JP7500555B2 - ヒト免疫不全ウイルス複製の阻害剤 - Google Patents
ヒト免疫不全ウイルス複製の阻害剤 Download PDFInfo
- Publication number
- JP7500555B2 JP7500555B2 JP2021521995A JP2021521995A JP7500555B2 JP 7500555 B2 JP7500555 B2 JP 7500555B2 JP 2021521995 A JP2021521995 A JP 2021521995A JP 2021521995 A JP2021521995 A JP 2021521995A JP 7500555 B2 JP7500555 B2 JP 7500555B2
- Authority
- JP
- Japan
- Prior art keywords
- chloro
- indazol
- ethyl
- mmol
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003112 inhibitor Substances 0.000 title claims description 10
- 241000725303 Human immunodeficiency virus Species 0.000 title description 14
- 230000029812 viral genome replication Effects 0.000 title 1
- 239000000203 mixture Substances 0.000 claims description 157
- 150000001875 compounds Chemical class 0.000 claims description 153
- 150000003839 salts Chemical class 0.000 claims description 72
- 239000003814 drug Substances 0.000 claims description 17
- 208000031886 HIV Infections Diseases 0.000 claims description 13
- 208000037357 HIV infectious disease Diseases 0.000 claims description 13
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 claims description 13
- 208000030507 AIDS Diseases 0.000 claims description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 claims description 6
- 239000003795 chemical substances by application Substances 0.000 claims description 5
- 108010078851 HIV Reverse Transcriptase Proteins 0.000 claims description 4
- 239000002777 nucleoside Substances 0.000 claims description 4
- 150000003833 nucleoside derivatives Chemical class 0.000 claims description 4
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 claims description 3
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 238000002560 therapeutic procedure Methods 0.000 claims description 3
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 claims description 2
- 229940099797 HIV integrase inhibitor Drugs 0.000 claims description 2
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 claims description 2
- 230000034303 cell budding Effects 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 239000002835 hiv fusion inhibitor Substances 0.000 claims description 2
- 239000003084 hiv integrase inhibitor Substances 0.000 claims description 2
- 239000004030 hiv protease inhibitor Substances 0.000 claims description 2
- 238000010255 intramuscular injection Methods 0.000 claims description 2
- 239000007927 intramuscular injection Substances 0.000 claims description 2
- 230000035800 maturation Effects 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 238000010254 subcutaneous injection Methods 0.000 claims description 2
- 239000007929 subcutaneous injection Substances 0.000 claims description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 295
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 200
- 239000007787 solid Substances 0.000 description 142
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 141
- 239000000243 solution Substances 0.000 description 137
- 235000019439 ethyl acetate Nutrition 0.000 description 124
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 113
- 238000002360 preparation method Methods 0.000 description 106
- 239000011541 reaction mixture Substances 0.000 description 105
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 101
- 238000006243 chemical reaction Methods 0.000 description 100
- 238000000034 method Methods 0.000 description 93
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 74
- 238000005160 1H NMR spectroscopy Methods 0.000 description 63
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 58
- 230000002829 reductive effect Effects 0.000 description 50
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 48
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 44
- 239000012044 organic layer Substances 0.000 description 44
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical group CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 42
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 42
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 42
- 239000002904 solvent Substances 0.000 description 42
- 239000000047 product Substances 0.000 description 41
- 239000012267 brine Substances 0.000 description 39
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 39
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 36
- 239000007832 Na2SO4 Substances 0.000 description 33
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 33
- 229910052938 sodium sulfate Inorganic materials 0.000 description 33
- 235000011152 sodium sulphate Nutrition 0.000 description 33
- 238000001914 filtration Methods 0.000 description 31
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 30
- 230000014759 maintenance of location Effects 0.000 description 30
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 28
- 150000002500 ions Chemical class 0.000 description 27
- 125000001153 fluoro group Chemical group F* 0.000 description 26
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 26
- 238000010898 silica gel chromatography Methods 0.000 description 25
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 24
- 229910052796 boron Inorganic materials 0.000 description 24
- 235000019000 fluorine Nutrition 0.000 description 24
- 238000004128 high performance liquid chromatography Methods 0.000 description 24
- 239000000463 material Substances 0.000 description 24
- 239000011734 sodium Substances 0.000 description 24
- 238000002474 experimental method Methods 0.000 description 23
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- 239000000706 filtrate Substances 0.000 description 21
- 238000000746 purification Methods 0.000 description 21
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 21
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 21
- 238000003756 stirring Methods 0.000 description 20
- 229910004298 SiO 2 Inorganic materials 0.000 description 19
- 239000012299 nitrogen atmosphere Substances 0.000 description 19
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 239000007788 liquid Substances 0.000 description 18
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 16
- 239000007819 coupling partner Substances 0.000 description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 239000012071 phase Substances 0.000 description 15
- 238000003828 vacuum filtration Methods 0.000 description 15
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- 239000010410 layer Substances 0.000 description 14
- 229920006395 saturated elastomer Polymers 0.000 description 14
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 13
- 235000019253 formic acid Nutrition 0.000 description 13
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 12
- 239000012300 argon atmosphere Substances 0.000 description 12
- 239000012043 crude product Substances 0.000 description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 12
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 229940079593 drug Drugs 0.000 description 11
- 229910052739 hydrogen Inorganic materials 0.000 description 11
- 239000002245 particle Substances 0.000 description 11
- -1 while the other Proteins 0.000 description 11
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 10
- 125000000217 alkyl group Chemical group 0.000 description 10
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 10
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 10
- 238000002347 injection Methods 0.000 description 10
- 239000007924 injection Substances 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- 239000000377 silicon dioxide Substances 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 8
- 241000700605 Viruses Species 0.000 description 8
- 229910000024 caesium carbonate Inorganic materials 0.000 description 8
- 229910052681 coesite Inorganic materials 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- 229910052906 cristobalite Inorganic materials 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 229910052731 fluorine Inorganic materials 0.000 description 8
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 8
- 239000002244 precipitate Substances 0.000 description 8
- 235000012239 silicon dioxide Nutrition 0.000 description 8
- 229910052682 stishovite Inorganic materials 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- 229910052905 tridymite Inorganic materials 0.000 description 8
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 7
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 7
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 7
- FXRMFOLZFQCWNG-UHFFFAOYSA-N N-(7-amino-4-chloro-1-methylindazol-3-yl)-N-[(4-methoxyphenyl)methyl]methanesulfonamide Chemical compound NC=1C=CC(=C2C(=NN(C=12)C)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC)Cl FXRMFOLZFQCWNG-UHFFFAOYSA-N 0.000 description 7
- 125000002947 alkylene group Chemical group 0.000 description 7
- 229910052786 argon Inorganic materials 0.000 description 7
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 7
- 238000004296 chiral HPLC Methods 0.000 description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 7
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 7
- 239000012074 organic phase Substances 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- HRLPDQGPXDMJPL-UHFFFAOYSA-N N-[7-amino-4-chloro-1-(2,2-difluoroethyl)indazol-3-yl]-N-[(4-methoxyphenyl)methyl]cyclopropanesulfonamide Chemical compound NC=1C=CC(=C2C(=NN(C=12)CC(F)F)N(S(=O)(=O)C1CC1)CC1=CC=C(C=C1)OC)Cl HRLPDQGPXDMJPL-UHFFFAOYSA-N 0.000 description 6
- XIMVKBDIIFQNDZ-UHFFFAOYSA-N N-[7-amino-4-chloro-1-(2,2-difluoroethyl)indazol-3-yl]-N-[(4-methoxyphenyl)methyl]methanesulfonamide Chemical compound NC=1C=CC(=C2C(=NN(C=12)CC(F)F)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC)Cl XIMVKBDIIFQNDZ-UHFFFAOYSA-N 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- 239000012298 atmosphere Substances 0.000 description 6
- 239000013058 crude material Substances 0.000 description 6
- 238000001514 detection method Methods 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 125000004353 pyrazol-1-yl group Chemical group [H]C1=NN(*)C([H])=C1[H] 0.000 description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 6
- 235000017557 sodium bicarbonate Nutrition 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 6
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 5
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 5
- 229940124522 antiretrovirals Drugs 0.000 description 5
- 239000008346 aqueous phase Substances 0.000 description 5
- 229910052801 chlorine Inorganic materials 0.000 description 5
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 5
- WMSPXQIQBQAWLL-UHFFFAOYSA-N cyclopropanesulfonamide Chemical compound NS(=O)(=O)C1CC1 WMSPXQIQBQAWLL-UHFFFAOYSA-N 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 5
- JRHPOFJADXHYBR-HTQZYQBOSA-N (1r,2r)-1-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CN[C@@H]1CCCC[C@H]1NC JRHPOFJADXHYBR-HTQZYQBOSA-N 0.000 description 4
- CZBNUDVCRKSYDG-NSHDSACASA-N (2s)-3-(3,5-difluorophenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CC1=CC(F)=CC(F)=C1 CZBNUDVCRKSYDG-NSHDSACASA-N 0.000 description 4
- 125000006645 (C3-C4) cycloalkyl group Chemical group 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- NSKVWZIEYFSHIM-UHFFFAOYSA-N 2,6-dichloro-3-nitrobenzonitrile Chemical compound [O-][N+](=O)C1=CC=C(Cl)C(C#N)=C1Cl NSKVWZIEYFSHIM-UHFFFAOYSA-N 0.000 description 4
- VQEPOQSIXGAOMF-IUYQGCFVSA-N 2-[(2S,4R)-9-(difluoromethyl)-5,5-difluoro-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetic acid Chemical compound FC([C@@H]1C[C@@H]11)(F)C2=C1C(C(F)F)=NN2CC(=O)O VQEPOQSIXGAOMF-IUYQGCFVSA-N 0.000 description 4
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 4
- QSVBVBFCFAOLAG-UHFFFAOYSA-N 4-chloro-1-(2,2-difluoroethyl)-7-nitroindazol-3-amine Chemical compound ClC1=C2C(=NN(C2=C(C=C1)[N+](=O)[O-])CC(F)F)N QSVBVBFCFAOLAG-UHFFFAOYSA-N 0.000 description 4
- IUSPKTIVYGCXMZ-UHFFFAOYSA-N 4-chloro-7-nitro-1h-indazol-3-amine Chemical compound C1=CC(Cl)=C2C(N)=NNC2=C1[N+]([O-])=O IUSPKTIVYGCXMZ-UHFFFAOYSA-N 0.000 description 4
- WSUZWTPRZQLWHI-UHFFFAOYSA-N 7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)indazol-3-amine Chemical compound BrC=1C=CC(=C2C(=NN(C=12)CC(F)(F)F)N)Cl WSUZWTPRZQLWHI-UHFFFAOYSA-N 0.000 description 4
