CN1284961A - 红霉素衍生物 - Google Patents
红霉素衍生物 Download PDFInfo
- Publication number
- CN1284961A CN1284961A CN 98813554 CN98813554A CN1284961A CN 1284961 A CN1284961 A CN 1284961A CN 98813554 CN98813554 CN 98813554 CN 98813554 A CN98813554 A CN 98813554A CN 1284961 A CN1284961 A CN 1284961A
- Authority
- CN
- China
- Prior art keywords
- replace
- substituting group
- alkyl
- compound
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及下面通式所示的新的红霉素衍生物,(式中,R1代表氢原子或低级烷基,R2代表烷基,环烷基,芳基,芳烷基,或式-X-R4所示的基团,X代表氧原子或氨基,R4代表烷基或芳基,R3代表烷基,环烷基,烯基,或式-(CH2)n-Y-R5所示的基团,Y代表亚甲基,氧原子,硫原子,亚硫酰基,或羰基,R5代表芳基,n代表1—5的整数。),以及含有该衍生物作为有效成分的对非典型耐酸细菌感染症治疗有用的药物。
Description
技术领域
本发明涉及作为抗菌剂有用的红霉素衍生物,特别涉及对非典型耐酸细菌感染症(非结核性耐酸细菌感染)有用的新的红霉素衍生物或其盐。
背景技术
由于非典型耐酸细菌对于以抗结核剂为首的各种抗菌剂的敏感性低,因此,非典型耐酸细菌感染是非常难治的疾病。作为与本发明化合物有类似适应症的化合物,已知有利福平[rifampicin;默克索引第12版,8382],另外,作为与本发明化合物有近似化学结构的大环内酯衍生物,已知有克拉霉素[clarithromycin;默克索引第12版,2400],9位肟型化合物的罗红霉素[roxithromycin;默克索引第12版,8433]等。另外,作为3位克拉定糖衍生物,国际公开WO 93/13116号等中公开了酯型化合物,国际公开WO 93/13115号中公开了氨基甲酸酯型化合物。在美国等,克拉霉素作为这些大环内酯衍生物中被视为有望作为现在最优秀的非典型耐酸细菌感染治疗剂临床应用已得到认可。但是,对于克拉霉素的抗菌力还不能说令人满意,因此需要开发更优秀的抗菌剂。
发明的描述
近年来,日和见感染症的增加已成为社会的重大问题。作为该日和见感染症增加的原因,有人类免疫缺陷病毒(HIV)感染者,癌症患者,糖尿病患者或老年人等体内防御机构衰退的易感染者增加,有对甲氧西林有耐药性的金黄色葡萄球菌(Methicillin-resistantStaphylococcus aureus)等为代表的对多种抗菌剂有耐药性的细菌增加等原因,或由于细菌交叉感染使得日和见感染症的化学疗法变得更加困难。由非典型耐酸细菌引起的日和见感染症也逐渐成为问题。由于非典型耐酸细菌繁殖速度慢,而且即使被巨噬细胞吞食后也可在该细胞中长期生存,因此,对于该细菌引起的感染症必须进行长时间的化学治疗。
特别是,几乎没有对非典型耐酸细菌中的鸟分支杆菌复合体(Mycobacterium avium complex:MAC)有效的抗菌剂,现在也在进行对该感染症的外科疗法的研究。另外,即使是上述记载的克拉霉素,作为非典型耐酸细菌感染治疗剂也缺乏选择性,并存在出现对克拉霉素有耐药性的MAC的问题。这样,对于非典型耐酸细菌感染的化学疗法,存在细菌对药剂缺乏敏感性,有容易出现细菌交叉感染或耐药菌的环境等各种问题。本发明的课题就是提供对非典型耐酸细菌有选择性,且具有优秀的抗菌活性的化合物。
本发明者为解决上述课题,进行了认真研究,其结果发现,本发明涉及的新的红霉素衍生物或其盐作为具有上述特征的抗菌剂是有用的,特别是,发现了对于非典型耐酸细菌具有优秀的选择性和抗菌力的化合物,并因此完成了本发明。
即,本发明涉及下述通式(Ⅰ)所示的新的红霉素衍生物及其盐:(式中,R1代表氢原子或低级烷基,R2代表可有取代基取代的烷基,可有取代基取代的环烷基,可有取代基取代的(环烷基)烷基,可有取代基取代的芳基,可有取代基取代的芳烷基,可有取代基取代的苯乙烯基,或式-X-R4所示的基团,X代表氧原子或氨基,R4代表可有取代基取代的烷基或可有取代基取代的芳基,R3代表羧基,烷氧羰基,芳氧羰基或芳烷氧羰基取代的烷基,可有取代基取代的环烷基,可有取代基取代的(环烷基)烷基,可有取代基取代的烯基,或式-(CH2)n-Y-R5所示的基团,Y代表可有取代基取代的亚甲基,氧原子,硫原子,亚硫酰基,磺酰基,烷基取代或未取代的氨基,或羰基,R5代表可有取代基取代的芳基,n代表1~5的整数。)
本发明的另一方案,提供了上述通式(Ⅰ)所示化合物中,R1是氢原子的化合物或其盐。
从另一观点看,本发明提供了含有上述通式(Ⅰ)所示化合物或其盐作为有效成分的药物。该药物作为抗菌剂是有用的,例如,它可作为各种微生物引起的感染症的治疗剂。优选非典型耐酸细菌感染症,进一步优选对于鸟分支杆菌复合体感染症的治疗是有用的。
再从另一观点看,本发明提供了以制造上述药物为目的的上述通式(Ⅰ)所示化合物或其盐。另外,本发明提供了感染症,优选非典型耐酸细菌感染症,特别优选鸟分支杆菌复合体感染症的治疗方法,包括使用有效量的上述通式(Ⅰ)所示化合物或其盐对患者给药。发明实施的最佳方式
本发明的上述通式(Ⅰ)中,作为R1所示的低级烷基,可列举的有,例如,甲基,乙基,正丙基,正丁基等。另外,作为R2,R3,R4所示的烷基,或Y所示的氨基中可有取代的烷基表示碳原子数1~10的直链或支链烷基,该烷基,可含有一个或多个选自氧原子,硫原子,和氮原子的杂原子。可列举的有,例如,甲基,乙基,正丙基,异丙基,正丁基,异丁基,仲丁基,叔丁基,正戊基,异戊基,新戊基,叔戊基,正己基,正庚基,正辛基,正壬基,正癸基,甲氧乙基,乙氧乙基,甲氧丙基,甲氧丁基,甲氧戊基,甲氧己基,甲硫乙基,乙硫乙基,甲硫丙基,甲硫丁基,甲硫戊基,甲硫己基,甲基氨基乙基,乙基氨基乙基,甲基氨基丙基,甲基氨基丁基,甲基氨基戊基,甲基氨基己基,二甲基氨基乙基,二甲基氨基丙基,二甲基氨基丁基,二甲基氨基戊基,二甲基氨基己基等。R2或R3所示的环烷基表示碳原子数3~7的环烷基,可列举的有,例如,环丙基,环丁基,环戊基,环己基,环庚基等。R2或R3所示的(环烷基)烷基,表示单环或多环的环烷基在任意位置取代的可含有氧原子,硫原子或氮原子的烷基。可列举的有,例如,(环丙基)甲基,(环丁基)甲基,(环戊基)甲基,(环己基)甲基,(环庚基)甲基,(环丙基)乙基,(环丁基)乙基,(环戊基)乙基,(环己基)乙基,(环庚基)乙基,(环己基)丙基,(环己基)丁基,(环己基)戊基,(环己基)己基,(环己基)庚基,(环己基)辛基,(环己基)壬基,(环己基)癸基,(2,3-二氢苯并呋喃-2-基)甲基,(2,3-二氢苯并呋喃-3-基)甲基,(3,4-二氢苯并[b]吡喃-2-基)甲基,(3,4-二氢苯并[b]吡喃-3-基)甲基,(3,4-二氢苯并[b]吡喃-4-基)甲基,(2,3-二氢-1,4-苯并二噁烯-2-基)甲基等。R3所示的烯基表示碳原子数3~7的烯基,可列举的有,例如,烯丙基,丁烯基,戊烯基,己烯基,庚烯基等。
作为R3所示的烷氧羰基,芳氧羰基或芳烷氧羰基取代的烷基中的烷氧羰基,可列举的有,例如,甲氧羰基,乙氧羰基,正丙氧羰基,异丙氧羰基,正丁氧羰基,异丁氧羰基,仲丁氧羰基,叔丁氧羰基,正戊氧羰基,异戊氧羰基,新戊氧羰基等,作为芳氧羰基,可列举的有,例如,苯氧羰基,吡啶氧羰基,噻吩氧羰基等,作为芳烷氧羰基,可列举的有,例如,苄氧羰基,吡啶甲基氧羰基,噻吩甲基氧羰基等。
