CN1304406A - 作为玻连蛋白拮抗剂的杂环甘氨酰β-丙氨酸衍生物 - Google Patents
作为玻连蛋白拮抗剂的杂环甘氨酰β-丙氨酸衍生物 Download PDFInfo
- Publication number
- CN1304406A CN1304406A CN99807091A CN99807091A CN1304406A CN 1304406 A CN1304406 A CN 1304406A CN 99807091 A CN99807091 A CN 99807091A CN 99807091 A CN99807091 A CN 99807091A CN 1304406 A CN1304406 A CN 1304406A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- group
- aryl
- product
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 *c(cc1Br)cc(C(CC(O)=O)NC(CNC(C(C=C2NC(NC3)=NCC3O)=CNC2=O)=O)=O)c1O Chemical compound *c(cc1Br)cc(C(CC(O)=O)NC(CNC(C(C=C2NC(NC3)=NCC3O)=CNC2=O)=O)=O)c1O 0.000 description 21
- UFEUGPZTCZLOSF-UHFFFAOYSA-N C=C(NC1)NCC1O Chemical compound C=C(NC1)NCC1O UFEUGPZTCZLOSF-UHFFFAOYSA-N 0.000 description 1
- WPKQCUROLWGTOT-UHFFFAOYSA-N CC(C)(C)OC(N(CCN1C)C1=S)=O Chemical compound CC(C)(C)OC(N(CCN1C)C1=S)=O WPKQCUROLWGTOT-UHFFFAOYSA-N 0.000 description 1
- SZMUJHRXSMTABS-NCWAPJAISA-N CCC(C)N[C@@H](CC(OCC)=O)c(cc(cc1Br)Cl)c1O Chemical compound CCC(C)N[C@@H](CC(OCC)=O)c(cc(cc1Br)Cl)c1O SZMUJHRXSMTABS-NCWAPJAISA-N 0.000 description 1
- BUZOMNLQSBOVAB-UHFFFAOYSA-N CCC(CN(C)C(C(C(OC(C)(C)C)=O)=C)=S)OC(OC(C)(C)C)=O Chemical compound CCC(CN(C)C(C(C(OC(C)(C)C)=O)=C)=S)OC(OC(C)(C)C)=O BUZOMNLQSBOVAB-UHFFFAOYSA-N 0.000 description 1
- KMNIVAYJOWHWEI-UHFFFAOYSA-N CCC(NC(CC(OCC)=O)c(cc(cc1Br)Cl)c1O)=O Chemical compound CCC(NC(CC(OCC)=O)c(cc(cc1Br)Cl)c1O)=O KMNIVAYJOWHWEI-UHFFFAOYSA-N 0.000 description 1
- JKYNZXCIABTGIP-UHFFFAOYSA-N CCOC(CC(c(cc(cc1Cl)Cl)c1O)NC(CNC(C(C=C1N)=CN(C)C1=O)=O)=O)=O Chemical compound CCOC(CC(c(cc(cc1Cl)Cl)c1O)NC(CNC(C(C=C1N)=CN(C)C1=O)=O)=O)=O JKYNZXCIABTGIP-UHFFFAOYSA-N 0.000 description 1
- MVKXHTLZHSTQMY-UHFFFAOYSA-N CCOC(CC(c(cc(cc1Cl)Cl)c1O)NC(CNC(C(C=C1[N+]([O-])=O)=CN(C)C1=O)=O)=O)=O Chemical compound CCOC(CC(c(cc(cc1Cl)Cl)c1O)NC(CNC(C(C=C1[N+]([O-])=O)=CN(C)C1=O)=O)=O)=O MVKXHTLZHSTQMY-UHFFFAOYSA-N 0.000 description 1
- TXCOOHVPZCNEDJ-YLHCSOALSA-N CCOC(CC(c(cc(cc1Cl)Cl)c1O)NC(CNC(c1cc(/N=C(/N(C)CC2)\N2C(OC(C)(C)C)=O)cnc1OC)=O)=O)=O Chemical compound CCOC(CC(c(cc(cc1Cl)Cl)c1O)NC(CNC(c1cc(/N=C(/N(C)CC2)\N2C(OC(C)(C)C)=O)cnc1OC)=O)=O)=O TXCOOHVPZCNEDJ-YLHCSOALSA-N 0.000 description 1
- YIOPTXTXTDMGET-UHFFFAOYSA-N CCOC(CC(c(cc(cc1Cl)Cl)c1O)NC(CNC(c1cc([N+]([O-])=O)cnc1Cl)=O)=O)=O Chemical compound CCOC(CC(c(cc(cc1Cl)Cl)c1O)NC(CNC(c1cc([N+]([O-])=O)cnc1Cl)=O)=O)=O YIOPTXTXTDMGET-UHFFFAOYSA-N 0.000 description 1
- IEUZDJAYTUGMFB-UHFFFAOYSA-N CN(C=C(C=C1NC2=NCCN2)C(NCC(NC(CC(O)=O)c(cc(cc2Cl)Cl)c2O)=O)=O)C1=O Chemical compound CN(C=C(C=C1NC2=NCCN2)C(NCC(NC(CC(O)=O)c(cc(cc2Cl)Cl)c2O)=O)=O)C1=O IEUZDJAYTUGMFB-UHFFFAOYSA-N 0.000 description 1
- VECUQHDZPCINQA-UHFFFAOYSA-N CN(C=C(C=C1[N+]([O-])=O)C(OC)=O)C1=O Chemical compound CN(C=C(C=C1[N+]([O-])=O)C(OC)=O)C1=O VECUQHDZPCINQA-UHFFFAOYSA-N 0.000 description 1
- ILGFGEIQCLRIBI-UHFFFAOYSA-N COC(c(cn1)cc([N+]([O-])=O)c1O)=O Chemical compound COC(c(cn1)cc([N+]([O-])=O)c1O)=O ILGFGEIQCLRIBI-UHFFFAOYSA-N 0.000 description 1
- MACLWDXRXQZYQB-UHFFFAOYSA-N COc(c(C(NCC(NC(CC(O)=O)c(cc(cc1Cl)Cl)c1O)=O)=O)c1)ncc1N=C1NCCN1 Chemical compound COc(c(C(NCC(NC(CC(O)=O)c(cc(cc1Cl)Cl)c1O)=O)=O)c1)ncc1N=C1NCCN1 MACLWDXRXQZYQB-UHFFFAOYSA-N 0.000 description 1
- XIHQBNINFNUFFG-BYPYZUCNSA-N C[C@H](C(O)=O)c(cc(cn1)[N+]([O-])=O)c1Cl Chemical compound C[C@H](C(O)=O)c(cc(cn1)[N+]([O-])=O)c1Cl XIHQBNINFNUFFG-BYPYZUCNSA-N 0.000 description 1
- QMJIMNGWYFBQKB-UHFFFAOYSA-N Nc(cc1C(O)=O)cnc1O Chemical compound Nc(cc1C(O)=O)cnc1O QMJIMNGWYFBQKB-UHFFFAOYSA-N 0.000 description 1
- JTHPGYYQHRWGBR-UHFFFAOYSA-N OC(CC1)CNC1=S Chemical compound OC(CC1)CNC1=S JTHPGYYQHRWGBR-UHFFFAOYSA-N 0.000 description 1
- BLHCMGRVFXRYRN-UHFFFAOYSA-N OC(c(cc1)cnc1O)=O Chemical compound OC(c(cc1)cnc1O)=O BLHCMGRVFXRYRN-UHFFFAOYSA-N 0.000 description 1
- UEYQJQVBUVAELZ-UHFFFAOYSA-N OC(c(cccn1)c1O)=O Chemical compound OC(c(cccn1)c1O)=O UEYQJQVBUVAELZ-UHFFFAOYSA-N 0.000 description 1
- SGCLUZVJDJBDPU-UHFFFAOYSA-N [O-][N+](c(cn1)cc(C(Cl)=O)c1Cl)=O Chemical compound [O-][N+](c(cn1)cc(C(Cl)=O)c1Cl)=O SGCLUZVJDJBDPU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
- A61P3/14—Drugs for disorders of the metabolism for electrolyte homeostasis for calcium homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0202—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-X-X-C(=0)-, X being an optionally substituted carbon atom or a heteroatom, e.g. beta-amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Crystallography & Structural Chemistry (AREA)
- Cardiology (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Heart & Thoracic Surgery (AREA)
- Biophysics (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Hematology (AREA)
- Immunology (AREA)
- Obesity (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Communicable Diseases (AREA)
- Ophthalmology & Optometry (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
本发明涉及一类由式(Ⅰ)表示的化合物或者它们的药学上可接受的盐,含有这类化合物的药用组合物和治疗由αvβ3整联蛋白介导的疾病的方法。
Description
本申请按照35USC§119(e),要求在1998年4月10日呈交的美国临时专利申请系列第60/081,394号的优先权。
本发明领域
本发明涉及用作αvβ3整联蛋白拮抗剂的和如此通过抑制或者拮抗αvβ3整联蛋白用于治疗由αvβ3介导的疾病的药用组合物和方法中的药物(化合物)。
本发明背景
整联蛋白是一组介导细胞粘附的细胞表面糖蛋白并由此是在多种生物过程中发生的细胞粘附相互作用的有用介质。整联蛋白为由非共价键连接的α和β多肽亚基组成的异二聚体。目前已经鉴定了十一种不同的α亚基并且已经鉴定六种不同的β亚基。各种α亚基能够与各种β亚基结合形成独特的整联蛋白。
鉴定为αvβ3(也称作玻连蛋白受体)的整联蛋白已被鉴定为在多种疾病或疾病状态包括肿瘤转移、实体肿瘤生长(瘤形成)、骨质疏松症、培吉特病、恶性肿瘤体液高钙血症、血管发生包括肿瘤血管发生、视网膜病包括黄斑变性、关节炎包括类风湿性关节炎、牙周病、牛皮癣和平滑肌细胞迁移(例如再狭窄)中起作用的整联蛋白。另外,已发现这样的药物将用作抗病毒剂、抗真菌剂和抗菌药。因此,选择性抑制或拮抗αvβ3的化合物将有利于治疗这样的疾病。
已显示αvβ3整联蛋白和其它含有αv的整联蛋白结合多种含有Arg-Gly-Asp(RGD)的基质大分子。含有RGD序列的化合物模拟细胞外基质配体以便结合于细胞表面受体。然而,人们也已知RGD肽对RGD依赖的整联蛋白一般是非选择性的。例如,大多数结合αvβ3的RGD肽也结合αvβ5、αvβ1和αⅡbβ3。已知血小板αⅡbβ3(也称作血纤蛋白原受体)拮抗作用阻断在人体中的血小板聚集。当治疗与整联蛋白αvβ3有关的病症或疾病状态时,为避免出血副作用,开发为抗αⅡbβ3的选择性拮抗剂的化合物将是有益的。
肿瘤细胞侵入的发生经过三个步骤的过程:1)肿瘤细胞附着于细胞外基质;2)蛋白水解下基质溶解和3)细胞运动穿过溶解的屏障。这个过程能够重复发生并且能够导致转移至远离原来肿瘤的位置。
Seftor等(Proc.Natl.Acad.Sci.USA,第89卷(1992)第1557-1561页)已显示αvβ3整联蛋白在黑素瘤细胞侵入中具有生物功能。Montgomery等(Proc.Natl.Acad.Sci.USA,第91卷(1994),第8856-60页)已证实在人黑素瘤细胞上表达的整联蛋白αvβ3促进生存信号,保护细胞免于编程性细胞死亡。通过αvβ3整联蛋白细胞粘附受体的干扰作用以阻抑肿瘤转移的肿瘤细胞转移途径的介导作用将是有益的。
Brooks等(细胞,第79卷(1994)第1157-1164页)已证实αvβ3拮抗剂提供用于治疗瘤形成(抑制实体肿瘤生长)的治疗方法,因为全身性给予αvβ3拮抗剂引起多种组织学上不同的人肿瘤的出人意料的退化。
粘附受体整联蛋白αvβ3被鉴定为在小鸡和人中的新生成血管的标记物,因此这样的受体在血管发生或者新血管形成中起着至关重要的作用。血管发生的特征在于平滑肌细胞和内皮细胞的侵入、迁移和增殖。αvβ3拮抗剂通过选择性促进新血管形成中细胞的编程性细胞死亡来抑制这个过程。新的血管生长或者血管发生也有助于病理性状况例如糖尿病性视网膜病包括黄斑变性(Adamis等,Amer.J.Ophthal,第118卷(1994),第445-450页)和类风湿性关节炎(Peacock等,J.Exp.Med.,第175卷(1992),第1135-1138页)。因此,αvβ3拮抗剂将是用于治疗与新血管生成有关疾病的有用的治疗药物(Brooks等,科学,第264卷(1994),第569-571页)。
已报道细胞表面受体αvβ3为负责粘附骨头的破骨细胞上的主要的整联蛋白。破骨细胞引起骨吸收,并且当这样的骨吸收活性超过骨形成活性时,这导致骨质疏松症(骨丧失),该疾病导致增加的骨折、使残废的人数和增加的死亡率。已显示αvβ3拮抗剂为体外[Sato等,J.Cell Biol.,第111卷(1990),第1713-1723页]和体内[Fisher等,Endocrinology,第132卷(1993),第1411-1413页]有效的破骨细胞活性的抑制剂。αvβ3的拮抗作用导致减少的骨吸收,因此恢复骨形成和骨吸收活性的正常平衡。因此,提供为骨吸收的有效抑制剂并且由此用于治疗或阻止骨质疏松症的破骨细胞αvβ3拮抗剂是有益的。
αvβ3整联蛋白在平滑肌细胞迁移中的作用也使其成为用于阻止或抑制新内膜增生的治疗靶,其为血管手术后再狭窄的首要因素(Choi等,J.Vasc.Surg.第19卷(1)(1994),第125-34页)。经药物阻止或抑制新内膜增生来阻止或抑制再狭窄将是有利的。
White(Current Biology,第3卷(9)(1993),第596-599页)已报道腺病毒使用αvβ3进入宿主细胞。整联蛋白似乎被病毒颗粒的胞吞作用所需要并且可被用于病毒基因组侵入宿主细胞胞质所需要。因此,将发现抑制αvβ3的化合物作为抗病毒药是有用的。
本发明概述
本发明涉及一类由式Ⅰ表示的化合物或者它们的药学上可接受的盐:其中 为含有1至4个选自O、N或S的杂原子的、任选不饱和的5-8元单环杂环,其中X1选自CH、CH2、N、NH、O和S;
为含有1至4个选自O、N或S的杂原子的、任选不饱和的5-8元单环杂环,其中X1选自CH、CH2、N、NH、O和S;
A为 其中Y1选自N-R2、O和S;
其中Y1选自N-R2、O和S;
R2选自H、烷基、芳基、羟基、烷氧基、氰基、硝基、氨基、链烯基、链炔基、酰氨基、烷基羰基、芳基羰基、烷氧基羰基、芳氧基羰基、卤代烷基羰基、卤代烷氧基羰基、烷硫基羰基、芳硫基羰基、酰氧基甲氧基羰基、以一个或多个选自以下的取代基任选取代的烷基:低级烷基、卤素、羟基、卤代烷基、氰基、硝基、羧基、氨基、烷氧基、芳基或以一个或多个卤素、卤代烷基、低级烷基、烷氧基、氰基、烷基磺酰基、烷硫基、硝基、羧基、氨基、羟基、磺酸、磺酰胺、芳基、稠合的芳基、单环杂环或稠合的单环杂环任选取代的芳基,以一个或多个选自以下的取代基任选取代的芳基:卤素、卤代烷基、羟基、低级烷基、烷氧基、亚甲二氧基、亚乙二氧基、氰基、硝基、烷硫基、烷基磺酰基、磺酸、磺酰胺、羧基衍生物、氨基、芳基、稠合的芳基、单环杂环和稠合的单环杂环,单环杂环和以一个或多个选自以下的取代基任选取代的单环杂环:卤素、卤代烷基、低级烷基、烷氧基、氨基、硝基、羟基、羧基衍生物、氰基、烷硫基、烷基磺酰基、磺酸、磺酰胺、芳基或稠合的芳基;或者
R2与R7一起形成以一个或多个选自低级烷基、硫代烷基、烷基氨基、羟基、酮基、烷氧基、卤代、苯基、氨基、羧基或羧酸酯和稠合的苯基的取代基任选取代的含有两个氮的4-12元杂环;或
R2与R7一起形成含有一个或多个选自O、N和S的杂原子的、任选不饱和的4-12元杂环;或
R2与R7一起形成以一个或多个选自低级烷基、苯基、烷氧基和羟基的取代基任选取代的5-9元杂芳环;或
R2与R7一起形成与芳基或杂芳基环稠合的5元杂芳环;
R7(当不与R2一起时)和R8独立选自H,烷基,链烯基,链炔基,芳烷基,氨基,烷基氨基,羟基,烷氧基,芳基氨基,酰氨基,烷基羰基,芳基羰基,烷氧基羰基,芳氧基,芳氧基羰基,卤代烷基羰基,卤代烷氧基羰基,烷硫基羰基,芳硫基羰基,酰氧基甲氧基羰基,环烷基,双环烷基,芳基,酰基,苯甲酰基,以一个或多个选自以下的取代基任选取代的烷基:低级烷基、卤素、羟基、卤代烷基、氰基、硝基、羧基衍生物、氨基、烷氧基、硫代、烷硫基、磺酰基、芳基、芳烷基、以一个或多个选自以下的取代基任选取代的芳基:卤素、卤代烷基、低级烷基、烷氧基、亚甲二氧基、亚乙二氧基、烷硫基、卤代烷硫基、硫代、羟基、氰基、硝基、羧基衍生物、芳氧基、酰氨基、酰基氨基、氨基、烷基氨基、二烷基氨基、三氟代烷氧基、三氟代甲基、磺酰基、烷基磺酰基、卤代烷基磺酰基、磺酸、磺酰胺、芳基、稠合的芳基、单环杂环、稠合的单环杂环,以一个或多个选自以下的取代基任选取代的芳基:卤素、卤代烷基、低级烷基、烷氧基、亚甲二氧基、亚乙二氧基、烷硫基、卤代烷硫基、硫代、羟基、氰基、硝基、羧基衍生物、芳氧基、酰氨基、酰基氨基、氨基、烷基氨基、二烷基氨基、三氟烷氧基、三氟甲基磺酰基、烷基磺酰基、磺酸、磺酰胺、芳基、稠合的芳基、单环杂环或稠合的单环杂环,单环杂环,以一个或多个选自卤素、卤代烷基、低级烷基、烷氧基、芳氧基、氨基、硝基、羟基、羧基衍生物、氰基、烷硫基、烷基磺酰基、芳基、稠合的芳基的取代基任选取代的单环杂环,单环和二环杂环烷基,其中R10选自烷基、芳基和单环杂环的-SO2R10,所有基团以一个或多个选自卤素、卤代烷基、烷基、烷氧基、氰基、硝基、氨基、酰基氨基、三氟烷基、酰氨基、烷基氨基磺酰基、烷基磺酰基、烷基磺酰基氨基、烷基氨基、二烷基氨基、三氟甲基硫代、三氟烷氧基、三氟甲基磺酰基、芳基、芳氧基、硫代、烷硫基和单环杂环的取代基任选取代;和其中R10如上定义的 ;或者NR7和R8一起形成含有单氮的4-12元单环或双环,其以一个或多个选自低级烷基、羧基衍生物、芳基或羟基的取代基任选取代并且其中所述环任选含有一个选自O、N和S的杂原子;
;或者NR7和R8一起形成含有单氮的4-12元单环或双环,其以一个或多个选自低级烷基、羧基衍生物、芳基或羟基的取代基任选取代并且其中所述环任选含有一个选自O、N和S的杂原子;
R5选自H、烷基、链烯基、链炔基、苄基和苯乙基;或者
A为其中Y2选自烷基,环烷基,双环烷基,芳基,单环杂环,由也能以一个或多个选自卤代、卤代烷基、烷基、硝基、羟基、烷氧基、芳氧基、芳基或稠合的芳基的取代基任选取代的芳基所任选取代的烷基,以一个或多个选自卤代、卤代烷基、羟基、烷氧基、芳氧基、芳基、稠合的芳基、硝基、亚甲二氧基、亚乙二氧基或烷基的取代基任选取代的芳基,链炔基,链烯基,-S-R9和-O-R9,其中R9选自H、烷基、芳烷基、芳基、链烯基和链炔基,或者R9与R7一起形成以低级烷基、羟基、酮基、苯基、羧基或羧酸酯和稠合的苯基任选取代的、含有单氮和单硫或单氧的4-12元杂环,或者R9与R7一起为噻唑、噁唑、苯并噁唑或苯并噻唑;和
R5和R7如上定义;或者
Y2(当Y2为碳时)与R7一起形成以烷基、芳基、酮基或羟基任选取代的、含有单氮或二氮的4-12元环;或者
A为其中R2和R7一起形成以一个或多个选自低级烷基、羟基、烷氧基、酮基、苯基或羧基衍生物的取代基任选取代的含有二氮的5-8元杂环;并且R8选自烷基羰基、芳基羰基、烷氧基羰基、芳氧基羰基、卤代烷基羰基、卤代烷氧基羰基、烷硫基羰基、芳硫基羰基或酰氧基甲氧基羰基;和
R5如上定义;或者
R2和R7一起形成杂芳环例如咪唑或嘧啶酮;或者
A为 其中R2和R7一起形成以羟基、酮基、苯基或烷基任选取代的含有二氮的5-8元杂环;和
其中R2和R7一起形成以羟基、酮基、苯基或烷基任选取代的含有二氮的5-8元杂环;和
R8选自烷基羰基、芳基羰基、烷氧基羰基、芳氧基羰基、卤代烷基羰基、卤代烷氧基羰基、烷硫基羰基、芳硫基羰基和酰氧基甲氧基羰基;
Z1为一个或多个选自H、烷基、羟基、烷氧基、芳氧基、卤素、卤代烷基、卤代烷氧基、硝基、氨基、烷基氨基、酰基氨基、二烷基氨基、氰基、烷硫基、烷基磺酰基、羧基衍生物、三卤代乙酰胺、乙酰胺、酰基、芳基、稠合的芳基、环烷基、硫代、单环杂环、稠合的单环杂环和A的取代基,其中A如上定义;
V选自-N-(R6)-,其中R6选自H、低级烷基、环烷基、芳烷基、芳基和单环杂环;或者R6与Y一起形成含有单氮的4-12元环;
Y、Y3、Z和Z3独立选自氢、烷基、芳基和环烷基;或者Y与Z一起形成环烷基;或者Y3和Z3一起形成环烷基;
n为整数1、2或3;
t为整数0、1或2;
p为整数0、1、2或3;
R为其中X选自O、S和NR4的X-R3,其中R3和R4独立选自氢、烷基、链烯基、链炔基、卤代烷基、芳基、芳基烷基、糖类、甾体化合物、聚烷基醚、烷基酰氨基、烷基N,N-二烷基酰氨基、新戊酰氧基甲基;和在游离酸的情况下所有它们的药学上可接受的盐;
R1选自氢、烷基、链烯基、链炔基、芳基、羧基衍生物、卤代烷基、环烷基、单环杂环、以烷基、卤素、卤代烷基、氰基、羟基、芳基、稠合的芳基、硝基、烷氧基、芳氧基、烷基磺酰基、芳基磺酰基、磺酰胺、硫代、烷硫基、羧基衍生物、氨基、酰氨基任选取代的单环杂环;
以一个或多个以下基团任选取代的烷基:卤代、卤代烷基、羟基、烷氧基、芳氧基、硫代、烷硫基、链炔基、链烯基、烷基、芳硫基、烷基亚砜、烷基磺酰基、芳基亚砜、芳基磺酰基、氰基、硝基、氨基、烷基氨基、二烷基氨基、烷基磺酰胺、芳基磺酰胺、酰基酰胺、羧基衍生物、磺酰胺、磺酸、膦酸衍生物、次膦酸衍生物、在芳基环上均用卤代、烷基、卤代烷基、氰基、硝基、羟基、羧基衍生物、烷氧基、芳氧基、氨基、烷基氨基、二烷基氨基、酰氨基、芳基、稠合的芳基、单环杂环任选取代的芳基、芳硫基、芳基亚砜或芳基砜;和稠合的单环杂环、单环杂环硫代、单环杂环亚砜和单环杂环砜,其能够用卤代、卤代烷基、硝基、羟基、烷氧基、稠合的芳基或烷基任选取代;
烷基羰基、卤代烷基羰基和芳基羰基;
在一个或多个位置上用以下基团任选取代的芳基,包括:卤代、卤代烷基、烷基、烷氧基、芳氧基、亚甲二氧基、亚乙二氧基、烷硫基、卤代烷硫基、硫代、羟基、氰基、硝基、酰氧基、羧基衍生物、羧基烷氧基、酰氨基、酰基氨基、氨基、烷基氨基、二烷基氨基、三氟烷氧基、三氟甲基磺酰基、烷基磺酰基、磺酸、磺酰胺、芳基、稠合的芳基、单环杂环和稠合的单环杂环和,其中R7和R8如上定义并且条件是与所述氮一起,R7和R8包括氨基酸;
和
R11选自H、烷基、芳烷基、链烯基、链炔基、卤代烷基或卤代链炔基或者R11与Y一起形成含有单氮的4-12元环。
本发明另一个目的是提供含有式Ⅰ化合物的药用组合物。这样的化合物和组合物用于选择性抑制或者拮抗αvβ3整联蛋白,由此在另一个实施方案中本发明涉及选择性抑制或者拮抗αvβ3整联蛋白的方法。本发明另外包括在需要这样治疗的哺乳动物上治疗或抑制与此有关的病理性疾病例如骨质疏松症、恶性肿瘤体液高钙血症、培吉特病、肿瘤转移、实体肿瘤生长(瘤形成)、血管发生包括肿瘤血管发生、视网膜病包括黄斑变性和糖尿病性视网膜病、关节炎包括类风湿性关节炎、牙周病、牛皮癣、平滑肌细胞迁移和再狭窄。另外,这样的药物用作抗病毒剂和抗菌药。详细描述
本发明涉及一类由以上描述的式Ⅰ表示的化合物。
本发明的一个优选实施方案为式Ⅱ化合物 其中其中R32为H、烷基、烷氧基烷基、氨基烷基、二烷基氨基烷基,其中所述烷基由一个或多个选自羟基、烷氧基、氨基、烷基氨基、二烷基氨基、芳基-或烷基-磺酰基、羧基和羧基衍生物的取代基任选取代并且其它的变量如同在式Ⅰ中描述。
其中其中R32为H、烷基、烷氧基烷基、氨基烷基、二烷基氨基烷基,其中所述烷基由一个或多个选自羟基、烷氧基、氨基、烷基氨基、二烷基氨基、芳基-或烷基-磺酰基、羧基和羧基衍生物的取代基任选取代并且其它的变量如同在式Ⅰ中描述。
本发明的另一个优选实施方案为式Ⅲ化合物其中后者被任选取代并且其它的变量如同在式Ⅰ中定义。
本发明的另一个优选实施方案为式Ⅳ化合物 其中后者被任选取代并且其它的变量如同在式Ⅰ中定义。
其中后者被任选取代并且其它的变量如同在式Ⅰ中定义。
本发明另一个优选实施方案为式Ⅴ化合物 其中后者被任选取代并且其它的变量如同在式Ⅰ中定义。
其中后者被任选取代并且其它的变量如同在式Ⅰ中定义。
本发明的另一个优选实施方案为式Ⅵ化合物其中 后者被任选取代并且其它的变量如同在式Ⅰ中定义。
后者被任选取代并且其它的变量如同在式Ⅰ中定义。
本发明另外涉及含有治疗有效量的式Ⅰ-Ⅵ化合物的药用组合物。
本发明也涉及选择性抑制或拮抗αvβ3整联蛋白的方法,并且更具体地讲,涉及通过给予与药学上可接受的载体一起的、治疗有效量的式Ⅰ-Ⅵ化合物(以达到这样的抑制作用)来抑制骨吸收、牙周病、骨质疏松症、恶性肿瘤体液高钙血症、培吉特病、肿瘤转移、实体肿瘤生长(瘤形成)、血管发生包括肿瘤血管发生、视网膜病包括黄斑变性和糖尿病性视网膜病、关节炎包括类风湿性关节炎、平滑肌细胞迁移和再狭窄的方法
以下列出在此使用的多种术语的定义:
如同在此使用的术语“烷基”或“低级烷基”指的是具有大约1至大约10个碳原子并且更优选为1至大约6个碳原子的直链的或分枝链的烃基。这样的烷基实例为甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、戊基、新戊基、己基、异己基等。
如同在此使用的术语“链烯基”或“低级链烯基”指的是含有至少一个双键和2至大约6个碳原子的不饱和无环烃基,相对于在所述双键碳上取代的基团,其在链烯基部分中的碳-碳双键可具有顺式或者反式几何构型。这样基团的实例为乙烯基、丙烯基、丁烯基、异丁烯基、戊烯基、己烯基等。
如同在此使用的术语“链炔基”或“低级链炔基”指的是含有一个或多个叁键和2至大约6个碳原子的无环烃基。这样基团的实例为乙炔基、丙炔基、丁炔基、戊炔基、己炔基等。
如同在此使用的术语“环烷基”意指饱和的或部分不饱和的含有3至大约8个碳原子并且更优选为4至大约6个碳原子的环碳基团。这样的环烷基的实例包括环丙基、环丙烯基、环丁基、环戊基、环己基、2-环己烯-1-基等。