- HSQISZWGASOOBB-UHFFFAOYSA-N 7-bromo-4-chloro-1-(2,2-difluoroethyl)indazol-3-amine Chemical compound BrC=1C=CC(=C2C(=NN(C=12)CC(F)F)N)Cl HSQISZWGASOOBB-UHFFFAOYSA-N 0.000 description 4
- IQJRJSKDZJKCIW-UHFFFAOYSA-N 7-bromo-4-chloro-1h-indazol-3-amine Chemical compound C1=CC(Cl)=C2C(N)=NNC2=C1Br IQJRJSKDZJKCIW-UHFFFAOYSA-N 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 108060001084 Luciferase Proteins 0.000 description 4
- PMPAFZLVEIWZPO-UHFFFAOYSA-N N-[7-bromo-4-chloro-1-(2,2-difluoroethyl)indazol-3-yl]-N-[(4-methoxyphenyl)methyl]methanesulfonamide Chemical compound BrC=1C=CC(=C2C(=NN(C=12)CC(F)F)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC)Cl PMPAFZLVEIWZPO-UHFFFAOYSA-N 0.000 description 4
- SFQGKMUQZGPZHN-UHFFFAOYSA-N N-[7-bromo-4-chloro-1-(2,2-difluoroethyl)indazol-3-yl]methanesulfonamide Chemical compound BrC=1C=CC(=C2C(=NN(C=12)CC(F)F)NS(=O)(=O)C)Cl SFQGKMUQZGPZHN-UHFFFAOYSA-N 0.000 description 4
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- WQQZKEFUOHKGII-UHFFFAOYSA-N bicyclo[3.1.0]hexan-3-one Chemical compound C1C(=O)CC2CC21 WQQZKEFUOHKGII-UHFFFAOYSA-N 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 4
- KUMNEOGIHFCNQW-UHFFFAOYSA-N diphenyl phosphite Chemical compound C=1C=CC=CC=1OP([O-])OC1=CC=CC=C1 KUMNEOGIHFCNQW-UHFFFAOYSA-N 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- MMXUNPIBPOFYMN-UHFFFAOYSA-N ethyl 2-[5-cyclopropyl-3-(difluoromethyl)pyrazol-1-yl]acetate Chemical compound CCOC(=O)CN1N=C(C(F)F)C=C1C1CC1 MMXUNPIBPOFYMN-UHFFFAOYSA-N 0.000 description 4
- 150000002431 hydrogen Chemical class 0.000 description 4
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 4
- 238000006396 nitration reaction Methods 0.000 description 4
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 4
- 235000010378 sodium ascorbate Nutrition 0.000 description 4
- 229960005055 sodium ascorbate Drugs 0.000 description 4
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- HEYONDYPXIUDCK-UHFFFAOYSA-L (5-diphenylphosphanyl-9,9-dimethylxanthen-4-yl)-diphenylphosphane;palladium(2+);dichloride Chemical compound Cl[Pd]Cl.C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 HEYONDYPXIUDCK-UHFFFAOYSA-L 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 3
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 3
- ZZMKJXLUGCPEFK-UHFFFAOYSA-N 1-bromo-2-nitro-4-[1-(trifluoromethyl)cyclopropyl]benzene Chemical compound C1(Br)=C(N(=O)=O)C=C(C2(CC2)C(F)(F)F)C=C1 ZZMKJXLUGCPEFK-UHFFFAOYSA-N 0.000 description 3
- UOFKQNOBCDNGOW-UHFFFAOYSA-N 1-cyclopropyl-4,4-difluorobutane-1,3-dione Chemical compound FC(F)C(=O)CC(=O)C1CC1 UOFKQNOBCDNGOW-UHFFFAOYSA-N 0.000 description 3
- FGABAUDZUVWFPM-UHFFFAOYSA-N 1-methyl-2-nitro-4-[1-(trifluoromethyl)cyclopropyl]benzene Chemical compound CC1=C(C=C(C=C1)C1(CC1)C(F)(F)F)[N+](=O)[O-] FGABAUDZUVWFPM-UHFFFAOYSA-N 0.000 description 3
- CQOQHGBSJIWOCA-UHFFFAOYSA-N 2,6-dichloro-3-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC=C(Cl)C(C=O)=C1Cl CQOQHGBSJIWOCA-UHFFFAOYSA-N 0.000 description 3
- IMJHQTMWQWGHCX-UHFFFAOYSA-N 2-(2,2-difluoroacetyl)bicyclo[3.1.0]hexan-3-one Chemical compound C1C(=O)C(C(=O)C(F)F)C2CC21 IMJHQTMWQWGHCX-UHFFFAOYSA-N 0.000 description 3
- DOSGEBYQRMBTGS-UHFFFAOYSA-N 2-(3,6-dihydro-2h-pyran-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CCOCC1 DOSGEBYQRMBTGS-UHFFFAOYSA-N 0.000 description 3
- VQEPOQSIXGAOMF-UHFFFAOYSA-N 2-(3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-1h-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid Chemical compound C12CC2C(F)(F)C2=C1C(C(F)F)=NN2CC(=O)O VQEPOQSIXGAOMF-UHFFFAOYSA-N 0.000 description 3
- TXIOULNLSCTYNR-UHFFFAOYSA-N 2-[3,5-bis(difluoromethyl)pyrazol-1-yl]acetic acid Chemical compound OC(=O)CN1N=C(C(F)F)C=C1C(F)F TXIOULNLSCTYNR-UHFFFAOYSA-N 0.000 description 3
- LHZSMGINLGGLEQ-UHFFFAOYSA-N 2-amino-4-[1-(trifluoromethyl)cyclopropyl]benzoic acid Chemical compound NC1=C(C(=O)O)C=CC(=C1)C1(CC1)C(F)(F)F LHZSMGINLGGLEQ-UHFFFAOYSA-N 0.000 description 3
- BNNICQAVXPXQAH-UHFFFAOYSA-N 2-amino-4-bromobenzoic acid Chemical compound NC1=CC(Br)=CC=C1C(O)=O BNNICQAVXPXQAH-UHFFFAOYSA-N 0.000 description 3
- AKXGJPCVOPWTAY-UHFFFAOYSA-N 2-nitro-4-[1-(trifluoromethyl)cyclopropyl]benzoic acid Chemical compound [N+](=O)([O-])C1=C(C(=O)O)C=CC(=C1)C1(CC1)C(F)(F)F AKXGJPCVOPWTAY-UHFFFAOYSA-N 0.000 description 3
- WFFSWRXTSBYYKP-UHFFFAOYSA-N 3-bromo-6-chloro-2-fluorobenzaldehyde Chemical compound FC1=C(Br)C=CC(Cl)=C1C=O WFFSWRXTSBYYKP-UHFFFAOYSA-N 0.000 description 3
- TWYUOUVQRHKSFE-UHFFFAOYSA-N 3-bromo-6-chloro-2-fluorobenzonitrile Chemical compound FC1=C(Br)C=CC(Cl)=C1C#N TWYUOUVQRHKSFE-UHFFFAOYSA-N 0.000 description 3
- XQIZZXZPBIMBGY-UHFFFAOYSA-N 4-chloro-1-methyl-7-nitroindazol-3-amine Chemical compound C1=CC([N+]([O-])=O)=C2N(C)N=C(N)C2=C1Cl XQIZZXZPBIMBGY-UHFFFAOYSA-N 0.000 description 3
- QOGBWRRHJQSLCY-UHFFFAOYSA-N 5-cyclopropyl-3-(difluoromethyl)-1h-pyrazole Chemical compound N1N=C(C(F)F)C=C1C1CC1 QOGBWRRHJQSLCY-UHFFFAOYSA-N 0.000 description 3
- JKVZDIFRPVCPJI-UHFFFAOYSA-N 7-bromo-4-chloro-1-methylindazol-3-amine Chemical compound C1=CC(Br)=C2N(C)N=C(N)C2=C1Cl JKVZDIFRPVCPJI-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 3
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 206010059866 Drug resistance Diseases 0.000 description 3
- AVNYEBJZDPZOKJ-UHFFFAOYSA-N N-(4-chloro-1-methyl-7-nitroindazol-3-yl)-N-[(4-methoxyphenyl)methyl]methanesulfonamide Chemical compound ClC1=C2C(=NN(C2=C(C=C1)[N+](=O)[O-])C)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC AVNYEBJZDPZOKJ-UHFFFAOYSA-N 0.000 description 3
- RJGHLBMMMIVMBJ-UHFFFAOYSA-N N-(4-chloro-1-methyl-7-nitroindazol-3-yl)methanesulfonamide Chemical compound ClC1=C2C(=NN(C2=C(C=C1)[N+](=O)[O-])C)NS(=O)(=O)C RJGHLBMMMIVMBJ-UHFFFAOYSA-N 0.000 description 3
- ZIYBIEZYLPJPKG-UHFFFAOYSA-N N-(7-bromo-4-chloro-1-methylindazol-3-yl)-N-[(4-methoxyphenyl)methyl]methanesulfonamide Chemical compound BrC=1C=CC(=C2C(=NN(C=12)C)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC)Cl ZIYBIEZYLPJPKG-UHFFFAOYSA-N 0.000 description 3
- IMCFAWQUNZCDHB-UHFFFAOYSA-N N-(7-bromo-4-chloro-1-methylindazol-3-yl)methanesulfonamide Chemical compound BrC=1C=CC(=C2C(=NN(C=12)C)NS(=O)(=O)C)Cl IMCFAWQUNZCDHB-UHFFFAOYSA-N 0.000 description 3
- MZBLAEJZKQDJKA-UHFFFAOYSA-N N-[4-chloro-1-(2,2-difluoroethyl)-7-nitroindazol-3-yl]-N-[(4-methoxyphenyl)methyl]cyclopropanesulfonamide Chemical compound ClC1=C2C(=NN(C2=C(C=C1)[N+](=O)[O-])CC(F)F)N(S(=O)(=O)C1CC1)CC1=CC=C(C=C1)OC MZBLAEJZKQDJKA-UHFFFAOYSA-N 0.000 description 3
- PCMKRLYEGGMIMY-UHFFFAOYSA-N N-[4-chloro-1-(2,2-difluoroethyl)-7-nitroindazol-3-yl]-N-[(4-methoxyphenyl)methyl]methanesulfonamide Chemical compound ClC1=C2C(=NN(C2=C(C=C1)[N+](=O)[O-])CC(F)F)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC PCMKRLYEGGMIMY-UHFFFAOYSA-N 0.000 description 3
- BTQCYIMPHCTVCC-UHFFFAOYSA-N N-[4-chloro-1-(2,2-difluoroethyl)-7-nitroindazol-3-yl]cyclopropanesulfonamide Chemical compound ClC1=C2C(=NN(C2=C(C=C1)[N+](=O)[O-])CC(F)F)NS(=O)(=O)C1CC1 BTQCYIMPHCTVCC-UHFFFAOYSA-N 0.000 description 3
- BNBJJEJNKSLOAO-UHFFFAOYSA-N N-[4-chloro-1-(2,2-difluoroethyl)-7-nitroindazol-3-yl]methanesulfonamide Chemical compound ClC1=C2C(=NN(C2=C(C=C1)[N+](=O)[O-])CC(F)F)NS(=O)(=O)C BNBJJEJNKSLOAO-UHFFFAOYSA-N 0.000 description 3
- DAMLIPMODAZWMK-UHFFFAOYSA-N N-[7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)indazol-3-yl]-N-[(4-methoxyphenyl)methyl]methanesulfonamide Chemical compound BrC=1C=CC(=C2C(=NN(C=12)CC(F)(F)F)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC)Cl DAMLIPMODAZWMK-UHFFFAOYSA-N 0.000 description 3
- KHNWRPJGEJTXFT-UHFFFAOYSA-N N-[7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)indazol-3-yl]methanesulfonamide Chemical compound BrC=1C=CC(=C2C(=NN(C=12)CC(F)(F)F)NS(=O)(=O)C)Cl KHNWRPJGEJTXFT-UHFFFAOYSA-N 0.000 description 3
- KVAPLICGHKBWAQ-UHFFFAOYSA-N N-[7-bromo-4-chloro-1-(2,2-difluoroethyl)indazol-3-yl]cyclopropanesulfonamide Chemical compound BrC=1C=CC(=C2C(=NN(C=12)CC(F)F)NS(=O)(=O)C1CC1)Cl KVAPLICGHKBWAQ-UHFFFAOYSA-N 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- JAGLUFIJRSKTHI-NTSWFWBYSA-N [(2R,6S)-2,6-dimethyl-3,6-dihydro-2H-pyran-4-yl] trifluoromethanesulfonate Chemical compound FC(S(=O)(=O)OC=1C[C@H](O[C@H](C=1)C)C)(F)F JAGLUFIJRSKTHI-NTSWFWBYSA-N 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 238000011225 antiretroviral therapy Methods 0.000 description 3
- 239000003903 antiretrovirus agent Substances 0.000 description 3
- YKXMZSYOSBDLMM-UHFFFAOYSA-N bicyclo[3.1.0]hexan-3-ol Chemical compound C1C(O)CC2CC21 YKXMZSYOSBDLMM-UHFFFAOYSA-N 0.000 description 3
- 230000002051 biphasic effect Effects 0.000 description 3
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 3
- 239000003610 charcoal Substances 0.000 description 3
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical compound CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 3
- 229910001873 dinitrogen Inorganic materials 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- OFRIPBLMPRXVMH-UHFFFAOYSA-N ethyl 2-[3,5-bis(difluoromethyl)pyrazol-1-yl]acetate Chemical compound CCOC(=O)CN1N=C(C(F)F)C=C1C(F)F OFRIPBLMPRXVMH-UHFFFAOYSA-N 0.000 description 3
- RULJEGNYRJHGJJ-UHFFFAOYSA-N ethyl 2-[9-(difluoromethyl)-5,5-difluoro-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetate Chemical compound C12CC2C(F)(F)C2=C1C(C(F)F)=NN2CC(=O)OCC RULJEGNYRJHGJJ-UHFFFAOYSA-N 0.000 description 3
- YMINJWQOSFPOLL-UHFFFAOYSA-N ethyl 2-[9-(difluoromethyl)-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetate Chemical compound C12CC2CC2=C1C(C(F)F)=NN2CC(=O)OCC YMINJWQOSFPOLL-UHFFFAOYSA-N 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 235000011056 potassium acetate Nutrition 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 230000001566 pro-viral effect Effects 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000000171 quenching effect Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- JPUCIUAPPVFCKF-PMERELPUSA-N tert-butyl N-[(1S)-1-[7-bromo-3-[4-chloro-1-(2,2-difluoroethyl)-3-[(4-methoxyphenyl)methyl-methylsulfonylamino]indazol-7-yl]-4-oxoquinazolin-2-yl]-2-(3,5-difluorophenyl)ethyl]carbamate Chemical compound BrC1=CC=C2C(N(C(=NC2=C1)[C@H](CC1=CC(=CC(=C1)F)F)NC(OC(C)(C)C)=O)C=1C=CC(=C2C(=NN(C=12)CC(F)F)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC)Cl)=O JPUCIUAPPVFCKF-PMERELPUSA-N 0.000 description 3
- MJBMQEOEAFETAK-PMERELPUSA-N tert-butyl N-[(1S)-1-[7-bromo-3-[4-chloro-3-[(4-methoxyphenyl)methyl-methylsulfonylamino]-1-methylindazol-7-yl]-4-oxoquinazolin-2-yl]-2-(3,5-difluorophenyl)ethyl]carbamate Chemical compound BrC1=CC=C2C(N(C(=NC2=C1)[C@H](CC1=CC(=CC(=C1)F)F)NC(OC(C)(C)C)=O)C=1C=CC(=C2C(=NN(C=12)C)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC)Cl)=O MJBMQEOEAFETAK-PMERELPUSA-N 0.000 description 3
- MZLJLANIXKQBPU-YTTGMZPUSA-N tert-butyl N-[(1S)-1-[7-bromo-3-[4-chloro-3-[cyclopropylsulfonyl-[(4-methoxyphenyl)methyl]amino]-1-(2,2-difluoroethyl)indazol-7-yl]-4-oxoquinazolin-2-yl]-2-(3,5-difluorophenyl)ethyl]carbamate Chemical compound BrC1=CC=C2C(N(C(=NC2=C1)[C@H](CC1=CC(=CC(=C1)F)F)NC(OC(C)(C)C)=O)C=1C=CC(=C2C(=NN(C=12)CC(F)F)N(S(=O)(=O)C1CC1)CC1=CC=C(C=C1)OC)Cl)=O MZLJLANIXKQBPU-YTTGMZPUSA-N 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 2
- NKULBUOBGILEAR-UHFFFAOYSA-N 2,2-difluoroethyl trifluoromethanesulfonate Chemical compound FC(F)COS(=O)(=O)C(F)(F)F NKULBUOBGILEAR-UHFFFAOYSA-N 0.000 description 2
- VQEPOQSIXGAOMF-DMTCNVIQSA-N 2-[(2R,4S)-9-(difluoromethyl)-5,5-difluoro-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetic acid Chemical compound FC([C@H]1C[C@H]11)(F)C2=C1C(C(F)F)=NN2CC(=O)O VQEPOQSIXGAOMF-DMTCNVIQSA-N 0.000 description 2
- KFYMLAWTVBTSLH-UHFFFAOYSA-N 2-[5-cyclopropyl-3-(difluoromethyl)pyrazol-1-yl]acetic acid Chemical compound OC(=O)CN1N=C(C(F)F)C=C1C1CC1 KFYMLAWTVBTSLH-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 2
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 208000033962 Fontaine progeroid syndrome Diseases 0.000 description 2
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- 239000005089 Luciferase Substances 0.000 description 2
- AMACRZNQWUGVSQ-UHFFFAOYSA-N N-(4-chloro-1-methyl-7-nitroindazol-3-yl)-N-methylsulfonylmethanesulfonamide Chemical compound ClC1=C2C(=NN(C2=C(C=C1)[N+](=O)[O-])C)N(S(=O)(=O)C)S(=O)(=O)C AMACRZNQWUGVSQ-UHFFFAOYSA-N 0.000 description 2
