WO2018146641A1 - Novel inhibitors of cellular signalling - Google Patents
Novel inhibitors of cellular signalling Download PDFInfo
- Publication number
- WO2018146641A1 WO2018146641A1 PCT/IB2018/050839 IB2018050839W WO2018146641A1 WO 2018146641 A1 WO2018146641 A1 WO 2018146641A1 IB 2018050839 W IB2018050839 W IB 2018050839W WO 2018146641 A1 WO2018146641 A1 WO 2018146641A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- compound
- cancer
- composition
- alcohol
- Prior art date
Links
- 0 CC(C)C(CC1CCCCC1)=* Chemical compound CC(C)C(CC1CCCCC1)=* 0.000 description 13
- MIYUBMONQAVOHI-UHFFFAOYSA-N CC(C)(CC(C)(C)C(O)=O)CC(N)=O Chemical compound CC(C)(CC(C)(C)C(O)=O)CC(N)=O MIYUBMONQAVOHI-UHFFFAOYSA-N 0.000 description 1
- AYQBNMYQPOEJOR-XVCHOOAUSA-N CC(C)CCC[C@@H](C)[C@@H](CC1)[C@@](C)(CC2)C1C1C2[C@@](C)(CC[C@@H](C2)C(C)C)C2=CC1 Chemical compound CC(C)CCC[C@@H](C)[C@@H](CC1)[C@@](C)(CC2)C1C1C2[C@@](C)(CC[C@@H](C2)C(C)C)C2=CC1 AYQBNMYQPOEJOR-XVCHOOAUSA-N 0.000 description 1
- HVYWMOMLDIMFJA-HVNCNQDNSA-N CC(C)CCC[C@@H](C)[C@@H](CC1)[C@@](C)(CC2)C1C1C2[C@@](C)(CC[C@@H](C2)O)C2=CC1 Chemical compound CC(C)CCC[C@@H](C)[C@@H](CC1)[C@@](C)(CC2)C1C1C2[C@@](C)(CC[C@@H](C2)O)C2=CC1 HVYWMOMLDIMFJA-HVNCNQDNSA-N 0.000 description 1
- ZIWQPHPYLTUOBU-UHFFFAOYSA-N CNC(c1nccc(Oc(cc2)cc3c2nc(SCCNC(c2nccc(Oc(cc4)cc([s]5)c4nc5S(C)=O)c2)=O)[s]3)c1)=O Chemical compound CNC(c1nccc(Oc(cc2)cc3c2nc(SCCNC(c2nccc(Oc(cc4)cc([s]5)c4nc5S(C)=O)c2)=O)[s]3)c1)=O ZIWQPHPYLTUOBU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/554—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being a steroid plant sterol, glycyrrhetic acid, enoxolone or bile acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6905—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion
- A61K47/6907—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion the form being a microemulsion, nanoemulsion or micelle
- A61K47/6909—Micelles formed by phospholipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6905—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion
- A61K47/6911—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion the form being a liposome
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
- A61K9/1075—Microemulsions or submicron emulsions; Preconcentrates or solids thereof; Micelles, e.g. made of phospholipids or block copolymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes, liposomes coated with polymers
Definitions
- the present disclosure relates generally to Cellular Signalling inhibitors, compositions and formulations comprising the same, and uses thereof.
- the present disclosure provides CSF-IR inhibitors of BLZ-945-lipids conjugates, GW2580-lipid conjugates and PLX-3397-lipid conjugates demonstrating sustained inhibition of CSF/CSF-1R signalling pathway with decreased toxicity.
- the present disclosure also provides supramolecular combinatorial therapeutics, wherein the CSF-IR inhibitor is combined with one or more of a chemotherapeutic agent, a kinase inhibitor, and an immunoregulator, each of which is optionally conjugated with a lipid.
- Targeted therapies have been in the limelight as cancer therapeutics for the last few years. They have resulted in high response rates and improved overall survival in patients with cancer. However, consistent with other oncogene-targeted therapies, initial patient response is of limited durability and tumors eventually relapse. 1
- the tumor microenvironment is increasingly recognized to play an important role in tumor proliferation, invasion, metastasis, and chemoresistance. It provides a conducive niche to the tumor through immunosuppression. Overcoming this immunosuppressive nature of the tumor microenvironment has been of particular interest in cancer therapy. Tumor cells manipulate the surrounding environment by producing cytokines that suppress cytolytic T-cells and recruit immunosuppressive cells.
- Colony stimulating factor 1 is one such cytokine secreted by several cancer cell types. It induces the proliferation and differentiation of immunosuppressive myeloid cells such as M2 polarized macrophages and myeloid derived suppressor cells (MDSC) by binding to the CSF-1 receptor (CSF-IR) on cell surface.
- CSF-1 receptor CSF-1 receptor
- 111 Cellular signalling mediated by colony-stimulating factor 1 (CSF1) and its receptor CSF-IR plays a critical role in monocyte differentiation and generation and activity of tissue-resident macrophages. The overexpression of CSF1 is associated with poor prognosis in breast, ovarian, and prostate cancer.
- TAMs tumor-associated macrophages
- CSF1-CSF1R axis may have an important role towards activity of TAMs.
- lv ' v vl The CSF1/CSF1R signalling pathway is targeted in the treatment against numerous malignancies, including breast, leukaemia, and glioblastoma.
- CSF-IR regulates the functioning of macrophages impacting tumor progression
- inhibiting the CSF-IR pathway has emerged as a major therapeutic goal in cancer.
- some of the CSF-IR inhibitors had shown promising results in terms of potency, selectivity and bioavailability of cFMS kinase activity vu
- some of the well-known inhibitors which are in the clinical phase trials are BLZ-945, GW2580, PLX-3397 etc. Although high in potency, these inhibitors fail to achieve a sustained inhibition of CSF-IR and are associated with toxicity to normal cells. Accordingly, there remains an urgent need for CSF-IR inhibitors with an improved activity profile while exhibiting decreased toxicity.
- the present application describes the design of novel CSF-IR inhibitors that can preferentially accumulate in the tumor microenvironment and exert sustained inhibition of the target molecule, overcoming the dose-limiting toxic effects and achieving superior antitumor efficacy compared to currently available CSF-IR inhibitors.
- the present invention is directed to hydrophobic, lipophilic prodrugs that lead to increased effective drug accumulation in tumor via enhanced permeability and retention (EPR) phenomenon leading to improved efficacy.
- the lipids used in these prodrugs include phospholipids, cholesterol, fatty acids etc.
- the present invention describes prodrugs of CSF-IR inhibitors that can assemble into supramolecular structure with improved pharmacokinetic profile such as long circulation time, enhanced uptake and sustained release of drug inside tumor.
- compositions of prodrug of a CSF-IR inhibitor comprise a linker wherein CSF-IR is coupled through ester, ether, amide, or other covalent conjugation with the linker.
- the lipid molecule can be cholesterol, oleic acid, alpha tocopherol, fatty acid or another naturally occurring lipid molecule which is conjugated to drug molecule through a suitable linker/spacer.
- the spacer can be composed of aliphatic dicarboxylic acid, unsaturated dicarboxylic acid, aldaric acid, fumaric acid, propargylic acid, acetylene dicarboxylic acid, aromatic/hetero aromatic dicarboxylic acid, ethylene glycol, diethylene glycol, natural or unnatural amino acids individually or in any combinations.
- Certain exemplary embodiments provide supramolecular combinatorial therapeutics, wherein a CSF-IR inhibitor is combined with one or more of a kinase inhibitor, or a chemotherapeutic drug, or a tumor-targeting antibody, each of which is optionally conjugated with a lipid.
- the antibody can be useful for therapeutic purposes (i.e., a therapeutic antibody) or for targeting the supramolecular combinatorial therapeutic to a desired site (i.e., a targeting antibody).
- the supramolecular combinatorial therapeutic further comprises an immunomodulator, which can include another CSF-IR inhibitor (s). Immunomodulators are active agents of immunotherapy, and can either activate or suppress an immune response.
- the immunomodulator activates and stimulates an immune response against cancer cells, non-limiting examples of which include immune cells (e.g., natural killer cells, lymphokine-activated killer cells, cytotoxic T cells and dendritic cells), antibodies (e.g., anti-PD-Ll and anti-PD-1 antibodies, anti-CD52, anti-VEGF-A, anti- CD30, anti-EGFR, anti- CD33, anti-CD20, anti-CTLA4, and anti-HER-2 antibodies), and cytokines (e.g., interferons and interleukins).
- the immunomodulator is conjugated with a lipid.
- the present disclosure relates to compounds of CSF-IR inhibitors and lipid conjugates of CSF- IR inhibitors of Formula I
- 'Ligand' is selected from the group consisting of:
- E C, O, NH, S;
- G C, O, NH, S;
- 'L' is a linker moiety connecting 'Ligand' and 'R' moieties; and 'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
- CSF-IR inhibitors are the inhibitors which inhibits the CSFI/CSFIR signalling pathway.
- CSF-IR inhibitors include compounds which target, decrease or inhibit the activity of Colony stimulating factor 1 receptor (CSF-IR).
- lipid conjugates of CSF-IR inhibitors BLZ-945-lipid conjugate, GW2580-lipid conjugate and PLX3397-lipid conjugate.
- the present invention is directed to lipid conjugates of CSF-IR inhibitors, such as BLZ-945, GW2580 and PLX3397, and that lipid conjugates of CSF-IR inhibitors.
- CSF-IR inhibitors such as BLZ-945, GW2580 and PLX3397
- the present disclosure relates to compounds of BLZ-945 series/Formula IA (BLZ-945-lipid conjugates), GW2580 series/ Formula IB (GW2580-lipid conjugates), PLX-3397 series/ Formula IC (PLX-3397-lipid conjugates).
- A H, O, NH, S;
- E C, O, NH, S;
- G C, O, NH, S;
- 'L' is a linker moiety connecting 'Ligand' and R' moieties
- 'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
- 'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
- the linker group(s) in the BLZ-945 series (BLZ-945-lipid conjugates), GW2580 series (GW2580-lipid conjugates), PLX-3397 series (PLX-3397-lipid conjugates) of the present disclosure is selected from a group comprising a direct bond or an atom such as oxygen or sulfur, a unit such as R 1 , C(O), C(0)0, C(0) R ⁇ SO, SO2, SO2 H or a chain of atoms, such as substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, heterocycly
- the linker group(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising a direct bond, ester, ether, amide or other covalent conjugation with linker.
- the linker group(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising one or more of succinic acid, fumaric acid, propargylic acid, ethylene glycol, diethylene glycol, and natural or unnatural amino acids.
- the linker group(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising at least one of oxalic acid, malonic acid, succinic acid, glutaric acid, succinic acid, ethylene diamine, natural or unnatural amino acid, ethylene glycol, diethylene glycol, acetic acid, propionic acid, butyric acid, valeric acid, acrylic acid, but-2-enoic acid, pent-2-enoic acid, hex-2-enoic acid, 2-propynoic acid, but-2-ynoic acid, pent-2-ynoic acid, hex-2-ynoic acid, ethylene, propylene, 1-butene, 1-pentene, 1-hexene, acetylene, propyne, but-
- the linker group(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising -C(0)CH 2 CH 2 C(0)-; -C(0)(CH 2 CH 2 ) m (C(0)CH 2 CH 2 )n- wherein 'n' is 1 to 10 and 'm' is 1 to 4; -C(0)(CH 2 ) x CH 2 C(0)NH(CH 2 CH 2 ) n -, wherein 'n' is 1 to 10 and 'x' is 0 to 3; -C(0)(CH 2 )mCH 2 C(0)NH(CH 2 CH 2 ) n HC(0)-, wherein 'n' is 1 to 10 and 'm' is 1 to 4; -C(0)CH 2 (CH 2 ) m C(0) H- wherein 'm' is 1 to 4; -C(0)(CH 2 )nCH 2 (R
- the lipid(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising cholesterol, cholesterol derivatives, oleic acid, oleic acid derivative, alpha tocopherol, alpha tocopherol derivatives, phospholipid, phospholipid derivatives, fatty acid, naturally occurring lipid molecule which is conjugated to drug molecules, 1,3-Propanediol Dicaprylate/Dicaprate, 10-undecenoic acid, 1-dotriacontanol, 1-heptacosanol, 1-nonacosanol,
- the lipid(s) or lipid derivative(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising cholesterol, cholesterol derivatives, oleic acid, oleic acid derivative, alpha tocopherol, alpha tocopherol derivatives, phospholipid, phospholipid derivatives, fatty acid or naturally occurring lipid molecule which is conjugated to drug molecules via a spacer, wherein the spacer is selected from a group comprising aliphatic dicarboxylic acid, unsaturated dicarboxylic acid, aldaric acid, fumaric acid, propargylic acid, acetylene dicarboxylic acid, aromatic/hetero aromatic dicarboxylic acid, ethylene glycol, diethylene glycol, natural or unnatural amino acids, or their derivatives individually or any combinations thereof.
- the lipid conjugate(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates is the lipid or the lipid derivative conjugated with a compound selected from a group comprising a kinase inhibitor, a chemotherapeutic drug, an immunomodulator or an antibody or any combinations thereof.
- the compound of Formula I is BLZ- 945-lipid conjugate(s).
- the compound of Formula I is GW2580-lipid conjugate(s).
- the compound of Formula I is PLX- 3397-lipid conjugate(s).
- the supramolecular combinatorial therapeutic comprises a BLZ- 945-lipid conjugate.
- the supramolecular combinatorial therapeutic comprises a GW2580-lipid conjugate.
- the supramolecular combinatorial therapeutic comprises a PLX- 3397-lipid conjugate.
- the supramolecular combinatorial therapeutic can further comprise a lipid conjugated kinase inhibitor.
- the supramolecular combinatorial therapeutic further comprises an antibody (or an antigen binding fragment thereof), optionally conjugated with a lipid.
- the supramolecular combinatorial therapeutic further comprises an antibody (or an antigen binding fragment thereof) conjugated with a lipid.
- the antibody can be useful for thereaputic purposes (i.e., a therapeutic antibody) or for targeting the the supramolecular combinatorial therapeutic to a desired site (i.e., a targeting antibody).
- the supramolecular combinatorial therapeutic further comprises an immunomodulator. Immunomodulators are active agents of immunotherapy, and can either activate or suppress an immune response.
- the immunomodulator activates and stimulates an immune response against cancer cells, non-limiting examples of which include immune cells (e.g., natural killer cells, lymphokine-activated killer cells, cytotoxic T cells and dendritic cells), antibodies (e.g., anti-PD-Ll and anti-PD-1 antibodies, anti-CD52, anti-VEGF-A, anti- CD30, anti-EGFR, anti-CD33, anti-CD20, anti-CTLA4, and anti-HER-2 antibodies), and cytokines (e.g., interferons and interleukins).
- the immunomodulator is conjugated with a lipid.
- described herein is a method of treating cancer, comprising, administering a supramolecular combinatorial therapeutic as described herein to a patient in need of treatment for cancer.
- the cancer is selected from the group consisting of: breast cancer; ovarian cancer; glioma; gastrointestinal cancer; prostate cancer; carcinoma, lung carcinoma, hepatocellular carcinoma, testicular cancer; cervical cancer; endometrial cancer; bladder cancer; head and neck cancer; lung cancer; gastro-esophageal cancer, and gynecological cancer.
- the method further comprises co-administering one or more additional anti-cancer therapy to the patient.
- the additional therapy is selected from the group consisting of surgery, chemotherapy, radiation therapy, thermotherapy, immunotherapy, hormone therapy, laser therapy, anti-angiogenic therapy, and any combinations thereof.
- the additional therapy comprises administering an anti-cancer agent to the patient.
- the method further comprises coadministration of one or more immunomodulators to the subject.
- described herein is a method of treating allergy, comprising, administering a supramolecular combinatorial therapeutic as described herein to a patient in need of treatment for allergy, Systemic lupus erythematosus, Chronic Obstructive Pulmonary Disease and abnormal macrophage functions.
- a composition comprises a compound of BLZ-945-lipid conjugate along with pharmaceutically acceptable excipient.
- the composition comprises a compound of GW2580-lipid conjugate along with pharmaceutically acceptable excipient. In a non-limiting embodiment of the present disclosure, the composition comprises a compound of PLX-3397-lipid conjugate along with pharmaceutically acceptable excipient.
- the pharmaceutically acceptable excipient is selected from the group comprising adjuvant, diluent, carrier, granulating agents, binding agents, lubricating agents, disintegrating agent, sweetening agents, glidant, anti- adherent, anti-static agent, surfactant, anti-oxidant, gum, coating agent, coloring agent, flavouring agent, coating agent, plasticizer, preservative, suspending agent, emulsifying agent, plant cellulosic material, spheronization agent, and other conventionally known pharmaceutically acceptable excipient, or any combination of excipients thereof.
- the composition inhibits colony stimulating factor-1 receptor (CSF-1R) and is administered to a subject in need thereof through modes selected from a group comprising intravenous administration, intramuscular administration, intraperitoneal administration, hepatoportal administration, intra articular administration and pancreatic duodenal artery administration, or any combination thereof.
- CSF-1R colony stimulating factor-1 receptor
- the present disclosure provides a method of treating cancer, comprising, administering the compounds of BLZ-945-lipid conjugate or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition to a subject in need of treatment for cancer.
- the present disclosure provides a method of treating cancer, comprising, administering the compounds of GW2580-lipid conjugate or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition to a subject in need of treatment for cancer.
- the present disclosure provides a method of treating cancer, comprising, administering the compounds of PLX-3397-lipid conjugate or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition to a subject in need of treatment for cancer.
- the cancer is selected from the group consisting of breast cancer; ovarian cancer; glioma; gastrointestinal cancer; prostate cancer; carcinoma, lung carcinoma, hepatocellular carcinoma, testicular cancer; cervical cancer; endometrial cancer; bladder cancer; head and neck cancer; lung cancer; gastro-esophageal cancer, and gynecological cancer, or any combination thereof.
- the method comprises coadministering one or more additional anti-cancer therapy to the subject.
- the additional anti-cancer therapy is selected from the group consisting of surgery, chemotherapy, radiation therapy, thermotherapy, immunotherapy, hormone therapy, laser therapy, and anti-angiogenic therapy, or any combination thereof.
- the additional anti-cancer therapy comprises administering a kinase inhibitor, a chemotherapeutic agent, an immunomodulator or any combination thereof, to the subject.
- the supramolecular combinatorial therapeutic comprises a BLZ-945- lipid conjugate and a platinum-lipid conjugate.
- the supramolecular combinatorial therapeutic comprises a BLZ-945- lipid conjugate and an antibody (or an antigen binding fragment thereof) lipid conjugate.
- the antibody, or the antigen binding fragment thereof can be a therapeutic agent or a targeting ligand.
- the supramolecular combinatorial therapeutic comprises a BLZ-945- lipid conjugate and an antibody (or an antigen binding fragment thereof) lipid conjugate, wherein the antibody is a therapeutic antibody or a targeting antibody or a combination thereof.
- BLZ-945-lipid conjugate is compound of formula 23.
- BLZ-945-lipid conjugate is compound of formula 24.
- 'n' is 1 to 10 and 'x' is 0 to 3.
- BLZ-945-lipid conj compound of formula 25 In yet another non-limiting embodiment of the present disclosure, BLZ-945-lipid conj compound of formula 25.
- 'n' is 1 to 10 and 'm' is 1 to 4.
- BLZ-945-lipid conjugate is compound of formula 26.
- BLZ-945-lipid conjugate is compound of formula 27.
- 'n' is 1 to 10;
- R is H, alkyl, acid, amine, aryl or thiol;
- Y is C, O, NH, S; and
- X is C, O, NH, S.
- the present disclosure further provides a BLZ-945 - lipid conjugate compound selected from:
- GW-2580 is a potent and selective inhibitor of cFMS receptor kinase, inhibits cFMS in vitro at 60 nM with no activity against 26 other kinases. lx It also completely inhibits CSF-1 induced growth of mouse M- FS-60 myeloid cells and human monocytes (at 1 ⁇ ). GW-2580 completely inhibits bone degradation in human osteoclasts, rat calvaria and fetal long bone. x
- GW-2580-lipid conjugate is compound of formula .
- 'n' is 1 to 10 and 'm' is 1 to 4.
- GW-2580-lipid conj compound of formula 55 In another non-limiting embodiment of the present disclosure, GW-2580-lipid conj compound of formula 55.
- 'n' is 1 to 10 and 'x' is 0 to 3.
- GW-2580-lipid conj compound of formula 56 In yet another non-limiting embodiment of the present disclosure, GW-2580-lipid conj compound of formula 56.
- 'n' is 1 to 10 and 'm' is 1 to 4.
- GW-2580-lipid conjugate is compound of formula 57.
- GW-2580-lipid conjugate is compound of formula 58.
- 'n' is 1 to 10; R is H, alkyl, acid, amine, aryl or thiol; Y is C, O, H, S; and X is C, O, NH, S.
- PLX3397 is an orally administered a small-molecule receptor tyrosine kinase (RTK) inhibitor of KIT, CSF-1R and FLT3, with anti-canecr potential.
- RTK receptor tyrosine kinase
- the FDA recently approved this molecule for patients with tenosynovial giant cell tumor (TGCT) for which surgical removal is contraindicated. It binds to and inhibits phosphorylation of stem cell factor receptor (KIT), colony-stimulating factor-1 receptor (CSFIR) and FMS-like tyrosine kinase 3 (FLT3), inhibiting tumor cell proliferation, down-modulation of macrophages, osteoclasts and mast cells involved in the metastatic disease. CSFIR and FLT3 are overexpressed or mutated in many cancer cell types and play major roles in tumor cell proliferation and metastasis 541
- the present disclosure further provides a PLX-3397- lipid conjugate compound selected from:
- 'n' is 1 to 10 and 'x' is 0 to 3;
- 'n' is 1 to 10;
- R is H, alkyl, acid, amine, aryl or thiol;
- Y is C, O, NH or S; and
- X is C, O, NH or S.
- composition(s) comprising the compound of Formula I
- 'Ligand' is selected from the group consisting of:
- A H, O, NH, S;
- E C, O, NH, S;
- G C, O, NH, S;
- 'L' is a linker moiety connecting 'Ligand' and 'R' moieties; and 'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
- the composition comprises from about 1% to about 99% (w/w) of the compound of Formula I.
- the composition further comprises a kinase inhibitor, a chemotherapeutic agent or an immunomodulator or any combination thereof.
- the composition further comprises a co-lipid.
- the co-lipid is selected from the group consisting of HSPC, DSPC, DPPC, DOPC, POPC, SOPC, Egg- PC, and DSPE-PEG, DPPE-PEG, DMPE-PEG or any combination thereof.
- the composition comprises from about 1% to about 99% (w/w) of the kinase inhibitor.
- the composition comprises from about 1%) to about 99% (w/w) of the chemotherapeutic agent.
- the chemotherapeutic agent is selected from the group consisting of PI3K inhibitors; platinum compounds; inhibitors of topoisomerase I and II; alkylating agents; microtubule inhibitors; and angiogenesis inhibitors; or any combination thereof.
- the chemotherapeutic agent is selected from the group consisting of germicitibine; Aldesleukin; Alemtuzumab; alitretinoin; allopurinol; altretamine; amifostine; anastrozole; arsenic trioxide; Asparaginase; BCG Live; bexarotene capsules; bexarotene gel; bleomycin; busulfan intravenous; busulfanoral; calusterone; capecitabine; platinate; carmustine; carmustine with Polifeprosan Implant; celecoxib; chlorambucil; cladribine; cyclophosphamide; cytarabine; cytarabine liposomal; dacarbazine; dactinomycin; actinomycin D; Darbepoetin alfa; daunorubicin liposomal; daunorubicin, daunomycin; Den
- VM-26 testolactone
- thioguanine (6-TG) thiotepa
- topotecan toremifene
- Tositumomab Tositumomab
- trastuzumab tretinoin (ATRA); Uracil Mustard; valrubicin; valtorcitabine (monoval LDC); vinblastine; vinorelbine; and zoledronate; or any mixture thereof
- the PI3K inhibitor is selected from the group consisting of PI103; P1828; LY294002; wortmannin; demethoxyviridin; IC486068; IC87114; GDC-0941; perifosine; CALIOI; PX-866; IPI-145;
- the chemotherapeutic agent is conjugated with a component of the composition.
- the chemotherapeutic agent is conjugated with Polyethylene glycol (PEG).
- PEG Polyethylene glycol
- the composition comprises from about 1% to about 99% (w/w) of the immunomodulator.
- the composition is a liposome, emulsion, or micelle.
- the composition is a nanoparticle, wherein the nanoparticle is about 1 nm to about 400 nm in diameter.
- the composition further comprises a pharmaceutically acceptable carrier.
- the composition further comprises a pharmaceutically acceptable excipient.
- the pharmaceutically acceptable excipient is selected from the group comprising adjuvant, diluent, carrier, granulating agents, binding agents, lubricating agents, disintegrating agent, sweetening agents, glidant, anti- adherent, anti-static agent, surfactant, anti-oxidant, gum, coating agent, coloring agent, flavouring agent, coating agent, plasticizer, preservative, suspending agent, emulsifying agent, plant cellulosic material, spheronization agent, and other conventionally known pharmaceutically acceptable excipient, or any combination of excipients thereof.
- the composition inhibits colony stimulating factor or colony stimulating factor-1 receptor (CSF-1R) signaling pathway and is administered to a subject in need thereof through modes selected from a group comprising intravenous administration, intramuscular administration, intraperitoneal administration, hepatoportal administration, intra articular administration and pancreatic duodenal artery administration, or any combination thereof.
- CSF-1R colony stimulating factor-1 receptor
- the present disclosure also provides a method of treating cancer, comprising, administering the compound of Formula I or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition thereof to a subject in need of treatment for cancer.
- the cancer is selected from the group consisting of breast cancer; ovarian cancer; glioma; gastrointestinal cancer; prostate cancer; carcinoma, lung carcinoma, hepatocellular carcinoma, testicular cancer; cervical cancer; endometrial cancer; bladder cancer; head and neck cancer; lung cancer; gastro-esophageal cancer, and gynecological cancer, or any combination thereof.
- the method comprises coadministering one or more additional anti-cancer therapy to the subject.
- the additional anti-cancer therapy is selected from the group consisting of surgery, chemotherapy, radiation therapy, thermotherapy, immunotherapy, hormone therapy, laser therapy, and anti-angiogenic therapy, or any combination thereof.
- the additional anti-cancer therapy comprises administering a kinase inhibitor, a chemotherapeutic agent, an immunomodulator or any combination thereof, to the subject.
- the immunomodulator activates an immune response against cancer cells, wherein the immunomodulator is selected from the group consisting of antibody, natural killer cells, lymphokine-activated killer cells, cytotoxic T cells and dendritic cells, anti-PD-Ll antibodies, anti-PD-1 antibodies, anti-CD52 antibodies, anti-VEGF-A antibodies, anti-CD30 antibodies, anti-EGFR antibodies, anti-CD33 antibodies, anti-CD20 antibodies, anti-CTLA4 antibodies, anti-HER-2 antibodies, interferons, and interleukins, or any combination thereof.
- the antibody is a therapeutic antibody or a targeting antibody or a combination thereof.
- the present disclosure also provides a method for inhibition of CSF or CSF-1R signalling pathway in a cell, wherein said method comprises act of contacting the cell with the compound of formula I, derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition thereof.
- the technology described herein relates to a pharmaceutical composition
- a pharmaceutical composition comprising a supramolecular combinatorial therapeutic and a pharmaceutically acceptable carrier.
- Pharmaceutically acceptable carriers and diluents include saline, aqueous buffer solutions, solvents and/or dispersion media. The use of such carriers and diluents is well known in the art.
- materials which can serve as pharmaceutically- acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, methylcellulose, ethyl cellulose, microcrystalline cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) lubricating agents, such as magnesium stearate, sodium lauryl sulfate and talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol (PEG); (12) esters, such as ethylene glycol;
- wetting agents, coloring agents, release agents, coating agents, sweetening agents, flavoring agents, perfuming agents, preservative and antioxidants can also be present in the formulation.
- the terms such as “excipient”, “carrier”, “pharmaceutically acceptable carrier” or the like are used interchangeably herein.
- the carrier inhibits the degradation of the active agent, e.g. a composition as described herein.
- the pharmaceutical composition comprising a supramolecular combinatorial therapeutic can be a parenteral dose form. Since administration of parenteral dosage forms typically bypasses the patient's natural defenses against contaminants, parenteral dosage forms are preferably sterile or capable of being sterilized prior to administration to a patient. Examples of parenteral dosage forms include, but are not limited to, solutions ready for injection, dry products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection, suspensions ready for injection, and emulsions. In addition, controlled- release parenteral dosage forms can be prepared for administration of a patient, including, but not limited to, DUROS ® -type dosage forms and dose-dumping.
- Suitable vehicles that can be used to provide parenteral dosage forms of a composition as described herein are well known to those skilled in the art. Examples include, without limitation: sterile water; water for injection USP; saline solution; glucose solution; aqueous vehicles such as but not limited to, sodium chloride injection, Ringer's injection, dextrose Injection, dextrose and sodium chloride injection, and lactated Ringer's injection; water- miscible vehicles such as, but not limited to, ethyl alcohol, polyethylene glycol, and propylene glycol; and non-aqueous vehicles such as, but not limited to, corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate.
- Compounds that alter or modify the solubility of a pharmaceutically acceptable salt can also be incorporated into the parenteral dosage forms of the disclosure, including conventional and controlled-release parenteral dosage forms.
- compositions can also be formulated to be suitable for oral administration, for example as discrete dosage forms, such as, but not limited to, tablets (including without limitation scored or coated tablets), pills, caplets, capsules, chewable tablets, powder packets, cachets, troches, wafers, aerosol sprays, or liquids, such as but not limited to, syrups, elixirs, solutions or suspensions in an aqueous liquid, a non-aqueous liquid, an oil-in-water emulsion, or a water-in-oil emulsion.
- Such compositions contain a predetermined amount of the pharmaceutically acceptable salt of the disclosed compounds, and may be prepared by methods of pharmacy well known to those skilled in the art. See generally, Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams, and Wilkins, Philadelphia PA. (2005).
- the present disclosure further provides a formulation comprising the compound of Formula I
- A H, O, H, S;
- E C, O, NH, S;
- G C, O, NH, S;
- 'L' is a linker moiety connecting 'Ligand' and 'R' moieties
- 'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
- the phospholipid or PEGylated phospholipid is selected from a group comprising HSPC, DSPC, DPPC, DOPC, POPC, SOPC, Egg PC , DPPE-PEG, DMPE-PEG and DSPE-PEG or any combination thereof; wherein the composition comprises about 1% to about 99% (w/w) of these phospholipid and PEGylated phospholipid or any combination thereof.
- the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 5 :55 :35 :5.
- the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 10:50:35 :5.
- the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 15 :50:30:5.
- compositions and methods described herein can be administered to a subject having or diagnosed as having cancer.
- the methods described herein comprise administering an effective amount of compositions described herein to a subject in order to alleviate a symptom of a cancer.
- "alleviating a symptom of a cancer” is ameliorating any condition or symptom associated with the cancer. As compared with an equivalent untreated control, such reduction is by at least 5%, 10%, 20%, 40%, 50%, 60%, 80%), 90%), 95%), 99% or more as measured by any standard technique.
- a variety of means for administering the compositions described herein to subjects are known to those of skill in the art.
- Such methods can include, but are not limited to oral, parenteral, intravenous, intramuscular, subcutaneous, transdermal, airway (aerosol), pulmonary, cutaneous, topical, injection, or intratumoral administration. Administration can be local or systemic.
- an effective amount refers to the amount of a composition described herein needed to alleviate at least one or more symptom of the disease or disorder, and relates to a sufficient amount of pharmacological composition to provide the desired effect.
- the term "therapeutically effective amount” therefore refers to an amount of a composition described herein that is sufficient to provide a particular anti-tumor effect when administered to a typical subject.
- An effective amount as used herein, in various contexts, would also include an amount sufficient to delay the development of a symptom of the disease, alter the course of a symptom disease (for example but not limited to, slowing the progression of a symptom of the disease), or reverse a symptom of the disease. Thus, it is not generally practicable to specify an exact "effective amount”. However, for any given case, an appropriate "effective amount” can be determined by one of ordinary skill in the art using only routine experimentation.
- an effective dose of a composition as described herein can be administered to a patient once. In certain embodiments, an effective dose of a composition as described herein can be administered to a patient repeatedly.
- subjects can be administered a therapeutic amount of a composition as described herein, such as, e.g. 0.1 mg/kg, 0.5 mg/kg, 1.0 mg/kg, 2.0 mg/kg, 2.5 mg/kg, 5 mg/kg, 10 mg/kg, 15 mg/kg, 20 mg/kg, 25 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, or more.
- the treatments after an initial treatment regimen, the treatments can be administered on a less frequent basis.
- treatment can be repeated once per month, for six months or a year or longer.
- Treatment according to the methods described herein can reduce levels of a marker or symptom of a condition, e.g. tumor size and/or growth by at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80 % or at least 90% or more.
- the dosage of a composition as described herein can be determined by a physician and adjusted, as necessary, to suit observed effects of the treatment. With respect to duration and frequency of treatment, it is typical for skilled clinicians to monitor subjects in order to determine when the treatment is providing therapeutic benefit, and to determine whether to increase or decrease dosage, increase or decrease administration frequency, discontinue treatment, resume treatment, or make other alterations to the treatment regimen.
- the dosing schedule can vary from once a week to daily depending on a number of clinical factors, such as the subject's sensitivity a composition as described herein.
- the desired dose or amount of activation can be administered at one time or divided into subdoses, e.g., 2-4 subdoses and administered over a period of time, e.g., at appropriate intervals through the day or other appropriate schedule.
- administration can be chronic, e.g., one or more doses and/or treatments daily over a period of weeks or months.
- dosing and/or treatment schedules are administration daily, twice daily, three times daily or four or more times daily over a period of 1 week, 2 weeks, 3 weeks, 4 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, or 6 months, or more.
- a composition as described herein can be administered over a period of time, such as over a 5 minute, 10 minute, 15 minute, 20 minute, or 25 minute period.
- the dosage ranges for the administration of a composition as described herein, according to the methods described herein depend upon, for example, the form of a composition as described herein, its potency, and the extent to which symptoms, markers, or indicators of a condition described herein are desired to be reduced, for example the percentage reduction desired for tumor growth.
- the dosage should not be so large as to cause adverse side effects.
- the dosage will vary with the age, condition, and sex of the patient and can be determined by one of skill in the art.
- the dosage can also be adjusted by the individual physician in the event of any complication.
- Efficacy of a composition as described herein in, e.g. the treatment of a condition described herein, or to induce a response as described herein can be determined by the skilled clinician. However, a treatment is considered "effective treatment," as the term is used herein, if one or more of the signs or symptoms of a condition described herein are altered in a beneficial manner, other clinically accepted symptoms are improved, or even ameliorated, or a desired response is induced e.g., by at least 10% following treatment according to the methods described herein. Efficacy can be assessed, for example, by measuring a marker, indicator, symptom, and/or the incidence of a condition treated according to the methods described herein or any other measurable parameter appropriate, e.g. tumor size and/or growth.
- Treatment includes any treatment of a disease in an individual or an animal (some non-limiting examples include a human or an animal) and includes: (1) inhibiting the disease, e.g., preventing a worsening of symptoms (e.g. pain or inflammation); or (2) relieving the severity of the disease, e.g., causing regression of symptoms.
- An effective amount for the treatment of a disease means that amount which, when administered to a subject in need thereof, is sufficient to result in effective treatment as that term is defined herein, for that disease.
- Efficacy of an agent can be determined by assessing physical indicators of a condition or desired response, (e.g. tumor size and/or growth). It is well within the ability of one skilled in the art to monitor efficacy of administration and/or treatment by measuring any one of such parameters, or any combination of parameters. Efficacy can be assessed in animal models of a condition described herein, for example treatment of cancer. When using an experimental animal model, efficacy of treatment is evidenced when a statistically significant change in a marker is observed, e.g. a decreased in tumor size and/or growth.
- a "subject” means a human or animal. Usually the animal is a vertebrate such as a primate, rodent, domestic animal or game animal. Primates include chimpanzees, cynomologous monkeys, spider monkeys, and macaques, e.g., Rhesus. Rodents include mice, rats, woodchucks, ferrets, rabbits and hamsters.
- Domestic and game animals include cows, horses, pigs, deer, bison, buffalo, feline species, e.g., domestic cat, canine species, e.g., dog, fox, wolf, avian species, e.g., chicken, emu, ostrich, and fish, e.g., trout, catfish and salmon.
- the subject is a mammal, e.g., a primate, e.g., a human.
- the terms, "individual,” “patient” and “subject” are used interchangeably herein.
- the subject is a mammal.
- the mammal can be a human, non-human primate, mouse, rat, dog, cat, horse, or cow, but is not limited to these examples. Mammals other than humans can be advantageously used as subjects that represent animal models of cancer.
- a subject can be male or female.
- a subject can be one who has been previously diagnosed with or identified as suffering from or having a condition in need of treatment (e.g. cancer) or one or more complications related to such a condition, and optionally, have already undergone treatment for cancer or the one or more complications related to cancer.
- a subject can also be one who has not been previously diagnosed as having cancer or one or more complications related to cancer.
- a subject can be one who exhibits one or more risk factors for cancer or one or more complications related to cancer or a subject who does not exhibit risk factors.
- a "subject in need" of treatment for a particular condition can be a subject having that condition, diagnosed as having that condition, or at risk of developing that condition.
- agent refers generally to any entity which is normally not present or not present at the levels being administered to a cell, tissue or subject.
- An agent can be selected from a group including but not limited to: polynucleotides; polypeptides; small molecules; and antibodies or antigen-binding fragments thereof.
- a polynucleotide can be RNA or DNA, and can be single or double stranded, and can be selected from a group including, for example, nucleic acids and nucleic acid analogues that encode a polypeptide.
- a polypeptide can be, but is not limited to, a naturally-occurring polypeptide, a mutated polypeptide or a fragment thereof that retains the function of interest.
- agents include, but are not limited to a nucleic acid aptamer, peptide-nucleic acid (PNA), locked nucleic acid (LNA), small organic or inorganic molecules; saccharide; oligosaccharides; polysaccharides; biological macromolecules, peptidomimetics; nucleic acid analogs and derivatives; extracts made from biological materials such as bacteria, plants, fungi, or mammalian cells or tissues and naturally occurring or synthetic compositions.
- PNA peptide-nucleic acid
- LNA locked nucleic acid
- An agent can be applied to the media, where it contacts the cell and induces its effects.
- an agent can be intracellular as a result of introduction of a nucleic acid sequence encoding the agent into the cell and its transcription resulting in the production of the nucleic acid and/or protein environmental stimuli within the cell.
- the agent is any chemical, entity or moiety, including without limitation synthetic and naturally-occurring non-proteinaceous entities.
- the agent is a small molecule having a chemical moiety selected, for example, from unsubstituted or substituted alkyl, aromatic, or heterocyclyl moieties including macrolides, leptomycins and related natural products or analogues thereof. Agents can be known to have a desired activity and/or property, or can be selected from a library of diverse compounds.
- small molecule can refer to compounds that are "natural product-like,” however, the term “small molecule” is not limited to "natural product-like” compounds. Rather, a small molecule is typically characterized in that it contains several carbon— carbon bonds, and has a molecular weight more than about 50, but less than about 5000 Daltons (5 kD). Preferably the small molecule has a molecular weight of less than 3 kD, still more preferably less than 2 kD, and most preferably less than 1 kD. In some cases it is preferred that a small molecule have a molecular mass equal to or less than 700 Daltons.
- the term "inhibitor” refers to an agent which can decrease the expression and/or activity of the targeted expression product (e.g. mRNA encoding the target or a target polypeptide), e.g. by at least 10% or more, e.g. by 10% or more, 50% or more, 70% or more, 80% or more, 90% or more, 95% or more, or 98 % or more.
- the efficacy of an inhibitor of, for example, PI3K e.g. its ability to decrease the level and/or activity of PI3K can be determined, e.g.
- PI3K polypeptide by measuring the level of a PI3K polypeptide (and/or mRNA encoding such a polypeptide) and/or the activity of PI3K.
- Methods for measuring the level of a given mRNA and/or polypeptide are known to one of skill in the art, e.g. RTPCR with primers can be used to determine the level of RNA and Western blotting with an antibody can be used to determine the level of a polypeptide.
- the activity of, e.g. PI3K can be determined using methods known in the art and described above herein. .
- the terms “treat,” “treatment,” “treating,” or “amelioration” refer to therapeutic treatments, wherein the object is to reverse, alleviate, ameliorate, inhibit, slow down or stop the progression or severity of a condition associated with a disease or disorder, e.g. cancer.
- the term “treating” includes reducing or alleviating at least one adverse effect or symptom of a condition, disease or disorder associated with a cancer.
- Treatment is generally “effective” if one or more symptoms or clinical markers are reduced. Alternatively, treatment is “effective” if the progression of a disease is reduced or halted.
- treatment includes not just the improvement of symptoms or markers, but also a cessation of, or at least slowing of, progress or worsening of symptoms compared to what would be expected in the absence of treatment.
- Beneficial or desired clinical results include, but are not limited to, alleviation of one or more symptom(s), diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, remission (whether partial or total), and/or decreased mortality, whether detectable or undetectable.
- treatment also includes providing relief from the symptoms or side-effects of the disease (including palliative treatment).
- composition or “pharmaceutical composition” are used interchangeably and refers to the active agent in combination with a pharmaceutically acceptable carrier e.g. a carrier commonly used in the pharmaceutical industry.
- a pharmaceutically acceptable carrier e.g. a carrier commonly used in the pharmaceutical industry.
- pharmaceutically acceptable is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- administering refers to the placement of a compound as disclosed herein into a subject by a method or route which results in at least partial delivery of the agent at a desired site.
- Pharmaceutical compositions comprising the compounds disclosed herein can be administered by any appropriate route which results in an effective treatment in the subject.
- amphiphilic refers to a molecule that has both a hydrophobic portion and a lipophobic portion, i.e. at least one a polar, water-soluble group and at least one a nonpolar, water- insoluble group.
- an amphiphilic molecule will partition to the interface of the two phases.
- an amphiphile is a molecule that is soluble in both an aqueous environment and a non-aqueous environment.
- the term "amphiphile" refers to an amphiphilic molecule.
- compositions, methods, and respective component(s) thereof are used in reference to compositions, methods, and respective component(s) thereof, that are essential to the method or composition, yet open to the inclusion of unspecified elements, whether essential or not.
- the amine salt (16- 20) thus produced was treated with freshly prepared sulphoxide derivative (Scheme 7) using diisopropyl ethyl amine in N-methyl morpholine at elevated temperature for 3 days.TM The compound formation was monitored by using TLC method. (Scheme 8) The crude reaction mixture was then evaporated and then purified by column chromatography to get the final compound.
- R alkyl groups, acids, amines, aromatic, thiols
- R cholesterol acid, saturated fatty acid, unsaturated fatty acid, branched unsaturated fatty acid.
- R alkyl groups, acids, amines, aromatic, thiols
- R alkyl groups, acids, amines, aromatic, thiols
- R alkyl groups, acids, amines, aromatic, thiols
- the PLX-3397 series of compound was also prepared in two steps starting from the same acids 4, 8 11, 14 and 15. Initially it was treated with N-Boc protected 5-((5-chloro-lH-pyrrolo[2,3- b]pyridin-3-yl)methyl)pyridin-2-amine to form the coupled at the indole -NH position. Then the N-Boc was deprotected using the TFA condition followed by reductive amination with 6- (trifluoromethyl)nicotinaldehyde to get the final compound.
- Scheme 19 Scheme 19
- R cholesterol acid, saturated fatty acid, unsaturated fatty acid, branched unsaturated fatty acid.
- R alkyl groups, acids, amines, aromatic, thiols
- R alkyl groups, acids, amines, aromatic, thiols
- lipid conjugates of CSF1R inhibitor lipid conjugates are prepared using the process described above for BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates.
- IO-801_01 3 ⁇ 4 NMR (500 MHz, CDCh) ⁇ 8.45-8.31 (d, 1H), 8.12-7.95 (m, 1H), 7.77-7.66 (m 1H), 7.63-7.51 (d, 1H), 7.36-7.30 (d, 1H), 7.13-7.02 (m, 1H), 6.98-6.90 (m, 1H), 5.38-5.34 (m, 1H), 4.89-4.79 (m, 1H), 4.24-4.17 (t, 2H), 3.74-3.60 (m, 3H), 3.26-3.14 (m, 1H), 3.05-2.98 (d, 3H), 2.69-2.49 (m, 4H), 2.40-2.30 (m, 2H), 2.27-1.77 (m, 12H), 1.60-1.41 (m, 10H), 1.40- 1.31 (m, 4H), 1.27-1.26 (m, 2H), 1.20-1.03(m, 7H), 1.01 (s, 3H), 0.94-0.91 (
- IO-801_02 ⁇ NMR (500 MHz, CDCh) ⁇ 8.50-8.38 (d, 1H), 8.10-8.01 (m, 1H), 7.73-7.67 (m 1H), 7.41-7.33 (m, 1H), 7.22-7.15 (m,lH), 7.03-6.95 (m, 1H), 5.39-5.33 (m, 1H), 4.91-4.81 (m, 1H), 3.62-3.52 (t, 2H), 3.44-3.33 (m, 2H), 3.23-3.11 (m, 1H), 3.06-2.98 (d, 3H), 2.67-2.55 (m, 2H), 2.54-2.45 (m, 2H), 2.39-2.28 (m, 2H), 2.25-2.12 (m, 3H), 2.10-1.93 (m, 3H), 1.92- 1.79 (m, 5H), 1.60-1.39 (m, 12H), 1.35 (m, 4H), 1.21-1.03 (m, 10H), 1.01 (s, 3H
- IO-801_03 3 ⁇ 4 NMR (500 MHz, CDCh) ⁇ 8.47-8.37 (d, 1H), 8.11-8.01 (m, 1H), 7.83-7.60 (m
- IO-803_01 3 ⁇ 4 MR (500 MHz, CDCb) ⁇ 8.49-8.37 (d, 1H), 8.10-7.97 (m, 1H), 7.81-7.62 (m 2H), 7.64-7.52 (d, 1H), 7.35(s, 1H), 7.19-7.10 (m, 1H), 7.05-6.92 (m, 1H), 6.01-5.77 (m, 1H), 5.38-5.28 (m, 1H), 4.95-4.78 (m, 1H), 3.87-3.43 (m, 2H), 2.63-2.54 (m, 2H), 2.50-2.33 (m, 4H), 2.31-2.24 (m, 1H), 2.21-2.06 (m, 2H), 2.04-1.91 (m, 2H), 1.90-1.80 (m, 5H), 1.65-1.31 (m, 15H), 1.29-1.25 (m, 2H), 1.21-1.03 (m, 8H), 1.00 (s, 3H), 0.95-0.91 (d, 3
- IO-806_01 3 ⁇ 4 NMR (500 MHz, CDCb) ⁇ 8.45-8.33 (d, 1H), 8.09-7.96 (m, 1H), 7.76-7.66 (m 1H), 7.65-7.55(m, 1H), 7.36-7.31 (m, 1H), 7.09-7.05 (m, lH), 6.98-6.92 (m, 1H), 5.38-5.32 (m, 1H), 4.89-4.76 (m, 1H), 4.24-4.17 (m, 2H), 3.85-3.75 (m,lH), 3.71-3.60 (m, 14H) , 3.24- 3.11 (m, 1H), 3.05-2.94 (d, 3H), 2.70-2.49 (m, 4H), 2.41-2.32 (m, 2H), 2.25-2.16 (m, 1H), 2.15-2.07 (m, 1H), 2.05-1.74 (m, 7H), 1.68-1.40(m, 11H), 1.38-1.31 (m, 4H
- IO-806_02 3 ⁇ 4 NMR (500 MHz, CDCb) ⁇ 8.44-8.36 (d, 1H), 8.09-7.96 (m, 1H), 7.75-7.56 (m, 2H), 7.37-7.30 (m, 1H), 7.10-7.03 (m, 1H), 7.00-6.93 (m, 1H), 5.42-5.21 (m, 2H), 4.87- 4.74 (m, 1H), 4.52-4.38 (m, 1H), 3.91-3.73 (m, 1H), 3.49-3.24 (m, 2H), 3.07-2.94 (d, 3H), 2.52-2.42 (m, 2H), 2.41-2.30 (m, 2H), 2.27-2.10 (m, 2H), 2.04-1.90 (m, 2H), 1.88-1.78 (m, 5H), 1.60-1.41 (m, 10H), 1.38-1.32 (m, 3H), 1.31-1.21 (m, 6H), 1.19-1.05 (m, 7H), 0.99
- IO-806_03 3 ⁇ 4NMR (500 MHz, CDCh) ⁇ 8.44-8.34 (d, 1H), 8.10-7.95 (m, 1H), 7.75-7.66 (d, 1H), 7.64-7.52 (d, 1H), 7.39-7.30 (m, 1H), 7.13-7.02 (m, 1H), 6.99-6.91 (m, 1H), 5.41-5.27 (m, 1H), 5.00-4.89 (m, 1H), 4.16-4.01 (m, 2H), 3.89-3.75 (m, 1H), 3.67-3.54 (m, 8H), 3.23- 3.11 (m, 1H), 3.06-2.97 (d, 3H), 2.41-2.27 (m, 2H), 2.24-2.08 (m, 2H), 2.06-1.93 (m, 2H), 1.92-1.79 (m, 5H), 1.62-1.41 (m, 11H), 1.40-1.32 (m, 3H), 1.29-1.26 (m, 5H), 1.20
- Supramolecular nanostructures were formulated using thin film hydration method with varying mole ratios of phospholipids (such as HSPC, POPC, SOPC, Egg PC, etc.), PEGylated- phospholipids (such as DSPE-PEG) and CSF1R inhibitor conjugates.
- % Encapsulation efficiency of the CSF1R inhibitors in the supramolecules was determined by UV spectrophotometry. Average size, polydispersity index (PDI) and surface potential of the nanoparticles were measured by Dynamic light scattering (DLS).
- phospholipids such as HSPC, POPC, SOPC, Egg PC, etc.
- PEGylated- phospholipids such as DSPE-PEG
- CSF1R inhibitor conjugates % Encapsulation efficiency of the CSF1R inhibitors in the supramolecules was determined by UV spectrophotometry. Average size, polydispersity index (PDI) and surface potential of the nanoparticles were measured by Dynamic light scattering
- HSPC, POPC, IO-806 03 and DSPE-PEG taken in 55:35:5:5 mol% ratio, were dissolved in Chloroform. All lipid solutions were mixed homogeneously in round bottom flask and organic solvent was evaporated by rotary evaporator resulting in a thin lipid film. The lipid film was kept under high vacuum for 3-4 hr. It was, then, hydrated (by adding 5% Lactose solution to it) for 1.0 h at 60°C in hot water bath. Next, hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at 60°C using Avanti extruder supported over hot plate.
- HSPC, POPC, IO-806 02 and DSPE-PEG taken in 55:35:5:5 mol% ratio, were dissolved in Chloroform. All lipid solutions were mixed homogeneously in round bottom flask and organic solvent was evaporated by rotary evaporator resulting in a thin lipid film. The lipid film was kept under high vacuum for 3-4 hr. It was, then, hydrated (by adding 5% Lactose solution to it) for 1.0 h at 60°C in hot water bath. Next, hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at 60°C using Avanti extruder supported over hot plate.
- hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at 60°C using Avanti extruder supported over hot plate. It is evident from UV measurement that 4.1 mol% of IO-803_01was encapsulated in the supramolecule. DLS measurement reveals average size of the nanoparticles to be 137.9 nm, PDI 0.058 and surface potential -24.5 mV. The resulting solution was lyophilized and stored at 4°C.
- HSPC, POPC, IO-801 02 and DSPE-PEG taken in 50:30: 15:5 mol% ratio, were dissolved in Chloroform. All lipid solutions were mixed homogeneously in round bottom flask and organic solvent was evaporated by rotary evaporator resulting in a thin lipid film. The lipid film was kept under high vacuum for 3-4 hr. It was, then, hydrated (by adding 5% Lactose solution to it) for 1.0 h at 60°C in hot water bath. Next, hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at 60°C using Avanti extruder supported over hot plate.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Dispersion Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Nanotechnology (AREA)
- Botany (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present disclosure relates generally to Cellular Signalling inhibitors of compound of Formula I, compositions and formulations comprising the same, methods, processes, and uses thereof. In particular, the present disclosure provides CSF-1R inhibitors of BLZ-945-lipids conjugates, GW2580-lipid conjugates and PLX-3397-lipid conjugates demonstrating sustained inhibition of CSF/CSF1R signalling pathway with decreased toxicity. The present disclosure also provides supramolecular combinatorial therapeutics, wherein a CSF-1R inhibitor is combined with one or more of a chemotherapeutic agent, a kinase inhibitor, and an immunoregulator, each of which is optionally conjugated with a lipid. The present disclosure also provides a method for treating cancer, allergy, Systemic lupus erythematosus, nephritis, Chronic Obstructive Pulmonary Disease, and abnormal macrophage functions or any combinations thereof.
Description
NOVEL INHIBITORS OF CELLULAR SIGNALLING
TECHNICAL FIELD
The present disclosure relates generally to Cellular Signalling inhibitors, compositions and formulations comprising the same, and uses thereof. In particular, the present disclosure provides CSF-IR inhibitors of BLZ-945-lipids conjugates, GW2580-lipid conjugates and PLX-3397-lipid conjugates demonstrating sustained inhibition of CSF/CSF-1R signalling pathway with decreased toxicity. The present disclosure also provides supramolecular combinatorial therapeutics, wherein the CSF-IR inhibitor is combined with one or more of a chemotherapeutic agent, a kinase inhibitor, and an immunoregulator, each of which is optionally conjugated with a lipid. Background
Targeted therapies have been in the limelight as cancer therapeutics for the last few years. They have resulted in high response rates and improved overall survival in patients with cancer. However, consistent with other oncogene-targeted therapies, initial patient response is of limited durability and tumors eventually relapse.1 The tumor microenvironment is increasingly recognized to play an important role in tumor proliferation, invasion, metastasis, and chemoresistance. It provides a conducive niche to the tumor through immunosuppression. Overcoming this immunosuppressive nature of the tumor microenvironment has been of particular interest in cancer therapy. Tumor cells manipulate the surrounding environment by producing cytokines that suppress cytolytic T-cells and recruit immunosuppressive cells.11 Colony stimulating factor 1 (CSF-1) is one such cytokine secreted by several cancer cell types. It induces the proliferation and differentiation of immunosuppressive myeloid cells such as M2 polarized macrophages and myeloid derived suppressor cells (MDSC) by binding to the CSF-1 receptor (CSF-IR) on cell surface.111 Cellular signalling mediated by colony-stimulating factor 1 (CSF1) and its receptor CSF-IR plays a critical role in monocyte differentiation and generation and activity of tissue-resident macrophages. The overexpression of CSF1 is associated with poor prognosis in breast, ovarian, and prostate cancer. Coincidentally, increased TAMs (tumor-associated macrophages) density also designates poor prognostic value, suggesting that CSF1-CSF1R axis may have an important role towards activity of TAMs.lv' v vl
The CSF1/CSF1R signalling pathway is targeted in the treatment against numerous malignancies, including breast, leukaemia, and glioblastoma. Studies have demonstrated that TAMs undergo turnover in a CSF-IR dependent manner, with continuous inhibition of the CSF-IR pathway being essential for depletion of TAMs and serving as an anticancer therapy. Therefore, the immunosuppressive tumor environment mediated by CSF-1 helps tumor cells escape killing by immune cells and assists them to metastasize. Since CSF-IR regulates the functioning of macrophages impacting tumor progression, inhibiting the CSF-IR pathway has emerged as a major therapeutic goal in cancer. Recently, some of the CSF-IR inhibitors had shown promising results in terms of potency, selectivity and bioavailability of cFMS kinase activity vu Among them some of the well-known inhibitors which are in the clinical phase trials are BLZ-945, GW2580, PLX-3397 etc. Although high in potency, these inhibitors fail to achieve a sustained inhibition of CSF-IR and are associated with toxicity to normal cells. Accordingly, there remains an urgent need for CSF-IR inhibitors with an improved activity profile while exhibiting decreased toxicity.
Summary The present application describes the design of novel CSF-IR inhibitors that can preferentially accumulate in the tumor microenvironment and exert sustained inhibition of the target molecule, overcoming the dose-limiting toxic effects and achieving superior antitumor efficacy compared to currently available CSF-IR inhibitors. The present invention is directed to hydrophobic, lipophilic prodrugs that lead to increased effective drug accumulation in tumor via enhanced permeability and retention (EPR) phenomenon leading to improved efficacy. The lipids used in these prodrugs include phospholipids, cholesterol, fatty acids etc. The present invention describes prodrugs of CSF-IR inhibitors that can assemble into supramolecular structure with improved pharmacokinetic profile such as long circulation time, enhanced uptake and sustained release of drug inside tumor. The CSF-IR uptake into tumor can be achieved in higher amounts by making supramolecular assemblies in aqueous buffer along with the addition of co-lipids to form supramolecules with an average particle size below 200 nm. Degradation of the supramolecular assembly as well as the prodrug releases effective drug inside the cell. Pharmaceutical compositions of prodrug of a CSF-IR inhibitor comprise a linker wherein CSF-IR is coupled through ester, ether, amide, or other covalent conjugation with the linker. The lipid molecule can be cholesterol, oleic acid, alpha tocopherol, fatty acid or another naturally occurring lipid molecule which is conjugated to drug molecule through a
suitable linker/spacer. The spacer can be composed of aliphatic dicarboxylic acid, unsaturated dicarboxylic acid, aldaric acid, fumaric acid, propargylic acid, acetylene dicarboxylic acid, aromatic/hetero aromatic dicarboxylic acid, ethylene glycol, diethylene glycol, natural or unnatural amino acids individually or in any combinations.
Certain exemplary embodiments provide supramolecular combinatorial therapeutics, wherein a CSF-IR inhibitor is combined with one or more of a kinase inhibitor, or a chemotherapeutic drug, or a tumor-targeting antibody, each of which is optionally conjugated with a lipid. Without limitations, the antibody can be useful for therapeutic purposes (i.e., a therapeutic antibody) or for targeting the supramolecular combinatorial therapeutic to a desired site (i.e., a targeting antibody). In some embodiments, the supramolecular combinatorial therapeutic further comprises an immunomodulator, which can include another CSF-IR inhibitor (s). Immunomodulators are active agents of immunotherapy, and can either activate or suppress an immune response. In certain embodiments, the immunomodulator activates and stimulates an immune response against cancer cells, non-limiting examples of which include immune cells (e.g., natural killer cells, lymphokine-activated killer cells, cytotoxic T cells and dendritic cells), antibodies (e.g., anti-PD-Ll and anti-PD-1 antibodies, anti-CD52, anti-VEGF-A, anti- CD30, anti-EGFR, anti- CD33, anti-CD20, anti-CTLA4, and anti-HER-2 antibodies), and cytokines (e.g., interferons and interleukins). In certain exemplary embodiments, the immunomodulator is conjugated with a lipid.
Detailed Description of the Invention
The present disclosure relates to compounds of CSF-IR inhibitors and lipid conjugates of CSF- IR inhibitors of Formula I


Formula I
wherein 'Ligand' is selected from the group consisting of:

wherein selected from a group consisting of
C=hydroxy, alkyl group, aryl group, cycloalkyl group;
A=H, O, ML S;
B=CH, N;
D=C, O, NH, S;
E=C, O, NH, S;
F=CH, N;
G=C, O, NH, S;
J=NH2, OH, SH; and
X=halogen.
'L' is a linker moiety connecting 'Ligand' and 'R' moieties; and
'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
or any derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates of compound of Formula I thereof. In non-limiting embodiment of the present disclosure, CSF-IR inhibitors are the inhibitors which inhibits the CSFI/CSFIR signalling pathway. CSF-IR inhibitors include compounds which target, decrease or inhibit the activity of Colony stimulating factor 1 receptor (CSF-IR). e.g., AC710, ARRY-382, AZD6495, BLZ945, CC- 223, cediranib, cerdulatinib, crenolanib, dovitinib, GTP-14564,GW-2580,JNJ-28312141, JNJ-40346527, Ki-20227, linifanib, OSI-930, pazopanib, pexidartinib, quizartinib, tandutinib,TG02, etc
In non-limiting embodiment of the present disclosure, lipid conjugates of CSF-IR inhibitors BLZ-945-lipid conjugate, GW2580-lipid conjugate and PLX3397-lipid conjugate.
The present invention is directed to lipid conjugates of CSF-IR inhibitors, such as BLZ-945, GW2580 and PLX3397, and that lipid conjugates of CSF-IR inhibitors. The present disclosure relates to compounds of BLZ-945 series/Formula IA (BLZ-945-lipid conjugates), GW2580 series/ Formula IB (GW2580-lipid conjugates), PLX-3397 series/ Formula IC (PLX-3397-lipid conjugates).
Figure 1. Representative Markush structures of CSF-IR inhibitor series
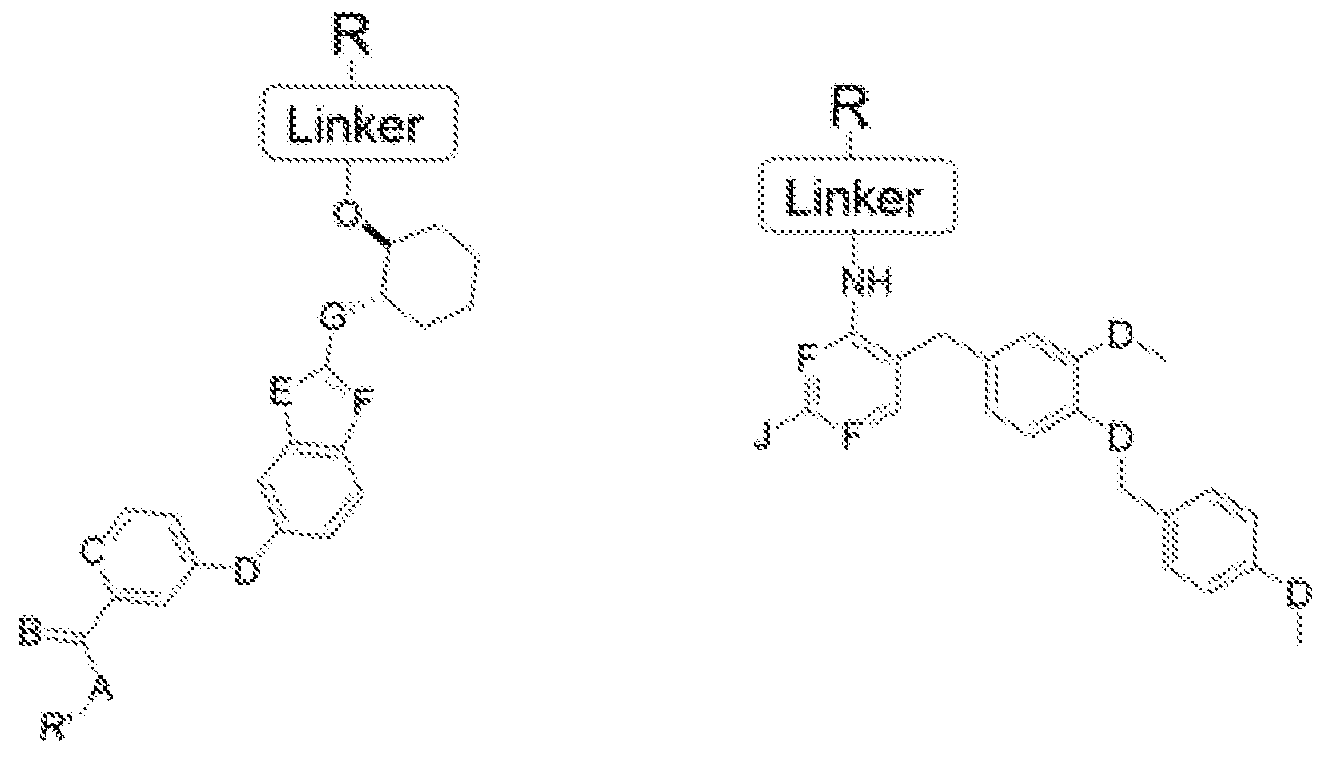
Formula IA Formula IB
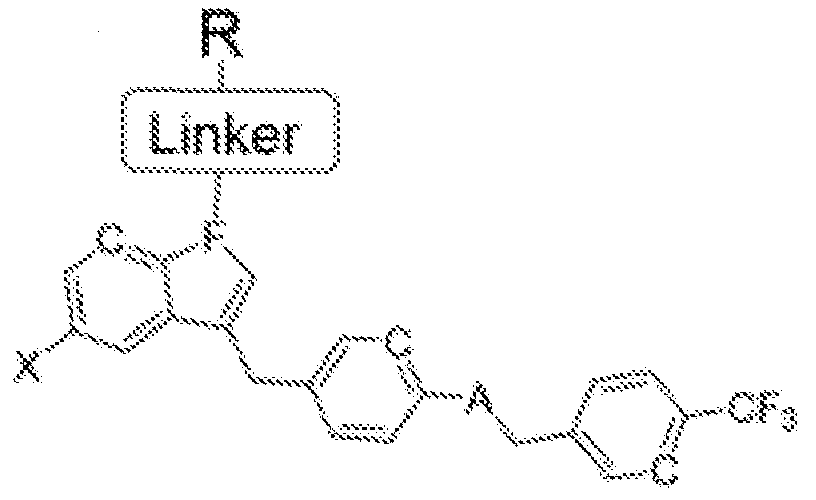
Formula IC
C=hydroxy, alkyl group, aryl group, cycloalkyl group;
A=H, O, NH, S;
B=CH, N;
D=C, O, NH, S;
E=C, O, NH, S;
F=CH, N;
G=C, O, NH, S;
J=NH2, OH, SH; and
X=halogen.
'L' is a linker moiety connecting 'Ligand' and R' moieties
'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
or
any derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates of compound of Formula I thereof.
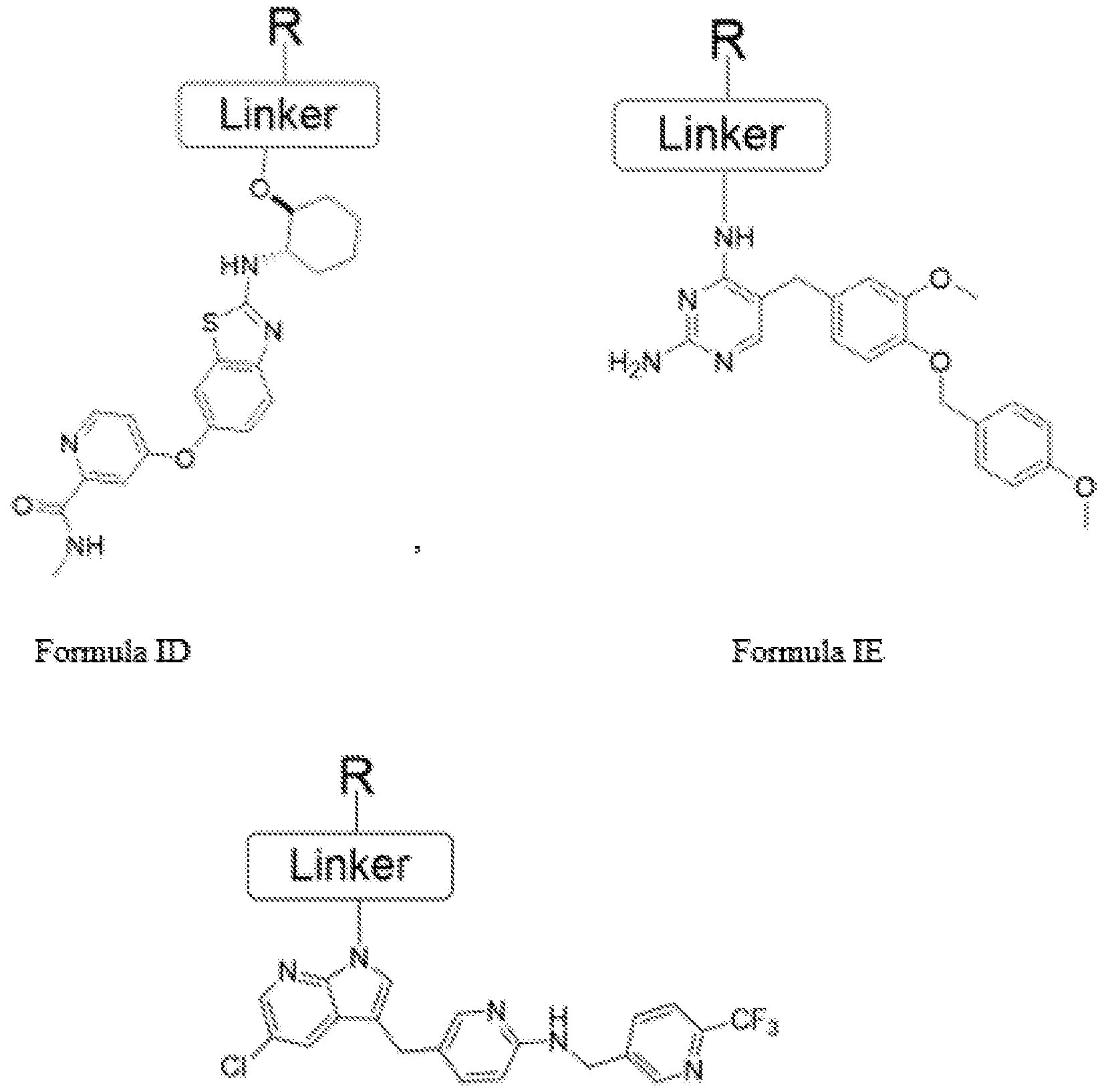
Fcsmula IF wherein 'L' is a linker moiety connecting 'Ligand' and 'R' moieties
'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
or
any derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates of compound of Formula I thereof.
In a non-limiting embodiment, the linker group(s) in the BLZ-945 series (BLZ-945-lipid conjugates), GW2580 series (GW2580-lipid conjugates), PLX-3397 series (PLX-3397-lipid conjugates) of the present disclosure is selected from a group comprising a direct bond or an atom such as oxygen or sulfur, a unit such as R1, C(O), C(0)0, C(0) R\ SO, SO2, SO2 H or a chain of atoms, such as substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, heterocyclylalkyl, heterocyclylalkenyl, heterocyclylalkynyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, cycloalkenyl, alkylarylalkyl, alkylarylalkenyl, alkylarylalkynyl, alkenylarylalkyl, alkenylarylalkenyl, alkenylarylalkynyl, alkynylarylalkyl, alkynylarylalkenyl, alkynylarylalkynyl, alkylheteroarylalkyl, alkylheteroarylalkenyl, alkylheteroarylalkynyl, alkenylheteroarylalkyl, alkenylheteroarylalkenyl, alkenylheteroarylalkynyl, alkynylheteroarylalkyl, alkynylheteroarylalkenyl, alkynylheteroarylalkynyl, alkylheterocyclylalkyl, alkylheterocyclylalkenyl, alkylhererocyclylalkynyl, alkenylheterocyclylalkyl, alkenylheterocyclylalkenyl, alkenylheterocyclylalkynyl, alkynylheterocyclylalkyl, alkynylheterocyclylalkenyl, alkynylheterocyclylalkynyl, alkylaryl, alkenylaryl, alkynylaryl, alkylheteroaryl, alkenylheteroaryl, alkynylhereroaryl, where one or more methylenes can be interrupted or terminated by O, S, S(O), SO2, C(O), cleavable linking group, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclic; where R1 is hydrogen, acyl, aliphatic or substituted aliphatic. In another non-limiting embodiment, the linker group(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising a direct bond, ester, ether, amide or other covalent conjugation with linker.
In yet another non-limiting embodiment, the linker group(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising one or more of succinic acid, fumaric acid, propargylic acid, ethylene glycol, diethylene glycol, and natural or unnatural amino acids.
In still another non-limiting embodiment, the linker group(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising at least one of oxalic acid, malonic acid, succinic acid, glutaric acid, succinic acid, ethylene diamine, natural or unnatural amino acid, ethylene glycol, diethylene glycol, acetic acid, propionic acid, butyric acid, valeric acid, acrylic acid, but-2-enoic acid,
pent-2-enoic acid, hex-2-enoic acid, 2-propynoic acid, but-2-ynoic acid, pent-2-ynoic acid, hex-2-ynoic acid, ethylene, propylene, 1-butene, 1-pentene, 1-hexene, acetylene, propyne, but-
1- yne, pent-l-yne, and any combinations thereof.
In still another non-limiting embodiment, the linker group(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising -C(0)CH2CH2C(0)-; -C(0)(CH2CH2)m(C(0)CH2CH2)n- wherein 'n' is 1 to 10 and 'm' is 1 to 4; -C(0)(CH2)xCH2C(0)NH(CH2CH2)n-, wherein 'n' is 1 to 10 and 'x' is 0 to 3; -C(0)(CH2)mCH2C(0)NH(CH2CH2)n HC(0)-, wherein 'n' is 1 to 10 and 'm' is 1 to 4; -C(0)CH2(CH2)mC(0) H- wherein 'm' is 1 to 4; -C(0)(CH2)nCH2(R) HC(0)- , wherein 'n' is 1 to 10 and R is H, alkyl, acid, amine, aryl, thiols; - C(0)(CH2)nCH2(R) HC(0)-, wherein R is H, alkyl, acid, amine, aryl, thiols; C(0)(CH2(R)CH2)nC(Y)X- wherein R is H, alkyl, acid, amine, aryl, thiols, wherein Y is C, O, NH and S, wherein X is C, O, NH and S; -C(0)CH2CH2C(0) HCH2CH2 HC(0)-; - C(0)CH2CH2C(0) HCH2 HC(0)-; -C(0)CH2OCH2CH2-; -C(0)CH2CH2OCH2CH2-; - C(0)CH2OCH2CH2OCH2CH2-; -C(0)CH(R)NHC(0)CH2- wherein R is H, CH3, CH(CH3)2, CH2CH(CH3)2, C(CH3)CH2CH3, or CH2-Phenyl; -C(0)CH(R)NHC(0)CH2CH2- wherein R is H, CH3, CH(CH3)2, CH2CH(CH3)2, C(CH3)CH2CH3, or CH2-Phenyl; - C(0)CH(R) HC(0)(CH2)nC(0)-, wherein R is H, CH3, CH(CH3)2, CH2CH(CH3)2, C(CH3)CH2CH3, or CH2-Phenyl, and n is 1 , 2, or 3; -C(0)CH(R)NHC(0)CH2OCH2CH2-, wherein R is H, CH3, CH(CH3)2, CH2CH(CH3)2, C(CH3)CH2CH3, or CHz-Phenyl; - C(0)C≡C(CH2)n-C(0)-, wherein n is 1, 2 or 3; -C(0)C≡C(CH2)n- wherein n is 0, 1, or 2; - C(0)CH=CH(CH2)nC(0)-, wherein n is 0, 1, 2, or 3; -C(0)CH=CH(CH2)n- wherein n is 1, 2, or 3; and -C(0)CH2CH2C(0)NHCH2C(0)-.
In a non-limiting embodiment, the lipid(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising cholesterol, cholesterol derivatives, oleic acid, oleic acid derivative, alpha tocopherol, alpha tocopherol derivatives, phospholipid, phospholipid derivatives, fatty acid, naturally occurring lipid molecule which is conjugated to drug molecules, 1,3-Propanediol Dicaprylate/Dicaprate, 10-undecenoic acid, 1-dotriacontanol, 1-heptacosanol, 1-nonacosanol,
2- ethyl hexanol, Androstanes, Arachidic acid, Arachidonic acid, arachidyl alcohol, Behenic acid, behenyl alcohol, Capmul MCM CIO, Capric acid, capric alcohol, capryl alcohol, Caprylic acid, Caprylic/Capric Acid Ester of Saturated Fatty Alcohol C12-C18, Caprylic/Capric Triglyceride, Caprylic/Capric Triglyceride, Ceramide phosphorylcholine (Sphingomyelin, SPH), Ceramide phosphorylethanolamine (Sphingomyelin, Cer-PE), Ceramide
phosphorylglycerol, Ceroplastic acid, Cerotic acid, Cerotic acid, ceryl alcohol, Cetearyl alcohol, Ceteth-10, cetyl alcohol, Cholanes, Cholestanes, cholesterol, cis-l l-eicosenoic acid, cis-l l-octadecenoic acid, cis-13-docosenoic acid, cluytyl alcohol, Dihomo-y-linolenic, Docosahexaenoic acid, egg lecithin, Eicosapentaenoic acid, Eicosenoic acid, Elaidic acid, elaidolinolenyl alcohol, elaidolinoleyl alcohol, elaidyl alcohol, Erucic acid, erucyl alcohol, Estranes, Ethylene glycol distearate (EGDS), Geddic acid, geddyl alcohol, glycerol distearate (type I) EP (Precirol ATO 5), Glycerol Tricaprylate/Caprate; Glycerol Tricaprylate/Caprate (CAPTEX® 355 EP/NF); glyceryl monocaprylate (Capmul MCM C8 EP), Glyceryl Triacetate, Glyceryl Tricaprylate, Glyceryl Tricaprylate/Caprate/Laurate, Glyceryl Tricaprylate/Tricaprate, glyceryl tripalmitate (Tripalmitin), Henatriacontylic acid, Heneicosyl alcohol, Heneicosylic acid, Heptacosylic acid, Heptadecanoic acid, Heptadecyl alcohol, Hexatriacontylic acid, isostearic acid, isostearyl alcohol, Lacceroic acid, Laurie acid, Lauryl alcohol, Lignoceric acid, lignoceryl alcohol, Linoelaidic acid, Linoleic acid, linolenyl alcohol, linoleyl alcohol, Margaric acid, Mead, Melissic acid, melissyl alcohol, Montanic acid, montanyl alcohol, myricyl alcohol, Myristic acid, Myristoleic acid, Myristyl alcohol, neodecanoic acid, neoheptanoic acid, neononanoic acid, Nervonic, Nonacosylic acid, Nonadecyl alcohol, Nonadecylic acid, Nonadecylic acid, Oleic acid, oleyl alcohol, Palmitic acid, Palmitoleic acid, palmitoleyl alcohol, Pelargonic acid, pelargonic alcohol, Pentacosylic acid, Pentadecyl alcohol, Pentadecylic acid, Phosphatidic acid (phosphatidate, PA), Phosphatidylcholine (lecithin, PC), Phosphatidylethanolamine (cephalin, PE), Phosphatidylinositol (PI), Phosphatidylinositol bisphosphate (PIP2), Phosphatidylinositol phosphate (PIP), Phosphatidylinositol triphosphate (PIP3), Phosphatidylserine (PS), polyglyceryl-6-distearate, Pregnanes, Propylene Glycol Dicaprate, Propylene Glycol Dicaprylocaprate, Propylene Glycol Dicaprylocaprate, Psyllic acid, recinoleaic acid, recinoleyl alcohol, Sapienic acid, soy lecithin, Stearic acid, Stearidonic, stearyl alcohol, Tricosylic acid, Tridecyl alcohol, Tridecylic acid, Triolein, Undecyl alcohol, undecylenic acid, Undecylic acid, Vaccenic acid, a-Linolenic acid, and γ-Linolenic acid via a spacer, wherein the spacer is selected from a group comprising aliphatic dicarboxylic acid, unsaturated dicarboxylic acid, aldaric acid, fumaric acid, propargylic acid, acetylene dicarboxylic acid, aromatic/hetero aromatic dicarboxylic acid, ethylene glycol, diethylene glycol, natural or unnatural amino acids, or their derivatives individually or in any combinations thereof.
In another non-limiting embodiment, the lipid(s) or lipid derivative(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates of the present disclosure is selected from a group comprising cholesterol, cholesterol derivatives, oleic acid, oleic acid
derivative, alpha tocopherol, alpha tocopherol derivatives, phospholipid, phospholipid derivatives, fatty acid or naturally occurring lipid molecule which is conjugated to drug molecules via a spacer, wherein the spacer is selected from a group comprising aliphatic dicarboxylic acid, unsaturated dicarboxylic acid, aldaric acid, fumaric acid, propargylic acid, acetylene dicarboxylic acid, aromatic/hetero aromatic dicarboxylic acid, ethylene glycol, diethylene glycol, natural or unnatural amino acids, or their derivatives individually or any combinations thereof.
In a non-limiting embodiment, the lipid conjugate(s) in the BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates is the lipid or the lipid derivative conjugated with a compound selected from a group comprising a kinase inhibitor, a chemotherapeutic drug, an immunomodulator or an antibody or any combinations thereof.
In a non-limiting embodiment of the present disclosure, the compound of Formula I is BLZ- 945-lipid conjugate(s).
In a non-limiting embodiment of the present disclosure, the compound of Formula I is GW2580-lipid conjugate(s).
In a non-limiting embodiment of the present disclosure, the compound of Formula I is PLX- 3397-lipid conjugate(s).
In a non-limiting embodiment, the supramolecular combinatorial therapeutic comprises a BLZ- 945-lipid conjugate.
In a non-limiting embodiment, the supramolecular combinatorial therapeutic comprises a GW2580-lipid conjugate.
In a non-limiting embodiment, the supramolecular combinatorial therapeutic comprises a PLX- 3397-lipid conjugate.
In a non-limiting embodiment, the supramolecular combinatorial therapeutic can further comprise a lipid conjugated kinase inhibitor. In some embodiments, the supramolecular combinatorial therapeutic further comprises an antibody (or an antigen binding fragment thereof), optionally conjugated with a lipid.
In some embodiments, the supramolecular combinatorial therapeutic further comprises an antibody (or an antigen binding fragment thereof) conjugated with a lipid. Without limitations, the antibody can be useful for thereaputic purposes (i.e., a therapeutic antibody) or for targeting the the supramolecular combinatorial therapeutic to a desired site (i.e., a targeting antibody). In some embodiments, the supramolecular combinatorial therapeutic further comprises an immunomodulator. Immunomodulators are active agents of immunotherapy, and can either activate or suppress an immune response. In certain embodiments, the immunomodulator
activates and stimulates an immune response against cancer cells, non-limiting examples of which include immune cells (e.g., natural killer cells, lymphokine-activated killer cells, cytotoxic T cells and dendritic cells), antibodies (e.g., anti-PD-Ll and anti-PD-1 antibodies, anti-CD52, anti-VEGF-A, anti- CD30, anti-EGFR, anti-CD33, anti-CD20, anti-CTLA4, and anti-HER-2 antibodies), and cytokines (e.g., interferons and interleukins). In certain exemplary embodiments, the immunomodulator is conjugated with a lipid.
In another aspect, described herein is a method of treating cancer, comprising, administering a supramolecular combinatorial therapeutic as described herein to a patient in need of treatment for cancer. In some embodiments, the cancer is selected from the group consisting of: breast cancer; ovarian cancer; glioma; gastrointestinal cancer; prostate cancer; carcinoma, lung carcinoma, hepatocellular carcinoma, testicular cancer; cervical cancer; endometrial cancer; bladder cancer; head and neck cancer; lung cancer; gastro-esophageal cancer, and gynecological cancer.
In some embodiments, the method further comprises co-administering one or more additional anti-cancer therapy to the patient. In some embodiments, the additional therapy is selected from the group consisting of surgery, chemotherapy, radiation therapy, thermotherapy, immunotherapy, hormone therapy, laser therapy, anti-angiogenic therapy, and any combinations thereof. In some embodiments, the additional therapy comprises administering an anti-cancer agent to the patient. In some embodiments, the method further comprises coadministration of one or more immunomodulators to the subject.
In another aspect, described herein is a method of treating allergy, comprising, administering a supramolecular combinatorial therapeutic as described herein to a patient in need of treatment for allergy, Systemic lupus erythematosus, Chronic Obstructive Pulmonary Disease and abnormal macrophage functions.
In a non-limiting embodiment of the present disclosure, a composition comprises a compound of BLZ-945-lipid conjugate along with pharmaceutically acceptable excipient.
In a non-limiting embodiment of the present disclosure, the composition comprises a compound of GW2580-lipid conjugate along with pharmaceutically acceptable excipient. In a non-limiting embodiment of the present disclosure, the composition comprises a compound of PLX-3397-lipid conjugate along with pharmaceutically acceptable excipient. In another non-limiting embodiment of the present disclosure, the pharmaceutically acceptable excipient is selected from the group comprising adjuvant, diluent, carrier, granulating agents,
binding agents, lubricating agents, disintegrating agent, sweetening agents, glidant, anti- adherent, anti-static agent, surfactant, anti-oxidant, gum, coating agent, coloring agent, flavouring agent, coating agent, plasticizer, preservative, suspending agent, emulsifying agent, plant cellulosic material, spheronization agent, and other conventionally known pharmaceutically acceptable excipient, or any combination of excipients thereof.
In a non-limiting embodiment of the present disclosure, the composition inhibits colony stimulating factor-1 receptor (CSF-1R) and is administered to a subject in need thereof through modes selected from a group comprising intravenous administration, intramuscular administration, intraperitoneal administration, hepatoportal administration, intra articular administration and pancreatic duodenal artery administration, or any combination thereof. The present disclosure provides a method of treating cancer, comprising, administering the compounds of BLZ-945-lipid conjugate or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition to a subject in need of treatment for cancer.
The present disclosure provides a method of treating cancer, comprising, administering the compounds of GW2580-lipid conjugate or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition to a subject in need of treatment for cancer.
The present disclosure provides a method of treating cancer, comprising, administering the compounds of PLX-3397-lipid conjugate or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition to a subject in need of treatment for cancer.
In a non-limiting embodiment of the present disclosure, the cancer is selected from the group consisting of breast cancer; ovarian cancer; glioma; gastrointestinal cancer; prostate cancer; carcinoma, lung carcinoma, hepatocellular carcinoma, testicular cancer; cervical cancer; endometrial cancer; bladder cancer; head and neck cancer; lung cancer; gastro-esophageal cancer, and gynecological cancer, or any combination thereof.
In another non-limiting embodiment of the present disclosure, the method comprises coadministering one or more additional anti-cancer therapy to the subject.
In yet another non-limiting embodiment of the present disclosure, the additional anti-cancer therapy is selected from the group consisting of surgery, chemotherapy, radiation therapy, thermotherapy, immunotherapy, hormone therapy, laser therapy, and anti-angiogenic therapy, or any combination thereof.
In still another non-limiting embodiment of the present disclosure, the additional anti-cancer therapy comprises administering a kinase inhibitor, a chemotherapeutic agent, an immunomodulator or any combination thereof, to the subject.
Figure 2. Representative examples of CSF-IR inhibitor series

Formula ID of BLZ-945 series Formula IE of GW-2580 series
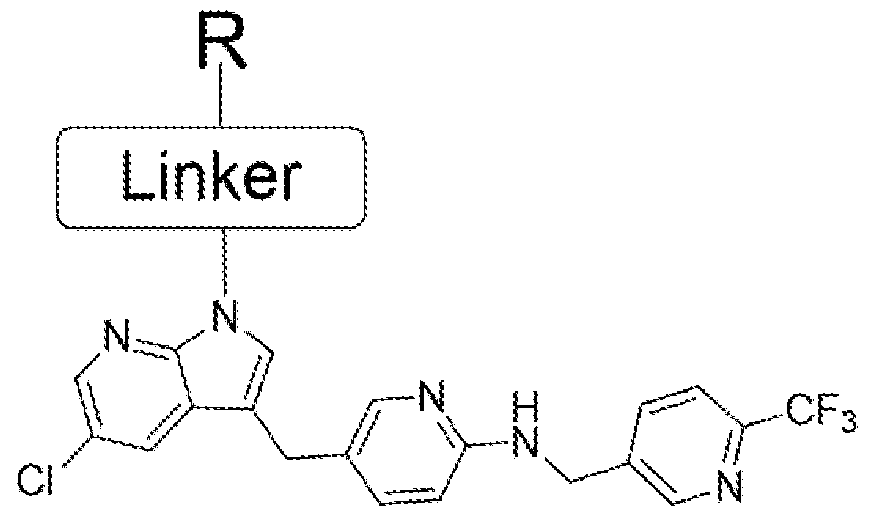
Formula IF of PLX-3397 series
BLZ-945, a small molecule inhibitor of CSF1R is highly selective with an IC50 value of 1 nM, thus attenuates the turnover rate of TAMs simultaneously increases the number of CD8+ T cells that infiltrate cervical and breast carcinomas vm. It is more than 1,000-fold selective against its closest receptor tyrosine kinase homologs. It has been shown to inhibit CSFl -dependent proliferation with an EC50 value of 67 nM in bone marrow-derived macrophages. In some embodiments, the supramolecular combinatorial therapeutic comprises a BLZ-945- lipid conjugate and a platinum-lipid conjugate.
In some embodiments, the supramolecular combinatorial therapeutic comprises a BLZ-945- lipid conjugate and an antibody (or an antigen binding fragment thereof) lipid conjugate. The antibody, or the antigen binding fragment thereof, can be a therapeutic agent or a targeting ligand.
In some embodiments, the supramolecular combinatorial therapeutic comprises a BLZ-945- lipid conjugate and an antibody (or an antigen binding fragment thereof) lipid conjugate, wherein the antibody is a therapeutic antibody or a targeting antibody or a combination thereof. In a non-limiting embodiment of the present disclosure, BLZ-945-lipid conjugate is compound of formula 23.
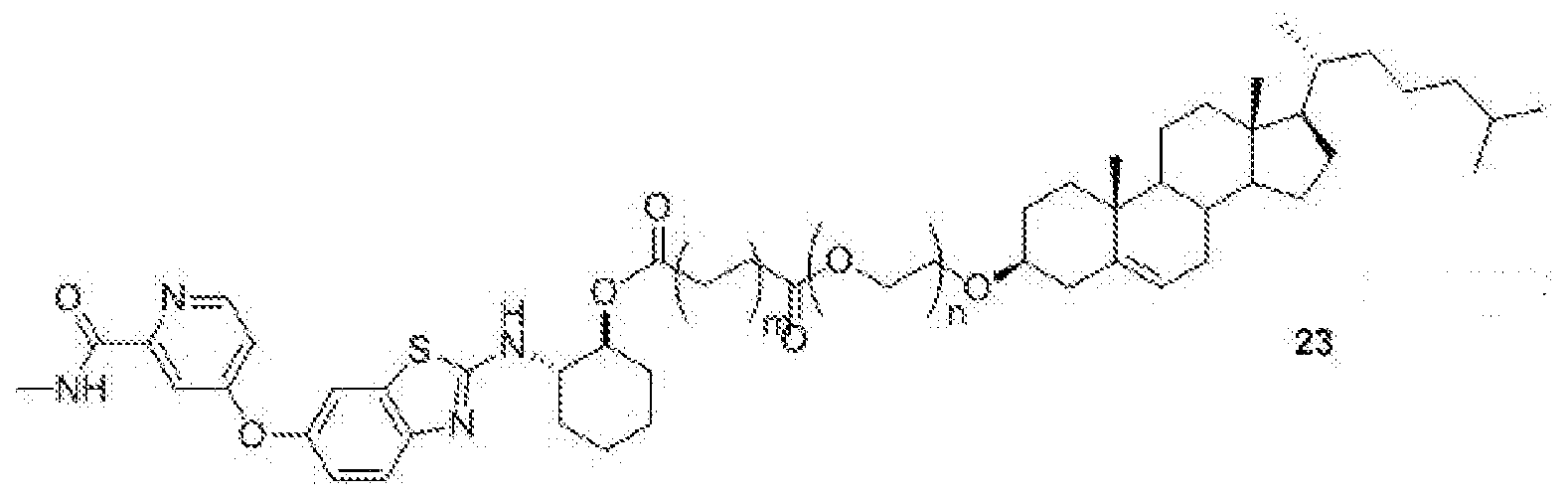
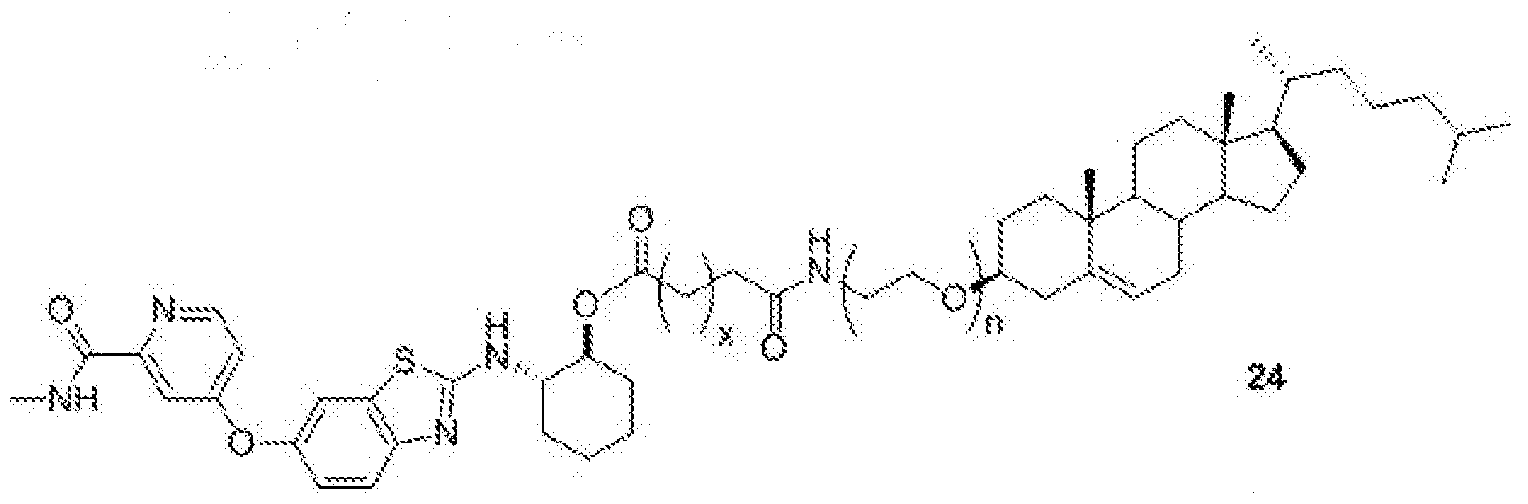
wherein 'n' is 1 to 10 and 'x' is 0 to 3.
In yet another non-limiting embodiment of the present disclosure, BLZ-945-lipid conj compound of formula 25.
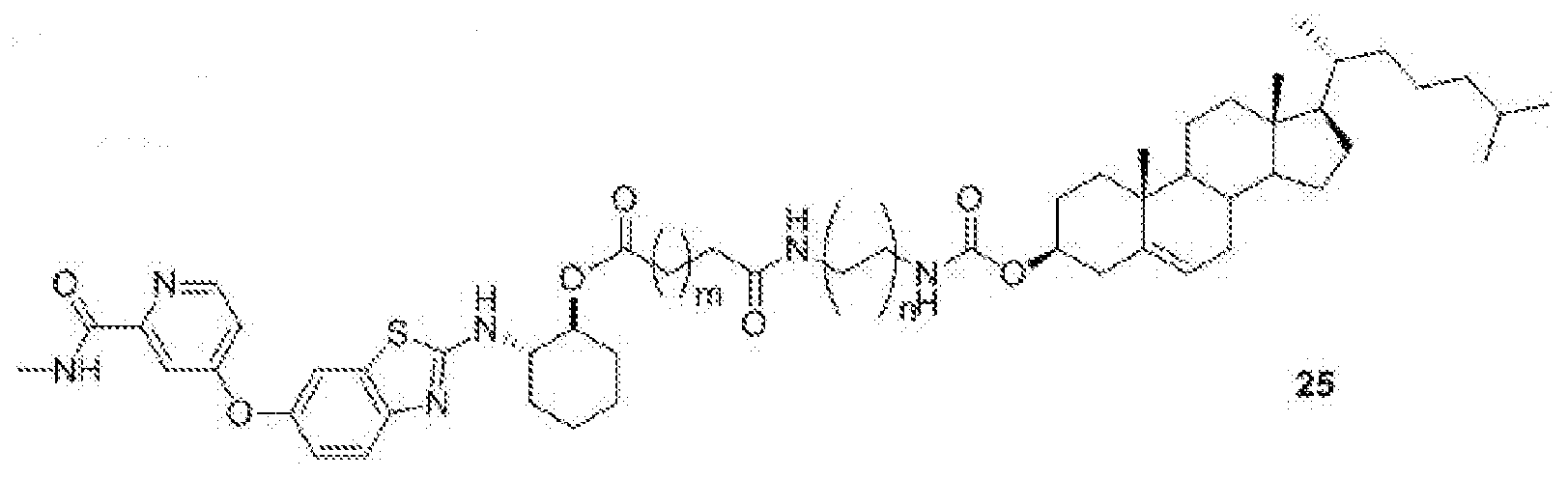
wherein 'n' is 1 to 10 and 'm' is 1 to 4.
In still another non-limiting embodiment of the present disclosure, BLZ-945-lipid conjugate is compound of formula 26.
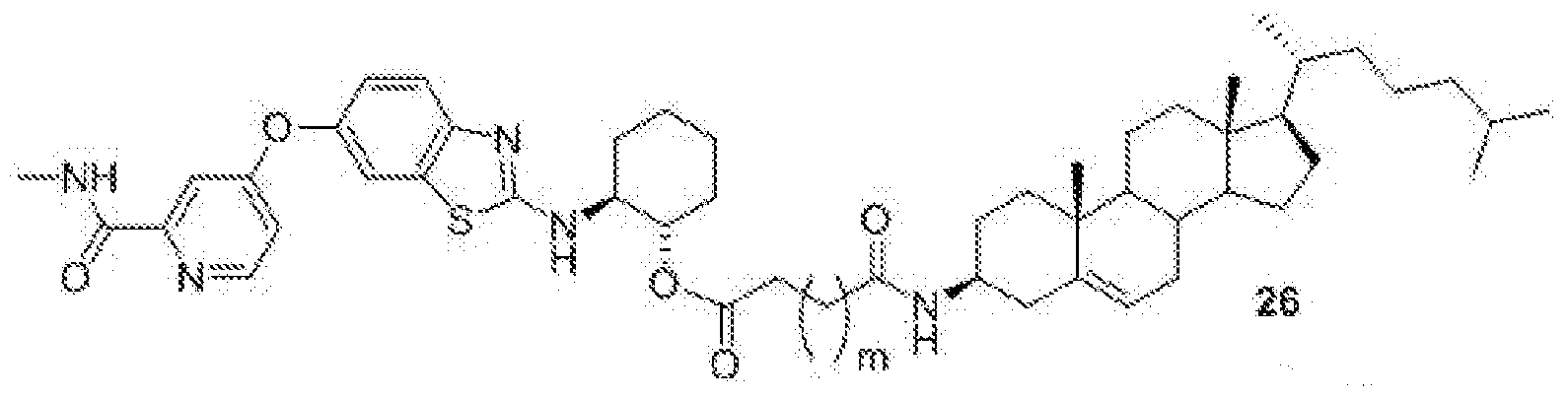
wherein and 'm' is 1 to 4.
In still another non-limiting embodiment of the present disclosure, BLZ-945-lipid conjugate is compound of formula 27.

wherein 'n' is 1 to 10; R is H, alkyl, acid, amine, aryl or thiol; Y is C, O, NH, S; and X is C, O, NH, S.
The present disclosure further provides a BLZ-945 - lipid conjugate compound selected from:
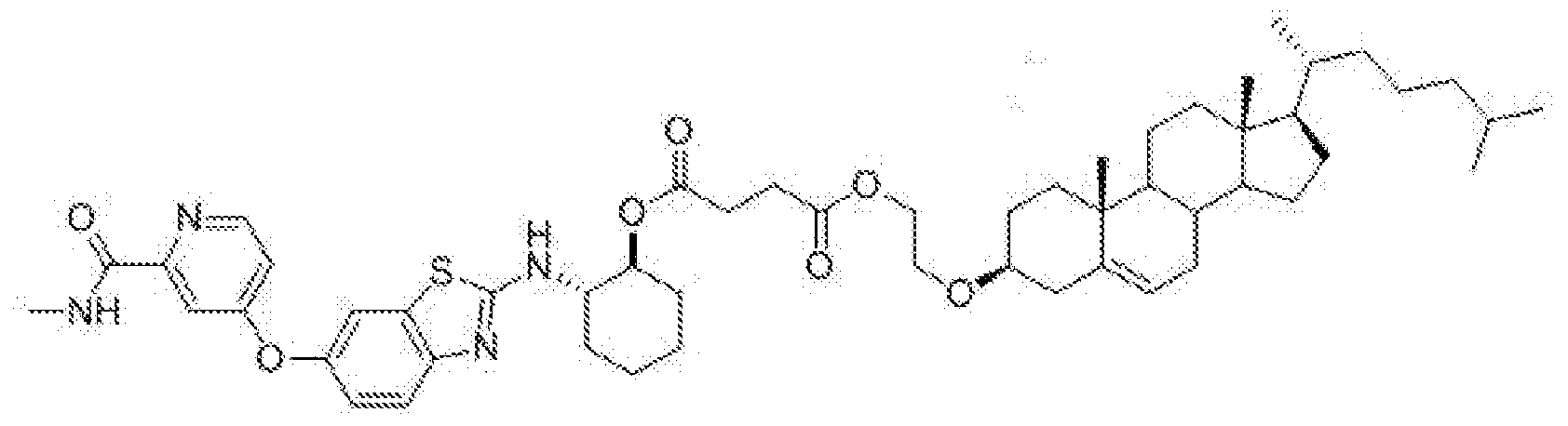
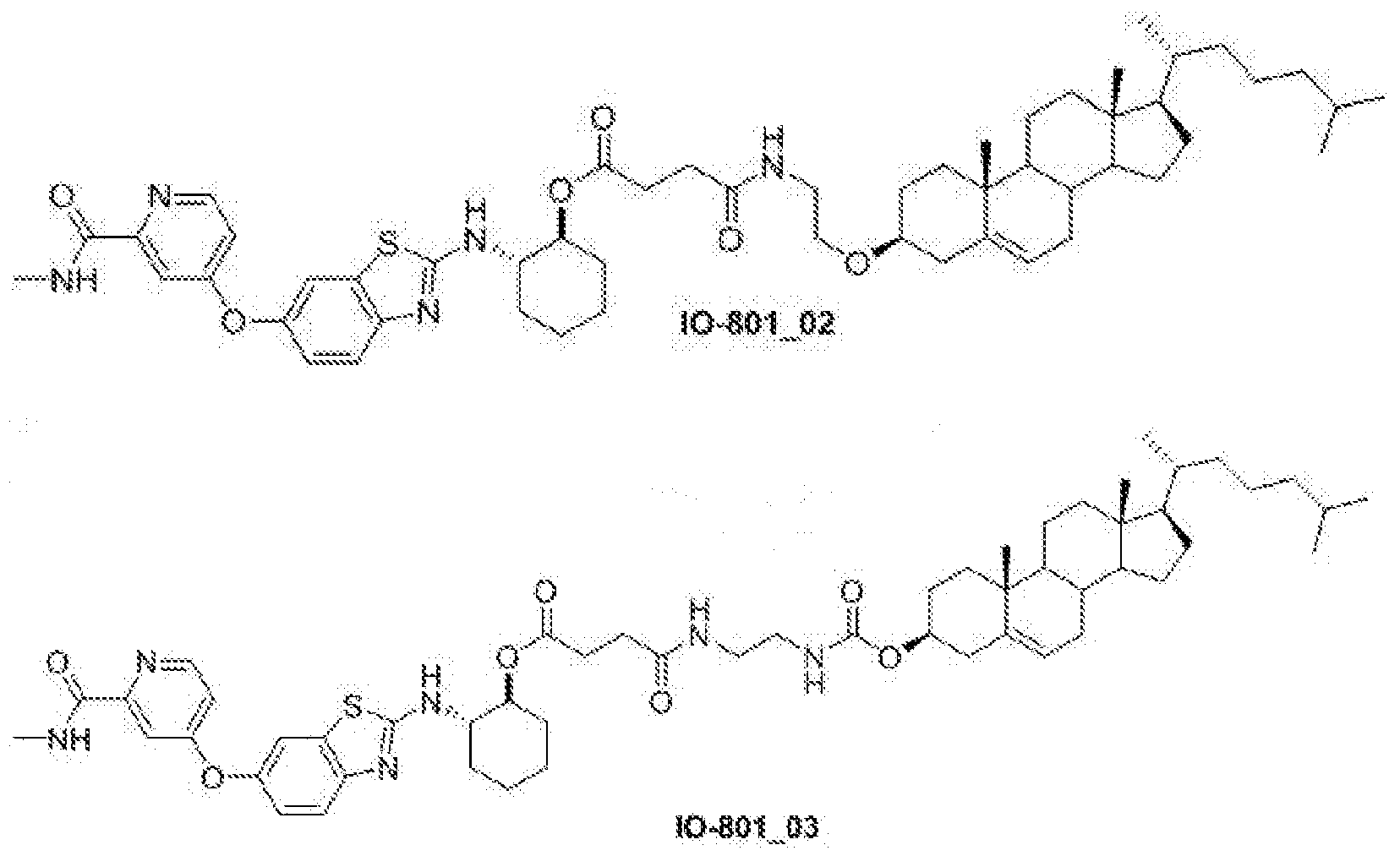
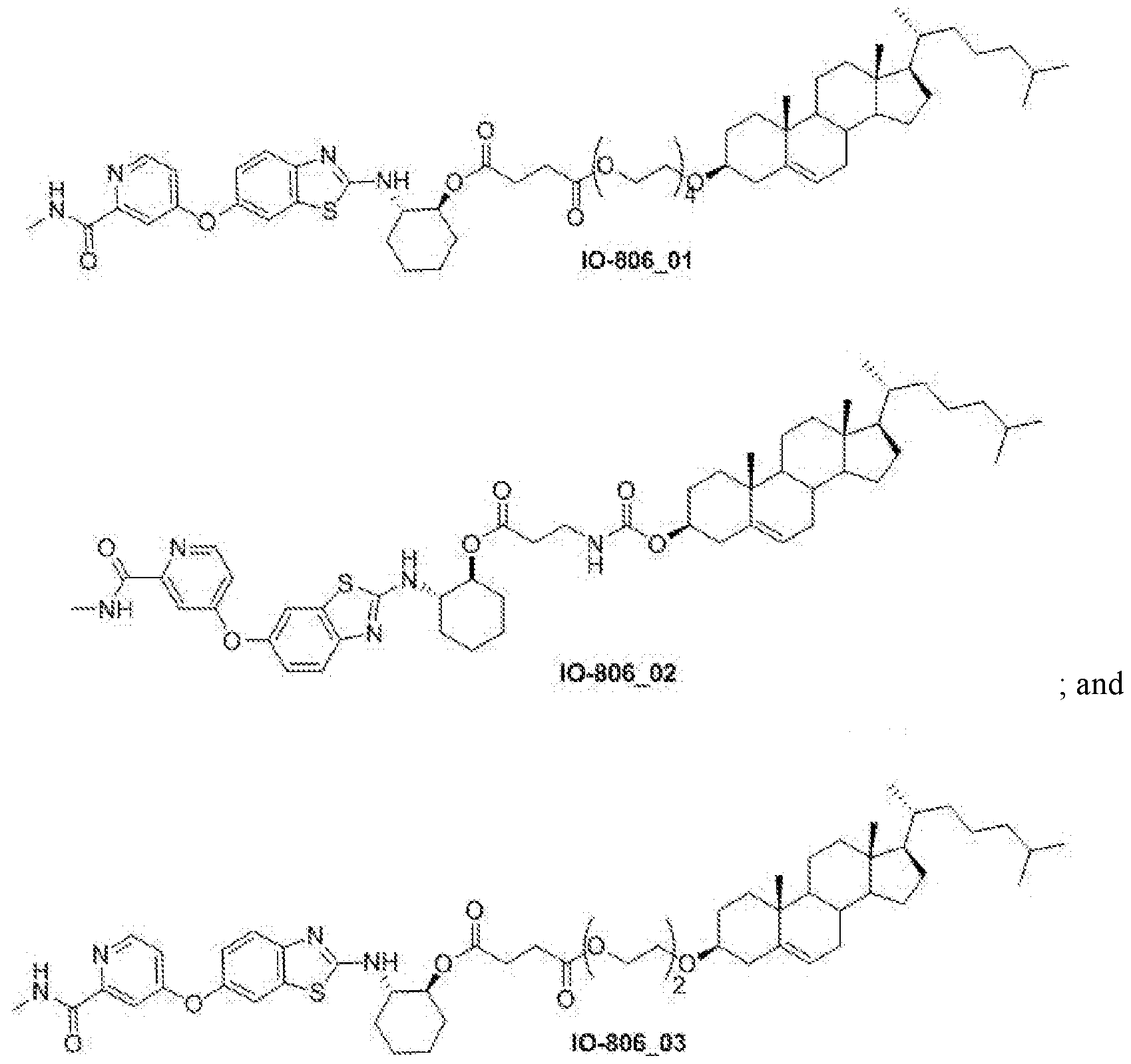
GW-2580 is a potent and selective inhibitor of cFMS receptor kinase, inhibits cFMS in vitro at 60 nM with no activity against 26 other kinases. lx It also completely inhibits CSF-1 induced growth of mouse M- FS-60 myeloid cells and human monocytes (at 1 μΜ). GW-2580 completely inhibits bone degradation in human osteoclasts, rat calvaria and fetal long bone.x
In a non-limiting embodiment of the present disclosure, GW-2580-lipid conjugate is compound of formula .

In another non-limiting embodiment of the present disclosure, GW-2580-lipid conj compound of formula 55.
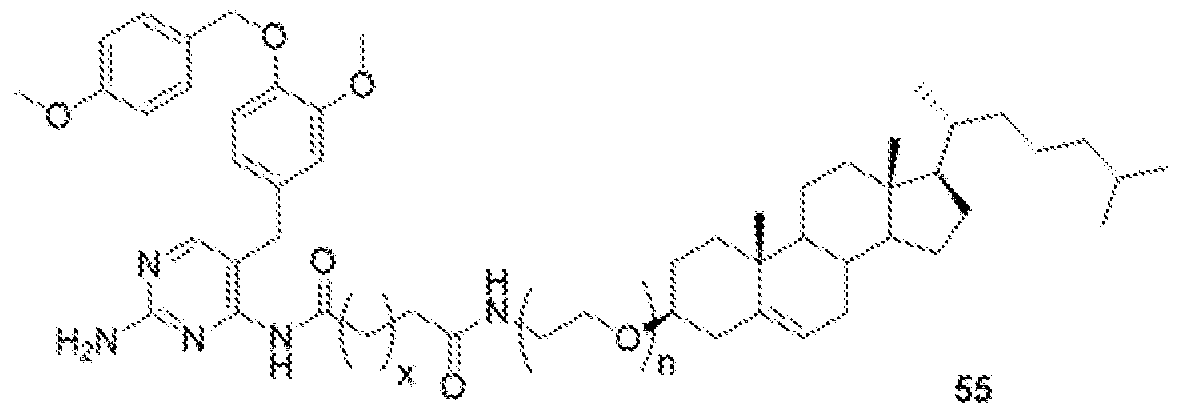
wherein 'n' is 1 to 10 and 'x' is 0 to 3.
In yet another non-limiting embodiment of the present disclosure, GW-2580-lipid conj compound of formula 56.

In still another non-limiting embodiment of the present disclosure, GW-2580-lipid conjugate is compound of formula 57.

wherein and 'm' is 1 to 4.
In still another non-limiting embodiment of the present disclosure, GW-2580-lipid conjugate is compound of formula 58.
wherein 'n' is 1 to 10; R is H, alkyl, acid, amine, aryl or thiol; Y is C, O, H, S; and X is C, O, NH, S.
PLX3397, is an orally administered a small-molecule receptor tyrosine kinase (RTK) inhibitor of KIT, CSF-1R and FLT3, with anti-canecr potential. . The FDA recently approved this molecule for patients with tenosynovial giant cell tumor (TGCT) for which surgical removal is contraindicated. It binds to and inhibits phosphorylation of stem cell factor receptor (KIT), colony-stimulating factor-1 receptor (CSFIR) and FMS-like tyrosine kinase 3 (FLT3), inhibiting tumor cell proliferation, down-modulation of macrophages, osteoclasts and mast cells involved in the metastatic disease. CSFIR and FLT3 are overexpressed or mutated in many cancer cell types and play major roles in tumor cell proliferation and metastasis541
The present disclosure further provides a PLX-3397- lipid conjugate compound selected from:
2.

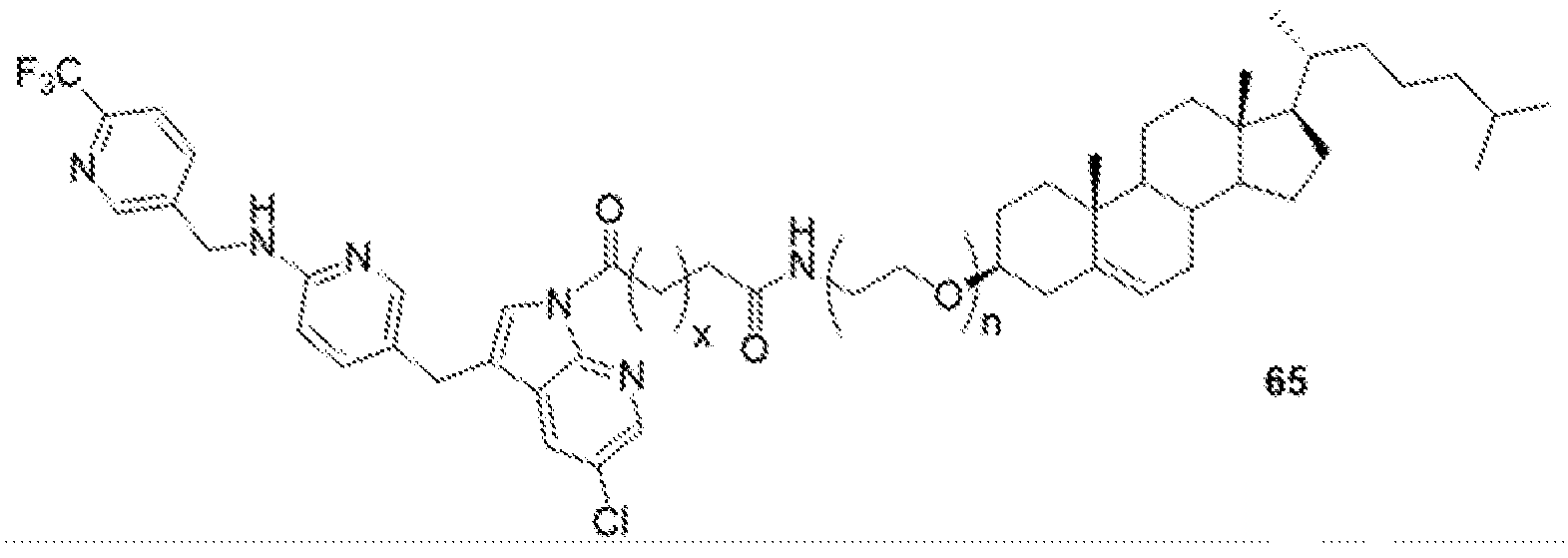
wherein 'n' is 1 to 10 and 'x' is 0 to 3;
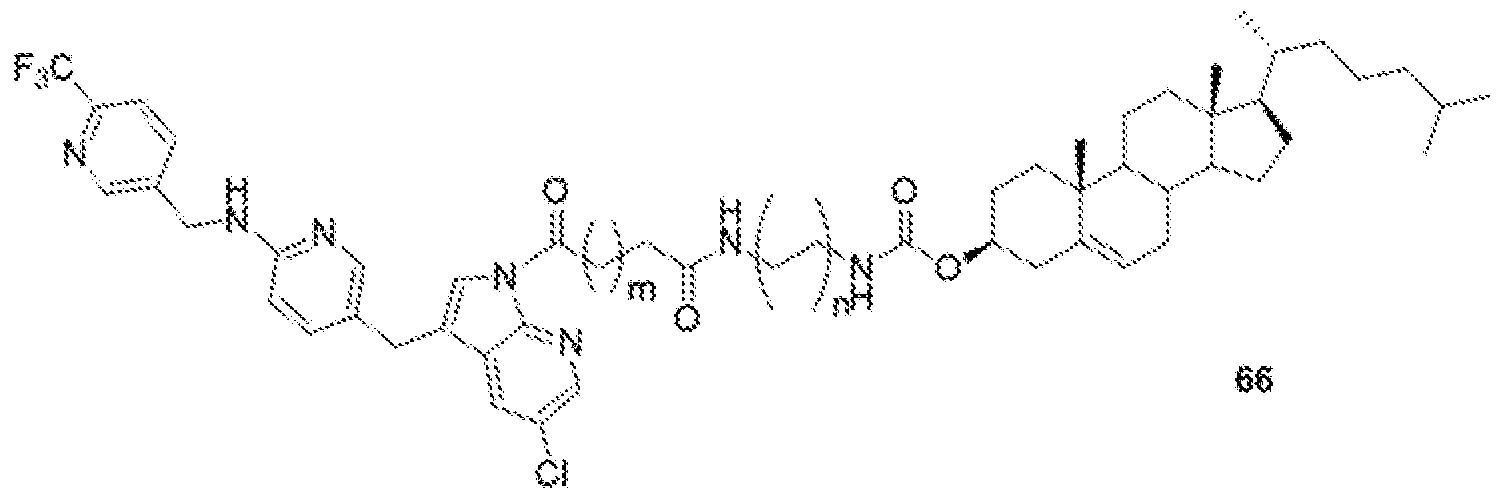
wherein 'n' is 1 to 10 and 'm' is 1 to 4;

wherein 'm' is 1 to 4: and
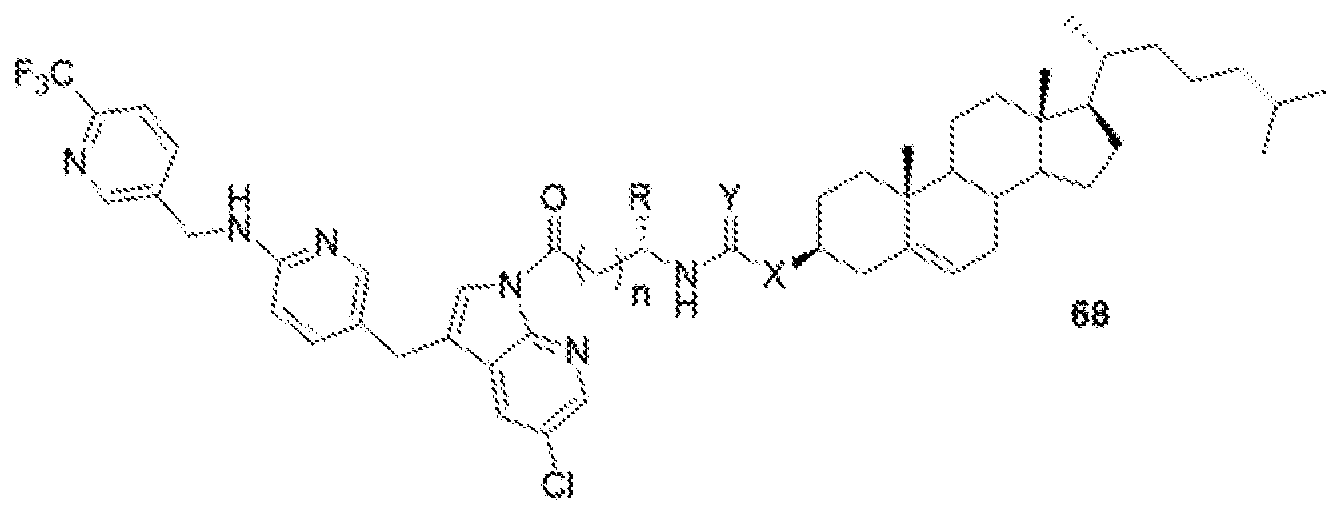
The present disclosure also provides composition(s) comprising the compound of Formula I

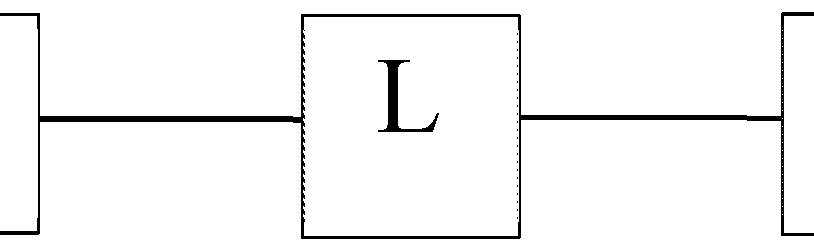
Formula I
wherein 'Ligand' is selected from the group consisting of:
;and
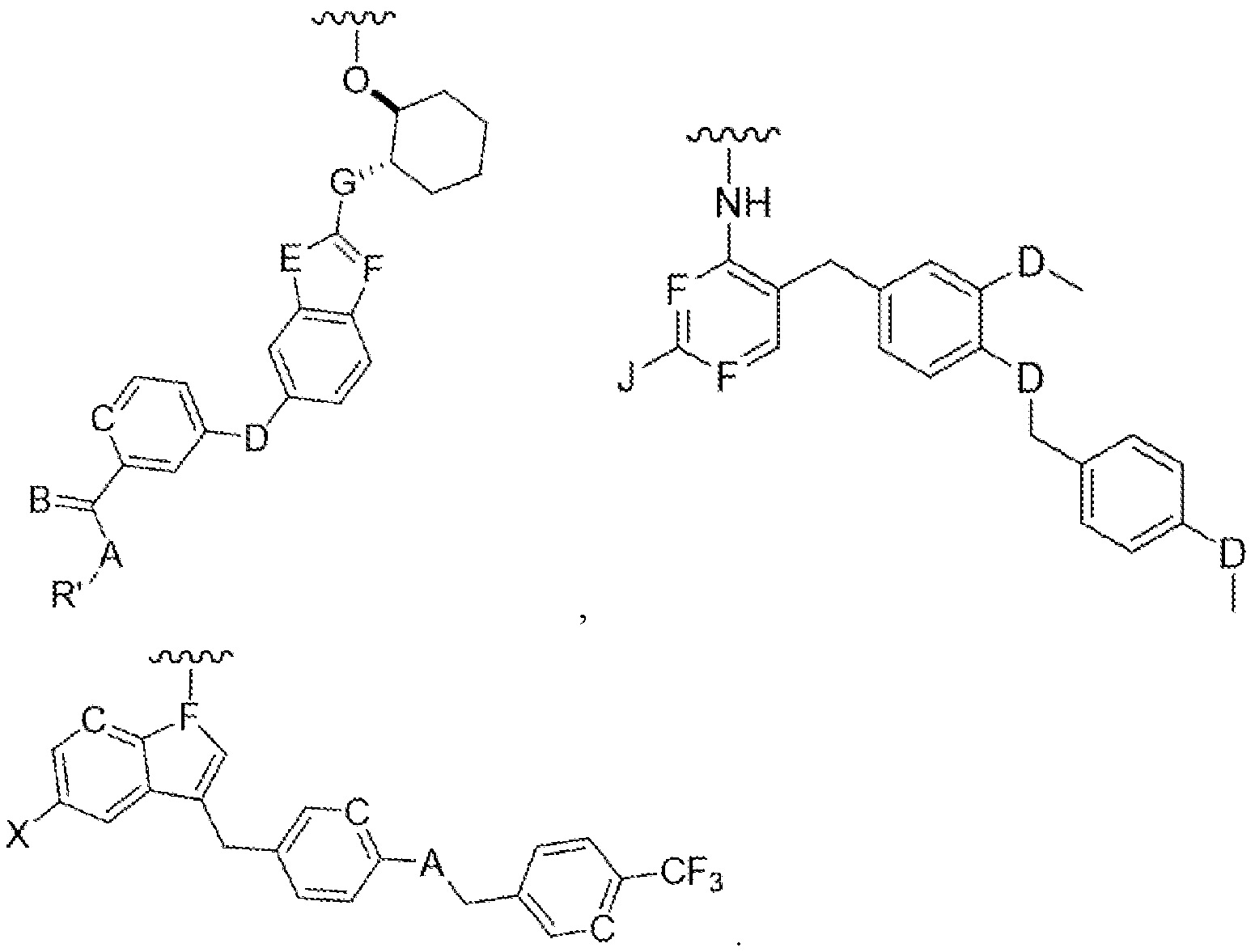
wherein:
C=hydroxy, alkyl group, aryl group, cycloalkyl group;
A=H, O, NH, S;
B=CH, N;
D=C, O, NH, S;
E=C, O, NH, S;
F=CH, N;
G=C, O, NH, S;
J=NH2, OH, SH; and
X=halogen.
'L' is a linker moiety connecting 'Ligand' and 'R' moieties; and
'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
or
any derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates of compound of Formula I thereof.
In a non-limiting embodiment of the present disclosure, the composition comprises from about 1% to about 99% (w/w) of the compound of Formula I.
In another non-limiting embodiment of the present disclosure, the composition further comprises a kinase inhibitor, a chemotherapeutic agent or an immunomodulator or any combination thereof.
In yet another non-limiting embodiment of the present disclosure, the composition further comprises a co-lipid.
In still another non-limiting embodiment of the present disclosure, the co-lipid is selected from the group consisting of HSPC, DSPC, DPPC, DOPC, POPC, SOPC, Egg- PC, and DSPE-PEG, DPPE-PEG, DMPE-PEG or any combination thereof.
In a non-limiting embodiment of the present disclosure, the composition comprises from about 1% to about 99% (w/w) of the kinase inhibitor.
In a non-limiting embodiment of the present disclosure, the composition comprises from about 1%) to about 99% (w/w) of the chemotherapeutic agent.
In another non-limiting embodiment of the present disclosure, the chemotherapeutic agent is selected from the group consisting of PI3K inhibitors; platinum compounds; inhibitors of topoisomerase I and II; alkylating agents; microtubule inhibitors; and angiogenesis inhibitors; or any combination thereof.
In yet another non-limiting embodiment of the present disclosure, the chemotherapeutic agent is selected from the group consisting of germicitibine; Aldesleukin; Alemtuzumab; alitretinoin; allopurinol; altretamine; amifostine; anastrozole; arsenic trioxide; Asparaginase; BCG Live; bexarotene capsules; bexarotene gel; bleomycin; busulfan intravenous; busulfanoral; calusterone; capecitabine; platinate; carmustine; carmustine with Polifeprosan Implant; celecoxib; chlorambucil; cladribine; cyclophosphamide; cytarabine; cytarabine liposomal; dacarbazine; dactinomycin; actinomycin D; Darbepoetin alfa; daunorubicin liposomal; daunorubicin, daunomycin; Denileukin diftitox, dexrazoxane; docetaxel; doxorubicin; doxorubicin liposomal; Dromostanolone propionate; Elliott's B Solution; epirubicin; Epoetin alfa estramustine; etoposide phosphate; etoposide (VP- 16); exemestane; Filgrastim; floxuridine (intraarterial); fludarabine; fluorouracil (5-FU); fulvestrant; gemtuzumab
ozogamicin; goserelin acetate; hydroxyurea; Ibritumomab Tiuxetan; idarubicin; ifosfamide; imatinib mesylate; Interferon alfa-2a; Interferon alfa-2b; irinotecan; letrozole; leucovorin; levamisole; lomustine (CCNU); mechlorethamine (nitrogenmustard); megestrol acetate; melphalan (L-PAM); mercaptopurine (6-MP); mesna; methotrexate; methoxsalen; mitomycin
C; mitotane; mitoxantrone; nandrolone phenpropionate; Nofetumomab; LOddC; Oprelvekin; pamidronate; pegademase; Pegaspargase; Pegfilgrastim; pentostatin; pipobroman; plicamycin; mithramycin; porfimer sodium; procarbazine; quinacrine; Rasburicase; Rituximab;
Sargramostim; streptozocin; talbuvidine (LDT); talc; tamoxifen; temozolomide; teniposide
(VM-26); testolactone; thioguanine (6-TG); thiotepa; topotecan; toremifene; Tositumomab;
Trastuzumab; tretinoin (ATRA); Uracil Mustard; valrubicin; valtorcitabine (monoval LDC); vinblastine; vinorelbine; and zoledronate; or any mixture thereof
In still another non-limiting embodiment of the present disclosure, the PI3K inhibitor is selected from the group consisting of PI103; P1828; LY294002; wortmannin; demethoxyviridin; IC486068; IC87114; GDC-0941; perifosine; CALIOI; PX-866; IPI-145;
BAY 80-6946; BEZ235; P6503; TGR1202; SF1126; INK1117; BKM120; IL147; XL765; Palomid 529; GSK1059615; ZSTK474; PWT33597; TG100-115; CAL263; G E-447;
CUDC-907; and AEZS-136, or any combination thereof.
In a non-limiting embodiment of the present disclosure, the chemotherapeutic agent is conjugated with a component of the composition.
In a non-limiting embodiment of the present disclosure, the chemotherapeutic agent is conjugated with Polyethylene glycol (PEG).
In a non-limiting embodiment of the present disclosure, the composition comprises from about 1% to about 99% (w/w) of the immunomodulator.
In a non-limiting embodiment of the present disclosure, the composition is a liposome, emulsion, or micelle.
In another non-limiting embodiment of the present disclosure, the composition is a nanoparticle, wherein the nanoparticle is about 1 nm to about 400 nm in diameter.
In a non-limiting embodiment of the present disclosure, the composition further comprises a pharmaceutically acceptable carrier.
In a non-limiting embodiment of the present disclosure, the composition further comprises a pharmaceutically acceptable excipient.
In another non-limiting embodiment of the present disclosure, the pharmaceutically acceptable excipient is selected from the group comprising adjuvant, diluent, carrier, granulating agents, binding agents, lubricating agents, disintegrating agent, sweetening agents, glidant, anti-
adherent, anti-static agent, surfactant, anti-oxidant, gum, coating agent, coloring agent, flavouring agent, coating agent, plasticizer, preservative, suspending agent, emulsifying agent, plant cellulosic material, spheronization agent, and other conventionally known pharmaceutically acceptable excipient, or any combination of excipients thereof.
In a non-limiting embodiment of the present disclosure, the composition inhibits colony stimulating factor or colony stimulating factor-1 receptor (CSF-1R) signaling pathway and is administered to a subject in need thereof through modes selected from a group comprising intravenous administration, intramuscular administration, intraperitoneal administration, hepatoportal administration, intra articular administration and pancreatic duodenal artery administration, or any combination thereof.
The present disclosure also provides a method of treating cancer, comprising, administering the compound of Formula I or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition thereof to a subject in need of treatment for cancer. In a non-limiting embodiment of the present disclosure, the cancer is selected from the group consisting of breast cancer; ovarian cancer; glioma; gastrointestinal cancer; prostate cancer; carcinoma, lung carcinoma, hepatocellular carcinoma, testicular cancer; cervical cancer; endometrial cancer; bladder cancer; head and neck cancer; lung cancer; gastro-esophageal cancer, and gynecological cancer, or any combination thereof.
In another non-limiting embodiment of the present disclosure, the method comprises coadministering one or more additional anti-cancer therapy to the subject.
In yet another non-limiting embodiment of the present disclosure, the additional anti-cancer therapy is selected from the group consisting of surgery, chemotherapy, radiation therapy, thermotherapy, immunotherapy, hormone therapy, laser therapy, and anti-angiogenic therapy, or any combination thereof.
In still another non-limiting embodiment of the present disclosure, the additional anti-cancer therapy comprises administering a kinase inhibitor, a chemotherapeutic agent, an immunomodulator or any combination thereof, to the subject.
In a non-limiting embodiment of the present disclosure, the immunomodulator activates an immune response against cancer cells, wherein the immunomodulator is selected from the group consisting of antibody, natural killer cells, lymphokine-activated killer cells, cytotoxic T cells and dendritic cells, anti-PD-Ll antibodies, anti-PD-1 antibodies, anti-CD52 antibodies, anti-VEGF-A antibodies, anti-CD30 antibodies, anti-EGFR antibodies, anti-CD33 antibodies, anti-CD20 antibodies, anti-CTLA4 antibodies, anti-HER-2 antibodies, interferons, and interleukins, or any combination thereof.
In another non-limiting embodiment of the present disclosure, the antibody is a therapeutic antibody or a targeting antibody or a combination thereof.
The present disclosure also provides a method for inhibition of CSF or CSF-1R signalling pathway in a cell, wherein said method comprises act of contacting the cell with the compound of formula I, derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition thereof.
In some embodiments, the technology described herein relates to a pharmaceutical composition comprising a supramolecular combinatorial therapeutic and a pharmaceutically acceptable carrier. Pharmaceutically acceptable carriers and diluents include saline, aqueous buffer solutions, solvents and/or dispersion media. The use of such carriers and diluents is well known in the art. Some non-limiting examples of materials which can serve as pharmaceutically- acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, methylcellulose, ethyl cellulose, microcrystalline cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) lubricating agents, such as magnesium stearate, sodium lauryl sulfate and talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol (PEG); (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminium hydroxide; (15) alginic acid; (16) pyrogen-free water; (17) isotonic saline; (18) Ringer's solution; (19) ethyl alcohol; (20) pH buffered solutions; (21) polyesters, polycarbonates and/or polyanhydrides; (22) bulking agents, such as polypeptides and amino acids (23) serum component, such as serum albumin, HDL and LDL; (22) C2-C12 alcohols, such as ethanol; and (23) other non-toxic compatible substances employed in pharmaceutical formulations. Wetting agents, coloring agents, release agents, coating agents, sweetening agents, flavoring agents, perfuming agents, preservative and antioxidants can also be present in the formulation. The terms such as "excipient", "carrier", "pharmaceutically acceptable carrier" or the like are used interchangeably herein. In some embodiments, the carrier inhibits the degradation of the active agent, e.g. a composition as described herein.
In some embodiments, the pharmaceutical composition comprising a supramolecular combinatorial therapeutic can be a parenteral dose form. Since administration of parenteral dosage forms typically bypasses the patient's natural defenses against contaminants, parenteral dosage forms are preferably sterile or capable of being sterilized prior to administration to a
patient. Examples of parenteral dosage forms include, but are not limited to, solutions ready for injection, dry products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection, suspensions ready for injection, and emulsions. In addition, controlled- release parenteral dosage forms can be prepared for administration of a patient, including, but not limited to, DUROS®-type dosage forms and dose-dumping.
Suitable vehicles that can be used to provide parenteral dosage forms of a composition as described herein are well known to those skilled in the art. Examples include, without limitation: sterile water; water for injection USP; saline solution; glucose solution; aqueous vehicles such as but not limited to, sodium chloride injection, Ringer's injection, dextrose Injection, dextrose and sodium chloride injection, and lactated Ringer's injection; water- miscible vehicles such as, but not limited to, ethyl alcohol, polyethylene glycol, and propylene glycol; and non-aqueous vehicles such as, but not limited to, corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate. Compounds that alter or modify the solubility of a pharmaceutically acceptable salt can also be incorporated into the parenteral dosage forms of the disclosure, including conventional and controlled-release parenteral dosage forms.
Pharmaceutical compositions can also be formulated to be suitable for oral administration, for example as discrete dosage forms, such as, but not limited to, tablets (including without limitation scored or coated tablets), pills, caplets, capsules, chewable tablets, powder packets, cachets, troches, wafers, aerosol sprays, or liquids, such as but not limited to, syrups, elixirs, solutions or suspensions in an aqueous liquid, a non-aqueous liquid, an oil-in-water emulsion, or a water-in-oil emulsion. Such compositions contain a predetermined amount of the pharmaceutically acceptable salt of the disclosed compounds, and may be prepared by methods of pharmacy well known to those skilled in the art. See generally, Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams, and Wilkins, Philadelphia PA. (2005).
The present disclosure further provides a formulation comprising the compound of Formula I

Formula I
wherein, 'Ligand' is selected from the group consisting
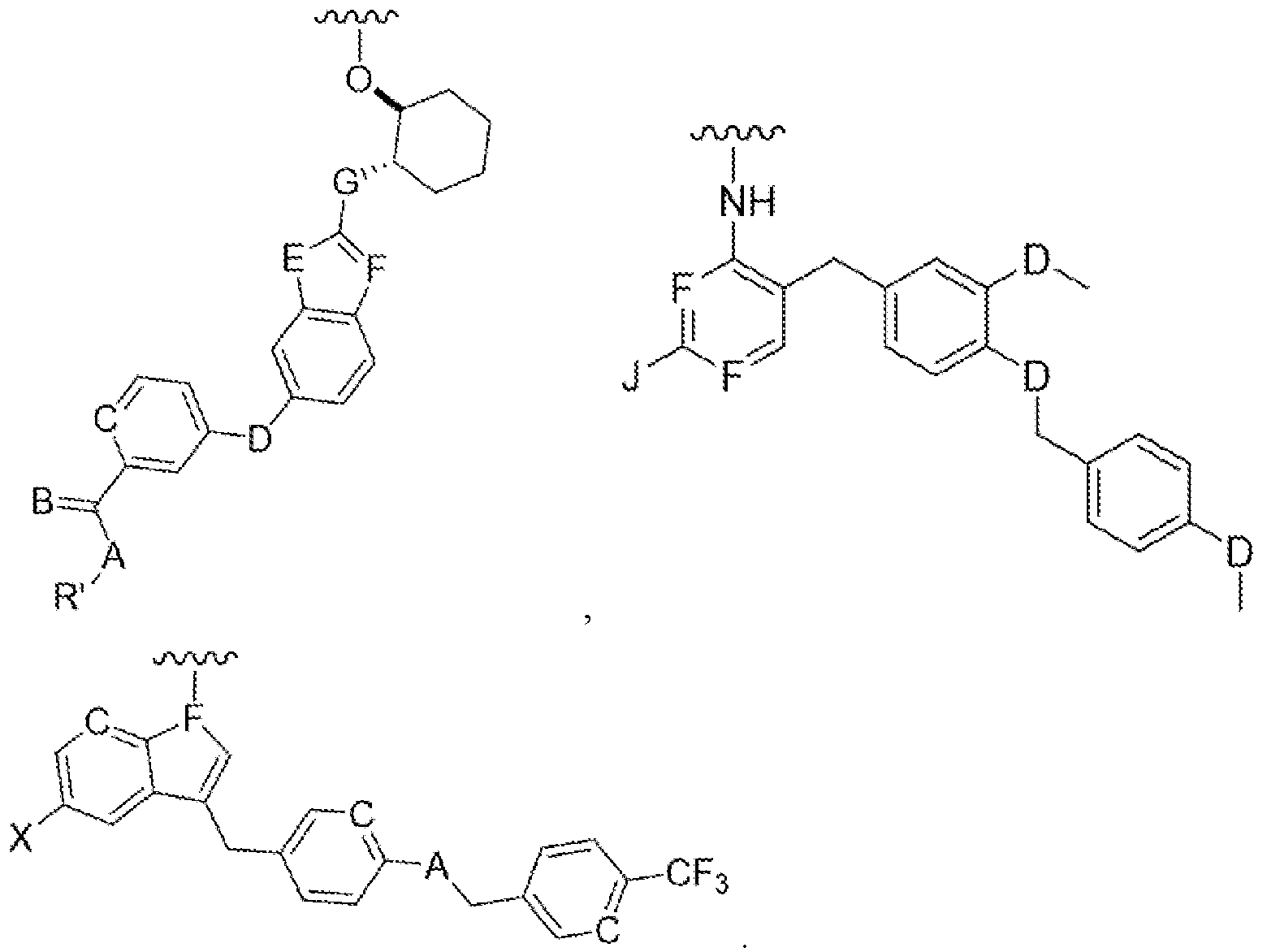
wherein:
C=hydroxy, alkyl group, aryl group, cycloalkyl group;
A=H, O, H, S;
B=CH, N;
D=C, O, NH, S;
E=C, O, NH, S;
F=CH, N;
G=C, O, NH, S;
J=NH2, OH, SH; and
X=halogen.
'L' is a linker moiety connecting 'Ligand' and 'R' moieties; and
'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
or
any derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates of compound of Formula I thereof.
In a non-limiting embodiment of the present disclosure, the phospholipid or PEGylated phospholipid is selected from a group comprising HSPC, DSPC, DPPC, DOPC, POPC, SOPC, Egg PC , DPPE-PEG, DMPE-PEG and DSPE-PEG or any combination thereof; wherein the composition comprises about 1% to about 99% (w/w) of these phospholipid and PEGylated phospholipid or any combination thereof.
In a non-limiting embodiment of the present disclosure, the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 5 :55 :35 :5.
In a non-limiting embodiment of the present disclosure, the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 10:50:35 :5.
In a non-limiting embodiment of the present disclosure, the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 15 :50:30:5.
The compounds, compositions and methods described herein can be administered to a subject having or diagnosed as having cancer. In some embodiments, the methods described herein comprise administering an effective amount of compositions described herein to a subject in order to alleviate a symptom of a cancer. As used herein, "alleviating a symptom of a cancer" is ameliorating any condition or symptom associated with the cancer. As compared with an equivalent untreated control, such reduction is by at least 5%, 10%, 20%, 40%, 50%, 60%, 80%), 90%), 95%), 99% or more as measured by any standard technique. A variety of means for administering the compositions described herein to subjects are known to those of skill in the art. Such methods can include, but are not limited to oral, parenteral, intravenous, intramuscular, subcutaneous, transdermal, airway (aerosol), pulmonary, cutaneous, topical, injection, or intratumoral administration. Administration can be local or systemic.
The term "effective amount" as used herein refers to the amount of a composition described herein needed to alleviate at least one or more symptom of the disease or disorder, and relates to a sufficient amount of pharmacological composition to provide the desired effect. The term "therapeutically effective amount" therefore refers to an amount of a composition described herein that is sufficient to provide a particular anti-tumor effect when administered to a typical subject. An effective amount as used herein, in various contexts, would also include an amount sufficient to delay the development of a symptom of the disease, alter the course of a symptom disease (for example but not limited to, slowing the progression of a symptom of the disease), or reverse a symptom of the disease. Thus, it is not generally practicable to specify an exact "effective amount". However, for any given case, an appropriate "effective amount" can be determined by one of ordinary skill in the art using only routine experimentation.
In certain embodiments, an effective dose of a composition as described herein can be administered to a patient once. In certain embodiments, an effective dose of a composition as described herein can be administered to a patient repeatedly. For systemic administration, subjects can be administered a therapeutic amount of a composition as described herein, such as, e.g. 0.1 mg/kg, 0.5 mg/kg, 1.0 mg/kg, 2.0 mg/kg, 2.5 mg/kg, 5 mg/kg, 10 mg/kg, 15 mg/kg, 20 mg/kg, 25 mg/kg, 30 mg/kg, 40 mg/kg, 50 mg/kg, or more.
In some embodiments, after an initial treatment regimen, the treatments can be administered on a less frequent basis. For example, after treatment biweekly for three months, treatment can be repeated once per month, for six months or a year or longer. Treatment according to the methods described herein can reduce levels of a marker or symptom of a condition, e.g. tumor size and/or growth by at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80 % or at least 90% or more.
The dosage of a composition as described herein can be determined by a physician and adjusted, as necessary, to suit observed effects of the treatment. With respect to duration and frequency of treatment, it is typical for skilled clinicians to monitor subjects in order to determine when the treatment is providing therapeutic benefit, and to determine whether to increase or decrease dosage, increase or decrease administration frequency, discontinue treatment, resume treatment, or make other alterations to the treatment regimen. The dosing schedule can vary from once a week to daily depending on a number of clinical factors, such as the subject's sensitivity a composition as described herein. The desired dose or amount of activation can be administered at one time or divided into subdoses, e.g., 2-4 subdoses and administered over a period of time, e.g., at appropriate intervals through the day or other appropriate schedule. In some embodiments, administration can be chronic, e.g., one or more doses and/or treatments daily over a period of weeks or months. Examples of dosing and/or treatment schedules are administration daily, twice daily, three times daily or four or more times daily over a period of 1 week, 2 weeks, 3 weeks, 4 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, or 6 months, or more. A composition as described herein can be administered over a period of time, such as over a 5 minute, 10 minute, 15 minute, 20 minute, or 25 minute period.
The dosage ranges for the administration of a composition as described herein, according to the methods described herein depend upon, for example, the form of a composition as described herein, its potency, and the extent to which symptoms, markers, or indicators of a condition described herein are desired to be reduced, for example the percentage reduction desired for tumor growth. The dosage should not be so large as to cause adverse side effects. Generally, the dosage will vary with the age, condition, and sex of the patient and can be determined by one of skill in the art. The dosage can also be adjusted by the individual physician in the event of any complication.
The efficacy of a composition as described herein in, e.g. the treatment of a condition described herein, or to induce a response as described herein can be determined by the skilled clinician. However, a treatment is considered "effective treatment," as the term is used herein, if one or
more of the signs or symptoms of a condition described herein are altered in a beneficial manner, other clinically accepted symptoms are improved, or even ameliorated, or a desired response is induced e.g., by at least 10% following treatment according to the methods described herein. Efficacy can be assessed, for example, by measuring a marker, indicator, symptom, and/or the incidence of a condition treated according to the methods described herein or any other measurable parameter appropriate, e.g. tumor size and/or growth. Efficacy can also be measured by a failure of an individual to worsen as assessed by hospitalization, or need for medical interventions (i.e., progression of the disease is halted). Methods of measuring these indicators are known to those of skill in the art and/or are described herein. Treatment includes any treatment of a disease in an individual or an animal (some non-limiting examples include a human or an animal) and includes: (1) inhibiting the disease, e.g., preventing a worsening of symptoms (e.g. pain or inflammation); or (2) relieving the severity of the disease, e.g., causing regression of symptoms. An effective amount for the treatment of a disease means that amount which, when administered to a subject in need thereof, is sufficient to result in effective treatment as that term is defined herein, for that disease. Efficacy of an agent can be determined by assessing physical indicators of a condition or desired response, (e.g. tumor size and/or growth). It is well within the ability of one skilled in the art to monitor efficacy of administration and/or treatment by measuring any one of such parameters, or any combination of parameters. Efficacy can be assessed in animal models of a condition described herein, for example treatment of cancer. When using an experimental animal model, efficacy of treatment is evidenced when a statistically significant change in a marker is observed, e.g. a decreased in tumor size and/or growth.
As used herein, a "subject" means a human or animal. Usually the animal is a vertebrate such as a primate, rodent, domestic animal or game animal. Primates include chimpanzees, cynomologous monkeys, spider monkeys, and macaques, e.g., Rhesus. Rodents include mice, rats, woodchucks, ferrets, rabbits and hamsters. Domestic and game animals include cows, horses, pigs, deer, bison, buffalo, feline species, e.g., domestic cat, canine species, e.g., dog, fox, wolf, avian species, e.g., chicken, emu, ostrich, and fish, e.g., trout, catfish and salmon. In some embodiments, the subject is a mammal, e.g., a primate, e.g., a human. The terms, "individual," "patient" and "subject" are used interchangeably herein.
Preferably, the subject is a mammal. The mammal can be a human, non-human primate, mouse, rat, dog, cat, horse, or cow, but is not limited to these examples. Mammals other than humans can be advantageously used as subjects that represent animal models of cancer. A subject can be male or female.
A subject can be one who has been previously diagnosed with or identified as suffering from or having a condition in need of treatment (e.g. cancer) or one or more complications related to such a condition, and optionally, have already undergone treatment for cancer or the one or more complications related to cancer. Alternatively, a subject can also be one who has not been previously diagnosed as having cancer or one or more complications related to cancer. For example, a subject can be one who exhibits one or more risk factors for cancer or one or more complications related to cancer or a subject who does not exhibit risk factors.
A "subject in need" of treatment for a particular condition can be a subject having that condition, diagnosed as having that condition, or at risk of developing that condition.
The term "agent" refers generally to any entity which is normally not present or not present at the levels being administered to a cell, tissue or subject. An agent can be selected from a group including but not limited to: polynucleotides; polypeptides; small molecules; and antibodies or antigen-binding fragments thereof. A polynucleotide can be RNA or DNA, and can be single or double stranded, and can be selected from a group including, for example, nucleic acids and nucleic acid analogues that encode a polypeptide. A polypeptide can be, but is not limited to, a naturally-occurring polypeptide, a mutated polypeptide or a fragment thereof that retains the function of interest. Further examples of agents include, but are not limited to a nucleic acid aptamer, peptide-nucleic acid (PNA), locked nucleic acid (LNA), small organic or inorganic molecules; saccharide; oligosaccharides; polysaccharides; biological macromolecules, peptidomimetics; nucleic acid analogs and derivatives; extracts made from biological materials such as bacteria, plants, fungi, or mammalian cells or tissues and naturally occurring or synthetic compositions. An agent can be applied to the media, where it contacts the cell and induces its effects. Alternatively, an agent can be intracellular as a result of introduction of a nucleic acid sequence encoding the agent into the cell and its transcription resulting in the production of the nucleic acid and/or protein environmental stimuli within the cell. In some embodiments, the agent is any chemical, entity or moiety, including without limitation synthetic and naturally-occurring non-proteinaceous entities. In certain embodiments the agent is a small molecule having a chemical moiety selected, for example, from unsubstituted or substituted alkyl, aromatic, or heterocyclyl moieties including macrolides, leptomycins and related natural products or analogues thereof. Agents can be known to have a desired activity and/or property, or can be selected from a library of diverse compounds. As used herein, the term "small molecule" can refer to compounds that are "natural product-like," however, the term "small molecule" is not limited to "natural product-like" compounds. Rather, a small molecule is typically characterized in that it contains several carbon— carbon bonds, and has a
molecular weight more than about 50, but less than about 5000 Daltons (5 kD). Preferably the small molecule has a molecular weight of less than 3 kD, still more preferably less than 2 kD, and most preferably less than 1 kD. In some cases it is preferred that a small molecule have a molecular mass equal to or less than 700 Daltons.
As used herein, the term "inhibitor" refers to an agent which can decrease the expression and/or activity of the targeted expression product (e.g. mRNA encoding the target or a target polypeptide), e.g. by at least 10% or more, e.g. by 10% or more, 50% or more, 70% or more, 80% or more, 90% or more, 95% or more, or 98 % or more. The efficacy of an inhibitor of, for example, PI3K, e.g. its ability to decrease the level and/or activity of PI3K can be determined, e.g. by measuring the level of a PI3K polypeptide (and/or mRNA encoding such a polypeptide) and/or the activity of PI3K. Methods for measuring the level of a given mRNA and/or polypeptide are known to one of skill in the art, e.g. RTPCR with primers can be used to determine the level of RNA and Western blotting with an antibody can be used to determine the level of a polypeptide. The activity of, e.g. PI3K can be determined using methods known in the art and described above herein. .
As used herein, the terms "treat," "treatment," "treating," or "amelioration" refer to therapeutic treatments, wherein the object is to reverse, alleviate, ameliorate, inhibit, slow down or stop the progression or severity of a condition associated with a disease or disorder, e.g. cancer. The term "treating" includes reducing or alleviating at least one adverse effect or symptom of a condition, disease or disorder associated with a cancer. Treatment is generally "effective" if one or more symptoms or clinical markers are reduced. Alternatively, treatment is "effective" if the progression of a disease is reduced or halted. That is, "treatment" includes not just the improvement of symptoms or markers, but also a cessation of, or at least slowing of, progress or worsening of symptoms compared to what would be expected in the absence of treatment. Beneficial or desired clinical results include, but are not limited to, alleviation of one or more symptom(s), diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, remission (whether partial or total), and/or decreased mortality, whether detectable or undetectable. The term "treatment" of a disease also includes providing relief from the symptoms or side-effects of the disease (including palliative treatment).
As used herein, the term "composition" or "pharmaceutical composition" are used interchangeably and refers to the active agent in combination with a pharmaceutically acceptable carrier e.g. a carrier commonly used in the pharmaceutical industry. The phrase "pharmaceutically acceptable" is employed herein to refer to those compounds, materials,
compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
As used herein, the term "administering," refers to the placement of a compound as disclosed herein into a subject by a method or route which results in at least partial delivery of the agent at a desired site. Pharmaceutical compositions comprising the compounds disclosed herein can be administered by any appropriate route which results in an effective treatment in the subject. As used herein, the term "amphiphilic" refers to a molecule that has both a hydrophobic portion and a lipophobic portion, i.e. at least one a polar, water-soluble group and at least one a nonpolar, water- insoluble group. Typically, in a two phase system having a polar, aqueous phase and a non-polar, non-aqueous phase, an amphiphilic molecule will partition to the interface of the two phases. In simpler non limiting terms, an amphiphile is a molecule that is soluble in both an aqueous environment and a non-aqueous environment. The term "amphiphile" refers to an amphiphilic molecule.
As used herein the term "comprising" or "comprises" is used in reference to compositions, methods, and respective component(s) thereof, that are essential to the method or composition, yet open to the inclusion of unspecified elements, whether essential or not.
The singular terms "a," "an," and "the" include plural referents unless context clearly indicates otherwise. Similarly, the word "or" is intended to include "and" unless the context clearly indicates otherwise. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of this disclosure, suitable methods and materials are described below. The abbreviation, "e.g." is derived from the Latin exempli gratia, and is used herein to indicate a non-limiting example. Thus, the abbreviation "e.g." is synonymous with the term "for example."
The description of embodiments of the disclosure is not intended to be exhaustive or to limit the disclosure to the precise form disclosed. While specific embodiments of, and examples for, the disclosure are described herein for illustrative purposes, various equivalent modifications are possible within the scope of the disclosure, as those skilled in the relevant art will recognize. For example, while method steps or functions are presented in a given order, alternative embodiments may perform functions in a different order, or functions may be performed substantially concurrently. The teachings of the disclosure provided herein can be applied to other procedures or methods as appropriate. The various embodiments described herein can be combined to provide further embodiments. Aspects of the disclosure can be modified, if
necessary, to employ the compositions, functions and concepts of the above references and application to provide yet further embodiments of the disclosure. These and other changes can be made to the disclosure in light of the detailed description. All such modifications are intended to be included within the scope of the appended claims.
Specific elements of any of the foregoing embodiments can be combined or substituted for elements in other embodiments. Furthermore, while advantages associated with certain embodiments of the disclosure have been described in the context of these embodiments, other embodiments may also exhibit such advantages, and not all embodiments need necessarily exhibit such advantages to fall within the scope of the disclosure.
The scheme for the preparation of the final compound was divided into 2 parts:
1. Preparation of the ligand
2. Attaching of other part to the molecule to make the final compound. Scheme 1
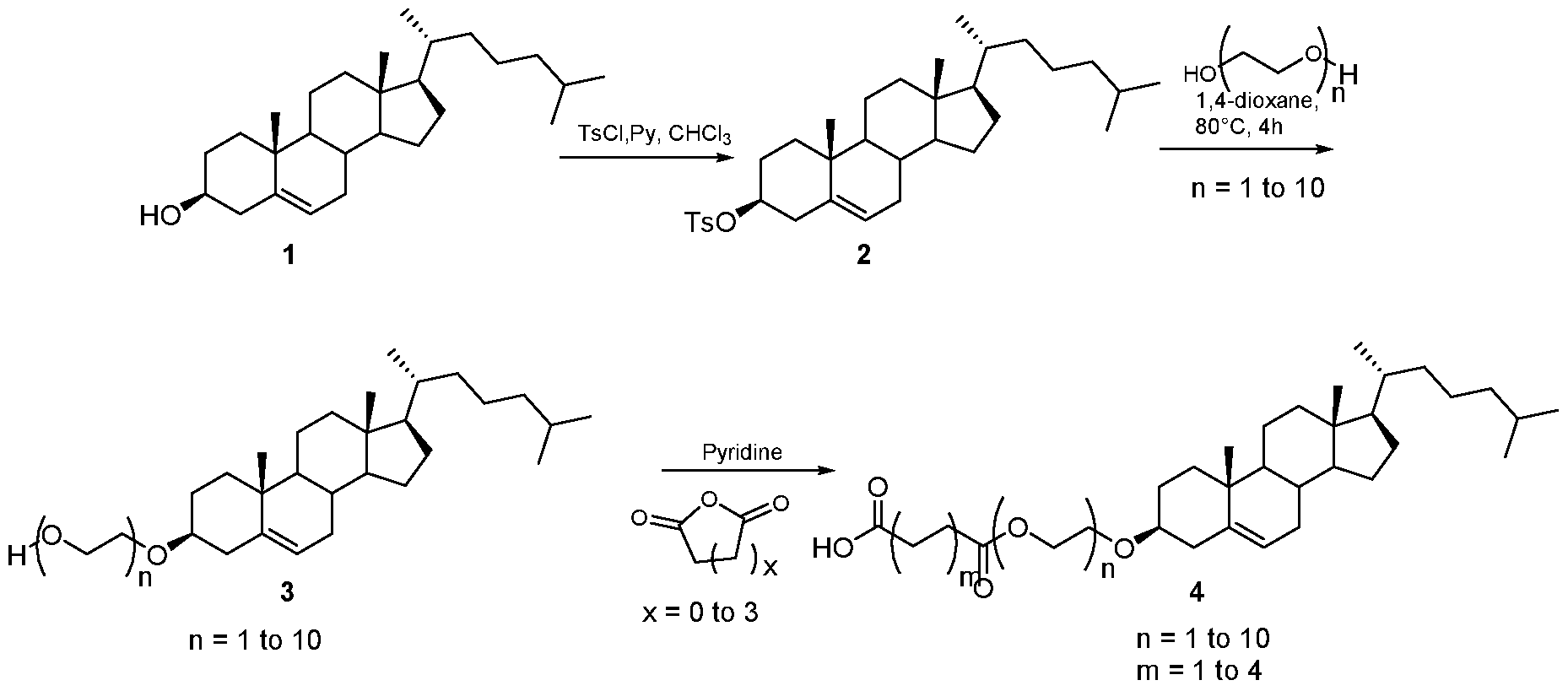
Preparation of the ligand
In scheme 1, cholesterol was converted to compound 3 using the reported procedure. Compound 3 was further treated with cyclic acid anhydride in presence of pyridine at room temperature for overnight to yield compound 4.
Starting from previously reported compound 3, tosylation of the hydroxyl group was performed using known procedure and then converted to compound 6 using sodium azide at room
temperature. (Scheme 2). The azide thus prepared was reduced to amine, which was again treated with cyclic acid anhydride to give carboxylic acid derivative 8.
Scheme 2

x = 0 to 3
In scheme 3, commercially available cholesteroyl chloride was initially treated with diamine of different chain length to give compound 10. Amine 10 was again treated with cyclic acid anhydride to give carboxylic acid derivative 11 of different chain length.
Scheme 3

Scheme 4
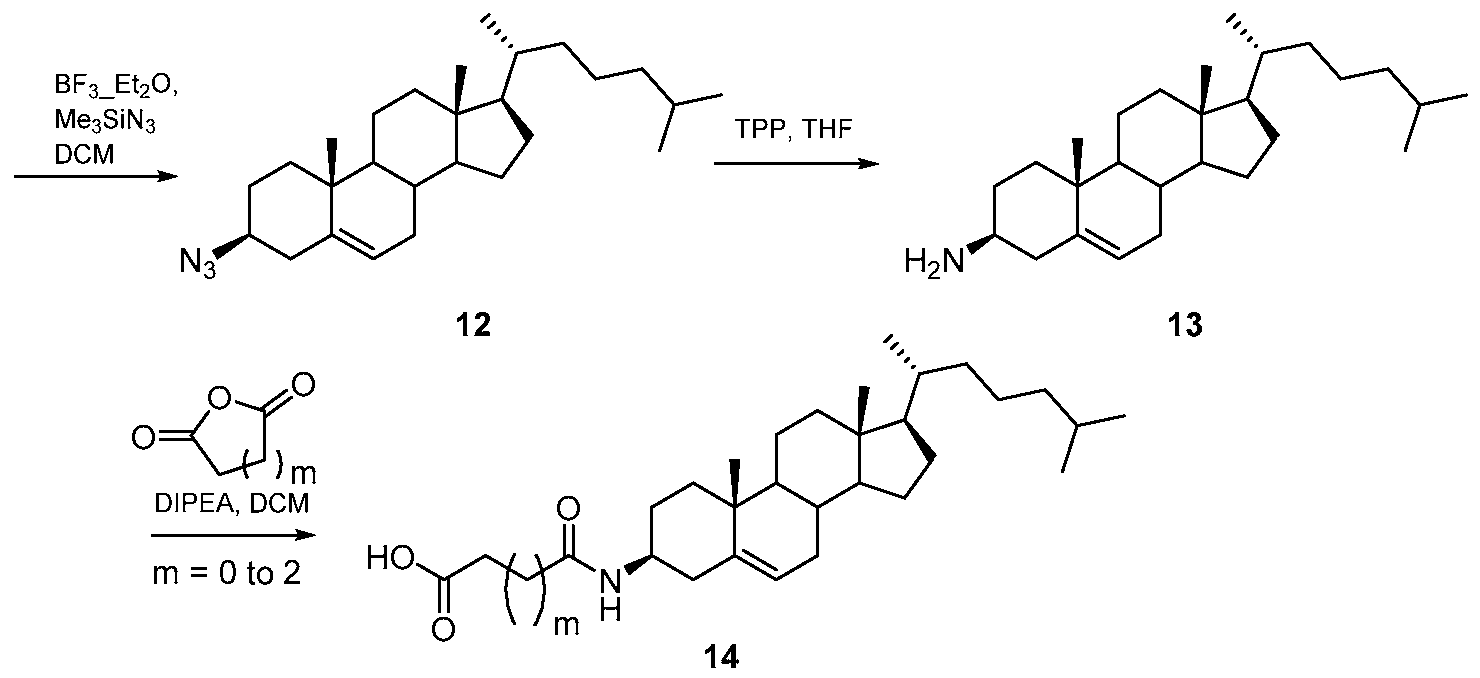
By following a known procedure compound 2 was transformed to azide derivative 12 in presence of BF3.Et20 and trimethyl silyl azide. (Scheme 4) The azide thus produced was successively reduced and then treated with cyclic acid anhydride of different chain length to prepare compound 14.
Scheme 5
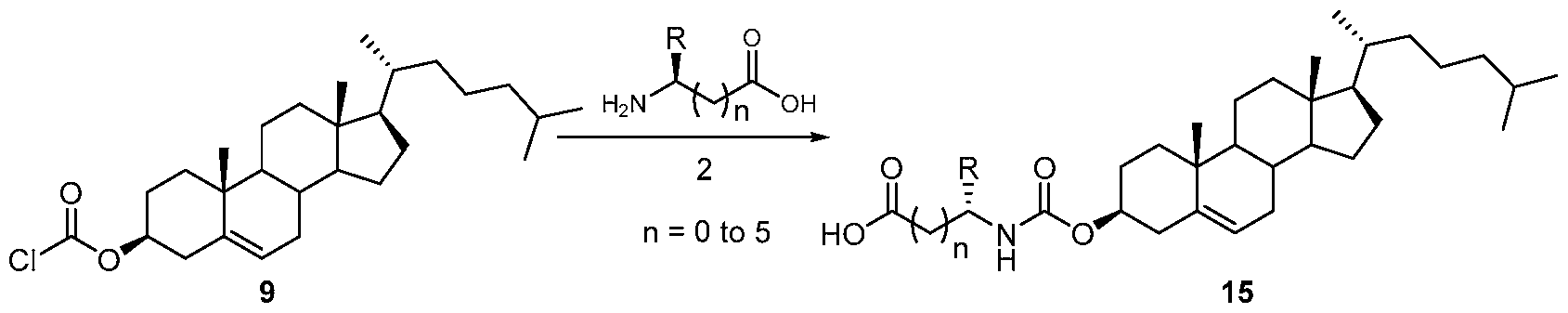
Different amino acid with varying chain lengths as well as di/tripeptide of various combination was directly attached with commercially available cholesteroyl chloride in known alkaline condition to give different cholesterol acid derivatives 15. (Scheme 5)
The acid thus prepared was then coupled with commercially available alicyclic derivative of N-Boc amine-alcohols and subsequently deprotected to get the amines. (Scheme 6)
Scheme 6
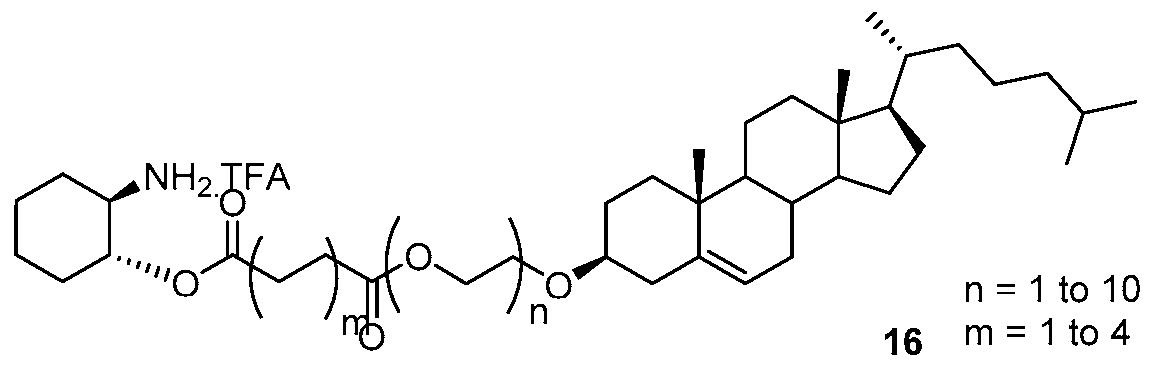

The amine salt (16- 20) thus produced was treated with freshly prepared sulphoxide derivative (Scheme 7) using diisopropyl ethyl amine in N-methyl morpholine at elevated temperature for 3 days.™ The compound formation was monitored by using TLC method. (Scheme 8) The crude reaction mixture was then evaporated and then purified by column chromatography to get the final compound.
Scheme 7
Scheme 8
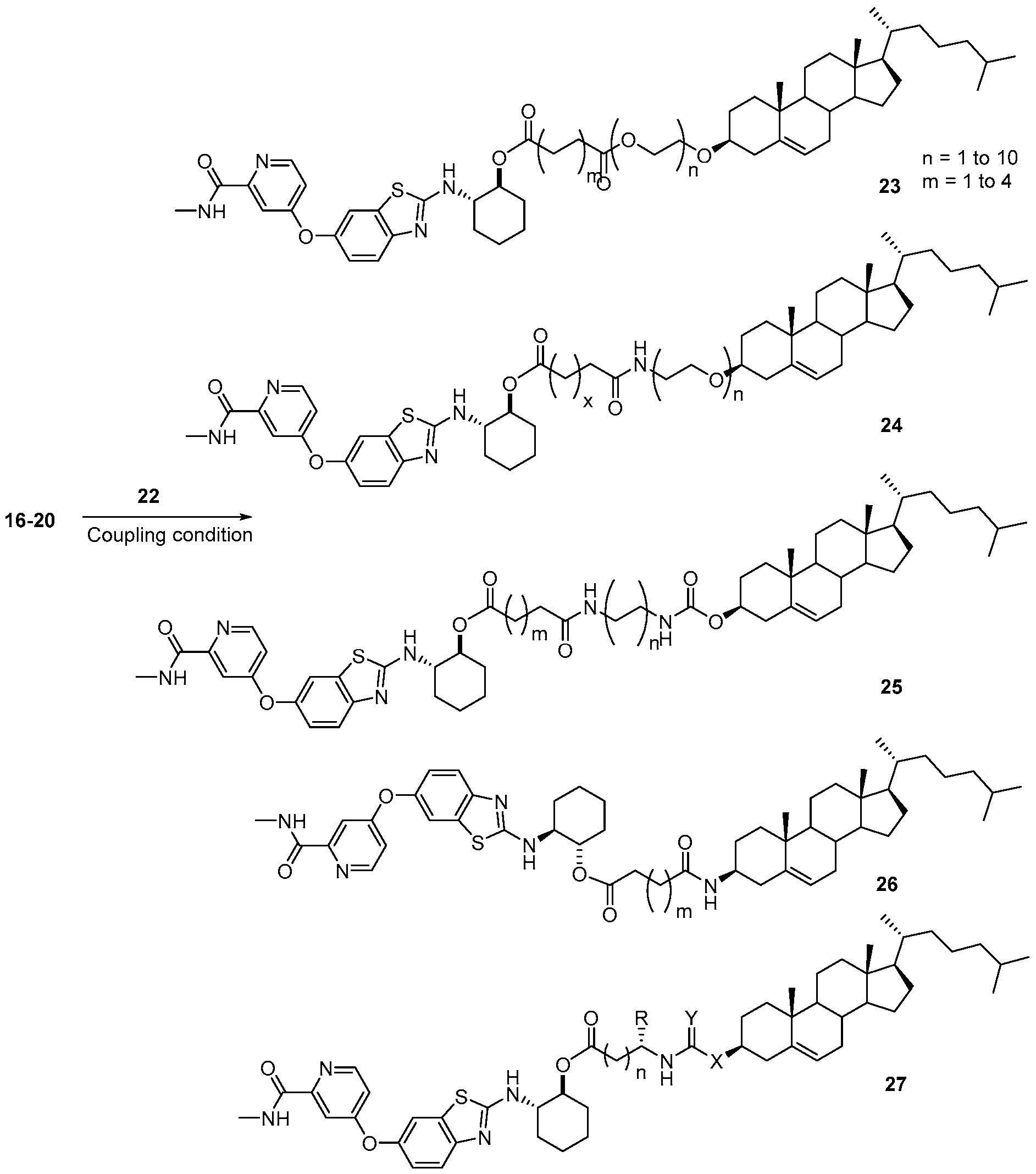
R = alkyl groups, acids, amines, aromatic, thiols
According to the scheme described above, we had synthesized compound IO-801_01 by following the below mentioned scheme. (Scheme 9) Staring from commercially available cholesterol the known intermediate 28xm was prepared. The alcohol 28 was further treated with succinic anhydride to give corresponding acid 29 which was further coupled with commercially available tert-butyl ((l S,2S)-2-hydroxycyclohexyl)carbamate to yield compound 30. Compound 30 thus formed was then treated with 1 : 1 DCM: TFA for Boc group deprotection,
the crude product thus obtained was consecutively used for the last replacement reaction with freshly prepared compound 22 to produce the final compound in moderate yield.
Scheme 9

IO-801 01
Other schemes have also been tried for the preparation of the final compound IO-801_01. Starting from the intermediate acid 29. Commercially available (l S,2S)-cyclohexane-l,2-diol was first coupled to acid under usual DCC coupling condition to get the compound 31. The alcohol was then oxidised to ketone and then successively reductive amination with amine 32 to give the final compound IO-801_01. (Scheme 10)
Scheme 10
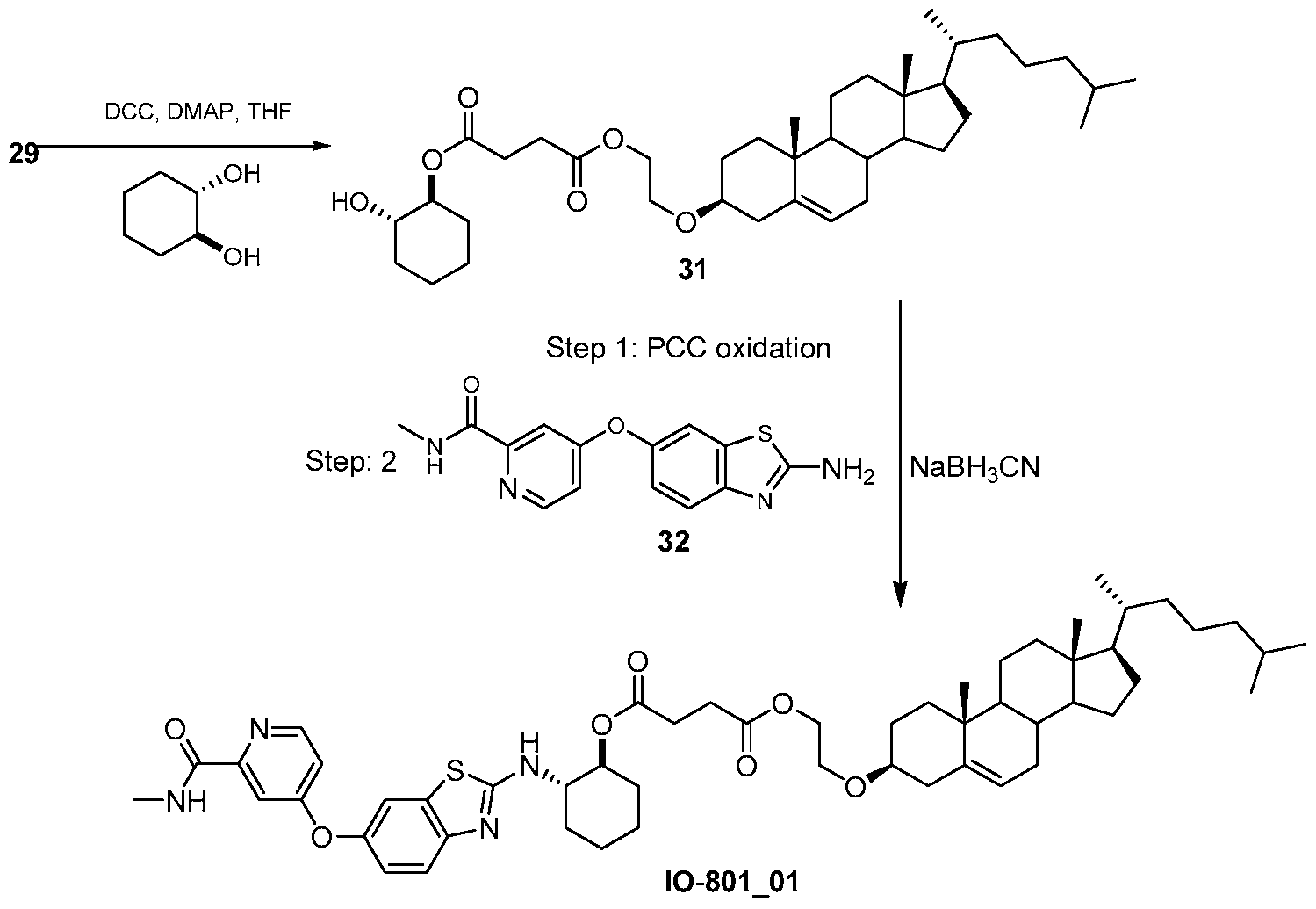
In scheme 11, compound 30 was first treated with TFA: MC to deprotect the Boc group and further used for the reaction with 2-chlorobenzo[d]thiazol-6-ol to produce the compound in moderate yield. Compound 30 thus produced was treated with 4-chloro-N-methylpicolinamide to give the compound 23 in poor yield.
Scheme 11
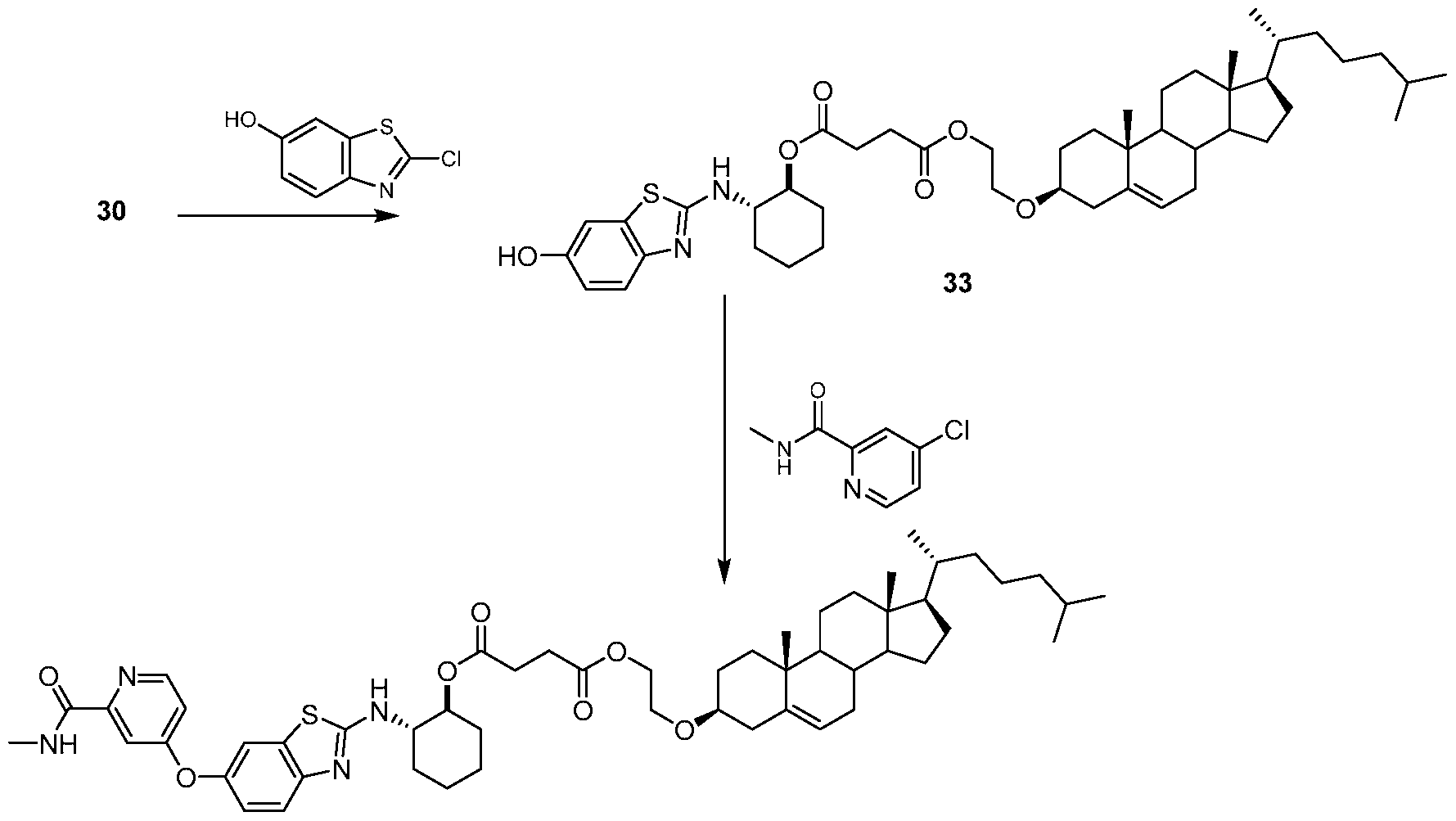
IO-801 01
Scheme 12
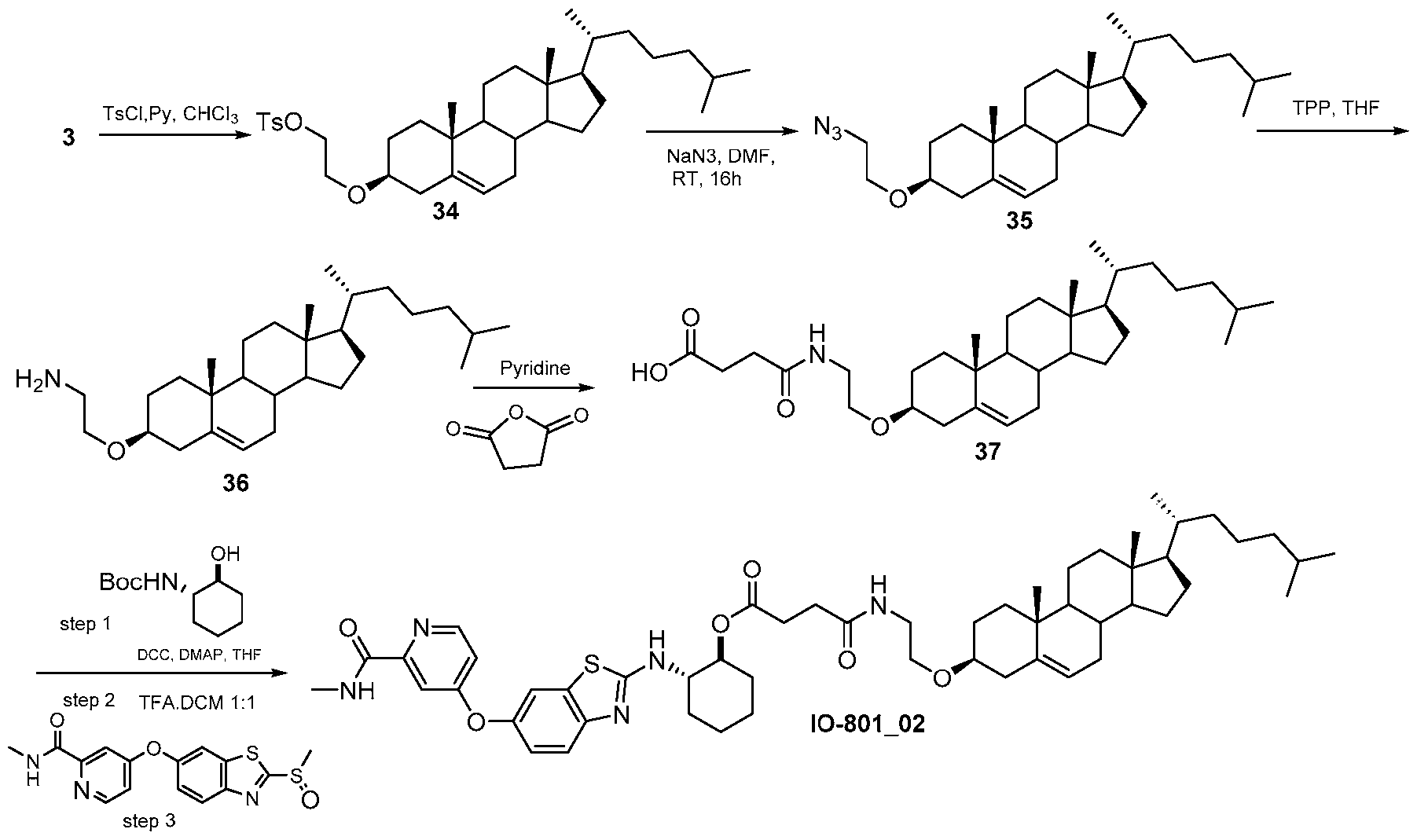
For the preparation of the compound IO-801_02 intermediate acid 37, we followed the procedure that we had described previously. (Scheme 13) And then the acid was coupled with the commercially available tert-butyl ((l S,2S)-2-hydroxycyclohexyl)carbamate, compound thus formed was then treated with 1 : 1 DCM: TFA for Boc group deprotection, the crude product thus obtained was consecutively used for the last replacement reaction with freshly prepared compound 22 to produce the final compound in moderate yield.

43
Scheme 15



For GW-2580 series of compounds we prepared the final compound in two steps starting from the same acids 4, 8 11, 14 and 15. Initially the lipid conjugated linker moiety was coupled with commercially available 4-((2,4-diaminopyrimidin-5-yl)methyl)-2-methoxyphenol and the resultant phenolic derivative was coupled with commercially available benzyl bromide derivatives to synthesize the final compounds. Scheme 18
Scheme 18
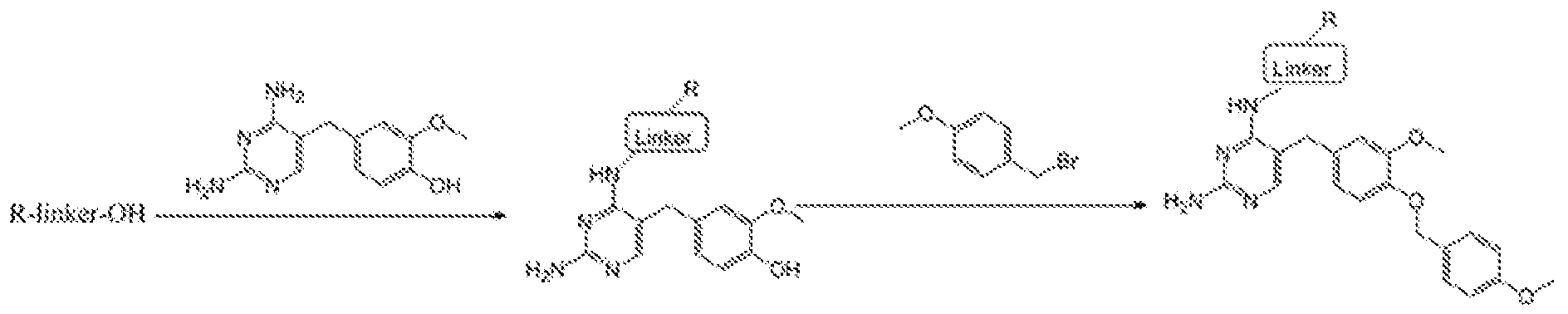
The cholesterol derived acids 4, 8 11, 14 and 15 which were synthesized earlier were initially coupled with commercially available 4-((2,4-diaminopyrimidin-5-yl)methyl)-2- methoxyphenol to give compound 49-53. Scheme 18a
Scheme 18a

R = alkyl groups, acids, amines, aromatic, thiols
The resultant phenolic derivatives 49-53 were coupled with commercially available benzyl bromide derivatives to synthesize the final compounds. Scheme 18b
Scheme 18b

R = alkyl groups, acids, amines, aromatic, thiols
The compounds 54-58 were also directly synthesized from the sets of acids 4, 8 11, 14 and 15 by coupling with commercially available GW-2580. Scheme 18c
Scheme 18c

R = alkyl groups, acids, amines, aromatic, thiols
The PLX-3397 series of compound was also prepared in two steps starting from the same acids 4, 8 11, 14 and 15. Initially it was treated with N-Boc protected 5-((5-chloro-lH-pyrrolo[2,3- b]pyridin-3-yl)methyl)pyridin-2-amine to form the coupled at the indole -NH position. Then the N-Boc was deprotected using the TFA condition followed by reductive amination with 6- (trifluoromethyl)nicotinaldehyde to get the final compound. Scheme 19
Scheme 19

The cholesterol derived acids 4, 8 11, 14 and 15 which were synthesized earlier were initially coupled with commercially available 4-((2,4-diaminopyrimidin-5-yl)methyl)-2- methoxyphenol to give compound 59-63. Scheme 19a
Scheme 19a
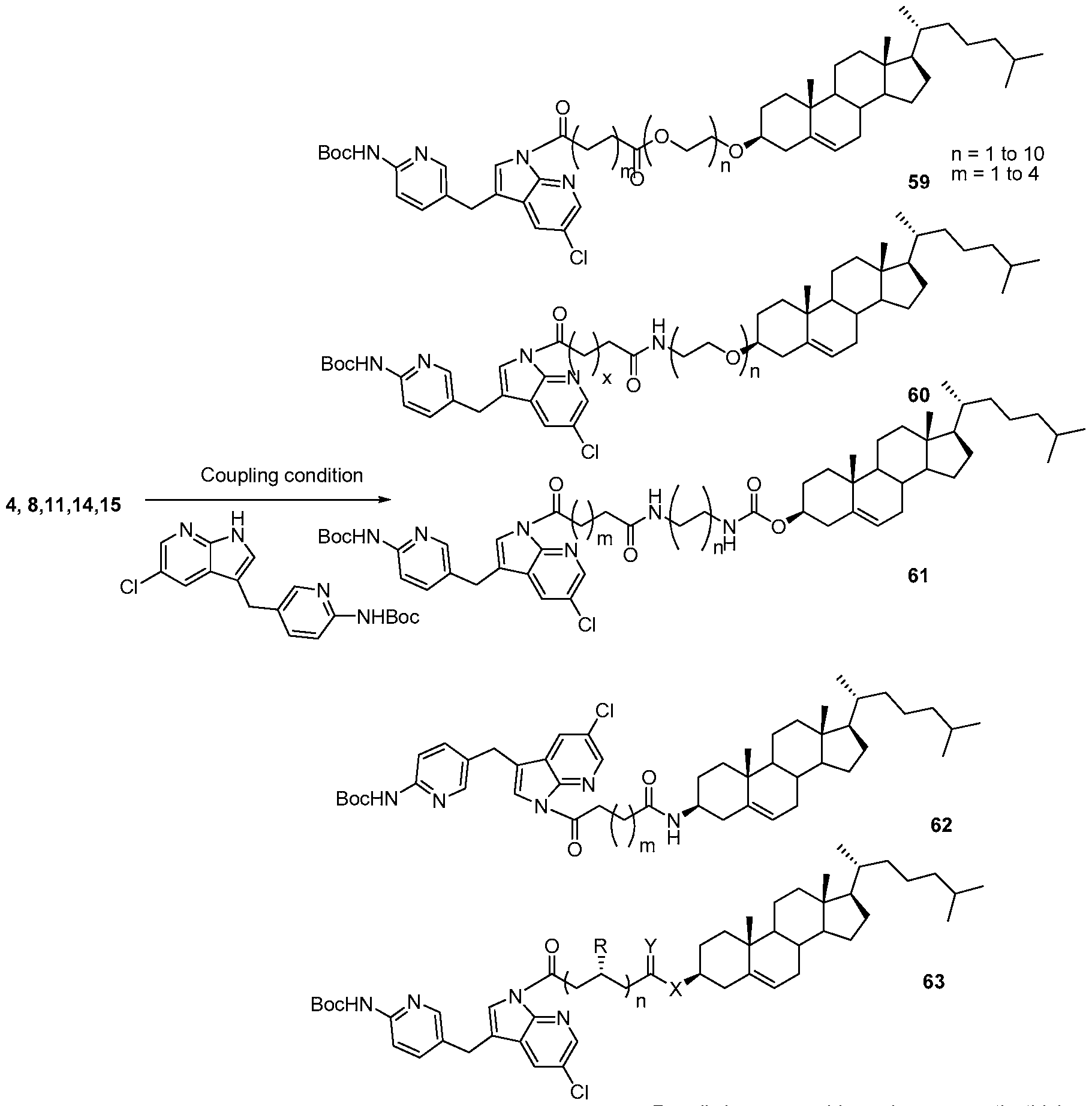
R = alkyl groups, acids, amines, aromatic, thiols
Then the N-Boc compounds 59-63 were deprotected using the TFA condition followed by reductive amination with 6-(trifluoromethyl)nicotinaldehyde to get the final compounds 64-68. Scheme 19b.
Scheme 19b
59-63
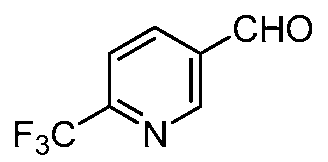
R = alkyl groups, acids, amines, aromatic, thiols The final compounds 64-68 were also directly synthesized from the sets of acids 4, 8 11, 14 and 15 by coupling with commercially available PLX-3397. Scheme 19c
Scheme 19c
4,8,11,14,15
R = alkyl groups, acids, amines, aromatic, thiols
In a non-limiting embodiment of the present disclosure, lipid conjugates of CSF1R inhibitor lipid conjugates are prepared using the process described above for BLZ-945-lipids conjugates, GW2580-lipid conjugates, PLX-3397-lipid conjugates.
General procedure for the preparation of Compound 28 :
To a solution of cholesterol (1 eq) in chloroform (8 mL) was added pyridine (8 mL) followed by tosyl chloride (1.2 eq) at 0°C. The solution was stirred at same temperature for 6h. Reaction was monitored by TLC. Solvent was evaporated under reduced pressure, water was added and extracted with chloroform, separate the layer. The organic layer was washed with IN HC1 solution (100 mL), dried the organic layer over anhydrous sodium sulfate and evaporated. The crude compound was dissolved in 40 mL CHCh and MeOH (300 mL) was added to get a white precipitate, filter it and dried to get intermediate tosylate in good yield.
¾ NMR (500 MHz, CDCh) δ 7.87-7.80 (d, 2H), 7.41-7.32 (d, 2H), 5.37-5.28 (m, 1H), 4.21- 4.12 (t, 2H), 3.70-3.63 (m, 2H), 3.17-3.05 (m, 1H), 2.47 (s, 3H), 2.28-2.23 (m, 1H), 2.16-2.07 (m, 1H), 2.05-1.94 (m, 2H), 1.88-1.78 (m, 5H), 1.63-1.22 (m, 12H), 1.21-1.06 (m, 6H), 1.04- 1.00 (m, 2H), 0.99 (s, 3H), 0.95-0.92 (m, 3H), 0.90-0.87 (m, 6H), 0.69 (s, 3H).
To a solution of intermediate tosylate (1 eq) in 1,4-dioxane (80 mL) was added mono/di/ tri/ poly ethylene glycol (1.2 eq) and contents were heated at 80°C for 16h. Cool the reaction mass, solvent was evaporated on reduced pressure. Water was added and extracted with chloroform (100 mL). Layer was separated, organic layer was washed with sat. sodium bicarbonate solution (100 mL), brine, dried over anhydrous sodium sulfate and evaporated. The crude compound was purified by column chromatography using 10-15% ethyl acetate: hexane to get compound of moderate yield
Compound 28: ¾ NMR (500 MHz, CDCh): δ 5.42-5.33 (m, 1H), 3.76-3.72 (m, 2H), 3.64- 3.59 (m, 2H), 3.26-3.18 (m, 1H), 2.43-2.36 (m, 1H), 2.29-2.18 (m, 1H), 2.08-1.77 (m, 8H), 1.63-1.42 (m, 8H), 1.40-1.22 (m, 6H), 1.21-1.05 (m, 8H), 1.03 (s, 3H), 1.01-0.96 (m, 2H), 0.95- 0.92 (m, 3H), 0.90-0.87 (m, 6H), 0.70 (s, 3H).
Compound 44: ¾ NMR (500 MHz, CDCh) δ ), 5.39-5.29 (m, 1H), 3.77-3.63 (m, 16H), 3.26-3.73 (m, 2H), 3.71-3.67 (m, 10H), 3.66-3.64 (m,4H), 3.25-3.16 (m, 1H), 2.07-1.80 (m, 5H), 1.63-1.42 (m, 8H), 1.40-1.23 (m, 5H), 1.20-1.05 (m, 7H), 1.01 (s, 3H), 0.95-0.92 (m, 3H), 0.90-0.85 (m, 6H), 0.69 (s, 3H). MS (ES-MS) [M+Na]+ calcd for CssHeiOsNa m/z 585.45, found m/z 585.46.
General procedure for the azide formation.
Experimental procedure: To a solution of tosylate (1 eq.) in dry DMF was added sodium azide (1.2 eq.) and stirred at room temperature for 16h, water was added and extracted with ethyl acetate, brine washing was given to the organic layer, dried over anhydrous sodium
sulfate and evaporated. The crude compound was purified by column chromatography to give azide of moderate yield.
Compound 35: Ή NMR (500 MHz, CDCh) δ 5.43-5.34 (m, 1H), 3.73-3.63 (m, 2H), 3.45- 3.34 (t, 2H), 3.28-3.18 (m, 1H), 2.44-2.36 (m, 1H), 2.29-2.19 (m, 1H), 2.06-1.78 (m, 5H), 1.63-1.24 (m, 13H), 1.23-1.04 (m, 8H), 1.03 (s, 3H), 0.95-0.93 (m, 3H), 0.90-0.87 (m, 6H), 0.70 (s, 3H).
Compound 41: To a solution of tosylate (1 eq.) in anhydrous CH2CI2, TMSN3 (1.1 eq.) was added, followed by boron trifluoride etherate (2 eq.). The reaction was stirred at 22°C. for 2 h. When the starting material was no longer visible by TLC analysis (hexanes), the reaction was slowly poured into saturated aqueous NaHCCb (100 mL) and vigorously stirred for 10 min. The organic layer was separated and the aqueous layer was extracted with diethyl ether (60 mLx2). The organic layers were combined, washed with deionized water (100 ml.), dried over anhydrous Na2S04 and concentrated under reduced pressure to give the crude product as light yellow solid. Flash colunm chromatography (hexanes) afforded the product as white solid.
Compound 41: ¾ NMR (500 MHz, CDCh) δ 5.45-5.38 (m, 1H), 3.28-3.16 (m, 1H), 2.35- 2.26 (m, 2H), 2.08-1.80 (m, 5H), 1.64-1.44 (m, 8H), 1.42-1.33 (m, 3H), 1.31-1.23 (m,3H), 1.19-1.07 (m, 7H), 1.03 (s, 3H), 0.95-0.92 (m, 3H), 0.91-0.87 (m, 6H), 0.70 (s, 3H).
General procedure for the azide reduction to amine.
Experimental procedure: To a solution of azide (1 eq.) in dry THF was added triphenyl phosphine (1.5 eq.) and refluxed for 2h. After monitoring the TLC to check the complete conversion of starting material, water was added to the reaction mixture and further refluxed for another 30 mins. Then solvent was evaporated under reduced pressure. The residue was taken in ethyl acetate and added HC1 in ethyl acetate drop wise under stirring. Solid was precipitated out, filter the solid, washed it with ethyl acetate and dried.
Compound 36: ¾ NMR (500 MHz, CDCh) δ 8.49-8.13 (m, 2H), 5.43-5.26 (m, 1H), 3.83- 3.71 (m, 2H), 3.31-3.13 (m, 3H), 2.49-2.29 (m, 4H), 2.26-2.17 (m, 1H), 2.05-1.80 (m, 5H), 1.62-1.22 (m, 12H), 1.21-1.03 (m, 6H), 1.01 (s, 3H), 0.95-0.92 (m, 3H), 0.90-0.86 (m, 6H), 0.69 (s, 3H). MS (ES-MS) [M+H]+ calcd for C29H51NO m/z 430.40, found m/z 430.22.
Compound 42: ¾ NMR (500 MHz, CDCh) δ 5.46-5.32 (m, 1H), 3.08-2.94 (m, 1H), 2.51- 2.37 (m, 2H), 2.04-1.65 (m, 6H), 1.62-1.42 (m, 5H), 1.40-1.29 (m, 3H), 1.19-0.95 (m, 12H), 0.93-0.89 (m, 3H), 0.88-0.82 (m, 6H), 0.67 (s, 3H).
General procedure for the carbamate formation with cholesteryl chloroformate.
Experimental procedure: Cholesteryl chloroformate (1 eq.) was dissolved in alkyl-diamine (80 mL) and the mixture stirred for 18 h. The reaction mixture was then quenched with water and extracted with dichloromethane. The organic extracts were dried (Na2S04) and the solvent was removed in vacuo to afford a residue, which was purified by flash column chromatography. Compound 39: ¾ NMR (500 MHz, CDCh) δ 6.74-6.54 (t, 1H), 5.19-5.05 (m, 1H), 4.22-4.05 (m, 1H), 2.91-2.80 (m, 2H), 2.47-2.39 (t, 2H), 2.14-1.93 (m, 2H), 1.83-1.53 (m, 6H), 1.39-1.10 (m, 11H), 1.07-1.01 (m, 1H), 0.99-0.83(m, 7H), 0.83-0.76(m, 5H), 0.73-0.70 (m, 3H), 0.66-0.62 (m, 6H), 0.47 (s, 3H).
Experimental procedure: To a solution of cholesteryl chloroformare (1 eq.) in THF was added amino acid (1.2 eq.) and 10% sodium carbonate solution and mixture was stirred at room temperature for 1.5h. The reaction was neutralized with 2N HCl solution and extracted with DCM, organic layer was separated, washed with water and brine, dried and evaporated.
Compound 46: ¾ NMR (500 MHz, CDCh) δ 7.12-6.99 (m, 1H), 5.38-5.27 (m, 1H), 4.34- 4.26 (m, 1H), 3.18-3.14 (m, 2H), 2.39-2.35 (m, 2H), 2.32-2.15 (m,4H), 1.99-1.89 (m, 4H), 1.87-1.74 (m, 5H), 1.55-1.46 (m, 8H), 1.43-1.37 (m, 4H), 1.17-1.03 (m, 12H), 0.98-0.95 (m, 3H), 0.91-0.89 (m, 3H), 0.86-0.83 (m, 6H), 0.66 (s, 3H). MS (ES-MS) [M+Na]+ calcd for C3iH5iNNa04 m/z 524.37, found m/z 524.39.
General procedure for the reaction with acid anhydride. Compound 29: To a solution of compound 28 (0.3 g, 0.69 mmol) in pyridine (3 mL) was added succinic anhydride (0.083 g, 0.69 mmol) and stirred at room temperature for 16h. Solvent was evaporated under reduced pressure, water was added and extracted with chloroform (25 mL), separate the layer. The organic layer was washed with IN HCl solution, dried the organic layer over anhydrous sodium sulfate and evaporated. The crude compound was purified by column chromatography to obtain compound 29. Yield: 0.25 g, 67.75%.
Compound 29: ¾ NMR (500 MHz, CDCh): δ 5.43-5.29 (m, 1H), 4.30-4.19 (t, 2H), 3.73-3.67 ( m, 2H), 3.26-3.15 (m, 1H), 2.75-2.64 (m, 5H), 2.41-2.34 (m, 1H), 2.28-2.19 (m, 1H), 2.06- 1.95 (m, 2H), 1.94-1.73 (m, 3H), 1.69-1.23 (m, 12H), 1.21-1.03 (m, 7H), 1.02, (s, 3H), 1.00- 0.96 (m, 1H), 0.95-0.91 (m, 3H), 0.90-0.84 (m, 6H), 0.70 (s, 3H). Compound 37: MS (ES-MS) [M+H]+ calcd for C33H56NO4 m/z 530.42, found m/z 530.28.
Compound 40: ¾ NMR (500 MHz, CDCh) δ 12.21-11.76 (m, 1H), 7.93-7.81 (t, 1H), 7.08- 6.97 (t, 1H), 5.41-5.27 (m, 1H), 4.36-4.25 (m, 1H), 3.12-2.93 (m, 4H), 2.45-2.36 (t, 2H), 2.33- 2.15 (m, 4H), 2.02-1.88 (m,2H), 1.86-1.72 (m, 3H), 1.59-1.45 (m, 5H), 1.43-1.28 (m, 5H),
1.26- 1.19 (m, 1H), 1.17-0.94 (m, 12H), 0.93-0.88 (m, 3H), 0.87-0.81 (m, 6H), 0.66 (s, 3H).
Compound 43: ¾ NMR (500 MHz, CDCh) δ 5.41-5.27 (m, 1H), 3.71-3.55 (m, 1H), 2.68- 2.64 (t, 2H), 2.47-2.40 (t, 2H), 2.30-2.22 (m, 1H), 2.13-2.04 (m, 1H), 2.03-1.89 (m, 2H), 1.87- 1.77 (m, 3H), 1.60-1.46 (m, 5H), 1.44-1.28 (m, 6H), 1.27-1.22 (m, 1H), 1.19-1.02 (m, 7H), 1.01-0.93 (m, 5H), 0.92-0.88 (m, 3H), 0.87-0.82 (m, 6H), 0.67 (s, 3H). MS (ES-MS) [M+H]+ calcd for C31H52NO3 m/z 486.39, found m/z 486.30.
Compound 45: ¾ NMR (500 MHz, CDCh) δ 11.28-11.08 (m, 1H), 5.39-5.32 (m, 1H), 4.31- 4.26 (m, 2H), 3.77-3.63 (m, 16H), 3.26-3.08 (m, 2H), 2.74-2.62 (m, 4H), 2.42-2.34 (m, 1H),
2.27- 2.17 (m, 1H), 2.07-1.80 (m, 5H), 1.60-1.53 (m, 5H), 1.48-1.45 (m, 3H), 1.38-1.34 (m, 2H), 1.21-1.06 (m, 6H), 1.01 (s, 3H), 0.95-0.92 (m, 3H), 0.90-0.87 (m, 6H), 0.70 (s, 3H). MS (ES-MS) [M+Na]+ calcd for CsgHeeOsNa m/z 685.47, found m/z 685.55.
General procedure for the preparation of sulphoxide. Compound 22: To a solution of commercially available N-methyl-4-((2- (methylthio)benzo[d]thiazol-6-yl)oxy)picolinamide (0.1 g, 0.30 mmol) in DCM (3 mL) was added m-CPBA (0.057 g, 0.33 mmol) and stirred at room temperature for 1.5h. Reaction quenched using sat. solution of aq. NaHCCb. Separate the layer, dried and evaporated to get desired product. Yield: 0.09 g, 86.53% and proceed as such for next step. Mass and 1H NMR spectra of the compound corroborate with the reported literarture. xu
General procedure for the coupling reaction 23-27.
Experimental procedure: To a solution of A (1.5 eq.) in DMF was added DMAP (0.5 eq.) followed by compound B (1.0 eq.). cooled to 0°C, then added DCC (1.5 eq.) and stirred at room temperature for 16h. Water was added and extracted with DCM, dried and evaporated. Crude compound was purified by column chromatography. Purified compound without any further characterization proceeded for deprotection reaction.
To a solution of N-Boc protected amine (1.0 eq.) in DCM was added TFA (2.0 eq.). and stirred at room temperature for 3h. Solvent was evaporated completely and proceed as such for next step.
To a solution of amine salt (1 eq.) in dry NMP was added DIPEA (5 eq.) followed by freshly prepared compound 22 (1.2 eq.) and heated at 110°C for 3 days. The crude mixture was concentrated to get dark brown sticky liquid which was further purified by column chromatography using methanol: dichlorom ethane to get the final compound.
IO-801_01: ¾ NMR (500 MHz, CDCh) δ 8.45-8.31 (d, 1H), 8.12-7.95 (m, 1H), 7.77-7.66 (m 1H), 7.63-7.51 (d, 1H), 7.36-7.30 (d, 1H), 7.13-7.02 (m, 1H), 6.98-6.90 (m, 1H), 5.38-5.34 (m, 1H), 4.89-4.79 (m, 1H), 4.24-4.17 (t, 2H), 3.74-3.60 (m, 3H), 3.26-3.14 (m, 1H), 3.05-2.98 (d, 3H), 2.69-2.49 (m, 4H), 2.40-2.30 (m, 2H), 2.27-1.77 (m, 12H), 1.60-1.41 (m, 10H), 1.40- 1.31 (m, 4H), 1.27-1.26 (m, 2H), 1.20-1.03(m, 7H), 1.01 (s, 3H), 0.94-0.91 (d, 3H), 0.89-0.87 (m, 6H), 0.69 (s, 3H). 13C NMR (126 MHz, CDCh) δ 175.07, 172.26, 171.14, 167.74, 166.62, 164.45, 152.31, 149.68, 148.76, 140.68, 121.78, 119.80, 118.74, 113.89, 113.71, 110.23, 79.63, 75.09, 65.78, 64.35, 60.39, 59.23, 56.76, 56.17, 50.17, 42.32, 39.78, 39.52, 39.01, 37.18, 36.84, 36.19, 35.78, 31.94, 31.89, 29.69, 29.43, 29.15, 28.33, 28.23, 28.01, 26.15, 24.29, 23.83, 23.52, 22.81, 22.56, 21.07, 21.04, 19.37, 18.72, 14.20, 11.86. MS (ES-MS) [M+H]+ calcd for C53H75N4O7S m/z 911.54, found m/z 911.19.
IO-801_02: Ή NMR (500 MHz, CDCh) δ 8.50-8.38 (d, 1H), 8.10-8.01 (m, 1H), 7.73-7.67 (m 1H), 7.41-7.33 (m, 1H), 7.22-7.15 (m,lH), 7.03-6.95 (m, 1H), 5.39-5.33 (m, 1H), 4.91-4.81 (m, 1H), 3.62-3.52 (t, 2H), 3.44-3.33 (m, 2H), 3.23-3.11 (m, 1H), 3.06-2.98 (d, 3H), 2.67-2.55 (m, 2H), 2.54-2.45 (m, 2H), 2.39-2.28 (m, 2H), 2.25-2.12 (m, 3H), 2.10-1.93 (m, 3H), 1.92- 1.79 (m, 5H), 1.60-1.39 (m, 12H), 1.35 (m, 4H), 1.21-1.03 (m, 10H), 1.01 (s, 3H), 0.95-0.91 (d, 3H), 0.90-0.87 (m, 6H), 0.69 (s, 3H). 13C NMR (126 MHz, CDCh) δ 175.33, 172.51, 166.26, 164.29, 156.78, 152.38, 149.79, 140.59, 121.85, 120.60, 114.12, 110.18, 79.35, 74.98, 66.36, 56.75, 56.17, 50.16, 49.17, 42.32, 39.83, 39.77, 39.52, 39.09, 37.15, 36.85, 36.19, 35.78, 33.94, 31.93, 31.89, 31.06, 30.06, 29.70, 28.41, 28.23, 28.02, 26.17, 25.62, 24.94, 24.29, 23.84, 22.81, 22.56, 21.07, 19.38, 18.72, 11.86. MS (ES-MS) [M+H]+ calcd for CssHveNsOeS m/z 910.55, found m/z 910.23.
IO-801_03: ¾ NMR (500 MHz, CDCh) δ 8.47-8.37 (d, 1H), 8.11-8.01 (m, 1H), 7.83-7.60 (m
2H), 7.43-7.32 (m, 1H), 7.22-7.08 (m,lH), 7.07-6.95 (m, 1H), 6.91-6.63 (m, 1H), 5.67-5.49 (m, 1H), 5.39-5.34 (m, 1H), 4.918-4.82 (m, 1H), 4.54-4.41 (m, 1H), 4.54-4.41 (m, 1H), .88-
3.42 9m, 2H), 3.38-3.17 (m, 4H), 3.08-2.95 (d, 3H), 2.66-2.23 (m, 8H), 2.19-1.77 (m, 8H),
1.68-1.22(m, 16H), 1.21-1.05 (m, 6H), 1.01 (s, 3H), 0.94-0.92 (d, 3H), 0.91-0.84 (m, 6H), 0.69
(s, 3H). 13C NMR (126 MHz, CDCh) δ 172.96, 172.21, 166.76, 164.58, 156.92, 152.17,
149.72, 148.39, 139.78, 122.53, 119.50, 119.15, 114.09, 113.62, 109.81, 75.51, 74.62, 59.10, 56.69, 56.15, 53.43, 50.04, 42.31, 40.66, 40.25, 39.74, 39.52, 38.60, 37.02, 36.56, 36.19, 35.79,
31.91, 31.86, 31.81, 31.16, 31.06, 30.32, 29.69, 28.22, 28.15, 28.01, 26.16, 24.28, 23.94, 23.83, 22.82, 22.69, 22.56, 21.04, 19.33, 18.72, 11.86. MS (ES-MS) [M+H]+ calcd for C54H77N6O7S m/z 953.56, found m/z 953.24.
IO-803_01: ¾ MR (500 MHz, CDCb) δ 8.49-8.37 (d, 1H), 8.10-7.97 (m, 1H), 7.81-7.62 (m 2H), 7.64-7.52 (d, 1H), 7.35(s, 1H), 7.19-7.10 (m, 1H), 7.05-6.92 (m, 1H), 6.01-5.77 (m, 1H), 5.38-5.28 (m, 1H), 4.95-4.78 (m, 1H), 3.87-3.43 (m, 2H), 2.63-2.54 (m, 2H), 2.50-2.33 (m, 4H), 2.31-2.24 (m, 1H), 2.21-2.06 (m, 2H), 2.04-1.91 (m, 2H), 1.90-1.80 (m, 5H), 1.65-1.31 (m, 15H), 1.29-1.25 (m, 2H), 1.21-1.03 (m, 8H), 1.00 (s, 3H), 0.95-0.91 (d, 3H), 0.91-0.86 (m, 6H), 0.69 (s, 3H). 13C NMR (126 MHz, CDCb) δ 173.16, 170.60, 166.77, 164.54, 152.23, 149.69, 148.35, 140.33, 121.84, 119.35, 119.32, 113.93, 113.47, 109.99, 75.54, 58.78, 56.68, 56.15, 50.08, 49.81, 42.30, 39.73, 39.52, 39.22, 37.87, 36.55, 36.19, 35.80, 33.95, 31.85, 31.83, 31.72, 31.33, 31.01, 30.26, 29.68, 29.05, 28.22, 28.00, 26.14, 24.26, 24.18, 23.95, 23.86, 22.81, 22.56, 20.96, 19.32, 18.72, 11.86. MS (ES-MS) [M+H]+ calcd for C51H72N5O5S m/z 866.53, found m/z 866.40.
IO-806_01: ¾ NMR (500 MHz, CDCb) δ 8.45-8.33 (d, 1H), 8.09-7.96 (m, 1H), 7.76-7.66 (m 1H), 7.65-7.55(m, 1H), 7.36-7.31 (m, 1H), 7.09-7.05 (m, lH), 6.98-6.92 (m, 1H), 5.38-5.32 (m, 1H), 4.89-4.76 (m, 1H), 4.24-4.17 (m, 2H), 3.85-3.75 (m,lH), 3.71-3.60 (m, 14H) , 3.24- 3.11 (m, 1H), 3.05-2.94 (d, 3H), 2.70-2.49 (m, 4H), 2.41-2.32 (m, 2H), 2.25-2.16 (m, 1H), 2.15-2.07 (m, 1H), 2.05-1.74 (m, 7H), 1.68-1.40(m, 11H), 1.38-1.31 (m, 4H), 1.29-1.22 (m, 4H), 1.20-1.03 (m, 7H), 1.00 (s, 3H), 0.94-0.91 (d, 3H), 0.89-0.85 (m, 6H), 0.69 (s, 3H). 13C NMR: (126 MHz, CDCb) δ 172.41, 172.20, 166.58, 164.44, 152.32, 149.70, 148.88, 140.95, 121.55, 119.80, 118.87, 113.93, 113.70, 110.19, 79.54, 79.50, 75.09, 70.89, 70.59, 70.54, 70.52, 69.05, 67.27, 63.89, 59.15, 56.78, 56.17, 50.20, 42.33, 39.79, 39.52, 39.07, 37.24, 36.87, 36.20, 35.78, 31.95, 31.90, 31.43, 30.70, 29.69, 29.41, 29.11, 28.37, 28.23, 28.01, 26.14, 24.29,
23.92, 23.83, 23.62, 22.81, 22.56, 21.07, 19.38, 18.72, 12.17, 11.86. MS (ES-MS) [M+H]+ calcd for C59H87N4O10S m/z 1043.61, found m/z 1043.47. IO-806_02: ¾ NMR (500 MHz, CDCb) δ 8.44-8.36 (d, 1H), 8.09-7.96 (m, 1H), 7.75-7.56 (m, 2H), 7.37-7.30 (m, 1H), 7.10-7.03 (m, 1H), 7.00-6.93 (m, 1H), 5.42-5.21 (m, 2H), 4.87- 4.74 (m, 1H), 4.52-4.38 (m, 1H), 3.91-3.73 (m, 1H), 3.49-3.24 (m, 2H), 3.07-2.94 (d, 3H),
2.52-2.42 (m, 2H), 2.41-2.30 (m, 2H), 2.27-2.10 (m, 2H), 2.04-1.90 (m, 2H), 1.88-1.78 (m, 5H), 1.60-1.41 (m, 10H), 1.38-1.32 (m, 3H), 1.31-1.21 (m, 6H), 1.19-1.05 (m, 7H), 0.99 (s, 3H), 0.94-0.91 (d, 3H), 0.90-0.85 (m, 6H), 0.71 (s, 3H). 13C NMR (126 MHz, CDCh) δ 172.17, 167.22, 166.57, 164.44, 152.31, 149.72, 148.95, 139.81, 122.48, 119.89, 118.92, 113.96, 113.76, 110.11, 75.36, 74.49, 59.24, 56.69, 56.15, 50.01, 42.31, 39.73, 39.52, 38.53, 36.97, 36.75, 36.55, 36.19, 35.79, 35.32, 31.90, 31.87, 31.54, 30.84, 29.69, 28.22, 28.08, 28.01, 26.15, 24.28, 24.00, 23.84, 23.69, 22.81, 22.69, 22.56, 21.03, 19.32, 18.72, 11.86. MS (ES-MS) [M+H]+ calcd for CsiHviNsOeS m/z 882.52, found m/z 882.55.
IO-806_03: ¾NMR (500 MHz, CDCh) δ 8.44-8.34 (d, 1H), 8.10-7.95 (m, 1H), 7.75-7.66 (d, 1H), 7.64-7.52 (d, 1H), 7.39-7.30 (m, 1H), 7.13-7.02 (m, 1H), 6.99-6.91 (m, 1H), 5.41-5.27 (m, 1H), 5.00-4.89 (m, 1H), 4.16-4.01 (m, 2H), 3.89-3.75 (m, 1H), 3.67-3.54 (m, 8H), 3.23- 3.11 (m, 1H), 3.06-2.97 (d, 3H), 2.41-2.27 (m, 2H), 2.24-2.08 (m, 2H), 2.06-1.93 (m, 2H), 1.92-1.79 (m, 5H), 1.62-1.41 (m, 11H), 1.40-1.32 (m, 3H), 1.29-1.26 (m, 5H), 1.20-1.04 (m, 7H), 0.99 (s, 3H), 0.94-0.91 (d, 3H), 0.90-0.85 (m, 6H), 0.68 (s, 3H). 13C NMR (126 MHz, CDCh) δ 170.69, 166.99, 166.57, 164.42, 152.32, 149.70, 148.90, 140.88, 121.60, 119.80, 118.94, 113.89, 113.71, 110.22, 79.53, 75.33, 70.85, 70.79, 70.54, 68.65, 67.22, 59.02, 56.78, 56.17, 50.18, 42.32, 39.79, 39.52, 39.06, 37.22, 36.86, 36.20, 35.78, 31.95, 31.93, 31.89, 29.69, 28.35, 28.23, 28.01, 26.14, 24.29, 23.98, 23.83, 23.61, 22.81, 22.56, 21.07, 19.38, 18.72, 11.86. MS (ES-MS) [M+H]+ calcd for C53H77N4O7S m/z 913.55, found m/z 913.59.
Formulation
Supramolecular nanostructures were formulated using thin film hydration method with varying mole ratios of phospholipids (such as HSPC, POPC, SOPC, Egg PC, etc.), PEGylated- phospholipids (such as DSPE-PEG) and CSF1R inhibitor conjugates. % Encapsulation efficiency of the CSF1R inhibitors in the supramolecules was determined by UV spectrophotometry. Average size, polydispersity index (PDI) and surface potential of the nanoparticles were measured by Dynamic light scattering (DLS).
These supramolecules were lyophilized (5% Lactose solution was used as cryo-protectant) over 16-20 hrs. The amorphous white solid powder formed thereafter was reconstituted by adding required volume of water. DLS study of this reconstituted supramolecular formulation reveals similar size, PDI, surface potential of supramolecules as it was before lyophilization.
To clarify the characteristics of these supramolecules, some examples of its implementation are described hereby:
EXAMPLE.1
HSPC, POPC, IO-806 03 and DSPE-PEG, taken in 55:35:5:5 mol% ratio, were dissolved in Chloroform. All lipid solutions were mixed homogeneously in round bottom flask and organic solvent was evaporated by rotary evaporator resulting in a thin lipid film. The lipid film was kept under high vacuum for 3-4 hr. It was, then, hydrated (by adding 5% Lactose solution to it) for 1.0 h at 60°C in hot water bath. Next, hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at 60°C using Avanti extruder supported over hot plate. It is evident from UV measurement that 4.2 mol% of IO-806 03 was encapsulated in the supramolecule. DLS measurement reveals average size of the nanoparticles to be 153.4 nm, PDI 0.100 and surface potential -20.9 mV. The resulting solution was lyophilized and stored at 4°C.
EX AMPLE.2
HSPC, POPC, IO-806 02 and DSPE-PEG, taken in 55:35:5:5 mol% ratio, were dissolved in Chloroform. All lipid solutions were mixed homogeneously in round bottom flask and organic solvent was evaporated by rotary evaporator resulting in a thin lipid film. The lipid film was kept under high vacuum for 3-4 hr. It was, then, hydrated (by adding 5% Lactose solution to it) for 1.0 h at 60°C in hot water bath. Next, hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at 60°C using Avanti extruder supported over hot plate. It is evident from UV measurement that 4.0 mol% of IO-806 02 was encapsulated in the supramolecule. DLS measurement reveals average size of the nanoparticles to be 131.5 nm, PDI 0.064 and surface potential -24.8 mV. The resulting solution was lyophilized and stored at 4°C.
EXAMPLE.3 HSPC, POPC, IO-803 01 and DSPE-PEG, taken in 55:35:5:5 mol% ratio, were dissolved in Chloroform. All lipid solutions were mixed homogeneously in round bottom flask and organic solvent was evaporated by rotary evaporator resulting in a thin lipid film. The lipid film was kept under high vacuum for 3-4 hr. It was, then, hydrated (by adding 5% Lactose solution to it) for 1.0 h at 60°C in hot water bath. Next, hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at
60°C using Avanti extruder supported over hot plate. It is evident from UV measurement that 4.1 mol% of IO-803_01was encapsulated in the supramolecule. DLS measurement reveals average size of the nanoparticles to be 137.9 nm, PDI 0.058 and surface potential -24.5 mV. The resulting solution was lyophilized and stored at 4°C.
EXAMPLE .4 HSPC, POPC, IO-801 03 and DSPE-PEG, taken in 50:35: 10:5 mol% ratio, were dissolved in Chloroform. All lipid solutions were mixed homogeneously in round bottom flask and organic solvent was evaporated by rotary evaporator resulting in a thin lipid film. The lipid film was kept under high vacuum for 3-4 hr. It was, then, hydrated (by adding 5% Lactose solution to it) for 1.0 h at 60°C in hot water bath. Next, hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at 60°C using Avanti extruder supported over hot plate. It is evident from UV measurement that 9.4 mol% of IO-801 03 was encapsulated in the supramolecule. DLS measurement reveals average size of the nanoparticles to be 146.0 nm, PDI 0.043 and surface potential -28.2 mV. The resulting solution was lyophilized and stored at 4°C. EXAMPLE 5
HSPC, POPC, IO-801 02 and DSPE-PEG, taken in 50:30: 15:5 mol% ratio, were dissolved in Chloroform. All lipid solutions were mixed homogeneously in round bottom flask and organic solvent was evaporated by rotary evaporator resulting in a thin lipid film. The lipid film was kept under high vacuum for 3-4 hr. It was, then, hydrated (by adding 5% Lactose solution to it) for 1.0 h at 60°C in hot water bath. Next, hydrated nanoparticles were sequentially extruded through 400nm, 200nm and lOOnm pore size membrane held by filter support for 11 times at 60°C using Avanti extruder supported over hot plate. It is evident from UV measurement that 6.3 mol% of IO-801 02 was encapsulated in the supramolecule. DLS measurement reveals average size of the nanoparticles to be 181.3 nm, PDI 0.137 and surface potential 3.46 mV. The resulting solution was lyophilized and stored at 4°C.
1 (a) Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-16. (b) Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J. Med. 2010;363(9):809-19. (c). Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced
melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707-14. (d). Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF- mutated melanoma. N Engl J Med. 2012;367(2): 107-14.
" (a) Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3): 162-74. (b). Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024): 1565-70. (c). Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cane Res. 2012;72(13):3125-30.
111 (a) Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al.Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99(1): 111-20. (b). Li J, Chen K, Zhu L, Pollard JW. Conditional deletion of the colony stimulating factor-1 receptor (c-fms proto-oncogene) in mice. Genesis. 2006;44(7):328-35. iv Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141 :39-51;
v Tang R, Beuvon F, Ojeda M, Mosseri V, Pouillart P, Scholl S. M-CSF (monocyte colony stimulating factor) and M-CSF receptor expression by breast tumour cells: M-CSF mediated recruitment of tumour infiltrating monocytes? J Cell Biochem 1992; 50:350-6;
vi Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J Mammary Gland Biol Neoplasia 2002; 7: 147-62) v" (a) Douglass TG, Driggers L, Zhang JG, Hoa N, Delgado C, et al. Macrophage colony stimulating factor: not just for macrophages anymore! A gateway into complex biologies. Int Immunopharmacol. 2008; 8: 1354-1376. (b). Lin H, Lee E, Hestir K, Leo C, Huang M, et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008; 320: 807-811. (c). Stanley ER, Chitu V CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. 2014; 6. (d) Rovida E, Sbarba PD Colony- Stimulating Factor-1 Receptor in the Polarization of Macrophages: A Target for Turning Bad to Good Ones? J Clin Cell Immunol. 2015; 6: 379. doi: 10.4172/2155- 9899.1000379.
viii Pyonteck. S. M., Akkari L, Schuhmacher A. J., et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013; 19(10), 1264-1272
ix Conway et al, Inhibition of colony-stimulating-factor- 1 signalling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580; Proc. Natl. Acad. Sci. USA, 2005; 102, 16078
x Shewchuk LM, Hassell AM, Holmes WD, Veal JM, Emmerson HK, Musso DL, Chamberlain SD, and Peckham GE (2004) inventors; GlaxoSmithKline Inc., assignee. Co-crystal structure of liganded
inhibitors of colony-stimulating factor 1 receptor kinase in treatment of disease. U.S. Patent 2004002145 Al . 2004 Jan 1.
Xl (a) Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA, et al. CSFIR inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015;16:949-56; (b) Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, et al. Structure-guided blockade of CSFIR kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015; 373:428-37. xu Sutton, James C; Wiesmann, Marion; Wang, Weibo; Lindvall, Mika K.; Lan, Jiong; Ramurthy, Savithri; Sharma, Anu; Mieuli, Elizabeth J.; Klivansky, Liana M.; Lenahan, William P.; et al. Preparation of 6-O-substituted benzoxazole and benzothiazole compounds for inhibiting CSF-IR signalling. PCT Int. Appl. (2007), WO 2007121484 A2 20071025. xm Sengupta, S.; Roy, M.; Sarkar, A.; Hossain, Sk S.; Sengupta, A.; Dutta, P. K.; Ansari, A.; Mandal, S. K. Preparation of lipid-based platinum compounds and nanoparticles useful in the treatment of cancer. PCT Int. Appl. (2014), WO 2014201376 A2 20141218.
Claims
WE CLAIM:
1. A compound of Formula I:
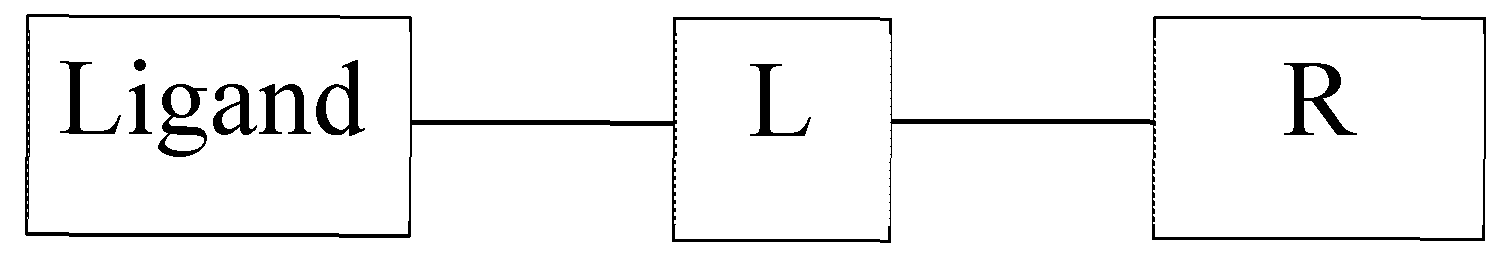
Formula I
wherein, 'Ligand' is selected from the group consisting of:
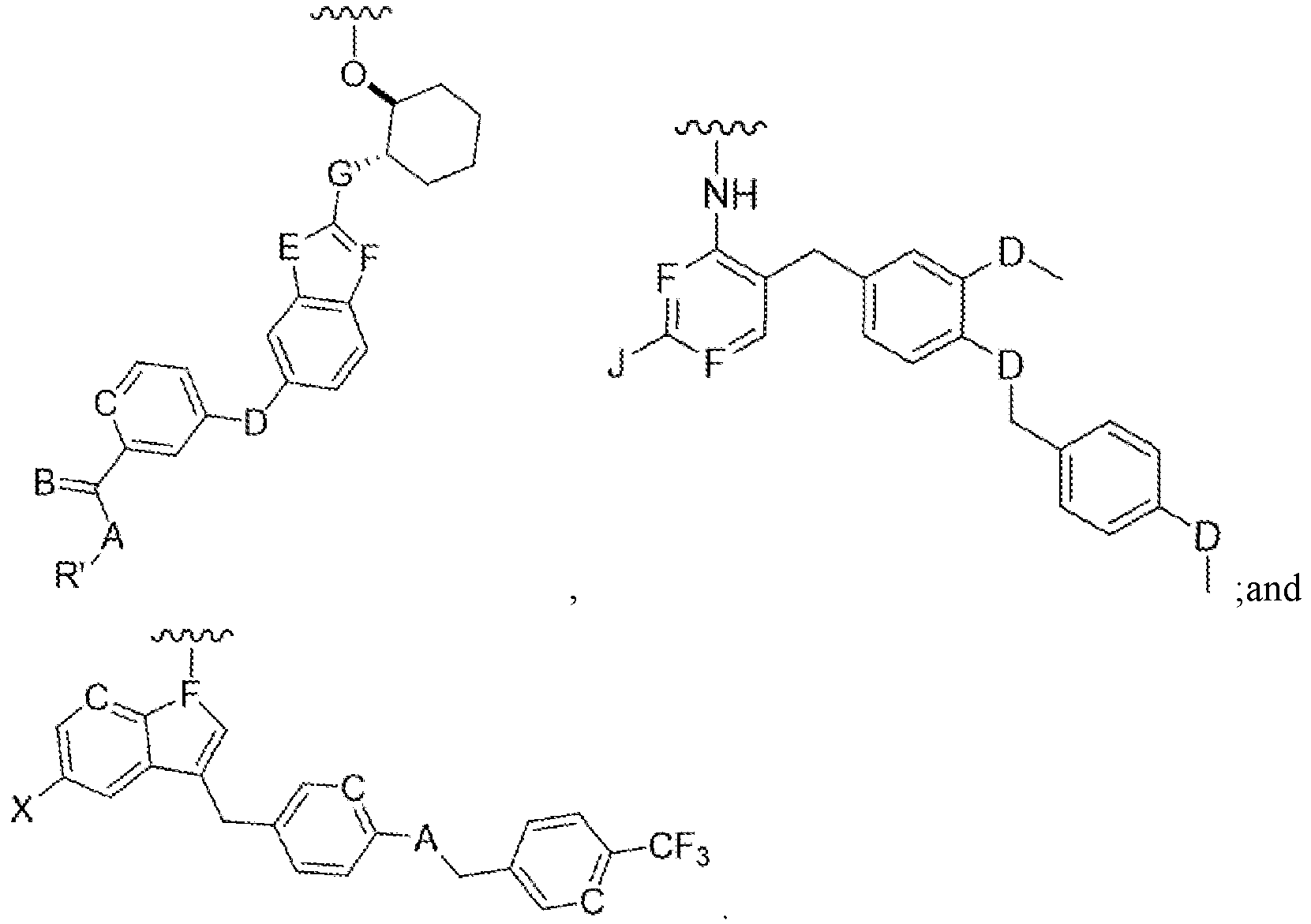
wherein:
C=hydroxy, alkyl group, aryl group, cycloalkyl group;
A=H, O, NH, S;
B=CH, N;
D=C, O, NH, S;
E=C, O, NH, S;
F=CH, N;
G=C, O, NH, S;
J=NH2, OH, SH; and
X=halogen.
'L' is a linker moiety connecting 'Ligand' and 'R' moieties; and
'R' is selected from the group consisting of a lipid, a lipid derivative, a lipid conjugate and combinations thereof;
or
any derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates of compound of Formula I thereof.
The compound of claim 1, wherein the 'L' (linker) is selected from the group consisting of a direct bond or an atom such as oxygen or sulfur, a unit such as R1, C(O), C(0)0, C(0) R1, SO, SO2, SO2 H, and a chain of atoms such as substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroaryl alkynyl, heterocyclylalkyl, heterocyclylalkenyl, heterocyclylalkynyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, cycloalkenyl, alkylarylalkyl, alkylarylalkenyl, alkylarylalkynyl, alkenylarylalkyl, alkenylarylalkenyl, alkenylarylalkynyl, alkynylarylalkyl, alkynylarylalkenyl, alkynylarylalkynyl, alkylheteroarylalkyl, alkylheteroarylalkenyl, alkylheteroarylalkynyl, alkenylheteroarylalkyl, alkenylheteroarylalkenyl, alkenylheteroarylalkynyl, alkynylheteroarylalkyl, alkynylheteroarylalkenyl, alkynylheteroarylalkynyl, alkylheterocyclylalkyl, alkylheterocyclylalkenyl, alkylhererocyclylalkynyl, alkenylheterocyclylalkyl, alkenylheterocyclylalkenyl, alkenylheterocyclylalkynyl, alkynylheterocyclylalkyl, alkynylheterocyclylalkenyl, alkynylheterocyclylalkynyl, alkylaryl, alkenylaryl, alkynylaryl, alkylheteroaryl, alkenylheteroaryl, or alkynylhereroaryl, where one or more methylenes can be interrupted or terminated by O, S, S(O), SO2, N(R1)2, C(O), cleavable linking group, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclic; where R1 is hydrogen, acyl, aliphatic or substituted aliphatic.
The compound of claim 1, wherein the linker is selected from the group consisting of a direct bond, ester, ether, amide and any functional group containing covalent linker. The compound of claim 1, wherein the linker comprises at least one cleavable group. The compound of claim 1, wherein the linker selected from the group consisting of succinic acid, fumaric acid, propargylic acid, ethylene glycol, diethylene glycol, or, natural or unnatural amino acids, and any combination thereof.
6. The compound of claim 1, wherein the linker selected from the group consisting of oxalic acid, malonic acid, glutaric acid, ethylene diamine, ethylene glycol, diethylene glycol, acetic acid, propionic acid, butyric acid, valeric acid, acrylic acid, but-2-enoic acid, pent-2-enoic acid, hex-2-enoic acid, 2-propynoic acid, but-2-ynoic acid, pent-2- ynoic acid, hex-2-ynoic acid, ethylene, propylene, 1-butene, 1-pentene, 1-hexene, acetylene, propyne, but-l-yne or pent-l-yne, and any combination thereof.
7. The compound of claim 1, wherein the linker is selected from the group consisting of- C(0)CH2CH2C(0)-; -C(0)(CH2CH2)mC(0)(OCH2CH2)n- wherein 'n' is 1 to 10 and 'm' is 1 to 4; -C(0)(CH2)xCH2C(0) H(CH2CH2)n-, wherein 'n' is 1 to 10 and 'x' is 0 to 3; -C(0)(CH2)mCH2C(0) H(CH2CH2)n HC(0)-, wherein 'n' is 1 to 10 and 'm' is 1 to 4; -C(0)CH2(CH2)mC(0) H- wherein 'm' is 1 to 4; - C(0)(CH2)nCH2(R) HC(0)-, wherein 'n' is 1 to 10 and R is H, alkyl, acid, amine, aryl, or thiol; -C(0)(CH2)nCH2(R) HC(0)-, wherein 'n' is 1 to 10 and R is H, alkyl, acid, amine, aryl or thiol; C(0)(CH2(R)CH2)nC(Y)X- wherein 'n' is 1 to 10, R is H, alkyl, acid, amine, aryl or thiol, Y is C, O, H, S, and X is C, O, H, S; - C(0)CH2CH2NHC(0)-; -C(0)CH2CH2C(0)NHCH2CH2 HC(0)-;
C(0)CH2CH2C(0) HCH2 HC(0)-; -C(0)CH2OCH2CH2-;
C(0)CH2CH2OCH2CH2-; -C(0)CH2OCH2CH2OCH2CH2-;
C(0)CH(R) HC(0)CH2- wherein R is H, CH3, CH(CH3)2, CH2CH(CH3)2, C(CH3)CH2CH3, or CH2-Phenyl; -C(0)CH(R) HC(0)CH2CH2- wherein R is H, CH3, CH(CH3)2, CH2CH(CH3)2, C(CH3)CH2CH3, or CH2-Phenyl; - C(0)CH(R) HC(0)(CH2)nC(0)-, wherein R is H, CH3, CH(CH3)2, CH2CH(CH3)2, C(CH3)CH2CH3, or CHz-Phenyl, and 'n' is 1, 2, or 3; - C(0)CH(R) HC(0)CH2OCH2CH2-, wherein R is H, CH3, CH(CH3)2, CH2CH(CH3)2, C(CH3)CH2CH3, or CH2-Phenyl; -C(0)C≡C(CH2)n-C(0)-, wherein 'n' is 1, 2 or 3; - C(0)C≡C(CH2)n- wherein 'n' is 0, 1, or 2; -C(0)CH=CH(CH2)nC(0)-, wherein 'n' is 0, 1, 2, or 3; -C(0)CH=CH(CH2)n- wherein 'n' is 1, 2, or 3; and - C(0)CH2CH2C(0) HCH2C(0)-.
8. The compound of claim 1, wherein the lipid, lipid derivative or lipid conjugate is selected from the group consisting of cholesterol, cholesterol derivatives, oleic acid, oleic acid derivative, alpha tocopherol, alpha tocopherol derivatives, phospholipid, phospholipid derivatives, fatty acid, naturally occurring lipid molecule which is conjugated to drug molecule(s), 1,3 -Propanediol Dicaprylate/Dicaprate, 10-undecenoic
acid, 1-dotriacontanol, 1-heptacosanol, 1-nonacosanol, 2-ethyl hexanol, Androstanes, Arachidic acid, Arachidonic acid, arachidyl alcohol, Behenic acid, behenyl alcohol, Capmul MCM CIO, Capric acid, capric alcohol, capryl alcohol, Caprylic acid, Caprylic/Capric Acid Ester of Saturated Fatty Alcohol C12-C18, Caprylic/Capric Triglyceride, Caprylic/Capric Triglyceride, Ceramide phosphorylcholine (Sphingomyelin, SPH), Ceramide phosphorylethanolamine (Sphingomyelin, Cer-PE), Ceramide phosphorylglycerol, Ceroplastic acid, Cerotic acid, Cerotic acid, ceryl alcohol, Cetearyl alcohol, Ceteth-10, cetyl alcohol, Cholanes, Cholestanes, cholesterol, cis-l l-eicosenoic acid, cis-l l-octadecenoic acid, cis-13-docosenoic acid, cluytyl alcohol, Dihomo-y-linolenic, Docosahexaenoic acid, egg lecithin, Eicosapentaenoic acid, Eicosenoic acid, Elaidic acid, elaidolinolenyl alcohol, elaidolinoleyl alcohol, elaidyl alcohol, Erucic acid, erucyl alcohol, Estranes, Ethylene glycol distearate (EGDS), Geddic acid, geddyl alcohol, glycerol distearate (type I) EP (Precirol ATO 5), Glycerol Tricaprylate/Caprate; Glycerol Tri capryl ate/Caprate (CAPTEX® 355 EP/NF); glyceryl monocaprylate (Capmul MCM C8 EP), Glyceryl Triacetate, Glyceryl Tricaprylate, Glyceryl Tricaprylate/Caprate/Laurate, Glyceryl Tricaprylate/Tricaprate, glyceryl tripalmitate (Tripalmitin), Henatriacontylic acid, Heneicosyl alcohol, Heneicosylic acid, Heptacosylic acid, Heptadecanoic acid, Heptadecyl alcohol, Hexatriacontylic acid, isostearic acid, isostearyl alcohol, Lacceroic acid, Laurie acid, Lauryl alcohol, Lignoceric acid, lignoceryl alcohol, Linoelaidic acid, Linoleic acid, linolenyl alcohol, linoleyl alcohol, Margaric acid, Mead, Melissic acid, melissyl alcohol, Montanic acid, montanyl alcohol, myricyl alcohol, Myristic acid, Myristoleic acid, Myristyl alcohol, neodecanoic acid, neoheptanoic acid, neononanoic acid, Nervonic, Nonacosylic acid, Nonadecyl alcohol, Nonadecylic acid, Nonadecylic acid, Oleic acid, oleyl alcohol, Palmitic acid, Palmitoleic acid, palmitoleyl alcohol, Pelargonic acid, pelargonic alcohol, Pentacosylic acid, Pentadecyl alcohol, Pentadecylic acid, Phosphatidic acid (phosphatidate, PA), Phosphatidylcholine (lecithin, PC), Phosphatidylethanolamine (cephalin, PE), Phosphatidylinositol (PI), Phosphatidylinositol bisphosphate (PIP2), Phosphatidylinositol phosphate (PIP), Phosphatidylinositol triphosphate (PIP3), Phosphatidylserine (PS), polyglyceryl-6- distearate, Pregnanes, Propylene Glycol Dicaprate, Propylene Glycol Di capryl ocaprate, Propylene Glycol Di capryl ocaprate, Psyllic acid, recinoleaic acid, recinoleyl alcohol, Sapienic acid, soy lecithin, Stearic acid, Stearidonic, stearyl alcohol, Tricosylic acid,
Tridecyl alcohol, Tridecylic acid, Triolein, Undecyl alcohol, undecylenic acid, Undecylic acid, Vaccenic acid, a-Linolenic acid, and γ-Linolenic acid via a spacer, wherein the spacer is selected from the group consisting of aliphatic dicarboxylic acid, unsaturated dicarboxylic acid, aldaric acid, fumaric acid, propargylic acid, acetylene dicarboxylic acid, aromatic/hetero aromatic dicarboxylic acid, ethylene glycol, diethylene glycol, natural or unnatural amino acids, and their derivatives individually and in any combinations thereof.
9. The compound of claim 1, wherein the lipid, lipid derivative or lipid conjugate is selected from the group consisting of cholesterol, cholesterol derivatives, oleic acid, oleic acid derivative, alpha tocopherol, alpha tocopherol derivatives, phospholipid, phospholipid derivatives, fatty acid or naturally occurring lipid molecule; which is conjugated to drug molecules via a spacer, wherein the spacer is selected from the group consisting of aliphatic dicarboxylic acid, unsaturated dicarboxylic acid, aldaric acid, fumaric acid, propargylic acid, acetylene dicarboxylic acid, aromatic/hetero aromatic dicarboxylic acid, ethylene glycol, diethylene glycol, natural or unnatural amino acids, and their derivatives individually or any combination thereof.
10. The compound of claim 1, wherein the lipid conjugate is the lipid or the lipid derivative conjugated with a compound selected from the group consisting of CSF-1R inhibitor, a kinase inhibitor, a chemotherapeutic drug, an immunomodulator and combinations thereof.
11. The compound of claim 10, wherein the immunomodulator is an antibody, a cytokine or a combination thereof.
12. The compound of claim 11, wherein the antibody is a therapeutic antibody, a targeting antibody or a combination thereof.
13. The compound of claim 11, wherein the cytokine is an interferon, an interleukin or a combination thereof.
14. The compound of claim 1 selected from:

Formula I A Formula IB

Formula IC wherein Ά', 'Β', 'C, D\ Έ', 'F', G\ , 'Χ', linker (L) and 'R' are as defined the preceding claims.
15. The compound of claim 1 selected from:

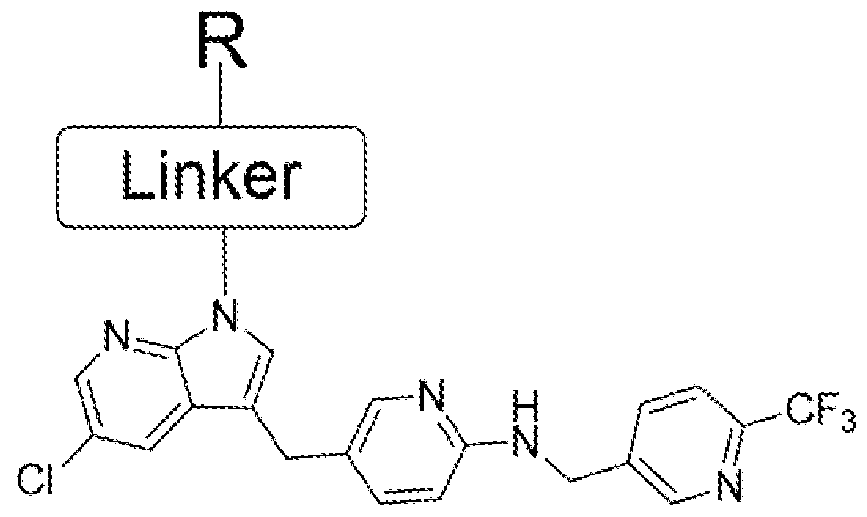
Formula IF
wherein linker (L) and 'R' are as defined in the preceding claims.
16. The compound of claim 1, wherein said compound is a compound of Formula 54:
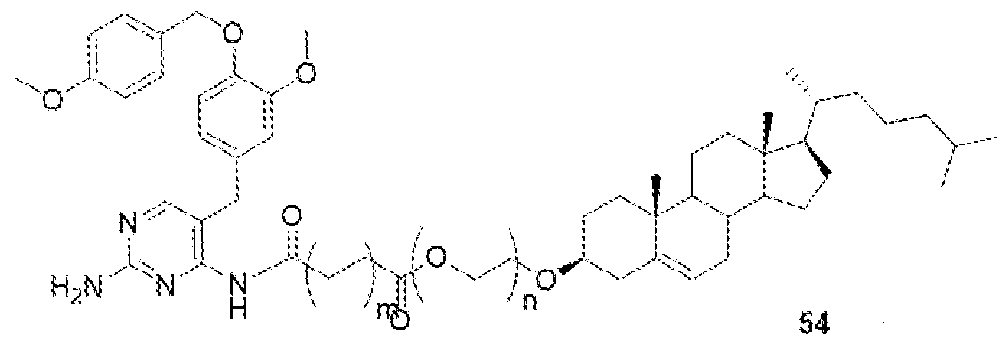
The com ound of claim 1, wherein said compound is a compound of Formula 55
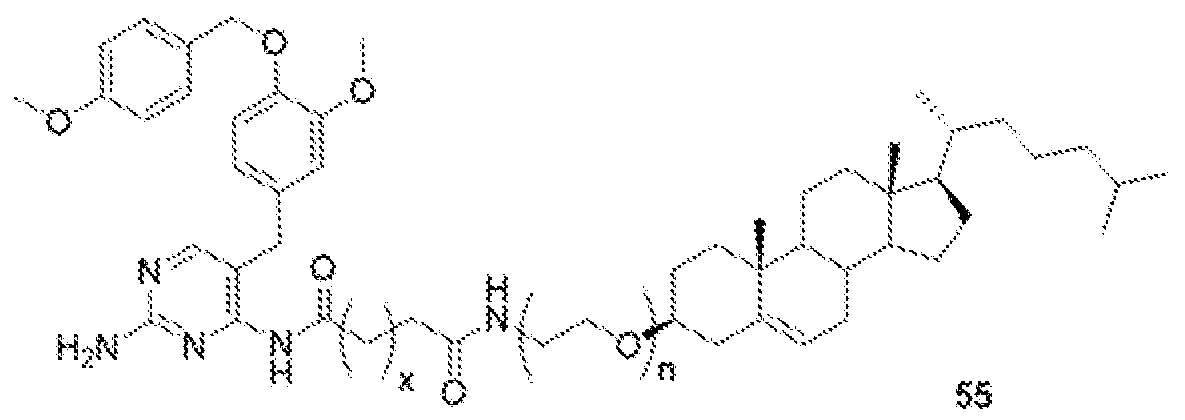
18. The compound of claim 1, wherein said compound is a compound of Formula 56:
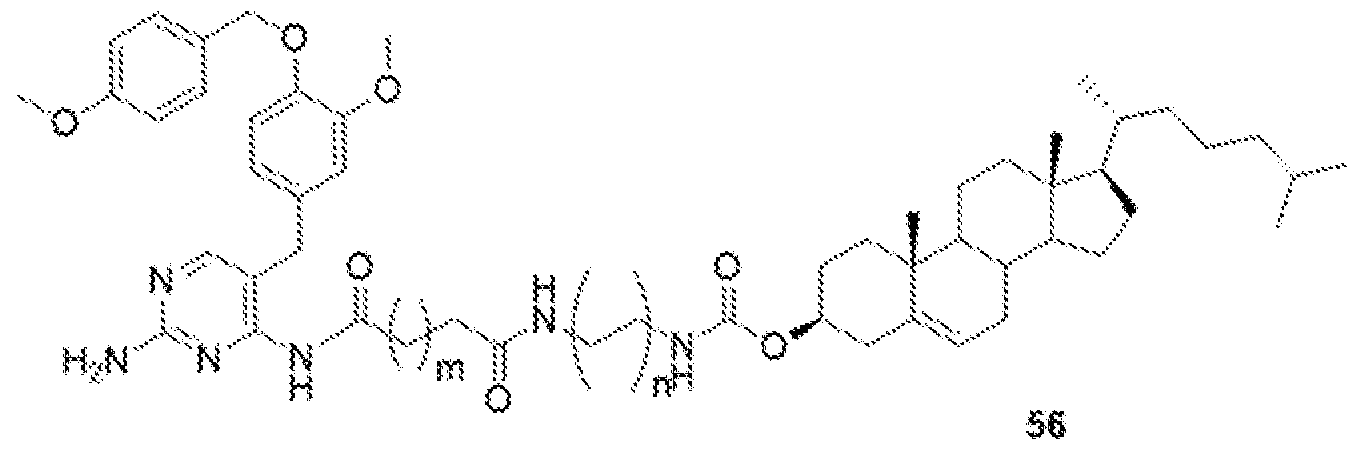 wherein 'n' is 1 to 10 and 'm' is 1 to 4.
wherein 'n' is 1 to 10 and 'm' is 1 to 4.
The com ound of claim 1, wherein said compound is a compound of Formula 57:
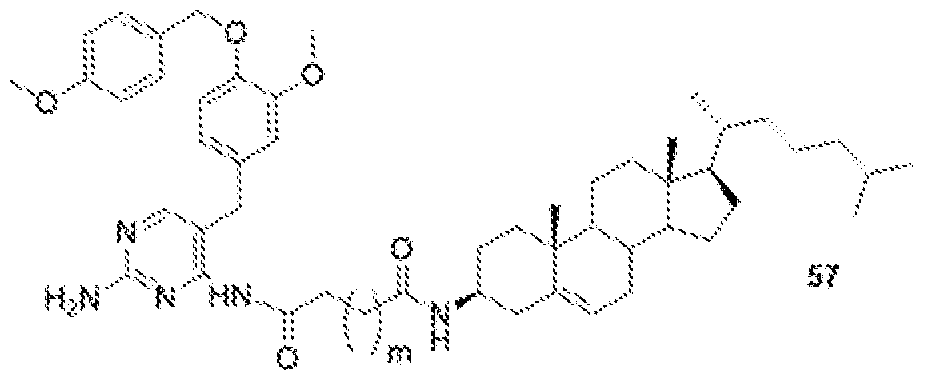
20. The compound of claim 1, wherein said compound is a compound of Formula 58:

21. The compound of claim 1 selected from the group consisting of:
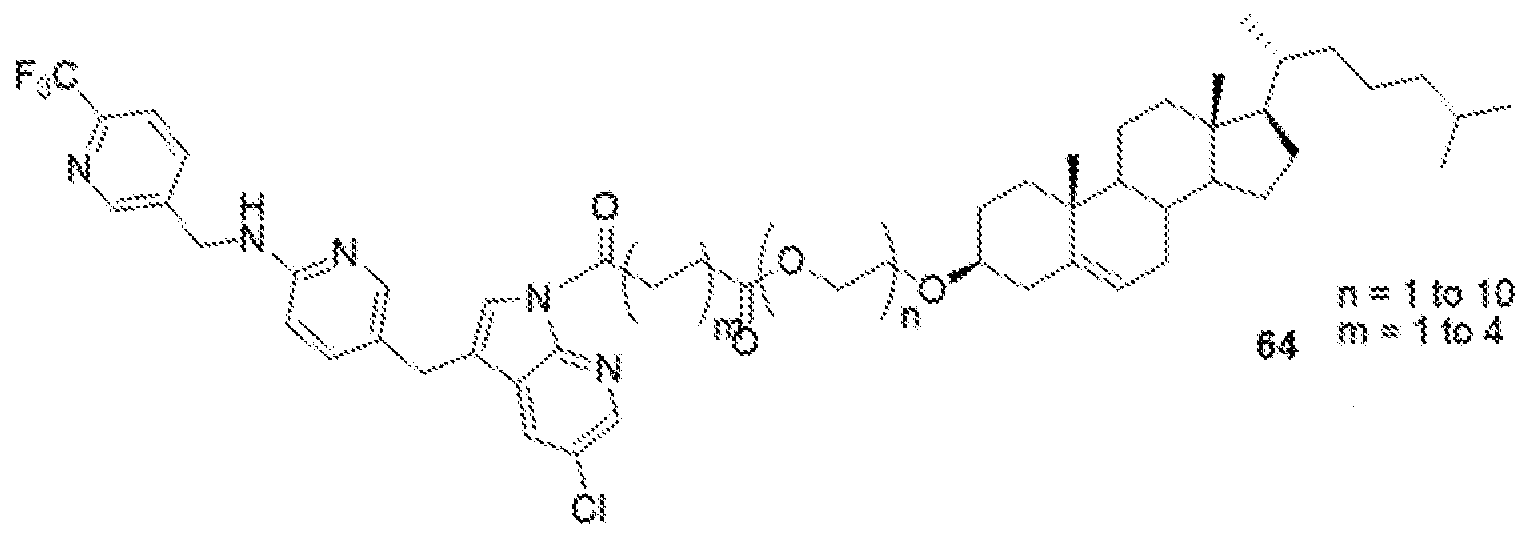
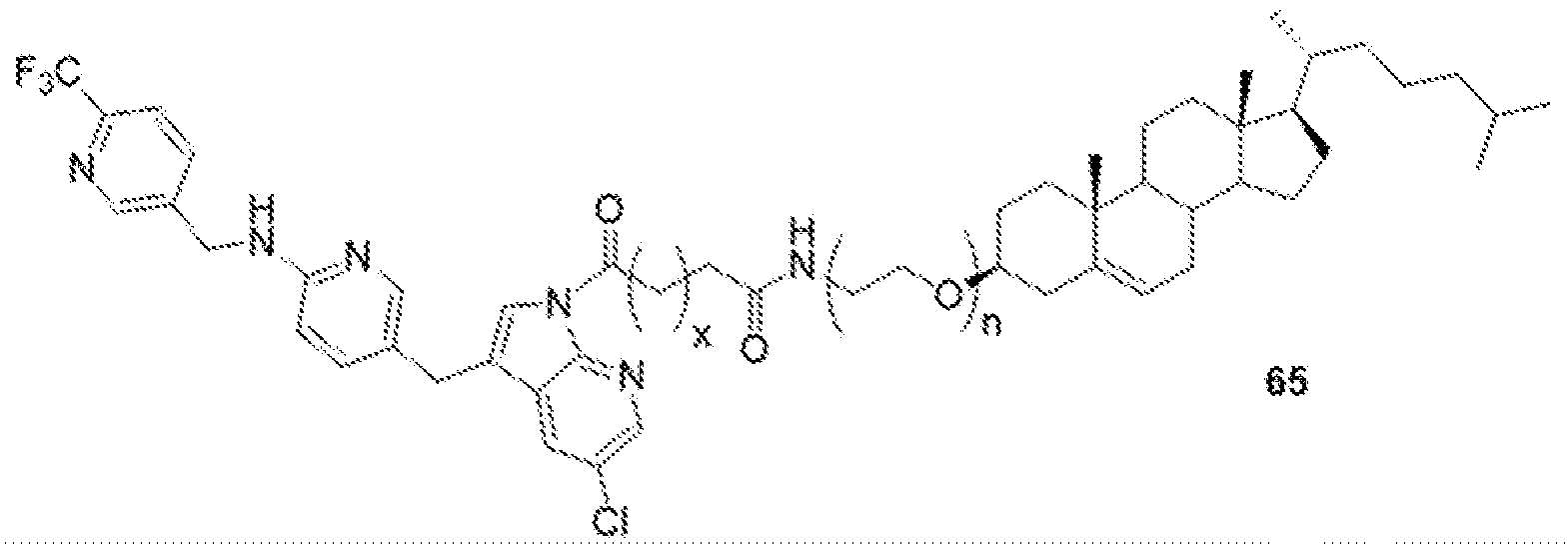
wherein 'n' is 1 to 10 and 'x' is 0 to 3:

wherein 'n' is 1 to 10 and 'm' is 1 to 4;
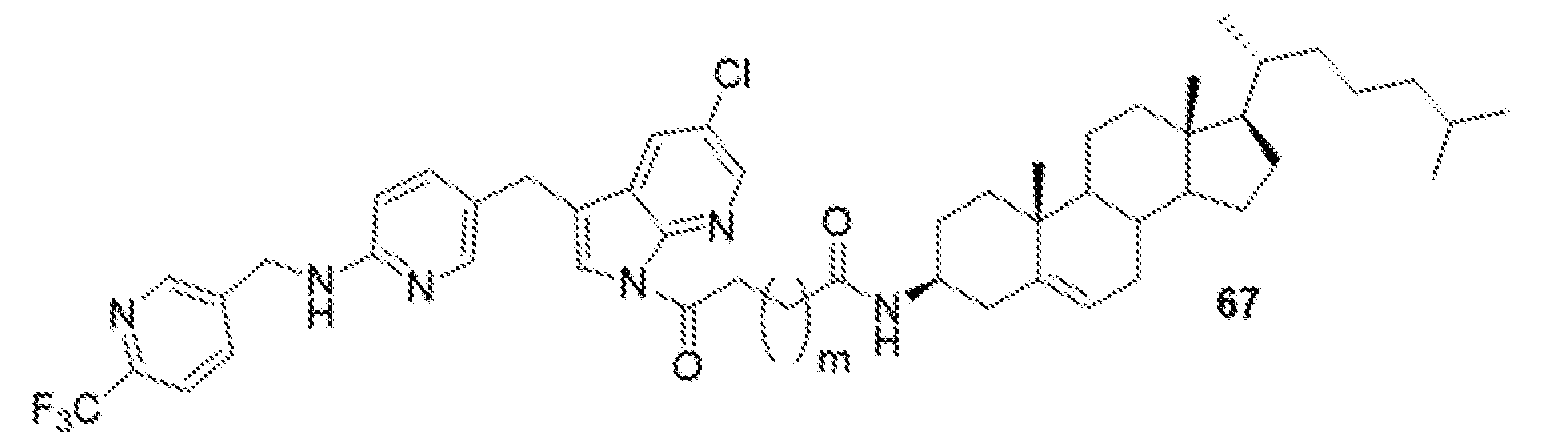
wherein 'm' is 1 to 4; and
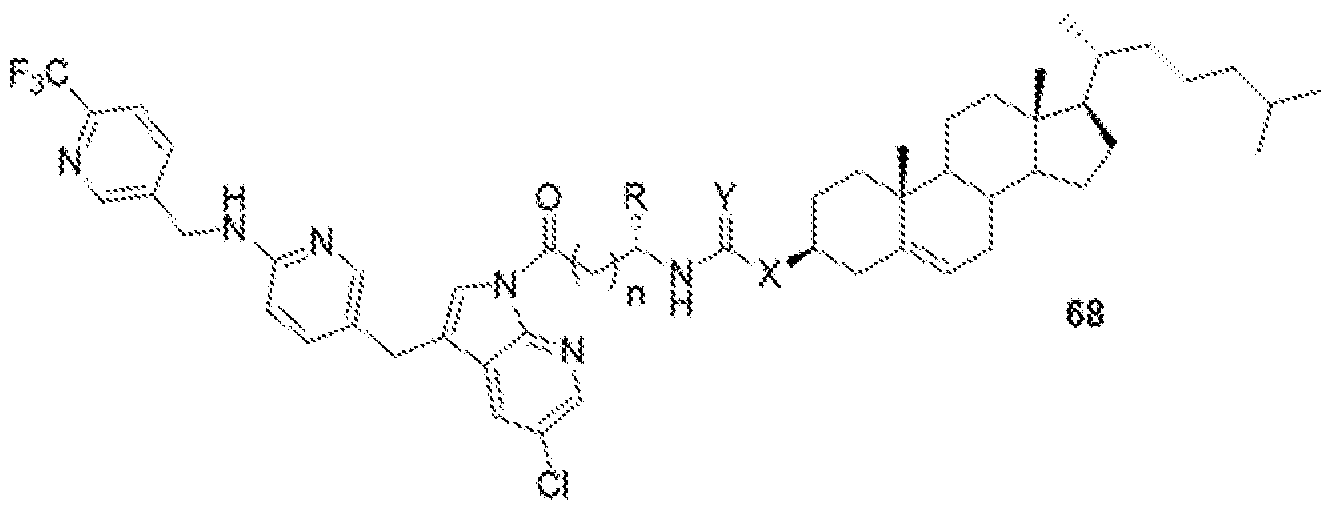
22. A composition comprising the compound of Formula I as claimed in any of the claims 1-21, or any derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates of compound of Formula I thereof; and a pharmaceutically acceptable excipient.
23. The composition of claim 22, wherein the composition comprises from about 1% to about 99% (w/w) of the compound of Formula I.
24. The composition of claim 22, wherein the composition further comprises a kinase inhibitor, a chemotherapeutic agent or an immunomodulator or any combination thereof.
25. The composition of claim 22, wherein the composition further comprises co-lipid.
26. The composition of claim 24, wherein the composition comprises about 1% to about 99% (w/w) of the kinase inhibitor.
27. The composition of claim 24, wherein the composition comprises about 1% to about 99% (w/w) of the chemotherapeutic agent.
28. The composition of claim 24, wherein the composition comprises about 1% to about 99% (w/w) of the immunomodulator.
29. The composition of claim 27, wherein the chemotherapeutic agent is selected from the group consisting of PI3K inhibitors; platinum compounds; inhibitors of topoisomerase I and II; alkylating agents; microtubule inhibitors; and angiogenesis inhibitors; or any combination thereof.
30. The composition of claim 27, wherein the chemotherapeutic agent is selected from the group consisting of germicitibine; aldesleukin; alemtuzumab; alitretinoin; allopurinol; altretamine; amifostine; anastrozole; arsenic trioxide; asparaginase; BCG Live; bexarotene capsules; bexarotene gel; bleomycin; busulfan intravenous; busulfanoral; calusterone; capecitabine; platinate; carmustine; carmustine with Polifeprosan Implant; celecoxib; chlorambucil; cladribine; cyclophosphamide; cytarabine; cytarabine liposomal; dacarbazine; dactinomycin; actinomycin D; darbepoetin alfa; daunorubicin liposomal; daunorubicin, daunomycin; denileukin diftitox, dexrazoxane; docetaxel; doxorubicin; doxorubicin liposomal; dromostanolone propionate; Elliott's B Solution; epirubicin; epoetin alfa estramustine; etoposide phosphate; etoposide (VP- 16);
exemestane; Filgrastim; floxuridine (intraarterial); fludarabine; fluorouracil (5-FU); fulvestrant; gemtuzumab ozogamicin; goserelin acetate; hydroxyurea; ibritumomab tiuxetan; idarubicin; ifosfamide; imatinib mesylate; Interferon alfa-2a; interferon alfa- 2b; irinotecan; letrozole; leucovorin; levamisole; lomustine (CCNU); mechlorethamine (nitrogenmustard); megestrol acetate; melphalan (L-PAM); mercaptopurine (6-MP); mesna; methotrexate; methoxsalen; mitomycin C; mitotane; mitoxantrone; nandrolone phenpropionate; nofetumomab; LOddC; Oprelvekin; pamidronate; pegademase; pegaspargase; pegfilgrastim; pentostatin; pipobroman; plicamycin; mithramycin; porfimer sodium; procarbazine; quinacrine; rasburicase; rituximab; sargramostim; streptozocin; talbuvidine (LDT); talc; tamoxifen; temozolomide; teniposide (VM-26); testolactone; thioguanine (6-TG); thiotepa; topotecan; toremifene; tositumomab; trastuzumab; tretinoin (ATRA); uracil mustard; valrubicin; valtorcitabine (monoval LDC); vinblastine; vinorelbine; and zoledronate; or any mixture thereof.
31. The composition of claim 29, wherein the PI3K inhibitor is selected from the group consisting of PI103; P1828; LY294002; wortmannin; demethoxyviridin; IC486068; IC87114; GDC-0941; perifosine; CAL101; PX-866; IPI-145; BAY 80-6946; BEZ235; P6503; TGR1202; SF1126; INK1117; BKM120; IL147; XL765; Palomid 529; GSK1059615; ZSTK474; PWT33597; TGI 00-115; CAL263; G E-447; CUDC-907; and AEZS-136, or any combination thereof.
32. The composition of claim 24, wherein the chemotherapeutic agent is conjugated with a component of the composition.
33. The composition of claim 32, wherein the chemotherapeutic agent is conjugated with PEG.
34. The composition of claim 32, wherein the chemotherapeutic agent is conjugated with a lipid or lipid derivative.
35. The composition of claim 25, wherein the co-lipid is selected from the group consisting of HSPC, DSPC, DPPC, DOPC, POPC, SOPC, Egg- PC, and DSPE-PEG, DPPE-PEG, DMPE-PEG and combinations thereof.
36. The composition of claim 22, wherein the composition is a liposome, emulsion or micelle.
37. The composition of claim 22, wherein the composition is a nanoparticle.
38. The composition of claim 37, wherein the nanoparticle is about 1 nm to about 400 nm in diameter.
39. The composition of claim 22, wherein the composition further comprises a pharmaceutically acceptable carrier.
40. The composition of claim 22, wherein the composition further comprises a pharmaceutically acceptable excipient.
41. The composition of claim 40, wherein the pharmaceutically acceptable excipient is selected from the group comprising adjuvant, diluent, carrier, granulating agents, binding agents, lubricating agents, disintegrating agent, sweetening agents, glidant, anti -adherent, anti-static agent, surfactant, anti-oxidant, gum, coating agent, coloring agent, flavouring agent, coating agent, plasticizer, preservative, suspending agent, emulsifying agent, plant cellulosic material, spheronization agent, and other conventionally known pharmaceutically acceptable excipient, or any combination of excipients thereof.
42. The composition of claim 22, wherein the composition inhibits colony stimulating factor- 1 receptor (CSF-1R) and is administered to a subject in need thereof through modes selected from a group comprising intravenous administration, intramuscular administration, intraperitoneal administration, hepatoportal administration, intra articular administration and pancreatic duodenal artery administration, or any combination thereof.
43. A method of treating cancer comprising administering a compound of Formula I of claim 1 or derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates thereof, or a composition of claim 23, to a subject in need of treatment for cancer.
44. The method of claim 43, wherein the cancer is selected from the group consisting of breast cancer; ovarian cancer; glioma; gastrointestinal cancer; prostate cancer; carcinoma, lung carcinoma, hepatocellular carcinoma, testicular cancer; cervical cancer; endometrial cancer; bladder cancer; head and neck cancer; lung cancer; gastroesophageal cancer, and gynecological cancer, or any combination thereof.
45. The method of claim 43, further comprising co-administering one or more additional anti-cancer therapy to the subject.
46. The method of claim 45, wherein the additional therapy is selected from the group consisting of surgery, chemotherapy, radiation therapy, thermotherapy, immunotherapy, hormone therapy, laser therapy, and anti-angiogenic therapy, or any combination thereof.
47. The method of claim 45, wherein the additional therapy comprises administering a kinase inhibitor, a chemotherapeutic agent, an immunomodulator or any combination thereof, to the subject.
48. The method of claim 47, wherein the immunomodulator activates an immune response against cancer cells.
49. The method of claim 47, wherein the immunomodulator is selected from the group consisting of antibody, natural killer cells, lymphokine-activated killer cells, cytotoxic T cells and dendritic cells, anti-PD-Ll antibodies, anti-PD-1 antibodies, anti-CD52 antibodies, anti-VEGF-A antibodies, anti-CD30 antibodies, anti-EGFR antibodies, anti-CD33 antibodies, anti-CD20 antibodies, anti-CTLA4 antibodies, anti-F£ER-2 antibodies, interferons, and interleukins, or any combination thereof.
50. The method of claim 49, wherein the antibody is a therapeutic antibody or a targeting antibody or a combination thereof.
51. A method for inhibition of CSF (colony stimulating factor) or CSF-1R (colony stimulating factor- 1 receptor) signalling pathway in a cell, said method comprising contacting the cell with the compound of Formula I or derivative, salt, tautomeric form, isomer, polymorph, solvate or intermediates thereof, or the composition as defined in any of the preceding claims.
52. Compound of Formula I or derivative, salt, tautomeric form, isomer, polymorph, solvate or intermediates thereof, or the composition as defined in any of the preceding claims, for use as a medicament.
53. Compound of Formula I or derivative, salt, tautomeric form, isomer, polymorph, solvate or intermediates thereof, or the composition as defined in any of the preceding claims, for use in the treatment of cancer, inhibition of CSF or CSF-1R signalling pathway, or a combination thereof.
54. Use of compound of Formula I or derivative, salt, tautomeric form, isomer, polymorph, solvate or intermediates thereof, or the composition as defined in any of the preceding claims, in the treatment of cancer, inhibition of CSF or CSF-1R signalling pathway, or a combination thereof.
55. The method or use of any of the preceding claims, wherein the cancer is selected from the group consisting of breast cancer; ovarian cancer; glioma; gastrointestinal cancer; prostate cancer; carcinoma, lung carcinoma, hepatocellular carcinoma, testicular cancer; cervical cancer; endometrial cancer; bladder cancer; head and neck cancer; lung
cancer; gastro-oesophageal cancer and gynecological cancer, or any combination thereof.
56. A formulation comprising the compound of Formula I as claimed in any of the claims 1-21, or any derivative, salt, tautomeric form, isomer, polymorph, solvate, or intermediates of compound of Formula I thereof; and a pharmaceutically acceptable excipient.
57. The formulation of claim 56, wherein the phospholipid or PEGylated phospholipid is selected from a group comprising HSPC, DSPC, DPPC, DOPC, POPC, SOPC, Egg PC , DPPE-PEG, DMPE-PEG and DSPE-PEG or any combination thereof; wherein the composition comprises about 1% to about 99% (w/w) of these phospholipid and PEGylated phospholipid or any combination thereof.
58. The formulation of claim 56, wherein the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 5:55:35:5.
59. The formulation of claim 56, wherein the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 10:50:35:5.
60. The formulation of claim 56, wherein the formulation comprises the compound along with HSPC, POPC and DSPE-PEG in a ratio 15:50:30:5.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201711004979 | 2017-02-11 | ||
| IN201711004979 | 2017-02-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018146641A1 true WO2018146641A1 (en) | 2018-08-16 |
Family
ID=63108014
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2018/050839 WO2018146641A1 (en) | 2017-02-11 | 2018-02-12 | Novel inhibitors of cellular signalling |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2018146641A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110054660A (en) * | 2018-12-25 | 2019-07-26 | 四川大学 | A kind of preparation and application of the breast cancer targeting lipids material of fructose modification |
| CN115381817A (en) * | 2022-08-15 | 2022-11-25 | 北京中医药大学 | Application of Sotuletinib in preparation of drugs for treating crescent nephritis |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007121484A2 (en) * | 2006-04-19 | 2007-10-25 | Novartis Ag | 6-o-substituted benzoxazole and benzothiazole compounds and methods of inhibiting csf-1r signaling |
| WO2013188763A1 (en) * | 2012-06-15 | 2013-12-19 | The Brigham And Women's Hospital, Inc. | Compositions for treating cancer and methods for making the same |
| WO2017137958A1 (en) * | 2016-02-11 | 2017-08-17 | Invictus Oncology Pvt. Ltd. | Cellular signalling inhibitors, their formulations and methods thereof |
-
2018
- 2018-02-12 WO PCT/IB2018/050839 patent/WO2018146641A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007121484A2 (en) * | 2006-04-19 | 2007-10-25 | Novartis Ag | 6-o-substituted benzoxazole and benzothiazole compounds and methods of inhibiting csf-1r signaling |
| WO2013188763A1 (en) * | 2012-06-15 | 2013-12-19 | The Brigham And Women's Hospital, Inc. | Compositions for treating cancer and methods for making the same |
| WO2017137958A1 (en) * | 2016-02-11 | 2017-08-17 | Invictus Oncology Pvt. Ltd. | Cellular signalling inhibitors, their formulations and methods thereof |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110054660A (en) * | 2018-12-25 | 2019-07-26 | 四川大学 | A kind of preparation and application of the breast cancer targeting lipids material of fructose modification |
| CN115381817A (en) * | 2022-08-15 | 2022-11-25 | 北京中医药大学 | Application of Sotuletinib in preparation of drugs for treating crescent nephritis |
| CN115381817B (en) * | 2022-08-15 | 2023-09-05 | 北京中医药大学 | Application of Sotuletinib in preparation of medicine for treating crescent nephritis |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2020125296A (en) | Lipid-based platinum compounds and nanoparticles | |
| JP2018188443A (en) | Compositions for treating cancer and methods for making those compositions | |
| US10342846B2 (en) | Nanoparticle drug delivery systems | |
| US10736968B2 (en) | Cellular signalling inhibitors, their formulations and methods thereof | |
| JP2022023183A (en) | Pentaaza macrocyclic ring complexes possessing oral bioavailability | |
| US10293021B2 (en) | Conjugates and prodrugs for treating cancer and inflammatory diseases | |
| WO2015117136A1 (en) | Boronic acid esters and pharmaceutical formulations thereof | |
| JP2019196380A (en) | Curcuphenol compounds for increasing mhc-i expression | |
| WO2018146641A1 (en) | Novel inhibitors of cellular signalling | |
| CN113336768B (en) | Multi-target tyrosine kinase inhibitor | |
| JP2019505537A5 (en) | ||
| JP7485311B2 (en) | Cancer cell proliferation inhibitors and cancer cell proliferation inhibitor enhancers | |
| WO2020014387A1 (en) | B-cell immunotherapy in cancer treatment | |
| WO2011147254A1 (en) | Phenylbutyryl curcumin derivatives and uses for preparing anti-tumor drugs thereof | |
| KR20160045668A (en) | Lipid-based platinum compounds and nanoparticles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18750697 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 18750697 Country of ref document: EP Kind code of ref document: A1 |