如同在此使用的术语“芳基”意指包括一个或多个芳香环的芳香环体系。优选芳基为那些含有一个、两个或三个芳香环的基团。该术语包括芳基例如苯基、吡啶基、萘基、噻吩、呋喃、联苯等。
如同在此使用的术语“氰基”由式 的基团表示。
的基团表示。
如同在此使用的术语“羟基”由式 的基团表示。下式的基团
的基团表示。下式的基团 指的是含有1至3个选自O、N和S的杂原子的、任选含有不饱和度的5-10元单环或双环杂环,其中X1为CH、N、O或S。所述基团代表性实例包括吡啶酮、吡啶、嘧啶、咪唑、噁唑、异噁唑、噻唑、哒嗪、噻吩、呋喃、吡唑和双环杂环如苯并咪唑、咪唑并吡啶、苯并呋喃等。
指的是含有1至3个选自O、N和S的杂原子的、任选含有不饱和度的5-10元单环或双环杂环,其中X1为CH、N、O或S。所述基团代表性实例包括吡啶酮、吡啶、嘧啶、咪唑、噁唑、异噁唑、噻唑、哒嗪、噻吩、呋喃、吡唑和双环杂环如苯并咪唑、咪唑并吡啶、苯并呋喃等。
如同在此使用的术语“低级亚烷基”或“亚烷基”指的是二价线形或分枝的饱和的1至大约6个碳原子的烃基团。
如同在此使用的术语“烷氧基”指的是直形的或分枝链的含有氧的式-OR20基团,其中R20为如上定义的烷基。所包括的烷氧基的实例包括甲氧基、乙氧基、正丙氧基、正丁氧基、异丙氧基、异丁氧基、仲丁氧基、叔丁氧基等。
如同在此使用的术语“芳基烷基”或“芳烷基”指的是式 的基团,其中R21为如上定义的芳基和R22为如上定义的亚烷基。芳烷基的实例包括苄基、吡啶基甲基、萘基丙基、苯乙基等。
的基团,其中R21为如上定义的芳基和R22为如上定义的亚烷基。芳烷基的实例包括苄基、吡啶基甲基、萘基丙基、苯乙基等。
如同在此使用的术语“硝基”由式 的基团表示。
的基团表示。
如同在此使用的术语“卤代”或“卤素”指的是溴代、氯代、氟代或碘代。
如同在此使用的术语“卤代烷基”指的是在一个或多个碳原子上以一个或多个相同的或不同的卤代基团取代的如上定义的烷基。卤代烷基的实例包括三氟代甲基、二氯代乙基、氟代丙基等。
如同在此使用的术语“羧基”指的是式-COOH的基团。
如同在此使用的术语“羧酸酯”指的是式-COOR23的基团,其中R23选自如上定义的H、烷基、芳烷基或芳基。
如同在此使用的术语“羧基衍生物”指的是式的基团,其中Y6和Y7独立选自O、N或S并且R23选自如上定义的H、烷基、芳烷基或芳基。
如同在此使用的术语“氨基”由式-NH2的基团表示。
如同在此使用的术语“烷基磺酰基”或“烷基砜”指的是式 的基团,其中R24为如上定义的烷基。
的基团,其中R24为如上定义的烷基。
如同在此使用的术语“烷硫基”指的是式-SR24的基团,其中R24为如上定义的烷基。
如同在此使用的术语“磺酸”指的是式 的基团,其中R25为如上定义的烷基。
的基团,其中R25为如上定义的烷基。
如同在此使用的术语“磺酰胺”指的是式 的基团,其中R7和R8如上定义。
的基团,其中R7和R8如上定义。
如同在此使用的术语“稠合的芳基”指的是芳香环例如以上定义的芳基稠合于一个或多个苯基环上。术语“稠合的芳基”包括的基团有萘基等。
如同在此使用的术语“单环杂环”或“单环杂环的”指的是含有4至大约12个原子并且更优选为5至大约10个原子的、其中的1至3个原子为选自氧、氮和硫的杂原子的单环,需要理解的是如果存在两个或多个不同的杂原子,至少所述杂原子之一必须为氮。这样的单环杂环的代表为咪唑、呋喃、吡啶、噁唑、吡喃、三唑、噻吩、吡唑、噻唑、噻二唑等。
如同在此使用的术语“稠合的单环杂环”指的是用苯稠合于其上面的如上定义的单环杂环。这样的稠合的单环杂环的实例包括苯并呋喃、苯并吡喃、苯并二氧杂戊环、苯并噻唑、苯并噻吩、苯并咪唑等。
如同在此使用的术语“亚甲二氧基”指的是式 的基团和术语“亚乙二氧基”指的是式的基团。
的基团和术语“亚乙二氧基”指的是式的基团。
如同在此使用的术语“含有二氮的4-12元杂环”指的是式 的基团,其中m为1或2并且R19为H、烷基、芳基或芳烷基且更优选为4-9元环并包括例如咪唑啉环。
的基团,其中m为1或2并且R19为H、烷基、芳基或芳烷基且更优选为4-9元环并包括例如咪唑啉环。
如同在此使用的术语“5-元任选取代的杂芳香环”包括例如式的基团并且“与苯基稠合的5-元杂芳香环”指的是这样的与苯基稠合的5-元杂芳香环。这样的与苯基稠合的5-元杂芳香环的代表为苯并咪唑。
如同在此使用的术语“二环烷基”指的是含有其为饱和的或者部分不饱和的、6至大约12个碳原子的二环烃基。
如同在此使用的术语“酰基”指的是式 的基团,其中R26为烷基、链烯基、链炔基、芳基或芳烷基并如上定义那样在其上任选取代。这样的基团包括乙酰基、苯甲酰基等。
的基团,其中R26为烷基、链烯基、链炔基、芳基或芳烷基并如上定义那样在其上任选取代。这样的基团包括乙酰基、苯甲酰基等。
如同在此使用的术语“硫代”指的是式 的基团。
的基团。
如同在此使用的术语“磺酰基”指的是式 的基团,其中R27为如上定义的烷基、芳基或芳烷基。
的基团,其中R27为如上定义的烷基、芳基或芳烷基。
如同在此使用的术语“卤代烷硫基”指的是式-S-R28的基团,其中R28为如上定义的卤代烷基。
如同在此使用的术语“芳氧基”指的是式 的基团,其中R29为如上定义的芳基。
的基团,其中R29为如上定义的芳基。
如同在此使用的术语“酰基氨基”指的是式 的基团,其中R30为如上定义的烷基、芳烷基或芳基。
的基团,其中R30为如上定义的烷基、芳烷基或芳基。
如同在此使用的术语“酰氨基”指的是式 的基团。
的基团。
如同在此使用的术语“烷基氨基”指的是式-NHR32的基团,其中R32为如上定义的烷基。
如同在此使用的术语“二烷基氨基”指的是式-NR33R34的基团,其中R33和R34为如上定义的相同的或不同的烷基。
如同在此使用的术语“三氟甲基”指的是式 的基团。
的基团。
如同在此使用的术语“三氟烷氧基”指的是式 的基团,其中R35为如上定义的键或亚烷基。
的基团,其中R35为如上定义的键或亚烷基。
如同在此使用的术语“烷基氨基磺酰基”指的是式的基团,其中R36为如上定义的烷基。
如同在此使用的术语“烷基磺酰氨基”指的是式 的基团,其中R36为如上定义的烷基。
的基团,其中R36为如上定义的烷基。
如同在此使用的术语“三氟甲基硫代”指的是式的基团。
如同在此使用的术语“三氟甲基磺酰基”指的是式 的基团。
的基团。
如同在此使用的术语“含有单氮的4-12元单环或双环”指的是饱和的或者部分不饱和的、4-12个原子并且更优选为其中一个原子为氮的4-9个原子的环的单环或双环。这样的环任选含有另外的选自氮、氧或硫的杂原子。在这个组中包括吗啉、哌啶、哌嗪、硫代吗啉、吡咯烷、脯氨酸、氮杂环庚烯等。
如同在此使用的术语“苄基”指的是基团
如同在此使用的术语“苯乙基”指的是基团
如同在此使用的术语“含有单氮的、含有单硫或单氧的4-12元杂环”指的是包括4-12个原子并且更优选为4-9个原子的环,其中至少一个原子为氮并且至少一个原子为氧或硫。在此定义内包括的环如噻唑啉等。
如同在此使用的术语“芳基磺酰基”或“芳基砜”指的是其中R37为如上定义的芳基的下式基团:
如同在此使用的术语“烷基亚砜”或“芳基亚砜”指的是其中R38分别为如上定义的烷基或芳基的下式基团:
如同在此使用的术语“膦酸衍生物”指的是其中R39和R40为相同的或者不同的H、烷基、芳基或芳烷基的下式基团:
如同在此使用的术语“次膦酸衍生物”指的是其中R41为如上定义的H、烷基、芳基或芳烷基的下式基团:
如同在此使用的术语“芳硫基”指的是其中R42为如上定义的芳基的下式基团:
如同在此使用的术语“单环杂环硫代”指的是其中R43为如上定义的单环杂环基团的下式基团:
如同在此使用的术语“单环杂环亚砜”和“单环杂环砜”分别指的是其中R43为如上定义的单环杂环基团的下式基团:
如同在此使用的术语“烷基羰基”指的是其中R50为如上定义的烷基的下式基团:
如同在此使用的术语“芳基羰基”指的是其中R51为如上定义的芳基的下式基团:
如同在此使用的术语“烷氧基羰基”指的是其中R52为如上定义的烷氧基的下式基团:
如同在此使用的术语“芳氧基羰基”指的是其中R51为如上定义的芳基的下式基团:
如同在此使用的术语“卤代烷基羰基”指的是其中R53为如上定义的卤代烷基的下式基团:
如同在此使用的术语“卤代烷氧基羰基”指的是其中R53为如上定义的卤代烷基的下式基团:
如同在此使用的术语“烷硫基羰基”指的是其中R50为如上定义的烷基的下式基团:
如同在此使用的术语“芳硫基羰基”指的是其中R51为如上定义的芳基的下式基团:
如同在此使用的术语“酰氧基甲氧基羰基”指的是其中R54为如上定义的酰基的下式基团:
如同在此使用的术语“芳基氨基”指的是其中R51为如上定义的芳基的式R51-NH-基团。
如同在此使用的术语“聚烷基醚”指的是经常使用的二元醇例如三甘醇、四甘醇、聚乙二醇等。
如同在此使用的术语“烷基酰氨基”指的是其中R50为如上定义的烷基的下式基团:
如同在此使用的术语“N,N-二烷基酰氨基”指的是下式基团,其中R50为相同的或不同的如上定义的烷基:
如同在此使用的术语“新戊酰氧基甲基”指的是下式基团:
如同在此使用的术语“酰氧基”指的是其中R55为如上定义的酰基的式R55-O-基团。
如同在此使用的术语“组合物”意指由将多于一种的要素或组分混合而得到的产物。
如同在此使用的术语“药学上可接受的载体”意指药学上可接受的材料、组成或溶媒,例如涉及携带或转运化学药物的液体或固体填充剂、稀释剂、赋形剂、溶剂或包封材料。
术语“治疗有效量”将意指引发组织、系统或研究者或临床医生研究的动物的生物或医学应答的药物或药剂的量。
以下列出如同在此互换使用的缩写与相应的含义:
1H-NMR=核磁共振氢谱
AcOH=乙酸
Ar=氩
BH3-THF=硼烷-四氢呋喃复合物
Bn=苄基
BOC=叔丁氧基羰基
ButLi=丁基锂
Cat=催化量
CDMT=2-氯-4,6-二甲氧基三嗪
CH2Cl2=二氯甲烷
CH3CN=乙腈
CH3I=碘甲烷
CHN分析=碳/氢/氮元素分析
CHNCl分析=碳/氢/氮/氯元素分析
CHNS分析=碳/氢/氮/硫元素分析
DAST=二乙基氨基三氟化硫
DCC=1,3-二环己基碳二亚胺
DCM=二氯甲烷
DIBAL=氢化二异丁基铝
DIEA=二异丙基乙基胺
DI水=去离子水
DMA=N,N-二甲基乙酰胺
DMAC=N,N-二甲基乙酰胺
DMAP=4-(N,N-二甲基氨基)吡啶
DMF=N,N-二甲基甲酰胺
DSC=二琥珀酰基碳酸酯
EDCI=1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐
Et=乙基
Et2O=乙醚
Et3N=三乙胺
EtOAc=乙酸乙酯
EtOH=乙醇
FABMS=快速原子轰击质谱
g=克
GIHA=间胍基马尿酸
GIHA HCl=间胍基马尿酸盐酸盐
G1y=甘氨酸
HMPA=六甲基磷酰胺
HOBT=1-羟基苯并三唑水合物
HPLC=高效液相色谱法
IBCF=氯甲酸异丁酯
i-Pr=异丙基
i-Prop=异丙基
K2CO3=碳酸钾
KMnO4=高锰酸钾
KOH=氢氧化钾
KSCN=硫氰酸钾
L=升
LiOH=氢氧化锂
MCPBA=间-氯过苯甲酸
Me=甲基
MeI=碘甲烷
MeOH=甲醇
MEMCl=甲氧基乙氧基甲基氯
MesCl=甲磺酰氯
mg=毫克
MgSO4=硫酸镁
ml=毫升
mL=毫升
MS=质谱
MTBE=甲基叔丁基醚
N2=氮
NaCNBH3=氰基硼氢化钠
NaH=氢化钠
NaHCO3=碳酸氢钠
NaOH=氢氧化钠
NaOMe=甲醇钠
Na2PO4=磷酸钠
Na2SO4=硫酸钠
NEt3=三乙胺
NH4HCO3=碳酸氢铵
NH4 +HCO2 -=甲酸铵
NH4OH=氢氧化铵
NMM=N-甲基吗啉
NMP=1-甲基-2-吡咯烷酮
NMR=核磁共振
Pd/C=披钯炭
Ph=苯基
Pt/C=披铂炭
RPHPLC=反相高效液相色谱法
RT=室温
t-BOC=叔丁氧基羰基
TFA=三氟乙酸
THF=四氢呋喃
TLC=薄层层析法
TMEDA=四甲基亚乙基二胺
TMS=三甲基甲硅烷基
△=加热反应混合物
如同在式Ⅰ-Ⅵ中显示的化合物能够以多种异构形式存在并且意指包括所有这样的异构形式。也包括互变异构形式以及这样的异构体和互变异构体的药学上可接受的盐。
在此所述结构和式中,穿过环的键所画出的键可为环上任何可利用的原子。
术语“药学上可接受的盐”指的是通过使式Ⅰ化合物与其一般考虑适宜于人体消耗的酸的阴离子接触来制备的盐。药学上可接受的盐的实例包括盐酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、磷酸盐、乙酸盐、丙酸盐、乳酸盐、马来酸盐、苹果酸盐、琥珀酸盐、酒石酸盐等。可通过常规方法制备所有的药学上可接受的盐。(用于药学上可接受的盐的另外的实例参见Berge等,J.Pharm.Sci.,66(1),1-19(1977))。
为选择性抑制或拮抗αvβ3整联蛋白,本发明化合物可以其包括常规药学上可接受的载体、辅助剂和溶媒的单位剂型经口服、胃肠外给药或通过吸入喷雾或局部给药。在此使用的术语胃肠外包括例如皮下、静脉、肌内、胸骨内、输注技术或腹膜内。
本发明化合物通过以适合于所述途径的药用组合物的形式和以用于治疗的有效剂量的任何适宜的途径给药。使用对医学领域熟悉的临床前和临床方法,本领域普通技术人员易于确定预防或阻止疾病的发展或治疗疾病所需要的治疗有效剂量的化合物。
因此,本发明提供治疗由选择性抑制或拮抗αvβ3细胞表面受体介导的疾病的方法,该方法包括给予治疗有效量的选自式Ⅰ-Ⅵ中阐明的这类化合物的化合物,其中一种或多种式Ⅰ-Ⅵ化合物与一种或多种非毒性的、药学上可接受的载体和/或稀释剂和/或辅剂(总称“载体”材料)和如果需要的话的其它活性成分一起给药。更准确地讲,本发明提供用于抑制αvβ3细胞表面受体的方法。本发明最优选提供用于抑制骨吸收、治疗骨质疏松症、抑制恶性肿瘤体液高钙血症、治疗培吉特病、抑制肿瘤转移、抑制瘤形成(实体肿瘤生长)、抑制血管发生包括肿瘤血管发生、治疗视网膜病包括黄斑变性和糖尿病性视网膜病、抑制关节炎、牛皮癣和牙周病和抑制平滑肌细胞迁移包括再狭窄的方法。
基于本领域技术人员熟知和理解的标准实验室实验技术和方法以及与已知用途的化合物相比较,式Ⅰ化合物能够用于治疗患有以上病理疾病的患者。本领域技术人员将认识到选择最适当的本发明化合物在具有本领域一般技术的人的能力范围内并且将依多种因素包括在标准试验和动物模型中得到的结果来确定。
治疗受到所述病理症状之一困扰的患者的方法包括给予这样的患者一定量的式Ⅰ化合物,其在所述疾病的控制或者延长患者的存活率中是有效的,超出不采用该治疗方法所能期待的存活率。如同在此使用的术语疾病的“抑制”指的是延缓、中断、阻止或停止疾症而不必表明全部消除所述疾病。人们相信除了自身的明显有利作用之外,延长患者的存活率也表明所述疾病被有利地控制到某种程度。
如同先前陈述的那样,能够在多种生物、预防或治疗领域中使用本发明化合物。打算把这些化合物用于预防或治疗其中αvβ3整联蛋白起作用的任何疾病或者症状。
用于化合物和/或含有这些化合物的组合物的剂量方案基于多种因素,包括患者的类型、年龄、体重、性别和医疗条件、疾病的严重程度、给药途径和所使用特定的化合物的活性。因此,剂量方案可变化很大。剂量水平从每天每公斤体重大约0.01至大约1000mg来用于治疗上述的疾病。
将经注射给予的活性成分配制为其中例如盐水、葡萄糖或水可用作适宜的载体的组合物。依以上列出的因素而定,适宜的每日剂量一般为每日以多剂量注射大约0.01至10mg/kg体重。
为给予需要这样治疗的哺乳动物,一般将治疗有效量的所述化合物与一种或多种适宜于所指明给药途径的辅剂混合。化合物可与乳糖、蔗糖、淀粉粉末、链烷酸纤维素酯、纤维素烷基酯、滑石粉、硬脂酸、硬脂酸镁、氧化镁、磷酸和硫酸的钠和钙盐、明胶、阿拉伯胶、藻酸钠、聚乙烯吡咯烷酮和/或聚乙烯醇混合,并且压片或包封以便于给药。或者可把化合物溶于水、聚乙二醇、丙二醇、乙醇、玉米油、棉籽油、花生油、芝麻油、苯甲醇、氯化钠和/或多种缓冲剂中。其它的辅剂和给药模式为药学领域中广泛熟知的。
用于本发明中的药用组合物可经受常规药用操作例如灭菌和/或可含有常规药用辅剂例如防腐剂、稳定剂、润湿剂、乳化剂、缓冲剂等。
制备用于本发明的化合物的一般合成顺序列于流程1-15中。对于本发明的多种方面的解释和实际方法两者被适当描述。以下流程和实施例仅打算阐明本发明,而不限制它们的范围或精神。本领域技术人员将易于理解在所述流程和实施例中描述的条件和方法的已知的变化形式能够用于合成本发明化合物。
除非另外指出,所使用的所有起始原料和装置可市售得到。
流程1
流程2
流程3
流程1-3阐明用于制备其用于偶合到甘氨酰-β-氨基酸部分的本发明胍基吡啶/环胍基吡啶羧酸部分的方法。这可以使用本领域技术人员已知的其它适当的胍基化(guanidating)试剂来完成。使用常规技术和方法能够改进流程1-3的方法以制备用于偶合到甘氨酰-β-氨基酸部分的另外的化合物。
流程4
流程4阐明用于制备其用于偶合甘氨酰-β-氨基酸部分的本发明的胍基噻唑羧酸部分的方法。使用本领域技术人员已知的常规技术能够改进流程4的方法以制备用于偶合到甘氨酰-β-氨基酸部分的这些和另外的化合物。
流程5
流程5阐明用于制备本发明化合物的N-甘氨酰-氨基-3-(3,5-二卤代-2-羟基)苯基丙酸乙酯部分的方法。简言之,通过直接卤代制备3,5-卤代的水杨醛(卤代-2-羟基苯甲醛)。例如,将5-溴代水杨醛在乙酸中浆化并加入等当量或更多量的氯,以生成3-氯-5-溴-2-羟基苯甲醛。
一些产物沉淀出来并经过滤回收。经用水稀释滤液并分离沉淀,回收剩余部分。合并固体并干燥,得到3-氯-5-溴-2-羟基苯甲醛。通过在DMF中使5-氯水杨醛与N-碘代琥珀酰亚胺反应并使该反应混合物经受常规处理条件,制备3-碘代-5-氯代水杨醛。能够通过在乙腈中使5-溴代水杨醛与碘化钾和氯胺T反应能够制备3-碘代-5-溴代水杨醛。常规处理得到物料,当用己烷处理时,得到要求的3-碘代-5-氯代水杨醛。
使用改进的佩金反应(例如,Vogel’s Textbook of Practical OrganicChemistry,第5版,1989,1040页),从水杨醛易于制备香豆素类。将卤代的香豆素类转化为3-氨基氢化香豆素类(参见J.G.Rico,四面体快报,1994,35,6599-6602),其在酸性醇中易于开环,得到3-氨基-3-(3,5-卤代-2-羟基)苯基丙酸酯。
通过Boc-N-甘氨酰-N-羟基琥珀酰亚胺的反应,把3-氨基-3-(3,5-卤代-2-羟基)苯基丙酸酯转化为N-甘氨酰-3-氨基-3(3,5-卤代-2羟基苯基丙酸酯,得到Boc-N-甘氨酰-3-氨基-3-(3,5-卤代-2-羟基苯基丙酸酯,将其转化为N-甘氨酰-3-氨基-3-(3,5-卤代-2-羟基)苯基丙酸酯的HX盐(其中X为Cl、Br或I)。
流程6
流程6阐明用于使杂环酸部分(A1-A13)偶合于本发明的甘氨酰-β-氨基部分(C)的方法。
在流程1-4中阐明A1-A13的合成并且在流程5(其中X和Y为相同的或不同的卤素)中阐明(C)的合成。
使用本领域普通技术人员已知的常规方法,能够进一步改进这样的方法。
流程7
步骤A
步骤B
流程7阐明以通法制备分子(F)的甘氨酰-β-氨基酸部分的方法。
在这个方法中使用的醛(R’CHO)可市售得到或者能够使用本领域普通技术人员通常已知的用于制备醛的方法由市售得到的试剂来制备。
所有其它的试剂可市售得到或者能够易于由本领域技术人员合成。
使用常规技术,能够进一步改进这样的方法和条件以生成相似要求的中间体。
流程8
流程8阐明用于使杂环部分(A1-13)偶合至甘氨酰-β-氨基酸部分(F)来制备本发明化合物的方法。
在流程1-4中阐明A1-13的合成并且在流程7中阐明(F)的合成。
使用本领域普通技术人员已知的常规方法,能够改进这样的方法。
流程9-12阐明制备本发明化合物的通法。
流程9
流程9阐明用于合成杂环衍生的甘氨酰-β-氨基酸偶合的目标化合物的通用方法。在肽偶合条件下,杂环羧酸与甘氨酰-β-氨基酸反应,得到中间体(2)。使用催化氢化(例如Pt/C,H2)还原硝基,得到氨基中间体(3)。使用SnCl2也可化学上实施这个转化。使用以上讨论的方法,能够将氨基精心制备以得到式Ⅰ的胍基或其它的官能团。
流程10
或者,可通过在与甘氨酰-β-氨基酸偶合之前,建立所述分子的左手部分来合成所述目标化合物(流程10)。杂环胺(1)的氨基官能度被官能化成为胍或其它基团(A,式1),然后在标准偶合条件下偶合于甘氨酰-β-氨基酸。
流程11
流程11显示取代的吡啶和吡啶酮衍生的目标化合物的合成通法。把6-羟基烟酸硝化随后经氯化,得到6-氯-5-硝基烟酸。使中间体3与甘氨酰-β-氨基酸偶合,得到产物4。在通用中间体4中的氯基团能易于由多种亲核试剂置换,得到5。如同在流程9中讨论的那样,还原4或5的硝基并进一步精心加工氨基,得到目标化合物。
流程12
流程12显示用2-氨基-6-羟基吡啶-4-甲酸起始进行取代的吡啶和吡啶酮衍生的目标化合物的合成。在本流程中使用的起始物料1可通过在高压条件下,使市售得到的2-氯-6-甲氧基-吡啶-4-甲酸与氢氧化铵反应来制备。如同在流程10中讨论的那样,精心加工氨基随后偶合于甘氨酰-β-氨基酸,得到目标化合物。
流程13
流程13显示用6-氨基-4-甲氧基-吡啶甲酸起始的异构吡啶和吡啶酮的合成。在流程13中使用的起始物料1可如同在文献(J.Am.Chem.Soc.,78,4130,1956)中描述的那样来制备。在1中的氨基进行官能化作用,随后经偶合反应和水解(如同在流程10中那样),得到目标化合物。
流程14
使用在流程14中显示的方法,可制备异构吡啶和吡啶酮衍生的化合物。通过用6-氯-吡啶甲酸开始,可制备关键中间体4。氧化随后通过硝化得到4-硝基吡啶衍生物(3)。将N-氧化物脱氧、还原硝基和亲核取代氯基团,得到中间体4。可使用在流程10中讨论的方法,以完成目标化合物的合成。
流程15
在流程10中显示的方法也能用于合成嘧啶衍生的目标化合物(流程15)。按照文献制备方法,可合成异构的嘧啶衍生物1和2(J.Org.Chem.,26,2755,1961)。
实施例A制备
将5-氨基烟酸(4.0g,0.021mole)(Helv.Chim.Acta,47,363(1964);JACS,70,2381[1948])、1H-吡唑-1-甲脒盐酸盐(4.6g,0.031mole)、二异丙基乙基胺(8.0g,0.062mole)、二噁烷(14mL)和H2O(7mL)于回流下加热2天。将反应物冷却至室温,过滤沉淀,用H2O/二噁烷(50∶50)洗涤并干燥。把该沉淀在H2O中浆化并用2NHCl酸化。真空除去溶剂,得到作为白色固体的以上化合物(750mg)。
MS和1H-NMR与要求的结构相一致。
实施例B制备步骤1
向在无水乙醇(75mL)中的3,4,5,6-四氢-2-嘧啶硫醇(10g,0.086mole)(Aldrich)中加入MeI(12.2g,0.086mole)。在回流下搅拌反应物2.5小时。真空除去溶剂并干燥产物,得到作为白色固体的2-甲基硫代-3,4,5,6-四氢-2-嘧啶氢碘酸盐(22g)。
MS和1H-NMR与要求的结构相一致。步骤2
在冰浴温度下,向在CH2Cl2(25mL)中的来自以上步骤1的产物(5.3g,O.021mole)和三乙胺(2.07g,0.021mole)中加入BOC酸酐(4.5g,0.021mole)。然后在室温下将反应物搅拌2天。用H2O(3X)洗涤CH2C12,经MgSO4干燥,并且真空除去,得到N-Boc-2-甲基硫代-3,4,5,6-四氢-2-嘧啶(4.14g)。
MS和1H-NMR与要求的结构相一致。步骤3
在80-85℃下,将在DMA(12mL)中的来自以上步骤2的产物(3.08g,0.0134mole)、5-氨基烟酸(1.8g,0.0134mole)加热两周。用CH3CN稀释该反应混合物,过滤沉淀,用CH3CN洗涤并干燥。将该沉淀在H2O中浆化并用浓HCl使pH降至1-2。把溶液冷冻并冷冻干燥,得到作为淡棕色固体的要求的产物(1.2g)。MS和1H-NMR与要求的结构相一致。
实施例C制备 步骤1
步骤1
在冰浴温度下,向在CH2Cl2(100mL)中的2-甲硫基-2-咪唑啉氢碘酸盐(20g,0.082mole)(Aldrich)和三乙胺(8.28g,0.082mole)中加入BOC酸酐(17.9g,0.082mole)。在室温下,将反应物搅拌过夜。以H2O(2x)洗涤CH2Cl2,经MgSO4干燥,并且真空除去,得到转化为蜡状白色固体的粘稠油的N-BOC-2-甲硫基-2-咪唑啉(15.93g)。
MS和1H-NMR与要求的结构相一致。步骤2
在100℃下,将在DMA(60mL)中的来自以上步骤1的产物(15.93g,0.0737mole)、5-氨基烟酸(9.6g,0.07mole)和三乙胺(7.1g,0.07mole)加热两天,然后在130℃下加热1天,并在150℃下加热另外2天。冷却后并用CH3CN稀释该反应混合物,过滤沉淀,用乙醚洗涤并干燥(两性离子产量为10.16g)。将其在H2O中浆化并用TFA酸化(pH1-2)。把溶液冷冻并冷冻干燥,得到淡黄色固体的要求的产物(20.15g)。
MS和1H-NMR与要求的结构相一致。
实施例D制备 步骤1制备
步骤1制备
向配置机械搅拌器和冷凝器的2L圆底烧瓶中加入3,5-二氯水杨醛(200.0g,1.05mol,1当量)、乙酸酐(356g,3.49mol)和三乙胺(95.0g,0.94mol,0.90当量)。将该反应溶液回流下加热过夜。把暗棕色反应混合物冷却至50℃并伴随搅拌下加入水(1L)。1小时后,过滤混合物并把滤液与EtOH(1L)混合。将该混合物加热至45℃一小时,冷却至室温,过滤并用EtOH(0.5L)洗涤固体(部分A)。经旋转蒸发浓缩合并的EtOH溶液至产生油(部分B)。将来自部分A的固体溶于二氯甲烷(1.5L)中并把生成的溶液通过硅胶垫(1300mL体积)。将生成的暗棕色溶液浓缩至油,该油用己烷(1.3L)研磨,得到固体,经过滤分离并洗涤(己烷),得到基本上纯的6,8-二氯香豆素(163g)。经用相似方法处理油(部分B),得到另外31g产物;将油溶于二氯甲烷(0.5L)中,通过硅胶垫(0.5L体积)并用己烷研磨。总分离收率为194g或86%的棕色固体。
MS和1H-NMR与要求的产物相一致。步骤2制备
向配置机械搅拌器的2L三口圆底烧瓶中加入在步骤1中制备的6,8-二氯香豆素(160g,0.74mol)和干燥THF(375mL,Aldrich SureSeal)。将生成的混合物冷却至-40℃(干冰-丙酮浴)并加入双(三甲基甲硅烷基)氨化锂(O.80mol,800ml的1M在THF中的溶液),其间维持温度在-40℃以下。加入完成后,撤除冷却浴。在0.5小时后,使该混合物温热至-5℃。通过加入在EtOH(1.25L)中的HCl溶液(0.5L的4M在二噁烷中的溶液)猝灭。把温度维持在0℃以下过夜。将反应混合物浓缩至其原来体积的大约一半并在EtOAc(3L)和水(2L)之间分配。用HCl水溶液(3×1L,0.5NHCl)洗涤有机层。经加入10%的NaOH水溶液把合并的水层的pH调至大约7并用二氯甲烷(3×2L)提取。干燥(MgSO4)合并的有机层,过滤,并伴随搅拌下加入HCl(210mL的4M在二噁烷中的溶液)。沉淀完全后,过滤除去固体。将滤液浓缩至小的体积并加入甲基叔丁基醚。将得到的固体与最初形成的固体合并,且用甲基叔丁基醚洗涤合并的产物,过滤分离并干燥(真空箱中放置过周末),得到要求的产物(172g,74%收率)。
MS和1H-NMR与要求的产物相一致。步骤3制备
在惰性气氛(Ar)中,向配置磁力搅拌棒的火焰干燥的圆底烧瓶(0.5L)中加入N-叔丁氧基羰基-甘氨酸N-羟基琥珀酰亚胺酯(Sigma,15.0g,0.055mol)、干燥的DMF(Aldrich Sure Seal,200mL)和来自步骤2的产物(21.67g,0.055mol)。将反应混合物冷却至大约0℃(盐-冰浴)并加入N-甲基吗啉(5.58g,0.056mole)和催化量的DMAP。使反应进行过夜。将反应混合物浓缩至脂膏并在EtOAc(0.4L)和碱水溶液(2×0.2L,饱和的NaHCO3水溶液)之间分配。有机层用枸橼酸水溶液(2×0.2L,10%w/v)、碳酸氢钠水溶液(2×0.2L)、盐水依次洗涤并干燥(Na2SO4)。在55℃下,真空下除去挥发物,得到油(22.5g,92%收率),其放置后固化。