本发明的上述通式(Ⅰ)中,作为R2,R4或R5所示的芳基,可列举的有,例如,苯基,吡啶基,嘧啶基,吡嗪基,咪唑基,萘基,呋喃基,苯并呋喃基,苯并[b]苯硫基,苯并咪唑基,吲哚基,噻吩基,吡咯基,喹啉基,异喹啉基,1,2,3,4-四氢萘-5-基,1,2,3,4-四氢萘-6-基等单环或多环的芳香环。另外,R2所示的芳烷基,表示在前述芳基的任意位置上被可含有氧原子,硫原子或氮原子的碳原子数1~6的直链或支链烷基取代的基团,可列举的有,例如,苄基,吡啶甲基,嘧啶甲基,吡嗪甲基,萘甲基,糠基,苯并呋喃甲基,噻吩甲基,吡咯甲基,喹啉甲基,异喹啉甲基,苯乙基,苯基丙基,苯基丁基,苯基戊基,苯基己基,苯氧甲基,吡啶氧甲基,嘧啶氧甲基,吡嗪氧甲基,萘氧甲基,四氢萘氧乙基,苯硫甲基,吡啶硫甲基,嘧啶硫甲基,吡嗪硫甲基,萘硫甲基,苯基氨基甲基等。
作为R2或R4所示的可有取代基取代的烷基,R2和R3所示的可有取代基取代的环烷基,可有取代基取代的(环烷基)烷基,R2,R4或R5所示可有取代基取代的芳基,R2所示的可有取代基取代的芳烷基和可有取代基取代的苯乙烯基,R3所示的可有取代基取代的链烯基,或Y所示的可有取代基取代的亚甲基的取代基,可以是任何可在这些基团上取代的基团。对取代基的个数和种类没有特别的限制,在存在2个或2个以上的取代基时,它们可相同或不同。可列举的有,例如,可有保护基的羟基,烷氧基,可有取代基取代的氨基,可有取代基取代的氨基甲酰基,卤原子,烷基,三氟甲基,烷酰基,环烷基,芳基,芳氧基,氰基,硝基,胍基,脒基,羧基,烷氧羰基,芳氧羰基,芳烷氧羰基等。作为该羟基保护基,可使用在羟基不会发生反应的体系中实质为惰性,且在特定的脱保护条件下容易断裂的任何基团,可列举的有,例如,烷酰基,卤代烷酰基,三烷基甲硅烷基,苄基等。作为羟基保护基的烷酰基,可列举的有,例如,甲酰基,乙酰基,丙酰基,丁酰基,三甲基乙酰基等,作为羟基保护基的卤代烷酰基,可列举的有,例如,三氟乙酰基,三氯乙酰基等。作为羟基保护基的三烷基甲硅烷基,可列举的有,例如,三甲基甲硅烷基,三乙基甲硅烷基等。另外,烷氧基表示碳原子数1~6的直链或支链烷氧基,可列举的有,例如,甲氧基,乙氧基,正丙氧基,异丙氧基,正丁氧基,异丁氧基,仲丁氧基,叔丁氧基,正戊氧基,异戊氧基,新戊氧基,叔戊氧基,正己氧基等。作为可有取代基取代的氨基,可列举的有,例如,氨基,甲基氨基,乙基氨基,正丙基氨基,异丙基氨基,正丁基氨基,异丁基氨基,仲丁基氨基,叔丁基氨基,正戊基氨基,异戊基氨基,新戊基氨基,叔戊基氨基,正己基氨基,N,N-二甲基氨基,N,N-二乙基氨基等,作为可有取代基取代的氨基甲酰基,可列举的有,氨基甲酰基,N-甲基氨基甲酰基,N-乙基氨基甲酰基,N-正丙基氨基甲酰基,N-异丙基氨基甲酰基,N-正丁基氨基甲酰基,N-异丁基氨基甲酰基,N-仲丁基氨基甲酰基,N-叔丁基氨基甲酰基,N-正戊基氨基甲酰基,N-异戊基氨基甲酰基,N-新戊基氨基甲酰基,N-正己基氨基甲酰基,N,N-二甲基氨基甲酰基,N,N-二乙基氨基甲酰基等。作为卤原子,可以是氟原子,氯原子,溴原子或碘原子中的任何一个。作为烷酰基,可列举的有,例如,乙酰基,丙酰基,丁酰基等,作为芳氧基,可列举的有,苯氧基,吡啶氧基,嘧啶氧基,吡嗪氧基,萘氧基,呋喃氧基,苯并呋喃氧基,噻吩氧基,吡咯氧基,喹啉氧基,异喹啉氧基等。
另外,作为上述可取代的烷基,烷酰基,环烷基,芳基,烷氧羰基,芳氧羰基,芳烷氧羰基,可列举的有,例如,前述举例中所列的基团。
本发明的前述通式(Ⅰ)所示化合物中,存在光学异构体,非对映异构体,几何异构体等立体异构体,这些异构体及它们的混合物和它们的盐都包括在本发明范围内。
本发明前述通式(Ⅰ)所示化合物,可转化为所需的盐,优选药学上可接受的盐,另外,也可从生成的盐转化为游离形式的化合物。作为本发明前述通式(Ⅰ)所示化合物的盐,可列举的有,酸加成盐或碱加成盐,作为酸加成盐,可列举的有,例如,盐酸盐,氢溴酸盐,硝酸盐,硫酸盐,氢碘酸盐,磷酸等无机酸盐,醋酸盐,丙酸盐,丁酸盐,甲酸盐,三氟乙酸盐,马来酸盐,酒石酸盐,柠檬酸盐,硬脂酸盐,琥珀酸盐,乳糖醛酸盐,葡糖酸盐,苯甲酸盐,甲磺酸盐,乙磺酸盐,2-羟基乙磺酸盐,苯磺酸盐,对甲苯磺酸盐,十二烷基硫酸盐,グルセプト酸盐,苹果酸盐,天冬氨酸盐,谷氨酸盐,己二酸盐,草酸盐,烟酸盐,苦味酸盐,硫氰酸盐,十一烷酸盐,扁桃酸盐,富马酸盐,lO-樟脑磺酸盐,乳酸盐,5-氧四氢呋喃-2-羧酸盐,2-羟基戊二酸盐等有机酸盐,作为碱加成盐,可列举的有,例如,钠盐,钾盐,钙盐,镁盐,铵盐等无机碱盐,乙醇胺盐,N,N-二烷基乙醇胺盐等有机碱盐。
本发明的前述通式(Ⅰ)所示化合物或其盐,可根据制备条件以各种结晶形式存在,另外,也有以水合物或与有机溶剂的溶剂化物形式存在的情况,它们任意的结晶形式,水合物,和溶剂化物,以及它们的混合物都包括在本发明的范围内。
作为本发明优选的化合物,可列举的有下列化合物,但本发明并不限于这些化合物。另外,表中所示Me表示甲基,Et表示乙基,n-Pr表示正丙基,i-Pr表示异丙基,n-Bu表示正丁基,n-Pent表示正戊基,n-Hex表示正己基,n-Hept表示正庚基,n-Oct表示正辛基,n-Non表示正壬基,n-Dec表示正癸基,Ph表示苯基,Bn表示苄基。 





本发明的前述通式(Ⅰ)所示的新的红霉素衍生物,例如,可按照下述方法制备,但本发明化合物的制备方法并不限于此。
如果按照本发明的第一种制备方法,制备前述通式(Ⅰ)所示化合物,可将下面通式(Ⅱ)所示化合物: (式中,R1和R3定义同前,R6表示氢原子或羟基保护基。)与下面通式(Ⅲ)所示的羧酸衍生物
(式中,R1和R3定义同前,R6表示氢原子或羟基保护基。)与下面通式(Ⅲ)所示的羧酸衍生物
R2-CO2H (Ⅲ)(式中R2定义同前。)和缩合剂在有或无碱存在下,在有或无溶剂条件下反应,进一步根据需要进行脱保护,制备通式(Ⅰ)所示化合物,或者,与下面通式(Ⅳ)所示的酸酐:
R2-CO2U (Ⅳ)(式中,R2定义同前,U表示酸酐残基。)或者,与下面通式(Ⅴ)所示的酰卤化物或卤化碳酸酯衍生物:
R2-COW (Ⅴ)(式中,R2定义同前,W表示卤素原子。)或者,与下面通式(Ⅵ)所示的异氰酸酯衍生物:
R4-N=C=O (Ⅵ)(式中,R4定义同前。)在有或无碱存在下,在有或无溶剂条件下反应,进一步根据需要进行脱保护,制备通式(Ⅰ)所示化合物。
作为本制备方法中使用的缩合剂,可列举的有,例如,1,3-二环己氧碳化二亚胺,1-乙基-3-(3-二甲基氨基丙基)碳化二亚胺盐酸盐,1,1’-羰基二咪唑,伍德沃德试剂K(2-乙基-5-苯基异噁唑-3’-磺酸)等,作为使用的碱,可列举的有,例如,三乙胺,吡啶,二异丙基乙胺,4-二甲基氨基吡啶,1,8-二氮杂双环[5,4,0]-7-十一碳烯,1,2,2,6,6-五甲基哌啶等有机碱,或碳酸钠,碳酸钾,碳酸氢钠,碳酸氢钾等无机碱。在本发明制造方法中使用的溶剂,可以是任何在该反应中是惰性,且不阻害反应的溶剂,可列举的有,例如,二氯甲烷,1,2-二氯乙烷,氯仿等卤代烃溶剂,苯,甲苯等芳香烃溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,N-甲基-2-吡咯烷酮,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリックトリアミド)等非质子极性溶剂,乙酸甲酯,乙酸乙酯,等酯类溶剂,四氢呋喃,乙醚,1,4-二嗯烷等醚类溶剂,吡啶,甲基吡啶,二甲基吡啶,三甲基吡啶等有机碱类溶剂或这些溶剂的混合物。另外,反应可在冰冷却下至200℃的温度范围内进行。
本发明制备方法中的脱保护反应,可根据羟基保护基R6的种类用各种方法进行脱保护。
例如,R6是烷酰基,卤代烷酰基,芳基羰基等形成酯的保护基时,可在有或无溶剂存在下,在有或无酸或碱存在下,水解脱保护。酯的水解是公知的方法,在酸性水解时,例如,可使用盐酸,硫酸等酸,在碱性水解时,例如可使用碳酸氢钠,碳酸钠,氢氧化钠,氢氧化锂,氢氧化钡,甲醇钠,乙醇钠,叔丁醇钠,叔丁醇钾等碱。可使用这些酸或碱的水溶液,也可使用甲醇,乙醇,正丙醇,异丙醇,正丁醇,仲丁醇,叔丁醇等醇类溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリッケトリアミド)等非质子极性溶剂,四氢呋喃,1,4-二嗯烷等醚类溶剂或这些溶剂的含水溶剂。另外,反应可在冰冷却下至200℃的温度范围内进行。
