WO2008018090A2 - An improved process for the preparation of zolmitriptan - Google Patents
An improved process for the preparation of zolmitriptan Download PDFInfo
- Publication number
- WO2008018090A2 WO2008018090A2 PCT/IN2007/000328 IN2007000328W WO2008018090A2 WO 2008018090 A2 WO2008018090 A2 WO 2008018090A2 IN 2007000328 W IN2007000328 W IN 2007000328W WO 2008018090 A2 WO2008018090 A2 WO 2008018090A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- process according
- zolmitriptan
- water
- solvent
- reduction
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- This invention in general related to a process for producing Zolmitriptan. More particularly, the present invention provides a process for producing pure Zolmitriptan employing novel reaction conditions as well as without using column.
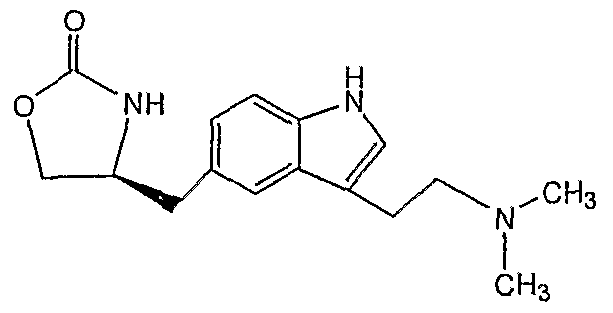
- Zolmitriptan has the chemical name (S)-4-[[3-[2-(dimethylamino)ethyl]-lH-indol-5- yl]methyl]-2-oxazolidinone (I). Zolmitriptan is a selective 5HTl -receptor agonist.
- 5HTl -receptor mediates vasoconstriction and thus modifies blood flow to the carotid vascular bed.
- 5HTl-receptor agonists are beneficial in the treatment (including prophylaxis) of disease conditions wherein vasoconstriction in the carotid vascular bed is indicated, for example migraine, cluster headache and headache associated with vascular disorders, herein after referred to collectively as migraine.
- Zolmitriptan has been developed for the acute treatment of migraine in the form of a 2.5 mg and 5 mg tablet intended to be taken up to maximum of 15 mg per day.
- the resultant compound was converted to (4S)-4-[3-(2-dimethylaminoethyl)-lH-5-indolyl methyl]- l,3-oxazolan-2-one by standard N-alkylation methods.
- the disadvantages lie in low overall yields and column chromatography as a part of process.
- PCT application WO 2001/34561 discloses the preparation of intermediate compounds, for use in preparing Zolmitriptan.
- the preparation involves a Fischer indole synthesis.
- PCT application WO 2004/014901 discloses the process for preparing
- Zolmitriptan in which the ⁇ -keto- ⁇ -valerolactone is used as starting material which is not a commonly used chemical for industrial purposes. Also, optical purity is not disclosed.
- the disclosed embodiment of the present invention deals with a process for producing Zolmitriptan by employing cheap and easily available raw materials and reagents.
- the present invention in its aspect is a new, improved, economical and industrially feasible method for preparing Zolmitriptan.
- the present invention is provided an improved process for the preparation of Zolmitriptan as hereinbefore defined, which process comprises the steps of
- process for the preparation of Zolmitriptan Step (a) includes the treatment of 4-nitrophenylalanine or its hydrate with a thionyl chloride to get methyl (S)-4-nitrophenylalaninate hydrochloride.
- the reaction is carried out in the presence of the solvent e.g. methanol.
- the reaction is conveniently carried out at room temperature to reflux temperature, preferably at reflux temperature.
- the reduction step (b) is conveniently carried out in the presence of polar protic solvent such as water, methanol, ethanol, isopropyl alcohol, n-butanol and/or mixture thereof.
- Step (b) is carried out by hydrogenation, preferably in the presence of sodium borohydride.
- the reaction is carried out at normal atmospheric pressure at -15 0 C to 6O 0 C, preferably at -1O 0 C to 5O 0 C, more preferably 0°C-25°C.
- the reaction mixture is filtered and the filtrate is concentrated under vacuum, followed by cooling to get precipitate.
- the obtained precipitate is filtered and washed with the solvent to get desired compound i.e. (S)-2-amino3 ⁇ (4-niuOphenyl)propanol.
- step (c) the above obtained compound i.e. (S) ⁇ 2- amino-3-(4-nitrophenyl)propanol is then treated with triphosgene in presence of base and a solvent to get (S)-4-(4-nitrobcnzyl)-l,3-oxazolidine-2-one.
- the used base in this reaction is selected from the group consisting of but not limited to such as alkali metal carbonates such as sodium carbonate, potassium carbonate, alkali metal hydrogen carbonates such as sodium bicarbonate, potassium bicarbonate, alkali metal hydroxide such as sodium hydroxide, potassium hydroxide, preferably sodium carbonate and potassium carbonate more preferably potassium carbonate.
- the solvent used herein is selected from the group consisting of aliphatic and aromatic hydrocarbon such as toluene, benzene, xylene, hexane, heptane, pentane, cyclohcxane, cyclopcntane, cycloheptane; water and/or mixture thereof.
- solvent used herein is toluene and toluene water mixture thereof.