MS和1H-NMR与要求的产物相一致。步骤4制备
使用以下方法,将在步骤3中得到的产物脱除保护,得到所述胺盐酸盐。向在配备搅拌棒的火焰干燥的圆底烧瓶(0.1L)中的步骤3中得到的产物(14.0g,0.032mole)中加入干燥的二噁烷(40mL)。在0℃下,向该溶液中加入HCl(4.0N在二噁烷中,2当量,6.32mL)。使反应进行直到气体释放停止并且反应完全。真空除去挥发物并且用乙醚(50mL)研磨残余物。过滤收集固体,用乙醚洗涤并干燥,得到要求的产物(12.5g)。
MS和1H-NMR与要求的产物相一致。实施例E制备步骤1制备
向3-溴-5-氯水杨醛(175g,743.2mmol)在乙酸酐(280.5mL,3mol)的悬浮液中加入三乙胺(103.6mL,743.2mmol)。在回流下,将该反应物溶液加热4.5小时。冷却溶液并真空浓缩。把棕色残余物加入到无水乙醇(730mL)中。在0℃下,将混合物贮存14小时。经过滤收集棕色固体并用冷的乙醇洗涤。真空干燥固体,得到要求的产物(123g,64%收率)。1H-NMR与提出的结构相一致。步骤2制备
在-76℃下,向步骤1中生成的香豆素(40.0g,154.1mmol)在THF(400mL)的悬浮液中伴随搅拌下滴加入双(三甲基甲硅烷基)氨化锂(154.1mL的1M在THF中的溶液)。10分钟内加入完成。然后,将该反应混合物搅拌5分钟,温热至-20℃并搅拌15分钟。在5分钟内,向该溶液中加入在THF(28mL)中的乙酸(9.25g,154.1mmol)。将混合物温热至室温并真空除去挥发物。把残余物溶于乙醚(850mL),用饱和的NaHCO3水溶液(2×100mL)、盐水(2×40mL)洗涤并干燥(MgSO4)。将乙醚溶液浓缩至大约160mL,并且冷却至0℃。向该悬浮液中加入HCl(4M在二噁烷中,56.3mL,225mmol)并在0℃下将该混合物搅拌30分钟。过滤悬浮液并用乙醚彻底洗涤滤饼。真空干燥固体,得到作为HCl盐的要求的产物,二噁烷溶剂合物(45g)。1H-NMR与提出的结构相一致。步骤3制备
于10分钟内向步骤2中生成的内酯(142.2g,354.5mmol)在无水乙醇(533mL)中的悬浮液中加入HCl(4M在二噁烷中,157.8mL,631.1mmol)。在室温下,将该反应混合物搅拌2.5小时。真空除去挥发物。把残余物溶于乙酸乙酯(450ml)中并在0℃下使该溶液保持15小时。经过滤收集褐色沉淀并用冷的乙酸乙酯洗涤。真空干燥固体,得到作为盐酸盐的要求的产物(100.4g,79%收率)。1H-NMR与提出的结构相一致。步骤4制备
在惰性气氛(Ar)中,向配置磁力搅拌棒的火焰干燥的圆底烧瓶(0.1L)中加入N-叔丁氧基羰基-甘氨酸N-羟基琥珀酰亚胺酯(Sigma,2.72g,0.010mol)、干燥的THF(Aldrich Sure Seal,50mL)和步骤3的产物(3.10g,0.01mol,经P2O5真空干燥过夜)。将反应混合物冷却至大约0℃(盐-冰浴)并加入三乙胺(1.01g,0.010mole)。使反应进行过夜。将反应混合物浓缩至半固体并用与实施例A步骤3相似的方法处理。在55℃下,从有机层中真空除去挥发物,得到油(4.0g,83%收率),其放置后固化。
MS和1H-NMR与要求的产物相一致。步骤5制备
使用以下方法,将在步骤4中得到的产物脱除保护,得到所述盐酸盐。向在配备搅拌棒的火焰干燥的圆底烧瓶(0.1L)中的步骤4中得到的产物(4.0g,0.0084mole)中加入干燥的二噁烷(20mL)。向该溶液中加入HCl(4N在二噁烷中,20mL),并使反应进行直到气体逸出停止并且反应完全(大约1小时)。真空除去挥发物并且用乙醚(50mL)研磨残余物。过滤收集固体,用乙醚洗涤并干燥,得到淡棕色固体(2.7g,78%收率)。
MS和1H-NMR与要求的产物相一致。
实施例F制备步骤1制备3-碘代-5-氯水杨醛
将N-碘代琥珀酰亚胺(144.0g,0.641mole)加入到5-氯水杨醛(100g,0.638mole)在二甲基甲酰胺(400mL)中的溶液中。在室温下,将该反应混合物搅拌2天。另外加入N-碘代琥珀酰亚胺(20.0g)并继续搅拌另外2天。用乙酸乙酯(1L)稀释反应混合物,用盐酸(300mL,0.1N)、水(300mL)、硫代硫酸钠(5%,300mL)、盐水(300mL)洗涤,干燥(MgSO4)并浓缩至干,得到浅黄色固体的要求的醛(162g,90%收率)。
1H-NMR和MS与要求的产物相一致。步骤2制备6-氯-8-碘代香豆素
在回流下,将3-碘代-5-氯水杨醛(100g,0.354mole)、乙酸酐(300mL)和三乙胺(54mL)的混合物加热18小时。冷却下,要求的香豆素作为深棕色结晶物料沉淀出来。过滤沉淀,用己烷/乙酸乙酯(4∶1,200mL)洗涤并且空气干燥[产量:60g(55%收率)]。贮存后,从滤液中能够得到另外量的要求的产物(10g,9%收率)。
1H-NMR和MS与要求的产物相一致。步骤3制备(R,S)-4-氨基-3,4-二氢-6-氯-8-碘代香豆素盐酸盐
在-78℃下,将六甲基二硅氮烷锂(21.62mL,1M,21.62mmol)加入到6-氯-8-碘代香豆素(6.63g,21.62mmol)在四氢呋喃(100mL)中的溶液中。在该温度下,将反应混合物搅拌30分钟,然后在0℃下搅拌1小时。将乙酸(1.3g,21.62mmol)加入到反应混合物中。把反应混合物倾入到乙酸乙酯(300mL)和饱和的碳酸钠溶液(200mL)中。分离有机层,在0℃下,用盐水(200mL)、二噁烷/HCl(4N,30mL)先后洗涤。在室温下,将反应混合物搅拌1小时,过滤,并真空干燥,得到作为粉末的要求的产物(4.6g,59%收率)。(反相高效液相色谱法:Rf6.8分钟。15分钟内梯度10%乙腈-90%乙腈,然后下一个6分钟100%乙腈。水和乙腈两者含有O.1%TFA。Vydac C18蛋白肽柱,2mL/min流速,在254nm下监控)。
1H-NMR和MS与要求的产物相一致。步骤4制备(R,S)-乙基3-氨基-3-(5-氯-2-羟基-3-碘代)苯基丙酸酯盐酸盐
将氯化氢气体鼓泡进入4-氨基-3,4-二氢-6-氯-8-碘代香豆素盐酸盐(22.0g,61.09mmol)在乙醇(250mL)中的溶液中,把该反应混合物维持在0-10℃下直到饱和。回流6小时后,经蒸馏除去大多数溶剂。将冷却的残余物加入到无水乙醚中并搅拌2小时。初始的胶状物转变为结晶物料。过滤结晶产物并干燥,得到作为灰白色结晶粉末的要求的产物(20g,81%收率)。(Rf.7.52分钟。条件如同步骤3)。
1H-NMR和MS与要求的产物相一致。步骤5制备(R,S)-乙基3-(N-叔丁氧基羰基-甘氨酰)-氨基-3-(5-氯-2-羟基-3-碘代)苯基丙酸酯
在0℃下,将叔丁氧基羰基-甘氨酸(2.16g,12.31mmol)、HOBT(1.67g,12.31)、EDCl(2.36g,12.31mmol)和DMF(50mL)的混合物搅拌1小时。将3-氨基-3-(5-氯-2-羟基-3-碘代)丙酸乙酯盐酸盐(5.0g,12.31mmol)加入到反应混合物中,随后加入三乙胺(3.5mL)。在室温下,将该反应混合物搅拌18小时。真空除去DMF并且残余物在乙酸乙酯(300mL)和碳酸氢钠(200mL)之间分配。用盐酸(1N,100mL)、盐水(200mL)洗涤有机层,干燥(MgSO4)并浓缩,得到作为固体的要求的产物(6g,93%收率)。
1H-NMR和MS与要求的产物相一致。步骤6制备(R,S)-乙基3-(N-甘氨酰)-氨基-3-(5-氯-2-羟基-3-碘代)苯基丙酸酯盐酸盐
在0℃下,将二噁烷/HCl(4N,20mL)加入到乙基3-(N-叔丁氧基羰基-甘氨酰)-氨基-3-(5-氯-2-羟基-3-碘代)丙酸酯(6.0g,11.30mmol)中并在室温下搅拌3小时。将该反应混合物浓缩并在加入甲苯(100mL)后再次浓缩。把得到的残余物悬浮于乙醚中,过滤并干燥,得到结晶粉末的要求的产物(5.0g,95%收率)。(反相高效液相色谱法:Rf8.3分钟。条件如同步骤3)。
1H-NMR和MS与要求的产物相一致。
实施例G制备 步骤1制备雷佛马茨基试剂
步骤1制备雷佛马茨基试剂
BrZnCH2CO2-t-Bu
把锌金属(180.0g,2.76mol,30-100目)和THF(1.25L)放入配备冷凝器、温度探头和机械搅拌器的4L烧瓶中。搅拌期间,借助加料管加入1,2-二溴乙烷(4.74mL,0.06mol)[或者,可替代为TMSCl(0.1当量)在室温下1小时]。在惰性气体(3N2/真空循环)清洗之后,将锌在THF中的悬浮液加热至回流(65℃)并维持这个温度达1小时。在将混合物冷却至50℃之后,于1.5小时内借助50mL加料管和加料泵(传递设置为4.1mL/min)加入溴代乙酸叔丁酯(488g,369mL,2.5mol)。整个加入期间维持反应温度50°+/-5℃。加入完成后,在50℃下,使反应混合物搅拌一小时。随后,使该混合物冷却至25℃并使沉淀出来的产物沉降。使用粗玻璃料的滤棒将THF母液倾析到2L圆底烧瓶中并且部分真空转移(20mmHg)。从该混合物中除去大约65%的THF。加入1-甲基-2-吡咯烷酮(NMP,800mL)并继续搅拌5分钟。过滤反应混合物以除去任何残留的锌。分析表明要求的雷佛马茨基试剂的滴定度为1.57M,具有摩尔收率94%。或者经过滤能够把固体试剂与原来的反应混合物分离。用THF洗涤滤饼直到得到白色固体并在N2下干燥,得到作为一THF溶剂合物的要求的产物,其可在-20℃(干燥)下贮存用于以下阶段。一般回收率为85-90%。步骤2A.制备 在室温下,将碳酸钾(粉末,烘箱中在100℃下真空干燥,8.82g,60mmoles)加入到3,5-氯水杨醛(11.46g,60moles)在DMF(40ml)中的溶液中,得到亮黄色浆状物。然后加入MEMCl(纯品,7.64g,61mmoles),其间维持浴温在20℃。在22℃下将混合物搅拌6小时并加入MEMCl(0.3g,2.4mmoles)。将混合物搅拌另外0.5小时并将反应混合物倾入到冷水(200mL)中,以沉淀出产物。在压力滤器上过滤该浆状物并用水(2×50mL)洗涤滤饼,并于N2下真空干燥,得到灰白色固体的产物(14.94g,89%收率)。1H-NMR(CDCl3,TMS)3.37(s,3H),3.54-3.56(m,2H),3.91-3.93(m,2H),5.30(s,2H),7.63(d,1H),7.73(d,1H),10.30(s,1H);13C NMR(CDCl3,TMS)δ(ppm):59.03,70.11,99.57,126.60,129.57,130.81,132.07,135.36,154.66,188.30。DSC:48.24℃(内90.51J/g);微量分析:对C11H12Cl2O4的
在室温下,将碳酸钾(粉末,烘箱中在100℃下真空干燥,8.82g,60mmoles)加入到3,5-氯水杨醛(11.46g,60moles)在DMF(40ml)中的溶液中,得到亮黄色浆状物。然后加入MEMCl(纯品,7.64g,61mmoles),其间维持浴温在20℃。在22℃下将混合物搅拌6小时并加入MEMCl(0.3g,2.4mmoles)。将混合物搅拌另外0.5小时并将反应混合物倾入到冷水(200mL)中,以沉淀出产物。在压力滤器上过滤该浆状物并用水(2×50mL)洗涤滤饼,并于N2下真空干燥,得到灰白色固体的产物(14.94g,89%收率)。1H-NMR(CDCl3,TMS)3.37(s,3H),3.54-3.56(m,2H),3.91-3.93(m,2H),5.30(s,2H),7.63(d,1H),7.73(d,1H),10.30(s,1H);13C NMR(CDCl3,TMS)δ(ppm):59.03,70.11,99.57,126.60,129.57,130.81,132.07,135.36,154.66,188.30。DSC:48.24℃(内90.51J/g);微量分析:对C11H12Cl2O4的
计算值:C:47.33%;H:4.33%;Cl:25.40%;
实测值:C:47.15%;H:4.26%;Cl:25.16%。B.制备
将来自步骤2A的产物(35.0g,0.125mol)置于配备机械搅拌器和加料漏斗的1L三口圆底烧瓶中,随后加入THF(200mL)。在22℃下,搅拌该溶液并立即加入(S)-苯基甘氨醇(17.20g,0.125mol)。在22℃下30分钟后,加入MgSO4(20g)。在22℃下,将该混合物搅拌1小时并在粗玻璃料滤器上过滤。减压下浓缩滤液,无需进一步纯化,粗品亚胺直接用于偶合反应步骤2的C中。C.制备
在氮气下,将在步骤1中得到的固体试剂(91.3g,0.275mol)和NMP(200mL)加入配备机械搅拌器和加料漏斗的1L三口圆底烧瓶中。然后将该溶液冷却至-10℃并在350rpm下搅拌。在氮气下,制备亚胺(在步骤B中制备)在NMP中的溶液,然后于20分钟内加入到以上反应混合物中,其间温度维持在-5℃(夹套温度-10℃)。加入完成后,在-8℃下,将该混合物搅拌另外1.5小时并在-5℃下搅拌1小时。冷却至-10℃之后,将浓HCl/饱和的NH4Cl溶液(100mL)、水(100mL)和盐水(100mL)的混合物加入到所述溶液中。加入MTBE(200mL)并在23℃下以200rpm的转速将该混合物搅拌15分钟。停止搅拌并分层。用MTBE(100ml)提取水层。合并两个有机层,依次用饱和的NH4Cl溶液(100ml)、水(100ml)和盐水(100ml)洗涤。用MgSO4(30g)干燥溶液,过滤并浓缩,如同质子和碳NMR测量的那样,得到含有作为单一非对映异构体的要求的产物的橙色的油(66.3g)(放置固化)。经从庚烷重结晶,得到作为灰白色固体的产物,将样品纯化用于分析。质子和碳NMR和IR谱与要求的产物一致。[α]D 25=+8.7°(c=1.057,MeOH)。微量分析:对C25H33Cl2NO6的计算值:C:58.77%;H:6.47%;N:2.72%;Cl:13.78%;实测值:C:58.22%;H:6.54%;N:2.70%;Cl:13.66%。步骤3制备A.将在步骤2中制备的粗品酯[17.40g,0.033mole(理论)]和EtOH(250mL)的溶液加入到1L的三口夹套反应器中。将该溶液冷却至0℃并立即加入Pb(OAc)4(14.63g,0.033mole)。2小时后,加入15%的NaOH溶液(30mL)并减压下除去乙醇。加入另一份15%的NaOH(100mL)并用MTBE(2×100mL)提取混合物,以水(2×100mL)和盐水(50mL)洗涤,用Na2SO4干燥,硅藻土过滤并减压下浓缩,得到橙色的油(12.46g),其经tlc测定为纯的。无须进一步纯化即可使用所述油。B.用EtOH(30mL)稀释来自A的油并加入对甲苯磺酸(1.3当量,0.043mole,8.18g)。将该溶液加热回流8小时,冷却至环境温度并减压下浓缩。用THF(20mL)处理残余物并加热回流以形成溶液。将该溶液冷却至室温并且该化合物结晶。加入庚烷(30mL)和THF(10mL)以形成流体浆状物,使其过滤。用THF/庚烷(40mL,1/1)洗涤滤饼,在氮气氛下,于压力滤器上真空干燥2小时,得到白色固体(7.40g)。
质子和碳NMR和IR谱与作为基本上单一的对映体的要求的产物相一致。
微量分析:对C18H21Cl2NO6S,0.25C4H8O的计算值:C:48.73%;H:4.95%;N:2.99%;Cl:15.14%;实测值:C:48.91%;H:4.95%;N:2.90%;Cl:14.95%。步骤4制备
向配备磁力搅拌棒和氮气通气管的500mL圆底烧瓶中加入来自步骤3的产物(21.7g,0.065mole)、N-叔丁氧基羰基-甘氨酸N-羟基琥珀酰亚胺酯(17.7g,0.065mole)和DMF(200mL)。在室温下,于氮气中将反应混合物搅拌3.25小时并形成浅橙色溶液。将反应混合物倾入到冰冷的乙酸乙酯(1.2L)中。用1M HCl(250mL)洗涤有机溶液,然后用盐水(500mL)洗涤,干燥(MgSO4)并真空浓缩几近至干,得到油,随后将其在50℃下干燥,得到无色油的产物(28.12g,99%收率)。从乙酸乙酯/己烷中制备晶种。把产物(大约28g)溶于乙酸乙酯(35mL)和己烷(125mL)中。以晶种接种该溶液并形成沉淀。过滤固体并在55℃下真空干燥过夜,得到无色固体(27.0g,95%收率)。1H-NMR与要求的产物相一致。步骤5制备
经P2O5和NaOH小片将在步骤4中制备的叔丁氧基羰基保护的甘氨酰胺(27.0g,0.062mole)干燥过夜。将该固体溶于二噁烷(40mL)中并把溶液冷却至0℃。加入等量体积的4N HCl/二噁烷(0.062mole)并将反应进行2小时。如同反相高效液相色谱法测量的那样,在此时的转化率为80%。4小时内使反应混合物温热至室温。在40℃下,将该反应混合物浓缩至泡沫,其用乙醚(200mL)研磨。过滤形成的白色固体并经P2O5干燥,得到作为盐酸盐的要求的甘氨酸β-氨基酸乙酯化合物(20.4%,88.5%分离收率)。
1H-NMR和MS与所述结构相一致。实施例H制备 步骤1制备
步骤1制备
在室温下,将碳酸钾(粉末,烘箱中在100℃下真空干燥,22.1g,0.16moles)加入到3-氯-5-溴代水杨醛(35g,0.15moles)在DMF(175ml)中的溶液中,得到亮黄色浆状物。然后维持浴温在20℃下,加入MEMCl(纯品,25.0g,0.2moles)。然后在22℃下将混合物搅拌6小时并倾入DI水(1200mL)中,沉淀出产物。在压力滤器上过滤该浆状物,用DI水(2×400mL)洗涤滤饼并在N2/真空下干燥,得到灰白色固体的产物(46g,95%收率)。1H-NMR(CDCl3,TMS)3.35(s,3H),3.54-3.56(m,2H),3.91-3.93(m,2H),5.30(s,2H),7.77(d,lH),7.85(d,1H),10.30(s,1H);13C NMR(CDCl3,TMS)(ppm):59.05,70.11,71.49,99.50,117.93,129.69,129.78,132.37,138.14,155.12,188.22。DSC:48.24℃(内90.51J/g);微量分析:对C11H12BrClO4的
计算值:C:40.82%;H:3.74%;Cl:10.95%;Br:24.69%;
实测值:C:40.64%;H:3.48%;Cl:10.99%;Br:24.67%。步骤2制备
将在步骤1中得到的产物(32.35g,0.1mol)加入配备机械搅拌器的500ml的三口圆底烧瓶中,随后加入THF(160ml)和(S)-苯基甘氨醇(13.71g,0.1mol)。在22℃下30分钟后,加入MgSO4(20g)。在22℃下,将该混合物搅拌1小时并在粗玻璃料滤器上过滤。减压下浓缩滤液,得到含有亚胺的浅黄色油(48.0g)。无需进一步纯化,该粗品产物直接用于偶合反应。微量分析:对C19H21BrClNO4的计算值:C:51.54%;H:4.78%;N:3.16%;Br:18.04%;Cl:8.00%;实测值:C:51.52%;H:5.02%;N:2.82%;Br:16.31%;Cl:7.61%。步骤3制备
在配备机械搅拌器的5L三口圆底烧瓶中,于氮气下,将来自实施例G的步骤1的试剂(332g,0.8mol)溶于NMP(660mL)。然后将该溶液冷却至-10℃。在氮气下,制备步骤2中生成的亚胺在NMP(320mL)中的溶液,然后在30分钟内将其加入到以上反应混合物中,其间温度维持在-5℃。将该混合物搅拌另外1小时,然后冷却至-10℃。于10分钟内加入浓HCl/饱和的NH4Cl溶液(30mL/720mL)的混合物。加入MTBE(760mL)并在23℃下将该混合物搅拌1小时。停止搅拌并分层。用MTBE(320ml)提取水层。合并两个有机层,依次用饱和的NH4Cl溶液(320ml)、DI水(320ml)和盐水(320ml)洗涤。用MgSO4(60g)干燥溶液,过滤并浓缩,得到含有作为单一对映异构体的要求的产物的黄色的油(228g)。DSC:227.54℃(内61.63J/g)微量分析:对C25H33BrClNO6的计算值:C:53.72%;H:5.95%;N:2.50%;Br:14.29%;Cl:6.33%;实测值:C:53.80%;H:6.45%;N:2.23%;Br:12.85%;Cl:6.12%。步骤4制备
于氮气氛下,将来自步骤3的粗品酯(~111g)在乙醇(1500mL)中的溶液加入到配备机械搅拌器的3L三口圆底烧瓶中。将该反应混合物冷却至0℃并一次性加入四乙酸铅(88.67g,0.2mol)。在0℃下,将该反应混合物搅拌3小时,然后向5℃下的反应混合物中加入15%NaOH水溶液(150mL)。在旋转蒸发器上减压下除去乙醇。加入另外的15%NaOH水溶液(600mL)并用乙酸乙酯(2×300mL)、MTBE(2×200mL)和乙酸乙酯(2×200mL)提取反应混合物。合并有机层,并用DI水(2×200mL)和盐水(2×100mL)洗涤,经无水MgSO4(30g)干燥。然后在硅藻土上过滤该溶液并减压下浓缩,得到作为橙色的油的产物(96g),其无需进一步纯化即可用于下一步反应。DSC:233.60℃(内67.85J/g)微量分析:对C24H29BrClNO5的计算值:C:54.71%;H:5.54%;N:2.65%;Br:15.16%;Cl:6.72%;实测值:C:52.12%;H:5.40%;N:2.47%;Br:14.77%;Cl:6.48%。步骤5制备
将来自步骤4的粗品产物(~94g)溶于无水乙醇(180mL)中并加入对甲苯磺酸一水合物(50.0g,0.26mol)。然后将该反应混合物加热至回流8小时,之后减压下除去溶剂。将残留的固体以THF(100mL)处理,然后减压下除掉THF。把残余物溶于乙酸乙酯(500mL)中并冷却至~5℃。过滤生成的固体并用庚烷(2×50ml)洗涤,得到白色固体。固体经空气干燥,得到作为单一异构体的白色固体的产物(38g)。1H-NMR(DMSO,TMS)(ppm)1.12(t,3H),2.29(s,3H),3.0(m,2H),4.05(q,2H),4.88(t,1H),7.11(d,2H),7.48(d,2H),7.55(d,1H),7.68(1H,d),8.35(br,s,3H);13C-NMR(DMSO,TMS)(ppm):13.82,20.75,37.13,45.59,60.59,110.53,122.47,125.44,127.87,128.06,129.51,131.95,137.77,145.33,150.14,168.98;DSC:69.86℃(内406.5J/g),165.72℃(内62.27J/g),211.24℃(外20.56J/g),[α]D 25=+4.2°(c=0.960,MeOH);IR(MIR)(cm-1)2922,1726,1621,1591,1494,1471,1413,1376,1324,1286,1237,1207;微量分析:对C18H21BrClNO6S的计算值:C:43.69%;H:4.27%;N:2.83%;Br:16.15%;Cl:7.16%;S:6.48%实测值:C:43.40%;H:4.24%;N:2.73%;Br:16.40%;Cl:7.20%;S:6.54%。步骤6制备
按照在实施例G步骤4和步骤5中概述的方法,制备以上化合物,其中在步骤5中制备的等量的中间体作为游离碱替代实施例G步骤4。
1H-NMR和MS与要求的产物相一致。实施例1制备 步骤1制备
步骤1制备
按照在实施例G步骤2A的方法,制备以上化合物,以等量的2-羟基-3,5-二溴苯甲醛替代3,5-二氯水杨醛。
收率:88%;浅黄色固体:m.p.46-47℃;Rf=0.6(EtOAc/己烷1∶1v/v);1H-NMR(CDCl3)δ3.37(s,3H),3.56(m,2H),3.92(m,2H),5.29(s,2H),7.91(d,1H,J=2.4Hz),7.94(d,1H,J=2.4Hz),10.27(s,1H);FAB-MS m/z367对C11H12Br2O4的(M+)HR-MS的计算值:367.9083实测值:367.9077。
1H-NMR和MS与要求的化合物相一致。步骤2
使用实施例G步骤2B和2C的方法,制备以上化合物,在所述方法中以等量的步骤1的化合物替代。
收率:90%;黄色固体:m.p.57-59℃;Rf=0.46(EtOAc/己烷1∶1v/v);1H-NMR(CDCl3)δ1.45(s,9H),2.1(br,1H,可交换),2.51(d,1H,J1=9.9Hz,J2=15.3Hz),2.66(d,1H,J1=4.2Hz,J2=15.3Hz),3.02(br,1H,可交换),3.39(s,3H),3.58-3.62(m,4H),3.81(m,1H),3.93(m,2H),4.63(dd,1H,J=4.2Hz),5.15(s,2H),7.17-7.25(m,6H),7.49(d,1H);FAB-MS m/z602(M+H)对C25H34NBr2O6的HR-MS的计算值:602.0753实测值:602.0749。
1H-NMR和MS与要求的化合物相一致。步骤3制备
按照在实施例G步骤3中的方法,制备以上化合物(对甲苯磺酸盐),以等量的实施例G步骤2中的产物替代步骤3A。收率:62%;白色固体;1H-NMR(DMSO-d6)δ1.09(t,3H,J=7.2Hz),2.27(s,3H),2.97(dd,2H,J1=3.0Hz,J2=7.2Hz),4.02(q,2H,J=7.2Hz),4.87(t,1H,J=7.2Hz),7.08(d,2H,J=4.8Hz),7.45(m,3H),7.57(d,1H,J=2.4Hz),8.2(br,3H);FAB-MS m/z365(M+H)对C11H14NBr2O3的HR-MS的计算值:365.9340实测值:365.9311。
1H-NMR和MS与要求的产物相一致。步骤4制备
使用实施例G步骤4的方法,制备以上化合物,并且替代在步骤3中制备的化合物以生成BOC保护的中间体。使用实施例G步骤5的方法,将生成的BOC保护的中间体转变为要求的化合物。
1H-NMR和MS与要求的化合物相一致。
实施例J制备β-[(2-氨基乙酰基)氨基]吡啶-3-丙酸乙酯双盐酸盐步骤1
向在2-丙醇(3升)中的3-吡啶甲醛(300ml)中加入乙酸铵(297g),随后加入丙二酸(398g)。在回流下将该反应混合物搅拌5小时。趁热过滤沉淀并用热的异丙醇(2升)洗涤。然后干燥生成的白色固体,得到作为白色固体的DL-3-氨基-3-(3-吡啶基)丙酸(220g)。
NMR和MS与要求的产物相一致。步骤2
将来自步骤1的DL-3-氨基-3-(3-吡啶基)丙酸(220g)在无水乙醇(3.6升)中浆化。使HCl气体(一个演示瓶-1b)鼓泡通入该反应物中,其间搅拌40分钟。然后将该浆化物加热回流4小时(1至1.5小时后溶液形成)。在冰浴上,把反应混合物冷却至5℃。在5℃下搅拌1.5小时后,过滤生成的白色沉淀并用乙醚彻底洗涤。在50℃下真空干燥后,得到作为白色固体的要求的产物DL-3-氨基-3-(3-吡啶基)丙酸乙酯二盐酸盐(331.3g)。
NMR和MS与要求的产物相一致。步骤3
在5-10℃下,向在无水THF(2升)和三乙胺(167.2g,1.65moles)中的来自步骤2的DL-3-氨基-3-(3-吡啶基)丙酸乙酯二盐酸盐(220.6g,0.83mole)中分批加入N-叔丁氧基羰基-甘氨酸N-羟基琥珀酰亚胺酯(225g,0.826moles)(Sigma)。在室温下,将该反应混合物搅拌过夜。过滤生成的沉淀并用THF洗涤。真空除去滤液中的溶剂。用乙酸乙酯(2.3升)处理残余物。用饱和的碳酸氢钠(2×900ml)和H2O(3×900ml)洗涤乙酸乙酯层,经MgSO4干燥并真空除去。该残余物在10%乙酸乙酯/己烷(2.5升)中浆化过夜。过滤沉淀,用10%乙酸乙酯/己烷(1升)洗涤,然后以己烷洗涤,然后干燥,得到作为白色固体的β-[[2-[[(1,1-二甲基乙氧基)羰基]氨基]乙酰基]-氨基]-吡啶-3-丙酸乙酯(233g)。
NMR和MS与要求的产物相一致。步骤4
