WO2007010557A2 - Process for the preparation of highly pure ropinirole - Google Patents
Process for the preparation of highly pure ropinirole Download PDFInfo
- Publication number
- WO2007010557A2 WO2007010557A2 PCT/IN2006/000256 IN2006000256W WO2007010557A2 WO 2007010557 A2 WO2007010557 A2 WO 2007010557A2 IN 2006000256 W IN2006000256 W IN 2006000256W WO 2007010557 A2 WO2007010557 A2 WO 2007010557A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ropinirole
- process according
- formula
- amino compound
- salt
- Prior art date
Links
- 0 **c1cccc(N2)c1CC2=O Chemical compound **c1cccc(N2)c1CC2=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/32—Oxygen atoms
- C07D209/34—Oxygen atoms in position 2
Definitions
- the field of the invention relates to highly pure ropinirole or salt thereof and a process for preparing highly pure ropinirole of structural Formula I,
- Ropinirole of Formula-I chemically known as 4-[2-(di-n-propylammo)-ethyl]- 2(3H)-indolone and is useful in the treatment of Parkinsons disease.
- Ropinirole has been first disclosed in US patent 4,452,808.
- the present invention provides a process for the preparation of highly pure ropinirole where starting material converts to final product in more than 97%.
- substantially pure ropinirole or a salt thereof having amino compound less than 0.05% in another general aspect there is provided substantially pure ropinirole or a salt thereof having amino compound less than 0.05%.
- a process for the preparation of substantially pure ropinirole or a salt thereof includes reducing nitro compound of Formula II with a reducing agent to produce amino compound of Formula III, cyclizing the resulting amino compound in situ using palladium on carbon in the presence of aqueous alcoholic medium and isolating the substantially pure ropinirole or a salt thereof by simple isolation method of extraction and acid base treatment.
- the reducing agent may be palladium on carbon.
- the inventors have developed an efficient process for the preparation of substantially pure ropinirole or a salt thereof, by reducing nitro compound of Formula II with a reducing agent to produce amino compound, 2-amino-6-(2-di- n-propylaminoethyl)-phenylacetic acid of Formula III and cyclizing in situ the amino compound of Formula III in aqueous alcoholic medium and isolating substantially pure ropinirole or a salt thereof by simple isolation method of extraction and acid base treatment.
- the 2 ⁇ nitro-6-(2-di-n-propylaminoethyl)-phenylacetic acid hydrochloride of formula II may be treated with a reducing agent in the presence of alcoholic solvent.
- the reaction is performed in an autoclave at a temperature between 25° -5O 0 C and preferably at 35° -4O 0 C under hydrogen pressure of 3.0 kg/cm 2 for 5 hours. It is advantageous to add some water in the reaction mass to complete cyclization of the resulting amino compound i.e. up to 98% conversion.
- the progress of reaction is monitored by HPLC.
- the reducing agent includes any reducing agent which is capable of carrying out the reduction of the nitro group, including, for example, palladium oh carbon, and the like and preferably palladium on carbon is used.
- the alcoholic solvent may include one or more of primary, secondary and tertiary alcohol having from one to six carbon atoms.
- the alcoholic solvent may include one or more of methanol, ethanol, n-propanol, isopropanol, isobutanol, n-butanol and t-butanol. Tn particular, the alcoholic solvent is preferably ethanol. Mixtures of all of these solvents are also contemplated.
- solvent aqueous alcohol
- product may be slurred in acetonitrile, isopropyl ether, ethyl acetate and chlorinated solvent such as methylene chloride, chloroform and the like.
- resulting solid may take in water and basified with sodium hydroxide and product is extracted in organic solvent such as isopropyl ether, ethyl acetate and chlorinated solvent such as methylene chloride, chloroform and the like.
- organic layer is optionally distilled to 60-70% with respect to its original volume.
- the hydrochloride salt of Ropinirole is prepared by treating the ; organic layer with ethanolic-HCl at ambient temperature.
- the product is isolated in high purity and high yield.
- the compound can be optionally recrystallized from alcohol to improve colour if required.
- the product so obtained is having purity greater than 99.5% by HPLC, and preferably greater than 99.8%.
- reaction mass was filtered and ethyl alcohol was recovered completely under vacuum at temperature 40°-45°C.
- Ethyl alcohol (300 ml) was added and recovered under same conditions.
- Ethyl alcohol (300 ml) was added to the dried mass and stirred for 30-45 min at 40° - 45 0 C then cooled at 15-2O 0 C and stirred at 15° - 20 0 C for 45 min. and filter.
- the wet cake was slurred in acetonitrile (500 ml) and stirred for 30 min at 35° - 4O 0 C then cooled to 15 - 2O 0 C.
- the cooled mass was filter and slurry washed with di- isopropyl ether (300 ml).
- the wet cake was added to D M water (120 ml), sodium hydroxide solution 2 % (400 ml) and isopropyl ether (900 ml) were added and stirred at 15° - 2O 0 C for 10 -15 min.
- the organic layer was separated and aqueous layer was extracted with isopropyl ether (2 x 250 ml).
- the combined organic layer was washed with chilled sodium hydroxide solution 1%, (200 ml) filtered by brine (200 ml) and heat to recover isopropyl ether (60 - 70%) was recovered under vacuum.
- reaction mass was cooled at 15° -20 ⁇ C and pH was adjusted to 1.5 - 2.5 with ethanolic hydrochloric acid (20 - 25%, 30 ml) and was stirred 30-40 min at 20° -25 0 C.
- the product obtained was centrifuged and slurry washed with absolute alcohol (200 ml).
- the wet material was dried under vacuum for 7 - 8 hr to obtain 57 g (66.80%) of title compound having purity of 99.30% by HPLC
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Indole Compounds (AREA)
Abstract
The present invention relates to highly pure Ropinirole or salt thereof and a process for preparing highly pure Ropinirole of structural Formula (I), by reducing nitro compound of formula (II), and cyclizing the resulting amino compound in situ using palladium on carbon in the presence of aqueous alcoholic medium.
Description
TITLE QF THE INVENTION
PROCESS FOR THE PREPARATION OF HIGHLY PURE ROPINIROLE
FIELD OF THE INVENTION
The field of the invention relates to highly pure ropinirole or salt thereof and a process for preparing highly pure ropinirole of structural Formula I,
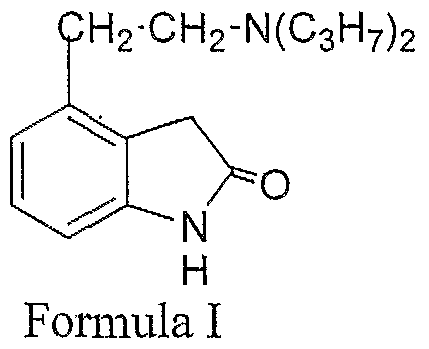
BACKGROUND OF THE INVENTION
Ropinirole of Formula-I chemically known as 4-[2-(di-n-propylammo)-ethyl]- 2(3H)-indolone and is useful in the treatment of Parkinsons disease. Ropinirole has been first disclosed in US patent 4,452,808.
In general, the synthetic approach reported in the literature for the preparation of Ropinirole involves the reduction of 2-nitro-6-(2-di-n-ρroρylaminoethyl)- phenylacetic acid hydrochloride of Formula II followed by in situ cyclization.

There are significant drawbacks to this approach as the reduction of nitro group of 2~nitro-6-(2-di-n-propylaminoethyl)-phenyl acid hydrochloride of formula II to
the corresponding amino compound 2-amino-6-(2-di-n-proρylaminoethyl)- phenylacetic acid of Formula III or salt thereof,

a) 4-[2-(n-propylamino)-ethyl]-2(3-H)-indolone or salt thereof

Formula IV b) 2-amino-6-(2-di-n-propylaminoethyl)-phenylacetic acid or salt thereof



Formula VI
These impurities are further carried into ropinirole.The prior art approach is not suitable from commercial point of view because the desired ropinirole is not obtained in high purity and requires purification by tedious and cumbersome purification processes. The presence of significant quantity of unreacted amino compound in final product even after using 100 times by volume of acetonitrile for recrystallization makes the process uneconomical and unviable.
The inventors have observed that during the cyclization of amino compound of formula III, only 80-85% conversion of amino compound is achieved. The unreacted amino compound leads to formation of impurities which are very difficult to remove from ropinirole. In order to achieve a high efficiency of the reaction for industrial synthesis of ropinirole, it is necessary to increase the conversion of amino compound and minimizes the formation of impurities.
Thus, the present invention provides a process for the preparation of highly pure ropinirole where starting material converts to final product in more than 97%.
SUMMARY QF THE INVENTION
In one general aspect there is provided a highly pure ropinirole or a salt thereof.
In another general aspect there is provided substantially pure ropinirole or a salt thereof having amino compound less than 0.05%.
In another general aspect there is provided highly pure ropinirole or a salt thereof having impurity (a) and (c) each less than 0.05%.
In another general aspect there is provided highly pure ropinirole or a salt thereof having total impurities less than 0.15 %.
In another general aspect there is provided a process for the preparation of substantially pure ropinirole or a salt thereof. The process includes reducing nitro compound of Formula II with a reducing agent to produce amino compound of Formula III, cyclizing the resulting amino compound in situ using palladium on carbon in the presence of aqueous alcoholic medium and isolating the substantially pure ropinirole or a salt thereof by simple isolation method of extraction and acid base treatment. The reducing agent may be palladium on carbon.
The details of one or more embodiments of the inventions are set forth in the description below. Other features, objects and advantages of the inventions will be apparent from the description and claims.
DETAILED DESCRIPTION OF THE INVENTION
The inventors have developed an efficient process for the preparation of substantially pure ropinirole or a salt thereof, by reducing nitro compound of Formula II with a reducing agent to produce amino compound, 2-amino-6-(2-di- n-propylaminoethyl)-phenylacetic acid of Formula III and cyclizing in situ the
amino compound of Formula III in aqueous alcoholic medium and isolating substantially pure ropinirole or a salt thereof by simple isolation method of extraction and acid base treatment.
Tn general, the 2~nitro-6-(2-di-n-propylaminoethyl)-phenylacetic acid hydrochloride of formula II may be treated with a reducing agent in the presence of alcoholic solvent. The reaction is performed in an autoclave at a temperature between 25° -5O0C and preferably at 35° -4O0C under hydrogen pressure of 3.0 kg/cm2 for 5 hours. It is advantageous to add some water in the reaction mass to complete cyclization of the resulting amino compound i.e. up to 98% conversion. The progress of reaction is monitored by HPLC.
The reducing agent includes any reducing agent which is capable of carrying out the reduction of the nitro group, including, for example, palladium oh carbon, and the like and preferably palladium on carbon is used.
The alcoholic solvent may include one or more of primary, secondary and tertiary alcohol having from one to six carbon atoms. The alcoholic solvent may include one or more of methanol, ethanol, n-propanol, isopropanol, isobutanol, n-butanol and t-butanol. Tn particular, the alcoholic solvent is preferably ethanol. Mixtures of all of these solvents are also contemplated.
After completion of reaction, solvent (aqueous alcohol) is removed under vacuum and product may be slurred in acetonitrile, isopropyl ether, ethyl acetate and chlorinated solvent such as methylene chloride, chloroform and the like. Further the resulting solid (crude ropinirole) may take in water and basified with sodium hydroxide and product is extracted in organic solvent such as isopropyl ether, ethyl acetate and chlorinated solvent such as methylene chloride, chloroform and the like. The organic layer is optionally distilled to 60-70% with respect to its original volume. The hydrochloride salt of Ropinirole is prepared by treating the ;
organic layer with ethanolic-HCl at ambient temperature. The product is isolated in high purity and high yield. The compound can be optionally recrystallized from alcohol to improve colour if required. The product so obtained is having purity greater than 99.5% by HPLC, and preferably greater than 99.8%. The major advantages realized in the present invention are:
• High purity
• Completion of cyclization reaction up to 97%
• Avoid use of costly solvent for recrystallization.
The present invention is further illustrated by the following examples which are provided merely to be exemplary of the inventions and is not intended to limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Examples
Preparation of 4-f2- (di-n-propylamino) ethyl] - 2 (3H)-indolone hydrochloride
2-Nitro-6(2-di-n-propylaminoethyl)-phenylacetic acid hydrochloride (10Og) was dissolved in absolute alcohol (4.0 lit.) and charged into hydrogenator under nitrogen. Palladium on carbon( 10%, 50% wet, 12.5 g) was charged to the filtered reaction mass at 35° - 4O0C and hydrogen pressure was maintained at 3.0 kg/cm2 and was stirred for 5 hours at temperature 35° - 4O0C. DM water (400 ml) was added and reaction mass was further stirred for 20 hours at hydrogen pressure 5 - 6 kg/cm2 at temperature 40°-45°C. After completion of reaction, the reaction mass was filtered and ethyl alcohol was recovered completely under vacuum at temperature 40°-45°C. Ethyl alcohol (300 ml) was added and recovered under same conditions. Ethyl alcohol (300 ml) was added to the dried mass and stirred for 30-45 min at 40° - 450C then cooled at 15-2O0C and stirred at 15° - 20 0C for
45 min. and filter. The wet cake was slurred in acetonitrile (500 ml) and stirred for 30 min at 35° - 4O0C then cooled to 15 - 2O0C. The cooled mass was filter and slurry washed with di- isopropyl ether (300 ml). The wet cake was added to D M water (120 ml), sodium hydroxide solution 2 % (400 ml) and isopropyl ether (900 ml) were added and stirred at 15° - 2O0C for 10 -15 min. The organic layer was separated and aqueous layer was extracted with isopropyl ether (2 x 250 ml). The combined organic layer was washed with chilled sodium hydroxide solution 1%, (200 ml) filtered by brine (200 ml) and heat to recover isopropyl ether (60 - 70%) was recovered under vacuum. The reaction mass was cooled at 15° -20υC and pH was adjusted to 1.5 - 2.5 with ethanolic hydrochloric acid (20 - 25%, 30 ml) and was stirred 30-40 min at 20° -250C. The product obtained was centrifuged and slurry washed with absolute alcohol (200 ml). The wet material was dried under vacuum for 7 - 8 hr to obtain 57 g (66.80%) of title compound having purity of 99.30% by HPLC
Purification of 4-[2-(Di-n-propylamino) ethyll 2(3H) indolone hydrochloride
Crude ropinirole hydrochloride (250 g) was added in the mixture of di-isopropyl ether (2.5 lit), purified water (500 ml) and sodium hydroxide solution (4%, 2.0 lit) at 20°-25°C. The reaction mass was stirred for 30-35 min. at 20°- 250C. The organic layer was separated and aqueous layer was extracted (two times) with di- isopropyl ether (2 x 650 ml). The combined organic layer was washed with water (500 ml) and the solvent was recovered under vacuum. The residue left was dissolved in absolute alcohol (300 ml), cooled at 5-1O0C and pH was adjusted to 1.0 - 2.0 using ethanolic hydrochloric acid (260 ml). The reaction mass was stirred at 15-2O0C for 30- 40 minutes and filtered. The wet cake was slurried in absolute alcohol (500 ml), filtered and dried under vacuum at 65-7O0C to obtain 222 g; (88.80%) of title compound having purity of 99.91 % by HPLC
Claims
1. Ail improved process for the preparation of Ropinirole of Formula I or salt thereof.

Formula I which comprises reducing iiitro compound of Formula II,


distilling the solvent completely, washing crude Ropinirole with organic solvent, dissolving in dilute sodium hydroxide solution, and treating with ethanolic-hydrochloric acid to prepare Ropinirole hydrochloride.
2. The process according to claim 1 wherein alcoholic aqueous medium is methanol/ ethanol/ isopropanol and water or mixture of
3. The process according to claim 2 wherein alcoholic aqueous medium is mixture of ethanol and water.
4. The process according to claim 1 wherein reaction is conducted at a temperature of 25° - 6O0C.
5. The process according to claim 4 wherein reaction is preferably conducted at a temperature of 30° -5O0C.
6. The process according to claim 1 wherein organic solvent is isopropyl ether, ethyl acetate and chlorinated solvents such as methylene chloride, chloroform or mixture thereof.
7. The process according to claim 1 wherein Ropinirole hydrochloride is recrytallized from alcohols.
8. The process according to claim 7 wherein ropinirole hydrochloride is further recrytallized from absolute ethanol.
Dated day 17th of July, 2006
Dr. Asha Aggarwal,
Head-IPR
Ind-Swift Laboratories Limited
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1920DE2005 | 2005-07-22 | ||
| IN1920/DEL/2005 | 2005-07-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007010557A2 true WO2007010557A2 (en) | 2007-01-25 |
| WO2007010557A3 WO2007010557A3 (en) | 2007-11-22 |
Family
ID=37669240
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2006/000256 WO2007010557A2 (en) | 2005-07-22 | 2006-07-19 | Process for the preparation of highly pure ropinirole |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007010557A2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010091128A2 (en) | 2009-02-04 | 2010-08-12 | Qualcomm Incorporated | Methods and systems for scheduling among nodes for a data slot in wireless communication networks |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4452808A (en) * | 1982-12-07 | 1984-06-05 | Smithkline Beckman Corporation | 4-Aminoalkyl-2(3H)-indolones |
-
2006
- 2006-07-19 WO PCT/IN2006/000256 patent/WO2007010557A2/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4452808A (en) * | 1982-12-07 | 1984-06-05 | Smithkline Beckman Corporation | 4-Aminoalkyl-2(3H)-indolones |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010091128A2 (en) | 2009-02-04 | 2010-08-12 | Qualcomm Incorporated | Methods and systems for scheduling among nodes for a data slot in wireless communication networks |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007010557A3 (en) | 2007-11-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5632279B2 (en) | Preparation method and polymorph of ivabradine hydrochloride | |
| EP1706113B1 (en) | A process for the preparation of 4-(2-dipropylaminoethyl)-1,3-dihydro-2h-indol-2-one hydrochloride | |
| WO2010140168A1 (en) | Improved process for preparing temozolomide | |
| JP2011508781A (en) | Method for the synthesis of tetrahydrobiopterin | |
| CN112062767B (en) | Preparation method and intermediate of rumepilone | |
| CA2619955A1 (en) | Process for preparing valsartan | |
| US20070249835A1 (en) | Process for Preparing Rebamipide | |
| EP1912975B1 (en) | A process for the preparation of telmisartan | |
| CA2773012A1 (en) | Process for the preparation of lenalidomide | |
| WO2016055015A1 (en) | Method for preparing sitagliptin intermediate via asymmetrical reduction method | |
| WO2016174685A1 (en) | Process for the enantiomeric resolution of apremilast intermediates | |
| WO2006061364A1 (en) | Process for the preparation of carvedilol and its enantiomers | |
| RU2320655C2 (en) | Improved method for preparing alpha-polymorphous eletriptane bromohydrate | |
| JP5192707B2 (en) | Manufacturing method of mirtazapine | |
| WO2015111085A2 (en) | Processes for the preparation of eltrombopag and pharmaceutically acceptable salts, solvates and intermediates thereof | |
| WO2007010557A2 (en) | Process for the preparation of highly pure ropinirole | |
| WO2019180547A1 (en) | A process for the preparation of vigabatrin | |
| EP1861367B1 (en) | An improved process for the purification of perindopril | |
| US20120253051A1 (en) | Process for the preparation of ropinirole and salts thereof | |
| US7943784B2 (en) | Process for the preparation of almotriptan | |
| US9006453B2 (en) | Process for preparation of zolmitriptan | |
| WO2008084499A1 (en) | An industrial process for the preparation of pure ropinirole | |
| US20120022292A1 (en) | Method for preparing eplivanserin hemifumarate | |
| WO2009084030A2 (en) | Improved process for the preparation of (1-benzyl-4-(5,6,- dimethoxyind anone-2-yl)methylpiperidine) hydrochloride-form iii | |
| WO2015087239A1 (en) | Processes for the preparation of darapladib and its intermediates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06780537 Country of ref document: EP Kind code of ref document: A2 |