- UXLFQONUWBTLSQ-UHFFFAOYSA-N N-[4-chloro-1-(2,2-difluoroethyl)-7-nitroindazol-3-yl]-N-methylsulfonylmethanesulfonamide Chemical compound CS(=O)(=O)N(C1=NN(CC(F)F)C2=C(C=CC(Cl)=C12)[N+]([O-])=O)S(C)(=O)=O UXLFQONUWBTLSQ-UHFFFAOYSA-N 0.000 description 2
- GLROOPIZDWVAMR-SFHVURJKSA-N N-[7-[2-[(1S)-1-amino-2-(3,5-difluorophenyl)ethyl]-7-bromo-4-oxoquinazolin-3-yl]-4-chloro-1-(2,2-difluoroethyl)indazol-3-yl]methanesulfonamide Chemical compound N[C@@H](CC1=CC(=CC(=C1)F)F)C1=NC2=CC(=CC=C2C(N1C=1C=CC(=C2C(=NN(C=12)CC(F)F)NS(=O)(=O)C)Cl)=O)Br GLROOPIZDWVAMR-SFHVURJKSA-N 0.000 description 2
- UYIXXDMFEDHPMF-UHFFFAOYSA-N N-[7-amino-4-chloro-1-(2,2,2-trifluoroethyl)indazol-3-yl]-N-[(4-methoxyphenyl)methyl]methanesulfonamide Chemical compound NC=1C=CC(=C2C(=NN(C=12)CC(F)(F)F)N(S(=O)(=O)C)CC1=CC=C(C=C1)OC)Cl UYIXXDMFEDHPMF-UHFFFAOYSA-N 0.000 description 2
- ONAYYKWSMXNXDV-UHFFFAOYSA-N N-[7-bromo-4-chloro-1-(2,2-difluoroethyl)indazol-3-yl]-N-[(4-methoxyphenyl)methyl]cyclopropanesulfonamide Chemical compound BrC=1C=CC(=C2C(=NN(C=12)CC(F)F)N(S(=O)(=O)C1CC1)CC1=CC=C(C=C1)OC)Cl ONAYYKWSMXNXDV-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229910002666 PdCl2 Inorganic materials 0.000 description 2
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 2
- 239000001099 ammonium carbonate Substances 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000005587 bubbling Effects 0.000 description 2
- 229940124765 capsid inhibitor Drugs 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- PFWWSGFPICCWGU-UHFFFAOYSA-N cyclopropanesulfonyl chloride Chemical compound ClS(=O)(=O)C1CC1 PFWWSGFPICCWGU-UHFFFAOYSA-N 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Substances CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 2
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 2
- LGIBDQRYOFBMTC-UHFFFAOYSA-N dnc010031 Chemical compound C1=CC(O)=CC=C1C1C(=O)NC2=CC=CC=C2C2=C3C1=CNC3=NC=C2 LGIBDQRYOFBMTC-UHFFFAOYSA-N 0.000 description 2
- GZKHDVAKKLTJPO-UHFFFAOYSA-N ethyl 2,2-difluoroacetate Chemical compound CCOC(=O)C(F)F GZKHDVAKKLTJPO-UHFFFAOYSA-N 0.000 description 2
- YXFKKLLBEIZKAO-UHFFFAOYSA-N ethyl 2-[9'-(difluoromethyl)spiro[1,3-dithiolane-2,5'-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-diene]-7'-yl]acetate Chemical compound C1=2N(CC(=O)OCC)N=C(C(F)F)C=2C2CC2C21SCCS2 YXFKKLLBEIZKAO-UHFFFAOYSA-N 0.000 description 2
- CMSIJCXFYYYCDW-UHFFFAOYSA-N ethyl 2-[9-(difluoromethyl)-5-oxo-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetate Chemical compound C12CC2C(=O)C2=C1C(C(F)F)=NN2CC(=O)OCC CMSIJCXFYYYCDW-UHFFFAOYSA-N 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 239000012456 homogeneous solution Substances 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 229940124525 integrase strand transfer inhibitor Drugs 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- 229940056360 penicillin g Drugs 0.000 description 2
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 239000012258 stirred mixture Substances 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 235000011149 sulphuric acid Nutrition 0.000 description 2
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- PBIMIGNDTBRRPI-UHFFFAOYSA-N trifluoro borate Chemical compound FOB(OF)OF PBIMIGNDTBRRPI-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- FTTDMWQQHZSARH-UHFFFAOYSA-N 1,1,5,5-tetrafluoropentane-2,4-dione Chemical compound FC(F)C(=O)CC(=O)C(F)F FTTDMWQQHZSARH-UHFFFAOYSA-N 0.000 description 1
- VYMPLPIFKRHAAC-UHFFFAOYSA-N 1,2-ethanedithiol Chemical compound SCCS VYMPLPIFKRHAAC-UHFFFAOYSA-N 0.000 description 1
- IMLSAISZLJGWPP-UHFFFAOYSA-N 1,3-dithiolane Chemical compound C1CSCS1 IMLSAISZLJGWPP-UHFFFAOYSA-N 0.000 description 1
- LJOJKFHNKMTEFS-UHFFFAOYSA-N 1-bromo-4-[1-(trifluoromethyl)cyclopropyl]benzene Chemical compound C=1C=C(Br)C=CC=1C1(C(F)(F)F)CC1 LJOJKFHNKMTEFS-UHFFFAOYSA-N 0.000 description 1
- FPNVMCMDWZNTEU-UHFFFAOYSA-N 1-bromo-4-chloro-2-fluorobenzene Chemical compound FC1=CC(Cl)=CC=C1Br FPNVMCMDWZNTEU-UHFFFAOYSA-N 0.000 description 1
- HVCFCNAITDHQFX-UHFFFAOYSA-N 1-cyclopropylethanone Chemical compound CC(=O)C1CC1 HVCFCNAITDHQFX-UHFFFAOYSA-N 0.000 description 1
- SQMVRFXDBRYXFQ-UHFFFAOYSA-N 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2h-pyridine Chemical compound C1N(C)CCC(B2OC(C)(C)C(C)(C)O2)=C1 SQMVRFXDBRYXFQ-UHFFFAOYSA-N 0.000 description 1
- RTMMSCJWQYWMNK-UHFFFAOYSA-N 2,2,2-trifluoroethyl trifluoromethanesulfonate Chemical compound FC(F)(F)COS(=O)(=O)C(F)(F)F RTMMSCJWQYWMNK-UHFFFAOYSA-N 0.000 description 1
- GBBSAMQTQCPOBF-UHFFFAOYSA-N 2,4,6-trimethyl-1,3,5,2,4,6-trioxatriborinane Chemical compound CB1OB(C)OB(C)O1 GBBSAMQTQCPOBF-UHFFFAOYSA-N 0.000 description 1
- DMIYKWPEFRFTPY-UHFFFAOYSA-N 2,6-dichlorobenzaldehyde Chemical compound ClC1=CC=CC(Cl)=C1C=O DMIYKWPEFRFTPY-UHFFFAOYSA-N 0.000 description 1
- GQAKLNMZZSCFDS-UHFFFAOYSA-N 2-(2-ethylhydrazinyl)acetic acid;hydrochloride Chemical compound Cl.CCNNCC(O)=O GQAKLNMZZSCFDS-UHFFFAOYSA-N 0.000 description 1
- QNZFUMVTUFOLRT-UHFFFAOYSA-N 2-(cyclohexen-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CCCCC1 QNZFUMVTUFOLRT-UHFFFAOYSA-N 0.000 description 1
- JFTZVYKESKQING-UHFFFAOYSA-N 2-(cyclopenten-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CCCC1 JFTZVYKESKQING-UHFFFAOYSA-N 0.000 description 1
- KZDCMKVLEYCGQX-UDPGNSCCSA-N 2-(diethylamino)ethyl 4-aminobenzoate;(2s,5r,6r)-3,3-dimethyl-7-oxo-6-[(2-phenylacetyl)amino]-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid;hydrate Chemical compound O.CCN(CC)CCOC(=O)C1=CC=C(N)C=C1.N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 KZDCMKVLEYCGQX-UDPGNSCCSA-N 0.000 description 1
- GBBLFSIRMZHIRY-UHFFFAOYSA-N 2-[3-cyclopropyl-5-(difluoromethyl)pyrazol-1-yl]acetic acid Chemical compound C1=C(C(F)F)N(CC(=O)O)N=C1C1CC1 GBBLFSIRMZHIRY-UHFFFAOYSA-N 0.000 description 1
- NXFFJDQHYLNEJK-UHFFFAOYSA-N 2-[4-[(4-chlorophenyl)methyl]-7-fluoro-5-methylsulfonyl-2,3-dihydro-1h-cyclopenta[b]indol-3-yl]acetic acid Chemical compound C1=2C(S(=O)(=O)C)=CC(F)=CC=2C=2CCC(CC(O)=O)C=2N1CC1=CC=C(Cl)C=C1 NXFFJDQHYLNEJK-UHFFFAOYSA-N 0.000 description 1
- ZYDXBIXQPLRRIK-UHFFFAOYSA-N 3-methoxy-3-methylbut-1-yne Chemical compound COC(C)(C)C#C ZYDXBIXQPLRRIK-UHFFFAOYSA-N 0.000 description 1
- ONHWZTRGTFWBIX-UHFFFAOYSA-N 3-methylbut-2-en-2-ylboronic acid Chemical compound CC(C)=C(C)B(O)O ONHWZTRGTFWBIX-UHFFFAOYSA-N 0.000 description 1
- CFBVWCHTNQHZLT-UHFFFAOYSA-N 4-methoxy-5-[3-(2-methoxy-4-nitro-5-sulfophenyl)-5-(phenylcarbamoyl)tetrazol-3-ium-2-yl]-2-nitrobenzenesulfonate Chemical compound COC1=CC([N+]([O-])=O)=C(S([O-])(=O)=O)C=C1N1[N+](C=2C(=CC(=C(C=2)S(O)(=O)=O)[N+]([O-])=O)OC)=NC(C(=O)NC=2C=CC=CC=2)=N1 CFBVWCHTNQHZLT-UHFFFAOYSA-N 0.000 description 1
- SVWCVXFHTHCJJB-UHFFFAOYSA-N 4-methylpentanoyl chloride Chemical compound CC(C)CCC(Cl)=O SVWCVXFHTHCJJB-UHFFFAOYSA-N 0.000 description 1
- PXRKCOCTEMYUEG-UHFFFAOYSA-N 5-aminoisoindole-1,3-dione Chemical compound NC1=CC=C2C(=O)NC(=O)C2=C1 PXRKCOCTEMYUEG-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 108010032976 Enfuvirtide Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 229940122440 HIV protease inhibitor Drugs 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- OTQYFZKDEYBJMF-UHFFFAOYSA-N N-[(2,6-dichloro-3-nitrophenyl)methylidene]hydroxylamine Chemical compound ClC1=C(C=NO)C(=CC=C1[N+](=O)[O-])Cl OTQYFZKDEYBJMF-UHFFFAOYSA-N 0.000 description 1
- WXEQKXFUSTXBGP-FQEVSTJZSA-N N-[7-[2-[(1S)-1-amino-2-(3,5-difluorophenyl)ethyl]-7-bromo-4-oxoquinazolin-3-yl]-4-chloro-1-(2,2-difluoroethyl)indazol-3-yl]cyclopropanesulfonamide Chemical compound C1(CC1)S(=O)(=O)NC1=NN(C2=C(C=CC(=C12)Cl)N1C(=NC2=CC(=CC=C2C1=O)Br)[C@H](CC1=CC(=CC(=C1)F)F)N)CC(F)F WXEQKXFUSTXBGP-FQEVSTJZSA-N 0.000 description 1
- UDSDYWQQYXTUTO-SFHVURJKSA-N N-[7-[2-[(1S)-1-amino-2-(3,5-difluorophenyl)ethyl]-7-bromo-4-oxoquinazolin-3-yl]-4-chloro-1-methylindazol-3-yl]methanesulfonamide Chemical compound N[C@@H](CC1=CC(=CC(=C1)F)F)C1=NC2=CC(=CC=C2C(N1C=1C=CC(=C2C(=NN(C=12)C)NS(=O)(=O)C)Cl)=O)Br UDSDYWQQYXTUTO-SFHVURJKSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 108010052090 Renilla Luciferases Proteins 0.000 description 1
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101800001690 Transmembrane protein gp41 Proteins 0.000 description 1
- 206010066901 Treatment failure Diseases 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- CIUQDSCDWFSTQR-UHFFFAOYSA-N [C]1=CC=CC=C1 Chemical class [C]1=CC=CC=C1 CIUQDSCDWFSTQR-UHFFFAOYSA-N 0.000 description 1
- LVZGQWKTUCVPBQ-UHFFFAOYSA-N acetic acid;trifluoroborane Chemical compound CC(O)=O.FB(F)F LVZGQWKTUCVPBQ-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- DQPBABKTKYNPMH-UHFFFAOYSA-N amino hydrogen sulfate Chemical compound NOS(O)(=O)=O DQPBABKTKYNPMH-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000002832 anti-viral assay Methods 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 239000011449 brick Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- ZCIGNRJZKPOIKD-CQXVEOKZSA-N cobicistat Chemical compound S1C(C(C)C)=NC(CN(C)C(=O)N[C@@H](CCN2CCOCC2)C(=O)N[C@H](CC[C@H](CC=2C=CC=CC=2)NC(=O)OCC=2SC=NC=2)CC=2C=CC=CC=2)=C1 ZCIGNRJZKPOIKD-CQXVEOKZSA-N 0.000 description 1
- 229960002402 cobicistat Drugs 0.000 description 1
- 238000007398 colorimetric assay Methods 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- JFWMYCVMQSLLOO-UHFFFAOYSA-N cyclobutanecarbonyl chloride Chemical compound ClC(=O)C1CCC1 JFWMYCVMQSLLOO-UHFFFAOYSA-N 0.000 description 1
- WEIMJSIRDZDHAH-UHFFFAOYSA-N cyclopent-3-en-1-ol Chemical compound OC1CC=CC1 WEIMJSIRDZDHAH-UHFFFAOYSA-N 0.000 description 1
- VRLDVERQJMEPIF-UHFFFAOYSA-N dbdmh Chemical compound CC1(C)N(Br)C(=O)N(Br)C1=O VRLDVERQJMEPIF-UHFFFAOYSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- HQWPLXHWEZZGKY-UHFFFAOYSA-N diethylzinc Chemical compound CC[Zn]CC HQWPLXHWEZZGKY-UHFFFAOYSA-N 0.000 description 1
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- PEASPLKKXBYDKL-FXEVSJAOSA-N enfuvirtide Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(C)=O)[C@@H](C)O)[C@@H](C)CC)C1=CN=CN1 PEASPLKKXBYDKL-FXEVSJAOSA-N 0.000 description 1
- 229960002062 enfuvirtide Drugs 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- HZZRIIPYFPIKHR-UHFFFAOYSA-N ethyl 2-hydrazinylacetate;hydron;chloride Chemical compound Cl.CCOC(=O)CNN HZZRIIPYFPIKHR-UHFFFAOYSA-N 0.000 description 1
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 150000003840 hydrochlorides Chemical group 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- KJDJPXUIZYHXEZ-UHFFFAOYSA-N hydrogen sulfate;methylaminoazanium Chemical compound CN[NH3+].OS([O-])(=O)=O KJDJPXUIZYHXEZ-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- RCBVKBFIWMOMHF-UHFFFAOYSA-L hydroxy-(hydroxy(dioxo)chromio)oxy-dioxochromium;pyridine Chemical compound C1=CC=NC=C1.C1=CC=NC=C1.O[Cr](=O)(=O)O[Cr](O)(=O)=O RCBVKBFIWMOMHF-UHFFFAOYSA-L 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- BAUYGSIQEAFULO-UHFFFAOYSA-L iron(2+) sulfate (anhydrous) Chemical compound [Fe+2].[O-]S([O-])(=O)=O BAUYGSIQEAFULO-UHFFFAOYSA-L 0.000 description 1
- 229910000359 iron(II) sulfate Inorganic materials 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 229960004710 maraviroc Drugs 0.000 description 1
- GSNHKUDZZFZSJB-QYOOZWMWSA-N maraviroc Chemical compound CC(C)C1=NN=C(C)N1[C@@H]1C[C@H](N2CC[C@H](NC(=O)C3CCC(F)(F)CC3)C=3C=CC=CC=3)CC[C@H]2C1 GSNHKUDZZFZSJB-QYOOZWMWSA-N 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000013264 metal-organic assembly Substances 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 108700004028 nef Genes Proteins 0.000 description 1
- 101150023385 nef gene Proteins 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 206010073131 oligoastrocytoma Diseases 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- CUQOHAYJWVTKDE-UHFFFAOYSA-N potassium;butan-1-olate Chemical compound [K+].CCCC[O-] CUQOHAYJWVTKDE-UHFFFAOYSA-N 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229960000311 ritonavir Drugs 0.000 description 1
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000008259 solid foam Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 239000012096 transfection reagent Substances 0.000 description 1
- REDSKZBUUUQMSK-UHFFFAOYSA-N tributyltin Chemical compound CCCC[Sn](CCCC)CCCC.CCCC[Sn](CCCC)CCCC REDSKZBUUUQMSK-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 230000017613 viral reproduction Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
Description