另外,羟基保护基R6是烷氧羰基,芳氧羰基,芳烷氧羰基等形成碳酸酯的保护基时,可在有或无溶剂存在下,在有或无阳离子清除剂存在下,使酸作用,或在溶剂中存在催化剂的条件下加氢分解,进行脱保护制备。
所使用的溶剂,可以是任何在该反应中是惰性,且不阻害反应的溶剂,可列举的有,例如,水,乙酸,甲醇,乙醇,正丙醇,异丙醇,正丁醇,仲丁醇,叔丁醇等醇类溶剂,二氯甲烷,1,2-二氯乙烷,氯仿等卤代烃类溶剂,苯,甲苯等芳香烃溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,N-甲基-2-吡咯烷酮,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリックトリアミド)等非质子极性溶剂,乙酸甲酯,乙酸乙酯等酯类溶剂,四氢呋喃,乙醚,l,4-二噁烷等醚类溶剂或这些溶剂的混合溶剂等。作为阳离子清除剂,可列举的有,例如,苯甲醚,苯甲硫醚,硫代乙醇等。另外,作为可使用的酸,可列举的有,例如,盐酸,氢溴酸,三氟乙酸,乙酸,硫酸等。另外,反应可在冰冷却下至200℃的温度范围内进行。
另外,作为水解时使用的催化剂,可列举的有,5%钯/炭,10%钯/,炭,20%氢氧化钯/炭等钯类催化剂或氧化铂等。作为氢的来源,除了氢气外,还可使用环己烯,1,3-环己二烯,甲酸,甲酸铵等。作为所使用的溶剂,可以是任何在该反应中是惰性,且不阻害反应的溶剂,可列举的有,例如,水,乙酸,甲醇,乙醇,正丙醇,异丙醇,正丁醇,仲丁醇,叔丁醇等醇类溶剂,二氯甲烷,1,2-二氯乙烷,氯仿等卤代烃类溶剂,苯,甲苯等芳香烃溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,N-甲基-2-吡咯烷酮,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリックトリアミド)等非质子极性溶剂,乙酸甲酯,乙酸乙酯等酯类溶剂,四氢呋喃,乙醚,1,4-二噁烷等醚类溶剂或这些溶剂的混合溶剂等。另外,反应可在室温至200℃的温度范围内进行,氢气压力为常压至200 kgf/cm2范围。
另外,羟基保护基R6是三烷基甲硅烷基型的保护基时,可在有或无溶剂存在下,与酸或氟化四丁基铵作用,进行脱保护制备。
所使用的溶剂,可以是任何在该反应中是惰性,且不阻害反应的溶剂,可列举的有,例如,水,乙酸,甲醇,乙醇,正丙醇,异丙醇,正丁醇,仲丁醇,叔丁醇等醇类溶剂,二氯甲烷,1,2-二氯乙烷,氯仿等卤代烃类溶剂,苯,甲苯等芳香烃溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,N-甲基-2-吡咯烷酮,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリックトリアミド)等非质子极性溶剂,乙酸甲酯,乙酸乙酯等酯类溶剂,四氢呋喃,乙醚,1,4-二噁烷等醚类溶剂或这些溶剂的混合溶剂等。作为可使用的酸,可列举的有,例如,氢氟酸,盐酸,氢溴酸,硫酸等无机酸,三氟乙酸,乙酸,对甲苯磺酸,柠檬酸,草酸等有机酸。另外,反应可在冰冷却下至200℃的温度范围内进行。
如果按照本发明的第二种制备方法,制备前述通式(Ⅰ)所示化合物,可将下面通式(Ⅶ)所示化合物:(式中,R1,R2和R6定义同前。)与下面通式(Ⅷ)所示的化合物
R3-Z (Ⅷ)(式中R3定义同前,Z代表卤原子,甲磺酰氧基或对甲苯磺酰氧基。)和碘化四丁基铵,在有或无碘化钠或碱存在下,在有或无溶剂条件下反应,进一步根据需要进行脱保护,制备通式(Ⅰ)所示化合物。
作为在本制备方法中使用的碱,可列举的有,例如,三乙胺,二异丙基乙胺,4-二甲基氨基吡啶,l,8-二氮杂双环[5,4,0]-7-十一碳烯,l,2,2,6,6-五甲基哌啶等有机碱,或碳酸钠,碳酸钾,碳酸氢钠,碳酸氢钾,氢化钠,氢氧化钾,氢氧化钾等无机碱。所使用的溶剂,可以是任何在该反应中是惰性,且不阻害反应的溶剂。可列举的有,例如,二氯甲烷,l,2-二氯乙烷,氯仿等卤代烃溶剂,苯,甲苯等芳香烃溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,N-甲基-2-吡咯烷酮,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリックトリアミド)等非质子极性溶剂,乙酸甲酯,乙酸乙酯等酯类溶剂,四氢呋喃,乙醚,1,4-二噁烷等醚类溶剂,吡啶,甲基吡啶,二甲基吡啶,三甲基吡啶等有机碱类溶剂或这些溶剂的混合溶剂。另外,反应可在冰冷却下至200℃的温度范围内进行。
本制备方法中的脱保护反应,可根据羟基保护基R6的种类用各种方法进行脱保护,可以按照前述第一种制备方法中记载的方法进行。
如果按照本发明的第三种制备方法,制备前述通式(Ⅰ)所示化合物中R3是可有取代基取代的环烷基的化合物,可将前述通式(Ⅶ)所示化合物与下面通式(Ⅸ)所示环烷化合物:(式中,R7和R8表示烷氧基,m表示1~5的整数。)或与下面通式(Ⅹ)所示环烯化合物: (式中,R9表示烷氧基,p表示l~5的整数。)在有或无酸催化剂存在下,在有或无溶剂条件下反应,进一步根据需要进行脱保护,制备化合物。
(式中,R9表示烷氧基,p表示l~5的整数。)在有或无酸催化剂存在下,在有或无溶剂条件下反应,进一步根据需要进行脱保护,制备化合物。
作为在本制备方法中使用的酸,可列举的有,例如,吡啶盐酸盐,吡啶三氟乙酸盐,吡啶对甲苯磺酸盐等。所使用的溶剂,可以是任何在该反应中是惰性,且不阻害反应的溶剂,可列举的有,例如,二氯甲烷,1,2-二氯乙烷,氯仿等卤代烃溶剂,苯,甲苯等芳香烃溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,N-甲基-2-吡咯烷酮,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリックトリアミド)等非质子极性溶剂,乙酸甲酯,乙酸乙酯等酯类溶剂,四氢呋喃,乙醚,1,4-二噁烷等醚类溶剂或这些溶剂的混合溶剂。另外,反应可在冰冷却下至200℃的温度范围内进行。
本制备方法中的脱保护反应,可根据羟基保护基R6的种类用各种方法进行脱保护,可以按照前述第一种制备方法中记载的方法进行。
如果按照本发明的第四种制备方法,制备前述通式(Ⅰ)所示化合物,可将下面通式(Ⅺ)所示化合物:(式中,R1,R2和R6定义同前。)与下面通式(ⅩⅡ)所示羟胺衍生物或其盐:
R3-0-NH2 (ⅩⅡ)(式中,R3定义同前。)在有或无碱存在下,在有或无溶剂条件下反应,进一步根据需要进行脱保护,制备化合物。
作为在本制备方法中使用的碱,可列举的有,例如,三乙胺,吡啶,咪唑,二异丙基乙胺,4-二甲基氨基吡啶,1,8-二氮杂双环[5,4,0]-7-十一碳烯,1,2,2,6,6-五甲基哌啶等有机碱,或碳酸钠,碳酸钾,碳酸氢钠,碳酸氢钾等无机碱。所使用的溶剂,可以是任何在该反应中是惰性,且不阻害反应的溶剂。可列举的有,例如,甲醇,乙醇,正丙醇,异丙醇,正丁醇,仲丁醇,叔丁醇等醇类溶剂,二氯甲烷,1,2-二氯乙烷,氯仿等卤代烃溶剂,苯,甲苯等芳香烃溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,N-甲基-2-吡咯烷酮,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリックトリアミド)等非质子极性溶剂,乙酸甲酯,乙酸乙酯等酯类溶剂,四氢呋喃,乙醚,1,4-二噁烷等醚类溶剂,吡啶,甲基吡啶,二甲基吡啶,三甲基吡啶等有机碱类溶剂或这些溶剂的混合溶剂。另外,反应可在冰冷却下至200℃的温度范围内进行。
本制备方法中的脱保护反应,可根据羟基保护基R6的种类用各种方法进行脱保护,可以按照前述第一种制备方法中记载的方法进行。
如果按照本发明的第五种制备方法,制备前述通式(Ⅰ)中R2是-X-R4所示化合物,其中X是氧原子,R4是可有取代基取代的烷基的化合物,可按照第一种方法制备X是氧原子,R4是可有取代基取代的芳基的化合物,然后将其与下面通式(ⅩⅢ)所示醇衍生物:
R10-OH (ⅩⅢ)(式中,R10表示可有取代基取代的烷基。)在有或无溶剂条件下反应,进一步根据需要进行脱保护,制备化合物。
作为在本制备方法中使用的溶剂,可以是任何在该反应中是惰性,且不阻害反应的溶剂,可列举的有,例如,二氯甲烷,1,2-二氯乙烷,氯仿等卤代烃溶剂,苯,甲苯等芳香烃溶剂,丙酮,乙腈,N,N-二甲基甲酰胺,N-甲基-2-吡咯烷酮,二甲基亚砜,环丁砜,四亚甲基亚砜,六亚甲基磷酰三胺(ホスホリックトリアミド)等非质子极性溶剂,乙酸甲酯,乙酸乙酯等酯类溶剂,四氢呋喃,乙醚,1,4-二噁烷等醚类溶剂,吡啶,甲基吡啶,二甲基吡啶,三甲基吡啶等有机碱类溶剂或这些溶剂的混合溶剂。另外,反应可在冰冷却下至200℃的温度范围内进行。
本制备方法中的脱保护反应,可根据羟基保护基R6的种类用各种方法进行脱保护,可以按照前述第一种制备方法中记载的方法进行。
另外,作为本发明制备方法中的起始原料,前述通式(Ⅱ)和(Ⅶ)所示的化合物,一部分是日本专利公开昭和63-264495号,日本专利公开平成8-104640号或国际公开WO 93/13116号等中公开的已知化合物,例如,可按照如下方式制备。另外,在下面的制备过程中,前述通式(Ⅱ)和(Ⅶ)所示化合物中,R6是羟基保护基的化合物,可通过向R6是氢原子的化合物中进一步导入保护基来制备。这其中,关于新的化合物,详细记载于参考例中。(式中,Ac表示乙酰基,R1,R2,R3和R6定义同前。)
含有如此制备的前述通式(Ⅰ)所示的新的红霉素衍生物或其盐中至少一种作为有效成分的药物,可用于感染症的治疗,优选非典型耐酸细菌感染症,特别优选用于鸟分支杆菌复合体(MAC)感染症的治疗。上述药物也可用于这些感染症的预防。上述药物通常制备成胶囊,片剂,细粒剂,颗粒剂,散剂,糖浆剂等口服药剂,或注射剂,栓剂,滴眼剂,眼用软膏,滴耳剂,或皮肤外用剂型等给药。作为有效成分,可使用上述红霉素衍生物或其盐的水合物或溶剂化物。这些药剂,可加入药理学,制剂学上允许的添加剂,按照常规方法制备。在口服药剂和栓剂的制备中,可使用赋形剂(乳糖,D-甘露糖醇,玉米淀粉,结晶纤维素等),崩解剂(羧甲基纤维素,羧甲基纤维素钙等),粘合剂(羟丙基纤维素,羟丙基甲基纤维素,聚乙烯吡咯烷酮等),润滑剂(硬脂酸镁,滑石粉等),包衣剂(羟丙基甲基纤维素,白糖,氧化钛等),增塑剂(聚乙二醇等),基质(聚乙二醇,硬脂等)等制剂用成分。在注射剂,或滴眼剂,滴耳剂等制备中,可使用水性或做成即用即溶型剂型的溶解剂或溶解辅助剂(注射用蒸馏水,生理盐水,丙二醇等),pH调节剂(无机或有机酸或碱等),等渗剂(食盐,葡萄糖,甘油等),稳定剂等制剂用成分。另外,在眼用软膏,外用药剂的制备中,可使用作为软膏剂,乳油剂,贴剂适用的制剂成分(白色凡士林,聚乙二醇,甘油,液体石蜡,棉布等)。
对本发明的药物给药量没有特别的限制,通常是,成人口服日给药量约为10~2000mg,非口服给药约1~1000mg,可以1日1次或分多次给药。不过,可根据治疗或预防的目的,感染的部位或病原菌的种类,患者的年龄或症状等,适当增减上述给药量。
实施例
下面,用参考例和实施例对本发明进行说明,但本发明的范围并不限于这些参考例和实施例。表中,Me代表甲基,Et代表乙基,n-Pr代表正丙基,i-Pr代表异丙基,n-Bu代表正丁基,n-Pent代表正戊基,n-Hept代表正庚基,n-Non代表正壬基,Bn代表苄基,Ac代表乙酰基,HR-MS表示高分辨质谱,Anal.Calcd.表示元素分析。参考例1
红霉素A9-[0-(苯乙基)肟]
将红霉素A9-肟5.00g,碘化四丁基铵0.13g和(2-溴乙基)苯1.49g的四氢呋喃30ml混合液在室温下搅拌,向其中加入粉末状氢氧化钾0.53g,将混合物在室温搅拌3.5小时。再加入(2-溴乙基)苯1.49g,粉末状氢氧化钾0.50g,在室温搅拌2.5小时。将反应液倒入冰水中,用饱和碳酸氢钠水溶液使之成碱性后,用乙酸乙酯萃取。萃取液用水洗涤,硫酸钠干燥后,减压蒸除溶剂。向残渣中加入异丙醚使之固化,得到黄褐色固体5.30g。 NMR谱δ(CDCl3)ppm:0.85(3H,t,J=7.5Hz),0.98-1.70(34H,m),1.88-2.00(2H,m),2.20(1H,d,J=10.5Hz),2.28(6H,s),2.37(1H,d,J=16Hz),2.37-2.46(1H,m),2.60-2.69(1H,m),2.85-2.99(3H,m),3.03(1H,t,J=10Hz),3.12(1H,s),3.20(1H,dd,J=10.5,7.5Hz),3.32(3H,s),3.37(1H,s),3.42-3.53(2H,m),3.54-3.63(1H,m),3.71(1H,s),3.95-4.05(2H,m),4.28(2H,t,J=6.5Hz),4.39 (1H,d,J=7.5Hz),4.46(1H,s),4.93(1H,d,J=5Hz),5.14(1H,dd,J=11,2.5Hz),7.17-7.34(5H,m)
按照参考例1同样的方法,得到参考例2~参考例37的化合物。 





 参考例385-0-德糖胺基(デソサミニル)红霉素(エリスロノライド)A9-[0-(苯乙基)肟]
参考例385-0-德糖胺基(デソサミニル)红霉素(エリスロノライド)A9-[0-(苯乙基)肟]
向红霉素A9-[0-(苯乙基)肟]4.80g的1N盐酸40ml悬浮液中,在室温搅拌下加入甲醇50ml,将混合物在室温搅拌3.5小时。将反应液减压浓缩,向残渣中加入冰水,再用10%氢氧化钠水溶液使之成碱性后,用乙醚萃取。萃取液用水洗涤,硫酸钠干燥后,减压蒸除溶剂。残渣用柱色谱纯化(硅胶,二氯甲烷∶甲醇=50∶1→25∶1),得到淡黄褐色非晶型固体3.43g。NMR谱δ(CDCl3)ppm:0.85(3H,t,J=7.5Hz),1.01(3H,d,J=6.5Hz),1.08(3H,d,J=7.5Hz),1.10-1.75(20H,m),1.88-1.99(1H,m),2.0l-2.1l(2H,m), 2.25(6H,s),2.42-2.52(1H,m),2.59-2.71(2H,m),2.90-3.00(2H,m),3.18(1H,s),3.23(1H,dd,J=10.5,7.5Hz),3.40-3.70(6H,m),3.83(1H,brs),4.21-4.3l(2H,m),4.38(1H,d,J=7.5Hz),4.38(1H,s),5.23(1H,dd,J=ll,2.5Hz),7.16-7.3l(5H,m)HR-MS m/z 694.43983[Calcd.forC37H62N2O10(M+):694.44045]
按照参考例38同样的方法,得到参考例39~参考例76的化合物。



 参考例775-0-德糖胺基-6-0-甲基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]
参考例775-0-德糖胺基-6-0-甲基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]
向5-0-德糖胺基-6-0-甲基红霉素(エリスロノライド)A9-肟1.10g和吡啶盐酸盐O.32g的二氯甲烷1lml混合液中,在室温搅拌下滴加入1,l-二甲基环己烷1.4m1的二氯甲烷4ml溶液,将混合物在室温搅拌19小时,然后再加热回流18小时。向反应液中加入水,用饱和碳酸氢钠水溶液使之成碱性后,用乙醚萃取。萃取液用饱和食盐水洗涤,硫酸钠干燥后,减压蒸除溶剂。残渣用柱色谱纯化(硅胶,乙酸乙酯∶甲醇∶氨水=20∶1∶0.1→10∶1∶0.1),所得固体用异丙醚洗净,得到无色非晶型固体0.37g。NMR谱δ(CDCl3)ppm:0.84(3H,t,J=7.5Hz),0.99(3H,d,J,7.5Hz),1.10(3H,d,J=7.5Hz),1.07-2.02(3lH,m),2.09-2.20(1H,m),2.25(6H,s),2.43-2.52(1H,m),2.57-2.72(2H,m),3.00(3H,s),3.2l(3H,s),3.24(1H,dd,J=10.5,7.5Hz),3.33(1H,s),3.48-3.60(3H,m),3.68-3.93(4H,m),4.38(1H,d,J=7.5Hz),4.54(1H,s),5.23(1H,dd,J=1l,2.5Hz)HR-MS m/z 716.47970[Calcd.forC37H68N2O11(M+):716.48231]
按照参考例77同样的方法,得到参考例78~参考例80的化合物。 参考例812’-0-乙酰基-5-0-德糖胺基红霉素(エリスロノライド)A9-[-(苯乙基)肟]
参考例812’-0-乙酰基-5-0-德糖胺基红霉素(エリスロノライド)A9-[-(苯乙基)肟]
向5-0-德糖胺基红霉素(エリスロノライド)A9-[0-(苯乙基)肟]3.25g的丙酮40ml溶液中,在室温搅拌下加入乙酸酐0.53ml,将混合物在室温搅拌4小时。将反应液减压浓缩,向残渣中加入水,用饱和碳酸氢钠水溶液使之成碱性后,用二氯甲烷萃取。萃取液用水洗涤,硫酸钠干燥后,减压蒸除溶剂。得到淡黄色非晶形固体3.45g。NMR谱δ(CDCl3)ppm:0.84(3H,t,J=7.5Hz),0.9l(3H,d,J=7.5Hz),0.99(3H,d,3=6.5Hz),1.12-1.52(19H,m),1.70-1.76(2H,m),1.90-2.00(1H,m),2.06(3H,s),2.05-2.14(1H,m),2.28(6H,s),2.6l-2.68(2H,m),2.73-2.83(1H,m),2.94(2H,t,J=6.5Hz),3.16(1H,brs),3.42=3.60(5H,m),3.68(1H,s),4.26(2H,m),4.46(1H,brs),4.56(1H,d,J=7.5Hz),4.77(1H,dd,J=lO.5,8Hz),5.24(1H,dd,J=ll,2.5Hz),7.17-7.3l(5H,m)HR-MS m/z 736.45008[Calcd.forC39H64N2O11(M+):736.45101]
按照参考例81同样的方法,得到参考例82-参考例123的化合物。 





 实施例15-0-德糖胺基-3-0-苯乙酰基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]
实施例15-0-德糖胺基-3-0-苯乙酰基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]
向乙酸苯酯1.11g和三乙胺1.2ml的二氯甲烷40ml溶液中,在冰冷却下,滴加入三甲基乙酰氯1.0ml后,在相同温度下,搅拌1小时。在冰冷却并搅拌下,向该反应液中,依次滴加入吡啶2.2ml和2’-0-乙酰基-5-0-德糖胺基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]2.00g的二氯甲烷15ml溶液,将混合物在室温搅拌1天。向反应液中加入冰水,用饱和碳酸氢钠水溶液使之成碱性后,用二氯甲烷萃取。萃取液用水洗涤,硫酸钠干燥后,减压蒸除溶剂。残渣用柱色谱纯化(硅胶,二氯甲烷∶甲醇=33∶1),得到黄褐色粘性液体1.38g。将该黄褐色粘性液体1.20g的甲醇50ml溶液在室温搅拌1天。将反应液减压浓缩,残渣用柱色谱纯化(硅胶,二氯甲烷∶甲醇=100∶1→50∶3),得到无色非晶型固体0.72g。NMR谱δ(CDCl3)ppm:0.80(3H,t,J=7.5Hz),0.86(3H,d,J=6.5Hz),1.03(3H,d,J=6.5Hz),1.12-1.98(32H,m),2.22-2.3l(1H,m),2.30(6H,s),2.33-2.42(1H,m),2.60(1H,brs),2.64-2.72(1H,m),2.75-2.85(1H,m),3.04-3.2l(3H,m),3.19(3H,s),3.50(1H,d,J=4.5Hz),3.64-3.78(2H,m),3.65(1H,d,J=14.5Hz),3.7l(1H,d,J=14.5Hz),3.96(1H,d,J=7.5Hz),4.57(1H,s),5.14(1H,d,J=lO.5Hz),5.20(1H,dd,J=11,2.5Hz),7.24-7.38(5H,m)HR-MS m/z 820.50550[Calcd.forC44H72N2O12(M+):820.50853]
按照实施例1同样的方法,得到实施例2~实施例3的化合物。 实施例45-0-德糖胺基-3-0-苯乙酰基-6-0-甲基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]
实施例45-0-德糖胺基-3-0-苯乙酰基-6-0-甲基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]
向乙酸苯酯0.05g和草酰氯0.03ml的二氯甲烷0.6ml溶液中,在室温搅拌下,加入N,N-二甲基甲酰胺1滴,在室温搅拌30分钟后,将反应液减压浓缩。在室温搅拌下,向残渣的二氯甲烷2.5ml溶液中,滴加入2’-0-乙酰基-5-0-德糖胺基-6-0-甲基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]0.10g和吡啶0.09ml的二氯甲烷0.8ml溶液,将混合物在室温搅拌1小时。向反应液中加入冰水,用饱和碳酸氢钠水溶液使之成碱性后,用乙醚萃取。萃取液用水洗涤,硫酸钠干燥后,减压蒸除溶剂。将所得残渣的甲醇4ml溶液在室温搅拌20小时。将反应液减压浓缩,向残渣中加入冰水,用饱和碳酸氢钠水溶液使之成碱性后,用乙醚萃取。萃取液用水洗涤,硫酸钠干燥后,减压蒸除溶剂。残渣用柱色谱纯化(硅胶,二氯甲烷∶甲醇=20∶1),得到无色非晶形固体0.05g。NMR谱δ(CDCl3)ppm:0.80(3H,t,J=7.5Hz),0.89(3H,d,J=6.5Hz),0.97(3H,d,3=7.5Hz),1.00=2.00(31H,m),2.10-2.39(2H,m),2.28(6H,s),2.52-2.64(1H,m),2.76-2.88(1H,m),2.90-3.35(4H,m),3.07(3H,s),3.2l(3H,s),3.37-3.50(1H,m),3.67(1H,d,J=15Hz),3.73(1H,d,J=15Hz),3.68-3.82(2H,m),3.90(1H,d,J=7.5Hz),4.58(1H,s),5.07(1H,d,J=11Hz),5.20(1H,dd,J=11,2Hz),7.17-7.40(5H,m)HR-MS m/z 721.42800[Calcd.forC38H61N2O11(M+-C7H13O):721.42754]
按照实施例4同样的方法,得到实施例5~实施例22的化合物。




 实施例235-0-德糖胺基-3-0-苯乙酰基红霉素(エリスロノライド)A9-[0-(苯乙基)肟]
实施例235-0-德糖胺基-3-0-苯乙酰基红霉素(エリスロノライド)A9-[0-(苯乙基)肟]
将2’-0-乙酰基-5-0-德糖胺基红霉素(エリスロノライド)A9-[0-(苯乙基)肟]0.5g,乙酸苯酯0.28g,l-乙基-3-0(3-二甲基氨基丙基)碳二亚胺盐酸盐0.40g和4-二甲基氨基吡啶0.11g的二氯甲烷5ml混合液,在室温下搅拌4小时。向反应液中加入水,用饱和碳酸氢钠水溶液使之成碱性后,用二氯甲烷萃取。萃取液用水洗涤,硫酸钠干燥后,减压蒸除溶剂。将所得残渣的甲醇40ml溶液在室温搅拌2.5天。将反应液减压浓缩,残渣用柱色谱纯化(硅胶,二氯甲烷∶甲醇=50∶1→25∶1),得到淡黄色非晶形固体O.32g。NMR谱δ(CDCl3)ppm:0.8l(3H,t,J=7.5Hz),0.88(3H,d,J=6.5Hz),0.97(3H,d.J=7.5Hz),1.05-1.65(2lH,m),1,87-1.97(1H,m),2.19-2.39(2H,m),2.27(6H,s),2.60-2.68(1H,m),2.76-2.85(1H,m),2.95(2H,t,J=7Hz),3.06-3.19(3H,m),3.26(1H,brs),3.39(1H,d,J=4.5Hz),3.52-3.61(1H,m),3.65(1H,d,J=14.5Hz),3.70(1H,d,J=14.5Hz),3.72(1H,s),3.94(1H,d,J=7.5Hz),4.27(2H,td,J=7.2Hz),4.47(1H,s),5.13(1H,d,J=llHz),5.22(1H,dd,J=11.2Hz),7.14-7.38(10H,m)HR-MS m/z 675.40315[Calcd.forC37H57NO10(M+-C8H11NO):675.39825]
按照实施例23同样的方法,得到实施例24~实施例202的化合物。







































 实施例2035-0-德糖胺基-3-0-咪唑基羰基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]
实施例2035-0-德糖胺基-3-0-咪唑基羰基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]
向2’-0-乙酰基-5-0-德糖胺基红霉素(エリスロノライド)A9-[0-(1-甲氧环己基)肟]2.00g的二氯甲烷30ml溶液中,在室温搅拌下,依次加入1,1’-羰基二咪唑2.18g和4-二甲基氨基吡啶0.36g,将混合物加热回流4天。将反应液冷却后,向其中加入冰水,用饱和碳酸氢钠水溶液使之成碱性后,用二氯甲烷萃取。萃取液用水洗涤,硫酸钠干燥后,减压蒸除溶剂。残渣用柱色谱纯化(硅胶,二氯甲烷∶甲醇=33∶1),得到淡黄色非晶形固体2.39g。将该淡黄色非晶型固体1.32g的甲醇50ml溶液在室温搅拌1天。将反应液减压浓缩,得到淡黄色非晶形固体1.20g。NMR谱δ(CDCl3)ppm:0.85(3H,t,J=7.5Hz),1.05(3H,d,J=7.5Hz),1.06(3H,d,J=6.5Hz),1.12-2.00(31H,m),2.08(1H,s),2.18(6H,s),2.37-2.44(1H,m),2.49-2.58(1H,m),2.67-2.77(1H,m),3.00-3.08(2H,m),3.18-3.24(1H,m),3.2l(3H,s),3.3l(1H,s),3.53(1H,d,J=3.5Hz),3.63-3.77(3H,m),3.80(1H,d,J=6.5Hz),4.64(1H,s),5.25-5.33(2H,m),7.1l(1H,s),7.50(1H,s),8.22(1H,s)HR-MS m/z 684.39400[Calcd.forC33H56N4O11(M++l-C7H13O):684.39456]实施例2045-0-德糖胺基-3-0-苯乙酰基红霉素(エリスロノライド)A9-[0-(2-甲氧苯乙基)肟]
将5-0-德糖胺基-3-0-苯乙酰基红霉素(エリスロノライド)A9-肟0.40g,碘化四丁基铵11mg,2-甲氧基苯乙基甲磺酸酯0.20g和粉末状氢氧化钾45mg的四氢呋喃4ml混合液在室温搅拌5天。向反应液中加入水,用乙醚萃取。萃取液用饱和食盐水洗净,硫酸钠干燥后,减压蒸除溶剂。残渣用柱色谱纯化(硅胶,二氯甲烷∶甲醇∶氨水=100∶3∶1.5),得到无色非晶形固体0.50g。 NMR谱δ(CDCl3)ppm:0.81(3H,t,J=7.5Hz),0.87(3H,d,J=6.5Hz),0.97(3H,d,J=7.5Hz),1.10-1.67(21H,m),1.88-1.99(1H,m),2.22-2.30(1H,m),2.27(6H,s),2.31-2.38(1H,m),2.59-2.68(1H,m),2.77-2.85(1H,m),2.87-3.04(2H,m),3.06-3.20(3H,m),3.28(1H,brs),3.41(1H,d,J=5Hz),3.54-3.63(1H,m),3.66(1H,d,J=14.5Hz),3.70(1H,d,J=14.5Hz),3.73(1H,s),3.84(3H,s),3.95(1H,d,J=7.5Hz),4.20-4.28(2H,m),4.51(1H,s),5.14(1H,d,J=11Hz),5.22(1H,dd,J=11.5,2Hz),6.81-6.93(2H,m),7.08-7.14(1H,m),7.17-7.40(6H,m)HR-MS m/z 668.38103[Calcd.forC38H54NO9(M++1-C8H16NO3):668.37986]
按照实施例204同样的方法,得到实施例205~实施例207的化合物。 实施例2085-0-德糖胺基-3-O-甲氧羰基红霉素(エリスロノライド)A9-[0-(3-苯基丙基)肟]
向2’-0-乙酰基-5-0-德糖胺基红霉素(エリスロノライド)A9-[0-(3-苯基丙基)肟]0.80g的吡啶8ml溶液中,滴入氯甲酸苯酯1.3ml后,将混合物在室温搅拌24小时。向反应液中加入水,用乙醚萃取。萃取液用饱和食盐水洗净,硫酸钠干燥后,减压蒸除溶剂。残渣用柱色谱纯化(硅胶,乙酸乙酯),得到无色非晶形固体0.38g。将该无色非晶型固体0.38g的甲醇5ml溶液在室温搅拌160小时。将反应液减压浓缩,残渣用柱色谱纯化(硅胶,乙酸乙酯),得到无色非晶型固体0.22g。NMR谱δ(CDCl3)ppm:0.84(3H,t,J=7.5Hz),1.04(3H,d,J=6.5Hz),1.19-1.80(23H,m),1.90-2.03(4H,m),2.20-2.40(7H,m),2.47-2.60(1H,m),2.60-2.75(3H,m),2.85-2.95(1H,m),3.12(1H,s),3.15-3.30(2H,m),3.30-3.40(1H,m),3.52(1H,d,J=3.5Hz),3.62-3.72(2H,m),3.80(3H,s),4.06(2H,t,J=6.5Hz),4.19(1H,d,J=7.5Hz),4.45(1H,s),4.89(1H,d,J=11Hz),5.26(1H,dd,J=ll,2Hz),7.10-7.3l(5H,m)HR-MS m/z 766.46354[Calcd.forC40H66N2O12(M+):766.46158]
按照实施例208同样的方法,得到实施例209的化合物。实施例2095-0-德糖胺基-3-0-甲氧羰基红霉素(エリスロノライド)A9-[0-(2-苯氧乙基)肟]性状无色非晶形固体NMR谱δ(CDCl3)ppm:0.85(3H,t,J=7.5Hz),1.00(3H,d,f=6.5Hz),1.05-1.70(23H,m),1.90-2.00(1H,m),2.12(1H,s),2.20-2.35(1H,m),2.27(6H,s),2.40-2.50(1H,m),2.62-2.75(1H,m),2.85-2.95(1H,m),3.12(1H,s),3.15(1H,dd,J=10.5,7.5Hz),3.24(1H,brs),3.30-3.40(1H,m),3.46(1H,d,J=4.5Hz),3.65-3.85(2H,m),3.79(3H,s),4.13-4.20(3H,m),4.37-4.43(3H,m),4.87(1H,d,J=10Hz),5.26(1H,dd,J=1l,2.5Hz),6.90-7.00(3H,m),7.25-7.35(2H,m)HR-MS m/z 768.43949[Calcd.forC39H64N2O13(M+):768.44084]实施例2105-0-德糖胺基-3-0-苯基氨基甲酰基红霉素(エリスロノライド)A9-[0-(3-苯基丙基)肟]
将2,-0-乙酰基-5-0-德糖胺基红霉素(エリスロノライド)A9-[0-(3-苯基丙基)肟]0.40g,异氰酸苯酯0.34ml和吡啶0.12ml的四氢呋喃4ml溶液,在室温搅拌28小时。向反应液中加入水,用乙醚萃取。萃取液用饱和食盐水洗净,硫酸钠干燥后,减压蒸除溶剂。将残渣的甲醇15ml溶液在室温搅拌24小时。将反应液减压浓缩,残渣用柱色谱纯化(硅胶,乙酸乙酯),得到无色非晶形固体0.31g。NMR谱δ(CDCl3)ppm:0.85(3H,t,J=7.5Hz),0.97-1.68(27H,m),1.90-2.03(4H,m),2.13-2.38(2H,m),2.19(6H,s),2.62-2.77(3H,m),2.82-2.95(1H,m),3.05-3.25(3H,m),3.55(1H,d,J=3.5Hz),3.63-3.79(2H,m),3.98-4.17(3H,m),4.48(1H,s),5.03(1H,d,J=10.5Hz),5.27(1H,dd,J=11.5,2Hz),7.05(1H,t,J=7.5Hz),7.11(1H,brs),7.16-7.22(3H,m),7.25-7.34(4H,m),7.40-7.48(2H,m)HR-MS m/z 827.49261[Calcd.forC45H69N3O11(M+):827.49321]
按照实施例210同样的方法,得到实施例211~实施例212的化合物。
下面,为确认本发明化合物的优良效果,进行对非典型耐酸细菌(MAC)抗菌谱的测定。另外,作为对照化合物,使用克拉霉素和利福平。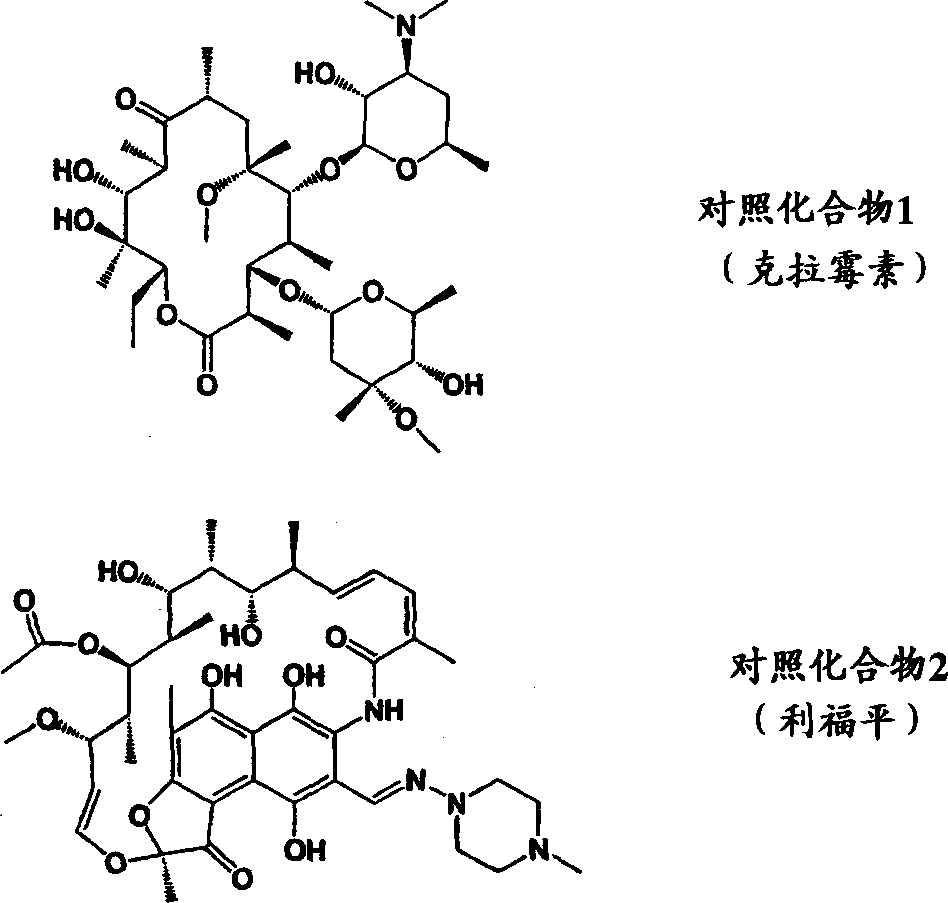 对非典型耐酸细菌的抗菌谱
对非典型耐酸细菌的抗菌谱
抗菌力(最小生长抑制浓度:MIC)的测定,按照日本化学疗法学会标准法[日本化学疗法学会志,29卷,76页(1981年)],使用临床分离菌株的非典型耐酸细菌,以活菌数为106CFU/ml进行试验。结果列于表126和表127。本发明化合物对包括耐红霉素的非典型耐酸细菌(M.avium20092等)在内的非典型耐酸细菌,与对照化合物相比,显示出优秀的抗菌力。另外,表中的细菌名如下。鸟分支杆菌(M.avium)胞内分支杆菌(M.intracellulare)
抗菌谱(最小生长抑制浓度μg/ml)
| 试验菌 | 实施例23 | 实施例40 | 实施例78 | 对照化合物1 | 对照化合物2 |
| M.avium 20034 | 3.13 | 3.13 | 3.13 | 3.13 | 12.5 |
| M.avium 20045 | 3.13 | 3.13 | 1.56 | 1.56 | 3.13 |
| M.avium 20092 | 6.25 | 3.13 | 3.13 | >50 | 50 |
| M.avium 20096 | 3.13 | 3.13 | 1.56 | >50 | 3.13 |
| M.intracellulare 20066 | 3.13 | 3.13 | 0.78 | 3.13 | 3.13 |
| M.intracellulare 20067 | 1.56 | 1.56 | 0.78 | 1.56 | 1.56 |
| M.intracellulare 20073 | 3.13 | 1.56 | 0.78 | 1.56 | 3.13 |
| M.intracellulare 20075 | 3.13 | 3.13 | 0.78 | 3.13 | 3.13 |
敏感性分布(最小生长抑制浓度μg/ml)
| 试验菌(菌株数) | 化合物 | 最小生长抑制浓度 μg/ml | |||
| 范围 | 50% | 80% | 90% | ||
| M.avium(27) | 实施例23实施例40实施例78对照化合物1对照化合物2 | 1.56 ~ 6.251.56 ~ 3.131.56 ~ 3.130.78 ~ >1000.39 ~ 50 | 3.133.131.563.1325 | 3.133.131.566.2550 | 3.133.133.1312.550 |
Claims (8)
1、下述通式所示的红霉素衍生物或其盐:(式中,R1代表氢原子或低级烷基,R2代表可有取代基取代的烷基,可有取代基取代的环烷基,可有取代基取代的(环烷基)烷基,可有取代基取代的芳基,可有取代基取代的芳烷基,可有取代基取代的苯乙烯基,或式-X-R4所示的基团,X代表氧原子或氨基,R4代表可有取代基取代的烷基或可有取代基取代的芳基,R3代表羧基,烷氧羰基,芳氧羰基或芳烷氧羰基取代的烷基,可有取代基取代的环烷基,可有取代基取代的(环烷基)烷基,可有取代基取代的烯基,或式-(CH2)n-Y-R5所示的基团,Y代表可有取代基取代的亚甲基,氧原子,硫原子,亚硫酰基,磺酰基,烷基取代或未取代的氨基,或羰基,R5代表可有取代基取代的芳基,n代表1~5的整数)。
2、权利要求1中记载的化合物或其盐,其中R1是氢原子。
3、含有权利要求1或2中记载的化合物或其生理上可接受的盐作为有效成分的药物。
4、作为感染症治疗剂使用的权利要求3中记载的药物。
5、作为非典型耐酸细菌感染症治疗剂使用的权利要求4中记载的药物。
6、权利要求5中记载的药物,其中非典型耐酸细菌是鸟分支杆菌复合体。
7、权利要求1中记载的通式(Ⅰ)所示化合物或其盐在制备权利要求3-6中任一项中记载的药物中的应用。
8、感染症的治疗方法,包括对患者使用治疗有效量的权利要求1中记载的通式(Ⅰ)所示化合物或其生理上可接受的盐。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP362634/97 | 1997-12-11 | ||
| JP36263497 | 1997-12-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1284961A true CN1284961A (zh) | 2001-02-21 |
Family
ID=18477360
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 98813554 Pending CN1284961A (zh) | 1997-12-11 | 1998-12-09 | 红霉素衍生物 |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1044985A4 (zh) |
| CN (1) | CN1284961A (zh) |
| AU (1) | AU1504899A (zh) |
| WO (1) | WO1999029709A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110018266A (zh) * | 2019-02-15 | 2019-07-16 | 广州市妇女儿童医疗中心 | 一种快速定量分析48种氨基酸的方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2373117C (en) | 1999-05-24 | 2006-02-28 | Pfizer Products Inc. | 13-methyl-erythromycin derivatives |
| US20020115621A1 (en) | 2000-08-07 | 2002-08-22 | Wei-Gu Su | Macrolide antibiotics |
| JP2002173498A (ja) * | 2000-09-27 | 2002-06-21 | Hokuriku Seiyaku Co Ltd | エリスロマイシン誘導体 |
| WO2005030786A1 (en) * | 2003-09-25 | 2005-04-07 | Ranbaxy Laboratories Limited | 3'-n-substituted-3-o-substituted erythronolide a derivatives |
| JP4737495B2 (ja) * | 2004-01-14 | 2011-08-03 | 塩野義製薬株式会社 | エリスロマイシン誘導体 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2260242A1 (de) * | 1972-12-08 | 1974-06-12 | Polska Akademia Neuk | 8-hydroxyerythromycin und derivate davon, ihre herstellung und solche verbindungen enthaltende mittel |
| US3923784A (en) * | 1973-09-10 | 1975-12-02 | Hoffmann La Roche | Erythromycin a derivatives |
| IL99995A (en) * | 1990-11-21 | 1997-11-20 | Roussel Uclaf | Erythromycin derivatives, their preparation and pharmaceutical compositions containing them |
| US5523399A (en) * | 1991-12-27 | 1996-06-04 | Taisho Pharmaceutical Co., Ltd. | 5-O-desosaminylerythronolide derivatives |
-
1998
- 1998-12-09 CN CN 98813554 patent/CN1284961A/zh active Pending
- 1998-12-09 EP EP98959141A patent/EP1044985A4/en not_active Withdrawn
- 1998-12-09 WO PCT/JP1998/005570 patent/WO1999029709A1/ja not_active Application Discontinuation
- 1998-12-09 AU AU15048/99A patent/AU1504899A/en not_active Abandoned
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110018266A (zh) * | 2019-02-15 | 2019-07-16 | 广州市妇女儿童医疗中心 | 一种快速定量分析48种氨基酸的方法 |
| CN110018266B (zh) * | 2019-02-15 | 2022-03-04 | 广州市妇女儿童医疗中心 | 一种快速定量分析48种氨基酸的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO1999029709A1 (fr) | 1999-06-17 |
| EP1044985A1 (en) | 2000-10-18 |
| EP1044985A4 (en) | 2001-04-18 |
| AU1504899A (en) | 1999-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5319080A (en) | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates | |
| JP5764168B2 (ja) | 大環状多形、そのような多形を含む組成物、ならびにその使用方法および製造方法 | |
| JP5153329B2 (ja) | (r/s)リファマイシン誘導体、その調製、および医薬組成物 | |
| ES2312023T3 (es) | Fenilos sustituidos con d-piranosilo, farmacos que contienen estos compuestos, su empleo y metodo para prepararlos. | |
| RU2453546C2 (ru) | Производные 5-гидроксиметилоксазолидин-2-она | |
| EP2734535A2 (en) | 2',3'-DIDEOXY-2'-alpha-FLUORO-2'-beta-C-METHYLNUCLEOSIDES AND PRODRUGS THEREOF | |
| CN108864192A (zh) | 功能改性的寡核苷酸及其亚单元 | |
| PT749432E (pt) | Compostos de 9-desoxotaxano | |
| UA70298C2 (uk) | Похідні с-4"-заміщених макролідів, спосіб їх одержання, проміжні сполуки, фармацевтична композиція та спосіб лікування бактеріальної інфекції або протозойної інфекції | |
| CN102459296B (zh) | 具有抗微生物活性的酮内酯类化合物 | |
| CN101855219B (zh) | 6,11-桥联的联芳大环内酯类 | |
| JP2005536465A (ja) | 6,11二環式エリスロマイシン誘導体 | |
| CN101801923A (zh) | 截短侧耳素衍生物及其作为抗微生物剂的应用 | |
| KR19990067492A (ko) | 트리사이클릭 에리트로마이신 유도체 | |
| CN101166749B (zh) | 6-11桥联的肟乙基琥珀酸红霉素酯衍生物 | |
| EP3450450A1 (en) | Vancomycin derivative, preparation method, pharmaceutical composition and use thereof | |
| CN107530365A (zh) | 具有修饰的脱氧糖胺糖的大环内酯及其用途 | |
| JP2002502366A (ja) | 3―デスクラジノース―2,3―無水エリスロマイシン誘導体 | |
| WO2012175047A1 (zh) | 一种含氟水溶性铂配合物在制备防治肿瘤药物的用途 | |
| WO2021146536A1 (en) | Therapeutic agents and methods of treatment | |
| CN1284961A (zh) | 红霉素衍生物 | |
| KR20060091050A (ko) | 항균 활성을 갖는 신규 린코마이신 유도체 | |
| CN1343216A (zh) | 红霉素衍生物 | |
| WO2021055933A1 (en) | Novel mimetics of heparin oligosaccharides | |
| WO2023014927A1 (en) | Prodrugs of tapinarof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |