Title of the invention
Novel Λ/-alkoxyadenine derivatives acting as cytokine inhibitors.
Field of the invention
The present invention relates to novel Λ/,9-disubstituted adenine derivatives and novel aristeromycin analogues which are further substituted at the adenine 2-position, and which have a hydroxymethyl group, or an ester or ester isostere at a position corresponding to the ribose 4-position, and pharmaceutically acceptable addition salts thereof. The compounds act as cytokine inhibitors. Also covered are processes for preparation of the above derivatives and their pharmaceutical compositions as well as methods for using the compounds and compositions as drugs for the treatment of disorders involving cytokines in humans.
Background of the Invention
Adenosine receptors represent a subclass (P.) of the group of purine nucleotide and nucleoside receptors known as purinoreceptors. This subclass has been further classified into distinct receptor types which are now known as A^ A^, A2B and A3.
Extensive research has been carried out in a quest to identify selective ligands at these receptors [see, for example, Fredholm, B.B.; Abbracchio, M.P.; Bumstock, G.; Daly, J.W.; Harden, T.K.; Jacobson, K.A.; Williams, M. Nomenclature and Classification of Purinoceptors. Pharmacol. Rev. 1994, 46, 143-156; Van Galen, P.J.M.; Stiles, G.L.; Michaels, G. Jacobson, K.A. Adenosine A., and A2 Receptors: Structure Activity Relationships. Med Chem. Rev. 1992, 12, 423-471; Linden, J. Cloned Adenosine A3 receptors: Pharmacological Properties, Species Differences and Receptor Functions. TIPS, 1994, 15, 298-306; Jacobson, K.A.; Kim, H.A.; Siddiqi, S.M.; Olah, M.E.; Stiles, G.L; von Lubitz, D..K.J.E.; A3 Adenosine Receptors: Design of Selective Ligands and Therapeutic Propects. Drugs of the
Future 1995, 20, 689-699; Collis, M.G.; Hourani, S.M.O.; Adenosine Receptor Subtypes, TIPS, 1993, 360-366].
Several purine derivatives are known to modulate the release and or action of cytokines. For example the naturally occurring 5'-methylthioadenosine has been shown to inhibit cytokine production (Cerri, M.A.; Beltran-Nunez, A.; Bernasconi, S.; Dejana, E.; Bassi, L.; Bazzoni, G. Inhibition of Cytokine Production and Endothelial Expression of Adhesion Antigens by 5'-Methylthioadenosine. Eur. J. Pharmacol. 1993, 232, 291-294).
Furthermore, the adenosine agonists R-PIA, NECA, CPCA, CGS 21680, 2- chloroadenosine and CHA as well as the adenosine uptake inhibitor dipyridamole have all been shown to have an inhibitory effect on Tumour Necrosis Factor (TNF) production (Le Vraux, V.; Chen, Y.L.; Masson, M.; De Sousa, M.; Giroud, J.P.; Florentin, I.; Chauvelot-Moachon, L. Inhibition of Human Monocyte TNF production by Adenosine Receptor Agonists. Life Sci. 1993, 52, 1917 - 1924) and these investigators conclude that it is the A2 adenosine receptor which is involved in this aspect of TNF inhibition. The post-receptor event is presumably upregulation of adenylate cyclase. In a separate study, the specific adenosine A- agonists 2-chloro- adenosine and CCPA as well as the A2 agonist CPCA have been identified as inhibitors of lipopolysaccharide-stimulated TNF-α production, with the A2 agonist CPCA being 1000-fold more potent than CCPA, giving further evidence of the probable involvement of the adenosine A2 receptor. These findings have been confirmed by others (Prabhakar, U.; Brooks, D.P. Lipschlitz, D.; Esser, K.M. Inhibition of LPS-induced TNF-α Production in Human Monocytes by Adenosine (A2) Receptor Selective Agonists. Int. J. Immunopharmac. 1995, 17, 221-224). This effect of adenosine receptor agonists on TNF biosynthesis have been reviewed recently (Lee, J.C.; Prabhakar, U.; Griswold, D.E.; Dunnington, D.; Young, P.R.; Badger, A. Low-Molecular-Weight TNF Biosynthesis Inhibitors: Strategies and Prospectives. Circulatory Shock 1995, 44, 97-103) as has their therapeutic potential
(Giroud, J.P.; Lian Chen, Y.; Le Vraux, V.; Chauvelot-Moachon, L. Therapeutic Aspects of Adenosine in Relation to its anti-TNF properties. Bull. Acad. Natl. Med. (Paris) 1995, 779, 79 -101).
A very recent article has confirmed the involvement of both adenosine and A2 and A3 receptors in the inhibition of human neutrophil degranulation (Bouma, M.G.; Jeunhomme, T.M.M.A.; Boyle, D.L.; Dentener, M.A.; Voitenok, N.N.; Vandenwildenberg, F.A.J.M; Buurman, W.A. Adenosine inhibits neutrophil degranulation in activated human whole-blood - involvement of adenosine A2 and A3 receptors. J. Immunol., 1997, 158, 5400-5408). Endogenous adenosine has been shown to inhibit lipopolysaccharide-stimulated tumor necrosis factor synthesis (Eigler, A.; Greten, T.F.; Sinha, B.; Haslberger, C; Sullivan, G.W.; Endres, S. Endogenous adenosine curtails lipopolysaccharide-stimulated tumor-necrosis-factor synthesis. Scand. J. Immunol. 1997, 45, 132 -139. Other recent scientific articles serve to underline the importance of adenosine A3 receptors in the modulation of TNF-α levels in mammals since the first published observation (Sajjadi, F.G.; Takabayashi, K.; Foster, A.C.; Domingo, R.C.; Firestein, G.S. Inhibition of TNF-alpha expression by adenosine - role of A3 adenosine receptors. J. Immunol., 1996, 156, 3435-3442. Adenosine agonists or adenosine regulating agents have potential therapeutic uses in acute and chronic inflammatory diseases. This has been demonstrated in macrophages (Bowlin, T.L.; Borcherding, D.R.; Edwards, C.K.; Mcwhinney, CD. Adenosine A3 receptor agonists inhibit macrophage tumor- necrosis-factor-alpha production. Drug Dev. Res., 1996, 39, 388-392; McWhinney, CD.; Dudley, M.W.; Bowlin, T.L.; Peet, N.P.; Schook, L; Bradshaw, M.; De, M.; Borcherding, D.R.; Edwards, C.K. Activation of adenosine A3 receptors on macrophages inhibits tumor-necrosis-factor-alpha. Eur. J. Pharmacol. 1996, 310, 209-216; Ritchie, P.K.; Spangelo, B.L.; Krzymowski, D.K.; Rossiter, T.B.; Kurth, E; Judd, A.M. Adenosine increases interleukin-6 release and decreases tumor- necrosis-factor release from rat adrenal zona glomerulosa cells, ovarian-cells, anterior-pituitary-cells, and peritoneal-macrophages. Cytokine, 1997, 9, 187- 98).
Disease states involving TNF-α
The suggestion has been made that TNF-α inhibitors are useful in the treatment of diabetes (Argiles, J.M., Lopez-Soriano, J. and Lopez-Soriano, F.J. Cytokines and Diabetes: The Final Step. Involvement of TNF-α in both Type I and Type II Diabetes Mellitus. Horm. Metab. Res., 1994, 26, 447 - 449).
It is now clear that TNF-α levels are increased in obese rodents (Yamakawa, T., Tanaka, S-l., Yamakawa, Y., Kiuchi, Y., Isoda, F., Kawamoto, S, Okuda, K. and Sekihara, H. Augmented Production of Tumor Necrosis Factor-α Production in Obese Mice. Clin. Immunol, and Immunopath., 1995, 75, 51-56). A clinical study of the expression pattern of TNF-α in adipose tissue of obese and normal premenopausal women has been carried out (Hotamisligil, G.S., Arner, P. Caro, J.F., Atkinson, R.L. and Spielgelman, B. Increased Adipose Tissue Expression of Tumor Necrosis Factor-α in Human Obesity and Insulin Resistance. J. Clin. Invest., 1995, 95, 2409 - 2415). Obese individuals express 2.5-fold more TNF-α mRNA in fat tissue relative to lean controls, and there was a correlation to the level of hyperinsulinemia, suggesting a role for TNF-α in the pathogenesis of obesity related insulin resistance. The topic of TNF-α in insulin resistance has been reviewed by the same group (Hotamisligil, G.S. and Spielgelman, B. Perspectives in Diabetes. Tumor Necrosis Factor-α: A Key Component of the Obesity-Diabetes Link. Diabetes, 1994, 43, 1271 - 1278).
Some investigators suggest that adenosine and adenosine agonists, acting via A2 receptors can be of benefit in for example septic shock or ischaemia-reperfusion injury, where cytokine production by mononuclear phagocytes can be inhibited by these agents (Bouma, M.G., Stad, R.K., van den Wildenberg, A.J.M. and Buurman, W.A. Differential Regulatory Effects of Adenosine on Cytokine Release by Activated Human Monocytes. J. Immunol., 1994, 153, 4159 - 4168). These effects by A2 receptor agonists have also been demonstrated on human polymorphonuclear leukocytes (Thiel, M. and Chouker, A. Acting Via A2 Receptors, Adenosine Inhibits
the Production of Tumor Necrosis Factor-α of Endotoxin-stimulated Human Polymorphonuclear Leucocytes. J. Lab Clin. Med. 1995, 275 - 282).
A massive release of TNF-α in the host produces severe damage to a range of tissues. It is therefore clear that TNF-α inhibitors have application in disorders which involve an inflammatory response, but this cytokine has multiple inflammatory, metabolic and immunological activities (Jirillo, E. Pellegrino, N.M. and Antonaci, S. Role of Tumor Necrosis Factor-α in Physiological and Pathological Conditions. Med. Sci. Res., 1995, 23, 75-79). Adenosine derivatives have potential in the treatment of Rheumatoid Arthritis (Firestein, G.S. Antiinflammatory effects of adenosine kinase inhibitors in acute and chronic inflammation. Drug Dev. Res., 1996, 39, 371-376).
Many patented TNF-α inhibitors such as rolipram, pentoxyfylline and denbufylline are phosphodiesterase (PDE) inhibitors and exert their effects on TNF-α via control of cAMP (Davidsen, S.K. and Summers, J.B. Inhibitors of TNF-α Synthesis. Exp. Opin. Ther. Patents 1995, 5, 1087 - 1100). Evidence is also available for some synergism between the effects of rolipram and adenosine in reduction of T primed neutrophil oxidative activity, thereby offering protection against inflammatory tissue damage (Sullivan, G., Carper, H.T. and Mandell, G.L. Int. J. Immunopharmac. 1995, 17, 793-803).
There is also good evidence for TNF inhibitors in the prevention of neuronal damage following cerebral ischaemia (Firestein, G.Z., Liu, T and Barone, F.C. Cytokines, Inflammation, and Brain Injury: Role of Tumor Necrosis Factor-α. Cerebrovascular and Brain Metabolism Reviews 1994, 6, 341-360).
Background art
Examples of adenosine derivatives in the chemical literature with the heteroatoms, oxygen or nitrogen bonded directly to the 6-amino substituent are summarised below.
Examples with hydrogen at the purine 2-position include /V-aminoadenosine, N-[(N- methyl-/V-phenyl)amino]adenosine, Λ/-hydroxyadenosine, /V-methoxyadenosine and Λ/-benzyloxyadenosine (Kusachi, S., Thompson, R.D. Bugni, W.J., Yamada, N. and Olsson, R.A. Dog Coronary Artery Adenosine Receptor: Structure of the Λ/6-Alkyl Subregion. J. Med. Chem., 1985, 28, 1636 - 1643); /V-ethoxyadenosine (Fujii, T., Wu, C.C., itaya, T., Moro, S. and Saito, T. Purines. XI. The Synthesis of N- Alkoxyadenosines and Their 2',3'-O-lsopropylidene Derivatives. Chem. Pharm. Bull., 1973, 21, 1676 - 1682); (Fuji, T., W, C.C. and Itaya, T. Purines. XII. Catalytic Hydrogenolysis of Alkoxyaminopurines and Related Derivatives, ibid., 1973, 21, 1835-1838); Λ/-(methylamino)adenosine and V-[(Λ/-hydroxy-/V-methyl)amino]adeno- sine (Giner-Sorolla, A., O'Bryant, S.A., Nanos, C, Dollinger M.R., Bendich, A. and Burchenal, J.H. The Synthesis and Biological Properties of Hydroxylaminopurines and Related Derivatives. J. Med. Chem., 1968, 11, 521 - 523).
Examples of adenosine derivatives with oxygen or nitrogen atoms bonded to the 6- amino substituent, containing an additional purine 2-substituent are 2-amino-Λ/- hydroxyadenosine (Kikugawa, K., lizuka, K., Higuchi, Y., Hirayama, H. and lchino, M. Platelet Aggregation Inhibitors. 2. Inhibition of Platelet Aggregation by 5'-,2-,6-, and 8-substituted Adenosines. J. Med. Chem., 1972, 15, 387 - 390); 2-amino-/V- aminoadenosine (Saneyoshi, M. and Terashima, K. Synthetic Nudeosides and Nucleotides. VII. A Direct Replacement of 6-Thiol Group of 6-Thioinosine and 6- Thioguanosine with Hydrazine Hydrate. Chem. Pharm. Bull., 1969, 17, 2373 - 2376); 2-amino-Λ/-methoxyadenosine (Chem. Pharm. Bull., 1975, 23, 464 - 466); (Ueda, T., Miura, K. and Kasai, T., Synthesis of 6-Thioguanosine and 2,6-diaminopurine Nudeosides and Nucleotides from Adenosine Counterparts via a facile Rearrangement in the Base Portion (Nudeosides and Nucleotides XIX). Chem. Pharm. Bull., 1978, 26, 2122 - 2127); 2-chloro-/V-hydroxyadenosine (Cristalli, G., Sauro, V., Eleuteri, A., Grifantini, M., Volpini, R., Lupidi, G., Capolongo, L and Pesenti, E. Purine and 1-Deazapurine Ribonucleosides and Deoxyribonucleosides: Synthesis and Biological Activity. J. Med. Chem., 1991 , 34, 2226 - 2230); (Uzerman,
A.P. von Frijtag Drabbe Kunzel, J.K., Vittori, S. and Cristalli, G. Purine-Substituted Adenosine Derivatives with Small Λ^-Substituents as Adenosine Receptor Agonists. Nudeosides and Nucleotides, 1994, 13, 2267 - 2281): 2-fluoro-/V-hydroxyadenosine and 2-fluoro-Λ/-aminoadenosine (Montgomery, J.A. and Hewson, K. 2-Fluoropurine Ribonucleosides. J. Med. Chem., 1970, 13, 427 - 430) and 2-fluoro-/\/- methoxyadenosine (Giner-Sorolla, A. and Burchenal, J.H., Substituted Hydroxylaminopurines and Related Derivatives. Synthesis and Screening Tests. J. Med. Chem., 1971, 14, 816 - 819).
(1R,4S)-9-[4-(Dimethylthexylsilyloxymethyl)cyclopent-2-enyl]-Λ/-methoxyadenine-2- amine is featured as an intermediate in the synthesis of (-)-carbovir (Exall A.M., Jones, M.F., Mo, C-L, Myer, P.L., Paternoster, I.L., Singh, H., Storer, R., Weingarten, G.G., Williamson, C, Brodie, A.C., Cook, J., Lake, D.E., Meerholz, C.A., Turnbull, P.J. and Highcock, R.M. Synthesis from (-)-Aristeromycin and X-Ray structure of (-)-Carbovir. J. Chem. Soc. Perkin Trans. I, 1991, 2467 - 2477). A related purin-2-amine is featured as an intermediate in the synthesis of the antiherpetic agent carbocyclic 9-(2'-deoxy-2'-fluoro- -D-arabinofuranosyl)guanine (Borthwick, A.D., Biggadike, K., Holman, S. and Mo, C-L., Enantiospecific Synthesis of the Potent Antiherpetic Carbocyclic 9-(2'-deoxy-2'-fluoro-β-D-arabinofuranosyl)- guanine (+)-C-APG from Aristeromycin. Tetrahedron Lett., 1990, 31 , 767-770).
Some of the biological effects of adenosine derivatives on the central nervous system have been published, in particular a series of Λ/-alkoxy adenosine derivatives which possess anticonvulsant activity in mice (Knutsen, L.J.S., Lau, J., Eskesen, K., Sheardown, M.J., Thomsen, C, Weis, J.U., Judge, M.E. and Klitgaard, H. Anticonvulsant Actions of Novel and Reference Adenosine Agonists. In Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology, Belardinelli, L. and Pelleg, A„ Eds.; Kluwer: Boston, MA; 1995, 479-487. See also Knutsen, L.J.S., Lau, J., Sheardown, M.J., Thomsen, C; The Synthesis and Biochemical Evaluation of New A, Selective Adenosine Receptor Agonists
Containing a 6-Hydrazinopurine Moiety; BioMed. Chem. Lett., 1993, 3, 2661-2666)
The TNF-inhibiting effects of some related compounds have been revealed (Bowler, A.N., Olsen, U.B., Thomsen, C. and Knutsen, L.J.S. New adenosine A3 ligands controlling cytokines. Drug Dev. Res., 1996, 173 and PCT applications WO DK97/00107 and WO DK97/00108).
Recent patent literature within novel purinergic agents has been reviewed (Bhagwhat, S.S.; Williams, M. Recent Progress in Modulators of Purinergic Activity. Exp. Opin. Ther. Patents 1995, 5, 547 - 558), as have patents covering TNF inhibitors (Davidsen, S.K. and Summers, J.B. Inhibitors of TNF-α Synthesis. Exp. Opin. Ther. Patents 1995, 5, 1087 - 1100).
Marion Merrell Dow Inc. has claimed some cyclopentyl substituted adenines which appear to exert their effects by control of TNF-α (US 5,244,896). A follow-up application by Merrell Dow Pharmaceuticals Inc. (WO 95/03304) disclosed some novel bicyclic 9-bicyclic nudeosides useful as selective inhibitors of proinflammatory cytokines. Selected compounds have been published (Borcherding, D.R.; Peet, N.P.; Munson, H.R.; Zhang, H.; Hoffman, P.F.; Bowlin, T.L.; Edwards, C.K. Carbocyclic nudeosides as inhibitors of human tumor-necrosis-factor-alpha production - effects of the stereoisomers of (3-hydroxycyclopentyl)adenines. J. Med. Chem., 1996, 39, 2615-2620).
Merck & Co. Inc. has filed a patent application claiming a method for inhibiting TNF- α production comprising contacting the A2B subtype of the adenosine receptor with an adenosine agonist (GB 2289218-A).
Some other purine derivatives which are TNF inhibitors, presumably owing to their activity as PDE inhibitors, have been reviewed (This Years Drug News. Cardiovascular Drugs. Treatment of Septic Shock 1994 pp 185 - 189).
In WO 9533750-A1 , Pfizer Inc. claim a range of heterocycles, including some purine
derivatives, as CRF antagonists.
In US Patent 3,819,613, substituted adenosine analogues with hydrazone derivatives on the 6-amino function are disclosed as hypotensive agents. In GB 1 ,351,501 , adenosine and 2-aminoadenosine derivatives having a -NH-R2 group joined to the 6-amino function are disclosed as coronary dilators and platelet aggregation inhibitors. In EP A 152,944, a series of 2-, 6- and 8-substituted adenosine derivatives are described having activity as anti-allergy agents. In EP 402.752A, derivatives of adenosine unsubstituted in the 2-position are described which have a substituted heteroaromatic 1-pyrrolyl moiety attached to the 6-amino group.
There are only relatively few examples where the ribose moiety in adenosine is chemically modified, and many of those known have poor affinity for the adenosine receptor (Taylor, M.D., Moos, W.H., Hamilton, H.W. Szotek, D.S. PAtt, W.C. Badger, E.W. Bristol, J.W. Bruns, R.F. Heffner, T.G. Mertz, T.E. Ribose-Modified Adenosine Analogues as Adenosine Receptor Agonists. J. Med. Chem., 1986, 29, 346-353).
However, minor modifications at 3'- and 5'- appear to be allowed and amongst these the 5'-chloro-5'-deoxy adenosines show particularly good receptor affinity (Trivedi, B.K., Bridges, A.J., Part, W.C. Priebe, S.R., Bruns, R.F. Λ -Bicycloalkyladenosines with Unusually High Potency and Selectivity for the Adenosine A, Receptor J. Med. Chem., 1989, 32, 8-11). Other scientific articles also describe 5'-modifications of adenosine derivatives (Olsson, R.A. Kusachi, S., Thompson, R.D., Ukena, D., Padgett, W. and Daly, J.W. N6-Substituted N-Alkyladenosine-5'-uronamides: Bifunctional Ligands Having Recognition Groups for A→ and A2 Adenosine Receptors. J. Med. Chem., 1986, 29, 1683-1689).
Many of the new adenosine A3 receptor agonists are 5'-modifιed adenosine derivatives (Jacobson, K.A.; Kim, H.A.; Siddiqi, S.M.; Olah, M.E.; Stiles, G.L.; von Lubitz, D..K.J.E.; A3 Adenosine Receptors: Design of Selective Ligands and
Therapeutic Propects. Drugs of the Future 1995, 20, 689-699; Baraldi, P.G., Cacciari, B., Spalluto, G. Ji, X-d, Olah, M.E. Stiles, G., Dionsiotti, S., Zocchi, C, Ongini, E. and Jacobson, K.A. Novel /v*-(Substituted-phenylcarbamoyl)adenosine-5'- uronamides as Potent Agonists for A3 Receptors. J. Med. Chem., 1996, 39, 802- 806).
EP-A-181 ,128 and EP-A-181,129 disclose 5'-deoxy adenosine derivatives containing δ'-hydrogen, 5'-halogen and 5'-methylthio, which are claimed to have desirable anti- inflammatory, analgesic as well as CNS and antihypertensive properties respectively.
EP-A-232,813 discloses Λ/-substituted adenosines including a larger range of 5'- modified compounds. In WO 88/03147 5'-substituted adenosine derivatives with selectivity for the adenosine A2 receptor are disclosed.
In US 4,962,194 methods for preparing 5', N6-disubstituted adenosine derivatives are revealed. GB 1,101 ,108 discloses 5', N6-disubstituted adenosine analogues which possess cardiovascular activity. US Patent No. 3,910,885 reveals 4'-alkoxy and 4'- haloalkoxy nudeosides. PCT publication WO 94/06348 discloses a number of pyrrolo[3,4-d]pyrimidine structures which are formally isosteric with adenosine and which are modified at the sugar 5'-position. US Patent No. 5,308,837 covers the use of 5'-amine substituted adenosine analogues as immunosupressants.
In EP Publication No. 0 423 777 A2 a method for treating gastrointestinal motility disorders using N(6) (substituted aminoalkyl) adenosine derivatives is disclosed. EP Publication No. 0 490 818 A1 describes a new use of 2-O-methyl adenosine derivatives for a range of ailments including neurodegenerative disorders.
In WO 93/23417 (corresponds to US patent No. 5,430,027) and WO 95/07921 adenosine derivatives having central nervous system (CNS) properties are disclosed.
Description of the invention
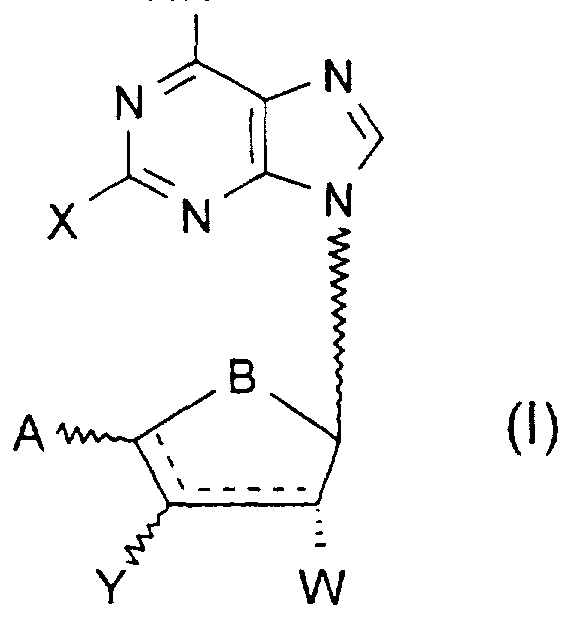
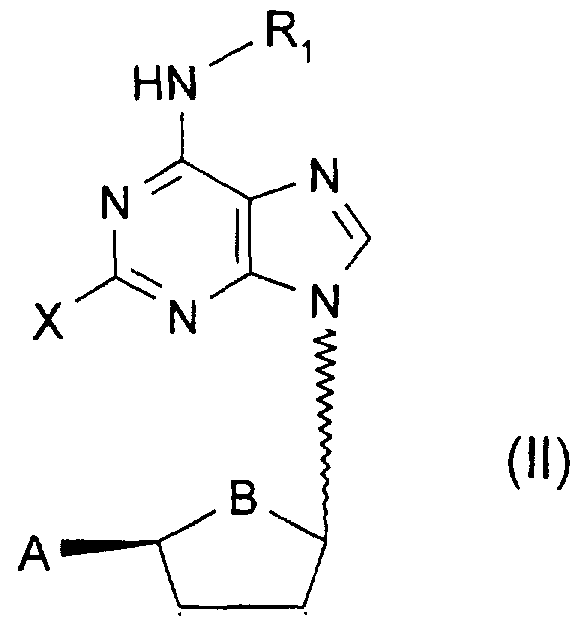
A compound of the general formula I, or pharmaceutically acceptable salts thereof:
Wherein
X represents hydrogen, halogen, C^-alky!, C^-alkoxy, C^-alkylthio, C^-alkyl- amino;
A is hydroxy, C -alkanoyloxy, C -aikanoyloxymethyl, hydroxymethyl, 1 ,2- dihydroxy-C -alkyl, oxazolyl or isoxazolyl;
B is oxygen or methylene;
W is hydrogen, hydroxy or O-C -alkyl or O-C -alkanoyl;
Y is hydrogen, hydroxy, O-C -alkyl or O-C -alkanoyl or halogen;
R→ is linear C<ι_6-alkyl. branched C 3-8 -alkyl, C 2-8 -alkenyl or C 3-8 -cydoalkyl, ail o which may be substituted by carboxyl, branched or linear C -alkoxycarbonyl, phenyl, phenoxy or halophenyl, or
R- is -OR2, wherein:
R «?→ is linear C ι_g -alkyl, branched C 3-8 -alkyl, C Z-o -alkenyl or C 3-B -cydoalkyl, all of which may be substituted by carboxyl, branched or linear C -alkoxycarbonyl, aminocarbonyl, phenyl, phenoxy or halophenyl;
wherein the solid/dotted lines both represent single bonds or one of the solid/dotted lines represent a single bond and the other a double bond;
provided that when A is hydroxymethyl, B is oxygen, W is hydroxy and Y is hydroxy, then R, is OR2 wherein R2 is linear C1 6-alkyl, branched C -alkyl or C -cydoalkyl, all substituted with carboxyl or branched or linear C1 6-alkoxycarbonyl or aminocarbonyl; and provided that when A is Cι_6-alkanoyloxymethyl, B is oxygen and W is hydroxy and Y is hydroxy, then R, is OR2 wherein R2 is linear C1 -alkyl, branched C -alkyl, C 2-8 -alkenyl or C 3-8 -cydoalkyl, all of which may be substituted with carboxyl or branched or linear C -alkoxycarbonyl or aminocarbonyl.
The compounds of the invention show potent binding to adenosine receptors and have been found to be cytokine inhibitors, for example inhibitors of TNF-α, and are found to be useful in the treatment of disorders related to cytokines in mammals, including humans. These conditions include inflammation, arthritis, type I and type II diabetes, autoimmune disorders, multiple schlerosis, stroke, osteoporosis, septic shock and menstrual complications.
Various salts of compounds of formula (I) can be prepared which can be considered physiologically acceptable. These include addition salts derived from inorganic or organic acids, for example, acetates, fumarates, glutarates, glutaconates, lactates, maleates, methanesulphonates, phosphates, salicylates, succinates, sulphates, sulphamates, tartrates and paratoluenesulphonates. In some cases, solvates of either the free nudeosides or the acid addition salts can be isolated and these solvates may, for example, be hydrates or alcoholates.
In a preferred embodiment, the compounds of the invention are selected from compounds of formula I wherein A is alkanoyloxymethyl.
In another preferred embodiment, the compounds of the invention are selected from compounds of formula I wherein A is isoxazolyl.
In another preferred embodiment, the compounds of the invention are selected from compounds of formula I wherein A is hydroxymethyl and B is methylene and/or W is hydrogen.
In another preferred embodiment, the compounds of the invention are selected from compounds of formula I wherein A is hydroxymethyl and R. is OR2 wherein R2 is linear C 1-6 -alkyl, branched C 3-8 -alkyl or C 3-9 -cydoalkyl, all substituted with carboxyl or branched or linear C -alkoxycarbonyl.
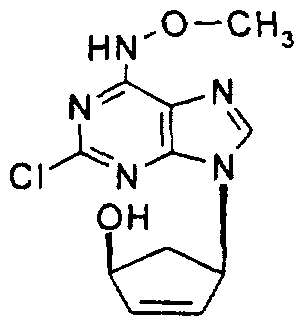
In yet another preferred embodiment, the compounds of the invention are selected from:
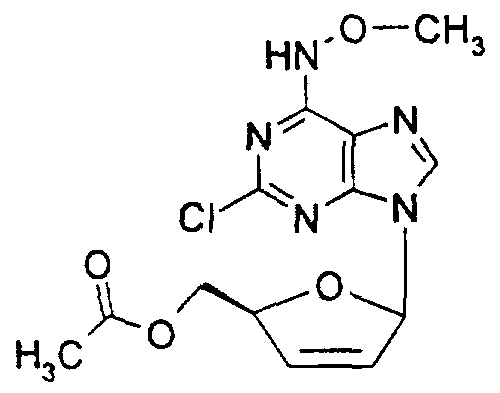
5'-0-Acetyl-2-chloro-Λ/-methoxyadenosine,
5'-0-Acetyl-2-chlorQ-Λ/-ethoxyadenosine,
5'-0-Acetyl-2-chloro-/V-(2-methyl-1-propyloxy)adenosine,
5'-O-Acetyl-2-chloro-/\/-(1-propen-3-yl)oxyadenosine,
5'-O-Acetyl-2-chloro-/\/-(tet -butyloxycarbonylmethoxy)adenosine,
2-Chloro-Λ/-(tetτ-butyloxycarbonylmethoxy)adenosine,
5'-0-Acetyl-2-chloro-Λ/-(methoxycarbonylmethoxy)adenosine,
2-Chloro-Λ/-(methoxycarbonylmethoxy)adenosine,
2-Chloro-Λ/-(aminocarbonylmethoxy)adenosine,
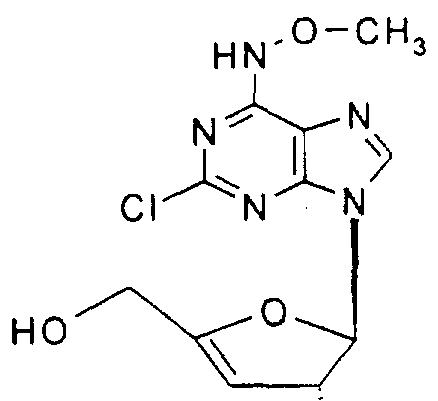
2-Chloro-2'-deoxy-/V-methoxyadenosine,
2-Chloro-9-(2'-deoxy-α-D-ribofuranosyl)-/v-methoxyadenine,
2',5'-Di-0-acetyl-3',4'didehydro-3'-deoxy-2-chloro-/V-methoxyadenosine,
3',4'-Didehydro-3'-deoxy-2-chloro-/\/-methoxyadenosine,
2'-O-Acetyl-3'-bromo-3'-deoxy-2-chloro-/\/-methoxyadenosine,
2'-0-Acetyl-3'-deoxy-2-chloro-/V-methoxyadenosine,
3'-Deoxy-2-chloro-N-methoxyadenosine,
5'-0-Acetyl-2',3'-didehydro-2',3'-dideoxy-2-chloro-/\/-methoxyadenosine,
5'-0-Acetyl-2',3'-dideoxy-2-chloro-/V-methoxyadenosine,
2',3'-Didehydro-2',3'-dideoxy-2-chloro-Λ/-methoxyadenosine,
2-Chloro-9-(4-isoxazol-3-yl-β-D-ety /?rofuranos-1-yl)-Λ/-methoxyadenine,
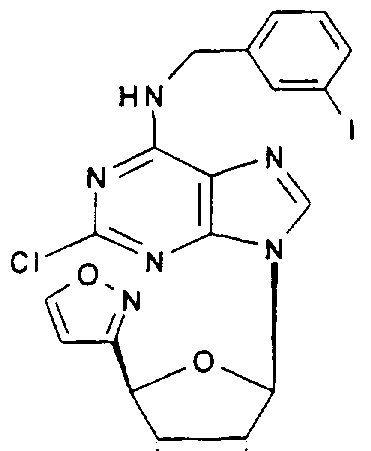
2-Chloro-Λ/-3-iodobenzyl-9-(4-isoxazol-3-yl-β-D-etyf 7rofuranos-1-yl)adenine,
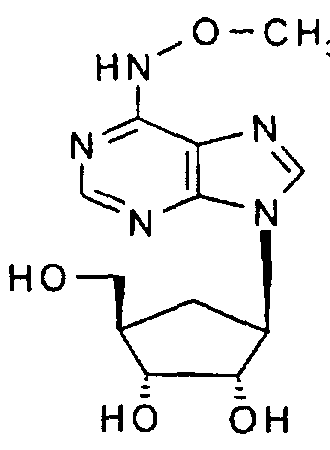
tV-Methoxyaristeromycin,
(±)-(1 S,3,R,5'S)-2-Chloro-/V-methoxy-9-[5'-(1 ,2-dihydroxyethyl)cyclopent-en-3'- yljadenine,
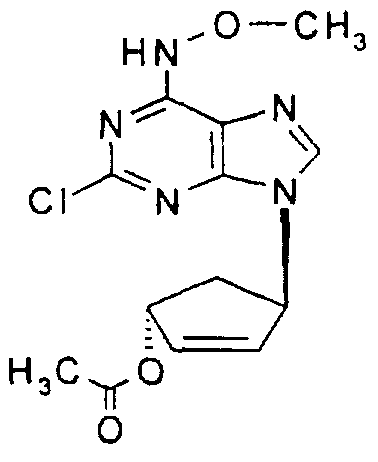
(1S, 4S)-Acetic acid, 4-(2-chloro-Λ/-methoxyadenin-9-yl)cyclopent-2-enyl ester,
(1S, 4S)-4-(2-Chloro-Λ/-methoxyadenin-9-yl)cyclopent-2-enol, and
(1 R, 4S)- -(2-Chloro-Λ/-methoxyadenin-9-yl)cyclopent-2-enol.
The present invention is furthermore concerned with a pharmaceutical composition comprising as active component a compound of formula I and a pharmaceutically
acceptable carrier or diluent.
In addition, the present invention relates to a method of treating disorders related to cytokines in mammals, preferably autoimmune disorders, inflammation, arthritis, type I or type II diabetes, multiple schlerosis, stroke, osteoporosis, septic shock, ocular ailments or menstrual complications, more preferably type I type II diabetes, even more preferably type II diabetes, comprising administering to a mammal in need thereof an effective amount of a compound of formula I, and to the use of these compounds for manufacturing a medicament for treating one or more of said disorders.
Pharmacological effects
It has been found that the compounds of formula (I) have affinity for subtypes of adenosine receptors, modulate cyclic AMP and act as cytokine inhibitors. Moreover, these compounds are found to be useful as drugs in the treatment of disorders where damaging effects of cytokines are observed in humans.
Evaluation of these compounds in established animal models has indicated that the compounds according to the invention possess desirable pharmacological properties which can be ascribed to cytokine modulation. For example they inhibit TNF-α release, indicated by lowering of plasma TNF-α following LPS challenge in rats.
Evaluation of in vitro binding to adenosine A^ A and A, receptors:
The affinity of the compounds of this invention for the adenosine A, receptor was determined essentially as described in the literature using [3H]-R-PIA as a radioligand (Naunyn-Schmiedeberg's Archives of Pharmacology, 1980, 313, 179- 187). Affinity for the A^ receptor was measured using the radioligand [3H]-CGS 21680 (European Journal of Pharmacology, 1989, 168, 243-246). An assay for the human adenosine A3 receptor is described in Jacobson, M. Cloning and Expression
of Human Adenosine Receptor Subtypes. In Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology, Belardinelli, L. and Pelleg, A., Eds.; Kluwer: Boston, MA; 1995, pp 5-13.
In vitro assay of TNF-α
The test compound is dissolved in dimethyl sulphoxide (DMSO) at 8 mg/mL and is diluted with Cremophor/5% saline to 160, 16 and 1.6 mg/mL. 25 mL is added to each tube, and 350 mL heparinised (50 iE/mL) rat blood, 25 mL LPS and finally 1.6 mg/mL saline are introduced, i.e the concentrations of the test compound is 10, 1 and 0.1 mg/mL respectively. The samples are shaken carefully and and are placed in a water bath for 5 h at 37°C. The samples are centrifuged for 10 min. at 3000 r.p.m. at 4°C. The plasma is removed by pipette in plastic tubes and is frozen. TNF- α levels are determined using a Genzyme ELISA kit.
Pharmaceutical compositions
The compounds of the invention, together with a conventional adjuvant, carrier or diluent, and if desired in the form of a pharmaceutically acceptable acid addition salt thereof, may be placed into the form of pharmaceutical compositions and unit dosages thereof, and in such form may be employed as solids, such as tablets of filled capsules, or liquids, such as solutions, suspensions, emulsions, elixirs, or capsules filled with the same, all for oral use, in the form of suppositories for rectal administration; or in the form of sterile injectable solutions for parenteral use (including subcutaneous administration and infusion). Such pharmaceutical compositions and unit dosage forms thereof may comprise conventional ingredients in conventional proportions, with or without additional active compounds or principles, and such unit dosage forms may contain any suitable effective amount of the adenosine receptor agonist commensurate with the intended daily dosage range to be employed. Tablets containing ten (10) milligrams of active ingredient or, more broadly, ten (10) to hundred (100) milligrams, per tablet, are accordingly suitable
representative unit dosage forms.
The compounds of this invention can thus be used for the formulation of pharmaceutical preparation, e.g. for oral and parenteral administration to mammals including humans, in accordance with conventional methods of galenic pharmacy.
Conventional excipients are such pharmaceutically acceptable organic or inorganic carrier substances suitable for parenteral or enteral application which do not dele- teriously react with the active compounds.
Examples of such carriers are water, salt solutions, alcohols, polyethylene glycols, polyhyroxyethoxylated castor oil, gelatine, lactose amylose, magnesium stearate, talc, silicic acid, fatty acid monoglycerides and diglycerides, pentaerythritol fatty acid esters, hydroxymethylcellulose and polyvinylpyrrolidone.
The pharmaceutical preparations can be sterilised and mixed, if desired, with auxiliary agents, emulsifiers, salt for influencing osmotic pressure, buffers and/or colouring substances and the like, which do not deleteriously react with the active compounds.
For parenteral application, particularly suitable are injectable solutions or suspensions, preferably aqueous solutions with the active compound dissolved in polyhydroxylated castor oil.
Ampoules are convenient unit dosage forms.
Tablets, dragees, or capsules having talc and/or carbohydrate carrier or binder or the like, the carrier preferably being lactose and/or corn starch and/or potato starch, are particularly suitable for oral application. A syrup, elixir or the like can be used in cases where a sweetened vehicle can be employed.
Generally, the compounds of this invention are dispensed in unit form comprising 0.05-100 mg in a pharmaceutically acceptable carrier per unit dosage.
The dosage of the compounds according to this invention is 0.1-300 mg/day, preferably 10-100 mg/day, when administered to patients, e.g. humans, as a drug.
A typical tablet which may be prepared by conventional tabletting techniques contains:
Active compound 5.0 mg
Lactosum 67.0 mg Ph. Eur.
AvicelTM 31.4 mg
AmberliteTMIRP 88 1.0 mg
Magnesii stearas 0.25 mg Ph. Eur.
Owing to activity against inflammation, arthritis, diabetes, multiple schlerosis, stroke, osteoporosis, septic shock, menstrual complications and autoimmune disorders the compounds of the invention are extremely useful in the treatment of related symptoms in mammals, when administered in an amount effective for agonist activity of compounds of the invention. The compounds of the invention may accordingly be administered to a subject, e.g., a living animal body, including a human, in need of adenosine receptor agonist, and if desired in the form of a pharmaceutically acceptable acid addition salt thereof (such as the hydrobromide, hydrochloride, or sulphate), in any event prepared in the usual or conventional manner, e.g., evaporation to dryness of the free base in solution together with the acid), ordinarily concurrently, simultaneously, or together with a pharmaceutically acceptable carrier or diluent, especially and preferably in the form of a pharmaceutical composition thereof, whether by oral, rectal, or parenteral (including subcutaneous) route, in an effective amount of adenosine receptor agonist, and in any event an amount which is effective for the treatment diseases related to cytokines, owing to their adenosine receptor agonist activity. Suitable dosage ranges are 1-200 milligrams daily, 10-100 milligrams daily, and especially 30-70 milligrams daily, depending as usual upon the exact mode of administration, form in which administered, the indication toward
which the administration is directed, the subject involved and the body weight of the subject involved, and the preference and experience of the physician or veterinarian in charge.
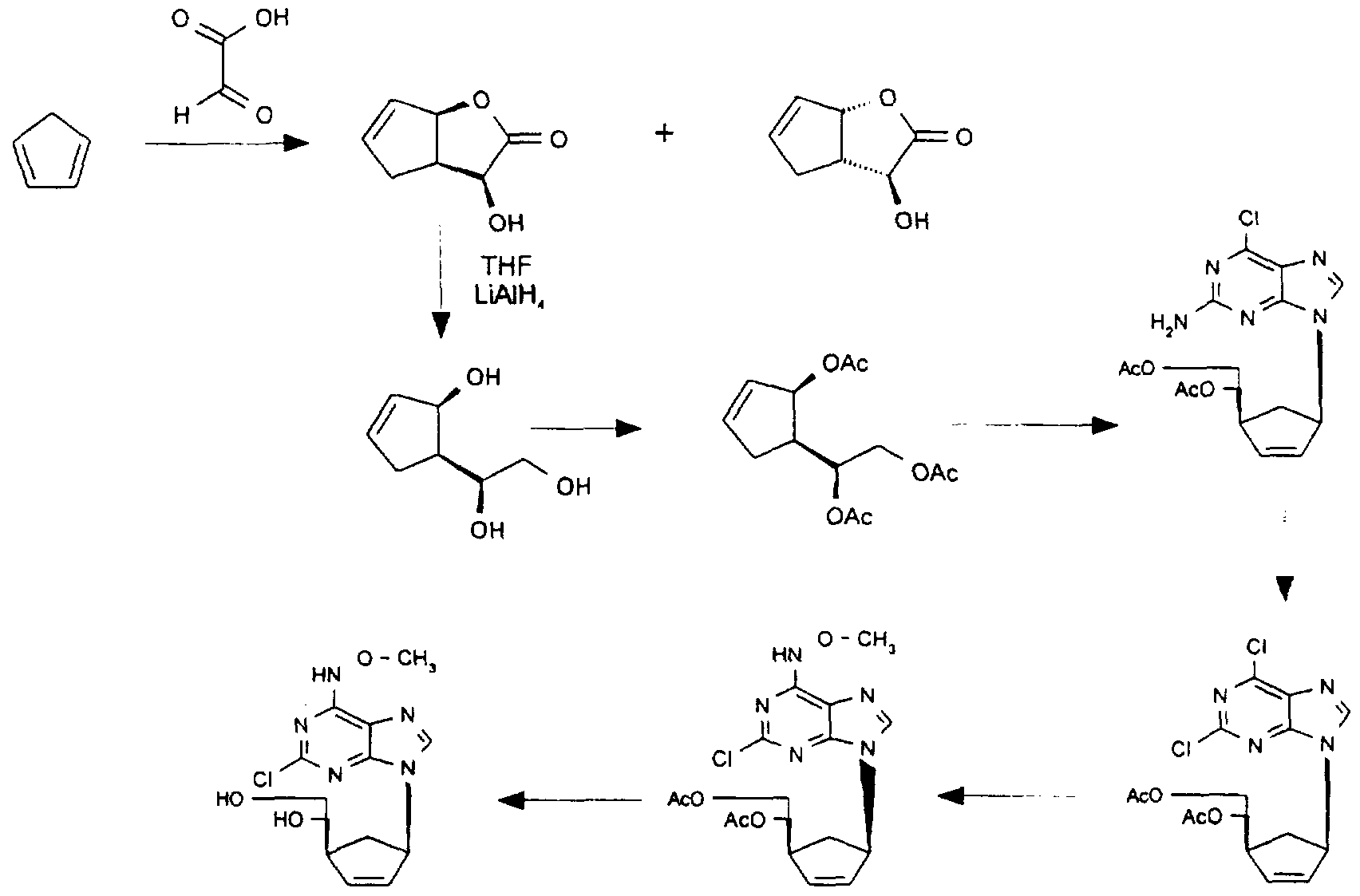
Synthesis methods The compounds of the invention can be prepared by resorting to the teaching obtainable from the general synthesis methods described in the above-cited documents (cf. in particular Knutsen, L.J.S., Lau, J., Eskesen, K., Sheardown, M.J., Thomsen, C, Weis, J.U., Judge, M.E. and Klitgaard, H. Anticonvulsant Actions of Novel and Reference Adenosine Agonists. In Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology, Belardinelli, L. and Pelleg, A., Eds.; Kluwer: Boston, MA; 1995, 479-487, and Knutsen, L.J.S., Lau, J., Sheardown, M.J., Thomsen, O; The Synthesis and Biochemical Evaluation of New A, Selective Adenosine Receptor Agonists Containing a 6-Hydrazinopurine Moiety; BioMed. Chem. Lett., 1993, 3, 2661-2666, and WO 93/23417) and applying the common general knowledge of those skilled in the art of purine and adenosine medicinal chemistry.
The preparation of compounds of the invention is further illustrated in the following examples:
TLC is thin layer chromatography, DMF is Λ/,Λ/-dimethylformamide, THF is tetra- hydrofuran, TFA is trifluoroacetic acid and m.p. is melting point. Where melting points are given, these are uncorrected. The structures of the compounds are confirmed by assignment of NMR spectra (from which representative peaks are quoted) and by mass spectroscopy MS and/or microanalysis where appropriate. Compounds used as starting materials are either known compounds or compounds which can be prepared by methods known per se. Flash
chromatography was carried out using the technique described by Still et al M on Merck silica gel 60 (Art 9385). HPLC was carried out on a Merck Hitachi model L6200A Intelligent chromatograph interfaced to a Merck Hitachi L4000A UV detector to a LiChrospher 100 reversed phase C18 column (5 mm, 250 x 4 mm, 5mm, 100A; eluent flow rate 1 mUmin). Retention times are given in minutes.
Example 1
5'-Q-Acetyl-2-chloro-Λ-methoxyadenosine
9-(2',3',5'-Tri-0-acetyl-β-D-ribofuranosyl)-2,6-dichloro-9H-purine (Robins, M.J.; Uznanski, B. Improved acetylation and deoxygenative chlorination of guanosine; nonaqueous diazotisation-dediazotisation of a 2-aminopurine nucleoside. In Nucleic Acid Chemistry, Volume 3, Townsend, L.B. II.; Tipson, R.S., Eds: John Wiley and Sons: New York 1986; pp 144-148) (4.92 g, 11.0 mmol) was dissolved in dioxan (50 mL). Λ/-Ethyldiisopropylamine (6.59 mL, 4.98 g, 38.5 mmol) and O-methylhydroxyla- mine hydrochloride (1.38 g, 16.5 mmol) were introduced, the reaction mixture was heated at 50°C for 20h, filtered and evaporated in vacuo. The crude product was purified by flash chromatography eluting with EtOAc to give 2',3',5'-tri-0-acetyl-2-chloro-Λ/-methoxy-adenosine as a foam (4.02 g, 80%), 1H NMR (400 MHz, DMSO-d6) δ 2.03, 2.06, 2.13 (9H, 3s, 3 x OCOCH3), 3.79 (3H, s, OCH3), 4.26 (1 H, dd, H-5'a), 4.38 - 4.43 (2H, m, H-5'B and H-4'), 5.59 (1 H, t, H-3'), 5.89 (1H, t, H-2'), 6.20 (1 H, d, H-1'), 8.47 (1H, s, H-8).
The above 2,,3',5'-tri-0-acetyl-2-chloro-Λ/-methoxy-adenosine (2.91 g, 6.36 mmol) was dissolved in CH3OH (25mL) and added to a previously prepared solution of sodium (0.015 g, 0.64 mmol) in CH3OH (25mL). The reaction mixture was stirred at ambient temperature for 45 h, after which time the starting material was not present by TLC. The reaction mixture was neutralised by addition of a solution of citric acid in CH3OH, evaporated in vacuo, and the residue was purified by flash chromatography, eluting
with a mixture of CH2CI2 and EtOH (20/1) initially, then with a 5/1 mixture of these solvents, to provide 5'-0-acetyl-2-chloro-/V-methoxyadenosine (0.51 g, 21%) as a foam, 1H NMR (400 MHz, DMSO-d6) δ 2.01 (3H, s, OCOCH3), 3.77 (3H, s, OCH3), 4.06 - 4.11 (1H, m, H-4'), 4.13 - 4.23 (2H, m, H-3' and H-5'a), 4.25 - 4.37 (1 H, m, H-5' ), 4.60 (1H, dd, H-2'), 5.40 (1H, d, 3'-OH), 5.61 (1H, d, 2'-OH), 5.88 (1 H, d, H-1'), 8.42 (1 H. S, H-8), 11.55 (1 H, s, N-H).
Elution was continued to provide 2-chloro-Λ/-methoxyadenosine (1.26 g, 59%) (PCT applications WO DK97/00107 and WO DK97/00108) as a white solid, mp 123 - 125°C. Η NMR (400 MHz, DMSO-d6) δ 3.52 - 3.59 (1H, m, H-5'a), 3.63 - 3.70 (1H, m, H-5'b), 3.78 (3H, s, -OCH3), 3.96 (1H, q, H-4'), 4.14 (1 H, q, H-3"), 4.52 (1H, q, H- 2'), 5.06 (1H, t, 5'-OH), 5.22, 5.51 (2H, 2d, 2'-and 3'-OH), 5.87 (1H, d, H-1'), 8.50 (1H, s, H-8), 11.60 (1H, s, N-H). Anal. Calcd. for C^H^CINA. 1.33 H2O requires C, 37.1 ; H, 4.7 ; N, 19.7. Found: C, 37.4; H, 4.4; N, 19.3%.
Example 2
5'-Q-Acetyl-2-chloro-Λ-ethoxyadenosine
This compound was prepared using the procedure described in Example 1. 9-(2\3\5'-Tri-O-acetyl^-D-ribofuranosyl)-2,6-dichloro-9H-purine (1.79 g, 4.0 mmol) was dissolved in dioxan (25 mL). Triethylamine (1.78 mL, 1.30 g, 12.8 mmol) and O-ethylhydroxylamine hydrochloride (0.305 g, 3.1 mmol) were introduced, the reaction mixture was heated at 50°C for 42 h, filtered and evaporated in vacuo. The crude product was purified by flash chromatography eluting initially with a mixture of EtOAc and n-heptane (2/1), followed by EtOAc to provide
2',3',5,-tri-O-acetyl-2-chloro-/V-ethoxyadenosine as a foam (1.03 g, 70%), 1H NMR (400 MHz, DMSO-d6) δ 1.24 (3H, t, Ci^CH- ), 2.03, 2.06, 2.12 (9H, 3s, 3 x OCOCH3), 4.01 (3H, s, CH3Ch20), 4.27 (1H, dd, H-5'a), 4.37 - 4.42 (2H, m, H-5'b and H-4'), 5.59 (1H, t,
H-3'), 5.90 (1 H, t, H-2'), 6.18 (1H, d, H-1'), 8.46 (1H, s, H-8), 11.52 (1 H, s, NH).
The above 2',3',5'-tri-0-acetyl-2-chloro-Λ/-ethoxyadenosine (1.03 g, 2.18 mmol) was dissolved in CH3OH (25 mL) and added to a previously prepared solution of sodium (0.005 g, 0.22 mmol) in CH3OH (5 mL). The reaction mixture was stirred at ambient temperature for 2 h, and the reaction mixture was neutralised by addition of a solution of citric acid in CH3OH, evaporated in vacuo, and the residue was purified by flash chromatography as described for Example 1 , to provide 5'-0-acetyl- 2-chloro-/V-ethoxyadenosine (0.275 g, 32%) as a foam, Η NMR (400 MHz, DMSO-d6) δ 1.25 (3H, t, CH3CH2O), 2.03 (3H, s, OCOCH3), 4.00 (3H, s, CH3CH.20), 4.09 (1H, q, H-4'), 4.14 - 4.22 (2H, m, H-3' and H-5'a), 4.31 (1H, dd, H-5'b), 4.58 (1 H, q, H-2'), 5.40 (1H, t, 3'-OH), 5.61 (1H, d, 2'-OH), 5.87 (1H, d, H-1'), 8.42 (1 H, s, H-8), 11.48 (1 H, s, N-H).
Elution was continued to provide 2-chloro-Λ/-ethoxyadenosine (1.26 g, 59%) 2-chloro- Λ/-ethoxyadenosine (0.161 g, 21%) as a foam, 1H NMR (400 MHz, DMSO-d6) d 1.25 (3H, t, -CH.Cty, 3.52 - 3.58 (1H, m, H-5'a), 3.63 - 3.70 (1H, m, H-5'b), 3.94 (1 H, q, H-4'), 4.00 (2H, q, -CH2CH3), 4.13 (1 H, q, H-3'), 4.51 (1 H, q, H-2'), 5.04 (1 H, t, 5-OH), 5.22, 5.49 (2H, 2d, 2'- and 3'-OH), 5.86 (1 H, d, H-1'), 8.48 (1H, s, H-8), 11.45 (1 H, s, N-H).
Example 3
5'-0-AceWI-2-chloro-N-(2-methyl-1-propyloxytedenosine
This compound was prepared using the procedure described in Example 1. 9-(2\3\5'-Tri-O-acetyl-β-D-ribofuranosyl)-2,6-dichloro-9H-purine (1.79 g, 4.0 mmol) was dissolved in dioxan (25 mL). Triethylamine (1.33 mL, 0.97 g, 9.6 mmol) and isobutoxyamine hydrochloride (0.603 g, 4.8 mmol) were introduced and the reaction mixture was heated at 50°C for 72 h before being filtered and evaporated in vacuo.
The residue was dissolved in EtOAc (100 mL), and washed with water (3 x 30 mL). The combined aqueous phases were extracted with EtOAc (50 mL), combined with the earlier EtOAc solution and dried (MgSOJ. The crude product was purified by flash chromatography eluting initially with EtOAc/n-heptane (1/2) and later with EtOAc to give 2',3',5'-tri-0-acetyl-2-chloro-Λ/-(2-methyl-1-propyloxy)adenosine as a foam (0.98 g, 49%). This product (1.96 mmol) was dissolved in CH3OH (15mL) and added to a previously prepared solution of sodium (0.0045 g, 0.196 mmol) in CH3OH (5mL). The reaction mixture was left at ambient temperature for 5 h, and at 4°C for 72 h, after which time the starting material was not present by TLC. The reaction mixture was neutralized by addition of a solution of citric acid in CH3OH, evaporated in vacuo, and the residue was purified by flash chromatography, eluting with a mixture of CH2CI2 and EtOH (10/1) initially, then with a 5/1 mixture of these solvents, to provide 5'-O-acetyl- 2-chloro-/V-(2-methyl-1-propyloxy)adenosine (0.26 g, 32%) as a foam, Η NMR (400 MHz, DMSO-d6) δ 0.95, 0.97 (6H, 2s, CH Oby,), 2.00 (1 H, h, CjH(CH3)2), 2.02 (3H, s, OCOCH3), 3.53 (2H, d, CH2CH(CH3)2), 4.09 (1H, m, H-4'), 4.15 - 4.23 (2H, m, H-3' and H-5'a), 4.30 (1 H, dd, H-5'b), 4.58 (1H, dd, H-2'), 5.39 (1 H, t, 3'-OH), 5.60 (1H, t, 2'-OH), 5.88 (1H, d, H-t), 8.42 (1H, s, H-8), 11.50 (1H, s, N-H).
Elution was continued to provide 2-chloro-Λ--(2-methyl-1-propyloxy)adenosine (0.31 g, 42%).
Example 4
5'-Q-Acetyl-2-chloro-Λ/-(1-propen-3-yl)oxyadenosine
This compound was prepared using the procedure described in Example 1. 9-(2,,3',5'-Tri-O-acetyl-β-D-ribofuranosyl)-2,6-dichloro-9H-purine (1.79 g, 4.0 mmol) was dissolved in dioxan (25 mL). Triethylamine (1.33 mL, 0.97 g, 9.6 mmol) and O-allylhydroxylamine hydrochloride (0.53 g, 4.8 mmol) were introduced and the reaction mixture was heated at 50°C for 20h, and stirred at ambient temperature for
48h before being filtered and evaporated in vacuo. The residue was dissolved in EtOAc (100 mL), and washed with water (3 x 30 mL). The combined aqueous phases were extracted with EtOAc (50 mL), combined with the earlier EtOAc solution and dried (MgSO . The crude product was purified by flash chromatography eluting initially with EtOAc/n-heptane (1/2) and later with EtOAc to give
2',3',5'-tri-O-acetyl-2-chloro- /-(1-propen-3-yl)oxyadenosine as a foam (0.88 g, 45%). This product (1.82 mmol) was dissolved in CH3OH (5mL) and added to a previously prepared solution of sodium (0.0042 g, 0.182 mmol) in CH3OH (5mL). The reaction mixture was stirred at ambient temperature for 27 h, after which time the starting material was not present by TLC. The reaction mixture was neutralized by addition of a solution of citric acid in CH3OH, evaporated in vacuo, and the residue was purified by flash chromatography, eluting with a mixture of CH2CI2 and EtOH (10/1) initially, then with a 5/1 mixture of these solvents, to provide 5'-O-acetyl-2-chloro-Λ/-(1-propen-3- yl)adenosine (0.043 g, 6%) as a foam, 1H NMR (400 MHz, DMSO-d6) δ 2.02 (3H, s, OCOCH3), 4.09 (1 H, m, H-4'), 4.15 - 4.23 (2H, m, H-3' and H-5'a), 4.30 (1H, dd, H-5'b), 4.49 (1 H, d, CH2CH), 4.58 (1H, dd, H-2'), 5.26, 5.36 (2H, 2 dd, CH=CH2), 5.41 (1H, t, 3'-OH), 5.62 (1H, t, 2'-OH), 5.88 (1 H, d, H-1'), 6.04 (1 H, dq, CH=CH2), 8.42 (1H, s, H- 8), 11.51 (1H, s, N-H).
Elution was continued to provide 2-chloro-Λ/-(1-propen-3-yl)oxyadenosine (0.392 g, 60%).
Example 5
5'-Q-Acetyl-2-chloro-/\/-ιfetτ-butyloxycarbonylmethoxy^adenosine
Aminooxyacetic acid ferf-butyl ester
To carboxymethoxyamine hemihydrochloride (4.04 g, 37 mmol) was added tert-butyl acetate (35 mL), followed by perchloric acid (1.60 mL, 18.5 mmol). This was allowed to
stir at room temperature for 24 h, by which time a large amount of white precipitate was observed. This dissolved during work-up, when the reaction mixture was poured carefully onto 5% NaHC03aq solution (300 mL). Further NaHC03 was added to adjust the pH to 9, before extraction with EtOAc (3 x 200 mL). The combined organic phase was washed with water (2 x 200 mL), brine (1 x 100 mL) and then dried (Na2S04). Evaporation of solvent afforded a clear colourless liquid (0.54 g, 11%). 1H NMR (200 MHz, CDCI3) δ 1.50 (9H, s, CMe3), 4.15 (2H, s, CH2), 5.88 (2H, br s, NH2).
5'-O-Acetyl-2-chloro-Λ/-(fetf-butyloxycarbonylmethoxy)adenosine
This compound was prepared using the procedure described in Example 1. θ^'.S'.S'-Tri-O-acetyl-β-D-ribofuranosyl^^-dichloro-ΘH-purine (1.35 g, 3.02 mmol) was dissolved in dry dioxane (25 mL). N-Ethyldiisopropylamine (0.57 mL, 3.32 mmol) and aminooxy-acetic acid terf-butyl ester (0.49 g, 3.32 mmol) were introduced and the reaction mixture was heated at 50°C for 3 days. Since TLC was showing only slow progress, the temperature was increased to 80°C and the reaction left for a further week. After this time, solvent was evaporated and EtOAc added to precipitate the N- ethyldiisopropylamine salt. This was filtered off and the filtrate evaporated in vacuo. The crude product (1.70 g) was purified by flash chromatography eluting with EtOAc/π- heptane (1:1) to give 2\3\5'-tri-O-acetyl-2-chloro-Λ/-(tetτ-butyloxycarbonylmethoxy)- adenosine as a foam (0.87 g, 52%), 1H NMR (200 MHz, CDCI3) δ 1.50 (9H, s, CMe3), 2.10, 2.15, 2.17 (9H, 3s, 3 x OCOCH3), 4.35 - 4.48 (3H, m, H-4', H-5'a & H-5'b), 4.60 (2H, s, OCH2), 5.58 (1H, t, H-3'), 5.78 (1H, t, H-2'), 6.20 (1H, d, H-1'), 8.05 (1H, s, H-8), 9.40 (1H, s, NH).
5'-O-Acetyl-2-chloro-Λ/-(tett-butyloxycarbonylmethoxy)adenosine
2',3,,5'-Tri-0-acetyl-2-chloro-/\/-(terf-butyloxycarbonylmethoxy)-adenosine (0.67 g, 1.20 mmol) was dissolved in CH3OH (13 mL) and added to a previously prepared solution of sodium (0.003 g, 0.13 mmol) in CH3OH (10 mL). The reaction mixture was stirred at ambient temperature for 5 days, after which time the starting material was not present
by TLC. The reaction mixture was neutralized by addition of acetic acid, evaporated in vacuo, and the residue was purified by flash chromatography, eluting with CH2CI2/EtOH (14:1), to provide 5'-0-acetyl-2-chloro-/V-(terf- butyloxycarbonylmethoxy)adenosine (0.100 g, 17%) as a foam, 1H NMR (400 MHz, DMSO-d6) δ 1.45 (9H, s, C(CH3)3), 2.02 (3H, s, OCOCH3), 4.09 (1H, m, H-4'), 4.15 - 4.33 (3H, m, H-3', H-5'a and H-5'b), 4.53 (2H, s, OCH2), 4.58 (1H, dd, H-2'), 5.41 (1H, d, 3'-OH), 5.62 (1H, d, 2'-OH), 5.88 (1H, d, H-1'), 8.45 (1H, s, H-8), 11.12 and 11.62 (1H, 2 x s, NH), MS (El) : m/z 475 and 473 [M+].
Example 6
2-Chloro-N-(tert-butvloxycarbonylmethoxy denosine
The chromatography elution in Example 5 was continued to provide 2-chloro-Λ/-(tert- butyloxycarbonylmethoxy)adenosine (0.325 g, 63%). 1H NMR (400 MHz, DMSO-d6) δ 1.45 (9H, s, C(CH3)3), 3.62 (2H, ABX, H-5'a and H-5V), 3.96 (1H, dd, H-4'), 4.15 (1H, dd, H-3'), 4.52 (1H, dd, H-2'), 4.54 (1H, s, OCH2), 5.07 (1H, t, 5'-OH) 5.41 (1H, d, 3'- OH), 5.23 (1 H, d, 2'-OH), 5.51 (1 H, d, 1'-OH), 5.88 (1 H, d, H-1'), 8.53 (1H, s, H-8), 11.58 (1 H, s, NH), MS (+FAB) : m/z 434 and 432 [M++1].
Example 7
5'-O-Acetvl-2-chloro-/V-(methoxycarbonylmethoxy)adenosine
Aminooxyacetic acid methyl ester
Aminooxyacetic acid hemihydrochloride (4.0 g, 37 mmol) was partially dissolved in methanol (70 mL) and cooled to 0°C. Thionyl chloride (5.30 mL, 73 mmol) was added dropwise at such a rate as to maintain the temperature below 10°C. Addition was
complete after 20 min and the reaction kept around 0°C for a further 30 min. The reaction was then stirred at room temperature for 30 min, then heated to 60°C for 2 h. After this time the solvent was carefully evaporated to leave an oily residue (4.70 g, 90%). Η NMR (200 MHz, DMSO-dβ) δ 3.72 (3H, s, OCH3), 4.71 (2H, s, OCH2), 7.23, 7.47 & 7.72 (3H, 3 x s, NH2.HCI).
5'-0-Acetyl-2-chloro-/\/-(tert-butyloxycarbonylmethoxy)adenosine
This compound was prepared using a similar procedure to that described in Example 1. 9-(2\3\5'-Tri-O-acetyl-β-D-ribofuranosyl)-2,6-dichloro-9H-purine (5.83 g, 13.0 mmol) was dissolved in dry dioxan (100 mL). W-Ethyldiisopropylamine (5.4 mL, 31.7 mmol) and aminooxyacetic acid methyl ester (2.03 g, 14.4 mmol) were introduced and the reaction mixture was stirred at room temperature under nitrogen for 2 days. Since the reaction was progressing only slowly, the mixture was heated at 60°C for 3 days. After this time, more W-ethyldiisopropylamine was added (2.7 mL, 16 mmol) and heating continued for a further 2 days. The solvent was evaporated and EtOAc added to precipitate the Λ/-ethyldiisopropylamine salt. This was filtered off and the filtrate evaporated in vacuo. The crude product (6.60 g) was purified by flash chromatography eluting with
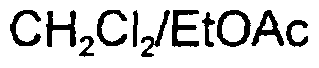
(7:3) initially, then with a 1:4 ratio of these solvents to give 2',3',5'-tri-O-acetyl-2-chloro-/\/-(methoxycarbonylmethoxy)-adenosine as a foam (0.38 g, 6%),
1H NMR (400 MHz, DMSO-d
5) δ 2.03, 2.06, 2.12 (9H, 3s, 3 x OCOCH
3), 3.70 (3H, s, OCH
3), 4.26 (1H, dd, H-5'
a), 4.36 - 4.42 (2H, m, H-4' & H-5'
b), 4.68 (2H, s, OCH
2), 5.58 (1 H, t, H-3'), 5.88 (1 H, t, H-2'), 6.18 (1H, d, H-1'), 8.50 (1H, s, H-8), 11.77 (1H, s, NH). Some earlier column fractions (1.15 g) were repurified by column chromatography on silica gel eluting with EtOAc/n-heptane (7:3) to provide a further sample of 2',3',5'-tri-0-acetyl-2-chloro- W-(methoxycarbonylmethoxy)adenosine (0.56 g, 8%).
5'-0-Acetyl-2-chloro-Λ/-(methoxycarbonylmethoxy)adenosine
The above 2',3',5'-tri-0-acetyl-2-chloro-/ /-(methoxycarbonylmethoxy)adenosine (0.37
g, 0.72 mmol) was dissolved in CH3OH (8 mL) and added to 3.2 mL of a previously prepared solution of sodium in CH3OH (0.01 g in 20 L). The reaction mixture was stirred at ambient temperature under nitrogen for 4 days, after which time the starting material was not present by TLC. The reaction mixture was neutralized by addition of acetic acid, evaporated in vacuo, and the orange residue was purified by flash chromatography, eluting with CH2Cl2/EtOH (14:1), to provide 5'-O-acetyl- 2-chloro-/V-(methoxycarbonylmethoxy)adenosine (0.082 g, 30%) as a white foam, 1H NMR (400 MHz, DMSO-d6) δ 2.03 (3H, s, OCOCH3), 3.70 (3H, s, OCH3), 4.09 (1H, m, H-4'), 4.15 - 4.24 (2H, m, H-3" & H-5'a), 4.31 (1H, dd, H-5'b), 4.59 (1H, dd, H-2'), 4.68 (2H, s, OCH2), 5.42 (1H, d, 3'-OH), 5.62 (1 H, d, 2'-OH), 5.89 (1H, d, H-1'), 8.47 (1H, s, H-8), 11.67 (1H, s, NH), C15H18N50BCI requires C, 41.73; H, 4.20; N, 16.22. Found C, 41.56; H, 4.36; N, 15.83%.
Example 8
2-Chloro-Λ/-(methoxycaιtjonylmethoxy denosine
The chromatography elution in Example 7 was continued using CH2CI2/EtOH (12:1), to provide 2-chloro-Λ/-(methoxycarbonylmethoxy)adenosine (0.13 g, 48%). 1H NMR (400 MHz, DMSO-dβ) δ 3.62 (2H, ABX, H-5'a and H-5'b), 3.70 (3H, s, OCH3), 3.96 (1H, dd, H-4'), 4.13 (1H, dd, H-3'), 4.51 (1H, dd, H-2'), 4.67 (1H, s, OCH2), 5.07 (1H, t, 5'-OH), 5.23 (1H, d, 3'-OH), 5.52 (1H, d, 2'-OH), 5.86 (1H, d, H-1'), 8.53 (1 H, s, H-8), 11.67 (1H, s, NH), C13H16N507CI. 0.4 H20 requires C, 39.33; H, 4.27; N, 17.64. Found C, 39.73; H, 4.39; N, 17.21%.
Example 9
2-Chloro-Λ/-.'aminocarbonylmethoxv denosine
The above 2-chloro-Λ/-(methoxycarbonylmethoxy)adenosine (0.050 g, 0.129 mmol) was treated with 11% w/w ammonia in methanol (1.50 mL) and allowed to stir at room temperature for 24 h. Evaporation of solvent provided an off-white foam, which was redissolved in ethanol and became a white crystalline material after evaporation. 1H NMR (400 MHz, DMSO-d6) δ 3.62 (2H, ABX, H-5'a and H-5'b), 3.96 (1H, dd, H-4'), 4.14 (1 H, dd, H-3'), 4.37 (1H, s, OCH2), 4.51 (1 H, dd, H-2'), 5.06 (1H, t, 5'-OH), 5.21 (1 H, d, 3'-OH), 5.50 (1 H, d, 2'-OH), 5.87 (1 H, d, H-1'), 7.52 and 8.03 (2H, 2 x br s, CONH2), 8.57 (1H, s, H-8), 11.60 (1 H, br s, NH),
Example 10
2-Chloro-2'-deoxy-/V-methoxyadenosine Method A^
2-Chloro-3',5'-0-(1 ,1 ,3,3-tetraisopropyldisiloxan-1 ,3-diyl)-Λ/-methoxyadenosine
2-Chloro-Λ/-methoxyadenosine (see experimental description of Example 1) (0.93 g, 2.80 mmol) was azeotropically dried using pyridine. The residue was then dissolved in pyridine (15 mL) and treated with 1,3-dichloro-1,1,3,3-tetraisopropytdisiloxane (0.96 mL, 3.00 mmol). The reaction mixture was stirred at ambient temperature under nitrogen for 3 h, after which time the starting material was not present by TLC. The pyridine was evaporated and the residue was partitioned between EtOAc (80 mL) and water (50 mL). The aqueous phase was run off and re-extracted with EtOAc (2 x 80 mL). The combined organic phase was washed with water (2 x 60 mL) then brine (60mL) and dried (Na2S04). Evaporation of solvent afforded a yellow oil (1.67 g, 100%) which was purified by column chromatography (n-heptane/EtOAc 5:4) to give a yellow foam (1.175 g, 73%). 1H NMR (200 MHz; CDCI3) δ 1.05 - 1.16 (24H, d, 2 x Si(CHMe2)2, 3.25 (1H, d, 2'-OH), 3.98 (3H, s, OCH3), 4.02 - 4.14 (3H, m, H-4\ H-5'a & H-5'b), 4.63 (1H, dd, H-3'), 5.08 (1H, dt, H-2"), 5.93 (1H, d, H-1'), 7.96 (1H, s, H-8), 8.80 (1H, br s, NH).
2-Chloro-2'-O-(imidazol-1-yl)thiocarbonyl-3',5'-0-(1 ,1 ,3,3-tetraisopropyldisiloxan-1 ,3- diyl)-Λ/-methoxyadenosine
2-Chloro-3',5'-0-(1 ,1,3,3-tetraisopropyldisiloxan-1 ,3-diyl)-/\/-methoxyadenosine (0.9 g, 1.56 mmol) was dissolved in dry DMF (10 mL) and treated with thiocarbonyldiimidazole (0.72 g, 4.07 mmol). The yellow reaction mixture was stirred at room temperature under nitrogen for 16 h. The solvent was evaporated off under reduced pressure and the residue was partitioned between EtOAc (100 mL) and water (50 mL). The aqueous phase was run off and re-extracted with EtOAc (2 x 60 mL). The combined organic phase was washed with water (2 x 60 mL) then brine (60mL) and dried (Na2S04). Evaporation of solvent afforded a yellow residue (1.1 g, 100%) which was purified by column chromatography (EtOAc/π-heptane 4:1) to give a yellow foam (0.664 g, 62%). Η NMR (200 MHz, DMSO-d6) δ 0.97 - 1.16 (24H, m, 2 x Si(CHMe2)2, 3.77 (3H, s, OCH3), 4.00 - 4.24 (3H, m, H-4', H-5'a & H-5'b), 5.34 (1H, dd, H-3'), 6.43 (1 H, dd, H-2'), 6.61 (1H, d, H-1'), 7.13 (1H, br s, imidazole 4-H), 8.40 (1H, br s, imidazole 4-H), 8.36 (1 H, s, imidazole 2-H), 8.63 (1H, s, H-8), 11.71 (1 H, br s, NH).
2-Chloro-2'-deoxy-3',5'-0-(1 ,1 ,3,3-tetraisopropyldisiloxan-1 ,3-diyl)-/V- methoxyadenosine
A solution of 2-chloro-2'-0-(imidazol-1-yl)thiocarbonyl-3',5'-0-(1 , 1,3,3- tetraisopropyldisiloxan-1 ,3-diyl)-Λ/-methoxyadenosine (0.55 g, 0.8 mmol) in dry toluene (10 mL) was heated to 90°C under nitrogen. A solution of azobisisobutyronitrile (AIBN) (0.14 g, 0.86 mmol) and tri-n-butyltinhydride (0.85 mL) in toluene (10 mL) was added dropwise during 10 min. After 3 h, the reaction mixture was concentrated to ca. 3 mL and applied to a column of silica gel. Flash chromatography, eluting with EtOAc/π- heptane (1:1) afforded the title compound as a foam (0.170 g, 38%). Η NMR (200 MHz; CDCI3) δ 0.98 - 1.16 (24H, m, 2 x Si(CHMe2)2, 2.60 - 2.73 (2H, m, H-2'a & H-2'b), 3.88 (1H, dt, H-4'), 3.95 (3H, s, OCH3), 4.04 (2H, ABX, H-5'a & H-5'b), 4.90 (1H, dd, H- 3'), 6.26 (1H, dd, H-1'), 8.06 (1H, s, H-8), 8.83 (1H, br s, NH).
2-Chloro-2'-deoxy-Λ/-methoxyadenosine
A solution of 2-chloro-2'-deoxy-3',5'-0-(1,1,3,3-tetraisopropyldisiloxan-1,3-diyl)-Λ/- methoxyadenosine (0.17 g, 0.30 mmol) in THF (7 mL) was treated with tetra-n- butylammonium fluoride (0.175 g, 0.67 mmol). The solution was kept at room temperature for 1 hour, and then evaporated to dryness. The residue was purified by flash chromatography with CH2CI2/ethanol (9:1) as eluent, to provide the title compound as a white foam (0.058 g, 66%). Η NMR (400 MHz, DMSO-d6) δ 2.29 (1H, dq, H-2'β), 2.65 (1H, p, H-2'b), 3.55 (2H, ABX, H-5, aand H-5, b), 3.77 (3H, s, OCH3), 3.86 (1H, q, H-4'), 4.39 (1H, m, H-3'), 4.96 (1H, t, 5'-OH), 5.34 (1H, d, 3'-0H), 6.29 (1H, t, H-1'), 8.40 (1H, s, H-8), 11.52 (1H, s, NH), MS (El): m/z 318 and 316 [M*], C-H^ -ACI. 0.8 H20 requires C, 40.02; H, 4.76; N, 21.21. Found C, 40.35; H, 4.68; N, 20.85%.
Example 10 (Method B^
2-Chloro-2'-deoxy-/\/-methoxyadenosine
9-(2'-Deoxy-di-O-p-toluoyl-β-D-ribofuranosyl)-2,6-dichloro-9H-purine
2,6-Dichloro-9H-purine (0.95 g, 5.0 mmol) was heated at reflux in 1,1,1 ,3,3,3- hexamethyldislazane (15 mL) for 2 h. The reaction mixture was evaporated in vacuo, and co-evaporated with xylene ((25 mL). The resultant white solid was dissolved in dry CH3CN (20 mL), 1-0-methyl-3,5-di-0-p-toluoyl-D-ribofuranose (1.92 g, 5 mmol) was introduced, and the mixture was cooled to -20°C. Trimethylsilyl trifluorornethanesulphonate (1.06 mL) in 1 ,2-dichloroethane (10 mL) was then added, and the temperature was maintained at -20°C for 16 h. The reaction mixture was diluted with CH2CI2 (100 mL), treated with cold saturated NaHC03 solution, and the organic phase was separated and dried (MgS04). Purification by flash chromatography eluting with a mixture of EtOAc and n-heptane (initially 4/1 , later with a 1/1
mixture of these solvents) provided 9-(2'-deoxy-3',5'-di-0-p-toluoyl-β-D-ribofuranosyl)- 2,6-dichloro-9H-purine (1.14 g, 42%) as a foam, Η NMR (400 MHz, DMSO-d6) δ 2.38, 2.42 (6H, 2s, 2 x ArCH3), 2.36 (1H, dq, H-2'a), 3.37 (1H, p, H-2'b), 4.54 (1H, q, H-5'a), 4.62 - 4.68 (2H, m, H-5'b and H-4'), 5.81 (1H, q, H-3'), 6.59 (1H, t, H-1'), 7.26, 7.37 (4H, 2d, Ar-H), 7.76, 7.95 (4H, 2d, Ar-H), 8.93 (1H, s, H-8), some mixed α,β fractions followed by pure 9-(2'-deoxy-3',5'-di-0-p-toluoyl-α-D-ribofuranosyl)-2,6- dichloro-9H-purine (0.1 g, 4%), as a foam, 1H NMR (400 MHz, DMSO-d6) δ 2.38, 2.42 (6H, 2s, 2 x ArCH3), 3.02 - 3.12 (2H, m, H-2'a and H-2'b), 4.54 (1H, d, H-5'a and H-5'b), 5.08 (1H, dd, H-4'), 5.62 - 5.65 (1H, m, H-3'), 6.15 (1H, dd, H-1'), 7.25, 7.37 (4H, 2d, Ar-H), 7.58, 7.93 (4H, 2d, Ar-H), 8.95 (1H, s, H-8).
2-Chloro-2'-deoxy-Λ/-methoxyadenosine
9-(2'-Deoxy-di-O-p-toluoyl-β-D-ribofuranosyl)-2,6-dichloro-9H-purine (1.13 g, 2.11 mmol) was readed with O-methylhydroxylamine hydrochloride (0.35 g, 4.2 mmol) in 1 ,4-dioxan (15 mL) in the presence of /V-ethyldisopropylamine (1.44 mL, 1.09 g, 8.4 mmol). The reaction mixture was stirred at ambient temperture for 96h, filtered and evaporated in vacuo. The crude product was purified by flash chromatography, eluting with a mixture of EtOAc and π-heptane (initially 4/1 , later with a 1/1 mixture of these solvents) to afford 2-chloro-2'deoxy-Λ/-methoxy-3',5'-di-0-p-toluoyladenosine (0.55 g, 47%). Deprotection of this product using methanolic ammonia at ambient temperature, followed by column chromatography provided the title 2-chloro-2'-deoxy- W-methoxyadenosine (0.26 g, 90%), Η NMR (400 MHz, DMSO-d6) δ 2.29 (1H, dq, H- 2'a), 2.65 (1H, p. H-2'b), 3.55 (2H, ABX, H-5'a and H-5'b), 3.78 (3H, s, -OCH3), 3.87 (1H, q, H-4'), 4.37 - 4.42 (1H, m, H-3'), 4.96 (1 H, t, 5'-OH), 5.34 (1H, d, 3'-0H), 6.30 (1H, t, H-1 '), 8.45 (1 H, s, H-8), 11.52 (1 H, s, N-H).
Example 11
2-Chloro-9-(2'-deoxy-α-D-ribofuranosyh-/V-methoxyadenine
This example was prepared from 9-(2'-deoxy-3',5'-di-0-p-toluoyl-α-D-ribofuranosyl)- 2,6-dichloro-9H-purine (0.10 g) (see experimental procedure for Example 10, method B) using O-methylhydroxylamine hydrochloride (0.035 g, 0.42 mmol) in 1,4-dioxan (10 mL) in the presence of Λ/-ethyldisopropylamine (0.14 mL, 0.11 g, 0.84 mmol), giving 2- chloro-9-(2'-deoxy-3',5'-di-0-p-toluoyl-α-D-ribofuranosyl)-Λ/-methoxyadenine (0.08 g, 88%). Deprotection of this product using methanolic ammonia at ambient temperature, followed by column chromatography provided the title 2-chloro-9-(2'-deoxy-α-D- ribofuranosyl)-Λ/-methoxyadenine (0.023 g, 53%), 1H NMR (400 MHz, DMSO-d6) δ 2.32 (1H, dt, H-2'a), 2.72 (1H, p, H-2'b), 3.44 (2H, dt, H-5'a and H-5'b), 3.77 (3H, s, - OCH3), 4.12 (1H, dt, H-A'), 4.29 - 4.34 (1H, m, H-3"), 4.87 (1H, t, 5'-OH), 5.51 (1H, br d, 3'-0H), 6.30 (1H, dd, H-1'), 8.45 (1H, s, H-8), 11.45 (1H, s, N-H).
Example 12
2'.5'-Di-0-acetyl-3'.4'didehydro-3'-deoxy-2-chloro-Λ/-methoxyadenosine
2',5'-Di-0-acetyl-3'-bromo-3,-deoxy-2-chloro-/\/-methoxyadenosine
5'-0-Acetyl-2-chloro-Λ/-methoxyadenosine (Example 1) (0.320 g, 0.86 mmol) was dissolved in dry acetonitrile (25 mL) and cooled to 0°C. α-Acetoxyisobutyryl bromide
(0.50 mL, 3.44 mmol) was added dropwise with vigorous stirring under nitrogen. The reaction was maintained at 0°C for 0.5 h, then stirred at room temperature for a further 2.5 h, before evaporation of the solvent. The oily residue was crystallised from CH2CI2/ether (1 :1), (0.32 g, 78%), Η NMR (200 MHz, DMSO-d6) δ 2.04 (3H, s, 5'- OCOCH3), 2.12 (3H, s, 2'-OCOCH3), 3.78 (3H, s, OCH3), 4.34 (2H, ABX, H-5'a and H- 5'b), 4.57 (1 H, q, H-4'), 4.94 (1H, dd, H-3'), 5.84 (1 H, t, H-2') 6.16 (1H, d, H-1'), 8.42 (1 H, s, H-8), 11.64 (1H, br s, NH).
2,,5'-Di-0-acetyl-3',4'-didehydro-3'-deoxy-2-chloro-/V-methoxyadenosine
The above 2',5'-di-0-acetyl-3'-bromo-3'-deoxy-2-chloro-/v'-methoxyadenosine (0.30 g, 0.63 mmol) was dissolved in dry acetonitrile (7 mL) and treated with 1 ,5-diazabicyclo- (4,3,0)-non-5-ene (DBN) (0.15 mL, 1.26 mmol). The reaction was stirred for 18 h under nitrogen, then treated with acetic acid to give a pH of around 4. Water (25 mL) was added and the aqueous phase extracted with EtOAc (3 x 15 mL). The combined organic phase was washed with water (2 x 15 mL) and brine (1 x 15 mL) then dried (MgS04). The solvent was evaporated and the residue was purified by column chromatography on silica gel eluting with EtOAc/EtOH (19:1) as eluent to afford almost pure product (0.45 g, 18%). This was repurified by column chromatography eluting with EtOAc/EtOH (99:1) to afford the title compound as a foam (0.02 g, 8%), Η NMR (200 MHz, CDCI3) δ 2.11 (3H, s, 5'-OCOCH3), 2.14 (3H, s, 2'-OCOCH3), 3.99 (3H, s, OCH3), 4.73 (2H, s, H-5'a and H-5'b), 5.48 (1H, dd, H-2'), 5.96 (1H, m, H-3'), 6.61 (1H, d, H-1'), 7.88 (1H, s, H-8), 8.35 (1H, br s, NH), MS (+FAB): m/z 398 [M++1j.
Example 13
3'.4'-Didehydro-3'-deoxy-2-chloro-,Λ/-methoxyadenosine
OH
2',5'-Di-0-acetyl-3',4'-didehydro-3'-deoxy-2-chloro-Λ/-methoxyadenosine (Example 12) (0.055 g, 0.14 mmol) was dissolved in CH3OH (5 mL) and treated with 0.3 L of a previously prepared solution of sodium in CH3OH (0.01 g in 10 mL). The reaction mixture was stirred at 50°C under nitrogen for 5 days. The pH was adjusted to ca. 5 by addition of acetic acid, and the reaction mixture evaporated in vacuo. The residue was purified by column chromatography on silica gel eluting with EtOAc/EtOH (19:1), and crystallised from diethyl ether to give the title compound (0.015 g, 34%), 1H NMR (200 MHz, DMSO-d6) δ 3.77 (3H, s, OCH3), 4.03 (2H, d, H-5'a & H-5'b), 5.15 (1H, br s, H-2'), 5.21-5.33 (2H, br m, H-3' & 2'-OH), 5.75 (1H, br d, 5'-OH), 6.21 (1 H, d, H-1'), 8.19 (1H, s, H-8), MS (+EI): m/z 314 [M+].
Example 14
2'-O-Acetyl-3'-bromo-3'-deoxy-2-chloro-Λ/-methoxyadenosine
2-chloro-V-methoxyadenosine (0.10 g, 0.3 mmol) was dissolved in dry acetonitrile (5
mL) and cooled to 0°C. α-Acetoxyisobutyryl bromide (0.178 mL,1.20 mmol) was added dropwise with vigorous stirring under nitrogen. The reaction was maintained at 0°C for 1 hour, then stirred at room temperature for a further 2 h, before evaporation of the solvent. The residue was purified by column chromatography on silica gel eluting with CH-jCI^EtOH (9:1) to afford a clear oil (45 mg), which crystallised from CH2CI2/π- heptane (2:1) as white microcrystals (0.035 g, 27%), mp 95-110°C dec. Η NMR (200 MHz, DMSO-d6) δ 2.10 (3H, s, COCH3), 3.66-3.86 (5H, m, OCH3, H-5'a and H-5'b), 4.34 (1 H, dd, H-4'), 4.38 (1H, dd, H-3'), 5.26 (1H, t, 5'-OH), 5.86 (1 H, t, H-2") 6.08 (1H, d, H-1'), 8.39 (1H, s, H-8), 11.58 (1 H, s, NH).
Example 15
2'-Q-Acetyl-3'-deoxy-2-chloro-Λ/-methoxyadenosine
The above 2'-0-acetyl-3'-bromo-3'-deoxy-2-chloro-/V-methoxyadenosine (0.105 g, 0.24 mmol) was dissolved in dry toluene (20 mL) under nitrogen and tri-π-butyl tin hydride (0.19 mL, 0.72 mmol) and 2,2'-azobis[2-methylpropionitrile] (0.004 g, 0.024 mmol) were added. The stirred reactants were then heated to 95°C for 1.5 h before cooling to room temperature and adding pentane (100 mL). The resulting solid was filtered and washed with more pentane to give the title compound (0.039 g, 45%), 1H NMR (200 MHz, CDCI3) δ 2.10 (3H, s, COCH3), 2.20 (1H, p, H-3'a), 2.91 (1H, p, H-3'b), 3.65-4.14 (5H, overlapping signals, OCH3, H-5'a and H-5'b), 4.33 (1 H, t, 5'-OH), 4.56
(1H, t, H-4'), 5.53 (1H, p, H-2') 5.92 (1H, d, H-1'), 8.46 (1H, s, H-8), 8.87 (1 H, s, NH), MS (+FAB): m/z 358 [M++1].
Example 16
3'-Deoxy-2-chloro-/\-methoxyadenosine
The above 2'-0-acetyl-3'-deoxy-2-chloro-Λ/-methoxyadenosine (0.080 g, 0.22 mmol) was dissolved in CH3OH (10 mL) and treated with 0.5 mL of a previously prepared solution of sodium in CH3OH (0.01 g in 10 mL). The reaction mixture was stirred at 50°C under nitrogen for 16 h, after which time the starting material was not present by TLC. The pH of the reaction mixture was adjusted to ca. 5 by addition of acetic acid, before evaporation in vacuo. The residue was purified by column chromatography on silica gel eluting with CH2CI;>/EtOH (9/1), and crystallised from n-heptane to give the title compound (0.010 g, 14%), 1H NMR (200 MHz, CDCI3) δ 2.37 (1H, m, H-3'a), 2.66 (1H, m, H-3'b), 3.63 (1H, dt, H-5'a). 3.84 (3H, s, OCH3), 3.95 (1H, br dd, H-5'b), 4.52 (1H, br dd, H-2'), 4.66 (1 H, br dd, H-4'), 4.76 (1H, br s, 5'-OH), 5.41 (1H, br s, 3'-OH), 5.63 (1H, d, H-1'), 8.00 (1H, s, H-8), MS (+EI): m/z 316 [M*].
Example 17
5'-Q-Acetyl-2'.3'-didehydro-2'.3'-dideoxy-2-chloro-/ /-methoxyadenosine
5'-0-Acetyl-2',3'-0-thionocarbonyl-2-chloro-/V-methoxyadenosine
5'-0-Acetyl-2-chloro-/V-methoxyadenosine (4.80 g, 12.8 mmol) was dissolved in dry DMF (50 mL) and treated with 1 ,1'-thiocarbonyldiimidazole (3.2 g, 18.0 mmol). The reaction was stirred at room temperature under nitrogen for 4 h and then poured onto water (300 mL). This was then extracted with EtOAc (3 x 150 mL) and the combined organic phase washed with water (2 x 150 mL), brine 1 x 100 mL) and dried (MgS04). After concentration in vacuo, the residue was purified by column chromatography on silica gel eluting with CH2CI2/EtOH (14:1) to afford a yellow powder (3.78 g, 71%), 1H NMR (200 MHz, DMSO-d6) δ 1.94 (3H, s, COCH3), 3.77 (3H, s, OCH3), 4.26 (2H, ddd, H-5'. and H-5'b), 4.75 (1H, m, H-4'), 5.81 (1H, dd, H-3'), 6.28 (1H, dd, H-2'), 6.60 (1H, d, H-1'), 8.38 (1H, s, H-8), 11.63 (1H, s, NH). C14H14CIN505S requires C, 40.44; H, 3.44; N, 16.85. Found C, 40.8; H, 3.5; N, 16.6%.
5'-0-Acetyl-2',3'-didehydro-2',3'-dideoxy-2-chloro-Λ/-methoxyadenosine
A solution of 5'-0-acetyl-2\3'-0-thionocarbonyl-2-chloro-/V-methoxyadenosine (0.416 g, 1.0 mmol) in anhydrous THF (10 mL) was cooled to 0°C and treated with 1 ,3- dimethyl-2-phenyl-1,3,2-diazaphospholidine (0.55 mL, 3.0 mmol) added dropwise over a period of 10 min. After complete addition, the reaction mixture was allowed to stir at room temperature for 30 min before heating to 40°C for 24 h. The solvent was evaporated and the residue dissolved in EtOAc (40 mL). This was washed with water (2 x 20 mL), brine (1 x 15 mL) and dried (MgS04). After concentration the residue was purified by column chromatography on silica gel with CH^I^EtOH (19:1) as eluent to afford the title compound (0.166 g, 49%) as white crystals, mp 145-155°C. Η NMR
(200 MHz, DMSO-d6) δ 1.97 (3H, s, COCH3), 3.77 (3H, s, OCH3), 4.19 (2H, d, H-5'a and H-5'b), 5.10 (1H, m, H-4'), 6.24 (1H, ddd, H-2'), 6.53 (1H, dt, H-3'), 6.90 (1 H, m, H-1'), 8.18 (1H, s, H-8), 11.58 (1H, s, NH), MS (+FAB): m/z 340 [M*+1].
Example 18
5'-Q-Acetyl-2'.3'-dideoxy-2-chloro-Λ/-methoxyadenosine
5'-0-acetyl-2\3'-didehydro-2\3'-dideoxy-2-chloro-W-methoxyadenosine (Example 17) (0.10 g, 0.29 mmol) was dissolved in methanol (20 mL) and hydrogenated in the presence of 5% palladium on charcoal (10 mg). After 16 h, the reaction mixture was filtered and evaporated. Purification by flash chromatography using CH2Cl2/CH3OH (19:1) as eluent provided the title compound (0.014 g, 14%) as a white foam. 1H NMR (200 MHz, DMSO-d6) δ 1.97 (3H, s, COCH3), 2.05-2.20 (2H, m, H-3'), 2.40-2.48 (2H, m, H-2'), 3.77 (3H, s, OCH3), 4.09-4.38 (3H, m, H-4', H-5'a and H-5'b), 6.21 (1H, t, H-1'), 8.38 (1H, s, H-8), 11.52 (1H, s, NH), MS (+FAB): m/z 342 [M*+1].
Example 19
2'.3'-Didehydro-2'.3'-dideoxy-2-chloro-Λ/-methoxyadenosine
5'-0-Acetyl-2',3'-didehydro-2',3'-dideoxy-2-chloro-Λ/-methoxyadenosine (0.20 g, 0.5 mmol) was dissolved in CH3OH (40 mL) and treated with 1.5 mL of a previousl
prepared solution of sodium in CH3OH (0.01 g in 10 mL). The reaction mixture was stirred at 50°C under nitrogen for 18 h, after which time the starting material was not present by TLC. On cooling to room temperature, some product cyrstallised out from solution. The reaction mixture was neutralized by addition of acetic acid and evaporated in vacuo to ca. 20 mL. The solid was filtered off and washed well with CH3OH giving white crystals of the title compound (0.097 g, 55%), mp 195°C dec. Η NMR (200 MHz, DMSO-d6) δ 3.57 (2H, m, H-5'a and H-5'b), 3.77 (3H, s, OCH3), 4.84- 4.97 (2H, m, 5'-OH and H-4'), 6.13 (1H, m, H-2'), 6.49 (1H, m, H-3'), 6.88 (1H, m, H-1'), 8.25 (1H, s, H-8), 11.52 (1H, s, NH), MS (+FAB): m/z 298 [M++1]. C-.H^CINsO^. 0.1 AcOH requires C, 44.29; H, 4.12; N, 23.06. Found C, 44.04; H, 4.14; N, 22.98%.
2-Chloro-9-(4-isoxazol-3-yl-β-D-ety^/7tOfuranos-1-vn-Λ/-methoxyadenine
3-(2,3-0-lsopropylidene-1-0-methyl-β-D-etyf/7rofuranos-4-yl)-5- trimethylsilylisoxazole
To E Z-1-O-methyl-5-aldoxime-2,3-0-isopropylidene-β-D-t7'6ofuranoside (1.02 g, 4.68 mmol) dissolved in DMF (10 mL) at 0°C, a solution of NBS (0.92 g, 5.15 mmol) in DMF (3 mL) was added over 5 min. After stirring for 30 min, trimethylsilylacetylene (689 mg, 7.02 mmol) was introduced, followed by TEA (1.42 g, 14.04 mmol) in DMF (7 mL) over 20 h using a motorised syringe pump. The
98/01459 . ..
reaction was diluted with CH2CI2 (100 mL) and washed with water (2 x 50 mL), brine (25 mL), dried (Na2S04), filtered and concentrated in vacuo to give the title compound as an oil (0.67 g, 44%) containing two isomers. Column chromatography (eluent CH2CI2) provided an a analytical sample which contained only one of the isomers as an oil. Analytical data is for this isomer, 3-(2,3-0-isopropylidene-1-0- methyl-b-D-etyt/7ro-furanos-4-yl)-5-trimethylsilyl isoxazole, δH (300MHz, CDCI3 ) 0.32 (9H, s, TMS), 1.35 (3H, s, one of (Cϋ)2C ), 1.52 (3H, s, one of (CH^C ), 3.20 (3H, s, CH30 ), 4.68 (1H, d, J 6, H-3 ), 5.04 (1 H, s, H-4 ), 5.28 (1H, dd, J 1 and 6 , H-2 ), 5.32 (1H, d, J 1, H-1), 6.47 (1H, s, H-4 in isoxazole); δc(75MHz, CDCI3) -1.95 ( TMS ), 24.98 (one of (£H3)2C ), 26.46 (one of (CH3)2C), 54.84 ( £H30), 80.81 (C-3), 83.44 (C-4), 85.45 (C-2), 110.13 (C-1), 111.50 ( C-4 in isoxazole ), 112.66 ( (CH3)2C ), 161.54 (C-5 in isoxazole), 178.36 (C-3 in isoxazole).
3-(2,3-0-lsopropylidene-1-0-methyl-β-D-etyt 7rofuranos-4-yl)isoxazole
A solution of the 3-(2,3-0-isopropylidene-1-0-methyl-β-D-etytf7rofuranos-4-yl)-5- trimethylsilylisoxazole isomers (0.45 g, 1.44 mmol) in CH3OH (10 mL) was treated with 1M NaOH (2 mL) and stirred for 4 h. The solution was diluted with CH2CI2 (150 mL), washed with water (2 x 20 mL), brine (25 mL), dried (Na2S04) and finally concentrated in vacuo to give the title compound as a pale yellow oil (0.23 g, 57%) n^ (thin film) 2990, 2941, 2838, 1672, 1613, 1560, 1422, 1383, 1374; m/z 241.09810 (M+), C^H^NOj requires 241.09502; δH (300MHz, CDCI3); 1.35 (3H, s, one of (CH3)2C ), 1.52 (3H, s, one of (CJ 3)2C), 3.20 (3H, s, CH30), 4.68 (1H, d, J 5,
H-3), 5.06 (1H, s, H-4), 5.29 (1H, dd, J 5, H-2 ), 5.31 (1H, s, H-1), 6.40 (1H, s, J 1, isoxazole H-4), 8.34 (1H, s, J 1 , isoxazole H-5); δc (75MHz, CDCl3) 24.96 (one of
(CH3)2C), 26.48 (one of (£H3)2C), 55.00 (£H30), 80.75 (C-3), 83.14 (C-4), 85.38 (C- 2), 103.45 (C-4 in isoxazole), 110.22 (C-1), 112.75 ((CH3)2C_), 158.49 (C-5 in isoxazole), 162.42 (C-3 in isoxazole).
3-( 1 -0-Methyl-β-D-etythro-furanos-4-yi)isoxazole
A solution of 3-(2,3-0-isopropylidene-1-0-methyl-β-D-etyf/7rofuranos-4-yl)-isoxazole (0.25 g, 1.03 mmol) in CH
3OH/H
20 (1/1) (100 mL) was passed through a column of Dowex 50 H
+ ion exchange resin (15 g) and concentrated in vacuo. After concentration the residue was purified by column chromatograpy using
(8:2) as eluent to provide the title compound (146mg, 70%), [a]
D 27 -65.01 (c = 1.00; CH
3OH); m/z 201.06369 (M+), C
11H
15N0
5 requires 241.06372; δ
H ( 400MHz, CDCI
3 ) 3.49 (3H, s, Cl aO), 3.82 (1H, br s, OH-2), 4.05 (1H, br s, OH-3), 4.15 (1 H, d, J 4.5, H-2 ), 4.55 (1H, dd, J 4.5 and 7 H-3), 5.01 (1H, s . H-1 ), 5.11 (1H, d, J 1 H-4 ), 6.40 (1H, d, J 1.5, H-4 in isoxazole), 8.34 (1H, d, J 1, H-5 in isoxazole); δ
c (100MHz, CDCI
3) 55.06 (£H
30), 74.64 (C-2), 75.19 (C-3), 76.59 (C-4), 102.62 (C-4 in isoxazole), 108.32 (C-1), 158.35 (C-5 in isoxazole), 163.10 ( C-3 in isoxazole).
3-(2,3-Di-0-benzoyl-1-0-methyl-β-D-etytf)rofuranos-4-yl)isoxazole
A solution of 3-(1-O-methyl-β-D-et #7rofuranos-4-yl)-isoxazole (2.01 g, 10 mmol) in a mixture of pyridine (10 mL) and dimethylaminopyridine (5 mg) was cooled to 0°C. Benzoyl chloride (3.08 g, 22 mmol) was added to the cooled solution with stirring the reaction mixture was kept at 0 for 12 h and poured onto ice (100 g) before extraction with CH
2CI
2 (2 x 100 mL). The combined organic extracts were washed with ice water (2 x 50 mL), brine (50 mL) and dried (Na
2S0
4). After concentration in vacuo the residue was purified by column chromatograpy (π-heptane: EtOAc 7:3) to yield the title compound (3.4 g, 83%) as a clear oil, m/z 409.11570 (M+),
requires 409.11615; δ
H (400MHz,CDCI
3) 3.52 (3H, s, CHgO ). 5.22 (1H, d, J 1Hz, H- 1), 5.59 (1 H, dd, J 1 and 7.5, H-2 ), 5.74 (1H, d, J 5Hz , H-4 ), 5.95 ( 1H, dd, J 5 and 7.5, H-3 ), 6.50 ( 1H, d, J 2, H-4 in isoxazole), 7.5 (6H, m, H-3.H-4 and H-5 in phenyl), 7.90 (2H,dd, J 1 and 7, H-2 and H-6 in one phenyl), 8.02 (2H,dd, J 1 and 7, H-2 and H-6 in one phenyl), 8.45 (1 H, d, J 2, H-4 in isoxazole); δ
c (100MHz, CDCI
3); 55.29 (£H
30), 74.57 (C-3), 74.77 (C-2), 74.98 (C-4), 102.33 (C-4 in isoxazole), 106.45 (C-1), 127.88, 127.98, 128.05, 128.28 (2 x C-3 and 2 x C-5 in phenyl), 128.33, 128.59, 129.32, 129.35 (2 x C-2 and 2 x C-6 in phenyl), 129.66 (2 x C-1 in
phenyl), 132.98, 133.11 (2 x C-4 in phenyl) 158.75 (C-5 in isoxazole), 161.79 (C-3 in isoxazole), 164.74 (2 x C=0).
2,6-Dichloro-9-(2,3-di-0-benzoyl-4-isoxazo-3-yl-β-D-ety//ιro-furanos-1-yl)-9H-purine
A solution of 2,6-dichloro-9H-purine (2 g, 10.5 mmol) in 1,1,1,3,3,3-hexamethyl disilazane (20 mL) was heated at reflux for 12 h and after cooling to room temperature was concentrated in vacuo. The resultant residue was dissolved in 1 ,2- dichloroethane (20 L), added to a solution of 3-(2,3-O-dibenzoyl-1-0-methyl-β-D- erytr?ro~furanos-4-yl)-isoxazole (1.8 g, 4.3 mmol) in 1 ,2-dichloroethane (20 mL) and TMS-triflate (1.07 g, 4.8 mmol) was introduced over 10 min. The reaction mixture was heated at reflux for 12 h, cooled and concentrated in vacuo. Purification by column chromatography eluting with n-heptane/EtOAc (7/3) produced the title compound (0.20 g, 12%) as an foam, m/z 565.05813 (M+), C26H17N506CI2 requires 565.05559; δH(400Mhz, CDCI3) 5.71 (1H, d, J 3.5Hz, H-1), 6.25 (2H, m, H-2 and H- 3), 6.21 (1 H, d, J 5Hz , H-4), 6.70 ( 1H, d, J 1.5Hz, H-4 in isoxazole), 7.5 (6H, m, H- 3, H-4 and H-5 in 2 x phenyl), 7.90 (2H, dd, J 1 and 7.5, H-2 and H-6 in one phenyl), 8.10 (2H, dd, J 1 and 7.5, H-2 and H-6 in one phenyl), 8.52 (1H, d, J 1.5Hz, H-4 in isoxazole), 8.60 (1H, s, H-8 in purine); δc (100MHz, CDCI3) 73.80 (C-3), 74.01 (C- 2), 77.23 (C-4), 87.12 (C-1), 102.76 (C-4 in isoxazole), 128.01 , 128.10, 128.13, 128.21 (2 x C-3 and 2 x C-5 in phenyl), 129.39, 129.44, 129.80, 130.01 (2 x C-2 and 2 x C-6 in phenyl), 133.52, 133.58 (2 x C-4 in phenyl), 144.31 (C-8 in purine), 151.87, 152.33, 152.36, 152.80 (C2, C-4, C-5, C-6 in purine), 159.45 (C-5 in isoxazole), 159.54 (C-3 in isoxazole), 164.67, 164.77 (2 x C=0).
2-Chloro-9-(2,3-di-0-benzoyl-4-isoxazol-3-yl-β-D-ery hrofuranos-1-yl)-Λ/- methoxyadenine
O-Methyl hydroxylamine hydrochloride (0.17 mg, 0.2 mmol) and TEA (40 mg, 0.4 mmol) were added to a solution of 2,6-dichloro-9-(2,3-di-0-benzoyl-4-isoxazol-3-yl- β-D-eryf/7rofuranos-1-yl)-9H-purine (0.056 g, 0.1 mmol) in 1 ,4 dioxane (5 mL). The
reaction mixture was refluxed for 20 h and concentrated in vacuo. The residue was purified by column chromatography eluting with a mixture of π-heptane and EtOAc (1/1) to give the title compound (10 mg, 17%), m/z 576.11702 (M+), C27H21N607CI requires 576.11603; δH (400MHz, CDCI3) 3.70 (0-CH3), 5.65 (1H, d, J 5, H-1 ), 6.21 (1H, dd, J 5 and 7.5, H-3), 6.33 (1H, dd, J 5 and 7.5, H-2), 6.54 (1H, d, J 5, H-4), 6.79 (1H, d, J 1.5, H-4 in isoxazole), 7.5 (6H, m, H-3.H-4 and H-5 in 2 x phenyl), 7.95 (2H, dd, J 1 and 7.5, H-2 and H-6 in one phenyl), 8.05 (2H,dd, J 1 and 7.5, H-2 and H-6 in one phenyl), 8.25 (1H, s, adenine H-8), 8.50 (1H, d, J 1.5, H-4 in isoxazole), 8.91 (1H, br s, NH); δc (100MHz, CDCI3), 73.80 (C-3), 74.01 (C-2), 77.23 (C-4), 87.12 (C-1), 102.76 (C-4 in isoxazole), 128.01 , 128.10, 128.13, 128.21 (2 x C-3 and 2 x C-5 in phenyl), 129.39, 129.44, 129.80, 130.01 (2 x C-2 and 2 x C-6 in phenyl), 133.52, 133.58 (2 x C-4 in phenyl), 144.31 (C-8 in adenine), 151.87, 152.33, 152.36, 152.80 (C2, C-4, C-5, C-6 in adenine), 159.45 (C-5 in isoxazole), 159.54 (C-3 in isoxazole), 164.67, 164.77 (2 x C=0).
2-Chloro-9-(4-isoxazol-3-yl-β-D-et f/7røfuranos-1 -yl)-Λ-methoxyadenine
To a solution of 2-chloro-9-(2,3-di-O-benzoyl-4-isoxazol-3-yl-β-D-etyfΛtOfuranos-1- yl)-/V-methoxy-9H-adenine (0.09 mg, 0.016 mmol) in CH3OH (2 mL) was added 10% ammonia in CH3OH (0.001 mL) and stirred for 4 h. The mixture was concentrated in vacuo and purified by column chromatograpy, eluting with a mixture of CHjCiyCHaOH (9/1) to provide the title compound (0.002 g, 34%) as a foam, m/z
368.06289 (M+), C13H13Ne05CI requires 368.06360. δH(400MHz, DMSO-d6) 3.72 (3H, s, CM30 ), 4.51 (1H, m, H'-3), 4.72 (1H, m, H'-2), 5.05 (1H, dd, J 5 , H'-4 ), 5.74
(1H, d, J 6, 3'-OH ), 5.78 ( 1H, d, J6, 2'-OH ), 6.02 (1H, d, J 5 , H'-1), 6.81 (1H, s, J
2, H-4 in isoxazole), 8.41 (1H, s, N-H), 8.96 (1H, s, J 2, H-5 in isoxazole); δc (100MHz, DMSO-d6) 56.42 (CH30), 74.71 (C-4 ), 76.11 (C-2), 76.90 ( C-3 ), 84.75 (C-1), 101.89 ( C-4 in isoxazole ),146.01 (C-δ in adenine), 150.33, 151.83, 152.78, 153.65 (C2, C-4, C-5, C-6 in adenine) 160.34 (C-5 in isoxazole), 161.24 (C-3 in isoxazole).
Example 21
2-Chloro-Λ/-3-iodobenzyl-9-(4-isoxazol-3-yl-β-D-etvf/?tOfuranos-1-vnadenine
HO O H
2-Chloro-9-(2,3-di-0-benzoyl-4-isoxazol-3-yl-β-D-etyf/7rofuranos-1-yl)-/\/-3- iodobenzyl adenine
3-lodobenzylamine hydrochloride (26.9 mg, 0.1 mmol) and Et3N (40 mg, 0.4 mmol) were introduced to a solution of 2,6-dichloro-9-(2,3-di-0-benzoyl-4-isoxazol-3-yl-β- D-ety /?ro-furanos-1-yl)-9H-purine (see Example 21) (0.056 g, 0.1 mmol) in 1 ,4- dioxane (5 mL). The solution was heated at reflux for 10 h and concentrated in vacuo. The resultant residue was purified by a short path column chromatography on silica gel eluting with a mixture of n-heptane and EtOAc (1/1) to produce the title compound (0.025 g, 32%), δH (400MHz, CDCI3), 4.85 (2H, br s, NHCH2), 5.68 (1H, d, J 5, H-1), 6.21 (1H, dd, J 5 and 7.5, H-3), 6.37 (1H, dd, J 5 and 7.5, H-2), 6.49 (1H, d, J 5, H-4), 6.85 (1 H, d, J 1.5, H-4 in isoxazole), 7.09 (1H, t, J 5, H-5 in 3- iodophenyl) 7.4 (6H, m, H-3.H-4 and H-5 in 2 x phenyl), 7.6 (3H, m, H-2, H^ and H- 6 in 3-iodophenyl), 7.95 (2H, dd, J 1 and 7.5, H-2 and H-6 in one phenyl), 8.05 (2H,dd, J 1 and 7.5, H-2 and H-6 in one phenyl), 8.09 (1H br s, NH), 8.11 (1H, s, adenine H-8), 8.50 (1H, d, J 1.5, H^ in isoxazole); δc (100MHz, CDCI3); 73.80 (C-3), 74.01 (C-2), 77.23 (C-4), 87.12 (C-1), 102.76 (C-4 in isoxazole), 128.01, 128.10, 128.13, 128.21 (2 x C-3 and 2 x C-5 in phenyl), 129.39, 129.44, 129.80, 130.01 (2 x
C-2 and 2 x C-6 in phenyl), 133.52, 133.58 (2 x C-4 in phenyl), 144.31 (C-8 in adenine), 151.87, 152.33, 152.36, 152.80 (C2, C-4, C-5, C-6 in adenine), 159.45 (C- 5 in isoxazole), 159.54 (C-3 in isoxazole), 164.67, 164.77 (2 x C=0).
2-Chloro-/V-3-iodobenzyl-9-(4-isoxazol-3-yl-β-D-ety Λrofuranos-1-yl)adenine
A solution of 2-chloro-Λ/-3-iodobenzyl-9-(2,3-di-0-benzoyl-4-isoxazol-3-yl-β-D- ety /7tofuranos-1-yl)adenine (0.010 g, 0.013 mmol) in CH3OH (2 mL) was added 10% ammonia in CH3OH (0.001 mL) and stirred for 6 h. After concentration the crude mixture was purified by column chromatography (CH-jC C^OH 9:1) to produce the title compound (0.004 g, 55%) as a foam, m/z 553.99893 (M+), C19H16N604ICI requires 553.99663; δH(400MHz, CDCI3) 4.85 (5H, m, N-CH2, H-2', H- 3' and 3'-OH), 5.43 (1H, d, J4, H-4'), 6.05 (1H, d, J 5.5, H-1 *), 6.49 ( 1H, d, J 2, H-4 in isoxazole), 7.09 (1H, dd, 8, H-5 in phenyl), 7.35 (1H, d, J 8, H-6 in phenyl), 7.63 (1H, d, J 8, H-4 in phenyl), 7.72 (1H, s, H-2 in phenyl), 7.91 (1H, s, H-8 in purine), 8.50 (1H, d, J2, H-5 in isoxazole).
Example 22
/V-Methoxyaristeromycin. MR. 2S. 3R. 4ffl-1-[6'-methoxyadenin-9'-yl]-4-hydroxymethyl cydρpentane-2,3-d.iQl)
,
(1R,2S,3R,4R)-3-[(5,-Amino-6'-chloropyrimidin-4-yl)amino]-4-hydroxymethyl cyclopentane-2,3-diol
A mixture of 5-amino-4,6-dichloropyrimidine (3.25 g, 19.8 mmol), (1S, 2R, 3R, 5S)-3- amino-5-hydroxymethylcyclopentane-1,2-diol (Bindu Madhavan, GN., Martin, J.C. A novel and stereospecific synthesis of (±) and (-)-Aristeromycin. J. Org. Chem., 1986, 51, 1287-1293) (2.2 g, 15 mmol) and Et3N (4.16 mL, 30 mmol) in dry n-BuOH (100 mL) was heated at 90°C for 24 h; product TLC R, 0.14 (Si02; CH2Clj EtOH/aq. NH3 40/10/1). The cooled reaction mixture was evaporated and the residue was treated with H20 (200 mL) and EtOH (200 mL). The mixture was evaporated until only an aqueous suspension was present and some starting material was removed by filtration, and the filtrate was extracted with CHCI3 (2 x 80 mL) to remove remaining starting material. The aqueous filtrate was evaporated to a solid, dissolved in CH3OH (50 mL) and impregnated onto silica gel prior to column chromatography. Elution initially with CH2CI2, followed by CH2C.2/EtOH/aq. NH3 (20/10/1) afforded (1 , 2S, 3R, 4R)-3-[(5'-amino-6'-chloropyrimidin-4-yl)amino]-4-hydroxymethyl cyclopentane-2,3-diol as a solid (1.47 g, 36%): mp 228-231 °C (EtOAc/CH3OH); Η NMR (400 MHz, DMSO- d6) δ 1.02 - 1.13 (1H, m, H-4), 1.87 - 1.98 (1H, m, H-5), 2.20 (1H, dt, H-5), 3.29 - 3.45 (4H, m, -NH2 & -CH_2OH), 4.23 (1H, m, H-1), 4.44, 4.63 (2H, 2d, 2- & 3-OH), 4.57 (1H, t, HOCH2-), 5.08 (2H, br s, -NH2), 6.73 (1H, d, NHCH), 7.71 (1H, s, pyrimidine C-H).
(1R, 2S, 3R, 4R)-1-[6'-Chloropurin-9'-yl]-4-hydroxymethylcyclopentane-2,3-diol
Freshly distilled triethylorthoformate (25 mL) and (1R, 2S, 3R, 4R)-3-[(5'-amino-6'- chloropyrimidin-4-yl)amino]-4-hydroxymethyl cyclopentane-2,3-diol (1.12 g, 4.1 mmol) were mixed and concentrated HCI (37%, 0.96 mL) was added. The reaction mixture was stirred at room temperature for 18 h, treated with 0.5 N aqueous HCI (37 mL) and stirred for a further 1 h. The resultant solution was neutralized with 1N aqueous NaOH and evaporated. The residue was stirred in CH
3OH (50 mL), filtered and impregnated onto silica gel prior to column chromatography. Elution initially with
19/1 followed by CH
2CI
2/CH
3OH 9/1 afforded (1R, 2S, 3R, 4R)-1-[6'-chloropurin-9'-yl]- 4-hydroxymethylcyclopentane-2,3-diol as a solid (0.68 g, 60%): Η NMR (400 MHz, DMSO-d
6) δ 1.80 (1H, dq, H-4), 2.08 (1H, p, H-5), 2.28 (1H, dt, H-5), 3.42 - 3.57 (2H,
m, -CH
2OH), 3.85 (1 H, br s, H-3), 4.35 - 4.43 (1 H, m, H-2), 4.70 - 4.78 (2H, br, 2- & 3- OH), 4.87 (1H, q, H-1), 5.01 (1 H, br d, -CH
2OH), 8.28, 8.31 (2H, 2s, H-2' and H-8') Anal. Calcd. for C^H^CINA. 0.25 H
20 C, 45.7; H, 4.7; N, 19.4. Found: C, 46.0; H, 4.6; N, 19.0%.
Λ/-Methoxyaristeromycin
(1R, 2S, 3R, 4R)-1-[6'-Chloropurin-9,-yl]-4-hydroxymethylcyclopentane-2,3-diol (0.80 g, 0.28 mmol), triethylamine (0.20 mL, 0.146 g, 1.4 mmol) and O-methylhydroxylamine hydrochloride (0.070 g, 0.84 mmol) were mixed in a glass vessel and sealed. The reaction mixture was heated at 70°C for 72h, filtered and evaporated in vacuo. The residue was purified by flash chromatography eluting with CH2CI2 and CH3OH (4/1) to provide Λ/-methoxyaristeromycin (1R, 2S, 3R, 4R)-1-[6'-methoxyadenin-9'-yl]-4- hydroxymethyl cyclopentane-2,3-diol) (0.047 g, 57%) as a foam, Η NMR (400 MHz, DMSO-d6) δ 1.59 (1H, dt, H-4), 1.97 - 2.06 (1H, m, H-5), 2.20 (1H, dt, H-5), 3.38 - 3.53 (2H, m, -CH2OH), 3.76 (3H, s, -OCH3), 3.76 - 3.82 (1H, m, H-3), 4.21 (1H, dt, H-2), 4.53 - 4.75 (3H, m, 2- & 3-OH, H-1), 4.90 (1H, d, -CH2OJ ), 7.54 (1 H, d, N-H), 7.92, 8.30 (2H, 2s, H-2' and H-8').
Example 23
(^-MS.3'R.5,S 2-Chloro-Λ/-methoxy-9-r5,-M .2-dihydroxyethyhcvclopent-en-3'- yijadeniπe
(±)-et7 o-4-Hydroxy-2-oxabicyclo[3.3.0]oct-7-en-3-one and (±)-e o-4-hydroxy-2- oxabicyclo[3.3.0joct-7-en-3-one
An aqueous solution of glyoxylic acid (50% w/w , 148.1 g, 1.0 mol) was diluted with water (220 mL) to give a 2.25 M solution. The solution was cooled to 0°C and newly cracked cydopentadiene (92.5 g, 1.4 mol) was added. The reaction mixture was stirred vigourously for 24 h at ambient temperature. The mixture was ex- tracted with petroleum ether (4 x 100 mL) to remove unreacted cydopentadiene. The aqueous layer was saturated with sodium chloride and extracted with EtOAc (15 x 200 mL). The combined organic layers were concentrated to 500 mL, cooled to 0°C and washed with cold saturated sodium bicarbonate solution (2 x 100 mL) to remove unreacted glyoxylic acid. The organic layer was dried (MgS04), filtered and the solvent evaporated in vacuo to give the crude material as an yellow mobile oil. A small portion of the crude oil was purified twice by column chromatography on silica gel eluting with EtOAc/petroleum ether (1/1) giving first the exo-epimer as a colourless oil (Rf 0.28); δH(200 MHz; CDCl3) 6.09 (1H, m, H-8), 5.91 (1 H, m, H-7), 5.55 (1H, m, H-1), 4.16 (1H, d, J = 7 Hz, H-4), 3.61 (1H, br s, OH), 3.06 (1H, m, H- 5), 2.78 (1H, m, H-6), 2.57 (1 H, m, H-6).
Later fractions provided the corresponding endo-epimer as white crystals (Rf 0.23); mp 65-67°C, vMax(CHCI3)/cπv1 3432, 1771 , 1189, 1137; δH(200 MHz; CDCI3) 6.27 (1H, ddd, J = 6, 2 and 2 Hz, H-8), 5.93 (1H, dddd, J = 6, 2, 2 and 2 Hz, H-7), 5.35 (1H, ddd, J = 6, 2 and 2 Hz, H-1), 4.75 (1H, d, J = 9 Hz, H-4), 3.77 (1 H, br s, OH), 3.23 (1H, dddd, J = 9, 9, 6 and 6 Hz, H-5), 2.73 (1H, dddd, J = 18, 6, 3 and 2 Hz, H-6), 2.46 (1 H, dddd, J = 18, 9, 3 and 2 Hz, H-6); δc(75 MHz; CDCI3) 177.8 (C, C- 3), 141.1 (CH, C-8), 127.7 (CH, C-7), 86.4 (CH, C-1), 69.2 (CH, C-4), 40.5 (CH, C- 5), 30.6 (CH2, C-6); mlz (Cl) : 158 [(M+NH4)+, 100%]; Accurate mass [(Cl) required for C7H12N03 (M+NH4)+ 158.08172, found 158.08164.].
(±)-(1 S,3'R,4'R)-(3'-Hydroxycyclopenten-4'-yl)-1 ,2-ethanediol
A suspension of LiAIH4 (1.80 g, 47.1 mmol) in anhydrous THF (20 L) was refluxed for 0.5 h. Heating was stopped and a solution of the above (±)-endo-4- hydroxy-2-oxabicyclo[3.3.0]oct-7-en-3-one (3.00 g, 21.4 mmol) in anhydrous THF
(40 mL) was added dropwise to maintain gentle reflux and the mixture was heated at reflux. The reaction mixture was quenched by adding a saturated aqueous solution of Na2S04 at 0°C until the mixture turned white. The viscous mixture was filtered through a glass sintered funnel rinsing with EtOAc and the filtrate was concentrated in vacuo to give the crude (±)-(1S,3'R,4'R)-(3'-hydroxycyclopenten- 4'-yl)-1 ,2-ethanediol as a yellow oil. This crude oil was reacted further without purification.
(±)-(1S,3'R,4'R)-(3'-Acetoxycyclopenten-4,-yl)-1 ,2-diacetoxyethane
(1 S,3'R,4'R)-(3'-Hydroxycyclopenten-4'-yl)-1 ,2-ethanediol (0.50 g, 3.47 mmol) was dissolved in a mixture of Ac20 (5 mL) and pyridine (5 mL) and stirred at room temperature 17 h. The reaction mixture was quenched with a saturated ammonium chloride solution (20 mL). Et20 (50 mL) was added to the mixture and the layers were separated. The organic layer was washed successively with saturated Na2C03 solution (20 mL) and brine (20 mL). Drying (MgS04) of the organic layer was proceeded with evaporation of solvent in vacuo giving a slightly yellow crude oil. The crude oil was recrystallised from petroleum ether, giving the title compound as white crystals (0.56 g, 60%); mp 83-86°C; vMax(CHCI3)/cnτ1 1741 , 1373, 1253, 1047, 1016; δH(200 MHz; CDCI3) 6.10 (1 H, m, H-5), 5.88 (1 H, m, H-4), 5.68 (1 H, m, H-1 ), 5.24 (1 H, ddd, J = 11 , 5 and 3 Hz, H-2), 4.40 (1 H, dd, J = 12 and 2 Hz, H-3), 3.98 (1H, dd, J = 12 and 5 Hz, H-3), 2.67-2.48 (1H, m, H-6), 2.31 (2H, m, H-7), 2.04 (3H, s, CH3 of Ac), 1.99 (3H, s, CH3 of Ac), 1.92 (3H, s, CH3 of Ac); δc(75 MHz; CDCI3) 170.8 (C, C=0), 170.5 (C, C=0), 170.3 (C, CO), 137.3 (H, C-5), 129.7 (CH, C-4), 77.0 (CH, C-1), 70.2 (CH, C-6), 64.4 (CH2, C-7), 41.8 (CH, C-2), 33.2 (CH2, C-3), 20.8 (CH3, £H3 of Ac), 20.7 (CH3l £H3 of Ac), 20.5 (CH3, C_H3 of Ac); Accurate mass [(Cl) required for C13H22NOβ (M+NH4)* 288.14471 , found 288.14517].
(±)-(1S,3,R,5,S)-2-Amino-6-chloro-9-[5,-(1 ,2-diacetoxyethyl)cyclopenten-3,-yi]-(9H)- purine
A suspension of 2-amino-6-chloro-(9H)-purine (207 mg, 1.22 mmol) and NaH (60% dispersion in mineral oil, 49 mg, 1.221 mmol) in anhydrous DMF (5 mL) was stirred for 20 min at room temperature followed by heating to 50°C for 10 min to give an almost clear solution. Tetrakis(triphenylphosphine)palladium (0.128 g, 0.111 mmol) and (±)-(1S,3'R,4'R)-(3'-acetoxycyclopenten-4,-yl)-1,2-diacetoxyethane were suspended in THF (10 mL) and added to the warm solution of 2-amino-6- chloro-(9H)-purine using a cannula and rinsing with DMF (3 x 1 mL). After 3 h stirring at 50°C, the reaction mixture was cooled to room temperature and water (20 mL) was added. The mixture was extracted with CH2CI2 (3 x 30 mL) and the combined organic layers were dried (MgS04) and concentrated in vacuo to a yellow oil. The oil was purified by column chromatography on silica gel, eluting initially with EtOAc/petroleum ether (4/1) to afford recovered unreacted triacetate (70 mg, 25%). Continued elution with EtOAc/petroleum ether (2/1) provided the title compound as a clear oil (0.17 g, 43%,); δH(300 MHz; CDCI3) 7.79 (1H, s, H-8), 6.11 (1H, ddd, J = 5.5, 2 and 2 Hz H-3'), 5.88 (1H, ddd, J = 5.5, 2 and 2 Hz, H-2'), 5.48 (4H, m, H-V, H-5' and NH2), 4.38 (1H, dd, J = 12 and 3 Hz, H-7'), 4.14 (1H, dd, J = 12 and 7 Hz, H-7'), 3.20 (1H, m, H-4'), 2.79 (1H, ddd, J = 14, 9 and 9 Hz, H-6'), 2.11 (3H, s, CH3 of Ac ); 2.07 (3H, s, CH3 of Ac ), 1.86 (1 H, ddd, J = 14, 6 and 6 Hz, H-6'); δc(75 MHz; CDCI3) 170.8 (C, C=0), 170.6 (C, C=0), 159.1 (C, C- 2), 153.6 (C, C-6), 151.6 (C, C-4), 141.0 (C, C-8), 136.0 (CH, C-3'), 130.2 (CH, C- 2'), 125.8 (CH, C-5), 73.0 (CH2, C-7'), 64.1 (CH, C-5'), 60.4 (CH, C-1'), 45.6 (CH, C-4'), 33.0 (CH2, C-6'), 20.8 (CH3, CH3 of Ac), 20.5 (CH3, CH3 of Ac); Accurate mass [(El): required for C16H18N504CI (M+) 379.10474, found 379.10531].
(±)-(1S,3'R,5'S)-2,6-Dichloro-9-[5,-(1,2-diacetoxyethyl)cyclopenten-3'-yl]-(9H)- purine
(±)-(1S,3'R,5'S)-2-Amino-6-chloro-9-[5'-(1,2-diacetoxyethyl)cyclopenten-3'-yl]-(9H)- purine (80 mg, 0.21 mmol) was dissolved in CH2CI2 (5 mL) and the solution was cooled to -10 °C. Trimethylchlorosilane (79 μl, 0.622 mmol) was added as quickly
as possible giving a slightly red and unclear solution. Isopentylnitrite (83 μl, 0.622 mmol) was added slowly to maintain a temperature at -10 °C. The mixture was stirred for 2 h at -10°C and at room temperature for 5 h before being quenched with water (4 mL) and the phases were separated, followed by extraction of the water layer with CH2CI2 (2 x 5 mL). The combined organic layers were washed with saturated NaHC03 solution (10 mL) and brine (10 mL) followed by drying (MgS04) and evaporation of solvent in vacuo to give a yellow crude oil. The oil was purified by column chromatography using silica gel and EtOAc/Petroleum ether (2:1) giving the title compound (48 mg, 57%); δH(200 MHz; CDCI3) 8.19 (1H, s, H-8), 6.27 (1H, ddd, J = 6, 2 and 2 Hz, H-3'), 5.99 (1H, ddd, J = 6, 2 and 2 Hz, H-2'), 5.79 (1H, dddd, J = 6, 6, 2 and 2 Hz H-1'), 5.08 (1H, ddd, J = 6, 6 and 4 Hz, H-5'), 4.36 (1H, dd, J = 12 and 4 Hz, H-7'), 4.08 (1H, dd, J = 12 and 6 Hz, H-7'), 3.21 (1H, m, H-4'), 2.96 (1H, ddd, = 14, 9 and 9 Hz, H-6'), 2.10 (3H, s, CH3 of Ac ); 2.07 (3H, s, CH3 of Ac ), 1.67(1H, ddd, J = 14, 6 and 6 Hz, H-6'); δc(75 MHz; CDCI3) 170.5 (C, C=0), 170.3 (C, C=0), 153.0 (C, C-2), 152.8 (C, C-6), 151.9 (C, C-4), 143.9 (C, C-8), 137.6 (CH, C-3'), 129.4 (CH, C-2'), 117.8 (CH, C-5), 72.8 (CH2, C-7'), 63.6 (CH, C-5'), 59.8 (CH, C-1'), 45.7 (CH, C-4'), 34.9 (CH2, C-6'), 20.7 (CHj, CH3 of Ac), 20.5 (CH3, CH3 of Ac); m/z Accurate mass [(Cl): required for C16H14N404CI2(M+NH4)+ 399.06269, found 399.06306.].
(±)-(1S,3'R,5'S)-2-Chloro-Λ/-methoxy-9-[5,-(1 ,2-diacetoxyethyl)cyclopent-en-3'- yl]adenine
(±)-(1 S,3,R,5'S)-2,6-Dichloro-9-[5'-(1 ,2-diacetoxyethyl)cyclopenten-3'-yl]-(9H)- purine (36 mg, 0.090 mmol), triethylamine (28 μl, 0.198 mmol) and methoxylamine hydrochloride (8.3 mg, 0.099 mmol) were dissolved in 1 ,4-dioxane (5 mL) and stirred at 75°C for 3 days. The reaction mixture was cooled to room temperature and filtered. The solvent was evaporated in vacuo to give a crystalline suspension which was purified by column chromatography using silica gel and CH2CI2/EtOH (29:1). The title compound was obtained as a clear oil (17 mg, 46%); δH(200 MHz;
CDCI3) 9.63 (1H, br s, NH), 7.92 (1H, s, H-8), 6.20 (1H, ddd, J = 6, 2 and 2 Hz, H- 3'), 5.96 (1H, ddd, J = 6, 2 and 2 Hz, H-2'), 5.79 (1H, m, H-V), 5.06 (1 H, ddd, J = 6,
6 and 4 Hz, H-51), 4.35 (1 H, dd, J = 12 and 4 Hz, H-7'), 4.07 (1 H, dd, J = 12 and 6 Hz, H-7'), 4.00 (3H, s, OCH3), 3.15 (1 H, m, H-4'), 2.92 (1H, ddd, J = 14, 9 and 9 Hz, H-6'), 2.09 (3H, s, CH3 of Ac ); 2.07 (3H, s, CH3 of Ac ), 1.60 (1 H, ddd, J = 14,
7 and 7 Hz, H-6'); δc(75 MHz; CDCI3) 170.6 (C, C=0), 170.4 (C, C=0), 156.2(C, C- 6), 154.2 (C, C-2), 151.8 (C, C-4), 139.6 (C, C-8), 136.8 (CH, C-3'), 120.0 (CH, C- 2'), 117.5 (CH, C-5), 73.0 (CH2, C-7'), 64.9 (CH3, OCH3), 63.6 (CH, C-5'), 59.0 (CH, C-1'), 45.7 (CH, C-4"), 35.8 (CH2, C-6'), 20.7 (CH3, CH3 of Ac), 20.5 (CH3, CH3 of Ac); Accurate mass [(El): required for C17H20N5O5CI (M+) 409.11530, found 409.11477]
(±)-(1S,3*R,5,S)-2-Chloro-/S/-methoxy-9-[5'-(1 ,2-dihydroxyethyl)cyclopent-en-3'- yl]adenine
(±)-(1S,3'R,5'S)-2-Chloro-Λ/-methoxy-9-[5'-(1 ,2-diacetoxyethyl)cyclopent-en-3'- yljadenine (40 mg, 0.097 mmol) and potassium carbonate (1.4 mg, 0.0098 mmol) was dissolved in methanol (10 mL) and the mixture was stirred at room temperature for 18 h. Quenching with solid ammonium chloride to neutral pH was followed by evaporation of solvent in vacuo giving an colourless oil which goes purple on standing. Purification of the crude oil by column chromatography on silica gel, eluting with a mixture of CH2CI2/CH3OH (9/1) provided (±)-(1 S,3'R,5'S)-2-chloro-/v- methoxy-9-[5'-(1 ,2-dihydroxyethyl)cyclopenten-3'-yl]adenine as a white powder (22 mg, 68%,Rf 0.32); δH(300 MHz; CD3OD) 8.25 (1H, s, H-8), 6.34 (1H, ddd, J = 6, 2 and 2 Hz, H-3'), 5.95 (1H, ddd, J = 6, 2 and 2 Hz, H-2'), 5.66 (1H, m, H-1'), 3.87 (3H, s, OCH3), 3.64-3.57 (3H, m, H-5' and H-7'), 3.06 (1H, m, H-4'), 2.84 (1H, ddd, J = 14, 9 and 9 Hz, H-6'), 1.79 (1H, ddd, J = 14, 6 and 6 Hz, H-6'); δc(100 MHz, DMSO-d6) 155.2 (C, C-6), 152.5 (C, C-2), 150.9 (C, C-4), 140.7 (C, C-8), 137.9 (CH, C-3'), 129.1 (CH, C-2'), 116.3 (CH, C-5), 73.8 (CH2, C-7'), 64.4 (CH3, OCH3), 63.7 (CH, C-5'), 59.2 (CH, C-1'), 47.6 (CH, C-4'), 34.4 (CH2, C-6'); Accurate mass
[(El): required for C13H1βNs03CI (M+) 325.09415, found 325.09405.].
Synthesis of Example 23
Example 24
MS. 4SΪ-Acetic acid. 4-(2-chloro-Λ/-methoxyadenin-9-ylPyclopent-2-enyl ester
(1 S, 4S)-Acetic acid, 4-(2,6-dichloro-9H-purin-9-yl)cyclopent-2-enyl ester
Acetic acid, (1S, 4R)-5-hydroxycyclopent-2-yl ester (0.76 g, 5.3 mmol), 2,6-dichloro-
(9H)-purine (1.01 g, 5.3 mmol) and triphenylphosphine (2.09 g, 8.0 mmol) were dissolved in THF (50 mL). Diethylazodicarboxylate (1.50 g, 8.0 mmol) was introduced dropwise under a nitrogen atmosphere, and the reaction mixture was evaporated to a residue which was purified by flash chromatography, eluting with a mixture of n- heptane and EtOAc (4/1) to provide the desired acetic acid, (1S, 4S)-4-(2,6-dichloro- 9H-ρurin-9-yl)cyclopent-2-enyl ester (0.95 g, 57%) as a foam, Η NMR (400 MHz, DMSO-d6) δ 2.05 (3H, s, OCOCH3), 2.43 (1H, o, H-5a), 2.58 (1H, o, H-5b), 5.85 - 5.90 (1H, m), 5.96 (1H, m), 6.25 - 6.31 (2H, m, CH=CH), 8.68 (1H, s, H-8).
(1S, 4S)-Acetic acid, 4-(2-chloro-Λ/-methoxyadenin-9-yl)cyclopent-2-enyl ester
Acetic acid, (1 S, 4S)-4-(2,6-dichloro-9H-purin-9-yl)cyclopent-2-enyl ester (0.72 g, 2.3 mmol) was dissolved in 1,4-dioxan (50 mL). O-Methylhydroxylamine hydrochloride (1.54 g, 18.39 mmol) and N.N-diisopropylethylamine (4.0 mL, 2.97 g, 23.0 mmol) were introduced and the reaction mixture was stirred at 50°C for 20h, and evaporated to a residue which was purified by flash chromatography. Elution with a mixture of n- heptane and EtOAc (1/1) provided (1S, 4S)-4-(2-chloro-/V-methoxyadenin-9- yl)cyclopent-2-enyl acetate (0.47 g, 73%) as a foam, 1H NMR (400 MHz, DMSO-d6) δ 2.04 (3H, s, OCOCH3), 2.39 - 2.50 (2H, m, H-5aand H-5b), 3.76 (3H, s, HNOCH3), 5.72 - 5.77 (1H, m), 5.93 - 5.97 (1H, m), 6.25 (2H, s, CH=CH), 8.18 (1H, s, H-8), 11.48 (1H, s, NH).
Example 25
MS. 4S -(2-Chloro-/S/-methoxyadenin-9-y cyclopent-2-enol
H,
(1S, 4S)-Acetic acid, 4-(2-chloro-6-methoxyadenin-9-yl)cyclopent-2-enyl ester (0.57 g, 2 mmol) was dissolved in methanolic ammonia (30 mL) and allowed to stand at room temperature for 18 h. The reaction mixture was evaporated and purified by flash chromatography. Elution initially with EtOAc, followed by EtOAc/CH3OH (19/1) provided (1S,4S)-4-(2-chloro-Λ/-methoxyadenin-9-yl)cyclopent-2-enol (0.24 g, 42%) as a foam, 1H NMR (400 MHz, DMSO-d6) δ 2.19 (1H, o, H-5a), 2.29 (1H, o, H-5b), 3.77 (3H, s, HNOCH3), 5.02 - 5.08 (1H, m), 5.08 (1H, t), 5.67 - 5.72 (1H, m), 6.00 (1 H, dd, C=CH), 6.20 (1H, dt, C=CH), 8.11 (1H, s, H-8), 11.45 (1H, s, NH).
Example 26
M R. 4S)-4-(2-Chloro-Λ/-methoxyadenin-9-ylPyclopent-2-enol
(1 , 4S)-4-(2-chloro-/V-methoxyadenin-9-yl)cyclopent-2-enol, 4-nitrobenzoate
(1S, 4S)-4-(2-Chloro-Λ/-methoxyadenin-9-yl)cyclopent-2-enol (0.20 g, 0.71 mmol), triphenylphosphine (0.28 g, 10.7 mmol) and 4-nitrobenzoic acid (0.12 g, 0.71 mmol) were dissolved in THF (20 mL). Diethyazo icarboxylate (0.19 g, 10.9 mmol) was introduced dropwise under a nitrogen atmosphere and after 0.25 h the reaction mixture was evaporated and purified by flash chromatography. Elution initially with a mixture of EtOAc and π-heptane (4/1), followed by a 1/1 mixture of these solvents
afforded (1R, 4S)-4-(2-chloro-/V-methoxyadenin-9-yl)cyclopent-2-enol, 4-nitrobenzoate (0.26 g, 84%) as a foam, Η NMR (400 MHz, DMSO-d6) δ 2.17 (1H, dt, H-5a), 3.18 (1H, dt, H-5b), 3.77 (3H, s, NOCH3), 5.58 - 5.62 (1H, m), 5.92 - 5.97 (1H, m), 6.90 (2H, dq, HC=CH), 8.18 (1H, s, H-8), 8.21 , 8.34 (4H, 2d, Ar-H), 11.48 (1H, s, NH).
(1R, 4S)-4-(2-Chloro-/V-methoxyadenin-9-yl)cyclopent-2-enol
(1R, 4S)-4-(2-chloro-Λ/-methoxyadenin-9-yl)cyclopent-2-enol, 4-nitrobenzoate (0.253 g, 0.59 mmol) was dissolved in methanolic ammonia (10 mL) in a sealed vessel and the mixture was heated at 40°C for 3 h. The reaction mixture was evaporated and purified by flash chromatography. Elution initially with a mixture of EtOAc and n- heptane (1/1) followed by EtOAc, then with EtOAc/CH3OH (50/1) provided (1 , 4S)- 4-(2-chloro-Λ/-methoxyadenin-9-yl)cyciopent-2-enol (0.108 g, 42%), Η NMR (400 MHz, DMSO-d6) δ 1.68 (1H, dt, H-5a), 2.90 (1 H, dt, H-5b), 3.77 (3H, s, NOCH3), 4.68 - 4.75 (1H, m), 5.31 (1H, d), 5.38 - 5.43 (1H, m), 6.00 (1H, dd, C=CH), 6.20 (1H, dt, HC=C), 8.17 (1H, s, H-8), 11.48 (1H, s, NH).