EP0000128A1 - Sulfamoyl-Arylketone und Verfahren zu ihrer Herstellung sowie ihre Verwendung als Arzneimittel - Google Patents
Sulfamoyl-Arylketone und Verfahren zu ihrer Herstellung sowie ihre Verwendung als Arzneimittel Download PDFInfo
- Publication number
- EP0000128A1 EP0000128A1 EP78100118A EP78100118A EP0000128A1 EP 0000128 A1 EP0000128 A1 EP 0000128A1 EP 78100118 A EP78100118 A EP 78100118A EP 78100118 A EP78100118 A EP 78100118A EP 0000128 A1 EP0000128 A1 EP 0000128A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- chloro
- sulfamoylbenzoyl
- compounds
- furan
- benzo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/79—Benzo [b] furans; Hydrogenated benzo [b] furans with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
- C07D307/80—Radicals substituted by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/12—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/54—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
- C07D333/56—Radicals substituted by oxygen atoms
Definitions
- the invention relates to compounds of general formula I. in which R 1 is hydrogen, halogen, alkyl having 1 to 3 C atoms, methoxy or ethoxy, R 2 and R 3 can be identical or different and is hydrogen or alkyl having 1 to 4 C atoms, X is a halogen atom, methyl or trifluoromethyl and Y is oxygen, sulfur or NR 4 , where R 4 is hydrogen or alkyl having 1 to 4 carbon atoms.
- the alkyl radicals for R 1 to R 4 can be straight-chain or branched.
- halogen-substituted benzene derivatives such as fluoro-, difluoro-, chloro- or dichlorobenzene, among others, have proven to be particularly suitable.
- Aluminum chloride, tin and titanium tetrachloride are preferably used as Friedel-Krafts catalysts, which include both Lewis acids and protonic acids, although other catalysts, such as HF, BF 3 , ZnCl 2 , GaCl 3 , J 2 , are also used can.
- HF, HClO 4 or polyphosphoric acid are suitable as protonic acids.
- A is hydroxy, aluminum chloride, zinc chloride, boron trifluoride, but also hydrogen fluoride and perchloric acid, and also polyphosphoric acid or phosphorus oxychloride are preferably used as catalysts, while POCl 3 is used as a catalyzing Lewis acid, especially when reacting III with the acid amide derivatives of the formula II , both-.
- NEN Y stands for -NR 2 , advantageously used.
- Polar solvents such as water, lower alcohols with 1 to 5 carbon atoms, dioxane, tetrahydrofuran, dimethylacetamide, mono-, di- or triethylene glycol dimethyl ether have proven to be particularly suitable as reaction media, the reaction being between 0 and 100 ° C. , preferably between 10 and 50 ° C.
- the reaction time is between 1/2 and 70 hours, preferably 4 to 14 hours.
- the sulfochlorides of the formula IV can be obtained in various ways in a manner known per se. They are preferably obtained from the amino derivatives of the formula X by the Meerwein reaction (Chem. Ber. 90, 841 (1957)) wherein the substituents have the meaning given.
- the compounds of formula X are, for example, compounds XI in which X and A have the meaning given, by a reaction analogous to procedure a) with compounds III and subsequent reduction of the nitro compound XII shown.
- compounds of the general formula VI are converted into the compounds of the formula I using an oxidizing agent.
- organic and inorganic oxidizing agents such as salts and complex compounds of Fe +3 , nickel peroxide, potassium permanganate, chromium-VI compounds, copper-II salts, halogen, nitrogen oxides such as N 2 0 3 in situ or N0 2 , ENT are suitable 3 , oxygen, inorganic and organic peroxo compounds such as H 2 0 2 , Behzoper- and m-chlorobenzoperic acid, N-chloro- and N-bromosuccinimide, dimethyl sulfoxide, aliphatic nitro compounds, ketones in the presence of an aluminum alcoholate in the sinus of an Oppenauer oxidation.
- active manganese IV oxide has proven to be a mild and particularly suitable oxidizing agent (cf., for example, AJ Fatiadi, Synthesis 1976 , 65; DE-OS 2 436 263), the solvent used preferably being acetonitrile or halogenated hydrocarbons, such as methylene chloride, chloroform, tetrachloroethane, and the reaction at temperatures between 0 ° and 40 ° C., preferably between 20 ° and 30 ° C. , over a period of 6 to 60 hours.
- oxidizing agent cf., for example, AJ Fatiadi, Synthesis 1976 , 65; DE-OS 2 436 263
- the solvent used preferably being acetonitrile or halogenated hydrocarbons, such as methylene chloride, chloroform, tetrachloroethane, and the reaction at temperatures between 0 ° and 40 ° C., preferably between 20 ° and 30 ° C. , over a
- ketimines of the formula VII which can also be present in the form of their acid addition salts, are hydrolyzed.
- the nitriles of the formula XIV are brought with the compounds of formula III in the sense of a Houben-Hoesch reaction for the reaction, cf. Organic Reactions 5, 387 (1949).
- the two reactants are preferably reacted in a molar ratio of 1: 1 in an inert polar and, as far as possible, water-free organic solvent, such as diethyl ether, diisopropyl ether, tetrahydrofuran, glacial acetic acid, dioxane, a halogenobenzene, preferably chlorobenzene, advantageously using mono- , Dier triethylene glycol dimethyl or diethyl ether as a solvent.
- the reaction mixture is passed over a period of 2 to 20 hours a dry stream of HCI gas until saturation, at temperatures between -30 ° and + 40 ° C, advantageously between -5 ° and + 15 ° C. Subsequently, the mixture is advantageously left at -5 ° to + for 1 to 3 days Stand at 15 ° C. It is also possible to work in the presence of an additional Lewis acid, such as, in particular, anhydrous zinc or aluminum chloride.
- ketimine hydrochlorides of the formula VII are obtained by adding a less polar solvent by precipitation, in particular by means of diisopropyl ether, ether, but also lower alkyl acetate, acetone and mixtures of the stated solvents with petroleum ether or cyclohexane.
- the hydrolysis of the ketimine hydrochloride can be carried out either in an alkaline or acidic medium, the compounds VII being heated in water or ethanol / water mixtures, if appropriate in the presence of small amounts of ammonia, sodium hydroxide solution, calcium carbonate, dilute hydrochloric acid or sulfuric acid, and the resulting mixture Filtered ketone or isolated after extraction with an organic solvent, preferably with ethyl acetate.
- the ketimines of the formula VII can also be obtained by reacting compounds of the formula VIII with the nitriles of the formula XIV by the method of Blaise, cf. Houben-Weyl, 4th edition, volume 7/2 a, page 603 (1973) become.
- the solvents used are the inert and anhydrous solvents customary for organometallic reactions, preferably ethers such as diethyl ether, dibutyl ether, but particularly advantageously tetrahydrofuran or mono-, di-, tri- ethylene glycol dimethyl or diethyl ether.
- Inert aromatic hydrocarbons such as toluene or xylene can be used.
- the process is preferably carried out between 30 ° and 130 ° C., between 3 and 50 hours; the reaction mixture is usually worked up by stirring with water for 10 to 24 hours.
- the imines obtained are converted into the compounds of formula I by hydrolysis in an acidic or basic medium.
- reaction is carried out in an inert and anhydrous solvent customary for organometallic reactions, preferably in ether, tetrahydrofuran, dioxane or in a mono-, di- or triethylene glycol dimethyl or -diethyl ether, preferably at temperatures between -0100 ° and + 100 ° C.
- the reaction products are hydrolyzed in a customary manner, for example by introducing the reaction mixture into an aqueous saturated ammonium chloride solution at temperatures between -5 ° and + 20 ° C. while maintaining a pH range from 6 to 11.
- the starting materials of the formula IX are obtained by reacting a halogen ketone of the formula XV where Hal is preferably bromine or iodine, with compounds of the formula XVI known from the literature wherein Z is preferably S or O.
- the process is carried out using the bases described in the cyclocondensation in the solvents mentioned there, advantageously under milder temperature conditions between -10 ° and + 60 ° C, but preferably between + 10 ° and + 40 ° C.
- the preparation of IX and whose cyclocondensation to I can proceed without isolation of the compounds IX in a one-pot reaction.
- R 3 denotes lower alkyl
- R 3 -X conventional alkylating agents of the formula R 3 -X are used, in which X is, for example, bromine, iodine, chlorine, -O-SO 2 CH 3 , -O-SO 2 OR 3 or stands..
- the alkylation is carried out in water, but preferably in polar organic solvents such as a lower alcohol having 1 to 4 carbon atoms, in dioxane, tetrahydrofuran, dimethylformamide, dimethylacetamide or a mono-, di-, triethylene glycol mono- or dimethyl- or -ethyl ether at temperatures. between -20 ° and + 50 ° C, preferably between + 15 ° and + 35 ° C, being left to regress for a period of 5 to 72 hours.
- a base for Acid binding is preferably carried out using carbonates, alcoholates or hydroxides of sodium or potassium.
- the most important compounds according to the invention are those of the general formula I in which the substituent X is bromine or chlorine, preferably chlorine, R 3 is hydrogen, methyl or ethyl, preferably hydrogen, R 2 is hydrogen, methyl or ethyl, R1 is hydrogen , Chlorine, methyl or methoxy in position 4 or 5 of the heterocycle, but is preferably hydrogen and Y is oxygen or sulfur.
- the process products are valuable medicinal products and cause a very good effect, which lowers the uric acid level of the blood, which is caused in particular by a uricosuric effect.
- the compounds according to the invention are distinguished by a desired good diuretic and saluretic activity, and are therefore superior to the previously known uricosuric agents.
- the uricosuric effect of the new process products was determined on the oxonate-treated rat in a unit dose of 50 mg / kg per os. They show the antiuricopathic efficacy of known commercial preparations of the probencid type and the benzbromarone type.
- the salidiuretic activity of the compounds according to the invention was determined on the rat in a unit dose of 50 mg / kg per os. You will achieve the salidiuretic activity of well-known commercial products like that of chlorothalidone.
- the new process products are characterized by a long-lasting effect, which means that the preparations are also suitable for the treatment of hypertensive conditions in humans. You can combine them with an antihypertensive.
- Tablets, dragees, capsules, suppositories and ampoules for parenteral administration are particularly suitable as therapeutic preparations for the new compounds.
- the therapeutic unit dose is between 5 and 1000 mg, preferably 10 to 500 mg per tablet.
- these preparations can also contain an antihypertensive, such as, for example, reserpine, hydralazine, guanethidine, ⁇ -methyldopa, a ⁇ -sympathicolytic or chloridine.
- an antihypertensive such as, for example, reserpine, hydralazine, guanethidine, ⁇ -methyldopa, a ⁇ -sympathicolytic or chloridine.
- potassium-retaining compounds such as aldosterone antagonists, for example spironolactone, or pseudoaldosterol antagonists, such as triamterene or amiloride
- aldosterone antagonists for example spironolactone
- pseudoaldosterol antagonists such as triamterene or amiloride
- K + substitution can also be used in various forms of application, for example coated tablets, tablets, effervescent tablets, juices and others.
- Combinations of the compounds according to the invention with another antihyperuricemic active agent can also be of therapeutic interest, which agent leads to an increase in the antiuricopathic effects, particularly by inhibiting xanthine oxidase.
- Any desired desired enhancement of the salidiuretic activity can be achieved by combining the compounds according to the invention with a salidiuretic.
- Example 10 a is obtained analogously to the procedure given in Example 10 a) from 2.68 g of 4-chloro-3-methylsulfamoylbenzoyl chloride and 1.3 g of 2-methylindole in the presence of 2.7 g of aluminum chloride in dichloroethane. Colorless crystals, mp 246 ° C.
- Example 10 a is obtained analogously to the procedure given in Example 10 a) from 10 g of 4-chloro-3-sulfamoylbenzoyl chloride and 7.8 g of 2,5-dimethylindole in dichloroethane in the presence of 12 g of aluminum chloride. Mp 248 ° - 250 ° C.
- Example 10 a is obtained analogously to the procedure given in Example 10 a) from 10 g of 4-chloro-3-sulfamoylbenzoyl chloride and 6.53 g of 1,2-dimethylindole in dichloroethane in the presence of 12 g of aluminum chloride. After treatment with isopropanol, crystals with a melting point of 247 ° -249 ° C. are obtained.
- Example 2 is obtained analogously to the procedure given in Example 1 from 2.5 g of 4-chloro-3-sulfamoylbenzoyl chloride, 1.6 g of 3-methyl-benzo [b] thiophene and 3 g of aluminum chloride in 50 ml of chlorobenzene. Colorless crystals, mp 210 ° C.
- Example 10 a is obtained analogously to the procedure given in Example 10 a) from 10.0 g of 4-chloro-3-sulfamoylbenzoyl chloride, 5.1 g of 3-methylindole in the presence of 10.4 g of aluminum chloride. Colorless crystals of isopropanol, mp. 205 ° C.
- Example 10 a is obtained analogously to the procedure given in Example 10 a) from 4-chloro-3-sulfamoylbenzoyl chloride and 2-methylbenzo [b] thiophene in dichloroethane in the presence of aluminum chloride.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Furan Compounds (AREA)
- Indole Compounds (AREA)
Abstract
Heterocyclische 3-Sulfamoylarylketone, der allgemeinen Formel <IMAGE> worin R¹ Wasserstoff, Halogen, Alkyl mit 1 bis 3 C-Atomen, Methoxy oder Aethoxy, R² und R³ gleich oder verschieden, Wasserstoff oder Alkyl mit 1 bis 4 C-Atomen, X ein Halogenatom, Methyl oder Trifluormethyl und Y Sauerstoff, Schwefel oder NR<4> bedeuten, wobei R<4> für Wasserstoff oder Alkyl mit 1 bis 4 C-Atomen steht, mit urikosurischer, hypourikämischer und salidiuretischer Wirkung und ein Verfahren zur Herstellung dieser Verbindungen sowie pharmazeutische Präparate, die diese Verbindungen enthalten.
Description
- Gegenstand der Erfindung sind Verbindungen der allgemeinen Formel I

