EP0241003B1 - 4H-1-Benzopyran-4-on-Derivate, ein Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel - Google Patents
4H-1-Benzopyran-4-on-Derivate, ein Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel Download PDFInfo
- Publication number
- EP0241003B1 EP0241003B1 EP87105180A EP87105180A EP0241003B1 EP 0241003 B1 EP0241003 B1 EP 0241003B1 EP 87105180 A EP87105180 A EP 87105180A EP 87105180 A EP87105180 A EP 87105180A EP 0241003 B1 EP0241003 B1 EP 0241003B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- alkyl
- formula
- methyl
- hydroxy
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000002360 preparation method Methods 0.000 title claims description 9
- 239000003814 drug Substances 0.000 title claims 3
- 150000004777 chromones Chemical class 0.000 title abstract description 6
- 238000000034 method Methods 0.000 title description 15
- 150000001875 compounds Chemical class 0.000 claims abstract description 62
- 239000002253 acid Substances 0.000 claims abstract description 22
- 150000003839 salts Chemical class 0.000 claims abstract description 18
- -1 nitro, amino Chemical group 0.000 claims abstract description 17
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 11
- 239000001257 hydrogen Substances 0.000 claims abstract description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 10
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims abstract description 7
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 7
- 150000002367 halogens Chemical class 0.000 claims abstract description 7
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 5
- 230000003110 anti-inflammatory effect Effects 0.000 claims abstract description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract 8
- 230000003287 optical effect Effects 0.000 claims description 6
- 238000006243 chemical reaction Methods 0.000 claims description 5
- 239000003153 chemical reaction reagent Substances 0.000 claims description 5
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 4
- 230000002378 acidificating effect Effects 0.000 claims description 4
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 claims description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 4
- 230000002519 immonomodulatory effect Effects 0.000 claims description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- 125000004076 pyridyl group Chemical group 0.000 claims description 3
- 229910052783 alkali metal Inorganic materials 0.000 claims description 2
- 150000001340 alkali metals Chemical class 0.000 claims description 2
- 125000005594 diketone group Chemical group 0.000 claims description 2
- 150000004820 halides Chemical class 0.000 claims description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 claims description 2
- 239000011707 mineral Substances 0.000 claims description 2
- 125000005490 tosylate group Chemical group 0.000 claims description 2
- 229910000102 alkali metal hydride Inorganic materials 0.000 claims 1
- 150000008046 alkali metal hydrides Chemical class 0.000 claims 1
- 125000005907 alkyl ester group Chemical group 0.000 claims 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims 1
- 150000003841 chloride salts Chemical class 0.000 claims 1
- 125000005843 halogen group Chemical group 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 abstract description 5
- 125000003118 aryl group Chemical group 0.000 abstract description 5
- 125000004432 carbon atom Chemical group C* 0.000 abstract description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 abstract description 3
- 229940035676 analgesics Drugs 0.000 abstract description 2
- 239000000730 antalgic agent Substances 0.000 abstract description 2
- 230000003266 anti-allergic effect Effects 0.000 abstract description 2
- 239000000043 antiallergic agent Substances 0.000 abstract description 2
- 229960003444 immunosuppressant agent Drugs 0.000 abstract description 2
- 239000003018 immunosuppressive agent Substances 0.000 abstract description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 abstract 2
- 125000002861 (C1-C4) alkanoyl group Chemical group 0.000 abstract 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 abstract 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 abstract 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 abstract 1
- 239000002260 anti-inflammatory agent Substances 0.000 abstract 1
- 229940121363 anti-inflammatory agent Drugs 0.000 abstract 1
- 125000003435 aroyl group Chemical group 0.000 abstract 1
- 125000004104 aryloxy group Chemical group 0.000 abstract 1
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 abstract 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 75
- 125000003386 piperidinyl group Chemical group 0.000 description 46
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 33
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 28
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- 238000002844 melting Methods 0.000 description 18
- 230000008018 melting Effects 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 15
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 239000000243 solution Substances 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- 239000000203 mixture Substances 0.000 description 12
- 239000011541 reaction mixture Substances 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 7
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- BWKXKKCHHWEDFS-UHFFFAOYSA-N 1-methyl-2-phenylpiperidine Chemical compound CN1CCCCC1C1=CC=CC=C1 BWKXKKCHHWEDFS-UHFFFAOYSA-N 0.000 description 6
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 6
- 0 CN(CC1)CC(*)C1c1c2OC(*)=C(*)C3OC3c2c(*)cc1* Chemical compound CN(CC1)CC(*)C1c1c2OC(*)=C(*)C3OC3c2c(*)cc1* 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- OTAFHZMPRISVEM-UHFFFAOYSA-N benzo-gamma-pyrone Natural products C1=CC=C2C(=O)C=COC2=C1 OTAFHZMPRISVEM-UHFFFAOYSA-N 0.000 description 5
- 239000002026 chloroform extract Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 239000012279 sodium borohydride Substances 0.000 description 5
- 229910000033 sodium borohydride Inorganic materials 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 206010030113 Oedema Diseases 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 238000009835 boiling Methods 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- MGNNDUKLPNLAFW-NWDGAFQWSA-N (3s,4r)-1-methyl-4-(2,4,6-trimethoxyphenyl)piperidin-3-ol Chemical compound COC1=CC(OC)=CC(OC)=C1[C@@H]1[C@H](O)CN(C)CC1 MGNNDUKLPNLAFW-NWDGAFQWSA-N 0.000 description 3
- MGNNDUKLPNLAFW-VXGBXAGGSA-N (3s,4s)-1-methyl-4-(2,4,6-trimethoxyphenyl)piperidin-3-ol Chemical compound COC1=CC(OC)=CC(OC)=C1[C@H]1[C@H](O)CN(C)CC1 MGNNDUKLPNLAFW-VXGBXAGGSA-N 0.000 description 3
- VWRZNLHDQXFHDR-UHFFFAOYSA-N 1-methyl-4-(2,4,6-trimethoxyphenyl)-3,6-dihydro-2h-pyridine Chemical compound COC1=CC(OC)=CC(OC)=C1C1=CCN(C)CC1 VWRZNLHDQXFHDR-UHFFFAOYSA-N 0.000 description 3
- VSYLKOXFYGLIPZ-UHFFFAOYSA-N 2,3-dimethoxychromen-4-one Chemical compound C1=CC=C2C(=O)C(OC)=C(OC)OC2=C1 VSYLKOXFYGLIPZ-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 235000010418 carrageenan Nutrition 0.000 description 3
- 229920001525 carrageenan Polymers 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 210000002683 foot Anatomy 0.000 description 3
- 238000001640 fractional crystallisation Methods 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 239000012442 inert solvent Substances 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 229940095064 tartrate Drugs 0.000 description 3
- YONLFQNRGZXBBF-ZIAGYGMSSA-N (2r,3r)-2,3-dibenzoyloxybutanedioic acid Chemical compound O([C@@H](C(=O)O)[C@@H](OC(=O)C=1C=CC=CC=1)C(O)=O)C(=O)C1=CC=CC=C1 YONLFQNRGZXBBF-ZIAGYGMSSA-N 0.000 description 2
- YONLFQNRGZXBBF-KBPBESRZSA-N (2s,3s)-2,3-dibenzoyloxybutanedioic acid Chemical compound O([C@H](C(=O)O)[C@H](OC(=O)C=1C=CC=CC=1)C(O)=O)C(=O)C1=CC=CC=C1 YONLFQNRGZXBBF-KBPBESRZSA-N 0.000 description 2
- CRUILBNAQILVHZ-UHFFFAOYSA-N 1,2,3-trimethoxybenzene Chemical compound COC1=CC=CC(OC)=C1OC CRUILBNAQILVHZ-UHFFFAOYSA-N 0.000 description 2
- LKUDPHPHKOZXCD-UHFFFAOYSA-N 1,3,5-trimethoxybenzene Chemical compound COC1=CC(OC)=CC(OC)=C1 LKUDPHPHKOZXCD-UHFFFAOYSA-N 0.000 description 2
- RFAWITYVAHWPNN-UHFFFAOYSA-N 1-methyl-4-(2,4,6-trimethoxyphenyl)piperidin-3-one Chemical compound COC1=CC(OC)=CC(OC)=C1C1C(=O)CN(C)CC1 RFAWITYVAHWPNN-UHFFFAOYSA-N 0.000 description 2
- HUUPVABNAQUEJW-UHFFFAOYSA-N 1-methylpiperidin-4-one Chemical compound CN1CCC(=O)CC1 HUUPVABNAQUEJW-UHFFFAOYSA-N 0.000 description 2
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 2
- VSWICNJIUPRZIK-UHFFFAOYSA-N 2-piperideine Chemical class C1CNC=CC1 VSWICNJIUPRZIK-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 150000001805 chlorine compounds Chemical class 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 230000017858 demethylation Effects 0.000 description 2
- 238000010520 demethylation reaction Methods 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 239000002024 ethyl acetate extract Substances 0.000 description 2
- FKRCODPIKNYEAC-UHFFFAOYSA-N ethyl propionate Chemical compound CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 150000004678 hydrides Chemical class 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 150000003840 hydrochlorides Chemical class 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 150000003892 tartrate salts Chemical class 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 229940030010 trimethoxybenzene Drugs 0.000 description 2
- HNAGHMKIPMKKBB-UHFFFAOYSA-N 1-benzylpyrrolidine-3-carboxamide Chemical compound C1C(C(=O)N)CCN1CC1=CC=CC=C1 HNAGHMKIPMKKBB-UHFFFAOYSA-N 0.000 description 1
- HVCFCNAITDHQFX-UHFFFAOYSA-N 1-cyclopropylethanone Chemical compound CC(=O)C1CC1 HVCFCNAITDHQFX-UHFFFAOYSA-N 0.000 description 1
- RWGFWNPSJBJNGF-UHFFFAOYSA-N 1-methyl-3-(2,4,6-trimethoxyphenyl)-3,6-dihydro-2h-pyridine Chemical compound COC1=CC(OC)=CC(OC)=C1C1C=CCN(C)C1 RWGFWNPSJBJNGF-UHFFFAOYSA-N 0.000 description 1
- GGYVTHJIUNGKFZ-UHFFFAOYSA-N 1-methylpiperidin-2-one Chemical compound CN1CCCCC1=O GGYVTHJIUNGKFZ-UHFFFAOYSA-N 0.000 description 1
- 125000004201 2,4-dichlorophenyl group Chemical group [H]C1=C([H])C(*)=C(Cl)C([H])=C1Cl 0.000 description 1
- CILPHQCEVYJUDN-VWYCJHECSA-N 2-[(1s,2s,5r)-5-methyl-2-propan-2-ylcyclohexyl]oxyacetic acid Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1OCC(O)=O CILPHQCEVYJUDN-VWYCJHECSA-N 0.000 description 1
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- XGAUYROUAAJVEH-RVDMUPIBSA-N CN(CC/C1=C2\C(OC)=CC(OC)=CC2OC)CC1=O Chemical compound CN(CC/C1=C2\C(OC)=CC(OC)=CC2OC)CC1=O XGAUYROUAAJVEH-RVDMUPIBSA-N 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241001671252 Dysoxylum binectariferum Species 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 description 1
- 239000012445 acidic reagent Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000001741 anti-phlogistic effect Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- OBNCKNCVKJNDBV-UHFFFAOYSA-N butanoic acid ethyl ester Natural products CCCC(=O)OCC OBNCKNCVKJNDBV-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 239000000679 carrageenan Substances 0.000 description 1
- 229940113118 carrageenan Drugs 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- ZVTQWXCKQTUVPY-UHFFFAOYSA-N chloromethylcyclopropane Chemical compound ClCC1CC1 ZVTQWXCKQTUVPY-UHFFFAOYSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 1
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 239000012067 demethylated product Substances 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 239000012259 ether extract Substances 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 210000000548 hind-foot Anatomy 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000002440 hydroxy compounds Chemical class 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229940035429 isobutyl alcohol Drugs 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/42—Oxygen atoms attached in position 3 or 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Definitions
- the present invention relates to new 4H-1-benzopyran-4-one derivatives, processes for their preparation and their use as antiphlogistics, analgesics, immunosuppressants and antiallergics.
- the present invention relates to new ones
- the compounds according to the invention have two asymmetric centers, one at the point of attachment of the nitrogen heterocycle to the benzopyr portion (C-4 ') and the other at the carbon atom substituted by R 4 (C-3'), so that two optical isomer pairs are possible.
- all possible stereoisomers and their mixtures are included in the definition of the compounds according to the invention.
- both the racemic forms and the isolated optical isomers possessing the stated activity are included.
- the two racemates can be separated using physical methods such as fractional crystallization.
- the individual optical isomers can be obtained from the racemates by standard methods such as salt formation with an optically active acid and subsequent crystallization.
- Suitable alkyl groups for R 1 and R 5 are, for example, straight-chain or branched radicals having up to 6 and preferably up to 5 carbon atoms, for example methyl, ethyl, propyl, isopropyl, t-butyl, pentyl or isopentyl groups.
- Suitable substituted alkyl groups for R 1 are, for example, haloalkyl such as trifluoromethyl, hydroxyalkyl such as hydroxyethyl or carboxyalkyl such as carboxyethyl.
- the cycloalkylalkyl group R 5 is, for example, cyclopropylmethyl.
- Suitable examples of salts of the compounds according to the invention with inorganic or organic acids are the hydrochloride, hydrobromide, sulfate, phosphate, acetate, oxalate, tartrate, citrate, maleinate or fumarate.
- the present invention also relates to a process for the preparation of compounds of the general formula I, which comprises the stages outlined in the diagram in the attached FIG. 1. If desired, further chromone derivatives according to the invention can be obtained by treating compounds of the formula I 'in FIG. 1 by known methods.
- the general scheme depicted in FIG. 1 is explained and described in more detail by the reaction sequence depicted in FIG. 2 ben relating to the preparation of one of the preferred compounds of the invention; it goes without saying that the scope of the invention is not restricted thereby.
- 1,3,5-trimethoxybenzene is stirred under acidic conditions with 1-methyl-4-piperidone in solvents such as water, acetic acid, alcoholic solvents or a suitable mixture thereof, glacial acetic acid being particularly preferred.
- the reaction temperature is kept between 20 and 90 ° C, but a temperature range of 50-60 ° C is preferred.
- the resulting organoborane complex is first treated with hydrochloric acid and then oxidized by adding alkali and hydrogen peroxide.
- the compound thus obtained is a trans alcohol of the formula IXA and is converted into the cis alcohol of the formula XIA by oxidation and subsequent reduction.
- the trans alcohol of the formula IXA is oxidized by means of a reagent combination, namely acid chlorides, with oxalkyl chloride being preferred, dimethyl sulfoxide and triethylamine and is known to the person skilled in the art as oxidation according to Swern.
- the ketone of the formula XA formed by oxidation of the compound of the formula IXA is reduced by means of hydride reagents, preferably diborane, lithium borohydride or sodium borohydride.
- a wide range of solvents are compatible with sodium borohydride, but proton-containing solvents such as methanol, ethanol and isopropanol are preferred. By maintaining a higher reaction temperature, the cis isomer can be obtained stereoselectively.
- the cis isomer can also be obtained by fractional crystallization of its acid addition salts, which are formed with optically active acids such as. B. (-) - and / or (+) - dibenzoyl tartaric acid.
- the cis isomer can optionally also be esterified with an optically active acid such as (-) - menthyloxyacetic acid and the resulting diastereomeric esters can then be separated by conventional methods such as fractional crystallization or chromatography.
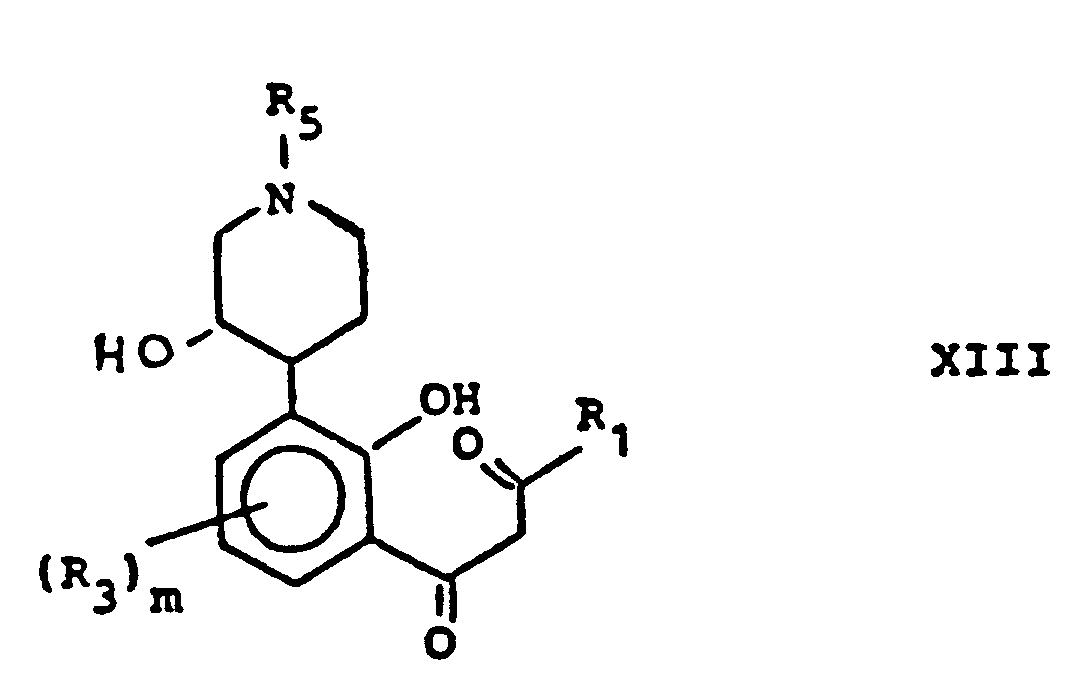
- the compound of the formula XIIA is then converted into the chromone by methods known per se, two of which are described here.
- the dimethoxychromone of the formula IA can also be demethoxylated using other acidic reagents.
- the demethoxylation is carried out by heating the dimethoxychromone derivatives with pyridine hydrochloride for a period of 2 to 10 hours at 180 ° C.
- the addition of high-boiling amines to the pyridine hydrochloride can be advantageous.
- the compounds according to the invention represented by the formula I have pharmacological properties.
- they show an anti-inflammatory and immunomodulating effect on laboratory animals. These properties are confirmed by the results of the subsequent pharmacological tests which were carried out to assess the compounds according to the invention and their salts.
- mice Male Charles Foster rats (120-150 g) were fasted for 18 hours, with water ad libitum. The test compound dissolved in distilled water was administered orally. The control group received distilled water. 0.05 ml of 0.5% carrageenin suspension was injected subcutaneously into the sole area of the left hind paw. The paw volume was determined using a Maclab differential volume meter from the carrageenan injection and 3 and 6 hours afterwards. The percent paw volume decrease was calculated using the following equation: The ED 50 value was calculated from the dose / effect curve. Six animals were used per group.
- the test compounds were administered orally one hour before induction of the Arthus response.
- the RPA response was induced by intradermal injection of 0.1 ml of appropriately diluted rabbit anti-BSA serum. Immediately after the intradermal injections, the rats received 0.5 ml of 0.4% bovine serum albumin intravenously. Four hours after the intradermal exposure, the animal groups were each killed by a broken neck.
- the entire thickness of the Skin was removed from the back of each animal and a 12 mm diameter disc was punched out with a metal punch at the site of the antiserum injection.
- the wet weight of the skin area was determined as soon as possible. Edema caused by the RPA was measured as the difference (expressed in mg) between the wet weight of the antibody-injected site and the normal rabbit serum-injected site.
- results are expressed as a percentage inhibition or potentiation of the edema by the compound compared to the edema induced in the untreated control animals.
- N-Methylpiperidone (2.8 mol) is added with stirring to a solution of trimethoxybenzene (2.38 mol) in glacial acetic acid (750 ml), the temperature of the reaction mixture being kept below 25 ° C.
- hydrogen chloride is bubbled through the reaction mixture, heated to 95-100 ° C. for 3 hours, then concentrated and the residue is diluted with water.
- the aqueous solution is extracted with ether, the ether separated and the aqueous layer made alkaline with concentrated sodium hydroxide solution. The precipitate thus obtained is filtered off, washed with water and dried.
- a solution of BF 3 etherate (42 ml) in diethylene glycol dimethyl ether (42 ml) is added dropwise to a cooled mixture of 1-methyl-4- (2,4,6-trimethoxyphenyl) -1,2,3,6-tetrahydropyridine ( 20 g) and sodium borohydride (12 g) in diethylene glycol dimethyl ether (140 ml).
- the mixture is heated at 50 ° C. for one hour and the cooled reaction mixture is then treated with water (20 ml) and then with concentrated HCl (116 ml).
- the mixture is stirred at 50-60 ° C for two hours, cooled and made alkaline with sodium hydroxide solution.
- Hydrogen peroxide solution (30%, 20 ml) is then added and the mixture is heated to 50-60 ° C. for two hours with stirring.
- the solution is cooled and extracted with ethyl acetate.
- the ethyl acetate extract is concentrated in vacuo.
- the residue is acidified with 2N HCl, extracted with ethyl acetate and the organic layer is separated off.
- the aqueous layer is then made alkaline with sodium hydroxide solution and extracted with ether.
- Dimethyl sulfoxide 35 ml is added dropwise to a solution of oxyalkyl chloride (20 ml) in dry methylene chloride (500 ml), cooled to -60 ° C., and the mixture is stirred for 5-10 minutes.
- a solution of ( ⁇ ) -trans-3-hydroxy-4- (2,4,6-trimethoxyphenyl) -1-methylpiperidine (62 g) in methylene chloride (300 ml) is then added while the temperature of the reaction mixture is at -60 ° C holds. After the addition, stir for 15 minutes and add triethylamine (155 ml). Then the reaction mixture is allowed to warm to a temperature of -30 ° C., diluted with water and made alkaline with sodium carbonate.
- BF 3 etherate (107.6 ml) is added dropwise to a solution of cis-3-hydroxy-4- (2,4,6-trimethoxyphenyl) -1-methylpiperidine (35 g) in methylene chloride (500 ml) with cooling in Ice bath. Then 76.2 ml of acetic anhydride are added dropwise. Then the reaction mixture is stirred for 24 hours at room temperature, diluted with water, made alkaline with sodium carbonate and extracted with methylene chloride. The extract is concentrated and the residue (37 g) is dissolved in methanol (200 ml) and stirred for 2 hours with 5% aqueous potassium hydroxide solution (500 ml).
- the chloroform extract is dried over anhydrous Na 2 S0 4 , concentrated in vacuo and purified with the aid of column chromatography (over silica gel). Thin layer chromatography (5% methanol in CHCl 3 + 1% by volume NH 4 0H: Rf value 0.5-0.7) can then be carried out.
- the chloroform extract is dried over anhydrous Na 2 S0 4 , concentrated in vacuo and purified by column chromatography on silica gel to give the desired product (8 g). Recrystallized from chloroform / petroleum ether, melting point 236-238 ° C (HCl salt).
- Dimethoxychromone (1.0 g), pyridine hydrochloride (5-10 g) and quinoline (0.5 ml) are mixed and heated to 180-190 ° C for 2-3 hours.
- the reaction mixture is then allowed to cool, water (1 ml) is added and the mixture is made basic by adding solid sodium bicarbonate.
- the semi-solid product is extracted thoroughly with 20% methanol in chloroform, the organic phase is concentrated and with the aid of column chromatography (silica gel; 15% by volume methanol in chloroform with the addition of 1% by volume NH 4 0H as element; Rf: 0, 4 - 0.7) cleaned.
- the hydrochloride salt is obtained by treatment with ethereal HCl.
- reaction mixture is extracted with ethyl acetate (5 times 100 ml each).
- the tartaric acid is recovered from the ethyl acetate extract.
- the aqueous layer is made alkaline with sodium carbonate and extracted with chloroform.
- Optically pure isomers were obtained from optically pure ( ⁇ ) - or (-) - cis-3-hydroxy-4- (3-acetyl-4,6-dimethoxy-2-hydroxy) -phenyl-1-methyl-piperidine as in the following examples 9 and 10:
- Cis-3-hydroxy-4- (3'-acetyl-4 ', 6'-dimethoxy-2'-hydroxy) -phenyl-1-methylpiperidine is treated as in Example 6 with ethyl propionate instead of ethyl acetate and the product is demethoxylated as in Example 7 described what cis-5,7-dihydroxy-2-ethyl-8- [4 '- (3'-hydroxy-1'-methyl) piperidinyl] -4H-1-benzopyran-4-one hydrochloride dated Delivers melting point 230-33 °.
- Cis-3-hydroxy-4- (3'-acetyl-4 ', 6'-dimethoxy-2'-hydroxy) -phenyl-1-methylpiperidine is treated as in Example 6 with ethyl butyrate instead of ethyl acetate and the product is demethoxylated as in Example 7 described what cis-5,7-dihydroxy-2-n-propyl-8- [4 '- (3'-hydroxy-1'-methyl) -piperidinyl] -4H-1-benzopyran-4- one- supplies hydrochloride with a melting point of 190-92 ° C.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Pulmonology (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
- Vorliegende Erfindung betrifft neue 4H-1-Benzopyran-4-on-derivate, Verfahren zu deren Herstellung sowie deren Verwendung als Antiphlogistika, Analgetika, Immunsuppressiva und Antiallergika. Insbesondere betrifft vorliegende Erfindung neue
- Verbindungen der Formel I in cis-Form

- R1 unsubstituiertes oder mit Halogen, Hydroxy- oder Carboxygruppen substituiertes C1-C6-Alkyl oder Pyridyl oder Phenyl, gegebenenfalls einfach oder mehrfach substituiert mit Halogen, C1-C4-Alkoxy, Nitro, Hydroxy, Carbonyl, Amino, Pyridyl oder Trifluormethyl,
- R3 Hydroxy oder C1-C4-Alkoxy,
- R5 Wasserstoff, C1-C6-Alkyl oder C3-C6-Cycloalkyl-C1-C4-alkyl,
- m eine ganze Zahl zwischen 0 und 3,
- Die erfindungsgemäßen Verbindungen besitzen zwei asymmetrische Zentren, eines an der Verknüpfungsstelle des Stickstoffheterocyclus mit dem Benzopyranteil (C-4') und das andere am durch R4 substituierten Kohlenstoffatom (C-3'), so daß zwei optische Isomerenpaare möglich sind. Es versteht sich, daß sämtliche möglichen Stereoisomeren und deren Gemische in der Definition der erfindungsgemäßen Verbindungen inbegriffen sind. Insbesondere sind dabei sowohl die racemischen Formen als auch die isolierten, die angegebene Aktivität besitzenden optischen Isomeren inbegriffen. Die beiden Racemate lassen sich nach physikalischen Methoden wie fraktionierte Kristallisation trennen. Die einzelnen optischen Isomeren sind aus den Racematen nach Standardmethoden wie Salzbildung mit einer optisch aktiven Säure und nachfolgende Kristallisation erhältlich.
- In der EP-A2-0 137 193 ist bereits die Verbindung (+)-cis-5,7-Dihydroxy-2-methyl-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyrano-4-on, deren Isolierung aus der Pflanze Dysoxylum binectariferum sowie ihre Verwendung als Mittel zur Immunmodulation beschrieben. Diese Verbindung ist daher von der vorliegenden Erfindung ausgenommen.
- Als Alkylgruppen für R1 und R5 eignen sich beispielsweise geradkettige oder verzweigte Reste mit bis zu 6 und vorzugsweise bis zu 5 Kohlenstoffatomen, z.B. Methyl-, Ethyl-, Propyl-, Isopropyl-, t-Butyl, Pentyl-oder Isopentylgruppen.
- Als substituierte Alkylgruppen für R1 eignen sich beispielsweise Halogenalkyl wie Trifluormethyl, Hydroxyalkyl wie Hydroxyäthyl oder Carboxyalkyl wie Carboxyäthyl.
- Die Cycloalkylalkylgruppe R5 ist beispielsweise Cyclopropylmethyl.
- Geeignete Beispiele für Salze der erfindungsgemäßen Verbindungen mit anorganischen oder organischen Säuren sind das Hydrochlorid, Hydrobromid, Sulfat, Phosphat, Acetat, Oxalat, Tartrat, Citrat, Maleinat oder Fumarat.
- Bevorzugte Verbindungen entsprechen der Formella,