G1は1~3個のフッ素で場合により置換されているC6~C8アルキルであるか、又はG1は-(C2~C3アルキレニル)-で置換されており、G2で1回場合により置換されており、1~3個のフッ素で場合により置換されているC2~C5アルキルであるか、又はG1はG2で置換されているC1~C5アルキルであるか、又はG1は

Z1及びZ3は独立に、-CH3又は-CH2CH3で1回場合により置換されている-C1~C3アルキレンであり、
Z2は-O-、-S(O2)-、-NH-又は-N(G4)であり、
X1、X2及びX3は独立に、H、F又はClであり、ここで、X1、X2及びX3のうちの1つは-CN、-OCH3、-CH3、-CH2F、-CHF2及び-CF3から場合により選択されてもよく、
E4及びE5は独立に、-CH3又は-CH2CH3で1回場合により置換されている-C1~C2アルキレンであり、
E6は-C(H2)-、-O-、-S(O2)-、-NH-又は-N(G4)であり、
E7、E8及びE9は独立に、H又は-CH3であり、
Y1及びY2は独立に、-N(H)-又は-O-であり、
G2はフェニル、-OH、1~3個のフッ素で場合により置換されている-O(C1~C3アルキル)、1~2個のフッ素で場合により置換されているC3~C4シクロアルキルであるか、又はG2は

G4は1~3個のフッ素で場合により置換されているC1~C2アルキル又は1~2個のフッ素で場合により置換されているC3~C4シクロアルキルであり、
R1は水素、1~3個のフッ素で場合により置換されているC1~C3アルキル又は1~3個のフッ素で場合により置換されているC1~C3シクロアルキルであり、
R2は1~3個のフッ素で場合により置換されているC1~C6アルキル又は1~3個のフッ素で場合により置換されているC3~C6シクロアルキルであり、
R3は水素、Cl、F、CH3又はOCH3であり、
Wは

を開示する。

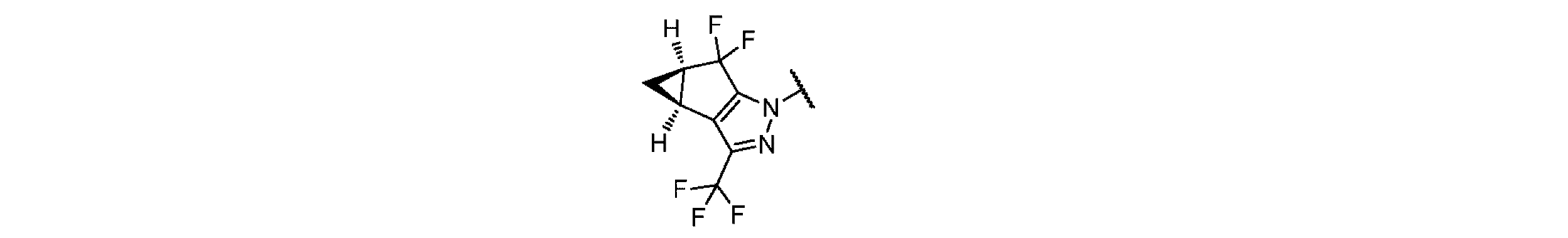


ここで、Z1及びZ3が独立に、-CH3で1回場合により置換されている-C1~C2アルキレンから選択され、Z2が-O-である、式Iの化合物及びその薬学的に許容される塩を開示する。


ここで、E4が、-CH3で1回場合により置換されている-C1~C2アルキレンであり、E5が、-CH3で1回場合により置換されている-C1~C2アルキレンであり、E6が-O-である、式Iの化合物及びその薬学的に許容される塩を開示する。









撹拌子を備えた適切な大きさのガラスバイアル(典型的には、5mLのマイクロ波バイアル)に、N-((S)-1-(7-ブロモ-(3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミド(1当量、典型的には50~100mg)、K3PO4(3当量)及びジクロロ[9,9-ジメチル-4,5-ビス(ジフェニルホスフィノ)キサンテン]パラジウム(II)(0.05~0.1当量)を添加した。バイアルに、示されたボロン酸又はボロン酸ピナコール(3当量)を添加した。バイアルをセプタムキャップで密封し、次いでアルゴン雰囲気下に置いた(真空/充填×3)。バイアルに、臭化物中0.05Mの濃度を達成するのに必要な量でTHF:水(4:1)を添加した。混合物を再びアルゴン雰囲気下に置いた(真空/充填×3)。反応物を周囲温度又は60℃のいずれかで終夜(約18時間)撹拌した。周囲温度に冷却したら、反応混合物を濃縮し、残留物をHPLC精製に供して、示された生成物を得た。
撹拌子を備えた適切な大きさのガラスバイアル(典型的には、5mLのマイクロ波バイアル)に、N-((S)-1-(7-ブロモ-(3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミド(1当量、典型的には50~100mg)に、示されたトリフルオロボレート(2.5当量)、炭酸セシウム(3当量)及びRuPhos Pd G3(0.2当量)を添加した。バイアルに、臭化物中0.05Mの濃度を達成するのに必要な量でトルエン:水(10:1)を添加した。バイアルをセプタムキャップで密封し、次いでアルゴン雰囲気下に置いた(真空/充填×3)。混合物を100℃で2時間撹拌した。周囲温度に冷却したら、反応混合物を濃縮し、得られた残留物をHPLC精製に供して、示された生成物を得た。「RuPhos Pd G3」(CAS:1445085-77-7)の構造は、

撹拌子を備えた適切な大きさのガラスバイアル(典型的には、5mLのマイクロ波バイアル)に、N-((S)-1-((3P)-3-(4-クロロ-3-(シクロプロパンスルホンアミド)-1-(2,2-ジフルオロエチル)-1H-インダゾール-7-イル)-4-オキソ-7-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミド(1当量、典型的には50~100mg)、K3PO4(3当量)及びジクロロ[9,9-ジメチル-4,5-ビス(ジフェニルホスフィノ)キサンテン]パラジウム(II)(0.05当量)を添加した。バイアルに、示されたトリフルオロメタンスルホネート(3当量)を添加した。バイアルに、ボロン酸エステル中0.05Mの濃度を達成するのに必要な量でTHF:水(4:1)を添加した。バイアルをセプタムキャップで密封し、次いでバイアルをアルゴン雰囲気下に置いた(真空/充填×3)。反応混合物を周囲温度、45℃又は60℃のいずれかで終夜(約18時間)撹拌した。周囲温度に冷却したら、反応混合物を濃縮し、残留物をHPLC精製に供して、示された生成物を得た。代替的に、N-((S)-1-((3P)-3-(4-クロロ-3-(シクロプロパンスルホンアミド)-1-(2,2-ジフルオロエチル)-1H-インダゾール-7-イル)-4-オキソ-7-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドを、標的生成物により決定されるようにN-((S)-1-((3P)-3-(4-クロロ-1-(2,2-ジフルオロエチル)-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-7-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミド又はN-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-7-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドに置き換えてもよい。
示されたアルケン(1当量、典型的には50mg)をメタノール:酢酸(1:1)に0.05Mの濃度に溶解した。溶液を、アルゴンを使用して脱気した。溶液に、Pd-C(0.5当量)(10% Degussa)を添加した。反応容器を排気し、次いでバルーンにより導入されたH2(g)で再充填した。反応物をH2(g)バルーン圧雰囲気下、室温で3~5時間撹拌した。次いで雰囲気をAr(g)に置き換え、次いでセライトを反応混合物に添加して、スラリーをDCMで洗浄しながらセライトのパッドに通して濾過した。濾液を濃縮して、得られた残留物をHPLC精製に供して、示された生成物を得た。
撹拌子を備えた適切な大きさのガラスバイアル(典型的には、5mLのマイクロ波バイアル)に、N-((S)-1-(7-ブロモ-(3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミド(1当量、典型的には50mg)、Pd(OAc)2(0.2当量)、ジシクロヘキシル(2',6'-ジメトキシ-[1,1'-ビフェニル]-2-イル)ホスファン(0.4当量)及びK3PO4を添加した。バイアルに、示されたボロン酸又はボロン酸エステル(3当量)を添加した。バイアルをセプタムキャップで冠着し、次いでアルゴン雰囲気下に置いた(真空/充填×3)。バイアルに、臭化物中0.05Mの濃度を達成するのに必要な量でTHF:水(4:1)を添加した。バイアルを再びアルゴン雰囲気下に置いた(真空/充填×3)。反応混合物を周囲温度、45℃又は60℃のいずれかで終夜(約18時間)撹拌した。周囲温度に冷却したら、反応混合物を濃縮し、得られた残留物をHPLC精製に供して、示された生成物を得た。
撹拌子を備えた適切な大きさのガラスバイアル(典型的には、5mLのマイクロ波バイアル)に、N-((S)-1-(7-ブロモ-(3P)-3-(4-クロロ-1-(2,2-ジフルオロエチル)-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミド(1当量、典型的には50mg)、K3PO4(3当量)、ジクロロ[9,9-ジメチル-4,5-ビス(ジフェニルホスフィノ)キサンテン]パラジウム(II)(0.05当量)及び示されたボロン酸エステル又はボロン酸(3当量)を添加した。バイアルをセプタムキャップで密封し、次いでアルゴン雰囲気下に置いた(真空/充填×3)。バイアルに、臭化物中0.05Mの濃度を達成するのに必要な量でTHF:水(4:1)を添加した。バイアルを再びアルゴン雰囲気下に置いた(真空/充填×3)。反応混合物を周囲温度、45℃又は60℃のいずれかで終夜(約18時間)撹拌した。周囲温度に冷却したら、反応混合物を濃縮し、得られた残留物をHPLC精製に供して、示された生成物を得た。代替的に、N-((S)-1-(7-ブロモ-3-(4-クロロ-1-(2,2-ジフルオロエチル)-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドを、標的生成物により決定されるようにN-((S)-1-(7-ブロモ-(3P)-3-(4-クロロ-3-(シクロプロパンスルホンアミド)-1-(2,2-ジフルオロエチル)-1H-インダゾール-7-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミド又はN-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-7-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドに置き換えてもよい。
HPLC精製は以下に示す条件のうちの1つを使用して行い、場合により続いて、以下に示す異なる条件を使用する第2のHPLC精製を行った。粗反応混合物で得られた分析HPLCデータに基づいて、最初の溶媒A:溶媒B比、勾配時間、最終の溶媒A:溶媒B比、及び最終の溶媒A:溶媒B濃度での保持時間を修正することにより、精製条件を各標的化合物に対して最適化した。
HPLC条件A:カラム:Zorbax Eclipse Plus C18、21.2×100mm、5μm粒子;溶媒A=100%水中0.1%ギ酸。溶媒B=アセトニトリル。流速=40mL/分。波長=215及び254nm。ESI+範囲:150~1500ダルトン。
HPLC条件B:カラム:Sunfire prep C18 OBD、30×100mm、5μm粒子;溶媒A:水:MeCN 95:5 0.1%TFA含有、溶媒B:MeCN:水 95:5 0.1%TFA含有。流速=42mL/分。波長=220及び254nm。
HPLC条件C:カラム:Waters Xterra C18、19×100mm、10μm粒子;溶媒A=100%水中0.1%NH4OH。溶媒B=アセトニトリル。流速=40mL/分。波長=215及び254nm。ESI+範囲:150~1500ダルトン。
LCMS方法A:
波長1:220nm、波長2:254nm、注入量:5.00μl、停止時間:4.00。勾配時間:3.0、開始%B:0、最終%B:100、総流速:0.80ml/分、溶媒A:95:5 水:MeCN 0.1%TFA、溶媒B:5:95 水:MeCN 0.1%TFA、カラム:Acquity UPLC BEH C18、2.1×50mm、1.7μm粒子。
LCMS方法B:
カラム:Acquity UPLC BEH C18、2.1×100mm、1.7μm粒子;溶媒A=95:5 水:MeCN中0.1%ギ酸。溶媒B=5:95 水:MeCN中0.1%ギ酸;流速=0.8mL/分;開始%B=0、最終%B=100;勾配時間=3.5分、次いで100%Bで1分保持。検出=220及び254nm。
LCMS方法C:
カラム:Acquity UPLC BEH C18、2.1×30mm、1.7μm粒子;溶媒A=100%水中0.1%ギ酸。溶媒B=100%アセトニトリル中0.1%ギ酸。流速=0.8mL/分。開始%B=5。最終%B=95。勾配時間=1.6分、次いで95%Bで0.25分保持。波長=215nm。
LCMS方法E:
カラム:Zorbax Eclipse Plus C18、2.1×50mm、1.7μm粒子;溶媒A=100%水中0.1%ギ酸。溶媒B=100%アセトニトリル中0.1%ギ酸。流速=1mL/分。開始%B=5。最終%B=95。勾配時間=2.1分、次いで95%Bで0.3分保持。波長=215及び254nm。
LCMS方法F:
カラム:Acquity BEH C18、2.1×30mm、1.7μm粒子;溶媒A=100%水中0.1%ギ酸。溶媒B=100%アセトニトリル中0.1%ギ酸。流速=0.8mL/分。開始%B=5。最終%B=95。勾配時間=1.7分、次いで95%Bで0.2分保持。波長=215及び254nm。















































N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(3-モルホリノ-3-オキソプロピル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(3,3-ジメチルブチル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-7-フェネチル-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-オクチル-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-(7-ベンジル-(3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(シクロヘキサ-1-エン-1-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製
N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(3-メチルブタ-2-エン-2-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(シクロペンタ-1-エン-1-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(3,6-ジヒドロ-2H-ピラン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((1S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(3-メチルピペラジン-2-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-7-(テトラヒドロ-2H-ピラン-4-イル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(シクロプロピルメチル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(1-メチル-1,2,3,6-テトラヒドロピリジン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(1-メチルピペリジン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-(2,2-ジフルオロエチル)-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(3,6-ジヒドロ-2H-ピラン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-3-(シクロプロパンスルホンアミド)-1-(2,2-ジフルオロエチル)-1H-インダゾール-7-イル)-7-(3,6-ジヒドロ-2H-ピラン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-(2,2-ジフルオロエチル)-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-7-(テトラヒドロ-2H-ピラン-4-イル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-3-(シクロプロパンスルホンアミド)-1-(2,2-ジフルオロエチル)-1H-インダゾール-7-イル)-4-オキソ-7-(テトラヒドロ-2H-ピラン-4-イル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-((2R,6S)-2,6-ジメチル-3,6-ジヒドロ-2H-ピラン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-(2,2-ジフルオロエチル)-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-((2R,6S)-2,6-ジメチル-3,6-ジヒドロ-2H-ピラン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-3-(シクロプロパンスルホンアミド)-1-(2,2-ジフルオロエチル)-1H-インダゾール-7-イル)-7-((2R,6S)-2,6-ジメチル-3,6-ジヒドロ-2H-ピラン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製





(ステップ2b) 0~5℃のDCM(9.04L、8.0V)中の粗オキシム(上記の調製物、1.13kg、4.80mol、1.0当量)の撹拌溶液に、トリエチルアミン(「TEA」、1.02kg、10.09mol、2.1当量)を添加した。5分間撹拌した後、メタンスルホニルクロリド(0.60kg、5.29mol、1.1当量)を15℃でゆっくりと添加した(観察:添加中に発熱が認められる)。次いで、反応塊を室温で30~45分間撹拌した。反応(反応の進行をTLC;移動相:ヘキサン中20%酢酸エチルによりモニターした)の完了後、反応塊を水(6.78L、6.0V)で希釈し、有機層を分離し、水層をDCM(3.4L、3.0V)で抽出した。合わせた有機層をブライン(5.65L、5.0V)で洗浄し、Na2SO4で乾燥させ、真空下で濃縮した。得られた粗固体を室温にてヘキサン(4.50L、4.0V)で摩砕した。湿潤物質を熱風オーブン内にて50~55℃で5~6時間乾燥させて、乾燥生成物、2,6-ジクロロ-3-ニトロベンゾニトリル(0.95kg、収率91%)を黄色固体として得た。1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 8.8 Hz, 1H), 7.63 (d, J = 8.8 Hz, 1H).



(ii) 室温のエタノール(10.5L、20.0V)中のN-(4-クロロ-1-メチル-7-ニトロ-1H-インダゾール-3-イル)-N-(メチルスルホニル)メタンスルホンアミド(上記で調製)の撹拌溶液に、5%NaOH水溶液(4.38L、7.0V)をゆっくりと添加した[注記:滴下漏斗によるゆっくりとした添加が好ましい]。反応塊を同じ温度で3時間撹拌した。反応(TLCによりモニターした)[TLC分析のための試料調製:約1.0mlの試料を2.0N HCl水溶液で酸性化してpH:2~3に到達させ、これを酢酸エチルで抽出し、有機層をTLCにより分析した]の完了後、反応塊を0~5℃に冷却し、反応温度を10℃未満に維持しながらpHを2.0N HCl水溶液(3.13L、5.0V)の添加により2~3に調整した[注記:HClを添加すると沈殿が生じ、撹拌とともに増加した]。反応混合物を室温に加温し、次いで1.5~2.0時間撹拌した。得られた固体を濾過により単離し、次いで水(1.25L、2.0V)で洗浄し、続いてヘキサン(1.25L、2.0V)で洗浄した。真空濾過を60~90分間維持することにより、バルク残留水を固体から除去した。湿潤物質を熱風オーブン内にて50℃で6~7時間(水分含有率が1.0%未満になるまで)乾燥させて、乾燥生成物、N-(4-クロロ-1-メチル-7-ニトロ-1H-インダゾール-3-イル)メタンスルホンアミド(640.0g、76%)を黄色固体として得た。1H NMR (400 MHz, CDCl3): δ 8.05 (d, J = 8.32 Hz, 1H), 7.32 (bs, 1H), 7.17 (d, J = 8.28 Hz, 1H), 4.15 (s, 3H), 3.45 (s, 3H).






ステップ2b: 室温のエタノール(1.7L、10.0V)中のN-(4-クロロ-1-(2,2-ジフルオロエチル)-7-ニトロ-1H-インダゾール-3-イル)-N-(メチルスルホニル)メタンスルホンアミド(上記で調製した物質の全体)の撹拌溶液に、5%NaOH水溶液(1.19L、7.0V)をゆっくりと添加した[注記:滴下漏斗によるゆっくりとした添加が好ましい]。反応塊を同じ温度で3時間撹拌した。反応[TLC分析のための試料調製:反応溶液のアリコート(約1mL)を2.0N HCl水溶液で酸性化してpH2~3に到達させ、次いで混合物を酢酸エチルで抽出し、有機層をTLCにより分析した]の完了後、反応塊を0~5℃に冷却し、pHを10℃未満で2.0N HCl水溶液(約850mL、5.0V)の添加により2~3に調整した[注記:HClを添加すると沈殿が生じ、撹拌とともに固体が徐々に増加した]。反応混合物を室温に加温し、次いで1.5~2.0時間撹拌した。得られた固体を濾過により単離し、次いで水(340mL、2.0V)で洗浄し、続いてヘキサン(340mL、2.0V)で洗浄した。真空濾過を60~90分間維持することにより、バルク残留水を固体から除去した。湿潤物質を熱風オーブン内にて50℃で6~7時間(水分含有率が1.0%未満になるまで)乾燥させて、乾燥生成物、N-(4-クロロ-1-(2,2-ジフルオロエチル)-7-ニトロ-1H-インダゾール-3-イル)メタンスルホンアミド(170.0g、75%)を黄色固体として得た。1H NMR (400MHz, CDCl3): δ 8.15 (d, J = 8.3 Hz, 1H), 7.52 (bs, 1H), 7.24 (d, J = 8.3 Hz, 1H), 6.04 (tt, J1 = 3.7 Hz, J2 = 7.9 Hz, 1H), 5.02 (td, J1 = 3.9 Hz, J2 = 14.3 Hz, 2H), 3.42 (s, 4H).







N-((1S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-((2R,6S)-2,6-ジメチルテトラヒドロ-2H-ピラン-4-イル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製


N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(1,1-ジフルオロ-4-メチルペンチル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(3-メトキシ-3-メチルブチル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製


N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-7-(シクロブチルジフルオロメチル)-4-オキソ-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製







N-((S)-1-((3P)-3-(4-クロロ-1-メチル-3-(メチルスルホンアミド)-1H-インダゾール-7-イル)-4-オキソ-7-(1-(トリフルオロメチル)シクロプロピル)-3,4-ジヒドロキナゾリン-2-イル)-2-(3,5-ジフルオロフェニル)エチル)-2-((3bS,4aR)-3-(ジフルオロメチル)-5,5-ジフルオロ-3b,4,4a,5-テトラヒドロ-1H-シクロプロパ[3,4]シクロペンタ[1,2-c]ピラゾール-1-イル)アセトアミドの調製

各実施例のIUPAC化学名を以下に列挙する。この際、これらの名称は、一般のソフトウェア、ツール、例えばChemDraw又はJChemにより認識されない。したがって、上記実施例セクション全体にわたって使用される化学名は、ChemDrawを用いて生成され、P/M命名法は手動で挿入された。この化学名は、P/M命名法、例えば「(3P)-」を除去した後にChemDrawを使用して化学構造に変換することができる。



HIV細胞培養アッセイ- MT-2細胞、293T細胞及びNL4-3ウイルスのプロウイルスDNAクローンを、アメリカ国立衛生研究所、AIDS研究及び標準試薬プログラム(NIH AIDS Research and Reference Reagent Program)から入手した。MT-2細胞を、10%の熱不活性化ウシ胎児血清(FBS)、100mg/mlのペニシリンG及び最大100単位/mLのストレプトマイシンを補充したRPMI1640培地中で増殖させた。293T細胞を、10%の熱不活性化FBS、100mg/mLのペニシリンG及び100mg/mLのストレプトマイシンを補充したDMEM培地中で増殖させた。nef遺伝子の一部分をウミシイタケルシフェラーゼ(Renilla luciferase)遺伝子に置き換えた組換えNL4-3プロウイルスクローンを使用して、これらの研究において使用される参照ウイルスを作製した。組換えウイルスを、Mirus Bio LLC(Madison、WI)から入手したTransit-293トランスフェクション試薬を使用して、293T細胞への組換えNL4-3プロウイルスクローンのトランスフェクションにより調製した。2~3日後に上清を回収し、ルシフェラーゼ酵素活性を測定することによりルシフェラーゼ酵素活性をマーカーとして使用して、存在するウイルスの量をMT-2細胞において力価測定した。ルシフェラーゼは、Promega(Madison、WI)から入手したEnduRen Live Cell Substrateを使用して定量化した。化合物の系列希釈の存在下で組換えウイルスに4~5日間感染させたMT-2細胞におけるルシフェラーゼ活性を測定することにより、組換えウイルスに対する化合物の抗ウイルス活性を定量化した。

(付記)
(付記1)
式Iの化合物、又はその薬学的に許容される塩:

G1は1~3個のフッ素で場合により置換されているC6~C8アルキルであるか、又はG1は-(C2~C3アルキレニル)-で置換されており、G2で1回場合により置換されており、1~3個のフッ素で場合により置換されているC2~C5アルキルであるか、又はG1はG2で置換されているC1~C5アルキルであるか、又はG1は

Z1及びZ3は独立に、-CH3又は-CH2CH3で1回場合により置換されている-C1~C3アルキレンであり、
Z2は-O-、-S(O2)-、-NH-又は-N(G4)であり、
X1、X2及びX3は独立に、H、F又はClであり、ここで、X1、X2及びX3のうちの1つは-CN、-OCH3、-CH3、-CH2F、-CHF2及び-CF3から場合により選択されてもよく、
E4及びE5は独立に、-CH3又は-CH2CH3で1回場合により置換されている-C1~C2アルキレンであり、
E6は-C(H2)-、-O-、-S(O2)-、-NH-又は-N(G4)であり、
E7、E8及びE9は独立に、H又は-CH3であり、
Y1及びY2は独立に、-N(H)-又は-O-であり、
G2はフェニル、-OH、1~3個のフッ素で場合により置換されている-O(C1~C3アルキル)、1~2個のフッ素で場合により置換されているC3~C4シクロアルキルであるか、又はG2は

G4は1~3個のフッ素で場合により置換されているC1~C2アルキル、又は1~2個のフッ素で場合により置換されているC3~C4シクロアルキルであり、
R1は水素、1~3個のフッ素で場合により置換されているC1~C3アルキル、又は1~3個のフッ素で場合により置換されているC1~C3シクロアルキルであり、
R2は1~3個のフッ素で場合により置換されているC1~C6アルキル、又は1~3個のフッ素で場合により置換されているC3~C6シクロアルキルであり、
R3は水素、Cl、F、CH3又はOCH3であり、
Wは

(付記2)
Wが

(付記3)
Wが

(付記4)
R1が-CH3、-CH2CHF2又は-CH2CF3であり、R2が-CH3又はシクロプロピルであり、R3がH、Cl又はCH3である、付記1から3のいずれか一項に記載の化合物又は塩。
(付記5)
R1が-CH3であり、R2が-CH3であり、R3がClである、付記1から3のいずれか一項に記載の化合物又は塩。
(付記6)
X1、X2及びX3が独立に、H又はFである、付記1から5のいずれか一項に記載の化合物又は塩。
(付記7)
X1がFであり、X2がHであり、X3がFである、付記1から5のいずれか一項に記載の化合物又は塩。
(付記8)
G1が1~3個のフッ素で場合により置換されているC6~C8アルキルである、付記1から7のいずれか一項に記載の化合物又は塩。
(付記9)
G1が-(C2~C3アルキレニル)-で置換されており、G2で1回場合により置換されており、1~3個のフッ素で場合により置換されているC2~C5アルキルである、付記1から7のいずれか一項に記載の化合物又は塩。
[付記9]
G1がG2で置換されているC1~C5アルキルである、付記1から7のいずれか一項に記載の化合物又は塩。
(付記10)
G1が

(付記11)
G1が

(付記12)
G1が

(付記13)
G1が

(付記14)
G1が

(付記15)
G1がフッ素原子を含有する、付記1から14のいずれか一項に記載の化合物又は塩。
(付記16)
立体化学構造が、

(付記17)
立体化学構造が、

(付記18)


(付記19)

(付記20)
付記1から19のいずれか一項に記載の化合物又は塩を含む、医薬組成物。
(付記21)
薬学的に許容される担体、賦形剤及び/又は希釈剤をさらに含む、付記20に記載の組成物。
(付記22)
付記20又は21に記載の組成物を患者に投与することを含む、HIV感染を処置する方法。
(付記23)
前記投与が経口である、付記22に記載の方法。
(付記24)
前記投与が、筋肉内注射又は皮下注射によるものである、付記22に記載の方法。
(付記25)
ヌクレオシドHIV逆転写酵素阻害剤、非ヌクレオシドHIV逆転写酵素阻害剤、HIVプロテアーゼ阻害剤、HIV融合阻害剤、HIV付着阻害剤、CCR5阻害剤、CXCR4阻害剤、HIV出芽又は成熟阻害剤及びHIVインテグラーゼ阻害剤からなる群から選択される、AIDS又はHIV感染の処置に使用される少なくとも1つの他の薬剤の投与をさらに含む、付記22、23又は24に記載の方法。
(付記26)
治療において使用するための、付記1から19のいずれか一項に記載の化合物又はその薬学的に許容される塩。
(付記27)
HIV感染の処置において使用するための、付記1から19のいずれか一項に記載の化合物又はその薬学的に許容される塩。
(付記28)
HIV感染の処置のための医薬の製造において使用するための、付記1から19のいずれか一項に記載の化合物又はその薬学的に許容される塩。
Claims (11)
-
 から選択される化合物、又はその薬学的に許容される塩。
から選択される化合物、又はその薬学的に許容される塩。
-

- 請求項1又は2に記載の化合物又は塩を含む、医薬組成物。
- 薬学的に許容される担体、賦形剤及び/又は希釈剤をさらに含む、請求項3に記載の組成物。
- HIV感染を処置するための、請求項3又は4に記載の組成物。
- 経口で投与される、請求項5に記載の組成物。
- 筋肉内注射又は皮下注射により投与される、請求項5に記載の組成物。
- ヌクレオシドHIV逆転写酵素阻害剤、非ヌクレオシドHIV逆転写酵素阻害剤、HIVプロテアーゼ阻害剤、HIV融合阻害剤、HIV付着阻害剤、CCR5阻害剤、CXCR4阻害剤、HIV出芽又は成熟阻害剤及びHIVインテグラーゼ阻害剤からなる群から選択される、AIDS又はHIV感染の処置に使用される少なくとも1つの他の薬剤と組み合わせて使用される、請求項5~7のいずれか一項に記載の組成物。
- 治療において使用するための、請求項1又は2に記載の化合物又はその薬学的に許容される塩。
- HIV感染の処置において使用するための、請求項1又は2に記載の化合物又はその薬学的に許容される塩。
- HIV感染の処置のための医薬の製造において使用するための、請求項1又は2に記載の化合物又はその薬学的に許容される塩。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862750395P | 2018-10-25 | 2018-10-25 | |
| US62/750,395 | 2018-10-25 | ||
| PCT/IB2019/059004 WO2020084480A1 (en) | 2018-10-25 | 2019-10-22 | Inhibitors of human immunodeficiency virus replication |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2022505597A JP2022505597A (ja) | 2022-01-14 |
| JPWO2020084480A5 JPWO2020084480A5 (ja) | 2022-10-11 |
| JP7500555B2 true JP7500555B2 (ja) | 2024-06-17 |
Family
ID=68344929
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021521995A Active JP7500555B2 (ja) | 2018-10-25 | 2019-10-22 | ヒト免疫不全ウイルス複製の阻害剤 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20210393633A1 (ja) |
| EP (1) | EP3870575B1 (ja) |
| JP (1) | JP7500555B2 (ja) |
| ES (1) | ES2942998T3 (ja) |
| PT (1) | PT3870575T (ja) |
| WO (1) | WO2020084480A1 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2962736T3 (es) * | 2018-04-11 | 2024-05-24 | Viiv Healthcare Uk No 5 Ltd | Compuestos de 4-oxo-3,4-dihidroquinazolina como inhibidores de la replicación del virus de la inmunodeficiencia humana |
| US20210395262A1 (en) * | 2018-10-24 | 2021-12-23 | VIIV Healthcare UK (No.5) Limited | Inhibitors of human immunodeficiency virus replication |
| US11541055B2 (en) | 2018-10-24 | 2023-01-03 | VIIV Healthcare UK (No.5) Limited | Inhibitors of human immunodeficiency virus replication |
| US20210403465A1 (en) * | 2018-10-29 | 2021-12-30 | VIIV Healthcare UK (No.5) Limited | Quinazolinyl-indazole derivatives and their use as inhibitors of human immunodeficiency virus replication |
| EP3962603A1 (en) | 2019-04-30 | 2022-03-09 | ViiV Healthcare UK (No.5) Limited | Inhibitors of human immunodeficiency virus replication |
| JP2022548690A (ja) * | 2019-09-20 | 2022-11-21 | アイディアヤ バイオサイエンシーズ,インコーポレイティド | Parg阻害剤としての4-置換インドールおよびインダゾールスルホンアミド誘導体 |
| WO2021107066A1 (ja) | 2019-11-28 | 2021-06-03 | 塩野義製薬株式会社 | インテグラーゼ阻害剤及び抗hiv薬を組み合わせることを特徴とするhiv感染症の予防及び治療用医薬 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016504392A (ja) | 2013-01-09 | 2016-02-12 | ギリアード サイエンシーズ, インコーポレイテッド | 抗レトロウイルス剤としての(ヘテロ)アリールアセトアミド誘導体 |
| JP2016510038A (ja) | 2013-03-01 | 2016-04-04 | ギリアード サイエンシーズ, インコーポレイテッド | Hivの処置のためのアミド化合物 |
| JP2017525722A (ja) | 2014-08-29 | 2017-09-07 | ギリアード サイエンシーズ, インコーポレイテッド | 抗レトロウイルス剤 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102464654B (zh) | 2010-11-12 | 2016-01-13 | 上海泓博智源医药技术有限公司 | 抗病毒化合物 |
| CA2840095A1 (en) | 2011-07-06 | 2013-01-10 | Gilead Sciences, Inc. | Compounds for the treatment of hiv |
| CN102863512B (zh) | 2011-07-07 | 2016-04-20 | 上海泓博智源医药技术有限公司 | 抗病毒化合物 |
| CA2896244C (en) | 2013-01-09 | 2017-07-04 | Gilead Sciences, Inc. | 5-membered heteroaryls and their use as antiviral agents |
| NZ631726A (en) | 2013-01-09 | 2017-01-27 | Gilead Sciences Inc | Therapeutic compounds for the treatment of viral infections |
| WO2015130964A1 (en) | 2014-02-28 | 2015-09-03 | Gilead Sciences, Inc. | Therapeutic compounds |
| WO2015130966A1 (en) | 2014-02-28 | 2015-09-03 | Gilead Sciences, Inc. | Antiviral agents |
| MA42795B1 (fr) | 2016-08-19 | 2019-08-30 | Gilead Sciences Inc | Composés thérapeutiques utiles pour le traitement prophylactique ou thérapeutique d'une infection par le virus du vih |
| TW201906834A (zh) * | 2017-05-02 | 2019-02-16 | 英商Viiv醫療保健英國(No.5)有限公司 | 人類免疫不全病毒複製之抑制劑 |
| JP7083398B2 (ja) | 2018-02-15 | 2022-06-10 | ギリアード サイエンシーズ, インコーポレイテッド | ピリジン誘導体およびhiv感染を処置するためのその使用 |
| CN116854630A (zh) | 2018-02-16 | 2023-10-10 | 吉利德科学公司 | 用于制备可用于治疗逆转录病毒科病毒感染的治疗性化合物的方法和中间体 |
| ES2962736T3 (es) * | 2018-04-11 | 2024-05-24 | Viiv Healthcare Uk No 5 Ltd | Compuestos de 4-oxo-3,4-dihidroquinazolina como inhibidores de la replicación del virus de la inmunodeficiencia humana |
| US20210379071A1 (en) * | 2018-11-05 | 2021-12-09 | VIIV Healthcare UK (No.5) Limited | Inhibitors of human immunodeficiency virus replication |
-
2019
- 2019-10-22 JP JP2021521995A patent/JP7500555B2/ja active Active
- 2019-10-22 WO PCT/IB2019/059004 patent/WO2020084480A1/en unknown
- 2019-10-22 EP EP19794679.1A patent/EP3870575B1/en active Active
- 2019-10-22 ES ES19794679T patent/ES2942998T3/es active Active
- 2019-10-22 PT PT197946791T patent/PT3870575T/pt unknown
- 2019-10-22 US US17/284,855 patent/US20210393633A1/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016504392A (ja) | 2013-01-09 | 2016-02-12 | ギリアード サイエンシーズ, インコーポレイテッド | 抗レトロウイルス剤としての(ヘテロ)アリールアセトアミド誘導体 |
| JP2016510038A (ja) | 2013-03-01 | 2016-04-04 | ギリアード サイエンシーズ, インコーポレイテッド | Hivの処置のためのアミド化合物 |
| JP2017525722A (ja) | 2014-08-29 | 2017-09-07 | ギリアード サイエンシーズ, インコーポレイテッド | 抗レトロウイルス剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20210393633A1 (en) | 2021-12-23 |
| WO2020084480A1 (en) | 2020-04-30 |
| EP3870575B1 (en) | 2023-03-29 |
| JP2022505597A (ja) | 2022-01-14 |
| ES2942998T3 (es) | 2023-06-08 |
| EP3870575A1 (en) | 2021-09-01 |
| PT3870575T (pt) | 2023-05-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7500555B2 (ja) | ヒト免疫不全ウイルス複製の阻害剤 | |
| JP7526174B2 (ja) | キナゾリニル-インダゾール誘導体及びそのヒト免疫不全ウイルス複製の阻害剤としてのその使用 | |
| JP7433303B2 (ja) | ヒト免疫不全ウイルス複製の阻害剤 | |
| JP7398448B2 (ja) | ヒト免疫不全ウイルス複製の阻害剤 | |
| TW201906834A (zh) | 人類免疫不全病毒複製之抑制劑 | |
| JP7361766B2 (ja) | ヒト免疫不全ウイルス複製の阻害剤 | |
| JP2022506399A (ja) | ヒト免疫不全ウイルス複製の阻害剤 | |
| JP7532354B2 (ja) | ヒト免疫不全ウイルス複製の阻害剤 | |
| JP2021521131A (ja) | ヒト免疫不全ウイルス複製の阻害剤としての4−オキソ−3,4−ジヒドロキナゾリン化合物 | |
| JP2022509919A (ja) | ヒト免疫不全ウイルス複製の阻害剤 | |
| JP7545414B2 (ja) | ヒト免疫不全ウイルス複製阻害剤 | |
| TW202229244A (zh) | 新化合物 | |
| CN115397424A (zh) | 人类免疫缺陷病毒复制的抑制剂 | |
| TW202128648A (zh) | 人類免疫不全病毒複製之抑制劑 | |
| US20240374598A1 (en) | Inhibitors of human immunodeficiency virus replication | |
| EA043730B1 (ru) | Ингибиторы репликации вируса иммунодефицита человека |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220928 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220928 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20230928 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20231010 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20231222 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20240222 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240409 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20240507 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20240605 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7500555 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |