CN101484445A - 吡咯烷取代的黄酮的对映选择性合成 - Google Patents
吡咯烷取代的黄酮的对映选择性合成 Download PDFInfo
- Publication number
- CN101484445A CN101484445A CNA2006800552570A CN200680055257A CN101484445A CN 101484445 A CN101484445 A CN 101484445A CN A2006800552570 A CNA2006800552570 A CN A2006800552570A CN 200680055257 A CN200680055257 A CN 200680055257A CN 101484445 A CN101484445 A CN 101484445A
- Authority
- CN
- China
- Prior art keywords
- formula
- compound
- methyl
- acid
- tetramethyleneimine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- -1 pyrrolidine-substituted flavones Chemical class 0.000 title claims description 19
- 230000015572 biosynthetic process Effects 0.000 title abstract 2
- 238000003786 synthesis reaction Methods 0.000 title abstract 2
- 150000003839 salts Chemical class 0.000 claims abstract description 19
- 229930003944 flavone Natural products 0.000 claims abstract description 5
- 150000002213 flavones Chemical group 0.000 claims abstract description 5
- 235000011949 flavones Nutrition 0.000 claims abstract description 5
- 150000001875 compounds Chemical class 0.000 claims description 69
- OKKJLVBELUTLKV-UHFFFAOYSA-N methyl alcohol Substances OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 66
- 238000000034 method Methods 0.000 claims description 57
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 52
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical group CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 36
- 239000002904 solvent Substances 0.000 claims description 34
- 239000000203 mixture Substances 0.000 claims description 31
- 239000003513 alkali Substances 0.000 claims description 27
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 26
- 239000011541 reaction mixture Substances 0.000 claims description 25
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 claims description 21
- 239000002253 acid Substances 0.000 claims description 21
- 239000003795 chemical substances by application Substances 0.000 claims description 20
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical group [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 18
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 claims description 18
- 230000002829 reductive effect Effects 0.000 claims description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 13
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 13
- 239000002585 base Substances 0.000 claims description 13
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 claims description 12
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 claims description 12
- 229910052751 metal Inorganic materials 0.000 claims description 10
- 239000002184 metal Substances 0.000 claims description 10
- 150000002918 oxazolines Chemical class 0.000 claims description 10
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical group [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 claims description 10
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 claims description 9
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 9
- 125000003118 aryl group Chemical group 0.000 claims description 9
- 238000006243 chemical reaction Methods 0.000 claims description 9
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 claims description 9
- 150000002148 esters Chemical class 0.000 claims description 9
- 239000011777 magnesium Substances 0.000 claims description 9
- 229910052749 magnesium Inorganic materials 0.000 claims description 9
- 238000002360 preparation method Methods 0.000 claims description 9
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 claims description 9
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 claims description 8
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 claims description 8
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 claims description 8
- 239000003054 catalyst Substances 0.000 claims description 8
- 239000002131 composite material Substances 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 8
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 claims description 8
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 7
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical group [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 claims description 7
- 229910021626 Tin(II) chloride Inorganic materials 0.000 claims description 7
- 239000012299 nitrogen atmosphere Substances 0.000 claims description 7
- 235000011150 stannous chloride Nutrition 0.000 claims description 7
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical group [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 claims description 7
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 claims description 6
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 6
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 claims description 6
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 6
- 229940125782 compound 2 Drugs 0.000 claims description 6
- 229910052802 copper Inorganic materials 0.000 claims description 6
- 239000010949 copper Substances 0.000 claims description 6
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 claims description 6
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 claims description 6
- 229960000890 hydrocortisone Drugs 0.000 claims description 6
- 150000004702 methyl esters Chemical class 0.000 claims description 6
- 239000012312 sodium hydride Substances 0.000 claims description 6
- 229910000104 sodium hydride Inorganic materials 0.000 claims description 6
- 229910052725 zinc Inorganic materials 0.000 claims description 6
- 239000011701 zinc Substances 0.000 claims description 6
- VNDYJBBGRKZCSX-UHFFFAOYSA-L zinc bromide Chemical compound Br[Zn]Br VNDYJBBGRKZCSX-UHFFFAOYSA-L 0.000 claims description 6
- UAYWVJHJZHQCIE-UHFFFAOYSA-L zinc iodide Chemical compound I[Zn]I UAYWVJHJZHQCIE-UHFFFAOYSA-L 0.000 claims description 6
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 5
- 230000017858 demethylation Effects 0.000 claims description 5
- 238000010520 demethylation reaction Methods 0.000 claims description 5
- 239000002808 molecular sieve Substances 0.000 claims description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 5
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 claims description 5
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 4
- 229910015900 BF3 Inorganic materials 0.000 claims description 4
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 claims description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 4
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 4
- DPDMMXDBJGCCQC-UHFFFAOYSA-N [Na].[Cl] Chemical compound [Na].[Cl] DPDMMXDBJGCCQC-UHFFFAOYSA-N 0.000 claims description 4
- 229910000102 alkali metal hydride Inorganic materials 0.000 claims description 4
- 150000008046 alkali metal hydrides Chemical class 0.000 claims description 4
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims description 4
- 150000003851 azoles Chemical class 0.000 claims description 4
- 239000000460 chlorine Substances 0.000 claims description 4
- BEPAFCGSDWSTEL-UHFFFAOYSA-N dimethyl malonate Chemical compound COC(=O)CC(=O)OC BEPAFCGSDWSTEL-UHFFFAOYSA-N 0.000 claims description 4
- 229910052736 halogen Inorganic materials 0.000 claims description 4
- 150000002367 halogens Chemical class 0.000 claims description 4
- 125000005842 heteroatom Chemical group 0.000 claims description 4
- 239000012022 methylating agents Substances 0.000 claims description 4
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 claims description 4
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 claims description 3
- ZRNSSRODJSSVEJ-UHFFFAOYSA-N 2-methylpentacosane Chemical compound CCCCCCCCCCCCCCCCCCCCCCCC(C)C ZRNSSRODJSSVEJ-UHFFFAOYSA-N 0.000 claims description 3
- LJUQGASMPRMWIW-UHFFFAOYSA-N 5,6-dimethylbenzimidazole Chemical compound C1=C(C)C(C)=CC2=C1NC=N2 LJUQGASMPRMWIW-UHFFFAOYSA-N 0.000 claims description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 3
- 229910021590 Copper(II) bromide Inorganic materials 0.000 claims description 3
- MPCRDALPQLDDFX-UHFFFAOYSA-L Magnesium perchlorate Chemical compound [Mg+2].[O-]Cl(=O)(=O)=O.[O-]Cl(=O)(=O)=O MPCRDALPQLDDFX-UHFFFAOYSA-L 0.000 claims description 3
- 238000006845 Michael addition reaction Methods 0.000 claims description 3
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 claims description 3
- 229910021588 Nickel(II) iodide Inorganic materials 0.000 claims description 3
- 238000007171 acid catalysis Methods 0.000 claims description 3
- 238000005904 alkaline hydrolysis reaction Methods 0.000 claims description 3
- 239000007864 aqueous solution Substances 0.000 claims description 3
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 3
- 229910052801 chlorine Inorganic materials 0.000 claims description 3
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 claims description 3
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 3
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 3
- 229940043279 diisopropylamine Drugs 0.000 claims description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 3
- 150000002460 imidazoles Chemical class 0.000 claims description 3
- 229910052746 lanthanum Inorganic materials 0.000 claims description 3
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 claims description 3
- OTCKOJUMXQWKQG-UHFFFAOYSA-L magnesium bromide Chemical compound [Mg+2].[Br-].[Br-] OTCKOJUMXQWKQG-UHFFFAOYSA-L 0.000 claims description 3
- 229910001623 magnesium bromide Inorganic materials 0.000 claims description 3
- BLQJIBCZHWBKSL-UHFFFAOYSA-L magnesium iodide Chemical compound [Mg+2].[I-].[I-] BLQJIBCZHWBKSL-UHFFFAOYSA-L 0.000 claims description 3
- 229910001641 magnesium iodide Inorganic materials 0.000 claims description 3
- NBVXSUQYWXRMNV-UHFFFAOYSA-N monofluoromethane Natural products FC NBVXSUQYWXRMNV-UHFFFAOYSA-N 0.000 claims description 3
- 229910052759 nickel Inorganic materials 0.000 claims description 3
- BMGNSKKZFQMGDH-FDGPNNRMSA-L nickel(2+);(z)-4-oxopent-2-en-2-olate Chemical compound [Ni+2].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O BMGNSKKZFQMGDH-FDGPNNRMSA-L 0.000 claims description 3
- IPLJNQFXJUCRNH-UHFFFAOYSA-L nickel(2+);dibromide Chemical compound [Ni+2].[Br-].[Br-] IPLJNQFXJUCRNH-UHFFFAOYSA-L 0.000 claims description 3
- BFSQJYRFLQUZKX-UHFFFAOYSA-L nickel(ii) iodide Chemical compound I[Ni]I BFSQJYRFLQUZKX-UHFFFAOYSA-L 0.000 claims description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 3
- 230000033912 thigmotaxis Effects 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 229940102001 zinc bromide Drugs 0.000 claims description 3
- 239000002841 Lewis acid Substances 0.000 claims description 2
- GARJITDFQFOYKD-UHFFFAOYSA-N [N-]=[N+]=[N-].[Li+].[Si+4].[N-]=[N+]=[N-].[N-]=[N+]=[N-].[N-]=[N+]=[N-].[N-]=[N+]=[N-] Chemical class [N-]=[N+]=[N-].[Li+].[Si+4].[N-]=[N+]=[N-].[N-]=[N+]=[N-].[N-]=[N+]=[N-].[N-]=[N+]=[N-] GARJITDFQFOYKD-UHFFFAOYSA-N 0.000 claims description 2
- 230000002378 acidificating effect Effects 0.000 claims description 2
- 150000007517 lewis acids Chemical class 0.000 claims description 2
- 239000011592 zinc chloride Substances 0.000 claims description 2
- 235000005074 zinc chloride Nutrition 0.000 claims description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims 2
- 102000016736 Cyclin Human genes 0.000 abstract description 7
- 108050006400 Cyclin Proteins 0.000 abstract description 7
- 239000003112 inhibitor Substances 0.000 abstract description 5
- 230000002062 proliferating effect Effects 0.000 abstract description 4
- 206010028980 Neoplasm Diseases 0.000 abstract description 3
- 108091000080 Phosphotransferase Proteins 0.000 abstract description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 3
- 102000020233 phosphotransferase Human genes 0.000 abstract description 3
- 201000011510 cancer Diseases 0.000 abstract description 2
- 150000003235 pyrrolidines Chemical group 0.000 abstract 1
- 238000005481 NMR spectroscopy Methods 0.000 description 18
- 239000000243 solution Substances 0.000 description 17
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 17
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 14
- 238000000524 positive electrospray ionisation mass spectrometry Methods 0.000 description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- 239000012044 organic layer Substances 0.000 description 10
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- 125000000217 alkyl group Chemical group 0.000 description 8
- 230000006837 decompression Effects 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 239000012141 concentrate Substances 0.000 description 6
- 238000010438 heat treatment Methods 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 238000001035 drying Methods 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 150000002431 hydrogen Chemical class 0.000 description 4
- 239000010813 municipal solid waste Substances 0.000 description 4
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 235000017550 sodium carbonate Nutrition 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 238000010189 synthetic method Methods 0.000 description 4
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- HVBSAKJJOYLTQU-UHFFFAOYSA-N 4-aminobenzenesulfonic acid Chemical compound NC1=CC=C(S(O)(=O)=O)C=C1 HVBSAKJJOYLTQU-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229910052500 inorganic mineral Inorganic materials 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 239000011707 mineral Substances 0.000 description 3
- 235000010755 mineral Nutrition 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 238000011914 asymmetric synthesis Methods 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 150000004678 hydrides Chemical class 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000011968 lewis acid catalyst Substances 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 238000013507 mapping Methods 0.000 description 2
- ALBSWLMUHHZLLR-UHFFFAOYSA-N methyl 2-nitroacetate Chemical class COC(=O)C[N+]([O-])=O ALBSWLMUHHZLLR-UHFFFAOYSA-N 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 239000003880 polar aprotic solvent Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 229950000244 sulfanilic acid Drugs 0.000 description 2
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Chemical compound O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- LKUDPHPHKOZXCD-UHFFFAOYSA-N 1,3,5-trimethoxybenzene Chemical compound COC1=CC(OC)=CC(OC)=C1 LKUDPHPHKOZXCD-UHFFFAOYSA-N 0.000 description 1
- KHEVPDFAQFJIGK-UHFFFAOYSA-N 2-sulfooxyethanesulfonic acid Chemical compound OS(=O)(=O)CCOS(O)(=O)=O KHEVPDFAQFJIGK-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- XYZZKVRWGOWVGO-UHFFFAOYSA-N Glycerol-phosphate Chemical class OP(O)(O)=O.OCC(O)CO XYZZKVRWGOWVGO-UHFFFAOYSA-N 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 239000005864 Sulphur Substances 0.000 description 1
- 230000004308 accommodation Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 125000001204 arachidyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- CUBCNYWQJHBXIY-UHFFFAOYSA-N benzoic acid;2-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=CC=C1.OC(=O)C1=CC=CC=C1O CUBCNYWQJHBXIY-UHFFFAOYSA-N 0.000 description 1
- AZWXAPCAJCYGIA-UHFFFAOYSA-N bis(2-methylpropyl)alumane Chemical compound CC(C)C[AlH]CC(C)C AZWXAPCAJCYGIA-UHFFFAOYSA-N 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000000151 deposition Methods 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N diisobutylaluminium hydride Substances CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- LBAQSKZHMLAFHH-UHFFFAOYSA-N ethoxyethane;hydron;chloride Chemical compound Cl.CCOCC LBAQSKZHMLAFHH-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229950006191 gluconic acid Drugs 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 229940045996 isethionic acid Drugs 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- RXUUYDXKHLUSHV-UHFFFAOYSA-N magnesium;trifluoromethanesulfonic acid Chemical compound [Mg].OS(=O)(=O)C(F)(F)F RXUUYDXKHLUSHV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- RLLPVAHGXHCWKJ-UHFFFAOYSA-N permethrin Chemical compound CC1(C)C(C=C(Cl)Cl)C1C(=O)OCC1=CC=CC(OC=2C=CC=CC=2)=C1 RLLPVAHGXHCWKJ-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- RPACBEVZENYWOL-XFULWGLBSA-M sodium;(2r)-2-[6-(4-chlorophenoxy)hexyl]oxirane-2-carboxylate Chemical compound [Na+].C=1C=C(Cl)C=CC=1OCCCCCC[C@]1(C(=O)[O-])CO1 RPACBEVZENYWOL-XFULWGLBSA-M 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pyrrole Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
















| 文件名称 | 替换页 | 被替换页 |
| 权利要求书 | 4 | 5 |

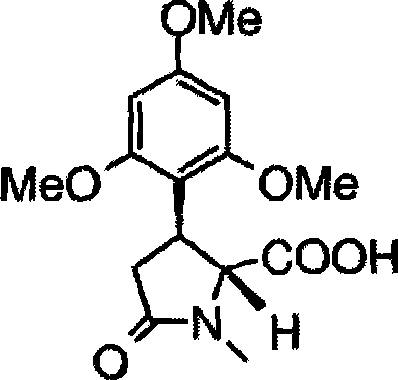



Claims (13)








Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2006/052294 WO2008007169A1 (en) | 2006-07-07 | 2006-07-07 | An enantioselective synthesis of pyrrolidine-substituted flavones |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101484445A true CN101484445A (zh) | 2009-07-15 |
| CN101484445B CN101484445B (zh) | 2012-09-05 |
Family
ID=37546592
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN200680055257.0A Expired - Fee Related CN101484445B (zh) | 2006-07-07 | 2006-07-07 | 吡咯烷取代的黄酮的对映选择性合成 |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US7951961B2 (zh) |
| EP (1) | EP2041121B1 (zh) |
| JP (1) | JP5006396B2 (zh) |
| KR (1) | KR101285957B1 (zh) |
| CN (1) | CN101484445B (zh) |
| AR (1) | AR061964A1 (zh) |
| AT (1) | ATE517892T1 (zh) |
| AU (1) | AU2006346193C1 (zh) |
| BR (1) | BRPI0621851A2 (zh) |
| CA (1) | CA2658215C (zh) |
| DK (1) | DK2041121T3 (zh) |
| HK (1) | HK1126762A1 (zh) |
| IL (1) | IL196298A0 (zh) |
| MX (1) | MX2009000109A (zh) |
| NZ (1) | NZ573841A (zh) |
| PT (1) | PT2041121E (zh) |
| TW (1) | TWI391388B (zh) |
| WO (1) | WO2008007169A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104487429A (zh) * | 2012-07-27 | 2015-04-01 | 爱默蕾大学 | 杂环黄酮衍生物、组合物及与其相关的方法 |
| CN104725279A (zh) * | 2015-02-12 | 2015-06-24 | 威海迪素制药有限公司 | 一种N-Boc-联苯丙氨酸衍生物的制备方法 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7884127B2 (en) | 2002-07-08 | 2011-02-08 | Pirimal Life Sciences Ltd. | Inhibitors of cyclin dependent kinases and their use |
| LV14179B (lv) * | 2008-12-03 | 2010-08-20 | Ivars Kalvins | 2,2'-ciklopropilidēn-bis-oksazolīnu reģenerēšana |
| TWI461194B (zh) | 2009-05-05 | 2014-11-21 | Piramal Entpr Ltd | 吡咯啶取代黃酮作為輻射致敏劑 |
| WO2012069972A1 (en) | 2010-11-19 | 2012-05-31 | Piramal Life Sciences Limited | A pharmaceutical combination for the treatment of breast cancer |
| KR101264012B1 (ko) * | 2010-12-31 | 2013-05-13 | 주식회사 코리아나화장품 | 플라바논 화합물 및 그 제조방법, 이로부터 제조된 화장료 조성물 |
| WO2012123889A1 (en) | 2011-03-14 | 2012-09-20 | Piramal Healthcare Limited | A synergistic pharmaceutical combination for the treatment of pancreatic cancer |
| EP2723449A2 (en) | 2011-06-24 | 2014-04-30 | Piramal Enterprises Limited | Compounds for the treatment of cancers associated with human papillomavirus |
| WO2013105056A1 (en) | 2012-01-13 | 2013-07-18 | Piramal Enterprises Limited | Pyrrolidine- substituted flavone derivatives for prevention or treatment of oral mucositis |
| JP5988305B2 (ja) * | 2013-03-29 | 2016-09-07 | 国立大学法人山口大学 | 光学活性α−ブロモベンゾイル酢酸エステル類の製造方法 |
| EP3019166B1 (en) | 2013-07-12 | 2019-05-08 | Piramal Enterprises Limited | A pharmaceutical combination for the treatment of melanoma |
| US10442807B2 (en) * | 2015-05-12 | 2019-10-15 | Fmc Corporation | Aryl substituted bicyclic compounds as herbicides |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5849733A (en) * | 1996-05-10 | 1998-12-15 | Bristol-Myers Squibb Co. | 2-thio or 2-oxo flavopiridol analogs |
| CN1433410A (zh) * | 2000-05-03 | 2003-07-30 | 株式会社Lg生命科学 | 具有3-羟基色烯-4-酮结构的cdk抑制剂 |
| US6784167B2 (en) * | 2000-09-29 | 2004-08-31 | Bayer Pharmaceuticals Corporation | 17-beta-hydroxysteroid dehydrogenase-II inhibitors |
| TWI331034B (en) * | 2002-07-08 | 2010-10-01 | Piramal Life Sciences Ltd | Inhibitors of cyclin-dependent kinases and their use |
| BRPI0621777A2 (pt) | 2006-06-21 | 2013-03-12 | Piramal Life Sciences Ltd | derivados de flavona enantiomericamente puros para o tratamento de distérbios proliferativos e processos para sua preparaÇço |
-
2006
- 2006-07-07 JP JP2009518985A patent/JP5006396B2/ja not_active Expired - Fee Related
- 2006-07-07 MX MX2009000109A patent/MX2009000109A/es active IP Right Grant
- 2006-07-07 DK DK06766035.7T patent/DK2041121T3/da active
- 2006-07-07 EP EP06766035A patent/EP2041121B1/en not_active Not-in-force
- 2006-07-07 NZ NZ573841A patent/NZ573841A/en not_active IP Right Cessation
- 2006-07-07 WO PCT/IB2006/052294 patent/WO2008007169A1/en active Application Filing
- 2006-07-07 PT PT06766035T patent/PT2041121E/pt unknown
- 2006-07-07 CA CA2658215A patent/CA2658215C/en active Active
- 2006-07-07 US US12/307,645 patent/US7951961B2/en active Active
- 2006-07-07 BR BRPI0621851-2A patent/BRPI0621851A2/pt not_active IP Right Cessation
- 2006-07-07 CN CN200680055257.0A patent/CN101484445B/zh not_active Expired - Fee Related
- 2006-07-07 KR KR1020097002349A patent/KR101285957B1/ko not_active IP Right Cessation
- 2006-07-07 AT AT06766035T patent/ATE517892T1/de active
- 2006-07-07 AU AU2006346193A patent/AU2006346193C1/en not_active Ceased
-
2007
- 2007-06-22 TW TW096122651A patent/TWI391388B/zh not_active IP Right Cessation
- 2007-07-06 AR ARP070103023A patent/AR061964A1/es not_active Application Discontinuation
-
2008
- 2008-12-31 IL IL196298A patent/IL196298A0/en unknown
-
2009
- 2009-06-22 HK HK09105577.1A patent/HK1126762A1/xx not_active IP Right Cessation
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104487429A (zh) * | 2012-07-27 | 2015-04-01 | 爱默蕾大学 | 杂环黄酮衍生物、组合物及与其相关的方法 |
| CN104487429B (zh) * | 2012-07-27 | 2017-07-14 | 爱默蕾大学 | 杂环黄酮衍生物、组合物及与其相关的方法 |
| CN104725279A (zh) * | 2015-02-12 | 2015-06-24 | 威海迪素制药有限公司 | 一种N-Boc-联苯丙氨酸衍生物的制备方法 |
| CN104725279B (zh) * | 2015-02-12 | 2018-03-02 | 威海迪素制药有限公司 | 一种N‑Boc‑联苯丙氨酸衍生物的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| DK2041121T3 (da) | 2011-11-14 |
| NZ573841A (en) | 2011-11-25 |
| AU2006346193A1 (en) | 2008-01-17 |
| KR101285957B1 (ko) | 2013-07-12 |
| AU2006346193C1 (en) | 2014-02-20 |
| CA2658215A1 (en) | 2008-01-17 |
| CA2658215C (en) | 2013-08-27 |
| MX2009000109A (es) | 2009-03-16 |
| HK1126762A1 (en) | 2009-09-11 |
| ATE517892T1 (de) | 2011-08-15 |
| AU2006346193A2 (en) | 2009-02-12 |
| BRPI0621851A2 (pt) | 2013-01-29 |
| JP5006396B2 (ja) | 2012-08-22 |
| IL196298A0 (en) | 2009-09-22 |
| JP2009542796A (ja) | 2009-12-03 |
| US7951961B2 (en) | 2011-05-31 |
| EP2041121A1 (en) | 2009-04-01 |
| EP2041121B1 (en) | 2011-07-27 |
| US20100113803A1 (en) | 2010-05-06 |
| CN101484445B (zh) | 2012-09-05 |
| AR061964A1 (es) | 2008-08-10 |
| TW200815413A (en) | 2008-04-01 |
| WO2008007169A1 (en) | 2008-01-17 |
| PT2041121E (pt) | 2011-09-12 |
| TWI391388B (zh) | 2013-04-01 |
| KR20090033465A (ko) | 2009-04-03 |
| AU2006346193B2 (en) | 2012-11-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101484445B (zh) | 吡咯烷取代的黄酮的对映选择性合成 | |
| DE602005000187T2 (de) | Verfahren zur Herstellung von (1S)-4,5-Dimethoxy-1-(methylaminomethyl)-benzocyclobutan und deren Säureadditionssalze, sowie ihre Verwendung für die Synthese von Ivabradin und deren Säureadditionssalze von pharmazeutischen verträglichen Säure | |
| US20110130463A1 (en) | Deuterium-enriched bupropion | |
| BRPI0612138B1 (pt) | compostos éter aminocicloexílicos e método para fazer os mesmos | |
| CN102875537A (zh) | 一种新的抗血栓药物的制备方法 | |
| DE69532849T2 (de) | Verfahren zum Epoxidieren prochiraler Olefine | |
| CN102757431A (zh) | 一种合成西他列汀的新方法 | |
| EP3063154B1 (en) | Cross-coupling of unactivated secondary boronic acids | |
| EP0413667B1 (de) | Halogenalkylphenyl-Alkohole, -Ketone und deren Hydrate | |
| CN109912569B (zh) | 艾司奥美拉唑镁三水合物及其中间体的新制备方法 | |
| DE10155018A1 (de) | Deuterierte Pyrazolopyrimidinone sowie diese Verbindungen enthaltende Arzneimittel | |
| JPS61171490A (ja) | グリセリルホスホリルコリンのジアシル誘導体 | |
| DE60004850T2 (de) | Verfahren zur herstellung von (r) und (s)-aminocarnitin und deren derivative aus d- und l-aspargin säure | |
| WO2008157565A1 (en) | Deuterium-enriched ezetimibe | |
| CN101979378B (zh) | 一种手性γ-内酰胺化合物的合成方法 | |
| US20080318904A1 (en) | Deuterium-enriched tenofovir | |
| JP2021531331A (ja) | チアジアゾール誘導体及びgls1阻害剤としてのその使用 | |
| WO2008157271A1 (en) | Deuterium-enriched escitalopram | |
| CN101384546A (zh) | 用于制备酰胺的方法 | |
| JPH03246277A (ja) | オクタヒドロイソキノリン誘導体のラセミ化法 | |
| US20080318964A1 (en) | Deuterium-enriched eszopiclone | |
| JP2012001454A (ja) | サルメテロールの簡便製造法 | |
| DE3735641A1 (de) | (+)-1-isopropylamino-3-(o-(pyrrol-1-yl) -phenoxy)-propanol, verfahren zu seiner herstellung sowie pharmazeutische praeparate und deren verwendung | |
| JPS63115858A (ja) | 医薬組成物 | |
| EP3015472A1 (en) | Compound, manufacturing method therefor, and method for manufacturing optically active -aminophosphonate derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| ASS | Succession or assignment of patent right |
Owner name: PIRAMAL HEALTHCARE UK LTMITED Free format text: FORMER OWNER: NICHOLAS PIRAMAL INDIA LTD. Effective date: 20130617 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| C56 | Change in the name or address of the patentee |
Owner name: PIRAMAL ENTERPRISES LTD. Free format text: FORMER NAME: PIRAMAL HEALTHCARE UK LTMITED |
|
| CP03 | Change of name, title or address |
Address after: Mumbai Patentee after: PIRAMAL ENTERPRISES LTD. Address before: Mumbai Patentee before: Piramal healthcare Ltd. |
|
| TR01 | Transfer of patent right |
Effective date of registration: 20130617 Address after: Mumbai Patentee after: Piramal healthcare Ltd. Address before: Maharashtra Patentee before: PIRAMAL LIFE SCIENCES LTD. |
|
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20120905 Termination date: 20160707 |
|
| CF01 | Termination of patent right due to non-payment of annual fee |