- the above said reaction is carried out at 0°C-35°C, preferably 1O 0 C- 35 0 C, more preferably 10°C-25°C.
- the reduction step (d) of the present invention is conveniently carried out in the presence of solvent e.g. methanol, ethanol, isopropyl alcohol preferably methanol.
- Step (d) is carried out by hydrogenation, preferably in the presence of a catalyst such as Raney Ni and Pd/C, preferably Raney Ni.
- the reaction may be carried out under an atmosphere of hydrogen at a pressure of 2.0-8.0 Kg/m 2 , preferably 5.0-7.0 Kg/m 2 , more preferably 6.0 Kg/m 2 .
- the reaction is carried out at 25 0 C to 4O 0 C, preferably al 3O 0 C.
- reaction mixture is filtered and the filtrate is concentrated under vacuum, followed by addition of a solvent such as ethyl acetate, methanol, ethanol, isopropyl alcohol or mixture thereof, to the residue and heated at 40-60 0 C preferably at 50-55 0 C, followed by cooling to get precipitate.
- a solvent such as ethyl acetate, methanol, ethanol, isopropyl alcohol or mixture thereof.
- the obtained precipitate is filtered and washed with the solvent to get desired compound i.e. (S)-4-(4-aminobenzyl)-l ,3-oxazolidine-2-one.
- the reduced compound from step (d) is then diazotized using aqueous sodium nitrite solution, preferably in the presence of concentrated hydrochloric acid, at a reduced temperature.
- the salt formation is carried out at a reduced temperature e.g. 0- 5 0 C.
- Hydrazine formation is then carried out on the diazonium salt solution by using stannous chloride.
- the stannous chloride is suitably in the form of an aqueous solution.
- the reduction is advantageously carried out at temperature below O 0 C during the addition and followed by increase in the temperature to room temperature. Water is added to the reaction mixture after the stirring at room temperature for 6-15 hrs, preferably 10-12 hrs.
- reaction mixture is then cooled and pH of the reaction mixture is adjusted to 1.0-2.5, preferably 1.5-2.0, more preferably 1.8-2.0 with 75% alkali metal hydroxide preferably sodium hydroxide.
- the reaction mixture is then heated to 80-100 0 C, preferably 90-95 0 C under nitrogen atmosphere followed by the addition of N,N-dimethylaminobutyraldehyde diethyl acetal.
- the reaction mixture is then cooled and pH of the reaction mixture is adjusted to 6.0-8.0, preferably 6.9-7.0 using 75% alkali metal hydroxide such as sodium hydroxide.
- the reaction mixture is the filtered to remove all inorganic salts such as tin oxide and/or other tin salts, if not then it creates the problem in getting the solid product as well as it carries forward to the final product as an impurity where it is very difficult to remove.
- the filtrate is washed with an organic solvent such as toluene, hexane, dichloromethane or chloroform.
- the filtrate is treated with activated carbon at 50-65 0 C preferably 55-6O 0 C and filtered.
- the organic solvent is added to the reaction mixture and then pH is adjusted to 10.0-12.0 preferably 10.5-11.0 using 75% alkali metal hydroxide preferably sodium hydroxide.
- the layers are separated and aqueous layer is extracted with ethyl acetate and combined both the organic layers.
- the organic solvent solution is evaporated to dryness to get residue which is then dissolved in alcohol and/or water and stirred.
- the reaction mixture is then concentrated to get the precipitate of crude Zolmitriptan.
- the organic solvent used herein is selected from the group consisting of but nol limited to ethyl acetate, methyl acetate, n-butanol, dichloromelhane, chloroform and/or mixture thereof, whereas alcohol used herein is selected from the group consisting of but not limited to methanol and ethanol.
- the obtained crude Zolmitriptan is then purified using water, isopropyl alcohol or mixture thereof as a solvent.
- Ethyl acetate (1.331ts) was added to the filtrate and pH adjusted to 10.5 - 1 1.0 with 75% sodium hydroxide. Stirred for 30 min. and the layers were separated. Aqueous layer was again extracted with ethyl acetate (660 ml) and the combined organic extracts washed with water (340 ml). The ethyl acetate layer was concentrated under reduced pressure to recover ethyl acetate completely and added isopropyl alcohol (500 ml) to the residue. The reaction mixture was heated to 65 0 C and treated with activated carbon for 30 min. Filtered through hyflo and washed bed with isopropyl alcohol (200 ml).
- the filtrate was concentrated under reduced pressure at 45-5O 0 C to recover -500 ml of isopropyl alcohol.
- the remaining thick mass was slowly cooled to room temperature and stirred for 5 hrs.
- the slurry obtained was filtered and the solids washed with chilled isopropyl alcohol (150 ml) to obtain crude Zolmitriptan.
- Zolmitriptan (crude) (25 g) was slurred with D. M. water (100 ml) at room temperature for 4 hrs. The slurry was filtered and solid washed with cold D. M. water (25 ml). The wet material is dried to get pure Zolmitriptan, which is found to be Form A.
- Zolmitriptan (crude) (7 g) was slurred with a solution of 20% V/V isopropyl alcohol and D. M. water mixture (10.5 ml) at room temperature for 4 hrs. The slurry was filtered and solid washed with cold 20% v/v isopropyl alcohol and D. M. water (3.5 ml). The wet ' material is dried to get pure Zolmitriptan, which is found to be Form A.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Disclosed herein the process for producing pure' Zolmitriptan employing improved reaction conditions which excluded the use of column chromatography.
Description
Title: An Improved Process for the Preparation of Zolmitriptan
Field of the Invention
This invention in general related to a process for producing Zolmitriptan. More particularly, the present invention provides a process for producing pure Zolmitriptan employing novel reaction conditions as well as without using column.
Background of the Invention
Zolmitriptan has the chemical name (S)-4-[[3-[2-(dimethylamino)ethyl]-lH-indol-5- yl]methyl]-2-oxazolidinone (I). Zolmitriptan is a selective 5HTl -receptor agonist. The
5HTl -receptor mediates vasoconstriction and thus modifies blood flow to the carotid vascular bed. 5HTl-receptor agonists are beneficial in the treatment (including prophylaxis) of disease conditions wherein vasoconstriction in the carotid vascular bed is indicated, for example migraine, cluster headache and headache associated with vascular disorders, herein after referred to collectively as migraine. Zolmitriptan has been developed for the acute treatment of migraine in the form of a 2.5 mg and 5 mg tablet intended to be taken up to maximum of 15 mg per day.

Although Zolmitriptan is a successful drug of considerable benefit to migraine sufferers, there is continuing need for alternative methods for the direct treatment of migraine and the prophylactic treatment of migraine. In particular, patients suffering from migraine or the onset of migraine need fast relief of their suffering.
US 5,466,699 discloses Zolmitriptan as a product along with other related compounds and the methods for their preparation. The synthetic route as disclosed in US'699 is shown below in scheme 1.

4-dimelhylaminobutanol-diethyl acelal Acetic acid, water, IPA
Scheme 1
Earlier disclosures for the preparation of optically pure Zolmitriptan have disadvantages, particularly for scale-up to produce large quantities. For e.g. PCT application WO 91/18897 discloses the preparation of Zolmitriptan by diazotization of (4S)-4-(4- aminobenzyl)-l,3-oxazolan-2-one, followed by reduction to the corresponding hydrazine intermediate and coupling with 1-butyraldehyde (or its acetal) substituted at C4 with an easy leaving group (such as chlorine or protected amine) by Fischer reaction. The resultant compound was converted to (4S)-4-[3-(2-dimethylaminoethyl)-lH-5-indolyl methyl]- l,3-oxazolan-2-one by standard N-alkylation methods. The disadvantages lie in low overall yields and column chromatography as a part of process.
Another PCT application WO 97/06162 discloses the preparation of Zolmitriptan by diazotization of (4S)-4-(4-aminobenzyl)-l,3-oxazolan-2-one, followed by reduction of the corresponding hydrazine intermediate and coupling with 4,4-methoxy-N,N-
dimelhylbulylamine by Fischer reaction. The disadvantage lies in extraction at elevated temperature leading to the hydrolysis as well as loss of the solvent.
PCT application WO 2001/34561 discloses the preparation of intermediate compounds, for use in preparing Zolmitriptan. The preparation involves a Fischer indole synthesis.
Whereas, PCT application WO 2004/014901 discloses the process for preparing
Zolmitriptan, in which the α-keto-δ-valerolactone is used as starting material which is not a commonly used chemical for industrial purposes. Also, optical purity is not disclosed.
Strong alkali reagents are used in the hydrolysis stage that may open the oxazolidinone ring when following the described process. Also, the decarboxylation process is more complicated in terms of the reagents and conditions used in the process disclosed in this publication.
The process disclosed in the prior art have several limitations like overall yields, side product formation, use of expensive or hazardous reagents and expensive starting materials. There remains a need of a process that is easily conducted on a commercial scale, to removal of inorganic salts impurities specially tin salts, to produce 'a product with high purity. Moreover, the isolation of crude Zolmitriptan from the reaction mixture has been achieved in high purity and yield without the use of column chromatography.
Summary of the Invention
It is, therefore, a principal aspect of the present invention to provide an improved way for producing Zolmitriptan over limitations in the prior art. These and other objects are attained in accordance with the present invention wherein there is provided several embodiments of the process for producing Zolmitriptan by employing cheap and easily available raw materials and reagents.
Another with one preferred embodiment of the present invention, there is provided a novel process for producing pure Zolmitriptan without using column chromatography. Accordance with another preferred embodiment of the present invention, there is provided a novel process for producing Zolmitriptan in a way to improve the yield and purity and obviates the formation of byproducts.
Detailed Description of the Invention
The disclosed embodiment of the present invention deals with a process for producing Zolmitriptan by employing cheap and easily available raw materials and reagents.
The present invention in its aspect is a new, improved, economical and industrially feasible method for preparing Zolmitriptan. The present invention is provided an improved process for the preparation of Zolmitriptan as hereinbefore defined, which process comprises the steps of
(a) treating 4-nitrophenylalanine monohydrate with thionyl chloride to get methyl
(S)-4-nitrophenylalaninate hydrochloride; (b) reducing methyl (S)-4-nitrophenylalaninate hydrochloride with sodium borohydride in a polar protic solvent to gel (S)-2-amino-3-(4- nitrophenyl)propanol ; (c) treating (S)-2-amino-3-(4-nitrophenyl)propanol with triphosgene in presence of base and solvent to get (S)-4-(4-nitrobenzyl)-l,3-oxazolidine-2-one; (d) reducing the (S)-4-(4-nitrobenzyl)-l.,3-oxazolidme-2-one in presence of catalyst under hydrogen pressure and a solvent to obtain (S)-4-(4-aminobenzyl)-l ,3- oxazolidine-2-one; (e) diazotization of (S)-4-(4-aminobenzyl)-l,3-oxazolidine-2-one with NaNO2ZHCl followed by reduction to obtain corresponding hydrazine salt which on condensation with N,N-dimethylaminobutyraldehyde diethyl acetal yielded crude
Zolmitriptan without using column;
(f) purification of crude Zolmitriptan in water or a mixture of isopropyl alcohol and water to obtain pure Zolmitriptan.
According to the present invention, process for the preparation of Zolmitriptan Step (a) includes the treatment of 4-nitrophenylalanine or its hydrate with a thionyl chloride to get methyl (S)-4-nitrophenylalaninate hydrochloride. The reaction is carried out in the presence of the solvent e.g. methanol. The reaction is conveniently carried out at room temperature to reflux temperature, preferably at reflux temperature.
The reduction step (b) is conveniently carried out in the presence of polar protic solvent such as water, methanol, ethanol, isopropyl alcohol, n-butanol and/or mixture thereof. Suitably Step (b) is carried out by hydrogenation, preferably in the presence of sodium borohydride. The reaction is carried out at normal atmospheric pressure at -150C to 6O0C, preferably at -1O0C to 5O0C, more preferably 0°C-25°C. The reaction mixture is filtered and the filtrate is concentrated under vacuum, followed by cooling to get precipitate. The obtained precipitate is filtered and washed with the solvent to get desired compound i.e. (S)-2-amino3~(4-niuOphenyl)propanol.
According to the present invention in step (c), the above obtained compound i.e. (S)~2- amino-3-(4-nitrophenyl)propanol is then treated with triphosgene in presence of base and a solvent to get (S)-4-(4-nitrobcnzyl)-l,3-oxazolidine-2-one. The used base in this reaction is selected from the group consisting of but not limited to such as alkali metal carbonates such as sodium carbonate, potassium carbonate, alkali metal hydrogen carbonates such as sodium bicarbonate, potassium bicarbonate, alkali metal hydroxide such as sodium hydroxide, potassium hydroxide, preferably sodium carbonate and potassium carbonate more preferably potassium carbonate. The solvent used herein is selected from the group consisting of aliphatic and aromatic hydrocarbon such as toluene, benzene, xylene, hexane, heptane, pentane, cyclohcxane, cyclopcntane, cycloheptane; water and/or mixture thereof. Preferably solvent used herein is toluene and toluene water mixture thereof. The above said reaction is carried out at 0°C-35°C, preferably 1O0C- 350C, more preferably 10°C-25°C.
The reduction step (d) of the present invention, is conveniently carried out in the presence of solvent e.g. methanol, ethanol, isopropyl alcohol preferably methanol. Suitably Step (d) is carried out by hydrogenation, preferably in the presence of a catalyst such as Raney Ni and Pd/C, preferably Raney Ni. The reaction may be carried out under an atmosphere of hydrogen at a pressure of 2.0-8.0 Kg/m2, preferably 5.0-7.0 Kg/m2, more preferably 6.0 Kg/m2. The reaction is carried out at 250C to 4O0C, preferably al 3O0C. The reaction mixture is filtered and the filtrate is concentrated under vacuum, followed by addition of
a solvent such as ethyl acetate, methanol, ethanol, isopropyl alcohol or mixture thereof, to the residue and heated at 40-600C preferably at 50-550C, followed by cooling to get precipitate. The obtained precipitate is filtered and washed with the solvent to get desired compound i.e. (S)-4-(4-aminobenzyl)-l ,3-oxazolidine-2-one.
The reduced compound from step (d) is then diazotized using aqueous sodium nitrite solution, preferably in the presence of concentrated hydrochloric acid, at a reduced temperature. Preferably the salt formation is carried out at a reduced temperature e.g. 0- 50C. Hydrazine formation is then carried out on the diazonium salt solution by using stannous chloride. The stannous chloride is suitably in the form of an aqueous solution. The reduction is advantageously carried out at temperature below O0C during the addition and followed by increase in the temperature to room temperature. Water is added to the reaction mixture after the stirring at room temperature for 6-15 hrs, preferably 10-12 hrs. The reaction mixture is then cooled and pH of the reaction mixture is adjusted to 1.0-2.5, preferably 1.5-2.0, more preferably 1.8-2.0 with 75% alkali metal hydroxide preferably sodium hydroxide. The reaction mixture is then heated to 80-1000C, preferably 90-950C under nitrogen atmosphere followed by the addition of N,N-dimethylaminobutyraldehyde diethyl acetal. The reaction mixture is then cooled and pH of the reaction mixture is adjusted to 6.0-8.0, preferably 6.9-7.0 using 75% alkali metal hydroxide such as sodium hydroxide. The reaction mixture is the filtered to remove all inorganic salts such as tin oxide and/or other tin salts, if not then it creates the problem in getting the solid product as well as it carries forward to the final product as an impurity where it is very difficult to remove. The filtrate is washed with an organic solvent such as toluene, hexane, dichloromethane or chloroform. The filtrate is treated with activated carbon at 50-650C preferably 55-6O0C and filtered. The organic solvent is added to the reaction mixture and then pH is adjusted to 10.0-12.0 preferably 10.5-11.0 using 75% alkali metal hydroxide preferably sodium hydroxide. The layers are separated and aqueous layer is extracted with ethyl acetate and combined both the organic layers. The organic solvent solution is evaporated to dryness to get residue which is then dissolved in alcohol and/or water and stirred. The reaction mixture is then concentrated to get the precipitate of crude Zolmitriptan. The organic solvent used herein is selected from the group consisting of but
nol limited to ethyl acetate, methyl acetate, n-butanol, dichloromelhane, chloroform and/or mixture thereof, whereas alcohol used herein is selected from the group consisting of but not limited to methanol and ethanol.
The obtained crude Zolmitriptan is then purified using water, isopropyl alcohol or mixture thereof as a solvent.
In conclusion, this is a novel, economical and high yielding process for the industrial production of Zolmitriptan using cheaply available reagents and starting materials.
The following non-limiting examples illustrate specific embodiments of the present invention. They are, however, not intended to be limiting the scope of present invention in any way.
Example 1
Preparation of Methyl (S)-(4-nitrophcnyI)alanina(e hydrochloride
To a suspension of 4-nitrophenyl alanine monohydrate (300.0 g, 1.315 moles) in chilled methanol (2.55 It.) thionyl chloride (234.8 g, 1.974 moles) was added. The temperature was raised to reflux and maintained for 6-7 hrs. After the completion of reaction, the clear solution was set for distillation to recover — 1.4It of methanol. The slurry was cooled, stirred for 10 min and filtered to obtain methyl (S)-(4-nitrophenyl)alaninate hydrochloride (314 g).
Example 2
Preparation of (S)-2-Amino-3-[4-nitrophenyI]propanol
To a pre cooled solution of methanol (96 ml) and DM water (84 ml) sodium borohydride (5.8 g, 0.153 moles) was added. Methyl (S)-(4-nitrophenyl)alaninate hydrochloride (1O g,
0.038 moles) was added while maintaining the temperature below O0C. After complete addition the temperature was gradually raised to 5-80C and maintained for 1 hr.
Thereafter the temperature was raised to room temperature and maintained for 2 hrs followed by heating at 5O0C and maintained. Then reaction mixture was cooled, filtered and washed the inorganic solids with methanol (50 ml). The filtrate was concentrated under reduced pressure to get the slurry followed by cooling to 250C to get the precipitate. The precipitated solid was filtered and washed to obtain the required (S)-2- Amino-3-[4-nitrophenyl]propanol (7.8 g).
Example 3
Preparation of (S)-2-Amino-3-[4-nitrophenyI]propanoI
To a pre-cooled (-50C) DM water (720 ml) sodium borohydride (23.22 g) was added and stirred. Hydrochloride salt of methyl (S)-4-nitrophenylalaninate (40 g) was charged in small portions to the reaction mixture while maintaining the temperature at O0C. The temperature was gradually raised to 45-5O0C and maintained for 3 hrs. The reaction mixture was cooled to 3O0C and stirred. The reaction mixture was filtered and the wet cake was washed with water (20 ml). The wet solid Avas again taken in water (120 ml) and warmed to 450C, maintained for 30 min followed by cooling to 3O0C. Reaction mixture was Filtered and washed with water (40 ml) to obtain (S)-2-Amino-3-[4- nitrophenyl]propanol (25 g).
Example 4
Preparation of (S)-4-(4-nilrobenzyI)-l,3-oxazolidinc-2-one
A suspension of (S)-2-Amino-3-(4-nitrophenyl)propanol (65.0 g, 0.331 moles) in toluene • (357.5 ml) was cooled to 10-15°C. Potassium carbonate (68.3 g, 0.494 moles) was added in lots followed by addition of a solution of triphosgene (38.3 g, 0.129 moles) in toluene
( 195 ml) while maintaining the temperature between 10-15°C. After the completion of the reaction water (552.5 ml) was added and the temperature of the reaction mixture was raised to 20-250C. The obtained solid was filtered, washed with water (195 ml) and dried. The solid obtained again slurred in water (650 ml) at 50-55°C for 1 hr. The solid obtained was filtered and washed with water (195 ml) to get the desired compound.
Example 5
Preparation of (S)-4-(4-nitrobenzyl)-l,3-oxazoIidinc-2-onc
A solution of potassium carbonate (89.3 g, 0.646 moles) in water (722.5 ml) was cooled to 10-150C and to this toluene (467.5 ml) was added followed by the addition of (S)-2-
Amino-3-(4-nitrophenyl) propanol (85 g, 0.433 moles). A solution of lriphosgene (51 g,
0.172 moles) in toluene (255 ml) was slowly added to the above mixture while maintaining the temperature between 10-150C. The temperature was raised to 20-250C and maintained. The solid obtained was filtered and washed with water (255 ml) and dried. The obtained solid was again slurred in water (850 ml) and toluene (255 ml) at 50-
550C for 1 hr. Filtered and washed the solid with water (840 ml) to get the desired compound.
Example 6
Preparation of (S)-4-(4-nitrobenzyI)-l,3-oxazoIidine-2-one
A solution of potassium carbonate (294 g, 2.12 moles) in water (2.38 It) was cooled to 1 0-150C and to this solution of (S)-2-Amino-3-(4-nitrophenyl) propanol (280 g, 1.428 moles) in toluene (1.54 It) was added. A solution of lriphosgene (165.26 g, 0.556 moles) in toluene (840 ml) was slowly added to the above mixture while maintaining the temperature between 10-150C. The temperature was raised to 20-250C and maintained. Filtered and washed the solid with water (840 ml) and dried. The obtained solid again slurred in water (2.8 It) at 50-550C for 1 hr. Filtered and washed the solid with water (840 ml) to get the desired compound.
Example 7
Preparation of (S)~4-(4-aminobcnzyl)-l,3-oxazoIidinc-2-one
(S)-4-(4-Nitrobenzyl)-l,3-oxazolidine-2-one (65 g, 0.293 moles) was dissolved in methanol (0.65 It) followed by the addition of Raney nickel (13 c.c.) and hydrogenated in the presence of hydrogen at a pressure of 6.0 Kg at -3O0C for 6-7 hrs. The reaction mass was filtered through a hyflo bed and washed residue with methanol (195 ml). The filtrate
was concentrated under reduced pressure to distil off methanol. Ethyl acetate (130 ml) was added to residue and heated to 50-550C for lhr to obtain thick slurry. The reaction mixture was cooled to 1O0C and maintained. The solid was filtered and the precipitates washed with chilled ethyl acetate (65 ml) to get the desired compound.
Example 8
Preparation of (S)-N,N-Dimcthyl-2-[5-[2-oxo-l,3-oxazoIidinylmcthyI]-lH- indoI-3- ylj ethyl amine (Zoltnitriptan -crude) (S)-4-(4-Aminobenzyl)-l,3-oxazolidine-2-one (100 g, 0.52 moles) was charged to a cooled solution of water (600 ml) and concentrated hydrochloric acid (500 ml) at -1 O0C. To this a solution of sodium nitrite (39.8 g, 0.577 moles) dissolved in DM water (200 ml) was slowly added. The reaction was maintained for 1 hr at -5 to O0C. The above cold solution was added to a pre cooled solution of stannous chloride (479.7 g, 2.132 moles) in cone, hydrochloric acid (600 ml). The temperature of the reaction mass was slowly raised to room temperature and maintained for overnight. Added water (2 It) to the above reaction mixture, cooled and adjusted the pH of the reaction mass to 1 .8-2.0 with 75% sodium hydroxide solution (~900 ml). The temperature was raised to 90-950C under nitrogen atmosphere and maintained for 30 min. (N,N-dimethyl)aminobutyraldehyde diethyl acetal (147.4 g, 0.78 moles) was added to the reaction mixture and maintained the temperature at 90-950C for another 2 hrs. The reaction mixture was cooled and the pH was adjusted to 6.9 — 7.0 with 75% sodium hydroxide solution (666 ml). After stirring of 30 min the inorganic salts were filtered through hyflo bed. The filtrate was then washed twice with methylene chloride (500 ml) followed by treatment with activated carbon (15 g) at 55-6O0C. Ethyl acetate (1.331ts) was added to the filtrate and pH adjusted to 10.5 - 1 1.0 with 75% sodium hydroxide. Stirred for 30 min. and the layers were separated. Aqueous layer was again extracted with ethyl acetate (660 ml) and the combined organic extracts washed with water (340 ml). The ethyl acetate layer was concentrated under reduced pressure to recover ethyl acetate completely and added isopropyl alcohol (500 ml) to the residue. The reaction mixture was heated to 650C and treated with activated carbon for 30 min. Filtered through hyflo and washed bed with isopropyl alcohol (200
ml). The filtrate was concentrated under reduced pressure at 45-5O0C to recover -500 ml of isopropyl alcohol. The remaining thick mass was slowly cooled to room temperature and stirred for 5 hrs. The slurry obtained was filtered and the solids washed with chilled isopropyl alcohol (150 ml) to obtain crude Zolmitriptan.
Example 9
Preparation of Pure Zolmitriptan
Zolmitriptan (crude) (25 g) was slurred with D. M. water (100 ml) at room temperature for 4 hrs. The slurry was filtered and solid washed with cold D. M. water (25 ml). The wet material is dried to get pure Zolmitriptan, which is found to be Form A.
Example 10
Preparation of Pure Zolmitriptan
Zolmitriptan (crude) (7 g) was slurred with a solution of 20% V/V isopropyl alcohol and D. M. water mixture (10.5 ml) at room temperature for 4 hrs. The slurry was filtered and solid washed with cold 20% v/v isopropyl alcohol and D. M. water (3.5 ml). The wet ' material is dried to get pure Zolmitriptan, which is found to be Form A.
Certain modifications and improvements of the disclosed invention will occur to those skilled in the art without departing the scope of the invention, which is limited only by the appended claims.
Claims
1. An improved process for the preparation of pure Zolmitriptan, which comprises:
(a) treating 4-nilrophenylalanine monohydrate with thionyl chloride to get methyl
(S)-4-nitrophenylalaninate hydrochloride;
(b) reducing methyl (S)-4-nitrophenylalaninate hydrochloride with sodium borohydride in a polar protic solvent to get (S)-2-amino-3-(4- nitrophenyl)propanol; (c) treating (S)-2-amino-3-(4-nitrophenyl)propanol with triphosgene in presence of base and solvent to get (S)-4-(4-nitrobenzyl)-l ,3-oxazolidine-2-one; (d) reducing the (S)-4-(4-nitrobenzyl)-l,3-oxazolidine-2-onc in presence of catalyst under hydrogen pressure and a solvent to obtain (S)-4-(4- aminobenzyl)-l,3-oxazolidine-2-one; (e) diazotization of (S)-4-(4-aminobenzyl)-l,3-oxazolidine-2-one with
NaNO2/HCl followed by reduction to obtain corresponding hydrazine salt which on condensation with N,N-dimethylaminobutyraldehyde diethyl acetal yielded crude Zolmitriptan without using column;
(f) purification of crude Zolmitriptan in water or a mixture of isopropyl alcohol and water to obtain pure Zolmitriptan.
2. The process according to claim 1 step (b), wherein polar protic solvent used is water, methanol, ethanol, isopropyl alcohol and n-butanol.
3. The process according to claim 1 step (b), wherein reduction is done at normal pressure and at -1O0C to 5O0C.
4. The process according to claim 3, reduction is done at normal atmospheric pressure and O0C to 250C.
5. The process according to claim 1 step (c), wherein base used herein is alkali metal carbonates such as sodium carbonate, potassium carbonate, alkali metal hydrogen carbonates such as sodium bicarbonate, potassium bicarbonate and alkali metal hydroxide such as sodium hydroxide, potassium hydroxide.
6. The process according to claim 5, wherein base used is potassium carbonate.
7. The process according to claim 1 step (c), wherein solvent used is aliphatic and aromatic hydrocarbon such as toluene, benzene, xylene, hexane, heptane pentane, cyclohexane, cyclopentane, cycloheptane; water and/or mixture thereof.
8. The process according to claim 7, wherein solvent used is toluene and mixture of toluene and water.
9. The process according to claim 1 step (d), wherein reduction is done by using catalyst such as Raney Ni, Pd/C.
10. The process according to claim 9, wherein catalyst used is Raney Ni.
1 1. The process according to claim 1 step (d), wherein hydrogen pressure is 2.0 to 8.0
Kg/m2.
12. The process according to claim 11, wherein hydrogen pressure is 5.0 to 7.0 Kg/m2.
13. The process according to claim 1 step (d), wherein solvent used is methanol, ethanol, isopropyl alcohol.
14. The process according to claim 1 step (e), wherein reduction is done by using SnCl2/HCl.
15. The process according to claim 1 step (e) further comprises, wherein pH is adjusted to 6.0 to 8.0 after the condensation of hydrazine salt with N,N- dimelhylaminobutyraldehyde diethyl acetal followed by treatment with activated carbon and filtered.
16. The process according to claim 15 wherein pH is adjusted to 6.9 to 7.0.
17. The process according to claim 15 further comprises, wherein organic solvent is added to the filtrate and pH is adjusted to 10.0 to 12.0.
18. The process according to claim 17, wherein organic solvent used herein is ethyl acetate, methyl acetate, n-butanol, dichloromethane, chloroform and/or mixture thereof.
19. The process according to claim 17, wherein pH is adjusted to 10.5 to 1 1.0.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1422CH2006 | 2006-08-09 | ||
| IN1422/CHE/2006 | 2006-08-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008018090A2 true WO2008018090A2 (en) | 2008-02-14 |
| WO2008018090A3 WO2008018090A3 (en) | 2009-09-24 |
Family
ID=39033393
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2007/000328 WO2008018090A2 (en) | 2006-08-09 | 2007-08-06 | An improved process for the preparation of zolmitriptan |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008018090A2 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009044211A1 (en) * | 2007-10-03 | 2009-04-09 | Generics [Uk] Limited | Process for the preparation of zolmitriptan, salts and solvates thereof |
| WO2013057739A3 (en) * | 2011-09-02 | 2013-06-20 | Emcure Pharmaceuticals Limited | An improved process for preparation of zolmitriptan |
| CN103275075A (en) * | 2013-06-24 | 2013-09-04 | 成都天台山制药有限公司 | Zolmitriptan and preparation method thereof |
| CN113620897A (en) * | 2021-09-06 | 2021-11-09 | 南京杰运医药科技有限公司 | Preparation method of oxazolidinone compound |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5466699A (en) * | 1990-06-07 | 1995-11-14 | Burroughs Wellcome Co. | Indolyl compounds for treating migraine |
| WO1997006162A1 (en) * | 1995-08-07 | 1997-02-20 | Zeneca Limited | One pot synthesis of 2-oxazolidinone derivatives |
| WO2006055964A2 (en) * | 2004-11-19 | 2006-05-26 | Teva Pharmaceutical Industries Ltd. | Zolmitriptan crystal forms |
| WO2008007390A2 (en) * | 2006-07-10 | 2008-01-17 | Natco Pharma Limited | An improved process for purification of zolmitriptan |
-
2007
- 2007-08-06 WO PCT/IN2007/000328 patent/WO2008018090A2/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5466699A (en) * | 1990-06-07 | 1995-11-14 | Burroughs Wellcome Co. | Indolyl compounds for treating migraine |
| WO1997006162A1 (en) * | 1995-08-07 | 1997-02-20 | Zeneca Limited | One pot synthesis of 2-oxazolidinone derivatives |
| WO2006055964A2 (en) * | 2004-11-19 | 2006-05-26 | Teva Pharmaceutical Industries Ltd. | Zolmitriptan crystal forms |
| WO2008007390A2 (en) * | 2006-07-10 | 2008-01-17 | Natco Pharma Limited | An improved process for purification of zolmitriptan |
Non-Patent Citations (1)
| Title |
|---|
| AMMAZZALORSO, A. ET AL.: 'Synthesis and antibacterial evaluation of oxazolidin-2-ones structurally related to linezolid' IL FARMACO vol. 59, 2004, pages 685 - 690 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009044211A1 (en) * | 2007-10-03 | 2009-04-09 | Generics [Uk] Limited | Process for the preparation of zolmitriptan, salts and solvates thereof |
| CN101883766A (en) * | 2007-10-03 | 2010-11-10 | 基因里克斯(英国)有限公司 | Process for the preparation of zolmitriptan, salts and solvates thereof |
| WO2013057739A3 (en) * | 2011-09-02 | 2013-06-20 | Emcure Pharmaceuticals Limited | An improved process for preparation of zolmitriptan |
| US9006453B2 (en) | 2011-09-02 | 2015-04-14 | Emcure Pharmaceuticals Limited | Process for preparation of zolmitriptan |
| CN103275075A (en) * | 2013-06-24 | 2013-09-04 | 成都天台山制药有限公司 | Zolmitriptan and preparation method thereof |
| CN103275075B (en) * | 2013-06-24 | 2015-01-07 | 成都天台山制药有限公司 | Zolmitriptan and preparation method thereof |
| CN113620897A (en) * | 2021-09-06 | 2021-11-09 | 南京杰运医药科技有限公司 | Preparation method of oxazolidinone compound |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008018090A3 (en) | 2009-09-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9765032B2 (en) | Method for producing (R)-1,1,3-trimethyl-4-aminoindane | |
| EP2158197B1 (en) | Method for the preparation of high purity almotriptan | |
| WO2008018090A2 (en) | An improved process for the preparation of zolmitriptan | |
| JPH11513023A (en) | One-pot synthesis of 2-oxazolidinone derivatives | |
| US11117862B2 (en) | Methods of producing molindone and its salts | |
| US20110112157A1 (en) | Process for the preparation of zolmitriptan, salts and solvates thereof | |
| WO2009082913A1 (en) | Process for isolation of a mixture of rrrs and sssr configurations of nebivolol intermediates | |
| US20130109853A1 (en) | Preparation method of 5-[[2(r)-[1(r)-[3,5-bis(trifluoromethyl) phenyl]ethoxy]-3(s)-4-fluorophenyl-4-morpholinyl]methyl]-1,2-dihydro-3h-1,2,4-triazole-3-one | |
| US20060100443A1 (en) | Novel process for preparation of valsartan | |
| US11518733B2 (en) | Process for preparation of highly pure Fingolimod hydrochloride | |
| WO2015085827A1 (en) | Method for preparing silodosin and intermediate thereof | |
| EP1608371A1 (en) | Process for the preparation of donepezil and derivatives thereof | |
| WO2005105792A1 (en) | Process for preparing optically pure zolmitriptan | |
| EP2751098B1 (en) | An improved process for preparation of zolmitriptan | |
| CN110615768A (en) | A crystal form of GnRHR medicine and preparation method thereof | |
| WO2009086705A1 (en) | Preparation method of rivastigmine, its intermediates and preparation method of the intermediates | |
| US20050245585A1 (en) | Process for preparing optically pure zolmitriptan | |
| WO2011004391A2 (en) | An improved process for the preparation of eletriptan and its salt thereof | |
| CN112300150B (en) | Preparation method of milpitant and intermediate thereof | |
| CN108727351B (en) | Refining method of prucalopride | |
| WO2019167058A1 (en) | An improved process for the preparation of propiomazine maleate | |
| WO2015087239A1 (en) | Processes for the preparation of darapladib and its intermediates | |
| WO2007010557A2 (en) | Process for the preparation of highly pure ropinirole | |
| US20100168385A1 (en) | Process for preparing enantiomerically enriched amino-alcohols | |
| EP2917189B1 (en) | Process for making linezolid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07827529 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| NENP | Non-entry into the national phase in: |
Ref country code: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07827529 Country of ref document: EP Kind code of ref document: A2 |