将β-[[2-[[(1,1-二甲基乙氧基)羰基]氨基]乙酰基]氨基]-吡啶-3-丙酸乙酯(来自步骤3)(232g,0.66mole)溶于热的二噁烷(1升)中。在冷却至室温后,缓慢加入在二噁烷(1.6升)(Aldrich)中的4MHCl。几分钟后白色沉淀形成,然后转变为稠粘的物料。2小时后,倾析溶剂。将该残余物在乙醚中浆化并倾析乙醚直到生成白色固体。使其真空干燥,得到作为白色吸湿性固体的β-[(2-氨基乙酰基)氨基]吡啶-3-丙酸乙酯双盐酸盐(224.2g)。
NMR和MS与要求的产物相一致。
实施例K制备2-胍基-4-羧基噻唑盐酸盐 步骤1
步骤1
向在无水EtOH(100mL)中的2-亚氨基-4-硫代缩二脲(11.1g,0.094mole)(Aldrich)中,在回流下分批加入溴代丙酮酸乙酯(20.0g,0.102mole)(Aldrich)。在回流下,将该溶液搅拌2小时。加入另外的溴代丙酮酸乙酯(2.0g)并且把该反应物继续回流另外2小时。将反应物冷却至10℃并加入浓NH4OH直到pH=10。过滤生成的沉淀,用乙醚洗涤并干燥,得到作为黄色固体的2-胍基-4-羧基乙基噻唑(15.1g)。
MS和1H-NMR与要求的结构相一致。步骤2
向在H2O(100mL)和乙醇(40mL)中浆化的步骤1的产物(5.0g,0.023mole)中加入NaOH(0.93g,0.023mole)。在室温下,将该溶液搅拌过夜。将另外的乙醇(20mL)和NaOH(0.93g)加入到反应混合物中并在室温下将该溶液搅拌另外1小时。用1NHCl使pH降至7并过滤生成的沉淀,用H2O和乙醚洗涤,然后干燥,得到作为两性离子的产物(4.1g)。
将该两性离子产物在H2O中浆化并用浓HCl酸化直到生成溶液。将该溶液冷冻并冷冻干燥,得到作为淡黄色固体的2-胍基-4-羧基噻唑盐酸盐(4.33g)。
MS和1H-NMR与要求的结构相一致。
按照所描述的流程,分别从市售2-氨基-6-甲基吡啶和2-氨基-4-甲基-吡啶起始,制备实施例L和M。
实施例L制备2-氨基吡啶-6-羧酸步骤A
在75℃下,向2-乙酰氨基-6-甲基吡啶(10.0g)在水(125mL)中的溶液中分批加入KMnO4(21.0g)并且继续加热2小时。将该反应混合物冷却,过滤并用热水(2×25mL)洗涤残余物。用二氯甲烷(3×40mL)洗涤合并的滤液水溶液和洗涤液,并以冷的3N HCl酸化。过滤生成的沉淀,用水洗涤至pH呈中性,真空干燥,得到2-乙酰氨基吡啶-6-甲酸(3.8g,48%收率)。将二氯甲烷提取液浓缩至干,得到未反应的2-乙酰氨基-6-甲基吡啶(3.5g)。1H-NMR(CD3OD)δ8.32(m,1H),7.94(t,1H),7.87(m,1H),2.2(s,3H),FAB MS m/z181(M+H)。步骤B
在80℃下,于氮气氛下将2-乙酰氨基吡啶-6-甲酸(4.1g)在2.5N的NaOH(36.5ml)中的悬浮液加热1.5小时。将生成的溶液冷却并用冷的3N HCl酸化。过滤得到的沉淀,用水和乙腈连续洗涤。真空干燥生成的白色固体,得到2-氨基吡啶-6-甲酸(2.4g,76%收率)。1H-NMR(DMSO-d6)δ7.57(t,1H,J=8.1Hz),7.14(d,1H,J=7.2Hz),6.67(1H,J=8.1Hz),6.51(br,2H);FAB MS m/z139(M+H)。步骤C
在无水条件下,将2-氨基吡啶-6-甲酸(2g)在EtOH(10.0mL)和4NHCl/二噁烷(10.0mL)中的悬浮液加热回流16小时。然后将反应混合物浓缩至干并且真空下在干燥器中干燥残余物,得到作为白色粉末的2-氨基-6-乙酯基吡啶盐酸盐(1.6g)。1H-NMR(DMSO-d6)δ8.3(br),7.94(m,1H),7.37(d,1H,J=7.2Hz),7.22(dd,1H,J=8.7Hz),4.37(q,2H,J=7.2Hz),1.32(t,3H,J=7.2Hz);FAB MS m/z167(M+H)。
实施例M制备2-氨基吡啶-4-乙酯基吡啶盐酸盐
从2-乙酰氨基-4-甲基吡啶开始,通过相似于制备实施例L的2-氨基吡啶-6-乙酯基吡啶盐酸盐的方法,制备该化合物(M)。
1H-NMR和MS与所述的结构相一致。实施例1制备
在冰浴温度下,向在无水DMA(5mL)中的实施例A的产物(0.5g,0.002mole)、实施例E的产物(0.83g,0.002mole)、三乙胺(0.2g,0.002mole)和DMAP(24mg)中加入EDCl(0.38g,0.002mole)。在室温下,将反应物搅拌2天。经反相制备型HPLC分离酯产物。向在H2O(3mL)和CH3CN(3mL)中的酯中加入LiOH(0.51g,0.012mole)。在室温下,将其搅拌1小时。用TFA使pH降至2并经反相制备型HPLC分离该产物,得到(冷冻干燥后)作为白色固体的要求的产物(350mg)。
MS和1H-NMR与要求的结构相一致。实施例2制备
按照实施例1的方法,制备标题化合物,用等量的实施例D的产物替代实施例E的产物,得到白色固体的要求的产物(210mg)。
MS和1H-NMR与要求的结构相一致。实施例3制备
在N2下,在冰浴温度下,向在火焰干燥的烧瓶中的无水DMA(85mL)中的实施例C的产物(19.5g,0.045mole)和N-甲基吗啉(9.1g,0.09mole)中缓慢加入氯甲酸异丁酯(6.2g,0.045mole)。在冰浴温度下,将该溶液搅拌15分钟。然后在冰浴温度下加入实施例D的产物(15g,0.04mole),随后缓慢加入N-甲基吗啉(4.1g,0.04mole)。在室温下,将反应混合物搅拌过夜。向在H2O(50mL)/CH3CN(30mL)中的酯中加入LiOH(16g,0.38mole)。在室温下,将其搅拌1小时。用TFA使pH降至2并经反相制备型HPLC分离产物,得到(冷冻干燥之后)作为白色固体的要求的产物(13.7g)。
MS和1H-NMR与要求的结构相一致。实施例4制备
按照实施例3的方法,制备以上化合物,用等量的实施例E的产物替代实施例D的产物。
MS和1H-NMR与要求的结构相一致。实施例5制备
按照实施例3的方法,制备以上化合物,用等量的实施例F的产物替代实施例D的产物。
MS和1H-NMR与要求的结构相一致。实施例6制备
按照实施例3的方法,制备以上化合物,用等量的实施例B的产物替代实施例C的产物。
MS和1H-NMR与要求的结构相一致。实施例7制备
在冰浴温度下,向在无水DMA(4mL)中的实施例9步骤3的产物(0.6g,0.0019mole)中加入氯甲酸异丁酯(0.27g,0.002mole),随后加入N-甲基吗啉(0.4g,0.0038mole)。在冰浴温度下,将该溶液搅拌15分钟。在冰浴温度下,加入实施例H的产物(0.71g,0.0017mole),随后加入N-甲基吗啉(0.17g,0.0017mole)。在室温下,将反应物搅拌过夜。HPLC分析指明产物含有明显量的起始物料。将EDCl(0.38g,0.002mole)和DMAP(15mg)加入到反应混合物中并在室温下将该反应物搅拌过夜。经反相制备型HPLC分离酯产物。向在H2O(10mL)和CH3CN(5mL)中的酯中加入LiOH(700mg,0.017mole)。在室温下,将其搅拌1小时。用TFA使pH降至2并经反相制备型HPLC分离产物,得到(冷冻干燥之后)作为白色固体的要求的产物(270mg)。
MS和1H-NMR与要求的结构相一致。实施例8制备
在冰浴温度下,向在无水DMF(8mL)中的实施例K的产物(0.5g,0.0022mole)和N-甲基吗啉(0.23g,0.0022mole)中加入氯甲酸异丁酯(0.31g,0.0022mole)。在冰浴温度下搅拌5分钟后,于冰浴温度下一次性加入在无水DMF(8mL)中的实施例J的产物(0.73g,0.0022mole)和N-甲基吗啉(0.45g,0.0045mole)。在室温下,将反应混合物搅拌过夜。经反相制备型HPLC分离酯(530mg)。
向在H2O(10mL)中的该酯(400mg)中加入LiOH(91mg)。在室温下,将其搅拌1小时。用TFA把pH降至3并且该产物经反相制备型HPLC分离,得到(冷冻干燥之后)白色固体的要求的产物(350mg)。
MS和1H-NMR与要求的结构相一致。
如同在实施例N和O中描述的流程中阐明的那样,使用三-叔丁氧基羰基试剂,进行2-氨基-6-乙酯基吡啶盐酸盐和2-氨基-4-乙酯基吡啶盐酸盐的胍基化(Kim.S.K.,Qian,L.,四面体快报,34,7677,1993)。实施例N
向2-氨基-6-乙酯基吡啶盐酸盐(0.56g,2.8mmol)和三-叔丁氧基羰基试剂(91.2g,2.8mmol)在脱气的DMF(7.0mL)中的溶液中加入氯化汞(0.76g,2.8mmol)和三乙胺(0.79g,7.8mmol)。在70℃下,于氮气氛中将生成的混合物加热24小时并且过滤。真空蒸馏DMF,用乙酸乙酯研磨残余物并过滤。浓缩橙色滤液并经硅胶快速层析法纯化生成的物料,使用含有1%三乙胺的乙酸乙酯作为洗脱液。合并适当的部分(蓝色荧光)且减压下浓缩至干,得到黄色粉末。MS分析[m/z565(M+H)]证实形成要求的三-叔丁氧基羰基产物N。
实施例O
使用与实施例N相似的方法,制备化合物(O)。MS分析[m/z565(M+H)]证实形成要求的三-叔丁氧基羰基产物O。
按照在实施例9步骤3-5中阐述的条件,能够制备以下化合物P和Q。
实施例P
实施例Q
实施例R
使用在实施例G中描述的方法,制备以上化合物。以步骤2中使用的试剂3-溴-5-氯代水杨醛替代3,5-二氯代水杨醛。 步骤1
步骤1
在0℃下,于30分钟内向机械搅拌着的试剂1(18.68g)在THF(1L)中的悬浮液中加入氢化钠(19g在矿物油中的60%悬浮液)。在30分钟后,加入纯的二碳酸二叔丁酯(95g)并在温和回流下将反应混合物加热16小时。将该混合物冷却至0℃并用饱和NaHCO3溶液猝灭。用乙酸乙酯提取混合物。用水洗涤有机提取液,干燥(Na2SO4)并浓缩。使用小硅胶柱(60mm×180mm)快速层析残余物,得到作为粘稠黄色液体的要求的产物2(68g)。NMR与指明的结构相一致。步骤2
在0℃下,将搅拌着的3-氨基-吡啶-6-甲酸(25g)在乙醇(300mL)中的悬浮液用氯化氢气体饱和。使该混合物温热至23℃并加热至回流2小时。得到透明的溶液。在冷却至23℃之后,真空浓缩混合物,用NaHCO3水溶液中和并以乙酸乙酯提取。用水洗涤有机相,经MgSO4干燥,并真空浓缩,得到作为浅黄色固体的3-氨基-吡啶-6-甲酸乙酯。
将步骤1的产物(40.0g)、HgCl2(25.0g)、三乙胺(27mL)和3-氨基-吡啶-6-甲酸乙酯(13g)和DMF(200mL)的混合物搅拌并加热至55℃30小时。将反应混合物冷却至23℃,用乙酸乙酯(500mL)稀释并通过硅藻土过滤。用水(2X)洗涤滤液,经MgSO4干燥并真空浓缩。使用在己烷中的乙酸乙酯作为洗脱剂,经硅胶层析残余物,得到要求的中间体3。NMR与要求的结构相一致。步骤3
在23℃下,将步骤2的产物(13g)在5N盐酸(100mL)中的悬浮液搅拌16小时。真空浓缩该混合物。用甲醇研磨残余物并过滤白色固体,得到要求的产物4。NMR和MS与要求的结构相一致。步骤4
将步骤3的产物(0.308g)悬浮于DMF(10mL)中并把4-甲基吗啉(0.2mL)加入到悬浮液中。在23℃下,把该混合物搅拌1小时。在将反应混合物冷却至0℃后,加入IBCF(0.129mL)。在1/2小时后,加入实施例R的产物(0.416g)和4-甲基吗啉(0.11mL)在DMF(3mL)中的溶液。2小时内使该混合物温热至23℃。然后过滤混合物并真空浓缩滤液。经HPLC纯化残余物,得到5。微量分析数据、NMR和MS与要求的结构相一致。步骤5
在23℃下,使步骤4的产物在3.5N HCl中的溶液放置3小时,真空浓缩并且经HPLC纯化残余物,得到要求的产物。微量分析数据、NMR和MS与要求的产物相一致。
对C21H22BrClN6O6·2CF3CO2H·H2O的分析计算值:C,36.80;H,3.21;N,10.30;实测值:C,36.52;H,2.90;N,9.91。
实施例10
除了使用实施例G的产物替代实施例R在步骤4中的产物以外,使用在实施例9中描述的方法,制备该化合物。
对C21H22Cl2N6O6·2CF3CO2H·H2O的分析计算值:C,38.93;H,3.40;N,10.89;实测值:C,39.27;H,3.12;N,11.09。
实施例11
使用实施例Ⅰ的产物替代实施例R在步骤4中的产物,如同实施例9描述的那样,制备该化合物。
对C21H22Br2N6O6·2CF3COOH·H2O的分析计算值:C,35.65;H,2.87;N,9.98;实测值:C,35.81;H,2.79;N,10.14。
实施例12
制备(3S)-N-[[4-[(1,4,5,6-四氢-5-羟基-2-嘧啶基)氨基]-2-噻吩基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸三氟乙酸盐 步骤1
步骤1
将5-溴-2-噻吩甲酸(5.0g)在浓硫酸(30ml)中的溶液冷却至-30℃并滴加在浓硫酸(30ml)中的70%硝酸溶液(7.7ml)(d=1.40)来处理。在-25℃下,将该反应混合物搅拌30分钟然后倾入冰水中浆化并搅拌几分钟。过滤沉淀的固体,从水中重结晶并干燥,得到白色粉末(2.3g)。1H-NMR与提出的结构相一致。步骤2
在室温下,于5磅/平方英寸(psi)的氢气下,用催化量的5%Pd/C将来自步骤1的产物(1.7g)在THF(25ml)中的溶液处理16小时。过滤反应混合物并浓缩。用乙酸乙酯加热残余物并过滤,得到棕色固体(750mg)。1H-NMR与提出的结构相一致。步骤3
将来自步骤2的产物(725mg)和在THF(15ml)中的胍基化试剂G1和二甲基乙酰胺(10ml)的溶液回流16小时。浓缩反应混合物并用TFA(5ml)和CH2Cl2(5ml)的溶液处理残余物。在室温下搅拌1小时后,浓缩反应混合物并经反相HPLC纯化残余物,使用水(0.5%TFA)和乙腈梯度作为洗脱剂,得到淡黄色的粘稠液体(300mg)。1H-NMR与提出的结构相一致。步骤4
在火焰干燥的烧瓶中,在N2下,将来自步骤3的产物(275mg)溶于8ml二甲基乙酰胺和N-甲基吗啉(81mg)中。将搅拌着的溶液冷却至0℃并滴加入氯甲酸异丁酯(109mg)在二甲基乙酰胺(1ml)中的溶液处理。在0℃下,将反应混合物搅拌45分钟,然后滴加入实施例G的产物(286mg)和N-甲基吗啉(81mg)在二甲基乙酰胺(2ml)中的溶液处理。使反应混合物温热至室温并继续搅拌16小时。浓缩反应混合物,并如同在步骤3中描述的那样使用反相HPLC纯化残余物,得到粘稠的无色液体(262mg)。1H-NMR与提出的结构相一致。步骤5
在室温下,将来自步骤4的产物(250mg)在甲醇(8ml)中的溶液和1N氢氧化钠溶液(8ml)搅拌16小时。用TFA(0.7ml)猝灭反应并浓缩。如同在步骤3中描述的那样,使用反相HPLC纯化残余物,得到白色固体(144mg)。1H-NMR与提出的结构相一致。
对C20H21N5O6Cl2S·1.50TFA·0.25H2O的分析计算值:C,39.13;H,3.28;N,9.92;S,4.54;Cl,10.04;实测值:C,38.81;H,3.17;N,9.90;S,4.86;Cl,10.25。
实施例13
制备N-[[4-[(4,5-二氢-1H-咪唑-2-基)氨基]-2-噻吩基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸三氟乙酸盐
以如同在实施例12中描述的化合物的制备中所使用的类似的反应顺序,制备以上化合物。使用在实施例C(步骤1)中描述的胍基化试剂替代在实施例12步骤3中的G1。相似地用实施例D的产物来替代在实施例G步骤4中的产物。1H-NMR与提出的结构相一致。
对C19H19N5O5Cl2S·1.50TFA·0.50H2O的分析计算值:C,38.84;H,3.18;N,10.29;S,4.71;Cl,10.42;实测值:C,38.84;H,3.01;N,10.50;S,5.08;Cl,11.01。
实施例14
制备(3S)-N-[[5-甲基-4-[(1,4,5,6-四氢-5-羟基-2-嘧啶基)氨基]-2-噻吩基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸三氟乙酸盐 步骤1
步骤1
使用5-甲基-2-噻吩甲酸(5.2g),如同在实施例13的步骤1中描述的那样,制备(A)。通过用稀氢氧化钠溶液提取,用稀盐酸酸化并用乙酸乙酯提取,纯化该产物。干燥(Na2SO4)有机提取液并浓缩,得到黄橙色固体(2.2g)。1H-NMR与提出的结构相一致。步骤2
使用步骤1的产物(2.2g)并按照如同在实施例12的步骤4中描述的偶合条件,制备(B)。浓缩该反应物并在乙酸乙酯和水之间分配残余物。用饱和的氯化钠溶液洗涤有机提取液。干燥(Na2SO4)并浓缩。将残余物从乙酸乙酯中重结晶,得到褐色固体(3.5g)。1H-NMR与提出的结构相一致。步骤3
在室温下,滴加入氯化锡(Ⅱ)二水合物在浓盐酸中的溶液(1.0g/2ml)处理步骤2的产物(2.8g)在THF(25ml)中的溶液,直到TLC表明起始原料完全消失。除去有机溶剂并用碳酸氢钠溶液中和水溶液部分。用CH2Cl2提取水溶液部分,干燥(Na2SO4)并浓缩,得到黄色粉末(1.1g)。1H-NMR与提出的结构相一致。步骤4
向在步骤3中描述的产物(375mg)和实施例9步骤1的产物(519mg)在DMF(15ml)中的溶液中加入氯化汞(326mg)和三乙胺(182mg)。在95-100℃下加热混合物3小时。将反应混合物冷却,用乙酸乙酯(30ml)处理,搅拌30分钟,然后通过硅藻土垫过滤。浓缩滤液并使用快速层析法(用97.5%CHCl3-2.5%CH3OH洗脱)纯化残余物,得到黄棕色固体(415mg)。1H-NMR与提出的结构相一致。步骤5
在室温下,将步骤4的产物(400mg)在CH2Cl2(7.5ml)和TFA(7.5ml)中的溶液搅拌1小时。浓缩反应物并在室温下于CH3OH(10ml)和1N氢氧化钠的溶液(10ml)中把残余物搅拌16小时,使其溶解。用TFA(0.8ml)猝灭反应物并浓缩。如同先前描述的那样,使用反相HPLC纯化残余物,得到白色固体(80mg)。1H-NMR与提出的结构相一致。
对C21H23N5O6Cl2·1.50TFA·0.50H2O的分析计算值:C,39.79;H,3.55;N,9.67;S,4.43;Cl,9.79;实测值:C,39.50;H,3.24;N,9.58;S,4.71;Cl,10.22。
实施例15
制备(3S)-N-[[4-(4,5-氢-1H-咪唑-2-基)氨基]-5-甲基-2-噻吩基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸三氟乙酸盐
除了使用在实施例C(步骤1)中描述的胍基化试剂替代在实施例14步骤4中的试剂以外,以如同在实施例14中描述的化合物的制备中使用的类似的反应顺序,制备以上化合物。1H-NMR与提出的结构相一致。
对C20H21N5O5Cl2S·1.50TFA·0.25H2O的分析计算值:C,40.04;H,3.36;N,10.15;S,4.65;Cl,10.28;实测值:C,39.82;H,3.19;N,10.17;S,4.86;Cl,10.69。
实施例16
N-[[5-(4,5-二氢-1H-咪唑-2-基)氨基]-2-噻吩基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸三氟乙酸盐
使用如同在实施例15中描述的相同的反应顺序,以2-硝基-5-噻吩甲酸开始,合成标题化合物。对于胍基化反应,按照在实施例17步骤G中描述的条件进行。
对C19H19N5O5Cl2S·1.50TFA的分析计算值:C,39.36;H,3.08;N,10.43;S,4.78;实测值:C,39.05;H,2.79;N,10.37;S,4.90。
实施例17
N-[[5-(4,5-二氢-1H-咪唑-2-基)氨基]-6-甲氧基-3-吡啶基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸双(三氟乙酸)盐步骤1
将6-羟基烟酸(20g)和无色发烟硝酸(60ml)(d=1.50)的混合物加热到90-95℃反应3小时。把反应物冷却至室温并在搅拌下倾入到1.5升冰-水浆状物中且于15分钟后过滤。用水洗涤沉淀并干燥,得到淡黄色粉末(9.3g)。1H-NMR与提出的结构相一致。
对C6H4N2O5的分析计算值:C,39.14;H,2.19;N,15.22;实测值:C,39.08;H,2.17;N,15.19。步骤2
将步骤1的产物(5.0g)和三氯氧化磷(15ml)的混合物回流3小时。将反应混合物冷却至室温并加入到冰水混合物中且搅拌30分钟。加入另外的冰以保持混合物在此期间冷却。用THF-乙醚(1∶2)的混合物提取反应混合物并用饱和的氯化钠溶液洗涤有机提取物,干燥(Na2SO4)并浓缩。残余物从1∶1乙醚-己烷中重结晶,得到黄色粉末(4.0g)。1H-NMR与提出的结构相一致。步骤3
将步骤2的产物(1.6g)在亚硫酰氯(5.0ml)中的溶液回流3小时。将该反应物冷却至室温并在氮气流下蒸发。减压下干燥残余物并未经进一步纯化使用。在氮气下,于火焰干燥的烧瓶中,将实施例D的产物(3.0g)溶于二甲基乙酰胺(30ml)和N,N-异丙基乙基胺(2.3g)中。在冰浴上冷却溶液并滴加入酰氯(如同以上得到的)在THF(20ml)中的溶液。搅拌该反应物并使其温热至室温,然后用水(25ml)猝灭。用乙酸乙酯提取混合物并用饱和的氯化钠溶液洗涤有机提取液,干燥(Na2SO4)并浓缩。生成的黄色固体从乙酸乙酯中重结晶,得到淡黄色粉末(3.4g)。1H-NMR与提出的结构相一致。步骤4
在0℃下,向甲醇钠(2.2g)在甲醇(30ml)中的悬浮液中滴加入步骤3的产物(4.5g)在甲醇(30ml)中的溶液。将该反应物搅拌30分钟,然后用乙酸(2.3ml)猝灭。浓缩反应物并在乙酸乙酯和水之间分配。干燥(Na2SO4)有机提取液,浓缩并在硅胶柱(用80%乙酸乙酯-20%己烷洗脱)上纯化,得到淡黄色固体(3.5g)。1H-NMR与提出的结构相一致。步骤5
在室温下,于5磅/平方英寸的氢气压力下,用以硫毒化的催化量的3%Pt/C将在步骤4中描述的产物(1.07g)在乙醇(25ml)中的溶液处理4小时。过滤反应混合物并浓缩,得到白色固体(930mg),其无需进一步纯化即可使用。1H-NMR与提出的结构相一致。步骤6
向步骤5的产物(330mg)、G2(276mg)在DMF(10ml)中的溶液中加入三乙胺(150mg)和氯化汞(258mg)并在氮气中、于85℃下把该混合物加热16小时。将反应物冷却,用乙酸乙酯(25ml)处理并过滤。浓缩滤液并且在硅胶柱上纯化残余物,使用98%CH2Cl2-2%甲醇洗脱,得到白色固体(325mg)。1H-NMR与提出的结构相一致。步骤7
在室温下,将步骤6的产物(114mg)、TFA(8ml)和二氯甲烷(8ml)的溶液搅拌90分钟。蒸发溶剂并在室温下用1N氢氧化钠的溶液(8ml)和甲醇(8ml)将残余物处理16小时。用TFA(1ml)猝灭反应并浓缩。使用先前描述的反相HPLC纯化残余物,得到白色固体(76mg)。1H-NMR与提出的结构相一致。
对C21H22N6O6Cl2·2TFA的分析计算值:C,41.74;H,3.88;N,12.70;实测值:C,41.47;H,3.49;N,12.85。
实施例18
N-[[5-(4,5-二氢-1H-咪唑-2-基)氨基]-1,6-二氢-1-甲基-6-氧代-3-吡啶基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸(三氟乙酸)盐 步骤1
步骤1
用浓硫酸(1ml)处理实施例17步骤1的产物(12.0g)在甲醇(200ml)中的悬浮液并使其回流。回流16小时后,反应混合物变成均匀的溶液,并使其冷却至室温。除去大约一半的溶剂,此时产物从溶液中结晶出来。在冰浴上冷却该混合物,过滤并干燥,得到黄色粉末(16.7g)。1H-NMR与提出的结构相一致。步骤2
在N2下,将来自步骤1的产物(2.6g)在DMF(40ml)中的溶液冷却至0℃并用60%氢化钠在矿物油中的分散液655mg)处理。在0℃下,将该混合物搅拌45分钟,然后用碘甲烷(2.8g)一次性处理。在室温下,将反应物搅拌16小时,然后在乙酸乙酯和水之间分配。用另外的乙酸乙酯将水部分提取几次,然后用饱和的氯化钠溶液洗涤合并的有机提取液,干燥(Na2SO4)并浓缩。在硅胶柱(2%甲醇-98%二氯甲烷洗脱)上纯化残余物,得到黄色固体(1.9g)。1H-NMR与提出的结构相一致。步骤3
在室温下,将步骤2的产物(2.6g)、甲醇(60ml)和2N氢氧化钠溶液(60ml)的溶液搅拌16小时。用冰乙酸(6.9ml)猝灭反应物并浓缩。高真空下干燥残余物。然后在氮气下将该物料与亚硫酰氯(100ml)回流3小时。将该反应物冷却,浓缩并在高真空下彻底干燥,得到褐色固体(2.2g)。1H-NMR与提出的结构相一致。在氮气下,于火焰干燥的烧瓶中把实施例D的产物(3.5g)溶于N,N二甲基乙酰胺(35ml)和二异丙基乙基胺(2.9ml)中。将该溶液冷却至0℃并用酰氯(1.9g)(如同以上得到的)在THF(35ml)中的溶液处理。将该反应物搅拌30分钟,然后高真空下浓缩以除去溶剂。把残余物在乙酸乙酯和水之间分配并用另外的乙酸乙酯把水部分提取几次。用水洗涤合并的有机提取液几次,然后用饱和的氯化钠溶液洗涤,干燥(Na2SO4)并浓缩,生成黄色固体(1.3g)。1H-NMR与提出的结构相一致。步骤4
在室温下,于5磅/平方英寸的氢气压力下,用催化量的5%Pt/C将来自步骤3的产物(1.0g)在THF(10ml)和DMF(10ml)中的溶液处理4小时。过滤反应混合物并浓缩。用最少量的THF处理残余物并使其在室温下缓慢结晶。把等体积的乙酸乙酯加入到结晶出来的混合物中并消化15分钟。将该混合物冷却,过滤并用冷的乙酸乙酯洗涤。干燥产物,得到白色固体(730mg)。1H-NMR与提出的结构相一致。步骤5
向步骤4的产物(720mg)和G2(581mg)在DMF(25ml)中的溶液中加入三乙胺(304mg)和氯化汞(544mg)并在氮气下于85℃下把该混合物加热反应1小时。将反应混合物冷却并通过硅藻土垫过滤。浓缩滤液并在硅胶柱(用10%甲醇-90%氯仿洗脱)上纯化,得到白色固体(375mg)。1H-NMR与提出的结构相一致。步骤6
使用在实施例14(步骤5)中描述的方法,由在步骤5中制备的化合物(360mg)来制备以上化合物。使用先前描述的反相HPLC纯化粗品产物,得到白色固体(223mg)。1H-NMR与提出的结构相一致。
对C21H22N6O6Cl2·2.5TFA·0.5H2O的分析计算值:C,38.11;H,3.14;N,10.26;Cl,8.65实测值:C,38.18;H,3.05;N,10.79;Cl,9.34。
实施例19
N-[[6-氯-5-[(4,5-氢-1H-咪唑-2-基)氨基]-3-吡啶基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸(三氟乙酸)盐一水合物步骤1
制备氯化锡(Ⅱ)(6.3g)在浓盐酸(10ml)中的溶液并滴加入实施例17步骤3的产物(1.0g)在THF(15ml)中的溶液中。将该反应物搅拌15分钟,其间使反应物冷却至室温。除去有机溶剂,得到胶状沉淀。倾析水溶液部分并把胶状沉淀在乙酸乙酯和稀的碳酸氢钠溶液之间分配。用另外的乙酸乙酯把水溶液部分提取几次,然后以水和饱和的氯化钠溶液洗涤合并的有机提取液,干燥(Na2SO4)并浓缩。真空干燥残余物,得到褐色粉末(375mg)。1H-NMR与提出的结构相一致。步骤2
使用在实施例18步骤5中概述的方法,将步骤1中描述的化合物(365mg)胍基化。在硅胶柱(用2%甲醇-98%氯仿洗脱)上纯化粗品产物,得到棕色粘稠的液体(205mg)。1H-NMR与提出的结构相一致。步骤3
使用步骤2的产物(200mg)并按照在实施例17步骤7中描述的方法,制备标题化合物。如前所述,使用反相HPLC纯化粗品产物,得到白色固体(53mg)。1H-NMR与提出的结构相一致。
对C20H19N6O5Cl3·1.7TFA.1.0H2O的分析计算值:C,37.90;H,3.09;N,11.33;Cl,14.34实测值:C,37.53;H,2.71;N,11.33;Cl,15.01。
实施例20
N-[[5-[(4,5-氢-1H-咪唑-2-基)氨基]-1,6-氢-6-氧代-3-吡啶基]羰基]甘氨酰-3-(3,5-氯-2-羟基苯基)-β-丙氨酸(三氟乙酸)盐
在室温下,通过将来自实施例17的步骤6的产物(353mg)溶于6N盐酸溶液(50ml)中进行48小时,制备以上化合物。浓缩该反应物并如同先前描述的那样使用反相HPLC纯化残余物,得到白色固体(115mg)。1H-NMR与提出的结构相一致。
对C20H20N6O6Cl2·1.25TFA的分析计算值:C,41.33;H,3.28;N,12.85;实测值:C,41.57;H,3.29;N,12.92。
实施例21
制备(3S)-N-[[1,6-氢-6-氧代-5-[(1,4,5,6-四氢-5-羟基-2-嘧啶基)氨基]-3-吡啶基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸三氟乙酸盐
使用在实施例17(步骤1-3)中描述的反应顺序,合成用在制备标题化合物中的起始原料。替代实施例D产物的实施例G的手性中间体被用于在步骤3中详述的偶合反应中。步骤1
使用实施例17步骤4中给出的方法,制备(A)。1H-NMR与提出的结构相一致。步骤2
在N2下,将步骤1的物料(2.7g)在4N的无水HCl/二噁烷(100ml)中的溶液温热至45℃反应72小时。将反应物浓缩至干,得到淡棕色固体(2.6g)。1H-NMR与提出的结构相一致。步骤3
在室温下,于5磅/平方英寸的氢气氛下,用催化量的5%Pt/C将来自步骤2的产物(1.85g)在乙醇(50ml)中的溶液处理2小时。过滤反应混合物并浓缩,且使用反相HPLC纯化残余物,得到白色固体(507mg)。1H-NMR与提出的结构相一致。步骤4
向步骤3的产物(500mg)和实施例4步骤1的产物(650mg)在DMF(15ml)中的溶液中加入三乙胺(304mg)和氯化汞(408mg)并在氮气下把该混合物加热至100℃反应2小时。将反应混合物冷却至室温并与乙酸乙酯(30ml)一起搅拌15分钟,然后过滤。浓缩滤液并且在室温下用TFA(7ml)和CH2Cl2(7ml)的溶液处理残余物1小时。浓缩混合物并使用先前描述的反相HPLC纯化残余物,得到白色固体(400mg)。1H-NMR与提出的结构相一致。步骤5
在室温下,将步骤4的产物(200mg)、1N氢氧化钠(8ml)和甲醇(8ml)的溶液搅拌16小时。用TFA(1ml)猝灭该反应物并浓缩。使用先前描述的反相HPLC纯化残余物,得到白色固体(79mg)。1H-NMR与提出的结构相一致。
对C21H22N6O7Cl2·1.5TFA的分析计算值:C,40.46;H,3.32;N,11.80;实测值:C,40.12;H,3.57;N,12.26。
实施例22
制备N-[[5-(4,5-二氢-1H-咪唑-2-基)氨基]-2-甲氧基-3-吡啶基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸(三氟乙酸)盐步骤1
向2-羟基烟酸(10.0g,71.9mmol)在浓硫酸(28.6ml)中的溶液中以滴加方式加入发烟硝酸(7.1ml)。在55℃下,将该反应混合物加热4小时。把冷却的反应混合物倾入到冰水上。产物作为黄色固体沉淀出来。过滤收集沉淀的固体,用水洗涤并空气干燥,得到要求的产物(7.2g,54%收率)。NMR与提出的结构相一致。步骤2
将步骤1的产物(5.0g,27.2mmol)在三氯氧化磷(13.5g)中的溶液回流4.5小时。将冷却的反应混合物倾入到冰水(200g)上。将生成的混合物搅拌30分钟并用四氢呋喃/乙酸乙酯(2/1)提取。用盐水洗涤有机层,经Na2SO4干燥并过滤。浓缩滤液得到油。用己烷/乙醚(4/1)的混合物处理油,得到作为细黄色粉末的要求的产物(5.0g,91%收率)。NMR与提出的结构相一致。步骤3
将步骤2的产物(3.1g,15.3mmol)在亚硫酰氯(8.1ml)中的溶液回流4.5小时。减压下从该反应混合物中除去剩余的亚硫酰氯,得到浅棕色的油。真空干燥油,得到作为黄色固体的产物(2.7g,79%收率)。NMR与提出的结构相一致。步骤4
将二异丙基乙基胺(3.7g,4.9ml)加入到实施例D的产物(5.5g,14.8mmol)在N,N-甲基乙酰胺(40ml)和四氢呋喃(15ml)中的溶液中并把该反应混合物冷却至-5℃。于15分钟内把步骤3的产物(3.1g,14.1mmol)在四氢呋喃(25ml)中的溶液加入到反应液中。在-5℃下,将反应物搅拌30分钟并使其温热至室温。3小时后,减压下从该反应混合物中除去四氢呋喃。将反应混合物倾入到冰水上。过滤收集沉淀的固体,用水洗涤并空气干燥,得到作为黄色固体的产物(6.2g,85%收率)。NMR与提出的结构相一致。步骤5
在0℃下,将步骤4的产物(4.0g,7.7mmol)和甲醇钠(1.67g,30.8mmol)在甲醇(45ml)中的溶液搅拌4小时。用乙酸中和反应物。从反应混合物中除去甲醇并经层析法(硅胶,乙酸乙酯-甲苯,6/4)纯化粗品混合物,得到作为黄色固体的要求的产物(2.5g,67%收率)。NMR与提出的结构相一致。步骤6
在室温下,于5磅/平方英寸压力下,用H2将来自步骤5的产物(0.99g,1.9mmol)、3%披铂炭和甲醇的混合物处理16小时。过滤除去铂催化剂。减压下除去甲醇,得到作为黄色固体的要求的产物(0.62g,70%收率)。其无需进一步纯化即可用于下一步反应。NMR与提出的结构相一致。步骤7
在90-5℃下,将步骤6的产物(0.26g,0.54mmol)、G2(0.18g,0.59mmol)、三乙胺(0.25g,1.78mmol)、氯化汞(Ⅱ)(0.16g,0.59mmol)和N,N-二甲基甲酰胺(15ml)的混合物加热16小时。将冷却的反应混合物通过短硅藻土柱过滤,用乙酸乙酯(40ml)和二氯甲烷(30ml)先后洗脱。把粗品混合物层析(硅胶,CH2Cl2-CH3OH-NH4OH,95/5/0.5),得到黄色固体的产物(0.31g,76%收率)。NMR与提出的结构相一致。步骤8
在室温下,把步骤7的产物(0.31g,0.41mmol)、三氟乙酸(3.5ml)和二氯甲烷(7.0ml)的溶液搅拌2小时。减压除去三氟乙酸和二氯甲烷并在室温下用甲醇(5ml)和氢氧化钠溶液(2N,2.5ml)把该粗品混合物处理18小时。加入乙酸以中和氢氧化钠。除去甲醇,得到粗品混合物。使用反相HPLC纯化粗品混合物,得到作为白色固体的以上化合物(0.10g,36%收率)。
对C21H22N6O6Cl2·1.3CF3COOH·0.25H2O的分析计算值:C,41.80;H,3.54;N,12.39;Cl,10.46;实测值:C,42.12;H,3.84;N,11.87;Cl,10.99。
实施例23
制备N-[[5-(4,5-二氢-1H-咪唑-2-基)氨基]-1,2-氢-2-氧代-3-吡啶基]-羰基]甘氨酰-3-(3,5-氯-2-羟基苯基)-β-丙氨酸(三氟乙酸)盐
在室温下,用6N HCl(6ml)将实施例22步骤7的产物(0.21g,0.28mmol)在二噁烷(4ml)中的溶液处理72小时。减压下除去溶剂,并使用乙腈-水作为洗脱剂,于反相HPLC上纯化粗品混合物,得到作为灰白色固体的要求的化合物(0.052g,27%收率)。
对C20H20N6O6Cl2·1.6CF3COOH的分析计算值:C,40.17;H,3.14;N,12.11;实测值:C,40.10;H,3.22;N,11.84。
实施例24
制备N-[[5-[(4,5-氢-1H-咪唑-2-基)氨基]-1-甲基-1H-吡唑-3-基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸(三氟乙酸)盐
按照文献方法, 实施合成目标化合物的步骤1和2(分别为JustusLiebigs.Ann.Chem,512,97,1934和J.Hetercyclic Chem.21,737,1994)。步骤3
向步骤2的产物(0.9g,5.3mmol)和G2(l.9g,5.9mmol)在DMF(25ml)中的溶液中加入三乙胺(2.5ml,17.6mmol)和氯化汞(1.6g,5.9mmol)。在80-85℃下把该混合物加热18小时后,使该反应混合物冷却并通过硅藻土柱过滤。用二氯甲烷洗涤残余物并浓缩合并的部分。将残余物重新溶解于二氯甲烷中并用水和盐水洗涤。在经硫酸钠干燥后,过滤混合物并浓缩。使用硅胶层析法(己烷/乙酸乙酯1/1)纯化粗品橙色固体(1.8g),得到作为灰白色固体的N-叔丁氧基羰基保护的产物(1.4g)。步骤4
向步骤3的产物(2.4g,5.5mmol)在乙醇(50ml)中的溶液中加入氢氧化锂水溶液(526mg在5ml水中)。在室温下,将反应混合物搅拌24小时。向反应物中加入乙酸(22ml)并把混合物搅拌15分钟。减压下除去溶剂后,在反相HPLC(含有三氟乙酸的乙腈/水)上纯化该粗品混合物,得到作为粘稠白色液体的TFA盐的产物。用氯化氢(4M在二噁烷中的溶液)处理残余物并搅拌10分钟。浓缩反应物以除去溶剂并将该过程重复两次,得到作为白色固体的要求的产物(3.9g)。无需进一步纯化,将所述产物用于下一步。步骤5
在0℃下,向步骤4的产物(0.9g,3.7mmol)在DMF(15ml)中的溶液中依次加入N-甲基吗啉(605ml,5.5mmol)和氯甲酸异丁酯(475ml,3.7mmol)。在搅拌5分钟后,将步骤D的产物在含有N-甲基吗啉(605ml,5.5mmol)的DMF(10ml)中的溶液注射到反应液中。在0℃下,将混合物搅拌1小时并在室温下搅拌36小时。减压下除去溶剂并在反相HPLC(含有三氟乙酸的乙腈/水)上纯化残余物,得到作为TFA盐的产物(525mg)。步骤6
向步骤5的产物(540mg,1mmol)在乙醇(50ml)中的溶液中加入氢氧化锂水溶液(144mg在10ml水中)。在室温下,将反应混合物搅拌24小时。向反应物中加入乙酸(305ml)并把混合物搅拌15分钟。减压下除去溶剂后,在反相HPLC(含有三氟乙酸的乙腈/水)上纯化该粗品混合物,得到作为TFA盐的要求的产物(486mg,75%收率)。
对C19H21N7O5Cl2·1.5CF3CO2H的分析计算值:C,39.48;H,3.39;N,14.65;实测值:C,39.29;H,3.14;N,14.72。
NMR和MS与要求的结构相一致。
实施例25
制备N-[[5-[(4,5-氢-1H-咪唑-2-基)氨基]-1H-吡唑-3-基]羰基]甘氨酰-3-(3,5-二氯-2-羟基苯基)-β-丙氨酸(三氟乙酸)盐
使用苄基肼替代在步骤2中的甲基肼,如同在实施例24中描述的那样,合成所述化合物。在最后一步使用催化氢化条件(5%Pd/C,20磅/平方英寸,室温,26小时)将生成的N-苄基吡唑化合物脱除保护。使用乙酸-三氟乙酸(4/1)作为溶剂,进行这个反应。
对C18H19N7O5Cl2·1.75CF3CO2H·1.25H2O的分析计算值:C,36.56;H,3.32;N,13.88;实测值:C,36.35;H,2.96;N,14.28。NMR和MS与要求的结构相一致。
实施例26
除了使用实施例S的产物替代实施例R步骤4的产物以外,如同在实施例9中描述的那样,制备以上化合物。
对C21H22BrIN6O6·2CF3COOH·0.25H2O的分析计算值:C,33.60;H,2.76;N,9.40;实测值:C,34.23;H,2.35;N,9.72。
实施例27
使用实施例21步骤1的方法,合成在这个制备中所需要的起始原料。步骤1
在室温下,于5磅/平方英寸压力的氢气下,用催化量的5%Pt/C将起始原料(1.4g)在乙醇(50ml)中的溶液处理5小时。过滤该反应混合物,浓缩并干燥,得到白色固体(1.1g)。1H-NMR与提出的结构相一致。步骤2
按照在实施例21步骤4中给出的实验方法,制备化合物2。1H-NMR与提出的结构相一致。步骤3
按照在实施例21步骤5中给出的实验方法,使产物2的酯基团水解。1H-MR与提出的结构相一致。
对C22H24N6O7Cl2·1.50TFA·0.25H2O的分析计算值:C,41.08;H,3.59;N,11.50;实测值:C,41.03;H,3.69;N,11.45。
实施例28
如同在实施例18步骤1中描述的那样,制备在这个制备中所需要的起始原料。步骤1
除了2-甲氧基乙氧基甲基氯替代碘甲烷用作烷基化试剂以外,按照在实施例18步骤2中给出的方法,制备化合物1。1H-NMR与提出的结构相一致。步骤2
在室温下,于5磅/平方英寸的氢气下,以催化量的5%Pt/C将步骤1中描述的产物(4.6g)在甲醇(50ml)中的溶液处理2小时。过滤该反应混合物,浓缩并干燥,得到粘性的油(4.3g)。1H-NMR与提出的结构相一致。步骤3
使用在实施例21步骤4中描述的反应条件,使一份步骤2的产物(2.0g)胍基化,得到作为粘性金黄色油的产物3(890mg)。1H-NMR与提出的结构相一致。步骤4
使用在实施例21步骤5中给出的实验方法,使产物3的酯基团水解。1H-NMR与提出的结构相一致。步骤5
按照实施例12步骤4中给出的方法,将步骤4的产物偶合于实施例1的产物。1H-NMR与提出的结构相一致。步骤6
按照在实施例21步骤5中给出的方法,使步骤5产物的酯基团水解。1H-NMR与提出的结构相一致。
对C25H31N6O9Br2·1.75TFA·0.25H2O的分析计算值:C,37.07;H,3.63;N,9.10;实测值:C,36.86;H,3.66;N,9.39。
实施例29
在这个方法中,实施例28步骤5的产物用作起始原料。步骤1
在室温下,用无水HCl在乙醇(10ml)中的饱和溶液将一份起始原料(425mg)处理4小时。将该反应混合物浓缩至干并且不经纯化把该产物用于下一步。步骤2
按照实施例21步骤5中的方法,将来自步骤1的粗品产物的酯基团水解。1H-NMR与提出的结构相一致。
对C21H22N6O7Br2·1.75TFA·0.25H2O的分析计算值:C,35.27;H,2.93;N,10.07;实测值:C,35.21;H,3.16;N,10.27。
实施例S
按照在实施例G中描述的方法,制备以上化合物。在步骤2A中,使用3-碘代-5-溴代水杨醛替代3,5-二氯水杨醛。
实施例T
按照在实施例G中描述的方法,制备以上化合物。在步骤2A中,使用3-溴-5-氯代水杨醛替代3,5-二氯代水杨醛。相似地,在步骤2B中,使用R-苯基甘氨醇替代S-苯基甘氨醇。
实施例U
按照在实施例D中描述的方法,制备以上化合物。如同以下描述的那样,拆分在步骤2中得到的外消旋混合物。 步骤1
步骤1
向在CH2Cl2(500mL)和水(380mL)中的从实施例D步骤2中得到的外消旋氨基酯盐酸盐(50.0g,158.9mmol)中加入NaHCO3(38.2g,454.5mmol)。在室温下,将该混合物搅拌10分钟,伴随剧烈气体逸出。伴随快速搅拌下,于20分钟内加入氯甲酸苄酯(43.4g,222.8mmol)在CH2Cl2(435mL)中的溶液。40分钟后,把反应混合物倾入到分液漏斗中并收集有机溶液。用CH2Cl2(170mL)洗涤水相。干燥(MgSO4)合并的有机溶液并真空浓缩。用己烷研磨生成的胶状固体并经过滤收集。真空干燥褐色固体,得到要求的外消旋产物(62.9g,96%收率)。使用手性柱使该物料经受反相HPLC,得到每一个纯的对映体A和B。所使用的柱为10微米粒度的Whelk-O(R,R),使用90∶10庚烷∶乙醇的流动相。使用具有相似的溶剂和条件的分析型HPLC测量光学纯度是>98%。1H NMR与提出的结构相一致。步骤2A
向步骤1的对映体A(57.9g,140.4mmol)在CH2Cl2(600ml)中的溶液中借助加料管加入在CH2Cl2(125ml)中的三甲基甲硅烷基碘(33.7g,168.5mmol)。在室温下,将该橙色溶液搅拌1小时。滴加入甲醇(27.3mL,674.1mmol)并把该溶液搅拌15分钟。真空浓缩反应溶液,得到橙色的油。将残余物溶于甲基叔丁基(670ml)中并用1M HCl(420mL)和水(1×230mL,1×130mL)提取。用MTBE(130mL)回洗水提取液。向该水溶液中分小批加入固体NaHCO3(52.9g,630mmol)。用MTBE(1×1.2L,2×265mL)提取碱化的含水混合物。用盐水洗涤合并的有机溶液并真空浓缩,得到要求的产物,(28.6g,73%收率)。1H NMR与提出的结构相一致。步骤2B
向步骤1的对映体B、实施例1化合物(38.5g,93.4mmol)在CH2Cl2(380ml)中的溶液中借助加料管加入在CH2Cl2(80ml)中的三甲基甲硅烷基碘(25.0g,125.0mmol)。在室温下,将该橙色溶液搅拌1.5小时,滴加入甲醇(20.0mL,500.0mmol)并把该溶液搅拌20分钟。真空浓缩反应物溶液,得到橙色的油。将残余物溶于乙醚(450ml)中并用1M HCl(320mL)和水(1×200mL,1×100mL)提取。用乙醚(100mL)回洗水提取液。向该水溶液中分小批加入固体NaHCO3(40.1g,478mmol)。用乙醚(1×1.0L,2×200mL)提取碱化的含水混合物。用盐水洗涤合并的有机溶液并真空浓缩,得到要求的产物(20.8g,80%收率)。
对C11H13Cl2NO3的分析计算值:C,47.50;H,4.71;N,5.04;实测值:C,47.11;H,4.66;N,4.93。
通过使用在实施例G的步骤4和5中描述的试剂和条件,将步骤2B的产物转化为目标中间体。
实施例V
按照在实施例G中描述的方法,制备以上化合物,在步骤2A中,使用5-溴-3-碘代苯甲醛替代3,5-二氯水杨醛。
实施例30 步骤1制备
步骤1制备
向搅拌着的4-甲氧基-2-氨基吡啶甲酸甲酯(602mg,3.3mmol)(JACS,78,4130,1956)在CH2Cl2(20ml)中的溶液中加入苯甲酰基异硫氰酸酯(1ml,Aldrich),并在室温下将该反应混合物搅拌90分钟。用乙醚(20ml)稀释混合物。在冰浴上冷却混合物并过滤固体且用乙醚洗涤,干燥,得到作为白色固体的要求的化合物(828mg,73.27%收率):1H NMR:300MHz与提出的结构相一致。步骤2制备
在氮气氛下,向搅拌着的步骤1的产物(750mg,2.17mmol)在甲醇(30ml)中的溶液中加入NaOMe(538mg)。在室温下,把该反应混合物搅拌2小时。用冰乙酸(0.575ml)猝灭该混合物,并在室温下搅拌15分钟。真空过滤固体,用冷的甲醇洗涤并干燥,得到作为白色固体的要求的化合物(385mg,73%收率):1H NMR:300MHz与提出的结构相一致。步骤3制备
向搅拌着的步骤2的产物(364mg,1.5mmol)在甲醇(20ml)中的溶液中加入碘甲烷(0.25ml)。将该反应混合物加热回流2小时。把该混合物冷却至室温并浓缩,得到作为白色固体的要求的化合物(560mg):1H NMR:300MHz与提出的结构相一致。步骤4制备
向搅拌着的步骤3的产物(500mg,1.30mmol)在N,N-甲基乙酰胺(10ml)中的溶液中加入1,3-二氨基-2-羟基丙烷(123mg,1.37mmol)。将该反应混合物加热至90℃反应2小时。真空浓缩混合物,得到粗品产物,其经反相HPLC纯化,得到作为白色固体的要求的化合物(228mg):1H NMR:300MHz与提出的结构相一致。步骤5制备
向搅拌着的步骤4的产物(200mg)在甲醇(3ml)和THF(3ml)中的溶液中加入1N NaOH溶液(3ml)。在室温下,将该反应混合物搅拌1小时。真空浓缩混合物,得到油状胶,用水(2ml)处理,并用1N HCl(3ml)中和。真空浓缩混合物,得到粗品产物,其经反相HPLC纯化,得到作为白色固体的要求的化合物(228mg)。1H NMR:300MHz与提出的结构相一致。步骤6制备
向搅拌着的步骤5的产物(151.25mg,0.5mmol)在N,N-二甲基乙酰胺(5ml)中的溶液中加入4-甲基吗啉(202mg,2mmol)和1-羟基苯并三唑(67mg,0.5mmol)及实施例R的氨基酯产物(208.5mg,0.5mmol)。在室温下,将该混合物搅拌5分钟。用1-[3-(二甲基氨基)丙基-3-乙基碳二亚胺(96mg,0.5mmol)和4-(二甲基氨基)吡啶(10mg)处理反应混合物。在氮气氛下,在室温下把反应混合物搅拌72小时。用水(1ml)猝灭反应混合物并真空浓缩,得到粗品产物,其经反相HPLC纯化,得到作为白色固体的要求的化合物(42mg):1H NMR:400MHz与提出的结构相一致。步骤7制备
向搅拌着的步骤6的产物(35mg)在甲醇(3ml)和THF(3ml)中的溶液中加入1N NaOH溶液(3ml)。在室温下,将该反应混合物搅拌1小时。真空浓缩混合物,得到油状胶,用水(2ml)处理,并用1N HCl(3ml)中和。真空浓缩混合物,得到粗品产物,其经反相HPLC纯化,得到作为白色固体的要求的化合物(22mg)。1H NMR:400MHz与提出的结构相一致。
对C22H24N6O7BrCl·2TFA,0.5H2O的分析计算值:C,37.22;H,3.25;N,10.04;实测值:C,36.91;H,3.17;N,10.02。
实施例31 步骤1制备
步骤1制备
向搅拌着的实施例30步骤4的产物(900mg)在冰乙酸(10ml)中的溶液中加入48%HBr(10ml)。将该反应混合物加热回流3小时。把混合物冷却至室温并在室温下搅拌18小时。真空浓缩混合物,得到粗品产物,其经反相HPLC纯化,得到作为油状胶的要求的化合物(720mg)。1H NMR:300MHz与提出的结构相一致。步骤2制备
向搅拌着的步骤1的产物(720mg,1.7mmol)在N,N-二甲基乙酰胺(10ml)中的溶液中加入4-甲基吗啉(520mg,5.2mmol)和1-羟基苯并三唑(221mg,1.71mmol)及实施例R的氨基酯产物(710mg,1.71mmol)。在室温下,将该混合物搅拌5分钟。用1-[3-(二甲基氨基)丙基-3-乙基碳二亚胺(211mg,1.7mmol)和4-(二甲基氨基)吡啶(10mg)处理反应混合物。在氮气氛下,在室温下把反应混合物搅拌72小时。用水(1ml)猝灭反应混合物并真空浓缩,得到粗品产物,其经反相HPLC纯化,得到作为白色固体的要求的化合物(182mg)。1H NMR:300MHz与提出的结构相一致。步骤3制备
向搅拌着的步骤2的产物(100mg)在甲醇(3ml)和THF(3ml)中的溶液中加入1N NaOH溶液(3ml)。在室温下,将该反应混合物搅拌1小时。真空浓缩混合物,得到油状胶,用水(2ml)处理,并用1N HCl(3ml)中和。真空浓缩混合物,得到粗品产物,其经反相HPLC纯化,得到作为白色固体的要求的化合物(42mg)。1H NMR:300MHz与提出的结构相一致。
对C21H22N6O7BrCl·1.5TFA,0.5H2O的分析计算值:C,37.64;H,3.22;N,10.97;实测值:C,37.56;H,3.05;N,10.99。
实施例32
在0℃下,向实施例9步骤3的产物(860mg,2.45mmol)和实施例T的产物(1.0g,2.41mmol)在二甲基乙酰胺(24mL)中的溶液中加入HOBT(358mg,2.64mmol)随后加入N-甲基吗啉(0.6mL,5.15mmol)。将该反应物溶液搅拌15分钟并加入EDC(470mg,2.45mmol)。把该混合物温热至室温过夜。真空浓缩混合物并将残余物在乙酸乙酯和水之间分配。用乙酸乙酯提取水溶液,干燥(Na2SO4)合并的有机层并且浓缩。经反相HPLC(90∶10水/TFA∶MeCN开始梯度洗脱,保留时间22.5min)纯化残余物,得到含有回收的化合物B(480mg)的要求的偶合产物。用1M NaOH水溶液(6mL)处理物料(化合物B)并搅拌3小时。用TFA将溶液酸化至pH4。经反相HPLC(95∶5水/TFA∶MeCN开始梯度洗脱,保留时间24.5min)纯化该混合物,得到要求的产物(160mg,8%收率对于这两个步骤)。对C21H22BrClN5O6+2.4TFA的分析计算值:C,36.74;H,2.92;N,9.96;实测值:C,36.83;H,3.07;N,9.88。
1H NMR与提出的结构相一致。
实施例33 步骤1
步骤1
在室温下,于5磅/平方英寸的氢气压力下,用催化量的5%Pd/C把实施例18步骤2的物料(5.72g)在THF(80ml)中的溶液处理2小时。过滤反应混合物并用DMF(250ml)洗涤残余物。浓缩滤液并用乙醚处理残余物,过滤并空气干燥,得到棕色粉末(4.9g),其无需进一步纯化直接用于下一步。1H-NMR与提出的结构相一致。步骤2
使用三BOC试剂和在实施例21步骤4中描述的条件,使步骤1的物料(500mg)胍基化,得到作为金黄色玻璃状物的要求的产物(128mg)。1H-NMR与提出的结构相一致。步骤3
使用在实施例21步骤5中给出的实验方法,将步骤2产物的酯基团水解。1H-NMR与提出的结构相一致。对C11H14N4O4.1.5TFA的分析计算值:C,38.45;H,3.57;N,12.81;实测值:C,38.32;H,3.77;N,12.80。步骤4
使用与在实施例12步骤4中描述的相同的方法,通过将在步骤3中描述的产物与实施例R的产物偶合,制备要求的化合物。1H-NMR与提出的结构相一致。步骤5
使用如同在实施例12步骤5中描述的相同方法,将步骤4产物的酯基团水解。1H-NMR与提出的结构相一致。对C21H22N6O7BrCl.1.5TFA.0.25H2O的分析计算值:C,37.86;H,3.18;N,11.04;Cl,4.66;Br,10.50.实测值:C,37.60;H,3.26;N,11.20;Cl,4.79;Br,10.19。
实施例34
使用在实施例33中描述的方法,制备以上化合物。在步骤4中,实施例H的产物被用于替代实施例R的产物。对C21H22N6O7BrCl.2.0TFA.0.5H2O的分析计算值:C,36.49;H,3.06;N,10.21;实测值:C,36.10;H,2.83;N,10.29。
实施例35
使用在实施例33中描述的方法,制备以上化合物。在步骤4中,实施例V的产物被用于替代实施例R的产物。对C21H22N6O6Br21.75TFA的分析计算值:C,35.76;H,3.03;N,10.21;实测值:C,35.58;H,3.07;N,10.61。
实施例36步骤1
使用双-BOC试剂G2和在实施例18步骤5中描述的反应条件,将实施例33步骤1的产物(261mg)胍基化,得到作为白色固体的产物(207mg)。然后使用在实施例21步骤5中描述的实验条件把该物料皂化。1H-NMR与提出的结构相一致。步骤2
使用与实施例12步骤4中描述的相同的方法,制备要求的化合物。将在步骤1中描述的产物与实施例1的产物偶合。1H-NMR与提出的结构相一致。步骤3
使用与实施例12的步骤5中描述的相同的方法,将步骤2产物的酯基团水解。1H-NMR与提出的结构相一致。对C20H20N6O6Br21.75TFA0.25H2O的分析计算值:C,35.10;H,2.79;N,10.45;实测值:C,34.85;H,2.59;N,10.62。
实施例37步骤1
在室温下,将2-硝基噻吩-4-甲醛(5.0g)悬浮于水(100ml)中并且一次性加入氢氧化钠(5.2g)在水(50ml)中的溶液。在室温下,将黑色的反应混合物搅拌1小时,然后经过硅藻土垫过滤。用1N HCl溶液使滤液酸化,然后用乙酸乙酯提取。用饱和的氯化钠溶液洗涤合并的有机提取液,干燥(Na2SO4)并浓缩,得到淡黄色固体(5.0g)。其无需进一步纯化即可使用。1H-NMR与提出的结构相一致。步骤2
在室温下,将步骤1的产物(2.5g)与在DMF(30ml)中的碳酸钾(2.0g)和碘甲烷(2.2g)一起搅拌16小时。把反应混合物在乙酸乙酯和水之间分配并分层。用另外的乙酸乙酯提取含水部分,然后用水、饱和的氯化钠溶液洗涤合并的有机提取液,干燥(Na2SO4)并浓缩。在硅胶柱上纯化深色残余物,用15%乙酸乙酯-85%己烷洗脱,得到黄色固体(950mg)。1H-NMR与提出的结构相一致。步骤3
将步骤2中得到的产物(2.0g)溶于甲醇(50ml)中,并在60℃下,于50磅/平方英寸的氢气压力下用催化量的5%Pd/C处理32小时。将反应物冷却并过滤,浓缩滤液。在硅胶柱上纯化残余物,用25%乙酸乙酯-75%己烷洗脱,得到淡黄色固体(920mg)。1H-NMR与提出的结构相一致。步骤4
将步骤3的产物(900mg)的溶液溶于乙酸乙酯(20ml)中并在室温下用一份苯甲酰基异硫氰酸酯(947mg)处理。将反应物搅拌30分钟并过滤沉淀,用乙酸乙酯洗涤且空气干燥,得到淡黄色固体(1.51g)。1H-NMR与提出的结构相一致。步骤5
将实施例35步骤4中得到的产物溶于甲醇(70ml)中并在室温下分批用甲醇钠(1.3g)处理。将反应物搅拌30分钟,然后用冰乙酸(1.4g)猝灭。浓缩反应物并且残余物在乙酸乙酯和水之间分配。用另外的乙酸乙酯提取含水部分并用饱和的氯化钠溶液洗涤合并的有机提取液,干燥(Na2SO4)并浓缩,得到淡黄色粉末(900mg)。1H-NMR与提出的结构相一致。步骤6
将步骤5的产物(900mg)溶于甲醇(20ml)中并用碘甲烷(1.4g)处理。将该反应物回流2小时,然后冷却并浓缩。用乙醚处理残余物,过滤并空气干燥,生成白色固体(1.27g)。1H-NMR与提出的结构相一致。步骤7
在95-100℃下,将步骤6的产物(1.2g)与1,3-二氨基-2-羟基丙烷(469mg)和DMA(20ml)一起加热8小时。将该反应物冷却并高真空下除去溶剂。借助反相HPLC纯化残余物,以水(0.5%TFA)-乙腈梯度洗脱,得到要求的产物(255mg)。1H-NMR与提出的结构相一致。步骤8
使用如同在实施例21步骤5中描述的条件,使用步骤7中生成的产物,制备要求的化合物。1H-NMR与提出的结构相一致。步骤9
使用与实施例12步骤4中描述的相同的方法,通过使步骤8的产物与实施例G的物料偶合,制备要求的化合物。1H-NMR与提出的结构相一致。步骤10
使用与实施例12步骤5中描述的相同的方法,将步骤9产物的酯基团水解。1H-NMR与提出的结构相一致。对C20H21N5O6Cl2S.1.75TFA的分析计算值:C,38.43;H,3.19;N,9.54;S,4.37;实测值:C,38.43;H,3.39;N,9.73;S,4.27。
实施例38步骤1
在室温下,用70%硝酸(4ml)处理乙酸酐(20ml)。然后在-30℃下将该溶液滴加到N-甲基吡咯-2-甲酸(5.0g)在乙酸酐(30ml)中的搅拌着的混合物中。将该反应物搅拌并使其温热至室温30分钟。然后把反应物重新冷却至-25℃并通过冷的滤器过滤,用冷的己烷洗涤并空气干燥,得到黄色粉末(1.5g)。1H-NMR与提出的结构相一致。步骤2
使用在步骤1中制备的产物,使用在实施例37步骤2中描述的方法,制备要求的化合物。经硅胶柱上的层析法纯化粗品产物,使用40%乙酸乙酯-己烷洗脱。1H-NMR与提出的结构相一致。步骤3
将步骤2的产物(1.0g)溶于甲醇(25ml)中,并在室温下,于5磅/平方英寸的氢气压力下用催化量的5%Pd/C处理3小时。过滤反应物并浓缩,得到深红色液体(1.0g),其无需进一步纯化即可使用。1H-NMR与提出的结构相一致。步骤4
使用实施例37中描述的步骤4-8的顺序,使用在步骤3中描述的产物,制备要求的化合物。1H-NMR与提出的结构相一致。步骤5
在氮气下,将步骤4的产物(425mg)、2-氯-4,6-二甲氧基三嗪(201mg)和N-甲基吗啉(263mg)在DMA(10ml)中的溶液于冰浴上搅拌。将该反应物温热至室温,并继续搅拌3小时。制备实施例G的产物(386mg)和N-甲基吗啉(105mg)在DMA(5ml)中的溶液并在室温下一次性加入到反应物中。将反应物搅拌15小时,用TFA(1.5ml)猝灭,然后高真空浓缩。借助反相HPLC纯化残余物,以水(0.5%TFA)-乙腈梯度洗脱,得到白色固体(706mg)。1H-NMR与提出的结构相一致。步骤6
使用与实施例12步骤5中描述的相同的方法,将步骤5产物的酯基团水解。1H-NMR与提出的结构相一致。对C21H24N6O6Cl21.5TFA·0.5H2O的分析计算值:C,40.75;H,3.78;N,11.88;Cl,10.02;实测值:C,40.40;H,3.68;N,12.10;Cl,10.20。
实施例39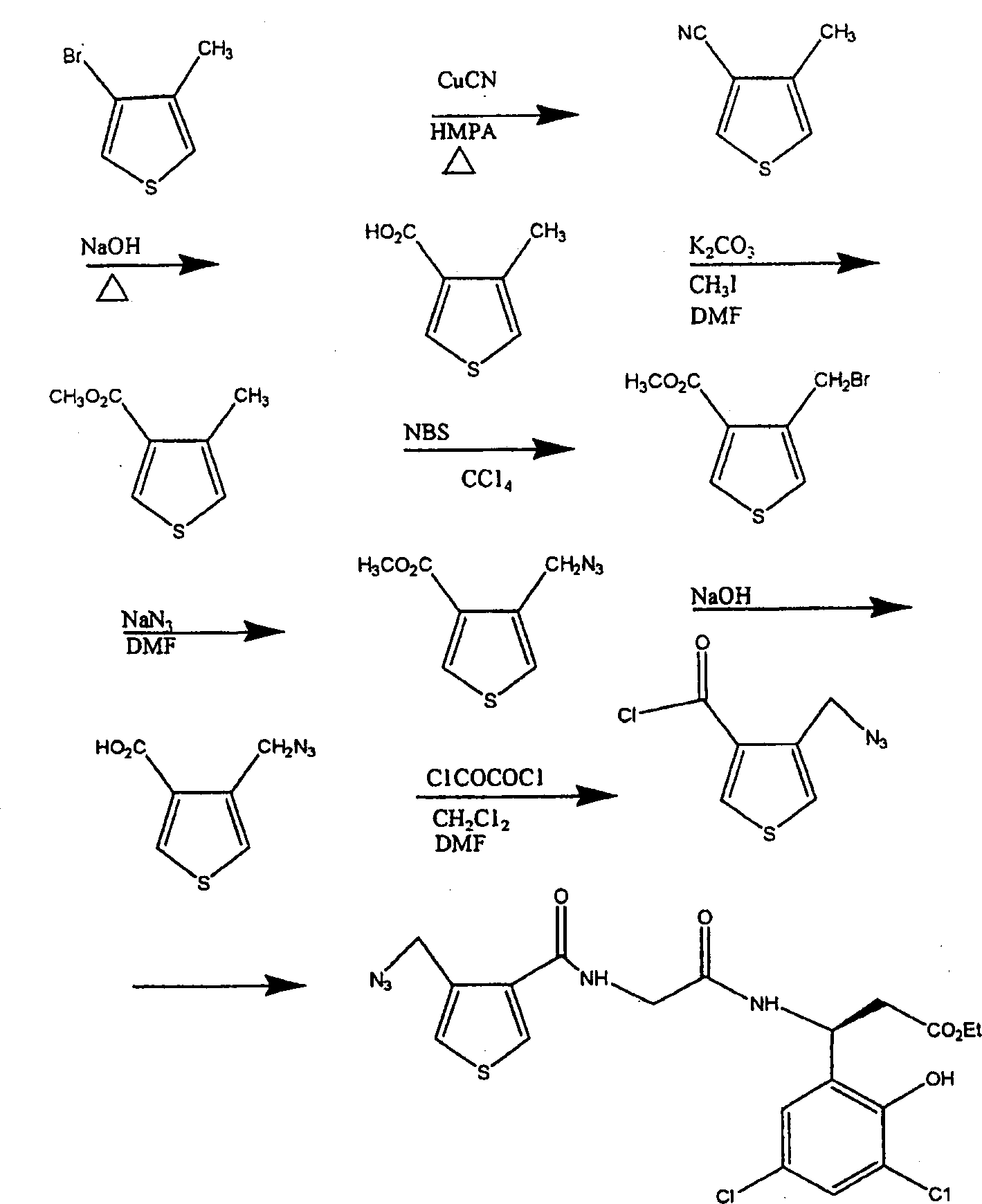
 步骤1
步骤1
在130-140℃下,将3-溴-4-甲基噻吩(10g)、氰化铜(Ⅰ)(11.3g)和HMPA(15ml)的混合物加热18小时。将混合物冷却并倾入到搅拌着的氰化钠(18.8g)在水(28ml)中的溶液中并搅拌1小时。用一些另外的水稀释粘稠的棕色混合物并以乙醚提取。用水、饱和的氯化钠溶液洗涤合并的乙醚提取液,干燥(Na2SO4)并浓缩。残余物在硅胶柱上纯化,以90%己烷-10%乙酸乙酯洗脱,得到浅黄色液体(5.2g)。1H-NMR与提出的结构相一致。步骤2
将在步骤1中制备的腈(1.5g)和2N氢氧化钠溶液(150ml)的混合物回流1小时。使该反应物冷却并用2N盐酸酸化直到pH~2且以乙醚提取。干燥(Na2SO4)合并的乙醚提取液并浓缩,得到白色固体(16.4g),其无需进一步纯化即可使用。1H-NMR与提出的结构相一致。步骤3
如同在实施例37步骤2中描述的那样,将步骤2的产物(16.4g)酯化,硅胶柱上纯化,用10%乙酸乙酯-90%己烷洗脱后,得到要求的产物(13.6g)。1H-NMR与提出的结构相一致。步骤4
将N-溴代琥珀酰亚胺(6.3g)和过氧化二苯甲酰(100mg)在四氯化碳(25ml)中的混合物于90分钟内加入到步骤3的产物(5.0g)和过氧化二苯甲酰(100mg)在CCl4(25ml)中的回流着的溶液中。在回流2小时后,使反应混合物冷却,过滤并浓缩,且残余物在硅胶柱上纯化,使用10%乙酸乙酯-90%己烷洗脱,得到要求的化合物(4.3g)。1H-NMR与提出的结构相一致。步骤5
在氮气氛下,于55℃下将步骤4中描述的产物(4.2g)、叠氮化钠(2.9g)和DMF(50ml)的混合物加热5小时。使反应混合物冷却并在水和乙酸乙酯之间分配。分层并用另外的乙酸乙酯提取含水部分。用饱和的氯化钠溶液洗涤合并的有机提取液,干燥(Na2SO4)并浓缩,得到金黄色的油(3.5g),其无需进一步纯化即可使用。1H-NMR与提出的结构相一致。步骤6
在室温下,将步骤5中描述的产物(1.0g)、1N氢氧化钠溶液(15ml)和甲醇(15ml)的溶液搅拌16小时。用冰乙酸(2ml)处理反应物并浓缩。将残余物在水和乙酸乙酯之间分配。分层,并用另外的乙酸乙酯提取含水部分。用水、饱和的氯化钠溶液洗涤合并的有机提取液,干燥(Na2SO4)并浓缩。在61℃下,高真空下干燥残余物2小时,得到白色固体(860mg)。1H-NMR与提出的结构相一致。步骤7
在冰浴上使步骤6中描述的产物(850mg)在二氯甲烷(10ml)中的溶液冷却并用2N草酰氯在二氯甲烷中的溶液(5ml)一次性处理,随后加入2滴DMF。将反应物搅拌2小时,其间使其温热至室温。浓缩溶液并在室温下高真空下干燥16小时,得到黄棕色固体(785mg)。1H-NMR与提出的结构相一致。步骤8
在氮气下,于火焰干燥的烧瓶中加入二异丙基乙基胺(1.3ml)、实施例G的产物(1.4g)和DMA(10ml)的溶液。在冰浴上使该溶液冷却并滴加入步骤7的酰氯(770mg)在DMA(5ml)中的溶液。加入完成后,将该反应物搅拌30分钟,然后在乙酸乙酯和水之间分配。分层并用另外的乙酸乙酯提取含水部分。在硅胶柱上纯化合并的有机提取液,用50%乙酸乙酯-50%己烷洗脱,得到白色固体(1.0g)。1H-NMR与提出的结构相一致。步骤9
在室温下,于5磅/平方英寸的氢气下,将步骤8的产物(806mg)在乙醇(20ml)中的溶液与催化量的5%Pt/C一起加热21小时。过滤反应物并浓缩,且残余物经反相HPLC纯化,用水(.5%TFA)-乙腈梯度洗脱,得到白色固体(480mg)。1H-NMR与提出的结构相一致。步骤10
使用在实施例21步骤4中描述的条件,将步骤9中描述的产物(550mg)胍基化并纯化,得到白色固体(206mg)。1H-NMR与提出的结构相一致。步骤11
使用在实施例12步骤5中阐述的相同方法,将步骤10的产物的酯基团水解。1H-NMR与提出的结构相一致。对C21H23N5O5Cl2S·1.5TFA,0.5H2O的分析计算值:C,39.79;H,3.55;N,9.67;Cl,9.79;S,4.43实测值:C,39.91;H,3.58;N,9.94;Cl,10.06;S,4.52。
实施例40步骤1
使用在实施例38步骤5中描述的方法,将实施例33步骤3中制备的产物(1.0g)与甘氨酸乙酯盐酸盐偶合,类似纯化后得到黄色固体(1.1g)。1H-NMR与提出的结构相一致。步骤2
使用在实施例21步骤5中描述的方法,将步骤1中描述的产物(1.0g)水解,得到白色固体(910mg)。1H-NMR与提出的结构相一致。步骤3
使用在实施例38步骤5中描述的方案,将步骤2的产物与3-氨基-4,4,4-三氟-丁酸乙酯盐酸盐偶合。1H-NMR与提出的结构相一致。步骤4
使用实施例12步骤5中描述的方法,将步骤3的产物中的酯基团水解。1H-NMR与提出的结构相一致。对C17H20N5O6F3·1.5TFA的分析计算值:C,38.01;H,3.67;N,11.08;实测值:C,37.88;H,3.64;N,11.02。
实施例41 步骤1
步骤1
在氩气下,将实施例33步骤3的产物(527mg,1.3mmol)和CDMT(244mg,1.4mmol)溶于DMAC(8ml)中。把该溶液冷却至0℃下反应3小时并滴加入NMM(0.15ml,1.3mmol)。在0℃下,把该溶液搅拌3小时,然后加入在DMAC(10ml)中的实施例U的产物和NMM(0.15ml)。把反应物温热至室温并在室温下搅拌过夜。用TFA(2ml)猝灭反应物并真空浓缩。经反相HPLC纯化粗品黄色固体,使用90∶10的H2O/TFA∶CH3CN(入=254nm)梯度洗脱。分离得到作为白色固体的产物(725mg,75%收率)。对C23H26N6O7Cl2·1.5TFA的分析计算值:C,42.12;H,3.74;N,11.35;实测值:C,42.26;H,3.87;N,11.45。
H.R.M.S.M+1对C23H27N6O7Cl2的计算值569.1318。实测值569.1323。步骤2
将步骤1的产物(725mg,0.98mmol)溶于THF(5ml)/lCH3OH)中并加入1M NaOH(6.5ml)。在室温下,将该反应物搅拌过夜,然后加入1M HCl(6.5ml)。在真空下浓缩后,经反相HPLC纯化粗品黄色固体,使用95∶5的H2O/TFA∶CH3CN(入=254nm)梯度洗脱。分离得到作为白色固体的产物(589mg,79%收率)。对C21H22N6O7Cl2·1.9TFA的分析计算值:C,39.30;H,3.18;N,11.09;实测值:C,39.22;H,3.16;N,11.39。
H.R.M.S.M+1对C21H23N6O7Cl2的计算值541.1005。实测值541.1000。
实施例42步骤1
在高压反应器中,将2-甲氧基-6-氯吡啶甲酸(10g,0.053mol)溶于过量的浓氢氧化铵水溶液中并在175℃下加热24小时,达到500磅/平方英寸的压力。把该溶液减压下蒸发至干并在25℃下经2N HCl处理2天转化为酸,然后在高真空下蒸发至于,其间加热到60℃。12小时后,用无水乙醇共沸除去残留的水,并且用甲醇处理生成的残余物且减压下蒸发三次。将生成的残余物溶于甲醇(250ml)并把在二噁烷中的4N HCl(25mol)加入到该溶液中,然后回流2.5天。减压下蒸发除去溶剂并经反相HPLC(梯度,95/5在H2O中的0.1%TFA/CH3CN-60/40在H2O中的0.1%TFA/CH3CN)纯化生成的粗品残余物。将要求的产物溶于甲醇中并且用固体碳酸氢钠处理,过滤,并蒸发溶剂。将生成的固体悬浮于50/50乙酸乙酯/己烷中并过滤收集固体,得到要求的棕色固体(1.7g,20%收率)。步骤2
将步骤1的化合物(300mg,1.8mmol)溶于DMF(7.1ml)中并加入实施例9步骤1的产物(927mg,2.1mmol),随后加入三乙胺(0.44ml,325mg,3.2mmol)和氯化汞(326mg,1.2mmol)。在25℃下,将反应混合物搅拌3.5小时,然后在60℃下加热16小时。在冷却至室温后,向反应物中加入乙酸乙酯(7ml)并通过硅藻土过滤不均匀的溶液。用97/3的乙酸乙酯/乙醇彻底洗涤硅藻土。减压下除去溶剂并把生成的红色粗品油溶于二氯甲烷(30ml)中并用三氟乙酸(2.77ml)处理且回流1小时。将反应物冷却并减压下除去溶剂,得到棕色的油。经反相HPLC(Cl8,梯度90/10 0.1%TFA/H2O)纯化样品,得到不纯的化合物,使其溶于甲醇(2ml)中并用1N NaOH(0.15ml)处理且解吸至干,得到深棕色的油。经反相HPLC(Cl8,梯度99/1 0.1%TFA/H2O/CH3CN)纯化该油,得到褐色的固体(36mg)。步骤3
将先前步骤的化合物(74mg;0.18mmol)在真空烘箱中干燥过夜并且溶于DMF(1ml)(经分子筛贮存)中,并冷却至0℃。然后加入N-甲基哌啶(27ml;22mg;0.22mmol),随后加入氯甲酸异丁酯(47ml;49mg,0.36mmol)并搅拌5分钟。加入实施例G的产物在DMF中的溶液(0.2mL)并用另外的DMF(0.2mL)洗涤残留的β-氨基酯。加入下一份N-甲基哌啶(22ml;18mg;0.18mmol)。使该反应混合物缓慢温热至室温。在25℃下搅拌12小时后,高真空下除去溶剂,得到红棕色的油,其经反相HPLC(Cl8,梯度80/20 0.1%TFA/H2O/CH3CN)纯化,得到所述偶合产物(22mg)和具有结合到吡啶酮的氧原子上的异丁基甲酰基的偶合产物(47mg)。合并这些产物并溶于甲醇(2ml)中,向其中加入1N氢氧化钠水溶液(240ml)。在25℃下,将该反应物搅拌过夜并加入三氟乙酸(84.7ml)。减压下除去溶剂并经反相HPLC(Cl8,梯度95/5 0.1%TFA/H2O/CH3CN)纯化,得到要求的产物(37mg)。对C21H22N6O7Cl2·2.1TFA·0.2H2O的微量分析计算值:C,38.58;H,3.15;N,10.71;实测值:C,38.12;H,3.36;N,10.71。
实施例43
按照实施例42步骤3中描述的方法,通过使实施例42步骤2的产物与实施例R的产物反应,制备以上化合物。对C21H22N6O7ClBr.1.8TFA的微量分析计算值:C,37.35;H,3.03;N,10.62;实测值:C,37.31;H,3.23;N,10.65。
实施例44步骤1
将实施例17步骤3的产物(3.0g,5.77mmol)溶于异丙醇(25ml)中并向该溶液中加入浓氢氧化铵(45ml)。在25℃下搅拌3小时后,该溶液减压下蒸发至干。在25℃下,在5磅/平方英寸下,于帕尔振摇器中,把粗品样品在乙醇(3A)(50ml)中经5%披铂炭氢化2.5小时。过滤样品并减压下将滤液浓缩至干。经反相层析法(Cl8,80/20CH3CN/H2O梯度)纯化化合物,得到要求的产物(1.5g,39%收率)。步骤2
将步骤1的产物(200mg)溶于甲醇(2ml)中并加入碳酸氢钠(133mg)。在25℃下,将溶液搅拌30分钟,过滤并使生成的溶液蒸发至干。把生成的样品(205mg;0.43mmol)溶于DMF(1.72ml)中,然后加入实施例9步骤1的产物(282.8mg,0.65),随后加入三乙胺(99mg;0.97mmol,0.14ml),然后加入氯化汞(177mg;0.65mmol)。将溶液加热至60℃下进行30分钟,然后加热至90℃ 12小时。冷却反应混合物,然后用乙酸乙酯(1.7ml)稀释并经硅藻土过滤。把生成的滤液蒸发至干。经快速层析法纯化粗品,以除去未反应的原料。用50/50的TFA/CH2Cl2处理所述物料,得到不纯的要求的产物。将该物料溶于甲醇(2ml)和1N氢氧化钠(2ml)中。在25℃下搅拌12小时后,用TFA(77ml)中和该溶液,然后减压下蒸发至干。经反相层析法(Cl8,95/5 H2O/CH3CN)纯化粗品产物,得到要求的产物(6.8mg)。对C21H23N7O6Cl2·3.3TFA的微量分析计算值:C,36.17;H,2.89;N,10.70;实测值:C,36.57;H,3.21;N,10.37。
实施例45 步骤1
步骤1
向3-甲基-2-噻吩甲酸(1.0g,7.0mmol)、N,N-二甲基甲酰胺(4.0ml)和碳酸钾(1.93g,14mmol)的混合物中加入碘甲烷(1.49g,10.5mmol)。在室温下,将反应物搅拌80小时,并用乙酸乙酯(150ml)稀释,用H2O(100ml)和盐水(100ml)洗涤。干燥(Mg2SO4)有机层并浓缩至作为浅棕色油的纯的产物(0.87g,81%收率)。1H NMR与提出的结构相一致。步骤2
在回流下,于30分钟内向步骤1的产物(5.0g,32.0mmol)、过氧化二苯甲酰(0.08g)和四氯化碳(20ml)的溶液中加入N-溴代琥珀酰亚胺(6.3g,35.3mmol)和过氧化二苯甲酰(0.08g)和四氯化碳(20ml)的混合物。将生成的反应混合物加热回流18小时。过滤固体并用四氯化碳(2×10ml)洗涤。浓缩滤液,得到油和固体的混合物(8.1g,80%收率)。1H NMR与提出的结构相一致。步骤3
在58℃下,将步骤2的产物(8.1g,34.7mmol)、叠氮化钠(5.6g,87mmol)和N,N-二甲基甲酰胺(25ml)的混合物加热2.5小时。用乙酸乙酯(800ml)稀释反应物并用H2O(500ml)洗涤。用乙酸乙酯(100ml)提取水层。干燥(Mg2SO4)合并的有机层并浓缩,得到浅棕色的油(6.0g,85%收率)。1H NMR与提出的结构相一致。步骤4
将步骤3的产物(6.0g,25.9mmol)、NaOH(1N,50ml)和MeOH(50ml)的溶液搅拌过夜。用乙酸(2.5ml)处理反应物。用乙酸乙酯(300ml)提取产物。以Na2SO4干燥有机层并浓缩,得到黄色的油。真空下得到黄色固体(2.0g,42%收率)。1H NMR与提出的结构相一致。步骤5
在室温下,将步骤4的产物(0.5g,2.7mmol)、草酰氯(2M在二氯甲烷中,2.7ml,5.4mmol)和二氯甲烷(20ml)的混合物搅拌2.5小时。从反应物中除去溶剂,得到作为棕色油的产物(0.51g,85%收率)。1H NMR与提出的结构相一致。步骤6
在0℃下,将实施例G的产物(0.91g,2.43mmol)、二异丙基乙基胺(0.63g,4.86mmol)和N,N-二甲基乙酰胺(20ml)的溶液加入到步骤5的产物(0.46g,2.43mmol)和THF(10ml)的混合物中。在0℃下,将反应混合物搅拌10分钟并使其温热至室温。16小时后,真空下从该反应物中除去溶剂。用乙酸乙酯(300ml)提取产物。用饱和的NaHCO3溶液(100ml)、H2O(100ml)洗涤有机溶液,用Na2SO4干燥并浓缩,得到粗品产物。经柱层析法(在硅胶上,CH2Cl2/MeOH/NH4OH,98/2/0.2)纯化粗品产物,得到作为胶状固体的纯的产物(0.7g,58%收率)。1H NMR与提出的结构相一致。步骤7
在室温下,于5磅/平方英寸下将步骤6的产物(0.78g,1.56mmol)、5%Pt/C和EtOH的混合物搅拌20小时。过滤催化剂。用三氟乙酸(0.6ml)处理滤液并浓缩,得到粗品产物。经HPLC纯化粗品产物得到白色固体(0.4g,54%收率)。1H NMR与提出的结构相一致。步骤8
在室温下,将氯化汞(Ⅱ)(0.51g,1.87mmol)加入到步骤7的产物(0.55g,1.25mmol)、实施例9步骤1的产物(0.81g,1.87mmol)、三乙胺(0.38g,3.75mmol)和N,N-二甲基甲酰胺(15ml)的混合物中。在95-100℃下,将反应物加热16小时。通过硅藻土垫(2”)过滤冷却的反应物并用乙酸乙酯洗涤。真空下从滤液中除去溶剂,得到油。用CH2Cl2/三氟乙酸(10ml/10ml)稀释该油并在室温下搅拌1小时。经过滤收集形成的固体。浓缩滤液并用CH2Cl2/三氟乙酸(15ml/15ml)处理1.5小时。减压下除去溶剂,得到棕色的油。经HPLC纯化该油,得到要求的产物及其乙酯的混合物。从乙腈中重结晶得到作为白色固体的要求的化合物(0.068g)。用NaOH/乙醇(7.0ml/7.0ml)把酯产物处理18小时,并经HPLC纯化,得到另外的化合物(0.055g)。
对C21H23N5O6Cl2S·2.0CF3COOH的计算值:C,38.87;H,3.26;N,9.07;实测值:C,38.78;H,3.6;N,9.16。
实施例46 步骤1
步骤1
在室温下,将苯甲酰基异硫氰酸酯(3.34g,21.3mmol)加入到实施例33步骤1的产物(3.5g,20.8mmol)和N,N-甲基甲酰胺(45ml)的溶液中。将该反应物搅拌2小时并倾入到乙醚(300ml)中。将生成的混合物搅拌15分钟。经过滤收集产物,用乙醚(2×40ml)洗涤并空气干燥为黄色固体(6.95g,100%收率)。1H NMR与提出的结构相一致。步骤2
在室温下,把甲醇钠(2.64g,48.9mmol)分小批量加入到步骤1产物(6.90g,20.8mmol)和甲醇(200ml)的悬浮液中。完成加入后,将该反应物搅拌1小时。把乙酸(2.8ml,48.9mmol)加入到该反应物中。将生成的混合物搅拌15分钟。经过滤收集产物,用甲醇(2×30ml)洗涤并减压下干燥,得到灰白色固体(3.8g,81%收率)。1H NMR与提出的结构相一致。步骤3
在回流下,将步骤2的产物(1.90g,8.4mmol)、碘甲烷(3.56,25.1mmol)和甲醇(60ml)的混合物加热3.5小时,并冷却至室温。减压下从反应物溶液中除去溶剂,得到黄色的油。用乙醚处理油状残余物,得到作为黄色固体的纯的产物(3.1g,100%收率)。1H NMR与提出的结构相一致。步骤4
在85℃下,将步骤3的产物(0.5g,1.36mmol)、2-甲氧基乙基胺(0.12ml,1.36mmol)和N,N-二甲基乙酰胺(5.0ml)的溶液加热2.5小时。加入另外的2-甲氧基乙基胺(0.12ml,1.36mmol)。在85℃下,将反应物加热2小时。减压下从冷却的反应物中除去N,N-甲基乙酰胺,得到粗品产物。经反相HPLC纯化粗品产物,得到要求的纯的产物(0.38g,76%收率)。1H NMR谱与提出的结构相一致。步骤5
在室温下,将步骤1的产物(0.38g,1.42mmol)、NaOH溶液(1N,15ml)和MeOH(15ml)的溶液搅拌16小时,然后用三氟乙酸(1.2ml)处理。减压下从反应物溶液中除去溶剂,得到粗品产物。经HPLC纯化粗品产物,得到作为白色固体的要求的产物(0.55g,100%收率)。1HNMR与提出的结构相一致。步骤6
将N-甲基吗啉(0.1ml,1.42mmol)加入到步骤5的产物(0.54g,1.42mmol)在N,N-二甲基乙酰胺(16ml)中的溶液中。在-5℃下,于5分钟内将氯甲酸异丁酯(0.18g,1.35mmol)加入到反应物中,并搅拌15分钟。将实施例G的产物(0.396g,1.07mmol)和N-甲基吗啉(0.075ml,1.07mmol)和N,N-二甲基乙酰胺(10ml)的溶液加入到该反应物中。使生成的反应物溶液温热到室温并搅拌16小时。减压下从反应物溶液中除去溶剂,得到粗品产物。经HPLC纯化粗品产物,得到作为白色固体的要求的产物(0.145g,14%收率)。1H NMR与提出的结构相一致。步骤7
将步骤6的产物(0.145g,0.20mmol)、NaOH(1N,25ml)和MeOH(25ml)的溶液搅拌16小时,减压下从反应混合物中除去溶剂,得到粗品产物。用三氟乙酸(2ml)处理粗品产物并经HPLC纯化,得到作为灰白色固体的要求的产物(0.11g,70%收率)。对C21H24N6O7Cl2·2CF3COOH的计算值:C,38.93;H,3.40;N,10.89;实测值:C,39.29;H,3.47;N,11.15。
实施例47 步骤1
步骤1
在92-95℃下,将实施例46步骤3的产物(1.56g,4.22mmol)、2,2-二甲基-1,3-丙二胺(0.45g,4.40mmol)和N,N-二甲基乙酰胺(15ml)的混合物加热6.5小时,然后使之冷却至室温。过夜形成固体。过滤收集产物,用N,N-二甲基乙酰胺(5ml)和乙醚(5ml)洗涤,得到作为白色固体的纯的产物(0.50g,43%收率)。1H NMR与提出的结构相一致。步骤2
在室温下,将步骤1的产物(0.48g,1.7mmol)、NaOH(1N,10ml)和MeOH(10ml)的溶液搅拌过夜。减压下从反应混合物中除去溶剂,得到粗品产物。用三氟乙酸(2ml)处理粗品产物并经HPLC纯化,得到作为要求的产物的白色固体(0.38g,58%收率)。步骤3
在0℃下,将4-甲基吗啉(0.14g,1.41mmol)在N,N-二甲基乙酰胺(1.5ml)中的溶液加入到步骤2的产物(0.36g,0.94mmol)、2-氯-4,6-二甲氧基三嗪(0.18g,1.03mmol)和N,N-甲基乙酰胺(10ml)的溶液中。使生成的亮橙色溶液温热至室温并搅拌3小时。将实施例G的产物(0.350g,0.94mmol)、4-甲基吗啉(0.93g,0.94mmol)和N,N-二甲基乙酰胺(4ml)的溶液加入到反应物溶液中。在室温下,将生成的溶液搅拌过夜,并用三氟乙酸(1.0ml)处理。减压下从反应物溶液中除去溶剂,得到粗品产物。经HPLC纯化粗品产物,得到作为要求的产物的黄色固体(0.35g,54%收率)。步骤4
将步骤3的产物(0.33g,0.47mmol)、NaOH(1N,9ml)和MeOH(9ml)的溶液搅拌18小时。用三氟乙酸(1.0ml)处理反应物。减压下从反应物溶液中除去溶剂,得到粗品产物。粗品产物经HPLC纯化,得到作为浅黄色固体的要求的化合物(0.285g,84%收率)。对C23H26N6O6Cl2·1.25CF3COOH·1.0H2O的计算值:C,42.90;H,4.13;N,11.77;实测值:C,43.03;H,4.03;N,11.26。
实施例48
使用在实施例47中描述的方法,合成以上化合物。在步骤1中,用乙二胺替代2,2-二甲基-1,3-二氨基丙二胺。在步骤3中,用实施例U的产物替代实施例G的产物。对C20H20N6O6Cl2·1.25CF3COOH·0.5H2O的计算值:C,40.77;H,3.38;N,12.68;实测值:C,40.98;H,3.17;N,12.57。
实施例X制备5-溴-3-碘代水杨醛
如同在文献(J.Org.Chem.1990,55,5287-5291)中描述的那样,通过5-溴代水杨醛的碘化,制备以上化合物。实施例Y和Z流程A1 步骤1制备
步骤1制备
在无水条件下,伴随剧烈搅拌下将5-氨基烟酸(10.0g,0.072mole)、苯甲酰基异硫氰酸酯(11.8g,0.072mole)和DMAP(催化量)在无水乙腈(250mL)中的混合物加热回流过夜(流程A1)。将生成的黄色悬浮液冷却并过滤。用水洗涤残余物,随后用乙腈洗涤,并且真空干燥过夜,得到作为浅黄色固体的要求的产物(21.4g,98%收率)。
MS和1H-NMR与要求的结构相一致。步骤2制备
向步骤1的产物(11.1g,0.037mole)在无水MeOH(230mL)中的悬浮液中加入NaOMe(25wt%在甲醇中的溶液,21.1mL,0.092mole),此时所述反应剂进入溶液,得到橙棕色的溶液(流程A1)。在室温下,把该溶液搅拌3小时,在冰浴上冷却并加入碘甲烷(3.45mL,0.055mole)。在10℃下,将生成的混合物搅拌30分钟并在室温下搅拌1.5小时。然后用乙酸(2mL)猝灭反应混合物,在冰浴上冷却并过滤,用冷的MeOH洗涤固体并真空干燥,得到作为米黄色固体的要求的产物(2.66g,37%收率)。
MS和1H-NMR与要求的结构相一致。步骤3制备实施例Y和Z
向1,3-二氨基-2-羟基丙烷(11.2g,0.124mol)在无水DMF(80mL)中的溶液中加入步骤2的产物(8.7g,0.041mol)。在无水条件下,于85℃下将该混合物加热3小时(流程A1)。在加热1-2小时后,溶液变得混浊并且在加热期间浊度增加。然后在冰浴上冷却反应混合物并过滤。用乙腈、水、乙腈洗涤固体,并真空干燥,得到作为米黄色固体的要求的产物(实施例Y)(3.7g,38%收率)。
MS和1H-NMR与要求的结构相一致。
使用对实施例Y描述的方法,合成实施例Z,以4当量的2,2-甲基-1,3-丙二胺替代1,3-二氨基-2-羟基丙烷。
通过在10℃下,将在无水THF(对1.0g底物10mL)和TFA(1当量)或4NHCl/二噁烷(2当量)中的溶液搅拌1小时,将步骤3的每一个产物转化为它们相应的TFA或者HCl盐。
实施例49制备 流程A2流程A2
流程A2流程A2
在-20℃下,向实施例Y(0.40g,0.00125mole,流程A2)在无水DMF(10mL)中的悬浮液中加入氯甲酸异丁酯(0.17g,0.00125mole),随后滴加入N-甲基吗啉(0.14g,0.00137mole)。于-20℃下,在氩气氛下把该混合物搅拌20分钟之后,加入另外量的N-甲基吗啉(0.14g,0.00137mole),随后加入实施例G的产物(0.46g,0.00125mole)。在-20℃下,将生成的混合物搅拌15分钟,然后在室温下搅拌2小时。真空蒸馏DMF并经反相HPLC纯化残余物,得到(冷冻干燥后)作为白色固体的要求的酯(0.20g,21%收率)。
MS和1H-NMR与要求的结构相一致。
在室温下,将该酯(0.2g)与1M LiOH(2mL)一起搅拌1小时。以三氟乙酸将pH调至2并通过反相HPLC纯化产物,提供(冷冻干燥后)作为白色固体的要求的酸(0.11g)。
MS和1H-NMR与要求的结构相一致。
实施例50制备
使用在实施例49中描述的方法,以等量实施例R替代实施例G,制备以上化合物(流程A2)。
MS和1H-NMR与要求的结构相一致。
在室温下,将酯(0.19g,0.00023mole)与1M LiOH(2mL)一起搅拌1小时。以三氟乙酸将pH调至2并通过反相HPLC纯化产物,提供(冷冻干燥后)作为白色固体的要求的酸(0.13g,72%收率)。
MS和1H-NMR与要求的结构相一致。
实施例51制备流程B流程B步骤1制备双-N-苄氧基羰基-2-羟基-1,3-二氨基丙烷
在10℃下,向2-羟基-1,3-二氨基丙烷(5.8g,0.064mol)在含有N-甲基吗啉(14mL)的二氯甲烷(150mL)中的悬浮溶液中分批加入苄氧基羰基琥珀酰亚胺(32g,0.129mol)(流程B)。在室温下,把反应混合物搅拌16小时,并用二氯甲烷(100mL)稀释生成的透明溶液,用10%柠檬酸(2×50mL)、水连续洗涤并干燥(Na2SO4)。过滤后,减压下除去溶剂,并且生成的白色固体从二氯甲烷中结晶,得到要求的化合物(20.0g,87%)。
1H-NMR和MS与该结构相一致。步骤2制备双-N-苄氧基羰基-1,3-二氨基丙-2-酮
于30分钟内,向步骤1的产物(21.0g,0.058mol)和EDC(33.0g)在含有DMSO(24.0mL)的二氯甲烷(120mL)中的悬浮液中滴加入三氟乙酸吡啶鎓盐(33.0g)在二氯甲烷(50mL)中的溶液并在10℃下搅拌。加入后,得到透明的黄色溶液。在室温下搅拌3小时后,从反应混合物中分离白色固体。冷却,过滤,并且用冷的二氯甲烷和水洗涤该固体。于干燥器中真空干燥得到的白色固体,得到要求的产物(15.5g,74%收率)。1H-NMR和MS与该结构相一致。步骤3制备
在无水条件下,将步骤2的产物(2.5g,0.007mol)、对甲苯磺酸(0.3g,0.0016mol)和DMSO(1.0mL)在含有乙二醇(2.0mL)的二氯乙烷(25mL)中的混合物加热至回流(流程B)。24小时后,冷却反应混合物,用二氯甲烷(25mL)稀释并用10%碳酸氢钠、水连续洗涤并干燥(Na2SO4)。减压下除去溶剂后,残余物从二氯甲烷/己烷中结晶,得到要求的化合物(2.5g,89%收率)。1H-NMR和MS与该结构相一致。步骤4
将步骤3的产物(3.0g,0.0075mol)溶于乙醇(50.0mL)和乙酸乙酯(50.0mL)的溶剂混合物中,并且在室温下,在Pd/C(5%,1.5g)存在下于50磅/平方英寸下氢化16小时(流程B)。过滤除去催化剂,用含有40%水的乙醇溶液(50mL)洗涤并过滤。将合并的滤液和水洗涤液减压下浓缩至干,得到糖浆状物。将该物料溶于DMF(10.0mL)中,与三乙胺(0.7mL)和DMAP(0.05g)一起加入到实施例1步骤2的产物(1.0g,0.0047mol)中。在无水条件下,于90℃下将生成的混合物加热。3小时后,真空蒸馏DMF,用水研磨残余物并过滤。其用水、乙腈洗涤,并在干燥器中真空干燥,得到作为粉末的实施例5(0.4g,30%)。在步骤B中,该物料无需进一步纯化即可使用。1H-NMR和MS与该结构相一致。
实施例52制备流程C流程C
将实施例51的化合物(0.38g,0.0014mol)悬浮于干燥的THF(5.0ml)中并加入三氟乙酸(0.1ml)且在无水条件下于10℃搅拌(流程C)。30分钟后,减压下蒸馏THF并把残余物真空干燥3小时。将生成的物料溶于干燥的DMF(4.0ml)中,冷却至-15℃,并加入氯甲酸异丁酯(0.18mL),随后加入N-甲基吗啉(0.17mL)。在氩气氛下将该溶液搅拌30分钟。在0℃下,向该混合物中加入通过将N-甲基吗啉(0.17mL)加入到实施例R(0.51g)在DMF(3.0mL)中的溶液中生成的胺的溶液。在-15℃下,将生成的混合物搅拌30分钟,然后在室温下搅拌16小时。然后经真空蒸馏除去溶剂,并通过反相HPLC纯化残余物,使用10-90%乙腈/水梯度(40分钟)以70mL/min的流速洗脱。
合并适当的部分并冷冻干燥,得到作为松散白色粉末的要求的酯(0.4g)。在室温下,将该物料与氢氧化锂(1M,2.0mL)一起搅拌。45分钟后,冷却反应混合物,用水稀释,用三氟乙酸酸化,并使用如同以上描述的10-90%乙腈/水洗脱,经反相HPLC分离要求的酸(0.25g)。1H-NMR和MS与该结构相一致。
实施例53制备流程D流程D 步骤1制备双-N-苄氧基羰基-2-氟-1,3-二氨基丙烷
步骤1制备双-N-苄氧基羰基-2-氟-1,3-二氨基丙烷
在-50℃下,向搅拌着的双-N-苄氧基羰基-2-羟基-1,3-二氨基丙烷(6.0g,0.017mol)在二氯甲烷(50mL)和吡啶(2.7mL)中的悬浮液中滴加入DAST(2.5mL)在二氯甲烷(7.5mL,流程D)中的溶液。在氩气氛下,于16小时内把反应混合物逐渐温热至室温。得到透明黄色溶液。冷却该溶液并倾入到冰、水(100mL)和二氯甲烷(50mL)的混合物中。用水(2×50mL)洗涤有机相并干燥(Na2SO4)。除去溶剂后,经硅胶快速层析法纯化残余物,使用30%在己烷中的EtOAc洗脱。合并适宜的部分,浓缩至干,并且产物从二氯甲烷/己烷中结晶,得到作为白色松散粉末的要求的氟代中间体(2.0g)。1H-NMR和MS与该结构相一致。步骤2
在室温下,在Pd/C(10%,2.7g)存在下,将步骤1中得到的双-N-苄氧基羰基-2-氟-1,3-二氨基丙烷(3.3g,0.0092mol)在EtOAc(30mL)和EtOH(30mL)中的溶液在50磅/平方英寸下氢化16小时(流程D)。过滤后,将催化剂与含有40%水的EtOH(50mL)一起搅拌并再次过滤。将滤液浓缩至干,得到糖浆状物(0.7g)。将该糖浆状物悬浮于DMF(8.0mL)中。加入实施例1步骤2的产物(0.7g,0.0033mol)、催化量的DMAP(0.01g)并在无水条件下于90℃下加热3小时。真空蒸馏DMF,将残余物悬浮于水(25mL)中并经加入1N HCl调节pH至4.5。冷却生成的混合物。过滤分离的固体并用水、乙腈彻底洗涤,在干燥器中真空干燥,得到作为棕色粉末的要求的化合物(0.24g)。1H-NMR和MS与该结构相一致。
实施例54制备 流程E流程E
流程E流程E
将以上得到的实施例53的化合物(0.22g)悬浮于干燥的THF(4.0mL)中。加入三氟乙酸(0.1mL)并在10℃下将该溶液搅拌30分钟,并减压下浓缩。在干燥器中真空干燥残余物。将得到的物料悬浮于干燥的DMF(5mL)中。加入氯甲酸异丁酯(0.12mL),随后加入N-甲基吗啉(0.11mL)。在氩气氛下,于-15℃下搅拌该溶液(流程E)。30分钟后,加入通过把N-甲基吗啉(0.095mL)加入到实施例R的产物(0.37mg)在DMF(3.0mL)中的溶液中生成的胺的溶液。在-15℃下,将生成的混合物搅拌30分钟,并且在室温下搅拌16小时。真空蒸馏DMF并经反相HPLC纯化残余物,使用10-90%乙腈/水洗脱。合并适宜的部分并冷冻干燥,得到作为浅黄色粉末的要求的酯产物(0.35g)。1H-NMR和MS与该结构相一致。
在室温下,将生成的产物(0.3g,流程E)与1M LiOH(3.0mL)一起搅拌。1小时后,用水(3.0mL)稀释该溶液,冷却并用三氟乙酸酸化。然后,生成的混合物经反相HPLC纯化,使用10-90%乙腈/水(30分钟的梯度)以流速70mL/min洗脱。合并适宜的部分并冷冻干燥,得到作为白色粉末的要求的化合物(0.22g)。
1H-NMR和MS与该结构相一致。
实施例55制备流程F流程F 步骤1制备
步骤1制备
向1,4-氨基-2,3-二羟基丁烷二盐酸盐[2.21g,0.012mole][如同在J.Carbohydrate Chemistry,5(2),183-197[1986]中描述的那样,由二甲基-L-酒石酸酯合成]在水(6mL)和无水DMF(10mL)中的溶液中加入碳酸钠(1.83g,0.017mole)。向该混合物中加入实施例1步骤2的产物(1.21g,0.006mole)并在85℃下把该混合物加热3小时。在冰浴上冷却后,真空蒸馏DMF并将生成的残余物悬浮于水中。调节pH至5.6。冷冻干燥该溶液,得到要求的产物(0.907g,59%收率)。
MS与要求的结构相一致(M+H267)。通过在10℃下与在THF(10mL)中的4N HCl/二噁烷(2当量)一起搅拌1小时,将所得到的化合物转化为其HCl盐。步骤2
在-20℃下,向步骤1的产物(0.11g,0.00023mole)在无水DMF(10mL)中的悬浮液中加入氯甲酸异丁酯(0.016g,0.00012mole),随后滴加入N-甲基吗啉(0.013g,0.00013mole,流程F)。在氩气氛下,于-20℃下将该混合物搅拌20分钟后,加入另外量的N-甲基吗啉(0.013g,0.00013mole),随后加入实施例R的产物(0.048g,0.00012mole)。在-20℃下,将生成的混合物搅拌15分钟。在室温下搅拌2小时后,真空蒸馏DMF并经反相HPLC纯化残余物,得到(冷冻干燥后)作为白色固体的要求的酯(0.03g,33%收率)。
MS(M+H627M+H629)和1H-NMR与要求的结构相一致。
1H-NMR(400MHz,Cd3OD):δ8.8(s,1H),8.5(s,1H),8.1(s,1H),7.4(s,1H),7.2(s,1H),5.6(m,1H),4.1(m,4H),3.7(m,2H),3.6(m,2H),3.3(m,2H),2.9(m,2H),1.2(m,3H)。
将生成的酯(0.03g,0.000035mole)与1N LiOH(2mL)一起搅拌。在室温下搅拌1小时后,用三氟乙酸调节pH至2,并且经反相HPLC分离产物,得到(冷冻干燥后)作为白色固体的要求的酸(0.001g,3.5%收率)。
MS(M+H599M+H601)和1H-NMR与要求的结构相一致。
1H-NMR(400MHz,CD3OD):δ8.8(s,1H),8.5(s,1H),8.0(s,1H),7.4(s,1H),7.2(s,1H),5.6(m,1H),4.1(m,2H),3.8(m,2H),3.5(m,2H),3.3(m,2H),2.8(m,2H)。
实施例56
按照在以下流程1中概述的合成顺序,制备二氟代类似物。流程1
使用DAST二乙基氨基三氟化硫,将苄酯基保护的酮转化为相应的二氟代中间体。随后经催化氢化除去苄酯基(Z)并使生成的二胺与5-氨基烟酸衍生物的S-甲基异硫脲衍生物偶合,提供相应的二氟取代的含有烟酸前体的胍。该化合物与甘氨酰-β-氨基酸酯反应随后水解和酸化,得到最终要求的化合物。
实施例57
按照以下流程,从已知的二异丙基-L-酒石酸酯开始,制备含有七元氟代/羟基取代的胍的化合物(J.Am.Chem.Soc.1988,110,7538)。流程2流程2
经过酰胺化、甲硅烷基化和N-脱甲硅烷基化,合成单甲硅烷基化中间体。乙硼烷还原该单甲硅烷基衍生物提供相应的二胺二盐酸盐。随后所述游离的二胺与S-甲基异硫脲5-氨基烟酸衍生物缩合,提供相应的七元胍衍生物。该胍进一步与甘氨酰-β-氨基酸酯反应,随后水解和酸化,得到最终要求的化合物。
实施例58
按照在以下流程中概述的反应,制备二烷基/二醇取代的七元环状胍化合物。由已知的苄酯基保护的二胺(J.Med.Chem.1996,39,2156)开始,通过使游离的二胺前体与S-甲基异硫脲5-氨基烟酸衍生物反应,合成相应的七元环状胍。生成的胍羧酸与甘氨酰-β-氨基酸酯缩合,随后经过水解和酸化,得到最终要求的化合物。流程3流程3 实施例59
实施例59
如同在以下流程中显示的那样,经酸催化下的环缩酮前体裂解,制备二羟基化合物。流程4流程4
实施例60
如同在实施例F中概述的那样,由D或者内消旋-酒石酸衍生物起始制备含有二醇取代的七元环状胍的化合物。
本发明化合物的活性在以下试验中测试。在所述试验中的测试结果列于表1中。玻连蛋白粘附试验材料
如同以前描述的那样[Pytela等,酶学中的方法,144:475-489(1987)],自人胎盘纯化人玻连蛋白受体(αvβ3)。如同以前描述的那样[Yatohgo等,细胞结构与功能,13:281-292(1988)],自新鲜冷冻的血浆纯化人玻连蛋白。如同以前描述的那样[Charo等,J.Biol.Chem.,266(3):1415-1421(1991)],通过将Pierce化学公司(Rockford,IL)的NHS-生物素与纯化的玻连蛋白偶合来制备生物素化的人玻连蛋白。从Sigma(St.Louis,MO)得到试验缓冲液、OPD底物片和RIA级别的BSA。从Calbiochem(La Jolla,CA)得到抗生物素抗体。从FlowLabs(McLean,VA)得到Linbro微量滴定板。从Sigma(St.Louis,MO)得到ADP试剂。方法固相受体试验
这个试验与先前报道[Niiya等,血液,70:475-483(1987)]的基本相同。在含有1.0mM的Ca++、Mg++和Mn++,pH7.4的Tris缓冲的盐水(TBS+++)中,将纯化的人玻连蛋白受体(αvβ3)从贮备液稀释至1.0μg/mL。将稀释的受体以100μL/孔(100ng受体/孔)立即转移至Linbro微量滴定板中。把这些板密封并在4℃下孵育过夜以使受体结合到孔上。所有其余的步骤在室温下进行。把这些试验板排空并加入200μL在TBS+++中的1%RIA级别的BSA(TBS+++/BSA),以封闭暴露的塑料表面。随后孵育2小时,使用96孔板洗涤器,用TBS+++洗涤这些试验板。在贮备液浓度2mM下开始并且使用在TBS+++/BSA作为稀释液中的2nM生物素化的玻连蛋白,制备受试化合物和对照组的对数级数稀释液。用CETUS Propette自动机进行,使受试(或者对照组)配基与标记的配基预先混合,并且随后将50μL等分试样转移至试验板上;所述标记的配基的最终浓度为1nM和受试化合物的最高浓度为1.0×10-4M。竞争作用发生两小时后,如同以前那样用板洗涤器洗涤所有的孔。在TBS+++/BSA中,将亲合纯化的辣根过氧化物酶标记的山羊抗生物素抗体稀释为1∶3000并且将125μL加入到每一个孔中。30分钟后,洗涤这些板并在100mM/L枸橼酸缓冲液(pH5.0)中与OPD/H2O2底物一起孵育。在450nm波长下,用微量滴定板读数器读取这些板并且当最大结合对照组孔达到大约1.0的吸收度时,记录最终的A450用于分析。使用以EXCELJ电子表格程序写出的宏分析这些数据。测定平均值、标准偏差和%CV用于重复试验浓度。将平均A450值标准化为四个最大结合对照组的平均值(不加入竞争剂)(B-MAX)。使标准化的数值经四个参数曲线拟合算法[Rodbard等,Int.Atomic Energy Agency,Vienna,第469页(1977)],在半-log标度上作图表示,并且计算相应于生物素化的玻连蛋白的最大结合的50%抑制率的浓度(IC50)和相应的R2被报道用于在最高试验浓度下呈现大于50%抑制率的那些化合物;另外,据报道所述IC50大于最高试验浓度。为有效的αvβ3拮抗剂(IC50在3-10nM范围内)的β-[[2-[[5-[(氨基亚氨基甲基)氨基]-1-氧代戊基]氨基]1-氧代乙基]氨基]-3-吡啶丙酸[USSN08/375,338,实施例1]作为阳性对照包含在每一个板中。纯化的Ⅱb/Ⅲa受体试验材料
从过时的血小板纯化人血纤蛋白原受体(αvβ3)(Pytela,R.,Pierschbacher,M.D.,Argraves,S.,Suzuki,S.和Rouslahti,E.“精氨酸-甘氨酸-天冬氨酸粘附受体”,酶学中的方法,144∶475-489(1987)]。如同在Yatohgo,T.,Izumi,M.,Kashiwagi,H.和Hayashi,M.,“通过肝素亲合层析法对来自人血浆中玻连蛋白的新的纯化方法”,细胞结构与功能,13∶281-292(1988)中描述的那样,将来自新鲜冷冻的血浆的人玻连蛋白纯化。如同以前描述的那样(Charo,I.F.,Nannizzi,L.,Phillips,D.R.,Hsu,M.A.,Scarborough,R.M.,“通过GPⅢa肽抑制结合于GPⅡb/Ⅲa的血纤蛋白原”,J.Biol.Chem.266(3)(1991):1415-1421),通过将Pierce化学公司(Rockford,IL)的NHS-生物素与纯化的玻连蛋白偶合来制备生物素化的人玻连蛋白。从Sigma(St.Louis,MO)得到试验缓冲液、OPD底物片和RIA级别BSA。从Calbiochem(La Jolla,CA)得到抗生物素抗体。从Flow Labs(McLean,VA)得到Linbro微量滴定板。从Sigma(St.Louis,MO)得到ADP试剂。方法固相受体试验
这个试验与Niiya,K.,Hodson,E.,Bader,R.,Byers-Ward,V.Koziol,J.A.,Plow,E.F.和Ruggeri,Z.M.,“由血小板活化引起的膜糖蛋白Ⅱb/Ⅲa复合物的增加的表面表达:血纤蛋白原结合与血小板聚集的关联”,血液,70(1987):475-483中报道的基本相同。在含有1.0mM的Ca++、Mg++和Mn++,pH7.4的Tris缓冲的盐水(TBS+++)中,将纯化的人血纤蛋白原受体(αvβ3)从贮备液稀释至1.0μg/mL。将稀释的受体以100μL/孔(100ng受体/孔)的量立即转移至Linbro微量滴定板中。把这些板密封并在4℃下孵育过夜以使受体结合到孔上。所有其余的步骤在室温下进行。把这些试验板排空并加入200μL在TBS+++中的1%RIA级别的BSA(TBS+++/BSA),以封闭暴露的塑料表面。随后孵育2小时,使用96孔板洗涤器,用TBS+++洗涤这些试验板。在贮备液浓度2mM下开始并且使用在TBS+++/BSA作为稀释液中的2nM生物素化的玻连蛋白,制备受试化合物和对照组的对数级数稀释液。用CETUS Propette自动机进行使标记的配基与受试(或者对照组)配基预先混合,并且随后将50μL等分试样转移至试验板上;所述标记的配基的最终浓度为1nM并且受试化合物的最高浓度为1.0×10-4M。竞争作用发生两小时后,如同以前那样用板洗涤器洗涤所有的孔。在TBS+++/BSA中,将亲合纯化的辣根过氧化物酶标记的山羊抗生物素抗体稀释为1∶3000并且将125μL加入到每一个孔中。30分钟后,洗涤这些板并在100mM/L枸橼酸缓冲液(pH5.0)中与ODD/H2O2底物一起孵育。在450nm波长下,用微量滴定板读数器读取这些板并且当最大结合对照组孔达到大约1.0的吸收度时,记录最终的A450用于分析。使用以EXCELJ电子表格程序写出的宏分析这些数据。测定平均值、标准偏差和%CV用于重复试验浓度。将平均A450值标准化为四个最大结合对照组的平均值(不加入竞争剂)(B-MAX)。使标准化的数值经四个参数曲线拟合算法[Rodbard等,Int.Atomic Energy Agency,Vienna,第469页(1977)],在半-log标度上作图表示,并且计算相应于生物素化的玻连蛋白的最大结合的50%抑制率的浓度(IC50)和相应的R2被报道用于在最高试验浓度下呈现大于50%抑制率的那些化合物;另外,据报道所述IC50大于最高试验浓度。为有效的αvβ3拮抗剂(IC50在3-10nM范围内)的β-[[2-[[5-[(氨基亚氨基甲基)氨基]-1-氧代戊基]氨基]-1-氧代乙基)氨基]-3-吡啶丙酸[USSN08/375,338,实施例1]作为阳性对照包含在每一个板中。人血小板富集血浆试验
从志愿者中选择健康的不服用阿司匹林供血者。如同在Zucker,M.B.“通过光量法测量血小板聚集”,酶学中的方法169(1989):117-133中描述的那样,收集富血小板血浆并随后实施ADP诱导的血小板聚集试验。使用翼形胶粘带的标准静脉穿刺术的技术使抽取45mL全血进入到含有5mL的3.8%枸橼酸三钠盐的60mL注射器中。随后在注射器中彻底混合,将抗凝全血转移至50mL锥形聚乙烯试管中。在室温下,于200xg下将该血离心12分钟,以沉淀非血小板细胞。将富血小板血浆转移到聚乙烯试管中并在室温下贮存备用。于2000xg下将剩余的血第二次离心15分钟得到贫血小板血浆。血小板计数一般为每微量滴定管300,000至500,000。将富血小板血浆(0.45mL)等分加入硅化杯中并在37℃下搅拌(1100rpm)1分钟,然后加入50μL预先稀释的受试化合物。在混合1分钟后,通过加入50μL的200μM ADP引发聚集。于Payton双道聚集仪(Payton Scientific,Buffalo,NY)上记录聚集3分钟。对于一系列受试化合物稀释液的最大应答(盐水对照组)抑制百分率用于测定剂量应答曲线。所有化合物重复试验并对于在试验的最高浓度下呈现50%或者更大抑制作用的化合物的剂量应答曲线作图计算半数最大抑制作用浓度(IC50),另外据报道所述IC50比最高试验浓度更大。
| 实施例编号 | αvβ3(IC50,nm) | Ⅱb/Ⅲa(IC50,nm) |
| 1 | 1.58 | 313 |
| 2 | 1.32 | 272 |
| 3 | 0.41 | 298 |
| 4 | 0.74 | 581 |
| 5 | 0.42 | 884 |
| 6 | 0.27 | 1260 |
| 7 | 0.21 | 361 |
| 8 | 95.1 | 857 |
| 9 | 0.18 | 244 |
| 10 | 0.14 | 161 |
| 11 | 0.34 | 462 |
| 12 | 4.5 | 442 |
| 13 | 9.32 | 780 |
| 14 | 12.4 | 772 |
| 15 | 25.4 | 1040 |
| 16 | 22000 | 6180 |
| 17 | 0.46 | 724 |
| 18 | 0.43 | 798 |
| 19 | 2.96 | 2490 |
| 20 | 0.32 | 2020 |
| 21 | 0.2 | 551 |
| 22 | 6.96 | 1710 |
| 23 | 3600 | 10100 |
| 24 | 91200 | 15800 |
| 25 | 1870 | 3910 |
| 26 | 0.71 | 831 |
| 27 | 0.26 | 482 |
| 28 | 0.43 | 984 |
| 29 | 0.46 | 1385 |
| 30 | 155 | 29.5 |
| 31 | 3942 | 1803 |
| 32 | 2.1 | 49 |
| 33 | 0.2 | 520 |
| 34 | 0.34 | 1203 |
| 35 | 0.14 | 340 |
| 36 | 1.13 | 970 |
| 37 | 63.8 | 267 |
| 38 | 278 | 115 |
| 39 | 1917 | 2458 |
| 40 | 1.5 | 20630 |
| 41 | 10.2 | 35400 |
| 42 | 3.1 | 5954 |
| 43 | 6.5 | 7527 |
| 44 | 42.1 | 1061 |
| 45 | 1094 | 1678 |
| 46 | 1.97 | 2693 |
| 47 | 0.99 | 6709 |
| 48 | 4.3 | 27910 |
Claims (22)
1.下式化合物或者它们的药学上可接受的盐: 其中为含有1至4个选自O、N或S的杂原子的、任选不饱和的5-8元单环杂环,其中X1选自CH、CH2、N、NH、O和S;
其中为含有1至4个选自O、N或S的杂原子的、任选不饱和的5-8元单环杂环,其中X1选自CH、CH2、N、NH、O和S;
A为其中Y1选自N-R2、O和S;
R2选自H、烷基、芳基、羟基、烷氧基、氰基、硝基、氨基、链烯基、链炔基、酰氨基、烷基羰基、芳基羰基、烷氧基羰基、芳氧基羰基、卤代烷基羰基、卤代烷氧基羰基、烷硫基羰基、芳硫基羰基、酰氧基甲氧基羰基、以一个或多个选自以下的取代基任选取代的烷基:低级烷基、卤素、羟基、卤代烷基、氰基、硝基、羧基、氨基、烷氧基、芳基或以一个或多个卤素、卤代烷基、低级烷基、烷氧基、氰基、烷基磺酰基、烷硫基、硝基、羧基、氨基、羟基、磺酸、磺酰胺、芳基、稠合的芳基、单环杂环或稠合的单环杂环任选取代的芳基,以一个或多个选自以下的取代基任选取代的芳基:卤素、卤代烷基、羟基、低级烷基、烷氧基、亚甲二氧基、亚乙二氧基、氰基、硝基、烷硫基、烷基磺酰基、磺酸、磺酰胺、羧基衍生物、氨基、芳基、稠合的芳基、单环杂环和稠合的单环杂环,单环杂环和以一个或多个选自以下的取代基任选取代的单环杂环:卤素、卤代烷基、低级烷基、烷氧基、氨基、硝基、羟基、羧基衍生物、氰基、烷硫基、烷基磺酰基、磺酸、磺酰胺、芳基或稠合的芳基;或者
R2与R7一起形成以一个或多个选自低级烷基、硫代烷基、烷基氨基、羟基、酮基、烷氧基、卤代、苯基、氨基、羧基或羧酸酯、螺二氧戊环和稠合的苯基的取代基任选取代的含有两个氮的4-12元杂环;或
R2与R7一起形成含有一个或多个选自O、N和S的杂原子的、任选不饱和的4-12元杂环;或
R2与R7一起形成以一个或多个选自低级烷基、苯基、烷氧基和羟基的取代基任选取代的5-9元杂芳环;或者
R2与R7一起形成与芳基或杂芳基环稠合的5元杂芳环;
R7(当不与R2一起时)和R8独立选自H,烷基,链烯基,链炔基,芳烷基,氨基,烷基氨基,羟基,烷氧基,芳基氨基,酰氨基,烷基羰基,芳基羰基,烷氧基羰基,芳氧基,芳氧基羰基,卤代烷基羰基,卤代烷氧基羰基,烷硫基羰基,芳硫基羰基,酰氧基甲氧基羰基,环烷基,双环烷基,芳基,酰基,苯甲酰基,以一个或多个选自以下的取代基任选取代的烷基:低级烷基、卤素、羟基、卤代烷基、氰基、硝基、羧基衍生物、氨基、烷氧基、硫代、烷硫基、磺酰基、芳基、芳烷基、以一个或多个选自以下的取代基任选取代的芳基:卤素、卤代烷基、低级烷基、烷氧基、亚甲二氧基、亚乙二氧基、烷硫基、卤代烷硫基、硫代、羟基、氰基、硝基、羧基衍生物、芳氧基、酰氨基、酰基氨基、氨基、烷基氨基、二烷基氨基、三氟代烷氧基、三氟代甲基、磺酰基、烷基磺酰基、卤代烷基磺酰基、磺酸、磺酰胺、芳基、稠合的芳基、单环杂环、稠合的单环杂环,以一个或多个选自以下的取代基任选取代的芳基:卤素、卤代烷基、低级烷基、烷氧基、亚甲二氧基、亚乙二氧基、烷硫基、卤代烷硫基、硫代、羟基、氰基、硝基、羧基衍生物、芳氧基、酰氨基、酰基氨基、氨基、烷基氨基、二烷基氨基、三氟烷氧基、三氟甲基磺酰基、烷基磺酰基、磺酸、磺酰胺、芳基、稠合的芳基、单环杂环或稠合的单环杂环,单环杂环,以一个或多个选自卤素、卤代烷基、低级烷基、烷氧基、芳氧基、氨基、硝基、羟基、羧基衍生物、氰基、烷硫基、烷基磺酰基、芳基、稠合的芳基的取代基任选取代的单环杂环,单环和二环杂环烷基,其中R10选自烷基、芳基和单环杂环的-SO2R10,所有基团以一个或多个选自卤素、卤代烷基、烷基、烷氧基、氰基、硝基、氨基、酰基氨基、三氟烷基、酰氨基、烷基氨基磺酰基、烷基磺酰基、烷基磺酰基氨基、烷基氨基、二烷基氨基、三氟甲基硫代、三氟烷氧基、三氟甲基磺酰基、芳基、芳氧基、硫代、烷硫基和单环杂环的取代基任选取代;和其中R10如上定义的 ;或者
