WO2022268230A1 - 作为kif18a抑制剂的化合物 - Google Patents
作为kif18a抑制剂的化合物 Download PDFInfo
- Publication number
- WO2022268230A1 WO2022268230A1 PCT/CN2022/101694 CN2022101694W WO2022268230A1 WO 2022268230 A1 WO2022268230 A1 WO 2022268230A1 CN 2022101694 W CN2022101694 W CN 2022101694W WO 2022268230 A1 WO2022268230 A1 WO 2022268230A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- mmol
- formula
- ethyl acetate
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 443
- 239000003112 inhibitor Substances 0.000 title abstract description 11
- 101001091231 Homo sapiens Kinesin-like protein KIF18A Proteins 0.000 claims abstract description 33
- 102100034895 Kinesin-like protein KIF18A Human genes 0.000 claims abstract description 33
- 201000010099 disease Diseases 0.000 claims abstract description 17
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 17
- 230000000694 effects Effects 0.000 claims abstract description 15
- 150000003839 salts Chemical class 0.000 claims abstract description 15
- 239000012453 solvate Substances 0.000 claims abstract description 8
- 230000003287 optical effect Effects 0.000 claims abstract description 7
- 229940002612 prodrug Drugs 0.000 claims abstract description 7
- 239000000651 prodrug Substances 0.000 claims abstract description 7
- 229940126262 KIF18A Drugs 0.000 claims abstract 3
- 125000000217 alkyl group Chemical group 0.000 claims description 95
- 125000000623 heterocyclic group Chemical group 0.000 claims description 54
- 239000012634 fragment Substances 0.000 claims description 52
- -1 NR d R d Chemical group 0.000 claims description 49
- 229910052739 hydrogen Inorganic materials 0.000 claims description 44
- 239000001257 hydrogen Substances 0.000 claims description 44
- 229910052757 nitrogen Inorganic materials 0.000 claims description 41
- 125000000304 alkynyl group Chemical group 0.000 claims description 38
- 150000002431 hydrogen Chemical class 0.000 claims description 38
- 229910052736 halogen Inorganic materials 0.000 claims description 36
- 150000002367 halogens Chemical class 0.000 claims description 36
- 125000003342 alkenyl group Chemical group 0.000 claims description 35
- 125000003118 aryl group Chemical group 0.000 claims description 29
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 26
- 125000001072 heteroaryl group Chemical group 0.000 claims description 26
- 125000004432 carbon atom Chemical group C* 0.000 claims description 25
- 229910052760 oxygen Inorganic materials 0.000 claims description 25
- 229920006395 saturated elastomer Polymers 0.000 claims description 25
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 19
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 18
- 229910052799 carbon Inorganic materials 0.000 claims description 17
- 229910052717 sulfur Inorganic materials 0.000 claims description 17
- 125000001424 substituent group Chemical group 0.000 claims description 16
- 125000005842 heteroatom Chemical group 0.000 claims description 15
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 15
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 claims description 14
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 14
- 239000008194 pharmaceutical composition Substances 0.000 claims description 14
- 239000000126 substance Substances 0.000 claims description 14
- 125000003545 alkoxy group Chemical group 0.000 claims description 12
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 9
- 239000001301 oxygen Substances 0.000 claims description 9
- 206010028980 Neoplasm Diseases 0.000 claims description 7
- 229910052731 fluorine Inorganic materials 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 125000005330 8 membered heterocyclic group Chemical group 0.000 claims description 5
- 206010006187 Breast cancer Diseases 0.000 claims description 5
- 208000026310 Breast neoplasm Diseases 0.000 claims description 5
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 5
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 5
- 201000010881 cervical cancer Diseases 0.000 claims description 5
- 125000004122 cyclic group Chemical group 0.000 claims description 5
- 150000004677 hydrates Chemical class 0.000 claims description 5
- 206010005003 Bladder cancer Diseases 0.000 claims description 4
- 206010009944 Colon cancer Diseases 0.000 claims description 4
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 4
- 206010033128 Ovarian cancer Diseases 0.000 claims description 4
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 4
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 4
- 206010060862 Prostate cancer Diseases 0.000 claims description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 4
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 4
- 208000029742 colonic neoplasm Diseases 0.000 claims description 4
- 229910052805 deuterium Inorganic materials 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 239000011737 fluorine Substances 0.000 claims description 4
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 4
- 201000002528 pancreatic cancer Diseases 0.000 claims description 4
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 4
- 241000712461 unidentified influenza virus Species 0.000 claims description 4
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 4
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 claims description 3
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 3
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 3
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 3
- 208000032612 Glial tumor Diseases 0.000 claims description 3
- 206010018338 Glioma Diseases 0.000 claims description 3
- 208000005234 Granulosa Cell Tumor Diseases 0.000 claims description 3
- 208000017604 Hodgkin disease Diseases 0.000 claims description 3
- 208000021519 Hodgkin lymphoma Diseases 0.000 claims description 3
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 3
- 208000028018 Lymphocytic leukaemia Diseases 0.000 claims description 3
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 3
- 208000015634 Rectal Neoplasms Diseases 0.000 claims description 3
- 206010038389 Renal cancer Diseases 0.000 claims description 3
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 3
- 206010041067 Small cell lung cancer Diseases 0.000 claims description 3
- 206010041660 Splenomegaly Diseases 0.000 claims description 3
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 claims description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 3
- 206010042971 T-cell lymphoma Diseases 0.000 claims description 3
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 claims description 3
- 206010043515 Throat cancer Diseases 0.000 claims description 3
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 3
- 208000036142 Viral infection Diseases 0.000 claims description 3
- 201000006491 bone marrow cancer Diseases 0.000 claims description 3
- 229910052794 bromium Inorganic materials 0.000 claims description 3
- 239000000460 chlorine Substances 0.000 claims description 3
- 229910052801 chlorine Inorganic materials 0.000 claims description 3
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 3
- 201000009409 embryonal rhabdomyosarcoma Diseases 0.000 claims description 3
- 201000004101 esophageal cancer Diseases 0.000 claims description 3
- 206010017758 gastric cancer Diseases 0.000 claims description 3
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 claims description 3
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 claims description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 3
- 201000007450 intrahepatic cholangiocarcinoma Diseases 0.000 claims description 3
- 201000010982 kidney cancer Diseases 0.000 claims description 3
- 201000005249 lung adenocarcinoma Diseases 0.000 claims description 3
- 201000005243 lung squamous cell carcinoma Diseases 0.000 claims description 3
- 208000003747 lymphoid leukemia Diseases 0.000 claims description 3
- 201000001441 melanoma Diseases 0.000 claims description 3
- 206010028537 myelofibrosis Diseases 0.000 claims description 3
- 208000025113 myeloid leukemia Diseases 0.000 claims description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 3
- 206010038038 rectal cancer Diseases 0.000 claims description 3
- 201000001275 rectum cancer Diseases 0.000 claims description 3
- 201000000849 skin cancer Diseases 0.000 claims description 3
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 3
- 201000011549 stomach cancer Diseases 0.000 claims description 3
- 208000011580 syndromic disease Diseases 0.000 claims description 3
- 201000002510 thyroid cancer Diseases 0.000 claims description 3
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 3
- 230000009385 viral infection Effects 0.000 claims description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 2
- 206010048643 Hypereosinophilic syndrome Diseases 0.000 claims description 2
- 206010024305 Leukaemia monocytic Diseases 0.000 claims description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 2
- 150000001335 aliphatic alkanes Chemical class 0.000 claims description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 2
- 125000000262 haloalkenyl group Chemical group 0.000 claims description 2
- 125000000232 haloalkynyl group Chemical group 0.000 claims description 2
- 201000006894 monocytic leukemia Diseases 0.000 claims description 2
- 125000004437 phosphorous atom Chemical group 0.000 claims description 2
- 125000004434 sulfur atom Chemical group 0.000 claims description 2
- 206010025323 Lymphomas Diseases 0.000 claims 1
- 208000024891 symptom Diseases 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 664
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 251
- 239000000243 solution Substances 0.000 description 235
- 238000006243 chemical reaction Methods 0.000 description 227
- 235000019439 ethyl acetate Nutrition 0.000 description 221
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 218
- 230000002829 reductive effect Effects 0.000 description 201
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 192
- 239000011541 reaction mixture Substances 0.000 description 149
- 239000012299 nitrogen atmosphere Substances 0.000 description 141
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 138
- 239000012043 crude product Substances 0.000 description 138
- 239000007787 solid Substances 0.000 description 127
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 118
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 114
- 239000003208 petroleum Substances 0.000 description 108
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 108
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 104
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 99
- 238000010898 silica gel chromatography Methods 0.000 description 98
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 90
- 239000012044 organic layer Substances 0.000 description 83
- 238000005481 NMR spectroscopy Methods 0.000 description 80
- 230000015572 biosynthetic process Effects 0.000 description 73
- 238000003786 synthesis reaction Methods 0.000 description 73
- 239000000203 mixture Substances 0.000 description 71
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 64
- 238000000746 purification Methods 0.000 description 64
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 56
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 56
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 56
- 238000000926 separation method Methods 0.000 description 56
- 229910000160 potassium phosphate Inorganic materials 0.000 description 54
- 235000011009 potassium phosphates Nutrition 0.000 description 54
- 108010077895 Sarcosine Proteins 0.000 description 52
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 49
- 229940043230 sarcosine Drugs 0.000 description 48
- 239000010410 layer Substances 0.000 description 43
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 42
- 238000012746 preparative thin layer chromatography Methods 0.000 description 41
- 239000012074 organic phase Substances 0.000 description 35
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 26
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 26
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 25
- 239000012265 solid product Substances 0.000 description 25
- 238000000034 method Methods 0.000 description 24
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 23
- 239000000706 filtrate Substances 0.000 description 23
- 239000000047 product Substances 0.000 description 21
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 19
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 19
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 18
- 210000004027 cell Anatomy 0.000 description 18
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 18
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 16
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 16
- 238000003756 stirring Methods 0.000 description 16
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 14
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 14
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 14
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 235000019198 oils Nutrition 0.000 description 13
- VAVHMEQFYYBAPR-ITWZMISCSA-N (e,3r,5s)-7-[4-(4-fluorophenyl)-1-phenyl-2-propan-2-ylpyrrol-3-yl]-3,5-dihydroxyhept-6-enoic acid Chemical compound CC(C)C1=C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)C(C=2C=CC(F)=CC=2)=CN1C1=CC=CC=C1 VAVHMEQFYYBAPR-ITWZMISCSA-N 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 10
- 125000003367 polycyclic group Chemical group 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 9
- 239000004202 carbamide Substances 0.000 description 9
- HBENZIXOGRCSQN-VQWWACLZSA-N (1S,2S,6R,14R,15R,16R)-5-(cyclopropylmethyl)-16-[(2S)-2-hydroxy-3,3-dimethylpentan-2-yl]-15-methoxy-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11-trien-11-ol Chemical compound N1([C@@H]2CC=3C4=C(C(=CC=3)O)O[C@H]3[C@@]5(OC)CC[C@@]2([C@@]43CC1)C[C@@H]5[C@](C)(O)C(C)(C)CC)CC1CC1 HBENZIXOGRCSQN-VQWWACLZSA-N 0.000 description 8
- MITGKKFYIJJQGL-UHFFFAOYSA-N 9-(4-chlorobenzoyl)-6-methylsulfonyl-2,3-dihydro-1H-carbazol-4-one Chemical compound ClC1=CC=C(C(=O)N2C3=CC=C(C=C3C=3C(CCCC2=3)=O)S(=O)(=O)C)C=C1 MITGKKFYIJJQGL-UHFFFAOYSA-N 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- 239000012295 chemical reaction liquid Substances 0.000 description 8
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 8
- 239000011259 mixed solution Substances 0.000 description 8
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 8
- 125000006413 ring segment Chemical group 0.000 description 8
- 238000006467 substitution reaction Methods 0.000 description 8
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 7
- 102000010638 Kinesin Human genes 0.000 description 7
- 108010063296 Kinesin Proteins 0.000 description 7
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 7
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 7
- 230000002401 inhibitory effect Effects 0.000 description 7
- HBEDNENASUYMPO-LJQANCHMSA-N n-hydroxy-4-[[(2r)-3-oxo-2-(thiophen-2-ylmethyl)-2,4-dihydroquinoxalin-1-yl]methyl]benzamide Chemical compound C1=CC(C(=O)NO)=CC=C1CN1C2=CC=CC=C2NC(=O)[C@H]1CC1=CC=CS1 HBEDNENASUYMPO-LJQANCHMSA-N 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 239000011593 sulfur Chemical group 0.000 description 7
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 6
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 6
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 6
- HIHOEGPXVVKJPP-JTQLQIEISA-N 5-fluoro-2-[[(1s)-1-(5-fluoropyridin-2-yl)ethyl]amino]-6-[(5-methyl-1h-pyrazol-3-yl)amino]pyridine-3-carbonitrile Chemical compound N([C@@H](C)C=1N=CC(F)=CC=1)C(C(=CC=1F)C#N)=NC=1NC=1C=C(C)NN=1 HIHOEGPXVVKJPP-JTQLQIEISA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 6
- 229910000024 caesium carbonate Inorganic materials 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 229940125758 compound 15 Drugs 0.000 description 6
- 229940125877 compound 31 Drugs 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 5
- FJZNNKJZHQFMCK-LRDDRELGSA-N 1-[(3S,4R)-4-(2,6-difluoro-4-methoxyphenyl)-2-oxopyrrolidin-3-yl]-3-phenylurea Chemical compound C1(=CC(=CC(=C1[C@H]1[C@@H](C(=O)NC1)NC(=O)NC1=CC=CC=C1)F)OC)F FJZNNKJZHQFMCK-LRDDRELGSA-N 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 230000004663 cell proliferation Effects 0.000 description 5
- 239000012230 colorless oil Substances 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- XAYGBKHKBBXDAK-UHFFFAOYSA-N n-[4-[4-(cyclopropylmethyl)piperazine-1-carbonyl]phenyl]quinoline-8-sulfonamide Chemical compound C=1C=C(NS(=O)(=O)C=2C3=NC=CC=C3C=CC=2)C=CC=1C(=O)N(CC1)CCN1CC1CC1 XAYGBKHKBBXDAK-UHFFFAOYSA-N 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 238000010791 quenching Methods 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 5
- 210000004881 tumor cell Anatomy 0.000 description 5
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 4
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 4
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 4
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- WCDLCPLAAKUJNY-UHFFFAOYSA-N 4-[4-[3-(1h-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidin-6-yl]phenyl]morpholine Chemical compound C1COCCN1C1=CC=C(C2=CN3N=CC(=C3N=C2)C2=CNN=C2)C=C1 WCDLCPLAAKUJNY-UHFFFAOYSA-N 0.000 description 4
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 4
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 208000037051 Chromosomal Instability Diseases 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 235000019270 ammonium chloride Nutrition 0.000 description 4
- 235000011114 ammonium hydroxide Nutrition 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 229910052796 boron Inorganic materials 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 230000032823 cell division Effects 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 229940125878 compound 36 Drugs 0.000 description 4
- 229940125807 compound 37 Drugs 0.000 description 4
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 4
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 4
- 239000006187 pill Substances 0.000 description 4
- TZSZZENYCISATO-WIOPSUGQSA-N rodatristat Chemical compound CCOC(=O)[C@@H]1CC2(CN1)CCN(CC2)c1cc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)nc(N)n1 TZSZZENYCISATO-WIOPSUGQSA-N 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 230000032258 transport Effects 0.000 description 4
