WO2012043651A1 - ミエロイド系血液細胞の製造方法 - Google Patents
ミエロイド系血液細胞の製造方法 Download PDFInfo
- Publication number
- WO2012043651A1 WO2012043651A1 PCT/JP2011/072234 JP2011072234W WO2012043651A1 WO 2012043651 A1 WO2012043651 A1 WO 2012043651A1 JP 2011072234 W JP2011072234 W JP 2011072234W WO 2012043651 A1 WO2012043651 A1 WO 2012043651A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- gene
- human
- myeloid blood
- Prior art date
Links
- 210000000601 blood cell Anatomy 0.000 title claims abstract description 163
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 23
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 94
- 101100239628 Danio rerio myca gene Proteins 0.000 claims abstract description 72
- 230000035755 proliferation Effects 0.000 claims abstract description 28
- 210000004027 cell Anatomy 0.000 claims description 352
- 238000000034 method Methods 0.000 claims description 85
- 241000282414 Homo sapiens Species 0.000 claims description 84
- 230000004069 differentiation Effects 0.000 claims description 49
- 210000001616 monocyte Anatomy 0.000 claims description 43
- 210000001778 pluripotent stem cell Anatomy 0.000 claims description 42
- 210000005259 peripheral blood Anatomy 0.000 claims description 39
- 239000011886 peripheral blood Substances 0.000 claims description 39
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 claims description 31
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 claims description 30
- 210000004443 dendritic cell Anatomy 0.000 claims description 26
- 239000003814 drug Substances 0.000 claims description 26
- 208000015181 infectious disease Diseases 0.000 claims description 24
- 210000004263 induced pluripotent stem cell Anatomy 0.000 claims description 21
- 230000001939 inductive effect Effects 0.000 claims description 19
- 238000012258 culturing Methods 0.000 claims description 18
- 102000004388 Interleukin-4 Human genes 0.000 claims description 15
- 108090000978 Interleukin-4 Proteins 0.000 claims description 15
- 229940028885 interleukin-4 Drugs 0.000 claims description 15
- 101150090105 Ezh2 gene Proteins 0.000 claims description 11
- 101150064607 HIF1A gene Proteins 0.000 claims description 11
- 101150007128 MDM4 gene Proteins 0.000 claims description 11
- 206010028980 Neoplasm Diseases 0.000 claims description 11
- 101150024228 mdm2 gene Proteins 0.000 claims description 11
- 101150062914 BMI1 gene Proteins 0.000 claims description 10
- 208000035473 Communicable disease Diseases 0.000 claims description 8
- 208000024827 Alzheimer disease Diseases 0.000 claims description 7
- 208000023275 Autoimmune disease Diseases 0.000 claims description 7
- 206010002022 amyloidosis Diseases 0.000 claims description 7
- 208000024777 Prion disease Diseases 0.000 claims description 6
- 208000032839 leukemia Diseases 0.000 claims description 5
- 210000000056 organ Anatomy 0.000 claims description 4
- 238000002054 transplantation Methods 0.000 claims description 4
- 101100058549 Homo sapiens BMI1 gene Proteins 0.000 claims description 3
- 101100389970 Homo sapiens EZH2 gene Proteins 0.000 claims description 3
- 101100506763 Homo sapiens HIF1A gene Proteins 0.000 claims description 3
- 101100236863 Homo sapiens MDM2 gene Proteins 0.000 claims description 3
- 101100129741 Homo sapiens MDM4 gene Proteins 0.000 claims description 3
- 210000003714 granulocyte Anatomy 0.000 claims description 3
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 claims description 2
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 claims description 2
- 230000002265 prevention Effects 0.000 claims description 2
- 230000014509 gene expression Effects 0.000 abstract description 46
- 108050002772 E3 ubiquitin-protein ligase Mdm2 Proteins 0.000 abstract description 14
- 101000882127 Homo sapiens Histone-lysine N-methyltransferase EZH2 Proteins 0.000 abstract description 11
- 108050005300 MDM4 Proteins 0.000 abstract description 6
- 101001046870 Homo sapiens Hypoxia-inducible factor 1-alpha Proteins 0.000 abstract description 5
- 239000000243 solution Substances 0.000 description 37
- 241000713666 Lentivirus Species 0.000 description 31
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 28
- 102100022338 Integrin alpha-M Human genes 0.000 description 28
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 27
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 27
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 27
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 27
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 24
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 24
- 210000002540 macrophage Anatomy 0.000 description 23
- 210000001744 T-lymphocyte Anatomy 0.000 description 21
- 239000002609 medium Substances 0.000 description 21
- 101000872170 Homo sapiens Polycomb complex protein BMI-1 Proteins 0.000 description 19
- 102100033566 Polycomb complex protein BMI-1 Human genes 0.000 description 19
- 230000006698 induction Effects 0.000 description 17
- 102000012199 E3 ubiquitin-protein ligase Mdm2 Human genes 0.000 description 16
- 230000008672 reprogramming Effects 0.000 description 16
- 239000013598 vector Substances 0.000 description 16
- 230000002062 proliferating effect Effects 0.000 description 15
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 14
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 14
- 230000000694 effects Effects 0.000 description 14
- 229920000392 Zymosan Polymers 0.000 description 13
- 238000000338 in vitro Methods 0.000 description 13
- 230000007774 longterm Effects 0.000 description 13
- 238000001000 micrograph Methods 0.000 description 13
- 239000013603 viral vector Substances 0.000 description 13
- 238000004458 analytical method Methods 0.000 description 12
- 201000011510 cancer Diseases 0.000 description 12
- 210000001671 embryonic stem cell Anatomy 0.000 description 12
- 239000002245 particle Substances 0.000 description 12
- 238000010186 staining Methods 0.000 description 12
- 102100038970 Histone-lysine N-methyltransferase EZH2 Human genes 0.000 description 11
- 230000010261 cell growth Effects 0.000 description 11
- 238000003501 co-culture Methods 0.000 description 11
- 210000001082 somatic cell Anatomy 0.000 description 11
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 10
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 10
- 241000700605 Viruses Species 0.000 description 10
- 239000013604 expression vector Substances 0.000 description 10
- 239000003550 marker Substances 0.000 description 10
- 238000004806 packaging method and process Methods 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- 241000699666 Mus <mouse, genus> Species 0.000 description 9
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 9
- 239000007758 minimum essential medium Substances 0.000 description 9
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 8
- 230000012010 growth Effects 0.000 description 8
- 238000000926 separation method Methods 0.000 description 8
- 230000000638 stimulation Effects 0.000 description 8
- 102000017274 MDM4 Human genes 0.000 description 7
- 241000124008 Mammalia Species 0.000 description 7
- 210000002950 fibroblast Anatomy 0.000 description 7
- 244000005700 microbiome Species 0.000 description 7
- 239000013612 plasmid Substances 0.000 description 7
- 230000004936 stimulating effect Effects 0.000 description 7
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 6
- 108020004414 DNA Proteins 0.000 description 6
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 6
- 230000001464 adherent effect Effects 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 238000004113 cell culture Methods 0.000 description 6
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 210000000265 leukocyte Anatomy 0.000 description 6
- 239000002953 phosphate buffered saline Substances 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 230000001737 promoting effect Effects 0.000 description 6
- 102000004169 proteins and genes Human genes 0.000 description 6
- 210000002966 serum Anatomy 0.000 description 6
- 239000006285 cell suspension Substances 0.000 description 5
- 238000002659 cell therapy Methods 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 238000007667 floating Methods 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 210000005087 mononuclear cell Anatomy 0.000 description 5
- 210000000066 myeloid cell Anatomy 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 101000746373 Homo sapiens Granulocyte-macrophage colony-stimulating factor Proteins 0.000 description 4
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 206010057249 Phagocytosis Diseases 0.000 description 4
- 101150086694 SLC22A3 gene Proteins 0.000 description 4
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 4
- -1 TGFb-1 Proteins 0.000 description 4
- 229940030156 cell vaccine Drugs 0.000 description 4
- 238000005119 centrifugation Methods 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 102000046157 human CSF2 Human genes 0.000 description 4
- 230000028993 immune response Effects 0.000 description 4
- 210000001704 mesoblast Anatomy 0.000 description 4
- 208000030159 metabolic disease Diseases 0.000 description 4
- 229960004857 mitomycin Drugs 0.000 description 4
- 230000008782 phagocytosis Effects 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 210000000130 stem cell Anatomy 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 102000003390 tumor necrosis factor Human genes 0.000 description 4
- 241000282693 Cercopithecidae Species 0.000 description 3
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 101001027128 Homo sapiens Fibronectin Proteins 0.000 description 3
- 101001139134 Homo sapiens Krueppel-like factor 4 Proteins 0.000 description 3
- 102100020677 Krueppel-like factor 4 Human genes 0.000 description 3
- 108700021430 Kruppel-Like Factor 4 Proteins 0.000 description 3
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 3
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 3
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 3
- 102000004142 Trypsin Human genes 0.000 description 3
- 108090000631 Trypsin Proteins 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 229960002424 collagenase Drugs 0.000 description 3
- 238000012136 culture method Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 238000000684 flow cytometry Methods 0.000 description 3
- 239000007850 fluorescent dye Substances 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 238000001638 lipofection Methods 0.000 description 3
- 239000013600 plasmid vector Substances 0.000 description 3
- 238000003345 scintillation counting Methods 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 229940104230 thymidine Drugs 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 239000012588 trypsin Substances 0.000 description 3
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 3
- 230000006820 DNA synthesis Effects 0.000 description 2
- 102000009109 Fc receptors Human genes 0.000 description 2
- 108010087819 Fc receptors Proteins 0.000 description 2
- 102100037362 Fibronectin Human genes 0.000 description 2
- 108010067306 Fibronectins Proteins 0.000 description 2
- 229920001917 Ficoll Polymers 0.000 description 2
- 208000009329 Graft vs Host Disease Diseases 0.000 description 2
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 2
- 101000762379 Homo sapiens Bone morphogenetic protein 4 Proteins 0.000 description 2
- 101000687905 Homo sapiens Transcription factor SOX-2 Proteins 0.000 description 2
- 102100022875 Hypoxia-inducible factor 1-alpha Human genes 0.000 description 2
- 101150039798 MYC gene Proteins 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 2
- 239000004677 Nylon Substances 0.000 description 2
- 206010036790 Productive cough Diseases 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 230000006052 T cell proliferation Effects 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 102100024270 Transcription factor SOX-2 Human genes 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 230000037429 base substitution Effects 0.000 description 2
- 210000005208 blood dendritic cell Anatomy 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 239000006143 cell culture medium Substances 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000002299 complementary DNA Substances 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 230000001605 fetal effect Effects 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 230000002538 fungal effect Effects 0.000 description 2
- 208000024908 graft versus host disease Diseases 0.000 description 2
- 229960002897 heparin Drugs 0.000 description 2
- 229920000669 heparin Polymers 0.000 description 2
- 102000046148 human BMP4 Human genes 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 208000021601 lentivirus infection Diseases 0.000 description 2
- 210000004698 lymphocyte Anatomy 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 210000001161 mammalian embryo Anatomy 0.000 description 2
- 239000011325 microbead Substances 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 230000000921 morphogenic effect Effects 0.000 description 2
- 108700024542 myc Genes Proteins 0.000 description 2
- 229920001778 nylon Polymers 0.000 description 2
- 230000007310 pathophysiology Effects 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 230000009696 proliferative response Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 210000003802 sputum Anatomy 0.000 description 2
- 208000024794 sputum Diseases 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 238000010600 3H thymidine incorporation assay Methods 0.000 description 1
- 101150033839 4 gene Proteins 0.000 description 1
- 101150111510 BMY1 gene Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 1
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 1
- 101100342337 Caenorhabditis elegans klf-1 gene Proteins 0.000 description 1
- 101100257372 Caenorhabditis elegans sox-3 gene Proteins 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 101001015963 Homo sapiens E3 ubiquitin-protein ligase Mdm2 Proteins 0.000 description 1
- 101000608935 Homo sapiens Leukosialin Proteins 0.000 description 1
- 101001094700 Homo sapiens POU domain, class 5, transcription factor 1 Proteins 0.000 description 1
- 101100137155 Homo sapiens POU5F1 gene Proteins 0.000 description 1
- 101000980354 Homo sapiens Protein Mdm4 Proteins 0.000 description 1
- 101100094846 Homo sapiens SLC22A3 gene Proteins 0.000 description 1
- 101000713275 Homo sapiens Solute carrier family 22 member 3 Proteins 0.000 description 1
- 101150072501 Klf2 gene Proteins 0.000 description 1
- 102100039564 Leukosialin Human genes 0.000 description 1
- 239000012097 Lipofectamine 2000 Substances 0.000 description 1
- 241000711408 Murine respirovirus Species 0.000 description 1
- 101100341513 Mus musculus Itgam gene Proteins 0.000 description 1
- 101100510267 Mus musculus Klf4 gene Proteins 0.000 description 1
- 101100310657 Mus musculus Sox1 gene Proteins 0.000 description 1
- 101100310648 Mus musculus Sox17 gene Proteins 0.000 description 1
- 101100310650 Mus musculus Sox18 gene Proteins 0.000 description 1
- 101100257376 Mus musculus Sox3 gene Proteins 0.000 description 1
- 101100043062 Mus musculus Sox7 gene Proteins 0.000 description 1
- 108700026495 N-Myc Proto-Oncogene Proteins 0.000 description 1
- 102000055056 N-Myc Proto-Oncogene Human genes 0.000 description 1
- 101150012532 NANOG gene Proteins 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 101150001847 Sox15 gene Proteins 0.000 description 1
- 101150037203 Sox2 gene Proteins 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 108010023082 activin A Proteins 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 238000002617 apheresis Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 239000012888 bovine serum Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 239000002771 cell marker Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 229960004106 citric acid Drugs 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 210000002308 embryonic cell Anatomy 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 238000011223 gene expression profiling Methods 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 102000058013 human BMI1 Human genes 0.000 description 1
- 102000056255 human EZH2 Human genes 0.000 description 1
- 102000049131 human HIF1A Human genes 0.000 description 1
- 102000056819 human MDM4 Human genes 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000003308 immunostimulating effect Effects 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 101150072261 large T gene Proteins 0.000 description 1
- 101150111214 lin-28 gene Proteins 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000003761 preservation solution Substances 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 239000004017 serum-free culture medium Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000004565 tumor cell growth Effects 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images





























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/15—Cells of the myeloid line, e.g. granulocytes, basophils, eosinophils, neutrophils, leucocytes, monocytes, macrophages or mast cells; Myeloid precursor cells; Antigen-presenting cells, e.g. dendritic cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0639—Dendritic cells, e.g. Langherhans cells in the epidermis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0645—Macrophages, e.g. Kuepfer cells in the liver; Monocytes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/22—Colony stimulating factors (G-CSF, GM-CSF)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2304—Interleukin-4 (IL-4)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/606—Transcription factors c-Myc
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2502/00—Coculture with; Conditioned medium produced by
- C12N2502/13—Coculture with; Conditioned medium produced by connective tissue cells; generic mesenchyme cells, e.g. so-called "embryonic fibroblasts"
- C12N2502/1394—Bone marrow stromal cells; whole marrow
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/45—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from artificially induced pluripotent stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
Definitions
- the present invention relates to production of human myeloid blood cells having the ability to proliferate in vitro, and a method for culturing the same. More specifically, the present invention relates to myeloid blood cells collected in vitro from a myeloid blood cell collected from a human body or a pluripotent stem cell such as an induced pluripotent stem cell using a predetermined differentiation induction method. The present invention relates to a method for allowing a cell to acquire the ability to proliferate in vitro while retaining its function. Furthermore, the present invention relates to a method for differentiating myeloid blood cells having the proliferation ability into dendritic cell-like cells having a stronger T cell stimulation ability.
- the myeloid blood cells produced by the method of the present invention and capable of proliferating in vitro have the activity of phagocytosing microorganisms and the like.
- the proliferative ability myeloid blood cells or dendritic cell-like cells derived therefrom are useful in the treatment of Alzheimer's disease, cancer, infectious diseases, prion diseases, amyloidosis, autoimmune diseases, etc. It is expected.
- the proliferative ability myeloid blood cells and dendritic cell-like cells derived therefrom produced by the present invention are also expected to be useful in the treatment of rejection in organ transplantation and graft-versus-host disease (GVHD). Is done.
- GVHD graft-versus-host disease
- myeloid blood cells have the proliferative ability produced by the present invention because they have an important role in vivo in pathophysiology such as cancer, immune-related diseases, metabolic diseases, and vascular diseases.
- Myeloid blood cells are expected to be useful as evaluation cells for examining drug efficacy and toxicity of drugs and the like.
- Myeloid blood cells are a group of cells classified into leukocytes, and include macrophages, dendritic cells, granulocytes, and the like. Macrophages are main foreign body processing cells in the living body, and have a role of protecting the living body from infectious diseases by phagocytosing infectious microorganisms and the like that have entered the living body and decomposing them. In vivo, a large amount of cells die every day, and macrophages phagocytose and decompose the debris present in the tissues in the living body. In addition, macrophages play an essential role in maintaining homeostasis by phagocytosing and decomposing various metabolites generated in the living body.
- Macrophages are often infiltrated locally in malignant tumors. It is believed that macrophages present in the tumor are both attacking tumor cells and promoting tumor cell growth. Until now, attempts have been made to treat malignant tumors by utilizing the ability of macrophages to attack tumor cells.
- Dendritic cells are cells that strongly stimulate and activate T lymphocytes, and are cells that control the immune response in vivo. Upon invasion of infectious microorganisms, dendritic cells engulf the microorganisms and present antigenic substances derived from them to T lymphocytes, stimulating and activating antigen-specific T lymphocytes, thereby enhancing the immune response. Induce. Attempts have been made to use dendritic cells as cellular vaccines in immunotherapy for cancer and infections by taking advantage of the ability of dendritic cells to strongly stimulate T lymphocytes.
- Macrophages and dendritic cells are cells having an important role in pathophysiology such as cancer, immune-related diseases, metabolic diseases, and vascular diseases. In developing various pharmaceuticals for treating these diseases, it is necessary to evaluate the effects of the drugs on macrophages and dendritic cells. In order to compare the effects of various kinds of chemical substances as drug candidates under the same conditions, a method of supplying large amounts of macrophages and dendritic cells having uniform properties is required.
- Pluripotent stem cells such as embryonic stem cells (ES cells) and induced pluripotent stem cells (induced pluripotent stem cells; iPS cells) are cells that have the ability to differentiate into various cells, And it has the ability to grow almost infinitely.
- ES cells embryonic stem cells
- induced pluripotent stem cells induced pluripotent stem cells
- group blood cell which has a certain fixed similarity with the macrophage or dendritic cell which exists in the living body from a pluripotent stem cell is reported (for example, patent document 1 and a nonpatent literature) 1-9).
- Patent Document 1 includes (A) a step of co-culturing human embryonic stem cells and cells having the property of inducing differentiation and proliferation of blood cells to obtain cell group A, (B) the above step Cell group A obtained in (A) and cells having the property of inducing differentiation and proliferation of blood cells are classified into granulocyte macrophage colony stimulating factor (GM-CSF) and macrophage colony stimulating factor (M-CSF). And (C) the cell group B obtained in the step (B) in the presence of GM-CSF and interleukin-4 (IL-4).
- GM-CSF granulocyte macrophage colony stimulating factor
- M-CSF macrophage colony stimulating factor
- IL-4 interleukin-4
- a method for differentiating human embryonic stem cells into dendritic cells comprising
- the differentiation-inducing culture methods reported so far including the differentiation method described in Patent Document 1, require considerable labor and time (one month or more) and are therefore intended for use in cell therapy.
- the cost and time are excessive.
- myeloid blood cells prepared by inducing differentiation from pluripotent stem cells have been proliferated over a long period (over one month or more), and a large amount (for example, 10 5 of pluripotent stem cells used as materials). No method has been reported that can produce myeloid blood cells (more than double).
- monocytes monocytes
- monocytes monocytes
- monocytes are isolated from human peripheral blood using the expression of CD14 molecule as an index, and are used for dendritic cells and Macrophages can be made.
- 10 10 monocytes are required to produce 10 10 dendritic cells or macrophages. To do so, it is necessary to separate monocytes from around 20 liters of peripheral blood.
- Senju S Suemori H, Zembutsu H, Uemura Y, Hirata S, Fukuma D, Matsuyoshi H, Shimomura M, Haruta M, Fukushima S, Matsunaga Y, Katagiri T, Nakamura Y, Furuya N manipulated human embryonic stem cell-derived dendritic cells with immune regulatory function.
- An object of the present invention is to provide a method for producing myeloid blood cells having proliferation ability, a method for growing the myeloid blood cells, a myeloid blood cell obtained by the method, and a cell medicine containing the myeloid blood cell. There is to do. More specifically, an object of the present invention is to provide a method for producing a large amount of human myeloid blood cells useful for use as cell therapy. In addition, it stably maintains myeloid blood cells that are useful as evaluation cells in tests to examine the effects of various drugs on myeloid blood cells, which retain the same functions as myeloid blood cells present in the human body. It is to provide a method of manufacturing.
- the present inventor has tried various methods to produce a large amount of myeloid blood cells from pluripotent stem cells at the lowest possible cost.
- a myeloid blood cell derived from a pluripotent stem cell was introduced with a BMY1, EZH2, MDM2, MDM4, or HIF1A gene in addition to the cMYC gene and forced to express it, and a culture solution supplemented with M-CSF was used.
- peripheral blood monocytes which are myeloid blood cells present in the human body, can be proliferated by introducing and forcibly expressing these genes.
- the present invention has been completed based on such findings.
- the aspect of the present invention relates to the following.
- (A) has a proliferation ability, including forcibly expressing at least one gene selected from the group consisting of cMYC gene and (B) BMI1 gene, EZH2 gene, MDM2 gene, MDM4 gene, and HIF1A gene
- a method for producing myeloid blood cells (2) The method according to (1), wherein the gene is forcibly expressed in the myeloid blood cell by introducing the gene into the myeloid blood cell.
- the method according to (1) or (2), wherein the myeloid blood cells are derived from pluripotent stem cells.
- the pluripotent stem cell is an induced pluripotent stem cell.
- the method according to (4), wherein the induced pluripotent stem cell is a human induced pluripotent stem cell.
- the method according to (6), wherein the peripheral blood monocytes are human peripheral blood monocytes.
- the cMYC gene, BMI1 gene, EZH2 gene, MDM2 gene, MDM4 gene, and HIF1A gene are the human cMYC gene, human BMI1 gene, human EZH2 gene, human MDM2 gene, human MDM4 gene, and human HIF1A gene, respectively.
- a myeloid blood cell comprising culturing a myeloid blood cell produced by the method according to any one of (1) to (8) in the presence of a macrophage colony stimulating factor (M-CSF). How to grow.
- M-CSF macrophage colony stimulating factor
- myeloid blood cells produced according to the present invention have the activity of phagocytosing microorganisms as well as macrophages present in the living body, and provide cell medicines for performing cell therapy for infectious diseases and malignant tumors. It is possible to do.
- myeloid blood cells produced according to the present invention it is possible to provide a cell medicine for diseases caused by accumulation of a large amount of a specific substance in the body, such as Alzheimer's disease, amyloidosis, or certain metabolic diseases.
- dendritic cell-like cells that can be used as cell vaccines against malignant tumors, infections, and the like. Further, according to the present invention, it becomes possible to produce dendritic cell-like cells or macrophages as cellular medicines for controlling immune responses for the purpose of treating autoimmune diseases or rejection associated with organ transplantation. . Furthermore, according to the present invention, it becomes possible to stably produce myeloid blood cells useful as evaluation cells in tests and studies for examining the effects of various pharmaceuticals on myeloid blood cells.
- FIG. 1 is a diagram outlining the production method of the present invention.
- FIG. 2 is a micrograph (photograph of phase contrast lens) of myeloid blood cells (iPS-MC) derived from human iPS cells.
- FIG. 3 shows the results of analyzing the expression of CD45, CD11b, and CD33 molecules of human iPS cell-derived myeloid blood cells (iPS-MC) using a flow cytometer.
- FIG. 4 is a micrograph (photograph of phase contrast lens) of a human iPS cell-derived myeloid blood cell line (iPS-ML) produced by forced expression of cMYC and BMI1.
- FIG. 1 is a diagram outlining the production method of the present invention.
- FIG. 2 is a micrograph (photograph of phase contrast lens) of myeloid blood cells (iPS-MC) derived from human iPS cells.
- FIG. 3 shows the results of analyzing the expression of CD45, CD11b, and CD33 molecules of human iPS cell-derived my
- FIG. 5 shows the results of analyzing the expression of CD45, CD11b, and CD33 molecules in a myeloid blood cell line (iPS-ML) prepared by forced expression of cMYC and BMI1, using a flow cytometer.
- FIG. 6 shows the results of investigating the dependency on M-CSF and GM-CSF regarding the growth of myeloid blood cell line (iPS-ML).
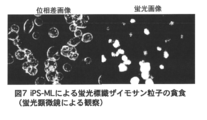
- FIG. 7 shows the results of analyzing the phagocytosis of fluorescent dye-labeled zymosan particles by a myeloid blood cell line (iPS-ML).
- FIG. 8 shows a change with time regarding phagocytosis of fluorescent dye-labeled zymosan particles by a myeloid blood cell line (iPS-ML).
- FIG. 9 is a photomicrograph of dendritic cell-like cells (ML-DC) derived from a myeloid blood cell line (iPS-ML).
- FIG. 10 shows the expression of HLA class II, CD80, and CD86 on the cell surface using flow cytometry for dendritic cell-like cells (ML-DC) derived from the myeloid blood cell line (iPS-ML). The analysis result is shown.
- FIG. 11 shows the results of investigating the activity of inducing the proliferation response of allo-T cells with respect to the myeloid blood cell line (iPS-ML) and dendritic cell-like cells (ML-DC) derived therefrom.
- FIG. 12 is a photomicrograph (phase contrast lens image) of a human iPS cell-derived myeloid blood cell line (iPS-ML) sputum produced by forced expression of cMYC and EZH2.
- FIG. 13 shows the results of analyzing the expression of CD45, CD11b, CD33 and CD14 molecules in a myeloid blood cell line (iPS-ML) prepared by forced expression of cMYC and EZH2, using a flow cytometer.
- FIG. 14 is a micrograph (photograph of phase contrast lens) of a human iPS cell-derived myeloid blood cell line (iPS-ML) produced by forced expression of cMYC and MDM2.
- FIG. 15 shows the results of analyzing the expression of CD45, CD11b, and CD33 molecules in a myeloid blood cell line (iPS-ML) prepared by forced expression of cMYC and MDM2 using a flow cytometer.
- FIG. 16 is a photomicrograph (phase contrast lens image) of a myeloid blood cell line derived from human iPS cells (iPS-ML) produced by forced expression of cMYC and MDM4.
- FIG. 17 shows the results of analyzing the expression of CD45, CD11b, and CD33 molecules in a myeloid blood cell line (iPS-ML) prepared by forced expression of cMYC and MDM4 using a flow cytometer.
- FIG. 18 is a photomicrograph (phase contrast lens image) of a human iPS cell-derived myeloid blood cell line (iPS-ML) sputum produced by forced expression of cMYC and HIF1A.
- FIG. 19 shows the results of analyzing the expression of CD45, CD11b, and CD33 molecules in a myeloid blood cell line (iPS-ML) prepared by forced expression of cMYC and HIF1A using a flow cytometer.
- FIG. 20 is a photomicrograph (photograph of phase contrast lens) of myeloid blood cells (iPS-MC) derived from human iPS cells prepared by a differentiation induction method that does not use feeder cells.
- FIG. 21 shows the results of analyzing the expression of CD45, CD11b, and CD33 molecules of myeloid blood cells derived from human iPS cells (iPS-MC) produced by a differentiation induction method without using feeder cells, using a flow cytometer.
- FIG. 22 is a photomicrograph of iPS-ML prepared by forcibly expressing cMYC and BMI1 in iPS-MC derived from human iPS cells prepared by a differentiation induction method that does not use feeder cells (taken by phase contrast lens). ).
- FIG. 23 shows CD45, CD11b, and CD33 molecules of iPS-ML prepared by forcibly expressing cMYC and BMI1 in iPS-MC derived from human iPS cells prepared by a differentiation induction method that does not use feeder cells. The result of having analyzed the expression of this with the flow cytometer is shown.
- FIG. 24 is a micrograph (photograph of phase contrast lens) of a myeloid blood cell line (Mo-ML) derived from human peripheral blood monocytes prepared by forced expression of cMYC and BMI1.
- FIG. 25 shows the results of analysis of the expression of CD45, CD11b, CD33 and CD14 molecules in a myeloid blood cell line (Mo-ML) derived from human peripheral blood monocytes prepared by forced expression of cMYC and BMI1 using a flow cytometer.
- FIG. 26 shows the expression of HLA class II, CD80, and CD86 on the cell surface for dendritic cell-like cells (ML-DC) derived from myeloid blood cell line (Mo-ML) derived from human peripheral blood monocytes. The result of having been analyzed using flow cytometry is shown.
- FIG. 27 shows the results of examining the activity of inducing the proliferation response of allo-T cells in relation to the myeloid blood cell line (Mo-ML) and dendritic cell-like cells (ML-DC) derived therefrom.
- FIG. 28 is a micrograph (photograph of phase contrast lens) of a myeloid blood cell line (Mo-ML) derived from human peripheral blood monocytes prepared by forced expression of cMYC and MDM2.
- FIG. 29 shows the results of analyzing the expression of CD45 and CD11b molecules of a myeloid blood cell line (Mo-ML) derived from human peripheral blood monocytes prepared by forced expression of cMYC and MDM2 using a flow cytometer.
- FIG. 30 is a micrograph (photograph of phase contrast lens) of a myeloid blood cell line (Mo-ML) derived from human peripheral blood monocytes prepared by forced expression of cMYC, EZH2, and MDM2.
- FIG. 31 shows the results of analyzing the expression of CD45 and CD11b molecules in a myeloid blood cell line (Mo-ML) derived from human peripheral blood monocytes prepared by forced expression of cMYC, EZH2, and MDM2, using a flow cytometer. .
- the present invention is defined as a cell that expresses a CD11b molecule or a CD33 molecule with respect to a myeloid blood cell derived from a pluripotent stem cell or a myeloid blood cell collected directly from a living body.
- the myeloid blood cells have the ability to proliferate in vitro.
- the present invention comprises (A) cMYC gene and (B) BMI1 gene, EZH2 gene, MDM2 gene, MDM4 gene, and HIF1A gene in the myeloid blood cells in order to acquire the ability of the cells to proliferate. Forcibly expressing at least one gene selected from the group.
- a “myeloid blood cell” as a starting material is defined as a cell that expresses a CD11b molecule or a CD33 molecule, and its origin is not particularly limited, but is derived from, for example, a pluripotent stem cell. Examples include myeloid blood cells or myeloid blood cells (eg, peripheral blood monocytes) collected from a living body (eg, human body).
- the term “pluripotent stem cell” refers to a cell that has the ability to grow under artificially constructed conditions (in vitro) such as in vitro and can differentiate into all the cells constituting the living body. .
- an embryonic stem cell or an induced pluripotent stem cell induced pluripotent stem cell (induced pluripotent stem cell iPS cell) is preferably used as the pluripotent stem cell, and an induced pluripotent stem cell is more preferably used.
- induced pluripotent stem cell iPS cell induced pluripotent stem cell
- the embryonic (ES) cell used in the present invention is not particularly limited as long as it is an ES cell derived from a mammal.
- an ES cell derived from mouse, monkey or human can be used.
- human embryonic (ES) cells are stem cells established from early human embryos and maintain the ability to differentiate into all cells existing in the living body (differentiation pluripotency). Cells that can grow in vitro.
- the iPS cells used in the present invention are cells that have acquired differentiation pluripotency similar to embryonic stem cells by artificially manipulating somatic cells.
- the type of somatic cell used here is not particularly limited, and includes all cells constituting the living body.
- the iPS cells referred to in the present invention have a self-replicating ability over a long period of time under predetermined culture conditions (for example, conditions under which ES cells are cultured). This refers to stem cells that have pluripotency.
- the induced pluripotent stem cell in the present invention may be a stem cell capable of forming a teratoma when transplanted to a test animal such as a mouse.
- the reprogramming gene is a gene encoding a reprogramming factor that has the action of reprogramming somatic cells into iPS cells.
- Specific examples of the combination of reprogramming genes include the following combinations, but are not limited thereto.
- (I) Oct gene, Klf gene, Sox gene, Myc gene ii) Oct gene, Sox gene, NANOG gene, LIN28 gene (iii) Oct gene, Klf gene, Sox gene, Myc gene, hTERT gene, SV40 large T gene
- Iv Oct gene, Klf gene, Sox gene
- Each of the Oct gene, Klf gene, Sox gene and Myc gene includes a plurality of family genes.
- family gene those described in pages 11 to 13 of the specification of International Publication No. WO2007 / 069666 can be used. Specifically, it is as follows.
- Oct3 / 4 is preferable.
- Oct3 / 4 is a transcription factor belonging to the POU family, is known as an undifferentiated marker, and has been reported to be involved in maintaining pluripotency.
- genes belonging to the Klf gene include Klf1 (NM_006563), Klf2 (NM_016270), Klf4 (NM_004235), Klf5 (NM_001730), etc. (in parentheses indicate NCBI accession numbers of human genes) ).
- Klf4 is preferred.
- Klf4 (Kruppel like factor-4) has been reported as a tumor suppressor.
- genes belonging to the Sox gene include, for example, Sox1 (NM_005986), Sox2 (NM_003106), Sox3 (NM_005634), Sox7 (NM_031439), Sox15 (NM_006942), Sox17 (NM_0022454), and Sox18 (NM_018419).
- Sox1 NM_005986
- Sox2 NM_003106
- Sox3 NM_005634
- Sox7 NM_031439
- Sox15 NM_006942
- Sox17 NM_0022454
- Sox18 NM_018419.
- Sox2 is a gene that is expressed during early development and encodes a transcription factor.
- genes belonging to the Myc gene include c-Myc (NM_002467), N-Myc (NM_005378), and L-Myc (NM_005376) (in parentheses the NCBI accession number of the human gene). Show).
- c-MycMy a transcriptional regulator involved in cell differentiation and proliferation, and has been reported to be involved in maintaining pluripotency.
- genes described above are genes that exist in common in mammals including humans, and genes derived from any mammal (eg, derived from mammals such as humans, mice, rats, monkeys) can be used in the present invention. .
- genes derived from any mammal eg, derived from mammals such as humans, mice, rats, monkeys
- the method for introducing the reprogramming gene into the somatic cell is not particularly limited as long as the introduced reprogramming gene is expressed and the reprogramming of the somatic cell can be achieved.
- the reprogramming gene can be introduced into a somatic cell using an expression vector containing at least one or more reprogramming genes.
- the expression vector may be introduced into a somatic cell by incorporating two or more reprogramming genes into one expression vector.
- Two or more types of expression vectors incorporating one type of reprogramming gene may be prepared and introduced into somatic cells.
- the type of expression vector is not particularly limited, and may be a viral vector or a plasmid vector.
- a viral vector that can be used to produce induced pluripotent stem cells include retroviral vectors (including lentiviral vectors), adenoviral vectors, and adenovirus vectors.
- retroviral vectors including lentiviral vectors
- adenoviral vectors include associated virus vectors, Sendai virus vectors, and the like.
- a recombinant viral vector can be produced by introducing a recombinant viral vector plasmid into a packaging cell.
- the method for introducing the viral vector plasmid into the packaging cell is not particularly limited, and can be performed by a known gene introduction method such as a calcium phosphate method, a lipofection method, or an electroporation method.
- a medium capable of maintaining the undifferentiation and pluripotency of ES cells is known in the art, and the artificial pluripotent stem cell of the present invention can be isolated and cultured by using a suitable medium in combination. That is, as a medium for culturing the induced pluripotent stem cells of the present invention, an ES medium, a supernatant obtained by culturing mouse embryonic fibroblasts for 24 hours after adding FGF-2 at a concentration of 10 ng / ml to the ES medium And MEF-conditioned ES medium (hereinafter referred to as MEF-conditioned ES medium).
- MEF-conditioned ES medium MEF-conditioned ES medium
- the medium for culturing the induced pluripotent stem cells of the present invention includes various growth factors, cytokines, hormones (eg, FGF-2, TGFb-1, activin A, Nanoggin, BDNF, NGF, NT -1, NT-2, NT-3, and other components involved in the growth and maintenance of human ES cells may be added.
- hormones eg, FGF-2, TGFb-1, activin A, Nanoggin, BDNF, NGF, NT -1, NT-2, NT-3, and other components involved in the growth and maintenance of human ES cells may be added.
- the differentiation ability and proliferation ability of the isolated induced pluripotent stem cells can be confirmed by using confirmation means known for ES cells.
- myeloid blood cells derived from pluripotent stem cells are cells produced by inducing differentiation while culturing pluripotent stem cells in vitro, and myeloid blood on the cell surface.
- Methods for differentiating human pluripotent stem cells into myeloid blood cells are known in the art. For example, Non-Patent Documents 6, 7, 8, and 9 describe methods for producing dendritic cells and macrophages that are myeloid blood cells from human pluripotent stem cells.
- the present invention relates to pluripotent stem cell-derived myeloid blood cells produced by induction of differentiation by the following method. It is not limited.
- a group of cells containing mesodermal cells by co-culturing pluripotent stem cells and the feeder cells with cells having the property of inducing differentiation and proliferation of blood cells as feeder cells Can be differentiated into.
- OP9 cells RIKEN BioResource Center deposit number: RCB1124
- RCB1124 OP9 cells
- Cells having the property of inducing the differentiation and proliferation of blood cells are cultured in a culture container containing an appropriate medium under culture conditions according to the feeder cells, and almost cover the bottom surface of the culture container. The cell growth is lost by treatment with a mitomycin C solution or irradiation, and then transplanted to a separately prepared cell culture container to form a feeder cell layer. The pluripotent stem cells can be seeded on the feeder cells thus prepared, and co-culture can be performed.
- a medium used for the production and co-culture of the feeder cells a medium suitable for culturing adherent mammalian cells is used, and is appropriately selected according to the type of feeder cells. Examples include ⁇ MEM, DMEM (Dulbecco's modified Eagle medium), IMDM (Iscob's modified Dulbecco medium), and the like.
- the culture conditions for the feeder cells can be appropriately set according to the type of cells used as the feeder cells. For example, in the case of OP9 cells and the like, conditions for culturing on a culture vessel coated with a 0.1 wt% gelatin solution or the like can be mentioned.
- Gas phase conditions for the co-culture the type of pluripotent stem cells used, depending on the composition of the culture medium, but may be set appropriately, for example, 37 ° C. before and after (in particular, 37 ° C.), 5 vol% CO 2 And the like.
- the cell group obtained by the above co-culture exhibits the properties of mesodermal cells and can be obtained as a cell group containing a mass of cells exhibiting a nearly spherical shape.
- mesodermal cells that are weakly adherent cells are removed by leaving the cells collected after co-culture in a culture vessel and removing the highly adherent cells.
- the method of recovering can be mentioned.
- the co-culture is treated with an enzyme such as trypsin or collagenase, and all cells are collected and diluted with an appropriate amount of an appropriate medium such as DMEM, and then the cell solution is seeded in a newly prepared culture vessel.
- the cells attached to the culture vessel can be discarded, and the non-attached cells in the medium can be collected as a cell population containing a lot of mesodermal cells.
- the cell group containing mesodermal cells obtained as described above is cultured in the presence of granulocyte macrophage colony stimulating factor (GM-CSF) and / or macrophage colony stimulating factor (M-CSF).
- GM-CSF granulocyte macrophage colony stimulating factor
- M-CSF macrophage colony stimulating factor
- the mesodermal cells can be differentiated into myeloid blood cells.
- Medium that can be used for differentiating a cell group containing mesoderm cells into myeloid blood cells, culture conditions and the like are not particularly limited, but the same medium as the culture and co-culture of the feeder cells described above, Culture conditions and the like can be employed.
- the content of granulocyte macrophage colony stimulating factor (GM-CSF) in the medium is 50 to 200 ng / ml, preferably 75 to 150 ng / ml, from the viewpoint of promoting differentiation of mesodermal cells into myeloid blood cells. Range.
- the content of macrophage colony stimulating factor (M-CSF) in the medium is 10 to 100 ng / ml, preferably 25 to 75 ng / ml, from the viewpoint of promoting the differentiation of mesodermal cells into myeloid blood cells. Range.
- the culture period required for the differentiation of mesoderm cells into myeloid blood cells is not limited by the culture conditions and the like, but is, for example, about 1 to 20 days, preferably about 2 to 15 days.
- Non-Patent Document 6 or 7 As a culture method for inducing differentiation from pluripotent stem cells into myeloid blood cells, as described in Non-Patent Document 6 or 7, a method that does not use feeder cells, or does not contain animal-derived serum It is also possible to use a method using a culture solution. Hereinafter, an example of a method for differentiating pluripotent stem cells into myeloid blood cells using a culture solution that does not use feeder cells and does not contain animal-derived serum will be specifically described. In addition, as described in Non-Patent Document 6 or 7, as a culture method for inducing differentiation from pluripotent stem cells to myeloid blood cells without using feeder cells, other than the method described here, Are known.
- the myeloid blood cells used in the present invention are not limited to myeloid blood cells derived from pluripotent stem cells produced by differentiation induction by any method.
- a culture vessel coated with fibronectin or the like can be used to help adhere the cells to the culture vessel.
- Fibronectin used for coating the culture vessel can be purified from human plasma, or can be a human fibronectin fragment prepared as a recombinant protein.
- KSR Life Technology
- Peprogrow III Peprogrow III
- D-MEM Dulbecco's Modified Eagle Medium
- ⁇ -MEM® Alpha-Minimum Essential Medium
- AIM-V OpTmizer: Life Technologies, Inc. Stemline: Sigma
- human pluripotent stem cells are cultured for 15-20 days using a culture solution not containing animal-derived serum.
- human BMP-4® (Bone® Morphogenic® Protein® 4) is added to a culture solution not containing serum of non-human animals for the purpose of promoting differentiation of pluripotent stem cells.
- Differentiated cells of various cell lineages appear when differentiation induction culture is performed. From these cells, cells that have differentiated into mesodermal cells are separated, and the separated cells are later used as cell groups containing mesodermal cells. It is preferable to use in the process.
- As a method of separating differentiated mesoderm cells as in the case of the differentiation induction method using feeder cells, by removing the adherent cells by leaving the cells collected after co-culture in a culture vessel, A method for recovering a cell population containing a lot of mesodermal cells that are floating cells can be mentioned.
- the cell group containing a lot of mesodermal cells obtained as described above is cultured in the presence of granulocyte macrophage colony stimulating factor (GM-CSF) and / or macrophage colony stimulating factor (M-CSF).
- GM-CSF granulocyte macrophage colony stimulating factor
- M-CSF macrophage colony stimulating factor
- the mesodermal cells can be differentiated into myeloid blood cells.
- the medium, culture conditions, and the like that can be used when differentiating a cell group containing mesoderm cells into myeloid blood cells are not particularly limited, and various serum-free culture solutions can be used.
- myeloid blood cells present in the living body for example, the human body.
- myeloid blood cells present in the living body for example, monocytes in the peripheral blood (monosite) can be used, and it is preferable to use human peripheral blood monocytes.
- a method for separating monocytes from human peripheral blood will be described as an example of a method for obtaining myeloid blood cells present in the living body.
- the method for obtaining myeloid blood cells used in the present invention is described below. The method is not limited.
- Human peripheral blood is collected.
- As the anticoagulant heparin or citric acid is used.
- the collected blood is diluted with an equal amount of physiological saline, phosphate buffered saline, Hanks buffer solution, or the like.
- the diluted blood is layered on Ficoll solution (GE Healthcare) previously dispensed in a centrifuge tube (BD-Falcon 352070 etc.). Then, after centrifugation for 20 minutes at a centrifugal force of 500 g using a centrifugal separator, a mononuclear cell fraction (including lymphocytes and monocytes) existing in the vicinity of the interface is collected.
- Monocytes can be separated from mononuclear cells by the magnetic bead method using the expression of CD14 molecule as an index. For example, it can be separated by using CD14 microbeads (Milte 2 Co., Ltd. 130-050-201).
- monocytes or macrophages derived therefrom are obtained by culturing the mononuclear cell fraction for about 6-16 hours using a cell culture vessel that has been surface-treated for cell culture, and collecting the cells attached to the vessel. It is also possible to obtain Usually, 200,000-500,000 monocytes can be collected from 10 ml of healthy adult peripheral blood.
- the present invention relates to a myeloid blood cell derived from a pluripotent stem cell or a myeloid blood cell collected from a living body, and at least selected from the group consisting of a cMYC gene and BMI1, EZH2, MDM2, MDM4, and HIF1A genes.
- a cMYC gene derived from a pluripotent stem cell or a myeloid blood cell collected from a living body
- BMI1, EZH2, MDM2, MDM4, and HIF1A genes By forcibly expressing one gene, these cells are given long-term proliferative ability.
- an endogenous gene present on the genome of the myeloid blood cell may be forcibly expressed, or a foreign gene may be introduced into the myeloid blood cell. By doing so, these genes may be forcibly expressed.
- the “myeloid blood cell having proliferation ability” means a myeloid blood cell to which long-term proliferation ability is imparted by forcibly expressing the gene in the myeloid blood cell as described above.
- the “myeloid blood cell having proliferation ability” of the present invention is longer than the control myeloid blood cell into which the above gene is not introduced (that is, the myeloid blood cell used as a starting material). For example, it is possible to proliferate for 2 weeks or more from the time when the gene is forcibly expressed (the time when the gene is introduced into the cell).
- cMYC gene examples include a human cMYC gene (NM_002467) used for the preparation of the above-mentioned induced pluripotent stem cells (the NCBI accession number is shown in parentheses).
- BMI1 gene, EZH2 gene, MDM2 gene, MDM4 gene, and HIF1A gene include human BMI1 gene (NM_005180), human EZH2 gene (NM_004456), human MDM2 gene (NM_002392), and human MDM4 gene, respectively.
- NM — 002393 human HIF1A gene (NM — 001530) can be mentioned (in parentheses indicate NCBI accession numbers).
- the cMYC gene, the BMI1 gene, the EZH2 gene, the MDM2 gene, the MDM4 gene, and the HIF1A gene are genes that are commonly present in mammals including humans.
- any gene derived from any mammal eg, human, mouse, rat
- Genes derived from mammals such as monkeys can be used.
- a mutated gene having a base substitution, insertion and / or deletion and having a function similar to that of a wild-type gene.
- the gene may be artificially modified so that the product of the gene is expressed as a fusion protein with another protein or peptide as long as it has a function equivalent to or higher than that of the wild-type gene.
- the method for introducing the cMYC, BMI1, EZH2, MDM2, MDM4, or HIF1A gene into the above myeloid blood cells is particularly limited as long as the introduced gene can be expressed to give the myeloid blood cells long-term proliferation ability. It is not something.
- the gene can be introduced into myeloid blood cells using an expression vector containing the transgene.
- a plurality of genes may be incorporated into one expression vector, and the expression vector may be introduced into myeloid blood cells, or an expression vector into which each gene is separately incorporated is prepared, and these are expressed as myeloid blood cells. May be introduced.
- the type of expression vector is not particularly limited, and may be a viral vector or a plasmid vector, but is preferably a viral vector, and particularly preferably a viral vector in which the introduced gene is integrated into the chromosome of a myeloid blood cell.
- virus vectors that can be used in the present invention include retrovirus vectors, lentivirus vectors, and adeno-associated virus vectors.
- a packaging cell used for producing a recombinant viral vector a cell capable of supplying the defective protein of a recombinant viral vector plasmid lacking at least one gene encoding a protein required for viral packaging Any cell can be used.
- packaging cells based on human kidney-derived HEK293 cells and mouse fibroblasts NIH3T3 can be used.
- a recombinant viral vector can be produced by introducing a recombinant viral vector plasmid into a packaging cell.
- the method for introducing the viral vector plasmid into the packaging cell is not particularly limited, and can be performed by a known gene introduction method such as a calcium phosphate method, a lipofection method, or an electroporation method.
- a solution enriched with the recombinant virus can be recovered by centrifugation or a method of concentrating using a commercially available column for virus purification. .
- a solution containing the recombinant virus prepared as described above is added in a culture container. Infect the virus and introduce the target gene.
- the myeloid blood cells produced as described above and capable of proliferating in vitro can be cultured using a cell culture medium containing M-CSF.
- the content of macrophage colony stimulating factor (M-CSF) in the culture can be in the range of 25-100 ng / ml.
- M-CSF can be produced in the myeloid blood cells themselves by introducing the M-CSF gene into the myeloid blood cells themselves using a lentiviral vector or the like. In this case, it is possible to grow and grow using a cell culture medium to which M-CSF is not added.
- Dendritic cell-like cells can be produced from the myeloid blood cells having the ability to proliferate in vitro according to the present invention.
- dendritic cell-like cells can be produced by culturing the myeloid blood cells having long-term proliferation ability of the present invention in the presence of GM-CSF and interleukin 4 (IL-4).
- the content of GM-CSF in the culture solution can be in the range of 50 to 200 ng / ml, and the content of IL-4 can be in the range of 5 to 20 ng / ml.
- a “dendritic cell-like cell” is a cell having properties similar to those of monocyte-derived dendritic cells in terms of morphology, cell surface molecules, and T cell stimulating ability.
- the myeloid blood cell of the present invention has an activity of phagocytosing microorganisms as well as macrophages and the like present in the living body, and provides a cell medicine for performing immune cell therapy for infectious diseases and malignant tumors. Can do.
- the myeloid blood cells of the present invention may be used for diseases such as Alzheimer's disease, prion disease, amyloidosis, cancer, leukemia, or certain metabolic diseases caused by the accumulation of a large amount of a specific substance in the body.
- Cellular medicine for use in therapy can also be provided.
- the dendritic cell-like cells derived from myeloid blood cells having long-term proliferative ability of the present invention can be used as a cell vaccine for use in the treatment of malignant tumors and infectious diseases. Furthermore, dendritic cell-like cells derived from the myeloid blood cells of the present invention provide a cell medicine for use in controlling immune responses for the purpose of treating rejection reactions associated with autoimmune diseases and organ transplantation. Can do.
- an aid other than those described above is used.
- An agent such as a medium may be used as appropriate.
- Example 1 Preparation of lentiviral vector CDNAs of reprogramming factors, human OCT3 / 4, SOX2, KLF4, and c-MYC, were synthesized by PCR, inserted into a plasmid vector (pENTR-D-TOPO, Gibco-Invitrogen), and cloned. Thereafter, the base sequence of the cloned plasmid DNA was confirmed by sequence analysis. Human BMI1, EZH2, MDM2, MDM4, and HIF1A cDNA clones were obtained from RIKEN BRC Genetic Material Development Office or Product Evaluation Technology Infrastructure.
- the above-mentioned cDNA fragment was inserted into a lentiviral vector (CSII-EF-RfAl, distributed by Dr. Hiroyuki Miyoshi, RIRI) using LR clonase (Gibco-Invitrogen).
- the cell culture solution was collected, passed through a 0.45 ⁇ m filter, and then virus particles were precipitated and collected by centrifugation (50,000 G, 2 hours).
- the recovered recombinant virus particles were suspended in a DMEM solution, dispensed into a freezing tube, and stored in a freezer ( ⁇ 80 ° C.) until use.
- Example 2 Production of Human Artificial Pluripotent Stem (iPS) Cells Human abdominal skin pieces were collected and cultured in a cell culture plate using a culture solution (DMEM / 10% bovine serum). Since the first week after the start of culture, fibroblasts migrated and proliferated from the skin pieces. Trypsin / EDTA-containing phosphate-interfering saline (trypsin-EDTA) was used as appropriate for fibroblasts. Collected and stored frozen.
- DMEM fetal bovine serum
- a colony showing an ES cell-like morphology using a microchip under microscopic observation as an induced pluripotent stem (iPS) cell clone Co-culture with mouse embryo-derived feeder cells that had been isolated and prepared separately was performed. Thereafter, the culture was continued while expanding the culture container according to the proliferation of the cells.
- iPS induced pluripotent stem
- Human iPS cells are maintained in culture for human embryonic stem cells (DMEM-F12 (WakoWChemicals) / 20% 20KSR (Gibco-Invitrogen) / bFGF (basic fibroblast growth factor: 10 ng / ml) / 2-ME 2-mercaptoethanol, 50 ⁇ M)), and mitomycin C-treated mouse fetal fibroblasts were used as feeder cells in a polystyrene culture dish.
- DMEM-F12 WiwakoWChemicals
- 20KSR Gibco-Invitrogen
- bFGF basic fibroblast growth factor: 10 ng / ml
- 2-ME 2-mercaptoethanol 50 ⁇ M
- CTK solution collagenase-trypsin-KSR solution, Biochemical and Biophysical Research Communications 345: 926-932, 2006
- the cells were collected and inoculated into an appropriately sized culture vessel, and the culture was continued.
- Example 3 Induction of differentiation of human induced pluripotent stem cells into myeloid blood cells (iPS-MC) (1) Preparation of OP9 feeder cells Mice-derived cultured cell line OP9 was treated with mitomycin C (0.01 mg / ml 60 minutes) was sown on a gelatin-coated dish and used after the next day.
- iPS-MC myeloid blood cells
- cells were treated with trypsin-EDTA (ethylenediaminetetraacetic acid) -collagenase solution (37 ° C. for 60 minutes), dissociated and collected, and a cell suspension was prepared by pipetting. Then, cells derived from one 10 cm diameter dish were suspended in 10 ⁇ m DMEM / 10% FCS, and seeded on two 10 cm diameter dishes without feeder cells and without gelatin coating. After 2-5 hours, the cells that did not adhere to the dish were collected and passed through a 100 micron mesh (BD-Falcon Co. cell strainer) to obtain a cell suspension from which aggregated cell mass had been removed.
- trypsin-EDTA ethylenediaminetetraacetic acid
- FIG. 2 shows a photomicrograph of differentiated cells derived from this iPS cell.
- CD45 which is a leukocyte marker molecule
- CD11b and CD33 which are myeloid cell markers.
- Fc receptor blocking reagent (Miltenyi Biotec) was used for 10 minutes.
- staining was carried out for 40 minutes at room temperature using fluorescein isothiocyanate (FITC) -labeled anti-human CD45 monoclonal antibody, phycoerythrosin (PE) -labeled anti-human / mouse CD11b antibody, or PE-labeled anti-human CD33 antibody.
- FITC fluorescein isothiocyanate
- PE phycoerythrosin
- PE PE-labeled anti-human / mouse CD11b antibody
- PE-labeled anti-human CD33 antibody As a negative control, staining was performed using an isotype-matched control antibody labeled with the same fluorescent dye.
- the cells were washed twice with PBS / 2% FCS, and the washed cells were analyzed using a flow cytometer analyzer (trade name: FACScan, manufactured by Becton Dickinson) equipped with CellQuest software.
- a flow cytometer analyzer (trade name: FACScan, manufactured by Becton Dickinson) equipped with CellQuest software.
- FIG. 3 shows the results of examining molecules expressed on the cell surface of the cells by flow cytometer analysis.
- a histogram is shown in which the pattern of specific staining and the staining pattern using the isotype-matched control antibody are overlaid.
- CD45 which is a pan-leukocyte marker molecule.
- CD11b or CD33 which are marker molecules of myeloid blood cells.
- This cell derived from iPS cells and expressing a marker molecule of myeloid blood cells was named iPS-MC (iPS-cell-derived myeloid cells: eloid blood cells derived from iPS cells).
- Example 4 Giving long-term proliferation ability by introducing cMYC and BMI1 into iPS-MC (production of iPS-ML)
- the iPS-MC prepared in the previous section was cultured in a 24-well culture plate and infected by adding a lentivirus suspension expressing cMYC or BMI1 alone or simultaneously. From the day after the gene transfer, the culture scale was expanded while adding a culture solution according to the cell growth. As a culture solution, ⁇ -MEM / 20% FCS / human GM-CSF (100 ng / ml) / human M-CSF (50 ng / ml) was continuously used.
- iPS-MC When infected only with lentivirus expressing cMYC, iPS-MC grew at a rate of about 2 days doubling time. However, growth stopped after approximately 2 weeks after lentivirus infection. As a result, the forced expression of cMYC by lentivirus stopped iPS-MC from growing 30 to 100 times and then stopped growing.
- iPS-MC After infection with cMYC lentivirus alone, iPS-MC, which had stopped growing for more than 2 weeks, was further infected with cMYC lentivirus, and subsequent cell growth was not observed.
- iPS-MC proliferated slowly and reached about 2-3 times the number of cells, and then stopped proliferating.
- iPS-MC When simultaneously infected with lentivirus expressing cMYC and BMI1, iPS-MC grew at a faster rate than when infected with cMYC lentivirus alone. Moreover, unlike the case where only cMYC lentivirus was infected, the cells continued to proliferate after the second week after the infection.
- IPS-MC iPS-MC line, iPS cell-derived myeloid cell line, a myeloid blood cell line derived from iPS cells with long-term proliferative potential
- iPS-MC that has acquired the ability to proliferate over a long period of time in this way Named.
- FIG. 4 shows a micrograph of iPS-ML.
- FIG. 5 shows the results of flow cytometer analysis in which the expression of CD45, CD11b, and CD33 in iPS-ML was examined. From this result, it was confirmed that iPS-ML expresses CD45, which is a leukocyte marker molecule, and CD11b and CD33, which are marker molecules of myeloid cells, on the cell surface.
- Example 5 Examination of the necessity of M-CSF and GM-CSF in the proliferation of iPS-ML After infecting with lentivirus expressing cMYC and BMI1, iPS-ML was collected after 2 months of culture and seeded in 96-well culture plate (FALSCON 353072) (5 x 10 3 cells / well) Went. The growth rate was compared between the case where GM-CSF and M-CSF were contained in the culture solution (50 ng / ml or 100 ng / ml) and the case where GM-CSF and M-CSF were not contained.
- 3 H-methylthymidine was added (37 Kbq / well), and 18 hours later, high molecular weight DNA in the cells was filtered with a cell filter using a cell harvester (Wallac). Captured. Then, 3 H-thymidine incorporation into high molecular weight DNA was measured by scintillation counting (using a Wallac microbeta system). The incorporation of 3 H-thymidine into high molecular weight DNA is proportional to the rate of DNA synthesis, ie the rate of cell growth.
- FIG. 6 shows the results of scintillation measurement. From this result, it can be seen that the growth of iPS-ML requires that the culture medium contains about 50 ng / ml of M-CSF. On the other hand, GM-CSF is not necessarily required, but it can be seen that it has an effect of promoting proliferation.
- Example 6 Analysis of Fungal Cell Particle (Zymosan) Phagocytosis by iPS-ML
- myeloid blood cells have a strong activity of phagocytosing microorganisms such as bacteria and fungi. Therefore, the discoloration activity of fungal cell particles (zymosan) by iPS-ML was examined.
- the iPS-ML during maintenance culture was recovered from the culture flask by pipetting, and seeded (2 ⁇ 10 5 / well) in a 24-well culture plate (FALCON 353047) coated with cell culture. After standing for 3 hours and confirming that most of iPS-ML was attached to the bottom of the culture plate, FITC-labeled zymosan (Molecular Probe Z2841) was added. After further 3 hours, observation with a fluorescence microscope was performed.
- FIG. 7 shows a photomicrograph.
- the left-side bright-field image (taken with a phase contrast lens) shows iPS-ML adhering to the culture plate.
- the image on the right is taken from the same field of view as the image on the left, under the conditions for detecting the fluorescence emitted by FITC.
- FITC fluorescence emitted by FITC.
- signals indicating the localization of FITC-labeled zymosan particles are accumulated in accordance with iPS-ML. From this result, it was shown that iPS-ML phagocytoses FITC-labeled zymosan particles.
- FIG. 8 shows the results of analysis using a flow cytometer. From this result, it was shown that the frequency of iPS-ML that phagocytosed zymosan particles increased with time. In this experiment, about half of iPS-MLs phagocytosed zymosan particles within 30 minutes after adding zymosan particles.
- Example 7 Differentiation induction of iPS-ML into dendritic cell-like cells (ML-DC) iPS- after 40 days have elapsed after simultaneously infecting a lentivirus expressing cMYC and a lentivirus expressing BMI1 ML was cultured for 4 days in the presence of GM-CSF (100 ng / ml) and IL-4 (10 ng / ml) to induce differentiation into dendritic cell-like cells (ML-DC).
- FIG. 9 shows cell morphology (phase contrast micrograph) when TNF (tumor necrosis factor) - ⁇ (10 ng / ml) was added and cultured for 2 days. It can be seen that irregularly shaped cells having protrusions form clusters.
- ML-DC produced as described above was collected by pipetting, and stained with anti-HLA class II antibody, anti-human 80 antibody, or anti-human 86 antibody. Alternatively, staining was performed with an isotype matched control antibody. Thereafter, the cells were washed twice with PBS / 2% FCS. The washed cells were analyzed using a flow cytometer analyzer (trade name: FACScan, manufactured by Becton Dickinson) equipped with CellQuest software.
- FIG. 10 shows the results of flow cytometer analysis after antibody staining.
- the pattern of specific staining (solid line) and the staining pattern using the isotype-matched control antibody (dashed line) are shown superimposed.
- the results of this analysis indicate that ML-DC expresses CD80, CD86, and HLA class II, which are related to T lymphocyte activation, on the cell surface as well as physiologically present dendritic cells. found.
- Example 8 Examination of T-cell stimulating ability of ML-DC iPS-ML was cultured in the presence of GM-CSF (100 ng / ml) and IL-4 (10 ng / ml) for 4 days to obtain ML-DC. Then, TNF (tumor necrosis factor) - ⁇ (10 ng / ml) was added and cultured for 2 days, and then the cells were collected. These cells were irradiated with 45 Gy X-rays to stop cell growth, and then seeded in a 96-well round bottom culture plate (FALCON 353077) (1 x 10 2 cells-1 x 10 4 cells / well) for stimulation Cells were used.
- GM-CSF 100 ng / ml
- IL-4 10 ng / ml
- ML-DC without TNF- ⁇ addition or iPS-ML without GM-CSF and IL-4 were also irradiated with 45 Gy X-rays to stop cell growth and cultured.
- the cells were seeded and used as stimulator cells.
- peripheral blood T cells derived from allo donors were added as reaction cells (5 ⁇ 10 4 cells / well) and cultured.
- Peripheral blood T cells were isolated using a human T cell separation kit (Milte-Biotech 130-091-156).
- 3 H-methylthymidine was added (37 Kbq / well). After 18 hours, high molecular weight DNA in the cells was captured on a glass filter using a cell harvester (Wallac). By measuring the radioactivity of 3 H-thymidine trapped on the glass filter by scintillation counting (using a Wallac microbeta system), the rate of macromolecular DNA synthesis, ie, the proliferation rate of T cells, was quantified.
- FIG. 11 shows the results of T cell proliferation response analysis. All of the three types of stimulating cells (MLPS with or without iPS-ML or TNF- ⁇ treatment) have the activity of stimulating allo-T cells and inducing proliferative responses found. And it turns out that ML-DC which performed TNF- (alpha) treatment has the strongest T cell stimulation capability among three types of stimulation cells. From the above results, it was shown that ML-DC derived from iPS-ML has a strong ability to stimulate T cells.
- Example 9 Preparation of iPS-ML by introduction of cMYC and EZH2 into iPS-MC
- the iPS-MC prepared in the previous section was cultured in a 24-well culture plate, and lentivirus suspension expressing cMYC and EZH2 therein IPS-ML was produced by simultaneously infecting.
- FIG. 12 shows a photomicrograph of this cell.
- FIG. 13 shows the results of examining the expression of CD45, CD11b, CD33 and CD14 using a flow cytometer.
- IPS-ML obtained in this example proliferated at a faster rate than cells into which cMYC alone was introduced.
- the obtained iPS-ML proliferated over a longer period of time than cells into which only cMYC had been introduced, and continued to proliferate after the second week after infection with lentivirus.
- Example 10 Production of iPS-ML by introducing cMYC and MDM2 into iPS-MC
- the iPS-MC produced in the previous section was cultured in a 24-well culture plate, and a lentiviral suspension expressing cMYC and MDM2 therein.
- IPS-ML was produced by simultaneously infecting.
- FIG. 14 shows a micrograph of this cell.
- FIG. 15 shows the results of examining the expression of CD45, CD11b, and CD33 using a flow cytometer.
- IPS-ML obtained in this example proliferated at a faster rate than cells into which cMYC alone was introduced.
- the obtained iPS-ML proliferated over a longer period of time than cells into which only cMYC had been introduced, and continued to proliferate after the second week after infection with lentivirus.
- Example 11 Preparation of iPS-ML by introducing cMYC and MDM4 into iPS-MC
- the iPS-MC prepared in the previous section was cultured in a 24-well culture plate, and a lentiviral suspension expressing cMYC and MDM4 therein.
- IPS-ML was produced by simultaneously infecting.
- FIG. 16 shows a micrograph of this cell.
- FIG. 17 shows the results of examining the expression of CD45, CD11b, and CD33 using a flow cytometer.
- IPS-ML obtained in this example proliferated at a faster rate than cells into which cMYC alone was introduced.
- the obtained iPS-ML proliferated over a longer period of time than cells into which only cMYC had been introduced, and continued to proliferate after the second week after infection with lentivirus.
- Example 12 Preparation of iPS-ML by introducing cMYC and HIF1a into iPS-MC
- the iPS-MC prepared in the previous section was cultured in a 24-well culture plate, and lentivirus suspension expressing cMYC and HIF1a therein IPS-ML was produced by simultaneously infecting.
- FIG. 18 shows a micrograph of this cell.
- FIG. 19 shows the results of examining the expression of CD45, CD11b, and CD33 using a flow cytometer.
- IPS-ML obtained in this example proliferated at a faster rate than cells into which cMYC alone was introduced.
- the obtained iPS-ML proliferated over a longer period of time than cells into which only cMYC had been introduced, and continued to proliferate after the second week after infection with lentivirus.
- Example 13 Differentiation of human induced pluripotent stem cells without feeder cells into myeloid blood cells (iPS-MC) and production of iPS-ML Undifferentiated human iPS cells were collected using CTK solution. The cells were cultured in a culture vessel coated with human fibronectin. As the culture solution, a mixture of OpTmizer TM T-Cell Expansion SFM (Life Technoilogies) and Stemline II Hematopoietic Stem Cell Expansion Medium (SIGMA) at 1: 1 and Peprogrow III (Peprotech) were sequentially used. Furthermore, human BMP-4 (Bone Morphogenic Protein 4) was added to the culture solution at a concentration of 5 ng / ml with the intention of promoting differentiation into mesodermal cells.
- iPS-MC myeloid blood cells
- cells 25 days after the start of the culture, cells were treated with a trypsin-EDTA-collagenase solution (37 ° C. for 60 minutes), dissociated and collected, and a cell suspension was prepared by pipetting. Then, cells derived from one 10 cm diameter dish were suspended in 10 ⁇ m DMEM / 10% FCS, and seeded on two 10 cm diameter dishes without feeder cells and without gelatin coating. After 2-5 hours, the cells that did not adhere to the dish were collected and passed through a 100 micron mesh (BD-Falcon Co. cell strainer) to obtain a cell suspension from which aggregated cell mass had been removed. Thereafter, the cells were stored frozen using a cell preservation solution (Cell Banker Toji Field).
- a trypsin-EDTA-collagenase solution 37 ° C. for 60 minutes
- FCS 10 ⁇ m DMEM / 10% FCS
- FIG. 20 shows a micrograph of this cell.
- FIG. 21 shows the results of examining the expression of CD45, CD11b, and CD33 using a flow cytometer. From these results, it was confirmed that CD45, which is a leukocyte marker molecule, and CD11b and CD33, which are marker molecules for myeloid blood cells, were expressed on the cell surface. Therefore, this differentiated cell derived from iPS cells was also determined to be iPS-MC.
- this iPS-MC was collected and cultured in a 24-well culture plate. Infections were made by simultaneously adding a lentivirus suspension expressing cMYC and BMI1 to prepare iPS-ML.
- FIG. 22 shows a micrograph of this cell.
- FIG. 23 shows the results of examining the expression of CD45, CD11b, and CD33 using a flow cytometer.
- IPS-ML obtained in this example proliferated at a faster rate than cells into which cMYC alone was introduced.
- the obtained iPS-ML proliferated over a longer period of time than cells into which only cMYC had been introduced, and continued to proliferate after the second week after infection with lentivirus.
- Example 14 Providing long-term proliferative ability by introducing cMYC and BMI1 into human peripheral blood monocytes (production of Mo-ML) Using a 50 ml injection syringe aspirated with a small amount (about 0.5 ml) of heparin, 50 ml of peripheral blood was collected from a healthy donor. The blood was dispensed in 25 ml aliquots into two 50 ml centrifuge tubes and diluted with an equal volume of phosphate buffered saline (PBS).
- PBS phosphate buffered saline
- the collected mononuclear cell fraction was washed with a RPMI-1640 culture solution and then suspended in a magnetic bead separation buffer (PBS containing 2 mM EDTA and 2% FCS). Then, magnetic microbeads bound with anti-human CD14 antibody (Milte-Biotech 130-050-201) were added and allowed to stand at 6 ° C. for 15 minutes. Then, after washing with a magnetic bead separation buffer, cells expressing CD14 on the cell surface, that is, monocytes, were separated using a cell separation column (MS column 130-042-201 manufactured by Milte 2-Biotech). separated. In addition, human peripheral blood monocytes (Lonza 2W-400C) that were separated by the same method and were commercially available for research were also used.
- a cell separation column MS column 130-042-201 manufactured by Milte 2-Biotech
- a lentivirus suspension expressing cMYC and a lentivirus suspension expressing BMI1 are simultaneously added thereto. Infected.
- ⁇ -MEM / 20% FCS / human GM-CSF (100 ng / ml) / human M-CSF (50 ng / ml) was used.
- FIG. 24 shows a photomicrograph of the cells about 40 days after the infection with the lentivirus. Moreover, the cells in the same period were collected, and the results of examining the expression of CD45, CD11b, CD33, and CD14 with a flow cytometer are shown in FIG. From these results, it was confirmed that monocyte-derived cells that acquired long-term proliferative ability by introduction of cMYC and BMI1 expressed myeloid blood cell markers. Therefore, this cell was named Mo-ML (Monocyte-derived myeloid cell line, a myeloid blood cell line derived from monocytes).
- Mo-ML Monocyte-derived myeloid cell line, a myeloid blood cell line derived from monocytes.
- Example 15 Induction of differentiation of Mo-ML into dendritic cell-like cells (ML-DC) After 40 days of simultaneous infection with lentivirus expressing cMYC and BMI1, Mo-ML was treated with GM-CSF (100 ng / ml) and IL-4 (10 ng / ml) for 4 days. The cells were cultured and induced to differentiate into dendritic cell-like cells (ML-DC). Further, TNF- ⁇ (10 ng / ml) was added and cultured for 2 days.
- GM-CSF 100 ng / ml
- IL-4 10 ng / ml
- ML-DC The expression of marker molecules on the cell surface of ML-DC was examined by flow cytometry. First, it processed for 10 minutes using Fc receptor block reagent (made by Miltenyi Biotec). Next, it was stained for 40 minutes at room temperature with FITC-anti-human 80 antibody, anti-human 86 antibody, or anti-HLA class II antibody. In addition, staining was performed using an isotype-matched control antibody labeled with FITC or PE. Stained cells were analyzed using FACScan (BD).
- FIG. 26 shows the results of flow cytometer analysis after antibody staining.
- the pattern of specific staining (solid line) and the staining pattern using the isotype-matched control antibody (broken line) are shown superimposed.
- the results of this analysis indicate that ML-DC expresses CD80, CD86, and HLA class II, which are related to T lymphocyte activation, on the cell surface as well as physiologically present dendritic cells. found.
- Example 16 Examination of T-cell stimulation ability of Mo-ML-derived ML-DC Mo-ML was cultured for 4 days in the presence of GM-CSF (100 ng / ml) and IL-4 (10 ng / ml). ML-DC was prepared, and TNF- ⁇ (10 ng / ml) was further added and cultured for 2 days, and then the cells were collected. These cells were irradiated with 45 Gy X-rays to stop cell growth, and then seeded in a 96-well round bottom culture plate (FALCON 353077) (1 x 10 2 cells-1 x 10 4 cells / well) for stimulation Cells were used.
- FALCON 353077 96-well round bottom culture plate
- ML-DC without TNF- ⁇ addition, or Mo-ML without GM-CSF and IL-4 were similarly cultured after 45 Gy X-rays were irradiated to stop cell growth.
- the cells were seeded and used as stimulator cells.
- peripheral blood T cells derived from allo donors were added as reaction cells (5 ⁇ 10 4 cells / well) and cultured.
- 3 H-methylthymidine was added (37 Kbq / well). After 18 hours, high molecular weight DNA in the cells was captured on a glass filter using a cell harvester (Wallac). The proliferation rate of T cells was quantified by measuring the radioactivity of 3 H-thymidine trapped on the glass filter by scintillation counting.
- FIG. 27 shows the results of T cell proliferation response analysis. All of the three types of stimulating cells (MLPS with or without iPS-ML or TNF- ⁇ treatment) have the activity of stimulating allo-T cells and inducing proliferative responses found. And it turns out that ML-DC which performed TNF- (alpha) treatment has the strongest T cell stimulation capability among three types of stimulation cells. From the above results, it was shown that ML-DC derived from Mo-ML has a strong ability to stimulate T cells.
- Example 17 Production of Mo-ML by introduction of cMYC and MDM2 into peripheral blood monocytes
- Human peripheral blood monocytes (CD14 positive cells) were cultured in 24-well culture plates, and wrench expressing cMYC and MDM2 there. Infections were made by simultaneously adding the virus suspension.
- As the culture solution ⁇ -MEM / 20% FCS / human GM-CSF (100 ng / ml) / human M-CSF (50 ng / ml) was used.
- the cells showed a clear tendency to proliferate after about 3 weeks after infection with lentivirus.
- FIG. 28 shows a photomicrograph of cells about 1 month after infection with lentivirus.
- FIG. 29 shows the results of examining the expression of CD45, CD11b, CD33 and CD14 using a flow cytometer at the same time.
- Example 18 Production of Mo-ML by introduction of cMYC, EZH2 and MDM2 into peripheral blood monocytes
- Human peripheral blood monocytes were cultured in 24-well culture plates, and lentivirus expressing cMYC, EZH2 and MDM2 there. Infections were made by adding the suspension simultaneously. The cells showed a clear tendency to proliferate after about 3 weeks after infection with lentivirus.
- FIG. 30 shows a photomicrograph of cells about 1 month after infection with lentivirus.
- FIG. 31 shows the results of examining the expression of CD45, CD11b, CD33 and CD14 using a flow cytometer at the same time.
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Hematology (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- General Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cell Biology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Developmental Biology & Embryology (AREA)
- Gastroenterology & Hepatology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Oncology (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Transplantation (AREA)
- Cardiology (AREA)
- Communicable Diseases (AREA)
Abstract
Description
(1) ミエロイド系血液細胞において、
(A)cMYC遺伝子、並びに
(B)BMI1遺伝子、EZH2遺伝子、MDM2遺伝子、MDM4遺伝子、及びHIF1A遺伝子からなる群から選択される少なくとも一つの遺伝子
を強制的に発現させることを含む、増殖能力を有するミエロイド系血液細胞を製造する方法。
(2) 前記ミエロイド系血液細胞に前記遺伝子を導入することにより、該ミエロイド系血液細胞において該遺伝子を強制的に発現させる、(1)に記載の方法。
(3) 前記ミエロイド系血液細胞が、多能性幹細胞に由来する、(1)又は(2)に記載の方法。
(4) 前記多能性幹細胞が、人工多能性幹細胞である、(3)に記載の方法。
(5) 前記人工多能性幹細胞が、ヒト人工多能性幹細胞である、(4)に記載の方法。
(6) 前記ミエロイド系血液細胞が、末梢血単球である、(1)又は(2)に記載の方法。
(7) 前記末梢血単球が、ヒト末梢血単球である、(6)に記載の方法。
(8) 前記cMYC遺伝子、BMI1遺伝子、EZH2遺伝子、MDM2遺伝子、MDM4遺伝子、及びHIF1A遺伝子が、それぞれ、ヒトcMYC遺伝子、ヒトBMI1遺伝子、ヒトEZH2遺伝子、ヒトMDM2遺伝子、ヒトMDM4遺伝子、及びヒトHIF1A遺伝子である、(1)~(7)のいずれかに記載の方法。
(9) マクロファージコロニー刺激因子(M-CSF)の存在下で、(1)~(8)のいずれかに記載の方法で製造したミエロイド系血液細胞を培養することを含む、ミエロイド系血液細胞を増殖させる方法。
(10) (1)~(9)のいずれかに記載の方法により製造される細胞。
(11) (10)に記載の細胞を樹状細胞様細胞に分化誘導することを含む、樹状細胞様細胞を製造する方法。
(12) 顆粒球・マクロファージコロニー刺激因子(GM-CSF)およびインターロイキン-4(IL-4)を含む培養液を用いて、前記ミエロイド系血液細胞を培養することにより、該ミエロイド系血液細胞を樹状細胞様細胞に分化誘導する、(11)に記載の方法。
(13) (11)又は(12)に記載の方法により製造される細胞。
(14) (10)又は(13)に記載の細胞を含む、細胞医薬。
(15) 前記細胞医薬は、感染症、腫瘍、アルツハイマー病、プリオン病、アミロイドーシス、白血病、及び/又は自己免疫疾患の治療又は予防に用いるための細胞医薬である、(14)に記載の細胞医薬。
(16) (10)若しくは(13)に記載の細胞、又は(14)に記載の細胞医薬を製造するための、
(A)cMYC遺伝子、並びに
(B)BMI1遺伝子、EZH2遺伝子、MDM2遺伝子、MDM4遺伝子、及びHIF1A遺伝子からなる群から選択される少なくとも一つの遺伝子
の使用。
(17) (14)に記載の細胞医薬を用いて、感染症、腫瘍、アルツハイマー病、プリオン病、アミロイドーシス、白血病、及び/又は自己免疫疾患を治療する方法。
(B)BMI1遺伝子、EZH2遺伝子、MDM2遺伝子、MDM4遺伝子、及びHIF1A遺伝子からなる群から選択される少なくとも一つの遺伝子を強制的に発現させることを含むことを特徴とする。
本発明で用いる胚性(ES)細胞は、哺乳動物由来のES細胞であればよく、その種類などは特に限定されず、例えば、マウス、サル又はヒト由来のES細胞などを使用することができる。例えば、ヒトの胚性(ES) 細胞は、ヒトの初期胚から樹立された幹細胞であり、生体内に存在する全ての細胞へ分化できる能力(分化多能性)を維持したまま、長期に渡って生体外で増殖できる細胞である。
本発明で用いるiPS細胞とは、体細胞に人為的な操作を加えることにより、胚性幹細胞に類似した分化多能性を獲得させた細胞である。ここで用いる体細胞の種類は特に限定されず、生体を構成する全ての細胞を包含する。
(i)Oct遺伝子、Klf遺伝子、Sox遺伝子、Myc遺伝子
(ii)Oct遺伝子、Sox遺伝子、NANOG遺伝子、LIN28遺伝子
(iii)Oct遺伝子、Klf遺伝子、Sox遺伝子、Myc遺伝子、hTERT遺伝子、SV40 large T遺伝子
(iv)Oct遺伝子、Klf遺伝子、Sox遺伝子
本発明において、「多能性幹細胞に由来するミエロイド系血液細胞」とは、多能性幹細胞を生体外で培養しつつ分化誘導することにより作製された細胞であり、細胞表面上にミエロイド系血液細胞のマーカー分子であるCD11bあるいはCD33分子を発現する細胞を言う。ヒト多能性幹細胞をミエロイド系血液細胞へ分化させる方法は、当業界では公知である。例えば、非特許文献6、7、8、9には、ヒトの多能性幹細胞から、ミエロイド系血液細胞である樹状細胞やマクロファージを作製する方法が記載されている。以下、多能性幹細胞をミエロイド系血液細胞へ分化させる方法の例について具体的に説明するが、本発明は、以下の方法による分化誘導により製造された多能性幹細胞由来のミエロイド系血液細胞に限定されるものではない。
血液細胞の分化と増殖とを誘導する性質を有する細胞をフィーダー細胞として、多能性幹細胞と、該フィーダー細胞とを共培養することにより、多能性幹細胞を、中胚葉系細胞を含む細胞群に分化させることができる。
フィーダー細胞と非ヒト動物の血清のいずれをも用いずに分化誘導を行う場合、細胞の培養容器への付着を助けるために、フィブロネクチンなどによるコーティングを施した培養容器を用いることができる。培養容器のコーティングに用いるフィブロネクチンは、ヒト血漿から精製したもの、あるいは、遺伝子組換えタンパク質として作製されたヒトフィブロネクチン断片等を用いることが可能である。
ヒトの末梢血を採血する。抗凝固剤としては、ヘパリンあるいはクエン酸などを用いる。採血した血液を等量の生理的食塩水、リン酸緩衝生理的食塩水、あるいは、ハンクス緩衝溶液などを用いて希釈する。次に、希釈した血液を、あらかじめ遠心チューブ(BD-Falcon 352070等)中に分注しておいたフィコール液(GE ヘルスケア社)の上に重層する。そして、遠心分離装置を用いて、遠心力500gで20分間遠心した後、界面付近に存在する単核細胞分画(リンパ球と単球を含む)を回収する。
本発明は、多能性幹細胞由来のミエロイド系血液細胞、あるいは、生体から採取したミエロイド系血液細胞において、cMYC遺伝子、並びにBMI1、EZH2、MDM2、MDM4、及びHIF1A遺伝子からなる群から選択される少なくとも一つの遺伝子を強制的に発現させることにより、これらの細胞に長期増殖能を付与する。前記遺伝子を強制的に発現させるための方法として、ミエロイド系血液細胞のゲノム上に存在する内在性の遺伝子を強制的に発現させても良いし、あるいは外来の遺伝子を該ミエロイド系血液細胞に導入することにより、これら遺伝子の強制的な発現を行っても良い。遺伝子の効率的かつ高レベルの発現を達成し、ミエロイド系血液細胞に長期増殖能を確実に付与する観点から、遺伝子工学技術を用いて外来遺伝子を該ミエロイド系血液細胞に導入する方法が好ましい。
本発明において、「増殖能力を有するミエロイド系血液細胞」とは、上記の通り、ミエロイド系血液細胞において上記遺伝子を強制的に発現させることにより長期増殖能が付与されたミエロイド系血液細胞を意味する。本発明の「増殖能力を有するミエロイド系血液細胞」は、上記遺伝子が導入されていない、対照となるミエロイド系血液細胞(つまり、出発材料として用いたミエロイド系血液細胞)と比較して、より長期に渡り増殖することが可能となり、例えば、上記遺伝子を強制的に発現させた時点(上記遺伝子を細胞に導入した時点)から2週間以上増殖することが可能である。
上記の通り製造した生体外で増殖する能力を有するミエロイド系血液細胞は、M-CSFを含む細胞培養液を用いて培養することができる。培養液中のマクロファージコロニー刺激因子(M-CSF)の含有量は、25~100ng/mlの範囲とすることができる。あるいは、レンチウイルスベクター等を用いてM-CSF遺伝子をミエロイド系血液細胞自身に導入することによりミエロイド系血液細胞自身にM-CSFを産生させることも可能である。この場合は、M-CSFを添加していない細胞培養液を用いて培養し、増殖させることが可能である。
本発明の生体外で増殖する能力を有するミエロイド系血液細胞から、樹状細胞様細胞を製造することが可能である。例えば、本発明の長期増殖能を有するミエロイド系血液細胞を、GM-CSF及びインターロイキン4(IL-4)の存在下で培養することにより、樹状細胞様細胞を製造することができる。培養液中のGM-CSFの含有量は、50~200ng/mlの範囲、IL-4の含有量は、5~20ng/mlの範囲とすることができる。本発明において、「樹状細胞様細胞」とは、形態、細胞表面分子、T細胞刺激能力という点で単球由来樹状細胞等と類似した性質を有する細胞である。
本発明のミエロイド系血液細胞は、生体内に存在するマクロファージ等と同様に微生物などを貪食する活性を有しており、感染症や悪性腫瘍に対する免疫細胞療法を行うための細胞医薬を提供することができる。また、本発明のミエロイド系血液細胞は、アルツハイマー病、プリオン病、アミロイドーシス、がん、白血病、或いはある種の代謝性疾患など、体内に特定の物質が大量に蓄積することに起因する疾患等の治療に用いるための細胞医薬を提供することもできる。また、本発明の長期増殖能を有するミエロイド系血液細胞に由来する樹状細胞様細胞は、悪性腫瘍や感染症の治療に用いるための細胞ワクチンとして使用することができる。さらに、本発明のミエロイド系血液細胞に由来する樹状細胞様細胞は、自己免疫疾患や臓器移植に伴う拒絶反応等を治療する目的で、免疫応答の制御に用いるための細胞医薬を提供することができる。
PCR法により初期化因子であるヒトのOCT3/4, SOX2, KLF4,および c-MYCのcDNAを合成し、プラスミドベクター(pENTR-D-TOPO, Gibco-Invitrogen社)へ挿入しクローニングを行った。その後、クローニングを行ったプラスミドDNAの塩基配列をシークエンス解析により確認した。ヒトBMI1、EZH2、MDM2、MDM4、及びHIF1AのcDNAクローンは、理化学研究所BRC遺伝子材料開発室あるいは製品評価技術基盤機構より入手した。
ヒト腹部の皮膚片を採取し、細胞培養用プレート中で培養液(DMEM / 10 % 牛血清)を用いて培養した。培養開始1週間目以降より、皮膚片からの線維芽細胞の遊走と増殖が認められたので、トリプシン/EDTA含有リン酸干渉生理食塩水(トリプシン-EDTA)を用いて、適宜、線維芽細胞を回収し凍結保存した。
(1)OP9フィーダー細胞の調整
マウス由来の培養細胞株OP9をマイトマイシンCにて処理(0.01 mg/ml、60分)したものを、ゼラチンをコートしたディッシュに播種し、翌日以降に使用した。
未分化なiPS細胞を、CTK液を用いて5-10分間処理し、牛胎児(FCS)血清入りのDMEM培養液で回収した。細胞をα-MEM/20%FCSに懸濁し、OP9フィーダー細胞上へ播種し、分化誘導培養を開始した。以降、3日に1度、培養液(α-MEM/20%FCS)を交換しつつ培養を継続した。
前項で作製したiPS-MCを24穴培養プレート中で培養し、そこへcMYCあるいはBMI1を発現するレンチウイルス懸濁液を単独で、あるいは同時に加えることにより、感染させた。遺伝子導入の翌日より、細胞の増殖に応じて培養液を追加しつつ、培養スケールの拡大を行った。培養液としては、継続的に、α-MEM / 20%FCS / ヒト GM-CSF (100 ng/ml) / ヒト M-CSF (50 ng/ml、)を用いた。
cMYCとBMI1を発現するレンチウイルスを感染させた後、2か月間培養を継続した後のiPS-MLを回収し96穴培養プレート(FALSCON 353072)に播種し(5 x 103細胞 / well)培養を行った。そして、培養液中にGM-CSFとM-CSFが含まれている(50ng/mlあるいは100ng/ml)場合と含まれていない場合とで増殖の速度を比較した。
一般に、ミエロイド系血液細胞は、細菌や真菌などの微生物を貪食する強い活性を有している。そこで、iPS-MLによる真菌の菌体粒子(ザイモサン)の貪色活性を検討した。維持培養中のiPS-MLをピペッティング操作により培養フラスコから回収し、細胞培養用のコーティングがなされている24穴培養プレート(FALCON 353047)へ播種した(2 x 105/well)。3時間静置しiPS-MLの多くが培養プレートの底面に付着したのを確認した後、FITC標識ザイモサン(Molecular Probe社 Z2841)を加えた。さらに3時間が経過した後、蛍光顕微鏡による観察を行った。
cMYCを発現するレンチウイルスとBMI1を発現するレンチウイルスを同時に感染させた後、40日経過した後のiPS-MLをGM-CSF(100 ng/ml)とIL-4 (10 ng/ml)の存在下で4日間培養し、樹状細胞様細胞(ML-DC)への分化誘導を行った。さらにTNF(腫瘍壊死因子)-α(10 ng/ml)を添加して2日間培養した時の細胞の形態(位相差顕微鏡写真)を、図9に示す。突起を有する不規則な形態の細胞がクラスターを形成しているのがわかる。
iPS-MLをGM-CSF(100 ng/ml)とIL-4 (10 ng/ml)の存在下で4日間培養してML-DCを作製し、さらにTNF(腫瘍壊死因子)-α(10 ng/ml)を添加して2日間培養した後、細胞を回収した。この細胞に45 GyのX線を照射して細胞増殖を停止させた後、96穴丸底培養プレート(FALCON 353077)に播種し(1 x 102細胞-1 x 104細胞/well)、刺激細胞とした。TNF-αの添加を行っていないML-DC、あるいは、GM-CSFとIL-4を加えていないiPS-MLも、同様に45GyのX線を照射して細胞増殖を停止させた後、培養プレートに播種し、刺激細胞とした。そして、反応細胞として、アロのドナー由来の末梢血T細胞を加え(5 x 104細胞/well)、培養を行った。末梢血T細胞は、ヒトT細胞分離キット(ミルテ二-バイオテク社 130-091-156)を用いて分離したものを用いた。
前項で作製したiPS-MCを24穴培養プレート中で培養し、そこへcMYCとEZH2を発現するレンチウイルス懸濁液を同時に加えることにより感染させ、iPS-MLを作製した。図12に、この細胞の顕微鏡写真を示す。図13に、フローサイトメーターによりCD45、CD11b、CD33およびCD14の発現を検討した結果を示す。
本実施例により得られたiPS-MLは、cMYCのみを導入した細胞よりも速い速度で増殖した。さらに、得られたiPS-ML は、cMYC のみ導入した細胞よりも長期に渡り増殖し、レンチウイルスを感染させてから2週間目以降も増殖を続けた。
前項で作製したiPS-MCを24穴培養プレート中で培養し、そこへcMYCとMDM2を発現するレンチウイルス懸濁液を同時に加えることにより感染させ、iPS-MLを作製した。図14に、この細胞の顕微鏡写真を示す。図15に、フローサイトメーターによりCD45、CD11b、およびCD33の発現を検討した結果を示す。
本実施例により得られたiPS-MLは、cMYCのみを導入した細胞よりも速い速度で増殖した。さらに、得られたiPS-ML は、cMYC のみ導入した細胞よりも長期に渡り増殖し、レンチウイルスを感染させてから2週間目以降も増殖を続けた。
前項で作製したiPS-MCを24穴培養プレート中で培養し、そこへcMYCとMDM4を発現するレンチウイルス懸濁液を同時に加えることにより感染させ、iPS-MLを作製した。図16に、この細胞の顕微鏡写真を示す。図17に、フローサイトメーターによりCD45、CD11b、およびCD33の発現を検討した結果を示す。
本実施例により得られたiPS-MLは、cMYCのみを導入した細胞よりも速い速度で増殖した。さらに、得られたiPS-ML は、cMYC のみ導入した細胞よりも長期に渡り増殖し、レンチウイルスを感染させてから2週間目以降も増殖を続けた。
前項で作製したiPS-MCを24穴培養プレート中で培養し、そこへcMYCとHIF1aを発現するレンチウイルス懸濁液を同時に加えることにより感染させ、iPS-MLを作製した。図18に、この細胞の顕微鏡写真を示す。図19に、フローサイトメーターによりCD45、CD11b、およびCD33の発現を検討した結果を示す。
本実施例により得られたiPS-MLは、cMYCのみを導入した細胞よりも速い速度で増殖した。さらに、得られたiPS-ML は、cMYC のみ導入した細胞よりも長期に渡り増殖し、レンチウイルスを感染させてから2週間目以降も増殖を続けた。
未分化なヒトiPS細胞を、CTK液を用いて回収し、ヒトフィブロネクチンをコートした培養容器中で培養した。培養液は、OpTmizerTM T-Cell Expansion SFM (Life Technoilogies社)とStemline II Hematopoietic Stem Cell Expansion Medium(SIGMA社)を1:1で混合したもの、および、Peprogrow III(Peprotech社)を順次用いた。さらに、培養液には、中胚葉系細胞への分化を促進することを意図して、ヒトBMP-4 (Bone Morphogenic Protein 4)を5ng/mlの濃度で添加した。
本実施例により得られたiPS-MLは、cMYCのみを導入した細胞よりも速い速度で増殖した。さらに、得られたiPS-ML は、cMYC のみ導入した細胞よりも長期に渡り増殖し、レンチウイルスを感染させてから2週間目以降も増殖を続けた。
少量(0.5ml程度)のヘパリンを吸引した50mlの注射シリンジを用いて、健常人ドナーより末梢血を50ml採血した。血液は、50mlの遠心チューブ2本に25mlずつ分注し、等量のリン酸緩衝生理的食塩水(PBS)で希釈した。次に、この希釈した血液25mlを、あらかじめ遠心チューブ(BD-Falcon 352070等)中に分注しておいたフィコール液(GE ヘルスケア社 17-1440-03)15 mlの上にゆっくりと重層した。そして、遠心分離装置を用いて、遠心力500gで20分間遠心した後、界面付近に存在する単核細胞分画(リンパ球と単球を含む細胞集団)を回収した。
cMYCとBMI1を発現するレンチウイルスを同時に感染させた後、40日経過した後のMo-MLをGM-CSF(100 ng/ml)とIL-4 (10 ng/ml)の存在下で4日間培養し、樹状細胞様細胞(ML-DC)への分化誘導を行った。さらにTNF-α(10 ng/ml)を添加して2日間培養した。
まず、Fcレセプターブロック試薬(Miltenyi Biotec社製)を用いて10分間処理した。次に、FITC-抗ヒト80抗体、抗ヒト86抗体、あるいは抗HLAクラスII抗体を用いて室温中で40分間染色した。また、FITCあるいはPEで標識されたアイソタイプ適合対照抗体を用いて染色した。染色した細胞を、FACScan(BD社)を用いて解析した。
Mo-MLをGM-CSF(100 ng/ml)とIL-4 (10 ng/ml)の存在下で4日間培養してML-DCを作製し、さらにTNF-α(10 ng/ml)を添加して2日間培養した後、細胞を回収した。この細胞に45 GyのX線を照射して細胞増殖を停止させた後、96穴丸底培養プレート(FALCON 353077)に播種し(1 x 102細胞-1 x 104細胞/well)、刺激細胞とした。TNF-αの添加を行っていないML-DC、あるいは、GM-CSFとIL-4を加えていないMo-MLも、同様に45GyのX線を照射して細胞増殖を停止させた後、培養プレートに播種し、刺激細胞とした。そして、反応細胞として、アロのドナー由来の末梢血T細胞を加え(5 x 104細胞/well)、培養を行った。
ヒト末梢血単球(CD14陽性細胞)を24穴培養プレート中で培養し、そこへcMYCおよびMDM2を発現するレンチウイルス懸濁液を同時に加えることにより、感染させた。培養液としては、α-MEM / 20%FCS / ヒト GM-CSF (100 ng/ml) / ヒト M-CSF (50 ng/ml、)を用いた。細胞は、レンチウイルスを感染させた後、3週間目頃よりはっきりとした増殖傾向を示した。図28に、レンチウイルスを感染させた約1月後の細胞の顕微鏡写真を示す。図29に、同時期にフローサイトメーターによりCD45、CD11b、CD33およびCD14の発現を検討した結果を示す。
ヒト末梢血単球を24穴培養プレート中で培養し、そこへcMYC、EZH2およびMDM2を発現するレンチウイルス懸濁液を同時に加えることにより、感染させた。細胞は、レンチウイルスを感染させた後、3週間目頃よりはっきりとした増殖傾向を示した。図30に、レンチウイルスを感染させた約1月後の細胞の顕微鏡写真を示す。図31に、同時期にフローサイトメーターによりCD45、CD11b、CD33およびCD14の発現を検討した結果を示す。
Claims (17)
- ミエロイド系血液細胞において、
(A)cMYC遺伝子、並びに
(B)BMI1遺伝子、EZH2遺伝子、MDM2遺伝子、MDM4遺伝子、及びHIF1A遺伝子からなる群から選択される少なくとも一つの遺伝子
を強制的に発現させることを含む、増殖能力を有するミエロイド系血液細胞を製造する方法。 - 前記ミエロイド系血液細胞に前記遺伝子を導入することにより、該ミエロイド系血液細胞において該遺伝子を強制的に発現させる、請求項1に記載の方法。
- 前記ミエロイド系血液細胞が、多能性幹細胞に由来する、請求項1又は2に記載の方法。
- 前記多能性幹細胞が、人工多能性幹細胞である、請求項3に記載の方法。
- 前記人工多能性幹細胞が、ヒト人工多能性幹細胞である、請求項4に記載の方法。
- 前記ミエロイド系血液細胞が、末梢血単球である、請求項1又は2に記載の方法。
- 前記末梢血単球が、ヒト末梢血単球である、請求項6に記載の方法。
- 前記cMYC遺伝子、BMI1遺伝子、EZH2遺伝子、MDM2遺伝子、MDM4遺伝子、及びHIF1A遺伝子が、それぞれ、ヒトcMYC遺伝子、ヒトBMI1遺伝子、ヒトEZH2遺伝子、ヒトMDM2遺伝子、ヒトMDM4遺伝子、及びヒトHIF1A遺伝子である、請求項1~7のいずれか1項に記載の方法。
- マクロファージコロニー刺激因子(M-CSF)の存在下で、請求項1~8のいずれか1項に記載の方法で製造したミエロイド系血液細胞を培養することを含む、ミエロイド系血液細胞を増殖させる方法。
- 請求項1~9のいずれか1項に記載の方法により製造される細胞。
- 請求項10に記載の細胞を樹状細胞様細胞に分化誘導することを含む、樹状細胞様細胞を製造する方法。
- 顆粒球・マクロファージコロニー刺激因子(GM-CSF)およびインターロイキン-4(IL-4)を含む培養液を用いて、前記ミエロイド系血液細胞を培養することにより、該ミエロイド系血液細胞を樹状細胞様細胞に分化誘導する、請求項11に記載の方法。
- 請求項11又は12に記載の方法により製造される細胞。
- 請求項10又は13に記載の細胞を含む、細胞医薬。
- 前記細胞医薬は、感染症、腫瘍、アルツハイマー病、プリオン病、アミロイドーシス、白血病、臓器移植に伴う拒絶反応、及び/又は自己免疫疾患の治療又は予防に用いるための細胞医薬である、請求項14に記載の細胞医薬。
- 請求項10若しくは13に記載の細胞、又は請求項14に記載の細胞医薬を製造するための、
(A)cMYC遺伝子、並びに
(B)BMI1遺伝子、EZH2遺伝子、MDM2遺伝子、MDM4遺伝子、及びHIF1A遺伝子からなる群から選択される少なくとも一つの遺伝子
の使用。 - 請求項14に記載の細胞医薬を用いて、感染症、腫瘍、アルツハイマー病、プリオン病、アミロイドーシス、白血病、及び/又は自己免疫疾患を治療する方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012536516A JP5861191B2 (ja) | 2010-09-30 | 2011-09-28 | ミエロイド系血液細胞の製造方法 |
| US13/825,883 US10034900B2 (en) | 2010-09-30 | 2011-09-28 | Method of producing myeloid blood cells |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-221809 | 2010-09-30 | ||
| JP2010221809 | 2010-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012043651A1 true WO2012043651A1 (ja) | 2012-04-05 |
Family
ID=45893090
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/072234 WO2012043651A1 (ja) | 2010-09-30 | 2011-09-28 | ミエロイド系血液細胞の製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US10034900B2 (ja) |
| JP (1) | JP5861191B2 (ja) |
| WO (1) | WO2012043651A1 (ja) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140037600A1 (en) * | 2011-02-08 | 2014-02-06 | Cellular Dynamics International, Inc. | Hematopoietic precursor cell production by programming |
| JP2017131136A (ja) * | 2016-01-27 | 2017-08-03 | 国立大学法人 熊本大学 | 血液由来単球の増殖誘導方法 |
| JPWO2017010544A1 (ja) * | 2015-07-15 | 2018-05-24 | テルモ株式会社 | 多能性幹細胞または脂肪組織もしくは骨髄由来の間葉系幹細胞由来の心筋細胞の凍結保存方法 |
| WO2018181903A1 (ja) * | 2017-03-31 | 2018-10-04 | 国立大学法人熊本大学 | インターフェロンβ産生細胞の作製方法 |
| JP2018171005A (ja) * | 2017-03-31 | 2018-11-08 | 国立大学法人 熊本大学 | ヒトミエロイド系血液細胞及び該細胞の作製方法 |
| WO2020045368A1 (ja) | 2018-08-27 | 2020-03-05 | マイキャン・テクノロジーズ株式会社 | 不死化単球細胞および誘導細胞を用いた抗感染症薬,ワクチンなどの評価方法 |
| US10718781B2 (en) | 2014-07-14 | 2020-07-21 | Chugai Seiyaku Kabushiki Kaisha | Method for identifying epitope on protein |
| WO2021100828A1 (ja) | 2019-11-19 | 2021-05-27 | マイキャン・テクノロジーズ株式会社 | ヒト不死化ミエロイド系細胞を使用したin vitroにおける物質の安全性評価方法 |
| WO2021230304A1 (ja) * | 2020-05-13 | 2021-11-18 | Agc株式会社 | ヒトプロフェッショナル抗原提示細胞の製造方法 |
| WO2022075384A1 (ja) | 2020-10-07 | 2022-04-14 | マイキャン・テクノロジーズ株式会社 | コロナウイルス感染性細胞及びその調製方法 |
| WO2023277153A1 (ja) * | 2021-06-30 | 2023-01-05 | 国立大学法人千葉大学 | 骨髄系共通前駆細胞(cmp)又は骨髄球系前駆細胞の増殖性を向上させる方法 |
| WO2023229013A1 (ja) * | 2022-05-27 | 2023-11-30 | 北京天一方生物科技▲発▼展有限公司 | IL-12p70を恒常的に産生する増殖性ミエロイド細胞の製造方法 |
| WO2024034656A1 (ja) * | 2022-08-09 | 2024-02-15 | Agc株式会社 | 増殖性マクロファージ様細胞(pMAC)の製造方法 |
| WO2024143555A1 (ja) * | 2022-12-28 | 2024-07-04 | 国立大学法人千葉大学 | 細胞分化度の調節方法 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR3018819A1 (fr) * | 2014-03-19 | 2015-09-25 | Univ Bourgogne | Traitement de la reponse inflammatoire et dysimmunitaire |
| US20170151281A1 (en) | 2015-02-19 | 2017-06-01 | Batu Biologics, Inc. | Chimeric antigen receptor dendritic cell (car-dc) for treatment of cancer |
| WO2020097193A1 (en) | 2018-11-06 | 2020-05-14 | The Regents Of The University Of California | Chimeric antigen receptors for phagocytosis |
| US11013764B2 (en) | 2019-04-30 | 2021-05-25 | Myeloid Therapeutics, Inc. | Engineered phagocytic receptor compositions and methods of use thereof |
| JP2022546592A (ja) | 2019-09-03 | 2022-11-04 | マイエロイド・セラピューティクス,インコーポレーテッド | ゲノム組込みのための方法および組成物 |
| US10980836B1 (en) | 2019-12-11 | 2021-04-20 | Myeloid Therapeutics, Inc. | Therapeutic cell compositions and methods of manufacturing and use thereof |
| EP4240367A4 (en) | 2020-11-04 | 2024-10-16 | Myeloid Therapeutics Inc | MODIFIED CHIMERIC FUSION PROTEIN COMPOSITIONS AND METHODS OF USE THEREOF |
| KR20240013094A (ko) | 2021-03-17 | 2024-01-30 | 마이얼로이드 테라퓨틱스, 인크. | 조작된 키메라 융합 단백질 조성물 및 그의 사용 방법 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003521439A (ja) * | 1998-03-05 | 2003-07-15 | メルクレ、ゲーエムベーハー | 末梢単球の増殖を促進するためのcd137の使用 |
| JP2005528899A (ja) * | 2002-05-28 | 2005-09-29 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | 抗原提示細胞の生成方法 |
| WO2007047583A2 (en) * | 2005-10-18 | 2007-04-26 | National Jewish Medical And Research Center | Conditionally immortalized long-term stem cells and methods of making and using such cells |
| WO2008056734A1 (fr) * | 2006-11-08 | 2008-05-15 | National University Corporation Kumamoto University | Procédé de production de cellules dendritiques à partir de cellules souches d'embryons humains |
| WO2010011644A2 (en) * | 2008-07-21 | 2010-01-28 | Taiga Biotechnologies, Inc. | Differentiated anucleated cells and method for preparing the same |
| WO2011034073A1 (ja) * | 2009-09-15 | 2011-03-24 | 国立大学法人東京大学 | 分化細胞の新規製造法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050130302A1 (en) * | 2003-09-29 | 2005-06-16 | Reprocell Inc. | Method and composition for regulating expansion of stem cells |
| JP2010268789A (ja) | 2009-04-24 | 2010-12-02 | Kumamoto Univ | 細胞医薬の製造方法 |
| KR20120060201A (ko) * | 2009-07-08 | 2012-06-11 | 인쎄름 (엥스띠뛰 나씨오날 드 라 쌍떼 에 드 라 흐쉐르슈 메디깔) | 기능적으로 분화된 체세포의 연장된 자기재생을 유도하는 방법 |
-
2011
- 2011-09-28 JP JP2012536516A patent/JP5861191B2/ja active Active
- 2011-09-28 US US13/825,883 patent/US10034900B2/en active Active
- 2011-09-28 WO PCT/JP2011/072234 patent/WO2012043651A1/ja active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003521439A (ja) * | 1998-03-05 | 2003-07-15 | メルクレ、ゲーエムベーハー | 末梢単球の増殖を促進するためのcd137の使用 |
| JP2005528899A (ja) * | 2002-05-28 | 2005-09-29 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | 抗原提示細胞の生成方法 |
| WO2007047583A2 (en) * | 2005-10-18 | 2007-04-26 | National Jewish Medical And Research Center | Conditionally immortalized long-term stem cells and methods of making and using such cells |
| WO2008056734A1 (fr) * | 2006-11-08 | 2008-05-15 | National University Corporation Kumamoto University | Procédé de production de cellules dendritiques à partir de cellules souches d'embryons humains |
| WO2010011644A2 (en) * | 2008-07-21 | 2010-01-28 | Taiga Biotechnologies, Inc. | Differentiated anucleated cells and method for preparing the same |
| WO2011034073A1 (ja) * | 2009-09-15 | 2011-03-24 | 国立大学法人東京大学 | 分化細胞の新規製造法 |
Non-Patent Citations (7)
| Title |
|---|
| BRACKEN, A.P. ET AL.: "EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer", EMBO J, vol. 22, no. 20, 2003, pages 5323 - 5335 * |
| CHOI KD ET AL.: "Generation of mature human myelomonocytic cells through expansion and differentiation of pluripotent stem cell- derived lin-CD34+CD43+CD45+ progenitors", J CLIN INVEST., vol. 119, no. 9, 2009, pages 2818 - 2829, XP008154584, DOI: doi:10.1172/JCI38591 * |
| GOETZ, A.W. ET AL.: "Requirement for Mdm2 in the Survival Effects of Bcr-Abl and Interleukin 3 in Hematopoietic Cells.", CANCER RES, vol. 61, no. 20, 2001, pages 7635 - 7641 * |
| HIDEYUKI OGURO ET AL.: "'Mijika na Wadai - Sekai no Wadai'(33) Zoketsu Kansaibo no Jiko Fukusei ni Okeru Polycomb Idenshi Bmi-1 no Kino", HEMATOLOGY FRONTIER, vol. 16, no. 4, 2006, pages 608 - 612 * |
| NAOYA TAKAYAMA ET AL.: "c-MYC Sai Kasseika ni Tomonau Hito iPS Saibo kara no Koritsu no Yoi Kyokakukyu/Kesshoban Sanseiho no Kakuritsu", DAI 71 KAI THE JAPANESE SOCIETY OF HEMATOLOGY GAKUJUTSU SHUKAI PROGRAM - SHOROKUSHU, 2009, pages 914 * |
| SATORU SENJU: "Anti-Cancer Therapy with Pluripotent Stem Cell-Derived Dendritic Cells", BIOTHERAPY, vol. 24, no. 2, March 2010 (2010-03-01), (TOKYO), pages 87 - 94 * |
| SATORU SENJU: "Jujo Saibo Ryoho to Tanosei Kansaibo", DOJIN NEWS, no. 133, January 2010 (2010-01-01), pages 1 - 5 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140037600A1 (en) * | 2011-02-08 | 2014-02-06 | Cellular Dynamics International, Inc. | Hematopoietic precursor cell production by programming |
| US9574179B2 (en) * | 2011-02-08 | 2017-02-21 | Cellular Dynamics International, Inc. | Hematopoietic precursor cell production by programming |
| EP3929302A1 (en) | 2014-07-14 | 2021-12-29 | Chugai Seiyaku Kabushiki Kaisha | Method for identifying epitope on protein |
| US10718781B2 (en) | 2014-07-14 | 2020-07-21 | Chugai Seiyaku Kabushiki Kaisha | Method for identifying epitope on protein |
| JPWO2017010544A1 (ja) * | 2015-07-15 | 2018-05-24 | テルモ株式会社 | 多能性幹細胞または脂肪組織もしくは骨髄由来の間葉系幹細胞由来の心筋細胞の凍結保存方法 |
| JP2017131136A (ja) * | 2016-01-27 | 2017-08-03 | 国立大学法人 熊本大学 | 血液由来単球の増殖誘導方法 |
| WO2018181903A1 (ja) * | 2017-03-31 | 2018-10-04 | 国立大学法人熊本大学 | インターフェロンβ産生細胞の作製方法 |
| JP2018171005A (ja) * | 2017-03-31 | 2018-11-08 | 国立大学法人 熊本大学 | ヒトミエロイド系血液細胞及び該細胞の作製方法 |
| WO2020045368A1 (ja) | 2018-08-27 | 2020-03-05 | マイキャン・テクノロジーズ株式会社 | 不死化単球細胞および誘導細胞を用いた抗感染症薬,ワクチンなどの評価方法 |
| JPWO2020045368A1 (ja) * | 2018-08-27 | 2021-08-10 | マイキャン・テクノロジーズ株式会社 | 不死化単球細胞および誘導細胞を用いた抗感染症薬,ワクチンなどの評価方法 |
| JP7376013B2 (ja) | 2018-08-27 | 2023-11-08 | マイキャン・テクノロジーズ株式会社 | 不死化単球細胞および誘導細胞を用いた抗感染症薬,ワクチンなどの評価方法 |
| WO2021100828A1 (ja) | 2019-11-19 | 2021-05-27 | マイキャン・テクノロジーズ株式会社 | ヒト不死化ミエロイド系細胞を使用したin vitroにおける物質の安全性評価方法 |
| EP4063493A1 (en) | 2019-11-19 | 2022-09-28 | Mican Technologies Inc. | Method for evaluating safety of substance in vitro using human immortalized myeloid cells |
| CN114729321A (zh) * | 2019-11-19 | 2022-07-08 | 迈凯恩技术有限公司 | 使用人永生化髓样细胞体外评价物质的安全性的方法 |
| WO2021230304A1 (ja) * | 2020-05-13 | 2021-11-18 | Agc株式会社 | ヒトプロフェッショナル抗原提示細胞の製造方法 |
| EP4151723A4 (en) * | 2020-05-13 | 2024-06-19 | Agc Inc. | METHOD FOR PRODUCING HUMAN PROFESSIONAL ANTIGEN PRESENTING CELLS |
| WO2022075384A1 (ja) | 2020-10-07 | 2022-04-14 | マイキャン・テクノロジーズ株式会社 | コロナウイルス感染性細胞及びその調製方法 |
| WO2023277153A1 (ja) * | 2021-06-30 | 2023-01-05 | 国立大学法人千葉大学 | 骨髄系共通前駆細胞(cmp)又は骨髄球系前駆細胞の増殖性を向上させる方法 |
| WO2023229013A1 (ja) * | 2022-05-27 | 2023-11-30 | 北京天一方生物科技▲発▼展有限公司 | IL-12p70を恒常的に産生する増殖性ミエロイド細胞の製造方法 |
| WO2024034656A1 (ja) * | 2022-08-09 | 2024-02-15 | Agc株式会社 | 増殖性マクロファージ様細胞(pMAC)の製造方法 |
| WO2024143555A1 (ja) * | 2022-12-28 | 2024-07-04 | 国立大学法人千葉大学 | 細胞分化度の調節方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US10034900B2 (en) | 2018-07-31 |
| JPWO2012043651A1 (ja) | 2014-02-24 |
| US20130195818A1 (en) | 2013-08-01 |
| JP5861191B2 (ja) | 2016-02-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5861191B2 (ja) | ミエロイド系血液細胞の製造方法 | |
| JP6937821B2 (ja) | 生体組織から単離できる多能性幹細胞 | |
| JP2022163165A (ja) | 抗原特異的t細胞の製造方法 | |
| CN107075484B (zh) | 从多能性干细胞诱导细胞免疫治疗用t细胞的方法 | |
| EP2336303B1 (en) | NKT CELL-DERIVED iPS CELLS AND NKT CELLS DERIVED THEREFROM | |
| CN110506110B (zh) | 产生t细胞祖细胞的方法 | |
| WO2012133948A1 (ja) | 生体組織から単離できるssea-3陽性の多能性幹細胞を含む他家移植用細胞治療用組成物 | |
| WO2016010155A1 (ja) | 抗原特異的t細胞受容体遺伝子を有する多能性幹細胞の製造方法 | |
| JPWO2003080817A1 (ja) | 細胞傷害性リンパ球の製造方法 | |
| EP4410965A1 (en) | Method for producing t cell | |
| WO2020045368A1 (ja) | 不死化単球細胞および誘導細胞を用いた抗感染症薬,ワクチンなどの評価方法 | |
| WO2016010153A1 (ja) | 免疫細胞療法用t細胞の誘導方法 | |
| KR20230004589A (ko) | 조건부 불멸화를 위한 제어가능한 트랜스진을 포함하는 유도된 만능 세포 | |
| US8551472B2 (en) | Method of making macrophage expressing an antibody directed against β-amyloid | |
| TW202345878A (zh) | 控制性t細胞之製造方法 | |
| JP2023550100A (ja) | セクレトーム含有組成物を分析するための方法およびアッセイ | |
| JP2023513632A (ja) | 血小板由来ミトコンドリア治療及び多能性細胞の生成方法 | |
| Botó | ZBTB46 and RUNX3 Regulated Blood Cell Differentiation from Pluripotent Embryonic Stem Cells | |
| WO2024168160A2 (en) | Cd4 + and cd8 + single positive t cells and compositions and methods for generating the same | |
| WO2016168586A1 (en) | Cd133+ cells and method for expanding |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11829204 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2012536516 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13825883 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11829204 Country of ref document: EP Kind code of ref document: A1 |