- Gegenstand der Erfindung ist weiterhin ein Verfahren zur Herstellung von Verbindungen der Formel I, das dadurch gekennzeichnet ist, dass man
- a) Verbindungen der allgemeinen Formel II

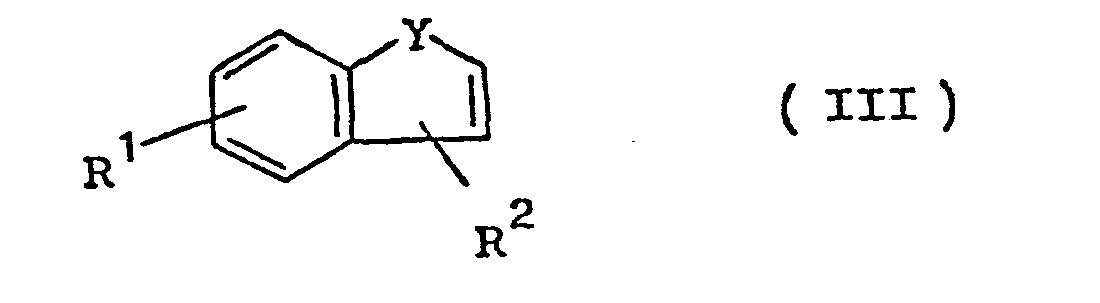
- b) Verbindungen der allgemeinen Formel IV
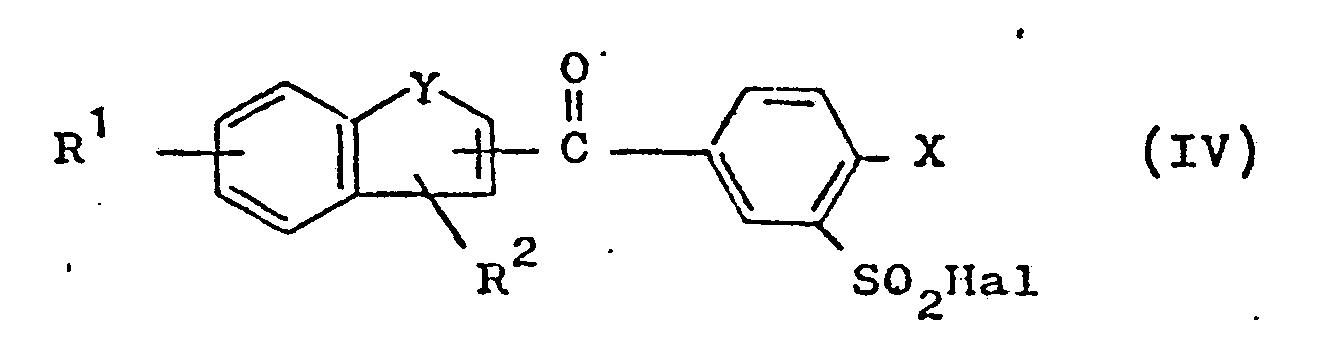
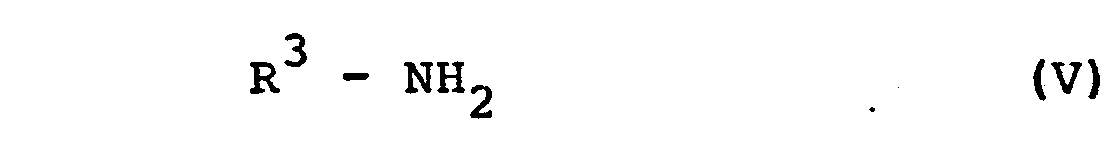
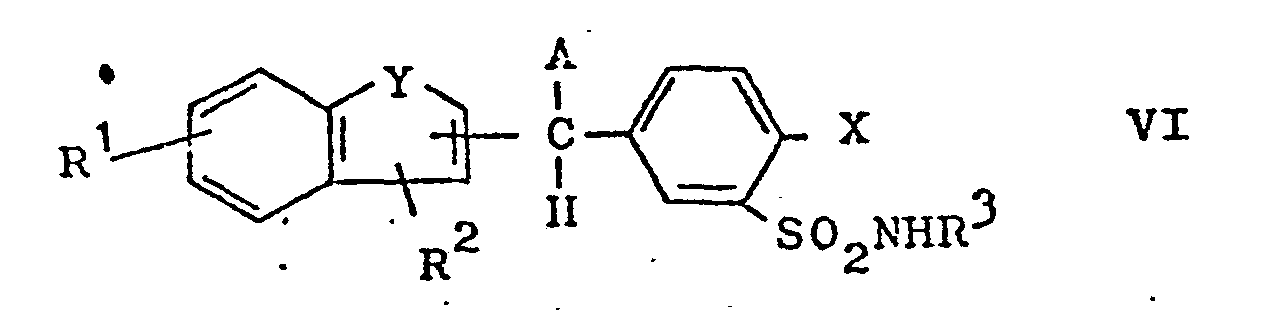
- c) Verbindungen der allgemeinen Formel VI
- d) Verbindungen der allgemeinen Formel VII
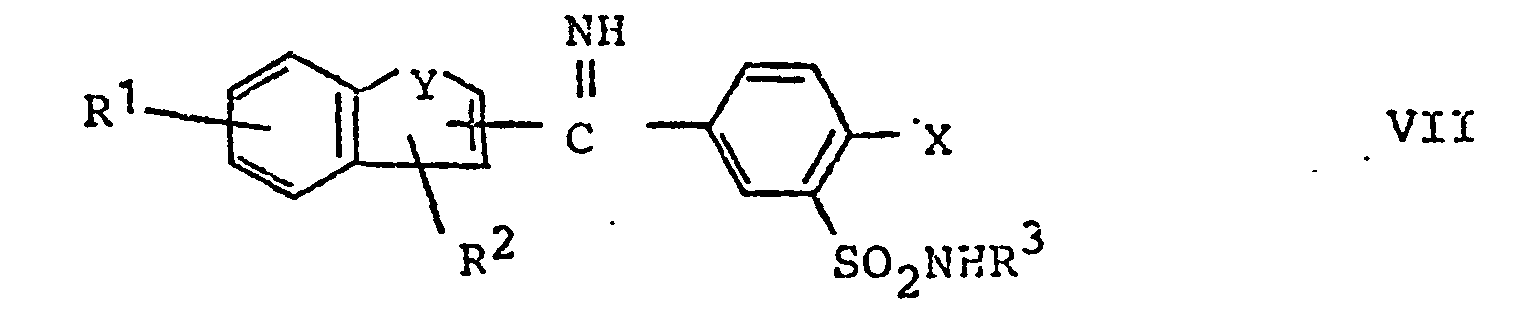
- e) Verbindungen der allgemeinen Formel VIII

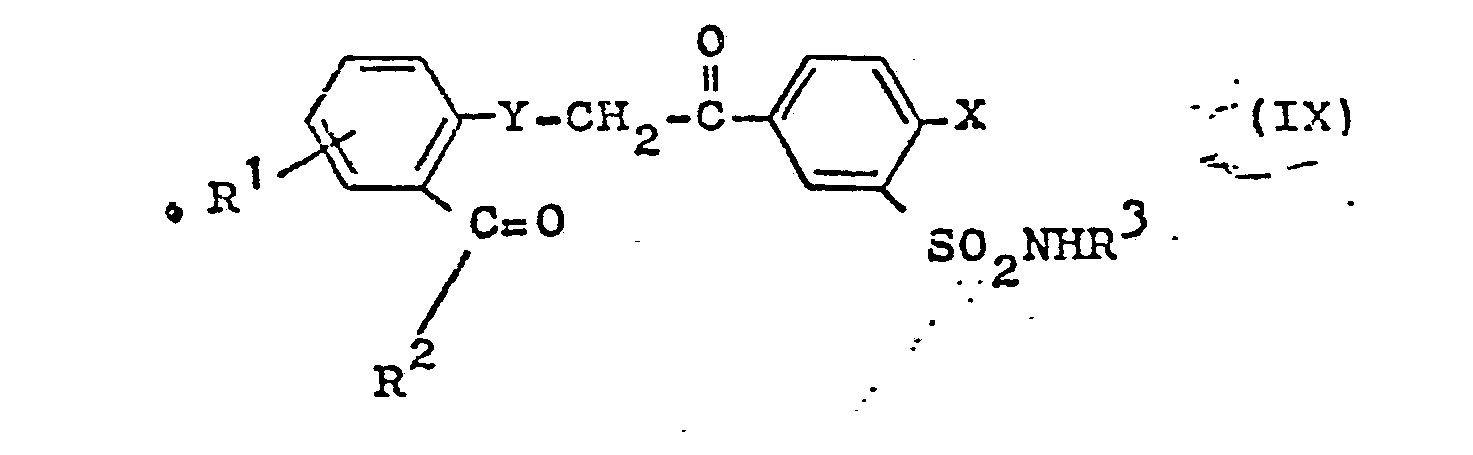
- f) Verbindungen der allgemeinen Formel IX

und gegebenenfalls die nach a) - f) erhaltenen Verbindungen der Formel I, in der R3 für Wasserstoff steht, anschliessend alkyliert. - Die Alkylreste für R 1 bis R4 können geradkettig oder verzweigt sein.
- Die unter a) bezeichnete Verfahrensweise wird so ausgeführt, dass man Verbindungen II mit den Verbindungen III bevorzugt im molaren Verhältnis 1 : 1 umsetzt, wobei die Reaktionsdurchführung hinsichtlich Katalysator, Reaktionstemperatur, Reaktionsdauer, Lösungsmittel und Aufarbeitung gemäss der Vorschrift für vergleichbare Beispiele z.B. nach Houben-Weyl, "Methoden der Organischen Chemie", 4. Auflage, Band 7/2 a, Seiten 15 - 375 (1973), erfolgt.
- Wenn A für Hal oder Acyloxy steht, haben sich unter den für Friedel-Crafts-Reaktionen verwendeten Lösungsmitteln neben den in der Literatur beschriebenen üblichen Lösungsmitteln u.a. halogensubstituierte Benzolderivate, wie z.B. Fluor-, Difluor-, Chlor- oder Dichlorbenzol als besonders geeignet erwiesen. Als Friedel-Krafts-Katalysatoren, die sowohl Lewis-Säuren wie Protonensäuren umfassen, kommen bevorzugt Aluminiumchlorid, Zinn- und Titantetrachlorid zur Anwendung, wobei jedoch auch andere Katalysatoren, wie beispielsweise HF, BF3, ZnCl2, GaCl3, J2 verwendet werden können. Als Protonensäuren kommen z.B. HF, HClO4 oder Polyphosphorsäure in Betracht. Insbesondere bei Anwendung von Aluminiumchlorid wie auch bei den anderen Katalysatoren muss berücksichtigt werden, dass die Sulfamoylfunktion mindestens 1 Mol AlCl3 bzw. Lewis-Säure infolge Komplexbildung desaktiviert, also ein überschuss von mindestens 2 Mol an Lewis-Säure eingesetzt wird.
- Wenn A für Hydroxy steht, verwendet man bevorzugt Aluminiumchlorid, Zinkchlorid, Bortrifluorid, aber auch Fluorwasserstoff und Perchlorsäure, sowie Polyphosphorsäure oder Phosphoroxychlorid als Katalysatoren, dagegen wird POCl3 als katalysierende Lewis-Säure insbesondere bei Umsetzungen von III mit den Säureamid-Derivaten der Formel II, bei de-. nen Y für -NR2 steht, vorteilhaft verwendet.
- Nach Verfahrensweise b) bringt man Sulfochloride der Formel IV mit Ammoniak oder einem Amin in an sich bekannter Weise zur Reaktion, vergl. Houben-Weyl, "Methoden der Organischen Chemie", 4. Auflage, Band 9 (1955), Seiten 605-627.
- Als Reaktionsmedien erwiesen sich polare Lösungsmittel,, wie Wasser, niedere Alkohole mit 1 bis 5 C-Atomen, Dioxan, Tetrahydrofuran, Dimethylacetamid, Mono-, Di- oder Tri- äthylenglykoldimethyläther als besonders geeignet, wobei man die Reaktion zwischen 0 und 100° C, bevorzugt zwischen 10 und 50° C durchführt. Die Reaktionsdauer liegt zwischen 1/2 und 70 Stunden, bevorzugt bei 4 bis 14 Stunden.
- Die Sulfochloride der Formel IV können in an sich bekannter Weise auf verschiedenen Wegen erhalten werden. Bevorzugt erhält man sie durch Meerwein-Reaktion (Chem. Ber. 90, 841 (1957)) aus den Aminoderivaten der Formel X

- Die Verbindungen der Formel X werden beispielsweise aus Verbindungen XI


- Gemäss Verfahrensweise c) werden Verbindungen der allgemeinen Formel VI mit einem Oxidationsmittel in die Verbindungen der Formel I übergeführt. Es eignen sich sowohl organische wie auch anorganische Oxidationsmittel wie beispielsweise Salze und Komplexverbindungen des Fe+3, Nickelperoxid, Kaliumpermanganat, Chrom-VI-verbindungen, Kupfer-II-salze, Halogen, Stickoxide wie N203 in situ oder N02, HNO3, Sauerstoff, anorganische und organische Peroxoverbindungen wie H202, Behzoper- und m-Chlorbenzopersäure, N-Chlor- und N-Brom-succinimid, Dimethylsulfoxid, aliphatische Nitroverbindungen, Ketone in Gegenwart eines Aluminiumalkoholates im Sinine einer Oppenauer-Oxidation. Hierbei hält man sich in Durchführung und Aufarbeitung an vergleichbare,in der Literatur erwähnte Beispiele, z.B. Houben-Weyl, "Methoden der Organischen Chemie", 4. Auflage, Band 4/1 b, (1975), Seiten 425, 465, 67.3, 901, und Band 7/2 a, (1973), Seiten 677 - 788. Wenn A eine OH-Gruppe bedeutet, hat sich als mildes und besonders geeignetes Oxidationsmittel aktives Mangan-IV-oxid erwiesen (vergl. z.B. A. J. Fatiadi, Synthesis 1976, 65; DE-OS 2 436 263), wobei man als Lösungsmittel vorzugsweise Acetonitril oder halogenierte Kohlenwasserstoffe, wie z.B. Methylenchlorid, Chloroform, Tetrachloräthan verwendet und die Reaktion bei Temperaturen zwischen 0° und 40° C, vorzugsweise zwischen 20° und 30° C, über eine Dauer von 6 bis 60 Stunden durchführt.
- Zu den Verbindungen VI mit A = OH gelangt man in üblicher Weise z.B. durch Addition eines Aldehyds der allgemeinen Formel XIII

- Bei der Cxiäation von Verbindungen VI, worin A Acyloxy bedeutet, wendet man bevorzugt Chromsäure an, vergl. Org. Synth. 42, 79 (1962). Wenn A für Halogen steht, verwendet man neben Chromsäure bzw. Natriumbichromat i.m sauren Medium bevorzugt Dimethylsulfoxid, die Oxide tertiärer Amine wie z.B. Pyridin-N-oxid oder Trimethylaminoxid und aliphatische Nitroverbindungen wie z.B. 2-Nitropropan. Die Verbindungen VI, worin A für N(R)2 steht, werden bevorzugt in die Verbindungen I übergeführt, vergl. Org. Prep. Proced. Int. 8, 33 (1976); J. Am. Chem. Soc. 97, 5927 (1975).
- Man erhält die Verbindungen VI, worin A Halogen oder Acyloxy bedeutet, z.B. aus den entsprechenden Verbindungen mit A = OH durch Acylierung oder Chlorierung, wenn A eine N(R)2-Gruppe bedeutet, beispielsweise aus den entsprechenden Verbindungen mit A = Halogen durch Umsetzung mit Aminen in üblicher Weise.
- Gemäss Verfahrensweise d) werden Ketimine der Formel VII, die auch in Form ihrer Säureadditionssalze vorliegen können, hydrolysiert. Zur Herstellung der Verbindungen der Formel VII bringt man die Nitrile der Formel XIV

- Um Verunreinigungen abzutrennen, empfiehlt es sich, die intermediär entstehenden Ketimin-hydrochloride der Formel VII zu isolieren, obwohl prinzipiell auch eine Hydrolyse ohne weitere Isolierungs- und Reinigungsoperationen möglich ist. Die Ketimin-hydrochloride werden durch Zusatz eines weniger polaren Lösungsmittels durch Ausfällung, erhalten, insbesondere durch Diisopropyläther, Äther, aber auch Essigsäureniederalkylester, Aceton sowie Gemische der angegebenen Lösungsmittel mit Petroläther oder Cyclohexan.
- Die Hydrolyse des Ketimin-hydrochlorides kann sowohl im alkalischen wie sauren Medium durchgeführt werden, wobei man die Verbindungen VII in Wasser oder Äthanol-Wasser-Gemischen gegebenenfalls in Gegenwart geringer Mengen an Ammoniak, Natronlauge, Calciumcarbonat, verdünnter Salzsäure oder Schwefelsäure erhitzt und das sich bildende Keton abfiltriert oder nach Extraktion mit einem organischen Lösungsmittel, bevorzugt mit Essigester, isoliert.
- Die Ketimine der Formel VII können auch durch Umsetzung von Verbindungen der Formel VIII mit den Nitrilen der Formel XIV nach der Methode von Blaise, vergl. Houben-Weyl, 4. Aufl., Band 7/2 a, Seite 603 (1973), erhalten werden.
- Als Lösungsmittel verwendet man die für metallorganische Reaktionen üblichen inerten und wasserfreien Lösungsmittel, bevorzugt Äther wie Diäthyläther, Dibutyläther, besonders vorteilhaft aber Tetrahydrofuran oder Mono-, Di-, Tri- äthylenglykoldimethyl- oder -diäthyläther. Aucr inerte aromatische Kohlenwasserstoffe wie Toluol oder Xylol können verwendet werden. Pro Mol XIV müssen, wenn R3 nicht Wasserstoff bedeutet, mindestens 2, bei R3 = H mindestens 3 Mol der Verbindungen VIII eingesetzt werden. Man arbeitet bevorzugt zwischen 30° und 130° C, zwischen 3 und 50 Stunden, meist wird das Reaktionsgemisch nach 10 bis 24stündigem Rühren durch Zersetzung mit Wasser aufgearbeitet. Die dabei erhaltenen Imine werden im sauren oder basischen Milieu in die Verbindungen der Formel I durch Hydrolyse überführt.
- Gemäss Verfahrensweise e) bringt man Verbindungen der allgemeinen Formel II mit metallorganischen Verbindungen VIII zur Reaktion, wobei insbesondere die entsprechende Lithium-und Magnesium-organischen Verbindungen eingesetzt werden. Bei dieser Verfahrensweise kann A in den Verbindungen der Formel II auch den Mercaptopyridylrest bedeuten, vergl. Bull. Chem. Soc. Japan 47, 1777 (1974).
- Dabei verwendet man pro Mol der Verbindungen II etwa 2 bis 2,5 Mole der Verbindung VIII, wenn R3 nicht für Wasserstoff steht, und etwa 3 bis 3,5 Mole VIII, wenn R3 Wasserstoff bedeutet. Die Umsetzung wird in einem für metallorganische Reaktionen üblichen inerten und wasserfreien Lösungsmittel, vorzugsweise in Äther, Tetrahydrofuran, Dioxan oder in einem Mono-, Di- oder Triäthylenglykoldimethyl- oder -diäthyläther durchgeführt, bevorzugt bei Temperaturen zwischen -0100° und +100° C. Nach Beendigung der Umsetzung werden die Reaktionsprodukte in üblicher Weise hydrolysiert, indem man beispielsweise das Reaktionsgemisch bei Temperaturen zwischen -5° und +20° C unter Aufrechterhaltung eines pH-Bereiches von 6 bis 11 in eine wässrige gesättigte Ammoniumchlorid-Lösung einträgt.
- Die in Verfahrensweise e) bevorzugt verwendeten Verbindungen der Formel VIII mit M = Li oder Mg Hal gewinnt man dadurch, dass man auf Halogenverbindungen der Formel XIV

- Gemäss Verfahrensweise f) werden Verbindungen der allgemeinen Formel IX unter üblichen Bedingungen cyclisiert (siehe z.B. Advances Het. Chem., Band 18, Seite 338 (1975), - oder Band 11, Seite 178 (1970). Die Cyclokondensation kann sowohl sauer wie alkalisch katalysiert werden; letzteres ist bevorzugt. Man arbeitet vorteilhaft mit den Alkalisalzen schwacher Säuren, beispielsweise mit dem Salz einer schwachen organischen Säure wie Natrium- oderKaliumacetat in Eisessig als geeignetem Lösungsmittel, oder mit Natrium- bzw. Kaliumcarbonat, Natrium- oder Kaliumalkoholaten wie Äthylat oder Methylat sowie mit Metallhydroxiden z.B. NaOH oder KOH in einem geeigneten polaren organischen Lösungsmittel, wie Aceton, Methyläthylketon, Dimethylformamid, Dimethylacetamid oder niedere Alkohole mit 1 bis 4 C-Atomen oder Gemischen der angegebenen Lösungsmittel. Man arbeitet über 1/2 bis 12 Stunden bei Temperaturen zwischen 0° und 140° C, bevorzugt 50° bis 100° C. Bei der anschliessenden Behandlung des Reaktionsproduktes mit Wasser soll möglichst ein pH-Wert unter 9 eingestellt werden, um Salzbildung der Sulfamoylfunktion zu vermeiden.
- Die Ausgangsstoffe der Formel IX erhält man durch Umsetzung eines Halogenketons der Formel XV


- Die erfindungsgemässen Verbindungen der Formel I, beidenen R 3 niederes Alkyl bedeutet, können auch durch Alkylierung der unsubstituierten Sulfamoylgruppe in üblicher Weise erhalten werden. Für die Alkylierungsreaktion verwendet man übliche Alkylierungsmittel der Formel R3-X, worin X beispielsweise für Brom, Jod, Chlor, -O-SO2CH3, -O-SO2OR3 oder

- Bei der Alkylierung arbeitet man in Wasser, vorzugsweise jedoch in polaren organischen Lösungsmitteln wie einem niederen Alkohol mit 1 bis 4 C-Atomen, in Dioxan, Tetrahydrofuran, Dimethylformamid, Dimethylacetamid oder einem Mono-, Di-, Triäthylenglykolmono- oder -dimethyl- bzw. -äthyläther bei Temperaturen.zwischen -20° und +50° C, vorzugsweise zwischen +15° und +35° C, wobei man über einen Zeitraum von 5 bis 72 Stunden reägieren lässt. Als Base zur Säurebindung verwendet man vorzugsweise Karbonate, Alkoholate oder Hydroxide des Natriums oder Kaliums.
- Die wichtigsten erfindungsgemässen Verbindungen sinddiejenigen der allgemeinen Formel I, in denen der SubstituentX für Brom oder Chlor, bevorzugt für Chlor, steht, R3 Wasserstoff, Methyl oder Äthyl, bevorzugt Wasserstoff, bedeutet, R2 für Wasserstoff, Methyl oder Äthyl steht, R1 Wasserstoff, Chlor, Methyl oder Methoxy in Position 4 oder 5 des Heterocyclus, bevorzugt aber Wasserstoff bedeutet und Y für Sauerstoff oder Schwefel steht.
- Erfindungsgemäss können ausser den in den Ausführungsbeispielen beschriebenen Substanzen auch die nachfolgend aufgeführten Verbindungen der allgemeinen Formel I dargestellt werden:
- 1. 3-(4-Chlor-3-sulfamoylbenzoyl)-benzo[b]furan.
- 2. 3-(4-Brom-3-sulfamoylbenzoyl)-benzo[b]furan.
- 3. 3-(4-Chlor-3-methylsulfamoylbenzoyl)-benzo[b]furan.
- 4. 3-(4-Chlor-3-methylsulfamoylbenzoyl)-2-methyl- benzo[b]furan.
- 5. 3-(4-Brom-3-sulfamoylbenzoyl)-2-methylbenzo[b]furan.
- 6. 2-Methyl-3-(3-sulfamoyl-4-trifluormethylbenzoyl)-benzo[b]furan.
- 7. 2-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl-5-methoxy- benzo[b]furan.
- 8. 2-Äthyl-3-(3-sulfamoyl-4-trifluormethylbenzoyl)-benzo[b]furan.
- 9. 3-(4-Chlor-3-sulfamoylbenzoyl)-2-propylbenzo[b]furan.
- 10.. 3-(4-Chlor-3-sulfamoylbenzoyl)-2-isopropyl- benzo[b]furan.
- 11. 2-Butyl-3-(4-chlor-3-sulfamoylberizoyl)-benzo[b]furan.
- 12. 2-(4-Chlor-3-sulfamoylbenzoyl)-3-raethylbenzo[b]furan.
- 13. 2-(4-Brom-3-sulfamoylbenzoyl)-benzo[b]furan.
- 14. 2-(3-Sulfamoyl-4-trifluormethylbenzoyl)-benzo[b]furan.
- 15. 3-Äthyl-2-(4-chlor-3-sulfamoylbenzoyl)-benzo[b]furan.
- 16. 2-(4-Chlor-3-methylsulfamoylbenzoyl)-3-methyl- benzo[b]thiophen.
- 17. 2-(4-Chlor-3-propylsulfamoylbenzoyl)-3-methyl- benzo[b]thiophen.
- 18. 2-(4-Chlor-3-isopropylsulfamoylbenzoyl)-3-methyl- benzo[b]thiophen.
- 19. 2-(3-Butylsulfamoyl-4-chlorbenzoyl)-3-methyl- benzo[b]thiophen.
- 20. 3-Äthyl-2-(4-chlor-3-sulfamoylbenzoyl)-benzo[b]thiophen.
- 21. 3-Äthyl-2-(4-chlor-3-sulfamoylbenzoyl)-5-methoxy- benzo[b]thiophen.
- 22. 3-Methyl-2-(3-sulfamoyl-4-trifluormethylbenzoyl)-benzo[b]thiophen.
- 23. 3-Äthyl-2-(4-chlor-3-isopropylsulfamoylbenzoyl)-benzo[b]thiophen.
- 24. 3-(4-Chlor-3-sulfamoylbenzoyl)-benzo[b]thiophen.
- 25. 3-(4-Chlor-3-sulfamoylbenzoyl)-2-methylbenzo[b]thiophen.
- 26. 2-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl)-benzo[b]thiophen.
- 27. 3-(4-Chlor-3-sulfamoylbenzoyl)-2-isopropyl- benzo[b]thiophen.
- 28. 3-(4-Chlor-3-methylsulfamoylbenzoyl)-2-methyl- benzo[b]thiophen.
- 29. 2-Äthyl-3-(4-chlor-3-methylsulfamoylbenzoyl)-benzo[b]thiophen.
- 30. 5-Chlor(-3-(4-chlor-3-sulfamoylbenzoyl)-2-methyl- benzo[b]thiophen.
- 31. 3-(4-Brom-3-sulfamoylbenzoyl)-2-methylbenzo[b]thiophen.
- 32. 2-Methyl-3-(3-sulfamoyl-4-trifluormethylbenzoyl)-benzo[b]thiophen.
- 33. 3-(3-Butylßulfamoyl-4-chlorbenzoyl)-2-methyl- benzo[b]thiophen.
- 34. 3-(4-Chlor-3-sulfamoylbenzoyl)-5-methoxy-3-methyl- benzo[b]thiophen.
- 35. 2-Äthyl-3-(4-brom-3-sulfamoylbenzoyl)-benzo[b]thiophen.
- 36. 2-(4-Chlor-3-sulfamoylbenzoyl)-indol
- 37. 2-(4-Chlor-3-sulfamoylbenzoyl)-3-methylindol
- 38. 2-(4-Chlor-3-sulfaraoylbenzoyl)-1-methylindol
- 39. 2-(4-Chlor-3-sulfamoylbenzoyl)-1,3-dimethylindol.
- 40. 1-Äthyl-2-(4-chlor-3-sulfamoylbenzoyl)-3-methylindol.
- 41. 3-Äthyl-2-(4-chlor-3-sulfamoylbenzoyl)-1-methylindol.
- 42. 3-Äthyl-2-(4-chlor-3-sulfamoylbenzoyl)-indol.
- 43. 2-(4-Brom-3-sulfamoylbenzoyl)-1,3-dimethylindol.
- 44. 2-(4-Chlor-3-sulfamoylbenzoyl)-5-methoxy-3-methylindol.
- 45. 5-Äthoxy-2-(4-chlor-3-sulfamoylbenzoyl)-3-methylindol.
- 46. 2-(4-Chlor-3-methylsulfamoylbenzoyl)-1,3-dimethylindol.
- 47. 2-(4-Chlor-3-propylsulfamoylbenzoyl)-1,3-dimethylindol.
- 48. 2-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl)-indol.
- 49. 1-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl)-2-methylindol.
- 50. 3-(4-Brom-3-sulfamoylbenzoyl)-2-methylindol.
- 51. 3-(4-Brom-3-sulfamoylbenzoyl)-1,2-dimethylindol.
- 52. 3-(4-Chlor-3-sulfamoylbenzoyl)-5-methoxy-2-methylindol.
- 53. 5-Äthoxy-3-(4-chlor-3-sulfamoylbenzoyl-1,3-dimethylindol.
- 54. 3-(4-Chlor-3-sulfamoylbenzoyl)-2-methyl-1-propylindol.
- 55. 3-(4-Chlor-3-methylsulfamoylbenzoyl)-2-methylindol.
- 56. 3-(4-Chlor-3-methylsulfamoylbenzoyl)-1,2-dimethylindol.
- 57. 1,2-Dimethyl-3-(3-sulfamoyl-4-trifluormethylbenzoyl)-indol.
- 58. 5-Äthoxy-3-(4-chlor-3-sulfamoylbenzoyl)-2-methylindol.
- 59. 2-(4-Chlor-3-sulfamoylbenzoyl)-7-methoxy- benzo[b]furan.
- 60. 2-(4-Chlor-3-sulfamoylbenzoyl)-5-methoxy- benzo[b]furan.
- 61. 2-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl)-5-methoxy- benzo[b]furan.
- 62.. 2-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl)-7-methoxy- benzo[b]furan.
- 63. 2-(4-Chlor-3-sulfamoylbenzoyl)-6-methoxybenzo[b]furan.
- 64. 2-(4-Chlor-3-methylsulfamoylbenzoyl)-6-methoxy- benzo[b]furan.
- 65. 2-(4-Chlor-3-sulfamoylbenzoyl)-5-methoxybenzo[b]furan.
- 66. 2-(4-Chlor-3-methylsulfamöylbenzoyl)-5-methoxy- benzo[b]furan.
- 67. 2-(4-Chlor-3-sulfamoylbenzoyl)-4-methoxy- benzo[b]furan.
- 68. 5-Äthoxy-2-(4-chlor-3-sulfamcylbenzoyl)-benzo[b]furan.
- 69. 6-Äthoxy-2-(4-chlor-3-sulfamoylbenzoyl)-benzo[b]furan.
- 70. 2-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl)-7-methoxy- benzo[b]furan.
- 71. 2-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl)-6-methoxy- benzo[b]furan.
- 72. 2-Äthyl-5-äthoxy-l-(4-chlor-3-sulfamoylbenzoyl)-benzo[b]furan.
- 73. 2-Äthyl-3-(4-chlor-3-methylsulfamoylbenzoyl)-7-methoxybsnzo[b]furan.
- Die Verfahrensprodukte sind wertvolle Arzneimittel und verursachen eine sehr gute, den Harnsäurespiegel des Blutes senkende Wirkung, die insbesondere durch eine urikosurische Wirkung hervorgerufen wird.
- Es ist bekannt, dass die meisten der zur Behandlung urikopathischer Krankheitsbilder verwendeten Arzneimittel über keine salidiuretische Wirkkomponente verfügen. Die erfindungsgemässen Verbindungen zeichnen sich dagegen durch eine erwünschte gute diuretische und saluretische Wirksamkeit aus, und sind somit den vorbekannten urikosurischen Mitteln überlegen.
- Die urikosurische Wirkung der neuen Verfahrensprodukte wurde an der Oxonat behandelten Ratte in einer Einheitsdosis von 50 mg/kg per os bestimmt. Sie zeigen dabei die antiurikopathische Wirksamkeit bekannter Handelspräparate des Probencid-Typs und des Benzbromaron-Typs.
- Die salidiuretische Wirkung der erfindungsgemässen Verbindungen wurde an der Ratte in einer Einheitsdosis von 50 mg/kg per os bestimmt. Sie erreichen dabei die salidiuretische Aktivität bekannter Handelspräparate wie die des Chlorthalidons. Darüber hinaus zeichnen sich die neuen Verfahrenserzeugnisse durch eine lang anhaltende Wirkungsdauer aus, wodurch die Präparate ebenfalls.zur Behandlung hypertoner Zustände am Menschen geeignet sind. Dabei kann man sie mit einem Antihypertonikum kombinieren.
- Als therapeutische Zubereitung der neuen Verbindungen kommen vor allem Tabletten, Dragees, Kapseln, Suppositorien sowie auch Ampullen zur parenteralen Verabreichung (i.v., s.c. und i.m.) in Frage.
- Die therapeutische Einheitsdosis liegt zwischen 5 und 1000 mg, vorzugsweise 10 bis 500 mg pro Tablette.
- Diese Zubereitungen können speziell bei der Behandlung des Bluthochdrucks ausser den üblichen Füll- und Trägerstoffen noch ein Antihypertensivum, wie beispielsweise Reserpin, Hydralazin, Guanethidin, α-Methyldopa, ein ß-Sympathikolytikum oder Chloridin enthalten.
- Ausserdem sind therapeutische Kombinationspräparate mit kaliumretinierenden Verbindungen, wie Aldosteronantagonisten, z.B. Spironolacton, oder Pseudoaldosterinantagonisten, wie Triamteren oder Amilorid, von Interesse. Weiterhin kommt K+-Substitution in verschiedenen Anwendungsformen, z.B. Dragees, Tabletten, Brausetabletten, Säften u.a. in Frage.
- Von therapeutischem Interesse können ebenfalls Kombinationen der erfindungsgemässen Verbindungen mit einem anderen antihyperurikämisch wirksamen Mittel sein, das besonders über eine Hemmung der Xanthinoxidase zu einer Verstärkung der antiurikopathischen Effekte führt. Eine gegebenenfalls erwünschte Verstärkung der salidiuretischen Wirksamkeit kann durch Kombination der erfindungsgemässen Verbindungen mit einem Salidiuretikum erzielt werden.
- In den nachfolgenden Beispielen sind die Schmelz- und Zersetzungspunkte der Ausführungsbeispiele nicht korrigiert.
- 10 g gepulvertes 4-Chlor-3-sulfamoylbenzoylchlorid, dargestellt aus 4-Chlor-3-sulfamoylbenzoesäure und Thionylchlorid (Schmp. 166° C), werden in 70 ml wasserfreies Chlorbenzol eingetragen, anschliessend 6,32 g 2-Äthyl- benzo[b]furan zugesetzt und das Reaktionsgemisch auf 0° C abgekühlt. Nach Zugabe von 11,4 g wasserfreiem Aluminiumchlorid hält man die Reaktion durch Aussenkühlung auf 5° bis 10° C, rührt sodann 5 Stunden bei 15° C und giesst das Gemisch in eine Suspension aus 200 g Eis und 10 ml konz. Salzsäure. Nach Extraktion mit Essigester und Waschen mit Wasser rührt man die organische Phase 6 Stunden mit einer verdünnten NaHCO3-Lösung vom pH 8,5, trocknet über Magnesiumsulfat und erhält nach Verdampfen ein hellgelbes bis farbloses viskoses öl, das unter Petroläther zur Kristallisation kommt. Farblose Kristalle, Schmp. 170° - 172° C (aus wenig Methanol).
- erhält man analog der in Beispiel 1 angegebenen Vorschrift aus 10 g 3-Butylsulfamoyl-4-chlorbenzoylchlorid und 5,1 g 2-Äthylbenzo[b]furan in Gegenwart von 9,4 g Aluminiumchlorid als farbloses bis schwach gelbes viskoses öl.
- erhält man analog der in Beispiel 1 angegebenen Vorschrift aus 10 g 4-Chlor-3-sulfamoylbenzoylchlorid und 6,2 g 2- Äthyl-5-methylbenzo[b]furan in Gegenwart von 11,4 g Aluminiumchlorid. Farblose Kristalle vom Schmp. 147° - 150° C (aus Methanol).
- erhält man analog der in Beispiel 1 angegebenen Vorschrift aus 10 g 4-Chlor-3-sulfamoylbenzoylchlorid und 7,06 g 2- Äthyl-5-chlorbenzo[b]furan in Gegenwart von 11,4 g Aluminiumchlorid. Farblose Kristalle, Schmp. 130° - 133° C.
- erhält man analog der in Beispiel 1 angegebenen Vorschrift aus 10,1 g 4-Chlor-3-sulfamoylbenzoylchlorid und 5,8 g 2-Methylbenzo[b]furan in Gegenwart von 11,4 g Aluminium- .chlorid. Nach Verdampfen des Extraktionsmittels rührt man den Rückstand unter Diisopropyläther und filtriert den Feststoff ab. Farblose Kristalle, Schmp. 183° C (aus Methanol).
- erhält man analog der in Beispiel 1 angegebenen Vorschrift aus 10,7 g 4-Chlor-3-methylsulfamoylbenzoylchlorid und 6,4 g 2-Äthylbenzo[b]furan als amorphen Feststoff vom Erweichungspunkt 63° C.
- erhält nian analog der in Beispiel 1 angegebenen Vorschrift aus 10,9 g 4-Brom-3-sulfamoylbenzoylchlorid und 6 g 2-Äthyl- benzo[b]furan in Gegenwart von 11,7 g Aluminiumchlorid. Farblose Kristalle, Schmp. 202° - 206° C (Äther).
- Zu einer Natriummethylatlösung, dargestellt aus 3,13 g Natrium und 170 ml Methanol,tropft man unter N2-Schutz rasch 16,6 g Salicylaldehyd, destilliert sodann das Lösungsmittel ab und schlämmt den Rückstand in 150 ml wasserfreiem Toluol auf. Zu dem Gemisch gibt man 38 g 2--Brom-4'-chlor-3'-nitroacetoplaenon in 100 ml Toluol unter Rührung zu und kocht anschliessend 3 Stunden am Rückflusskühler. Nach dem Stehenlassen bei Raumtemperatur über Nacht destilliert man das Lösungsmittel ab und kristallisiert den Rückstand aus Äthanol um. Hellgelbe Kristalle, Schmp. 132° C.
- 37 g 2-(4-Chlor-3-nitrobenzoyl)-benzo[b]furan werden unter Rührung in einer Mischung aus 500 ml 50%iger wässriger Essigsäure, 250 ml Äthanol und 90 g gepulverter Nickel-Aluminium-Legierung (1 : 1) 4 Stunden am Rückflusskühler gekocht. Nach Filtration des Metallpulvers destilliert man das Lösungsmittel ab, versetzt den - Rückstand mit Wasser und extrahiert mit Essigsäureäthyl-ester. Nach Vertreiben des Lösungsmittels erhält man farblose Kristalle vom Schmp. 137° C (aus Isopropanol);
- 3 g 2-(3-Amine-4-chlorbenzoyl)-benzo[b]furan werden in 10 ml Eisessig und 5 ml H2O aufgeschlämmt-und sodann mit 10 ml konz. Salzsäure versetzt. Bei 0° bis 5° C tropft man unter Rührung eine Lösung von 1 g Natriumnitrit in 4 ml Wasser unter die Oberfläche. Das Reaktionsgemisch wird sodann portionsweise in eine Mischung aus 2,3 g CuCl2 x 2 H20 in 70 ml SO2-gesättigte Eisessiglösung eingetragen und nach 20 Minuten Rührung das Volumen mit Wasser verdoppelt. Man rührt 45 Minuten nach, filtriert die Kristalle ab und trocknet.
Schmp. 131° C (Zers.) - 3,3 g 2-(4-Chlor-3-chlorsulforsylbenzoyl)-benzo[b]furan werden in 19 ml 25%ige wässrige Ammoniaklösung eingetragen und nach Stehenlassen über Nacht die Flüssigkeit abdestilliert. Nach Zugabe von Wasser stellt man mit verdünnter HC1 auf pH 6, rührt 30 Minuten nach und kristallisiert aus wenig Eisessig. Farblose Kristalle, Schmp. 181° C.
- 5,1 g gepulvertes 4-Chlor-3-sulfamoylbenzoylchlorid wer- den in eine Lösung aus 3 g Benzo[b]thiophen in 50 ml wasserfreiem Toluol eingetragen und anschliessend unter Rührung mit 12 g Titantetrachlorid versetzt. Man rührt 15 Min. bei Raumtemperatur, erhitzt sodann etwa 15 Minuten zum Sieden, kühlt ab und giesst auf eine Wasser-Eis-Suspension. Nun extrahiert man mit 70 ml Essigester, trennt die organische Phase ab und rührt diese 5 Stunden mit einer wässrigen Natriumbicarbonatlösung vom pH 8,5. Nach Abtrennen der organischen Phase und deren Trocknung über Magnesiumsulfat wird das Lösungsmittel abdestilliert und der Rückstand unter Toluol zur Kristallisation gebracht. Schmp. 154° - 156° C.
-
- a) 2,5 g 4-Chlor-3-sulfamoylbenzoylchlorid werden in 25 ml Dichloräthan aufgeschlämmt und portionsweise unter Rührung zu einer Suspension von 2,7 g Aluminiumchlorid in 25 ml Dichloräthan gegeben, wobei die Temperatur durch Aussenkühlung zwischen -10° C und 0° C gehalten wird. Die erhaltene klare Lösung rührt man 1 Stunde bei +5° C, erwärmt sodann auf 30° C und gibt sodann eine Lösung von 1,3 g 2-Methylindol in 25 ml Dichloräthan zu, wobei die Temperatur zwischen 30° und 40° C gehalten wird. Die Reaktionsmischung wird auf etwa 10° C gekühlt, sodann mit Eiswasser zersetzt und zweimal mit Essigester extrahiert. Die vereinigten organischen Phasen rührt man 8 Stunden mit wässriger Natriumbicarbonatlösung vom pH 8 - 9, wäscht diese mit Wasser, trocknet über Natriumsulfat und vertreibt das Lösungsmittel unter vermindertem Druck. Der Rückstand wird mit Diisopropyläther gerührt und der kristalline Feststoff abfiltriert. Schmp. 258° - 260° C.
- b) 3,1 g 4-Chlor-3-sulfamoylbenzoesäure-N,N-dimethylamid, dargestellt aus 4-Chlor-3-sulfamoylbenzoylchlorid und wässriger Dimethylaminlösung (Schmp. 143° C) werden zusammen mit 0,8 g 2-Methylindol und 0,7 ml Phosphoroxichlorid unter Rührung und Ausschluss von Luftfeuchtigkeit über 2 Stunden auf 80° C erhitzt. Man stellt mit 2N NaOH alkalisch, rührt 40 Stunden bei Raumtemperatur, bringt sodann auf pH 7 bis 8 und filtriert die Kristalle ab. Schmp. 257° - 260° C.
- c) In eine Lösung von 10 g 4-Chlor-3-sulfamoylbenonitril, dargestellt durch Rückfluss von 4-Chlor-3-sulfamoyl- benzamid in Phosphoroxichlorid (Schmp. 199° C) und 6,06 g 2-Methylindol in 80 ml wasserfreien Diäthylenglykoldimethyläther leitet man 5 Stunden bei 15° - 20° C einen Strom Chlorwasserstoffgas ein und lässt das Gemisch 72 Stunden bei 10° - 15° C stehen. Durch Eingies- .sen des Reaktionsgemisches in Essigester scheidet sich das 4'-Chlor-3'-sulfamoylphenyl-2-methyl-3-indolyl- ketonimin-hydrochlorid kristallin ab. Schmp. 320° C. 9 g Ketonimin-hydrochlorid werden in 100 ml heissem Wasser gelöst, nach Zugabe von wässrigem Ammoniak unter Rührung bei 50° - 60° C hydrolysiert und das kristalline 3-(4-Chlor-3-sulfamoylbenzoyl)-2-methylindol abfiltriert. Schmp. 252° - 257° C.
- d) Zu einer Lösung von Methylmagnesiumjodid, dargestellt aus 3,28 g Magnesiumspänen in 80 ml wasserfreiem Äther und 22,5 g Methyljodid, fügt man rasch eine Lösung von 17 g 2-Methylindol in 200 ml Tetrahydrofuran und erhitzt ca. 15 Minuten zum Rückfluss. Sodann lässt man rasch eine Lösung von 15,75 g 4-Chlor-3-sulfamoylben- zoylchlorid in 100 ml Tetrahydrofuran zufliessen, erhitzt weitere 14 Stunden am Rückflusskühler, kühlt ab und giesst in eine Lösung aus 20 g Ammoniumchlorid in 200 ml Wasser. Nach Zugabe von 350 ml Essigester rührt man 10 Minuten, filtriert das in homogene Gemisch über eine Klärschicht, trennt sodann die organische Phase ab und trocknet nach einmaligem Waschen mit Wasser über Magnesiumsulfat. Nach dem Verdampfen des Lösungsmittels suspendiert man den kristallinen Rückstand in Diisopropyläther und filtriert die Kristalle ab. Schmp. 255° - 259° C.
- e) Die nach den Methoden a) bis d) dargestellten Proben des 3-(4-Chlor-3-sulfamoylbenzoyl)-2-methylindols zeigen identische IR-Spektren und depressionslose Mischschmelzpunkte.
- erhält man analog der in Beispiel 10 a) angegebenen Vorschrift aus 2,68 g 4-Chlor-3-methylsulfamoylbenzoyl- chlorid und 1,3 g 2-Methylindol in Gegenwart von 2,7 g Aluminiumchlorid in Dichloräthan. Farblose Kristalle, Schmp. 246° C.
- erhält man analog der in Beispiel 10 a) angegebenen Vorschrift aus 10 g 4-Chlor-3-sulfamoylbenzoylchlorid und 7,8 g 2,5-Dimethylindol in Dichloräthan in Gegenwart von 12 g Aluminiumchlorid. Schmp. 248° - 250° C.
- erhält man analog der in Beispiel 10 a) angegebenen Vorschrift aus 10 g 4-Chlor-3-sulfamoylbenzoylchlorid und 6,53 g 1,2-Dimethylindol in Dichloräthan in Gegenwart von 12 g Aluminiumchlorid. Nach Behandeln mit Isopropanol erhält man Kristalle vom Schmp. 247° - 249° C.
-
- I a) 8 g 4-Chlor-3-sulfamoylbenzonitril und 12 g Nickel-Aluminium-Legierung (1 : 1) werden in 120 ml 75%iger wässriger Ameisensäure 2 Stunden am Rückflusskühler gekocht, sodann heiss abfiltriert und das unumgesetzte Metallpulver mehrfach mit heissem Methanol nachgewaschen. Nach dem Einengen versetzt man mit Wasser und filtriert den kristallinen 4-Chlor-3-sulfamoyl- benzaldehyd (Schmp. 162° - 164° C) ab.
- .b) 6 g (0,03 Mol) 2-Brombenzo[b[furan. werder in 20 ml Diäthyläther gelöst und auf einmal zu einer auf -70° abgekühlten Lösung von 0,033 Mol n-Butyllithium in 150 ml wasserfreiem Tetrahydrofuran gegeben. Man rührt etwa 5 Minuten nach und gibt nun in kleinen Portionen eine Mischung von 2,2 g (0,01 Mol) 4-Chlor-3-sulfamoyl- benzaldehyd in 30 ml wasserfreiem Tetrahydrofuran zu, wobei das Reaktionsgemisch bei -40° bis -70° C gehalten wird. Man rührt 20 Min. bei -40° C, rührt sodann 30 Stunden bei Zimmertemperatur und 6 weitere Stunden bei +50° C und behandelt das Gemisch anschliessend unter Eiskühlung mit 10 ml einer gesättigten Ammoniumchloridlösung. Der Niederschlag wird abfiltriert, mehrfach mit Essigester gewaschen, die vereinigten organischen Phasen über Magnesiumsulfat getrocknet und das Lösungsmittel unter vermindertem Druck verdampft. Farblose Kristalle, Zersetzungspunkt 148° C.
- c) 1,69 g (0,05 Mol) 4-Chlor-3-sulfamoylphenyl-2'-benzo[b]furan-carbinol werden in 40 ml Acetonitril gelöst und nach Zugabe von 6 g aktivem Mangandioxid 60 Stunden bei Raumtemperatur gerührt. Man filtriert das anorganische Material ab, wäscht einmal mit Acetonitril nach, engt unter vermindertem Druck ein und kristalisiert den Rückstand aus wenig Eisessig um. Farblose Kristalle, Schmp. 170° - 172° C.
- II) Zu einer wie unter I b) dargestellten Lösung von 2-Benzo[b]furyllithium in Tetrahydrofuran fügt man eine Mischung aus 2,16 g 4-Chlor-3-sulfamoylbenzonitril in 50 ml absol. Tetrahydrofuran und erhitzt unter Stickstoffschutz und guter Rührung 18 Stunden amRückflusskühler. Sodann wird das Lösungsmittel unter vermindertem Druck abdestilliert, der Rückstand mit Eiswasser versetzt und mit 2N HCl sauer gestellt. Man rührt 2 Stunden bei Raumtemperatur, extrahiert das öl mit 100 ml Essigester, wäscht die organische Phase zweimal mit Wasser und trocknet über Natriumsulfat. Nach dem Abdestillieren des Lösungsmittels unter vermindertem Druck erhält man ein schwach gelbes Öl, das in 20%iger wässriger KOH 20 Minuten bei Raumtemperatur gerührt und sodann in eine gute gerührte, gesättigte, wässrige Ammoniumchloridlösung getropft. Man saugt ab und kristallisiert aus wenig Eisessig um. Farblose Kristalle, Schmp. 167° - 170° C.
- III, 4 2-Brom-4'-chlor-3'-sulfamoylacetophenon werden zusammen mit 1,63 g Salizylaldehyd in Gegenwart von 2,9 g unter wasserfreien Bedingungen gemahlenen Kaliumcarbonat 2 Stunden unter Rührung und unter Ausschluss von Feuchtigkeit in 50 ml wasserfreiem Dimethylformamid auf 80° C gehalten. Nach dem Abkühlen giesst man in ein Gemisch aus Eiswasser und überschüssiger Salzsäure, extrahiert mit Essigester und trocknet über Natriumsulfat. Nach Abdestillieren des Lösungsmittels bringt man unter Diisopropyläther und unter Isopropanol zur Kristallisation, Schmp. 152° - 156° C. Umkristallisation aus Eisessig nach Klärung mit Aktivkohle liefert farblose Kristalle vom Schmp. 167° - 170° C.
- erhält man analog der in Beispiel 1 angegebenen Vorschrift aus 2,5 g 4-Chlor-3-sulfamoylbenzoylchlorid, 1,6 g 3-Methyl-benzo[b]thiophen und 3 g Aluminiumchlorid in 50 ml Chlorbenzol. Farblose Kristalle, Schmp. 210° C.
- erhält man analog der in Beispiel 10 a) angegebenen Vorschrift aus 10,0 g 4-Chlor-3-sulfamoylbenzoylchlorid, 5,1 g 3-Methylindol in Gegenwart von 10,4 g Aluminiumchlorid. Farblose Kristalle aus Isopropanol, Schmp. 205° C.
-
- a) 3-(4-Chlor-3-nitrobenzoyl)-2-methylbenzo/b7furan 4,4 g 4-Chlor-3-nitrobenzoylchlorid und 2,6 g 2-Methyl- benzo[b]furan werden in 50 ml Chlorbenzol gelöst, auf 0° C gekühlt, und nach Zugabe von 4 g wasserfreiem Aluminiumchlorid 1 Stunde bei 0° bis 5° C gerührt. Nach dem Stehenlassen über Nacht bei Raumtemperatur giesst man auf Eiswasser, extrahiert mit mehrfach Essigester, wäscht die vereinigten organischen Phasen mit Wasser und rührt intensiv mit einer wässrigen NaHCO3-Lösung (pH 8 - 9) bis zum Verschwinden der geringen Anteile an 4-Chlor-3-nitrobenzoylchlorid. Man trocknet über Natriumsulfat, destilliert das Lösungsmittel unter vermindertem Druck ab und bringt den Rückstand unter Äther zur Kristallisation. Schmp. 122° C.
- b) 3-(3-Amino-4-chlorbenzoyl)-2-methylbenzo[b]furan erhält man analog der in Beispiel 8 b) angegebenen Vorschrift aus 3,2 g 3-(4-Chlor-3-nitrobenzoyl)-2-methyl- benzo[b]furan mit 9,6 g Nickel-Aluminium-Legierung (1 : 1). Schmp. 103° C.
- c) 3-(4-Chlor-3-chlorsulfamoylbenzoyl)-2-methyl- benzo[b]furan
erhält man analog der in Beispiel 8 c) angegebenen Vorschrift aus 3-(3-Amino-4-chlorbenzoyl)-2-methyl- benzo[b]furan. Farblose Kristalle, Schmp. 138° C. - d) 3-(4-Chlor-3-sulfamoylbenzoyl)-2-methylbenzo[b]furan erhält man durch Umsetzung von 2,3 g 3-(4-Chlor-3- chlorsulfonylbenzoyl)-2-methylbenzo[b]furan in einer Mischung aus 20 ml Methanol und 10 ml wasserfreiem Ammoniak. Nach dem Stehenlassen über Nacht destilliert man das Lösungsmittel ab, nimmt den Rückstand in Wasser auf und filtriert ab. Farblose Kristalle aus wenig Äthanol, Schmp. 180° - 183° C.
-
- a) 2-(4-Chlor-3-nitrobenzoyl)-7-methoxybenzo[b]furan 2 g KOH werden in 50 ml Methanol gelöst, versetzt sodann mit einer Lösung aus 5,4 g 2-Hydroxy-3-methoxybenzaldehyd und anschliessend mit einer Suspension aus 10 g 2-Brom-4'-chlor-3'-nitroacetophenon in 75 ml Methanol. Man kocht 6 Stunden am Rückflusskühler, rührt über Nacht bei Raumtemperatur und saugt die Kristalle ab. Schmp. 136° C (aus Essigester).
- b) 2-(3-Amino-4-chlorbenzoyl)-7-methoxyberzo[b]furan erhält man analog der in Beispiel 8 b) angegebenen Vorschrift aus 2,2 g 2-(4-chlor-3-nitrobenzoyl-7-methoxy- benzo[b]furan und 6,6 g Nickel-Aluminium-Legierung (1 : 1). Farblose Kristalle, Schmp. 180° C(aus Äthanol).
- c) 2-(4-Chlor-3-chlorsulfonylbenzoyl)-7-methoxy- benzo[b]furan
erhält man analog der in Beispiel 8 c) angegebenen Vorschrift aus 2-(3-Amino-4-chlorbenzoyl)-7-methoxy- benzo[b]furan. Schmp. 143° - 145° C. - d) 2-(4-Chlor-3-sulfamoylbenzoyl)-7-methoxy- benzo[b]furan
erhält man analog der in Beispiel 8 d) angegebenen Vorschrift aus 2-(4-Chlor-3-chlorsulfamoylbenzoyl)-7-methoxybenzo[b]furan und 25%iger wässriger Ammoniaklösung. Schmp. 167° - 171° C. - erhält man durch Behandlung von 4,5 g 3-(4-Chlor-3-chlor- sulfonylbenzoyl)-2-methylbenzo[b]furan mit einem Gemisch aus 20 ml Methanol und 30 ml 40%iger wässriger Methylaminlösung über 12 Stunden bei Raumtemperatur, anschliessendem Abdampfen des Lösungsmittels, Zugabe von Wasser und Filtration der Kristalle. Schmp. 146° C.
- erhalt man analog der in Beispiel 10 a) angegebenen Vorschrift aus 4-Chlor-3-sulfamoylbenzoylchlorid und 2-Methylbenzo[b]thiophen in Dichloräthan in Gegenwart von Aluminiumchlorid.
Claims (13)
1. Verbindungen der allgemeinen Formel I

worin R1 Wasserstoff, Halogen, Alkyl mit 1 bis 3 C-Atomen,Methoxy oder Äthoxy, R2 und R3 gleich oder verschieden sein können und Wasserstoff oder Alkyl mit 1 bis 4 C-Atomen, X ein Halogenatom, Methyl oder Trifluormethyl und Y Sauerstoff, Schwefel oder NR4 bedeuten, wobei R4 für Wasserstoff oder Alkyl mit 1 bis 4 C-Atomen steht.
2. 2-Äthyl-3-(4-chlor-3-sulfamoylbenzoyl)-benzo[b]furan.
3. 3-(4-Chlor-3-sulfamoylbenzoyl)-2-methylbenzo[b]furan.
4. 3-(4-Chlor-3-sulfamoylbenzoyl)-2-methylindol.
5. 3-(4-Chlor-3-merhylsulfamoylbenzoyl)-2-methylindol.
6. 2-(4-Chlor-3-sulfamoylbenzoyl)-benzo[b]furan.
7. 2-(4-Chlor-3-sulfamoylbenzoyl)-7-methoxy-benzo[b]furan.
8. 3-(4-Chlor-3-sulfamoylbenzoyl)-2-methylbenzo[b]thiophen.
9. 2-(4-Chlor-3-sulfamoylbenzoyl)-3-methylbenzo[b]thiophen.
10. Verfahren zur Herstellung von Verbindungen gemäss Anspruch 1, dadurch gekennzeichnet, dass man
a) Verbindungen der allgemeinen Formel II

worin R3 und X die angegebene Bedeutung besitzen und A für Halogen, Acyloxy, Hydroxy oder NR2 steht und R Wasserstoff, niederes Alkyl oder Phenyl bedentet, mit einer heterocyclischen Verbindung der allgemeinen Formel III,

worin R1, R2 und Y die angegebene Bedeutung besitzen, in Gegenwart einer Lewis-Säure oder Protonensäure umsetzt,
b) Verbindungen der allgemeinen Formel IV

worin R1, R2, X und Y die angegebene Bedeutung besitzen und Hal für Halogen steht, mit einem Amin der allgemeinen Formel V

worin R3 die angegebene Bedeutung hat, umsetzt oder.
c) Verbindungen der allgemeinen Formel VI

worin R1 bis R 3, A, X und Y die angegebene Bedeutung besitzen, mit einem Oxidationsmittel behandelt,
d) Verbindungen der allgemeinen Formel VII
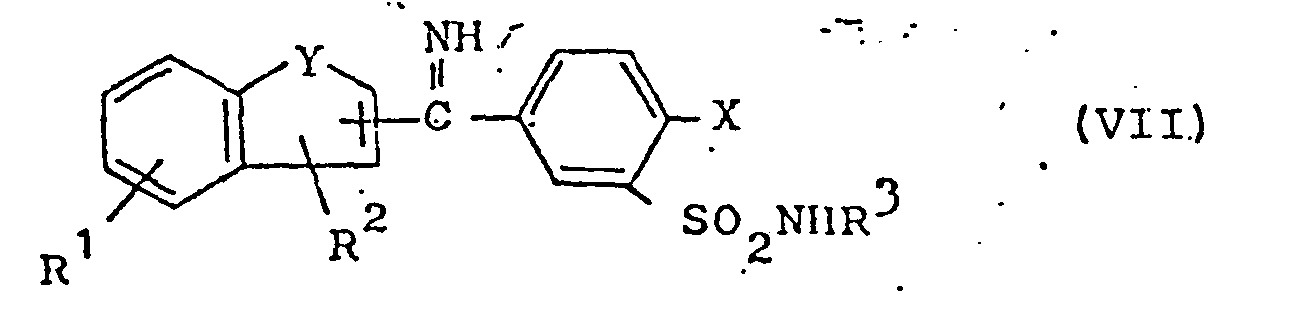
worin R1 bis R3, X und Y die obige Bedeutung haben, gegebenenfalls in Form ihrer Säureadditionssalze der Hydrolyse unterwirft,
e) Verbindungen der allgemeinen Formel VIII

worin R1 bis R2 die angegebene Bedeutung besitzen, M für Li, MgHal, CdHal, HgHal, of HgOCOCH3 steht, und Y neben der angegebenen Bedeutung auch diejenige von NM besitzen kann,
mit Verbindungen der allgemeinen Formel II umsetzt, worin A neben der angegebenen Bedeutung auch für einen 2-Mercaptopyridylrest 
stehen kann, oder
mit Verbindungen der allgemeinen Formel II umsetzt, worin A neben der angegebenen Bedeutung auch für einen 2-Mercaptopyridylrest
f) Verbindungen der allgemeinen Formel IX
cyclisiert und gegebenenfalls die nach a) bis f) erhaltenen Verbindungen der allgemeinen Formel I,worin R3 Wasserstoff bedeutet, anschliessend alkyliert.
11. Pharmazeutische Präparate mit urikosurischer, hypourikämischer und salidiuretischer Wirkung, bestehend aus bzw. enthaltend eine Verbindung gemäss Anspruch 1.
12. Verfahren zur Herstellung pharmazeutischer Präparate mit urikosurischer, hypourikämischer und salidiuretischer Wirkung, dadurch gekennzeichnet, dass man eine Verbindung gemäss Anspruch 1 gegebenenfalls mit pharmazeutischen Trägern und/oder Stabilisatoren in eine für therapeutische Zwecke geeignete Anwendungsform bringt.
13. Verwendung von Verbindungen der Formal I zur Senkung des Harnsäurespiegels des Blutes und zur Bekämpfung des Bluthochdrucks.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE2727802 | 1977-06-21 | ||
| DE19772727802 DE2727802A1 (de) | 1977-06-21 | 1977-06-21 | Sulfamoyl-arylketone und verfahren zu ihrer herstellung |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| EP0000128A1 true EP0000128A1 (de) | 1979-01-10 |
Family
ID=6011950
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP78100118A Withdrawn EP0000128A1 (de) | 1977-06-21 | 1978-06-08 | Sulfamoyl-Arylketone und Verfahren zu ihrer Herstellung sowie ihre Verwendung als Arzneimittel |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US4156732A (de) |
| EP (1) | EP0000128A1 (de) |
| JP (1) | JPS549259A (de) |
| AU (1) | AU3728978A (de) |
| DE (1) | DE2727802A1 (de) |
| DK (1) | DK278178A (de) |
| IL (1) | IL54945A0 (de) |
| IT (1) | IT1096492B (de) |
| ZA (1) | ZA783511B (de) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002000210A2 (en) * | 2000-06-28 | 2002-01-03 | Merck & Co., Inc. | Use of agents capable of reducing uric acid levels for the treatment of cardiovascular disease |
| US8841333B2 (en) | 2005-05-09 | 2014-09-23 | Takeda Pharmaceuticals U.S.A., Inc. | Methods for treating nephrolithiasis |
| US9107912B2 (en) | 2010-09-10 | 2015-08-18 | Takeda Pharmaceuticals U.S.A., Inc. | Methods for concomitant treatment of theophylline and febuxostat |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5835653U (ja) * | 1981-09-03 | 1983-03-08 | 三國工業株式会社 | 始動装置のワツクス加熱調整回路 |
| JPS5848963U (ja) * | 1981-09-28 | 1983-04-02 | 本田技研工業株式会社 | 電熱式オ−トチヨ−ク付気化器 |
| JPS58186152U (ja) * | 1982-06-07 | 1983-12-10 | 本田技研工業株式会社 | 多連式気化器におけるア−ス装置 |
| DE3317884A1 (de) * | 1983-05-17 | 1984-11-22 | Hoechst Ag, 6230 Frankfurt | 5-(4-chlor-3-sulfamoylbenzoyl)-2,3-dihydro-2-benzofurancarbonsaeuren und verfahren zu ihrer herstellung |
| US4745222A (en) * | 1983-05-25 | 1988-05-17 | Merrell Dow Pharmaceuticals Inc. | Novel aryloxycycloalkanolaminoalkylene aryl ketones |
| JP3157882B2 (ja) * | 1991-11-15 | 2001-04-16 | 帝国臓器製薬株式会社 | 新規なベンゾチオフエン誘導体 |
| EP1940397A4 (de) * | 2005-08-03 | 2010-01-20 | Takeda Pharmaceuticals North A | Verfahren zur behandlung von hypertonie |
| US20090124623A1 (en) * | 2006-11-13 | 2009-05-14 | Christopher Lademacher | Methods for preserving and/or increasing renal function using xanthine oxidoreductase inhibitors |
| KR20090103879A (ko) * | 2006-11-13 | 2009-10-01 | 다케다 파마슈티칼스 노쓰 어메리카, 인코포레이티드 | 크산틴 옥시도리덕타아제 억제제를 사용하여 신장 기능을 보존하는 방법 |
| KR20090127870A (ko) * | 2007-01-19 | 2009-12-14 | 다케다 파마슈티칼스 노쓰 어메리카, 인코포레이티드 | 크산틴 산화환원효소 저해제들 및 항염증제들을 사용하여 통풍 발열을 방지하거나 또는 통풍 발열의 회수를 감소시키는 방법 |
| AU2009228765B2 (en) | 2008-03-24 | 2012-05-31 | Novartis Ag | Arylsulfonamide-based matrix metalloprotease inhibitors |
| US20100311756A1 (en) * | 2009-01-22 | 2010-12-09 | Takeda Pharmaceuticals North America, Inc. | Methods for delaying the progression of at least one of cardiac hypertrophy, cardiac remodeling or left ventricular function or the onset of heart failure in subjects in need of treatment thereof |
| CN106008421A (zh) * | 2016-06-12 | 2016-10-12 | 南方医科大学 | 一种能促进尿酸排泄的人体尿酸盐转运体-1抑制剂及其制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL108331C (de) * |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1521932A (en) * | 1976-03-08 | 1978-08-16 | Labaz | Sulphonamide derivatives and process for preparing the same |
-
1977
- 1977-06-21 DE DE19772727802 patent/DE2727802A1/de not_active Withdrawn
-
1978
- 1978-06-08 EP EP78100118A patent/EP0000128A1/de not_active Withdrawn
- 1978-06-19 IL IL7854945A patent/IL54945A0/xx unknown
- 1978-06-19 IT IT24695/78A patent/IT1096492B/it active
- 1978-06-20 US US05/917,194 patent/US4156732A/en not_active Expired - Lifetime
- 1978-06-20 ZA ZA00783511A patent/ZA783511B/xx unknown
- 1978-06-20 AU AU37289/78A patent/AU3728978A/en active Pending
- 1978-06-20 DK DK278178A patent/DK278178A/da unknown
- 1978-06-21 JP JP7432778A patent/JPS549259A/ja active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL108331C (de) * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002000210A2 (en) * | 2000-06-28 | 2002-01-03 | Merck & Co., Inc. | Use of agents capable of reducing uric acid levels for the treatment of cardiovascular disease |
| WO2002000210A3 (en) * | 2000-06-28 | 2002-10-24 | Merck & Co Inc | Use of agents capable of reducing uric acid levels for the treatment of cardiovascular disease |
| US7799794B2 (en) | 2000-06-28 | 2010-09-21 | Merck Sharp & Dohme Corp. | Treatment for cardiovascular disease |
| US8841333B2 (en) | 2005-05-09 | 2014-09-23 | Takeda Pharmaceuticals U.S.A., Inc. | Methods for treating nephrolithiasis |
| US9107912B2 (en) | 2010-09-10 | 2015-08-18 | Takeda Pharmaceuticals U.S.A., Inc. | Methods for concomitant treatment of theophylline and febuxostat |
Also Published As
| Publication number | Publication date |
|---|---|
| IL54945A0 (en) | 1978-08-31 |
| US4156732A (en) | 1979-05-29 |
| ZA783511B (en) | 1979-06-27 |
| DK278178A (da) | 1978-12-22 |
| JPS549259A (en) | 1979-01-24 |
| IT7824695A0 (it) | 1978-06-19 |
| AU3728978A (en) | 1980-01-03 |
| DE2727802A1 (de) | 1979-04-19 |
| IT1096492B (it) | 1985-08-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0000128A1 (de) | Sulfamoyl-Arylketone und Verfahren zu ihrer Herstellung sowie ihre Verwendung als Arzneimittel | |
| CA1254216A (en) | Benzothiazole and benzothiophene derivatives | |
| CH650782A5 (de) | Carbostyrilverbindungen, verfahren zu deren herstellung und arzneimittel, welche diese enthalten. | |
| DE1939809C3 (de) | ||
| WO1995032970A1 (de) | [a]-ANNELIERTE PYRROLDERIVATE UND DEREN ANWENDUNG IN DER PHARMAZIE | |
| DE3419009A1 (de) | Neue substituierte bis(4-aminophenyl)sulfone, ihre herstellung und ihre verwendung als arzneimittel | |
| EP0071935B1 (de) | 1-Phenyl-2-aminocarbonylindol-Verbindungen sowie Verfahren und Zwischenprodukte zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| US4196292A (en) | 6-Substituted amiloride derivatives | |
| EP0064027B1 (de) | Neue Benzodioxolderivate, Verfahren zu deren Herstellung und diese enthaltende pharmazeutische Zusammensetzungen | |
| US4086353A (en) | Certain azolinylamino (azolidinylimino) indazoles | |
| DE2351411C2 (de) | (1-Oxo-2,3-hydrocarbylen-6,7-dichlor-5- indanyloxy)-essigsäuren, Verfahren zu ihrer Herstellung und sie enthaltende pharmazeutische Mittel | |
| US4134900A (en) | 5,6-Dihydro-4-oxo-4H-thieno[2,3-b]thiopyran-5-carboxamides, and process for the preparation thereof | |
| DE2436263C2 (de) | Thiazolidinderivate und Verfahren zu ihrer Herstellung | |
| DE3784835T2 (de) | Chinoxalinon-derivate. | |
| DE2740836A1 (de) | Anellierte indolderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP0107622B1 (de) | Neue Benzodioxolderivate, Verfahren zu deren Herstellung und entsprechende pharmazeutische Zusammensetzungen | |
| DE2737407A1 (de) | Verfahren zur herstellung neuer benzopyranderivate | |
| US4154734A (en) | Amides of 4-hydroxy-6H-thieno[2,3-b]thiopyran-5-carboxylic acid-7,7-dioxide | |
| EP0072961B1 (de) | 1-Phenylindazol-3-on-Verbindungen sowie Verfahren und Zwischenprodukte zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| Catsoulacos et al. | Thiazo compounds. Derivatives of 4, 5-dihydro-7, 8-dimethoxybenzothiazepin-3-one 1, 1-dioxides | |
| DE2106038C3 (de) | 2-Imidazolin-2-ylamino-benzo [b] thiophene und diese enthaltende pharmazeutische Präparate | |
| CH625217A5 (de) | ||
| DE2804894A1 (de) | Halogen-benzofuranon-carbonsaeuren | |
| EP0064026B1 (de) | Neue dioxaheterocyclische Verbindungen, Verfahren zu deren Herstellung und diese enthaltende pharmazeutische Zusammensetzungen | |
| US3697517A (en) | 1,3-benzoxazines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): BE CH DE FR GB NL SE |
|
| 17P | Request for examination filed | ||
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION IS DEEMED TO BE WITHDRAWN |
|
| 18D | Application deemed to be withdrawn | ||
| RIN1 | Information on inventor provided before grant (corrected) |
Inventor name: MUSCHAWECK, ROMAN, DR. Inventor name: MUSIL, JOSEF, DR. Inventor name: LANG, HANS JOCHEN, DR. |