- JRHPOFJADXHYBR-HTQZYQBOSA-N (1r,2r)-1-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CN[C@@H]1CCCC[C@H]1NC JRHPOFJADXHYBR-HTQZYQBOSA-N 0.000 description 3
- SLTBMTIRYMGWLX-XMMPIXPASA-N (2r)-2-[(4-chloroanilino)carbamoylamino]-3-(1h-indol-3-yl)-n-(2-phenylethyl)propanamide Chemical compound C1=CC(Cl)=CC=C1NNC(=O)N[C@@H](C(=O)NCCC=1C=CC=CC=1)CC1=CNC2=CC=CC=C12 SLTBMTIRYMGWLX-XMMPIXPASA-N 0.000 description 3
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 3
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 3
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 3
- MHSLDASSAFCCDO-UHFFFAOYSA-N 1-(5-tert-butyl-2-methylpyrazol-3-yl)-3-(4-pyridin-4-yloxyphenyl)urea Chemical compound CN1N=C(C(C)(C)C)C=C1NC(=O)NC(C=C1)=CC=C1OC1=CC=NC=C1 MHSLDASSAFCCDO-UHFFFAOYSA-N 0.000 description 3
- WGFNXGPBPIJYLI-UHFFFAOYSA-N 2,6-difluoro-3-[(3-fluorophenyl)sulfonylamino]-n-(3-methoxy-1h-pyrazolo[3,4-b]pyridin-5-yl)benzamide Chemical compound C1=C2C(OC)=NNC2=NC=C1NC(=O)C(C=1F)=C(F)C=CC=1NS(=O)(=O)C1=CC=CC(F)=C1 WGFNXGPBPIJYLI-UHFFFAOYSA-N 0.000 description 3
- MVXAKOGJWVQPKC-UHFFFAOYSA-N 5-(3-ethynyl-5-fluorophenyl)-2-pyridin-2-yl-4,6,7,8-tetrahydro-[1,3]oxazolo[4,5-c]azepine Chemical compound FC1=CC(C#C)=CC(N2CC=3N=C(OC=3CCC2)C=2N=CC=CC=2)=C1 MVXAKOGJWVQPKC-UHFFFAOYSA-N 0.000 description 3
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 3
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- PKMUHQIDVVOXHQ-HXUWFJFHSA-N C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O Chemical compound C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O PKMUHQIDVVOXHQ-HXUWFJFHSA-N 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- IOSLINNLJFQMFF-XMMPIXPASA-N [(2R)-1-[[4-[[3-[(4-fluorophenyl)methylsulfanyl]phenoxy]methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound FC1=CC=C(CSC=2C=C(OCC3=CC=C(CN4[C@H](CCC4)CO)C=C3)C=CC=2)C=C1 IOSLINNLJFQMFF-XMMPIXPASA-N 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- OSVHLUXLWQLPIY-KBAYOESNSA-N butyl 2-[(6aR,9R,10aR)-1-hydroxy-9-(hydroxymethyl)-6,6-dimethyl-6a,7,8,9,10,10a-hexahydrobenzo[c]chromen-3-yl]-2-methylpropanoate Chemical compound C(CCC)OC(C(C)(C)C1=CC(=C2[C@H]3[C@H](C(OC2=C1)(C)C)CC[C@H](C3)CO)O)=O OSVHLUXLWQLPIY-KBAYOESNSA-N 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 229940126209 compound 43b Drugs 0.000 description 3
- 229940125900 compound 59 Drugs 0.000 description 3
- 229940126179 compound 72 Drugs 0.000 description 3
- LCSNDSFWVKMJCT-UHFFFAOYSA-N dicyclohexyl-(2-phenylphenyl)phosphane Chemical group C1CCCCC1P(C=1C(=CC=CC=1)C=1C=CC=CC=1)C1CCCCC1 LCSNDSFWVKMJCT-UHFFFAOYSA-N 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- BJXYHBKEQFQVES-NWDGAFQWSA-N enpatoran Chemical compound N[C@H]1CN(C[C@H](C1)C(F)(F)F)C1=C2C=CC=NC2=C(C=C1)C#N BJXYHBKEQFQVES-NWDGAFQWSA-N 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 238000003197 gene knockdown Methods 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- JFOZKMSJYSPYLN-QHCPKHFHSA-N lifitegrast Chemical compound CS(=O)(=O)C1=CC=CC(C[C@H](NC(=O)C=2C(=C3CCN(CC3=CC=2Cl)C(=O)C=2C=C3OC=CC3=CC=2)Cl)C(O)=O)=C1 JFOZKMSJYSPYLN-QHCPKHFHSA-N 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 239000008247 solid mixture Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- 239000000080 wetting agent Substances 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 2
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- FANCTJAFZSYTIS-IQUVVAJASA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-4-(phenylsulfonimidoyl)butan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C([C@@H](C)[C@@H]1[C@]2(CCCC(/[C@@H]2CC1)=C\C=C\1C([C@@H](O)C[C@H](O)C/1)=C)C)CS(=N)(=O)C1=CC=CC=C1 FANCTJAFZSYTIS-IQUVVAJASA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 2
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 2
- PHDIJLFSKNMCMI-ITGJKDDRSA-N (3R,4S,5R,6R)-6-(hydroxymethyl)-4-(8-quinolin-6-yloxyoctoxy)oxane-2,3,5-triol Chemical compound OC[C@@H]1[C@H]([C@@H]([C@H](C(O1)O)O)OCCCCCCCCOC=1C=C2C=CC=NC2=CC=1)O PHDIJLFSKNMCMI-ITGJKDDRSA-N 0.000 description 2
- FNHHVPPSBFQMEL-KQHDFZBMSA-N (3S)-5-N-[(1S,5R)-3-hydroxy-6-bicyclo[3.1.0]hexanyl]-7-N,3-dimethyl-3-phenyl-2H-1-benzofuran-5,7-dicarboxamide Chemical compound CNC(=O)c1cc(cc2c1OC[C@@]2(C)c1ccccc1)C(=O)NC1[C@H]2CC(O)C[C@@H]12 FNHHVPPSBFQMEL-KQHDFZBMSA-N 0.000 description 2
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 2
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 2
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 2
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 2
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 2
- OMBVEVHRIQULKW-DNQXCXABSA-M (3r,5r)-7-[3-(4-fluorophenyl)-8-oxo-7-phenyl-1-propan-2-yl-5,6-dihydro-4h-pyrrolo[2,3-c]azepin-2-yl]-3,5-dihydroxyheptanoate Chemical compound O=C1C=2N(C(C)C)C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C(C=3C=CC(F)=CC=3)C=2CCCN1C1=CC=CC=C1 OMBVEVHRIQULKW-DNQXCXABSA-M 0.000 description 2
- QKLXBIHSGMPUQS-FGZHOGPDSA-M (3r,5r)-7-[4-(4-fluorophenyl)-2,5-dimethyl-1-phenylpyrrol-3-yl]-3,5-dihydroxyheptanoate Chemical compound CC1=C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)C(C=2C=CC(F)=CC=2)=C(C)N1C1=CC=CC=C1 QKLXBIHSGMPUQS-FGZHOGPDSA-M 0.000 description 2
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 2
- RXNPEQZHMGFNAY-GEALJGNFSA-N (5R)-4-[(1S,6R)-5-[(2S)-2-(4-chlorophenyl)-3-(propan-2-ylamino)propanoyl]-2,5-diazabicyclo[4.1.0]heptan-2-yl]-5-methyl-6,8-dihydro-5H-pyrido[2,3-d]pyrimidin-7-one Chemical compound C[C@@H]1CC(=O)NC2=C1C(=NC=N2)N3CCN([C@H]4[C@@H]3C4)C(=O)[C@H](CNC(C)C)C5=CC=C(C=C5)Cl RXNPEQZHMGFNAY-GEALJGNFSA-N 0.000 description 2
- QPAGOTNPJABYCP-FITNPZAZSA-N (E)-N-(oxan-4-yl)-N'-[[(3R)-1,2,3,4-tetrahydroisoquinolin-3-yl]methyl]-N'-[(8S)-5,6,7,8-tetrahydroquinolin-8-yl]but-2-ene-1,4-diamine Chemical compound O1CCC(CC1)NC\C=C\CN([C@H]1CCCC=2C=CC=NC1=2)C[C@@H]1NCC2=CC=CC=C2C1 QPAGOTNPJABYCP-FITNPZAZSA-N 0.000 description 2
- IGVKWAAPMVVTFX-BUHFOSPRSA-N (e)-octadec-5-en-7,9-diynoic acid Chemical compound CCCCCCCCC#CC#C\C=C\CCCC(O)=O IGVKWAAPMVVTFX-BUHFOSPRSA-N 0.000 description 2
- JNPGUXGVLNJQSQ-BGGMYYEUSA-M (e,3r,5s)-7-[4-(4-fluorophenyl)-1,2-di(propan-2-yl)pyrrol-3-yl]-3,5-dihydroxyhept-6-enoate Chemical compound CC(C)N1C(C(C)C)=C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)C(C=2C=CC(F)=CC=2)=C1 JNPGUXGVLNJQSQ-BGGMYYEUSA-M 0.000 description 2
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 2
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- MXPCZHSTOIKVST-UHFFFAOYSA-N 1-[3-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-[3-(2-hydroxyethoxy)phenyl]-2-oxo-1,8-naphthyridin-3-yl]urea;hydrochloride Chemical compound Cl.CC(C)C=1C=CC(N)=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OCCO)=C1 MXPCZHSTOIKVST-UHFFFAOYSA-N 0.000 description 2
- OXTVBHDILDPYAS-UHFFFAOYSA-N 1-[4-(aminomethyl)-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-[3-(3-hydroxypropoxy)phenyl]-2-oxo-1,8-naphthyridin-3-yl]urea;hydrochloride Chemical compound Cl.CC(C)C=1C=C(CN)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OCCCO)=C1 OXTVBHDILDPYAS-UHFFFAOYSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 2
- VCUXVXLUOHDHKK-UHFFFAOYSA-N 2-(2-aminopyrimidin-4-yl)-4-(2-chloro-4-methoxyphenyl)-1,3-thiazole-5-carboxamide Chemical compound ClC1=CC(OC)=CC=C1C1=C(C(N)=O)SC(C=2N=C(N)N=CC=2)=N1 VCUXVXLUOHDHKK-UHFFFAOYSA-N 0.000 description 2
- MSSQOQPKGAMUSY-LEAFIULHSA-N 2-[1-[2-[(4r,6s)-8-chloro-6-(2,3-dimethoxyphenyl)-4,6-dihydropyrrolo[1,2-a][4,1]benzoxazepin-4-yl]acetyl]piperidin-4-yl]acetic acid Chemical compound COC1=CC=CC([C@@H]2C3=CC(Cl)=CC=C3N3C=CC=C3[C@@H](CC(=O)N3CCC(CC(O)=O)CC3)O2)=C1OC MSSQOQPKGAMUSY-LEAFIULHSA-N 0.000 description 2
- AXHVEQQLKVIZLO-UHFFFAOYSA-N 2-[4-[2-[5-(2,2-dimethylbutyl)-1h-imidazol-2-yl]ethyl]phenyl]benzoic acid Chemical compound CCC(C)(C)CC1=CNC(CCC=2C=CC(=CC=2)C=2C(=CC=CC=2)C(O)=O)=N1 AXHVEQQLKVIZLO-UHFFFAOYSA-N 0.000 description 2
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 2
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 2
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 2
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 2
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 2
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 2
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 2
- LXBGSDVWAMZHDD-UHFFFAOYSA-N 2-methyl-1h-imidazole Chemical compound CC1=NC=CN1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 2
- DFRAKBCRUYUFNT-UHFFFAOYSA-N 3,8-dicyclohexyl-2,4,7,9-tetrahydro-[1,3]oxazino[5,6-h][1,3]benzoxazine Chemical compound C1CCCCC1N1CC(C=CC2=C3OCN(C2)C2CCCCC2)=C3OC1 DFRAKBCRUYUFNT-UHFFFAOYSA-N 0.000 description 2
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 2
- UNSHMXUHOHBLIQ-UHFFFAOYSA-N 3-[4-chloro-3-(2-methylphenoxy)naphthalen-1-yl]-6-(trifluoromethyl)-1H-pyrimidine-2,4-dione Chemical compound ClC1=C(C=C(C2=CC=CC=C12)N1C(NC(=CC1=O)C(F)(F)F)=O)OC1=C(C=CC=C1)C UNSHMXUHOHBLIQ-UHFFFAOYSA-N 0.000 description 2
- HCLQARMRCPEALF-DNQXCXABSA-N 3-[[(2r)-2-[(1r)-2-[[1-(1-benzothiophen-2-yl)-2-methylpropan-2-yl]amino]-1-hydroxyethyl]pyrrolidin-1-yl]methyl]benzonitrile Chemical compound C([C@@H]1[C@H](O)CNC(C)(CC=2SC3=CC=CC=C3C=2)C)CCN1CC1=CC=CC(C#N)=C1 HCLQARMRCPEALF-DNQXCXABSA-N 0.000 description 2
- MWDVCHRYCKXEBY-LBPRGKRZSA-N 3-chloro-n-[2-oxo-2-[[(1s)-1-phenylethyl]amino]ethyl]benzamide Chemical compound N([C@@H](C)C=1C=CC=CC=1)C(=O)CNC(=O)C1=CC=CC(Cl)=C1 MWDVCHRYCKXEBY-LBPRGKRZSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- OVDGUTHABMXVMI-UHFFFAOYSA-N 3-nitro-4-(propylamino)benzoic acid Chemical compound CCCNC1=CC=C(C(O)=O)C=C1[N+]([O-])=O OVDGUTHABMXVMI-UHFFFAOYSA-N 0.000 description 2
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 2
- RXCVUHMIWHRLDF-HXUWFJFHSA-N 5,8-dichloro-2-[(4-methoxy-6-methyl-2-oxo-1H-pyridin-3-yl)methyl]-7-[(R)-methoxy(oxetan-3-yl)methyl]-3,4-dihydroisoquinolin-1-one Chemical compound ClC1=C2CCN(C(C2=C(C(=C1)[C@@H](C1COC1)OC)Cl)=O)CC=1C(NC(=CC=1OC)C)=O RXCVUHMIWHRLDF-HXUWFJFHSA-N 0.000 description 2
- GDUANFXPOZTYKS-UHFFFAOYSA-N 6-bromo-8-[(2,6-difluoro-4-methoxybenzoyl)amino]-4-oxochromene-2-carboxylic acid Chemical compound FC1=CC(OC)=CC(F)=C1C(=O)NC1=CC(Br)=CC2=C1OC(C(O)=O)=CC2=O GDUANFXPOZTYKS-UHFFFAOYSA-N 0.000 description 2
- TYYCNSUODPEVQP-UHFFFAOYSA-N 8-[(4-fluorophenyl)sulfonylamino]-4-(3-pyridin-3-ylpropyl)octanoic acid Chemical compound C=1C=CN=CC=1CCCC(CCC(=O)O)CCCCNS(=O)(=O)C1=CC=C(F)C=C1 TYYCNSUODPEVQP-UHFFFAOYSA-N 0.000 description 2
- LPWKFUVWWQYLOD-UHFFFAOYSA-N 9-(dimethylamino)-3-(4-methylphenyl)pyrido[2,3]thieno[2,4-d]pyrimidin-4-one Chemical compound C1=2C(N(C)C)=CN=CC=2SC(C2=O)=C1N=CN2C1=CC=C(C)C=C1 LPWKFUVWWQYLOD-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 2
- ISMDILRWKSYCOD-GNKBHMEESA-N C(C1=CC=CC=C1)[C@@H]1NC(OCCCCCCCCCCCNC([C@@H](NC(C[C@@H]1O)=O)C(C)C)=O)=O Chemical compound C(C1=CC=CC=C1)[C@@H]1NC(OCCCCCCCCCCCNC([C@@H](NC(C[C@@H]1O)=O)C(C)C)=O)=O ISMDILRWKSYCOD-GNKBHMEESA-N 0.000 description 2
- 125000004649 C2-C8 alkynyl group Chemical group 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- 229940126639 Compound 33 Drugs 0.000 description 2
- 229940127007 Compound 39 Drugs 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 101001008993 Dictyostelium discoideum Kinesin-related protein 10 Proteins 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 239000005909 Kieselgur Substances 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- 102000029749 Microtubule Human genes 0.000 description 2
- 108091022875 Microtubule Proteins 0.000 description 2
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 2
- TZYWCYJVHRLUCT-VABKMULXSA-N N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal Chemical compound CC(C)C[C@@H](C=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)OCC1=CC=CC=C1 TZYWCYJVHRLUCT-VABKMULXSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- TZCCKCLHNUSAMQ-DUGSHLAESA-N NC(=O)C[C@H](NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](Cc2ccc(F)cc2)NC(=O)[C@H](Cc3c[nH]c4ccccc34)NC(=O)Cc5cccs5)C(=O)N Chemical compound NC(=O)C[C@H](NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](Cc2ccc(F)cc2)NC(=O)[C@H](Cc3c[nH]c4ccccc34)NC(=O)Cc5cccs5)C(=O)N TZCCKCLHNUSAMQ-DUGSHLAESA-N 0.000 description 2
- QOVYHDHLFPKQQG-NDEPHWFRSA-N N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O Chemical compound N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O QOVYHDHLFPKQQG-NDEPHWFRSA-N 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- KZVWEOXAPZXAFB-BQFCYCMXSA-N Temocaprilat Chemical compound C([C@H](N[C@H]1CS[C@@H](CN(C1=O)CC(=O)O)C=1SC=CC=1)C(O)=O)CC1=CC=CC=C1 KZVWEOXAPZXAFB-BQFCYCMXSA-N 0.000 description 2
- 102000004243 Tubulin Human genes 0.000 description 2
- 108090000704 Tubulin Proteins 0.000 description 2
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 2
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 2
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229940044683 chemotherapy drug Drugs 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125876 compound 15a Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125810 compound 20 Drugs 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940126208 compound 22 Drugs 0.000 description 2
- 229940125961 compound 24 Drugs 0.000 description 2
- 229940125846 compound 25 Drugs 0.000 description 2
- 229940125851 compound 27 Drugs 0.000 description 2
- 229940127204 compound 29 Drugs 0.000 description 2
- 229940127573 compound 38 Drugs 0.000 description 2
- 229940126540 compound 41 Drugs 0.000 description 2
- 229940125936 compound 42 Drugs 0.000 description 2
- 229940125844 compound 46 Drugs 0.000 description 2
- 229940127271 compound 49 Drugs 0.000 description 2
- 229940126545 compound 53 Drugs 0.000 description 2
- 229940127113 compound 57 Drugs 0.000 description 2
- 229940125873 compound 60b Drugs 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 235000008504 concentrate Nutrition 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- GWNFQAKCJYEJEW-UHFFFAOYSA-N ethyl 3-[8-[[4-methyl-5-[(3-methyl-4-oxophthalazin-1-yl)methyl]-1,2,4-triazol-3-yl]sulfanyl]octanoylamino]benzoate Chemical compound CCOC(=O)C1=CC(NC(=O)CCCCCCCSC2=NN=C(CC3=NN(C)C(=O)C4=CC=CC=C34)N2C)=CC=C1 GWNFQAKCJYEJEW-UHFFFAOYSA-N 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000003301 hydrolyzing effect Effects 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- ZBELDPMWYXDLNY-UHFFFAOYSA-N methyl 9-(4-bromo-2-fluoroanilino)-[1,3]thiazolo[5,4-f]quinazoline-2-carboximidate Chemical compound C12=C3SC(C(=N)OC)=NC3=CC=C2N=CN=C1NC1=CC=C(Br)C=C1F ZBELDPMWYXDLNY-UHFFFAOYSA-N 0.000 description 2
- XLSZMDLNRCVEIJ-UHFFFAOYSA-N methylimidazole Natural products CC1=CNC=N1 XLSZMDLNRCVEIJ-UHFFFAOYSA-N 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 210000004688 microtubule Anatomy 0.000 description 2
- 230000011278 mitosis Effects 0.000 description 2
- XONPDZSGENTBNJ-UHFFFAOYSA-N molecular hydrogen;sodium Chemical compound [Na].[H][H] XONPDZSGENTBNJ-UHFFFAOYSA-N 0.000 description 2
- YCJZWBZJSYLMPB-UHFFFAOYSA-N n-(2-chloropyrimidin-4-yl)-2,5-dimethyl-1-phenylimidazole-4-carboxamide Chemical compound CC=1N(C=2C=CC=CC=2)C(C)=NC=1C(=O)NC1=CC=NC(Cl)=N1 YCJZWBZJSYLMPB-UHFFFAOYSA-N 0.000 description 2
- NCNJHAHPOUUEQX-LDVROUIZSA-N n-[(2s,3r)-3-hydroxy-1-phenyl-4-[[(1r)-1-phenylethyl]amino]butan-2-yl]-4-(4-methylpiperazine-1-carbonyl)benzamide Chemical compound C([C@@H]([C@H](O)CN[C@H](C)C=1C=CC=CC=1)NC(=O)C=1C=CC(=CC=1)C(=O)N1CCN(C)CC1)C1=CC=CC=C1 NCNJHAHPOUUEQX-LDVROUIZSA-N 0.000 description 2
- NFPUARSXIMWASK-GOEBONIOSA-N n-[(5r,7s)-2-(3-chlorophenyl)-1-oxa-3-azaspiro[4.5]dec-2-en-7-yl]acetamide Chemical compound C1[C@@H](NC(=O)C)CCC[C@@]11OC(C=2C=C(Cl)C=CC=2)=NC1 NFPUARSXIMWASK-GOEBONIOSA-N 0.000 description 2
- YGBMCLDVRUGXOV-UHFFFAOYSA-N n-[6-[6-chloro-5-[(4-fluorophenyl)sulfonylamino]pyridin-3-yl]-1,3-benzothiazol-2-yl]acetamide Chemical compound C1=C2SC(NC(=O)C)=NC2=CC=C1C(C=1)=CN=C(Cl)C=1NS(=O)(=O)C1=CC=C(F)C=C1 YGBMCLDVRUGXOV-UHFFFAOYSA-N 0.000 description 2
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 235000013772 propylene glycol Nutrition 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000022983 regulation of cell cycle Effects 0.000 description 2
- 239000012363 selectfluor Substances 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000001509 sodium citrate Substances 0.000 description 2
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 230000007046 spindle assembly involved in mitosis Effects 0.000 description 2
- 125000003003 spiro group Chemical group 0.000 description 2
- 229940124530 sulfonamide Drugs 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- JYRWUSXRTGACLY-UHFFFAOYSA-N tert-butyl 4-[[3-(4-methylsulfonylphenyl)-[1,2]oxazolo[4,5-d]pyrimidin-7-yl]oxy]piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1OC1=NC=NC2=C1ON=C2C1=CC=C(S(C)(=O)=O)C=C1 JYRWUSXRTGACLY-UHFFFAOYSA-N 0.000 description 2
- FRACPXUHUTXLCX-BELIEFIBSA-N tert-butyl N-{1-[(1S)-1-{[(1R,2S)-1-(benzylcarbamoyl)-1-hydroxy-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]carbamoyl}-2-cyclopropylethyl]-2-oxopyridin-3-yl}carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CN(C1=O)[C@@H](CC2CC2)C(=O)N[C@@H](C[C@@H]3CCNC3=O)[C@H](C(=O)NCC4=CC=CC=C4)O FRACPXUHUTXLCX-BELIEFIBSA-N 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 1
- LAJAFFLJAJMYLK-CVOKMOJFSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[[(7s)-4-methoxy-7-morpholin-4-yl-6,7,8,9-tetrahydro-5h-benzo[7]annulen-3-yl]amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound N1([C@H]2CCC3=CC=C(C(=C3CC2)OC)NC=2N=C(C(=CN=2)Cl)N[C@H]2[C@H]([C@@]3([H])C[C@@]2(C=C3)[H])C(N)=O)CCOCC1 LAJAFFLJAJMYLK-CVOKMOJFSA-N 0.000 description 1
- SHAHPWSYJFYMRX-GDLCADMTSA-N (2S)-2-(4-{[(1R,2S)-2-hydroxycyclopentyl]methyl}phenyl)propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1C[C@@H]1[C@@H](O)CCC1 SHAHPWSYJFYMRX-GDLCADMTSA-N 0.000 description 1
- DIXMBHMNEHPFCX-MCMMXHMISA-N (2r)-2-[5-[6-amino-5-[(1r)-1-[5-fluoro-2-(triazol-2-yl)phenyl]ethoxy]pyridin-3-yl]-4-methyl-1,3-thiazol-2-yl]propane-1,2-diol Chemical compound O([C@H](C)C=1C(=CC=C(F)C=1)N1N=CC=N1)C(C(=NC=1)N)=CC=1C=1SC([C@](C)(O)CO)=NC=1C DIXMBHMNEHPFCX-MCMMXHMISA-N 0.000 description 1
- WLWNRAWQDZRXMB-YLFCFFPRSA-N (2r,3r,4r,5s)-n,3,4,5-tetrahydroxy-1-(4-phenoxyphenyl)sulfonylpiperidine-2-carboxamide Chemical compound ONC(=O)[C@H]1[C@@H](O)[C@H](O)[C@@H](O)CN1S(=O)(=O)C(C=C1)=CC=C1OC1=CC=CC=C1 WLWNRAWQDZRXMB-YLFCFFPRSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- HPJGEESDHAUUQR-SKGSPYGFSA-N (2s)-2-[[(2s)-5-(diaminomethylideneamino)-2-[[(2s)-1-[(2s)-5-(diaminomethylideneamino)-2-[[(2s)-2-[[(2s)-3-naphthalen-2-yl-2-(3-pyridin-3-ylpropanoylamino)propanoyl]amino]-3-phenylpropanoyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]amino]buta Chemical compound NC(=O)C[C@@H](C(N)=O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=C2C=CC=CC2=CC=1)NC(=O)CCC=1C=NC=CC=1)CC1=CC=CC=C1 HPJGEESDHAUUQR-SKGSPYGFSA-N 0.000 description 1
- LJIOTBMDLVHTBO-CUYJMHBOSA-N (2s)-2-amino-n-[(1r,2r)-1-cyano-2-[4-[4-(4-methylpiperazin-1-yl)sulfonylphenyl]phenyl]cyclopropyl]butanamide Chemical compound CC[C@H](N)C(=O)N[C@]1(C#N)C[C@@H]1C1=CC=C(C=2C=CC(=CC=2)S(=O)(=O)N2CCN(C)CC2)C=C1 LJIOTBMDLVHTBO-CUYJMHBOSA-N 0.000 description 1
- AEVBPXDFDKBGLT-YOUFYPILSA-N (2s,3s,4r,5r)-n-[2-[4-(diethoxyphosphorylmethyl)anilino]-2-oxoethyl]-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolane-2-carboxamide Chemical compound C1=CC(CP(=O)(OCC)OCC)=CC=C1NC(=O)CNC(=O)[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C(NC(=O)C=C2)=O)O1 AEVBPXDFDKBGLT-YOUFYPILSA-N 0.000 description 1
- TWYYFYNJOJGNFP-CUXYNZQBSA-N (2s,4r,5s,6s)-2-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-2-carbamoyl-4-[[(e,4s,6s)-4,6-dimethyloct-2-enoyl]oxymethyl]-5-hydroxy-1,3-dioxane-4,5,6-tricarboxylic acid Chemical compound O1[C@H](C(O)=O)[C@](C(O)=O)(O)[C@](COC(=O)/C=C/[C@@H](C)C[C@@H](C)CC)(C(O)=O)O[C@]1(C(N)=O)CCC(=C)[C@@H](OC(C)=O)[C@H](C)CC1=CC=CC=C1 TWYYFYNJOJGNFP-CUXYNZQBSA-N 0.000 description 1
- VUDZSIYXZUYWSC-DBRKOABJSA-N (4r)-1-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4-hydroxy-1,3-diazinan-2-one Chemical compound FC1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N[C@H](O)CC1 VUDZSIYXZUYWSC-DBRKOABJSA-N 0.000 description 1
- STPKWKPURVSAJF-LJEWAXOPSA-N (4r,5r)-5-[4-[[4-(1-aza-4-azoniabicyclo[2.2.2]octan-4-ylmethyl)phenyl]methoxy]phenyl]-3,3-dibutyl-7-(dimethylamino)-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-4-ol Chemical compound O[C@H]1C(CCCC)(CCCC)CS(=O)(=O)C2=CC=C(N(C)C)C=C2[C@H]1C(C=C1)=CC=C1OCC(C=C1)=CC=C1C[N+]1(CC2)CCN2CC1 STPKWKPURVSAJF-LJEWAXOPSA-N 0.000 description 1
- BSGIKRCNMBTGEB-UHFFFAOYSA-N (5-bromo-1-methylpyrrolo[2,3-c]pyridin-2-yl)-[6-[(dimethylamino)methyl]-4-methyl-3,4-dihydro-1h-isoquinolin-2-yl]methanone Chemical compound BrC1=NC=C2N(C)C(C(=O)N3CC(C4=CC(CN(C)C)=CC=C4C3)C)=CC2=C1 BSGIKRCNMBTGEB-UHFFFAOYSA-N 0.000 description 1
- LENAVORGWBTPJR-UHFFFAOYSA-N (5-pyridin-3-ylfuran-2-yl)methanamine Chemical compound O1C(CN)=CC=C1C1=CC=CN=C1 LENAVORGWBTPJR-UHFFFAOYSA-N 0.000 description 1
- JHLIGYPHPBLDDL-UHFFFAOYSA-N (5-pyridin-3-ylthiophen-2-yl)methanamine Chemical compound S1C(CN)=CC=C1C1=CC=CN=C1 JHLIGYPHPBLDDL-UHFFFAOYSA-N 0.000 description 1
- UVNPEUJXKZFWSJ-LMTQTHQJSA-N (R)-N-[(4S)-8-[6-amino-5-[(3,3-difluoro-2-oxo-1H-pyrrolo[2,3-b]pyridin-4-yl)sulfanyl]pyrazin-2-yl]-2-oxa-8-azaspiro[4.5]decan-4-yl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H]1COCC11CCN(CC1)c1cnc(Sc2ccnc3NC(=O)C(F)(F)c23)c(N)n1 UVNPEUJXKZFWSJ-LMTQTHQJSA-N 0.000 description 1
- VICOOSNNZUPVHM-IGPZRPDBSA-M (e,3r,5s)-7-[2-(4-fluorophenyl)-4-(3-phenylpentan-3-yl)phenyl]-3,5-dihydroxyhept-6-enoate Chemical compound C=1C=C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)C(C=2C=CC(F)=CC=2)=CC=1C(CC)(CC)C1=CC=CC=C1 VICOOSNNZUPVHM-IGPZRPDBSA-M 0.000 description 1
- FTTATHOUSOIFOQ-UHFFFAOYSA-N 1,2,3,4,6,7,8,8a-octahydropyrrolo[1,2-a]pyrazine Chemical compound C1NCCN2CCCC21 FTTATHOUSOIFOQ-UHFFFAOYSA-N 0.000 description 1
- ZXSQEZNORDWBGZ-UHFFFAOYSA-N 1,3-dihydropyrrolo[2,3-b]pyridin-2-one Chemical compound C1=CN=C2NC(=O)CC2=C1 ZXSQEZNORDWBGZ-UHFFFAOYSA-N 0.000 description 1
- HBKPDEWGANZHJO-UHFFFAOYSA-N 1-(4-methoxyphenyl)-n-[(4-methoxyphenyl)methyl]methanamine Chemical compound C1=CC(OC)=CC=C1CNCC1=CC=C(OC)C=C1 HBKPDEWGANZHJO-UHFFFAOYSA-N 0.000 description 1
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 1
- SZCBDIVMCGFVPW-UHFFFAOYSA-N 1-[4-(aminomethyl)-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-(3-methoxyphenyl)-2-oxo-1,8-naphthyridin-3-yl]urea;hydrochloride Chemical compound Cl.CC(C)C=1C=C(CN)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OC)=C1 SZCBDIVMCGFVPW-UHFFFAOYSA-N 0.000 description 1
- YRTFLDFDKPFNCJ-UHFFFAOYSA-N 1-[4-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-2-oxo-4-[3-(3-pyrrolidin-1-ylpropoxy)phenyl]-1,8-naphthyridin-3-yl]urea;dihydrochloride Chemical compound Cl.Cl.CC(C)C=1C=C(N)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C(C=1)=CC=CC=1OCCCN1CCCC1 YRTFLDFDKPFNCJ-UHFFFAOYSA-N 0.000 description 1
- ZFNNBIMQDHBELV-UHFFFAOYSA-N 1-[4-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-[3-(3-cyclohexylpropoxy)phenyl]-2-oxo-1,8-naphthyridin-3-yl]urea;dihydrochloride Chemical compound Cl.Cl.CC(C)C=1C=C(N)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C(C=1)=CC=CC=1OCCCC1CCCCC1 ZFNNBIMQDHBELV-UHFFFAOYSA-N 0.000 description 1
- YKYWUHHZZRBGMG-JWTNVVGKSA-N 1-methyl-2-[[(1r,5s)-6-[[5-(trifluoromethyl)pyridin-2-yl]methoxymethyl]-3-azabicyclo[3.1.0]hexan-3-yl]methyl]benzimidazole Chemical compound C1([C@@H]2CN(C[C@@H]21)CC=1N(C2=CC=CC=C2N=1)C)COCC1=CC=C(C(F)(F)F)C=N1 YKYWUHHZZRBGMG-JWTNVVGKSA-N 0.000 description 1
- 238000004791 1D NOESY Methods 0.000 description 1
- JYWKEVKEKOTYEX-UHFFFAOYSA-N 2,6-dibromo-4-chloroiminocyclohexa-2,5-dien-1-one Chemical compound ClN=C1C=C(Br)C(=O)C(Br)=C1 JYWKEVKEKOTYEX-UHFFFAOYSA-N 0.000 description 1
- HLAOENZTDMPYNY-UHFFFAOYSA-N 2,6-dichloropyrimidine-4-carbonitrile Chemical compound ClC1=CC(C#N)=NC(Cl)=N1 HLAOENZTDMPYNY-UHFFFAOYSA-N 0.000 description 1
- QEBYEVQKHRUYPE-UHFFFAOYSA-N 2-(2-chlorophenyl)-5-[(1-methylpyrazol-3-yl)methyl]-4-[[methyl(pyridin-3-ylmethyl)amino]methyl]-1h-pyrazolo[4,3-c]pyridine-3,6-dione Chemical compound C1=CN(C)N=C1CN1C(=O)C=C2NN(C=3C(=CC=CC=3)Cl)C(=O)C2=C1CN(C)CC1=CC=CN=C1 QEBYEVQKHRUYPE-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- XZORQADPEUSNQJ-UHFFFAOYSA-N 2-(3-piperidin-4-yloxy-1-benzothiophen-2-yl)-5-[(1,3,5-trimethylpyrazol-4-yl)methyl]-1,3,4-oxadiazole Chemical compound CC1=NN(C)C(C)=C1CC1=NN=C(C2=C(C3=CC=CC=C3S2)OC2CCNCC2)O1 XZORQADPEUSNQJ-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- WGABOZPQOOZAOI-UHFFFAOYSA-N 2-[4-[[(3,5-dimethoxy-4-methylbenzoyl)-(3-phenylpropyl)amino]methyl]phenyl]acetic acid Chemical compound COC1=C(C)C(OC)=CC(C(=O)N(CCCC=2C=CC=CC=2)CC=2C=CC(CC(O)=O)=CC=2)=C1 WGABOZPQOOZAOI-UHFFFAOYSA-N 0.000 description 1
- BCSHRERPHLTPEE-NRFANRHFSA-N 2-[[5-chloro-2-[[(6s)-6-[4-(2-hydroxyethyl)piperazin-1-yl]-1-methoxy-6,7,8,9-tetrahydro-5h-benzo[7]annulen-2-yl]amino]pyrimidin-4-yl]amino]-n-methylbenzamide Chemical compound CNC(=O)C1=CC=CC=C1NC1=NC(NC=2C(=C3CCC[C@@H](CC3=CC=2)N2CCN(CCO)CC2)OC)=NC=C1Cl BCSHRERPHLTPEE-NRFANRHFSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- FBEIDYLEFVIOEY-UHFFFAOYSA-N 2-chloro-6-methylpyrimidin-4-amine Chemical compound CC1=CC(N)=NC(Cl)=N1 FBEIDYLEFVIOEY-UHFFFAOYSA-N 0.000 description 1
- YRSNXUYQRUULNJ-UHFFFAOYSA-N 2-chloro-6-nitropyridine Chemical compound [O-][N+](=O)C1=CC=CC(Cl)=N1 YRSNXUYQRUULNJ-UHFFFAOYSA-N 0.000 description 1
- VGOALPIDEXVYQI-UHFFFAOYSA-N 3-(2-imidazo[1,2-b]pyridazin-3-ylethynyl)-n-[3-imidazol-1-yl-5-(trifluoromethyl)phenyl]-4-methylbenzamide Chemical compound C1=C(C#CC=2N3N=CC=CC3=NC=2)C(C)=CC=C1C(=O)NC(C=C(C=1)C(F)(F)F)=CC=1N1C=CN=C1 VGOALPIDEXVYQI-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- SKJCWIFHLDWPEO-UHFFFAOYSA-N 3-benzyl-4-phenylbutan-2-one Chemical compound C=1C=CC=CC=1CC(C(=O)C)CC1=CC=CC=C1 SKJCWIFHLDWPEO-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- KBEIFKMKVCDETC-UHFFFAOYSA-N 3-iodooxetane Chemical compound IC1COC1 KBEIFKMKVCDETC-UHFFFAOYSA-N 0.000 description 1
- GPPSQLLIFNWNSB-UHFFFAOYSA-N 3-phenylmethoxycyclobutan-1-one Chemical compound C1C(=O)CC1OCC1=CC=CC=C1 GPPSQLLIFNWNSB-UHFFFAOYSA-N 0.000 description 1
- WDBQJSCPCGTAFG-QHCPKHFHSA-N 4,4-difluoro-N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclohexane-1-carboxamide Chemical compound FC1(CCC(CC1)C(=O)N[C@@H](CCN1CCC(CC1)N1C(=NN=C1C)C(C)C)C=1C=NC=CC=1)F WDBQJSCPCGTAFG-QHCPKHFHSA-N 0.000 description 1
- BWGRDBSNKQABCB-UHFFFAOYSA-N 4,4-difluoro-N-[3-[3-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl]-1-thiophen-2-ylpropyl]cyclohexane-1-carboxamide Chemical compound CC(C)C1=NN=C(C)N1C1CC2CCC(C1)N2CCC(NC(=O)C1CCC(F)(F)CC1)C1=CC=CS1 BWGRDBSNKQABCB-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- JOFDSYLCZIHGGO-UHFFFAOYSA-N 4-[(4-cyclohexylphenyl)methyl-[2-[[5-(dimethylamino)naphthalen-1-yl]sulfonyl-methylamino]acetyl]amino]-2-hydroxybenzoic acid Chemical compound C1=CC=C2C(N(C)C)=CC=CC2=C1S(=O)(=O)N(C)CC(=O)N(C=1C=C(O)C(C(O)=O)=CC=1)CC(C=C1)=CC=C1C1CCCCC1 JOFDSYLCZIHGGO-UHFFFAOYSA-N 0.000 description 1
- TXEBWPPWSVMYOA-UHFFFAOYSA-N 4-[3-[(1-amino-2-chloroethyl)amino]propyl]-1-[[3-(2-chlorophenyl)phenyl]methyl]-5-hydroxyimidazolidin-2-one Chemical compound NC(CCl)NCCCC1NC(=O)N(Cc2cccc(c2)-c2ccccc2Cl)C1O TXEBWPPWSVMYOA-UHFFFAOYSA-N 0.000 description 1
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 1
- ZDHQVQVNOIWLNX-UHFFFAOYSA-N 5-(7-chloro-10h-benzo[1,2]cyclohepta[2,4-c][1,3]thiazol-10-yl)-1-methyl-4-sulfanylidenepyrimidin-2-one Chemical compound S=C1NC(=O)N(C)C=C1C1C2=CC=C(Cl)C=C2C=CC2=C1N=CS2 ZDHQVQVNOIWLNX-UHFFFAOYSA-N 0.000 description 1
- VKLKXFOZNHEBSW-UHFFFAOYSA-N 5-[[3-[(4-morpholin-4-ylbenzoyl)amino]phenyl]methoxy]pyridine-3-carboxamide Chemical compound O1CCN(CC1)C1=CC=C(C(=O)NC=2C=C(COC=3C=NC=C(C(=O)N)C=3)C=CC=2)C=C1 VKLKXFOZNHEBSW-UHFFFAOYSA-N 0.000 description 1
- UZALKVXCOUSWSL-UHFFFAOYSA-N 6-fluoropyridin-2-amine Chemical compound NC1=CC=CC(F)=N1 UZALKVXCOUSWSL-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- MCMSJVMUSBZUCN-UHFFFAOYSA-N 9,10-dimethoxy-3-methyl-2-(2,4,6-trimethylphenyl)imino-6,7-dihydropyrimido[6,1-a]isoquinolin-4-one Chemical compound C1=2C=C(OC)C(OC)=CC=2CCN(C(N2C)=O)C1=CC2=NC1=C(C)C=C(C)C=C1C MCMSJVMUSBZUCN-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- QUMCIHKVKQYNPA-RUZDIDTESA-N C1(CCCCC1)CN1[C@@H](C=2N(C=3C=NC(=NC1=3)NC1=C(C=C(C(=O)NC3CCN(CC3)C)C=C1)OC)C(=NN=2)C)CC Chemical compound C1(CCCCC1)CN1[C@@H](C=2N(C=3C=NC(=NC1=3)NC1=C(C=C(C(=O)NC3CCN(CC3)C)C=C1)OC)C(=NN=2)C)CC QUMCIHKVKQYNPA-RUZDIDTESA-N 0.000 description 1
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 1
- QCMHGCDOZLWPOT-FMNCTDSISA-N COC1=C(CC[C@@H]2CCC3=C(C2)C=CC(=C3)[C@H]2CC[C@](N)(CO)C2)C=CC=C1 Chemical compound COC1=C(CC[C@@H]2CCC3=C(C2)C=CC(=C3)[C@H]2CC[C@](N)(CO)C2)C=CC=C1 QCMHGCDOZLWPOT-FMNCTDSISA-N 0.000 description 1
- VJQALSOBHVEJQM-UHFFFAOYSA-N COCCOC1CCC(CC1)Nc1cc(=O)n(C)c2ccc(cc12)-c1cncs1 Chemical compound COCCOC1CCC(CC1)Nc1cc(=O)n(C)c2ccc(cc12)-c1cncs1 VJQALSOBHVEJQM-UHFFFAOYSA-N 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 1
- 108700039887 Essential Genes Proteins 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 239000001116 FEMA 4028 Substances 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 102000007474 Multiprotein Complexes Human genes 0.000 description 1
- 108010085220 Multiprotein Complexes Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- NUGPIZCTELGDOS-QHCPKHFHSA-N N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclopentanecarboxamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CC[C@@H](C=1C=NC=CC=1)NC(=O)C1CCCC1)C NUGPIZCTELGDOS-QHCPKHFHSA-N 0.000 description 1
- LFZAGIJXANFPFN-UHFFFAOYSA-N N-[3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-thiophen-2-ylpropyl]acetamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CCC(C=1SC=CC=1)NC(C)=O)C LFZAGIJXANFPFN-UHFFFAOYSA-N 0.000 description 1
- 206010029350 Neurotoxicity Diseases 0.000 description 1
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 208000008601 Polycythemia Diseases 0.000 description 1
- 102000052575 Proto-Oncogene Human genes 0.000 description 1
- 108700020978 Proto-Oncogene Proteins 0.000 description 1
- 239000004280 Sodium formate Substances 0.000 description 1
- 239000005708 Sodium hypochlorite Substances 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- 206010044221 Toxic encephalopathy Diseases 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000003080 antimitotic agent Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- GMWFCJXSQQHBPI-UHFFFAOYSA-N azetidin-3-ol Chemical compound OC1CNC1 GMWFCJXSQQHBPI-UHFFFAOYSA-N 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- AJRQIXBBIDPNGK-BVSLBCMMSA-N benzyl n-[(2s)-1-[[(2s)-1-(1,3-benzothiazol-2-yl)-1-oxo-3-[(3s)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]carbamate Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H](C[C@H]1C(NCC1)=O)C(=O)C=1SC2=CC=CC=C2N=1)C(=O)OCC1=CC=CC=C1 AJRQIXBBIDPNGK-BVSLBCMMSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 1
- 235000011175 beta-cyclodextrine Nutrition 0.000 description 1
- 229960004853 betadex Drugs 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- LKZMBDSASOBTPN-UHFFFAOYSA-N carbonic acid;silver Chemical compound [Ag].OC(O)=O LKZMBDSASOBTPN-UHFFFAOYSA-N 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 230000016593 centrosome cycle Effects 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940127206 compound 14d Drugs 0.000 description 1
- 229940126212 compound 17a Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-N dichloropalladium;triphenylphosphanium Chemical compound Cl[Pd]Cl.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000003209 gene knockout Methods 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 210000005061 intracellular organelle Anatomy 0.000 description 1
- 230000010189 intracellular transport Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 206010024378 leukocytosis Diseases 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 230000021121 meiosis Effects 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- KPJWVJURYXOHOO-UHFFFAOYSA-N methyl 1-hydroxycyclopropane-1-carboxylate Chemical compound COC(=O)C1(O)CC1 KPJWVJURYXOHOO-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 230000036456 mitotic arrest Effects 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 1
- QAPTWHXHEYAIKG-RCOXNQKVSA-N n-[(1r,2s,5r)-5-(tert-butylamino)-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](NC(C)(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 QAPTWHXHEYAIKG-RCOXNQKVSA-N 0.000 description 1
- VMFMUJZRXZXYAH-UHFFFAOYSA-N n-[5-[[5-chloro-4-[2-[2-(dimethylamino)-2-oxoacetyl]anilino]pyrimidin-2-yl]amino]-4-methoxy-2-(4-methylpiperazin-1-yl)phenyl]prop-2-enamide Chemical compound C=CC(=O)NC=1C=C(NC=2N=C(NC=3C(=CC=CC=3)C(=O)C(=O)N(C)C)C(Cl)=CN=2)C(OC)=CC=1N1CCN(C)CC1 VMFMUJZRXZXYAH-UHFFFAOYSA-N 0.000 description 1
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 230000007135 neurotoxicity Effects 0.000 description 1
- 231100000228 neurotoxicity Toxicity 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- PFGVNLZDWRZPJW-OPAMFIHVSA-N otamixaban Chemical compound C([C@@H](C(=O)OC)[C@@H](C)NC(=O)C=1C=CC(=CC=1)C=1C=C[N+]([O-])=CC=1)C1=CC=CC(C(N)=N)=C1 PFGVNLZDWRZPJW-OPAMFIHVSA-N 0.000 description 1
- GPRIERYVMZVKTC-UHFFFAOYSA-N p-quaterphenyl Chemical group C1=CC=CC=C1C1=CC=C(C=2C=CC(=CC=2)C=2C=CC=CC=2)C=C1 GPRIERYVMZVKTC-UHFFFAOYSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 1
- RWPGFSMJFRPDDP-UHFFFAOYSA-L potassium metabisulfite Chemical compound [K+].[K+].[O-]S(=O)S([O-])(=O)=O RWPGFSMJFRPDDP-UHFFFAOYSA-L 0.000 description 1
- 229940043349 potassium metabisulfite Drugs 0.000 description 1
- 235000010263 potassium metabisulphite Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- HNDXKIMMSFCCFW-UHFFFAOYSA-M propane-2-sulfonate Chemical compound CC(C)S([O-])(=O)=O HNDXKIMMSFCCFW-UHFFFAOYSA-M 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000034394 regulation of mitosis Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000011257 shell material Substances 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229910001958 silver carbonate Inorganic materials 0.000 description 1
- LKZMBDSASOBTPN-UHFFFAOYSA-L silver carbonate Substances [Ag].[O-]C([O-])=O LKZMBDSASOBTPN-UHFFFAOYSA-L 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- HLBBKKJFGFRGMU-UHFFFAOYSA-M sodium formate Chemical compound [Na+].[O-]C=O HLBBKKJFGFRGMU-UHFFFAOYSA-M 0.000 description 1
- 235000019254 sodium formate Nutrition 0.000 description 1
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 230000020347 spindle assembly Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000001119 stannous chloride Substances 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- BUUPQKDIAURBJP-UHFFFAOYSA-N sulfinic acid Chemical compound OS=O BUUPQKDIAURBJP-UHFFFAOYSA-N 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- WGRQANOPCQRCME-PMACEKPBSA-N teneligliptin Chemical compound O=C([C@H]1NC[C@H](C1)N1CCN(CC1)C1=CC(=NN1C=1C=CC=CC=1)C)N1CCSC1 WGRQANOPCQRCME-PMACEKPBSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- UAEJRRZPRZCUBE-UHFFFAOYSA-N trimethoxyalumane Chemical compound [Al+3].[O-]C.[O-]C.[O-]C UAEJRRZPRZCUBE-UHFFFAOYSA-N 0.000 description 1
- HHBXWXJLQYJJBW-UHFFFAOYSA-M triphenyl(propan-2-yl)phosphanium;iodide Chemical compound [I-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(C(C)C)C1=CC=CC=C1 HHBXWXJLQYJJBW-UHFFFAOYSA-M 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- JQSHBVHOMNKWFT-DTORHVGOSA-N varenicline Chemical compound C12=CC3=NC=CN=C3C=C2[C@H]2C[C@@H]1CNC2 JQSHBVHOMNKWFT-DTORHVGOSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000012224 working solution Substances 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4453—Non condensed piperidines, e.g. piperocaine only substituted in position 1, e.g. propipocaine, diperodon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/10—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/70—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/72—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/20—Spiro-condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/59—Hydrogenated pyridine rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
Definitions
- the present invention relates to the field of medicinal chemistry; in particular, the present invention relates to a novel compound, its synthesis method and its application as a KIF18A inhibitor in the preparation of medicines for treating tumors and other related diseases.
- Tumors often have uncontrolled cell proliferation. Disruption of one or more genes involved in cell signaling pathways that regulate the cell cycle and centrosome cycle can affect normal cell division. These abnormal genes encode a variety of different tumor suppressor genes or proto-oncogenes, which are involved in a series of physiological processes in the body, resulting in cell cycle progression and cell division that are not controlled by checkpoints. A variety of kinases and kinesins have been identified, which play key roles in the regulation of cell cycle and mitosis in normal dividing cells and tumor cells.
- Intracellular trafficking is critical for maintaining normal cell morphology and function.
- Members of the kinesin superfamily are a class of molecular motors that obtain energy by hydrolyzing ATP and transport intracellular organelles, protein complexes, and mRNA. Kinesins are also involved in the movement of chromosomes and spindles during mitosis and meiosis. According to sequence homology, the human kinesin superfamily can be divided into 14 subfamilies. The globular domain of about 360 amino acid residues is the hallmark of the kinesin family members. This globular domain contains the catalytic pocket for hydrolyzing ATP and the region for binding microtubules, also known as the motor domain.
- the non-motor domain of the protein is responsible for connecting the intracellular "cargo" transported by the protein, which includes the above-mentioned organelles, proteins and chromosomes.
- Kinesins use the energy gained from the hydrolysis of ATP to transport "cargo" along polar tubulin. Therefore, kinesins are also known as positive or negative transport motors.
- KIF18A Kinesin Family Member 18A belongs to the kinesin-8 (Kinesin-8) subfamily and is a positive transport motor.
- Knockdown of KIF18A in Hela cervical cancer cells elongated the cells' spindles, promoted chromosomal instability, and activated the mitotic spindle assembly checkpoint.
- knockdown of KIF18A triggered activation of the mitotic spindle assembly checkpoint in a series of chromosomally unstable tumor cells, interfering with tumor cell proliferation, but not in normal mammary epithelial cells and near-normal karyotype tumors.
- knocking down KIF18A does not significantly affect cell proliferation.
- mice knocked out of KIF18A were viable but defective in germ cell division, suggesting that KIF18A is not an essential gene for normal somatic cell division.
- KIF18A is overexpressed in various types of cancers, such as breast cancer, cervical cancer, colon cancer, lung cancer, pancreatic cancer, prostate cancer, bladder cancer and head and neck cancer.
- Gene knockout or knockdown of KIF18A will affect the mitotic spindle of tumor cells, leading to cell mitotic arrest and promoting cell apoptosis.
- KIF18A was significantly increased in influenza virus-infected host cells. Inhibiting the ATP hydrolysis activity of KIF18A can hinder the replication of influenza virus.
- KIF18A inhibitors has very good antitumor and antiviral prospects.
- the purpose of the present invention is to provide a new class of KIF18A inhibitors.
- the first aspect of the present invention provides a compound with the structure shown in the following formula (I), or its optical isomer, pharmaceutically acceptable salt, prodrug, deuterated derivative, hydrate, solvate :
- A is selected from -YR 1 ; wherein, Y is selected from chemical bonds, -S(O)-, -S(O) 2 -, -S(O)(NH)-, -S(O) 2 NR a -, - NR a S(O) 2 -, -NR a S(O) 2 NR a -, -P(O)(R q )-, -P(O)(R q )NR a -, -NR a P( O)(R q )-, -P(O)(R q )O-, -OP(O)(R q )-, -NR a -, -O-, -S-, -C(O)NR a -, -NR a C(O)-, -C(O)O-, -OC(O)-, -NR a C(O)NR a -, -OC(O)-
- L is selected from -C(O) NRb- , -NRbC (O)-, -NRbC (O) NRb- , -OC(O) NRb- , -C(O)O-, - OC(O)-, -NR b C(O)O-, -S(O) 2 NR b -, -NR b S(O) 2 -, -NR b -, -O-, 3- to 6- Member heterocyclic group or heteroaryl;
- each R b is independently selected from hydrogen or C 1-4 alkyl;
- B is selected from formula (Ib) or (Id):
- X 1 , X 2 , X 3 and X 4 are each independently selected from N or CR c ; wherein R c is selected from hydrogen, halogen, C 1-4 alkyl, C 1-4 haloalkyl , C 2- 4 alkynyl, C 1-4 alkoxy, hydroxyl, C 1-4 haloalkoxy, CN, NR d R d , C 3-6 cycloalkyl, or 3- to 8-membered heterocyclic group; wherein, Each R is independently selected from hydrogen, C 1-4 alkyl, or C 1-4 haloalkyl;
- U is selected from N or CR e ;
- Re is selected from hydrogen, halogen, C 1-4 alkyl;
- Z 1 , Z 2 , and Z 3 are each independently selected from N or CR m ; wherein, R m is selected from hydrogen, halogen, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkane Oxygen, hydroxyl, C 1-4 haloalkoxy, CN, NR d R d , C 3-8 cycloalkyl, or 3- to 8-membered heterocyclyl; wherein, the definition of each R d is as above; D is selected from chemical bonds, -O-, -S-, NR d -, C 1-2 alkyl, or C 2-4 alkynyl; G is selected from 3- to -10 membered saturated or unsaturated ring structures, where The ring structure optionally contains 0, 1, or 2 heteroatoms selected from N, O, S, or is optionally substituted by R and R ;
- R 1 is selected from the group consisting of C 1-4 alkyl, C 2-4 alkenyl, C 2-4 alkynyl, C 3-6 cycloalkyl, 3- to 10-membered heterocyclyl; Alkenyl, alkynyl, cycloalkyl or heterocyclyl is optionally substituted by one or more groups selected from the group consisting of halogen, C 1-4 alkyl, C 1-4 haloalkyl, C 2 -4 alkenyl, C 2-4 haloalkenyl, C 2-4 alkynyl, C 2-4 haloalkynyl, C 3-6 cycloalkyl, 3- to 6-membered heterocyclyl, CN, OR f , SR f , NR d R d , -OC 1-4 alkyl OR f , -OC 1-4 alkyl NR d R d , -NR d C 1-4 alkyl OR f , -NR d C 1
- R 2 and R 3 are each independently selected from the following group: hydrogen, fluorine, or C 1-4 alkyl; wherein, the alkyl is optionally substituted by one or more groups selected from the group: halogen , C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkoxy, C 1-4 haloalkoxy, hydroxyl, NR d R d , C 3-8 cycloalkyl, 3- to 8 -membered heterocyclyl, aryl, or heteroaryl; wherein, each R is as defined above;
- Each R is independently selected from hydrogen, halogen, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkoxy, C 1-4 alkoxy C 1-4 alkyl , hydroxyl, Hydroxy C 1-4 alkyl, C 1-4 haloalkoxy, C 1-4 haloalkoxy C 1-4 alkyl, or CN;
- R 5 is selected from -QR 8 ; wherein, Q is selected from chemical bonds, -S(O)-, -S(O) 2 -, -S(O)(NH)-, -S(O) 2 NR a -, -NR a S(O) 2 -, -NR a S(O) 2 NR a -, -NR a -, -O-, -S-, -C(O)NR a -, -NR a C(O )-, -C(O)O-, -OC(O)-, -NR a C(O)NR a -, -OC(O)NR a -, -NR a C(O)O-, C 2 -4 alkenyl, or C 2-4 alkynyl;
- Q is selected from chemical bonds, -S(O)-, -S(O) 2 -, -S(O)(NH)-, -S(
- R is selected from the group consisting of C 1-4 alkyl, C 2-4 alkenyl, C 2-4 alkynyl, C 3-6 cycloalkyl, 3- to 10 -membered heterocyclyl; Alkenyl, alkynyl, cycloalkyl or heterocyclyl is optionally substituted by one or more groups selected from the group consisting of halogen, C 1-4 alkyl, C 1-4 haloalkyl, C 2 -4 alkenyl, C 2-4 alkynyl, C 3-6 cycloalkyl, 3- to 6-membered heterocyclyl, CN, OR f , SR f , NR d R d , -OC 1-4 alkyl OR f , -OC 1-4 alkyl NR d R d , -NR d C 1-4 alkyl OR f , -NR d C 1-4 alkyl NR d R d , -C(O)R g
- p and q are each independently selected from 0, 1, 2, 3, 4, 5, or 6; the prerequisite is that p and q cannot be selected from 0 at the same time;
- n is selected from 0, 1, 2, 3, or 4;
- k and n are selected from 1, 2, 3, 4, or 5;
- each of the above-mentioned alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aryl and heteroaryl groups is optionally and independently substituted by 1-3 substituents selected from the group consisting of: halogen , C 1-4 alkyl, C 1-4 haloalkyl, C 2-4 alkenyl, C 2-4 alkynyl, C 3-8 cycloalkyl, 3- to 8-membered heterocyclyl, aryl, Heteroaryl, CN, NO 2 , OR f , SR f , NR d R d , C(O)R g , C(O)OR f , C(O)NR d R d , NR d C(O)R g , NR d S(O) 2 R g , or S(O) 2 R g , provided that the formed chemical structure is stable and meaningful; where, the definitions of R d
- the above-mentioned aryl group is an aromatic group containing 6-12 carbon atoms;
- the heteroaryl group is a 5- to 15-membered (preferably 5- to 12-membered) heteroaromatic group;
- the ring structure is Saturated or unsaturated, heteroatom-containing or heteroatom-free cyclic groups.
- formula (I) is formula (XIa) or formula (XIb):
- fragment or fragment Groups selected from the following groups:
- A is selected from the following groups:
- X 1 , X 2 , X 3 and X 4 are as defined above;
- fragment Groups selected from the following groups:
- fragment Groups selected from the following groups:
- L selection represents -C(O)NH-, -NHC(O)-, -S(O) 2 NH-, or -NHS(O) 2 -;
- formula (I) is formula (XII):
- R j and R k are each independently selected from the following group: hydrogen, deuterium, fluorine, chlorine, bromine, or C 1-4 alkyl;
- k and n are selected from 1, 2, 3, 4, or 5;
- formula (I) is formula (XIII):
- k and n are selected from 1, 2, 3, or 4;
- A is selected from the following groups:
- fragment Groups selected from the following groups:
- fragment Groups selected from the following groups:
- fragment Groups selected from the following groups:
- L is selected from -C(O)NH-, -NHC(O)-, -S(O) 2 NH-, or -NHS(O) 2 -;
- formula (I) is formula (XIV):
- formula (I) is formula (XV):
- formula (I) is formula (XVII):
- k and n are each independently selected from 1 or 2;
- X is selected from N, CH, or CF
- X is selected from N, CH , CF, C-CN, or C-Me;
- X is selected from CH, CF, or N;
- Z is selected from CH, CF, or N ;
- R is selected from hydrogen, halogen, C 1-4 alkyl, or CN;
- L is selected from -C(O)NH- or -NHC(O)-;
- formula (I) is formula (XVIII):
- X is selected from N, CH, or CF
- X is selected from N, CH , CF, C-CN, or C-Me;
- X is selected from CH, CF, or N;
- Z is selected from CH, CF, or N ;
- Rc is selected from hydrogen, halogen, methyl, ethyl, or CN;
- A is selected from the following groups:
- fragment Groups selected from the following groups:
- fragment A in formula (XIa) or formula (XIb) is selected from the following groups:
- the compound of formula (I) is selected from the following group:
- the second aspect of the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising the compound described in the first aspect of the present invention, or its optical isomer, pharmaceutically acceptable salt, prodrug, deuterated Derivatives, hydrates, solvates, and pharmaceutically acceptable carriers.
- the third aspect of the present invention provides a compound as described in the first aspect of the present invention, or its optical isomer, pharmaceutically acceptable salt, prodrug, deuterated derivative, hydrate, solvate
- the disease, disease or condition is selected from the group consisting of non-small cell lung cancer, small cell lung cancer, lung adenocarcinoma, lung squamous cell carcinoma, pancreatic cancer, colon cancer, thyroid cancer, embryonal rhabdomyosarcoma, Cutaneous granulosa cell tumor, melanoma, hepatocellular carcinoma, intrahepatic cholangiocarcinoma, rectal cancer, bladder cancer, throat cancer, breast cancer, prostate cancer, brain tumor, glioma, ovarian cancer, head and neck squamous cell carcinoma, Cervical cancer, esophageal cancer, kidney cancer, skin cancer, gastric cancer, myeloid leukemia, lymphoid leukemia, myelofibrosis, B-cell lymphoma, T-cell lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, single Various solid tumors and blood tumors such as nuclear cell
- the inventors unexpectedly discovered a class of KIF18A inhibitors with novel structures, as well as their preparation methods and applications.
- the compound of the present invention can be applied to the treatment of various diseases related to the activity of the ATP hydrolase. Based on the above findings, the inventors have accomplished the present invention.
- each chiral carbon atom may optionally be in R configuration or S configuration, or a mixture of R configuration and S configuration.
- alkyl refers to a straight chain (i.e., unbranched) or branched chain saturated hydrocarbon group containing only carbon atoms, or a combination of straight chain and branched chain groups .
- the alkyl group is defined by the number of carbon atoms (such as C 1-10 ), it means that the said alkyl group contains 1-10 carbon atoms.
- C 1-8 alkyl refers to an alkyl group containing 1-8 carbon atoms, including methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, or Similar groups.
- alkenyl refers to a straight or branched, carbon chain group having at least one carbon-carbon double bond. Alkenyl groups can be substituted or unsubstituted. When the number of carbon atoms (such as C 2-8 ) is limited before the alkenyl group, it means that the alkenyl group contains 2-8 carbon atoms.
- C 2-8 alkenyl refers to alkenyl groups containing 2-8 carbon atoms, including vinyl, propenyl, 1,2-butenyl, 2,3-butenyl, butadienyl, or similar groups group.
- alkynyl alone or as part of another substituent, refers to an aliphatic hydrocarbon group having at least one carbon-carbon triple bond.
- the alkynyl group can be straight or branched, or a combination thereof. When the number of carbon atoms is limited before the alkynyl group (such as C 2-8 alkynyl group), it means that the alkynyl group contains 2-8 carbon atoms.
- C2-8 alkynyl refers to a straight or branched chain alkynyl group having 2-8 carbon atoms, including ethynyl, propynyl, isopropynyl, butynyl, isobutynyl, sec-butynyl, tert-butynyl, or similar groups.
- cycloalkyl alone or as part of another substituent, refers to a monocyclic or polycyclic (fused, bridged or spiro) ring system group having a saturated or partially saturated ring .
- a cycloalkyl group is preceded by a restriction on the number of carbon atoms (such as C 3-10 ), it means that the cycloalkyl group contains 3-10 carbon atoms.
- C 3-8 cycloalkyl refers to a saturated or partially unsaturated monocyclic or bicyclic alkyl group with 3-8 carbon atoms, including cyclopropyl, cyclobutyl, cyclo Pentyl, cycloheptyl, or similar groups.
- Spirocycloalkyl means a bicyclic or polycyclic group in which the single rings share a single carbon atom (called a spiro atom), these may contain one or more double bonds, but none of the rings has fully conjugated pi electrons system.
- “Fused cycloalkyl” means an all-carbon bicyclic or polycyclic group in which each ring of the system shares an adjacent pair of carbon atoms with other rings in the system, one or more of which may contain one or more bicyclic bonds, but none of the rings have a fully conjugated ⁇ -electron system.
- “Bridged cycloalkyl” refers to an all-carbon polycyclic group in which any two rings share two carbon atoms not directly attached, these may contain one or more double bonds, but none of the rings has a fully conjugated pi-electron system .
- the atoms contained in the cycloalkyl group are all carbon atoms.
- the following are some examples of cycloalkyl groups, and the present invention is not limited only to the cycloalkyl groups described below.
- Aryl refers to an all-carbon monocyclic or fused polycyclic (ie, rings sharing adjacent pairs of carbon atoms) group having a conjugated pi-electron system, eg, phenyl and naphthyl.
- the aryl ring can be fused to other cyclic groups (including saturated and unsaturated rings), but cannot contain heteroatoms such as nitrogen, oxygen, or sulfur, and the point of connection to the parent must be in a conjugated ⁇ -electron system on the carbon atoms in the ring.
- Aryl groups can be substituted or unsubstituted. The following are some examples of aryl groups, and the present invention is not limited only to the aryl groups described below.
- Heteroaryl refers to an aromatic monocyclic or polycyclic group containing one to more heteroatoms (optionally selected from nitrogen, oxygen and sulfur), or a heterocyclic group (containing one to more heteroatoms optionally A polycyclic group formed by condensing nitrogen, oxygen and sulfur) with an aryl group, and the connection point is located on the aryl group. Heteroaryl groups can be optionally substituted or unsubstituted. The following are some examples of heteroaryl groups, and the present invention is not limited only to the heteroaryl groups described below.
- Heterocyclyl means a saturated or partially unsaturated monocyclic or polycyclic cyclic hydrocarbon substituent in which one or more ring atoms are selected from nitrogen, oxygen or sulfur, and the remaining ring atoms are carbon.
- monocyclic heterocyclyl groups include pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, homopiperazinyl.
- a polycyclic heterocyclic group refers to a heterocyclic group including spiro rings, fused rings and bridged rings.
- “Spirocyclic heterocyclic group” refers to a polycyclic heterocyclic group in which each ring in the system shares an atom (called a spiro atom) with other rings in the system, wherein one or more ring atoms are selected from nitrogen, oxygen or sulfur, with the remaining ring atoms being carbon.
- “fused ring heterocyclyl” means a polycyclic heterocyclic group in which each ring in the system shares an adjacent pair of atoms with other rings in the system, one or more rings may contain one or more double bonds, but none A ring has a fully conjugated pi-electron system, and one or more ring atoms are selected from nitrogen, oxygen or sulfur, and the remaining ring atoms are carbon.
- “Bridged heterocyclyl” means a polycyclic heterocyclic group in which any two rings share two atoms not directly connected, these may contain one or more double bonds, but none of the rings has a fully conjugated pi-electron system , and wherein one or more ring atoms are selected from nitrogen, oxygen or sulfur, and the remaining ring atoms are carbon. If there are both saturated and aromatic rings in the heterocyclic group (for example, the saturated ring and the aromatic ring are fused together), the point of attachment to the parent must be on the saturated ring. Note: When the point of connection to the parent is on the aromatic ring, it is called heteroaryl, not heterocyclic. The following are some examples of heterocyclic groups, and the present invention is not limited only to the following heterocyclic groups.
- halogen refers to F, Cl, Br and I, alone or as part of another substituent.
- substituted refers to the replacement of one or more hydrogen atoms on a specified group with a specified substituent.
- the specific substituents are the corresponding substituents described above, or the substituents appearing in each embodiment.
- an optionally substituted group may have a substituent selected from a specific group at any substitutable position of the group, and the substituents may be the same or different at each position.
- a cyclic substituent, such as a heterocyclyl may be attached to another ring, such as a cycloalkyl, to form a spirobicyclic ring system, ie, two rings that share a single carbon atom.
- substituents contemplated by this invention are those that are stable or chemically feasible.
- the substituents are for example (but not limited to): C 1-8 alkyl, C 2-8 alkenyl, C 2-8 alkynyl, C 3-8 cycloalkyl, 3- to 12-membered heterocyclyl , Aryl, heteroaryl, halogen, hydroxyl, carboxyl (-COOH), C 1-8 aldehyde, C 2-10 acyl, C 2-10 ester, amino.
- a pharmaceutically acceptable salt of a compound of the present invention refers to a salt that is suitable for contact with the tissues of a subject (eg, a human) without undue side effects.
- a pharmaceutically acceptable salt of a compound of the present invention includes a salt of a compound of the present invention having an acidic group (e.g., potassium salt, sodium salt, magnesium salt, calcium salt) or having a basic Salts (eg, sulfates, hydrochlorides, phosphates, nitrates, carbonates) of compounds of the invention.
- the present invention provides a class of compounds of formula (I), or their deuterated derivatives, their salts, isomers (enantiomers or diastereoisomers, if present), hydrates , the use of a pharmaceutically acceptable carrier or excipient for inhibiting KIF18A.
- the compounds of the present invention are useful as a KIF18A inhibitor.
- the invention is a single inhibitor of KIF18A, which can prevent, alleviate or cure diseases by regulating the activity of KIF18A.
- the diseases referred to include but are not limited to: non-small cell lung cancer, small cell lung cancer, lung adenocarcinoma, lung squamous cell carcinoma, pancreatic cancer, colon cancer, thyroid cancer, embryonal rhabdomyosarcoma, granulosa cell tumor of the skin, melanoma, hepatocellular carcinoma , intrahepatic cholangiocarcinoma, rectal cancer, bladder cancer, throat cancer, breast cancer, prostate cancer, brain tumor, glioma, ovarian cancer, head and neck squamous cell carcinoma, cervical cancer, esophagus cancer, kidney cancer, skin cancer , gastric cancer, myeloid leukemia, lymphoid leukemia, myelofibrosis, B-cell lymphoma, T-cell lymphoma, Hodgkin's lymphoma
- compositions can be mixed with pharmaceutically acceptable excipients or The carriers are formulated together and the resulting compositions can be administered in vivo to mammals, such as men, women and animals, for the treatment of conditions, symptoms and diseases.
- Compositions can be: tablets, pills, suspensions, solutions, emulsions, capsules, aerosols, sterile injections. Sterile powder etc.
- pharmaceutically acceptable excipients include microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, calcium hydrogen phosphate, mannitol, hydroxypropyl- ⁇ -cyclodextrin, ⁇ -cyclodextrin (increase), glycine, disintegrants (such as starch, croscarmellose sodium, complex silicate and macromolecular polyethylene glycol), granulation binders (such as polyvinylpyrrolidone, sucrose, gelatin and gum arabic) and lubricants (such as magnesium stearate, glycerin, and talc).
- disintegrants such as starch, croscarmellose sodium, complex silicate and macromolecular polyethylene glycol
- granulation binders such as polyvinylpyrrolidone, sucrose, gelatin and gum arabic
- lubricants such as magnesium stearate, glycerin, and talc
- the pharmaceutical composition is in a dosage form suitable for oral administration, including but not limited to tablets, solutions, suspensions, capsules, granules, and powders.
- the amount of the compound or pharmaceutical composition of the present invention administered to the patient is not fixed, and is usually administered in a pharmaceutically effective amount.
- the amount of the compound to be actually administered can be determined by the physician according to the actual situation, including the disease to be treated, the route of administration selected, the actual compound administered, the individual condition of the patient, and the like. Dosage of the compounds of this invention will depend on the particular use for the treatment, the mode of administration, the state of the patient, and the judgment of the physician.
- the ratio or concentration of the compound of the present invention in the pharmaceutical composition depends on various factors, including dosage, physicochemical properties, route of administration and the like.
- compositions and methods of administration are provided.
- the compound of the present invention has excellent inhibitory activity on KIF18A, the compound of the present invention and its various crystal forms, pharmaceutically acceptable inorganic or organic salts, hydrates or solvates, and compounds containing the compound of the present invention as the main active ingredient
- the pharmaceutical composition can be used for treating, preventing and alleviating diseases related to KIF18A activity or expression.
- the pharmaceutical composition of the present invention comprises the compound of the present invention or a pharmacologically acceptable salt thereof within a safe and effective amount range and a pharmaceutically acceptable excipient or carrier.
- safe and effective dose refers to: the amount of the compound is sufficient to obviously improve the condition without causing severe side effects.
- the pharmaceutical composition contains 1-2000 mg of the compound of the present invention per dose, more preferably 5-200 mg of the compound of the present invention per dose.
- the "one dose” is a capsule or tablet.
- “Pharmaceutically acceptable carrier” refers to: one or more compatible solid or liquid fillers or gel substances, which are suitable for human use, and must have sufficient purity and low enough toxicity. "Compatibility” herein means that the components of the composition can be blended with the compound of the present invention and with each other without significantly reducing the efficacy of the compound.
- Examples of pharmaceutically acceptable carrier parts include cellulose and derivatives thereof (such as sodium carboxymethylcellulose, sodium ethylcellulose, cellulose acetate, etc.), gelatin, talc, solid lubricants (such as stearic acid , magnesium stearate), calcium sulfate, vegetable oil (such as soybean oil, sesame oil, peanut oil, olive oil, etc.), polyols (such as propylene glycol, glycerin, mannitol, sorbitol, etc.), emulsifiers (such as Tween ), wetting agent (such as sodium lauryl sulfate), coloring agent, flavoring agent, stabilizer, antioxidant, preservative, pyrogen-free water, etc.
- cellulose and derivatives thereof such as sodium carboxymethylcellulose, sodium ethylcellulose, cellulose acetate, etc.
- gelatin such as talc
- solid lubricants such as stearic acid , magnesium stearate
- the mode of administration of the compound or pharmaceutical composition of the present invention is not particularly limited, and representative modes of administration include (but are not limited to): oral, intratumoral, rectal, parenteral (intravenous, intramuscular or subcutaneous), and topical administration .
- Solid dosage forms for oral administration include capsules, tablets, pills, powders and granules.
- the active compound is admixed with at least one conventional inert excipient (or carrier), such as sodium citrate or dicalcium phosphate, or with (a) fillers or extenders, for example, Starch, lactose, sucrose, glucose, mannitol and silicic acid; (b) binders such as hydroxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose and acacia; (c) humectants, For example, glycerol; (d) disintegrants, such as agar, calcium carbonate, potato starch or tapioca starch, alginic acid, certain complex silicates, and sodium carbonate; (e) slow agents, such as paraffin; (f) Absorption accelerators such as quaternary ammonium compounds; (g) wetting agents such as cetyl alcohol and glyceryl monostea, or
- Solid dosage forms such as tablets, dragees, capsules, pills, and granules can be prepared with coatings and shell materials, such as enteric coatings and others well known in the art. They may contain opacifying agents and, in such compositions, the release of the active compound or compounds may be in a certain part of the alimentary canal in a delayed manner.
- coatings and shell materials such as enteric coatings and others well known in the art. They may contain opacifying agents and, in such compositions, the release of the active compound or compounds may be in a certain part of the alimentary canal in a delayed manner.
- Examples of usable embedding components are polymeric substances and waxy substances.
- the active compounds can also be in microencapsulated form, if desired, with one or more of the above-mentioned excipients.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups or tinctures.
- liquid dosage forms may contain inert diluents conventionally used in the art, such as water or other solvents, solubilizers and emulsifiers, for example, ethanol, isopropanol, ethyl carbonate, ethyl acetate, propylene glycol, 1 , 3-butanediol, dimethylformamide and oils, especially cottonseed oil, peanut oil, corn germ oil, olive oil, castor oil and sesame oil or mixtures of these substances, etc.
- inert diluents conventionally used in the art, such as water or other solvents, solubilizers and emulsifiers, for example, ethanol, isopropanol, ethyl carbonate, ethyl acetate, propylene glycol, 1 , 3-butanediol, dimethylformamide and
- compositions can also contain adjuvants, such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- Suspensions in addition to the active compounds, may contain suspending agents, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum methoxide and agar, or mixtures of these substances, and the like.
- suspending agents for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum methoxide and agar, or mixtures of these substances, and the like.
- compositions for parenteral injection may comprise physiologically acceptable sterile aqueous or anhydrous solutions, dispersions, suspensions or emulsions, and sterile powders for reconstitution into sterile injectable solutions or dispersions.
- Suitable aqueous and non-aqueous carriers, diluents, solvents or vehicles include water, ethanol, polyols, and suitable mixtures thereof.
- Dosage forms for topical administration of a compound of this invention include ointments, powders, patches, sprays and inhalants.
- the active ingredient is mixed under sterile conditions with a physiologically acceptable carrier and any preservatives, buffers, or propellants which may be required, if necessary.
- the compounds of the present invention may be administered alone or in combination with other pharmaceutically acceptable compounds.
- a safe and effective amount of the compound of the present invention is applied to a mammal (such as a human) in need of treatment, wherein the dosage is a pharmaceutically effective dosage when administered, for a person with a body weight of 60kg, the daily
- the dosage is usually 1-2000 mg, preferably 5-500 mg.
- factors such as the route of administration and the health status of the patient should also be considered for the specific dosage, which are within the skill of skilled physicians.
- KIF18A inhibitor with a novel structure, and its preparation and application.
- the inhibitor can inhibit the activity of KIF18A at a very low concentration.
- a KIF18A inhibitor with good oral absorption is provided.
- a pharmaceutical composition for treating diseases related to KIF18A activity is provided.
- Some representative compounds of the present invention can be prepared by the following synthetic methods.
- the reagents and conditions of each step can be selected from the conventional reagents or conditions of this type of preparation method in the art.
- the above selection can be made by those skilled in the art based on the knowledge in the art
- Boc tert-butoxycarbonyl
- DIPEA or DIEA N,N-Diisopropylethylamine
- DIBAL-H diisobutylaluminum hydride
- HATU N,N,N',N'-tetramethyl-O-(7-azabenzotriazol-1-yl)urea hexafluorophosphate
- LiHMDS lithium bistrimethylsilylamide
- NMP N-methylpyrrolidone
- Ph phenyl
- TBAF tetra-n-butylammonium fluoride
- TIPS Triisopropylsilyl
- Embodiment 1 the synthesis of compound 1
- Compound 1f was synthesized by the method in patent WO2020/132648.
- Compound 1e (67mg, 0.28mmol) was dissolved in acetonitrile (2mL), and compound 1f (100mg, 0.28mmol), N,N-diisopropylethylamine (108mg, 0.84mmol) and HATU (160mg, 0.42mmol) were added ) at room temperature for 12 hours.
- Embodiment 2 the synthesis of compound 2
- Embodiment 3 the synthesis of compound 3 and compound 4
- Embodiment 4 the synthesis of compound 5
- Embodiment 5 the synthesis of compound 6
- Compound 6a was synthesized by the method in patent WO2021/026101. Compound 6 was obtained from compound 6a through ring-closing reaction and coupling reaction.
- Embodiment 6 the synthesis of compound 7
- Compound 7a and compound 1b were obtained under basic conditions to obtain compound 7b, and then deprotected under acidic conditions to obtain compound 7c.
- Compound 7d was synthesized by the method in patent WO2021/026098.
- Compound 7 was obtained through coupling reaction between compound 7d and compound 7c.
- Embodiment 7 the synthesis of compound 8
- compound 8d was obtained through substitution reaction and oxidation reaction, and compound 8e was obtained through coupling reaction with compound 7d, compound 8g was obtained through deprotection reaction and oxidation reaction, and compound 8 was obtained by reacting with compound 1b.
- Embodiment 8 the synthesis of compound 9
- Embodiment 9 the synthesis of compound 10
- Embodiment 11 the synthesis of compound 12
- Compound 12a was synthesized by the method in patent WO2020132648.
- Embodiment 12 the synthesis of compound 13
- Compound 13a was synthesized by the method in US9527885.
- Embodiment 13 the synthesis of compound 14
- Embodiment 14 the synthesis of compound 15
- Compound 12a was synthesized by the method in patent WO2020132648.
- Embodiment 15 the synthesis of compound 16
- Embodiment 16 the synthesis of compound 17
- Embodiment 17 the synthesis of compound 18
- Embodiment 18 the synthesis of compound 19
- Embodiment 19 Synthesis of Compound 20
- Embodiment 20 the synthesis of compound 21
- Embodiment 21 the synthesis of compound 22
- Embodiment 22 the synthesis of compound 23
- the crude compound 23a was dissolved in tetrahydrofuran (1.5 mL), and stirred at -78°C under a nitrogen atmosphere. A solution of diisobutylaluminum hydride in n-hexane (1M, 0.4 mL) was then added. The mixture was stirred at -78°C and slowly warmed to 0°C for 1 hour. After the reaction was completed, it was quenched with ice water. The mixture was extracted with ethyl acetate (3 x 5 mL). The combined organic phases were washed with saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- Embodiment 23 the synthesis of compound 24
- Embodiment 24 the synthesis of compound 25
- Embodiment 25 the synthesis of compound 26
- Embodiment 26 Synthesis of compound 27
- Embodiment 27 the synthesis of compound 28
- Embodiment 28 Synthesis of compound 29
- Embodiment 29 Synthesis of compound 30
- Embodiment 30 the synthesis of compound 31
- Embodiment 31 the synthesis of compound 32
- Embodiment 32 the synthesis of compound 33
- Embodiment 33 the synthesis of compound 34
- Embodiment 34 the synthesis of compound 35 and compound 35'
- Embodiment 35 the synthesis of compound 36 and compound 36'
- Embodiment 36 the synthesis of compound 37 and compound 37'
- Embodiment 37 the synthesis of compound 38
- Dissolve compound 38e (5mg, 0.03mmol) in N,N-dimethylformamide (1mL), add sarcosine (0.5mg, 0.01mmol), cuprous iodide (0.9mg, 0.01mmol), phosphoric acid Potassium (13 mg, 0.06 mmol) was reacted at 50° C. for 5 minutes under a nitrogen atmosphere in a sealed tube. Then compound 12a (12 mg, 0.02 mmol) was added and reacted overnight at 100° C. under nitrogen atmosphere.
- Embodiment 38 the synthesis of compound 39
- Embodiment 39 the synthesis of compound 40
- Embodiment 40 the synthesis of compound 41
- Embodiment 41 the synthesis of compound 42
- Embodiment 42 the synthesis of compound 43
- Embodiment 43 the synthesis of compound 44
- Compound 44a was synthesized by the method in literature (Helvetica Chimica Acta, 1981, 64, 1849-1853).
- Compound 43b (75mg, 0.28mmol) was dissolved in dichloromethane (3mL), and compound 44a (70mg, 0.31mmol), N,N-diisopropylethylamine (150mg, 1.16mmol), N,N, N',N'-tetramethyl-O-(7-azabenzotriazol-1-yl)urea hexafluorophosphate (162 mg, 0.43 mmol) was reacted at room temperature for 96 hours.
- Embodiment 44 the synthesis of compound 45
- Embodiment 45 the synthesis of compound 46
- Compound 46a was synthesized by literature method (Chemical Communications (Cambridge, United Kingdom) 2021, 57(27), 3335-3338), compound 46a was nitrated to obtain compound 46b, compound 46b was subjected to catalytic hydrogenation to obtain compound 46c, compound 46c and compound Compound 46d was obtained by condensation reaction of 42a, and compound 46 was obtained by substitution reaction.
- Embodiment 46 the synthesis of compound 47
- Embodiment 47 the synthesis of compound 48
- Embodiment 48 the synthesis of compound 49
- Embodiment 49 the synthesis of compound 50
- Embodiment 50 the synthesis of compound 51
- Embodiment 51 the synthesis of compound 52
- Embodiment 52 the synthesis of compound 53
- Embodiment 53 the synthesis of compound 54
- Embodiment 54 the synthesis of compound 55
- Embodiment 55 the synthesis of compound 56
- Embodiment 56 the synthesis of compound 57
- Embodiment 57 the synthesis of compound 58 and compound 59
- Embodiment 58 the synthesis of compound 60
- Embodiment 59 the synthesis of compound 61 and compound 62
- Embodiment 60 the synthesis of compound 63
- reaction mixture was placed in a sealed tube, heated and stirred at 85° C. for 3 hours under a nitrogen atmosphere.
- the reaction mixture was cooled to room temperature, tert-butylamine (10 mg, 0.14 mmol) and sodium hypochlorite (10 mg, 0.14 mmol) were added and stirred overnight at room temperature.
- the reaction mixture was added with water, extracted with ethyl acetate (10mL ⁇ 3), the combined organic layers were washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- Embodiment 61 the synthesis of compound 64
- Embodiment 62 the synthesis of compound 65
- Embodiment 63 the synthesis of compound 66 and compound 67
- Embodiment 64 the synthesis of compound 68
- Embodiment 65 the synthesis of compound 69
- Embodiment 66 the synthesis of compound 70
- Embodiment 67 the synthesis of compound 71
- Dissolve compound 1h (20 mg, 0.16 mmol), sarcosine (4 mg, 0.05 mmol), cuprous iodide (19 mg, 0.10 mmol), potassium phosphate (64 mg, 0.03 mmol) in N,N-dimethylformamide (1 mL) was placed in a sealed tube under nitrogen atmosphere at 50°C for 5 minutes. Then compound 71d (60 mg, 0.10 mmol) was added and reacted at 100° C. for 4 hours under nitrogen atmosphere.
- Embodiment 68 the synthesis of compound 72
- Embodiment 69 the synthesis of compound 73
- Embodiment 70 the synthesis of compound 74
- Embodiment 71 the synthesis of compound 75
- Embodiment 72 the synthesis of compound 76
- Embodiment 73 the synthesis of compound 77
- Embodiment 74 the synthesis of compound 78
- Embodiment 75 the synthesis of compound 79, compound 80, compound 81
- Embodiment 76 the synthesis of compound 82
- Embodiment 77 the synthesis of compound 83
- Embodiment 78 the synthesis of compound 84
- Embodiment 79 the synthesis of compound 85
- Example 81 Compounds Inhibit the Proliferation of Ovary Cancer Cell Line OVCAR-3, Breast Cancer Cell Lines HCC1954 and HCC1806
- OVCAR-3, HCC1954 and HCC1806 cells in good growth state were selected and digested with trypsin. Add fresh medium, mix well, and centrifuge at 800rpm for 3 minutes. According to the seeding density of 3300 OVCAR-3 cells, 2500 HCC1954 cells and 3000 HCC1806 cells per well, they were seeded in 96-well plates and cultured overnight in a 37°C incubator. On the second day, the culture plate was taken out, and the compound was diluted in a four-fold gradient for administration. Among them, the blank control wells only added normal medium, no cells, and no DMSO; the DMSO wells contained cells and contained 0.5% DMSO. The 96-well plate was placed in a 37°C incubator for 96 hours.
- Table 1 Inhibitory activity of compounds on OVCAR-3, HCC1954 and HCC1806 cell proliferation
- the enzyme activity of KIF18A was tested by ADP-Glo method.
- the compound was diluted with DMSO according to the initial concentration of 3000nM, 3-fold gradient dilution, a total of ten concentrations, repeated hole test.
- Use Echo to transfer 25nL of the diluted compound to a 384-well plate, add 2.5 ⁇ L of enzyme working solution, centrifuge at 1000rpm for 1min, and incubate at 25°C for 10min. Add 2.5 ⁇ L of ATP and substrate solution to initiate the reaction, and incubate at 25° C. for 60 min.
- Inhibition rate (%) (control reading of DMSO well-reading of compound well)/(control reading of DMSO well-reading of blank control) ⁇ 100%.
- the enzyme inhibitory activity data of some representative compounds are shown in Table 2.
- Embodiment 83 Pharmacokinetic study in rat body
- Chromatographic column ACQUITY UPLC BEH C18 (2.1mm ⁇ 50mm, 1.7 ⁇ m); column temperature 40°C; mobile phase A is water (0.1% formic acid), mobile phase B is acetonitrile, the flow rate is 0.350 ml/min, using gradient elution , the elution gradient is 0.50min: 10%B; 1.50min: 90%B; 2.50min: 90%B; 2.51min: 10%B; 3.50min: stop. Injection volume: 1 ⁇ L.
- Animals 3 SD male rats, with a weight range of 200-220g, were used after being raised in the laboratory of the Experimental Animal Center for 2 days after purchase, fasted for 12 hours before administration and within 4 hours after administration, and free to drink water during the test. After the rats were gavaged, blood samples were collected within a predetermined period of time.
- Drug samples generally take multiple samples with similar structures (the difference in molecular weight is more than 2 units), weigh them accurately, and administer them together (cassette PK). This allows multiple compounds to be screened simultaneously and their oral absorption rates compared. Pharmacokinetics of drug samples in rats were also studied using single administration.
- Blood was collected from the orbit at 0.25, 0.5, 1, 2, 4, 8, 10 and 24 hours after intragastric administration. Take 50 ⁇ L of plasma sample, add 200 ⁇ L of acetonitrile (containing internal standard verapamil 2ng/mL), vortex for 3 min, centrifuge at 20000 rcf, 4 °C for 10 min, and take the supernatant for LC-MS/MS analysis.
- acetonitrile containing internal standard verapamil 2ng/mL
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Virology (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
















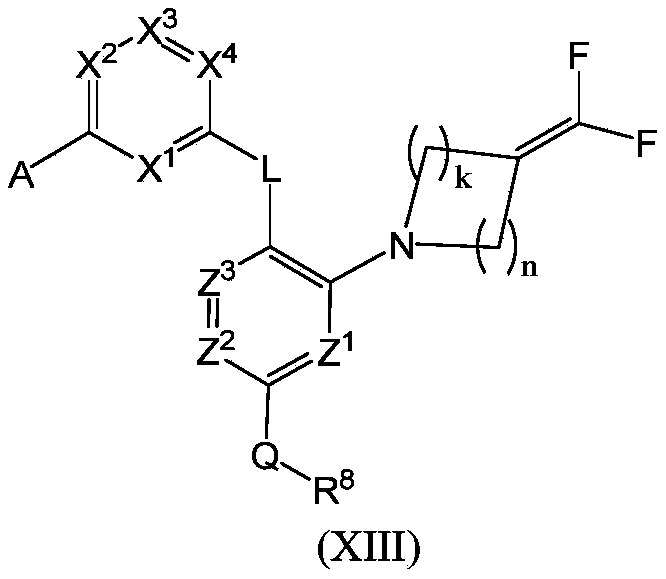
















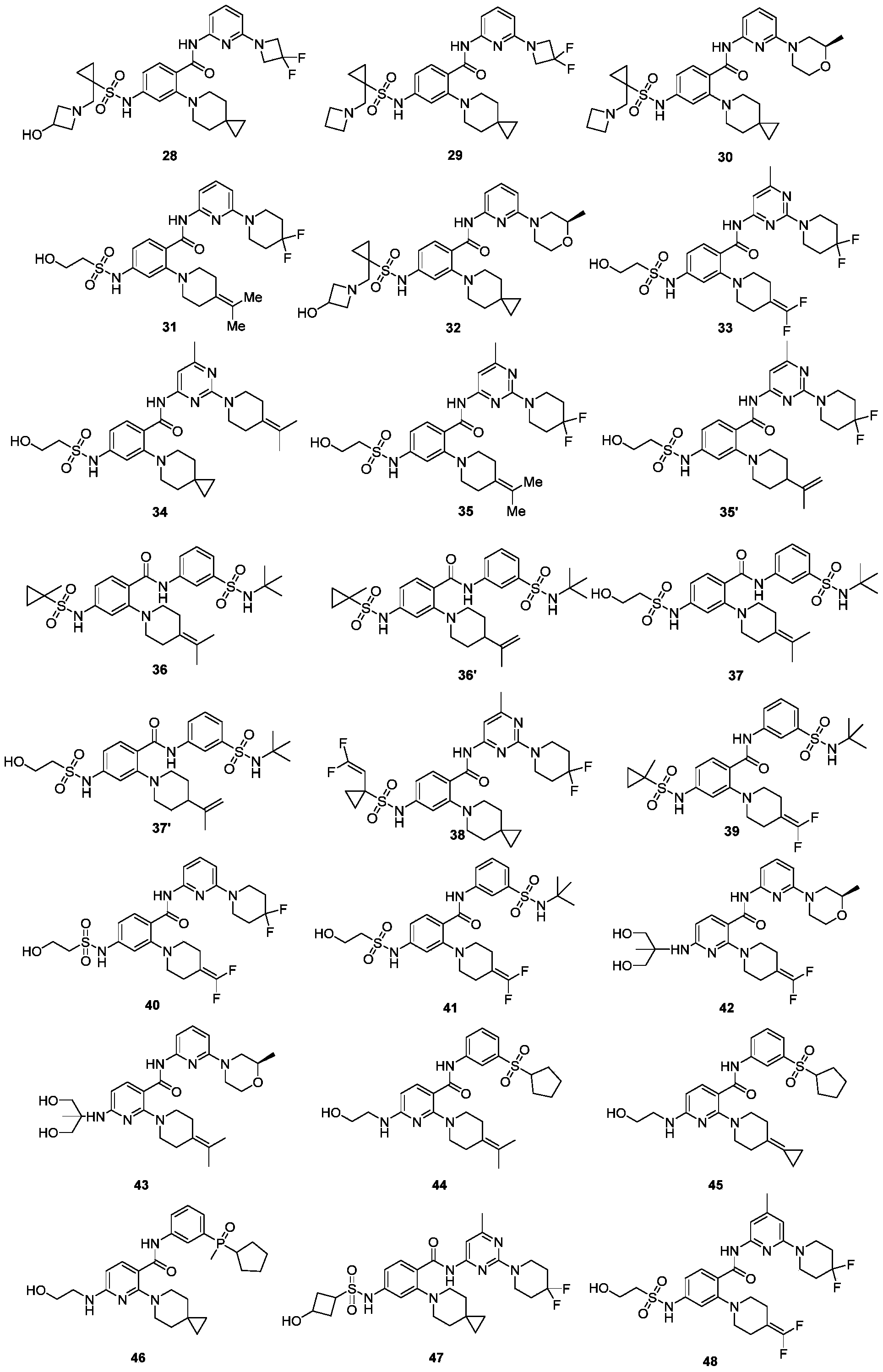


















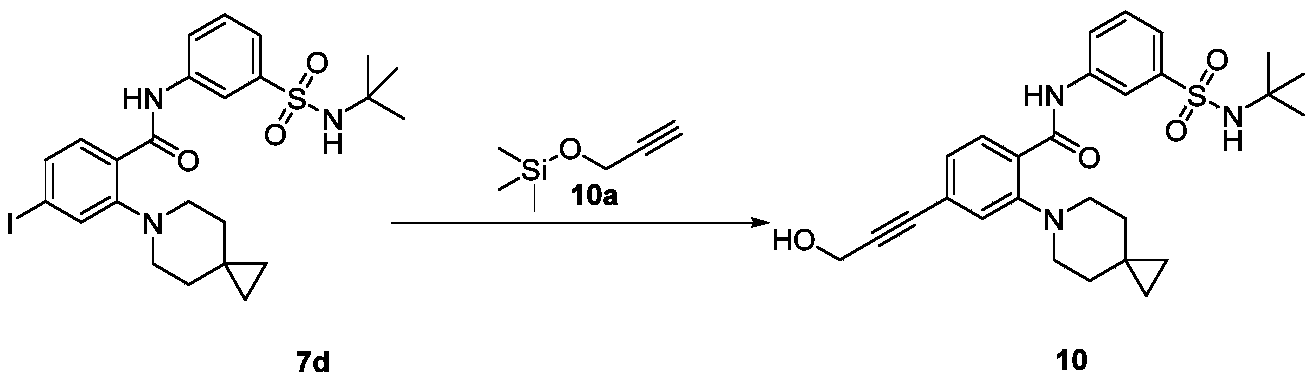





















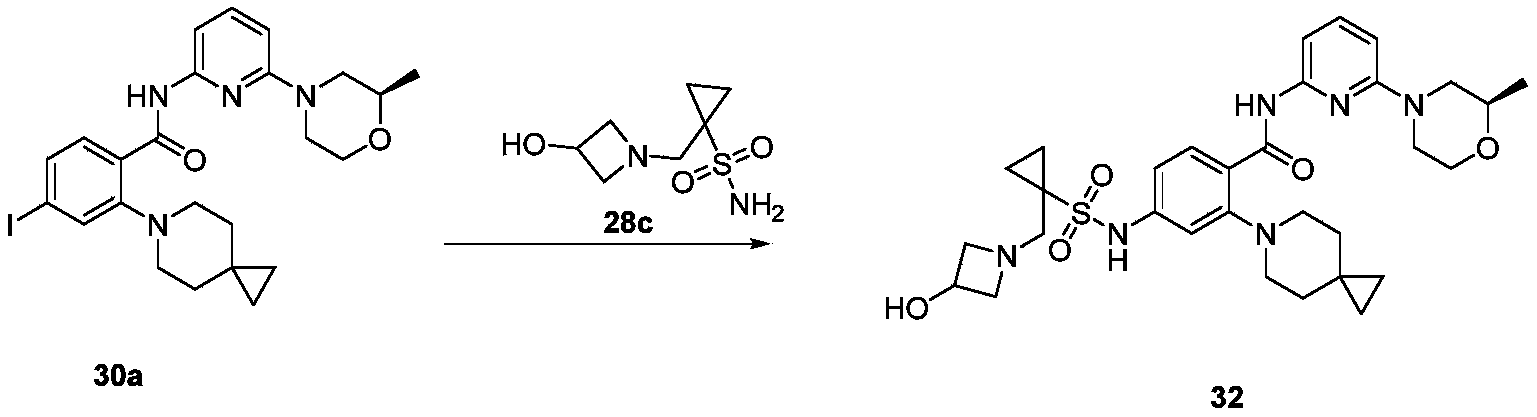














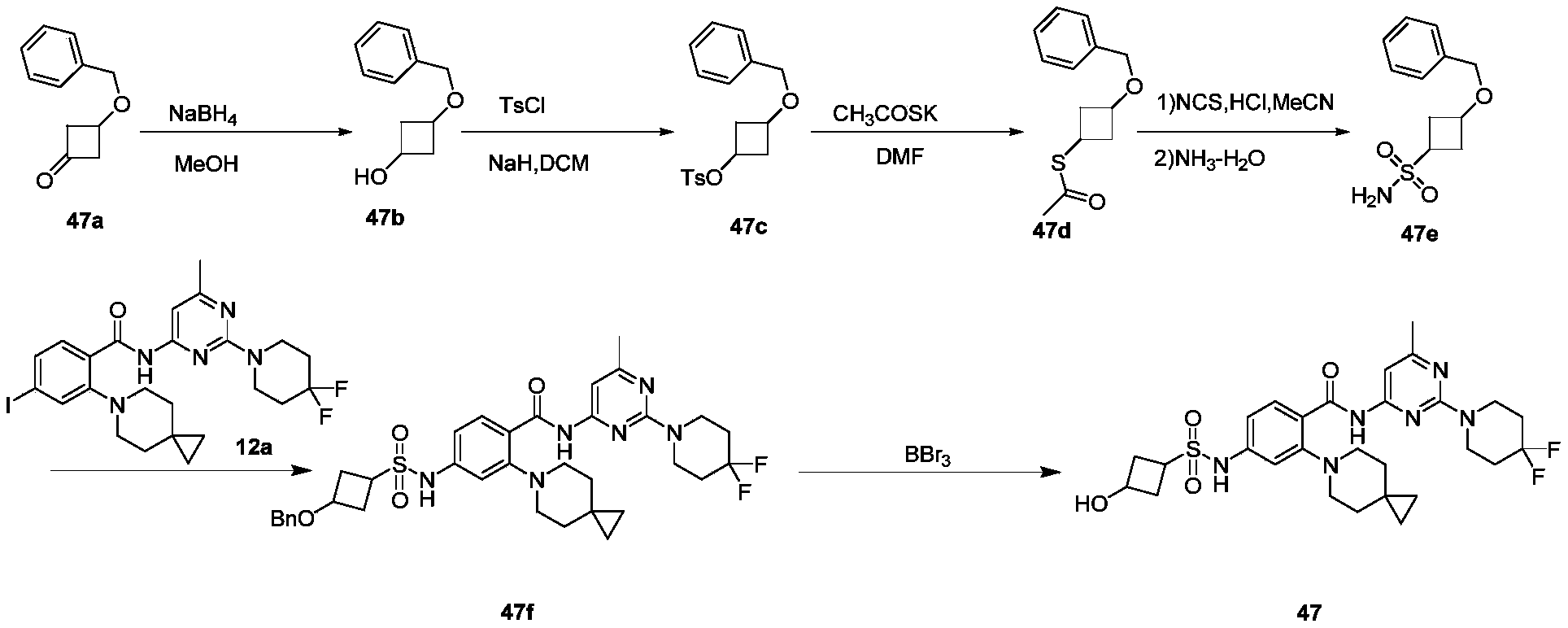











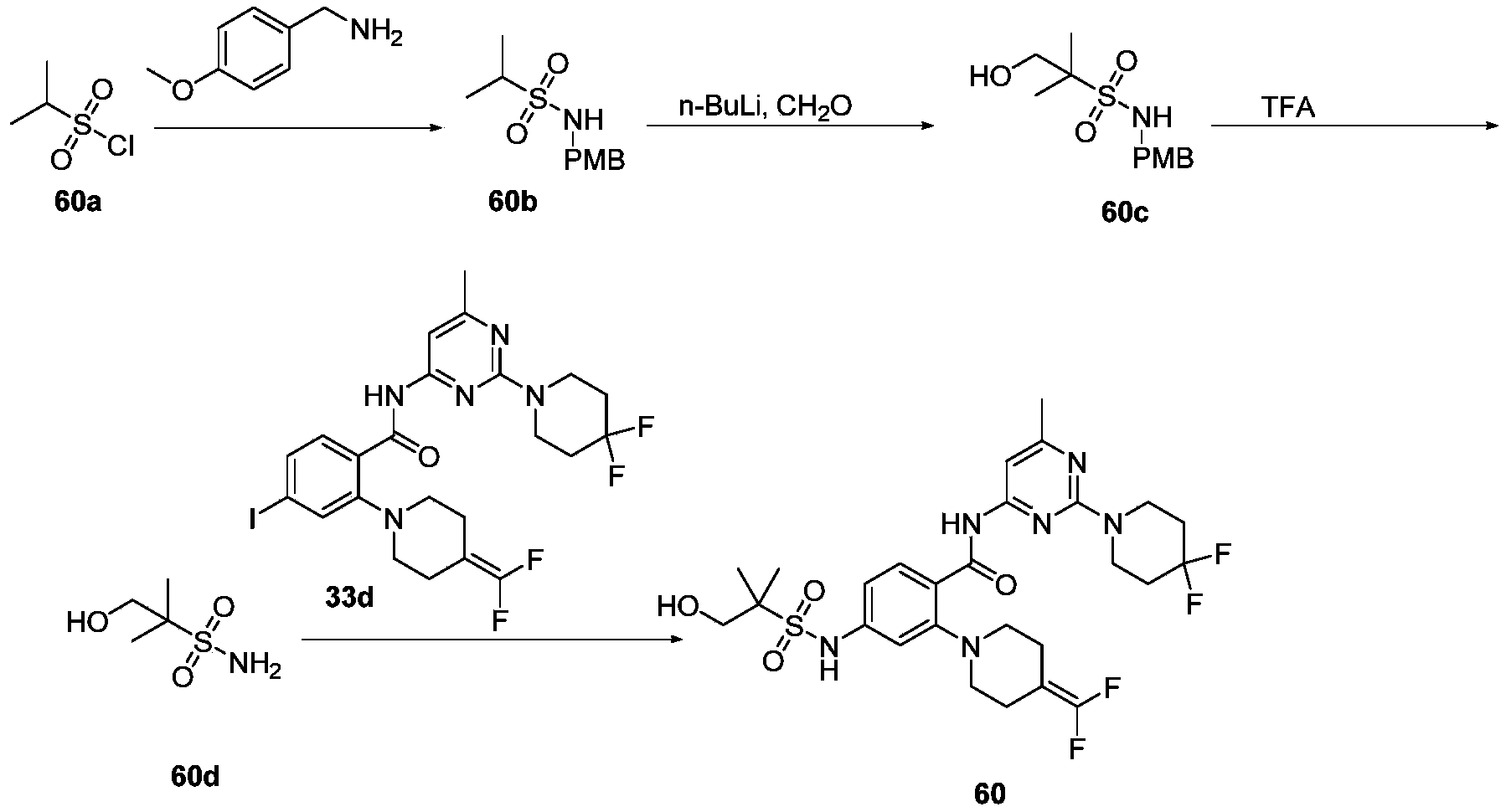








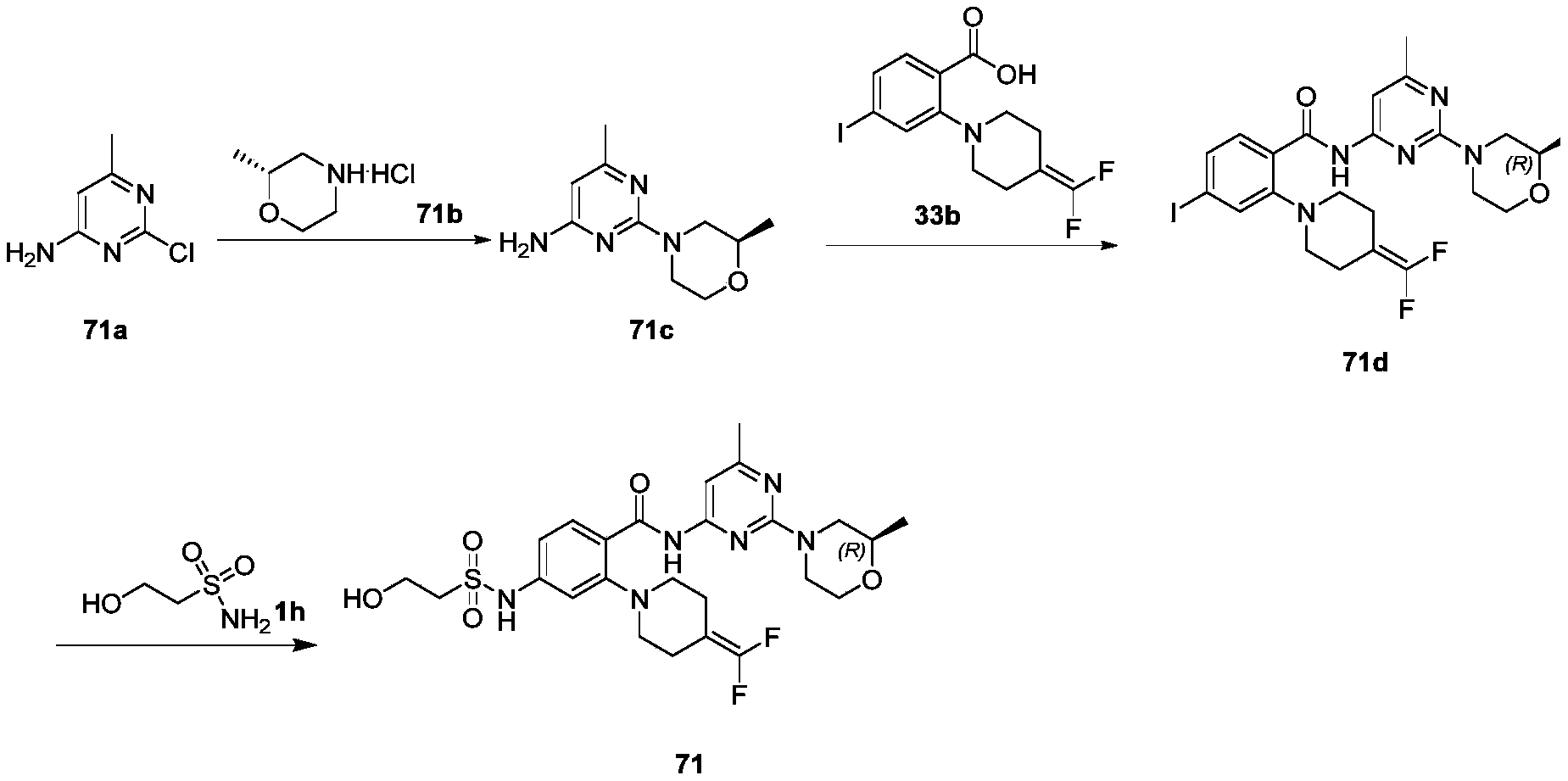




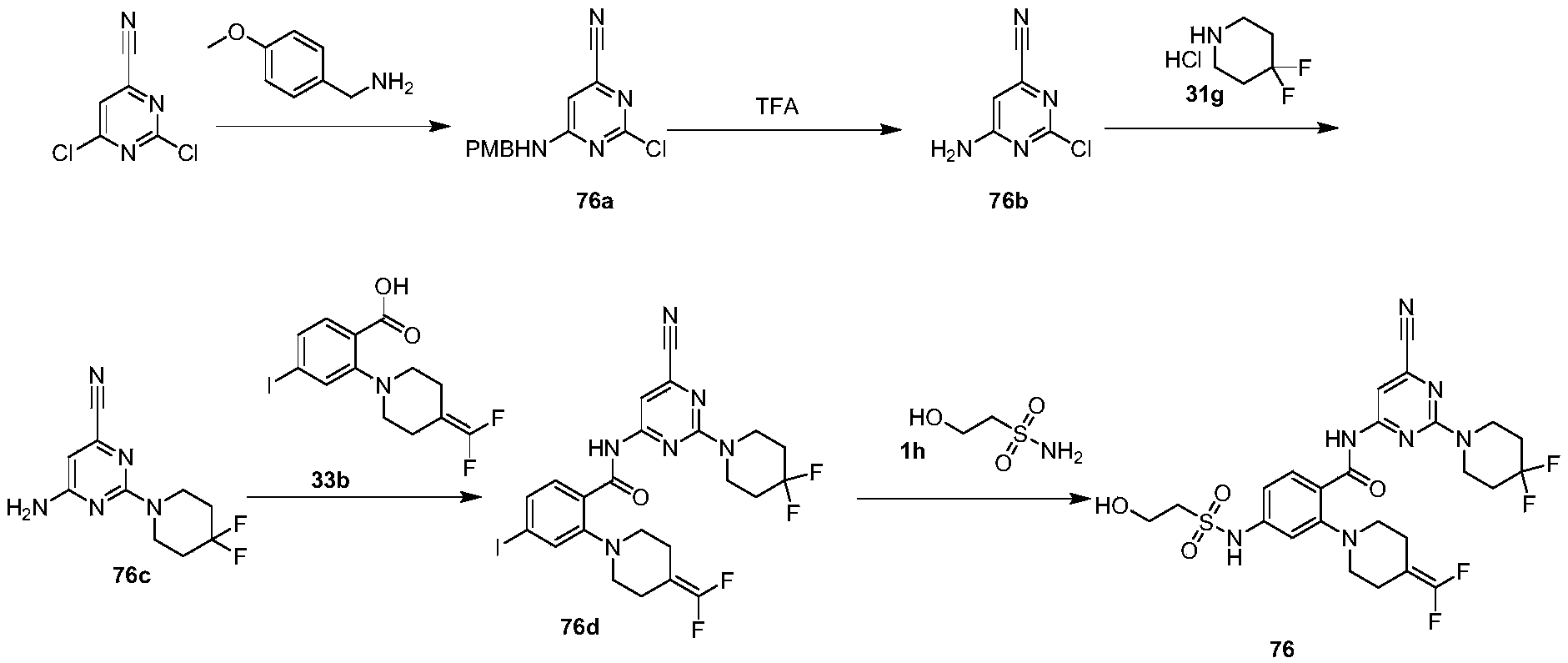















Claims (15)
- 一种如下式(I)所示结构的化合物,或其光学异构体,药学上可接受的盐,前药,氘代衍生物,水合物,溶剂合物:
 式(I)中:A选自-Y-R 1;其中,Y选自化学键、-S(O)-、-S(O) 2-、-S(O)(NH)-、-S(O) 2NR a-、-NR aS(O) 2-、-NR aS(O) 2NR a-、-P(O)(R q)-、-P(O)(R q)NR a-、-NR aP(O)(R q)-、-P(O)(R q)O-、-OP(O)(R q)-、-NR a-、-O-、-S-、-C(O)NR a-、-NR aC(O)-、-C(O)O-、-OC(O)-、-NR aC(O)NR a-、-OC(O)NR a-、-NR aC(O)O-、C 2-4烯基、或C 2-4炔基;其中,各个R a各自独立地选自氢或C 1- 4烷基;各个R q各自独立地选自C 1-4烷基、C 3-6环烷基、或3-至6-元杂环基;或R q和R 1与连接的磷原子一起共同形成任意取代的4-至8-元的环状结构,此环状结构可额外含有0-1个任选自N、O、S的杂原子;或A选自式(Ia):
式(I)中:A选自-Y-R 1;其中,Y选自化学键、-S(O)-、-S(O) 2-、-S(O)(NH)-、-S(O) 2NR a-、-NR aS(O) 2-、-NR aS(O) 2NR a-、-P(O)(R q)-、-P(O)(R q)NR a-、-NR aP(O)(R q)-、-P(O)(R q)O-、-OP(O)(R q)-、-NR a-、-O-、-S-、-C(O)NR a-、-NR aC(O)-、-C(O)O-、-OC(O)-、-NR aC(O)NR a-、-OC(O)NR a-、-NR aC(O)O-、C 2-4烯基、或C 2-4炔基;其中,各个R a各自独立地选自氢或C 1- 4烷基;各个R q各自独立地选自C 1-4烷基、C 3-6环烷基、或3-至6-元杂环基;或R q和R 1与连接的磷原子一起共同形成任意取代的4-至8-元的环状结构,此环状结构可额外含有0-1个任选自N、O、S的杂原子;或A选自式(Ia): 其中,表示式(Ia)片段与式(I)化合物其它部分连接的位点;
其中,表示式(Ia)片段与式(I)化合物其它部分连接的位点; L选自-C(O)NR b-、-NR bC(O)-、-NR bC(O)NR b-、-OC(O)NR b-、-C(O)O-、-OC(O)-、-NR bC(O)O-、-S(O) 2NR b-、-NR bS(O) 2-、-NR b-、-O-、3-至6-元杂环基、或杂芳基;其中,各个R b各自独立地选自氢或C 1-4烷基;B选自式(Ib)或(Id):
L选自-C(O)NR b-、-NR bC(O)-、-NR bC(O)NR b-、-OC(O)NR b-、-C(O)O-、-OC(O)-、-NR bC(O)O-、-S(O) 2NR b-、-NR bS(O) 2-、-NR b-、-O-、3-至6-元杂环基、或杂芳基;其中,各个R b各自独立地选自氢或C 1-4烷基;B选自式(Ib)或(Id): 其中,表示式(Ib)或(Id)片段与式(I)化合物中L的连接位点;
其中,表示式(Ib)或(Id)片段与式(I)化合物中L的连接位点; X 1、X 2、X 3和X 4各自独立地选自N或CR c;其中,R c选自选自氢、卤素、C 1-4烷基、C 1- 4卤代烷基、C 2-4炔基、C 1-4烷氧基、羟基、C 1-4卤代烷氧基、CN、NR dR d、C 3-6环烷基、或3-至8-元杂环基;其中,各个R d各自独立地选自氢、C 1-4烷基、或C 1-4卤代烷基;U选自N或CR e;其中,R e选自选自氢、卤素、C 1-4烷基;Z 1、Z 2、和Z 3各自独立地选自N或CR m;其中,R m选自选自氢、卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、羟基、C 1-4卤代烷氧基、CN、NR dR d、C 3-8环烷基、或3-至8-元杂环基; 其中,各个R d的定义如上所述;D选自化学键、-O-、-S-、NR d-、C 1-2烷基、或C 2-4炔基;G选自3-至-10元饱和或不饱和环状结构,此环状结构任选地含有0、1、或2个选自N、O、S的杂原子,或任选地被R 6和R 7取代;R 1选自下组:C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-6环烷基、3-至10-元杂环基;所述的烷基、烯基、炔基、环烷基或杂环基任选地被一个或多个选自下组的基团取代:卤素、C 1-4烷基、C 1-4卤代烷基、C 2-4烯基、C 2-4卤代烯基、C 2-4炔基、C 2-4卤代炔基、C 3-6环烷基、3-至6-元杂环基、CN、OR f、SR f、NR dR d、-OC 1-4烷基OR f、-OC 1-4烷基NR dR d、-NR dC 1-4烷基OR f、-NR dC 1-4烷基NR dR d、-C(O)R g、-C(O)OR f、-OC(O)R g、-C(O)NR dR d、-NR dC(O)R g、-NR dC(O)NR dR d、-OC(O)NR dR d、-NR dC(O)OR f、-OC(O)OR f、-S(O) 2NR dR d、-NR dS(O) 2R g、-NR dS(O) 2NR dR d;或所述的环烷基或杂环基任选地被=M取代;其中,R f选自氢、C 1-4烷基、或C 1-4卤代烷基;R g选自氢、C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-8环烷基、4-至8-元杂环基、芳基、或杂芳基;M选自O或CR hR i;其中,R h和R i各自独立地选自氢、卤素、或C 1-4烷基;其中,R h或R i中所述的烷基任选地被一个或多个选自下组的基团取代:氢、卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、C 1-4卤代烷氧基、羟基、NR dR d、C 3-8环烷基、3-至8-元杂环基、芳基、或杂芳基;或R h和R i与其连接的碳原子一起形成3-至-6元环状结构,此环状结构任选地含有0、1、或2个选自N、O、S的杂原子;各个R d的定义如上所述;R 2和R 3各自独立地选自下组:氢、氟、或C 1-4烷基;其中,所述的烷基任选地被一个或多个选自下组的基团取代:卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、C 1-4卤代烷氧基、羟基、NR dR d、C 3-8环烷基、3-至8-元杂环基、芳基、或杂芳基;其中,各个R d的定义如上所述;各个R 4各自独立地选自氢、卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、C 1-4烷氧基C 1- 4烷基、羟基、羟基C 1-4烷基、C 1-4卤代烷氧基、C 1-4卤代烷氧基C 1-4烷基、或CN;R 5选自-Q-R 8;其中,Q选自化学键、-S(O)-、-S(O) 2-、-S(O)(NH)-、-S(O) 2NR a-、-NR aS(O) 2-、-NR aS(O) 2NR a-、-NR a-、-O-、-S-、-C(O)NR a-、-NR aC(O)-、-C(O)O-、-OC(O)-、-NR aC(O)NR a-、-OC(O)NR a-、-NR aC(O)O-、C 2-4烯基、或C 2-4炔基;其中,各个R a的定义如上所述;R 6和R 7各自独立地选自氢、卤素、C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-8环烷基、4-至8-元杂环基;或R 6和R 7与其连接的碳原子一起形成3-至-6元环状结构,此环状结构任选地含有0、1、或2个选自N、O、S的杂原子;或R 6和R 7与其连接的碳原子共同形成C=T,其中,T选自CR jR k;其中,R j和R k各自独立地选自下组:氢、氘、卤素、或C 1-4烷基;所述的烷基任选地被一个或多个选自下组的基团取代:卤素、C 1-4卤代烷基、C 2-4烯基、C 2-4炔基、C 3-6环烷基、3-至6-元杂环基、CN、OR f、SR f、或NR dR d;或R j和R k与其连接的碳原子一起形成3-至-6元环状结构,此环状结构任选地含有0、或1个选自N、O、S的杂原子;R d和R f的定义如上所述;R 8选自下组:C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-6环烷基、3-至10-元杂环基;所述的烷基、烯基、炔基、环烷基或杂环基任选地被一个或多个选自下组的基团取代:卤素、C 1-4烷基、C 1-4卤代烷基、C 2-4烯基、C 2-4炔基、C 3-6环烷基、3-至6-元杂环基、CN、OR f、SR f、NR dR d、-OC 1-4烷基OR f、-OC 1-4烷基NR dR d、-NR dC 1-4烷基OR f、-NR dC 1-4烷基NR dR d、-C(O)R g、-C(O)OR f、-OC(O)R g、-C(O)NR dR d、-NR dC(O)R g、-NR dC(O)NR dR d、-OC(O)NR dR d、-NR dC(O)OR f、-OC(O)OR f、-S(O) 2NR dR d、-NR dS(O) 2R g、-NR dS(O) 2NR dR d;或所述的环烷基或杂环基被=M取代;其中,R f选自氢、C 1-4烷基、或C 1-4卤代烷基;R g选自氢、C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-8环烷基、4-至8-元杂环基、芳基、或杂芳基;M选自O或CR hR i; 其中,R h和R i各自独立地选自氢、卤素、或C 1-4烷基;其中,R h或R i中所述的烷基任选地被一个或多个选自下组的基团取代:氢、卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、C 1-4卤代烷氧基、羟基、NR dR d、C 3-8环烷基、3-至8-元杂环基、芳基、或杂芳基;或R h和R i与其连接的碳原子一起形成3-至-6元环状结构,此环状结构任选地含有0、1、或2个选自N、O、S的杂原子;各个R d的定义如上所述;p和q各自独立地选自0、1、2、3、4、5、或6;前提条件是p和q不能同时选自0;m选自0、1、2、3、或4;k和n选自1、2、3、4、或5;其中,各个上述的烷基、烯基、炔基、环烷基、杂环基、芳基和杂芳基任选地且各自独立地被1-3个选自下组的取代基取代:卤素、C 1-4烷基、C 1-4卤代烷基、C 2-4烯基、C 2-4炔基、C 3-8环烷基、3-至8-元杂环基、芳基、杂芳基、CN、NO 2、OR f、SR f、NR dR d、C(O)R g、C(O)OR f、C(O)NR dR d、NR dC(O)R g、NR dS(O) 2R g、或S(O) 2R g,前提条件是所形成的化学结构是稳定的和有意义的;其中,R d、R f、和R g的定义如上所述。除非特别说明,上述的芳基为含有6-12个碳原子的芳香基团;杂芳基为5-至15-元(优选为5-至12-元)杂芳香基团;环状结构为饱和的或不饱和的、含杂原子或不含杂原子的环状基团。
X 1、X 2、X 3和X 4各自独立地选自N或CR c;其中,R c选自选自氢、卤素、C 1-4烷基、C 1- 4卤代烷基、C 2-4炔基、C 1-4烷氧基、羟基、C 1-4卤代烷氧基、CN、NR dR d、C 3-6环烷基、或3-至8-元杂环基;其中,各个R d各自独立地选自氢、C 1-4烷基、或C 1-4卤代烷基;U选自N或CR e;其中,R e选自选自氢、卤素、C 1-4烷基;Z 1、Z 2、和Z 3各自独立地选自N或CR m;其中,R m选自选自氢、卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、羟基、C 1-4卤代烷氧基、CN、NR dR d、C 3-8环烷基、或3-至8-元杂环基; 其中,各个R d的定义如上所述;D选自化学键、-O-、-S-、NR d-、C 1-2烷基、或C 2-4炔基;G选自3-至-10元饱和或不饱和环状结构,此环状结构任选地含有0、1、或2个选自N、O、S的杂原子,或任选地被R 6和R 7取代;R 1选自下组:C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-6环烷基、3-至10-元杂环基;所述的烷基、烯基、炔基、环烷基或杂环基任选地被一个或多个选自下组的基团取代:卤素、C 1-4烷基、C 1-4卤代烷基、C 2-4烯基、C 2-4卤代烯基、C 2-4炔基、C 2-4卤代炔基、C 3-6环烷基、3-至6-元杂环基、CN、OR f、SR f、NR dR d、-OC 1-4烷基OR f、-OC 1-4烷基NR dR d、-NR dC 1-4烷基OR f、-NR dC 1-4烷基NR dR d、-C(O)R g、-C(O)OR f、-OC(O)R g、-C(O)NR dR d、-NR dC(O)R g、-NR dC(O)NR dR d、-OC(O)NR dR d、-NR dC(O)OR f、-OC(O)OR f、-S(O) 2NR dR d、-NR dS(O) 2R g、-NR dS(O) 2NR dR d;或所述的环烷基或杂环基任选地被=M取代;其中,R f选自氢、C 1-4烷基、或C 1-4卤代烷基;R g选自氢、C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-8环烷基、4-至8-元杂环基、芳基、或杂芳基;M选自O或CR hR i;其中,R h和R i各自独立地选自氢、卤素、或C 1-4烷基;其中,R h或R i中所述的烷基任选地被一个或多个选自下组的基团取代:氢、卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、C 1-4卤代烷氧基、羟基、NR dR d、C 3-8环烷基、3-至8-元杂环基、芳基、或杂芳基;或R h和R i与其连接的碳原子一起形成3-至-6元环状结构,此环状结构任选地含有0、1、或2个选自N、O、S的杂原子;各个R d的定义如上所述;R 2和R 3各自独立地选自下组:氢、氟、或C 1-4烷基;其中,所述的烷基任选地被一个或多个选自下组的基团取代:卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、C 1-4卤代烷氧基、羟基、NR dR d、C 3-8环烷基、3-至8-元杂环基、芳基、或杂芳基;其中,各个R d的定义如上所述;各个R 4各自独立地选自氢、卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、C 1-4烷氧基C 1- 4烷基、羟基、羟基C 1-4烷基、C 1-4卤代烷氧基、C 1-4卤代烷氧基C 1-4烷基、或CN;R 5选自-Q-R 8;其中,Q选自化学键、-S(O)-、-S(O) 2-、-S(O)(NH)-、-S(O) 2NR a-、-NR aS(O) 2-、-NR aS(O) 2NR a-、-NR a-、-O-、-S-、-C(O)NR a-、-NR aC(O)-、-C(O)O-、-OC(O)-、-NR aC(O)NR a-、-OC(O)NR a-、-NR aC(O)O-、C 2-4烯基、或C 2-4炔基;其中,各个R a的定义如上所述;R 6和R 7各自独立地选自氢、卤素、C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-8环烷基、4-至8-元杂环基;或R 6和R 7与其连接的碳原子一起形成3-至-6元环状结构,此环状结构任选地含有0、1、或2个选自N、O、S的杂原子;或R 6和R 7与其连接的碳原子共同形成C=T,其中,T选自CR jR k;其中,R j和R k各自独立地选自下组:氢、氘、卤素、或C 1-4烷基;所述的烷基任选地被一个或多个选自下组的基团取代:卤素、C 1-4卤代烷基、C 2-4烯基、C 2-4炔基、C 3-6环烷基、3-至6-元杂环基、CN、OR f、SR f、或NR dR d;或R j和R k与其连接的碳原子一起形成3-至-6元环状结构,此环状结构任选地含有0、或1个选自N、O、S的杂原子;R d和R f的定义如上所述;R 8选自下组:C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-6环烷基、3-至10-元杂环基;所述的烷基、烯基、炔基、环烷基或杂环基任选地被一个或多个选自下组的基团取代:卤素、C 1-4烷基、C 1-4卤代烷基、C 2-4烯基、C 2-4炔基、C 3-6环烷基、3-至6-元杂环基、CN、OR f、SR f、NR dR d、-OC 1-4烷基OR f、-OC 1-4烷基NR dR d、-NR dC 1-4烷基OR f、-NR dC 1-4烷基NR dR d、-C(O)R g、-C(O)OR f、-OC(O)R g、-C(O)NR dR d、-NR dC(O)R g、-NR dC(O)NR dR d、-OC(O)NR dR d、-NR dC(O)OR f、-OC(O)OR f、-S(O) 2NR dR d、-NR dS(O) 2R g、-NR dS(O) 2NR dR d;或所述的环烷基或杂环基被=M取代;其中,R f选自氢、C 1-4烷基、或C 1-4卤代烷基;R g选自氢、C 1-4烷基、C 2-4烯基、C 2-4炔基、C 3-8环烷基、4-至8-元杂环基、芳基、或杂芳基;M选自O或CR hR i; 其中,R h和R i各自独立地选自氢、卤素、或C 1-4烷基;其中,R h或R i中所述的烷基任选地被一个或多个选自下组的基团取代:氢、卤素、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基、C 1-4卤代烷氧基、羟基、NR dR d、C 3-8环烷基、3-至8-元杂环基、芳基、或杂芳基;或R h和R i与其连接的碳原子一起形成3-至-6元环状结构,此环状结构任选地含有0、1、或2个选自N、O、S的杂原子;各个R d的定义如上所述;p和q各自独立地选自0、1、2、3、4、5、或6;前提条件是p和q不能同时选自0;m选自0、1、2、3、或4;k和n选自1、2、3、4、或5;其中,各个上述的烷基、烯基、炔基、环烷基、杂环基、芳基和杂芳基任选地且各自独立地被1-3个选自下组的取代基取代:卤素、C 1-4烷基、C 1-4卤代烷基、C 2-4烯基、C 2-4炔基、C 3-8环烷基、3-至8-元杂环基、芳基、杂芳基、CN、NO 2、OR f、SR f、NR dR d、C(O)R g、C(O)OR f、C(O)NR dR d、NR dC(O)R g、NR dS(O) 2R g、或S(O) 2R g,前提条件是所形成的化学结构是稳定的和有意义的;其中,R d、R f、和R g的定义如上所述。除非特别说明,上述的芳基为含有6-12个碳原子的芳香基团;杂芳基为5-至15-元(优选为5-至12-元)杂芳香基团;环状结构为饱和的或不饱和的、含杂原子或不含杂原子的环状基团。 - 如权利要求1所述的化合物,其特征在于,式(I)为式(XIa)或式(XIb):
 其中,片段或片段
其中,片段或片段 选自下组基团:
选自下组基团:
 表示片段与式(XIa)或式(XIb)化合物其它部分连接的位点;
表示片段与式(XIa)或式(XIb)化合物其它部分连接的位点; A选自下组基团:
A选自下组基团: 表示A与式(XIa)或式(XIb)化合物其它部分连接的位点;
表示A与式(XIa)或式(XIb)化合物其它部分连接的位点; X 1、X 2、X 3和X 4的定义如利要求1所述;片段选自下组基团:
X 1、X 2、X 3和X 4的定义如利要求1所述;片段选自下组基团:
 表示片段与L连接的位点;
表示片段与L连接的位点; 表示片段与Q连接的位点;
表示片段与Q连接的位点; 表示片段 与
表示片段 与 连接的位点;
连接的位点; 片段选自下组基团:
片段选自下组基团:
 表示片段
表示片段 与式(XIa)或式(XIb)化合物其它部分连接的位点;
与式(XIa)或式(XIb)化合物其它部分连接的位点; L选自-C(O)NH-、-NHC(O)-、-S(O) 2NH-、或-NHS(O) 2-;“*”表示手性中心。
L选自-C(O)NH-、-NHC(O)-、-S(O) 2NH-、或-NHS(O) 2-;“*”表示手性中心。 - 如权利要求2所述的化合物,其特征在于:式(XIa)或式(XIb)中片段或片段
 选自下组基团:
选自下组基团:
 表示片段与式(XIa)或式(XIb)化合物其它部分连接的位点。
表示片段与式(XIa)或式(XIb)化合物其它部分连接的位点。
- 如权利要求2所述的化合物,其特征在于:式(XIa)中片段选自下组基团:

 表示片段与式(XIa)化合物其它部分连接的位点。
表示片段与式(XIa)化合物其它部分连接的位点。
- 如权利要求1所述的化合物,其特征在于,式(I)为式(XII):
 其中,R j和R k各自独立地选自下组:氢、氘、氟、氯、溴、或C 1-4烷基;k和n选自1、2、3、4、或5;其余各基团的定义如利要求2所述。
其中,R j和R k各自独立地选自下组:氢、氘、氟、氯、溴、或C 1-4烷基;k和n选自1、2、3、4、或5;其余各基团的定义如利要求2所述。 - 如权利要求1所述的化合物,其特征在于,式(I)为式(XIII):
 k和n选自1、2、3、或4;A选自下组基团:
k和n选自1、2、3、或4;A选自下组基团: 表示A与式(XIII)化合物其它部分连接的位点;
表示A与式(XIII)化合物其它部分连接的位点; 片段选自下组基团:
片段选自下组基团:
 表示片段与A连接的位点;
表示片段与A连接的位点; 表示与L连接的位点;
表示与L连接的位点; 片段选自下组基团:
片段选自下组基团:
 表示片段与L连接的位点;
表示片段与L连接的位点; 表示片段与Q连接的位点;
表示片段与Q连接的位点; 表示片段 与
表示片段 与 连接的位点;
连接的位点;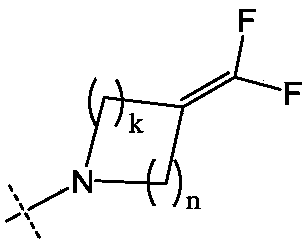 片段
片段

 表示片段
表示片段 与式(XIII)化合物其它部分连接的位点;
与式(XIII)化合物其它部分连接的位点; L选自-C(O)NH-、-NHC(O)-、-S(O) 2NH-、或-NHS(O) 2-;“*”表示手性中心。
L选自-C(O)NH-、-NHC(O)-、-S(O) 2NH-、或-NHS(O) 2-;“*”表示手性中心。 - 如权利要求1所述的化合物,其特征在于,式(I)为式(XIV):
 各基团的定义如利要求6所述。
各基团的定义如利要求6所述。 - 如权利要求1所述的化合物,其特征在于,式(I)为式(XV):
 各基团的定义如利要求1所述。
各基团的定义如利要求1所述。 - 如权利要求1所述的化合物,其特征在于,式(I)为式(XVII):
 k和n各自独立地选自1或2;X 1选自N、C-H、或C-F;X 2选自N、C-H、C-F、C-CN、或C-Me;X 4选自C-H、C-F、或N;Z 1选自C-H、C-F、或N;R c选自氢、卤素、C 1-4烷基、或CN;L选自-C(O)NH-或-NHC(O)-;其余各基团的定义如利要求6所述。
k和n各自独立地选自1或2;X 1选自N、C-H、或C-F;X 2选自N、C-H、C-F、C-CN、或C-Me;X 4选自C-H、C-F、或N;Z 1选自C-H、C-F、或N;R c选自氢、卤素、C 1-4烷基、或CN;L选自-C(O)NH-或-NHC(O)-;其余各基团的定义如利要求6所述。 - 如权利要求1所述的化合物,其特征在于,式(I)为式(XVIII):
 X 1选自N、C-H、或C-F;X 2选自N、C-H、C-F、C-CN、或C-Me;X 4选自C-H、C-F、或N;Z 1选自C-H、C-F、或N;R c选自氢、卤素、甲基、乙基、或CN;A选自下组基团:
X 1选自N、C-H、或C-F;X 2选自N、C-H、C-F、C-CN、或C-Me;X 4选自C-H、C-F、或N;Z 1选自C-H、C-F、或N;R c选自氢、卤素、甲基、乙基、或CN;A选自下组基团: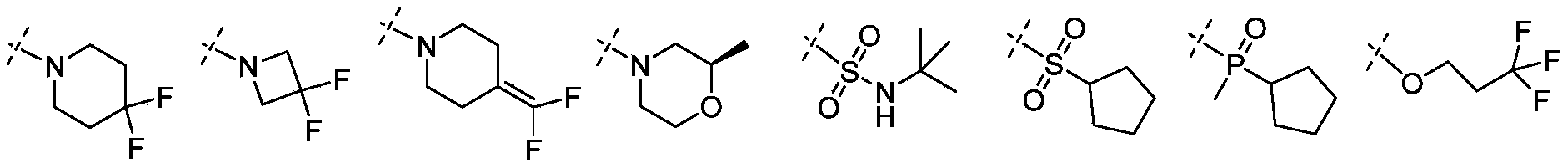 片段选自下组基团:
片段选自下组基团:

- 如权利要求2所述的化合物,其特征在于:式(XIa)或式(XIb)中片段A选自下组基团:
 表示A与式(XIa)或式(XIb)化合物其它部分连接的位点;
表示A与式(XIa)或式(XIb)化合物其它部分连接的位点;
- 如权利要求1所述的化合物,其特征在于,所述的式(I)化合物选自下组:
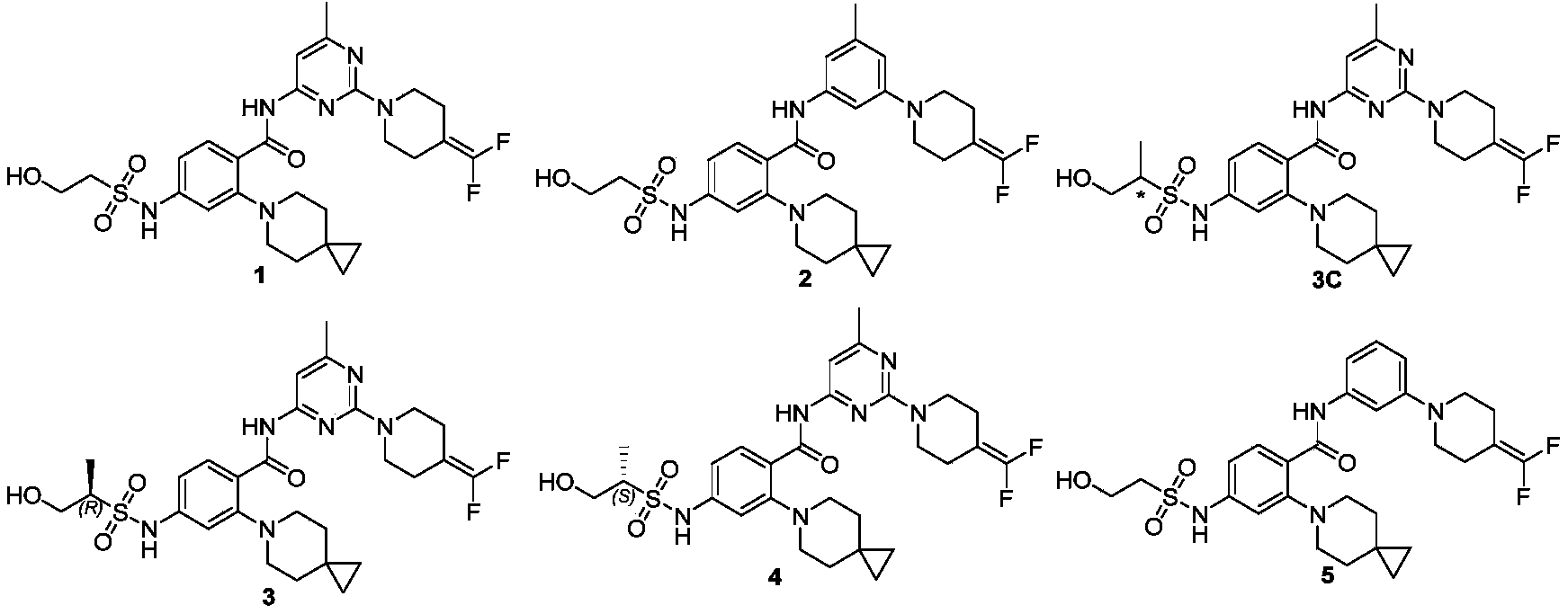







 “*”表示手性中心。
“*”表示手性中心。 - 一种药物组合物,其特征在于,包含权利要求1至12中任一项所述的化合物,或其光学异构体,药学上可接受的盐,前药,氘代衍生物,水合物,溶剂合物,以及药学上可接受的载体。
- 一种权利要求1至12中任一项的化合物,或其光学异构体,药学上可接受的盐,前药,氘代衍生物,水合物,溶剂合物的用途,其特征在于,用于制备治疗与KIF18A活性或表达量相关的疾病,病症或病状的药物组合物。
- 如权利要求14所述的用途,其特征在于,所述疾病,病症或病状选自下组:非小细胞肺癌、小细胞肺癌、肺腺癌、肺鳞癌、胰腺癌、结肠癌、甲状腺癌、胚胎性横纹肌肉 瘤、皮肤颗粒细胞肿瘤、黑色素瘤、肝细胞癌、肝内胆管癌、直肠癌、膀胱癌、咽喉癌、乳腺癌、前列腺癌、脑瘤、神经胶质细胞瘤、卵巢癌、头颈部鳞癌、宫颈癌、食管癌、肾癌、皮肤癌、胃癌、髓系白血病、淋巴系白血病、骨髓纤维化、B细胞淋巴瘤、T细胞淋巴瘤、霍奇金淋巴瘤、非霍奇金淋巴瘤、单核细胞白血病、脾大性红细胞增多、嗜酸性白细胞增多综合征多发性、骨髓癌等各种实体瘤和血液瘤,以及流感病毒等病毒的感染。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22827722.4A EP4361142A1 (en) | 2021-06-25 | 2022-06-27 | Compound as kif18a inhibitor |
| KR1020247003072A KR20240040742A (ko) | 2021-06-25 | 2022-06-27 | Kif18a 억제제로서의 화합물 |
| AU2022298761A AU2022298761A1 (en) | 2021-06-25 | 2022-06-27 | Compound as kif18a inhibitor |
| CA3224249A CA3224249A1 (en) | 2021-06-25 | 2022-06-27 | Compound as kif18a inhibitor |
| CN202280045351.7A CN117980298A (zh) | 2021-06-25 | 2022-06-27 | 作为kif18a抑制剂的化合物 |
| JP2023579635A JP2024526218A (ja) | 2021-06-25 | 2022-06-27 | Kif18a阻害剤としての化合物 |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110711270 | 2021-06-25 | ||
| CN202110711270.3 | 2021-06-25 | ||
| CN202110963377.7 | 2021-08-20 | ||
| CN202110963377 | 2021-08-20 | ||
| CN202111493904.9 | 2021-12-08 | ||
| CN202111493904 | 2021-12-08 | ||
| CN202210307789 | 2022-03-25 | ||
| CN202210307789.X | 2022-03-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022268230A1 true WO2022268230A1 (zh) | 2022-12-29 |
Family
ID=84544202
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2022/101694 WO2022268230A1 (zh) | 2021-06-25 | 2022-06-27 | 作为kif18a抑制剂的化合物 |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP4361142A1 (zh) |
| JP (1) | JP2024526218A (zh) |
| KR (1) | KR20240040742A (zh) |
| CN (1) | CN117980298A (zh) |
| AU (1) | AU2022298761A1 (zh) |
| CA (1) | CA3224249A1 (zh) |
| WO (1) | WO2022268230A1 (zh) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115947717A (zh) * | 2023-03-09 | 2023-04-11 | 英矽智能科技(上海)有限公司 | 一类kif18a抑制剂 |
| CN116535400A (zh) * | 2023-04-26 | 2023-08-04 | 上海湃隆生物科技有限公司 | 氮杂螺环化合物及其应用 |
| WO2023212240A1 (en) * | 2022-04-28 | 2023-11-02 | Volastra Therapeutics, Inc. | Compounds for inhibiting kif18a |
| WO2024002328A1 (zh) * | 2022-06-30 | 2024-01-04 | 勤浩医药(苏州)有限公司 | 一种含氮化合物及其应用 |
| WO2024083208A1 (zh) * | 2022-10-21 | 2024-04-25 | 杭州邦顺制药有限公司 | Kif18a蛋白抑制剂 |
| WO2024083137A1 (zh) * | 2022-10-19 | 2024-04-25 | 海南先声再明医药股份有限公司 | 稠合杂芳烃类化合物、其组合物及用途 |
| WO2024114787A1 (zh) * | 2022-12-02 | 2024-06-06 | 深圳众格生物科技有限公司 | 酰胺或脲类化合物 |
| WO2024140799A1 (en) * | 2022-12-28 | 2024-07-04 | Insilico Medicine Ip Limited | Inhibitors of kif18a and uses thereof |
| WO2024146593A1 (zh) * | 2023-01-05 | 2024-07-11 | 浙江海正药业股份有限公司 | 芳香酰胺类衍生物及其制备方法和医药上的用途 |
| WO2024156288A1 (en) * | 2023-01-29 | 2024-08-02 | Shanghai Kygent Pharmaceutical Co., Ltd | Dnmt1 inhibitors, pharmaceutical compositions, and therapeutic applications |
| US12054476B2 (en) | 2018-12-20 | 2024-08-06 | Amgen Inc. | KIF18A inhibitors |
| US12084420B2 (en) | 2021-08-26 | 2024-09-10 | Volastra Therapeutics, Inc. | Indoline compounds for inhibiting KIF18A |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9527885B2 (en) | 2011-05-05 | 2016-12-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20200140411A1 (en) * | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-heteroaryl-nicotinamides as nav1.8 inhibitors |
| WO2020092187A1 (en) * | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-phenyl-nicotinamides as nav1.8 inhibitors |
| WO2020132648A1 (en) | 2018-12-20 | 2020-06-25 | Amgen Inc. | Kif18a inhibitors |
| WO2020132653A1 (en) | 2018-12-20 | 2020-06-25 | Amgen Inc. | Heteroaryl amides useful as kif18a inhibitors |
| WO2020132651A1 (en) | 2018-12-20 | 2020-06-25 | Amgen Inc. | Kif18a inhibitors |
| WO2020132649A1 (en) * | 2018-12-20 | 2020-06-25 | Amgen Inc. | Heteroaryl amides useful as kif18a inhibitors |
| WO2021026099A1 (en) * | 2019-08-02 | 2021-02-11 | Amgen Inc. | Kif18a inhibitors |
| WO2021026101A1 (en) | 2019-08-02 | 2021-02-11 | Amgen Inc. | Kif18a inhibitors |
| WO2021026098A1 (en) | 2019-08-02 | 2021-02-11 | Amgen Inc. | Kif18a inhibitors |
| WO2021026100A1 (en) * | 2019-08-02 | 2021-02-11 | Amgen Inc. | Pyridine derivatives as kif18a inhibitors |
-
2022
- 2022-06-27 EP EP22827722.4A patent/EP4361142A1/en active Pending
- 2022-06-27 CN CN202280045351.7A patent/CN117980298A/zh active Pending
- 2022-06-27 KR KR1020247003072A patent/KR20240040742A/ko active Search and Examination
- 2022-06-27 CA CA3224249A patent/CA3224249A1/en active Pending
- 2022-06-27 JP JP2023579635A patent/JP2024526218A/ja active Pending
- 2022-06-27 WO PCT/CN2022/101694 patent/WO2022268230A1/zh active Application Filing
- 2022-06-27 AU AU2022298761A patent/AU2022298761A1/en active Pending
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9527885B2 (en) | 2011-05-05 | 2016-12-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20200140411A1 (en) * | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-heteroaryl-nicotinamides as nav1.8 inhibitors |
| WO2020092187A1 (en) * | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-phenyl-nicotinamides as nav1.8 inhibitors |
| WO2020132648A1 (en) | 2018-12-20 | 2020-06-25 | Amgen Inc. | Kif18a inhibitors |
| WO2020132653A1 (en) | 2018-12-20 | 2020-06-25 | Amgen Inc. | Heteroaryl amides useful as kif18a inhibitors |
| WO2020132651A1 (en) | 2018-12-20 | 2020-06-25 | Amgen Inc. | Kif18a inhibitors |
| WO2020132649A1 (en) * | 2018-12-20 | 2020-06-25 | Amgen Inc. | Heteroaryl amides useful as kif18a inhibitors |
| WO2021026099A1 (en) * | 2019-08-02 | 2021-02-11 | Amgen Inc. | Kif18a inhibitors |
| WO2021026101A1 (en) | 2019-08-02 | 2021-02-11 | Amgen Inc. | Kif18a inhibitors |
| WO2021026098A1 (en) | 2019-08-02 | 2021-02-11 | Amgen Inc. | Kif18a inhibitors |
| WO2021026100A1 (en) * | 2019-08-02 | 2021-02-11 | Amgen Inc. | Pyridine derivatives as kif18a inhibitors |
Non-Patent Citations (3)
| Title |
|---|
| CHEMICAL COMMUNICATIONS, vol. 57, no. 27, 2021 |
| HELVETICA CHIMICA ACTA, vol. 64, 1981, pages 1849 - 1853 |
| ORGANIC LETTERS, vol. 16, 2014, pages 6248 - 6251 |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12054476B2 (en) | 2018-12-20 | 2024-08-06 | Amgen Inc. | KIF18A inhibitors |
| US12084420B2 (en) | 2021-08-26 | 2024-09-10 | Volastra Therapeutics, Inc. | Indoline compounds for inhibiting KIF18A |
| WO2023212240A1 (en) * | 2022-04-28 | 2023-11-02 | Volastra Therapeutics, Inc. | Compounds for inhibiting kif18a |
| WO2024002328A1 (zh) * | 2022-06-30 | 2024-01-04 | 勤浩医药(苏州)有限公司 | 一种含氮化合物及其应用 |
| WO2024083137A1 (zh) * | 2022-10-19 | 2024-04-25 | 海南先声再明医药股份有限公司 | 稠合杂芳烃类化合物、其组合物及用途 |
| WO2024083208A1 (zh) * | 2022-10-21 | 2024-04-25 | 杭州邦顺制药有限公司 | Kif18a蛋白抑制剂 |
| WO2024114787A1 (zh) * | 2022-12-02 | 2024-06-06 | 深圳众格生物科技有限公司 | 酰胺或脲类化合物 |
| WO2024140799A1 (en) * | 2022-12-28 | 2024-07-04 | Insilico Medicine Ip Limited | Inhibitors of kif18a and uses thereof |
| WO2024146593A1 (zh) * | 2023-01-05 | 2024-07-11 | 浙江海正药业股份有限公司 | 芳香酰胺类衍生物及其制备方法和医药上的用途 |
| WO2024156288A1 (en) * | 2023-01-29 | 2024-08-02 | Shanghai Kygent Pharmaceutical Co., Ltd | Dnmt1 inhibitors, pharmaceutical compositions, and therapeutic applications |
| CN115947717A (zh) * | 2023-03-09 | 2023-04-11 | 英矽智能科技(上海)有限公司 | 一类kif18a抑制剂 |
| CN115947717B (zh) * | 2023-03-09 | 2023-05-30 | 英矽智能科技(上海)有限公司 | 一类kif18a抑制剂 |
| CN116535400A (zh) * | 2023-04-26 | 2023-08-04 | 上海湃隆生物科技有限公司 | 氮杂螺环化合物及其应用 |
| CN116535400B (zh) * | 2023-04-26 | 2024-08-16 | 上海湃隆生物科技有限公司 | 氮杂螺环化合物及其应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4361142A1 (en) | 2024-05-01 |
| CA3224249A1 (en) | 2022-12-29 |
| CN117980298A (zh) | 2024-05-03 |
| AU2022298761A1 (en) | 2024-02-15 |
| JP2024526218A (ja) | 2024-07-17 |
| KR20240040742A (ko) | 2024-03-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2022268230A1 (zh) | 作为kif18a抑制剂的化合物 | |
| CN109153636B (zh) | 作为双重lsd1/hdac抑制剂的环丙基-酰胺化合物 | |
| TWI476199B (zh) | 用於治療疾病之巨環衍生物 | |
| JP5425057B2 (ja) | Hsp90阻害剤としてのキナゾリン−オキシム誘導体 | |
| WO2022214102A1 (zh) | 作为kras g12d抑制剂的杂环化合物 | |
| WO2021000885A1 (zh) | 喹唑啉酮类衍生物、其制备方法及其在医药上的应用 | |
| CN113200981A (zh) | 作为sos1抑制剂的杂环化合物 | |
| WO2021027911A1 (zh) | 新型螺环类K-Ras G12C抑制剂 | |
| WO2023174175A1 (zh) | Kif18a抑制剂 | |
| WO2020238791A1 (zh) | 氢化吡啶并嘧啶类衍生物、其制备方法及其在医药上的应用 | |
| WO2016169421A1 (zh) | 咪唑并异吲哚类衍生物、其制备方法及其在医药上的应用 | |
| WO2020244518A1 (zh) | 一种具有苄氧基芳环结构的化合物,其制备方法和用途 | |
| WO2019223715A1 (zh) | 苯并哌啶或杂芳基并哌啶类衍生物、其制备方法及其在医药上的应用 | |
| CN108191861B (zh) | N-[5-(嘧啶-2-氨基)-2,4-二取代苯基]-反式-2,4-戊二烯酰胺 | |
| WO2021249475A1 (zh) | 稠合喹唑啉类衍生物、其制备方法及其在医药上的应用 | |
| WO2023280182A1 (zh) | 作为kat6抑制剂的化合物 | |
| WO2021185256A1 (zh) | 取代的嘧啶或吡啶胺衍生物、其组合物及医药上的用途 | |
| WO2021208918A1 (zh) | 作为egfr抑制剂的三环化合物 | |
| WO2022166860A1 (zh) | Pim激酶抑制剂 | |
| TW202214631A (zh) | 作為Akt激酶抑制劑的化合物 | |
| WO2022258023A1 (zh) | 用作cdk激酶抑制剂的化合物及其应用 | |
| WO2023141852A1 (zh) | Cdk2抑制剂及其制备方法和用途 | |
| CN103304571B (zh) | 螺环化合物、其制备方法、中间体、药物组合物和应用 | |
| WO2023274280A1 (zh) | 一种联苯类衍生物抑制剂的晶型及其制备方法 | |
| WO2023025324A1 (zh) | 作为p53调节剂的化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22827722 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2023579635 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280045351.7 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3224249 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022827722 Country of ref document: EP Ref document number: 2024101702 Country of ref document: RU Ref document number: AU2022298761 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022827722 Country of ref document: EP Effective date: 20240125 |
|
| ENP | Entry into the national phase |
Ref document number: 2022298761 Country of ref document: AU Date of ref document: 20220627 Kind code of ref document: A |