WO2019062435A1 - 三氮唑并嘧啶、三氮唑并吡啶化合物及其组合物用于治疗prc2介导的疾病 - Google Patents
三氮唑并嘧啶、三氮唑并吡啶化合物及其组合物用于治疗prc2介导的疾病 Download PDFInfo
- Publication number
- WO2019062435A1 WO2019062435A1 PCT/CN2018/102833 CN2018102833W WO2019062435A1 WO 2019062435 A1 WO2019062435 A1 WO 2019062435A1 CN 2018102833 W CN2018102833 W CN 2018102833W WO 2019062435 A1 WO2019062435 A1 WO 2019062435A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- independently selected
- compound
- substituted
- Prior art date
Links
- 201000010099 disease Diseases 0.000 title claims abstract description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title claims abstract description 14
- 230000001404 mediated effect Effects 0.000 title claims abstract description 12
- 239000000203 mixture Substances 0.000 title description 18
- YWBFPKPWMSWWEA-UHFFFAOYSA-O triazolopyrimidine Chemical compound BrC1=CC=CC(C=2N=C3N=CN[N+]3=C(NCC=3C=CN=CC=3)C=2)=C1 YWBFPKPWMSWWEA-UHFFFAOYSA-O 0.000 title description 5
- 150000008523 triazolopyridines Chemical class 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 443
- 101100465401 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) SCL1 gene Proteins 0.000 claims abstract description 25
- 150000003839 salts Chemical class 0.000 claims abstract description 16
- 239000003814 drug Substances 0.000 claims abstract description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 14
- 238000002360 preparation method Methods 0.000 claims abstract description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 630
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 330
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 160
- 229910052760 oxygen Inorganic materials 0.000 claims description 127
- 238000006243 chemical reaction Methods 0.000 claims description 119
- 125000005842 heteroatom Chemical group 0.000 claims description 114
- -1 -O-(C 1 -C 4 alkyl) Chemical group 0.000 claims description 110
- 229910052799 carbon Inorganic materials 0.000 claims description 106
- 125000004432 carbon atom Chemical class C* 0.000 claims description 96
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 84
- 125000003118 aryl group Chemical group 0.000 claims description 82
- 229910020008 S(O) Inorganic materials 0.000 claims description 66
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 62
- 229910052736 halogen Inorganic materials 0.000 claims description 62
- 150000002367 halogens Chemical class 0.000 claims description 62
- 229910052757 nitrogen Inorganic materials 0.000 claims description 60
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 56
- 229910052717 sulfur Inorganic materials 0.000 claims description 54
- 239000002904 solvent Substances 0.000 claims description 51
- 125000001072 heteroaryl group Chemical group 0.000 claims description 46
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims description 44
- 150000001412 amines Chemical class 0.000 claims description 39
- 229910052739 hydrogen Inorganic materials 0.000 claims description 39
- 125000004404 heteroalkyl group Chemical group 0.000 claims description 38
- 238000006467 substitution reaction Methods 0.000 claims description 36
- 125000000217 alkyl group Chemical group 0.000 claims description 30
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 30
- SNOOUWRIMMFWNE-UHFFFAOYSA-M sodium;6-[(3,4,5-trimethoxybenzoyl)amino]hexanoate Chemical compound [Na+].COC1=CC(C(=O)NCCCCCC([O-])=O)=CC(OC)=C1OC SNOOUWRIMMFWNE-UHFFFAOYSA-M 0.000 claims description 28
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 26
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 22
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 22
- 229910052794 bromium Inorganic materials 0.000 claims description 22
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 20
- 238000000034 method Methods 0.000 claims description 19
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 16
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 12
- 239000003153 chemical reaction reagent Substances 0.000 claims description 12
- 150000003852 triazoles Chemical class 0.000 claims description 12
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 claims description 12
- 125000001246 bromo group Chemical group Br* 0.000 claims description 11
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 claims description 10
- 239000003795 chemical substances by application Substances 0.000 claims description 10
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 claims description 9
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 claims description 9
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims description 8
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 8
- 230000009471 action Effects 0.000 claims description 8
- 239000003054 catalyst Substances 0.000 claims description 8
- 125000006165 cyclic alkyl group Chemical group 0.000 claims description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 8
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 claims description 7
- 238000006722 reduction reaction Methods 0.000 claims description 7
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 claims description 6
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 claims description 6
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 6
- 238000006069 Suzuki reaction reaction Methods 0.000 claims description 6
- WREOTYWODABZMH-DTZQCDIJSA-N [[(2r,3s,4r,5r)-3,4-dihydroxy-5-[2-oxo-4-(2-phenylethoxyamino)pyrimidin-1-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O[C@H]1N(C=C\1)C(=O)NC/1=N\OCCC1=CC=CC=C1 WREOTYWODABZMH-DTZQCDIJSA-N 0.000 claims description 6
- 125000000304 alkynyl group Chemical group 0.000 claims description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 6
- 229940125797 compound 12 Drugs 0.000 claims description 6
- 229940125758 compound 15 Drugs 0.000 claims description 6
- 125000004122 cyclic group Chemical group 0.000 claims description 6
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 6
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 claims description 6
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims description 6
- 229910052763 palladium Inorganic materials 0.000 claims description 6
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 claims description 6
- 125000006239 protecting group Chemical group 0.000 claims description 6
- 229920006395 saturated elastomer Polymers 0.000 claims description 6
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 claims description 6
- 206010025323 Lymphomas Diseases 0.000 claims description 5
- 150000001408 amides Chemical class 0.000 claims description 5
- 239000012948 isocyanate Substances 0.000 claims description 5
- 150000002513 isocyanates Chemical class 0.000 claims description 5
- NONOKGVFTBWRLD-UHFFFAOYSA-N isocyanatosulfanylimino(oxo)methane Chemical compound O=C=NSN=C=O NONOKGVFTBWRLD-UHFFFAOYSA-N 0.000 claims description 5
- 230000009467 reduction Effects 0.000 claims description 5
- 125000006716 (C1-C6) heteroalkyl group Chemical group 0.000 claims description 4
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 claims description 4
- XVRYJQACDDVZEI-UHFFFAOYSA-N 5-bromo-4-chloro-2-methylsulfanylpyrimidine Chemical compound CSC1=NC=C(Br)C(Cl)=N1 XVRYJQACDDVZEI-UHFFFAOYSA-N 0.000 claims description 4
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 claims description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 4
- 150000008064 anhydrides Chemical class 0.000 claims description 4
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 claims description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 4
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 claims description 4
- 238000006482 condensation reaction Methods 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 4
- 229940124597 therapeutic agent Drugs 0.000 claims description 4
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 claims description 3
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 claims description 3
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 claims description 3
- ZLJGWRQGNGPMBV-UHFFFAOYSA-N 5-bromo-2-methylsulfanyl-1h-pyrimidine-6-thione Chemical compound CSC1=NC=C(Br)C(=S)N1 ZLJGWRQGNGPMBV-UHFFFAOYSA-N 0.000 claims description 3
- 206010003571 Astrocytoma Diseases 0.000 claims description 3
- 206010004593 Bile duct cancer Diseases 0.000 claims description 3
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 3
- 206010005003 Bladder cancer Diseases 0.000 claims description 3
- SKBIXVHFUZJOTK-UHFFFAOYSA-N BrC=1C(=NC(=NC1)SC)C=1NC2=CC=CC=C2C1 Chemical compound BrC=1C(=NC(=NC1)SC)C=1NC2=CC=CC=C2C1 SKBIXVHFUZJOTK-UHFFFAOYSA-N 0.000 claims description 3
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 3
- 206010006187 Breast cancer Diseases 0.000 claims description 3
- 208000026310 Breast neoplasm Diseases 0.000 claims description 3
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 3
- 206010009944 Colon cancer Diseases 0.000 claims description 3
- 229940126657 Compound 17 Drugs 0.000 claims description 3
- 206010014733 Endometrial cancer Diseases 0.000 claims description 3
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 3
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 3
- 208000022072 Gallbladder Neoplasms Diseases 0.000 claims description 3
- 208000032612 Glial tumor Diseases 0.000 claims description 3
- 206010018338 Glioma Diseases 0.000 claims description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 3
- 206010027406 Mesothelioma Diseases 0.000 claims description 3
- 208000034578 Multiple myelomas Diseases 0.000 claims description 3
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 claims description 3
- 206010061306 Nasopharyngeal cancer Diseases 0.000 claims description 3
- 206010029260 Neuroblastoma Diseases 0.000 claims description 3
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 3
- 206010033128 Ovarian cancer Diseases 0.000 claims description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 3
- 206010033963 Parathyroid tumour Diseases 0.000 claims description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 3
- 206010060862 Prostate cancer Diseases 0.000 claims description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 3
- 208000015634 Rectal Neoplasms Diseases 0.000 claims description 3
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 3
- 208000008938 Rhabdoid tumor Diseases 0.000 claims description 3
- 206010039491 Sarcoma Diseases 0.000 claims description 3
- 208000021712 Soft tissue sarcoma Diseases 0.000 claims description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 3
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 3
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 3
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 3
- 150000008065 acid anhydrides Chemical class 0.000 claims description 3
- 210000000013 bile duct Anatomy 0.000 claims description 3
- 208000026900 bile duct neoplasm Diseases 0.000 claims description 3
- 125000005620 boronic acid group Chemical class 0.000 claims description 3
- 230000031709 bromination Effects 0.000 claims description 3
- 238000005893 bromination reaction Methods 0.000 claims description 3
- 201000010881 cervical cancer Diseases 0.000 claims description 3
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 3
- 208000029742 colonic neoplasm Diseases 0.000 claims description 3
- 229940125846 compound 25 Drugs 0.000 claims description 3
- DGJMPUGMZIKDRO-UHFFFAOYSA-N cyanoacetamide Chemical compound NC(=O)CC#N DGJMPUGMZIKDRO-UHFFFAOYSA-N 0.000 claims description 3
- 201000004101 esophageal cancer Diseases 0.000 claims description 3
- FMVJYQGSRWVMQV-UHFFFAOYSA-N ethyl propiolate Chemical compound CCOC(=O)C#C FMVJYQGSRWVMQV-UHFFFAOYSA-N 0.000 claims description 3
- 201000003444 follicular lymphoma Diseases 0.000 claims description 3
- 201000010175 gallbladder cancer Diseases 0.000 claims description 3
- 206010017758 gastric cancer Diseases 0.000 claims description 3
- 208000005017 glioblastoma Diseases 0.000 claims description 3
- 201000010536 head and neck cancer Diseases 0.000 claims description 3
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 3
- 208000032839 leukemia Diseases 0.000 claims description 3
- 201000005202 lung cancer Diseases 0.000 claims description 3
- 208000020816 lung neoplasm Diseases 0.000 claims description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 3
- 208000007538 neurilemmoma Diseases 0.000 claims description 3
- 201000002528 pancreatic cancer Diseases 0.000 claims description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 3
- 206010038038 rectal cancer Diseases 0.000 claims description 3
- 201000001275 rectum cancer Diseases 0.000 claims description 3
- 201000011549 stomach cancer Diseases 0.000 claims description 3
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 claims description 3
- 201000002510 thyroid cancer Diseases 0.000 claims description 3
- 208000017997 tumor of parathyroid gland Diseases 0.000 claims description 3
- 208000025421 tumor of uterus Diseases 0.000 claims description 3
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 3
- 206010046766 uterine cancer Diseases 0.000 claims description 3
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 claims description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 claims description 2
- 239000007868 Raney catalyst Substances 0.000 claims description 2
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 claims description 2
- 229910000564 Raney nickel Inorganic materials 0.000 claims description 2
- 125000003342 alkenyl group Chemical group 0.000 claims description 2
- 125000003282 alkyl amino group Chemical group 0.000 claims description 2
- 125000003277 amino group Chemical group 0.000 claims description 2
- 230000003474 anti-emetic effect Effects 0.000 claims description 2
- 239000000043 antiallergic agent Substances 0.000 claims description 2
- 239000002111 antiemetic agent Substances 0.000 claims description 2
- 229940125683 antiemetic agent Drugs 0.000 claims description 2
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 claims description 2
- 229940126086 compound 21 Drugs 0.000 claims description 2
- 229940126208 compound 22 Drugs 0.000 claims description 2
- 229940125833 compound 23 Drugs 0.000 claims description 2
- 230000001120 cytoprotective effect Effects 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 125000000623 heterocyclic group Chemical group 0.000 claims description 2
- 239000002955 immunomodulating agent Substances 0.000 claims description 2
- 229940121354 immunomodulator Drugs 0.000 claims description 2
- 201000001441 melanoma Diseases 0.000 claims description 2
- 229910052751 metal Inorganic materials 0.000 claims description 2
- 239000002184 metal Substances 0.000 claims description 2
- 229940127084 other anti-cancer agent Drugs 0.000 claims description 2
- 238000007254 oxidation reaction Methods 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 claims 1
- 238000005859 coupling reaction Methods 0.000 claims 1
- 229940079593 drug Drugs 0.000 abstract description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 165
- 230000015572 biosynthetic process Effects 0.000 description 133
- 238000003786 synthesis reaction Methods 0.000 description 133
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 130
- 238000004440 column chromatography Methods 0.000 description 98
- 238000005481 NMR spectroscopy Methods 0.000 description 82
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 62
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 61
- 239000007787 solid Substances 0.000 description 51
- 239000000243 solution Substances 0.000 description 46
- 238000003756 stirring Methods 0.000 description 43
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 36
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 32
- 230000002829 reductive effect Effects 0.000 description 31
- 239000011734 sodium Substances 0.000 description 30
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 30
- 210000004027 cell Anatomy 0.000 description 28
- XDBWCOGKBXTANE-UHFFFAOYSA-N dicyclohexyl-[2-(2,6-dimethoxyphenyl)phenyl]phosphane;sodium Chemical compound [Na].COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 XDBWCOGKBXTANE-UHFFFAOYSA-N 0.000 description 28
- 239000007821 HATU Substances 0.000 description 26
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 24
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 22
- 0 *CNc([n]1c(c(I*)c2*)nnc1)*2I Chemical compound *CNc([n]1c(c(I*)c2*)nnc1)*2I 0.000 description 17
- 239000000543 intermediate Substances 0.000 description 17
- 239000011541 reaction mixture Substances 0.000 description 17
- 239000000047 product Substances 0.000 description 16
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 15
- 101000866766 Homo sapiens Polycomb protein EED Proteins 0.000 description 10
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 10
- 241000700159 Rattus Species 0.000 description 9
- DYIRSNMPIZZNBK-UHFFFAOYSA-N N-(furan-2-ylmethyl)-8-(4-methylsulfonylphenyl)-[1,2,4]triazolo[4,3-c]pyrimidin-5-amine Chemical compound CS(=O)(=O)c1ccc(cc1)-c1cnc(NCc2ccco2)n2cnnc12 DYIRSNMPIZZNBK-UHFFFAOYSA-N 0.000 description 8
- 238000003821 enantio-separation Methods 0.000 description 8
- 239000003960 organic solvent Substances 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 102100038970 Histone-lysine N-methyltransferase EZH2 Human genes 0.000 description 6
- 101000882127 Homo sapiens Histone-lysine N-methyltransferase EZH2 Proteins 0.000 description 6
- FFDGPVCHZBVARC-UHFFFAOYSA-N N,N-dimethylglycine Chemical compound CN(C)CC(O)=O FFDGPVCHZBVARC-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- 238000010790 dilution Methods 0.000 description 6
- 239000012895 dilution Substances 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 210000001853 liver microsome Anatomy 0.000 description 6
- 239000011550 stock solution Substances 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 6
- 239000007983 Tris buffer Substances 0.000 description 5
- 238000012054 celltiter-glo Methods 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- IZDROVVXIHRYMH-UHFFFAOYSA-N methanesulfonic anhydride Chemical compound CS(=O)(=O)OS(C)(=O)=O IZDROVVXIHRYMH-UHFFFAOYSA-N 0.000 description 5
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 4
- 230000009036 growth inhibition Effects 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- NOGFHTGYPKWWRX-UHFFFAOYSA-N 2,2,6,6-tetramethyloxan-4-one Chemical compound CC1(C)CC(=O)CC(C)(C)O1 NOGFHTGYPKWWRX-UHFFFAOYSA-N 0.000 description 3
- CLLLODNOQBVIMS-UHFFFAOYSA-N 2-(2-methoxyethoxy)acetic acid Chemical compound COCCOCC(O)=O CLLLODNOQBVIMS-UHFFFAOYSA-N 0.000 description 3
- IPXNXMNCBXHYLQ-UHFFFAOYSA-N 2-pyrrolidin-1-ylacetic acid Chemical compound OC(=O)CN1CCCC1 IPXNXMNCBXHYLQ-UHFFFAOYSA-N 0.000 description 3
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 230000003698 anagen phase Effects 0.000 description 3
- 239000004327 boric acid Substances 0.000 description 3
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 3
- 239000006143 cell culture medium Substances 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- 239000008358 core component Substances 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 229910000071 diazene Inorganic materials 0.000 description 3
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 3
- 108700003601 dimethylglycine Proteins 0.000 description 3
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- RMIODHQZRUFFFF-UHFFFAOYSA-N methoxyacetic acid Chemical compound COCC(O)=O RMIODHQZRUFFFF-UHFFFAOYSA-N 0.000 description 3
- 229940078490 n,n-dimethylglycine Drugs 0.000 description 3
- UWOTZNQZPLAURK-UHFFFAOYSA-N oxetane-3-carboxylic acid Chemical compound OC(=O)C1COC1 UWOTZNQZPLAURK-UHFFFAOYSA-N 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 239000012224 working solution Substances 0.000 description 3
- PDVFSPNIEOYOQL-UHFFFAOYSA-N (4-methylphenyl)sulfonyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OS(=O)(=O)C1=CC=C(C)C=C1 PDVFSPNIEOYOQL-UHFFFAOYSA-N 0.000 description 2
- ADAKRBAJFHTIEW-UHFFFAOYSA-N 1-chloro-4-isocyanatobenzene Chemical compound ClC1=CC=C(N=C=O)C=C1 ADAKRBAJFHTIEW-UHFFFAOYSA-N 0.000 description 2
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 2
- QRWZFUBHOQWUGH-UHFFFAOYSA-N 2,5-dimethylpyrazole-3-carboxylic acid Chemical compound CC=1C=C(C(O)=O)N(C)N=1 QRWZFUBHOQWUGH-UHFFFAOYSA-N 0.000 description 2
- MGOLNIXAPIAKFM-UHFFFAOYSA-N 2-isocyanato-2-methylpropane Chemical compound CC(C)(C)N=C=O MGOLNIXAPIAKFM-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- HIHGQEFNUAJUMC-UHFFFAOYSA-N Fc(cc1)c(CNc([n]2c3nnc2)ncc3C2=CCCNCC2)c2c1OCC2 Chemical compound Fc(cc1)c(CNc([n]2c3nnc2)ncc3C2=CCCNCC2)c2c1OCC2 HIHGQEFNUAJUMC-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 108010033040 Histones Proteins 0.000 description 2
- 102000006947 Histones Human genes 0.000 description 2
- 101000584499 Homo sapiens Polycomb protein SUZ12 Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- XUMBMVFBXHLACL-UHFFFAOYSA-N Melanin Chemical compound O=C1C(=O)C(C2=CNC3=C(C(C(=O)C4=C32)=O)C)=C2C4=CNC2=C1C XUMBMVFBXHLACL-UHFFFAOYSA-N 0.000 description 2
- VIWZVFVJPXTXPA-UHFFFAOYSA-N N-(2-Carboxymethyl)-morpholine Chemical compound OC(=O)CN1CCOCC1 VIWZVFVJPXTXPA-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- 102100030702 Polycomb protein SUZ12 Human genes 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- MEFKEPWMEQBLKI-AIRLBKTGSA-N S-adenosyl-L-methioninate Chemical compound O[C@@H]1[C@H](O)[C@@H](C[S+](CC[C@H](N)C([O-])=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 MEFKEPWMEQBLKI-AIRLBKTGSA-N 0.000 description 2
- 229960001570 ademetionine Drugs 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000003115 biocidal effect Effects 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000012295 chemical reaction liquid Substances 0.000 description 2
- 238000009509 drug development Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- WUDNUHPRLBTKOJ-UHFFFAOYSA-N ethyl isocyanate Chemical compound CCN=C=O WUDNUHPRLBTKOJ-UHFFFAOYSA-N 0.000 description 2
- HBNYJWAFDZLWRS-UHFFFAOYSA-N ethyl isothiocyanate Chemical compound CCN=C=S HBNYJWAFDZLWRS-UHFFFAOYSA-N 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 239000012737 fresh medium Substances 0.000 description 2
- VANNPISTIUFMLH-UHFFFAOYSA-N glutaric anhydride Chemical compound O=C1CCCC(=O)O1 VANNPISTIUFMLH-UHFFFAOYSA-N 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- YDNLNVZZTACNJX-UHFFFAOYSA-N isocyanatomethylbenzene Chemical compound O=C=NCC1=CC=CC=C1 YDNLNVZZTACNJX-UHFFFAOYSA-N 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- RZWZRACFZGVKFM-UHFFFAOYSA-N propanoyl chloride Chemical compound CCC(Cl)=O RZWZRACFZGVKFM-UHFFFAOYSA-N 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- QCGMEWVZBGQOFN-UHFFFAOYSA-N 1,3-oxazole-5-carboxylic acid Chemical compound OC(=O)C1=CN=CO1 QCGMEWVZBGQOFN-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- HCKNAJXCHMACDN-UHFFFAOYSA-N 1-methylpiperidine-4-carboxylic acid Chemical compound CN1CCC(C(O)=O)CC1 HCKNAJXCHMACDN-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- PMWGIVRHUIAIII-UHFFFAOYSA-N 2,2-difluoropropanoic acid Chemical compound CC(F)(F)C(O)=O PMWGIVRHUIAIII-UHFFFAOYSA-N 0.000 description 1
- KWPQTFXULUUCGD-UHFFFAOYSA-N 3,4,5,7,8,9,10,10a-octahydropyrido[1,2-a][1,4]diazepine Chemical compound C1CCN=CC2CCCCN21 KWPQTFXULUUCGD-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- MGYGFNQQGAQEON-UHFFFAOYSA-N 4-tolyl isocyanate Chemical compound CC1=CC=C(N=C=O)C=C1 MGYGFNQQGAQEON-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- MSSOYRMNKPKEGK-UHFFFAOYSA-N CC(C)C(C(C)C)=N Chemical compound CC(C)C(C(C)C)=N MSSOYRMNKPKEGK-UHFFFAOYSA-N 0.000 description 1
- OEEAMCUIBZPOFU-UHFFFAOYSA-N CC(CC(CC1C)c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)N1C(CN(C)C)=O Chemical compound CC(CC(CC1C)c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)N1C(CN(C)C)=O OEEAMCUIBZPOFU-UHFFFAOYSA-N 0.000 description 1
- UMYPWWQTIZUKBN-UHFFFAOYSA-N CC(CC(c1cnc(NCc(c2c(cc3)OCC2)c3F)[n]2c1nnc2)=CC1C)N1C(CN(C)C)=O Chemical compound CC(CC(c1cnc(NCc(c2c(cc3)OCC2)c3F)[n]2c1nnc2)=CC1C)N1C(CN(C)C)=O UMYPWWQTIZUKBN-UHFFFAOYSA-N 0.000 description 1
- PCTHPQISJZRLGB-UHFFFAOYSA-N CN(CC1)CCC1C(N(CCC1)CCC1c1cnc(NCc2c(CCO3)c3ccc2F)[n]2c1nnc2)=O Chemical compound CN(CC1)CCC1C(N(CCC1)CCC1c1cnc(NCc2c(CCO3)c3ccc2F)[n]2c1nnc2)=O PCTHPQISJZRLGB-UHFFFAOYSA-N 0.000 description 1
- PCTHPQISJZRLGB-GOSISDBHSA-N CN(CC1)CCC1C(N(CCC1)CC[C@@H]1c1cnc(NCc2c(CCO3)c3ccc2F)[n]2c1nnc2)=O Chemical compound CN(CC1)CCC1C(N(CCC1)CC[C@@H]1c1cnc(NCc2c(CCO3)c3ccc2F)[n]2c1nnc2)=O PCTHPQISJZRLGB-GOSISDBHSA-N 0.000 description 1
- PCTHPQISJZRLGB-SFHVURJKSA-N CN(CC1)CCC1C(N(CCC1)CC[C@H]1c1cnc(NCc(c2c(cc3)OCC2)c3F)[n]2c1nnc2)=O Chemical compound CN(CC1)CCC1C(N(CCC1)CC[C@H]1c1cnc(NCc(c2c(cc3)OCC2)c3F)[n]2c1nnc2)=O PCTHPQISJZRLGB-SFHVURJKSA-N 0.000 description 1
- MCQCMUQCRNUZKM-UHFFFAOYSA-N CN(CC1)CCC1C(N1CCC(c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)=CCC1)=O Chemical compound CN(CC1)CCC1C(N1CCC(c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)=CCC1)=O MCQCMUQCRNUZKM-UHFFFAOYSA-N 0.000 description 1
- JOZKOZOOMSDJKQ-UHFFFAOYSA-N COCC(N1CCC(c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)=CCC1)=O Chemical compound COCC(N1CCC(c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)=CCC1)=O JOZKOZOOMSDJKQ-UHFFFAOYSA-N 0.000 description 1
- DMVFYQIOVMSVCZ-QGZVFWFLSA-N COCCOCC(N(CCC1)CC[C@@H]1c1cnc(NCc2c(CCO3)c3ccc2F)[n]2c1nnc2)=O Chemical compound COCCOCC(N(CCC1)CC[C@@H]1c1cnc(NCc2c(CCO3)c3ccc2F)[n]2c1nnc2)=O DMVFYQIOVMSVCZ-QGZVFWFLSA-N 0.000 description 1
- DMVFYQIOVMSVCZ-KRWDZBQOSA-N COCCOCC(N(CCC1)CC[C@H]1c1cnc(NCc(c2c(cc3)OCC2)c3F)[n]2c1nnc2)=O Chemical compound COCCOCC(N(CCC1)CC[C@H]1c1cnc(NCc(c2c(cc3)OCC2)c3F)[n]2c1nnc2)=O DMVFYQIOVMSVCZ-KRWDZBQOSA-N 0.000 description 1
- YVHXDKFMORVYQC-UHFFFAOYSA-N COCCOCC(N1CCC(c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)=CCC1)=O Chemical compound COCCOCC(N1CCC(c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)=CCC1)=O YVHXDKFMORVYQC-UHFFFAOYSA-N 0.000 description 1
- PYXSQRIHTWWWIN-UHFFFAOYSA-N CSc([n]1c2nnc1)ncc2Br Chemical compound CSc([n]1c2nnc1)ncc2Br PYXSQRIHTWWWIN-UHFFFAOYSA-N 0.000 description 1
- WHXIJUFFJQVVDB-UHFFFAOYSA-N CSc(nc1NN)ncc1Br Chemical compound CSc(nc1NN)ncc1Br WHXIJUFFJQVVDB-UHFFFAOYSA-N 0.000 description 1
- PMNFISMZZAVMSE-UHFFFAOYSA-N Cc1n[n](C)c(C(N(CC2)CC=C2c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)=O)c1 Chemical compound Cc1n[n](C)c(C(N(CC2)CC=C2c2cnc(NCc(c3c(cc4)OCC3)c4F)[n]3c2nnc3)=O)c1 PMNFISMZZAVMSE-UHFFFAOYSA-N 0.000 description 1
- OZNZCKRIYZQPPD-UHFFFAOYSA-N Cc1nnc[n]1/C(/NCc(c1c(cc2)OCC1)c2F)=N\C Chemical compound Cc1nnc[n]1/C(/NCc(c1c(cc2)OCC1)c2F)=N\C OZNZCKRIYZQPPD-UHFFFAOYSA-N 0.000 description 1
- GIZJELUKZWKFBH-UHFFFAOYSA-N Cl[ClH]C(Cl)(Cl)Cl Chemical compound Cl[ClH]C(Cl)(Cl)Cl GIZJELUKZWKFBH-UHFFFAOYSA-N 0.000 description 1
- YIFZTYMYYDQEER-UHFFFAOYSA-N Fc(cc1)c(CNc([n]2c3nnc2)ncc3C2=CCCOCC2)c2c1OCC2 Chemical compound Fc(cc1)c(CNc([n]2c3nnc2)ncc3C2=CCCOCC2)c2c1OCC2 YIFZTYMYYDQEER-UHFFFAOYSA-N 0.000 description 1
- AAMTZISHVXYYHH-UHFFFAOYSA-N Fc1ccc2OCCc2c1CNc1ncc(C2CCOCCC2)c2nnc[n]12 Chemical compound Fc1ccc2OCCc2c1CNc1ncc(C2CCOCCC2)c2nnc[n]12 AAMTZISHVXYYHH-UHFFFAOYSA-N 0.000 description 1
- 108010036115 Histone Methyltransferases Proteins 0.000 description 1
- 102000011787 Histone Methyltransferases Human genes 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- 102000016397 Methyltransferase Human genes 0.000 description 1
- RXQOSISJLCVABE-UHFFFAOYSA-N N#Cc(c(O)n1)cc(Br)c1O Chemical compound N#Cc(c(O)n1)cc(Br)c1O RXQOSISJLCVABE-UHFFFAOYSA-N 0.000 description 1
- BFHKYHMCIAMQIN-UHFFFAOYSA-N N#Cc(c(O)n1)ccc1O Chemical compound N#Cc(c(O)n1)ccc1O BFHKYHMCIAMQIN-UHFFFAOYSA-N 0.000 description 1
- FSLHBMXZVCGMSM-UHFFFAOYSA-N N#Cc(cc(c(Cl)n1)Br)c1Cl Chemical compound N#Cc(cc(c(Cl)n1)Br)c1Cl FSLHBMXZVCGMSM-UHFFFAOYSA-N 0.000 description 1
- DWFBUDOZFLAQPE-UHFFFAOYSA-N N#Cc(cc(c1nnc[n]11)Br)c1Cl Chemical compound N#Cc(cc(c1nnc[n]11)Br)c1Cl DWFBUDOZFLAQPE-UHFFFAOYSA-N 0.000 description 1
- SGXDXUYKISDCAZ-UHFFFAOYSA-N N,N-diethylglycine Chemical compound CCN(CC)CC(O)=O SGXDXUYKISDCAZ-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- WKHNJDDEPDBXFX-UHFFFAOYSA-N NNc(nc(c(C#N)c1)Cl)c1Br Chemical compound NNc(nc(c(C#N)c1)Cl)c1Br WKHNJDDEPDBXFX-UHFFFAOYSA-N 0.000 description 1
- RFLOHLBFKJWRIN-UHFFFAOYSA-N O=C(C1COC1)N1CCC(c2cnc(NCc3c(CCO4)c4ccc3F)[n]3c2nnc3)=CCC1 Chemical compound O=C(C1COC1)N1CCC(c2cnc(NCc3c(CCO4)c4ccc3F)[n]3c2nnc3)=CCC1 RFLOHLBFKJWRIN-UHFFFAOYSA-N 0.000 description 1
- 108010000597 Polycomb Repressive Complex 2 Proteins 0.000 description 1
- 102000002272 Polycomb Repressive Complex 2 Human genes 0.000 description 1
- 102100031338 Polycomb protein EED Human genes 0.000 description 1
- 108010022429 Polycomb-Group Proteins Proteins 0.000 description 1
- 102000012425 Polycomb-Group Proteins Human genes 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- CPEKAXYCDKETEN-UHFFFAOYSA-N benzoyl isothiocyanate Chemical compound S=C=NC(=O)C1=CC=CC=C1 CPEKAXYCDKETEN-UHFFFAOYSA-N 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 238000000006 cell growth inhibition assay Methods 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- PFWWSGFPICCWGU-UHFFFAOYSA-N cyclopropanesulfonyl chloride Chemical compound ClS(=O)(=O)C1CC1 PFWWSGFPICCWGU-UHFFFAOYSA-N 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- FRYHCSODNHYDPU-UHFFFAOYSA-N ethanesulfonyl chloride Chemical compound CCS(Cl)(=O)=O FRYHCSODNHYDPU-UHFFFAOYSA-N 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000013213 extrapolation Methods 0.000 description 1
- 210000003191 femoral vein Anatomy 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000003304 gavage Methods 0.000 description 1
- 210000001102 germinal center b cell Anatomy 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- AVPKHOTUOHDTLW-UHFFFAOYSA-N oxane-4-carboxylic acid Chemical compound OC(=O)C1CCOCC1 AVPKHOTUOHDTLW-UHFFFAOYSA-N 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 230000000079 pharmacotherapeutic effect Effects 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- ARJOQCYCJMAIFR-UHFFFAOYSA-N prop-2-enoyl prop-2-enoate Chemical compound C=CC(=O)OC(=O)C=C ARJOQCYCJMAIFR-UHFFFAOYSA-N 0.000 description 1
- DRINJBFRTLBHNF-UHFFFAOYSA-N propane-2-sulfonyl chloride Chemical compound CC(C)S(Cl)(=O)=O DRINJBFRTLBHNF-UHFFFAOYSA-N 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical group COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 1
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to the field of medicinal chemistry and pharmacotherapeutics, and generally relates to a class of triazolopyrimidine, triazolopyridine compounds and pharmaceutical compositions thereof and their use in the treatment of neoplastic diseases.
- such compounds may be useful in the manufacture of a medicament for the treatment of a disease or condition mediated by PRC2.
- Polycomb Repressive Complex 2 is a core member of the Polycomb Group, which has histone methyltransferase activity and specifically catalyzes the top three lysine of histone H3.
- the base is modified (H3K27me3) to inhibit the expression of a specific gene.
- the methyltransferase activity of PRC2 is derived from its catalytic member EZH2, whereas EZH2 has no catalytic activity when present alone, which requires at least a complex with the other two members of PRC2, EED and SUZ12, to catalyze methylation modification.
- EZH2, EED and SUZ12 are considered to be core components of the PRC2 complex.
- PRC2 is a very promising anticancer drug development target, and the discovery of inhibitors targeting PRC2 is currently a hot spot in the pharmaceutical industry.
- Novartis and Abbott have invented a small molecule that inhibits PRC2 activity by targeting EED (Reference: Novartis EED226, US 2016/0176682, J. Med. Chem. 2017, 60, 2215-2226, J. Med. Chem. 2017, 60, 415-427, Nat. Chem. Biol. 2017, 13, 381-388; Aberdeen, A-395, Nat. Chem. Biol. 2017, 13, 389-395),
- the compounds show strong inhibitory activity at the molecular level, at the cellular level, and in animal experiments.
- the PRC2 complex is considered to be a key driver of the development of a variety of malignancies, and the development of inhibitors that inhibit the activity of PRC2 by targeting EED is currently highly competitive in the industry and is beneficial for use in New drug development related to it.
- the present invention relates to a triazolopyrimidine, triazolopyridine compound of the formula I and combinations thereof, and for binding activity evaluation and related biological experiments, which can be used for the preparation of the treatment by EED and/or Or a drug that is mediated by PRC2.
- Another object of the present invention is to provide a process for the preparation of the above compounds.
- It is still another object of the present invention to provide a pharmaceutical composition comprising a therapeutically effective amount of one or more of the above compounds or a pharmaceutically acceptable salt thereof.
- a further object of the invention is to provide the use of a compound as described above for the manufacture of a medicament for the treatment of a disease or condition mediated by EED and/or PRC2.
- a further object of the invention is to provide a method of treating a disease or condition mediated by EED and/or PRC2, characterized in that a therapeutically effective amount of one or more of the above compounds or a medicament thereof is administered to a subject Salt used.
- X 1 is independently selected from N and C-CN;
- R 2 is independently selected from the group consisting of H, halogen, C 1 -C 4 haloalkyl and C 1 -C 4 alkyl;
- A is independently selected from the following structures:
- R 3 , R 4 and R 5 are independently selected from H, halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, -O-(C 1 -C 4 alkyl), C 1 -C 4 Haloalkoxy, C 3 -C 6 cycloalkyl;
- R 7 is independently selected from the group consisting of H, OH, halogen, CN, and C 1 -C 4 alkyl;
- n are each independently selected from 0, 1 and 2;
- X 2 is independently selected from the group consisting of O, NR a and S(O) p heteroatoms;
- p are each independently selected from 0, 1 and 2;
- R 1 is independently selected from the following structures:
- p are each independently selected from 0, 1 and 2;
- p are each independently selected from 0, 1 and 2;
- p are each independently selected from 0, 1 and 2;
- R 1X is independently selected from halogen, OH, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, C 1 -C 4 haloalkoxy, C 3 -C 8 cyclic alkyl and cyclic heteroalkyl;
- An alkyl group, -C( O)H, an aryl group, a 5- to 6-membered heteroaryl group containing a carbon atom and 1 to 2 heteroatoms selected from N, O and S; wherein the aryl group and the
- R f is independently selected from a C 3 -C 8 cyclic alkyl group, a heteroalkyl group containing a carbon atom and 1 to 4 heteroatoms selected from O, N, S(O) p , an aryl group, and a heteroaryl group comprising a carbon atom and 1 to 2 hetero atoms selected from the group consisting of N, NR a , O and S(O) p ; wherein the aryl group and the heteroaryl group are substituted by 0-2 R 1X ;
- M is independently selected from a 3 to 7 membered saturated or unsaturated cycloalkyl group, a heterocycloalkyl group containing a carbon atom and 1 to 4 hetero atoms selected from O, N, S(O) p , an aryl group, and the like a carbon atom and 1 to 2 5- to 6-membered heteroaryl groups selected from N, O and S heteroatoms;
- R 1A in the section 4a corresponds to the corresponding definition in R 1A in the section 4a);
- n are each independently selected from 0, 1 and 2;
- n are each independently selected from 0-4;
- p are each independently selected from 0-2;
- q are each independently selected from 0-3;
- z are each independently selected from 0 and 1;
- R 1 is independently selected from the following structures: among them
- R c is independently selected from OH, halogen, CN, C 1 -C 6 alkyl, carboxy, C 1 -C 6 haloalkyl, C 1 -C 6 alkoxy, C 1 -C 6 haloalkoxy, C 3 -C 8 cyclic alkyl;
- R c is independently selected from OH, halogen, CN, C 1 -C 6 alkyl, carboxy, C 1 -C 6 haloalkyl, C 1 -C 6 alkoxy, C 1 -C 6 haloalkoxy, C 3 -C 8 cyclic alkyl;
- M is independently selected from a 3 to 7 membered saturated or unsaturated cycloalkyl group, a heterocycloalkyl group containing a carbon atom and 1 to 4 hetero atoms selected from O, N, S(O) p , an aryl group, and the like a carbon atom and 1 to 2 5- to 6-membered heteroaryl groups selected from N, O and S heteroatoms;
- n are each independently selected from 0, 1 and 2;
- n are each independently selected from 0-4;
- p are each independently selected from 0-2;
- q are each independently selected from 0-3;
- z are each independently selected from 0 and 1;
- R 1A is independently selected from H, hydroxy, halogen, C 1 -C 6 alkyl, C 1 -C 6 alkoxy, C 1 -C 6 haloalkyl, C 1 -C 6 haloalkoxy, C 3 -C 8 cyclic alkyl;
- R 1B and R 1C , R 2B and R 2C , and R 3B and R 3C are independently selected from H, OH, halogen, C 1 -C 6 alkyl, C 1 -C 6 alkoxy, C 1 -C 6 Haloalkyl, C 1 -C 6 haloalkoxy, C 3 -C 8 cyclic alkyl;
- R 1D is independently selected from H, -OH, halogen, C 1 -C 6 alkyl, C 1 -C 6 alkoxy, C 1 -C 6 haloalkyl, C 1 -C 6 haloalkoxy, C 3 - C 8 cyclic alkyl group;
- M is independently selected from a 5- to 6-membered saturated or unsaturated cycloalkyl group, a heterocycloalkyl group containing a carbon atom and 1 to 4 hetero atoms selected from O, N, S(O) p , an aryl group, and the like a carbon atom and 1 to 2 5- to 6-membered heteroaryl groups selected from N, O and S heteroatoms;
- n are each independently selected from 0, 1 and 2;
- n are each independently selected from 0-4;
- p are each independently selected from 0-2;
- q are each independently selected from 0-3;
- z are each independently selected from 0 and 1;
- R 1X is independently selected from halogen, OH, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, C 1 -C 4 haloalkoxy, C 3 -C 8 cyclic alkyl and cyclic heteroalkyl;
- R c as defined in the above defined 4a) of the same portion of R c;
- R j and R k are independently selected from the group consisting of H, CN, C 1 -C 10 alkyl group having 0-3 R s substitutions, C 1 -C 6 haloalkyl group, C 3 -C 10 cyclic alkyl group, and a carbon atom and a heteroalkyl group and a heterocycloalkyl group selected from the group consisting of 1-4 selected from O, N, S(O) p heteroatoms, an alkenyl group or an alkynyl group substituted by R y a 6 to 10 membered aryl group, a carbon atom and 1 to 2 5- to 10-membered heteroaryl groups selected from N, O and S heteroatoms; wherein the aryl group and the heteroaryl group may be substituted by 0 to 2 R 1Y ;
- R y is independently selected from H, C 3 -C 10 alkyl group having 0-3 R c substitutions, C 1 -C 6 haloalkyl group, C 3 -C 10 cyclic alkyl group, containing carbon atom and 1-4 a heteroalkyl group and a heterocycloalkyl group selected from the group consisting of O, N, S(O) p heteroatoms, NR d R e , OR d , aryl, containing carbon atoms and 1 to 2 selected from N, O and S a 5- to 6-membered heteroaryl group of a hetero atom; wherein the aryl group and the heteroaryl group are substituted by 0-2 R 1X ; R 1X is independently selected from halogen, OH, CN, C 1 -C 4 alkyl, C 1- C 4 haloalkyl, C 1 -C 4 alkoxy, C 1 -C 4 haloalkoxy, C 3 -C 8 cyclic alky
- R c as defined in the above defined 4a) of the same portion of R c;
- R i is independently selected from the group consisting of H, CN, C1-C4 alkyl
- n are each independently selected from 0-4;
- Y is independently selected from the group consisting of O, NR g , S(O) p , -CR i (CH 2 ) m NR g R h and -CR i (CH 2 ) m OR g ;
- R j and R k are independently selected from H, CN, C 1 -C 10 alkyl substituted with 0-3 R s , C 1 -C 6 haloalkyl, C 3 -C 10 cyclic alkyl, containing carbon Atom and 1-4 heteroalkyl and heterocycloalkyl selected from O, N, S(O) p heteroatoms, C 2 -C 10 alkenyl or alkynyl, 6 to 10 membered aryl, containing carbon atoms And 1 to 2 5- to 10-membered heteroaryl groups selected from N, O and S heteroatoms; wherein the aryl and heteroaryl groups may be substituted by 0-2 R 1Y ;
- p are each independently selected from 0, 1 and 2;
- R 1X is independently selected from halogen, OH, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, C 1 -C 4 haloalkoxy, C 3 -C 8 cyclic alkyl and cyclic heteroalkyl;
- R 1Y is independently selected from C 1 -C 10 alkyl, halogen, CN, C 1 -C 6 haloalkyl, C 1 -C 6 haloalkoxy, C 3 -C 8 cyclic alkyl;
- p are each independently 0, 1 and 2;
- R c as defined in the above defined 4a) of the same portion of R c;
- p are each independently selected from 0, 1 and 2;
- R i is independently selected from the group consisting of H, CN, and C 1 -C 4 alkyl;
- n are each independently selected from 0-4;
- Y is independently selected from the group consisting of O, NR g , S, -CR i NR g R h and -CR i OR g ;
- R j and R k are independently selected from H, CN, C 1 -C 10 alkyl substituted with 0-3 R s , C 1 -C 6 haloalkyl, C 3 -C 10 cyclic alkyl, containing carbon Atom and 1-4 heteroalkyl and heterocycloalkyl selected from O, N, S heteroatoms, C 2 -C 10 alkenyl or alkynyl, 6 to 10 membered aryl, containing carbon atoms and 1 to 2 a 5- to 10-membered heteroaryl group selected from the group consisting of N, O and S heteroatoms; wherein the aryl group and the heteroaryl group may be substituted by 0-2 R 1Y ;
- R s is independently selected from OH, CN, halogen, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, C 1 -C 6 alkoxy, C 1 -C 6 haloalkoxy, C 3 -C 8 cyclic alkyl, -(OCH 2 CH 2 ) m OR d , (OCH 2 CH 2 ) m NR d R e , a heteroalkyl group containing a carbon atom and 1 to 2 heteroatoms selected from N, O and S And a heterocycloalkyl group, an aryl group, and a heteroaryl group containing a carbon atom and 1 to 2 hetero atoms selected from N, O and S, wherein the aryl group and the heteroaryl group may be substituted by 0-2 R 1Y ;
- R d, R e and defined in the above 4b) portion R d, R e is the same as defined;
- R 1Y is independently selected from C 1 -C 10 alkyl, halogen, CN, C 1 -C 6 haloalkyl, C 1 -C 6 haloalkoxy, C 3 -C 8 cyclic alkyl;
- R c as defined in the above defined 4a) of the same portion of R c;
- p are each independently selected from 0, 1 and 2;
- R i is independently selected from the group consisting of H, CN, and C 1 -C 4 alkyl;
- n are each independently selected from 0-4;
- the compound of formula I has the formula Ia-1 or Ia-2:
- R 1A , R 1B and R 1C , R 2B and R 2C , R 3B and R 3C , R 1D , n, m, q, Y, M are the same as corresponding definitions in part 4) of formula I ;
- the compound of formula I has formula Ia-3 or Ia-4:
- R 1A , R 1B and R 1C , R 2B and R 2C , R 3B and R 3C , R 1D , n, m, q, Y, M are the same as corresponding definitions in part 4) of formula I ;
- the compound of formula I has formula Ia-5 or Ia-6:
- the compound of formula I has formula Ia-7 or Ia-8:
- R g is the same as the corresponding definition in the 4b) part of the 4) part of the formula I;
- the compound of formula I has formula Ia-9:
- R g is the same as the corresponding definition in the 4b) part of the 4) part of the formula I;
- the compound of formula I has one of the following structural formulae:
- R g is the same as the corresponding definition in the 4b) part of the 4) part of the formula I;
- the compound of formula I is selected from the group consisting of:
- the compound further includes stereoisomers, tautomers, atropisomers, isotopically labeled compounds (including hydrazine substituted), medically acceptable salts, polymorphs, solvents
- the composition can be used to treat a disease or condition mediated by EED and/or PRC2.
- the method comprises the following steps:
- compound 12 is subjected to a Suzuki coupling reaction with various types of boronic acid having an R 1 group or its equivalent under the action of a palladium catalyst to obtain a product 13.
- the amines described in Schemes 1 and 2 can be prepared by the preparation of US2016/0176682A1 (for example, the preparation of A1 in the following reaction formula), or purchased by a reagent company (the following reaction formula, indole A2, purchased from BEHRINGER TECHNOLOGY CO., LTD.
- the boric acid or its equivalent B can be purchased from a reagent company or prepared according to conventional literature.
- a reagent or compound having an Rg group such as, but not limited to, an acid anhydride, a sulfonic acid anhydride, an isocyanate, a thioisocyanate, an acid chloride Sulfonyl chloride, carbonate, chloroformate, carbamate, etc., such as, but not limited to, triethylamine, diisopropylethylamine, DMAP, potassium carbonate, sodium hydroxide, potassium hydroxide, Potassium tert-butoxide, NaH, the organic solvent such as, but not limited to, dichloromethane, tetrahydrofuran, acetonitrile, 1,4-dioxane,
- a reagent or compound having an Rg group such as, but not limited to, an acid anhydride, a sulfonic acid anhydride, an isocyanate, a thioisocyanate, an acid chloride Sulfonyl chloride, carbonate,
- A, X 1 , R g , R 1A , R 1B , R 1C , R 2B , R 2C , R 3B , R 3C , q , m have the same meanings as defined above.
- the product 15 obtained by removing the protecting group in the step (3a) of the scheme 3 is condensed with a carboxylic acid having a R j group by a condensing agent to obtain an amide compound 17, for example, but not Limited to carbonyl diimidazole, dicyclohexylcarbodiimide, diisopropylcarbodiimide, 1-(-3-dimethylaminopropyl)-3-ethylcarbodiimide, 1-hydroxybenzotriene Oxazole, 2-(7-azobenzotriazole)-N,N,N',N'-tetramethylurea hexafluorophosphate, benzotriazole-N,N,N',N' -tetramethylurea hexafluorophosphate, 6-chlorobenzotriazole-1,1,3,3-tetramethyluronium hexafluorophosphate, O-benzotriazole-N,N,N' , N'
- (7a) 20 is oxidized with mCPBA or hydrogen peroxide to give compound 23.
- reaction 15 reduction of a double bond (such as, but not limited to, under hydrogenation reduction conditions) to give 24, followed by reaction with a reagent or compound bearing an Rg group in the presence of a base to give 25, such as but not limited to Anhydride, sulfonic anhydride, isocyanate, thioisocyanate, acid chloride, sulfonyl chloride, carbonate, chloroformate, carbamate, such as, but not limited to, triethylamine, diisopropylethylamine, DMAP, Potassium carbonate, sodium hydroxide, potassium hydroxide, potassium t-butoxide, NaH, the reaction can be carried out in an organic solvent such as, but not limited to, dichloromethane, tetrahydrofuran, acetonitrile, 1,4-dioxane Hexacyclic ring; or 24 may also be condensed with various carboxylic acids in the presence of a condensing agent to give an amide compound
- the method comprises the following steps:
- (3a) 14' is removed from the Boc protecting group by trifluoroacetic acid using dichloromethane as a solvent to give the amine compound 15'.
- X 1 and Y are the same as defined above.
- the above-mentioned compounds 14, 14', 18, 18' and 20 can be obtained by a Suzuki reaction according to the step (1d) or the step (2g) in the first or second embodiment.
- optically active forms of the compounds of the invention are desired, they can be obtained using optically active starting materials, or by resolution of compounds or intermediates using standard procedures known to those skilled in the art, such as separation by chiral chromatography columns. A mixture of stereoisomers is obtained.
- a pure geometric isomer of a compound of the invention when desired, it can be obtained by using pure geometric isomers as starting materials, or by geometrical isomerization of compounds or intermediates by standard procedures, such as chromatographic separation. The body mixture is obtained.
- a pharmaceutical composition comprising a triazolopyrimidine, a triazolopyridine compound as described above, a pharmaceutically acceptable salt thereof, an enantiomer, and a non- One or more of the enantiomers or racemates.
- the pharmaceutical composition further comprises at least one other therapeutic agent.
- the at least one additional therapeutic agent included in the pharmaceutical composition is selected from the group consisting of other anticancer agents, immunomodulators, anti-allergic agents, antiemetics, pain relieving agents, cytoprotective agents, and combinations thereof.
- the pharmaceutical composition comprises at least one compound of the invention and at least one pharmaceutically acceptable carrier, diluent or excipient,
- the disease or condition comprises diffuse large B-cell lymphoma, follicular lymphoma, other lymphoma, leukemia, multiple myeloma, mesothelioma, gastric cancer, malignant rhabdoid tumor, hepatocellular carcinoma, prostate Cancer, breast cancer, bile duct and gallbladder cancer, bladder cancer; brain tumors, including neuroblastoma, schwannomas, glioma, glioblastoma and astrocytoma; cervical cancer, colon cancer, melanin Tumor, endometrial cancer, esophageal cancer, head and neck cancer, lung cancer, nasopharyngeal cancer, ovarian cancer, pancreatic cancer, renal cell carcinoma, rectal cancer, thyroid cancer, parathyroid tumor, uterine tumor and soft tissue sarcoma.
- brain tumors including neuroblastoma, schwannomas, glioma, glioblastoma and astrocytoma
- cervical cancer
- a method of treating a disease or condition mediated by EED and/or PRC2 comprising
- a therapeutically effective amount of a compound of formula I or a pharmaceutical composition thereof is provided to a subject in need thereof.
- the disease or condition is selected from the group consisting of diffuse large B-cell lymphoma, follicular lymphoma, other lymphoma, leukemia, multiple myeloma, mesothelioma, gastric cancer, malignant rhabdoid tumor, hepatocellular carcinoma, Prostate cancer, breast cancer, bile duct and gallbladder cancer, bladder cancer; brain tumors, including neuroblastoma, schwannomas, glioma, glioblastoma and astrocytoma; cervical cancer, colon cancer, Melanoma, endometrial cancer, esophageal cancer, head and neck cancer, lung cancer, nasopharyngeal cancer, ovarian cancer, pancreatic cancer, renal cell carcinoma, rectal cancer, thyroid cancer, parathyroid tumor, uterine tumor and soft tissue sarcoma.
- diffuse large B-cell lymphoma follicular lymphoma, other lymphoma
- leukemia multiple myel
- Bromine 4-1 (72 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-1 (84 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-1 (72 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-2 (90 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-1 (72 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-3 (123 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-2 (58 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-1 (84 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-2 (58 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2 M aqueous Na 2 CO 3 solution, and borate B-2 (90 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-2 (58 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-3 (123 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-2 (58 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-4 (83 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-2 (58 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-5 (102 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-2 (58 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-6 (129 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-1 (72 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-7 (137 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-3 (62 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-3 (123 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- the bromine 13-2 (63 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2 M aqueous Na 2 CO 3 solution, and borate B-3 (123 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- the bromine 13-1 (77 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-1 (84 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 13-1 (77 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-3 (123 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-1 (72 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-8 (89 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Example 56 Synthesis of Compound SL-ZYE-08, SL-ZYE-08-S, SL-ZYE-08-R
- SL-ZYE-08 can be further separated by chiral column to obtain a pair of optically pure compounds SL-ZYE-08-S and SL-ZYE-08-R.
- the chiral column separation method is a conventional method known to those skilled in the art.
- Bromine 4-4 (69 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-1 (84 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- the bromide 4-5 (68mg, 0.2mmol) was dissolved in 6 mL 1,4-dioxane and 2mL of concentration of Na 2M 2 CO 3 solution was added boric acid ester B-1 (84mg, 0.4mmol) , Ar substitution protection, stirring at room temperature for 10 minutes.
- the bromide 4-5 (68mg, 0.2mmol) was dissolved in 6 mL 1,4-dioxane and 2mL of concentration of Na 2M 2 CO 3 solution was added boric acid ester B-3 (123mg, 0.4mmol) , Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-1 (72 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-10 (101 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- Bromine 4-1 (72 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-11 (95 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- the bromo 4-1 (72 mg, 0.2 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-12 (103 mg, 0.4 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- SL-E2 was isolated by chiral chromatography to obtain optically pure compounds SL-E2-S and SL-E2-R.
- SL-E12 (10 mg) was dissolved in 1 mL of dichloromethane and 0.1 mL of triethylamine (TEA), dissolved by stirring, and methanesulfonic anhydride (Ms 2 O, 8 mg) was added at room temperature for 1 hour, and TLC showed the reaction was completed.
- TAA triethylamine
- Ms 2 O methanesulfonic anhydride
- the bromo 4-1 (36 mg, 0.1 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate B-14 (44 mg, 0.2 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- the bromo 4-1 (36 mg, 0.1 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2 M aqueous Na 2 CO 3 solution, and borate B-15 (47 mg, 0.2 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- SL-E29 (10 mg) was dissolved in 1 mL of dichloromethane and 0.1 mL of triethylamine (TEA), dissolved by stirring, and acetic anhydride (Ac 2 O, 8 mg) was added at room temperature for 1 hour, and TLC showed the reaction was completed.
- SL-E29 (10 mg) was dissolved in 1 mL of dichloromethane and 0.1 mL of triethylamine (TEA), dissolved by stirring, and methanesulfonic anhydride (Ms 2 O, 7 mg) was added at room temperature for 1 hour, and TLC showed the reaction was completed.
- TAA triethylamine
- Ms 2 O methanesulfonic anhydride
- SL-E43 was separated by chiral chromatography to obtain optically pure compounds SL-E43-S and SL-E43-R.
- Example 110 Synthesis of Compound SL-E44, SL-E44-S, SL-E44-R
- SL-E44 was isolated by chiral chromatography to obtain optically pure compounds SL-E44-S and SL-E44-R.
- Example 111 Synthesis of Compound SL-E45, SL-E45-S, SL-E45-R
- SL-E45 was separated by chiral chromatography to obtain optically pure compounds SL-E45-S and SL-E45-R.
- Example 112 Synthesis of Compound SL-E46, SL-E46-S, SL-E46-R
- SL-E46 was separated by chiral column to obtain optically pure compounds SL-E46-S and SL-E46-R.
- SL-E47 was separated by chiral column to obtain optically pure compounds SL-E47-S and SL-E47-R.
- SL-E48 was separated by chiral column to obtain optically pure compounds SL-E48-S and SL-E48-R.
- SL-E49 was separated by chiral column to obtain optically pure compounds SL-E49-S and SL-E49-R.
- SL-E50 was separated by chiral chromatography to obtain optically pure compounds SL-E50-S and SL-E50-R.
- SL-E51 was separated by chiral chromatography to obtain optically pure compounds SL-E51-S and SL-E51-R.
- SL-E52 was separated by chiral chromatography to obtain optically pure compounds SL-E52-S and SL-E52-R.
- the bromo 4-1 (36 mg, 0.1 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2 M aqueous Na 2 CO 3 solution, and the boronic acid ester SL-B3 (48 mg, 0.2 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes. Add 10% allyl palladium (II) dimer (3.5 mg, 0.01 mmol), 20% 2'-dicyclohexylphosphino-2,6-dimethoxy-1,1'-biphenyl Sodium 3-sulfonate hydrate (11 mg, 0.02 mmol) was kept at 90 ° C for 40 minutes under Ar protection.
- the bromo 4-1 (36 mg, 0.1 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and borate SL-B4 (48 mg, 0.2 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes.
- SL-ZYE-119 (10 mg) was dissolved in 2 mL of methanol, and reacted with 10% Pd/C (4 mg) at room temperature for 6 hours, filtered, and separated by column chromatography to give the title compound SL-ZYE-121 (4 mg).
- 1 H NMR 400MHz, DMSO- d 6) ⁇ 9.32 (s, 1H), 8.32 (s, 1H), 7.38 (m, 1H), 6.85 (m, 1H), 6.62 (m, 1H), 4.56- 4.47 (m, 4H), 3.68 (m, 2H), 3.20 (m, 3H), 1.70 (m, 4H), 1.09 (m, 6H).
- LC-MS: [M+H] + 398.2.
- the bromo 4-1 (36 mg, 0.1 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2 M aqueous Na 2 CO 3 solution, and the boronic acid ester SL-B5 (50 mg, 0.2 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes. Add 10% allyl palladium (II) dimer (3.5 mg, 0.01 mmol), 20% 2'-dicyclohexylphosphino-2,6-dimethoxy-1,1'-biphenyl Sodium 3-sulfonate hydrate (11 mg, 0.02 mmol) was kept at 90 ° C for 40 minutes under the protection of Ar.
- the bromo 4-1 (36 mg, 0.1 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2M aqueous Na 2 CO 3 solution, and the boronic acid ester SL-B6 (49 mg, 0.2 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes. Add 10% allyl palladium (II) dimer (3.5 mg, 0.01 mmol), 20% 2'-dicyclohexylphosphino-2,6-dimethoxy-1,1'-biphenyl Sodium 3-sulfonate hydrate (11 mg, 0.02 mmol) was kept at 90 ° C for 40 minutes under the protection of Ar.
- the bromo 4-1 (36 mg, 0.1 mmol) was dissolved in 6 mL of 1,4-dioxane and 2 mL of a 2 M aqueous Na 2 CO 3 solution, and the boronic acid ester SL-B7 (49 mg, 0.2 mmol) was added. Ar substitution protection, stirring at room temperature for 10 minutes. Add 10% allyl palladium (II) dimer (3.5 mg, 0.01 mmol), 20% 2'-dicyclohexylphosphino-2,6-dimethoxy-1,1'-biphenyl Sodium 3-sulfonate hydrate (11 mg, 0.02 mmol) was kept at 90 ° C for 40 minutes under the protection of Ar. After cooling, it was concentrated under reduced pressure and purified by column chromatography to give the title compound SL-ZYE-197 (4.1. Mg).
- SL-E12 (30 mg) was dissolved in 3 mL of dichloromethane and 0.1 mL of triethylamine (TEA), dissolved by stirring, and methyl chloroformate (15 mg) was added at room temperature to continue the reaction. TLC showed the end of the reaction, directly by column chromatography. Separation and preparation gave the title compound SL-ZYE-183 (5 mg).
- the activity of the PRC2 enzyme was detected using the TR-FRET method.
- the enzyme was mixed with different concentrations of the compound and incubated for 30 minutes at room temperature.
- the biotinylated histone H3 polypeptide substrate and the cofactor S-adenosylmethionine (SAM) were added to initiate the enzymatic reaction.
- SAM cofactor S-adenosylmethionine
- the receptor Acceptor and donor Donor were added and incubated for half an hour. Fluorescence signals were detected using a multi-function microplate reader EnVision (Perkin Elmer). Data was analyzed using GraphPad Prism 5.0 software, the value of 50 obtained IC.
- Example 135 Cell growth inhibition test (11 days)
- Pfeiffer cells in the exponential growth phase were seeded in 24-well plates at a cell density of 1*10E5 cells/mL. Cells were treated with different concentrations of compounds on the day. Fresh medium and compound were replaced at 4 and 7 days of compound treatment. After 11 days of compound treatment, cell viability was measured using CellTiter-Glo reagent (Promega). Data was analyzed using GraphPad Prism 5.0 software, get 50 values GI.
- Example 136 Long-term growth inhibition test of pfeiffer cells (14 days)
- DLBCL Human diffuse large B-cell lymphoma (DLBCL) cell line pfeiffer (from ATCC, CRL-2632) containing 10% fetal bovine serum (Gibco, purchased from Life Technologies, 10099-141) and 1% antibiotic (penicillin and chain) RPMI 1640 medium (Gibco, available from Life Technologies, Inc., 22400-089) purchased from Life Technologies, Inc., 10378016) was cultured in a CO 2 cell incubator (37 ° C, 5% CO 2 ).
- pfeiffer cells of exponential growth phase were plated in a 24-well plate (purchased from Corning, 3524) in a volume of 1 mL/well and a cell density of 2*10E5 cells/mL. The cells were seeded and placed in a CO 2 incubator for 1 hour. 2 ⁇ L of 9 different concentrations of 3-fold gradient dilutions of compound or DMSO were added to each well in a 24-well plate containing cells at a final concentration ranging from 0.003 to 20 ⁇ M or 0.3 to 2000 nM with a final concentration of 0.2% DMSO.
- Example 137 Long-term growth inhibition test of cells Karpas-422 and SU-DHL-4 (11 days)
- DLBCL Human diffuse large B-cell lymphoma (DLBCL) cell lines Karpas-422, SU-DHL-4 (ATCC, CRL-2957) containing 10% fetal bovine serum (Gibco, purchased from Life Technologies, 10099-141) and 1% antibiotic (penicillin and streptomycin, purchased from Life Technologies, 10378016) in RPMI 1640 medium (Gibco, purchased from Life Technologies, Inc., 22400-089) in a CO 2 cell incubator (37 ° C, 5% CO 2 ) ) cultured.
- RPMI 1640 medium Gibco, purchased from Life Technologies, Inc., 22400-089
- CO 2 cell incubator 37 ° C, 5% CO 2
- the cell culture medium volume was 1 mL. After the cells were cultured for 1 hour in a 24-well plate, 2 ⁇ L of the compound or DMSO was added to each well. Each compound has 9 different concentrations, with a final concentration in the cell culture medium ranging from 0.003 to 20 [mu]M or 0.3 to 2000 nM and a final concentration of 0.2% DMSO. At 4 and 7 days of compound treatment, fresh cell culture medium and compound were replaced, and the cell density of the DMSO control well was diluted to 1*10E5/mL, and the cell dilution ratio of the compound well was the same as that of the DMSO control well.
- Cell viability was determined using CellTiter-Glo reagent (purchased from Promega, G7572): cells treated with compound for 11 days were transferred to a white 384-well plate (OptiPlate-384, available from PerkinElmer, 6007299) at 40 ⁇ L/well. Add an equal volume of CellTiter-Glo reagent. After incubation for 10 minutes at room temperature, the cold luminescence signal was detected using a multi-plate reader EnVision (available from PerkinElmer) at a wavelength of 400-700 nm. Data was analyzed using GraphPad Prism 5.0 software, the value of 50 obtained IC.
- the IC 50 value of the partial compound of the present invention to the PRC2 enzyme can be up to nM order, which is significantly higher than that of the positive control group EED226 compound; similarly, for Pfeiffer, Karpas-422 and In the SU-DHL-4 cell long-term growth inhibition experiment, the IC 50 values of the various compounds of the present invention also reached the single digit nM order of magnitude, which was significantly higher than the positive control group EED226 compound.
- test animals were healthy adult male SD rats, 3 in each group, purchased from Shanghai Xipuer-Beikai Experimental Animal Co., Ltd.
- Rats were administered EED226, E-Y1, E-Y13, E-Y47, and SL-ZYE-07 by intragastric administration (3mg/kg) at 0.25, 0.5, 1, 2, 4, 8 after administration. 45 ⁇ L of blood was taken from the femoral vein for 24 hours, centrifuged in a heparinized centrifuge tube for 5 min, and plasma samples were separated for analysis. We used liquid chromatography-tandem mass spectrometry (LC-MS/MS) to determine the plasma of rats after intragastric administration. The content of the test compound.
- LC-MS/MS liquid chromatography-tandem mass spectrometry
- the pharmacokinetic parameters of the compounds of the invention are as follows:
- AUC last AUC from the time of administration to the last time
- AUC INF_obs AUC from the time of administration to the time when the theoretical extrapolation is infinitely long
- the compounds of the present invention have good pharmacological absorption and have obvious pharmacokinetic advantages.
- Example 139 Liver microsome stability test (mouse, rat, human):
- Tris pH 7.4 buffer (0.1 M) was prepared: 12.12 g of Tris was dissolved in 1000 mL of H 2 O, adjusted to pH 7.4 with 2N HCl, dispensed at 50 mL/tube, and frozen at -20 °C.
- VIVID stock solution Dissolve 1 mg of VIVID in 1 mL of acetonitrile, dispense 50 ⁇ L/tube, and store at -20 °C.
- MgCl 2 solution 100 mM: 1.016 g of MgCl 2 was dissolved in 50 mL of Tris pH 7.4 buffer, dispensed in 1 mL/tube, and frozen at -20 °C.
- NADPH solution (10 mM): 355 mg of NADPH was dissolved in 42.6 mL of Tris pH 7.4 buffer, dispensed at 1.8 mL/tube, and frozen at -20 °C.
- Dilution Step 3 Take 245 ⁇ L of working solution into a 96-well compound plate and add 5 ⁇ L of VIVID stock solution.
- the 96-well compound plate was placed on an shaker for 5 minutes.
- the logarithm of the residual rate of the drug in the incubation system was plotted against the incubation time, and linear regression was performed to obtain the slope k.
- the intrinsic clearance rate (Clint, mL/min/g) and the in vivo clearance rate (Cl, mL) were estimated according to the following formula. /min), liver clearance (Clhep, mL/min), metabolic bioavailability (%MF):
- the compounds of the present invention have good stability in liver microsomes of human, rat and mouse, and have obvious advantages.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pulmonology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明提供了一种由通式I表示的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,其制备方法,包含其的药物组合物,以及其在制备用于治疗由EED和/或PRC2介导的疾病或病症的药物中的用途。本发明所述的化合物可用于治疗由PRC2介导的疾病或病症。(I)
Description
本发明涉及药物化学和药物治疗学领域,总体上涉及一类三氮唑并嘧啶、三氮唑并吡啶化合物以及药物组合物及其在肿瘤疾病治疗中的应用。特别地,该类化合物可制备用于治疗由PRC2介导的疾病或病症的药物。
多梳抑制复合物PRC2(Polycomb Repressive Complex 2)是多梳家族蛋白(Polycomb Group)的核心成员,具有组蛋白甲基转移酶活性,可特异性催化组蛋白H3第27位赖氨酸的三甲基化修饰(H3K27me3),从而抑制特定基因的表达。PRC2的甲基转移酶活性来源于其催化成员EZH2,然而EZH2在单独存在时并没有催化活性,其至少需要与PRC2的另外两个成员EED和SUZ12形成复合物后才能催化甲基化修饰。因而,EZH2,EED和SUZ12被认为是PRC2复合物的核心组分。近来研究发现,PRC2的核心组分在多种肿瘤细胞中过表达,其活性异常是导致多种恶性肿瘤发病及恶化的直接原因。同时,最近对淋巴瘤病人的基因测序结果表明,EZH2在生发中心B细胞淋巴瘤(GCB-DLBCL)病人中出现激活性突变,突变后的EZH2改变PRC2的底物特异性,从而提高细胞中H3K27me3水平。通过siRNA方法下调EZH2或其他核心组分的表达,将显著抑制淋巴瘤细胞的增殖,这表明GCB-DLBCL的发生发展与PRC2的过度激活密切相关。因而,PRC2是一个非常有前景的抗癌药物开发靶标,靶向PRC2的抑制剂发现是目前制药界研究的热点。近期,诺华及艾伯维两大制药公司发明了一类通过借助靶向EED来抑制PRC2活性的小分子(参考文献:诺华的EED226,US 2016/0176682,J.Med.Chem.2017,60,2215–2226,J.Med.Chem.2017,60,415–427,Nat.Chem.Biol.2017,13,381–388;艾伯维的A-395,Nat.Chem.Biol.2017,13,389–395),该类化合物在分子水平、细胞水平以及动物实验上都显示极强的抑制活性。综上所述,PRC2复合物被认为是导致多种恶性肿瘤发生发展的关键驱动因子,而借助靶向EED来抑制PRC2活性的抑制剂的开发目前在业界具有很高的热度,有利于用于与之相关的新药研发。
发明内容
本发明的涉及一种如通式I所示三氮唑并嘧啶、三氮唑并吡啶化合物及其组合物,并且结合活性评价和相关生物学实验,可以用于制备用以治疗由EED及/或PRC2介导的疾病或病症的药物。
本发明的一个目的是提供一类三氮唑并嘧啶、三氮唑并吡啶化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体。
本发明的另一个目的在于提供一种上述化合物的制备方法。
本发明的再一个目的在于提供一种包含治疗有效量的一种或多种上述化合物或其可药用的盐的药物组合物。
本发明的又一个目的在于提供上述化合物在制备用于治疗由EED和/或PRC2介导的疾病或病症的药物中的用途。
本发明的又一个目的在于提供一种治疗由EED和/或PRC2介导的疾病或病症的方法,其特征在于,向受试者施用治疗有效量的一种或多种上述化合物或其可药用的盐。
具体地,根据本发明的一个方面,其提供了一种通式I的化合物:

其中
1)X
1独立地选自N及C-CN;
2)R
2独立地选自H、卤素、C
1-C
4卤代烷基及C
1-C
4烷基;
3)A独立地选自以下结构:

R
3、R
4及R
5独立地选自H、卤素、C
1-C
4烷基、C
1-C
4卤代烷基、-O-(C
1-C
4烷基)、C
1-C
4卤代烷氧基、C
3-C
6环烷基;
R
6独立地选自H、OH、=O及C
1-C
4烷基;
R
7独立地选自H、OH、卤素、CN及C
1-C
4烷基;
n各自独立地选自0,1及2;
X
2独立地选自O、NR
a及S(O)
p杂原子;
每一R
a独立地选自H、O、由0-2个R
b取代的C
1-C
10烷基、C
1-C
6卤代烷基、-O-(C
1-C
6烷基)、C
1-C
6卤代烷氧基、C
3-C
6环状烷基、-C(=O)(C
1-C
4烷基)、-CO
2(C
1-C
4烷基)、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;
R
b独立地选自卤素、OH、NH
2、NHC(=O)(C
1-C
4烷基)、NHS(=O)
2(C
1-C
4烷基)、=O、CN、C
1-C
4烷基及C
1-C
4烷氧基;
p各自独立地选自0,1及2;
4)R
1独立地选自以下结构:

4a)R
1A独立地选自H、羟基、卤素、CN、-(O)
z-(包含0-2个R
c取代C
1-C
10烷基)、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、C
3-C
8环状烷基、-C(=O)(C
1-C
4烷基)、-C(=O)NH(C
1-C
4烷基)、氨基、C
1-C
6直链、支链以及环状的烷氨基、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R
1X取代;
p各自独立地选自0,1及2;
R
c独立地选自OH、卤素、CN、C
1-C
6烷基、C
1-C
6卤代烷基、C
1-C
6烷氧基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基、-(OCH
2CH
2)
mOR
d、NHC(=O)NR
dR
e、NHC(=S)NR
dR
e、-NHC(=NH)NR
dR
e、(OCH
2CH
2)
mNR
dR
e、-C(=O)R
d、-S(=O)R
d、-C(=O)NR
dR
e、-S(=O)
2R
d、-NHC(=O)R
d、-NHC(=S)R
d、-NHS(=O)
2R
d、-S(=O)
2NHR
d、包含碳原子及1至2个选自N、NR
a、O及S(O)
p杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选自N、NR
a、O及S(O)
p杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R
1X取代;
R
d及R
e独立地选自H、含0-2个R
b取代的C
1-C
6烷基、C
1-C
6卤代烷基、C
3-C
6环状烷基、-C(=O)(C
1-C
4烷基)、-CO
2(C
1-C
4烷基)、-C(=O)NH(C
1-C
4烷基)、含0-2个O、N、S(O)
p杂原子的支链或环状的C
1-C
6杂烷基、-C(=O)H、芳基及包含碳原子及1至2个选自N、NR
a、O及S(O)
p杂原子的杂芳基,其中该芳基以及杂芳基可由0-2个R
1X取代;
R
a独立地选自H、O、由0-2个R
b取代的C
1-C
10烷基、C
1-C
6卤代烷基、-O-(C
1-C
6烷基)、C
1-C
6卤代烷氧基、C
3-C
6环状烷基、-C(=O)(C
1-C
4烷基)、-CO
2(C
1-C
4烷基)、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;
R
b独立地选自卤素、OH、NH
2、-NHC(=O)(C
1-C
4烷基)、-NHS(=O)
2(C
1-C
4烷基)、=O、CN、C
1-C
4烷基及C
1-C
4烷氧基;
p各自独立地选自0,1及2;
R
d与R
e可通过R
d——R
e或
 的方式连接,其中Z
1可选自含0-2个R
b取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6杂烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p;R
b独立地选自卤素、OH、NH
2、-NHC(=O)(C
1-C
4烷基)、-NHS(=O)
2(C
1-C
4烷基)、=O、CN、C
1-C
4烷基及C
1-C
4烷氧基;p各自独立地选自0,1及2;
的方式连接,其中Z
1可选自含0-2个R
b取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6杂烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p;R
b独立地选自卤素、OH、NH
2、-NHC(=O)(C
1-C
4烷基)、-NHS(=O)
2(C
1-C
4烷基)、=O、CN、C
1-C
4烷基及C
1-C
4烷氧基;p各自独立地选自0,1及2;
R
1X独立地选自卤素、OH、CN、C
1-C
4烷基、C
1-C
4卤代烷基、C
1-C
4烷氧基、C
1-C
4卤代烷氧基、C
3-C
8环状烷基和环状杂烷基;
R
1B及R
1C独立地选自H、OH、卤素、CN、-(O)
z-(包含0-2个R
c取代C
1-C
10烷基)、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、C
3-C
8环状烷基、-C(=O)(C
1-C
4烷基)、-C(=O)NH(C
1-C
4烷基)、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R
1X取代;p各自独立地 为0,1及2;
R
2B及R
2C独立地选自H、OH、卤素、CN、-(O)
z-(包含0-2个R
c取代C
1-C
10烷基)、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、C
3-C
8环状烷基、-C(=O)(C
1-C
4烷基)、-C(=O)NH(C
1-C
4烷基)、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R
1X取代;p各自独立地选自0,1及2;
R
3B及R
3C独立地选自H、-OH、卤素、CN、-(O)
z-(包含0-2个R
c取代C
1-C
10烷基)、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、C
3-C
8环状烷基、-C(=O)(C
1-C
4烷基)、-C(=O)NH(C
1-C
4烷基)、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R
1X取代;p各自独立地选自0,1及2;
或者,R
1B与R
1C、R
2B与R
2C、R
3B与R
3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);
R
1D独立地选自H、-OH、卤素、CN、-C(=O)H、-(O)
z-(包含0-2个R
c取代C
1-C
6烷基)、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、R
f、-OR
f、-C(=O)R
c、NR
dR
e、-C(=O)NR
dR
e、-NHC(=O)R
c、-S(=O)
2R
c、-S(=O)
2NR
dR
e、-NHS(=O)
2R
d、-(OCH
2CH
2)
mOR
d、-(OCH
2CH
2)
mNR
dR
e;
R
f独立地选自C
3-C
8环状烷基、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、芳基及包含碳原子及1至2个选自N、NR
a、O及S(O)
p杂原子的杂芳基;其中,芳基及杂芳基由0-2个R
1X取代;
M独立地选自3至7元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;
在R
1B及R
1C、R
2B及R
2C、R
3B及R
3C、R
1D以及R
f的定义中,R
a、R
c、R
d、R
e、p、z、m、R
1X的定义与在4a)部分中的R
1A中的相应的定义相同;
n各自独立地选自0,1及2;
m各自独立地选自0-4;
p各自独立地选自0-2;
q各自独立地选自0-3;
z各自独立地选自0和1;
4a’)优选地,R
1独立地选自以下结构:
 其中
其中
R
1A独立地选自H、羟基、卤素、CN、-(O)
z-(包含0-2个R
c取代C
1-C
10烷基)、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、C
3-C
8环状烷基、-C(=O)(C
1-C
4烷基)、-C(=O)NH(C
1-C
4烷基)、-C(=O)H;
R
c独立地选自OH、卤素、CN、C
1-C
6烷基、羧基、C
1-C
6卤代烷基、C
1-C
6烷氧基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基;
R
1B及R
1C、R
2B及R
2C、以及R
3B及R
3C独立地选自H、OH、卤素、CN、-(O)
z-(包含0-2个R
c取代C
1-C
10烷基)、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、C
3-C
8环状烷基、-C(=O)(C
1-C
4烷基)、-C(=O)NH(C
1-C
4烷基);
或者,R
1B与R
1C、R
2B与R
2C、R
3B与R
3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);
R
1D独立地选自H、-OH、卤素、CN、-C(=O)H、-(O)
z-(包含0-2个R
c取代C
1-C
6烷基)、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、C
3-C
8环状烷基;
R
c独立地选自OH,卤素、CN、C
1-C
6烷基、羧基、C
1-C
6卤代烷基、C
1-C
6烷氧基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基;
M独立地选自3至7元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;
n各自独立地选自0,1及2;
m各自独立地选自0-4;
p各自独立地选自0-2;
q各自独立地选自0-3;
z各自独立地选自0和1;
4a”)更优选地,在R
1中,
R
1A独立地选自H、羟基、卤素、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基;
R
1B及R
1C、R
2B及R
2C、以及R
3B及R
3C独立地选自H、OH、卤素、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基;
或者,R
1B与R
1C、R
2B与R
2C、R
3B与R
3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);
R
1D独立地选自H、-OH、卤素、C
1-C
6烷基、C
1-C
6烷氧基、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基;
M独立地选自5至6元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;
n各自独立地选自0,1及2;
m各自独立地选自0-4;
p各自独立地选自0-2;
q各自独立地选自0-3;
z各自独立地选自0和1;
4b)Y独立地选自O、NR
g、S(O)
p等杂原子以及CH
2、C=O、-CR
i(CH
2)
mNR
gR
h以及-CR
i(CH
2)
mOR
g;
R
g及R
h独立地选自H、O、含0-3个R
s取代的C
1-C
10烷基、C
1-C
6卤代烷基、C
3-C
6环状烷基、

 -C(=S)NHC(=O)-R
j、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1X取代;
-C(=S)NHC(=O)-R
j、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1X取代;
R
s独立地选自OH,CN、卤素、C
1-C
6烷基、C
1-C
6卤代烷基、C
1-C
6烷氧基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基、-(OCH
2CH
2)
mOR
d、NHC(=O)NR
dR
e、NHC(=S)NR
dR
e、-NHC(=NH)NR
dR
e、(OCH
2CH
2)
mNR
dR
e、-C(=O)R
d、-C(=S)R
d、-S(=O)R
d、-C(=O)NR
dR
e、-S(=O)
2R
d、-NHC(=O)R
d、-NHC(=S)R
d、-NHS(=O)
2R
d、-S(=O)
2NR
dR
e、-NHS(=O)
2NR
dR
e、-C(=S)NR
dR
e、NHC(=O)OR
d、NHC(=S)OR
d、-NHS(=O)
2OR
d、NHC(=O)SR
d、NHC(=S)SR
d、-NHC(=NH)OR
d、-C(=O)OR
d、-C(=O)SR
d、-S(=O)
2OR
d、包含碳原子及1至2个选自N、NR
a、O及S(O)
p杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选自N、NR
a、O及S(O)
p杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R
1Y取代;
R
d及R
e独立地选自H、含0-2个R
b取代的C
1-C
6烷基、C
1-C
6卤代烷基、C
3-C
6环状烷基、-C(=O)(C
1-C
4烷基)、-CO
2(C
1-C
4烷基)、-C(=O)NH(C
1-C
4烷基)、含0-2个O、N、S(O)
p杂原子的支链或环状的C
1-C
6杂烷基、-C(=O)H、芳基及包含碳原子及1至2个选自N、NR
a、O及S(O)
p杂原子的杂芳基,其中该芳基以及杂芳基可由0-2个R
1X取代;
R
d与R
e可通过以下方式R
d——R
e或
 连接形成一个环,其中Z
1可选自含0-2个R
b取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p,
连接形成一个环,其中Z
1可选自含0-2个R
b取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p,
R
a独立地选自H、O、由0-2个R
b取代的C
1-C
10烷基、C
1-C
6卤代烷基、-O-(C
1-C
6烷基)、C
1-C
6卤代烷氧基、C
3-C
6环状烷基、-C(=O)(C
1-C
4烷基)、-CO
2(C
1-C
4烷基)、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;
R
b独立地选自卤素、OH、NH
2、-NHC(=O)(C
1-C
4烷基)、-NHS(=O)
2(C
1-C
4烷基)、=O、CN、C
1-C
4烷基及C
1-C
4烷氧基;p各自独立地选自0,1及2;
R
1X独立地选自卤素、OH、CN、C
1-C
4烷基、C
1-C
4卤代烷基、C
1-C
4烷氧基、C
1-C
4卤代烷氧基、C
3-C
8环状烷基和环状杂烷基;
R
1Y独立地选自C
1-C
10烷基、卤素、CN、-(O)
z-(包含0-2个R
c取代C
1-C
10烷基)、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、SCF
3、C
3-C
8环状烷基、-C(=O)(C
1-C
4烷基)、-CO
2 (C
1-C
4烷基)、NR
dR
e、-C(=O)NR
dR
e、-S(=O)
2R
d、-NHC(=O)R
d、-NHC(=S)R
d、-NHS(=O)
2R
d、-NHC(=O)NR
dR
e、-NHC(=S)NR
dR
e、-NHS(=O)
2NR
dR
e、-C(=S)NR
dR
e、-S(=O)
2NHR
d、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、-C(=O)H、p各自独立地为0,1及2;
其中,R
c与在上述4a)部分中限定的R
c的定义相同;
其中R
d与R
e可通过R
d——R
e或
 的方式连接,其中Z
1可选自含0-2个R
b取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6杂烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p,
的方式连接,其中Z
1可选自含0-2个R
b取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6杂烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p,
R
b独立地选自卤素、OH、NH
2、-NHC(=O)(C
1-C
4烷基)、-NHS(=O)
2(C
1-C
4烷基)、=O、CN、C
1-C
4烷基及C
1-C
4烷氧基;p各自独立地选自0,1及2;
R
j及R
k独立地为选自H、CN、含0-3个R
s取代的C
1-C
10烷基、C
1-C
6卤代烷基、C
3-C
10环状烷基、包含碳原子以及选自和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、由R
y取代的烯基或者炔基
 6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1Y取代;
6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1Y取代;
其中,R
1Y与在本4b)部分中的上述R
s中限定的R
1Y的定义相同;
R
y独立地选自H、含0-3个R
c取代的C
1-C
10烷基、C
1-C
6卤代烷基、C
3-C
10环状烷基、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、NR
dR
e、OR
d、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1X取代;R
1X独立地选自卤素、OH、CN、C
1-C
4烷基、C
1-C
4卤代烷基、C
1-C
4烷氧基、C
1-C
4卤代烷氧基、C
3-C
8环状烷基和环状杂烷基;
其中,R
c与在上述4a)部分中限定的R
c的定义相同;R
d和R
e与在本4b)部分中的上述R
s中限定的R
d和R
e的定义相同;
特别地,R
g与R
h以及R
j及R
k可通过以下方式R
g——R
h、
 R
j——R
k、
R
j——R
k、
 连接,其中Z
1可选自含0-2个R
c取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p;p各自独立地选自0,1及2;
连接,其中Z
1可选自含0-2个R
c取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p;p各自独立地选自0,1及2;
其中,R
c与在上述4a)部分中限定的R
c的定义相同;
R
i独立地选自H、CN、C1-C4烷基;
m各自独立地选自0-4;
4b’)优选地,Y独立地选自O、NR
g、S(O)
p、-CR
i(CH
2)
mNR
gR
h以及-CR
i(CH
2)
mOR
g;
R
g及R
h独立地选自H、C
1-C
6卤代烷基、

 -C(=S)NHC(=O)-R
j、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1X取代;
-C(=S)NHC(=O)-R
j、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1X取代;
R
j及R
k独立地选自H、CN、含0-3个R
s取代的C
1-C
10烷基、C
1-C
6卤代烷基、C
3-C
10环状烷基、包含碳原子和1-4个选自O、N、S(O)
p杂原子的杂烷基和杂环烷基、C
2-C
10烯基或者炔基、6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1Y取代;
其中,R
s的定义与在上述4b)部分中的R
s的定义相同;
p各自独立地选自0,1及2;
R
1X独立地选自卤素、OH、CN、C
1-C
4烷基、C
1-C
4卤代烷基、C
1-C
4烷氧基、C
1-C
4卤代烷氧基、C
3-C
8环状烷基和环状杂烷基;
R
1Y独立地选自C
1-C
10烷基、卤素、CN、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基;
p各自独立地为0,1及2;
特别地,R
g与R
h以及R
j及R
k可通过以下方式R
g——R
h、
 R
j——R
k、
R
j——R
k、
 连接,其中Z
1可选自含0-2个R
c取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p;
连接,其中Z
1可选自含0-2个R
c取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p;
其中,R
c与在上述4a)部分中限定的R
c的定义相同;
p各自独立地选自0,1及2;
R
i独立地选自H、CN及C
1-C
4烷基;
m各自独立地选自0-4;
4b”)更优选地,Y独立地选自O、NR
g、S、-CR
iNR
gR
h以及-CR
iOR
g;
R
g及R
h独立地选自H、C
1-C
6卤代烷基、

 -C(=S)NHC(=O)-R
j、包含碳原子和1-4个选自O、N、S杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1X取代;
-C(=S)NHC(=O)-R
j、包含碳原子和1-4个选自O、N、S杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1X取代;
R
j及R
k独立地选自H、CN、含0-3个R
s取代的C
1-C
10烷基、C
1-C
6卤代烷基、C
3-C
10环状烷基、包含碳原子和1-4个选自O、N、S杂原子的杂烷基和杂环烷基、C
2-C
10烯基或者炔基、6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R
1Y取代;
R
s独立地选自OH,CN、卤素、C
1-C
6烷基、C
1-C
6卤代烷基、C
1-C
6烷氧基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基、-(OCH
2CH
2)
mOR
d、(OCH
2CH
2)
mNR
dR
e、包含碳原子及1至2个选自N、O及S杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选 自N、O及S杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R
1Y取代;
其中,R
d、R
e与在上述4b)部分中限定的R
d、R
e的定义相同;
R
1Y独立地选自C
1-C
10烷基、卤素、CN、C
1-C
6卤代烷基、C
1-C
6卤代烷氧基、C
3-C
8环状烷基;
特别地,R
g与R
h以及R
j及R
k可通过以下方式R
g——R
h、
 R
j——R
k、
R
j——R
k、
 连接,其中Z
1可选自含0-2个R
c取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p;
连接,其中Z
1可选自含0-2个R
c取代的C
1-C
6烷基、含0-2个O、N、S(O)
p杂原子的C
1-C
6烷基、O、-N(C
1-C
6烷基)、-NH、-N(C=O)C
1-C
6烷基、-NS(=O)
2(C
1-C
6烷基)、S(O)
p;
其中,R
c与在上述4a)部分中限定的R
c的定义相同;
p各自独立地选自0,1及2;
R
i独立地选自H、CN及C
1-C
4烷基;
m各自独立地选自0-4;
优选地,所述通式I所述的化合物具有式Ia-1或Ia-2:

其中,X
1与通式I中的1)部分中的定义相同;
X
2、R
3-R
5、R
6以及n与通式I中的3)部分中的定义相同;
R
1A、R
1B及R
1C、R
2B及R
2C、R
3B及R
3C、R
1D、n、m、q、Y、M的定义与通式I中的4)部分中的相应的定义相同;
优选地,所述通式I所述的化合物具有式Ia-3或Ia-4:

其中,X
1与通式I中的1)部分中的定义相同;
X
2、R
7以及n与通式I中的3)部分中的定义相同;
R
1A、R
1B及R
1C、R
2B及R
2C、R
3B及R
3C、R
1D、n、m、q、Y、M的定义与通式I中的4)部分中的相应的定义相同;
优选地,所述通式I所述的化合物具有式Ia-5或Ia-6:

其中,X
1与通式I中的1)部分中的定义相同;
Y的定义与通式I中的4)部分中的相应的定义相同;
优选地,所述通式I所述的化合物具有式Ia-7或Ia-8:

其中,X
1与通式I中的1)部分中的定义相同;
R
g的定义与通式I中的4)部分中的4b)部分中的相应的定义相同;
优选地,所述通式I所述的化合物具有式Ia-9:

其中,
R
g的定义与通式I中的4)部分中的4b)部分中的相应的定义相同;
优选地,所述通式I所述的化合物具有以下结构式之一:

其中,
M的定义与通式I中的4)部分中的相应的定义相同;
R
g的定义与通式I中的4)部分中的4b)部分中的相应的定义相同;
优选地,所述通式I所述的化合物选自如下化合物:








优选地,所述化合物还包括其立体异构体,互变异构体,阻转异构体,经同位素标记之化合物(包括氘取代),医学上可接受的盐,多晶型物,溶剂合物,可以用于治疗由EED及/或PRC2介导的疾病或病症。
根据本发明的另一方面,提供了用于制备本发明化合物的方法及中间体。
其中,所述方法包括如下步骤:
方案一

(1a)用水合肼处理5-溴-4-氯-2-(甲硫基)嘧啶1,生成5-溴-4-肼基-2-(甲硫基)嘧啶2,
(1b)再用原甲酸三甲酯将5-溴-4-肼基-2-(甲硫基)嘧啶2转化为三氮唑产物3,
(1c)将三氮唑产物3与胺NH
2CH
2A发生取代反应生成化合物4,
(1d)将化合物4在钯催化剂作用下与各类具有R
1基团的硼酸或其等效物发生铃木(Suzuki)偶联反应,得到产物5。
其中,A、R
1的定义与上文中定义的相同。
方案二

(2a)将氰乙酰胺6与丙炔酸乙酯反应生成中间体7,
(2b)将中间体7用溴素处理发生溴代反应得到溴代物8,
(2c)将溴代物8与三氯氧磷反应得到中间体9,
(2d)将中间体9由水合肼处理,生成中间体10,
(2e)将中间体10用原甲酸三甲酯将其转化为三氮唑中间体11,
(2f)将三氮唑中间体11与各种胺发生取代反应生成化合物12,
(2g)最后将化合物12在钯催化剂作用下与各类具有R
1基团的硼酸或其等效物发生铃木(Suzuki)偶联反应,得到产物13。
其中,A、R
1的定义与上文中定义的相同。
在方案一及方案二中所述胺可参见专利US2016/0176682A1制备而得(例如下反应式中A1的制备),或由试剂公司购买(如下反应式,糠胺A2,购于百灵威科技有限公司),所述硼酸或其等效物B可由试剂公司购买或根据常规文献制备所得。

方案三

(3a)14以二氯甲烷为溶剂,在三氟乙酸作用下,去除Boc保护基,得到胺基化合物15,
(3b)将胺基化合物15在碱性条件下,与带有R
g基团的试剂或化合物反应得到16,所述试剂或化合物例如但不限于酸酐、磺酸酐、异氰酸酯、硫代异氰酸酯、酰氯、磺酰氯、碳酸酯、氯甲酸酯、氨基甲酸酯等,所述碱例如但不限于三乙胺、二异丙基乙基胺、DMAP、碳酸钾、氢氧化钠、氢氧化钾、叔丁醇钾、NaH,所述有机溶剂例如但不限于二氯甲烷、四氢呋喃、乙腈、1,4-二氧六环,
其中,A、X
1、R
g、R
1A、R
1B、R
1C、R
2B、R
2C、R
3B、R
3C、q、m的定义与上文中定义的相同。
方案四
(4a)将在方案三的步骤(3a)中去除保护基所得的产物15与含有R
j基团的羧酸在缩合剂的作用下发生缩合反应得到酰胺化合物17,所述缩合剂例如但不限于羰基二咪唑、二环己基碳二亚胺、二异丙基碳二亚胺、1-(-3-二甲氨基丙基)-3-乙基碳二亚胺、1-羟基苯并三唑、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯、苯并三氮唑-N,N,N',N'-四甲基脲六氟磷酸盐、6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸盐、O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯、6-氯苯并三氮唑-1,1,3,3-四甲基脲四氟硼酸酯、2-琥珀酰亚胺基-1,1,3,3-四甲基脲四氟硼酸酯和2-(5-降冰片烯-2,3-二甲酰亚胺基)-1,1,3,3-四甲基脲四氟硼酸季铵盐,所述缩合反应可以在碱存在下在有机溶剂中进行,所述碱例如但不限于三乙胺、二异丙基乙基胺、1,5-二氮杂二环[5.4.0]十一-5-烯,所述有机溶剂例如但不限于二氯甲烷、氯仿、N,N-二甲基甲酰胺、四氢呋喃,
其中,A、X
1、R
j、R
1A、R
1B、R
1C、R
2B、R
2C、R
3B、R
3C、q、m的定义与上文中 定义的相同。
方案五

(5a)将18溶于溶剂中,所述溶剂例如但不限于甲醇、乙醇、乙酸乙酯、四氢呋喃,加入金属催化剂,所述金属催化剂例如但不限于10%钯碳、Pd(OH)
2、雷尼镍、RhCl(PPh
3)
3,通入氢气,室温下反应,得到双键还原的化合物19,
其中,A、X
1、Y、R
1A、R
1B、R
1C、R
2B、R
2C、R
3B、R
3C、q、m的定义与上文中定义的相同,
方案六

(6a)20通过还原反应得到化合物21,随后与mCPBA(间氯过氧苯甲酸)或者双氧水发生氧化反应得到化合物22。
其中,A、X
1、R
1A、R
1B、R
1C、R
2B、R
2C、R
3B、R
3C、q、m的定义与上文中定义的相同
方案七

(7a)20与mCPBA或者双氧水发生氧化反应得到化合物23。
其中,A、X
1、R
1A、R
1B、R
1C、R
2B、R
2C、R
3B、R
3C、q、m的定义与上文中定义的相同。
方案八

15将双键还原(例如但不限于在氢化还原的条件下)得到24,随后与带有R
g基团的试剂或者化合物在碱存在下发生反应得到25,所述试剂或化合物例如但不限于酸酐、磺酸酐、异氰酸酯、硫代异氰酸酯、酰氯、磺酰氯、碳酸酯、氯甲酸酯、氨基甲酸酯,所述碱例如但不限于三乙胺、二异丙基乙基胺、DMAP、碳酸钾、氢氧化钠、氢氧化钾、叔丁醇钾、NaH,所述反应可以在有机溶剂中进行,所述有机溶剂例如但不限于二氯甲烷、四氢呋喃、乙腈、1,4-二氧六环;或者24也可与各类羧酸在缩合剂的存在下发生缩合反应得到酰胺化合物25,所述缩合剂例如但不限于羰基二咪唑、二环己基碳二亚胺、二异丙基碳二亚胺、1-(-3-二甲氨基丙基)-3-乙基碳二亚胺、1-羟基苯并三唑、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯、苯并三氮唑-N,N,N',N'-四甲基脲六氟磷酸盐、6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸盐、O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯、6-氯苯并三氮唑-1,1,3,3-四甲基脲四氟硼酸酯、2-琥珀酰亚胺基-1,1,3,3-四甲基脲四氟硼酸酯和2-(5-降冰片烯-2,3-二甲酰亚胺基)-1,1,3,3-四甲基脲四氟硼酸季铵盐,所述缩合反应可以在碱存在下在有机溶剂中进行,所述碱为三乙胺、二异丙基乙基胺、1,5-二氮杂二环[5.4.0]十一-5-烯,所述有机溶剂为二氯甲烷、氯仿、N,N-二甲基甲酰胺、四氢呋喃,
其中,A、X
1、R
1A、R
1B、R
1C、R
2B、R
2C、R
3B、R
3C、q、m的定义与上文中定义的相同。
优选地,该方法包括如下步骤:
方案一

(1a)用水合肼处理5-溴-4-氯-2-(甲硫基)嘧啶1,生成5-溴-4-肼基-2-(甲硫基) 嘧啶2,
(1b)再用原甲酸三甲酯将5-溴-4-肼基-2-(甲硫基)嘧啶2转化为三氮唑产物3,
(1c)将三氮唑产物3与胺NH
2CH
2A发生取代反应生成化合物4,
(1d)将化合物4在钯催化剂作用下与各类具有R
1基团的硼酸或其等效物发生铃木(Suzuki)偶联反应,得到产物5,
其中,A、R
1的定义与上文中定义的相同;
方案二

(2a)将氰乙酰胺6与丙炔酸乙酯反应生成中间体7,
(2b)将中间体7用溴素处理发生溴代反应得到溴代物8,
(2c)将溴代物8与三氯氧磷反应得到中间体9,
(2d)将中间体9由水合肼处理,生成中间体10,
(2e)将中间体10用原甲酸三甲酯将其转化为三氮唑产物11,
(2f)将三氮唑产物11与各种胺发生取代反应生成化合物12,
(2g)最后将化合物12在钯催化剂作用下与各类具有R
1基团的硼酸或其等效物发生铃木(Suzuki)偶联反应,得到产物13,
其中,A、R
1的定义与上文中定义的相同;
方案三

(3a)14’以二氯甲烷为溶剂,在三氟乙酸作用下,去除Boc保护基,得到胺基化合物15’,
(3b)将胺基化合物15’在碱性条件下进一步与带有R
g基团的酸酐、磺酸酐、异氰酸酯、硫代异氰酸酯发生反应得到化合物16’,所述碱为三乙胺、二异丙基乙基胺,所 述反应可以在有机溶剂中进行,所述有机溶剂为二氯甲烷、四氢呋喃、乙腈、1,4-二氧六环,
其中,A、X
1、R
g的定义与上文中定义的相同;
方案四

(4a)将在方案三的步骤(3b)中去除保护基所得的产物15’与含有R
j基团的羧酸在缩合剂的作用下发生缩合反应得到酰胺化合物17’,所述缩合剂选自羰基二咪唑、二环己基碳二亚胺、二异丙基碳二亚胺、1-(-3-二甲氨基丙基)-3-乙基碳二亚胺、1-羟基苯并三唑、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯、苯并三氮唑-N,N,N',N'-四甲基脲六氟磷酸盐、6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸盐、O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯、6-氯苯并三氮唑-1,1,3,3-四甲基脲四氟硼酸酯、2-琥珀酰亚胺基-1,1,3,3-四甲基脲四氟硼酸酯和2-(5-降冰片烯-2,3-二甲酰亚胺基)-1,1,3,3-四甲基脲四氟硼酸季铵盐,所述缩合反应可以在碱存在下在有机溶剂中进行,所述碱为三乙胺、二异丙基乙基胺、1,5-二氮杂二环[5.4.0]十一-5-烯,所述有机溶剂为二氯甲烷、氯仿、N,N-二甲基甲酰胺、四氢呋喃,
其中,A、X
1、R
j的定义与上文中定义的相同;
方案五

(5a)18’以甲醇为溶剂,在10%钯碳的催化下,通入氢气,室温下反应6小时,得到双键还原的化合物19’,
其中,X
1、Y的定义与上文中定义的相同。上述的化合物14、14’、18、18’和20可以按照在方案一或方案二中的步骤(1d)或步骤(2g)经铃木偶联(Suzuki)反应得到。
当需要本发明化合物的光学活性形式时,其可以使用光学活性起始材料获得,或者通过使用本领域技术人员已知的标准步骤(例如通过手性色谱柱进行分离)拆分化合物或中间体的立体异构体混合物获得。
类似地,当需要本发明化合物的纯几何异构体时,其可以由使用纯几何异构体作为起始材料获得,或者通过使用标准步骤,例如色谱分离拆分化合物或中间体的几何异构体混合物获得。
根据本发明的另一方面,提供了一种药物组合物,其包含如上所述的三氮唑并嘧啶、三氮唑并吡啶类化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体中的一 种或多种。
优选地,所述药物组合物进一步包含至少一种其他治疗剂。
优选地,所述药物组合物中包含的所述至少一种其他治疗剂选自其他抗癌剂、免疫调节剂、抗过敏剂、止吐剂、疼痛缓解剂、细胞保护剂及其组合。
优选地,所述药物组合物包含至少一种本发明化合物及至少一种药学上可接受的载体、稀释剂或赋形剂,
根据本发明的另一方面,提供了一种所述化合物或药物组合物在制备用于治疗由EED和/或PRC2介导的疾病或病症的药物中的用途。
优选地,所述疾病或病症包括弥漫性大B细胞淋巴瘤、滤泡性淋巴瘤、其他淋巴瘤、白血病、多发性骨髓瘤、间皮瘤、胃癌、恶性横纹肌样瘤、肝细胞癌、前列腺癌、乳腺癌、胆管及胆囊癌、膀胱癌;脑瘤、包括神经母细胞瘤、神经鞘瘤、神经胶质瘤、神经胶质母细胞瘤及星细胞瘤;子宫颈癌、结肠癌、黑色素瘤、子宫内膜癌、食道癌、头颈癌、肺癌、鼻咽癌、卵巢癌、胰腺癌、肾细胞癌、直肠癌、甲状腺癌、副甲状腺肿瘤、子宫肿瘤及软组织肉瘤。
根据本发明的另一方面,提供了治疗由EED及/或PRC2介导的疾病或病症的方法,该方法包括
向有需要的受试者提供治疗有效量的通式I所述的化合物或其药物组合物。
优选地,所述疾病或病症选自弥漫性大B细胞淋巴瘤、滤泡性淋巴瘤、其他淋巴瘤、白血病、多发性骨髓瘤、间皮瘤、胃癌、恶性横纹肌样瘤、肝细胞癌、前列腺癌、乳腺癌、胆管及胆囊癌、膀胱癌;脑瘤、包括神经母细胞瘤、神经鞘瘤、神经胶质瘤、神经胶质母细胞瘤及星细胞瘤;子宫颈癌、结肠癌、黑色素瘤、子宫内膜癌、食道癌、头颈癌、肺癌、鼻咽癌、卵巢癌、胰腺癌、肾细胞癌、直肠癌、甲状腺癌、副甲状腺肿瘤、子宫肿瘤及软组织肉瘤。
本说明书中公开的所有特征,或公开的所有方法或过程中的步骤,除了互相排斥的特征和/或步骤以外,均可以以任何方式组合。以下实施例证实本发明的部分范围,且并非特意限制本发明的保护范围。
实施例1:化合物E-Y1的合成

将溴代物4-1(72mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-1(84mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦 基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y1,为白色固体(41mg,57%)。
1H NMR(400MHz,MeOD-d
4)δ9.33(s,1H),7.83(s,1H),6.96(s,1H),6.90–6.84(m,1H),6.69–6.63(m,1H),4.81(s,2H),4.59(t,J=8.7Hz,2H),4.39(m,2H),3.99(t,J=5.5Hz,2H),3.38(m,2H),2.62(m,2H).LC-MS:[M+H]
+=368.1。
实施例2:化合物E-Y2的合成

将溴代物4-1(72mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-2(90mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y2,为白色固体(34mg,45%)。
1H NMR(400MHz,DMSO-d
6)δ9.49(s,1H),8.70(t,J=4.7Hz,1H),7.67(s,1H),7.26(s,1H),6.94(t,J=9.5Hz,1H),6.70(dd,J=8.6,3.9Hz,1H),4.68(d,J=4.6Hz,2H),4.53(t,J=8.7Hz,2H),3.36(m,2H),3.29(t,J=8.6Hz,2H),2.86(t,J=5.6Hz,2H),2.74(m,2H).LC-MS:[M+H]
+=384.1。
实施例3:化合物E-Y3的合成

将溴代物4-1(72mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-3(123mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y3,为白色固体(67mg,72%)。
1H NMR(400MHz,MeOD-d
4)δ9.36(s,1H),7.88(s,1H),6.91–6.82(m,1H),6.73(s,1H),6.66(dd,J=8.7,3.9Hz,1H),4.81(s,2H),4.59(t,J=8.7Hz,2H),4.16(s,2H),3.76–3.66(m,2H),3.38(t,J=8.7Hz,2H),2.62(s,2H),1.52(s,9H).LC-MS:[M+H]
+=467.2。
实施例4:化合物E-Y4的合成

将溴代物4-2(58mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-1(84mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y4,为白色固体(37mg,64%)。
1H NMR(400MHz,MeOD-d
4)δ9.24(s,1H),7.70(s,1H),7.46(d,J=0.9Hz,1H),7.06(s,1H),6.44–6.31(m,2H),4.77(s,2H),4.37(m,2H),3.96(t,J=5.5Hz,2H),2.60(m,2H).LC-MS:[M+H]
+=298.1。
实施例5:化合物E-Y5的合成

将溴代物4-2(58mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-2(90mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y5,为白色固体(37mg,60%)。
1H NMR(400MHz,MeOD-d
4)δ9.25(s,1H),7.68(s,1H),7.48(d,J=0.9Hz,1H),6.85–6.81(m,1H),6.42–6.36(m,2H),4.79(s,2H),3.43–3.37(m,2H),2.93(t,J=5.7Hz,2H),2.81(dd,J=5.7,2.0Hz,2H).LC-MS:[M+H]
+=314.1。
实施例6:化合物E-Y6的合成

将溴代物4-2(58mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的 Na
2CO
3水溶液中,加入硼酸酯B-3(123mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y6,为白色固体(55mg,70%)。
1H NMR(400MHz,MeOD-d
4)δ9.24(s,1H),7.68(s,1H),7.47(dd,J=1.8,0.8Hz,1H),6.94(s,1H),6.40(ddd,J=5.1,3.2,2.2Hz,2H),4.78(s,2H),4.15(m,2H),3.69(m,2H),2.62(m,2H),1.51(s,9H).LC-MS:[M+H]
+=397.1。
实施例7:化合物E-Y7的合成

将溴代物4-2(58mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-4(83mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y7,为白色固体(38mg,64%)。
1H NMR(400MHz,MeOD-d
4)δ9.24(s,1H),7.66(s,1H),7.49(s,1H),6.83(s,1H),6.44–6.36(m,2H),4.79(s,2H),2.52(s,2H),2.31(s,2H),1.87(dd,J=11.7,6.1Hz,2H),1.79–1.70(m,2H).LC-MS:[M+H]
+=296.1。
实施例8:化合物E-Y8的合成

将溴代物4-2(58mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-5(102mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y8,为白色固体(39mg,57%)。
1H NMR(400MHz,MeOD-d
4)δ9.24(s,1H),7.65(s,1H),7.48(d,J=1.0Hz,1H),7.20(d,J=6.9Hz,1H),7.12(t,J=6.9Hz,1H),7.03(t,J=7.0Hz,1H),6.80(d,J=7.3Hz,1H),6.46–6.38(m,2H),6.34(t,J=4.6Hz,1H),4.81(s,2H),2.91(t,J=8.0Hz,2H),2.46 (td,J=8.0,4.7Hz,2H).LC-MS:[M+H]
+=344.1。
实施例9:化合物E-Y9的合成

将溴代物4-2(58mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-6(129mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y9,为白色固体(42mg,51%)。
1H NMR(400MHz,MeOD-d
4)δ9.23(s,1H),7.62(s,1H),7.47(dd,J=1.8,0.9Hz,1H),6.76(s,1H),6.39(ddd,J=5.1,3.2,1.3Hz,2H),4.77(s,2H),3.75(m,1H),2.68–2.51(m,3H),2.25–2.14(m,1H),2.04(m,1H),1.80–1.68(m,1H),1.46(s,9H).LC-MS:[M+H]
+=411.2。
实施例10:化合物E-Y10的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,浓缩后直接经HPLC分离,流动相:40%乙腈/60%水(含0.1%TFA),保留时间4.7分钟,产率70%。
LC-MS:[M+H]
+=367.1。
实施例11:化合物E-Y11的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,浓缩后直接经HPLC分离,流动相:40%乙腈/60%水(含0.1%TFA),保留时间5.4分钟。产率82%。
1H NMR(400MHz,MeOD-d
4)δ9.27(s,1H),7.77(s,1H),7.45(dd,J=1.8,0.8Hz,1H),7.02(s,1H),6.38(dt,J=3.2,2.2Hz,2H),4.79(s,2H),3.92(m,2H),3.50(t,J=6.1Hz,2H),2.95–2.85(m,2H).LC-MS:[M+H]
+=297.1。
实施例12:化合物E-Y12的合成

将化合物E-Y9(41mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,浓缩后直接经HPLC分离,流动相:40%乙腈/60%水(含0.1%TFA),保留时间5.4分钟。产率69%。
1H NMR(400MHz,MeOD-d
4)δ9.29(s,1H),7.79(s,1H),7.46(s,1H),6.61(s,1H),6.45–6.34(m,2H),4.79(s,2H),3.51(m,1H),2.70(m,2H),2.41–2.32(m,1H),2.20(m,1H),1.91(m,2H).LC-MS:[M+H]
+=311.1。
实施例13:化合物E-Y13的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入甲磺酸酐(Ms
2O,26mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y13,为白色固体(24mg,两步收率51%)。
1H NMR(400MHz,DMSO-d
6)δ9.46(s,1H),8.67(t,J=4.9Hz,1H),7.71(s,1H),7.32(s,1H),7.01–6.88(m,1H),6.70(dd,J=8.6,4.0Hz,1H),4.69(d,J=4.8Hz,2H),4.54(t,J=8.8Hz,2H),3.94(m,2H),3.40(m,2H),3.29(m,2H),2.95(s,3H),2.70(m,2H).LC-MS:[M+H]
+=445.1。
实施例14:化合物E-Y14的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入对甲苯磺酸酸酐(Ts
2O,48mg,0.15mmol),继 续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y14,为白色固体(16mg,两步收率32%)。
1H NMR(400MHz,MeOD-d
4)δ9.45(s,1H),7.89–7.64(m,3H),7.44(d,J=7.9Hz,2H),6.85(t,J=9.4Hz,1H),6.76(s,1H),6.64(dd,J=8.5,3.6Hz,1H),4.77(s,2H),4.57(t,J=8.6Hz,2H),3.79(s,2H),3.44–3.33(m,4H),2.67(s,2H),2.45(s,3H).LC-MS:[M+H]
+=521.2。
实施例15:化合物E-Y15的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入乙酸酐(Ac
2O,15mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y15,为白色固体(14mg,两步收率35%)。
1H NMR(400MHz,DMSO-d
6)δ9.46(d,J=1.1Hz,1H),8.65(s,1H),7.70(d,J=8.3Hz,1H),7.27(d,J=21.1Hz,1H),6.97–6.89(m,1H),6.71(dd,J=8.7,3.8Hz,1H),4.69(d,J=4.2Hz,2H),4.54(t,J=8.7Hz,2H),4.19(d,J=25.2Hz,2H),3.67(dt,J=11.2,5.7Hz,2H),3.28(t,J=8.7Hz,2H),2.64(s,1H),2.52(s,1H),2.06(d,J=15.7Hz,3H).LC-MS:[M+H]
+=409.1。
实施例16:化合物E-Y16的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入苯甲酸酐((PhCO)
2O,34mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y16,为白色固体(14mg,两步收率30%)。
1H NMR(400MHz,DMSO-d
6)δ9.46(s,1H),8.66(s,1H),7.70(s,1H),7.47(d,J=3.9Hz,5H),7.01–6.90(m,1H),6.71(dd,J=8.7,3.9Hz,1H),4.69(d,J=4.9Hz,2H),4.54(t,J=8.8Hz,2H),4.35(s,1H),4.15(s,1H),3.87(s,1H),3.56(s,1H),3.28(t,J=8.6Hz,2H),2.64(s,2H).LC-MS:[M+H]
+=471.1。
实施例17:化合物E-Y17的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入异氰酸乙酯(11mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y17,为白色固体(17mg,两步收率40%)。
1H NMR(400MHz,DMSO-d
6)δ9.45(s,1H),8.64(s,1H),7.69(s,1H),7.27(s,1H),6.95(t,J=9.2Hz,1H),6.78–6.64(m,1H),6.51(s,1H),4.69(s,2H),4.54(t,J=8.8Hz,2H),4.03(s,2H),3.54(s,2H),3.27(m,2H),3.15–2.98(m,2H),1.03(t,J=7.1Hz,3H).LC-MS:[M+H]
+=438.2。
实施例18:化合物E-Y18的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入叔丁基异氰酸酯(11mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y18,为白色固体(15mg,两步收率33%)。
1H NMR(400MHz,MeOD-d
4)δ9.38(s,1H),7.93(s,1H),6.93–6.83(m,1H),6.69(s,1H),6.66(dd,J=8.7,3.9Hz,1H),4.82(s,2H),4.59(t,J=8.7Hz,2H),4.10(d,J=2.9Hz,2H),3.66(t,J=5.6Hz,2H),3.39(t,J=7.7Hz,2H),2.62(s,2H),1.38(s,9H).LC-MS:[M+H]
+=466.2。
实施例19:化合物E-Y19的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟 乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入异硫氰酸乙酯(13mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y19,为白色固体(14mg,两步收率31%)。
1H NMR(400MHz,DMSO-d
6)δ9.46(s,1H),8.66(d,J=5.0Hz,1H),7.71(d,J=9.9Hz,2H),7.27(s,1H),7.02–6.90(m,1H),6.71(dd,J=8.5,3.8Hz,1H),4.70(d,J=4.7Hz,2H),4.54(t,J=8.7Hz,2H),4.41(s,2H),4.07(t,J=5.4Hz,2H),3.55(dd,J=12.3,6.8Hz,2H),3.27(d,J=8.7Hz,2H),2.61(s,2H),1.12(t,J=7.1Hz,3H).LC-MS:[M+H]
+=454.1。
实施例20:化合物E-Y20的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入乙酸酐(Ac
2O,15mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y20,为白色固体(14mg,两步收率42%)。
1H NMR(400MHz,MeOD-d
4)δ9.33(s,1H),7.91(s,1H),7.49(s,1H),6.73(brs,1H),6.48–6.36(m,2H),4.83(s,2H),4.30(m,2H),3.84(m,2H),2.67(m,2H),2.20(s,3H).LC-MS:[M+H]
+=339.1。
实施例21:化合物E-Y21的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入苯甲酸酐((PhCO)
2O,15mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y21,为白色固体(13mg,两步收率32%)。
1H NMR(400MHz,MeOD-d
4)δ9.30(s,1H),7.82(s,1H),7.51(m,5H),7.48(s,1H),6.92(s,1H),6.47–6.36(m,2H),4.82(s,2H),4.46(m,1H),4.24(m,1H),4.06(m,1H),3.71(m,1H),2.74(m,2H).LC-MS:[M+H]
+=401.1
实施例22:化合物E-Y22的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入甲磺酸酐(Ms
2O,26mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y22,为白色固体(18mg,两步收率47%)。
1H NMR(400MHz,DMSO-d
6)δ9.43(s,1H),8.86(s,1H),7.71(s,1H),7.63(s,1H),7.30(s,1H),6.43(m,2H),4.73(s,2H),3.94(m,2H),3.40(m,2H),2.95(s,3H),2.69(m,2H).LC-MS:[M+H]
+=375.1。
实施例23:化合物E-Y23的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入对甲苯磺酸酸酐(Ts
2O,48mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y23,为白色固体(16mg,两步收率35%)。
1H NMR(400MHz,MeOD-d
4)δ9.24(s,1H),7.76(d,J=7.9Hz,2H),7.67(s,1H),7.47(s,1H),7.46(d,J=8.4Hz,2H),6.93(s,1H),6.40(m,2H),4.78(s,2H),3.82(m,2H),3.36(m,2H),2.71(m,2H),2.46(s,3H).LC-MS:[M+H]
+=451.1。
实施例24:化合物E-Y24的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入异氰酸乙酯(11mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y24,为白色固体(20mg,两步收率55%)。
1H NMR(400MHz,MeOD-d
4)δ9.25(s,1H),7.70(s,1H),7.47(d,J=1.0Hz,1H),6.98(m,1H),6.44–6.36(m,2H),4.79(s,2H),4.11(d,J=2.8Hz,2H),3.69(t,J=5.6Hz,2H),3.25(q,J=7.2Hz,2H),2.66(d,J=16.7Hz,2H),1.16(t,J=7.2Hz,3H).LC-MS:[M+H]
+=368.1。
实施例25:化合物E-Y25的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入叔丁基异氰酸酯(11mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y25,为白色固体(14mg,两步收率37%)。
1H NMR(400MHz,MeOD-d
4)δ9.28(s,1H),7.76(s,1H),7.48(m,1H),6.90(s,1H),6.45–6.34(m,2H),4.80(s,2H),4.10(d,J=2.5Hz,2H),3.65(t,J=5.6Hz,2H),2.63(m,2H),1.38(s,9H).LC-MS:[M+H]
+=396.1。
实施例26:化合物E-Y26的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入对甲苯异氰酸酯(20mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y26,为白色固体(20mg,两步收率46%)。
1H NMR(400MHz,DMSO-d
6)δ9.44(s,1H),8.81(m,1H),8.46(s,1H),7.72(s,1H),7.63(s,1H),7.37(d,J=8.4Hz,2H),7.32(s,1H),7.05(d,J=8.2Hz,2H),6.43(s,2H),4.74(d,J=4.8Hz,2H),4.22(s,2H),3.69(t,J=5.4Hz,2H),2.62(s,2H),2.24(s,3H).LC-MS:[M+H]
+=430.1。
实施例27:化合物E-Y27的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入对氯苯异氰酸酯(23mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y27,为白色固体(23mg,两步收率51%)。
1H NMR(400MHz,DMSO-d
6)δ9.44(s,1H),8.81(s,1H),8.70(s,1H),7.72(s,1H),7.63(s,1H),7.54(d,J=8.9Hz,2H),7.32(s,1H),7.29(d,J=8.9Hz,2H),6.43(s,2H),4.74(s,2H),4.23(s,2H),3.71(t,J=5.6Hz,2H),2.63(s,2H).LC-MS:[M+H]
+=450.1。
实施例28:化合物E-Y28的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入异氰酸苄酯(20mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y28,为白色固体(20mg,两步收率47%)。
1H NMR(400MHz,DMSO-d
6)δ9.43(s,1H),8.79(s,1H),7.69(s,1H),7.63(s,1H),7.37–7.25(m,5H),7.21(t,J=6.6Hz,1H),7.16(dd,J=14.8,9.1Hz,1H),6.43(d,J=1.7Hz,2H),4.74(s,2H),4.29(d,J=5.8Hz,2H),4.11(s,2H),3.60(t,J=5.6Hz,2H),2.55(s,2H).LC-MS:[M+H]
+=430.1。
实施例29:化合物E-Y29的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入戊二酸酐(17mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y29,为白色固体(20mg,两步收率47%)。
1H NMR(400MHz,MeOD-d
4)δ9.32(s,1H),7.89(d,J=4.5Hz,1H),7.49(s,1H),6.76(d,J=26.4Hz,1H),6.46–6.34(m,2H),4.83(s,2H),4.31(m,2H),3.92–3.79(m,2H),2.67(m,2H),2.60–2.49(m,2H),2.46–2.36(m,2H),1.95(m,2H).LC-MS:[M+H]
+=411.1。
实施例30:化合物E-Y30的合成

将化合物E-Y9(41mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入乙酸酐(Ac
2O,15mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y30,为白色固体(12mg,两步收率33%)。
1H NMR(400MHz,MeOD-d
4)δ9.33(s,1H),7.94(s,1H),7.49(dd,J=1.8,0.8Hz,1H),6.49(s,1H),6.47–6.37(m,2H),4.84(s,2H),4.06(m,1H),2.62(m,3H),2.32–2.15(m,1H),2.07(m,1H),1.99(s,3H),1.80(m,1H).LC-MS:[M+H]
+=353.1。
实施例31:化合物E-Y31的合成

将化合物E-Y9(41mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入苯甲酸酐((PhCO)
2O,15mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y31,为白色固体(12mg,两步收率27%)。
1H NMR(400MHz,MeOD-d
4)δ9.29(s,1H),7.90–7.83(m,2H),7.81(s,1H),7.56(m,1H),7.51–7.44(m,3H),6.73(m,1H),6.47–6.37(m,2H),4.81(s,2H),4.30(m,1H),2.73(m,3H),2.21(m,1H),2.05(m,1H),1.97(m,1H).LC-MS:[M+H]
+=415.1。
实施例32:化合物E-Y32的合成

将化合物E-Y9(41mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入甲磺酸酐(Ms
2O,26mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y32,为白色固体(11mg,两步收率29%)。
1H NMR(400MHz,MeOD-d
4)δ9.32(s,1H),7.89(s,1H),7.49(dd,J=1.8,0.8Hz,1H),6.51(m,1H),6.45–6.38(m,2H),4.83(s,2H),3.66(m,1H),3.03(s,3H),2.67(m,3H), 2.38–2.25(m,1H),2.23–2.14(m,1H),1.94–1.79(m,1H).LC-MS:[M+H]
+=389.1。
实施例33:化合物E-Y33的合成

将化合物E-Y9(41mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入戊二酸酐(17mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y33,为白色固体(16mg,两步收率38%)。
1H NMR(400MHz,DMSO-d
6)δ12.21(brs,1H),9.41(s,1H),8.75(s,1H),7.85(d,J=6.9Hz,1H),7.64(d,J=4.0Hz,2H),7.19(s,1H),6.42(s,2H),4.72(s,2H),3.85(m,1H),2.60(m,2H),2.48–2.33(m,2H),2.21(m,2H),2.11(m,2H),1.91(m,1H),1.73(m,2H),1.61(m,1H).LC-MS:[M+H]
+=425.1。
实施例34:化合物E-Y34的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(76mg,0.2mmol),2,2-二氟丙酸(18mg,0.16mmol),反应6h,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y34,为白色固体(10mg,两步收率25%)。
1H NMR(400MHz,MeOD-d
4)δ9.29(s,1H),7.82(m,1H),7.49(dd,J=1.8,0.8Hz,1H),6.91(m,1H),6.50–6.35(m,2H),4.81(s,2H),4.51(m,1H),4.34(m,1H),4.03(t,J=5.6Hz,1H),3.93(t,J=5.6Hz,1H),2.76(m,2H),1.87(td,J=19.9,6.4Hz,3H).LC-MS:[M+H]
+=389.1。
实施例35:化合物E-Y35的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(76mg,0.2mmol),N,N-二甲基甘氨酸(17mg,0.16mmol),反应6h,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y35,为白色固体(11mg,两步收率28%)。
1H NMR(400MHz,MeOD-d
4)δ9.29(s,1H),7.77(d,J=8.5Hz,1H),7.48(m,1H),6.92(d,J=30.7Hz,1H),6.47–6.32(m,2H),4.81(s,2H),4.38(s,1H),4.35(m,1H),4.32(s,1H),4.20(m,1H),3.93(t,J=5.8Hz,1H),3.70(t,J=5.7Hz,1H),3.00(d,J=3.4Hz,6H).LC-MS:[M+H]
+=382.1。
实施例36:化合物E-Y36的合成

将化合物E-Y6(39mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(76mg,0.2mmol),吗啉-4-基乙酸(23mg,0.16mmol),反应6h,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y36,为白色固体(9mg,两步收率21%)。
LC-MS:[M+H]
+=424.2。
实施例37:化合物E-Y37的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入异氰酸苄酯(20mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y37,为白色固体(12mg,两步收率23%)。
1H NMR(400MHz,DMSO-d
6)δ9.45(s,1H),8.64(t,J=4.9Hz,1H),7.69(s,1H),7.30(dd,J=12.0,5.0Hz,6H),7.25–7.12(m,2H),7.00–6.91(m,1H),6.70(dd,J=8.6,3.8Hz,1H),4.69(d,J=4.9Hz,2H),4.54(t,J=8.7Hz,2H),4.28(d,J=5.6Hz,2H),4.10(s,2H),3.28(t,J=8.8Hz,3H),2.55(s,2H).LC-MS:[M+H]
+=500.1。
实施例38:化合物E-Y38的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入对氯苯异氰酸酯(23mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y38,为白色固体(18mg,两步收率34%)。
1H NMR(400MHz,DMSO-d
6)δ9.46(s,1H),8.67(d,J=21.7Hz,2H),7.72(s,1H),7.54(d,J=8.1Hz,2H),7.29(d,J=8.4Hz,3H),6.95(s,1H),6.72(s,1H),4.70(s,2H),4.53(d,J=8.2Hz,2H),4.23(s,2H),3.70(s,2H),3.29(s,2H),2.63(s,2H).LC-MS:[M+H]
+=520.1。
实施例39:化合物E-Y39的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入苯甲酰基异硫氰酸酯(24mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y39,为白色固体(10mg,两步收率19%)。
1H NMR(400MHz,DMSO-d
6)δ10.89(d,J=17.5Hz,1H),9.47(d,J=5.7Hz,1H),8.69(s,1H),7.98(t,J=8.1Hz,2H),7.75(d,J=25.1Hz,1H),7.63(s,1H),7.53(d,J=8.1Hz,2H),7.21(s,1H),7.01–6.91(m,1H),6.79–6.63(m,1H),4.76(s,1H),4.70(d,J=5.0Hz,2H),4.54(t,J=8.6Hz,2H),4.36(s,2H),3.82(s,1H),3.29(t,J=8.7Hz,3H),2.79(s,2H).LC-MS:[M+H]
+=530.1。
实施例40:化合物E-Y40的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(CAS号:148893-10-1)(76mg,0.2mmol),吗啉-4-基乙酸(23mg,0.16mmol),反应6h,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y40,为白色固体(5mg,两步收率10%)。
1H NMR(400MHz,DMSO-d
6)δ9.48(d,J=2.5Hz,1H),8.72(s,1H),7.73(d,J=9.0Hz,1H),7.31(d,J=17.2Hz,1H),6.95(t,J=9.4Hz,1H),6.71(dd,J=8.6,3.7Hz,1H),4.70(m,2H),4.54(t,J=8.4Hz,2H),4.44(d,J=25.8Hz,2H),4.23(d,J=30.0Hz,2H),3.96(m,2H),3.78(m,2H),3.61(m,2H),3.44(m,2H),3.29(m,2H),3.17(m,2H),2.67(m,2H).LC-MS:[M+H]
+=494.2。
实施例41:化合物E-Y41的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(76mg,0.2mmol),噁唑-5-羧酸(18mg,0.16mmol),反应6h,TLC显示反应结675F(DCM:MeOH=10:1,Rf=0.5),反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y41,为白色固体(10mg,两步收率22%)。
1H NMR(400MHz,MeOD-d
4)δ9.32(s,1H),8.41(s,1H),7.81(s,1H),7.77(s,1H),6.95(s,1H),6.91–6.81(m,1H),6.66(dd,J=8.6,3.9Hz,1H),4.80(s,2H),4.59(t,J=8.7Hz,3H),4.43(m,1H),4.05(m,2H),3.38(t,J=8.7Hz,2H),2.93–2.65(m,2H).LC-MS:[M+H]
+=462.2。
实施例42:化合物E-Y42的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(76mg,0.2mmol),1,3-二甲基-1H-吡唑-5-甲酸(22mg,0.16mmol),反应6h,TLC显示反应结束(DCM:MeOH =10:1,Rf=0.5),反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y42,为白色固体(10mg,两步收率19%)。
1H NMR(400MHz,MeOD-d
4)δ9.40(s,1H),7.98(s,1H),6.95–6.80(m,1H),6.76–6.52(m,2H),6.37(s,1H),4.82(s,2H),4.59(t,J=8.7Hz,2H),4.40(d,J=24.2Hz,2H),4.01(d,J=10.7Hz,1H),3.87(s,4H),3.39(t,J=8.5Hz,2H),2.72(s,2H),2.28(s,3H).LC-MS:[M+H]
+=489.2。
实施例43:化合物E-Y43的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(76mg,0.2mmol),四氢吡喃-4-甲酸(21mg,0.16mmol),反应6h,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y43,为白色固体(10mg,两步收率21%)。
1H NMR(400MHz,MeOD-d
4)δ9.36(s,1H),7.86(d,J=5.7Hz,1H),6.91–6.58(m,3H),4.79m,2H),4.58(m,2H),4.38(s,1H),4.27(s,1H),3.99(m,2H),3.87(m,2H),3.55(m,2H),3.37(m,2H),3.04(m,1H),2.65(m,2H),1.83(m,2H),1.68(m,2H).LC-MS:[M+H]
+=479.2。
实施例44:化合物E-Y44的合成

将溴代物4-1(72mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-7(137mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y44,为白色固体(18mg,36%)。
1H NMR(400MHz,DMSO-d
6)δ9.46(s,1H),8.67(s,1H),7.69(s,1H),7.38(dd,J=7.5,4.3Hz,6H),7.24(s,1H),7.03–6.88(m,1H),6.70(dd,J=8.7,3.9Hz,1H),5.12(d,J=12.1Hz,2H),4.68(s,2H),4.61–4.46(m,2H),4.17(m,2H),3.65(m,2H),3.28(t,J=8.7Hz,2H),2.58(m,2H).LC-MS:[M+H]
+=501.1。
实施例45:化合物E-Y45的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入丙烯酸酐(20mg,0.15mmol),继续反应1小时,TLC显示反应结束(DCM:MeOH=10:1,Rf=0.5),直接由柱层析分离制备(DCM:MeOH=30:1),得到目标化合物E-Y45,为白色固体(14mg,两步收率35%)。
1H NMR(400MHz,MeOH-d
4)δ9.38(s,1H),7.92(s,1H),6.94–6.80(m,2H),6.79–6.68(m,1H),6.65(dd,J=8.6,3.9Hz,1H),6.27(d,J=16.7Hz,1H),5.81(d,J=10.6Hz,1H),4.81(s,2H),4.59(t,J=8.7Hz,2H),4.37(m,2H),3.92(m,2H),3.38(m,2H),2.68(m,2H).LC-MS:[M+H]
+=421.1。
实施例46:化合物E-Y46的合成

将溴代物4-3(62mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-3(123mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y46,为白色固体(32mg,40%)。
1H NMR(400MHz,MeOD-d
4)δ9.43(s,1H),7.71(s,1H),7.47(m,1H),7.16(m,1H),6.99(m,2H),4.91(s,2H),4.08(m,2H),3.57(m,2H),2.78(m,2H),1.44(s,9H).LC-MS:[M+H]
+=412.1。
实施例47:化合物E-Y47的合成

化合物E-Y1(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y47,为白色固体(8mg,80%)。
1H NMR(400MHz,MeOD-d
4)δ9.33(s,1H),7.74(s,1H),6.93–6.80(m,1H),6.66(dd,J=8.7,3.8Hz,1H),4.78(s,2H),4.59(t,J=8.7Hz,2H),4.09(d,J=11.2Hz,2H),3.71–3.57(m,2H),3.38(t,J=8.7Hz,2H),3.21(m,1H),1.96(m,4H).LC-MS:[M+H]
+=370.1。
实施例48:化合物E-Y48的合成

化合物E-Y17(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y48,为白色固体(5mg,50%)。
1H NMR(400MHz,MeOD-d
4)δ9.27(s,1H),7.58(s,1H),6.99–6.77(m,1H),6.66(dd,J=8.5,3.7Hz,1H),4.76(s,2H),4.59(t,J=8.7Hz,2H),4.20(m,2H),3.37(m,2H),3.23(dd,J=14.2,7.1Hz,2H),2.96(t,J=11.7Hz,2H),2.04(m,4H),1.15(t,J=7.2Hz,3H).LC-MS:[M+H]
+=440.2。
实施例49:化合物E-Y49的合成

化合物E-Y18(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y49,为白色固体(6mg,60%)。
1H NMR(400MHz,DMSO-d
6)δ9.40(s,1H),8.44(s,1H),7.49(s,1H),7.02–6.87(m,1H),6.70(dd,J=8.6,3.9Hz,1H),4.64(d,J=4.8Hz,2H),4.54(t,J=8.8Hz,2H),4.10(d,J=12.9Hz,2H),3.29(t,J=8.7Hz,2H),2.96(s,1H),2.77–2.64(m,2H),1.90–1.68(m,4H),1.27(s,9H).LC-MS:[M+H]
+=468.2。
实施例50:化合物E-Y50的合成

化合物E-Y15(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y50,为白色固体(7mg,72%)。
1H NMR(400MHz,MeOD)δ9.28(s,1H),7.63(s,1H),6.92–6.78(m,1H),6.66(dd,J=8.7,3.9Hz,1H),4.77(s,2H),4.71(m,1H),4.59(t,J=8.7Hz,2H),4.09(m,1H),3.37(t,J=8.6Hz,2H),3.30–3.14(m,1H),2.80(m,1H),2.25–2.18(m,1H),2.17(s,3H),2.06(m,2H),1.91(m,1H),1.81(m,1H).LC-MS:[M+H]
+=411.2。
实施例51:化合物E-Y51的合成

将溴代物13-2(63mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-3(123mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y51,为白色固体(17mg,20%)。
1H NMR(400MHz,MeOD-d
4)δ9.48(s,1H),7.50(s,1H),7.34(s,1H),6.95(s,1H),6.47(m,2H),5.08(s,2H),4.18(m,2H),3.72(m,2H),2.62(m,2H),1.48(s,9H).LC-MS:[M+H]
+=421.2。
实施例52:化合物E-Y52的合成

将溴代物13-1(77mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-1(84mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH= 20:1),得到目标化合物E-Y52,为白色固体(20mg,21%)。LC-MS:[M+H]
+=392.2。
实施例53:化合物E-Y53的合成

将溴代物13-1(77mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-3(123mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y53,为白色固体(13mg,14%)。LC-MS:[M+H]
+=491.2。
实施例54:化合物E-Y54的合成

将化合物E-Y3(46mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后再加入1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(76mg,0.2mmol),N,N-二甲基甘氨酸(17mg,0.16mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物E-Y54(6mg)。
1H NMR(400MHz,DMSO-d
6)δ9.54(d,J=3.3Hz,1H),8.79(s,1H),7.70(d,J=6.1Hz,1H),7.30(s,1H),6.95(t,J=9.4Hz,1H),6.70(dd,J=8.5,3.7Hz,1H),4.69(d,J=4.6Hz,2H),4.53(t,J=8.7Hz,2H),4.24(d,J=41.1Hz,2H),3.71(s,2H),3.48(s,2H),3.29(t,J=8.7Hz,2H),2.60(d,J=38.4Hz,2H),2.37(s,6H).LC-MS:[M+H]
+=452.2。
实施例55:化合物SL-ZYE-07的合成

将溴代物4-1(72mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-8(89mg,0.4mmol),Ar置换保护,室温下搅拌10 分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-07(47mg,62%)。
1H NMR(400MHz,DMSO-d
6)δ9.39(s,1H),8.51(s,1H),7.56(s,1H),6.91(t,J=9.3Hz,1H),6.69(m,2H),4.65(d,J=3.8Hz,2H),4.51(t,J=8.5Hz,2H),3.71(m,2H),3.62(m,2H),3.26(m,2H),2.79(m,2H),2.45(m,2H).LC-MS:[M+H]
+=382.2。
实施例56:化合物SL-ZYE-08,SL-ZYE-08-S,SL-ZYE-08-R的合成

化合物SL-ZYE-07(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-08(8mg,82%)。
1H NMR(400MHz,CDCl
3+MeOD-d
4)δ9.05(s,1H),7.41(s,1H),6.82–6.64(m,1H),6.56(dd,J=8.6,3.9Hz,1H),4.60(s,2H),4.52(t,J=8.7Hz,2H),3.91–3.77(m,2H),3.69(ddd,J=19.0,10.7,6.3Hz,2H),3.27(t,J=8.7Hz,2H),3.16(d,J=7.8Hz,1H),2.05–1.91(m,4H),1.84(dd,J=9.0,5.1Hz,2H).LC-MS:[M+H]
+=384.2。
SL-ZYE-08可进一步通过手性色谱柱分离获得一对光学纯的化合物SL-ZYE-08-S和SL-ZYE-08-R,分离条件为:(手性柱:Chiralcel OD-3,4.6mm x 250mm,particle size:3μm,流动相:正己烷:异丙醇=50:50,流速=1ml/min,得到两个流分,流分1(Rt=17.085分钟);流分2(Rt=18.627分钟)。所述手性色谱柱分离方法为本领域技术人员已知的常规方法。
实施例57:化合物SL-ZYE-09的合成

将E-Y2(5mg)溶解于1mL二氯甲烷中,加入mCPBA(间氯过氧苯甲酸)(2mg), Ar置换保护,室温下搅拌120分钟。减压浓缩,由柱层析分离制备,得到目标化合物SL-ZYE-09(2mg)。
1H NMR(400MHz,DMSO-d
6)δ9.46(s,1H),8.70(t,J=5.1Hz,1H),7.76(s,1H),7.12(t,J=4.6Hz,1H),7.01–6.85(m,1H),6.70(dd,J=8.7,3.9Hz,1H),4.69(d,J=4.9Hz,2H),4.54(t,J=8.7Hz,2H),3.99(s,2H),3.37(t,J=6.2Hz,2H),3.28(t,J=8.7Hz,2H),3.17(d,J=5.3Hz,2H).LC-MS:[M+H]
+=416.1。
实施例58:化合物SL-ZYE-11的合成

将溴代物4-4(69mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-1(84mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-11(17mg)。
1H NMR(400MHz,CDCl
3+MeOD-d
4)δ9.08(s,1H),7.56(s,1H),7.14(s,1H),7.02(d,J=7.4Hz,1H),6.79(d,J=8.0Hz,1H),6.66(d,J=7.9Hz,1H),4.53(t,J=8.7Hz,2H),4.35(s,2H),3.92(t,J=5.3Hz,2H),3.30(s,2H),3.19(t,J=8.4Hz,2H),2.53(s,2H).LC-MS:[M+H]
+=350.2。
实施例59:化合物SL-ZYE-14的合成

化合物SL-ZYE-11(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-14(5mg)。
1H NMR(400MHz,DMSO-d
6)δ9.41(s,1H),8.55(t,J=5.4Hz,1H),7.45(s,1H),7.06(t,J=7.8Hz,1H),6.85(d,J=7.5Hz,1H),6.69(d,J=7.9Hz,1H),4.63(d,J=5.3Hz,2H),4.54(t,J=8.7Hz,2H),3.96(dd,J=10.8,3.6Hz,2H),3.51–3.44(m,2H),3.22(t,J=8.7Hz,2H),3.09(ddd,J=15.4,7.9,3.7Hz,1H),2.01–1.87(m,2H),1.82(d,J=12.5Hz,2H).LC-MS:[M+H]
+=352.2。
实施例60:化合物SL-ZYE-17的合成

将溴代物4-5(68mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-1(84mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-17(17mg)。
1H NMR(400MHz,MeOD-d
4)δ9.26(s,1H),7.78(d,J=2.3Hz,1H),7.74(s,1H),7.47(d,J=7.9Hz,1H),7.30(d,J=7.6Hz,2H),7.08(s,1H),7.03(s,1H),5.07(s,2H),4.39(d,J=2.6Hz,2H),3.98(t,J=5.5Hz,2H),2.63(s,2H).LC-MS:[M+H]
+=348.1。
实施例61:化合物SL-ZYE-18的合成

将溴代物4-5(68mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-3(123mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-18(15mg)。
1H NMR(400MHz,CDCl
3)δ8.77(s,1H),7.71(s,1H),7.67(d,J=2.3Hz,1H),7.51(s,1H),7.31–7.29(m,4H),6.90(s,1H),5.08(d,J=5.3Hz,2H),4.18(s,2H),3.70(s,2H),2.64(s,2H),1.51(s,9H).LC-MS:[M+H]
+=447.1。
实施例62:化合物E-Y20-H的合成

化合物E-Y20(10mg)溶解于2mL甲醇中,加入10%Pd/C(3mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y20-H(4mg,40%)。
1H NMR(400MHz,MeOD-d
4)δ9.23(s,1H),7.57(d,J=0.7Hz,1H),7.47(dd,J=1.8,0.9Hz,1H),6.42–6.36(m,2H),4.77(s,2H),4.73(m,1H),4.09(m,1H),3.35–3.17(m,2H),2.80(dd,J=12.9,10.2Hz,1H),2.24–2.18(m,1H),2.17(s,3H),2.07(m,1H),1.87(m,2H).LC-MS:[M+H]
+=341.1。
实施例63:化合物E-Y13-H的合成

化合物E-Y13(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y13-H(7mg,71%)。
1H NMR(400MHz,DMSO-d
6)δ9.41(s,1H),8.48(t,J=5.2Hz,1H),7.50(s,1H),6.98–6.90(m,1H),6.70(dd,J=8.6,3.9Hz,1H),4.64(d,J=5.0Hz,2H),4.54(t,J=8.7Hz,2H),3.69(d,J=11.6Hz,2H),3.29(t,J=8.7Hz,2H),3.01(m,1H),2.91(s,3H),2.85(dd,J=8.7,6.0Hz,2H),2.07–1.85(m,4H).LC-MS:[M+H]
+=447.2。
实施例64:化合物E-Y2-H与SL-ZYE-34的合成

化合物E-Y2(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,旋干溶剂得到E-Y2-H,LC-MS:[M+H]
+=386.1。
将5mg E-Y2-H溶于1mL二氯甲烷溶液,往溶液中加入mCPBA(5mg),室温反应3小时,柱层析分离制备,得到目标化合物SL-ZYE-34(1.5mg)。
1H NMR(400MHz,MeOD-d
4)δ9.27(s,1H),7.63(s,1H),6.92–6.78(m,1H),6.65(dd, J=8.6,3.7Hz,1H),4.76(s,2H),4.59(t,J=8.7Hz,2H),3.45–3.33(m,5H),3.17(d,J=11.8Hz,2H),2.55–2.33(m,4H).LC-MS:[M+H]
+=418.2。
实施例65:化合物SL-ZYE-23的合成

将溴代物4-1(72mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-10(101mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-23(14mg)。
1H NMR(400MHz,DMSO-d
6)δ9.46(s,1H),8.67(s,1H),7.72(s,1H),7.27(s,1H),7.00–6.87(m,1H),6.70(dd,J=8.7,3.8Hz,2H),4.70(d,J=4.8Hz,2H),4.54(t,J=8.6Hz,2H),4.30(d,J=5.7Hz,2H),3.27(t,J=8.7Hz,2H),2.68(s,2H),1.45(s,9H),1.29(d,J=6.4Hz,3H),1.09(d,J=6.4Hz,3H).LC-MS:[M+H]
+=495.3。
实施例66:化合物SL-ZYE-24的合成

将溴代物4-1(72mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-11(95mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-24(21mg)。
1H NMR(400MHz,CDCl
3)δ8.93(s,1H),7.66(s,1H),7.24(s,1H),6.88–6.70(m,1H),6.61(dd,J=8.7,4.0Hz,1H),6.35(s,1H),4.79(d,J=5.5Hz,2H),4.61(t,J=8.7Hz,2H),4.51(s,1H),3.94–3.82(m,1H),3.50(m,1H),3.38(t,J=8.8Hz,2H),2.45(t,J=16.4Hz,2H),1.38(d,J=6.2Hz,3H),1.35(d,J=6.8Hz,3H).LC-MS:[M+H]
+=396.2。
实施例67:化合物SL-ZYE-28的合成

将溴代物4-1(72mg,0.2mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-12(103mg,0.4mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(7.3mg,0.02mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(21.2mg,0.04mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备(DCM:MeOH=20:1),得到目标化合物SL-ZYE-28(27mg,32%)。
1H NMR(400MHz,MeOD-d
4)δ9.44(s,1H),8.07(s,1H),7.23–7.14(m,1H),6.94–6.79(m,4H),6.68(dd,J=8.6,3.9Hz,1H),6.17(t,J=3.9Hz,1H),4.92(d,J=3.9Hz,2H),4.88(s,2H),4.61(t,J=8.7Hz,2H),3.43(t,J=8.7Hz,2H).LC-MS:[M+H]
+=416.2。
实施例68:化合物E-Y54-H的合成

化合物E-Y54(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备(DCM:MeOH=20:1),得到目标化合物E-Y54-H(5mg)。
1H NMR(400MHz,MeOD-d
4)δ9.27(s,1H),7.57(s,1H),6.91–6.81(m,1H),6.65(dd,J=8.6,3.9Hz,1H),4.76(s,2H),4.70(d,J=13.5Hz,1H),4.58(t,J=8.7Hz,2H),4.10(d,J=13.9Hz,1H),3.60(dd,J=16.9,6.6Hz,2H),3.36(dd,J=10.0,7.6Hz,2H),3.25(d,J=16.0Hz,2H),2.84(m,1H),2.52(s,6H),2.07(m,2H),1.91(m,2H).LC-MS:[M+H]
+=454.2。
实施例69:化合物SL-E1的合成

将溴代物4-1(36mg,0.1mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-13(64mg,0.2mmol),Ar置换保护,室温下搅拌10 分钟。加入10%氯化烯丙基钯(II)二聚物(3.5mg,0.01mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(11mg,0.02mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备得到目标化合物SL-E1(11mg)。LC-MS:[M+H]
+=481.2。
实施例70:化合物SL-E2的合成

化合物SL-E1(10mg)溶解于2mL甲醇中,加入10%Pd/C(5mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E2(7mg)。LC-MS:[M+H]
+=483.2。
参考实施例56,SL-E2可通过手性色谱柱分离获得光学纯化合物SL-E2-S和SL-E2-R。
实施例71:化合物SL-E3的合成

化合物SL-E3(9mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E3(5mg)。LC-MS:[M+H]
+=497.2。
实施例72:化合物SL-E4的合成

化合物SL-E3(9mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E4(5mg)。LC-MS:[M+H]
+=398.1。
实施例73:化合物SL-E5的合成

将化合物E-Y10(20mg)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg)以及2-(1-吡咯烷基)乙酸(CAS:37386-15-5)(13mg),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E5(3mg)。LC-MS:[M+H]
+=478.2。
实施例74:化合物SL-E6的合成

将化合物E-Y10(20mg)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及N,N-二乙基甘氨酸(13mg,0.1mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E6(6mg)。LC-MS:[M+H]
+=480.2。
实施例75:化合物SL-E7的合成

将化合物E-Y10(20mg)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg)以及甲氧基乙酸(CAS:625-45-6)(9mg),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E7(2mg)。LC-MS:[M+H]
+=439.2。
实施例76:化合物SL-E8的合成

将化合物E-Y10(20mg)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA), 搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及酸(oxetane-3-carboxylic acid)(10mg,0.1mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E8(2.5mg)。LC-MS:[M+H]
+=451.2。
实施例77:化合物SL-E9的合成

将化合物E-Y10(20mg)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg)以及2-(2-甲氧基乙氧基)乙酸(CAS号:16024-56-9)(14mg),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E9(5mg)。LC-MS:[M+H]
+=483.2。
实施例78:化合物SL-E10的合成

将化合物E-Y10(20mg)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及1-甲基哌啶-4-甲酸(15mg,0.1mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E10(6mg)。LC-MS:[M+H]
+=492.2。
实施例79:化合物SL-E11的合成

将化合物E-Y10(20mg)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及酸(CAS号:1158712-36-7)(22mg,0.1mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E11(7mg)。LC-MS:[M+H]
+=562.2。
实施例80:化合物SL-E12的合成

将化合物SL-E1(96mg,0.2mmol)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,浓缩后直接经HPLC分离得到产物SL-E12(50mg),LC-MS:[M+H]
+=381.2。
实施例81:化合物SL-E13的合成

将SL-E12(25mg)溶解于1mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入乙酸酐(Ac
2O,15mg),反应1小时,TLC显示反应结束后,由柱层析分离制备得到目标化合物SL-E13(6mg)。LC-MS:[M+H]
+=423.2。
实施例82:化合物SL-E14的合成

将SL-E12(10mg)溶解于1mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入甲磺酸酐(Ms
2O,8mg),反应1小时,TLC显示反应结束后,由柱层析分离制备得到目标化合物SL-E14(3mg)。
1H NMR(400MHz,DMSO-d
6)δ9.44(s,1H),8.63(m,1H),7.67(s,1H),6.99–6.90(m,1H),6.82(t,J=5.9Hz,1H),6.70(dd,J=8.7,3.8Hz,1H),4.68(d,J=4.9Hz,2H),4.54(t,J=8.7Hz,2H),4.03(d,J=6.0Hz,2H),3.57–3.48(m,2H),3.31–3.24(m,2H),2.91(s,3H),2.83(m,2H),1.91(m,2H).LC-MS:[M+H]
+=459.2。
实施例83:化合物SL-E15的合成

将化合物SL-E12(19mg,0.05mmol)溶解于1mL DMF和0.1mL二异丙基乙基 胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及N,N-二甲基甘氨酸(11mg,0.1mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E15(2mg)。LC-MS:[M+H]
+=466.3。
实施例84:化合物SL-E16的合成

将化合物SL-E12(19mg,0.05mmol)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及2-(1-吡咯烷基)乙酸(CAS:37386-15-5)(14mg),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E16(2mg)。LC-MS:[M+H]
+=492.2。
实施例85:化合物SL-E17的合成

将化合物SL-E12(19mg,0.05mmol)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及N,N-二乙基甘氨酸(13mg),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E17(2mg)。LC-MS:[M+H]
+=494.4。
实施例86:化合物SL-E18的合成

将化合物SL-E12(19mg,0.05mmol)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg)以及甲氧基乙酸(CAS:625-45-6)(11mg),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E18(3mg)。LC-MS:[M+H]
+=453.2。
实施例87:化合物SL-E19的合成

将化合物SL-E12(19mg,0.05mmol)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及酸(oxetane-3-carboxylic acid)(10mg,0.1mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E19(1.5mg)。LC-MS:[M+H]
+=465.2。
实施例88:化合物SL-E20的合成

将化合物SL-E12(19mg,0.05mmol)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg)以及2-(2-甲氧基乙氧基)乙酸(CAS号:16024-56-9)(15mg),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E20(2mg)。LC-MS:[M+H]
+=497.2。
实施例89:化合物SL-E21的合成

将化合物SL-E12(19mg,0.05mmol)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及1-甲基哌啶-4-甲酸(15mg,0.1mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E21(3mg)。LC-MS:[M+H]
+=506.2。
实施例90:化合物SL-E22的合成

将化合物SL-E12(19mg,0.05mmol)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(38mg,0.1mmol)以及酸(CAS号:1158712-36-7)(22mg,0.1mmol),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E22(0.8mg)。LC-MS:[M+H]
+=576.2。
实施例91:化合物SL-E23的合成

将溴代物4-1(36mg,0.1mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-14(44mg,0.2mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(3.5mg,0.01mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(11mg,0.02mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备得到目标化合物SL-E23,为白色固体(6mg)。
1H NMR(400MHz,DMSO-d
6)δ9.47(d,J=6.2Hz,1H),8.67(d,J=4.9Hz,1H),7.71(d,J=8.4Hz,1H),7.30(m,1H),6.94(t,J=9.4Hz,1H),6.70(dd,J=8.5,3.8Hz,1H),4.69(d,J=4.8Hz,2H),4.57(m,2H),3.47(m,2H),3.28(t,J=8.8Hz,2H),2.97(m,2H),2.69(m,2H),2.56(m,3H).LC-MS:[M+H]
+=381.2。
实施例92:化合物SL-E24的合成

将溴代物4-1(36mg,0.1mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯B-15(47mg,0.2mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(3.5mg,0.01mmol),20%2′-二环己基膦基 -2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(11mg,0.02mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备得到目标化合物SL-E24(3.5mg)。
1H NMR(400MHz,CD
3OD+CDCl
3)δ9.31(s,1H),7.94(s,1H),7.18(s,1H),6.83(m,1H),6.65(m,1H),4.79(s,2H),4.60(t,J=8.8Hz,2H),3.64(t,J=7.3Hz,2H),3.37(m,2H),3.06(s,3H),2.99(m,2H).LC-MS:[M+H]
+=395.1。
实施例93:化合物SL-E25的合成

化合物SL-E23(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E25(5mg)。
1H NMR(400MHz,CD
3OD)δ9.29(s,1H),7.61(s,1H),6.90–6.80(m,1H),6.63(m,1H),4.76(s,2H),4.66–4.51(m,2H),3.58–3.44(m,2H),3.40–3.34(m,2H),3.23–3.11(m,1H),3.09–2.95(m,2H),2.83(s,3H),2.27–2.17(m,4H).LC-MS:[M+H]
+=383.2。
实施例94:化合物SL-E26的合成

化合物SL-E24(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E26(2mg)。
1H NMR(400MHz,CDCl
3)δ8.94(s,1H),7.45(s,1H),6.84(t,J=9.4Hz,1H),6.66(dd,J=8.6,3.9Hz,1H),6.37(m,1H),4.77(d,J=5.3Hz,2H),4.63(t,J=8.7Hz,2H),3.49(m,2H),3.39(m,3H),2.99(s,3H),2.75(m,2H),2.33(m,2H).LC-MS:[M+H]
+=397.2。
实施例95:化合物SL-E29的合成

将化合物SL-ZYE-23(50mg,0.1mmol)溶解于3mL二氯甲烷(DCM)和1mL三 氟乙酸(TFA)中,室温搅拌30min,浓缩后直接经HPLC分离得到产物SL-E29(38mg),LC-MS:[M+H]
+=395.2。
实施例96:化合物SL-E30的合成

将SL-E29(10mg)溶解于1mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入乙酸酐(Ac
2O,8mg),反应1小时,TLC显示反应结束后,由柱层析分离制备得到目标化合物SL-E30(4mg)。LC-MS:[M+H]
+=437.2。
实施例97:化合物SL-E31的合成

将SL-E29(10mg)溶解于1mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入甲磺酸酐(Ms
2O,7mg),反应1小时,TLC显示反应结束后,由柱层析分离制备得到目标化合物SL-E31(3mg)。LC-MS:[M+H]
+=473.2。
实施例98:化合物SL-E32的合成

将化合物SL-E29(12mg)溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA),搅拌溶解,室温下加入HATU(30mg)以及N,N-二甲基甘氨酸(9mg),反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E32(4mg)。LC-MS:[M+H]
+=480.2。
实施例99:化合物SL-E33的合成

化合物SL-E30(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E33(4mg)。LC-MS:[M+H]
+=439.2。
实施例100:化合物SL-E34的合成

化合物SL-E31(9mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E34(4mg)。LC-MS:[M+H]
+=475.2。
实施例101:化合物SL-E35的合成

化合物SL-E32(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E35(4mg)。LC-MS:[M+H]
+=482.2。
实施例102:化合物SL-E36的合成

化合物SL-E5(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E36(5mg)。LC-MS:[M+H]
+=480.2。
实施例103:化合物SL-E37的合成

化合物SL-E6(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E37(5mg)。LC-MS:[M+H]
+=482.2。
实施例104:化合物SL-E38的合成

化合物SL-E7(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E38(5mg)。LC-MS:[M+H]
+=441.2。
实施例105:化合物SL-E39的合成

将化合物E-Y3(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应3小时,硅藻土过滤,将滤液旋干后,溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后,再次溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA)中,室温下加入HATU(15mg)以及酸(oxetane-3-carboxylic acid)(9mg),反应1h,将反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E39(0.7mg)。LC-MS:[M+H]
+=453.4。
实施例106:化合物SL-E40的合成

化合物SL-E9(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E40(5mg)。LC-MS:[M+H]
+=485.2。
实施例107:化合物SL-E41的合成

SL-E10(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E41(5mg)。LC-MS:[M+H]
+=494.3。
实施例108:化合物SL-E42的合成

SL-E11(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E42(5mg)。LC-MS:[M+H]
+=564.2。
实施例109:化合物SL-E43,SL-E43-S,SL-E43-R的合成

SL-E13(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E43(5mg)。LC-MS:[M+H]
+=425.2。
参考实施例56,SL-E43通过手性色谱柱分离获得光学纯化合物SL-E43-S和SL-E43-R。
实施例110:化合物SL-E44,SL-E44-S,SL-E44-R的合成

SL-E14(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E44(5mg)。LC-MS:[M+H]
+=461.2。
参考实施例56,SL-E44通过手性色谱柱分离获得光学纯化合物SL-E44-S和SL-E44-R。
实施例111:化合物SL-E45,SL-E45-S,SL-E45-R的合成

SL-E15(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E45(5mg)。LC-MS:[M+H]
+=468.2。
参考实施例56,SL-E45通过手性色谱柱分离获得光学纯化合物SL-E45-S和SL-E45-R。
实施例112:化合物SL-E46,SL-E46-S,SL-E46-R的合成

SL-E16(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E46(4mg)。LC-MS:[M+H]
+=494.2。
参考实施例56,SL-E46通过手性色谱柱分离获得光学纯化合物SL-E46-S和 SL-E46-R。
实施例113:化合物SL-E47的合成

SL-E17(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E47(4mg)。LC-MS:[M+H]
+=496.2。
参考实施例56,SL-E47通过手性色谱柱分离获得光学纯化合物SL-E47-S和SL-E47-R。
实施例114:化合物SL-E48的合成

SL-E18(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E48(4mg)。LC-MS:[M+H]
+=455.2。
参考实施例56,SL-E48通过手性色谱柱分离获得光学纯化合物SL-E48-S和SL-E48-R。
实施例115:化合物SL-E49的合成

将化合物SL-E2(10mg)溶解于3mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌30min,将溶剂浓缩干后,溶解于1mL DMF和0.1mL二异丙基乙基胺(DIEPA)中,室温下加入HATU(30mg)以及酸(oxetane-3-carboxylic acid)(9mg), 反应1h,TLC显示反应结束,反应液倒入水中,大量乙酸乙酯萃取,干燥浓缩,经柱层析分离制备,得到目标化合物SL-E49(1.2mg)。LC-MS:[M+H]
+=467.2。
参考实施例56,SL-E49通过手性色谱柱分离获得光学纯化合物SL-E49-S和SL-E49-R。
实施例116:化合物SL-E50的合成

SL-E20(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E50(4mg)。LC-MS:[M+H]
+=499.2。
参考实施例56,SL-E50通过手性色谱柱分离获得光学纯化合物SL-E50-S和SL-E50-R。
实施例117:化合物SL-E51的合成

SL-E21(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E51(4mg)。LC-MS:[M+H]
+=508.3。
参考实施例56,SL-E51通过手性色谱柱分离获得光学纯化合物SL-E51-S和SL-E51-R。
实施例118:化合物SL-E52的合成

SL-E22(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E52(4mg)。LC-MS:[M+H]
+=578.2。
参考实施例56,SL-E52通过手性色谱柱分离获得光学纯化合物SL-E52-S和SL-E52-R。
实施例119:化合物SL-E53的合成

SL-ZYE-28(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-E53(4mg)。LC-MS:[M+H]
+=418.3。
实施例120:化合物SL-ZYE-119的合成

将溴代物4-1(36mg,0.1mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯SL-B3(48mg,0.2mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(3.5mg,0.01mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(11mg,0.02mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备得到目标化合物SL-ZYE-119(4.1mg)。1H NMR(400MHz,DMSO-d
6)δ9.44(s,1H),8.61(m,1H),7.66(s,1H),7.30(m,1H),6.94(m,1H),6.70(m,1H),4.69(m,2H),4.54(m,2H),4.30(m,2H),3.29(m,2H),2.40(m,2H),1.23(s,6H).LC-MS:[M+H]
+=396.2。
实施例121:化合物SL-ZYE-120的合成

将溴代物4-1(36mg,0.1mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯SL-B4(48mg,0.2mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(3.5mg,0.01mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(11mg,0.02mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备得到目标化合物SL-ZYE-120(4.1mg)。
1H NMR(400MHz,DMSO-d
6)δ9.44(s,1H),8.62(m,1H),7.67(m,1H),7.27(m,1H),6.93(m,1H),6.70(m,1H),4.69(m,2H),4.52(m,2H),3.84(m,2H),3.26(m,2H),2.42(m,2H),1.28(s,6H).LC-MS:[M+H]
+=396.2。
实施例122:化合物SL-ZYE-121的合成

SL-ZYE-119(10mg)溶解于2mL甲醇中,加入10%Pd/C(4mg)室温下反应6小时,过滤,柱层析分离制备,得到目标化合物SL-ZYE-121(4mg)。
1H NMR(400MHz,DMSO-d
6)δ9.32(s,1H),8.32(s,1H),7.38(m,1H),6.85(m,1H),6.62(m,1H),4.56-4.47(m,4H),3.68(m,2H),3.20(m,3H),1.70(m,4H),1.09(m,6H).LC-MS:[M+H]
+=398.2。
实施例123:化合物SL-ZYE-195的合成

将溴代物4-1(36mg,0.1mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯SL-B5(50mg,0.2mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(3.5mg,0.01mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(11mg,0.02mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备得到目标化合物SL-ZYE-195(4.1mg)。
1H NMR(400MHz,DMSO-d
6)δ9.45(s,1H),8.63(m,1H),7.68(m,1H),7.28(m,1H),6.93(m,1H),6.71(m,1H),4.68(d,J=4.0Hz,2H),4.54(J=8.0Hz,2H),3.27(J=8.0Hz,2H),2.38(m,2H),1.29(s,6H),1.24(s,6H).LC-MS:[M+H]
+=424.2。
实施例124:化合物SL-ZYE-196的合成

将溴代物4-1(36mg,0.1mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯SL-B6(49mg,0.2mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(3.5mg,0.01mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(11mg,0.02mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备得到目标化合物SL-ZYE-196(4.1mg)。
1H NMR(400MHz,DMSO-d
6)δ9.44(s,1H),8.61(m,1H),7.66(m,1H),7.32(m,1H),6.94(m,1H),6.70(m,1H),4.68(d,J=4.0Hz,2H),4.54(t,J=8.0Hz,2H),4.34(m,2H),3.70(m,1H),3.29(J=8.0Hz,2H),2.56-2.21(m,2H),1.25(d,J=4.0Hz,3H).LC-MS:[M+H]
+=382.2。
实施例125:化合物SL-ZYE-197的合成

将溴代物4-1(36mg,0.1mmol)溶解于6mL 1,4-二氧六环和2mL浓度为2M的Na
2CO
3水溶液中,加入硼酸酯SL-B7(49mg,0.2mmol),Ar置换保护,室温下搅拌10分钟。加入10%氯化烯丙基钯(II)二聚物(3.5mg,0.01mmol),20%2′-二环己基膦基-2,6-二甲氧基-1,1′-联苯基-3-磺酸钠水合物(11mg,0.02mmol),在Ar保护下保持在90℃反应40分钟,冷却后减压浓缩,由柱层析分离制备得到目标化合物SL-ZYE-197(4.1mg)。
1H NMR(400MHz,DMSO-d
6)δ9.44(s,1H),8.63(m,1H),7.67(m,1H),7.28(m,1H),6.94(m,1H),6.71(m,1H),4.69(d,J=4.0Hz,2H),4.53(t,J=8.0Hz,2H),4.37(m,1H),4.06(m,1H),3.68(m,1H),3.28(J=8.0Hz,2H),2.60-2.32(m,2H),1.26(d,J=4.0Hz,3H).LC-MS:[M+H]
+=382.2。
实施例126:化合物SL-ZYE-144的合成

将化合物E-Y3(46mg)溶解于5mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入乙基磺酰氯(20mg),继续反应,TLC显示反应结束,直接由柱层析分离制备,得到目标化合物SL-ZYE-144(7mg)。
1H NMR(400MHz,CDCl
3)δ9.00(s,1H),7.64(s,1H),7.24(m,1H),6.78(m,1H),6.61(m,2H),4.80(s,2H),4.61(t,J=8.7Hz,2H),4.06(m,2H),3.61(t,J=5.6Hz,2H),3.38(t,J=8.7Hz,2H),3.04(q,J=7.4Hz,2H),2.72(m,2H),1.40(t,J=7.4Hz,3H).LC-MS:[M+H]
+=459.1。
实施例127:化合物SL-ZYE-146的合成

将化合物E-Y3(46mg)溶解于5mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入环丙基磺酰氯(20mg),继续反应,TLC显示反应结束,直接由柱层析分离制备,得到目标化合物SL-ZYE-146(6mg)。
1H NMR(400MHz,CDCl
3)δ9.03(s,1H),7.64(s,1H),7.23(m,1H),6.77(m,2H),6.58(dd,J=8.7,3.9Hz,1H),4.78(d,J=5.3Hz,2H),4.60(t,J=8.7Hz,2H),4.08(m,2H),3.60(t,J=5.7Hz,2H),3.37(t,J=8.7Hz,2H),2.74(m,2H),2.42–2.27(m,1H),1.32–1.14(m,2H),1.11–0.88(m,2H).LC-MS:[M-H]
+=469.2。
实施例128:化合物SL-ZYE-147的合成

将化合物E-Y3(46mg)溶解于5mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入氯甲酸甲酯(15mg),继续反应,TLC显示反应结束,直接由柱层析分离制备,得到目标化合物SL-ZYE-147(5mg)。
1H NMR(400MHz,DMSO-d
6)δ9.45(s,1H),8.64(m,1H),7.69(s,1H),7.26(s,1H),6.93(m,1H),6.71(m,1H),4.69(m,2H),4.54(m,2H),4.12(m,2H),3.64(s,3H),3.61(m,2H),3.28(m,2H),2.57(m,2H).LC-MS:[M+H]
+=425.2。
实施例129:化合物SL-ZYE-148的合成

将化合物E-Y3(46mg)溶解于5mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入氯甲酸乙酯(15mg),继续反应,TLC显示反应结束,直接由柱层析分离制备,得到目标化合物SL-ZYE-148(4mg)。
1H NMR(400MHz,CDCl
3)δ9.12(s,1H),7.64(s,1H),7.26–7.09(m,1H),7.00(m,1H),6.74(t,J=9.2Hz,1H),6.57(dd,J=8.6,3.9Hz,1H),4.78(d,J=4.9Hz,2H),4.58(t,J=8.8Hz,2H),4.23–4.10(m,4H),3.72(d,J=5.3Hz,2H),3.35(t,J=9.0Hz,2H),2.63(m,2H),1.29(t,J=7.0Hz,3H).LC-MS:[M+H]
+=439.2。
实施例130:化合物SL-ZYE-161的合成

将化合物E-Y3(46mg)溶解于5mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入丙酰氯(10mg),继续反应,TLC显示反应结束,直接由柱层析分离制备,得到目标化合物SL-ZYE-161(4mg)。
1H NMR(400MHz,CDCl
3)δ9.76(m,1H),8.19(m,1H),7.62(m,1H),7.34-7.13(m,1H),6.82–6.67(m,1H),6.58(m,1H),4.79(m,2H),4.57(m,2H),4.26(m,2H),3.82(m,2H),3.39(m,2H),2.65(m,2H),2.50–2.29(m,2H),1.17(m,3H).LC-MS:[M+H]
+=423.2。
实施例131:化合物SL-ZYE-162的合成

将化合物E-Y3(46mg)溶解于5mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入丙酰氯(10mg),继续反应,TLC显示反应结束,直接由柱层析分离制备,得到目标化合物SL-ZYE-162(4mg)。
1H NMR(400MHz,CDCl
3)δ9.35(m,1H),7.81(m,1H),7.64(m,1H),7.33-7.10(m,1H),6.74(m,1H),6.57(m,1H),4.77(m,2H),4.57(m,2H),4.29(m,2H),3.85(m,2H),3.34(m,2H),2.77-2.60(m,2H),1.84(m,1H),0.85(m,4H).LC-MS:[M+H]
+=435.2。
实施例132:化合物SL-ZYE-145的合成

将化合物E-Y3(46mg)溶解于5mL二氯甲烷(DCM)和1mL三氟乙酸(TFA)中,室温搅拌,将溶剂浓缩干后再加入3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入异丙基磺酰氯(20mg),继续反应,TLC显示反应结束,直接由柱层析分离制备,得到目标化合物SL-ZYE-145(7mg)。
1H NMR(400MHz,DMSO-d
6)δ9.46(s,1H),8.69(m,1H),7.70(s,1H),7.33(m,1H),6.96(d,J=8.7Hz,1H),6.71(d,J=3.9Hz,1H),4.69(m,2H),4.55(d,J=8.7Hz,2H),4.04(m,2H),3.53(d,J=5.7Hz,2H),3.47–3.37(m,1H),3.32–3.24(m,2H),2.66(m,2H),1.25(s,3H),1.23(s,3H).LC-MS:[M+H]
+=473.2。
实施例133:化合物SL-ZYE-183的合成

将SL-E12(30mg)溶于3mL二氯甲烷和0.1mL三乙胺(TEA),搅拌溶解,室温下加入氯甲酸甲酯(15mg),继续反应,TLC显示反应结束,直接由柱层析分离制备, 得到目标化合物SL-ZYE-183(5mg)。
1H NMR(400MHz,DMSO-d
6)δ9.43(s,1H),8.60(m,1H),7.62(s,1H),6.98–6.90(m,1H),6.82(m,1H),6.69(m,1H),4.67(d,J=5.0Hz,2H),4.53(t,J=8.7Hz,2H),4.07(m,2H),3.59(m,5H),3.28(t,J=8.7Hz,2H),2.72(m,2H),1.86(m,2H).LC-MS:[M+H]
+=439.2。
实施例134:多梳抑制复合物2(PRC2)酶活性测定实验
使用TR-FRET方法检测PRC2酶的活性。首先,酶与不同浓度的化合物混合,室温孵育30分钟。加入生物素标记的组蛋白H3多肽底物和辅因子S-腺苷甲硫氨酸(SAM)起始酶促反应。室温反应4小时后,加入受体Acceptor和供体Donor,孵育半小时。用多功能酶标仪EnVision(Perkin Elmer公司)检测荧光信号。用GraphPad Prism 5.0软件分析数据,获得IC
50值。
表1中所述化合物可由上述实施例所述方法制备而成,EED226为阳性化合物(Nat.Chem.Biol.2017,13,381–388),N/A代表未分析。









实施例135:细胞长时生长抑制实验(11天)
将指数生长期的Pfeiffer细胞种在24孔板中,细胞密度为1*10E5cells/mL。当天加入不同浓度的化合物处理细胞。在化合物处理4天和7天时,更换新鲜的培养基和化合物。化合物处理11天后,用CellTiter-Glo试剂(Promega公司)测定细胞存活率。用GraphPad Prism 5.0软件分析数据,获得GI
50值。
表2中所述化合物可由上述实施例所述方法制备而成,EED226为阳性化合物(Nat.Chem.Biol.2017,13,381–388)。




实施例136:pfeiffer细胞长时生长抑制实验(14天)
人弥漫性大B细胞淋巴瘤(DLBCL)细胞株pfeiffer(来自ATCC,CRL-2632)用含10%胎牛血清(Gibco,购自Life Technologies公司,10099-141)及1%抗生素(青霉素和链霉素,购自Life Technologies公司,10378016)的RPMI 1640培养基(Gibco,购自Life Technologies公司,22400-089)于CO
2细胞培养箱(37℃,5%CO
2)中培养。在细胞长时生长抑制实验中,将指数生长期的pfeiffer细胞铺种在24孔板(购自Corning公司,3524)中,体积为1mL/孔,细胞密度为2*10E5个细胞/mL。细胞种板后置于CO
2培养箱中静置培养1小时。在含有细胞的24孔板中每孔加入2μL 9种不同浓度的3倍梯度稀释的化合物或DMSO,化合物的终浓度范围为0.003~20μM或0.3~2000nM, DMSO的终浓度为0.2%。在化合物处理4、7和11天时,更换新鲜的培养基和化合物,将DMSO对照孔的细胞密度稀释到2*10E5个细胞/mL,其他化合物孔的细胞稀释比例与DMSO对照孔相同。使用CellTiter-Glo试剂(购自Promega公司,G7572)测定细胞存活率:将化合物处理14天的细胞按40μL/孔转移到白色384孔板(OptiPlate-384,购自PerkinElmer公司,6007299)中,再加入等体积的CellTiter-Glo试剂。在室温孵育10分钟后用多功能酶标仪EnVision(PerkinElmer公司)在400~700nm波长下检测冷发光信号。用GraphPad Prism 5.0软件分析数据,获得IC
50值。
表3中所述化合物可由上述实施例所述方法制备而成,EED226为阳性化合物(Nat.Chem.Biol.2017,13,381–388)。



实施例137:细胞Karpas-422、SU-DHL-4长时生长抑制实验(11天)
人弥漫性大B细胞淋巴瘤(DLBCL)细胞株Karpas-422、SU-DHL-4(ATCC,CRL-2957)用含10%胎牛血清(Gibco,购自Life Technologies公司,10099-141)及1%抗生素(青霉素和链霉素,购自Life Technologies公司,10378016)的RPMI 1640培养基(Gibco,购自Life Technologies公司,22400-089)于CO
2细胞培养箱(37℃,5%CO
2)中培养。在细胞长时生长抑制实验中,将指数生长期的Karpas-422、SU-DHL-4细胞铺种在24孔板(购自Corning公司,3524)中,细胞密度为1*10E5个/mL,细胞培养基体积为1mL。细胞在24孔板中培养1小时后,每孔加入2μL化合物或DMSO。每个化合物有9个不同的浓度,在细胞培养基中的终浓度范围为0.003~20μM或0.3~2000nM,DMSO的终浓度为0.2%。在化合物处理4、7天时,更换新鲜的细胞培养基和化合物,将DMSO对照孔的细胞密度稀释到1*10E5个/mL,化合物孔的细胞稀释比例与DMSO对照孔相同。使用CellTiter-Glo试剂(购自Promega公司,G7572)测定细胞存活率:将化合物处理11天的细胞按40μL/孔转移到白色384孔板(OptiPlate-384,购自PerkinElmer公司,6007299)中,再加入等体积的CellTiter-Glo试剂。在室温孵育10分钟后用多功能酶标仪EnVision(购自PerkinElmer公司)在400~700nm波长下检测冷发光信号。用GraphPad Prism 5.0软件分析数据,获得IC
50值。
表4中所述化合物可由上述实施例所述方法制备而成,EED226为阳性化合物(Nat.Chem.Biol.2017,13,381–388)。



由以上表1至表4中的数据可以看出,本发明的部分化合物对PRC2酶的IC
50值可达nM数量级,显著高于阳性对照组EED226化合物;同样的,对于Pfeiffer,Karpas-422以及SU-DHL-4细胞长时生长抑制实验,本发明的多个化合物的IC
50值也达到了个位数nM数量级,显著高于阳性对照组EED226化合物。
实施例138:大鼠口服药代动力学研究
1、以健康雄性SD大鼠为受试动物,灌胃给予EED226,E-Y1,E-Y13,E-Y47,SL-ZYE-07(3mg/kg),应用LC/MS/MS法测定给药后不同时间点大鼠血浆中的药物浓度。研究本发明的化合物在大鼠体内的药代动力学行为,评价其药动学特性。
2、试验动物为健康成年雄性SD大鼠,每组3只,购自上海西普尔-必凯实验动物有限公司。
3、药物配制:化合物EED226,E-Y1,E-Y13,E-Y47溶于DMSO/0.5%HPMC(5/95,v/v)配制。而化合物SL-ZYE-07则溶于0.5%HPMC(含0.5%Tween 80),涡旋振荡,超声,使固体物质分散均匀,得淡白色混悬液。
4、操作:大鼠灌胃(3mg/kg)给药EED226,E-Y1,E-Y13,E-Y47,SL-ZYE-07于给药后0.25,0.5,1,2,4,8,24小时经股静脉取血45μL,置肝素化的离心管中离心5min,分离血浆样品分析,我们采用液相色谱串联质谱(LC-MS/MS)法测定不同化合物灌胃给药后大鼠血浆的待测化合物含量。
本发明化合物的药代动力学参数如下:


AUC
last:从给药时间开始到最后一个时间点的这段时间的AUC
AUC
INF_obs:从给药时间开始到理论外推无穷远的时间点的这段时间的AUC
结论:本发明的化合物药代吸收良好,具有明显的药代动力学优势。
实施例139:肝微粒体稳定性实验(小鼠、大鼠、人):
试剂和材料:
| 名称 | 供应商 | 货号/批号 |
| 人肝微粒体 | BD | H34 |
| 小鼠肝微粒体 | Rild | M11 |
| 大鼠肝微粒体 | BD | R40/R42 |
| NADPH | Roche | N8100-1000 |
| VIVID BOMCC | Life | P2980 |
| 384孔黑板 | Greiner | 781209 |
| 96孔孵育板 | Corning | 3957 |
| 96孔化合物板 | Apricotdesigns | DWP-16-96-SQ-C |
| MgCl 2 | Sigma | M9272-100G |
| TRIZMA BASE | Sigma | T1503-1KG |
| BSA | Roche分装 | A8020-100 |
| DMSO | Merck | K42958652 225 |
| 甲醇 | Merck | I622207203 |
化合物信息
化合物E-Y1,E-Y13,E-Y15,E-Y40,E-Y43,E-Y47,E-Y50,E-Y54,E-Y54-H,SL-E23的贮备液(10mM in DMSO)
实验步骤
1.开启JANUS及温控装置,初始化后冲洗管道至管道内无气泡。
2.准备实验用缓冲液
准备Tris pH 7.4缓冲液(0.1M):将12.12g Tris溶解在1000mL H
2O中,用2N HCl调pH至7.4,分装50mL/管,-20℃冻存。
准备H
2O/0.1%BSA缓冲液:将200μL 25%BSA加入到50mL H
2O中。
准备VIVID贮备液:将1mg VIVID溶解在1mL乙腈中,分装50μL/管,-20℃冻存。
准备MgCl
2溶液(100mM):将1.016g MgCl
2溶解在50mL Tris pH 7.4缓冲液中,分装1mL/管,-20℃冻存。
准备NADPH溶液(10mM):将355mg NADPH溶解在42.6mL Tris pH 7.4缓冲液中,分装1.8mL/管,-20℃冻存。
准备空白对照:取7.937mL Tris、163μL RLM、450μL MgCL
2溶液、450μL NADPH溶液和9mL甲醇混匀,分装1mL/管,-20℃冻存。
3.准备化合物工作溶液
稀释步骤1:取10μL化合物贮备液加入90μL DMSO=1mM贮备液。
稀释步骤2:取2μL 1mM贮备液加入1mL H2O/0.1%BSA缓冲液=0.2μM工作溶液。
稀释步骤3:取245μL工作溶液到96孔化合物板中,加入5μL VIVID贮备液。
将96孔化合物板放在震荡器上震荡5分钟。
4.打开JANUS电脑里的STM运行EXCEL文件,按EXCEL文件指示操作并运行JANUS程序。
5.JANUS程序运行结束后,向384孔黑板的20列加入乙腈/水50:50(V/V),向384孔黑板的21列加入空白对照。
6.在酶标仪上读板(420nm-465nm),确定VIVID荧光强度。
7.封板并震荡均匀离心后,将样品板提交至LC/MS仪进行样品分析。
数据分析:
以孵育体系中药物的剩余率的对数值对孵育时间作图,进行线性回归得到斜率k,按以下公式推测体内固有清除率(Clint,mL/min/g)值、体内清除率(Cl,mL/min)、肝清除率(Clhep,mL/min)、代谢生物利用度(%MF):
化合物肝微粒体稳定性结果:


结论:本发明的化合物在人、大鼠、小鼠的肝微粒体稳定性良好,具有明显的优势。
Claims (13)
- 一种由通式I表示的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体:
 1)X 1独立地选自N及C-CN;2)R 2独立地选自H、卤素、C 1-C 4卤代烷基及C 1-C 4烷基;3)A独立地选自以下结构:
1)X 1独立地选自N及C-CN;2)R 2独立地选自H、卤素、C 1-C 4卤代烷基及C 1-C 4烷基;3)A独立地选自以下结构: 为单键或双键;
为单键或双键; R 3、R 4及R 5独立地选自H、卤素、C 1-C 4烷基、C 1-C 4卤代烷基、-O-(C 1-C 4烷基)、C 1-C 4卤代烷氧基、C 3-C 6环烷基;R 6独立地选自H、OH、=O及C 1-C 4烷基;R 7独立地选自H、OH、卤素、CN及C 1-C 4烷基;n各自独立地选自0,1及2;X 2独立地选自O、NR a及S(O) p杂原子;每一R a独立地选自H、O、由0-2个R b取代的C 1-C 10烷基、C 1-C 6卤代烷基、-O-(C 1-C 6烷基)、C 1-C 6卤代烷氧基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;4)R 1独立地选自以下结构:
R 3、R 4及R 5独立地选自H、卤素、C 1-C 4烷基、C 1-C 4卤代烷基、-O-(C 1-C 4烷基)、C 1-C 4卤代烷氧基、C 3-C 6环烷基;R 6独立地选自H、OH、=O及C 1-C 4烷基;R 7独立地选自H、OH、卤素、CN及C 1-C 4烷基;n各自独立地选自0,1及2;X 2独立地选自O、NR a及S(O) p杂原子;每一R a独立地选自H、O、由0-2个R b取代的C 1-C 10烷基、C 1-C 6卤代烷基、-O-(C 1-C 6烷基)、C 1-C 6卤代烷氧基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;4)R 1独立地选自以下结构: 为单键或双键;
为单键或双键; 4a)R 1A独立地选自H、羟基、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、氨基、C 1-C 6直链、支链以及环状的烷 氨基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R 1X取代;p各自独立地选自0,1及2;R c独立地选自OH、卤素、CN、C 1-C 6烷基、C 1-C 6卤代烷基、C 1-C 6烷氧基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基、-(OCH 2CH 2) mOR d、NHC(=O)NR dR e、NHC(=S)NR dR e、-NHC(=NH)NR dR e、(OCH 2CH 2) mNR dR e、-C(=O)R d、-S(=O)R d、-C(=O)NR dR e、-S(=O) 2R d、-NHC(=O)R d、-NHC(=S)R d、-NHS(=O) 2R d、-S(=O) 2NHR d、包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R 1X取代;R d及R e独立地选自H、含0-2个R b取代的C 1-C 6烷基、C 1-C 6卤代烷基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、含0-2个O、N、S(O) p杂原子的支链或环状的C 1-C 6杂烷基、-C(=O)H、芳基及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基,其中该芳基以及杂芳基可由0-2个R 1X取代;R a独立地选自H、O、由0-2个R b取代的C 1-C 10烷基、C 1-C 6卤代烷基、-O-(C 1-C 6烷基)、C 1-C 6卤代烷氧基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;R d与R e可通过R d——R e或的方式连接,其中Z 1可选自含0-2个R b取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6杂烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p;R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;
4a)R 1A独立地选自H、羟基、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、氨基、C 1-C 6直链、支链以及环状的烷 氨基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R 1X取代;p各自独立地选自0,1及2;R c独立地选自OH、卤素、CN、C 1-C 6烷基、C 1-C 6卤代烷基、C 1-C 6烷氧基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基、-(OCH 2CH 2) mOR d、NHC(=O)NR dR e、NHC(=S)NR dR e、-NHC(=NH)NR dR e、(OCH 2CH 2) mNR dR e、-C(=O)R d、-S(=O)R d、-C(=O)NR dR e、-S(=O) 2R d、-NHC(=O)R d、-NHC(=S)R d、-NHS(=O) 2R d、-S(=O) 2NHR d、包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R 1X取代;R d及R e独立地选自H、含0-2个R b取代的C 1-C 6烷基、C 1-C 6卤代烷基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、含0-2个O、N、S(O) p杂原子的支链或环状的C 1-C 6杂烷基、-C(=O)H、芳基及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基,其中该芳基以及杂芳基可由0-2个R 1X取代;R a独立地选自H、O、由0-2个R b取代的C 1-C 10烷基、C 1-C 6卤代烷基、-O-(C 1-C 6烷基)、C 1-C 6卤代烷氧基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;R d与R e可通过R d——R e或的方式连接,其中Z 1可选自含0-2个R b取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6杂烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p;R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2; R 1X独立地选自卤素、OH、CN、C 1-C 4烷基、C 1-C 4卤代烷基、C 1-C 4烷氧基、C 1-C 4卤代烷氧基、C 3-C 8环状烷基和环状杂烷基;R 1B及R 1C独立地选自H、OH、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R 1X取代;p各自独立地为0,1及2;R 2B及R 2C独立地选自H、OH、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p 杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R 1X取代;p各自独立地选自0,1及2;R 3B及R 3C独立地选自H、-OH、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R 1X取代;p各自独立地选自0,1及2;或者,R 1B与R 1C、R 2B与R 2C、R 3B与R 3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);R 1D独立地选自H、-OH、卤素、CN、-C(=O)H、-(O) z-(包含0-2个R c取代C 1-C 6烷基)、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、R f、-OR f、-C(=O)R c、NR dR e、-C(=O)NR dR e、-NHC(=O)R c、-S(=O) 2R c、-S(=O) 2NR dR e、-NHS(=O) 2R d、-(OCH 2CH 2) mOR d、-(OCH 2CH 2) mNR dR e;R f独立地选自C 3-C 8环状烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、芳基及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基;其中,芳基及杂芳基由0-2个R 1X取代;M独立地选自3至7元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;在R 1B及R 1C、R 2B及R 2C、R 3B及R 3C、R 1D以及R f的定义中,R a、R c、R d、R e、p、z、m、R 1X的定义与在4a)部分中的R 1A中的相应的定义相同;n各自独立地选自0,1及2;m各自独立地选自0-4;p各自独立地选自0-2;q各自独立地选自0-3;z各自独立地选自0和1;4a’)优选地,R 1独立地选自以下结构:其中
R 1X独立地选自卤素、OH、CN、C 1-C 4烷基、C 1-C 4卤代烷基、C 1-C 4烷氧基、C 1-C 4卤代烷氧基、C 3-C 8环状烷基和环状杂烷基;R 1B及R 1C独立地选自H、OH、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R 1X取代;p各自独立地为0,1及2;R 2B及R 2C独立地选自H、OH、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p 杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R 1X取代;p各自独立地选自0,1及2;R 3B及R 3C独立地选自H、-OH、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中该芳基以及杂芳基可由0-2个R 1X取代;p各自独立地选自0,1及2;或者,R 1B与R 1C、R 2B与R 2C、R 3B与R 3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);R 1D独立地选自H、-OH、卤素、CN、-C(=O)H、-(O) z-(包含0-2个R c取代C 1-C 6烷基)、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、R f、-OR f、-C(=O)R c、NR dR e、-C(=O)NR dR e、-NHC(=O)R c、-S(=O) 2R c、-S(=O) 2NR dR e、-NHS(=O) 2R d、-(OCH 2CH 2) mOR d、-(OCH 2CH 2) mNR dR e;R f独立地选自C 3-C 8环状烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、芳基及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基;其中,芳基及杂芳基由0-2个R 1X取代;M独立地选自3至7元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;在R 1B及R 1C、R 2B及R 2C、R 3B及R 3C、R 1D以及R f的定义中,R a、R c、R d、R e、p、z、m、R 1X的定义与在4a)部分中的R 1A中的相应的定义相同;n各自独立地选自0,1及2;m各自独立地选自0-4;p各自独立地选自0-2;q各自独立地选自0-3;z各自独立地选自0和1;4a’)优选地,R 1独立地选自以下结构:其中 为单键或双键;
为单键或双键; R 1A独立地选自H、羟基、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、-C(=O)H;R c独立地选自OH、卤素、CN、C 1-C 6烷基、羧基、C 1-C 6卤代烷基、C 1-C 6烷氧基、 C 1-C 6卤代烷氧基、C 3-C 8环状烷基;R 1B及R 1C、R 2B及R 2C、以及R 3B及R 3C独立地选自H、OH、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基);或者,R 1B与R 1C、R 2B与R 2C、R 3B与R 3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);R 1D独立地选自H、-OH、卤素、CN、-C(=O)H、-(O) z-(包含0-2个R c取代C 1-C 6烷基)、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基;R c独立地选自OH,卤素、CN、C 1-C 6烷基、羧基、C 1-C 6卤代烷基、C 1-C 6烷氧基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;M独立地选自3至7元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;n各自独立地选自0,1及2;m各自独立地选自0-4;p各自独立地选自0-2;q各自独立地选自0-3;z各自独立地选自0和1;4a”)更优选地,在R 1中,R 1A独立地选自H、羟基、卤素、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;R 1B及R 1C、R 2B及R 2C、以及R 3B及R 3C独立地选自H、OH、卤素、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;或者,R 1B与R 1C、R 2B与R 2C、R 3B与R 3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);R 1D独立地选自H、-OH、卤素、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;M独立地选自5至6元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;n各自独立地选自0,1及2;m各自独立地选自0-4;p各自独立地选自0-2;q各自独立地选自0-3;z各自独立地选自0和1;4b)Y独立地选自O、NR g、S(O) p等杂原子以及CH 2、C=O、-CR i(CH 2) mNR gR h以及-CR i(CH 2) mOR g;R g及R h独立地选自H、O、含0-3个R s取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 6环状烷基、
R 1A独立地选自H、羟基、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、-C(=O)H;R c独立地选自OH、卤素、CN、C 1-C 6烷基、羧基、C 1-C 6卤代烷基、C 1-C 6烷氧基、 C 1-C 6卤代烷氧基、C 3-C 8环状烷基;R 1B及R 1C、R 2B及R 2C、以及R 3B及R 3C独立地选自H、OH、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基);或者,R 1B与R 1C、R 2B与R 2C、R 3B与R 3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);R 1D独立地选自H、-OH、卤素、CN、-C(=O)H、-(O) z-(包含0-2个R c取代C 1-C 6烷基)、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基;R c独立地选自OH,卤素、CN、C 1-C 6烷基、羧基、C 1-C 6卤代烷基、C 1-C 6烷氧基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;M独立地选自3至7元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;n各自独立地选自0,1及2;m各自独立地选自0-4;p各自独立地选自0-2;q各自独立地选自0-3;z各自独立地选自0和1;4a”)更优选地,在R 1中,R 1A独立地选自H、羟基、卤素、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;R 1B及R 1C、R 2B及R 2C、以及R 3B及R 3C独立地选自H、OH、卤素、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;或者,R 1B与R 1C、R 2B与R 2C、R 3B与R 3C可和与之连接的碳原子形成羰基(=O)或者硫羰基(=S);R 1D独立地选自H、-OH、卤素、C 1-C 6烷基、C 1-C 6烷氧基、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;M独立地选自5至6元饱和的或不饱和的环烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂环烷基、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;n各自独立地选自0,1及2;m各自独立地选自0-4;p各自独立地选自0-2;q各自独立地选自0-3;z各自独立地选自0和1;4b)Y独立地选自O、NR g、S(O) p等杂原子以及CH 2、C=O、-CR i(CH 2) mNR gR h以及-CR i(CH 2) mOR g;R g及R h独立地选自H、O、含0-3个R s取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 6环状烷基、 -C(=S)NHC(=O)-R j、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1X取代;
-C(=S)NHC(=O)-R j、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1X取代; R s独立地选自OH,CN、卤素、C 1-C 6烷基、C 1-C 6卤代烷基、C 1-C 6烷氧基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基、-(OCH 2CH 2) mOR d、NHC(=O)NR dR e、NHC(=S)NR dR e、-NHC(=NH)NR dR e、(OCH 2CH 2) mNR dR e、-C(=O)R d、-C(=S)R d、-S(=O)R d、-C(=O)NR dR e、-S(=O) 2R d、-NHC(=O)R d、-NHC(=S)R d、-NHS(=O) 2R d、-S(=O) 2NR dR e、-NHS(=O) 2NR dR e、-C(=S)NR dR e、NHC(=O)OR d、NHC(=S)OR d、-NHS(=O) 2OR d、NHC(=O)SR d、NHC(=S)SR d、-NHC(=NH)OR d、-C(=O)OR d、-C(=O)SR d、-S(=O) 2OR d、包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R 1Y取代;R d及R e独立地选自H、含0-2个R b取代的C 1-C 6烷基、C 1-C 6卤代烷基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、含0-2个O、N、S(O) p杂原子的支链或环状的C 1-C 6杂烷基、-C(=O)H、芳基及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基,其中该芳基以及杂芳基可由0-2个R 1X取代;R d与R e可通过以下方式R d——R e或连接形成一个环,其中Z 1可选自含0-2个R b取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p,
R s独立地选自OH,CN、卤素、C 1-C 6烷基、C 1-C 6卤代烷基、C 1-C 6烷氧基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基、-(OCH 2CH 2) mOR d、NHC(=O)NR dR e、NHC(=S)NR dR e、-NHC(=NH)NR dR e、(OCH 2CH 2) mNR dR e、-C(=O)R d、-C(=S)R d、-S(=O)R d、-C(=O)NR dR e、-S(=O) 2R d、-NHC(=O)R d、-NHC(=S)R d、-NHS(=O) 2R d、-S(=O) 2NR dR e、-NHS(=O) 2NR dR e、-C(=S)NR dR e、NHC(=O)OR d、NHC(=S)OR d、-NHS(=O) 2OR d、NHC(=O)SR d、NHC(=S)SR d、-NHC(=NH)OR d、-C(=O)OR d、-C(=O)SR d、-S(=O) 2OR d、包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R 1Y取代;R d及R e独立地选自H、含0-2个R b取代的C 1-C 6烷基、C 1-C 6卤代烷基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、-C(=O)NH(C 1-C 4烷基)、含0-2个O、N、S(O) p杂原子的支链或环状的C 1-C 6杂烷基、-C(=O)H、芳基及包含碳原子及1至2个选自N、NR a、O及S(O) p杂原子的杂芳基,其中该芳基以及杂芳基可由0-2个R 1X取代;R d与R e可通过以下方式R d——R e或连接形成一个环,其中Z 1可选自含0-2个R b取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p, R a独立地选自H、O、由0-2个R b取代的C 1-C 10烷基、C 1-C 6卤代烷基、-O-(C 1-C 6烷基)、C 1-C 6卤代烷氧基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;R 1X独立地选自卤素、OH、CN、C 1-C 4烷基、C 1-C 4卤代烷基、C 1-C 4烷氧基、C 1-C 4卤代烷氧基、C 3-C 8环状烷基和环状杂烷基;R 1Y独立地选自C 1-C 10烷基、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、NR dR e、-C(=O)NR dR e、-S(=O) 2R d、-NHC(=O)R d、-NHC(=S)R d、-NHS(=O) 2R d、-NHC(=O)NR dR e、-NHC(=S)NR dR e、-NHS(=O) 2NR dR e、-C(=S)NR dR e、-S(=O) 2NHR d、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、p各自独立地为0,1及2;其中,R c与在上述4a)部分中限定的R c的定义相同;其中R d与R e可通过R d——R e或的方式连接,其中Z 1可选自含0-2个R b取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6杂烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p,
R a独立地选自H、O、由0-2个R b取代的C 1-C 10烷基、C 1-C 6卤代烷基、-O-(C 1-C 6烷基)、C 1-C 6卤代烷氧基、C 3-C 6环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;R 1X独立地选自卤素、OH、CN、C 1-C 4烷基、C 1-C 4卤代烷基、C 1-C 4烷氧基、C 1-C 4卤代烷氧基、C 3-C 8环状烷基和环状杂烷基;R 1Y独立地选自C 1-C 10烷基、卤素、CN、-(O) z-(包含0-2个R c取代C 1-C 10烷基)、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、SCF 3、C 3-C 8环状烷基、-C(=O)(C 1-C 4烷基)、-CO 2(C 1-C 4烷基)、NR dR e、-C(=O)NR dR e、-S(=O) 2R d、-NHC(=O)R d、-NHC(=S)R d、-NHS(=O) 2R d、-NHC(=O)NR dR e、-NHC(=S)NR dR e、-NHS(=O) 2NR dR e、-C(=S)NR dR e、-S(=O) 2NHR d、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、-C(=O)H、p各自独立地为0,1及2;其中,R c与在上述4a)部分中限定的R c的定义相同;其中R d与R e可通过R d——R e或的方式连接,其中Z 1可选自含0-2个R b取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6杂烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p, R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;R j及R k独立地为选自H、CN、含0-3个R s取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 10环状烷基、包含碳原子以及选自和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、由R y取代的烯基或者炔基6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1Y取代;
R b独立地选自卤素、OH、NH 2、-NHC(=O)(C 1-C 4烷基)、-NHS(=O) 2(C 1-C 4烷基)、=O、CN、C 1-C 4烷基及C 1-C 4烷氧基;p各自独立地选自0,1及2;R j及R k独立地为选自H、CN、含0-3个R s取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 10环状烷基、包含碳原子以及选自和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、由R y取代的烯基或者炔基6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1Y取代; 其中,R 1Y与在本4b)部分中的上述R s中限定的R 1Y的定义相同;R y独立地选自H、含0-3个R c取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 10环状烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、NR dR e、OR d、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1X取代;R 1X独立地选自卤素、OH、CN、C 1-C 4烷基、C 1-C 4卤代烷基、C 1-C 4烷氧基、C 1-C 4卤代烷氧基、C 3-C 8环状烷基和环状杂烷基;其中,R c与在上述4a)部分中限定的R c的定义相同;R d和R e与在本4b)部分中的上述R s中限定的R d和R e的定义相同;特别地,R g与R h以及R j及R k可通过以下方式R g——R h、R j——R k、
其中,R 1Y与在本4b)部分中的上述R s中限定的R 1Y的定义相同;R y独立地选自H、含0-3个R c取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 10环状烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、NR dR e、OR d、芳基、包含碳原子及1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1X取代;R 1X独立地选自卤素、OH、CN、C 1-C 4烷基、C 1-C 4卤代烷基、C 1-C 4烷氧基、C 1-C 4卤代烷氧基、C 3-C 8环状烷基和环状杂烷基;其中,R c与在上述4a)部分中限定的R c的定义相同;R d和R e与在本4b)部分中的上述R s中限定的R d和R e的定义相同;特别地,R g与R h以及R j及R k可通过以下方式R g——R h、R j——R k、 连接,其中Z 1可选自含0-2个R c取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p;p各自独立地选自0,1及2;
连接,其中Z 1可选自含0-2个R c取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p;p各自独立地选自0,1及2; 其中,R c与在上述4a)部分中限定的R c的定义相同;R i独立地选自H、CN、C1-C4烷基;m各自独立地选自0-4;4b’)优选地,Y独立地选自O、NR g、S(O) p、-CR i(CH 2) mNR gR h以及-CR i(CH 2) mOR g;R g及R h独立地选自H、C 1-C 6卤代烷基、
其中,R c与在上述4a)部分中限定的R c的定义相同;R i独立地选自H、CN、C1-C4烷基;m各自独立地选自0-4;4b’)优选地,Y独立地选自O、NR g、S(O) p、-CR i(CH 2) mNR gR h以及-CR i(CH 2) mOR g;R g及R h独立地选自H、C 1-C 6卤代烷基、 -C(=S)NHC(=O)-R j、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1X取代;
-C(=S)NHC(=O)-R j、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1X取代;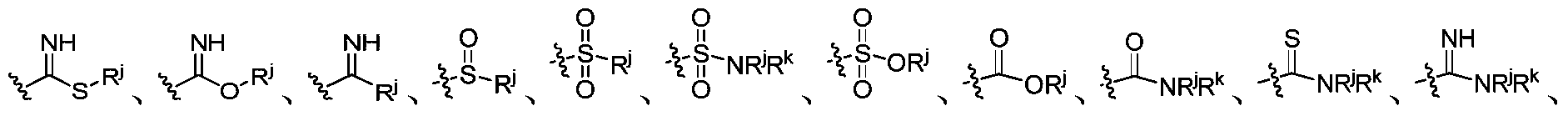 R j及R k独立地选自H、CN、含0-3个R s取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 10环状烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、C 2-C 10 烯基或者炔基、6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1Y取代;其中,R s的定义与在上述4b)部分中的R s的定义相同;p各自独立地选自0,1及2;R 1X独立地选自卤素、OH、CN、C 1-C 4烷基、C 1-C 4卤代烷基、C 1-C 4烷氧基、C 1-C 4卤代烷氧基、C 3-C 8环状烷基和环状杂烷基;R 1Y独立地选自C 1-C 10烷基、卤素、CN、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;p各自独立地为0,1及2;特别地,R g与R h以及R j及R k可通过以下方式R g——R h、R j——R k、
R j及R k独立地选自H、CN、含0-3个R s取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 10环状烷基、包含碳原子和1-4个选自O、N、S(O) p杂原子的杂烷基和杂环烷基、C 2-C 10 烯基或者炔基、6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1Y取代;其中,R s的定义与在上述4b)部分中的R s的定义相同;p各自独立地选自0,1及2;R 1X独立地选自卤素、OH、CN、C 1-C 4烷基、C 1-C 4卤代烷基、C 1-C 4烷氧基、C 1-C 4卤代烷氧基、C 3-C 8环状烷基和环状杂烷基;R 1Y独立地选自C 1-C 10烷基、卤素、CN、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;p各自独立地为0,1及2;特别地,R g与R h以及R j及R k可通过以下方式R g——R h、R j——R k、 连接,其中Z 1可选自含0-2个R c取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p;
连接,其中Z 1可选自含0-2个R c取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)(C 1-C 6烷基)、-NS(=O) 2(C 1-C 6烷基)、S(O) p; 其中,R c与在上述4a)部分中限定的R c的定义相同;p各自独立地选自0,1及2;R i独立地选自H、CN及C 1-C 4烷基;m各自独立地选自0-4;4b”)更优选地,Y独立地选自O、NR g、S、-CR iNR gR h以及-CR iOR g;R g及R h独立地选自H、C 1-C 6卤代烷基、
其中,R c与在上述4a)部分中限定的R c的定义相同;p各自独立地选自0,1及2;R i独立地选自H、CN及C 1-C 4烷基;m各自独立地选自0-4;4b”)更优选地,Y独立地选自O、NR g、S、-CR iNR gR h以及-CR iOR g;R g及R h独立地选自H、C 1-C 6卤代烷基、 -C(=S)NHC(=O)-R j、包含碳原子和1-4个选自O、N、S杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1X取代;
-C(=S)NHC(=O)-R j、包含碳原子和1-4个选自O、N、S杂原子的杂烷基和杂环烷基、芳基、包含碳原子和1至2个选自N、O及S杂原子的5至6元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1X取代; R j及R k独立地选自H、CN、含0-3个R s取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 10环状烷基、包含碳原子和1-4个选自O、N、S杂原子的杂烷基和杂环烷基、C 2-C 10烯基或者炔基、6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1Y取代;R s独立地选自OH,CN、卤素、C 1-C 6烷基、C 1-C 6卤代烷基、C 1-C 6烷氧基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基、-(OCH 2CH 2) mOR d、(OCH 2CH 2) mNR dR e、包含碳原子及1至2个选自N、O及S杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选自N、O及S杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R 1Y取代;其中,R d、R e与在上述4b)部分中限定的R d、R e的定义相同;R 1Y独立地选自C 1-C 10烷基、卤素、CN、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;特别地,R g与R h以及R j及R k可通过以下方式R g——R h、R j——R k、
R j及R k独立地选自H、CN、含0-3个R s取代的C 1-C 10烷基、C 1-C 6卤代烷基、C 3-C 10环状烷基、包含碳原子和1-4个选自O、N、S杂原子的杂烷基和杂环烷基、C 2-C 10烯基或者炔基、6至10元芳基、包含碳原子及1至2个选自N、O及S杂原子的5至10元杂芳基;其中,该芳基以及杂芳基可由0-2个R 1Y取代;R s独立地选自OH,CN、卤素、C 1-C 6烷基、C 1-C 6卤代烷基、C 1-C 6烷氧基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基、-(OCH 2CH 2) mOR d、(OCH 2CH 2) mNR dR e、包含碳原子及1至2个选自N、O及S杂原子的杂烷基和杂环烷基、芳基以及包含碳原子及1至2个选自N、O及S杂原子的杂芳基、其中该芳基以及杂芳基可由0-2个R 1Y取代;其中,R d、R e与在上述4b)部分中限定的R d、R e的定义相同;R 1Y独立地选自C 1-C 10烷基、卤素、CN、C 1-C 6卤代烷基、C 1-C 6卤代烷氧基、C 3-C 8环状烷基;特别地,R g与R h以及R j及R k可通过以下方式R g——R h、R j——R k、 连接,其中Z 1可选自含0-2个R c取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)C 1-C 6烷基、NS(=O) 2(C 1-C 6烷基)、S(O) p;
连接,其中Z 1可选自含0-2个R c取代的C 1-C 6烷基、含0-2个O、N、S(O) p杂原子的C 1-C 6烷基、O、-N(C 1-C 6烷基)、-NH、-N(C=O)C 1-C 6烷基、NS(=O) 2(C 1-C 6烷基)、S(O) p; 其中,R c与在上述4a)部分中限定的R c的定义相同;p各自独立地选自0,1及2;R i独立地选自H、CN及C 1-C 4烷基;m各自独立地选自0-4。
其中,R c与在上述4a)部分中限定的R c的定义相同;p各自独立地选自0,1及2;R i独立地选自H、CN及C 1-C 4烷基;m各自独立地选自0-4。 - 根据权利要求1所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,其中,所述通式I所述的化合物具有式Ia-1或Ia-2:
 其中,X 1与权利要求1中所述通式I中的1)部分中的定义相同;为单键或双键;
其中,X 1与权利要求1中所述通式I中的1)部分中的定义相同;为单键或双键; X 2、R 3-R 5、R 6以及n与权利要求1中所述通式I中的3)部分中的定义相同;R 1A、R 1B及R 1C、R 2B及R 2C、R 3B及R 3C、R 1D、n、m、q、Y、M的定义与权利要求1中所述通式I中的4)部分中的相应的定义相同。
X 2、R 3-R 5、R 6以及n与权利要求1中所述通式I中的3)部分中的定义相同;R 1A、R 1B及R 1C、R 2B及R 2C、R 3B及R 3C、R 1D、n、m、q、Y、M的定义与权利要求1中所述通式I中的4)部分中的相应的定义相同。 - 根据权利要求1所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,其中所述通式I所述的化合物具有式Ia-3或Ia-4:
 其中,X 1与权利要求1中所述通式I中的1)部分中的定义相同;为单键或双键;
其中,X 1与权利要求1中所述通式I中的1)部分中的定义相同;为单键或双键; X 2、R 7以及n与权利要求1中所述通式I中的3)部分中的定义相同;R 1A、R 1B及R 1C、R 2B及R 2C、R 3B及R 3C、R 1D、n、m、q、Y、M的定义与权利要求1中所述通式I中的4)部分中的相应的定义相同。
X 2、R 7以及n与权利要求1中所述通式I中的3)部分中的定义相同;R 1A、R 1B及R 1C、R 2B及R 2C、R 3B及R 3C、R 1D、n、m、q、Y、M的定义与权利要求1中所述通式I中的4)部分中的相应的定义相同。 - 根据权利要求1所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,其中所述通式I所述的化合物具有式Ia-5或Ia-6:
 其中,X 1与权利要求1中所述通式I中的1)部分中的定义相同;为单键或双键;
其中,X 1与权利要求1中所述通式I中的1)部分中的定义相同;为单键或双键; Y的定义与权利要求1中所述通式I中的4)部分中的相应的定义相同。
Y的定义与权利要求1中所述通式I中的4)部分中的相应的定义相同。 - 根据权利要求1所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,其中所述通式I所述的化合物具有式Ia-7或Ia-8:
 其中,X 1与权利要求1中所述通式I中的1)部分中的定义相同;为单键或双键;
其中,X 1与权利要求1中所述通式I中的1)部分中的定义相同;为单键或双键; R g的定义与权利要求1中所述通式I中的4)部分中的4b)部分中的相应的定义相同。
R g的定义与权利要求1中所述通式I中的4)部分中的4b)部分中的相应的定义相同。 - 根据权利要求1所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,其中所述通式I所述的化合物具有式Ia-9:
 其中,为单键或双键;
其中,为单键或双键; R g的定义与权利要求1中所述通式I中的4)部分中的4b)部分中的相应的定义相同。
R g的定义与权利要求1中所述通式I中的4)部分中的4b)部分中的相应的定义相同。 - 根据权利要求1所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,其中所述通式I所述的化合物具有以下结构式之一:
 其中,M的定义与权利要求1中所述通式I中的4)部分中的相应的定义相同;R g的定义与权利要求1中所述通式I中的4)部分中的4b)部分中的相应的定义相同。
其中,M的定义与权利要求1中所述通式I中的4)部分中的相应的定义相同;R g的定义与权利要求1中所述通式I中的4)部分中的4b)部分中的相应的定义相同。 - 根据权利要求1所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,其中所述通式I所述的化合物选自如下化合物:








- 一种用于制备根据权利要求1所述的由通式I表示的化合物的方法,所述方法包括如下步骤:方案一
 (1a)用水合肼处理5-溴-4-氯-2-(甲硫基)嘧啶1,生成5-溴-4-肼基-2-(甲硫基)嘧啶2,(1b)再用原甲酸三甲酯将5-溴-4-肼基-2-(甲硫基)嘧啶2转化为三氮唑产物3,(1c)将三氮唑产物3与胺NH 2CH 2A发生取代反应生成化合物4,(1d)将化合物4在钯催化剂作用下与各类具有R 1基团的硼酸或其等效物发生铃木偶联反应,得到产物5,其中,A、R 1的定义与在权利要求1中定义的相同;方案二
(1a)用水合肼处理5-溴-4-氯-2-(甲硫基)嘧啶1,生成5-溴-4-肼基-2-(甲硫基)嘧啶2,(1b)再用原甲酸三甲酯将5-溴-4-肼基-2-(甲硫基)嘧啶2转化为三氮唑产物3,(1c)将三氮唑产物3与胺NH 2CH 2A发生取代反应生成化合物4,(1d)将化合物4在钯催化剂作用下与各类具有R 1基团的硼酸或其等效物发生铃木偶联反应,得到产物5,其中,A、R 1的定义与在权利要求1中定义的相同;方案二 (2a)将氰乙酰胺6与丙炔酸乙酯反应生成中间体7,(2b)将中间体7用溴素处理发生溴代反应得到溴代物8,(2c)将溴代物8与三氯氧磷反应得到中间体9,(2d)将中间体9由水合肼处理,生成中间体10,(2e)将中间体10用原甲酸三甲酯将其转化为三氮唑产物11,(2f)将三氮唑产物11与各种胺发生取代反应生成化合物12,(2g)最后将化合物12在钯催化剂作用下与各类具有R 1基团的硼酸或其等效物发生铃木偶联(Suzuki)反应,得到产物13,其中,A、R 1的定义与在权利要求1中定义的相同;方案三
(2a)将氰乙酰胺6与丙炔酸乙酯反应生成中间体7,(2b)将中间体7用溴素处理发生溴代反应得到溴代物8,(2c)将溴代物8与三氯氧磷反应得到中间体9,(2d)将中间体9由水合肼处理,生成中间体10,(2e)将中间体10用原甲酸三甲酯将其转化为三氮唑产物11,(2f)将三氮唑产物11与各种胺发生取代反应生成化合物12,(2g)最后将化合物12在钯催化剂作用下与各类具有R 1基团的硼酸或其等效物发生铃木偶联(Suzuki)反应,得到产物13,其中,A、R 1的定义与在权利要求1中定义的相同;方案三 (3a)14以二氯甲烷为溶剂,在三氟乙酸作用下,去除Boc保护基,得到胺基化合物15,(3b)将胺基化合物15在碱性条件下进一步与带有R g基团的试剂或化合物反应得到16,所述试剂或化合物例如但不限于酸酐、磺酸酐、异氰酸酯、硫代异氰酸酯、酰氯、磺酰氯、碳酸酯、氯甲酸酯、氨基甲酸酯,其中,A、X 1、R g、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案四
(3a)14以二氯甲烷为溶剂,在三氟乙酸作用下,去除Boc保护基,得到胺基化合物15,(3b)将胺基化合物15在碱性条件下进一步与带有R g基团的试剂或化合物反应得到16,所述试剂或化合物例如但不限于酸酐、磺酸酐、异氰酸酯、硫代异氰酸酯、酰氯、磺酰氯、碳酸酯、氯甲酸酯、氨基甲酸酯,其中,A、X 1、R g、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案四 (4a)将在方案三的步骤(3a)中去除保护基所得的产物15与含有R j基团的羧酸在缩合剂的作用下发生缩合反应得到酰胺化合物17,其中,A、X 1、R j、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案五
(4a)将在方案三的步骤(3a)中去除保护基所得的产物15与含有R j基团的羧酸在缩合剂的作用下发生缩合反应得到酰胺化合物17,其中,A、X 1、R j、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案五 (5a)将18溶于溶剂中,所述溶剂例如但不限于甲醇、乙醇、乙酸乙酯、四氢呋喃,加入金属催化剂,所述金属催化剂例如但不限于10%钯碳、Pd(OH) 2、雷尼镍、RhCl(PPh 3) 3,通入氢气,室温下反应,得到双键还原的化合物19,其中,A、X 1、Y、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案六
(5a)将18溶于溶剂中,所述溶剂例如但不限于甲醇、乙醇、乙酸乙酯、四氢呋喃,加入金属催化剂,所述金属催化剂例如但不限于10%钯碳、Pd(OH) 2、雷尼镍、RhCl(PPh 3) 3,通入氢气,室温下反应,得到双键还原的化合物19,其中,A、X 1、Y、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案六 (6a)20通过还原反应得到化合物21,随后与mCPBA(间氯过氧苯甲酸)或者双氧水发生氧化反应得到化合物22,其中,A、X 1、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案七
(6a)20通过还原反应得到化合物21,随后与mCPBA(间氯过氧苯甲酸)或者双氧水发生氧化反应得到化合物22,其中,A、X 1、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案七 (7a)20与mCPBA或者双氧水发生氧化反应得到化合物23,其中,A、X 1、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案八
(7a)20与mCPBA或者双氧水发生氧化反应得到化合物23,其中,A、X 1、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同;方案八 15将双键还原得到24,随后与带有R g基团的试剂或化合物在碱存在下反应得到化合物25,所述试剂或化合物例如但不限于酸酐、磺酸酐、异氰酸酯、硫代异氰酸酯、酰氯、磺酰氯、碳酸酯、氯甲酸酯、氨基甲酸酯;或者24与各类羧酸在缩合剂的存在下发生缩合反应得到酰胺化合物25,其中,A、X 1、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同。
15将双键还原得到24,随后与带有R g基团的试剂或化合物在碱存在下反应得到化合物25,所述试剂或化合物例如但不限于酸酐、磺酸酐、异氰酸酯、硫代异氰酸酯、酰氯、磺酰氯、碳酸酯、氯甲酸酯、氨基甲酸酯;或者24与各类羧酸在缩合剂的存在下发生缩合反应得到酰胺化合物25,其中,A、X 1、R 1A、R 1B、R 1C、R 2B、R 2C、R 3B、R 3C、q、m的定义与在权利要求1中定义的相同。 - 一种药物组合物,其包含根据权利要求1-8中任一项所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体中的一种或多种,以及至少一种药学上可接受的载体、稀释剂或赋形剂。
- 根据权利要求10所述的药物组合物,其中所述药物组合物进一步包含至少一种其他治疗剂,优选地,所述药物组合物中包含的所述至少一种其他治疗剂选自其他抗癌剂、免疫调节剂、抗过敏剂、止吐剂、疼痛缓解剂、细胞保护剂及其组合。
- 根据权利要求1-8中任一项所述的化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体或根据权利要求10所述的药物组合物在制备用于治疗由EED和/或PRC2介导的疾病或病症的药物中的用途。
- 根据权利要求12所述的用途,其中所述由EED和/或PRC2介导的疾病或病症包括弥漫性大B细胞淋巴瘤、滤泡性淋巴瘤、其他淋巴瘤、白血病、多发性骨髓瘤、间皮瘤、胃癌、恶性横纹肌样瘤、肝细胞癌、前列腺癌、乳腺癌、胆管及胆囊癌、膀胱癌;脑瘤、包括神经母细胞瘤、神经鞘瘤、神经胶质瘤、神经胶质母细胞瘤及星细胞瘤;子宫颈癌、结肠癌、黑色素瘤、子宫内膜癌、食道癌、头颈癌、肺癌、鼻咽癌、卵巢癌、胰腺癌、肾细胞癌、直肠癌、甲状腺癌、副甲状腺肿瘤、子宫肿瘤及软组织肉瘤。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020529171A JP6876875B2 (ja) | 2017-09-28 | 2018-08-29 | Prc2介在の疾患を治療するためのトリアゾロピリミジン、トリアゾロピリジン化合物及びその組成物 |
| US16/651,510 US11013745B2 (en) | 2017-09-28 | 2018-08-29 | Triazolopyrimidine, triazolopyridine compounds, and the composition thereof for treating PRC2-mediated diseases |
| CN201880002779.7A CN109843890B (zh) | 2017-09-28 | 2018-08-29 | 三氮唑并嘧啶、三氮唑并吡啶化合物及其组合物用于治疗prc2介导的疾病 |
| EP18860160.3A EP3689875B1 (en) | 2017-09-28 | 2018-08-29 | Use of triazolopyrimidine, triazolopyridine compounds and composition thereof for treating prc2-mediated diseases |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710898099 | 2017-09-28 | ||
| CN201710898099.5 | 2017-09-28 | ||
| CN201711408714.6 | 2017-12-22 | ||
| CN201711408714.6A CN109575013A (zh) | 2017-09-28 | 2017-12-22 | 三氮唑并嘧啶、三氮唑并吡啶化合物及其组合物用于治疗prc2介导的疾病 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019062435A1 true WO2019062435A1 (zh) | 2019-04-04 |
Family
ID=65900657
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2018/102833 WO2019062435A1 (zh) | 2017-09-28 | 2018-08-29 | 三氮唑并嘧啶、三氮唑并吡啶化合物及其组合物用于治疗prc2介导的疾病 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2019062435A1 (zh) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021032004A1 (zh) * | 2019-08-22 | 2021-02-25 | 上海青煜医药科技有限公司 | 氮杂芳基化合物及其应用 |
| WO2021083380A1 (zh) * | 2019-11-01 | 2021-05-06 | 上海科技大学 | Eed抑制剂及其制备方法和用途 |
| CN113004233A (zh) * | 2019-12-18 | 2021-06-22 | 南京优氟医药科技有限公司 | 一种用于制备prc2抑制剂的化合物、其制备方法和用途 |
| US11091495B2 (en) | 2018-01-31 | 2021-08-17 | Mirati Therapeutics, Inc. | Substituted imidazo[1,2-c]pyrimidines as PRC2 inhibitors |
| JP7541538B2 (ja) | 2019-06-05 | 2024-08-28 | ミラティ セラピューティクス,インク. | 癌を処置するためのprc2阻害剤としてのイミダゾ[1,2-c]ピリミジン誘導体 |
| JP7546780B2 (ja) | 2021-02-10 | 2024-09-06 | シャンハイ ブルーレイ バイオファーマ カンパニー,リミティド | アザヘテロアリール化合物、その調製方法及び使用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160176682A1 (en) | 2014-12-19 | 2016-06-23 | Airbus Defence And Space, S.A. | Device for hoisting and controlling loads |
| WO2016103155A1 (en) * | 2014-12-23 | 2016-06-30 | Novartis Ag | Triazolopyrimidine compounds and uses thereof |
-
2018
- 2018-08-29 WO PCT/CN2018/102833 patent/WO2019062435A1/zh unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160176682A1 (en) | 2014-12-19 | 2016-06-23 | Airbus Defence And Space, S.A. | Device for hoisting and controlling loads |
| WO2016103155A1 (en) * | 2014-12-23 | 2016-06-30 | Novartis Ag | Triazolopyrimidine compounds and uses thereof |
Non-Patent Citations (6)
| Title |
|---|
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 1158712-36-7 |
| HUANG, YING ET AL.: "Discovery of First-in-Class, Potent, and Orally Bioavailable Embryonic Ectoderm Development (EED) Inhibitor with Robust Anticancer Efficacy", J. MED. CHEM., vol. 60, 16 January 2017 (2017-01-16), pages 2215 - 2226, XP002772151, DOI: doi:10.1021/acs.jmedchem.6b01576 * |
| J. MED. CHEM., vol. 60, 2017, pages 2215 - 2226 |
| NAT. CHEM. BIOL., vol. 13, 2017, pages 389 - 395 |
| QI, WEI ET AL.: "An Allosteric PRC2 Inhibitor Targeting the H3K27me3 Binding Pocket of EED", NATURE CHEMICAL BIOLOGY, vol. 13, 30 January 2017 (2017-01-30), pages 381 - 388, XP055560226, DOI: doi:10.1038/nchembio.2304 * |
| YANG, CHAO-YIE ET AL.: "Allosteric Inactivation of Polycomb Repressive Complex 2 (PRC2) by Inhibiting Its Adapter Protein: Embryonic Ectodomain Development (EED", J. MED. CHEM., vol. 60, no. 6, 3 March 2017 (2017-03-03), pages 2212 - 2214, XP055587874 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11091495B2 (en) | 2018-01-31 | 2021-08-17 | Mirati Therapeutics, Inc. | Substituted imidazo[1,2-c]pyrimidines as PRC2 inhibitors |
| US11220509B2 (en) | 2018-01-31 | 2022-01-11 | Mirati Therapeutics, Inc. | Substituted imidazo[1,2-c]pyrimidines as PRC2 inhibitors |
| US11485738B2 (en) | 2018-01-31 | 2022-11-01 | Mirati Therapeutics, Inc. | Substituted imidazo[1,2-c]pyrimidines as PRC2 inhibitors |
| JP7541538B2 (ja) | 2019-06-05 | 2024-08-28 | ミラティ セラピューティクス,インク. | 癌を処置するためのprc2阻害剤としてのイミダゾ[1,2-c]ピリミジン誘導体 |
| EP4019521A4 (en) * | 2019-08-22 | 2023-05-03 | Blueray Therapeutics (Shanghai) Co., Ltd | AZAHETEROARYL COMPOUND AND USE |
| CN112409385A (zh) * | 2019-08-22 | 2021-02-26 | 上海青煜医药科技有限公司 | 氮杂芳基化合物及其应用 |
| CN112409385B (zh) * | 2019-08-22 | 2024-08-13 | 上海青煜医药科技有限公司 | 氮杂芳基化合物及其应用 |
| JP7357146B2 (ja) | 2019-08-22 | 2023-10-05 | ブルーレイ セラピューティクス (シャンハイ) カンパニー,リミティド | アザヘテロアリール化合物及びその使用 |
| JP2022547815A (ja) * | 2019-08-22 | 2022-11-16 | ブルーレイ セラピューティクス (シャンハイ) カンパニー,リミティド | アザヘテロアリール化合物及びその使用 |
| AU2020332462B2 (en) * | 2019-08-22 | 2023-08-17 | Shanghai Blueray Biopharma Co., Ltd. | Azaheteroaryl compound and application thereof |
| WO2021032004A1 (zh) * | 2019-08-22 | 2021-02-25 | 上海青煜医药科技有限公司 | 氮杂芳基化合物及其应用 |
| JP2023502889A (ja) * | 2019-11-01 | 2023-01-26 | 上海科技大学 | Eed阻害剤、その製造方法およびその使用 |
| CN114929707A (zh) * | 2019-11-01 | 2022-08-19 | 上海科技大学 | Eed抑制剂及其制备方法和用途 |
| EP4056570A4 (en) * | 2019-11-01 | 2023-12-20 | ShanghaiTech University | EED INHIBITOR AND PRODUCTION METHOD AND USE THEREOF |
| AU2020374041B2 (en) * | 2019-11-01 | 2024-02-08 | Jing Medicine Technology (Shanghai) Ltd. | EED inhibitor, and preparation method therefor and use thereof |
| CN114929707B (zh) * | 2019-11-01 | 2024-04-09 | 上海科技大学 | Eed抑制剂及其制备方法和用途 |
| JP7481689B2 (ja) | 2019-11-01 | 2024-05-13 | 上海科技大学 | Eed阻害剤、その製造方法およびその使用 |
| WO2021083380A1 (zh) * | 2019-11-01 | 2021-05-06 | 上海科技大学 | Eed抑制剂及其制备方法和用途 |
| CN113004233B (zh) * | 2019-12-18 | 2022-12-20 | 南京优氟医药科技有限公司 | 一种用于制备prc2抑制剂的化合物、其制备方法和用途 |
| CN113004233A (zh) * | 2019-12-18 | 2021-06-22 | 南京优氟医药科技有限公司 | 一种用于制备prc2抑制剂的化合物、其制备方法和用途 |
| JP7546780B2 (ja) | 2021-02-10 | 2024-09-06 | シャンハイ ブルーレイ バイオファーマ カンパニー,リミティド | アザヘテロアリール化合物、その調製方法及び使用 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019062435A1 (zh) | 三氮唑并嘧啶、三氮唑并吡啶化合物及其组合物用于治疗prc2介导的疾病 | |
| CN110156787B (zh) | 一种三氮唑并嘧啶衍生化合物、包含其的药物组合物及其用途 | |
| EP3573983B1 (en) | N-[4-fluoro-5-[[(2s,4s)-2-methyl-4-[(5-methyl-1,2,4-oxadiazol-3-yl)methoxy]-1-piperidyl]methyl]thiazol-2-yl]acetamide as oga inhibitor | |
| EP3630755B1 (en) | 5-methyl-1,3,4-oxadiazol-2-yl compounds | |
| CN109843890B (zh) | 三氮唑并嘧啶、三氮唑并吡啶化合物及其组合物用于治疗prc2介导的疾病 | |
| JP6457623B2 (ja) | 2,4−二置換7H−ピロロ[2,3−d]ピリミジン誘導体、その製造方法および医薬における使用 | |
| EP2665706B1 (en) | Novel 4-amino-n-hydroxy-benzamides as hdac inhibitors for the treatment of cancer | |
| ZA200701266B (en) | Method of preparing poly (ADP-ribose) polymerases inhibitors | |
| WO2014134774A1 (en) | Compounds inhibiting leucine-rich repeat kinase enzyme activity | |
| IL305046A (en) | Pyridopyrimidinone derivative, the method of preparation thereof and its use | |
| TW201702226A (zh) | 尿素衍生物或其醫藥上可接受鹽 | |
| EA012295B1 (ru) | Конденсированные пирролокарбазолы | |
| AU2019391279A1 (en) | Heteroaromatic compounds as Vanin inhibitors | |
| JP2021500334A (ja) | Ehmt2阻害剤としてのアミン置換複素環化合物、その塩、及びそれらの合成方法 | |
| WO2022135610A1 (zh) | 四并环化合物及其药物组合物和应用 | |
| MXPA06002347A (es) | Compuestos y composiciones como inhibidores de quinasa de proteina. | |
| CN103833756A (zh) | 一类哒嗪酮类化合物及其制备方法和用途 | |
| WO2018001332A1 (zh) | 具有突变型异柠檬酸脱氢酶抑制活性的化合物、其制备方法及用途 | |
| Wang et al. | Discovery of a new class of valosine containing protein (VCP/P97) inhibitors for the treatment of non-small cell lung cancer | |
| AU2003217373A1 (en) | Novel tyloindicines and related processes, pharmaceutical compositions and methods | |
| CN111205244B (zh) | 噻唑并环类化合物、其制备方法、中间体和应用 | |
| WO2019149128A1 (zh) | 作为抗肿瘤药物的5-氯-2,4-嘧啶衍生物 | |
| CN115806560B (zh) | 氮杂四并环化合物及其药物组合物和应用 | |
| WO2022174803A1 (zh) | 含s构型的氨基苯甲酰胺基哒嗪酮类化合物、其制备方法、药物组合物及应用 | |
| EP4157263A1 (en) | Mitochondrial targeting compounds for the treatment of associated diseases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18860160 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020529171 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018860160 Country of ref document: EP Effective date: 20200428 |