CN1170842C - 新的脒基苄基胺衍生物及其作为凝血酶抑制剂的用途 - Google Patents
新的脒基苄基胺衍生物及其作为凝血酶抑制剂的用途 Download PDFInfo
- Publication number
- CN1170842C CN1170842C CNB008048401A CN00804840A CN1170842C CN 1170842 C CN1170842 C CN 1170842C CN B008048401 A CNB008048401 A CN B008048401A CN 00804840 A CN00804840 A CN 00804840A CN 1170842 C CN1170842 C CN 1170842C
- Authority
- CN
- China
- Prior art keywords
- compound
- represent
- formula
- alkyl
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biochemistry (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明提供式(I)化合物,其中R1、R2、Y和R3和R4具有说明书中给出的意义,式(I)化合物用作胰蛋白酶样蛋白酶例如凝血酶的竞争性抑制剂或其前药,尤其用于治疗其中需要抑制凝血酶的病症(例如血栓形成)或者用作抗凝药。
Description
本发明的领域
本发明涉及新的药用化合物,尤其涉及为胰蛋白酶样丝氨酸蛋白酶特别是凝血酶的竞争性抑制剂或其前药的化合物,涉及它们作为药物的用途、含有它们的药用组合物和其制备的合成路线。
背景
血凝固为涉及止血(即预防血液自损伤的血管中流失)和血栓形成(即在血管中形成血凝块,有时导致血管阻塞)的关键过程。
血凝固为一系列复杂的酶反应的结果。这一系列反应的最终步骤之一为酶原凝血酶原转化为活化的酶凝血酶。
已知凝血酶在血凝固中起着重要的作用。它激活血小板导致血小板聚集,将血纤蛋白原转化为血纤蛋白单体,该单体自发聚合成血纤蛋白聚合物并且激活因子XIII,该因子依次交联所述聚合物形成不溶性血纤蛋白。此外,凝血酶激活因子V和因子VIII导致“正反馈”自凝血酶原产生凝血酶。
通过抑制血小板的聚集以及血纤蛋白的形成和交联,可以期待有效的凝血酶抑制剂将显示抗血栓形成活性。此外,可以期待通过有效地抑制正反馈机制将加强抗血栓形成活性。
此外,已知给予凝血酶抑制剂前药可以产生在以下方面的改善:
(a)给予那些抑制剂后某些药物动力学性质;和
(b)与那些抑制剂有关的某些副作用的流行。
先有技术
Claesson在Blood Coagul.Fibrinol.(1994)5,411中已经介绍了早期开发的低分子量凝血酶抑制剂。
Blombǎck等(J.Clin.Lab.Invest.24,增补107,59(1969))报道基于位于血纤蛋白原Aα链的裂解位点周围的氨基酸序列的凝血酶抑制剂。在所讨论的氨基酸序列中,这些作者建议三肽序列Phe-Val-Arg(P9-P2-P1,此后称为P3-P2-P1序列)将是最有效的抑制剂。
从美国专利第4,346,078号和国际专利申请WO 93/11152中得知基于带有在P1-位上α,ω-氨基烷基胍的二肽基衍生物的凝血酶抑制剂。也已经报道了类似的、结构相关的二肽基衍生物。例如,国际专利申请WO 94/29336公开了在P1-位上具有例如氨基甲基苄脒、环状氨基烷基脒和环状氨基烷基胍的化合物(国际专利申请WO 97/23499公开了某些这类化合物的前药);欧洲专利申请0 648 780公开了在P1-位上具有例如环状氨基烷基胍的化合物。
从欧洲专利申请0 468 231、0 559 046和0 641 779中得知基于肽基衍生物并在P1-位上也具有环状氨基烷基胍(例如3-或4-氨基甲基-1-脒基-哌啶)的凝血酶抑制剂。
在欧洲专利申请0 185 390中首次公开了基于在P1-位上具有精氨醛的三肽基衍生物的凝血酶抑制剂。
最近,已经报道了基于精氨醛的在P3位修饰的肽基衍生物。例如,国际专利申请WO 93/18060公开了在P3位的羟基酸,欧洲专利申请0 526 877公开了在P3位的脱氨基酸以及欧洲专利申请0 542 525公开了在P3位的O-甲基扁桃酸的基于精氨醛的肽基衍生物。
也已知基于在P1位的亲电酮的丝氨酸蛋白酶(例如凝血酶)抑制剂。例如,欧洲专利申请0 195 212公开了P1位的肽基α-酮基酯和酰胺,欧洲专利申请0 362 002公开了P1位的氟代烷基酰胺酮,欧洲专利申请0 364 344公开了P1位的α,β,δ-三酮基化合物以及欧洲专利申请0 530 167公开了P1位的精氨酸的α-烷氧基酮衍生物。
从欧洲专利申请0 293 881中得知其它基于精氨酸和异硫脲鎓类似物的碳端硼酸衍生物的胰蛋白酶样丝氨酸蛋白酶的结构不同的抑制剂。
最近,欧洲专利申请0 669 317和国际专利申请WO 95/35309、WO 95/23609、WO 96/25426、WO 97/02284、WO 97/46577、WO96/32110、WO 96/31504、WO 96/03374、WO 98/06740和WO97/49404已经公开了基于肽基衍生物的凝血酶抑制剂。
然而,仍然需要有效的胰蛋白酶样丝氨酸蛋白酶例如凝血酶的抑制剂。也需要口服生物利用率好并且选择性抑制凝血酶优先于其它丝氨酸蛋白酶,尤其那些涉及止血的酶的化合物。可以期待显示对凝血酶竞争性抑制活性的化合物尤其可以用作抗凝血药,因此用于治疗血栓形成和相关的疾病。
本发明的公开
根据本发明提供式I化合物或其药学上可接受的盐:

其中
R1代表N(R5)R6或S(O)mR7取代基;
R2和R3独立代表选自卤素、C1-4烷基或C1-4烷氧基(后两个基团可任选被卤素取代)的任选取代基;
Y代表C1-3亚烷基,任选被C1-4烷基、亚甲基、=O或羟基取代;
R4代表H、OH、OR8a、C(O)OR8b或R8c;
R5代表C1-6烷基(任选被卤素取代)或者与R6以及R5和R6所连接的氮原子一起代表3-7元的含氮环,该环任选包括氧原子和/或任选被=O基团取代;
R6代表C1-6烷基(任选被卤素取代)、C(O)R9或者与R5以及R5和R6所连接的氮原子一起代表3-7元的含氮环,该环任选包括氧原子和/或任选被=O基团取代;
或者基团N(R5)R6代表结构片段Ia:
R6a代表一个或多个选自卤素、C1-4烷基和C1-4烷氧基(后两个基团可任选被卤素取代)的任选取代基;
X代表CH或N;
m代表0、1或2;
R7代表H、NH2或C1-6烷基;
R8a和R8b独立代表C1-10烷基、C1-3烷基苯基或C6-10芳基,或者R8a代表C(R10a)(R10b)OC(O)R11、C(R10a)(R10b)N(H)C(O)OR12或C(R10a)(R10b)OC(O)N(H)R12;
R8c代表C(R10a)(R10b)OC(O)R11、C(R10a)(R10b)N(H)C(O)OR12或C(R10a)(R10b)OC(O)N(H)R12;
R10a和R10b在各种情况下,独立代表H或C1-4烷基;
R11在各种情况下,代表C6-10芳基、OR12或C1-7烷基(后一个基团任选被选自OH、CO2H和C6-10芳基的取代基取代);
R12在各种情况下,代表C6-10芳基或C1-6烷基(后一个基团任选被选自OH、CO2H和C6-10芳基的取代基取代);
R9代表C1-8烷基、Het1、C6-10芳基或被C6-10芳基取代的C1-4烷基;和
Het1代表4-12元的杂环,该杂环含有一个或多个选自氧、氮和/或硫的杂原子,并且该环可以为完全饱和、部分饱和或芳族的和/或任选单环、双环和/或苯并稠合;其中每一个芳基/苯基以及如上所定义的Het1基团任选被一个或多个卤素、C1-4烷基和/或C1-4烷氧基(后两个基团本身任选被一个或多个卤素基团取代)取代;条件为:
(a)当m代表1或2时,则R7不代表H;和
(b)当m代表0时,则R7不代表NH2;
所述化合物此后称为“本发明化合物”。
药学上可接受的盐包括无机酸(例如氢卤酸)和有机酸(例如乙酸、甲磺酸或三氟乙酸)的加成盐。
本发明化合物可以显示互变异构现象。所有的互变异构形式及其混合物均包括在本发明的范围内。可以提及的具体互变异构形式包括那些连接在式I化合物的脒官能度中的双键位置和所述取代基R4的位置(当其不代表H时)的互变异构形式。
式I化合物也含有至少两个不对称碳原子,因此可以显示旋光性和/或非对映异构现象。使用常规技术例如层析或分步结晶可以分离所有的非对映异构体。通过使用常规的技术例如分步结晶或HPLC析解所述化合物的外消旋体或其它混合物可以分离各种立体异构体。或者,通过合适的旋光性原料在不引起外消旋化或差向异构化的条件下反应或者通过例如用纯手性的酸衍生化,然后通过常规方法(例如HPLC、硅胶层析)分离非对映衍生物可以制备所需要的旋光性异构体。所有的立体异构体均包括在本发明的范围内。
如在此所使用的术语“芳基”包括苯基、萘基等。
R2、R3、R5、R6、R6a、R7、R8a、R8b、R9、R10a、R10b、R11和R12可以代表的烷基以及可用于取代Y和芳基/苯基和Het1基团的烷基;R2、R3和R6a可以代表的烷氧基以及可用于取代芳基/苯基和Het1的烷氧基;R8a、R8b、R9、R11和R12可代表的烷基苯基或烷基芳基的烷基部分;以及Y可代表的亚烷基,当具有足够的碳原子数目时,可以为直链或支链、饱和或不饱和、环状、无环或部分环状/无环,和/或任选被O原子所间断。技术人员将可以理解:当R2、R3、R5、R6、R6a、R7、R8a、R8b、R9、R10a、R10b、R11和R12可以代表的烷基以及可用于取代Y和芳基/苯基和Het1基团的烷基为环状的并且被氧所间断时,则它们可以代表含氧的杂环例如四氢呋喃基或(当合适时)四氢吡喃基。
R2、R3和R6a可以代表的卤素以及可用于取代R2、R3、R5、R6、R6a和芳基/苯基和Het1基团的卤素包括氟、氯、溴和碘。
缩写列在本说明书结尾处。
当R5和R6与它们所连接的氮原子一起代表3-7元的含氮环(例如吡咯烷)时,该环任选包含氧原子和/或被=O取代,该环优选在所述氮原子α位的碳原子上被取代。为了避免产生疑问,R5和R6所连接的氮原子为必定存在于所述环上的氮原子。
可以提及的本发明化合物包括具有以下结构特征的那些化合物,其中
R2和R3独立代表选自卤素或C1-4烷基(任选被卤素取代)的任选的取代基;
R5代表C1-6烷基或者与R6以及R5和R6所连接的氮原子一起代表3-7元的含氮环,该环任选被=O基团取代;
R6代表C1-6烷基、C(O)R9或者与R5以及R5和R6所连接的氮原子一起代表3-7元的含氮环,任选被=O基团取代;
当R4代表OR8a或C(O)OR8b时,R8a和R8b在每种情况下独立代表C1-10烷基、C1-3烷基苯基或C6-10芳基,后两个基团任选被一个或多个卤素、C1-4烷基和/或C1-4烷氧基取代;
R9代表C1-6烷基;和
另外,所有其它取代基如此前所定义。
可以提及的其它本发明的化合物包括其中R4不代表R8c的那些化合物。
本发明优选的化合物包括具有以下结构特征的那些化合物,其中:
R2如果存在,代表直链或支链的C1-4烷基或C1-4烷氧基(二者均任选被卤素取代)或者卤素(例如氯);
R3不存在或者如果存在,代表直链或支链的C1-4烷基或卤素;
R5代表直链、支链或环状的C1-6烷基或者与R6以及R5和R6所连接的氮原子一起代表4-6元的含氮环,任选被=O基团取代;
R6代表直链、支链或环状的C1-6烷基、C(O)-C1-6烷基或者与R5以及
R5和R6所连接的氮原子一起代表4-6元的含氮环,任选被=O基团取代;
R7代表直链、支链或环状的C1-6烷基;
Y代表CH2或(CH2)2。
当R4代表OR8a时,本发明优选的化合物包括那些化合物,其中R8a代表直链或支链的C1-6烷基、C4-5环烷基(后两个基团任选被氧所间断)或者苯基或C1-2烷基苯基(例如苄基)(后两个基团如此前定义被任选取代),或者R8a代表CH2OC(O)R11,其中R11代表苯基、直链、支链或环状的C1-6烷基(后一个基团任选被选自OH、CO2H和苯基的取代基取代)或OR12(其中R12代表苯基或者直链、支链或环状的C1-6烷基(后一个基团任选被选自OH、CO2H和苯基的取代基取代))。
当R4代表C(O)OR8b时,本发明优选的化合物包括那些化合物,其中R8b代表直链或支链的C1-2烷基苯基或苯基(后两个基团如此前定义被任选取代)。
本发明优选的化合物包括那些化合物,其中R1在相对也连接在所述苯环上的-CH(OH)-的3位上与所述苯环连接。任选的取代基R2优选在相对也连接在所述苯环上的-CH(OH)-的5位上与所述苯环连接。
当基团N(R5)R6代表结构片段Ia时,所述片段优选为未取代的。
本发明更优选的化合物包括那些化合物,其中:
R1代表N(R5)R6;
R3不存在或者如果存在,代表甲基或氯,优选在相对于也连接所述苯环的-CH2-基团的2位上;
R8a代表直链或支链的C1-4烷基(任选被氧间断)或被氧间断的C4-5环烷基;
R5代表C1-4烷基或者与R6以及R5和R6所连接的氮原子一起代表5-6元的含氮环,任选被=O基团取代;
R6代表C1-4烷基、C(O)-C1-6烷基(例如C(O)-C1-4烷基)或者与R5以及
R5和R6所连接的氮原子一起代表5或6元的含氮环,任选被=O基团取代。
其中所述片段
为S构型的式I化合物为优选的化合物。
其中所述片段
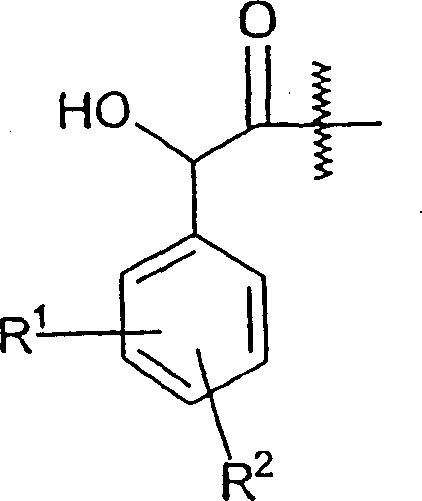
为R构型的式I化合物为优选的化合物。
在以上两个片段中的键上的波浪线表示所述片段的连接位置。
优选的式I化合物包括此后介绍的实施例中的化合物。
制备
根据本发明也提供制备式I化合物的方法,该方法包括:
(i)式II的化合物
其中R1和R2如此前所定义,与式III化合物
其中Y、R3和R4如此前所定义,例如在偶合剂(例如EDC、DCC、HBTU、HATU、TBTU、PyBOP或在DMF中的草酰氯)、合适的碱(例如吡啶、2,4,6-三甲基吡啶、2,4,6-可力丁、DMAP、TEA或DIPEA)和合适的有机溶剂(例如二氯甲烷、乙腈或DMF)存在下偶合;
(ii)式IV的化合物

其中R1、R2和Y如此前所定义,与式V化合物

其中R3和R4如此前所定义,例如在偶合剂(例如在DMF中的草酰氯、EDC、DCC、HBTU、HATU、PyBOP或TBTU)、合适的碱(例如吡啶、2,4,6-三甲基吡啶、DMAP、TEA、2,4,6-可力丁或DIPEA)和合适的有机溶剂(例如二氯甲烷、乙腈或DMF)存在下偶合;
(iii)对于其中R4代表OH或OR8a的式I化合物而言,式VI的化合物
其中R1、R2、Y和R3如此前所定义,与式VII化合物
H2NORa VII
其中Ra代表H或R8a,R8a如此前所定义,例如在40-60℃,在合适的碱(例如TEA)和合适的有机溶剂(例如THF、CH3CN、DMF或DMSO)存在下,任选通过用气态的HCl在低级烷基(例如C1-6烷基)醇(例如乙醇)存在下,在例如0℃预处理式VI化合物,反应形成式VIII的化合物

其中Rc代表低级(例如C1-6)烷基,例如乙基,R1、R2、Y和R3如此前所定义,如果需要可以分离该化合物;
(iv)对于其中R4代表OH或OR8a的式I化合物而言,相应于式I化合物的化合物,其中代替R4,存在保护基团C(O)ORb1,其中Rb1代表基团例如2-三甲基甲硅烷基乙基、C1-6烷基或烷基苯基(例如苄基)与如此前所定义的式VII化合物例如在类似于此前所述的用于制备式I化合物(步骤(iii))的反应条件下反应(技术人员可以理解在此类反应中,在以下情况下,如果需要可以分离所述二保护的脒衍生物(即C(O)ORb1和ORa保护),然后使用常规技术除去C(O)ORb1基团);
(v)对于其中R4代表C(O)OR8b的式I化合物而言,其中R4代表H的式I化合物与式IX的化合物
L1-C(O)OR8b IX
其中L1代表合适的离去基团,例如卤素或对硝基苯氧基,R8b如此前所定义,例如在0℃,在合适的碱(例如氢氧化钠)和合适的有机溶剂(例如THF)和/或水的存在下反应;
(vi)对于其中R4代表OR8a的式I化合物而言,其中R4代表OH的相应的式I化合物与式IXA的化合物
L1-R8a IXA
其中R8a和L1如此前所定义,例如在0℃和回流温度之间,任选在合适的溶剂(例如DCM、THF、CH3CN或DMF)和合适的碱(例如Et3N或吡啶)存在下反应;
(vii)对于其中R4代表R8c,其中R8c代表C(R10a)(R10b)OC(O)R11或C(R10a)(R10b)OC(O)N(H)R12的式I化合物而言,式IXB的相应的化合物
其中R1、R2、Y、R3、R10a和R10b如此前所定义,与式IXC的化合物
L1C(O)R13 IXC
其中R13代表R11或N(H)R12,L1、R11和R12如此前所定义,例如在此前所述的条件(方法步骤(vi))下反应;
(viii)对于其中R4代表R8c的式I化合物而言,其中R4代表H的相应的式I化合物与式IXD的化合物
L1C(R10a)(R10b)R14 IXD
其中R14代表OC(O)R11、NHC(O)OR12或OC(O)N(H)R12,L1、R10a、R10b、R11和R12如此前所定义,例如在此前所述的条件(方法步骤(vi))下反应;
(ix)对于其中R1包括S(O)或S(O)2基团的式I化合物而言,在合适量的合适的氧化剂(例如mCPBA或过氧化单硫酸钾)和合适的有机溶剂(例如二氯甲烷、甲醇、水或其混合物(例如甲醇/水))存在下氧化其中R1包括S基团的相应的式I化合物。
使用已知和/或标准技术可以得到式II化合物。
例如通过式X的醛

其中R1和R2如此前所定义,与下列化合物反应可以制备式II的化合物:
(a)式XI的化合物
R”CN XI
其中R”代表H或(CH3)3Si,例如在室温或高温(例如低于100℃),在合适的有机溶剂(例如氯仿或二氯甲烷)存在下,如果需要,在合适的碱(例如TEA)和/或合适的催化剂体系(例如氯化苄铵或碘化锌)存在下进行,然后在对于本领域技术人员来说熟知的条件下(例如照此前所述)水解;
(b)氢化钠或氢化钾,例如在亚硫酸氢钠和水的存在下进行,然后水解;
(c)氯仿,例如在高温(例如室温以上,但低于100℃)下,在合适的有机溶剂(例如氯仿)存在下,如果需要,在合适的催化剂体系(例如氯化苄基铵)存在下进行,然后水解;
(d)式XII化合物

其中M代表Mg或Li,然后在对于本领域技术人员熟知的条件(例如臭氧解或锇或钌催化)下氧化裂解;或者
(e)三(甲硫基)甲烷,在对于本领域技术人员熟知的条件下进行,然后在如HgO和HBF4存在下水解。
通过对映选择性衍生化步骤可以分离式II化合物的对映体形式(即那些具有在与所述CO2H基团为α位的C原子周围的不同构型的取代基的化合物)。这例如可以通过酶催化过程完成。该酶催化过程包括例如在室温和回流温度之间(即45-55℃),在合适的酶(例如脂酶PSAmano)、合适的酯(即乙酸乙烯酯)和合适的溶剂(例如甲基叔丁基醚)存在下的α-OH的酯交换。然后,通过常规的分离技术(例如层析)可以从未反应的异构体中分离所述衍生化的异构体。
在该衍生化步骤中加到式II化合物中的基团可以在任何其它反应之前或者在合成式I化合物的任何以后的阶段除去。使用常规的技术可以除去所附加的基团(例如对于α-OH的酯而言,在本领域技术人员已知的条件下水解(例如在室温和回流温度之间,在合适的碱(例如氢氧化钠)和合适的溶剂(例如甲醇、水或其混合物)存在下进行))。
通过式XIII化合物

其中Y如此前所定义,与如此前所定义的式V化合物,例如在此前所述用于合成式I化合物的条件下(例如见方法步骤(i)和(ii))反应可以制备式III化合物。
使用已知的技术可以容易地得到式IV的化合物。例如,通过如此前所定义的式II化合物与如此前所定义的式XIII化合物,例如在如此前所述用于合成式I化合物的条件下(例如参见方法步骤(i)和(ii))反应,可以制备式IV化合物。
式V化合物为文献中已知的和/或可以使用已知技术制备的。例如,通过在本领域技术人员熟知的条件下,还原式XIV化合物
其中R3和R4如此前所定义,可以制备式V的化合物。
根据肽偶合技术,例如以与此前所述用于式I化合物的方法(例如参见方法步骤(i)和(ii))类似的方式可以制备式VI的化合物。如果需要,也可以以该方式制备式VIII化合物。
通过其中R4代表H的相应的式I化合物与过量的式IVA化合物
R10aC(O)R10b XIVA
其中R10a和R10b如此前所定义,例如在本领域技术人员已知的条件下反应可以制备式IXB的化合物。
式X的化合物可以购买得到,在文献中众所周知或者使用已知的和/或标准技术可以得到。
例如,通过在合适的还原剂(例如DIBAL-H)存在下,还原式XV化合物可以制备式X的化合物

其中R1和R2如此前所定义。
或者,通过在合适的氧化剂(例如氯铬酸吡啶鎓或DMSO和草酰氯的组合)的存在下,氧化式XVI的化合物

其中R1和R2如此前所定义,可以制备式X的化合物。
例如,如此前所述,其中R1包括S(O)或S(O)2
基团的式II、IV、VI、VIII、X、XV和XVI的化合物可以通过氧化其中R1包括S基团的相应的式II、IV、VI、VIII、X、XV或XVI的化合物(适合时)制备。
式VII、IX、IXA、IXC、IXD、XI、XII、XIII、XIV、XIVA、XV和XVI的化合物和其衍生物可以购买得到、文献中已知或者通过类似于在此所述的方法,或者通过常规合成方法,根据标准技术,从容易得到的原料,使用合适的试剂和反应条件(例如如此前所述的反应条件)得到。
使用本领域技术人员熟知的技术可以导入和/或互变式I、II、III、IV、V、VI、VII、VIII、IX、IXA、IXB、IXC、IXD、X、XIII、XIV、XV和XVI化合物中芳族和/或非芳族、碳环和杂环上的取代基。例如,可以将硝基还原为氨基,氨基可以烷基化或酰基化得到烷基-和/或酰基氨基,氨基可以转化为吡咯并(在催化剂例如五氧化二磷存在下与2,5-二甲氧基四氢呋喃缩合),氨基可以(经重氮化)转化为卤素或(例如经与1,4-或1,5-二卤代烷基化合物或者β-或γ-卤代酯反应)转化为含氮环(任选被=O基团取代),碘代可以转化为含氮的杂环(例如咪唑基和哌啶基,通过在Buchwald条件下,用咪唑或哌啶处理),氮羟基可以烷基化得到烷氧基,烷氧基可以水解为羟基,烯烃可以氢化为烷烃,卤代可以氢化为H等。在这方面,其中R1代表-N(CH3)2和R2代表氯或甲基的式XV化合物采用例如按照Wolfe等在Tetrahedron Lett.38,6367(1997)中所述的方法Pd催化胺化,然后还原胺化(例如使用HCHO和还原剂例如Na(CN)BH3或氧化铂(IV)和氢的组合)或对得到的苯胺进行烷基化(例如使用碘甲烷和合适的碱),可从购买得到的碘-氯或碘-甲基二取代的苯甲酸甲酯中得到。例如,按照Tarbell等在“有机合成”Coll.Vol.III第809-11(1955)中所述的方法,其中R1代表-S(O)mCH3(其中m如此前所定义)和R2代表氯或甲基的式XV化合物可由以上所述得到的苯胺(或由相应的苯甲酸)经重氮化、然后用乙基黄原酸钾处理重氮盐,然后水解所述中间体得到相应的苯硫酚而获得。然后,可以将所得到的苯硫酚烷基化(例如使用合适的烷基碘,在乙醇中的合适的碱存在下进行),然后(如果需要)氧化形成砜或亚砜(例如使用在二氯甲烷中的mCPBA或甲醇/水中的过氧单硫酸钾)。
使用常规的技术可以自其反应混合物中分离式I化合物。
本领域技术人员可以理解在上述方法中,中间体化合物的官能基团可能需要用保护基来保护。
需要保护的官能基团包括羟基、氨基、醛、2-羟基羧酸和羧酸。对于羟基的合适的保护基包括三烷基甲硅烷基或二芳基烷基甲硅烷基(例如叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基或三甲基甲硅烷基)和四氢吡喃基。对于羧酸的合适的保护基包括C1-6烷基或苄基酯。对于氨基和脒基的合适的保护基包括叔丁氧基羰基、苄氧基羰基或2-三甲基甲硅烷基乙氧基羰基(Teoc)。脒基氮也可以用羟基或烷氧基保护并可以被一-或二保护。通过与例如乙二醇反应可以作为缩醛保护醛。通过与例如丙酮缩合可以保护2-羟基羧酸。
官能团的保护和脱保护可以在偶合前或偶合后或者在上述流程中的任何其它反应之前或之后进行。
按照对于本领域技术人员为熟知的技术并且如此后所述的方法可以除去保护基团。
本领域内的技术人员可以理解为了以另一种并且在某些场合下更便利的方式得到式I化合物,此前提及的各方法步骤可以以不同的次序进行和/或各反应可以在总的路线中不同的阶段进行(即结合具体的反应,取代基可以加到此前提及的化合物的不同中间体上和/或化学转化可以在其上发生)。该过程可能需要也可能不需要保护基团。
例如,对于其中R4不代表氢的式I化合物的合成尤其如此。在这种情况下,采用此前所述的方法步骤(例如参见方法步骤(iii)-(viii)),在总的合成中的最初阶段可以导入OH、OR8a、C(O)OR8b和/或R8c。此外,式II和IV化合物的扁桃酸OH基团可能需要在此前所述偶合步骤之前保护。
因此,所涉及化学的次序和类型将决定对于保护基的需要和类型以及用于完成该合成的顺序。
保护基的用途在由JWF McOmie编辑的“有机化学中的保护基团”(Plenum Press(1973))和“有机合成中的保护基团”(第2版,TWGreene & PGM Wutz,Wiley-Interscience(1991))中被全面描述。
采用标准的脱保护技术(例如氢化)可以将式I化合物的保护衍生物在化学上转化为式I化合物。技术人员也可以理解某些式I化合物也可以称作其它式I化合物的“被保护的衍生物”。
医学和药学用途
本发明化合物本身可以具有药理活性。可以具有该活性的本发明化合物包括(但不限于)其中R4为氢的那些化合物。
然而,其它式I化合物(包括其中R4不为氢的那些化合物)可能不具有此类活性,但可以胃肠外或口服给予,此后在体内代谢形成药理活性的化合物(包括但不限于其中R4为氢的相应化合物)。这类化合物(也包括可以具有某些药理活性,但该活性适当低于它们代谢产生的“活性”化合物的那些化合物)因此可以称作所述活性化合物的“前药”。
因此,本发明的化合物是有用的,因为它们具有药理活性和/或口服或胃肠外给药后在体内代谢形成具有药理活性的化合物。因此,本发明化合物适合作为药物。
因此,根据本发明的另一个方面,提供用作药物的本发明化合物。
特别是,本发明化合物是本身是有效的凝血酶抑制剂和/或(例如在前药的情况下)在给药后代谢形成有效的凝血酶抑制剂,例如以下所述试验中所显示的。
所谓“凝血酶抑制剂的前药”包括在口服或胃肠外给药后,在实验可检测量下并在预定的时间(例如约1小时)内形成凝血酶抑制剂的化合物。
因此,期待本发明化合物用于那些需要抑制凝血酶的疾病中。
因此,本发明的化合物适用于治疗和/或预防动物包括人类的血液和组织中血栓形成和凝固性过高。
已知凝固性过高可以导致血栓栓塞疾病。可以提及的与凝固性过高有关的症状和血栓栓塞疾病包括遗传性或获得性激活蛋白C抗性例如因子V-突变(因子V Leiden)以及抗凝血酶III、蛋白C、蛋白S或肝素辅因子II方面遗传性或获得性缺乏。其它已知与凝固性过高有关的症状和血栓栓塞疾病包括循环抗磷脂抗体(Lupus抗凝药)、高胱氨酸尿症(homocysteinemi)、肝素诱导的血小板减少症和纤维蛋白溶解缺陷。因此,本发明化合物适用于这些症状的治疗性和/或预防性处理。
本发明化合物另外适用于治疗其中有不合乎需要的过量的凝血酶而无凝固性过高迹象的病症,例如在神经变性疾病例如阿尔茨海莫氏病中的症状。
可以提及的具体的病症包括治疗性和/或预防性处理静脉血栓形成和肺栓塞、动脉血栓形成(例如在心肌梗塞、不稳定性心绞痛、基于血栓形成的中风和外周动脉血栓形成)以及通常来自于房颤期间心房或来自于透壁心肌梗塞后左心室的系统性栓塞。
此外,期待本发明化合物具有在预防血栓溶解、经皮经腔血管成形术(PTA)和冠脉旁路手术后的再阻塞(即血栓形成);防止在显微手术和普通血管手术后再次血栓形成中的用途。
其它适应症包括治疗性/或预防性处理由细菌、多发性损伤、中毒或任何其它机制引起的播散性血管内凝血;当血液与体内的异质表面例如血管移植物、血管斯滕特固定膜、血管导管、机械和生物的修复瓣膜或任何其它的医疗装置接触时抗凝药处理;和当血液与体外的医疗装置例如在心血管手术期间使用心-肺机或在血液透析中接触时抗凝药处理。
除了其在血凝固过程中的作用外,已知凝血酶激活大量的细胞(例如中性白细胞、成纤维细胞、内皮细胞和平滑肌细胞)。因此,本发明化合物也可用于治疗性和/或预防性处理特发性和成人呼吸窘迫综合征、采用放疗或化疗治疗后肺纤维化、败血症性休克、败血症、炎性反应(包括但不限于水肿)、急性或慢性动脉粥样硬化症例如冠状动脉疾病、脑动脉疾病、外周动脉疾病、再灌注损伤和经皮经腔血管成形术(PTA)后再狭窄。
抑制胰蛋白酶/或凝血酶的本发明化合物也可以用于治疗胰腺炎。
根据本发明的另一个方面,提供治疗其中需要抑制凝血酶的症状的方法,该方法包括给予患有或者易感该症状的人治疗有效量的本发明的化合物或或其药学上可接受的盐。
本发明的化合物一般可以口服、静脉内、皮下、向颊、直肠、经皮、鼻腔、气管、支气管、通过任何其它的胃肠外的途径或经吸入,以药学上可接受的剂型中含有游离碱或药学上可接受的、非毒性有机酸或无机酸加成盐形式的活性化合物的药用制剂形式给予。根据所述疾病、所治疗的患者和给药途径,可以按不同的剂量给予所述组合物。
本发明化合物也可以与任何具有不同的作用机制的抗血栓形成药联合和/或同时给药,例如抗血小板药乙酰水杨酸、噻氯匹定、氯吡格雷、血栓烷受体和/或合成酶抑制剂、血纤蛋白原受体拮抗剂、前列环素模拟物和磷酸二酯酶抑制剂和ADP-受体(P2T)拮抗剂。
本发明化合物还可以与血栓溶解剂例如组织溶酶原激活物(天然、重组或修饰)、链激酶、尿激酶、尿激酶原、茴香酰化溶酶原-链激酶激活物复合物(APSAC)、动物唾腺溶酶原激活物等,在治疗血栓形成疾病尤其是心肌梗塞中联合和/或同时给药。
因此,根据本发明的另一个方面,提供包含以与药学上可接受的辅助剂、稀释剂或载体的混合物形式的本发明的化合物的药用制剂。
在人类治疗中,本发明化合物的合适的日剂量为:经口给药大约0.001-100mg/kg体重,胃肠外给药0.001-50mg/kg体重。
本发明的化合物本身或其所代谢产生的化合物具有更有效、毒性更低、作用更长久的优点,具有比先有技术中已知的化合物更广泛的活性、更有效、产生更少的副作用、更容易吸收或者具有其它有用的药理学、物理学或化学性质。
生物学试验
试验A
测定凝血酶凝固时间(TT)
将所述抑制剂溶液(25μl)与血浆(25μl)温育3分钟。然后,加入在缓冲液,pH 7.4中的人凝血酶(T 6769;Sigma Chem.Co.或Hematologic Technologies)(25μl,4.0NIH单位/ml)并在自动装置(KC10;Amelung)上测定该凝固时间。
所述凝血酶凝固时间(TT)表达为绝对值(秒)以及不含有抑制剂的TT(TT0)与含有抑制剂的TT(TTi)的比值。后面的比值(范围1-0)对抑制剂的浓度(log转换)作图并且根据以下的方程式拟合为S形的剂量-应答曲线
y=a/[1+(x/IC50)s]
其中:a=最大范围,即1;s=该剂量-应答曲线的斜率;和IC50=使凝固时间加倍的抑制剂浓度。在PC上进行计算,使用GraFit第3版的软件程序,设定方程等于:于0开始,规定结束=1(ErithacusSoftware,Robin Leatherbarrow,Imperial College of Science,London,UK)。
试验B
采用生色、自动(Robotic)测试测定凝血酶抑制
采用生色底物方法,在Plato 3300自动微量滴定板处理器(RosysAG,CH-8634 Hombrechtikon,Switzerland)上,使用96孔、半体积微量滴定板(Costar,Cambridge,MA,USA;目录号3690)测定所述凝血酶抑制剂的效能。采用DMSO以1∶3(24+48μl)系列稀释试验物质在DMSO(72μl)中的贮备液(0.1-1mmol/L)得到十种不同的浓度,作为样品在本测试中分析。用124μl测试缓冲液稀释2μl的试验样品,加入12μl生色底物在测试缓冲液中的溶液(S-2366,Chromogenix,Mǒlndal,Sweden)和最终12μlα-凝血酶在测试缓冲液中的溶液(人α-凝血酶,Sigma Chemical Co.或Hematologic Technologies)并且混合样品。其最终测试浓度为:试验物质0.00068-13.3μmol/L,S-2366 0.30mmol/L,α-凝血酶0.020NIHU/ml。使用在40分钟的37℃温育期间线性吸光度递增,以便于计算当与无抑制剂的空白对比时的试验样品的百分抑制率。由log浓度对%抑制率曲线计算相应于引起凝血酶活性50%抑制率的抑制剂浓度的IC50-自动测定值。
试验C
对于人凝血酶的抑制常数Ki的测定
使用生色底物方法,于37℃在Cobas Bio离心分析仪(Roche,Basel,Switzerland)上进行,完成Ki测定。三种不同的底物浓度下,测定在人α-凝血酶与各种浓度的试验化合物温育后残余酶活性,测定结果以在405nm下吸光度变化表示。
将试验化合物溶液(100μl;通常在含有BSA 10g/L的缓冲液或盐水中)与200μl人α-凝血酶(Sigma Chemical Co)在含有BSA(10g/L)的测试缓冲液(0.05mol/L Tris-HCl pH 7.4,用氯化钠调节离子强度为0.15)中混合并且作为在Cobas Bio中的样品分析。将60μl样品以及20μl水加入到320μl的测试缓冲液中的底物S-2238(Chromogenix AB,Mǒlndal,Sweden)中,并且监测吸光度变化(ΔA/分钟)。S-2238的最终浓度为16、24和50μmol/L和凝血酶的最终浓度为0.125NIH U/mL。
使用稳定态反应速率构建Dixon曲线即抑制剂浓度对1/(ΔA/分钟)的图。对于可逆、竞争性抑制剂而言,对于不同的底物浓度的数据点一般形成直线,其在x处的截距为-Ki。
试验D
测定激活部分促凝血酶原激酶时间(APTT)
用由Stago制备的试剂PTT Automated 5,在收集的正常人柠檬酸化的血浆中测定APTT。将所述抑制剂加入到该血浆(10μl抑制剂溶液比90μl血浆)中,与所述APTT试剂温育3分钟,然后加入100μl氯化钙溶液(0.025M),并且根据该试剂制造商的说明使用血凝固分析仪KC10(Amelung)测定APTT。
所述凝固时间表达为绝对值(秒)以及不加抑制剂的APTT(APTT0)与加入抑制剂的APTT(APTTi)的比值。后面的比值(范围1-0)对抑制剂的浓度(log转换)作图并且根据以下方程拟合为S形的剂量-应答曲线
y=a/[1+(x/IC50)s]
其中:a=最大范围,即1;s=该剂量-应答曲线的斜率;和IC50=使凝固时间加倍的抑制剂浓度。在PC上进行计算,使用GraFit第3版的软件程序,设定方程等于:于0开始,规定结束=1(ErithacusSoftware,Robin Leatherbarrow,Imperial College of Science,London,UK)。
IC50APTT定义为使激活部分促凝血酶原激酶时间加倍的人血浆中抑制剂的浓度。
试验E
测定体内凝血酶时间
检测清醒大鼠在口服或胃肠外给予溶解在乙醇∶SolutolTM∶水(5∶5∶90)中的式I化合物后凝血酶的抑制,为该大鼠在实验前一或两天安放导管以便从颈动脉采血。在实验的当天,在给予化合物后的固定时间采取血样到含有1份柠檬酸钠溶液(0.13mol/L)和9份血的塑料试管中。离心该试管以便得到贫血小板血浆。使用该血浆按如下所述测定凝血酶时间或ecarin凝固时间(ECT)。
用100μl 0.9%盐水稀释100μl柠檬酸化的大鼠血浆,通过加入在100μl缓冲溶液pH7.4中的人凝血酶(T 6769,Sigma Chem.Co.USA或Hematologic Technologies)或ecarin(Pentapharm)引起血浆凝固。在自动装置(KC 10,Amelung,Germany)中测定所述凝固时间。
当给予式I的“前药”化合物时,通过使用使收集的柠檬酸化的大鼠血浆中的凝血酶时间或ecarin凝固时间与相应于溶解在盐水中的“活性”凝血酶抑制剂的已知浓度相关的标准曲线,评估大鼠血浆中式I的合适活性凝血酶抑制剂(例如游离脒化合物)的浓度。
基于评估的大鼠中活性凝血酶抑制剂的血浆浓度(假定凝血酶时间或ECT延长是由上述化合物引起的),使用梯形法则并外推数据到无穷大,计算口服和/或胃肠外给予相应的式I前体化合物后曲线下的面积(AUCpd)。
如下计算口服或胃肠外给予所述前药后活性凝血酶抑制剂的生物利用率:
[(AUCpd/剂量)/(AUC活性,胃肠外/剂量)]×100
其中AUC活性,胃肠外代表如上所述胃肠外给予清醒大鼠相应的活性凝血酶抑制剂后获得的AUC。
试验F
测定来自体内尿中凝血酶时间
通过测定来自体内尿中凝血酶时间(假定凝血酶时间延长是由上述化合物引起的),估计口服或胃肠外给予溶解在乙醇∶SolutoliTM∶水(5∶5∶90)中的本发明“前药”化合物后,尿中所排泄的“活性”凝血酶抑制剂的量。
将清醒大鼠置于代谢笼中,在口服给予本发明的化合物后分别收集尿和粪便24小时。按如下所述方法测定所收集尿中凝血酶时间。
将收集的正常柠檬酸化的人血浆(100μl)与浓的大鼠尿或其盐水稀释液保温1分钟。然后,通过给予缓冲溶液(pH7.4;100μl)中的人凝血酶(T 6769,Sigma Chem Company)引发血浆凝固。在自动装置(KC 10;Amelung)中测定该凝固时间。
通过使用使收集的正常柠檬酸化的人血浆中的凝血酶时间与溶解在浓的大鼠尿(或其盐水稀释液)中的上述活性凝血酶抑制剂的已知浓度相关的标准曲线估计大鼠尿中活性凝血酶抑制剂的浓度。通过用估计的大鼠尿中的上述活性抑制剂的平均浓度乘以24小时内总的大鼠排尿量,可以计算在所述尿中排泄的活性抑制剂的量(量pd)。
如下计算口服或胃肠外给予所述前药后活性凝血酶抑制剂的生物利用率:
[(量pd/剂量)/(量活性,胃肠外/剂量)]×100
其中量活性,胃肠外代表如上所述胃肠外给予清醒大鼠相应的活性凝血酶抑制剂后在尿中所排泄的量。
试验G
体外前药化合物的代谢活化
于37℃下,将式I前药化合物与肝微粒体或自人或大鼠肝匀浆制备的10000g(指离心速度)上清液部分(即s9部分)温育。在所述温育液中总的蛋白质浓度为1或3mg/ml(溶解在0.05mol/L TRIS缓冲液(pH7.4)中),同时存在辅因子NADH(2.5mmol/L)和NADPH(0.8mmol/L)。温育液的总体积为1.2ml。初始的前药浓度为5或10μmol/L。在开始温育后超过60分钟的定期间隔内自温育液中收集样品。将来自所述温育液的样品(25μl)与等体积的人或大鼠血浆和合适量的凝血酶混合,在凝固仪(KC 10;Amelung)上测定凝固时间(即凝血酶时间)。通过使用使收集的柠檬酸化的人或大鼠血浆中凝血酶时间与相应的“活性凝血酶抑制剂”的已知浓度相关的标准曲线估计所形成的“活性”凝血酶抑制剂的量。
或者,除了使用上述方法,通过使用LC-MS估计“活性”凝血酶抑制剂的量。
实施例
通过以下实施例说明本发明。如果没有另外说明,氨基酸Pro和Aze定义为S-异构体。如果没有另外说明,得到的实施例为非对映异构体。
实施例1
Ph(3-N(Me)2)-(R)-或-(S)CH(OH)-C(O)-Aze-PabxHOAc
(i)Ph(3-N(Me)2)-CHO
将Ph(3-N(Me)2)-CH2OH(1.9g;12.6mmol)和MnO2(8.8g;100mmol)的CH2Cl2混合物在室温下搅拌2.5天。将混合物经Celite过滤并蒸发滤液。将粗制产物在硅胶上经快速层析,用异丙基醚∶三甲基戊烷(7∶3)作为洗脱剂。产量0.93g(50%)。
1H-NMR(400MHz;CDCl3):δ9.89(s,1H),7.37(m,1H),7.17-7.25(m,2H),7.05(m,1H),2.98(s,6H)。
(ii)Ph(3-N(Me)2)-(R,S)CH(OSiMe3)CN
将TMS-CN(0.75mL;6.0mmol)滴加入到Ph(3-N(Me)2)-CHO(0.9g;6.0mmol;来自以上步骤(i))和Et3N(0.08mL;6.0mmol)的CH2Cl2(15mL)的混合物中。将反应混合物在室温下搅拌24小时。加入附加量的Et3N(0.08mL;6.1mmol)和TMS-CN(0.75mL;6.0mmol)并继续再搅拌24小时。蒸发反应混合物,得到小标题化合物1.35g(90%)。
1H-NMR(400MHz;CDCl3):δ7.27(t,1H),6.78-6.84(m,2H),6.74(dd,1H),5.47(s,1H),3.00(s,6H)。
(iii)Ph(3-N(Me)2)-(R,S)CH(OH)-C(O)OH
将Ph(3-N(Me)2)-(R,S)CH(OSiMe3)CN(1.35g;5.43mmol;来自以上步骤(ii))和HCl(20mL;浓的)的混合物在室温下搅拌10分钟,然后在90℃和100℃之间(在油浴中)搅拌3小时。将反应混合物蒸发并加入水。用Et2O洗涤酸化的水层并放在阳离子交换剂(IR-120,10-15g;通过将阳离子交换剂悬浮在NaOH(2M)中预制备)上,然后将所述淤桨倒进柱中。随后用HCl(2M;2×50mL)、H2O(2×50mL)洗涤阳离子交换剂,然后用H2O洗涤直到pH为中性并用NH4OH/水溶液(1M)洗脱产物。蒸发生成物的水层并冻干,得到小标题化合物0.78g(74%)。
LC-MS:(M-1)194m/z
1H-NMR(500MHz;CD3OD):δ7.15(t,1H),6.94(s,1H),6.84(d,1H),6.69(dd,1H),4.85(s,1H),2.92(s,6H)。
(iv)Ph(3-N(Me)2)-(R)-或-(S)CH(OH)-C(O)OHxHCl
用ChiralcelTMOD作为固定相和n-庚烷∶2-丙醇∶甲酸(80∶20∶1)作为流动相,经制备型HPLC分离Ph(3-N(Me)2)-(R,S)CH(OH)-C(O)OH(以上步骤(iii))的对映体。蒸发后洗脱的对映体并冻干,然后重溶于水中,并加入3当量1M的HCl。将该溶液冻干得到其盐酸盐,其[α]D 20为-63.7°(c=1.0,MeOH)。用分析型手性HPLC测定,对映体过量为97%。
(v)Ph(3-N(Me)2)-(R)-或-(S)CH(OH)-C(O)-Aze-Pab(Z)
在0℃下,将DIPEA(1.03mL;6.15mmol)加入到Ph(3-N(Me)2)-(R)-或-(S)CH(OH)-C(O)OHxHCl(0.36g;1.54mmol;以上步骤(iv)中分离的产物)、H-Aze-Pab(Z)x2HCl(0.743g;1.69mmol;参见国际专利申请WO 97/02284)和TBTU(0.543g;1.69mmol)的DMF(10mL)中的混合物中。将反应混合物在室温下搅拌4天,倒入H2O(400mL)中并经加入NaHCO3/水溶液将pH调到10。用EtOAc萃取水层,然后用NaHCO3/水溶液、H2O和NaCl/水溶液洗涤有机层,干燥(Na2SO4)并蒸发。经在硅胶上快速层析纯化粗制产物,用CH2Cl2∶MeOH(95∶5)作为洗脱剂。经制备型HPLC进一步纯化该产物,得到小标题化合物203mg(24%)。
LC-MS:(M+1)544;(M-1)542m/z
1H-NMR(400MHz;CDCl3):δ8.20(t,1H),7.75(d,2H),7.43(d,2H),7.18-7.38(m,6H),6.61-6.72(m,3H),5.20(s,2H),4.88(s,1H),4.84(dd,1H),4.36-4.52(m,2H),4.03(m,1H),3.63(m,1H),2.93(s,6H),2.54(m,1H),2.30(m,1H)。
(vi)Ph(3-N(Me)2)-(R)-或-(S)CH(OH)-C(O)-Aze-PabxHOAc
将Ph(3-N(Me)2)-(R)-或-(S)CH(OH)-C(O)-Aze-Pab(Z)(112mg;0.206mmol;来自以上步骤(v))、HOAc(0.41mL)和10%Pd/C的EtOH(7mL)中的混合物在常压、室温下氢化3小时。将反应混合物经Celite过滤,蒸发滤液并冻干(x2),得到90mg(93%)白色结晶。
LC-MS:(M+1)410;(M-1)408m/z
1H-NMR(500MHz;CD3OD):δ7.74(d,2H),7.54(d,2H),7.21(t,1H),6.85(s,1H),6.73-6.77(m,2H),5.11(s,1H),4.77(dd,1H),4.52(dd,2H),4.30(m,1H),3.92(m,1H),2.92(s,6H),2.46(m,1H),2.27(m,1H)。
13C-NMR(125MHz;CDCl3):(羰基和/或脒碳)δ173.3,171.9,167.0。
实施例2
Ph(3-N(Me)2)-R)-或-(S)CH(OH)-CO-Aze-Pab(OMe)
(i)4-(氨基,甲氧基亚氨基甲基)苄基叠氮化物
将O-甲基羟胺盐酸盐(10.5g;125mmol)、三乙胺(56mL)和甲醇(200mL)的混合物加入到4-乙基imidato苄基叠氮化物盐酸盐(22.5g;110mmol;根据在WO 94/29336中所述方法制备)的乙醚溶液中。将反应混合物在室温下搅拌3到4天。在真空下除去大部分甲醇并用乙酸乙酯置换。用H2O、HOAc/水溶液(1.5%;pH 4)、NaHCO3/水溶液洗涤有机层并干燥(Na2SO4)。用乙酸乙酯稀释生成物溶液至500mL并浓缩25mL该稀释溶液以便估计产量。总产量大约为20g。
1H-NMR(400MHz;CD3OD):δ7.66(d,2H),7.36(d,2H),4.37(s,2H),3.83(s,3H)。
(ii)H-Pab(OMe)
将氧化铂(200mg)加入到4-(氨基,甲氧基亚氨基甲基)苄基叠氮化物(10g;0.049mol;来自以上步骤(i))的200mL乙醇的溶液中。在常压下将该混合物氢化8小时,经CeliteTM过滤并浓缩。所述粗制产物直接用于以下步骤中。
1H-NMR(400MHz;CD3OD):δ7.60(d,2H),7.37(d,2H),3.81(s,3H),3.80(s,2H)。
(iii)Boc-Aze-Pab(OMe)
将DIPEA(17.5mL;105mmol)加入到Boc-Aze-OH(9.7g;48mmol;参见国际专利申请WO 97/02284)和H-Pab(OMe)(9.4g;52mmol;来自以上步骤(ii))和TBTU(18.5g;58mmol)的DMF(100mL)冰冷的溶液中,在室温下将该混合物搅拌过夜。将得到的混合物倒入水(50mL)中,将pH调到大约为9并将该混合物用EtOAc萃取三次。用NaHCO3(水溶液)、水和盐水洗涤合并的有机层,干燥(Na2SO4)并浓缩。用快速层析(Si-gel;EtOAc)纯化粗制产物。产量为11.9g(69%)。
1H-NMR(400MHz;CDCl3):δ7.60(d,2H),7.31(d,2H),4.78(b,2H),4.69(t,1H),4.50(b,2H),3.92(s+m,4H),3.79(m,1H),2.46(b,2H),2.04(s,3H)。
(iv)Aze-Pab(OMe)x2HCl
将Boc-Aze-Pab(OMe)(9.4g;26mmol;来自以上步骤(iii))的EtOAc(250mL)溶液用HCl(g)饱和。将EtOH(无水;125mL)加入到生成的乳状液中并将该混合物声处理10分钟。加入EtOAc直至所述溶液成为混浊,此后小标题产物立即结晶。产量6.7g(77%)。
LC-MS:(M+1)263m/z
1H-NMR(400MHz;CD3OD):δ7.74(d,2H),7.58(d,2H),5.13(t,1H),4.57(m,2H),4.15(m,2H),3.97(s+m,4H),2.87(m,1H),2.57(m,1H)。
13C-NMR(75MHz;CDCl3):(羰基和/或脒碳)δ168.9;168.8;161.9。
(v)Ph(3-N(Me)2)-(R)-或-(S)CH(OH)-C(O)-Aze-Pab(OMe)
将Ph(3-N(Me)2)-(R)-或-(S)CH(OH)-C(O)OHxHCl(118mg;0.51mmol;参见以上实施例1(iv))和HATU(214mg;0.56mmol)的DMF(3mL)中的混合物在0℃下搅拌1.5小时。在0℃下,将H-Aze-Pab(OMe)x2HCl(189mg,0.56mmol;来自以上步骤(iv))、2,4,6-三甲基吡啶(0.3mL,2.25mmol)和DMF(3mL)在滴加入第一混合物前分别混合。将该反应混合物在0℃搅拌3小时,放入冰箱中3天并蒸发。将粗制产物经制备型HPLC纯化,得到标题化合物140mg(62%)。
LC-MS:(M+1)440;(M-1)438m/z
1H-NMR(500MHz;CD3OD):δ8.60(t,1H),7.61(d,2H),7.37(d,2H),7.22(t,1H),6.87(s,1H),6.77(d,2H),5.08(s,1H),4.75(dd,1H),4.46(dd,2H),4.26(m,1H),3.90(m,1H),3.84(s,3H),2.94(s,6H),2.44(m,1H),2.26(m,1H)。
13C-NMR(125MHz;CD3OD):(羰基和/或脒碳)δ173.3,171.8,154.9。
实施例3
Ph(3-SMe)-(R)-或-(S)CH(OH)C(O)-Aze-PabxTFA
(i)Ph(3-SMe)-(R,S)CH(OTMS)CN
在0℃、氮气氛下,向Ph(3-SMe)-CHO(19.8g,130mmol)和ZnI2(2.1g,6.50mmol)的CH2Cl2(450mL)溶液中滴加入三甲基甲硅烷基氰化物(14.2g,143mmol)。在25℃下搅拌过夜后,用H2O(450mL)猝灭所述有机混合物。分离有机层并用饱和盐水(300mL)洗涤,干燥(Na2SO4),过滤并在真空下浓缩,得到小标题化合物32.0g(98%粗品),为橙色油状物,其没有进一步纯化而使用。
1H-NMR(300MHz;CDCl3):δ7.20-7.41(m,4H),5.50(s,1H),2.51(s,3H),0.23(s,9H)。
(ii)Ph(3-SMe)-(R,S)CH(OH)C(O)OH
将Ph(3-SMe)-(R,S)CH(OTMS)CN(32.0g,130mmol;参见以上步骤(i))的浓HCl(250mL)中的溶液回流2.5小时。用6N NaOH(450mL)使该混合物成碱性并用Et2O(3×300mL)洗涤以除去有机杂质。用6NHCl(150mL)酸化含水层并用EtOAc(4×500mL)萃取。干燥(Na2SO4)合并的萃取液,过滤并在真空下浓缩,得到小标题化合物22.6g(90%粗品收率),为橙色油状物,其储存时结晶成黄-褐色固体。
1H-NMR(300MHz;CD3OD):δ7.20-7.40(m,4H),5.12(s,1H),2.50(s,3H)。
(iii)Ph(3-SMe)-(R)或-(S)CH(OH)C(O)OH(a)和Ph(3-SMe)-(S)或-(R)CH(OAc)C(O)OH(b)
将Ph(3-SMe)-(R,S)CH(OH)C(O)OH(2.0g,10.1mmol;参见以上步骤(ii))、脂酶PS Amano(1.0g)、乙酸乙烯酯(5.0mL)和MTBE(5.0mL)的混合物在45℃下加热24小时。将反应物过滤并用EtOAc(100mL)洗涤滤饼。在真空下浓缩滤液并在硅胶上层析,用CHCl3∶MaOH∶NH3(饱和溶液)(6∶3∶1)洗脱,得到为黄色油状物的小标题化合物(a)630mg(32%)和为褐色固体的小标题化合物(b)850mg(35%)。
对于小标题化合物(a):
1H-NMR(300MHz;CD3OD):δ7.38(s,1H),7.10-7.25(m,3H),5.08(s,1H),2.40(s,3H)。
13C NMR(75MHz;CD3OD):δ178.4,142.6,140.2,130.0,127.3,126.4,125.2,75.5,15.8。
HPLC分析:98.9%,96.0%ee
[α]D 25=-119.8°(c=1.0,MeOH)
CI-MS:(M+1)199m/z
对于小标题化合物(b):
1H-NMR(300MHz;CD3OD):δ7.62(s,1H),7.32-7.44(m,3H),5.82(s,1H),2.62(s,3H),2.30(s,3H)。
(iv)Boc-Aze-PabxHCOOH
将甲酸铵(3.0g;50mmol)和Pd/C(5%;1.0g)加入到Boc-Aze-Pab(Z)(4.7g;10mmol;参见国际专利申请WO 94/29336)的50mLMeOH中的溶液中。加入甲酸(1.0g;22mmol)并将混合物搅拌30分钟。将该反应混合物经Hyflo过滤并将溶液浓缩。将粗制产物悬浮在CH2Cl2(50mL)中,过滤并再用附加的CH2Cl2洗涤。将该固体物质干燥并没有进一步纯化而在下一步骤中使用。
(v)Boc-Aze-Pab(Teoc)
将Teoc-对-硝基苯基碳酸酯(3.5g;12.3mmol)加入到Boc-Aze-PabxHCOOH(3.7g;10mmol;参见以上步骤(iv))的THF(100mL)中的溶液中,以后用2分钟加入K2CO3(1.8g;13mmol)的水(20mL)溶液。将生成物溶液搅拌3天,浓缩并将残余物溶于EtOAc(150mL)和NaOH(水溶液;0.5M;50mL)中。用盐水(2×50mL)洗涤有机层,干燥(Na2SO4)并浓缩。用快速层析(Si-gel;二氯甲烷∶丙酮;4∶1)纯化粗制产物。产量4.6g(96%)。
1H-NMR(500MHz;CDCl3):δ7.86(d,2H),7.39(d,2H),4.72(bt,1H),4.7-4.5(br,2H),3.93(m,1H),3.81(m,1H),2.48(br,2H),1.43(s,9H),0.09(s,9H)。
(vi)H-Aze-Pab(Teoc)xHCl
将Boc-Aze-Pab(Teoc)(4.6g;9.6mmol;参见以上步骤(v))的二氯甲烷(150mL)中的溶液用干燥的HCl饱和。将该溶液放置在室温下塞好的烧瓶中10分钟,以后将其浓缩。产量4.2g(97%)。
1H-NMR(400MHz;CD3OD):δ7.80(d,2H),7.60(d,2H),5.10(m,1H),4.60(bs,2H),4.15(m,1H),3.97(q,1H),2.86(m,1H),2.57(m,1H),0.11(s,9H)。
(vii)Ph(3-SMe)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)
将Ph(3-SMe)-(R)或-(S)CH(OH)C(O)OH(300mg,1.51mmol;参见以上步骤(iii)(a))、H-Aze-Pab(Teoc)(627mg,1.66mmol;参见以上步骤(vi))、TBTU(632mg,1.66mmol)和DIPEA(391mg,3.03mmol)的DMF(8.0mL)中的混合物在0℃下搅拌,然后在25℃下搅拌过夜。用H2O(50mL)猝灭该反应物并用EtOAc(3×50mL)萃取。干燥(Na2SO4)合并的萃取液,过滤并在真空下浓缩。在硅胶上层析残余物,用CH2Cl2∶MeOH(9∶1)洗脱,得到小标题化合物150mg(18%),为白色固体。
1H-NMR(300MHz;CD3OD):δ7.74-7.86(m,2H),7.10-7.45(m,6H),5.10-5.15(m,2H),4.70-4.81(m,1H),3.90-4.44(m,6H),2.50(s,3H),2.10-2.32(m,2H),1.02-1.18(m,2H),0.10(s,9H)。
API-MS:(M+1)557m/z
(viii)Ph(3-SMe)-(R)或-(S)CH(OH)C(O)-Aze-PabxTFA
将Ph(3-SMe)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(80mg,0.19mmol;参见以上步骤(vii))和TFA(2.0mL)的CH2Cl2(2mL)中的混合物在0℃下搅拌3小时。将该溶液在真空下浓缩,将残余物溶于水中并冻干,得到标题化合物90mg(87%)。
LC-MS:(M+1)413;(M-1)411m/z
1H-NMR(400MHz;CD3OD;旋转异构体的混合物):δ7.74(m,2H),7.52(m,2H),7.38-7.13(m,4H),5.2-5.0(m,1H),4.79(m,1H),4.62-3.94(m,4H),2.68,2.49(2m,1H),2.28,2.14(2m,1H),2.45(s,3H)。
13C NMR(100MHz):δ185.0,172.8,171.8,167.0。
实施例4
Ph(3-SO2Me)-(R)或-(S)CH(OH)C(O)-Aze-PabxTFA
(i)Ph(3-SO2Me)-(R)或-(S)CH(OH)C(O)OH
将Ph(3-SMe)-(R)或-(S)CH(OH)C(O)OH(890mg,4.49mmol;参见以上实施例3(iii)(a))和Oxone(8.3g,13.5mmol)的MeOH(40mL)和H2O(25mL)的混合物在0℃下搅拌,然后在25℃下搅拌过夜。将固体滤出并用EtOAc(200mL)洗涤。在真空下浓缩滤液,用H2O(50mL)稀释,然后用EtOAc(4×60mL)萃取。干燥(Na2SO4)合并的有机萃取液,过滤并在真空下浓缩。在硅胶上层析残余物,用CHCl3∶MeOH∶NH3(饱和水溶液)(6∶3∶1)洗脱,得到小标题化合物150mg(15%),为白色固体。
1H-NMR(300MHz;CD3OD):δ8.10(s,1H),7.80-7.88(m,2H),7.55(t,J=7.5Hz,1H),5.02(s,1H),3.10(s,3H)。
13C NMR(75MHz;CD3OD):δ178.4,145.6,142.2,133.2,130.3,127.4,126.2,75.5,42.4。
HPLC分析:94.8%,>99%ee
[α]D 25=-86.2°(c=1.0,MeOH)
API-MS:(M-1)229m/z
(ii)Ph(3-SO2Me)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)
将Ph(3-SO2Me)-(R)或-(S)CH(OH)C(O)OH(400mg,1.74mmol;参见以上步骤(i))、H-Aze-Pab(Teoc)(720mg,1.91mmol;参见以上实施例3(vi))、PyBOP(995mg,1.91mmol)和2,4,6-可力丁(463mg,3.83mmol)的DMF(10mL)中的混合物在0℃下搅拌,然后在25℃下搅拌过夜。用H2O(50mL)猝灭该混合物并用EtOAc(3×50mL)萃取。干燥(Na2SO4)合并的萃取液,过滤,然后在真空下浓缩。在硅胶上层析残余物,用CHCl3∶MeOH(15∶1)洗脱,得到小标题化合物570mg(57%),为白色固体。
1H-NMR(300MHz;CD3OD):δ7.58-8.10(m,6H),7.40-7.50(m,2H),5.32(s,1H),5.25(s,1H),4.70-4.81(m,1H),3.97-4.54(m,6H),3.20(s,3H),2.10-2.82(m,2H),1.02-1.18(m,2H),0.10(s,9H)。
API-MS:(M+1)589m/z
(iii)Ph(3-SO2Me)-(R)或-(S)CH(OH)C(O)-Aze-PabxTFA
向Ph(3-SO2Me)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(65mg,0.11mmol;参见以上步骤(ii))的二氯甲烷(0.5mL)中的冷溶液中加入TFA(3mL)并将该溶液搅拌100分钟。浓缩生成物溶液,加入水并将含水溶液冻干,得到标题化合物60mg(96%)。
LC-MS:(M+1)445;(M-1)443m/z
1H-NMR(400MHz;CD3OD):δ8.10-7.45(m,8H),5.34,5.25(2m,1H),4.81(m,1H),4.62-3.93(m,4H),3.10(s,3H),2.70,2.54(m,1H),2.28,2.17(m,1H)。
13C NMR(羰基和/或脒碳;100MHz):δ172.2,171.7,167.0,161.0。
实施例5
Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-PabxTFA
(i)Ph(3-Cl,5-NO2)-(R,S)CH(OTMS)CN
向3-氯代-5-硝基苯甲醛(24.1g,0.13mol)的CH2Cl2(1.0L)中的溶液中加入ZnI2(2.1g,6.5mmol)。将得到的悬浮液冷却至0℃并用5分钟加入三甲基甲硅烷基氰化物(13.9g,0.14mol)。将该溶液在0℃下搅拌3小时,温热至25℃并搅拌18小时。用H2O稀释反应物并将有机物分离,干燥(Na2SO4),过滤,然后在真空下浓缩,得到为油状物的小标题化合物36.8g(99%)。
1H-NMR(300MHz;CDCl3):δ8.21-8.29(m,2H),7.83(s,1H),5.59(s,1H),0.36(s,9H),
(ii)Ph(3-Cl,5-NO2)-(R,S)CH(OH)C(O)OH
将Ph(3-Cl,5-NO2)(R,S)CH(OTMS)CN(59.0g,0.209mol;参见以上步骤(i))的浓HCl(600mL)中的溶液加热至回流3小时。将该溶液冷却并在真空下浓缩至500mL。用Et2O(4x)萃取该酸性溶液,用盐水(2x)洗涤有机物,干燥(Na2SO4),过滤,然后在真空下浓缩,得到小标题化合物48.4g(93%),为固体,没有进一纯化而使用。
1H-NMR(300MHz,CD3OD):δ8.33(m,1H),8.23(m,1H),7.94(m,1H),5.34(s,1H)。
(iii)Ph(3-Cl,5-NO2)-(R)或-(S)CH(OH)C(O)OH(a)和Ph(3-Cl,5-NO2)-(S)或-(R)CH(OAc)C(O)OH(b)
将Ph(3-Cl,5-NO2)-(R,S)CH(OH)C(O)OH(17.1g,73.84mmol;参见以上步骤(ii))和脂酶PS Amano(8.5g)的乙酸乙烯酯(300mL)和MTBE(300mL)中的混合物在55℃下搅拌24小时。将反应物经Celite过滤并用Et2O洗涤滤饼。在真空下浓缩滤液,然后在硅胶上快速层析,用CHCl3∶CH3CN∶TFA(180∶20∶1)洗脱,得到为固体的小标题化合物(a)7.1g(42%)和为固体的小标题化合物(b)10.7g(52%)。
对于小标题化合物(a):
1H NMR(300MHz;CD3OD):δ8.33(s,1H),8.22(s,1H),7.95(s,1H),5.34(s,1H)。
13C NMR(75MHz;CD3OD):δ174.6,150.2,145.2,136.3,133.8,124.1,121.1,72.7。
API-MS:(M-1)230m/z
[α]D 25=-101.2°(c=1.0,MeOH)
HPLC分析:99.6%,99%ee
对于小标题化合物(b):
1H NMR(300MHz;CD3OD):δ8.32(m,1H),8.28(m,1H),7.96(m,1H),6.10(s,1H),2.21(s,3H)。
(iv)Ph(3-Cl,5-NH2)-(R)或-(S)CH(OH)C(O)OH
将Ph(3-Cl,5-NO2)-(R)或-(S)CH(OH)C(O)OH(3.9g,16.8mmol;参见以上步骤(iii)(a))和氧化铂(IV)(0.4g)的EtOH(200mL)中的混合物在40℃、氢气氛下搅拌4小时。将混合物经Celite的滤垫过滤并用EtOH洗涤滤饼。在真空下浓缩滤液,得到小标题化合物3.5g(约100%),为可压碎的泡沫状物,其没有进一步纯化而使用。
1H NMR(300MHz;CD3OD):δ6.77(m,1H),6.71(m,1H),6.57(m,1H),4.78(s,1H)。
(v)Ph(3-Cl,5-NHMe)-(R)或-(S)CH(OH)C(O)OH
方法A:
将Ph(3-Cl,5-NH2)-(R)或-(S)CH(OH)C(O)OH(3.5g,16.8mmol;参见以上步骤(iv)和甲醛(1.8mL 37重量%水溶液,23.9mmol)的EtOH(400mL)中的混合物在25℃下搅拌18小时。在真空下浓缩该溶液,得到可压碎的泡沫状物,将其与氧化铂(IV)(0.35g)在EtOH(400mL)中混合并在氢气氛下搅拌48小时。将该混合物经Celite的滤垫过滤并用EtOH洗涤滤饼。在真空下浓缩有机物并在硅胶上快速层析,用CHCl3∶MeOH∶NH3(饱和的水溶液)(14∶5∶1)洗脱,得到小标题化合物的铵盐1.0g(28%),为可压碎的泡沫状物。通过用CH3CN∶MeOH(3∶1)冲洗相应的铵盐经过AmberliteCG-50的滤垫,得到小标题化合物。
方法B:
将Ph(3-Cl,5-NH2)-(R)或-(S)CH(OH)C(O)OH(8.67g,43.0mmol;参见以上步骤(iv))和甲基碘(6.10g,43.0mmol)的CH3CN(500mL)和MeOH(100mL)中的混合物加热至50℃24小时。在真空下浓缩该溶液,在硅胶上快速层析,用CHCl3∶MeOH∶NH3(饱和的水溶液)(14∶5∶1)洗脱,得到小标题化合物的铵盐2.9g(31%),为固体。通过用CH3CN∶MeOH(3∶1)冲洗相应的铵盐经过AmberliteCG-50的滤垫,得到小标题化合物。
1H NMR(300MHz;CD3OD):δ6.68(m,1H),6.61(m,1H),6.50(m,1H),4.98(s,1H),2.75(s,3H)。
13C NMR(75MHz;CD3OD):δ176.8,153.4,144.1,136.7,116.3,113.2,111.0,74.7,31.3。
API-MS:(M+1)216m/z
HPLC分析:97.2%,97.9%ee
[α]D 25=-81.6°(c=1.0,MeOH)
(vi)Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)OH
将Ph(3-Cl,5-NHMe)-(R)或-(S)CH(OH)C(O)OH(1.0g,4.64mmol;参见以上步骤(v))的MeOH(100mL)中的溶液,用72小时内,用四部分乙酐(每一部分40.47g,4.64mmol)处理。用2NNaOH使该溶液成碱性,搅拌3小时,用2N HCl中和,然后在真空下浓缩。在硅胶上快速层析(2x),用CHCl3∶MeOH∶NH3(饱和的水溶液)(6∶3∶1)洗脱,得到小标题化合物的铵盐0.83g(69%)为可压碎的泡沫状物。通过用CH3CN∶MeOH(3∶1)冲洗相应的铵盐经过AmberliteCG-50的滤垫,得到小标题化合物。
1H NMR(300MHz;CD3OD):δ7.54(s,1H),7.35(s,2H),5.19(s,1H),3.26(s,3H),1.88(s,3H)。
13C NMR(75MHz;CD3OD):δ175.3,172.8,146.8,145.2,136.2,128.0,127.5,125.4,73.2,37.6,22.5。
API-MS:(M+1)258m/z
HPLC分析:98.5%,97.4%ee
[α]D 25=-97.5°(c=1.0,MeOH)
(vii)Ph(3-Cl,5-NMeAc)(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)
在0℃下,向Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)OH(0.34g,1.32mmol;参见以上步骤(vi))和H-Aze-Pab(Teoc)(0.52g,1.39mmol;参见以上实施例3(vi))在DMF(15mL)中的混合物中加入可力丁(0.35g,2.90mmol)和PyBOP(0.75g,1.45mmol)。在0℃下将该溶液搅拌2小时,加热至25℃并搅拌2小时,然后在真空下浓缩。在硅胶上快速层析(2x),用CHCl3∶EtOH(95∶5)洗脱,得到小标题化合物0.36g(44%),为可压碎的泡沫状物。
1H NMR(300MHz;CD3OD;旋转异构体的混合物):δ7.78(d,2H,J=9Hz),7.25-7.55(m,5H),5.25和4.78(2m,1H),5.22和5.15(2s,1H),3.93-4.56(m,6H),3.23(s,3H),2.12-2.78(m,2H),1.87(s,3H),1.04-1.11(m,1H),0.06(s,9H)。
API-MS:(M+1)616m/z
(viii)Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-PabxTFA
将Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(73mg,0.12mmol;参见以上步骤(vii))的TFA(5.0mL)中的溶液在室温下搅拌80分钟,此后将得到的溶液蒸发至干。将残余的固体溶在水中并将溶液冻干,得到标题化合物70mg(98%),为泡沫状物。
LC-MS:(M+1)472m/z
1H-NMR(400MHz;D2O):δ7.74(dd,2H),7.55-7.10(m,5H),5.36,5.20(2s,1H),5.23,4.88(2m,1H),4.60-4.05(m,4H),3.38,3.20(2s,3H),2.80,2.60(2m,1H),2.38-2.20(m,1.5H),1.87(2.5H)。
13C NMR(羰基和/或脒碳;100MHz):δ173.9,173.3,172.6,166.5,163.3。
实施例6
Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pabx2TFA
(i)3-氯代-5-N,N-二甲基氨基苯甲醇
向3-氯代-5-硝基苯甲醇(12.5g,66.6mmol)的EtOH(750mL)中的溶液中加入氧化铂(IV)(1.25g)。将得到的悬浮液用氢气清洗3小时。加入甲醛溶液(37重量%,在H2O中,97ml,1.3mol)并在氢气氛下将混合物搅拌18小时。将溶液经Celite的滤垫过滤并在真空下浓缩,得到粗制产物。在硅胶上快速层析,用己烷∶EtOAc(7∶3)洗脱,得到小标题化合物8.2g(66%),为油状物。
1H NMR(300MHz;CDCl3):δ6.67(s,1H),6.55-6.63(m,2H),4.58(d,2H,J=7Hz),2.96(s,6H),1.74(t,1H,J=7Hz)。
CI-MS:(M+1)185m/z
(ii)3-氯代-5-N,N-二甲基氨基苯甲醛
在-78℃下用10分钟时间,向DMSO(7.58g,97.0mmol)的CH2Cl2(100mL)溶液中加入草酰氯(6.16g,48.5mmol)。在-78℃下另外15分钟后,用15分钟加入3-氯代-5-N,N-二甲基氨基苯甲醇(8.18g,44.1mmol;参见以上步骤(i))的CH2Cl2(100mL)中的溶液。将得到的溶液在-78℃下搅拌1小时,然后加入DIPEA(28.5g,220.5mmol)。将该溶液温热到25℃并搅拌18小时,然后在真空下浓缩,得到粗制产物。在硅胶上快速层析,用己烷∶EtOAc(5∶1)洗脱,得到小标题化合物7.50g(93%),为黄色固体。
1H NMR(300MHz;CDCl3):δ9.88(s,1H),7.15(m,1H),7.05(m,1H),6.87(m,1H),3.04(s,6H)。
(iii)Ph(3-Cl,5-NMe2)-(R,S)CH(OTMS)CN
向3-氯代-5-N,N-二甲基氨基苯甲醛(7.5g,40.8mmol;参见以上步骤(ii))的CH2Cl2(300mL)溶液中加入ZnI2(0.65g,2.04mmol)。将得到的悬浮液冷却至0℃并用5分钟加入三甲基甲硅烷基氰化物(4.5g,44.9mmol)。将该溶液在0℃下搅拌1小时,然后温热至25℃并搅拌2小时。用H2O稀释得到的混合物并分离有机物,干燥(Na2SO4),过滤,然后在真空下浓缩,得到小标题化合物11.7g(100%),为油状物。
1H NMR(300MHz;CDCl3):δ6.75(m,1H),6.60-6.68(m,2H),5.39(s,1H),2.97(s,6H),0.28(s,9H)。
(iv)Ph(3-Cl,5-NMe2)-(R,S)CH(OH)C(O)OH
将Ph(3-Cl,5-NMe2)-(R,S)CH(OTMS)CN(11.7g,41.4mmol;参见以上步骤(iii))溶于浓HCl(300mL)中并加热至回流1.5小时。将该溶液冷却并在真空下浓缩。将残余物溶于H2O中,用NaHCO3中和并在真空下浓缩。将有机物和盐的混合物在MeOH中成为淤浆,过滤,然后浓缩得到粗制产物。在硅胶上快速层析,用CHCl3∶MeOH∶浓NH4OH(水溶液)(6∶3∶1)洗脱,得到小标题化合物的铵盐9.0g(95%),为固体。
1H NMR(300MHz;CD3OD):δ6.77-6.82(m,2H),6.58(m,1H),4.80(s,1H),2.94(s,6H)。
(v)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)OH(a)和Ph(3-Cl,5-NMe2)-(S)或-(R)CH(OAc)C(O)OH(b)
将Ph(3-Cl,5-NMe2)-(R,S)CH(OH)C(O)OH(1.0g;参见以上步骤(iv))和脂酶PS Amano(0.5g)的乙酸乙烯酯(10mL)和MTBE(10mL)中的混合物在45℃下搅拌48小时。将反应物经Celite过滤并用MeOH洗涤滤饼。在真空下浓缩滤液并在硅胶上快速层析,用CHCl3∶MeOH∶NH3(饱和溶液)(6∶3∶1)洗脱,得到可压碎泡沫状物的小标题化合物(a)0.40g(40%)和可压碎泡沫状物的小标题化合物(b)0.45g(38%)。小标题化合物(a)可以从CH2Cl2和MeOH中结晶进一步纯化。对于小标题化合物(a):
1H NMR(300MHz;CD3OD):δ6.81(m,1H),6.74(m,1H),6.57(m,1H),4.98(s,1H),2.87(s,6H)。
13C NMR(75MHz;CD3OD):δ180.0,152.9,144.8,135.6,116.1,112.2,110.9,76.9,40.5。
API-MS:(M+1)230m/z
HPLC分析:98.5%,97.9%ee
[α]D 25=-73.5°(c=0.5,DMSO)
对于小标题化合物(b):
1H NMR(300MHz;CD3OD):δ6.77-6.83(m,2H),6.64(m,1H),5.67(s,1H),2.94(s,6H),2.14(s,3H)。
(vi)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)
在0℃下,向Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)OH(0.11g,0.48mmol;参见以上步骤(v)(a))和H-Aze-Pab(Teoc)(0.20g,0.53mmol;参见实施例3(vi))的DMF(15mL)中的混合物中加入DIPEA(0.12g,0.96mmol)和TBTU(0.17g,0.53mmol)。将该溶液在0℃下搅拌2小时,温热至25℃并搅拌18小时,然后在真空下浓缩。在硅胶上快速层析,用CH2Cl2∶MeOH(从100∶0到95∶5)梯度洗脱,得到0.25g小标题化合物,将该化合物在硅胶上第二次快速层析,用EtOAc∶MeOH(30∶1)洗脱,得到小标题化合物0.22g(78%),为可压碎的泡沫状物。
1H NMR(300MHz;CD3OD,旋转异构体的混合物):δ7.78(d,2H,J=9Hz),7.42(d,2H,J=9Hz),6.62-6.75(m,3H),5.14和4.78(2m,1H),5.07(m,1H),4.15-4.57(m,4H),3.94-4.12(m,2H),2.96(s,6H),2.05-2.75(m,2H),1.04-1.13(m,2H),0.08(s,9H)。
API-MS:(M+1)588m/z
(vii)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pabx2TFA
向Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(84mg,0.14mmol;参见以上步骤(vi))的冰冷的溶液中加入TFA(4mL),并将生成物溶液在0℃下搅拌2小时。将该溶液浓缩得到残余物,将其于水中,然后冻干。得到标题化合物78mg(81%),为白色粉末状物。
LC-MS:(M-1)442m/z
1H-NMR(400MHz;CD3OD;旋转异构体的混合物):δ7.78-7.49(m,4H),6.94-6.79(m,4H),5.15,5.08(m,1H),5.20,4.79(2m,1H),4.51(ABX光谱的AB部分,2H),4.41-3.95(m,2H),2.98(s,6H),2.69,2.52(2m,1H),2.28,2.14(2m,1H)。
13C NMR(羰基和/或脒碳;100MHz):δ172.5,171.7,166.9,161.0,160.7。
实施例7
Ph(3-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-PabxHOAc
(i)Ph(3-NO2)-(R)或-(S)CH(OH)C(O)OH(a)和Ph(3-NO2)-(S)或-(R)CH(OAc)C(O)OH(b)
将Ph(3-NO2)-(R,S)CH(OH)C(O)OH(25g,126mmol)、脂酶PSAmano(12.5g)、乙酸乙烯酯(150mL)和MTBE(375mL)的混合物在45℃下加热24小时。将反应物过滤并用EtOAc(500mL)洗涤滤饼。在真空下浓缩滤液并在硅胶上层析,用CHCl3∶MeOH∶NH3(饱和水溶液)(6∶3∶1)的混合物洗脱,得到黄色油状物的小标题化合物(a)9.0g(36%)和为褐色固体的小标题化合物(b)6.5g(21%)。
对于小标题化合物(a):
1H NMR(300MHz;CD3OD):δ8.34(s,1H),8.25(d,J=7.5Hz,1H),7.82(d,J=7.5Hz,1H),7.62(t,J=7.5Hz,1H),5.30(s,1H)。
对于小标题化合物(b):
1H NMR(300MHz;CD3OD):δ8.34(s,1H),8.25(d,J=7.5Hz,1H),7.82(d,J=7.5Hz,1H),7.62(t,J=7.5Hz,1H),5.82(s,1H),2.20(s,3H)。
(ii)Ph(3-NH2)-(R)或-(S)CH(OH)C(O)OH
将Ph(3-NO2)-(R)或-(S)CH(OH)C(O)OH(8.0g,40.6mmol;参见以上步骤(i)(a))和10%披钯碳(800mg)的MeOH(200mL)中的混合物在25℃、一个大气压的氢气氛下搅拌过夜。将该混合物经Celite的滤垫过滤,用EtOAc(250mL)洗涤。在真空下浓缩滤液,得到小标题化合物7.0g(100%),为白色泡沫状物。
1H NMR(300MHz;CD3OD):δ7.0-7.12(m,1H),6.75-6.90(m,2H),6.60-6.70(m,1H),4.80(s,1H)。
(iii)Ph(3-NHMe)-(R)或-(S)CH(OH)C(O)OH
将Ph(3-NH2)-(R)或-(S)CH(OH)C(O)OH(2.9g,17.3mmol;参见以上步骤(ii))和甲基碘(2.95g,20.8mmol)的MeOH(50mL)中的混合物在55℃下加热过夜。将反应混合物在真空下浓缩并在硅胶上层析,用CHCl3∶MeOH∶NH3(饱和水溶液)(6∶3∶1)洗脱,得到小标题化合物616mg(20%),为棕色油状物。
1H NMR(300MHz;CD3OD):δ7.00-7.12(m,1H),6.70-6.80(m,2H),6.50-6.55(m,1H),4.80(s,1H),2.80(s,3H)。
(iv)Ph(3-NMeAc)-(R)或-(S)CH(OH)C(O)OH
将Ph(3-NHMe)-(R)或-(S)CH(OH)C(O)OH(540mg,2.99mmol;参见以上步骤(iii))和乙酐(612mg,5.98mmol)的MeOH(15mL)中的混合物在25℃、氮气氛下搅拌过夜。将该混合物在真空下浓缩并在硅胶上层析,用CHCl3∶MeOH∶浓NH4OH(水溶液)(6∶3∶1)洗脱,得到小标题化合物380mg(57%),为白色泡沫状物。
1H NMR(300MHz;CD3OD):δ7.51-7.60(m,1H),7.38-7.49(m,2H),7.15-7.25(m,1H),5.04(s,1H),3.22(s,3H),1.85(s,3H)。
13C NMR(75MHz;CD3OD):δ178.2,173.6,145.8,142.8,131.5,127.8,126.5,126.2,75.5,37.8,22.5。
HPLC分析:95.7%,95.3%ee
[α]D 25=-4.32°(c=0.5,MeOH)
CI-MS:(M+1)224m/z
(v)Ph(3-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)
将Ph(3-NMeAc)-(R)或-(S)CH(OH)C(O)OH(301mg,1.35mmol;参见以上步骤(iv))、H-Aze-Pab(Teoc)(560mg,1.48mmol;参见以上实施例3(vi))、PyBOP(774mg,1.48mmol)和2,4,6-可力丁(360mg,2.97mmol)的DMF(10mL)中的混合物在0℃下搅拌,然后在25℃下搅拌过夜。将该混合物用H2O(50mL)猝灭并用EtOAc(3×50mL)萃取。干燥(Na2SO4)合并的有机萃取液,过滤并在真空下浓缩。将残余物在硅胶上层析,用CHCl3∶MeOH(9∶1)洗脱,得到小标题化合物175mg(23%),为白色固体。
1H NMR(CD3OD):δ7.82-7.90(m,2H),7.20-7.50(m,6H),5.32(s,1H),5.25(s,1H),4.70-4.81(m,1H),3.97-4.54(m,6H),3.20(s,3H),2.10-2.82(m,2H),1.85(s,3H),1.02-1.18(m,2H),0.10(s,9H)。
API-MS:(M+1)582m/z
(vi)Ph(3-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-PabxHOAc
将Ph(3-NMeAc)-(R,S)CH(OH)C(O)-Aze-Pab(Teoc)(65mg,0.11mmol;参见以上步骤(v))和TFA(2.0mL)的CH2Cl3(2mL)中的混合物在0℃下搅拌3小时。将该溶液在真空、室温下浓缩并用制备型HPLC(CH3CN:0.1M NH4OAc,梯度:0-50%CH3CN)纯化残余物,并将所需部分浓缩。将该产物溶于水/HOAc中并冻干,得到标题化合物55mg(100%)。
LC-MS:(M+1)438;(M-1)436m/z
1H-NMR(400MHz;D2O;旋转异构体的混合物):δ7.74(m,3H),7.61-7.20(m,5H),5.36,5.24(2m,1H),4.84(m,1H),4.58-3.94(m,4H),3.42-3.08(m,3H),2.80,2.57(2m,1H),2.36-1.98(m,4H),1.84(s,3H)。
13C NMR(100MHz):δ174.2,173.1,172.7,166.7。
实施例8
Ph(3-NMe2,5-CF3)-(R)或-(S)CH(OH)C(O)-Aze-PabxTFA
(i)Ph(3-NO2,5-CF3)CH2OH
在氮气氛下,用1小时将硼烷-四氢呋喃复合物(170mL的1M于THF中的溶液,170mmol)滴加入到Ph(3-NO2,5-CF3)CH2H(10.0g,42.6mmol)的THF(50mL)的冷却至0℃的溶液中。将该溶液温热至室温并搅拌4小时。经缓慢加入H2O将该溶液猝灭,倒入EtOAc(200mL)中,然后用H2O(150mL)和盐水(150mL)顺序洗涤。干燥(Na2SO4)有机相,过滤并在真空下浓缩,得到小标题化合物6.9g(73%),为橙色油状物。
1H NMR(300MHz;CDCl3):δ8.44(s,1H),8.46(s,1H),8.01(s,1H),4.92(d,J=5.5Hz,2H),2.10(br s,1H)。
(ii)Ph(3-NO2,5-CF3)-CHO
在氮气氛下,将草酰氯(3.0mL,34mmol)滴加入DMSO(4.86mL,68.6mmol)的冷却至-78℃的70mL干燥的CH2Cl2溶液中。在-78℃下15分钟后,用30分钟滴加入Ph(3-NO2,5-CF3)CH2OH(6.9g,31mmol;参见以上步骤(i))在75ml CH2Cl2中的溶液。在-78℃下45分钟后,用20分钟加入DIPEA(27.2mL,156mmol)。在-78℃下将该溶液再搅拌1小时,同时,将该溶液温热至室温并搅拌15小时。用1M HCl(2×150mL)、盐水(150mL)顺序洗涤所述溶液,干燥(Na2SO4),过滤并在真空下浓缩,得到小标题化合物6.9g(99%),为橙色油状物。
1H NMR(300MHz;CDCl3):δ10.19(s,1H),8.94(s,1H),8.76(s,1H),8.51(s,1H)。
(iii)Ph(3-NO2,5-CF3)-(R,S)CH(OTMS)CN
向Ph(3-NO2,5-CF3)-CHO(6.52g,29.7mmol;来自以上步骤(ii))的220mL CH2Cl2中的溶液中加入ZnI2(474mg,1.49mmol)。用氮气清洗该溶液并冷却至0℃。用10分钟加入三甲基甲硅烷基氰化物(3.25g,32.7mmol),此后将该溶液搅拌2小时。然后将该溶液温热至室温并再搅拌5.5小时,此后用H2O(250mL)将反应物猝灭。分离有机相并用CH2Cl2(125mL)萃取含水相。干燥(Na2SO4)合并的有机层,过滤并在真空下浓缩,得到小标题化合物9.1g(96%),为橙色油状物。
1H NMR(300MHz;CDCl3):δ8.64(s,1H),8.58(s,1H),8.23(s,1H),6.14(s,1H),0.80(s,9H)。
(iv)Ph(3-NO2,5-CF3)-(R,S)CH(OH)C(O)OH
将Ph(3-NO2,5-CF3)-(R,S)CH(OTMS)CN(9.1g,29mmol;参见以上步骤(iii))溶于浓HCl(83mL,1000mmol)中并加热至回流3小时。将该溶液用H2O(200mL)稀释并用Et2O(3×150mL)萃取。用盐水(200mL)洗涤合并的有机物,干燥(Na2SO4),过滤并在真空下浓缩,得到棕色油状物。将粗制产物在硅胶上快速层析,用CHCl3∶MeOH∶NH3(饱和水溶液)(14∶5∶1)洗脱。将得到的白色固体悬浮在Et2O中并加入2M HCl(100mL)。分离各层并用Et2O(3×200mL)萃取含水层。干燥(Na2SO4)合并的有机层,过滤并浓缩,得到小标题化合物5.9g(78%),为棕色固体。
1H NMR(300MHz;CD3OD):δ8.65(s,1H),8.47(s,1H),8.23(s,1H),5.43(s,1H)。
(v)Ph(3-NH2,5-CF3)-(R,S)CH(OH)C(O)OH
向Ph(3-NO2,5-CF3)-(R,S)CH(OH)C(O)OH(5.9g,22mmol;参见以上步骤(iv))的无水EtOH(350mL)中的溶液中加入氧化铂(IV)(590mg)。将该溶液用氢气清洗5小时,此后将混合物经Celite过滤,然后在真空下浓缩,得到小标题化合物5.8g(100%),为橙色油状物。
1H NMR(300MHz;CD3OD):δ7.00(s,2H),6.86(s,1H),5.06(s,1H)。
(vi)Ph(3-NMe2,5-CF3)-(R,S)CH(OH)C(O)OH
向溶于无水EtOH(250mL)中的Ph(3-NH2,5-CF3)-(R,S)CH(OH)C(O)OH(5.27g,22.4mmol;参见以上步骤(v))的溶液中加入37%甲醛水溶液(54mL,720mmol)。加入氧化铂(IV)(520mg)并用氢气清洗该溶液。在氢气氛下搅拌22小时后将该溶液经Celite过滤并在真空下浓缩。在硅胶上快速层析,用CHCl3∶MeOH∶NH3(饱和水溶液)(6∶3∶1)洗脱,得到小标题化合物2.7g(46%),为白色固体。
1H NMR(300MHz;CD3OD):δ7.10(s,1H),7.07(s,1H),6.80(s,1H),4.88(s,1H),2.98(s,6H)。
(vii)Ph(3-NMe2,5-CF3)-(R)或-(S)CH(OH)C(O)OH(a)和Ph(3-NMe2)(5-CF3)-(S)或-(R)CH(OAc)C(O)OH(b)
将Ph(3-NMe2,5-CF3)-(R,S)CH(OH)C(O)OH(2.7g,10mmol;参见以上步骤(vi))、脂酶PS Amano(1.4g)、乙酸乙烯酯(56mL)和MTBE(120mL)的混合物回流1天。将反应物经Celite过滤并用Et2O洗涤滤饼。在真空下浓缩滤液并在硅胶上快速层析,用CHCl3∶MeOH∶NH3(饱和水溶液)(14∶5∶1)洗脱,得到为白色固体的小标题化合物(a)的铵盐727mg(27%)和为白色固体的小标题化合物(b)的铵盐1.53g(49%)。将小标题化合物(a)的铵盐流过AmberliteCG-50滤垫,用CH3CN∶MeOH(3∶1)作为洗脱剂,得到小标题化合物(a),为白色固体。
对于小标题化合物(a):
1H NMR(300MHz;CD3OD):δ7.03-7.09(m,2H),6.79(s,1H),4.95(s,1H),2.88(s,6H)。
13C NMR(75MHz;CD3OD):δ180.2,152.9,146.0,133.0(q,J=32.2Hz),125.2(t,J=284.0Hz),116.3,113.4,109.3,77.4,41.4。
HPLC分析:98.8%,>99%ee
[α]D 25=-59.5°(c=1.0,MeOH)
API-MS:(M+1)264m/z
对于小标题化合物(b):
1H NMR(300MHz;CD3OD):δ7.12(s,1H),7.09(s,1H),6.83(s,1H),5.73(s,1H),3.00(s,6H),2.14(s,3H)。
(viii)Ph(3-NMe2,5-CF3)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)
向Ph(3-NMe2,5-CF3)-(R)或-(S)CH(OH)C(O)OH(290mg,1.10mmol;参见以上步骤(vii)(a))和H-Aze-Pab(Teoc)(436mg,1.16mmol;参见实施例3(vi))的混合物中加入10mL干燥的DMF。将该溶液冷却至0℃,然后加入PyBOP(630mg,1.21mmol)和可力丁(295mg,2.42mmol)将该溶液在氮气氛、0℃下搅拌2小时并在室温下搅拌15小时。将混合物浓缩并在硅胶上快速层析,用EtOAc∶EtOH(20∶1)洗脱,得到小标题化合物383mg(56%),为白色固体。
1H NMR(300MHz;CD3OD):δ7.76-7.83(d,J=8.0Hz,2H),7.38-7.44(d,J=8.0Hz,2H),7.12(m,2H),6.86-6.87(m,1H),5.15-5.17(m,1H),4.75-4.81(m,1H),3.98-4.56(m,6H),3.00(s,6H),2.48-2.58(m,1H),2.24-2.33(m,1H),1.03-1.13(m,2H),0.08(s,9H)。
API-MS:(M+1)622m/z
(ix)Ph(3-NMe2,5-CF3)-(R)或-(S)CH(OH)C(O)-Aze-PabxTFA
向Ph(3-NMe2,5-CF3)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(87mg,0.14mmol;参见以上步骤(viii))的二氯甲烷冰冷的溶液中加入TFA(4 mL)并将该混合物在0℃下搅拌100分钟。将生成物溶液浓缩至干燥,将得到的残余物溶于水/CH3CN中,然后冻干,得到标题化合物81mg(80%),为白色粉末状物。
LC-MS:(M+1)478;(M-1)476m/z
1H-NMR(400MHz;CD3OD;旋转异构体的混合物):δ7.78-7.50(m,4H),7.09-7.04(m,2H),6.92(br s,1H),5.21,5.17(2s,1H),4.80(m,1H),4.52(ABX光谱的AB部分;2H),4.41-3.95(m,2H),3.00(s,3H),2.70,2.52(2m,1H),2.30,2.15(2m,1H)。
13C NMR(羰基和/或脒碳;100MHz):δ172.6,171.7,167.0,161.5,161.2。
实施例9
Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-PabxHOAc
(i)(R,S)-5-Ph(3-Cl,5-NO2)-2,2-二甲基-4-氧代-1,3-二氧戊环
向Ph(3-Cl,5-NO2)-(R,S)CH(OH)C(O)OH(18.8g,81.2mmol;参见以上实施例5(ii))的丙酮(300mL)中加入对-甲苯磺酸一水合物(750mg,3.94mmol)和2,2-二甲氧基丙烷(75mL,514mmol)。将该溶液回流6小时并在真空下浓缩。将残余物溶于EtOAc(200mL)中,然后用H2O(100mL)、饱和NaHCO3(150mL)和盐水(150mL)洗涤。干燥(Na2SO4)有机相,过滤并浓缩,得到棕色固体,将其在硅胶上快速层析,用己烷∶EtOAc(7∶3)洗脱。将得到的固体从EtOAc/己烷(1∶10)中重结晶进一步纯化,得到小标题化合物14.7g(67%),为白色固体。
1H NMR(300MHz;CDCl3):δ8.29(m,1H),8.24(m,1H),7.86(m,1H),5.45(s,1H),1.78(s,3H),1.72(s,3H)。
(ii)(R,S)-5-Ph(3-Cl,5-NH2)-2,2-二甲基-4-氧代-1,3-二氧戊环
向(R,S)-5-Ph(3-Cl,5-NO2)-2,2-二甲基-4-氧代-1,3-二氧戊环(14.7g,54.1mmol;参见以上步骤(i))的EtOH(400mL)溶液中加入氧化铂(IV)(1.5g)。在室温、一个大气压的氢气下将该悬浮液搅拌27小时。将该悬浮液经Celite过滤并用EtOH洗涤滤饼。在真空下浓缩滤液,得到黄色油状物,将其在硅胶上快速层析,用己烷∶EtOH(4∶1)洗脱,得到小标题化合物6.5g(50%),为黄色油状物。
1H NMR(300MHz;CDCl3):δ6.76(m,1H),6.56(m,2H),5.18(s,1H),3.74(br s,2H),1.64(s,3H),1.68(s,3H)。
(iii)(R,S)-5-Ph(3-Cl,5-(1-吡咯烷基-2-酮))-2,2-二甲基-4-氧代-1,3-二氧戊环
向(R,S)-5-Ph(3-Cl,5-NH2)-2,2-二甲基-4-氧代-1,3-二氧戊环(6.5g,26.9mmol;参见以上步骤(ii))的DMF(100mL)溶液中加入4-溴代丁酸乙酯(10.5g,53.8mmol)和Et3N(5.4g,53.8mmol)。在氩气氛下将该溶液在95℃下加热21小时。浓缩反应混合物,然后溶于EtOAc(200mL)中,用H2O(150mL)和盐水(150mL)洗涤得到的溶液。干燥(Na2SO4)有机相,过滤并浓缩,得到橙色油状物9.6g。将粗制产物溶于对-二甲苯(250mL)中并于回流下加热。三天后,浓缩混合物得到橙色油状物并在硅胶上快速层析,用EtOAc∶己烷(1∶1)洗脱,得到小标题化合物4.5g(54%),为黄色固体。
1H NMR(300MHz;CDCl3):δ7.74(m,1H),7.69(m,1H),7.26(m,1H),5.38(s,1H),3.80-3.93(m,2H),2.60-2.68(t,J=7.5Hz,2H),2.15-2.25(m,2H),1.77(s,3H),1.70(s,3H)。
(iv)Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R,S)CH(OH)C(O)OH
向(R,S)-5-Ph(3-Cl,5-(1-吡咯烷-2-酮))-2,2-二甲基-4-氧代-1,3-二氧戊环(4.5g,14.5mmol;参见以上步骤(iii))的THF(300mL)中的溶液中加入1N NaOH(145mL)。将该溶液搅拌30分钟,然后在真空下将生成物溶液部分还原。用2N HC1将该溶液酸化并用EtOAc(2×150mL)萃取。用盐水(200mL)洗涤有机相,干燥(Na2SO4),过滤并浓缩,得到小标题化合物3.2g(82%),为白色固体。
1H NMR(300MHz;CD3OD):δ7.81(m,1H),7.59(m,1H),7.30(m,1H),5.15(s,1H),3.89-3.96(t,J=7.5Hz,2H),2.57-2.65(t,J=7.5Hz,2H),2.12-2.22(m,2H)。
(v)Ph(3-Cl,5-(1-吡咯烷-2-酮))-(S)或-(R)-CH(OAc)C(O)OH(b)和Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)或-(S)-CH(OH)C(O)OH(a)
将Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R,S)CH(OH)C(O)OH(3.2g,11.9mmol;参见以上步骤(iv))和脂酶PS Amano(1.6g)的乙酸乙烯酯(65mL)和MTBE(130mL)中的混合物在55℃下搅拌24小时。将反应物经Celite过滤并用THF,然后用MeOH顺序洗涤滤饼。在真空下浓缩滤液并在硅胶上快速层析,用CHC3∶MeOH∶NH3(饱和水溶液)(14∶5∶1)洗脱,得到为白色固体的小标题化合物(b)的铵盐1.3g(33%)。另外得到小标题化合物(a)的铵盐800mg(20%)。将该物质溶于H2O(40mL)中,用1N HCl酸化并用EtOAc(2×150mL)萃取。干燥(Na2SO4)有机相,过滤并浓缩,得到小标题化合物(a),为白色固体。由于低光学纯度,使小标题化合物(b)再经历以上酶催化析解条件(0.5g脂酶PS Amano;35mL乙酸乙烯酯;60mL MTBE;55℃;24小时)。如上所述分离和纯化得到小标题化合物(a)470mg,为白色固体。
对于小标题化合物(a):
1H NMR(300MHz;CD3OD):δ7.80(m,1H),7.59(m,1H),7.29(m,1H),5.15(s,1H),3.88-3.92(t,J=7.1Hz,2H),2.57-2.62(t,J=8.1Hz,2H),2.11-2.21(m,2H)。
13C NMR(75MHz;CD3OD):δ179.7,177.8,146.1,144.4,137.9,126.3,123.5,120.2,75.9,52.7,36.0,21.2。
API-MS:(M+1)270m/z
HPLC分析:95.3%,96.5%ee
[α]D 25=-64.5°(c=1.0,MeOH)
(vi)Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)-或(S)-CH(OH)C(O)-Aze-Pab(Teoc)
在0℃下,向Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)-或-(S)-CH(OH)C(O)OH(250mg,0.927mmol;来自以上步骤(v)(a))和H-Aze-Pab(Teoc)(367mg,0.973mmol;参见实施例3(vi))的DMF(9mL)中的混合物中加入PyBop(531mg,1.02mmol)和可力丁(250mg,2.04mmol)。在氮气氛、0℃下将该溶液搅拌2小时,然后温热至室温15小时。将该混合物浓缩并在硅胶上快速层析,用EtOAc∶EtOH(20∶1)洗脱,随后经EtOH冲洗柱,得到白色固体。进一步在硅胶上快速层析,用CHCl3∶EtOH(9∶1)洗脱,得到小标题化合物420mg(72%),为白色固体。
1H NMR(300MHz;CD3OD):δ7.73-7.85(m,3H),7.51-7.65(m,1H),7.36-7.47(m,2H),7.22-7.31(m,1H),5.11-5.23(m,1H),4.76-4.86(m,1H),3.95-4.55(m,6H),3.84-3.94(t,J=7.5Hz,2H),2.46-2.74(m,3H),2.08-2.47(m,3H),1.02-1.14(m,2H),0.09(s,9H)。
API-MS:(M+1)629m/z
(vii)Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-PabxHOAc
向Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R或S)CH(OH)C(O)-Aze-Pab(Teoc)(90mg,0.14mmol;参见以上步骤(vi))的二氯甲烷(0.5mL)中的溶液中加入TFA(4mL)。在室温下将该混合物搅拌100分钟。在真空下浓缩生成物溶液并用PHPLC(CH3CN∶0.1M乙酸铵20∶80)纯化固体粗制产物。合并所需部分并冻干两次过夜。产量51mg(67%)。纯度99.8%。
LC-MS:(M+1)484,486m/z
1H NMR(400MHz;CD3OD):δ7.73(m,3H),7.62(m,1H),7.52(m,2H),7.28,7.23(2s,1H),5.19(s,1H),4.80(dd,1H),4.51(ABX光谱的AB部分;2H),4.38(m,1H),4.19(m,1H),4.02(m,1H),3.88(t,2H),2.61-2.48(m,3H),2.29(m,1H),2.14(m,2H),1.90(s,3H)。
13C NMR(100MHz)(羰基和/或脒碳):δ176.0,172.3,171.7,167.0。
实施例10
Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-PabxHOAc
(i)(R,S)-5-Ph(3-NO2)-2,2-二甲基-4-氧代-1,3-二氧戊环
将间-硝基扁桃酸(6.0g,30.4mmol)、2,2-二甲氧基丙烷(15.1mL)、对-甲苯磺酸一水合物(0.29g,1.52mmol)和丙酮(60mL)的混合物在室温下搅拌12小时。将该混合物在真空下浓缩并将粗制产物溶于EtOAc中用饱和NaHCO3水液和盐水顺序洗涤有机相,干燥(MgSO4),然后在真空下浓缩。将残余物在硅胶上层析,用庚烷/EtOAc(80∶20-70∶30)洗脱,得到小标题化合物5.7g(79%)。(该粗制产物很难溶于少量EtOAc中,所以层析柱的上样是通过使用已吸附所述样品的硅胶来完成的。)
FAB-MS:(M+1)238m/z
1H NMR(400MHz;CDCl3):δ8.36(br s,1H),8.22(dd,1H),7.84(dd 1H),7.60(dd,1H),5.44(s,1H),1.76(s,3H),1.71(s,3H)。
(ii)(R,S)-5-Ph(3-NH2)-2,2-二甲基-4-氧代-1,3-二氧戊环
将(R,S)-5-Ph(3-NO2)-2,2-二甲基-4-氧代-1,3-二氧戊环(3.1g,13.1mmol;参见以上步骤(i))、5%Pd/C(1.7g)和HOAc(0.75mL,13.1mmol)在EtOH(250mL)中的混合物在氢气氛下搅拌4小时。将该混合物经Celite的滤垫过滤并用EtOH洗涤滤饼。在真空下浓缩滤液并将形成的无色固体分配在EtOAc和饱和NaHCO3水溶液之间。用EtOAc萃取水相并用盐水洗涤合并的有机相,干燥(Na2SO4)并在真空下浓缩,得到小标题化合物2.3g(85%)。
LC-MS:(M+1)208m/z
1H NMR(400MHz;CDCl3):δ7.16(dd,1H),6.84(dd,1H),6.76(br s,1H),6.67(dd,1H),5.30(s,1H),1.70(s,3H),1.65(s,3H)。
(iii)(R,S)-5-Ph(3-NH(CH2)3C(O)OEt)-2,2-二甲基-4-氧代-1,3-二氧戊环
将(R,S)-5-Ph(3-NH2)-2,2-二甲基-4-氧代-1,3-二氧戊环(1.63g,7.87mmol;参见以上步骤(ii))、4-溴代丁酸乙酯(3.4mL,23.6mmol)和Et3N(3.3mL,23.6mmol)的CH2Cl2中的混合物回流过夜。加入附加量的4-溴代丁酸乙酯(2.3mL,15.7mmol)和Et3N(2.2mL,15.7mmol)并将该混合物再回流一夜。将溶剂除去并将粗制产物分配在EtOAc和水之间。用EtOAc萃取水相并用盐水洗涤合并的有机相,干燥(Na2SO4),然后在真空下浓缩。将其残余物在硅胶上层析,用庚烷∶EtOAc(90∶10-80∶20)洗脱,得到小标题化合物2.1g(84%)。
FAB-MS:(M+1)322m/z
1H NMR(400MHz;CDCl3):δ7.17(dd,1H),6.77(br d,1H),6.66(br s,1H),6.59(dd,1H),5.30(s,1H),4.12(q,2H),3.16(t,2H),2.40(t,2H),1.93(m,2H),1.70(s,3H),1.64(s,3H),1.24(t,2H)。
(iv)(R,S)-5-Ph(3-(1-吡咯烷-2-酮))-2,2-二甲基-4-氧代-1,3-二氧戊环
将(R,S)-5-Ph(3-NH(CH2)3C(O)OEt)-2,2-二甲基-4-氧代-1,3-二氧戊环(2.2g,6.85mmol;参见以上步骤(iii))的甲苯(15mL)中的溶液回流两夜。除去溶剂并将粗制产物经快速层析,用庚烷∶EtOAc(80∶20-60∶40)洗脱,得到小标题化合物1.4g(74%)。
1H NMR(400MHz;CDCl3):δ7.78(br s,1H),7.60(br d,1H),7.38(dd,1H),7.23(br d,1H),5.40(s,1H),3.85(m,2H),2.59(t,2H),2.14(m,2H),1.70(s,3H),1.65(s,3H)。
(v)Ph(3-(1-吡咯烷-2-酮))-(R,S)CH(OH)C(O)OH
将(R,S)-5-Ph(3-(1-吡咯烷-2-酮))-2,2-二甲基-4-氧代-1,3-二氧戊环(1.4g,5.1mmol;参见以上步骤(iv))和1M NaOH(10mL)的THF(15mL)中的混合物在室温下剧烈搅拌过夜。除去THF并用CH2Cl2将水相洗涤一次,然后在真空下浓缩。将残余物用制备型RPLC(CH3CN∶0.1MHOAc(16∶84))纯化。浓缩所需部分并冻干,得到小标题化合物0.94g(79%)。
LC-MS:(M+1)236;(M-1)234m/z
1H NMR(400MHz;CD3OD):δ7.64(br s,1H),7.59(br d,1H),7.35(dd,1H),7.28(br d,1H),5.10(s,1H),3.92(t,2H),2.58(t,2H),2.16(m,2H)。
(vi)Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)OH
用ChiralpakTMAD作为固定相和庚烷∶2-丙醇∶乙腈∶甲酸(160∶30∶10∶1)作为流动相,经制备型HPLC分离Ph(3-(1-吡咯烷-2-酮))-(R,S)CH(OH)C(O)OH(0.94g,4.0mmol;参见以上步骤(v))的对映体。真空下蒸发先洗脱的对映体,得到小标题化合物0.37g(39%),98.6%ee,其[α]D 20为-90.9°(c=1.0,MeOH)。
(vii)Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)
将PyBop(365mg,0.70mmol),随后将DIPEA(0.5mL,2.8mmol)加入到冷(-20℃)的Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)OH(150mg,0.64mmol;参见以上步骤(vi))和H-Aze-Pab(Teoc)(264mg,0.70mmol,参见以上实施例3(vi))的DMF(8mL)中的溶液中。使该混合物缓慢升至室温并搅拌过夜。真空下除去DMF并将残余物在硅胶上层析,用CH2Cl2∶MeOH(95∶5)洗脱,得到小标题化合物310mg(82%)。
LC-MS:(M+1)594;(M-1)592m/z
1H NMR(400MHz;CDCl3;旋转异构体的混合物):δ8.09(br dd,1H),7.66(br d,2H),7.47(br d,1H),7.34(dd,1H),7.25(br d,2H),7.12(d,1H),5.02(s,1H),4.83(dd,1H),4.43(d,2H),4.25(t,2H),4.17(m,1H),3.82(m,3H),3.65(m,1H),3.10(m,1H),2.55(dd,2H),2.50(m,1H),2.38(m,1H),2.13(m,2H),1.10(t,2H),0.05(s,9H)。
(viii)Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-PabxHOAc
将Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(70mg,0.12mmol;参见以上步骤(vii))和TFA(2.0mL)的CH2Cl2(2mL)中的混合物在0℃下搅拌2小时。将该溶液真空下浓缩并将残余物用制备型HPLC(CH3CN∶0.1M NH4OAc(梯度:0-50%CH3CN))纯化。浓缩所需部分并将得到的产物溶于水/HOAc中,冻干,得到标题化合物52mg(87%)。
LC-MS:(M+1)450m/z
1H NMR(400MHz;CD3OD;旋转异构体的混合物):δ7.72(m,3H),7.52(m,3H),7.42-7.20(m,2H),5.22-5.12(m,1H),4.80(m,1H),4.50(ABX谱的AB部分,2H),4.14(m,1H),4.07(m,1H),3.90(m,2H),2.74-2.44(m,3H),2.34-2.08(m,3H),1.90(s,3H)。
13C NMR(羰基和/或脒碳;100MHz):δ176.0,172.4,171.8,167.0。
实施例11
Ph(3-(1-吡咯烷))-(R)-或-(S)-CH(OH)C(O)-Aze-Pabx2TFA
(i)(R,S)-5-Ph(3-(1-吡咯烷))-2,2-二甲基-4-氧代-1,3-二氧戊环
将(R,S)-5-Ph(3-NH2)-2,2-二甲基-4-氧代-1,3-二氧戊环(450mg,2.17mmol;参见以上实施例10(ii))、1,4-二溴代-丁烷(0.30mL,3.26mmol)和Cs2CO3(2.1g,6.5mmol)的丙酮中的混合物回流3天。除去溶剂并将粗制产物分配在CH2Cl2和水之间。用CH2Cl2萃取水相并用盐水洗涤合并的有机相,干燥(Na2SO4)并在真空下浓缩。将残余物在硅胶上层析,用庚烷∶EtOAc(100∶0-90∶10)洗脱,得到小标题化合物140mg(25%)。
LC-MS:(M+1)413;(M-1)411m/z
1H NMR(400MHz;CDCl3):δ7.23(dd,1H),6.74(d,1H),6.61(br s,1H),6.55(br d,1H),5.35(s,1H),3.29(m,4H),2.00(m,4H),1.72(s,3H),1.66(s,3H)。
(ii)Ph(3-(1-吡咯烷))-(R,S)CH(OH)C(O)OHxHCl
将(R,S)-5-Ph(3-(1-吡咯烷))-2,2-二甲基-4-氧代-1,3-二氧戊环(640mg,2.45mmol;参见以上步骤(i))和1M NaOH(10mL)的THF(10mL)中的混合物在室温下剧烈搅拌过夜。除去THF并用CH2Cl2洗涤水相一次,然后在真空下浓缩。将残余物在硅胶上层析,用CH2Cl2∶MeOH∶NH2OH(6∶3∶1)洗脱。先用水/HOAc,然后用水/2M HCl,将产物冻干两次成为盐。得到小标题化合物0.62g(98%)。
LC-MS:(M+1)222;(M-1)220m/z
1H NMR(400MHz;CD3OD):δ7.40(m,1H),7.20-7.55(m,3H),5.28(s,1H),3.80(m,4H),2.30(m,4H)。
(iii)Ph(3-(1-吡咯烷))-(R,S)CH(OH)C(O)-Aze-Pab(Teoc)
将PyBop(355mg,0.68mmol)、然后将可力丁(0.4mL,3.35mmol)加入到冷(-20℃)的Ph(3-(1-吡咯烷))-(R,S)CH(OH)C(O)OH(160mg,0.62mmol;参见以上步骤(ii))和H-Aze-Pab(Teoc)x2HCl(307mg,0.68mmol,参见以上实施例3(vi))的DMF(8mL)中的溶液中。使反应物缓慢达到室温并搅拌过夜。除去DMF并将粗品产物分配在EtOAc和水之间。用EtOAc萃取水相并干燥(Na2SO4)有机相,并在真空下浓缩。将残余物在硅胶上层析,用CH2Cl2∶MeOH(95∶5)洗脱,得到小标题化合物50mg(14%)。
LC-MS:(M+1)580;(M-1)578m/z
1H NMR(400MHz;CDCl3;旋转异构体的混合物):δ7.82(m,2H),7.46-7.30(m,2H),7.18-7.08(m,1H),6.70-6.45(m,3H),5.12-5.00(m,1H),4.75(m,1H),4.45(m,2H),4.23(m,2H),4.1-3.85(m,2H),3.22(m,4H),2.69-2.34(m,1H),2.25,2.11(2m,1H),1.07(m,2H),0.08(s,9H)。
(iv)Ph(3-(1-吡咯烷))-(R,S)CH(OH)C(O)-Aze-Pabx2TFA
将Ph(3-(1-吡咯烷))-(R,S)CH(OH)C(O)-Aze-Pab(Teoc)(100mg,0.17mmol;参见以上步骤(iii))和TFA(2.0mL)的CH2Cl2(2mL)中的混合物在0℃下搅拌2小时。在真空下浓缩溶液,将得到的残余物溶于水中并冻干,得到标题化合物70mg(58%)。
LC-MS:(M+1)580m/z
1H NMR(400MHz;CD3OD;旋转异构体的混合物):δ7.75(m,2H),7.50(m,2H),7.15(m,1H),6.75-6.50(m,3H),5.14,5.08(2s,1H),4.76(m,1H),4.60-4.42(m,2H),4.28(m,1H),4.11-3.87(m,2H),3.26(m,4H),2.75-2.40(m,1H),2.26,2.13(2m,1H),1.89(m,4H)。
13C NMR(羰基和/或脒碳;100MHz):δ173.8,173.3,171.8,167.0。
实施例12
Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(OMe)
(i)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)
向Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(92mg,0.16mmol;参见以上实施例6(vi))的THF(6mL)中的溶液中加入O-甲基羟胺(78mg,0.92mmol),将得到的混合物在60℃搅拌过夜。在真空下除去溶剂并将得到的固体在硅胶上层析,用EtOAc洗脱。浓缩所需部分,得到小标题化合物(82mg,85%),为白色固体。
1H NMR(400MHz;CDCl3):δ8.0(bt,1H),7.57(b,1H),7.50(d,2H),7.33(d,2H),6.64(m,2H),6.51(s,1H),4.90(dd,1H),4.82(s,1H),4.51(ABX谱的AB部分,2H),4.16(m,2H),4.07(m,1H),3.97(s,3H),3.65(m,1H),2.97(s,6H),2.70(m,1H),2.40(m,1H),0.99(m,2H),0.03(s,12H)。
LC-MS:(M+1)618m/z
(ii)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(OMe)
将Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)(78mg,0.13mmol,参见以上步骤(i))的TFA(3mL)中的溶液在0℃下搅拌2小时。将该溶液在真空下低温浓缩并将所得固体在制备型HPLC(CH3CN∶0.1M乙酸铵(40∶60))上层析。将所需部分部分浓缩。将残余物冻干(CH3CN∶水)三次,得到标题化合物40mg(30%)。纯度99.4%。
1H NMR(400MHz;CDCl3):δ7.59(d,2H),7.32(d,2H),6.72(s 2H),6.66(m,1H),5.05(s,1H),4.84(s,4H),4.76(dd,1H),4.44(ABX谱的AB部分,2H),4.30(m,1H),4.00(m,1H),3.82(s,3H),2.93(s,6H),2.49(m,1H),2.39(m,1H)。
13C NMR(羰基和/或脒碳;100MHz):δ172.7,171.8,171.7,158.7。
LC-MS:(M+1)474m/z
实施例13
Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-Et)
(i)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-Et)
向Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(40mg,0.07mmol,参见以上实施例6(vi))的THF(3mL)中的溶液中加入O-乙基羟胺xHCl(40mg,0.41mmol),并将该溶液在60℃下搅拌过夜。将该溶液浓缩并将生成物用制备型HPLC(CH3CN∶0.1M乙酸铵(60∶40))纯化。将所需部分部分浓缩并将残余物用EtOAc(3x)萃取。用水洗涤有机层并在真空下浓缩,得到小标题化合物16mg(37%)。
1H NMR(400MHz;CDCl3):δ8.00(b,1H),7.58(b,1H),7.49(d,2H),7.32(d,2H),6.65(s,1H),6.63(s,1H),6.51(s,1H),4.90(dd,1H),4.82(s,1H),4.50(ABX谱的AB部分,2H),4.25-4.15(m,5H),4.06(m,1H),3.65(q,1H),2.97(s,6H),2.69(m,1H),2.39(m,1H),1.34(t,3H),0.99(t,2H),0.05(s,9H)。
LC-MS:(M+1)633m/z
(ii)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-Et)
向Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-Et)(16mg,0.03mmol,参见以上步骤(i))的二氯甲烷(0.5mL)中的溶液中加入TFA(1mL),并将该混合物在0℃下搅拌2小时。将得到的混合物在真空下浓缩得到固体残余物,将其溶于水/CH3CN中并冻干两次,得到标题化合物14mg(92%)。纯度94.4%。
1H NMR(400MHz;CD3OD):δ8.73(bt,1H),7.66(d,2H),7.53(d,2H),6.73(s,2H),6.67(s,2H),5.07(s,2H),4.78(dd,1H),4.51(ABX谱的AB部分,2H),4.32(m,1H),4.16(m,1H),4.04(m,1H),2.94(s,6H),2.50(m,1H),2.29(m,1H),1.39(t,3H)。
13C NMR(羰基和/或脒碳;100MHz):δ172.7,171.7,160.6,152.0。
LC-MS:(M+1)489m/z
实施例14
Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)
(i)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-n-Pr)
向Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(40mg,0.07mmol;参见以上实施例6(vi))的THF(5mL)中的溶液中加入O-n-丙基羟胺xHCl(46mg,0.41mmol),并将该溶液在60℃下搅拌过夜。将该溶液浓缩至干并将残余物用制备型HPLC(CH3CN∶0.1M乙酸铵(60∶40))纯化。将所需部分部分浓缩并用EtOAc(3x)萃取所述水溶液。用水洗涤有机层,干燥(Na2SO4)并浓缩,得到小标题化合物16mg(36%)。
1H NMR(400MHz;CDCl3):δ8.00(bt,1H),7.58(bs,1H),7.48(d,2H),7.32(d,2H),6.65(s,1H),6.63(s,1H),6.51(s,1H),4.89(dd,1H),4.82(s,1H),4.50(ABX谱的AB部分,2H),4.16(dd,1H),4.11(t,1H),4.06(m,1H),3.65(q,1H),2.96(s,6H),2.68(m,1H),2.39(m,1H),1.75(m,2H),0.98(t,5H),0.05(s,9H)。
LC-MS:(M+1)647m/z
(ii)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)
将TFA(2mL)加入到冰冷的Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-n-Pr)(16mg,0.02mmol;参见以上步骤(i))的二氯甲烷(0.5mL)中的溶液中,并将得到的混合物低温搅拌2小时。将得到的溶液在真空下浓缩并将得到的固体残余物溶于水/CH3CN中并冻干。将该产物用快速层析(EtOAc∶MeOH(9∶1))纯化。将所需部分浓缩,得到标题化合物14mg(92%)。纯度98%。
1H NMR(400MHz;CDCl3):δ8.71(bt,1H),7.65(d,2H),7.50(d,2H),6.73(m,2H),6.67(s,1H),5.07(s,1H),4.78(dd,1H),4.50(ABX谱的AB部分,2H),4.32(m,1H),4.06(m,3H),2.94(s,6H),2.49(m,1H),2.29(m,1H),1.80(m,2H),1.03(m,3H)。
LC-MS:(M+1)503m/z
实施例15
Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-i-Pr)
(i)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-i-Pr)
将O-异丙基羟胺xHCl(46mg,0.41mmol)加入到Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(40mg,0.07mmol;参见以上实施例6(vi))的THF(3mL)中的溶液中,并将得到的混合物在60℃下搅拌过夜。浓缩生成物溶液并将粗制产物用制备型HPLC(CH3CN∶0.1M乙酸铵(60∶40))纯化。将所需部分部分浓缩,然后用EtOAc(3x)萃取。用水洗涤合并的有机物,干燥(Na2SO4)并浓缩,得到小标题化合物16mg(36%)。
1H NMR(400MHz;CDCl3):δ8.00(b,1H),7.57(s,1H),7.50(d,2H),7.32(d,2H),6.65(s,1H),6.63(s,1H),6.51(s,1H),4.90(dd,1H),4.82(s,1H),4.58-4.40(m,3H),4.17(m,3H),4.07(m,1H),3.65(m,1H),2.97(s,2H),2.69(m,1H),2.39(m,1H),1.31(d,6H),1.00(m,2H),0.05(s,9H)。
LC-MS:(M+1)647m/z
(ii)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-i-Pr)
将冰冷的Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-i-Pr)(16mg,0.02mmol;参见以上步骤(i))的TFA(1.5mL)中的溶液低温搅拌2小时。将生成物溶液在真空下蒸发并加入水/CH3CN,将该溶液冻干。将粗制产物用快速层析(EtOAc∶MeOH(9∶1))纯化。然后将所需部分浓缩,得到标题化合物14mg(92%)。纯度(HPLC)96%。
1H NMR(400MHz;CDCl3):δ8.73(bt,1H),7.66(d,2H),7.50(d,2H),6.73(d,2H),6.67(t,1H),5.08(s,1H),4.78(dd,1H),4.51(ABX谱的AB部分,2H),4.4-4.3(m,2H),4.02(m,1H),2.95(s,6H),2.51(m,1H),2.30(m,1H),1.39(d,6H)。
LC-MS:(M+1)503m/z
实施例16
Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-CH2-CH2-O-CH3)
(i)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-CH2-CH2-O-CH3)
将O-(2-甲氧基)乙基羟胺和HOAc(23.3μL)的THF(2mL)中的溶液加入到Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(40mg,0.07mmol;参见以上实施例6(vi))的THF(1mL)中的溶液中,将该混合物在60℃下搅拌3.5天。将生成物溶液浓缩至干燥并将粗制产物用制备型HPLC(CH3CN∶0.1M乙酸铵(60∶40))纯化。将所需部分部分浓缩并用EtOAc(3x)萃取。用水洗涤合并的有机层,干燥(Na2SO4),然后浓缩至干燥,得到小标题化合物20mg(44%)。
1H NMR(400MHz;CDCl3):δ8.00(bt,1H),7.71(b,1H),7.48(d,2H),7.32(d,2H),6.65(s,1H),6.63(s,1H),6.51(s,1H),4.89(dd,1H),4.81(s,1H),4.49(ABX谱的AB部分,2H),4.29(m,2H),4.14(m,2H),4.07(m,2H),3.74-3.60(m,3H),3.42(s,3H),2.96(s,6H),2.68(m,1H),2.39(m,1H),1.79(b,2H),0.97(m,1H),0.02(s,9H)。
LC-MS:(M+1)663m/z
(ii)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-CH2-CH2-O-CH3)
将TFA(2mL,26mmol)加入到冰冷的Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-CH2-CH2-O-CH3)(20mg,0.03mmol;来自以上步骤(i))的二氯甲烷(0.5mL)中的溶液中,并将得到的混合物低温搅拌21/2小时。将生成物溶液蒸发至干燥,并将粗制产物经快速层析(EtOAc∶MeOH(9∶1))纯化。将所需部分浓缩得到残余物,向其中加入水/CH3CN。将得到的溶液冻干过夜,得到标题化合物13mg(65%)。纯度(HPLC)96%。
1H NMR(400MHz;CD3OD):δ8.69(bt,1H),7.65(d,2H),7.49(d,2H),6.73(d,2H),6.67(t,1H),5.07(s,2H),4.78(dd,1H),4.51(ABX谱的AB部分,2H),4.32(m,1H),4.22(m,2H),4.03(m,1H),3.72(m,2H),3.41(s,3H),2.94(s,6H),2.48(m,1H),2.29(m,1H)。
13C NMR(羰基和/或脒碳;100MHz;CD3OD):δ172.7,171.8,171.7,159.3,152.0。
LC-MS:(M+1)519m/z
实施例17
Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-THP)
(i)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-THP)
将O-(四氢吡喃-2-基)-羟胺(51mg,0.44mmol)和HOAc(25μL)的THF(1mL)中的溶液加入到Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(43mg,0.07mmol;参见以上实施例6(vi))的THF(2mL)中的溶液中,并将得到的混合物在60℃下搅拌22小时,然后在室温下搅拌过夜。浓缩生成物溶液并将粗制产物经制备型HPLC(CH3CN∶0.1M乙酸铵(60∶40))纯化。将所需部分部分浓缩并用EtOAc(3x)萃取含水的残余物。用水洗涤合并的有机物,干燥(Na2SO4),然后浓缩,得到小标题化合物26mg(52%)。
1H NMR(400MHz;CDCl3):δ8.00(bt,1H),7.61(b,1H),7.52(d,2H),7.32(d,2H),6.66(s,1H),6.63(s,1H),6.51(s,1H),5.30(m,1H),4.90(dd,1H),4.82(s,1H),4.50(ABX谱的AB部分,2H),4.17(m,2H),4.07(m,1H),3.95(m,1H),3.66(m,2H),2.97(s,6H),2.69(m,1H),2.39(m,1H),2.00-1.55(m,7H),0.98(m,2H),0.04(s,9H)。
LC-MS:(M+1)689m/z
(ii)Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-THP)
将AmberlystA-26上的氟化物(140mg)加入到Ph(3-Cl,5-NMe2)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-THP)(34mg,0.05mmol;参见以上步骤(i))的CH3CN(3mL)中的溶液中,并将该混合物在60℃下放置过夜。冷却后,经过滤除去树脂,然后用多份CH3CN和EtOH(95%)洗涤。浓缩合并的有机物得到粗制产物,使其经制备型HPLC(CH3CN∶0.1M乙酸铵(50∶50))纯化。浓缩所需部分,溶于水/CH3CN中,然后冻干,得到标题化合物18mg(60%)。
1H NMR(400MHz;CD3OD):δ7.62(d,2H),7.32(d,2H),6.72(s,2H),6.66(s,1H),5.15(m,1H),5.05(s,1H),4.77(dd,1H),4.44(ABX谱的AB部分,2H),4.29(m,1H),3.95(m,2H),3.57(m,1H),2.93(s,6H),2.48(m,1H),2.27(m,1H),1.94(m,2H),1.82(m,1H),1.75(m,1H),1.61(m,3H)。
13C NMR(羰基和/或脒碳;100MHz;CD3OD):δ172.7,171.5,154.7,152.0。
LC-MS:(M+1)545m/z
实施例18
Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-Pab(OMe)xTFA(i)Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)
将Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(38mg,0.06mmol;参见以上实施例5(vii))和O-甲基羟胺(62mg,0.74mmol)的THF(3mL)中的混合物在60℃下加热30小时,然后除去溶剂并将反应混合物经制备型HPLC(CH3CN∶0.1M乙酸铵(50∶50))纯化。将所需部分部分浓缩并用EtOAc(3x)萃取含水的残余物。干燥(Na2SO4)合并的有机物并在真空下浓缩,得到小标题化合物22mg(50%)。
1H NMR(600MHz;CDCl3):δ7.81(br,1H),7.55(br,1H),7.45(d,2H),7.28(d,2H),7.18(br,1H),7.09(br,1H),4.89(s,1H),4.86(dd,1H),4.46(ABX谱的AB部分,2H),4.11(m,2H),3.92(s,3H),3.67(br,1H),3.22(br,3H),2.68(br,1H),2.41(m,1H),1.87(br,2H),1.71(m,4H),0.95(m,2H),-0.02(s,9H)。
(ii)Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-Pab(OMe)xTFA
将Ph(3-Cl,5-NMeAc)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)(22mg,0.03mmol,参见以上步骤(i))的TFA(3mL)中的溶液放置在室温下1小时,然后在真空下除去溶剂。将固体残余物溶于水中并将该溶液冻干过夜,得到标题化合物20mg(76%)。
1H NMR(400MHz;D2O)(由于几何异构而变得复杂):δ8.79(bt,1H),7.67(t,2H),7.51(d,2H),7.46(d,2H),7.17(s,1H),5.35(s,1H),5.20(s+m,1H),4.88(dd,1H),4.55(m,1H),4.40(m,1H),4.11(m,2H),3.98(2xs,3H),3.38(s,1H),3.19(2s,2H),2.80(m,0.5H),2.60(m,0.5H),2.28(m,2H),1.88(s,2H)。
13C NMR(100MHz,CD3OD)(羰基和/或脒碳):δ174.0,173.3,172.6,172.5,163.4,163.0。
LC-MS:(M+1)502m/z
实施例19
Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(OMe)xTFA
(i)Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)
将O-甲基羟胺xHCl(42mg,0.50mmol)加入到Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(50mg,0.08mmol;参见以上实施例10(vii))的THF(3mL)溶液中,并将该混合物在60℃下搅拌过夜。除去溶剂并将残余物分配在水和EtOAc之间。用EtOAc萃取水相并干燥(Na2SO4)合并的有机相,然后在真空下浓缩,得到小标题化合物大约52mg(大约100%),为固体,其没有进一步纯化而使用。
LC-MS:(M+1)624;(M-1)622m/z
(i)Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(OMe)xTFA
将Ph(3-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)(53mg,0.08mmol;参见以上步骤(i))和TFA(2.0mL)的CH2Cl2(1mL)中的混合物在室温下搅拌4小时。除去溶剂并将残余物分配在水和EtOAc之间。用EtOAc萃取水相并干燥(Na2SO4)合并的有机相,然后在真空下浓缩,将残余物在硅胶上层析,用CH2Cl2∶MeOH(98∶2-95∶5)洗脱,得到标题化合物22mg(44%)。
LC-MS:(M+1)480;(M-1)478m/z
1H NMR(400MHz;CD3OD):δ7.76-7.63(m,3H),7.59-7.50(m,3H),7.44-7.20(m,2H),5.20-5.14(m,1H),4.61-4.02(m,4H),3.97-3.87(m,5H),2.74-2.44(m,3H),2.34-2.09(m,3H)。
实施例20
Ph(3-Cl,5-吡咯并)-(R)或-(S)CH(OH)C(O)-Aze-Pab
(i)(R,S)-5-Ph(3-Cl,5-吡咯并)-2,2-二甲基-4-氧代-1,3-二氧戊环
向(R,S)-5-Ph(3-Cl,5-NH2)-2,2-二甲基-4-氧代-1,3-二氧戊环(6.0g,24.8mmol;参见以上实施例9(ii))和五氧化二磷(3.5g,24.8mmol)的干燥甲苯(50mL)中的溶液中滴加入2,5-二甲氧基四氢呋喃(4.9g,37.3mmol)。将反应物加热至回流30分钟,然后冷却至室温。用2N NaOH(10mL)猝灭反应物,转移到分液漏斗中并分离含水相,用甲苯(100mL)萃取。接着用盐水(20mL)洗涤合并的有机相,干燥(MgSO4),过滤并在真空下浓缩,得到浅橙色的油状物(5.0g)。在硅胶上快速层析,用CH2Cl2洗脱,得到小标题化合物2.8g(39%),为黄色固体。
1H NMR(300MHz;CD3OD):δ7.28-7.42(m,3H),7.18(t,J=2.1Hz,2H),6.38(t,J=2.1Hz,2H),5.40(s,1H),1.72(d,J=8.2Hz,6H)。
(ii)Ph(3-Cl,5-吡咯并)-(R,S)CH(OH)C(O)OH
在室温下,向(R,S)-5-Ph(3-Cl,5-吡咯并)-2,2-二甲基-4-氧代-1,3-二氧戊环(3.1g,10.7mmol;参见以上步骤(i))的THF(40mL)中的溶液中与溴化四丁基铵(0.35g,1.07mmol)一起加入3N NaOH(36mL,107.3mmol)。然后,将反应混合物在室温下再搅拌2小时。将反应混合物在真空下浓缩以除去THF。将剩下的含水相冷却至0℃并用浓HCl酸化至pH为2,用EtOAc(2×150mL)萃取。用盐水洗涤合并的有机物,干燥(MgSO4),过滤并在真空下浓缩,得到橙色泡沫状物。在硅胶上快速层析,用CHCl3∶MeOH∶浓氢氧化铵(85∶15∶5)洗脱,得到小标题化合物的铵盐(2.0g),为白色固体。接着用2N HCl将其酸化至pH为1,随后用EtOAc萃取,在真空下浓缩并干燥,得到小标题化合物1.8g(68%),为白色固体。
1H NMR(300MHz;CD3OD):δ7.52(s,1H),7.46(s,1H),7.36(s,1H),7.22(t,J=3Hz,2H),6.32(t,J=3Hz,2H),5.4(s,1H)。
(iii)Ph(3-Cl,5-吡咯并)-(R)或-(S)CH(OH)C(O)OH(a)和Ph(3-Cl,5-吡咯并)-(S)或-(R)CH(OAc)C(O)OH(b)
将Ph(3-Cl,5-吡咯并)-(R,S)CH(OH)C(O)OH(1.8g,7.3mmol;参见以上步骤(ii))、脂酶PS‘Amano’(1.0g)、乙酸乙烯酯(5.0mL)和MTBE(5.0mL)的混合物于45℃下加热24小时。将反应物过滤并用EtOAc(100mL)洗涤滤饼。在真空下浓缩滤液并在硅胶上层析,用CHCl3∶HOAc(95∶5)洗脱,得到为白色固体的小标题化合物(a)710mg(38%)和为乳色固体的小标题化合物(b)910mg(42%)。
对于小标题化合物(a):
1H NMR(300MHz;CD3OD):δ7.54(s,1H),7.47(s,1H),7.36(s,1H),7.19(d,J=3Hz,2H),6.30(d,J=3Hz,2H),5.21(s,1H)。
13C NMR(75MHz;CD3OD):δ175.2,142.9,136.1,124.4,120.3,119.9,117.3,112.0,73.2。
HPLC分析:98.3%,98.0%ee
[α]D 25=-99°(c=1.0,甲醇)
API-MS:(M+1)252m/z
对于小标题化合物(b):
1H NMR(300MHz;CD3OD):δ7.52(s,1H),7.46(s,1H),7.38(s,1H),7.20(br s,2H),6.30(br s,2H),5.22(s,1H),1.98(s,3H)。
(iv)Ph(3-Cl,5-吡咯并)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)
将Ph(3-Cl,5-吡咯并)-(R)或-(S)CH(OH)C(O)OH(285mg,1.14mmol;参见以上步骤(iii))、HAze-Pab(Teoc)(470mg,1.25mmol)、PyBOP(650mg,1.25mmol)和2,4,6-可力丁(0.33mL,2.49mmol)的DMF(14mL)中的混合物在0℃下搅拌2小时,然后在25℃下搅拌30分钟。将反应物用H2O(50mL)猝灭并用EtOAc(3×50mL)萃取。干燥(Na2SO4)合并的有机物,过滤并在真空下浓缩。在硅胶上快速层析(2x),用EtOAc洗脱,得到小标题化合物180mg(26%),为白色固体。
1H NMR(300MHz;CD3OD):δ7.74-7.86(m,2H),7.14-7.58(m,7H),6.28(br s,2H),5.14-5.28(m,2H),4.76-4.82(m,1H),3.92-4.58(m,7H),2.40-2.68(m,2H),2.10-2.38(m,2H),1.02-1.16(m,2H),0.09(s,9H)。
API-MS:(M+1)610m/z
(v)Ph(3-Cl,5-吡咯并)-(R)或-(S)CH(OH)C(O)-Aze-Pab
向Ph(3-Cl,5-吡咯并)-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(38mg,0.06mmol;参见以上步骤(iv))的乙腈中的溶液中加入结合氟离子的聚合物(AmberlystA-26)(170mg)并将该混合物在60℃下加热过夜,随后在70℃下加热4小时。将反应混合物过滤,用乙腈、乙醇和THF洗涤该聚合物和溶液并在真空下浓缩溶液。将粗制产物用制备型HPLC(CH3CN∶0.1M乙酸铵,40∶60和CH3CN∶0.1M乙酸铵,30∶70,分别地)纯化两次。将所需部分冻干(3x),得到标题化合物8mg(28%)。
1H NMR(400MHz;CD3OD):δ7.77(m,2H),7.61-7.48(m,3H),7.40(d,1H),7.26(m,2H),6.33(m,2H),5.30(d,1H),4.68-4.25(m,2H),4.09(m,1H),2.60(m,1H),2.35(m,1H),1.94(s,3H)。
LC-MS:(M+1)466m/z
实施例21
Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)(i)Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(O-n-Pr)
向Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(40mg,0.064mol;参见以上实施例9(vi))的THF(3mL)中的溶液中加入O-正丙基羟胺xHCl(43mg,0.38mmol),并将该溶液在60℃下加热4.5小时。将该溶液在真空下浓缩并将得到的粗制产物经快速层析(Sigel,EtOAc∶MeOH 9∶1)纯化。将所需部分浓缩,得到小标题化合物43mg(98%)。
1H NMR(400MHz;CD3OD):δ7.75(2s,1H),7.59(br,1H),7.43(d,2H),7.23(m,3H),5.18(br,1H),4.50-4.30(m,3H),4.20-4.05(m,5H),3.99(t,3H),3.84(m,2H),2.55(t,2H),2.28(m,1H),2.12(m,2H),1.71(m,2H),0.99(m,4H),0.02(s,9H)。
(ii)Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)
向Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)或-(S)CH(OH)C(O)-Aze-Pab(O-n-Pr)(Teoc)(43mg,0.063mmol;来自以上步骤(i))的二氯甲烷(0.5mL)冰冷的溶液中加入TFA(2.5mL),并将该溶液在0℃下搅拌100分钟,然后将该溶液在真空下浓缩并将得到的粗制产物用制备型HPLC(CH3CN∶0.1M乙酸铵30∶70)纯化。将所需部分合并并冻干,得到23mg(68%),纯度99.9%。
LC-MS:(M+1)542m/z
1H NMR(400MHz;CD3OD)(由于几何异构而变得复杂):δ7.75(br,1H),7.57(m,2.5H),7.49(s,0.5H),7.36-7.22(m,3H),5.16(s,1H),4.78(dd,1H),4.48-4.32(m,3H),4.17(m,1H),3.97(m,2H),3.86(m,2H),2.55(t,3H),2.52(m,0.5H),2.28(m,0.5H),2.13(m,3H),1.71(m,2H),0.98(m,2H)。
实施例22
Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(OMe)
(i)Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)
向Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(80mg,0.13mmol;参见以上实施例9(vi))的THF(6mL)中的溶液中加入O-甲基羟胺xHCl(64mg,0.77mmol)。将该混合物在60℃下搅拌5小时,然后蒸发。将残余物在硅胶上层析,用乙酸乙酯∶甲醇(9∶1)洗脱,得到粗制产物75mg。将粗制产物用制备型HPLC(CH3CN∶0.1M乙酸铵,60∶40)进一步纯化。浓缩所需部分。在真空下除去CH3CN。用EtOAc(3x)萃取含水相。用盐水洗涤合并的乙酸乙酯相,然后干燥(Na2SO4)并浓缩,得到小标题化合物65.8mg(78%)。
1H NMR(400MHz;CDCl3):δ7.97-7.90(m,1H),7.60-7.55(m,2H),7.46(d,2H),7.29(d,2H),7.13(s,1H),4.95-4.81(m,2H),4.55-4.30(m,3H),4.18-4.09(m,3H),3.95(s,3H),3.87-3.75(m,3H),2.69-2.55(m,3H),2.45-2.34(m,1H),2.20-2.10(m,2H),2.00(br,1H),1.01-0.92(m,2H),0.00(s,9H)。
(ii)Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(OMe)
向Ph(3-Cl,5-(1-吡咯烷-2-酮))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(Teoc)(OMe)(55.9mg,0.08mmol;来自以上步骤(i))的二氯甲烷(0.5mL)中的冰冷溶液中加入TFA(3.0mL)。将该溶液在0℃下搅拌130分钟,然后将该溶液在真空下浓缩并将生成的粗制产物用制备型HPLC(CH3CN∶0.1M乙酸铵,30∶70)纯化。将所需部分合并并冻干(2x),得到标题化合物38mg(87%)。
LC-MS:(M+1)514m/z
1H NMR(400MHz;CD3OD):δ7.75(s,1H),7.62-7.47(m,3H),7.36-7.21(m,3H),5.19-5.10(m,1H),4.48-3.93(m,4H),3.89-3.79(m,5H),2.72-2.45(m,3H),2.33-2.06(m,3H)。
实施例23
Ph(3-Cl,5-NMe(3-甲基丁酰基))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(OMe)(i)(R,S)-5-Ph(3-Cl,5-NHMe)-2,2-二甲基-4-氧代-1,3-二氧戊环
将(R,S)-5-Ph(3-Cl,5NH2)-2,2-二甲基-4-氧代-1,3-二氧戊环(3.0g,12.4mmol;参见以上实施例9(ii))、甲醛(0.81mL 37重量%的水溶液,9.9mmol)和氧化铂(IV)(330mg)的EtOAc(100mL)中的混合物在氢气氛、25℃下搅拌5小时。将该混合物经Celite滤垫过滤并用EtOAc(200mL)洗涤滤饼。将有机物在真空下浓缩并在硅胶上快速层析,用EtOAc∶己烷(1∶4)洗脱,得到小标题化合物1.63g(52%),为黄色油状物。
1H NMR(300MHz;CDCl3):δ6.75(s,1H),6.54(s,2H),5.25(s,1H),3.90(br s,1H),2.80(s,3H),1.68(s,3H),1.64(s,3H)。
(ii)(R,S)-5-Ph(3-Cl,5-NMe(3-甲基丁酰基))-2,2-二甲基-4-氧代-1,3-二氧戊环
在0℃下,向(R,S)-5-Ph(3-Cl,5-NHMe)-2,2-二甲基-4-氧代-1,3-二氧戊环(945mg,3.70mmol;参见以上步骤(i))和三乙胺(560mg,5.54mmol)的丙酮(20mL)中的溶液中滴加入异戊酰氯(623mg,5.17mmol)。将该混合物搅拌0.5小时并分配在EtOAc(3×30mL)和H2O(30mL)之间。用NaHCO3(30mL)水溶液洗涤合并的有机萃取液,干燥(Na2SO4),过滤并在真空下浓缩,得到小标题化合物1.35g(>100%),为黄色油状物,其没有纯化直接使用。
1H NMR(300MHz;CDCl3):δ7.50(s,1H),7.20(s,2H),5.38(s,1H),3.28(s,3H),1.90-2.22(m,3H),1.70(s,3H),1.68(s,3H),0.70-0.92(m,6H)。
(iii)Ph(3-Cl,5-NMe(3-甲基丁酰基))-(R,S)CH(OH)C(O)OH
将(R,S)-5-Ph(3-Cl,5-NMe(3-甲基丁酰基))-2,2-二甲基-4-氧代-1,3-二氧戊环(1.35g,3.97mmol;参见以上步骤(ii))和NaOH(1.60g,39.7mmol)的MeOH(20mL)中的混合物在25℃下搅拌1小时。将该混合物在真空下浓缩并将残余物用H2O(30mL)稀释,用2N HCl(20mL)酸化并用EtOAc(3×50mL)萃取。干燥(Na2SO4)合并的有机萃取液,过滤并在真空下浓缩,得到小标题化合物1.0g(100%),为黄色油状物,其没有纯化直接使用。
1H NMR(300MHz;CD3OD):δ7.55(s,1H),7.34(s,2H),5.21(s,1H),3.23(s,3H),1.90-2.10(m,3H),0.70-0.92(m,6H)。
(iv)Ph(3-Cl,5-NMe(3-甲基丁酰基))-(R)-或-(S)-CH(OH)C(O)OH(a)和Ph(3-Cl,5-NMe(3-甲基丁酰基))-(S)-或-(R)-CH(OAc)C(O)OH(b)
将Ph(3-Cl,5-NMe(3-甲基丁酰基))-(R,S)CH(OH)C(O)OH(1.0g,3.34mmol;参见以上步骤(iii))和脂酶PS Amano(510mg)的乙酸乙烯酯(25mL)和MTBE(25mL)中的混合物在55℃下加热14小时。将反应物经Celite过滤并用MeOH(200mL)洗涤滤饼。将滤液在真空下浓缩并在硅胶上层析,用CHCl3∶MeOH∶浓NH4OH(6.5∶3.0∶0.5)洗脱,得到为可压碎泡沫状物的小标题化合物(a)的铵盐285mg和为白色泡沫状物的小标题化合物(b)的铵盐370mg(32%)。将小标题化合物(a)的铵盐溶于EtOAc(25mL)中并用2M HCl的Et2O溶液(0.60mL)中和。加入水(25mL)并分离各层。用EtOAc(2×25mL)萃取含水层,干燥(Na2SO4)有机萃取物,过滤并在真空下浓缩,得到小标题化合物(a)230mg(23%),为可压碎的白色泡沫状物。
对于小标题化合物(a):
1H NMR(300MHz;CD3OD):δ7.55(s,1H),7.34(s,1H),7.30(s,1H),5.21(s,1H),3.23(s,3H),1.90-2.10(m,3H),0.70-0.92(m,6H)。
13C NMR(75MHz;CD3OD):δ175.4,175.0,146.6,145.2,136.2,128.1,127.4,125.6,73.6,44.0,37.7,27.4,22.8。
HPLC分析:96.0%,>99%ee
[α]D 25=-85.1°(c=0.5,MeOH)
API-MS:(M+1)=300m/z
对于小标题化合物(b):
1H NMR(300MHz;CD3OD):δ7.55(s,1H),7.34(s,1H),7.30(s,1H),5.75(s,1H),3.23(s,3H),2.11(s,3H),1.90-2.10(m,3H),0.70-0.92(m,6H)。
(v)Ph(3-Cl,5-NMe(3-甲基丁酰基))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(OMe)
向Ph(3-Cl,5-NMe(3-甲基丁酰基))-(R)-或-(S)-CH(OH)C(O)OH(119mg,0.40mmol;参见以上步骤(iv))和H-Aze-Pab(OMe)x2HCl(146mg,0.44mmol;参见以上实施例2(iv))的DMF(5mL)中的混合物中加入PyBOP(227mg,0.44mmol)和可力丁(168mg,1.39mmol)。在0℃、氮气氛下将该溶液搅拌3小时。将混合物分配在EtOAc(3×30mL)和H2O(30mL)之间,干燥(Na2SO4),过滤并在真空下浓缩。在硅胶上快速层析,用CHCl3∶MeOH(15∶1)洗脱,得到小标题化合物125mg(58%),为可压碎的白色泡沫状物。
1H NMR(300MHz;CD3OD,旋转异构体的混合物):δ7.60(d,J=8Hz,2H),7.42-7.54(m,1H),7.20-7.50(m,4H),5.20和5.14(s,1H),4.72-4.81(m,1H),4.30-4.48(m,3H),3.90-4.20(m,2H),3.80(s,3H),3.22(s,3H),2.46-2.72(m,2H),2.10-2.36(m,1H),1.94-2.10(m,2H),0.80-0.94(m,6H)。
API-MS(M+1)=545m/z
实施例24
Ph(3-Cl,5-NMe(环戊基羰基))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(OMe)
(i)(R,S)-5-Ph(3-Cl,5-NMe(环戊基羰基))-2,2-二甲基-4-氧代-1,3-二氧戊环
在0℃下,向(R,S)-5-Ph(3-Cl,5-NHMe)-2,2-二甲基-4-氧代-1,3-二氧戊环(945mg,3.70mmol;参见以上实施例23(i))和三乙胺(635mg,5.86mmol)的丙酮(20mL)中的溶液中滴加入环戊烷碳酰氯(776mg,5.86mmol)。将该混合物搅拌1小时并分配在EtOAc(3×30mL)和H2O(30mL)之间。用NaHCO3(30mL)水溶液洗涤合并的有机萃取液,干燥(Na2SO4),过滤并在真空下浓缩,得到小标题化合物1.58g(>100%),为黄色油状物,其没有纯化而直接使用。
1H NMR(300MHz;CDCl3):δ7.50(s,1H),7.20(s,2H),5.38(s,1H),3.28(s,3H),2.50-2.60(m,1H),1.70(s,3H),1.68(s,3H),1.40-1.90(m,8H)。
(ii)Ph(3-Cl,5-NMe(环戊基羰基))-(R,S)CH(OH)C(O)OH
将(R,S)-5-Ph(3-Cl,5-NMe(环戊基羰基))-2,2-二甲基-4-氧代-1,3-二氧戊环(1.58g,5.07mmol;参见以上步骤(i))和NaOH(2.03g,50.7mmol)的MeOH(25mL)中的混合物在25℃下搅拌1小时。将该混合物在真空下浓缩,用H2O(30mL)稀释并用2N HCl(20mL)酸化。用EtOAc(3×50mL)萃取含水层,干燥(Na2SO4)合并的有机萃取液,过滤并在真空下浓缩,得到小标题化合物1.17g(100%),为白色泡沫状物,其没有纯化而直接使用。
1H NMR(300MHz;CD3OD):δ7.55(s,1H),7.32(s,2H),5.20(s,1H),3.20(s,3H),2.50-2.62(m,1H),1.40-1.80(m,8H)。
(iii)Ph(3-Cl,5-NMe(环戊基羰基))-(R)-或-(S)-CH(OH)C(O)OH(a)和Ph(3-Cl,5-NMe(环戊基羰基))-(S)-或-(R)-CH(OAc)C(O)OH(b)
将Ph(3-Cl,5-NMe(环戊基羰基))-(R,S)CH(OH)C(O)OH(1.17g,3.75mmol;参见以上步骤(ii))和脂酶PS Amano(600mg)的乙酸乙烯酯(25mL)和MTBE(25mL)中的混合物在55℃下加热14小时。将反应物经Celite过滤并用MeOH(100mL)洗涤滤饼。在真空下浓缩滤液并在硅胶上层析,用CHCl3∶MeOH∶浓NH4OH(6.5∶3.0∶0.5)洗脱,得到为可压碎的泡沫状物的小标题化合物(a)的铵盐336mg和为白色泡沫状物的小标题化合物(b)的铵盐557mg(50%)。将小标题化合物(a)的铵盐溶于EtOAc(25mL)中并用2M HCl的Et2O溶液(0.70mL)中和。加入水(25mL)并分离各层。用EtOAc(2×25mL)萃取含水层并干燥(Na2SO4)有机萃取液,过滤并浓缩,得到小标题化合物(a)290mg(29%),为可压碎的白色泡沫状物。
对于小标题化合物(a):
1H NMR(300MHz;CD3OD):δ7.55(s,1H),7.32(s,2H),5.20(s,1H),3.20(s,3H),2.50-2.62(m,1H),1.40-1.80(m,8H)。
13C NMR(75MHz;CD3OD):δ178.8,175.1,146.5,144.9,136.2,128.2,127.5,125.7,73.0,43.4,38.0,32.2,27.3。
HPLC分析:91.9%,>99%ee
[α]D 25=-77.9°(c=1.0,MeOH)
CI-MS:(M+1)=312m/z
对于小标题化合物(b):
1H NMR(300MHz;CD3OD):δ7.55(s,1H),7.40(s,1H),7.34(s,1H),5.75(s,1H),3.20(s,3H),2.50-2.62(m,1H),2.18(s,3H),1.40-1.80(m,8H)。
(iv)Ph(3-Cl,5-NMe(环戊基羰基))-(R)-或-(S)CH(OH)C(O)-Aze-Pab(OMe)
向Ph(3-Cl,5-NMe(环戊基羰基))-(R,S)CH(OH)C(O)OH(120mg,0.39mmol;参见以上步骤(iii))和H-Aze-Pab(OMe)x2HCl(142mg,0.42mmol;参见以上实施例2(iv))的DMF(5mL)中的混合物中加入PyBOP(220mg,0.42mmol)和可力丁(161mg,1.35mmol)。将该溶液在0℃、氮气氛下搅拌6小时。将该混合物分配在EtOAc(3×30mL)和H2O(30mL)之间,干燥(Na2SO4),过滤并在真空下浓缩。在硅胶上快速层析,用CHCl3∶MeOH(15∶1)洗脱,随后用EtOAc∶EtOH(20∶1)层析,得到标题化合物85mg(40%),为可压碎的白色泡沫状物。
1H NMR(300MHz;CD3OD,旋转异构体的混合物):δ7.60(d,J=8Hz,2H),7.42-7.54(m,1H),7.20-7.50(m,4H),5.20和5.14(s,1H),4.72-4.81(m,1H),4.30-4.48(m,3H),3.90-4.20(m,2H),3.80(s,3H),3.22(s,3H),2.46-2.72(m,2H),2.10-2.36(m,1H),1.40-1.80(m,8H)。
API-MS(M+1)=556m/z
实施例25
按以上试验A的方法,检测实施例1、3-11和20的标题化合物并发现其显示小于0.5μM的IC50TT值。
实施例26
按以上试验E中的方法,检测实施例2、12、13、15、18、19和21的标题化合物并发现其在大鼠中显示如相应的活性抑制剂(游离的脒)的口服和/或肠胃外的生物利用率。
实施例27
按以上试验G中的方法,检测实施例2、12-19和21的标题化合物并显示形成相应的活性抑制剂(游离的脒)。
缩写
AC=乙酰基
AcOH=乙酸
API=常压电离(与MS有关)
Aze=氮杂环丁烷-2-羧酸酯
AzeOH=氮杂环丁烷-2-羧酸
Bzl=苄基
CI=化学电离(与MS有关)
DIPEA=二异丙基乙胺
DMAP=4-(N,N-二甲基氨基)吡啶
DMF=二甲基甲酰胺
DMSO=二甲基亚砜
EDC=1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐
Et=乙基
ether=乙醚
EtOAc=乙酸乙酯
EtOH=乙醇
h=小时
HATU=O-(氮杂苯并三唑-1-基)-N,N,N’,N’-四甲基脲鎓六氟磷酸盐
HBTU=[N,N,N’,N’-四甲基-O-(苯并三唑-1-基)脲鎓六氟磷酸盐]
HCl=盐酸
HCl(g)=氯化氢气体
Hex=己烷
HOAc=乙酸
HPLC=高效液相层析
LC=液相层析
Me=甲基
MeOH=甲醇
MS=质谱
MTBE=甲基叔-丁基醚
Pab=对-脒基苄基氨基
H-Pab=对-脒基苄胺
PyBOP=(苯并三唑-1-基氧基)三吡咯烷基磷鎓六氟磷酸盐
RPLC=反相高效液相层析
RT=室温
TBTU=[N,N,N’,N’-四甲基-O-(苯并三唑-1-基)脲鎓四氟硼酸盐]
TEA=三乙胺
Teoc=2-(三甲基甲硅烷基)乙氧基羰基
TFA=三氟乙酸
THF=四氢呋喃
THP=四氢吡喃基
TLC=薄层层析
TMSCN=三甲基甲硅烷基氰化物
Z=苄氧基羰基
前缀n、s、i和t具有其通常的含义:正、仲、异和叔。
Claims (25)
1.一种式I化合物或其药学上可接受的盐:
其中
R1代表N(R5)R6或S(O)mR7取代基;
R2和R3独立代表选自卤素或任选被卤素取代的C1-4烷基的任选取代基;
Y代表CH2或(CH2)2;
R4代表H、OH或OR8a;
R5代表C1-6烷基或者与R6以及R5和R6所连接的氮原子一起代表3-7元的含氮环,该环任选被=O基团取代;
R6代表C1-6烷基、C(O)R9或者与R5以及R5和R6所连接的氮原子一起代表3-7元的含氮环,该环任选被=O基团取代;
m代表0、1或2;
R7代表直链、支链或环状的C1-6烷基;
R8a代表任选被氧间断的直链或支链的C1-4烷基或被氧间断的C4-5环烷基;和
R9代表C1-6烷基。
2.根据权利要求1所要求的化合物,其中当R5和R6与它们所连接的氮原子一起代表被=O取代的3-7元环时,该环在所述氮原子α位的碳原子上被取代。
3.根据权利要求1或权利要求2所要求的化合物,其中R2代表直链或支链的C1-4烷基或者卤素,其中支链的C1-4烷基任选被卤素取代。
4.根据上述权利要求中任何一项所要求的化合物,其中R3不存在或者代表直链或支链的C1-4烷基或卤素。
5.根据权利要求4所要求的化合物,其中R3代表甲基或氯。
6.根据权利要求4或权利要求5所要求的化合物,其中所述取代基在相对于也连接在所述苯环上的-CH2-基团的2位。
7.根据上述权利要求中任何一项所要求的化合物,其中R1代表N(R5)R6。
8.权利要求1或3-7中任何一项所要求的化合物,其中R5代表直链、支链或环状的C1-6烷基或者与R6以及R5和R6所连接的氮原子一起代表4-6元的含氮环,任选被=O基团取代。
9.根据权利要求8所要求的化合物,其中R5代表C1-4烷基或者与R6以及R5和R6所连接的氮原子一起代表5或6元的含氮环,任选被=O基团取代。
10.根据权利要求1或3-9中任何一项所要求的化合物,其中R6代表直链、支链或环状的C1-6烷基、C(O)-C1-6烷基或者与R5以及R5和R6所连接的氮原子一起代表4-6元的含氮环,任选被=O基团取代。
11.根据权利要求10所要求的化合物,其中R6代表甲基、C(O)-C1-6烷基或者与R5以及R5和R6所连接的氮原子一起代表5或6元的含氮环,任选被=O基团取代。
12.根据上述权利要求中任何一项所要求的化合物,其中R1在相对也连接在所述苯环上的-CH(OH)-基团的3位上与所述苯环连接。
13.根据上述权利要求中任何一项所要求的化合物,其中当R2存在时,它在相对也连接在所述苯环上的-CH(OH)-基团的5位上与所述苯环连接。
14.一种根据上述权利要求中任何一项所定义的式I化合物,其中所述片段

为S-构型。
15.一种根据上述权利要求中任何一项所定义的式I化合物,其中所述片段
为R-构型。
16.一种药用制剂,其包含一种与药学上可接受的辅助剂、稀释剂或载体混合的、根据权利要求1-15中任何一项所定义的化合物或其药学上可接受的盐。
17.一种用作药物的、根据权利要求1-15中任何一项所定义的化合物或其药学上可接受的盐。
18.一种用于治疗其中需要抑制凝血酶的病症的、根据权利要求1-15中任何一项所定义的化合物或其药学上可接受的盐。
19.一种用于治疗血栓形成的、根据权利要求1-15中任何一项所定义的化合物或其药学上可接受的盐。
20.一种用作抗凝药的、根据权利要求1-15中任何一项所定义的化合物或其药学上可接受的盐。
21.一种作为活性成分的、根据权利要求1-15中任何一项所定义的化合物或其药学上可接受的盐在制备用于治疗其中需要抑制凝血酶的病症的药物中的用途。
22.根据权利要求21所要求的用途,其中所述病症为血栓形成。
23.根据权利要求21所要求的用途,其中所述病症为血液和组织中凝固性过高。
24.一种作为活性成分的、根据权利要求1-15中任何一项所定义的化合物或其药学上可接受的盐在制备抗凝药中的用途。
25.一种制备式I化合物的方法,该方法包括:
(i)使式II的化合物

其中R1和R2如权利要求1所定义,与式III化合物偶合
其中Y、R3和R4如权利要求1所定义;
(ii)使式IV的化合物

其中R1、R2和Y如权利要求1所定义,与式V化合物偶合

其中R3和R4如权利要求1所定义;
(iii)对于其中R4代表OH或OR8a的式I化合物而言,使式VI的化合物

其中R1、R2、Y和R3如权利要求1所定义,与式VII化合物反应
H2NORa VII
其中Ra代表H或R8a,R8a如权利要求1所定义,任选通过用气态的HCl在低级烷基醇存在下预处理式VI化合物,形成式VIII的化合物
其中Rc代表低级烷基,R1、R2、Y和R3如权利要求1所定义;
(iv)对于其中R4代表OH或OR8a的式I化合物而言,使相应于式I化合物的化合物,其中代替R4,存在保护基团C(O)ORb1,其中Rb1代表2-三甲基甲硅烷基乙基、C1-6烷基或烷基苯基,与如此前所定义的式VII化合物反应;
(v)对于其中R4代表OR8b的式I化合物而言,使其中R4代表OH的式I化合物与式IXA的化合物反应
L1-R8a IXA
其中L1代表离去基团,R8a如权利要求1中所定义;
(vi)将如权利要求1所定义的式I化合物的保护的衍生物脱除保护;或者
(vii)导入或相互转化如权利要求1所定义的式I化合物中芳族、非芳族、碳环或杂环上的取代基。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SE9900071A SE9900071D0 (sv) | 1999-01-13 | 1999-01-13 | New compounds |
| SE99000713 | 1999-01-13 | ||
| SE99042285 | 1999-11-12 | ||
| SE9904228A SE9904228D0 (sv) | 1999-11-22 | 1999-11-22 | New compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1343217A CN1343217A (zh) | 2002-04-03 |
| CN1170842C true CN1170842C (zh) | 2004-10-13 |
Family
ID=26663479
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB008048401A Expired - Fee Related CN1170842C (zh) | 1999-01-13 | 2000-01-13 | 新的脒基苄基胺衍生物及其作为凝血酶抑制剂的用途 |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US6599894B1 (zh) |
| EP (1) | EP1150996B1 (zh) |
| JP (1) | JP2002535250A (zh) |
| KR (1) | KR100639274B1 (zh) |
| CN (1) | CN1170842C (zh) |
| AU (1) | AU764121B2 (zh) |
| BR (1) | BR0007453A (zh) |
| CA (1) | CA2355792A1 (zh) |
| CY (1) | CY1107149T1 (zh) |
| CZ (1) | CZ20012529A3 (zh) |
| DE (1) | DE60037183T2 (zh) |
| DK (1) | DK1150996T3 (zh) |
| EE (1) | EE200100371A (zh) |
| ES (1) | ES2295004T3 (zh) |
| IL (1) | IL143987A (zh) |
| IS (1) | IS5990A (zh) |
| NO (1) | NO20013319L (zh) |
| NZ (1) | NZ512714A (zh) |
| PL (1) | PL349769A1 (zh) |
| PT (1) | PT1150996E (zh) |
| TR (1) | TR200102037T2 (zh) |
| WO (1) | WO2000042059A1 (zh) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE0001803D0 (sv) * | 2000-05-16 | 2000-05-16 | Astrazeneca Ab | New compounds i |
| US6433186B1 (en) * | 2000-08-16 | 2002-08-13 | Astrazeneca Ab | Amidino derivatives and their use as thormbin inhibitors |
| IL154077A0 (en) * | 2000-08-16 | 2003-07-31 | Astrazeneca Ab | New amidino derivatives and their use as thrombin inhibitors |
| US7129233B2 (en) | 2000-12-01 | 2006-10-31 | Astrazeneca Ab | Mandelic acid derivatives and their use as thrombin inhibitors |
| AR035216A1 (es) * | 2000-12-01 | 2004-05-05 | Astrazeneca Ab | Derivados de acido mandelico ,derivados farmaceuticamente aceptables, uso de estos derivados para la fabricacion de medicamentos, metodos de tratamiento ,procesos para la preparacion de estos derivados, y compuestos intermediarios |
| SE0102921D0 (sv) * | 2001-08-30 | 2001-08-30 | Astrazeneca Ab | Pharmaceutically useful compounds |
| AR034517A1 (es) * | 2001-06-21 | 2004-02-25 | Astrazeneca Ab | Formulacion farmaceutica |
| UA78195C2 (uk) * | 2001-08-30 | 2007-03-15 | Astrazeneca Ab | Похідні мигдалевої кислоти та їх застосування як інгібіторів тромбіну, фармацевтична композиція на їх основі |
| AU2002367752A1 (en) * | 2001-10-03 | 2003-11-17 | Pharmacia Corporation | Substituted 5-membered polycyclic compounds useful for selective inhibition of the coagulation cascade |
| SE0201659D0 (sv) * | 2002-05-31 | 2002-05-31 | Astrazeneca Ab | Modified release pharmaceutical formulation |
| SE0201658D0 (sv) * | 2002-05-31 | 2002-05-31 | Astrazeneca Ab | Immediate release pharmaceutical formulation |
| SE0201661D0 (sv) | 2002-05-31 | 2002-05-31 | Astrazeneca Ab | New salts |
| US7781424B2 (en) * | 2003-05-27 | 2010-08-24 | Astrazeneca Ab | Modified release pharmaceutical formulation |
| US7524354B2 (en) * | 2005-07-07 | 2009-04-28 | Research Foundation Of State University Of New York | Controlled synthesis of highly monodispersed gold nanoparticles |
| TW200827336A (en) * | 2006-12-06 | 2008-07-01 | Astrazeneca Ab | New crystalline forms |
| US20090061000A1 (en) * | 2007-08-31 | 2009-03-05 | Astrazeneca Ab | Pharmaceutical formulation use 030 |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HU178398B (en) | 1979-06-12 | 1982-04-28 | Gyogyszerkutato Intezet | Process for producing new agmatine derivatives of activity against haemagglutination |
| HU192646B (en) | 1984-12-21 | 1987-06-29 | Gyogyszerkutato Intezet | Process for preparing new n-alkyl-peptide aldehydes |
| IL77748A (en) | 1985-02-04 | 1991-11-21 | Merrell Dow Pharma | Amino acid and peptide derivatives as peptidase inhibitors |
| US5187157A (en) | 1987-06-05 | 1993-02-16 | Du Pont Merck Pharmaceutical Company | Peptide boronic acid inhibitors of trypsin-like proteases |
| EP0362002B1 (en) | 1988-09-01 | 1995-07-26 | Merrell Dow Pharmaceuticals Inc. | HIV protease inhibitors |
| ZA897514B (en) | 1988-10-07 | 1990-06-27 | Merrell Dow Pharma | Novel peptidase inhibitors |
| TW201303B (zh) | 1990-07-05 | 1993-03-01 | Hoffmann La Roche | |
| CA2075154A1 (en) | 1991-08-06 | 1993-02-07 | Neelakantan Balasubramanian | Peptide aldehydes as antithrombotic agents |
| SE9102462D0 (sv) | 1991-08-28 | 1991-08-28 | Astra Ab | New isosteric peptides |
| ZA928581B (en) | 1991-11-12 | 1994-05-06 | Lilly Co Eli | Antithrombotic agents |
| SE9103612D0 (sv) | 1991-12-04 | 1991-12-04 | Astra Ab | New peptide derivatives |
| WO1993018060A1 (en) | 1992-03-04 | 1993-09-16 | Gyógyszerkutató Intézet Kft. | New anticoagulant peptide derivatives and pharmaceutical compositions containing the same as well as a process for the preparation thereof |
| TW223629B (zh) | 1992-03-06 | 1994-05-11 | Hoffmann La Roche | |
| SE9301916D0 (sv) | 1993-06-03 | 1993-06-03 | Ab Astra | New peptides derivatives |
| EP0648780A1 (en) | 1993-08-26 | 1995-04-19 | Bristol-Myers Squibb Company | Heterocyclic thrombin inhibitors |
| TW394760B (en) | 1993-09-07 | 2000-06-21 | Hoffmann La Roche | Novel Carboxamides, process for their preparation and pharmaceutical composition containing the same |
| AU1025795A (en) | 1994-01-27 | 1995-08-03 | Mitsubishi Chemical Corporation | Prolineamide derivatives |
| US5705487A (en) | 1994-03-04 | 1998-01-06 | Eli Lilly And Company | Antithrombotic agents |
| IL112795A (en) | 1994-03-04 | 2001-01-28 | Astrazeneca Ab | Derivatives of peptides as antithrombotic drugs, their preparation, and pharmaceutical preparations containing them |
| US5707966A (en) | 1994-03-04 | 1998-01-13 | Eli Lilly And Company | Antithrombotic agents |
| DE4421052A1 (de) | 1994-06-17 | 1995-12-21 | Basf Ag | Neue Thrombininhibitoren, ihre Herstellung und Verwendung |
| US5510369A (en) | 1994-07-22 | 1996-04-23 | Merck & Co., Inc. | Pyrrolidine thrombin inhibitors |
| EA199700186A1 (ru) | 1995-02-17 | 1998-02-26 | Басф Акциенгезельшафт | Новые дипептидные амидины, используемые в качестве ингибиторов тромбина |
| US5710130A (en) | 1995-02-27 | 1998-01-20 | Eli Lilly And Company | Antithrombotic agents |
| CA2216859A1 (en) | 1995-04-04 | 1996-10-10 | Merck & Co., Inc. | Thrombin inhibitors |
| US5629324A (en) | 1995-04-10 | 1997-05-13 | Merck & Co., Inc. | Thrombin inhibitors |
| SA96170106A (ar) * | 1995-07-06 | 2005-12-03 | أسترا أكتيبولاج | مشتقات حامض أميني جديدة |
| AR005245A1 (es) | 1995-12-21 | 1999-04-28 | Astrazeneca Ab | Prodrogas de inhibidores de trombina, una formulación farmaceutica que las comprende, el uso de dichas prodrogas para la manufactura de un medicamento y un procedimiento para su preparacion |
| AU1990197A (en) | 1996-03-12 | 1997-10-01 | Bristol-Myers Squibb Company | Carbamyl guanidine and amidine prodrugs |
| SE9602263D0 (sv) | 1996-06-07 | 1996-06-07 | Astra Ab | New amino acid derivatives |
| WO1997049404A1 (en) | 1996-06-25 | 1997-12-31 | Eli Lilly And Company | Anticoagulant agents |
| DE19632772A1 (de) | 1996-08-14 | 1998-02-19 | Basf Ag | Neue Benzamidine |
| AR013084A1 (es) | 1997-06-19 | 2000-12-13 | Astrazeneca Ab | Derivados de amidino utiles como inhibidores de la trombina, composicion farmaceutica, utilizacion de dichos compuestos para la preparacion demedicamentos y proceso para la preparacion de los compuestos mencionados |
-
2000
- 2000-01-13 AU AU23362/00A patent/AU764121B2/en not_active Ceased
- 2000-01-13 EP EP00902233A patent/EP1150996B1/en not_active Expired - Lifetime
- 2000-01-13 CZ CZ20012529A patent/CZ20012529A3/cs unknown
- 2000-01-13 WO PCT/SE2000/000052 patent/WO2000042059A1/en active IP Right Grant
- 2000-01-13 US US09/508,742 patent/US6599894B1/en not_active Expired - Fee Related
- 2000-01-13 TR TR2001/02037T patent/TR200102037T2/xx unknown
- 2000-01-13 EE EEP200100371A patent/EE200100371A/xx unknown
- 2000-01-13 CN CNB008048401A patent/CN1170842C/zh not_active Expired - Fee Related
- 2000-01-13 NZ NZ512714A patent/NZ512714A/en not_active IP Right Cessation
- 2000-01-13 PL PL00349769A patent/PL349769A1/xx not_active Application Discontinuation
- 2000-01-13 CA CA002355792A patent/CA2355792A1/en not_active Abandoned
- 2000-01-13 PT PT00902233T patent/PT1150996E/pt unknown
- 2000-01-13 DE DE60037183T patent/DE60037183T2/de not_active Expired - Lifetime
- 2000-01-13 ES ES00902233T patent/ES2295004T3/es not_active Expired - Lifetime
- 2000-01-13 DK DK00902233T patent/DK1150996T3/da active
- 2000-01-13 KR KR1020017008826A patent/KR100639274B1/ko not_active IP Right Cessation
- 2000-01-13 JP JP2000593626A patent/JP2002535250A/ja active Pending
- 2000-01-13 BR BR0007453-5A patent/BR0007453A/pt not_active IP Right Cessation
- 2000-01-13 IL IL14398700A patent/IL143987A/xx not_active IP Right Cessation
-
2001
- 2001-07-04 NO NO20013319A patent/NO20013319L/no unknown
- 2001-07-05 IS IS5990A patent/IS5990A/is unknown
-
2008
- 2008-01-18 CY CY20081100076T patent/CY1107149T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP1150996A1 (en) | 2001-11-07 |
| IS5990A (is) | 2001-07-05 |
| ES2295004T3 (es) | 2008-04-16 |
| NO20013319L (no) | 2001-09-12 |
| CY1107149T1 (el) | 2012-10-24 |
| EE200100371A (et) | 2002-02-15 |
| IL143987A (en) | 2005-12-18 |
| IL143987A0 (en) | 2002-04-21 |
| BR0007453A (pt) | 2001-10-30 |
| EP1150996B1 (en) | 2007-11-21 |
| DE60037183D1 (de) | 2008-01-03 |
| PL349769A1 (en) | 2002-09-09 |
| NO20013319D0 (no) | 2001-07-04 |
| TR200102037T2 (tr) | 2001-10-22 |
| US6599894B1 (en) | 2003-07-29 |
| WO2000042059A1 (en) | 2000-07-20 |
| KR100639274B1 (ko) | 2006-10-27 |
| DK1150996T3 (da) | 2008-02-11 |
| KR20010101493A (ko) | 2001-11-14 |
| CN1343217A (zh) | 2002-04-03 |
| JP2002535250A (ja) | 2002-10-22 |
| PT1150996E (pt) | 2008-01-15 |
| AU764121B2 (en) | 2003-08-07 |
| AU2336200A (en) | 2000-08-01 |
| DE60037183T2 (de) | 2008-10-09 |
| CA2355792A1 (en) | 2000-07-20 |
| CZ20012529A3 (cs) | 2001-11-14 |
| NZ512714A (en) | 2003-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1170842C (zh) | 新的脒基苄基胺衍生物及其作为凝血酶抑制剂的用途 | |
| CN1129604C (zh) | 新的氨基酸衍生物及其作为凝血酶抑制剂的用途 | |
| CN1298703C (zh) | 氰基吡咯烷衍生物 | |
| CN1205187C (zh) | 新的脒基衍生物及其作为凝血酶抑制剂的用途 | |
| CN1043762C (zh) | 用作兴奋性氨基酸受体拮抗剂的化合物及其药物组合物 | |
| CN1057522C (zh) | 作为前列腺素i2激动剂的萘衍生物 | |
| CN1114598C (zh) | 芳族化合物和含有它们的药物组合物 | |
| CN1170409A (zh) | 新的2,3-二氧代哌嗪衍生物及其盐 | |
| CN1175953A (zh) | 作为凝血酶抑制剂的新的二肽脒类 | |
| CN1079224A (zh) | 抗菌素化合物 | |
| CN1568323A (zh) | 过氧化物酶体增殖剂应答性受体δ的活化剂 | |
| CN1069971A (zh) | 新型氨磺酰血纤维蛋白原受体对抗剂 | |
| CN1072180A (zh) | 取代的联苯吡啶酮 | |
| CN1274719A (zh) | 取代的吡啶化合物的制备方法 | |
| CN1542002A (zh) | 金属蛋白酶抑制剂,含有它们的药物组合物和其药物用途,其制备方法和中间体 | |
| CN1078968A (zh) | 2-哌嗪酮化合物及它们的制备和使用 | |
| CN1094035A (zh) | 羧酸衍生物,含有这些化合物的药物组合物及其制备方法 | |
| CN1443173A (zh) | 作为TAFIa抑制剂的取代的咪唑 | |
| CN1135212A (zh) | 极性取代的烃类 | |
| CN1159303C (zh) | 单环β-内酰胺类抗生素衍生物的中间体的制备方法 | |
| CN1196696C (zh) | 新的二氢苯并噻喃衍生物和它们作为凝血酶抑制剂的用途 | |
| CN1319987C (zh) | γ-分泌酶抑制剂 | |
| CN1187360C (zh) | 有羧基肽酶b抑制活性的膦酸衍生物 | |
| CN1878768A (zh) | 作为ppar激活剂的杂芳基衍生物 | |
| CN1205183C (zh) | 新的脒基衍生物及其作为凝血酶抑制剂的用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20041013 Termination date: 20110113 |