CN1022914C - 新的1,3-二取代哌啶衍生物的制备方法 - Google Patents
新的1,3-二取代哌啶衍生物的制备方法 Download PDFInfo
- Publication number
- CN1022914C CN1022914C CN89104933A CN89104933A CN1022914C CN 1022914 C CN1022914 C CN 1022914C CN 89104933 A CN89104933 A CN 89104933A CN 89104933 A CN89104933 A CN 89104933A CN 1022914 C CN1022914 C CN 1022914C
- Authority
- CN
- China
- Prior art keywords
- formula
- alkyl
- compound
- reaction
- definition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000002360 preparation method Methods 0.000 title claims description 86
- 238000000034 method Methods 0.000 title claims description 78
- -1 1, 3-disubstituted piperidine Chemical class 0.000 title claims description 58
- 230000008569 process Effects 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 151
- 150000003839 salts Chemical class 0.000 claims abstract description 13
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 135
- 238000006243 chemical reaction Methods 0.000 claims description 59
- 239000000203 mixture Substances 0.000 claims description 48
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 21
- 239000002253 acid Substances 0.000 claims description 20
- 229910052739 hydrogen Inorganic materials 0.000 claims description 18
- 229910052799 carbon Inorganic materials 0.000 claims description 15
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 claims description 15
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 14
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 13
- 229910021529 ammonia Inorganic materials 0.000 claims description 10
- 239000012280 lithium aluminium hydride Substances 0.000 claims description 10
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 claims description 9
- 230000002829 reductive effect Effects 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 8
- 239000000460 chlorine Substances 0.000 claims description 7
- 229910052801 chlorine Inorganic materials 0.000 claims description 7
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- 150000002367 halogens Chemical class 0.000 claims description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 7
- 150000001412 amines Chemical class 0.000 claims description 6
- 229910052794 bromium Inorganic materials 0.000 claims description 6
- 239000003795 chemical substances by application Substances 0.000 claims description 6
- 239000003513 alkali Substances 0.000 claims description 5
- NDKBVBUGCNGSJJ-UHFFFAOYSA-M benzyltrimethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)CC1=CC=CC=C1 NDKBVBUGCNGSJJ-UHFFFAOYSA-M 0.000 claims description 5
- 229910002091 carbon monoxide Inorganic materials 0.000 claims description 5
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 5
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 5
- 239000000376 reactant Substances 0.000 claims description 5
- 230000009467 reduction Effects 0.000 claims description 5
- 235000009518 sodium iodide Nutrition 0.000 claims description 5
- FDDDEECHVMSUSB-UHFFFAOYSA-N sulfanilamide Chemical compound NC1=CC=C(S(N)(=O)=O)C=C1 FDDDEECHVMSUSB-UHFFFAOYSA-N 0.000 claims description 5
- 229940124530 sulfonamide Drugs 0.000 claims description 5
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 claims description 4
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 claims description 4
- OWIKHYCFFJSOEH-UHFFFAOYSA-N Isocyanic acid Chemical compound N=C=O OWIKHYCFFJSOEH-UHFFFAOYSA-N 0.000 claims description 4
- 150000008065 acid anhydrides Chemical class 0.000 claims description 4
- 150000001263 acyl chlorides Chemical class 0.000 claims description 4
- 125000003545 alkoxy group Chemical group 0.000 claims description 4
- XLJMAIOERFSOGZ-UHFFFAOYSA-N anhydrous cyanic acid Natural products OC#N XLJMAIOERFSOGZ-UHFFFAOYSA-N 0.000 claims description 4
- 229910052740 iodine Inorganic materials 0.000 claims description 4
- 125000002346 iodo group Chemical group I* 0.000 claims description 4
- 239000003054 catalyst Substances 0.000 claims description 3
- IHLVCKWPAMTVTG-UHFFFAOYSA-N lithium;carbanide Chemical compound [Li+].[CH3-] IHLVCKWPAMTVTG-UHFFFAOYSA-N 0.000 claims description 3
- 125000005905 mesyloxy group Chemical group 0.000 claims description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 3
- 235000007715 potassium iodide Nutrition 0.000 claims description 3
- 229960004839 potassium iodide Drugs 0.000 claims description 3
- NVBFHJWHLNUMCV-UHFFFAOYSA-N sulfamide Chemical compound NS(N)(=O)=O NVBFHJWHLNUMCV-UHFFFAOYSA-N 0.000 claims description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 2
- BBOPZLFBGQCZHF-UHFFFAOYSA-N [Br].C[Mg] Chemical compound [Br].C[Mg] BBOPZLFBGQCZHF-UHFFFAOYSA-N 0.000 claims description 2
- FEDYKTWNKOVRSK-UHFFFAOYSA-N [Cl].[Mg]C Chemical compound [Cl].[Mg]C FEDYKTWNKOVRSK-UHFFFAOYSA-N 0.000 claims description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 2
- 230000010933 acylation Effects 0.000 claims description 2
- 238000005917 acylation reaction Methods 0.000 claims description 2
- 150000001351 alkyl iodides Chemical class 0.000 claims description 2
- 239000001569 carbon dioxide Substances 0.000 claims description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 claims description 2
- 239000003638 chemical reducing agent Substances 0.000 claims description 2
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 claims description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 2
- 150000002431 hydrogen Chemical class 0.000 claims 3
- DVNYTAVYBRSTGK-UHFFFAOYSA-N 5-aminoimidazole-4-carboxamide Chemical class NC(=O)C=1N=CNC=1N DVNYTAVYBRSTGK-UHFFFAOYSA-N 0.000 claims 2
- 125000004185 ester group Chemical group 0.000 claims 2
- GNLJBJNONOOOQC-UHFFFAOYSA-N $l^{3}-carbane;magnesium Chemical compound [Mg]C GNLJBJNONOOOQC-UHFFFAOYSA-N 0.000 claims 1
- 125000001589 carboacyl group Chemical group 0.000 claims 1
- 150000002148 esters Chemical class 0.000 claims 1
- 229950010911 orazamide Drugs 0.000 claims 1
- 150000003573 thiols Chemical class 0.000 claims 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 11
- 201000010099 disease Diseases 0.000 abstract description 8
- 230000004899 motility Effects 0.000 abstract description 4
- 210000002460 smooth muscle Anatomy 0.000 abstract description 2
- 238000004519 manufacturing process Methods 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 105
- 238000001704 evaporation Methods 0.000 description 80
- 230000008020 evaporation Effects 0.000 description 79
- 150000003053 piperidines Chemical class 0.000 description 70
- 239000000243 solution Substances 0.000 description 63
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 60
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 58
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 53
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 46
- GJUMZNFPQSOVCL-UHFFFAOYSA-N 1-phenylmethoxypiperidine Chemical class C=1C=CC=CC=1CON1CCCCC1 GJUMZNFPQSOVCL-UHFFFAOYSA-N 0.000 description 38
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 38
- 239000012230 colorless oil Substances 0.000 description 36
- 239000000377 silicon dioxide Substances 0.000 description 29
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 27
- 238000000746 purification Methods 0.000 description 27
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 26
- 239000012044 organic layer Substances 0.000 description 21
- 238000010992 reflux Methods 0.000 description 21
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 239000007787 solid Substances 0.000 description 18
- 239000002585 base Substances 0.000 description 17
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 17
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 239000007864 aqueous solution Substances 0.000 description 13
- 235000017550 sodium carbonate Nutrition 0.000 description 13
- 229910000029 sodium carbonate Inorganic materials 0.000 description 13
- 238000003756 stirring Methods 0.000 description 13
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 13
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- 239000003960 organic solvent Substances 0.000 description 12
- 239000002994 raw material Substances 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 241000786363 Rhampholeon spectrum Species 0.000 description 11
- 238000005406 washing Methods 0.000 description 11
- 239000002904 solvent Substances 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000000047 product Substances 0.000 description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 7
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 7
- 235000017557 sodium bicarbonate Nutrition 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 7
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 6
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 239000001301 oxygen Substances 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 229910052938 sodium sulfate Inorganic materials 0.000 description 6
- 235000011152 sodium sulphate Nutrition 0.000 description 6
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Chemical group O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 6
- 238000007796 conventional method Methods 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 238000010791 quenching Methods 0.000 description 5
- 230000000171 quenching effect Effects 0.000 description 5
- 238000006722 reduction reaction Methods 0.000 description 5
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 4
- 206010062575 Muscle contracture Diseases 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- 239000002168 alkylating agent Substances 0.000 description 4
- 229940100198 alkylating agent Drugs 0.000 description 4
- 208000006111 contracture Diseases 0.000 description 4
- QPJORFLSOJAUNL-UHFFFAOYSA-N dibenzo[a,d][7]annulene Chemical compound C1=CC2=CC=CC=C2CC2=CC=CC=C21 QPJORFLSOJAUNL-UHFFFAOYSA-N 0.000 description 4
- YNLAOSYQHBDIKW-UHFFFAOYSA-M diethylaluminium chloride Chemical compound CC[Al](Cl)CC YNLAOSYQHBDIKW-UHFFFAOYSA-M 0.000 description 4
- QILSFLSDHQAZET-UHFFFAOYSA-N diphenylmethanol Chemical compound C=1C=CC=CC=1C(O)C1=CC=CC=C1 QILSFLSDHQAZET-UHFFFAOYSA-N 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 210000003405 ileum Anatomy 0.000 description 4
- 208000002551 irritable bowel syndrome Diseases 0.000 description 4
- 210000003205 muscle Anatomy 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 235000015320 potassium carbonate Nutrition 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- LISAIUHOJQKVHK-UHFFFAOYSA-N 1-(3-bromopropyl)-3-methoxybenzene Chemical compound COC1=CC=CC(CCCBr)=C1 LISAIUHOJQKVHK-UHFFFAOYSA-N 0.000 description 3
- 206010013554 Diverticulum Diseases 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical compound SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- 206010046543 Urinary incontinence Diseases 0.000 description 3
- 125000000217 alkyl group Chemical group 0.000 description 3
- GETQZCLCWQTVFV-UHFFFAOYSA-N anhydrous trimethylamine Natural products CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 3
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 210000005245 right atrium Anatomy 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- 238000004611 spectroscopical analysis Methods 0.000 description 3
- UAIGESSCTLSFEB-UHFFFAOYSA-N 1-[3-(4-methoxyphenyl)propyl]piperidine Chemical class C1=CC(OC)=CC=C1CCCN1CCCCC1 UAIGESSCTLSFEB-UHFFFAOYSA-N 0.000 description 2
- JADSGOFBFPTCHG-UHFFFAOYSA-N 2-(1,3-benzodioxol-5-yl)ethanol Chemical compound OCCC1=CC=C2OCOC2=C1 JADSGOFBFPTCHG-UHFFFAOYSA-N 0.000 description 2
- ZWKJGSFAORXDED-UHFFFAOYSA-N 2-(2,3-dihydro-1,4-benzodioxin-6-yl)ethanol Chemical compound O1CCOC2=CC(CCO)=CC=C21 ZWKJGSFAORXDED-UHFFFAOYSA-N 0.000 description 2
- KANZWHBYRHQMKZ-UHFFFAOYSA-N 2-ethenylpyrazine Chemical compound C=CC1=CN=CC=N1 KANZWHBYRHQMKZ-UHFFFAOYSA-N 0.000 description 2
- WRMNZCZEMHIOCP-UHFFFAOYSA-N 2-phenylethanol Chemical compound OCCC1=CC=CC=C1 WRMNZCZEMHIOCP-UHFFFAOYSA-N 0.000 description 2
- CFFZDZCDUFSOFZ-UHFFFAOYSA-N 3,4-Dihydroxy-phenylacetic acid Chemical compound OC(=O)CC1=CC=C(O)C(O)=C1 CFFZDZCDUFSOFZ-UHFFFAOYSA-N 0.000 description 2
- CCARPEUWUWIJPX-UHFFFAOYSA-N 5-(2-bromoethyl)-1,3-benzodioxole Chemical compound BrCCC1=CC=C2OCOC2=C1 CCARPEUWUWIJPX-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- UQOFGTXDASPNLL-XHNCKOQMSA-N Muscarine Chemical compound C[C@@H]1O[C@H](C[N+](C)(C)C)C[C@H]1O UQOFGTXDASPNLL-XHNCKOQMSA-N 0.000 description 2
- 229940121948 Muscarinic receptor antagonist Drugs 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical class CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 2
- 206010061876 Obstruction Diseases 0.000 description 2
- 229910000831 Steel Inorganic materials 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- BNBQRQQYDMDJAH-UHFFFAOYSA-N benzodioxan Chemical compound C1=CC=C2OCCOC2=C1 BNBQRQQYDMDJAH-UHFFFAOYSA-N 0.000 description 2
- 229960004217 benzyl alcohol Drugs 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000001273 butane Substances 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000005660 chlorination reaction Methods 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzylether Substances C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000003292 glue Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 239000003149 muscarinic antagonist Substances 0.000 description 2
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- 229940067107 phenylethyl alcohol Drugs 0.000 description 2
- 210000001747 pupil Anatomy 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000010959 steel Substances 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000009834 vaporization Methods 0.000 description 2
- 230000008016 vaporization Effects 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- 238000013022 venting Methods 0.000 description 2
- CPHLODVMQBMDNC-UHFFFAOYSA-N 1-(3-bromopropyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCCBr)C=C1 CPHLODVMQBMDNC-UHFFFAOYSA-N 0.000 description 1
- HXQKJEIGFRLGIH-UHFFFAOYSA-N 1-ethenyl-2h-pyrazine Chemical compound C=CN1CC=NC=C1 HXQKJEIGFRLGIH-UHFFFAOYSA-N 0.000 description 1
- VUFRIRUMGMOOJQ-UHFFFAOYSA-N 2,3-dihydro-1,4-benzodioxin-6-yl acetate Chemical compound O1CCOC2=CC(OC(=O)C)=CC=C21 VUFRIRUMGMOOJQ-UHFFFAOYSA-N 0.000 description 1
- IPSIYKHOHYJGMO-UHFFFAOYSA-N 2-(2,3-dihydro-1-benzofuran-5-yl)ethanol Chemical class OCCC1=CC=C2OCCC2=C1 IPSIYKHOHYJGMO-UHFFFAOYSA-N 0.000 description 1
- QXHDYMUPPXAMPQ-UHFFFAOYSA-N 2-(4-aminophenyl)ethanol Chemical compound NC1=CC=C(CCO)C=C1 QXHDYMUPPXAMPQ-UHFFFAOYSA-N 0.000 description 1
- GGRLKHMFMUXIOG-UHFFFAOYSA-M 2-acetyloxyethyl(trimethyl)azanium;hydroxide Chemical compound [OH-].CC(=O)OCC[N+](C)(C)C GGRLKHMFMUXIOG-UHFFFAOYSA-M 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical compound BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- KGIGUEBEKRSTEW-UHFFFAOYSA-N 2-vinylpyridine Chemical compound C=CC1=CC=CC=N1 KGIGUEBEKRSTEW-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 208000000884 Airway Obstruction Diseases 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- YZCRICMBBCLNNL-UHFFFAOYSA-N C(C)N1CCCCC1.IC=1C=CC=CC1 Chemical compound C(C)N1CCCCC1.IC=1C=CC=CC1 YZCRICMBBCLNNL-UHFFFAOYSA-N 0.000 description 1
- LRPBANGBMZKCBF-UHFFFAOYSA-N C(C)O.C1(=CC=CC=C1)I Chemical compound C(C)O.C1(=CC=CC=C1)I LRPBANGBMZKCBF-UHFFFAOYSA-N 0.000 description 1
- 0 CC1=C(C(c2ccccc2)O*C(CCC2)CI2(C)#C)C=CCC1 Chemical compound CC1=C(C(c2ccccc2)O*C(CCC2)CI2(C)#C)C=CCC1 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Divinylene sulfide Natural products C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 1
- 239000004278 EU approved seasoning Substances 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 1
- RKOTXQYWCBGZLP-UHFFFAOYSA-N N-[(2,4-difluorophenyl)methyl]-2-ethyl-9-hydroxy-3-methoxy-1,8-dioxospiro[3H-pyrido[1,2-a]pyrazine-4,3'-oxolane]-7-carboxamide Chemical compound CCN1C(OC)C2(CCOC2)N2C=C(C(=O)NCC3=C(F)C=C(F)C=C3)C(=O)C(O)=C2C1=O RKOTXQYWCBGZLP-UHFFFAOYSA-N 0.000 description 1
- XGEGHDBEHXKFPX-UHFFFAOYSA-N N-methylthiourea Natural products CNC(N)=O XGEGHDBEHXKFPX-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- RSDOPYMFZBJHRL-UHFFFAOYSA-N Oxotremorine Chemical compound O=C1CCCN1CC#CCN1CCCC1 RSDOPYMFZBJHRL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 1
- 206010044565 Tremor Diseases 0.000 description 1
- VAHXQYYBOYZVLM-UHFFFAOYSA-N [I].C[Mg] Chemical compound [I].C[Mg] VAHXQYYBOYZVLM-UHFFFAOYSA-N 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- UGAPHEBNTGUMBB-UHFFFAOYSA-N acetic acid;ethyl acetate Chemical class CC(O)=O.CCOC(C)=O UGAPHEBNTGUMBB-UHFFFAOYSA-N 0.000 description 1
- DORMTBIPKNPJPY-UHFFFAOYSA-N acetic acid;iodobenzene Chemical compound CC(O)=O.IC1=CC=CC=C1 DORMTBIPKNPJPY-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical compound [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000001078 anti-cholinergic effect Effects 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 230000002921 anti-spasmodic effect Effects 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229940125717 barbiturate Drugs 0.000 description 1
- HNYOPLTXPVRDBG-UHFFFAOYSA-N barbituric acid Chemical compound O=C1CC(=O)NC(=O)N1 HNYOPLTXPVRDBG-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Substances N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 1
- TXQVTGMFRXWDOB-UHFFFAOYSA-N bromoethane;iodobenzene Chemical compound CCBr.IC1=CC=CC=C1 TXQVTGMFRXWDOB-UHFFFAOYSA-N 0.000 description 1
- QNOJNOBJMMNCFZ-UHFFFAOYSA-N bromoethane;toluene Chemical compound CCBr.CC1=CC=CC=C1 QNOJNOBJMMNCFZ-UHFFFAOYSA-N 0.000 description 1
- 230000007885 bronchoconstriction Effects 0.000 description 1
- AIXAANGOTKPUOY-UHFFFAOYSA-N carbachol Chemical compound [Cl-].C[N+](C)(C)CCOC(N)=O AIXAANGOTKPUOY-UHFFFAOYSA-N 0.000 description 1
- 229960004484 carbachol Drugs 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000002027 dichloromethane extract Substances 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- RCFKEIREOSXLET-UHFFFAOYSA-N disulfamide Chemical compound CC1=CC(Cl)=C(S(N)(=O)=O)C=C1S(N)(=O)=O RCFKEIREOSXLET-UHFFFAOYSA-N 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- FRYHCSODNHYDPU-UHFFFAOYSA-N ethanesulfonyl chloride Chemical compound CCS(Cl)(=O)=O FRYHCSODNHYDPU-UHFFFAOYSA-N 0.000 description 1
- UESSEMPSSAXQJC-UHFFFAOYSA-N ethanol;methanamine Chemical compound NC.CCO UESSEMPSSAXQJC-UHFFFAOYSA-N 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 239000005308 flint glass Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000011194 food seasoning agent Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- SPRIOUNJHPCKPV-UHFFFAOYSA-N hydridoaluminium Chemical compound [AlH] SPRIOUNJHPCKPV-UHFFFAOYSA-N 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- SHFJWMWCIHQNCP-UHFFFAOYSA-M hydron;tetrabutylazanium;sulfate Chemical compound OS([O-])(=O)=O.CCCC[N+](CCCC)(CCCC)CCCC SHFJWMWCIHQNCP-UHFFFAOYSA-M 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical class CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 1
- 150000002597 lactoses Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 125000004492 methyl ester group Chemical group 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- HAMGRBXTJNITHG-UHFFFAOYSA-N methyl isocyanate Chemical compound CN=C=O HAMGRBXTJNITHG-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- LKPFBGKZCCBZDK-UHFFFAOYSA-N n-hydroxypiperidine Chemical compound ON1CCCCC1 LKPFBGKZCCBZDK-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000012053 oil suspension Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 230000010355 oscillation Effects 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000003444 phase transfer catalyst Substances 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 230000007096 poisonous effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 230000001624 sedative effect Effects 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 230000002557 soporific effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- NBNBICNWNFQDDD-UHFFFAOYSA-N sulfuryl dibromide Chemical compound BrS(Br)(=O)=O NBNBICNWNFQDDD-UHFFFAOYSA-N 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 150000003577 thiophenes Chemical class 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 125000005951 trifluoromethanesulfonyloxy group Chemical group 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/42—Oxygen atoms attached in position 3 or 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Pulmonology (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
- Hydrogenated Pyridines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Steroid Compounds (AREA)
Abstract
本文介绍(3R,S)-型或(3R)-型(I)化合物或其药物上可接受的盐的制备方法,或(I)如下:
这些化合物用于治疗与改变平滑肌运动型和/或紧张有关疾病。
Description
本发明涉及某些1,3-二取代哌啶衍生物。
更具体地讲,本发明涉及用作选择性毒蕈硷受体拮抗剂的1-芳基乙基-3-取代哌啶衍生物。
英国专利780,027号公开的一些化合物中,用作催化剂的3-(二苯甲氧基)-和3-(咕吨氧基)-N-芳烷基哌啶,其所用治疗剂量不起解痉作用。在该专利范围内没有合成或举例说明N-苯乙基取代的实例。
美国专利2,974,146号提供的N-芳烷基-3-哌啶基二苯甲基醚具有镇静活性,并延长巴比妥盐的安眠作用,指出只有相应的季铵盐具有胃肠解痉作用。虽然把N-苯乙基-3-哌啶基二苯甲基醚列为“由该发明提供的具体化合物”,但没有提供制备方法或药理数据,显然,该化合物实际上从没制得过。
现在,意想不到地发现由本发明提供的1-(苯乙基)和1-(2-杂芳基乙基)-3-取代哌啶衍生物是毒蕈碱受体拮抗剂,它选择性作用于贲门毒蕈碱位点之上的平滑肌毒蕈碱位点,并不具有任何明显的抗组胺活性。因此,这些化合物用于治疗与改变平滑肌的能动性和/或紧张程度有关的疾病,例如可在肠、气管和膀胱处出现平滑肌紧张。这类病包括肠过敏综合症,憩室病,尿失禁,食道紧缩症和慢性气管阻塞症。
本发明提供式(Ⅰ)化合物的3R,S-(外消旋)体和3R-(旋光活性)化合物及它们在药物上可接受的盐。式(Ⅰ)如下:

*=不对称中心
式中R1为下述基团:

式中每个Y可以相同或不同,它选自氢、卤素和C1-4烷基;
X是-(CH2)2-,-CH=CH-,CH2-S-,-CH2-O-,-S-或-O-;
R是下述基团:

式中或者R2和R3各自分别为氢,C1-4烷基,羟基-(C1-4烷基),羟基,C1-4烷氧基,卤素,三氟甲基,硝基,氰基,氨磺酰,-CO(C1-4烷基),-OCO(C1-4烷基),-CO2-(C1-4烷基),-(CH2)nCONR6R7,-(CH2)nOCONR6R7,-(CH2)nNR8R9或-NHSO2NH2,其中,R6和R7各自分别为H或C1-4烷基,n是0、1或2,或者R8和R9各自分别为H或C1-4烷基,或者R8是氢,R9
是-SO2(C1-4烷基),-CO(C1-4烷基)或-CONH(C1-4烷基);
R2和R3连在一起与相邻碳原子连接时,表示下述基团:-O(CH2)mO-(m是1、2或3)、-O(CH2)2-或-(CH2)3-;
R4是H,C1-4烷基或-CONH2;
R5是H,C1-4烷基或C1-4烷氧基。
“卤素”指的是F、Cl、Br或I。碳原子为3或4的烷基和烷氧基可以直链或支链烷基。优选的烷基和烷氧基是甲基,乙基,甲氧基和乙氧基。
R1的优选基团包括:

一种情况,每个Y最好是相同的。
然后,当R是任意取代苯基时,Y最好是下式:

式中R2和R3的定义同式(Ⅰ)。
当R是任意取代噻吩基时,Y最好是下式:

式中R4的定义同式(Ⅰ)。
R的优选基团如下所述:
(a)
式中R2和R3各自分别为氢,C1-2烷基,羟基-(C1-3烷基),羟基,C1-2烷氧基,卤素,氨磺酰,-CO(C1-2烷基),-OCO(C1-2烷基),-CONH2,-CONH(C1-2烷基),-OCONH(C1-2烷基),-NH2,-CH2NH2,-CH2NH(C1-2烷基),-NHSO2(C1-2烷基),-NHCO(C1-2烷基),-CH2NHCO(C1-2烷基),-CH2NHCONH(C1-2烷基)或-NHSO2NH2;或者R2和R3连接在一起,表示-O(CH2)mO-(其中m是1、2或3);-O(CH2)2-或-(CH2)3-;
b)
 ,式中R4是H或-CONH2;
,式中R4是H或-CONH2;
c)
;或
d)
R1的最佳基团为:

R的最佳基团为:

式(Ⅰ)化合物含至少一个不对称中心,因此以一对对映体或一对非对映体形式存在。这种对映体或非对映体可通过物理方法拆分,例如,将式(Ⅰ)化合物的外消旋混合物或其适宜的盐或衍生物通过分步重结晶,色谱法或H.P.L.C.拆分。最好由光学纯中间体制备含一个不对称中心的式(Ⅰ)化合物的各个对映体。
本发明化合物的抗胆碱能活性主要出现于3R构型化合物,即,该化合物哌啶环3位立体构型为R型。在此,本发明只限于式(Ⅰ)化合物的3R和3R,S-(外消旋)型。
本发明特别优选的单一化合物是(3R)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶或其药物上可接受的盐。
本发明还包括某些合成中间体,即下述各式化合物的3R,S-和3R-型;
(a)
 -(Ⅳ),
-(Ⅳ),
(b)
 -(Ⅵ),
-(Ⅵ),
式中R和R1的定义同式(Ⅰ)。
式(Ⅰ)化合物在药物上可接受的盐包括酸成盐,例如盐酸盐,氢溴酸盐,硫酸盐或硫氢酸盐,磷酸盐或磷酸氢盐,乙酸盐,苯磺酸盐,柠檬酸盐,富马酸盐,葡糖酸盐,乳酸盐,马来酸盐,甲磺酸盐,琥珀酸盐和酒石酸盐。为更详细说明药物上可接受的盐,请参见Journal of Pharmaceutical Scicnces,Vol.66,No.1,January1977,Pages1-19。这些盐可按常规方法制备,例如在适宜溶剂如乙醇中,混合游离碱和酸溶液,通过沉淀回收酸成盐或将溶液蒸发。
可通过包括下述反应的多种途径制备式(Ⅰ)化合物:
路线A
这一路线说明如下:
R和R1的定义同式(Ⅰ),Q是离去基团,例如Br,Cl,I,C1-4链烷磺酰氧基(例如甲磺酰氧基),苯磺酰氧基,甲苯磺酰氧基(例如对-甲苯磺酰氧基)或三氟甲磺酰氧基。最好Q为Cl,Br,I或甲磺酰氧基。
该反应最好在回流温度下,于酸受体存在的适宜溶剂如乙腈中进行。所述酸受体如碳酸钠或碳酸钾,碳酸氢钠,三乙胺或吡啶。一般要求反应温度为60-120℃,在回流下最便于实施该反应。碘代通常是最适宜离去基团,但由于原料(Ⅲ)一般最容易得到氯化物或溴化物,因此常常用化合物(Ⅲ)的氯化物或溴化物,但是在碘化物(如碘化钠或碘化钾)存在下,最适于实施该反应。就最佳工艺而言,在碳酸钠和碘化钠存在下,于乙腈中将化合物(Ⅱ)和(Ⅲ)一起回流,其中化合物(Ⅲ)为溴化物或氯化物。产品(Ⅰ)可用常规方法分离和纯化。
根据所要求产品是3R,S-还是3R-型而决定应使用3R,S-或3R-型起始原料(Ⅱ)。
式(Ⅱ)原料可用常规方法得到,如制备方法1至7所介绍的。式(Ⅲ)原料是众所周知的化合物,可用常规技术制备。在下文制备部分介绍用于实施例的式Ⅲ新颖原料的制备。
路线B
这一路线说明如下:

R和R的定义同式(Ⅰ):按照需要再次使用3R,S-或3R-型化合物(Ⅳ)。
按常规,一般用无机还原剂例如氢化铝锂、氢化铝(由氢化铝锂和
浓硫酸直接制取AlH3)或乙硼烷,在适宜的有机溶剂如四氢呋喃、醚或二噁烷中进行该还原反应。该反应温度最好在0℃至室温。虽然一般不必加热,如有必要,可在反应混合物的回流温度下进行该反应。产品(Ⅰ)可按常规方法分离和纯化。
式(Ⅳ)原料(3R,S-或3R-)可用常规技术制备,其中包括下文制备部分介绍的那些方法(特别请参见制备8至10)。
路线C
这一路线说明如下:

R和R1的定义同式(Ⅰ),Q是离去基团(例如路线A所述)。在本路线中,Q最好是Cl或Br。其中式Ⅴ化合物颇具活性,进而在室温下完成该反应。必要时,该反应混合物可加热至160℃,以加速反应。尽管在某些情况下,如实施例8(B),个别有机溶剂的存在是没必要的,但该反应可以在适宜有机溶剂如二氯甲烷中进行。化合物(Ⅰ)按常规分离和纯化。
原料(Ⅴ)可是已知化合物,也可按常规方法制得。化合物(Ⅵ)可按常规方法制备,例如在制备15-16中介绍的技术。
根据所要求化合物(Ⅰ)是3R,S-还是3R-型而决定应使用3R,S-或3R型原料(Ⅵ)。
路线D
这一路线用于制备式中R是2或4-吡啶基或吡嗪基的化合物,如下所述:

R1和R5的定义同式(Ⅰ)。很清楚,乙烯基必须连接于吡啶环的2-或4-位。
该反应一般在适宜的有机溶剂如1-丁醇中,加热至160℃,最好是80-140℃下进行。使用碱性(最好是可溶于有机溶剂的强碱如N-苄基三甲基氢氧化铵〔商标“Triton B”〕)催化剂或酸性(最好是C1-4链烷酸)催化剂是有效的。最佳方法是在碱性催化剂如“Triton B”的存在下,于有机溶剂中回流该反应物。
路线E
式中R是5-氨基甲酰基-2-噻吩基的化合物也可按如下路线制备:


R1的定义同式(Ⅰ)
式(Ⅰ)中R是取代苯基的某些化合物可以转化为式(Ⅰ)的其它化合物,详述如下:
(a)苯基的-CO2(C1-4烷基)取代基可还原为-CH2OH,氢化铝锂是最适宜还原剂。该反应一般在0℃至室温下,于适宜的有机溶剂如醚中进行,一般使用其甲酯形式的原料最为方便。
(b)苯基的羟基取代基可以通过使用C1-4链烷酰氯或链烷酰溴或C1-4链烷酐的酰化作用,转化为-OCO(C1-4烷基)。有酸受体存在为最佳。该反应一般在约室温下,于适宜有机溶剂如二噁烷中进行。
(c)苯基的-CO(C1-3烷基)取代基可还原为-CH(OH)(C1-3烷基)的取代基。适宜的还原剂包括氢硼化钠和氢化铝锂。该反应一般在0℃至
室温下,于适宜有机溶剂如甲醇中进行。氢硼化钠是最佳还原剂。
(d)-CO2(C1-4烷基)取代基,最好是-CO2CH3可通过与氨或适当的胺R6R7NH反应,转化为-CONR6R7。当R6和R7都为H时,虽然可用溶于有机溶剂(如甲醇或乙醇)的氨;或用高压气体贮罐中的净氨进行该反应,但使用氨水(0.880)最为方便。其中与甲胺的反应最便于在乙醇中进行。虽然在某些情况下,该反应可在室温下以令人满意的速度进行,但加热至120℃,最好为60至100℃是必要的。至于挥发性胺,该反应最好在高压釜中进行。
(e)苯基的硝基取代基可按常规方法还原为氨基。最佳还原剂是氯化亚锡二水合物,该反应一般在有机溶剂如乙醇中回流进行。
(f)苯基的氨基取代基可以通过与C1-4链烷磺酰氯或C1-4链烷磺酰溴或C1-4链烷磺酸酐反应,转化为-NHSO2(C1-4烷基)。酸受体,如吡啶、三乙胺、碳酸氢钠或碳酸钠或碳酸钾的存在为最佳。尤其当使用磺酰氯时,在吡啶中进行该反应常常是最方便的,该吡啶起溶剂和酸受体作用。通常不必加热,一般在室温下,就能以令人满意的速度进行该反应。
(g)式中n是0、1或2的-(CH2)nNH2取代基可通过与C1-4链烷酰氯或链烷酰溴或C1-4链烷酸酐反应,转化为-(CH2)nNHCO(C1-4烷基)。可按类似于上述(f)进行该反应。将碳酸氢钠用作酸受体,在乙酸乙酯/水中使用乙酐是最佳反应。
(h)苯基的氨基取代基一般可在有机溶剂如二噁烷中,与硫酰胺回流反应,转化为氨磺酰。
(i)羟基取代基可先与强碱如氢化钠反应,再与C1-4烷基碘反应,转化为C1-4烷氧基。该反应最好在约室温下,于溶剂如二甲基甲酰胺中进行。
(j)式中n是0、1或2的-(CH2)nOH的羟基取代基通过与异氰酸
C1-4烷基酯反应,可转化为-(CH2)nOCONH(C1-4烷基)。该反应一般在约室温下,于溶剂如二氯甲烷中进行。
(k)苯基的羟甲基取代基通过先与亚硫酰氯反应,再与氨或适当的胺R8R9NH反应,可转化为式中R8和R9各自分别为H或C1-4烷基的-CH2NR8R9。与亚硫酰氯的反应一般在溶剂如二氯甲烷中加热进行,最好回流加热。与氨或胺的反应一般在溶剂如乙醇中,在约室温下进行。
(l)乙酰取代基可通过与甲基锂、甲基镁化溴、甲基镁化碘或甲基镁化氯反应,转化为-C(OH)(CH3)2。该反应一般在0℃至室温下,于溶剂如醚中进行。
(m)碘代取代基一般可在约室温下,通过在含碱〔例如碳酸钾〕和钯(Ⅱ)催化剂〔例如双(三苯膦)氯化钯(Ⅱ)〕的C1-4烷醇中与一氧化碳反应,转化为C1-4烷氧基羰基。
(n)苯基的氰基取代基一般可用H2/Pd/C溶于含少量浓盐酸的乙醇溶液,通过催化氢化作用,还原为氨甲基。
(o)式中n是0、1或2的-(CH2)nNH2取代基通过与异氰酸C1-4烷酯反应,转化为式-(CH2)nNHCONH(C1-4烷基)取代基。该反应一般在约室温下,于溶剂如二氯甲烷中进行。
(p)C1-4烷氧基取代基,最好是甲氧基可以在强碱如氢化钠存在下,通过与C1-4链烷硫醇反应,转化为羟基。一般在适宜溶剂如二甲基甲酰胺中回流反应物,进行该反应。丁烷硫醇为最佳硫醇。
可按如下所述测定用作毒蕈碱受体拮抗化合物的选择性。
处死雄豚鼠,取出回肠、气管、膀胱和右心房,并在保持1g拉力下悬挂于生理盐水溶液中,在32℃下充气95%O2和5%CO2。用等张(回肠)或等张转换器(膀胱和气管)记录回肠、膀胱和气管的挛缩。由等张记录挛缩得到右心房自然跳动的挛缩频率。
对每种兴奋剂剂量采用1-5分钟接触时间,直至获得最大应答值由
此确定对乙酰胆碱(回肠)或碳酰胆碱(气管、膀胱和右心房)的剂量应答曲线。排去器官浴的水,再灌满含最低剂量试验化合物的生理盐水溶液。使该试验化合物与组织平衡20分钟,重复兴奋剂剂量应答曲线,直至得到最大应答值。排去器官浴的水,再灌满含第二种浓度试验化合物的生理盐水溶液,并重复上述方法。一般对每种组织评价该试验化合物四种浓度。
测定使兴奋剂浓度加倍以产生最初应答的试验化合物浓度(pA值,Arunlakshana and Schild(1959),Brit.J.Pharmacol.,14,48-58)。采用上述分析技术测定毒蕈受体拮抗剂的组织选择性。
通过与心率的变化相比较,在麽醉狗体内测定对于兴奋剂诱发的支气管收缩,肠和膀胱的挛缩作用的对抗作用。用神志清醒的狗确定化合物对心率、瞳孔直径和肠运动性影响的口服活性。
给鼠静脉或腹膜内施药后,确定化合物对其他胆碱能位点的亲合作用。从而测定引起双倍瞳孔尺寸的剂量,以及抑制50%静脉氧化震颤素引起的多涎和震颤应答的剂量。
就施药于人而言,在治疗或预防与改变平滑肌能动性和/或紧张程度有关疾病中,该化合物口服剂量对一般成年患者(体重70kg)每天一般为3.5至350mg。所述疾病如肠过敏综合症,憩室疾病,尿失禁,食管紧缩和慢性阻塞气管病。因此,对典型的成年患者来说,每个片剂或胶囊一般含活性化合物1至250mg,置于适宜的药物可接受的赋形剂或载体内,以单次剂量或多次剂量形式,每天一次或多次给药。静脉注射的剂量范围按要求每单剂量为0.35至35mg。实际上,由医师确定最适合于每个患者的实际剂量,它随具体患者的年龄、体重和反应而改变。上述剂量用于一般病例,当然特殊病例应得较高或较低剂量,并且这也在本发明范围内。
对人类应用而言,式(Ⅰ)化合物可单独给药,但一般与药用载体
混合施用,该载体是根据预期给药途径和标准的制药工艺所选出的。例如,这些化合物可以下述形式口服给药:片剂(含淀粉或乳糖类赋形剂)或胶囊或小丸(单独给药或与赋形剂混合)给药,或者以含调味剂或着色剂的剂或混悬液形式给药。它们可经非肠道注射,如静脉内、肌内或皮下注射。就非肠道给药而言,它们最好以无菌水溶液形式使用,该水溶液可含其它物质,例如足够的盐或葡萄糖,使该溶液与血液等渗。
在另一方面,本发明提供一种含式(Ⅰ)化合物或其在药物上可接受的盐,以及药物上可接受稀释剂或载体的药用组合物。
本发明还包括一种式(Ⅰ)化合物或其药物上可接受的盐,用作药剂,尤其用于治疗肠过敏综合症。
本发明进一步包括式(Ⅰ)化合物或其药物上可接受盐的用途,即用于制备治疗与改变平滑肌能动性和/或紧张程度有关疾病的药物。所述疾病如肠过敏综合症,憩室疾病,尿失禁,食管紧缩和慢性阻塞气管病。
下述实施例说明本发明,其中所有温度以℃表示。
实施例1
(3R,S)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶
方法A

在10分钟内将(3R,S)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙酰)哌啶(1.08g)(见制备8)的四氢呋喃(10ml)溶液滴加到氢化铝锂(0.20g)悬浮于四氢呋喃(15ml)的冰冷却搅拌悬浮液中,该混合物在室温下搅拌3小时,通过加入饱和氯化铵水溶液骤冷,直至形成白色沉淀,然后过滤。滤液蒸发,残余物经硅石色谱纯化,用含0-5%甲醇的二氯甲烷作洗脱液。合并适当组分,蒸发,得到淡黄色油状所要求产品(0.54g,52%),其中含0.25摩尔当量水。
分析%:
实验值:C,77.2;H,7.2;N,3.3;
C27H29NO3;0.25HO计算值:C,77.2;H,7.0;N,3.3;
方法B

将(3R,S)-二苯甲氧基哌啶(2.67g)(见制备1),3,4-亚甲二氧基苯乙基溴化物(2.29g)(见制备20),碳酸钠(2.10g)和碘化钠(0.25g)溶于乙腈(50ml)的混合物回流加热68小时,用乙酸乙酯和水稀释,分层。有机层用水洗涤,硫酸镁干燥,蒸发。剩余物经硅石(50g)色谱纯化,用含0-5%甲醇的二氯甲烷作洗脱液,合并适当组分,蒸发,得到淡黄色油
状本题目化合物(3.20g,77%),它的光谱数据和由方法A得到的产品数据等同。
1H n.m.r.(CDCl3):δ=7.22-7.64(10H,m);7.61-7.80(3H,m);5.93(2H,s);5.58(1H,s);3.52-3.64(1H,m);3.13(1H,dd,J=6 and 2Hz);2.54-2.85(5H,m) and 1.30-2.17(6H,m).
实施例2-7
按实施例1方法A所述,用氢化铝锂还原适当的(3R,S)原材料,制备下述化合物(R,S-型)。得到的所有化合物如无色油状,并照此赋予特征。
实施例2的特征在于H-n.m.r.;(CDCl3):δ=7.2-7.5(10H,m);6.60-6.88(3H,m);5.60(1H,s);4.24(4H,s);3.52-3.65(1H,m);3.09(1H,d,J=6Hz);2.54-2.88(5H,m)1.25-2.15(6H,m).

哌啶原材料在制备9-14中介绍。

实施例8
(3R,S)-二苯甲氧基-1-(3-甲氧基苯乙基)哌啶
方法A

用3-甲氧苯乙基溴化物代替3,4-亚甲二氧基苯乙基溴化物,按实施例1方法B进行制备,得到无色油状本题目化合物(1.37g,68%)。
分析%:
实验值:C,80.5;H,7.8;N,3.3;
C27H31NO2计算值:C,80.8;H,7.8;N,3.5。
方法B

将(3R,S)-羟基-1-(3-甲氧苯乙基)哌啶(1.00g)(见制备16)和溴二甲苯(0.95g)的均匀混合物在140℃加热1小时,冷却至室温,溶于二氯甲烷,用10%碳酸氢钠水溶液洗涤,硫酸镁干燥和蒸发。剩余物经硅石(10g)色谱纯化,用二氯甲烷作洗脱液。合并适当组分,蒸发,得到无色油状本题目化合物(0.45g,27%),它的光谱数据和由方法A得到的材料数据相同。
1H-n.m.r.(CDCl3):δ=7.14-7.44(11H,m);6.72-6.90(3H,m);5.59(1H,s);3.80(3H,s);3.50-3.63(1H,m);3.15(1H,dd,J=6 and 2Hz);2.58-2.86(5H,m) and 1.27-2.16(8H,m).
实施例9-25
按实施例1方法B所述,使(3R,S)-二苯甲氧基哌啶与适当的烷化剂反应,制备下述化合物(R,S-型)。该烷化剂是已知化合物,或是制备24、25和27所述化合物。得到的所有化合物如无色油状,除指明外,照此赋予特征。
实施例17和23的特征在于1H-n.m.r.:
实施例17(CDCl3):δ=7.99(2H,d,J=8Hz);7.22-7.46(12H,m);5.58(1H,s);3.96(3H,s);3.52-3.62(1H,m);3.10(1H,d,J=6Hz);2.60-2.90(5H,m) and 1.25-2.20(6H,m).
实施例23(CDCl3):δ=6.98-7.63(14H,m);5.60(1H,s);3.52-3.64(1H,m);3.10(1H,d,J=6Hz);2.56-2.80(5H,m) and 1.26-2.18(6H,m).




实施例26
(3R,S)-二(4-氟苯基)甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶

用(3R,S)-二(4-氟苯基)甲氧基哌啶(见制备2)代替(3R,S)-二苯甲氧基哌啶,按实施例1方法B所述进行制备,得到无色油状本题目化合物(0.70g,78%)。
分析%:
实验值:C,72.4;H,6.2;N,3.0;
C27H27F2NO3计算值:C,71.8;H,6.0;N,3.1。
实施例27
1-〔2-(苯并二噁烷-6-基)乙基〕-(3R,S)-二(4-氟苯基)甲氧基哌啶

按实施例1方法B所述方法,使(3R,S)-二(4-氟苯基)甲氧基哌啶(见制备2)和6-(2-溴乙基)苯并二噁烷(见制备23)反应,制得无色油状本题目化合物,它是水合物(0.44g,51%)。
分析%:
实验值:C,70.0;H,6.3;N,2.8;
C28H29F2NO3·H2O计算值:C,69.6;H,6.4;N,2.9。
实施例28
(3R)-二(4-氟苯基)甲氧基-1-(3-甲氧苯乙基)哌啶
按实施例1方法B所述方法,由(3R)-二(4-氟苯基)甲氧基哌啶(见制备3)和3-甲氧苯乙基溴化物进行制备,得到无色油状本题目化合物(0.33g,38%)。
分析%:
实验值:C,73.9;H,6.6;N,3.1;
C27H29F2NO2计算值:C,74.1;H,6.6;N,3.2。
实施例29
(3R)-〔(R,S)-1-(2-甲苯基)-1-苯基甲氧基〕-1-(4-甲基苯乙基)
哌啶

按实施例1方法B所述方法,由(3R)-〔(R,S)-1-(2-甲基苯基)-1-苯基甲氧基〕哌啶(见制备4)和4-甲基苯乙基溴化物进行制备。得到淡黄色油状本题目化合物(0.51g,70%),〔α〕25 D+19.7°(甲醇中C11.6)。
分析%:
实验值:C,84.1;H,8.6;N,3.5;
C28H33NO计算值:C,84.2;H,8.3;N,3.5。
实施例30
(3R)-〔1-(2-叔-丁苯基)-1-苯氧基〕-1-(4-甲氧苯乙基)哌啶;非对映体A和B
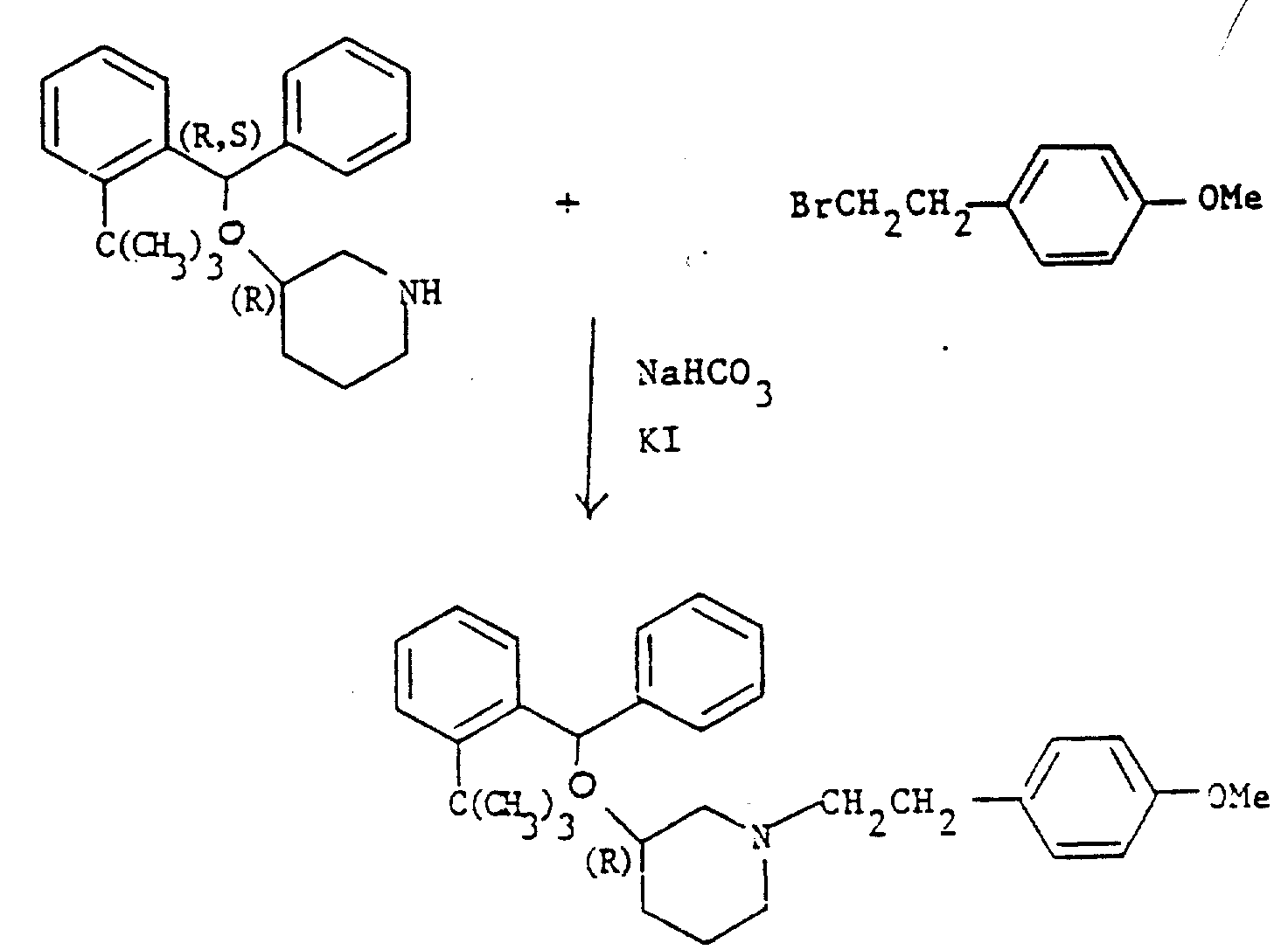
将(3R)-〔(R,S)-1-(2-叔-丁苯基)-1-苯基甲氧基〕哌啶(见制备5)(420mg)、4-甲氧苯乙基溴化物(288mg)、碘化钾(108g)和碳酸氢钠(169mg)的混合物溶于乙腈(50ml)中,回流加热16小时,过滤,蒸发。残余物经硅石(10g)色谱纯化,用二氯甲烷加0-4%甲醇作洗脱液。合并含初始产品组分,蒸发,得到无色固体状本题目化合物,非对映体A(300mg,50%),m.p.117℃。
分析%:
实验值:C,81.3;H,8.9;N,2.9;
C31H39NO2计算值:C,81.4;H,8.6;N,3.1;
合并和蒸发适当组分后,进一步洗脱,得到无色固体状本题目化合物,非对映体B(253mg,43%),m.p.108-113℃。
分析%:
实验值:C,79.8;H,8.9;N,2.7;
C31H39NO2·0.5H2O计算值:C,79.8;H,8.6;N,3.0。
实施例31
(3R,S)-〔(11H)-6,11-二氢二苯并〔b,e〕噻庚英-11-基氧基〕-1-(3-甲氧苯乙基)〕哌啶

按实施例1方法B所述方法,通过(3R,S)-〔(11H)-6,11-二氢二苯并〔b,e〕噻庚英-11-基氧基〕哌啶(见制备6)和3-甲氧苯乙基溴化物反应,制得无色油状本题目化合物(0.12g,11%)。
分析%:
实验值:C,75.4;H,6.8;N,2.8;
C28H31NO2S计算值:C,75.5;H,7.0;N,3.1。
实施例32
(3R)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶

用(3R)-二苯甲氧基哌啶{〔α〕25 D-3.3°(乙醇中C1.5)}(见制备7)代替(3R,S)-二苯甲氧基哌啶,按实施例1方法B所述进行制备。用己烷重结晶后,得到无色油状本题目化合物(1.25g,78%),m.p.52-55℃,〔α〕25+22.5°(乙醇中C1.5)。
分析%:
实验值:C,78.4;H,7.2;N,3.3;
C H NO 计算值:C,78.0;H,7.0;N,3.4。
实施例33-52
按实施例1方法B所述,使(3R)-二苯甲氧基哌啶(见制备7)〔α〕25 D-3.0°(乙醇中C1.5)与适当烷化剂反应,制备下述化合物(R-型)。该烷化剂是已知化合物或制备21、23、29和31所述化合物。这些化合物是所示型式的游离碱。所示型式如下:



实施例53
1-(4乙酰氧基苯乙基)-(3R)-二苯甲氧基哌啶
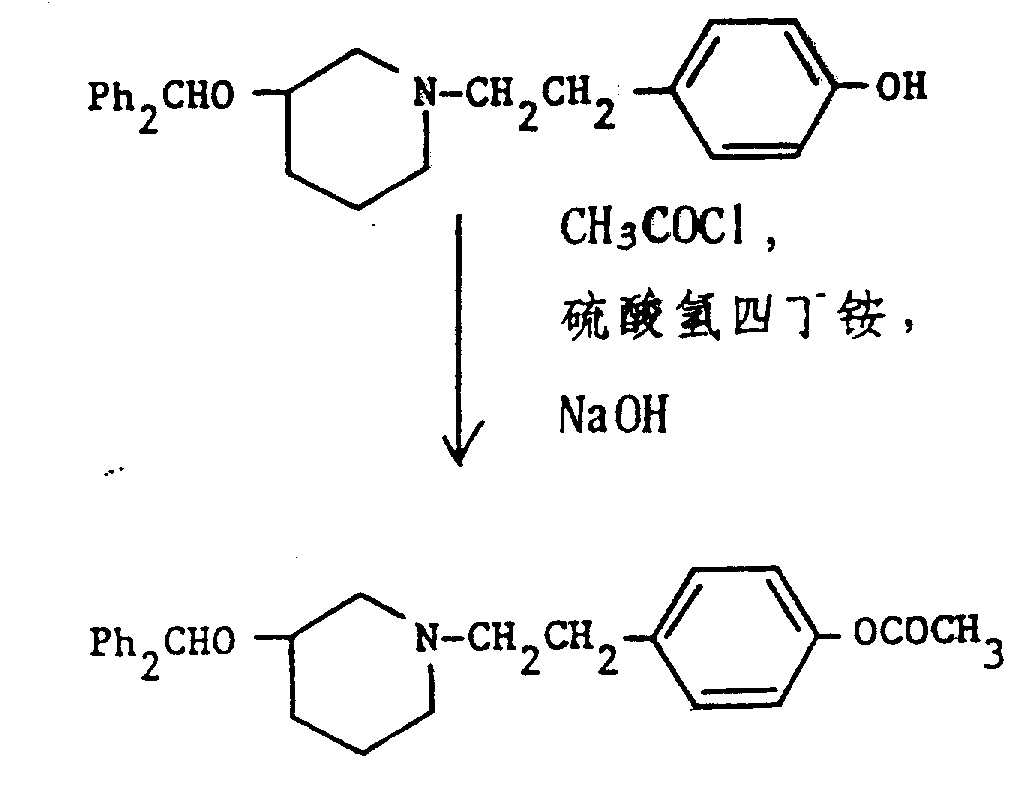
在30分钟内,将乙酰氯(33ml)的二噁烷(5ml)溶液滴加到(3R)-二苯甲氧基-1-(4-羟基苯乙基)哌啶(150mg)(见实施例35)、硫酸氢四丁铵(2.7mg)(相转移催化剂)和粉状氢氧化钠(50mg)混合物的二噁烷(15ml)溶液中,该混合物在室温搅拌4天,过滤和蒸发。剩余物经硅石(5g)色谱纯化,用二氯甲烷加0-5%乙醇作洗脱液。合并适当组分,蒸发,得到无色油状本题目化合物(128mg,74%),〔α〕25 D+24.4°(甲醇中C0.775)。
分析%:
实验值:C,78.3;H,7.3,N,3.5;
C28H31NO3计算值:C,78.3;H,7.3,N,3.3。
实施例54
(3R,S)-二苯甲氧基-1-〔2-(4-吡啶基)乙基〕哌啶

将(3R,S)-二苯甲氧基哌啶(534mg)(见制备1)、4-乙烯基吡啶(630mg)和40%N-氢氧化苄基三甲铵(“Triton B”-商品名)水溶液(5滴)混合物溶于1-丁醇(20ml)中,回流加热70小时,然后蒸发。剩余物分布在乙酸乙酯和水之间,用水洗涤有机层,硫酸钠干燥和蒸发。该剩余物经硅石(8g)色谱法纯化,用二氯甲烷加0-5%甲醇作洗脱液。合并适当组分,蒸发,得到淡棕色胶状标题目化合物(310mg,42%)。
分析%:
实验值:C,80.3;H,7.5,N,7.8;
C25H28N2O计算值:C,80.6;H,7.6;N,7.5。
实施例55
(3R,S)-二苯甲氧基-1-〔2-(2-吡啶基)乙基〕哌啶

用2-乙烯基吡啶代替4-乙烯基吡啶,按实施例54所述进行制备,得到淡棕色胶状本题目化合物(240mg,32%)。
分析%:
实验值:C,80.9;H,7.6;N,7.5;
C25H28N2O计算值:C,80.6;H,7.6;N,7.5。
实施例56
(3R,S)-二苯甲氧基-1-〔2-(2-吡嗪基)乙基〕哌啶

用2-乙烯基吡嗪代替4-乙烯基吡嗪,按实施例54所述进行制备,得到淡棕色胶状本题目化合物(185mg,50%)。
分析%:
实验值:C,77.2;H,7.5;N,11.2;
C24H27NO3计算值:C,77.2;H,7.3;N,11.2。
实施例57
(3R)-〔(5H)-二苯并〔a,d〕环庚烯-5-基氧基〕-1-(4-甲氧苯乙基)哌啶

将5-氯-(5H)-二苯并〔a,d〕环庚烯(0.49g)〔用亚硫酰氯氯化市售得到的5-羟基化合物制得〕和(3R)-羟基-1-(4-羟基苯乙基)哌啶(见制备15)(0.47g){〔α〕-3.0°(乙醇中C1.5)}溶于二氯甲烷,该溶液在室温搅拌4小时,用2M碳酸氢钠水溶液和饱和盐水洗涤,硫酸镁干燥和蒸发。剩余物经硅石(10g)色谱纯化,用二氯甲烷加0-2%甲醇作洗脱液。合并适当组分,蒸发,得到淡澄色胶状本题目化合物,它是半水合物(300mg,35%),〔α〕25 D+17.4°(甲醇中C 0.995)。
分析%:
实验值:C,79.6;H,7.2;N,3.1;
C29H31NO2;0.5H2O计算值:C,80.1;H,7.4;N,3.2。
实施例58
(3R)-〔(5H)-10,11-二氢二苯并〔a,d〕环庚烯-5-基〕-1-(4-甲氧苯乙基)哌啶

用5-氯-(5H)-10,11-二氢二苯并〔a,d〕环庚烯(市售得到)代替5-氯-(5H)-二苯并〔a,d〕环庚烯,按实施例57进行制备,得到无色油状本题目化合物,它是半水合物(491mg,58%),〔α〕25 D+23.8°(甲醇中C0.95)。
分析%:
实验值:C,79.7;H,7.6;N,3.2;
C29H33NO2;0.5H2O计算值:C,79.8;H,7.8;N,3.2。
实施例59
(3R)-〔(11H)-6,11-二氢二苯并〔b,e〕噻庚英-11-基氧基〕-1-(4-甲氧苯乙基)哌啶
用11-氯-(11H)-6,11-二氢二苯并〔b,e〕噻庚英(用亚硫酰氯氯化市售得到的11-羟基化合物制得)代替5-氯-(5H)-二苯并〔a,d〕环庚烯,按实施例57所述进行制备。得到无色油状本题目化合物,它是半水合物(0.80g,90%),〔α〕25 D+18.6°(甲醇中C0.81)。
分析%:
实验值:C,74.0;H,7.0;N,2.9;
C28H31NO2S;0.5H2O计算值:C,74.0;H,7.1;N,3.1。
实施例60
(3R,S)-二苯甲氧基-1-(4-羟甲基苯乙基)哌啶

将(3R,S)-二苯甲氧基-1-(4-甲氧羰基苯乙基)哌啶(0.43g)(见实施例17)的乙醚(5ml)溶液滴加到氢化铝锂(80mg)的乙醚(10ml)搅拌悬浮液中,该混合物在室温搅拌4小时,通过依次加入水(0.2ml),15%氢氧化钠水溶液(0.2ml)和水(0.6ml)骤冷,过滤。滤液经蒸发,得到无色固体状本题目化合物(270mg,67%),m.p.93-95°。
分析%:
实验值:C,80.2;H,7.9;N,3.3;
C27H31NO2计算值:C,80.8;H,7.7;N,3.5。
实施例61
(3R)-二苯甲氧基-1-(4-羟甲基苯乙基)哌啶
用(3R)-二苯甲氧基-1-(4-甲氧羰基苯乙基)哌啶(见实施例44)代替(3R,S)-二苯甲氧基-1-(4-羟羰基苯乙基)哌啶,按实施例60所述进行制备。用甲苯/60-80°石油醚重结晶后,得到无色固体状本题目化合物(358mg,89%),m.p.94.5-95°,〔α〕25 D+26.3°(甲醇中C0.955)
分析%:
实验值:C,80.4;H,7.8;N,3.2;
C27H31NO2计算值:C,80.8;H,7.7;N,3.5。
实施例62
(3R,S)-二苯甲氧基-1-〔4-(1-羟乙基)苯乙基〕哌啶

将硼氢化钠(40mg)加到1-(4-乙酰苯乙基)-(3R,S)-二苯甲氧基哌啶(250mg)(见实施例18)的甲醇(5ml)搅拌溶液中,该混合物在室温下搅拌14小时,用乙酸乙酯稀释,用水两次洗涤,硫酸镁干燥,得到无色油状本题目化合物(202mg,81%)。
分析%:
实验值:C,80.3;H,8.1;N,3.6;
C28H33NO2计算值:C,81.0;H,7.9;N,3.4。
实施例63
(3R)-二苯甲氧基-1-〔4-(1-羟乙基)苯乙基〕哌啶
用1-(4-乙酰苯乙基)-(3R)-二苯甲氧基哌啶(见实施例42)代替1-(4-乙酰苯乙基)-(3R,S)-二苯甲氧基哌啶,按实施例62所述进行制备,得到黄色油状本题目化合物(131mg,33%)。
分析%:
实验值:C,81.2;H,8.0;N,3.3;
C28H33NO2计算值:C,81.0;H,8.0;N,3.4。
实施例64
1-(4-氨基甲酰基-(3R)-二苯甲氧基哌啶
用0.880氨水(10ml)处理(3R)-二苯甲氧基-1-(4-甲氧羰基苯乙基)哌啶(0.43g)(见实施例44)的甲醇(10ml)溶液,在钢制反应釜中,将该混合物于80℃加热16小时,然后蒸发。剩余物分布在乙酸乙酯和水之中,有机层用水洗涤,硫酸镁干燥和蒸发。剩余物通过硅石(5g)色谱纯化,用二氯甲烷加0-5%甲醇作洗脱液。合并适当组分,蒸发,得到无色固体状本题目化合物,它是半水合物(75mg,16%),m.p.144.5-145.5°,〔α〕25 D+25.1°(甲醇中C0.475)。
分析%:
实验值:C,76.6;H,7.4;N,6.5;
C27H30N2O2;0.5H2O计算值:C,76.6;H,7.4;N,6.6。
实施例65
1-(4-氨基甲酰基苯乙基)-(3R,S)-二苯甲氧基哌啶

在钢制反应釜中,将(3R,S)-二苯甲氧基-1-(4-甲氧羰基苯乙基)哌啶(0.43g)(见实施例17)和0.880氨水溶液(5ml)混合物的四氢呋喃(5ml)溶液于80℃加热19小时,然后蒸发。剩余物分布在乙酸乙酯和水之中,有机层用水洗涤,硫酸镁干燥和蒸发。剩余物经硅石(8g)色谱分离,用二氯甲烷∶乙酸乙酯(4∶1)加1-20%甲醇作洗脱液。合并适当组分,蒸发,得到无色固体状本题目化合物,它是半水合物(150mg,37%),m.p.165-166°。
分析%:
实验值:C,76.7;H,7.1;N,6.6;
C27H30N2O2;0.5H2O计算值:C,76.6;H,7.4;N,6.6。
实施例66
(3R)-二苯甲氧基-1-〔4-(N-甲氨基甲酰基)苯乙基〕哌啶
用33%乙醇甲胺溶液代替0.880氨水溶液,按实施例64所述进行制备,得到无色油状本题目化合物(327mg,97%),〔α〕25 D+24.9°(甲醇中C1.005)。
分析%:
实验值:C,78.5;H,7.4;N,6.4;
C28H32N2O2计算值:C,78.5;H,7.5;N,6.5。
实施例67
1-(3-氨基甲酰基苯乙基)-(3R,S)-二苯甲氧基哌啶
用(3R,S)-二苯甲氧基-1-(3-甲氧羰基苯乙基)哌啶(见实施例87)代替(3R)-二苯甲氧基-1-(4-甲氧羰基苯乙基)哌啶,按实施例64所述进行制备,得到无色油状本题目化合物(27mg,23%)。
分析%:
实验值:C,78.4;H,7.3;N,6.5;
C27H30N2O2计算值:C,78.2;H,7.3;N,6.8。
实施例68
1-(3-氨基甲酰基苯乙基)-(3R)-二苯甲氧基哌啶

用(3R)-二苯甲氧基-1-(3-甲氧羰基苯乙基)哌啶(见实施例88)代替(3R)-二苯甲氧基-1-(4-甲氧羰基苯乙基)哌啶,按实施例64所述进行制备,得到无色油状本题目化合物(0.12g,24%)。
分析%:
实验值:C,74.4;H,7.2;N,5.6;
C27H30N2O2·H2O计算值:C,75.0;N,7.4;N,6.5。
实施例69
(3R,S)-二苯甲氧基-1-〔3-(N-甲氨基甲酰基)苯乙基〕哌啶

按实施例64所述方法,由(3R,S)-二苯甲氧基-1-(3-甲氧羰基苯乙基)哌啶(见实施例87)和33%甲胺的乙醇溶液进行制备,得到无色油状本题目化合物(64mg,38%)。
分析%:
实验值:C,78.0;H,7.7;N,6.4;
C28H32N2O2计算值:C,78.5;H,7.5;N,6.5。
实施例70 1-(3-氨基苯乙基)-(3R,S)-二苯基甲氧基哌啶

将(3R,S)-二苯甲氧基-1-(3-硝基苯乙基)-哌啶(4.1g)(见实施例12)和氯化亚锡二水合物(10.8g)混合物的乙醇(50ml)溶液回流加热1小时,用乙酸乙酯和饱和碳酸氢钠水溶液稀释,分层。有机层用水洗涤,硫酸镁干燥和蒸发。剩余物经硅石(25g)色谱纯化,用二氯甲烷加0-2%甲醇作洗脱液。合并相当相分,蒸发,得到油状本题目化合物(3.26g),93%)。
分析%:
实验值:C,80.8;H,7.8,N,7.2;
C26H30N2O计算值:C,80.8;H,7.8;N,7.2。
实施例71-74
按实施例70所述方法,用氯化亚锡二水合物还原适当的硝基取代原料(R,S-型)(分别见实施例13、19、11和24),制备下述化合物(R,S-型)。
实施例72的特征在于H-n.m.r.(CDCl):δ=7.20-7.50(10H,m);6.48-6.66(3H,m);5.59(1H,S);3.55-3.70(3H,m);3.12(2H,d,J=6Hz):2.51-2.87(5H,m)和1.24-2.15(6H,m)。
在实施例71中,将用氯化氢饱和的醚溶液加到粗制游离碱的醚溶液中,该混合物在室温下保持16小时,从沉淀油去上清液,干燥该油,得到无色泡沫状盐酸盐。


实施例75
(3R,S)-二苯甲氧基-1-(4-甲亚磺酰氨基苯乙基)哌啶

将甲磺酰氯(0.165g)滴入1-(4-氨基苯乙基)-(3R,S)-二苯甲氧基哌啶(0.50g)(见实施例71)的吡啶(5ml)搅拌溶液中,该混合物在室温下搅拌16小时,然后蒸发。剩余物分布在二氯甲烷和饱和碳酸氢钠水溶液之中,有机层用水洗涤,硫酸镁干燥和蒸发。剩余物经硅石(8g)色谱纯化,用二氯甲烷加D-1%甲醇作洗脱液,合并适宜组分,蒸发,得到无色油状本题目化合物(0.32g,53%)。
分析%:
实验值:C,69.7;H,6.9;N,6.0;
C27H32N2O3S计算值:C,69.8;H,6.9;N,6.0;
实施例76
(3R,S)-二苯甲氧基-1-(3-甲磺酰氨基苯乙基)哌啶

用1-(3-氨基苯乙基)-(3R,S)-二苯甲氧基哌啶(见实施例70)代替1-(4-氨基苯乙基)-(3R,S)-二苯甲氧基哌啶,按实施例75所述进行制备,得到无色油状本题目化合物(0.21g,35%)。
分析%:
实验值:C,69.9;H,7.1;N,6.2;
C27H32N2O3S计算值:C,69.8;H,6.9;N,6.0;
实施例77
(3R,S)-二苯甲氧基-1-(4-乙磺酰氨基苯乙基)哌啶

用乙磺酰氯代替甲磺酰氯,按实施例75所述进行制备,得到淡黄色油状本题目化合物(0.40g,67%)。
分析%:
实验值:C,70.0;H,7.4;N,5.8;
C28H34N2O3S计算值:C,70.3;H,7.2;N,5.8;
实施例78
1-(4-乙酰氨基苯乙基)-(3R,S)-二苯甲氧基哌啶

将1-(4-氨基苯乙基)-(3R,S)-二苯甲氧基哌啶(0.50g)(见实施例71)、碳酸氢钠(1.0g)、乙酸乙酯(5ml)和水(5ml)混合,加入乙酐(154mg),得到的混合物经用力振荡20秒后,在室温下静置10分钟,分层,有机层用饱和盐水洗涤,硫酸镁干燥和蒸发。剩余物经硅石(8g)色谱纯化,用二氯甲烷0-5%甲醇作洗脱液。合并适当组分,蒸发,得到无色油状本题目化合物(0.33g,60%)。
分析%:
实验值:C,78.8;H,7.7;N,6.8;
C28H32N2O2计算值:C,78.5;H,7.5;N,6.5;
实施例79
1-(3-乙酰氨基苯乙基)-(3R,S)-二苯甲氧基哌啶
用1-(3-氨基苯乙基)-(3R,S)-二苯甲氧基哌啶(见实施例70)代替1-(4-氨基苯乙基)-(3R,S)-二苯甲氧基哌啶,按实施例78所述进行制备,得到淡黄色油状本题目化合物,其特征带有0.25摩尔当量水(0.35g,63%)。
分析%:
实验值:C,77.8;H,7.6;N,6.6;
C28H32N2O2,0.25H2O:计算值:C,77.6;H,7.6;N,6.5;
实施例80
1-(4-乙酰氨基甲基苯乙基)-(3R)-二苯甲氧基哌啶

将乙酐(115mg)和1-(4-氨基-甲基苯乙基)-(3R)-二苯甲氧基哌啶(407mg)(见实施例89)混合物溶于二氯甲烷(35ml),在室温下搅拌18小时,用饱和碳酸氢钠水溶液洗涤,硫酸镁干燥和蒸发。剩余物经硅石(10g)色谱纯化,用二氯甲烷加0-5%甲醇作洗脱液。合并适当组分,蒸发,得到黄色油状化合物本题目化合物,它是半水合物(349mg,76%),〔α〕+20.6°(甲醇中C0.925)。
分析%:
实验值:C,76.8;H,8.0;N,5.9;
C29H34N2O2;0.5H2O计算值:C,77.1;H,7.8;N,6.2;
实施例81
(3R,S)-二苯甲氧基-1-(4-氨磺酰氨基苯乙基)哌啶
将1-(4-氨基苯乙基)-(3R,S)-二苯甲氧基哌啶(0.45g)(见实施例71)和硫酰胺(1.0g)的二噁烷(10ml)溶液回流加热1小时,蒸发。剩余物分布在乙酸乙酯和水之中,有机层用硫酸钠干燥,蒸发。剩余物经硅石(13g)色谱纯化,用二氯甲烷加0-5%甲醇作洗脱液。合并适当组分,蒸发,得到无色玻璃状本题目化合物(0.24g,44%)。
分析%:
实验值:C,66.7;H,6.8;N,8.8;
C26H31N3O3S计算值:C,67.1;H,6.7;N,9.0;
实施例82
(3R,S)-二苯甲氧基-1-(3-乙氧苯乙基)哌啶
将氢化钠(64mg;50%油悬浮液)加到(3R,S)-二苯甲氧基-1-(3-羟苯乙基)哌啶(0.46g)(见实施例15)的二甲基甲酰胺(10ml)溶液中,该混合物在室温下搅拌30分钟,用碘乙烷(0.19g)处理,然后在室温下搅拌3小时,该混合物分布在乙酸乙酯和水之中,有机层用水洗涤,硫酸钠干燥和蒸发。剩余物经硅石(9g)色谱纯化,用二氯甲烷加0-1%甲醇作
洗脱液。合并适当组分,蒸发,得到淡黄色油状本题目化合物(0.38g,77%)。
分析%:
实验值:C,80.9;H,8.1;N,3.3;
C28H33NO2计算值:C,80.9;H,8.0;N,3.4;
实施例83
(3R)-二苯甲氧基-1-[4-(N-甲氨基甲酰氧基)苯乙基]哌啶
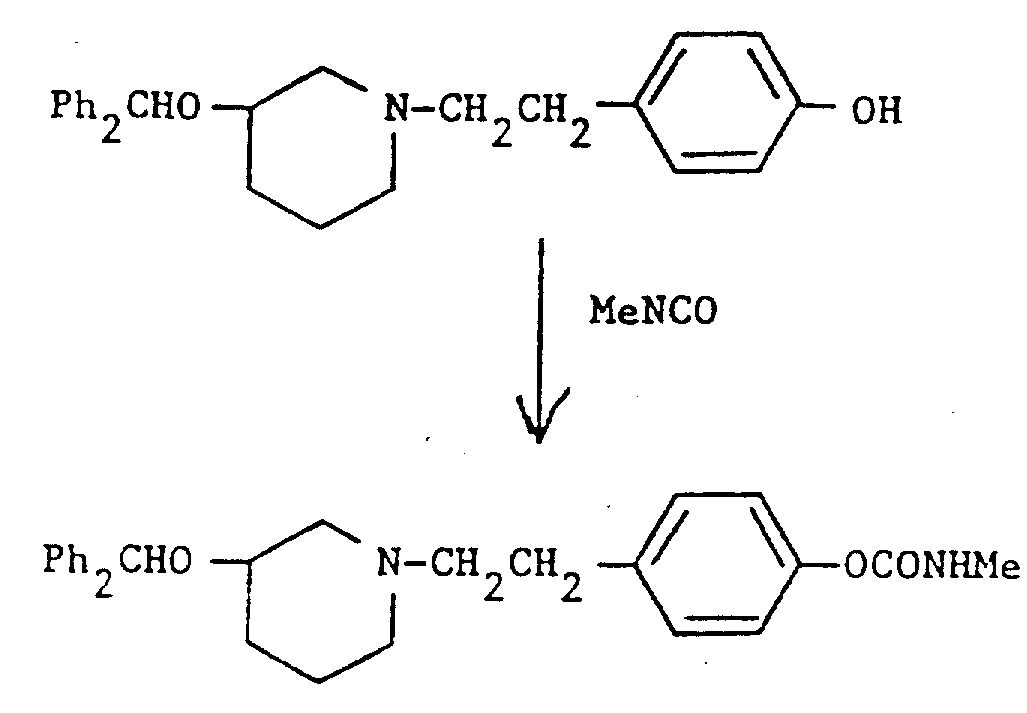
将异氰酸甲酯(1.0ml)加到(3R)-二苯甲氧基-(4-羟苯乙基)哌啶(0.50g)(见实施例35)的二氯甲烷(25ml)溶液中,该溶液在室温下搅拌65小时,然后蒸发。剩余物经硅石(4g)色谱纯化,用二氯甲烷加0-2%甲醇作洗脱液。合并适当组分,蒸发,得到无色油状本题目化合物,它是半水合物(401mg),70%)。
分析%:
实验值:C,74.0;H,7.7;N,6.5;
C28H32N2O3;0.5H2O 计算值:C,74.1;H,7.3;N,6.2;
实施例84
1-[2-(5-氨基甲酰基-2-噻吩基)乙基]-(3R,S)-二苯甲氧基哌啶
将1-[2-(5-羧基-2-噻吩基)乙基]-(3R,S)-二苯甲氧基哌啶(126mg)(见制备17)和1,1′-羰基二咪唑(“CDI”)(49mg)混合物溶于四氢呋喃(20ml),在室温下搅拌4小时,然后用饱和氨的四氢呋喃溶液(10ml)处理。该混合物在室温下搅拌22小时,蒸发。剩余物分布在乙酸乙酯和水之中,有机层用水洗涤,硫酸镁干燥,蒸发。剩余物经硅石(7g)色谱纯化,用二氯甲烷加0-2%甲醇作洗脱液。合并适当组分,蒸发,得到无色胶状本题目化合物(50mg,40%)。
分析%:
实验值:C,71.6;H,6.7;N,7.0;
C25H28N2O2S计算值:C,71.4;H,6.7;N,6.7;
实施例85
(3R)-二苯甲氧基-1-(4-甲氨基甲基苯乙基)哌啶

将亚硫酰氯(0.2ml)和(3R)-二苯甲氧基-1-(4-羟甲基苯乙基)哌啶(300mg)(见实施例61)的二氯甲烷(25ml)溶液回流加热2小时,蒸发。剩余物用正-己烷共沸3次,用33%甲胺的乙醇(25ml)溶液处理,于室温搅拌16小时,蒸发。剩余物分布在二氯甲烷和10%碳酸钠水溶液之中,有机层用10%碳酸钠水溶液和饱和盐水洗涤,硫酸镁干燥,蒸发。剩余物经硅石(10g)色谱纯化,用二氯甲烷加0-10%甲醇作洗脱液。合并当组分,蒸发,得到黄色油状本题目化合物(55mg,18%),〔α]+20.0°(甲醇中C0.52)。
分析%:
实验值:C,80.8;H,8.3;N,7.0;
C28H34N2O计算值:C,81.1;H,8.3;N,6.8;
实施例86
(3R)-二苯甲氧基-1-(2-羟基丙-2-基苯乙基)哌啶

在5分钟内,将1.4M甲基锂的己烷(0.90ml)溶液滴加到1-(4-乙酰苯乙基)-(3R)-二苯甲氧基哌啶(0.50g)(见实施例42)溶于醚(5ml)的水冷却搅拌溶液中,该混合物在室温下搅拌4小时,用水骤冷,用水稀释,分层,有机层用硫酸镁干燥,蒸发。剩余物经硅石(10g)色谱纯化,
用二氯甲烷加0-4%甲醇作洗脱液。合并适当组分,蒸发,用正-己烷重结晶后,得到无色固体状本题目化合物,它是半水合物(21mg,4%),m.p.83-85°。
分析%:
实验值:C,79.6;H,8.3;N,3.3;
C29H35NO2;0.5H2O计算值:C,79.4;H,8.3;N,3.2;
实施例87
(3R,S)-二苯甲氧基-1-(3-甲氧羰基苯乙基)哌啶
将一氧化碳吹过(3R,S)-二苯甲氧基-1-(3-碘苯乙基)哌啶(1.00g)(见实施例23)的甲醇(35ml)搅拌溶液,然后加入碳酸钾(0.70g)和双(三苯膦)氯化钯(Ⅱ)(30mg)。该混合物随着连续通过一氧化碳,于室温下搅拌3小时,过滤和蒸发。剩余物分布在乙酸乙酯和饱和碳酸氢钠水溶液之中,有机层用硫酸镁干燥,蒸发。剩余物经硅石(15g)色谱纯化,用二氯甲烷加0-2%甲醇作洗脱液。合并适当组分,蒸发,得到淡黄色油状本题目化合物(0.31g),36%),其特征在于′H-n.m.r.;(CDCl);δ=7.91(2H,S);
7.20-7.46(12H,m);5.59(1H,s);3.97(3H,s);3.52-3.63(1H,m);3.14(1H,dd,J=6 and 2Hz);2.54-2.90(5H,m) and 1.28-2.20(6H,m).
实施例88
(3R)-二苯甲氧基-1-(3-甲氧羰基苯乙基)哌啶

用(3R)-二苯甲氧基-1,3-碘苯乙基哌啶(见实施例52)代替(3R,S)-二苯甲氧基-1-(3-碘苯乙基)哌啶,按实施例87所述进行制备,得到无色油状本题目化合物(0.52g,60%),其特征在于
1H-n.m.r.(CDCl3)δ=7.91(2H,s);7.20-7.55(12H,m);5.60(1H,s);3.97(3H,s);3.52-3.64(1H,m);3.13(1H,dd,J=7 and 2Hz);and 1.25-2.90(11H,m)ppm.
实施例89
1-(4-氨甲基苯乙基)-(3R)-二苯甲氧基哌啶

在10%钯-炭(0.40g)存在下,于氢气压45p.s.i.(310.3kpa)中,将1-(4-氰苯乙基)-(3R)-二苯甲氧基哌啶(3.18g)(见实施例43)和浓盐酸(3.0ml)的乙醇(155ml)溶液搅拌44小时,过滤和蒸发。剩余物分布在二氯甲烷和饱和碳酸氢钠水溶液之中,有机层用水洗涤,硫酸镁干燥和蒸发。剩余物经硅石(50g)色谱纯化,用二氯甲烷加0-15%甲醇作洗脱液。合并当组分,蒸发,得到无色油状本题目化合物(2.14g,60%)。
分析%:
实验值:C,79.5;H,8.1;N,6.5;
C27H32N2O 计算值:C,79.2;H,8.1;N,6.8;
实施例90
(3R)-二苯甲氧基-1-[4-(3-甲脲基甲基)苯乙基]哌啶

将异氰甲酯加到1-(4-氨甲基苯乙基)-(3R)-二苯甲氧基哌啶(400mg)(见实施例89)的二氯甲烷(30ml)溶液中,该混合物在室温下搅拌19小时,蒸发。剩余物硅石(10g)色谱纯化,用二氯甲烷加0-10%甲醇作洗脱液。合并适当组分,蒸发,得到无色油状本题目化合物(320mg,70%),〔α〕+21.1°(甲醇中C0.835)。
分析%:
实验值:C,75.8;H,7.8;N,9.4;
C29H35N3O2计算值:C,76.1;H,7.7;N,9.2;
实施例91
(3R,S)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶鎓富马酸盐
将富马酸(0.87g)的热乙醇(15ml)溶液加到(3R,S)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶(3.11g)(见实施例1)的溶液中,该混合物在室温搅拌64小时,收集得到的固体,用醚洗涤,干燥,得到无色固体状本题目化合物(3.12g,78%),m.p.171-173°。
分析%:
实验值:C,70.4;H,6.4;N,2.6;
C27H29NO3C4H4O4计算值:C,70.1;H,6.2;N,2.6;
实施例92
(3R)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶鎓富马酸盐。
用(3R)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶(见实施例32)代替(3R,S)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶,按实施例91所述进行制备,得到无色固体状本题目化合物(0.53g,75%),m.p.167-169°。
分析%:
实验值:C,70.0;H,6.3;N,2.5;
C27H29NO3.C4H4O4计算值:C,70.1;H,6.2;N,2.6;
实施例93
(3R,S)-二苯甲氧基-1-(3-甲氧苯乙基)哌啶鎓富马酸盐
用(3R,S)-二苯甲氧基-1-(3-甲氧苯乙基)哌啶(见实施例8)代替(3R,S)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶,按实施例91所述进行制备,得到无色油状本题目化合物(1.80g,50%),m.p.148-150°。
分析%:
实验值:C,71.6;H,6.7;N,2.7;
C27H31NO2.C4H4O4计算值:C,71.9;H,6.8;N,2.7;
实施例94
1-[2-(苯并二噁烷-6-基)乙基]-(3R,S)-二苯甲氧基哌啶鎓富马酸盐
用1-[2-(苯并二噁烷-6-基)乙基]-(3R,S)-二苯甲氧基哌啶(见实施例2)代替(3R,S)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙基)哌啶,按实施例91所述进行制备,得到无色固体状本题目化合物(2.53g,60%),m.p.213-214°。
分析%:
实验值:C,70.3;H,6.5;N,2.6;
C28H31NO3.C4H4O4计算值:C,70.4;H,6.5;N,2.6;
实施例95
(3R,S)-二苯甲氧基-1-(2-羟苯乙基)哌啶

将氢化钠(0.16g,60%分散于油中)加到丁烷硫醇(0.31g)的二甲基甲酰胺(15ml)溶液中,该混合物在室温搅拌2小时,用(3R,S)-二苯甲氧基-1-(2-甲氧苯乙基)哌啶(0.50g)(见实施例25)的二甲基甲酰胺(5ml)溶液处理,回流加热3.5小时,然后,该混合物分布在乙酸乙酯和水之中,有机层用水洗涤,硫酸钠干燥,蒸发。剩余物经硅石(5g)色谱纯化,用二氯甲烷加0-2%甲醇作洗脱流。合并适当组分,蒸发,得到无色油状本题目化合物(0.21g)。
分析%:
实验值:C,80.4;H,7.6;N,3.3;
C26H29NO2计算值:C,80.6;H,7.5;N,3.6;
下述各制备说明用于上述实施例的新颖原料的制备方法。所有温度以℃表示。
制备1
(3R,S)-二苯甲氧基哌啶
方法A
将(3R,S)-羟基哌啶(50.5g),二苯基甲醇(92.0g)和对-甲苯磺酸一水合物(“PTSA”)(114.0g)的甲苯(600ml)溶液回流加热4小时,用迪安斯达克装置除去形成的水。该混合物分布在2M氢氧化钠水溶液和乙酸乙酯之中,有机层用水洗涤,蒸发。剩余物分布在醚和10%柠檬酸水溶液之中,酸性层用醚洗涤,用过量固体碳酸钠碱化,提入醚中,有机层用水洗涤,硫酸镁干燥,蒸发,得到无色油状本题目化合物(89.2g 67%),其特征在于′H-n.m.r光谱;(CDCl3):δ=7.22-7.45(10H,m);5.59(1H,s);3.38-3.48(1H,m);3.07(1H,dd,J=6 and 2Hz);2.40-2.88(3H,m) and 1.30-2.05(5H,m)。
方法B

将三氟乙酸(“TFA”)(20ml)谨慎地加到(3R,S)-羟基哌啶(5.05g)的二氯甲烷(20ml)搅拌溶液中,分批用二苯基甲醇(9.20g)处理,在室温搅拌2小时,蒸发。剩余物溶于二噁烷(50ml),用2M氢氧化钠水溶液(50ml)处理,在室温搅拌2小时,分布在醚和水之中。有机层用水洗涤,提入2M盐酸中,用醚洗涤,用过量固体碳酸钠碱化,提入醚中,有机层用水洗涤,硫酸镁干燥,蒸发得到无色油状本题目化合物
(5.30g,40%),它的光谱数据与由方法A得到的产品数据等同。
制备2
(3R,S)-二(4-氟苯基)甲氧基哌啶

用二(4-氟苯基)甲醇(市售得到)代替二苯基甲醇,按制备1方法A所述进行制备,得到无色油状本题目化合物(4.01g,66%),其特征在于H-n.m.r.光谱;(CDCl3):δ=7.31(4H,dt,J=8 and 10Hz);7.02(4H,t,J=8Hz);5.50(1H,s);3.34-3.42(1H,m);3.08(1H,dd,J=6 and 2Hz);2.60-2.89(3H,m)and 1.32-2.00(5H,m).
制备3
(3R)-二(4-氟苯基)甲氧基哌啶

在迪安-斯达克装置中,将二(4-氟苯基)甲醇(2.20g)、(3R)-羟基哌啶鎓(IS)-樟脑-10-磺酸盐(见制备7)(3.30g)和苯磺酸(1.58g)的甲苯(60ml)混合物回流加热3小时,用10%础酸钠水溶液洗涤,蒸发。剩余物溶于醚,用10%柠檬酸水溶液提取,酸性提取液用醚洗涤,用固体碳酸钠碱化,用醚提取。有机提取液用水洗涤,MgSO干燥,蒸发,得到无色油状本题目化合物(2.40g,79%)。分析%:
实验值:C,71.7;H,6.3;N,4.7;
C18H19F2NO计算值: C,71.3;H,6.3;N,4.6;
制备4
(3R)-[(R,S)-1-(2-甲苯基)-1-苯甲氧基]哌啶

用1-(2-甲苯基)-1-苯甲醇和对-甲苯磺酸代替二(4-氟苯基)甲醇和苯磺酸,按制备3所述进行制备,得到黄色油将本题目化合物(5.76g,51%),它直接用于实施例29,无需进一步纯化。
制备5
(3R)-[R,S)-1-(2-叔-丁苯基)-1-苯甲氧基]哌啶

用1-(2-叔-丁苯基)-1-苯甲醇(见J.Med、Chem.,2,57(1960)和对-甲苯磺酸代替二(4-氟苯基)甲醇和苯磺酸,按制备3进行制备,得到无色油状本题目化合物(847mg,72%),它直接用于实施例30,无需进一步纯化。
制备6
(3R,S)-[(11H)-6,11-二氢二苯并[b,e]噻庚英-11-基氧基]哌啶
用(11H)-6,11-二氢二苯并[b,e]噻庚英-11-醇(市售得到)代替二苯基甲醇,按制备1方法A进行制备,得到谈橙色油状本题目化合物(1.00g,12%),它直接用于实施例31,无需进一步纯化或检定。
制备7
(3R)-二苯甲氧基哌啶
用(3R)-羟基哌啶鎓(IS)-樟脑-10-磺酸盐[按B.Ringdahl,V.F.W.Ohnsorge和J.C.Craig在[J.Chem,Soc,PerkinⅡ,(1981),697]中介绍的方法制得,[α]+23.1°(50%乙醇水溶液中c 1.5)]代替(3R,S)-羟基哌啶,按制备1方法A所述进行制备,得到无色油状本题目化合物(2.7g,50%),[α]-3.3°(乙醇中C1.5)。
分析%:
实验值:C,80.2;H,7.9;N,5.1;
C18H21NO计算值:C,80.9;H,7.9;N,5.2;
制备8
(3R,S)-二苯甲氧基-1-(3,4-亚甲二氧基苯乙酰基)哌啶
将(3R,S)-二苯甲氧基哌啶(0.80g)(见制备1),3,4-亚甲二氧基苯乙酸(0.54g)、1-羟基苯并三唑(0.51g)和1-(3-二甲基氨丙基)-3-乙基碳化二亚胺盐酸盐(1.20g)混合物溶于二氯甲烷(50ml),把1-甲基吗啉(1.50g)加入其中,该溶液在室温搅拌40小时,依次用2M盐酸、水、10%碳酸钠水溶液和水洗涤,硫酸镁干燥、蒸发,得到无色油状本题目化合物(1.19g,92%),其特征在于′H-n.m.r.光谱
(CDCl3):=7.2-7.5(10H,m);6.60-6.83(3H,m);5.98(2H,s);5.38(s)and 5.64(s)(1H);3.20-3.95(7H,m)and 1.25-2.00(4H,m).
制备9-14
通过适当的芳乙酸与(3R,S)-二苯甲氧基哌啶(见制备1)结合,按制备8所述方法制备下述化合物(R,S-型)。芳乙酸一般是已知化合物。用于制备14的原料在制备18中介绍。
由制备12和13得到的产品,其特征在于它们的′H-n.m.r.光谱;制备12(CDCl):δ=7.2-7.6
(14H,m);5.42(s)and 5.63(s)(1H);3.30-3.92(7H,m)and 1.25-2.00(4H,m):制备 13(CDCl3):δ=7.22-7.50(10H,m);6.61-6.88(3H,m);5.36(s)and 5.64(s)(1H);4.25(4H,s);3.16-4.02(7H,m)and 1.23-2.00(4H,m).


制备15
(3R)-羟基-1-(4-羟基苯乙基)哌啶
将(3R)-羟基哌啶鎓樟脑-10-磺酸盐(8.30g)[按B.Ringdahl,V.F.W.Ohnsorge和J.C.Craig在J.Chem.Soc.PerkinⅡ,(1981),697中介绍的方法制得][α]+23.1°(在50%乙醇水溶液中C1.5)、4-甲氧基苯乙基溴化物(5.4g)、碳酸钠(5.30g)和碘化钠(250mg)的混合物溶于乙腈(125ml),回流加热84小时,蒸发。剩余物分布在二氯甲烷和水之中,有机层用饱和盐水洗涤,硫酸镁干燥,蒸发。剩余物经硅石(60g)色谱纯化,用二氯甲烷加0-3%甲醇作洗脱液。合并当组分,蒸发,得到无色油状本题目化合物(3.80g,65%),[α]+1.6°(甲醇中C1.0),其特征在于′H-n.m.r.光谱:(CDCl):δ=7.15(2H,d,J=8Hz);6.83(2H,d,J=8Hz);3.80-3.88(1H,m);3.79(3H,s);2.32-2.80(9H,m)and 1.48-1.92(4H,m)。
制备16
(3R,S)-羟基-1-(3-甲氧基苯乙基)哌啶
按制备15所述方法,用(3R,S)-羟基哌啶和3-甲氧基苯苯乙基溴化物进行制备,得到淡黄色油状本题目化合物(16.3g,72%),其特征在于′H-n.m.r.光谱:(CDCl):δ=7.21(1H,dd,J=8 and 7Hz);6.72-6.83(3H,m);3.78-3.88(1H,m);3.81(3H,s);2.30-2.84(9H,m)and 1.47-1.90(4H,m).
制备17
1-[2-(5-羧基-2-噻吩基)乙基]-(3r,s)-二苯甲氧基哌啶
在-20°下,10分钟内,将2.6M正-丁基锂的己烷(1.28ml)溶液滴加到(3R,S)-二苯甲氧基-1-[2-噻吩基)乙基]哌啶(378mg)(见实施例21)的醚(25ml)溶液中,该混合物在-20°搅拌1小时,倒在固体二氧化碳和醚混合物上,用水稀释,分层,水层用冰乙酸酸化至pH7,提入乙酸乙酯。该乙酸乙酯提取液用水洗涤,硫酸钠干燥,蒸发。剩余物经硅石(7g)色谱纯化,用二氯甲烷加0-20%甲醇作洗脱液。合并相当组分,蒸发,得到无色胶状本题目化合物(70mg,17%)。
分析%:
实验值:C,70.9;H,6.3;N,3.3;
C25H27NO3S计算值:C,71.2;H,6.5;N,3.3;
制备18
(苯并二氧杂庚烷-7-基)乙酸
将3,4-二羟基苯乙酸(5.0g)、1,3-二溴丙烷(72.g)和氢氧化钾(7.3g)混合物溶于水(25ml)中,回流加热17小时,用2M盐水酸化至pH1,经几次提取,提入二氯甲烷。合并有机提取流,用硫酸镁干燥,蒸发。剩余物经硅石(75g)色谱纯化,用二氯甲烷加0-2%乙酸作洗脱液。合并相当组分,蒸发,剩余物吸收在二氯甲烷中,提入5%碳酸钠水溶液中。碱性提取液用二氯甲烷洗涤,用5M盐酸酸化至pH1,提入二氯甲烷。有机提取液用硫酸镁干燥,蒸发,得到无色固体状本题目化合物(1.4g,23%)m.p.99-101°。
分析%:
实验值:C,63.4;H,5.9;N,0.0;
C11H12O4计算值: C,63.4;H,5.8;N,0.0;
制备19
3,4-亚甲二氧基苯乙醇

在30分钟内,将3,4-亚甲二氧基苯乙酸(18.0g)分批加到氢化铝锂(4.0g)的醚(400ml)冰冷却搅拌悬浮液中,该混合物在室温搅拌2小时,通过谨慎地加入饱和氯化铵水溶液骤冷,过滤。滤液用10%碳酸钠水溶液洗涤,用硫酸镁干燥,蒸发,得到淡黄色油状本题目化合物(15.01g,
90%)其特征在于′H-n.m.r.光谱。
1H-n.m.r.(CDCl3)δ=6.69-6.83(3H,m);5.98(2H,s);3.82(2H,dt,J=7 and 6Hz);2.81(2H,t,J=7Hz)and 1.44(1H,t,J=6Hz,可用D2O交换)。
制备20
3,4-亚甲二氧基苯乙基溴化物

在30分钟内,将三溴化磷(8.1g)的四氯化碳(50ml)溶液分批加到3,4-亚甲二氧基苯乙醇(15.0g)(见制备19)的四氯化碳(200ml)搅拌溶液中,该混合物回流加热3小时,依次用水(二次)、5M氢氧化钠水溶液和水洗涤,硫酸镁干燥和蒸发。剩余物经硅石(100g)色谱纯化,用四氯化碳作洗脱液。合并适当组分,蒸发,得到淡黄色油状本题目化合物(8.3g,40%),其特征在于′H-n.m.r.光谱。
1H-n.m.r.(CDCl3)δ=6.80(1H,d,J=8Hz),6.75(1H,s);6.71(1H,d,J=8Hz);6.00(2H,s);3.56(2H,t,J=7Hz)and 3.13(2H,t,J=7Hz).
制备21
3-羟基-4-甲氧基苯乙基氯化物

将3-羟基-4-甲氧基苯乙醇(2.25g)和亚硫酰氯(5ml)的混合物溶于二氯甲烷(120ml),回流加热16小时,蒸发。剩余物与己烷共沸两次,经硅石(30g)色谱纯化,用二氯甲烷加0-6%甲醇作洗脱液。合并相当组分,蒸发,得到无色固体状本题目化合物(0.82g,33%),m.p.53-54°,其特征在于′H-n.m.r.光谱。
1H-n.m.r.(CDCl3)δ=6.86(1H,d,J=8Hz);6.82(1H,d,J=2Hz);6.73(1H,dd,J=8 and 2Hz);5.61(1H,s,exchangeable with D2O);3.92(3H,s);3.70(2H,t,J=7Hz)and 3.01(2H,t,J=7Hz).
制备22
6-(2-羟乙基)苯并二噁烷

用(苯并二噁烷6-基)乙酸代替3,4-亚甲二氧基苯乙酸,按制备19所述进行制备,得到无色油状本题目化合物(19.8g,92%),其特征在于′H-n.m.r.光谱。
1H-n.m.r.(CDCl3)δ=6.84(1H,d,J=8Hz);6.77(1H,d,J=2Hz);6.73(1H,dd,J=8 and 2Hz);4.28(4H,s);3.59(2H,t,J=7Hz)and 3.08(2H,t,J=7Hz).
制备23
6-(2-溴乙基)苯并二噁烷
用6-(2-羟乙基)苯并二噁烷(见制备22)代替3,4-亚甲二氧基苯乙醇,按制备20所述进行制备,得到淡黄色油状本题目化合物(21.4g,80%),其特征在于′H-n.m.r.光谱。
1H-n.m.r.(CDCl3)δ=6.83(1H,d,J=8Hz);6.77(1H,d,J=2Hz);6.72(1H,dd,J=8 and 2Hz);4.28(4H,s);3.59(2H,t,J=7Hz)and 3.10(2H,t,J=7Hz).
制备24
4-羟基-3-硝基苯乙基氯化物

将浓硝酸(1.8ml)的乙酸(4ml)溶液加到4-羟基苯乙基氯化物(4.5g)的乙酸(25ml)搅拌溶液中,保持温度在15°以下。然后,该混合物在10°搅拌3.5小时,倒入水中,提入乙酸乙酯。有机提取液用5%碳酸钠水溶液洗涤,硫酸镁干燥和蒸发。剩余物经硅石(50g)色谱纯化,用己烷加0-10%乙酸乙酯作洗脱液。合并适当组分,蒸发,得到无色固体状本题目化合物(3.2g,55%),m.p53-55°。
分析%:
实验值:C,48.0;H,3.7;N,6.9;
C8H8ClNO3计算值:C,47.6;H,4.0;N,6.9;
制备25
4-甲氧基-3-硝基苯乙基氯化物

用4-甲氧基苯乙基氯化物代替4-羟基苯乙基氯化物,按制备24所述进行制备,得到淡黄色油状本题目化合物(1.9g,18%)。
分析%:
实验值:C,50.4;H,4.6;N.6.5;
C9H10ClNO3计算值:C,50.1;H,4.7;N,6.5;
制备26
3-碘苯乙醇
用3-碘苯乙酸(市售得到)代替3,4-亚甲二氧基苯乙酸,按制备19所述进行制备,得到无色油状本题目化合物(2.2g,58%),其特征在于′H-n.mr光谱。
1H-n.m.r.(CDCl3)δ=7.58-7.70(2H,m);7.23(1H,d,J=8Hz);7.04(1H,d,J=8Hz);3.91(2H,t,J=7Hz);2.84(2H,t,J=7Hz)and 1.43(1H,broad s,exchangeable with D2O).
制备27
3-碘苯乙基溴化物

将3-苯乙醇(见制备26)(1.2g)和48%氢溴酸水溶液(20ml)的混合物在室温下搅拌8小时,倒入水中,提入二氯甲烷。有机提取液用碳酸氢钠水溶液和水洗涤,硫酸钠干燥和蒸发,得到淡棕色油状本题目化合物(1.1g,73%),其特征在于′H-n.m.r.光谱。
1H-n.m.r.(CDCl3)δ=7.60-7.70(2H,m);7.22(1H,d,J=8Hz);7.07(1H,t,J=8Hz);3.58(2H,t,J=7Hz)and 3.16(2H,t,J=7Hz).
制备28
N-[4-(2-甲磺酰氧基乙基)苯基]甲磺酰胺
在0℃下,把甲磺酰氯(50.4g)滴加到4-氨基苯乙醇(27.44g)的无水吡啶(300ml)搅拌溶液中,该溶液在0℃搅拌30分钟,然后,在室温搅拌2.5小时,倒入水中,滤去固体,用水洗涤,干燥,经乙酸乙酯结晶,得到本题目化合物(39.0g,66%),m.p.136-137℃。
分析%
实验值 C,40.6;H,5.2;N,4.9;
C10H15NO5S2计算值:C,40.9;H,5.1;N.4.8;
制备29
5-(2-溴乙基)1,2-二氢化茚。

将三溴化磷(3.5ml)滴加到5-(2-羟乙基)1,2-二氢化茚(按FR-2,139,628所述制得)(14.0g)的四氯化碳(100ml)冰冷却溶液中,该混合物回流加热2小时,用冰水骤冷,分布在二氯甲烷和10%碳酸钠水溶液之中,有机层用水洗涤,硫酸镁干燥,减压蒸发。剩余物经硅胶色谱纯化,用二氯甲烷作洗脱液。合并相当组分,减压蒸发,得到淡黄色油状本题目化合物(10.5g,54%)。
1H-n.m.r.(300MHz.CDCl3)δ=7.20(dd,1H,J=8 and 1.5Hz);7.10(d,1H,J=1.5Hz);6.99(d,1H,J=8Hz);3.58(t,2H,J=7Hz);3.17(t,2H,J=7Hz);2.80-3.02(m,4H);and 2.02-2.18(m,2H)ppm.
制备30
5-(2-羟乙基)-2,3-二氢苯并呋喃

在0℃下,于10分钟内,将(2,3-二氢苯并呋喃-5-基)乙酸(4.9g-见欧洲专利申请132130号)的无水四氢呋喃(50ml)溶液滴加到氢化铝锂(1.5g)的无水四氢呋喃(50ml)搅拌悬浮液中。该混合物升温至室温,搅拌1小时。谨慎地滴入水(1.5ml)接着加入10%氢氧化钠水溶液(1.5ml),最后加入水(4.5ml)。过滤该混合物,用乙酸乙酯(2×50ml)洗涤无机盐,合并滤液和洗液,真空浓缩,得到油状本题目化合物(3.3g)。
1H-n.m.r.(CDCl3)δ=7.10(s,1H);7.00(d,1H);6.75(m,1H);4.65-4.55(m,2H);3.90-3.75(m,2H);3.30-3.15(m,2H);2.90-2.80(m,2H);1.85-1.75(broad s,1H)ppm.
制备31
5-(2-溴乙基)-2,3-二氢苯并呋喃

将三溴化磷(0.37g)加到5-(2-羟乙基)-2,3-二氢苯并呋喃(0.612g-见制备30)的四氯化碳(3ml)溶液中,该混合物回流加热3小时,冷却至室温,将该混合物分布在10%碳酸钠水溶液(20ml)和二氯甲烷(20ml)之中,分层,水层用二氯甲烷(20×10ml)提取。合并的二氯甲烷提取液用MgSO干燥,真空浓缩,静置结晶,得到油状本题目化合物(0.584g),m.p.60-62℃。
1H-n.m.r.(CDCl3)δ=7.10(s,1H);7.00-6.95(d,1H);6.80-6.70(d,1H);4.65-4.55(t,2H);3.60-3.50(t,2H);3.25-3.15(t,2H);3.15-3.10(t,2H)ppm.
Claims (16)
1、一种制备(3R,S)-或(3R)-型式(Ⅰ)化合物或它们在药物上可接受的盐的方法,式Ⅰ如下:
(Ⅰ)
式中R1为下述基团:

式中每个Y可以相同或不同,它选自取代基氢、卤素和C1-4烷基;
X是-(CH2)2-,-CH=CH-,-CH2-S-, -CH2-O-,-S-或-O-;
R是下述基团:

式中R2和R3各自分别为氢,C1-4烷基,羟基-(C1-4烷基),羟基,C1-4烷氧基,卤素,三氟甲基,硝基,氰基,氨磺酰,-CO(C1-4烷基),-OCO(C1-4烷基),-CO2-(C1-4烷基),-(CH2)nCONR6R7,-(CH2)nOCONR6R7,-(CH2)nNR8R9或-NHSO2NH2,其中,R6和R7各自分别为H或C1-4烷基,n是0、1或2,R8和R9各自分别为H或C1-4烷基,或者R8是氢,R9是-SO2(C1-4烷基),-CO(C1-4烷基)或-CONH(C1-4烷基);或
R2和R3连在一起与相邻碳原子连接时,表示下述基团-O(CH2)mO-、-O(CH2)2或-(CH2)3-,其中m是1、2或3;
R4是H,C1-4烷基或-CONH2;
R5是H,C1-4烷基或C1-4烷氧基,
其特征在于,该方法包括:
(i)当制备按式(Ⅰ)、其中R同R1按前述式(Ⅰ)定义的化合物,将按式(Ⅱ)的(3R,S)-或(3R)-型化合物分别与式(Ⅲ)Q CH2CH2R的化合物进行反应,

式(Ⅱ)、(Ⅲ)中、R1和R的定义同式(Ⅰ),Q代表离去基团;
(ii)当制备按式(Ⅰ)、其中R和R1按前述式(Ⅰ)定义的化合物,将按式(Ⅳ)的(3R,S)-或(3R)-型化合物分别用一种无机还原剂进行还原,

式(Ⅳ)中R和R1的定义同式(Ⅰ);
(iii)当制备按式(Ⅰ)、其中R和R1按前述式(Ⅰ)定义的化合物,将按式(Ⅵ)的(3R,S)-或(3R)-型化合物分别与式R1Q的化合物反应,

式(Ⅵ)中,R的定义同式(Ⅰ),
式R1Q中,R1的定义同式(Ⅰ),Q代表离去基团;
(iv)当制备按式(Ⅰ)、其中R1按前述式(Ⅰ)定义、R是:

其中R5按式(Ⅰ)中定义的化合物,将按式(Ⅱ)的(3R,S)-或(3R)-型化合物分别与RCH=CH2的化合物进行反应,反应时,任选在酸性或碱性催化剂存在下进行,

式(Ⅱ)中R1的定义同式(Ⅰ),
式RCH=CH2中,R按本方法(iv)中的定义;
(v)当制备按式(Ⅰ)、其中R1按前述式(Ⅰ)定义、R是:

的化合物,先将按式(Ⅰ)、其中R1按前述式(Ⅰ)的定义、R是

的(3R,S)-或(3R)-型化合物分别与一种强碱反应,第二步与二氧化碳反应,得到按式(Ⅰ)、其中R1的定义同前述式(Ⅰ)、R是
的(3R,S)-或(3R)-型化合物,然后形成所制得羧基化合物的活性酯或咪唑酰胺衍生物,随后用氨外理;
上述方法(i)可以任选继续进行下述的一个或多个步骤:
(a),当R2和R3中至少其一表示-CO2(C1-4烷基)时,将所述酯基团还原为-CH2OH;
(b),当R2和R3中至少其一表示羟基时,将所述羟基通过使用C1-4链烷酰氯或链烷酰溴或C1-4链烷酐的酰化作用,转化为-OCO(C1-4烷基);
(c),当R2和R3中至少其一表示-CO(C1-3烷基)时,将所述链烷酰基还原为-CH(OH)(C1-3烷基);
(d),当R2和R3中至少其一表示-CO2(C1-4烷基)时,将所述酯基团与氨或适当的胺R6R7NH反应,转化为-CONR6R7,其中R6和R7的定义同式(Ⅰ);
(e),当R2和R3中至少其一表示硝基时,将所述硝基还原为-NH2;
(f),当R2和R3中至少其一表示氨基时,将所述氨基通过与C1-4链烷磺酰氯或链烷磺酰溴或C1-4链烷磺酸酐反应,转化为-NHSO2(C1-4烷基);
(g),当R2和R3中至少其一表示n是0、1或2的-(CH2)nNH2时,将所述基团通过与C1-4链烷酰氯或链烷酰溴或C1-4链烷酸酐反应,转化为-(CH2)nNHCO(C1-4烷基);
(h),当R2和R3中至少其一表示氨基时,将所述氨基通过与硫酰胺反应,转化为-NHSO2NH2;
(i),当R2和R3中至少其一表示羟基时,将所述羟基通过先与强碱反应,再与C1-4烷基碘反应,转化为C1-4烷氧基;
(j),当R2和R3中至少其一表示n是0、1或2的-(CH2)nOH时,将所述基团通过与异氰酸C1-4烷酯反应,转化为-(CH2)nOCONH(C1-4烷基);
(k),当R2和R3中至少其一表示-CH2OH时,将所述羟甲基先与亚硫酰氯反应,再与氨或适当的胺R8R9NH反应,转化为R8和R9各自分别为H或C1-4烷基的-CH2NR8R9;
(l),当R2和R3中至少其一表示-COCH3时,将所述乙酰基通过与甲基锂或甲基镁溴或甲基镁氯或甲基镁碘反应,转化为-C(OH)(CH3)2;
(m),当R2和R3中至少其一表示碘代时,将所述碘代取代基通过与一氧化碳、碱、C1-4链烷醇和钯(Ⅱ)催化剂的混合物反应,转化为-CO2(C1-4烷基);
(n),当R2和R3中至少其一表示氰基时,将所述氰基还原为-CH2NH2;
(o),当R2和R3中至少其一表示n是0、1或2的-(CH2)nNH2时,将所述基团通过与异氰酸C1-4烷酯反应,转化为-(CH2)nNHCONH(C1-4烷基);
(p),当R2和R3中至少其一表示C1-4烷氧基时,将所述烷氧基在强碱存在下,通过用C1-4链烷硫醇处理,转化为羟基;
(q),将式(Ⅰ)化合物转化为药物上可接受的盐;
上述方法(ii)可以任选继续进行上述(b)、(f)、(g)、(h)、(i)、(j)、(k)、(m)、(o)、(p)或(q)步骤中的一步或多步反应;
上述方法(iii)可以任选继续进行上述(a)、(c)、(d)、(e)、(l)、(m)、(n)、(p)或(q)步骤中的一步或多步反应;
上述方法(iv)和(v)可以任选继续进行上述的(q)步骤的反应。
2、根据权利要求1中方法(ⅰ)所述方法,其中Q是Cl、Br、I或甲磺酰氧基。
3、根据权利要求1中方法(ⅰ)或权利要求2所述的方法,其中该方法在酸受体存在下实施。
4、根据权利要求1中方法(ⅰ)或权利要求2所述方法,其中Q是Cl或Br,该方法在碘化钠或碘化钾存在下实施。
5、根据权利要求1中方法(ⅱ)所述方法,其中用氢化铝锂作为还原剂实施该方法。
6、根据权利要求1中方法(ⅲ)所述方法,其中Q是Cl或Br。
7、根据权利要求1中方法(ⅳ)所述方法,其中用N-苄基三甲基铵氢氧化物(″Triton B″)作为催化剂实施该方法。
8、根据权利要求1中方法(ⅴ)所述方法,其中在第一步用正-丁基锂作为所述的强碱,其后用1,1′-羰基二咪唑形成咪唑酰胺。
9、根据权利要求1中方法(ⅰ)、(ⅱ)或(ⅲ)或权利要求2所述方法,其中R是下述基团:

式中R2、R3、R4和R5的定义同权利要求1的式(Ⅰ)
10、根据权利要求1或2所述方法,其中R1是如下基团:


11、根据权利要求1中方法(ⅰ)、(ⅱ)或(ⅲ)或权利要求2所述方法,其中R是下述基团:
(a)

式中R2和R3各自分别为H,C1-2烷基,羟基-(C1-3烷基),羟基,C1-2烷氧基,卤素,氨磺酰,-CO(C1-2烷基),-OCO(C1-2烷基),-CONH2,-CONH(C1-2烷基),-OCONH(C1-2烷基),-NH2,-CH2NH2,-CH2NH(C1-2烷基),-NHSO2(C1-2烷基),-NHCO(C1-2烷基),-CH2NHCO(C1-2烷基),-CH2NHCONH(C1-2烷基)或-NHSO2NH2;或R2和R3连接在一起表示-O(CH2)mO-,其中m是1、2或3,-O(CH2)2-,或-(CH2)3-;
(b)
 其中R4是H或-CONH2;
其中R4是H或-CONH2;
(c)
 或
或
(d)

12、根据权利要求1或2所述方法,其中R1是中R1是
13、根据权利要求1中方法(ⅰ)、(ⅱ)或(ⅲ)或权利要求2所述方法,其中R是
14、根据权利要求1或2所述方法,其中得到下式所示(3R)-构型的式(Ⅰ)化合物:

15、根据权利要求1所述方法,其特征在于每个Y是相同的。
16、根据权利要求1中方法(ⅰ)或权利要求2所述方法,其特征在于R1是

以及R是

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB8816365.4 | 1988-07-08 | ||
| GB888816365A GB8816365D0 (en) | 1988-07-08 | 1988-07-08 | Therapeutic agents |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1039415A CN1039415A (zh) | 1990-02-07 |
| CN1022914C true CN1022914C (zh) | 1993-12-01 |
Family
ID=10640168
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN89104933A Expired - Fee Related CN1022914C (zh) | 1988-07-08 | 1989-07-08 | 新的1,3-二取代哌啶衍生物的制备方法 |
Country Status (28)
| Country | Link |
|---|---|
| US (1) | US5089505A (zh) |
| EP (1) | EP0350309B1 (zh) |
| JP (2) | JPH0739388B2 (zh) |
| KR (1) | KR930002728B1 (zh) |
| CN (1) | CN1022914C (zh) |
| AT (1) | ATE82262T1 (zh) |
| AU (1) | AU602453B2 (zh) |
| CA (1) | CA1336833C (zh) |
| DD (1) | DD284011A5 (zh) |
| DE (1) | DE68903437T2 (zh) |
| DK (1) | DK337589A (zh) |
| EG (1) | EG18853A (zh) |
| ES (1) | ES2052917T3 (zh) |
| FI (1) | FI94241C (zh) |
| GB (1) | GB8816365D0 (zh) |
| GR (1) | GR3006595T3 (zh) |
| HU (1) | HUT50771A (zh) |
| IE (1) | IE63438B1 (zh) |
| IL (1) | IL90792A0 (zh) |
| MX (1) | MX16728A (zh) |
| MY (1) | MY106976A (zh) |
| NO (1) | NO892828L (zh) |
| NZ (1) | NZ229865A (zh) |
| PL (2) | PL161887B1 (zh) |
| PT (1) | PT91081B (zh) |
| RU (2) | RU1838307C (zh) |
| YU (1) | YU135389A (zh) |
| ZA (1) | ZA895175B (zh) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5231104A (en) * | 1988-07-08 | 1993-07-27 | Pfizer Inc. | 1-arylethyl-3-substituted piperidines |
| GB9000301D0 (en) * | 1990-01-06 | 1990-03-07 | Pfizer Ltd | Piperidine & pyrrolidine derivatives |
| GB9208230D0 (en) * | 1992-04-14 | 1992-05-27 | Pfizer Ltd | Treatment of delayed gastric emptying |
| US5340826A (en) * | 1993-02-04 | 1994-08-23 | Pfizer Inc. | Pharmaceutical agents for treatment of urinary incontinence |
| GB9400600D0 (en) * | 1994-01-14 | 1994-03-09 | Pfizer Ltd | Treatment of motion seckness |
| US6492400B1 (en) | 1998-12-18 | 2002-12-10 | Bristol-Myers Squibb Pharma Company | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US6525069B1 (en) | 1998-12-18 | 2003-02-25 | Bristol-Myers Squibb Pharma Co. | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US6331541B1 (en) | 1998-12-18 | 2001-12-18 | Soo S. Ko | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| EP1161240B1 (en) * | 1998-12-18 | 2005-08-17 | Bristol-Myers Squibb Pharma Company | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| AU2482100A (en) * | 1998-12-18 | 2000-07-03 | Du Pont Pharmaceuticals Company | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US6897234B2 (en) | 1999-12-17 | 2005-05-24 | Bristol-Myers Squibb Pharma Company | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| IL153123A0 (en) | 2000-06-30 | 2003-06-24 | Bristol Myers Squibb Co | N-ureidoheterocycloalkyl-piperidine derivatives and pharmaceutical compositions containing the same |
| CA2422055A1 (en) * | 2000-09-11 | 2002-03-21 | Sepracor, Inc. | Ligands for monoamine receptors and transporters, and methods of use thereof (neurotransmission) |
| US6508872B2 (en) * | 2001-02-26 | 2003-01-21 | Hewlett-Packard Company | Lightfast additive molecule for inkjet ink |
| US20090005309A1 (en) * | 2007-05-18 | 2009-01-01 | Auspex Pharmaceuticals, Inc. | Substituted piperidines |
| EP2578569B1 (en) * | 2010-05-27 | 2015-10-28 | ASKA Pharmaceutical Co., Ltd. | Heterocyclic ring compound and h1 receptor antagonist |
| CN107129453B (zh) * | 2016-02-26 | 2019-10-11 | 中国科学院大连化学物理研究所 | 化合物、毒蕈碱m受体拮抗剂、组合物及应用 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA541598A (en) * | 1957-05-28 | Levy Joseph | N-substituted-4-benzhydryl etherpiperidines and method for obtaining the same | |
| GB688345A (en) * | 1951-02-19 | 1953-03-04 | Greenwood And Hughes Ltd | Improvements in sanitary liquid traps |
| US2745837A (en) * | 1954-01-21 | 1956-05-15 | Schering Corp | Benzhydryl ethers of alkyl piperidinols |
| GB780027A (en) * | 1955-01-20 | 1957-07-31 | Schering Corp | 3-piperidyl ethers and thioethers |
| US2974146A (en) * | 1956-02-24 | 1961-03-07 | Lakeside Lab Inc | 3-piperidyl benzhydryl ethers |
| NL237664A (zh) * | 1959-04-01 |
-
1988
- 1988-07-08 GB GB888816365A patent/GB8816365D0/en active Pending
-
1989
- 1989-06-29 IL IL90792A patent/IL90792A0/xx not_active IP Right Cessation
- 1989-07-05 MY MYPI89000908A patent/MY106976A/en unknown
- 1989-07-06 CA CA000604882A patent/CA1336833C/en not_active Expired - Fee Related
- 1989-07-06 PT PT91081A patent/PT91081B/pt not_active IP Right Cessation
- 1989-07-06 ES ES89306881T patent/ES2052917T3/es not_active Expired - Lifetime
- 1989-07-06 DD DD89330509A patent/DD284011A5/de not_active IP Right Cessation
- 1989-07-06 US US07/376,263 patent/US5089505A/en not_active Expired - Fee Related
- 1989-07-06 EP EP89306881A patent/EP0350309B1/en not_active Expired - Lifetime
- 1989-07-06 DE DE8989306881T patent/DE68903437T2/de not_active Expired - Fee Related
- 1989-07-06 EG EG32789A patent/EG18853A/xx active
- 1989-07-06 YU YU01353/89A patent/YU135389A/xx unknown
- 1989-07-06 AT AT89306881T patent/ATE82262T1/de not_active IP Right Cessation
- 1989-07-07 PL PL89280474A patent/PL161887B1/pl unknown
- 1989-07-07 RU SU894614534A patent/RU1838307C/ru active
- 1989-07-07 DK DK337589A patent/DK337589A/da not_active Application Discontinuation
- 1989-07-07 PL PL89283209A patent/PL162318B1/pl unknown
- 1989-07-07 NO NO89892828A patent/NO892828L/no unknown
- 1989-07-07 HU HU893449A patent/HUT50771A/hu unknown
- 1989-07-07 KR KR1019890009671A patent/KR930002728B1/ko not_active IP Right Cessation
- 1989-07-07 ZA ZA895175A patent/ZA895175B/xx unknown
- 1989-07-07 MX MX1672889A patent/MX16728A/es unknown
- 1989-07-07 FI FI893326A patent/FI94241C/fi not_active IP Right Cessation
- 1989-07-07 NZ NZ229865A patent/NZ229865A/en unknown
- 1989-07-07 AU AU37971/89A patent/AU602453B2/en not_active Ceased
- 1989-07-07 IE IE220289A patent/IE63438B1/en not_active IP Right Cessation
- 1989-07-08 CN CN89104933A patent/CN1022914C/zh not_active Expired - Fee Related
- 1989-07-10 JP JP1177854A patent/JPH0739388B2/ja not_active Expired - Fee Related
-
1990
- 1990-09-17 RU SU904831151A patent/RU1836364C/ru active
-
1992
- 1992-12-21 GR GR920403003T patent/GR3006595T3/el unknown
-
1994
- 1994-07-26 JP JP6173882A patent/JPH07157469A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1022914C (zh) | 新的1,3-二取代哌啶衍生物的制备方法 | |
| CN1146540C (zh) | 具有镇痛作用的新化合物及其用途 | |
| CN1024547C (zh) | 一类环胺化合物的制备方法 | |
| CN1114596C (zh) | 联苯基衍生物 | |
| CN1046721C (zh) | 杂环化合物及其制备方法和用途 | |
| CN1297550C (zh) | 苯并呋喃衍生物及其制备和用途 | |
| CN1226888A (zh) | 含氟-1,4-二取代哌啶衍生物 | |
| CN1665502A (zh) | Mchir拮抗剂 | |
| CN1210521A (zh) | 异喹啉衍生物及医药 | |
| CN1077953A (zh) | 乙酸衍生物 | |
| CN1802350A (zh) | 氮杂环丁烷羧酰胺衍生物以及它们在治疗cb1受体介导的病症中的应用 | |
| CN1906180A (zh) | 新的长效β-2-激动剂及其作为药物的用途 | |
| CN1791568A (zh) | Hsp90家族蛋白质阻断剂 | |
| CN1022185C (zh) | 镇痛羧酸酰胺衍生物的制备方法 | |
| CN1774245A (zh) | 用于视网膜神经疾病的含有烷基醚衍生物或其盐的预防剂/治疗剂 | |
| CN1599722A (zh) | 作为毒蕈碱受体拮抗剂的4-哌啶基烷基胺衍生物 | |
| CN1028527C (zh) | 香豆素衍生物的制备方法 | |
| CN1183907C (zh) | 新的咪唑啉化合物、其制备方法和含有它们的药物组合物 | |
| CN1041583A (zh) | 蝇蕈碱受体拮抗剂 | |
| CN1064350C (zh) | 芳基和杂芳基烷氧基萘衍生物 | |
| CN1273463C (zh) | (1-苯甲酰甲基-3-苯基-3-哌啶基乙基)哌啶衍生物,制备它们的方法以及包含它们的药物组合物 | |
| CN1305871C (zh) | 用于治疗与5-ht2a受体有关疾病的新哌嗪基-吡嗪酮衍生物 | |
| CN1202103A (zh) | 兴奋性的氨基酸受体拮抗剂 | |
| CN1231466C (zh) | 新型哌啶衍生物 | |
| CN1264836C (zh) | 苯并噁噻哌衍生物及其在制备药物中的用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |