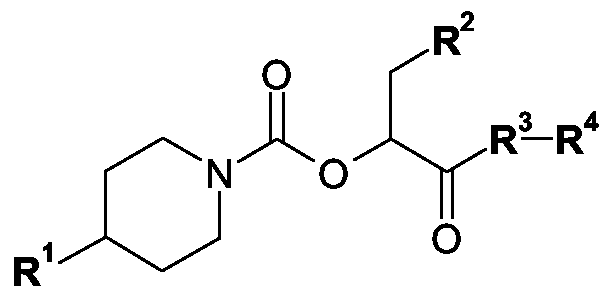
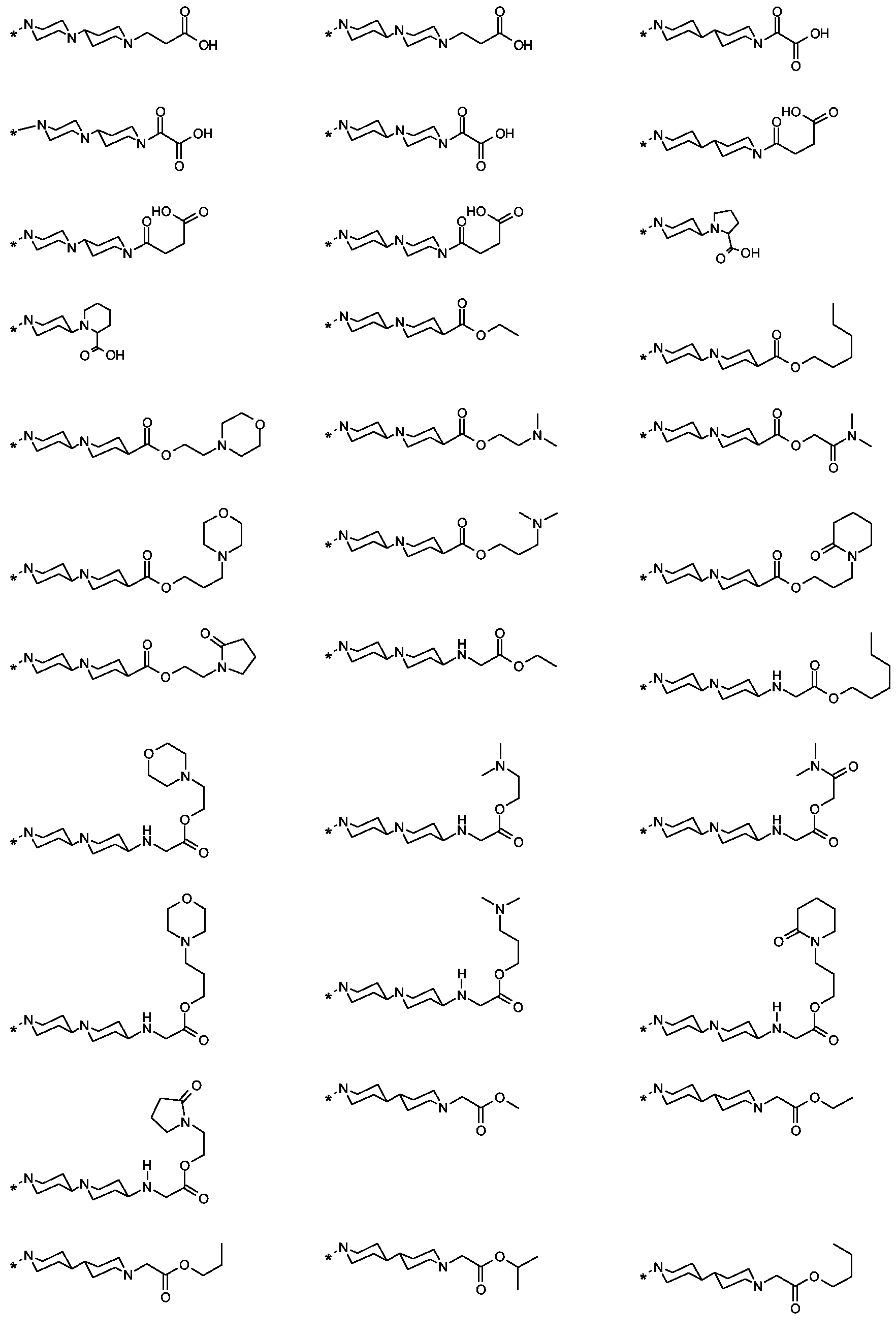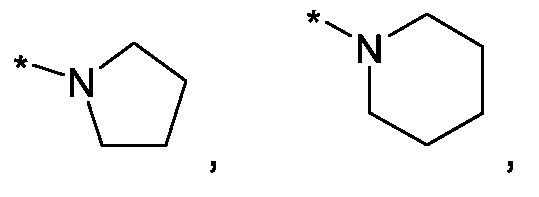AUSGEWÄHLTE CGRP-ANTAGONISTEN, VERFAHREN ZU DEREN HERSTELLUNG SOWIE DEREN VERWENDUNG ALS ARZNEIMITTEL
Gegenstand der vorliegenden Erfindung sind die CGRP-Antagonisten der allgemeinen Formel I
in der R1, R2, R3 und R4 wie in Anspruch 1 definiert sind, deren Tautomere, deren Isomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen, sowie diejenigen Verbindungen der allgemeinen Formel I, in denen ein oder mehrere Wasserstoffatome durch Deuterium ausgetauscht sind, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung.
STAND DER TECHNIK
In den internationalen Patentanmeldungen PCT/EP97/04862 und PCT/EP03/11762 werden bereits CGRP-Antagonisten zur Behandlung von Migräne beschrieben.
DETAILLIERTE BESCHREIBUNG DER ERFINDUNG
In der obigen allgemeinen Formel I bedeuten in einer ersten Ausführungsform
R1 eine Gruppe ausgewählt aus
R H oder H3C-O- darstellt,
R2 eine Gruppe ausgewählt aus
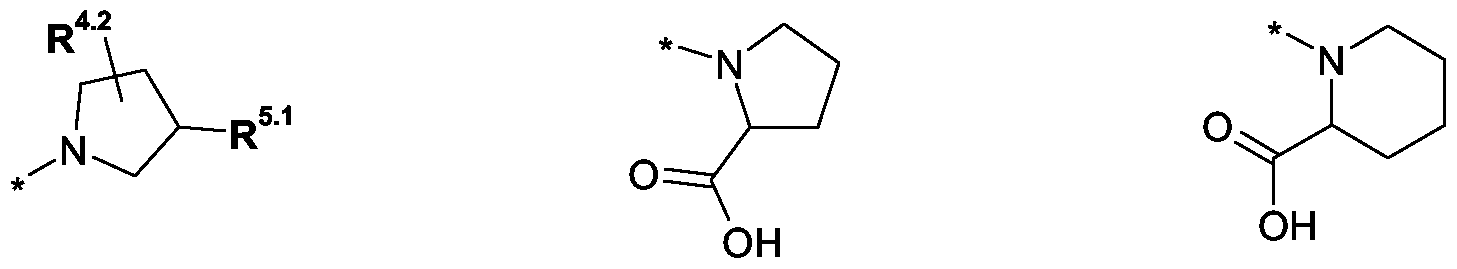
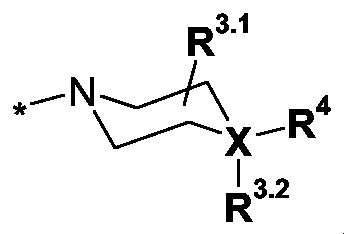
R3 eine Gruppe der allgemeinen Formel Il
worin
X N oder C,
R3.1 H, C1-3-Alkyl- oder R3.1.1-O-C(O)-,
R3.1.1 H, C1-6-Alkyl-, H2N-C2-4-alkylen-, (C1-3-Alkyl)-NH-C2-4-alkylen-, (C1-3-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-3-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder
R3.1.1.1-C2-4-alkylen-,
R3.1.1.1 eine Gruppe ausgewählt aus
R3.2 ein freies Elektronenpaar, wenn X = N ist, oder
R3.2 H, C1-3-Alkyl- oder R3.2.1-O-C(O)-, wenn X = C ist,
R3.2.1 H, C1-6-Alkyl-, H2N-C2-4-alkylen-, (C1-3-Alkyl)-NH-C2-4-alkylen-, (C1-3-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-3-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R3.2.1.1-C2-4-alkylen-,
eine Gruppe ausgewählt aus
darstellt,
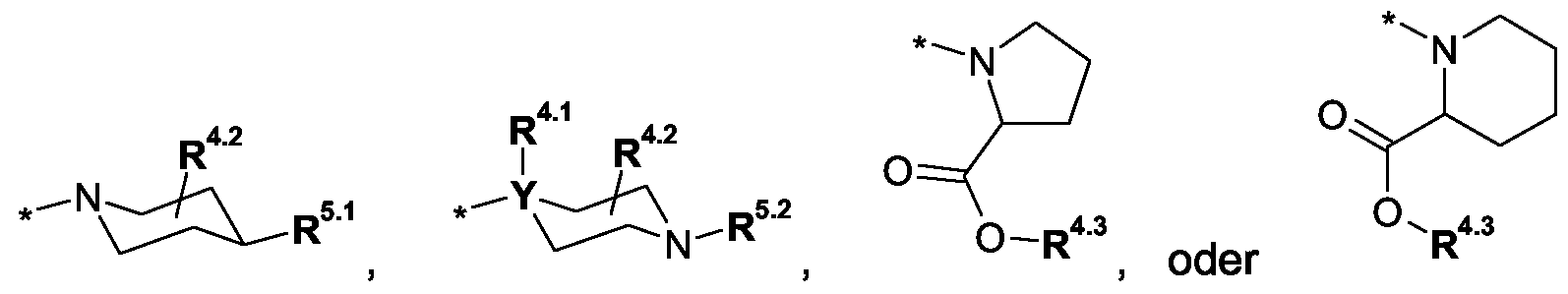
R4 eine Gruppe der allgemeinen Formeln
worin
Y C und
R4.1 H oder d-3-Alkyl-, oder
Y N und
R4.1 ein freies Elektronenpaar darstellt,
Maßgabe, dass X und Y nicht gleichzeitig N bedeuten,
R4.2 H, C1-3-Alkyl- oder R4^-O-C(O)-,
R4.2.1 H, C1-6-Alkyl-, H2N-C2-4-alkylen-, (C1-3-Alkyl)-NH-C2-4-alkylen-, (C1-3-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-3-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R4.2.1.1-C2-4-alkylen-,
R4.2.1.1 eine Gruppe ausgewählt aus
darstellt, und
R4.3 H, C1-6-Alkyl-, H2N-C2-4-alkylen-, (C1-3-Alkyl)-NH-C2-4-alkylen-, (C1-3-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-3-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R4.3.1-C2-4-alkylen-,
R4.3.1 eine Gruppe ausgewählt aus
R
5.1 H, C
1-3-Alkyl-, R
5.I.I-O-C(O)-, R
5.M-O-C(O)-C
1-3-alkylen-NH-,
R5.1.1-O-C(O)-C1-3-alkylen-N(C1-3-alkyl)-, R5.1.1-O-C(O)-C1-3-alkylen-O-, R5.1.1-O-C(O)-C1-3-alkylen-, R5.1.1-O-C(O)-C(O)-, R5.1.1-O-C(O)-C(O)-O-, R5.1.1-O-C(O)-C1-3-alkylen-C(O)- oder R5.1.1-O-C(O)-C1-3-alkylen-C(O)-O-,
R5.1.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, Pyridyl-C1-3-alkylen-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Al kyl)-NH-C2-4-alkylen-, (C1-6-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Al kyl)-NH-CCOJ-C1-3-alkylen-, (C1-6-Alkyl)2N-CCOJ-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-,
R5.1.1.1-C(O)-C1-3-alkylen- oder R5.1.1.2.c2.4-alkylen-,
R5 eine Gruppe ausgewählt aus
R 5.i.i.2 ejne Qruppe ausgewählt aus
R5.2 H, C1-3-Alkyl-, R5.2.1-O-C(O)-C1-3-alkylen-NH-, R5.2.1-O-C(O)-C1-3-alkylen-O-, R5.2.1-O-C(O)-C1-3-alkylen-, R5.2.I-O-C(O)-C(O)- oder
R5.2.1-O-C(O)-C1-3-alkylen-C(O)-,
R5.2.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, Pyridyl-C1-3-alkylen-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Alkyl)-NH-C2-4-alkylen-, (C1-6-Alkyl)2N-C2-4-alkylen-,
^N-C^J-C1-3-alkylen-, (C1-6-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-, R5.2.1.1-C(O)-C1-3-alkylen- oder R^ '^-C2-4-alkylen-,
R eine Gruppe ausgewählt aus
)5.Z.1.Z eine Gruppe ausgewählt aus
eine Gruppe der Formel
darstellt mit der Maßgabe, dass mindestens einer der Reste R3 1, R32, R42, R51 oder R52 einen anderen Rest als H oder d-3-Alkyl- bedeuten,
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine bevorzugte erste Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
R eine Gruppe ausgewählt aus
eine Gruppe der allgemeinen Formel Il
worin
X N oder C,
R3.1 H, C1-3-Alkyl- oder HO-C(O)-,
R3.2 ein freies Elektronenpaar, wenn X = N ist, oder
R3.2 H, C1-3-Alkyl- oder HO-C(O)-, wenn X = C ist, darstellt,
R4 eine Gruppe der allgemeinen Formeln III
worin
Y C und
R4.1 H oder C1-3-Alkyl-, oder
Y N und
R4.1 ein freies Elektronenpaar darstellt,
mit der Maßgabe, dass X und Y nicht gleichzeitig N bedeuten,
R4.2 H, C1-3-Alkyl- oder HO-C(O)-,
l5.1 H, C1-3-Alkyl-, HO-C(O)-, HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-N(C1-3-alkyl)-, HO-C(O)-C1-3-alkylen-O-, HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)-,HO-C(O)-C(O)-O-, HO-C(O)-C1-3-alkylen-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-O-,
»52 H, C1-3-Alkyl-, HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-O-, HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-,
darstellt mit der Maßgabe, dass mindestens einer der Reste R3 1, R32, R42, R51 oder R52 einen anderen Rest als H oder d-3-Alkyl- bedeuten,
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine zweite Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
R eine Gruppe ausgewählt aus
eine Gruppe der allgemeinen Formel Il
worin
N oder C,
13.1 H, C1-3-Alkyl- oder R3 1 MO)C-,
»3.1.1 HO, C1-6-Alkyl-O-,
R ein freies Elektronenpaar, wenn X = N ist, oder
R3.2 H oder C1-3-Alkyl-, wenn X = C ist, darstellt,
R4 eine Gruppe der allgemeinen Formeln III
worin
Y C und
R4-1 H oder Ci-3-Alkyl-, oder
Y N und
R4.1 ein freies Elektronenpaar darstellt,
mit der Maßgabe, dass X und Y nicht gleichzeitig N bedeuten,
R4.2 H, C1-3-Alkyl- oder R4^-(O)C-,
R -,44-.22-.11 HO, C1-6-Alkyl-O- darstellt, und
R4.3 H, C1-6-Alkyl-, H2N-C2-4-alkylen-, (C1-3-Alkyl)-NH-C2-4-alkylen-,
(C1-3-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-3-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder
R4.3.1-C2-4-alkylen-,
R
4.3.1 eine Gruppe ausgewählt aus
R5.I RM-^o-C(O)-, R5.1.1-O-C(O)-C1-3-alkylen-NH-, R5.M-O-C(O)-C1-3-alkylen-O-,
R5.1.1-O-C(O)-C1-3-alkylen-, R5.I.I-O-C(O)-C(O)- oder R5.1.1-O-C(O)-C1-3-alkylen-C(O)-,
R5.1.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, Pyridyl-C1-3-alkylen-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Alkyl)-NH-C2-4-alkylen-, (C1-6-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-CCOJ-O-C1-3-alkylen-, C1-6-Alkyl-O-CCOJ-O-C1-3-alkylen-, R5.1.1.1-C(O)-C1-3-alkylen- oder RM-^-c^-alkylen-,
R5.1.1.1 ejne Gruppe ausgewählt aus
R eine Gruppe ausgewählt aus
R5.2 R^-O-CfCO-C1-3-alkylen-NH-, R^-'-O-CCOJ-C1-3-alkylen-O-, R5.2.1-O-C(O)-C1-3-alkylen-, R5 Z1-O-C(O)-C(O)- oder R5.2.1-O-C(O)-C1-3-alkylen-C(O)-,
R5.2.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, Pyridyl-C1-3-alkylen-, HO-C2-4-alkylen-, C1-e-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Alkyl)-NH-C2-4-alkylen-,
(C1-e-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Al kyl)-NH-CCOJ-C1-3-alkylen-, (C1-6-Alkyl)2N-CCOJ-C1-3-alkylen-,
C1-6-Alkyl-CCOJ-O-C1-3-alkylen-, C1-6-Alkyl-O-CCOJ-O-C1-3-alkylen-, RS-2.1.1.Q(O)-C1.3-alkylen- oder R5.2.1.2-C2-4-alkylen-,
R eine Gruppe ausgewählt aus
eine Gruppe ausgewählt aus
bedeuten,
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine bevorzugte zweite Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
eine Gruppe ausgewählt aus
eine Gruppe der allgemeinen Formel Il
worin
N oder C,
13.1 H, C1-3-Alkyl- oder HO-C(O)-,
ein freies Elektronenpaar, wenn X = N ist, oder
»3 2 H oder C1-3-Alkyl-, wenn X = C ist, darstellt,
R4 eine Gruppe der allgemeinen Formeln
worin
C und
H oder d-3-Alkyl-, oder
N und
R4.1 ein freies Elektronenpaar darstellt,
mit der Maßgabe, dass X und Y nicht gleichzeitig N bedeuten,
R4.2 H, C1-3-Alkyl- oder HO-C(O)-,
f" HO-C(O)-, HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-O-,
HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-,
R5.2 HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-O-, HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-d-3-alkylen-C(O)-,
bedeuten,
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine dritte Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
eine Gruppe ausgewählt aus
R3-R4 zusammen eine Gruppe der allgemeinen Formeln IV
worin
R3.1 H, H3C- oder F?ΛΛ-O-C(O)-,
R3.1.1 H, C1-6-Alkyl-, (C1-3-Alkyl)2N-C2-4-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R3.1.1.1-C2-4-alkylen-,
R3.1.1.1 eine Gruppe ausgewählt aus
R3.2 H, C1-3-Alkyl- oder R32^-O-C(O)-,
R3.2.1 H, C1-6-Alkyl-, (C1-3-Alkyl)2N-C2-4-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R3.2.1.1-C2-4-alkylen-,
R3.2.1.1 eine Gruppe ausgewählt aus
R4.1 H oder C1-3-Alkyl-,
R4.2 H, C1-3-Alkyl- oder R4^-O-C(O)-,
R4.2.1 H, C1-6-Alkyl-, (C1-3-Alkyl)2N-C2-4-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R4.2.1.1-C2-4-alkylen-,
R4.2.1.1 eine Gruppe ausgewählt aus
R4.3 H, C1-6-Alkyl-, H2N-C2-4-alkylen-, (C1-3-Alkyl)-NH-C2-4-alkylen-,
(C1-3-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-,
(C1-3-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R4.3.1-C2-4-alkylen-,
r eine Gruppe ausgewählt aus
R5.1 H, H3C-, R5.I.I-O-C(O)-, R5.M-O-C(O)-C1-3-alkylen-NH-,
R5.1.1-O-C(O)-C1-3-alkylen-N(C1-3-alkyl)-, R5.M-O-C(O)-C1-3-alkylen-O-, R5.1.1-O-C(O)-C1-3-alkylen-, R5.1.1-O-C(O)-C(O)-, R5.1.1-O-C(O)-C(O)-O-, R5.1.1-O-C(O)-C1-3-alkylen-C(O)- oder R5.1.1-O-C(O)-C1-3-alkylen-C(O)-O-,
R5.1.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, Pyridyl-C1-3-alkylen-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Alkyl)-NH-C2-4-alkylen-, (C1-6-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-, R5.1.1.1-C(O)-C1-3-alkylen- oder R^'-'^-C2-4-alkylen-,
R5 eine Gruppe ausgewählt aus
R 5.i.i.2 ejne Qruppe ausgewählt aus
R5.2 H, H3C-, R5.2.1-O-C(O)-C1-3-alkylen-NH-, R5.2.1-O-C(O)-C1-3-alkylen-O-, R5.2.1-O-C(O)-C1-3-alkylen-, R5.2.I-O-C(O)-C(O)- oder R5.2.1-O-C(O)-C1-3-alkylen-C(O)-,
R5.2.1 H, d-s-Alkyl-, Phenyl-, Indanyl-, Pyridyl-C1-3-alkylen-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Al kyl)-NH-C2-4-alkylen-, (C1-6-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Alkyl)-NH-C(O)-C1-3-alkylen-,
(C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-, R5.2.1.1-C(O)-C1-3-alkylen- oder R5.2.1.2-C2-4-alkylen-,
R52.1.1 eine Gruppe ausgewählt aus
R eine Gruppe ausgewählt aus
R5.2.1 eine Gruppe der Formel
bedeutet, mit der Maßgabe, dass mindestens einer der Reste R3 1, R32, R42, R51 oder R52 einen anderen Rest als H oder C1-3-Alkyl- bedeuten,
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine bevorzugte dritte Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
eine Gruppe ausgewählt aus
R3-R4 zusammen eine Gruppe der allgemeinen Formeln IV
worin
>3.1 H, H3C- oder HO-C(O)-,
,3.2 H, C1-3-Alkyl- oder HO-C(O)-,
R4.1 H oder C1-3-Alkyl-,
R4.2 H, C1-3-Alkyl- oder HO-C(O)-,
R5.1 H, H3C-, HO-C(O)-, HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-N(C1-3-alkyl)-, HO-C(O)-C1-3-alkylen-O-, HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)-, HO-C(O)-C(O)-O- HO-C(O)-C1-3-alkylen-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-O-,
R5.2 H, H3C-, HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-O-,
HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-,
bedeutet, mit der Maßgabe, dass mindestens einer der Reste R3-1, R32, R42, R51 oder R52 einen anderen Rest als H, H3C- oder C1-3-Alkyl- bedeutet,
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine vierte Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
R2 eine Gruppe ausgewählt aus
R3-R4 zusammen eine Gruppe der allgemeinen Formeln IV
worin
R3.1 H1
R3.2 H oder C1-3-Alkyl-,
R4.1 H oder C1-3-Alkyl-,
R4.2 H,
R4.3 H, C1-6-Alkyl-, H2N-C2-4-alkylen-, (C1-3-Alkyl)-NH-C2-4-alkylen-, (C1-3-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-3-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R4.3.1-C2-4-alkylen-,
j4.3.1 eine Gruppe ausgewählt aus
darstellt,
R5.I RM-^o-C(O)-, R5.1.1-O-C(O)-C1-3-alkylen-NH-, R5.1.1-O-C(O)-C1-3-alkylen-O-, R5.1.1-O-C(O)-C1-3-alkylen-, R5.I.I-O-C(O)-C(O)- oder R5.1.1-O-C(O)-C1-3-alkylen-C(O)-,
R5.1.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, Pyridyl-C1-3-alkylen-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Alkyl)-NH-C2-4-alkylen-,
(C1-6-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Al kyl)-NH-CCOJ-C1-3-alkylen-, (C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-, R5.1.1.1-C(O)-C1-3-alkylen- oder R5.1.1.2-C2-4-alkylen-,
»5.1.1.1 eine Gruppe ausgewählt aus
R5 eine Gruppe ausgewählt aus
R5.2 R5.2.1.o-C(O)-C1-3-alkylen-NH-, R5.2.1-O-C(O)-C1-3-alkylen-O-, R5.2.1-O-C(O)-C1-3-alkylen-, R5.2.I-O-C(O)-C(O)- oder R5.2.1-O-C(O)-C1-3-alkylen-C(O)-,
R5.2.1 H, d-s-Alkyl-, Phenyl-, Indanyl-, Pyridyl-C1-3-alkylen-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Alkyl)-NH-C2-4-alkylen-, (C1-6-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-,
R5.2.1.1-C(O)-C1-3-alkylen- oder R".1.2-C2-4-alkylen-,
R5 eine Gruppe ausgewählt aus
R 5.
2.
i.
2 ejne Q
rup
pe ausgewählt aus
bedeutet, deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine bevorzugte vierte Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
eine Gruppe ausgewählt aus
R3-R4 zusammen eine Gruppe der allgemeinen Formeln IV
worin
,3.1 H1
j3.Z H oder C1-3-Alkyl-,
R4.1 H oder C1-3-Alkyl-,
R4.2 H1
R5.1 HO-C(O)-, HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-O-,
HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-,
R5^ HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-O-, HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-,
bedeutet, deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine fünfte Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R
1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
eine Gruppe ausgewählt aus
R3-R4 zusammen eine Gruppe der allgemeinen Formeln IV
worin
R3.1 H oder R3.1.1-O-C(O)-,
R3.1.1 H, C1-6-Alkyl-, (C1-3-Alkyl)2N-C2-4-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R3.1.1.1-C2-4-alkylen-,
R3.1.1.1 eine Gruppe
R3.2 H, H3C- oder R32^-O-C(O)-,
R3.2.1 H, C1-6-Alkyl-, (C1-3-Alkyl)2N-C2-4-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R3.2.1.1-C2-4-alkylen-,
R42 H oder H3C-,
R4.3 H, C1-6-Alkyl-, (C1-3-Alkyl)2N-C2-4-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R4.3.1-C2-4-alkylen-,
R4.3.1 eine Gruppe ausgewählt aus
R5.1 H, H3C-, R5.I.I-O-C(O)-, R5.1.1-O-C(O)-C1-3-alkylen-NH-,
R5.1.1-O-C(O)-C1-3-alkylen-N(C1-3-alkyl)-, R5.1.1-O-C(O)-C1-3-alkylen-O-, R5.1.1-O-C(O)-C1-3-alkylen-, R5.1.1-O-C(O)-C(O)-, R5.1.1-O-C(O)-C(O)-O- R5.1.1-O-C(O)-C1-3-alkylen-C(O)- oder R5.1.1-O-C(O)-C1-3-alkylen-C(O)-O-,
R5.1.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, PyHdYl-CH2-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Alkyl)-NH-C2-4-alkylen-, (C1-e-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-,
R5.1.1.1-C(O)-C1-3-alkylen- oder R5.1.1.2-C2-4-alkylen-,
R5.1.1.1 eine Gruppe ausgewählt aus
R
5.1.1.2 eine Gruppe ausgewählt aus
R5.2 H, H3C-, R^-O-CfCO-C1-3-alkylen-, R5 Z1-O-C(O)-C(O)- oder
R5.2.1-O-C(O)-C1-3-alkylen-C(O)-,
R5-2-1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, PyHdYl-CH2-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Al kyl)-NH-C2-4-alkylen-, (C1-6-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Alkyl)-NH-C(0)-C1-3-alkylen-, (C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-, R5.2.1.1-C(O)-C1-3-alkylen- oder R5.2.1.2-C2-4-alkylen-,
,5.2.1.1 eine Gruppe ausgewählt aus
R eine Gruppe ausgewählt aus
R eine Gruppe der Formel
bedeutet, mit der Maßgabe, dass mindestens einer der Reste R3 1, R32, R42, R51 oder R52 einen anderen Rest als H, H3C- oder d-3-Alkyl- bedeutet,
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine bevorzugte fünfte Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
R eine Gruppe ausgewählt aus
R3-R4 zusammen eine Gruppe der allgemeinen Formeln IV
worin
R3.1 H oder HO-C(O)-,
R3.2 H, H3C- oder HO-C(O)-,
R42 H oder H3C-,
»5.1 HO-C(O)-, HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-N(C1-3-alkyl)-, HO-C(O)-C1-3-alkylen-O-, HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)-, HO-C(O)-C(O)-O-, HO-C(O)-C1-3-alkylen-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-O-,
»52 HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-,
bedeuten, mit der Maßgabe, dass mindestens einer der Reste R3-1, R32, R42, R51 oder R5.2 einen anderen Rest als H, H3C- oder d-3-Alkyl- bedeutet,
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine sechste Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
R eine Gruppe ausgewählt aus
R
3-R
4 zusammen eine Gruppe der allgemeinen Formeln IV
worin
R4.3 H, C1-6-Alkyl-, (C1-3-Alkyl)2N-C2-4-alkylen-, (C1-3-Alkyl)2N-C(O)-C1-3-alkylen- oder R4.3.1-C2-4-alkylen-,
j4.3.1 eine Gruppe ausgewählt aus
R5.I RM-^o-C(O)-, R5.1.1-O-C(O)-C1-3-alkylen-NH-, R5.1.1-O-C(O)-C1-3-alkylen-O-, R5.1.1-O-C(O)-C1-3-alkylen-, R5.I.I-O-C(O)-C(O)- oder R5.1.1-O-C(O)-C1-3-alkylen-C(O)-,
R5.1.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, PyHdYl-CH2-, HO-C2-4-alkylen-, C1-6-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (C1-6-Alkyl)-NH-C2-4-alkylen-, (C1-e-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Alkyl)-NH-C(O)-C1-3-alkylen-, (C1-6-Alkyl)2N-C(O)-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-, R5.1.1.1-C(O)-C1-3-alkylen- oder R5.1.1.2-C2-4-alkylen-,
,5.1.1.1 eine Gruppe ausgewählt aus
j5.i.i.2 ejne Gruppe ausgewählt aus
R5.2 R5.2.1.O-C(O)-C1-3-alkylen-, R5 Z1-O-C(O)-C(O)- oder R5.2.1-O-C(O)-C1-3-alkylen-C(O)-,
R5.2.1 H, C1-8-Alkyl-, Phenyl-, Indanyl-, PyHdYl-CH2-, HO-C2-4-alkylen-, C1-e-Alkyl-O-C2-4-alkylen-, H2N-C2-4-alkylen-, (d-e-Alkyl)-NH-C2-4-alkylen-,
(C1-6-Alkyl)2N-C2-4-alkylen-, H2N-C(O)-C1-3-alkylen-, (C1-6-Al kyl)-NH-CCOJ-C1-3-alkylen-, (C1-6-Alkyl)2N-CCOJ-C1-3-alkylen-, C1-6-Alkyl-C(O)-O-C1-3-alkylen-, C1-6-Alkyl-O-C(O)-O-C1-3-alkylen-, R5.2.1.1-C(O)-C1-3-alkylen- oder R^-^-C2-4-alkylen-,
R 5.2.i.i ejne Qruppe ausgewählt aus
R 5.2.i.2 ejne Qruppe ausgewählt aus
bedeutet, deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine bevorzugte sechste Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
eine Gruppe ausgewählt aus
R3-R4 zusammen eine Gruppe der allgemeinen Formeln IV
R5.1 HO-C(O)-, HO-C(O)-C1-3-alkylen-NH-, HO-C(O)-C1-3-alkylen-O-,
HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-,
l5.2 HO-C(O)-C1-3-alkylen-, HO-C(O)-C(O)- oder HO-C(O)-C1-3-alkylen-C(O)-,
bedeutet, deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
Eine siebte Ausführungsform der vorliegenden Erfindung besteht in den Verbindungen der obigen allgemeinen Formel I, in denen
R1 eine Gruppe ausgewählt aus
worin
H oder H3C-O- darstellt,
eine Gruppe ausgewählt aus
R -R zusammen eine Gruppe der Formeln IV
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
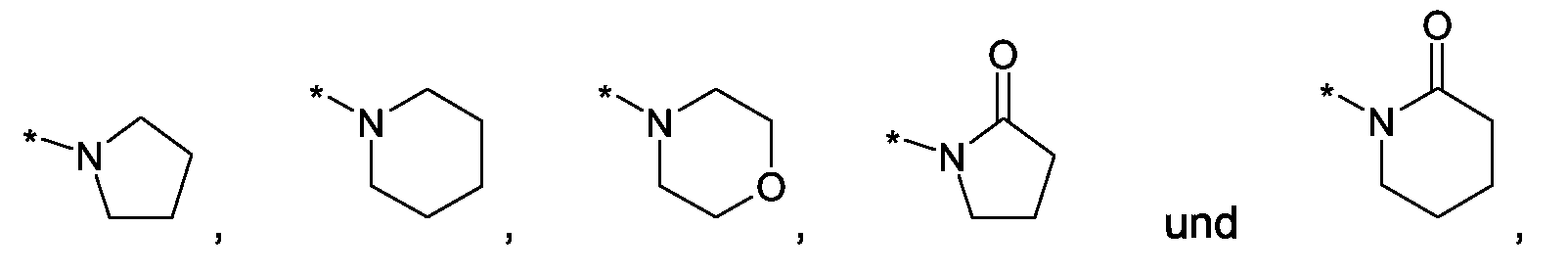
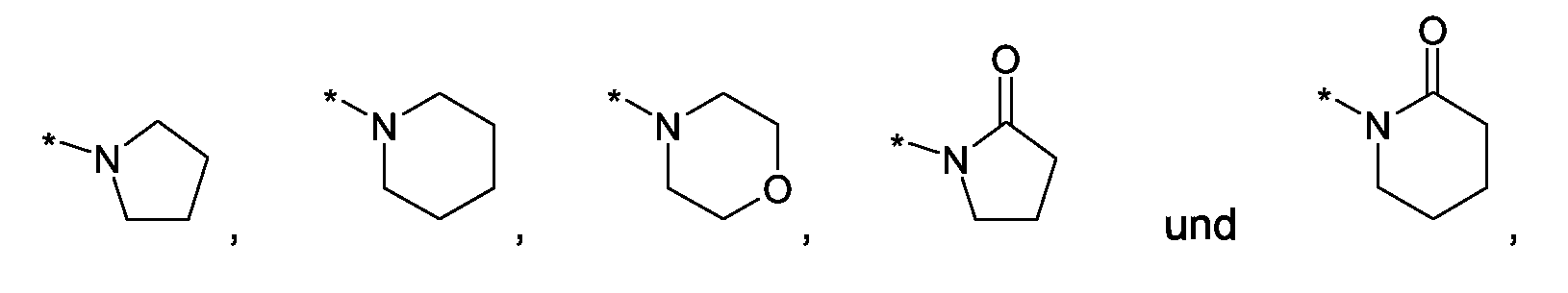
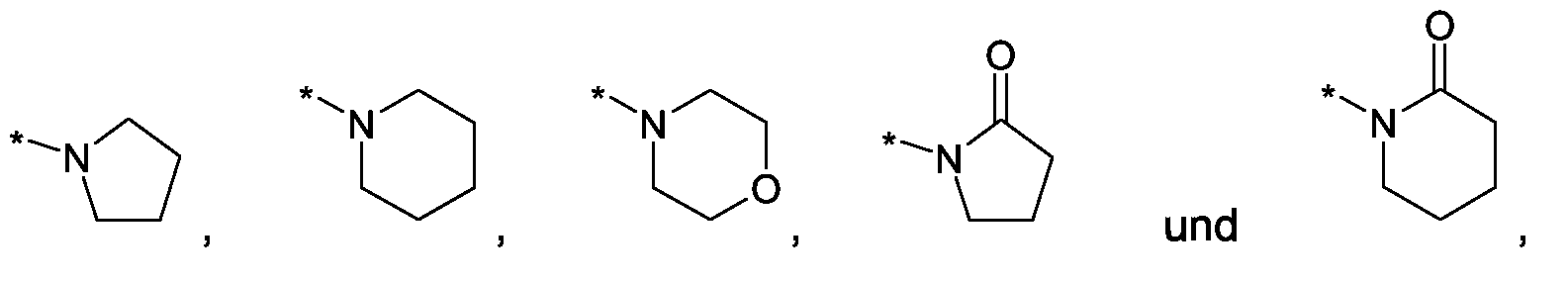
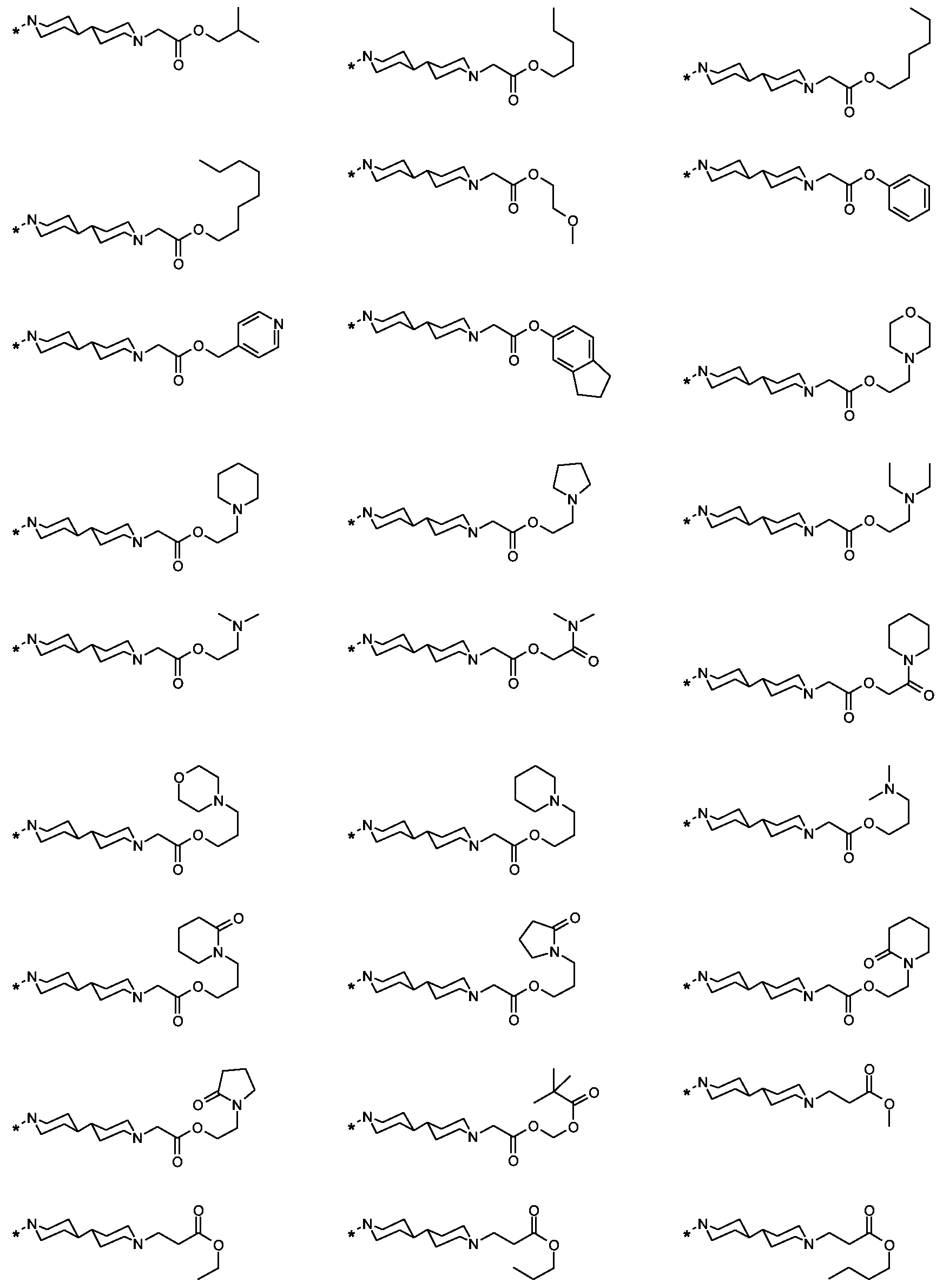
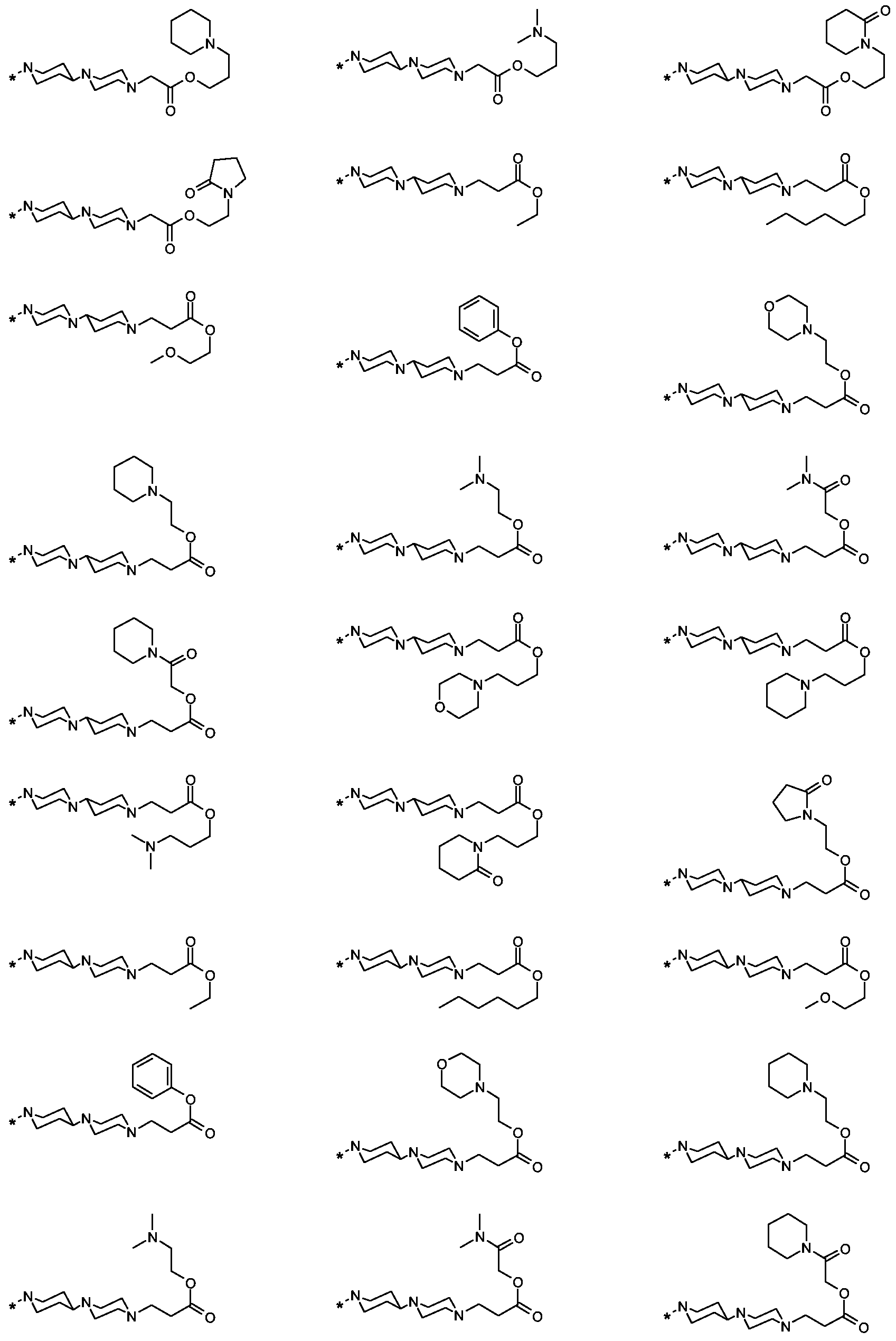
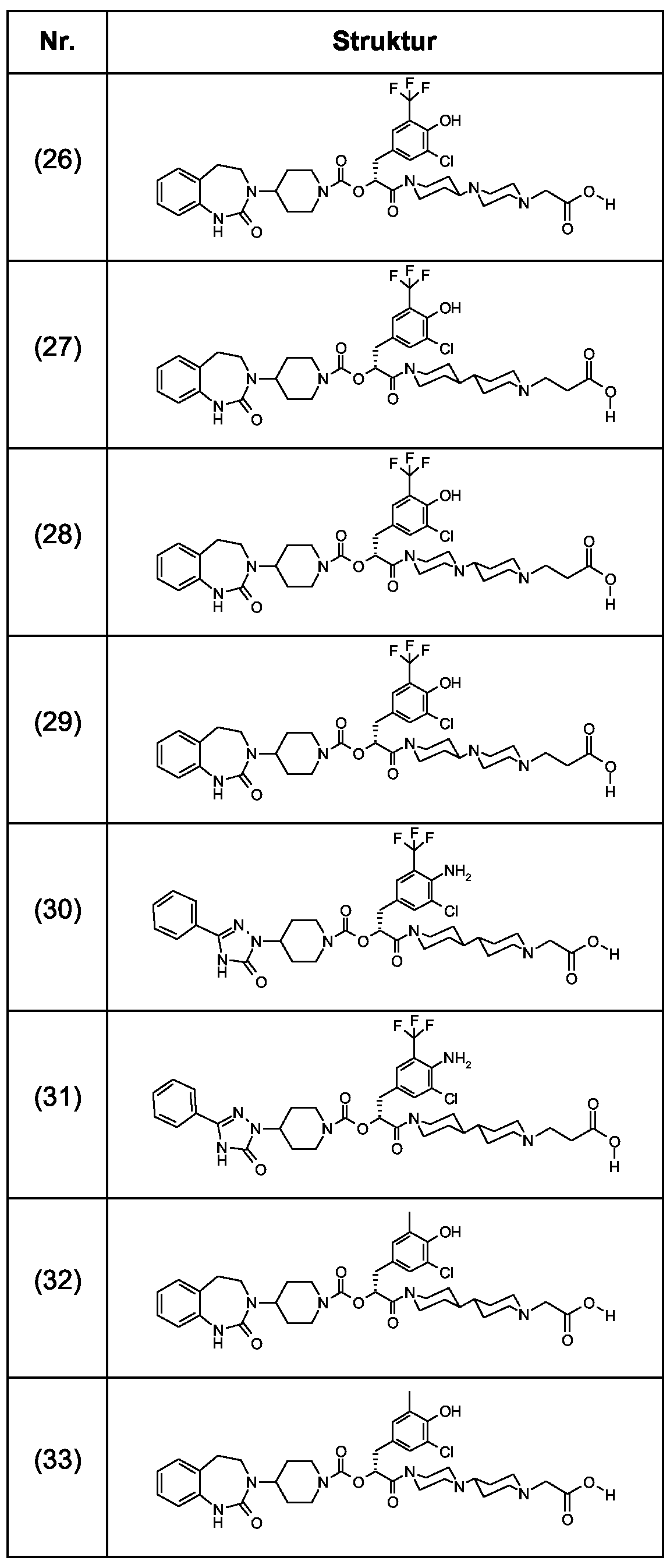
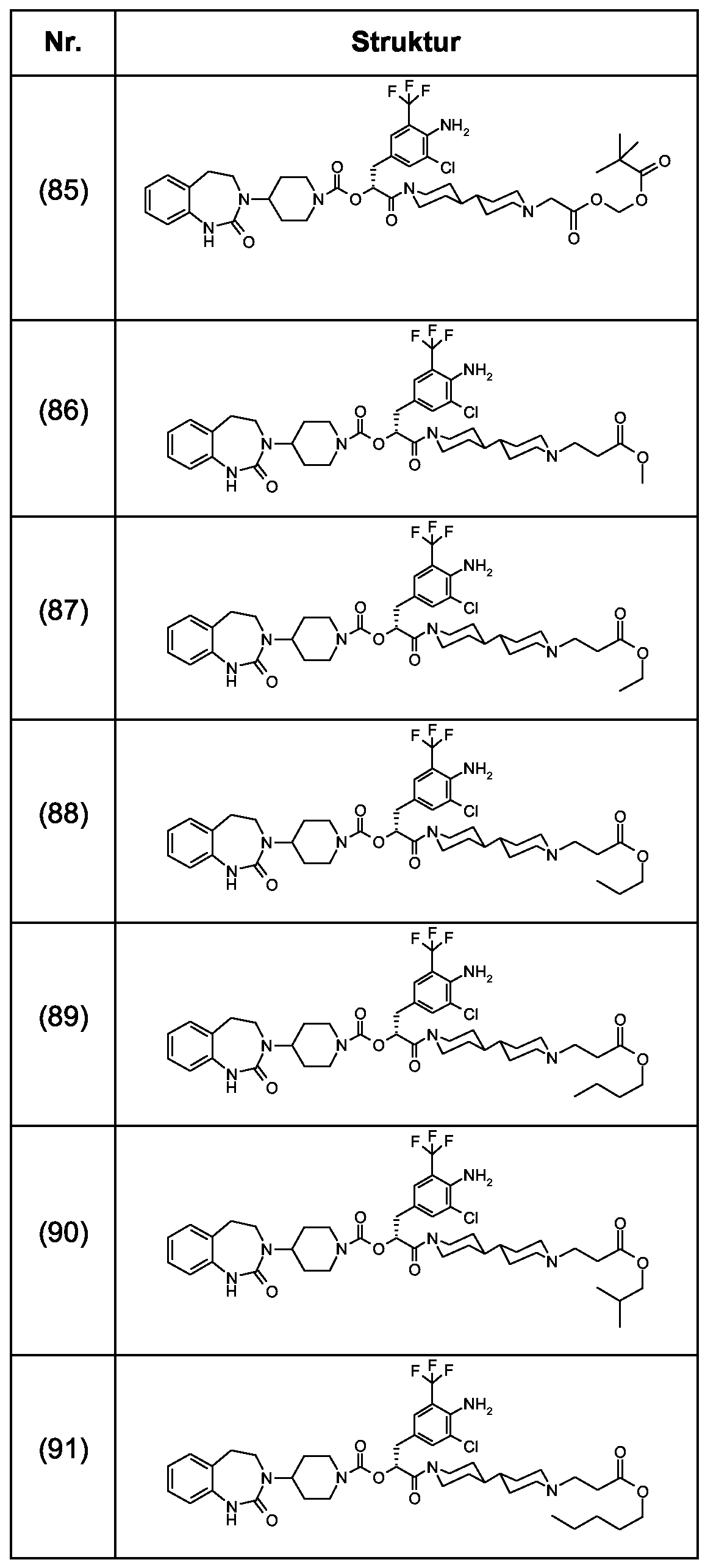
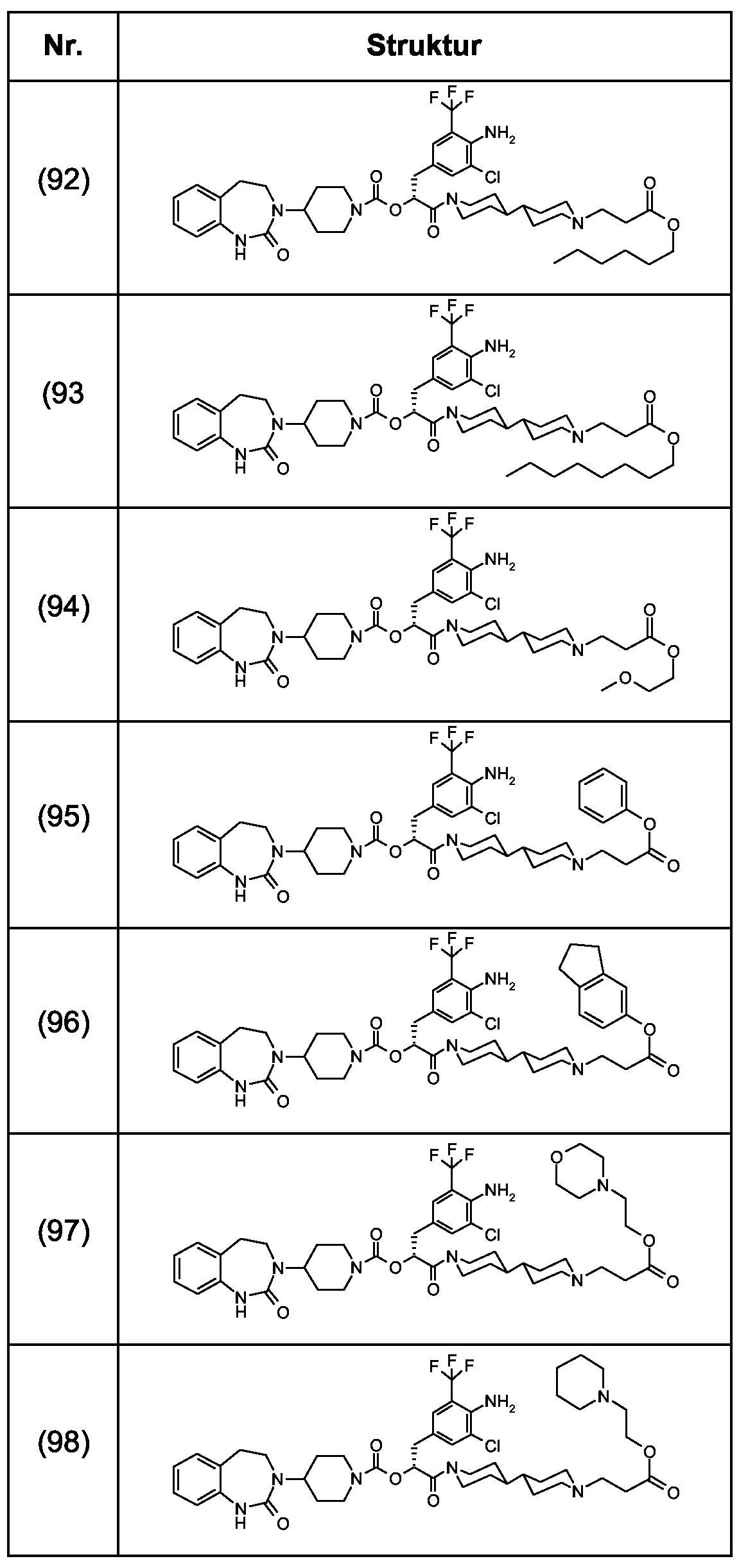
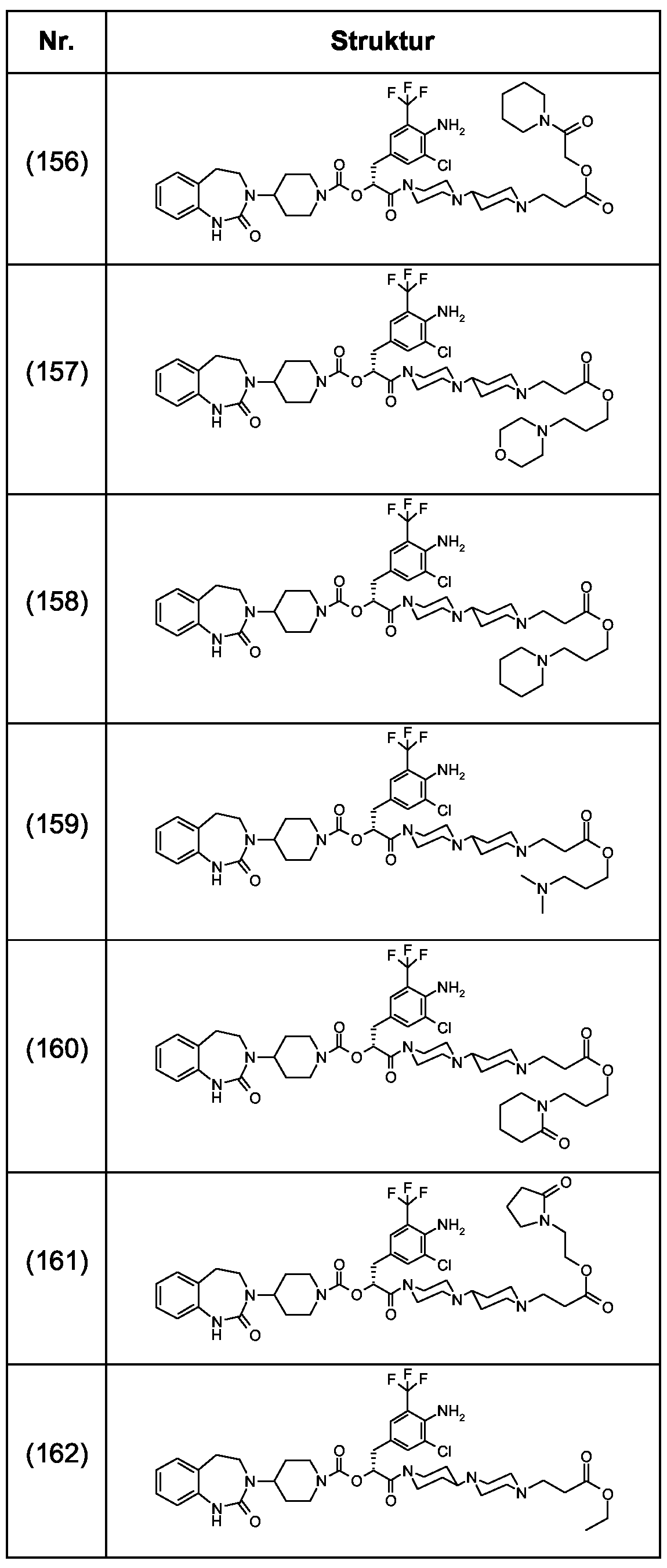
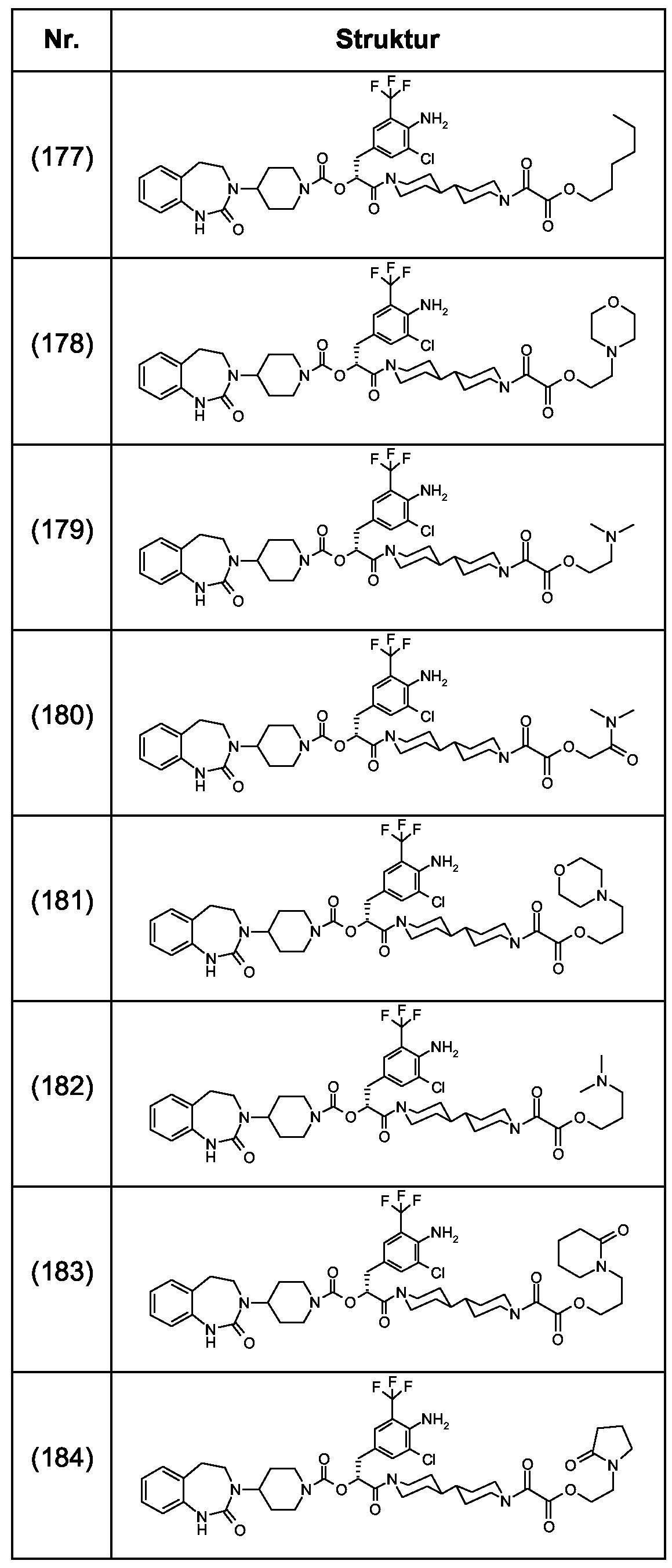
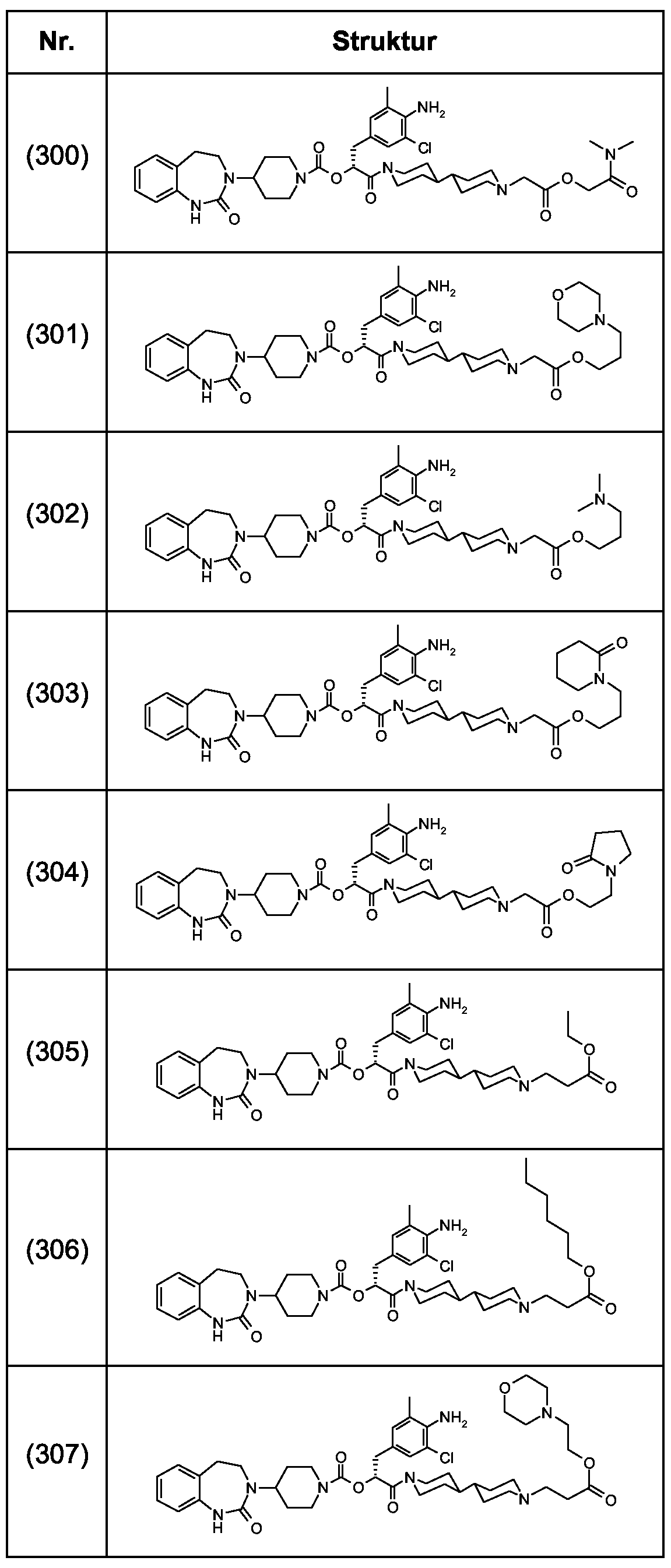
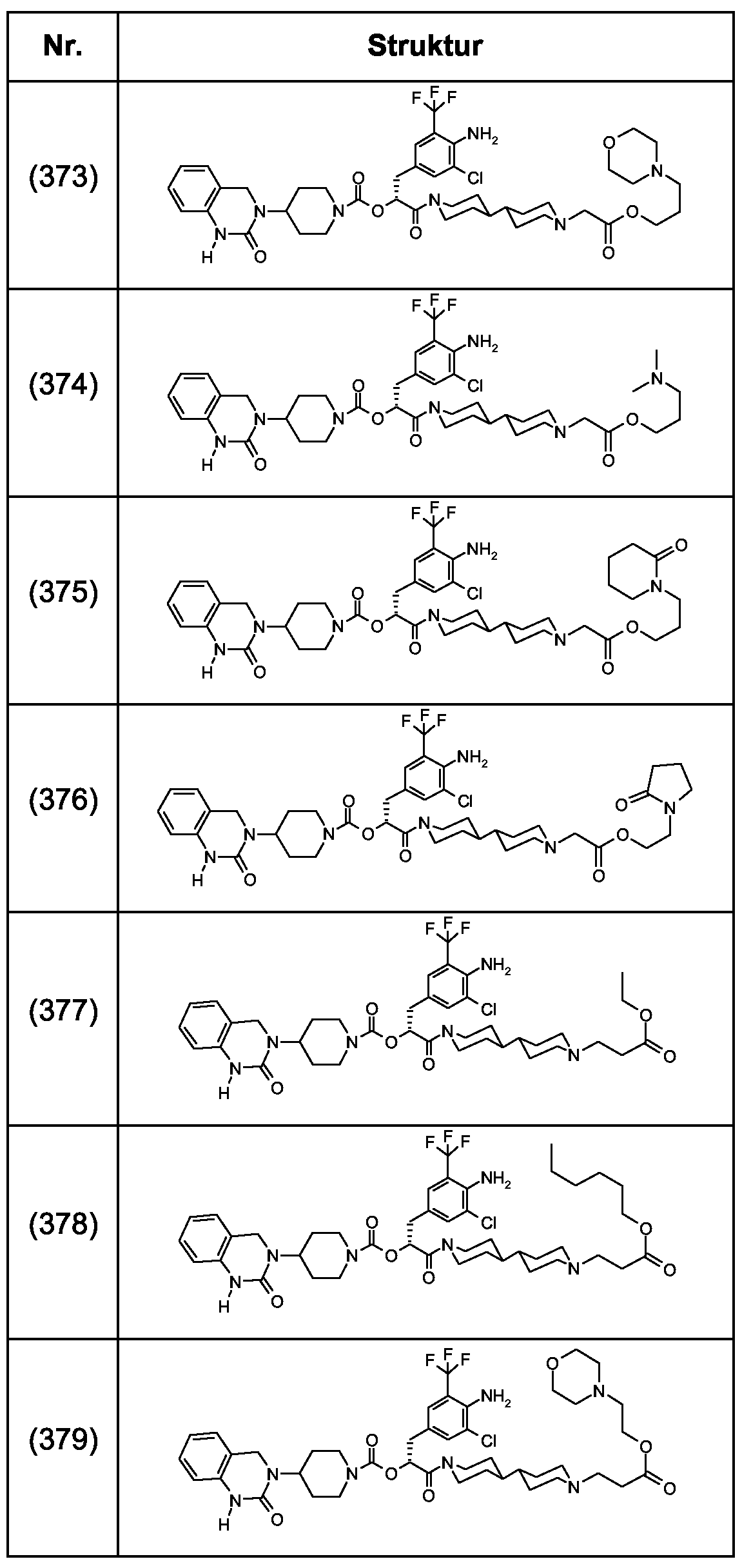
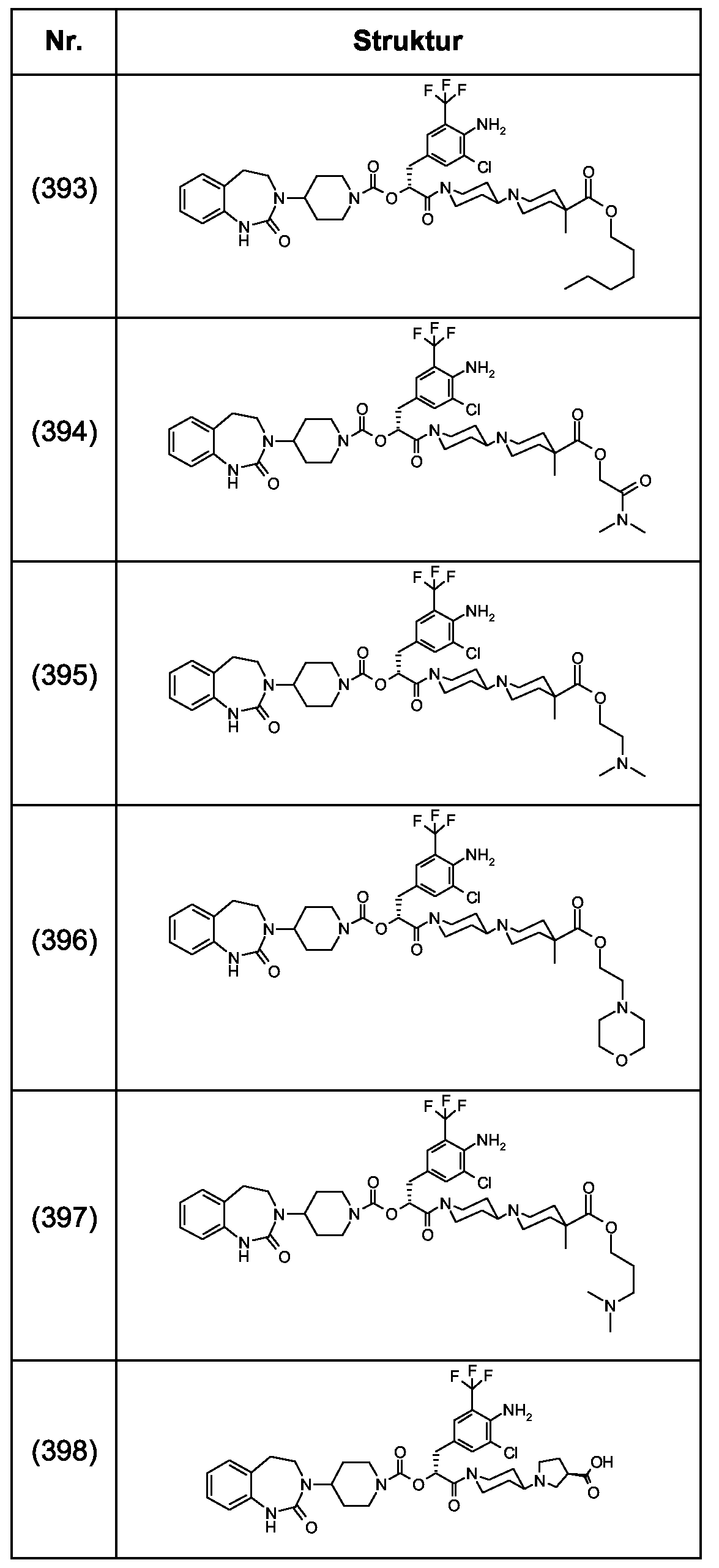
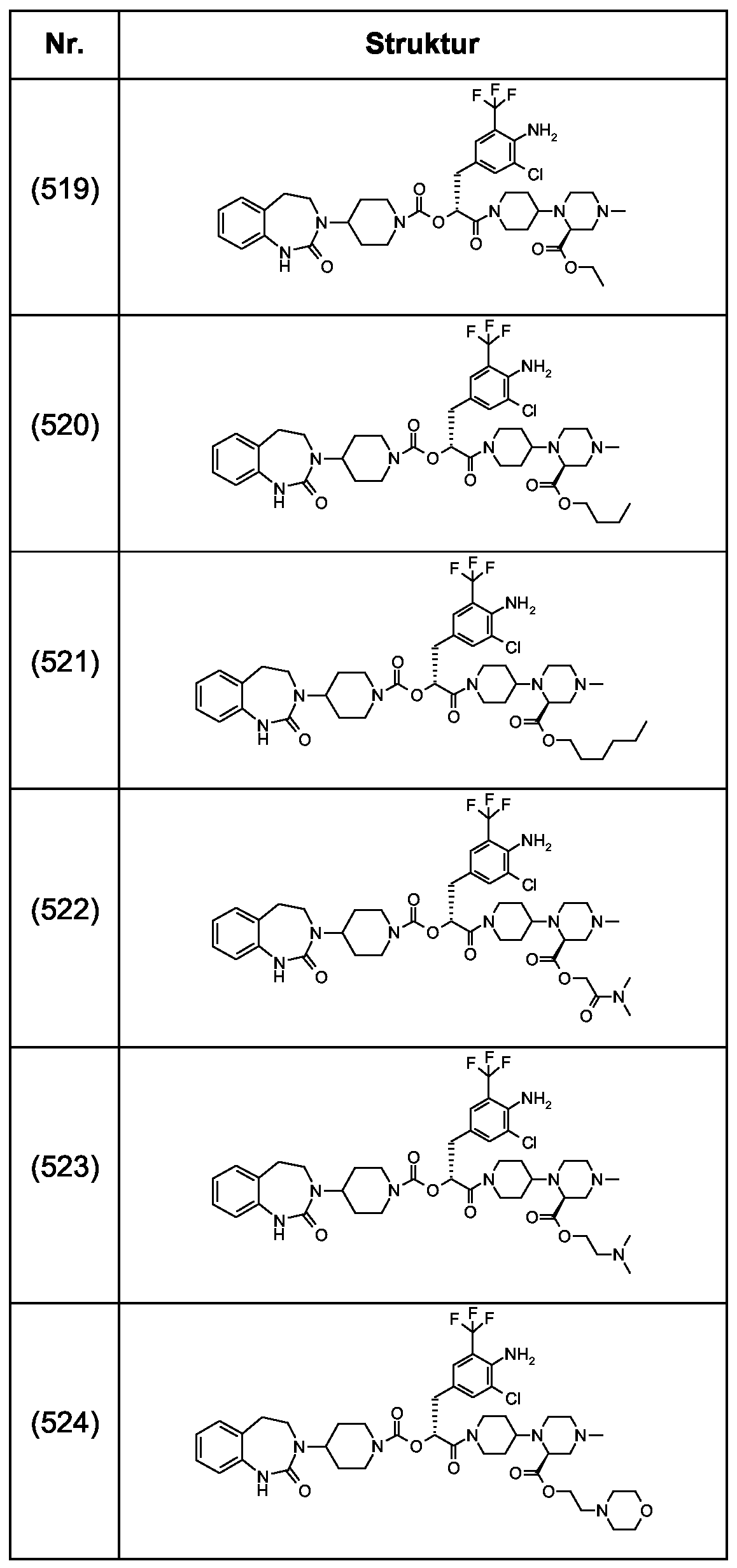
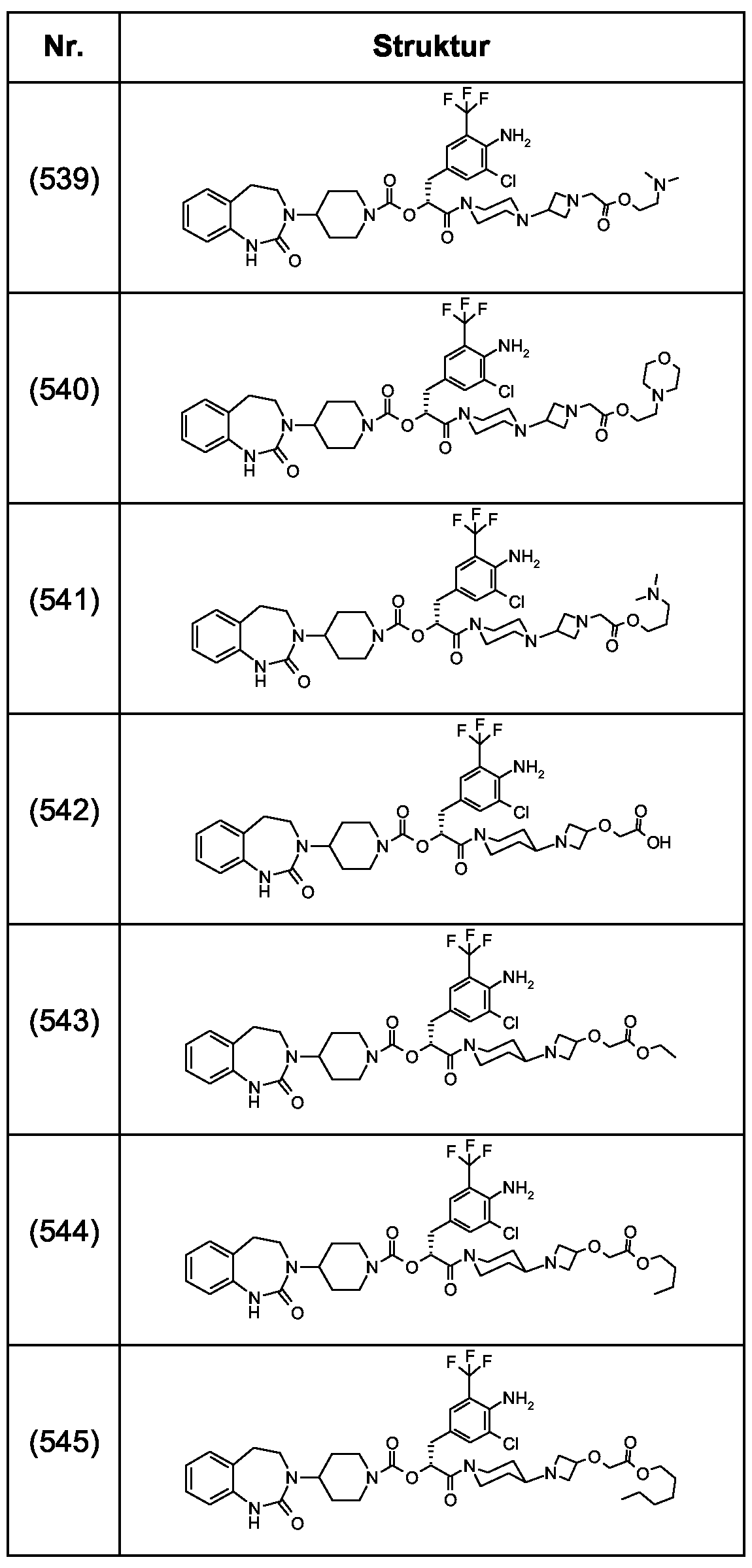
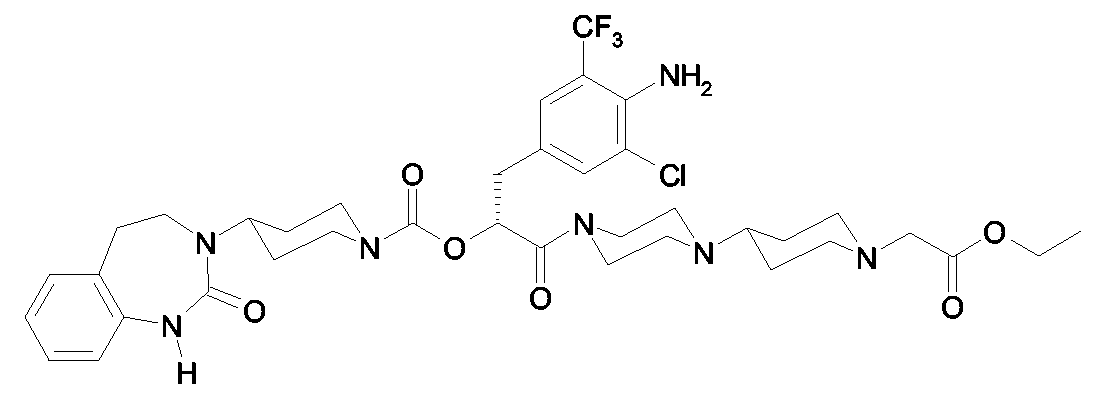
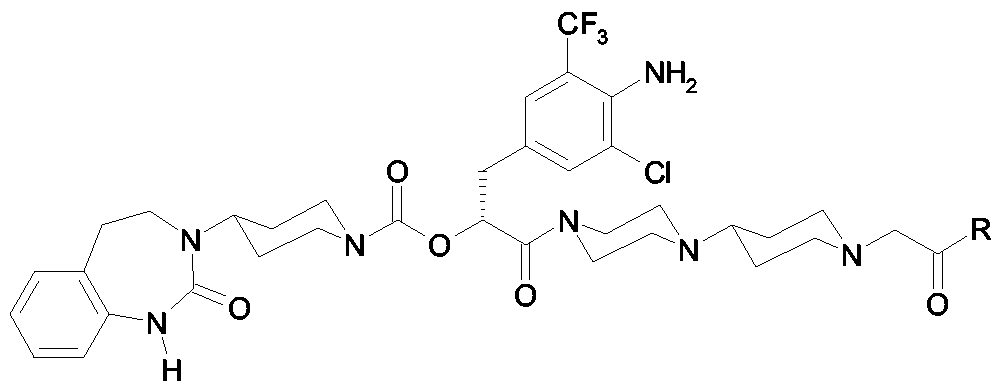
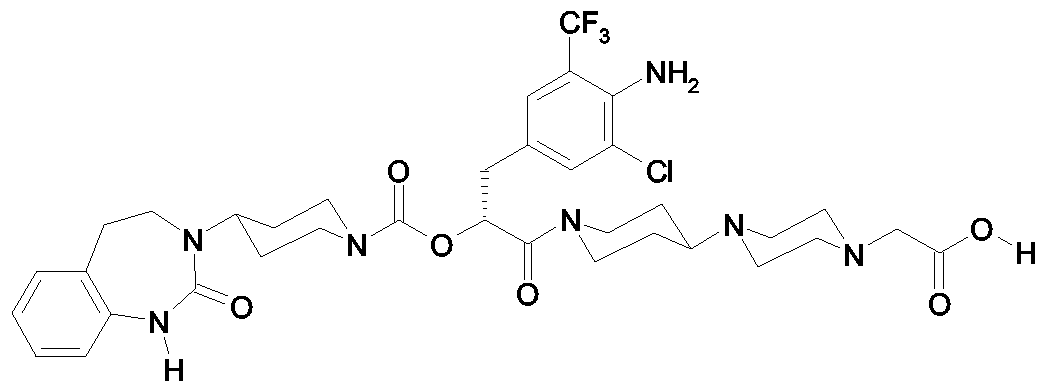
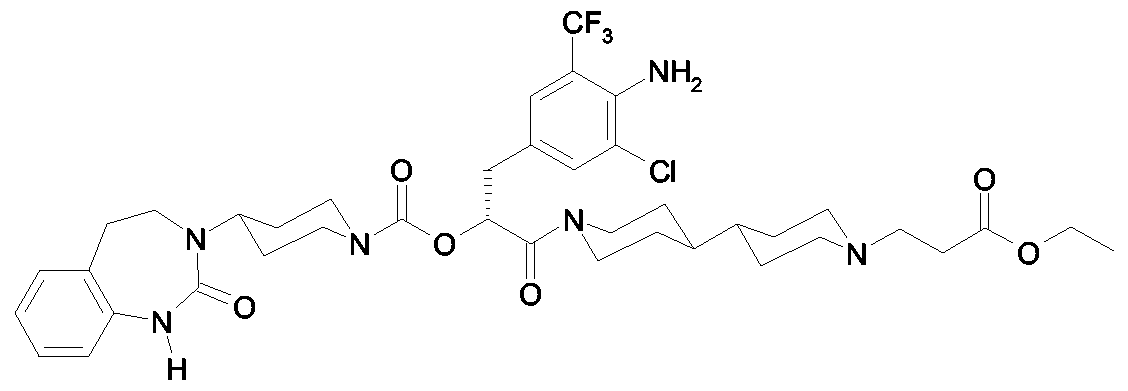
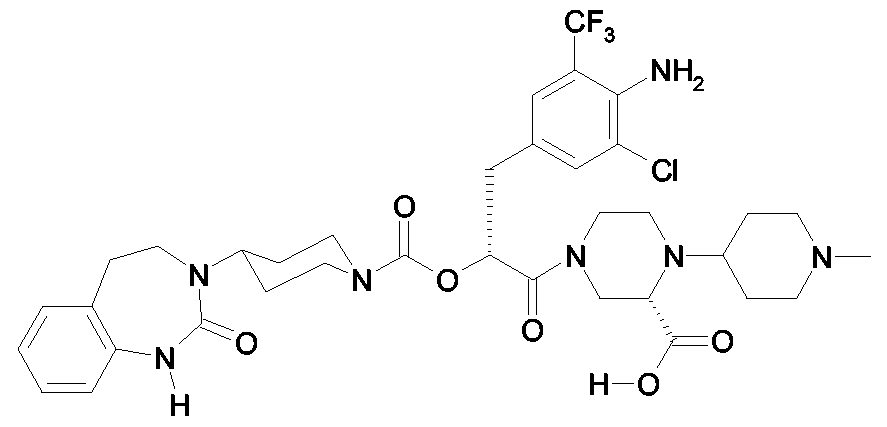
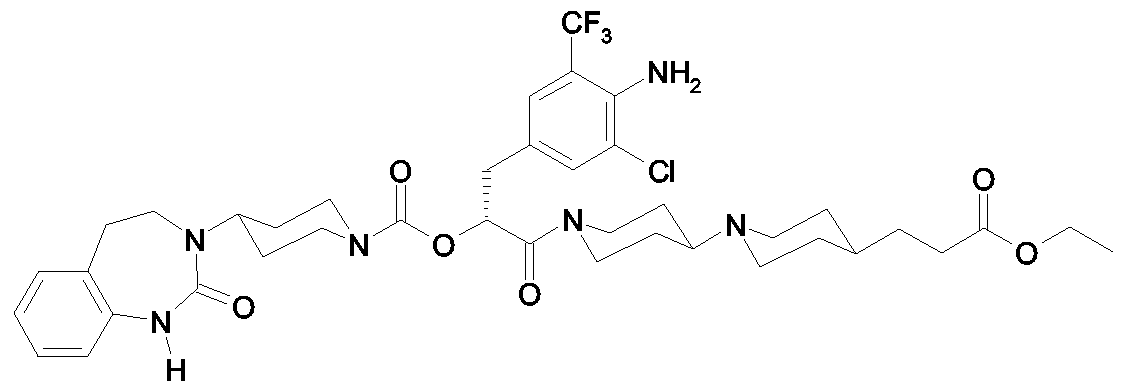
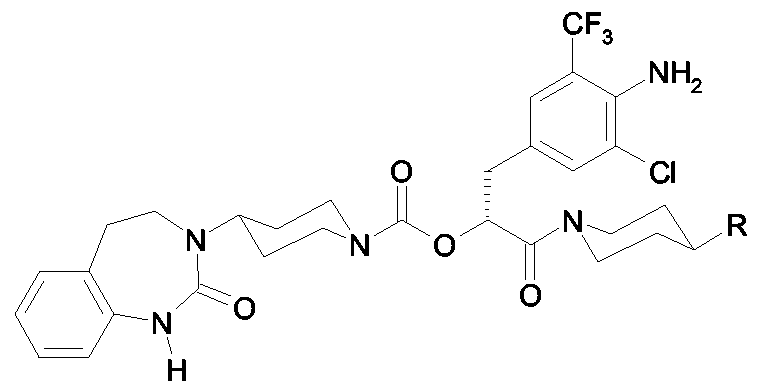
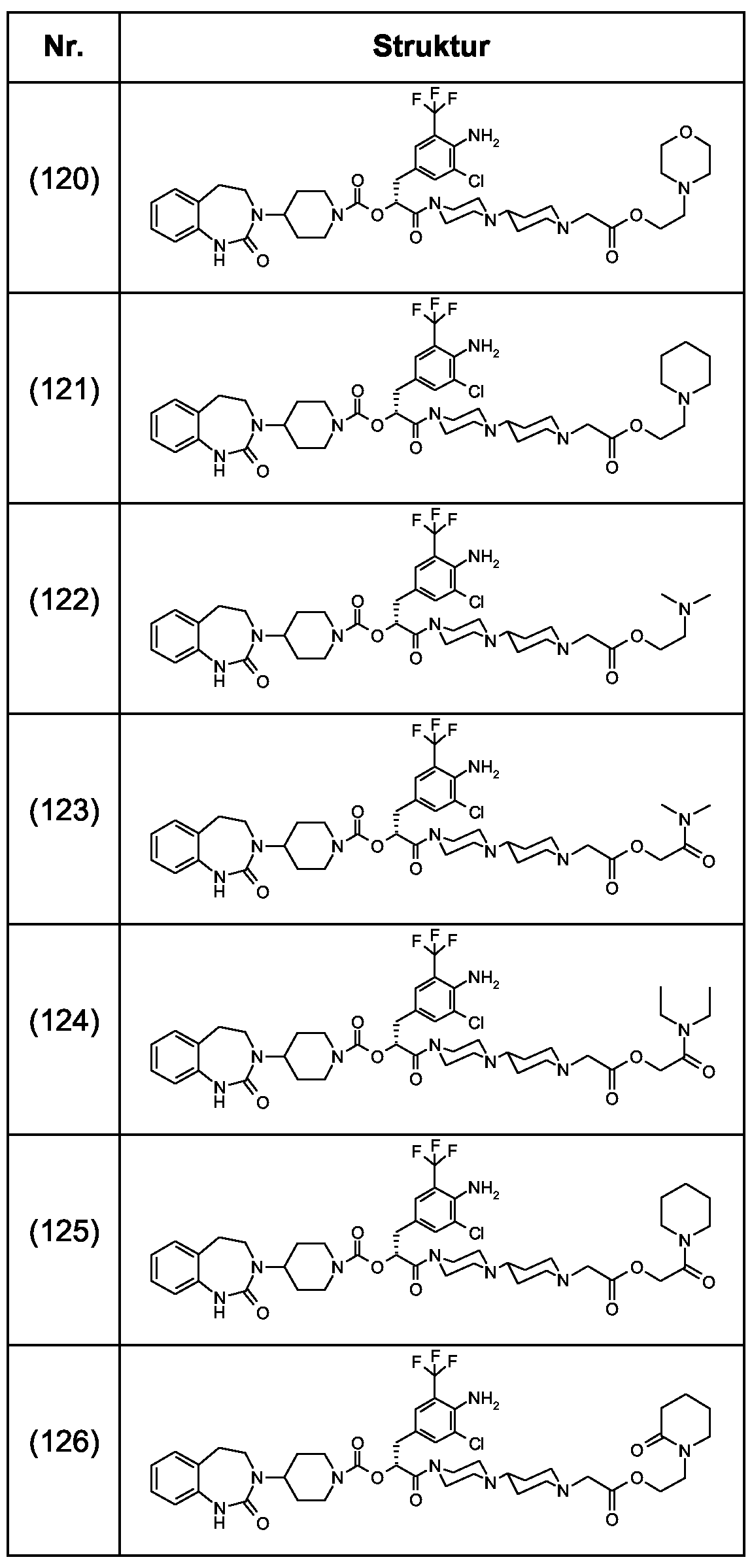
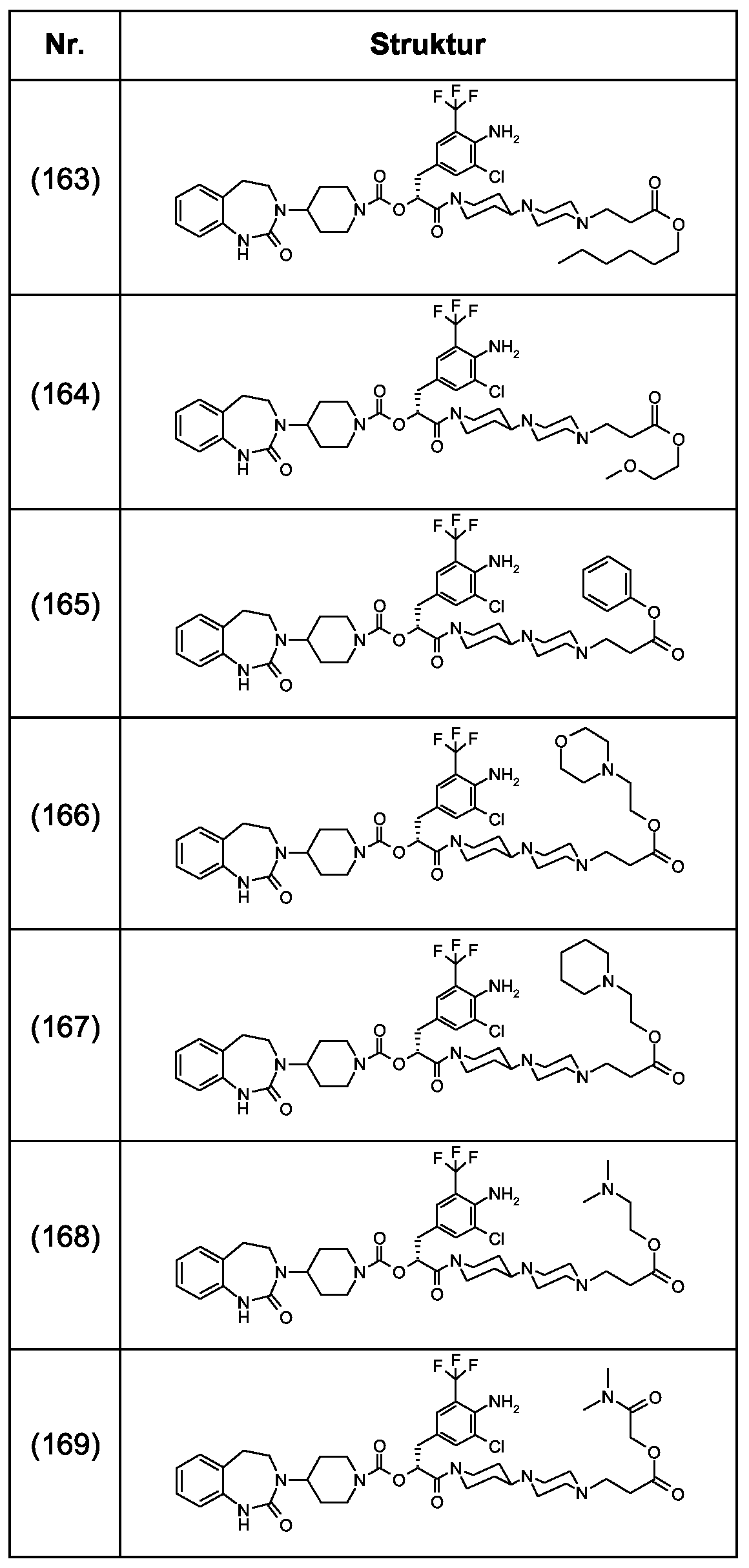
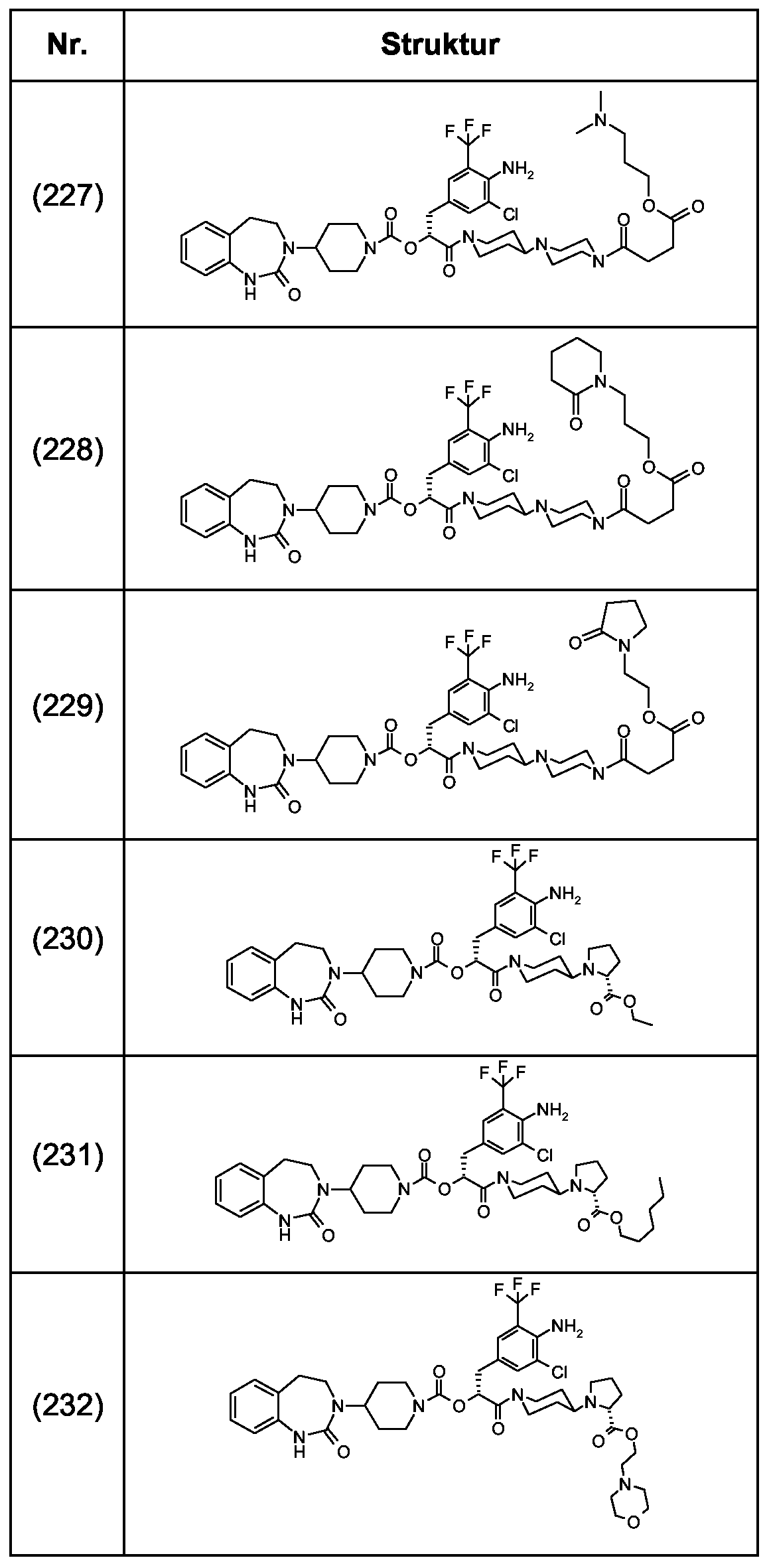
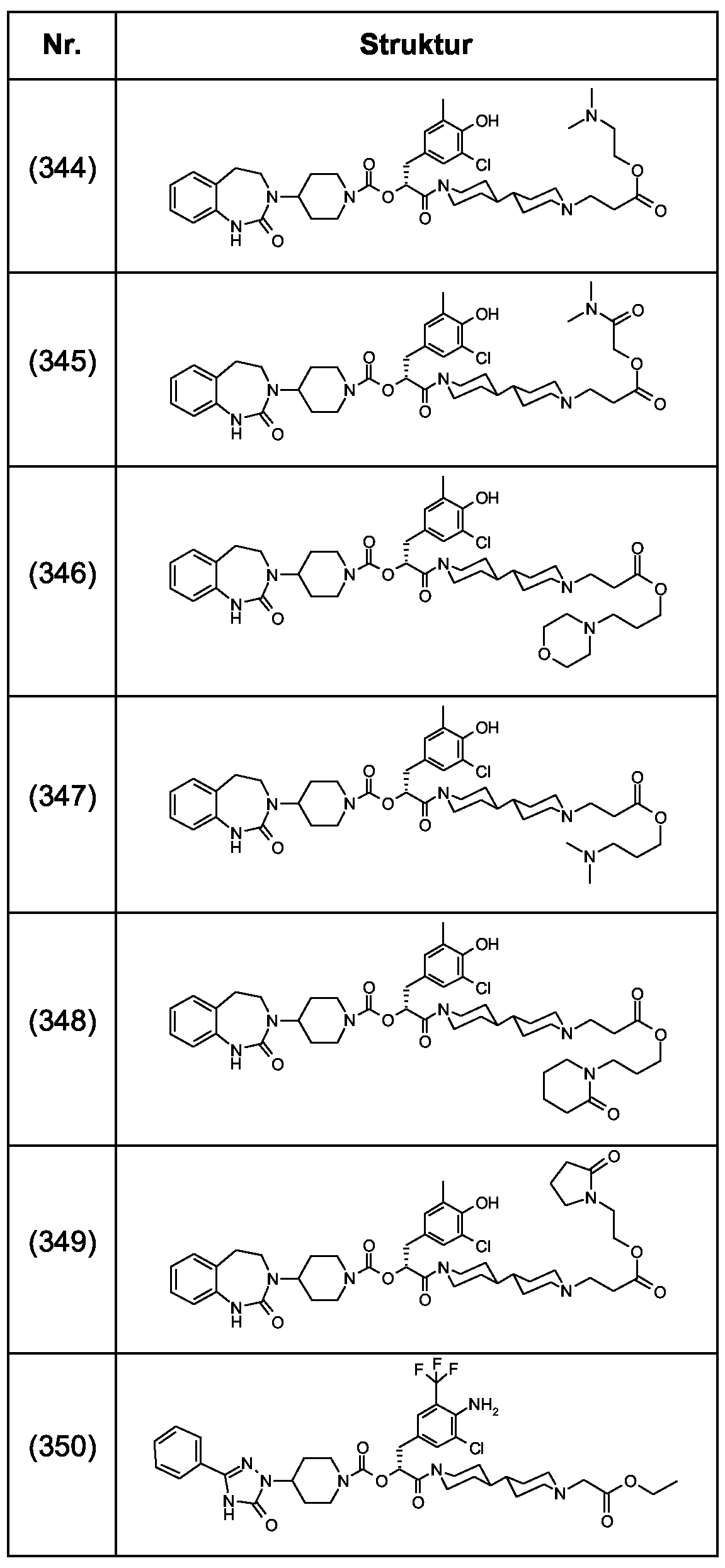
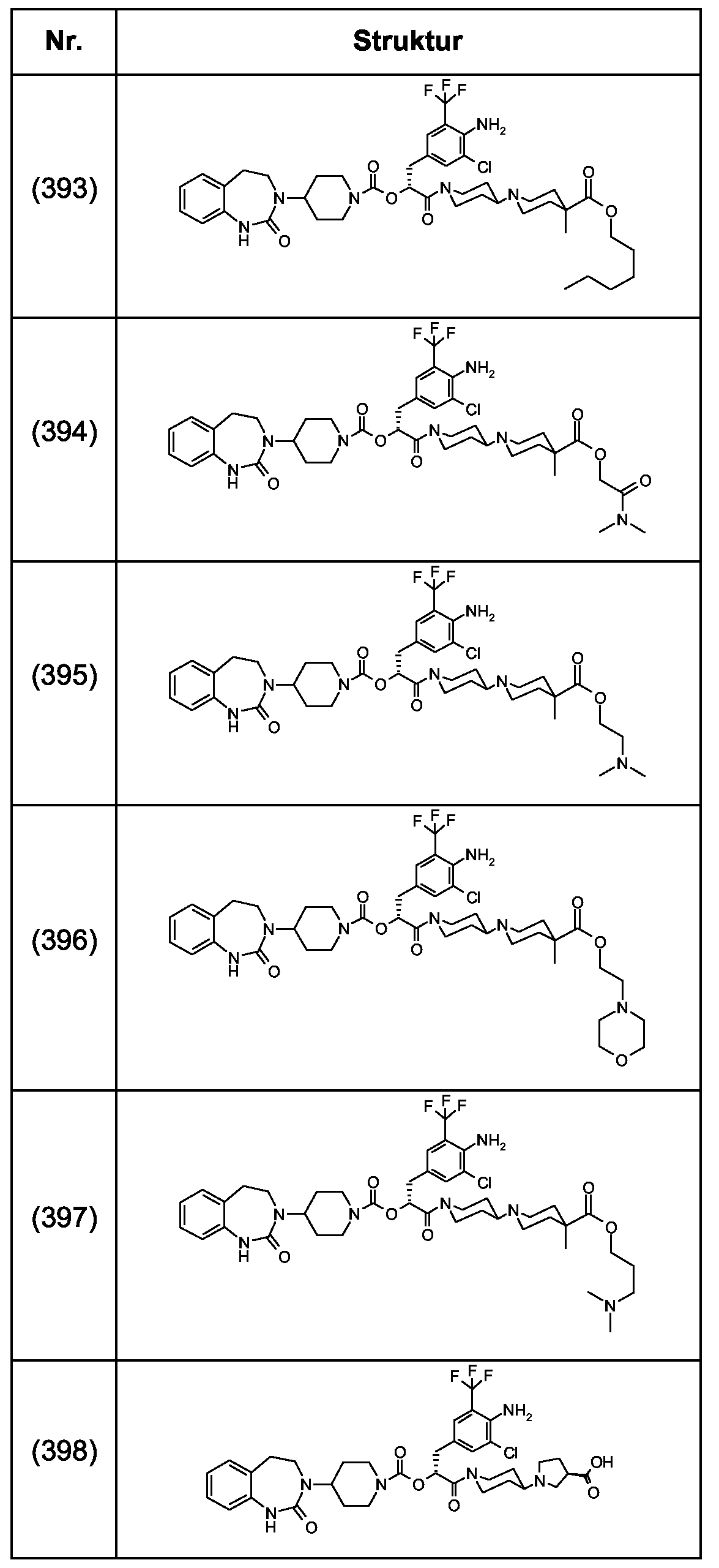
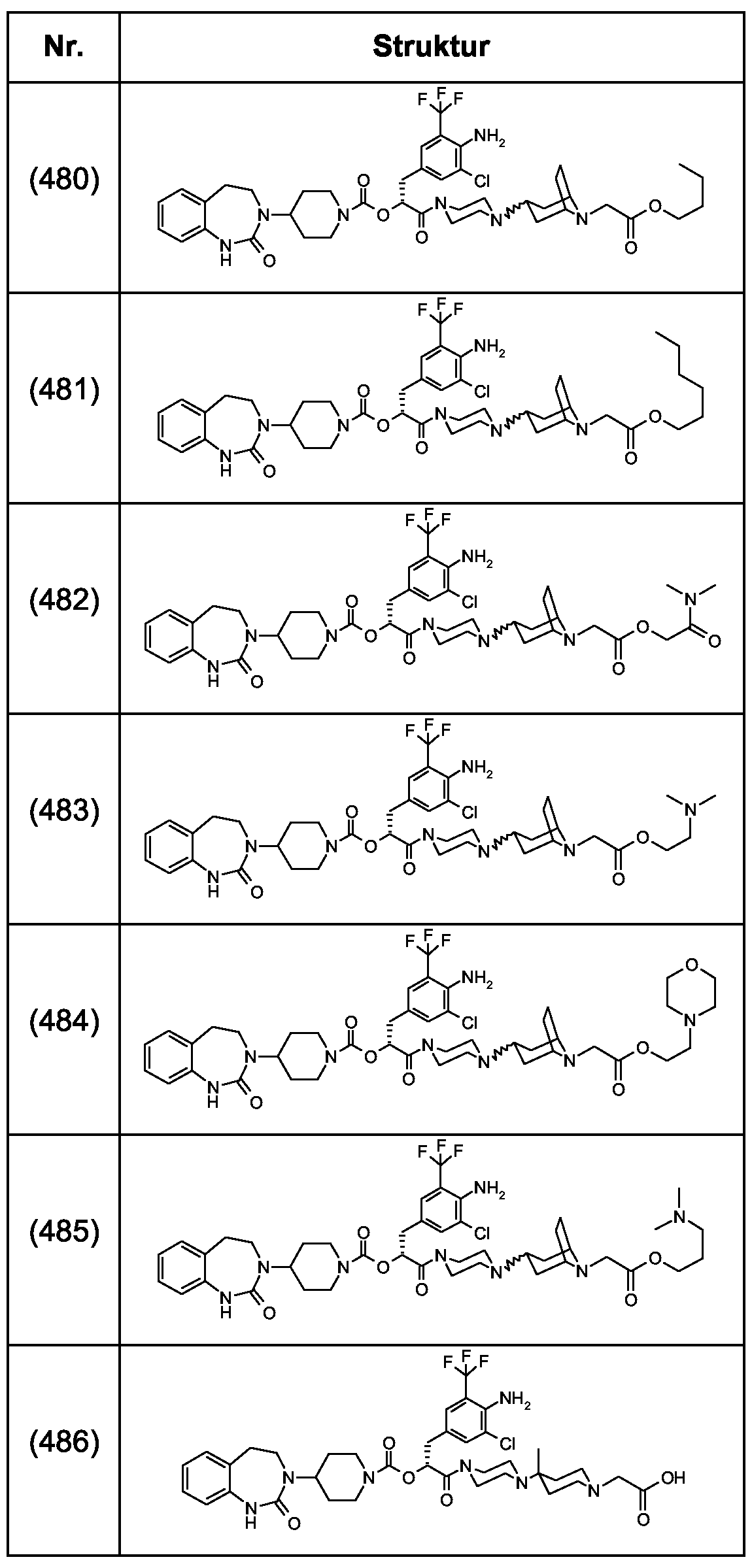
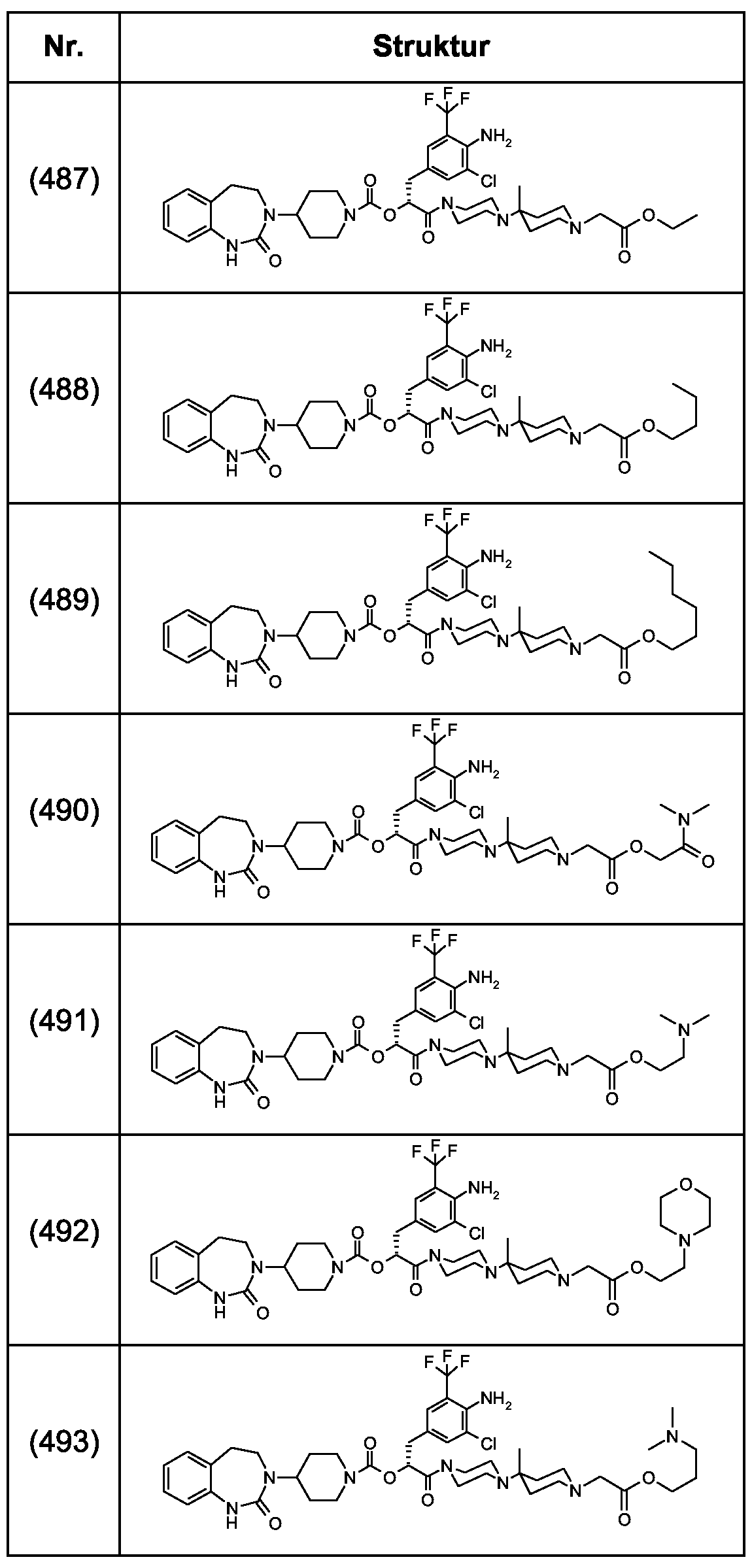
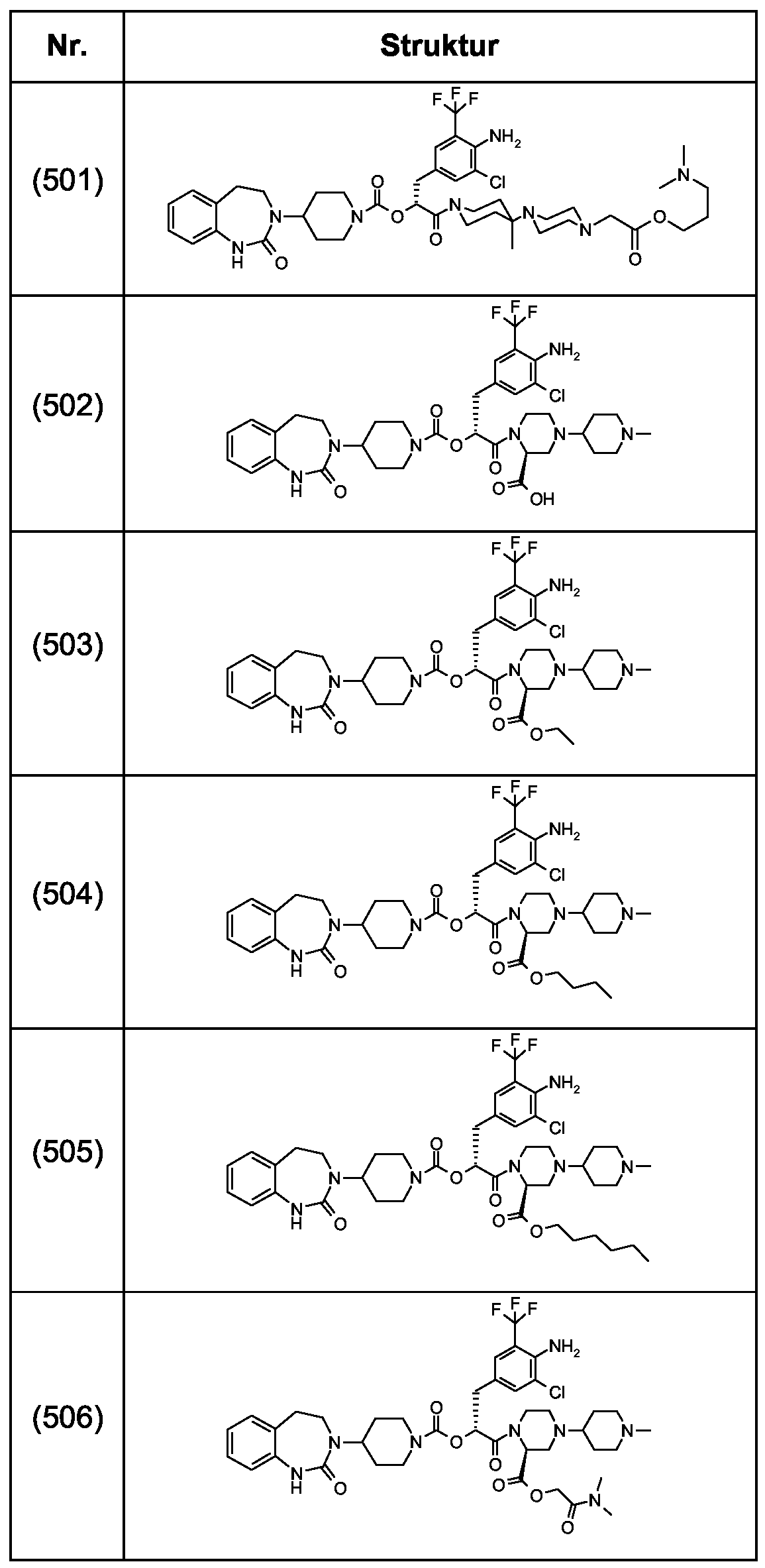
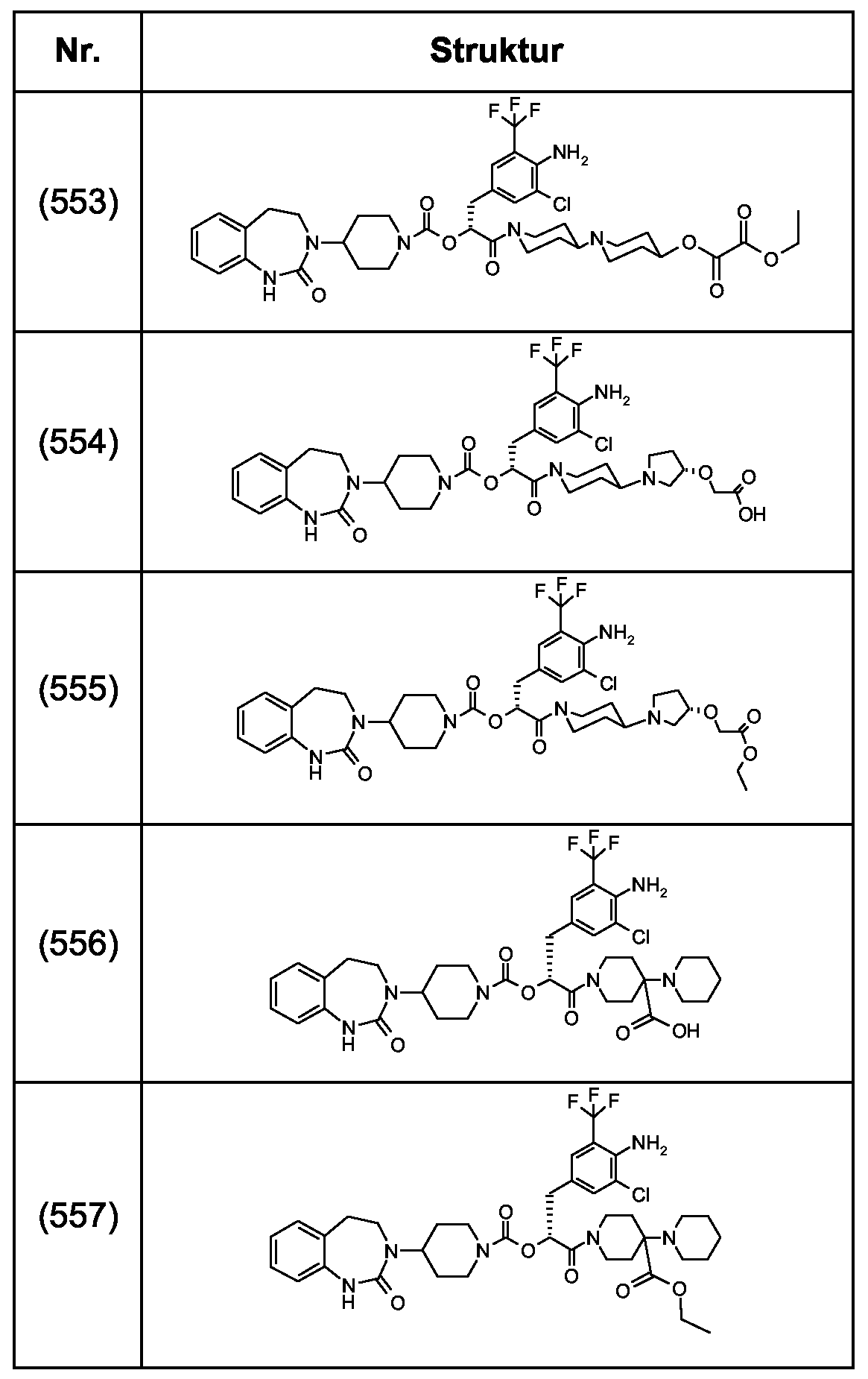
Als ganz besonders bevorzugte Verbindungen der obigen allgemeinen Formel I seien beispielsweise weiterhin folgende Verbindungen genannt:
deren Tautomere, deren Diastereomere, deren Enantiomere, deren Hydrate, deren Gemische und deren Salze sowie die Hydrate der Salze, insbesondere deren physiologisch verträgliche Salze mit anorganischen oder organischen Säuren oder Basen.
VERWENDETE BEGRIFFE UND DEFINITIONEN
Soweit nicht anders angegeben, sind alle Substituenten voneinander unabhängig. Sollten an einer Gruppe z.B. mehrere C1-6-Alkylgruppen als Substituenten sein, so könnte im Fall von drei Substituenten d-6-Alkyl unabhängig voneinander einmal Methyl, einmal n-Propyl und einmal fert-Butyl bedeuten.
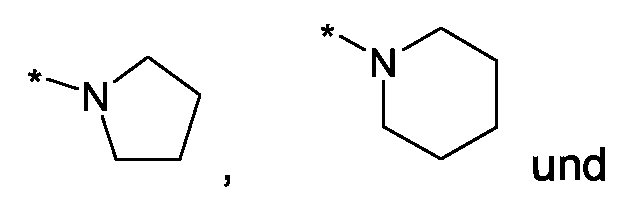
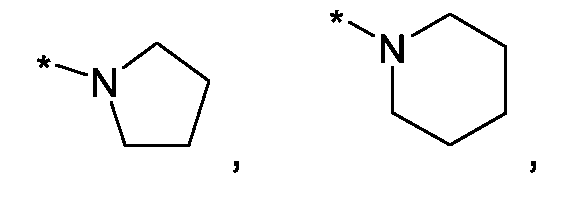
Im Rahmen dieser Anmeldung können bei der Definition von möglichen Substituenten, diese auch in Form einer Strukturformel dargestellt werden. Dabei wird, falls vorhanden, ein Stern (*) in der Strukturformel des Substituenten als der Verknüpfungspunkt zum Rest des Moleküls verstanden.
Ebenfalls mit vom Gegenstand dieser Erfindung umfasst sind die erfindungsgemäßen Verbindungen, einschließlich deren Salze, in denen ein oder mehrere Wasserstoffatome, beispielsweise ein, zwei, drei, vier oder fünf Wasserstoffatome, durch Deuterium ausgetauscht sind.
Unter dem Begriff "C1-3-AIkVl" (auch soweit sie Bestandteil anderer Reste sind) werden verzweigte und unverzweigte Alkylgruppen mit 1 bis 3 Kohlenstoffatomen verstanden, unter dem Begriff "C1-6-AIkVl" verzweigte und unverzweigte Alkylgruppen mit 1 bis 6 Kohlenstoffatomen und unter dem Begriff "C1-8-AIkVl" verzweigte und unverzweigte Alkylgruppen mit 1 bis 8 Kohlenstoffatomen verstanden. Beispielsweise werden hierfür genannt: Methyl, Ethyl, n-Propyl, /so-Propyl, n-Butyl, /so-Butyl, sec-Butyl, fert-Butyl, n-Pentyl, /so-Pentyl, neo-Pentyl, Hexyl, Heptyl und Octyl. Gegebenenfalls werden für vorstehend genannten Gruppen auch die Abkürzungen Me, Et, n-Pr, /-Pr, n-Bu, /-Bu, f-Bu, etc. verwendet. Sofern nicht anders beschrieben, umfassen die Definitionen Propyl, Butyl,
Pentyl, Hexyl, Heptyl und Octyl alle denkbaren isomeren Formen der jeweiligen Reste. So umfasst beispielsweise Propyl n-Propyl und /so-Propyl, Butyl umfasst /so-Butyl, sec-Butyl und fert-Butyl etc.
Unter dem Begriff "C1-3-Alkylen" (auch soweit sie Bestandteil anderer Reste sind) werden verzweigte und unverzweigte Alkylengruppen mit 1 bis 3 Kohlenstoffatomen und unter dem Begriff "C2-4-Alkylen" verzweigte und unverzweigte Alkylengruppen mit 2 bis 4 Kohlenstoffatomen verstanden. Beispielsweise werden hierfür genannt: Methylen, Ethylen, Ethan-1,1-diyl, Propylen, Propan-2,2-diyl, 1-Methylethylen, Butylen, 1- Methyl propylen, 1,1-Dimethylethylen, 1,2-Dimethylethylen. Sofern nicht anders beschrieben, umfassen die Definitionen Propylen und Butylen alle denkbaren isomeren Formen der gleichen Kohlenstoffanzahl. So umfasst beispielsweise Propylen auch 1- Methylethylen und Butylen umfasst 1-Methylpropylen, 1,1-Dimethylethylen, 1,2- Dimethylethylen.
Es sei weiterhin erwähnt, dass im Rahmen der vorliegenden Erfindung die Begriffe "Alkylen" und "Alkylenyl" synonym verwendet werden.
Verbindungen der allgemeinen Formel I können Säuregruppen besitzen, hauptsächlich Carboxylgruppen, und/oder basische Gruppen wie z.B. Aminofunktionen. Verbindungen der allgemeinen Formel I können deshalb als innere Salze, als Salze mit pharmazeutisch verwendbaren anorganischen Säuren wie beispielsweise Bromwasserstoffsäure, Phosphorsäure, Salpetersäure, Salzsäure, Schwefelsäure, Methansulfonsäure, Ethan- sulfonsäure, Benzolsulfonsäure, p-Toluolsulfonsäure oder organischen Säuren wie beispielsweise Äpfelsäure, Bernsteinsäure, Essigsäure, Fumarsäure, Maleinsäure,
Mandelsäure, Milchsäure, Weinsäure, Zitronensäure oder als Salze mit pharmazeutisch verwendbaren Basen wie Alkali- oder Erdalkalimetallhydroxiden, beispielsweise Natriumhydroxid oder Kaliumhydroxid, oder Carbonaten, Ammoniak, Zink- oder Ammoniumhydroxiden oder organischen Aminen wie z.B. Diethylamin, Triethylamin, Ethanolamin, Diethanolamin, Triethanolamin, Cyclohexylamin, Dicyclohexylamin u.a. vorliegen.
Gegenstand der Erfindung sind die jeweiligen Verbindungen gegebenenfalls in Form der einzelnen optischen Isomeren, Mischungen der einzelnen Enantiomeren oder Racemate, in Form der Tautomere sowie in Form der freien Basen oder der entsprechenden Säure- additionssalze mit pharmakologisch unbedenklichen Säuren - wie beispielsweise Säureadditionssalze mit Halogenwasserstoffsäuren - beispielsweise Chlor- oder Bromwasserstoffsäure - oder organische Säuren - wie beispielsweise Oxal-, Fumar-, Diglykol- oder Methansulfonsäure
Die erfindungsgemäßen Verbindungen können als Racemate vorliegen, sofern sie nur ein Chiralitätselement besitzen, sie können aber auch als reine Enantiomere, d.h. in (R)- oder (S)-Form gewonnen werden. Bevorzugt sind Verbindungen die als Racemate bzw. als (R)-Form vorliegen.
Die Anmeldung umfasst jedoch auch die einzelnen diastereomeren Antipodenpaare oder deren Gemische, die dann vorliegen, wenn mehr als ein Chiralitätselement in den Verbindungen der allgemeinen Formel I vorhanden ist, sowie die einzelnen optisch aktiven Enatiomeren, aus denen sich die erwähnten Racemate zusammensetzen.
Gegenstand der Erfindung sind die jeweiligen Verbindungen gegebenenfalls in Form der einzelnen optischen Isomeren, Mischungen der einzelnen Enantiomeren oder Racemate, in Form der Tautomere sowie in Form der freien Basen oder der entsprechenden Säureadditionssalze mit pharmakologisch unbedenklichen Säuren - wie beispielsweise Säure- additionssalze mit Halogenwasserstoffsäuren - beispielsweise Chlor- oder Bromwasserstoffsäure - oder organische Säuren - wie beispielsweise Oxal-, Fumar-, Diglycol- oder Methansulfonsäure
HERSTELLVERFAHREN
Die Verbindungen der allgemeinen Formel I werden nach prinzipiell bekannten Methoden hergestellt. Die folgenden Verfahren haben sich zur Herstellung der erfindungsgemäßen Verbindungen der allgemeinen Formel I besonders bewährt:
(a) Zur Herstellung von Verbindungen der allgemeinen Formel I, in der alle Reste wie eingangs erwähnt definiert sind:
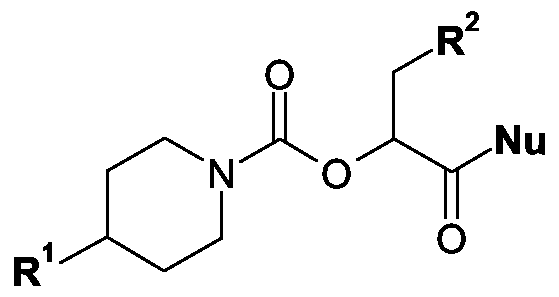
Kupplung einer Carbonsäure der allgemeinen Formel V
in der R1 und R2 wie eingangs erwähnt definiert sind, mit einem Amin der allgemeinen Formel VI
H-R3-R4
in der R3 und R4 wie eingangs definiert sind, wobei die Verknüpfung über das
Stickstoffatom von R3 erfolgt.
Vor Durchführung der Reaktion können in den Resten des Amins der Formel H-R3-R4 gegebenenfalls vorhandene Carbonsäurefunktionen, primäre oder sekundäre Amino- funktionen oder Hydroxyfunktionen durch übliche Schutzreste geschützt und gegebenen-
falls verwendete Schutzreste nach Durchführung der Reaktion nach für den Fachmann geläufigen Methoden wieder abgespalten werden.
Die Kupplung wird bevorzugt unter Verwendung von aus der Peptidchemie bekannten Verfahren (siehe z. B. Houben-Weyl, Methoden der Organischen Chemie, Bd. 15/2) durchgeführt, wobei zum Beispiel Carbodiimide, wie z. B. Dicyclohexylcarbodiimid (DCC), Diisopropylcarbodiimid (DIC) oder Ethyl-(3-dimethylamino-propyl)-carbodiimid, O-(1 H-Benzotriazol-1-yl)-N,N-NI,NI-tetramethyluronium-hexa-fluorphosphat (HBTU) oder -tetrafluorborat (TBTU) oder 1H-Benzotriazol-1-yl-oxy-tris-(dimethylamino)-phosphonium- hexafluorphosphat (BOP) eingesetzt werden. Durch Zugabe von 1-Hydroxybenzotriazol (HOBt) oder von 3-Hydroxy-4-oxo-3,4-dihydro-1 ,2,3-benzotriazin (HOObt) kann die
Reaktionsgeschwindigkeit gesteigert werden. Die Kupplungen werden normalerweise mit äquimolaren Anteilen der Kupplungskomponenten sowie des Kupplungsreagenz in Lösungsmitteln wie Dichlormethan, Tetrahydrofuran, Acetonitril, Dimethylformamid (DMF), Dimethylacetamid (DMA), N-Methylpyrrolidon (NMP) oder Gemischen aus diesen und bei Temperaturen zwischen -30°C und +30°C, bevorzugt -20°C und +25°C, durchgeführt. Sofern erforderlich wird als zusätzliche Hilfsbase N-Ethyldiisopropylamin (Hünig-Base) bevorzugt.
Als weiteres Kupplungsverfahren zur Synthese von Verbindungen der allgemeinen Formel I wird das sogenannte "Anhydridverfahren" (siehe auch: M. Bodanszky, "Peptide Chemistry", Springer-Verlag 1988, S. 58-59; M. Bodanszky, "Principles of Peptide Synthesis", Springer-Verlag 1984, S. 21-27) eingesetzt. Bevorzugt wird das "gemischte Anhydridverfahren" in der Variante nach Vaughan (J. R. Vaughan Jr., J. Amer. Chem.Soc. 73, 3547 (1951)), bei der unter Verwendung von Chlorkohlensäureisobutylester in Gegen- wart von Basen, wie 4-Methylmorpholin oder 4-Ethylmorpholin, das gemischte Anhydrid aus der zu kuppelnden Carbonsäure der allgemeinen Formel V und dem Kohlensäure- monoisobutylester erhalten wird. Die Herstellung dieses gemischten Anhydrids und die Kupplung mit den Aminen der allgemeinen Formel VI erfolgt im Eintopfverfahren unter Verwendung der voranstehend genannten Lösungsmittel und bei Temperaturen zwischen -20°C und +25°C, bevorzugt zwischen 0°C und +25°C.
(b) Zur Herstellung von Verbindungen der allgemeinen Formel I, in der alle Reste wie eingangs erwähnt definiert sind:
Kupplung einer Verbindung der allgemeinen Formel VII
in der R1 und R2 wie eingangs erwähnt definiert sind und Nu eine Austrittsgruppe, beispielsweise ein Halogenatom, wie das Chlor-, Brom- oder lodatom, eine Alkylsulfonyl- oxygruppe mit 1 bis 10 Kohlenstoffatomen im Alkylteil, eine gegebenenfalls durch Chloroder Bromatome, durch Methyl- oder Nitrogruppen mono-, di- oder trisubstituierte Phenyl- sulfonyloxy- oder Naphthylsulfonyloxygruppe, wobei die Substituenten gleich oder verschieden sein können, eine 1H-lmidazol-1-yl-, eine gegebenenfalls durch eine oder zwei Methylgruppen im Kohlenstoffgerüst substituierte 1H-Pyrazol-1-yl-, eine 1H-1,2,4- Triazol-1-yl-, 1H-1,2,3-Triazol-1-yl-, 1H-1 ,2,3,4-Tetrazol-1-yl-, eine Vinyl-, Propargyl-, p-Nitrophenyl-, 2,4-Dinitrophenyl-, Trichlorphenyl-, Pentachlorphenyl-, Pentafluorphenyl-, Pyranyl- oder Pyridinyl-, eine Dimethylaminyloxy-, 2(1H)-Oxopyridin-1-yl-oxy-, 2,5-Dioxo- pyrrolidin-1-yloxy-, Phthalimidyloxy-, 1H-Benzotriazol-1-yloxy- oder Azidgruppe darstellt, mit einem Amin der allgemeinen Formel VI
H-R3-R4
in der alle Reste wie eingangs erwähnt definiert sind und wobei die Verknüpfung über das Stickstoffatom des Amins R3 erfolgt.
Vor Durchführung der Reaktion können in den Resten des Amins der allgemeinen Formel VI gegebenenfalls vorhandene Carbonsäurefunktionen, primäre oder sekundäre Amino- funktionen oder Hydroxyfunktionen durch übliche Schutzreste geschützt und gegebenenfalls verwendete Schutzreste nach Durchführung der Reaktion nach für den Fachmann geläufigen Methoden wieder abgespalten werden.
Die Umsetzung wird unter Schotten-Baumann- oder Einhorn-Bedingungen durchgeführt, das heißt, die Komponenten werden in Gegenwart von wenigstens einem Äquivalent einer Hilfsbase bei Temperaturen zwischen -50°C und +120°C, bevorzugt -10°C und +30°C,
und gegebenenfalls in Gegenwart von Lösungsmitteln zur Reaktion gebracht. Als Hilfs- basen kommen bevorzugt Alkali- und Erdalkalihydroxide, beispielsweise Natriumhydroxid, Kaliumhydroxid oder Bariumhydroxid, Alkalicarbonate, z. B. Natriumcarbonat, Kalium- carbonat oder Cäsiumcarbonat, Alkaliacetate, z.B. Natrium- oder Kaliumacetat, sowie tertiäre Amine, beispielsweise Pyridin, 2,4,6-Trimethylpyridin, Chinolin, Triethylamin, N-Ethyldiisopropylamin, N-Ethyldicyclohexylamin, 1 ,4-Di-azabicyclo[2,2,2]octan oder 1 ,8-Diaza-bicyclo[5,4,0]undec-7-en, als Lösungsmittel beispielsweise Dichlormethan, Tetrahydrofuran, 1,4-Dioxan, Acetonitril, Dimethylformamid, Dimethylacetamid, N-Methyl- pyrrolidon oder Gemische davon in Betracht; werden als Hilfsbasen Alkali- oder Erdalkali- hydroxide, Alkalicarbonate oder -acetate verwendet, kann dem Reaktionsgemisch auch Wasser als Cosolvens zugesetzt werden.
Die erfindungsgemäßen neuen Verbindungen der allgemeinen Formel I enthalten ein oder mehrere Chiralitätszentren. Sind beispielsweise zwei Chiralitätszentren vorhanden, dann können die Verbindungen in Form zweier diastereomerer Antipoden paare auftreten. Die Erfindung umfasst die einzelnen Isomeren ebenso wie ihre Gemische.
Die Trennung der jeweiligen Diastereomeren gelingt auf Grund ihrer unterschiedlichen physikochemischen Eigenschaften, z.B. durch fraktionierte Kristallisation aus geeigneten Lösemitteln, durch Hochdruckflüssigkeits- oder Säulenchromatographie unter Verwendung chiraler oder bevorzugt achiraler stationärer Phasen.
Die Trennung von unter die allgemeine Formel I fallenden Racematen gelingt beispielsweise durch HPLC an geeigneten chiralen stationären Phasen (z. B. Chiral AGP, Chiralpak AD). Racemate, die eine basische oder saure Funktion enthalten, lassen sich auch über die diastereomeren, optisch aktiven Salze trennen, die bei Umsetzung mit einer optisch aktiven Säure, beispielsweise (+)- oder (-)-Weinsäure, (+)- oder (-)-Diacetylwein- säure, (+)- oder (-)-Monomethyltartrat oder (+)- oder (-)-Camphersulfonsäure, bzw. einer optisch aktiven Base, beispielsweise mit (R)-(+)-1-Phenylethylamin, (S)-(-)-1-Phenylethyl- amin oder (S)-Brucin, entstehen.
Nach einem üblichen Verfahren zur Isomerentrennung wird das Racemat einer Verbindung der allgemeinen Formel I mit einer der vorstehend angegebenen optisch aktiven Säuren bzw. Basen in äquimolarer Menge in einem Lösemittel umgesetzt und die
erhaltenen kristallinen, diastereomeren, optisch aktiven Salze unter Ausnutzung ihrer verschiedenen Löslichkeit getrennt. Diese Umsetzung kann in jeder Art von Lösemitteln durchgeführt werden, solange sie einen ausreichenden Unterschied hinsichtlich der Löslichkeit der Salze aufweisen. Vorzugsweise werden Methanol, Ethanol oder deren Gemische, beispielsweise im Volumenverhältnis 50:50, verwendet. Sodann wird jedes der optisch aktiven Salze in Wasser gelöst, mit einer Base, wie Natriumcarbonat oder Kaliumcarbonat, oder mit einer geeigneten Säure, beispielsweise mit verdünnter Salzsäure oder wässeriger Methansulfonsäure, vorsichtig neutralisiert und dadurch die entsprechende freie Verbindung in der (+)- oder (-)-Form erhalten.
Jeweils nur das (R)- oder (S)-Enantiomer bzw. ein Gemisch zweier optisch aktiver, unter die allgemeine Formel I fallender, diastereomerer Verbindungen wird auch dadurch erhalten, dass man die oben beschriebenen Synthesen mit jeweils einer geeigneten (R)- bzw. (S)-konfigurierten Reaktionskomponente durchführt.
Die als Ausgangsverbindungen benötigten Hydroxycarbonsäuren der allgemeinen Formel V sind durch Umsetzung von Piperidinen der allgemeinen Formel VIII
in der R1 wie eingangs erwähnt definiert ist, mit Kohlensäurederivaten der allgemeinen Formel IX
in der Y1 und Y2 nucleofuge Gruppen bedeuten, die gleich oder verschieden sein können, bevorzugt das Chloratom, die p-Nitrophenoxy- oder Trichlormethoxy-Gruppe,
und mit Verbindungen der allgemeinen Formel X
in der R2 wie eingangs erwähnt definiert ist und Z1 eine Schutzgruppe für eine Carboxy- gruppe darstellt, beispielsweise eine d-6-Alkyl- oder eine gegebenenfalls substituierte Benzylgruppe, wobei die Alkylgruppen linear oder verzweigt sein können und die Benzyl- gruppe durch ein oder zwei Methoxygruppen substituiert sein kann, erhältlich. Bevorzugt ist für Z1 die Methyl-, Ethyl-, fert-Butyl oder Benzylgruppe. Vor Durchführung der Reaktion kann im Rest R2 einer Verbindung der Formel (VI) gegebenenfalls vorhandene Hydroxyfunktionen durch übliche Schutzreste geschützt und gegebenenfalls verwendete Schutzreste nach Durchführung der Reaktion nach dem Fachmann geläufigen Methoden wieder abgespalten werden.
In einer ersten Stufe werden die Verbindungen der allgemeinen Formel VIII in einem Lösungsmittel, beispielsweise in Dichlormethan, THF, Pyridin oder deren Mischungen, bei einer Temperatur zwischen -20°C bis 50°C in Gegenwart einer Base, beispielsweise Triethylamin, Pyridin oder Ethyldiisopropylamin, mit den Kohlensäurederivaten der allgemeinen Formel IX zur Reaktion gebracht. Die dabei entstehende Zwischenstufe kann aufgereinigt oder ohne Reinigung weiter umgesetzt werden. Die Umsetzung dieser Zwischenstufen mit Verbindungen der allgemeinen Formel X erfolgt ebenfalls in einem der oben genannten Lösungsmittel, und bei den oben genannten Temperaturen, in Gegenwart einer Base, wie Triethylamin oder Pyridin, mit oder ohne Zusatz eines Aktivierungsreagenz, wie z.B. 4-Dimethylaminopyridin. Zur Aktivierung können die Verbindungen der allgemeinen Formel X auch mittels eines Metallhydrides, wie z.B. NaH oder KH, deprotoniert werden, wobei in diesem Fall auf die Gegenwart der Base oder des Aktivierungsreagenzes verzichtet werden kann.
Die Ausgangsverbindungen der Formel VIII und IX sind entweder käuflich, literaturbekannt oder können nach literaturbekannten Methoden hergestellt werden.
Ein Zugang zu Verbindungen der allgemeinen Formel X besteht in der Umsetzung von Aldehyden der allgemeinen Formel Xl
in der R2 wie eingangs erwähnt definiert ist, mit N-Acetylglycin in Acetanhydrid als Lösungsmittel in Gegenwart von Alkaliacetat, bevorzugt Natrium- oder Kaliumacetat, bei geeigneter Temperatur, bevorzugt bei 80 bis 130°C.
Die primär entstehenden Azlactone werden ohne Isolierung zu den Verbindungen der allgemeinen Formel XII
in der R2 wie eingangs erwähnt definiert ist, hydrolysiert. Durch weitere Umsetzung in Gegenwart von wässrigen Mineralsäuren, wie beispielsweise Schwefel-, Phosphor- oder Chlorwasserstoffsäure, bevorzugt jedoch von Chlorwasserstoffsäure, werden Verbindungen der allgemeinen Formel XIII
in der R2 wie eingangs erwähnt definiert ist, erhalten. Diese werden dann mit geeigneten Reduktionsmitteln in die Verbindungen der allgemeinen Formel XIV
in der R
2 wie eingangs erwähnt definiert ist, überführt.
Als Reduktionsmittel können Alkaliborhydride, wie Natrium- oder Kaliumborhydrid verwendet werden. Weitere Reduktionsmittel stellen Chlordialkylborane, wie Chlordicyclohexyl- boran, dar. Werden chirale Chlordialkylborane, wie z.B. B-Chlordiisopinocampheylboran benutzt, können die Verbindungen der allgemeinen Formel XIV in enantiomerenreiner Form isoliert werden. Die weitere Umsetzung von Verbindungen der allgemeinen Formel XIV zu Verbindungen der allgemeinen Formel X erfolgt im alkoholischen Milieu, bevorzugt in Methanol oder Ethanol, in Gegenwart einer geeigneten Säure, wie Chlorwasserstoffsäure. Die Reaktion kann alternativ durch Umsetzung in alkoholischen Lösungsmitteln, bevorzugt Methanol, mit Thionychlorid erfolgen.
Alle Verbindungen der allgemeinen Formel I, die primäre oder sekundäre Amino-, Hydroxy- oder Hydroxycarbonylfunktionen enthalten, werden bevorzugt aus mit Schutzgruppen versehenen Vorstufen gewonnen. Als Schutzgruppen für Aminofunktionen kommen beispielsweise eine Benzyloxycarbonyl-, 2-Nitrobenzyloxycarbonyl-, 4-Nitro- benzyloxycarbonyl-, 4-Methoxy-benzyloxycarbonyl-, 2-Chlor-benzyloxycarbonyl-, 3-Chlor- benzyloxycarbonyl-, 4-Chlor-benzyloxycarbonyl-, 4-Biphenylyl-α,α-dimethyl-benzyloxy- carbonyl- oder 3,5-Dimethoxy-α,α-dimethyl-benzyloxycarbonylgruppe, eine Alkoxy- carbonylgruppe mit insgesamt 1 bis 5 Kohlenstoffatomen im Alkylteil, beispielsweise die Methoxycarbonyl-, Ethoxycarbonyl-, n-Propoxycarbonyl-, Isopropoxycarbonyl-, n-Butoxy- carbonyl-, 1 -Methyl propoxycarbonyl-, 2-Methylpropoxy-carbonyl- oder fert-Butyloxy- carbonylgruppe, die Allyloxycarbonyl-, 2,2,2-Trichlor-(1 ,1-dimethylethoxy)carbonyl- oder 9-Fluorenylmethoxycarbonyl-Gruppe oder eine Formyl-, Acetyl- oder Trifluoracetylgruppe in Frage. Als Schutzgruppe für Hydroxyfunktionen kommt beispielsweise eine Trimethylsilyl-,
Triethylsilyl-, Triisopropyl-, fert-Butyldimethylsilyl- oder fert-Butyldiphenylsilylgruppe, eine fert-Butyl-, Benzyl-, 4-Methoxybenzyl- oder 3,4-Dimethoxybenzylgruppe in Frage. Als Schutzgruppe für Hydroxycarbonylfunktionen kommt beispielsweise eine Alkylgruppe mit insgesamt 1 bis 5 Kohlenstoffatomen, beispielsweise die Methyl-, Ethyl-, n-Propyl-, Isopropyl-, n-Butyl-, fert-Butyl, AIIyI-, 2,2,2-Trichlorethyl-, Benzyl- oder 4-Methoxybenzyl- gruppe in Frage.
Die erhaltenen Verbindungen der allgemeinen Formel I können, sofern sie geeignete basische Funktionen enthalten, insbesondere für pharmazeutische Anwendungen in ihre
physiologisch verträglichen Salze mit anorganischen oder organischen Säuren übergeführt werden. Als Säuren kommen hierfür beispielsweise Salzsäure, Bromwasserstoffsäure, Phosphorsäure, Salpetersäure, Schwefelsäure, Methansulfonsäure, Ethansulfon- säure, Benzolsulfonsäure, p-Toluolsulfonsäure, Essigsäure, Fumarsäure, Bernsteinsäure, Milchsäure, Mandelsäure, Äpfelsäure, Zitronensäure, Weinsäure oder Maleinsäure in Betracht.
Außerdem lassen sich die neuen Verbindungen der Formel I, falls sie Carbonsäurefunktion enthalten, in ihre Additionssalze mit anorganischen oder organischen Basen, insbesondere für die pharmazeutische Anwendung in ihre physiologisch verträglichen Additionssalze überführen. Als Basen kommen hierfür beispielsweise Natriumhydroxid, Kaliumhydroxid, Ammoniak, Cyclohexylamin, Dicyclohexylamin, Ethanolamin, Diethanolamin und Triethanolamin in Betracht.
Die vorliegende Erfindung betrifft Racemate, sofern die Verbindungen der allgemeinen Formel I nur ein Chiralitätselement besitzen. Die Anmeldung umfasst jedoch auch die einzelnen diastereomeren Antipodenpaare oder deren Gemische, die dann vorliegen, wenn mehr als ein Chiralitätselement in den Verbindungen der allgemeinen Formel I vorhanden ist, sowie die einzelnen optisch aktiven Enantiomeren, aus denen sich die erwähnten Racemate zusammensetzen.
Ebenfalls mit vom Gegenstand dieser Erfindung umfasst sind die erfindungsgemäßen Verbindungen, einschließlich deren Salze, in denen ein oder mehrere Wasserstoffatome, beispielsweise ein, zwei, drei, vier oder fünf Wasserstoffatome, durch Deuterium ausge- tauscht sind.
Die neuen Verbindungen der allgemeinen Formel I und deren physiologisch verträgliche Salze weisen wertvolle pharmakologische Eigenschaften auf, die auf ihre selektiven CGRP-antagonistischen Eigenschaften zurückgehen. Ein weiterer Gegenstand der Erfindung sind diese Verbindungen enthaltende Arzneimittel, deren Verwendung und deren Herstellung.
Die voranstehend genannten neuen Verbindungen und deren physiologisch verträgliche Salze besitzen CG RP-antagon istische Eigenschaften und zeigen gute Affinitäten in
CGRP-Rezeptorbindungsstudien. Die Verbindungen weisen in den nachstehend beschriebenen pharmakologischen Testsystemen CGRP-antagonistische Eigenschaften auf.
Zum Nachweis der Affinität der voranstehend genannten Verbindungen zu humanen CGRP-Rezeptoren und ihrer antagonistischen Eigenschaften wurden die folgenden Versuche durchgeführt:
A. Bindungsstudien mit (den humanen CGRP-Rezeptor exprimierenden) SK-N-MC- Zellen
SK-N-MC-Zellen werden in "Dulbecco's modified Eagle Medium" kultiviert. Das Medium konfluenter Kulturen wird entfernt. Die Zellen werden zweimal mit PBS-Puffer (Gibco 041- 04190 M) gewaschen, durch Zugabe von PBS-Puffer, versetzt mit 0.02% EDTA, abgelöst und durch Zentrifugation isoliert. Nach Resuspension in 20 ml "Balanced Salts Solution" [BSS (in mM): NaCI 120, KCl 5.4, NaHCO3 16.2, MgSO4 0.8, NaHPO4 1.0, CaCI2 1.8, D- Glucose 5.5, HEPES 30, pH 7.40] werden die Zellen zweimal bei 100 x g zentrifugiert und in BSS resuspendiert. Nach Bestimmung der Zellzahl werden die Zellen mit Hilfe eines Ultra-Turrax homogenisiert und für 10 Minuten bei 3000 x g zentrifugiert. Der Überstand wird verworfen und das Pellet in Tris-Puffer (10 mM Tris, 50 mM NaCI, 5 mM MgCI2, 1 mM EDTA, pH 7.40), angereichert mit 1% Rinderserum-Albumin und 0.1% Bacitracin, rezentrifugiert und resuspendiert (1 ml / 1000000 Zellen). Das Homogenat wird bei -80°C eingefroren. Die Membranpräparationen sind bei diesen Bedingungen für mehr als 6 Wochen stabil.
Nach Auftauen wird das Homogenat 1 : 10 mit Assay-Puffer (50 mM Tris, 150 mM NaCI, 5 mM MgCI2, 1 mM EDTA, pH 7.40) verdünnt und 30 Sekunden lang mit einem Ultra-Turrax homogenisiert. 230 μl des Homogenats werden für 180 Minuten bei Raumtemperatur mit 50 pM 125l-lodotyrosyl-Calcitonin-Gene-Related Peptide (Amersham) und ansteigenden Konzentrationen der Testsubstanzen in einem Gesamtvolumen von 250 μl inkubiert. Die Inkubation wird durch rasche Filtration durch mit Polyethylenimin (0.1%) behandelte GF/B-Glasfaserfilter mittels eines Zellharvesters beendet. Die an Protein gebundene Radioaktivität wird mit Hilfe eines Gammacounters bestimmt. Als nichtspezifische Bindung wird die gebundene Radioaktivität nach Gegenwart von 1 μM humanem CGRP-alpha während der Inkubation definiert.
Die Analyse der Konzentrations-Bindungskurven erfolgt mit Hilfe einer computer- gestützten nichtlinearen Kurvenanpassung.
Die eingangs erwähnten Verbindungen zeigen in dem beschriebenen Test IC50-Werte < 10000 nM.
B. CGRP-Antagonismus in SK-N-MC-Zellen
SK-N-MC-Zellen (1 Mio. Zellen) werden zweimal mit 250 μl Inkubationspuffer (Hanks' HEPES, 1 rtiM 3-lsobutyl-i-methylxanthin, 1% BSA, pH 7.4) gewaschen und bei 37°C für 15 Minuten vorinkubiert. Nach Zugabe von CGRP (10 μl) als Agonist in steigenden Konzentrationen (10~11 bis 10~6 M) bzw. zusätzlich von Substanz in 3 bis 4 verschiedenen Konzentrationen wird nochmals 15 Minuten inkubiert.
Intrazelluläres cAMP wird anschließend durch Zugabe von 20 μl 1M HCl und Zentrifugation (2000 x g, 4°C für 15 Minuten) extrahiert. Die Überstände werden in flüssigem Stickstoff eingefroren und bei -20°C gelagert.
Die cAMP-Gehalte der Proben werden mittels Radioimmunassay (Fa. Amersham) bestimmt und die pA2-Werte antagonistisch wirkender Substanzen graphisch ermittelt.
Die erfindungsgemäßen Verbindungen zeigen in dem beschriebenen in wfro-Testmodell CGRP-antagonistische Eigenschaften in einem Dosisbereich zwischen 10~12 bis 10~5 M.
INDIKATIONSGEBIETE
Aufgrund ihrer pharmakologischen Eigenschaften eignen sich die erfindungsgemäßen Verbindungen und deren Salze mit physiologisch verträglichen Säuren somit zur akuten und prophylaktischen Behandlung von Kopfschmerzen, insbesondere Migräne-, Cluster- Kopfschmerz sowie Spannungskopfschmerzen. Weiterhin beeinflussen die erfindungsgemäßen Verbindungen auch die folgenden Erkrankungen positiv:
Nicht-insulinabhängigen Diabetes mellitus ("NIDDM"), cardiovaskuläre Erkrankungen, Morphintoleranz, Clostridiumtoxin-bedingte Durchfallerkrankungen, Erkrankungen der Haut, insbesondere thermische und strahlenbedingte Hautschäden inklusive Sonnenbrand, Liehen, Prurigo, pruriginöse Toxidermien sowie schwerer Juckreiz, entzündliche Erkrankungen, z.B. entzündliche Gelenkerkrankungen (Osteoarthritis, rheumatoide Arthritis, neurogene Arthritis), generalisierter Weichteilrheumatismus (Fibromyalgie), neurogene Entzündungen der oralen Mucosa, entzündliche Lungenerkrankungen, allergische Rhinitis, Asthma, COPD, Erkrankungen, die mit einer überschießenden Gefäßerweiterung und dadurch bedingter verringerter Gewebedurchblutung einhergehen, z.B. Schock und Sepsis, chronische Schmerzerkrankungen, wie z.B. diabetische Neuropathien, durch Chemotherapien induzierte Neuropathien, HlV-induzierte Neuropathien, postherpetische Neuropathien durch Gewebetrauma induzierte Neuropathien, trigeminale Neuralgien, temporomandibuläre Dysfunktionen, CRPS (complex regional pain Syndrome), Rückenschmerzen, und viszerale Erkrankungen, wie z.B. irritable bowel Syndrome (IBS), inflammatory bowel Syndrome. Darüber hinaus zeigen die erfindungsgemäßen Verbindungen eine lindernde Wirkung auf Schmerzzustände im allgemeinen. Die Symptomatik menopausaler, durch Gefäßerweiterung und erhöhten Blutfluss verursachter Hitzewallungen östrogendefizienter Frauen sowie hormonbehandelter Prostatakarzinompatienten und Kastraten wird durch die CGRP-Antagonisten der vorliegenden Anwendung präventiv und akut-therapeutisch günstig beeinflusst, wobei sich dieser Therapieansatz vor der Hormonsubstitution durch Nebenwirkungsarmut auszeichnet.
Vorzugsweise eignen sich die erfindungsgemäßen Verbindungen zur akuten und prophylaktischen Behandlung von Migräne- und Cluster-Kopfschmerz, zur Behandlung des irritable bowel Syndroms (IBS) und zur präventiven und akut-therapeutischen Behandlung von Hitzewallungen östrogendefizienter Frauen.
Die zur Erzielung einer entsprechenden Wirkung erforderliche Dosierung beträgt zweckmäßigerweise bei intravenöser oder subkutaner Gabe 0.0001 bis 3 mg/kg Körpergewicht, vorzugsweise 0.01 bis 1 mg/kg Körpergewicht, und bei oraler, nasaler oder inhalativer Gabe 0.01 bis 10 mg/kg Körpergewicht, vorzugsweise 0.1 bis 10 mg/kg Körpergewicht, jeweils 1 bis 3 x täglich.
Sofern die Behandlung mit CGRP-Antagonisten oder/und CGRP-Release-Hemmem in Ergänzung zu einer üblichen Hormonsubstitution erfolgt, empfiehlt sich eine Verringerung der vorstehend angegebenen Dosierungen, wobei die Dosierung dann 1/5 der vorstehend angegebenen Untergrenzen bis zu 1/1 der vorstehend angegebenen Obergrenzen betragen kann.
CGRP wird von sensorischen Nerven freigesetzt, beispielsweise vom Nervus Trigeminus, der einen Teil der Gesichtshaut innerviert. Es wurde bereits gezeigt, daß die Reizung des Trigeminalganglions bei Menschen zu einer Erhöhung des CGRP Plasmaspiegels führt und Gesichtsröte hervorruft ([4]: PJ. Goadsby et al., Annais of Neurology, Vol. 23, No. 2, 1988, 193-196,).
Zum Nachweis, dass mit Hilfe von CGRP-Antagonisten der allgemeinen Formel I Hitzewallungen erfolgreich behandelt werden können, wurde eine erhöhte Freisetzung von endogenem CGRP bei Marmosets durch Stimulierung des Trigeminusganglions herbeigeführt, was zu einer erhöhten Durchblutung der Hautgefäße führte. Die Wirksamkeit wurde durch Bestimmung derjenigen i.v. -applizierten Dosis charakterisiert, welche die durch endogenes CGRP hervorgerufene, erhöhte Durchblutung der Gesichtshaut um 50% reduziert. Eine detaillierte Methodenbeschreibung ist in der europäischen Patentschrift EP 1 207 884 B1 offenbart.
Die erfindungsgemäßen CGRP-Antagonisten sind weiterhin aktiv in einem Model für viszerale Schmerzen in Nagetieren. In diesem Model wird eine Überempfindlichkeit des viszeralen Systems durch Reizung des Darms mittels der Einträufelung chemischer Stoffe wie z.B. Butyrat, Trinitrobenzensulfonsäure oder Essigsäure erreicht. Die Überempfindlichkeit des Darms wird z.B. über die Anzahl von abdominalen Kontraktionen ermittelt. Diese entstehen nach Dehnung eines in den Darm eingeführten Ballons und sind in einem überempfindlichen Darm erhöht (Bourdu et al., Gastroenterology 2005, 128, 1996-2008; Diop et al., J. Phamacol. Exp. Ther. 2002, 302, 1013-1022; Plourde et al. Am. J. Physiol. 1997, 273, G191-G196).
Weil die Substanzen die Darmüberempfindlichkeit in dem beschriebenen Modell rückgängig machen, können die erfindungsgemäßen CGRP-Antagonisten für die Behandlung von IBS (irritable bowel Syndrome, Reizdarm) eingesetzt werden.
Ein weiterer Gegenstand der Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen als wertvolle Hilfsmittel zur Erzeugung und Reinigung (Affinitätschromatographie) von Antikörpern sowie, nach geeigneter radioaktiver Markierung, beispielsweise durch Tritiierung geeigneter Vorstufen, beispielsweise durch katalytische Hydrierung mit Trithium oder Ersatz von Halogenatomen durch Tritium, in RIA- und ELISA-Assays und als diagnostische bzw. analytische Hilfsmittel in der Neurotransmitter-Forschung.
KOMBINATIONEN
Als Kombinationspartner denkbare Wirkstoffklassen sind z.B. Antiemetica, Prokinetica, Neuroleptica, Antidepressiva, Neurokinin-Antagonisten, Anticonvulsiva, Histamin-H1- Rezeptorantagonisten, ß-Blocker, α-Agonisten und α-Antagonisten, Ergotalkaloiden, schwachen Analgetica, nichtsteroidalen Antiphlogistica, Corticosteroiden, Calcium- Antagonisten, 5-HT1B/iD-Agonisten oder andere Antimigränemitteln, die zusammen mit einem oder mehreren inerten üblichen Trägerstoffen und/oder Verdünnungsmitteln, z.B. mit Maisstärke, Milchzucker, Rohrzucker, mikrokristalliner Zellulose, Magnesiumstearat, Polyvinylpyrrolidon, Zitronensäure, Weinsäure, Wasser, Wasser/Ethanol, Wasser/- Glycerin, Wasser/Sorbit, Wasser/Polyethylenglykol, Propylenglykol, Cetylstearylalkohol, Carboxymethylcellulose oder fetthaltigen Substanzen wie Hartfett oder deren geeigneten Gemischen, in übliche galenische Zubereitungen wie Tabletten, Dragees, Kapseln, Pulver, Suspensionen, Lösungen, Dosieraerosole oder Zäpfchen eingearbeitet werden können.
Für die oben erwähnten Kombinationen kommen somit als weitere Wirksubstanzen beispielsweise die nicht-steroidalen Antiphlogistika Aceclofenac, Acemetacin, Acetylsalicylsäure, Acetaminophen (Paracetamol), Azathioprin, Diclofenac, Diflunisal, Fenbufen, Fenoprofen, Flurbiprofen, Ibuprofen, Indometacin, Ketoprofen, Leflunomid, Lomoxicam, Mefenaminsäure, Naproxen, Phenylbutazon, Piroxicam, Sulfasalazin, Zomepirac oder deren pharmazeutisch verträgliche Salze sowie Meloxicam und andere selektive COX2-lnhibitoren, wie beispielsweise Rofecoxib, Valdecoxib, Parecoxib, Etoricoxib und Celecoxib, in Betracht sowie Substanzen, die frühere oder spätere Schritte in der Prostaglandin-Synthese inhibieren oder Prostaglandinrezeptor Antagonisten wie z.B. EP2-Rezeptor Antagonisten und I P-Rezeptor Antagonisten.
Weiterhin können Ergotamin, Dihydroergotamin, Metoclopramid, Domperidon, Diphenhydramin, Cyclizin, Promethazin, Chlorpromazin, Vigabatrin, Timolol, Isomethepten, Pizotifen, Botox, Gabapentin, Pregabalin, Duloxetin, Topiramat, Riboflavin, Montelukast, Lisinopril, Micardis, Prochlorperazin, Dexamethason, Flunarizin,
Dextropropoxyphen, Meperidin, Metoprolol, Propranolol, Nadolol, Atenolol, Clonidin, Indoramin, Carbamazepin, Phenytoin, Valproat, Amitryptilin, Imipramine, Venlafaxine, Lidocain oder Diltiazem und andere 5-HT1B/iD-Agonisten wie z.B. Almotriptan, Avitriptan, Eletriptan, Frovatriptan, Naratriptan, Rizatriptan, Sumatriptan und Zolmitriptan verwendet werden.
Außerdem können CGRP-Antagonisten mit Vanilloid-Rezeptor Antagonisten, wie z.B. VR- 1 Antagonisten, Glutamatrezeptor Antagonisten, wie z.B. mGlu5-Rezeptor Antagonisten, mGlui -Rezeptor Antagonisten, iGluδ-Rezeptor Antagonisten, AM PA-Rezeptor Antago- nisten, Purinrezeptor Blockern, wie z.B. P2X3 Antagonisten, NO-Synthase Inhibitoren, wie z.B. iNOS Inhibitoren, Calciumkanal-Blockern, wie z.B. PQ-typ Blockern, N-typ Blockern, Kaliumkanalöffnern, wie z.B. KCNQ Kanalöffnern, Natriumkanal Blockern, wie z.B. PN3 Kanal Blockern, NMDA-Rezeptor Antagonisten, Acid-sensing lonenkanal Antagonisten, wie z.B. ASIC3 Antagonisten, Bradykinin Rezeptor Antagonisten wie z.B. B1-Rezeptor Antagonisten, Cannabinoid-Rezeptoren Agonisten, wie z.B. CB2 Agonisten, CB1
Agonisten, Somatostatin-Rezeptor Agonisten, wie z.B. sst2 Rezeptor Agonisten gegeben werden.
Die Dosis für diese Wirksubstanzen beträgt hierbei zweckmäßigerweise 1/5 der üblicherweise empfohlenen niedrigsten Dosierung bis zu 1/1 der normalerweise empfohlenen Dosierung, also beispielsweise 20 bis 100 mg Sumatriptan.
DARREICHUNGSFORMEN
Die erfindungsgemäß hergestellten Verbindungen können entweder alleine oder gegebenenfalls in Kombination mit anderen Wirksubstanzen zur Behandlung von Migräne intravenös, subkutan, intramuskulär, intraartikulär, intrarektal, intranasal, durch Inhalation, topisch, transdermal oder oral erfolgen, wobei zur Inhalation insbesondere Aerosol-
formulierungen geeignet sind. Die Kombinationen können entweder simultan oder sequentiell verabreicht werden.
Geeignete Anwendungsformen sind beispielsweise Tabletten, Kapseln, Lösungen, Säfte, Emulsionen oder Inhalationspulver oder -aerosole. Hierbei soll der Anteil der pharmazeutisch wirksamen Verbindung(en) jeweils im Bereich von 0.1 bis 90 Gew.-%, bevorzugt 0.5 bis 50 Gew.-% der Gesamtzusammensetzung liegen, d.h. in Mengen die ausreichend sind, um den voranstehend angegebenen Dosierungsbereich zu erreichen.
Die orale Gabe kann in Form einer Tablette, als Pulver, als Pulver in einer Kapsel (z.B. Hartgelatinekapsel), als Lösung oder Suspension erfolgen. Im Fall einer inhalativen Gabe kann die Wirkstoffkombination als Pulver, als wässrige oder wässrig-ethanolische Lösung oder mittels einer Treibgasformulierung erfolgen.
Bevorzugt sind deshalb pharmazeutische Formulierungen gekennzeichnet durch den
Gehalt an einer oder mehrerer Verbindungen der Formel I gemäß der obigen bevorzugten Ausführungsformen.
Besonders bevorzugt ist es, wenn die Verbindungen der Formel I oral verabreicht werden, besonders bevorzugt ist es, wenn die Verabreichung ein oder zweimal täglich erfolgt. Entsprechende Tabletten können beispielsweise durch Mischen des oder der Wirkstoffe mit bekannten Hilfsstoffen, beispielsweise inerten Verdünnungsmitteln, wie Calciumcarbonat, Calciumphosphat oder Milchzucker, Sprengmitteln, wie Maisstärke oder Alginsäure, Bindemitteln, wie Stärke oder Gelatine, Schmiermitteln, wie Magnesium- stearat oder Talk, und/oder Mitteln zur Erzielung des Depoteffektes, wie
Carboxymethylcellulose, Celluloseacetatphthalat, oder Polyvinylacetat erhalten werden. Die Tabletten können auch aus mehreren Schichten bestehen.
Entsprechend können Dragees durch Überziehen von analog den Tabletten hergestellten Kernen mit üblicherweise in Drageeüberzügen verwendeten Mitteln, beispielsweise Kollidon oder Schellack, Gummi arabicum, Talk, Titandioxid oder Zucker, hergestellt werden. Zur Erzielung eines Depoteffektes oder zur Vermeidung von Inkompatibilitäten kann der Kern auch aus mehreren Schichten bestehen. Desgleichen kann auch die
Drageehülle zur Erzielung eines Depoteffektes aus mehreren Schichten bestehen wobei die oben bei den Tabletten erwähnten Hilfsstoffe verwendet werden können.
Säfte der erfindungsgemäßen Wirkstoffe beziehungsweise Wirkstoffkombinationen können zusätzlich noch ein Süßungsmittel, wie Saccharin, Cyclamat, Glycerin oder Zucker sowie ein Geschmack verbesserndes Mittel, z.B. Aromastoffe, wie Vanillin oder Orangenextrakt, enthalten. Sie können außerdem Suspendierhilfsstoffe oder Dickungsmittel, wie Natriumcarboxymethylcellulose, Netzmittel, beispielsweise Kondensationsprodukte von Fettalkoholen mit Ethylenoxid, oder Schutzstoffe, wie p-Hydroxybenzoate, enthalten.
Die eine oder mehrere Wirkstoffe beziehungsweise Wirkstoffkombinationen enthaltenden Kapseln können beispielsweise hergestellt werden, indem man die Wirkstoffe mit inerten Trägern, wie Milchzucker oder Sorbit, mischt und in Gelatinekapseln einkapselt. Geeignete Zäpfchen lassen sich beispielsweise durch Vermischen mit dafür vorgesehenen Trägermitteln, wie Neutralfetten oder Polyäthylenglykol beziehungsweise dessen Derivaten, herstellen.
Als Hilfsstoffe seien beispielsweise Wasser, pharmazeutisch unbedenkliche organische Lösemittel, wie Paraffine (z.B. Erdölfraktionen), öle pflanzlichen Ursprungs (z.B. Erdnuss- oder Sesamöl), mono- oder polyfunktionelle Alkohole (z.B. Ethanol oder Glycerin), Trägerstoffe wie z.B. natürliche Gesteinsmehle (z.B. Kaoline, Tonerden, Talkum, Kreide) synthetische Gesteinsmehle (z.B. hochdisperse Kieselsäure und Silikate), Zucker (z.B. Rohr-, Milch- und Traubenzucker) Emulgiermittel (z.B. Lignin, Sulfitablaugen, Methylcellulose, Stärke und Polyvinylpyrrolidon) und Gleitmittel (z.B. Magnesiumstearat, Talkum, Stearinsäure und Natriumlaurγlsulfat) erwähnt.
Im Falle der oralen Anwendung können die Tabletten selbstverständlich außer den genannten Trägerstoffen auch Zusätze, wie z.B. Natriumeitrat, Calciumcarbonat und Dicalciumphosphat zusammen mit verschiedenen Zuschlagstoffen, wie Stärke, vorzugsweise Kartoffelstärke, Gelatine und dergleichen enthalten. Weiterhin können Gleitmittel, wie Magnesiumstearat, Natriumlaurγlsulfat und Talkum zum Tablettieren mit verwendet werden. Im Falle wässriger Suspensionen können die Wirkstoffe außer den
oben genannten Hilfsstoffen mit verschiedenen Geschmacksaufbesserem oder Farbstoffen versetzt werden.
Ebenfalls bevorzugt ist es, wenn die Verbindungen der Formel I inhalativ verabreicht werden, besonders bevorzugt ist es, wenn die Verabreichung ein oder zweimal täglich erfolgt. Hierzu müssen die Verbindungen der Formel I in inhalierbaren Darreichungsformen bereitgestellt werden. Als inhalierbare Darreichungsformen kommen Inhalationspulver, treibgashaltige Dosieraerosole oder treibgasfreie Inhalationslösungen in Betracht, die gegebenenfalls im Gemisch mit gebräuchlichen physiologisch verträglichen Hilfsstoffen vorliegen.
Im Rahmen der vorliegenden Erfindung sind von dem Begriff treibgasfreie Inhalationslösungen auch Konzentrate oder sterile, gebrauchsfertige Inhalationslösungen umfasst. Die im Rahmen der vorliegenden Erfindung einsetzbaren Darreichungsformen werden im nachfolgenden Teil der Beschreibung detailliert beschrieben.
Inhalationspulver
Sind die Verbindungen der Formel I im Gemisch mit physiologisch unbedenklichen Hilfsstoffen enthalten, können zur Darstellung der erfindungsgemäßen Inhalationspulver die folgenden physiologisch unbedenklichen Hilfsstoffe zur Anwendung gelangen:
Monosaccharide (z.B. Glucose oder Arabinose), Disaccharide (z.B. Lactose, Saccharose, Maltose), Oligo- und Polysaccharide (z.B. Dextrane), Polyalkohole (z.B. Sorbit, Mannit, Xylit), Salze (z.B. Natriumchlorid, Calciumcarbonat) oder Mischungen dieser Hilfsstoffe miteinander. Bevorzugt gelangen Mono- oder Disaccharide zur Anwendung, wobei die Verwendung von Lactose oder Glucose, insbesondere, aber nicht ausschließlich in Form ihrer Hydrate, bevorzugt ist. Als besonders bevorzugt im Sinne der Erfindung gelangt Lactose, höchst bevorzugt Lactose monohyd rat als Hilfsstoff zur Anwendung. Verfahren zur Herstellung der erfindungsgemäßen Inhalationspulver durch Mahlen und Mikronisieren sowie durch abschließendes Mischen der Bestandteile sind aus dem Stand der Technik bekannt.
Treibgashaltige Inhalationsaerosole
Die im Rahmen der erfindungsgemäßen Verwendung einsetzbaren treibgashaltigen Inhalationsaerosole können I im Treibgas gelöst oder in dispergierter Form enthalten. Die
zur Herstellung der Inhalationsaerosole einsetzbaren Treibgase sind aus dem Stand der Technik bekannt. Geeignete Treibgase sind ausgewählt aus der Gruppe bestehend aus Kohlenwasserstoffen wie n-Propan, n-Butan oder Isobutan und Halogenkohlenwasserstoffen wie bevorzugt fluorierten Derivaten des Methans, Ethans, Propans, Butans, Cyclopropans oder Cyclobutans. Die vorstehend genannten Treibgase können dabei allein oder in Mischungen derselben zur Verwendung kommen. Besonders bevorzugte Treibgase sind fluorierte Alkanderivate ausgewählt aus TG 134a (1 , 1, 1, 2-Tetrafluorethan), TG227 (1 , 1 , 1, 2, 3, 3, 3-Heptafluorpropan) und Mischungen derselben. Die im Rahmen der erfindungsgemäßen Verwendung einsetzbaren treibgashaltigen Inhalationsaerosole können ferner weitere Bestandteile wie
Co-Solventien, Stabilisatoren, oberflächenaktive Mittel (Surfactants), Antioxidantien, Schmiermittel sowie Mittel zur Einstellung des pH-Werts enthalten. All diese Bestandteile sind im Stand der Technik bekannt.
Treibgasfreie Inhalationslösungen
Die erfindungsgemäße Verwendung von Verbindungen der Formel I erfolgt bevorzugt zur Herstellung von treibgasfreien Inhalationslösungen und Inhalationssuspensionen. Als Lösungsmittel kommen hierzu wässrige oder alkoholische, bevorzugt ethanolische Lösungen in Betracht. Das Lösungsmittel kann ausschließlich Wasser sein oder es ist ein Gemisch aus Wasser und Ethanol. Die Lösungen oder Suspensionen werden mit geeigneten Säuren auf einen pH-Wert von 2 bis 7, bevorzugt von 2 bis 5 eingestellt. Zur Einstellung dieses pH-Werts können Säuren ausgewählt aus anorganischen oder organischen Säuren Verwendung finden. Beispiele für besonders geeignete anorganische Säuren sind Salzsäure, Bromwasserstoffsäure, Salpetersäure, Schwefelsäure und/oder Phosphorsäure. Beispiele für besonders geeignete organische Säuren sind:
Ascorbinsäure, Zitronensäure, Äpfelsäure, Weinsäure, Maleinsäure, Bernsteinsäure, Fumarsäure, Essigsäure, Ameisensäure und/oder Propionsäure und andere. Bevorzugte anorganische Säuren sind Salzsäure, Schwefelsäure. Es können auch die Säuren verwendet werden, die bereits mit einem der Wirkstoffe ein Säureadditionssalz bilden. Unter den organischen Säuren sind Ascorbinsäure, Fumarsäure und Zitronensäure bevorzugt. Gegebenenfalls können auch Gemische der genannten Säuren eingesetzt werden, insbesondere in Fällen von Säuren, die neben ihren Säuerungseigenschaften auch andere Eigenschaften, z.B. als Geschmackstoffe, Antioxidantien oder Komplexbildner besitzen, wie beispielsweise Zitronensäure oder Ascorbinsäure.
Erfindungsgemäß besonders bevorzugt wird Salzsäure zur Einstellung des pH-Werts verwendet.
Den im Rahmen der erfindungsgemäßen Verwendung einsetzbaren treibgasfreien Inhalationslösungen können Cosolventien und/oder weitere Hilfsstoffe zugesetzt werden. Bevorzugte Cosolventien sind solche, die Hydroxylgruppen oder andere polare Gruppen enthalten, beispielsweise Alkohole - insbesondere Isopropylalkohol, Glykole - insbesondere Propylenglykol, Polyethylenglykol, Polypropylenglykol, Glykolether, Glycerol, Polyoxyethylenalkohole und Polyoxyethylen-Fettsäureester. Unter Hilfs- und Zusatzstoffen wird in diesem Zusammenhang jeder pharmakologisch verträgliche Stoff verstanden, der kein Wirkstoff ist, aber zusammen mit dem (den) Wirkstoffen) in dem pharmakologisch geeigneten Lösungsmittel formuliert werden kann, um die qualitativen Eigenschaften der Wirkstoffformulierung zu verbessern. Bevorzugt entfalten diese Stoffe keine oder im Kontext mit der angestrebten Therapie keine nennenswerte oder zumindest keine unerwünschte pharmakologische Wirkung. Zu den Hilfs- und Zusatzstoffen zählen z.B. oberflächenaktive Stoffe, wie z.B. Sojalecithin, Ölsäure, Sorbitanester, wie Polysorbate, Polyvinylpyrrolidon sonstige Stabilisatoren, Komplexbildner, Antioxidantien und/oder Konservierungsstoffe, die die Verwendungsdauer der fertigen Arzneimittelformulierung gewährleisten oder verlängern, Geschmackstoffe, Vitamine und/oder sonstige dem Stand der Technik bekannte
Zusatzstoffe. Zu den Zusatzstoffen zählen auch pharmakologisch unbedenkliche Salze wie beispielsweise Natriumchlorid als Isotonantien. Zu den bevorzugten Hilfsstoffen zählen Antioxidantien, wie beispielsweise Ascorbinsäure, sofern nicht bereits für die Einstellung des pH-Werts verwendet, Vitamin A, Vitamin E, Tocopherole und ähnliche im menschlichen Organismus vorkommende Vitamine oder Provitamine. Konservierungsstoffe können eingesetzt werden, um die Formulierung vor Kontamination mit Keimen zu schützen. Als Konservierungsstoffe eignen sich die dem Stand der Technik bekannten, insbesondere Cetylpyridiniumchlorid, Benzalkoniumchlorid oder Benzoesäure bzw. Benzoate wie Natriumbenzoat in der aus dem Stand der Technik bekannten Konzentration.
EXPERIMENTELLER TEIL
Für die hergestellten Verbindungen liegen in der Regel 1H-NMR und Massenspektren vor.
Wenn nicht anders angegeben, werden RrWerte unter Verwendung von DC-Fertigplatten
Kieselgel 60 F254 (E. Merck, Darmstadt, Artikel-Nr. 1.05714) ohne Kammersättigung bestimmt. Die unter der Bezeichnung Polygram-Kieselgel ermittelten Rf-Werte werden unter Verwendung von DC-Fertigfolien Polygram SIL G/UV254 (beschichtet mit 0.2 mm
Kieselgel) der Firma Macherey-Nagel (Düren, Artikel-Nr. 805 021) erhoben.
Die unter der Bezeichnung Polygram-Alox ermittelten Rf-Werte werden unter Verwendung von DC-Fertigfolien Polygram Alox N/UV254 (beschichtet mit 0.2 mm Aluminiumoxid) der
Firma Macherey-Nagel (Düren, Artikel-Nr. 802 021) erhoben.
Die bei den Fliessmitteln angegebenen Verhältnisse beziehen sich auf Volumeneinheiten der jeweiligen Lösungsmittel. Die angegebenen Volumeneinheiten bei NH3 beziehen sich auf eine konzentrierte Lösung von NH3 in Wasser.
Soweit nicht anders vermerkt sind die bei den Aufarbeitungen der Reaktionslösungen verwendeten Säure-, Basen- und Salzlösungen wässrige Systeme der angegebenen
Konzentrationen.
Zu chromatographischen Reinigungen wird Kieselgel der Firma Millipore (MATREX™, 35-
70 μm) verwendet.
Zu chromatographischen Reinigungen wird Aluminiumoxid (Alox) der Firma ICN
Biomedicals (Eschwege, Artikelnummer 02090) verwendet. Gemäss Herstellerangaben wird vor Verwendung die benötigte Aktivitätsstufe (Aktivitätsstufe M-III) erzeugt.
Die angegebenen HPLC-Daten werden unter nachstehend angeführten Parametern gemessen:
Methode A:
Analytische Säule: Merck Chromolith Speed ROD, RP18e; 4.6 x 50 mm; Säulentemperatur: 30°C; Fluss: 1.5 mL / min; Injektionsvolumen: 5 μL; Detektion bei 254 nm
Methode B:
Analytische Säule: Zorbax-Säule (Agilent Technologies), SB (Stable Bond) - C18; 3.5 μm; 4.6 x 75 mm; Säulentemperatur: 30°C; Fluss: 1.6 mL / min; Injektionsvolumen: 5 μL; Detektion bei 254 nm
Methode C:
Analytische Säule: Zorbax-Säule (Agilent Technologies), SB (Stable Bond) - C18; 3.5 μm; 4.6 x 75 mm; Säulentemperatur: 30°C; Fluss: 0.8 mL / min; Injektionsvolumen: 5 μL; Detektion bei 254 nm
Bei präparativen HPLC-Reinigungen werden in der Regel die gleichen Gradienten verwendet, die bei der Erhebung der analytischen HPLC-Daten benutzt wurden. Die Sammlung der Produkte erfolgt massengesteuert, die Produkt enthaltenden Fraktionen werden vereinigt und gefriergetrocknet.
Falls nähere Angaben zur Konfiguration fehlen, bleibt offen, ob es sich um reine Enantiomere handelt oder ob partielle oder gar völlige Racemisierung eingetreten ist.
In den Versuchsbeschreibungen werden die folgenden Abkürzungen verwendet:
Boc fert-Butoxycarbonyl
Cyc Cyclohexan
DCM Dichlormethan
DIPE Diisopropylether DMF N,N-Dimethylformamid
EtOAc Essigsäureethylester
EtOH Ethanol h Stunde
HCl Salzsäure HOAc Essigsäure i. vac. in vacuo (im Vakuum) min Minute
MeOH Methanol
MTBE Methyl-fert-Butylether NaOH Natriumhydroxid
PE Petrolether
RT Raumtemperatur
TBTU 2-(1H-Benzotriazol-1-yl)-1 ,1 ,3,3-tetramethyluronium-Tetrafluorborat
TFA Trifluoressigsäure THF Tetrahydrofuran
Im Folgenden wird die Herstellung von Ausgangsverbindungen beschrieben:
Amin 1
S-μ^'jBipiperidinyl-i-yl-propionsäureethylester
A1a) 1'-(2-Ethoxycarbonyl-ethyl)-r4,4'lbipiperidinyl-1-carbonsäure-fe/f-butylester Zu einer Lösung von 10.0 g (37.3 mmol) ^^'JBipiperidinyl-i-carbonsäure-fert-butylester in 100 mL EtOH wurden 4.4 mL (40.6 mmol) Acrylsäureethylester gegeben und das Reaktionsgemisch 2 h unter Rückfluss erhitzt. Zur Vervollständigung der Reaktion wurden weitere 1 mL (9.2 mmol) Acrylsäureethylester zugegeben, 1 h unter Rückfluss erhitzt und über Nacht bei RT belassen. Man entfernte i.vac. das Lösungsmittel und setzte das Rohprodukt ohne Reinigung weiter um. Ausbeute: 14.0 g (100% der Theorie) ESI-MS: (M+H)
+ = 369
A1 b) 3-r4,4'lBipiperidinyl-1-yl-propionsäureethylester
Zu einer Lösung von 14.0 g des Rohproduktes aus Beispiel A1a in 250 mL DCM wurden 28 mL TFA zugetropft und das Reaktionsgemisch 4 h bei RT gerührt. Man engte i.vac. ein, nahm den Rückstand in 200 mL DCM auf und gab diese Lösung portionenweise zu einer Lösung von 20 g Na2CO3 in 120 mL Wasser. Die organische Phase wurde abgetrennt, die wässrige Phase noch zweimal mit DCM extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand getrocknet und ohne weitere Reinigung umgesetzt. Ausbeute: 8.8 g (88% der Theorie) ESI-MS: (M+H)+ = 269
Amin 2
3-(4-Piperazin-1-yl-piperidin-1-yl)-propionsäureethylester
A2a) 3-|4-(4-Benzyl-piperazin-1-yl)-piperidin-1-vπ-propionsäureethylester Zu einer Lösung von 11.7 g (44.9 mmol) 1-Benzyl-4-piperidin-4-yl-piperazin in 120 mL trockenem EtOH wurden 5.5 mL (50.8 mmol) Acrylsäureethylester gegeben und das Reaktionsgemisch 1 h unter Rückfluss erhitzt und dann über Nacht bei RT gerührt. Man entfernte das Lösungsmittel i.vac. und trocknete den Rückstand 1 h im ölpumpenvakuum. Das Rohprodukt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 16.5 g (99% der Theorie)
ESI-MS: (M+H)+ = 360
A2b) 3-(4-Piperazin-1 -yl-piperidin-1 -vD-propionsäureethylester Eine Suspension von 16.5 g des Rohproduktes aus Beispiel A2a und 1.6 g 10 % Pd/C in 200 mL EtOH und wurde 4 h bei 50°C und 50 psi Wasserstoffdruck hydriert. Der Katalysator wurde abgesaugt, das Filtrat auf ca. 120 mL eingeengt und mit 72 mL ethanolischer HCl (1.3 M) versetzt. Der ausgefallene Niederschlag wurde abgesaugt und i.vac. getrocknet.
Das Produkt wurde als Bis-Hydrochlorid Salz erhalten. Ausbeute: 12.6 g (83% der Theorie) ESI-MS: (M+H)+ = 270
Amin 3
3-(4-Piperidin-4-yl-piperazin-1-yl)-propionsäureethylester
A3a) 3-r4-(1-Benzyl-piperidin-4-yl)-piperazin-1-yll-propionsäureethylester
Zu einer Lösung von 11.0 g (33.2 mmol) 1-(1-Benzyl-piperidin-4-yl)-piperazin (eingesetzt als Bis-Hydrochlorid Salz) in 40 mL EtOH wurden 12.5 mL (73.0 mmol) Ethyldiisopropyl- amin und 5.0 mL (46.1 mmol) Acrylsäureethylester gegeben und das Reaktionsgemisch 3 h auf 90°C (Badtemperatur) erhitzt. Nach dem Abkühlen wurde mit Wasser versetzt, erschöpfend mit EtOAc extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Kieselgel, DCM/EtOH/NH3 100:10:1) gereinigt. Ausbeute: 6.8 g (56% der Theorie) ESI-MS: (M+H)+ = 360
Rf = 0.64 (Kieselgel, DCM/MeOH/NH390:9: 1 )
A3b) 3-(4-Piperidin-4-yl-piperazin-1-yl)-propionsäureethylester
Eine Suspension von 5.13 g (14.3 mmol) 3-[4-(1-Benzyl-piperidin-4-yl)-piperazin-1-yl]- propionsäureethylester und 1.0 g 10% Pd/C in 100 mL EtOH wurde 2 h bei 50°C und 50
psi Wasserstoffdruck hydriert. Der Katalysator wurde abfiltriert und das Filtrat zur Trockene eingeengt. Das ölige Produkt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 3.6 g (93% der Theorie) ESI-MS: (M+H)+ = 270
Amin 4
μ^'JBipiperidinyl-i-yl-oxo-essigsäureethylester
A4a) 1'-Ethoxyoxalyl-r4,4'lbipiperidinyl-1-carbonsäure-ferf-butylester Zu einer auf 0°C gekühlten Lösung von 4.0 g (14.9 mmol) [4,4']Bipiperidinyl-1-carbon- säure-fert-butylester und 2.15 mL (15.4 mmol) Triethylamin in 80 mL DCM wurden 1.68 mL (15.0 mmol) Chlor-oxo-essigsäureethylester zugetropft. Nach beendeter Zugabe wurde das Kältebad entfernt und 1 h bei RT gerührt. Das Reaktionsgemisch wurde mit Wasser versetzt, die organische Phase abgetrennt und über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand in EtOAc aufgenommen, die Lösung über Kieselgel filtriert und i.vac. eingeengt. Ausbeute: 3.1 g (57% der Theorie) ESI-MS: (M+H)+ = 386
A4b) r4,4'lBipiperidinyl-1-yl-oxo-essigsäureethylester
Zu einer Lösung von 3.1 g (8.36 mmol) i'-Ethoxyoxalyl-^^'Jbipiperidinyl-i-carbonsäure- fert-butylester in 40 mL DCM wurden 5.0 mL TFA zugetropft und das Reaktionsgemisch 4 h bei RT gerührt. Man engte i.vac. ein, nahm den Rückstand in 50 mL DCM auf und gab diese Lösung portionenweise zu einer eiskalten Lösung von 4.0 g Na2CO3 in 20 mL Wasser. Die organische Phase wurde abgetrennt, die wässrige Phase noch zweimal mit DCM extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde das Produkt als öl erhalten, welches ohne Reinigung weiter umgesetzt wurde. Ausbeute: 2.3 g (84% der Theorie) ESI-MS: (M+H)+ = 269
Amin 5
4-[4,4l]Bipiperidinyl-1-yl-4-oxo-buttersäureethylester
A5a) 1'-(3-Carboxy-propionyl)-r4,4'lbipiperidinyl-1-carbonsäure-fe/f-butylester
Zu einer Lösung von 10.0 g (37.3 mmol) ^^'JBipiperidinyl-i-carbonsäure-fert-butylester in
100 mL THF wurde bei RT eine Lösung von 4.1 g (40.7 mmol) Bernsteinsäureanhydrid in 50 mL THF zugetropft und das Reaktionsgemisch über Nacht bei RT gerührt. Zur
Vervollständigung der Reaktion wurden weitere 2.0 g (19.9 mmol) Bernsteinsäureanhydrid zugegeben, 4 h bei 50°C und über Nacht bei RT gerührt. Man versetzte mit 200 mL 7.5% K2CO3-Lösung und wusch die wässrige Phase mit 200 mL EtOAc. Die organische Phase wurde mit 200 mL 7.5% K2CO3-Lösung extrahiert und die vereinigten wässrigen Phasen mit Zitronensäure sauer gestellt. Man extrahierte erschöpfend mit EtOAc und engte die vereinigen organischen Phasen i.vac. ein. Ausbeute: 11.7 g (85% der Theorie) ESI-MS: (M+H)+ = 369
A5b) 4-r4,4'lBipiperidinyl-1-yl-4-oxo-buttersäureethylester
Eine Lösung von 11.7 g (31.7 mmol) i'-ß-Carboxy-propionylJ-^'ppiperidinyl-i-carbon- säure-fert-butylester in 250 mL ethanolischer HCl (1.25 M) wurde über Nacht bei RT gerührt. Man entfernte i.vac. das Lösungsmittel und erhielt das Produkt als Hydrochlorid- SaIz, welches ohne Reinigung weiter umgesetzt wurde. Ausbeute: 4.3 g (46% der Theorie) ESI-MS: (M+H)+ = 297
Amin 6
([i ^'JBipiperidinyl^-yl-fert-butoxycarbonyl-aminoJ-essigsäureethylester
A6a) (1'-Benzyl-ri ,4'lbipiperidinyl-4-yl)-carbanninsäure-fe/f-butylester Eine Lösung von 5.0 g (25.0 mmol) Piperidin-4-yl-carbaminsäure-fert-butylester und 4.46 mL (25.0 mmol) 1-Benzyl-piperidin-4-on in 150 mL THF wurde mit AcOH auf einen pH- Wert von 5 eingestellt und dann unter Eiskühlung portionenweise mit 5.61 g (26.5 mmol) NaBH(OAc)3 innerhalb von 3 h versetzt. Das Reaktionsgemisch wurde über Nacht bei RT nachgerührt, dann mit 500 mL 30% K2CO3-Lösung alkalisch gestellt, 1 h bei RT nachgerührt, dreimal mit je 100 mL EtOAc extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt. Ausbeute: 7.0 g (75% der Theorie)
A6b) 1 '-Benzyl-H ,4'lbipiperidinyl-4-ylamin Eine Lösung von 7.0 g (18.7 mmol) (i'-Benzyl-II^'Jbipiperidinyl^-ylJ-carbaminsäure-fert- butylester und 14.3 mL (185 mmol) TFA in 80 mL DCM wurde über Nacht unter Rückfluss erhitzt. Man engte i.vac. ein, versetzte den Rückstand mit 200 mL 30% K2CO3-Lösung und extrahierte mit dreimal mit je 100 mL EtOAc, wobei der erste 100 mL-Extrakt verworfen wurde. Die vereinigten organischen Phasen wurden über Na2SO4 getrocknet, filtriert und eingeengt. Das Produkt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 5.1 g (100% der Theorie)
A6c) (1'-Benzyl-ri ,4'lbipiperidinyl-4-ylamino)-essigsäureethylester
Zu einer auf 0°C gekühlten Lösung von 1.8 g (6.58 mmol) 1 '-Benzyl-[1 ,4']bipiperidinyl-4- ylamin, 2.69 mL (13.0 mmol) Oxo-essigsäureethylester (eingesetzt als 50% Lösung in Toluol) und 1 mL (17.45 mmol) AcOH in 250 mL THF wurden portionenweise 2.79 g (13.17 mmol) NaBH(OAc)3 gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man engte i.vac. ein, nahm den Rückstand in EtOAc auf, wusch die organische Phase mit gesättigter K2CO3-Lösung und trocknete über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt, das organische Lösungsmittel (Acetonitril)
i.vac. entfernt, der wässrige Rückstand erschöpfend mit DCM extrahiert und die vereinigten organischen Phasen über MgSO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels erhielt man das Produkt als gelbes öl. Ausbeute: 1.25 g (53% der Theorie) ESI-MS: (M+H)+ = 360
Rf = 0.35 (Kieselgel, DCM/MeOH/NH390: 10:1)
A6d) r(1'-Benzyl-ri ,4'lbipiperidinyl-4-yl)-fe/f-butoxycarbonyl-aminol-essigsäureethylester Zu einer Lösung von 1.20 g (3.34 mmol) (i'-Benzyl-ti ^bipiperidinyM-ylaminoJ-essig- säureethylester in 15 mL DCM wurden 3.34 mL (30.0 mmol) Triethylamin zugetropft und dann portionenweise mit 0.73 g (3.34 mmol) Boc-Anhydrid versetzt. Das Reaktionsgemisch wurde 70 h bei RT gerührt und dann i.vac. eingeengt. Der Rückstand wurde in EtOAc aufgenommen, mit 15% K2CO3-Lösung gewaschen und über Na2CO3 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt.
Ausbeute: 1.3 g (85% der Theorie)
A6e) (ri ,4'lBipiperidinyl-4-yl-fe/f-butoxycarbonyl-amino)-essigsäureethylester
Eine Suspension von 1.30 g (2.83 mmol) [(i'-Benzyl-II^'Jbipiperidinyl^-ylJ-fert-butoxy- carbonyl-aminoj-essigsäureethylester und 0.16 g 10% Pd/C in 25 mL EtOH wurde 5 h bei 50°C und 50 psi Wasserstoffdruck hydriert. Der Katalysator wurde abgesaugt und das Filtrat zur Trockene eingeengt. Man erhielt das Produkt als farbloses öl, welches ohne Reinigung weiter umgesetzt wurde. Ausbeute: 1.00 g (96% der Theorie) ESI-MS: (M+H)+ = 370
Amin 7
(4-Methyl-4-piperazin-1-yl-piperidin-1-yl)-essigsäureethylester
A7a) 1 -Benzyl-4-piperazin-1 -yl-piperidin-4-carbonitril
Eine Mischung aus 11.0 g (49.8 mmol) 1-Benzyl-4-hydroxy-piperidin-4-carbonitril und 22.0 g (255 mmol) Piperazin in 200 mL MeOH wurde 2 h unter Rückfluss erhitzt. Der Niederschlag wurde abgesaugt, das Filtrat i.vac. eingeengt, der Rückstand in wenig Wasser aufgenommen, erschöpfend mit DCM extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Alox, DCM/MeOH 30:1) gereinigt. Ausbeute: 2.38 g (16% der Theorie) ESI-MS: (M+H)+ = 285 Rf = 0.37 (Polygram-Alox, DCM/MeOH 25:1)
A7b) 1 -(1 -Benzyl-4-methyl-piperidin-4-yl)-piperazin
Zu 2.37 g (7.92 mmol) 1-Benzyl-4-piperazin-1-yl-piperidin-4-carbonitril in 100 mL trockenem THF wurden bei RT 15 mL Methylmagnesiumchlorid-Lösung (45 mmol, 3 M in THF) zugegeben und das Reaktionsgemisch 3 h gerührt. Man versetzte mit gesättigter NH4CI-Lösung, rührte 10 min nach, wusch die wässrige Phase mit EtOAc, versetzte mit 4 M NaOH-Lösung bis zur alkalischen Reaktion, extrahierte erschöpfend mit DCM und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt. Ausbeute: 0.64 g (30% der Theorie) ESI-MS: (M+H)+ = 274
A7c) 4-(1-Benzyl-4-methyl-piperidin-4-yl)-piperazin-1-carbonsäure-fe/f-butylester Zu 1.63 g (5.66 mmol) 1-(1-Benzyl-4-methyl-piperidin-4-yl)-piperazin in 50 mL THF wurden 1.35 g (6.00 mmol) Boc-Anhydrid gegeben und das Reaktionsgemisch 3 h bei RT gerührt. Anschließend wurde i.vac. zur Trockene eingeengt und der Rückstand ohne Reinigung weiter umgesetzt. Ausbeute: 2.10 g (100% der Theorie) ESI-MS: (M+H)+ = 374
A7d) 4-(4-Methyl-piperidin-4-yl)-piperazin-1-carbonsäure-fe/f-butyl-ester Eine Suspension von 2.28 g (5.62 mmol) 4-(1-Benzyl-4-methyl-piperidin-4-yl)-piperazin- 1-carbonsäure-fert-butylester und 300 mg 10% Pd/C in 50 mL MeOH wurde 3 h bei 50°C und 3447 hPa Wasserstoffdruck hydriert. Zur Vervollständigung der Reaktion wurden 0.47
mL konz. HCl zugegeben und weitere 3 h bei 50°C und 3447 hPa Wasserstoffdruck hydriert. Der Katalysator wurde abfiltriert, das Filtrat i.vac. eingeengt, der Rückstand mit Diethylether verrührt, abgesaugt und getrocknet. Ausbeute: 1.54 g (86% der Theorie) ESI-MS: (M+H)+ = 284
A7e) 4-(1-Ethoxycarbonylmethyl-4-methyl-piperidin-4-yl)-piperazin-1-carbonsäure-fe/f- butylester Eine Mischung von 1.53 g (4.78 mmol) 4-(4-Methyl-piperidin-4-yl)-piperazin-1-carbon- säure-fert-butyl-ester und 1.1 mL (5.55 mmol) Oxo-essigsäureethylester (50% in Toluol) in 50 mL THF wurde 1 h bei RT gerührt. Das Reaktionsgemisch wurde auf 0°C gekühlt, portionsweise mit 1.25 g (5.90 mmol) Natriumtriacetoxyborhydrid versetzt und nach Entfernen des Kältebades über Nacht bei RT gerührt. Man versetzte mit 10 mL 20% NaHCO3-Lösung, extrahierte erschöpfend mit EtOAc und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Alox, DCM/EtOH 100:1) gereinigt. Ausbeute: 0.47 g (27% der Theorie) ESI-MS: (M+H)+ = 370
A7f) (4-Methyl-4-piperazin-1-yl-piperidin-1-yl)-essigsäureethylester
Zu 0.46 g (1.25 mmol) 4-(1-Ethoxycarbonylmethyl-4-methyl-piperidin-4-yl)-piperazin- 1-carbonsäure-fert-butylester in 5 mL DCM wurden bei 0°C 2 mL TFA zugegeben und das Reaktionsgemisch 2 h bei RT gerührt. Dieses wurde i.vac. eingeengt und das Rohprodukt, welches als Bis-trifluoracetat-Salz anfiel, ohne Reinigung weiter umgesetzt. Ausbeute: 0.65 g (100% der Theorie) ESI-MS: (M+H)+ = 270
Amin 8
[4-(4-Methyl-piperidin-4-yl)-piperazin-1-yl]-essigsäureethylester
A8a) r4-(1-Benzyl-4-methyl-piperidin-4-yl)-piperazin-1-yll-essigsäureethylester Hergestellt analog Beispiel A7e aus 0.62 g (2.27 mmol) 1-(1-Benzyl-4-methyl-piperidin-4- yl)-piperazin (Beispiel A7b) und 0.55 mL (2.77 mmol) Oxo-essigsäureethylester (50% in Toluol). Das Rohprodukt wurde chromatographisch (Alox, Gradient PE/EtOAc 2:1 zu 1 :1) gereinigt.
Ausbeute: 0.45 g (50% der Theorie)
ESI-MS: (M+H)+ = 360
Rf = 0.56 (Polygram-Alox, PE/EtOAc 1 :1)
A8b) r4-(4-Methyl-piperidin-4-yl)-piperazin-1-yll-essigsäureethylester
Eine Suspension von 0.44 g (1.10 mmol) [4-(1-Benzyl-4-methyl-piperidin-4-yl)-piperazin- 1-yl]-essigsäureethylester und 100 mg 10% Pd/C in 20 mL EtOH wurde 12 h bei 50°C und 3447 hPa Wasserstoffdruck hydriert. Der Katalysator wurde abfiltriert und das Filtrat zur Trockene eingeengt. Das Produkt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 0.29 g (97% der Theorie) ESI-MS: (M+H)+ = 270
Amin 9
(S)-4-Methyl-1-piperidin-4-yl-piperazin-2-carbonsäureethylester
A9a) (S)-Piperazin-1 ,2,4-tricarbonsäure-1-benzylester-4-fe/f-butylester-2-ethylester Zu einer auf 0°C gekühlten Mischung aus 12.2 g (32.8 mmol) (S)-Piperazin-1 ,2,4-tri- carbonsäure-i-benzylester-4-ferf-butylester und 30 mL EtOH in 150 mL THF wurden 11.2 g (34.9 mmol) TBTU und 5.5 mL (39.6 mmol) Triethylamin zugegeben, weitere 30 min bei dieser Temperatur und anschließend 68 h bei RT gerührt. Zum Reaktionsgemisch wurden 600 mL Diethylether gegeben, mit 200 mL gesättigter NaHCO3-Lösung versetzt, die wässrige Phase abgetrennt, die organische Phase mit gesättigter NaCI-Lösung gewaschen und über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Kieselgel, Gradient PE/EtOAc 4:1 zu 7:3) gereinigt.
Ausbeute: 11.35 g (88% der Theorie)
ESI-MS: (M+H)+ = 393
Rf = 0.38 (Polygram-Kieselgel, PE/EtOAc 3:1)
A9b) (S)-Piperazin-I ,2-dicarbonsäure-1 -benzylester-2-ethylester
Zu einer auf 0°C gekühlten Mischung von 3.27 g (8.33 mmol) (S)-Piperazin-1 ,2,4-tri- carbonsäure-1-benzylester-4-ferf-butylester-2-ethylester und 30 mL DCM wurden 10 mL TFA zugegeben, 10 min unter Eiskühlung und 2 h bei RT gerührt. Man engte i.vac. bei 30°C ein, nahm den Rückstand erneut in EtOAc auf und engte erneut i.vac. ein. Das Rohprodukt, welches als Trifluoracetat-Salz anfiel, wurde ohne Reinigung weiter umgesetzt.
A9c) (S)-4-Methyl-piperazin-1 ,2-dicarbonsäure-1-benzylester-2-ethylester
Zu einer Mischung aus 4.4 g des Rohproduktes aus A9b) und 1.2 mL (16.0 mmol) Form- aldehyd (37% in Wasser) in 80 mL THF wurden 1.00 g (12.2 mmol) NaOAc und 10 g Molsieb A3 zugegeben und das Reaktionsgemisch 2 h bei RT gerührt. Nach Kühlen auf 0°C wurde portionenweise mit 3.39 g (16.0 mmol) Natriumtriacetoxyborhydrid versetzt, 30 min bei dieser Temperatur und 68 h bei RT gerührt. Man filtrierte von unlöslichen Bestandteilen ab, gab zum Filtrat gesättigte K2CO3-Lösung, rührte 15 min, versetzte mit EtOAc, trennte die organische Phase ab und trocknete diese über Na2SO4. Nach
Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Alox, PE/EtOAc 2:1) gereinigt. Ausbeute: 2.04 g (81 % der Theorie) ESI-MS: (M+H)+ = 307 Rf = 0.73 (Polygram-Alox, PE/EtOAc 1 :1)
A9d) (S)-4-Methyl-piperazin-2-carbonsäureethylester
Eine Suspension von 2.04 (6.66 mmol) (S)-4-Methyl-piperazin-1,2-dicarbonsäure- 1 -benzylester-2-ethylester und 200 mg 10% Pd/C in 100 mL EtOH wurde bei 50°C und 3447 hPa Wasserstoffdruck 6 h hydriert. Der Katalysator wurde abfiltriert, das Filtrat i.vac. zur Trockene eingeengt und der Rückstand ohne Reinigung weiter umgesetzt. Ausbeute: 1.06 g (92% der Theorie) ESI-MS: (M+H)+ = 173
A9e) (SVI-fi-fe/f-ButoxycarbonvI-piperidin^-vIM-methyl-piperazin^-carbonsäure- ethylester
Eine Mischung von 0.90 g (5.23 mmol) (S)-4-Methyl-piperazin-2-carbonsäureethylester und 1.20 g (6.02 mmol) 4-Oxo-piperidin-i-carbonsäure-ferf-butylester in 20 mL EtOH wurde mit 50 μL Ameisensäure und 3 g Molsieb A3 versetzt und das Reaktionsgemisch 66 h bei RT belassen. Man filtrierte das Molsieb ab, versetzte mit 100 mg 10% Pd/C und hydrierte bei 50°C und 3447 hPa Wasserstoffdruck 4 h. Der Katalysator wurde abgesaugt, das Filtrat i.vac. eingeengt und der Rückstand chromatographisch (Kieselgel, Gradient DCM/EtOH 98:2 zu 75:25) gereinigt. Ausbeute: 0.35 g (19% der Theorie) ESI-MS: (M+H)+ = 356
Rf = 0.5 (Polygram-Alox, DCM/MeOH 50:1 )
A9f) (S)-4-Methyl-1-piperidin-4-yl-piperazin-2-carbonsäureethylester Eine Mischung von 0.34 g (0.96 mmol) (S)-1-(1-fert-Butoxycarbonyl-piperidin-4-yl)-4- methyl-piperazin-2-carbonsäure-ethylester in 6 mL ethanolischer HCl (1.25 M) wurde 1 h unter Rückfluss erhitzt. Nach Abkühlen des Reaktionsgemisches wurde der Niederschlag abgesaugt und getrocknet. Das Produkt fiel als Tris-Hydrochlorid-Salz an. Ausbeute: 0.33 g (95% der Theorie) ESI-MS: (M+H)+ = 256
Amin 10
(S)-1-(1-Methyl-piperidin-4-yl)-piperazin-2-carbonsäureethylester
A10a) (S)-Piperazin-1 ,3-dicarbonsäure-1-fe/f-butyl-ester-3-ethylester Eine Suspension von 4.00 g (10.2 mmol) (S)-Piperazin-1 ,2,4-tricarbonsäure-1-benzyl- ester-4-fert-butylester-2-ethylester (Beispiel A9a) und 200 mg 10% Pd/C in 100 mL EtOH wurde 2 h bei 50°C und 3447 hPa Wasserstoffdruck hydriert. Der Katalysator wurde abfiltriert, das Filtrat i.vac. eingeengt und der Rückstand ohne Reinigung weiter umgesetzt.
Ausbeute: 2.61 g (98% der Theorie) ESI-MS: (M+H)+ = 259
A10b) (S)-4-(1 -Methyl-piperidin-4-yl)-piperazin-1 ,3-dicarbonsäure-1 -fe/f-butylester-3- ethylester
Eine Mischung aus 2.65 g (10.05 mmol) (S)-Piperazin-1,3-dicarbonsäure-1-ferf-butyl- ester-3-ethylester und 1.36 mL (11.06 mmol) 1-Methyl-piperidin-4-on in 100 mL THF wurde 1 h bei RT gerührt. Nach Kühlen auf 0°C wurden portionenweise 3.00 g (14.16 mmol) Natriumtriacetoxyborhydrid zugegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Zur Vervollständigung der Reaktion wurde mit weiteren 1.00 g (4.72 mmol) Natriumtriacetoxyborhydrid und 0.3 mL AcOH versetzt und erneut 48 h bei RT gerührt. Man versetzte mit 40 mL gesättigter K2CO3-Lösung, rührte 15 min, extrahierte erschöpfend mit EtOAc und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatogra- phisch (Alox, DCM/EtOH 100:1) gereinigt. Ausbeute: 1.93 g (53% der Theorie) ESI-MS: (M+H)+ = 356
A10c) (S)-1-(1-Methyl-piperidin-4-yl)-piperazin-2-carbonsäureethylester Eine Mischung aus 1.88 (5.18 mmol) (S)-4-(1-Methyl-piperidin-4-yl)-piperazin-1 ,3-di- carbonsäure-1-fert-butylester-3-ethylester, 3 mL EtOH und 15 mL ethanolische HCl (1.25 M) wurde über Nacht bei RT gerührt. Zur Vervollständigung der Reaktion wurde 1 h unter Rückfluss erhitzt. Nach Abkühlen des Reaktionsgemisches wurde der Niederschlag abgesaugt und getrocknet. Das Produkt fiel als Bis-Hydrochlorid-Salz an. Ausbeute: 1.35 g (79% der Theorie) ESI-MS: (M+H)+ = 256
Amin 11
(S)-4-(1-Methyl-piperidin-4-yl)-piperazin-2-carbonsäureethylester
A11a) (S)-4-(1 -Methyl-piperidin-4-yl)-piperazin-1 ,2-dicarbonsäure-1 -benzylester-2- ethylester
Eine Mischung von 4.20 g (10.13 mmol) (S)-Piperazin-1,2-dicarbonsäure-1-benzylester- 2-ethylester (Beispiel A9b, eingesetzt als Trifluoracetat-Salz) und 1.37 mL 1-Methyl- piperidin-4-on in 100 mL THF wurde 1 h bei RT gerührt. Nach Kühlen auf 0°C wurden portionenweise 3.00 g (14.16 mmol) Natriumtriacetoxyborhydrid zugegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man versetzte mit 40 mL gesättigter K2CO3-Lösung, rührte weitere 15 min, extrahierte erschöpfend mit EtOAc und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Alox, DCM/EtOH 100:1) gereinigt.
Ausbeute: 2.17 g (55% der Theorie) ESI-MS: (M+H)+ = 390
Rf = 0.43 (Polygram-Alox, DCM/MeOH 50: 1 )
A11b) (S)-4-(1-Methyl-piperidin-4-yl)-piperazin-2-carbonsäureethylester Eine Suspension von 2.15 g (5.52 mmol) (S)-4-(1-Methyl-piperidin-4-yl)-piperazin-1,2-di- carbonsäure-1-benzylester-2-ethylester und 100 mg 10% Pd/C in 50 mL EtOH wurde 2 h bei 50°C und 3447 hPa Wasserstoffdruck hydriert. Der Katalysator wurde abfiltriert und das Filtrat zur Trockene eingeengt. Das Produkt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 1.35 g (96% der Theorie) ESI-MS: (M+H)+ = 256
Amin 12
(S-Piperazin-i-yl-δ-aza-bicyclop^.iJoct-δ-yl)-essigsäureethylester
A12a) 3-(4-Benzyloxycarbonyl-piperazin-1-yl)-8-aza-bicvclor3.2.1loctan-8-carbonsäure- ferf-butylester
Eine Mischung aus 5.00 g (22.2 mmol) 3-Oxo-8-aza-bicyclo[3.2.1]octan-8-carbonsäure- fert-butylester und Piperazin-1-carbonsäurebenzylester in 60 mL THF wurde mit AcOH
auf einen pH-Wert von 5 eingestellt und 1 h bei RT gerührt. Man versetzte portionenweise unter Eiskühlung mit 5.64 g (26.6 mmol) Natriumtriacetoxyborhydrid und rührte das Reaktionsgemisch über Nacht bei RT. Man gab mit 150 mL 15% K2CO3-Lösung zu, trennte die organische Phase ab, extrahierte die wässrige Phase erschöpfend mit EtOAc und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Kieselgel, EtOAc) gereinigt.
Es konnten 2 isomere Produkte isoliert werden: Isomer A: Ausbeute: 2.10 g (22% der Theorie) ESI-MS: (M+H)+ = 430
Rf = 0.55 (Kieselgel, EtOAc) Retentionszeit (HPLC): 3.2 min (Methode B)
Isomer B:
Ausbeute: 2.20 g (23% der Theorie) ESI-MS: (M+H)+ = 430
Rf = 0.68 (Kieselgel, EtOAc)
A12b) 4-(8-Aza-bicvclor3.2.1loct-3-yl)-piperazin-1-carbonsäurebenzylester
Zu einer Mischung aus 2.10 g (4.89 mmol) 3-(4-Benzyloxycarbonyl-piperazin-1-yl)-8-aza- bicyclo[3.2.1]octan-8-carbonsäure-fert-butylester (Isomer A) in 60 mL DCM wurden langsam 6.01 mL (78.0 mmol) TFA gegeben und das Reaktionsgemisch 2 bei RT gerührt. Man engte i.vac. ein, nahm den Rückstand in 15% K2CO3-Lösung auf, extrahierte erschöpfend mit DCM und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde das Produkt ohne Reinigung weiter umgesetzt.
Ausbeute: 1.50 g (93% der Theorie) ESI-MS: (M+H)+ = 330 Rf = 0.16 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
A12c) 4-(8-Ethoxycarbonylmethyl-8-aza-bicvclor3.2.1loct-3-yl)-piperazin-1-carbonsäure- benzylester
Zu 1.50 g (4.55 mmol) 4-(8-Aza-bicyclo[3.2.1]oct-3-yl)-piperazin-1-carbonsäure-benzyl- ester (A12b) in 10 mL DMF wurden 1.29 g (9.30 mmol) K2CO3 zugegeben, dann langsam 0.56 mL (5.00 mmol) Bromessigsäureethylester zugetropft und das Reaktionsgemisch weitere 4 h bei RT gerührt. Man filtrierte von unlöslichen Bestandteilen ab, versetzte mit EtOAc, wusch die organische Phase zweimal mit gesättigter NaHCO3-Lösung und trocknete über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt. Ausbeute: 1.75 g (92% der Theorie) ESI-MS: (M+H)+ = 416 Rf = 0.72 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
A12d) O-Piperazin-i-yl-S-aza-bicvcloß^.iloct-δ-vD-essigsäureethvIester
Eine Suspension von 1.70 g (4.09 mmol) 4-(8-Ethoxycarbonylmethyl-8-aza-bicyclo[3.2.1]- oct-3-yl)-piperazin-1-carbonsäurebenzylester (A12c) und 200 mg 10% Pd/C in 30 mL EtOH wurde bei RT und 3000 hPa Wasserstoffdruck 3 h geschüttelt. Der Katalysator wurde abgesaugt und das Filtrat zur Trockene eingeengt. Das Produkt wurde ohne
Reinigung weiter umgesetzt.
Ausbeute: 1.10 g (96% der Theorie)
ESI-MS: (M+H)+ = 282 Rf = 0.21 (Kieselgel, DCM/MeOH/NH3 80:20:2)
Amin 13
([1 ,4']Bipiperidinyl-4-yloxy)-essigsäureethylester
A13a) 4-ferf-Butoxycarbonylmethoxy-ri ,4'lbipiperidinyl-1'-carbonsäurebenzylester Zu einer Mischung aus 2.58 g (11.06 mmol) 4-Oxo-piperidin-1-carbonsäurebenzylester und 2.80 g (12.36 mmol) (Piperidin-4-yloxy)-essigsäure-fert-butylester in 30 mL THF wurden portionenweise 2.90 g (13.27 mmol) Natriumtriacetoxyborhydrid gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man versetzte mit 50 mL 1 M NaOH, rührte 1 h bei RT, versetzte mit EtOAc, trennte die organische Phase ab und trocknete diese
über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand Chromatograph isch (Alox, DCM/MeOH 100:1) gereinigt. Ausbeute: 3.10 g (65% der Theorie) ESI-MS: (M+H)+ = 433
A13b) (ri ,4'lBipiperidinyl-4-yloxy)-essigsäure-fe/f-butylester
Eine Suspension von 3.08 g (7.12 mmol) 4-fert-Butoxycarbonylmethoxy-[1,4l]bipiperidinyl- r-carbonsäurebenzylester und 300 mg 10% Pd/C in 60 mL MeOH wurde 2 h bei 50°C und 3447 hPa Wasserstoffdruck hydriert. Der Katalysator wurde abgesaugt und das Filtrat zur Trockene eingeengt. Das Produkt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 2.15 g (99% der Theorie) ESI-MS: (M+H)+ = 299
A13c) (ri ,4'lBipiperidinyl-4-yloxy)-essigsäureethylester Zu 2.02 g (6.63 mmol) ([1 ^'JBipiperidinyl^-yloxyJ-essigsäure-fert-butylester in 20 mL EtOH wurden 20 mL ethanolische HCl (1.25M) gegeben und das Reaktionsgemisch 3 h unter Rückfluss erhitzt. Nach Abkühlen auf 0°C wurde der ausgefallene Niederschlag abgesaugt und im Hochvakuum getrocknet. Das Produkt, das als Bis-Hydrochlorid-Salz anfiel, wurde ohne Reinigung weiter umgesetzt. Ausbeute: 1.74 g (76% der Theorie) ESI-MS: (M+H)+ = 271
Amin 14
[1 ^'JBipiperidinyl^-yl-essigsäureethylester
A14a) (1 '-Benzyl-[1 ,4']bipiperidinyl-4-yl)-essigsäureethylester
Zu 11.8 g (56.8 mmol) Piperidin-4-yl-essigsäureethylester in 100 mL THF wurden 50 mL DCM und 11.7 mL (62.5 mmol) 1-Benzyl-piperidin-4-on gegeben und das Reaktionsgemisch 2 h bei RT gerührt. Man versetzte portionenweise mit 13.7 g (62.5 mmol) Natriumtriacetoxyborhydrid und rührte weitere 36 h bei RT. Zum Reaktionsgemisch wurden 100 mL 10% NaOH gegeben, zweimal mit je 100 mL MTBE extrahiert und die
vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand mittels HPLC gereinigt. Ausbeute: 3.27 g (17% der Theorie) ESI-MS: (M+H)+ = 345 Rf = 0.55 (Kieselgel, EtOAc/MeOH/NH3 90:10:1)
A14b) ri ,4'lBipiperidinyl-4-yl-essigsäureethylester
Eine Suspension von 3.24 g (9.41 mmol) (i'-Benzyl-II^'Jbipiperidinyl^-ylJ-essigsäure- ethylester und 300 mg 10% Pd/C in 50 mL EtOH wurde bei RT und 3000 hPa Wasser- stoffdruck bis zur theoretischen Aufnahme an Wasserstoff hydriert. Der Katalysator wurde abgesaugt, das Filtrat zur Trockene eingeengt, der Rückstand mit 50 mL EtOH und ethanolischer HCl (1.25 M) versetzt. Man engte i.vac. zur Trockene ein, verrührte den Rückstand mit 100 mL DIPE/Isopropanol (2:1), filtrierte den Niederschlag ab und trocknete diesen bei 35°C im Umlufttrockenschrank. Das Produkt fiel als Bis-Hydrochlorid- Salz an.
Ausbeute: 2.90 g (94% der Theorie)
ESI-MS: (M+H)+ = 255
Rf = 0.05 (Kieselgel, EtOAc/MeOH/NH3 70:30:3)
Amin 15
3-[1,4']Bipiperidinyl-4-yl-propionsäureethylester
A15a) 4-(2-Methoxycarbonyl-ethyl)-ri ,4'lbipiperidinyl-1'-carbonsäure-fe/f-butylester
Unter Stickstoffatmosphäre wurde eine Mischung aus 4.00 g (19.3 mmol) 3-Piperidin-4-yl- propionsäuremethylester und 3.85 g (19.3 mmol) 4-Oxo-piperidin-1-carbonsäure-fert- butylester in 50 mL THF mit AcOH auf einen pH-Wert von 5 eingestellt und 1 h bei RT nachgerührt. Nach Abkühlen auf 0°C wurde portionenweise mit 5.15 g (24.3 mmol) Natriumtriacetoxyborhydrid versetzt und das Reaktionsgemisch über Nacht bei RT gerührt. Man tropfte innerhalb 10 min 90 mL 30% K2CO3-Lösung zu, extrahierte dreimal mit EtOAc und trocknete die vereinigten organischen Phasen über Na2SO4. Nach
Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt.
Ausbeute: 5.40 g (79% der Theorie) ESI-MS: (M+H)+ = 355 Rf = 0.63 (Kieselgel, DCM/MeOH/NH3 80:20:2)
A15b) 3-ri ,4'lBipiperidinyl-4-yl-propionsäureethylester
5.40 g (15.2 mmol) 4-(2-Methoxycarbonyl-ethyl)-[1,4l]bipiperidinyl-1'-carbonsäure-fert- butylester in 150 mL ethanolischer HCl (1.25 M) wurde über Nacht bei RT gerührt. Man zog i.vac. das Lösungsmittel weitgehend ab, filtrierte den entstandenen Niederschlag ab und trocknete diesen. Das Produkt, das als Bis-Hydrochlorid-Salz anfiel, wurde ohne
Reinigung weiter umgesetzt.
Ausbeute: 2.30 g (79% der Theorie)
ESI-MS: (M+H)+ = 269 Retentionszeit (HPLC): 1.2 min (Methode B)
Amin 16
4-(4-Piperazin-1-yl-piperidin-1-yl)-buttersäureethylester
A16a) 4-r4-(4-Benzyl-piperazin-1-yl)-piperidin-1-vH-buttersäureethylester
Unter Stickstoffatmosphäre wurde eine Mischung aus 3.11 g (12.0 mmol) 1-Benzyl-4- piperidin-4-yl-piperazin und 7.50 mL (12.0 mmol, 15% in Wasser) 4-Oxo-buttersäure in 70 mL THF mit AcOH auf einen pH-Wert von 5 eingestellt und 1 h bei RT nachgerührt. Nach Abkühlen auf 0°C wurde portionenweise mit 5.35 g (24.0 mmol) Natriumtriacetoxybor- hydrid versetzt und das Reaktionsgemisch über Nacht bei RT gerührt. Man tropfte innerhalb von 15 min 80 mL 30% K2CO3-Lösung zu, wusch die wässrige Phase zweimal mit EtOAc und engte diese i.vac. zur Hälfte ein. Man versetzte mit 1 M KHSO4-Lösung, saugte den entstandenen Niederschlag ab, wusch das Filtrat mit EtOAc und engte die wässrige Phase i.vac zur Trockene ein. Man nahm den Rückstand in 150 mL ethanolischer HCl (1.25 M) auf und rührte das Reaktionsgemisch über Nacht bei RT. Man engte i.vac. ein, nahm den Rückstand in wenig 15% K2CO3-Lösung auf, extrahierte erschöpfend
mit EtOAc und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt.
Ausbeute: 2.90 g (65% der Theorie) ESI-MS: (M+H)+ = 374
A16b) 4-(4-Piperazin-1 -yl-piperidin-1 -vD-buttersäureethylester
Eine Suspension von 2.90 g (7.76 mmol) 4-[4-(4-Benzyl-piperazin-1-yl)-piperidin-1-yl]- buttersäureethylester und 300 mg 10% Pd/C in 60 mL MeOH wurde 24 h bei RT und 3447 hPa Wasserstoffdruck hydriert. Der Katalysator wurde abgesaugt, das Filtrat zur Trockene eingeengt, der Rückstand in DIPE und wenig Isopropanol aufgenommen und mit 4 M HCl in 1,4-Dioxan versetzt. Der Niederschlag wurde abgesaugt und getrocknet. Das Produkt fiel als Bis-Hydrochlorid-Salz an. Ausbeute: 2.50 g (90% der Theorie) ESI-MS: (M+H)+ = 284
Retentionszeit (HPLC): 0.7 min (Methode B)
Amin 17
4-Piperazin-1-yl-piperidin-1-carbonsäurebenzylester
A17a) 4-(1-Benzyloxycarbonyl-piperidin-4-yl)-piperazin-1-carbonsäure-fe/f-butylester Zu 12.3 g (45.7 mmol) 4-Piperidin-4-yl-piperazin-1-carbonsäure-fert-butylester und 8.2 mL (50 mmol) N-Ethyldiisopropylamin in 200 mL DCM wurde unter Eiskühlung eine Lösung von 7.18 mL (48.0 mmol) Chlorameisensäurebenzylester in 50 mL DCM zugetropft und das Reaktionsgemisch über Nacht bei RT nachgerührt. Dieses wurde mit 200 mL 15% K2CO3-Lösung gewaschen, die organische Phase abgetrennt und über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt.
Ausbeute: 16.0 g (87% der Theorie)
A17b) 4-Piperazin-1 -yl-piperidin-1 -carbonsäurebenzylester
Zu 16.0 g (39.7 mmol) 4-(1-Benzyloxycarbonyl-piperidin-4-yl)-piperazin-1-carbonsäure- fert-butylester in 200 mL DCM wurden bei RT 25 mL TFA zugegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man engte i.vac. ein, nahm den Rückstand in 200 mL Wasser und 200 mL EtOAc auf, trennte die wässrige Phase ab, versetzte diese mit 50 mL 15% K2CO3-Lösung, extrahierte zweimal mit je 200 mL EtOAc und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne weitere Reinigung umgesetzt. Ausbeute: 4.00 g (33% der Theorie) ESI-MS: (M+H)+ = 304
Im Folgenden wird die Herstellung der Endverbindungen beschrieben:
Beispiel 1
1 '-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-S-yl)-piperidin-i-carbonyloxyJ-propionylJ-i ^'-bipiperidinyl^-carbonsäure ethylester
1a) (Z,E)-2-Acetylamino-3-(4-amino-3-chlor-5-trifluormethyl-phenyl)-acrylsäure Zu einer Suspension von 50.0 g (224 mmol) 4-Amino-3-chlor-5-trifluormethyl-benzaldehyd und 27.5 g (335 mmol) NaOAc in 202 mL Acetanhydrid wurden 39.7 g (335 mmol) N-Acetylglycin gegeben und das Reaktionsgemisch 1 h auf 115°C erhitzt. Nach Abkühlen auf 80°C wurden 100 mL Wasser zugetropft, wobei die Temperatur des Gemisches bei 80°C gehalten wurde. Die Suspension wurde erneut 40 min auf 95°C erhitzt und dann auf eine Mischung aus 250 mL Toluol und 500 mL Wasser gegeben. Die Suspension wurde bei RT nachgerührt, der Niederschlag abgesaugt und bei 60°C im Umlufttrockenschrank getrocknet. Ausbeute: 48.8 g (68% der Theorie)
ESI-MS: (M+H)+ = 321/323 (Cl)
Rf = 0.37 (Kieselgel, DCM/MeOH/AcOH 90:10:1)
1 b) 3-(4-Amino-3-chlor-5-trifluornnethyl-phenyl)-2-oxo-propionsäure Eine Suspension von 97.0 g (300 mmol) (Z,£)-2-Acetylamino-3-(4-amino-3-chlor-5-tri- fluormethyl-phenyl)-acrylsäure in 900 mL 1,4-Dioxan und 1050 mL 4 M HCl wurde 8 h auf 100°C erhitzt. Man engte i.vac auf ca. 600 mL ein, kühlte auf RT ab, filtrierte die ausgefallene Substanz ab, wusch diese mit zweimal je 100 mL Wasser und trocknete bei 50°C. Der Rückstand wurde in 850 mL Toluol aufgenommen, unter Rückfluss erhitzt und anschließend im Eisbad gekühlt. Der entstandene Niederschlag wurde filtriert, mit PE gewaschen und im Umlufttrockenschrank bei 50°C getrocknet. Ausbeute: 63.0 g (74% der Theorie) ESI-MS: (M-H)- = 280/282 (Cl)
Rf = 0.21 (Kieselgel, DCM/MeOH/NH3 80:20:2)
1c) (f?)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-hvdroxy-propionsäure Zu einer auf ca. -30°C gekühlten Lösung von 63.0 g (224 mmol) 3-(4-Amino-3-chlor-5- trifluormethyl-phenyl)-2-oxo-propionsäure und 31.2 mL (224 mmol) Triethylamin in 300 mL THF wurde eine Lösung von 100.0 g (312 mmol) (1R)-B-Chlordiisopinocampheylboran in 150 mL THF zugetropft und das Reaktionsgemisch 1.5 h bei dieser Temperatur gehalten und dann innerhalb einer weiteren Stunde auf RT erwärmt. Zum Reaktionsgemisch wurden mit 80 mL 4 M NaOH gegeben, 5 min nachgerührt, auf 0°C gekühlt, mit 300 mL MTBE versetzt, erneut 20 min bei dieser Temperatur nachgerührt und anschließend die Phasen getrennt. Die organische Phase wurde erschöpfend mit Wasser extrahiert, die vereinigten wässrigen Phasen mit 4 M HCl sauer gestellt, erschöpfend mit MTBE extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet. Die THF/MTBE/NaOH Phase wurde mit 4 M HCl sauer gestellt, die Phasen getrennt und die organische Phase i.vac. eingeengt. Beide Rückstände wurden vereinigt und ohne Reinigung weiter umgesetzt. Rf = 0.20 (Kieselgel, DCM/MeOH/NH3 80:20:2)
1d) (f?)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-hvdroxy-propionsäuremethylester Das Rohprodukt aus Beispiel 1c (62 g) wurde in 300 mL MeOH gelöst und zu dieser Lösung langsam 3.65 mL (50 mmol) SOCI2 zugetropft. Das Reaktionsgemisch wurde
weitere 3 h bei RT gerührt, dann i.vac. eingeengt, der Rückstand in DCM aufgenommen und über Kieselgel filtriert. Die Lösung wurde i.vac. eingeengt und der Rückstand chromatographisch (Kieselgel, DCM/MeOH/NH3 80:20:2) gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt, i.vac. eingeengt, der Rückstand mit PE versetzt, abgesaugt und getrocknet.
Ausbeute: 43.1 g (65% der Theorie über 2 Stufen)
ESI-MS: (M+H)+ = 298/300 (Cl)
Rf = 0.86 (Kieselgel, DCM/MeOH/NH3 80:20:2)
1e) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-2-
(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-methoxycarbonyl-ethylester Unter Stickstoffatmosphäre wurden bei 60°C (Badtemperatur) zu 100 mL Pyridin innerhalb von 10 min eine Lösung von 13.5 g (65.0 mmol) Chlorameisensäure-4-nitrophenyl- ester in 40 mL THF zudosiert, 10 min nachgerührt, dann eine Lösung von 18.0 g (60.5 mmol) (R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-hydroxy-propionsäure-methyl- ester in 50 ml Pyridin zugetropft und das Reaktionsgemisch 1.5 h bei dieser Temperatur gehalten. Dann erfolgte portionenweise die Zugabe von 15.9 g (65.0 mmol) 3-Piperidin-4- yl-1 ,3,4,5-tetrahydro-1,3-benzdiazepin-2-on. Die Temperatur des Reaktionsgemisches wurde auf 100 °C erhöht, dieses 6 h bei dieser Temperatur gehalten und anschließend über Nacht bei RT nachgerührt. Man engte i.vac. ein, nahm den Rückstand in 200 mL EtOAc auf, wusch die organische Phase zweimal mit je 100 mL 1 M KHSO4-Lösung, zehnmal mit je 50 mL 15% K2CO3-Lösung und trocknete diese über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt. Ausbeute: 33.1 g (96% der Theorie) ESI-MS: (M+H)+ = 569/571 (Cl)
Rf = 0.72 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
1f) 4-(2-Oxo-1 ,2,4,5-tetrahvdro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-2- (4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy-ethylester
Zu einer Lösung von 33.0 g (58.0 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-methoxy- carbonyl-ethylester in 200 mL THF wurde eine Lösung von 2.11 g (88.0 mmol) LiOH in 100 mL Wasser zugegeben und die Reaktionslösung 3.5 h bei RT gerührt. THF wurde
i.vac. entfernt, der wässrige Rückstand zweimal mit MTBE gewaschen, mit 2 M HCl sauer gestellt, erschöpfend mit DCM extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand bei 65°C in 80 mL Isopropanol gelöst und über Nacht langsam auf RT abgekühlt. Die Suspension wurde im Eisbad gekühlt, abgesaugt, mit wenig Isopropanol und DIPE gewaschen und getrocknet.
Ausbeute: 26.2 g (81 % der Theorie)
ESI-MS: (M+H)+ = 555/557 (Cl)
Rf = 0.18 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2) Retentionszeit (HPLC): 4.0 min (Methode B)
1g) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-1 - (4-amino-3-chlor-5-trifluormethyl-benzyl)-2-oxo-2-(4-oxo-piperidin-1-yl)-ethylester
Zu einer Lösung von 10.0 g (18.0 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 50 mL DMF wurden 7.40 g (23.0 mmol) TBTU und 5.84 mL (40.0 mmol)
Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 2.77 g (18.0 mmol) Piperidin-4-on (eingesetzt als Hydrat des Hydrochlorid-Salzes) gegeben und dieses über Nacht bei RT gerührt. Die Reaktionslösung wurde auf 1 L 7% K2CO3- Lösung gegossen, die ausgefallene Substanz abfiltriert, mit Wasser gewaschen und bei
60°C 6 h getrocknet. Weitere Reinigung erfolgte durch Säulenchromatographie (Kieselgel,
EtOAc)
Ausbeute: 7.5 g (65% der Theorie)
ESI-MS: (M+H)+ = 636/638 (Cl) Rf = 0.25 (Kieselgel, EtOAc)
1 h) 1'-Uffl-3-(4-Amino-3-chlor-5-trifluormethyl-phenvn-2-r4-(2-oxo-1 ,2,4,5-tetrahvdro- 1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonyloxyl-propionyll-i ,4'-bipiperidinyl-4- carbonsäureethylester Zu einer Lösung von 127 mg (0.2 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-oxo-2-(4- oxo-piperidin-1-yl)-ethylester in 2 mL THF/MeOH (2:1) wurden 62.9 mg (0.4 mmol) Piperidin-4-carbonsäureethylester und 11 μL (0.2 mmol) AcOH gegeben und das Reaktionsgemisch 2 h bei RT gerührt. Anschließend wurde auf 0°C gekühlt, nach 2 h mit
10.6 mg (0.16 mmol) NaBH3CN versetzt und über Nacht bei O°C gerührt. Man ließ das Lösungsmittel verdunsten, nahm den Rückstand in 2 mL DMF auf und reinigte das Rohprodukt via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 68 mg (44% der Theorie)
ESI-MS: (M+H)+ = 777/779 (Cl)
Retentionszeit (HPLC): 7.0 min (Methode C)
Beispiel 1.1
1 l-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-S-yl)-piperidin-i-carbonyloxyl-propionylJ-i ^'-bipiperidinyM-carbonsäure
Zu einer Lösung von 140 mg (0.18 mmol) 1l-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl- phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonyloxy]- propionyl}-1 ,4'-bipiperidinyl-4-carbonsäureethylester in 1 mL THF wurde eine Lösung von 7.2 mg (0.3 mmol) LiOH in 1 mL Wasser gegeben und das Reaktionsgemisch 3 h bei RT gerührt. Man versetzte mit 1 mL HCl (1 M) und reinigte das Rohprodukt via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 53 mg (39% der Theorie)
ESI-MS: (M+H)+ = 749/751 (Cl)
Retentionszeit (HPLC): 3.4 min (Methode B)
Beispiel 2
4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(ethoxycarbonylmethyl-amino)-1 ,4
1- bipiperidinyl-1'-yl]-2-oxo-ethylester
2a) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-1 - (4-amino-3-chlor-5-trifluornnethyl-benzyl)-2-r4-(fe/f-butoxycarbonyl-ethoxycarbonyl- methylamino)-1 ,4'-bipiperidinyl-1 '-yll-2-oxo-ethylester
Zu einer Lösung von 200 mg (0.36 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 2 mL DMF wurden 128 mg (0.40 mmol) TBTU und 56 μL (0.40 mmol) Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 148 mg (0.40 mmol) ([1,4']Bipiperidinyl-4-yl-fert-butoxycarbonyl-amino)-essigsäureethylester (Amin A6) gegeben und dieses über Nacht bei RT gerührt. Die Reaktionslösung wurde i.vac. eingeengt, der Rückstand in EtOAc aufgenommen, der ausgefallene Niederschlag abgesaugt und getrocknet. Das Produkt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 330 mg (100% der Theorie)
2b) 4-(2-Oxo-1 ,2,4,5-tetrahvdro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-1 - (4-amino-3-chlor-5-trifluormethyl-benzyl)-2-r4-(ethoxycarbonylmethyl-amino)-1 ,4'- bipiperidinyl-1'-yll-2-oxo-ethylester Zu einer auf 0°C gekühlten Lösung von 330 mg (0.36 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-(fert-butoxycarbonyl-ethoxycarbonyl-methylamino)-1 ,4l-bipiperidinyl-1l-yl]-2- oxo-ethylester in 5 mL DCM wurden 0.5 mL TFA gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man engte i.vac. ein und reinigte den Rückstand via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 263 mg (90% der Theorie)
ESI-MS: (M+H)+ = 806/808 (Cl)
Retentionszeit (HPLC): 2.6 min (Methode A)
Beispiel 2.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-S-chlor-S-trifluormethyl-benzyl^-^-^arboxynnethyl-anninoJ-i ^'-bipiperidinyl-i'-yl]-
2-oxo-ethylester
Zu einer Lösung von 212 mg (0.26 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(ethoxy- carbonylmethylaminoj-i ^'-bipiperidinyl-i'-ylj^-oxo-ethylester in 9 mL THF wurde eine Lösung von 9.4 mg (0.39 mmol) LiOH gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Zur Vervollständigung der Reaktion wurde erneut mit 6.3 mg (0.26 mmol) LiOH versetzt und erneut über Nacht bei RT gerührt. Das Reaktionsgemisch wurde ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet, wobei das Produkt als Trifluoracetat-Salz anfiel. Ausbeute: 130 mg (55% der Theorie) ESI-MS: (M+H)+ = 778/780 (Cl)
Retentionszeit (HPLC): 2.7 min (Methode A)
Beispiel 3
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(1l-ethoxycarbonylmethyl-4,4l-bipiperidinyl-1-yl)- 2-oxo-ethylester
Zu einer Lösung von 1.0 g (1.80 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1.S-benzdiazepin-S- yl)-piperidin-i-carbonsäure-^^-^-amino-S-chlor-S-trifluormethyl-phenyl)-^^ ethylester in 10 mL THF wurden 632 mg (1.97 mmol) TBTU und 0.34 mL (1.97 mmol) Ethyldiisopropylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 500 mg (1.97 mmol) [4,4']Bipiperidinyl-1-yl-essigsäure-ethylester gegeben und dieses über Nacht bei RT gerührt. Man engte i.vac. ein, nahm den Rückstand in DCM auf, wusch die organische Phase mit 1 M KHSO
4-Lösung und 15% K
2CO
3-Lösung und trocknete über Na
2SO
4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt, i.vac. eingeengt, der Rückstand mit DIPE verrieben, abgesaugt und getrocknet. Ausbeute: 150 mg (11% der Theorie)
ESI-MS: (M+H)+ = 791/793 (Cl)
Rf = 0.46 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 3.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(1l-carboxymethyl-4,4l-bipiperidinyl-1-yl)-2-oxo- ethylester
Zu einer Lösung von 30 mg (0.04 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(1 '-ethoxy- carbonylmethyl^^'-bi-piperidinyl-i-yl^-oxo-ethylester in 2 mL THF wurde eine Lösung von 1 mg (0.04 mmol) LiOH in 1 mL Wasser gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man engte i.vac. ein, versetzte den Rückstand mit wenig Wasser und 1 M HCl bis zur sauren Reaktion. Der entstandene Niederschlag wurde abgesaugt und getrocknet. Ausbeute: 20 mg (66% der Theorie) ESI-MS: (M+H)+ = 763/765 (Cl)
Rf = 0.31 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 3.2
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(1l-nnethoxycarbonylnnethyl-4,4l-bipiperidinyl-1-yl)- 2-oxo-ethylester
Zu einer Lösung von 200 mg (0.26 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(1 '- carboxymethyl-4,4'-bipiperidinyl-1-yl)-2-oxo-ethylester in 15 mL THF wurden 100 mg (0.31 mmol) TBTU und 0.06 mL (0.34 mmol) Ethyldiisopropylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurde 1 mL (24.6 mmol) MeOH gegeben und dieses über Nacht bei RT gerührt. Man engte i.vac. ein, nahm den Rückstand in 2 mL DMF auf und reinigte das Rohprodukt via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 80 mg (39% der Theorie) ESI-MS: (M+H)+ = 777/779 (Cl) Rf = 0.60 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Analog wurden folgende Verbindungen aus jeweils 200 mg 4-(2-Oxo-1,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-(1
l-carboxymethyl-4,4
l-bipiperidinyl-1-yl)-2-oxo-ethylester und 1 mL des jeweiligen Alkohols hergestellt:
Beispiel 3.7
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-oxo-2-(1l-phenoxycarbonylnnethyl-4,4l-bipiperi- dinyl-1-yl)-ethylester
Zu einer Lösung von 100 mg (0.13 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(1 '- carboxymethyl-4,4'-bipiperidinyl-1-yl)-2-oxo-ethylester in 1.5 mL DMF wurden 50 mg (0.16 mmol) TBTU und 37 μL (0.26 mmol) Triethylamin zugegeben und 1 h bei RT gerührt. Es wurden 14.8 mg (0.16 mmol) Phenol zugegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man filtrierte die Reaktionslösung über einen Spritzenfilter und reinigte das Rohprodukt via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Der Rückstand wurde in DCM gelöst, i.vac. eingeengt, mit DIPE verrieben, abgesaugt und getrocknet. Ausbeute: 48 mg (44% der Theorie)
ESI-MS: (M+H)+ = 839/841 (Cl)
Rf = 0.47 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Retentionszeit (HPLC): 3.2 min (Methode B)
Analog wurden folgende Verbindungen aus jeweils 100 mg (Beispiele 3.8 bis 3.12), 120 mg (Beispiel 3.13) oder 90 mg (Beispiele 3.14 und 3.15) 4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-(1l-carboxymethyl-4,4l-bipiperidinyl-1-yl)-2-oxo-ethylester und der jeweils nötigen Menge der Alkoholkomponente hergestellt:
Beispiel 3.16
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[1l-(2,2-dimethylpropionyloxymethoxycarbonyl- methyl)-4,4'-bipiperidinyl-1-yl]-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.13 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(1 '- carboxymethyl-4,4'-bipiperidinyl-1-yl)-2-oxo-ethylester in 2 mL DMF wurden 27 mg (0.20 mmol) K
2CO
3 und 28 μL (0.20 mmol) 2,2-Dimethyl-propionsäurechlormethylester gegeben und das Reaktionsgemisch 2 h bei RT gerührt. Man engte i.vac. ein, nahm den Rückstand in 50 mL DCM auf, wusch die organische Phase mit 20 mL Wasser und trocknete über Na
2SO
4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Kieselgel, DCM/MeOH/NH
3 97:3:0.3) gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt, eingeengt, der Rückstand mit wenig Diethylether/DIPE verrieben, abgesaugt und getrocknet. Ausbeute: 39 mg (34% der Theorie)
ESI-MS: (M+H)+ = 877/879 (Cl)
Rf = 0.47 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Retentionszeit (HPLC): 3.3 min (Methode B)
Beispiel 3.17
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(1l-dimethylcarbamoylmethoxycarbonylmethyl- 4,4'-bipiperidinyl-1-yl)-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.13 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(1 '- carboxymethyl-4,4'-bipiperidinyl-1-yl)-2-oxo-ethylester in 1 mL DMF wurden 48 mg (0.15 mmol) TBTU, 21 μL (0.15 mmol) Triethylamin und 15 mg (0.15 mmol) 2-Hydroxy-N,N- dimethyl-acetamid gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man filtrierte die Reaktionslösung über einen Spritzenfilter und reinigte das Rohprodukt via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und i.vac. eingeengt. Ausbeute: 23 mg (21% der Theorie)
ESI-MS: (M+H)+ = 848/850 (Cl)
Retentionszeit (HPLC): 2.9 min (Methode B)
Beispiel 4
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1-ethoxycarbonylnnethyl-piperidin-4-yl)- piperazin-1-yl]-2-oxo-ethylester
Zu einer Lösung von 0.80 g (1.44 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 30 mL THF wurden 0.52 g (1.62 mmol) TBTU und 0.28 mL (1.61 mmol) Ethyldiisopropylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 0.40 g (1.57 mmol) (4-Piperazin-1-yl-piperidin-1-yl)-essigsäureethylester gegeben und dieses über Nacht bei RT gerührt. Man versetzte mit 40 mL EtOAc, wusch die organische Phase mit 15% K2CO3-Lösung und trocknete über MgSO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Kieselgel, Gradient DCM zu DCM/EtOH/NH3 70:30:3) gereinigt. Ausbeute: 0.68 g (60% der Theorie) ESI-MS: (M+H)+ = 792/794 (Cl)
Rf = 0.70 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 4.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1-carboxymethyl-piperidin-4-yl)-piperazin-1- yl]-2-oxo-ethylester
Zu einer Lösung von 380 mg (0.48 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1 - ethoxycarbonylmethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester in 20 mL THF wurde eine Lösung von 25 mg (1.02 mmol) LiOH in 20 mL Wasser gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man entfernte i.vac. das organische Lösungsmittel und versetzte mit 20 mL Wasser und 1.1 mL 1 M HCl. Man engte i.vac. ein, nahm den Rückstand in 5 mL DMF auf und reinigte das Rohprodukt via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 114 mg (31 % der Theorie) ESI-MS: (M+H)+ = 764/766 (Cl)
Rf = 0.07 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 4.2
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1-butoxycarbonylmethyl-piperidin-4-yl)- piperazin-1-yl]-2-oxo-ethylester
Zu einer Lösung von 80 mg (0.11 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1 - carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester in 1.5 mL DMF wurden 51 mg (0.16 mmol) TBTU und 29 μL (0.34 mmol) Triethylamin gegeben und 30 min bei RT
geschüttelt. Zum Reaktionsgemisch wurden 100 μL (1.09 mmol) 1-Butanol gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde das
Rohprodukt via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet.
Ausbeute: 46 mg (54% der Theorie)
ESI-MS: (M+H)+ = 820/822 (Cl)
Rf = 0.67 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Analog wurden folgende Verbindungen aus jeweils 80 mg 4-(2-Oxo-1 ,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-(1-carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester und 100 μL der jeweiligen Alkoholkomponente hergestellt:
Analog wurden folgende Verbindungen aus jeweils 80 mg 4-(2-Oxo-1 ,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-(1-carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester und 1.5 Äquivalenten (Beispiele 4.5 und 4.6) bzw. 1.6 Äquivalenten (Beispiele 4.7 und 4.8) der jeweiligen Alkoholkomponente hergestellt:
Beispiel 4.9
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzodiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(2-nnorpholin-4-yl-ethoxycarbonylnnethyl)- piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.13 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1 - carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester in 5 mL THF wurden 50 mg (0.16 mmol) TBTU und 37 μL (0.34 mmol) Triethylamin gegeben und 30 min bei RT gerührt. Zum Reaktionsgemisch wurden 19 μL (0.16 mmol) 2-Morpholin-4-yl-ethanol gegeben und die Suspension 1.5 h bei RT gerührt. Man gab 3 mL DMF zu und rührte weitere 4 h bei RT. Das Reaktionsgemisch wurde i.vac. eingeengt, der Rückstand in 1.5 mL MeOH gelöst, über einen Spritzenfilter filtriert und via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden i.vac. eingeengt, der Rückstand mit DIPE verrieben, abgesaugt und bei 50°C im Vakuum getrocknet.
Ausbeute: 47 mg (41 % der Theorie)
ESI-MS: (M+H)+ = 877/879 (Cl)
Rf = 0.31 (Kieselgel, DCM/MeOH/NH3 90:10:1)
Retentionszeit (HPLC): 2.7 min (Methode B)
Beispiel 4.10
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-oxo-2-(4-{1-[2-(2-oxo-pyrrolidin-1-yl)-ethoxy- carbonylmethyl]-piperidin-4-yl}-piperazin-1-yl)-ethylester
Hergestellt analog zu Beispiel 4.9 aus 80 mg (0.11 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-(1-carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester und 18 μL
(0.16 mmol) 1-(2-Hydroxy-ethyl)-pyrrolidin-2-on, wobei als Lösungsmittel 1.5 mL DMF verwendet wurde.
Ausbeute: 39 mg (43% der Theorie)
ESI-MS: (M+H)+ = 875/877 (Cl) Rf = 0.40 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Retentionszeit (HPLC): 2.9 min (Methode B)
Beispiel 4.11
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chloro-5-trifluoromethyl-benzyl)-2-{4-[1-(3-nnorpholin-4-yl-propoxycarbonyl- methyl)-piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester
Hergestellt analog zu Beispiel 4.9 aus 90 mg (0.12 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-(1-carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester und 22 mg (0.15 mmol) 3-Morpholin-4-yl-propan-1-ol, wobei als Lösungsmittel 1.5 mL DMF verwen- det wurde. Nach der Reinigung via HPLC wurde das Produkt in DCM aufgenommen, die organische Phase mit 5% NaHCO3-Lösung extrahiert und über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand mit DIPE verrührt, abgesaugt und an der Luft getrocknet. Ausbeute: 48 mg (46% der Theorie) ESI-MS: (M+H)+ = 891/893 (Cl)
Rf = 0.17 (Kieselgel, DCM/MeOH/NH3 90:10:1)
Retentionszeit (HPLC): 2.7 min (Methode B)
Beispiel 4.12
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzodiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(2,2-dimethyl-propionyloxymethoxycarbonyl- methyl)-piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester
Zu einer Lösung von 150 mg (0.20 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1 - carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester in 5 mL DMF wurden 42 mg (0.30 mmol) K2CO3 und 43 μL (0.30 mmol) 2,2-Dimethyl-propionsäurechlormethylester gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man engte i.vac. ein, versetzte den Rückstand mit 30 mL 15% K2CO3-Lösung, saugte das ausgefallene Produkt ab und reinigte dieses via Säulenchromatographie (Kieselgel, Gradient DCM zu DCM/MeOH/NHs 50:47:3).
Ausbeute: 50 mg (29% der Theorie)
ESI-MS: (M+H)+ = 878/880 (Cl)
Rf = 0.63 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 4.13
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(1-ethoxycarbonyloxy-ethoxycarbonyl- methyl)-piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester
Hergestellt analog zu Beispiel 4.12 aus 150 mg (0.20 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-
benzyl)-2-[4-(1-carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester und 40 μL (0.30 mmol) 1-Chlorethyl-ethylcarbonat. Ausbeute: 50 mg (29% der Theorie) ESI-MS: (M+H)+ = 880/882 (Cl) Rf = 0.68 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 4.14
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1-diethylcarbamoylmethoxycarbonylmethyl- piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.13 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluor-methyl-benzyl)-2-[4-(1 - carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester in 1 mL DMF wurden 50 mg (0.16 mmol) TBTU, 25 μL (0.18 mmol) Triethylamin und 30 mg (0.23 mmol) N,N-Diethyl-2- hydroxy-acetamid gegeben und das Reaktionsgemisch 20 h bei RT gerührt. Die Reaktionslösung wurde auf gesättigte NaHCO3-Lösung gegossen, der ausgefallene
Niederschlag abgesaugt und getrocknet. Das Rohprodukt wurde via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 36 mg (31 % der Theorie)
ESI-MS: (M+H)+ = 877/879 (Cl) Retentionszeit (HPLC): 3.2 min (Methode B)
Analog wurden folgende Verbindungen aus jeweils 100 mg 4-(2-Oxo-1,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-(1 -carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester und der entsprechenden Menge der jeweiligen Alkoholkomponente hergestellt:
Beispiel 5
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1-yl)- piperidin-1-yl]-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 1 mL DMF wurden 64 mg (0.20 mmol) TBTU und 28 μL (0.20 mmol) Triethyl- amin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 51 mg (0.20 mmol) (4-Piperidin-4-yl-piperazin-1-yl)-essigsäureethylester gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktions-
lösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 91 mg (64% der Theorie) ESI-MS: (M+H)+ = 792/794 (Cl) Rf = 0.48 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Beispiel 5.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4-carboxymethyl-piperazin-1-yl)-piperidin-1- yl]-2-oxo-ethylester
Zu einer Lösung von 70 mg (0.09 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4-ethoxy- carbonylmethyl-piperazin-1-yl)-piperidin-1-yl]-2-oxo-ethylester in 10 mL THF wurde eine Lösung von 3.6 mg (0.15 mmol) LiOH in 1 mL Wasser gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man entfernte das THF im Stickstoffstrom, versetzte mit wenig Wasser, mit Ameisensäure bis zur sauren Reaktion und mit Acetonitril und unterzog das Produkt einer Gefriertrocknung. Ausbeute: 52 mg (76% der Theorie) ESI-MS: (M-H). = 762/764 (Cl)
Rf = 0.14 (Kieselgel, DCM/MeOH/NH3 90:10:1)
Beispiel 5.2
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4-dimethylcarbamoylmethoxycarbonyl-methyl- piperazin-1-yl)-piperidin-1-yl]-2-oxo-ethylester
Zu einer Lösung von 190 mg (0.25 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-
3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4- carboxymethyl-piperazin-1-yl)-piperidin-1-yl]-2-oxo-ethylester in 5 mL DMF wurden 90 mg
(0.28 mmol) TBTU, 39 μL (0.15 mmol) Triethylamin und 29 mg (0.28 mmol) 2-Hydroxy- N,N-dimethyl-acetamid gegeben und das Reaktionsgemisch über Nacht bei RT gerührt.
Man filtrierte die Reaktionslösung über einen Spritzenfilter und reinigte das Rohprodukt via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet.
Ausbeute: 106 mg (50% der Theorie)
ESI-MS: (M+H)+ = 849/851 (Cl)
Retentionszeit (HPLC): 3.3 min (Methode B)
Beispiel 6
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[1l-(2-ethoxycarbonyl-ethyl)-4,4l-bipiperidinyl-1- yl]-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 1 mL DMF wurden 64 mg (0.20 mmol) TBTU und 28 μL (0.20 mmol) Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 54 mg (0.20
mmol) S-^^'JBipiperidinyl-i-yl-propionsäureethylester (Amin A1) gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 42 mg (29% der Theorie) ESI-MS: (M+H)+ = 805/807 (Cl)
Rf = 0.58 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Beispiel 6.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[1l-(2-carboxy-ethyl)-4,4l-bipiperidinyl-1-yl]-2-oxo- ethylester
Zu einer Lösung von 30 mg (0.04 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[1 '-(2-ethoxy- carbonyl-ethyl)-4,4'-bipiperidinyl-1-yl]-2-oxo-ethylester in 10 mL THF wurde eine Lösung von 1.4 mg (0.06 mmol) LiOH in 1 mL Wasser gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man entfernte das THF im Stickstoffstrom, versetzte mit wenig Wasser, mit Ameisensäure bis zur sauren Reaktion und mit Acetonitril und reinigte das Rohprodukt via HPLC. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 28 mg (97% der Theorie) ESI-MS: (M+H)+ = 777/779 (Cl)
Rf = 0.15 (Kieselgel, DCM/MeOH/NH3 90:10:1)
Beispiel 7
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(2-ethoxycarbonyl-ethyl)-piperidin-4-yl]- piperazin-1-yl}-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 1 mL DMF wurden 64 mg (0.20 mmol) TBTU und 111 μL (0.80 mmol) Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 122 mg (0.20 mmol) 3-(4-Piperazin-1-yl-piperidin-1-yl)-propionsäureethylester (Amin A2, eingesetzt als Tris-Trifluoracetat-Salz) gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 45 mg (31% der Theorie) ESI-MS: (M+H)+ = 806/808 (Cl)
Rf = 0.57 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Beispiel 7.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(2-carboxy-ethyl)-piperidin-4-yl]-piperazin-1- yl}-2-oxo-ethylester
Hergestellt analog zu Beispiel 6.1 aus 30 mg (0.04 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-{4-[1-(2-ethoxycarbonyl-ethyl)-piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester und 1.4 mg (0.06 mmol) LiOH. Ausbeute: 15 mg (51% der Theorie) ESI-MS: (M-H)
. = 776/778 (Cl)
Rf = 0.13 (Kieselgel, DCM/MeOH/NH3 90:10:1)
Beispiel 7.2
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-{1-[2-(2-dimethylamino-ethoxycarbonyl)-ethyl]- piperidin-4-yl}-piperazin-1-yl)-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.13 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1 -(2- carboxy-ethyl)-piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester in 1 mL DMF wurden 48 mg (0.15 mmol) TBTU und 21 μL (0.15 mmol) Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 15 μL (0.15 mmol) 2-Dimethylamino-ethanol gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 22 mg (20% der Theorie) ESI-MS: (M+H)+ = 849/851 (Cl)
Retentionszeit (HPLC): 2.8 min (Methode B)
Analog wurden folgende Verbindungen aus jeweils 100 mg (Beispiele 7.3 und 7.4) bzw. aus 95 mg (Beispiel 7.5) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1- carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1 -(2-carboxy-ethyl)-
piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester und der entsprechenden Menge der jeweiligen Alkoholkomponente hergestellt:
Beispiel 8
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[4-(2-ethoxycarbonyl-ethyl)-piperazin-1-yl]- piperidin-1-yl}-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy-
ethylester in 1 mL DMF wurden 64 mg (0.20 mmol) TBTU und 28 μL (0.20 mmol) Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 54 mg (0.20 mmol) 3-(4-Piperidin-4-yl-piperazin-1-yl)-propionsäureethylester (Amin A3) gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzen- filter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 77 mg (53% der Theorie) ESI-MS: (M+H)+ = 806/808 (Cl)
Rf = 0.58 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Beispiel 8.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[4-(2-carboxy-ethyl)-piperazin-1-yl]-piperidin-1- yl}-2-oxo-ethylester
Hergestellt analog zu Beispiel 6.1 aus 60 mg (0.07 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-{4-[4-(2-ethoxycarbonyl-ethyl)-piperazin-1-yl]-piperidin-1-yl}-2-oxo-ethylester und 2.9 mg (0.12 mmol) LiOH.
Ausbeute: 40 mg (70% der Theorie)
ESI-MS: (M-H). = 776/778 (Cl)
Rf = 0.14 (Kieselgel, DCM/MeOH/NH3 90:10:1)
Beispiel 9
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-((R)-2-ethoxycarbonyl-pyrrolidin-1-yl)-piperidin- 1-yl]-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 1 mL DMF wurden 64 mg (0.20 mmol) TBTU und 28 μL (0.20 mmol)
Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 45 mg (0.20 mmol) (R)-1-Piperidin-4-yl-pyrrolidin-2-carbonsäureethylester gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 77 mg (56% der Theorie)
ESI-MS: (M+H)+ = 763/765 (Cl)
Retentionszeit (HPLC): 3.4 min (Methode B)
Beispiel 9.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-((R)-2-carboxy-pyrrolidin-1-yl)-piperidin-1-yl]-2- oxo-ethylester
Hergestellt analog zu Beispiel 6.1 aus 60 mg (0.08 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-((R)-2-ethoxycarbonyl-pyrrolidin-1-yl)-piperidin-1-yl]-2-oxo-ethylester und 2.9 mg (0.12 mmol) LiOH.
Ausbeute: 42 mg (73% der Theorie)
ESI-MS: (M+H)+ = 735/737 (Cl)
Retentionszeit (HPLC): 3.3 min (Methode B)
Beispiel 9.2
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-((R)-2-dimethylcarbamoylmethoxycarbonyl- pyrrolidin-1-yl)-piperidin-1-yl]-2-oxo-ethylester
Zu einer Lösung von 50 mg (0.07 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-((R)-2- carboxy-pyrrolidin-1-yl)-piperidin-1-yl]-2-oxo-ethylester in 1 mL DMF wurden 25 mg (0.08 mmol) TBTU und 11 μL (0.08 mmol) Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 8.1 mg (0.08 mmol) 2-Hydroxy-N,N-dimethyl-acetamid gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 38 mg (68% der Theorie)
ESI-MS: (M+H)+ = 820/822 (Cl)
Retentionszeit (HPLC): 2.9 min (Methode A)
Beispiel 9.3
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[(R)-2-(2-morpholin-4-yl-ethoxycarbonyl)- pyrrolidin-1-yl]-piperidin-1-yl}-2-oxo-ethylester
Hergestellt analog zu Beispiel 9.2 aus 50 mg (0.07 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-((R)-2-carboxy-pyrrolidin-1-yl)-piperidin-1-yl]-2-oxo-ethylester und 10.3 mg (0.08 mmol) 2-Morpholin-4-yl-ethanol. Ausbeute: 35 mg (61 % der Theorie)
ESI-MS: (M+H)+ = 848/850 (Cl)
Retentionszeit (HPLC): 2.6 min (Methode A)
Beispiel 10
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-((S)-2-methoxycarbonyl-pyrrolidin-1-yl)- piperidin-1-yl]-2-oxo-ethylester
Hergestellt analog zu Beispiel 9 aus 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl- phenyl)-1-carboxy-ethylester und 42 mg (0.20 mmol) (S)-1-Piperidin-4-yl-pyrrolidin-2- carbonsäuremethylester.
Ausbeute: 80 mg (59% der Theorie)
ESI-MS: (M+H)+ = 749/751 (Cl)
Retentionszeit (HPLC): 3.3 min (Methode B)
Beispiel 10.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-((S)-2-carboxy-pyrrolidin-1-yl)-piperidin-1-yl]-2- oxo-ethylester
Zu einer Lösung aus 60 mg (0.08 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-((S)-2- methoxycarbonyl-pyrrolidin-1-yl)-piperidin-1-yl]-2-oxo-ethylester in 3 mL THF wurde eine Lösung von 2.9 mg (0.12 mmol) LiOH in 1 mL Wasser gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Das Lösungsmittel wurde im Stickstoffstrom entfernt, der Rückstand in 1 mL Wasser aufgenommen, mit Ameisensäure bis zur sauren Reaktion versetzt und erschöpfend mit EtOAc extrahiert. Die vereinigten organischen Extrakte wurden über Na2SO4 getrocknet, filtriert und i.vac. eingeengt. Der Rückstand wurde in wenig Wasser und Acetonitril aufgenommen und gefriergetrocknet. Ausbeute: 51 mg (87% der Theorie)
ESI-MS: (M+H)+ = 735/737 (Cl)
Retentionszeit (HPLC): 3.3 min (Methode B)
Beispiel 10.2
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-((S)-2-dimethylcarbamoylmethoxycarbonyl- pyrrolidin-1-yl)-piperidin-1-yl]-2-oxo-ethylester
Hergestellt analog zu Beispiel 9.2 aus 70 mg (0.10 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-((S)-2-carboxy-pyrrolidin-1-yl)-piperidin-1-yl]-2-oxo-ethylester und 11 mg (0.11 mmol) 2-Hydroxy-N,N-dimethyl-acetamid. Ausbeute: 45 mg (58% der Theorie)
ESI-MS: (M+H)+ = 820/822 (Cl)
Retentionszeit (HPLC): 3.0 min (Methode A)
Beispiel 10.3
4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[(S)-2-(2-morpholin-4-yl-ethoxycarbonyl)- pyrrolidin-1-yl]-piperidin-1-yl}-2-oxo-ethylester
Hergestellt analog zu Beispiel 9.2 aus 70 mg (0.10 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-((S)-2-carboxy-pyrrolidin-1-yl)-piperidin-1-yl]-2-oxo-ethylester und 13.2 mg
(0.11 mmol) 2-Morpholin-4-yl-ethanol.
Ausbeute: 54 mg (59% der Theorie)
ESI-MS: (M+H)+ = 848/850 (Cl)
Retentionszeit (HPLC): 2.6 min (Methode A)
Beispiel 11
(K)- 1 '-{(R)-3-(4-Annino-3-chlor-5-trifluornnethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-S-yl)-piperidin-i-carbonyloxyl-propionylJ-i ^'-bipiperidinyl^-carbonsäure- methylester
Zu einer Lösung von 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 1 mL DMF wurden 64 mg (0.20 mmol) TBTU und 56 μL (0.40 mmol)
Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 57 mg (0.20 mmol) (R)-[I ^'JBipiperidinyl^-carbonsäuremethylester (eingesetzt als Acetat- SaIz) gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 59 mg (43% der Theorie) ESI-MS: (M+H)+ = 763/765 (Cl)
Rf = 0.50 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Beispiel 11.1
(K)- 1 l-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzodiazepin-S-yl)-piperidin-i-carbonyloxyJ-propionylJ-i ^'-bipiperidinyl^-carbonsäure
Hergestellt analog zu Beispiel 6.1 aus 40 mg (0.05 mmol) (R)-1'-{(R)-3-(4-Amino-3-chlor- 5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1- carbonyloxyj-propionylj-i ^'-bipiperidinyl^-carbonsäuremethylester und 1.9 mg (0.08 mmol) LiOH. Ausbeute: 10 mg (25% der Theorie) ESI-MS: (M-H)
. = 747/749 (Cl)
Rf = 0.27 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Beispiel 12
(S)-1 '-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-S-yl)-piperidin-i-carbonyloxyl-propionylJ-i ^'-bipiperidinyl^-carbonsäure- methylester
Hergestellt analog zu Beispiel 11 aus 100 mg (0.18 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl- phenyl)-1-carboxy-ethylester und 45 mg (0.20 mmol) (S)-[1 ,4']Bipiperidinyl-2- carbonsäuremethylester. Ausbeute: 33 mg (24% der Theorie) ESI-MS: (M+H)+ = 763/765 (Cl)
Rf = 0.44 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Beispiel 12.1
(S)-1
l-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzodiazepin-S-yl)-piperidin-i-carbonyloxyJ-propionylJ-i ^'-bipiperidinyl^-carbonsäure
Hergestellt analog zu Beispiel 6.1 aus 20 mg (0.03 mmol) (S)-1'-{(R)-3-(4-Amino-3-chlor- 5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1- carbonyloxy]-propionyl}-1 ^'-bipiperidinyl^-carbonsäuremethylester und 1.2 mg (0.05 mmol) LiOH.
Ausbeute: 3 mg (15% der Theorie) ESI-MS: (M-H). = 747/749 (Cl)
Rf = 0.29 (Kieselgel, DCM/MeOH/NH3 90: 10:1)
Beispiel 13
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(1l-ethoxyoxalyl-4,4l-bipiperidinyl-1-yl)-2-oxo- ethylester
Zu einer Lösung von 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 2 mL DMF wurden 64 mg (0.20 mmol) TBTU und 70 μL (0.50 mmol)
Triethylamin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 54 mg (0.20 mmol) μ^'JBipiperidinyl-i-yl-oxo-essigsäureethylester (Amin A4) gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet.
Ausbeute: 88 mg (61 % der Theorie)
ESI-MS: (M+H)+ = 805/807 (Cl)
Retentionszeit (HPLC): 4.5 min (Methode B)
Beispiel 13.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(1l-oxalyl-4,4l-bipiperidinyl-1-yl)-2-oxo-ethylester
Hergestellt analog zu Beispiel 6.1 aus 40 mg (0.05 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-(1'-ethoxyoxalyl-4,4l-bipiperidinyl-1-yl)-2-oxo-ethylester und 3.6 mg (0.15 mmol) LiOH. Ausbeute: 26 mg (67% der Theorie)
ESI-MS: (M+H)+ = 777/779 (Cl)
Retentionszeit (HPLC): 5.0 min (Methode C)
Beispiel 13.2
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(1l-dimethylcarbamoylmethoxyoxalyl-4,4l-bi- piperidinyl-1-yl)-2-oxo-ethylester
Hergestellt analog zu Beispiel 7.2 aus 100 mg (0.13 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-(1'-oxalyl-4,4
l-bipiperidinyl-1-yl)-2-oxo-ethylester und 15 mg (0.15 mmol) 2- Hydroxy-N,N-dimethyl-acetamid. Ausbeute: 70 mg (63% der Theorie)
ESI-MS: (M+H)+ = 862/864 (Cl)
Retentionszeit (HPLC): 4.1 min (Methode B)
Beispiel 13.3
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[1l-(2-morpholin-4-yl-ethoxyoxalyl)-4,4l-bipiperi- dinyl-1-yl]-2-oxo-ethylester
Hergestellt analog zu Beispiel 9.2 aus 100 mg (0.13 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzylj^i'-oxalyl^^'-bipiperidinyl-i-yl^-oxo-ethylester und 18 μL (0.15 mmol) 2- Morpholin-4-yl-ethanol. Ausbeute: 69 mg (60% der Theorie)
ESI-MS: (M+H)+ = 890/892 (Cl)
Retentionszeit (HPLC): 3.6 min (Methode B)
Beispiel 14
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[1
l-(3-ethoxycarbonyl-propionyl)-4,4
l-bipiperidinyl- 1 -yl]-2-oxo-ethylester
Zu einer Lösung von 100 mg (0.18 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester in 1 mL DMF wurden 64 mg (0.20 mmol) TBTU und 28 μL (0.50 mmol) Triethyl- amin zugegeben und 10 min bei RT gerührt. Zum Reaktionsgemisch wurden 59 mg (0.20 mmol) 4-[4,4l]Bipiperidinyl-1-yl-4-oxo-buttersäureethylester (Amin A5) gegeben und dieses über Nacht bei RT geschüttelt. Nach Filtration über einen Spritzenfilter wurde die Reaktionslösung ohne weitere Aufarbeitung via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 61 mg (41% der Theorie) ESI-MS: (M+H)+ = 833/835 (Cl)
Rf = 0.61 (Kieselgel, DCM/MeOH/NH3 90:10:1)
Beispiel 14.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[1l-(3-carboxy-propionyl)-4,4l-bipiperidinyl-1-yl]-2- oxo-ethylester
Zu einer Lösung aus 40 mg (0.05 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[1 '-(3-ethoxy- carbonyl-propionyl)-4,4'-bipiperidinyl-1-yl]-2-oxo-ethylester in 10 mL THF wurde eine Lösung von 1.9 mg (0.08 mmol) LiOH in 1 mL Wasser gegeben und das Reaktions-
gemisch über Nacht bei RT gerührt. Das Lösungsmittel wurde im Stickstoffstrom entfernt, der Rückstand in 1 mL Wasser aufgenommen, mit Ameisensäure bis zur sauren Reaktion und mit 1 mL Acetonitril versetzt und gefriergetrocknet. Der Rückstand wurde in 1 mL DMF aufgenommen und via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 19 mg (39% der Theorie) ESI-MS: (M+H)+ = 805/807 (Cl)
Rf = 0.16 (Kieselgel, DCM/MeOH/NH3 90:10:1)
Beispiel 14.2
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{1l-[3-(2-morpholin-4-yl-ethoxycarbonyl)- propionyl]-4,4'-bipiperidinyl-1-yl}-2-oxo-ethylester
Hergestellt analog zu Beispiel 9.2 aus 100 mg (0.12 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl^-II'^S-carboxy-propionyl^^'-bipiperidinyl-i-ylJ^-oxo-ethylester und 17 μL (0.14 mmol) 2-Morpholin-4-yl-ethanol.
Ausbeute: 16 mg (14% der Theorie)
ESI-MS: (M+H)2+ = 459/460 (Cl)
Retentionszeit (HPLC): 3.7 min (Methode B)
Beispiel 15
4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure- (R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(1
l-ethoxycarbonylmethyl-4,4
l-bipiperi- dinyl-1-yl)-2-oxo-ethylester
15a) 4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbon- säure-(f?)-2-(4-amino-3-chlor-5-trifluornnethyl-phenyl)-1-ethoxycarbonyl-ethylester Zu einer Lösung von 0.79 g (6.42 mmol) DMAP in 50 mL Pyridin wurden 1.29 g (6.42 mmol) Chlorameisensäure-4-nitrophenylester zugegeben und 1 h bei RT gerührt. Eine Lösung von 2.00 g (6.42 mmol) (R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-hydroxy- propionsäureethylester in 15 mL Pyridin wurde zugetropft und die Reaktionsmischung 2 h bei RT gerührt. Dann erfolgte portionenweise die Zugabe von 1.77 g (6.42 mmol) 7-Methoxy-3-piperidin-4-yl-1 ,3,4,5-tetrahydro-1,3-benzdiazepin-2-on. Die Suspension wurde 72 h bei RT gerührt und dann i.vac. eingeengt. Der Rückstand wurde mit 200 mL EtOAc versetzt, mit 200 mL 15% K2CO3-Lösung gewaschen, die organische Phase abgetrennt und i.vac. eingeengt. Der Rückstand wurde chromatographisch (Kieselgel, Gradient DCM zu MeOH/NH3 95:5) gereinigt. Ausbeute: 1.80 g (46% der Theorie) ESI-MS: (M+H)+ = 613/615 (Cl)
Rf = 0.50 (Kieselgel, DCM/MeOH 9:1)
15b) 4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 - carbonsäure-(f?)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy-ethylester
Zu einer Lösung von 1.80 g (2.94 mmol) 4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1 ,3- benzdiazepin-3-yl)-piperidin-1-carbon-säure-(R)-2-(4-amino-3-chlor-5-trifluormethyl- phenyl)-1-ethoxycarbonyl-ethylester in 50 mL THF wurde eine Lösung von 0.11 g (4.50 mmol) LiOH in 50 mL Wasser gegeben und das Reaktionsgemisch 3 h bei RT gerührt. Man entfernte i.vac. das THF, verdünnte mit 100 mL Wasser und säuerte mit 1 M HCl an. Die ausgefallene Substanz wurde abgesaugt, mit 50 mL Wasser gewaschen und im Vakuumtrockenschrank bei 65°C getrocknet. Ausbeute: 1.60 g (93% der Theorie) ESI-MS: (M+H)+ = 585/587 (Cl)
15c) 4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbon- säure-(f?)-1-(4-amino-3-chlor-5-trifluornnethyl-benzyl)-2-(1'-ethoxycarbonylnnethyl- 4,4'-bipiperidinyl-1-yl)-2-oxo-ethylester
Hergestellt analog zu Beispiel 9 aus 100 mg (0.17 mmol) 4-(7-Methoxy-2-oxo-1 ,2,4,5- tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-tri- fluormethyl-phenyl)-1-carboxy-ethylester und 48 mg (0.19 mmol) μ^'JBipiperidinyl-i-yl- essigsäureethylester.
Ausbeute: 58 mg (41 % der Theorie)
ESI-MS: (M+H)+ = 821/823 (Cl) Retentionszeit (HPLC): 3.0 min (Methode B)
Beispiel 15.1
4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure- (R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(1l-carboxymethyl-4,4l-bipiperidinyl-1-yl)- 2-oxo-ethylester
Zu einer Lösung von 20 mg (0.02 mmol) 4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benz- diazepin-3-yl)-piperidin-1-carbon-säure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)- 2-(1'-ethoxycarbonylmethyl-4,4l-bipiperidinyl-1-yl)-2-oxo-ethylester in 1 mL THF wurde eine Lösung von 1 mg (0.04 mmol) LiOH in 1 mL Wasser zugegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Zur Vervollständigung der Reaktion wurde erneut mit einer Lösung von 1 mg LiOH in 1 mL Wasser versetzt und weitere 3 h bei RT gerührt. Man entfernt die Lösungsmittel im Stickstoffstrom, nahm den Rückstand im einem
Gemisch aus Acetontril und Wasser auf und unterzog das Produkt einer Gefriertrocknung. Ausbeute: 14 mg (72% der Theorie)
ESI-MS: (M+H)+ = 793/795 (Cl)
Retentionszeit (HPLC): 2.6 min (Methode B)
Beispiel 15.2
4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure- (R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[1l-(2-nnorpholin-4-yl-ethoxycarbonyl- methyl^^'-bipiperidinyl-i -yl]-2-oxo-ethylester
Zu einer Lösung von 230 mg (0.29 mmol) 4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-(1l-carboxymethyl-4,4l-bipiperidinyl-1-yl)-2-oxo-ethylester in 5 mL DMF wurden 113 mg (0.35 mmol) TBTU, 84 μL (0.60 mmol) Triethylamin und 39 mg (0.30 mmol) 2-Morpholin-4-yl-ethanol gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Die Reaktionslösung wurde auf eine gesättigte NaHCO3-LOSu ng gegossen, das ausgefallene Produkt abgesaugt und bei 40°C getrocknet. Das Rohprodukt wurde in 25 mL trockenem Isopropanol gelöst und mit 0.5 M HCl in Isopropanol als Salz gefällt. Man filtrierte den Niederschlag ab, wusch diesen mit 5 mL Isopropanol und 30 mL DIPE und trocknete im Vakuumtrockenschrank über Nacht bei 30°C. Ausbeute: 90 mg (34% der Theorie)
ESI-MS: (M+H)+ = 906/908 (Cl) Retentionszeit (HPLC): 3.1 min (Methode B)
Beispiel 16
4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure- (R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1- yl)-piperidin-1-yl]-2-oxo-ethylester
Hergestellt analog zu Beispiel 9 aus 100 mg (0.17 mmol) 4-(7-Methoxy-2-oxo-1 ,2,4,5- tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5- trifluormethyl-phenyl)-1-carboxy-ethylester und 49 mg (0.19 mmol) (4-Piperidin-4-yl- piperazin-1-yl)-essigsäureethylester. Ausbeute: 62 mg (44% der Theorie)
ESI-MS: (M+H)+ = 822/824 (Cl)
Retentionszeit (HPLC): 2.9 min (Methode B)
Beispiel 16.1
4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure- (R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4-carboxymethyl-piperazin-1-yl)- piperidin-1-yl]-2-oxo-ethylester
Hergestellt analog zu Beispiel 15.1 aus 20 mg (0.02 mmol) 4-(7-Methoxy-2-oxo-1 ,2,4,5- tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-tri- fluormethyl-benzyl)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1-yl)-piperidin-1-yl]-2-oxo- ethylester und 2 mg (0.08 mmol) LiOH.
Ausbeute: 19 mg (99% der Theorie)
ESI-MS: (M+H)+ = 794/796 (Cl)
Retentionszeit (HPLC): 2.6 min (Methode B)
Beispiel 17
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-methyl-benzyl)-2-(1l-ethoxycarbonylnnethyl-4,4l-bipiperidinyl-1-yl)-2-oxo- ethylester
17a) (Z,E)-2-Acetylamino-3-(4-amino-3-chlor-5-nnethyl-phenyl)-acrylsäurennethylester Unter Stickstoffatmosphäre wurde zu einer Mischung aus 72.8 g (330 mmol) 4-Brom- 2-chlor-6-methyl-phenylamin und 58.0 g (397 mmol) 2-Acetylamino-acrylsäuremethylester in 970 mL Triethylamin und 1.2 L Acetonitril 15.4 g (49.1 mmol) Tri-o-tolyl-phosphan und 11.0 g (49.0 mmol) Pd(OAc)2 zugegeben und die Reaktionsmischung 18 h bei 80°C gerührt. Nach dem Abkühlen wurde die Reaktionslösung filtriert, i.vac. eingeengt, der Rückstand mit 350 mL Wasser und 350 mL EtOAc verrührt. Die Lösungsmittel wurden abdekantiert, der Rückstand erneut mit 300 mL EtOAc verrührt, abgesaugt, mit wenig EtOAc und MTBE gewaschen und bei 60°C getrocknet. Ausbeute: 40.6 g (44% der Theorie) ESI-MS: (M+H)+ = 283/285 (Cl)
Rf = 0.47 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
17b) 3-(4-Amino-3-chlor-5-methyl-phenyl)-2-oxo-propionsäure Unter Stickstoffatmosphäre wurde eine Lösung von 28.0 g (99.0 mmol) (Z,E)-2-Acetyl- amino-3-(4-amino-3-chlor-5-methyl-phenyl)-acrylsäuremethylester in 250 mL 1 ,4-Dioxan und 125 mL HCl (4 M) 6 h bei 80°C gerührt. Man entfernte i.vac. die Lösungsmittel, verrieb den Rückstand mit Isopropanol und DIPE, saugte den Niederschlag ab und trocknete diesen bei 60°C. Das Produkt fiel als Hydrochlorid-Salz an, welches ohne Reinigung weiter umgesetzt wurde. Ausbeute: 26.0 g (99% der Theorie) ESI-MS: (M-H). = 226/228 (Cl) Rf = 0.15 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
17c) (R)-3-(4-Amino-3-chlor-5-methyl-phenyl)-2-hvdroxy-propionsäuremethylester Unter Stickstoffatmosphäre wurde zu einer auf ca. -30°C gekühlten Lösung von 13.0 g (49.2 mmol) 3-(4-Amino-3-chlor-5-methyl-phenyl)-2-oxo-propionsäure und 17.0 mL (122.3 mmol) Triethylamin in 175 mL THF eine Lösung von 27.0 g (84.2 mmol) (1R)-B-Chlordi- isopinocampheylboran in 75 mL THF innerhalb von 15 min zugetropft. Nach beendeter Zugabe wurde das Kältebad entfernt und das Reaktionsgemisch über Nacht bei RT gerührt. Zum Reaktionsgemisch wurden unter Eiskühlung 150 mL 1 M NaOH zugetropft. Man versetzte mit 200 mL EtOAc, rührte 15 min nach, trennte die wässrige Phase ab und wusch die organische Phase zweimal mit je 50 mL 1 M NaOH, einmal mit 40 mL Wasser und stellte diese mit 4 M HCl sauer. Die organische Phase wurde abgetrennt, über MgSO4 getrocknet und i.vac. eingeengt. Der Rückstand wurde mit 250 mL methanolischer HCl (1.25 M) versetzt und über Nacht bei RT gerührt. Das Reaktionsgemisch wurde i.vac. eingeengt, der Rückstand in wenig PE und EtOAc gelöst, auf Kieselgel gegeben und mit PE/EtOAc (2:1) eluiert. Die das Produkt enthaltenden Fraktionen wurden vereinigt und eingeengt.
Ausbeute: 6.0 g (50% der Theorie) ESI-MS: (M+H)+ = 244/246 (Cl)
Rf = 0.74 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
17d) 4-(2-Oxo-1 ,2,4,5-tetrahvdro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-
(R)-2-(4-amino-3-chlor-5-methyl-phenyl)-1-carboxy-ethylester Hergestellt analog Beispiel 1e aus 6.0 g (24.6 mmol) (R)-3-(4-Amino-3-chlor-5-methyl- phenyl)-2-hydroxy-propionsäuremethylester und 6.1 g (24.9 mmol) 3-Piperidin-4-yl- 1 ,3,4,5-tetrahydro-1 ,3-benzdiazepin-2-on.
Das Methylester-Rohprodukt wurde in 100 mL THF gelöst und mit einer Lösung von 1.0 g (40.9 mmol) LiOH in 50 mL Wasser versetzt. Das Reaktionsgemisch wurde 15 h bei RT gerührt, mit Wasser verdünnt und das organische Lösungsmittel i.vac. entfernt. Die wässrige Phase wurde mit 60 mL EtOAc gewaschen, mit 21 mL 4 M HCl angesäuert und 15 min bei RT nachgerührt. Man extrahierte dreimal mit je 150 mL DCM und trocknete die vereinigten organischen Phasen über MgSO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Kieselgel, Gradient DCM zu DCM/MeOH/NH3 70:27:3) gereinigt. Ausbeute: 0.88 g (7% der Theorie)
ESI-MS: (M+H)+ = 501/503 (Cl)
Rf = 0.17 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
17e) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-1 - (4-amino-3-chlor-5-methyl-benzyl)-2-(1'-ethoxycarbonylnnethyl-4,4'-bipiperidinyl-1- yl)-2-oxo-ethylester
Hergestellt analog Beispiel 5 aus 100 mg (0.20 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-methyl-phenyl)-1- carboxy-ethylester und 56 mg (0.22 mmol) [4,4']Bipiperidinyl-1-yl-essigsäureethylester Ausbeute: 19 mg (13% der Theorie) ESI-MS: (M+H)+ = 737/739 (Cl)
Rf = 0.72 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 17.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-methyl-benzyl)-2-(1l-carboxymethyl-4,4l-bipiperidinyl-1-yl)-2-oxo-ethyl- ester
Zu einer Lösung von 80 mg (0.11 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-methyl-benzyl)-2-(1 '-ethoxycarbonyl- methyl^^'-bipiperidinyl-i-yl^-oxo-ethylester in 10 mL THF wurde eine Lösung von 7.0 mg (0.29 mmol) LiOH in 5 mL Wasser gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man entfernte i.vac. das THF, versetzte den wässrigen Rückstand mit 0.35 mL 1 M HCl und engte i.vac. ein. Der Rückstand wurde via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 8 mg (10% der Theorie) ESI-MS: (M+H)+ = 709/711 (Cl) Rf = 0.21 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 18
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(3- chlor-4-hydroxy-5-methyl-benzyl)-2-(1 '-ethoxycarbonylmethyl^^'-bipiperidinyl-i -yl)-2-oxo- ethylester
18a) 2-Benzyloxy-5-brom-1-chlor-3-nnethyl-benzol Zu einer Suspension von 10.2 g (46.1 mmol) 4-Brom-2-chlor-6-methyl-phenol und 30.0 g (217 mmol) K2CO3 in 130 mL DMF wurden 7.0 mL (57.7 mmol) Benzylbromid gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Man filtrierte von unlöslichen Bestandteilen ab, engte das Filtrat i.vac. ein, versetzte mit Wasser und extrahierte erschöpfend mit EtOAc. Die vereinigten organischen Phasen wurden über Na2SO4 getrocknet, filtriert und eingeengt. Das Produkt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 14.0 g (98% der Theorie) Rf = 0.91 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
18b) (Z,E)-2-Acetylamino-3-(4-benzyloxy-3-chlor-5-methyl-phenyl)-acrylsäuremethyl- ester
Unter Stickstoffatmosphäre wurde zu einer Mischung aus 28.0 g (89.9 mmol) 2-Benzyl- oxy-5-brom-1-chlor-3-methyl-benzol und 15.0 g (103 mmol) 2-Acetylamino-acrylsäure- methylester in 260 mL Triethylamin und 400 mL Acetonitril 4.4 g (14.0 mmol) Tri-o-tolyl- phosphan und 3.2 g (14.3 mmol) Pd(OAc)2 zugegeben und die Reaktionsmischung 18 h bei 80°C gerührt. Nach dem Abkühlen wurde die Reaktionslösung i.vac. eingeengt, der Rückstand mit 100 mL Wasser, 50 mL EtOAc und 50 mL PE verrührt und von unlöslichen Bestandteilen filtriert. Der Rückstand wurde in DCM/MeOH (5:1) aufgenommen, mit Aktivkohle versetzt, filtriert und i.vac. eingeengt. Das Rohprodukt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 12.5 g (37% der Theorie)
ESI-MS: (M+H)+ = 374/376 (Cl)
Rf = 0.67 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
18c) 3-(4-Benzyloxy-3-chlor-5-nnethyl-phenyl)-2-oxo-propionsäure Zu einer Lösung von 18.4 g (49.2 mmol) (Z,£)-2-Acetylamino-3-(4-benzyloxy-3-chlor-5- methylphenyl)-acrylsäuremethylester in 150 mL 1,4-Dioxan wurden 75 mL 4 M HCl gegeben und das Reaktionsgemisch über Nacht unter Rückfluss erhitzt. Man entfernte i.vac. das 1,4-Dioxan, filtrierte das ausgefallene Produkt ab, löste dieses erneut in DIPE und trocknete über MgSO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand ohne Reinigung weiter umgesetzt. Ausbeute: 15.5 g (99% der Theorie) ESI-MS: (M-H). = 317/319 (Cl)
Rf = 0.20 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
18d) (f?)-3-(4-Benzyloxy-3-chlor-5-methyl-phenyl)-2-hvdroxy-propionsäuremethylester Unter Stickstoffatmosphäre wurde zu einer auf ca. -30°C gekühlten Lösung von 15.5 g (48.6 mmol) 3-(4-Benzyloxy-3-chlor-5-methyl-phenyl)-2-oxo-propionsäure und 9.2 mL (66.2 mmol) Triethylamin in 200 mL THF eine Lösung von 27.6 g (86.0 mmol) (1K)-B- Chlordiisopinocampheylboran in 100 mL THF innerhalb von 15 min zugetropft. Nach beendeter Zugabe wurde das Kältebad entfernt und das Reaktionsgemisch über Nacht bei RT gerührt. Zum Reaktionsgemisch wurden unter Eiskühlung 240 mL 1 M NaOH zugetropft. Man versetzte mit 400 mL EtOAc, rührte 15 min nach, trennte die wässrige Phase ab und wusch die organische Phase zweimal mit je 100 mL 1 M NaOH und einmal mit 100 mL Wasser. Die vereinigten wässrigen Phasen wurden mit halbkonzentrierter HCl sauer gestellt, zweimal mit je 150 mL EtOAc extrahiert und die vereinigten organischen Phasen über MgSO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der ölige Rückstand mit 150 mL methanolischer HCl (1.25 M) versetzt und das Reaktionsgemisch über Nacht bei RT gerührt. Man engte i.vac. ein, versetzte den Rückstand mit 70 mL 15% K2CO3-Lösung und extrahierte zweimal mit je 50 mL EtOAc. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet, filtriert und i.vac. eingeengt. Das Produkt wurde ohne Reinigung weiter umgesetzt. Ausbeute: 7.0 g (43% der Theorie) ESI-MS: (M+NH4)+ = 352/354 (Cl)
Rf = 0.87 (Kieselgel, DCM/Cyc/MeOH/NHs 70:15:15:2)
18e) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-2- (4-benzyloxy-3-chlor-5-methyl-phenyl)-1-carboxy-ethylester
Hergestellt analog Beispiel 1e aus 7.0 g (20.9 mmol) (R)-3-(4-Benzyloxy-3-chlor-5-methyl- phenyl)-2-hydroxy-propionsäuremethylester und 5.2 g (21.2 mmol) 3-Piperidin-4-yl-
1 ,3,4,5-tetrahydro-i ,3-benzdiazepin-2-on.
Das Methylester-Rohprodukt wurde in 150 mL THF gelöst und mit einer Lösung von 0.5 g
(20.7 mmol) LiOH in 50 mL Wasser versetzt. Das Reaktionsgemisch wurde über Nacht bei RT gerührt, mit Wasser verdünnt und das organische Lösungsmittel i.vac. entfernt. Die wässrige Phase wurde zweimal mit je 60 mL EtOAc gewaschen, mit 21 mL 4 M HCl angesäuert und das sich bildende öl erschöpfend mit EtOAc extrahiert. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet, filtriert und das Lösungsmittel i.vac. eingeengt. Der Rückstand wurde mit DIPE verrieben und abgesaugt.
Ausbeute: 3.3 g (26% der Theorie) ESI-MS: (M+H)+ = 592/594 (Cl)
Rf = 0.35 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
18f) 4-(2-Oxo-1 ,2,4,5-tetrahvdro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-1 - carboxy-2-(3-chlor-4-hvdroxy-5-methyl-phenyl)-ethylester Eine Suspension von 2.75 g (4.65 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-2-(4-benzyloxy-3-chlor-5-methyl-phenyl)-1-carboxy- ethylester und 1.0 g Rhodium auf Aluminiumoxid in 150 mL MeOH wurde 18 h bei 40°C und 50 psi Wasserstoffdruck hydriert. Zur Vervollständigung der Reaktion wurde erneut mit 0.5 g Rhodium auf Aluminiumoxid versetzt und weitere 6 h hydriert. Der Katalysator wurde abgesaugt und das Filtrat i.vac. eingeengt. Das Rückstand, der mit ca. 50% des entsprechenden Methylesters verunreinigt war, wurde in 25 mL THF gelöst und mit einer Lösung von 250 mg (10.23 mmol) LiOH in 15 mL Wasser versetzt und das Reaktionsgemisch 3 h bei RT gerührt. Man entfernte i.vac. das THF, versetzte mit Wasser und 10.5 mL 1 M HCl, saugte das ausgefallene Produkt ab, wusch dieses mit wenig Wasser und trocknete es bei 60°C. Ausbeute: 2.1 g (90% der Theorie) ESI-MS: (M+H)+ = 502/504 (Cl)
Rf = 0.12 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
18g) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-1 - Q-chloM-hvdroxy-δ-nnethvI-benzvπ^-d'-ethoxycarbonvInnethvI^^'-bipiperidinvI- 1 -yl)-2-oxo-ethylester
Hergestellt analog Beispiel 5 aus 100 mg (0.20 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-carboxy-2-(3-chlor-4-hydroxy-5-methyl- phenyl)-ethylester und 51 mg (0.20 mmol) μ^'JBipiperidinyl-i-yl-essigsäureethylester Ausbeute: 14 mg (9% der Theorie)
ESI-MS: (M+H)+ = 738/740 (Cl)
Retentionszeit (HPLC): 3.2 min (Methode B)
Beispiel 18.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(1 '- carboxymethyl-4,4'-bipiperidinyl-1-yl)-1-(3-chlor-4-hydroxy-5-methyl-benzyl)-2-oxo-ethyl- ester
Zu einer Lösung von 38 mg (0.05 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(3-chlor-4-hydroxy-5-methyl-benzyl)-2-(1 '-ethoxy- carbonylmethyl-4,4'-bipiperidinyl-1-yl)-2-oxo-ethylester in 1 mL THF wurde eine Lösung von 2.0 mg (0.08 mmol) LiOH in 1 mL Wasser zugegeben und das Reaktionsgemisch 3 h bei RT geschüttelt. Die Reaktionslösung wurde mit 1 M HCl sauer gestellt und via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt und gefriergetrocknet. Ausbeute: 30 mg (82% der Theorie) ESI-MS: (M+H)+ = 710/712 (Cl)
Rf = 0.20 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 19
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1-ethoxycarbonylnnethyl-4-nnethyl-piperidin-4- yl)-piperazin-1-yl]-2-oxo-ethylester
Zu einer Mischung von 250 mg (0.45 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdi- azepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1- carboxy-ethylester (Beispiel 1f), 250 mg (0.50 mmol) (4-Methyl-4-piperazin-1-yl-piperidin- 1-yl)-essigsäureethylester (Beispiel A7f) und 160 mg (0.50 mmol) TBTU in 2 mL DMF wurden 0.25 mL (1.18 mmol) Triethylamin gegeben und das Reaktionsgemisch 4 h bei RT gerührt. Man engte i.vac. ein und reinigte den Rückstand via HPLC; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 225 mg (62% der Theorie)
ESI-MS: (M+H)+ = 806/808 (Cl) Retentionszeit (HPLC): 3.2 min (Methode B)
Beispiel 19.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1-carboxymethyl-4-methyl-piperidin-4-yl)- piperazin-1-yl]-2-oxo-ethylester
Zu 100 mg (0.12 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1- carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1 -ethoxy-carbonyl-
methyl-4-methyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester in 10 mL THF wurde eine Lösung von 10.0 mg (0.42 mmol) LiOH in 5 mL Wasser gegeben und das Reaktionsgemisch 3 h bei RT gerührt. Man versetzte mit 0.42 mL (0.42 mmol) 1 M HCl und engte i.vac. ein. Der Rückstand wurde in wenig DCM/MeOH (1 :1) aufgenommen, über wenig Kieselgel filtriert und mit DCM/MeOH (1 :1) eluiert. Das Filtrat wurde i.vac. eingeengt und im Hochvakuum getrocknet.
Ausbeute: 95 mg (98% der Theorie)
ESI-MS: (M+H)+ = 778/780 (Cl)
Retentionszeit (HPLC): 2.9 min (Methode B)
Beispiel 20
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1-yl)-4- methyl-piperidin-1-yl]-2-oxo-ethylester
Zu einer Mischung von 250 mg (0.45 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdi- azepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1- carboxy-ethylester (Beispiel 1f), 140 mg (0.49 mmol) [4-(4-Methyl-piperidin-4-yl)- piperazin-1-yl]-essigsäureethylester (Beispiel A8b) und 160 mg (0.50 mmol) TBTU in 2 mL DMF wurden 0.10 mL (0.72 mmol) Triethylamin gegeben und das Reaktionsgemisch 18 h bei RT gerührt. Man engte i.vac. ein und reinigte den Rückstand chromatographisch (Alox, Gradient DCM/MeOH 40:1 nach 30:1). Die das Produkt enthaltenden Fraktionen wurden vereinigt, i.vac. eingeengt und via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 178 mg (49% der Theorie)
ESI-MS: (M+H)+ = 806/808 (Cl)
Retentionszeit (HPLC): 3.8 min (Methode B)
Beispiel 20.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(4-carboxynnethyl-piperazin-1-yl)-4-nnethyl- piperidin-1-yl]-2-oxo-ethylester
Hergestellt analog Beispiel 19.1 aus 100 mg (0.12 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-[4-(4-ethoxycarbonylmethyl-piperazin-1-yl)-4-methyl-piperidin-1-yl]-2-oxo- ethylester und 10.0 mg (0.42 mmol) LiOH.
Ausbeute: 42 mg (44% der Theorie)
ESI-MS: (M+H)+ = 778/780 (Cl)
Retentionszeit (HPLC): 3.3 min (Methode B)
Beispiel 21
(S)-1-(1 -{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro- I .S-benzdiazepin-S-yl)-piperidin-i-carbonyloxyl-propionylJ-piperidin^-ylH-methyl- piperazin-2-carbonsäureethylester
Zu einer Mischung von 260 mg (0.46 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdi- azepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1- carboxy-ethylester (Beispiel 1f), 210 mg (0.58 mmol) (S)-4-Methyl-1-piperidin-4-yl-
piperazin-2-carbonsäureethylester (Beispiel A9f) und 170 mg (0.53 mmol) TBTU in 2.4 mL DMF wurden 0.40 mL (2.88 mmol) Triethylamin gegeben und das Reaktionsgemisch 18 h bei RT gerührt. Das Reaktionsgemisch wurde ohne weitere Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 227 mg (61% der Theorie)
ESI-MS: (M+H)+ = 792/794 (Cl)
Retentionszeit (HPLC): 3.4 min (Methode B)
Beispiel 21.1
(S)-1-(1 -{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-
1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonyloxy]-propionyl}-piperidin-4-yl)-4-methyl- piperazin-2-carbonsäure
Zu 80.0 mg (0.10 mmol) (S)-1-(1-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2- oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonyloxy]-propionyl}- piperidin-4-yl)-4-methyl-piperazin-2-carbonsäureethylester in 1 mL THF wurden 0.5 mL (1.00 mmol) 2 M LiOH-Lösung gegeben und das Reaktionsgemisch 20 h bei RT gerührt. Dieses wurde ohne Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 50 mg (65% der Theorie)
ESI-MS: (M+H)+ = 764/766 (Cl)
Retentionszeit (HPLC): 3.0 min (Methode B)
Beispiel 22
(S)-4-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonyloxy]-propionyl}-1-(1 -methyl-piperidin-4-yl)-pipera- zin-2-carbonsäureethylester
Zu einer Mischung von 200 mg (0.36 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdi- azepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1- carboxy-ethylester (Beispiel 1f), 150 mg (0.58 mmol) (S)-1-(1-Methyl-piperidin-4-yl)- piperazin-2-carbonsäureethylester (Beispiel A10c) und 140 mg (0.44 mmol) TBTU in 2.0 mL DMF wurden 0.25 mL (2.88 mmol) Triethylamin gegeben und das Reaktionsgemisch 5 h bei RT gerührt. Dieses wurde ohne weitere Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 84 mg (29% der Theorie)
ESI-MS: (M+H)+ = 792/794 (Cl)
Retentionszeit (HPLC): 3.5 min (Methode B)
Beispiel 22.1
(S)-4-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonyloxy]-propionyl}-1-(1 -methyl-piperidin-4-yl)- piperazin-2-carbonsäure
Zu 50.0 mg (0.06 mmol) (S)-4-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2- oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonyloxy]-propionyl}-1-(1 - methyl-piperidin-4-yl)-piperazin-2-carbonsäureethylester in 0.8 mL THF wurden 0.26 mL (0.13 mmol) 0.5 M LiOH-Lösung gegeben und das Reaktionsgemisch über Nacht bei RT
gerührt. Zur Vervollständigung der Reaktion wurden weitere 50 μL (0.1 mmol) 0.5 M LiOH-Lösung zugegeben, weitere 4 h bei RT gerührt und anschließend das Reaktionsgemisch ohne Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 22 mg (46% der Theorie)
ESI-MS: (M+H)+ = 764/766 (Cl)
Retentionszeit (HPLC): 2.9 min (Methode B)
Beispiel 23
(S)-1-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-3-yl)-piperidin-1-carbonyloxy]-propionyl}-4-(1-methyl-piperidin-4-yl)- piperazin-2-carbonsäureethylester
Zu einer Mischung von 700 mg (1.26 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdi- azepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1- carboxy-ethylester (Beispiel 1f), 430 mg (1.41 mmol) (S)-4-(1-Methyl-piperidin-4-yl)- piperazin-2-carbonsäureethylester (Beispiel A11b) und 450 mg (1.40 mmol) TBTU in 7 mL DMF wurden 0.30 mL (2.16 mmol) Triethylamin gegeben und das Reaktionsgemisch 18 h bei RT gerührt. Man engte i.vac. ein, verrührte den Rückstand mit gesättigter NaHCO3- Lösung, extrahierte erschöpfend mit EtOAc und trocknete die vereinigten organischen Phasen über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert.
Ausbeute: 670 mg (67% der Theorie)
ESI-MS: (M+H)+ = 792/794 (Cl)
Retentionszeit (HPLC): 3.4 min (Methode B)
Beispiel 23.1
(S)-1-{(R)-3-(4-Amino-3-chlor-5-trifluornnethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-3-yl)-piperidin-1-carbonyloxy]-propionyl}-4-(1-methyl-piperidin-4-yl)-pipera- zin-2-carbonsäure
Hergestellt analog zu Beispiel 22.1 aus 80.0 mg (0.10 mmol) (S)-1-{(R)-3-(4-Amino-3- chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)- piperidin-1-carbonyloxy]-propionyl}-4-(1-methyl-piperidin-4-yl)-piperazin-2-carbonsäure- ethylester und 0.46 mL (0.23 mmol) 0.5 M LiOH-Lösung.
Ausbeute: 53 mg (69% der Theorie)
ESI-MS: (M+H)+ = 764/766 (Cl)
Retentionszeit (HPLC): 2.9 min (Methode B)
Beispiel 24
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(8-ethoxycarbonylmethyl-8-aza-bicyclo[3.2.1]- oct-3-yl)-piperazin-1-yl]-2-oxo-ethylester
Zu einer Mischung von 200 mg (0.45 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdi- azepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1- carboxy-ethylester (Beispiel 1f), 128 mg (0.40 mmol) TBTU und 56 μL (0.40 mmol)
Triethylamin in 2 mL DMF wurden 113 mg (0.40 mmol) (3-Piperazin-1-yl-8-aza-bicyclo- [3.2.1]oct-8-yl)-essigsäureethylester (Beispiel A12d) gegeben und das Reaktionsgemisch 2 h bei RT gerührt. Dieses wurde ohne weitere Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und i.vac. eingeengt. Ausbeute: 156 mg (53% der Theorie)
ESI-MS: (M+H)+ = 818/820 (Cl)
Retentionszeit (HPLC): 3.1 min (Methode B)
Beispiel 24.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(8-carboxymethyl-8-aza-bicyclo[3.2.1]oct-3-yl)- piperazin-1-yl]-2-oxo-ethylester
Zu 40.0 mg (0.05 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1- carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(8-ethoxycarbonyl- methyl-δ-aza-bicyclop^.iJoct-S-yl)-piperazin-i-ylJ^-oxo-ethylester in 1 mL THF wurde eine Lösung von 1.92 mg (0.08 mmol) LiOH in 1 mL Wasser gegeben und das Reaktions- gemisch über Nacht bei RT geschüttelt. Die Lösungsmittel wurden im Stickstoffstrom entfernt und der Rückstand via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert.
Ausbeute: 13 mg (34% der Theorie)
ESI-MS: (M+H)+ = 790/792 (Cl) Retentionszeit (HPLC): 2.7 min (Methode B)
Beispiel 25
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-ethoxycarbonylnnethoxy-1 ,4l-bipiperidinyl-1l-yl)-
2-oxo-ethylester
Zu einer Mischung von 300 mg (0.54 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdi- azepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1- carboxy-ethylester (Beispiel 1f), 220 mg (0.64 mmol) ([1,4']Bipiperidinyl-4-yloxy)-essig- säureethylester (Beispiel A13c) und 200 mg (0.62 mmol) TBTU in 3 mL DMF wurden 0.30 mL (2.16 mmol) Triethylamin gegeben und das Reaktionsgemisch 4 h bei RT gerührt. Man versetzte mit Eis und gesättigter NaHCO3-Lösung und filtrierte den Niederschlag ab. Dieser wurde in DCM und wenig EtOH aufgenommen und über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 170 mg (39% der Theorie)
ESI-MS: (M+H)+ = 807/809 (Cl)
Retentionszeit (HPLC): 3.7 min (Methode B)
Beispiel 25.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-carboxymethoxy-1 ,4l-bipiperidinyl-1l-yl)-2-oxo- ethylester
Zu 100 mg (0.12 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1- carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-ethoxycarbonyl- methoxy-i ^'-bipiperidinyl-i'-yl^-oxo-ethylester in 8 mL THF wurde eine Lösung von 8.00 mg (0.33 mmol) LiOH in 2.5 mL Wasser gegeben und das Reaktionsgemisch 3 h bei RT gerührt. Man versetzte mit 0.34 mL (0.34 mmol) 1 M HCl und engte i.vac. ein. Der Rückstand wurde in DCM/MeOH aufgenommen, über wenig Kieselgel filtriert und mit DCM/MeOH (7:3) eluiert. Man engte i.vac. ein und trocknete den Rückstand im Hochvakuum.
Ausbeute: 61 mg (63% der Theorie) ESI-MS: (M+H)+ = 779/781 (Cl)
Retentionszeit (HPLC): 3.2 min (Methode B)
Beispiel 26
4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-ethoxycarbonylmethyl-1 ,4l-bipiperidinyl-1l-yl)- 2-oxo-ethylester
Zu 650 mg (1.17 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1- carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy-ethylester (Beispiel 1f) in 30 mL THF und 5 mL DMF wurden 200 μL (1.43 mmol) Triethylamin und 414 mg (1.29 mmol) TBTU gegeben und das Reaktionsgemisch 30 min bei RT gerührt. Dann erfolgte die Zugabe von 422 mg (1.29 mmol) [1 ,4']Bipiperidinyl-4-yl-essigsäureethylester (A14b) und 330 μL (2.38 mmol) Triethylamin in 10 mL DCM und weiteres Rühren bei RT für 18 h. Man versetzte mit 50 mL halbgesättigter NaHCO3-Lösung, extrahierte zweimal mit je 50 mL EtOAc, wusch die vereinigten organischen Phasen mit 50 mL gesättigter NaCI-Lösung und trocknete über Na2SO4. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand chromatographisch (Kieselgel, EtOAc/MeOH/NH3 95:5:0.5)
gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt, i.vac. eingeengt, der Rückstand mit DIPE verrieben, abgesaugt und getrocknet. Ausbeute: 646 mg (70% der Theorie)
ESI-MS: (M+H)+ = 791/793 (Cl) Rf = 0.33 (Kieselgel, EtOAc/MeOH/NH3 90:10:1)
Beispiel 26.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-carboxymethyl-1 ,4l-bipiperidinyl-1l-yl)-2-oxo- ethylester
Zu 100 mg (0.13 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1- carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-ethoxycarbonyl-methyl- 1 ,4'-bipiperidinyl-1 '-yl)-2-oxo-ethylester in 10 mL THF wurde eine Lösung von 5.00 mg (0.21 mmol) LiOH in 3 mL Wasser gegeben und das Reaktionsgemisch 2 Tage bei RT gerührt. Man engte i.vac. zur Trockene ein, nahm den Rückstand in 1 mL DMF auf und reinigte diesen via HPLC; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert.
Ausbeute: 22 mg (22% der Theorie)
ESI-MS: (M+H)+ = 763/765 (Cl)
Retentionszeit (HPLC): 3.2 min (Methode B)
Beispiel 27
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(2-ethoxycarbonyl-ethyl)-1 , 4'-bipiperidinyl-1 '- yl]-2-oxo-ethylester
Hergestellt analog Beispiel 24 aus 500 mg (0.90 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl- phenyl)-1-carboxy-ethylester (Beispiel 1f) und 341 mg (1.00 mmol) 3-[1,4']Bipiperidinyl-4- yl-propionsäureethylester (Beispiel A15b). Ausbeute: 340 mg (47% der Theorie)
ESI-MS: (M+H)+ = 805/807 (Cl)
Retentionszeit (HPLC): 3.6 min (Methode B)
Beispiel 27.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(2-carboxy-ethyl)-1 ,4l-bipiperidinyl-1l-yl]-2-oxo- ethylester
Zu 50 mg (0.06 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1- carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(2-ethoxycarbonyl-ethyl)- i ^'-bipiperidinyl-i'-ylj^-oxo-ethylester in 3 mL THF wurde eine Lösung von 2.40 mg (0.10 mmol) LiOH in 1 mL Wasser gegeben und das Reaktionsgemisch 4 h bei RT gerührt. THF wurde im Stickstoffstrom entfernt, der Rückstand in wenig Wasser aufgenommen und via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 38 mg (79% der Theorie)
ESI-MS: (M+H)+ = 777/779 (Cl)
Retentionszeit (HPLC): 3.4 min (Methode B)
Beispiel 28
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(3-ethoxycarbonyl-propyl)-piperidin-4-yl]- piperazin-1-yl}-2-oxo-ethylester
Zu einer Mischung von 500 mg (0.90 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3-benzdi- azepin-3-yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1- carboxy-ethylester (Beispiel 1f), 321 mg (1.00 mmol) TBTU und 0.14 mL (1.00 mmol) Triethylamin in 10 mL DMF wurden 283 mg (1.00 mmol) 4-(4-Piperazin-1-yl-piperidin-1- yl)-buttersäureethylester (Beispiel A16b) gegeben und das Reaktionsgemisch 2 h bei RT gerührt. Dieses wurde ohne weitere Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt, i.vac. eingeengt, der Rückstand mit gesättigter NaHCO3-Lösung verrührt, filtriert und getrocknet. Ausbeute: 165 mg (22% der Theorie) ESI-MS: (M+H)+ = 820/822 (Cl)
Retentionszeit (HPLC): 3.1 min (Methode B)
Beispiel 28.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(3-carboxy-propyl)-piperidin-4-yl]-piperazin- 1 -yl}-2-oxo-ethylester
Hergestellt analog Beispiel 27.1 aus 50 mg (0.06 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-{4-[1 -(3-ethoxycarbonyl-propyl)-piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester und 2.40 mg (0.10 mmol) LiOH. Ausbeute: 29 mg (60% der Theorie)
ESI-MS: (M+H)+ = 792/794 (Cl)
Retentionszeit (HPLC): 3.0 min (Methode B)
Beispiel 29
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(1-ethoxycarbonyl-ethyl)-piperidin-4-yl]- piperazin-1-yl}-2-oxo-ethylester
29a) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-1 - (4-amino-3-chlor-5-trifluormethyl-benzyl)-2-oxo-2-(4-piperidin-4-yl-piperazin-1-yl)- ethylester
Eine Mischung von 1.00 g (1.80 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-2-(4-amino-3-chlor-5-trifluormethyl-phenyl)-1-carboxy- ethylester (Beispiel 1f), 546 mg (1.80 mmol) 4-Piperazin-1-yl-piperidin-1-carbonsäure- benzylester (Beispiel A17b), 643 mg (2.00 mmol) TBTU und 0.35 mL (2.50 mmol) Triethylamin in 10 mL DMF wurde über Nacht bei RT gerührt. Man versetzte mit 150 mL
15% K2CO3-LOSUrIg, saugte den Niederschlag ab, wusch diesen mit 30 mL Wasser und trocknete das Rohprodukt bei 40°C im Trockenschrank. Ausbeute: 1.50 g (99% der Theorie)
ESI-MS: (M+H)+ = 840/842 (Cl) Retentionszeit (HPLC): 3.7 min (Methode B)
Eine Suspension von 750 mg (0.89 mmol) des obigen Produktes und 600 mg Raney- Nickel in 50 mL MeOH wurde 30 h bei RT und 3447 hPa hydriert. Der Katalysator wurde abgesaugt, i.vac. eingeengt und der Rückstand via HPLC gereinigt. Die das Produkt enthaltenden Fraktionen wurden vereinigt, i.vac. eingeengt, der Rückstand mit 15% K2CO3-Lösung alkalisch gestellt, mit 100 mL EtOAc extrahiert, die organische Phase abgetrennt und über Na2SO4 getrocknet. Nach Entfernen des Trocken- und Lösungsmittels wurde der Rückstand mit 30 mL DIPE verrieben, abgesaugt und getrocknet. Ausbeute: 280 mg (44% der Theorie) ESI-MS: (M+H)+ = 706/708 (Cl)
Retentionszeit (HPLC): 2.8 min (Methode B)
29b) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1 -carbonsäure-(f?)-1 - (4-amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-ri-(1-ethoxycarbonyl-ethyl)-piperidin- 4-yll-piperazin-1-yl}-2-oxo-ethylester
Eine Mischung von 140 mg (0.20 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-oxo-2-(4- piperidin-4-yl-piperazin-1-yl)-ethylester, 60.8 mg (0.44 mmol) K2CO3 und 29 μL (0.22 mmol) 2-Brompropionsäureethylester in 1.8 mL DMF wurde bei 50°C 2 h geschüttelt. Das Reaktionsgemisch wurde ohne Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert.
Ausbeute: 111 mg (69% der Theorie)
ESI-MS: (M+H)+ = 806/808 (Cl)
Retentionszeit (HPLC): 3.1 min (Methode B)
Beispiel 29.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1 -(1 -carboxy-ethyl)-piperidin-4-yl]-piperazin-1 ■ yl}-2-oxo-ethylester
Zu 50 mg (0.06 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1- carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1 -(1 -ethoxycarbonyl- ethyl)-piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester in 1 mL THF wurde eine Lösung von 2.4 mg (0.10 mmol) LiOH in 0.8 mL Wasser gegeben und das Reaktionsgemisch 2 h bei RT und 4 h bei 50°C geschüttelt. Dieses wurde anschließend ohne Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert.
Ausbeute: 42 mg (87% der Theorie)
ESI-MS: (M+H)+ = 778/780 (Cl) Retentionszeit (HPLC): 3.0 min (Methode B)
Beispiel 30
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1 -(1 -ethoxycarbonyl-1-methyl-ethyl)-piperidin- 4-yl]-piperazin-1-yl}-2-oxo-ethylester
Eine Mischung von 140 mg (0.20 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-oxo-2-(4-
piperidin-4-yl-piperazin-1-yl)-ethylester (Beispiel 29a), 60.8 mg (0.44 mmol) K2CO3 und 45 μL (0.30 mmol) 2-Brom-2-methyl-propionsäureethylester in 1.8 mL DMF wurde bei 50°C 12 h und anschließend 48 h bei RT geschüttelt. Der Niederschlag wurde abfiltriert und das Filtrat ohne Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt, i.vac. eingeengt, der Rückstand mit gesättigter NaHCO3-Lösung alkalisch gestellt, der Niederschlag abgesaugt, mit 20 mL Wasser gewaschen und bei 40°C getrocknet.
Ausbeute: 85 mg (52% der Theorie)
ESI-MS: (M+H)+ = 820/822 (Cl) Retentionszeit (HPLC): 3.0 min (Methode B)
Beispiel 31
1 l-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-S-yl)-piperidin-i-carbonyloxyJ-propionyl^-methyl-i ^'-bipiperidinyl^- carbonsäureethylester
Eine Mischung von 127 mg (0.20 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-oxo-2-(4- oxo-piperidin-1-yl)-ethylester (Beispiel 1g), 68.5 mg (0.40 mmol) 4-Methyl-piperidin-4- carbonsäureethylester und 11 μL (0.2 mmol) AcOH in 2 mL DCM wurde 2 h bei RT gerührt, auf 0°C gekühlt, 2 h gerührt, dann mit 57.7 mg (0.26 mmol) Natriumtriacetoxy- borhydrid versetzt und über Nacht bei 0°C nachgerührt. Das Reaktionsgemisch wurde ohne Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert.
Ausbeute: 85 mg (54% der Theorie)
ESI-MS: (M+H)+ = 791/793 (Cl)
Retentionszeit (HPLC): 7.7 min (Methode C)
Analog wurden folgende Verbindungen aus jeweils 127 mg 4-(2-Oxo-1,2,4,5-tetrahydro- 1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-oxo-2-(4-oxo-piperidin-1-yl)-ethylester (Beispiel 1g) und der jeweils nötigen Menge der Aminkomponente hergestellt:
Beispiel 32
1
l-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo-1 ,2,4,5-tetrahydro-i ,3- benzdiazepin-S-yl)-piperidin-i-carbonyloxyj-propionyl^-methyl-i ^'-bipiperidinyl^- carbonsäure
Zu 40 mg (0.05 mmol) 1l-{(R)-3-(4-Amino-3-chlor-5-trifluormethyl-phenyl)-2-[4-(2-oxo- 1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonyloxy]-propionyl}-4-methyl- 1 ,4'-bipiperidinyl-4-carbonsäureethylester in 0.8 mL THF wurde eine Lösung von 2.00 mg (0.08 mmol) LiOH in 1 mL Wasser gegeben und das Reaktionsgemisch 1 h bei RT und 8 h bei 50°C gerührt. Dieses wurde ohne Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 18 mg (47% der Theorie)
ESI-MS: (M+H)+ = 763/765 (Cl)
Retentionszeit (HPLC): 3.5 min (Methode B)
Analog wurden folgende Verbindungen aus jeweils 40 mg des entsprechenden Ethylesters hergestellt, wobei zur Hydrolyse nur 1 h bei RT nötig war:
Beispiel 33
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-{4-[1-(5-methyl-2-oxo-1 ,3-dioxol-4-ylmethoxy- carbonylmethyl)-piperidin-4-yl]-piperazin-1-yl}-2-oxo-ethylester
Eine Mischung aus 185 mg (0.24 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3- yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1 - carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester (Beispiel 4.1), 25 mg (0.25 mmol) KHCO3 und 50 mg (0.26 mmol) 4-Brommethyl-5-methyl-1 ,3-dioxol-2-on in 1 mL DMF wurde über Nacht bei RT geschüttelt. Das Reaktionsgemisch wurde über einen Spritzenfilter filtriert und via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 14 mg (7% der Theorie) ESI-MS: (M+H)+ = 876/878 (Cl)
Rf = 0.54 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 33.1
4-(2-Oxo-1 ,2,4,5-tetrahydro-i ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-[1l-(5-nnethyl-2-oxo-1,3-dioxol-4-ylnnethoxy- carbonylmethyl^^'-bipiperidinyl-i-ylj^-oxo-ethylester
Hergestellt analog Beispiel 33 aus 150 mg (0.20 mmol) 4-(2-Oxo-1,2,4,5-tetrahydro-1,3- benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl- benzyl)-2-(1l-carboxymethyl-4,4l-bipiperidinyl-1-yl)-2-oxo-ethylester (Beispiel 3.1) und 50 mg (0.26 mmol) 4-Brommethyl-5-methyl-1,3-dioxol-2-on. Ausbeute: 8 mg (5% der Theorie) ESI-MS: (M+H)+ = 875/877 (Cl)
Rf = 0.74 (Kieselgel, DCM/Cyc/MeOH/NH3 70:15:15:2)
Beispiel 34
4-(2-Oxo-1 ,2,4,5-tetrahydro-1 ,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4- amino-3-chlor-5-trifluormethyl-benzyl)-2-(4-{1-[2-(2-methoxy-ethoxy)-ethoxycarbonyl- methyl]-piperidin-4-yl}-piperazin-1-yl)-2-oxo-ethylester
Zu einer Mischung aus 75 mg (0.10 mmol) 4-(2-Oxo-1 ,2,4,5-tetrahydro-1,3-benzdiazepin- 3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[4-(1 - carboxymethyl-piperidin-4-yl)-piperazin-1-yl]-2-oxo-ethylester (Beispiel 4.1), 35 mg (0.11 mmol) TBTU und 28 μL (0.20 mmol) Triethylamin in 1 mL DMF wurden 13 μL (0.11 mmol)
2-(2-Methoxy-ethoxy)-ethanol zugegeben und das Reaktionsgemisch 1 h bei RT gerührt. Dieses wurde ohne Aufarbeitung via HPLC gereinigt; die das Produkt enthaltenden Fraktionen wurden vereinigt und lyophilisiert. Ausbeute: 22 mg (26% der Theorie) ESI-MS: (M+H)+ = 866/868 (Cl)
Retentionszeit (HPLC): 3.2 min (Methode B)
Beispiel 35
4-(7-Methoxy-2-oxo-1,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl)-piperidin-1-carbonsäure- (R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2-[1l-(2-morpholin-4-yl-ethoxycarbonyl- methyl)-4,4'-bipiperidinyl-1-yl]-2-oxo-ethylester
Eine Mischung aus 230 mg (0.29 mmol) 4-(7-Methoxy-2-oxo-1 ,2,4,5-tetrahydro-1 ,3-benz- diazepin-3-yl)-piperidin-1-carbonsäure-(R)-1-(4-amino-3-chlor-5-trifluormethyl-benzyl)-2- (i'-carboxymethyl^^'-bipiperidinyl-i-yl^-oxo-ethylester (Beispiel 15.1), 113 mg (0.35 mmol) TBTU, 84 μL (0.60 mmol) Triethylamin und 39.3 mg (0.30 mmol) 2-Morpholin-4-yl- ethanol in 5 mL DMF wurde über Nacht bei RT gerührt. Das Reaktionsgemisch wurde auf gesättigte NaHCO3-Lösung gegossen, das ausgefallene Produkt abgesaugt und bei 40°C getrocknet. Dieses wurde in 25 mL Isopropanol gelöst und durch Zugabe von 0.5 M HCl in Isopropanol als Hydrochlorid Salz gefällt. Dieses wurde abgesaugt, mit 5 mL Isopropanol und 30 mL DIPE gewaschen und bei 30°C im Vakuumtrockenschrank getrocknet. Ausbeute: 90 mg (34% der Theorie) ESI-MS: (M+H)+ = 906/908 (Cl)
Retentionszeit (HPLC): 3.1 min (Methode B)
Die nachfolgenden Beispiele beschreiben die Herstellung pharmazeutischer Anwendungsformen, die als Wirkstoff eine beliebige Verbindung der allgemeinen Formel I enthalten:
Beispiel I
Kapseln zur Pulverinhalation mit 1 mg Wirkstoff
Zusammensetzung: 1 Kapsel zur Pulverinhalation enthält: Wirkstoff 1.0 mg
Milchzucker 20.0 mg Hartgelatinekapseln 50.0 mg
71.0 mg
Herstellungsverfahren:
Der Wirkstoff wird auf die für Inhalative erforderliche Korngröße gemahlen. Der gemahlene Wirkstoff wird mit dem Milchzucker homogen gemischt. Die Mischung wird in Hartgelatinekapseln abgefüllt.
Beispiel
Inhalationslösung für Respimat® mit 1 mg Wirkstoff
Zusammensetzung: 1 Hub enthält:
Wirkstoff 1.0 mg Benzalkoniumchlorid 0.002 mg
Dinatriumedetat 0.0075 mg
Wasser gereinigt ad 15.0 μl
Herstellungsverfahren: Der Wirkstoff und Benzalkoniumchlorid werden in Wasser gelöst und in Respimaf Kartuschen abgefüllt.
Beispiel IM
Inhalationslösung für Vernebler mit 1 mg Wirkstoff
Zusammensetzung:
1 Fläschchen enthält:
Wirkstoff 0.1 g
Natriumchlorid 0.18 g
Benzalkoniumchlorid 0.002 g
Wasser gereinigt ad 20.0 ml
Herstellungsverfahren:
Wirkstoff, Natriumchlorid und Benzalkoniumchlorid werden in Wasser gelöst.
Beispiel IV
Treibgas-Dosieraerosol mit 1 mg Wirkstoff
Zusammensetzung:
1 Hub enthält:
Wirkstoff 1.0 mg Lecithin 0.1 %
Treibgas ad 50.0 μl
Herstellungsverfahren:
Der mikronisierte Wirkstoff wird in dem Gemisch aus Lecithin und Treibgas homogen suspendiert. Die Suspension wird in einen Druckbehälter mit Dosierventil abgefüllt.
Beispiel V
Nasalspray mit 1 mg Wirkstoff
Zusammensetzung:
Wirkstoff 1.0 mg
Natriumchlorid 0.9 mg
Benzalkoniumchlorid 0.025 mg
Dinatriumedetat 0.05 mg
Wasser gereinigt ad 0.1 ml
Herstellunαsverfahren: Der Wirkstoff und die Hilfsstoffe werden in Wasser gelöst und in ein entsprechendes Behältnis abgefüllt.
Beispiel VI
Injektionslösung mit 5 mg Wirksubstanz pro 5 ml
Zusammensetzung:
Wirksubstanz 5 mg
Glucose 250 mg Human-Serum-Albumin 10 mg
Glykofurol 250 mg
Wasser für Injektionszwecke ad 5 ml
Herstellung: Glykofurol und Glucose in Wasser für Injektionszwecke auflösen (WfI); Human-Serum- Albumin zugeben; Wirkstoff unter Erwärmen auflösen; mit WfI auf Ansatzvolumen auffüllen; unter Stickstoff-Begasung in Ampullen abfüllen.
Beispiel VII
Injektionslösung mit 100 mg Wirksubstanz pro 20 ml
Zusammensetzung:
Wirksubstanz 100 mg Monokaliumdihydrogenphosphat = KH2PO4 12 mg
Dinatriumhydrogenphosphat = Na2HPO4 *2H2O 2 mg
Natriumchlorid 180 mg
Human-Serum-Albumin 50 mg
Polysorbat 80 20 mg
Wasser für Injektionszwecke ad 20 ml
Herstellung:
Polysorbat 80, Natriumchlorid, Monokaliumdihydrogenphosphat und Dinatriumhydro- genphosphat in Wasser für Injektionszwecke (WfI) auflösen; Human-Serum-Albumin zugeben; Wirkstoff unter Erwärmen auflösen; mit WfI auf Ansatzvolumen auffüllen; in Ampullen abfüllen.
Beispiel VIII
Lyophilisat mit 10 mg Wirksubstanz
Zusammensetzung:
Wirksubstanz 10 mg Mannit 300 mg
Human-Serum-Albumin 20 mg
Wasser für Injektionszwecke ad 2 ml
Herstellung: Mannit in Wasser für Injektionszwecke (WfI) auflösen; Human-Serum-Albumin zugeben; Wirkstoff unter Erwärmen auflösen; mit WfI auf Ansatzvolumen auffüllen; in Vials abfüllen; gefriertrocknen.
Lösungsmittel für Lyophilisat: Polysorbat 80 = Tween 80 20 mg
Mannit 200 mg
Wasser für Injektionszwecke ad 10 ml
Herstellung: Polysorbat 80 und Mannit in Wasser für Injektionszwecke (WfI) auflösen; in Ampullen abfüllen.
Beispiel IX
Tabletten mit 20 mg Wirksubstanz
Zusammensetzung:
Wirksubstanz 20 mg
Lactose 120 mg
Maisstärke 40 mg
Magnesiumstearat 2 mg
Povidon K 25 18 mg
Herstellung:
Wirksubstanz, Lactose und Maisstärke homogen mischen; mit einer wässerigen Lösung von Povidon granulieren; mit Magnesiumstearat mischen; auf einer Tablettenpresse abpressen; Tablettengewicht 200 mg.
Beispiel X
Kapseln mit 20 mg Wirksubstanz
Zusammensetzung:
Wirksubstanz 20 mg
Maisstärke 80 mg
Kieselsäure, hochdispers 5 mg
Magnesiumstearat 2.5 mg
Herstellung:
Wirksubstanz, Maisstärke und Kieselsäure homogen mischen; mit Magnesiumstearat mischen; Mischung auf einer Kapselfüllmaschine in Hartgelatine-Kapseln Größe 3 abfüllen.
Beispiel Xl
Zäpfchen mit 50 mg Wirksubstanz
Zusammensetzung:
Wirksubstanz 50 mg
Hartfett (Adeps solidus) q.s. ad 1700 mg
Herstellung: Hartfett bei ca. 38°C aufschmelzen; gemahlene Wirksubstanz im geschmolzenen Hartfett homogen dispergieren; nach Abkühlen auf ca. 35°C in vorgekühlte Formen ausgießen.
Beispiel XII
Injektionslösung mit 10 mg Wirksubstanz pro 1 mL
Zusammensetzung:
Wirksubstanz 10 mg
Mannitol 50 mg Human-Serum-Albumin 10 mg
Wasser für Injektionszwecke ad 1 ml
Herstellung:
Mannitol in Wasser für Injektionszwecke auflösen (WfI); Human-Serum-Albumin zugeben; Wirkstoff unter Erwärmen auflösen; mit WfI auf Ansatzvolumen auffüllen; unter Stickstoff- Begasung in Ampullen abfüllen.