WO1992012718A1 - Adenosine kinase inhibitors - Google Patents
Adenosine kinase inhibitors Download PDFInfo
- Publication number
- WO1992012718A1 WO1992012718A1 PCT/US1992/000515 US9200515W WO9212718A1 WO 1992012718 A1 WO1992012718 A1 WO 1992012718A1 US 9200515 W US9200515 W US 9200515W WO 9212718 A1 WO9212718 A1 WO 9212718A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrogen
- amino
- halogen
- compound according
- compound
- Prior art date
Links
- 0 *C(C(C1O)O)NC1*(*=*1)C(*2)=C1C(N)=*C2N Chemical compound *C(C(C1O)O)NC1*(*=*1)C(*2)=C1C(N)=*C2N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/14—Pyrrolo-pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
Definitions
- This invention relates to adenosine kinase inhibitors and to novel nucleoside analogs, specifically to purine, pyrrolo [2,3-d]pyrimidine and pyrazolo[3,4-d]pyrimidine nucleoside analogs having activity as adenosine kinase inhibitors.
- the invention also relates to the preparation and use of these and other adenosine kinase inhibitors in the treatment of cardiovascular, and cerebrovascular diseases, inflammation and other diseaseswhich can be regulated by increasing the local concentration of adenosine.
- Adenosine has been reported to have cardioprotective
- adenosine has a short half life ( ⁇ 1 sec) in human blood (Moser, et al., Am. J. Physiol.. 1989, 256:C799-C806), and therefore high doses of adenosine would need to be administered continuously to achieve effective levels.
- Adenosine has been reported to exhibit negative inotropic, chronotropic and dromotropic effects
- Adenosine kinase is a cytosolic enzyme which catalyzes the phosphorylation of adenosine to AMP.
- adenosine kinase can potentially reduce the ability of the cell to utilize adenosine, leading to increased adenosine outside of the cell where it is pharmacologically active.
- the regulation of adenosine concentration is complex and involves other adenosine- metabolizing enzymes each with different kinetic properties and mechanisms of regulation.
- Adenosine can also be deaminated to inosine by adenosine deaminase (ADA) and condensed with L-horoocysteine to S-adenosylhomocysteine (SAH) by SAH hydrolase.
- ADA adenosine deaminase
- SAH S-adenosylhomocysteine
- concentration is dependent on the prevailing physiological conditions, is tissue specific and is not well understood.
- nucleosides including purine, pyrrolo[2,3- djpyrimidine and pyrazolo[3,4-d]pyrimidine analogs have been evaluated for inhibition of adenosine kinase but were reported to have K i 's of greater than 800 nM (Caldwell and Henderson Cancer Chemother. Rep., 1971 2:237-246; Miller et al., J. Biol. Chem., 1979, 254:2346-2352). A few compounds have been reported as potent inhibitors of adenosine kinase with K-'s of less than 100 nM.
- adenosine kinase adenosine kinase (AK).
- the AK cells used in this study were said to release adenosine at an accelerated rate; the concentration of adenosine in the growth medium was reported to be elevated compared to the normal cells (Green, J. Supramol.
- adenosine kinase inhibitors 5-iodotubercidin and 5'-deoxy-5-iodotubercidin (Davis et al., Biochem. Pharmacol., 1984, 33:347-355).
- inhibition of uptake and intracellular trapping via phosphorylation does not necessarily result in increased extracellular adenosine, since the adenosine could enter other metabolic pathways or the percentage of adenosine being phosphorylated could be insignificant compared to the total adenosine removed.
- pyrrolo[2,3-d]pyrimidines 5-iodotubercidi and 5'-deoxy-5-iodotubercidin have been reported to cause pronounced general flaccidity and much-reduced spontaneous locomoto activity in mice, interpreted to be skeletal muscle relaxation; to cause hypothermia in mice; and to decrease blood pressure and heart rate in anesthetized rats (Daves et al., Biochem.
- the present invention is directed to novel compounds which are potent and selective adenosine kinase inhibitors.
- the present invention is directed to certain novel compounds which inhibit adenosine kinase, to the preparation of these compounds, and to the in vitro and in vivo adenosine kinase inhibition activity of these compounds.
- Another aspect of the present invention is directed to the clinical use of adenosine kinase inhibitors as a method of increasing adenosine concentrations in biological systems. In vivo inhibition of adenosine kinase prevents phosphorylation of adenosine resulting in higher local concentrations of endogenous adenosine. As a result of the very short half-life of adenosine and very low quantities of adenosine in tissues, this effect is most
- adenosine is pronounced in regions producing the most adenosine such as ischemic regions. Hence, the beneficial effects of adenosine are enhanced in a site and event specific manner and toxic systemic effects are reduced.
- the present invention in one preferred aspect, the present invention
- novel nucleoside analogs which comprise a 5'-modified ribose linked to a substituted purine, pyrrolo[2,3-d]pyrimidine, or pyrazolo[3,4-d]pyrimidine base.
- Certain preferred compounds within these groups possess potencies many times greater than previously described inhibitors of adenosine kinase.
- the compounds of the present invention possess advantages for pharmaceutical use such as enhanced pharmacological selectivity, efficacy, bioavailability, ease of manufacture and compound stability. This invention also discloses novel processes for the preparation of these compounds.
- novel compounds of the present invention and other adenosine kinase inhibitors may be used clinically to treat medical conditions where an increased localized adenosine
- the present invention is directed to the prophylactic and affirmative treatment of ischemic conditions such as myocardial infarction, angina, percutaneous transluminal coronary angiography (PTCA), stroke, otherthrombotic and embolic conditions, neurological conditions such as seizures and psychosis, and other conditions benefitted by enhanced adenosine levels such as inflammation, arthritis, autoimmune diseases, cardiac arrhythmias, ulcers and irritable bowel syndrome.
- ischemic conditions such as myocardial infarction, angina, percutaneous transluminal coronary angiography (PTCA), stroke, otherthrombotic and embolic conditions, neurological conditions such as seizures and psychosis, and other conditions benefitted by enhanced adenosine levels such as inflammation, arthritis, autoimmune diseases, cardiac arrhythmias, ulcers and irritable bowel syndrome.
- PTCA percutaneous transluminal coronary angiography
- the present invention is also directed to prodrugs and pharmaceutically acceptable salts of the compounds described herein and to pharmaceutical compositions suitable for different routes of drug administration and which comprise a
- hydrocarbyl refers to an organic radical
- alkyl alkenyl, alkynyl and aryl groups, groups which have a mixture of saturated and unsaturated bonds, carbocyclic rings and includes combinations of such groups. It may refer to straight-chain, branched-chain cyclic structures or combinations thereof.
- aryl refers aromatic groups which have at least one ring having a conjugated pi electron system and includes carbocyclic aryl, heterocyclic aryl and biaryl groups, all of which may be optionally substituted.
- Carbocyclic aryl groups are groups wherein the ring atoms on the aromatic ring are carbon atoms.
- Carbocyclic aryl groups include monocyclic carbocyclic aryl groups and optionally
- monocyclic carbocyclic aryl refers to optionally substituted phenyl, being preferably phenyl or phenyl substituted by one to three substituents, such being advantageously lower alkyl, hydroxy, lower alkoxy, lower alkanoyloxy, halogen, cyano, trihalomethyl, lower acylamino or lower alkoxycarbonyl .
- Optionally substituted naphthyl refers to 1- or 2- naphthyl or 1- or 2-naphthyl preferably substituted by lower alkyl, lower alkoxy or halogen.
- Heterocyclic aryl groups are groups having from 1 to 3 heteroatoms as ring atoms in the aromatic ring and the remainder of the ring atoms carbon atoms. Suitable heteroatoms include oxygen, sulfur, and nitrogen, and include furanyl, thienyl, pyridyl, pyrrolyl, N-lower alkyl pyrrolo, pyrimidyl, pyrazinyl, imidazolyl, and the like, all optionally substituted.
- Optionally substituted furanyl represents 2- or 3-furanyl or 2- or 3-furanyl preferably substituted by lower alkyl or halogen.
- Optionally substituted pyridyl represents 2-, 3- or 4- pyridyl or 2-, 3- or 4-pyridyl preferably substituted by lower alkyl or halogen.
- Optionally substituted thienyl represents 2- or 3-thienyl, or 2- or 3-thienyl preferably substituted by lower alkyl or halogen.
- biasing represents phenyl substituted by
- aralkyl refers to an alkyl group substituted wit an aryl group. Suitable aralkyl groups include benzyl, picolyl, and the like, and may be optionally substituted.
- Such groups may be straight chain or branched.
- aralkylamino refers to the groups -NRR' wherein respectively, (a) R is alkyl and R 1 is hydrogen or alkyl; (b) R is aryl and R 1 is hydrogen or aryl, and (c) R is aralkyl and R' is hydrogen or aralkyl.
- acyl refers to hydrocarbyl-CO- or HCO-.
- acylamino refers to RC(O)NCR)- and (RCO) 2 N-respectively, wherein each R is independently hydrogen or hydrocarbyl.
- ⁇ -alkoxyalkylidene refers to hydrocarbyl-O-CR (an orthoester) wherein R is hydrogen or hydrocarbyl.
- hydrocarbyloxycarbonyloxy refers to the group ROC(O)O- wherein R is hydrocarbyl.
- hydrocarbyloxycarbonymethyl refers to hydrocarbyl-OC(O)CH 2 - with the hydrocarbyl group containing ten or less carbon atoms.
- carbonyl refers to -C(O)-.
- carboxylate or “carboxamido” refers to -CONR 2 wherein each R is independently hydrogen or hydrocarbyl.
- lower hydrocarbyl refers to any hydrocarbyl grou of ten or less carbon atoms.
- alkyl refers to saturated aliphatic groups including straight-chain, branched chain and cyclic groups.
- alkenyl refers to unsaturated hydrocarbyl groups which contain at least one carbon-carbon double bond and include straight-chain, branched-chain and cyclic groups.
- alkynyl refers to unsaturated hydrocarbyl groups which contain at least one carbon-carbon triple bond and include straight-chain, branched-chain and cyclic groups.
- halogen refers to fluorine, chlorine, bromine or iodine.
- hydrocarbyloxycarbonylamino refers to a urethane, hydrocarbyl-O-CONR- wherein R is H or hydrocarbyl and wherein each hydrocarbyl is independently selected.
- di(hydrocarbyloxycarbonyl)amino refers to di(hydrocarbyloxycarbonyl)amino
- hydrocarbyl-O-CO (hydrocarbyl-O-CO) 2 N- wherein each hydrocarbyl is independently selected.
- hydrocarbylamino refers to -NRR' wherein R is hydrocarbyl and R' is independently selected hydrocarbyl or hydrogen.
- mercapto refers to SH or a tautomeric form.
- metal refers to .
- methylene refers to -CH 2 -.
- alkylene refers to a divalent straight chain or branched chain saturated aliphatic radical.
- oxy refers to -O- (oxygen).
- thio refers to -S- (sulfur).
- prodrug refers to any compound that has less intrinsic activity than the "drug” but when administered to a biological system generates the "drug” substance either as a result of spontaneous chemical reaction or by enzyme catalyzed or metabolic reaction.
- prodrugs such as acyl esters, carbonates, and urethanes, included herein as examples.
- the groups illustrated are exemplary, not exhaustive and one skilled in the art could prepare other known varieties of prodrugs.
- pharmaceutically acceptable salt includes salts of compounds of Formula I derived from the combination of a compound of this invention and an organic or inorganic acid.
- the compounds of Formula I are useful in both free base and salt form. In practice the use of salt form amounts to use of base form; both forms are within the scope of the present invention.
- Figure 1 depicts the effects of the adenosine kinase inhibitor GP-1-238 on mean arterial pressure, heart rate and bod temperature following intravenous administration to anesthetized or conscious rats.
- Figure 2 depicts the dose-dependent inhibition of neutrophil adhesion to endothelial cells by the adenosine kinase inhibitors GP-1-272 and GP-1-456 and the reversal of this inhibition by co- treatment with adenosine deaminase ("ADA").
- ADA adenosine deaminase
- Figure 4 depicts (A) the dose-dependent inhibition of pentylenetetrazole (PTZ) induced seizures by the adenosine kinase inhibitor GP-1-456, and (B) the reversal of this inhibition by the central adenosine receptor antagonist theophylline but not the peripheral antagonist 8-sulfophenyltheophylline.
- PTZ pentylenetetrazole
- Figures 5 to 9 depict reaction schemes for preparing certain of these adenosine kinase inhibitors.
- Figure 10 depicts the structures of certain preferred intermediates useful in the synthesis of adenosine kinase inhibitors.
- the present invention relates to novel adenosine kinase inhibitors which comprise compounds of the general formula I.
- A is oxygen, methylene or sulfur
- B' is -(CH 2 ) n -B wherein n is 1, 2, 3 or 4 and B is hydrogen, alkyl, alkoxy, amino, alkylamino, acylamino, hydrocarbyloxycarbonylamino, mercapto, alkylthio, azido, cyano, halogen, or B' is alkenyl or alkynyl;
- C 1 and C 2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein c, is a single bond to C 2 and C 2 is carbonyl or ⁇ - alkoxyalkylidene;
- D is hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl, alkynyl, haloalkyl, cyano, cyanoalkyl, acyl, carboxamido, a carboxylic acid or carboxylic acid ester group, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, amino, alkylamino arylamino, aralkylamino, acylamino, or nitro;
- E is hydrogen, halogen, alkyl, or alkylthio
- F is alkyl, aryl, aralkyl, halogen, amino
- G is hydrogen, halogen, lower alkyl, lower alkoxy, lower alkylamino or lower alkylthio; and pharmaceutically acceptable salts thereof; with the proviso that:
- novel adenosine kinase inhibitors which have a 5'- group which comprises a hydroxyl or hydroxyl derivative.
- adenosine kinase inhibitors comprise compounds of the formula:
- A is oxygen, methylene or sulfur
- B' is -(CH 2 ) n B wherein n is 1, 2, 3 or 4 and B is hydroxy, acyloxy, hydrocarbyloxycarbonyloxy, or -OCONR 2 wherein R is independently hydrocarbyl;
- C 1 and C 2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C 1 is a single bond to C 2 and C 2 is carbonyl or ⁇ - alkoxyalkylidene;
- D is halogen, aryl or aralkyl
- F is alkyl, aryl, aralkyl, halogen, amino
- G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio; and pharmaceutically acceptable salts
- A is oxygen, methylene or sulfur
- B' is -(CH 2 ) n B wherein n is 1, 2, 3 or 4 and B is hydroxy, acyloxy, hydrocarbyloxycarbonyloxy, or -OCONR 2 wherein R is hydrocarbyl;
- C 1 and C 2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C 1 is a single bond to C 2 and C 2 is carbonyl or ⁇ -alkoxyalkylidene;
- D is aryl or aralkyl
- E is hydrogen, halogen, alkyl, or alkylthio;
- F is alkyl, aryl, aralkyl, halogen, amino, alkylamino, arylamino, aralkylamino, cyano, cyanoalkyl, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, optionall substituted indolinyl or indolyl, pyrrolidinyl or piperazinyl; and
- G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio; and pharmaceutically acceptable salts thereof; with the proviso that: when A is oxygen, D is
- adenosine kinase inhibitors which comprise modified purine nucleosides of the formula:
- B' is -CH 2 B wherein and B is amino, alkylamino, or acylamino;
- E is hydrogen, halogen, alkyl, amino, alkylamino, azido, acylamino, alkoxy or alkylthio;
- F is halogen, amino, alkylamino, arylamino,
- aralkylamino cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio, alkyl, aryl, aralkyl, optionally
- G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio and pharmaceutical acceptable salts thereof; with the proviso that:
- novel adenosine kinase inhibitors comprise dimeric compounds of the formula:
- a and A' are independently oxygen, methylene or sulfur;
- B' and B" are independently -(CH 2 ) n B wherein n is independently 1, 2, 3 or 4 and B is independently hydrogen, hydroxy, alkyl, alkoxy, amino, alkylamino, acylamino,
- hydrocarbyloxycarbonylamino, mercapto, alkylthio, azido, or either or both of B' or B" is independently alkenyl or alkynyl;
- D is independently hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl, alkynyl, haloalkyl, cyano, cyanoalkyl, acyl, carboxamido, a carboxylic acid or corresponding carboxylic acid ester group, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, amino, alkylamino, arylamino, aralkylamino acylamino or nitro;
- (f) E is independently hydrogen, halogen, alkyl, or
- (g) L is an optionally substituted piperazinyl divalent radical or -NH(ALKL)NH- wherein ALKL is a divalent alkylene radical of 2 to 24 carbon atoms;
- G and G' are each independently hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkoxy; or pharmaceutically
- G is hydrogen, halogen, alkyl or alkylthio.
- G groups include hydrogen.
- Preferred C 1 and C 2 groups include hydrogen and acetyl.
- E groups include hydrogen or halogen, especially preferred are compounds where E is hydrogen. Preferred are compounds where A is oxygen.
- D is hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl or alkynyl, cyano, cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio, amino, alkylamino, arylamino, aralkylamino, carboxamido, or
- D groups include hydrogen, halogen, alkyl, aryl, aralkyl, cyano, alkoxy, aryloxy, aralkoxy, alkenyl or alkynyl, more preferably hydrogen, halogen, aryl, cyano, alkoxy or aryloxy.
- a particularly preferred group of compounds include those wherein D is hydrogen, halogen or aryl.
- D is aryl such as heterocyclic aryl or monocyclic carbocyclic aryl, such as
- B is -(CH 2 ) n B, and n is 1 or 2, more preferably n is 1.
- B may preferably include hydrogen, halogen, alkyl, amino, alkylamino, alkoxy, mercapto, alkylthio, azido or cyano; more preferably B is hydrogen,
- B groups include hydrogen, amino or azido. Also preferred are compounds wherein B' is vinyl, ethynyl, or propargyl.
- Preferred F groups include halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl, more preferably amino or arylamino.
- preferred F groups include optionally substituted anilino.
- the compounds of the present invention contain asymmetric carbon atoms and hence can exist as stereoisomers, both enantio- mers and diastereomers.
- the individual preferred stereoisomers and mixtures thereof are considered to fall within the scope of the present invention.
- the compounds described by Formula I contain a 5-modified 1- ⁇ -D-ribofuranosyl group and that isomer comprises a particularly preferred diastereomeric and enantiomeric form for compounds of the present invention.
- the synthetic examples cited herein provide the most preferred isomer. It is evident that in addition to the sugar moiety, additional asymmetric carbons may be present in compounds of Formula I, being present in moieties B', C 1 or C 2 or the
- compounds of formula I where B is hydroxy are in many cases potent inhibitors of adenosine kinase.
- the use of compounds having formula I wherein B' replaced by -CH 2 OH, as adenosine kinase inhibitors are included in the scope of this invention.
- some of these compounds may be phosphorylated in vivo and since the resulting 5'-phosphates may be toxic, mutagenic or teratogenic, 5'-hydroxy compounds which can serve as substrates for
- phosphorylation enzymes may not comprise preferred compounds for clinical or therapeutic use.
- An important aspect of the novel compounds of the present invention is that these preferred compounds are either non-phosphorylatable at the 5' position or are not substrates of enzymes that lead to phosphorylation.
- Preferred adenosine kinase inhibitor compounds of the present invention include certain pyrazolo[3,4-d]pyrimidine compounds of Formulas I and II.
- Preferred pyrazolo[3,4-d]pyrimidine compunds of Formula I include those where G is hydrogen and A is oxygen.
- Preferred D groups include hydrogen, alkyl, aryl, aralkyl, cyano, alkoxy, aryloxy, aralkoxy, alkenyl or alkynyl, more preferably hydrogen, halogen, aryl, cyano, alkoxy or aryloxy, more particularly hydrogen, halogen or aryl.
- An especially preferred group of compounds includes those where D is aryl, especially heterocyclic aryl or monocyclic carbocyclic aryl, more preferably optionally substituted phenyl.
- Preferred B' groups include -(CH 2 ) n B wherein B is hydrogen, halogen, alkyl, amino, alkylamino, alkoxy,
- Examples of preferred pyrazolo[3,4-d]pyrimidine compounds include those noted as GP-1-515, GP-1-547, GP-1-560, GP-1-665, GP-1-666 GP-1-667, GP-1-695, and GP-1-704.
- Preferred pyrazolo[3,4-d]pyrimidine compunds of Formula II include those where G is hydrogen and A is oxygen.
- Preferred D groups include aryl.
- Prefered aryl groups include heterocyclic carbocyclic aryl groups, especially optionally substituted phenyl.
- Preferred F groups include halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl, or aralkyl, more preferably amino or arylamino.
- Certain preferred compounds of Formula II may include F groups which comprise optionally substituted anilino groups.
- inventions include pyrrolo[2,3-d]pyrimidine compounds of Formulas I and II.
- Preferred pyrrolo[2,3-d]pyrimidine compounds of Formula I include those wherein G is hydrogen. Preferred are compounds wherein E is hydrogen or halogen; more preferably E is hydrogen. Preferred are compounds where A is oxygen. Preferred compounds include those where D is hydrogen, halogen, alkyl, aryl, aralkyl, cyano, alkenyl or alkynyl, more preferably hydrogen, halogen or aryl. An especially preferred group of compounds includes those where D is aryl, especially heterocyclic aryl or monocyclic carbocyclic aryl, especially optionally substituted phenyl.
- Preferred B' groups include -(CH 2 ) n B wherein n is 1 or 2,
- B is hydrogen, halogen, alkyl, amino, alkylamino, alkoxy, mercapto, alkylthio, azido or cyano, more preferably B is hydrogen, halogen, lower alkyl, amino, lower alkylamino, lower alkoxy, lower alkylthio, or azido, more
- B groups particularly hydrogen, lower alkyl, amino, lower alkylamino, or azido.
- Especially preferred B groups include hydrogen, amino or azido.
- Other preferred B' groups include vinyl and ethynyl.
- Preferred pyrrolo[2,3-d]pyrimidine compounds of Formula I include those wherein F is halogen, amino, alkylamino, arylamino,
- aralkylamino alkylthio, aralkylthio, alkyl, aryl or aralkyl, more preferably amino or arylamino.
- Certain preferred compounds include F groups which comprise optionally substituted anilino.
- Examples of preferred pyrrolo[2, 3-d]pyrimidine compounds include those noted as GP-1-448, GP-1-606, GP-1-608, GP-1-639, GP-1-683, GP-1-684, GP-1-691, GP-1-711, GP-1-714, and GP-1-718.
- Preferred pyrrolo[2, 3-d]pyrimidines of Formula II include those where G is hydrogen and A is oxygen.
- E is hydrogen or halogen, more preferably hydrogen.
- Preferred D groups include aryl.
- Preferred aryl groups include heterocyclic aryl groups and monocyclic carbocyclic aryl groups, especially optionally substitued phenyl.
- Preferred heterocyclic aryl groups include 2-furanyl, 2-thienyl and 3-thienyl.
- Preferred purine compounds include those where G is
- Preferred E groups include hydrogen, halogen or
- alkylthio Preferred are compounds wherein B is amino.
- Preferred F groups include halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl, more preferably amino or arylamino.
- Preferred dimeric compounds include those which comprises dimers of the above-described pyrazolo[3,4-d]pyrimidines, the pyrrolo[2,3-d]-pyrimidines and purines. These dimers may comprise monomeric units which are the same or different.
- the present invention also directed to processes for preparing compounds of Formula I.
- Disclosed herein are general synthetic routes for preparing variously substituted purine nucleosides or pyrrolo[2,3-d]pyrimidine nucleosides, including a novel and improved synthesis of 5'-deoxy- 5-iodotubercidin; and pyrazolo[3,4-d]pyrimidine nucleosides of the present invention.
- N 6 -substituted azide (4a) is deblocked using an aqueous acid suc as 50% formic acid, to provide the N 6 -substituted
- 5'-azido-5'-deoxyadenosine (5a). Reduction of the azide (5a) to the amine (6) is effected by catalytic hydrogenation with a catalyst such as platinum oxide, palladium on carbon and the like. For molecules containing other functional groups sensitiv to hydrogenation, triphenylphosphine is used to selectively reduce the azide moiety to the amine.
- a catalyst such as platinum oxide, palladium on carbon and the like.
- triphenylphosphine is used to selectively reduce the azide moiety to the amine.
- N-acylamin (7a) and hydrocarbyloxycarbonylamino (7b) compounds the azide 4a is reduced to the amine and treated with an acyl anhydride or acyl chloride or alkyl chloroformate and deblocked to give (7a) or (7b) respectively.
- Analogous processes are used to prepare th 2- and 8- substituted analogs beginning with appropriately substituted intermediates.
- hydrocarbylamine hydrocarbylamine
- nucleosides The appropriately substituted 5'-deoxy-2',3'-O- isopropylideneinosine (2b) is chlorinated or activated using other reagents described above, aminated to 4b and subsequently deblocked to afford the 5'-deoxy nucleoside 5b.
- pyrrolo[2,3-d]pyrimidine riboside compounds of Formula I is depicted in Figure 8.
- a key step comprises the sodium salt glycosylation method (K. Ramasamy et al., Tetrahedron Letters, 1987, 28: 5107) using the anion of a substituted
- sodium salt glycosylation method is a solid-liquid phase transfer reaction using the same substrates and potassium hydroxide in place of sodium hydride as described by Rosemeyer H., and Seela, F, Helvetica Chimica Acta, 1988, 71:1573.
- the 5-substituted-5-deoxy ribose analogs 10 are prepared by tosylation of the protected ribose 8, displacement of the tosylate by appropriate nucleophiles and subsequent deblocking (Synder, J.; Serianni, A.; Carbohydrate Res., 1987, 163: 169).
- the ribose homologs ( Figure 7) are prepared by oxidation of the protected ribose (8) to the aldehyd (11) (Moorman, A.; Borchaedt, R.; Nucleic Acid Chemistry-Part III.
- the aldehyde is homologated via the appropriate Wittig reagent to give the key intermediate protected vinyl sugar (12).
- the protected intermediate is deblocked to give the vinyl ribose homolog (16a) or reduced to (13) and then deblocked to give the saturated deoxy analog (16b).
- the vinylated intermediate (12) is hydroborated and oxidized affording the protected homologous ribose (14a) which is deblocked to the ribose homolog or converted to the azide (14b) via tosylation and displacement with azide. Deblocking of (14b) then affords the homologous azido ribose (16d).
- the protected 5-aldehyde (11) was also methylated to ultimately afford 6-deoxy-D-allofuranose
- nucleosides and the corresponding deblocked compounds are versatile intermediates and comprise an aspect of the present invention.
- the 4-chloro substituent of 19 can be displaced by sulfur (such as thiourea or mercaptide anions) leading to thionated and hydrocarbylthio compounds. More importantly, displacement of the 4-chloro substituent by ammonia or amines leads to 4-amino- and 4-arylaminopyrrolo[2,3- d]pyrimidine nucleosides.
- an improved synthesis of the adenosine kinase inhibitor, 5'deoxy-5-iodotubercidin is described.
- pyrrolo[2,3-d]pyrimidine nucleosides having a 4-chloro and a 5-iodo or bromo substituent.
- Another aspect of the present invention is directed to the use of arylboronic acids to prepare 4- and 5-arylated
- halogenated nucleoside such as .19 or the corresponding base was heated with an arylboronic acid and a palladium-phosphine catalyst such as palladium tetrakis(triphenylphosphine) to prepare the analogous arylated compound by displacement of halogen.
- a palladium-phosphine catalyst such as palladium tetrakis(triphenylphosphine)
- reactions may be deblocked at appropriate points with aqueous acids such as 50% formic acid or trifluoroacetic acid.
- Still another aspect of this invention is the preparation of 5'-substituted pyrazolo[3,4-d]pyrimidine ribosides of Formula I as depicted in Figure 9.
- a substituted pyrazolo[3,4-d]pyrimidine is ribosylated with an esterified 5- hydroxy, 5-azido or 5-deoxyribofuranoside in the presence of a Lewis acid such as boron trifluoride (Cottam, H., Petrie, C.; McKernan, P.; Goebel, R.; Dalley, N.; Davidson, R.; Robins, R.; Revankar, G.; J. Med. Chem. , 1984, 27: 1120).
- boron trifluoride Cottam, H., Petrie, C.; McKernan, P.; Goebel, R.; Dalley, N.; Davidson, R.; Robins, R.; Revankar, G.; J. Med. Che
- the 5-substituted sugar is prepared by esterification of the deblocked sugar 10a to 10c or 16a to 16e (See Figures 6 and 7). Suitable esters include the acetate, benzoate, toluate, anisoate and the like.
- the substituted pyrazolo[3,4-d]pyrimidine base (22) may be prepared by a variety of procedures as illustrated in the Examples. Two general routes to the compounds of the present invention are described below.
- the first general route comprises coupling an esterified ribose (21), prepared from 10 or 16, with a 3-substituted
- pyrimidone riboside (24a) may be activated by chlorination with thionyl chloride/dimethylformamide or other reagents previously described and then reacted with ammonia or an amine to provide a variety of substituted 5'-modified N 4 -substituted-amino-pyrazolo[3,4-d]pyrimidine nucleosides (24b). Examples of this aspect of the invention, 3-iodopyrazolo[3,4-d]pyrimidone
- nucleosides are prepared by nonaqueous diazotization-iodmation of the 3-amino compounds using a nitrite ester such as isoamyl nitrite and methylene iodide.
- a nitrite ester such as isoamyl nitrite and methylene iodide.
- Previous attempts to diazotize the 3-aminopyrazolo[3,4-d]pyrimidones using aqueous nitrous acid gave only N-nitrosated pyrazolo[3,4-d]pyrimidin-3,4-diones (Cottam, H.; Petrie, C.; McKernan, P.; Goebel, R.; Dalley, N.; Davidson, R.; Robins, R.; Revankar, G.; J. Med. Chem.. 1984, 27:1119).
- the second general route for preparation of substituted pyrazolo[3,4-d]pyrimidine nucleosides comprises coupling the esterified ribose (21) with various substituted 4-amino or 4- hydrocarbylaminopyrazolo[3,4-d]pyrimidines.
- the resulting products are then further modified or deblocked to afford the desired compounds.
- the utility of this procedure is demonstrated in the Examples, by the preparation of 3-phenyl-4- (phenylamino)pyrazolo[3,4-d]pyrimidine 5'-modified ribosides from 3-phenyl-4-(phenylamino)pyrazolo[3,4-d]pyrimidine and various 5'-modified sugars.
- halogenated pyrazolo[3,4]pyrimidine ribosides can be arylated using arylboronic acids and palladium catalysts as described for the pyrrolo[2,3-d]pyrimidines.
- the base can be boronated and then coupled with an aryl halide.
- One preferred method of the present invention is a novel procedure for preparing C-6 alkylated purine nucleosides and C-4 alkylated pyrazolo[3,4-d]pyrimidine nucleosides from the 6-chloropurine and 4-chloropyrazolo[3,4-d]pyrimidine nucleosides, respectively, using various carbanions (enolates, cyanide anion, etc.) and trimethylamine as a specific catalyst.
- Another preferred method of the present invention is a process for preparing arylated bases and nucleosides by reaction of a halogenated pyrrolo[2,3-d]pyrimidine or pyrazolo[3,4- d]pyrimidine with an aryl boronic acid in the presence of a palladium-phosphine catalyst.
- the halogen atom of a brominated or preferably, iodinated pyrrolo[2,3-d]pyrimidine or pyrazolo[3,4-d]pyrimidine base or nucleoside is replaced by an aryl moiety such as phenyl, substituted phenyl or a heteroaryl moiety such as furanyl.
- a catalyst consisting of a metal such as palladium, complexed to an arylphosphine such as
- triphenylphosphine must be present as well as a base such as sodium carbonate.
- a base such as sodium carbonate.
- Still another preferred method of the present invention is a process for preparing the previously unknown 3-iodo- and 3-chloropyrazolo[3,4-d]pyrimidine nucleoside by nonaqueous
- a suitably substituted 3- aminopyrazolo[3,4-d]pyrimidine nucleoside is diazotized by heating with an alkyl nitrite such as isoamyl nitrite in the presence of an iodine source (such as methylene iodide) resulting in replacement of the 3-amino moiety with an iodine atom.
- an alkyl nitrite such as isoamyl nitrite
- an iodine source such as methylene iodide
- methylene iodide can be replaced by a chlorine source such as carbon tetrachloride resulting in replacement of the amino moiety by a chlorine atom.
- a chlorine source such as carbon tetrachloride
- a previously reported attempt to effect replacement of the amino moiety in a 3- aminopyrazolo[3,4-d]pyrimidine riboside with other moieties using nitrous acid resulted only in replacement of the amino moiety by a hydroxyl group.
- the resulting 3-chloro- and particularly 3-iodopyrazolo[3,4-d]pyrimidine nucleoside are an important subject of adenosine kinase inhibitors disclosed in the present
- Certain intermediates useful in the preparation of certain preferred adenosine kinase inhibitors which comprise substituted pyrrolo[2,3-d]pyrimidine nucleosides include compounds of the formula:
- B' is lower alkyl or 1 to 3 carbon atoms optionally substituted with azido or hydroxy, or lower alkenyl of 1 to 3 carbon atoms; D is bromo or iodo, E is hydrogen, F is chloro, mercapto, arylamino and G is hydrogen.
- certain of these preferred intermediates exhibit activity as adenosine kinase inhibitors themselves.
- Certain intermediates useful in the preparation of certain preferred adenosine kinase inhibitor compounds comprise substituted pyrazolo[3,4-d]pyrimidines of the formula:
- aryl groups include heterocyclic aryl groups and monocyclic carbocyclic aryl groups, including optionally
- adenosine kinase inhibitors of the present invention may be used in the treatment of a variety of clinical situations where increasing local levels of adenosine are beneficial.
- these compounds may be used in treating cardiovascular disorders in which injury or dysfunction is caused by
- heart attack a situation that arises from obstruction of one or more of the coronary arteries supplying blood to the heart muscle, and which, if prolonged, leads to irreversible tissue damage
- angina pectoris a clinical condition in which the blood supply to the heart is sufficient to meet the normal needs of the heart but insufficient when the needs of the heart increase (e.g. during exercise), and/or when the blood supply becomes more limited (e.g. during coronary artery spasm); (3) unstable angina associated with pain at rest; and (4) silent ischemia.
- treatment with adenosine kinase inhibitors will increase local levels of adenosine, and thereby blood flow to the ischemic tissue would be increased and tissue damage reduced and function improved.
- adenosine kinase inhibitors may also be used to treat or prevent congestive heart failure.
- PTCA percutaneous transluminal coronary angioplasty
- percutaneous transluminal directional coronary atherectomy percutaneous transluminal directional coronary atherectomy
- laser atherectomy intra-vascular stents
- coronary artery bypass graft surgery a number of clinical procedures are currently used to improve blood supply to the heart.
- the compounds of this invention will also be useful as adjunctive therapies to these techniques.
- ischemia Other clinical settings that involve ischemia would also be ameliorated by agents effecting regional blood flow including organ transplantation, skin flap grafting and other reconstructive surgery, peripheral vascular disease, endotoxemia, hemorrhagic shock, pulmonary emboli, pul monary injury secondary to burns (thermal injury) or septicemia, pulmonary hypertension, microembolization, glomerulonephritis or progressive glomerulosclerosis, atherosclerosis, myocarditis, vasculitis, cardiomyopathies, intestinal ischemia,. peripheral vascular disease, transient ischemic attacks, stroke and cardio- pulmonary arrest. Adenosine kinase inhibitors will enhance protection afforded by preconditioning a tissue with a brief period of ischemia, before a more prolonged period of ischemia.
- Thrombolytic therapy has been limited by a number of factors including the resistance of some thrombi to lysis, delays in reperfusion, and reocclusion following successful thrombolysis. These limitations are believed to be mediated, in part, by platelet aggregation (Born and Cross J. Physiol., 1963, 166:29- 30) and, since adenosine inhibits platelet aggregation in
- adenosine kinase inhibitors may comprise a useful adjunctive therapy for thrombolytic therapy or for the treatment or prevention of thrombotic diseases such as myocardial infarction, stroke, angina, deep vein thrombosis, transient ischemic attacks, and pulmonary embolus.
- Adenosine has been reported to be an endogenous modulator of inflammation by virtue of its effects on stimulated granulocyte function (Cronstein et al., J. Clin. Invest.. 1986, 78:760-770) and on macrophage, lymphocyte and platelet function.
- the compounds of this invention may therefore be used in treating conditions in which inflammatory processes are prevalent such as arthritis, osteoarthritis, autoimmune disease, adult respiratory distress syndrome (ARDS), inflammatory bowel disease, necrotizing enterocolitis, chronic obstructive pulmonary disease (COPD), psoriasis, conjunctivitis, iridocyditis, myositis, cerebritis, meningitis, dermitis, renal inflammation, ischemia, reperfusion injury, peripheral vascular disease, atherosclerosis and other inflammatory disorders.
- ARDS adult respiratory distress syndrome
- COPD chronic obstructive pulmonary disease
- psoriasis conjunctivitis, iridocyditis, myositis, cerebritis, meningitis, dermitis, renal inflammation, ischemia, reperfusion injury, peripheral vascular disease, atherosclerosis and other inflammatory disorders.
- Adenosine receptor agonists have been reported to be beneficial in an experimental
- adenosine has been reported to inhibit excitatory amino acid release (Burke and Nadler J. Neurochem.. 1988, 51:1541) and excitatory amino acid responsiveness.
- the compounds of this invention which increase adenosine levels, may also be used in the treatment of conditions where release of or sensitivity to excitatory amino acids is implicated such as Parkinson's disease, Amyotrophic Lateral
- adenosine kinase inhibitors may be useful in treating schizophrenia.
- adenosine kinase inhibitors may also be useful in reducing anxiety, as skeletal muscle relaxants and in preventing skeletal muscle spasm.
- Adenosine has been proposed to serve as a natural
- Agents that enhance adenosine levels may be used in the treatment of seizure disorders.
- Adenosine kinase inhibitors may be used in the
- excitatory neuromuscular tissues such as smooth muscle and cardiac muscle may be treated using these adenosine kinase inhibitors.
- these adenosine kinase inhibitors may be used to decrease contraction in smooth muscle such as in the gastrointestinal tract, or in vascular tissue such as an artery to prevent vasospasm which may limit blood supply to a tissue.
- these adenosine kinase inhibitors may be used to treat conditions such as Buerger's disease, Raynaud's disease,
- thromboangiitis obliterans angina, unstable angina, silent ischemia, or transient ischemic attacks.
- Other conditions suitable for such therapy include cardiac arrhythmias (including supraventricular tachycardia), irritable bowel syndrome, and impotence.
- These compounds include pyrrolo[2,3- d]pyrimidine nucleosides modified at the 5'-position or at other positions such that it is less likely to serve as a substrate for phosphorylation enzymes and that, in contrast to 5-iodotubercidin (GP-1-202), these compounds are unlikely to be phosphorylated at the 5'-position, incorporated into nucleotides or DNA, which may cause toxicity to cells or animals.
- GP-1-202 5-iodotubercidin
- adenosine kinase inhibitor GP-1-515 is beneficial in an experimental model of stable angina in dogs.
- intravenous infusion of the compound attenuated the decline in function associated with repeated episodes of pacing-induced ischemia (Example E).
- the potential antithrombotic activity of adenosine kinase inhibitors is supported by the ability of GP-1-515 to abolish cyclic flow reductions (CFR's) in 3 out of 8 dogs examined in the Folts model of coronary artery thrombosis (Example F).
- CFR's cyclic flow reductions
- Example G the ability of selected adenosine kinase inhibitors (GP-1-272 and GP-1-456) to inhibit neutrophil adherence to endothelial cells, an inflammatory response mediated at the cellular level was evaluated. Certain adenosine kinase inhibitors were found to exhibit anti-inflammatory activity in animal models of inflammation.
- adenosine kinase inhibitors GP-1-515, GP-1-547) to attenuate contraction in the isolated ileum (Example H) supports the utility of these compounds in gastrointestinal disorders especially irritable bowel syndrome, in the central nervous system (CNS), the potent effects of selected adenosine kinase inhibitors (such as GP-1-456, GP-1-560) in attenuating chemical and electroshock induced seizures in experimental animal models demonstrates that these compounds will be useful as anticonvulsants in epilepsy (Example I), as well as in other CNS diseases treatable by local increases in adenosine levels.
- CNS central nervous system
- Compounds of the invention are administered to the affected tissue at the rate of from 0.1 to 200 nmole/min/kg, preferably from 1 to 20 nmol/min/kg. Such rates are easily maintained when these compounds are intravenously administered as discussed below. When other methods are used (e.g., oral administration), use of time-release preparations to control the rate of release of the active ingredient may be preferred. These compounds are administered in a dose of about 0.01 mg/kg/day to about 100 mg/kg/day, preferably from about 0.1 mg/kg/day to about 10 mg/kg/day.
- the compounds of the invention may be administered by a variety of means including orally, parenterally, by inhalation spray, topically, or rectally in formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, and intraarterial injections with a variety of infusion techniques.
- Intraarterial and intravenous injection as used herein includes administration through
- catheters Preferred for certain indications are methods of administration which allow rapid access to the tissue or organ being treated, such as intravenous injections for the treatment of myocardial infarction.
- intravenous injections for the treatment of myocardial infarction.
- perfusion is preferred.
- compositions containing the active ingredient may be in any form suitable for the intended method of
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents including those from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation. Tablets containing the active ingredient in
- excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, such as maize starch, or alginic acid; binding agents, such as starch, gelatin or acacia; and lubricating agents, such as magnesium stearate, stearic acid or talc. Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and adsorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- Formulations for oral use may be also presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, such as peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example calcium phosphate or kaolin
- an oil medium such as peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions of the invention contain the active materials in admixture with excipients suitable for the
- Such excipients include a suspending agent, such as sodium carboxymethylcellulose,
- methylcellulose hydroxypropylmethylcelluose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia, and dispersing or wetting agents such as a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene
- stearate a condensation product of ethylene oxide with a long chain aliphatic alcohol (e.g., heptadeaethyleneoxycetanol), a condensation product of ethylene oxide with a partial ester derived from a fatty acid and a hexitol anhydride (e.g.,
- the aqueous suspension may also contain one or more preservative such as ethyl of n-propyl p-hydroxybenzoate, one or more coloring agent, one or more flavoring agent and one or more sweetening agent, such as sucrose or saccharin.
- Oil suspensions may be formulated by suspending the active ingredient in a vegetable oil, such as arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oral suspensions may contain a thickening agent, such as beeswax, hard paraffin or cetyl alcohol. Sweetening agents, such as those set forth above, and flavoring agents may be added to provide a palable oral preparation.
- compositions may be preserved by the addition of an antioxidant such as ascorbic acid.
- Dispersible powders and granules of the invention suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, a suspending agent, and one or more preservatives.
- a dispersing or wetting agent e.g., sodium tartrate
- suspending agent e.g., sodium EDTA
- preservatives e.g., sodium bicarbonate, sodium bicarbonate, sodium bicarbonate, sodium bicarbonate
- compositions of the invention may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, such as olive oil or arachis oil, a mineral oil, such as liquid paraffin, or a mixture of these.
- emulsifying agents include naturally-occurring gums, such as gum acacia and gum tragacanth, naturally occurring phosphatides, such as soybean lecithin, esters or partial esters derived from fatty acids and hexitol anhydrides, such as sorbitan mono-oleate, and condensation products of these partial esters with ethylene oxide, such as polyoxyethylene sorbitan mono-oleate.
- naturally-occurring gums such as gum acacia and gum tragacanth
- naturally occurring phosphatides such as soybean lecithin
- esters or partial esters derived from fatty acids and hexitol anhydrides such as sorbitan mono-oleate
- condensation products of these partial esters with ethylene oxide such as polyoxyethylene sorbitan mono-oleate.
- emulsion may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, such as glycerol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, a flavoring or a
- compositions of the invention may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous or oleaginous suspension.
- a sterile injectable preparation such as a sterile injectable aqueous or oleaginous suspension.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non- toxic parenterally-acceptable diluent or solvent, such as a solution in 1,3-butanediol or prepared as a lyophyl-ized powder.
- a non- toxic parenterally-acceptable diluent or solvent such as a solution in 1,3-butanediol or prepared as a lyophyl-ized powder.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride
- sterile fixed oils may conventionally be employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid may likewise be used in the preparation of injectables.
- a time-release formulation intended for oral administration to humans may contain 20 to 200 moles of active material compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95% of the total compositions. It is preferred that
- an aqueous solution intended for intravenous infusion should contain from about 20 to about 50 moles of the active ingredient per milliliter of solution in order that infusion of a suitable volume at a rate of about 30 ml/hr can occur.
- formations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion.
- the active ingredient may also be administered as a bolus, electuary or paste.
- a tablet may be made by compression or moulding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder (e.g., povidone, gelatin,
- Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydroxypropylmethyl cellulose in varying proportions to provide the desired release profile.
- Tablets may optionally be provided with an enteric coating, to provide release in parts of the gut other than the stomach. This is particularly advantageous with the compounds of formula (I) as such compounds are susceptible to acid hydrolysis.
- Formulations suitable for topical administration in the mouth include lozenges comprising the active ingredient in a flavored basis, usually sucrose and acacia or tragacanth;
- pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- Formulations for rectal administration may be presented as a suppository with a suitable base comprising for example cocoa butter or a salicylate.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the ddPN ingredient such carriers as are known in the art to be appropriate.
- Formations suitable for parenteral administration include aqueous and non-aqueous isotonic sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile
- suspensions which may include suspending agents and thickening agents.
- the formulations may be presented in unit-dose or multi- dose sealed containers, for example, ampoules and vials, and may be sorted in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- sterile liquid carrier for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Preferred unit dosage formulations are those containing a daily dose or unit, daily sub-dose, or an appropriate fraction thereof, of an adenosine kinase inhibitor compound.
- the specific dose level for any particular patient will depend on a variety of factors including the activity of the specific compound employed; the age, body weight, general health, sex and diet of the individual being treated; the time and route of administration; the rate of excretion; other drugs which have previously been administered; and the severity of the particular disease undergoing therapy, as is well understood by those skilled in the art.
- the method may be used following thrombolysis for coronary occlusion.
- the compound would be given as a sterile injectable preparation with water or isotonic sodium chloride as the solvent.
- the solution can be administered intravenously or directly into the coronary artery at the time of left heart catheter!zation or into a carotid artery. The rate of
- administration could vary from 1 to 20 nmole/min/kg with, for example, an infusion volume of 30 ml/hr. Duration of therapy would typically be about 96 hours.
- Angina and early myocardial infarcts can be treated by intravenous administration using a sterile injectable preparation using the rates discussed above.
- Capsules comprising adenosine kinase inhibitors suitable for oral administration according to the methods of the present invention may be prepared as follows: (1) for a 10,000 capsule preparation: 1500g of adenosine kinase inhibitor is blended with other ingredients (as described above) and filled into capsules which are suitable for administration depending on dose, from about 4 capsules per day (1 per 6 hours) to about 8 capsules per day (2 capsules per 6 hours), to an adult human.
- This material was prepared by tosylation of 2', 3'-(1- methylethylidene)iniosine and subsequent reaction with sodium azide in DMSO as described by Hampton, A.; J. Org. Chem., 1968, 11:1220.
- Example 3 The N 6 -substituted isopropylidene azide (Example 3) (1.0 g) was dissolved in HCO 2 H (10-20 ml) and diluted with an equal volume of H 2 0. The reaction was followed by TLC. After disappearance of the starting material (12-48 hr), the reaction mixture was evaporated under vacuum, coevaporated with H 2 O (3x) then ETOH (2x). The residue was crystallized from H 2 O, alcohol or mixtures.
- the above-identified compound may be prepared as described: Ikshara, M.; Kaneko, M.; Sagi, M.; Tetrahedron, 1970, 26:5757.
- N 6 -formyl derivative was deformylated by slurrying in MeOH, adding saturated methanolic ammonia (80 ml) and warming until homogenous. After 15 minutes the solution was evaporated, the residue recrystallized from EtOH and dried to give the title compound; 0.900 g (60% yield); m.p. 166-168°C.
- Example 26 The isopropylidene diazide of Example 26 (1.00 g, 3.15 mmol) was deblocked as described under Example 4 and recrystallized from H 2 O; 760 mg (85% yield); m.p. 128-130°.
- Example 30 To a cold (0oC) solution of the alcohol (Example 30) (6.0 g, 0.015 mol) in dry pyridine (40 ml) was added with stirring, p-toluenesulfonyl chloride (6.96 g, 0.36 mol). The solution was sealed and stored at 0-10oC for 72 hours then poured with
- This compound may also be prepared as described: Tollman et al., J. Amer. Chem. Soc.. 1969, 91:2102.
- Example 35 (232 mg; 1 mmol) in anhydrous THF (5 mL) was cooled to -78 °C under argon and a solution of n-butyl lithium (1.3 mL of 2.31 M) was added at such a rate that the temperature of the reaction mixture remained below -72°C. After stirring the reaction mixture at -78 °C for 45 minutes, a solution of ethyl chloroformate (0.15 mL) in THF (2 ml) was added slowly,
- 5-Deoxy-D-ribofuranose (8.g, 60 mmole) was dissolved in DMF (25 ml) and to the solution was added dimethoxypropane (10 ml) and p-toluenesulfonic acid (150 mg). The reaction was stirred overnight then neutralized with Amberlite 400 (OH resin. The mixture was filtered, concentrated and the residue chromatographed on SiO 2 gel using 15:1 CH 2 Cl 2 -MeOH. The
- Example 51 The crude 5-azido-5-deoxyribose (Example 51) was dissolved in dry DMF (10 mL) and treated with 2,2- dimethoxypropane (10 mL and p-toluenesulfonic acid (100 mg). The solution was stirred at room temperature for 20 hours then evaporated under high vacuum. The residue was chromatographed over SiO 2 gel using 3:1
- the sugar aldehyde from Example 53 (100 mmole) was dissolve in anhydrous THF and treated with a commercially available solution of methyl magnesium bromide (100 m mol) under anhydrous conditions. After 2 hours of stirring at room temperature, a saturated solution of ammonium chloride in water (180 mL) was added. The organic layer was separated and the aqueous layer wa extracted with ether (2 x 100 mL). The combined organic layers were dried and evaporated to obtain an oily product whose NMR was consistent with methyl-6-deoxy-2,3-isopropylidene-D- allofuranoside. The crude product was dissolved in pyridine (50 ml) and treated with benzoic anhydride (120 mmole).
- the benzoylated sugar (Example 54) was dissolved in a mixture of dry DMF (20 ml), 2,2-dimethoxypropane (20 ml) and p-toluenesulfonic acid (200 mg) and stirred at room temperature with the exclusion of moisture. The reaction was complete within two hours as evidenced by the absence of the starting material (TLC). The acid was neutralized by strongly basic ion exchange resin and the resin removed by filtration and washed. The combined washings and filtrate were evaporated under high vacuum and the residue was purified by chromatography. The pure product obtained was a glassy solid. IR and NMR were consistent with the title compound.
- the protected nucleoside was deblocked by dissolving in 90% trifluoroacetic acid and stirring for 2 hours. The solvent was evaporated and chased with methanol (3x). The product was crystallized from EtOH or EtOAc.
- Example 54 (50 mg), in dry pyridine (5 ml), acetic anhydride (0.5 ml) was added with stirring. The reaction mixture was allowed to warm to room temperature over a period of 1 hour at which time the reaction was complete. The flask was reimmersed into the cooling bath and 15 ml of methanol was added to the reaction mixture to neutralize unreacted acetic anhydride. The solvent was evaporated under reduced pressure and the residue was purified by a short column chromatography to give the above- identified product, m.p. 160-163°C.
- phenylboronic acid 250 mg in dry diglyme (10 ml) was added palladium-tetrakis-triphenylphosphine (30 mg), followed by aqueous Na 2 CO 3 solution (0.2 ml of 2M solution).
- the reaction mixture was heated to 90 °C under anhydrous conditions for 6 hours.
- the solvent was evaporated under high vacuum and the residue was purified by HPLC on a reverse phase C-18 column.
- the purified intermediate was treated with 2 ml of trifluoracetic acid (80%) and stirred for 15 minutes then evaporated under high vacuum and the residue crystallized from ethanol; yield 20 mg, m.p. 163-164°C.
- Example 127 The 4-chloro compound, Example 127, was heated in a steel bomb with methanolic ammonia at 120oC for 12 hours followed by the usual work.up (see Example 85). The product was obtained as a white crystalline solid; m.p. 206-208°C.
- the above-identified compounds may be prepared from the corresponding pyrazolo[3,4-d]-pyrimidones analogously to the procedure described in Example 2.
- Example 171 To a solution of the isopropylidene alcohol (Example 171) (3.0 g, 7.74 mmol) in pyridine (18 ml) at 0°C was added p- toluenesulfonyl chloride (1.77 g, 9.30 mmol). The reaction was held at 0°C for 3 hours then poured into 160 ml of cold H 2 O with stirring. The mixture was allowed to settle, the H 2 O decanted and the residue dissolved in CH 2 Cl 2 . The CH 2 Cl 2 solution was washed with 0.5 N H 2 SO 4 , 5% aqueous K 2 CO 3 and dried (Na 2 SO 4 ).
- Example 172 To a warm stirred solution of NaN 3 (7.69 g, 0.12 moles) in DMSO (70 ml) was added the tosylate (Example 172) (16.0 g, 0.03 mol). The solution was rapidly heated to 80 °C and maintained at this temperature for 45 minutes. After cooling, the reaction mixture was added with stirring to H 2 O (600 ml). The mixture was extracted 4x with CHCl 3 (75 ml) and the combined CHCl 3 extracts were washed with H 2 O, dilute brine, dried (Na 2 SO 4 ) and
- Example 173 or 177 The above-identified compounds were prepared analogously to the procedure described for Example 2 from the pyrimidin-4-one (Example 173 or 177). The title compounds were obtained as unstable yellow oils and used immediately in the next step.
- the reactions using amines were worked up in the following manner.
- the reaction mixture was evaporated, the residue dissolved in CH 2 Cl 2 and the solution washed with aqueous NaHCO 3 , then H 2 O and dried (Na 2 SO 4 ).
- Concentration of the CH 2 Cl 2 solution and chromatography of the residue over Si0 2 gel using CH 2 Cl 2 - Me 2 CO mixtures gave the purified isopropylidene N 4 -substituted compounds.
- the isopropylidene 4-amino compounds were isolated by evaporating the reaction mixture and recrystallizing the residue from EtOH.
- the above-identified compounds were prepared from the 5'-azides (Examples 205-221) by catalytic hydrogenation as described in Examples 15-20 (method A) or triphenylphosphine followed by ammonium hydroxide as described in Examples 82-83 (method B).
- the salts were prepared by standard methods.
- SiO 2 gel was loaded on a column of SiO 2 gel and the product eluted with CH 2 Cl 2 -MeOH mixtures. The appropriate fractions more combined and evaporated to yield the title compounds.
- the above-identified compounds may be prepared by reduction of the 5-azido ribosides listed in Examples 238-239 by catalytic hydrogenation as described in Examples 15-20 or by treatment with triphenylphosphine and ammonium hydroxide as described in
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Novel compounds which selectively inhibit adenosine kinase and methods of preparing adenosine kinase inhibitors are provided. Also provided are methods of treating various conditions which may be ameliorated by increased local concentrations of adenosine using adenosine kinase inhibitors.
Description
ADENOSINE KINASE INHIBITORS
CROSS-REFERENCE TO RELATED APPLICATIONS
This application is a continuation-in-part of Serial No. 07/647,117, filed January 23, 1991, which is a continuation-in-part of Serial No. 466,979, filed January 18, 1990; which is a continuation-in-part of Serial No. 408,707, filed September 15, 1989; the disclosures of these applications are incorporated herein by reference.
FIELD OF INVENTION
This invention relates to adenosine kinase inhibitors and to novel nucleoside analogs, specifically to purine, pyrrolo [2,3-d]pyrimidine and pyrazolo[3,4-d]pyrimidine nucleoside analogs having activity as adenosine kinase inhibitors. The invention also relates to the preparation and use of these and other adenosine kinase inhibitors in the treatment of cardiovascular, and cerebrovascular diseases, inflammation and other diseaseswhich can be regulated by increasing the local concentration of adenosine.
BACKGROUND OF THE INVENTION
Adenosine has been reported to have cardioprotective
(Olafsson et al., Circulation, 1987, 76:1135-1145) and neuroprotective properties (Dragunow and Faull, Trends in Pharmacol. Sci., 1988, 9:193; Marangos, Medical Hypothesis, 1990, 32:45). It is reportedly released from cells in response to alterations in the supply of or demand for oxygen (Schrader, Circulation, 1990, 81: 389-391), is said to be a potent vasodilator, and is believed to be involved in the metabolic regulation of blood flo (Berne, Circ. Res., 1980, 47:808-813). However, adenosine has a short half life (< 1 sec) in human blood (Moser, et al., Am. J. Physiol.. 1989, 256:C799-C806), and therefore high doses of adenosine would need to be administered continuously to achieve effective levels. Adenosine has been reported to exhibit negative inotropic, chronotropic and dromotropic effects
(Belardinelli et al., Prog, in Cardiovasc. Diseases, 1989, 32:73-97) and to cause coronary steal by preferentially dilating vessels in nonischemic regions. Consequently, high doses of adenosine are toxic and severely limit its therapeutic potential. However, it is believed that by increasing adenosine concentration locally, i.e. at the target site within the target tissue, the beneficial effects of adenosine can be provided without the toxic systemic effects.
Adenosine kinase is a cytosolic enzyme which catalyzes the phosphorylation of adenosine to AMP. Inhibition of adenosine kinase can potentially reduce the ability of the cell to utilize adenosine, leading to increased adenosine outside of the cell where it is pharmacologically active. However, the regulation of adenosine concentration is complex and involves other adenosine- metabolizing enzymes each with different kinetic properties and mechanisms of regulation. Adenosine can also be deaminated to inosine by adenosine deaminase (ADA) and condensed with L-horoocysteine to S-adenosylhomocysteine (SAH) by SAH hydrolase. The role of each of these enzymes in modulating adenosine
concentration is dependent on the prevailing physiological conditions, is tissue specific and is not well understood.
A number of nucleosides including purine, pyrrolo[2,3- djpyrimidine and pyrazolo[3,4-d]pyrimidine analogs have been evaluated for inhibition of adenosine kinase but were reported to have Ki's of greater than 800 nM (Caldwell and Henderson Cancer Chemother. Rep., 1971 2:237-246; Miller et al., J. Biol. Chem., 1979, 254:2346-2352). A few compounds have been reported as potent inhibitors of adenosine kinase with K-'s of less than 100 nM. These are the purine nucleosides, δ'-amino-S'-deoxyadenosine (Miller et al., J. Biol. Chem., 1979, 154:2346-2352) and 1,12-bis(adenosin-N6-yl)dodecane (Prescott et al., Nucleosides &
Nucleotides, 1989, 8:297), and the pyrrolopyrimidine nucleosides,
5-iodotubercidin (Henderson et al., Cancer Chemotherapy Rep. Part 2, 1972, 3:71-85; Bontemps et al., Proc. Natl. Acad. Sci. USA, 1983, 80: 2829-2833; Davies et al., Biochem. Pharmacol., 1986, 35:3021-3029) and 5'-deoxy-5-iodotubercidin (Davies et al.,
Biochem. Pharmacol., 1984, 33:347-355; Davies et al., Biochem. Pharmacol., 1986, 35:3021-3029).
Some of these compounds have been used to evaluate whether adenosine kinase inhibition might lead to increased extracellular adenosine concentrations. In rat cardiomyocytes, inhibition of adenosine deaminase by 2'-deoxycoformycin was reported to have no effect on adenosine release from the cells. In contrast,
inhibition of ADA together with adenosine kinase by 5'-amino-5'- deoxyadenosine resulted in a 6-fold increase in adenosine release (Zoref-Shani et al., J. Mol. Cell. Cardiol., 1988, 20:23-33).
The effects of the adenosine kinase inhibitor alone were not reported. Similar results were reported in isolated guinea pig hearts; in these studies addition of 5'-amino-5'-deoxyadenosine to the perfusion medium, in the presence of EHNA to inhibit deamination, was reported to result in a 15-fold increase of adenosine release (Schrader in Regulatory Function of Adenosine; (Berne et al.) eds. pp. 133-156, 1983). These effects were not apparent in the absence of ADA inhibition and other studies using isolated rat hearts perfused with 5-iodotubercidin alone, have reported no increase in perfusate adenosine concentration under
normoxic conditions (Newby et al., Biochem. J.. 1983, 214:317- 323) or under hypoxic, anoxic or ischemic conditions (Achtenberg et al., Biochem. J., 1986, 235:13-17). In other studies, adenosine release has been measured in neuroblastoma cells in culture and compared with that of a variant deficient in
adenosine kinase (AK). The AK cells used in this study were said to release adenosine at an accelerated rate; the concentration of adenosine in the growth medium was reported to be elevated compared to the normal cells (Green, J. Supramol.
Structure, 1980, 13: 175-182). In rat and guinea pig brain slices, adenosine uptake was reportedly inhibited by the
adenosine kinase inhibitors, 5-iodotubercidin and 5'-deoxy-5-iodotubercidin (Davis et al., Biochem. Pharmacol., 1984, 33:347-355). However, inhibition of uptake and intracellular trapping via phosphorylation does not necessarily result in increased extracellular adenosine, since the adenosine could enter other metabolic pathways or the percentage of adenosine being phosphorylated could be insignificant compared to the total adenosine removed.
The effects of adenosine and certain inhibitors of adenosine catabolism, including 5-iodotubericidin were evaluated in an experiemental model in which dog hearts were subjected to
ischemia and reperfusion; 5-iodotubericidin was reported to have inconsistent effects. (Wu, et al., Cytobios, 1987, 50:7-12).
Although the adenosine kinase inhibitors, 5'-amino-5'- deoxyadenosine and 5-iodotubercidin have been widely used in experimental models, the susceptibility of 5'-amino-5'- deoxyadenosine to deamination, and hence its potentially short half life, and the cytotoxicity of 5-iodotubercidin make their clinical utility limited and may limit interpretations based on these compounds. The pyrrolo[2,3-d]pyrimidines, 5-iodotubercidi and 5'-deoxy-5-iodotubercidin have been reported to cause pronounced general flaccidity and much-reduced spontaneous locomoto activity in mice, interpreted to be skeletal muscle relaxation; to cause hypothermia in mice; and to decrease blood pressure and heart rate in anesthetized rats (Daves et al., Biochem.
Pharmacol., 1984, 33:347-355; Daves et al., Biochem. Pharmacol., 1986, 35:3021-3029; U.S. Patent No. 4,455,420). The skeletal muscle effects of these compounds have been poorly documented, while the other effects were considered significant toxicities. It is believed that studies using these compounds were curtailed due to these toxicities and also because of their limited availability.
SUMMARY OF THE INVENTION
The present invention is directed to novel compounds which are potent and selective adenosine kinase inhibitors.
In one aspect, the present invention is directed to certain novel compounds which inhibit adenosine kinase, to the preparation of these compounds, and to the in vitro and in vivo adenosine kinase inhibition activity of these compounds. Another aspect of the present invention is directed to the clinical use of adenosine kinase inhibitors as a method of increasing adenosine concentrations in biological systems. In vivo inhibition of adenosine kinase prevents phosphorylation of adenosine resulting in higher local concentrations of endogenous adenosine. As a result of the very short half-life of adenosine and very low quantities of adenosine in tissues, this effect is most
pronounced in regions producing the most adenosine such as ischemic regions. Hence, the beneficial effects of adenosine are enhanced in a site and event specific manner and toxic systemic effects are reduced.
In particular, in one preferred aspect, the present
invention is directed to novel nucleoside analogs which comprise a 5'-modified ribose linked to a substituted purine, pyrrolo[2,3-d]pyrimidine, or pyrazolo[3,4-d]pyrimidine base. Certain preferred compounds within these groups possess potencies many times greater than previously described inhibitors of adenosine kinase. The compounds of the present invention possess advantages for pharmaceutical use such as enhanced pharmacological selectivity, efficacy, bioavailability, ease of manufacture and compound
stability. This invention also discloses novel processes for the preparation of these compounds.
The novel compounds of the present invention and other adenosine kinase inhibitors may be used clinically to treat medical conditions where an increased localized adenosine
concentration is beneficial. Accordingly, the present invention is directed to the prophylactic and affirmative treatment of ischemic conditions such as myocardial infarction, angina, percutaneous transluminal coronary angiography (PTCA), stroke, otherthrombotic and embolic conditions, neurological conditions such as seizures and psychosis, and other conditions benefitted by enhanced adenosine levels such as inflammation, arthritis, autoimmune diseases, cardiac arrhythmias, ulcers and irritable bowel syndrome. These compounds are useful as muscle relaxants and also in inducing sleep and in treating anxiety.
The present invention is also directed to prodrugs and pharmaceutically acceptable salts of the compounds described herein and to pharmaceutical compositions suitable for different routes of drug administration and which comprise a
therapeutically effective amount of an adenosine kinase inhibitor compound described herein admixed with a pharmacologically
acceptable carrier.
Definitions
In accordance with the present invention and as used herein, the following terms, are defined with the following meanings, unless explicitly stated otherwise.
The term "hydrocarbyl" refers to an organic radical
comprised of carbon chains to which hydrogen and other elements are attached. The term includes alkyl, alkenyl, alkynyl and aryl groups, groups which have a mixture of saturated and unsaturated bonds, carbocyclic rings and includes combinations of such groups. It may refer to straight-chain, branched-chain cyclic structures or combinations thereof.
The term "aryl" refers aromatic groups which have at least one ring having a conjugated pi electron system and includes carbocyclic aryl, heterocyclic aryl and biaryl groups, all of which may be optionally substituted.
Carbocyclic aryl groups are groups wherein the ring atoms on the aromatic ring are carbon atoms. Carbocyclic aryl groups include monocyclic carbocyclic aryl groups and optionally
substituted naphthyl groups.
The term "monocyclic carbocyclic aryl" refers to optionally substituted phenyl, being preferably phenyl or phenyl substituted by one to three substituents, such being advantageously lower alkyl, hydroxy, lower alkoxy, lower alkanoyloxy, halogen, cyano, trihalomethyl, lower acylamino or lower alkoxycarbonyl .
"Optionally substituted naphthyl" refers to 1- or 2- naphthyl or 1- or 2-naphthyl preferably substituted by lower alkyl, lower alkoxy or halogen.
Heterocyclic aryl groups are groups having from 1 to 3 heteroatoms as ring atoms in the aromatic ring and the remainder of the ring atoms carbon atoms. Suitable heteroatoms include oxygen, sulfur, and nitrogen, and include furanyl, thienyl, pyridyl, pyrrolyl, N-lower alkyl pyrrolo, pyrimidyl, pyrazinyl, imidazolyl, and the like, all optionally substituted.
Optionally substituted furanyl represents 2- or 3-furanyl or 2- or 3-furanyl preferably substituted by lower alkyl or halogen.
Optionally substituted pyridyl represents 2-, 3- or 4- pyridyl or 2-, 3- or 4-pyridyl preferably substituted by lower alkyl or halogen.
Optionally substituted thienyl represents 2- or 3-thienyl, or 2- or 3-thienyl preferably substituted by lower alkyl or halogen.
The term "biaryl" represents phenyl substituted by
carbocyclic aryl or heterocyclic aryl as defined herein, ortho, meta or para to the point of attachment of the phenyl ring, advantageously para; biaryl is also represented as the -C6H4-Ar substituent where Ar is aryl.
The term "aralkyl" refers to an alkyl group substituted wit an aryl group. Suitable aralkyl groups include benzyl, picolyl, and the like, and may be optionally substituted.
The term "lower" referred to herein in connection with organic radicals or compounds respectively defines such with up to and including 7, preferably up to and including 4 and
advantageously one or two carbon atoms. Such groups may be straight chain or branched.
The terms (a) "alkyl amino", (b) "arylamino", and (c)
"aralkylamino", respectively, refer to the groups -NRR' wherein respectively, (a) R is alkyl and R1 is hydrogen or alkyl; (b) R is aryl and R1 is hydrogen or aryl, and (c) R is aralkyl and R' is hydrogen or aralkyl.
The term "acyl" refers to hydrocarbyl-CO- or HCO-.
The terms "acylamino" refers to RC(O)NCR)- and (RCO)2N-respectively, wherein each R is independently hydrogen or hydrocarbyl.
The term "α-alkoxyalkylidene" refers to hydrocarbyl-O-CR (an orthoester) wherein R is hydrogen or hydrocarbyl.
The term "hydrocarbyloxycarbonyloxy" refers to the group ROC(O)O- wherein R is hydrocarbyl.
The term "lower carboalkoxymethyl" or "lower
hydrocarbyloxycarbonymethyl" refers to hydrocarbyl-OC(O)CH2- with the hydrocarbyl group containing ten or less carbon atoms.
The term "carbonyl" refers to -C(O)-.
The term "carboxamide" or "carboxamido" refers to -CONR2 wherein each R is independently hydrogen or hydrocarbyl.
The term "lower hydrocarbyl" refers to any hydrocarbyl grou of ten or less carbon atoms.
The term "alkyl" refers to saturated aliphatic groups including straight-chain, branched chain and cyclic groups.
The term "alkenyl" refers to unsaturated hydrocarbyl groups which contain at least one carbon-carbon double bond and include straight-chain, branched-chain and cyclic groups.
The term "alkynyl" refers to unsaturated hydrocarbyl groups which contain at least one carbon-carbon triple bond and include straight-chain, branched-chain and cyclic groups.
The term "halogen" refers to fluorine, chlorine, bromine or iodine.
The term "hydrocarbyloxycarbonylamino" refers to a urethane, hydrocarbyl-O-CONR- wherein R is H or hydrocarbyl and wherein each hydrocarbyl is independently selected.
The term "di(hydrocarbyloxycarbonyl)amino" refers to
(hydrocarbyl-O-CO)2N- wherein each hydrocarbyl is independently selected.
The term "hydrocarbylamino" refers to -NRR' wherein R is hydrocarbyl and R' is independently selected hydrocarbyl or hydrogen.
The term "mercapto" refers to SH or a tautomeric form.
The term "methine" refers to .

The term "methylene" refers to -CH2-.
The term "alkylene" refers to a divalent straight chain or branched chain saturated aliphatic radical.
The term "oxy" refers to -O- (oxygen).
The term "thio" refers to -S- (sulfur).
The term "prodrug" as used herein refers to any compound that has less intrinsic activity than the "drug" but when administered to a biological system generates the "drug" substance either as a result of spontaneous chemical reaction or by enzyme catalyzed or metabolic reaction. Reference is made to various prodrugs such as acyl esters, carbonates, and urethanes, included herein as examples. The groups illustrated are exemplary, not exhaustive and one skilled in the art could prepare other known varieties of prodrugs. Such prodrugs of the compounds of
Formula I, fall within the scope of the present invention.
The term "pharmaceutically acceptable salt" includes salts of compounds of Formula I derived from the combination of a compound of this invention and an organic or inorganic acid. The compounds of Formula I are useful in both free base and salt form. In practice the use of salt form amounts to use of base form; both forms are within the scope of the present invention.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts the effects of the adenosine kinase inhibitor GP-1-238 on mean arterial pressure, heart rate and bod temperature following intravenous administration to anesthetized or conscious rats.
Figure 2 depicts the dose-dependent inhibition of neutrophil adhesion to endothelial cells by the adenosine kinase inhibitors GP-1-272 and GP-1-456 and the reversal of this inhibition by co- treatment with adenosine deaminase ("ADA").
Figure 3 depicts the dose-dependent inhibition of
contraction in the isolated ileum by the adenosine kinase
inhibitors (A) GP-1-515 and (B) GP-1-547 and the reversal of this inhibition by co-treatment with the adenosine receptor
antagonist, 8-sulfophenyltheophylline, or adenosine deaminase.
Figure 4 depicts (A) the dose-dependent inhibition of pentylenetetrazole (PTZ) induced seizures by the adenosine kinase inhibitor GP-1-456, and (B) the reversal of this inhibition by the central adenosine receptor antagonist theophylline but not the peripheral antagonist 8-sulfophenyltheophylline.
Figures 5 to 9 depict reaction schemes for preparing certain of these adenosine kinase inhibitors.
Figure 10 depicts the structures of certain preferred intermediates useful in the synthesis of adenosine kinase inhibitors.
DETAILED DESCRIPTION OF THE INVENTION
NOVEL ADENOSINE KINASE INHIBITORS
The present invention relates to novel adenosine kinase inhibitors which comprise compounds of the general formula I.

(I) wherein:
(a) A is oxygen, methylene or sulfur;
(b) B' is -(CH2)n-B wherein n is 1, 2, 3 or 4 and B is hydrogen, alkyl, alkoxy, amino, alkylamino, acylamino,
hydrocarbyloxycarbonylamino, mercapto, alkylthio, azido, cyano, halogen, or B' is alkenyl or alkynyl;
(c) C1 and C2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein c, is a single bond to C2 and C2 is carbonyl or α- alkoxyalkylidene;
(d) X is
 and Y is -N= or
and Y is -N= or
 ;
;
(e) D is hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl, alkynyl, haloalkyl, cyano, cyanoalkyl, acyl, carboxamido, a carboxylic acid or carboxylic acid ester group, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, amino, alkylamino arylamino, aralkylamino, acylamino, or nitro;
(f) E is hydrogen, halogen, alkyl, or alkylthio;
(g) F is alkyl, aryl, aralkyl, halogen, amino,
alkylamino, arylamino, aralkylamino, cyano, cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio; optionally substituted indolinyl or indolyl; pyrrolidinyl or piperazinyl; and
(h) G is hydrogen, halogen, lower alkyl, lower alkoxy, lower alkylamino or lower alkylthio; and pharmaceutically acceptable salts thereof; with the proviso that:
when A is oxygen and
(i) X is
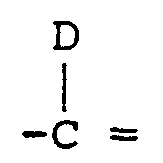 and Y is
and Y is
 , then if B' is methyl, D is halogen, cyano or carboxamido, F is amino, then G is not hydrogen; or if is hydrogen, then F is not amino; or (ii) X is
, then if B' is methyl, D is halogen, cyano or carboxamido, F is amino, then G is not hydrogen; or if is hydrogen, then F is not amino; or (ii) X is
 and Y is -N=, if B is hydrogen or halogen, D and G are hydrogen, then F is not amino; or when A is methylene, X is
and Y is -N=, if B is hydrogen or halogen, D and G are hydrogen, then F is not amino; or when A is methylene, X is
 , Y is
, Y is
 , B, D, E and G are hydrogen, then F is not amino.
, B, D, E and G are hydrogen, then F is not amino.

According to an alternative aspect of the present invention, novel adenosine kinase inhibitors are provided which have a 5'- group which comprises a hydroxyl or hydroxyl derivative.
However, it is believed due to their overall structures, those compounds which have a 5'-hydroxyl would not act as substrates for phosphorylation enzymes and, thus, would be unlikely to undergo 5'-phosphorylation or would be phosphorylated at an extremely slow rate.
One preferred group of these adenosine kinase inhibitors comprise compounds of the formula:

(a) A is oxygen, methylene or sulfur;
(b) B' is -(CH2)nB wherein n is 1, 2, 3 or 4 and B is hydroxy, acyloxy, hydrocarbyloxycarbonyloxy, or -OCONR2 wherein R is independently hydrocarbyl;
(c) C1 and C2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C1 is a single bond to C2 and C2 is carbonyl or α- alkoxyalkylidene;
(d) X is
 and Y is -N=;
and Y is -N=;
(e) D is halogen, aryl or aralkyl;
(f) F is alkyl, aryl, aralkyl, halogen, amino,
alkylamino, arylamino, aralkylamino, cyano, cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio, optionally substituted indolinyl or indolyl, pyrrolidinyl or piperazinyl; and
(g) G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio; and pharmaceutically acceptable salts
thereof; with the proviso that when A is oxygen and D is halogen, then F is not amino.
Another preferred group of these adenosine kinase inhibitor comprise compounds of the formula:

wherein:
(a) A is oxygen, methylene or sulfur;
(b) B' is -(CH2)nB wherein n is 1, 2, 3 or 4 and B is hydroxy, acyloxy, hydrocarbyloxycarbonyloxy, or -OCONR2 wherein R is hydrocarbyl;
(c) C1 and C2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C1 is a single bond to C2 and C2 is carbonyl or α-alkoxyalkylidene;
D
(d) X is
 and Y is
and Y is
 ;
;
(e) D is aryl or aralkyl;
(f) E is hydrogen, halogen, alkyl, or alkylthio;
(g) F is alkyl, aryl, aralkyl, halogen, amino, alkylamino, arylamino, aralkylamino, cyano, cyanoalkyl, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, optionall substituted indolinyl or indolyl, pyrrolidinyl or piperazinyl; and
(h) G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio; and pharmaceutically acceptable salts thereof; with the proviso that: when A is oxygen, D is
oxadiazolyl, triazolyl or triazinyl, E and G are both hydrogen, then F is not amino.
Also included within the present invention are adenosine kinase inhibitors which comprise modified purine nucleosides of the formula:

wherein
(a) A is oxygen, methylene or sulfur;
(b) B' is -CH2B wherein and B is amino, alkylamino, or acylamino;
(c) C, and C2 are each independently hydrogen, acyl,
hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C1 is a single bond to C2 and C2 is carbonyl or α- alkoxyalkylidene;
(d) X is -N= and Y is

(e) E is hydrogen, halogen, alkyl, amino, alkylamino, azido, acylamino, alkoxy or alkylthio;
(f) F is halogen, amino, alkylamino, arylamino,
aralkylamino, cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio, alkyl, aryl, aralkyl, optionally
substituted indolinyl or indolyl, pyrrolidinyl or piperazinyl; and
(g) G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio and pharmaceutical acceptable salts thereof; with the proviso that:
when A is oxygen, B is amino or hydrocarbylamino, E and G are hydrogen, then F is not amino.
According to a further aspect of the present invention, novel adenosine kinase inhibitors are provided that comprise dimeric compounds of the formula:

wherein
(a) A and A' are independently oxygen, methylene or sulfur;
(b) B' and B" are independently -(CH2)nB wherein n is independently 1, 2, 3 or 4 and B is independently hydrogen, hydroxy, alkyl, alkoxy, amino, alkylamino, acylamino,
hydrocarbyloxycarbonylamino, mercapto, alkylthio, azido, or either or both of B' or B" is independently alkenyl or alkynyl;
(c) C1 and C1' and C2 and C2' are each independently
hydrogen, acyl, hydrocarbyloxycarbonyl, or C1 and C2 or C1' and C2' taken together form a 5-membered ring wherein C, or C1' is a single bond to C2 or C2' and C2 or C2' is carbonyl or α-alkoxyalkylidene;
(d) X and X' are each independently
 or -N=; and Y and Y'
are each independently -N= or
or -N=; and Y and Y'
are each independently -N= or
 provided that either of and X and Y or X' and Y' are not both -N=;
provided that either of and X and Y or X' and Y' are not both -N=;
(e) D is independently hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl, alkynyl, haloalkyl, cyano, cyanoalkyl, acyl, carboxamido, a carboxylic acid or corresponding carboxylic acid ester group, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, amino, alkylamino, arylamino, aralkylamino acylamino or nitro;
(f) E is independently hydrogen, halogen, alkyl, or
alkylthio;
(g) L is an optionally substituted piperazinyl divalent radical or -NH(ALKL)NH- wherein ALKL is a divalent alkylene radical of 2 to 24 carbon atoms; and
(h) G and G' are each independently hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkoxy; or pharmaceutically
acceptable salts thereof; with the proviso that if B is OH, then X and X' are not both -N=.
In general, preferred are compounds where G is hydrogen, halogen, alkyl or alkylthio. Especially preferred G groups include hydrogen. Preferred C1 and C2 groups include hydrogen and acetyl.
Preferred E groups include hydrogen or halogen, especially preferred are compounds where E is hydrogen.
Preferred are compounds where A is oxygen.
Preferred are compounds where D is hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl or alkynyl, cyano, cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio, amino, alkylamino, arylamino, aralkylamino, carboxamido, or
hydrocarbyloxycarbonyl. Especially preferred D groups include hydrogen, halogen, alkyl, aryl, aralkyl, cyano, alkoxy, aryloxy, aralkoxy, alkenyl or alkynyl, more preferably hydrogen, halogen, aryl, cyano, alkoxy or aryloxy. A particularly preferred group of compounds include those wherein D is hydrogen, halogen or aryl. According to one preferred aspect, D is aryl such as heterocyclic aryl or monocyclic carbocyclic aryl, such as
optionally substitued phenyl.
Preferred compounds include those where B' is -(CH2)nB, and n is 1 or 2, more preferably n is 1. B may preferably include hydrogen, halogen, alkyl, amino, alkylamino, alkoxy, mercapto, alkylthio, azido or cyano; more preferably B is hydrogen,
halogen, lower alkyl, amino, lower alkylamino, azido or cyano. Particularly preferred B groups include hydrogen, amino or azido. Also preferred are compounds wherein B' is vinyl, ethynyl, or propargyl.
Preferred F groups include halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or
aralkyl, more preferably amino or arylamino. Especially, preferred F groups include optionally substituted anilino.
A. Preferred Compounds
The compounds of the present invention contain asymmetric carbon atoms and hence can exist as stereoisomers, both enantio- mers and diastereomers. The individual preferred stereoisomers and mixtures thereof are considered to fall within the scope of the present invention. The compounds described by Formula I contain a 5-modified 1-β-D-ribofuranosyl group and that isomer comprises a particularly preferred diastereomeric and enantiomeric form for compounds of the present invention. Aptly, the synthetic examples cited herein provide the most preferred isomer. It is evident that in addition to the sugar moiety, additional asymmetric carbons may be present in compounds of Formula I, being present in moieties B', C1 or C2 or the
substituted heterocyclic purine, pyrrolo[2,3-d]pyrimidine or pyrazolo[3,4-d]pyrimidine ring. In this event, both of the resulting diastereomers are considered to fall within the scope of the present invention.
It is noted that compounds of formula I where B is hydroxy (i.e. a 5'-hydroxyl moiety) are in many cases potent inhibitors of adenosine kinase. The use of compounds having formula I wherein B' replaced by -CH2OH, as adenosine kinase inhibitors are
included in the scope of this invention. However, since some of these compounds may be phosphorylated in vivo and since the resulting 5'-phosphates may be toxic, mutagenic or teratogenic, 5'-hydroxy compounds which can serve as substrates for
phosphorylation enzymes may not comprise preferred compounds for clinical or therapeutic use. An important aspect of the novel compounds of the present invention is that these preferred compounds are either non-phosphorylatable at the 5' position or are not substrates of enzymes that lead to phosphorylation.
(i) Preferred Pyrazolo[S,4-dlpyrimidines
Preferred adenosine kinase inhibitor compounds of the present invention include certain pyrazolo[3,4-d]pyrimidine compounds of Formulas I and II.
Preferred pyrazolo[3,4-d]pyrimidine compunds of Formula I include those where G is hydrogen and A is oxygen. Preferred D groups include hydrogen, alkyl, aryl, aralkyl, cyano, alkoxy, aryloxy, aralkoxy, alkenyl or alkynyl, more preferably hydrogen, halogen, aryl, cyano, alkoxy or aryloxy, more particularly hydrogen, halogen or aryl. An especially preferred group of compounds includes those where D is aryl, especially heterocyclic aryl or monocyclic carbocyclic aryl, more preferably optionally substituted phenyl. Preferred B' groups include -(CH2)nB wherein B is hydrogen, halogen, alkyl, amino, alkylamino, alkoxy,
mercapto, alkylthio, azido or cyano; more preferably B is
hydrogen, halogen, lower alkyl, amino, lower alkylamino, azido or cyano. Particularly preferred B groups include hydrogen, amino or azido. Preferably, n is 1 or 2, more preferably 1. Other preferred B* groups include vinyl and ethynyl. Preferred are compounds of formula I wherein F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl, more preferably amino or arylamino. Certain preferred compounds include F groups which comprise optionally substituted anilino.
Examples of preferred pyrazolo[3,4-d]pyrimidine compounds include those noted as GP-1-515, GP-1-547, GP-1-560, GP-1-665, GP-1-666 GP-1-667, GP-1-695, and GP-1-704.
Preferred pyrazolo[3,4-d]pyrimidine compunds of Formula II include those where G is hydrogen and A is oxygen. Preferred D groups include aryl. Prefered aryl groups include heterocyclic carbocyclic aryl groups, especially optionally substituted phenyl. Preferred F groups include halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl, or aralkyl, more preferably amino or arylamino. Certain preferred compounds of Formula II may include F groups which comprise optionally substituted anilino groups.
(ii) Preferred Pyrrolo[2,3-d]pyrimidines
Preferred adenosine kinase compounds of the present
invention include pyrrolo[2,3-d]pyrimidine compounds of Formulas I and II.
Preferred pyrrolo[2,3-d]pyrimidine compounds of Formula I include those wherein G is hydrogen. Preferred are compounds wherein E is hydrogen or halogen; more preferably E is hydrogen. Preferred are compounds where A is oxygen. Preferred compounds include those where D is hydrogen, halogen, alkyl, aryl, aralkyl, cyano, alkenyl or alkynyl, more preferably hydrogen, halogen or aryl. An especially preferred group of compounds includes those where D is aryl, especially heterocyclic aryl or monocyclic carbocyclic aryl, especially optionally substituted phenyl.
Preferred B' groups include -(CH2)nB wherein n is 1 or 2,
preferably 1. Preferably, B is hydrogen, halogen, alkyl, amino, alkylamino, alkoxy, mercapto, alkylthio, azido or cyano, more preferably B is hydrogen, halogen, lower alkyl, amino, lower alkylamino, lower alkoxy, lower alkylthio, or azido, more
particularly hydrogen, lower alkyl, amino, lower alkylamino, or azido. Especially preferred B groups include hydrogen, amino or azido. Other preferred B' groups include vinyl and ethynyl.
Preferred pyrrolo[2,3-d]pyrimidine compounds of Formula I include those wherein F is halogen, amino, alkylamino, arylamino,
aralkylamino, alkylthio, aralkylthio, alkyl, aryl or aralkyl,
more preferably amino or arylamino. Certain preferred compounds include F groups which comprise optionally substituted anilino. Examples of preferred pyrrolo[2, 3-d]pyrimidine compounds include those noted as GP-1-448, GP-1-606, GP-1-608, GP-1-639, GP-1-683, GP-1-684, GP-1-691, GP-1-711, GP-1-714, and GP-1-718.
Preferred pyrrolo[2, 3-d]pyrimidines of Formula II include those where G is hydrogen and A is oxygen. Preferably E is hydrogen or halogen, more preferably hydrogen. Preferred D groups include aryl. Preferred aryl groups include heterocyclic aryl groups and monocyclic carbocyclic aryl groups, especially optionally substitued phenyl. Preferred heterocyclic aryl groups include 2-furanyl, 2-thienyl and 3-thienyl.
(iii) Preferred Purines
Preferred purine compounds include those where G is
hydrogen, halogen, lower alkyl or lower alkylthio, more
preferably hydrogen. Preferred are compounds wherein A is oxygen. Preferred E groups include hydrogen, halogen or
alkylthio. Preferred are compounds wherein B is amino.
Preferred F groups include halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl, more preferably amino or arylamino.
(iv) Preferred Dimer Compounds
Preferred dimeric compounds include those which comprises dimers of the above-described pyrazolo[3,4-d]pyrimidines, the pyrrolo[2,3-d]-pyrimidines and purines. These dimers may comprise monomeric units which are the same or different.
SYNTHESIS OF PREFERRED COMPOUNDS
A. General Synthetic Methods
The present invention also directed to processes for preparing compounds of Formula I. Disclosed herein are general synthetic routes for preparing variously substituted purine nucleosides or pyrrolo[2,3-d]pyrimidine nucleosides, including a novel and improved synthesis of 5'-deoxy- 5-iodotubercidin; and pyrazolo[3,4-d]pyrimidine nucleosides of the present invention.
A process for preparing 5'-azido, 5'-amino and 5'-deoxy analogs of N6- substituted purine ribosides is depicted in Figure 5. The protected azide (2a), prepared from 2',3'-O-isopropylideneinosine, is activated for nucleophilic attack at position six by chlorination with thionyl chloride/
dimethylformamide. Other standard reagents may also be used to activate position six of 2. such as thionyl bromide, phosphorous oxychloride, triphenylphosphine dibromide-thiophenol-potassium permanganate or hexamethyldisilazane-ammonium sulfate. The chloride (3) or other activated intermediate (Br, RSO2, R3SiO,
etc.) is then reacted with ammonia or an appropriate amine such as aniline, piperazine or indoline in solvents such as water, alcohols, THF and the like. The resulting protected
N6-substituted azide (4a) is deblocked using an aqueous acid suc as 50% formic acid, to provide the N6-substituted
5'-azido-5'-deoxyadenosine (5a). Reduction of the azide (5a) to the amine (6) is effected by catalytic hydrogenation with a catalyst such as platinum oxide, palladium on carbon and the like. For molecules containing other functional groups sensitiv to hydrogenation, triphenylphosphine is used to selectively reduce the azide moiety to the amine. To prepare the N-acylamin (7a) and hydrocarbyloxycarbonylamino (7b) compounds, the azide 4a is reduced to the amine and treated with an acyl anhydride or acyl chloride or alkyl chloroformate and deblocked to give (7a) or (7b) respectively. Analogous processes are used to prepare th 2- and 8- substituted analogs beginning with appropriately substituted intermediates. An alternative synthesis of 5'-amino and 5'-hydrocarbylamino compounds comprises deblocking a
2 ',3'-isopropylidene-5'-tosylate with aqueous acid and then reacting the deblocked tosylate with ammonia or a lower
hydrocarbylamine. Further description of these procedures is set forth in the Examples.
A similar process is used to prepare 5'-deoxy purine
nucleosides. The appropriately substituted 5'-deoxy-2',3'-O-
isopropylideneinosine (2b) is chlorinated or activated using other reagents described above, aminated to 4b and subsequently deblocked to afford the 5'-deoxy nucleoside 5b.
The overall process for preparing 5'-modified
pyrrolo[2,3-d]pyrimidine riboside compounds of Formula I, is depicted in Figure 8. A key step comprises the sodium salt glycosylation method (K. Ramasamy et al., Tetrahedron Letters, 1987, 28: 5107) using the anion of a substituted
4-chloropyrrolo[2,3-d]pyrimidine (18) and 1-chloro-2,3-O- isopropylidene-5-O-tert-butyldimethylsilyl-α-D-ribofuranoside (17). This method is also suitable for direct preparation of ribofuranosides wherein the 5-hydroxy group has been replaced with substituents such as hydrogen or azido or extended with additional carbons (Figure 8). The azide sugars further provide for facile synthesis of 5'-amino nucleosides by reductions of the azide function after ribosylation. An alternative to the sodium salt glycosylation method is a solid-liquid phase transfer reaction using the same substrates and potassium hydroxide in place of sodium hydride as described by Rosemeyer H., and Seela, F, Helvetica Chimica Acta, 1988, 71:1573.
Preparation of the 5-substituted ribose analogs and homologs is outlined in Figures 6 and 7. The 5-substituted-5-deoxy ribose analogs 10 are prepared by tosylation of the protected ribose 8, displacement of the tosylate by appropriate nucleophiles and
subsequent deblocking (Synder, J.; Serianni, A.; Carbohydrate Res., 1987, 163: 169). The ribose homologs (Figure 7) are prepared by oxidation of the protected ribose (8) to the aldehyd (11) (Moorman, A.; Borchaedt, R.; Nucleic Acid Chemistry-Part III. Townsend, L.; Tipson, R.; John Wiley & Sons, 1986). The aldehyde is homologated via the appropriate Wittig reagent to give the key intermediate protected vinyl sugar (12). The protected intermediate is deblocked to give the vinyl ribose homolog (16a) or reduced to (13) and then deblocked to give the saturated deoxy analog (16b). Alternatively, the vinylated intermediate (12) is hydroborated and oxidized affording the protected homologous ribose (14a) which is deblocked to the ribose homolog or converted to the azide (14b) via tosylation and displacement with azide. Deblocking of (14b) then affords the homologous azido ribose (16d). The protected 5-aldehyde (11) was also methylated to ultimately afford 6-deoxy-D-allofuranose
(16e). The various 5- substituted riboses are then converted to the corresponding 2,3-0-isopropylidine ketals (Figure 8) which are chlorinated stereoselectively to 5-modified 1-chloro-α-D-ribofuranosides (17) using carbon tetrachloride and
hexamethylphosphorous triamide (Wilcox, C.; Otaski, R.;
Tetrahedron Lett., 1986, 27:1011).
The preparation of various substituted 4-chloro-pyrrolo[2,3-d]pyrimidines is described in the Examples. The
initial products from the ribosylation reactions, ribosyl protected 5-substituted-4-chloropyrrolo[2,3-d]pyrimidine
nucleosides and the corresponding deblocked compounds are versatile intermediates and comprise an aspect of the present invention. As examples, the 4-chloro substituent of 19 can be displaced by sulfur (such as thiourea or mercaptide anions) leading to thionated and hydrocarbylthio compounds. More importantly, displacement of the 4-chloro substituent by ammonia or amines leads to 4-amino- and 4-arylaminopyrrolo[2,3- d]pyrimidine nucleosides. As further example, and an aspect of the present invention, an improved synthesis of the adenosine kinase inhibitor, 5'deoxy-5-iodotubercidin is described.
According to this novel method, coupling of the sodium salt of 4- chloro-5-iodopyrrolo[2,3-d]pyrimidine with 1-chloro-5-deoxy-2,3- isopropylidene-α-D-ribofuranoside (3/7, B'=CH3) in acetonitrile gives the protected 4-chloro compound. Amination of this product with ammonia, followed by deblocking affords 5'-deoxy-5- iodotubercidin.
Especially preferred intermediates are protected
pyrrolo[2,3-d]pyrimidine nucleosides having a 4-chloro and a 5-iodo or bromo substituent.
Another aspect of the present invention is directed to the use of arylboronic acids to prepare 4- and 5-arylated
pyrrolo[2,3-d]pyrimidine bases and nucleosides from the
corresponding 4- and 5-halogenated compounds. Thus, a
halogenated nucleoside such as .19 or the corresponding base was heated with an arylboronic acid and a palladium-phosphine catalyst such as palladium tetrakis(triphenylphosphine) to prepare the analogous arylated compound by displacement of halogen. Various 4- and 5- arylated pyrrolo[2,3-d]pyrimidines also can be prepared using arylstannyl compounds in place of the arylboronic acids (Flynn, B.; Macolino, B.; Crisp, G.
Nucleosides & Nucleosides. 1991, 10:763). Synthesis of 5-arylpyrrolo[2,3-d]pyrimidines can also be effected by
condensation of arylamino ketones and malononitrile to arylated pyrroles and subsequent ring closure to 5-arylpyrrolo[2,3-d]pyrimidines. (Taylor, E.; Hendess, R., J. Am. Chem. Soc., 1965, 87:1995).
The various above-mentioned products of ribosylation
reactions may be deblocked at appropriate points with aqueous acids such as 50% formic acid or trifluoroacetic acid.
Preparation of 5'-amino compounds consists of reducing an
appropriate azide. The 5'-amides and urethanes are prepared analogously to those described previously for purine analogs. Further description of these procedures is set forth in the
Examples.
Still another aspect of this invention is the preparation of 5'-substituted pyrazolo[3,4-d]pyrimidine ribosides of Formula I
as depicted in Figure 9. Accordingly, a substituted pyrazolo[3,4-d]pyrimidine is ribosylated with an esterified 5- hydroxy, 5-azido or 5-deoxyribofuranoside in the presence of a Lewis acid such as boron trifluoride (Cottam, H., Petrie, C.; McKernan, P.; Goebel, R.; Dalley, N.; Davidson, R.; Robins, R.; Revankar, G.; J. Med. Chem. , 1984, 27: 1120). The 5-substituted sugar is prepared by esterification of the deblocked sugar 10a to 10c or 16a to 16e (See Figures 6 and 7). Suitable esters include the acetate, benzoate, toluate, anisoate and the like. The substituted pyrazolo[3,4-d]pyrimidine base (22) may be prepared by a variety of procedures as illustrated in the Examples. Two general routes to the compounds of the present invention are described below.
The first general route comprises coupling an esterified ribose (21), prepared from 10 or 16, with a 3-substituted
pyrazolo[3,4-d]pyrimidin-4-one. After ribosylation the
pyrimidone riboside (24a) may be activated by chlorination with thionyl chloride/dimethylformamide or other reagents previously described and then reacted with ammonia or an amine to provide a variety of substituted 5'-modified N4-substituted-amino-pyrazolo[3,4-d]pyrimidine nucleosides (24b). Examples of this aspect of the invention, 3-iodopyrazolo[3,4-d]pyrimidone
nucleosides, are prepared by nonaqueous diazotization-iodmation of the 3-amino compounds using a nitrite ester such as isoamyl
nitrite and methylene iodide. Previous attempts to diazotize the 3-aminopyrazolo[3,4-d]pyrimidones using aqueous nitrous acid gave only N-nitrosated pyrazolo[3,4-d]pyrimidin-3,4-diones (Cottam, H.; Petrie, C.; McKernan, P.; Goebel, R.; Dalley, N.; Davidson, R.; Robins, R.; Revankar, G.; J. Med. Chem.. 1984, 27:1119).
Further modifications of 23 or 24 include reduction of the 5'-azido moiety to afford the 5'-amino compounds or the 5'-amides and urethanes as described in Figure 5. Ester prodrugs (C1 and C2) of various 5'-amino nucleosides are prepared by reduction of the 5'-azide esters (23) using previously described reagents.
Various C-4 alkylated pyrazolo[3,4-d]pyrimidine nucleosides are prepared by reaction of the above mentioned suitably
protected 4-chloropyrazolo[3,4-d]pyrimidine nucleosides with carbanion nucleophiles. A specific catalyst for this alkylation reaction was found to be trimethylamine; these reactions either do not occur or proceed very slowly and in poor yield in the absence of trimethylamine. Suitable carbanions include those derived from diethyl malonate, ethyl cyanoacetate, malononitrile, nitromethane, cyanide salts and the like. This procedure is also used to prepare C-6 alkylated purine ribosides. The initial C-alkylated products were deblocked and optionally further modified by hydrolysis and decarboxylation to afford the desired products.
An alternative process for synthesis of 5'-azido- and 5'-amino-5'-deoxypyrazolo[3,4-d]pyrimidine ribosides is also
described. Accordingly, a substituted allopurinol riboside (24a) is protected by conversion to the 2',3'-isopropylidene
derivative, tosylated and reacted with sodium azide in DMSO or DMF to form the azide. Activation of position four by
chlorination with thionyl chloride/dimethylformamide or other reagents as described, followed by displacement of the activating group by ammonia or an amine results in a protected
5'-azido-5'-deoxy riboside. The azide is deblocked to afford (24b, B=N3) and subsequently reduced to the 5'-amino riboside using the previously described procedures.
The second general route for preparation of substituted pyrazolo[3,4-d]pyrimidine nucleosides comprises coupling the esterified ribose (21) with various substituted 4-amino or 4- hydrocarbylaminopyrazolo[3,4-d]pyrimidines. The resulting products are then further modified or deblocked to afford the desired compounds. The utility of this procedure is demonstrated in the Examples, by the preparation of 3-phenyl-4- (phenylamino)pyrazolo[3,4-d]pyrimidine 5'-modified ribosides from 3-phenyl-4-(phenylamino)pyrazolo[3,4-d]pyrimidine and various 5'-modified sugars. In another aspect of the present invention, halogenated pyrazolo[3,4]pyrimidine ribosides can be arylated using arylboronic acids and palladium catalysts as described for the pyrrolo[2,3-d]pyrimidines. Alternatively, the base can be
boronated and then coupled with an aryl halide. Further
description of these procedures is set forth in the Examples.
B. Preferred Methods of Synthesis
According to another aspect of the present invention, certain preferred methods of preparing the adenosine kinase inhibiting compounds of Formula I are provided.
One preferred method of the present invention is a novel procedure for preparing C-6 alkylated purine nucleosides and C-4 alkylated pyrazolo[3,4-d]pyrimidine nucleosides from the 6-chloropurine and 4-chloropyrazolo[3,4-d]pyrimidine nucleosides, respectively, using various carbanions (enolates, cyanide anion, etc.) and trimethylamine as a specific catalyst. Previous methodology for C-alkylation of 6-chloropurines consisted of a multistep route involving alkylthiolation and oxidation to a sulfone followed by nucleophilic displacement with a carbanion (Yame, A.; Matsuda, A.; Veda, T.; Chem. Pharm. Bull. (Jap.), 1980, 28:150). This multistep route can be accomplished in one step using the specific catalyst trimethylamine which reacts to form a quaternary salt and in turn is displaced by a carbanion in situ, regenerating trimethylamine. The reactions are
specifically catalyzed by unhindered trialkylamines.
Another preferred method of the present invention is a process for preparing arylated bases and nucleosides by reaction
of a halogenated pyrrolo[2,3-d]pyrimidine or pyrazolo[3,4- d]pyrimidine with an aryl boronic acid in the presence of a palladium-phosphine catalyst. In this process, the halogen atom of a brominated or preferably, iodinated pyrrolo[2,3-d]pyrimidine or pyrazolo[3,4-d]pyrimidine base or nucleoside, is replaced by an aryl moiety such as phenyl, substituted phenyl or a heteroaryl moiety such as furanyl. A catalyst consisting of a metal such as palladium, complexed to an arylphosphine such as
triphenylphosphine must be present as well as a base such as sodium carbonate. The resulting arylated nucleosides are
important examples of the present invention and this method is shorter and more versatile than alternative syntheses of arylated nucleosides.
Still another preferred method of the present invention is a process for preparing the previously unknown 3-iodo- and 3-chloropyrazolo[3,4-d]pyrimidine nucleoside by nonaqueous
diazotization of 3-aminopyrazolo[3,4-d]pyrimidine nucleosides. According to this invention, a suitably substituted 3- aminopyrazolo[3,4-d]pyrimidine nucleoside is diazotized by heating with an alkyl nitrite such as isoamyl nitrite in the presence of an iodine source (such as methylene iodide) resulting in replacement of the 3-amino moiety with an iodine atom.
Alternatively, methylene iodide can be replaced by a chlorine source such as carbon tetrachloride resulting in replacement of
the amino moiety by a chlorine atom. A previously reported attempt to effect replacement of the amino moiety in a 3- aminopyrazolo[3,4-d]pyrimidine riboside with other moieties using nitrous acid resulted only in replacement of the amino moiety by a hydroxyl group. The resulting 3-chloro- and particularly 3-iodopyrazolo[3,4-d]pyrimidine nucleoside are an important subject of adenosine kinase inhibitors disclosed in the present
invention.
C. Preferred Intermediates
According to a further aspect of the present invention, certain novel intermediates are provided which are useful in the synthesis of the adenosine kinase inhibitors of the present invention.
(i) Intermediates for Pyrrolo[2,3-d]pyrimidines
Certain intermediates useful in the preparation of certain preferred adenosine kinase inhibitors which comprise substituted pyrrolo[2,3-d]pyrimidine nucleosides include compounds of the formula:

These preferred intermediates include the following
compounds:
5-Bromo-4-chloro-7-(5-deoxy-1-β-D-ribofuranosyl)pyrrolo[2,3- d]pyrimidine;
4-Chloro-5-iodo-7-(5-deoxy-1-β-D-ribofuranosyl)pyrrolo- [2,3-d]pyrimidine;
5-Iodo-7-(5-deoxy-1-β-D-ribofuranosyl)pyrrolo[2,3-d]-pyrimidin-4(3H)-thione;
5-Bromo-4-chloro-7-(5,6-dideoxy-l)-β-D-allo¬furanosyl)pyrrolo[2,3-d]pyrimidine;
4-Chloro-5-iodo-7-(5,6-dideoxy-1-β-D-ribofuranosyl)pyrrolo- [2,3-d]pyrimidine;
5-Bromo-4-chloro-7-(5,6-dideoxy-5,6-didehydro-1-β-D-allofuranosyl)pyrrolo[2,3-d]pyrimidine;
4-Chloro-5-iodo-7-(5,6-dideoxy-5,6-didehydro-1-β-D-allofuranosyl)pyrrolo[2,3-d]pyrimidine;
4-Chloro-5-iodo-7-(5-azido-5-deoxy-1-β-D- ribofuranosyl)pyrrolo[2,3-d]pyrimidine;
5-Bromo-4-chloro-7-(5-azido-5-deoxy-1-β-D- ribofuranosyl)pyrrolo[2,3-d]pyrimidine;
4-Chloro-5-iodo-7-(6-azido-5,6-dideoxy-1-β-D-allofuranosyl)pyrrolo[2,3-d]pyrimidine;
5-Bromo-4-arylamino-7-(5-deoxy-1-β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine;
5-Iodo-4-arylamino-7-(5-deoxy-1-β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine;
5-Iodo-4-arylamino-7-(1-β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine; and
5-Bromo-4-arylamino-7-(1-β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine.
In addition to being useful in the preparation of certain preferred adenosine kinase inhibitors, certain of these preferred intermediates exhibit activity as adenosine kinase inhibitors themselves.
(ii) Intermediates for Pyrazolo[3,4-dlpyrimidines
Certain intermediates useful in the preparation of certain preferred adenosine kinase inhibitor compounds comprise substituted pyrazolo[3,4-d]pyrimidines of the formula:

substituted phenyl groups. These preferred intermediates include the following compounds:
4-chloro-3-phenylpyrazolo[3,4-d]pyrimidine;
4-chloro-3-(2-thienyl)pyrazolo[3,4-d]pyrimidine;
4-chloro-3-(4-methoxyphenyl)pyrazolo[3,4-d]pyrimidine; and
4-chloro-3-(4-chlorophenyl)pyrazolo[3,4-d]pyrimidine.
UTILITY
The adenosine kinase inhibitors of the present invention may be used in the treatment of a variety of clinical situations where increasing local levels of adenosine are beneficial. In particular, these compounds may be used in treating cardiovascular disorders in which injury or dysfunction is caused by
ischemia and/or reperfusion (following a period of ischemia).
These include (1) heart attack, a situation that arises from obstruction of one or more of the coronary arteries supplying blood to the heart muscle, and which, if prolonged, leads to irreversible tissue damage; (2) angina pectoris, a clinical
condition in which the blood supply to the heart is sufficient to meet the normal needs of the heart but insufficient when the needs of the heart increase (e.g. during exercise), and/or when the blood supply becomes more limited (e.g. during coronary artery spasm); (3) unstable angina associated with pain at rest; and (4) silent ischemia. In each of these conditions, treatment with adenosine kinase inhibitors will increase local levels of adenosine, and thereby blood flow to the ischemic tissue would be increased and tissue damage reduced and function improved.
Therefore, according to the present invention adenosine kinase inhibitors may also be used to treat or prevent congestive heart failure.
In advanced coronary artery disease or persistent chest pain at rest, a number of clinical procedures are currently used to improve blood supply to the heart. These include percutaneous transluminal coronary angioplasty (PTCA) , percutaneous transluminal directional coronary atherectomy, laser atherectomy, intra-vascular stents and coronary artery bypass graft surgery. The compounds of this invention will also be useful as adjunctive therapies to these techniques. Other clinical settings that involve ischemia would also be ameliorated by agents effecting regional blood flow including organ transplantation, skin flap grafting and other reconstructive surgery, peripheral vascular disease, endotoxemia, hemorrhagic shock, pulmonary emboli, pul
monary injury secondary to burns (thermal injury) or septicemia, pulmonary hypertension, microembolization, glomerulonephritis or progressive glomerulosclerosis, atherosclerosis, myocarditis, vasculitis, cardiomyopathies, intestinal ischemia,. peripheral vascular disease, transient ischemic attacks, stroke and cardio- pulmonary arrest. Adenosine kinase inhibitors will enhance protection afforded by preconditioning a tissue with a brief period of ischemia, before a more prolonged period of ischemia.
Thrombolytic therapy has been limited by a number of factors including the resistance of some thrombi to lysis, delays in reperfusion, and reocclusion following successful thrombolysis. These limitations are believed to be mediated, in part, by platelet aggregation (Born and Cross J. Physiol., 1963, 166:29- 30) and, since adenosine inhibits platelet aggregation in
addition to its other effects on preventing ischemic injury, use of these adenosine kinase inhibitors may comprise a useful adjunctive therapy for thrombolytic therapy or for the treatment or prevention of thrombotic diseases such as myocardial infarction, stroke, angina, deep vein thrombosis, transient ischemic attacks, and pulmonary embolus.
Adenosine has been reported to be an endogenous modulator of inflammation by virtue of its effects on stimulated granulocyte function (Cronstein et al., J. Clin. Invest.. 1986, 78:760-770) and on macrophage, lymphocyte and platelet function. The
compounds of this invention may therefore be used in treating conditions in which inflammatory processes are prevalent such as arthritis, osteoarthritis, autoimmune disease, adult respiratory distress syndrome (ARDS), inflammatory bowel disease, necrotizing enterocolitis, chronic obstructive pulmonary disease (COPD), psoriasis, conjunctivitis, iridocyditis, myositis, cerebritis, meningitis, dermitis, renal inflammation, ischemia, reperfusion injury, peripheral vascular disease, atherosclerosis and other inflammatory disorders. Adenosine receptor agonists have been reported to be beneficial in an experimental model of
inflammation. (Schrier, et al., J. Immunol., 1990, 145: 1874-1879).
Stroke and central nervous system ("CNS") trauma are
conditions where tissue injury results from reduced blood supply to the CNS and are thus amenable to an intervention that provides increased levels of adenosine to the compromised tissue. It is reported that a significant component of the neurodegeneration resulting from stroke or CNS trauma or neurodegenerative diseases is caused by increased excitatory amino acid release and sensitivity, which results in neurons being stimulated to death. In addition to its vasodilatory properties, adenosine has been reported to inhibit excitatory amino acid release (Burke and Nadler J. Neurochem.. 1988, 51:1541) and excitatory amino acid responsiveness. The compounds of this invention, which increase
adenosine levels, may also be used in the treatment of conditions where release of or sensitivity to excitatory amino acids is implicated such as Parkinson's disease, Amyotrophic Lateral
Sclerosis, Huntington's chorea or Alzheimer's disease (Maragos et al., Trends Neurosci.. 1987, 10: 65 and Sonsella et al. Science, 1989, 243:398). These studies, together with results from experimental models of memory (Harris et al. Brain Res., 1984, 323:132) suggest additional utilities of these compounds in the treatment of disorders related to the effects of the aging process on CNS function such as Alzheimer's disease. Other studies have also linked excitatory amino acids with the
pathophysiology of schizophrenia. (Komhuber and Fischer,
Neurosci. Lett., 1982, 34:32; Kim et al. Eur. Neurol., 1983, 22:367). This suggests that adenosine kinase inhibitors may be useful in treating schizophrenia.
These adenosine kinase inhibitors may also be useful in reducing anxiety, as skeletal muscle relaxants and in preventing skeletal muscle spasm.
Adenosine has been proposed to serve as a natural
anticonvulsant (Lee et al., Brain Res.. 1984, 321:160-164;
Dunwiddie, Int. Rev. Neurobiol.. 1985, 27:63-139). Agents that enhance adenosine levels may be used in the treatment of seizure disorders. Adenosine kinase inhibitors may be used in the
treatment of patients with seizures or epilepsy or who might have
chronic low or insufficient adenosine levels or might benefit from increased adenosine such as those suffering from autism, cerebral palsy, insomnia or other neuropsychiatric symptoms.
Other excitatory neuromuscular tissues such as smooth muscle and cardiac muscle may be treated using these adenosine kinase inhibitors. In particular, these adenosine kinase inhibitors may be used to decrease contraction in smooth muscle such as in the gastrointestinal tract, or in vascular tissue such as an artery to prevent vasospasm which may limit blood supply to a tissue. Thus, these adenosine kinase inhibitors may be used to treat conditions such as Buerger's disease, Raynaud's disease,
thromboangiitis obliterans, angina, unstable angina, silent ischemia, or transient ischemic attacks. Other conditions suitable for such therapy include cardiac arrhythmias (including supraventricular tachycardia), irritable bowel syndrome, and impotence.
To assist in understanding the present inventions and especially their properties and utilities, the results of a series of experiments are also included. These experiments demonstrated that a number of compounds of Formula I were potent inhibitors of a purified cardiac adenosine kinase with IC50's of less than 1 M. Moreover, we have shown that these compounds are specific inhibitors of adenosine kinase with low affinity at the A1 adenosine receptor and no significant adenosine deaminase
(ADA) inhibition (Example A) . We have demonstrated that a number of these compounds are also inhibitors of adenosine kinase in intact cells (Example B) . These compounds include pyrrolo[2,3- d]pyrimidine nucleosides modified at the 5'-position or at other positions such that it is less likely to serve as a substrate for phosphorylation enzymes and that, in contrast to 5-iodotubercidin (GP-1-202), these compounds are unlikely to be phosphorylated at the 5'-position, incorporated into nucleotides or DNA, which may cause toxicity to cells or animals. We have demonstrated that inhibition of the cardiac adenosine kinase was achieved in vivo following systemic administration or, in some cases, oral administration of these compounds.
We have demonstrated the ability of these compounds to reduce damage resulting from ischemia and/or reperfusion in an experimental isolated heart model as shown in Example C. A more detailed study demonstrates that functional benefit may be achieved without vasodilatory effects on basal coronary flow which reflects increases in non-ischemic flow (indicative of the potential for coronary steal) or on heart rate. This result was unexpected given the previous descriptions by Newby et al
(Biochem. J. 1983, 214:317-323) and Schrader (Regulatory
Functions of Adenosine, Berne et al. eds., pp. 133-156 1983) of the increases in basal coronary flow caused by adenosine kinase inhibitors. Selected compounds, such as GP-1-238, were also
evaluated to determine the potential for toxic hemodynamic effects or hypothermia associated with administration of adenosine kinase inhibitors. No effects were observed in conscious animals on blood pressure, heart rate or temperature with doses of inhibitor greatly in excess of that required to inhibit the cardiac adenosine kinase (Example D).
Further experiments demonstrated that the adenosine kinase inhibitor GP-1-515 is beneficial in an experimental model of stable angina in dogs. In this study intravenous infusion of the compound attenuated the decline in function associated with repeated episodes of pacing-induced ischemia (Example E). The potential antithrombotic activity of adenosine kinase inhibitors is supported by the ability of GP-1-515 to abolish cyclic flow reductions (CFR's) in 3 out of 8 dogs examined in the Folts model of coronary artery thrombosis (Example F). These results support potential utility of these compounds in thrombotic diseases, such as angina and myocardial infarction.
In other experimental models, the ability of selected adenosine kinase inhibitors (GP-1-272 and GP-1-456) to inhibit neutrophil adherence to endothelial cells, an inflammatory response mediated at the cellular level was evaluated (Example G). Certain adenosine kinase inhibitors were found to exhibit anti-inflammatory activity in animal models of inflammation. The ability of adenosine kinase inhibitors (GP-1-515, GP-1-547) to
attenuate contraction in the isolated ileum (Example H) supports the utility of these compounds in gastrointestinal disorders especially irritable bowel syndrome, in the central nervous system (CNS), the potent effects of selected adenosine kinase inhibitors (such as GP-1-456, GP-1-560) in attenuating chemical and electroshock induced seizures in experimental animal models demonstrates that these compounds will be useful as anticonvulsants in epilepsy (Example I), as well as in other CNS diseases treatable by local increases in adenosine levels.
FORMULATIONS
Compounds of the invention are administered to the affected tissue at the rate of from 0.1 to 200 nmole/min/kg, preferably from 1 to 20 nmol/min/kg. Such rates are easily maintained when these compounds are intravenously administered as discussed below. When other methods are used (e.g., oral administration), use of time-release preparations to control the rate of release of the active ingredient may be preferred. These compounds are administered in a dose of about 0.01 mg/kg/day to about 100 mg/kg/day, preferably from about 0.1 mg/kg/day to about 10 mg/kg/day.
For the purposes of this invention, the compounds of the invention may be administered by a variety of means including orally, parenterally, by inhalation spray, topically, or rectally
in formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles. The term parenteral as used herein includes subcutaneous, intravenous, intramuscular, and intraarterial injections with a variety of infusion techniques. Intraarterial and intravenous injection as used herein includes administration through
catheters. Preferred for certain indications are methods of administration which allow rapid access to the tissue or organ being treated, such as intravenous injections for the treatment of myocardial infarction. When an organ outside a body is being treated, perfusion is preferred.
Pharmaceutical compositions containing the active ingredient may be in any form suitable for the intended method of
administration. When used for oral use for example, tablets, troches, lozenges, aqueous or oil suspensions, dispersible powders or granules, emulsions, hard or soft capsules, syrups or elixirs may be prepared. Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents including those from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation. Tablets containing the active ingredient in
admixture with non-toxic pharmaceutically acceptable excipient
which are suitable for manufacture of tablets are acceptable. These excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, such as maize starch, or alginic acid; binding agents, such as starch, gelatin or acacia; and lubricating agents, such as magnesium stearate, stearic acid or talc. Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and adsorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
Formulations for oral use may be also presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, such as peanut oil, liquid paraffin or olive oil.
Aqueous suspensions of the invention contain the active materials in admixture with excipients suitable for the
manufacture of aqueous suspensions. Such excipients include a suspending agent, such as sodium carboxymethylcellulose,
methylcellulose, hydroxypropylmethylcelluose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia, and
dispersing or wetting agents such as a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene
stearate), a condensation product of ethylene oxide with a long chain aliphatic alcohol (e.g., heptadeaethyleneoxycetanol), a condensation product of ethylene oxide with a partial ester derived from a fatty acid and a hexitol anhydride (e.g.,
polyoxyethylene sorbitan mono-oleate). The aqueous suspension may also contain one or more preservative such as ethyl of n-propyl p-hydroxybenzoate, one or more coloring agent, one or more flavoring agent and one or more sweetening agent, such as sucrose or saccharin.
Oil suspensions may be formulated by suspending the active ingredient in a vegetable oil, such as arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. The oral suspensions may contain a thickening agent, such as beeswax, hard paraffin or cetyl alcohol. Sweetening agents, such as those set forth above, and flavoring agents may be added to provide a palable oral preparation. These
compositions may be preserved by the addition of an antioxidant such as ascorbic acid.
Dispersible powders and granules of the invention suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or
wetting agent, a suspending agent, and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those disclosed above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present.
The pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions. The oily phase may be a vegetable oil, such as olive oil or arachis oil, a mineral oil, such as liquid paraffin, or a mixture of these. Suitable
emulsifying agents include naturally-occurring gums, such as gum acacia and gum tragacanth, naturally occurring phosphatides, such as soybean lecithin, esters or partial esters derived from fatty acids and hexitol anhydrides, such as sorbitan mono-oleate, and condensation products of these partial esters with ethylene oxide, such as polyoxyethylene sorbitan mono-oleate. The
emulsion may also contain sweetening and flavoring agents.
Syrups and elixirs may be formulated with sweetening agents, such as glycerol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, a flavoring or a
coloring agent.
The pharmaceutical compositions of the invention may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to the known art using those suitable
dispersing or wetting agents and suspending agents which have been mentioned above. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non- toxic parenterally-acceptable diluent or solvent, such as a solution in 1,3-butanediol or prepared as a lyophyl-ized powder. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution. In addition, sterile fixed oils may conventionally be employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid may likewise be used in the preparation of injectables.
The amount of active ingredient that may be combined with the carrier material to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. For example, a time-release formulation intended for oral administration to humans may contain 20 to 200 moles of active material compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95% of the total compositions. It is preferred that
pharmaceutical composition be prepared which provides easily measurable amounts for administration. For example, an aqueous solution intended for intravenous infusion should contain from about 20 to about 50 moles of the active ingredient per
milliliter of solution in order that infusion of a suitable volume at a rate of about 30 ml/hr can occur.
As noted above, formations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion. The active ingredient may also be administered as a bolus, electuary or paste.
A tablet may be made by compression or moulding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder (e.g., povidone, gelatin,
hydroxypropylmethyl cellulose), lubricant, inert diluent,
preservative, disintegrant (e.g., sodium starch glycolate, cross- linked povidone, cross-linked sodium carboxymethyl cellulose) surface-active or dispersing agent. Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydroxypropylmethyl cellulose in
varying proportions to provide the desired release profile.
Tablets may optionally be provided with an enteric coating, to provide release in parts of the gut other than the stomach. This is particularly advantageous with the compounds of formula (I) as such compounds are susceptible to acid hydrolysis.
Formulations suitable for topical administration in the mouth include lozenges comprising the active ingredient in a flavored basis, usually sucrose and acacia or tragacanth;
pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
Formulations for rectal administration may be presented as a suppository with a suitable base comprising for example cocoa butter or a salicylate.
Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the ddPN ingredient such carriers as are known in the art to be appropriate.
Formations suitable for parenteral administration include aqueous and non-aqueous isotonic sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile
suspensions which may include suspending agents and thickening
agents. The formulations may be presented in unit-dose or multi- dose sealed containers, for example, ampoules and vials, and may be sorted in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
Preferred unit dosage formulations are those containing a daily dose or unit, daily sub-dose, or an appropriate fraction thereof, of an adenosine kinase inhibitor compound.
It will be understood, however, that the specific dose level for any particular patient will depend on a variety of factors including the activity of the specific compound employed; the age, body weight, general health, sex and diet of the individual being treated; the time and route of administration; the rate of excretion; other drugs which have previously been administered; and the severity of the particular disease undergoing therapy, as is well understood by those skilled in the art.
Examples of use of the method of the invention includes the following. It will be understood that these examples are
exemplary and that the method of the invention is not limited solely to these examples.
The method may be used following thrombolysis for coronary occlusion. The compound would be given as a sterile injectable
preparation with water or isotonic sodium chloride as the solvent. The solution can be administered intravenously or directly into the coronary artery at the time of left heart catheter!zation or into a carotid artery. The rate of
administration could vary from 1 to 20 nmole/min/kg with, for example, an infusion volume of 30 ml/hr. Duration of therapy would typically be about 96 hours.
Angina and early myocardial infarcts can be treated by intravenous administration using a sterile injectable preparation using the rates discussed above.
Capsules comprising adenosine kinase inhibitors suitable for oral administration according to the methods of the present invention may be prepared as follows: (1) for a 10,000 capsule preparation: 1500g of adenosine kinase inhibitor is blended with other ingredients (as described above) and filled into capsules which are suitable for administration depending on dose, from about 4 capsules per day (1 per 6 hours) to about 8 capsules per day (2 capsules per 6 hours), to an adult human.
The compounds of this invention and their preparation can be understood further by the examples which illustrate some of the processes by which these compounds are prepared. These examples should not however be construed as specifically limiting the invention and variations of the invention, now known or later
developed, are considered to fall within the scope of the present invention as herein after claimed.
EXAMPLES
EXAMPLE 1
Preparation of 5'-Azido-5'-deoxy
2', 3'-O-(1-methylethylidene) inosine
This material was prepared by tosylation of 2', 3'-(1- methylethylidene)iniosine and subsequent reaction with sodium azide in DMSO as described by Hampton, A.; J. Org. Chem., 1968, 11:1220.
EXAMPLE 2
Preparation of 9-(5-Azido-5-deoxy-2,3-O-(1-methylethylidene)
-1-β-D-ribofuranosyl]-6-chloropurine
A solution of the azide (Example 1) (12.01 g, 0.036 mole) in dry CH2Cl2 (500ml) was added, during 1 hr, to a warm solution of SOCl2 (8.1 ml, 0.11 moles) and DMF (4.05 ml, 0.05 moles) in CH2Cl2 (50 ml). The resulting solution was refluxed
for 6 hrs while a continuous stream of argon gas was bubbled through the reaction mixture to remove HCl. The reaction was cooled and added to a cold, rapidly stirred aqueous solution of KHCO3. After stirring for 15 minutes the layers were separated and the organic layer was washed with cold aqueous K2CO3,
H2O(2x), dried (Na2SO4) and concentrated under vacuum. The residue was redissolved in CH2Cl2 and filtered through a plug of Si02 gel. Evaporation of the filtrate under vacuum gave 11.3 g (89% yield) of the title compound as a pale yellow oil.
EXAMPLE 3
General Procedure for the Preparation of N6-Substituted-5'- azido-5'-deoxy-2'.3'-O-(1-methylethylidene)adenosines
To a solution of chloride (Example 2) (1 mmole/5 ml) in EtOH or n-BuOH was added amine (1.3 equivalents) and Et3N (1.8
equivalents). The solution was heated to reflux for 12-24 hours under Ar until judged complete by TLC. The reaction mixture was evaporated under vacuum, dissolved in CH2Cl2 and washed with aq. K2CO3 and H2O. The CH2Cl2 solution was dried (Na2SO4) concentrated and used directly in the next step or chromatographed on SiO2 gel using CH2Cl2-MeOH mixtures.
EXAMPLES 4 to 14
General Procedure for the Preparation of N6- Substituted-5'-azido-5'deoxyadenosines
The N6-substituted isopropylidene azide (Example 3) (1.0 g) was dissolved in HCO2H (10-20 ml) and diluted with an equal volume of H20. The reaction was followed by TLC. After disappearance of the starting material (12-48 hr), the reaction mixture was evaporated under vacuum, coevaporated with H2O (3x) then ETOH (2x). The residue was crystallized from H2O, alcohol or mixtures.
The compounds in Table I (Examples 4-14) were prepared by this procedure:
TABLE I
GPl-# Example F MP ( ºC)
266 4 ϕNH 121-126 ° - - - 5 CH3NH 180 ° (d)
317 6 4-C1J3NH 209-210 °
299 7 ϕCH2CH2NH 144-146 °337 8 N-indolinyl 157-159 °346 9 4- (HOCH2CH2)ϕNH 147-151 º
- - - 10 -NH (CH2) 12NH- foam
(dimer)
385 11 N-indolyl1 186-188°
391 12 N-(5-bromoindolinyl) 194-196°
421 13 N-(5-methoxyindolinyl) 184-185°
557 14 1,4-piperazinyl 198-204°
1/ Prepared from indole and sodium hydride in DMF.
EXAMPLES 15-20
General Procedure for the Preparation of N6-Substituted- 5'-amino-5'-deoxyadenosines and Hydrochloride Salts
A solution of the azide in EtOH or MeOH containing 10% Pd-C (25-50% weight of azide) was hydrogenated on a Parr shaker at 25 psi for 4-8 hours. The mixture was filtered, the catalyst rinsed well with solvent and the filtrate evaporated. The residue was recrystallized to give the free base or converted to the salt. The hydrochloride salt was prepared by slurrying or dissolving the free base in a small volume of EtOH, adding dry ethanolic HCl to a pH of 4-6, warming the mixture and then chilling to crystalize out the salt (in some cases Et2O was added
to precipitate the salt). The compounds in Table II (Examples 15-20) were prepared by this procedure:
TABLE II
GPl-## Example F M.P.ºC (salt)
272 15 CH3NH 170-172°
(HCO2H)
286 16 ϕNH 169-173° (HCl)
328 17 ϕCH2CH2NH 130° (d) (HCl)
345 18 N-indolinyl 202-203° (HCl)
373 19 -HN(CH2)12NH- 151-153° (HCl)
(dimer)
565 20 1,4-piperazinyl 140-145° (HCl)
EXAMPLE 21
Preparation Of 5'-deoxy-2 ',3'-O-(1-methylethylidene)inosine
A solution of 5'-deoxy-5'-iodo-2',3'-O-(1-methylethylidene)inosine (5.45 g, 0.013 mol) in 80 ml of methanol containing triethylamine (2.0g) and 10% palladium on charcoal (737 mg) was hydrogenated for 2 hr under 50 psi H2. The reaction mixture was filtered and the filtrate concentrated and allowed to crystalize. The product was collected by filtration and dried under vacuum to give 2.45 g (82% yield) of the title compound.
EXAMPLE 22
Preparation of 6-Chloro-9-[5-deoxy-2,3-O- (1-methylethylidene)-1-β-D-ribofuranosyllpurine
A solution of the blocked 5'-deoxyinosine (1.4 g, 4.8 mmol) tetraethylammonium chloride (1.9 g, 11.5 mmol), diethylaniline (1.2 ml, 7.2 mmol) and phosphorous oxychloride (3.35 ml, 36 mmole) in CH3CN (24 ml) was refluxed for 10 minutes then
evaporated. The residue was dissolved in CH2Cl2, washed with water, aqueous KHCO3 solution, water and dried (Na2SO4). The solution was concentrated and filtered through a plug of SiO2 gel. The filtrate was evaporated to give 860 mg (65% yield) of title compound as a yellow oil.
EXAMPLE 23
General Procedure for Preparation of
N6-substituted 5'-deoxyadenosines
The above identified compounds were prepared using the procedures described in Example 3 and Examples 4-14.
The compounds listed in Table III were prepared by this procedure.
TABLE III
GPI-# EXAMPLE F M. P. ( ºC)
595 23 1,4-piperazinyl 220-225 °
EXAMPLE 24
Preparation of 8-Bromo-2',3'-O-(1-methylidene)- 5'-O-(4-methylbenzenesulfonyl)adenosine
The above-identified compound may be prepared as described: Ikshara, M.; Kaneko, M.; Sagi, M.; Tetrahedron, 1970, 26:5757.
EXAMPLE 25
Preparation of N-Formyl-8-bromo-2',3'-O- (1-methylethylidene)-5'-O-(4-methylbenzenesulfonyl)
adenosine
To acetic-formic anhydride (prepared by stirring 25 ml of acetic anhydride and 12.5 ml of formic acid for 15 minutes at 45°C) at 0°C, was added the tosylate (Example 24) (4.0 g, 7.30
mmol). The resulting solution was allowed to warm to 22 °C and stirred for 48 hours. The reaction mixture was evaporated, chased 2x with toluene and the residue dissolved in CHCl3 and filtered through a plug of silica gel. Evaporation of the filtrate and crystallization of the residue from ethanol gave 4.0 g (96%) of the title compound.
EXAMPLE 26
Preparation of 5'-Deoxy-5'.8-diazido-2'.3'-O- (1-methylethylidene)adenosine
To a hot (75°C), well stirred slurry of NaN3 (2.23 g, 34.3 mmol) in dimethylsulfoxide (35 ml) was added the formyl tosylate (Example 25) (4.00 g, 6.9 mmoles). The reaction temperature was held at 75 °C for one hour then cooled to 25 °C. The mixture was poured into stirring H2O (90 ml), slurried for ten minutes, the solid was collected by filtration, rinsed with H2O (3x), cold ethanol and dried. The crude product was dissolved in CHCl3, filtered through silica gel and the filtrate evaporated to give the N6-formyl derivative 1.20 g; m.p. 107-110°C.
The N6-formyl derivative was deformylated by slurrying in MeOH, adding saturated methanolic ammonia (80 ml) and warming until homogenous. After 15 minutes the solution was evaporated,
the residue recrystallized from EtOH and dried to give the title compound; 0.900 g (60% yield); m.p. 166-168°C.
EXAMPLE 27 Preparation of 5'-Deoxy-5',8-diazidoadenosine
The isopropylidene diazide of Example 26 (1.00 g, 3.15 mmol) was deblocked as described under Example 4 and recrystallized from H2O; 760 mg (85% yield); m.p. 128-130°.
EXAMPLE 28
Preparation of 5'-Deoxy-5'-8-diaminoadenosine Formate
The diazide of Example 27 (0.660 g, 2.0 mmol) was
hydrogenated as described under Example 15 and recrystallized from abs. EtOH to give after drying, 400 mg (61% yield). This material was further purified by conversion to the formate salt (HCO2/EtOH/Et2O); m.p. 98°C(d).
EXAMPLE 29
Preparation of 5'-Deoxy-5'-formylaminoadenosine
To cold (5°C) acetic-formic anhydride (10 ml acetic
anhydride and 5 ml formic acid) was added 5'-amino-5'-deoxy-2', 3'-O-(1-methylethylidene)adenosine (670 mg, 2.0 mmol). The solution was stirred for 24 hours then evaporated, and
coevaporated with toluene (2x) then EtOH. The residual foam was dissolved in methanolic ammonia containing CH2Cl2 and stirred overnight. TLC (SiO2 gel, 9:1 CHCl3-MeOH) indicated the initial product was converted to a more polar product. The solution was evaporated, the residue dissolved in CHCl3 with 3% MeOH and filtered through a plug of SiO2 gel. Evaporation of the filtrate gave 520 mg of a white foam. This material (500 mg) was
deblocked with HCO2H as described for Example 4 and
recrystallized from EtOH-H2O to give the title compound: yield 0.35 g (55%); m.p. 212-213ºC.
EXAMPLE 30
Preparation of 6-(N-Indolinyl)-9-[2,3-O-(1-methylethylidene)
-1-β-D-ribofuranosyl]purine
A mixture of the isopropylidene of 6-chloropurine riboside (11.5 g, 0.035 mol), indoline (5.13 ml, 0.046 mole) and
triethylamine (8.82 ml, 0.063 mole) in n-butanol (60 ml) was stirred and heated to reflux for 24 hours. The reaction was cooled and the solid collected by filtration, rinsed with EtOH and dried to give the title compound: 10.70 g (75% yield); m.p. 119-125°C.
EXAMPLE 31
Preparation of 6-(N-Indolinyl)-9-[2,3-O-(1-methylethylidene) -5-O-(4-methylbenzenesulfonyl)-1-β-D-ribofuranosyl)]purine
To a cold (0ºC) solution of the alcohol (Example 30) (6.0 g, 0.015 mol) in dry pyridine (40 ml) was added with stirring, p-toluenesulfonyl chloride (6.96 g, 0.36 mol). The solution was sealed and stored at 0-10ºC for 72 hours then poured with
stirring into cold H2O (30 ml). The solid was collected by filtration and rinsed 3x with H2O. After drying at 25°C under
vacuum, the title compound was obtained: 6.85 g (83% yield); m.p 195°C (d).
EXAMPLE 32
Preparation of 6-(N-Indolinyl)-9-[5-O-(4- methylbenzenesulfonyl)-1-β-D-ribofuranosyl)]purine
A slurry of the protected tosylate (Example 31) (5.80 g, 10.0 mmole) in hydrochloric acid (23 ml) and EtOH (255 ml) was heated until homogenous then refluxed for 15 minutes. The solution was cooled in an ice bath and neutralized with KHCO3. The solid was collected by filtration, washed with H2O, EtOH then MeOH. After drying, 3.38 g (65% yield) of the title compound were obtained; m.p. 112°C(d).
EXAMPLE 33
Preparation of 6-(N-Indolinyl)-9-(5-methylamino-5- deoxy-1-β-D-ribofuranosyl)purine Hydrochloride
To 40% aqueous methylamine (40 ml) was added the tosylate (Example 32) (2.0 g, 3.8 mmol) and sufficient MeOH to give a clear solution. The solution was stirred for one week then
concentrated under vacuum. The residue was coevaporated 3x with MeOH then recrystallized from MeOH to give the free base, 0.310 (21% yield). A portion of this material was converted (ethanoli HC1) to the hydrochloride salt, m.p. 170-172°.
EXAMPLE 34
Preparation of 4-Chloro-7H-pyrrolo[2,3-d]pyrimidine
The above-identified compound was prepared as described: Davoll, J.; J. Chem. Soc., 1960, 131.
EXAMPLE 35
Preparation of 5-Bromo-4-chloro-7H-pyrrolo[2,3-d]pyrimidine
The above identified compound was prepared as described: Hinshaw, B. ; Gerster, J.; Robins, R.; Townsend, L.; J.
Heterocyclic Chem., 1969, 215.
EXAMPLE 36
Preparation of 4-Chloro-5-iodo-7H-pyrrolo[2,3-d]pyrimidine
The above-identified compound was prepared as described: Pudlo, J.; Nassiri, M. ; Kern, E.; Wartiny, L.; Drach, J.;
Townsend, L.; J. Med. Chem.. 1990, 33, 1984.
EXAMPLE 37
Preparation of 4-Chloro-5-methyl-7H-pyrrolo[2,3-dlpyrimidine
The above-identified compounds was prepared as described: Pudlo, J.; Nassir, M.; Kern, E.; Wotring, L.; Drach, J.;
Townsend, L.; J. Med. Chem., 1990, 33, 1984.
EXAMPLE 38
Preparation of 4-Chloro-2-methylthio-7H-pyrrolo[2,3-d] pyrimidine
The above-identified compound was prepared as described: Noel, C., Robins, R.; J. Heterocyclic Chem.. 1964, 1, 34.
EXAMPLE 39
Preparation of 2-Amino-4-chloro-7H-pyrrolo[2,3-dlpyrimidine
The above-identified compound was prepared as described: Pudlo, J.; Nassiri, M.; Kern, E.; Wotrlng, L.; Drach, J.;
Townsend, L.; J. Med. Chem.. 1990, 33, 1984.
EXAMPLE 40
Preparation of 2-Amino-4-chloro-7H-pyrrolo[2,3-dlpyrimidine
The above-identified compound was prepared as described. Seela, F.; Stiker, H.; Driller, H.; Binding, N.; Liebigs Ann. Chem., 1987, 15.
EXAMPLE 41
Preparation of 4-Chloro-5-methylthio-7H-pyrrolo[2,3-d]pyrimidine
A solution of 5-bromo-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (Example 35) (2.53 g, 10 mmol) in anhydrous THF (30 mL) was cooled to -78 ºC under argon and a solution of n-butyl lithium (12 mL of 2.3 M solution, 25 mmol) was added at such a rate that the
reaction temperature remained below -72 ºC. After the addition, the reaction mixture was stirred at -78 °C for 45 min, a solution of methyl disulfide (0.95 ml, 10 mmol) in tetrahydrofuran (10 mL) was added over a period of 30 minutes maintaining the temperature below -72 °C. The reaction mixture was stirred at -78ºC for 2.5 hours then allowed to warm to room temperature. A saturated solution of ammonium chloride (40 mL) was added to the reaction with stirring. The organic layer was separated, the aqueous layer extracted with ethyl acetate (2×40 mL) and the combined organic extracts were dried (MgSO4), filtered and evaporated to obtain a pale yellow solid which was crystallized from EtOH:
yield 1.65 g; (70%) m.p. 166-167°C.
EXAMPLE 42
Preparation of 4-Chloro-5-cyano-7H-pyrrolo[2,3-d]pyrimidine
A 2.31 M solution of n-butyllithium in hexane (6.1 mL, 14.0 mmol) was added dropwise to a solution of 4-chloro-5-bromopyrrolopyrimidine (Example 35) (1.481 g, 6.37 mmol) in 65 mL THF at -78°C and the resulting light yellow suspension stirred at this temperature for 1 hour. A cold (-78°C) solution of p-tolylsulfonylcyanide (2.08 g, 11.5 mmol) in 35 mL THF was added dropwise via cannula and the resulting mixture stirred at this
temperature for 1 hour. Aqueous NH4Cl was added and the
resulting solution was diluted with 100 mL CH2Cl2. The organic layer was separated, washed with water, brine, dried (MgSO4) and evaporated to provide a tan solid (1.3 g) which appeared to be a 1:1 mixture of the title nitrile and 4-chloropyrrolo-pyrimidine by 1H NMR. This material was recrystallized from 25 mL ethanol to provide 435 mg (38%) of the title nitrile as a tan solid: m.p.
300"C.
This compound may also be prepared as described: Tollman et al., J. Amer. Chem. Soc.. 1969, 91:2102.
EXAMPLE 43
Preparation of 4-Chloro-5-ethoxycarbonyl-7H- pyrrolo[2,3-d]pyrimidine
A solution of 5-bromo-4-chloropyrrolo[2,3-d]pyrimidine
(Example 35) (232 mg; 1 mmol) in anhydrous THF (5 mL) was cooled to -78 °C under argon and a solution of n-butyl lithium (1.3 mL of 2.31 M) was added at such a rate that the temperature of the reaction mixture remained below -72°C. After stirring the reaction mixture at -78 °C for 45 minutes, a solution of ethyl chloroformate (0.15 mL) in THF (2 ml) was added slowly,
maintaining the reaction temperature below -72°C. The reaction
mixture was stirred at -78 °C for 2 hours then allowed to warm to room temperature. A saturated solution of NH4Cl (20 mL) was added to the reaction mixture. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2x25 mL) The combined organic extracts were dried and evaporated to a white solid, yield 210 mg (92%): m.p. 140-141ºC.
EXAMPLE 44
Preparation of 5-O-[(1,1-Dimethylethyl)dimethylsilyl]-2,3-β- (1-methylethylidene)-D-ribofuranose
The above-identified compound was prepared as described: H. Rosemeyer, H.; Seela, Helv. Chim. Acta., 1988, 71, 1573.
EXAMPLE 45
Preparation of 5-Deoxy-D-ribofuranose
The above-identified compound was prepared as described:
Snyder J . ; Serianni , A. ; Carbohydrate Research , 1987 , 163 , 169.
EXAMPLE 46
Preparation of 5-O-Methyl-D-ribofuranose
The above-identified compound was prepared as described: Snyder, J.; Serianni, A.; Carbohydrate Research. 1987, 163. 169.
EXAMPLE 47
Preparation of 1-O-Methyl-2 , 3-0-(1-methylethylidene) -5-O- (4-methylbenzenesulfonyl) -D-ribofuranoεide
The above-identified compound waε prepared as described: Snyder, J. ; Serianni, A. ; Carbohydrate Reεearch, 1987 , 163 , 169.
EXAMPLE 48
Preparation of 5-Deoxy-2,3-O-(1-methylethylidene)-D-ribofuranose
5-Deoxy-D-ribofuranose (8.g, 60 mmole) was dissolved in DMF (25 ml) and to the solution was added dimethoxypropane (10 ml) and p-toluenesulfonic acid (150 mg). The reaction was stirred overnight then neutralized with Amberlite 400 (OH resin. The mixture was filtered, concentrated and the residue
chromatographed on SiO2 gel using 15:1 CH2Cl2-MeOH. The
appropriate fractions were collected and evaporated to yield 4.1 g (39% yield) of viscous liquid.
EXAMPLE 49
Preparation of 5-β-Methyl-2.3-O- (l-methylethylidene)-D-ribofuranose
To a solution of 5-O-methyl-D-ribofuranose (6.0 g, 36 mmole) in dry DMF (25 mL), 2,2-d-methoxypropane (25 mL) and p-toluenesulfonic acid (250 mg) were added and the solution was stirred at room temperature for 20 hours. Volatile materials were evaporated under reduced pressure and the residue was chromatographed over SiO2 gel using 3:1 hexane:ethyl acetate.
Appropriate fractions were pooled and evaporated to give the title compound as an oily product, yield: 5.0 g (68%).
EXAMPLE 50
5-Azido-5-deoxy-1-O-methyl-2,3-β-(1-methylethylidene) -D-ribofuranoside
A mixture of 1-O-methyl-2,3-O-(1-methylethylidene)-5-0-(4- methylbenzenesulfonyl)-D-ribofuranoside (8.0 g, 22 mmol), dry DMF (40 mL) and NaN3 (4.0 g, 62 mmol) was heated at 80°C under anhydrous conditions for 12 hours. The solvent was evaporated under high vacuum and the residue was chromatographed over silica gel using CH2Cl2. The fractions containing the faster moving product were pooled and evaporated to obtain 4.8 g (94% yield) of a syrupy product.
EXAMPLE 51
Preparation of 5-Azido-5-deoxy-D-ribofuranose
A solution of 5-azido-5-deoxy-1-0-methyl-2,3-O-(1-methylethylidene)-D-ribofuranoside (4.6 g, 20 mmol) in 0.1% H2SO4 (300 mL) was gently refluxed for 3 hours. The acid was
neutralized (pH ~5) with Amberlite 400 (OH form) and the resin filtered and washed with ethanol (2x20 mL). The filtrate was evaporated to dryness under high vacuum to give the title
compound as a syrupy residue; 1H and 13C NMR confirmed the identity of the product as a mixture of α and β anomers.
EXAMPLE 52
Preparation of 5-Azido-5-deoxy-2.3-β-(l-methylethylidene)-β-D-ribofuranose
The crude 5-azido-5-deoxyribose (Example 51) was dissolved in dry DMF (10 mL) and treated with 2,2- dimethoxypropane (10 mL and p-toluenesulfonic acid (100 mg). The solution was stirred at room temperature for 20 hours then evaporated under high vacuum. The residue was chromatographed over SiO2 gel using 3:1
hexane: ethyl acetate. The appropriate fractions were pooled and evaporated to obtain the title compound, yield 2.4 g (56% yield).
EXAMPLE 53
Preparation of 1-O-Methyl-2,3-O-(1-methylethylidene)
D-pentodialdo-1,4-furanoside
The above-identified compound was prepared as described:
Moorman, A.,; Borchardt, R.; Nucleic Acid Chemistry-Part III, Ed. Towsend, L., Tipson, R.; John Wiley and Sons, N.Y.; 1986, pages 38-41.
EXAMPLE 54
Preparation of 5-Benaoyl-D-allofuranose
The sugar aldehyde from Example 53 (100 mmole) was dissolve in anhydrous THF and treated with a commercially available solution of methyl magnesium bromide (100 m mol) under anhydrous conditions. After 2 hours of stirring at room temperature, a saturated solution of ammonium chloride in water (180 mL) was added. The organic layer was separated and the aqueous layer wa extracted with ether (2 x 100 mL). The combined organic layers were dried and evaporated to obtain an oily product whose NMR was consistent with methyl-6-deoxy-2,3-isopropylidene-D- allofuranoside. The crude product was dissolved in pyridine (50 ml) and treated with benzoic anhydride (120 mmole). After stirring for 18 hours, methanol (2 ml) was added and the reaction mixture was evaporated under high vacuum. The residue was dissolved in ethyl acetate (300 ml) and washed successively with water, saturated bicarbonate solution, and brine. The organic layer was dried and evaporated to obtain a glassy product which was purified by column chromatography. Identity of the product was confirmed by IR and NMR spectroscopy. The intermediate protected sugar was then heated with aqueous sulfuric acid solution (0.01 N in water, 300 ml) to @ 80°C for 2 hours and neutralized with strongly basic ion exchange resin. The aqueous
layer was separated and evaporated under high vacuum to obtain the title compound as a sticky mass. The product was confirmed by NMR and used in the next step without further purification.
EXAMPLE 55
Preparation of 5-Benzoyl-6-deoxy-2.3-β-(1-methylethylidene)- D-allofuranose
The benzoylated sugar (Example 54) was dissolved in a mixture of dry DMF (20 ml), 2,2-dimethoxypropane (20 ml) and p-toluenesulfonic acid (200 mg) and stirred at room temperature with the exclusion of moisture. The reaction was complete within two hours as evidenced by the absence of the starting material (TLC). The acid was neutralized by strongly basic ion exchange resin and the resin removed by filtration and washed. The combined washings and filtrate were evaporated under high vacuum and the residue was purified by chromatography. The pure product obtained was a glassy solid. IR and NMR were consistent with the title compound.
EXAMPLE 56
Preparation of 5,6-Dideoxy-5.6-didehvdro-1-O-methyl-2,3-β-(1- methylethyldene)-D-allofuranoside
To a suspension of potassium-tert-butoxide (9.36 g) in anhydrous ether (300 ml), methyltriphenylphosphonium bromide (29.6 g) was added in small portions over 5 minute period with stirring under anhydrous conditions. The bright yellow colored solution (with some solid separated) was stirred for 1 1/2 hours then a solution of methyl-2,3-isopropylidene-D-pentodialdo-1,4- furanoside (8.0 g), Example 53, in anhydrous ether (75 ml) was added over 5 minute period. The reaction mixture was stirred overnight at room temperature. The solid material that formed in the reaction mixture was removed by filtration and washed repeatedly with ether. The combined washings and filtrate were evaporated and the residue purified by chromatography to obtain 6.5 g of product as an oil; TLC Rf + 0.5 (Silica gel, 97:3 hexane: EtoAc).
EXAMPLE 57
Preparation of 5, 6-Dideoxy-5.6-didehydro-2.3-O- (1-methylethylidene)-D-allofuranose
A mixture of 5, 6-dideoxy-5, 6 didehydro-1-O-methyl-2,3-O-(1- methylethylidene)-D-allofuranose (Example 56) (2.0 g), and aqueous H2SO4 (0.1%, 50 ml) was heated to 90º for 3 hours at which time the indicated the starting material was consumed. The pH of the solution was adjusted to 7.5 with 1N NaOH solution, and evaporated to dryness. The residue was evaporated with DMF (2 X 20 ml) and the resulting semi solid product was slurried in methanol (25 ml). The undissolved solid was removed by
filtration and washed with methanol (2 X 20 ml). The combined washings and the filtrate were evaporated to dryness and the resulting residue was dissolved in a mixture of dry DMF (10 ml), 2,2-dimethoxypropane (6 ml) and p-toluenesulfonic acid (50 mg). After stirring for 2 hours at room temperature the acid was neutralized with ion exchange resin (strongly basic type) and the resin was removed by filtration. Evaporation of the filtrate gave a product which was purified by chromatography over silica gel using 97:3 hexane:EtOAc as the a mobile phase; Rf of the product 0.8 (silica gel, 9:1 hexane;EtOAc).
EXAMPLE 58
Preparation of 5.6-Dideoxy-1-O-methyl-2,3-O- (1-methylethylidene)-D-allofuranoside
A solution of the vinylic sugar (6.2 g), Example 56, in methanol (55 ml) was purged with argon and hydrogenated using 10% platinum on carbon as catalyst at 80 psi for 100 hours. The catalyst was removed by filtration and washed with methanol. The combined washings and filtrate were evaporated to obtain a colorless oil used directly for the next step. Yield:
quantitative. TLC Rf = 0.48 (Silica gel, 97:3 Hexane:EtOAc).
EXAMPLE 59
Preparation of 5.6-Dideoxy-2,3-O- (1-methylethylidene)-D-allofuranose
A mixture of 5,6-dideoxy-1-methyl-2,3-O-(1-methylethylidene)-D-allofuranoside (5.8 g), (Example 58), and water (160 ml) containing 0.16 ml of cone H2SO4 was heated to 90°C for 3 hours. The pH of the reaction mixture was adjusted to 7.5 with 1N NaOH solution and evaporated to dryness under high vacuum. The residue was coevaporated with DMF (2 x 50 ml) then dissolved in methanol (25 ml) and filtered. The filtrate was
evaporated, dissolved in a mixture of dry DMF (10 ml), 2,2- dimethoxypropane (10 ml) and p-toluenesulfonic acid (200 mg) and stirred for 2 hours. The acid was* neutralized with strongly basic ion exchange resin and the resin removed by filtration. The filtrate was evaporated and the residue chromatographed to obtain the title product; yield: 4.3 g.; TLC, Rf = 0.6 (Silica gel, 2:1 hexane: EtOAc).
EXAMPLE 60
Preparation of 5-deoxy-1-β-methyl-2.3-β- (1-methylethylidene)-D-allofuranoside
To a solution of the vinylic sugar Example 56 (4.1 g) in THF (20 ml), borane:THF solution (10.65 ml of 1 M solution in THF) was added over 10 minutes. After stirring 2 hours at room temperature the reaction vessel was immersed in a cooling bath (ice-water) and an aqueous solution of NaOH (8 ml of 3M solution) was added with stirring. After 15 minutes a solution of H2O2 (30% aq. 4 ml) was added dropwise, and stirring was continued for an additional 15 minutes. Then the flask was immersed for 30 minutes in a water bath maintained at 55 °C and cooled. The contents were extracted with methylene chloride (3 X 150 ml) and the organic layer was dried over anhydrous MgSO4. The solvent was evaporated and the residue was chromatographed over silica
gel using a hexane:ethyl acetate gradient as solvent. The fast moving minor product, Rf=0.7, (10%) was identified as 6-deoxy-1- methyl-2,3-O-(1-methylethylidene)-D-allofuranoside. The slower moving major product (90%), Rf=0.3 was identified by proton NMR to be the title compound; yield 3.7 g; Rf=0.3 (silica gel, 2:1 hexane:EtOAc).
EXAMPLE 61
Preparation of 6-O-(t-Butyldimethylsilyl)- 2.3-β-(1-methylethylidene)-D-allofuranose This compound may be prepared by t-butyldi- methylsilylation of the corresponding 6-hydroxy sugar, using t-butyldimethylsilyl chloride and imidazole in DMF.
EXAMPLE 62
Preparation of 5-deoxy-1-methyl-2,3-O- (1-methylethylidene)-6-p-toluenesulfonyl-D-allofuranoside
To an ice-cold solution of the hydroxy sugar Example 60, (3.69 g) in anhydrous pyridine (25 ml), p-toluenesulfonyl
chloride (3.7 g) was added in small portions. The reaction mixture was stirred and allowed to warm to room temperature over 2 hours, at which time the reaction was complete (TLC).
Unreacted p-toluenesulfonyl-chloride was quenched by adding 1 ml of methanol and the volatile components were evaporated under high vacuum. The residue was evaporated with DMF (2 x 20 ml), then dissolved in ethyl acetate (350 ml). The solution was washed successively with water and bicarbonate solution and the organic layer was dried over anhydrous MgS04. Evaporation of the solvent gave a residue which was purified by silica gel column chromatography; yield 4.1 g; Rf=0.8 (silica gel 2:1
hexane:EtOAc). Proton NMR indicated the product to be a mixture of α and β anomers.
EXAMPLE 63
Preparation of 6-azido-5,6-dideoxy-1-O-methyl- 2.3-β-(1-methylethylidene)-D-allofuranoside A mixture of the tosyl sugar, Example 62, (4.0 g), dry DMF (20 ml) and sodium azide (1.5 g) was heated to 100 °C in an oil bath under anhydrous conditions for 24 hours. The solvents were evaporated under high vacuum and the residue was dissolved in ethyl acetate (200 ml) and washed with water. The organic layer was dried (MgSO4) and evaporated to obtain a colorless oil which was sufficiently pure by TLC and NMR for use in the next
reaction; yield 2.09 g; Rf=0.35 (silica gel, 93:7 hexane:EtOAc).
EXAMPLE 64
Preparation of 6-azido-5 ,6-dideoxy-2.3-β- (1-methylethylidene)-D-allofuranose
A mixture of the azido sugar Example 63 (2.8 g), and aqueo sulfuric acid solution (100 ml of 0.1% by volume) was heated to 90*C for 3 1/2 hours at which time the starting material was found (TLC) to be consumed. The pH of the reaction mixture was adjusted to at 7.5 with 1N NaOH and evaporated to dryness under high vacuum. The residue was coevaporated with DMF (2 x 20 ml) and treated with methanol (25 ml). The insoluble solids were removed by filtration and washed with methanol (2 X 20 ml). The combined filtrates were evaporated to dryness. The oily product thus obtained was dissolved in a mixture of dry DMF (10 ml), 2,2 dimethoxy propane (6 ml) and p-toluene sulfonic acid (50 mg) and stirred for 2 hours at room temperature. The solvents were evaporated under high vacuum and the residue was chromatographed over a silica gel column using a 4:1 hexane:EtOAc mixture as the mobile phase. After a fast moving spot, fractions containing th main product were combined and evaporated to obtain the title compound as a colorless oil; Yield 1.89 g; Rf=0.5 (silica gel, 2:1 hexane:EtOAc).
EXAMPLES 65-81
General procedure for the preparation of 5'-substituted -4-chloropyrrolo[2,3-d]Pyriιftidine-7-(1-β-D-ribosides)
A solution of the 5-substituted (H, OCH3, N3 or TBDMS-O-) 5-deoxy-isopropylideneribose (1 eq) in CCl4 (1.4 eq) and THF was cooled to -78 °C. Hexamethylphosphorous triamide (1.2 eq) was added dropwise and the reaction mixture stirred for 2 hours at -78 °C. This solution of 1-α-chloro sugar was used directly in the next step.
To a slurry or solution of the substituted 4-chloropyrrolo[2,3-d]pyrimidine (1.4 eq corresponding to the sugar) in DMF, was added in four portions, NaH (1.4 eq) over 10 minutes. The solution was stirred 30 minutes then the above solution of chloro sugar (-25°C) was added and the reaction was stirred for 24 hours. The mixture was concentrated, diluted wit EtOAc, filtered and the filtrate concentrated under vacuum. The residue was chromatographed on SiO2 gel using 2:1 hexane-EtOAc. The appropriate fractions were collected and evaporated to yield the protected nucleoside.
The protected nucleoside was deblocked by dissolving in 90% trifluoroacetic acid and stirring for 2 hours. The solvent was
evaporated and chased with methanol (3x). The product was crystallized from EtOH or EtOAc.
The compounds in Table IV (Examples 65-81) were prepared by this procedure:
TABLE IV Gpl-# EXAMPLE B ' D G m.p (ºC)
475 65 CH2OH I H 183-181°
66 CH2N3 I NH2 203-205°
406 67 CH2OH Br H >230°
448 68 CH3 I H 180-181° 449 69 CH3 CH3 H 155-157°
462 70 CH2OCH3 CH3H 142-144°
460 71 CH2OCH3 I H 179-180°
464 72 CH2OCH3 H H 122-124°
692 73 -CH2CH3 Br H 163-165°
690 74 -CH2CH3 I H 181-183º
529 75 CH2N3 I H 203-205°
554 76 CH3 Br H 174-175°
555 77 CH3 Br H 140-142°
569 78 CH3 SCH3 H 147-148°
605 79 CH2N3 Br H 147-148°
- - - 80 CH2CH2N3 I H foam
713 81 CH=CH2 I H 183-185 º
EXAMPLES 82 - 83
Preparation of 4-Amino-7-(5-amino-5-deoxy-1-)3
-D-ribofuranosy)-5-halopyrrolo[2,3-d]pyrimidines
A mixture of 4-chloro-5-iodo-7-[5-azido-5-deoxy-1-β-D-ribofuranosyl]pyrrolo[2>3-d]pyrimidine (Example 75 or 79) (500 mg), triphenylphosphine (550 mg) and pyridine (6 ml) was stirred under an argon atmosphere at room temperature for 24 hours.
Pyridine was evaporated under high vacuum and the residue was triturated with ether. The residual semi-solid was treated with ammonium hydroxide (5 ml). A small amount of ethanol was added to cause complete dissolution of the compound. After stirring for 5 hours at room temperature the mixture was evaporated under vacuum and the residue was triturated with water (10 ml). The insoluble material was removed by filtration and the pH of the filtrate was adjusted to 5.5 with dilute HCl. The solution was refiltered and lyophilized to obtain a hygroscopic solid, whose NMR was compatible with the structure.
The above hygroscopic solid was dissolved in methanol, saturated with dry ammonia at at -15°C, then heated in a steel bomb at 80 °C for 24 hours. The bomb was cooled and opened.
Ammonia and methanol were evaporated and the residue was dissolved in water, charcoaled and filtered. The filtrate was lyophilized to obtain the title compound as a hygroscopic solids The compounds listed in Table V were obtained by this procedure.
TABLE V
GP l-# EXAMPLE D F m.p (°C)
550 82 I H 166-206°
649 83 Br H 217-219° EXAMPLE 84
Preparation of 4-Amino-5-iodo-7-(5-acetylamino-5-deoxy-1-β- D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine
To an ice cold solution of the 5 '-amino compound from
Example 54 (50 mg), in dry pyridine (5 ml), acetic anhydride (0.5 ml) was added with stirring. The reaction mixture was allowed to warm to room temperature over a period of 1 hour at which time the reaction was complete. The flask was reimmersed into the cooling bath and 15 ml of methanol was added to the reaction mixture to neutralize unreacted acetic anhydride. The solvent was evaporated under reduced pressure and the residue was
purified by a short column chromatography to give the above- identified product, m.p. 160-163°C.
EXAMPLES 85 TO 113B
General Procedure for the Preparation of N4- Substituted-4-aminopyrrolo[2,3-d]pyrimidine Nucleosides
A suspension of the substituted 4-Cl-pyrrolo[2,3-d]pyrimidine nucleoside (1 eq) in EtOH containing the amine (3 eq) and triethylamine (5 eq) was added to a small stainless steel bomb (in the case of diamines a 25% excess of chloride was used) . The bomb was heated overnight (bath temperature 70-120°C), cooled, opened and the reaction mixture evaporated.
The product was crystallized from ethanol or ethyl acetate. The compounds in Table VI (Examples 85 to 113B) were prepared by this procedure:
TABLE VI
GPl-# Example B' D F G m. p . ( ° C)
334 85 CH2OH H NH2 H 249-250 °
394 86 CH2OH H N-inH Foam
dolinyl
393 87 CH2OH H N-proH Foam
linyl
296 88 CH2OH H cycloH Foam
pentyl-NH
376 89 CH2OH Br NH2 H Foam
321 90 CH2OH H NHϕ H Foam
476 91 CH2OH I N-Indolinyl H 185-188°
456 92 CH3 I NH2 H 245-246°
470 93 CH3 I N-indolinyl H 188-190°
457 94 CH3 I CH3NH H 226-228° 485 95 CH3 I N3 H 223-214°
498 96 CH3 CH3 NH2 H 212-214°
H 171-173°
461 97 CH3 CH3 N-indolinyl
463 98 CH2OCH3 I NH2 H 216-218°
465 99 CH2OCH3 I CH3NH H 188-189°
474 100 CH2OCH3 H N-indolinyl H 205-208°
48o 101 CH2OCH3 H CH3NH H 163-164°
513 102 CH3 H N-piperazinyl H 216-219°
499 103 CH3 I N,N'-piperH 220-223° azinyl1
500 104 CH3 I -NH(CH2)6NH-1/ H 227-229°
512 105 CH3 I -NH(CH2)2NH-1/ H > 230°
559 106 CH3 I NH2 CH3S 200-202°
H foam
561 107 CH3 H 1,4-piperazinyl
606 108 CH2N3 Br NH2 H 182-184°
639 109 CH3 I NHφ H >230°
581 110 CH3 CO2C2H5 NH2 H 162-1688°
681 111 CH2OH I NHϕ H 224 -225 °
680 112 CH2OH I NH (4-Clϕ) H 234 -235 °
689 113 CH2OH I NH (4-CH30-ϕ) H 212-214 °
711 113A CH2CH2N3 I NH2 H 151-153 °
714 113B CH=CH2 I NH2 H 224-226 °
1/ Dimers having two pyrrolo[2,3-d]pyrimidine riboside moieties linked by the listed diamine.
2/ Dimer with purine riboside.
EXAMPLE 114
Preparation of 5-Iodo-7-f5-deoxy-1-β-D-ribofuranosyl)
pyrrolo[ 2 ,3-d]pyrimidin-4(3H)-thione
A solution of 4-chloro-5-iodo-7-[5-deoxy-1-β-D-ribofuranosyl]pyrrolo[2,3-d]pyrimidine (Example 68) (250 mg, 0.60 mmol) and thiourea (250 mg) in absolute EtOH was refluxed gently under an argon atmosphere for 16 hours. The solvent was
evaporated under reduced pressure and the residue was triturated with water (10 ml). The solid was collected by filtration, washed with water and dried in air: Yield 200 mg (81%); m.p. 161-163°C.
EXAMPLES 115 TO 120
General Procedure for S-Alkylation of 5-Iodo-7-(5-deoxy- 1-β-D-rlbofuranosyl)pyrrolof2.3-d]pyrimidin-4(3H)-thione
To a solution of 5-iodo-7-(5-deoxy-1-β-D-ribofuranosyl) pyrrolo[2,3-d]pyrimidine-4-thione (Example 114) (50
mg) in concentrated NH4OH (10 ml), the appropriate alkylating agent (e.g. methyl iodide, alkyl or substituted benzyl bromide) was added and the mixture stirred at room temperature for 20 hours. Volatile material was evaporated under reduced pressure and the residue triturated with ether. To the residue, water (5 ml) was added and the solid was collected by filtration and washed with water.
The products obtained by this procedure (Examples 115 to 120) are listed in Table VII:
TABLE VII
GPl-# Example Bi D F m . p .
482 115 CH3 I SCH3 212-233 °
493 116 CH3 I SCH2CH=CH2 192-193 °494 117 CH3 I SCH2ϕNO2 (4 ) 224-226 º 502 118 CH3 I SC4H9 186-187 °
503 119 CH3 I SCH2ϕ 212-213
511 120 CH2OH I SCH3 214-150
EXAMPLE 121
Preparation of 4-Phenyl-7-(1-β-D-ribofuranosyl)pyrrolo
[2,3-d]pyrimidine
To a solution of 4-chloro-7-[2,3-O-(1-methylethylidene)-1-β-D-ribofuranosyl]pyrrolo[2,3-d]pyrimidine (200 mg) and
phenylboronic acid (250 mg) in dry diglyme (10 ml) was added palladium-tetrakis-triphenylphosphine (30 mg), followed by aqueous Na2CO3 solution (0.2 ml of 2M solution). The reaction mixture was heated to 90 °C under anhydrous conditions for 6 hours. The solvent was evaporated under high vacuum and the residue was purified by HPLC on a reverse phase C-18 column. The purified intermediate was treated with 2 ml of trifluoracetic acid (80%) and stirred for 15 minutes then evaporated under high vacuum and the residue crystallized from ethanol; yield 20 mg, m.p. 163-164°C.
EXAMPLES 122 TO 124
General Procedure for the Preparation of 4-Amino- and
4-Arylamino-5-aryl-7-(1-β-D-ribofuranosyl)pyrrolo- [2,3-d]pyrimidines
To stirred mixture of the 4-amino- or 4-arylamino-5- iodopyrrolo[2,3-d]pyrimidine riboside (or corresponding hydroxyl protected compound) (0.1 m mole), Pd(PPh3)4 (10 mg, 0.01 mole) in diglyme was added a solution of the arylboronic acid (0.4 mmol) in EtOH and 0.4 ml of aqueous 2 M Na2CO3. The mixture was heated to 100ºC and the reaction monitored by TLC. After the reaction was complete, the cooled mixture was filtered and concentrated under vacuum. The residue was chromatographed over SiO2, eluting with CH2Cl2-MeOH mixtures or by HPLC on a Bondapak C-18 column with a MeOH-H2O gradient.
The compounds in Table VIII may be prepared by this
procedure:
TABLE VIII
Example D F M.P. C O
122 0 ϕNH
123 0 NH2
718 124 2-furanyl ϕNH foam
EXAMPLES 125-126
General Procedure for the Preparation of 4-Amino- and 4-Arylamino-5-aryl-7- (5-deoxy-l-/3-d- ribofuranosyl) pyrrolo[2.3-d1pyrimidines
The above-identified compounds were prepared as described in Example 122-124 from the 4-amino- or 4-arylamino-5-iodo-7-(5- deoxy-1-β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine and an
arylboronic acid.
The compounds in Table IX were prepared by this procedure:
TABLE IX
GPl # Example B' D F G M.P. ( ºC)
684 125 CH3 Phenyl NH2 H 106-109
683 126 CH3 Phenyl NH-ϕ H 207-208
EXAMPLE 127
Preparation of 4-Chloro-5-iodo-7-f6-deoxy-1-β- D-allofuranosyl)pyrrolo[2,3-dlpyrimidine
The above-identified compound was prepared according to the general procedure used for Examples 65-81; m.p. 211-213°C.
EXAMPLE 128
Preparation of 4-Amino-5-iodo-7-(6-deoxy-1-β-D- allofuranosyl)pyrrolo[2,3-dlpyrimidine
The 4-chloro compound, Example 127, was heated in a steel bomb with methanolic ammonia at 120ºC for 12 hours followed by the usual work.up (see Example 85). The product was obtained as a white crystalline solid; m.p. 206-208°C.
EXAMPLES 129-130
General Procedure for the Preparation of 4-Amino-5-halo- -7-(5,6-dideoxy-1-β-D-allofuranosyl)pyrrolo[2.3-d]pyrimidine The above-described compounds were prepared by the general procedures for Examples 65 to 81.
The compounds obtained by this procedure are listed in Table X.
TABLE X
GPl # Example B' D F G M. P. ( ºC)
693 129 CH2CH3 Br NH2 H 229-230
691 130 CH2CH3 I NH2 H 233-234
EXAMPLE 131
Preparation of 4-Amino-3-bromopyrazolo[3.4-d1pyrimidine
The above-identified compound was prepared as described: Leonova, T.; Yashunskii, V.; Khim. Get. Soed.. 1982, 982.
EXAMPLE 132
Preparation of 4-Amino-3- (cvanomethyl)pyrazolo[3.4-d]pyrimidine
The above-identified compound was prepared as described: Carboni, R.,; Coffman, D.,; Howard, E.; J.Am. Chem. Soc., 1958 80:2838.
EXAMPLE 133
Preparation of 4-Amino-3-cvanopyrazolo[3.4-d]pyrimidine
The above-identified compound was prepared as described: Taylor, E.; Abul-Hsan, A.; J. Org. Chem.. 1966, 31:342.
EXAMPLE 134
Preparation of 4-Amino-3-phenylpyrazolo[3.4-dlpyrimidine The above-identified compound was prepared from trimethyl orthobenzoate as described: Kobayashi, S.; Chem. Pharm. Bull.
(Jap.) 1973, 21:941.
EXAMPLES 135-139
General Procedure for the Preparation of Aryl Thiomorpholides A mixture of the aromatic carboxaldehyde (0.1 mole), sulfur (4.8 g, 0.15 mole) and morpholine (18 mL, 0.15 mole) was heated at 180°C for 3-5 hours then cooled and diluted with H2O. The solid was collected by filtration or, if oily, extracted with CH2Cl2, dried (Na2SO4) and concentrated. The crude product was recrystallized from alcohol, alcohol-H2O mixtures or
chromatographed over SiO2.
The compounds in Table XI were prepared by this procedure:
TABLE XI
Example Aryl M. P. ( ºC)
135 4-CH3Oϕ 95-98 °
136 4-Clϕ 137-140 °
137 2-Brϕ - - - -
138 2 -thienyl 75-77 °
3 -thienyl 84-87 °
139 3-CH3Oϕ 134-139 °
EXAMPLES 140-144
General Procedure for the Preparation of
5-Amino-3-aryl-4-cyanopyrazoles
The above-identified compounds were prepared from the corresponding aryl thiomorpholides (Examples 135-139) following the general procedure described: Tominaga, Y.,; et al.; J. Heterocyclic Chem.. 1990, 27:647.
The compounds listed in Table XII were prepared by this procedure:
TABLE XII
Example Aryl M.P. ( ºC)
140 4-CH3Oϕ 155-160°
141 4-Clϕ 218-222°
142 2-Brϕ - - - - 143 2-thienyl 260-265'
144 3-thienyl 229-231º
EXAMPLES 145-148
General Procedure for the Preparation of 5-Amino-3-aryl-4- carboxamidopyrazoles
The above-identified compounds were obtained from the corresponding cyano compounds (Example 140-144) following the general procedure described: Kobayashi, S.; Chem. Pharm. Bull (Jap.). 1973, 21:941.
The compounds listed in Table XIII were prepared by this procedure:
TABLE XIII
Example Aryl M.P. CO
145 ϕ 203-205°
146 4-CH3Oϕ - - - -
147 4-Clϕ 210-215°
148 2-Brϕ - - - -
EXAMPLE 149-154
General Procedure for the Preparation of 4-Amino-3- arylpyrazolo[3.4-dlpyrimidines
A mixture of the 5-amino-3-aryl-4-cyanopyrazole and formamide (5 ml/g) under N2, was refluxed (190-200°C) with stirring for 4 hours. The cooled mixture was diluted with H2O and the solid collected by filtration. The crude products were
used directly for subsequent steps or purified py recrystallization. The compounds listed in Table XIV were prepared by this procedure.
TABLE XIV
Example Aryl M.P. ( ºC)
149 ϕ >220
150 4-CH3Oϕ >220
151 4-Clϕ >220
152 2-Brϕ >220
153 2-Thienyl >220
154 3-Thienyl >283 (dec.)
EXAMPLES 155-158
General Procedure for the Preparation of 3-Arylpyrazolo- T3 , 4-d1pyrimidin-4-ones from 5-Amino-3-aryl-4- carboxamidopyrazoles
A mixture of the 5-amino-3-aryl-4-carboxamidopyrazole and formamide (5 ml/g) was refluxed at 190°-200°C for 2 hours, cooled and diluted with H2O. The solid was collected by filtration and dried under vacuum. Further purification was effected by dissolving the compound in dilute sodium hydroxide, followed by charcoal treatment and precipitation with acetic acid.
The compounds listed in Table XV were prepared by this procedure:
TABLE XV
Example Arvl M.P. ( ºC)
155 ϕ >200°
156 4-CH3Oϕ >220°
157 4-Clϕ >220°
158 2-Thienyl >220°
EXAMPLES 159-160
General Procedure for Preparation of 3-Arylpyrazolo
[3 ,4-d]pyrimidin-4-ones from 4-Amino-3-aryl
pyrazolo[3,4-d]pyrimidines
To a well stirred slurry of the 3-aryl-4-aminopyrazolo[3,4-d]pyrimidine (25 mmoles) in 175 ml of 9% HCl at 0 to 5°C, was added dropwise, over 45 minutes, an aqueous solution of sodium nitrite (15.0 g in 30 ml). The mixture was allowed to warm to room temperature and solid sodium nitrite (5.0 g) was added. After 15 minutes the mixture was cautiously heated to boiling (foaming!), then cooled. The product was collected by
filtration, rinsed with H2O and dried at 50 °C under vacuum.
The compounds listed in Table XVI were prepared by this procedure:
TABLE XVI
Example Aryl M.P. ( ºC)
159 ϕ >220º
160 2-thienyl >220º
EXAMPLES 161-163
General Procedure for the Preparation of N4-Aryl and N4-Alkyl substituted 4-amino-3-aryl-pyrazolo[3,4-d]pyrimidines
A mixture of the 3-arylpyrazolo[3,4-d]pyrimidin-4-one (15 mmoles), POCl3(18 ml, 195 mmoles) and diethylaniline (5 ml, 31 mmoles) was refluxed under N, for 4 hours then concentrated under vacuum. The residue was decomposed by addition of ice and extracted (4x) with 3:1 ether-ethyl acetate. The combined organic extracts were washed with water and dried (Na2SO4). The solution was concentrated under vacuum and the crude 4-chloro-3-arylpyrazolo[3,4-d]pyrimidine (50-70% yield) was added to a solution of amine (2.2 equivalents) in EtOH (25 ml/mmole chloro compound). The mixture was heated to reflux for 30 minutes then cooled and the product collected by filtration and rinsed with
EtOH. Recrystallization from alcohol-ethyl acetate mixtures gave the title compounds.
The compounds listed in Table XVII were prepared by this procedure:
TABLE XVII
Example 3- Aryl 4-Arylamino M. P. ( ºC)
161 ϕ ϕ 229-232 °
162 ϕ 4-ClϕNH 232-233 °
163 ϕ 4-CH3OϕNH 218-220 °
EXAMPLE 164
Preparation of 3-Bromopyrazolo[3 ,4-d]pyrimidin-4-one The above-identified compound was prepared as described:
Chu, I.; Lynch, B.; J. Med. Chem.. 1975, 18:161.
EXAMPLE 165
Preparation of 3-Bromo-1-(2,3,5-β-tribenzoyl-1-β-D- ribofuranosyl)pyrazolo[3,4-d]pyrimidin-4-one
The above-identified compound was prepared as described:
Cottam, H.; Petrie, C,; McKernan, P.; Goebel, R.; Dailey, N.;
Davidson, R.; Robins, R.; Revankar, G.; J. Med. Chem.. 1984, 22:1120.
EXAMPLE 166
Preparation of 3-Substituted-4-chloro-1-(2,3.5,-O-tribenzo -1-β-D-ribofuranosyl)pyrazolo[3,4-d]pyrimidin-4-ones
The above-identified compounds may be prepared from the corresponding pyrazolo[3,4-d]-pyrimidones analogously to the procedure described in Example 2.
EXAMPLES 167 TO 169
General Procedure for Preparation of 3-Substituted 4-amino- and 4-(arylamino)-1-(1-β-D-ribofuranosyl)pyrazolo[3,4-d]pyrimidines
To a slurry of the 3-substituted-4-chloropyrazolo[3,4-d]pyrimidine nucleoside tribenzoate (1.0 eq) (Example 166) in a mixture of EtOH and THF, was added ethanolic ammonia or the amine (1.5 eq) and Et3N (3.5 eq). The reaction rapidly became
homogenous and after 0.5-12 hours, was evaporated. The residue was dissolved in CH2Cl2, washed with aqueous K2CO3 then H2O and the solution dried (Na2SO4) . After evaporation, the residue was
recrystallized or chromatographed on SiO2 gel using CH2Cl2-MeOH mixtures. The resulting tribenzoate of the title compound was deblocked by stirring in methanolic NaOMe. The mixture was neutralized with amberlite IR-120(+) resin, filtered and evaporated. The residue was recrystallized to give the title compounds.
Examples 167 to 169 listed in Table XVIII were prepared by this procedure:
TABLE XVIII
GPl-# Example 3- 4- m.p. ( ºC)
596 167 I NH2 180-185°
469 168 Br N-in195-196°
dolinyl
536 169 CH, NH2 241-242°
EXAMPLE 170
Preparation of 4-(N-indolinyl)-1-(1-β-D- ribofuranosyl)pyrazolo[3,4-d]pyrimidine
A solution of the bromide (Example 168) (351 mg, 0.78 mmol) in methanol containing 300mg 10% Pd-C and Raney Ni was
hydrogenated at 40 psi until the reaction was complete as judged by TLC. The mixture was filtered, the filtrate concentrated and the product collected by filtration to give the title compound: 120 mg (42%); m.p. 215-219°.
EXAMPLE 171
Preparation of 3-Bromo-1-[2,3-β-(1-methylethylidene) -1-β-D-ribofuranosyl]Pyrazolo[3,4-d]pyrimidin-4-one Crude 3-bromoallopurinol riboside (prepared from 33.0 g of tribenzoate and NaOMe/MeOH (Example 83) was added to a cold (5°C), stirred solution of 1 M ethanolic HCl (6.5 ml) and dimethoxypropane (20 ml) in 1.1 L of Me2CO. The mixture rapidly became homogenous and was stirred 45 minutes until complete (TLC). To the solution was added Na2CO3 (5.0 g), concentrated NH4OH (5 ml) and the mixture was stirred until the pH reached 6-7. The reaction was filtered and evaporated to a solid. The residual solid was dissolved in 300 ml of boiling EtOH and the solution concentrated by distilling EtOH to a final volume of 150 ml. The solution was chilled overnight and the solid collected by filtration and rinsed with cold EtOH. After drying (50°C), 16.7 g (86%) of the title compound were obtained; m.p. 221-224°C.
EXAMPLE 172
Preparation of 3-Bromo-1-[2,3-β-(1-methylethylidene)-5-O- (4-methylbenzenesulfonyl)-1-β-D-ribofuranosyl]pyrazolo
[3,4-d]pyrimidin-4-one
To a solution of the isopropylidene alcohol (Example 171) (3.0 g, 7.74 mmol) in pyridine (18 ml) at 0°C was added p- toluenesulfonyl chloride (1.77 g, 9.30 mmol). The reaction was held at 0°C for 3 hours then poured into 160 ml of cold H2O with stirring. The mixture was allowed to settle, the H2O decanted and the residue dissolved in CH2Cl2. The CH2Cl2 solution was washed with 0.5 N H2SO4, 5% aqueous K2CO3 and dried (Na2SO4).
After evaporation under vacuum, 4.03g (96% yield) of the title compound were obtained as a foam.
EXAMPLE 173
Preparation of 1-[5-Azido-5-deoxy-2,3-β- (1-methylethylidene)-1-β-D-ribofuranosyl]-3-bromopyrazolo-
[3.4-d]pyrimidin-4-one
To a warm stirred solution of NaN3 (7.69 g, 0.12 moles) in DMSO (70 ml) was added the tosylate (Example 172) (16.0 g, 0.03 mol). The solution was rapidly heated to 80 °C and maintained at this temperature for 45 minutes. After cooling, the reaction mixture was added with stirring to H2O (600 ml). The mixture was extracted 4x with CHCl3 (75 ml) and the combined CHCl3 extracts
were washed with H2O, dilute brine, dried (Na2SO4) and
concentrated to give 11.0 g of a white foam. TLC (9:1 CH2Cl2- MeOH on SiO2) indicated a mixture of three products in the approximate ratio of 1:2:1. The middle spot was subsequently determined to be the desired azide.
The mixture was purified by chromatography on a 10 x 15 cm column of SiO2 eluting with 1% Me2CO in CH2Cl2 then increasing concentrations of Me2CO. The first product eluted with 4% acetone. 13C-NMR indicated a lack of tosyl and azide functions at C-5'. Chemical shifts of the ribose C5' suggested a
cyclonucleoside. The desired azide eluted with 6% Me2CO. Its identity was confirmed by 1H and 13C NMR. Further elution afforded the third product which appeared by 13C-NMR to also be a cyclonucleoside.
The fractions containing the desired product (middle TLC spot) were combined and evaporated to give the title compound; 5.90 g (48% yield), m.p. 168°C (d).
EXAMPLE 174
Preparation of 3-Amino-1-[2,3-β-(1-methylethylidene) -1-β-D-ribofuranosvπpyazolo[3,4-dlpyrimidin-4-one
A mixture of bromide (Example 171) (2.35 g, 6.1 mmol) CuCl
(88 mg) and Cu (101 mg) in MeOH (45 ml) was placed in a bomb and saturated with gaseous NH3. The bomb was sealed and heated to
110 °C for 10 hours. After cooling, the bomb was opened, the contents filtered and the filtrate evaporated. The residue was chromatographed on SiO2 gel using 9:1 CH2Cl2-MeOH. The appropriate fractions were combined and evaporated to yield the. title compound as a solid; 2.1 g (98% yield); m.p. 142-144'C.
EXAMPLE 175
Preparation of 3-Iodo-1-[2,3-β-(1-methylethylidene) -1-β-D-ribofuranosylIpyrazolo[3,4-dlpyrimidin-4-one A mixture of the amine (Example 174) (2.58 g, 8.0 mmole), isoamyl nitrite (30 ml), methylene iodide (20 ml) and CH3CN was refluxed under argon for 10 minutes. The cooled mixture was evaporated and chromatographed on SiO2 gel using 2% methanol in methylene chloride. Appropriate fractions were combined and evaporated to give the title compound: 1.08 g (66% yield); m.p. > 220°C.
EXAMPLE 176
Preparation of 3-Iodo-1-[2,S-O-d-methylethylidene)-S-O- M-methylbenzenesulfonyl)-1-β-D-ribofuranosyl]pyrazoIo- [3.4-d]pyrimidin-4-one
The above identified compound was prepared analogously to the procedure described for Example 172.
EXAMPLE 177
Preparation of 3-Iodo-1-[5-azido-5-deoxy-2,3-β-(1- methylethylidene)-1-g-D-ribofuranosyl]pyrazolo- [3,4-d]pyrimidin-4-one
The above identified compound was prepared analogously to the procedure described for Example 173 in 45% yield; m.p.
203°C(d).
EXAMPLE 178
Preparation of 3-Halo-4-chloro-1-[5-azido-5-deoxy-2,3-β- (1-methylethylidene)-1-β-D-ribofuranosyl)]pyrazolo(3,4-d] pyrimidine
The above-identified compounds were prepared analogously to the procedure described for Example 2 from the pyrimidin-4-one (Example 173 or 177). The title compounds were obtained as unstable yellow oils and used immediately in the next step.
EXAMPLES 179 to 181
General Procedure for the Preparation of 4-Amino and
4-hvdrocarbyl-amino-1-(5-azido-5- deoxy-1-β-D-ribofuranosyl)pyrazolo[3,4-d]pyrimidines
To a solution of the chloro azide (Example 178) (1 eq) in 1:1 THF-EtOH (10% w/v) was added to the amine (1.2-2.0 eq) and excess Et3N (for the 4-amino compounds the solution was saturated
with NH3 gas). The resulting solution was stirred for 2-24 hour and checked for complete reaction by TLC (SiO2; 9:1
CH2Cl2:Me2CO).
The reactions using amines were worked up in the following manner. The reaction mixture was evaporated, the residue dissolved in CH2Cl2 and the solution washed with aqueous NaHCO3, then H2O and dried (Na2SO4). Concentration of the CH2Cl2 solution and chromatography of the residue over Si02 gel using CH2Cl2- Me2CO mixtures gave the purified isopropylidene N4-substituted compounds. The isopropylidene 4-amino compounds were isolated by evaporating the reaction mixture and recrystallizing the residue from EtOH.
The isopropylidene compounds were deblocked using the procedure described under Example 3.
The compounds in Table XIX (Examples 179 to 181) were prepared by this procedure:
TABLE XIX
GPl-# Example D F m.p. ( ºC)
507 179 Br NH2 169-170°
501 180 Br N-indolinyl 133-138º - - - 181 I NH2 193-195°
EXAMPLES 182 TO 185
General Procedure for the Preparation of 4-Amino- and
4-Substituted-amino-1-(5-amino-5-deoxy-1-β-D- ribofuranosyl)-3-halo-pyrazolo[3,4-dlpyrimidines
and Their Hydrochloride Salts
A solution of the azide (Examples 179 to 181) (1.0
equivalent) and triphenylphosphine (1.5 equivalents) in pyridine (5 ml/g of azide) was stirred for 2 hours and checked by TLC (9:1 CH2Cl2-MeOH) for completion. To the reaction mixture was added concentrated NH4OH (1.25 ml/g of azide) and the solution stirred overnight. The solution was evaporated to dryness, slurried in Et2O, filtered (3x) and the insoluble residue dried under vacuum. The resulting solid was recrystallized or converted to its HCl salt (0.1 N HCl, EtOH) and crystallized to give the title
compounds.
The compounds in Table XX (Examples 182-185) may be prepared by this procedure:
TABLE XX
GPl-# Example # D F m.p. (°C (HCl salt))
515 182 Br NH2 >230°
516 183 Br N-indolinyl 170° (broad)
547 184 I NH2 188°
558 185 Br 1,4- piperazinyl1 195° (broad)
1/ a dimer
EXAMPLE 186
Preparation of 1,2.3-O-Triacetyl-5-deoxy-D-ribofuranoside The above-identified compound was prepared as described: Snyder, J.; Serianni, A.; Carbohydrate Research, 1987, 163 : 169.
EXAMPLE 187
Preparation of 1,2,3-β-Triacetyl-5-azido-5-deoxy-D-ribofuranoside
To a cooled solution of 5-azido-5-deoxyribose (6.2 g, 0.035 mole) (Example 51) in 10 ml of pyridine was added acetic
anhydride (18 ml) and the mixture stirred for 24 hours at room temperature. The mixture was concentrated under vacuum, the residue dissolved in CH2Cl2 and the solution washed with 5%
NaHCO3. The organic layer was then washed with 0.5 N H2SO4, dried (Na2SO4) and evaporated. The residue was filtered through a plug of SiO2 gel (CH2Cl2) and the filtrate concentrated to afford the
title compound, 9.0 g (98% yield) as a semisolid mixture of α and β isomers.
EXAMPLES 188-203
General Procedure for the Preparation of
5'-Substituted-3.4-disubstituted- pyrazolo[3,4-dlpyrimidine Nucleosides
To a slurry of the 3,4-disubstituted pyrazolo[3,4-d]pyrimidine (5.0 mmol) in nitromethane, nitroethane or
benzonitrile under N2, was added the acyl-protected ribose (5.0- 7.0 mmoles). To the stirred mixture, was added BF3.Et2O (7.0 mmoles) and the mixture was refluxed for 90 minutes, then cooled and evaporated under vacuum. If a 5'-deoxy derivative was used, Et3N was added prior to the evaporation of the solvent (to complex BF3. Et2O).
The residue was taken up in CH2Cl2, filtered and
chromatographed over SiO2 gel using CH2Cl2-MeOH gradients. Later fractions contained the N-2 isomer. Fractions containing the desired N-l isomer were combined and evaporated to yield the title compounds as foams.
The compounds in Table XXI (Examples 188-203) were prepared by this procedure:
TABLE XXI
Example # B' D F M. P. (ºC)
188 ϕCO2CH2 CN NH2 foam
189 ϕCO2CH2 CH2CN NH2 foam
190 ϕCO2CH2 ϕ NHϕ foam
191 CH2N3 Br NH2 foam
192 CH2N3 CN NH2 foam
193 CH2N3 CH2CN NH2 foam
194 CH2N3 ϕ NH2 foam
195 CH2N3 4-Clϕ NH2 foam
196 CH2N3 4-CH3Oϕ NH2 foam
197 CH2N3 2-thienyl NH2 foam
198 CH3 ϕ NH2 foam
199 CH3 4-CH3Oϕ NH2 foam
200 CH3 4-Clϕ NH2 foam
201 CH3 2-thienyl NH2 foam
202 CH3 3-thienyl NH2 foam
203 CH3 ϕ NHϕ foam
EXAMPLE 204
General Procedure for the Preparation of 3-Substituted
1-(5-azido-5-deoxy-2,3-O-diacetyl-1-β-D-ribofuranosyl)-4-chloropyrazolo[3,4-d]pyrimidines,
5'-Deoxy Analogs and Protected 5'-Hydroxy Analogs
The above identified compounds were prepared from the pyrazolo[3,4-d]pyrimidone esters analogously to the procedure described in Example 2 and were used immediately in the next step.
EXAMPLES 205-221
General Procedure for the Preparation of 3,4-Disubstituted- 1-(5-azido-5-deoxy-1-β-D-ribofuranosyl))pyrazolo- [3,4-dlpyrimidines, 5'-Deoxy Analogs and
5'-Hydroxy Analogs
The above identified compounds were prepared from the diesters analogously to the procedure described in Example 167-169. Methanolic NH3 (method A) or NaOMe (method B) was used to deblock the acyl-protected nucleosides (Examples 188-204). In the case of the cyano-substituted compounds, these methods led to
different products by further reaction of the cyano group. The title compounds were isolated by conventional techniques.
The compounds listed in Table XXII (Examples 205-221) were prepared by this procedure.
TABLE XXII
Example GPl-f B' D F Method M.P. (ºC)
205 612 CH2OH CH2CN NH2 A 220º (dec)
206 613 CH2OH CH2C(=NH)OCH3 NH2 B 75º (dec)
207 695 CH2OH ϕ NHϕ B 220-224°
208 507 CH2N3 Br NH2 B 172° (d)
209 623 CH2N3 C(=NH)NH2 NH2 A 203-206º
210 624 CH2N3 CH2CN NH2 A 153-156°
211 641 CH2N3 ϕ NH2 B 203-205°
212 662 CH2N3 4-Clϕ NH2 B 175-177°
213 666 CH2N3 4-CH30ϕ NH2 B 153-155°
214 654 CH2N3 2 -Thienyl NH2 B 180-181°
215 667 CH2N3 ϕ NHϕ B 120-125°
216 663 CH3 ϕ NH2 B 223-224°
217 678 CH3 4-Clϕ NH2 B 130-133°
TABLE XXII
Example GPl-# B' D F Method M.P. (ºC)
218 679 CH3 4-CH3Oϕ NH2 B 175-176º
219 664 CH3 2-Thienyl NH2 B 174-175º
220 685 CH3 3-Thienyl NH2 B 153-154°
221 683 CH, ϕ NHϕ B 207-208"
EXAMPLES 222-229
General Procedure for the Preparation of 4-Amino- and
4-Arylamino-3-substituted-1-(5-amino-5-deoxy
-1-β-D-ribofuranosyl)pyrazolo[3,4-dlpyrimidines
and Their Salts
The above-identified compounds were prepared from the 5'-azides (Examples 205-221) by catalytic hydrogenation as described in Examples 15-20 (method A) or triphenylphosphine followed by ammonium hydroxide as described in Examples 82-83 (method B). The salts were prepared by standard methods.
The compounds listed in Table XXIII were be prepared by these methods:
TABLE XXIII
GPl-# Example 3 - 4- Method Salt M . P .(ºC)
515 222 Br NH2 B HCl >230 °
614 223 H NH2 A HBr 260-165 °
625 224 CH2CN NH2 B 1 175 ° (d)
642 225 ϕ NH2 A HCl 218-219 °
682 226 2-thi< ≥nyl NH, B HCl >220 °
694 227 4-CH3Oϕ NH2 A - - -1/ 222-225 °
701 228 4-Clϕ NH2 B HCl 189-194 °
704 229 ϕ NHΦ A CF3CO2H 185-190 °
EXAMPLES 230-231
General Procedure for the Preparation of
4-Amino- and 4-Arylamino-1-(5-amino-2.3-O-diacetyl- 5-deoxy-1-β-D-ribofuranosyl)-3-substitutedpyrazolo
[3,4-d]pyrimidines
A slurry of 10% Pd-C in a solution (MeOH or EtOH with THF, dioxane or EtOAc) of the 5'-azido-2',3'-diacetate nucleoside (Examples 191-198) is hydrogenated in a Parr shaker at 40 psi. After disappearance of the starting material (TLC), the mixture is filtered and concentrated under vacuum at a temperature of less than 40°C. The residual product is purified by
recrystallization or HPLC.
The compounds listed in Table XXIV may be prepared by this method:
1/ Not a salt.
TABLE XXIV
GPl-# Example D F C1,C2 M.P. (ºC)
- - - 230 Br NH2 OAC - - -
- - - 231 0 NHϕ OAc - - -
EXAMPLES 232-233
General Procedure for the Preparation of
3-Substituted-4-(1.1-dicarboethoxyalkyl) -
1- (2.3.5-O-tribenzoyl-l-g-D-ribofuranosyl) pyrazolo
T3.4-d]pyrimidines
To a stirred solution of the diethyl (alkyl)malonate (0.10 mol) in dry DMF (100 ml) under N2 was added 80% NaH in mineral oil (0.125 mol). After stirring (cooling) for 10 minutes, a solution of the 3-substituted-4-chloro-1-(2,3,5-tribenzoyl-1-β-D-ribofuranosyl)pyrazolo[3,4-d]pyrimidine (0.0.9 mol) (Example 178) in DMF (75 ml) was added dropwise. The solution was cooled and anhydrous trimethylamine was bubbled into the solution for 4 minutes. The solution was stirred for 3 hours at room
temperature (heating may be required in some cases) then quenched with dilute acetic acid. The mixture was extracted with ether-ethyl acetate (9:1) and the organic extract dried (Na2SO4), concentrated and pumped under vacuum. The residue was
chromatographed on SiO2 gel with CH2Cl2-acetone mixtures and the appropriate fractions combined and evaporated to yield the title compounds. The identity of the compounds were confirmed by 1H and 13C NMR.
The compounds listed in Table XXV were prepared by this procedure:
TABLE XXV
Example D F M.P. (°C)
232 Br CH(CO2C2H5)2 foam
233 Br Cϕ(CO2C2H5)2 foam
EXAMPLES 234-235
General Procedure for Preparation of 4-Alkyl,
4-Arylalkyl- and 3 ,4-disubstituted- 1-(1-β-D-ribofuranosyl)pyrazolo[3,4-d]pyrimidines The diester (Examples 232-233) was dissolved in aqueous ethanolic sodium hydroxide and heated. The solution was
neutralized with acetic acid, evaporated, extracted with hot ethanol and the extract then evaporated and recrystallized or evaporated on SiO2 gel. The SiO2 gel was loaded on a column of
SiO2 gel and the product eluted with CH2Cl2-MeOH mixtures. The appropriate fractions more combined and evaporated to yield the title compounds.
The compounds described in Table XXVI were prepared by the procedure:
TABLE XXVI
GP-1-# Example D F M. P .(ºC)
719 234 Br CH3 204-205
235 Br CH2ϕ - - - - -
EXAMPLES 236-237
General Procedure for Preparation of 3-Substituted-4- (1,1-dicarboethoxyalkyl)-1-(5-azido-5-deoxy-2.3-O- diacetyl-1-β-D-ribofuranosyl)pyrazolo[3,4-d]pyrimidines
The above-identified compounds are prepared analogously by the procedure described in Examples 232-233 using the 3-substituted-(5-azido-5-deoxy-2,3-O-diacetyl-1-β-D-ribofuranosyl)-4-chloropyrazolo[3,4-d]pyrimidine.
The compounds listed in Table XXVII may be prepared by this procedure:
TABLE XXVII
Example D F M.P. (ºC)
236 Br Cϕ(CO2C2H5)2 - - - -
237 Br CH(CO2C2H5)2 - - - -
EXAMPLES 238-239
General Procedure for Preparation of 4-Alkyl-, 4- Phenylalkyl-and 4-Substituted-3-Substituted-1-(5- azido-5-deoxy-1-β-D-ribofuranosyl)pyrazolo[3,4-d]- pyrimidines
The above identified compounds are prepared analogously by the procedure described in Examples 234-235 from the 5'-azide esters described in Examples 236-237.
The following compounds listed in Table XXVIII may be prepared by this procedure:
TABLE XXVIII
GPl-# Example D F M.P. (ºC)
- - - 238 Br CH3 - - - - - - - - 239 Br CH2O - - - - -
EXAMPLES 240-241
General Procedure for the Preparation of 4-Alkyl-.
4-Phenylalkyl- and 3.4-Disubstituted-1- (5-amino-5-deoxy-1-β-D-ribofuranosyl)
pyrazolo[3.4-d]pyrimidines
The above-identified compounds may be prepared by reduction of the 5-azido ribosides listed in Examples 238-239 by catalytic hydrogenation as described in Examples 15-20 or by treatment with triphenylphosphine and ammonium hydroxide as described in
Examples 182-185.
The compounds listed in Table XXIX are prepared by this procedure:
TABLE XXIX
GPl-# Example D M.P. (ºC)
- - - - 240 Br CH3 - - - - - - - - - 241 Br CH2ϕ - - - - -
EXAMPLES 242-243
General Procedures for the Preparation of 3-Substituted-1- (5-deoxy-2.3-O-diacetyl-1-β-D-ribofuranosyl)-4- (1,1-dicarboethoxyalkyl)pyrazolo[3,4-dlpyrimidines
The above-identified compounds are prepared analogously to the procedure described in Examples 232-233 using 3-substituted- 4-chloro-1-(5-deoxy-2,3-O-diacetyl-1-β-D- ribofuranosyl)pyrazolo[3,4-d]pyrimidines (Examples 198-203).
The compounds listed in Table XXX may be prepared by this procedure:
TABLE XXX
GPl-# Example D M.P. (ºC)
- - - 242 Br CH(CO2C2H5)2 - - - - - - - 243 Br C(CO2C2H5)2CHϕ - - - - -
EXAMPLES 244-245
General Procedure for Preparation of 4-Alkyl-, 4-Phenylalkyl-,
or 4-Substituted-3-substituted-1-(5-deoxy-1- β-D-ribofuranosyl)pyrazolo[3.4-d]pyrimidines
The above-identified compounds may be prepared from the esters (Examples 242-243) using the procedure described in
Examples 234-235.
The compounds listed in Table XXXI may be prepared by this method:
TABLE XXXI
GPl-# Example D F M.P. (ºC)
244 Br CH2CH2ϕ - - - - 245 Br CH2ϕ - - - -
By following the procedures described in the Detailed
Description of the Invention and Examples 1 to 245 and using the appropriate starting materials and reagents, the following compounds are made:
4-Amino-7-(5-deoxy-1-β-D-ribofuranosyl)-5-vinylpyrrolo[2,3-d]pyrimidine;
4-Amino-5-ethynyl-7-(5-deoxy-1-β-D- ribofuranosyl)pyrrolo[2,3-d]pyrimidine;
5-(2-Chlorophenyl)-7-(5-deoxy-1-β-D-ribofuranosyl)-4- phenylaminopyrrolo[2,3-d]pyrimidine;
5-(3-Chlorophenyl)-7-(5-deoxy-1-^-D-ribofuranosyl)-4- phenylaminopyrrolo[2,3-d]pyrimidine;
5-(4-Chlorophenyl)-7-(5-deoxy-1-β-D-ribofuranosyl)-4- phenylaminopyrrolo[2,3-d]pyrimidine;
5-(2-Methoxyphenyl)-7-(5-deoxy-1-β-D-ribofuranosyl)-4- phenylaminopyrrolo[2,3-d]pyrimidine;
5-(4-Methoxyphenyl)-7-(5-deoxy-1-β-D-ribofuranosyl)-4- phenylaminopyrrolo[2,3-d]pyrimdine;
5-(2-Furanyl)-7-(5-deoxy-1-β-D-ribofuranosyl)-4- phenylaminopyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-4-phenyamino-5-(2-pyridyl)pyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-4-phenylamino-5-(4-pyridyl)pyrrolo[2,3-d]pyrimidine.
7-(5-Deoxy-1-β-D-ribofuranosyl)-5-phenyl-4-(4-pyridylamino)pyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-5-phenyl-4-(2-pyridylamino)-pyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-5-phenyl-4-(1-piperazinyl)-pyrrolo[2,3-d]pyrimidine;
4-(2-Chlorophenyl)-7-(5-deoxy-1-β-D-ribofuranosyl)-5- phenylpyrrolo[2,3-d]pyrimidine;
4-(3-Chlorophenyl)-7-(5-deoxy-1-β-D-ribofuranosyl)-5- phenylpyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-5-phenyl-4-(2-thiazolyl-amino)pyrrolo[2,3-d]pyrimidine;
4-Cyclohexylamino-7-(5-deoxy-1-β-D-ribofuranosyl)-5-phenylpyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-5-phenyl-4-phenylthio-pyrrolo[2,3-d]pyrimidine;
4-Benzyl-7-(5-deoxy-1-β-D-ribofuranosyl)-5-phenyl-pyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-4-ethynyl-5-phenyl-pyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-4-methyl-5-phenyl-pyrrolo[2,3-d]pyrimidine;
4-Benzyl-7-(5-deoxy-1-β-D-ribofuranosyl)-5-iodopyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-5-iodo-4-methyl-pyrrolo[2,3d]pyrimidine;
7-(5-Deoxy-1-β-D-ribofuranosyl)-5-phenyl-4-phenylamino-pyrrolo[2,3-d]pyrimidine;
4-Amino-5-phenyl-7-(1-β-D-ribofuranosyl)pyrrolo[2,3-d]-pyrimidine;
4-Amino-7-(5-deoxy-5-mercapto-1-β-D-ribofuranosyl)-5- iodopyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-5-mercapto-1-β-D-ribofuranosyl)-5-iodo-4- phenylaminopyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-5-mercapto-1-β-D-ribofuranosyl)-5-phenyl-4- phenylaminopyrrolo[2,3-d]pyrimidine;
7-(5-Amino-5-deoxy-1-β-D-ribofuranosyl)-5-phenyl-4- phenylaminopyrrolo[2,3-d]pyrimidine;
7-(5,6-Dideoxy-1-β-D-allofuranosyl)-5-iodo-4-phenylamino- pyrrolo[2,3-d]pyrimidine;
7-(5,6-Dideoxy-1-β-D-allofuranosyl)-5-phenyl-4- phenylalminopyrrolo[2,3-d]pyrimidine;
4-Amino-7-(5,6-dideoxy-1-β-D-allofuranosyl)-5-phenyl- pyrrolo[2,3-d]pyrimidine;
4-Amino-7-(5-deoxy-5-fluoro-1-β-D-ribofuranosyl)-5- iodopyrrolo[2,3-d]pyrimidine;
4-Amino-7-(5-deoxy-5-chloro-1-β-D-ribofuranosyl)-pyrrolo[2,3-d]pyrimidine;
7-(5-Deoxy-5-fluoro-1-β-D-ribofuranosyl)-5-phenyl-4-phenylaminopyrrolo[2,3-d]pyrimidine;
4-Amino-7-(6-azido-5,6-dideoxy-1-β-D-allofuranosyl)-5-iodopyrrolo[2,3-d]pyrimidine;
7-(6-Azido-5,6-dideoxy-1-β-D-allofuranosyl)-5-phenyl-4-phenylaminopyrrolo[2,3-d]pyrimidine;
4-Amino-7-(6-amino-5,6-dideoxy-1-β-D-allofuranosyl)-5- iodopyrrolo[2,3-d]pyrimidine;
7-(6-Amino-5,6-dideoxy-1-β-D-allofuranosyl)-5-phenyl-4- phenylaminopyrrolo[2,3-d]pyrimidine;
5- (2-Methoxyphenyl)-7-[1-β-D-ribofuranosyl]-4-phenylamino- pyrrolo[2,3-d]pyrimidine;
4-Amino-5-bromo-7-(5,6-didehydro-5,6-dideoxy-1-β-D-allofruanosyl)pyrrolo[2,3-d]pyrimidine;
7-(5,6-Didehydro-5,6-dideoxy-1-β-D-allofuranosyl)-5-phenyl-4-phenylalminopyrrolo[2,3-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-methoxy-pyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-phenoxy-pyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-phenylthiopyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-methylthiorpyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-chloro-pyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-cyclopropylpyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-dimethylamino-pyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-fluoro- pyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5-amino-5-deoxy-1-β-D-ribofuranosyl)-3-(3- pyridyl)pyrazolo[3,4-d]pyrimidine;
1-(5-Amino-5-deoxy-1-β-D-ribofuranosyl)-4-(3-chlorophenyl)- 3-(4-methoxyphenyl)pyrazolo[3,4-d]pyrimidine;
1-(5-Amino-5-deoxy-1-β-D-ribofuranosyl)-4-(4-chlorophenyl)- 3-(4-methoxyphenyl)pyrazolo[3,4-d]pyrimidine;
1-(5-Am3no-5-deoxy-1-β-β-ribofuranosyl)-4-(4-ethoxyphenyl)- 3-(4-methoxyphenyl)pyrazolo[3,4-d]pyrimidine;
1-(5-Amino-5-deoxy-1-β-D-ribofuranosyl)-4-(3- carboxamidophenyl-amino)-3-(4-methoxyphenyl)pyrazolo[3,4- d]pyrimidine;
1-(5-Amino-5-Deoxy-1-β-D-ribofuranosyl)-4-(2-furanyl)-3-(4-methoxyphenyl)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(4-methoxyphenyl)-4- (phenylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(3-methoxyphenyl)-4- (phenylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(2-pyridyl)-4-(phenylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(4-methoxyphenyl)-4-(4-pyridylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(3-methoxyphenyl)-4-(4- pyridylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(2-pyridyl)-4-(4- pyridylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-deoxy-1-β-D-ribofuranosyl)-3-(4-methoxyphenyl)-4-(2-methoxyphenylamino)pyrazolo[3,4-d]pyridimine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(3-methoxyphenyl)-4-(2-methoxyphenylamino)pyrazolo[3,4-d]pyridimine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(4-pyridyl)-4-(2-methoxyphenylamino)pyrazolo[3,4-d]pyridimine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(4-methoxyphenyl)-4-(2-imidazolylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(3-methoxyphenyl)-4-(2-imidazolylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(2-pyridyl)-4-(2-imidazolylamino)pyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(2-pyrazinyl)-4-phenylaminopyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-(2-pyrazinyl)-4-(N-indolinyl)pyrazolo[3,4-d]pyrimidine;
1-(5,6-Dideoxy-1-β-D-allofuranosyl)-3-phenyl-4-pheny1aminopyrazolo[3,4-d]pyrimidine;
4-Amino-1-(5,6-dideoxy-1-/S-D-allofuranosyl)-3-iodopyrazolo[3,4-d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-phenyl-4- phenylthiopyrazolo[3,4-d]pyrimidine;
1-(5-Amino-5-deoxy-1-β-D-ribopuranosyl)-3-bromo-4- methylpyrazolo[3,4-d]pyrimidine;
1-(5-Amino-5-deoxy-1-β-D-ribofuranosyl)-4-methyl-3- iodopyrazolo[3,4-d]pyrimidine;
7-(5-Deoxy-β-D-ribofuranosyl)-5-iodo-4-methylpyrrolo[2,3- d]pyrimidine;
4-Methyl-3-phenyl-1-(1-β-D-ribofuranosyl)pyrazolo[3,4- d]pyrimidine;
1-(5-Deoxy-1-β-D-ribofuranosyl)-3-phenyl-4- (phenylmethyl)pyrazolo[3,4-d]pyrimidine; and
7-(5-Amino-5-deoxy-1-β-D-ribofuranosyl)-5-bromo-4- chloropyrrolo[2,3-d]pyrimidine(GP-1-608).
EXAMPLE A INHIBITION OF ADENOSINE KINASE ACTIVITY
Inhibition of enzyme activity was determined using a 0.1 ml assay mixture containing 50 mM Tris-maleate, pH 7.0, 0.1% (w/v) BSA, 1 mM ATP, 1 mM MgCl2, 0.5 M [U-14C] adenosine (500 mCi/mmol) and 0.1 g of purified pig heart adenosine kinase. Different concentrations of the test compounds were incubated in the assay mixture for 20 min. at 37ºC. From each reaction mixture, 20 1 portions were removed and spotted on 2 cm2 pieces of Whatman DE81
filter paper. The papers were then washed to remove [14C] adenosine in ImM ammonium formate followed by deionized water and finally 95% ethanol. The papers were dried, and [14C]AMP measured by scintillation counting. Activities were determined from the amount of [14C]AMP formed.
A, receptor binding affinity was determined using 0.5 ml mixture containing 50 mM Tris HCl, pH 7.4, 1 nM [3H]CHA and 0.5 mg of neuronal membrane incubated with different concentrations of the test compound for 60 min at 37 °C. The reaction was stopped and unbound [3H]CHA removed by rapid filtration through Whatman GF/B filters. The filter papers were then solubilized and bound [3H]CHA determined by scintillation counting.
Inhibition of adenosine deaminase activity was determined spectrophotometrically using a 1 ml assay mixture containing 50mM potassium phosphate, pH 7.0, 1 mM ADP, 2.5 mM alpha-ketoglutarate, 15 units glutamic dehydrogenase, 0.125 mM NADH, 80 M adenosine and 0.002 units of calf intestinal mucosa adenosine deaminase. Different concentrations of the test compounds were incubated in the assay mixture for 10 min at 37 ºC. The reaction was monitored continuously for oxidation of NADH from the change in absorbance at 340nm.
Illustrative of the invention, the compounds designated GP-1-515, GP-1-608, GP-1-683, GP-1-695, GP-1-718, GP-1-704, GP-1-665, and GP-1-667, were found to have an IC50 of less than 10 nM
in the adenosine kinase inhibition assay. The compound GP-1-515 was found to be much less potent in the A, receptor assay and in the adenosine deaminase inhibition assay, having an IC50 greater than 100 M in the A, receptor assay and an IC50 greater than 1000 M in the adenosine deaminase inhibition assay.
EXAMPLE B
ADENOSINE KINASE INHIBITION IN INTACT CELLS
Inhibition of adenosine kinase in intact cells was determined from the amount of incorporation of radioisotope from adenosine into the adenylates (AMP, ADP and ATP) in the presence of adenosine deaminase inhibition. Capillary endothelial cells from bovine heart were incubated for 60 min. with 20 M
2'-deoxycoformycin, a potent adenosine deaminase inhibitor. Different concentrations of the test compounds were then added to the cells and incubated for 15 min. after which 5 M [3H]adenosine was added and the cells incubated for a further 15 min. The media was then discarded and the cells were treated with 50 1 0.4 M perchloric acid, centrifuged and the supernatants neutralized with 100 1 alanine: freon (1:4). Radioisotope-labelled adenylates were separated by TLC on PEI cellulose plates developed in
methanol:water (1:1) and incorporation of 3H determined by scintillation counting.
Illustrative of the invention, the compounds designated GP- 1-515, GP-1-683 and GP-1-665 were shown to have an IC50 of 9nM, 73 nM and 4.5 nM, respectively, in the adenosine kinase
inhibition assay in intact cells.
EXAMPLE C
IMPROVED FUNCTIONAL RECOVERY IN ISOLATED HEARTS
The ability of a number of adenosine kinase inhibitors to improve the recovery of post-ischemic function was examined in an isolated guinea pig heart model.
Isolated guinea pig hearts were cannulated via the ascending aorta and attached to a perfusion apparatus according to the method of Langendorff. The hearts were perfused at a constant pressure of 60 cm of H2O with a modified Krebs-Hanseleit buffer (pH 7.4) at 37ºC. Left ventricular developed pressures (LVDP) were monitored continuously using a latex balloon attached to a pressure transducer. Coronary flows were measured
gravimetrically by timed collection of pulmonary effluent.
Following equilibration of the hearts for a period of 30 minutes, the hearts were subjected to 45 minutes of low flow ischemia, by
reducing the perfusion pressure to 10 cm of H2O, and then
reperfused for 30 minutes by restoring the pressure to its original level (60 cm of H2O). The adenosine kinase inhibitors GP-1-238, GP-1-515 and GP1-547 were added to the perfusion buffer at the final concentrations specified. The results of these experiments are shown in Table A and demonstrate that adenosine kinase inhibitors enhance recovery of post ischemic function without affecting basal coronary flow.
TABLE A
GP-1-# Cone. ( M) Functional Recovery Preischemic Flow
(% preischemic LVDP) (ml/min/g)
Control 66.0 ± 2.1 5.9 ± 0.2
(n=14)
238 5 81.3 ± 3.3* 5.8 ± 0.3
(n=6)
515 0.3 79.0 ± 3.4* 6.5 ± 0.3
(n-6)
547 0.3 79.0 ± 3.1* 6.2 ± 0.3
* p < 0.05 vs. Control
Cone. = Concentration of test compound added to perfusion medium.
EXAMPLE D
EFFECT OF ADENOSINE KINASE INHIBITION ON ACUTE I.V.
HEMODYNAMICS IN THE RAT
The ability of the adenosine kinase inhibitor GP-1-238 to show effects on blood pressure, heart rate or body temperature was compared in anesthetized and conscious rats. Sprague Dawley rats were anesthetized with pentobarbital and catheterized in the jugular vein and carotid artery. GP-1-238 (0.1-5 mg/kg/min) was infused intravenously in stepwise increments (0.2 ml/min x 5 minutes). The experiments in conscious rats were conducted in the same manner after rats had been catheterized and allowed to recover for 2 days following surgery. In conscious rats, in contrast to anesthetized animals, no hemodynamic effects were seen at doses which completely inhibited adenosine kinase in vivo See Figure 1.
EXAMPLE E
FUNCTIONAL BENEFIT OF GP-1-515 IN A PRECLINICAL MODEL
OF STABLE ANGINA
GP-1-515, was evaluated for its ability to prevent
cumulative cardiac dysfunction associated with repeated episodes of pacing-induced ischemia.
Anesthetized male dogs were instrumented to measure regional myocardial wall thickening during right atrial pacing (Young & Mullane, Am. J. Physio., 1991, 261:1570-1577). Animals were subjected to six repeated episodes of pacing. A continuous IV infusion of 1 g/kg/min of GP-1-515 or saline (control) was administered post pace #1. The results of these experiments are shown in Table B and reveal that the adenosine kinase inhibitor, GP-1-515, attenuates the decline in wall thickening associated with pacing induced ischemia.
TABLE B
% of Non-Ischemic Wall Thickening
Pace # Saline (n=3) GP-1-515 (n=4)
1 33.3 ± 9.3 39.8 ± 3.2
2 40.5 ± 11 43.6 ± 12.0
3 27.4 ± 16 34.0 ± 19.0
4 19.2 ± 23 50.5 + 21.3
5 22.6 ± 16 75.6 ± 19.4
6 13.7 ± 19 62.6 ± 17.1
EXAMPLE F EFFICACY OF GP-1-515 IN A PRECLINICAL MODEL OF UNSTABLE ANGINA
GP-1-515 was tested in a canine model of platelet-induced coronary thrombosis (Folts, J., Circulation. 1991, 83 fSuppl.
IV] :IV-3-IV-14). In this model, platelets cyclically aggregate and embolize causing cyclic flow reductions (CFRs) that are quantitated by the frequency and the change in coronary blood flow from low point to peak with each cycle. The results are shown in Table C. Administration of the adenosine kinase inhibitor, GP-1-515, abolished CFRs in 3 of 8 animals and reduced both the frequency and the change in flow in those animals where CFRs were not abolished.
TABLE C
Control 0.03 0.1 0.3
CFR 5.2 ± 0.2 3.0 ± 0.6 3.4 ± 0.8 4.2 ± 0.6 (freq.) (n=8) (n=7) (n=5) (n=5)
CFR 17 ± 1.7 10.1 ± 2.6 11.0 ± 2.1 14.1 ± 1.9
(D flow) (n=8) (n=7) (n=5) (n=5)
N abolished 0/8 1/8 3/8 3/8
N total
EXAMPLE G
INHIBITION OF NEUTROPHIL ADHERENCE TO FIBROBLASTS OR ENDOTHELIAL CELLS
The ability of an adenosine kinase inhibitor to affect neutrophil adherence to fibroblasts and endothelial cells was evaluated in a cell culture model.
Cultures of human dermal fibroblasts or human umbillical vein endothelial cells were washed and then incubated for 2 hours at 37°C in a 5% CO2 atmosphere in fresh medium containing
different concentrations of the adenosine kinase inhibitors GP-1-272 and GP-1-456. These incubations were carried out in the presence of fMLP-stimulated human neutrophils isolated from whole blood (1.25 × 106/ml) with or without adenosine deaminase (0.125
U/ml). At the end of the incubation, the medium was removed and the monolayers of fibroblasts or endothelial cells and adherent neutrophils were fixed by addition of formaldehyde (3.7%) and, after washing to remove non-adherent neutrophils, adherent neutrophils were stained with Weigart's hematoxylin and counted under a light microscope. The results depicted in Figure 2 show that the adenosine kinase inhibitors GP-1-272 and GD-1-456 inhibit neutrophil adhesion to endothelial cells and that this inhibition is reversed by adenosine deaminase treatment.
EXAMPLE H
INHIBITION OF CONTRACTION IN ISOLATED ILEUM
The ability of adenosine kinase inhibition to affect stimulated contraction of muscle strips from the isolated ileum has been investigated.
Segments (about 1 cm) of longitudinal muscle were stripped from the guinea pig ileum, connected to isotonic force
transducers and suspended in jacketed tissue baths containing Krebs-Ringer solution aerated with 95% O2/5% CO2. Parallel platinum electrodes were used to deliver electrical current at 1 minute intervals at a voltage adequate to induce contraction of 90% of maximal. The adenosine kinase inhibitors, GP-1-515 or
GP-1-547 were added to the tissue baths at different
concentrations, with or without either the adenosine receptor antagonist, 8-sulphophenyltheophylline (8SPT) or adenosine deaminase (ADA), and the effects on contraction monitored.
Inhibition of contraction by (A) GP-1-515 and (B) GP-1-547 together with reversal by both 8SPT and ADA are shown in Figure 3.
EXAMPLE I
INHIBITION OF SEIZURES BY ADENOSINE KINASE INHIBITORS
The ability of selected adenosine kinase inhibitors to influence PTZ-induced seizures was evaluated in an experimental animal model.
Male Swiss Webster mice in groups of 6-8 were preinjected intraperitoneally (IP) with either vehicle or an adenosine kinase inhibitor followed 1 hour later by 100 mg/kg pentylenetetrazol (PTZ) administered subcutaneously in the upper back of the animal. After injections of the convulsant, animals were isolated in separate cages and observed for the onset of seizure. Animals were scored as being fully protected from seizure if they failed to seize for a period of 1 hour after PTZ administration (vehicle
control animals seized after about 4 minutes). The results are shown in Figure 4.
In Panel A, the adenosine kinase inhibitor, GP-1-456, was administered IP at doses of 1, 5 and 10 mg/kg followed after 45 minutes by PTZ administration.
In Panel B, GP-1-456 was administered IP at a dose of 2.5 mg/kg alone or together with either 34 mg/kg of theophylline or 70 mg/kg of 8-sulfophenyltheophylline followed after 45 minutes by PTZ administration.
In additional studies, mice were given GP-1-456 or GP-1-560 by oral administration 45 minutes prior to PTZ administration. The results are given in Table D and demonstrate that the
adenosine kinase inhibitors exhibit oral anticonvulsant activity.
The ability of the adenosine kinase inhibitors to influence electroshock-induced seizures was also evaluated in an
experimental model.
The adenosine kinase inhibitor GP-1-456 or vehicle were administered orally to rats prior to electroshock treatment.
After 1 hour, corneal electrodes were applied to the eyes of eac animal and an electrical stimulus of 160 mA delivered for 0.2 seconds. Animals were isolated in separate cages and observed for seizure. Abolition of the hindleg tonic extensor component is taken as the end point for this test and reflects ability of the compound to prevent seizure spread.
From the data presented in Table E, an ED50 of 0.18 mg/kg was determined for protection of electroshock-induced seizures by the adenosine kinase inhibitor, GP-1-456.
TABLE D
Percent Seizure Latency To
Treatment N= Seizure Score First Seizure (minutes]
Control 13 100 3.6±1.2 3.3
GP-1-456 13 15 0.3+0.4* 8.2*
(2.5 mg/kg)
Control 8 100 2.5 ±1.6 4.5
GP-1-560 7 71 0.71±0.5 4.5
(30 mg/kg)
GP-1-560 43 1.0±0.9 7.0*
(50 mg/kg)
* p < 0.05 vs. Control
Data is presented as the mean ± standard deviation.
TABLE E
GP-1-456 No. of Animals
(mg/kg) (protected/tested)
0.00 0/8
0.05 1/8
0.10 2/8
0.15 2/8
0.19 6/8
0.38 6/8
0.75 7/8
1.50 8/8
Claims
1. A compound of the formula:

wherein:
(a) A is oxygen, methylene or sulfur;
(b) B' is -(CH2)n-B wherein n is 1, 2, 3 or 4 and B is hydrogen, alkyl, alkoxy, amino, alkylamino, acylamino,
hydrocarbyloxycarbonylamino, mercapto, alkylthio, azido, cyano, halogen, or B' is alkenyl or alkynyl;
(c) C1 and C2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C1 is a single bond to C2 and C2 is carbonyl or α- alkoxyalkylidene;
(d) X is  = and Y is -N= or
= and Y is -N= or  ;
;
(e) D is hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl, alkynyl, haloalkyl, cyano, cyanoalkyl, acyl,
carboxamido, a carboxylic acid or carboxylic acid ester group, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, amino, alkylamino arylamino, aralkylamino, acylamino, or nitro;
(f) E is hydrogen, halogen, alkyl, or alkylthio;
(g) F is alkyl, aryl, aralkyl, halogen, amino, alkylamino, arylamino, aralkylamino, cyano, cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio; optionally substituted indolinyl or indolyl; pyrrolidinyl or piperazinyl; and
(h) G is hydrogen, halogen, lower alkyl, lower alkoxy, lower alkylamino or lower alkylthio; and pharmaceutically
acceptable salts thereof; with the proviso that:
when A is oxygen and
(i) X is  = and Y is
= and Y is  , then if B' is methyl, D is halogen, cyano or carboxamido, F is amino, then G is not hydrogen; or if D is hydrogen, then F is not amino; or (ii) X is
, then if B' is methyl, D is halogen, cyano or carboxamido, F is amino, then G is not hydrogen; or if D is hydrogen, then F is not amino; or (ii) X is  and Y is -N=, if B is hydrogen or halogen, D and G are hydrogen, then F is not amino; or when A is methylene, X is
and Y is -N=, if B is hydrogen or halogen, D and G are hydrogen, then F is not amino; or when A is methylene, X is  Y is
Y is  B, D, E and G are hydrogen, then F is not amino.
B, D, E and G are hydrogen, then F is not amino.
2. A compound according to claim 1 wherein D is hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl, alkynyl, cyano,
cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio, amino, alkylamino, arylamino, aralkylamino, amido, hydrocarbyloxycarbonyl; and E is hydrogen, halogen, alkyl, alkylamino, azido or alkylthio.
3. A compound according to claim 2 wherein A is oxygen.
4. A compound according to claim 2 wherein B' is -(CH2)nB wherein B is hydrogen, halogen, alkyl, amino, alkylamino, alkoxy, mercapto, alkylthio, azido or cyano.
5. A compound according to claim 4 wherein G is hydrogen, halogen, lower alkyl or lower alkylthio.
6. A compound according to claim 5 wherein A is oxygen.
7. A compound according to claim 5 wherein G is hydrogen, halogen, alkyl or alkylthio.
8. A compound according to claim 7 wherein A is oxygen.
9. A compound according to claim 8 wherein G is hydrogen.
10. A compound according to claim 9 wherein E is hydrogen.
11. A compound according to claim 10 wherein F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl.
12. A compound according to claim 11 wherein D is halogen or aryl.
13. A compound according to claim 10 wherein D is halogen or aryl.
14. A compound according to claim 10 wherein Y is -N=.
15. A compound according to claim 14 wherein B is hydrogen, halogen, lower alkyl, amino, lower alkylamino, azido or cyano.
16. A compound according to claim 14 wherein D is hydrogen, halogen, alkyl, aryl, aralkyl, cyano, alkoxy, aryloxy, aralkoxy, alkenyl, or alkynyl.
17. A compound according to claim 14 wherein B is hydrogen, halogen, lower alkyl, amino, lower alkylamino, lower alkoxy, lower alkylthio, azido, or cyano; and D is hydrogen, halogen, aryl, cyano, alkoxy or aryloxy.
18. A compound according to claim 17 wherein D is hydrogen, halogen or aryl.
19. A compound according to claim 18 wherein B is hydrogen, amino or azido.
20. A compound according to claim 18 wherein F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl.
21. A compound according to claim 18 wherein B is hydrogen, amino or azido and F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl and n is 1.
22. A compound according to claim 21 wherein B is amino, D is bromo, F is amino, and C1 and C2 are both hydrogen or acetyl.
23. A compound according to claim 21 wherein B is amino, D is iodo, F is amino and C1 and C2 are both hydrogen.
24. A compound according to claim 21 wherein B is hydrogen, D is iodo, F is amino and C1 and C2 are both hydrogen.
25. A compound according to claim 21 wherein B is hydrogen, D is phenyl, F is anilino and C1 and C2 are both hydrogen.
26. A compound according to claim 21 wherein B is azido, D is p-methoxyphenyl, F is amino and C1 and C2 are both hydrogen.
27. A compound according to claim 21 wherein B is azido, D is phenyl, F is anilino and C1 and C2 are both hydrogen.
28. A compound according to claim 21 wherein D is aryl.
29. A compound according to claim 1 wherein Y is -N=, A is oxygen, B' is -(CH2)nB wherein B is hydrogen, amino or azido; D is halogen or aryl; F is amino, arylamino, alkyl or aralkyl; G is hydrogen; and C1 and C2 are both hydrogen or acetyl.
30. A compound according to claim 1 wherein Y is -N=, A is oxygen, B' is -(CH2)nB wherein B is hydrogen, amino, or azido; D is halogen or aryl; F is amino or arylamino; G is hydrogen; and C1 and C2 are both hydrogen or acetyl.
31. A compound according to claim 30 wherein D is aryl.
32. A compound according to claim 31 wherein F is
arylamino.
33. A compound according to claim 32 wherein D is
optionally substituted phenyl.
34. A compound according to claim 10 wherein Y is  .
.
35. A compound according to claim 34 wherein B is hydrogen, halogen, lower alkyl, amino, lower alkylamino, azido or cyano.
36. A compound according to claim 34 wherein D is hydrogen, alkyl, aryl, aralkyl, cyano, alkenyl or alkynyl.
37. A compound according to claim 34 wherein E is hydrogen or halogen.
38. A compound according to claim 37 wherein B is hydrogen, halogen, lower alkyl, amino, lower alkylamino, alkoxy, alkylthio, azido or cyano; and D is hydrogen, alkyl, aryl, aralkyl, cyano, alkenyl or alkynyl.
39. A compound according to claim 38 wherein D is hydrogen, halogen or aryl.
40. A compound according to claim 39 wherein B is hydrogen, amino or azido.
41. A compound according to claim 39 wherein F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl.
42. A compound according to claim 39 wherein B is hydrogen, amino or azido and F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl, and n is 1.
43. A compound according to claim 42 wherein B is azido, D is bromo, E is hydrogen, F is amino, and C1 and C2 are both hydrogen.
44. A compound according to claim 42 wherein B is hydrogen, D is iodo, E is hydrogen, F is anilino, and Ct and C2 are both hydrogen.
45. A compound according to claim 42 wherein B is hydrogen, D is phenyl, E is hydrogen and F is anilino, and C1 and C2 are both hydrogen.
46. A compound according to claim 42 wherein B is hydrogen, D is phenyl, E is hydrogen, F is amino, and C1 and C2 are both hydrogen.
47. A compound according to claim 42 wherein B is methyl, D is iodo, E is hydrogen, F is amino and C1 and C2 are both
hydrogen.
48. A compound according to claim 41 wherein n is 2, B is azido, D is iodo, E is hydrogen, F is amino, and C1 and C2 and both hydrogen.
49. A compound according to claim 3 wherein Y is  B' is vinyl, D is iodo, E is hydrogen, F is amino, G is hydrogen and C1 and C2 are both hydrogen.
B' is vinyl, D is iodo, E is hydrogen, F is amino, G is hydrogen and C1 and C2 are both hydrogen.
50. A compound according to claim 1 wherein A is oxygen; D is halogen or aryl; E is hydrogen or halogen and G is hydrogen.
51. A compound according to claim 46 wherein n is 2, B is hydrogen, D is iodo, E is hydrogen, F is amino and C1 and C2 are both hydrogen.
52. A compound according to claim 41 wherein n is 2, B is azido, D is iodo, E is hydrogen, F is amino and C1 and C2 are both hydrogen.
53. A compound according to claim 3 wherein B' is vinyl, C1 and C2 are both hydrogen, Y is  D is iodo, E is hydrogen, F is amino, and G is hydrogen.
D is iodo, E is hydrogen, F is amino, and G is hydrogen.
54. A compound according to any of claims 2, 10 or 18 wherein F is alkyl, aryl or aralkyl.
55. A compound of the formula:

wherein:
(a) A is oxygen, methylene or sulfur;
(b) B' is -(CH2)nB wherein n is 1, 2, 3 or 4 and B is hydroxy, acyloxy, hydrocarbyloxycarbonyloxy, or -OCONR2 wherein R is independently hydrocarbyl;
(c) C1 and C2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C1 is a single bond to C2 and C2 is carbonyl or α- alkoxyalkylidene;
(d) X is  and Y is -N=;
and Y is -N=;
(e) D is halogen aryl or aralkyl;
(f) F is alkyl, aryl, aralkyl, halogen, amino, alkylamino, arylamino, aralkylamino, cyano, cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio, optionally substituted indolinyl or indolyl, pyrrolidinyl or piperazinyl; and (g) G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio; and pharmaceutically acceptable salts
thereof; with the proviso that when A is oxygen and D is halogen, then F is not amino.
56. A compound according to claim 55 wherein A is oxygen and G is hydrogen.
57. A compound according to claim 56 wherein D is aryl.
58. A compound according to claim 57 wherein F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl.
59. A compound according to claim 58 wherein n is 1, B is hydroxy, D is phenyl, F is anilino and C1 and C2 are both
hydrogen.
60. A compound of the formula:

wherein:
(a) A is oxygen, methylene or sulfur; (b) B' is -(CH2)nB wherein n is 1, 2, 3 or 4 and B is hydroxy, acyloxy, hydrocarbyloxycarbonyloxy, or -OCONR2 wherein R is hydrocarbyl;
(c) C1 and C2 are each independently hydrogen, acyl, hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C1 is a single bond to C2 and C2 is carbonyl or α- alkoxyalkylidene;
(d) X is  and Y is
and Y is  ;
;
(e) D is aryl or aralkyl;
(f) E is hydrogen, halogen, alkyl, or alkylthio;
(g) F is alkyl, aryl, aralkyl, halogen, amino, alkylamino, arylamino, aralkylamino, cyano, cyanoalkyl, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, optionally substituted indolinyl or indolyl, pyrrolidinyl or piperazinyl; and
(h) G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio; and pharmaceutically acceptable salts
thereof; with the proviso that: when A is oxygen, D is
oxadiazolyl, triazolyl or triazinyl, E and G are both hydrogen, then F is not amino.
61. A compound according to claim 60 wherein A is oxygen, E is hydrogen or halogen, and G is hydrogen and n is 1 or 2.
62. A compound according to claim 61 wherein D is aryl.
63. A compound according to claim 62 wherein F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl.
64. A compound according to claim 63 wherein n is 1, B is hydroxy, D is 2-furanyl, E is hydrogen, F is anilino and C1 and C2 are both hydrogen.
65. A compound of the formula:

wherein
(a) A is oxygen, methylene or sulfur;
(b) B' is -CH2B wherein B is amino, alkylamino, or
acylamino;
(c) C1 and C2 are each independently hydrogen, acyl,
hydrocarbyloxycarbonyl or taken together form a 5-membered ring wherein C1 is a single bond to C2 and C2 is carbonyl or α-alkoxyalkylidene;
(d) X is -N= and Y is 
(e) E is hydrogen, halogen, a  lkyl, amino, alkylamino, azido, acylamino, alkoxy or alkylthio;
lkyl, amino, alkylamino, azido, acylamino, alkoxy or alkylthio;
(f) F is halogen, amino, alkylamino, arylamino,
aralkylamino, cyanoalkyl, alkoxy, aryloxy, aralkoxy, alkylthio, arylthio, aralkylthio, alkyl, aryl, aralkyl, optionally
substituted indolinyl or indolyl, pyrrolidinyl or piperazinyl; and
(g) G is hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkylthio and pharmaceutical acceptable salts thereof; with the proviso that:
when A is oxygen, B is amino or hydrocarbylamino, E and G are hydrogen, then F is not amino.
66. A compound according to claim 65 wherein G is hydrogen, halogen, lower alkyl or lower alkylthio.
67. A compound according to claim 66 wherein A is oxygen.
68. A compound according to claim 67 wherein E is halogen, alkyl or alkylthio.
69. A compound according to claim 68 wherein B is amino.
70. A compound according to claim 69 wherein F is halogen, amino, alkylamino, arylamino, aralkylamino, alkylthio, arylthio, alkyl, aryl or aralkyl.
71. A compound according to claim 70 wherein F is amino, arylamino, alkyl, aryl or aralkyl.
72. A compound according to claim 68 wherein B is amino, E is hydrogen or halogen, F is alkyl, aryl or aralkyl, and G is hydrogen.
73. A compound of the formula:

wherein
(a) A and A' are independently oxygen, methylene or sulfur; (b) B' and B" are independently -(CH2)nB wherein n is independently 1, 2, 3 or 4 and B is independently hydrogen, hydroxy, alkyl, alkoxy, amino, alkylamino, acylamino,
hydrocarbyloxycarbonylammo, mercapto, alkylthio, azido, or B' i independently alkenyl or alkynyl;
(c) C1 and C1' and C2 and C2' are each independently
hydrogen, acyl, hydrocarbyloxycarbonyl, or C, and C2 or C1' and C2' taken together form a 5-membered ring wherein C1 or C1' is a single bond to C2 or C2' and C2 or C2' is carbonyl or α-alkoxyalkylidene;
(d) X and X' are each independently  or -N=; and Y and Y' are each independently -N= or
or -N=; and Y and Y' are each independently -N= or  provided that either of and X and Y or X' and Y' are not both -N=;
provided that either of and X and Y or X' and Y' are not both -N=;
(e) D is independently hydrogen, halogen, alkyl, aryl, aralkyl, alkenyl, alkynyl, haloalkyl, cyano, cyanoalkyl, acyl, carboxamido, a carboxylic acid or corresponding carboxylic acid ester group, alkoxy, aryloxy, aralkyloxy, alkylthio, arylthio, aralkylthio, amino, alkylamino, arylamino, aralkylamino or nitro;
(f) E is independently hydrogen, halogen, alkyl, or
alkylthio; (g) L is an optionally substituted piperazinyl divalent radical or -NH(ALKL)NH- wherein ALKL is a divalent alkylene radical of 2 to 24 carbon atoms; and
(h) G and G' are each independently hydrogen, halogen, lower alkyl, lower alkoxy, or lower alkoxy; or pharmaceutically
acceptable salts thereof; with the proviso that when B is
hydroxy, then X and X' are not both -N=.
74. A compound according to claim 73 wherein at least one of X and X' or Y and Y' is different.
75. A method of enhancing adenosine levels in a mammal which comprises administering an effective amount of a compound which selectively inhibits adenosine kinase.
76. A method according to claim 75 wherein said compound selectively inhibits adenosine kinase and increases adenosine at a specific locus.
77. A method according to claim 76 wherein said locus is in the heart or brain.
78. A method according to claim 76 wherein said locus is an area of ischemia.
79. A method according to claim 75 wherein said compound comprises an orally bioavailable pyrrolo[2,3-d]pyrimidine riboside analog.
80. A method according to claim 76 wherein said compound comprises an orally bioavailable pyrrolo[2,3-d]pyrimidine riboside analog.
81. A method according to claim 80 wherein said pyrrolo
[2,3-d]pyrimidine riboside analog is 5'-deoxy-5-iodo-tubericidin.
82. A method according to claim 75 wherein said compound comprises a pyrrolo[2,3-d]pyrimidine which is capable of penetrating the central nervous system or blood-brain barrier.
83. A method according to claim 82 wherein said compound is 5'-deoxy-5-iodo-tubericidin.
84. A method according to claim 75 wherein said compound comprises a purine, pyrrolo[2,3]pyrimidine or pyrazolo[3,4-d] pyrimidine riboside analog which has decreased toxicity and is not phosphorylatable at the 5'-position of the ribosyl moiety.
85. A method according to claim 84 wherein said compound is 5'-deoxy-iodo-tubericidin.
86. A method of enhancing adenosine levels in a mammal which comprises administering and effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
87. A method of enhancing local levels of adenosine around cells having a decreased ratio of synthesis of adenosine triphosphate to breakdown of adenosine triphosphate which comprises administering an effective amount of a compound which selectively inhibits adenosine kinase.
88. A method according to claim 87 wherein said decreased ratio is caused by heart attack, angina, stroke, or transient ischemic attack.
89. A method of enhancing local levels of adenosine around cells having a decreased ratio of synthesis of adenosine triphosphate to breakdown of adenosine triphosphate which comprises administering an effective amount of a compound of any of
claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
90. A method of treating or preventing a cardiovascular disorder in a mammal where injury or dysfunction causes or is caused by ischemia, reperfusion or arrhythmia which comprises administering to said mammal an effective amount of a compound which selectively inhibits adenosine kinase.
91. A method according to claim 90 wherein said cardiovascular condition is heart attack, angina pectoris, unstable angina, silent ischemia, congestive heart failure or arrhythmia.
92. A method of treating or preventing a cardiovascular disorder in a mammal where injury or dysfunction is caused by ischemia or reperfusion which comprises administering an effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
93. A method of adjunctive therapy to protect the heart for use with a procedure which comprises administering therewith a therapeutically effective amount of an adenosine kinase
inhibitor.
94. A method according to claim 93 wherein said procedure comprises a procedure to improve blood flow or supply.
95. A method according to claim 94 wherein said procedure to improve blood flow or supply comprises percutaneous transluminal coronary angioplasty, percutaneous transluminal directional coronary atherectomy, laser atherectomy, intravascular stents, coronary artery bypass graft surgery, or thrombolysis therapy.
96. A method of adjunctive therapy to protect the heart for use with a procedure which comprises administering therewith a therapeutically effective amount of an adenosine kinase inhibitor which comprises a compound of any of claims 1, 55, 60, 65 or 73.
97. A method of preventing or decreasing reperfusion injury to a tissue in a mammal following interrupted or decreased blood flow to that tissue which comprises administering to that tissue or organism a compound which selectively inhibits adenosine kinase.
98. A method according to claim 97 wherein said interrupted or decreased blood flow causes or is believed to be caused by a stroke or heart attack.
99. A method of preventing or decreasing reperfusion injur in a mammal following interrupted or decreased flow in a tissue which comprises administering to that tissue an effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which
selectively inhibits adenosine kinase.
100. A method of preventing or decreasing platelet aggregation in a mammal which comprises administering to said mammal an effective amount of a compound which selectively inhibits adenosine kinase.
101. A method according to claim 100 wherein said method is used to treat conditions in which thrombosis or emboli are prevalent.
102. A method according to claim 101 wherein said
conditions are stroke, angina, myocardial infarct or pulmonary embolism.
103. A method of preventing or decreasing platelet aggregation in a mammal which comprises administering to said mammal an effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
104. A method of preventing or decreasing inflammatory responses in a mammal which comprises administering to the mammal a therapeutically effective amount of a compound which
selectively inhibits adenosine kinase.
105. A method according to claim 104 wherein said method is used to treat conditions in which inflammatory processes are prevalent.
106. A method according to claim 105 wherein said conditions are arthritis, rheumatoid arthritis, osteoarthritis, autoimmune disease, adult respiratory distress syndrome, inflammatory bowel syndrome, necrotizing enterocolitis, chronic obstructive
pulmonary disease, psoriasis, conjunctivitis, iridocyditis, ischemia, reperfusion or peripheral vascular disease, or
atherosclerosis.
107. A method according to claim 104 wherein said
inflammatory response causes arthritis, meningitis, autoimmune disease, inflammatory bowel disease, vasculitis, dermatitis, myositis, or renal inflammation.
108. A method of preventing or decreasing inflammatory responses in a mammal which comprises administering to the mammal a therapeutically effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
109. A method according to claim 108 wherein said
inflammatory response causes arthritis, meningitis, autoimmune disease, vasculitis, dermatitis, myositis or renal inflammation.
110. A method of treating patients having chronic low or insufficient adenosine levels or who would benefit from increased central nervous system adenosine levels which comprises administering an effective amount of a compound which selectively inhibits adenosine kinase.
111. A method according to claim 109 wherein said chronic low or insufficient adenosine levels are associated with
insomnia, autism, schizophrenia or other neuropsychiatric symptoms.
112. A method of treating patients having chronic low or insufficient adenosine levels or who would benefit from increased central nervous system adenosine levels which comprises administering an effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
113. A method of producing skeletal muscle relaxation or preventing skeletal muscle spasm in patients which comprises administering to said patient a muscle relaxant effective amount of a compound according to any of claims 1, 55, 60, 65 or 73.
114. A method of encouraging a sleep state in a patient which comprises administering to said patient a sleep-inducing effective amount of a compound which selectively inhibits
adenosine kinase.
115. A method of encouraging a sleep state in a patient which comprises administering to said patient a sleep-inducing effective amount of a compound of any of claims 1, 55, 60, 65, or 73.
116. A method of treating anxiety in a patient which comprises administering to said patient an anxiety-reducing effective amount of a comound which selectively inhibibts adenosine kinase.
117. A method of treating anxiety in a patient which comprises administering to said patient an anxiety-reducing effective amount of a compound according to any of claims 1, 55, 60, 65 or 73.
118. A method of reducing or preventing neural tissue damage caused by excitotoxicity in an affected mammal which comprises administering a therapeutically effective amount of a compound which selectively inhibits adenosine kinase.
119. A method according to claim 118 wherein said neural tissue is brain or spinal chord.
120. A method according to claim 119 wherein said excitotoxicity causes a neurodegenerative condition.
121. A method according to claim 119 wherein said excitotoxicity is caused by a stroke or CNS trauma.
122. A method according to claim 120 wherein said neurodegenerative condition is Parkinson's Disease, Alzheimer's
Disease, Amyotrophic Lateral Sclerosis or Huntington's Disease, or schizophrenia.
123. A method of reducing or preventing neural tissue damage caused by excitotoxicity in an affected mammal which comprises administering a therapeutically effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
124. A method of decreasing neural tissue damage associated with neurodegenerative diseases in an affected animal which comprises administering an effective amount of a compound which selectively inhibits adenosine kinase.
125. A method according to claim 124 wherein said neurodegenerative diseases are Parkinson's Disease, Alzheimer's
Disease, Amyotrophic Laterial Sclerosis or Huntington's Disease.
126. A method of decreasing a neural tissue damage associated with neurodegenerative disease in an effected animal which comprises administering an effective amount of a compound of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
127. A method of treating or preventing epilepsy or seizures in a mammal which comprises administering to the mammal a therapeutically effective amount of a compound which selectively inhibits adenosine kinase.
128. A method of treating or preventing epilepsy or seizures in a mammal which comprises administering to the mammal a therapeutically effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
129. A method of decreasing local contraction of smooth muscle in a mammal which comprises administering to said mammal an effective amount of a compound which selectively inhibits adenosine kinase.
130. A method according to claim 129 wherein said smooth muscle is in the gastrointestinal tract.
131. A method according to claim 130 wherein contraction of smooth muscle causes irritable bowel syndrome.
132. A method according to claim 129 wherein said smooth muscle is in an artery and its contraction causes vasospasm and limits blood supply to a tissue.
133. A method according to claim 132 wherein said mammal has angina pectoris, Beurger's disease, Raynaud's disease, transient ischemic attacks or thromboangiitis obliterans.
134. A method of decreasing local contraction of smooth muscle in a mammal which comprises administering to said mammal an effective amount of a compound of any of claims 1, 55, 60, 65 or 73 which selectively inhibits adenosine kinase.
135. A method according to any of claims 87, 90, 91, 93, 98, 100, 104, 106, 110, 114, 116, 118, 127, or 129 wherein said compound comprises a pyrrolo[2,3-d]pyrimidine riboside analog.
136. A method according to any of claims 87, 90, 91, 93, 98, 100, 104, 106, 110, 114, 116, 118, 127 or 129 wherein said compound comprises a pyrazolo[3,4-d]pyrimidine analog.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU13599/92A AU665184B2 (en) | 1991-01-23 | 1992-01-21 | Adenosine kinase inhibitors |
| NO932628A NO180418C (en) | 1991-01-23 | 1993-07-21 | adenosine kinase inhibitors |
| FI933303A FI933303A (en) | 1991-01-23 | 1993-07-22 | ADENOSINKINASINHIBITORER |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US812,916 | 1977-07-05 | ||
| US64711791A | 1991-01-23 | 1991-01-23 | |
| US647,117 | 1996-05-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1992012718A1 true WO1992012718A1 (en) | 1992-08-06 |
Family
ID=24595775
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1992/000515 WO1992012718A1 (en) | 1991-01-23 | 1992-01-21 | Adenosine kinase inhibitors |
Country Status (2)
| Country | Link |
|---|---|
| AU (1) | AU665184B2 (en) |
| WO (1) | WO1992012718A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1045636A1 (en) * | 1997-09-19 | 2000-10-25 | Arthur M. Feldman | Pharmaceutical compositions and methods for the treatment of failing myocardial tissue |
| EP0832091B1 (en) * | 1995-06-07 | 2004-01-14 | Gensia Sicor Inc. | C-4' modified adenosine kinase inhibitors |
| FR2851248A1 (en) * | 2003-02-18 | 2004-08-20 | Aventis Pharma Sa | New purine derivatives are protein kinase inhibitors used for treating e.g. fungal infections, psoriasis, cancers, neurodegenerative disorders, allergies, autoimmune diseases and cardiovascular disease |
| JP2005501122A (en) * | 2001-08-22 | 2005-01-13 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | Novel 4-aminofuropyrimidine compounds and their use |
| US7253284B2 (en) | 2001-07-17 | 2007-08-07 | Giaxo Group Limited | Chemical compounds |
| US7427630B2 (en) | 2003-04-09 | 2008-09-23 | Sb Pharmaco Puerto Rico Inc. | Condensed N-heterocyclic compounds and their use as CRF receptor antagonists |
| US7476670B2 (en) | 2003-02-18 | 2009-01-13 | Aventis Pharma S.A. | Purine derivatives, method for preparing, pharmaceutical compositions and novel use |
| US8513267B2 (en) | 2004-02-03 | 2013-08-20 | Universidade Estadual De Campinas-Unicamp | 4-anilinoquinazoline derivatives with adenosine-kinase inhibitor properties |
| US8871737B2 (en) | 2010-09-22 | 2014-10-28 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US8916538B2 (en) | 2012-03-21 | 2014-12-23 | Vertex Pharmaceuticals Incorporated | Solid forms of a thiophosphoramidate nucleotide prodrug |
| US8980865B2 (en) | 2011-12-22 | 2015-03-17 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US9012427B2 (en) | 2012-03-22 | 2015-04-21 | Alios Biopharma, Inc. | Pharmaceutical combinations comprising a thionucleotide analog |
| US9073960B2 (en) | 2011-12-22 | 2015-07-07 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| RU2593201C2 (en) * | 1998-12-04 | 2016-08-10 | Астразенека Аб | NOVEL INTERMEDIATE COMPOUNDS FOR PRODUCING TRIAZOLO(4,5-d)PYRIMIDINE |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| EP3445768A4 (en) * | 2016-04-19 | 2019-12-18 | The Regents of The University of California | Erbb inhibitors and uses thereof |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| CN115677704A (en) * | 2021-07-30 | 2023-02-03 | 山东大学 | Histone deacetylase 6 inhibitor containing 7H-pyrrolo [2,3-d ] pyrimidine structure, and preparation method and application thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW444018B (en) * | 1992-12-17 | 2001-07-01 | Pfizer | Pyrazolopyrimidines |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4912092A (en) * | 1986-03-27 | 1990-03-27 | The Regents Of The University Of California | Methods for increasing extracellular adenosine and for stabilizing mast cells |
| US5082829A (en) * | 1989-01-24 | 1992-01-21 | Gensia Pharmaceuticals | AICA riboside prodrugs |
| US5104859A (en) * | 1985-09-24 | 1992-04-14 | Solimedco Aktiebolag | Continuous administration of adenosine to reduce pulmonary vascular resistance |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6339868A (en) * | 1986-08-04 | 1988-02-20 | Otsuka Pharmaceut Factory Inc | Di (lower alkyl) phenol derivative |
-
1992
- 1992-01-21 AU AU13599/92A patent/AU665184B2/en not_active Ceased
- 1992-01-21 WO PCT/US1992/000515 patent/WO1992012718A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5104859A (en) * | 1985-09-24 | 1992-04-14 | Solimedco Aktiebolag | Continuous administration of adenosine to reduce pulmonary vascular resistance |
| US4912092A (en) * | 1986-03-27 | 1990-03-27 | The Regents Of The University Of California | Methods for increasing extracellular adenosine and for stabilizing mast cells |
| US5082829A (en) * | 1989-01-24 | 1992-01-21 | Gensia Pharmaceuticals | AICA riboside prodrugs |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0832091B1 (en) * | 1995-06-07 | 2004-01-14 | Gensia Sicor Inc. | C-4' modified adenosine kinase inhibitors |
| EP1045636A1 (en) * | 1997-09-19 | 2000-10-25 | Arthur M. Feldman | Pharmaceutical compositions and methods for the treatment of failing myocardial tissue |
| EP1045636A4 (en) * | 1997-09-19 | 2003-09-10 | Arthur M Feldman | Pharmaceutical compositions and methods for the treatment of failing myocardial tissue |
| USRE46276E1 (en) | 1998-12-04 | 2017-01-17 | Astrazeneca Uk Limited | Triazolo(4,5-D)pyrimidine compounds |
| RU2593201C2 (en) * | 1998-12-04 | 2016-08-10 | Астразенека Аб | NOVEL INTERMEDIATE COMPOUNDS FOR PRODUCING TRIAZOLO(4,5-d)PYRIMIDINE |
| US7253284B2 (en) | 2001-07-17 | 2007-08-07 | Giaxo Group Limited | Chemical compounds |
| JP2005501122A (en) * | 2001-08-22 | 2005-01-13 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | Novel 4-aminofuropyrimidine compounds and their use |
| WO2004073595A3 (en) * | 2003-02-18 | 2005-01-06 | Aventis Pharma Sa | Novel purine derivatives, preparation method thereof, application of same as medicaments, pharmaceutical compositions and novel use |
| US7476670B2 (en) | 2003-02-18 | 2009-01-13 | Aventis Pharma S.A. | Purine derivatives, method for preparing, pharmaceutical compositions and novel use |
| AU2004212733B2 (en) * | 2003-02-18 | 2009-12-24 | Aventis Pharma S.A. | Novel purine derivatives, preparation method thereof, application of same as medicaments, pharmaceutical compositions and novel use |
| FR2851248A1 (en) * | 2003-02-18 | 2004-08-20 | Aventis Pharma Sa | New purine derivatives are protein kinase inhibitors used for treating e.g. fungal infections, psoriasis, cancers, neurodegenerative disorders, allergies, autoimmune diseases and cardiovascular disease |
| WO2004073595A2 (en) * | 2003-02-18 | 2004-09-02 | Aventis Pharma S.A. | Novel purine derivatives, preparation method thereof, application of same as medicaments, pharmaceutical compositions and novel use |
| US7427630B2 (en) | 2003-04-09 | 2008-09-23 | Sb Pharmaco Puerto Rico Inc. | Condensed N-heterocyclic compounds and their use as CRF receptor antagonists |
| US8513267B2 (en) | 2004-02-03 | 2013-08-20 | Universidade Estadual De Campinas-Unicamp | 4-anilinoquinazoline derivatives with adenosine-kinase inhibitor properties |
| US9278990B2 (en) | 2010-09-22 | 2016-03-08 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US8871737B2 (en) | 2010-09-22 | 2014-10-28 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US10464965B2 (en) | 2011-12-22 | 2019-11-05 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US8980865B2 (en) | 2011-12-22 | 2015-03-17 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US11021509B2 (en) | 2011-12-22 | 2021-06-01 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9073960B2 (en) | 2011-12-22 | 2015-07-07 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9605018B2 (en) | 2011-12-22 | 2017-03-28 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US9856284B2 (en) | 2012-03-21 | 2018-01-02 | Alios Biopharma, Inc. | Solid forms of a thiophosphoramidate nucleotide prodrug |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US8916538B2 (en) | 2012-03-21 | 2014-12-23 | Vertex Pharmaceuticals Incorporated | Solid forms of a thiophosphoramidate nucleotide prodrug |
| US10485815B2 (en) | 2012-03-21 | 2019-11-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9394330B2 (en) | 2012-03-21 | 2016-07-19 | Alios Biopharma, Inc. | Solid forms of a thiophosphoramidate nucleotide prodrug |
| US9012427B2 (en) | 2012-03-22 | 2015-04-21 | Alios Biopharma, Inc. | Pharmaceutical combinations comprising a thionucleotide analog |
| EP3445768A4 (en) * | 2016-04-19 | 2019-12-18 | The Regents of The University of California | Erbb inhibitors and uses thereof |
| CN115677704A (en) * | 2021-07-30 | 2023-02-03 | 山东大学 | Histone deacetylase 6 inhibitor containing 7H-pyrrolo [2,3-d ] pyrimidine structure, and preparation method and application thereof |
| CN115677704B (en) * | 2021-07-30 | 2023-12-05 | 山东大学 | Histone deacetylase 6 inhibitor containing 7H-pyrrolo [2,3-d ] pyrimidine structure, and preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AU665184B2 (en) | 1995-12-21 |
| AU1359992A (en) | 1992-08-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0496617B1 (en) | Adenosine kinase inhibitors | |
| US5646128A (en) | Methods for treating adenosine kinase related conditions | |
| US5721356A (en) | Orally active adenosine kinase inhibitors | |
| AU665184B2 (en) | Adenosine kinase inhibitors | |
| US5763597A (en) | Orally active adenosine kinase inhibitors | |
| WO1994017803A9 (en) | Adenosine kinase inhibitors | |
| EP0682519A1 (en) | Adenosine kinase inhibitors | |
| US5506347A (en) | Lyxofuranosyl analogues of adenosine | |
| Cottam et al. | New adenosine kinase inhibitors with oral antiinflammatory activity: synthesis and biological evaluation | |
| US6211158B1 (en) | Desazapurine-nucleotide derivatives, processes for the preparation thereof, pharmaceutical compositions containing them and the use thereof for nucleic acid sequencing and as antiviral agents | |
| EP0832091B1 (en) | C-4' modified adenosine kinase inhibitors | |
| US5726302A (en) | Water soluble adenosine kinase inhibitors | |
| US5795977A (en) | Water soluble adenosine kinase inhibitors | |
| AU4510101A (en) | N6 heterocyclic 8-modified adenosine derivatives | |
| US5864033A (en) | Adenosine kinase inhibitors | |
| WO1991001325A1 (en) | Nucleoside derivatives and their use as medicaments | |
| Kini et al. | Synthesis and antiviral activity of certain guanosine analogs in the thiazolo [4, 5-d] pyrimidine ring system | |
| WO1997033591A1 (en) | A method of treating disorders related to cytokines in mammals | |
| IE912833A1 (en) | Aica riboside analogs | |
| WO1997033590A1 (en) | A method of treating disorders related to cytokines in mammals | |
| IE920190A1 (en) | Adenosine kinase inhibitors | |
| MXPA94000878A (en) | Inhibitors of the adenosin kinase, which include derivatives of lixofurans | |
| MXPA97009656A (en) | Soluble adenosin kinase inhibitors in a | |
| MXPA97009773A (en) | Inhibitors of adenosin cinasa oralmente acti |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA FI NO |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2100863 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 933303 Country of ref document: FI |