;或者
NR7和R8一起形成含有单氮的4-12元单环或双环,其以一个或多个选自低级烷基、羧基衍生物、芳基或羟基的取代基任选取代并且其中所述环任选含有一个选自O、N和S的杂原子;
R5选自H、烷基、链烯基、链炔基、苄基和苯乙基;或者
A为
其中Y2选自烷基,环烷基,双环烷基,芳基,单环杂环,由也能以一个或多个选自卤代、卤代烷基、烷基、硝基、羟基、烷氧基、芳氧基、芳基或稠合的芳基的取代基任选取代的芳基所任选取代的烷基,以一个或多个选自卤代、卤代烷基、羟基、烷氧基、芳氧基、芳基、稠合的芳基、硝基、亚甲二氧基、亚乙二氧基或烷基的取代基任选取代的芳基,链炔基,链烯基,-S-R9和-O-R9,其中R9选自H、烷基、芳烷基、芳基、链烯基和链炔基,或者R9与R7一起形成以低级烷基、羟基、酮基、苯基、羧基或羧酸酯和稠合的苯基任选取代的、含有单氮和单硫或单氧的4-12元杂环,或者R9与R7一起为噻唑、噁唑、苯并噁唑或苯并噻唑;和
R5和R7如上定义;或者
Y2(当Y2为碳时)与R7一起形成以烷基、芳基、酮基或羟基任选取代的、含有单氮或二氮的4-12元环;或者
A为 其中R2和R7一起形成以一个或多个选自低级烷基、羟基、烷氧基、酮基、苯基或羧基衍生物的取代基任选取代的含有二氮的5-8元杂环;并且R8选自烷基羰基、芳基羰基、烷氧基羰基、芳氧基羰基、卤代烷基羰基、卤代烷氧基羰基、烷硫基羰基、芳硫基羰基或酰氧基甲氧基羰基;和
其中R2和R7一起形成以一个或多个选自低级烷基、羟基、烷氧基、酮基、苯基或羧基衍生物的取代基任选取代的含有二氮的5-8元杂环;并且R8选自烷基羰基、芳基羰基、烷氧基羰基、芳氧基羰基、卤代烷基羰基、卤代烷氧基羰基、烷硫基羰基、芳硫基羰基或酰氧基甲氧基羰基;和
R5如上定义;或者
R2和R7一起形成杂芳环例如咪唑或嘧啶酮;或者
A为其中R2和R7一起形成以羟基、酮基、苯基或烷基任选取代的含有二氮的5-8元杂环;和
R8选自烷基羰基、芳基羰基、烷氧基羰基、芳氧基羰基、卤代烷基羰基、卤代烷氧基羰基、烷硫基羰基、芳硫基羰基和酰氧基甲氧基羰基;
Z1为一个或多个选自H、烷基、羟基、烷氧基、芳氧基、卤素、卤代烷基、卤代烷氧基、硝基、氨基、烷基氨基、酰基氨基、二烷基氨基、氰基、烷硫基、烷基磺酰基、羧基衍生物、三卤代乙酰胺、乙酰胺、酰基、芳基、稠合的芳基、环烷基、硫代、单环杂环、稠合的单环杂环和A的取代基,其中A如上定义;
V选自-N-(R6)-,其中R6选自H、低级烷基、环烷基、芳烷基、芳基和单环杂环;或者R6与Y一起形成含有单氮的4-12元环;
Y、Y3、Z和Z3独立选自氢、烷基、芳基和环烷基;或者Y与Z一起形成环烷基;或者Y3和Z3一起形成环烷基;
n为整数1、2或3;
t为整数0、1或2;
p为整数0、1、2或3;
R为其中X选自O、S和NR4的X-R3,其中R3和R4独立选自氢、烷基、链烯基、链炔基、卤代烷基、芳基、芳基烷基、糖类、甾体化合物、聚烷基醚、烷基酰氨基、烷基N,N-二烷基酰氨基、新戊酰氧基甲基;和在游离酸的情况下所有它们的药学上可接受的盐;
R1选自氢、烷基、链烯基、链炔基、芳基、羧基衍生物、卤代烷基、环烷基、单环杂环、以烷基、卤素、卤代烷基、氰基、羟基、芳基、稠合的芳基、硝基、烷氧基、芳氧基、烷基磺酰基、芳基磺酰基、磺酰胺、硫代、烷硫基、羧基衍生物、氨基、酰氨基任选取代的单环杂环;
以一个或多个以下基团任选取代的烷基:卤代、卤代烷基、羟基、烷氧基、芳氧基、硫代、烷硫基、链炔基、链烯基、烷基、芳硫基、烷基亚砜、烷基磺酰基、芳基亚砜、芳基磺酰基、氰基、硝基、氨基、烷基氨基、二烷基氨基、烷基磺酰胺、芳基磺酰胺、酰基酰胺、羧基衍生物、磺酰胺、磺酸、膦酸衍生物、次膦酸衍生物、在芳基环上均用卤代、烷基、卤代烷基、氰基、硝基、羟基、羧基衍生物、烷氧基、芳氧基、氨基、烷基氨基、二烷基氨基、酰氨基、芳基、稠合的芳基、单环杂环任选取代的芳基、芳硫基、芳基亚砜或芳基砜;和稠合的单环杂环、单环杂环硫代、单环杂环亚砜和单环杂环砜,其能够用卤代、卤代烷基、硝基、羟基、烷氧基、稠合的芳基或烷基任选取代;
烷基羰基、卤代烷基羰基和芳基羰基;
在一个或多个位置上用以下基团任选取代的芳基,包括:卤代、卤代烷基、烷基、烷氧基、芳氧基、亚甲二氧基、亚乙二氧基、烷硫基、卤代烷硫基、硫代、羟基、氰基、硝基、酰氧基、羧基衍生物、羧基烷氧基、酰氨基、酰基氨基、氨基、烷基氨基、二烷基氨基、三氟烷氧基、三氟甲基磺酰基、烷基磺酰基、磺酸、磺酰胺、芳基、稠合的芳基、单环杂环和稠合的单环杂环和 ,其中R7和R8如上定义并且条件是与所述氮一起,R7和R8包括氨基酸;和
,其中R7和R8如上定义并且条件是与所述氮一起,R7和R8包括氨基酸;和
R11选自H、烷基、芳烷基、链烯基、链炔基、卤代烷基或卤代链炔基或者R11与Y一起形成含有单氮的4-12元环;或者它们的药学上可接受的盐。
2.权利要求1的下式化合物其中其中R32为H、烷基、烷氧基烷基、氨基烷基、二烷基氨基烷基,其中所述烷基由一个或多个选自羟基、烷氧基、氨基、烷基氨基、二烷基氨基、芳基-或烷基-磺酰基、羧基和羧基衍生物的取代基任选取代。
3.权利要求2的化合物,其中所述化合物选自:


4.权利要求1的下式化合物 其中
其中
5.权利要求4的化合物,其中所述化合物选自:



6.权利要求1的下式化合物其中
7.权利要求6的化合物选自:
8.权利要求1的化合物,其中所述化合物为下式化合物:其中
9.权利要求8的下式化合物:
10.权利要求1的下式化合物: 其中
其中
11.权利要求10的下式化合物:
12.药用组合物,包括治疗有效量的权利要求1、2、3、4、5、6、7、8、9、10或11的化合物和药学上可接受的载体。
13.在需要这样的治疗的哺乳动物上治疗由αvβ3介导的疾病的方法,包括给予治疗上有效抑制αvβ3量的权利要求1、2、3、4、5、6、7、8、9、10或11的化合物。
14.权利要求13的方法,其中所治疗的疾病为肿瘤转移。
15.权利要求13的方法,其中所治疗的疾病为实体肿瘤的生长。
16.权利要求13的方法,其中所治疗的疾病为血管生成。
17.权利要求13的方法,其中所治疗的疾病为骨质疏松症。
18.权利要求13的方法,其中所治疗的疾病为恶性肿瘤体液高钙血症。
19.权利要求13的方法,其中所治疗的疾病为平滑肌细胞迁移。
20.权利要求13的方法,其中抑制再狭窄。
21.权利要求13的方法,其中所治疗的疾病为类风湿性关节炎。
22.权利要求13的方法,其中所治疗的疾病为黄斑变性。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8139498P | 1998-04-10 | 1998-04-10 | |
| US60/081,394 | 1998-04-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1304406A true CN1304406A (zh) | 2001-07-18 |
Family
ID=22163868
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN99807091A Pending CN1304406A (zh) | 1998-04-10 | 1999-04-09 | 作为玻连蛋白拮抗剂的杂环甘氨酰β-丙氨酸衍生物 |
Country Status (17)
| Country | Link |
|---|---|
| EP (1) | EP1070060A1 (zh) |
| JP (1) | JP2002511462A (zh) |
| KR (1) | KR20010042614A (zh) |
| CN (1) | CN1304406A (zh) |
| AR (1) | AR015759A1 (zh) |
| AU (1) | AU765294B2 (zh) |
| BR (1) | BR9910119A (zh) |
| CA (1) | CA2326665A1 (zh) |
| CZ (1) | CZ20003672A3 (zh) |
| IL (1) | IL138677A0 (zh) |
| MY (1) | MY133473A (zh) |
| NO (1) | NO20005084L (zh) |
| NZ (1) | NZ507292A (zh) |
| PL (1) | PL343406A1 (zh) |
| RU (1) | RU2215746C2 (zh) |
| TW (1) | TWI247008B (zh) |
| WO (1) | WO1999052896A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115626912A (zh) * | 2022-09-27 | 2023-01-20 | 中山大学 | 一种硫脲化合物及其制备方法与应用 |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6172256B1 (en) * | 1998-03-04 | 2001-01-09 | G.D. Searle & Co. | Chiral-β-amino acid compounds and derivatives thereof |
| GB9805655D0 (en) | 1998-03-16 | 1998-05-13 | Celltech Therapeutics Ltd | Chemical compounds |
| US6521626B1 (en) | 1998-03-24 | 2003-02-18 | Celltech R&D Limited | Thiocarboxamide derivatives |
| GB9814414D0 (en) | 1998-07-03 | 1998-09-02 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9821061D0 (en) | 1998-09-28 | 1998-11-18 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9826174D0 (en) | 1998-11-30 | 1999-01-20 | Celltech Therapeutics Ltd | Chemical compounds |
| CA2356459A1 (en) * | 1998-12-23 | 2000-07-06 | G.D. Searle & Co. | Method of using a matrix metalloproteinase inhibitor and radiation therapy as combination therapy in the treatment of neoplasia |
| WO2000047207A1 (en) | 1999-02-09 | 2000-08-17 | Bristol-Myers Squibb Company | LACTAM INHIBITORS OF FXa AND METHOD |
| GB9908355D0 (en) * | 1999-04-12 | 1999-06-09 | Rhone Poulenc Rorer Ltd | Chemical compounds |
| TR200102959T2 (tr) | 1999-04-12 | 2002-04-22 | Aventis Pharma Limited | İntegrin antagonistleri olarak ikame edilmiş bisiklik heteroaril bileşikleri |
| WO2000066618A1 (de) * | 1999-04-28 | 2000-11-09 | Basf Aktiengesellschaft | Integrinrezeptorantagonisten |
| US6518283B1 (en) | 1999-05-28 | 2003-02-11 | Celltech R&D Limited | Squaric acid derivatives |
| US6455539B2 (en) | 1999-12-23 | 2002-09-24 | Celltech R&D Limited | Squaric acid derivates |
| ES2295150T3 (es) | 2000-04-17 | 2008-04-16 | Ucb Pharma, S.A. | Derivados de enamina como moleculas de adhesion celular. |
| US6403608B1 (en) | 2000-05-30 | 2002-06-11 | Celltech R&D, Ltd. | 3-Substituted isoquinolin-1-yl derivatives |
| US6545013B2 (en) | 2000-05-30 | 2003-04-08 | Celltech R&D Limited | 2,7-naphthyridine derivatives |
| US6720315B2 (en) | 2000-06-15 | 2004-04-13 | Pharmacia Corporation | Dihydrostilbene alkanoic acid derivatives |
| US6511973B2 (en) | 2000-08-02 | 2003-01-28 | Bristol-Myers Squibb Co. | Lactam inhibitors of FXa and method |
| WO2002010136A1 (en) | 2000-08-02 | 2002-02-07 | Celltech R & D Limited | 3-substituted isoquinolin-1-yl derivatives |
| DE10204789A1 (de) * | 2002-02-06 | 2003-08-14 | Merck Patent Gmbh | Inhibitoren des Integrins alpha¶v¶beta6 |
| TW200307671A (en) | 2002-05-24 | 2003-12-16 | Elan Pharm Inc | Heteroaryl compounds which inhibit leukocyte adhesion mediated by α 4 integrins |
| EP1572210A1 (en) * | 2002-12-20 | 2005-09-14 | Pharmacia Corporation | The r-isomer of beta amino acid compounds as integrin receptor antagonists derivatives |
| UA87854C2 (en) | 2004-06-07 | 2009-08-25 | Мерк Энд Ко., Инк. | N-(2-benzyl)-2-phenylbutanamides as androgen receptor modulators |
| EP2065381A1 (de) | 2007-10-18 | 2009-06-03 | Boehringer Ingelheim Pharma GmbH & Co. KG | CGRP Antagonisten |
| MX2010003849A (es) | 2007-10-18 | 2010-04-27 | Boehringer Ingelheim Int | Antagonistas de cgrp. |
| EP2225223B1 (de) | 2007-11-22 | 2017-01-11 | Boehringer Ingelheim International GmbH | Organische verbindungen |
| JP2011504481A (ja) * | 2007-11-22 | 2011-02-10 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 有機化合物 |
| ES2647585T3 (es) | 2012-07-18 | 2017-12-22 | Saint Louis University | Derivados de aminoácido beta como antagonistas de integrina |
| US8716226B2 (en) | 2012-07-18 | 2014-05-06 | Saint Louis University | 3,5 phenyl-substituted beta amino acid derivatives as integrin antagonists |
| MX2016015467A (es) | 2014-05-30 | 2017-03-23 | Pfizer | Derivados de carbonitrilo como moduladores selectivos del receptor de androgenos. |
| EP3294716B1 (en) | 2015-12-30 | 2020-04-15 | Saint Louis University | Meta-azacyclic amino benzoic acid derivatives as pan integrin antagonists |
| SI3538528T1 (sl) * | 2016-11-08 | 2021-03-31 | Bristol-Myers Squibb Company | Pirol amidi kot alfa v integrin inhibitorji |
| AU2021270095A1 (en) * | 2020-05-14 | 2022-12-22 | Ube Corporation | 1, 4, 5, 6-tetrahydropyrimidine-2-amine derivative |
| WO2023275715A1 (en) | 2021-06-30 | 2023-01-05 | Pfizer Inc. | Metabolites of selective androgen receptor modulators |
| JPWO2023085396A1 (zh) * | 2021-11-12 | 2023-05-19 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5120859A (en) * | 1989-09-22 | 1992-06-09 | Genentech, Inc. | Chimeric amino acid analogues |
| US6028223A (en) * | 1995-08-30 | 2000-02-22 | G. D. Searle & Co. | Meta-guanidine, urea, thiourea or azacyclic amino benzoic acid compounds and derivatives thereof |
| DE19629816A1 (de) * | 1996-07-24 | 1998-01-29 | Hoechst Ag | Neue Cycloalkyl-Derivate als Inhibitoren der Knochenresorption und Vitronectinrezeptor-Antagonisten |
| JP2002511052A (ja) * | 1996-08-29 | 2002-04-09 | メルク エンド カンパニー インコーポレーテッド | インテグリンアンタゴニスト |
-
1999
- 1999-04-09 KR KR1020007011291A patent/KR20010042614A/ko not_active Application Discontinuation
- 1999-04-09 BR BR9910119-0A patent/BR9910119A/pt not_active IP Right Cessation
- 1999-04-09 IL IL13867799A patent/IL138677A0/xx unknown
- 1999-04-09 PL PL99343406A patent/PL343406A1/xx unknown
- 1999-04-09 CZ CZ20003672A patent/CZ20003672A3/cs unknown
- 1999-04-09 RU RU2000128033/04A patent/RU2215746C2/ru not_active IP Right Cessation
- 1999-04-09 WO PCT/US1999/004297 patent/WO1999052896A1/en not_active Application Discontinuation
- 1999-04-09 CN CN99807091A patent/CN1304406A/zh active Pending
- 1999-04-09 NZ NZ507292A patent/NZ507292A/en unknown
- 1999-04-09 CA CA002326665A patent/CA2326665A1/en not_active Abandoned
- 1999-04-09 TW TW088105718A patent/TWI247008B/zh active
- 1999-04-09 AR ARP990101636A patent/AR015759A1/es unknown
- 1999-04-09 JP JP2000543454A patent/JP2002511462A/ja active Pending
- 1999-04-09 AU AU34499/99A patent/AU765294B2/en not_active Ceased
- 1999-04-09 EP EP99916119A patent/EP1070060A1/en not_active Withdrawn
- 1999-04-10 MY MYPI99001398A patent/MY133473A/en unknown
-
2000
- 2000-10-09 NO NO20005084A patent/NO20005084L/no not_active Application Discontinuation
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115626912A (zh) * | 2022-09-27 | 2023-01-20 | 中山大学 | 一种硫脲化合物及其制备方法与应用 |
| CN115626912B (zh) * | 2022-09-27 | 2024-03-26 | 中山大学 | 一种硫脲化合物及其制备方法与应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU3449999A (en) | 1999-11-01 |
| CZ20003672A3 (cs) | 2001-08-15 |
| WO1999052896A1 (en) | 1999-10-21 |
| MY133473A (en) | 2007-11-30 |
| JP2002511462A (ja) | 2002-04-16 |
| EP1070060A1 (en) | 2001-01-24 |
| NO20005084D0 (no) | 2000-10-09 |
| BR9910119A (pt) | 2001-10-09 |
| PL343406A1 (en) | 2001-08-13 |
| AR015759A1 (es) | 2001-05-16 |
| TWI247008B (en) | 2006-01-11 |
| NO20005084L (no) | 2000-11-27 |
| AU765294B2 (en) | 2003-09-11 |
| CA2326665A1 (en) | 1999-10-21 |
| IL138677A0 (en) | 2001-10-31 |
| NZ507292A (en) | 2003-12-19 |
| KR20010042614A (ko) | 2001-05-25 |
| RU2215746C2 (ru) | 2003-11-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1304406A (zh) | 作为玻连蛋白拮抗剂的杂环甘氨酰β-丙氨酸衍生物 | |
| CN1237061C (zh) | 结合趋化因子受体的杂环化合物 | |
| CN1282640C (zh) | N-芳基苯乙酰胺衍生物及包含所述化合物的药物组合物 | |
| CN1291988C (zh) | 三氮杂螺[5,5]十一烷衍生物及包含其作为活性成分的药物组合物 | |
| CN1259307C (zh) | 酰化的二氢化茚基胺及其作为药物的用途 | |
| CN1097051C (zh) | N-杂芳基吡啶磺酰胺衍生物及其作为内皮素拮抗剂的用途 | |
| CN1078889C (zh) | 非肽类速激肽受体拮抗剂 | |
| CN1167680C (zh) | 稠环化合物及其药物用途 | |
| CN100338061C (zh) | 炔-芳基磷酸二酯酶-4抑制剂 | |
| CN1214011C (zh) | 作为整联蛋白拮抗剂的间-氮杂环氨基苯甲酸化合物及其衍生物 | |
| CN1703405A (zh) | 用作糖原合酶激酶3β抑制剂的氨基苯甲酰胺衍生物 | |
| CN1930126A (zh) | 吡啶酮衍生物 | |
| CN101031559A (zh) | 氨基环脲衍生物和其制法及作为激酶抑制剂的医药用途 | |
| CN1582285A (zh) | 用作糖原合酶激酶3β抑制剂(GSK3抑制剂)的杂芳胺化合物 | |
| CN1902177A (zh) | 新哌啶衍生物 | |
| CN1918128A (zh) | 稠环4-氧代-嘧啶衍生物 | |
| CN1646495A (zh) | 胺化合物及其用途 | |
| CN1350534A (zh) | 作为整联蛋白拮抗剂的取代双环杂芳基化合物 | |
| CN1867560A (zh) | 4-嘧啶酮衍生物及其作为肽基肽酶抑制剂的用途 | |
| CN1688573A (zh) | 糖原合成酶激酶3的吡咯基抑制剂 | |
| CN1656073A (zh) | 可用于治疗蛋白激酶依赖性疾病的二芳基脲衍生物 | |
| CN1662236A (zh) | 组蛋白脱乙酰基转移酶抑制剂 | |
| CN1350530A (zh) | 酰胺衍生物 | |
| CN1457339A (zh) | 趋化因子受体结合性杂环化合物 | |
| CN1993347A (zh) | 钾通道抑制剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: GR Ref document number: 1084123 Country of ref document: HK |
|
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |