 25℃에서 수소 1 기압하에서 밤새 교반하였다. 혼합물을 세리트 패드를 통해 여과시키고, 250mL의 EtOAc로 세척하였다. 여과액을 진공에서 농축시켜서 7.0g(100%)의 백색 포말로 부표제 화합물을 생성하였다.
패드를 통해 여과시키고, 250mL의 EtOAc로 세척하였다. 여과액을 진공에서 농축시켜서 7.0g(100%)의 백색 포말로 부표제 화합물을 생성하였다.

(iii)Ph(3-NHMe)-(R) 또는 -(S)CH(OH)C(O)OH
50mL의 MeOH 중의 2.9g(17.3mmol)의 Ph(3-NH2)-(R) 또는 -(S)CH(OH)C(O)OH(상기 단계 (ii) 참조) 및 2.95g(20.8mmol)의 요오드화 메틸의 혼합물을 55℃에서 밤새 가열하였다. 반응 혼합물을 진공에서 농축시키고 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 크로마토그래피하여서 616mg(20%)의 갈색 오일로 부표제 화합물을 얻었다.

(iv) Ph(3-NMeAc)-(R) 또는 -(S)CH(OH)C(O)OH
15mL의 MeOH 중의 540mg(2.99mmol)의 Ph(3-NHMe)-(R) 또는 -(S)CH(OH) C(O)OH(상기 단계 (iii) 참조) 및 612mg(5.98mmol)의 무수 아세트산의 혼합물을 25℃에서 밤새 질소 분위기하에서 교반하였다. 혼합물을 진공에서 농축시키고 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:진한 NH4OH 포화 수용액으로 용리하는 크로마토그 래피하여서 380mg(57%)의 백색 포말로 부표제 화합물을 얻었다.

(v) Ph(3-NMeAc)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
10mL의 DMF 중의 301mg(1.36mmol)의 Ph(3-NMeAc)-(R) 또는 -(S)CH(OH)C(O)OH (상기 단계 (iv) 참조), 560mg(1.48mmol)의 H-Aze-Pab(Teoc)(상기 실시예 3(vi) 참조), 774mg(1.48mmol)의 PyBOP 및 360mg(2.97mmol)의 2,4,6-콜리딘의 혼합물을 0℃에서 교반한 후 25℃에서 밤새 교반하였다. 혼합물을 50mL의 H20로 켄칭시키고 3 ×50mL의 EtOAc로 추출하였다. 합쳐진 유기 추출물을 Na2SO4 상에서 건조시키고 여과시키고 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 9:1의 CHCl3:MeOH로 용리하여 크로마토그래피하여서 175mg(23%)의 백색 고체로 부표제 화합물을 얻었다.

(vi) Ph(3-NMeAc)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x HOAc
2mL의 CH2Cl2 중의 65mg(0.11mmol)의 Ph(3-NMeAc)-(R,S)CH(OH)C(O)-Aze- Pab(Teoc) 및 2.0mL의 TFA의 혼합물을 0℃에서 3 시간 동안 교반하였다. 용액을 실온에서 진공에서 농축시키고 잔여물을 예비 HPLC(CH3CN:0.1 M NH4OAc, 농도구배 : 0-50% CH3CN)로 정제하고 목적하는 분획물을 농축시켰다. 생성물을 물/HOAc 중에 용해하고 냉동 건조시켜서 55mg(100%)의 표제 화합물을 생성하였다.

실시예 8:
Ph(3-NMe
2
, 5-CF
3
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x TFA
(i) Ph(3-NO
2
, 5-CF
3
)CH
2
OH
50mL의 THF 중의 10.0g(42.6mmol)의 Ph(3-NO2,5-CF3)CO2H의 용액에 170mL( 170mmol)의 보란-테트라히드로푸란 착물(THF 중의 1M 용액)을 1시간에 결쳐 적가하고, 질소 하에서 0℃로 냉각하였다. 용액을 실온으로 가온시키고 4시간 동안 교반하였다. H20를 천천히 첨가하여서 용액을 켄칭시키고, 200mL의 EtOAc로 부은 후, 150mL의 H20 및 150mL의 염수로 차례대로 세척하였다. 유기상을 Na2SO4에서 건조시키고 여과시키고 진공에서 농축시켜서 6.9g(73%)의 오렌지색 오일로 부표제 화합물 을 생성하였다.

(ii)Ph(3-NO
2
,5-CF
3
)-CHO
3.0mL(34mmol)의 옥살릴 클로라이드를 70mL의 건조 CH2Cl2 중의 4.86mL (68.6mmol)의 DMSO의 용액에 적가하고 질소 하에서 -78℃로 냉각시켰다. -78℃에서 15분 후, 75mL의 CH2Cl2 중의 6.9gm(31mmol)의 Ph(3-NO2,5-CF3
)CH2OH(상기 단계 (i) 참조)를 30분에 걸쳐 적가하였다. -78℃에서 45분 후, 27.2mL(156mmol)의 DIPEA를 20분에 걸쳐 첨가하였다. 그 후 용액을 -78℃에서 추가로 1시간 동안 교반하고, 용액을 실온으로 가온하고 15 시간 동안 교반하였다. 용액을 2 ×150mL의 1M HCl, 150mL의 염수로 차례대로 세척시키고, 여과시키고 진공에서 농축시켜서 6.9g(99%)의 오렌지색 오일로 부표제 화합물을 생성하였다.

(iii)Ph(3-NO
2
,5-CF
3
)-(R,S)CH(OTMS)CN
220mL의 CH2Cl2 중의 6.52g(29.7mmol)의 Ph(3-NO2,5-CF3)-CHO(상기 단계 (ii) 참조)의 용액에 474mg(1.49mmol)의 ZnI2를 첨가하였다. 용액을 질소로 세정하고 0℃로 냉각하였다. 3.25g(32.7mmol)의 트리메틸실릴 시아나이드를 10분에 걸쳐 첨 가한 후, 용액을 2시간 동안 교반하였다. 그 후, 용액을 실온으로 가온하고 추가로 5.5 시간 동안 교반한 후, 250mL의 H20로 반응물을 켄칭시켰다. 유기상을 분리하고 수상을 125mL의 CH2Cl2로 추출하였다. 합쳐진 유기층을 Na2SO4
상에서 건조시키고, 여과시키고 진공에서 농축시켜서 9.1g(96%)의 오렌지색 오일로 부표제 화합물을 생성하였다.

(iv) Ph(3-NO
2
, 5-CF
3
)-(R,S)CH(OH)C(O)OH
9.1g(29mmol)의 Ph(3-NO2,5-CF3)-(R,S)CH(OTMS)CN(상기 단계 (iii) 참조)을 83mL(1000mmol)의 진한 HCl 중에 용해시키고 3시간 동안 가열하여 환류시켰다. 용액을 200mL의 H20로 희석하고 3 ×150mL의 Et2O로 추출하였다. 합쳐진 유기물을 200mL의 염수로 세척하고, Na2SO4 상에서 건조시키고 여과시키고 진공에서 농축시켜서 갈색 오일을 생성하였다. 조 생성물을 실리카 겔 상에서 14:5:1의 CHCl3: MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하였다. 생성된 백색 고체를 Et2O 중에 현탁하고 100mL의 2M HCl을 첨가하였다. 층을 분리하고 수상을 3 ×200mL의 Et2O로 추출하였다. 합쳐진 유기층을 Na2SO4 상에서 건조시키고, 여과시 키고 농축시켜서 5.9g(78%)의 갈색 고체로 부표제 화합물을 생성하였다.

(v)Ph(3-NH
2
,5-CF
3
)-(R,S)CH(OH)C(O)OH
350mL의 무수 EtOH 중의 5.9g(22mmol)의 Ph(3-NO2,5-CF3)-(R,S)CH(OH) C(O)OH(상기 단계 (iv) 참조)의 용액에 590mg의 산화 백금(IV)을 첨가하였다. 용액을 수소로 5 시간 동안 세정한 후, 혼합물을 세리트 을 통해 여과시킨 후 진공에서 농축시켜서 5.8g(100%)의 오렌지색 오일로 부표제 화합물을 생성하였다.
을 통해 여과시킨 후 진공에서 농축시켜서 5.8g(100%)의 오렌지색 오일로 부표제 화합물을 생성하였다.
(vi)Ph(3-NMe
2
,5-CF
3
)-(R,S)CH(OH)C(O)OH
250mL의 무수 EtOH 중에 용해되어 있는 5.27g(22.4mmol)의 Ph(3-NH2,5-CF3)-(R,S)CH(OH)C(O)OH(상기 단계 (v) 참조)의 용액에 54mL(720mmol)의 37% 포름알데히드 수용액을 첨가하였다. 520mg의 산화 백금(IV)을 첨가하고, 용액을 수소로 정화하였다. 수소 하에서 22시간 동안 교반한 후, 용액을 세리트 을 통해 여과시키고 진공에서 농축시켰다. 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하여서 2.7g(46%)의 백색 고체의 부표제 화합물을 생성하였다.
을 통해 여과시키고 진공에서 농축시켰다. 실리카 겔 상에서 6:3:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하여서 2.7g(46%)의 백색 고체의 부표제 화합물을 생성하였다.
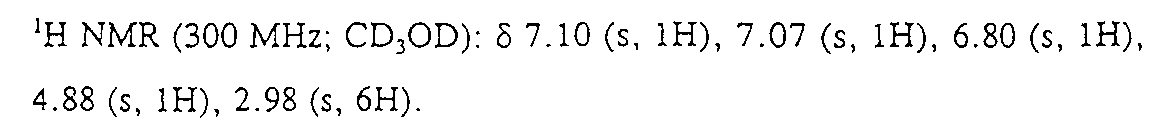
(vii)Ph(3-NMe
2
,5-CF
3
)-(R) 또는 -(S)CH(OH)C(O)OH (a) 및 Ph(3-NMe
2
)(5-CF
3
)-(S) 또는 -(R)CH(OAc)C(0)OH (b)
2.7g(10mmol)의 Ph(3-NMe2,5-CF3)-(R,S)CH(OH)C(O)OH(상기 단계 (vi) 참조), 1.4g의 리파아제 PS 아마노, 56mL의 비닐 아세테이트 및 120mL의 MTBE의 혼합물을 1일간 환류시켰다. 반응물을 세리트 을 통해 여과시키고 여과 케이크를 Et2O로 세척하였다. 여과액을 진공에서 농축시켜서 실리카 겔 상에서 14:5:1의 CHCl3: MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하여서 727mg(27%)의 백색 고체로 부표제 화합물(a)의 암모늄염 및 1.53g(49%)의 백색 고체로 부표제 화합물(b)의 암모늄염을 생성하였다. 부표제 화합물(a)의 암모늄염을 3:1의 CH3CN:MeOH를 용리액으로 사용하여 앰벌리트
을 통해 여과시키고 여과 케이크를 Et2O로 세척하였다. 여과액을 진공에서 농축시켜서 실리카 겔 상에서 14:5:1의 CHCl3: MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하여서 727mg(27%)의 백색 고체로 부표제 화합물(a)의 암모늄염 및 1.53g(49%)의 백색 고체로 부표제 화합물(b)의 암모늄염을 생성하였다. 부표제 화합물(a)의 암모늄염을 3:1의 CH3CN:MeOH를 용리액으로 사용하여 앰벌리트 CG-50 패드를 통해 플러시하여 백색 고체의 부표제 화합물(a)를 생성하였다.
CG-50 패드를 통해 플러시하여 백색 고체의 부표제 화합물(a)를 생성하였다.
부표제 화합물 (a):

부표제 화합물 (b):

(viii)Ph(3-NMe
2
,5-CF
3
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
290mg(1.10mmol)의 Ph(3-NMe2,5-CF3)-(R) 또는 -(S)CH(OH)C(O)OH(상기 단계 (vii)(a) 참조) 및 436mg(1.16mmol)의 H-Aze-Pab(Teoc)(상기 실시예 3(vi) 참조)의 혼합물에 10mL의 건조 DMF를 첨가하였다. 용액을 0℃로 냉각시킨 후, 630mg( 1.21mmol)의 PyBOP 및 295mg(2.42mmol)의 콜리딘을 첨가하였다. 용액을 질소 하에서 0℃에서 2시간 동안 교반하고 실온에서 15시간 동안 교반하였다. 혼합물을 농축시키고 실리카 겔 상에서 20:1의 EtOAc:EtOH로 용리하여 플래시 크로마토그래피하여서 383mg(56%)의 백색 고체로 부표제 화합물을 생성하였다.

(ix)Ph(3-NMe
2
, 5-CF
3
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x TFA
메틸렌 클로라이드 중의 87mg(0.14mmol)의 Ph(3-NMe2,5-CF3)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 단계 (viii) 참조)의 냉각된 용액에 4mL의 TFA를 첨가하고 혼합물을 0℃에서 100분간 교반하였다. 생성된 용액을 건조될 때까지 농축시켜서, 물/CH3CN 중에 용해된 잔여물을 생성한 후 냉동 건조시켜서, 81mg(80%)의 백색 분말로 표제 화합물을 생성하였다.

실시예 9
Ph(3-Cl,5-(1-피롤리딘-2-온)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x HOAc
(i) (R,S)-5-Ph(3-Cl,5-NO 2 )-2,2-디메틸-4-옥소-1,3-디옥솔란
Ph(3-Cl,5-(1-피롤리딘-2-온)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x HOAc
(i) (R,S)-5-Ph(3-Cl,5-NO 2 )-2,2-디메틸-4-옥소-1,3-디옥솔란
삭제
300mL의 아세톤 중의 18.8mg(81.2mmol)의 Ph(3-Cl,5-NO2)-(R,S)CH(OH)C(O) OH(상기 실시예 5(ii) 참조)의 용액에 750mg(3.94mmol)의 p-톨루엔술폰산 일수화물 및 75mL(514mmol)의 2,2-디메톡시프로판을 첨가하였다. 용액을 6 시간 동안 환류시키고 진공에서 농축시켰다. 잔여물을 200mL의 EtOAc에 용해시킨 후, 100mL의 H20, 150mL의 NaHCO3 포화 수용액 및 150mL의 염수로 세척시켰다. 유기상을 Na2
SO4 상에서 건조시키고, 여과시키고 농축시켜서, 갈색의 고체를 얻고, 이것을 실리카 겔 상에서 7:3의 Hex:EtOAc로 용리하여 플래시 크로마토그래피하였다. 생성된 고체를 1:10의 EtOAc/Hex로부터 재결정화하여 추가로 정제하여서 14.7g(67%)의 백색 고체로 부표제 화합물을 생성하였다.
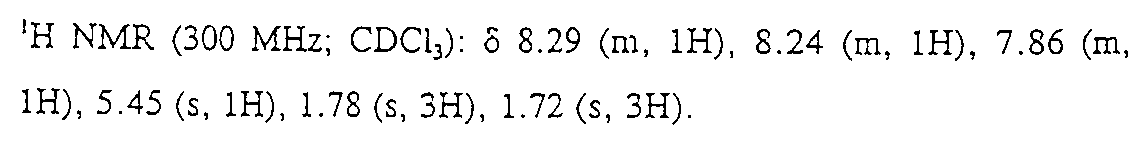
(ii)(R,S)-5-Ph(3-Cl,5-NH
2
)-2,2-디메틸-4-옥소-1,3-디옥솔란
400mL의 EtOH 중의 14.7g(54.1mmol)의 (R,S)-5-Ph(3-Cl,5-NO2)-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (i) 참조)의 용액에 1.5g의 산화 백금(IV)을 첨가하였다. 현탁액을 수소 1 기압하에서 실온에서 27 시간 동안 교반하였다. 현탁액을 세리트 을 통해 여과시키고 여과 케이크를 EtOH로 세척하였다. 여과액을 진공에서 농축시켜서 노란색 오일을 생성하고, 이것을 실리카 겔 상에서 4:1의 Hex:EtOAc로 용리하여 크로마토그래피하여서, 6.5g(50%)의 노란색 오일로 부표제 화합물을 생성하였다.
을 통해 여과시키고 여과 케이크를 EtOH로 세척하였다. 여과액을 진공에서 농축시켜서 노란색 오일을 생성하고, 이것을 실리카 겔 상에서 4:1의 Hex:EtOAc로 용리하여 크로마토그래피하여서, 6.5g(50%)의 노란색 오일로 부표제 화합물을 생성하였다.

(iii) (R,S)-5-Ph(3-Cl,5-(1-피롤리디닐-2-온))-2,2-디메틸-4-옥소-1,3-디옥솔란
100mL의 DMF 중의 6.5g(26.9mmol)의 (R,S)-5-Ph(3-Cl,5-NH2)-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (ii) 참조)의 용액에 10.5g(53.8mmol)의 에틸 4-브로모부티레이트 및 5.4g(53.8mmol)의 Et3N을 첨가하였다. 용액을 아르곤 하에서 21시간 동안 95℃에서 가열하였다. 반응 혼합물을 농축시킨 후 200mL의 EtOAc 중에 용해시켜서 용액을 생성하고, 이것을 150mL의 H20 및 150mL의 염수로 세척하였다. 유기상을 Na2SO4 상에서 건조시키고, 여과시키고 농축시켜서 9.6g의 오렌지색 오일을 생성하였다. 조 물질을 250mL의 p-크실렌 중에 용해시키고 환류하에 가열하였다. 3일 후, 혼합물을 오렌지색 오일로 농축시키고 실리카 겔 상에서 1:1의 EtOAc:헥산으로 용리하여 플래시 크로마토그래피하여서 4.5g(54%)의 노란색 고체로 부표제 화합물을 생성하였다.

(iv)Ph(3-Cl,5-(1-피롤리딘-2-온))-(R,S)CH(OH)C(O)OH
300mL의 THF 중의 4.5g(14.5mmol)의 (R,S)-5-Ph(3-Cl,5-(1-피롤리디닐-2- 온))-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (iii)참조)의 용액에 145mL의 1N NaOH를 첨가하였다. 용액을 30분간 교반한 후, 생성된 용액을 진공에서 일부 감소 시켰다. 용액을 2N HCl로 산성화시키고 2 ×150mL의 EtOAc로 추출하였다. 유기상을 200mL의 염수로 세척하고, Na2SO4 상에서 건조시키고 여과시키고 농축시켜서 3.2g(82%)의 백색 고체로 부표제 화합물을 생성하였다.

(v)Ph(3-Cl, 5-(1-피롤리딘-2-온))-(S) 또는 -(R)-CH(OAc)C(O)OH (b) 및 Ph(3-Cl,5-(1-피롤리딘-2-온))-(R) 또는 -(S)-CH(OH)C(O)OH (a)
65mL의 비닐 아세테이트 및 130mL의 MTBE 중의 3.2g(11.9mmol)의 Ph(3-Cl,5-(1-피롤리딘-2-온))-(R,S)CH(OH)C(O)OH(상기 단계 (iv) 참조) 및 1.6g의 리파아제 PS 아마노의 혼합물을 55℃에서 24시간 교반하였다. 반응액을 세리트 을 통해 여과시키고 여과 케이크를 THF 및 MeOH로 차례대로 세척시켰다. 여과액을 진공에서 농축시키고 실리카 겔 상에서 14:5:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하여서 1.3g(33%)의 백색 고체로 부표제 화합물 (b)의 암모늄염을 생성하였다. 추가로, 800mg(20%)의 부표제 화합물 (a)의 암모늄염을 얻었다. 이 물질을 40mL의 H20 중에 용해시키고, 1N HCl로 산성화시키고, 2 ×50mL의 EtOAc로 추출하였다. 유기상을 Na2SO4 상에서 건조시키고 여과시키고 농축시켜서 백색 고체의 부표제 화합물 (a)를 생성하였다. 낮은 광학 순도로 인해, 부표제 화합물 (b)를 상기 효소 분해 조건(0.5g 리파아제 PS 아마노; 35mL 비닐 아세테이트; 60mL MTBE; 55℃; 24h)으로 다시 처리하였다. 상기에서 언급한 단리 및 정제 방법으로 470mg의 백색 고체의 부표제 화합물(a)를 생성하였다.
을 통해 여과시키고 여과 케이크를 THF 및 MeOH로 차례대로 세척시켰다. 여과액을 진공에서 농축시키고 실리카 겔 상에서 14:5:1의 CHCl3:MeOH:NH3 포화 수용액으로 용리하여 플래시 크로마토그래피하여서 1.3g(33%)의 백색 고체로 부표제 화합물 (b)의 암모늄염을 생성하였다. 추가로, 800mg(20%)의 부표제 화합물 (a)의 암모늄염을 얻었다. 이 물질을 40mL의 H20 중에 용해시키고, 1N HCl로 산성화시키고, 2 ×50mL의 EtOAc로 추출하였다. 유기상을 Na2SO4 상에서 건조시키고 여과시키고 농축시켜서 백색 고체의 부표제 화합물 (a)를 생성하였다. 낮은 광학 순도로 인해, 부표제 화합물 (b)를 상기 효소 분해 조건(0.5g 리파아제 PS 아마노; 35mL 비닐 아세테이트; 60mL MTBE; 55℃; 24h)으로 다시 처리하였다. 상기에서 언급한 단리 및 정제 방법으로 470mg의 백색 고체의 부표제 화합물(a)를 생성하였다.
부표제 화합물 (a):

(vi)Ph(3-Cl,5-(1-피롤리딘-2-온)-(R)- 또는 (S)-CH(OH)C(O)-Aze-Pab(Teoc)
9mL의 DMF 중의 250mg(0.927mmol)의 Ph(3-Cl,5-(1-피롤리딘-2-온)-(R)- 또는 -(S)-CH(OH)C(O)OH(상기 단계 (v)(a) 참조) 및 367mg(0.973mmol)의 H-Aze-Pab(Teoc)(상기 실시예 3(vi)참조)의 혼합물에 0℃에서 531mg(1.02mmol)의 PyBOP 및 250mg(2.04mmol)의 콜리딘을 첨가하였다. 용액을 질소 하 0℃에서 2시간 교반한 후 15시간 동안 실온으로 가온하였다. 혼합물을 농축시키고 실리카 겔 상에서 20:1의 EtOAc:EtOH로 용리하여 플래시 크로마토그래피시키고, EtOH 컬럼 플러시를 하여서 백색 고체를 생성하였다. 추가로 9:1의 CHCl3:EtOH로 용리하는 실리카 겔 상에서 플래시 크로마토그래피하여서 420mg(72%)의 백색 고체로 부표제 화합물을 생성하였다.

(vii)Ph(3-Cl,5-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x HOAc
0.5mL의 메틸렌 클로라이드 중의 90mg(0.14mmol)의 Ph(3-Cl,5-(1-피롤리딘-2-온)-(RorS)CH(OH)C(O)-Aze-Pab(Teoc)(상기 단계 (vi) 참조)의 용액에 4mL의 TFA를 첨가하였다. 혼합물을 실온에서 100분간 교반하였다. 생성된 용액을 진공에서 농축시키고 고체 조 생성물을 HPLC(CH3CN:0.1M 암모늄 아세테이트 20:80)을 사용하여 정제하였다. 목적 분획물을 수집하고 밤새 2회 냉동 건조시켰다. 수득량 51mg(67%). 순도 99.8%.

실시예 10
Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x HOAc
(i)(R,S)-5-Ph(3-NO
2
)-2,2-디메틸-4-옥소-1,3-디옥솔란
6.0g(30.4mmol)의 m-니트로만델산, 15.1mL의 2,2-디메톡시프로판, 0.29g( 1.52mmol)의 p-톨루엔술폰산 일수화물 및 60mL의 아세톤의 혼합물을 실온에서 12시간 교반하였다. 혼합물을 진공에서 농축시키고 조 생성물을 EtOAc에 용해시켰다. 유기상을 포화 NaHCO3 및 염수로 차례대로 세척하고, MgSO4 상에서 건조시킨 후 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 80/20에서 70/30으로 변화하는 헵탄/EtOAc로 용리하여 크로마토그래피하여서 5.7g(79%)의 부표제 화합물을 생성하였다(조 생성물은 적은 양의 EtOAC에 용해시키기 어렵고, 따라서 크로마토그래피 상에 로딩하는 것은 생성물이 흡착된 실리카 겔을 이용하여 달성할 수 있었다).

(ii)(R,S)-5-Ph(3-NH
2
)-2,2-디메틸-4-옥소-1,3-디옥솔란
250mL의 EtOH 중의 3.1g(13.1mmol)의 (R,S)-5-Ph(3-NO2)-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (i)참조), 1.7g(5%)의 Pd/C, 및 0.75mL(13.1mmol)의 HOAc의 혼합물을 수소 분위기 하에서 4시간 교반하였다. 혼합물을 세리트 패드를 통해 여과시키고 여과 케이크를 EtOH로 세척하였다. 여과액을 진공에서 농축시키고 형성된 무색의 고체를 EtOAc와 포화 수성 NaHCO3 사이에 분배하였다. 수상을 EtOAc로 추출하고 합쳐진 유기상을 염수로 세척하고, Na2SO4 상에서 건조시키고 진공에서 농축시켜서 2.3g(85%)의 부표제 화합물을 생성하였다.
패드를 통해 여과시키고 여과 케이크를 EtOH로 세척하였다. 여과액을 진공에서 농축시키고 형성된 무색의 고체를 EtOAc와 포화 수성 NaHCO3 사이에 분배하였다. 수상을 EtOAc로 추출하고 합쳐진 유기상을 염수로 세척하고, Na2SO4 상에서 건조시키고 진공에서 농축시켜서 2.3g(85%)의 부표제 화합물을 생성하였다.

(iii)(R,S)-5-Ph(3-NH(CH
2
)
3
C(O)OEt)-2,2-디메틸-4-옥소-1,3-디옥솔란
CH2Cl2 중의 1.63g(7.87mmol)의 (R,S)-5-Ph(3-NH2)-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (ii) 참조), 3.4mL(23.6mmol)의 에틸 4-브로모부티레이트 및 3.3mL(23.6mmol)의 Et3N의 혼합물을 밤새 환류시켰다. 추가량의 2.3mL (15.7mmol)의 에틸 4-브로모부티레이트 및 2.2mL(15.7mmol)의 Et3N을 첨가하고 혼합물을 하룻밤 이상 환류시켰다. 용매를 제거하고 조 생성물을 EtOAc와 물 사이에 분배하였다. 수상을 EtOAc로 추출하고 합쳐진 유기상을 염수로 세척하고, Na2SO4 상에서 건조시킨 후 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 90:10에서 80:20으로 변화하는 헵탄:EtOAc로 용리하여 크로마토그래피하여서 2.1g(84%)의 부표제 화합물을 생성하였다.

(iv) (R,S)-5-Ph(3-(1-피롤리딘-2-온))-2,2-디메틸-4-옥소-1,3-디옥솔란
15mL의 톨루엔 중의 2.2g(6.85mmol)의 (R,S)-5-Ph(3-NH(CH2)3C(O)OEt)-2,2-디 메틸-4-옥소-1,3-디옥솔란(상기 단계 (iii) 참조)의 용액을 이틀 밤 동안 환류시켰다. 용매를 제거하고 조 생성물을 80:20 에서 60:40으로 변화하는 헵탄:EtOAc로 용리하여 플래시 크로마토그래피하여서 1.4g(74%)의 부표제 화합물을 생성하였다.

(v) Ph(3-(1-피롤리딘-2-온))-(R,S)CH(OH)C(O)OH
15mL의 THF 중의 1.4g(5.1mmol)의 (R,S)-5-Ph(3-(1-피롤리딘-2-온))-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (iv) 참조) 및 10mL의 1M NaOH의 혼합물을 실온에서 밤새 격렬하게 교반하였다. THF를 제거하고 수상을 CH2Cl2로 한 번 세척한 후 진공에서 농축시켰다. 잔여물을 예비 RPLC(CH3CN:0.1M HOAC(16:84))을 사용하여 정제하고 목적 분획물을 농축시키고 냉동 건조시켜서 0.94g(79%)의 부표제 화합물을 생성하였다.

(vi)Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)OH
0.94g(4.0mmol)의 Ph(3-(1-피롤리딘-2-온))-(R,S)CH(OH)C(O)OH(상기 단계 (v) 참조)의 거울상 이성질체를 정지상으로 키랄팩TM AD를 사용하고 이동상으로 헵 탄:2-프로판올:아세토니트릴:포름산(160:30:10:1)을 사용한 예비 HPLC로 분리하였다. 최초로 용리되는 거울상 이성질체를 진공에서 증발시켜서 98.6%ee 및 [α]D
20 =-90.9°(c=1.0, MeOH)인 0.37g(39%)의 부표제 화합물을 생성하였다.

(vii)Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
365mg(0.70mmol)의 PyBOP 및 이어서 0.5mL(2.8mmol)의 DIPEA를 8mL의 DMF 중의 150mg(0.64mmol)의 Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)OH(상기 단계 (vi) 참조)및 264mg(0.70mmol)의 H-Aze-Pab(Teoc)(상기 실시예 3(vi) 참조) 의 냉각된(-20℃) 용액에 첨가하였다. 혼합물이 서서히 실온에 도달하도록 하였으며 밤새 교반하였다. DMF를 진공에서 제거하고 잔여물을 실리카 겔 상에서 95:5의 CH2Cl2:MeOH로 용리하여 크로마토그래피하여서 310mg(82%)의 부표제 화합물을 생성하였다.

(viii)Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab x HOAc
2mL의 CH2Cl2 중의 70mg(0.12mmol)의 Ph(3-(1-피롤리딘-2-온)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 단계 (vii) 참조) 및 2.0mL의 TFA를 0℃에서 2시간 교반하였다. 용액을 진공에서 농축시키고 잔여물을 예비 HPLC(CH3CN:0.1M NH4OAc (농도구배:0-50% CH3CN)를 사용하여 정제하였다. 목적 분획물을 농축시키고 생성물을 물/HOAc 중에 용해시키고 냉동 건조시켜서 52mg(87%)의 표제 화합물을 생성하였다.

실시예 11
Ph(3-(1-피롤리딘))-(R)- 또는 -(S)-CH(OH)C(O)-Aze-Pab x 2TFA
(i)(R,S)-5-Ph(3-(1-피롤리딘))-2,2-디메틸-4-옥소-1,3-디옥솔란
아세톤 중의 450mg(2.17mmol)의 (R,S)-5-Ph(3-NH2)-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 실시예 10(ii)참조), 0.30mL(3.26mmol)의 1,4-디브로모-부탄 및 2.1g(6.5mmol)의 Cs2CO3의 혼합물을 3일 동안 환류시켰다. 용매를 제거하고 조 생 성물을 CH2Cl2와 물 사이에 분배하였다. 수상을 CH2Cl2로 추출하고 합쳐진 유기상을 염수로 세척하고 Na2SO4 상에서 건조시킨 후 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 100:0에서 90:10으로 변화하는 헵탄:EtOAc로 용리하여 크로마토그래피하여서 140mg(25%)의 부표제 화합물을 생성하였다.

(ii)Ph(3-(1-피롤리딘))-(R,S)CH(OH)C(O)OH x HCl
10mL의 THF 중의 640mg(2.45mmol)의 (R,S)-5-Ph(3-(1-피롤리딘)-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (i)참조) 및 10mL의 1M NaOH의 혼합물을 밤새 실온에서 격렬하게 교반하였다. THF를 제거하고 수상을 CH2Cl2로 한 번 세척한 후 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 6:3:1의 CH2Cl2:MeOH:NH2OH로 용리하여 크로마토그래피하였다. 생성물을 첫번째는 물/HAOc을, 두번째는 물/2M HCl을 사용하여서 두 번 냉동 건조시켜서 염을 교환하였다. 0.62g(98%)의 부표제 화합물을 생성하였다.

(iii)Ph(3-(1-피롤리딘))-(R,S)CH(OH)C(O)-Aze-Pab(Teoc)
8mL의 DMF 중의 160mg(0.62mmol)의 Ph(3-(1-피롤리딘))-(R,S)CH(OH)C(O)OH(상기 단계 (ii) 참조) 및 307mg(0.68mmol)의 H-Aze-Pab(Teoc) x 2HCl(상기 단계 3(vi) 참조)의 냉각된(-20℃) 용액에 355mg(0.68mmol)의 PyBOP를 첨가한 후 0.4mL(3.35mmol)의 콜리딘을 첨가하였다. 반응액이 천천히 실온에 도달하도록하고 밤새 교반하였다. DMF를 제거하고 조 생성물을 EtOAC와 물 사이에 분배하였다. 수상을 EtOAc로 추출하고 유기상을 Na2SO4 상에서 건조시키고 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 95:5의 CH2Cl2:MeOH로 용리하여 크로마토그래피하여서 50mg(14%)의 부표제 화합물을 생성하였다.

(iv)Ph(3-(1-피롤리딘))-(R,S)CH(OH)C(O)-Aze-Pab x 2TFA
2mL의 CH2Cl2 중의 100mg(0.17mmol)의 Ph(3-(1-피롤리딘))-(R,S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 단계 (iii) 참조) 및 2.0mL의 TFA의 혼합물을 0℃에서 2시간 교반하였다. 용액을 진공에서 농축시켜서 잔여물을 생성하고 물에 용해시키고 냉 동 건조시켜서 70mg(58%)의 표제 화합물을 생성하였다.

실시예 12
Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe)
(i) Ph(3-Cl,5-NMe 2 )-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)
(i) Ph(3-Cl,5-NMe 2 )-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)
6mL의 THF 중의 92mg(0.16mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)
C(O)- Aze-Pab(Teoc)(상기 실시예 6(vi) 참조)의 용액에 78mg(0.92mmol)의 O-메틸히드록실아민을 첨가하여 혼합물을 생성하고 60℃에서 밤새 교반하였다. 용매를 진공에서 제거하고 생성된 고체를 실리카 겔 상에서 EtOAc로 용리하여 크로마토그래피하였다. 목적 분획물을 농축시켜서 82mg(85%)의 백색 고체의 부표제 화합물을 생성하였다.

(ii)Ph(3-Cl,50NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe)
3mL의 TFA 중의 78mg(0.13mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH) C(O)-Aze-Pab(Teoc)(상기 단계 (i) 참조)의 용액을 0℃에서 2시간 교반하였다. 용액을 진공에서 차가운 상태에서 농축시키고 생성된 고체를 예비 HPLC (CH3CN:0.1 M 암모늄 아세테이트(40:60)) 상에서 크로마토그래피하였다. 목적 분획물을 부분 농축시켰다. 잔여물을 3번 냉동 건조(CH3CN:물)시켜서, 40mg(30%)의 표제 화합물을 생성하였다. 순도 99.4%.

실시예 13
Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-Et)
(i)Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-Et)
3mL의 THF 중의 40mg(0.07mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH) C(O)-Aze-Pab(Teoc)(상기 실시예 6(vi) 참조)의 용액에 40mg(0.41mmol)의 O-에틸히드록실아민 x HCl을 첨가하고, 용액을 60℃에서 밤새 교반하였다. 용액을 농축시키고, 생성된 물질을 예비 HPLC(CH3CN:0.1 M 암모늄 아세테이트(60:40))으로 정제하 였다. 목적 분획물을 부분 농축시키고 잔여물을 EtOAc로 3번 추출하였다. 유기상을 물로 세척하고 진공에서 농축시켜서 16mg(37%)의 부표제 화합물을 생성하였다.

(ii)Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-Et)
0.5mL의 메틸렌 클로라이드 중의 16mg(0.03mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-Et)(상기 단계 (i) 참조)의 용액에 1mL의 TFA를 첨가하고 혼합물을 0℃에서 2시간 교반하였다. 생성된 혼합물을 진공에서 농축시켜서 고체 잔여물을 생성하고, 물/CH3CN 중에 용해시키고 2번 냉동 건조시켜서 14mg(92%)의 표제 화합물을 생성하였다. 순도 94.4%.

실시예 14
Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)
(i) Ph(3-Cl,5-NMe 2 )-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-n-Pr)
(i) Ph(3-Cl,5-NMe 2 )-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-n-Pr)
5mL의 THF 중의 40mg(0.07mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C (O)-Aze-Pab(Teoc)(상기 실시예 6(vi)처럼)의 용액에 46mg(0.41mmol)의 O-n-프로필히드록실아민 x HCl를 첨가하고, 용액을 60℃에서 밤새 교반하였다. 용액을 농축시켜서 건조시키고, 나머지를 예비 HPLC(CH3CN:0.1M 암모늄 아세테이트 (60:40))을 사용하여 정제하였다. 목적 분획물을 부분 농축시키고, 수용액을 EtOAc로 3번 추출하였다. 유기상을 물로 세척하고, Na2SO4 상에서 건조시키고 농축시켜서 16mg(36%)의 부표제 화합물을 생성하였다.

(ii)Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)
0.5mL의 메틸렌 클로라이드 중의 16mg(0.02mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-n-Pr)(상기 단계 (i) 참조)의 얼음 냉각 용액에 2mL의 TFA를 첨가하고, 생성된 혼합물을 2시간 동안 차가운 상태에서 교반하였 다. 생성된 용액을 진공에서 농축시켜서 고체 잔여물을 생성하고 물/CH3CN 중에 용해시키고 냉동 건조시켰다. 생성물을 플래시 크로마토그래피(9:1의 EtOAc:MeOH )로 정제하였다. 목적 분획물을 농축시켜서 14mg(92%)의 표제 화합물을 생성하였다. 순도 98%.

실시예 15
Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-i-Pr)
(i) Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-i-Pr)
3mL의 THF 중의 40mg(0.07mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH) C(O)-Aze-Pab(Teoc)(상기 실시예 6(vi) 참조)의 용액에 46mg(0.41mmol)의 O-이소프로필히드록실아민 x HCl을 첨가하고, 생성된 혼합물을 60℃에서 밤새 교반하였다. 생성된 용액을 농축시키고, 조 생성물을 예비 HPLC(CH3CN:0.1M 암모늄 아세테이트 (60:40))을 사용하여 정제하였다. 목적 분획물을 부분 농축시킨 후 EtOAc로 3번 추출하였다. 합쳐진 유기물을 물로 세척하고, Na2SO4 상에서 건조시키고 농축시켜 서 16mg(36%)의 부표제 화합물을 생성하였다.

(ii)Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-i-Pr)
1.5mL의 TFA 중의 16mg(0.02mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH) C(O)-Aze-Pab(Teoc)(O-i-Pr)(상기 단계 (i) 참조)의 얼음 냉각 용액을 2시간 동안 차가운 상태에서 교반하였다. 생성된 용액을 물/CH3CN을 첨가하기 전에 진공에서 증발시키고, 용액을 냉동 건조시켰다. 조 생성물을 플래시 크로마토그래피 (EtOAc:MeOH(9:1))로 정제하였다. 그 후 목적 분획물을 농축시켜서 14mg(92%)의 표제 화합물을 생성하였다. 순도(HPLC) 96%.

실시예 16
Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-CH
2
-CH
2
-O-CH
3
)
(i)Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-CH
2
-CH
2
-O-CH
3
)
2mL의 THF 중의 O-(2-메톡시)에틸히드록실아민 및 23.3㎕의 HOAc의 용액을 1mL의 THF 중의 40mg(0.07mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 실시예 6(vi) 참조)의 용액에 첨가하고, 혼합물을 60℃에서 3.5일 동안 교반하였다. 생성된 용액을 농축시켜서 건조시키고, 조 생성물을 예비 HPLC(CH3CN:0.1M 암모눔 아세테이트(60:40))을 사용하여 정제시켰다. 목적 분획물을 부분 농축시키고 EtOAc로 3번 추출하였다. 합쳐진 유기층을 물로 세척하고, Na2SO4 상에서 건조시킨 후 농축시켜서 건조시켜서 20mg(44%)의 부표제 화합물을 생성하였다.

(ii)Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-CH
2
-CH
2
-O-CH
3
)
2mL(26mmol)의 TFA를 0.5mL의 메틸렌 클로라이드 중의 20mg(0.03mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(0-CH2-CH2-O-CH
3)(상기 단 계 (i) 참조)의 얼음 냉각된 용액에 첨가하고 생성된 혼합물을 2½시간 동안 차가운 상태에서 교반하였다. 생성된 용액을 증발시켜서 건조시키고, 조 생성물을 플래시 크로마토그래피(EtOAc:MeOH(9:1))로 정제하였다. 목적 분획물을 농축시켜서 잔여물을 생성하고 여기에 물/CH3CN을 첨가하였다. 생성된 용액을 밤새 냉동 건조시켜서 13mg(65%)의 표제 화합물을 생성하였다. 순도(HPLC) 96%.

실시예 17
Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-THP)
(i) Ph(3,Cl,5-NMe 2 )-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-THP)
(i) Ph(3,Cl,5-NMe 2 )-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-THP)
1mL의 THF 중의 51mg(0.44mmol)의 O-(테트라히드로피란-2-일)히드록실아민 및 25㎕의 HOAc의 용액을 2mL의 THF 중의 43mg(0.07mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 실시예 6(vi) 참조)의 용액에 첨가하고, 생성된 혼합물을 60℃에서 22시간 동안 교반한 후 실온에서 밤새 교반하였다. 생성된 용액을 농축시키고, 조 생성물을 예비 HPLC(CH3CN:0.1M 암모늄 아세테이트 (60:40))으로 정제하였다. 목적 분획물을 부분 농축시키고 수성 잔여물을 EtOAc로 3번 추출하였다. 합쳐진 유기물을 물로 세척하고, Na2SO4 상에서 건조시킨 후 농축시켜서 26mg(52%)의 부표제 화합물을 생성하였다.

(ii)Ph(3-Cl,5-NMe
2
)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-THP)
140mg의 앰벌리스트 A-26 상의 플루오라이드를 3mL의 CH3CN 중의 34mg(0.05mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-THP)(상기 단계 (i) 참조)의 용액에 첨가하고, 혼합물을 60℃에서 밤새 방치하였다. 냉각 후, 수지를 여과하여 제거한 후, CH3CN 및 EtOH(95%)로 수차례 세척하였다. 합쳐진 유기상을 농축시켜서 조 생성물을 생성하고 예비 HPLC(CH3CN:0.1M 암모늄 아세테이트(50:50))로 정제하였다. 목적 분획물을 농축시키고, 물/CH3CN 중에 용해시킨 후 냉동 건조시켜서 18mg(60%)의 표제 화합물을 생성하였다.
A-26 상의 플루오라이드를 3mL의 CH3CN 중의 34mg(0.05mmol)의 Ph(3-Cl,5-NMe2)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-THP)(상기 단계 (i) 참조)의 용액에 첨가하고, 혼합물을 60℃에서 밤새 방치하였다. 냉각 후, 수지를 여과하여 제거한 후, CH3CN 및 EtOH(95%)로 수차례 세척하였다. 합쳐진 유기상을 농축시켜서 조 생성물을 생성하고 예비 HPLC(CH3CN:0.1M 암모늄 아세테이트(50:50))로 정제하였다. 목적 분획물을 농축시키고, 물/CH3CN 중에 용해시킨 후 냉동 건조시켜서 18mg(60%)의 표제 화합물을 생성하였다.

실시예 18
Ph(3-Cl,5-NMeAC)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe) x TFA
(i) Ph(3-Cl,5-NMeAc)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)
3mL의 THF 중의 38mg(0.06mmol)의 Ph(3-Cl,5-NMeAc)-(R) 또는
-(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 실시예 5(vii) 참조) 및 62mg(0.74mmol)의 O-메틸히드록실아민의 혼합물을 60℃에서 30시간 동안 가열한 후, 용매를 제거하고 반응 혼합물을 예비 HPLC(CH3CN:0.1M 암모늄 아세테이트 50:50)로 정제하였다. 목적 분획물을 부분 농축시키고 수성 잔여물을 EtOAc로 3번 추출하였다. 합쳐진 유기상을 Na2SO4 상에서 건조시키고 진공에서 농축시켜서, 22mg(50%)의 부표제 화합물을 생성하였다.

(ii) Ph(3-Cl,5-NMeAc)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe) x TFA
3mL의 TFA 중의 22mg(0.03mmol)의 Ph(3-Cl,5-NMeAc)-(R) 또는 -(S)CH(OH) C(O)-Aze-Pab(Teoc)(OMe)의 용액을 실온에서 1시간 유지한 후 용매를 진공에서 제거하였다. 고체 잔여물을 물에 용해시키고 용액을 밤새 냉동 건조시켜서 20mg (76%)의 표제 화합물을 생성하였다.

실시예 19
Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe) x TFA
(i) Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)
O-메틸히드록실아민 x HCl을 3mL의 THF 중의 42mg(0.50mmol)의 50mg (0.08mmol)의 Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 실시예 10(vii) 참조) 용액에 첨가하고 혼합물을 60 ℃에서 밤새 교반하였다. 용매를 제거하고 잔여물을 물과 EtOAc 사이에 분배하였다. 수상을 EtOAc로 추출하고 합쳐진 상을 Na2SO4 상에서 건조시킨 후 진공에서 농축시켜서 약 52mg(약 100%)의 고체 부표제 화합물을 생성하였으며, 추가 정제 없이 사용하였다.
(ii) Ph(3-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe) x TFA
1mL의 CH2Cl2 중의 53mg(0.08mmol)의 Ph(3-(1-피롤리딘-2-온))-(R) 또는
-(S)CH(OH) C(O)-Aze-Pab(Teoc)(OMe)(상기 단계 (i) 참조) 및 2.0mL의 TFA의 혼합물을 실온에서 4 시간 동안 교반하였다. 용매를 제거하였고 잔여물을 물 및 EtOAc 사이에 분배하였다. 수상을 EtOAc로 추출하고 합쳐진 유기상을 Na2SO4 상에서 건조시킨 후 진공에서 농축시켰다. 잔여물을 실리카 겔 상에서 98:2에서 95:5로 변화하는 CH2Cl2:MeOH로 용리하여 크로마토그래피하여서 22mg(44%)의 표제 화합물을 생성하였다.

실시예 20
Ph(3-Cl,5-피롤로)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab
(i) (R,S)-5-pH(3-Cl,5-피롤로)-2,2-디메틸-4-옥소-1,3-디옥솔란
50mL의 건조 톨루엔 중의 6.0g(24.8mmol)의 (R,S)-5-Ph(3-Cl,5-NH2)-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 실시예 9(ii) 참조) 및 3.5g(24.8mmol)의 오산화인(V)의 용액에 4.9g(37.3mmol)의 2,5-디메톡시테트라히드로푸란를 적가하였다. 반응물을 환류로 30분간 가열한 후 실온으로 냉각되도록 하였다. 반응물을 10mL의 2N NaOH로 중단시키고, 분별 깔때기로 옮기고 수상을 분리하고 100mL의 톨루엔으로 추출하였다. 합쳐진 유기상을 이어서 20mL의 염수로 세척하고, MgSO4 상에서 건조시키고 여과시키고 진공에서 농축시켜서 5.0g의 옅은 오렌지색 오일을 생성하였다. 실리카 겔 상에서 CH2Cl2로 용리하여 플래시 크로마토그래피하여서 2.8g(39%)의 황색 고체로 부표제 화합물을 생성하였다.

(ii) Ph(3-Cl,5-피롤로)-(R,S)CH(OH)C(O)OH
40mL의 THF 중의 3.1g(10.7mmol)의 (R,S)-5-Ph(3-Cl,5-피롤로)-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (i) 참조)의 용액에 실온에서 36mL(107.3mmol)의 3N NaOH를 0.35g(1.07mmol)의 테트라부틸암모늄 브로마이드와 함께 첨가하였다. 그 후 반응 혼합물을 실온에서 추가 2시간 동안 교반하였다. 반응 혼합물을 진공에서 농축시켜서 THF를 제거하였다. 나머지 수성 상을 0℃로 냉각시키고 농축된 HCl로 pH 2로 산성화시키고 2 ×150mL의 EtOAc로 추출하였다. 합쳐진 유기상을 염수로 세척하고, MgSO4 상에서 건조시키고 여과시키고 진공에서 농축시켜서 오렌지색 포말을 생성하였다. 실리카 겔 상에서 85:15:5의 CHCl3:MeOH:농축된 암모늄 히드록사이드로 용리하여 플래시 크로마토그래피하여서 2.0g의 백색 고체로 부표제 화합물의 암모늄염을 생성하였다. 이어서 2N HCl로 pH 1로 산성화시키고, EtOAc로 추출하고 진공에서 농축시키고 건조시켜서 1.8g(68%)의 백색 고체로 부표제 화합물을 생성하였다.

(iii) Ph(3-Cl,5-피롤로)-(R) 또는 -(S)CH(OH)C(O)OH (a) 및 Ph(3-Cl,5-피롤로)-(S) 또는 -(R)CH(OAc)C(O)OH (b)
1.8g(7.3mmol)의 Ph(3-Cl,5-피롤로)-(R,S)CH(OH)C(O)OH(상기 단계 (ii) 참조), 1.0g의 리파아제 PS 아마노, 5.0mL의 비닐 아세테이트 및 5.0mL의 MTBE의 혼합물을 45℃에서 24시간 가열하였다. 반응액을 여과시키시고 여과 케이크를 100mL의 EtOAc로 세척하였다. 여과물을 진공에서 농축시키고 실리카 겔 상에서 95:5의 CHCl3:HOAc로 용리하여 크로마토그래피하여서 710mg(38%)의 백색 고체로 부표제 화합물 (a)를 생성하고 910mg(42%)의 크림색 고체로 부표제 화합물 (b)를 생성하였다.
부표제 화합물 (a):

부표제 화합물 (b):

(iv) Ph(3-Cl,5-피롤로)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
14mL의 DMF 중의 285mg(1.14mmol)의 Ph(3-Cl,5-피롤로)-(R) 또는 -(S)CH
(OH)C(O)OH(상기 단계 (iii)참조), 470mg(1.25mmol)의 HAze-Pab(Teoc), 650mg (1.25mmol)의 PyBOP, 및 0.33mL(2.49mmol)의 2,4,6-콜리딘의 혼합물을 0℃에서 2시간 교반한 후 25℃에서 30분 교반하였다. 반응물을 50mL의 H20로 켄칭시키고 3 ×50mL의 EtOAc로 추출하였다. 합쳐진 추출물을 Na2SO4상에서 건조시키고 여과시키고 진공에서 농축시켰다. 실리카 겔 상에서 EtOAc로 용리하여 2번 플래시 크로마토그래피하여서 180mg(26%)의 백색 고체의 부표제 화합물을 생성하였다.

(v)Ph(3-Cl,5-피롤로)-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab
아세토니트릴 중의 38mg(0.06mmol)의 Ph(3-Cl,5-피롤로)-(R) 또는 -(S)CH (OH)C(O)-Aze-Pab(Teoc)(상기 단계 (iv) 참조)의 용액에 170mg의 폴리머 결합된 플로라이드 이온(앰벌리스트 )A-26을 첨가하고 혼합물을 60℃에서 밤새 가열한 후 70℃에서 4시간 가열하였다. 생성된 혼합물을 여과시키고, 폴리머를 세척하고 용액을 아세토니트릴, 에탄올 및 THF로 세척하고, 용액을 진공에서 농축시켰다. 조 생성물을 예비 HPLC로 2회(각각 40:60의 CH3CN:0.1M 암모늄 아세테이트, 및 30:70의 CH3CN:0.1M 암모늄 아세테이트) 정제하였다. 목적 분획물을 3회 냉동 건조시켜서, 8mg(28%)의 표제 화합물을 생성하였다.
)A-26을 첨가하고 혼합물을 60℃에서 밤새 가열한 후 70℃에서 4시간 가열하였다. 생성된 혼합물을 여과시키고, 폴리머를 세척하고 용액을 아세토니트릴, 에탄올 및 THF로 세척하고, 용액을 진공에서 농축시켰다. 조 생성물을 예비 HPLC로 2회(각각 40:60의 CH3CN:0.1M 암모늄 아세테이트, 및 30:70의 CH3CN:0.1M 암모늄 아세테이트) 정제하였다. 목적 분획물을 3회 냉동 건조시켜서, 8mg(28%)의 표제 화합물을 생성하였다.

실시예 21
Ph(3-Cl,5-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)
(i) Ph(3-Cl,5-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)
(O-n-Pr)
3mL의 THF 중의 40mg(0.064mmol)의 Ph(3-Cl,5-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 실시예 9(vi) 참조)의 용액에 43mg(0.38mmol)의 O-n-프로필히드록실아민 x HCl을 첨가하고 용액을 60℃에서 4.5시간 동안 가열하였다. 용액을 진공에서 농축시키고, 생성된 조 물질을 플래시 크로마토그래피 (Si 겔, 9:1의 EtOAc:MeOH)로 정제하였다. 목적 분획물을 농축시켜서 43mg(98%)의 부표제 화합물을 생성하였다.

(ii)Ph(3-Cl,5-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)
0.5mL의 메틸렌 클로라이드 중의 43mg, 0.063mmol의 상기 단계 (i)로부터의 Ph(3-Cl, 5-(1-피롤리딘-2-온))-(R) 또는 -(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)(Teoc)의 냉각된 용액에 2.5mL의 TFA를 첨가하고, 용액을 0℃에서 100분 교반한 후, 용액을 진공에서 농축시키고 생성된 조 물질을 예비 HPLC(30:70의 CH3CN:0.1M 암모늄 아세테이트)를 사용하여 정제하였다. 목적 분획물을 수집하고 냉동 건조시켜서 23mg (68%)을 생성하였다. 순도 99.9%
실시예 22
Ph(3-Cl,5-(1-피롤리딘-2-온))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe)
(i) Ph(3-Cl,5-(1-피롤리딘-2-온))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc) (OMe)
6mL의 THF 중의 80mg(0.13mmol)의 Ph(3-Cl,5-(1-피롤리딘-2-온))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(상기 실시예 9(vi) 참조)의 혼합물에 64mg (0.77mmol)의 O-메틸히드록실아민 x HCl를 첨가하였다. 혼합물을 60℃에서 5시간 교반한 후 증발시켰다. 잔여물을 실리카 겔 상에서 9:1의 에틸 아세테이트:메탄올로 용리하여 크로마토그래피하여서 75mg의 조 생성물을 생성하였다. 조 생성물을 예비 HPLC(60:40의 CH3CN:0.1 M 암모늄 아세테이트)를 사용하여 추가 정제하였다. 목적 분획물을 농축시켰다. CH3CN을 진공에서 제거하였다. 수성 상을 EtOAc로 3회 추출하였다. 합쳐진 에틸 아세테이트를 염수로 세척한 후 Na2SO4 상에서 건조시키고 농축시켜서 65.8mg(78%)의 부표제 화합물을 생성하였다.

(ii) Ph(3-Cl, 5-(1-피롤리딘-2-온))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab
(OMe)
0.5mL의 메틸렌 클로라이드 중의 55.9mg(0.08mmol)의 상기 단계 (i)로부터의 Ph(3-Cl,5-(1-피롤리딘-2-온))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)의 얼음 냉각 용액에 3.0mL의 TFA를 첨가하였다. 용액을 0℃에서 130분간 교반한 후, 용액을 진공에서 농축시키고 생성된 조 물질을 예비 HPLC(30:70의 CH3CN:0.1M 암모늄 아세테이트)를 사용하여 정제하였다. 목적 분획물을 수집하고, 냉동 건조시키기를 2회 반복하여서, 38mg(87%)의 표제 화합물을 생성하였다.

실시예 23
Ph(3-Cl,5-NMe(3-메틸부타노일))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe)
(i) (R,S)-5-Ph(3-Cl,5-NHMe)-2,2-디메틸-4-옥소-1,3-디옥솔란
100mL의 EtOAc 중의 3.0g(12.4mmol)의 (R,S)-5-Ph(3-Cl,5NM2)-2,2-디메틸-4- 옥소-1,3-디옥솔란(상기 실시예 9(ii) 참조), 0.81mL(9.9mmol)의 포름알데히드(H20 중의 37 중량%) 및 330mg의 산화 백금(IV)의 혼합물을 수소 분위기 하에서 25℃에서 5시간 동안 교반하였다. 혼합물을 세리트 패드를 통해 여과시키고 여과 케이크를 200mL의 EtOAc로 세척하였다. 유기상을 진공에서 농축시키고 실리카 겔 상에서 1:4의 EtOAc:Hex로 용리하여 플래시 크로마토그래피하여서 1.63g(52%)의 황색 오일로 부표제 화합물을 생성하였다.

(ii) (R,S)-5-Ph(3-Cl,5-NMe(3-메틸부타노일))-2,2-디메틸-4-옥소-1,3-디옥솔란
20mL의 아세톤 중의 945mg(3.70mmol)의 (R,S)-5-Ph(3-Cl,5-NHMe)-2,2-디메틸 -4-옥소-1,3-디옥솔란(상기 단계 (i) 참조) 및 560mg(5.54mmol)의 트리에틸아민의 용액에 0℃에서 623mg(5.17mmol)의 이소발레릴 클로라이드를 적가하였다. 혼합물을 0.5 시간 동안 교반하고 3 ×30mL의 EtOAc와 30mL의 H20에 분배하였다. 합쳐진 유기 추출물을 30mL의 NaHCO3 수용액으로 세척하고, Na2SO4 상에서 건조시키고 여과시키고 진공에서 농축시켜서 1.35g(>100%)의 황색 오일로 부표제 화합물을 생성하였으며, 이 생성물을 정제하지 않고 직접 사용하였다.

(iii) Ph(3-Cl,5-NMe(3-메틸부타노일))-(R,S)CH(OH)C(O)OH
20mL의 MeOH 중의 1.35g(3.97mmol)의 (R,S)-5-Ph(3-Cl,5-NMe(3-메틸부타노일))-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (ii) 참조) 및 1.60g(39.7mmol)의 NaOH의 혼합물을 25℃에서 1시간 교반하였다. 혼합물을 진공에서 농축시키고, 잔여물을 30mL의 H20로 희석시키고 20mL의 2N HCl로 산성화시키고 3 ×50mL의 EtOAc로 추출하였다. 합쳐진 유기 추출물을 Na2SO4 상에서 건조시키고, 여과시키고 진공에서 농축시켜서 1.0g(100%)의 황색 오일로 부표제 화합물을 얻었으며, 이 생성물을 정제하지 않고 직접 사용하였다.
(iv) Ph(3-Cl,5-NMe(3-메틸부타노일))-(R)- 또는 -(S)-CH(OH)C(O)OH (a) 및 Ph(3-Cl,5-NMe(3-메틸부타노일))-(S)- 또는 -(R)-CH(OAc)C(O)OH (b)
25mL의 비닐 아세테이트 및 25mL의 MTBE 중의 1.0g(3.34mmol)의 Ph(3-Cl,5-NMe(3-메틸부타노일))-(R,S)CH(OH)C(O)OH (상기 단계 (iii) 참조) 및 510mg의 리파아제 PS 아마노의 혼합물을 55℃에서 14시간 동안 가열하였다. 반응물을 세리트를 통해 여과시키고 여과 케이크를 200mL의 MeOH로 세척하였다. 여액을 진공에서 농축시키고 실리카 겔 상에서 6.5:3.0:0.5의 CHCl3:MeOH:진한 NH4OH로 용리하여 크로마토그래피하여서 285mg의 부스러지는 포말로 부표제 화합물(a)의 암모늄염 및 370mg(32%)의 백색 포말로 부표제 화합물 (b)의 암모늄염을 생성하였다. 부표제 화합물 (a)의 암모늄염을 25mL의 EtOAc에 용해하고 0.60mL의 Et20 중의 2M HCl로 중화시켰다. 25mL의 물을 첨가하고, 층을 분리하였다. 수성 층을 2 ×25mL의 EtOAc로 추출하고, 유기 추출물을 Na2SO4 상에서 건조시키고 여과시키고 진공에서 농축시켜서 230mg(23%)의 부스러지는 백색 포말로 부표제 화합물(a)를 생성하였다.
부표제 화합물 (a):

부표제 화합물 (b):

(v) Ph(3-Cl,5-NMe(3-메틸부타노일))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab
(OMe)
5mL의 DMF 중의 119mg(0.40mmol)의 Ph(3-Cl,5-NMe(3-메틸부타노일)-(R)- 또는 -(S)-CH(OH)C(O)OH(상기 단계 (iv)참조) 및 146mg(0.44mmol)의 H-Aze-Pab(OMe) x 2HCl(상기 실시예 2(iv) 참조)의 혼합물에 227mg(0.44mmol)의 PyBOP 및 168mg(1.39mmol)의 콜리딘을 첨가하였다. 용액을 3시간 동안 0℃에서 질소 하에서 교반하고, Na2SO4 상에서 건조시키고 여과시키고 진공에서 농축시켰다. 실리카 겔 상에서 15:1의 CHCl3:MeOH로 용리하여 플래시 크로마토그래피하여서 125mg(58%)의 부스러지는 포말로 표제 화합물을 생성하였다.

실시예 24
Ph(3-Cl,5-NMe(시클로펜틸카르보닐))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab (OMe)
(i) (R,S)-5-Ph(3-Cl,5-NMe(시클로펜틸카르보닐))-2,2-디메틸-4-옥소-1,3-디옥솔란
20mL의 아세톤 중의 945mg(3.70mmol)의 (R,S)-5-Ph(3-Cl,5-NHMe)-2,2-디메틸
-4-옥소-1,3-디옥솔란(상기 실시예 23(i) 참조) 및 635mg(5.86mmol)의 트리에틸아민의 용액에 776mg(5.86mmol)의 시클로펜탄카르보닐 클로라이드를 0℃에서 적가하였다. 혼합물을 1시간 교반하고, 3 ×30mL의 EtOAc와 30mL의 H20 사이에 분배하였다. 합쳐진 유기 추출물을 30mL의 수성 NaHCO3로 세척하고, Na2SO4 상에서 건조시키고 여과시키고 진공에서 농축시켜서 1.58g(>100%)의 황색 오일로 부표제 화합물을 생성하였고, 정제하지 않고 직접 사용하였다.

(ii) Ph(3-Cl, 5-NMe(시클로펜틸카르보닐))-(R,S)CH(OH)C(O)OH
25mL의 MeOH 중의 1.58g(5.07mmol)의 (R,S)-5-Ph(3-Cl,5-NMe(시클로펜틸카르보닐))-2,2-디메틸-4-옥소-1,3-디옥솔란(상기 단계 (i) 참조) 및 2.03g(50.7mmol)의 NaOH의 혼합물을 25℃에서 1시간 교반하였다. 혼합물을 진공에서 농축시키고, 30mL의 H20로 희석시키고 20mL의 2N HCl로 산성화시켰다. 수성 층을 3 ×50mL의 EtOAc로 추출하고 합쳐진 유기 추출물을 Na2SO4 상에서 건조시키고 여과시키고 진공에서 농축시켜서 1.17g(100%)의 백색 포말로 부표제 화합물을 생성하고, 정제하지 않고 직접 사용하였다.

(iii) Ph(3-Cl,5-NMe(시클로펜틸카르보닐))-(R)- 또는 -(S)-CH(OH)C(O)0H (a) 및 Ph(3-Cl,5-NMe(시클로펜틸카르보닐))-(S)- 또는 -(R)-CH(OAc)C(O)OH (b)
25mL의 비닐 아세테이트 및 25mL의 MTBE 중의 1.17g(3.75mmol)의 Ph(3-Cl,5-NMe(시클로펜틸카르보닐))-(R,S)CH(OH)C(O)OH(상기 단계 (ii) 참조) 및 600mg의 리파아제 PS 아마노의 혼합물을 55℃에서 14시간 가열하였다. 반응 혼합물을 세리트를 통해서 여과시키고 여과 케이크를 100mL의 MeOH로 세척하였다. 여과물을 진공 에서 농축시키고 실리카 겔 상에서 6.5:3.0:0.5의 CHCl3:MeOH:농축 NH4OH로 용리하여 크로마토그래피하여서 336mg의 부스러지는 포말로 부표제 화합물 (a)의 암모늄염 및 557mg(50%)의 백색 포말로 부표제 화합물 (b)의 암모늄염을 생성하였다. 부표제 화합물 (a)의 암모늄염을 25mL의 EtOAc 중에 용해시키고, 0.70mL의 Et2O 중의 2M HCl로 중화시켰다. 25mL의 물을 첨가하고, 층을 분리하였다. 수성 층을 2 ×25mL의 EtOAc로 추출하고, 유기 추출물을 Na2SO4 상에서 건조시키고 농축시켜서 290mg(29%)의 부스러지는 백색 포말로 부표제 화합물 (a)를 생성하였다.
부표제 화합물(a):

부표제 화합물(b):

(iv)Ph(3-Cl,5-NMe(시클로펜틸카르보닐))-(R)- 또는 -(S)CH(OH)C(O)-Aze-Pab(OMe)
5mL의 DMF 중의 120mg(0.39mmol)의 Ph(3-Cl,5-NMe(시클로펜틸카르보닐))-(R,S)CH(OH)C(O)OH(상기 단계 (iii) 참조) 및 142mg(0.42mmol)의 H-Aze-Pab(OMe) x 2HCl(상기 실시예 2(iv) 참조)의 혼합물에 220mg(0.42mmol)의 PyBOP 및 161mg(1.35mmol)의 콜리딘을 첨가하였다. 용액을 0℃에서 6시간 동안 질소 하에서 교반하였다. 혼합물을 3 ×30mL의 EtOAc와 30mL의 H2O 사이에 분배하고 Na2SO4
상에서 건조시키고 여과시키고 진공에서 농축시켰다. 실리카 겔 상에서 15:1의 CHCl3:MeOH로 용리하여 플래시 크로마토그래피하고, 이어서 20:1의 EtOAc:EtOH로 용리하여 크로마토그래피하여서 85mg(40%)의 부스러지는 백색 포말로 표제 화합물을 생성하였다.

실시예 25
실시예 1, 3 내지 11 및 20의 표제 화합물을 상기 시험 A에서 시험하였고, 0.5uM 미만의 IC50TT 값을 나타내는 것을 발견하였다.
실시예 26
실시예 2, 12, 13, 15, 18, 19 및 21의 표제 화합물을 상기 시험 E에서 시험하였고, 상응하는 활성 억제제(유리 아미딘)로서 쥐에서 경구 및(또는) 비경구 생체이용성을 나타내는 것을 발견하였다.
실시예 27
실시예 2, 12 내지 19 및 21의 표제 화합물을 상기 시험 G에서 시험하였고, 대응 활성 억제제 (자유 아미딘)의 형성을 나타내었다.
약자
Ac = 아세틸
AcOH = 아세트산
API = 대기압 이온화(MS 관련)
Aze = 아제티딘-2-카르복실레이트
AzeOH = 아제티딘-2-카르복실산
Bzl = 벤질
CI = 화학 이온화(MS 관련)
DIPEA = 디이소프로필에틸아민
DMAP = 4-(N,N-디메틸 아미노)피리딘
DMF = 디메틸포름아미드
DMSO = 디메틸술폭사이드
EDC = 1-(3-디메틸아미노프로필)-3-에틸카르보디이미드
히드로클로라이드
Et = 에틸
에테르 = 디에틸 에테르
EtOAc = 에틸 아세테이트
EtOH = 에탄올
h = 시간
HATU = O-(아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄
헥사플루오로포스페이트
HBTU = [N,N,N',N'-테트라메틸-O-(벤조트리아졸-1-일)우로늄
헥사플루오르포스페이트
HCl = 염산
HCl(g) = 염화 수소 기체
Hex = 헥산
HOAc = 아세트산
HPLC = 고성능 액체 크로마토그래피
LC = 액체 크로마토그래피
Me = 메틸
MeOH = 메탄올
MS = 질량 분광법
MTBE = 메틸 tert-부틸 에테르
Pab = para-아미노벤질아미노
H-Pab = para-아미디노벤질아민
PyBOP = (벤조트리아졸-1-일옥시)트리피롤리디노포스포늄 헥사
플루오로포스페이트
RPLC = 역상 고성능 액체 크로마토그래피
RT = 실온
TBTU = [N,N,N',N'-테트라메틸-0-(벤조트리아졸-1-일)우로늄
테트라플루오로보레이트
TEA = 트리에틸아민
Teoc = 2-(트리메틸실릴)에톡시카르보닐
TFA = 트리플루오로아세트산
THF = 테트라히드로푸란
THP = 테트라히드로피라닐
TLC = 박층 크로마토그래피
TMSCN = 트리메틸실릴 시아나이드
Z = 벤질옥시카르보닐
접두사 n, s, i 및 t는 일반적인 의미를 가진다: 표준, 2차, 이소 및 3차.
Claims (31)
- 하기 화학식 I의 화합물 또는 그의 제약학적으로 허용되는 염.<화학식 I>
 (상기 식에서,R1은 N(R5)R6 또는 S(O)mR7 치환체이고,R2 및 R3는 독립적으로 할로, C1-4 알킬 또는 C1-4 알콕시 (후자의 두 개의 기는 임의로 할로에 의해 치환됨)로부터 선택되는 임의적 치환체이고,Y는 임의로 C1-4 알킬, 메틸렌, =O 또는 히드록시에 의해 치환되는 C1-3 알킬렌이고,R4는 H, OH, OR8a, C(O)OR8b 또는 R8c이고,R5는 C1-6 알킬(임의로 할로에 의해 치환됨)이거나, 또는 R6, 및 R5 및 R6이 부착된 질소 원자와 함께, 임의로 산소 원자를 포함하고(하거나) 임의로 =O기로 치환되는 3 내지 7원 질소 함유 고리이고,R6은 C1-6 알킬 (임의로 할로에 의해 치환됨), 또는 C(O)R9이거나, 또는 R5, 및 R5 및 R6이 부착된 질소 원자와 함께, 임의로 산소 원자를 포함하고(하거나) 임의로 =O기로 치환되는 3 내지 7원 질소 함유 고리이거나, 또는N(R5)R6기는 하기 화학식 Ia의 구조적 단편이고,<화학식 Ia>
(상기 식에서,R1은 N(R5)R6 또는 S(O)mR7 치환체이고,R2 및 R3는 독립적으로 할로, C1-4 알킬 또는 C1-4 알콕시 (후자의 두 개의 기는 임의로 할로에 의해 치환됨)로부터 선택되는 임의적 치환체이고,Y는 임의로 C1-4 알킬, 메틸렌, =O 또는 히드록시에 의해 치환되는 C1-3 알킬렌이고,R4는 H, OH, OR8a, C(O)OR8b 또는 R8c이고,R5는 C1-6 알킬(임의로 할로에 의해 치환됨)이거나, 또는 R6, 및 R5 및 R6이 부착된 질소 원자와 함께, 임의로 산소 원자를 포함하고(하거나) 임의로 =O기로 치환되는 3 내지 7원 질소 함유 고리이고,R6은 C1-6 알킬 (임의로 할로에 의해 치환됨), 또는 C(O)R9이거나, 또는 R5, 및 R5 및 R6이 부착된 질소 원자와 함께, 임의로 산소 원자를 포함하고(하거나) 임의로 =O기로 치환되는 3 내지 7원 질소 함유 고리이거나, 또는N(R5)R6기는 하기 화학식 Ia의 구조적 단편이고,<화학식 Ia> R6a는 할로, C1-4 알킬 및 C1-4 알콕시 (후자의 두 개의 기는 임의로 할로에 의해 치환됨)로부터 선택되는 하나 이상의 임의적 치환체이고,X는 CH 또는 N이고,m은 0, 1 또는 2이고,R7은 H, NH2 또는 C1-6 알킬이고,R8a 및 R8b는 독립적으로 C1-10 알킬, C1-3 알킬페닐 또는 C6-10 아릴이거나, 또는 R8a는 C(R10a)(R10b)OC(O)R11, C(R10a)(R10b)N(H)C(O)OR12 또는 C(R10a)(R10b)OC(O)N(H)R12이고,R8c는 C(R10a)(R10b)OC(O)R11, C(R10a)(R10b)N(H)C(O)OR12 또는 C(R10a)(R10b)OC(O) N(H)R12이고,R10a 및 R10b는 독립적으로 각 경우에서, H 또는 C1-4 알킬이고,R11은 각 경우에서, C6-10 아릴, OR12 또는 C1-7 알킬 (후자의 기는 임의로 OH, CO2H 및 C6-10 아릴로부터 선택되는 치환체에 의해 치환됨)이고,R12는 각 경우에서, C6-10 아릴 또는 C1-6 알킬 (후자의 기는 임의로 OH, CO2H 및 C6-10 아릴로부터 선택되는 치환체에 의해 치환됨)이고,R9는 C1-8 알킬, Het1, C6-10 아릴, 또는 C6-10 아릴에 의해 치환되는 C1-4 알킬이고,Het1은 산소, 질소 및(또는) 황으로부터 선택되는 하나 이상의 헤테로원자를 함유하고, 완전히 포화되거나 또는 부분적으로 포화되거나 또는 방향족일 수 있고 (있거나) 임의로 모노시클릭, 비시클릭 및(또는) 벤조 융합일 수 있는 4 내지 12원 헤테로시클릭 고리이고,상기 각 아릴/페닐기 및 Het1기는 임의로 하나 이상의 할로, C1-4알킬 및(또는) C1-4 알콕시기 (후자의 두 개의 기는 임의로 하나 이상의 할로기에 의해 치환됨)에 의해 치환되고,단, (a) m이 1 또는 2일 때, R7은 H가 아니고,(b) m이 0일 때, R7은 NH2가 아니다.)
R6a는 할로, C1-4 알킬 및 C1-4 알콕시 (후자의 두 개의 기는 임의로 할로에 의해 치환됨)로부터 선택되는 하나 이상의 임의적 치환체이고,X는 CH 또는 N이고,m은 0, 1 또는 2이고,R7은 H, NH2 또는 C1-6 알킬이고,R8a 및 R8b는 독립적으로 C1-10 알킬, C1-3 알킬페닐 또는 C6-10 아릴이거나, 또는 R8a는 C(R10a)(R10b)OC(O)R11, C(R10a)(R10b)N(H)C(O)OR12 또는 C(R10a)(R10b)OC(O)N(H)R12이고,R8c는 C(R10a)(R10b)OC(O)R11, C(R10a)(R10b)N(H)C(O)OR12 또는 C(R10a)(R10b)OC(O) N(H)R12이고,R10a 및 R10b는 독립적으로 각 경우에서, H 또는 C1-4 알킬이고,R11은 각 경우에서, C6-10 아릴, OR12 또는 C1-7 알킬 (후자의 기는 임의로 OH, CO2H 및 C6-10 아릴로부터 선택되는 치환체에 의해 치환됨)이고,R12는 각 경우에서, C6-10 아릴 또는 C1-6 알킬 (후자의 기는 임의로 OH, CO2H 및 C6-10 아릴로부터 선택되는 치환체에 의해 치환됨)이고,R9는 C1-8 알킬, Het1, C6-10 아릴, 또는 C6-10 아릴에 의해 치환되는 C1-4 알킬이고,Het1은 산소, 질소 및(또는) 황으로부터 선택되는 하나 이상의 헤테로원자를 함유하고, 완전히 포화되거나 또는 부분적으로 포화되거나 또는 방향족일 수 있고 (있거나) 임의로 모노시클릭, 비시클릭 및(또는) 벤조 융합일 수 있는 4 내지 12원 헤테로시클릭 고리이고,상기 각 아릴/페닐기 및 Het1기는 임의로 하나 이상의 할로, C1-4알킬 및(또는) C1-4 알콕시기 (후자의 두 개의 기는 임의로 하나 이상의 할로기에 의해 치환됨)에 의해 치환되고,단, (a) m이 1 또는 2일 때, R7은 H가 아니고,(b) m이 0일 때, R7은 NH2가 아니다.) - 제 1항에 있어서, R5 와 R6이 그들이 부착된 질소 원자와 함께 =O기에 의해 치환된 3 내지 7원 고리이며, 질소 원자에 대해 α위치의 탄소 원자가 치환된 것인 화합물.
- 제 1항 또는 2항에 있어서, R2가 존재하는 경우, R2가 직쇄형 또는 분지쇄형 C1-4 알킬 또는 C1-4 알콕시 (후자의 두 개의 기는 임의로 할로에 의해 치환됨) 또는 할로인 화합물.
- 제1항 또는 2항에 있어서, R3가 존재하지 않거나, 또는 존재하는 경우 R3가 직쇄형 또는 분지쇄형 C1-4 알킬 또는 할로인 화합물.
- 제 4항에 있어서, R3가 존재하는 경우, R3가 메틸 또는 클로로기인 화합물.
- 제 4항에 있어서, 상기 치환체가 (존재하는 경우), 페닐 고리가 부착되어 있는 -CH2-기에 대해 2-위치에 있는 화합물.
- 제1항 또는 2항에 있어서, R1이 N(R5)R6인 화합물.
- 제 1항에 있어서, R5가 직쇄형, 분지쇄형 또는 시클릭 C1-6알킬이거나, 또는 R6, 및 R5 및 R6이 부착된 질소 원자와 함께 임의로 =O기에 의해 치환되는 4 내지 6원 질소 함유 고리인 화합물.
- 제 8항에 있어서, R5가 C1-4 알킬이거나, 또는 R6, 및 R5 및 R6이 부착된 질소 원자와 함께 임의로 =O기에 의해 치환되는 5 내지 6원 질소 함유 고리인 화합물.
- 제 1항에 있어서, R6이 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬 또는 C(O)-C1-6 알킬이거나, 또는 R5, 및 R5 및 R6이 부착된 질소 원자와 함께 임의로 =O기에 의해 치환되는 4 내지 6원 질소 함유 고리인 화합물.
- 제 10항에 있어서, R6이 메틸 또는 C(O)-C1-6 알킬이거나, 또는 R5, 및 R5 및 R6이 부착된 질소 원자와 함께, 임의로 =O기에 의해 치환되는 5 내지 6원 질소 함유 고리인 화합물.
- 제 1항에 있어서, R7이 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬인 화합물.
- 제1항 또는 2항에 있어서, R1이, 페닐 고리가 부착되어 있는 -CH(OH)-기에 대해 3-위치에서 페닐 고리에 부착된 화합물.
- 제1항 또는 2항에 있어서, R2가 존재하는 경우, 페닐 고리가 부착되어 있는 -CH(OH)-기에 대해 R2가 5-위치에서 페닐 고리에 부착된 화합물.
- 제1항 또는 2항에 있어서, R4가 OR8a일 때, R8a가 직쇄형 또는 분지쇄형 C1-6알킬, C4-5 시클릭 알킬 (후자의 두 개의 기는 임의로 산소에 의해 단속됨) 또는 페닐 또는 C1-2 알킬페닐 (후자의 두 개의 기는 임의로 제 1항에서 정의한 바와 같이 치환됨)이거나, 또는 R8a가 CH2OC(O)R11 (여기서, R11은 페닐, 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬이고, 후자의 기는 임의로 OH, CO2H 및 페닐로부터 선택되는 치환체에 의해 치환됨) 또는 OR12 (여기서, R12는 페닐 또는 직쇄형, 분지쇄형 또는 시클릭 C1-6 알킬이고, 후자의 기는 임의로 OH, CO2H 및 페닐로부터 선택되는 치환체에 의해 치환됨)인 화합물.
- 제1항 또는 2항에 있어서, R4가 C(O)OR8b일 때, R8b가 직쇄형 또는 분지쇄형 C1-2 알킬페닐 또는 페닐 (후자의 두 개의 기는 임의로 제 1항에서 정의한 바와 같이 치환됨)인 화합물.
- 제1항 또는 2항에 있어서, 단편이 S-배열인 화학식 I의 화합물.

- 제1항 또는 2항에 있어서, 단편이 R-배열인 화학식 I의 화합물.

- 제1항 또는 2항에 정의된 화합물 또는 이의 제약학적으로 허용되는 염 및 제약학적으로 허용되는 보조제, 희석제 또는 담체를 포함하는, 혈전증; 혈액 및 조직에서의 응고항진; 선천성 또는 후천성 활성화 단백질 C 내성; 항혈전 III, 단백질 C, 단백질 S 또는 헤파린 보조인자 II의 선천성 또는 후천성 결핍; 순환하는 항인지질 항체 (루푸스 항응고제); 호모시스테인혈증; 헤파린 유도 혈소판감소증; 섬유소용해 결손; 알쯔하이머 질병; 정맥 혈전증; 폐 색전증; 동맥 혈전증; 전신성 색전증; 세균, 다중 외상, 중독 또는 임의의 다른 기전에 의해 유발된 파종성 혈관내 응고; 특발성 및 성인 호흡 곤란증; 방사선 또는 화학요법 치료 후 폐 섬유증; 패혈성 쇽; 패혈증; 염증 반응; 부종; 급성 또는 만성 동맥경화증; 관상 동맥 질병; 뇌 동맥 질환; 말초 동맥 질환; 재관류 손상; 경피적 경관 혈관성형술 (PTA) 후 재협착; 및 췌장염으로 이루어진 군으로부터 선택되는 질환을 치료하기 위한 제약 조성물.
- 삭제
- 삭제
- 제19항에 있어서, 혈전증의 치료에 사용하기 위한 것인 제약 조성물.
- 제1항 또는 2항에 정의된 화합물 또는 이의 제약학적으로 허용되는 염 및 제약학적으로 허용되는 보조제, 희석제 또는 담체를 포함하는, 항응고제로서 사용하기 위한 제약 조성물.
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- (i) 하기 화학식 II의 화합물을 하기 화학식 III의 화합물과 커플링하거나, 또는(ii) 하기 화학식 IV의 화합물을 하기 화학식 V의 화합물과 커플링하거나, 또는(iii) R4가 OH 또는 OR8a인 화학식 I의 화합물의 경우에는, 하기 화학식 VI의 화합물을, 임의로 하기 화학식 VI의 화합물을 C1-6 알킬 알콜의 존재 하에서 기체 HCl로 전처리하여 하기 화학식 VIII의 화합물을 형성함으로써, 하기 화학식 VII의 화합물과 반응시키거나, 또는(iv) R4가 OH 또는 OR8a인 화합물의 경우에는, R4 대신 보호기 C(O)ORb1 (여기서, Rb1은 2-트리메틸실릴에틸, C1-6 알킬 또는 알킬페닐임)이 존재하는 화학식 I의 화합물에 대응하는 화합물을 상기 정의한 화학식 VII의 화합물과 반응시키거나, 또는(v) R4가 C(O)OR8b인 화학식 I의 화합물의 경우에는, R4가 H인 화학식 I의 화합물을 하기 화학식 IX의 화합물과 반응시키거나, 또는(vi) R4가 OR8a인 화학식 I의 화합물의 경우에는, R4가 OH인 대응 화학식 I의 화합물을 하기 화학식 IXA의 화합물과 반응시키거나, 또는(vii) R4가 R8c (여기서, R8c는 C(R10a)(R10b)OC(O)R11 또는 C(R10a)(R10b)OC(O)N(H)R12임)인 화학식 I의 화합물의 경우에는, 대응 하기 화학식 IXB의 화합물을 하기 화학식 IXC의 화합물과 반응시키거나, 또는(viii) R4가 R8c인 화학식 I의 화합물의 경우에는, R4가 H인 대응 화학식 I의 화합물을 하기 화학식 IXD의 화합물과 반응시키거나, 또는(ix) R1이 S(O) 또는 S(O)2기인 화학식 I의 화합물의 경우에는, R1이 S기를 포함하는 대응 화학식 I의 화합물을 산화시키거나, 또는(x) 제 1항에서 정의한 화학식 I의 화합물의 보호된 유도체를 탈보호시키거나, 또는(xi) 제 1항에서 정의한 화학식 I의 화합물에서 방향족, 비방향족, 카르보시클릭 또는 헤테로시클릭 고리 상에서 치환체를 도입하거나 또는 상호전환하는 것을 포함하는 화학식 I의 화합물의 제조 방법.<화학식 II>
 (여기서, R1 및 R2는 제 1항에서 정의한 것임)<화학식 III>
(여기서, R1 및 R2는 제 1항에서 정의한 것임)<화학식 III> (여기서, Y, R3 및 R4는 제 1항에서 정의한 것임)<화학식 IV>
(여기서, Y, R3 및 R4는 제 1항에서 정의한 것임)<화학식 IV> (여기서, R1, R2 및 Y는 제 1항에서 정의한 것임)<화학식 V>
(여기서, R1, R2 및 Y는 제 1항에서 정의한 것임)<화학식 V> (여기서, R3 및 R4는 제 1항에서 정의한 것임)<화학식 VI>
(여기서, R3 및 R4는 제 1항에서 정의한 것임)<화학식 VI> (여기서, R1,R2,Y 및 R3는 제 1항에서 정의한 것임)<화학식 VII>H2NORa(여기서, Ra는 H 또는 R8a이고, R8a는 제 1항에서 정의한 것임)<화학식 VIII>
(여기서, R1,R2,Y 및 R3는 제 1항에서 정의한 것임)<화학식 VII>H2NORa(여기서, Ra는 H 또는 R8a이고, R8a는 제 1항에서 정의한 것임)<화학식 VIII> (여기서, Rc는 C1-6 알킬이고, R1, R2, Y 및 R3는 제 1항에서 정의한 것임)<화학식 IX>L1-C(O)OR8b(여기서, L1은 이탈기이고, R8b는 제 1항에서 정의한 것임)<화학식 IXA>L1-R8a(여기서, R8a는 제 1항에서 정의한 것이고, L1은 상기에서 정의한 것임)<화학식 IXB>
(여기서, Rc는 C1-6 알킬이고, R1, R2, Y 및 R3는 제 1항에서 정의한 것임)<화학식 IX>L1-C(O)OR8b(여기서, L1은 이탈기이고, R8b는 제 1항에서 정의한 것임)<화학식 IXA>L1-R8a(여기서, R8a는 제 1항에서 정의한 것이고, L1은 상기에서 정의한 것임)<화학식 IXB> (여기서, R1, R2, Y, R3, R10a및 R10b는 제 1항에서 정의한 것임)<화학식 IXC>L1C(O)R13(여기서, R13은 R11 또는 N(H)R12이고, R11 및 R12는 제 1항에서 정의한 것이고, L1은 상기에서 정의한 것임)<화학식 IXD>L1C(R10a)(R10b)R14(여기서, R14는 OC(O)R11, NHC(O)OR12 또는 OC(0)N(H)R12이고, R10a, R10b, R11 및 R12는 제 1항에서 정의한 것이고, L1은 상기에서 정의한 것임)
(여기서, R1, R2, Y, R3, R10a및 R10b는 제 1항에서 정의한 것임)<화학식 IXC>L1C(O)R13(여기서, R13은 R11 또는 N(H)R12이고, R11 및 R12는 제 1항에서 정의한 것이고, L1은 상기에서 정의한 것임)<화학식 IXD>L1C(R10a)(R10b)R14(여기서, R14는 OC(O)R11, NHC(O)OR12 또는 OC(0)N(H)R12이고, R10a, R10b, R11 및 R12는 제 1항에서 정의한 것이고, L1은 상기에서 정의한 것임) - 제19항에 있어서, 상기 질환이 혈액 및 조직에서의 응고항진인 제약 조성물.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SE9900071A SE9900071D0 (sv) | 1999-01-13 | 1999-01-13 | New compounds |
| SE9900071-3 | 1999-01-13 | ||
| SE9904228-5 | 1999-11-22 | ||
| SE9904228A SE9904228D0 (sv) | 1999-11-22 | 1999-11-22 | New compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20010101493A KR20010101493A (ko) | 2001-11-14 |
| KR100639274B1 true KR100639274B1 (ko) | 2006-10-27 |
Family
ID=26663479
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020017008826A KR100639274B1 (ko) | 1999-01-13 | 2000-01-13 | 신규 아미디노벤질아민 유도체 및 트롬빈 억제제로서의 용도 |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US6599894B1 (ko) |
| EP (1) | EP1150996B1 (ko) |
| JP (1) | JP2002535250A (ko) |
| KR (1) | KR100639274B1 (ko) |
| CN (1) | CN1170842C (ko) |
| AU (1) | AU764121B2 (ko) |
| BR (1) | BR0007453A (ko) |
| CA (1) | CA2355792A1 (ko) |
| CY (1) | CY1107149T1 (ko) |
| CZ (1) | CZ20012529A3 (ko) |
| DE (1) | DE60037183T2 (ko) |
| DK (1) | DK1150996T3 (ko) |
| EE (1) | EE200100371A (ko) |
| ES (1) | ES2295004T3 (ko) |
| IL (1) | IL143987A (ko) |
| IS (1) | IS5990A (ko) |
| NO (1) | NO20013319L (ko) |
| NZ (1) | NZ512714A (ko) |
| PL (1) | PL349769A1 (ko) |
| PT (1) | PT1150996E (ko) |
| TR (1) | TR200102037T2 (ko) |
| WO (1) | WO2000042059A1 (ko) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE0001803D0 (sv) * | 2000-05-16 | 2000-05-16 | Astrazeneca Ab | New compounds i |
| US6433186B1 (en) * | 2000-08-16 | 2002-08-13 | Astrazeneca Ab | Amidino derivatives and their use as thormbin inhibitors |
| IL154077A0 (en) * | 2000-08-16 | 2003-07-31 | Astrazeneca Ab | New amidino derivatives and their use as thrombin inhibitors |
| US7129233B2 (en) | 2000-12-01 | 2006-10-31 | Astrazeneca Ab | Mandelic acid derivatives and their use as thrombin inhibitors |
| AR035216A1 (es) * | 2000-12-01 | 2004-05-05 | Astrazeneca Ab | Derivados de acido mandelico ,derivados farmaceuticamente aceptables, uso de estos derivados para la fabricacion de medicamentos, metodos de tratamiento ,procesos para la preparacion de estos derivados, y compuestos intermediarios |
| SE0102921D0 (sv) * | 2001-08-30 | 2001-08-30 | Astrazeneca Ab | Pharmaceutically useful compounds |
| AR034517A1 (es) * | 2001-06-21 | 2004-02-25 | Astrazeneca Ab | Formulacion farmaceutica |
| UA78195C2 (uk) * | 2001-08-30 | 2007-03-15 | Astrazeneca Ab | Похідні мигдалевої кислоти та їх застосування як інгібіторів тромбіну, фармацевтична композиція на їх основі |
| AU2002367752A1 (en) * | 2001-10-03 | 2003-11-17 | Pharmacia Corporation | Substituted 5-membered polycyclic compounds useful for selective inhibition of the coagulation cascade |
| SE0201659D0 (sv) * | 2002-05-31 | 2002-05-31 | Astrazeneca Ab | Modified release pharmaceutical formulation |
| SE0201658D0 (sv) * | 2002-05-31 | 2002-05-31 | Astrazeneca Ab | Immediate release pharmaceutical formulation |
| SE0201661D0 (sv) | 2002-05-31 | 2002-05-31 | Astrazeneca Ab | New salts |
| US7781424B2 (en) * | 2003-05-27 | 2010-08-24 | Astrazeneca Ab | Modified release pharmaceutical formulation |
| US7524354B2 (en) * | 2005-07-07 | 2009-04-28 | Research Foundation Of State University Of New York | Controlled synthesis of highly monodispersed gold nanoparticles |
| TW200827336A (en) * | 2006-12-06 | 2008-07-01 | Astrazeneca Ab | New crystalline forms |
| US20090061000A1 (en) * | 2007-08-31 | 2009-03-05 | Astrazeneca Ab | Pharmaceutical formulation use 030 |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HU178398B (en) | 1979-06-12 | 1982-04-28 | Gyogyszerkutato Intezet | Process for producing new agmatine derivatives of activity against haemagglutination |
| HU192646B (en) | 1984-12-21 | 1987-06-29 | Gyogyszerkutato Intezet | Process for preparing new n-alkyl-peptide aldehydes |
| IL77748A (en) | 1985-02-04 | 1991-11-21 | Merrell Dow Pharma | Amino acid and peptide derivatives as peptidase inhibitors |
| US5187157A (en) | 1987-06-05 | 1993-02-16 | Du Pont Merck Pharmaceutical Company | Peptide boronic acid inhibitors of trypsin-like proteases |
| EP0362002B1 (en) | 1988-09-01 | 1995-07-26 | Merrell Dow Pharmaceuticals Inc. | HIV protease inhibitors |
| ZA897514B (en) | 1988-10-07 | 1990-06-27 | Merrell Dow Pharma | Novel peptidase inhibitors |
| TW201303B (ko) | 1990-07-05 | 1993-03-01 | Hoffmann La Roche | |
| CA2075154A1 (en) | 1991-08-06 | 1993-02-07 | Neelakantan Balasubramanian | Peptide aldehydes as antithrombotic agents |
| SE9102462D0 (sv) | 1991-08-28 | 1991-08-28 | Astra Ab | New isosteric peptides |
| ZA928581B (en) | 1991-11-12 | 1994-05-06 | Lilly Co Eli | Antithrombotic agents |
| SE9103612D0 (sv) | 1991-12-04 | 1991-12-04 | Astra Ab | New peptide derivatives |
| WO1993018060A1 (en) | 1992-03-04 | 1993-09-16 | Gyógyszerkutató Intézet Kft. | New anticoagulant peptide derivatives and pharmaceutical compositions containing the same as well as a process for the preparation thereof |
| TW223629B (ko) | 1992-03-06 | 1994-05-11 | Hoffmann La Roche | |
| SE9301916D0 (sv) | 1993-06-03 | 1993-06-03 | Ab Astra | New peptides derivatives |
| EP0648780A1 (en) | 1993-08-26 | 1995-04-19 | Bristol-Myers Squibb Company | Heterocyclic thrombin inhibitors |
| TW394760B (en) | 1993-09-07 | 2000-06-21 | Hoffmann La Roche | Novel Carboxamides, process for their preparation and pharmaceutical composition containing the same |
| AU1025795A (en) | 1994-01-27 | 1995-08-03 | Mitsubishi Chemical Corporation | Prolineamide derivatives |
| US5705487A (en) | 1994-03-04 | 1998-01-06 | Eli Lilly And Company | Antithrombotic agents |
| IL112795A (en) | 1994-03-04 | 2001-01-28 | Astrazeneca Ab | Derivatives of peptides as antithrombotic drugs, their preparation, and pharmaceutical preparations containing them |
| US5707966A (en) | 1994-03-04 | 1998-01-13 | Eli Lilly And Company | Antithrombotic agents |
| DE4421052A1 (de) | 1994-06-17 | 1995-12-21 | Basf Ag | Neue Thrombininhibitoren, ihre Herstellung und Verwendung |
| US5510369A (en) | 1994-07-22 | 1996-04-23 | Merck & Co., Inc. | Pyrrolidine thrombin inhibitors |
| EA199700186A1 (ru) | 1995-02-17 | 1998-02-26 | Басф Акциенгезельшафт | Новые дипептидные амидины, используемые в качестве ингибиторов тромбина |
| US5710130A (en) | 1995-02-27 | 1998-01-20 | Eli Lilly And Company | Antithrombotic agents |
| CA2216859A1 (en) | 1995-04-04 | 1996-10-10 | Merck & Co., Inc. | Thrombin inhibitors |
| US5629324A (en) | 1995-04-10 | 1997-05-13 | Merck & Co., Inc. | Thrombin inhibitors |
| SA96170106A (ar) * | 1995-07-06 | 2005-12-03 | أسترا أكتيبولاج | مشتقات حامض أميني جديدة |
| AR005245A1 (es) | 1995-12-21 | 1999-04-28 | Astrazeneca Ab | Prodrogas de inhibidores de trombina, una formulación farmaceutica que las comprende, el uso de dichas prodrogas para la manufactura de un medicamento y un procedimiento para su preparacion |
| AU1990197A (en) | 1996-03-12 | 1997-10-01 | Bristol-Myers Squibb Company | Carbamyl guanidine and amidine prodrugs |
| SE9602263D0 (sv) | 1996-06-07 | 1996-06-07 | Astra Ab | New amino acid derivatives |
| WO1997049404A1 (en) | 1996-06-25 | 1997-12-31 | Eli Lilly And Company | Anticoagulant agents |
| DE19632772A1 (de) | 1996-08-14 | 1998-02-19 | Basf Ag | Neue Benzamidine |
| AR013084A1 (es) | 1997-06-19 | 2000-12-13 | Astrazeneca Ab | Derivados de amidino utiles como inhibidores de la trombina, composicion farmaceutica, utilizacion de dichos compuestos para la preparacion demedicamentos y proceso para la preparacion de los compuestos mencionados |
-
2000
- 2000-01-13 AU AU23362/00A patent/AU764121B2/en not_active Ceased
- 2000-01-13 EP EP00902233A patent/EP1150996B1/en not_active Expired - Lifetime
- 2000-01-13 CZ CZ20012529A patent/CZ20012529A3/cs unknown
- 2000-01-13 WO PCT/SE2000/000052 patent/WO2000042059A1/en active IP Right Grant
- 2000-01-13 US US09/508,742 patent/US6599894B1/en not_active Expired - Fee Related
- 2000-01-13 TR TR2001/02037T patent/TR200102037T2/xx unknown
- 2000-01-13 EE EEP200100371A patent/EE200100371A/xx unknown
- 2000-01-13 CN CNB008048401A patent/CN1170842C/zh not_active Expired - Fee Related
- 2000-01-13 NZ NZ512714A patent/NZ512714A/en not_active IP Right Cessation
- 2000-01-13 PL PL00349769A patent/PL349769A1/xx not_active Application Discontinuation
- 2000-01-13 CA CA002355792A patent/CA2355792A1/en not_active Abandoned
- 2000-01-13 PT PT00902233T patent/PT1150996E/pt unknown
- 2000-01-13 DE DE60037183T patent/DE60037183T2/de not_active Expired - Lifetime
- 2000-01-13 ES ES00902233T patent/ES2295004T3/es not_active Expired - Lifetime
- 2000-01-13 DK DK00902233T patent/DK1150996T3/da active
- 2000-01-13 KR KR1020017008826A patent/KR100639274B1/ko not_active IP Right Cessation
- 2000-01-13 JP JP2000593626A patent/JP2002535250A/ja active Pending
- 2000-01-13 BR BR0007453-5A patent/BR0007453A/pt not_active IP Right Cessation
- 2000-01-13 IL IL14398700A patent/IL143987A/xx not_active IP Right Cessation
-
2001
- 2001-07-04 NO NO20013319A patent/NO20013319L/no unknown
- 2001-07-05 IS IS5990A patent/IS5990A/is unknown
-
2008
- 2008-01-18 CY CY20081100076T patent/CY1107149T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP1150996A1 (en) | 2001-11-07 |
| IS5990A (is) | 2001-07-05 |
| ES2295004T3 (es) | 2008-04-16 |
| NO20013319L (no) | 2001-09-12 |
| CY1107149T1 (el) | 2012-10-24 |
| EE200100371A (et) | 2002-02-15 |
| CN1170842C (zh) | 2004-10-13 |
| IL143987A (en) | 2005-12-18 |
| IL143987A0 (en) | 2002-04-21 |
| BR0007453A (pt) | 2001-10-30 |
| EP1150996B1 (en) | 2007-11-21 |
| DE60037183D1 (de) | 2008-01-03 |
| PL349769A1 (en) | 2002-09-09 |
| NO20013319D0 (no) | 2001-07-04 |
| TR200102037T2 (tr) | 2001-10-22 |
| US6599894B1 (en) | 2003-07-29 |
| WO2000042059A1 (en) | 2000-07-20 |
| DK1150996T3 (da) | 2008-02-11 |
| KR20010101493A (ko) | 2001-11-14 |
| CN1343217A (zh) | 2002-04-03 |
| JP2002535250A (ja) | 2002-10-22 |
| PT1150996E (pt) | 2008-01-15 |
| AU764121B2 (en) | 2003-08-07 |
| AU2336200A (en) | 2000-08-01 |
| DE60037183T2 (de) | 2008-10-09 |
| CA2355792A1 (en) | 2000-07-20 |
| CZ20012529A3 (cs) | 2001-11-14 |
| NZ512714A (en) | 2003-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100639274B1 (ko) | 신규 아미디노벤질아민 유도체 및 트롬빈 억제제로서의 용도 | |
| US5550131A (en) | 2-piperazinone compounds and their use | |
| US20070249578A1 (en) | New amidino derivatives and their use as thrombin inhibitors | |
| JP2000512631A (ja) | 新規なアミノ酸誘導体およびトロンビン阻害剤としてのその使用 | |
| JP2001525394A (ja) | 新規な化合物 | |
| EP1283837B1 (en) | New thiochromane derivatives and their use as thrombin inhibitors | |
| EP1311480B1 (en) | New amidino derivatives and their use as thrombin inhibitors | |
| AU2001280395A1 (en) | New amidino derivatives and their use as thrombin inhibitors | |
| KR100928285B1 (ko) | 신규한 만델산 유도체 및 이것의 트롬빈 억제제로서의 용도 | |
| US6433186B1 (en) | Amidino derivatives and their use as thormbin inhibitors | |
| RU2341516C2 (ru) | Новые производные миндальной кислоты и их применение в качестве ингибиторов тромбина | |
| MXPA01007004A (en) | New amidinobenzylamine derivatives and their use as thrombin inhibitors | |
| ZA200300258B (en) | New amidino derivatives and their use as thrombin inhibitors. | |
| MXPA01005937A (en) | New amidino derivatives and their use as thrombin inhibitors | |
| MXPA04001825A (es) | Derivados de acido mandelico nuevos y su uso como inhibidores de trombina. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| FPAY | Annual fee payment |
Payment date: 20090930 Year of fee payment: 4 |
|
| LAPS | Lapse due to unpaid annual fee |