- R1 Wasserstoff oder C1-C3-Alkyl
- R5 C1-C3-Alkyl oder C3-C5-Cycloalkyl-C1 -C3-alkyl.
- Besonders bevorzugte erfindungsgemäße Verbindungen sind:
- cis-(-)-5,7-Dihydroxy-2-methyl-8-[4'-(1'-cyclopropylmethyl-3'-hydroxy)-piperidinyl]-4H-1-benzopyran-4-on,
- cis-(±)-5,7-Dihydroxy-2-äthyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on und
- cis-(±)-5,7-Dihydroxy-2-n-propyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on.
- cis-( + )-5,7-Dihydroxy-2-n-propyl-8-[4'-(3'-hydroxy-1'-methyl)piperidingl-4H-1-benzopyran-4-on.
- cis-(-)-5,7-Dihydroxy-2-n-propyl-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl-]-4H-1-benzopyran-4-on.
- cis-(±)-2-n-Butyl-5,7-dihydroxy-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-5,7-Dihydroxy-2-phenyl-8-[4'-(3'-hydroxy-1'-methyl) piperidinyl]-4H-1-benzopyran-4-on.
- cis-(-)-5,7-Dihydroxy-2-phenyl-8-[4'-(3'-hydroxy-1'-methyl) piperidinylj-4H-1-benzopyran-4-on.
- cis-(±)-2-(2-Chlorophenyl)-5,7-dihydroxy-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-benzopyran-4-on.
- cis-(-)-2-(2-Chlorophenyl)-5,7-dihydroxy-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-2-(4-Aminophenyl)-5,7-dihydroxy-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-2-(4-Bromophenyl)-5,7-dihydroxy-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-2-(4-chlorophenyl)-5,7-dihydroxy-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-2-(2,4-Dichlorophenyl)-5,7-dihydroxy-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-5,7-Dihydroxy-2-(4-fluorophenyi)-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-5,7-Dihydroxy-2-(2-fluorphenyl)-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-5,7-Dihydroxy-2-(2-pyridyl)-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- cis-(±)-5,7-Dihydroxy-2-(4-pyridyl)-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on.
- Neue erfindungsgemäße 4H-1 benzopyran-4-on-derivate sind in den nachfolgenden Tabellen 1 bis 5 aufgeführt, wobei auf die folgenden Formeln Bezug genommen wird:












- Gegenstand der vorliegenden Erfindung ist auch ein Verfahren zur Herstellung von Verbindungen der allgemeinen Formel I, welches die in dem Schema auf der beigefügten Figur 1 skizzierten Stufen umfaßt. Gewünschtenfalls kann man weitere erfindungsgemäße Chromonderivate durch Behandlung von Verbindungen der Formel I' in Figur 1 nach bekannten Methoden erhalten. Das in Figur 1 abgebildete allgemeine Schema wird durch die auf Figur 2 abgebildete Reaktionsfolge mehr im einzelnen erläutert und beschrieben, das sich auf die Herstellung einer der bevorzugten erfindungsgemäßen Verbindung bezieht; dabei versteht es sich daß der Umfang der Erfindung dadurch nicht eingeschränkt wird.
- Die Herstellung der Verbindung der Formel VIII mit n = 1 ist dem Fachmann bekannt [S.M. McClavain und R.S. Berger, J. Am. Chem. Soc., 77, 2848 (1955); A. Ziering, L. Berger, S.D. Heineman und J. Lec., J. Org. Chem., 12, 894 (1947)]. Dort sind zwei Methoden beschrieben. Bei der ersten rührt man 1,3,5,-Trimethoxybenzol mit n-Butyllithium bei tiefen Temperaturen, vorzugsweise zwischen -60 und -90 ° C, in inerten Lösungsmitteln wie Kohlenwasserstoffen, z.B. Pentan oder Hexan, oder Ätherlösungsmitteln, z.B. Diäthyläther oder Tetrahydrofuran, zur Herstellung des Lithiosalzes, das beim Rühren mit 1-Methyl-4-piperidon und nachfolgendem Ansäuern das Tetrahydropyridinderivat ergibt. Bei der zweiten und besonders bevorzugten Methode rührt man 1,3,5-Trimethoxybenzol unter sauren Bedingungen mit 1-Methyl-4-piperi- don in Lösungsmitteln wie Wasser, Essigsäure, alkoholischen Lösungsmitteln oder einem geeigneten Gemisch daraus, wobei Eisessig besonders bevorzugt ist.
- Mit Bezug auf das Schema in Figur 2 wird das Tetrahydropyridinderivat der abgebildeten Formel VIIIA (d.h. Formel VIII mit n = 1) unter Verwendung von sich durch Zusatz von BF3-Ätherat zu einer Aufschlämmung von Natriumborhydrid in Diäthylenglykoldimethyläther unter wasserfreien Bedingungen und in einer durch laufendes Durchleiten von Stickstoff oder Argon aufrechterhaltenen inerten Atmosphäre direkt bildendem Diboran hydroboriert. Die Reaktionstemperatur hält man zwischen 20 und 90 ° C, doch wird ein Temperaturbereich von 50-60 °C bevorzugt. Der entstandene Organoborankomplex wird zunächst mit Salzsäure behandelt und dann durch Zusatz von Alkali und Wasserstoffperoxyd oxydiert. Die so erhaltene Verbindung ist ein trans-Alkohol der Formel IXA und wird durch Oxydation und nachfolgende Reduktion in den cis-Alkohol der Formel XIA umgewandelt. Die Oxydation des trans-Alkohols der Formel IXA erfolgt mittels einer Reagenzienkombination, nämlich Säurechloriden, wobei Oxalkylchlorid bevorzugt ist, Dimethylsulfoxid und Triäthylamin und ist dem Fachmann als Oxydation nach Swern bekannt. Das durch Oxydation der Verbindung der Formel IXA gebildete Keton der Formel XA wird mittels Hydridreagenzien, vorzugsweise Diboran, Lithiumborhydrid oder Natriumborhydrid, reduziert. Eine ganze Reihe von Lösungsmitteln ist mit Natriumborhydrid verträglich, doch werden protonenhaltige Lösungsmittel wie Methanol, Äthanol und Isopropanol bevorzugt. Durch Einhaltung einer höheren Reaktionstemperatur kann man das cis-Isomer stereoselektiv erhalten.
- Das cis-Isomere kann weiterhin gewonnen werden durch fraktionierte Kristallisation seiner Säureadditionssalze, die gebildet werden mit optisch aktiven Säuren wie z. B. (-)- und/oder (+)-Dibenzoylweinsäure. Das cis-Isomere kann gegebenenfalls auch verestert werden mit einer optisch aktiven Säure wie (-)-Menthyloxyessigsäure und die entstehenden diastereomeren Ester können anschließend durch konventionelle Methoden wie fraktionierte Kristallisation oder Chromatograpie getrennt werden.
- Die cis-Hydroxyverbindung der Formel XIA wird mit Essigsäureanhydrid und sauren Katalysatoren wie Aluminiumchlorid, Bortrifluoridätherat und Zinn(IV)-chlorid acetyliert, wobei als Reagenz Bortrifluoridätherat besonders bevorzugt ist. Bei Anwendung eines großen Überschusses an Bortrifluoridätherat erfolgt auch gleichzeitig eine Entmethylierung, und nur die gewünschte Methoxylgruppe wird regiospezifisch demethyliert, wobei eine Verbindung der Formel XIIA erhalten wird, worin R den Rest -COCH3 darstellt. Die Hydrolyse dieser Verbindung mit einem Alkalimetallhydroxid führt zu Verbindungen der Formel XIIA mit R = H. Die Verbindung der Formel XIIA wird dann nach an sich bekannten Methoden, von denen zwei hier beschrieben werden, in das Chromon umgewandelt. Bei der ersten Methode wird die Verbindung der Formel XIIA mit R = H bei Raumtemperatur und in inerten Lösungsmitteln wie Äther, Tetrahydrofuran, Dioxan oder Kohlenwasserstofflösungsmitteln wie Hexan mit Essigester und Alkalimetall oder NaH, bevorzugt Natriummetall gerührt; ist der Ester eine niedrigsiedende Flüssigkeit wie im vorliegenden Beispiel, so ist es auch als Lösungsmittel verwendbar. Die Reaktion ist normalerweise nach ein bis zehn Stunden beendet und ergibt das Diketon der Formel XIIIA mit R = H, das beim Rühren mit Mineralsäuren wie Salzsäure oder Schwefelsäure zum Chromon der Formel IA zyklisiert wird. Bei der zweiten Methode wird eine Verbindung der Formel XII A mit R = Ac verestert mit einer geeigneten Säure, z. B. Benzoesäure, und der erhaltene Ester mit einer Base, wie z. B. Alkalihydroxid, in einem inerten Lösungsmittel wie z. B. THF, Dioxan oder Pyridin gerührt, wobei das Chromon der Formel XIII A mit R = Ac und R1 = Phenyl gebildet wird. Verbindungen der Formel la mit R = H und R1 = CH3 werden mit Pyridinhydrochlorid entmethoxyliert, um die Hydroxyverbindung der abgebildeten Formel IB zu erhalten. Unter Verwendung der entsprechenden Ester anstelle von Essigester lassen sich verschiedene 2-substituierte Chromone herstellen.
- Das Dimethoxychromon der Formel IA kann außer mit AICb, BBr3 oder HBr/Essigsäure auch mittels anderer saurer Reagenzien entmethoxyliert werden. Die Entmethoxylierung erfolgt durch Erhitzen der Dimethoxychromonderivate mit Pyridinhydrochlorid für einen Zeitraum von 2 bis 10 Stunden auf 180°C. In manchen Fällen kann die Zugabe von hochsiedenden Aminen zum Pyridinhydrochlorid vorteilhaft sein.
- Das Syntheseschema nach Figur 2 läßt sich zur Herstellung von Verbindungen der Formel I mit R5 = H, Alkyl (von Methyl verschieden), Cycloalkyl, Aralkyl und Aryl anwenden. Verbindungen der Formel I, worin R5 dieselbe Bedeutung wie oben hat, lassen sich auch aus den entsprechenden N-Methylverbindungen, d.h. R5 = CH3 (Verbindungen der Formel IB) nach einer der bekannten Methoden herstellen. Eine typische Arbeitsweise ist aus dem Schema der Figur 3 ersichtlich, wo eine Verbindung der Formel IB mit R5 = CH3 nach Schutz der Hydroxylgruppen mit Bromcyan behandelt und danach sauer oder alkalisch zu Verbindungen mit R5 = H (Verbindung der Formel XIV) hydrolysiert wird. Diese Verbindung ergibt bei Behandlung mit geeigneten elektrophilen Reagenzien wie Halogeniden, Säurechloriden, Tosylaten oder Enonen Verbindungen mit R5 = Alkyl, Cycloalkyl, Aralkyl oder Aryl, wobei die Verbindung der abgebildeten Formel XVII ein spezielles Beispiel darstellt. Gemäß Figur 3 wird 5,7-Dihydroxy-2-methyl-8-[4'-(1'-cyclopropylmethyl-3'-hydroxy)-piperidinyl]-4H-1-benzopyran-4-on der abgebildeten Formel XVII durch Peracetylierung von 5,7-Dihydroxy-2-methyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on der Formel IB mit Essigsäureanhydrid und Natriumacetat bei 80-90 ° C hergestellt. Das peracetylierte Produkt der Formel XIV wird mit Bromcyan in Chloroform verrührt und ergibt in Gegenwart von Kaliumcarbonat 5,7-Diacetyl-2-methyl-8-[4'-(3'-acetoxy-1'-cyan)-piperidinyl]-4H-1-benzopyran-4-on der Formel XV, das bei 5 Stunden langem Erhitzen mit 2n-Salzsäure auf 110°C das hydrolysierte und N-entmethylierte Produkt 5,7-Dihydroxy-2-methyl-8-[4'-(3'-hydroxy)-piperidinyl]-4H-1-benzopyran-4-on der Formel XVI liefert. Beim Erhitzen mit Cyclopropylmethylchlorid in Isobutylalkohol erhält man daraus das N-Cyclopropylmethylderivat der abgebildeten Formel XVII.
- Verbindungen der Formel I, worin R2 Dialkylaminomethyl bedeutet, werden dadurch hergestellt, daß man das entsprechende Chromon mit R2 = H mit einem sekundären Aminhydrochlorid und Paraformaldehyd in Dioxan oder alkoholischen Lösungsmitteln zum Rückfluß erhitzt. Verbindungen der Formel I mit R2 = N02 werden zweckmäßig durch Verrühren des entsprechenden Chromons mit R2 = H mit Essigsäure und konzentrierter Salpetersäure hergestellt. Verbindungen der Formel I mit R2 = NH2 erhält man aus den entsprechenden Nitroderivaten durch Hydrierung über 10%igem Pd/C.
- Verbindungen der Formel I, worin eine der R3-Gruppen für Brom steht, stellt man durch Verrühren der entsprechenden Chromone mit R3 = H mit N-Bromsuccinimid in Dimethylformamid her.
- Ein weiteres Merkmal der Erfindung ist, daß die durch die Formel I dargestellten erfindungsgemäßen Verbindungen pharmakologische Eigenschaften besitzen. Insbesondere zeigen sie eine entzündungshemmende und immunomodulierende Wirkung an Laboratoriumstieren. Diese Eigenschaften werden durch die Ergebnisse der nachfolgenden pharmakologischen Prüfungen belegt, die zur Beurteilung der erfindungsgemäßen Verbindungen und ihrer Salze durchgeführt wurden.
- Männliche Charles Foster-Ratten (120-150 g) wurden 18 Stunden lang nüchtern gesetzt, mit Wasser ad libitum. Die in destilliertem Wasser gelöste Testverbindung wurde oral verabreicht. Die Kontrollgruppe erhielt destilliertes Wasser. 0,05 ml 0,5%ige Carrageeninsuspension wurde subkutan in die Sohlengegend der linken Hinterpfote injiziert. Das Pfotenvolumen wurde unter Verwendung eines Differentialvolumenmeßgeräts nach Maclab von der Carrageenininjektion sowie 3 und 6 Stunden danach bestimmt. Die prozentuale Pfotenvolumenabnahme wurde nach folgender Gleichung berechnet:

- Die Ergebnisse mit repräsentativen erfindungsgemäßen Verbindungen und deren Salzen sind in Tabelle 4 angeführt, wobei die Substituenten sich auf die folgende Formel la beziehen:


- Charles Foster-Ratten beider Geschlechter von 150-180 g Gewicht wurden in Gruppen von je sechs Tieren sortiert. 24 Stunden vor der Auslösung der RPA wurden die Ratten von der Mittelrückengegend geschoren und über Nacht nüchtern gesetzt. Die Testverbindungen wurden oral eine Stunde vor der Induzierung der Arthus-Reaktion verabreicht. Die RPA-Reaktion wurde durch intradermale Injektion von 0,1 ml entsprechend verdünntem Kaninchen-anti-BSA-Serum induziert. Sofort nach den intradermalen Injektionen erhielten die Ratten intravenös je 0,5 ml 0,4%iges Rinderserumalbumin. Vier Stunden nach der intradermalen Exposition wurden die Tiergruppen jeweils durch Halsbruch getötet. Die gesamte Dicke der Haut wurde vom Rücken jedes Tiers entfernt, und eine Scheibe von 12 mm Durchmesser wurde mit einer Metallstanze an der Stelle der Antiseruminjektion ausgestanzt. Das Naßgewicht der Hautstelle wurde jeweils so bald wie möglich bestimmt. Das durch die RPA verursachte Ödem wurde als die Differenz (in mg ausgedrückt) zwischen dem Naßgewicht der mit Antikörper injizierten Stelle und der mit normalem Kaninchenserum injizierten Stelle gemessen.
- Die Ergebnisse sind als prozentuale Hemmung bzw. Potenzierung des Ödems durch die Verbindung gegenüber dem in den unbehandelten Kontrolltieren induzierten Ödem ausgedrückt.
- Die Ergebnisse mit repräsentativen erfindungsgemäßen Verbindungen und deren Salzen sind in Tabelle 7 angeführt.



- Die Erfindung wird durch die nachfolgenden Beispiele erläutert, jedoch nicht eingeschränkt.
- Man gibt N-Methylpiperidon (2,8 Mol) unter Rühren zu einer Lösung von Trimethoxybenzol (2,38 Mol) in Eisessig (750 ml), wobei man die Temperatur des Reaktionsgemisches unter 25 °C hält. Nach beendeter Zugabe perlt man Chlorwasserstoff durch das Reaktionsgemisch, erhitzt 3 Stunden lang auf 95-100 °C, engt dann ein und verdünnt den Rückstand mit Wasser. Die wäßrige Lösung wird mit Äther extrahiert, der Äther abgetrennt und die wäßrige Schicht mit konzentrierter Natronlauge alkalisch gemacht. Der so erhaltene Niederschlag wird abfiltriert, mit Wasser gewaschen und getrocknet. Umkristallisieren aus Petroläther (60°-80 °C) ergibt 550 g 1-Methyl-3-(2,4,6-trimethoxyphenyl)-1,2,3,6-tetrahydropyridin vom Schmelzpunkt 118-122°C.

- Man tropft eine Lösung von BF3-Ätherat (42 ml) in Diäthylenglykoldimethyläther (42 ml) zu einem gekühlten Gemisch aus 1-Methyl-4-(2,4,6-trimethoxyphenyl)-1,2,3,6-tetrahydropyridin (20 g) und Natriumborhydrid (12 g) in Diäthylenglykoldimethyläther (140 ml). Man erhitzt eine Stunde lang auf 50°C und behandelt das abgekühlte Reaktionsgemisch dann mit Wasser (20 ml) und danach mit konzentrierter HCI (116 ml). Man rührt zwei Stunden lang bei 50-60 °C, kühlt ab und macht mit Natronlauge alkalisch. Dann wird Wasserstoffperoxydlösung (30 %, 20 ml) dazugegeben und unter Rühren zwei Stunden lang auf 50-60°C erhitzt. Die Lösung wird abgekühlt und mit Essigester extrahiert. Der Essigesterextrakt wird im Vakuum eingeengt. Den Rückstand säuert man mit 2n-HCI an, extrahiert mit Essigester und trennt die organische Schicht ab. Die wäßrige Schicht wird dann mit Natronlauge alkalisch gemacht und mit Äther extrahiert. Der Ätherextrakt wird mit Sole gewaschen, über Natriumsulfat getrocknet und eingeengt, wobei man einen festen Rückstand erhält, der aus heißem Wasser umkristallisiert wird, was trans-3-Hydroxy-4-(2,4,6-trimethoxyphenyl)-1-methylpiperidin (12 g) ergibt. Ausbeute: 12g; Schmelzpunkt 88°-89° C.

- Zu einer auf -60 °C gekühlten Lösung von Oxalkylchlorid (20 ml) in trockenem Methylenchlorid (500 ml) tropft man Dimethylsulfoxyd (35 ml) unter Stickstoff und rührt 5-10 Minuten lang. Dann versetzt man mit einer Lösung von (±)-trans-3-Hydroxy-4-(2,4,6-trimethoxyphenyl)-1-methylpiperidin (62 g) in Methylenchlorid (300 ml), während man die Temperatur des Reaktionsgemisches bei -60 ° C hält. Nach der Zugabe rührt man 15 Minuten lang und gibt Triäthylamin (155 ml) dazu. Dann läßt man das Reaktionsgemisch sich auf eine Temperatur von -30 °C erwärmen, verdünnt mit Wasser und macht mit Natriumcarbonat alkalisch.
- Die organische Schicht wird abgetrennt und die wäßrige Schicht mit Essigester extrahiert. Die organischen Schichten werden vereinigt, mit Sole gewaschen, über wasserfreiem Na2S04 getrocknet und zu einem festen Rückstand eingeengt, der beim Kristallisieren aus Isopropanol das gewünschte Produkt (47 g) vom Schmelzpunkt 110-112°C ergibt.

- Man gibt Natriumborhydrid (10 g) unter Rühren zu einer am Rückfluß siedenden Lösung von 1-Methyl-4-(2,4,6-trimethoxyphenyl)-piperidin-3-on in absolutem Äthanol. Dann wird noch eine Stunde weitergerührt und am Rückfluß erhitzt. Beim Abkühlen verdünnt man das Reaktionsgemisch mit Wasser, engt dann zur Entfernung des Äthanols ein und extrahiert mit Chloroform. Der Chloroformextrakt wird mit Wasser gewaschen, über wasserfreiem Natriumsulfat getrocknet und zu einem festen Rückstand eingeengt, der beim Kristallisieren aus Aceton das erwünschte Produkt (29,2 g) vom Schmelzpunkt 124-125 °C ergibt.

- Man tropft BF3-Ätherat (107,6 ml) zu einer Lösung von cis-3-Hydroxy-4-(2,4,6-trimethoxyphenyl)-1-methylpiperidin (35 g) in Methylenchlorid (500 ml) unter Kühlen im Eisbad. Danach werden 76,2 ml Acetanhydrid tropfenweise hinzugefügt. Dann rührt man das Reaktionsgemisch 24 Stunden lang bei Raumtemperatur, verdünnt mit Wasser, macht mit Natriumcarbonat alkalisch und extrahiert mit Methylenchlorid. Der Extrakt wird eingeengt und der Rückstand (37 g) in Methanol (200 ml) gelöst und 2 Stunden lang mit 5 %iger wäßriger Kalilauge (500 ml) gerührt. Dann engt man im Vakuum ein und extrahiert den Rückstand mit Chloroform. Der nach Einengen des Chloroformextrakts erhaltene Rückstand wird dann durch Chromatographie über Silicagel gereinigt, wobei man cis-3-Hydroxy-4-(3'-acetyl-4',6'-dimethoxy-2'-hydroxy)-phenyl-1-methylpiperidin (28 g) vom Schmelzpunkt 215-218 ° C (als HCI-Salz) erhält.

- Allgemeines Verfahren zur Herstellung von cis/trans5,7-Dimethoxy-2-(R1)-8-[4'-(3'-hydroxy-1-methyl)-piperidinyl]-4H-1-benzopyran-4-one
- Die Lösung von cis/trans-3-Hydroxy-4-(3'-acetyl-4,6-dimethoxy-2'-hydroxy)phenyl-1-methylpiperidin (1 Äquival.) wird mit einem geeigneten Ester (3 Äquival.) und Na-Metall (- 10 Äquival.) oder Na-Hydrid (- 5 Äquival.) in trockenem Dioxan oder Dimethylformamid bei Raumtemperatur bzw. bei 70 - 80 °C (siehe Tabelle 8) gerührt. Dann wird vorsichtig Wasser zugegeben und mit Chloroform extrahiert. Die organische Phase wird abgetrennt, etwas eingeengt, mit HCI-Gas gesättigt und anschließend eine Stunde gerührt. Die Lösung wird dann durch Zugabe von Na2C03 basisch gemacht und mit Chloroform extrahiert. Der Chloroformextrakt wird über wasserfreiem Na2S04 getrocknet, im Vakuum eingeengt und mit Hilfe von Säulenchromatographie (über Silicagel) gereinigt. Anschließend kann eine Dünnschichtchromatographie (5 % Methanol in CHCI3 + 1 Vol.-% NH40H: Rf-Wert 0,5 - 0,7) durchgeführt werden.
- Mit dem angegebenen allgemeinen Verfahren wurden die in der folgenden Tabelle 8 und im Beispiel 6a angeführten Verbindungen hergestellt:

- Man erhitzt eine Lösung von cis-3-Hydroxy-4-(3'-acetyl-4', 6'-dimethoxy-2'-hydroxy)-phenyl-1-methylpiperidin (10 g) in Essigester (500 ml) zum Rückfluß und gibt Natrium (7 g) in kleinen Anteilen dazu. Man rührt und erhitzt 2 bis 3 Stunden lang am Rückfluß. Nach dem Abkühlen wird das Gemisch mit Wasser verdünnt und die organische Schicht abgetrennt. Diese wird dann auf das halbe Volumen eingeengt, mit konzentrierter HCI (10 ml) behandelt und etwa eine Stunde lang gerührt. Dann verdünnt man mit Wasser, macht die wäßrige Schicht mit Na2C03 alkalisch und extrahiert mit Chloroform. Der Chloroformextrakt wird über wasserfreiem Na2S04 getrocknet, im Vakuum eingeengt und durch Säulenchromatographie über Silicagel gereinigt, wobei man das erwünschte Produkt (8 g) erhält. Umkristallisiert aus Chloroform/Petroläther, Schmelzpunkt 236-238 °C (HCI-Salz).

- Allgemeines Demethylierungsverfahren zur Herstellung von cis/trans-5,7-Dihydroxy-2-(R1)-8-[4'-(3'-hydroxy-1'-methyl)piperidinyl]-4H-1-benzopyran-4-on hydrochlorid:
- Dimethoxychromon (1,0 g), Pyridinhydrochlorid (5 - 10 g) und Chinolin (0,5 ml) werden gemischt und 2 - 3 Stunden auf 180 - 190°C erhitzt. Man läßt die Reaktionsmischung dann abkühlen, gibt Wasser (1 ml) hinzu und macht basisch durch Zugeben von festem Natriumbicarbonat. Das halbfeste Produkt wird gründlich mit 20 % Methanol in Chloroform extrahiert, die organische Phase eingeengt und mit Hilfe von Säulenchromatographie (Silicagel; 15 Vol.-% Methanol in Chloroform unter Zusatz von 1 Vol-% NH40H als Element; Rf: 0,4 - 0,7) gereinigt. Durch Behandlung mit ätherischem HCI erhält man das Hydrochlorid-Salz.
- Mit dem angegebenen allgemeinen Verfahren werden die in der folgenden Tabelle 9 aufgeführten Verbindungen hergestellt:


- Man löst die racemische cis-3-Hydroxyverbindung (90 g) in Methanol (300 ml), versetzt mit (-)-Dibenzoylweinsäure (126,4 g) in Methanol (200 ml) und erhitzt zum Sieden. Dann gibt man langsam Diisopropyläther (ca. 500 ml) dazu und läßt die klare Lösung abkühlen. Dabei kristallisiert das Tartratsalz langsam aus. Dieses wird abfiltriert und fünfmal aus Methanol/Diisopropyläther umkristallisiert, [α]


- Die Filtrate von den Tartratkristallisationen werden vereinigt und die freie Base wie oben beschrieben zurückgewonnen. Man löst die freie Base (20 g) in Methanol (110 ml), versetzt mit (+)-Dibenzoylweinsäure (29 g) und erhitzt die Lösung zum Sieden. Dann gibt man langsam Diisopropyläther (110 ml) dazu. Beim Stehen bei Raumtemperatur kristallisiert das Tartrat aus. Es wird abfiltriert und dreimal aus Methanol/Diisopropyläthergemisch umkristallisiert. Ausbeute: 20,2 g, [α]


- Optisch reine Isomere wurden aus optisch reinen (±)- oder (-)-cis-3-hydroxy-4-(3-Acetyl-4,6-dimethoxy-2-hydroxy)-phenyl-1-methyl-piperidin wie in den folgenden Beispielen 9 und 10 hergestellt:
- Man behandelt (-)-cis-3-Hydroxy-4-(2',4',6'-trimethoxyphenyl)-1-methylpiperidin auf die gleiche Weise wie in Beispiel 5, was (-)-cis-3-Hydroxy-4(3'-acetyl-4',6'-dimethoxy-2'-hydroxy)-phenyl-1-methylpiperidin vom Schmelzpunkt 184-86°C, [α]

- Man behandelt (+)-cis-3-Hydroxy-4-(2',4',6'-trimethoxyphenyl)-1-methylpiperidin auf die gleiche Weise wie in Beispiel 5, was (+)-cis-3-Hydroxy-4-(3'-acetyl-4',6'-dimethoxy-2'-hydroxy)-phenyl-1-methylpiperidin vom Schmelzpunkt 184-85°C, [α]

- Man behandelt (-)-cis-3-Hydroxy-4-(3'-acetyl-4',6'-dimethoxy-2'-hydroxy)-phenyl-1-methylpiperidin auf die gleiche Weise wie in Beispiel 6, was (-)-cis-5,7-di-methoxy-2-methyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on vom Schmelzpunkt 228-30 °C, [α]

- Man behandelt (+)-cis-3-Hydroxy-4-(3'-acetyl-4',6'-dimethoxy-2'-hydroxy)-phenyl-1-methylpiperidin auf die gleiche Weise wie in Beispiel 6, was (+)-cis-5,6-dimethoxy-2-methyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on vom Schmelzpunkt 228-29 °C, [α]

- Man behandelt (-)-cis-5,7-Dimethoxy-2-methyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on auf die gleiche Weise wie in Beispiel 7, was (-)-cis-5,7-Dihydroxy-2-methyl-8-[4'-(3'-hydroxy-I'-methyl)-piperidinyl]-4H-I-benzopyran-4-on-hydrochlorid vom Schmelzpunkt 242-45°C, [α]

- Man behandelt (+)-cis-5,7-Dimethoxy-2-methyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on auf die gleiche Weise wie in Beispiel 7, was (+)-cis-5,7-Dihydroxy-2-methyl-8-['-(3'-hydroxy-I'-methyl)-piperidinyl]-4H-I-benzopyran-4-on-hydrochlorid vom Schmelzpunkt 242-44 °C, [α]

- Man behandelt cis-3-Hydroxy-4-(3'-acetyl-4',6'-dimethoxy-2'-hydroxy)-phenyl-1-methylpiperidin wie in Beispiel 6 mit Propionsäureäthylester anstelle von Essigester und entmethoxyliert das Produkt wie in Beispiel 7 beschrieben, was cis-5,7-Dihydroxy-2-äthyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on-hydrochlorid vom Schmelzpunkt 230-33 ° liefert.

- Man behandelt cis-3-Hydroxy-4-(3'-acetyl-4',6'-dimethoxy-2'-hydroxy)-phenyl-1-methylpiperidin wie in Beispiel 6 mit Buttersäureäthylester anstelle von Essigester und entmethoxyliert das Produkt wie in Beispiel 7 beschrieben, was cis-5,7-Dihydroxy-2-n-propyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4- on-hydrochlorid vom Schmelzpunkt 190-92 °C liefert.

- Man erhitzt cis-(-)-5,7-Dihydroxy-2-methyl-8-[4'-(3'-hydroxy-1'-methyl)-piperidinyl]-4H-1-benzopyran-4-on (5 g) mit Essigsäureanhydrid (25 ml) und Natriumacetat (4,5 g) 12 Stunden lang auf 90°C. Das Essigsäureanhydrid wird im Hochvakuum abdestilliert und der Rückstand mit Essigester angerührt. Der in Essigester lösliche Anteil wird zur Trockne eingeengt. Den Rückstand löst man in trockenem Chloroform (27 ml), gibt wasserfreies Kaliumcarbonat (5 g) dazu und kühlt auf 0°C ab. Bromcyan (6 g) in trockenem Chloroform (25 ml) wird dazugetropft. Nach der Zugabe rührt man das Reaktionsgemisch 4-5 Stunden lang bei 40-50°C, filtriert und wäscht das Filtrat mit einer kleinen Menge Sole, trocknet über wasserfreiem Natriumsulfat und engt ein. Der Rückstand wird auf dem Dampfbad 7-8 Stunden lang mit 1 n Salzsäure (30 ml) erhitzt. Das Reaktionsgemisch wird durch Zugabe festen Natriumcarbonats alkalisch gemacht und eingeengt. Den Rückstand läßt man durch eine HP-20-Säule laufen und eluiert das Produkt mit 20%igem MeOH in H20. Das Produkt wird aus MeOH/Diisopropyläther kristallisiert, Schmelzpunkt 300°C, [α]



- Man vermischt cis-(-)-5,7-Dihydroxy-2-methyl-8-[4'-(3'-hydroxy)-piperidinyl]-4H-1-benzopyran-4-on (1,0 g), Cyclopropylmethylketon (1,5 ml), Isobutanol (15 ml) und Kaliumcarbonat (3 g) und erhitzt 15 Stunden lang auf 90°C. Das Reaktionsgemisch wird filtriert und der Rückstand mit Chloroform gewaschen. Das Filtrat wird eingeengt und über Silicagel säulenchromatographiert. Die Verbindung wird mit 6%igem MeOH in Chloroform eluiert. Durch Zusatz von ätherischer HCI stellt man das Hydrochlorid her, Ausbeute 0,7 g, Schmelzpunkt 249-51 °C, [α]


Claims (12)

, dadurch gekennzeichnet, daß man eine
Verbindung der Formel XII

worin Ri, R3, R5 und m die angegebene Bedeutung haben, , gegebenenfalls eine Verbindung der Formel I demethoxyliert zu einer Verbindung der Formel I, worin R3 die Hydroxygruppe bedeutet und m die genannte Bedeutung hat,
und gegebenenfalls eine Verbindung der Formel I, worin R5 CH3 bedeutet, nach Schutz der Hydroxylgruppen mit Bromcyan umgesetzt und die erhaltene Verbindung sauer oder alkalisch umgesetzt wird zu einer Verbindung der Formel I, worin R5 Wasserstoff bedeutet,
gegebenenfalls eine Verbindung der Formel I, worin R5 Wasserstoff bedeutet, mit geeigneten elektrophilen Reagenzien wie Halogeniden, Säurechloriden, Tosylaten oder Enonen zu Verbindungen der Formel I umsetzt, worin R5 unsubstituiertes oder substituiertes C1-C6-Alkyl, oder C3-C6-Cycloalkyl-C1-C4-alkyl bedeutet,
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT87105180T ATE95519T1 (de) | 1986-04-11 | 1987-04-08 | 4h-1-benzopyran-4-on-derivate, ein verfahren zu ihrer herstellung und ihre verwendung als arzneimittel. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE3612337 | 1986-04-11 | ||
| DE19863612337 DE3612337A1 (de) | 1986-04-11 | 1986-04-11 | 4h-1-benzopyran-4-on-derivate, ein verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0241003A2 EP0241003A2 (de) | 1987-10-14 |
| EP0241003A3 EP0241003A3 (en) | 1988-10-12 |
| EP0241003B1 true EP0241003B1 (de) | 1993-10-06 |
Family
ID=6298535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP87105180A Expired - Lifetime EP0241003B1 (de) | 1986-04-11 | 1987-04-08 | 4H-1-Benzopyran-4-on-Derivate, ein Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US4900727A (de) |
| EP (1) | EP0241003B1 (de) |
| JP (1) | JPH0686446B2 (de) |
| KR (1) | KR950009861B1 (de) |
| AT (2) | ATE95519T1 (de) |
| AU (1) | AU602891B2 (de) |
| CA (1) | CA1332238C (de) |
| DE (2) | DE3612337A1 (de) |
| DK (1) | DK169760B1 (de) |
| ES (1) | ES2060582T3 (de) |
| HK (1) | HK1006021A1 (de) |
| IE (1) | IE62244B1 (de) |
| IL (1) | IL82149A (de) |
| IN (1) | IN164232B (de) |
| PT (1) | PT84654B (de) |
| ZA (1) | ZA872555B (de) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6225473B1 (en) | 1998-01-23 | 2001-05-01 | Aventis Pharma Deutschland Gmbh | Method for producing (−)cis-3-hydroxy-1-methyl-4-(2,4,6-trimethoxyphenyl)-piperidine |
| US8119678B2 (en) | 2002-04-17 | 2012-02-21 | Cytokinetics, Incorporated | Compounds, compositions and methods |
| US10562925B2 (en) | 2015-05-18 | 2020-02-18 | Tolero Pharmaceuticals, Inc. | Alvocidib prodrugs having increased bioavailability |
| US10568887B2 (en) | 2015-08-03 | 2020-02-25 | Tolero Pharmaceuticals, Inc. | Combination therapies for treatment of cancer |
| US10624880B2 (en) | 2015-04-20 | 2020-04-21 | Tolero Pharmaceuticals, Inc. | Predicting response to alvocidib by mitochondrial profiling |
| US11034710B2 (en) | 2018-12-04 | 2021-06-15 | Sumitomo Dainippon Pharma Oncology, Inc. | CDK9 inhibitors and polymorphs thereof for use as agents for treatment of cancer |
| US11279694B2 (en) | 2016-11-18 | 2022-03-22 | Sumitomo Dainippon Pharma Oncology, Inc. | Alvocidib prodrugs and their use as protein kinase inhibitors |
| US11497756B2 (en) | 2017-09-12 | 2022-11-15 | Sumitomo Pharma Oncology, Inc. | Treatment regimen for cancers that are insensitive to BCL-2 inhibitors using the MCL-1 inhibitor alvocidib |
Families Citing this family (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5284856A (en) * | 1988-10-28 | 1994-02-08 | Hoechst Aktiengesellschaft | Oncogene-encoded kinases inhibition using 4-H-1-benzopyran-4-one derivatives |
| DE3836676A1 (de) * | 1988-10-28 | 1990-05-03 | Hoechst Ag | Die verwendung von 4h-1-benzopyran-4-on-derivaten, neue 4h-1-benzopyran-4-on-derivate und diese enthaltende arzneimittel |
| US5278174A (en) * | 1990-06-04 | 1994-01-11 | Scios Nova, Inc. | Sigma binding site agents |
| EP0508347A1 (de) * | 1991-04-10 | 1992-10-14 | Hoechst Aktiengesellschaft | 5,7-Dihydroxy-2-Methyl-8-[4-(3-Hydroxy-1-(1-Propyl))Piperidinyl]-4H-1-Benzopyran-4-on, seine Herstellung und Verwendung |
| GB9604709D0 (en) * | 1996-03-05 | 1996-05-01 | Imperial College | A compound |
| US6011024A (en) | 1991-08-28 | 2000-01-04 | Imperial College Of Science Technology & Medicine | Steroid sulphatase inhibitors |
| US6476011B1 (en) | 1991-08-28 | 2002-11-05 | Sterix Limited | Methods for introducing an estrogenic compound |
| US6903084B2 (en) * | 1991-08-29 | 2005-06-07 | Sterix Limited | Steroid sulphatase inhibitors |
| CZ285937B6 (cs) * | 1992-01-16 | 1999-12-15 | Hoechst Aktiengesellschaft | Arylcykloalkylové deriváty, způsob přípravy těchto derivátů a jejich použití |
| US5733920A (en) * | 1995-10-31 | 1998-03-31 | Mitotix, Inc. | Inhibitors of cyclin dependent kinases |
| US20060241173A1 (en) * | 1996-02-16 | 2006-10-26 | Sterix Ltd. | Compound |
| US6506792B1 (en) | 1997-03-04 | 2003-01-14 | Sterix Limited | Compounds that inhibit oestrone sulphatase and/or aromatase and methods for making and using |
| US6087366A (en) * | 1996-03-07 | 2000-07-11 | The Trustees Of Columbia University In The City Of New York | Use of flavopiridol or a pharmaceutically acceptable salt thereof for inhibiting cell damage or cell death |
| US5849733A (en) * | 1996-05-10 | 1998-12-15 | Bristol-Myers Squibb Co. | 2-thio or 2-oxo flavopiridol analogs |
| US5908934A (en) * | 1996-09-26 | 1999-06-01 | Bristol-Myers Squibb Company | Process for the preparation of chiral ketone intermediates useful for the preparation of flavopiridol and analogs |
| GB9807779D0 (en) * | 1998-04-09 | 1998-06-10 | Ciba Geigy Ag | Organic compounds |
| US6399633B1 (en) * | 1999-02-01 | 2002-06-04 | Aventis Pharmaceuticals Inc. | Use of 4-H-1-benzopryan-4-one derivatives as inhibitors of smooth muscle cell proliferation |
| DE19959546A1 (de) * | 1999-12-09 | 2001-06-21 | Rhone Poulenc Rorer Gmbh | Pharmazeutische Zubereitung zur Behandlung von Tumorerkrankungen |
| US7335650B2 (en) | 2000-01-14 | 2008-02-26 | Sterix Limited | Composition |
| BR0107726A (pt) * | 2000-01-18 | 2002-10-01 | Aventis Pharma Inc | Composto, composição farmacêutica, e, métodos de tratamento de um paciente canceroso, de inibição de proteìna quinases em um paciente, de inibição de quinases dependentes de ciclina em um paciente, e de preparação do composto |
| US6576647B2 (en) | 2000-01-18 | 2003-06-10 | Aventis Pharmaceuticals Inc. | Pseudopolymorph of (—)-cis-2-(2-chlorophenyl)-5,7-dihydroxy-8[4R-(3S-hydroxy -1-methyl)piperidinyl]-4H-1-benzopyran-4-one |
| SK287216B6 (sk) * | 2000-01-18 | 2010-03-08 | Aventis Pharmaceuticals Inc. | Hydrochloridová etanolová solvátová forma (-)-cis-2-(2- chlórfenyl)-5,7-dihydroxy-8[4R-(3S-hydroxy-1-metyl)piperidinyl]- 4H-1-benzopyran-4-ónu, spôsob jej prípravy, farmaceutická kompozícia, ktorá ju obsahuje, a jej použitie |
| US6821990B2 (en) | 2000-01-18 | 2004-11-23 | Aventis Pharma Deutschland Gmbh | Ethanol solvate of (-)-cis-2-(2-chlorophenyl)-5, 7-dihydroxy-8 [4R-(3S-hydroxy-1-M ethyl) piperidinyl]-4H-1-benzopyran-4-one |
| FR2805538B1 (fr) * | 2000-02-29 | 2006-08-04 | Hoechst Marion Roussel Inc | Nouveaux derives de flavones, leur procede de preparation, leur application a titre de medicaments, compositions pharmaceutiques et nouvelle utilisation |
| KR100423899B1 (ko) | 2000-05-10 | 2004-03-24 | 주식회사 엘지생명과학 | 세포 증식 억제제로 유용한 1,1-디옥소이소티아졸리딘을갖는 인다졸 |
| US7884127B2 (en) * | 2002-07-08 | 2011-02-08 | Pirimal Life Sciences Ltd. | Inhibitors of cyclin dependent kinases and their use |
| US7271193B2 (en) * | 2002-07-08 | 2007-09-18 | Nicholas Piramal India, Ltd. | Inhibitors of cyclin-dependent kinases and their use |
| US7915301B2 (en) * | 2002-07-08 | 2011-03-29 | Piramal Life Science Limited | Inhibitors of cyclin dependent kinases and their use |
| EP1539727B1 (de) * | 2002-07-17 | 2009-02-18 | Cytokinetics, Inc. | Verbindungen, zusammensetzungen und verfahren zur behandlung von zellulären proliferativen erkrankungen |
| EP1554265A4 (de) * | 2002-09-13 | 2008-05-07 | Cytokinetics Inc | Verbindungen, zusammensetzungen und verfahren |
| WO2005042697A2 (en) * | 2003-10-06 | 2005-05-12 | Cytokinetics, Inc. | Compounds, compositions and methods |
| WO2005056014A1 (en) * | 2003-12-09 | 2005-06-23 | The Government Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Methods for suppressing an immune response or a treating a proliferative disorder |
| WO2005058341A2 (en) * | 2003-12-11 | 2005-06-30 | Theravance, Inc. | Compositions for use in the treatment of mutant receptor tyrosine kinase driven cellular proliferative diseases |
| ES2552338T3 (es) | 2005-01-21 | 2015-11-27 | Astex Therapeutics Limited | Compuestos farmacéuticos |
| BRPI0621777A2 (pt) | 2006-06-21 | 2013-03-12 | Piramal Life Sciences Ltd | derivados de flavona enantiomericamente puros para o tratamento de distérbios proliferativos e processos para sua preparaÇço |
| WO2008044045A1 (en) | 2006-10-12 | 2008-04-17 | Astex Therapeutics Limited | Pharmaceutical combinations |
| JP5528806B2 (ja) | 2006-10-12 | 2014-06-25 | アステックス、セラピューティックス、リミテッド | 複合薬剤 |
| TWI461194B (zh) | 2009-05-05 | 2014-11-21 | Piramal Entpr Ltd | 吡咯啶取代黃酮作為輻射致敏劑 |
| AU2010346973A1 (en) | 2010-02-26 | 2012-10-18 | Piramal Enterprises Limited | Pyrrolidine substituted flavones for the treatment of inflammatory disorders |
| EP3399026B1 (de) | 2010-06-14 | 2024-06-26 | The Scripps Research Institute | Neuprogrammierung von zellen für ein neues zellschicksal |
| TW201300105A (zh) | 2011-05-31 | 2013-01-01 | Piramal Life Sciences Ltd | 治療頭頸鱗狀細胞癌之相乘藥物組合物 |
| US9776989B2 (en) | 2013-04-10 | 2017-10-03 | Council Of Scientific & Industrial Research | Chromone alkaloid dysoline for the treatment of cancer and inflammatory disorders |
| CA2908084C (en) | 2013-04-17 | 2022-05-03 | Council Of Scientific And Industrial Research | Rohitukine analogs as cyclin-dependent kinase inhibitors and a process for the preparation thereof |
| EP3019166B1 (de) | 2013-07-12 | 2019-05-08 | Piramal Enterprises Limited | Pharmazeutische zusammensetzungen zur behandlung von melanomen |
| DK3218005T5 (da) | 2014-11-12 | 2024-10-14 | Seagen Inc | Glycan-interagerende forbindelser og anvendelsesfremgangsmåder |
| CN116217729A (zh) | 2015-11-12 | 2023-06-06 | 思进公司 | 聚糖相互作用化合物及使用方法 |
| WO2018094143A1 (en) | 2016-11-17 | 2018-05-24 | Siamab Therapeutics, Inc. | Glycan-interacting compounds and methods of use |
| JP6619519B2 (ja) | 2016-12-19 | 2019-12-11 | トレロ ファーマシューティカルズ, インコーポレイテッド | プロファイリングペプチドおよび感受性プロファイリングのための方法 |
| CA3054939A1 (en) | 2017-03-03 | 2018-09-07 | Seattle Genetics, Inc. | Glycan-interacting compounds and methods of use |
| US11793802B2 (en) | 2019-03-20 | 2023-10-24 | Sumitomo Pharma Oncology, Inc. | Treatment of acute myeloid leukemia (AML) with venetoclax failure |
| WO2020213714A1 (ja) * | 2019-04-18 | 2020-10-22 | 大日本住友製薬株式会社 | シス-(-)-フロシノピペリドールの製造方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1223690A (en) * | 1967-10-17 | 1971-03-03 | Fisons Pharmaceuticals Ltd | Substituted chromon-2-carboxylic acids |
| DE2731566A1 (de) * | 1977-07-13 | 1979-02-01 | Bayer Ag | Verfahren zur herstellung von neuen chromon-derivaten, sowie ihre verwendung als pflanzenschutzmittel |
| GB2101115A (en) * | 1980-10-23 | 1983-01-12 | Pfizer Ltd | Thromboxane synthetase inhibitors |
| DE3311005A1 (de) * | 1983-03-25 | 1984-09-27 | Bayer Ag, 5090 Leverkusen | Chromon- und thiochromonsubstituierte 1,4-dihydropyridinderivate, mehrere verfahren zu ihrer herstellung sowie ihre verwendung in arzneimitteln |
| DE3329186A1 (de) * | 1983-08-12 | 1985-02-21 | Hoechst Ag, 6230 Frankfurt | Chromonalkaloid, verfahren zu seiner isolierung aus dysoxylum binectariferum, und seine verwendung als arzneimittel |
| DE3445852A1 (de) * | 1984-12-15 | 1986-06-19 | Bayer Ag, 5090 Leverkusen | Dihydropyridin-carbonsaeureamide, verfahren zu ihrer herstellung und ihre verwendung in arzneimitteln |
-
1985
- 1985-10-31 IN IN300/BOM/85A patent/IN164232B/en unknown
-
1986
- 1986-04-11 DE DE19863612337 patent/DE3612337A1/de not_active Withdrawn
-
1987
- 1987-04-08 AT AT87105180T patent/ATE95519T1/de not_active IP Right Cessation
- 1987-04-08 DE DE87105180T patent/DE3787661D1/de not_active Expired - Lifetime
- 1987-04-08 ES ES87105180T patent/ES2060582T3/es not_active Expired - Lifetime
- 1987-04-08 EP EP87105180A patent/EP0241003B1/de not_active Expired - Lifetime
- 1987-04-09 IL IL8214987A patent/IL82149A/en not_active IP Right Cessation
- 1987-04-09 ZA ZA872555A patent/ZA872555B/xx unknown
- 1987-04-10 KR KR87003427A patent/KR950009861B1/ko not_active IP Right Cessation
- 1987-04-10 IE IE94187A patent/IE62244B1/en not_active IP Right Cessation
- 1987-04-10 AU AU71397/87A patent/AU602891B2/en not_active Expired
- 1987-04-10 CA CA000534430A patent/CA1332238C/en not_active Expired - Lifetime
- 1987-04-10 DK DK185287A patent/DK169760B1/da not_active IP Right Cessation
- 1987-04-10 PT PT84654A patent/PT84654B/pt unknown
- 1987-04-10 JP JP62087252A patent/JPH0686446B2/ja not_active Expired - Lifetime
- 1987-10-08 AT AT0260587A patent/AT389875B/de active
-
1989
- 1989-01-26 US US07/302,084 patent/US4900727A/en not_active Expired - Lifetime
-
1998
- 1998-06-11 HK HK98105203A patent/HK1006021A1/xx not_active IP Right Cessation
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6225473B1 (en) | 1998-01-23 | 2001-05-01 | Aventis Pharma Deutschland Gmbh | Method for producing (−)cis-3-hydroxy-1-methyl-4-(2,4,6-trimethoxyphenyl)-piperidine |
| US8119678B2 (en) | 2002-04-17 | 2012-02-21 | Cytokinetics, Incorporated | Compounds, compositions and methods |
| US8329928B2 (en) | 2002-04-17 | 2012-12-11 | Cytokinetics, Incorporated | Compounds, compositions and methods |
| US8633236B2 (en) | 2002-04-17 | 2014-01-21 | Cytokinetics, Inc. | Compounds, compositions and methods |
| US10624880B2 (en) | 2015-04-20 | 2020-04-21 | Tolero Pharmaceuticals, Inc. | Predicting response to alvocidib by mitochondrial profiling |
| US10562925B2 (en) | 2015-05-18 | 2020-02-18 | Tolero Pharmaceuticals, Inc. | Alvocidib prodrugs having increased bioavailability |
| US10568887B2 (en) | 2015-08-03 | 2020-02-25 | Tolero Pharmaceuticals, Inc. | Combination therapies for treatment of cancer |
| US10682356B2 (en) | 2015-08-03 | 2020-06-16 | Tolero Pharmaceuticals, Inc. | Combination therapies for treatment of cancer |
| US10835537B2 (en) | 2015-08-03 | 2020-11-17 | Sumitomo Dainippon Pharma Oncology, Inc. | Combination therapies for treatment of cancer |
| US11279694B2 (en) | 2016-11-18 | 2022-03-22 | Sumitomo Dainippon Pharma Oncology, Inc. | Alvocidib prodrugs and their use as protein kinase inhibitors |
| US11497756B2 (en) | 2017-09-12 | 2022-11-15 | Sumitomo Pharma Oncology, Inc. | Treatment regimen for cancers that are insensitive to BCL-2 inhibitors using the MCL-1 inhibitor alvocidib |
| US11034710B2 (en) | 2018-12-04 | 2021-06-15 | Sumitomo Dainippon Pharma Oncology, Inc. | CDK9 inhibitors and polymorphs thereof for use as agents for treatment of cancer |
| US11530231B2 (en) | 2018-12-04 | 2022-12-20 | Sumitomo Pharma Oncology, Inc. | CDK9 inhibitors and polymorphs thereof for use as agents for treatment of cancer |
| US12077554B2 (en) | 2018-12-04 | 2024-09-03 | Sumitomo Pharma Oncology, Inc. | CDK9 inhibitors and polymorphs thereof for use as agents for treatment of cancer |
Also Published As
| Publication number | Publication date |
|---|---|
| JPS62242680A (ja) | 1987-10-23 |
| KR870010045A (ko) | 1987-11-30 |
| AU7139787A (en) | 1987-10-15 |
| AT389875B (de) | 1990-02-12 |
| HK1006021A1 (en) | 1999-02-05 |
| CA1332238C (en) | 1994-10-04 |
| IL82149A (en) | 1994-01-25 |
| DE3612337A1 (de) | 1987-10-15 |
| ZA872555B (en) | 1987-11-25 |
| IE62244B1 (en) | 1995-01-11 |
| KR950009861B1 (en) | 1995-08-29 |
| AU602891B2 (en) | 1990-11-01 |
| ATE95519T1 (de) | 1993-10-15 |
| DK185287A (da) | 1987-10-12 |
| DK185287D0 (da) | 1987-04-10 |
| PT84654A (de) | 1987-05-01 |
| IN164232B (de) | 1989-02-04 |
| DK169760B1 (da) | 1995-02-20 |
| EP0241003A2 (de) | 1987-10-14 |
| EP0241003A3 (en) | 1988-10-12 |
| DE3787661D1 (de) | 1993-11-11 |
| PT84654B (pt) | 1989-12-29 |
| IL82149A0 (en) | 1987-10-30 |
| JPH0686446B2 (ja) | 1994-11-02 |
| US4900727A (en) | 1990-02-13 |
| ES2060582T3 (es) | 1994-12-01 |
| ATA260587A (de) | 1989-07-15 |
| IE870941L (en) | 1987-10-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0241003B1 (de) | 4H-1-Benzopyran-4-on-Derivate, ein Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| DE60030883T2 (de) | Verfahren zur herstellung von alfentanil, sufentanil und remifentanil | |
| DE69011628T2 (de) | 1,2-Benzisoxazolderivate, Verfahren zu deren Herstellung, und sie enthaltende pharmazeutische Zusammensetzungen. | |
| DE69308029T2 (de) | 1,2,3,5,6,7,8,8a-OCTAHYDRO-5,5,8a-TRIMETHYL-(8aBETA)-6-ISOCHINOLINAMINE-DERIVATE, IHRE HERSTELLUNG UND VERWENDUNG ALS THERAPEUTIKA | |
| DE2243961C2 (de) | ||
| CH624102A5 (de) | ||
| CH625787A5 (de) | ||
| DE2002840A1 (de) | 2,3,5-trisubstit,-2'-Hydroxy-6,7-benzomorphane und Verfahren zu deren Herstellung | |
| DE2603600A1 (de) | Alpha-aminomethyl-5-hydroxy-2- pyridinmethanol-derivate | |
| CH642353A5 (de) | 2-methyl-dihydropyridin-verbindung, verfahren zu ihrer herstellung und sie enthaltendes pharmazeutisches mittel. | |
| DD154364A5 (de) | Verfahren zur herstellung von neuen derivaten des piperidylbenzimidazolinons | |
| DD147537A5 (de) | Verfahren zur herstellung von 1,2,4,5-tetra-alkyl-4-aryl-piperidinen | |
| CH543509A (de) | Verfahren zur Herstellung von aromatischen Äthern | |
| DE1445836A1 (de) | Verfahren zur Herstellung von neuen Derivaten des 1,2,3,6-Tetrahydro-pyridins | |
| EP0219055B1 (de) | Verfahren zur Herstellung von Clausenamid | |
| EP0064685A1 (de) | Dibenzo(de,g)chinolin-Derivate, Verfahren zu deren Herstellung und diese enthaltende Arzneimittel | |
| CH624673A5 (de) | ||
| EP0000013B1 (de) | 4-Phenyl-8-amino-tetrahydroisochinoline, diese enthaltende pharmazeutische Präparate und Verfahren zur Herstellung dieser Präparate | |
| DE2354327C3 (de) | 2-Amino-l,23>4-tetrahydronaphthalin-Derivate, Verfahren zu deren Herstellung und Verfahren zur Herstellung von 6,7-Benzomorphanen ausgehend von jenen | |
| DE2747987C2 (de) | ||
| DD231073A1 (de) | Verfahren zur herstellung von neuen azabicyclo (3.3.1) nonan-derivaten | |
| DE60213603T2 (de) | Verfahren zur herstellung von 4 methylamino 4 phenylpiperidin | |
| DE2229770A1 (de) | N-(heteroarylmethyl)-7alpha-acyl6,14-endoaethenotetrahydro-nororipavine und -thebaine, deren hydrierungsprodukte und saeureadditionssalze sowie verfahren zu deren herstellung | |
| DE2510831A1 (de) | Isonipecotinsaeurederivate enthaltende herbizidzubereitungen | |
| DE60213604T2 (de) | Verfahren zur herstellung von 4-amino-4-phenylpiperidinen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE CH DE ES FR GB GR IT LI LU NL SE |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AT BE CH DE ES FR GB GR IT LI LU NL SE |
|
| 17P | Request for examination filed |
Effective date: 19890325 |
|
| 17Q | First examination report despatched |
Effective date: 19890809 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE ES FR GB GR IT LI LU NL SE |
|
| REF | Corresponds to: |
Ref document number: 95519 Country of ref document: AT Date of ref document: 19931015 Kind code of ref document: T |
|
| REF | Corresponds to: |
Ref document number: 3787661 Country of ref document: DE Date of ref document: 19931111 |
|
| ITF | It: translation for a ep patent filed | ||
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 19940106 |
|
| ET | Fr: translation filed | ||
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: FG4A Free format text: 3010231 |
|
| EPTA | Lu: last paid annual fee | ||
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2060582 Country of ref document: ES Kind code of ref document: T3 |
|
| EAL | Se: european patent in force in sweden |
Ref document number: 87105180.1 |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: IF02 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20060208 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20060215 Year of fee payment: 20 Ref country code: AT Payment date: 20060215 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 20060222 Year of fee payment: 20 Ref country code: GB Payment date: 20060222 Year of fee payment: 20 Ref country code: CH Payment date: 20060222 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20060308 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GR Payment date: 20060317 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 20060403 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 20060407 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20060430 Year of fee payment: 20 Ref country code: IT Payment date: 20060430 Year of fee payment: 20 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20070408 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20070409 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: PE20 |
|
| NLV7 | Nl: ceased due to reaching the maximum lifetime of a patent |
Effective date: 20070408 |
|
| EUG | Se: european patent has lapsed | ||
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20070409 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20070407 |
|
| BE20 | Be: patent expired |
Owner name: *HOECHST A.G. Effective date: 20070408 |