RU2525392C2 - Соединения и фармацевтические композиции для лечения вирусных инфекций - Google Patents
Соединения и фармацевтические композиции для лечения вирусных инфекций Download PDFInfo
- Publication number
- RU2525392C2 RU2525392C2 RU2012132218/04A RU2012132218A RU2525392C2 RU 2525392 C2 RU2525392 C2 RU 2525392C2 RU 2012132218/04 A RU2012132218/04 A RU 2012132218/04A RU 2012132218 A RU2012132218 A RU 2012132218A RU 2525392 C2 RU2525392 C2 RU 2525392C2
- Authority
- RU
- Russia
- Prior art keywords
- alkyl
- compound
- substituted
- aryl
- benzyl
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 277
- 239000008194 pharmaceutical composition Substances 0.000 title claims description 48
- 208000036142 Viral infection Diseases 0.000 title description 12
- 230000009385 viral infection Effects 0.000 title description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 159
- 239000001257 hydrogen Substances 0.000 claims abstract description 146
- 239000000203 mixture Substances 0.000 claims abstract description 127
- 125000003118 aryl group Chemical group 0.000 claims abstract description 89
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 43
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 36
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 34
- 208000005176 Hepatitis C Diseases 0.000 claims abstract description 21
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 claims abstract description 20
- 208000002672 hepatitis B Diseases 0.000 claims abstract description 20
- 229960000329 ribavirin Drugs 0.000 claims abstract description 20
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 claims abstract description 20
- XQSPYNMVSIKCOC-NTSWFWBYSA-N Emtricitabine Chemical compound C1=C(F)C(N)=NC(=O)N1[C@H]1O[C@@H](CO)SC1 XQSPYNMVSIKCOC-NTSWFWBYSA-N 0.000 claims abstract description 17
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 17
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 claims abstract description 13
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 claims abstract description 10
- GBBJCSTXCAQSSJ-XQXXSGGOSA-N clevudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1[C@H](F)[C@@H](O)[C@H](CO)O1 GBBJCSTXCAQSSJ-XQXXSGGOSA-N 0.000 claims abstract description 10
- 229960001627 lamivudine Drugs 0.000 claims abstract description 9
- 229960005311 telbivudine Drugs 0.000 claims abstract description 9
- IQFYYKKMVGJFEH-CSMHCCOUSA-N telbivudine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1O[C@@H](CO)[C@H](O)C1 IQFYYKKMVGJFEH-CSMHCCOUSA-N 0.000 claims abstract description 9
- TVRCRTJYMVTEFS-ICGCPXGVSA-N [(2r,3r,4r,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-4-hydroxy-2-(hydroxymethyl)-4-methyloxolan-3-yl] (2s)-2-amino-3-methylbutanoate Chemical compound C[C@@]1(O)[C@H](OC(=O)[C@@H](N)C(C)C)[C@@H](CO)O[C@H]1N1C(=O)N=C(N)C=C1 TVRCRTJYMVTEFS-ICGCPXGVSA-N 0.000 claims abstract description 8
- RLAHNGKRJJEIJL-RFZPGFLSSA-N [(2r,4r)-4-(2,6-diaminopurin-9-yl)-1,3-dioxolan-2-yl]methanol Chemical compound C12=NC(N)=NC(N)=C2N=CN1[C@H]1CO[C@@H](CO)O1 RLAHNGKRJJEIJL-RFZPGFLSSA-N 0.000 claims abstract description 8
- NHKZSTHOYNWEEZ-AFCXAGJDSA-N taribavirin Chemical compound N1=C(C(=N)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NHKZSTHOYNWEEZ-AFCXAGJDSA-N 0.000 claims abstract description 8
- 229960000366 emtricitabine Drugs 0.000 claims abstract description 7
- 229950006081 taribavirin Drugs 0.000 claims abstract description 7
- HTJGLYIJVSDQAE-VWNXEWBOSA-N [(1s,6s,7s,8r,8ar)-1,7,8-trihydroxy-1,2,3,5,6,7,8,8a-octahydroindolizin-6-yl] butanoate Chemical compound O[C@H]1[C@H](O)[C@@H](OC(=O)CCC)CN2CC[C@H](O)[C@@H]21 HTJGLYIJVSDQAE-VWNXEWBOSA-N 0.000 claims abstract description 6
- VFCYZPOEGWLYRM-QCZKYFFMSA-N [(2s,3r,5s)-5-(4-amino-2-oxopyrimidin-1-yl)-2-(hydroxymethyl)oxolan-3-yl] (2s)-2-amino-3-methylbutanoate Chemical compound O1[C@@H](CO)[C@H](OC(=O)[C@@H](N)C(C)C)C[C@H]1N1C(=O)N=C(N)C=C1 VFCYZPOEGWLYRM-QCZKYFFMSA-N 0.000 claims abstract description 6
- 229950005846 amdoxovir Drugs 0.000 claims abstract description 6
- 229960005338 clevudine Drugs 0.000 claims abstract description 6
- 229960000980 entecavir Drugs 0.000 claims abstract description 6
- YXPVEXCTPGULBZ-WQYNNSOESA-N entecavir hydrate Chemical compound O.C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)C1=C YXPVEXCTPGULBZ-WQYNNSOESA-N 0.000 claims abstract description 6
- 229950002819 valtorcitabine Drugs 0.000 claims abstract description 6
- IRZRJANZDIOOIF-GAJNKVMBSA-N (2r,3r,4r,5r)-2-(4-aminopyrrolo[2,3-d]pyrimidin-7-yl)-5-(hydroxymethyl)-3-methyloxolane-3,4-diol Chemical compound C[C@@]1(O)[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(N)=C2C=C1 IRZRJANZDIOOIF-GAJNKVMBSA-N 0.000 claims abstract description 5
- NYPIRLYMDJMKGW-VPCXQMTMSA-N 4-amino-1-[(2r,3r,4r,5r)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-3-methyloxolan-2-yl]pyrimidin-2-one Chemical compound C[C@@]1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N=C(N)C=C1 NYPIRLYMDJMKGW-VPCXQMTMSA-N 0.000 claims abstract description 5
- 229950003414 celgosivir Drugs 0.000 claims abstract description 5
- 229950002810 valopicitabine Drugs 0.000 claims abstract description 5
- BXNMTOQRYBFHNZ-UHFFFAOYSA-N resiquimod Chemical compound C1=CC=CC2=C(N(C(COCC)=N3)CC(C)(C)O)C3=C(N)N=C21 BXNMTOQRYBFHNZ-UHFFFAOYSA-N 0.000 claims abstract description 4
- 229950010550 resiquimod Drugs 0.000 claims abstract description 4
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims abstract 4
- 125000000217 alkyl group Chemical group 0.000 claims description 354
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 185
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 87
- 238000011282 treatment Methods 0.000 claims description 79
- 239000003795 chemical substances by application Substances 0.000 claims description 75
- 241000700721 Hepatitis B virus Species 0.000 claims description 59
- 150000003839 salts Chemical class 0.000 claims description 43
- 241000700605 Viruses Species 0.000 claims description 42
- 210000004185 liver Anatomy 0.000 claims description 28
- 229940079322 interferon Drugs 0.000 claims description 25
- 102000014150 Interferons Human genes 0.000 claims description 23
- 108010050904 Interferons Proteins 0.000 claims description 23
- 239000003443 antiviral agent Substances 0.000 claims description 22
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 22
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 20
- 238000009472 formulation Methods 0.000 claims description 18
- 229910052757 nitrogen Inorganic materials 0.000 claims description 18
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 18
- 229910019142 PO4 Inorganic materials 0.000 claims description 17
- 125000003545 alkoxy group Chemical group 0.000 claims description 17
- 239000010452 phosphate Substances 0.000 claims description 17
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 16
- 239000003112 inhibitor Substances 0.000 claims description 13
- 239000000137 peptide hydrolase inhibitor Substances 0.000 claims description 13
- 108010047761 Interferon-alpha Proteins 0.000 claims description 12
- 102000006992 Interferon-alpha Human genes 0.000 claims description 12
- 239000003085 diluting agent Substances 0.000 claims description 12
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 claims description 12
- 239000003937 drug carrier Substances 0.000 claims description 11
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 claims description 7
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- 150000002367 halogens Chemical class 0.000 claims description 7
- 101800001838 Serine protease/helicase NS3 Proteins 0.000 claims description 6
- 108010010648 interferon alfacon-1 Proteins 0.000 claims description 6
- 125000003729 nucleotide group Chemical group 0.000 claims description 6
- 108090000994 Catalytic RNA Proteins 0.000 claims description 5
- 102000053642 Catalytic RNA Human genes 0.000 claims description 5
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 claims description 5
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 5
- 125000003282 alkyl amino group Chemical group 0.000 claims description 5
- 125000001769 aryl amino group Chemical group 0.000 claims description 5
- 125000001188 haloalkyl group Chemical group 0.000 claims description 5
- 108091092562 ribozyme Proteins 0.000 claims description 5
- 125000004104 aryloxy group Chemical group 0.000 claims description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 4
- LBQAJLBSGOBDQF-UHFFFAOYSA-N nitro azanylidynemethanesulfonate Chemical compound [O-][N+](=O)OS(=O)(=O)C#N LBQAJLBSGOBDQF-UHFFFAOYSA-N 0.000 claims description 4
- 229940046166 oligodeoxynucleotide Drugs 0.000 claims description 4
- 150000003548 thiazolidines Chemical class 0.000 claims description 4
- 238000013519 translation Methods 0.000 claims description 4
- GVEZIHKRYBHEFX-MNOVXSKESA-N 13C-Cerulenin Natural products CC=CCC=CCCC(=O)[C@H]1O[C@@H]1C(N)=O GVEZIHKRYBHEFX-MNOVXSKESA-N 0.000 claims description 3
- 108010063738 Interleukins Proteins 0.000 claims description 3
- 102000015696 Interleukins Human genes 0.000 claims description 3
- 229940123066 Polymerase inhibitor Drugs 0.000 claims description 3
- 230000000692 anti-sense effect Effects 0.000 claims description 3
- FIVPIPIDMRVLAY-UHFFFAOYSA-N aspergillin Natural products C1C2=CC=CC(O)C2N2C1(SS1)C(=O)N(C)C1(CO)C2=O FIVPIPIDMRVLAY-UHFFFAOYSA-N 0.000 claims description 3
- ZVSKZLHKADLHSD-UHFFFAOYSA-N benzanilide Chemical compound C=1C=CC=CC=1C(=O)NC1=CC=CC=C1 ZVSKZLHKADLHSD-UHFFFAOYSA-N 0.000 claims description 3
- GVEZIHKRYBHEFX-UHFFFAOYSA-N caerulein A Natural products CC=CCC=CCCC(=O)C1OC1C(N)=O GVEZIHKRYBHEFX-UHFFFAOYSA-N 0.000 claims description 3
- GVEZIHKRYBHEFX-NQQPLRFYSA-N cerulenin Chemical compound C\C=C\C\C=C\CCC(=O)[C@H]1O[C@H]1C(N)=O GVEZIHKRYBHEFX-NQQPLRFYSA-N 0.000 claims description 3
- 229950005984 cerulenin Drugs 0.000 claims description 3
- 230000001419 dependent effect Effects 0.000 claims description 3
- FIVPIPIDMRVLAY-RBJBARPLSA-N gliotoxin Chemical compound C1C2=CC=C[C@H](O)[C@H]2N2[C@]1(SS1)C(=O)N(C)[C@@]1(CO)C2=O FIVPIPIDMRVLAY-RBJBARPLSA-N 0.000 claims description 3
- 229940103893 gliotoxin Drugs 0.000 claims description 3
- 229930190252 gliotoxin Natural products 0.000 claims description 3
- 229960003358 interferon alfacon-1 Drugs 0.000 claims description 3
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims description 3
- 108010005716 Interferon beta-1a Proteins 0.000 claims description 2
- 108010080374 albuferon Proteins 0.000 claims description 2
- 235000010290 biphenyl Nutrition 0.000 claims description 2
- 239000004305 biphenyl Substances 0.000 claims description 2
- 108010045648 interferon omega 1 Proteins 0.000 claims description 2
- 125000001624 naphthyl group Chemical group 0.000 claims description 2
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 claims 2
- 101000749287 Clitocybe nebularis Clitocypin Proteins 0.000 claims 2
- 101000767029 Clitocybe nebularis Clitocypin-1 Proteins 0.000 claims 2
- 229940094664 Cysteine protease inhibitor Drugs 0.000 claims 2
- 229940121759 Helicase inhibitor Drugs 0.000 claims 2
- 150000002431 hydrogen Chemical class 0.000 claims 2
- 108010074328 Interferon-gamma Proteins 0.000 claims 1
- 102000008070 Interferon-gamma Human genes 0.000 claims 1
- 229960004461 interferon beta-1a Drugs 0.000 claims 1
- 229960003130 interferon gamma Drugs 0.000 claims 1
- 108010042414 interferon gamma-1b Proteins 0.000 claims 1
- 229940028862 interferon gamma-1b Drugs 0.000 claims 1
- 108700027921 interferon tau Proteins 0.000 claims 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 abstract description 121
- 239000003814 drug Substances 0.000 abstract description 103
- 229910052705 radium Inorganic materials 0.000 abstract description 56
- 229910052701 rubidium Inorganic materials 0.000 abstract description 56
- 230000000694 effects Effects 0.000 abstract description 24
- 239000000126 substance Substances 0.000 abstract description 11
- -1 nucleoside compounds Chemical class 0.000 description 257
- 125000000547 substituted alkyl group Chemical group 0.000 description 131
- 241000711549 Hepacivirus C Species 0.000 description 97
- 239000002777 nucleoside Substances 0.000 description 84
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 78
- 125000002252 acyl group Chemical group 0.000 description 77
- 238000000034 method Methods 0.000 description 76
- 125000003277 amino group Chemical group 0.000 description 71
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 63
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 58
- 229940079593 drug Drugs 0.000 description 56
- 239000002552 dosage form Substances 0.000 description 52
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 48
- 208000015181 infectious disease Diseases 0.000 description 48
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 46
- 239000000460 chlorine Substances 0.000 description 45
- 150000003833 nucleoside derivatives Chemical class 0.000 description 45
- 125000003342 alkenyl group Chemical group 0.000 description 41
- 125000000304 alkynyl group Chemical group 0.000 description 41
- 201000010099 disease Diseases 0.000 description 41
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 41
- 208000006454 hepatitis Diseases 0.000 description 39
- 229940024606 amino acid Drugs 0.000 description 38
- 239000000243 solution Substances 0.000 description 38
- 235000001014 amino acid Nutrition 0.000 description 37
- 229940124597 therapeutic agent Drugs 0.000 description 37
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 35
- BVMWIXWOIGJRGE-UHFFFAOYSA-N NP(O)=O Chemical class NP(O)=O BVMWIXWOIGJRGE-UHFFFAOYSA-N 0.000 description 35
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 34
- 125000005140 aralkylsulfonyl group Chemical group 0.000 description 33
- 150000001413 amino acids Chemical class 0.000 description 32
- 125000004103 aminoalkyl group Chemical group 0.000 description 31
- 229910052760 oxygen Inorganic materials 0.000 description 31
- 230000015572 biosynthetic process Effects 0.000 description 30
- 238000003786 synthesis reaction Methods 0.000 description 30
- 230000000069 prophylactic effect Effects 0.000 description 29
- 229910052717 sulfur Inorganic materials 0.000 description 28
- 125000003710 aryl alkyl group Chemical group 0.000 description 27
- 239000004480 active ingredient Substances 0.000 description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 26
- 239000011541 reaction mixture Substances 0.000 description 25
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 24
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 24
- 238000005516 engineering process Methods 0.000 description 24
- 125000003107 substituted aryl group Chemical group 0.000 description 24
- 241000710778 Pestivirus Species 0.000 description 22
- 239000002585 base Substances 0.000 description 22
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 21
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 21
- 230000000840 anti-viral effect Effects 0.000 description 21
- 230000002829 reductive effect Effects 0.000 description 20
- 241000710831 Flavivirus Species 0.000 description 19
- 125000004122 cyclic group Chemical group 0.000 description 19
- 125000000539 amino acid group Chemical group 0.000 description 18
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 18
- 125000003835 nucleoside group Chemical group 0.000 description 18
- 238000002360 preparation method Methods 0.000 description 18
- 125000000524 functional group Chemical group 0.000 description 17
- 150000002632 lipids Chemical class 0.000 description 17
- 229940127073 nucleoside analogue Drugs 0.000 description 17
- 238000005160 1H NMR spectroscopy Methods 0.000 description 16
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 16
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 16
- 150000001720 carbohydrates Chemical class 0.000 description 16
- 235000014633 carbohydrates Nutrition 0.000 description 16
- 231100000283 hepatitis Toxicity 0.000 description 16
- 150000003904 phospholipids Chemical class 0.000 description 16
- 150000003871 sulfonates Chemical class 0.000 description 16
- 229920002554 vinyl polymer Polymers 0.000 description 16
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 238000013270 controlled release Methods 0.000 description 15
- 239000000047 product Substances 0.000 description 15
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 14
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- 150000002148 esters Chemical class 0.000 description 14
- 229940002612 prodrug Drugs 0.000 description 14
- 239000000651 prodrug Substances 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- 239000001226 triphosphate Substances 0.000 description 14
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 13
- 125000001153 fluoro group Chemical group F* 0.000 description 13
- 208000019423 liver disease Diseases 0.000 description 13
- 125000005017 substituted alkenyl group Chemical group 0.000 description 13
- 125000004426 substituted alkynyl group Chemical group 0.000 description 13
- 208000024891 symptom Diseases 0.000 description 13
- 238000002560 therapeutic procedure Methods 0.000 description 13
- 235000011178 triphosphate Nutrition 0.000 description 13
- 229960004295 valine Drugs 0.000 description 13
- 229920002472 Starch Polymers 0.000 description 12
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 12
- 239000007924 injection Substances 0.000 description 12
- 238000002347 injection Methods 0.000 description 12
- 239000000843 powder Substances 0.000 description 12
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 12
- 235000019698 starch Nutrition 0.000 description 12
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 11
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 11
- 125000002877 alkyl aryl group Chemical group 0.000 description 11
- 239000007864 aqueous solution Substances 0.000 description 11
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 11
- 238000006243 chemical reaction Methods 0.000 description 11
- 230000001684 chronic effect Effects 0.000 description 11
- 125000004093 cyano group Chemical group *C#N 0.000 description 11
- 125000005843 halogen group Chemical group 0.000 description 11
- 230000002265 prevention Effects 0.000 description 11
- 239000003826 tablet Substances 0.000 description 11
- 206010012310 Dengue fever Diseases 0.000 description 10
- 241000282412 Homo Species 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 10
- 238000005481 NMR spectroscopy Methods 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- 239000000969 carrier Substances 0.000 description 10
- 229910052801 chlorine Inorganic materials 0.000 description 10
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 10
- 208000019425 cirrhosis of liver Diseases 0.000 description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 10
- 229910052731 fluorine Inorganic materials 0.000 description 10
- 125000000262 haloalkenyl group Chemical group 0.000 description 10
- 125000000232 haloalkynyl group Chemical group 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- 239000000463 material Substances 0.000 description 10
- 239000003921 oil Substances 0.000 description 10
- 239000012074 organic phase Substances 0.000 description 10
- 125000004430 oxygen atom Chemical group O* 0.000 description 10
- 239000003208 petroleum Substances 0.000 description 10
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- 229920006395 saturated elastomer Polymers 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 241000710780 Bovine viral diarrhea virus 1 Species 0.000 description 9
- 206010008909 Chronic Hepatitis Diseases 0.000 description 9
- 208000001490 Dengue Diseases 0.000 description 9
- 206010016654 Fibrosis Diseases 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- 229910052799 carbon Inorganic materials 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 208000025729 dengue disease Diseases 0.000 description 9
- 239000003480 eluent Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- 239000008107 starch Substances 0.000 description 9
- 229940032147 starch Drugs 0.000 description 9
- 125000001424 substituent group Chemical group 0.000 description 9
- 125000004434 sulfur atom Chemical group 0.000 description 9
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 241000710781 Flaviviridae Species 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 239000013543 active substance Substances 0.000 description 8
- 231100000354 acute hepatitis Toxicity 0.000 description 8
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 8
- 229910052794 bromium Inorganic materials 0.000 description 8
- 239000002775 capsule Substances 0.000 description 8
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 8
- 239000007884 disintegrant Substances 0.000 description 8
- 239000000839 emulsion Substances 0.000 description 8
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 239000006186 oral dosage form Substances 0.000 description 8
- 239000006201 parenteral dosage form Substances 0.000 description 8
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 8
- 238000010898 silica gel chromatography Methods 0.000 description 8
- 239000012453 solvate Substances 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 8
- 229960005486 vaccine Drugs 0.000 description 8
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 7
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 7
- PPUDLEUZKVJXSZ-VPCXQMTMSA-N 4-amino-1-[(2r,3r,4r,5r)-3,4-dihydroxy-5-(hydroxymethyl)-3-methyloxolan-2-yl]pyrimidin-2-one Chemical compound C[C@@]1(O)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N=C(N)C=C1 PPUDLEUZKVJXSZ-VPCXQMTMSA-N 0.000 description 7
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 7
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 7
- 101710144111 Non-structural protein 3 Proteins 0.000 description 7
- 108700026244 Open Reading Frames Proteins 0.000 description 7
- 125000005041 acyloxyalkyl group Chemical group 0.000 description 7
- 125000005078 alkoxycarbonylalkyl group Chemical group 0.000 description 7
- 125000005097 aminocarbonylalkyl group Chemical group 0.000 description 7
- 125000004181 carboxyalkyl group Chemical group 0.000 description 7
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 7
- 229920001577 copolymer Polymers 0.000 description 7
- 125000000392 cycloalkenyl group Chemical group 0.000 description 7
- 208000035475 disorder Diseases 0.000 description 7
- 238000001704 evaporation Methods 0.000 description 7
- 230000008020 evaporation Effects 0.000 description 7
- 208000016253 exhaustion Diseases 0.000 description 7
- 239000000284 extract Substances 0.000 description 7
- 239000006260 foam Substances 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 239000000314 lubricant Substances 0.000 description 7
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 7
- 239000008108 microcrystalline cellulose Substances 0.000 description 7
- 229940016286 microcrystalline cellulose Drugs 0.000 description 7
- 108010092853 peginterferon alfa-2a Proteins 0.000 description 7
- 230000002085 persistent effect Effects 0.000 description 7
- 210000002966 serum Anatomy 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 230000001052 transient effect Effects 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 6
- 239000005977 Ethylene Substances 0.000 description 6
- 206010054261 Flavivirus infection Diseases 0.000 description 6
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 6
- 108010078049 Interferon alpha-2 Proteins 0.000 description 6
- 108010076039 Polyproteins Proteins 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 6
- 108010022999 Serine Proteases Proteins 0.000 description 6
- 102000012479 Serine Proteases Human genes 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 6
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 6
- WOZSCQDILHKSGG-UHFFFAOYSA-N adefovir depivoxil Chemical compound N1=CN=C2N(CCOCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C)C=NC2=C1N WOZSCQDILHKSGG-UHFFFAOYSA-N 0.000 description 6
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 6
- 239000000443 aerosol Substances 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 6
- 239000011230 binding agent Substances 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 238000011161 development Methods 0.000 description 6
- 229940088598 enzyme Drugs 0.000 description 6
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 6
- 230000004761 fibrosis Effects 0.000 description 6
- 238000003818 flash chromatography Methods 0.000 description 6
- 208000018191 liver inflammation Diseases 0.000 description 6
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 6
- 238000004806 packaging method and process Methods 0.000 description 6
- 239000002243 precursor Substances 0.000 description 6
- 238000011321 prophylaxis Methods 0.000 description 6
- 235000018102 proteins Nutrition 0.000 description 6
- 102000004169 proteins and genes Human genes 0.000 description 6
- 238000003860 storage Methods 0.000 description 6
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 6
- 238000002054 transplantation Methods 0.000 description 6
- 230000003612 virological effect Effects 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 5
- 241000283690 Bos taurus Species 0.000 description 5
- 150000008575 L-amino acids Chemical class 0.000 description 5
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 5
- 108060004795 Methyltransferase Proteins 0.000 description 5
- ACFIXJIJDZMPPO-NNYOXOHSSA-N NADPH Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](OP(O)(O)=O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ACFIXJIJDZMPPO-NNYOXOHSSA-N 0.000 description 5
- 102000035195 Peptidases Human genes 0.000 description 5
- 108091005804 Peptidases Proteins 0.000 description 5
- 239000004698 Polyethylene Substances 0.000 description 5
- 229910004298 SiO 2 Inorganic materials 0.000 description 5
- 108020004459 Small interfering RNA Proteins 0.000 description 5
- 241000282898 Sus scrofa Species 0.000 description 5
- 229960001997 adefovir Drugs 0.000 description 5
- 239000002671 adjuvant Substances 0.000 description 5
- 230000002411 adverse Effects 0.000 description 5
- 239000000427 antigen Substances 0.000 description 5
- 150000007860 aryl ester derivatives Chemical class 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 239000012267 brine Substances 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 235000012000 cholesterol Nutrition 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 125000001651 cyanato group Chemical group [*]OC#N 0.000 description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 5
- 238000010828 elution Methods 0.000 description 5
- 206010014599 encephalitis Diseases 0.000 description 5
- 235000019439 ethyl acetate Nutrition 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- 208000010710 hepatitis C virus infection Diseases 0.000 description 5
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 5
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 5
- 230000001965 increasing effect Effects 0.000 description 5
- 238000001802 infusion Methods 0.000 description 5
- 238000001990 intravenous administration Methods 0.000 description 5
- 229910052740 iodine Inorganic materials 0.000 description 5
- 125000001261 isocyanato group Chemical group *N=C=O 0.000 description 5
- 239000008101 lactose Substances 0.000 description 5
- 210000005229 liver cell Anatomy 0.000 description 5
- 238000012423 maintenance Methods 0.000 description 5
- 150000004712 monophosphates Chemical class 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- 239000002773 nucleotide Substances 0.000 description 5
- 229940002988 pegasys Drugs 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 150000008298 phosphoramidates Chemical class 0.000 description 5
- 229920000573 polyethylene Polymers 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 239000004055 small Interfering RNA Substances 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 5
- 150000003413 spiro compounds Chemical class 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- 229960004556 tenofovir Drugs 0.000 description 5
- VCMJCVGFSROFHV-WZGZYPNHSA-N tenofovir disoproxil fumarate Chemical compound OC(=O)\C=C\C(O)=O.N1=CN=C2N(C[C@@H](C)OCP(=O)(OCOC(=O)OC(C)C)OCOC(=O)OC(C)C)C=NC2=C1N VCMJCVGFSROFHV-WZGZYPNHSA-N 0.000 description 5
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N thiocyanic acid Chemical compound SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 5
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- OAKPWEUQDVLTCN-NKWVEPMBSA-N 2',3'-Dideoxyadenosine-5-triphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1CC[C@@H](CO[P@@](O)(=O)O[P@](O)(=O)OP(O)(O)=O)O1 OAKPWEUQDVLTCN-NKWVEPMBSA-N 0.000 description 4
- YJQYHFMKGAVKDP-UHFFFAOYSA-N 3-butanoyl-1,8-dihydroxy-2-methylphenanthrene-9,10-dione Chemical compound C12=CC=CC(O)=C2C(=O)C(=O)C2=C1C=C(C(=O)CCC)C(C)=C2O YJQYHFMKGAVKDP-UHFFFAOYSA-N 0.000 description 4
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N 4-amino-1-[(2s,3s,4r,5s)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 4
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 241001118702 Border disease virus Species 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 241000282693 Cercopithecidae Species 0.000 description 4
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- 208000037319 Hepatitis infectious Diseases 0.000 description 4
- 102100040018 Interferon alpha-2 Human genes 0.000 description 4
- 108010079944 Interferon-alpha2b Proteins 0.000 description 4
- 229930195725 Mannitol Natural products 0.000 description 4
- 241000282579 Pan Species 0.000 description 4
- ZRWPUFFVAOMMNM-UHFFFAOYSA-N Patulin Chemical compound OC1OCC=C2OC(=O)C=C12 ZRWPUFFVAOMMNM-UHFFFAOYSA-N 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- 101800001554 RNA-directed RNA polymerase Proteins 0.000 description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 4
- 101710172711 Structural protein Proteins 0.000 description 4
- 229960000643 adenine Drugs 0.000 description 4
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 4
- 230000003441 anti-flavivirus Effects 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 4
- 239000007894 caplet Substances 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- 125000001309 chloro group Chemical group Cl* 0.000 description 4
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 4
- 238000002648 combination therapy Methods 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 230000008025 crystallization Effects 0.000 description 4
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 208000005252 hepatitis A Diseases 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 4
- 235000019359 magnesium stearate Nutrition 0.000 description 4
- 239000000594 mannitol Substances 0.000 description 4
- 235000010355 mannitol Nutrition 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 210000004379 membrane Anatomy 0.000 description 4
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 230000002035 prolonged effect Effects 0.000 description 4
- IGFXRKMLLMBKSA-UHFFFAOYSA-N purine Chemical compound N1=C[N]C2=NC=NC2=C1 IGFXRKMLLMBKSA-UHFFFAOYSA-N 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 229960001153 serine Drugs 0.000 description 4
- 235000004400 serine Nutrition 0.000 description 4
- 239000008223 sterile water Substances 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 230000002195 synergetic effect Effects 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 4
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 4
- XXJGBENTLXFVFI-UHFFFAOYSA-N 1-amino-methylene Chemical compound N[CH2] XXJGBENTLXFVFI-UHFFFAOYSA-N 0.000 description 3
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 3
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 3
- CKTSBUTUHBMZGZ-SHYZEUOFSA-N 2'‐deoxycytidine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-SHYZEUOFSA-N 0.000 description 3
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 3
- CKTSBUTUHBMZGZ-CHKWXVPMSA-N 4-amino-1-[(2s,4r,5s)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)[C@H](O)C1 CKTSBUTUHBMZGZ-CHKWXVPMSA-N 0.000 description 3
- STRZQWQNZQMHQR-UAKXSSHOSA-N 5-fluorocytidine Chemical compound C1=C(F)C(N)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 STRZQWQNZQMHQR-UAKXSSHOSA-N 0.000 description 3
- GWNHAOBXDGOXRR-HJFSHJIFSA-N 9-[2-[[(2r,4s)-4-(3-chlorophenyl)-2-oxo-1,3,2$l^{5}-dioxaphosphinan-2-yl]methoxy]ethyl]purin-6-amine Chemical compound C1([C@@H]2CCO[P@](O2)(=O)COCCN2C=3N=CN=C(C=3N=C2)N)=CC=CC(Cl)=C1 GWNHAOBXDGOXRR-HJFSHJIFSA-N 0.000 description 3
- 208000030507 AIDS Diseases 0.000 description 3
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- 206010008631 Cholera Diseases 0.000 description 3
- 241000710777 Classical swine fever virus Species 0.000 description 3
- 108091026890 Coding region Proteins 0.000 description 3
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 3
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 3
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- 230000005526 G1 to G0 transition Effects 0.000 description 3
- 241000531123 GB virus C Species 0.000 description 3
- 241000711557 Hepacivirus Species 0.000 description 3
- 241000710842 Japanese encephalitis virus Species 0.000 description 3
- 206010067125 Liver injury Diseases 0.000 description 3
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 101800001020 Non-structural protein 4A Proteins 0.000 description 3
- 101800001014 Non-structural protein 5A Proteins 0.000 description 3
- 235000019483 Peanut oil Nutrition 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- 229910005965 SO 2 Inorganic materials 0.000 description 3
- 108010067390 Viral Proteins Proteins 0.000 description 3
- 208000003152 Yellow Fever Diseases 0.000 description 3
- WVXRAFOPTSTNLL-RQJHMYQMSA-N [(2r,5s)-5-(6-aminopurin-9-yl)oxolan-2-yl]methanol Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1CC[C@H](CO)O1 WVXRAFOPTSTNLL-RQJHMYQMSA-N 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 229960005305 adenosine Drugs 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 3
- 108091007433 antigens Proteins 0.000 description 3
- 102000036639 antigens Human genes 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 125000001246 bromo group Chemical group Br* 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 3
- JRZBPELLUMBLQU-UHFFFAOYSA-N carbonazidic acid Chemical compound OC(=O)N=[N+]=[N-] JRZBPELLUMBLQU-UHFFFAOYSA-N 0.000 description 3
- 150000003857 carboxamides Chemical class 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 235000010980 cellulose Nutrition 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 230000007882 cirrhosis Effects 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- LXWYCLOUQZZDBD-LIYNQYRNSA-N csfv Chemical compound C([C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)[C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(O)=O)C1=CC=C(O)C=C1 LXWYCLOUQZZDBD-LIYNQYRNSA-N 0.000 description 3
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 3
- 239000008121 dextrose Substances 0.000 description 3
- 238000012377 drug delivery Methods 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 230000002255 enzymatic effect Effects 0.000 description 3
- 229940074057 epivir hbv Drugs 0.000 description 3
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 3
- 229940093471 ethyl oleate Drugs 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- VUWZPRWSIVNGKG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH2] VUWZPRWSIVNGKG-UHFFFAOYSA-N 0.000 description 3
- 125000004005 formimidoyl group Chemical group [H]\N=C(/[H])* 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 229940029575 guanosine Drugs 0.000 description 3
- 230000002489 hematologic effect Effects 0.000 description 3
- 231100000234 hepatic damage Toxicity 0.000 description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 229940090438 infergen Drugs 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 238000007918 intramuscular administration Methods 0.000 description 3
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 3
- 125000002346 iodo group Chemical group I* 0.000 description 3
- 239000008297 liquid dosage form Substances 0.000 description 3
- 230000008818 liver damage Effects 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- 239000006210 lotion Substances 0.000 description 3
- 231100000053 low toxicity Toxicity 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- AQIAIZBHFAKICS-UHFFFAOYSA-N methylaminomethyl Chemical compound [CH2]NC AQIAIZBHFAKICS-UHFFFAOYSA-N 0.000 description 3
- 210000004400 mucous membrane Anatomy 0.000 description 3
- SFDJOSRHYKHMOK-UHFFFAOYSA-N nitramide Chemical compound N[N+]([O-])=O SFDJOSRHYKHMOK-UHFFFAOYSA-N 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 230000007170 pathology Effects 0.000 description 3
- 239000000312 peanut oil Substances 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- 229920003023 plastic Polymers 0.000 description 3
- 239000004033 plastic Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 3
- 235000019833 protease Nutrition 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000008159 sesame oil Substances 0.000 description 3
- 235000011803 sesame oil Nutrition 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 230000001954 sterilising effect Effects 0.000 description 3
- 238000004659 sterilization and disinfection Methods 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- NZVYCXVTEHPMHE-ZSUJOUNUSA-N thymalfasin Chemical compound CC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O NZVYCXVTEHPMHE-ZSUJOUNUSA-N 0.000 description 3
- 229940104230 thymidine Drugs 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 229950002635 torcitabine Drugs 0.000 description 3
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 3
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 3
- 229940045145 uridine Drugs 0.000 description 3
- 230000029812 viral genome replication Effects 0.000 description 3
- 239000003039 volatile agent Substances 0.000 description 3
- 238000009736 wetting Methods 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- SCVHJVCATBPIHN-SJCJKPOMSA-N (3s)-3-[[(2s)-2-[[2-(2-tert-butylanilino)-2-oxoacetyl]amino]propanoyl]amino]-4-oxo-5-(2,3,5,6-tetrafluorophenoxy)pentanoic acid Chemical compound N([C@@H](C)C(=O)N[C@@H](CC(O)=O)C(=O)COC=1C(=C(F)C=C(F)C=1F)F)C(=O)C(=O)NC1=CC=CC=C1C(C)(C)C SCVHJVCATBPIHN-SJCJKPOMSA-N 0.000 description 2
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- JBWYRBLDOOOJEU-UHFFFAOYSA-N 1-[chloro-(4-methoxyphenyl)-phenylmethyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC(OC)=CC=1)C1=CC=CC=C1 JBWYRBLDOOOJEU-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- NVKAMPJSWMHVDK-GITKWUPZSA-N 2-amino-9-[(2r,3r,4r,5r)-3,4-dihydroxy-5-(hydroxymethyl)-3-methyloxolan-2-yl]-3h-purin-6-one Chemical compound C[C@@]1(O)[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC(N)=NC(O)=C2N=C1 NVKAMPJSWMHVDK-GITKWUPZSA-N 0.000 description 2
- GXYYUDQAGCVAGJ-HHGSPMIASA-M 217rji972k Chemical compound [Na+].O=C([C@@]12C[C@H]1\C=C/CCCCC[C@@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC(=O)N1CC2=C(F)C=CC=C2C1)=O)NC(=O)OC(C)(C)C)[N-]S(=O)(=O)C1CC1 GXYYUDQAGCVAGJ-HHGSPMIASA-M 0.000 description 2
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 2
- 229930024421 Adenine Natural products 0.000 description 2
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical class [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 241001167018 Aroa Species 0.000 description 2
- MRKCXBHWXHYQCZ-OPHTVVJGSA-N C[C@@]1([C@@H](O[C@@H]([C@H]1O)CO)N1C(=O)N=C(N)C=C1)O.C(C1=CC=CC=C1)NP(O)(O)=O Chemical compound C[C@@]1([C@@H](O[C@@H]([C@H]1O)CO)N1C(=O)N=C(N)C=C1)O.C(C1=CC=CC=C1)NP(O)(O)=O MRKCXBHWXHYQCZ-OPHTVVJGSA-N 0.000 description 2
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 208000003322 Coinfection Diseases 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 206010059866 Drug resistance Diseases 0.000 description 2
- 241000283073 Equus caballus Species 0.000 description 2
- 241001125671 Eretmochelys imbricata Species 0.000 description 2
- 240000000915 Fagraea fragrans Species 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 229940122604 HCV protease inhibitor Drugs 0.000 description 2
- 206010019759 Hepatitis chronic persistent Diseases 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 229920000881 Modified starch Polymers 0.000 description 2
- 241000288894 Myotis Species 0.000 description 2
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- 240000003492 Neolamarckia cadamba Species 0.000 description 2
- 101800001019 Non-structural protein 4B Proteins 0.000 description 2
- 101710144121 Non-structural protein 5 Proteins 0.000 description 2
- 241000725177 Omsk hemorrhagic fever virus Species 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- 241000009328 Perro Species 0.000 description 2
- 208000037581 Persistent Infection Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 241000282887 Suidae Species 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 208000002903 Thalassemia Diseases 0.000 description 2
- 108010078233 Thymalfasin Proteins 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 241000005757 Turkey meningoencephalitis virus Species 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 241000464917 Vieja Species 0.000 description 2
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 2
- 241000710886 West Nile virus Species 0.000 description 2
- 208000020329 Zika virus infectious disease Diseases 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- YQNQNVDNTFHQSW-UHFFFAOYSA-N acetic acid [2-[[(5-nitro-2-thiazolyl)amino]-oxomethyl]phenyl] ester Chemical compound CC(=O)OC1=CC=CC=C1C(=O)NC1=NC=C([N+]([O-])=O)S1 YQNQNVDNTFHQSW-UHFFFAOYSA-N 0.000 description 2
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 2
- 229960004150 aciclovir Drugs 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 229940054685 alinia Drugs 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 2
- 125000003275 alpha amino acid group Chemical group 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- 229960003805 amantadine Drugs 0.000 description 2
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 125000005124 aminocycloalkyl group Chemical group 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 239000003708 ampul Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 229960003121 arginine Drugs 0.000 description 2
- 125000005160 aryl oxy alkyl group Chemical group 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 238000011914 asymmetric synthesis Methods 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 230000036765 blood level Effects 0.000 description 2
- LHHCSNFAOIFYRV-DOVBMPENSA-N boceprevir Chemical compound O=C([C@@H]1[C@@H]2[C@@H](C2(C)C)CN1C(=O)[C@@H](NC(=O)NC(C)(C)C)C(C)(C)C)NC(C(=O)C(N)=O)CC1CCC1 LHHCSNFAOIFYRV-DOVBMPENSA-N 0.000 description 2
- 229960000517 boceprevir Drugs 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000013877 carbamide Nutrition 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000013626 chemical specie Substances 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000011284 combination treatment Methods 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 235000005687 corn oil Nutrition 0.000 description 2
- 239000002285 corn oil Substances 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- 239000002385 cottonseed oil Substances 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 125000000000 cycloalkoxy group Chemical group 0.000 description 2
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 2
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 238000006297 dehydration reaction Methods 0.000 description 2
- 201000002950 dengue hemorrhagic fever Diseases 0.000 description 2
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 2
- 229940043237 diethanolamine Drugs 0.000 description 2
- 239000001177 diphosphate Substances 0.000 description 2
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 2
- 235000011180 diphosphates Nutrition 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 229950000234 emricasan Drugs 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 229930195712 glutamate Natural products 0.000 description 2
- 229940049906 glutamate Drugs 0.000 description 2
- 150000002334 glycols Chemical class 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 208000034737 hemoglobinopathy Diseases 0.000 description 2
- 108700012707 hepatitis C virus NS3 Proteins 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 2
- 229960002885 histidine Drugs 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 229960003507 interferon alfa-2b Drugs 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 229940065638 intron a Drugs 0.000 description 2
- 125000000654 isopropylidene group Chemical group C(C)(C)=* 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 244000144972 livestock Species 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 229960003646 lysine Drugs 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000008384 membrane barrier Effects 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 230000008520 organization Effects 0.000 description 2
- 230000003204 osmotic effect Effects 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 229960003930 peginterferon alfa-2a Drugs 0.000 description 2
- 108010092851 peginterferon alfa-2b Proteins 0.000 description 2
- 229940106366 pegintron Drugs 0.000 description 2
- 239000003961 penetration enhancing agent Substances 0.000 description 2
- 239000008177 pharmaceutical agent Substances 0.000 description 2
- 229940127557 pharmaceutical product Drugs 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 150000003053 piperidines Chemical class 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920005597 polymer membrane Polymers 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- 208000020016 psychiatric disease Diseases 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 230000000405 serological effect Effects 0.000 description 2
- 208000007056 sickle cell anemia Diseases 0.000 description 2
- 230000002226 simultaneous effect Effects 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- RMLUKZWYIKEASN-UHFFFAOYSA-M sodium;2-amino-9-(2-hydroxyethoxymethyl)purin-6-olate Chemical compound [Na+].O=C1[N-]C(N)=NC2=C1N=CN2COCCO RMLUKZWYIKEASN-UHFFFAOYSA-M 0.000 description 2
- MIXCUJKCXRNYFM-UHFFFAOYSA-M sodium;diiodomethanesulfonate;n-propyl-n-[2-(2,4,6-trichlorophenoxy)ethyl]imidazole-1-carboxamide Chemical compound [Na+].[O-]S(=O)(=O)C(I)I.C1=CN=CN1C(=O)N(CCC)CCOC1=C(Cl)C=C(Cl)C=C1Cl MIXCUJKCXRNYFM-UHFFFAOYSA-M 0.000 description 2
- 239000008247 solid mixture Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 239000003549 soybean oil Substances 0.000 description 2
- 235000012424 soybean oil Nutrition 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000008174 sterile solution Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- HWCKGOZZJDHMNC-UHFFFAOYSA-M tetraethylammonium bromide Chemical compound [Br-].CC[N+](CC)(CC)CC HWCKGOZZJDHMNC-UHFFFAOYSA-M 0.000 description 2
- 125000004001 thioalkyl group Chemical group 0.000 description 2
- 229960004231 thymalfasin Drugs 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- DZRQMHSNVNTFAQ-IVGJVWKCSA-N (2r,3r)-2,3-dihydroxybutanedioic acid;(2r,3s,4r,5s)-1-(6-ethoxyhexyl)-2-methylpiperidine-3,4,5-triol Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.CCOCCCCCCN1C[C@H](O)[C@@H](O)[C@@H](O)[C@H]1C DZRQMHSNVNTFAQ-IVGJVWKCSA-N 0.000 description 1
- IVWWFWFVSWOTLP-YVZVNANGSA-N (3'as,4r,7'as)-2,2,2',2'-tetramethylspiro[1,3-dioxolane-4,6'-4,7a-dihydro-3ah-[1,3]dioxolo[4,5-c]pyran]-7'-one Chemical compound C([C@@H]1OC(O[C@@H]1C1=O)(C)C)O[C@]21COC(C)(C)O2 IVWWFWFVSWOTLP-YVZVNANGSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- AAWZDTNXLSGCEK-LNVDRNJUSA-N (3r,5r)-1,3,4,5-tetrahydroxycyclohexane-1-carboxylic acid Chemical class O[C@@H]1CC(O)(C(O)=O)C[C@@H](O)C1O AAWZDTNXLSGCEK-LNVDRNJUSA-N 0.000 description 1
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- WSWCOQWTEOXDQX-MQQKCMAXSA-M (E,E)-sorbate Chemical compound C\C=C\C=C\C([O-])=O WSWCOQWTEOXDQX-MQQKCMAXSA-M 0.000 description 1
- ZJDYSLGODIRURM-UHFFFAOYSA-N (benzylamino)phosphonic acid Chemical compound OP(O)(=O)NCC1=CC=CC=C1 ZJDYSLGODIRURM-UHFFFAOYSA-N 0.000 description 1
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical compound C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 description 1
- CXUAEBDTJFKMBV-UHFFFAOYSA-N 1-(chloromethyl)-2,3,4,5,6-pentamethylbenzene Chemical compound CC1=C(C)C(C)=C(CCl)C(C)=C1C CXUAEBDTJFKMBV-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- AMMPLVWPWSYRDR-UHFFFAOYSA-N 1-methylbicyclo[2.2.2]oct-2-ene-4-carboxylic acid Chemical compound C1CC2(C(O)=O)CCC1(C)C=C2 AMMPLVWPWSYRDR-UHFFFAOYSA-N 0.000 description 1
- CEHJYEXLKQVWOT-UHFFFAOYSA-N 2,4,6-trihydroxy-3-nitrobenzamide Chemical class NC(=O)C1=C(O)C=C(O)C([N+]([O-])=O)=C1O CEHJYEXLKQVWOT-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 108020005345 3' Untranslated Regions Proteins 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- YUDSCJBUWTYENI-VPCXQMTMSA-N 4-amino-1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)-2-methyloxolan-2-yl]pyrimidin-2-one Chemical compound C1=CC(N)=NC(=O)N1[C@]1(C)O[C@H](CO)[C@@H](O)[C@H]1O YUDSCJBUWTYENI-VPCXQMTMSA-N 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- SVXNJCYYMRMXNM-UHFFFAOYSA-N 5-amino-2h-1,2,4-triazin-3-one Chemical compound NC=1C=NNC(=O)N=1 SVXNJCYYMRMXNM-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- LZGICTBSBIWVFA-UHFFFAOYSA-N 5-bromo-2-ethenylpyrimidine Chemical compound BrC1=CN=C(C=C)N=C1 LZGICTBSBIWVFA-UHFFFAOYSA-N 0.000 description 1
- WTDWVLJJJOTABN-UHFFFAOYSA-N 5-cyclopropyl-2-(4-fluorophenyl)-6-[(2-hydroxyethyl)(methylsulfonyl)amino]-n-methyl-1-benzofuran-3-carboxamide Chemical compound C1=C2C(C(=O)NC)=C(C=3C=CC(F)=CC=3)OC2=CC(N(CCO)S(C)(=O)=O)=C1C1CC1 WTDWVLJJJOTABN-UHFFFAOYSA-N 0.000 description 1
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 1
- ZKBQDFAWXLTYKS-UHFFFAOYSA-N 6-Chloro-1H-purine Chemical compound ClC1=NC=NC2=C1NC=N2 ZKBQDFAWXLTYKS-UHFFFAOYSA-N 0.000 description 1
- PVRBGBGMDLPYKG-UHFFFAOYSA-N 6-benzyl-7h-purine Chemical compound N=1C=NC=2N=CNC=2C=1CC1=CC=CC=C1 PVRBGBGMDLPYKG-UHFFFAOYSA-N 0.000 description 1
- DBCMWACNZJYUHS-UHFFFAOYSA-N 6-ethenyl-7h-purine Chemical compound C=CC1=NC=NC2=C1NC=N2 DBCMWACNZJYUHS-UHFFFAOYSA-N 0.000 description 1
- LOSIULRWFAEMFL-UHFFFAOYSA-N 7-deazaguanine Chemical compound O=C1NC(N)=NC2=C1CC=N2 LOSIULRWFAEMFL-UHFFFAOYSA-N 0.000 description 1
- YYVYAPXYZVYDHN-UHFFFAOYSA-N 9,10-phenanthroquinone Chemical compound C1=CC=C2C(=O)C(=O)C3=CC=CC=C3C2=C1 YYVYAPXYZVYDHN-UHFFFAOYSA-N 0.000 description 1
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 1
- 208000004998 Abdominal Pain Diseases 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 229910002016 Aerosil® 200 Inorganic materials 0.000 description 1
- 206010001488 Aggression Diseases 0.000 description 1
- OLROWHGDTNFZBH-XEMWPYQTSA-N Alisporivir Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)N(CC)C(=O)[C@@H](C)N(C)C1=O OLROWHGDTNFZBH-XEMWPYQTSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 244000144725 Amygdalus communis Species 0.000 description 1
- 235000011437 Amygdalus communis Nutrition 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- OMSPNIGCBQFQFQ-UHFFFAOYSA-N BrC1=CC=NC(C=C)=N1 Chemical compound BrC1=CC=NC(C=C)=N1 OMSPNIGCBQFQFQ-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 239000004358 Butane-1, 3-diol Substances 0.000 description 1
- 101100129500 Caenorhabditis elegans max-2 gene Proteins 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241001227713 Chiron Species 0.000 description 1
- 239000004709 Chlorinated polyethylene Substances 0.000 description 1
- 208000006154 Chronic hepatitis C Diseases 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- 108090000227 Chymases Proteins 0.000 description 1
- 102000003858 Chymases Human genes 0.000 description 1
- 244000223760 Cinnamomum zeylanicum Species 0.000 description 1
- 235000005979 Citrus limon Nutrition 0.000 description 1
- 244000131522 Citrus pyriformis Species 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- HVXBOLULGPECHP-WAYWQWQTSA-N Combretastatin A4 Chemical compound C1=C(O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 HVXBOLULGPECHP-WAYWQWQTSA-N 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 101710118188 DNA-binding protein HU-alpha Proteins 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 101100083446 Danio rerio plekhh1 gene Proteins 0.000 description 1
- 206010054089 Depressive symptom Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- BXZVVICBKDXVGW-NKWVEPMBSA-N Didanosine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(NC=NC2=O)=C2N=C1 BXZVVICBKDXVGW-NKWVEPMBSA-N 0.000 description 1
- 208000003870 Drug Overdose Diseases 0.000 description 1
- 241001457989 Encephalus Species 0.000 description 1
- IMROMDMJAWUWLK-UHFFFAOYSA-N Ethenol Chemical compound OC=C IMROMDMJAWUWLK-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 239000001828 Gelatine Substances 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 229920002907 Guar gum Polymers 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 206010018910 Haemolysis Diseases 0.000 description 1
- 206010061192 Haemorrhagic fever Diseases 0.000 description 1
- 241001529916 Hepatitis GB virus B Species 0.000 description 1
- 206010019791 Hepatitis post transfusion Diseases 0.000 description 1
- 206010019799 Hepatitis viral Diseases 0.000 description 1
- 244000043261 Hevea brasiliensis Species 0.000 description 1
- 241000545744 Hirudinea Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 206010023126 Jaundice Diseases 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 101100170604 Mus musculus Dmap1 gene Proteins 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 101710144128 Non-structural protein 2 Proteins 0.000 description 1
- 101710199667 Nuclear export protein Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 108090001074 Nucleocapsid Proteins Proteins 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 206010033296 Overdoses Diseases 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 241000682990 Pegivirus A Species 0.000 description 1
- 241000228127 Penicillium griseofulvum Species 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 229920012485 Plasticized Polyvinyl chloride Polymers 0.000 description 1
- 239000005062 Polybutadiene Substances 0.000 description 1
- 229920002367 Polyisobutene Polymers 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102100038239 Protein Churchill Human genes 0.000 description 1
- 108010078762 Protein Precursors Proteins 0.000 description 1
- 102000014961 Protein Precursors Human genes 0.000 description 1
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 1
- 101000774655 Protobothrops mucrosquamatus Snake venom metalloproteinase TM-1 Proteins 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 108030002536 RNA-directed RNA polymerases Proteins 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 229920000297 Rayon Polymers 0.000 description 1
- 239000008156 Ringer's lactate solution Substances 0.000 description 1
- 241000015473 Schizothorax griseus Species 0.000 description 1
- 241000242583 Scyphozoa Species 0.000 description 1
- 208000009714 Severe Dengue Diseases 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 241000187180 Streptomyces sp. Species 0.000 description 1
- 108090000787 Subtilisin Proteins 0.000 description 1
- 206010042458 Suicidal ideation Diseases 0.000 description 1
- 235000019486 Sunflower oil Nutrition 0.000 description 1
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108020005202 Viral DNA Proteins 0.000 description 1
- 108020000999 Viral RNA Proteins 0.000 description 1
- 108010087302 Viral Structural Proteins Proteins 0.000 description 1
- 206010051511 Viral diarrhoea Diseases 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 239000005862 Whey Substances 0.000 description 1
- 102000007544 Whey Proteins Human genes 0.000 description 1
- 108010046377 Whey Proteins Proteins 0.000 description 1
- 241000710772 Yellow fever virus Species 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- ZWELIJXAKMASLK-UGKPPGOTSA-N [(2r,3r,4r,5r)-4-acetyloxy-5-(5-amino-2-oxo-[1,3]thiazolo[4,5-d]pyrimidin-3-yl)-2-(hydroxymethyl)oxolan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1[C@H](OC(=O)C)[C@@H](CO)O[C@H]1N1C(=O)SC2=CN=C(N)N=C21 ZWELIJXAKMASLK-UGKPPGOTSA-N 0.000 description 1
- MLESJYFEMSJZLZ-MAAOGQSESA-N [(2r,3r,4r,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-4-fluoro-4-methyl-3-(2-methylpropanoyloxy)oxolan-2-yl]methyl 2-methylpropanoate Chemical compound C[C@@]1(F)[C@H](OC(=O)C(C)C)[C@@H](COC(=O)C(C)C)O[C@H]1N1C(=O)N=C(N)C=C1 MLESJYFEMSJZLZ-MAAOGQSESA-N 0.000 description 1
- VKXWOLCNTHXCLF-DXEZIKHYSA-N [(2r,3s,4r,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-2-azido-3,4-bis(2-methylpropanoyloxy)oxolan-2-yl]methyl 2-methylpropanoate Chemical compound CC(C)C(=O)O[C@@H]1[C@H](OC(=O)C(C)C)[C@](COC(=O)C(C)C)(N=[N+]=[N-])O[C@H]1N1C(=O)N=C(N)C=C1 VKXWOLCNTHXCLF-DXEZIKHYSA-N 0.000 description 1
- JVMVGISGLJMHAW-UOERWJHTSA-N [C@@H]1(CC[C@@H](CO)O1)N1C=NC=2C(N)=NC=NC12.C(C1=CC=CC=C1)NP(O)(O)=O Chemical compound [C@@H]1(CC[C@@H](CO)O1)N1C=NC=2C(N)=NC=NC12.C(C1=CC=CC=C1)NP(O)(O)=O JVMVGISGLJMHAW-UOERWJHTSA-N 0.000 description 1
- 230000009102 absorption Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 238000007259 addition reaction Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- SUPKOOSCJHTBAH-UHFFFAOYSA-N adefovir Chemical compound NC1=NC=NC2=C1N=CN2CCOCP(O)(O)=O SUPKOOSCJHTBAH-UHFFFAOYSA-N 0.000 description 1
- 229960003205 adefovir dipivoxil Drugs 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000016571 aggressive behavior Effects 0.000 description 1
- 208000012761 aggressive behavior Diseases 0.000 description 1
- 229960003767 alanine Drugs 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 1
- 108010058359 alisporivir Proteins 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 229910001420 alkaline earth metal ion Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 125000003302 alkenyloxy group Chemical group 0.000 description 1
- 125000005360 alkyl sulfoxide group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 235000020224 almond Nutrition 0.000 description 1
- 108010027597 alpha-chymotrypsin Proteins 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 238000005576 amination reaction Methods 0.000 description 1
- 150000003862 amino acid derivatives Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 1
- 230000036436 anti-hiv Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 125000000254 aspartoyl group Chemical group 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- 230000000721 bacterilogical effect Effects 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- UPABQMWFWCMOFV-UHFFFAOYSA-N benethamine Chemical compound C=1C=CC=CC=1CNCCC1=CC=CC=C1 UPABQMWFWCMOFV-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- 150000001556 benzimidazoles Chemical class 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- DLRVVLDZNNYCBX-JZSVMVJISA-N beta-D-Galp-(1->6)-D-Galp Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OC[C@@H]1[C@H](O)[C@H](O)[C@@H](O)C(O)O1 DLRVVLDZNNYCBX-JZSVMVJISA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 239000003613 bile acid Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229920005549 butyl rubber Polymers 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229960003563 calcium carbonate Drugs 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- ZEWYCNBZMPELPF-UHFFFAOYSA-J calcium;potassium;sodium;2-hydroxypropanoic acid;sodium;tetrachloride Chemical compound [Na].[Na+].[Cl-].[Cl-].[Cl-].[Cl-].[K+].[Ca+2].CC(O)C(O)=O ZEWYCNBZMPELPF-UHFFFAOYSA-J 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229940114081 cinnamate Drugs 0.000 description 1
- 235000017803 cinnamon Nutrition 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 229960005537 combretastatin A-4 Drugs 0.000 description 1
- HVXBOLULGPECHP-UHFFFAOYSA-N combretastatin A4 Natural products C1=C(O)C(OC)=CC=C1C=CC1=CC(OC)=C(OC)C(OC)=C1 HVXBOLULGPECHP-UHFFFAOYSA-N 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000010411 cooking Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- MRKZAZMYXYSBDG-UHFFFAOYSA-N cyclopentyl propanoate Chemical compound CCC(=O)OC1CCCC1 MRKZAZMYXYSBDG-UHFFFAOYSA-N 0.000 description 1
- 229960002433 cysteine Drugs 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 239000002852 cysteine proteinase inhibitor Substances 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 235000013365 dairy product Nutrition 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 125000003074 decanoyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(*)=O 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 235000013870 dimethyl polysiloxane Nutrition 0.000 description 1
- KUMNEOGIHFCNQW-UHFFFAOYSA-N diphenyl phosphite Chemical compound C=1C=CC=CC=1OP([O-])OC1=CC=CC=C1 KUMNEOGIHFCNQW-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 231100000725 drug overdose Toxicity 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 241001493065 dsRNA viruses Species 0.000 description 1
- 238000010894 electron beam technology Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 229920005558 epichlorohydrin rubber Polymers 0.000 description 1
- 229940072253 epivir Drugs 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- IDGUHHHQCWSQLU-UHFFFAOYSA-N ethanol;hydrate Chemical compound O.CCO IDGUHHHQCWSQLU-UHFFFAOYSA-N 0.000 description 1
- 229940031098 ethanolamine Drugs 0.000 description 1
- HQQADJVZYDDRJT-UHFFFAOYSA-N ethene;prop-1-ene Chemical group C=C.CC=C HQQADJVZYDDRJT-UHFFFAOYSA-N 0.000 description 1
- 125000005678 ethenylene group Chemical group [H]C([*:1])=C([H])[*:2] 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 235000020375 flavoured syrup Nutrition 0.000 description 1
- 235000013312 flour Nutrition 0.000 description 1
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 1
- 229960004413 flucytosine Drugs 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229910021485 fumed silica Inorganic materials 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 108010074605 gamma-Globulins Proteins 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 229960002743 glutamine Drugs 0.000 description 1
- 235000004554 glutamine Nutrition 0.000 description 1
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N glutaric acid Chemical compound OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 235000010417 guar gum Nutrition 0.000 description 1
- 239000000665 guar gum Substances 0.000 description 1
- 229960002154 guar gum Drugs 0.000 description 1
- 230000008588 hemolysis Effects 0.000 description 1
- 125000000268 heptanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N hydrazine Substances NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- AFQIYTIJXGTIEY-UHFFFAOYSA-N hydrogen carbonate;triethylazanium Chemical compound OC(O)=O.CCN(CC)CC AFQIYTIJXGTIEY-UHFFFAOYSA-N 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 150000008624 imidazolidinones Chemical class 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000002584 immunomodulator Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 230000001861 immunosuppressant effect Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 208000018337 inherited hemoglobinopathy Diseases 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 229920000554 ionomer Polymers 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229940074928 isopropyl myristate Drugs 0.000 description 1
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 229960003136 leucine Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 229940059904 light mineral oil Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000008263 liquid aerosol Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- WMFOQBRAJBCJND-UHFFFAOYSA-M lithium hydroxide Inorganic materials [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 108010046177 locteron Proteins 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 108010026228 mRNA guanylyltransferase Proteins 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000014380 magnesium carbonate Nutrition 0.000 description 1
- 238000011418 maintenance treatment Methods 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 229960004452 methionine Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- KJRFTNVYOAGTHK-UHFFFAOYSA-N methyl 3-hydroxy-2,2-dimethylpropanoate Chemical compound COC(=O)C(C)(C)CO KJRFTNVYOAGTHK-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- LOTBYPQQWICYBB-UHFFFAOYSA-N methyl n-hexyl-n-[2-(hexylamino)ethyl]carbamate Chemical compound CCCCCCNCCN(C(=O)OC)CCCCCC LOTBYPQQWICYBB-UHFFFAOYSA-N 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- VKHAHZOOUSRJNA-GCNJZUOMSA-N mifepristone Chemical compound C1([C@@H]2C3=C4CCC(=O)C=C4CC[C@H]3[C@@H]3CC[C@@]([C@]3(C2)C)(O)C#CC)=CC=C(N(C)C)C=C1 VKHAHZOOUSRJNA-GCNJZUOMSA-N 0.000 description 1
- GVZFUVXPTPGOQT-UHFFFAOYSA-M mitoq Chemical compound CS([O-])(=O)=O.O=C1C(OC)=C(OC)C(=O)C(CCCCCCCCCC[P+](C=2C=CC=CC=2)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1C GVZFUVXPTPGOQT-UHFFFAOYSA-M 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 230000003020 moisturizing effect Effects 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 210000002200 mouth mucosa Anatomy 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- ZZKNRXZVGOYGJT-GSVOUGTGSA-N n-(phosphonacetyl)-l-aspartic acid Chemical compound OC(=O)C[C@H](C(O)=O)NC(=O)CP(O)(O)=O ZZKNRXZVGOYGJT-GSVOUGTGSA-N 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 208000004235 neutropenia Diseases 0.000 description 1
- 229940080607 nexavar Drugs 0.000 description 1
- YCWSUKQGVSGXJO-NTUHNPAUSA-N nifuroxazide Chemical group C1=CC(O)=CC=C1C(=O)N\N=C\C1=CC=C([N+]([O-])=O)O1 YCWSUKQGVSGXJO-NTUHNPAUSA-N 0.000 description 1
- 125000004355 nitrogen functional group Chemical group 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000002801 octanoyl group Chemical group C(CCCCCCC)(=O)* 0.000 description 1
- 125000001117 oleyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 229960005190 phenylalanine Drugs 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- NAYYNDKKHOIIOD-UHFFFAOYSA-N phthalamide Chemical class NC(=O)C1=CC=CC=C1C(N)=O NAYYNDKKHOIIOD-UHFFFAOYSA-N 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- PJPOCNJHYFUPCE-UHFFFAOYSA-N picen-1-ol Chemical compound C1=CC=CC2=C(C=CC=3C4=CC=C5C=CC=C(C=35)O)C4=CC=C21 PJPOCNJHYFUPCE-UHFFFAOYSA-N 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-M pivalate Chemical compound CC(C)(C)C([O-])=O IUGYQRQAERSCNH-UHFFFAOYSA-M 0.000 description 1
- 239000000419 plant extract Substances 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229920001490 poly(butyl methacrylate) polymer Polymers 0.000 description 1
- 229920001084 poly(chloroprene) Polymers 0.000 description 1
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 229920002857 polybutadiene Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920000139 polyethylene terephthalate Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 229920001195 polyisoprene Polymers 0.000 description 1
- 229920006254 polymer film Polymers 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920000379 polypropylene carbonate Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 229960003975 potassium Drugs 0.000 description 1
- 235000007686 potassium Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 229920003124 powdered cellulose Polymers 0.000 description 1
- 235000019814 powdered cellulose Nutrition 0.000 description 1
- 229950001452 pradefovir Drugs 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 229960002429 proline Drugs 0.000 description 1
- 235000013930 proline Nutrition 0.000 description 1
- 125000006410 propenylene group Chemical group 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 230000004853 protein function Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 239000002212 purine nucleoside Substances 0.000 description 1
- 239000002718 pyrimidine nucleoside Substances 0.000 description 1
- JAEIBKXSIXOLOL-UHFFFAOYSA-N pyrrolidin-1-ium-3-carboxylate Chemical compound OC(=O)C1CCNC1 JAEIBKXSIXOLOL-UHFFFAOYSA-N 0.000 description 1
- 150000004040 pyrrolidinones Chemical class 0.000 description 1
- 229940076788 pyruvate Drugs 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 229940038850 rebif Drugs 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 239000002342 ribonucleoside Substances 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- ILJOYZVVZZFIKA-UHFFFAOYSA-M sodium;1,1-dioxo-1,2-benzothiazol-3-olate;hydrate Chemical compound O.[Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 ILJOYZVVZZFIKA-UHFFFAOYSA-M 0.000 description 1
- 239000007905 soft elastic gelatin capsule Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- IVDHYUQIDRJSTI-UHFFFAOYSA-N sorafenib tosylate Chemical compound [H+].CC1=CC=C(S([O-])(=O)=O)C=C1.C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 IVDHYUQIDRJSTI-UHFFFAOYSA-N 0.000 description 1
- 229940075554 sorbate Drugs 0.000 description 1
- WSWCOQWTEOXDQX-MQQKCMAXSA-N sorbic acid group Chemical group C(\C=C\C=C\C)(=O)O WSWCOQWTEOXDQX-MQQKCMAXSA-N 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- 229940031439 squalene Drugs 0.000 description 1
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000009102 step therapy Methods 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-M sulfamate Chemical compound NS([O-])(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-M 0.000 description 1
- 239000002600 sunflower oil Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- BBAWEDCPNXPBQM-GDEBMMAJSA-N telaprevir Chemical compound N([C@H](C(=O)N[C@H](C(=O)N1C[C@@H]2CCC[C@@H]2[C@H]1C(=O)N[C@@H](CCC)C(=O)C(=O)NC1CC1)C(C)(C)C)C1CCCCC1)C(=O)C1=CN=CC=N1 BBAWEDCPNXPBQM-GDEBMMAJSA-N 0.000 description 1
- 229920001897 terpolymer Polymers 0.000 description 1
- WMOVHXAZOJBABW-UHFFFAOYSA-N tert-butyl acetate Chemical compound CC(=O)OC(C)(C)C WMOVHXAZOJBABW-UHFFFAOYSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000005207 tetraalkylammonium group Chemical class 0.000 description 1
- YMBCJWGVCUEGHA-UHFFFAOYSA-M tetraethylammonium chloride Chemical compound [Cl-].CC[N+](CC)(CC)CC YMBCJWGVCUEGHA-UHFFFAOYSA-M 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- 125000005309 thioalkoxy group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 229960002898 threonine Drugs 0.000 description 1
- 235000008521 threonine Nutrition 0.000 description 1
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 206010043554 thrombocytopenia Diseases 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 239000006208 topical dosage form Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M trans-cinnamate Chemical compound [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- 239000006211 transdermal dosage form Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 229940066528 trichloroacetate Drugs 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000002264 triphosphate group Chemical group [H]OP(=O)(O[H])OP(=O)(O[H])OP(=O)(O[H])O* 0.000 description 1
- 229960004418 trolamine Drugs 0.000 description 1
- 229960004799 tryptophan Drugs 0.000 description 1
- 125000005454 tryptophanyl group Chemical group 0.000 description 1
- 229960004441 tyrosine Drugs 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 235000002374 tyrosine Nutrition 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 229920011532 unplasticized polyvinyl chloride Polymers 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 201000001862 viral hepatitis Diseases 0.000 description 1
- 230000009265 virologic response Effects 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 229940051021 yellow-fever virus Drugs 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910021511 zinc hydroxide Inorganic materials 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/02—Heterocyclic radicals containing only nitrogen as ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/22—Amides of acids of phosphorus
- C07F9/24—Esteramides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/7056—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing five-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
- A61K31/7072—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid having two oxo groups directly attached to the pyrimidine ring, e.g. uridine, uridylic acid, thymidine, zidovudine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
- A61K31/708—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid having oxo groups directly attached to the purine ring system, e.g. guanosine, guanylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65586—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system at least one of the hetero rings does not contain nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Diabetes (AREA)
- Biotechnology (AREA)
- Cardiology (AREA)
- Virology (AREA)
- Genetics & Genomics (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Communicable Diseases (AREA)
- Hematology (AREA)
- Physical Education & Sports Medicine (AREA)
- Obesity (AREA)
- Oncology (AREA)
- Rheumatology (AREA)
- Ophthalmology & Optometry (AREA)
- Urology & Nephrology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Hospice & Palliative Care (AREA)
- Vascular Medicine (AREA)
Abstract
Настоящее изобретение относится к соединениям формул:



где Ry представляет собой гидрокси(C1-C10)алкил; Ra и Rb независимо представляют собой водород, необязательно замещенный C1-C10 алкил, арил, арил(C1-C10)алкил или (C1-C10)циклоалкил, или Ra и Rb вместе с атомом азота образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов; R1 выбран из рибавирина, вирамидина, валопицитабина, PSI-6130, МК-0608, резиквимода, целгосивира, ламивудина, энтекавира, телбивудина, рацивира, эмтрицитабина, клевудина, амдоксовира и валторцитабина; а также композициям на их основе для лечения гепатита B или C. Предложены новые эффективные соединения против гепатита В или С. 4 н. и 9 з.п. ф-лы, 2 ил., 28 пр.
Description
ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая патентная заявка устанавливает приоритет 1) предварительной заявки США № 60/877944, зарегистрированной 28 декабря 2006 года; 2) предварительной заявки США № 60/936290, зарегистрированной 18 июня 2007 года; и 3) предварительной заявки США № 60/985891, зарегистрированной 6 ноября 2007 года. Раскрытия вышеупомянутых заявок включены в настоящее изобретение в качестве ссылки во всей их полноте.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, способам и фармацевтическим композициям для применения в лечении вирусных инфекций, включающих в себя вирусные инфекции гепатит C и гепатит B у организма-хозяина, который нуждается в лечении. В конкретном варианте осуществления рассматриваются нуклеозидные соединения фосфорамидатов или фосфонамидатов, которые обеспечивают концентрацию препарата в печени.
УРОВЕНЬ ТЕХНИКИ
Вирусы семейства Flaviviridae
Семейство вирусов Flaviviridae содержит по меньшей мере три разных рода: пестивирусы (pestivirus), которые вызывают болезни крупного рогатого скота и свиней, флавивирусы (flavivirus), которые являются первичной причиной таких болезней, как лихорадка денге и желтая лихорадка; и гепацивирусы (hepacivirus), единственным представителем которых является HCV. Род флавивирусов включает в себя более 68 членов, подразделяемых на группы на основе серологического родства (Calisher et al., J. Gen. Virol, 1993, 70, 37-43). Клинические симптомы различны и включают в себя лихорадку, энцефалит и геморрагическую лихорадку (Fields Virology, Editors: Fields, B.N., Knipe, D.M., and Howley, P.M., Lippincott-Raven Publishers, Philadelphia, PA, 1996, Chapter 31, 931-959). Флавивирусы, связанные с болезнями человека, вызывающие глобальную озабоченность, включают в себя вирусы геморрагической лихорадки денге (DHF), вирус желтой лихорадки, синдром шока и вирус японского энцефалита (Halstead, S.B., Rev. Infect. Dis., 1984, 6, 251-264; Halstead, S.B., Science, 239:476-481, 1988; Monath, T.P., New Eng. J. Med., 1988, 319, 641-643).
Род пестивирусов включает в себя вирус бычьей вирусной диареи (BVDV), классический вирус лихорадки свиней (CSFV, также называемый вирусом холеры свиней) и вирусом пограничной болезни овец (BDV) (Moennig, V. et al Adv. Vir. Res. 1992, 41, 53-98). Пестивирусные инфекции домашнего скота (крупного рогатого скота, свиней и овец) приносят значительные экономические потери во всем мире. Вирус BVDV вызывает вирусную диарею крупного рогатого скота и в большой степени влияет на экономику животноводства (Meyers, G. and Thiel, H.- J., Advances in Virus Research, 1996, 47, 53-118; Moennig V., et al., Adv. Vir. Res. 1992, 41, 53-98). В отличие от пестивирусов животных пестивирусы человека не были так широко изучены. Вместе с тем, серологические обследования указывают на значительное распространение пестивирусов у людей.
Pestiviruses и hepaciviruses представляют собой близко родственные группы вирусов в семействе Flaviviridae. Другие близко родственные вирусы в этом семействе включают в себя GB-вирус A, GB А-вирусоподобные агенты, GB-вирус B и GB-вирус C (также называемый G-вирусом гепатита, HGV). Группа гепацивирусов (гепатит C вирус; HCV) состоит из множества близко родственных, но генотипически различимых вирусов, инфицирующих людей. Существует около 6 генотипов и более 50 подтипов HCV. По причине сходства пестивирусов и гепацивирусов, в сочетании со слабой способностью гепацивирусов к эффективному росту в клеточной культуре, вирус бычьей вирусной диареи (BVDV) часто используют в качестве заместителя для изучения вируса HCV.
Генетическая организация пестивирусов и гепацивирусов является очень сходной. Эти плюс-нитевые РНК-вирусы обладают единственной большой открытой рамкой считывания (ORF), кодирующий все вирусные белки, необходимые для репликации вируса. Эти белки экспрессируются как полипротеины, которые со-трансляционно и пост-трансляционно процессируются как клеточными, так и вирус-кодируемыми протеиназами, для получения зрелых вирусных белков. Вирусные белки, отвечающие за репликацию вирусного генома РНК, расположены вблизи карбокси-конца. Две трети ORF называют неструктурными белками (NS). Генетическая организация и процессирование полипротеина из участка неструктурного белка ORF у пестивирусов и гепацивирусов является очень сходной. И у пестивирусов и гепацивирусов зрелые неструктурные белки (NS) состоят из p7, NS2, NS3, NS4A, NS4B, NS5A, и NS5B, в порядке последовательности от амино-конца кодирующего участка неструктурного белка к карбокси-концу ORF.
NS белки пестивирусов и гепацивирусов имеют общие области последовательностей, которые характерны для некоторых функций белка. Например, белки NS3 обеих групп вирусов имеют мотивы аминокислотной последовательности сериновых протеиназ и геликаз (Gorbalenya et al (1988) Nature 333:22; Bazan and Fletterick (1989) Virology 171:637-639; Gorbalenya et al (1989) Nucleic Acid Res. 17.3889-3897). Аналогично, белки NS5B пестивирусов и гепацивирусов имеют мотивы, характерные для РНК-направленных РНК-полимераз (Koonin, E.V. and Dolja, V.V. (1993; Crit. Rev. Biochem. Molec. Biol. 28:375-430).
Фактически, в жизненном цикле вирусов роли и функции NS белков пестивирусов и гепацивирусов являются прямо аналогичными. В обоих случаях сериновая протеиназа NS3 отвечает за весь протеолитический процессинг предшественников полипротеина, расположенных ниже от ее положения в ORF (Wiskerchen and Collett (1991) Virology 184:341-350; Bartenschlager et al (1993) J. Virol. 67:3835-3844; Eckart et al (1993) Biochem. Biophys. Res. Comm. 192:399-406; Grakoui et al (1993) J. Virol. 67:2832-2843; Grakoui et al (1993) Proc. Natl. Acad. Sci. USA 90:10583-10587; Hijikata et al (1993) J. Virol. 67:4665-4675; Tome et al (1993) J. Virol. 67:4017-4026). В обоих случаях белок NS4A действует как кофактор с сериновой протеазой NS3 (Bartenschlager et al (1994) J. Virol. 68:5045-5055; Failla et al (1994) J. Virol. 68:3753-3760; Lin et al (1994) 68:8147-8157; Xu et al (1997) J. Virol. 71:5312-5322). Белок NS3 обоих вирусов также функционирует как геликаза (Kim et al (1995) Biochem. Biophys. Res. Comm. 215: 160-166; Jin and Peterson (1995; Arch. Biochem. Biophys., 323:47-53; Warrener and Collett (1995) J. Virol. 69:1720-1726). Наконец, белки NS5B пестивирусов и гепацивирусов обладают прогнозирумым действием РНК-направленной РНК-полимеразы (Behrens et al (1996) EMBO J. 15:12-22; Lchmann et al (1997) J. Virol. 71:8416-8428; Yuan et al (1997) Biochem. Biophys. Res. Comm. 232:231-235; Hagedorn, PCT WO 97/12033; патенты США US №№ 5981247; 6248589 и 6461845, Zhong et al (1998) J. Virol. 72.9365-9369).
Вирус гепатита C
Вирус гепатита C (HCV) является основной причиной хронического заболевания печени во всем мире. (Boyer, N. et al. J. Hepatol. 32:98-112, 2000). Вирус HCV вызывает медленно развивающуюся вирусную инфекцию и является главной причиной цирроза печени и гепатоцеллюлярной карциномы (Di Besceglie, A. M. and Bacon, B.R., Scientific American, Oct.: 80-85, (1999); Boyer, N. et al. J. Hepatol. 32:98-112, 2000). Около 170 миллионов человек во всем мире инфицированы вирусом HCV. (Boyer, N. et al J. Hepatol. 32:98-112, 2000). Цирроз печени, вызываемый хроническим инфекционным гепатитом C, ежегодно в США является причиной 8000-12000 смертельных исходов, и HCV-инфекция представляет собой ведущее показание для трансплантации печени.
Известно, что HCV вызывает по меньшей мере 80% случаев посттрансфузионного гепатита и значительный процент случаев спорадического острого гепатита. Ранее доказано, что HCV также вовлечен во многие случаи "идиопатического" хронического гепатита, "криптогенного" цирроза печени, и вероятно, гепатоцеллюлярной карциномы, которые не связаны с другими вирусами гепатита, такими как вирус гепатита B (HBV). Среди здоровых людей небольшой процент, отличающийся в зависимости от географических и других эпидемиологических факторов, указывает на хроническое носительство HCV. Количество может значительно превышать цифры для HBV, хотя информация является предварительной; остается неясным число таких людей, имеющих субклиническое хроническое заболевание печени. (The Merck Manual, ch. 69, p. 901, 16th ed., (1992)).
HCV представляет собой оболочечный вирус, содержащий позитивный смысловой одноцепочечный геном РНК размером около 9,4 т.п.н. Вирусный геном состоит из 5' нетранслируемой области (UTR), длинной открытой рамки считывания, кодирующей полипротеиновый предшественник размером около 3011 аминокислот, и короткой области 3' UTR. Область 5' UTR представляет собой наиболее высококонсервативную часть генома HCV и является важной для инициации и контроля трансляции полипротеина. Трансляция генома HCV инициируется кэп-независимым механизмом, известным как внутренний вход рибосом. Этот механизм охватывает связывание рибосом с последовательностью РНК, называемой участком внутреннего входа рибосом (IRES). Недавно было выявлено, что структура псевдоузла РНК является важным структурным элементом участка IRES у вируса HCV. Вирусные структурные белки включают в себя нуклеокапсидный белок сердцевины (C) и два оболочечных гликопротеина, E1 и E2. HCV также кодирует две протеиназы: цинк-зависимую металлопротеиназу, кодирующую NS2-NS3 область, и сериновую протеиназу, кодирующую область NS3. Указанные протеиназы необходимы для расщепления некоторых областей предшественника полипротеина в зрелые пептиды. Карбоксильная половина неструктурного белка 5, а именно NS5B, содержит РНК-зависимую РНК-полимеразу. Функция остальных неструктурных белков, NS4A и NS4B, и части NS5A (амино-концевая половина неструктурного белка 5) остается неизвестной.
Современные противовирусные исследования в большой степени направлены на разработку улучшенных способов лечения хронических HCV-инфекций у людей (Di Besceglie, A.M. and Bacon, B.R., Scientific American, Oct.: 80-85, (1999)).
В свете того, что инфекция HCV достигла эпидемического уровня во всем мире, и имеет трагические последствия для инфицированного больного, существует серьезная потребность в обеспечении новых эффективных фармацевтических средств для лечения гепатита C, при этом обладающих низкой токсичностью для организма-хозяина.
Дополнительно, учитывая возрастающую угрозу других флавивирусных инфекций, остается большая потребность в обеспечении новых эффективных фармацевтических средств, обладающих низкой токсичностью для организма-хозяина.
Гепатит B
Вирусный гепатит B достиг эпидемического уровня во всем мире. После инкубационного периода продолжительностью от двух до шести месяцев, в течение которых организм не знает об инфекции, инфицирование HBV может привести к развитию острого гепатита и поражению печени, которые вызывают боль в животе, желтуху и повышение уровня содержания в крови некоторых ферментов. Вирус HBV может вызывать скоротечный гепатит, быстро прогрессирующую, часто летальную форму болезни, при которой происходит массивное разрушение долей печени. Исходом острого вирусного гепатита обычно является выздоровление больных. Вместе с тем, у некоторых больных сохраняются высокие уровни в крови вирусного антигена в течение продолжительного, или неопределенного периода, что вызывает хроническую инфекцию. Хронические инфекции могут приводить к хроническому персистирующему гепатиту. Наибольшая частота больных, инфицированных хроническим персистирующим HBV, встречается в развивающихся странах. Хронический персистирующий гепатит может вызвать истощение, цирроз печени и гепатоцеллюлярную карциному, первичный рак печени. В западных промышленно развитых странах в группы высокого риска по инфекции HBV входят лица, имеющие контакт с носителями HBV или с образцами их крови. Эпидемиология HBV фактически очень сходна с эпидемиологией синдрома приобретенного иммунодефицита, который вносит свой вклад в причины распространенности HBV-инфекции у больных СПИДом или ВИЧ-ассоциированными инфекциями. При этом HBV является более контагиозным, чем ВИЧ.
Благоприятный прогноз дает ежедневное введение α-интерферона, белка, созданного генно-инженерной технологией. Также для иммунизации пациентов против HBV разработана вакцина, полученная из человеческой сыворотки. Вакцины были получены способом генной инженерии. Вакцина проявила эффективность, но вместе с тем ее производство имеет ряд проблем, поскольку получение человеческой сыворотки от хронических носителей ограничено, и процедура очистки является продолжительной и дорогой. Дополнительно, для гарантии безопасности каждую партию вакцины, изготовленную из разной сыворотки, необходимо проверять на шимпанзе. Кроме того, вакцина не эффективна у пациентов, уже инфицированных вирусом.
Важным этапом в схеме действия пуриновых и пиримидиновых нуклеозидов против вирусных болезней, и в частности, против HBV и HCV, является их метаболическая активация клеточными киназами, для получения моно-, ди- и три-производных фосфата. Биологически активным видом многих нуклеозидов является трифосфатная форма, которая ингибирует вирусную ДНК-полимеразу, РНК-полимеразу или обратную транскриптазу, или приводит к терминации цепи.
В свете того, что инфицирование вирусами гепатита B и C достигло эпидемического уровня во всем мире и имеет тяжелейшие последствия для инфицированного больного, существует серьезная необходимость обеспечения новых эффективных фармацевтических средств для лечения людей, зараженных вирусом, при этом обладающих низкой токсичностью для организма-хозяина.
В этой связи остается потребность в эффективных способах лечения инфекций HCV и HBV.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении рассмотрены фосфорамидатные и фосфонамидатные соединения ряда терапевтических средств, а также способы их изготовления и использования при лечении ряда патологий, включающих в себя заболевания печени. Указанные соединения можно использовать в некоторых вариантах осуществления для обеспечения концентрации терапевтического средства в печени. В одном варианте осуществления соединение представляет собой S-пивалоил-2-тиоэтилфосфорамидат, S-пивалоил-2-тиоэтилфосфонамидат, S-гидроксипивалоил-2-тиоэтилфосфорамидат или S-гидроксипивалоил-2-тиоэтилфосфонамидат.
В настоящем изобретении рассмотрены фосфорамидатные и фосфонамидатные соединения ряда терапевтических средств. Используемый в настоящем изобретении термин "фосфорамидатное или фосфонамидатное соединение терапевтического средства" включает в себя терапевтическое вещество, дериватизированное для включения в него фосфорамидатной или фосфонамидатной группы. Терапевтическое средство, например, представляет собой противовирусное средство, которое включает в себя, или было дериватизировано, чтобы включать в себя реакционно-способную группу, такую как гидроксильная группа, для присоединения фосфорамидатной или фосфонамидатной группы. Такие терапевтические средства включают в себя без ограничения нуклеозиды и аналоги нуклеозидов, включающих в себя нециклические нуклеозиды. В некоторых вариантах осуществления также рассматриваются фосфорамидаты нуклеотидов и аналогов нуклеотидов, такие как фосфорамидаты 1', 2', 3'-разветвленных и 4'-разветвленных нуклеозидов. Такие соединения можно вводить в эффективном количестве для лечения заболеваний печени, в число которых входят инфекционные болезни, такие как инфекции гепатита B и гепатита C, включающие в себя резистентные штаммы их вирусов.
В некоторых вариантах осуществления, без ограничения какой-либо теорией, возможно получение исходного лекарственного средства путем селективного метаболизма фосфорамидатного или фосфонамидатного соединения в печени, и таким образом, исходного лекарственного средства, способного к накоплению в печени организма-хозяина. Путем селективного направленного транспорта и активации соединений в печени можно уменьшать потенциальное нежелательное распространение активного соединения в желудочно-кишечном тракте. Кроме того, можно увеличивать терапевтическое количество активного соединения в участке печени, пораженном инфекцией.
В некоторых вариантах осуществления препарат 5'-монофосфата или фосфоната исходного нуклеозида (или нуклеозидного производного) создается путем метаболизма фосфорамидатного или фосфонамидатного соединения в печени, что дает возможность образования и накопления монофосфата или фосфоната в печени организма-хозяина. Таким образом, в некоторых вариантах осуществления, фосфорамидат эффективно обеспечивает устойчивый фосфат на нуклеозиде или нуклеозидном аналоге. В некоторых вариантах осуществления, когда для активации соединения необходимо его трифосфорилирование, преимущество дает устранение необходимости начального этапа фосфорилирования, и облегчение образования более зрелой формы активного трифосфата, который ингибирует фермент-мишень и может повышать общую активность нуклеозида или нуклеозидного аналога.
Без ограничения какой-либо теорией, в одном варианте осуществления рассмотрен фосфорамидат нуклеозида, такого как 2'-C-метил-рибонуклеозида, который после перорального введения селективно концентрируется в печени и метаболизируется клетками печени, с получением 5'-монофосфата, который можно энзиматически преобразовать в активную форму 5'-трифосфата, ингибирующего HCV-полимеразу. Таким образом, существует возможность уменьшить терапевтические дозы по сравнению с введением исходной нуклеозидной молекулы.
Таким образом, в некоторых вариантах осуществления, после перорального введения описываемых в настоящем изобретении фосфорамидатных и фосфонамидатных соединений, для получения терапевтического эффекта преимущество дает концентрация в клетках инфицированного участка печени указанных соединений и преобразование их в фосфат или фосфонат в клетках печени, и затем их необязательное дополнительное фосфорилирование.
Поскольку указанные способы позволяют накапливать в печени организма-хозяина фосфорамидатные или фосфонамидатные соединения, раскрытые в настоящем изобретении, указанные способы могут быть полезными, например, для лечения и/или профилактики болезней или патологии печени, таких как гепатит B или C.
В некоторых вариантах осуществления в настоящем изобретении рассматриваются соединения, полезные для профилактики и лечения флавивирусных инфекций и других связанных с ними состояний, таких как состояния с положительными анти-флавивирусными антителами и флавивирус-положительные состояния, хроническое воспаление печени, вызванное HCV, циррозом печени, фиброзом, острым гепатитом, скоротечным гепатитом, хроническим персистирующим гепатитом и истощением. Эти соединения или рецептуры можно также использовать с целью профилактики для предотвращения или замедления развития клинического проявления болезни у людей, являющихся позитивными на анти-флавивирусные антитела или флавивирус-антиген, или у людей, контактировавших с флавивирусом. В одном конкретном варианте осуществления флавивирус представляет собой вирус гепатита C. В некоторых вариантах осуществления указанное соединение используют для лечения инфекции любым вирусом, который реплицируется посредством РНК-зависимой РНК-полимеразы.
Также рассматривается способ лечения флавивирусной инфекции у организма-хозяина, включающего в себя человека, который включает в себя введение эффективного количества рассматриваемого в настоящем изобретении соединения, которое вводят единственным или в комбинации или чередовании с другим анти-флавивирусным средством, необязательно в фармацевтически приемлемом носителе.
В некоторых вариантах осуществления настоящего изобретения рассматривается способ лечения и/или профилактики инфекций гепатита B и других связанных с ним состояний, таких как состояния с положительными анти-HBV антителами и HBV-положительных состояний, хроническое воспаление печени, вызванное HBV, фиброзом, циррозом печени, острым гепатитом, скоротечным гепатитом, хроническим персистирующим гепатитом и истощением.
В некоторых вариантах осуществления можно изготавливать фосфорамидатные или фосфонамидатные соединения ряда фармацевтических средств и терапевтически применять их согласно настоящему изобретению для увеличения доставки лекарственного средства в печень. В одном варианте осуществления соединение представляет собой производное S-ацил-2-тиоэтилфосфорамидата или S-ацил-2-тиоэтилфосфонамидата, например, производное S-пивалоил-2-тиоэтилфосфорамидата или S-гидроксипивалоил-2-тиоэтилфосфонамидата.
Рассматриваемые в настоящем изобретении фосфорамидатные или фосфонамидатные соединения, а также их соли и композиции, содержащие указанные соединения, являются полезными для лечения заболеваний печени, таких как инфекции HBV и/или HCV. В одном варианте осуществления, соединение согласно настоящему изобретению представляет собой соединение формулы I:

или его фармацевтически приемлемую соль, сольват, стереоизомерную, таутомерную или полиморфную форму, в которой
Xa представляет собой

Z является O или S;
каждый из W независимо является O или S;
Ry и Ru каждый независимо представляет собой алкил, алкенил, алкинил, арил, алкиларил, циклоалкил, циклоалкенил, амино, аминоалкил, гидроксиалкил, алкокси, гетероциклил или гетероарил, все из которых необязательно замещаются;
Ra и Rb выбирают следующим образом:
i) Ra и Rb каждый независимо представляют собой водород, алкил, карбоксиалкил, гидроксиалкил, гидроксиарилалкил, ацилоксиалкил, аминокарбонилалкил, алкоксикарбонилалкил, арил, арилалкил, циклоалкил, арил, гетероарил или гетероциклил, все из которых необязательно замещаются; или
ii) Ra и Rb вместе с атомом азота, на который они замещены, образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов;
n равен от 0 до 3;
n2 равен 1-4; и
R1 является функциональной группой, получаемой удалением водорода из гидроксигруппы противовирусного препарата.
В другом варианте осуществления
Xa представляет собой

Z является O, S, NH или NRW, при этом Rw, например, представляет собой алкил, алкил, алкенил, алкинил, арил, алкиларил, циклоалкил, циклоалкенил, амино, аминоалкил, алкокси, гетероциклил или гетероарил, все из которых необязательно замещаются;
каждый из W является O, S, NH или NRW, где Rw, например, представляет собой алкил, алкил, алкенил, алкинил, арил, алкиларил, циклоалкил, циклоалкенил, амино, аминоалкил, алкокси, гетероциклил или гетероарил, все из которых необязательно замещаются;
Ry и Ru каждый независимо представляет собой алкил, алкенил, алкинил, арил, алкиларил, циклоалкил, циклоалкенил, амино, аминоалкил, алкокси, гетероциклил или гетероарил, все из которых необязательно замещаются;
Ra и Rb выбирают следующим образом:
i) Ra и Rb каждый независимо представляют собой водород, алкил, карбоксиалкил, гидроксиалкил, гидроксиарилалкил, ацилоксиалкил, аминокарбонилалкил, алкоксикарбонилалкил, арил, алкиларил, циклоалкил, гетероарил или гетероциклил, все из которых необязательно замещаются; или
ii) Ra и Rb вместе с атомом азота, на который они замещены, образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов;
n равен от 0 до 3;
n2 равен 1-4; и
R1 такой, как указано выше.
Специалистам в данной области техники известно, что соединения формулы I можно конструировать или изготавливать путем реакции, например, по гидроксигруппе указанного противовирусного препарата, например, посредством конденсации или дегидратирования. Для удобства, в описании настоящего изобретения, когда заместители, такие как приведенные в качестве примера R1 указаны как лекарственный препарат, специалистам в данной области техники будет очевидно, что соединение, например, формулы I, содержит производное, например, радикал противовирусного препарата. Такие производные можно изготовить, например, путем удаления водородного радикала из гидроксигруппы препарата, например, в реакции дегидратации. Если целесообразно, некоторые производные можно изготавливать модификацией фосфата или фосфоната противовирусного препарата, для получения соединения формулы I.
В некоторых вариантах осуществления формулы I R1 представляет собой нуклеозид, содержащий циклический или нециклический сахар или его аналог.
В некоторых вариантах осуществления R1 представляет собой противовирусный нуклеозидный аналог, пригодный для лечения вирусной HCV-инфекции, выбираемый из рибавирина, вирамидина, 2'-C-метилцитидина, 2'-C-метилгуанозина, валопицитабина (NM-283), МК-0608 и PSI-6130.
В некоторых вариантах осуществления R1 представляет собой противовирусный нуклеозидный аналог, пригодный для лечения вирусной HBV-инфекции, который выбирают из ламивудина (EPIVIR-HBV, Зеффикс или Гептодин), адефовира, энтекавира (Бараклуд), телбивудина (Тизека, Себиво), эмтрицитабина (FTC), клевудина (L-FMAU), виреда (Тенофовир), торцитабина, валторцитабина (моновал LdC), амдоксовира (DAPD) и RCV (Рацивир).
В некоторых вариантах осуществления R1 представляет собой ненуклеозидное противовирусное соединение, пригодное для лечения вирусной HBV-инфекции, выбираемое из резиквимода или целгосивира.
В некоторых вариантах осуществления согласно формуле I Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb каждый независимо представляет собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ry является -C(CH3)2CH2OH.
В некоторых вариантах осуществления соединения, рассматриваемые в настоящем изобретении, выбирают таким образом, что R1 не является 3'-азидо-2',3'-дидезокситимидином.
В другом варианте осуществления соединение, рассматриваемое в настоящем изобретении, является соединением формулы IIa или IIb:

или его фармацевтически приемлемой солью, сольватом, стереоизомерной, таутомерной или полиморфной формой, в котором:
Ry представляет собой алкил, алкенил, алкинил, арил, алкиларил, циклоалкил, циклоалкенил, амино, аминоалкил, гидроксиалкил, гетероциклил или гетероарил, все из которых необязательно замещаются;
Ra и Rb выбирают следующим образом:
i) Ra и Rb каждый независимо представляют собой водород, алкил, карбоксиалкил, гидроксиалкил, гидроксиарилалкил, ацилоксиалкил, аминокарбонилалкил, алкоксикарбонилалкил, арил, арилалкил, циклоалкил, арил, гетероарил или гетероциклил, все из которых необязательно замещаются; или
ii) Ra и Rb вместе с атомом азота, на который они замещены, образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов; и
R1 является противовирусным препаратом (согласно использованию в настоящем изобретении, где R1 представляет собой противовирусный препарат, в этом варианте осуществления в него включена функциональная группа, получаемая удалением водорода из гидроксигруппы противовирусного препарата), такого как нуклеозид или нуклеозидный аналог.
В некоторых вариантах осуществления согласно формуле IIa или IIb, Ry является замещенным алкилом, например, гидроксиалкилом или аминоалкилом; и Ra и Rb каждый независимо представляет собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ry является -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:

в которых Ra, Rb и Ry таковы, как описаны в формуле I, и
в которых R2 и R3 каждый независимо представляет собой H, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещается; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток, углевод; пептид; холестерин; или другая фармацевтически приемлемая уходящая группа, которая способна дать соединение, в которых R2 и/или R3 независимо являются H, например, при введении in vivo; или R2 и R3 соединяют для образования циклической группы посредством алкильной, эфирной или карбаматной связи; и в которых каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, Со-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещается; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; аминокислотный остаток, или углевод. В некоторых вариантах осуществления согласно этому параграфу каждый из R2 и R3 является H; Ry является замещенным алкилом, например, гидроксиалкилом или аминоалкилом; и Ra и Rb каждый независимо представляет собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый из R2 и R3 является H; Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый из R2 и R3 является H; Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления каждый из R2 и R3 является H; Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый из R2 и R3 является H; Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления согласно этому параграфу каждый из R2 и R3 является водородом, Ra является водородом, Rb является -СН2-С6Н5, и Ry является -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:

в которых описание Ra, Rb и Ry приведено в формуле I. Rd выбирают из группы, состоящей из водорода, алкила и алкокси. В некоторых вариантах осуществления Rd является водородом, метилом или метокси. В некоторых вариантах осуществления согласно этому параграфу Ry является замещенным алкилом, например, гидроксиалкилом или аминоалкилом; и Ra и Rb каждый независимо представляет собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо являются бензилом или замещенным алкилом. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления согласно этому параграфу каждый из R2 и R3 является водородом, Ra является водородом, Rb является -СН2-С6Н5, и Ry является -C(CH3)2CH2OH. В некоторых вариантах осуществления согласно этому параграфу Ra является водородом, Rb является -СН2-С6Н5, и Ry является -C(CH3)2CH2OH.
В одном варианте осуществления нуклеозиды, которые можно дериватизировать с целью включения фосфорамидата или фосфонамидата, например, в 5'-положении, включают в себя:

Примеры нуклеозидных соединений фосфорамидата или фосфонамидата включают в себя:



В одном варианте осуществления нуклеозиды, которые можно дериватизировать с целью включения фосфорамидата или фосфонамидата, например, в 5'-положении, включают в себя:

В одном варианте осуществления нуклеозидные фосфорамидатные или фосфонамидатные соединения включают в себя:



В одном варианте осуществления нуклеозиды, которые можно дериватизировать с целью включения фосфорамидата или фосфонамидата, например, в 5'-положении, включают в себя:

В одном варианте осуществления нуклеозидные фосфорамидатные или фосфонамидатные соединения включают в себя:



В одном аспекте соединения, описанные в настоящем изобретении, предоставляются или вводятся в комбинации со вторым терапевтическим средством, например, со средством, пригодным для лечения или профилактики HBV- и/или HCV-инфекций. Примеры терапевтических средств подробно описаны в нижеприведенных разделах.
В другом аспекте предоставлены фармацевтические композиции, монолитные лекарственные формы и наборы, подходящие для лечения или профилактики заболеваний, таких как HBV- и/или HCV-инфекций, которые содержат терапевтически или профилактически эффективное количество соединения, описанного в настоящем изобретении, например, соединения с формулами I, IIa или IIb, и терапевтически или профилактически эффективное количество второго препарата, например, средства, полезного для лечения или профилактики инфекций HBV и/или HCV.
В некоторых вариантах осуществления рассматривается способ лечения заболевания печени, содержащий введение человеку, нуждающемуся в таком лечении, эффективного количества фосфорамидатного или фосфонамидатного производного нуклеозида или нуклеозидного аналога, в котором необязательно производное представляет собой S-пивалоил-2-тиоэтилфосфорамидатное или S-пивалоил-2-тиоэтилфосфонамидатное производное. Производное необязательно выбирают из соединений, раскрытых в настоящем изобретении.
В некоторых вариантах осуществления к настоящему изобретению относятся:
(a) соединения согласно настоящему изобретению, например, соединения с формулами I, IIa или IIb, и их фармацевтически приемлемые соли и композиции;
(b) соединения согласно настоящему изобретению, например, соединения с формулами I, IIa или IIb, и их фармацевтически приемлемые соли и композиции для использования в лечении и/или профилактике заболевания печени, включающего в себя флавивирусную инфекцию, в особенности у людей с диагнозом флавивирусной инфекции или имеющих риск инфицирования гепатитом C;
(c) способы изготовления соединений, описанных в настоящем изобретении, например, соединений с формулами I, IIa или IIb, как более подробно рассмотрено ниже;
(d) фармацевтические рецептуры, содержащие соединение согласно настоящему изобретению, например, соединение с формулой I, IIa или IIb, и его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым носителем или разбавителем;
(e) фармацевтические рецептуры, содержащие соединение согласно настоящему изобретению, например, соединение с формулой I, IIa или IIb, или его фармацевтически приемлемую соль вместе с одним или более другими эффективными средствами против вируса HCV, необязательно в фармацевтически приемлемом носителе или разбавителе;
(f) способ лечения и/или профилактики организма-хозяина, инфицированного флавивирусом, который включает в себя введение эффективного количества соединения согласно настоящему изобретению, например, соединения с формулой I, IIa или IIb, его фармацевтически приемлемой соли или композиции;
(g) способ лечения и/или профилактики организма-хозяина, инфицированного флавивирусом, который включает в себя введение эффективного количества соединения согласно настоящему изобретению, например, соединения формулы I, IIa или IIb, его фармацевтически приемлемой соли или композиции в комбинации и/или чередовании с одним или более эффективным средством против вируса HCV;
(h) соединения согласно настоящему изобретению, например, соединения формулы I, IIa или IIb, и их фармацевтически приемлемые соли или композиции для использования в лечении и/или профилактике HBV-инфекции, в особенности у людей с диагнозом HBV-инфекции или имеющих риск инфицирования гепатитом В;
(i) фармацевтические рецептуры, содержащие соединение согласно настоящему изобретению, например, соединение формулы I, IIa или IIb, или его фармацевтически приемлемую соль вместе с одним или более другими эффективными средствами против вируса HBV, необязательно в фармацевтически приемлемом носителе или разбавителе;
(j) способ лечения и/или профилактики инфекционного гепатита B и других связанных с ним состояний, таких как состояний с положительными анти-HBV антителами и HBV-положительных состояний, хроническое воспаление печени, вызванное HBV, фиброзом, циррозом печени, острым гепатитом, скоротечным гепатитом, хроническим персистирующим гепатитом и истощением, который включает в себя введение эффективного количества соединения согласно настоящему изобретению, например, соединения формулы I, IIa или IIb, или его фармацевтически приемлемой соли или композиции;
(k) способ профилактики для предотвращения или замедления развития клинических симптомов заболевания у людей, положительных на анти-HBV антитела или HBV-антиген, или у людей, контактировавших с HBV.
Флавивирусы, поддающиеся лечению, например, в общем описаны в литературе: Fields Virology, Editors: Fields, B.N., Knipe, D.M. and Howley, P. M., Lippincott-Raven Publishers, Philadelphia, PA, Chapter 31, 1996. В конкретном варианте осуществления изобретения Flaviviridae представлены HCV. В дополнительном варианте осуществления Flaviviridae является флавивирусом или пестивирусом. Конкретные флавивирусы включают в себя без ограничения: Absettarov, Alfuy, Apoi, Aroa, Bagaza, Banzi, Bouboui, Bussuquara, Cacipacore, Carey Island, Dakar bat, денге 1, денге 2, денге 3, денге 4, Edge Hill, Entebbe bat, Gadgets Gully, Hanzalova, Hypr, Ilheus, израильский вирус менингоэнцефалита индеек, японского энцефалита, Jugra, Jutiapa, Kadam, Karshi, Kedougou, Kokobera, Koutango, Kumlinge, Kunjin, вирус Кьясанурской лесной болезни, Langat, болезни Louping, Meaban, Modoc, вирус лейкоэнцефалита Монтана миотис, энцефалита долины Мюррей, Naranjal, Negishi, Ntaya, вирус омской геморрагической лихорадки, Phnom-Penh bat, Powassan, Rio Bravo, Rocio, Royal Farm, вирус российского весеннее-летнего энцефалита, Saboya, энцефалита St.Louis, Sal Vieja, San Perlita, Saumarez Reef, Sepik, Sokuluk, Spondweni, Stratford, Tembusu, Tyuleniy, Uganda S, Usutu, Wesselsbron, вирус Западного Нила, Яунде, желтой лихорадки и Zika.
Пестивирусы, поддающиеся лечению, например, описаны в общем в литературе: Fields Virology, Editors: Fields, B.N., Knipe, D.M., and Howley, P.M., Lippincott-Raven Publishers, Philadelphia, PA, Chapter 33, 1996. Конкретные пестивирусы включают в себя без ограничения: вирус бычьей вирусной диареи ("BVDV"), классический вирус лихорадки свиней ("CSFV", также называемый вирусом холеры свиней) и вирус пограничной болезни ("BDV").
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг.1 изображает элиминацию NM108 гидроксиSATE фосфорамидата (B299) после инкубации с НАДФН (восстановленным никотинамидадениндинуклеотидфосфатом) и без НАДФН в печени обезьяны S9.
Фиг.2 изображает элиминацию NM107 гидроксиSATE фосфорамидата (B102) после инкубации с НАДФН и без НАДФН в печени обезьяны S9.
ОПИСАНИЕ ПРИМЕРОВ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Настоящее изобретение относится к соединениям, композициям и способам, пригодным для лечения заболеваний печени, таких как инфекция HBV и/или HCV у субъекта. Дополнительно обеспечиваются лекарственные формы, полезные для указанных способов.
Определения
Нижеприведенные термины, относящиеся к соединениям настоящего изобретения, имеют следующие значения, если не указано иначе.
Используемый в настоящем изобретении термин "алкил", если иначе не указано, включает в себя насыщенный прямой, разветвленный или циклический, первичный, вторичный или третичный углеводород, обычно от C1 до C10, и в частности, включает в себя метил, CF3, CCl3, CFCl2, CF2Cl, этил, CH2CF3, CF2CF3, пропил, изопропил, циклопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, циклогексилметил, 3-метилпентил, 2,2-диметилбутил и 2,3-диметилбутил. Термин включает в себя как замещенные, так и незамещенные алкильные группы, и в частности, включает в себя галогенизированные алкильные группы, и более конкретно, фторированные алкильные группы. Неограничивающие примеры функциональных групп, которыми можно замещать алкильную группу, выбирают из группы, состоящей из галогена (фтор, хлор, бром или иод), гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, как незащищенных, так и защищенных при необходимости, как известно специалистам в данной области техники, например, согласно теории Greene, et al., Protective Groups in Organic Synthesis, John Wiley and Sons, Second Edition, 1991, включенной в настоящее изобретение в качестве ссылки.
Используемый в настоящем изобретении термин "низший алкил", если не указано иначе, включает в себя C1-C4 насыщенную, прямую, разветвленную, или если подходит, циклическую (например, циклопропильную) алкильную группу, включающую в себя и замещенные и незамещенные функциональные группы.
Термин "алкилен" включает в себя двухвалентные насыщенные алифатические углеводородные группы, имеющие, в частности, до 11 атомов углерода и, более конкретно, от 1 до 6 атомов углерода, которые могут иметь прямую или разветвленную цепь. В качестве примеров указанного приведены такие группы, как метилен (-СН2-), этилен (-СН2СН2-), изомеры пропилена (например, -CH2CH2CH2- и -CH(CH3)CH2-) и тому подобные.
Термин "алкенил" включает в себя одновалентные олефиново ненасыщенные углеводородные группы, в некоторых вариантах осуществления имеющие до 11 атомов углерода, от 2 до 8 атомов углерода или от 2 до 6 атомов углерода, которые могут иметь прямую или разветвленную цепь и имеют по меньшей мере 1 или от 1 до 2 участков олефиновой ненасыщенности. Примеры алкенильных групп включают в себя этенил (-СН2=СН2), н-пропенил (-СН2СН=СН2), изопропенил (-C(CH3)=CH2), винил и замещенный винил, и тому подобные.
Термин "алкенилен" включает в себя двухвалентные олефиново ненасыщенные углеводородные группы, в некоторых вариантах осуществления имеющие до 11 атомов углерода или от 2 до 6 атомов углерода, которые могут иметь прямую или разветвленную цепь и имеют по меньшей мере 1 или от 1 до 2 участков олефиновой ненасыщенности. Указанный термин включает в себя в качестве примеров таких групп этенилен (-СН=СН-), изомеры пропенилена (например, -CH=CHCH2- и -C(CH3)=CH- и -CH=C(CH3)-) и тому подобное.
Термин "алкинил" включает в себя ацетиленово ненасыщенные углеводородные группы, в некоторых вариантах осуществления имеющие до 11 атомов углерода или от 2 до 6 атомов углерода, которые могут иметь прямую или разветвленную цепь и имеют по меньшей мере 1 или от 1 до 2 участков алкиниловой ненасыщенности. Неограничивающие примеры алкинильных групп включают в себя ацетиленовые, этинил (-С≡СН), пропаргил (-СН2С≡СН), и тому подобное.
Используемый в настоящем изобретении термин "арил", если не указано иначе, включает в себя фенил, бифенил или нафтил, и предпочтительно, фенил. Термин включает в себя и замещенные и незамещенные функциональные группы. Арильную группу можно замещать любой описанной функциональных группой, включающей в себя без ограничения одну или более функциональных групп, выбираемых из группы, состоящей из галогена (фтор, хлор, бром или иод), алкила, галоалкила, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, как незащищенной, так и защищенной при необходимости, как известно специалистам в данной области техники, например, согласно руководству Greene, et al., Protective Groups in Organic Synthesis, John Wiley and Sons, Second Edition, 1991.
Термин "алкокси" включает в себя группу -OR, в которой R является алкилом. Конкретнные алкокси группы включают в себя, в качестве примера, метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси, втор-бутокси, н-пентокси, н-гексокси, 1,2-диметилбутокси и тому подобное.
Термин "алкоксикарбонил" включает в себя радикал -C(O)-алкокси, где определение алкокси приведено в настоящем изобретении.
Термин "амино" включает в себя радикал -NH2.
Термин "карбоксил" включает в себя радикал -C(O)ОН.
Термин "алкиламино" или "ариламино" включает в себя аминогруппу, которая имеет один или два, соответственно, алкильных или арильных заместителя. Если в настоящей заявке конкретно не указано иначе, когда подходящая функциональная группа представляет собой алкил, предпочтительным является низший алкил. Аналогично, если подходящая функциональная группа представляет собой алкил или низший алкил, предпочтительным является незамещенный алкил или низший алкил.
Термины "галоген" или "гало" включают в себя хлор, бром, фтор или иод.
"Моноалкиламино" включает в себя группу алкил-NR`, в которой R' выбирают из водорода и алкила.
"Тиоалкокси" включает в себя группу -SR, где R является алкилом.
Используемый в настоящем изобретении термин "защищенный", если не указано иначе, относится к группе, которую добавляют к атомам кислорода, азота или фосфора, чтобы предотвратить их дальнейшую реакцию, или в других целях. Широкое разнообразие кислородных и азотных защитных групп известно специалистам в области техники органического синтеза.
Термин "фармацевтически приемлемая соль" включает в себя любую соль соединения настоящего изобретения, которая сохраняет его биологические свойства и которая не является токсичной или иным образом нежелательной для фармацевтического использования. Такие соли можно получать из ряда органических и неорганических противоионов, общеизвестных в данной области техники. Такие соли включают в себя: (1) кислотно-аддитивные соли, образованные органическими или неорганическими кислотами, такими как хлористоводородная, бромистоводородная, серная, азотная, фосфорная, сульфамовая, уксусная, трифторуксусная, трихлоруксусная, пропионовая, капроновая, циклопентилпропионовая, гликолевая, глутаровая, пировиноградная, молочная, малоновая, янтарная, сорбиновая, аскорбиновая, яблочная, малеиновая, фумаровая, виннокаменная, лимонная, бензойная, 3-(4-гидроксибензоил)бензойная, пикриновая, коричная, миндальная, фталевая, лауриновая, метансульфоновая, этансульфоновая, 1,2-этан-дисульфоновая, 2-гидроксиэтансульфоновая, бензолсульфоновая, 4-хлорбензолсульфоновая, 2-нафталинсульфоновая, 4-толуолсульфоновая, камфарная, камфаросульфоновая, 4-метилбицикло[2,2,2]-окт-2-ен-1-карбоновая, глюкогептоновая, 3-фенилпропионовая, триметилуксусная, трет-бутилуксусная, лаурилсерная, глюконовая, бензойная, глутаминовая, гидроксинафтойная, салициловая, стеариновая, циклогексилсульфамовая, хинная, муконовая кислота и тому подобные кислоты; или (2) соли, образованные в присутствии кислого протона в исходном соединении, при этом он или (a) замещен ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия, или гидроокиси щелочного металла или щелочноземельного металла, такого как натрий, калий, кальций, магний, алюминий, литий, цинк, и гидроокись бария, аммиак или (b) координирует с органическим основанием, таким как алифатические, ациклические или ароматические органические амины, например, аммиак, метиламин, диметиламин, диэтиламин, пиколин, этаноламин, диэтаноламин, триэтаноламин, этилендиамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, N-бензилфенэтиламин, N-метилглюкаминпиперазин, трис(гидроксиметил)аминометан, гидроокись тетраметиламмония и тому подобное.
Соли дополнительно включают в себя, исключительно в качестве примера, соли натрия, калия, кальция, магния, аммония, тетраалкиламмония и т.п., и если соединение содержит основные функциональные группы, соли нетоксичных органических или неорганических кислот, такие как гидрогалиды, например, гидрохлорид и гидробромид, сульфат, фосфат, сульфамат, нитрат, ацетат, трифторацетат, трихлорацетат, пропионат, гексаноат, циклопентилпропионат, гликолят, глутарат, пируват, лактат, малонат, сукцинат, сорбат, аскорбат, малат, малеат, фумарат, тартрат, соль лимонной кислоты, бензоат, 3-(4-гидроксибензоил)бензоат, пикрат, циннамат, манделат, фталат, лаурат, метансульфонат (месилат), этансульфонат, 1,2-этандисульфонат, 2-гидроксиэтансульфонат, бензолсульфонат (бесилат), 4-хлорбензолсульфонат, 2-нафталинсульфонат, 4-толуолсульфонат, камфорат, камфорсульфонат, 4-метилбицикло[2,2,2]-окт-2-ен-1-карбоксилат, глюкогептонат, 3-фенилпропионат, триметилацетат, трет-бутилацетат, лаурилсульфат, глюконат, бензоат, глутамат, гидроксинафтоат, салицилат, стеарат, циклогексилсульфамат, соль хинной кислоты, муконат и тому подобное.
Термин "алкарил" или "алкиларил" включает в себя арилзамещенную группу с алкильным заместителем. Термины «аралкил» или «арилалкил» охватывают алкильную группу с арильным заместителем.
Термин "пуриновое" или "пиримидиновое" основание включает в себя без ограничения аденин, N6-алкилпурины, N6-ацилпурины (в которых ацил представляет собой C(O)(алкил, арил, алкиларил или арилалкил), N6-бензилпурин, N6-галопурин, N6-винилпурин, N6-ацетиленовый пурин, N6-ацилпурин, N6-гидроксиалкилпурин, N6-алкиламинопурин, N6-тиоалкилпурин, N2-алкилпурины, N2-алкил-6-тиопурины, тимин, цитозин, 5-фторцитозин, 5-метилцитозин, 6-азапиримидин, включающий в себя 6-азацитозин, 2- и/или 4-меркаптопиримидин, урацил, 5-галоурацил, включающий в себя 5-фтороурацил, C5-алкилпиримидины, C5-бензилпиримидины, C5-галопиримидины, C5-винилпиримидин, C5-ацетиленовый пиримидин, C5-ацилпиримидин, C5-гидроксиалкилпурин, C5-амидопиримидин, C5-цианопиримидин, C5-иодопиримидин, C6-иодопиримидин, C5-Br-винилпиримидин, C6-Br-винилпиримидин, C5-нитропиримидин, C5-аминопиримидин, N2-алкилпурины, N2-алкил-6-тиопурины, 5-азацитидинил, 5-азаурацилил, триазолопиридинил, имидазолопиридинил, пирролопиримидинил и пиразолопиримидинил. Пуриновые основания включают в себя без ограничения, гуанин, аденин, гипоксантин, 7-деазагуанин, 7-деазааденин, 2,6-диаминопурин и 6-хлорпурин. Если это необходимо или желательно, кислородные и азотные функциональные группы в основании могут быть защищены. Подходящие защитные группы известны специалистам в данной области техники и включают в себя триметилсилил, диметилгексилсилил, трет-бутилдиметилсилил и трет-бутилдифенилсилил, тритил, алкильные группы и ацильные группы, такие как ацетил и пропионил, метансульфонил и п-толуолсульфонил.
Термин "ацил" или "O-связанный сложный эфир" включает в себя группу, имеющую формулу C(O)R', в которой R' представляет собой прямой, разветвленный или циклический алкил (включающий в себя низший алкил), карбоксильный остаток аминокислоты, арил, включающий в себя фенил, алкарил, арилалкил, включающий в себя бензил, алкоксиалкил, включающий в себя метоксиметил, арилоксиалкил, такой как феноксиметил; или замещенный алкил (включающий в себя низший алкил), арил, включающий в себя фенил, необязательно замещенный хлор, бром, фтор, иод, C1-C4алкил или C1-C4алкокси, сложный эфир сульфонат, такой как алкил или арилалкил сульфонил, включающий в себя метансульфонил, сложный эфир моно-, ди- или трифосфата, тритил или монометокси-тритил, замещенный бензил, алкарил, арилалкил, включающий в себя бензил, алкоксиалкил, включающий в себя метоксиметил, арилоксиалкил, такой как феноксиметил. Арильные группы в сложных эфирах оптимально содержат фенильную группу. В частности, ацильные группы включают в себя ацетил, трифторацетил, метилацетил, циклопропилацетил, пропионил, бутирил, гексаноил, гептаноил, октаноил, нео-гептаноил, фенилацетил, 2-ацетокси-2-фенилацетил, дифенилацетил, α-метокси-α-трифторметилфенилацетил, бромацетил, 2-нитробензолацетил, 4-хлорбензолацетил, 2-хлор-2,2-дифенилацетил, 2-хлор-2-фенилацетил, триметилацетил, хлордифторацетил, перфторацетил, фторацетил, бромдифторацетил, метоксиацетил, 2-тиофенацетил, хлорсульфонилацетил, 3-метоксифенилацетил, феноксиацетил, трет-бутилацетил, трихлорацетил, монохлорацетил, дихлорацетил, 7H-додекафторгептаноил, перфторгептаноил, 7H-додекафторгептаноил, 7-хлордодекафторгептаноил, 7-хлордодекафторгептаноил, 7H-додекафторгептаноил, 7H-додекафторгептаноил, нонафтор-3,6-диоксагептаноил, нонафтор-3,6-диоксагептаноил, перфторгептаноил, метоксибензоил, метил-3-амино-5-фенилтиофен-2-карбоксил, 3,6-дихлор-2-метоксибензоил, 4-(1,1,2,2-тетрафторэтокси)бензоил, 2-бромпропионил, омега-аминокаприл, деканоил, н-пентадеканоил, стеарил, 3-циклопентилпропионил, 1-бензолкарбоксил, O-ацетилманделил, ацетилпивалоил, 1-адамантанкарбоксил, циклогексанкарбоксил, 2,6-пиридиндикарбоксил, циклопропанкарбоксил, циклобутанкарбоксил, перфторциклогексилкарбоксил, 4-метилбензоил, хлорметилизоксазолилкарбонил, перфторциклогексилкарбоксил, кротонил, 1-метил-1H-индазол-3-карбонил, 2-пропенил, изовалерил, 1-пирролидинкарбонил, 4-фенилбензоил.
Термин "аминокислота" включает в себя α, β, γ или δ аминокислоты природного и синтетического происхождения и включает в себя без ограничения, аминокислоты, обнаруживаемые в белках, то есть в глицине, аланине, валине, лейцине, изолейцине, метионине, фенилаланине, триптофане, пролине, серине, треонине, цистеине, тирозине, аспарагине, глутамине, аспартате, глутамате, лизине, аргинине и гистидине. В предпочтительном варианте осуществления аминокислота находится в L-конфигурации. Альтернативно, аминокислота может быть производной аланила, валинила, лейцинила, изолейцинила, пролинила, фенилаланинила, триптофанила, метионинила, глицинила, серинила, треонинила, цистеинила, тирозинила, аспарагинила, глутаминила, аспартоила, глутароила, лизинила, аргининила, гистидинила, β-аланила, β-валинила, β-лейцинила, β-изолейцинила, β-пролинила, β-фенилаланинила, β-триптофанила, β-метионинила, β-глицинила, β-серинила, β-треонинила, β-цистеинила, β-тирозинила, β-аспарагинила, β-глутаминила, β-аспартоила, β-глутароила, β-лизинила, β-аргининила или β-гистидинила.
Используемое в настоящем изобретении понятие «по существу не содержит» или «по существу отсутствует» относительно нуклеозидной композиции, включает в себя нуклеозидную композицию, которая включает в себя определенный энантиомер указанного нуклеозида в количестве по меньшей мере 85 или 90% веса, предпочтительно 95%, 98%, 99% или 100% веса. В предпочтительном варианте осуществления, относящиеся к способам и соединениям настоящего изобретения, указанные соединения по существу не содержат энантиомеров.
Аналогично, термин "выделенная", относящийся к нуклеозидной композиции, включает в себя нуклеозидную композицию, которая включает в себя по меньшей мере от 85, 90%, 95%, 98%, 99% до 100% веса нуклеозида, и в оставшемся процентном количестве другие химические виды или энантиомеры.
Термин "сольват" включает в себя соединение согласно настоящему изобретению или его соль, и дополнительно включает в себя стехиометрическое или нестехиометрическое количество растворителя, связанного нековалентными межмолекулярными силами. Если растворителем является вода, то сольват представляет собой гидрат.
Термин "организм-хозяин", используемый в настоящем изобретении, включает в себя любой одноклеточный или многоклеточный организм, в котором может реплицироваться вирус, и включает в себя клеточные линии и животных, и предпочтительно, человека. Альтернативно, организм-хозяин может нести часть вирусного генома флавивируса, на репликацию или функции которого могут влиять соединения настоящего изобретения. Термин «организм-хозяин», в частности, включает в себя инфицированные клетки, клетки, трансфицированные полным геномом или частью генома флавивируса и животных, в частности, приматов (включающих шимпанзе) и людей. В большинстве случаев применения настоящего изобретения у животных организмом-хозяином является больной человек. Вместе с тем, по некоторым признакам, применение в ветеринарии (например, у шимпанзе) очевидно предусмотрено согласно настоящему изобретению.
Используемые в настоящем изобретении термины "субъект" и "больной/пациент" использованы попеременно. Термины "субъект" и "субъекты" относятся к животным, таким как млекопитающие, включающие в себя не-приматов (например, корова, свинья, лошадь, кошка, собака, крыса и мышь) и приматов (например, обезьяна, такая как обезьяна cynomolgous, шимпанзе и человек) и, например, человек. В одном варианте осуществления субъект является резистентным или невосприимчивым к текущему лечению гепатита C. В другом варианте осуществления субъектом является сельскохозяйственное животное (например, лошадь, корова, свинья и т.д.) или домашнее животное (например, собака или кошка). В одном варианте осуществления субъектом является человек.
Используемые в настоящем изобретении понятия "терапевтическое средство" и "терапевтические средства" относятся к любому средству (средствам), которые можно применять для лечения или профилактики заболевания или одного или более его симптомов. В некоторых вариантах осуществления термин "терапевтическое средство" включает в себя соединение согласно настоящему изобретении. В одном варианте осуществления терапевтическое средство представляет собой средство, которое известно в качестве полезного, или применялось, или применяется в настоящее время для лечения или профилактики заболевания или одного или более его симптомов.
Понятие "терапевтически эффективное количество" включает в себя количество соединения или композиции, которое при введении субъекту для лечения болезни является достаточным для осуществления такого лечения болезни. "Терапевтически эффективное количество" может варьировать в зависимости, среди прочего, от вида соединения, заболевания и степени его тяжести, и от возраста, веса и т.д. субъекта, который подвергается лечению.
Понятия "лечение" или "процесс лечения" любого заболевания или нарушения относится, в одном варианте осуществления, к улучшению состояния субъекта при заболевании или нарушении у субъекта. В другом варианте осуществления "лечение" или "процесс лечения" включает в себя улучшение по меньшей мере одного физического параметра, который субъект самостоятельно может не распознавать. Еще в одном варианте осуществления "лечение" или "процесс лечения" включает в себя изменение заболевания или нарушения, как физическое (например, стабилизация видимого симптома), так и физиологическое (например, стабилизация физического параметра), или оба указанных изменения. Еще в одном варианте осуществления "лечение" или "процесс лечения" включает в себя отсрочку начала заболевания или нарушения.
Используемые в настоящем изобретении понятия "профилактическое средство" и "профилактические средства" относятся к любому средству (средствам), которые можно применять для профилактики заболевания или одного или более его симптомов. В некоторых вариантах осуществления понятие "профилактическое средство" включает в себя соединение согласно настоящему изобретению. В некоторых других вариантах осуществления понятие "профилактическое средство" не относится к соединению, рассматриваемому в настоящем изобретении. Например, профилактическое средство представляет собой средство, которое известно в качестве полезного, или применялось или применяется в настоящее время для профилактики или сдерживания начала, развития, прогрессирования и/или тяжести заболевания.
Используемое в настоящем изобретении выражение "профилактически эффективное количество" включает в себя количество лекарства (например, профилактического средства), которое достаточно для предотвращения или уменьшения развития, рецидива или начала одного или более симптомов, связанных с заболеванием, или для увеличения или улучшения профилактического эффекта (эффектов) другого лекарства (например, другого профилактического средства).
Соединения
Фосфорамидатные и фосфонамидатные соединения ряда терапевтических средств можно изготавливать, используя способы, доступные в данной области техники и раскрытые в настоящем изобретении. Такие соединения можно использовать в некоторых вариантах осуществления для увеличения доставки лекарственного препарата в печень. В одном варианте осуществления соединение представляет собой S-ацил-2-тиоэтилфосфорамидатное или S-ацил-2-тиоэтилфосфонамидатное производное, например, S-пивалоил-2-тиоэтилфосфорамидат, S'-гидроксипивалоил-2-тиоэтилфосфорамидат, S-пивалоил-2-тиоэтилфосфонамидат или S-гидроксипивалоил-2-тиоэтилфосфонамидат. Терапевтические средства, которые можно дериватизировать до формы соединения, включают в себя противовирусное средство, которое включает в себя, или было дериватизировано, чтобы включать в себя реакционно-способную группу для присоединения фосфорамидатной или фосфонамидатной функциональной группы и включают в себя без ограничения нуклеозиды и нуклеозидные аналоги, включающие в себя нециклические нуклеозиды. Терапевтические средства, которые можно дериватизировать до формы соединения, также включают в себя противовирусное средство, которое включает в себя, или было дериватизировано, чтобы включать в себя фосфатную или фосфонатную группы, которые можно дериватизировать для образования фосфорамидатной или фосфонамидатной функциональной группы и включают в себя без ограничения нуклеозиды и нуклеозидные аналоги, включающие в себя нециклические нуклеозиды.
Нуклеозиды, которые можно дериватизировать, включают в себя любые из R1, раскрытые в настоящем изобретении. Примеры нуклеозидов, которые можно дериватизировать для включения в них фосфорамидата или фосфонамидата, например, в 5', 3' или 2' положениях, включают в себя:

Примеры фосфорамидатных или фосфонамидатных нуклеозидных соединений включают в себя:



Фосфорамидатные или фосфонамидатные соединения других нуклеозидов и нуклеозидных аналогов, описанных в настоящем изобретении и известных в данной области техники, можно создавать согласно описанию настоящего изобретения и использовать для лечения заболеваний печени. Фосфорамидатная или фосфонамидатная функциональная группа может находиться, например, в 5'-положении.
В одном варианте осуществления в настоящем изобретении рассматриваются соединения, а также их соли и композиции, содержащие указанные соединения, которые полезны для лечения заболеваний печени, таких как инфекции HBV и/или HCV. В одном варианте осуществления, фосфорамидатное или фосфонамидатное соединение согласно настоящему изобретению представляет собой соединение формулы IIa или IIb:

или его фармацевтически приемлемую соль, сольват, стереоизомерную, таутомерную или полиморфную форму, в которой:
Ry представляет собой алкил, алкенил, алкинил, арил, арилалкил, циклоалкил, циклоалкенил, амино, гетероциклил или гетероарил, все из которых необязательно замещаются;
Ra и Rb выбирают следующим образом:
i) Ra и Rb каждый независимо представляют собой водород, алкил, карбоксиалкил, гидроксиалкил, гидроксиарилалкил, ацилоксиалкил, аминокарбонилалкил, алкоксикарбонилалкил, арил, арилалкил, циклоалкил, гетероарил или гетероциклил, все из которых необязательно замещаются; или
ii) Ra и Rb вместе с атомом азота, на который они замещены, образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов; и
R1 является противовирусным препаратом (который включает в себя функциональную группу, получаемую удалением водорода из гидроксигруппы противовирусного препарата).
В некоторых вариантах осуществления соединение формулы IIa или IIb выбирают с условием, что, если Ry является трет-бутилом или гидрокси-трет-бутилом, тогда R1 не является 3'-азидо-2',3'-дидезокситимидином.
В некоторых вариантах осуществления R1, Ra, Rb и Ry необязательно замещены одним или более заместителями, как указано в разделе определений.
В некоторых вариантах осуществления соединения имеют формулу IIa или IIb, в которой Ry представляет собой алкил, алкенил, алкинил, арил, арилалкил, циклоалкил, циклоалкенил, амино, гетероциклил или гетероарил;
Ra и Rb каждый независимо представляют собой водород, алкил, карбоксиалкил, гидроксиалкил, гидроксиарилалкил, ацилоксиалкил, аминокарбонилалкил, алкоксикарбонилалкил, арил, арилалкил, циклоалкил, гетероарил или гетероциклил, все из которых необязательно замещены; и
R1 является противовирусным препаратом (что подразумевает включение в него функциональной группы, получаемой удалением водорода из гидроксигруппы противовирусного препарата).
В одном варианте осуществления, R1 является нуклеозидом, содержащим циклический или нециклический сахар или его аналог, включающий в себя любой нуклеозид или его аналог, описанный в настоящем изобретении или известный в данной области техники.
Ниже приведены примеры нуклеозидных лекарственных средств, пригодных для лечения гепатита C, которые можно дериватизировать согласно настоящему изобретению:
| Наименование | Структура |
| Рибавирин |
 |
| Вирамидин |
 |
| Валопицитабин (NM283) |
 |
| 2`-C-метилцитидин (NM107) |
 |
| PSI-6130 |
 |
| МК-0608 |
 |
| 7-фтор-МК-0608 |
 |
| NM108 |
 |
 |
Далее приведены примеры ненуклеозидных лекарственных средств, которые можно дериватизировать согласно настоящему изобретению:
| Наименование | Структура |
| Резиквимод |
 |
| Целгосивир |
 |
| Глиотоксин |
 |
Ниже приведены примеры нуклеозидных лекарственных средств, пригодных для лечения гепатита В, которые можно дериватизировать согласно настоящему изобретению:
| Наименование | Структура |
| Ламивудин или 3ТС или Эпивир® |
 |
| Энтекавир |
 |
| Телбивудин или L-dt |
 |
| Рацивир |
 |
| Эмтрицитабин или (-)FTC |
 |
| Клевудин или L-FMAU |
 |
| Амдоксовир |
 |
| Валторцитабин |
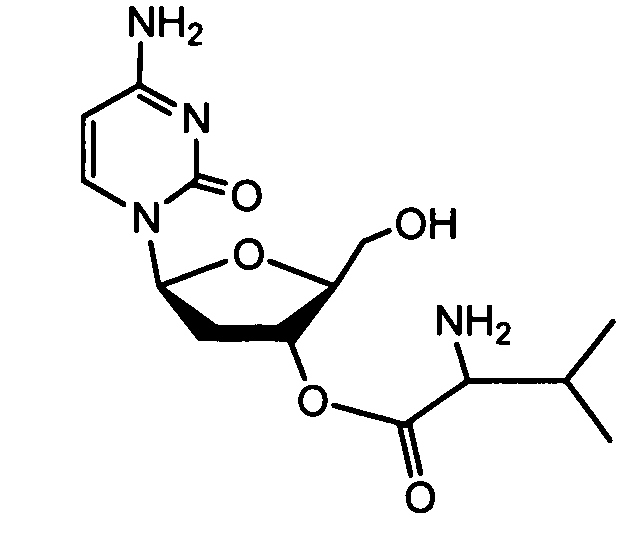 |
| Торцитабин (L-dC) |
 |
| Тенофовир или РМРА |
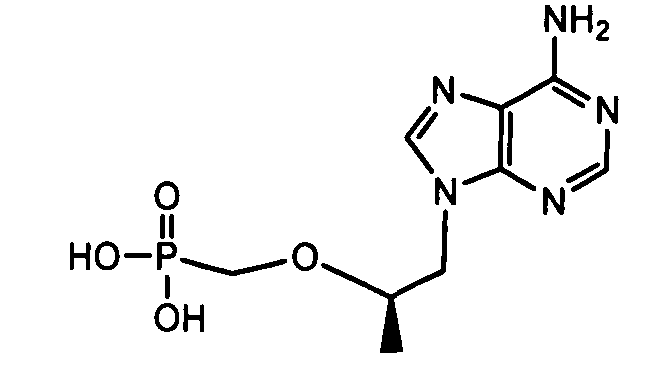 |
| Адефовир или РМЕА |
 |
| L-цитидин |
 |
Примеры фосфорамидатных или фосфонамидатных соединений ацикловира, L-ddA или D-ddA, которые можно вводить для лечения гепатита B, показаны ниже:

Если нуклеозидный аналог уже включает в себя фосфонат, указанная фосфонатная группа может быть включена в фосфонамидатную функциональную группу, показанную в формулах настоящего изобретения, и далее в качестве примера показан фосфонамидат адефовира:

Таким образом, в некоторых вариантах осуществления соединения формулы IIa показаны ниже:

с функциональной группой:

получают из лекарственного препарата, который представляет собой нециклический нуклеозид фосфоната, то есть:

который представляет собой, например, PMEA (9-[2-(фосфонметокси)этил]аденин (адефовир).
В некоторых вариантах осуществления, согласно формуле IIa или IIb, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси-, или амино-замещенный алкил или бензил. В другом варианте осуществления Ry предствляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо является алкилом, замещенным алкилом, арилом или замещенным арилом, например, гидрокси- или амино-замещенным алкилом или арилом; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил.
В дополнительном варианте осуществления Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления согласно этому параграфу каждый из R2 и R3 является водородом, Ra является водородом, Rb представляет собой -CH2-C6H5, и Ry представляет собой -C(CH3)2CH2OH.
В одном варианте осуществления Ry является алкилом или гидроксиалкилом. В одном варианте осуществления Ry представляет собой метил, трет-бутил, гидрокси-трет-бутил или гидроксиэтил. В некоторых вариантах осуществления Ry представляет собой -C(CH3)2CH2OH.
В одном варианте осуществления каждый из Ra и Rb независимо представляет собой водород, алкил, карбоксиалкил, гидроксиалкил, гидроксиарилалкил, ацилоксиалкил, аминокарбонилалкил, алкоксикарбонилалкил, арил или арилалкил, в которых алкильные группы можно дополнительно замещать одним или более заместителями. В одном варианте осуществления по меньшей мере один из Ra или Rb не является водородом. В одном варианте осуществления каждый из Ra и Rb независимо является водородом, метилом или бензилом.
В некоторых вариантах осуществления Ry представляет собой -C(CH3)2CH2OH и каждый из Ra и Rb независимо является водородом, метилом или бензилом. В некоторых вариантах осуществления Ry представляет собой -C(CH3)2CH2OH, и Ra является водородом и Rb является бензилом.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением формулы:

в которых R1 и Ry определены в формуле IIa или IIb.
В некоторых вариантах осуществления в формулах IIIa, b, c или d:
Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и
В другом варианте осуществления, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или гидрокси-, амино-, алкил-, галоалкил- или трифторметил-замещенный бензил. В некоторых вариантах осуществления Ra и Rb вместе с атомом азота, на который они замещены, образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов.
В одном варианте осуществления Ry является алкилом или гидроксиалкилом. В одном варианте осуществления Ry представляет собой метил, трет-бутил, гидрокси-трет-бутил или гидроксиэтил. В одном варианте осуществления Ry является -C(CH3)2CH2OH.
В некоторых вариантах осуществления согласно формуле IIIa или IIIb, Ry является замещенным алкилом, например, гидроксиалкилом или аминоалкилом. В другом варианте осуществления Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ry является -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением формулы:

в которой R1, Ra и Rb определены, например, как в формуле IIa или IIb.
В некоторых вариантах осуществления формулы IVa или IVb:
R1 является противовирусным препаратом, таким как нуклеозид или производное нуклеозида; и
Ra и Rb каждый независимо представляет собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или гидрокси-, амино-, алкил-, галоалкил- или трифторметил-замещенный бензил. В дополнительных вариантах осуществления Ra и Rb независимо являются H, бензилом или замещенным алкилом. В некоторых вариантах осуществления Ra и Rb вместе с атомом азота, на который они замещены, образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов.
В некоторых вариантах осуществления формулы IVa или IVb:
R1 представляет собой противовирусный препарат, такой как нуклеозид или производное нуклеозида; и
Ra и Rb каждый независимо представляет собой водород, алкил, карбоксиалкил, гидроксиалкил, гидроксиарилалкил, ацилоксиалкил, аминокарбонилалкил, алкоксикарбонилалкил, арил, арилалкил, циклоалкил, гетероарил или гетероциклил, все из которых необязательно замещены; и
в котором, в одном варианте осуществления, один из Ra и Rb является H, и другой представляет собой алкил, необязательно замещенный арилом, бензилом, или гетероарилом, каждый из которых необязательно замещен.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением формулы:

в которых R1 определен в формуле IIa или IIb.
В некоторых вариантах осуществления R1 является противовирусным нуклеозидным аналогом, пригодным для лечения вирусной инфекции HCV, выбираемым из следующего: рибавирин, вирамидин, 2'-C-метилцитидин, 2'-C-метилгуанозин, валопицитабин (NM283), МК-0608 и PSI-6130. Согласно применению в настоящем изобретении, если R1 является нуклеозидным аналогом, таким как ацикловир, который непосредственно включает в себя фосфонатную группу, то фосфонат может быть в форме фосфонамидата и иметь формулы, раскрытые в настоящем изобретении. Таким образом, например, в формуле Va или Vb фрагмент R1P(O)O- получают из нуклеозидного аналога, который включает в себя фосфонат.
В некоторых вариантах осуществления R1 является противовирусным нуклеозидным аналогом, пригодным для лечения вирусной инфекции HBV, который выбирают из ламивудина (Эпивир-HBV, Зеффикс или Гептодин), адефовира, энтекавира (Бараклуд), телбивудина (Тизека, Себиво), эмтрицитабина (FTC), клевудина (L-FMAU), виреада (Тенофовир), торцитабина, валторцитабина (моновал LdC), амдоксовира (DAPD) и RCV (Рацивир).
Дополнительно, примеры противовирусных нуклеозидных аналогов, которые можно использовать в качестве R1, раскрыты в международных патентных публикациях №№ WO 2005021568; WO 2006094347 и WO 2006093987 и патентной публикации США № US20050215510.
В некоторых вариантах осуществления R1 является ненуклеозидным противовирусным препаратом, пригодным для лечения вирусной инфекции HBV, выбираемым из резиквимода или целгосивира.
В одном варианте осуществления R1 представляет собой иммунодепрессант, такой как комбретастатин A-4, микофеноловая кислота, пентостатин, неларабин или митоксантрон.
В одном варианте осуществления R1 представляет собой противовирусный препарат на основе интерферирующей РНК (iРНК), включающий в себя противовирусный препарат на основе короткой интерферирующей РНК (siРНК). Такие соединения описаны в международных патентных публикациях №№ WO/03/070750 и WO 2005/012525, патентах США № 7176304; 7109165; 7041817; 7034009; 7022828; 6852535 и 6849726 и патентной публикации США № US 2004/0209831.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением формулы:

в которых Ra, Rb и Ry таковы, как описаны в формулах IIa или IIb, и в которых R2 и R3 каждый независимо представляет собой H; линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, аралкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток, углевод; пептид; холестерин; или другая фармацевтически приемлемая уходящая группа, которая способна дать соединение, в которых R2 и/или R3, независимо являются H или фосфатом (включающим в себя моно-, ди- или трифосфат), например, при введении in vivo; или R2 и R3 соединяют для образования циклической группы посредством алкильной, эфирной или карбаматной связи. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещается; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; аминокислотный остаток, или углевод. В некоторых вариантах осуществления каждый из R2 и R3 является водородом; Ra является водородом, Rb представляет собой -CH2-C6H5, и Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления согласно этому параграфу каждый R2 и R3 представляет собой H; Ry является замещенным алкилом, например, гидроксиалкилом или аминоалкилом; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый из R2 и R3 является H; Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый из R2 и R3 является H; Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления каждый из R2 и R3 является H; Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый из R2 и R3 является H; Ry представляет собой C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb и Re является водородом или алкилом. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, аралкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток; или углевод. В некоторых вариантах осуществления Re является метилом, этилом или пропилом, Ra является водородом, Rb представляет собой -CH2-C6H5, и Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления согласно этому параграфу каждый R2 и R3 представляет собой H; Ry является замещенным алкилом, например, гидроксиалкилом или аминоалкилом; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Re является метилом, этилом или пропилом; каждый из R2 и R3 представляет собой H; Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо являются водородом, алкилом, замещенным алкилом, бензилом или замещенным бензилом, например, гидрокси- или амино-замещенным алкилом или бензилом. В другом варианте осуществления Re является метилом, этилом или пропилом; каждый из R2 и R3 представляет собой Н; Ra и Rb независимо являются бензилом или алкилом. В дополнительном варианте осуществления каждый из R2 и R3 является H; Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Re является метилом, этилом или пропилом; каждый из R2 и R3 представляет собой H; Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления согласно этому параграфу R1 выбран из нуклеозидов, описанных в опубликованной патентной заявке США № US 2006/0111324 А1, содержание которой включено в настоящее изобретение в качестве ссылки во всей полноте.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток; или углевод. В некоторых вариантах осуществления согласно этому параграфу, RL является водородом, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый RL является водородом, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый RL является водородом; Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый RL является водородом и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:


в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток; или углевод. В некоторых вариантах осуществления согласно этому параграфу, каждый RL является водородом, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый RL является водородом, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый RL является водородом; Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый RL является водородом и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток; или углевод. В некоторых вариантах осуществления согласно этому параграфу, каждый RL является водородом, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый RL является водородом, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый RL является водородом; Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый RL является водородом и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; аминокислотный остаток; или углевод. В некоторых вариантах осуществления согласно этому параграфу, каждый RL является водородом, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый RL является водородом, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый RL является водородом, Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый RL является водородом и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток; или углевод. В некоторых вариантах осуществления согласно этому параграфу, каждый RL является водородом, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый RL является водородом, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый RL является водородом; Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый RL является водородом и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; аминокислотный остаток; или углевод. В некоторых вариантах осуществления согласно этому параграфу, каждый RL является водородом, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый RL является водородом, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый RL является водородом, Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый RL является водородом и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; аминокислотный остаток; или углевод. В некоторых вариантах осуществления согласно этому параграфу, каждый RL является водородом, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый RL является водородом, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый RL является водородом, Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый RL является водородом и Ry представляет собой -C(CH3)2CH2OH.
В некоторых вариантах осуществления 2-дезокси-2-фтор-2-C-этинил-β-D-нуклеозиды можно изготавливать и превращать в фосфорамидатные соединения для потенцирования доставки активного монофосфата в печень человека, пораженного HCV, например, в соединения, описанные в настоящем изобретении посредством примеров. В некоторых вариантах осуществления приводится соединение следующей формулы:

в которой:
T=O, S, CH2, CH(гал) или CH(гал)2, S(O)n;
n=1, 2;
гал = галоген;
R представляет собой H, ацил (с низшим линейным и нелинейным алкилом от С1 до 6-, аминокислотой), монофосфат, дифосфат, трифосфат, пролекарство монофосфата, такое как (алкил-O)2PO, (tBuSate-O)2PO, пролекарство циклического монофосфата, фосфорамидатное пролекарство (ароматический амин, аминокислота);
X и Y независимо представляют собой H, ОН, O-алкил (низший), O-ацил, F, NH2, NH-алкил, N-диалкил, NH-ацил, N-диацил или азидо;
Z представляет собой H, алкил, алкенил, алкинил, гидроксиметил, фторметил или азидо;
W представляет собой H, алкил, алкенил, алкинил, гидроксиметил, фторметил, азидо, карбоновую кислоту, CO2-алкил, циано или карбоксамид;
A представляет собой H, алкил, алкенил, алкинил, гидроксиметил, фторметил, азидо, карбоновую кислоту, CO2-алкил, циано или карбоксамид; и
Основание является модифицированным или природного происхождения.
Указанные соединения необязательно включают в себя атом хлора в 2'-положении.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb; A представляет собой H, алкил, алкенил, алкинил, гидроксиметил, фторметил, азидо, карбоновую кислоту, CO2-алкил, циано, или карбоксамид; и Rb1 является гало, алкокси или галоалкилом. Каждый RL независимо представляет собой H, карбамил, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; аминокислотный остаток; или углевод. В некоторых вариантах осуществления согласно этому параграфу, каждый RL является водородом, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления каждый RL является водородом, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления каждый RL является водородом; Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления каждый RL является водородом и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулы:

в которых описание Ra, Rb и Ry приведено в формуле IIa или IIb. В некоторых вариантах осуществления Ra является водородом, Rb представляет собой -CH2-C6H5, и Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления согласно этому параграфу, Ry представляет собой замещенный алкил, т.е. гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:

в которой X является галогеном, описание Ra, Rb и Ry приведено в формуле IIa или IIb и каждый из R2 и R3 независимо представляет собой H, линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, аралкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток, углевод; пептид; холестерин; или другая фармацевтически приемлемая уходящая группа, которая способна дать соединение, в котором R2 и/или R3, независимо являются H или фосфатом (включая моно-, ди- и трифосфат), например, при введении in vivo; или R2 и R3 соединяют для образования циклической группы посредством алкильной, эфирной или карбаматной связи. RL представляет собой водород или любую липофильную группу, известную специалистам в данной области техники. В некоторых вариантах осуществления каждый из R2 и R3 представляет собой водород, Ra является водородом, Rb представляет собой -CH2-C6H5 и Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления указанную липофильную группу выбирают из алкила, алкенила, циклоалкила, арила, гетероарила, арилалкила и гетероарил-алкила. В некоторых вариантах осуществления согласно этому параграфу X является фтором, RL является водородом, каждый из R2 и R3 представляет собой H, Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb каждый независимо представляет собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления X является фтором, RL является водородом, каждый из R2 и R3 является H, Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый из RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и каждый из Ra и Rb независимо представляет собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления X является фтором, RL является водородом, каждый из R2 и R3 представляет собой H, Ra и Rb каждый независимо представляет собой бензил или замещенный алкил. В дополнительном варианте осуществления X является фтором, RL является водородом, каждый из R2 и R3 представляет собой H, Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления X является фтором, RL является водородом, каждый R2 и R3 представляет собой H, Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:

в которой описание Ra, Rb и Ry приведено в формуле IIa или IIb. В некоторых вариантах осуществления Ry является замещенным алкилом, например, гидроксиалкилом или аминоалкилом; и Ra и Rb каждый независимо представляет собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо представляет собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ra является водородом, Rb представляет собой -CH2-C6H5 и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:

в которой описание Ra, Rb и Ry приведено в формуле IIa или IIb. В некоторых вариантах осуществления Ry является замещенным алкилом, то есть гидроксиалкилом или аминоалкилом; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ra является водородом, Rb представляет собой -CH2-C6H5 и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:

в которой описание Ra, Rb и Ry приведено в формуле IIa или IIb. В некоторых вариантах осуществления Ry является замещенным алкилом, то есть гидроксиалкилом или аминоалкилом; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ra является водородом, Rb представляет собой -CH2-C6H5 и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:

в которой описание Ra, Rb и Ry приведено в формуле IIa или IIb. В некоторых вариантах осуществления Ry является замещенным алкилом, то есть гидроксиалкилом или аминоалкилом; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой -ORC, -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ra является водородом, Rb представляет собой -CH2-C6H5 и Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:


или 
в которой описание Ra, Rb и Ry приведено в формуле IIa или IIb. RL представляет собой водород или любую липофильную группу, известную специалистам в данной области техники. В некоторых вариантах осуществления Ra является водородом, Rb представляет собой -CH2-C6H5 и Ry представляет собой -C(CH3)2CH2OH. В некоторых вариантах осуществления указанная липофильная группа выбрана из алкила, алкенила, циклоалкила, арила, гетероарила, арилалкила и гетероарилалкила. В некоторых вариантах осуществления согласно этому параграфу Ry представляет собой замещенный алкил, например, гидроксиалкил или аминоалкил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В другом варианте осуществления Ry представляет собой -C(RC)3 или -NHRC, где каждый RC независимо представляет собой алкил, замещенный алкил, арил или замещенный арил, например, гидрокси- или амино-замещенный алкил или арил; и Ra и Rb независимо представляют собой водород, алкил, замещенный алкил, бензил или замещенный бензил, например, гидрокси- или амино-замещенный алкил или бензил. В дополнительном варианте осуществления Ra и Rb независимо представляют собой бензил или замещенный алкил. В дополнительном варианте осуществления Ry выбирают из группы, состоящей из алкила и гидроксиалкила. В некоторых вариантах осуществления Ry представляет собой -C(CH3)2CH2OH.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:

для которых варианты осуществления описаны выше.
В другом варианте осуществления соединение согласно настоящему изобретению является соединением, имеющим формулу:
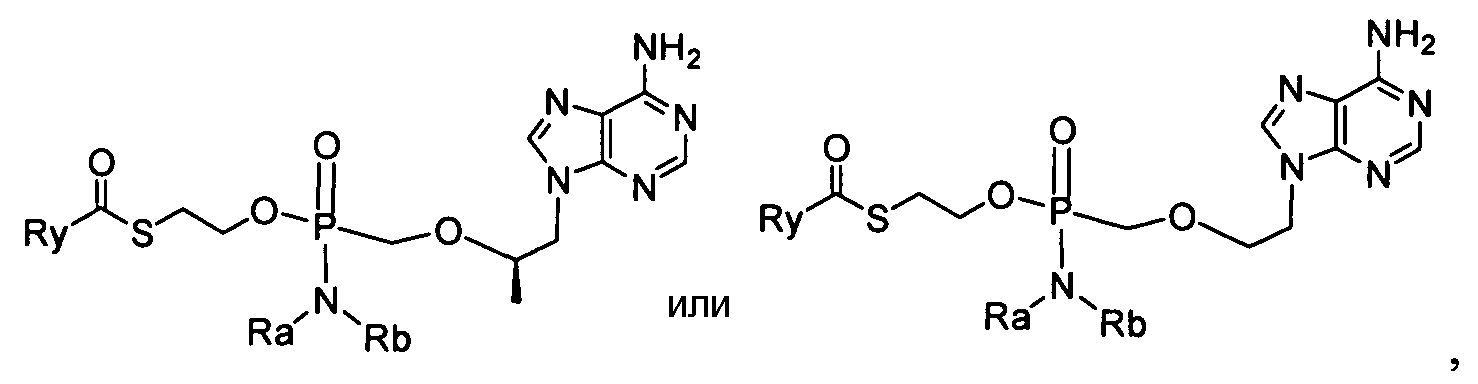
для которых варианты осуществления описаны выше.
В одном варианте осуществления R1 является нуклеозидом природного происхождения. В одном варианте осуществления R1 представляет собой 2'- или 3'-пролекарство биологически активного 1', 2', 3' или 4'-C-разветвленного β-D или β-L нуклеозида. Понятие 1', 2', 3' или 4'C-разветвленный, используемое в настоящем описании, включает в себя нуклеозид, имеющий дополнительный заместитель неприродного происхождения в указанном 1', 2', 3' или 4'-положениях (то есть, углерод связан с четырьмя неводородными заместителями). Термин 2`-пролекарство, используемый в настоящем изобретении, включает в себя 1', 2', 3' или 4'-C-разветвленный β-D или β-L нуклеозид, имеющий биологически расщепляемую функциональную группу в 2'-положении, включающий в себя без ограничения ацил, и в одном варианте осуществления, D- или L-аминокислоту природного или синтетического происхождения, и, в одном варианте осуществления, L-аминокислоту. Понятие 3'-пролекарство, используемое в настоящем изобретении, включает в себя 1', 2', 3' или 4' C-разветвленный β-D или β-L нуклеозид, имеющий биологически расщепляемую функциональную группу в 3'-положении, включающий в себя без ограничения ацил, и в одном варианте осуществления, D- или L-аминокислоту природного или синтетического происхождения, в одном варианте осуществления L-аминокислоту. В одном варианте осуществления аминокислотой является валин.
Примеры пролекарств (которые можно в дальнейшем дериватизировать согласно настоящему изобретению, для включения в них фосфорамидатной или фосфонамидатной функциональной группы, например, в 5' положениях) включают в себя 2'-L-валин-сложный эфир β-D-2'-C-метилцитидина; 2'-L-валин-сложный эфир β-D-2'-C-метилтимидина; 2'-L-валин-сложный эфир β-D-2'-C-метиладенозина; 2'-L-валин сложный эфир β-D-2'-C-метилгуанозина; 2'-L-валин сложный эфир β-D-2'-C-метил-5-фторцитидина; 2'-L-валин сложный эфир β-D-2'-C-метилуридина; 2'-ацетил-сложный эфир β-D-2'-C-метилцитидина; 2'-ацетил-сложный эфир β-D-2'-C-метилтимидина; 2'-ацетил-сложный эфир β-D-2'-C-метиладенозина; 2'-ацетил-сложный эфир β-D-2'-C-метилгуанозина; 2'-ацетил-сложный эфир β-D-2'-C-метил-5-фторцитидина; и 2'-сложные эфиры β-D-2'-C-метил-(цитидина, 5-фторцитидина, гуанозина, уридина, аденозина или тимидина), в которых (i) 2'-сложный эфир является сложным эфиром аминокислоты; или (ii) 2'-сложный эфир является сложным эфиром алкила или арила.
Дополнительными примерами пролекарств являются 3'-L-валин-сложный эфир β-D-2'-C-метилцитидина; 3'-L-валин-сложный эфир β-D-2'-C-метилтимидина; 3'-L-валин-сложный эфир β-D-2'-C-метиладенозина; 3'-L-валин-сложный эфир β-D-2'-C-метилгуанозина; 3'-L-валин-сложный эфир β-D-2'-C-метил-5-фторцитидина; 3'-L-валин-сложный эфир β-D-2'-C-метилуридина; 3'-ацетил-сложный эфир β-D-2'-C-метилцитидина; 3'-ацетил-сложный эфир β-D-2'-C-метилтимидина; 3'-ацетил-сложный эфир β-D-2'-C-метиладенозина; 3'-ацетил-сложный эфир β-D-2'-C-метилгуанозина; 3'-ацетил-сложный эфир β-D-2'-C-метил-5-фторцитидина; и 3'-сложные эфиры β-D-2'-C-метил-(цитидина, 5-фторцитидина, гуанозина, уридина, аденозина или тимидина), в которых (i) 3'-сложный эфир является сложным эфиром аминокислоты; или (ii) 3'-сложный эфир является сложным эфиром алкила или арила.
Дополнительные примеры пролекарств включают в себя 2', 3'-L-дивалин-сложный эфир β-D-2'-C-метилцитидина (дивал-2'-Me-L-dC); 2', 3'-L-дивалин-сложный эфир β-D-2'-C-метилтимидина; 2', 3'-L-дивалин-сложный эфир β-D-2'-C-метиладенозина; 2', 3'-L-дивалин-сложный эфир β-D-2'-C-метилгуанозина; 2', 3'-L-дивалин-сложный эфир β-D-2'-C-метил-5-фторцитидина; 2', 3'-L-дивалин-сложный эфир β-D-2'-C-метилуридина; 2',3'-диацетил-сложный эфир β-D-2'-C-метилцитидина; 2', 3'-диацетил-сложный эфир β-D-2'-C-метилтимидина; 2',3'-диацетил сложный эфир β-D-2'-C-метиладенозина; 2',3'-диацетил-сложный эфир β-D-2'-C-метилгуанозина; 2',3'-диацетил-сложный эфир β-D-2'-C-метил-5-фторцитидина; и 2',3'-ди-сложные эфиры β-D-2'-C-метил-(цитидина, 5-фторцитидина, гуанозина, уридина, аденозина или тимидина), в которых (i) 2'-сложный эфир является сложным эфиром аминокислоты и 3'-сложный эфир является сложным эфиром алкила или арила; (ii) оба сложных эфира являются сложными эфирами аминокислоты; (iii) оба сложных эфира независимо являются сложными эфирами алкила или арила; или (iv) 2'-сложный эфир является сложным эфиром алкила или арила и 3'-сложный эфир является сложным эфиром аминокислоты.
В одном варианте осуществления R1 представлен формулами:

в которых основанием является пуриновое или пиримидиновое основание природного или неприродного происхождения, как указано в настоящем изобретении;
R6 представляет собой водород, гидрокси, алкил, алкенил, алкинил, азидо, циано, бромвинил, алкокси, ацилокси, алкоксикарбонил, алкенилокси, гало, NO2 или NR6aR6b;
каждый из R6a и R6b независимо представляет собой водород, алкил, алкенил, алкинил, циклоалкил, ацил, арил, гетероарил или гетероциклил;
R7, R9, R8 и R10 выбирают следующим образом:
i) каждый из R7 и R9 независимо является водородом, OR7a, гидрокси, алкилом, алкенилом, алкинилом, азидо, циано, бромвинилом, алкилоксикарбонилом, ацилокси, гало, NO2 или NR6aR6b;
ii) каждый из R8 и R10 независимо является H, алкилом или гало; или
iii) каждый из R7 и R9, R7 и R10, R8 и R9 или R8 и R10 вместе образуют двойную связь;
R7a представляет собой H; линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена одним или более заместителями, согласно описанию арила в разделе определений настоящего изобретения; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, включающий в себя фосфолипид; аминокислота; и аминокислотный остаток, углевод; пептид; холестерин; или другую фармацевтически приемлемую уходящую группу, которая способна дать соединение, в котором R7a является H или фосфатом, (включающим в себя моно, ди или трифосфат), например, при введении in vivo; в котором в одном варианте осуществления R7a не является фосфатом (включающим в себя моно-, ди- или трифосфат или устойчивое фосфатное пролекарство), или две группы R7a соединены для образования циклической группы посредством алкильной, эфирной или карбаматной связи; и
X является O, S, SO2 или CH2.
В одном варианте осуществления R1 имеет формулу:

в которой каждый из R2 и R3 независимо представляет собой H; линейный, разветвленный или циклический алкил; ацил (включающий в себя низший ацил); СО-алкил, СО-арил, СО-алкоксиалкил, CO-арилоксиалкил, СО-замещенный арил, сложный эфир сульфоната, такой как алкил- или арилалкилсульфонил, включающий в себя метансульфонил и бензил, в котором фенильная группа необязательно замещена; алкилсульфонил, арилсульфонил, арилалкилсульфонил, липид, такой как фосфолипид; аминокислота; и аминокислотный остаток, углевод; пептид; холестерин; или другую фармацевтически приемлемую уходящую группу, которая способна дать соединение, в котором R2 и/или R3 независимо является H или фосфатом, (включающим в себя моно-, ди- или трифосфат), например, при введении in vivo; или R2 и R3 являются соединенными для образования циклической группы посредством алкильной, эфирной или карбаматной связи;
в которой Y1 является водородом, бромом, хлором, фтором, иодом, CN, ОН, OR4, NH2, NHR4, NR4R5, SH или SR4;
X1 представляет собой линейный, разветвленный или циклический необязательно замещенный алкил, CH3, CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, CH2OH, необязательно замещенный алкенил, необязательно замещенный алкинил, COOH, COOR4, COO-алкил, COO-арил, со-O-алкоксиалкил, CONH2, CONHR4, CON(R4)2, хлор, бром, фтор, иод, CN, N3, ОН, OR4, NH2, NHR4, NR4R5, SH или SR5; и
X2 представляет собой H, линейный, разветвленный или циклический необязательно замещенный алкил, CH3, CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, CH2OH, необязательно замещенный алкенил, необязательно замещенный алкинил, COOH, COOR4, COO-алкил, COO-арил, со-O-алкоксиалкил, CONH2, CONHR4, CON(R4)2, хлор, бром, фтор, иод, CN, N3, ОН, OR4, NH2, NHR4, NR4R5, SH или SR5; и
в котором каждый Y3 независимо является H, F, Cl, Br или I;
каждый R4 и R5 независимо представляют собой водород, ацил (включающий в себя низший ацил), алкил (включающий в себя без ограничения метил, этил, пропил и циклопропил), низший алкил, алкенил, алкинил или циклоалкил.
В вариантах осуществления, описанных в настоящем изобретении, R2 и/или R3 могут быть фармацевтически приемлемой уходящей группой, которая способна дать соединение, в котором R2 и/или R3 независимо являются H или фосфатом, (включающим в себя моно-, ди- или трифосфат), например, при введении in vivo.
В другом варианте осуществления каждый из R2 и R3 независимо является водородом или ацилом. В другом варианте осуществления R2 и R3 являются связанными для образования циклической группы посредством алкильной, эфирной или карбаматной связи.
В другом варианте осуществления R1 имеет формулу:

в которой определение R2, R3, Y1, Y3, X1 и X2 соответствует определению для формулы XIII.
В одном варианте осуществления R1 имеет формулу:

в которой основание выбирают из группы, состоящей из:








в которой каждый из W1, W2, W3 и W4 независимо представляет собой N, CH, CF, Cl, CBr, CCl, CCN, CCH3, CCF3, CCH2CH3, CC(O)NH2, CC(O)NHR4, CC(O)N(R4)2, CC(O)ОН, CC(O)OR4 или CX3;
каждый из W* независимо представляет собой O, S, NH или NR4;
X представляет собой O, S, SO2, CH2, CH2OH, CHF, CF2, C(Y3)2, CHCN, C(CN)2, CHR4 или C(R4)2;
X* представляет собой CH, CF, CY3 или CR4;
X2 представляет собой H, линейный, разветвленный или циклический необязательно замещенный алкил, CH3, CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, CH2OH, необязательно замещенный алкенил, необязательно замещенный алкинил, COOH, COOR4, COO-алкил, COO-арил, со-Oалкоксиалкил, CONH2, CONHR4, CON(R4)2, хлор, бром, фтор, иод, CN, N3, ОН, OR4, NH2, NHR4, NR4R5, SH или SR5;
каждый X3 независимо представляет собой линейный, разветвленный или циклический необязательно замещенный алкил (включающий в себя низший алкил), CH3, CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, необязательно замещенный алкенил, галоалкенил, Br-винил, необязательно замещенный алкинил, галоалкинил, N3, CN, -C(O)ОН, -C(O)OR4, -C(O)O (низший алкил), -C(O)NH2, -C(O)NHR4, -C(O)NH (низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2, ОН, OR4, -O(ацил), -O(низший ацил), -O(алкил), -O(низший алкил), -O(алкенил), -O(алкинил), -O(арилалкил), -O(циклоалкил), -S(ацил), -S(низший ацил), -S(R4), -S(низший алкил), -S(алкенил), -S(алкинил), -S(арилалкил), -S(циклоалкил), хлор, бром, фтор, иод, NH2, -NH(низший алкил), -NHR4, -NR4R5, -NH(ацил), -N(низший алкил)2, -NH(алкенил), -NH(алкинил), -NH(арилалкил), -NH(циклоалкил), -N(ацил)2;
каждый Y независимо выбирают из группы, состоящей из H, необязательно замещенного низшего алкила, циклоалкила, алкенила, алкинила, CH2OH, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2F, CH2Cl, CH2N3, CH2CN, CH2CF3, CF3, CF2CF3, CH2CO2R, (CH2)mCOOH, (CH2)mCOOR, (CH2)mCONH2, (CH2)mCONR2 и (CH2)mCONHR;
в котором R является H, алкилом или ацилом;
Y1 представляет собой водород, бром, хлор, фтор, иод, CN, ОН, OR4, NH2, NHR4, NR4R5, SH или SR4;
каждый Y2 независимо представляет собой O, S, NH или NR4;
каждый Y3 независимо представляет собой H, F, Cl, Br или I;
каждый R4 и R5 независимо представляет собой водород, ацил (включающий в себя низший ацил), алкил (включающий в себя без ограничения метил, этил, пропил и циклопропил), низший алкил, алкенил, алкинил или циклоалкил;
каждый R6 независимо представляет собой необязательно замещенный алкил (включающий в себя низший алкил), CH3, CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, необязательно замещенный алкенил, галоалкенил, Br-винил, необязательно замещенный алкинил, галоалкинил, -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, -(CH2)mC(O)ОН, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), - (CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), -(CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил)2, -C(O)ОН, -C(O)OR4, -C(O)O(низший алкил), -C(O)NH2, -C(O)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2 или циано;
каждый R7 независимо представляет собой H, ОН, OR2, необязательно замещенный алкил (включающий в себя низший алкил), CH3, CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, необязательно замещенный алкенил, галоалкенил, Br-винил, необязательно замещенный алкинил, галоалкинил, необязательно замещенный карбоцикл (например, карбоциклическое кольцо, содержащее от 3 до 7 членов), необязательно замещенный гетероцикл (например, 3-7-членное гетероциклическое кольцо, имеющее один или более атомов O, S и/или N), необязательно замещенный гетероарил (например, 3-7-членное гетероароматическое кольцо, имеющее один или более атомов O, S и/или N), -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)SH, -CH2C(O)SR4, -CH2C(O)S(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, -(CH2)mC(O)ОН, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), -(CH2)mC(O)SH, -(CH2)mC(O)SR4, -(CH2)mC(O)S(низший алкил), -(CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), - (CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил)2, -C(O)ОН, -C(O)OR4, -C(O)O(низший алкил), -C(O)SH, -C(O)SR4, -C(O)S(низший алкил), -C(O)NH2, -C(O)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2, -O(ацил), -O(низший ацил), -O(R4), -O(алкил), -О(низший алкил), -О(алкенил), -О(алкинил), -О(арилалки), -О(циклоалкил), -S(ацил), -S(низший ацил), -S(R4), -S(низший алкил), -S(алкенил), -S(алкинил), -S(арилалкил), -S(циклоалкил), NO2, NH2, -NH(низший алкил), -NHR4, -NR4R5, -NH(ацил), -N(низший алкил)2, -NH(алкенил), -NH(алкинил), -NH(арилалкил), -NH(циклоалкил), -N(ацил)2, азидо, циано, SCN, OCN, NCO или гало (фтор, хлор, бром, иод);
альтернативно, R6 и R7 можно объединять для образования спиросоединения, выбираемого из группы, состоящей из необязательно замещенного карбоцикла (например, 3-7-членного карбоциклического кольца), или необязательно замещенного гетероцикла (например, 3-7-членного гетероциклического кольца, имеющего один или более атомов O, S и/или N);
каждый m независимо равен 0, 1 или 2.
В одном варианте осуществления основание имеет формулу

В одном варианте осуществления основание имеет формулу

В другом варианте осуществления R1 имеет формулу

в которых определение каждого из R6 и R7 соответствует вышеприведенным формулам XX, XXI или XXII;
в которых каждый из R8 и R11 независимо представляют собой водород, необязательно замещенный алкил (включающий в себя низший алкил), CH3, CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, необязательно замещенный алкенил, галоалкенил, Br-винил, необязательно замещенный алкинил, галоалкинил, -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, -(CH2)mC(O)ОН, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), -(CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), -(CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил)2, -С(О)ОН, -С(О)OR4, -С(О)О(низший алкил), -С(О)NH2, -С(О)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2, циано, азидо, NH-ацил или N(ацил)2;
каждый из R9 и R10 независимо представляют собой водород, ОН, OR2, необязательно замещенный алкил (включающий в себя низший алкил), CH3, CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, необязательно замещенный алкенил, галоалкенил, Br-винил, необязательно замещенный алкинил, галоалкинил, необязательно замещенный карбоцикл (например, 3-7-членное карбоциклическое кольцо), необязательно замещенный гетероцикл (например, 3-7-членное гетероциклическое кольцо, имеющее один или более атомов O, S и/или N), необязательно замещенный гетероарил (например, 3-7-членное гетероароматическое кольцо, имеющее один или более атомов O, S и/или N), -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)SH, -CH2C(O)SR4, -CH2C(O)S(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, -(CH2)mC(O)ОН, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), - (CH2)mC(O)SH, -(CH2)mC(O)SR4, -(CH2)mC(O)S(низший алкил), -(CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), -(CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил)2, -C(O)ОН, -C(O)OR4, -C(O)O(низший алкил), -C(O)SH, -C(O)SR4, -C(O)S(низший алкил), -C(O)NH2, -C(O)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2, -O(ацил), -O(низший ацил), -O(R4), -O(алкил), -О(низший алкил), -О(алкенил), -О(алкинил), -О(арилалки), -О(циклоалкил), -S(ацил), -S(низший ацил), -S(R4), -S(низший алкил), -S(алкенил), -S(алкинил), -S(арилалкил), -S(циклоалкил), NO2, NH2, -NH(низший алкил), -NHR4, -NR4R5, -NH-(ацил), -N(низший алкил)2, -NH(алкенил), -NH(алкинил), -NH(арилалкил), -NH(циклоалкил), -N(ацил)2, азидо, циано, SCN, OCN, NCO или гало (фтор, хлор, бром, иод);
каждый m независимо равен 0, 1 или 2.
альтернативно, R6 и R10, R7 и R9, R8 и R7 или R9 и R11 можно объединять для образования мостикового соединения, выбираемого из группы, состоящей из необязательно замещенного карбоцикла (например, 3-7-членного карбоциклического кольца), или необязательно замещенного гетероцикла (например, 3-7-членного гетероциклического кольца, имеющего один или более атомов O, S и/или N); или
альтернативно, R6 и R7 или R9 и R10 можно объединять для образования спиросоединения, выбираемого из группы, состоящей из необязательно замещенного карбоцикла (например, 3-7-членного карбоциклического кольца), или необязательно замещенного гетероцикла (например, 3-7-членного гетероциклического кольца, имеющего один или более атомов O, S и/или N).
В другом варианте осуществления R1 имеет формулу:

в которых основание* представляет собой пуриновое или пиримидиновое основание согласно настоящему изобретению;
каждый из R12 независимо представляет собой замещенный алкил (включающий в себя низший алкил), CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, замещенный алкенил, галоалкенил (но не Br-винил), замещенный алкинил, галоалкинил, -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, -(CH2)mC(O)ОН, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), -(CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), -(CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил)2, -C(O)ОН, -С(О)OR4, -C(O)NH2, -C(O)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2;
каждый R13 независимо представляет собой замещенный алкил (включающий в себя низший алкил), CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, замещенный алкенил, галоалкенил (но не Br-винил), замещенный алкинил, галоалкинил, необязательно замещенный карбоцикл (например, 3-7-членное карбоциклическое кольцо), необязательно замещенный гетероцикл (например, 3-7-членное гетероциклическое кольцо, имеющее один или более атомов O, S и/или N), необязательно замещенный гетероарил (например, 3-7-членное гетероароматическое кольцо, имеющее один или более атомов O, S и/или N), -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)SH, -CH2C(O)SR4, -CH2C(O)S(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, (CH2)mC(О)OH, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), -(CH2)mC(O)SH, -(CH2)mC(O)SR4, -(CH2)mC(O)S(низший алкил), -(CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), -(CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил)2, -C(O)ОН, -C(O)OR4, -C(O)SH, -C(O)SR4, -C(O)S(низший алкил), -C(O)NH2, -C(O)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2, -О(R4), -O(алкинил), -O(арилалкил), -O(циклоалкил), -S(ацил), -S(низший ацил), -S(R4), -S(низший алкил), -S(алкенил), -S(алкинил), -S(арилалкил), -S(циклоалкил), -NHR4, -NR4R5, -NH(алкенил), -NH(алкинил), -NH(арилалкил), -NH(циклоалкил), SCN, OCN, NCO или фтор;
альтернативно, R12 и R13 можно объединять для образования спиросоединения, выбираемого из группы, состоящей из необязательно замещенного карбоцикла (например, 3-7-членного карбоциклического кольца), или необязательно замещенного гетероцикла (например, 3-7-членного гетероциклического кольца, имеющего один или более атомов O, S и/или N);
R2 и R3 соответствуют формуле XII; и
каждый m независимо равен 0, 1 или 2.
В другом варианте осуществления R1 имеет формулу:

в которых основание* представляет собой пуриновое или пиримидиновое основание согласно настоящему изобретению; и
каждый из R8 и R11 независимо представляет собой водород, необязательно замещенный алкил (включающий в себя низший алкил), CH3, CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, необязательно замещенный алкенил, галоалкенил, Br-винил, необязательно замещенный алкинил, галоалкинил, -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, -(CH2)mC(O)OH, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), -(CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), -(CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил), -С(О)ОН, -С(О)OR4, С(О)О(низший алкил), -C(O)NH2, -C(O)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2, циано, NH-ацил или N(ацил)2;
каждый из R9 и R10 независимо представляет собой водород, ОН, OR2, необязательно замещенный алкил (включающий в себя низший алкил), CH3, CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, необязательно замещенный алкенил, галоалкенил, Br-винил, необязательно замещенный алкинил, галоалкинил, необязательно замещенный карбоцикл (например, 3-7-членное карбоциклическое кольцо), необязательно замещенный гетероцикл (например, 3-7-членное гетероциклическое кольцо, имеющее один или более атомов O, S и/или N), необязательно замещенный гетероарил (например, 3-7-членное гетероароматическое кольцо, имеющее один или более атомов O, S и/или N), -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)SH, -CH2C(O)SR4, -CH2C(O)S(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, -(CH2)mC(О)OH, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), -(CH2)mC(O)SH, -(CH2)mC(O)SR4, -(CH2)mC(O)S(низший алкил), -(CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), -(CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил)2, -C(O)ОН, -C(O)OR4, -C(O)O(низший алкил), -C(O)SH, -C(O)SR4, -C(O)S(низший алкил), -C(O)NH2, -C(O)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2, -O(ацил), -O(низший ацил), -O(R4), -O(алкил), -О(низший алкил), -О(алкенил), -О(алкинил), -О(арилалки), -О(циклоалкил), -S(ацил), -S(низший ацил), -S(R4), -S(низший алкил), -S(алкенил), -S(алкинил), -S(арилалкил), -S(циклоалкил), NO2, NH2, -NH(низший алкил), -NHR4, -NR4R5, -NH(ацил), -N(низший алкил)2, -NH(алкенил), -NH(алкинил), -NH(арилалкил), -NH(циклоалкил), -N(ацил)2, азидо, циано, SCN, OCN, NCO или гало (фтор, хлор, бром, иод);
каждый R12 независимо представляет собой замещенный алкил (включающий в себя низший алкил), CH2CN, CH2N3, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2OH, галогенированный алкил (включающий в себя галогенированный низший алкил), CF3, C(Y3)3, 2-Br-этил, CH2F, CH2Cl, CH2CF3, CF2CF3, C(Y3)2C(Y3)3, замещенный алкенил, галоалкенил (но не Br-винил), замещенный алкинил, галоалкинил, -CH2C(O)ОН, -CH2C(O)OR4, -CH2C(O)O(низший алкил), -CH2C(O)NH2, -CH2C(O)NHR4, -CH2C(O)NH(низший алкил), -CH2C(O)N(R4)2, -CH2C(O)N(низший алкил)2, -(CH2)mC(O)ОН, -(CH2)mC(O)OR4, -(CH2)mC(O)O(низший алкил), -(CH2)mC(O)NH2, -(CH2)mC(O)NHR4, -(CH2)mC(O)NH(низший алкил), -(CH2)mC(O)N(R4)2, -(CH2)mC(O)N(низший алкил)2, -C(O)ОН, -С(О)OR4, -C(O)NH2, -C(O)NHR4, -C(O)NH(низший алкил), -C(O)N(R4)2, -C(O)N(низший алкил)2;
каждый m независимо равен 0, 1 или 2;
альтернативно, R8 и R13, R9 и R13, R9 и R11 или R10 и R12 можно объединять для образования мостикового соединения, выбираемого из группы, состоящей из необязательно замещенного карбоцикла (например, 3-7-членного карбоциклического кольца), или необязательно замещенного гетероцикла (например, 3-7-членного гетероциклического кольца, имеющего один или более атомов O, S и/или N); или
альтернативно, R12 и R13 или R9 и R10 можно объединять для образования спиросоединения, выбираемого из группы, состоящей из необязательно замещенного карбоцикла (например, 3-7-членного карбоциклического кольца), или необязательно замещенного гетероцикла (например, 3-7-членного гетероциклического кольца, имеющего один или более атомов O, S и/или N).
В одном аспекте R1 имеет формулу:

Буквой B обозначают спиросоединение, выбираемое из группы, состоящей из необязательно замещенного карбоцикла, (например, 3-7-членного карбоциклического кольца) или необязательно замещенного гетероцикла (например, 3-7-членного гетероциклического кольца, имеющего один или более атомов O, S и/или N);
Основание выбирают из группы, состоящей из:


в котором каждый из R', R", R'" и R"" независимо выбирают из группы, состоящей из H, ОН, замещенного или незамещенного алкила, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, циклоалкила, Br-винила, -O-алкила, O-алкенила, O-алкинила, O-арила, O-арилалкила, -O-ацила, O-циклоалкила, NH2, NH-алкила, N-диалкила, NH-ацила, N-арила, N-арилалкила, NH-циклоалкила, SH, S-алкила, S-ацила, S-арила, S-циклоалкила, S-арилалкила, F, Cl, Br, I, CN, COOH, CONH2, CO2-алкила, CONH-алкила, CON-диалкила, ОН, CF3, CH2OH, (CH2)mOH, (CH2)mNH2, (CH2)mCOOH, (CH2)mCN, (CH2)mNO2 и (CH2)mCONH2;
m равен 0 или 1;
каждый W независимо представляет собой C-R" или N;
T и V независимо представляет собой CH или N;
Q представляет собой CH, -CCl, -CBr, -CF, -CI, -CCN, -C-COOH,-C-CONH2 или N;
Q1 и Q2 независимо представляют собой N или C-R;
Q3, Q4, Q5 и Q6 независимо представляют собой N или CH; и
их таутомерные формы.
В другом аспекте R1 имеет формулы:

G и E независимо выбирают из группы, состоящей из CH3, CH2OH, CH2F, CH2N3, CH2CN, (CH2)mCOOH, (CH2)mCOOR, (CH2)mCONH2, (CH2)mCONR2, (CH2)mCONHR, N3 и N-ацила;
m равен 0 или 1;
R представляет собой H, алкил или ацил; и
Определение основания соответствует формуле (XIII).
В одном варианте осуществления в большинстве случаев один из G и E дополнительно может быть водородом.
В другом варианте осуществления R1 имеет формулу:

в которой М выбирают из группы, состоящей из O, S, SO и SO2; и определение основания соответствует формуле (XIII).
В некоторых вариантах осуществления R1 имеет формулы:


в которой A выбирают из группы, состоящей из необязательно замещенного низшего алкила, циклоалкила, алкенила, алкинила, CH2OH, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2F, CH2Cl, CH2N3, CH2CN, CH2CF3, CF3, CF2CF3, CH2CO2R, (CH2)mCOOH, (CH2)mCOOR, (CH2)mCONH2, (CH2)mCONR2 и (CH2)mCONHR;
Y выбирают из группы, состоящей из H, необязательно замещенного низшего алкила, циклоалкила, алкенила, алкинила, CH2OH, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2F, CH2Cl, CH2N3, CH2CN, CH2CF3, CF3, CF2CF3, CH2CO2R, (CH2)mCOOH, (CH2)mCOOR, (CH2)mCONH2, (CH2)mCONR2 и (CH2)mCONHR;
X выбирают из группы, состоящей из H, -ОН, необязательно замещенного алкила, циклоалкила, алкенила, алкинила, -O-алкила, -O-алкенила, -O-алкинила, О-арила, -O-арилалкила, -O-циклоалкил-, O-ацила, F, Cl, Br, I, CN, NC, SCN, OCN, NCO, NO2, NH2, N3, NH-ацила, NH-алкила, N-диалкила, NH-алкенила, NH-алкинила, NH-арила, NH-арилалкила, NH-циклоалкила, SH, S-алкила, S-алкенила, S-алкинила, S-арила, S-арилалкила, S-ацила, S-циклоалкила, CO2-алкила, CONH-алкила, CON-диалкила, CONH-алкенила, CONH-алкинила, CONH-арилалкила, CONH-циклоалкила, CH2OH, CH2NH2, CH2NHCH3, CH2N(CH3)2, CH2F, CH2Cl, CH2N3, CH2CN, CH2CF3, CF3, CF2CF3, CH2CO2R, (CH2)mCOOH, (CH2)mCOOR, (CH2)mCONH2, (CH2)mCONR2, (CH2)mCONHR, необязательно замещенного 3-7-членного карбоциклического и необязательно замещенного 3-7-членного гетероциклического кольца, независимо имеющих O, S и/или N в качестве гетероатомов, взятых единственными или в комбинации;
m равен 0 или 1;
R является H, алкилом или ацилом; и основание представляет собой основание синтетического происхождения, выбираемое из следующей группы:

в которой каждый из R', R", R'" и R"" независимо выбирают из группы, состоящей из H, ОН, замещенного или незамещенного алкила, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, циклоалкила, Br-винила, -O-алкила, O-алкенила, O-алкинила, O-арила, O-арилалкила, -O-ацила, O-циклоалкила, NH2, NH-алкила, N-диалкила, NH-ацила, N-арила, N-арилалкила, NH-циклоалкила, SH, S-алкила, S-ацила, S-арила, S-циклоалкила, S-арилалкила, F, Cl, Br, I, CN, COOH, CONH2, CO2-алкила, CONH-алкила, CON-диалкила, ОН, CF3, CH2OH, (CH2)mOH, (CH2)mNH2, (CH2)mCOOH, (CH2)mCN, (CH2)mNO2 и (CH2)mCONH2;
m равен 0 или 1;
каждый из W независимо является C-R" или N;
T и V независимо являются CH или N;
Q представляет собой CH, -CCl, -CBr, -CF, -CI, -CCN, -C-COOH, -C-CONH2 или N;
Q1 и Q2 независимо являются N или C-R""; и
Q3, Q4, Q5 и Q6 независимо являются N или CH;
при условии, что в основаниях (g) и (i) R', R"" не являются H, ОН или NH2; и Q, T, V, Q2, Q5 и Q6 не является N.
В одном варианте осуществления R1 представляет собой 2'-(алкил или арил)сложный эфир или 3'-(алкил или арил)сложный эфир 1', 2', 3' или 4'C-разветвленного-β-D или β-L нуклеозида с любым пуриновым или пиримидиновым основанием природного или синтетического происхождения. В одном варианте осуществления R1 представляет собой 2' или 3'-(D или L)-аминокислотный сложный эфир 1', 2', 3' или 4' C-разветвленного-β-D или β-L нуклеозида, в котором аминокислотой является аминокислота природного или синтетического происхождения. В другом варианте осуществления R1 представляет собой 3'-D или L-аминокислотный сложный эфир 1', 2', 3' или 4' C-разветвленного-β-D или β-L нуклеозида, в котором аминокислотой является аминокислота природного или синтетического происхождения. В одном варианте осуществления аминокислота представляет собой L-аминокислоту.
В одном варианте осуществления аминокислотный остаток имеет формулу
C(O)C(R11)(R12)(NR13R14),
в которой R11 является боковой цепью аминокислоты, и в которой R11 необязательно может быть присоединен к R13 для образования структуры кольца; или альтернативно, R11 представляет собой алкильную, арильную, гетероарильную или гетероциклическую функциональную группу;
R12 представляет собой водород, алкил (включающий в себя низший алкил) или арил; и
R13 и R14 независимо представляют собой водород, ацил (включающий в себя производное ацила, присоединенное к R11) или алкил (включающий в себя без ограничения метил, этил, пропил и циклопропил).
В другом варианте осуществления по меньшей мере один из R2 и R3 представляет собой аминокислотный остаток. В одном варианте осуществления, по меньшей мере один из R2 и R3 является L-валинилом.
В одном варианте осуществления R1 представлен формулами:

в которых R6, R7, R8, R9, R10 и основание* соответствуют определению из формул XXX, XXXI, XL, XLI или XLII.
В одном варианте осуществления R1 представлен формулами:

в которых R2, R3, R6 и основание* соответствуют определению из формул XXX, XXXI, XL, XLI или XLII.
В одном варианте осуществления R1 представлен формулами:

в которых каждый из X1 и X2 независимо представляет собой водород, алкил, гало или амино; Y представляет собой водород, амино, аминоалкил, аминоциклоалкил, алкил, циклоалкил, гидрокси, алкокси, циклоалкокси, SH или тиоалкил; X является O или S; и в которых R6, R7, R8, R9 соответствуют определениям из формул XXX, XXXI, XL, XLI или XLII.
В одном варианте осуществления R1 имеет формулы:

в которых X1 является водородом, алкилом, гало или амино; Y представляет собой водород, амино, аминоалкил, аминоциклоалкил, алкил, циклоалкил, гидрокси, алкокси, циклоалкокси, SH или тиоалкил; X является O или S; и в которой R6, R7, R8, R9 соответствуют определениям из формул XXX, XXXI, XL, XLI или XLII.
В одном варианте осуществления R1 имеет формулы:

в которых R2, R3 и основание* соответствуют определениям из формул XIII, XXX, XXXI, XL, XLI или XLII.
В одном варианте осуществления R1 имеет формулы:


в которых R2, R3, Y1, Y3, X1 и X2 соответствуют определению из формулы XIII.
В одном варианте осуществления R1 имеет формулы:

в которых R2, R3, Y1, Y3, X1 и X2 соответствуют определению из формулы XIII.
В одном варианте осуществления R1 имеет формулы:

в которых R2, R3, R6, Y и X1 к соответствуют определению из формул XIII, XX, XXI или XXII.
В одном варианте осуществления, R1 имеет формулы:

в которых R2, R3, R6, R7, X и основание* соответствуют определению из формул XIII, XX, XXI, XXII, XL, XLI или XLII.
В одном варианте осуществления R1 представляет собой

в которой R8 является алкилом, алкенилом или алкинилом; R7 является OR7a;
R9 является OR7a;
R7a является H или

Rm представляет собой боковую цепь любой аминокислоты природного или синтетического происхождения; и
Rp является водородом, гидрокси, алкилом или алкокси; и
определение основания* соответствует формулам XL, XLI или XLII.
В одном варианте осуществления R8 представляет собой метил, этил, винил или этинил; R7 представляет собой гидрокси или фтор; R9 представляет собой гидрокси и другие варианты групп, описанные в настоящем изобретении.
В одном варианте осуществления R1 представляет собой

В одном варианте осуществления R8 является метилом или этилом. В одном варианте осуществления R7a представляет собой H или

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:


или является его фармацевтически приемлемой солью, сольватом или гидратом.
В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:

В одном варианте осуществления фосфорамидатное соединение согласно настоящему изобретению имеет формулу:
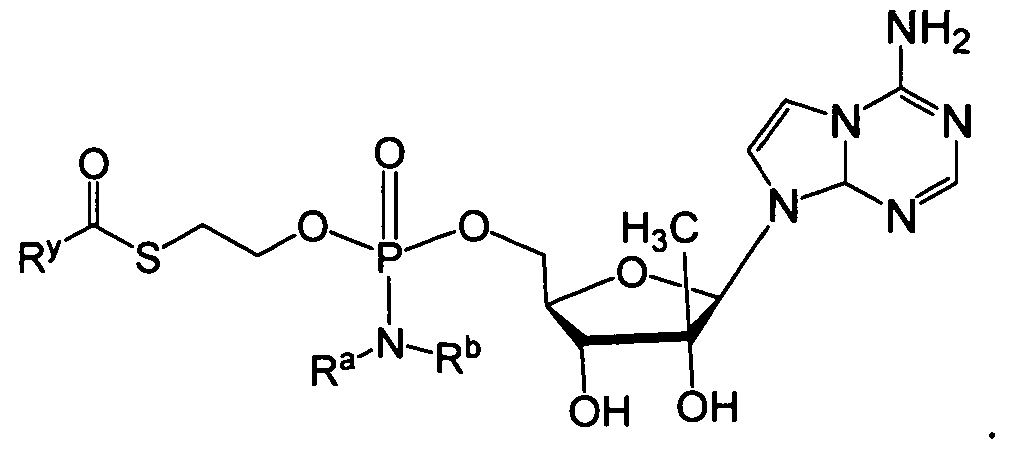
В одном варианте осуществления фосфонамидатное соединение согласно настоящему изобретению образует форму фосфонамидата PMPA (фосфонил-метоксипропил-аденин) или PMEA (фосфонил-метоксиэтил-аденин), например, имеющего формулу:

Оптически активные соединения
Предполагается, что соединения согласно настоящему изобретению имеют несколько хиральных центров и могут существовать и быть выделенными в оптически активной и рацемической формах. Некоторые соединения могут проявлять полиморфизм. Подразумевается, что любая рацемическая, оптически активная, диастереомерическая, полиморфная или стереоизомерная форма соединения согласно настоящему изобретению, или их смеси, которые обладают полезными свойствами, описанными в настоящем изобретении, входят в его объем. Способы приготовления оптически активных форм общеизвестны в данной области техники (например, приготовление посредством растворения рацемической формы способом рекристаллизации, посредством синтеза из оптически активных исходных материалов, посредством хирального синтеза или хроматографического разделения с использованием хиральной неподвижной фазы).
В частности, поскольку 1' и 4' атомы углерода в нуклеозиде являются хиральными, их неводородные заместители (соответственно, основание и группы CHOR) могут представлять собой как цис- (на одной стороне) или транс- (на противоположных сторонах) изомеры относительно кольцевой системы сахара. Таким образом, четыре оптических изомера представлены следующими конфигурациями (при ориентации сахарной функциональной группы в горизонтальной плоскости таким образом, что атом кислорода находится сзади): цис (с обеими группами «верх», что соответствует конфигурации β-D-нуклеозидов природного происхождения), цис (с обеими группами "низ", что является конфигурацией β-L синтетического происхождения), транс (с C2' заместителем «верх» и C4' заместителем "низ") и транс (с C2' заместителем "низ" и C4' заместителем «верх»). "D-нуклеозиды" представляют собой цис-нуклеозиды в конфигурации природного происхождения и "L-нуклеозиды" представляют собой цис-нуклеозиды в конфигурации синтетического происхождения.
Аналогично, большинство аминокислот являются хиральными (обозначаемыми буквами L или D, где L-энантиомер имеет конфигурацию природного происхождения), и могут существовать в виде раздельных энантиомеров.
Примеры способов получения оптически активных материалов известны в данной области техники и включают в себя по меньшей мере следующие способы:
i) физическое разделение кристаллов, что представляет собой технологию, посредством которой вручную разделяют макроскопические кристаллы отдельных энантиомеров. Эту технологию можно применять при существовании кристаллов отдельных энантиомеров, то есть, когда материал представляет собой конгломерат, и кристаллы визуально различимы;
ii) одновременная кристаллизация, что представляет собой технологию, посредством которой происходит раздельная кристаллизация отдельных энантиомеров из рацемических растворов, которая возможна только в случае, если твердая фаза рацемического раствора представляет собой конгломерат;
iii) способ ферментативных растворов, при котором происходит частичное или полное разделение рацемата на основании разной скорости реакции с ферментом для указанных энантиомеров;
iv) ферментативный асимметричный синтез, представляющий собой технологию синтеза, при котором по меньшей мере в одном этапе синтеза применяется ферментативная реакция для получения энантиомерно чистого или обогащенного синтетического предшественника желательного энантиомера;
v) химический асимметричный синтез, представляющий собой технологию синтеза, при котором желательный энантиомер синтезируют из нехирального предшественника при условиях, которые вызывают асимметрию (то есть, хиральность) продукта, что может достигаться при использовании хиральных катализаторов или хиральных вспомогательных веществ;
vi) диастереомерное разделение, представляющее собой технологию, посредством которой рацемическое соединение реагирует с энантиомерно чистым реагентом (хиральным вспомогательным веществом), который превращает раздельные энантиомеры в диастереомеры. Затем, достигнув большей степени различимости структурных отличий, получаемые диастереомеры разделяют хроматографией или кристаллизацией, после чего удаляют хиральное вспомогательное вещество для получения желательного энантиомера;
vii) асимметричные трансформации первого и второго порядка представляют собой технологию, посредством которой диастереомеры в рацемической смеси уравновешивают для достижения преобладания в растворе диастереомера желательного энантиомера, или когда предпочтительная кристаллизация диастереомера желательного энантиомера нарушает равновесие таким образом, что в результате по существу весь материал превращается в кристаллический диастереомер желательного энантиомера. Затем желательный энантиомер высвобождают из диастереомера;
viii) кинетические растворы, представляющие собой технологию, направленную на достижение частичного или полного растворения рацемата (или дальнейшего растворения частично растворенного соединения) с учетом разной скорости реакции энантиомеров с хиральным, нерацемическим реагентом или катализатором при кинетических условиях;
ix) энантиоселективный синтез из нерацемических предшественников, представляющий собой технологию синтеза, при которой желательный энантиомер получают из нехиральных исходных материалов, и при которой в ходе синтеза не происходит нарушения или только минимальное нарушение стереохимической целостности;
x) хиральная жидкостная хроматография, представляющая собой технологию, посредством которой происходит разделение энантиомеров в жидкой подвижной фазе в рацемате на основе разницы их взаимодействия с неподвижной фазой. Неподвижная фаза может быть сделана из хирального материала, или подвижная фаза может содержать дополнительный хиральный материал, чтобы вызвать различающиеся взаимодействия;
xi) хиральная газовая хроматография, представляющая собой технологию, посредством которой выпаривают рацематы и разделяют энантиомеры на основе разницы их взаимодействия в газообразной подвижной фазе при помощи колонки, содержащей неподвижную нерацемическую хиральную адсорбирующую фазу;
xii) экстракция хиральными растворителями, представляющая собой технологию, посредством которой энантиомеры разделяют на основе преимущественного растворения одного энантиомера в конкретном хиральном растворителе;
xiii) транспорт через хиральные мембраны, представляющий собой технологию, при которой рацемат располагают в контакте с тонким мембранным барьером. Барьер обычно разделяет две смешивающиеся жидкости, в одной из которых содержится рацемат, и преимущественный транспорт через мембранный барьер вызывается движущей силой, такой как разница концентрации или давления. Разделение происходит в результате нерацемического хирального строения мембраны, которое позволяет прохождение через нее только одного энантиомера из рацемата.
В некоторых вариантах осуществления рассматриваются композиции фосфонамидатных или фосфорамидатных соединений, которые по существу не содержат обозначенный энантиомер указанного нуклеозида. В предпочтительном варианте осуществления, в способах и соединениях настоящего изобретения соединения по существу не содержат энантиомеров. Некоторые варианты осуществления включают в себя композицию, которая включает в себя соединение, состоящее из по меньшей мере от 85, 90%, 95%, 98%, 99% до 100% веса указанного соединения, и остальное процентное содержание составляют другие химические виды или энантиомеры.
Получение соединений
Соединения согласно настоящему изобретению можно изготавливать, выделять или получать любым способом, очевидным для специалиста в данной области техники. Примеры способов приготовления подробно описаны ниже в разделе примеров.
В некоторых вариантах осуществления соединения согласно настоящему изобретению можно приготавливать посредством присоединения спиртов и H-фосфонатных моноэфиров, как показано ниже в схеме реакции:
Схема A

и любой реагент, действующий на Ry, R7, R8, R9, R10 или на основание, может быть защищен в ходе реакции присоединения. Можно использовать ряд веществ для присоединения, известных специалистам в данной области техники. Примеры веществ для присоединения, используемых в указанной реакции, включают в себя без ограничения HOBt (N-гидроксибензотриазол), HBTU (2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламиний гексафторфосфат), DCC (N,N'-дициклогексилкарбодиимид), BOP (бензотриазол-1-ил-окси-трис-(диметиламино)фосфонийгексафторфосфат), PyBOP (1H-бензотриазол-1-илокситрипирролидинфосфоний гексафторфосфат) и другие агенты, известные специалистам в данной области техники.
В схемах B1-B3 ниже приведена общая схема синтеза производных гидроксиtBuSATE N-бензилфосфорамидатнуклеозида, обозначенных буквой B.

в которой R является H, Tr, MMTr или DMTr в случае активного амина; R1, R2, R4, R6 является H, алкилом или гало и обе группы R3/R5 являются H или изопропилиденом.
Схема В1: Синтез H-фосфонат моноэфирного реагента

Схема B2: Синтез защищенных нуклеозидов (R=DMTr и/или R
3
/R
5
= изопропилиден)

Схема B3: Присоединение (не)защищенных нуклеозидов с реагентом 5, окислительное аминирование и этап снятия защиты

Дополнительно, некоторые нуклеозиды и их аналоги и пролекарства можно изготавливать согласно способам, описанным в патентах США №№ 6812219; 7105493; 7101861; 6914054; 6555676; 7202224; 7105499; 6777395; 6914054; 7192936; опубликованных заявках США №№ 2005203243; 2007087960; 2007060541; 2007060505; 2007060504; 2007060503; 2007060498; 2007042991; 2007042990; 2007042940; 2007042939 и 2007037735; международных опубликованных заявках №№ WO 04/003000; WO 04/022999; WO 04/002422; WO 01/90121 и WO 01/92282. Другие патенты/патентные заявки, раскрывающие нуклеозидные аналоги для лечения вируса гепатита C, которые можно получать согласно настоящему изобретению, включают в себя: PCT/CA00/01316 (WO 01/32153; зарегистрирована 3 ноября 2000 года) и PCT/CA01/00197 (WO 01/60315; зарегистрирована 19 февраля 2001 года) поданные компанией BioChem Pharma, Inc (в настоящее время Shire Biochem, Inc); PCT/US02/01531 (WO 02/057425; зарегистрирована 18 января 2002 года); PCT/US02/03086 (WO 02/057287; зарегистрирована 18 января 2002 года); заявки США №№ US 7202224; 7125855; 7105499 и 6777395 компании Merck & Co., Inc.; PCT/EP01/09633 (WO 02/18404; опубликована 21 августа 2001 года); США №№ 2006/0040890; 2005/0038240; 2004/0121980; 6846810; 6784166 и 6660721 компании Roche; опубликованные заявки РСТ №№ WO 01/79246 (зарегистрирована 13 апреля 2001 года), WO 02/32920 (зарегистрирована 18 октября 2001 года) и WO 02/48165; заявки США US 2005/0009737 и US 2005/0009737; 7094770 и 6927291 компании Pharmasset, Ltd. Содержание указанных ссылок включено в настоящее изобретение отсылочно во всей их полноте.
Способы анализа
Оценку соединений на HBV-активность можно проводить согласно любому исследованию, известному специалистам в данной области техники. Соединения можно оценивать на HCV-активность согласно любому исследованию, известному специалистам в данной области техники.
Дополнительно, можно проводить оценку соединения на его накопление в клетках печени субъекта согласно любому способу исследования, известному специалистам в данной области техники. В некоторых вариантах осуществления можно вводить соединение субъекту, и можно исследовать клетки печени субъекта на содержание соединения или его производного, например, нуклеозида, его нуклеозидфосфатного или нуклеозидтрифосфатного производного.
В одном варианте осуществления фосфорамидатное или фосфонамидатное нуклеозидное соединение вводят в клетки, такие как клетки печени, in vivo или in vitro, и для определения доставки соединения и трифосфорилирования в клетке измеряют достигнутые внутриклеточные уровни нуклеозидтрифосфата. Можно измерить уровни внутриклеточного нуклеозидтрифосфата, используя способы анализа, известные в данной области техники. Способы обнаружения ddАТФ описаны ниже в настоящем изобретении посредством примера, при этом, используя подходящие способы контроля, калибровочные пробы и методики анализа, можно легко обнаруживать другие нуклеозидтрифосфаты.
В одном варианте осуществления концентрации ddАТФ в пробе измеряют по сравнению с калибровочными стандартами, сделанным из контрольных образцов. Можно измерять уровни концентрации ddАТФ в образце, используя способ анализа, например, высокоэффективную жидкостную хроматографию - масс-спектрометрию (ВЭЖХ - LC MS). В одном варианте осуществления тестовую пробу сравнивают с калибровочной кривой, полученной с известными значениями концентрации ddАТФ, чтобы таким образом получить концентрацию в пробе.
В одном варианте осуществления пробы перед анализом обрабатывают для удаления примесей, таких как соли (Na+, K+ и т.д.). В одном варианте осуществления низшей границей количественного анализа в клеточных экстрактах гепатоцитов является концентрация около ~0,2 пмоль/мл, в частности, если присутствует восстановленная соль.
В одном варианте осуществления способ позволяет успешно измерять нуклеотидтрифосфаты, имеющие уровень концентрации от 1 до 10000 пмоль на миллион клеток, например, в культивированных гепатоцитах и клетках HepG2.
Способы применения
Можно образовывать фосфорамидатные и фосфонамидатные соединения ряда терапевтических средств, используя способы, доступные в данной области техники и раскрытые в настоящем изобретении. Такие соединения можно использовать в некоторых вариантах осуществления для увеличения доставки препарата в печень.
В одном варианте осуществления соединение включает в себя производное S-ацил-2-тиоэтилфосфорамидата или S-ацил-2-тиоэтилфосфонамидата, например, S-пивалоил-2-тиоэтилфосфорамидат или S-гидроксипивалоил-2-тиоэтилфосфонамидат. Терапевтические средства, которые можно превращать в форму фосфорамидатного или фосфонамидатного соединения, включают в себя любое противовирусное средство, которое включает в себя, или было дериватизировано, чтобы включать в себя активную группу для присоединения фосфорамидатной или фосфонамидатной функциональной группы, включающей в себя без ограничения нуклеозиды и нуклеозидные аналоги, включающие в себя нециклические нуклеозиды.
Преимущество состоит в том, что такие фосфорамидатные и фосфонамидатные соединения могут выгодно обладать увеличенной доставкой в печень. В некоторых вариантах осуществления соединения позволяют доставлять в печень активные 5'-монофосфаты нуклеозида, что способно увеличить образование активного трифосфорилированного соединения.
В одном варианте осуществления в настоящем изобретении рассматриваются способы для лечения и/или профилактики организма-хозяина, инфицированного флавивирусом, которые включают в себя введение эффективного количества соединений или их фармацевтически приемлемой соли согласно настоящему изобретению. В одном варианте осуществления настоящее изобретение обеспечивает способы лечения HCV-инфекции у субъекта. В некоторых вариантах осуществления эти способы охватывают этап введения субъекту, нуждающемуся в таком лечении, некоего количества соединения, эффективного для лечения или для профилактики HCV-инфекции в комбинации со вторым средством, эффективным для лечения или профилактики инфекции. Соединение может быть любым соединением, описанным в настоящем изобретении, и второе средство может быть любым вторым средством, описанным в настоящем изобретении или известным в данной области техники. В некоторых вариантах осуществления соединение находится в виде фармацевтической композиции или в лекарственной форме, как описано в разделах выше.
Вирусы Flaviviridae, поддающиеся лечению, в общих чертах рассмотрены в публикациях: Fields Virology, Editors: Fields, B.N., Knipe, D.M., and Howley, P.M., Lippincott-Raven Publishers, Philadelphia, PA, Chapter 31, 1996. В конкретном варианте осуществления настоящего изобретения Flaviviridae представлены вирусом HCV. В дополнительном варианте осуществления изобретения Flaviviridae являются флавивирусом или пестивирусом. Конкретно, флавивирусы включают в себя без ограничения: Absettarov, Alfuy, Apoi, Aroa, Bagaza, Banzi, Bouboui, Bussuquara, Cacipacore, Carey Island, Dakar bat, денге 1, денге 2, денге 3, денге 4, Edge Hill, Entebbe bat, Gadgets Gully, Hanzalova, Hypr, Ilheus, израильский вирус менингоэнцефалита индеек, вирус японского энцефалита, Jugra, Jutiapa, Kadam, Karshi, Kedougou, Kokobera, Koutango, Kumlinge, Kunjin, вирус Кьясанурской лесной болезни, Langat, болезни Louping, Meaban, Modoc, вирус лейкоэнцефалита Монтана миотис, энцефалита долины Мюррей, Naranjal, Negishi, Ntaya, вирус омской геморрагической лихорадки, Phnom-Penh bat, Powassan, Rio Bravo, Rocio, Royal, Farm, вирус российского весенне-летнего энцефалита, Saboya, энцефалита St-Louis, Sal Vieja, San Perlita, Saumarez Reef, Sepik, Sokuluk, Spondweni, Stratford, Tembusu, Tyuleniy, Uganda S, Usutu, Wesselsbron, вирус Западного Нила, Яунде, желтой лихорадки и Zika.
Пестивирусы, поддающиеся лечению, описаны в общих чертах в литературе: Fields Virology, Editors: Fields, B.N., Knipe, D.M., and Howley, P.M., Lippincott-Raven Publishers, Philadelphia, PA, Chapter 33, 1996. Конкретные пестивирусы включают в себя без ограничения: вирус бычьей вирусной диареи ("BVDV"), классический вирус лихорадки свиней ("CSFV", также называемый вирусом холеры свиней) и вирус пограничной болезни ("BDV").
В некоторых вариантах осуществления настоящее изобретение представляет способы лечения и/или профилактики инфекции гепатита B, которые включают в себя введение эффективного количества соединения согласно настоящему изобретению, например, соединения формулы I, IIa или IIb, его фармацевтически приемлемой соли или композиции. В другом варианте осуществления настоящее изобретение обеспечивает способы лечения и/или профилактики инфекционного гепатита B и других связанных с ним состояний, таких как состояния с положительными анти-HBV антителами и HBV-положительные состояния, хроническое воспаление печени, вызванное HBV, фиброзом, циррозом печени, острым гепатитом, скоротечным гепатитом, хроническим персистирующим гепатитом и истощением. В некоторых вариантах осуществления в настоящем изобретении рассмотрены способы профилактики для предотвращения или замедления развития клинических симптомов заболевания у людей, положительных на анти-HBV антитела или HBV-антиген, или у людей, контактировавших с HBV.
В некоторых вариантах осуществления субъект может быть любым субъектом, инфицированным или имеющим риск инфицирования вирусами HCV и/или HBV. Инфицированность или риск инфицирования можно определить с помощью любой технологии, которую практикующий специалист в данной области техники считает подходящей. В одном варианте осуществления субъектами являются люди, инфицированные HCV и/или HBV.
В некоторых вариантах осуществления субъект никогда ранее не подвергался лечению или профилактике инфекций HCV и/или HBV. В дополнительных вариантах осуществления субъект ранее получал лечение или профилактику инфекций HCV и/или HBV. Например, в некоторых вариантах осуществления субъект является резистентным к терапии против HCV и/или HBV. Например, до 50% или более субъектов с вирусом HCV являются резистентными к современной терапии интерфероном. В некоторых вариантах осуществления субъект может являться субъектом, получившим лечение, но продолжающим страдать вирусной инфекцией или одним или более ее симптомом. В некоторых вариантах осуществления субъект может представлять собой субъект, которому провели лечение, но не смогли достигнуть продолжительного вирологического ответа. В некоторых вариантах осуществления субъект получал лечение от инфекции HCV и/или HBV, но при этом не удалось показать, например, снижение 2 log10 уровня РНК HCV через 12 недель лечения. Считается, что субъекты, у которых через 12 недель лечения не выявлено снижение в сыворотке больше, чем 2 log10 уровня РНК HCV, имеют 97-100% шанс быть резистентными.
В некоторых вариантах осуществления субъектом является субъект, который прервал лечение HCV и/или HBV по причине одного или более неблагоприятных явлений, связанных с лечением. В некоторых вариантах осуществления субъектом является субъект, для которого не показано современное лечение. Например, некоторые способы лечения HCV связаны с нейропсихическими явлениями. Интерферон (ИФН)-альфа плюс рибавирин связан с высоким уровнем депрессии. Депрессивные симптомы связаны с худшим исходом ряда клинических нарушений. В процессе лечения HCV у субъектов, ранее имевших и не имевших психических нарушений, наблюдалась опасная для жизни или летальная нейропсихическая симптоматика, включающая в себя суицид, суицидальное мышление и идеи убийства, депрессию, рецидив наркомании/наркотическую передозировку и агрессивное поведение. Интерферон-индуцированная депрессия является ограничением при лечении хронического гепатита C, в частности, для субъектов с психическими нарушениями. Побочные эффекты со стороны психики часто наблюдаются при терапии интерфероном и обуславливают примерно от 10% до 20% случаев прекращения проводимого лечения инфекции HCV.
Соответственно, рассматриваются способы лечения или профилактики инфекции HCV у субъектов, у которых риск нейропсихических явлений, таких как депрессия, служит противопоказанием для современного лечения HCV. В одном варианте осуществления обеспечиваются способы лечения или профилактики инфекции HCV у субъектов, у которых нейропсихические явления, такие как депрессия, или риск такого явления, предусматривает прекращение современного лечения HCV. Дополнительно рассматриваются способы лечения или профилактики инфекции HCV у субъектов, у которых нейропсихические явления, такие как депрессия, или риск такого явления, предусматривает снижение дозировки современного лечения HCV.
Современная терапия также противопоказана субъектам, имеющим гиперчувствительность к интерферону или рибавирину, или к обоим препаратам, или к любому другому компоненту фармацевтического продукта для введения интерферона или рибавирина. Современная терапия не показана субъектам с гемоглобинопатиями (например, с большой талассемией, серповидноклеточной анемией) и другим субъектам группы риска гематологических побочных эффектов от современной терапии. Обычные гематологические побочные эффекты включают в себя супрессию костного мозга, нейтропению и тромбоцитопению. Кроме того, рибавирин проявляет токсичность по отношению к эритроцитам и связан с гемолизом. Соответственно, в одном варианте осуществления рассматриваются способы лечения или профилактики инфекции HCV у субъектов с гиперчувствительностью к интерферону или рибавирину, или к обоим препаратам, у субъектов с гемоглобинопатией, например, с большой талассемией, серповидноклеточной анемией и другим субъектам группы риска гематологических побочных эффектов от современной терапии.
В некоторых вариантах осуществления субъект получал лечение HCV и/или HBV и прекратил указанное лечение перед применением способа, рассмотренного в настоящем изобретении. В дополнительных вариантах осуществления субъект получал лечение и продолжает получать это лечение наряду с применением способа согласно настоящему изобретению. Способы можно применять совместно с другим лечением HBC и/или HCV в соответствии с мнением специалиста в данной области техники. В некоторых вариантах осуществления рассматриваемые в настоящем изобретении способы или композиции можно применять совместно с уменьшенной дозой другого препарата против HBC и/или HCV.
В некоторых вариантах осуществления рассматриваются способы лечения субъекта, резистентного к лечению интерфероном. Например, в некоторых вариантах осуществления, субъект может быть субъектом, способным реагировать на лечение одним или более средствами, выбираемыми из группы, состоящей из интерферона, интерферона α, пегилированного интерферона α, интерферона плюс рибавирин, интерферона α плюс рибавирин и пегилированного интерферона α плюс рибавирин. В некоторых вариантах осуществления субъект может быть субъектом, который слабо реагирует на лечение одним или более средствами, выбираемыми из группы, состоящей из интерферона, интерферона α, пегилированного интерферона α, интерферона плюс рибавирин, интерферона α плюс рибавирин и пегилированного интерферона α плюс рибавирин. Также можно использовать форму пролекарства рибавирина, такую как тарибавирин.
В некоторых вариантах осуществления субъект является ко-инфицированным или находится в группе риска по совместной инфекции HCV и ВИЧ. Например, в Соединенных Штатах Америки 30% субъектов с ВИЧ одновременно инфицированы HCV, и доказано, что течение инфекционного гепатита C у ВИЧ-инфицированных людей присходит намного быстрее (Maier and Wu, 2002, World J. Gastroenterol 8:577-57). Способы согласно настоящему изобретению можно использовать для лечения или профилактики HCV-инфекции у таких субъектов. Считается, что излечение HCV у таких субъектов снизит смертность по причине поражения печени в последней стадии. Доказано, что у субъектов с тяжелым СПИД-обусловленным иммунодефицитом риск прогрессирующего поражения печени выше по сравнению с субъектами без него. См., например, публикации Lesens et al., 1999, J. Infect Dis 179:1254-1258. В одном варианте осуществления показано, что соединения согласно настоящему изобретению подавляют ВИЧ у ВИЧ-инфицированных субъектов. Таким образом, в некоторых вариантах осуществления рассмотрены способы лечения или профилактики ВИЧ-инфекции и HCV-инфекции у субъектов, нуждающихся в таком лечении или профилактике.
В некоторых вариантах осуществления соединения или композиции вводят субъекту после пересадки печени. Гепатит C является ведущей причиной трансплантации печени в США, и многие субъекты, которые подвергаются трансплантации печени, остаются HCV-положительными после трансплантации. В одном варианте осуществления представлены способы лечения таких субъектов с рецидивом HCV с помощью соединения или композиции согласно настоящему изобретению. В некоторых вариантах осуществления рассмотрены способы лечения субъекта для профилактики рецидива HCV-инфекции перед трансплантацией печени, во время или после трансплантации печени.
В некоторых вариантах осуществления в настоящем изобретении рассматриваются способы лечения и/или профилактики инфекции гепатита B и других связанных с ним состояний, таких как состояний анти-HBV антитело положительной и HBV-положительной реакции, хронического воспаления печени, вызванного HCV, циррозом печени, фиброзом, острым гепатитом, скоротечным гепатитом, хроническим персистирующим гепатитом и истощением, которые включают в себя введение эффективного количества соединения или композиции согласно настоящему изобретению.
В одном варианте осуществления в настоящем изобретении рассматриваются способы лечения и/или профилактики инфекции гепатита B и других связанных с ним состояний, таких как состояний анти-HBV антитело положительной и HBV-положительной реакции, хронического воспаления печени, вызванного HCV, циррозом печени, фиброзом, острым гепатитом, скоротечным гепатитом, хроническим персистирующим гепатитом и истощением, которые включают в себя введение эффективного количества соединения или композиции согласно настоящему изобретению.
Вторые терапевтические средства
В некоторых вариантах осуществления соединения и композиции согласно настоящему изобретению являются полезными в способах лечения заболевания печени, которые содержат дополнительное введение второго средства, эффективного для лечения заболевания, такого как инфекция HCV и/или HBV у субъекта, нуждающегося в таком лечении. Второе средство может быть любым средством, известным специалисту в данной области техники в качестве эффективного для лечения указанного заболевания, включая в себя средства, одобренные в настоящее время FDA (Управлением по контролю за продуктами и лекарствами США).
В некоторых вариантах осуществления соединение согласно настоящему изобретению вводят в комбинации с одним вторым средством. В дополнительных вариантах осуществления второе средство вводят в комбинации с двумя вторыми средствами. В других дополнительных вариантах осуществления второе средство вводят в комбинации с двумя или более вторыми средствами.
Используемое в настоящем изобретении понятие "в комбинации" включает в себя использование больше чем одного лекарственного средства (например, одного или более профилактического и/или терапевтического средства). Использование понятия "в комбинации" не ограничивает порядок, в котором лекарственные средства (например, профилактические и/или терапевтические средства) вводятся больному субъекту. Первое лекарственное средство (например, профилактическое или терапевтическое средство, такое как соединение согласно настоящему изобретению) можно вводить перед введением второго лекарственного средства (например, профилактического или терапевтического средства) (например, за 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, за 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель, или за 12 недель до введения), одновременно или после введения через (например, 5 минут, 15 минут, 30 минут, 45 минут, через 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, через 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель, или через 12 недель) после введения второго лекарственного средства (например, профилактического или терапевтического средства) больному субъекту.
Используемый в настоящем изобретении термин "синергический" включает в себя комбинацию соединения согласно настоящему изобретению и другого лекарственного средства (например, профилактического или терапевтического средства), которое применяли ранее или применяют в настоящее время для профилактики, контроля или лечения болезни, и указанная комбинация более эффективна по сравнению с совокупными эффектами указанных лекарственных средств. Синергический эффект комбинации лекарственных средств (например, комбинации профилактических или терапевтических средств) позволяет применять более низкие дозы одного или более лекарственных средств и/или менее частое введение указанных лекарственных средств больному субъекту. Возможность применения более низких доз лекарственного препарата (например, профилактического или терапевтического средства) и/или менее частого введения указанного лекарственного препарата уменьшает токсичность, связанную с введением указанного препарата субъекту, не снижая эффективности указанного препарата для профилактики или лечения болезни). Дополнительно, синергический эффект может приводить к повышению эффективности препаратов в профилактике или лечении болезни. Наконец, синергический эффект комбинации лекарственных средств (например, комбинации профилактических или терапевтических средств) может избежать или уменьшить неблагоприятные или нежелательные побочные эффекты, связанные с применением только одного какого-либо лекарственного средства.
Активные соединения согласно настоящему изобретению можно вводить в комбинации или чередовании с другим терапевтическим средством, в частности, с препаратом против HCV или гепатита B. При комбинированной терапии эффективные дозы двух или более средств вводят вместе, тогда как при альтернирующей или при последовательной этапной терапии эффективные дозы каждого средства вводят периодически или последовательно. Назначаемые дозы будут зависеть от скорости абсорбции, расщепления и выделения препарата, а также от других факторов, известных специалистам в данной области техники. Необходимо отметить, что значения дозы будут также варьировать в зависимости от тяжести состояния, которое нужно облегчить. Дополнительно подразумевается, что для любого конкретного субъекта необходимо подбирать определенные режимы и схемы введения для периода времени согласно индивидуальным потребностям и профессиональному мнению человека, осуществляющему введение или контроль введения указанных композиций. В некоторых вариантах осуществления желательно введение соединения анти-HCV (или анти-пестивирусного или анти-флавивирусного) соединения, у которых значение EC50 составляет от 10 до 15 мкМ, или предпочтительно, менее чем от 1 до 5 мкМ.
Было показано, что после продолжительного лечения противовирусным средством могут возникать устойчивые к препарату варианты флавивирусов, пестивирусов или HCV-вирусов. Лекарственная устойчивость наиболее часто обусловлена мутацией гена, который кодирует фермент, используемый в репликации вируса. Эффективность лекарственного средства против вирусной инфекции можно пролонгировать, увеличивать или восстанавливать посредством применения соединения в комбинации или чередовании со вторым, и возможно, третьим противовирусным соединением, которое индуцирует другую мутацию, отличную от мутации, вызванной рассматриваемым лекарственным средством. Альтернативно, с помощью указанной комбинированной или альтернирующей терапии можно влиять на фармакокинетику, биораспределение или другой параметр лекарственного средства. В целом, обычно комбинированная терапия имеет предпочтение над альтернирующей терапией чередования, поскольку она вызывает множественные одновременные воздействия на вирус.
Любой из видов противовирусного лечения, описанных в разделе Уровень техники изобретения, можно применять в комбинации или чередовании с соединениями настоящего описания. Неограничивающие примеры вторых средств включают в себя:
Ингибиторы HCV-протеазы: Примеры включают в себя ингибитор HCV-протеазы Медивир (Медивир/Тоботек); ITMN-191 (InterMune), SCH 503034 (Schering) и VX950 (Vertex). Дополнительные примеры ингибиторов протеазы включают в себя ингибиторы NS3-протеазы на субстратной основе (Attwood et al., Antiviral peptide derivatives, PCT WO 98/22496, 1998; Attwood et al., Antiviral Chemistry and Chemotherapy 1999, 10, 259-273; Attwood et al., Preparation and use of amino acid derivatives as anti-viral agents, German Patent Pub. DE 19914474; Tung et al. Inhibitors of serine proteases, particularly hepatitis C virus NS3 protease, PCT WO 98/17679), включающие в себя альфакетонамиды и гидразинмочевины, и ингибиторы, имеющие электофильное окончание, такие как бороновая кислота или фосфонат (Llinas-Brunet et al., Hepatitis C inhibitor peptide analogues, PCT WO 99/07734); "не субстратные" ингибиторы NS3-протеазы, такие как производные 2,4,6-тригидрокси-3-нитробензамида (Sudo K. et al., Biochemical and Biophysical Research Communications, 1997, 238, 643-647; Sudo K. et al Antiviral Chemistry and Chemotherapy, 1998, 9, 186), включающие в себя RD3-4082 и RD3-4078, предварительно замещенных на амид с 14 углеродными цепочками и после этого процессированных пара-феноксифенильной группой, и фенантренхинон Sch 68631, ингибитор HCV-протеазы (Chu M. et al., Tetrahedron Letters 37:7229-7232, 1996).
В качестве ингибитора протеазы был идентифицирован SCH 351633, выделенный из гриба Penicillum griseofulvum (Chu M. et al., Bioorganic and Medicinal Chemistry Letters 9:1949-1952). Выделенный из пиявки Эглин С является мощным ингибитором ряда сериновых протеаз, таких как S. Griseus-протеаз A и B, α-химотрипсина, химазы и субтилизина. Qasim M.A. et al., Biochemistry 36:1598-1607, 1997.
Патенты США, раскрывающие ингибиторы протеазы для лечения HCV, включают в себя, например, патент США US № 6004933 авторов Spruce et al., в котором раскрыт класс ингибиторов цистеиновой протеазы для ингибирования HCV-эндопептидазы 2; патент США № 5990276 авторов Zhang et al., который раскрывает синтетические ингибиторы NS3-протеазы вируса гепатита C; патент США № 5538865 Reyes et al.; WO 02/008251 компании Corvas International, Inc, и US 7169760, US 2005/176648, WO 02/08187 и WO 02/008256 корпорации Schering. Ингибитор HCV-трипептидов раскрыт в патентах США № 6534523, 6410531 и 6420380 компании Boehringer Ingelheim, и в WO 02/060926 компании Bristol Myers Squibb. Диарил-пептиды в качестве ингибиторов сериновой NS3-протеазы HCV раскрыты в WO 02/48172 и US 6911428 корпорации Schering. Имидазолидиноны в качестве ингибиторов сериновой NS3-протеазы HCV раскрыты в WO 02/08198 и US 6838475 Корпорации Schering и WO 02/48157 и US 6727366 компании Bristol Myers Squibb. В WO 98/17679 и US 6265380 компании Vertex Pharmaceuticals и WO 02/48116 и US 6653295 компании Bristol Myers Squibb также раскрываются ингибиторы HCV-протеазы. Дополнительные примеры ингибиторов сериновой HCV-протеазы рассмотрены в US 6872805 (Bristol Myers Squibb); WO 2006000085 (Boehringer Ingelheim); US 7208600 (Vertex); US 2006/0046956 (Schering-Plough); WO 2007/001406 (Chiron); US 2005/0153877; WO 2006/119061 (Merck); WO 00/09543 (Boehringer Ingelheim), US № 6323180 (Boehringer Ingelheim), WO 03/064456 (Boehringer Ingelheim), US 6642204 (Boehringer Ingelheim), WO 03/064416 (Boehringer Ingelheim), US 7091184 (Boehringer Ingelheim), WO 03/053349 (Bristol-Myers Squibb), US 6867185, WO 03/099316 (Bristol-Myers Squibb), US 6869964, WO 03/099274 (Bristol-Myers Squibb), US 6995174, WO 2004/032827 (Bristol-Myers Squibb), US 7041698, WO 2004/043339 и US 6878722 (Bristol-Myers Squibb).
Производные тиазолидина, которые проявляют соответствующее ингибирование в анализе обращенно-фазовой ВЭЖХ со слитым белком NS3/4A и субстратом NS5A/5B (Sudo K. et al., Antiviral Research, 1996, 32, 9-18), в частности, соединение RD-1-6250, обладающее слитой циннамоил-группой, замещенной длинной алкильной цепью, RD4 6205 и RD4 6193;
Тиазолидины и бензанилиды, идентифицированные авторами Kakiuchi N. et al J. EBS Letters 421, 217-220; Takeshita N. et al Analytical Biochemistry, 1997, 247, 242-246;
Фенантренхинон, обладающий действием против протеазы в анализе SDS-PAGE (электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия) и в ауторадиографическом исследовании, выделенный из ферментативного культурального бульона Streptomyces sp., SCH 68631 (Chu M. et al., Tetrahedron Letters, 1996, 37, 7229-7232), и SCH 351633, выделенный из гриба Penicillium griseofulvum, который проявляет действие в анализе сцинтилляционной близости (Chu M. et al., Bioorganic and Medicinal Chemistry Letters 9, 1949-1952);
Ингибиторы геликазы (Diana G.D. et al., Compounds, compositions and methods for treatment of hepatitis C, U.S. Pat. No. 5633358; Diana G.D. et al., Piperidine derivatives, pharmaceutical compositions thereof and their use in the treatment of hepatitis C, PCT WO 97/36554);
Ингибиторы нуклеотид-полимеразы и глиотоксин (Ferrari R. et al Journal of Virology, 1999, 73, 1649-1654) и продукт природного происхождения церуленин (Lohmann V. et al., Virology, 1998, 249, 108-118);
Противовирусные препараты на основе интерферирующей РНК (iРНК), включающие в себя противовирусные препараты на основе короткой интерферирующей РНК (siRNA), такие как Sirna-034 и другие, описанные в международных патентных публикациях WO/03/070750 и WO 2005/012525 и патентной публикации США № 2004/0209831.
Антисмысловые фосфортиоатные олигодезоксинуклеотиды (S-ODN), комплементарные к участкам последовательностей в 5'-некодирующих областях (NCR) вируса (Alt M. et al., Hepatology, 1995, 22, 707-717), или нуклеотиды 326-348, содержащие 3'-конец NCR и нуклеотиды 371-388, расположенные в коровой кодирующей области РНК вируса HCV (Alt M. et al., Archives of Virology, 1997, 142, 589-599; Galderisi U. et al., Journal of Cellular Physiology, 1999, 181, 251-257);
Ингибиторы IRES-зависимой трансляции (Ikeda N et al., Agent for the prevention and treatment of hepatitis C, Japanese Patent Pub. JP-08268890; Kai Y. et al Prevention and treatment of viral diseases, Japanese Patent Pub. JP-10101591);
Рибозимы, такие как нуклеазо-устойчивые рибозимы (Maccjak, D.J. et al., Hepatology 1999, 30, abstract 995) и рибозимы, раскрытые в патенте США № 6043077 авторов Barber et al., и в патентах США № 5869253 и 5610054 авторов Draper et al; и
Нуклеозидные аналоги также были разработаны для лечения флавивирусных инфекций.
В некоторых вариантах осуществления соединения согласно настоящему изобретению можно вводить в комбинации с любым из соединений, описанных в международных патентных публикациях Idenix Pharmaceuticals №№ WO 01/90121, WO 01/92282, WO 2004/003000, 2004/002422 и WO 2004/002999.
Другие патентные заявки, раскрывающие использование некоторых нуклеозидных аналогов, которые можно применять в качестве вторых средств для лечения вирусного гепатита C, включают в себя: PCT/CA00/01316 (WO 01/32153; зарегистрированную 3 ноября 2000 года) и PCT/CA01/00197 (WO 01/60315; зарегистрированную 19 февраля 2001 года) зарегистрированную BioChem Pharma, Inc (в настоящее время Shire Biochem, Inc); PCT/US02/01531 (WO 02/057425; зарегистрированную 18 января 2002 года); PCT/US02/03086 (WO 02/057287; зарегистрированную 18 января 2002 года); US 7202224; 7125855; 7105499 и 6777395 компании Merck & Co., Inc.; PCT/EP01/09633 (WO 02/18404; опубликованную 21 августа 2001 года); US 2006/0040890; 2005/0038240; 2004/0121980; 6846810; 6784166 и 6660721 компании Roche; опубликованные PCT №№ WO 01/79246 (зарегистрированную 13 апреля 2001 года), WO 02/32920 (зарегистрированную 18 октября 2001 года) и WO 02/48165; US 2005/0009737; US 2005/0009737; 7094770 и 6927291 компании Pharmasset, Ltd.
Дополнительные соединения, которые можно использовать в качестве вторых средств для лечения вирусного гепатита C, раскрыты в опубликованной PCT № WO 99/43691 Университета Эмори, под названием "2'-фторнуклеозиды". Раскрыто применение некоторых 2'-фторнуклеозидов для лечения HCV.
Другие различные соединения, которые можно применять в качестве вторых средств, включают в себя 1-аминоалкилциклогексаны (патент США № 6034134 авторов Gold et al.), алкиллипиды (патент США № 5922757 авторов Chojkier et al.), витамин Е и другие антиоксиданты (патент США № 5922757 авторов Chojkier et al), сквален, амантадин, желчные кислоты (патент США № 5846964 авторов Ozeki et al.), N-(фосфонацетил)-L-аспарагиновую кислоту (патент США № 5830905 авторов Diana et al.), бензолдикарбоксамиды (патент США № 5633388 авторов Diana et al.), производные полиадениловой кислоты (патент США № 5496546 авторов Wang et al.), 2',3'-дидезоксиинозин (патент США № 5026687 авторов Yarchoan et al.), бензимидазолы (патент США № 5891874 авторов Colacino et al.), растительные экстракты (патент США № 5837257 авторов Tsai et al., патент США № 5725859 авторов Omer et al., и патент США № 6056961) и пиперидины (патент США № 5830905 авторов Diana et al.).
Примеры вторых средств для лечения HCV
В одном варианте осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с интерфероном против вируса гепатита C, таким как Интрон A® (интерферон-альфа-2b) и Пегасис® (Пегинтерферон-альфа-2a); Роферон A® (рекомбинантный интерферон-альфа-2a), Инферген® (консенсус-интерферон; интерферон альфакон-1), ПЕГ-Интрон® (пегилированный интерферон-альфа-2b) и Пегасис® (пегилированный интерферон-альфа-2a).
В одном варианте осуществления интерфероном против вируса гепатита C является инферген, ИЛ-29 (ПЕГ-интерферон лямбда), R7025 (Макси-альфа), Белерофон, пероральный интерферон-альфа, BLX-883 (Локтерон), интерферон-омега, мультиферон, медуза- интерферон, Альбуферон или REBIF®.
В одном варианте осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с ингибитором полимеразы против вируса гепатита C, таким как рибавирин, вирамидин, НМ 283 (валопицитабин), PSI-6130, R1626, HCV-796 или R7128.
В некоторых вариантах осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с рибавирином и интерфероном против вируса гепатита C, таким как Интрон A® (интерферон-альфа-2b) и Пегасис® (Пегинтерферон-альфа-2a); Роферон A® (рекомбинантный интерферон-альфа-2a), Инферген® (консенсус-интерферон; интерферон альфакон-1), ПЕГ-Интрон® (пегилированный интерферон-альфа-2b) и Пегасис® (пегилированный интерферон-альфа-2a).
В одном варианте осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с ингибитором протеазы против вируса гепатита C, таким как ингибитор протеазы ITMN-191, SCH 503034, VX950 (телапревир) или Медивир HCV.
В одном варианте осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с вакциной против вируса гепатита C, такой как TG4040, PeviPROTM, CGI-5005, HCV/MF59, GV1001, IC41 или INNO0101 (Е1).
В одном варианте осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с моноклональным антителом против вируса гепатита С, таким как AB68 или XTL-6865 (ранее называемым HepX-C); или с поликлональным антителом против вируса гепатита С, таким как цикавир (cicavir).
В одном варианте осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с иммуномодулятором против вируса гепатита C, таким как Задаксин® (тималфасин), NOV-205 или оглуфанид.
В одном варианте осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с такими препаратами как Нексавар, доксирубицин, PI-88, амантадин, JBK-122, VGX-410C, MX-3253 (Цеглосивир (Ceglosivir)), Сувус (Suvus) (BIVN-401 или виростат), PF-03491390 (ранее IDN-6556), G126270, UT-231B, DEBIO-025, EMZ702, ACH-0137171, MitoQ, ANA975, AVI-4065, Бавитуксинаб (Тарвацин), Алиниа (нитрозоксанид) или PYN17.
Примеры вторых средств для лечения HBV
Было выявлено, что после продолжительного лечения противовирусным средством могут появляться варианты HBV с лекарственной устойчивостью. Чаще всего лекарственная устойчивость возникает путем мутации гена, который кодирует фермент, задействованный в жизненном цикле вируса, и, в случае HBV, чаще всего ДНК-полимеразы. Эффективность лекарственного средства против HBV-инфекции можно пролонгировать, увеличивать или восстанавливать посредством применения соединения в комбинации или чередовании со вторым, и возможно, третьим противовирусным соединением, которое индуцирует другую мутацию, отличную от мутации, вызванной рассматриваемым лекарственным средством. Альтернативно, с помощью указанной комбинированной или альтернирующей терапии можно влиять на фармакокинетику, биораспределение или другой параметр лекарственного средства. В целом, обычно комбинированная терапия имеет предпочтение над альтернирующей терапией чередования, поскольку она вызывает множественные одновременные воздействия на вирус.
Эффективность соединений согласно настоящему изобретению против вируса гепатита B можно повышать путем применения одного или более указанных дополнительных средств в комбинации или чередовании. Альтернативно, например, одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании с любым другим известным средством против вируса гепатита B. Такие средства включают в себя интерфероны против вируса гепатита B, такие как Интрон A® (интерферон-альфа-2b) и Пегасис® (Пегинтерфен-альфа-2a); ингибиторы полимеразы, такие как Эпивир-HBV (ламивудин), Гепсера (адефовир дипивоксил), бараклуд (энтекавир), Тизека (телбивудин), эмтрицитабин (FTC), Клевудин (L-FMAU), Виред (тенофовир), Валторцитабин, Амдоксовир, ANA380, Прадефовир (ремофовир) и RCV (рацивир); вакцины, такие как Hi-8 HBV, HepaVaxx B и HBV-коровая антигенная вакцина; и другие средства, такие как HepX, SpecifEx-HepB, Задаксин, EHT899, Bay 41-4109, UT 231-B, HepeX-B и NOV-205 или любое другое соединение, EC50 которого составляет меньше 15 микромолей на 2.2.15 клеток; или их пролекарства или фармацевтически приемлемые соли. Ряд других примеров средств против HBV представлены в опубликованной заявке США № 20050080034 и международной публикации № WO 2004/096286, которые включены со ссылкой во всей их полноте.
В одном варианте осуществления одно или более соединений согласно настоящему изобретению можно вводить в комбинации или чередовании со средством против вируса гепатита B, таким как интерферон α-2b, пегинтерферон α-2a, ламивудин, гепсера, бараклуд, телбивудин, эмтрицитабин, клевудин, тенофовир, валторцитабин, амдоксовир, ANA 380, ремофовир, рацивир, алиниа, Hi-8 HBV и HepaVaxx B.
В другом варианте осуществления соединение согласно настоящему изобретению вводят в комбинации или чередовании с иммуномодулятором или другим фармацевтически активным модификатором вирусной репликации, включающим в себя биологический материал, такой как белок, пептид, олигонуклеотид, или гаммаглобулин, включающий в себя без ограничения интерферон, интерлейкин или антисмысловые олигонуклеотиды для генов, которые экспрессируют или регулируют репликацию вирусов гепатита B.
Можно применять любой способ чередования при проведении лечения больного. Неограничивающие примеры схем альтернирующей терапии включают в себя от 1 до 6 недель введения эффективного количества одного средства, с последующим введением в течение 1-6 недель лечения эффективного количества второго средства против HBV. Альтернативная схема может включать в себя периоды отсутствия лечения. Комбинированная терапия обычно включает в себя одновременное введение эффективного соотношения доз двух или более средств против HBV.
В свете того факта, что HBV часто выявляют у больных, которые также являются анти-ВИЧ антитело-положительными или ВИЧ-антиген-положительными или у лиц, находившихся в контакте с ВИЧ, эффективные соединения против HBV, раскрытые в настоящем изобретении, или их производные или пролекарства, можно вводить при подходящих обстоятельствах в комбинации или чередовании с лекарственными препаратами против ВИЧ.
Соединения согласно настоящему изобретению также можно вводить в комбинации с антибиотиками, другими противовирусными соединениями, противогрибковыми средствами или другими фармацевтическими веществами, которые применяют для лечения вторичных инфекций.
Фармацевтические композиции и способы введения
Фосфорамидатные и фосфонамидатные соединения ряда терапевтических средств можно создавать в виде рецептуры фармацевтических композиций, используя способы, доступные в данной области техники и раскрытые в настоящем изобретении. Такие соединения в некоторых вариантах осуществления можно использовать для увеличения доставки препарата в печень. В одном варианте осуществления соединение содержит производное S'-ацил-2-тиоэтилфосфорамидата или S-ацил-2-тиоэтилфосфонамидата, например, S-пивалоил-2-тиоэтилфосфорамидат или S-гидроксипивалоил-2-тиоэтилфосфонамидат. Терапевтические средства, которые можно дериватизировать до формы фосфорамидатного или фосфонамидатного соединения, включают в себя любое противовирусное средство, которое включает в себя, или было дериватизировано, чтобы включать в себя активную группу для присоединения фосфорамидатной или фосфонамидатной функциональной группы, включающий в себя без ограничения нуклеозиды и нуклеозидные аналоги, которые включают в себя нециклические нуклеозиды. Любое из фосфорамидатных или фосфонамидатных соединений, раскрытых в настоящем изобретении, можно предоставлять в подходящей фармацевтической композиции и вводить подходящим путем введения.
Способы согласно настоящему изобретению охватывают введение фармацевтических композиций, содержащих по меньшей мере одно соединение, описанное в настоящем изобретении, включающее в себя соединение с общей формулой I, IIa или IIb, в форме соли при соответствии, при этом применяемое единственным или в комбинированной форме с одним или более совместимыми и фармацевтически приемлемыми носителями, такими как разбавители или адъюванты, или с другим средством против HCV или против HBV.
В некоторых вариантах осуществления второе вещество может быть приготовлено или упаковано вместе с соединением согласно настоящему изобретению. Рецептура второго вещества, безусловно, будет создана вместе с соединением согласно настоящему изобретению, если, согласно мнению специалиста в данной области техники, такая комбинированная рецептура не нарушает или эффективность средства или способ введения. В некоторых вариантах осуществления рецептуру соединения согласно настоящему изобретению и второго средства создают раздельно. Они могут быть упакованы вместе, или упакованы раздельно, для удобства специалиста, работающего в данной области техники.
В клинической практике действующие средства согласно настоящему изобретению, можно вводить любым общепринятым путем, в частности, перорально, парентерально, ректально или путем ингаляции (например, в виде аэрозолей). В некоторых вариантах осуществления соединение согласно настоящему изобретению вводят перорально.
Можно осуществлять применение, в случае твердых композиций для перорального введения, используя таблетки, пилюли, твердые желатиновые капсулы, порошки или гранулы. В указанных композициях активный продукт смешан с одним или более инертными разбавителями или адъювантами, такими как сахароза, лактоза или крахмал.
Указанные композиции могут содержать другие вещества, кроме разбавителей, например, смазывающие вещества, такие как стеарат магния, или вещества для покрытия, предназначенные для регулируемого высвобождения.
Можно осуществлять применение, в случае жидких композиций для перорального введения, используя растворы, которые являются фармацевтически приемлемыми, суспензии, эмульсии, сиропы и эликсиры, содержащие инертные разбавители, такие как воду или жидкий вазелин. Указанные композиции могут также содержать другие вещества, кроме разбавителей, например, смачивающие, подслащивающие или ароматизирующие продукты.
Композиции для парентерального введения могут представлять собой эмульсии или стерильные растворы. Можно применять в качестве растворителя или носителя пропиленгликоль, полиэтиленгликоль, растительные масла, в частности, оливковое масло, или органические сложные эфиры для инъекций, например, этилолеат. Указанные композиции могут также содержать адъюванты, в частности, смачивающие, изотонизирующие, эмульгирующие, диспергирующие и стабилизирующие агенты. Можно осуществлять стерилизацию рядом способов, например, с использованием бактериологического фильтра, излучения или нагревания. Также указанные композиции можно изготавливать в стерильной твердой форме, и указанные композиции можно разводить для применения в стерильной воде или любой другой стерильной среде для инъекций.
Композиции для ректального введения представляют собой суппозитории или ректальные капсулы, которые содержат, в дополнение к действующему рассматриваемому соединению наполнители, такие как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Композиции могут также представлять собой аэрозоли. Композиции для применения в виде жидких аэрозолей могут представлять собой устойчивые стерильные растворы или твердые композиции, растворяемые при использовании в апирогенной стерильной воде, солевом растворе или любом другом фармацевтически приемлемом носителе. Для использования в форме сухих аэрозолей, предназначенных для непосредственной ингаляции, рассматриваемое действующее соединение подвергают тонкодисперсному измельчению и соединяют с водорастворимым твердым разбавителем или носителем, например, декстраном, маннитом или лактозой.
В одном варианте осуществления композиция согласно настоящему изобретению представляет собой фармацевтическую композицию или монолитную лекарственную форму. Фармацевтические композиции и монолитные лекарственные формы согласно настоящему изобретению содержат профилактически или терапевтически эффективное количество одного или более профилактических или терапевтических средств (например, соединение согласно настоящему изобретению или другое профилактическое или терапевтическое средство), и обычно один или более фармацевтически приемлемых носителей или наполнителей. В конкретном варианте осуществления и в этом контексте термин "фармацевтически приемлемый" означает вещества, одобренные регулирующим органом федерального правительства или правительства штата или перечисленные в Фармакопее США или другой общепринято признанной фармакопее, применяемые у животных, и более конкретно, у людей. Термин "носитель" включает в себя разбавитель, адъювант (например, адъювант Фрейнда (полный и неполный)), наполнитель или носитель, с которым вводят терапевтическое средство. Такие фармацевтические носители могут представлять собой стерильные жидкости, такие как вода и масла, включающие в себя масла нефтяного, животного, растительного или синтетического происхождения, такие как арахисовое масло, соевое масло, вазелиновое масло, кунжутное масло и тому подобные. Если фармацевтическую композицию вводят внутривенно, в качестве носителя может использоваться вода. Солевые растворы и водные растворы декстроз и глицерина также можно использовать в качестве жидких носителей, в частности, для инъекционных растворов. Примеры подходящих фармацевтических носителей описаны в издании "Remington's Pharmaceutical Sciences" автора E.W. Martin.
Обычные фармацевтические композиции и лекарственные формы содержат один или более наполнителей. Подходящие наполнители известны специалистам в области фармации; и неограничивающие примеры подходящих наполнителей включают в себя крахмал, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, мел, силикагель, стеарат натрия, моностеарат глицерина, тальк, натрия хлорид, сухое обезжиренное молоко, глицерин, пропилен, гликоль, воду, этанол и тому подобное. Приемлемость конкретного наполнителя для его включения в состав фармацевтической композиции или лекарственной формы зависит от множества факторов, известных в данной области техники, которые включают в себя без ограничений путь введения лекарственной формы субъекту и конкретные действующие компоненты лекарственной формы. Если желательно, композиция или лекарственная форма также может содержать незначительное количество смачивающих или эмульгирующих агентов, или pH-буферных агентов.
Безлактозные композиции согласно настоящему изобретению могут содержать наполнители, известные в данной области техники и перечисленные, например, в Фармакопее США (USP) SP(XXI)/NF (XVI). Обычно безлактозные композиции содержат активный компонент, связующий агент/наполнитель и лубрикант в фармацевтически совместимом и фармацевтически приемлемом количестве. Образцы лекарственных форм безлактозных композиций содержат активный компонент, микрокристаллическую целлюлозу, предварительно желатинизированный крахмал и стеарат магния.
Дополнительно в объем настоящего изобретения входят безводные фармацевтические композиции и лекарственные формы, содержащие активные компоненты, поскольку вода может облегчать расщепление некоторых соединений. Например, добавление воды (например, 5%) широко принято в фармацевтической технологии в качестве способа моделирования длительного хранения, чтобы определить такие характеристики, как срок годности или устойчивость рецептуры на протяжении времени. См., например, публикации Jens T. Carstensen, Drug Stability: Principles & Practice, 2d. Ed., Marcel Dekker, NY, NY, 1995, pp. 379 80. В действительности, вода и нагревание ускоряют разложение некоторых соединений. Таким образом, действие воды на рецептуру может иметь большое значение, поскольку влага и/или степень влажности обычно является препятствием во время изготовления, обработки, упаковки, хранения, отгрузки и использования рецептур.
Безводные фармацевтические композиции и лекарственные формы согласно настоящему изобретению можно изготавливать, используя безводные компоненты или компоненты с низким содержанием влаги, и условия с низким содержанием влаги и низкой влажностью. Фармацевтические композиции и лекарственные формы, которые содержат лактозу и, по меньшей мере, один активный компонент, который содержит первичный или вторичный амин, могут быть безводными, если предполагается значительный контакт с влагой и/или степенью влажности в ходе производства, упаковки и/или хранения.
Изготовление и хранение безводной фармацевтической композиции должно осуществляться таким образом, чтобы сохранять его безводные характеристики. Соответственно, можно упаковывать безводные композиции, используя материалы, предотвращающие воздействие воды, таким образом, чтобы было возможно включать их в подходящие наборы рецептур. Примеры подходящей упаковки включают в себя без ограничения герметично запечатанную фольгу, пластики, контейнеры для монолитных доз (например, флаконы), блистерные упаковки и упаковки ленточного типа.
Дополнительно рассматриваются фармацевтические композиции и лекарственные формы, которые содержат одно или более соединений, уменьшающих скорость разложения активного компонента. Такие соединения, которые в настоящем изобретении упоминаются, как "стабилизаторы", включают в себя без ограничения антиокислители, такие как аскорбиновая кислота, pH буферы или солевые буферы.
Фармацевтические композиции и монолитные лекарственные формы могут представлять собой растворы, суспензии, эмульсии, таблетки, пилюли, капсулы, порошки, рецептуры продолжительного высвобождения и тому подобное. Рецептура для перорального приема может включать в себя стандартные носители, такие как фармацевтические сорта маннита, лактозы, крахмала, стеарата магния, сахарина натрия, целлюлозы, карбоната магния и т.д. Такие композиции и лекарственные формы будут содержать профилактически или терапевтически эффективное количество профилактического или терапевтического средства, в некоторых вариантах осуществления, в очищенной форме, вместе с подходящим количеством носителя, с целью получения формы для надлежащего введения субъекту. Рецептура должна соответствовать способу введения. В определенном варианте осуществления фармацевтические композиции или монолитные лекарственные формы являются стерильными и имеют подходящую форму для введения субъекту, например, животному, такому, как млекопитающее, например, человек.
Рецептуру фармацевтической композиции создают таким образом, чтобы она была совместима с планируемым путем ее введения. Примеры путей введения включают в себя без ограничения парентеральное, например, внутривенное, внутрикожное, подкожное, внутримышечное, пероральное, буккальное, подъязычное введение, ингаляцию, интраназальное, чрескожное, местное, чресслизистое, внутриопухолевое, интрасиновиальное и ректальное введение. В определенном варианте осуществления рецептуру композиции создают в соответствии со стандартными процедурами в виде фармацевтической композиции, адаптированной для внутривенного, подкожного, внутримышечного, перорального, интраназального или местного введения людям. В одном варианте осуществления рецептуру фармацевтической композиции создают согласно стандартным процедурам для подкожного введения людям. Обычно, композиции для внутривенного введения представляют собой растворы в стерильном изотоническом водном буфере. При необходимости композиция может также включать в себя солюбилизирующий агент и местное анестезирующее средство, такое как лигнокаин, для ослабления боли в месте инъекции.
Примеры лекарственных форм включают в себя без ограничения: таблетки; каплеты; капсулы, такие как мягкие эластичные желатиновые капсулы; саше; пастилки; таблетки для рассасывания; дисперсии; суппозитории; мази; припарки (примочки); пасты; порошки; повязки; кремы; пластыри; растворы; наклейки; аэрозоли (например, назальные спреи или ингаляторы); гели; жидкие лекарственные формы, подходящие для перорального введения или нанесения на слизистые оболочки субъекта, включающие в себя суспензии (например, водные или неводные жидкие суспензии, эмульсии масло-в-воде или эмульсии вода-в-масле), растворы и эликсиры; жидкие лекарственные формы, подходящие для парентерального введения субъекту; и стерильные твердые формы (например, кристаллические или аморфные твердые формы), которые можно восстанавливать для получения жидких лекарственных форм, подходящих для парентерального введения субъекту.
Указанная композиция, форма и тип лекарственного препарата согласно настоящему изобретению обычно могут варьировать в зависимости от их применения. Например, лекарственная форма, применяемая на начальном этапе лечения вирусной инфекции, может содержать большее количество одного или более содержащихся в ней активных компонентов по сравнению с лекарственной формой, используемой для поддерживающего лечения при этой же инфекции. Аналогично, парентеральная лекарственная форма может содержать меньшее количество одного или более содержащихся в ней активных компонентов по сравнению с пероральной лекарственной формой, используемой для лечения этого же заболевания или нарушения. Эти и другие факторы, которые варьируют для конкретных лекарственных форм, входящих в объем настоящего изобретения, будут легко понятны специалистам в данной области техники. См., например, Remington's Pharmaceutical Sciences, 20th ed., Mack Publishing, Easton PA (2000).
В общем, компоненты композиций поставляются или раздельно или смешанными друг с другом в монолитной лекарственной форме, например, в виде сухого лиофилизированного порошка или безводного концентрата в герметичном контейнере, таком как ампула или саше с указанием количества действующего вещества. Если необходимо введение композиции путем вливания, она может быть расфасована в инфузионный флакон, содержащей стерильную воду или солевой раствор фармацевтического сорта. Если композицию вводят путем инъекции, можно обеспечивать ампулу стерильной воды или солевого раствора для инъекции с тем, чтобы была возможность смешивать компоненты перед введением.
Обычные лекарственные формы содержат соединение согласно настоящему изобретению или его фармацевтически приемлемую соль, сольват или гидрат в количестве в диапазоне от около 0,1 мг до около 1000 мг в день, вводимых в виде единственной однократной дозы один раз в день утром или в виде разделенных доз в течение дня, принимаемых с пищей. Конкретные лекарственные формы могут содержать около 0,1, 0,2, 0,3, 0,4, 0,5, 1,0, 2,0, 2,5, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 100, 200, 250, 500 или 1000 мг активного соединения.
Пероральные лекарственные формы
Фармацевтические композиции, которые подходят для перорального введения, могут представлять собой дискретные лекарственные формы, например, таблетки (например, жевательные таблетки), каплеты, капсулы и жидкости (например, ароматизированные сиропы), но не ограничены перечисленным. Такие лекарственные формы содержат заданное количество действующих компонентов и могут изготавливаться с помощью фармацевтических технологий, известных специалистам в данной области техники. Для общей информации см.: Remington's Pharmaceutical Sciences, 20th ed., Mack Publishing, Easton PA (2000).
В некоторых вариантах осуществления пероральные лекарственные формы являются твердыми и изготовляются при безводных условиях с безводными компонентами, как описано подробно в разделах выше. Вместе с тем, объем настоящего изобретения композиций охватывает не только безводные твердые пероральные лекарственные формы. То есть, в настоящем изобретении описаны дополнительные формы.
Обычные пероральные лекарственные формы изготавливают путем сочетания действующего компонента (компонентов), тщательно смешанного по меньшей мере с одним наполнителем согласно общепринятым фармацевтическим технологиям приготовления. В зависимости от желательной для введения формы приготовления можно применять широкое разнообразие наполнителей. Например, наполнители, подходящие для использования в лекарственных формах, таких как пероральные жидкие формы или аэрозоли, включают в себя без ограничения воду, гликоли, масла, спирты, ароматические агенты, консерванты и красители. Примеры наполнителей, подходящих для использования в твердых пероральных лекарственных формах (например, порошки, таблетки, капсулы и каплеты), включают в себя без ограничения крахмалы, сахар, микрокристаллическую целлюлозу, разбавители, гранулирующие агенты, лубриканты, связующие агенты и дезинтегрирующие агенты.
В силу простоты применения таблетки и капсулы обладают самым большим преимуществом в качестве пероральных монолитных лекарственных форм, и в этом случае используются твердые наполнители. Если желательно, таблетки можно покрывать с помощью стандартных водных или неводных технологий. Такие лекарственные формы можно изготавливать любой из фармацевтических технологий. В общем, фармацевтические композиции и лекарственные формы приготовляют путем однородного и тщательного смешивания активных компонентов с жидкими носителями, тонкодисперсными твердыми носителями, или с обоими видами носителей, и затем, при необходимости, создавая из них продукт с желательным внешним видом.
Например, таблетка может быть изготовлена путем прессования или формования. Прессованные таблетки можно изготавливать путем прессования в подходящей машине действующих компонентов в сыпучей форме, такой как порошок или гранулы, необязательно смешанные с наполнителем. Формованные таблетки можно изготавливать путем формования в подходящей машине смеси порошкового соединения, увлажненной инертным жидким разбавителем.
Примеры наполнителей, которые можно использовать в пероральных лекарственных формах, включают в себя без ограничения связующие агенты, наполнители, дезинтегрирующие агенты и лубриканты. Связующие агенты, подходящие для использования в фармацевтических композициях и лекарственных формах, включают в себя без ограничения кукурузный крахмал, картофельный крахмал или другие крахмалы, желатин, природную и синтетическую смолу, такую как гуммиарабик, альгинат натрия, альгиновую кислоту, другие альгинаты, порошок трагаканта, гуаровую смолу, целлюлозу и ее производные (например, этилцеллюлозу, ацетат целлюлозы, кальций карбоксиметилцеллюлозу, натрий карбоксиметилцеллюлозу), поливинилпирролидон, метилцеллюлозу, предварительно желатинизированный крахмал, гидроксипропилметилцеллюлозу (например, №№ 2208, 2906, 2910), микрокристаллическую целлюлозу и их смеси.
Примеры наполнителей, подходящих для использования в фармацевтических композициях и лекарственных формах, раскрытых в настоящем изобретении, включают в себя без ограничения тальк, кальция карбонат (например, гранулы или порошок), микрокристаллическую целлюлозу, порошковую целлюлозу, декстраты, каолин, маннит, кремниевую кислоту, сорбит, крахмал, предварительно желатинизированный крахмал и их смеси. Связующий агент или наполнитель в фармацевтических композициях обычно присутствуют в количестве от около 50 до около 99 процентов веса фармацевтической композиции или лекарственной формы.
Подходящие виды микрокристаллической целлюлозы включают в себя без ограничения материалы, имеющиеся в продаже под наименованиями AVICEL PH 101, AVICEL PH 103, AVICEL RC 581, AVICEL PH 105 (доступные в компании FMC Corporation, American Viscose Division, Avicel Sales, Marcus Hook, PA) и их смеси. Конкретный связующий агент представляет собой смесь микрокристаллической целлюлозы и натрий карбоксиметилцеллюлозы, продаваемой под названием AVICEL RC 581. Подходящие наполнители или добавки с низким содержанием влаги или безводные включают в себя AVICEL PH 103™ и крахмал Starch 1500 LM.
Дезинтегрирующие агенты используют в композициях для изготовления таблеток, которые распадаются при воздействии водной среды. Таблетки, которые содержат излишек дезинтегранта, могут распадаться при хранении, в то время как таблетки, содержащие слишком мало дезинтегранта, могут не распадаться с желательной скоростью или при желательных условиях. Таким образом, необходимо использовать достаточное количество дезинтегранта, которое не является ни чрезмерным, ни слишком малым, чтобы не нарушать высвобождение действующих компонентов для приготовления твердых пероральных лекарственных форм. Количество используемых дезинтегрантов варьирует в зависимости от типа рецептуры и является очевидным для рядового специалиста в данной области техники. Обычные фармацевтические композиции содержат от около 0,5 до около 15 процентов веса дезинтегранта, более конкретно, от около 1 до около 5 процентов веса дезинтегранта.
Дезинтегранты, которые можно использовать в фармацевтических композициях и лекарственных формах, включают в себя без ограничения, агар-агар, альгиновую кислоту, карбонат кальция, микрокристаллическую целлюлозу, натрия кроскармелозу, кросповидон, калия полакрилин, натрия крахмал гликолят, картофельный или тапиоковый крахмал, предварительно желатинизированный крахмал, другие крахмалы, глины, другие альгины, другие целлюлозы, смолы и их смеси.
Лубриканты, которые можно использовать в фармацевтических композициях и лекарственных формах, включают в себя без ограничения стеарат кальция, стеарат магния, минеральное масло, светлое минеральное масло, глицерин, сорбит, маннит, полиэтиленгликоль, другие гликоли, стеариновую кислоту, лаурилсульфат натрия, тальк, гидрогенированное растительное масло (например, арахисовое масло, хлопковое масло, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло), стеарат цинка, этилолеат, этиллаурат, агар и их смеси. Дополнительные лубриканты включают, например, силоидный силикагель (AEROSIL 200, производимый компанией W.R.Grace Co. of Baltimore, MD), коагулированный аэрозоль синтетической двуокиси кремния (выпускаемый Degussa Co. of Plano, TX), CAB O SIL (пирогенная двуокись кремния, продаваемая компанией Cabot Co., Boston, MA) и их смеси. Если используются лубриканты, они обычно вводятся в количестве менее чем около 1% вес. в расчете на фармацевтические композиции или лекарственные формы, в которые они вводятся.
Лекарственные формы с отсроченным высвобождением
Активные компоненты, такие как соединения согласно изобретению, можно доставлять посредством регулируемого высвобождения, или устройствами для доставки, известными рядовым специалистам в данной области техники. Их примеры включают в себя без ограничения способы, описанные в патентах США №№: 3845770; 3916899; 3536809; 3598123 и 4008719; 5674533; 5059595; 5591767; 5120548; 5073543; 5639476; 5354556; 5639480; 5733566; 5739108; 5891474; 5922356; 5972891; 5980945; 5993855; 6045830; 6087324; 6113943; 6197350; 6248363; 6264970; 6267981; 6376461; 6419961; 6589548; 6613358; 6699500 каждый из которых включен в настоящее изобретение в качестве ссылки. Такие лекарственные формы можно использовать для достижения замедленного или регулируемого высвобождения одного или более действующих компонентов, например, используя гидроксипропилметилцеллюлозу, другие полимерные матрицы, гели, проницаемые мембраны, осмотические системы, многослойные покрытия, микрочастицы, липосомы, микросферы или их комбинации, чтобы получить желательный профиль высвобождения с варьирующими соотношениями. Подходящие рецептуры с регулируемым высвобождением, известные рядовым специалистам в данной области техники, включают в себя рецептуры, описанные в настоящем изобретении, и их можно легко выбрать для использования с действующими компонентами согласно изобретению. Таким образом, в объем настоящего изобретения входят монолитные лекарственные формы, подходящие для перорального введения, например, таблетки, капсулы, желатиновые капсулы и каплеты, которые адаптированы для регулируемого высвобождения, но не ограничены вышеперечисленным.
Общей задачей всех фармацевтических продуктов с регулируемым высвобождением является улучшение лекарственной терапии по сравнению с их аналогами с нерегулируемым высвобождением. В идеале, использование оптимально разработанного препарата с регулируемым высвобождением в клинической практике отличается минимальным количеством лекарственной субстанции, используемой для излечения или контроля патологических состояний в течение минимального промежутка времени. Преимущества рецептур с регулируемым высвобождением включают в себя более широкое действие препарата, уменьшение частоты приема и повышение согласия субъекта. Дополнительно, рецептуры с регулируемым высвобождением можно использовать для влияния на время начала действия или другие характеристики, такие, как уровни в крови препарата, и таким образом можно влиять на возникновение побочных (например, неблагоприятных) эффектов.
Большинство рецептур с регулируемым высвобождением разработаны для начального высвобождения такого количества лекарственного средства (действующего компонента), которое быстро оказывает желательное терапевтическое воздействие, и постепенного и непрерывного высвобождения другого количества лекарственного средства для поддержания указанного уровня терапевтического или профилактического эффекта в течение длительного времени. Чтобы поддерживать указанный постоянный уровень лекарственного средства в организме, необходимо его высвобождение из лекарственной формы со скоростью, при которой количество метаболизируемого и выделяемого из организма лекарственного средства будет замещаться. Регулируемое высвобождение действующего компонента можно стимулировать разными условиями, включающими в себя без ограничения уровень pH, температуру, ферменты, воду или другие физиологические условия или соединения.
В некоторых вариантах осуществления можно вводить лекарственное средство, используя внутривенное вливание, имплантируемую осмотическую помпу, трансдермальный пластырь, липосомы или другие способы введения. В одном варианте осуществления можно использовать помпу (см. Sefton, CRC Crit. Ref. Biomed Eng. 14:201 (1987); Buchwald et ai, Surgery 88:507 (1980); Saudek et al., N. Engl. J. Med. 321:574 (1989)). В другом варианте осуществления можно использовать полимерные материалы. Еще в одном варианте осуществления систему с регулируемым высвобождением можно размещать у субъекта в подходящей локализации, которую определяет практикующий специалист, то есть, таким образом требуется только часть системной дозы (см., например, Goodson, Medical Applications of Controlled Release, vol. 2, pp. 115-138 (1984)). Другие системы с регулируемым высвобождением рассмотрены в издании Langer (Science 249:1527-1533 (1990)). Действующий компонент может быть диспергирован в твердой внутренней матрице, например, такой как полиметилметакрилат, полибутилметакрилат, пластифицированный или непластифицированный поливинилхлорид, пластифицированный нейлон, пластифицированный полиэтилентерефталат, каучук природного происхождения, полиизопрен, полиизобутилен, полибутадиен, полиэтилен, этиленвинилацетатные сополимеры, силиконовые каучуки, полидиметилсилоксаны, силиконкарбонатные сополимеры, гидрофильньные полимеры, такие как гидрогели сложных эфиров акриловой и метакриловой кислоты, коллаген, поперечно-сшитый поливинилалкоголь и поперечно-сшитый частично гидролизованный поливинилацетат, которая окружена внешней полимерной мембраной, например, такой как полиэтилен, полипропилен, сополимеры этилена/пропилена, сополимеры этилена/этилакрилата, сополимеры этилена/винилацетата, силиконовые каучуки, полидиметилсилоксаны, неопреновый каучук, хлорированный полиэтилен, поливинилхлорид, винилхлоридные сополимеры с винилацетатом, винилиденхлорид, этилен и пропилен, иономер полиэтилентерефталат, бутилкаучук эпихлоргидрин каучуки, сополимер этилена/винилалкоголя, терполимер этилена/винилацетата/винилалкоголя и сополимер этилен/винилоксиэтанола, которые нерастворимы в жидкостях организма. Затем на этапе регулируемого высвобождения действующий компонент диффундирует через внешнюю полимерную мембрану со скоростью высвобождения. Процент действующего компонента в таких парентеральных композициях в большой степени зависит от их конкретной природы, а также от потребностей субъекта.
Парентеральные лекарственные формы
В одном варианте осуществления рассматриваются парентеральные лекарственные формы. Парентеральные лекарственные формы можно вводить субъектам разными путями, включающими в себя без ограничения подкожный, внутривенный (включающий в себя болюсное вливание), внутримышечный и внутриартериальный пути введения. Поскольку такое введение обычно осуществляют сквозь естественные защитные оболочки субъектов против инфицирования, парентеральные лекарственные формы обычно являются стерильными или их можно стерилизовать перед введением субъекту. Примеры парентеральных лекарственных форм включают в себя без ограничения готовые для инъекции растворы, сухие продукты, готовые для растворения или суспендирования в фармацевтически приемлемом носителе для инъекции, готовые для инъекции суспензии и эмульсии.
Подходящие носители, которые можно использовать для парентеральных лекарственных форм, известны специалистам в данной области техники. Их примеры включают в себя без ограничения: воду для инъекции USP; водные носители, такие как натрия хлорид для инъекций, раствор Рингера для инъекций, декстрозу для инъекций, декстрозу и натрия хлорид для инъекций и раствор Рингера-лактат для инъекций, но не ограниченные перечисленным; смешивающиеся с водой носители, такие как этиловый спирт, полиэтиленгликоль и полипропиленгликоль, но не ограниченные перечисленным; и неводные носители, такие как кукурузное масло, хлопковое масло, арахисовое масло, кунжутное масло, этилолеат, изопропил миристат и бензилбензоат, но не ограниченные перечисленным.
Также в состав парентеральных лекарственных форм можно включать соединения, которые увеличивают растворимость одного или более действующих компонентов, раскрытых в настоящем изобретении.
Трансдермальные, местные лекарственные формы и формы для применения на слизистых оболочках
Также рассматриваются трансдермальные, местные лекарственные формы и формы для применения на слизистых оболочках. Трансдермальные, местные лекарственные формы и формы для слизистых включают в себя без ограничения растворы для глаз, спреи, аэрозоли, кремы, лосьоны, мази, гели, растворы, эмульсии, суспензии или другие формы, известные специалистам в данной области техники. См., например, Remington's Pharmaceutical Sciences, 16th, 18th and 20th eds., Mack Publishing, Easton PA (1980, 1990 & 2000); и Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985). Лекарственные формы, подходящие для лечения слизистой ткани в полости рта, могут представлять собой рецептуры жидкостей для полоскания рта или пероральных гелей. Дополнительно, трансдермальные лекарственные формы включают в себя пластыри "резервуарного типа" или "матричного типа", которые можно накладывать на кожу и носить в течение определенного промежутка времени для проникновения желательного количества действующих компонентов.
Подходящие наполнители (например, носители и разбавители) и другие материалы, которые можно использовать для изготовления трансдермальных, местных форм и форм для слизистых, входящие в объем настоящего изобретения, известны специалистам в области фармации и зависят от конкретной ткани, на которой будет применяться данная фармацевтическая композиция или лекарственная форма. С учетом этого факта обычные наполнители включают в себя без ограничения воду, ацетон, этанол, этиленгликоль, пропиленгликоль, бутан-1,3-диол, изопропил миристат, изопропил пальмитат, минеральное масло и их смеси для изготовления лосьонов, настоек, кремов, эмульсий, гелей или мазей, которые являются нетоксичными и фармацевтически приемлемыми. Увлажняющие или смачивающие агенты также можно добавлять к фармацевтическим композициям и лекарственным формам, если это желательно. Примеры таких дополнительных компонентов известны в данной области техники. См., например, Remington's Pharmaceutical Sciences, 16th, 18th and 20th eds., Mack Publishing, Easton PA (1980, 1990 & 2000).
В зависимости от конкретного вида ткани, предназначенной для лечения, можно применять дополнительные компоненты перед лечением, совместно или после лечения действующими рассматриваемыми компонентами. Например, можно использовать средства для усиления пенетрации, чтобы способствовать доставке действующих компонентов к тканям. Подходящие средства для усиления пенетрации включают в себя без ограничения: ацетон; разные спирты, такие как этанол, олеил и тетрагидрофурил; алкилсульфоксиды, такие как диметилсульфоксид; диметилацетамид; диметилформамид; полиэтиленгликоль; пирролидоны, такие как поливинилпирролидон; сорта Коллидона (Повидон, Поливидон); мочевину; и разные водорастворимые или нерастворимые сложные эфиры сахаров, такие как Твин 80 (полисорбат 80) и Спан 60 (сорбитан моностеарат).
Также для улучшения доставки одного или более действующих компонентов можно подбирать уровень pH фармацевтической композиции или лекарственной формы, или ткани, на которой применяется фармацевтическая композиции или лекарственная форма. Аналогично можно подбирать полярность растворителя-носителя, его ионную силу или тоничность для улучшения доставки. Также к фармацевтическим композициям или лекарственным формам можно добавлять соединения, такие как стеараты, чтобы выгодно влиять на гидрофильность или липофильность одного или более действующих компонентов для улучшения доставки. В этом отношении стеараты могут служить липидным носителем для препарата в качестве эмульгирующего средства или сурфактанта, и как средство, усиливающее пенетрацию или увеличивающее доставку. Для дополнительной адаптации свойств получаемой композиции можно использовать различные соли, гидраты или сольваты действующих компонентов.
Дозы и монолитные лекарственные формы
В клинической практике человека врач будет определять позологию исходя из наиболее подходящей и соответствующей, по его мнению, задачи профилактики или лечения и в соответствии с возрастом, весом, стадией инфекции и другими факторами, конкретными для субъекта, лечение которого планируется. В некоторых вариантах осуществления дозы составляют от около 1 до около 1000 мг в день для взрослого, или от около 5 до около 250 мг в день или от около 10 до 50 мг в день для взрослого. В некоторых вариантах осуществления дозы составляют от около 5 до около 400 мг в день или от 25 до 200 мг в день для взрослого. В некоторых вариантах осуществления также рассматриваются уровни дозы от около 50 до около 500 мг в день.
В дополнительных аспектах представлены способы лечения или профилактики HCV- и/или HBV-инфекции у субъекта посредством введения субъекту, нуждающемуся в таком лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Количество соединения или композиции, которое будет эффективно для профилактики или лечения заболевания или одного или более его симптомов, будет варьировать в зависимости от характера и степени тяжести болезни или состояния, и пути введения действующего компонента. Частота и доза также будет варьировать согласно факторам, конкретным для каждого субъекта в зависимости от конкретного вводимого препарата (например, терапевтические или профилактические средства), степени тяжести нарушения, болезни или состояния, пути введения, а также возраста, тела, веса, реакции и анамнеза субъекта. Эффективные дозы можно экстраполировать из кривых зависимости ответа от дозы, полученных in vitro или в тестовых системах на модели животных.
В некоторых вариантах осуществления примеры доз композиции включают в себя миллиграммы или микрограммы по количеству действующего соединения на килограмм веса субъекта или веса образца (например, от около 10 микрограмм на килограмм до около 50 миллиграмм на килограмм, от около 100 микрограмм на килограмм до около 25 миллиграмм на килограмм, или от около 100 микрограмм на килограмм до около 10 миллиграмм на килограмм). Для композиций согласно настоящему изобретению в некоторых вариантах осуществления вводимая субъекту доза составляет от 0,140 мг/кг до 3 мг/кг веса тела субъекта, в пересчете на вес действующего соединения. В некоторых вариантах осуществления доза, которую вводят субъекту, находится в диапазоне от 0,20 мг/кг до 2,00 мг/кг, или от 0,30 мг/кг до 1,50 мг/кг веса тела субъекта.
В некоторых вариантах осуществления рекомендуемый ежедневный диапазон дозы композиции согласно изобретению для состояний, описанных в настоящем изобретении, находится в диапазоне от около 0,1 мг до около 1000 мг в день, назначаемых в виде монолитной дозы один раз в день, или в виде разделенных доз в течение дня. В одном варианте осуществления ежедневную дозу вводят два раза в день в равных разделенных дозах. В некоторых вариантах осуществления ежедневная доза должна быть в диапазоне от около 10 мг до около 200 мг в день, в других вариантах осуществления, в диапазоне от около 10 мг до около 150 мг в день, в дополнительных вариантах осуществления в диапазоне от около 25 до около 100 мг в день. В некоторых случаях может быть необходимым применение дозировок действующего компонента вне диапазонов, раскрытых в настоящем изобретении, что будет очевидно рядовым специалистам в данной области техники. Кроме того, необходимо отметить, что клиницист или лечащий врач будет знать о том, каким образом и когда прервать, подобрать или завершить лечение в соответствии с реакцией субъекта.
Разное терапевтически эффективное количество может быть применимым для разных заболеваний и состояний, что общеизвестно рядовым специалистам в данной области техники. Аналогично, количество, достаточное для профилактики, контроля, лечения или улучшения указанных патологий, но недостаточное, чтобы вызвать отрицательные эффекты, или достаточное, чтобы уменьшить отрицательные эффекты, связанные с композицией согласно изобретению, также входят в объем описанных вышеупомянутых дозировок и схем частоты введения. Дополнительно, если субъекту вводят множественные дозы композиции согласно изобретению, не все дозы должны быть одинаковыми. Например, можно увеличивать вводимую субъекту дозу для улучшения профилактического или терапевтического эффекта композиции, или можно уменьшать дозу, чтобы уменьшить один или более побочных эффектов, которые испытывает конкретный субъект.
В определенном варианте осуществления доза композиции согласно настоящему изобретению, в пересчете на вес действующего соединения, которую вводят для профилактики, лечения, контроля или улучшения заболевания, или одного или более его симптомов у субъекта, составляет 0,1 мг/кг, 1 мг/кг, 2 мг/кг, 3 мг/кг, 4 мг/кг, 5 мг/кг, 6 мг/кг, 10 мг/кг или 15 мг/кг или более от веса тела субъекта. В другом варианте осуществления доза соединения или композиции согласно настоящему изобретению, вводимая для профилактики, лечения, контроля или улучшения заболевания, или одного или более его симптомов у субъекта, представляет собой монолитную дозу от 0,1 мг до 200 мг, от 0,1 мг до 100 мг, от 0,1 мг до 50 мг, от 0,1 мг до 25 мг, от 0,1 мг до 20 мг, от 0,1 мг до 15 мг, от 0,1 мг до 10 мг, от 0,1 мг до 7,5 мг, от 0,1 мг до 5 мг, от 0,1 до 2,5 мг, от 0,25 мг до 20 мг, от 0,25 до 15 мг, от 0,25 до 12 мг, от 0,25 до 10 мг, от 0,25 мг до 7,5 мг, от 0,25 мг до 5 мг, от 0,5 мг до 2,5 мг, от 1 мг до 20 мг, от 1 мг до 15 мг, от 1 мг до 12 мг, от 1 мг до 10 мг, от 1 мг до 7,5 мг, от 1 мг до 5 мг или от 1 мг до 2,5 мг.
В некоторых вариантах осуществления можно начинать лечение или профилактику с одной или более нагрузочной дозы соединения или композиции согласно изобретению с последующим применением одной или более поддерживающей дозы. В таких вариантах осуществления доза нагрузки может, например, составлять от около 60 до около 400 мг в день или от около 100 до около 200 мг в день в течение от одного дня до пяти недель. После дозы нагрузки может применяться одна или более поддерживающих доз. В некоторых вариантах осуществления каждая поддерживающая доза независимо составляет от около 10 мг до около 200 мг в день, в диапазоне от около 25 мг до около 150 мг в день или от около 25 до около 80 мг в день. Поддерживающие дозы можно вводить ежедневно и можно вводить в виде монолитной дозы или в виде разделенных доз.
В некоторых вариантах осуществления дозу соединения или композиции согласно изобретению можно вводить для достижения устойчивой концентрации действующего компонента в крови или сыворотке субъекта. Устойчивую концентрацию можно определять путем измерения согласно способам, доступным специалистам в данной области, или на основании физических характеристик субъекта, таких как рост, вес и возраст. В некоторых вариантах осуществления вводят достаточное количество соединения или композиции согласно изобретению для достижения устойчивой концентрации в крови или сыворотке субъекта от около 300 до около 4000 нг/мл, от около 400 до около 1600 нг/мл или от около 600 до около 1200 нг/мл. В некоторых вариантах осуществления можно вводить нагрузочную дозу для достижения постоянной концентрации в крови или сыворотке от около 1200 до около 8000 нг/мл, или от около 2000 до около 4000 нг/мл в течение от одного до пяти дней. В некоторых вариантах осуществления можно вводить поддерживающие дозы, чтобы достигнуть устойчивой концентрации в крови или сыворотке субъекта от около 300 до около 4000 нг/мл, от около 400 до около 1600 нг/мл или от около 600 до около 1200 нг/мл.
В некоторых вариантах осуществления введение одной и той же композиции можно повторять, и можно осуществлять введения с промежутком по меньшей мере в 1 день, 2 дня, 3 дня, 5 дней, 10 дней, 15 дней, 30 дней, 45 дней, 2 месяца, 75 дней, 3 месяца или 6 месяцев. В других вариантах осуществления можно повторно вводить одно и тоже профилактическое или терапевтическое средство, и введение можно проводить с промежутками по меньшей мере в 1 день, 2 дня, 3 дня, 5 дней, 10 дней, 15 дней, 30 дней, 45 дней, 2 месяца, 75 дней, 3 месяца или 6 месяцев.
В некоторых аспектах в настоящем изобретении рассматриваются монолитные лекарственные формы, содержащие соединение или его фармацевтически приемлемую соль в подходящей для введения форме. Подробное описание таких форм приведено выше. В некоторых вариантах осуществления монолитная доза содержит действующий компонент в количестве от 1 до 1000 мг, от 5 до 250 мг, от 10 до 50 мг. В конкретных вариантах осуществления монолитные дозы содержат около 1, 5, 10, 25, 50, 100, 125, 250, 500 или 1000 мг действующего компонента. Такие монолитные дозы можно изготавливать согласно способам, известным специалистам в данной области техники.
При комбинированном лечении согласно настоящему изобретению должны применяться дозы вторых средств. В некоторых вариантах осуществления применяют дозы ниже тех, которые применялись ранее или применяют в настоящее время для профилактики или лечения HCV- и/или HBV-инфекции, и их применяют при комбинированном лечении согласно настоящему изобретению. Информацию о рекомендованных дозах вторых средств можно получить из источников для специалистов в данной области. Для указанных вторых веществ, одобренных для клинического применения, рекомендованные дозы указаны, например, в изданиях: Hardman et al., eds., 1996, Goodman & Gilman's The Pharmacological Basis Of Basis Of Therapeutics 9th Ed, Mc-Graw-Hill, New York; Physician's Desk Reference (PDR) 57th Ed., 2003, Medical Economics Co., Inc., Montvale, NJ, которые включены в настоящее изобретение в качестве ссылки во всей их полноте.
В разных вариантах осуществления препараты (например, соединение согласно изобретению и второе средство) вводят с промежутком менее 5 минут, менее 30 минут, с промежутком 1 час, с промежутком около 1 часа, с промежутком от около 1 до около 2 часов, с промежутком от около 2 часов до около 3 часов, с промежутком от около 3 часов до около 4 часов, с промежутком от около 4 часов до около 5 часов, с промежутком от около 5 часов до около 6 часов, с промежутком от около 6 часов до около 7 часов, с промежутком от около 7 часов до около 8 часов, с промежутком от около 8 часов до около 9 часов, с промежутком от около 9 часов до около 10 часов, с промежутком от около 10 часов до около 11 часов, с промежутком от около 11 часов до около 12 часов, с промежутком от около 12 часов до 18 часов, с промежутком от 18 часов до 24 часов, с промежутком от 24 часов до 36 часов, с промежутком от 36 часов до 48 часов, с промежутком от 48 часов до 52 часов, с промежутком от 52 часов до 60 часов, с промежутком от 60 часов до 72 часов, с промежутком от 72 часов до 84 часов, с промежутком от 84 часов до 96 часов или с промежутком от 96 часов до 120 часов. В разных вариантах осуществления препараты вводят с промежутком не более 24 часов или с промежутком не более 48 часов. В некоторых вариантах осуществления два или более препарата вводят во время одного посещения больного. В других вариантах соединение согласно настоящему изобретению и второе средство вводят одновременно.
В других вариантах осуществления соединение согласно изобретению и второе средство вводят с промежутком около 2-4 дней, с промежутком около 4-6 дней, с промежутком около 1 недели, с промежутком около 1-2 недель или с промежутком больше 2 недель.
В некоторых вариантах осуществления можно повторно вводить один и тот же препарат, и введение можно проводить с промежутком по меньшей мере в 1 день, 2 дня, 3 дня, 5 дней, 10 дней, 15 дней, 30 дней, 45 дней, 2 месяца, 75 дней, 3 месяца или 6 месяцев. В других вариантах осуществления можно повторно вводить один и тот же препарат, и введение можно проводить с промежутком по меньшей мере в 1 день, 2 дня, 3 дня, 5 дней, 10 дней, 15 дней, 30 дней, 45 дней, 2 месяца, 75 дней, 3 месяца или 6 месяцев.
В некоторых вариантах осуществления соединение согласно изобретению и второе средство вводят пациенту, например, млекопитающему, такому как человек, последовательно и в пределах интервала времени таким образом, что соединение согласно изобретению может действовать совместно с другим средством, чтобы повысить преимущество по сравнению с введением другим способом. Например, второе активное вещество можно вводить одновременно или последовательно в любом порядке в разные моменты времени; вместе с тем, если их введение происходит не одновременно, их необходимо вводить достаточно близко по времени, чтобы обеспечить желательный терапевтический или профилактический эффект. В одном варианте осуществления соединение согласно изобретению и второе активное средство проявляют свои эффекты в перекрывающихся интервалах времени. Каждое из вторых активных веществ можно вводить отдельно, в любой подходящей форме и любым путем введения. В других вариантах осуществления соединение согласно изобретению вводят перед введением второго действующего средства, одновременно или после его введения.
В некоторых вариантах соединение согласно изобретению и второе средство вводят больному циклами. Циклическая терапия охватывает введение первого средства (например, первых профилактических или терапевтических средств) в течение периода времени, с последующим введением второго средства и/или третьего средства (например, вторых и/или третьих профилактических или терапевтических средств) в течение периода времени и повторение указанной последовательности введения. Циклическая терапия может уменьшать развитие резистентности к одному или более препаратам, избежать или уменьшить побочные эффекты одного из препаратов и/или повысить эффективность лечения.
В некоторых вариантах осуществления соединение согласно изобретению и второе действующее средство вводят циклами короче 3 недель, примерно однократно каждые две недели, примерно однократно каждые 10 дней или примерно однократно каждую неделю. Один цикл может содержать введение соединения согласно изобретению и второго средства путем вливания в течение около 90 минут в каждом цикле, около 1 часа в каждом цикле, около 45 минут в каждом цикле. Каждый цикл может содержать по меньшей мере 1 неделю перерыва, по меньшей мере 2 недели перерыва, по меньшей мере 3 недели перерыва. Количество циклов введения составляет от около 1 до около 12 циклов, более часто, от около 2 до около 10 циклов и более часто, от около 2 до около 8 циклов.
В других вариантах осуществления курсы препаратов вводят больному конкурентно, то есть, отдельные дозы второго средства вводят раздельно, но в пределах такого временного интервала, что соединение согласно настоящему изобретению может действовать совместно со вторым активным средством. Например, один компонент можно вводить однократно в неделю в комбинации с другими компонентами, которые можно вводить однократно каждые две недели или однократно каждые три недели. Другими словами, схемы введения являются конкурентными, даже если введение препаратов происходит не одновременно или в течение одного дня.
Второе средство может действовать аддитивно или синергически с соединением согласно настоящему изобретению. В одном варианте осуществления соединение согласно изобретению вводят одновременно с одним или более вторым средствам в одной фармацевтической композиции. В другом варианте осуществления соединение согласно настоящему изобретению вводят одновременно с одним или более вторым средствам в разных фармацевтических композициях. Еще в одном варианте осуществления соединение согласно настоящему изобретению вводят перед введением второго средства или после этого. Также рассматривается введение соединения согласно изобретению и второго средства одинаковыми или разными путями введения, например, перорально и парентерально. В некоторых вариантах осуществления, когда соединение согласно настоящему изобретению вводят конкурентно со вторым средством, что потенциально вызывает неблагоприятные побочные эффекты, включающие в себя без ограничения токсичность, будет обладать преимуществом введение второго действующего вещества в дозе ниже порога возникновения неблагоприятного побочного эффекта.
Наборы
Также рассматриваются наборы, применяемые для способов лечения заболеваний печени, таких как HCV- и/или HBV-инфекции. Наборы могут включать в себя соединение или композицию согласно настоящему изобретению, второе средство или композицию и инструкции, в которых представлена информация относительно использования при лечении заболевания для специалиста, осуществляющего медицинскую помощь. Инструкции могут быть предоставлены в печатной форме или в виде электронного носителя, такого как дискета, компакт-диск или DVD, или в виде адреса вебсайта, где можно найти указанные инструкции. Монолитная доза соединения или композиции согласно настоящему изобретению, или второго средства или композиции, может включать такую дозировку, которая при введении субъекту может поддерживать терапевтически или профилактически эффективный уровень соединения или композиции в плазме у субъекта в течение по меньшей мере 1 дня. В некоторых вариантах осуществления соединение или композиция могут входить в состав стерильной водной фармацевтической композиции или в состав фармацевтической композиции сухого порошка (например, лиофилизированного).
В некоторых вариантах осуществления предоставляется подходящая упаковка. Используемое в настоящем изобретении понятие "упаковка" включает в себя твердую матрицу или материал, общепринято используемый в системе и способный к удерживанию в пределах фиксированных границ соединение согласно настоящему изобретению и/или второе средство, подходящие для введения субъекту. Такие материалы включают в себя стеклянные и пластиковые (например, полиэтиленовые, полипропиленовые и поликарбонатные) флаконы, пробирки, конверты из бумаги, пластика и из ламинированной полимерной пленки и тому подобное. Если применяют способы электронно-лучевой стерилизации, упаковка должна иметь достаточно низкую плотность, позволяющую стерилизацию содержимого.
Далее приведены примеры, показывающие синтез примеров соединений согласно настоящему изобретению. Эти примеры не предназначены для ограничения объема заявленного объекта изобретения и их не следует толковать в этом аспекте. Очевидно, что заявленный объект изобретения в приведенном объеме можно осуществлять иным образом, чем описано, в частности, в настоящем изобретении. Возможно множество модификаций и вариантов объекта изобретения в свете его теории и, таким образом, они входят в объем объекта изобретения.
ПРИМЕРЫ
Пример 1
Приготовление A550 (NM204), производного гидрокси-tBuSATE N-бензилфосфорамидата L-2',3'-дидезоксиаденозина L-ddA

СХЕМА СИНТЕЗА:

Синтез карбоновой кислоты 2:

Метиловый сложный эфир 2,2-диметил-3-гидроксипропионовой кислоты (965 мкл, 7,57 ммоль) добавляли по капле к перемешиваемому раствору 4,4'-диметокситритил хлорида (2,82 г, 8,33 ммоль) в безводном пиридине (7,6 мл) при комнатной температуре. Реакционная смесь быстро превращалась в раствор красного цвета, затем в суспензию оранжевого цвета (около 30 минут), и ее оставляли для перемешивания в течение ночи. Смесь аккуратно вливали в насыщенный водный раствор NaHCO3 (30 мл) и продукт экстрагировали Et2O (3×20 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), высушивали (Na2SO4), и удаляли летучие компоненты при пониженном давлении. Получаемое масло выпаривали совместно с толуолом, и остаток быстро очищали колоночной флэш-хроматографией (SiO2, ⌀=4 см, H=20 см) с элюированием 5→10→20→30% Et2O в петролейном эфире (40-60). Испарение фракций (Rf=0,25, 30% Et2O в петролейном эфире (40-60)) давало эфир 1 в виде масла желтого цвета (3,11 г, 95%). Это соединение (3,00 г, 6,91 ммоль) растворяли в ТГФ (35 мл), и затем при комнатной температуре добавляли водный раствор NaOH (10%, 3,5 г в H2O, 35 мл). Цвет раствора становился темно-оранжевым, и его перемешивали в течение 2 дней. Затем тщательно нейтрализовали среду путем добавления по капле HCl (1M). Продукт экстрагировали Et2O (4×50 мл), и объединенные органические экстракты промывали солевым раствором (50 мл), высушивали (Na2SO4), и летучие компоненты удаляли при пониженном давлении. Сырое масло желтого цвета быстро очищали колоночной флэш-хроматографией (SiO2, ⌀=2 см, H=10 см) с элюированием 50% Et2O в петролейном эфире (40-60). Выпаривание фракций давало карбоновую кислоту 2 в виде пены белого цвета (2,23 г, 77%). Rf=0,50 (50% Et2O в петролейном эфире (40-60)); 1H-ЯМР (300 МГц, CDCl3) 1,10 (с, 6H, 2×CH3), 3,06 (с, 2H, CH2O), 3,65 (с, 6H, 2×OCH3), 6,62-6,79 (м, 4H, PhCH), 7,02-7,46 (стек, 8H, PhCH); 13C-ЯМР (75 МГц, CDCl3) 22,6 (2×CH3), 43,5 (C(CH3)2), 55,1 (2×OCH3), 85,9 (CPh3), [125,3, 126,7, 127,7, 128,2, 129,1, 130,0, 136,0, 144,9, 158,4 (Ph), некоторое наложение], 182,2 (C=O).
Синтез тиоэфира 3:

1,1'-карбонилдиимидазол (830 мг, 5,12 ммоль) добавляли к перемешиваемому раствору карбоновой кислоты 2 в безводном PhMe/ДМФ (2/1, об/об, 2,7 мл) при комнатной температуре и реакционная смесь немедленно становилась мутной. Через 30 минут среду разводили путем добавления безводного PhMe/ДМФ (93/7, об/об, 17 мл) и охлаждали до -10°C. Затем по капле добавляли 2-меркаптоэтанол (359 мкл, 5,12 ммоль), и раствор перемешивали в течение 1 часа при указанной температуре. Реакционную смесь разводили H2O (60 мл), и продукт экстрагировали Et2O (3×15 мл). Объединенные органические экстракты промывали солевым раствором (15 мл), высушивали (Na2SO4), и летучие компоненты удаляли при пониженном давлении (температура ванны не превышала 20°C). Остаток очищали колоночной флэш-хроматографией (SiO2, ⌀=4 см, H=15 см, 1% Et3N) с элюированием 60→70% Et2O в петролейном эфире (40-60). Выпаривание фракций давало тиоэфир 3 в виде сиропа белого цвета (1,74 г, 92%), который затвердевал при хранении при 4°C. Rf=0,35 (70% Et2O в петролейном эфире (40-60)); 1H-ЯМР (300 МГц, CDCl3) 1,16 (с, 6H, 2×CH3), 3,02 (т, J=6,0, 2H, CH2S), 3,09 (с, 2H, CH2O), 3,66 (т, J=6,0, 2H, CH2OH), 3,72 (с, 6H, 2×OCH3), 6,74-6,78 (м, 4H, PhCH), 7,09-7,36 (стек, 8H, PhCH); 13C-ЯМР (75 МГц, CDCl3) 22,9 (CH3, 2×CH3), 31,7 (CH2, CH2S), 51,0 (кв, C, C(CH3)2), 55,2 (CH3, 2×OCH3), 61,9 (CH2, CH2OH), 70,0 (CH2, CH2O), 85,8 (кв, C, CPh3), [113,0 (CH, Ph), 126,7 (CH, Ph), 127,7 (CH, Ph), 128,2 (CH, Ph), 130,1 (CH, Ph), некоторое наложение], [135,9 (кв, C, Ph), 144,8 (кв, C, Ph), 158,4 (кв, C, Ph), некоторое наложение], 205,0 (кв, C, C=O).
Синтез H-фосфонат-моноэфира 4:

β-L-ddA (1,00 г, 4,25 ммоль) совместно выпаривали с безводным пиридином (3×10 мл) и затем растворяли в безводном пиридине/ДМФ (1/1, об/об, 21 мл). Затем при комнатной температуре к этому раствору по капле добавляли дифенилфосфит (5,76 мл, 29,8 ммоль). Реакционную смесь перемешивали в течение 20 минут, после чего по капле добавляли смесь Et3N/H2O (1/1, об/об, 8,5 мл), и продолжали взбалтывать дополнительно еще 20 мин. Реакционную смесь выпаривали при пониженном давлении примерно до объема 15-20 мл, и указанный остаток непосредственно очищали колоночной флэш-хроматографией (SiO2, ⌀=4 см, H=15 см, 1% Et3N) с медленным элюированием CH2Cl2 (150 мл), затем 5% (200 мл) → 10% (200 мл) → 15%-ый MeOH (300 мл) в CH2Cl2. Выпаривание фракций давало H-фосфонат моноэфир 4 в виде пены белого цвета (1,36 г, 80%), которая могла сохраняться в течение нескольких недель при 4°C. Rf=0,10 (Et3N/MeOH/CH2Cl2, 1/10/89); 1H-ЯМР (300 МГц, CDCl3) 1,21 (т, J=7,4, 9H, 3×NCH2CH3), 1,92-2,50 (стек, 4H, 2×2'-H, 2×3'-H), 3,02 (кв, J=7,4, 6H, 3×NCH2CH3), [3,96-4,03 и 4,18-4,30 (стеки, 3H, 4'-H, 2×5'-H), 6,28 (м, 1'-H), 6,91 (д, J=623, 1H, P-H), 7,05 (ушир.с, 2H, NH2), 8,21 (с, 1H), 8,54 (ушир.с, 1H, OH), 8,57 (с, 1H).
Синтез фосфорамидатдиэфира 5

H-фосфонат моноэфир 4 (1,03 г, 2,57 ммоль) и спирт 3 (1,66 г, 3,45 ммоль) совместно выпаривали с безводным пиридином (3×5 мл) и затем растворяли в безводном пиридине (5 мл). Затем добавляли PyBOP (1H-бензотриазол-1-илокситрипирролидинфосфоний гексафторфосфат, 1,60 г, 3,08 ммоль) одной порцией, и реакционную смесь перемешивали в течение 15 минут при комнатной температуре. Раствор вливали в насыщенный водный раствор NaHCO3 (30 мл), и продукт экстрагировали CH2Cl2 (4×15 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), высушивали (Na2SO4) и концентрировали при пониженном давлении, чтобы остался соответствующий H-фосфонат диэфир в виде масла желтоватого цвета (1,84 г, принимая, что 2,41 ммоль). Его совместно выпаривали с безводным пиридином (3×5 мл; примечание: не выпаривать досуха, чтобы в дальнейшем облегчить солюбилизацию), и остаток растворяли в безводном CCl4 (24 мл). Бензиламин (791 мкл, 7,23 ммоль) добавляли по капле, и реакционная смесь немедленно становилась мутной (наблюдалось небольшое повышение температуры). Мутный раствор перемешивали в течение 1 часа при комнатной температуре и вливали в насыщенный водный раствор NaHCO3 (30 мл), и продукт экстрагировали CH2Cl2 (4×15 мл). Объединенные органические экстракты промывали солевым раствором (15 мл), высушивали (Na2SO4) и концентрировали при пониженном давлении, чтобы получить фосфорамидат диэфир 5 в виде масла желтого цвета (2,00 г, принимая количество 2,31 ммоль). Его использовали в следующем этапе без дополнительной очистки. Rf=0,29 (4% MeOH в CH2Cl2); 1H-ЯМР (300 МГц, CDCl3) 1,11 (с, 6H, 2×CH3), 1,91-2,05 (м, 2H), 2,31-2,59 (м, 2H), 3,06 (м, 2H, CH2S), 3,08 (с, 2H, CH2ODMTr), 3,69 (с, 6H, 2×OCH3), 3,83-4,28 (стеки, 7H, CH2O, NCH2Ph, 4'-H, 2×5'-H), 5,71 (ушир.с, 1H, NH), 6,18 (м, 1H, 1'-H), 6,69-6,80 (м, 4H, PhCH), 7,02-7,31 (стек, 13H, PhCH), 7,90 (с, 1H), 8,01 (с, 1H), 8,23 (с, 2H, NH2); 13P-ЯМР (61 МГц, CDCl3) 8,82, 8,99.
Синтез NM204 (A550), производного гидрокси-tBuSATE N-бензилфосфорамидата L-ddA:

Неочищенный фосфорамидат диэфир 5 (2,00 г, принимая, что 2,31 ммоль) растворяли в диоксане/AcOH/H2O (25/17/25, об/об/об, 462 мл), и раствор перемешивали в течение 3 дней при комнатной температуре. Выпаривание летучих компонентов при пониженном давлении давало остаток, который очищали колоночной флэш-хроматографией (SiO2, ⌀=3 см, H=15 см) с элюированием CH2Cl2 (100 мл), затем 2% (100 мл) → 4% (100 мл) → 6% (100 мл) → 8% MeOH (150 мл) в CH2Cl2. Выпаривание фракций давало остаток NM204 в виде пены белого цвета, которую растворяли в MeCN (5 мл). После добавления H2O (5 мл) раствор становился мутным, и перед лиофилизацией была необходима обработка ультразвуком. Получаемый белый порошок высушивали при комнатной температуре (с использованием в качестве осушителя P2O5) в вакууме в течение 1 дня. Получали названное соединение в виде высоко гигроскопичного порошка белого цвета (смесь диастереоизомеров 1:1, что выявляли с помощью 31P-ЯМР; 499 мг, 35% после 3 этапа). [a]20 D=+4,2° (c 1,0, CHCl3); Rf=0,29 (4% MeOH в CH2C12); 1H-ЯМР (300 МГц, ДМСО-d6) 1,10 (с, 6H, 2×CH3), 2,02-2,14 (м, 2H, 2×3'-H), 2,41-2,55 (м, 2H, 2×2'-H), 3,01 (т, J=6,4, 2H, CH2S), 3,43 (д, J=5,0, 2H, CH2OH), 3,75-4,07 и 4,18-4,29 (стеки, 7H, CH2O, NCH2Ph, 4'-H, 2×5'-H), 5,02 (т, J=5,0, 1H, OH), 5,62 (м, 1H, NH), 6,25 (т, J=5,1, 1H, 1'-H), 7,16-7,36 (стек, 7H, PhH, NH2), 8,14 (с, 1H), 8,26 (с, 1H); 13C-ЯМР (75 МГц, ДМСО-d6) 21,8 (2×CH3), 25,9 и 26,0 (CH2, 3'-C), 28,2 и 28,3 (CH2, CH2S), 30,9 и 31,0 (CH2, 2'-C), 44,2 (CH2, NCH2Ph), 51,7 (кв, C, C(CH3)2), 63,7 и 63,8 (CH2, CH2O), 66,8 (CH2, м, 5'-C), 68,3 (CH2, CH2OH), 78,9 (CH, м, 4'-C), 84,2 (CH, 1'-C), 118,9 (кв, C), [126,5 (CH, Ph), 127,2 (CH, Ph), 128,1 (CH, Ph), некоторое наложение], 138,8 и 138,9 (CH), 140,5 и 140,6 (кв, C), 148,9 (кв, C), 152,3 (CH), 155,0 (кв, C), 204,0 (кв, C, C=O); 13P-ЯМР (61 МГц, ДМСО-d6) 9,86, 9,95; m/z (FAB') 563 (2), 306 (76), 153 (100); HRMS 565,2034 ([M+H]+, C24H34O6N6PS требует 565,1998); ВЭЖХ tR=3,52 мин (20% TEAC 20 мМ в MeCN); УФ (EtOH 95%) λмакс=259 (εмакс 15900), λмин.=224 (εмин. 7200).
Пример 2
Приготовление B102, производного гидрокси-tBuSATE N-бензилфосфорамидата 2'-C-метилцитидина:

ПРОЦЕДУРА A:
Синтез H-фосфонат моноэфира 5

Синтез карбоновой кислоты 3:
К размешиваемому раствору метилэфира 2,2-диметил-3-гидроксипропионовой кислоты (1, 15 мл, 117,6 ммоль) в смеси безводного метиленхлорида (590 мл) и триэтиламина (23 мл), добавляли трифенилметиленхлорид (1,2 экв., 39,3 г) и 4-диметиламинопиридин (0,1 экв, 1,44 г). Реакционную смесь оставляли нагреваться с обратным холодильником в течение ночи. Смесь аккуратно вливали в насыщенный водный раствор NaHCO3, и продукт экстрагировали метиленхлоридом и промывали водой. Объединенные органические экстракты выпаривали при пониженном давлении, чтобы получить неочищенное соединение 2, которое будет использоваться в следующем этапе без дополнительной очистки. Получаемое масло растворяли в смеси диоксана (350 мл) и водного раствора NaOH (30%, 350 мл). Гетерогенную смесь нагревали с обратным холодильником в течение 16 часов. Реакционную смесь оставляли для остывания при комнатной температуре, разделяли две фазы, и органическую фазу аккуратно нейтрализовывали добавлением по капле HCl (1M). Продукт экстрагировали метиленхлоридом, и органические фазы выпаривали при пониженном давлении. Осуществляли рекристаллизацию неочищенного масла оранжевого цвета из метиленхлорида для получения карбоновой кислоты 3 в виде кристаллов белого цвета (92%). Rf=0,50 (70% диэтиловый эфир в петролейном эфире); 1H-ЯМР (400 МГц, CDCl3) 1,24 (с, 6H, 2×CH3), 3,19 (с, 2H, CH2O), 7,2-7,5 (м, 15Η, C6H5).
Синтез H-фосфонат моноэфира 5:
1,1'-карбонилдиимидазол (1,3 экв., 1,17 г) добавляли к перемешиваемому раствору карбоновой кислоты 3 (2 г, 5,56 ммоль) в безводной смеси толуола и диметилформамида (2/1, об/об, 4,5 мл) при комнатной температуре, и реакционная смесь быстро становилась мутной. Через 30 минут реакционную смесь разводили смесью толуола и диметилформамида (93/7, об/об, 28 мл), охлаждали до -10°C и добавляли 2-меркаптоэтанол (1,3 экв., 500 мкл). Раствор перемешивали в течение 3 часов при указанной температуре. Летучие компоненты удаляли при пониженном давлении (температура ванны не превышала 25°C). Остаток растворяли в метиленхлориде и промывали водой. Органические фазы объединяли, высушивали сульфатом натрия (Na2SO4), фильтровали и выпаривали досуха, чтобы получить соединение 4 в виде масла желтого цвета. Это соединение будут выпаривать совместно с безводным пиридином и использовать в следующем этапе без дополнительной очистки. Rf=0,71 (70% Et2O в петролейном эфире); 1H-ЯМР (400 МГц, CDCl3) 1,20 (с, 6H, 2×CH5), 3,05 (т, J=6,4 Гц, 2H, CH2S), 3,15 (с, 2Η, CH2OTr), 3,69 (т, J=6,4 Гц, 2H, CH2OH), 7,3-7,9 (м, 15Н, C6H5).
Осуществляли совместное выпаривание фосфорной кислоты (10 экв, 4,1 г) с безводным пиридином дважды, ее растворяли в указанном растворителе (25 мл) и добавляли к неочищенному 4. Реакционную смесь перемешивали при комнатной температуре, и через несколько минут появлялся осадок белого цвета. Реакционную смесь охлаждали до 0°C и добавляли пивалоилхлорид (5,5 экв, 3,4 мл). Реакционную смесь оставляли для нагревания до комнатной температуры и взбалтывали в течение 3 часов. Реакцию прекращали путем добавления раствора триэтиламмония бикарбоната (TEAB 1M, 10 мл) и разводили этилацетатом (EtOAc). После экстрагирования с EtOAc и 0,5M TEAB органические фазы объединяли, высушивали сульфатом натрия, фильтровали и выпаривали досуха (температура ванны не превышала 30°C). Остаток очищали колоночной флэш-хроматографией с элюированием 10% метанолом в метиленхлориде + 1% триэтиламине. Выпаривание фракций приводило к получению H-фосфонат моноэфира 5 в виде белого сиропа (90%). Rf=0,25 (70% Et2O в петролейном эфире); 1H-ЯМР (400 МГц, CDCl3) 1,17 (м, 2×CH3 + избыток (CH3CΗ2)3N), 2,9 (м, избыток (CH3CH2)3N), 3,12 (т, J=6,8 Гц, 2H, CH2S), 3,37 (с, 2Η, CH2OTr), 3,90 (м, 2 Η, CH2OP), 7,2-7,6 (м, 15 Η, C6H5), 9,9 (м, избыток (CΗ3CΗ2)3NH); 31P-ЯМР (161 МГц, CDCl3) 3,85 (с).
Синтез B102, производного гидрокси-tBuSATE N-бензилфосфорамидата 2'-C-метилцитидина:
Применяли следующие две стратегии:
Стратегия а:
Синтез защищенного нуклеозида 7

Смесь 2'C-метилцитидина (NM107) (10 г, 39,0 ммоль), триэтилортоформиата (8,3 экв., 54 мл) и п-толуолсульфоновой кислоты моногидрата (1 экв., 7,4 г) в безводном ацетоне (650 мл) нагревали с обратным холодильником в течение ночи в атмосфере азота. Реакционную смесь нейтрализовали водным раствором аммония (26%) и фильтровали осадок. Фильтрат выпаривали при пониженном давлении и совместно выпаривали с этанолом. Очистка сырой смеси колоночной хроматографией на силикагеле (элюент: поэтапный градиент [0-10%] метанола в метиленхлориде) давала соединение 6 в виде твердого вещества бледно-желтого цвета (86%). Rf=0,30 (20% MeOH в метиленхлориде), 1H-ЯМР (400 МГц, ДМСО-d6) 1,06 (с, 3H, CH3), 1,33 (с, 3H, CH3), 1,47 (с, 3H, CH3), 3,6 (м, 2H, H-5', H-5"), 4,1 (м, 1H, H-4'), 4,41 (д, 1H, H-3',J=3,2 Гц), 5,16 (т, 1H, OH-5', J=4,0 Гц, D2O заменяемый), 5,69 (д, 1H, H-5, J=8,0 Гц), 6,04 (с, 1H, H-1'), 7,14-7,19 (ушир.д, 2H, NH2, D2O заменяемый), 7,74 (д, 1H, H-6, J=8,0 Гц); LC/MS сканирование ES- 296 (M-H)', сканирование ES+ 298 (M+H)+, λмакс=280,7 нм.
Соединение 6 (4,4 г, 14,8 ммоль) растворяли в безводном пиридине (74 мл), и добавляли хлортриметилсилан (3 экв., 5,4 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 2 часов, затем последовательно добавляли 4,4'-диметокситритил хлорид (1,5 экв., 7,5 г) и 4-диметиламинопиридин (0,5 экв., 900 мг). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем гасили насыщенным водным раствором NaHCO3. Сырой продукт экстрагировали метиленхлоридом, промывали насыщенным водн. раствором NaHCO3 и водой. Объединенные органические фазы концентрировали при пониженном давлении, затем растворяли в смеси диоксана (160 мл) и водного аммония (28%, 29 мл). Раствор нагревали при 70°C в течение 3 часов и выпаривали досуха. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапный градиент метанола [1-5%] в метиленхлориде), с получением защищенного нуклеозида 7 в виде твердого вещества желтого цвета (81%). Rf=0,16 (30% EtOAc в CH2Cl2) 1H-ЯМР (400 МГц, ДМСО-d6) 1,03 (с, 3H, CH3), 1,30 (с, 3H, CH3), 1,42 (с, 3H, CH3), 3,5 (м, 2H, H-5', Н-5"), 3,71 (с, 6H, 2×OCH3), 4,0 (д, 1H, H-4', J=3,2 Гц), 4,36 (д, 1H, H-3', J=2,8 Гц), 5,1 (м, 1H, OH-5', D2O заменяемый), 5,90 (с, 1H, H-1'), 6,2 (м, 1H, H-5), 6,8-7,2 (м, 13H, DMTr), 7,6 (м, 1H, H-6), 8,32 (с, 1H, NH, D2O заменяемый); LC/MS сканирование ES-598 (M-H)-, λмакс1=231,7 нм, λмакс2=283,7.
Синтез B102 (соединение 10)

Соединения 7 (2,0 г, 3,34 ммоль) и 5 (2,2 экв., 4,3 г) совместно выпаривали с безводным пиридином и растворяли в указанном растворителе (50 мл). По капле добавляли пивалоилхлорид (2,5 экв., 1 мл), и раствор перемешивали при комнатной температуре в течение 2 часов 30 мин. Реакционную смесь разводили метиленхлоридом и нейтрализовали водным раствором аммония хлорида (NH4Cl 0,5M). После экстракции метиленхлоридом/водным NH4Cl 0,5M органические фазы объединяли, выпаривали при пониженном давлении (температура ванны не превышала 30°C) и совместно выпаривали с толуолом. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапный градиент [0-5%] метанола в метиленхлориде + 2% уксусной кислоты), для получения желаемого продукта 8, который совместно выпаривали с толуолом, чтобы получить пену бежевого цвета (94%). Rf=0,63 (5% MeOH в CH2Cl2); 1H-ЯМР (400 МГц, CDCl3) 1,21 (м, 9H, 3CH3), 1,42 (с, 3H, CH3), 1,60 (с, 3H, CH3), 3,13 (м, 2H, CH2S), 3,17 (м, 2H, CH2OTr), 3,79 (с, 6H, 2×OCH3), 4,1 (м, 2H, CH2OP), 4,2-4,3 (м, 3H, H-5', H-5", H-4'), 5,09 (д, 1H, H-3', J=7,6 Гц), 5,89 (д, 1H, H-5, J=5,6 Гц), 6,0 (м, 1H, H-1'), 6,8-7,7 (м, 29 H, Tr, DMTr, H-6); 13P-ЯМР (161 МГц, CDCl3) 7,92, 8,55; LC/MS сканирование ES+ 1066 (M+H)+, сканирование ES- 1064 (M-H)-.
В раствор соединения 8 (3,4 г, 3,15 ммоль) в безводном четыреххлористом углероде (30 мл) добавляли по капле бензиламин (10 экв., 3,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч 30 мин. Появлялся осадок белого цвета. Раствор разводили метиленхлоридом и нейтрализовали водным раствором хлористого водорода (HCl 1M). После последовательных экстракций CH2Cl2/HCl 1M и CH2Cl2/водный NaHCO3, органические фазы объединяли, высушивали Na2SO4, фильтровали и выпаривали досуха. Сырую смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапный градиент [0-5%] метанола в метиленхлориде), для получения 9 в виде пены желтого цвета (87%). Rf=0,35 (5% MeOH в метиленхлориде); 1H-ЯМР (400 МГц, CDCl3) 1,1-1,2 (м, 9H, 3CH3), 1,40 (с, 3H, CH3), 1,59 (с, 3H, CH3), 2,9-3,2 (м, 4H, CH2OTr, CH2OS), 3,76 (с, 6H, 2×OCH3), 3,9-4,4 (м, 8H, CH2OP, CH2N, H-3', H-4', H-5', H-5"), 5,0 (м, 1H, H-5), 6,0 (2с, 1H, H-1'), 6,7-7,7 (м, 34H, Tr, DMTr, C6H5CH2, H-6); 13P-ЯМР (161 МГц, CDCl3) 8,40, 8,8,68; LC/MS сканирование ES+ 1171 (M+H)+.
Наконец, соединение 9 (2,39 г, 2,04 ммоль) растворяли в смеси метиленхлорида (10 мл) и водного раствора трифторуксусной кислоты (90%, 10 мл). Реакционную смесь перемешивали при 35-40°C в течение 2 часов, затем разводили этанолом (140 мл). Летучие компоненты выпаривали при пониженном давлении и совместно выпаривали с этанолом. Сырую смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапный градиент метанола [0-30%] в метиленхлориде), с последующей очисткой обращенно-фазовой хроматографией (элюент: поэтапный градиент ацетонитрила [0-50%] в воде), для получения желаемого продукта 10 (B102) (смесь диастереоизомеров 1:1, что оценивали 31P-ЯМР, 36%), который лиофилизировали из смеси диоксан/вода. Rf=0,34 (15% MeOH в метиленхлориде); 1H-ЯМР (400 МГц, ДМСО-d6) 0,92 (с, 3H, CH3), 1,10 (с, 6H, 2×CH3), 3,0 (м, 2H, CH2S), 3,33 (м, 1H, H-3'), 3,56 (с, 2H, CH2OH), 3,8-4,0 и 4,05-4,25 (стеки, 7H, CH2OP, NCH2Ph, H-4', H-5', и H-5"), 4,9 (м, 1H, OH-3', J=5,4 Гц, D2O заменяемый), 5,07 (с, 1H, OH-2', D2O заменяемый), 5,3 (м, 1H, CH2OH, D2O заменяемый), 5,6-5,7 (м, 2H, H-5 и NH, D2O заменяемый), 5,91 (с, 1H, H-1'), 7,3-7,4 (стек, 7H, PhH, NH2, D2O заменяемый), 7,6 (м, 1H, H-6); 13P-ЯМР (161 МГц, ДМСО-d6) 9,71, 9,91; ВЭЖХ tR=4,67 мин (0-100% ацетонитрил в течение 8 мин), λмакс=274,9; LC/MS сканирование ES+ 587 (M+H)+.
Стратегия b:
Синтез защищенного нуклеозида 11

NM107 (10 г, 38,87 ммоль) растворяли в безводном пиридине (194 мл) и добавляли хлортриметилсилан (4,5 экв., 21,6 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 2 ч 30 мин, затем последовательно добавляли 4,4'-диметокситритилхлорид (1,5 экв., 19,8 г) и 4-диметиламинопиридин (0,5 экв., 2,37 г). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем гасили насыщенным водным раствором NaHCO3. Сырой продукт экстрагировали метиленхлоридом, промывали насыщенным водным раствором NaHCO3 и водой. Объединенные органические фазы выпаривали при пониженном давлении, затем разводили в тетрагидрофуране (110 мл). К этому раствору добавляли 1M тетрабутиламмония фторид в ТГФ (1 экв., 38,87 мл), и реакционную смесь перемешивали в течение 30 минут при комнатной температуре. После экстрагирования EtOAc и водой органические фазы собирали и выпаривали досуха. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапный градиент метанола [0-10%] в метиленхлориде), чтобы получить защищенный нуклеозид 11 в виде твердого вещества желтого цвета (93%). Rf=0,32 (10% MeOH в CH2Cl2) 1H-ЯМР (400 МГц, ДМСО-d6) 0,79 (с, 3H, CH3), 3,56 (м, 2H, H-5', H-5"), 3,71 (с, 7H, 2×OCH3, H-4'), 5,0 (м, 4H, H-3', OH-2', OH-3', OH-5', D2O заменяемый), 5,72 (с, 1H, H-1), 6,16 (м, 1H, H-5), 6,8-7,2 (м, 13H, DMTr), 7,82 (м, 1H, H-6), 8,24 (м, 1H, NH, D2O заменяемый); LC/MS сканирование ES- 560 (M+H)+, ES- 558 (M-H), λмакс=284,7 нм.
Синтез защищенного фосфорамидатного пронуклеотида 13, предшественника 10

Соединение 11 (7 г, 12,5 ммоль) и 5 (1,5 экв., 11,0 г) совместно выпаривали с безводным пиридином и растворяли в указанном растворителе (187 мл). Пивалоилхлорид (2,0 экв., 3,08 мл) добавляли по капле при -15°C, и раствор перемешивали при указанной температуре в течение 1 ч 30 мин. Реакционную смесь разводили метиленхлоридом и нейтрализовали водным раствором аммония хлорида (NH4Cl 0,5M). После экстрагирования метиленхлоридом/водным 0,5M NH4Cl объединяли органические фазы, выпаривали при пониженном давлении (температура ванны не превышала 30°C) и совместно выпаривали с толуолом. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапный градиент [0-5%] метанола в метиленхлориде + 0,2% уксусной кислоты), чтобы получить желаемый продукт 12, который выпаривали совместно с толуолом для получения пены белого цвета (3,5 г, 27%). Rf=0,44 (5% MeOH в CH2Cl2); 1H-ЯМР (400 МГц, ДМСО) 0,8 (м, 3H, CH3), 1,14 и 1,06 (2c, 6H, 2 CH3), 3,06 (м, 2H, CH2S), 3,16 (м, 2H, CH2OTr), 3,5 (м, 1H, H-3'), 3,70 (м, 6H, 2 OCH3), 3,90 (м, 1H, H-4'), 4,03 (м, 2H, CH2OP), 4,24 (м, 2H, H-5', H-5"), 5,30 и 5,04 (2мс, 2H, OH-2' и OH-3', D2O заменяемый), 5,78 (м, 1H, H-1'), 5,98 (м, 1H, P-H), 6,22 (м, 1H, H-5), 7,0-7,5 (м, 16H, Tr), 8,32 (м, 1H, H-6); 13P-ЯМР (161 МГц, ДМСО) 9,17, 9,65; LC/MS сканирование ES+ 1026 (M+H)+, λмакс=282,7 нм.
К раствору соединения 12 (500 мг, 0,49 ммоль) в безводном тетрахлориде углерода (4,9 мл), по капле добавляли бензиламин (5 экв., 0,266 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и удаляли растворитель при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапный градиент [0-5%] метанола в метиленхлориде), чтобы получить соединение 13 в виде пены (75%). Rf=0,25 (3% MeOH в метиленхлориде); 1H-ЯМР (400 МГц, ДМСО) 0,79 (с, 3H, CH3), 1,13 и 1,06 (2c, 6H, 2 CH3), 3,05 (м, 4H, CH2OTr, CH2OS), 3,51 (м, 1H, H-3'), 3,69 (с, 6H, 2×OCH3), 3,87 (м, 3H, CH2OP, CH2N, H-3'), 4,08 (м, 2H, H-5', H-5"), 5,19 и 5,0 (2м, 2H, OH-2' и OH-3', D2O заменяемый), 5,67 (м, 1H, NH, D2O заменяемый), 5,75 (2c, 1H, H-1'), 6,21 (м, 1H, H-5), 6,7-7,5 (м, 34H, Tr, DMTr, C6H5CH2, H-6); 13P-ЯМР (161 МГц, ДМСО) 9,84, 9,69; LC/MS сканирование ES+ 1132 (M+H)+.
Соединение 13 можно превращать в фосфорамидатное пролекарство 10 (B102), следуя условиям эксперимента, описанным для последнего этапа в примере 3 (процедура A) и в примере 4.
ПРОЦЕДУРА B:

СХЕМА СИНТЕЗА:
B102 синтезируют в виде смеси фосфорных диастереомеров в соотношении 1:1. Общий выход продукта B102 из NM107 составлял 31%, поскольку не весь связанный полученный материал использовали для снятия защиты.
Этап 1.1:

| Материал | Степень | FW (полный вес) гмоль-1 | Коли-чество | Плот-ность гмл-1 | Коли-чество в молях | Экв. |
| NM107 | 99% | 257,2 | 150 г | - | 0,583 | 1 |
| PhB(OH)2 | 98% | 122,1 | 78 г | - | 0,639 | 1,10 |
| Безводный пиридин | 98% | 79,1 | 2,5 л | 0,978 | - | - |
NM107 растворяли в пиридине в атмосфере аргона и добавляли бензолбороновую кислоту. Перемешиваемую смесь нагревали с обратным холодильником в течение 3 часов в аргоне. Затем проводили дистилляцию азеотропа, удаляя 1,2 л (пиридин/вода).
T головной фракции: 103°C→113°C; T смеси: 112°C→116°C
Смесь охлаждали до комнатной температуры, и под вакуумом выпаривали пиридин для получения масла золотистого цвета. Продукт сохраняли в вакууме в течение ночи для использования на следующем этапе. Соотношение продукта/исходного материала составляло 97:3 и подтверждалось 1H-ЯМР (d6-ДМСО (диметилсульфоксид)). Перед этапом 1.2 неочищенный продукт растворяли в 250 мл безводного пиридина. Альтернативно можно применять следующие условия:
- экв. бензолбороновой кислоты
- 5 экв. пиридина
- 1,5 экв. Na2SO4
- 5 мл CH3CN для 1 г NM107
- нагревание с обратным холодильником в течение от 1 ч до 1 ч 30 мин. Охлаждение до КТ. Использование для следующей реакции.
- 98-99% превращения, оцениваемое протонной ЯМР
Этап 1.2:

| Материал | Степень | FW (полный вес) гмоль-1 | Количест-во | Плот-ность гмл-1 | Коли-чество в молях | Экв. |
| 2,3-PhB-NM107 | - | 343,1 | Раствор | - | 0,583 | 1 |
| Фосфонат 3 | - | 585,7 | 615 г | - | 1,049 | 1,8 |
| EDCI,HC1 | 98% | 191,7 | 570 г | - | 2,973 | 5,1 |
| Безводный ацетонитрил | 98% | - | 3 л | - | - | - |
| Бензиламин | 98% | 107,2 | 445 мл | 0,98 | 4,0 | 7* |
| Тетрахлорид углерода | 98% | 153,8 | 260 мл | 1,59 | 2,6 | 4* |
| *Дополнительное эквивалентное количество указанных реактивов можно добавлять при необходимости (например, 15 экв.) (если P-OH выявляется при ВЭЖХ) | ||||||
Фосфонат 3 растворяли в 3 л ацетонитрила в атмосфере аргона. Добавляли раствор 2,3-PhB-NM107 из этапа 1.1 с последующим добавлением EDCI.HCl. Смесь перемешивали в атмосфере аргона при 41-46°C в течение 4 часов, и спустя указанное время анализ ВЭЖХ показывал соотношение продукта P-H к NM107 около 7:1. Смесь охлаждали до 18°C и по капле добавляли бензиламин, с последующим добавлением тетрахлорида углерода. Реакция была слегка экзотермической. Анализ ВЭЖХ показывал полное превращение P-H в продукт фосфорамидат. Этилацетат (1 л) добавляли к смеси, которую затем подкисляли до уровня pH 4 с помощью 3 л 20% лимонной кислоты. Водную фазу экстрагировали 2,5 л этилацетата. Органические фазы объединяли и промывали 3 л 10% лимонной кислоты. Органическую фазу подщелачивали до уровня pH 8 с помощью 5 л водного (насыщенного) бикарбоната натрия и промывали второй раз 2 л водного (насыщенного) бикарбоната натрия. Органическую фазу высушивали сульфатом натрия, фильтровали в вакууме и выпаривали для получения пены желтого цвета, 712 г.
Сырой остаток растворяли в дихлорметане (ДХМ) (1 л) и очищали на кварцевом устройстве (2,3 кг кварца). Элюирование: 5 л 4% метанола/ДХМ, 2*1 л 4%, 3*1 л 5%, 8*250 мл 6%, 4*250 мл 7%, 9*1 л 7%. Выпаривание соответствующих фракций давало 254 г фосфорамидата 4 (чистота по ВЭЖХ: 98,5%, выход: 52%) и 73 г (чистота по ВЭЖХ: 87,6%, выход: 13%).
Этап 2:

| Материал | Степень | FW (полный вес) гмоль-1 | Коли-чество | Плот-ность гмл-1 | Коли-чество в молях | Экв. |
| Фосфорамидат 4 | - | 828,9 | 246 г | - | 0,291 | 1 |
| AcCl | 99% | 78,5 | 62,6 мл | 1,105 | 1,049 | 3,0* |
| Безводный EtOH | 98% | - | 3,5 л* | - | - | - |
| *Последовательно использовали 2,0 экв. AcCl и соотношение 4:EtOH составляло 1:10 вес/об. | ||||||
Фосфорамидат 4 растворяли в безводном этаноле и к реакционной смеси добавляли ацетилхлорид (экзотермически: от 18°C до 27°C) в атмосфере аргона. Смесь перемешивали при 60°C в атмосфере аргона. Через 30 минут анализ ВЭЖХ указывал на полное превращение фосфорамидата 4 в продукт со снятой защитой 5. Смесь охлаждали до 25°C и добавляли твердый бикарбонат натрия (1,04 кг) несколькими порциями (вспенивание, уровень pH~5,5-6). Смесь фильтровали через Целит и промывали двумя объемами этанола. Фильтрат выпаривали под вакуумом при 35°C. Остаток растирали TBME (3 л) в течение 1 часа и затем фильтровали для удаления побочного продукта тритила. Полученное твердое вещество высушивали под вакуумом, чтобы получить 185 г продукта с чистотой 93% по ВЭЖХ 254 нм.
При необходимости можно удалять из продукта любую остаточную бензолбороновую кислоту путем растворения в воде и обработкой смолой IRA-743 Amberlite.
Возможно применение следующих альтернативных условий реакции (чтобы избежать возможности ацилирования 4):
- 2,0 экв. AcCl в EtOH 1:10 об:об, чтобы получить HCl и использовать полностью AcCl (экзотермически)
- Фосфорамидат 4 в EtOH (чтобы достичь соотношения 1:10 вес:об общего объема EtOH)
- Добавление раствора HCl/EtOH в реакционную смесь при 20°C
- 60°C в атмосфере аргона, 30-45 минут
Сырой продукт очищали обращенно-фазовой хроматографией (1,5 кг готового 40 мкм Bakerbond C-18 RP-silica - промывали 100% ацетонитрилом с градиентом до 100% H2O). Сырой продукт растворяли в ацетонитриле (58 мл), H2O (164 мл) и насыщенном водном растворе (170 мл) бикарбоната натрия.
Элюирование под мягким вакуумом с поэтапным градиентом от 3% MeCN/H2O, 10%, 15%, 25% (чистый элюированный продукт) и выпаривание соответствующих фракций давало 106 г продукта B102 (62% выхода) с чистотой 98,6% по данным ВЭЖХ, 254 нм.
Типичные данные анализов приведены ниже:
B102: C24H35N4O9PS - 586,59 гмоль-1
ВЭЖХ AUC (площадь под кривой) (Методика теста 20): 98,9% при 254 нм, Rt (время удержания) 3,34 минуты
m/z (ESI +): 587,12 [M+H]+ 100%; 1173,62 [2M+H]+ 80%
νмакс (диск KBr) (см-1): 3343,1 ушир. (O-H, N-H); 1647,2 ушир. (C=O основание, тиоэфир)
KF: содержание H2O на 2,02%
Определенное вращение: [α]D 20 +55,011 (c. 10,492 мг см-3 в ДМСО)
Элементный анализ: Рассчитано: C 49,14%; H 6,01%; N 9,55%; S 5,47%; P 5,28%;
Найдено: C 48,74%; H 5,83%; N 9,41%; S 5,81%; P 5,33%
ЯМР: Анализ с применением экспериментов 1H, 13C, 31P, COSY, DEPT, HSQC и HMBC.
1H ЯМР δH (400 МГц, d6-ДМСО): 0,94 (3H, д, J=1,8 Гц, CH3), 1,11 (6H, c, (CH3)2C), 3,04 (2H, м, J=6,4 Гц, CH2S), 3,44 (2H, д, J=5,0 Гц, CH2OH), 3,60 (1H, ушир.м, H-3'), 3,82-4,01 (5H, м, H-4', CH2O, CH2Ph), 4,07-4,12 (1H, м, H-5'), 4,13-4,24 (1H, м, H-5"), 4,94 (1H, т, J=5,0 Гц, CH2OH), 5,07 (1H, д, J=1,8 Гц, OH-2'), 5,26 (1H, т, J=6,8 Гц, OH-3'), 5,64-5,76 (1H, м, P-N-H), 5,69, 5,70 (1H, 2×д, 2×J=7,6 Гц, H-5), 5,93 (1H, ушир.с, H-1'), 7,13-7,20 (2H, 2×ушир.с, NH2), 7,20-7,25 (1H, м, Ar-H), 7,28-7,35 (4H, м, 4×Ar-H), 7,53, 7,57 (1H, 2×д, J=7,6 Гц, H-6).
13C ЯМР δc (100 МГц, d6-ДМСО): 19,81 (CH3), 21,79 (C(CH3)2), 28,17, 28,24 (CH2S), 44,18 (PhCH2), 51,62 (C(CH3)2), 63,74, 63,79 (CH2O), 64,21, 64,51 (C-5'), 68,29 (CH2OH), 72,41, 72,57 (C-3'), 77,80, 77,85 (C-2'), 79,47, (C-4'), 91,66, (C-1'), 93,82 (C-5), 126,68, 127,09, 128,08, 128,09 (5×Ar-C), 140,34, 140,38, 140,40 (Ar-Cipso, C-6), 155,12, 165,21 (C-2, C-4), 203,85 (C=OS).
31Р ЯМР δр (162 МГц, d6-ДМСО): 9,71, 9,91 (1Р, 2×с, соотношение 1,00:1,07).
Для синтеза нуклеозидных пролекарств, таких как B102, можно применять процедуру синтеза A. Если это предпочтительно, на нуклеозидном основании может присутствовать защита 2' и 3' гидроксильных групп, а также аминогруппы. В стратегии A 2' и 3' гидроксильные группы имеют такую защиту, например, как защищены производное ацетонида и аминогруппа, например, как производное ди-метокситритила. Гидролиз актеонида после связывания нуклеозида с промежуточным продуктом SATE осуществляют с использованием кислоты, такой как ТФУ. Указанная процедура гидролиза может потенциально давать побочные продукты и иметь низкий выход, и ди-метокситритилхлорид имеет недостаток высокой стоимости. Процедура B синтеза, приведенная ниже, способна преодолеть такие проблемы. Для защиты 2' и 3' гидроксильных групп на сахарной функциональных группе используют кислоту, например, бороновую кислоту, такую как фенилбороновая кислота. Связывание нуклеозидного производного фенилбороната с промежуточным продуктом SATE может дать хороший выход, и снятие защиты с фенилбороната удобно происходит во время работы с реакционной смесью, при промывании кислотой, такой как раствор водной лимонной кислоты. Окончательное удаление защитной группы, такой как тритильная группа (на функциональной группе SATE), мягко осуществляется с использованием системы органических растворителей, такой как смесь ацетилхлорида/этанола. Такая реакция снятия защиты может быть последовательно воспроизводимой, измеряемой и дает значительный высокий выход.
Пример 3
Приготовление B299, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метилгуанозина

ПРОЦЕДУРА A:
СХЕМА СИНТЕЗА:

2'-C-метилгуанозин (NM108) (3 г, 10,10 ммоль) и соединение 5 [синтез 5 см. пример 2] (6,48 г, 11,10 ммоль) совместно выпаривали с безводным пиридином и разводили в указанном растворителе (152 мл). Пивалоилхлорид (2,48 мл, 20,18 ммоль) добавляли по капле при -15°C, и раствор перемешивали при указанной температуре в течение 2 часов. Реакционную смесь растворяли в метиленхлориде и нейтрализовали водным раствором аммония хлорида (NH4Cl 0,5M). После экстрагирования метиленхлоридом/водн. 0,5M NH4Cl, органические фазы объединяли, высушивали Na2SO4, выпаривали при пониженном давлении (температура ванны не превышала 30°C) и дважды совместно выпаривали с толуолом. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапный градиент [0-10%] метанола в метиленхлориде + 0,2% уксусной кислоты), чтобы получить желательный продукт 6 (2,5 г, 32%). Rf= 0,34 (15% MeOH в CH2Cl2); 1H-ЯМР (400 МГц, ДМСО-d6) 0,80 (с, 3H, CH3), 1,13 (с, 6H, 2×CH3), 3,04 (м, 2H, CH2OTr), 3,14 (м, 2H, CH2S), 3,97-4,08 (м, 4H, Н-3', Н-4, CH2OP), 4,28-4,38 (м, 2H, Н-5', Н-5"), 5,10-5,35 (м, 2H, OH-2', OH-3', D2O заменяемый), 5,77 (с, 1H, H-1'), 6,52 (ушир.с, 2H, NH2, D2O заменяемый), 7,11-7,42 (м, 15H, Tr), 7,75 (с, 1H, Н-8), 10,67 (ушир.с, 1H, NH, D2O заменяемый); 13P-ЯМР (161 МГц, ДМСО-d6) 9,47, 9,20; LC/MS сканирование ES+ 764 (M+H)+, сканирование ES- 762 (M-H)-.
К раствору соединения 6 (2,5 г, 3,27 ммоль) в безводном тетрахлориде углерода (33 мл) по капле добавляли бензиламин (5 экв., 1,79 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и выпаривали при пониженном давлении (температура ванны не превышала 30°C). Неочищенную смесь очищали колоночной флэш-хроматографией на силикагеле (элюент: поэтапный градиент [0-10%] метанола в метиленхлориде), для получения соединения 7 в виде пены белого цвета (2,9 г, количественный выход). Rf=0,27 (10% MeOH в метиленхлориде); 1H-ЯМР (400 МГц, ДМСО-d6) 0,81 (с, 3H, CH3), 1,10 (с, 6H, 2×CH3), 2,99-3,08 (м, 4H, CH2OTr, CH2S), 3,87-4,30 (м, 8H, H-3', H-4', H-5', H-5'' CH2OP, NCH2Ph), 5,66 (м, 1H, NH, D2O заменяемый), 5,76 (с, 1H, H-1'), 6,60 (ушир.с, 2H, NH2, D2O заменяемый), 7,17-7,39 (м, 20H, Tr, C6H5CH2), 7,77 (с, 1H, H-8); 13P-ЯМР (161 МГц, ДМСО-d6) 9,93, 9,78; LC/MS сканирование ES+ 869 (M+H)+, сканирование ES- 867 (M-H)-.
Соединение 7 (2,84 г, 3,27 ммоль) растворяли в смеси трифторуксусной кислоты (1,1 мл) и метиленхлорида (11,4 мл). Реакционную смесь перемешивали в течение 0,5 часа при комнатной температуре. Раствор разводили этанолом, выпаривали при пониженном давлении (температура ванны не превышала 30°C) и выпаривали совместно с толуолом. Неочищенную смесь очищали колоночной флэш-хроматографией на силикагеле (элюент: поэтапный градиент [0-30%] метанола в метиленхлориде) и затем обращенно-фазовой хроматографией (элюент: поэтапный градиент [0-100%] ацетонитрила в воде), для получения желаемого продукта 8 (B299) (1:1 смесь диастереоизомеров согласно данным 31P-ЯМР, 800 мг, 39%), который лиофилизировали из смеси диоксан/вода. Rf=0,57 (20% MeOH в метиленхлориде); 1H-ЯМР (400 МГц, ДМСО-d6) 0,82 (с, 3H, CH3), 1,09 (с, 6H, 2×CH3), 3,01 (м, 2H, CH2S), 3,42 (д, 2H, CH2OH, J=8,0 Гц), 3,81-4,00 (м, 6H, H-3', H-4', CH2OP, NCH2Ph), 4,11-4,27 (м, 2H, H-5', H-5"), 4,92 (т, 1H, CH2OH, J=8,0 Гц, D2O заменяемый), 5,16 (с, 1H, OH-2', D2O заменяемый), 5,40 (м, 1H, OH-3', D2O заменяемый), 5,64 (м, 1H, NH, D2O заменяемый), 5,75 (с, 1H, H-1'), 6,50 (ушир.с, 2H, NH2, D2O заменяемый), 7,19-7,32 (м, 5H, PhH), 7,77 (с, 1H, H-8), 10,61 (ушир.с, 1H, NH, D2O заменяемый); 13P-ЯМР (161 МГц, ДМСО-d6) 9,91, 9,78; ВЭЖХ tR = 3,67 мин (0-100% ацетонитрил в течение 8 минут), λмакс=251,3; LC/MS сканирование ES+ 627 (M+H)+, сканирование ES- 625 (M-H)-.
ПРИМЕР 4
Приготовление B208, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метилтимидина

ПРОЦЕДУРА A:
СХЕМА СИНТЕЗА:

2'-C-метилтимидин (NM105) (700 мг, 2,57 ммоль) и 5 [синтез 5 см. в примере 2] (1,1 экв., 1,6 г) совместно выпаривали с безводным пиридином и растворяли в указанном растворителе (40 мл). Пивалоилхлорид (2,0 экв., 0,633 мл) добавляли по капле при -15°C и раствор перемешивали при указанной температуре в течение 1 ч 30 мин. Реакционную смесь разводили метиленхлоридом и нейтрализовали водным раствором аммония хлорида (0,5M NH4Cl). После экстрагирования метиленхлоридом/водн. 0,5M NH4Cl объединяли органические фазы, выпаривали при пониженном давлении (температура ванны не превышала 30°C) и выпаривали совместно с толуолом. Неочищенную смесь очищали колоночной флэш-хроматографией на силикагеле (элюент: поэтапный градиент [0-10%] метанола в метиленхлориде + уксусная кислота, 0,2%), чтобы получить желаемый продукт 6, который совместно выпаривали с толуолом для получения пены белого цвета (942 мг, 50%). Rf=0,56 (15% MeOH в CH2Cl2); 1H-ЯМР (400 МГц, ДМСО) 1,00 (с, 3H, CH3), 1,13 (с, 6H, 2 CH3), 1,77 (с, 3H, CH3), 3,16 (м, 2H, CH2S), 3,32 (м, 2H, CH2OTr), 3,6 (м, 1H, H-3'), 3,9 (м, 1H, H-4'), 4,0 (м, 2H, CH2OP), 4,2-4,3 (м, 2H, H-5', H-5"), 5,21 (с, 1H, OH-2', D2O заменяемый), 5,40 (т, 1H, OH-3', D2O заменяемый), 5,83 (с, 1H, H-1'), 6,0 (с, 1H, P-H), 7,0-7,5 (м, 16H, Tr, H-6); 13P-ЯМР (161 МГц, ДМСО) 9,29, 9,68; LC/MS сканирование ES+ 761 (M+Na)+.
К раствору соединения 6 (920 мг, 1,25 ммоль) в безводном тетрахлориде углерода (13 мл) добавляли по капле бензиламин (10 экв., 1,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Наблюдали осадок белого цвета. Раствор растворяли метиленхлоридом и нейтрализовали водным раствором хлористого водорода (HCl 1M). После последовательных экстракций с использованием CH2Cl2/1M HCl и CH2Cl2/водн. NaHCO3 органические фазы объединяли, высушивали Na2SO4, фильтровали и выпаривали досуха. Неочищенную смесь очищали колоночной флэш-хроматографией на силикагеле (элюент: поэтапный градиент [0-10%] метанола в метиленхлориде), чтобы получить соединение 7 в виде пены белого цвета (875 мг, 83%). Rf=0,56 (15% MeOH в CH2Cl2); 1H-ЯМР (400 МГц, ДМСО) 0,99 (с, 3H, CH3), 1,12 (с, 6H, 2 CH3), 1,75 (с, 3H, CH3), 3,04 (м, 4H, CH2OTr, CH2S), 3,69 (м, 1H, H-3'), 3,8-4,0 (м, 5H, CH2OP, CH2N, H-4'), 4,0-4,2 (м, 2H, H-5', H-5"), 5,17 (с, 1H, OH-T, D2O заменяемый), 5,3 (м, 1H, OH-3', D2O заменяемый), 5,7 (м, 1H, NH, D2O заменяемый), 5,82 (с, 1H, H-1'), 7,1-7,5 (м, 21H, Tr, C6H5CH2, H-6); 13P-ЯМР (161 МГц, ДМСО) 9,95, 9,86; ВЭЖХ tR=7,91 мин (0-100% ацетонитрил в течение 8 минут), λмакс=266,7 нм; LC/MS сканирование ES+ 866 (M+Na)+.
Наконец, соединение 7 (860 мг, 1,02 ммоль) растворяли в смеси метиленхлорида (15 мл) и трифторуксусной кислоты (0,51 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем разводили толуолом. Летучие компоненты выпаривали при пониженном давлении и выпаривали совместно с этанолом. Неочищенную смесь очищали колоночной флэш-хроматографией на силикагеле (элюент: поэтапный градиент метанола [0-10%] в метиленхлориде), с последующей очисткой обращенно-фазовой хроматографией (элюент: поэтапный градиент ацетонитрила [0-50%] в воде) для получения желаемого продукта 8 (B208) (257 мг, 42%). Rf=0,31 (10% MeOH в метиленхлориде); 1H-ЯМР (400 МГц, ДМСО-d6) 0,99 (с, 3H, CH3), 1,10 (с, 6H, 2×CH3), 1,75 (с, 3H, CH3), 3,0 (м, 2H, CH2S), 3,42 (д, 2H, CH2OH), 3,7 (м, 1H, H-3'), 3,8-4,0 (стек, 5H, CH2OP, NCH2Ph, H-4'), 4,0-4,3 (м, 2H, H-5' и H-5"), 4,9 (м, 1H, CH2OH, D2O заменяемый), 5,17 (с, 1H, OH-2', D2O заменяемый), 5,3 (м, 1H, OH-3', D2O заменяемый), 5,7 (м, 1H, NH, D2O заменяемый), 5,81 (с, 1H, H-1'), 7,2-7,4 (стек, 6H, PhH, H-6); 13P-ЯМР (161 МГц, ДМСО-d6) 9,84, 9,90; ВЭЖХ tR=4,98 мин (0-100% ацетонитрил в течение 8 минут), λмакс=269,0 нм; LC/MS сканирование ES+ 602 (M+H)+.
ПРИМЕР 5
Приготовление B261, гидрокси-tBuSATE N-бензилфосфонамидатного производного PMEA

ПРОЦЕДУРА A:
Синтез промежуточного продукта 4:

В трехгорловый флакон объемом 500 мл, снабженный конденсатором, помещали PMEA (2,00 г, 7,25 ммоль), CH2Cl2 (121 мл) и ДМФ (617 мкл, 7,98 ммоль). Полученную пасту энергично пермешивали, и по капле добавляли оксалилхлорид (2,21 мл, 25,4 ммоль) при 0°C в течение 10 минут (выделение газа). Паста превращалась в желтый раствор (10 минут) перед помутнением (10 минут). Его дополнительно перемешивали в течение 3 часов при нагревании с обратным холодильником и превращали в плотную пасту белого цвета. Продукты высушивали по способу schlenk в течение 1 часа in situ путем выпаривания всех летучих компонентов при пониженном давлении при комнатной температуре. Получаемое твердое вещество желтого цвета затем можно было частично растворять в CH2Cl2 (121 мл), и по капле добавлять пиридин (1,17 мл, 14,5 ммоль) в течение 10 мин при 0°C. Наблюдали превращение суспензии белого цвета в раствор синего цвета, который охлаждали до -78°C. Затем медленно по капле по внутренней стенке добавляли раствор спирта 3 [синтез 3 см. в примере 1] (3,480 г, 7,25 ммоль) и триэтиламина (6,37 мл, 45,7 ммоль) в CH2Cl2 (72 мл) (около 45 минут), и реакционную смесь перемешивали в течение 10 часов при -78°C. Затем по капле добавляли бензиламин (2,37 мл, 21,7 ммоль) при -78°C, и раствор оставляли, перемешивая и нагревая до КТ в течение 1 часа. NaHCO3 (водный, насыщенный, 200 мл) вливали в реакционную смесь и разделяли слои. Водную фазу экстрагировали с CH2Cl2 (2×100 мл), и объединенные органические экстракты высушивали солевым раствором (50 мл) и Na2SO4. Раствор фильтровали и концентрировали, чтобы получить около 6,5 г неочищенного сиропа желтого цвета. Проводили очистку колоночной флэш-хроматографией (SiO2, ⌀=3,5 см, H=11 см) с элюированием с 4→8→12% MeOH в CH2Cl2 (1% Et3N) с получением 3,70 г пены желтого цвета (0,15<Rf<0,30, 10% MeOH в CH2Cl2), которую подвергали второй очистке колоночной флэш-хроматографией (SiO2, ⌀=3,5 см, H=12 см) с элюированием 4→6 MeOH в CH2Cl2 (1% Et3N), чтобы получить 2,67 г пены желтого цвета (0,16<Rf<0,25, 10% MeOH в CH2Cl2). Этот продукт подвергали третьей очистке колоночной флэш-хроматографией (SiO2, ⌀=3,5 см, H=12 см) с элюированием 4→6 MeOH в CH2Cl2 (1% Et3N), чтобы получить 165 мг фосфонамидата 4 (около 2,7%) в виде пены белого цвета и 1,75 г смеси соединений. Их подвергали заключительной очистке колоночной флэш-хроматографией (SiO2, ⌀=3,5 см, H=12 см) с элюированием 4→6 MeOH в CH2Cl2 (1% Et3N), с получением 353 мг фосфонамидата 4 (около 5,9%) в виде пены белого цвета. Общий выход: 8,6%. Rf=0,21 (6% MeOH в CH2Cl2); 1H-ЯМР (300 МГц, CDCl3) 1,13 (с, 6H, 2CH3), 3,02-3,10 (м, 2H, CH2S), 3,59 (т, J=7,5, 2H, CH2), 3,58 (с, 6H, 2×OCH3), 3,73 (т, J=7,1, 2H, CH2), 3,88-4,09 (стеки, 4H, 2×CH2), 4,21 (т, J=7,0, 2H, CH2O), 5,50 (ушир.c, 2H, NH2), 6,67-6,78 (м, 4H, PhH), 7,04-7,38 (стек, 9H, PhH), 7,72 (с, 1H), 8,22 (с, 1H); 31P-ЯМР (121 МГц, CDCl3) 25,0; m/z (FAB+) 825 (1), 303 (100); HRMS 825,3171 ([M+H]+. C43H50O7N6PS требует 825,3199).
Синтез соединения 5 (B261):

Дихлоруксусную кислоту (20% раствор в CH2Cl2, около 140 капель) добавляли по капле к раствору эфира 4 (353 мг, 0,43 ммоль) в CH2Cl2 (4,3 мл) при 0°C, и все перемешивали в течение 55 мин. Затем добавляли твердый NaHCO3 (около 1,5 г) и перемешивали пасту в течение 10 минут перед фильтрацией и выпариванием. Осуществляли очистку колоночной флэш-хроматографией (SiO2, ⌀=1,5 см, H=10 см) с элюированием с 4%→10% MeOH в CH2Cl2 с получением чистого фосфонамидата 5 (130 мг после лиофилизации в ТГФ/H2O и 3 дней пребывания в сушильном шкафу P2O5, 58%). Указанную реакцию также проводили с 165 мг эфира 4, чтобы получить 51 мг фосфонамидата 5 (B261, 49%). Rf=0,20 (10% MeOH в CH2Cl2); 1H-ЯМР (300 МГц, ДМСО-d6) 1,10 (с, 6H, 2×CH3), 2,80 (т, J=7,0, 2H, CH2S), 3,43 (д, J=5,5, 2H, CH2OH), 3,69 (A из AB, J=4,8, 1H, 1×CH2P), 3,71 (B из AB, J=4,8, 1H, 1×CH2P), 3,75-3,88 (стеки, 4H, CH2O, NCH2), 3,88-4,07 (м, 2H, NCH2Ph), 4,30 (т, J=7,0, 2H, CH2O), 4,97 (т, J=6,1, 1H, OH), 5,31-5,42 (м, 1H, NH), 7,16-7,32 (стек, 7H, PhH, NH2), 8,09 (с, 1H), 8,13 (с, 1H); 13C-ЯМР (75 МГц, ДМСО-d6) 21,8 (2×CH3), 28,4 и 28,5 (CH2, CH2S), 42,4 (CH2, NCH2), 43,3 (CH2, NCH2), 51,7 (кват, C, C(CH3)2), 61,7 и 61,8 (CH2, CH2O), 64,6 (CH2, CH2O), 68,4 (CH2, CH2O), 118,5 (кват, C), [126,5 (CH, Ph), 127,0 (CH, Ph), 128,0 (CH, Ph), некоторое наложение], 140,5 и 140,6 (кват, C), 141,0 (CH), 149,4 (кват, C), 152,3 (CH), 155,9 (кват, C), 203,9 (кват, C, C=O); 31P-ЯМР (121 МГц, ДМСО-d6) 25,9; m/z (FAB+) 161 (32), 256 (42), 523 (100); HRMS (масс-спектрометрия с высоким разрешением) 523,1899 ([M+H]+. C22H32O5N6PS предусматривает 523,1892); ВЭЖХ (C 18, поток: 0,5 мл/минуты, раствор = TEAC 20 мМ, раствор B=20% TEAC 20 мМ): tR=5,04 минут (60% А в B), tR=27,24 минут (t=0→10 минут: 100% A; t=10→30 минут: 0→50% B в A; t=30→35 минут: 50→100% B в A); УФ (EtOH 95%) λмакс=205 (εмакс 23900), λмин=228 (εмин 5400).
ПРОЦЕДУРА B;
[Улучшенное приготовление гидрокси-tBuSATE N-бензилфосфорамидатного производного (B261, Соединение 5) PMEA]
СХЕМА СИНТЕЗА:

Этап 1: Синтез промежуточного продукта A
К суспензии PMEA (2 г, 7,3 ммоль) в 120 мл (безводного) ДХМ добавляли ДМФ (640 мг, 1,2 экв.), после этого добавляли оксалилхлорид (2,3 мл, 3,5 экв.) при комнатной температуре. Реакционную смесь нагревали с обратным холодильником в течение 1,5 часов, чтобы получить плотную суспензию желтого цвета. Реакционную смесь выпаривали досуха посредством роторного испарителя, чтобы получить сырой промежуточный продукт 2 в виде твердого вещества бледно-желтого цвета. Анализ LC-MS аликвотных количеств промежуточного продукта 2 в метанольном растворе подтвердил характеристику хорошей чистоты продукта.
Этап 2: Синтез промежуточного продукта B:
Неочищенный промежуточный продукт A (7,33 ммоль) суспендировали в 100 мл безводного ДХМ. Суспензию охлаждали до 0°C. К ней добавляли пиридин (1,2 мл, 14,6 ммоль, 2 экв.) при 0°C. После указанного добавления суспензия бледно-желтого цвета менялась до золотистого прозрачного раствора. Этот раствор охлаждали до -32°C на ACN/бане с сухим льдом. К нему по капле добавляли раствор 3 (3,52 г, 7,33 ммоль, 1 экв.) в 70 мл безводного ДХМ, который содержал триэтиламин (6,3 мл, 44 ммоль, 6 экв.). Температуру внутри реакции во время добавления поддерживали в диапазоне примерно от -35°C до -30°C. Яркий золотистый раствор превращался в раствор зеленого цвета с некоторым количеством осадка, выпадавшего из раствора во время добавления. Вероятно, осадком являлась триэтиламиновая соль HCl. Для завершения добавления требовалось 20 минут. После добавления смесь перемешивали при температуре примерно от -30°C до -10°C в течение 1 часа. Реакционную смесь обратно охлаждали до -20°C. К ней добавляли бензиламин (2,4 мл, 22 ммоль, 3 экв.). Смесь перемешивали при -20°C в течение 10 минут. К реакционной смеси добавляли насыщенный NaHCO3/H2O, и смесь перемешивали в течение 2 минут. Слой ДХМ отделяли, высушивали Na2SO4 и выпаривали досуха, чтобы получить сырой промежуточный продукт B в виде желтого вязкого масла. Анализ ВЭЖХ сырого промежуточного продукта показывал чистоту 62% при 272 нм.
Этап 3: Синтез промежуточного продукта 4:
Сырой промежуточной продукт B (7,33 ммоль) в виде вязкого масла бледно-желтого цвета растворяли в 200 мл MeOH. В течение ночи реакционную смесь нагревали с обратным холодильником. Анализ ВЭЖХ реакционной смеси показывал полное превращение амидина в амин. [Время удержания амидина (Rt=5,92 минут) близко к времени амина (Rt=5,98 мин), полученное способом ВЭЖХ, применяемым заявителями!]. Смесь охлаждали до КТ и фильтровали. Фильтрат выпаривали досуха посредством роторного испарителя. Полученный сырой продукт очищали колоночной флэш-хроматографией на силикагеле (120 г, использовали комби-флэш колонку с силикагелем, 3-8% MeOH в ДХМ в качестве элюента), чтобы получить 3,1 г очищенного продукта 4 в виде пены белого цвета с выделенным выходом из 2 г PMEA 51%. 1H-ЯМР анализ полученного продукта 4 соответствовал желаемой структуре. Анализ ВЭЖХ полученного продукта 4 показал 96% чистоту (AUC).
Этап 4: Синтез B261 (Соединение 5)
Промежуточный продукт 4 (300 мг, 0,36 ммоль) растворяли в EtOH (безводный, 5 мл). К нему одной порцией добавляли ацетилхлорид (43 мг, 1,5 экв.) при комнатной температуре. Реакцию необходимо проводить в закрытой реакционной колбе, чтобы избежать утечки газа HCl. Реакционную смесь перемешивали при КТ в течение 30 мин. К ней добавляли твердый NaHCO3 и реакционную смесь перемешивали в течение 15 минут. Уровень pH, выявленный в реакционной смеси, составлял около 7-8. Смесь фильтровали, и фильтрат концентрировали досуха. Сырой продукт очищали колоночной флэш-хроматографией на силикагеле (5-10% MeOH в ДХМ в качестве элюента) для получения 163 мг соединения 5 в виде прозрачного вязкого масла с выходом 86%. 1H-ЯМР полученного продукта был совместим с желаемой структурой. Анализ ВЭЖХ полученного продукта показывал чистоту 97,4% (AUC).
Пример 6
Приготовление B263, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метиладенозина

СХЕМА СИНТЕЗА:

Осуществляли синтез пронуклеотида B263 (94 мг, общий выход 6%) из его исходного нуклеозида 2'-C-метил-6-NH-диметокситритил-аденозина (1,59 г, 2,73 ммоль), следуя процедуре, сходной с процедурой, которая описана для синтеза пронуклеотида, приготовленного в примере 2 (процедура A, стратегия b), и выделяли его в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,80 (с, 3H), 0,97-0,98 (д, J=4,26 Гц, 6H), 3,02 (м, 2H), 3,34-3,35 (м, 2H), 3,76-3,96 (м, 4H), 4,03-4,05 (м, 2H), 4,15-4,17 (м, 2H), 4,76-4,79 (м, 1H), 5,32 (с, 1H), 5,34-5,36 (м, 1H), 5,45-5,55 (м, 1H), 5,93 (с, 1H), 7,1-7,4 (м, 7H), 8,14 (с, 1H), 8,21 (1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,75 и 9,86 (2c); сканирование ES+ 611 (M+H)+, λмакс=258 нм; ВЭЖХ (0-100% ацетонитрила (ACN) в течение 8 минут) tR=4,79 мин, λмакс=260,8 нм.
Пример 7
Приготовление B229, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метилуридина


Реактивы и условия: (i) pTsOH.H2O, CH(OEt)3, ацетон, КТ;
(ii) PivCl, пиримидин, КТ; (iii) бензиламин, CCl4, КТ;
(iv) водн. 90% ТФУ, ДХМ, КТ
Осуществляли синтез пронуклеотида 6 (446 мг, 0,76 ммоль, общий выход 9% после 4 этапов) из его исходного нуклеозида 1. следуя процедуре, сходной с процедурой, в которой описан синтез пронуклеотида, приготовленного в примере 2, стратегия A.
B229 6.
1H ЯМР (400 МГц, ДМСО-d6): δ 0,98 (с, 3H, CH3); 1,10 (с, 6H, 2×CH3); 3,03 (м, 2H, CH2S); 3,41 (м, 2H, CH 2OH, J=5,6 Гц); 3,61 (м, 1H, H-3'); 3,8-4,0 и 4,05-4,25 (стеки, 5H, NCH 2Ph, H-4', H-5' и H-5"); 4,05-4,25 (2×1H, 2×м, CH 2OP); 4,91 (т, 1H, 3'-OH, D2O заменяемый, J=5,62 Гц); 5,20 (ушир.с, 1H, 2'-OH, D2O заменяемый); 5,39 (a-t, 1H, CH2OH, D2O заменяемый, J=7,32 Гц); 5,52 (м, 1H, H-5); 5,65 (м, 1H, PhNH, D2O заменяемый); 5,8 (ушир.с, 1H, H-1'); 7,2-7,32 (м, 5H, ArH); 7,55 (a-dd, 1H, H-6); 11,37 (ушир.с, 1H, NH, D2O заменяемый).
31P ЯМР (161,8 МГц, ДМСО-d6): δ 9,73 и 9,98 (соотношение сигналов путем вычисления интеграла 52:48)
m/z (ES+) 588,11 (M+H)+.
ВЭЖХ (способ 20): химическая чистота 99,2%, 3,48 минут.
Анализ CHN (определение углерода, водорода и азота): Найдено: C, 49,29, H, 5,95, N, 6,88, P, 5,16; C24H34N3O10PS требует C, 49,06, H, 5,83, N, 7,15, P, 5,46.
[α]D 23 +26,3 (c, 0,571 в H2O).
νмакс (KBr): 3373 (Br, NH и О), 1682 (C=O).
Пример 8
Приготовление B186, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метилинозина

СХЕМА СИНТЕЗА:

Пронуклеотид B186 (314 мг, общий выход 8%), синтезировали из его родительского нуклеозида 2',3'-O-изопропилиден-2'-C-метилинозина (2,0 г, 6,26 ммоль), следуя процедуре, сходной с процедурой, в которой описан синтез пронуклеотида, приготовленного в примере 2 (процедура A, стратегия a), и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,79 (с, 3H), 1,09 (с, 6H), 3,01-3,04 (т, J=6,53 Гц, 2H), 3,42 (с, 2H), 3,84-3,91 (м, 2H), 3,94-4,03 (м, 3H), 4,05-4,09 (м, 1H), 4,15-4,26 (м, 2H), 4,92 (с, 1H), 5,36 (с, 1H), 5,43 (т, J=6,54 Гц, 1H), 5,62-5,71 (м, 1H), 5,94 (с, 1H), 7,18-7,22 (м, 1H), 7,25-7,30 (м, 4H), 8,08 (с, 1H), 8,10 (с, 1H), 12,15 (ушир.с, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) 5 (м.д.) 9,76-9,90 (2s); сканирование ES+ 612(M+H)+, λмакс=240,7 нм; ВЭЖХ (0-100% ацетонитрила (ACN) в течение 8 минут) tR=4,72 мин, λмакс=243,1 нм.
Пример 9
Приготовление B396, гидрокси-tBuSATE N-бензилфосфорамидатного производного 9-[2-C-метил-β-рибофуранозил]-6-хлорпурина

СХЕМА СИНТЕЗА:

Пронуклеотид B396 (75 мг, общий выход 10%) синтезировали из его исходного нуклеозида 9-[2-C-метил-β-рибофуранозил]-6-хлорпурина (571 мг, 1,90 ммоль), следуя процедуре, сходной с процедурой, в которой описан синтез пронуклеотида, приготовленного в примере 4, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,82 (д, J=2,63 Гц, 3H), 1,07 (с, 6H), 3,02 (м, 2H), 3,40-3,41 (кв, J=3,36 Гц и J=1,89 Гц, 2H), 3,85-3,98 (м, 4H), 4,12 (с, 2H), 4,25 (м, 2H), 4,89-4,90 (м, 1H), 5,47 (с, 1H), 5,50 (с, 1H), 5,62-5,70 (м, 1H), 6,10 (д, J=1,23 Гц, 1H), 7,17-7,29 (м, 5H), 8,76 (с, 1H), 8,82 (с, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,91 и 9,79 (2c); сканирование ES+ 630 (M+H)+, λмакс=260 нм; ВЭЖХ (0-100% ацетонитрила (ACN) в течение 8 минут) tR=4,42 мин, λмакс=265 нм.
Пример 10
Приготовление B307, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2',3'-О-карбонат-2'-C-метилгуанозина

СХЕМА СИНТЕЗА:

N-Бензиламинил-2',3'-O-карбонат-2'-C-метилгуанозин-5'-ил-О-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил) фосфат (С1):
Соединение B2 [см. соединение 7, пример 3, процедура A] (250 мг, 0,288 ммоль) растворяли в диметилформамиде (3,5 мл) и обрабатывали 1,1-карбонилдиимидазолом (186,60 мг, 1,15 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов 30 минут и выпаривали при пониженном давлении (температура ванны не превышала 30°C). Сырой остаток подвергали хроматографии на силикагеле, с элюированием метанолом в дихлорметане при градиенте 0-10% для получения С1 в виде бесцветного масла. (68 мг, 26%). Соединение С1: 1H ЯМР (400 МГц, ДМСО-d6) δ 10,80 (1с, 1H, NH), 7,80 (с, 1H, H-8), 7,33-7,18 (м, 20H, 4C6H5), 6,66 (с1, 2H, NH2), 6,30 (с, 1H, H-1'), 5,78 (м, 1H, PNH), 5,22 (м, 1H, H-3'), 4,47-4,30 (м, 2H, H-4' и H-5'a), 4,20-4,05 (м, 1H, H-5'b), 3,99-3,87 (м, 4H, CH2O и CH2N), 3,10-3,03 (м, 4H, CH2S и CH2OTr), 1,27 (с, 3H, CH3), 1,11 (с, 6H, 2CH3), 31P ЯМР (162 МГц, ДМСО-d6,) δ 10,42 (с), 10,18 (с). LR LC/MS (M+H+) 895,4 (5,57 мин), УФ: λмакс=253 нм.
N-Бензиламинил-O-(гидрокси-трет-бутил-S-ацил-2-тиоэтил)-2',3'-О-карбонат-2'-C-метилгуанозин-5'-илфосфат B307 (Соединение C2):
Соединение C1 (65 мг, 0,073 ммоль) растворяли в дихлорметане (260 мкл) и обрабатывали ТФУ (26 мкл). Смесь перемешивали при комнатной температуре в течение 15 минут, затем разводили этанолом, выпаривали досуха (температура ванны не превышала 30°C) и выпаривали совместно с толуолом. Полученный остаток очищали обращенно-фазовой хроматографией (C 18) на колонке с силикагелем, с элюированием ацетонитрила в воде с градиентом 0-100% и лиофилизировали из смеси воды/диоксана, чтобы получить B307 (Соединение C2) (34 мг, 72%, лиофилизированный порошок белого цвета). B307 (Соединение C2):
1H ЯМР (400 МГц, ДМСО-d6) δ 10,84 (1с, 1H, NH), 7,80 (с, 1H, H-8), 7,32-7,20 (м, 5H, C6H5), 6,69 (1с, 2H, NH2), 6,30 (с, 1H, H-1'), 5,77 (м, 1H, PNH), 5,25 (д, 1H, H-3', J3'-4'= 20,0 Гц), 4,92 (1с, 1H, OH), 4,50-4,41 (с, 2H, CH2OH), 3,03 (т, 2H, CH2S, JCH2s-CH2O=8,0 Гц), 1,30 (с, 3H, CH3), 1,10 (с, 3H, CH3), 1,08 (с, 3H, CH3). 13C ЯМР (100 МГц, ДМСО-d6): δ 204,4 (CO), 154,5 (C-4), 153,1 (C-2), 150,7 (C-6), 140,9 (C6H5), 135,6 (C-8), 128,7-127,3 (5C, C6H5), 117,0 (C-5), 89,7 (C-1'), 83,7 и 83,6 (2C, C-2' и C-3'), 81,8 (C-4'), 68,8 (CH2OH), 65,1 (CH2O), 64,5 (C-5'), 52,2 (C(CH3)2CH2OH), 44,7 (CH2N), 28,7 (CH2S), 22,3 (2C, 2CH3), 18,3 (CH3). 31P ЯМР (162 МГц, ДМСО-d6) δ 10,39 (с), 10,15 (с). LRLC/MX (2M+H+) 1305,4 (M+H+) 653,2 (2M-H-) 1303,8 (M-H-) 651,4 (5,57 мин), HRFAB-MC C26H34O10N6PS (M+H+). Рассчитано: 653,1795, Найдено: 653,1819. УФ: λмакс=251 нм, Rf=0,67 (MeOH/CH2Cl, 20/80 об/об).
Пример 11
Приготовление B242, гидрокси-tBuSATE N-(4-трифторметил)бензилфосфорамидатного производного 2'-C-метилгуанозина

СХЕМА СИНТЕЗА:

2'-C-Метилгуанозин-5'-ил-N-(4-трифторметил)бензиламинил-О-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)фосфат (D1):
К раствору соединения В1 [см. соединение 7, пример 3, процедура A] (355 мг, 0,465 ммоль) в безводном тетрахлориде углерода (4,65 мл) добавляли 4-трифторметилбензиламин (331 мкл, 2,324 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа 30 мин и выпаривали при пониженном давлении (температура ванны не превышала 30°C). Полученный остаток подвергали хроматографии на силикагеле, с элюированием с градиентом 0-10% метанолом в дихлорметане, для получения D1 в виде белого твердого вещества. (420 мг, 96%). Соединение D1: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,77-7,20 (м, 20H, 3 C6H5, C6H4CF3 и H-8), 6,57 (1с, 2H, NH2), 5,84-5,75 (м, 2H, H-1' и PNH), 5,50 (м, 1H, OH-3'), 4,26-3,86 (м, 8H, H-3', H-4', H-5', CH2O и CH2N), 3,10 (т, 2H, CH2S, JCH2S-CH2O=4,0 Гц), 3,03 (м, 2H, CH2OTr), 1,11 (с, 6H, 2CH3), 0,82 (с, 3H, CH3). 13C ЯМР (100 МГц, ДМСО-d6): δ 204,0 (C=O), 157,2 (C-4), 154,2 (C-2), 151,3 (C-6), 145,8-143,9 (4C, 3C6H5 и C6H4CF3), 135,6 (C-8), 129,0-120,0 (20C, 3 C6H5 и C6H4CF3), 117,0 (C-5), 91,0 (C-1'), 86,1 (C(C6H5)), 80,7 (C-3'), 78,7 (C-2'), 73,3 (C-4'), 70,0 (CH2OTr), 65,9 (CH2O), 64,4 (C-5'), 50,8 (C(CH3)2CH2OTr), 44,2 (CH2N), 28,8 (CH2S), 22,7 (2C, 2CH3), 20,4 (CH3). 31P ЯМР (162 МГц, ДМСО-d6): δ 9,80 (c), 9,64 (с), 19F ЯМР (376 МГц, ДМСО-d6): δ - 60,8 (c), LR LC/MS (M+H+) 937,3 (M-H-) 935,4 (5,47 мин), УФ: λмакс=254 нм, Rf=0,61 (MeOH/CH2Cl, 15/85 об/об).
O-(Гидрокси-трет-бутил-S-ацил-2-тиоэтил)-2'-C-метилгуанозин-5'-ил-N-(4-трифторметил)бензиламинилфосфат B242 (Соединение D2):
Соединение D1 (400 мг, 0,427 ммоль) растворяли в дихлорметане (1,6 мл) и обрабатывали ТФУ (160 мкл). Смесь перемешивали при комнатной температуре в течение 15 минут, затем разводили этанолом, выпаривали досуха (температура ванны не превышала 30°C) и выпаривали совместно с толуолом. Полученный остаток подвергали хроматографии на силикагеле, с элюированием метанолом в дихлорметане с градиентом 0-15%, и затем очищали обращенно-фазовой хроматографией (C 18) на колонке с силикагелем, элюировали ацетонитрилом в воде с градиентом 0-100% и лиофилизировали из смеси воды/диоксана, чтобы получить соединение B2742 (Соединение D2) (90 мг, 30%, лиофилизированный порошок белого цвета). B242 (Соединение D2): 1H ЯМР (400 МГц, ДМСО-d6) δ 10,54 (1с, 1H, NH), 7,75 (с, 1H, H-8), 7,75-7,52 (м, 4H, C6H4CF3), 6,50 (с1, 2H, NH2), 5,82-5,74 (м, 2H, H-1' и PNH), 5,40 (м, 1H, OH-3'), 5,17 (с, 1H, OH-2'), 4,92 (т, 1H, OH, JOH-CH2=4,0 Гц), 4,26-3,84 (м, 8H, H-3', H-4', H-5', CH2O и CH2N), 3,41 (д, 2H, CH2OH, JCH2-OH=4,0 Гц), 3,03 (т, 2H, C2S, JCH2S-CH2O=8,0 Гц), 1,07 (с, 6H, 2CH3), 0,82 (с, 3H, CH3). 13C ЯМР (100 МГц, ДМСО-d6): δ 204,4 (C=O), 157,2 (C-4), 154,1 (C-2), 151,2 (C-6), 145,9 (C6H4CF3), 135,8 (C-8), 128,3-125,4 (6C, C6H4CF3), 117,0 (C-5), 90,9 (C-1'), 80,5 (C-3'), 78,7 (C-2'), 73,2 (C-4'), 68,8 (CH2OH), 66,0 (CH2O), 64,4 (C-5'), 52,2 (C(CH3)2CH2OH), 44,3 (CH2N), 28,7 (CH2S), 22,3 (2C, 2 CH3), 20,4 (CH3), 31P ЯМР (162 МГц, ДМСО-d6): δ 9,62 (с), 9,77 (с), 19F ЯМР (376 МГц, ДМСО-d6): δ - 60,8 (с), LR LC/MS (M+H+) 695,2 (M-H-) 693,4 (4,25 мин), HRFAB-MS C26H35O9N6F3PS (M+H+). Рассчитано: 696,1876, Найдено: 695,1874. УФ: λмакс=253 нм, Rf=0,43 (MeOH/CH2Cl2, 20/80 об/об).
Пример 12
Приготовление B503, гидрокси-tBuSATE N-бензилфосфорамидатного производного 9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин

СХЕМА СИНТЕЗА:

{9-[(2R)2-Дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-О-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)Н-фосфонат (G1):
Соединение B5 [по неопубликованным результатам] (100 мг, 0,32 ммоль) и соединение F3 [см. соединение 5 из примера 2] (246 мг, 0,42 ммоль) выпаривали совместно с безводным пиридином и растворяли в указанном растворителе (4,8 мл). Пивалоилхлорид (80 мкл, 0,64 ммоль) добавляли по капле при -15°C, и раствор перемешивали при той же температуре в течение 2 часов. Реакционную смесь разводили дихлорметаном и нейтрализовали водным раствором 0,5M NH4Cl. Смесь разделяли между дихлорметаном и водной 0,5M NH4Cl, органические фазы объединяли, высушивали Na2SO4, выпаривали при пониженном давлении (температура ванны не превышала 30°C) и дважды выпаривали совместно с толуолом. Неочищенную смесь очищали колоночной флэш-хроматографией с элюированием метанолом в дихлорметане + 0,2% уксусной кислоты с градиентом 0-10%), чтобы получить желаемый продукт G1 в виде бесцветного масла (68 мг, 28%). Соединение G1: 1H ЯМР (400 МГц, ДМСО-d6): δ 10,72 (1c, 1H, NH), 7,83 (с, 1H, H-8), 7,35-7,11 (м, 15H, 3C6H5), 6,59 (м, 2H, NH2), 6,36 (д, 1H, ОН-3', JOH-3'=7,6 Гц), 6,14 (д, 1H, H-1', J1'-F=18,0 Гц), 4,65 (м, 1H, H-3'), 4,40-4,33 (м, 2H, H-5'), 4,10-4,01 (м, 3H, H-4' и CH2O), 3,93 (д, 1H, CCH, 4JH-F=5,6 Гц), 3,15-3,12 (м, 2H, CH2S), 3,04 (с, 2H, CH2OTr). 31P ЯМР (162 МГц, ДМСО-d6): δ 9,50 (с), 9,22 (c). 19F ЯМР (376 МГц, ДМСО-d6): δ -156,5 (м), LR LC/MS (B) (M+Na+) 798,2 (M-H-) 774,2 (4,93 мин), УФ: λмакс=254 нм, Rf=0,48 (MeOH/CH2Cl, 15/85, об/об).
N-Бензиламинил-{9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-O-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)фосфат (G2)
К раствору соединения G1 (68 мг, 0,088 ммоль) в безводном тетрахлориде углерода (880 мкл) по капле добавляли бензиламин (48 мкл, 0,44 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 2 часа и выпаривали досуха (температура ванны не превышала 30°C). Неочищенную смесь фильтровали на устройстве с силикагелем, с элюированием метанолом в дихлорметане с градиентом 0-10%, чтобы получить соединение G2 в виде твердого вещества белого цвета (80 мг, количественный выход). Соединение G2: 31P ЯМР (162 МГц, ДМСО-d6): δ 9,95 (с) 9,80 (с), 19F ЯМР (376 МГц, ДМСО-d6): δ -157,5 (м), LR LC/MS (B) (M+H+) 881,3 (M-H-) 879,4 (5,18 мин), УФ: λмакс=254 нм, Rf=0,31 (MeOH/CH2Cl, 15/85, об/об).
N-Бензиламинил-{9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-O-(гидрокси-трет-бутил-S-ацил-2-тиоэтил)фосфат B503 (Соединение G3):
Соединение G2 (80 мг, 0,09 ммоль) растворяли в дихлорметане (320 мкл) и обрабатывали ТФУ (32 мкл). Смесь взбалтывали при комнатной температуре в течение 10 минут, фильтровали с помощью твердофазной экстракционной колонки с элюированием метанолом в дихлорметане с градиентом 0-30%, затем очищали обращенно-фазовой (C 18) колоночной хроматографией на силикагеле, элюировали ацетонитрилом в воде с 0-100% градиентом и лиофилизировали из смеси воды/диоксана, чтобы получить соединение B503 (Соединение G3) (15 мг, 26%, лиофилизированный порошок белого цвета). B503 (Соединение G3): 1H ЯМР (400 МГц, ДМСО-d6): δ 10,61 (1с, 1H, NH), 7,83 (с, 1H, H-8), 7,30-7,18 (м, 5H, C6H5), 6,60 (1с, 2H, NH2), 6,32 (м, 1H, OH-3'), 6,11 и 6,12 (2д, 2×1H, 2H-1', J1'-F=18,0 Гц), 5,68 (м, 1H, PNH), 4,93 (т, 1H, OH, JOH-CH2=5,5 Гц), 4,61 (м, 1H, H-3'), 4,26-4,18 (м, 2H, H-5'), 4,08 (м, 1H, H-4', 3,98-3,82 (м, 5H, CH2O, CH2N и CCH), 3,42 (д, 2H, CH2OH, JCH2-OH=5,0 Гц), 3,01 (м, 2H, CH2S), 1,09 (с, 6H, 2CH3). 31P ЯМР (162 МГц, ДМСО-d6): δ 9,92 (с), 9,79 (с). 19F ЯМР (376 МГц, ДМСО-d6): δ -156,8 (м), LR LC/MS (B) (M+H+) 639,2 (M-H-) 637,3 (3,85 мин). HRFAB-MS C26H33O8N6FPS (M+H+). Рассчитано: 639,1802, Найдено: 639,1816. УФ: λмакс=253 нм, Rf=0,46 (MeOH/CH2Cl2, 20/80 об/об).
Стартовый нуклеозид синтезировали следующим образом:
Синтез 9-[(2R)2-дезокси-2-C-этинил-2-фтор-β-D-эритрофуранозил]гуанина} (D961, стартовый нуклеозид из примера 12) и синтез его трифосфатного производного B427)

СХЕМА СИНТЕЗА:

9-[3,5-О-(1,3-Диил-1,1,3,3-тетраизопропилдисилоксан)-рибофуранозил]-N 2 -изобутирилгуанин (В1): Hirao, L; Ishikawa, M.; Miura, K. Chem. Lett. 1986, 11, 1929-1932.
9-[3,5-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-арабинофуранозил]-N2-изобутирилгуанин (B2): К суспензии CrO3 (11,07 г, 110,76 ммоль) в дихлорметане (220 мл) добавляли при 0°C уксусный ангидрид (10,4 мл, 110,76 ммоль) и безводный пиридин (17,82 мл, 221,52 ммоль). По капле добавляли соединение В1 (22 г, 36,92 ммоль) в растворе дихлорметана (110 мл). Удаляли охлаждающую ванну, и полученный раствор перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь вливали в холодный этилацетат, подвергали гель-фильтрации на силикагеле и целите, концентрировали досуха и дважды выпаривали совместно с толуолом. Полученный остаток растворяли в дихлорметане и взбалтывали с избытком MgSO4 в течение ночи, фильтровали и выпаривали для получения кетона. Триметилсилилацетилен (12,5 мл, 88,60 ммоль) растворяли в безводном ТГФ (98 мл) в атмосфере аргона. Бутиллитий (55,4 мл, 1,6М р-р в гексанах) добавляли по капле при -78°C. Реакционную смесь взбалтывали в течение 30 минут при -78°C и затем оставляли для нагревания до -55°C. По капле при -78°C добавляли кетон в растворе ТГФ (49 мл). Реакционную смесь взбалтывали в течение 1 часа при -78°C, и затем оставляли для нагревания до -30°C и взбалтывали в течение 3 часов. Реакцию гасили осторожным добавлением водного насыщенного NH4Cl (72 мл) при -78°C. После нагревания до комнатной температуры реакционную смесь разводили этилацетатом, промывали дважды насыщенным солевым раствором, высушивали (Na2SO4) и концентрировали досуха. Сырой материал очищали, используя колоночную флэш-хроматографию с элюированием MeOH в 1,5% дихлорметана, чтобы получить соединение B2 (8,59 г, 34%, 2 этапа) в виде пены бледно-желтого цвета. Соединение B2: 1H ЯМР (250 МГц, ДМСО-d6): δ 12,10 (1c, 1H, NH), 11,69 (1с, 1H, NH), 7,91 (с, 1H, H-8), 6,69 (с, 1H, OH), 5,94 (с, 1H, H-1'), 4,29 (д, 1H, H-3', J3'-4'=5,5 Гц), 3,85-3,95 (м, 3H, H-4', H-5' и H-5"), 2,46 (м, 1H, CH(CH3)2), 0,90-1,08 (м, 30H, iPr и CH(CH3)3), 0,00 (с, 9H, Si(CH3)2). LC/MS (A): (M+H+) 692,4 (24,96 мин). УФ: λмакс1=254 нм, λмакс2=281 нм, Rf=0,34 (MeOH/CH2Cl, 15/85, об/об).
9-[(2R)-2-Дезокси-2-фтор-3,5-O(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритрофуранозил]-N
2
-изобутирилгуанин (B3):
Соединение B2 (2,00 г, 2,89 ммоль) растворяли в сухом ДХМ (60 мл) в атмосфере аргона и добавляли пиридин (1,45 мл, 18,06 ммоль). Реакционную смесь охлаждали до -20°C, и по капле добавляли DAST (4,11 мл, 31,35 ммоль). Охлаждающую ванну удаляли после завершения добавления. Перемешивание продолжали в течение 1 ч 15 мин, и реакционную смесь растворяли этилацетатом, и вливали в насыщенный NaHCO3 и взбалтывали в течение 5 мин. Органический слой промывали насыщенным солевым раствором, высушивали (Na2SO4), концентрировали и очищали хроматографией на силикагеле, с элюированием этилацетатом в ДХМ (2%), чтобы получить желаемое соединение B3 (1,41 г, 70%) в виде масла желтого цвета. Соединение B3: 1H (250 МГц, ДМСО-d6): δ 12,22 (с, 1H, NH), 8,09 (с, 1H, H-8), 6,21 (д, 1H, H-1', J1'-F=15,6 Гц), 4,54 (дд, 1H, H-3', J3'-F=23,6 Гц, J3'-4'=9,8 Гц), 4,33 (м, 1H, H-5', 2J5'-5''= 13,1 Гц), 4,16 (м, 1H, H-5"), 2,81 (м, 1H, CH(CH3)2), 1,13-1,03 (м, 34H, iPr и CH(CH3)2), 0,08 (с, 9H, Si(CH3)3, 3JH-H=6,9 Гц). ЯМР 19F (235 МГц, ДМСО-d6): δ - 160,26 (дд, JF-1'=16,1 Гц, JF-3'=23,3 Гц). LC/MS (A): (M+H+) 694,7 (24,02 мин). LRFAB-MS (GT): 694 (M+H)+, 692 (M-H)-, УФ: λмакс=256 нм. Rf=0,46 (MeOH/CH2Cl, 05/95, об/об).
9-[(2R)-2-Дезокси-2-C-этинил-2-фтор-β-D-эритрофуранозил]-N
2
-изобутирилгуанин (B4):
Соединение B3 (1,31 г, 1,89 ммоль) растворяли в метаноле (13,8 мл) и добавляли фторид аммония (908,9 мг, 24,54 ммоль). Получаемый раствор взбалтывали при нагревании с обратным холодильником в течение 1 ч и выпаривали досуха. Сырой материал очищали хроматографией на силикагеле, с элюированием метанолом в дихлорметане с поэтапным градиентом 6-10% для получения соединения B4 (344 мг, 48%) в виде масла бледно-желтого цвета. Соединение B4: 1H ЯМР (400 МГц, ДМСО-d6): δ 12,18 (1c, 1H, NH), 11,77 (1c, 1H, NH), 8,34 (с, 1H, H-8), 6,29 (д, 1H, OH-3', JOH-3'=7,5 Гц), 6,20 (д, 1H, H-1', J1'-F=16,2 Гц), 5,39 (т, 1H, OH-5', JOH-5'=5,1 Гц), 4,52 (дт, 1H, H-3', J3'-F=22,9 Гц), 3,98 (м, 1H, H-4'), 3,90-3,85 (м, 2H, H-5' и этинил), 3,72 (м, 1H, H-5"), 2,52 (м, 1H, CH(CH3)2), 1,14 (д, 6 H, CH(CH3)2, 3JH-H=6,9 Гц). 13C ЯМР (100 МГц, ДМСО-d6): δ 180,7 (C-6), 155,3 (C-2), 148,9 (C-4), 137,3 (C-8), 120,4 (C-5), 95,8 (д, C-2', 1J2'-F=182,1 Гц), 87,7 (д, C-1', 2J1'-F=39,2 Гц), 83,4 (д, CCH, 3JC-F=9,1 Гц), 82,6 (C-4'), 75,9 (д, CCH, 2JC-F=31,2 Гц), 72,9 (д, C-3', 2J3'-F=19,1 Гц), 59,3 (C-5'). 19F ЯМР (235 МГц, ДМСО-d6) δ - 158,9 (м). LC/MS (A): (M+H+) 380,3 (8,34 мин), УФ: λмакс1=260 нм, δмакс2=211 нм, Rf 0,40 (MeOH/CH2Cl, 15/85, об/об).
9-[(2R)-2-Дезокси-2-C-этинил-2-фтор-β-D-эритрофуранозил]гуанин D961 (Соединение B5):
Соединение B4 (0,78 г, 1,33 ммоль) растворяли в насыщенном метанольном растворе аммония (62 мл) и взбалтывали при комнатной температуре в течение 20 ч. Реакционную смесь затем выпаривали досуха при пониженном давлении. Остаток растворяли в воде и промывали дважды этилацетатом. Водный слой выпаривали и очищали обращенно-фазовой колоночной хроматографией (C 18) с элюированием ацетонитрилом в воде с градиентом 2-15%. Полученный остаток затем растворяли в горячем этилацетате, фильтровали и высушивали для получения D961 (Соединение B5) (134 мг, 33%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ 10,70 (1c, 1H, NH), 7,98 (с, 1H, H-8), 6,60 (1c, 2H, NH2), 6,21 (д, 1H, OH-3', JOH-3'=7,6 Гц), 5,83 (д, 1H, H-1', J1'-F = 16,9 Гц), 5,29 (т, 1H, ОН-5', JOH-5'=5,2 Гц), 4,50 (тд, 1H, H-3', J3'-F-22,8 Гц, J3'-4'=9,2 Гц), 3,93-3,81 (м, 3H, H-4', H-5' и этинил), 3,70 (м, 1H, H-5"). ЯМР 13C (100 МГц, ДМСО-d6): δ 157,2 (C-6), 154,3 (C-2), 151,05 (C-4), 135,1 (C-8), 116,7 (C-5), 96,4 (д, C-2', 1Jc-F=182,1 Гц), 87,4 (д, C-1', 2JC-F=39,2 Гц), 83,1 (д, CCH, JC-F=9,1 Гц), 82,4 (C-4'), 76,2 (д, CCH, 2JC-F=31,2 Гц), 73,2 (д, C-3',2JC-F=20,1 Гц), 59,5 (C-5'). 19F ЯМР (235 МГц, ДМСО-d6): δ -158,5 (м). LC/MS (A): (M+H+) 310,1 (5,55 мин), LRFAB-MS (GD: 619 (2M+H)+, 310 (M+H)+, 152 (B+H)+, 617 (2M-H)-, 308 (M-H)-. УФ: λмакс=254 нм.

СХЕМА СИНТЕЗА:

Стандартная процедура приготовления нуклеозида 5'-трифосфата: (Ludwig, J. Acta Biochim. Biophys. Acad. Sci. Hung. 1981, 16, 131-133.)
К раствору нуклеозида (0,286 ммоль) в триэтилфосфате (750 мкл) добавляли фосфорил хлорид (75 мкл, 0,807 ммоль) при 0°C. Указанную реакционную смесь A перемешивали в течение ночи при 5°C. Трибутиламмония пирофосфат (PPi/Bu3N 1/1,5, 1 г, 2,19 ммоль) растворяли в безводном ДМФ (2 мл). Трибутиламин (420 мкл, 1,76 ммоль) добавляли к PPi, и полученную смесь взбалтывали в течение 15 минут при 0°C. Указанный раствор в количестве 2,4 мл добавляли к реакционной смеси A. Реакционную смесь взбалтывали при 0°C в течение 1 мин. Реакционную смесь аккуратно гасили посредством 1М TEAB (уровень pH=7,5, 10 мл), перемешивали в течение 20 минут при 0°C, затем разводили этилацетатом и водой. Водную фазу выпаривали при пониженном давлении. Сырой материал подвергали хроматографии на колонке DEAE-Sephadex, с элюированием 1М TEAB с градиентом 10-3). Желаемые фракции объединяли, выпаривали при пониженном давлении и выпаривали совместно со смесью воды/метанола, и в конце выпаривали совместно с водой. Полученный остаток очищали на полупрепаративной ВЭЖХ. Фракции, содержащие ожидаемый продукт, выпаривали при пониженном давлении, выпаривали совместно со смесью воды/метанола и лиофилизировали из воды. Триэтиламмониевую соль трифосфата трижды элюировали водой на колонке Dowex на смоле Na+, чтобы после лиофилизации из воды получить соли натрия.
Натриевая соль 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритрофуранозил]гуанина 5'-трифосфата (B427): 1H ЯМР (400 МГц, D2O): δ 7,97 (с, 1H, H-8), 6,19 (д, 1H, H-1', 3J1'-F = 16,0 Гц), 4,70 (м, 1H в H2O, H-3'), 4,39 (м, 1H, H-5'), 4,29-4,22 (м, 2H, H-4' и H-5"), 2,98 (д, 1H, этинтил, 4JH-F=5,0 Гц). 31P ЯМР (162 МГц, D2O): -10,50 (д, 1P, Pγ, JPγ-Pβ=19,4 Гц), -11,03 (д, 1P, Pα, JPα-Pβ=19,4 Гц), -22,38 (т, 1P, Pβ, JPβ-Pγ=JPβ-Pα=19,4 Гц). l9F ЯМР (376 МГц, ДМСО-d6): δ -159,1 (м), LRFAB-MS (GT): 638 (M+Na)+, 616 (M+H)+, 594 (M-Na+2H)+, 572 (M-2Na+3H)+, 550 (M-3Na+4H)+, 592 (M-Na)-, 570 (M-2Na+H)-, 548 (M-3Na+2H)-.
Пример 13
Приготовление B306, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метил-5-аза-7-деазагуанозина

СХЕМА СИНТЕЗА:

Пронуклеотид B306 (25 мг, общий выход 6%) синтезировали из его исходного нуклеозида 2'-C-метил-5-аза-7-деазагуанозина (200 мг, 0,67 ммоль), следуя процедуре, сходной с процедурой, в которой описан синтез пронуклеотида, приготовленного в примере 4, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,90-0,91 (д, J=2,56 Гц, 3H), 1,09 (д, J=4,26 Гц, 6H), 3,07-3,10 (т, J=6,66 Гц, 2H), 3,42 (д, J=5,64 Гц, 2H), 3,86-3,99 (м, 6H), 4,10-4,15 (м, 1H), 4,15-4,20 (м, 1H), 4,90-4,93 (т, J=5,64 Гц, 1H), 5,28 (с, 1H), 5,46-5,50 (м, 1H) 5,62-5,69 (м, 1H), 5,80 (с, 1H), 7,00 (с, 2H), 7,18-7,21 (м, 2H), 7,26-7,33 (м, 5H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,80-9,95 (2c); сканирование ES+ 627 (M+H)+, λмакс=261,7 нм; ВЭЖХ (0-100% ACN в течение 8 минут) tR=3,18 мин. λмакс=258,4 нм.
Пример 14
Приготовление B389, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метил-7-деазагуанозина

СХЕМА СИНТЕЗА:

Синтез пронуклеотида B389 (80 мг, общий выход 21%) осуществляли из его исходного нуклеозида 2'-C-метил-7-деазагуанозина (200 мг, 0,67 ммоль), следуя процедуре, сходной с процедурой, в которой описан синтез пронуклеотида, приготовленного в примере 4, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6 400 МГц) δ (м.д.) 0,74 (с, 3H), 1,09 (с, 6H), 3,0 (т, J=6,10 Гц, 2H), 3,42 (д, J=5,49 Гц, 2H), 3,8-4,0 (2м, 6H), 4,04-4,11 (м, 1H), 4,24-4,17 (м, 1H), 4,90-4,93 (т, J=5,36 Гц, 1H), 4,96-4,98 (д, J=4,76 Гц, 1H), 5,31-5,36 (м, 1H), 5,57-5,67 (м, 1H), 5,93 (с, 1H), 6,21-6,26 (м, 3H), 6,76 (д, J=22 Гц, 1H), 7,19-7,23 (м, 1H), 7,27-7,32 (м, 4H), 10,34 (ушир.с, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,77 и 9,90 (2с); сканирование ES+ 626 (M+H)+, λмакс=258,7 нм; ВЭЖХ (0-100% ацетонитрила (ACN) в течение 8 минут) tR=3,84 мин λмакс=259,6 нм.
Пример 15
Приготовление B288, гидрокси-tBuSATE N-бензилфосфорамидатного производного 3'-C-метилуридина

СХЕМА СИНТЕЗА:

Пронуклеотид B288 (34 мг, общий выход 3%) синтезировали из его исходного нуклеозида 3'-C-метилуридина (513 мг, 1,99 ммоль), следуя процедуре, сходной с процедурой, в которой описан синтез пронуклеотида, приготовленного в примере 4, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,09 (с, 6H), 1,15 (с, 3H), 3,00-3,05 (м, 2H), 3,30 (с, 1H), 3,42 (д, J=6,13 Гц, 2H), 3,76-3,79 (м, 1H), 3,86-3,99 (м, 6H), 4,92-4,94 (т, J=5,40 Гц, 1H), 4,97 (с, 1H), 5,47 (м, 1H), 5,59-5,62 (м, 1H), 5,67-5,78 (м, 1H), 5,83-5,87 (м, 1H), 7,20-7,24 (м, 1H), 7,30 (м, 4H), 7,66-7,71 (м, 1H), 11,32 (ушир.с, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,66 и 9,95 (2c); сканирование ES+ 588 (M+H)+, λмакс=261,7 нм).
Пример 16
Приготовление B350, гидрокси-tBuSATE N-бензилфосфорамидатного производного 3'-C-метилгуанозина

СХЕМА СИНТЕЗА:

3'-C-Метилгуанозин-5'-ил-О-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)-Н-фосфонат (Е1):
3'-C-метилгуанозин (NM76) (233,7 мг, 0,79 ммоль) и соединение A3 [см. соединение 5 из примера 2] (504,9 мг, 0,87 ммоль) выпаривали совместно с безводным пиридином и растворяли в указанном растворителе (11,8 мл). Пивалоилхлорид (193,7 мкл, 1,57 ммоль) добавляли по капле при -15°C, и раствор перемешивали при той же температуре в течение 2 ч. Реакционную смесь разводили дихлорметаном и нейтрализовали водным раствором 0,5M NH4Cl. Реакционную смесь разделяли на части между дихлорметаном и водным 0,5M NH4Cl, органические фазы объединяли, высушивали Na2SO4, выпаривали при пониженном давлении (температура ванны не превышала 30°C) и дважды выпаривали совместно с толуолом. Неочищенную смесь фильтровали на устройстве с силикагелем, элюировали (метанол в дихлорметане + 0,2% уксусной кислоты) с градиентом 0-10%, чтобы получить желаемый продукт Е1 (250 мг, 42%). Соединение Е1: 31P ЯМР (162 МГц, ДМСО-d6): δ 9,93 (с), 9,13 (с). LR LC/MC (M+H+) 521,1 (5,88 мин). УФ: λмакс=262 нм. Rf=0,21 (MeOH/CH2Cl, 15/85, об/об).
N-Бензиламинил-3'-C-метилгуанозин-5'-ил-О-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)фосфат (E2):
К раствору соединения Е1 (250 мг, 0,33 ммоль) в безводном тетрахлориде углерода (3,3 мл) добавляли по капле бензиламин (178 мкл, 1,637 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 1 ч 30 мин и выпаривали досуха (температура ванны не превышала 30°C). Неочищенную смесь фильтровали на устройстве с силикагелем, с элюированием метанолом в дихлорметане с градиентом 0-30%, чтобы получить соединение E2 в виде белого твердого вещества (290 мг, количественный выход). Соединение E2: 31P ЯМР (162 МГц, ДМСО-d6) δ 9,91 (с), 9,74 (с). LR LC/MC (M+H+) 869,3 (M-H-) 867,7 (5,20 минут). УФ: λмакс=253 нм. Rf=0,13 (MeOH/CH2Cl, 10/90, об/об).
N-Бензиламинил-О-(гидроксил-трет-бутил-S-ацил-2-тиоэтил)-3'-C-метилгуанозин-5'-илфосфат B350 (Соединение E3):
Соединение E2 (290 мг, 0,33 ммоль) растворяли в дихлорметане (1,16 мл) и обрабатывали ТФУ (113 мкл). Смесь взбалтывали при комнатной температуре в течение 10 минут, затем разводили этанолом, выпаривали досуха (температура ванны не превышала 30°C) и выпаривали совместно с толуолом. Полученный остаток подвергали хроматографии на силикагеле с элюированием метанолом в дихлорметане с градиентом 0-30%, затем очищали обращенно-фазовой (C 18) колоночной хроматографией на силикагеле, с элюированием ацетонитрилом в воде с градиентом 0-100% и лиофилизировали из смеси воды/диоксана, для получения B350 (Соединения E3) (15 мг, 7%, лиофилизированный порошок белого цвета). B350 (Соединение E3): 1H ЯМР (400 МГц, ДМСО-d6) δ 10,60 (м, 1H, NH), 7,90 (с, 1H, H-8), 7,30-7,19 (м, 5H, C6H5), 6,47 (1c, 2H, NH2), 5,72-5,59 (м, 2H, H-1' и PNH), 5,51 (д, 1H, OH-2', JOH2'-1'=8,0 Гц), 4,94-4,92 (2H, OH-3' и OH), 4,28 (м, 1H, H-2'), 4,01-3,83 (м, 7H, H-4', H-5', CH2O и CH2N), 3,41 (м, 2H, CH2OH), 3,02 (т, 2H, CH2S, JCH2S-CH2O=6,0 Гц), 1,20 (с, 3H, CH3), 1,09 (с, 6H, 2CH3). 31P ЯМР (162 МГц, ДМСО-d6; δ 9,86 (c), 9,72 (c), LR LC/MS (M+H+) 627,2 (M-H-) 625,5 (3,87 мин). HRFAB-MS C25H36O9N6PS (M+H+). Рассчитано 627,2002, полученный 627,2014. УФ: λмакс=251 нм.
Пример 17
Приготовление B305, гидрокси-tBuSATE N-бензилфосфорамидатного производного 1-[2-C-метил-β-рибофуранозил]-3-карбоксамидо-4-фторпиразола

СХЕМА СИНТЕЗА:

Пронуклеотид B305 (28,3 мг, общий выход 8%) синтезировали из его исходного 1-[2-C-метил-β-рибофуранозил]пиразоло-3-карбоксамид-4-фтор нуклеозида (180 мг, 0,65 ммоль), следуя процедуре, сходной с процедурой, в которой описан синтез пронуклеотида, приготовленного в примере 4, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,75 (с, 3H), 1,08-1,09 (д, J=3,35 Гц, 6H), 2,98-3,02 (м, 2H), 3,40-3,42 (м, 2H), 3,85-4,03 (м, 5H), 4,16-4,19 (м, 2H), 4,89-4,92 (м, 1H), 5,25-5,29 (м, 2H), 5,55 (с, 1H), 5,56-5,64 (м, 1H), 7,19-7,22 (м, 1H), 7,26-7,50 (м, 7H), 8,05 (д, J=4,32 Гц, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,75 и 9,90 (2c); 19F ЯМР (d6-ДМСО, 235 МГц) δ (м.д.) -170,70 (д, J=61,74 Гц, 1F); сканирование ES+ 605 (M+H)+, λмакс=233,7 нм; ВЭЖХ (0-100% ACN в течение 10 минут) tR=4,56 мин λмакс=235,2 нм.
Пример 18
Приготовление B436, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метил-7-деаза-7-фтораденозина

СХЕМА СИНТЕЗА:

Пронуклеотид B436 (30 мг, общий выход 9%) синтезировали из его исходного нуклеозида 2'-C-метил-7-деаза-6-NH-диметокситритил-аденозина (320 мг, 0,53 ммоль), следуя процедуре, сходной с процедурой, в которой описан синтез пронуклеотида, приготовленного в примере 2 (процедура A, стратегия b), и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,66 (с, 3H), 1,02 (с, 6H), 2,95-2,98 (т, J=6,10 Гц, 2H), 3,35 (д, J=5,49 Гц, 2H), 3,77-3,85 (м, 3H), 3,88-3,95 (м, 3H), 4,03-4,18 (м, 2H), 4,83-4,86 (т, J=5,44 Гц, 1H), 5,14 (с, 1H), 5,21-5,25 (т, J=7,40 Гц, 1H), 5,55-5,66 (м, 1H), 6,14 (с, 1H), 6,9-7,3 (м, 8H), 8,01 (с, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,77 и 9,89 (2s); 19F ЯМР (d6-ДМСО, 235 МГц) δ (м.д.) -166,85 (д, J=14,16 Гц, 1F); сканирование ES+ 628(M+H)+, λмакс=280,7 нм; ВЭЖХ (0-100% ACN в течение 10 минут) tR=4,78 мин λмакс=280,8 нм.
Пример 19
Приготовление B589, гидрокси-tBuSATE N-бензилфосфорамидатного производного 4'-C-метилуридина

СХЕМА СИНТЕЗА:

Следуя методике, описанной в примере 4, с исходным 4'-C-метилуридином (A437) (200 мг, 0,77 ммоль), вначале получали промежуточный продукт P3 (62 мг, 11%. 31P ЯМР (ДМСО-d6, 162 МГц) δ (ppm) 9,15, 9,56 (2с); сканирование ES+ 747 (M+Na)+, λмакс=259,7 нм), затем соединение P4 во время второго этапа (28 мг, 39%. сканирование ES+ 852 (M+Na)+), и в конце после лиофилизации из диоксана получали желаемое пролекарство B589 в виде порошка белого цвета (21 мг, 58%). 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,10 (с, 6H), 1,13 (с, 3H), 3,03 (т, J=6,42 Гц, 2H), 3,15 (д, J=5,29 Гц, 1H), 3,42 (д, J=5,67 Гц, 2H), 3,72-4,16 (м, 7H), 4,06-4,15 (м, 1H), 4,92 (т, J=5,29 Гц, 1H), 5,22 (д, J=5,29 Гц, 1H), 5,36-5,38 (2д, 1H), 5,57-5,60 (2д, 1H), 5,64-5,70 (м, 1H), 5,78-5,80 (2д, 1H), 7,20-7,31 (м, 5H), 7,60-7,64 (2д, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,58, 9,77 (2с); сканирование ES+ 610 (M+Na)+, λмакс=260,7 нм.
Пример 20
Приготовление B678, гидрокси-tBuSATE N-бензилфосфорамидатного производного 4'-C-фторметилгуанозина

СХЕМА СИНТЕЗА:

Следуя методике, описанной в примере 3, с исходным 4'-C-фторметилгуанозином (A402) (69,4 мг, 0,22 ммоль), после первого этапа в качестве промежуточного продукта получали соединение Р1 (67,5 мг, 39%). Сканирование ES- 780 (M-H)-. Результатом второго этапа было образование промежуточного продукта P2 (57,5 мг; 76%). Соединение P2 (26,3 мг, 0,03 ммоль) растворяли в дихлорметане (1 мл), и обрабатывали монтмориллонитом K10 (150 мг) и взбалтывали при комнатной температуре в течение 1 ч. Реакционную смесь непосредственно помещали в трубку SPE (для твердофазной экстракции) с силикагелем и экстрагировали MeOH в дихлорметане с градиентом 0-100%, чтобы после лиофилизации из диоксана/воды получить соединение B678 в виде порошка белого цвета (7,7 мг, 40%). 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,09 (2 1с, 6H), 3,03-3,05 (м, 2H), 3,42 (м, 2H), 3,87-4,00 (м, 6H), 4,15-4,24 (м, 2H), 4,43-4,66 (м, 2H), 4,74 (м, 1H), 4,93 (м, 1H), 5,52 (д, J=4,36 Гц, 1H), 5,58 (м, 1H), 5,70-5,73 (м, 1H), 5,75-5,77 (д, J=8,05 Гц, 1H), 6,52 (1с, 2H), 7,24-7,35 (м, 5H), 7,92 (2с, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,70, 9,83 (2с); 19F ЯМР (d6-ДМСО, 235 МГц) 5 (м.д.) -235,92, -236,25 (2с); сканирование ES+ 645 (M+H)+, λмакс=250,7 нм. ВЭЖХ (0-100% ACN в течение 8 минут) tR=3,91 мин, λмакс=251,1 нм.
Пример 21
Приготовление B704, гидрокси-tBuSATE N-бензилфосфорамидатного производного ацикловира

СХЕМА СИНТЕЗА:

Схема 1
9-(2-гидрокси-этоксиметил)гуанин-5'-ил-О-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)H-фосфонат (F1):
Ацикловир (200 мг, 0,89 ммоль) и соединение A3 [см. соединение 5 из примера 2] (674,2 мг, 1,15 ммоль) выпаривали совместно с безводным пиридином и растворяли в указанном растворителе (13,3 мл). Добавляли пивалоилхлорид (162 мкл, 1,15 ммоль) по каплям при -15°C и раствор перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разводили дихлорметаном и нейтрализовали водным раствором 0,5M NH4Cl. Смесь разделяли между дихлорметаном и водным 0,5M NH4Cl, органические фазы объединяли, высушивали Na2SO4, выпаривали при пониженном давлении (температура ванны не превышала 30°C) и дважды выпаривали совместно с толуолом. Неочищенную смесь подвергали фильтрации на силикагеле с элюированием (метанол в дихлорметане + 0,2% уксусной кислоты) с градиентом 0-15%, для получения желаемого продукта F1 (602 мг, 98%). Соединение F1:LR LC/MS (M+H+) 691,9 (M-H-) 690,0 (4,82 мин). УФ: λмакс=254 нм.
N-бензиламинил-9-(2-гидроксиэтоксиметил)гуанин-5'-ил-O-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)фосфат (F2):
К раствору соединения F1 (602 мг, 0,87 ммоль) в безводном тетрахлориде углерода (8,7 мл) по капле добавляли бензиламин (475 мкл, 4,35 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 1 ч 30 мин и выпаривали досуха (температура ванны не превышала 30°C). Неочищенную смесь затем подвергали хроматографии на силикагеле, с элюированием метанолом в дихлорметане с градиентом 0-10%, чтобы получить соединение F2 в виде твердого вещества белого цвета (550 мг, 79%). Соединение F2: 1H ЯМР (400 МГц, ДМСО-d6) δ 7,77 (с, 1H, H-8), 7,58-7,17 (м, 20H, 4C6H5), 6,68 (1cs, 2H, NH2), 5,59 (м, 1H, PNH), 5,32 (с, 2H, OCH2N), 3,92-3,78 (м, 6H, CH2SCH2O, CH2N, POCH2CH2O), 3,51 (т, 2H, POCH2CH2O, JCH2-CH2=5,2 Гц), 3,16 (с, 2H, CH2OTr), 3,00 (т, 2H, CH2S, JCH2S-CH2O=5,6 Гц), 1,12 (с, 6H, 2 CH3). 13C ЯМР (100 МГц, ДМСО-d6): δ 204,0 (C=O), 157,2 (C-4), 154,6 (C-2), 151,8 (C-6), 143,9 (4C, 4C6H5), 136,9 (C-8), 128,9-127,2 (20C, 4C6H5), 117,0 (C-5), 86,3 (1C, C(C6H5)3), 72,3 (OCH2N), 70,0 (CH2OTr), 68,2 (POCH2CH2O), 64,8 (POCH2CH2O), 64,2 (CH2SCH2O), 50,8 (C(CH3)2), 44,7 (CH2N), 28,8 (CH2S), 22,8 (2C, C(CH3)2). 31P ЯМР (162 МГц, ДМСО-d6) δ 9,79 (c). LR LC/MS (M+H+) 797,2 (5,15 мин), УФ: λмакс=254 нм, Rf=0,57 (MeOH/CH2Cl, 15/85, об/об).
N-бензиламинил-9-(2-гидроксиэтоксиметил)гуанин-5'-ил-О-(гидрокси-трет-бутил-S-ацил-2-тиоэтил)фосфат B704 (Соединение F3):
Соединение F2 (550 мг, 0,69 ммоль) растворяли в дихлорметане (2,2 мл) и обрабатывали ТФУ (220 мкл). Смесь взбалтывали при комнатной температуре в течение 15 минут, фильтровали на колонке для твердофазной экстракции с элюированием метанолом в дихлорметане с градиентом 0-15%, затем очищали обращенно-фазовой (C 18) колоночной хроматографией на силикагеле, элюировали ацетонитрилом в воде с градиентом 0-100% и лиофилизировали из смеси воды/диоксана для получения соединения B704 (Соединение F3), (103 мг, 27%, лиофилизированный порошок белого цвета). B704 (Соединение F3): 1H ЯМР (400 МГц, ДМСО-d6): δ 10,57 (1c, 1H, NH), 7,79 (с, 1H, H-8), 7,29-7,18 (м, 5H, C6H3), 6,49 (1c, 2H, NH2), 5,55 (м, 1H, PNH), 5,33 (с, 2H, OCH2N), 4,92 (т, 1H, OH, JOH-CH2=5,2 Гц), 3,94-3,73 (м, 6H, CH2SCH2O, CH2N, POCH2CH2O), 3,60 (т, 2H, POCH2CH2O, JCH2-CH2=4,2 Гц), 3,42 (д, 2H, CH2OH, JCH2-OH=4,4 Гц), 3,00 (т, 2H, CH2S, JCH2S-CH2O=6,4 Гц), 1,10 (с, 6H, 2 CH3). 13C ЯМР (100 МГц, ДМСО-d6): δ 204,4 (C=O), 157,2 (C-4), 154,4 (C-2), 151,9 (C-6), 141,0 (1C, C6H5), 138,1 (C-8), 128,6-127,2 (5C, C6H5), 117,0 (C-5), 72,3 (OCH2N), 68,8 (CH2OH), 68,2 (POCH2CH2O), 64,7 (POCH2CH2O), 64,2 (CH2SCH2O), 52,2 (C(CH3)2), 44,7 (CH2N), 28,7 (CH2S), 22,3 (2C, C(CH3)2). 31P ЯМР (162 МГц, ДМСО-d6): δ 9,76 (c), LR LC/MS (M+H+) 555,9 (M-H-) 553,9 (3,77 мин). HRFAB-MS C22H32O7N6PS (M+Н+). Рассчитано 555,1791, найдено 555,1795. УФ: λмакс=250 нм.
Пример 22
Приготовление B390, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метил-2',3'-ди-О-ацетилцитидина

СХЕМА СИНТЕЗА:

К раствору пронуклеотида 13 (см. пример 2, процедура A, стратегия b) (300 мг, 0,27 ммоль) в безводном ацетонитриле последовательно добавляли триэтиламин (92 мкл), уксусный ангидрид (2,2 экв., 54 мкл) и 4-диметиламинопиридин (0,1 экв., 4 мг). Реакционную смесь взбалтывали при комнатной температуре в течение 2 ч, и повторно добавляли триэтиламин (92 мкл), уксусный ангидрид (2,2 экв., 54 мкл) и 4-диметиламинопиридин (0,1 экв., 4 мг). После удаления растворителей при пониженном давлении неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-5%]), чтобы получить полностью защищенный пронуклеотид (329 мг, количественный выход). Указанное соединение в конце обрабатывали смесью трифторуксусной кислоты (132 мкл) и метиленхлорида (3,9 мл). После взбалтывания в течение 1 ч 30 мин при комнатной температуре снова добавляли трифторуксусную кислоту (132 мкл), и смесь взбалтывали дольше 1 часа. Растворители выпаривали при пониженном давлении и выпаривали совместно с толуолом. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-10%]), чтобы получить лиофилизированное соединение B390 (36,4 мг, 21%) в виде порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,10 (с, 6H), 1,33 (д, J=2,60 Гц, 3H), 2,05 (с, 6H), 3,01-3,04 (т, J=6,54 Гц, 2H), 3,31 (д, J=5,45 H, 2H), 3,85-3,90 (м, 2H), 3,94-3,99 (м, 2H), 4,09-4,11 (м, 2H), 4,21-4,23 (м, 1H), 4,90-4,93 (т, J=5,71 Гц, 1H), 5,22 (м, 1H), 5,67-5,73 (м, 2H), 6,20(м, 1H), 7,21-7,27 (м, 7H), 7,54 (м, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,69 и 9,86 (2c); сканирование ES+ 671 (M+H)+. λмакс=273,7 нм; ВЭЖХ (0-100% ACN в течение 10 минут) tR=5,04 мин, λмакс=233,7 нм и 271,4 нм.
Пример 23
Приготовление B302

В302
Синтез гидрокси-tBuSATE N-бензилфосфорамидатного 2',3'-циклокарбонатного производного 2'-C-метилцитидина B302
Для синтеза применяли следующую стратегию:

Защищенный фосфорамидат 13 (1,72 г, 1,52 ммоль) растворяли в безводном дихлорметане (17 мл) в атмосфере аргона. Добавляли 1,1'-карбонилдиимидазол (251 мг, 1,55 ммоль), и реакционную смесь взбалтывали при комнатной температуре в атмосфере аргона в течение 1 часа.
Анализ тонкослойной хроматографией ТСХ (8% MeOH в ДХМ) показал неполное превращение стартового материала (Rf=0,35) в продукт (Rf=0,56). Анализ ВЭЖХ (способ Test20, 272 нм) подтвердил указанный профиль: 9% стартового материала (Rt=7,30 минут) и 91% продукта (Rt=7,97 минут).
Добавляли дополнительные порции CDI (общий выход 299 мг, 1,84 ммоль) и реакционную смесь оставляли для взбалтывания в течение дополнительных 24 часов при комнатной температуре, после чего анализ времени ВЭЖХ показывал 1,5% SM и 97,5% P.
Реакционную смесь выпаривали в вакууме для получения пены беловатого цвета (1,97 г). Очистка на колонке с силикагелем с элюированием этилацетатом и выпариванием соответствующих фракций приводила к получению защищенного циклического карбоната 14 (C65H65N4O12PS 1157,27 гмоль-1) в виде пены белого цвета (1,62 г, выход 92%). Результаты ТСХ (8% MeOH в ДХМ): Rf=0,56; ВЭЖХ Test20 AUC: 99,5% при 254 нм, Rt=7,97 минут; m/z (ESI-): 1155,9 [M-H]- 100%; m/z (ESI+): 1157,5 [M+H]+ 100%, 1179,5 [M+Na]+ 20%.
Защищенный циклический карбонат 14 (1,50 г, 1,30 ммоль) растворяли в безводном дихлорметане (15 мл) при комнатной температуре в атмосфере аргона. К реакционной смеси по капле добавляли беспримесную трифторуксусную кислоту (1,77 г, 15,5 ммоль), которую затем перемешивали в течение 45 минут при комнатной температуре. Анализ ВЭЖХ (способ Test20, 272 нм) показывал исчезновение стартового материала (Rt=7,97 минут) и образование продукта (Rt=3,80 минуты).
К реакционной смеси добавляли безводный метанол (5 мл), и растворители (10 мл) частично удаляли в вакууме при 25°C. К смеси дополнительно добавляли метанол (7 мл), после чего смесь выпаривали с получением оранжевого остатка. Растирание с гексаном/TBME 3:2 (12 мл) в течение 20 минут приводило к получению липкого остатка вместе с непрозрачным супернатантом, который сливали с осадка. Повторное растирание с гексаном/TBME 3:2 (5 мл) в течение 1 ч и удаление второго супернатанта после совместного выпаривания с метанолом (3 мл) приводило к получению бледной пены (1,18 г).
Неочищенную пену очищали обращенно-фазовой хроматографией (с погружением в 1 мл ацетонитрила и элюированием 0%, 10%, 15%, 20%, 25%, 30% раствором ацетонитрила в воде). Комбинация соответствующих фракций, выпаривание растворителей при 25°C и выдавливание этанолом (1 мл) приводили к получению циклического карбоната 15, B302, в виде белого вспененного твердого вещества (560 мг, выход 71%).
B302: C25H33N4O10PS 612,59 гмоль-1
ВЭЖХ AUC (Способ Test20): 99% при 254 нм, Rt=3,83 минуты
m/z (ESI +): 613,1 [M+H]+ 100%; 1225,5 [2M+H]+ 100%; 453,1 [N+H]+ 95%
m/z (ESI-): 611,4 [M-H]- 80%; 1223,9 [2M-H]- 50%; 451,3 [N-H]- 100%

Vмакс (диск KBr) (см-1): 3346,4, 3206,5 O-H, межмолекулярная H-связь; 1815,3 C=O циклический карбонат с 5 кольцами; 1650,9 ушир. основание C=O, тиоэфир
KF: содержание H2O 1,54%
Удельное вращение: [α]D 20 +9,289 (c 10,104 мг см-3 в ДМСО)
m.p.: уплотнение и размягчение 100-102°C, фазовый переход I 104-106°C, фазовый переход II до липкого стекла 127-135°C, частичное плавление до липкого осадка 140-150°C, разложение до коричневого липкого материала 200-210°C
Элементный анализ: Рассчитано: C 49,02%; H 5,43%; N 9,15%, Найдено: C 49,30%; H 5,26%; N 9,30% - пропущенный с 0,26% присутствующего F (из ТФУ)
ЯМР: Заданный с использованием 1H, 13C, 31P, COSY, TOCSY, DEPT, HSQC и HMBC
1H ЯМР δH (400 МГц, d6-ДМСО): 1,11 (6H, c, (CH3)2C), 1,30 (3H, ушир.с, CH3), 3,04 (2H, м, CH2S), 3,44 (2H, д, J=4 Гц, CH2OH), 3,87-3,92 (2H, м, CH2O), 3,94-4,01 (2H, м, CH2Ph), 4,15-4,25 (2H, м, H-5', H-5"), 4,37 (1H, ушир.с, H-4'), 4,95 (2H, ушир.с, H-3', CH2OH), 5,75-5,77 (2H, 2×д, J=7 Гц, H-5, P-N-H), 6,07 (1H, ушир.с, H-1'), 7,22-7,25 (1H, м, Ar-H), 7,29-7,33 (4H, м, 4 × Ar-H), 7,39, 7,44 (2H, 2 × ушир.с, NH2), 7,62 (1H, ушир.д, J=7 Гц, H-6).
13C ЯМР δc (100 МГц, d6-ДМСО): 17,72 (CH3), 21,78 (C(CH3)2), 28,13, 28,21 (CH2S), 44,17 (PhCH2), 51,62 (C(CH3)2), 63,84, 63,89 (CH2O), 64,55 (C-5'), 68,29 (CH2OH), 94,23 (C-5), 126,70 (Ar-Cpara), 127,08, 128,11 (2 × Ar-Cmeta, 2 × Ar-Cortho), 140,35, 140,38 (Ar-Cipso), 152,73, 154,45 (C-2, C-4), 165,69 (C-6), 203,87 (C=OS), C-1', C-2', C-3', C-4' и C=O расширялись до начального порога и не были выявлены.
31P ЯМР δP (162 МГц, d6-ДМСО): 9,80, 9,94 (1P, 2×с, соотношение 1,15:1,00).
Пример 24
Приготовление B234, гидрокси-tBuSATE N-бензилфосфорамидатного производного 3'-O-L-валинил-2'-C-метилцитидина

Для синтеза применяли следующую стратегию:


Boc-защищенный валин (6,72 г, 30,94 ммоль) растворяли в безводном ДХМ (50 мл), и при комнатной температуре в атмосфере аргона добавляли 1,1'-карбонилдиимидазол (4,87 г, 30,01 ммоль). Вначале во время этапа активации наблюдалось усиленное газообразование, и смесь взбалтывали при комнатной температуре в течение 30 мин.
Защищенный фосфорамидат 13 (10,0 г, 8,84 ммоль) растворяли в безводном дихлорметане (50 мл) в отдельной колбе в атмосфере аргона. Активированный раствор Boc-валина добавляли по капле к фосфорамидатному раствору, и полученную смесь нагревали до 40°C в атмосфере аргона в течение 24 часов.
Анализ ТСХ (8% MeOH в ДХМ) указывал на полное превращение стартового материала 13 (Rf=0,35) в продукт (Rf=0,50). Анализ ВЭЖХ (способ Test20, 272 нм) подтверждал указанный профиль: стартовый материал (Rt=7,30 минут) и продукт (Rt=9,46 минут).
Реакционную смесь выпаривали в вакууме для получения беловатой пены. Очистка колоночной хроматографией на силикагеле с ДХМ, с элюированием этилацетатом/гексаном 1:1, затем 100% этилацетатом, и выпаривание соответствующих фракций приводили к получению защищенного сложного эфира валина 16 (C74H84N5O14PS 1330,52 гмоль-1) в виде пены белого цвета (10,3 г, выход 88%). ТСХ (этилацетат): Rf=0,24; ВЭЖХ Test20 AUC: 97% при 272 нм, Rt=9,46 минут; m/z (ESI-): 1329,29 [M-H]- 100%; m/z (ESI+): 1331,68 [M+H]+ 25%, 303,16 [DMTr]+ 100%.
Защищенный сложный эфир валина 16 (3,0 г, 2,25 ммоль) растворяли в безводном дихлорметане (22,5 мл) при комнатной температуре в атмосфере аргона. Беспримесную трифторуксусную кислоту (4,5 мл, 58,4 ммоль) добавляли по капле к реакционной смеси в течение 3 минут, после чего смесь взбалтывали в течение 1 часа при комнатной температуре. Анализ ВЭЖХ (способ Test20, 272 нм) показывал исчезновение стартового материала (Rt=9,46 минут) и образование продукта (Rt=3,33 минуты) наряду со значительным количеством промежуточного Boc-продукта (Rt=4,60 минуты).
К реакционной смеси добавляли по капле дополнительное количество беспримесной трифторуксусной кислоты (1,0 мл, 13,0 ммоль), после чего смесь взбалтывали в течение дополнительного 1 часа при комнатной температуре. Анализ ВЭЖХ (способ Test20, 272 нм) показывал исчезновение промежуточного Boc-продукта (Rt=4,60 минуты) и образование продукта (Rt=3,33 минуты).
Реакционную смесь охлаждали до 5°C и добавляли безводный метанол (50 мл), перемешивали в течение 30 мин. Растворители удаляли в вакууме при 25°C. Остаток обрабатывали TBME (50 мл × 3) и растирали, сливая три жидкие фракции TBME.
Остаточный материал растворяли в безводном метаноле (5 мл) и безводном ДХМ (10 мл), и добавляли твердый бикарбонат натрия (5 г), перемешивали в течение 30 минут, чтобы достичь уровня pH 6. Прозрачную жидкость пропускали через шприцевой фильтр. Остаточный твердый бикарбонат промывали 25% метанолом в ДХМ (безводный, 10 мл), и раствор снова фильтровали. Объединенные фильтраты выпаривали в вакууме, чтобы получить неочищенный продукт 17 (2,15 г).
Сырой материал очищали обращенно-фазовой хроматографией (с водой 15 мл и ацетонитрилом 3 мл, и элюировали 0%, 5%, 20%, 30% раствором ацетонитрила в воде). Комбинация соответствующих фракций и выпаривание растворителей при 25°С приводили к получению сложного эфира валина 17, B234, в виде вспененного твердого вещества белого цвета (737 мг, выход 48%).
B234: C29H44N5O10PS 685,73 гмоль-1
ВЭЖХ AUC (Способ Test20): 99% при 254 нм, Rt=3,33 минуты
m/z (ESI+): 686,3 [M+H]+ 100%; 1371,6 [2M+H]+ 20%; 526,1 [N+H]+ 20%
m/z (ESI-): 744,4 [M+OAc]- 35%; 1369,8 [2M-H]- 35%; 1430,2 [2M+OAc]- 15%; 524,5 [N-H]- 100%

Vмакс (диск KBr) (см-1): 3350,7, 3211,9 O-H, N-H; 1757,8 C=O сложный эфир; 1673,9, 1652,0 C=O тиоэфир, основание
KF: содержание H2O 1,94%
Удельное вращение: [α]D 20 +44,370 (c 10,033 мг см-3 в ДМСО)
ЯМР: Заданный с использованием 1H, 13C, 31P, COSY, TOCSY, DEPT, HSQC и HMBC
H ЯМР δH (400 МГц, d6-ДМСО): 0,96, 0,98 (2×3H, 2×c, (CH3)2CH), 1,03 (3H, ушир.с, CH3), 1,11 (6H, c, (CH3)2C), 2,15 (1H, м, (CH3)2CH), 3,03 (2H, м, CH2S), 3,44 (2H, a-c, CH2OH), 3,85 (1H, a-d, J=4,8Гц, CHNH2), 3,85-3,92 (2H, м, CH2O), 3,92-4,00 (2H, м, CH2Ph), 4,06-4,11 (1H, ушир.м, H-5'), 4,17-4,20 (1H, ушир.м, H-5"), 4,27-4,29 (1H, ушир.м, H-4'), 5,08 (1H, ушир.с, H-3'), 5,73 (1H, a-т, J=7,3 Гц, H-5), 5,74-5,82 (1H, м, P-N-H), 5,92 (1H, ушир.с, H-1'), 7,22-7,25 (1H, м, Ar-H), 7,28-7,32 (4H, м, 4×Ar-H), 7,60, 7,63 (2×0,5H, 2×д, J=7,3 Гц, H-6), 2×OH и 2×NH2 не выявлено.
13C ЯМР δc (100 МГц, d6-ДМСО): 17,84, 17,96 (CH(CH3)2), 20,42, 20,48 (CH3), 21,78, (C(CH3)2), 28,09, 28,16 (CH2S), 29,72 (CH(CH3)2), 44,16 (PhCH2), 51,62 (C(CH3)2), 57,84 (CHNH2), 63,77, 63,81 (CH2O, C-5'), 68,26 (CH2OH), 74,67 (C-3'), 77,28 (C-4'), 78,09 (C-2'), 91,32 (C-1'), 94,22 (C-5), 126,71 (Ar-Cpara), 127,04, 127,08, 128,11 (2×Ar-Cmeta, 2×Ar-Cortho), 140,23, 140,27, 140,32 (Ar-Cipso, C-6), 155,06 (C-2), 165,32 (C-4), 169,65, 169,72 (CO2R), 203,84 (C=OS), C-1', C-3', C-4' расширено до начального порога и не выявлено.
31P ЯМР δP (162 МГц, d6-ДМСО): 9,63, 9,96 (1P, 2×с, соотношение 1,02:1,00).
Пример 25
Приготовление B183, гидрокси-tBuSATE N-бензилфосфорамидатного производного 2'-C-метил-NH-4-ацетилцитидина

СХЕМА СИНТЕЗА:

К раствору B102 (см. пример 2) (соединение 10, 200 мг, 0,34 ммоль) в безводном диметилформамиде (3,4 мл) по капле добавляли уксусный ангидрид (1,1 экв., 34 мкл). Реакционную смесь взбалтывали при комнатной температуре в течение 4 часов, и повторно добавляли 10 мкл уксусного ангидрида. Реакционную смесь взбалтывали в течение ночи, и растворитель выпаривали при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-10%]), чтобы получить желаемый ацетилированный пронуклеотид B183 (169 мг, 79%), выделяемый в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,93 (с, 3H), 1,09 (с, 6H), 2,09 (с, 3H), 3,01-3,04 (т, J=6,54 Гц, 2H), 3,40-3,42 (д, J=5,10 Гц, 1H), 3,53-3,62 (м, 2H), 3,83-3,91 (м, 1H), 3,94-4,01 (м, 4H), 4,10-4,15 (м, 1H), 4,20-4,25 (м, 1H), 4,88-4,91 (т, J=5,20 Гц, 1H), 5,23 (с, 1H), 5,33-5,37 (т, J=7,19 Гц, 1H), 5,67-5,78 (м, 1H), 5,93 (с, 1H), 7,18-7,21 (м, 1H), 7,27-7,32 (м, 5H), 7,96 и 8,03 (2д, J=7,59 Гц, 1H), 10,87 (с, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,74 и 9,98 (2c); сканирование ES+ 629(M+H)+, λмакс=300,7 нм; ВЭЖХ (0-100% ACN в течение 8 минут) tR=4,89 минут λмакс=302,1 нм.
Пример 26
Приготовление B187, гидрокси-tBuSATE N-(2-(трифторметил)бензил)фосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 8 (см. пример 2, процедура A, стратегия a) (1,4 г, 1,3 ммоль) в безводном тетрахлориде углерода (13 мл), добавляли по капле N-2-(трифторметил)бензиламин (10 экв., 2,3 г). Реакционную смесь взбалтывали при комнатной температуре в течение 3 часов, и растворитель удаляли при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-3%]), чтобы получить желаемый защищенный нуклеозид в виде пены (60%). Указанное соединение превращали в фосфорамидатное пролекарство B187 (245 мг, 35%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии A, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,92 (с, 3H), 1,09 (с, 6H), 3,05 (т, J=6,45 Гц, 2H), 3,29 (с, 1H), 3,41 (д, J=5,60 Гц, 2H), 3,91-3,93 (м, 3H), 4,17-4,21 (м, 4H), 4,91 (т, J=5,59 Гц, 1H), 5,06 (д, J=4,25 Гц, 1H), 5,23 (т,J=7,50 Гц, 1H), 5,65-5,67 (м, 1H), 5,76-5,83 (м, 1H), 5,91 (с, 1H), 7,08 и 7,16 (2c, 2H), 7,45-7,79 (м, 5H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,57-9,78 (2c, 1P); 19F ЯМР (d6-ДМСО, 235 МГц) δ (м.д.) -60,79 (с, 3F); сканирование ES+ 655 (M+H)+, λмакс=280,73 нм; ВЭЖХ (0-100% ACN в течение 10 минут) tR=5,08 минута λмакс=271,4 нм.
Пример 27
Приготовление B399, гидрокси-tBuSATE N-(4-(трифторметил)бензил)фосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 8 (см. пример 2, процедура A, стратегия a) (1,0 г, 0,94 ммоль) в безводном тетрахлориде углерода (10 мл) по капле добавляли N-4-трифторметилбензиламин (5 экв., 670 мкл). Реакционную смесь взбалтывали при комнатной температуре в течение 3 ч и растворитель удаляли при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-5%]), чтобы получить желаемый защищенный нуклеозид в виде пены (84%). Указанное соединение превращали в фосфорамидатное пролекарство B399 (204 мг, 40%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии A, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,91-0,92 (д, J=2,09 Гц, 3H), 1,09 (с, 6H), 3,02-3,06 (м, 2H), 3,41 (д, J=6,17 Гц, 2H), 3,53-3,57 (м, 1H), 3,84-3,94 (м, 3H), 4,03-4,13 (м, 3H), 4,18-4,23 (м, 1H), 4,91-4,94 (т, J=5,48 Гц, 1H), 5,06 (с, 1H), 5,23-5,27 (т, J=6,82 Гц, 1H), 5,65-5,67 (м, 1H), 5,79-5,87 (м, 1H), 5,90 (с, 1H), 7,09 и 7,16 (2s, 2H), 7,48-7,55 (м, 3H), 7,64-7,67 (м, 2H); 19F ЯМР (d6-ДМСО, 235 МГц) δ (м.д.) -60,79 (с, 3F); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,55 и 9,76 (2c); сканирование ES+ 655 (M+H)+, λмакс=270 нм; ВЭЖХ (0-100% ACN в течение 10 минут), tR=5,03 минут λмакс=271 нм.
Пример 28
Приготовление B204, гидрокси-tBuSATE N-(n-метил-n-октил-амин)фосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 8 (см. пример 2, процедура A, стратегия a) (950 мг, 0,89 ммоль) в безводном тетрахлориде углерода (9 мл) по капле добавляли н-метил-н-октиламин (10 экв., 1,28 г). Реакционную смесь взбалтывали при комнатной температуре в течение 3 ч и растворитель удаляли при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-3%]), чтобы получить желаемый защищенный нуклеозид в виде пены (88%). Указанное соединение превращали в фосфорамидатное пролекарство B204 (52 мг, 7%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии A, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,83 (м, 3H), 0,93-0,94 (д, J=3,75 Гц, 3H), 1,10 (с, 6H), 1,22 (с, 10H), 1,44 (м, 2H), 2,56 (д, J=8,2 Гц, 3H), 2,88-2,93 (м, 2H), 3,31 (м, 2H), 3,43 (д, J=5,60 Гц, 2H), 3,50-3,53 (м, 1H), 3,91-3,93 (м, 3H), 4,04-4,07 (м, 1H), 4,13-4,16 (м, 1H), 4,91 (т, J=5,59 Гц, 1H), 5,06 (с, 1H), 5,23 (м, 1H), 5,65-5,67 (м, 1H), 5,91 (с, 1H), 7,08-7,16 (м, 2H), 7,50-7,57 (м, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 10,52 и 10,66 (2c); сканирование ES+ 623 (M+H)+, λмакс=280,73 нм; ВЭЖХ (0-100% ACN в течение 8 минут), tR=6,07 минут, λмакс=274,9 нм.
Пример 29
Приготовление B244, гидрокси-tBuSATE N,N-(дибутиламин)фосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 12 (см. пример 2, процедура A, стратегия b) (1,5 г, 1,46 ммоль) в безводном тетрахлориде углерода (15 мл), добавляли по капле дибутиламин (10 экв., 2,5 мл). Реакционную смесь взбалтывали при комнатной температуре в течение 3 часов и удаляли растворитель при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-5%]), чтобы получить желаемый защищенный нуклеозид в виде пены (61%). Указанное соединение превращали в фосфорамидатное пролекарство B244 (21 мг, 4%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии B, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,76-0,81 (тд, J=2,40 Гц и J=7,43 Гц, 6H), 0,86-0,87 (д, J=5,51 Гц, 3H), 1,05 (с, 6H), 1,11-1,19 (м, 4H), 1,33-1,39 (м, 4H), 2,80-2,87 (кв, J=9,50 Гц, J=8,67 Гц, 4H), 3,01-3,04 (т, J=6,23 Гц, 2H), 3,42-3,43 (м, 2H), 3,50-3,60 (м, 1H), 3,81-3,88 (м, 3H), 3,97-4,01 (м, 1H), 4,07-4,10 (м, 1H), 4,84-4,87 (м, 1H), 5,06 (с, 1H), 5,23 и 5,29 (2д, J=8,0 Гц, 1H), 5,70 (с, 1H), 5,91 (ушир.с, 1H), 7,10 и 7,17 (2c, 2H), 7,49 и 7,55 (2д, J=8,0 Гц, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 10,44 и 10,56 (2c); сканирование ES+ 609 (M+H)+, λмакс=279,7 нм; ВЭЖХ (0-100% ACN в течение 8 минут), tR=5,59 минут, λмакс=274,9 нм.
Пример 30
Приготовление B308, гидрокси-tBuSATE N-метилбензилфосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 12 (см. пример 2, процедура A, стратегия b) (2,7 г, 2,6 ммоль) в безводном тетрахлориде углерода (26 мл) добавляли по капле N-бензилметиламин (5 экв., 1,67 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и при пониженном давлении удаляли растворитель. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-5%]), чтобы получить желаемый защищенный нуклеозид в виде пены (44%). Указанное соединение превращали в фосфорамидатное пролекарство B308 (43 мг, 2%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии B, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,93-0,94 (с, 3H), 1,10 (с, 6H), 2,43-2,45 (д, J=4,26 Гц, 3H), 3,13 (т, J=6,23 Гц, 2H), 3,36-3,37 (д, J=5,24 Гц, 2H), 3,56-3,60 (м, 2H), 3,97-4,01 (м, 3H), 4,07-4,21 (м, 3H), 4,92-4,94 (м, 1H), 5,08 (с, 1H), 5,30-5,32 (м, 1H), 5,59-5,67 (2д, J=8,0 Гц, 1H), 5,91 (с, 1H), 7,13 (м, 2H), 7,42-7,50 (м, 5H), 7,45-7,54 (2д, J=8,0 Гц, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 10,53 и 10,34 (2c); сканирование ES+ 601 (M+H)+, λмакс=268,7; ВЭЖХ (0-100% ACN в течение 8 минут), tR=3,37 мин, λмакс=274,9 нм.
Пример 31
Приготовление B353, гидрокси-tBuSATE N-пиперидинбензилфосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 12 (см. пример 2, процедура A, стратегия b) (300 мг, 0,29 ммоль) в безводном тетрахлориде углерода (3 мл) по капле добавляли пиперидин (5 экв., 145 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и растворитель удаляли при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-5%]) для получения желаемого защищенного нуклеозида в виде пены (55%). Указанное соединение превращали в фосфорамидатное пролекарство B353 (19 мг, 22%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии B, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,92 (д, J=2,56, 3H), 1,10 (с, 6H), 1,44-1,43 (м, 4H), 1,50-1,53 (м, 2H), 2,97-3,02 (м, 4H), 3,07-3,10 (т, J=6,66 Гц, 2H), 3,42 (д, J=5,64 Гц, 2H), 3,56-3,60 (м, 1H), 3,89-3,94 (м, 3H), 4,04-4,10 (м, 1H), 4,13-4,20 (м, 1H), 4,91-4,93 (т, J=5,64 Гц, 1H), 5,06 (с, 1H), 5,25-5,31 (2д, J=9,31 Гц, 1H), 5,68 (м, 1H), 5,90 (с, 1H), 7,17 и 7,10 (2c, 2H), 7,50-7,55 (2д, J=9,01 Гц, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 8,75 и 8,59 (2c); сканирование ES+ 565 (M+H)+, λмакс=275,7 нм; ВЭЖХ (0-100% ACN в течение 6 минут), tR=3,08 мин, λмакс=273,7 нм.
Пример 32
Приготовление B354, гидрокси-tBuSATE N-циклогексиламинфосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 12 (см. пример 2, процедура A, стратегия b) (300 мг, 0,29 ммоль) в безводном тетрахлориде углерода (3 мл) добавляли по капле циклогексиламин (5 экв., 170 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и растворитель удаляли при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-5%]) для получения желаемого защищенного нуклеозида в виде пены (55%). Указанное соединение превращали в фосфорамидатное пролекарство B354 (44 мг, 50%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии B, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,92 (д, 7=2,56, 3H), 1,10 (с, 6H), 1,13 (м, 5H), 1,46-1,47 (м, 1H), 1,62 (м, 2H), 1,76-1,78 (м, 2H), 2,80 (м, 1H), 3,07-3,10 (т, J=6,66 Гц, 2H), 3,42 (д, J=5,64 Гц, 2H), 3,56-3,60 (м, 1H), 3,89-3,94 (м, 3H), 4,04-4,10 (м, 1H), 4,13-4,20 (м, 1H), 4,91-4,93 (т, J=5,64 Гц, 1H), 5,06 (м, 2H), 5,25 и 5,31 (2д, J=7,2 Гц, 1H), 5,68-5,71 (м, 1H), 5,90 (с, 1H), 7,19 и 7,09 (2c, 2H), 7,50 и 7,55 (2д, J=7,2 Гц, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,05 и 8,91 (2c) сканирование ES+ 579 (M+H)+, λмакс=280,7 нм; ВЭЖХ (0-100% ACN в течение 6 минут), tR=3,23 мин, λмакс=274,9 нм.
Пример 33
Приготовление B391, гидрокси-tBuSATE N-морфолинофосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 12 (см. пример 2, процедура A, стратегия b) (350 мг, 0,34 ммоль) в безводном тетрахлориде углерода (3,4 мл), по капле добавляли морфолин (10 экв., 300 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, и растворитель удаляли при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанол в метиленхлориде с градиентом [0-5%]), чтобы получить желаемый защищенный нуклеозид в виде пены (70%). Указанное соединение превращали в фосфорамидатное пролекарство B391 (53 мг, 49%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии B, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,92 (д, J=2,56, 3H), 1,10 (с, 6H), 3,0 (м, 4H), 3,07-3,10 (т, J=6,66 Гц, 2H), 3,31 (с, 2H), 3,42 (д, J=5,64 Гц, 2H), 3,56-3,60 (м, 3H), 3,89-3,94 (м, 3H), 4,04-4,10 (м, 1H), 4,13-4,20 (м, 1H), 4,91-4,93 (т, J=5,64 Гц, 1H), 5,08 (с, 1H), 5,25-5,31 (м, 1H), 5,68-5,71 (д, J=7,2 Гц, 1H), 5,90 (с, 1H), 7,18 и 7,12 (2s, 2H), 7,52 и 7,50 (2д, J=7,6 Гц, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 7,76 и 7,61 (2c); сканирование ES+ 567(M+H)+, λмакс=279,7 нм; ВЭЖХ (0-100% ACN в течение 10 минут), tR=3,42 мин, λмакс=273,7 нм.
Пример 34
Приготовление B395, гидрокси-tBuSATE N-пирролидинфосфорамидатного производного 2'-C-метилцитидина

СХЕМА СИНТЕЗА:

К раствору соединения 12 (см. пример 2, процедура A, стратегия b) (500 мг, 0,49 ммоль) в безводном тетрахлориде углерода (5 мл), добавляли пирролидин по капле (5 экв., 200 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и при пониженном давлении удаляли растворитель. Неочищенную смесь очищали колоночной хроматографией на силикагеле (элюент: поэтапно метанолом в метиленхлориде с градиентом [0-5%]) для получения желаемого защищенного нуклеозида в виде пены (87%). Указанное соединение превращали в фосфорамидатное пролекарство B395 (48 мг, 21%) после осуществления эксперимента согласно условиям, описанным в примере 2 стратегии B, и выделяли в виде лиофилизированного порошка белого цвета. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,93-0,94 (д, J=3,75 Гц, 3H), 1,10 (с, 6H), 1,78-1,79 (кв, J=5,80 Гц, 4H), 3,09-3,09 (м, 6H), 3,42 (с, 2H), 3,57-3,59 (м, 1H), 3,92-3,93 (м, 3H), 4,09-4,11 (м, 1H), 4,16-4,18 (м, 1H), 4,93 (ушир.с, 1H), 5,10 (с, 1H), 5,28-5,32 (т, J=8,00 Гц, 1H), 5,70 (д, 7=8,0 Гц, 1H), 5,89 (с, 1H), 7,27 и 7,40 (2c, 2H), 7,55 и 7,61 (2д, J=8,0 Гц, 1H); 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 7,56 и 7,69 (2c); сканирование ES+ 551(M+H)+, λмакс=275,7 нм; ВЭЖХ (0-100% ACN в течение 10 минут), tR=3,88 мин, λмакс=273,7 нм.
Пример 35
Действие против HBV
Осуществляли контакт соединения из примера 1 (NM 204) (гидрокси-tBuSATE N-бензилфосфорамидатное производное L-ddA) (A550) с клетками, инфицированными HBV-HepG2. Значения EC50 измеряли согласно стандартным способам. Как показано в таблице ниже, соединение из примера 1 показало значительную активность по сравнению с исходной молекулой LddA.
| Препарат | N | Вес HBV (HepG2) |
| EC50 (мкМ) | ||
| LddA | 3 | >10 |
| Пример 1 (A550) | 3 | 0,062±0,018 |
| LdT | 3 | 0,26±0,048 |
| Ламивудин | 3 | 0,022±0,007 |
Пример 36
Подготовка калибровочной кривой для измерения ddАТФ
Измерения концентрации 2'-3'-дидезоксиаденозин-5'-трифосфата (ddАТФ) (трифосфатный нуклеотид 2'-3'-дидезоксиаденозина (ddA) осуществляли способом жидкостной хроматографии - тандемной масс-спектрометрии (LC/MS/MS), например, в метанольном экстракте гепатоцитов.
Концентрацию ddАТФ измеряли по сравнению со стандартной кривой.
Рабочие стоковые растворы TP-ddA готовили из стокового раствора 100 пмоль/мкл ddАТФ в деионизированной воде (тетранатриевая соль с чистотой >91%), закупаемого в компании Sigma Chemical Co, следующим образом:
Рабочие стоковые растворы ddАТФ и подготовка стандартной кривой для ddАТФ.
| 1. Рабочий стоковый раствор № 1 | ||||||
| Тестовое соединение | Стоковая концентрация пмоль/мкл | Взятый объем мкл | Объем DIH2O мкл | Общий объем мкл | Концентрация пмоль/мкл | моль на 10 мкл |
| ТФ-ddA | 100 | 2000 | 2000 | 4000 | 50,0 | 500 |
| 2. Рабочий стоковый раствор № 2 | ||||||
| Тестовое вещество | Стоковая концентрация пмоль/мкл | Взятый объем мкл | Объем DIH2O мкл | Общий объем мкл | Концентрация пмоль/мкл | |
| ТФ-ddA | 100 | 1000 | 3000 | 4000 | 25,0 | 250 |
| 3. Рабочий стоковый раствор №4 (приготовленный из стокового раствора № 1) | ||||||
| Тестовое вещество | Стоковая концентрация пмоль/мкл | Взятый объем мкл | Объем DIH2O мкл | Общий объем мкл | Концентрация пмоль/мкл | |
| ТФ-ddA | 100 | 500 | 3500 | 4000 | 12,5 | 125 |
| 4. Рабочий стоковый раствор №5 (приготовленный из стокового раствора № 1) | ||||||
| Тестовое вещество | Стоковая концентрация пмоль/мкл | Взятый объем мкл | Объем DIH2O мкл | Общий объем мкл | Концентрация пмоль/мкл | |
| ТФ-ddA | 100 | 200 | 3800 | 4000 | 5,0 | 50 |
| 5. Рабочий стоковый раствор №6 (приготовленный из стокового раствора № 1) | ||||||
| Тестовое вещество | Стоковая концентрация пмоль/мкл | Взятый объем мкл | Объем DIH2O мкл | Общий объем мкл | Концентрация пмоль/мкл | |
| ТФ-ddA | 100 | 100 | 3900 | 4000 | 2,5 | 25 |
| 6. Рабочий стоковый раствор №7 (приготовленный из стокового раствора № 1) | ||||||
| Тестовое вещество | Стоковая концентрация пмоль/мкл | Взятый объем мкл | Объем DIH2O мкл | Общий объем мкл | Концентрация пмоль/мкл | |
| ТФ-ddA | 100 | 40 | 3960 | 4000 | 1,0 | 10 |
Внутренний стандартный (ISTD) рабочий стоковый раствор готовили из стокового раствора 0,50 мг/мл 2-дезоксиаденозина 5-трифосфат, приобретенного в компании Sigma Chemical Co.
| Раствор ISTD | Стоковая концентрация пмоль/мкл | Взятый объем мкл | Объем МеОН мкл | Общий объем мкл | Концент-рация мкг/мл | Концент-рация пмоль/мл |
| dAТФ | 500 | 200 | 9800 | 10000 | 10,0 | 500 |
В некоторых вариантах осуществления калибровочные стандарты подготавливают следующим образом, используя:
| Приготовление калибровочных стандартов: | ||||||||
| Калибровочный стандарт № | Стандартная концентрация пмоль/мл | Вес пече-ни г |
Рабочий стоко-вый раствор № | Концент-рация рабочего стокового раствора пмоль/мкл | Объем рабочего стоко-вого раствора мкл | Объем ISTD мкл | Объем MeOH мкл | Общий объем мкл |
| Blk | 0 | 0,1 | 0 | 50 | 940 | 990 | ||
| #1 | 50 | 0,1 | #5 | 5,0 | 10 | 50 | 940 | 1000 |
| #2 | 125 | 0,1 | #4 | 12,5 | 10 | 50 | 940 | 1000 |
| #3 | 250 | 0,1 | #3 | 25,0 | 10 | 50 | 940 | 1000 |
| #4 | 500 | 0,1 | #2 | 50,0 | 10 | 50 | 940 | 1000 |
| #5 | 1000 | 0,1 | #1 | 100,0 | 10 | 50 | 940 | 1000 |
| Образцы печени: | ||||||||
В некоторых вариантах осуществления применяли следующие условия ВЭЖХ для MS-ВЭЖХ, например, тандемный способ инструментального анализа MS-ВЭЖХ:
ВЭЖХ проводили на устройстве Phenomenex Luna Amino 3 мкм 100A, колонка 30×2 мм, с мобильной фазой: A: 70% 10 мМ NH4OAc, 30% ACN pH 6,0; и B: 70% 1 мМ NH4OAc, 30% ACN pH 0,5 следующим образом:
Программа элюирования градиента:
| Этап | Время (минуты) | Поток (мкл/мин) | А (%) | B (%) |
| 0 | 0 | 400 | 60 | 40 |
| 1 | 1,1 | 400 | 60 | 40 |
| 2 | 1,11 | 400 | 40 | 60 |
| 3 | 2,11 | 400 | 30 | 70 |
| 4 | 2,6 | 400 | 20 | 80 |
| 5 | 3,1 | 400 | 0 | 100 |
| 6 | 5,5 | 400 | 0 | 100 |
| 7 | 5,51 | 400 | 60 | 40 |
| 8 | 10 | 400 | 60 | 40 |
| Объем инъекции: 50 мкл Скорость потока в MS: 0,400 мл/мин, отсутствие деления потока |
||||
Условия мониторинга множественных реакций (MRM): (API3000)
| Способ ионизации: | Положительная ионозация электрораспылением (ESI+) |
| Напряжение ионораспыления: (IS) | 5000 V |
| Температура (TEM): | 550°C |
| Турбо IS газ | 8 л/мин |
| Небулайзер (NEB): | 14 |
| Установка газа CAD (CAD): | 6 |
| Потенциал декластеризации (DP) | 68 V |
| Энергия столкновений (CE) | 27 eV |
| Потенциалы вход/выход (EP/CXP) | 10 V/11 V |
| Соединение | Ион-предшественник→ион продукта |
| ddA трифосфат | 476,2 → 135,9 |
| ddA дифосфат | 396,2 → 135,9 |
| dA трифосфат (ISTD) | 460,2 → 135,9 |
| Колонка *Luna Amino непосредственно связана с держателем кассеты «охрана безопасности» на входном конце, подходящем для колонок Phenomenex 2,1 мм, содержащих кассету С18. | |
Пример 37
Фосфорилирование in vitro в гепатоцитах
Первичные гепатоциты (крысы, обезьяны Cynomolgus или человека) высевали в концентрации 0,8×106 в покрытые коллагеном 12-луночные планшеты и оставляли для фиксирования на 4-6 часов, после чего среда высевания была замещена культуральной бессывороточно средой, и клетки оставляли для акклиматизации в новой среде в течение ночи. На следующий день клетки подвергали воздействию в течение 1, 4, 8 и 24 часов тестового соединения (NM204) (A550) в концентрации 10 и 50 мкМ, приготовленных в новой культуральной среде из стокового раствора в ДМСО (конечная концентрация ДМСО составляла 0,1%). В каждой точке времени собирали аликвотное количество (500 мкл), немедленно добавляли 500 мкл ацетонитрила и сохраняли при -20°C до анализа. Оставшуюся воздействующую среду удаляли, и клеточный монослой (прикрепившийся к планшете) промывали 2 раза ледяным фосфатно-буферным раствором ФБР. Какое-либо количество оставшегося ФБР тщательно удаляли путем аспирации, и клетки собирали посредством соскабливания в 1 мл 70% ледяного метанола. Клеточные пробы помещали на ночь при -20°C и на следующий день центрифугированием удаляли клеточный дебрис. Удаляли супернатанты и проводили фильтрацию перед анализом LC/MS. Стандартную кривую подготавливали с использованием необработанных клеток, с которыми провели сходные манипуляции, же за исключением манипуляций до их сбора в 70% метаноле, и к промытым монослоям добавляли 10 мкл стандартных растворов LddАТФ, приготовленных в метаноле. Эти контрольные пробы затем обрабатывали и анализировали так же, как это описано для тестовых проб.
Ниже приведены результаты:
| Образование LddA-ТФ в гепатоцитах | |||
| Пример 1 (A550) 10 мкМ | |||
| Уровни LddA ТФ (пмоль/миллион клеток) | |||
| Время (часы) | Крыса | Обезьяна | Человек |
| 1 | 159,5 | 287,5 | 161,5 |
| 4 | 388,0 | 978,0 | 312,5 |
| 8 | 468,5 | 1230,0 | 352,5 |
| 24 | 422,0 | 344,0 | 366,0 |
| Пример 1 (A550) 50 мкМ | |||
| Уровни LddA АТФ (пмоль/миллион клеток) | |||
| Время (часы) | Крыса | Обезьяна | Человек |
| 1 | 393,0 | 2085,0 | 682,5 |
| 4 | 1212,0 | 5690,0 | 1480,0 |
| 8 | 1590,0 | 6030,0 | 1930,0 |
| 24 | 1505,0 | 3030,0 | 2062,5 |
Как показывают приведенные данные, были выявлены значительные уровни L-ddАТФ в гепатоцитах. Было выявлено, что в гепатоцитах обезьяны уровни достигали максимального уровня через 8 часов, с быстрым последующим снижением. Напротив, обнаружено, что в гепатоцитах крысы и человека через 8 часов уровни выравниваются.
Пример 38
Исследования in vivo на крысах
Распределение NM-204 (соединения из примера 1 (гидрокси-tBuSATE N-бензилфосфорамидатного производного L-ddA) (A550) в печени крысы оценивали после однократного внутривенного (в/в) или перорального введения A550 (NM-204) в дозе 20 мг/кг (перорально) или 10 мг/кг (в/в) веса тела. Растворы дозировок готовили в тот же день перед введением дозы.
В определенное время (через 1 и 3 часа для животных с в/в введением или через 1, 3 и 8 часов для животных с пероральным введением) каждое животное подвергали эвтаназии газом CO2, с последующим обескровливанием через брюшную вену. Сразу после умерщвления печень немедленно забирали, быстро замораживали в жидком азоте, помещали на сухой лед и затем сохраняли при -70°C до анализа.
Подготовка калибровочных стандартов из экстрактов печени контроля:
Контрольные образцы печени крысы забирали из целой замороженной печени ((Bioreclamation, Inc. Hicksville, NY) при помощи инструмента для биопсии образцов тканей (Harris Unicore, 8,0 мм, VWR). Каждый образец весом примерно 0,1 г помещали в отдельный поли-флакон объемом 2 мл с 0,940 мл 80% MeOH/20% DIH2O и готовили гомогенаты, используя механический разрушитель тканей (Tissue Master, Omni-International, Inc, Marietta GA). Во флаконы вводили 10 мкл аликвотных количеств рабочего стокового раствора и 50 мкл аликвотных количеств ISTD перед перемешиванием на вортексе в течение ~30 секунд. Смеси сохраняли в течение ночи при -20°C и на следующий день на 10 мин извлекали для центрифугирования в настольной центрифуге. Каждый супернатант переносили в отдельную центрифужную фильтрационную единицу (0,45 мкм), и полученные фильтраты переносили в пробирки ВЭЖХ для анализа LC/MS/MS. Конечные концентрации ddАТФ в калибровочных стандартах составляли 1000, 500, 250, 125, 50 и 0 пмоль/мл. Каждый калибровочный стандарт непосредственно вносили в объеме 50 мкл в ионнообменную колонку для анализа. Проводили анализ стандартной кривой калибровочных стандартов из контрольных экстрактов печени.
Анализ ddАТФ проводили путем ионообменной хроматографии с онлайн обнаружением положительной ионизации ESI-MS/MS способом мониторинга множественных реакций (MRM). Пиковые области, полученные для калибровочных стандартов 4 из 5, позволяли построить стандартную кривую, которая демонстрировала хорошую линейность (R2=0,9996) в диапазоне концентрации от 50 до 1000 пмоль/мл. Это является эквивалентным диапазону от 5 до 100 пмоль/г печени при использовании препарата образца. Применяли условия ВЭЖХ MS/MS, описанные в примере 5. Более низкий предел количественного определения, показываемый методикой LC/MS/MS, составляет, например, около 0,2 пмоль/мл для клеточных экстрактов гепатоцитов, которые содержат намного меньше соли.
Ниже приведены результаты, показывающие внутриклеточные уровни A550 (NM204) (показывающие соединения, проникшие в клетки печени) и уровни LddАТФ (показывающие расщепление фосфорамидатной функциональной группы и трифосфорилирование ddA до активного трифосфата в печени):
| A550 (пример 1) и LddАТФ, измеренные в печени самцов крыс, которым в/в или перорально водили A550 (пример 1) | ||||
| Номер животного | Концентрация соединения (A550) (пример 1) Концентрация (пмоль/г печени) | Точка времени (часы) | Концентрация ddA-TP | |
| (пмоль/г печени) | (пмоль/106 клеток)* | |||
| В/в дозировка (10 мг/кг) | ||||
| 1М1 | 65,8 | 1 | 2025 | 17,8 |
| 1М2 | 89,1 | 1 | 1930 | 16,9 |
| 1М3 | 85,1 | 1 | 1355 | 11,9 |
| Среднее значение | 80,0 | 1770 | 15,5 | |
| В/в дозировка (10 мг/кг) | ||||
| 2М1 | 28,3 | 3 | 1345 | 11,8 |
| 2М2 | 26,0 | 3 | 1940 | 17,0 |
| 2М3 | 29,3 | 3 | 2990 | 26,2 |
| Среднее значение | 27,9 | 2092 | 18,3 | |
| Пероральная дозировка (20 мг/кг) | ||||
| 3М1 | 411 | 1 | 210 | 1,8 |
| 3М2 | 272 | 1 | 575 | 5,0 |
| 3М3 | 70,2 | 1 | 400 | 3,5 |
| Среднее значение | 251 | 395 | 3,5 | |
| Пероральная дозировка (20 мг/кг) | ||||
| 4М1 | 360 | 3 | 200 | 1,8 |
| 4М2 | 92,1 | 3 | 330 | 2,9 |
| 4М3 | 161 | 3 | 405 | 3,6 |
| Среднее значение | 204 | 312 | 2,7 | |
| Пероральная дозировка (20 мг/кг) | ||||
| 5М1 | 16,4 | 8 | 280 | 2,5 |
| 5М2 | 28 | 8 | 805 | 5,2 |
| 5М3 | 16,2 | 8 | 275 | 2,4 |
| Среднее значение | 20,1 | 382 | 3,3 | |
| *Гепатоцеллюлярное количество для крысы составляло 114×106 клеток на грамм печени (Toxicology in Vitro 20 (2005) 1582-1586). | ||||
Таким образом, приведенные результаты показывают, что соединение можно использовать для повышения концентрации препарата в печени. Эти результаты также показывают увеличенную концентрацию активного трифосфата, который образуется в клетках печени.
Пример 39
Исследование репликона HCV
Клетки Huh-7, содержащие субгеномный репликон HCV Con1 (клетки GS4.1), (C. Seeger; Fox Chase University, Philadelphia, PA, USA), выращивали в среде Игла, модифицированной по Дульбекко (DMEM) с добавлением 10% эмбриональной бычьей сыворотки (ЭБС), 2 мМ L-глутамина, 110 мг/л пирувата натрия, 1X не незаменимых аминокислот, 100 ед/мл пенициллина - стрептомицина и 0,5 мг/мл G418 (Invitrogen). Для анализа эффекта дозы клетки высевали в 96-луночные планшеты в количестве 7,5×103 клеток/на лунку в объеме 50 мкл и инкубировали при 37°C/5% CO2. Через три часа после высевания добавляли 50 мкл десяти 2-кратных серийных разведений соединений (самая высокая концентрация составляла 75 мкМ), и клеточные культуры инкубировали при 37°C/5% CO2 в присутствии 0,5% ДМСО. Альтернативно, соединения тестировали в единственной концентрации 15 мкМ. Во всех случаях отрицательным контролем были клетки Huh-7, не имеющие HCV репликона. Клетки инкубировали в присутствии соединений в течение 72 часов, после чего их анализировали на экспрессию белка NS4A посредством твердофазного иммуноферментного анализа (ELISA). Для этого планшеты фиксировали в течение 1 минуты ацетоном:метанолом 1:1, дважды промывали фосфатно-буферным раствором (ФБР), 0,1% Твин20, блокировали в течение 1 часа при комнатной температуре буфером TNE, содержащим 10% ЭБС и затем инкубировали в течение 2 часов при 37°C с анти-NS4A мышиным моноклональным антителом A-236 (ViroGen), разведенным указанным буфером. После трехкратного промывания ФБР, 0,1% Твин20 клетки инкубировали в течение 1 часа при 37°C с антимышиным иммуноглобулином G, конъюгированным с пероксидазой, в буфере TNE, 10% ЭБС. После промывания, как описано выше, реакцию продолжали с O-фенилендиамином (Zymed). Реакцию прекращали через 30 минут с помощью 2н H2SO4 и считывали спектральную поглощательную способность при 492 нм, используя спектрофотометр Sunrise Tecan. Значения EC50 определяли исходя из % ингибирования по отношению к данным концентрации, используя сигмоидальный нелинейный анализ регрессии, основанный на четырех параметрах, с программным обеспечением Tecan Magellan. При исследовании единственной концентрации результаты выражали как % ингибирования в 15 мкМ. Для оценки цитотоксичности клетки GS4.1 обрабатывали соединениями, как описано выше, и тестировали жизнеспособность клеток, используя анализ клеточной пролиферации с единственным раствором Cell Titer 96 AQueous One Solution Cell Proliferation Assay (Promega). Значения CC50 определяли из % цитотоксичности по отношению к данным концентрации, используя программное обеспечение Tecan Magellan, как описано выше.
Результаты
Провели оценку соединений, представленных в таблице ниже, согласно анализу репликона, описанному выше.




Значения EC50 в анализе ELISA 2 обозначены следующим образом:
+++ ≤1 мкм, ++>1-10 мкм и + >10 мкм
Значения CC50 обозначены следующим образом:
++ ≤75 мкм, + >75 мкм
Пример 40
Анализ лекарственной чувствительности к препарату против HBV
a) На культуральные планшеты с покрытием коллагеном-I высевали клетки с плотностью 0,25-0,5×106 клеток на лунку в 2 мл ростовой/селективной среды.
b) Стоковые свежие растворы лекарственного препарата готовили в 100% ДМСО как 200× исходный раствор. Готовили семь 4-кратных разведений тестового соединения в диапазоне от 2,5 мкМ до 0,0006 мкМ (конечное). Основные разведения препарата разделяли на 4 аликвотных количества и затем сохраняли при -20°C до использования.
c) Через один день после посева клеток начинали применение препарата путем добавления 10 мкл разведения препарата вместе с 2 мл свежей ростовой/селективной среды.
Таким образом, конечная концентрация ДМСО не превышала 0,5%. В контрольные лунки без препарата вводили 10 мкл ДМСО в свежей среде.
d) Клетки обрабатывали через один день 2 мл свежей комбинации среды/препарата в течение в общей сложности 8 дней. Затем на 10 день собирали клеточные лизаты, как описано ниже, и проводили анализ эндогенной полимеразы.
Приготовление нуклеокапсид-содержащих лизатов для анализа EPA
a) Через два дня после заключительного применения препарата осуществляли сбор клеток.
b) Среду аккуратно аспирировали, и клеточные монослои однократно ополаскивали 1 мл ФБР.
c) В каждую лунку добавляли 1 мл лизирующего буфера (50 мМ Трис-HCl с уровнем pH 7,5/150 мМ NaCl/5 мМ MgCl2/0,2% NP-40). Промывочное средство должно снимать внешнюю оболочку вирионов и позволять захватывать внутренние нуклеокапсиды. Планшеты сохраняли на льду в течение >30 мин.
d) Лизированные клетки переносили в микроцентрифужные пробирки объемом 1,5 мл.
e) Лизаты очищали центрифугированием при комнатной температуре в течение 5 минут при 14000 оборотов в минуту.
f) Очищенные лизаты переносили в новые пробирки и немедленно замораживали на сухом льду, затем сохраняли при -80°C, до получения возможности проведения анализа эндогенной полимеразы, как описано ниже.
Анализ эндогенной полимеразы (EPA) клеточных лизатов
a) EPA проводили по существу как описано авторами Seifer, et al (1998). J. Virol. 72: 2765-2776. Очищенные лизаты оттаивали при комнатной температуре.
b) Внутриклеточные HBV-нуклеокапсиды подвергали имунопреципитации из цитоплазматических лизатов в течение ночи при 4°C с поликлональным кроличьим анти-HBcAg антителом и иммобилизировали на протеин А сефарозных CL-4B шариках.
c) После 2 промываний иммобилизированных капсид 1 мл промывочного буфера EPA (75 мМ NH4Cl, 50 мМ Трис-HCl с уровнем pH 7,4, 1 мМ ЭДТА), начинали реакции эндогенной полимеразы путем добавления 50 мкл детергент-содержащего коктейля EPA (50 мм Трис-HCl, pH 7,4, 75 мМ NH4Cl, 1 мМ ЭДТА, 20 мМ MgCl2, 0,1 мМ β-МЕ, 0,5% NP-40, 100 мкМ холодной dGTP, TTP, dCTP и 50 нм 33P-dАТФ) и инкубировали в течение ночи при 37°C. Детергент должен увеличивать проницаемость нуклеокапсид.
d) Последующее расщепление эндогенно 33P-меченой HBV-ДНК в течение 1 часа при 37°C посредством 1 мг/мл протеиназы K проходило беспрепятственно с помощью фенол/хлороформной экстракции.
e) Затем осаждали нуклеиновые кислоты с одним объемом 5М NH4-ацетата и 2,5 объемами 100% EtOH, и сепарировали на 1% нативном агарозном геле в Трис-боратном буфере.
f) Гели блотировали на положительно заряженную нейлоновую мембрану в течение ночи при комнатной температуре посредством капиллярного переноса в 0,4н NaOH.
g) Проводили скрининг сухих мембран с помощью фосфоримиджера (PhosphorImager, GE Healthcare) в течение ночи при комнатной температуре, затем сканирование (Storm 860, GE Healthcare) и количественный анализ с программным обеспечением ImageQuant (GE Healthcare).
h) Создавали кривые эффекта с использованием программного обеспечения XLfit 4.1. Средние значения эффективной концентрации препарата, которые ингибируют активность эндогенной HBV-полимеразы на 50%, вычисляли по нескольким независимым экспериментам.
Определение цитотоксичности
Стандартное исследование цитотоксичности in vitro проводили в клетках HepG2. Клетки подвергали воздействию препарата в течение 9 дней. Жизнеспособность клетки определяли посредством MTS-окрашивания с использованием анализа клеточной пролиферации CellTiter 96 Aqueous One Solution согласно инструкциям фирмы-изготовителя.
a) Клетки HepG2 высевали в 96-луночные планшеты для тканевых культур в 100 мкл свежей ростовой среды в количестве 7×103 клеток на лунку.
b) Стоковые растворы препарата готовили в 100% ДМСО как 400× исходные растворы, и их сохраняли при -20°C до использования.
c) Через четыре часа после высевания клеток готовили разведения препарата, после чего их добавляли в клетки. Клетки получали до 100 мкМ препарата, с общим объемом 200 мкл свежей ростовой среды, содержащей 0,25% ДМСО. В контрольные лунки добавляли ростовую среду с ростовой средой с 0,25% ДМСО. Планшеты инкубировали при 37°C с 5% CO2.
d) Клетки обрабатывали через один день свежей ростовой средой и свежими разведениями препарата в течение в общей сложности 8 дней, как описано выше.
e) На 9 день определяли жизнеспособность клеток HepG2 путем добавления 20 мкл раствора CellTiter 96 Aqueous One Solution. После 4 часов инкубации при 37°C измеряли спектральную поглощательную способность при A490 нм в планшетном ридере Victor V (Perkin Elmer).
f) Концентрации CC50 определяли с использованием программного обеспечения XLfit 4.1.
Противовирусное действие in vitro PMEA, B261 (гидрокси-tBuSATE N-бензилфосфорамидатного производного PMEA, как показано в таблице из примера 10), как и LdT в качестве контроля, тестировали всего в 4 анализах лекарственной чувствительности препарата HBV. Ниже в таблице приведены результаты:
HBV клеточный анализ (Показания EPA)
| Цитотоксичность | Противовирусное действие* | ||
| Препарат | (CC50 в мкМ) | (EC50 в мкМ) | SI |
| РМЕА | >100 | 0,328±0,082 | >310 |
| В261 | 19,6 | 0,016±0,004 | 1225 |
| LdT | >100 | 0,366±0,056 | >273 |
| *Цитотоксичность и эффективность определены на коллагеновых планшетах. | |||
Пример 41
Определение общего метаболизма в субклеточных фракциях печени (деплеция исходного)
Инкубирование НАДФН (восстановленного никотинамидадениндинуклеотидфосфата). Микросомальное или S9 инкубирование проводили в конечном объеме 0,5 мл. Объединенные микросомальные или S9 белки печени (1,0 мг/мл), суспендированные в инкубационном буфере (100 мМ фосфата калия, уровень pH 7,4, 5 мМ MgCl2 и 0,1 мМ ЭДТА), предварительно инкубировали в течение 5 минут при 37°C с 10-50 мкМ OHSATE фосфорамидатным соединением из стокового раствора в ДМСО (конечная концентрация ДМСО составляла 0,1%); реакцию начинали путем добавления НАДФН (конечная концентрация 3 мМ). Инкубирование без НАДФН являлось контролем. В определенные точки времени (0-120 минут) забирали пробы в 0,1 мл, и реакцию прекращали путем добавления 1 объема останавливающего раствора (ацетонитрил). Образцы подвергали вортекс-смешиванию в течение 30 секунд и затем центрифугировали при 1500g в течение 10 мин. Супернатант переносили в стеклянные пробирки для ВЭЖХ и проводили ВЭЖХ без дополнительной обработки. Фиг.1 и 2 отображают деплецию NM108 SATE фосфорамидата (B299), и NM107 SATE фосфорамидата (B102), соответственно, после инкубации с НАДФН в печени обезьяны S9.
Система ВЭЖХ для пролекарства в образцах с неизменяемой средой
| ВЭЖХ: | Agilent 1100 |
| Колонка: | Phenomenex Luna C 18 (2), 20×2 мм |
| Мобильные фазы (MP): | MP (A) 10 мМ K2HPO4 pH 5, MP (B) ACN |
| Градиент элюирования: | 20-63% MP (B) действующий от 0 до 30 минут |
| Время выполнения: | 20 минут |
| Скорость потока: | 1 мл/минуту |
| Объем инъекции: | 10-20 мкл |
| УФ: | 252 нм производного NM108SATE (B299) 272 нм производного NM107SATE (B102) |
Таким образом, не будучи ограниченным какой-либо теорией, учитывая, что метаболизм является НАДФН-зависимым, возможно, что фосфорамидатное соединение предпочтительно активируется в печени Цитохромом P450.
Пример 42
Определение клеточного уровня трифосфата
Приготовление культур первичных гепатоцитов
Клетки, только что выделенные из печени животного и человека, находились в суспензии на льду. Следуя методике, клетки осаждали центрифугированием при 500 оборотах в минуту (клетки крысы) или при 700 оборотах в минуту (обезьяна и человек) и ресуспендировали в количестве 0,8 миллионов клеток на мл среды в планшете (HPM). Затем их высевали на многолуночные покрытые коллагеном планшеты (12-луночные) путем добавления 1 мл клеточной суспензии (0,8 млн клеток/мл). Планшеты аккуратно встряхивали для равномерного распределения клеток и помещали в инкубатор при 37°C в течение около 4-6 часов, чтобы позволить клеткам зафиксироваться. После фиксации клеток планшетную среду удаляли и замещали культуральной средой для гепатоцитов (HCM). Клетки оставляли на ночь в инкубаторе при 37°C для акклиматизации к культивированию и среде.
Инкубирование с тестовым соединением
Инкубирование гепатоцитов проводили в конечном объеме 1,0 мл HCM/на лунку (0,8 миллионов клеток/мл). После инкубации клеток в течение ночи удаляли HCM и замещали свежей HCM, предварительно нагретой до 37°C и содержащей 10 мкМ тестового соединения из стокового раствора в ДМСО (конечная концентрация ДМСО составляла 0,1%). В определенные точки времени (до 24 часов) удаляли инкубационную среду, и монослои клеток аккуратно промывали два раза ледяным ФБР. После последнего промывания тщательно полностью удаляли ФБР и добавляли 1 мл экстракционного буфера (ледяной 70% метанол). После добавления метанола каждую лунку сразу закрывали парфильмом. После обработки всей планшеты парафильмом дополнительно закрывали всю планшету, образуя двойное покрытие для предотвращения испарения во время процесса экстракции. Затем планшет закрывали крышкой и герметизировали лентой. После этого планшеты сохраняли при -20°C в течение минимум 24 часов, чтобы протекала экстракция внутриклеточного содержимого.
Приготовление культур Huh7 или HepG2
Клетки HepG2 или Huh7 высевали в количестве 0,4×106 клеток/на лунку в покрытые коллагеном 12-луночные планшеты. Клетки оставляли для фиксации в течение ночи. После инкубации клеток в течение ночи удаляли культуральную среду и замещали ее свежей культуральной средой, предварительно нагретой до 37°C, содержащей 10 мкМ тестового соединения из стокового раствора в ДМСО (конечная концентрация ДМСО составляла 0,1%). Через 24-72 часа удаляли инкубационную среду, и монослои клеток аккуратно промывали два раза ледяным ФБР. После последнего промывания тщательно полностью удаляли ФБР и добавляли 1 мл экстракционного буфера (ледяной 70% метанол). После добавления метанола каждую лунку сразу закрывали парфильмом. После обработки всей планшеты парафильмом дополнительно закрывали всю планшету, образуя двойное покрытие для предотвращения испарения во время процесса экстракции. Затем планшет закрывали крышкой и герметизировали лентой. После этого планшеты сохраняли при -20°C в течение минимум 24 часов, чтобы протекала экстракция внутриклеточного содержимого.
Приготовление образца для анализа
Клеточные экстракты готовили путем переноса 0,9 мл экстракта в микроцентрифужную пробирку объемом 2 мл, с последующим центрифугированием в течение 5 минут при 14000 оборотах в минуту. Около 100 мкл супернатанта переносили в пробирки ВЭЖХ, и определяли уровни трифосфата анализом LCMS-MS, как описано ниже.
Условия ВЭЖХ: NM107-трифосфат
| ВЭЖХ: | |
| Колонка: | Phenomenex Luna Amino, 3 мкм, 100 А, 30×2 мм |
| Мобильные фазы (MP): | A) 70% 10 мМ NH4OAc 30% ACN, pH 6,0 B) 70% 1 мМ NH4OAc 30% ACN, pH 10,5 |
Градиент элюирования:
| Этап | Время | Поток | А | В |
| 0 | 0,00 | 400 | 80 | 20 |
| 1 | 0,10 | 400 | 80 | 20 |
| 2 | 0,11 | 400 | 40 | 60 |
| 3 | 0,21 | 400 | 40 | 60 |
| 4 | 2,60 | 400 | 10 | 90 |
| 5 | 2,61 | 400 | 0 | 100 |
| 6 | 5,60 | 400 | 0 | 100 |
| 7 | 5,61 | 400 | 80 | 20 |
| 8 | 9,00 | 400 | 80 | 20 |
| Скорость потока в MS: | 0,400 мл/мин, без разделения | |||
| Объем инъекции: | 10 мкл | |||
| Соединение | ион-предшественник | ион-продукт | ||
| NM107 трифосфат | 498,0 | 112,0 | ||
Примеры условий ВЭЖХ: NM108-трифосфат
| ВЭЖХ: | |
| Колонка: | Phenomenex Luna Amino, 3 мкм, 100 А, 30×2 мм |
| Мобильные фазы (MP): | (A) 70% 10 мМ NH4OAc, 30% ACN, pH 6,0 (B) 70% 1 мМ NH4OAc, 30% ACN, pH 10,5 P |
Градиент элюирования:
| Этап | Время | Поток | А | В |
| 0 | 0,00 | 400 | 60 | 40 |
| 1 | 0,10 | 400 | 60 | 40 |
| 2 | 0,11 | 400 | 40 | 60 |
| 3 | 0,21 | 400 | 40 | 60 |
| 4 | 2,60 | 400 | 10 | 90 |
| 5 | 2,61 | 400 | 0 | 100 |
| 6 | 5,61 | 400 | 0 | 100 |
| 7 | 5,61 | 400 | 60 | 40 |
| 8 | 9,00 | 400 | 60 | 40 |
| Скорость потока в MS: | 0,400 мл/мин, без разделения | |||
| Объем инъекции: | 10 мкл | |||
| Соединение | ион-предшественник | ион-продукт | ||
| NM108 трифосфат | 538,0 | 152,0 | ||

Уровни NM107 трифосфата и B102 трифосфата в клеточных экстрактах выявляли следующим образом:
| Внутриклеточный трифосфат (пмоль/млн клеток) | ||||
| Лекарственный препарат в культуре | Человек | Обезьяна | HepG2* | Huh7* |
| B102 | 991 | 1838 | 1,5 | 9,2 |
| NM107 | 19 | 10 | 17 | 37 |
| 24 часа инкубации в 10 мкм препарата | ||||
| *72 часа инкубации в 10 мкм препарата | ||||
Как указано в данных выше, уровень внутриклеточного трифосфата у B102 был выше по сравнению с уровнем NM107.
Пример 43
Демонстрация мощного противовирусного действия нуклеозидных ингибиторов второго поколения B102 у HCV-инфицированных шимпанзе
Нуклеозидные аналоги, такие как NM107 (2'-метилцитидин, нуклеозидный компонент валопицитабина), показали эффективность против HCV в клинических условиях, и их 5'-трифосфаты (ТФ) могут быть мощными ингибиторами NS5B-полимеразы HCV. Вместе с тем, их широкое системное распределение и неэффективное превращение в ТФ в печени могут приводить к снижению безопасности и эффективности противовирусного действия. Проводили доклиническое исследование безопасности и противовирусного действия in vivo нуклеотидных пролекарств B102.
Способы: Фармакокинетическое (ФК) и токсикологическое исследование B102 заключалось в его пероральном введении крысам или обезьянам в дозах от 20 до 300 мг/кг/в день в течение до 14 дней. Определяли с помощью LC-MS/MS уровни нуклеозида ТФ в печени. Соединения (10 мг/кг/в день) вводили шимпанзе, хронически инфицированным HCV генотипом 1, один раз в день ежедневно в течение 4 дней перорально через зонд. Вирусные нагрузки HCV контролировали перед лечением, в ходе лечения и после, посредством количественной полимеразной цепной реакции в реальном времени RT-ПЦР.
Результаты: ФК исследования у крыс и обезьян показали, что B102 имеет первоначальную экстракцию в печени >95% с низким системным воздействием (<1%). Уровни ТФ в печени нуклеотидных пролекарств были в 10-50 раз выше по сравнению с нуклеозидной копией. Не выявлено какой-либо токсичности после введения A2 в дозе 50 мг/кг/в день обезьянам в течение 14 дней. Не выявлено какой-либо тошноты или токсичности для ЖКТ. У HCV-инфицированных шимпанзе B102 оказывал быстрое и мощное противовирусное воздействие, с последующим возвращением к исходному уровню после прекращения приема препарата. Среднее снижение вирусных нагрузок у B102 на протяжении 4 дней воздействия препарата находилось в диапазоне от 1,5 log10. Эквивалентная доза валопицитабина давала в результате снижение вирусной нагрузки 0,7 log10. У шимпанзе не выявлено каких-либо лабораторных отклонений или доказательст токсичности.
Таким образом, при пероральном введении B102 создает в печени высокие уровни трифосфатов наряду с низким системным воздействием, что приводит к быстрому и мощному ингибированию репликации HCV у шимпанзе, и таким образом показывает in vivo перспективный доклинический профиль безопасности и противовирусного действия.
Пример 44
Соединения исследовали на действие против HBV. Определяли действие против HBV на HBV-вирионах и нуклеокапсидах посредством анализа эндогенной полимеразы (EPA)
Испытание чувствительности лекарственного препарата с использованием HBV-продуцирующей клеточной линии дикого типа
1) На 12-луночные планшеты с покрытием коллагеном-I высевали продуцирующие клетки, экспрессирующие HBV дикого типа с плотностью 0,5-1×106 клеток на лунку в 2 мл ростовой/селективной среды.
2) Стоковые свежие растворы лекарственного препарата готовили в 100% ДМСО как 200× исходный раствор. Готовили пять 4-кратных разведений их указанного 200× стокового раствора в 100% ДМСО. Для каждого эксперимента 4 аликвотных количества каждого разведения препарата сохраняли при -20°C до использования.
3. Как только клетки достигали конфлюэнтности (через 1 день после посева клеток), начинали применение препарата путем добавления 10 мкл разведения препарата вместе с 2 мл свежей ростовой/селективной среды. Таким образом, конечная концентрация ДМСО не превышала 0,5%. В контрольные лунки без препарата вводили 10 мкл ДМСО в свежей среде.
4. Клетки обрабатывали через один день 2 мл свежей среды/препарата в течение в общей сложности 8 дней. Затем на 10 день собирали клеточные лизаты клетки, как описано ниже, и проводили анализ эндогенной полимеразы (EPA).
Приготовление нуклеокапсид-содержащих лизатов для анализа EPA
1. Клетки выращивали в течение 3-4 дней в 12-луночных покрытых коллагеном-I планшетах до достижения конфлюэнтности.
2. Среду аккуратно аспирировали, и клеточные монослои клетки однократно ополаскивали 1 мл ФБР.
3. В каждую лунку добавляли 1 мл лизирующего буфера (50 мМ Трис-HCl, pH 7,5/150 мМ NaCl/5 мМ MgCl2/0,2% NP-40). Планшеты сохраняли на льду в течение от 30 мин до 4 часов.
4. Лизированные клетки переносили в микроцентрифужные пробирки объемом 1,5 мл.
5. Лизаты очищали центрифугированием при комнатной температуре в течение 5 минут при 14000 оборотов в минуту.
6. Очищенные лизаты переносили в новые пробирки и немедленно замораживали на сухом льду, затем сохраняли при -80°C, до получения возможности проведения анализа эндогенной полимеразы, как описано ниже.
Приготовление секретируемых вирионов из супернатанта для анализа
EPA
1. Клетки выращивали в течение 3-4 дней в 12-луночных покрытых коллагеном-I планшетах до достижения конфлюэнтности.
2. Среду аккуратно аспирировали и переносили в микроцентрифужные пробирки объемом 1,5 мл.
5. Супернатанты очищали центрифугированием при комнатной температуре в течение 5 минут при 14000 оборотов в минуту.
6. Очищенные супернатанты переносили в новые пробирки и немедленно замораживали на сухом льду, затем сохраняли при -80°C, до получения возможности проведения анализа эндогенной полимеразы, как описано ниже.
Анализ эндогенной полимеразы (EPA) клеточных лизатов и супернатантов
1. Анализы EPA выполняли по существу как описано авторами Seifer, et al (1998). Внутриклеточные HBV-нуклеокапсиды подвергали имунопреципитации из цитоплазматических лизатов в течение ночи при 4°C с поликлональным кроличьим анти-HBcAg антителом и иммобилизировали на протеин А сефарозных CL-4B шариках. Секретируемые вирионы подвергали имунопреципитации из очищенных клеточных супернатантов в течение ночи при 4°C с моноклональным мышью анти-LS антителом (МА18/7) в отсутствии детергента.
2. После 3 промываний иммобилизированных капсид или вирионов с помощью 1 мл промывочного буфера EPA (75 мМ NH4Cl, 50 мМ Трис-HCl, pH 7,4, 1 мМ ЭДТА), начинали реакции эндогенной полимеразы путем добавления 50 мкл детергент-содержащего коктейля EPA (50 мм Трис-HCl, pH 7,4, 75 мМ NH4Cl, 1 мМ ЭДТА, 20 мМ MgCl2, 0,1 мМ β-МЕ, 0,5% NP-40, 100 мкМ холодной dGTP, TTP, dCTP и 50 нМ 33P-dАТФ) и инкубировали в течение ночи при 37°C. Детергент является необходимым для снятия наружной оболочки с иммунопреципитированных вирионов, а также для увеличения проницаемости нуклеокапсид.
3. Последующее расщепление эндогенно 33P-меченой HBV-ДНК в течение 1 часа при 37°C посредством 1 мг/мл протеиназы K проходило беспрепятственно с помощью фенол/хлороформной экстракции.
4. Затем осаждали нуклеиновые кислоты с одним объемом 5М NH4-ацетата и 2,5 объемами 100% EtOH, и сепарировали на 1% нативном агарозном геле в буфере Трис-борат-ЭДТА.
5. Гели блотировали на положительно заряженную нейлоновую мембрану в течение ночи при комнатной температуре посредством капиллярного переноса в 0,4н NaOH.
6. Проводили скрининг сухих мембран с помощью фосфоримиджера (PhosphorImager, GE Healthcare) в течение ночи при комнатной температуре, затем сканирование (Storm 860, GE Healthcare) и количественный анализ с программным обеспечением ImageQuant (GE Healthcare).
7. Значения 50% эффективной концентрации (EC50) вычисляли из уравнений с максимальным соответствием, полученных с использованием программного обеспечения XLfit версия 4.1 (IDBS).
Были получены следующие результаты.
| Рассматриваемое соединение № | Структура | EC50 (мкм) вириона | RI EC50 (мкм) |
| А348 (NM 48) |
 |
+++ | |
| А363 (NM 77) |
 |
++ | ++ |
| А616 (NM 128) |
 |
++ | |
| А819 (NM 177) |
 |
++ | ++ |
| А361 (NM 55) |
 |
+ | ++ |
| А550 (NM 204) |
 |
+++ | +++ |
| С791 |
 |
++ | |
| В261 |
 |
+++ | |
| PMEA |
 |
+++ | |
| L-dT | +++ | ||
| EC50 HBV вириона и RI обозначены следующим образом: +++ ≤1 мкм, ++ >1-10 мкм и + >10 мкм |
|||
Пример 45
Этинильные нуклеозиды для лечения HCV
Далее описаны примеры способа синтеза соединений:

29: {9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-О-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)H-фосфонат
К перемешиваемому раствору 7j (0,32 ммоль) и S-(2-фосфитэтил)2,2-диметил-3-трифенилметилокситиопропианата (0,42 ммоль) в пиридине (5 мл) при -15°C по капле добавляли пивалоилхлорид (0,64 ммоль) в атмосфере азота. Реакционную смесь перемешивали при -15°C в течение 2 часов. Добавляли дихлорметан и раствор NH4Cl. Органическую фазу отделяли и промывали раствором NH4Cl, высушивали Na2SO4, фильтровали и выпаривали при пониженном давлении. Сырой материал очищали колоночной флэш-хроматографией (ДХМ/MeOH), чтобы получить названное соединение. Порошок коричневого цвета. Молекулярная формула C38H39FN5O8PS. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,12 (с, 6H), 1,84 (м, 4H), 3,04 (с, 2H), 3,92 (д, J=5,60 Гц, 1H), 4,01-4,10 (м, 3H), 4,33-4,39 (м, 2H), 4,60-4,66 (м, 1H), 6,13 (д, J=18,00 Гц, 1H), 6,67 (с, 2H), 7,21-7,35 (м, 15H), 7,81 (с, 1H), 10,86 (ушир.с, 1H).
30a: N-(4-фторбензиламинил)-{9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-O-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)фосфат
К перемешиваемому раствору 29 (0,088 ммоль) в безводном тетрахлориде углероде (880 мкл) добавляли по капле 4-фторбензиламин (0,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и выпаривали досуха (температура ванны не превышала 30°C). Неочищенную смесь подвергали гельфильтрации на силикагеле с элюированием метанолом в дихлорметане с градиентом 0-10%, чтобы получить названное соединение. Твердое вещество белого цвета. Молекулярная формула C45H45F2N6O8PS. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,09 (с, 6H), 3,03 (с, 2H), 3,39-3,41 (м, 2H), 3,90-3,93 (м, 5H), 4,05-4,08 (м, 1H), 4,20-4,23 (м, 2H), 4,62-4,65 (м, 1H), 5,74 (м, 1H), 6,08-6,14 (дд, J=17,94 Гц и J=4,22 Гц, 1H), 6,32 (м, 1H), 6,67 (с, 2H), 7,21-7,35 (м, 19H), 7,81 (с, 1H), 10,86 (ушир.с, 1H). 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,83 (с, 1P). l9F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -116,24 (с, 1F), -158-,2 (с, 1F). Сканирование ES+ 899 (M-H)+, УФ λмакс=255 нм.
31a: N-(4-фторбензиламинил)-{9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-O-(гидрокси-трет-бутил-S-ацил-2-тиоэтил)фосфат
30a (0,09 ммоль) растворяли в дихлорметане (320 мкл) и обрабатывали муравьиной кислотой (32 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут, фильтровали твердофазную экстракцией на колонке, элюировали метанолом в дихлорметане с градиентом 0-30%, затем очищали обращенно-фазовой (C 18) колоночной хроматографией на силикагеле с элюированием ацетонитрилом в воде с градиентом 0-100% и лиофилизировали из смеси воды/диоксана, чтобы получить названное соединение. Твердое вещество белого цвета. Молекулярная формула C26H31F2N6O8PS 1H ЯМР (d6-ДМСО, 400 МГц) δ (м.д.) 1,09 (с, 6H), 3,03 (с, 2H), 3,39-3,41 (м, 2H), 3,90-3,93 (м, 5H), 4,05-4,08 (м, 1H), 4,20-4,23 (м, 2H), 4,62-4,65 (м, 1H), 4,92 (м, 1H), 5,74 (м, 1H), 6,08-6,14 (дд, J=17,94 Гц и J=4,22 Гц, 1H), 6,32 (м, 1H), 6,67 (с, 2H), 7,21-7,35 (м, 4H), 7,81 (с, 1H), 10,86 (ушир.с, 1H). 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,66 (с, 1P). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -116,24 (с, 1F), -158,44 (с, 1F). Сканирование ES+ 657 (M-H)+, УФ λмакс 254 нм.
30b: N-(4-метоксибензиламинил)-{9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-O-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)фосфат
30b синтезировали из 29 и 4-метоксибензиламина, как описано для 30a. Твердое вещество белого цвета. Молекулярная формула C46H48FN6O9PS 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,09 (с, 6H), 3,03 (м, 2H), 3,42 (д, 7=5,02 Гц, 2H), 3,71 (д, J=3,60 Гц, 3!:H), 3,85-3,90 (м, 5H), 4,06-4,10 (м, 1H), 4,23-4,29 (м, 2H), 4,60-4,66 (м, 1H), 5,54-5,57 (м, 1H), 6,08-6,14 (дд, J=17,94 Гц и J=4,22 Гц, 1H), 6,28-6,33 (м, 1H), 6,60 (с, 2H), 6,80-6,85 (м, 2H), 7,18-7,20 (м, 2H), 7,23-7,25 (м, 15H), 7,82 (с, 1H), 10,56 (ушир.с, 1H). 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,83 (с, 1P). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -116,24 (с, 1F), -158,2 (с, 1F). Сканирование ES+ 911 (M-H)+, УФ λмакс=255 нм.
31b : N-(4-метоксибензиламинил)-{9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-O-(гидрокси-трет-бутил-S-ацил-2-тиоэтил)фосфат
31b синтезировали из 30b как описано для 31a. Твердое вещество белого цвета. Молекулярная формула C27H34FN6O9PS 1H ЯМР (d6-ДМСО, 400 МГц) δ (м.д.) 1,09 (с, 6H), 3,03 (м, 2H), 3,42 (д, J=5,02 Гц, 2H), 3,71 (д, J=3,60 Гц, 3H), 3,85-3,90 (м, 5H), 4,06-4,10 (м, 1H), 4,23-4,29 (м, 2H), 4,60-4,66 (м, 1H), 4,92 (т, J=5,50 Гц, 1H), 5,54-5,57 (м, 1H), 6,08-6,14 (дд, J=17,94 Гц и J=4,22 Гц, 1H), 6,28-6,33 (м, 1H), 6,60 (с, 2H), 6,80-6,85 (м, 2H), 7,18-7,20 (м, 2H), 7,82 (с, 1H), 10,56 (ушир.с, 1H). 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,86 (с, 1P). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -158,24 (с, 1F). Сканирование ES+ 669 (M-H)+, УФ λмакс=254 нм.
30c: N-(4-трифторбензиламинил)-{9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-O-(трифенилметилокси-трет-бутил-S-ацил-2-тиоэтил)фосфат
30c синтезировали из 29 и 4-трифторметилбензиламина, как описано для 30a. Твердое вещество белого цвета. Молекулярная формула C46H45F4N6O8PS 1H ЯМР (d6-ДМСО, 400 МГц) δ (м.д.) 1,09 (с, 6H), 3,03 (т, J=6,44 Гц, 2H), 3,42 (с, 2H), 3,87-3,92 (м, 5H), 4,03-4,08 (м, 1H), 4,24-4,29 (м, 2H), 4,60-4,64 (м, 1H), 5,79-5,82 (м, 1H), 6,08-6,14 (дд, J=17,94 Гц и J=4,22 Гц, 1H), 6,28-6,33 (м, 1H), 6,60 (с, 2H), 7,23-7,25 (м, 15H), 7,50-7,70 (м, 4H), 8,25 (ушир.с, 1H), 10,76 (ушир.с, 1H). 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,86 (с, 1P). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -158,20 (с, 1F) сканирование ES+ 949 (M-H)+, УФ λмакс=254 нм.
31c: N-(4-трифторметилбензиламинил)-{9-[(2R)2-дезокси-2-фтор-2-C-этинил-β-D-эритрофуранозил]гуанин}-5'-ил-O-(гидрокси-трет-бутил-S-ацил-2-тиоэтил)фосфат
31c синтезировали из 30c, как описано для 31a. Твердое вещество белого цвета. Молекулярная формула C27H31F4N6O8PS 1H ЯМР (d6-ДМСО, 400 МГц) δ (м.д.) 1,09 (с, 6H), 3,03 (т, J=6,44 Гц, 2H), 3,42 (с, 2H), 3,87-3,92 (м, 5H), 4,03-4,08 (м, 1H), 4,24-4,29 (м, 2H), 4,60-4,64 (м, 1H), 4,91 (ушир.с, 1H), 5,79-5,82 (м, 1H), 6,08-6,14 (дд, J=17,94 Гц и J=4,22 Гц, 1H), 6,28-6,33 (м, 1H), 6,60 (с, 2H), 7,50-7,70 (м, 4H), 8,25 (ушир.с, 1H), 10,64 (ушир.с, 1H). 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) 9,86 (c, 1P). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -158,24 (c, 1F) сканирование ES+ 669 (M-H)+, УФ λмакс-254 нм.
Ниже перечислены дополнительные примеры соединений, синтезируемых согласно способам, описанным в настоящем изобретении. Отмечены следующие названия соединений, синтезируемых в качестве примеров.
6a: 6-хлор-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]пурин
6b: N2-изобутирил-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
6c: 1-[(2R)2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]урацил
6d: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]тимин
6e: N4-бензоил-1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]цитозин
6f: 5-фтор-1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]урацил
6g: 4-хлор-7-[(2R)2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]пирроло[2,3-d]пиримидин
7i: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]аденин
7j: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
7k: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]цитозин
7l: 4-амино-7-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]пирроло[2,3-d]пиримидин
11c: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]урацил-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
11f: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]-5-фтороурацил-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
11k: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]цитозин-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
11l: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]-4-аминопирроло[2,3-d]пиримидин-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
16: 9-[(2R)-2,3-дидезокси-2-C-этинил-2-фтор-β-D-глицеропентофуранозил]гуанин
17: 9-[(2R)-2,3-дидезокси-2-C-этинил-2-фтор-β-D-глицеропентофуранозил]гуанин-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
20: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
23: 9-[(2R)-2-дезокси-3,5-ди-O-изобутирил-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
24: N2-изобутирил-9-[(2R)-2-дезокси-3,5-ди-O-изобутирил-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
27i: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]аденин-5'-трифосфата натриевая соль
27j: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин 5'-трифосфата натриевая соль
28: 9-[(2R)-2,3-дидезокси-2-C-этинил-2-фтор-β-D-глицеропентофуранозил]гуанин 5'-трифосфата натриевая соль





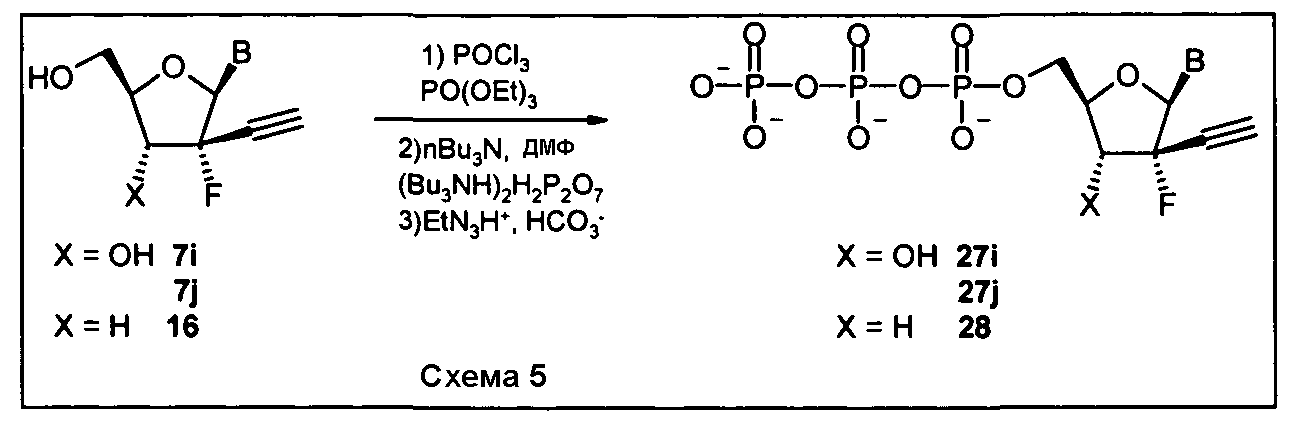
3a: 6-хлор-9-[2-оксо-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-β-D-рибофуранозил]пурин
6-Хлор-9-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-β-D-рибофуранозил]пурин (18,84 ммоль) выпаривали дважды совместно с ТГФ, затем растворяли в безводном ТГФ (50 мл). Добавляли безводный ДМСО (119,82 ммоль), и полученный раствор охлаждали до температуры от -40°C до -30°C. Добавляли по капле трифторуксусный ангидрид (36,17 ммоль), и раствор взбалтывали при температуре от -40°C до -30°C в течение 2 часов, после чего добавляли EtN3 (97,52 ммоль). Полученный раствор оставляли для нагревания до комнатной температуры в течение 30 минут, продолжая перемешивание, затем разводили диэтиловым эфиром и промывали H2O, высушивали (Na2SO4) и выпаривали досуха. Сырой материал очищали колоночной хроматографией с элюированием 1% этилацетатом в дихлорметане. Полученное масло желтого цвета растворяли в ДХМ и взбалтывали с избытком MgSO4 при комнатной температуре в течение 36 часов, фильтровали и концентрировали при пониженном давлении, чтобы получить названное соединение. Пена бледно-желтого цвета. Молекулярная формула C22H35ClN4O5Si2. 1H ЯМР (ДМСО-d6, 250 МГц) δ (м.д.) 9,01 (с, 1H, H-8), 8,61 (с, 1H, H-2), 6,35 (с, 1H, H-1'), 5,35 (д, 1H, H-3', J3'-4'=9,7 Гц), 4,31 (м, 1H, H-4'), 4,12-4,09 (м, 2H, H-5', H-5"), 1,22-0,94 (м, 28H, iPr). LRFAB-MS (GT): 527 (M+H)+, 525 (M-H)-. УФ λмакс=263 нм. Rf=0,17 (этилацетат/CH2Cl, 10/90 об/об).
4a и 4'a: 6-хлор-9-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-арабинофуранозил]пурин (4a) и 6-хлор-9-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-рибофуранозил]пурин (4'a)
Триметилсилилацетилен (59,20 ммоль) растворяли в безводном ТГФ (70 мл). Добавляли по капле при -78°C н-бутиллитий (37 мл, 1,6М в гексанах). Реакционную смесь взбалтывали в течение 30 минут при -78°C и затем оставляли до нагревания до -55°C. 3a (11,84 ммоль) в растворе ТГФ (34 мл) добавляли по капле при -78°C. Реакционную смесь взбалтывали в течение 1 часа при -78°C и затем оставляли для нагревания до -30°C. Реакцию гасили осторожным добавлением водного насыщенного NH4Cl (45 мл) при -78°C. После нагревания до комнатной температуры смесь разводили диэтиловым эфиром, промывали насыщенным солевым раствором, высушивали (Na2SO4) и концентрировали досуха. Сырой материал очищали хроматографией на силикагеле с элюированием 20% Et2O в петролейном эфире, чтобы получить два соединения: 4a (4,62 г, 62%). Пена бледно-желтого цвета. Молекулярная формула C27H45ClN4O5Si3 1H ЯМР (ДМСО-d6, 200 МГц) δ (м.д.) 8,81 (с, 1H, H-8), 8,64 (с, 1H, H-2), 6,64 (с, 1H, OH-2'), 6,33 (с, 1H, H-1'), 4,57 (д, 1H, H-3', J3'-4',=6,6 Гц), 4,20-3,97 (м, 3H, H-4', H-5' и H-5"), 1,20-1,00 (м, 28H, iPr), 0,14 (с, 9H, Si(CH3)3). LRFAB-MS (GT): 625 (M+H)+. Rf=0,72 (этилацетат/CH2Cl, 10/90, об/об);
и (4'a) (0,75 г, 10%). Масло желтого цвета. Молекулярная формула C27H45ClN4O5Si3 1H ЯМР (ДМСО-d6, 200 МГц) δ (м.д.) 8,80 (с, 1H, H-8), 8,73 (с, 1H, H-2), 6,64 (с, 1H, OH-2'), 6,55 (с, 1H, H-1'), 4,62 (д, 1H, H-3', 3J3'-4'=9,1 Гц), 4,39 (м, 1H, H-4'), 4,13 (дд, 1H, H-5', J=5'-4'=3,4 Гц, 2J5'-5"=13,2 Гц), 3,90 (дд, 1H, H-5", J5"-4'=2,6 Гц, 2J5"-5'=13,2 Гц), 1,15-1,00 (м, 28H, iPr), 0,10 (с, 9H, Si(CH3)3). LRFAB-MS (GT): 625 (M+H)+. Rf=0,64 (этилацетат/CH2Cl, 10/90, об/об).
5a: 6-хлор-9-[(2R)2-дезокси-2-фтор-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритропентофуранозил]пурин
4a (6,78 ммоль) растворяли в безводном толуоле (31,8 мл) в атмосфере аргона и охлаждали до -20°C. Добавляли по капле DAST (40,68 ммоль) и после завершения добавления удаляли охлаждающую ванну. Перемешивание продолжали в течение 1,5 часов, реакционную смесь растворяли этилацетатом, вливали в насыщенный NaHCO3 и взбалтывали в течение 5 мин. Органический слой промывали насыщенным солевым раствором, высушивали (Na2SO4), концентрировали и очищали хроматографией на силикагеле, с элюированием 20% Et2O в петролейном эфире, чтобы получить названное соединение (1,11 г, 26%). Масло желтого цвета. Молекулярная формула C27H44ClN4O4Si3. 1H ЯМР (CDCl3-d6, 200 МГц,) δ (м.д.) 8,79 (с, 1H, H-8), 8,48 (с, 1H, H-2), 6,48 (д, 1H, H-1', J1'-F=16,0 Гц), 4,74 (дд, 1H, H-3', J3'-4'=9,4 Гц, J3'-F=22,4 Гц), 4,36 (д, 1H, H-5', 2J5'-5"=13,4 Гц), 4,20 (м, 1H, H-4'), 4,10 (дд, 1H, H-5", 2J5"-5'=13,4 Гц, J5"-4'=2,6 Гц ), 1,30-1,10 (м, 28H, iPr), 0,00 (с, 9H, Si(CH3)3). LRFAB-MS (GT): 627 (M+H)+, УФ λмакс=263 нм, Rf=0,24 (диэтиловый эфир/петролейный эфир, 30/70, об/об).
6a: 6-хлор-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]пурин
Смесь 5a (3,65 ммоль) и фторид аммония (47,45 ммоль) в метаноле (12,5 мл) нагревали с обратным холодильником в течение 2 часов. После охлаждения при комнатной температуре смесь выпаривали досуха и очищали хроматографией на силикагеле, с элюированием метанолом в ДХМ с поэтапным градиентом 2-4%, чтобы получить названное соединение (0,89 г, 78%). Твердое вещество желтого цвета. Молекулярная формула C12H10ClFN4O3. 1H ЯМР (ДМСО-d6, 200 МГц) δ (м.д.) 9,02 (с, 1H, H-8), 8,89 (с, 1H, H-2), 6,55 (д, 1H, H-1', J1'-F=16,1 Гц), 6,34 (1д, 1H, OH-3'), 5,38 (1т, 1H, OH-5'), 4,64 (дт, 1H, H-3', J3'-4'=9,3 Гц, J3'-F=22,5 Гц), 4,07 (м, 1H, H-4'), 3,83 (м, 2H, H-5', H-5"), 3,76 (д, 1H, этинтил, 4JH-F=5,3 Гц). 13C ЯМР (ДМСО-d6, 75 МГц,) δ (м.д.) 152,0 (C-2), 151,2 (C-4), 149,5 (C-6), 144,7 (C-8), 130,9 (C-5), 95,1 (д, C-2', 1J2'-F=182,3 Гц), 88,0 (д, C-1', 2J1'-F=39,8 Гц), 82,9 (д, CCH, JC-F=8,2 Гц), 82,5 (C-4'), 75,3 (д, CCH, JC-F=31,5 Гц), 72,7 (д, C-3', 2J3'-F=19,5 Гц), 59,0 (C-5'). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -159,0 (м). LC/MS: (M+H+) 313,1 (8,29 мин), УФ λмакс 262 нм, Rf=0,21 (MeOH/CH2Cl, 7/93, об/об).
7i: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]аденин
6a (2,24 ммоль) растворяли в насыщенном аммонийном метаноле (80 мл) и нагревали в течение 4 часов в стальной бомбе при 90°C. После охлаждения до комнатной температуры смесь совместно выпаривали досуха и очищали хроматографией на силикагеле, с элюированием метанолом в ДХМ с градиентом 5-8%, чтобы получить названное соединение (305 мг, 46%). Твердое вещество желтого цвета. Молекулярная формула C12H12FN5O3. 1H ЯМР (ДМСО-d6, 200 МГц) δ (м.д.) 8,40 (с, 1H, H-8), 8,17 (с, 1H, H-2), 7,38 (1c, 2H, NH2) 6,35 (д, 1H, H-1', 3J1'-F=17,1 Гц), 6,25 (м, 1H, OH-3'), 5,33 (1т, 1H, OH-5'), 4,68 (м, 1H, H-3'), 4,00-3,69 (м, 3H, H-4', H-5', H-5"), 3,77 (д, 1H, этинил, 4JH-F=5,4 Гц). 13C ЯМР (ДМСО-d6, 75 МГц) δ (м.д.) 155,8 (C-4), 152, (C-2), 149,0 (C-6), 138,7 (C-8), 118,5 (C-5), 95,4 (д, C-2', 1J2'-F=180,8 Гц), 87,6 (д, C-1', 2J1'-F=40,5 Гц), 82,5 (д, CCH, 3JC-F=8,0 Гц), 82,0 (C-4'), 74,5 (д, CCH, 2JC-F=31,0 Гц), 72,8 (д, C-3', 2J3-F=19,5 Гц), 59,2 (C-5'). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -158,0 (т). LC/MS: (M+H+) 294,1 (5,74 мин). УФ λмакс 258 нм, Rf=0,33 (MeOH/CH2Cl, 15/85, об/об).
4b: N
2
-изобутирил-9-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-арабинофуранозил]гуанин
К суспензии CrO3 (110,76 ммоль) в ДХМ (220 мл) при 0°C добавляли уксусный ангидрид (110,76 ммоль) и безводный пиридин (221,52 ммоль). Добавляли по капле 9-[3,5-O-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)рибофуранозил]-N2-изобутирилгуанин (36,92 ммоль) в растворе ДХМ (110 мл). Удаляли охлаждающую ванну, и полученный раствор перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь вливали в холодный этилацетат, проводили гельфильтрацию на силикагеле и целите, концентрировали досуха и дважды выпаривали совместно с толуолом. Полученный остаток растворяли в ДХМ и смешивали с избытком MgSO4 в течение ночи, фильтровали и выпаривали для получения кетона. Триметилсилилацетилен (88,60 ммоль) растворяли в безводном ТГФ (98 мл) в атмосфере аргона. Добавляли по капле при -78°C н-бутиллитий (55,4 мл, 1,6М в гексанах). Реакционную смесь взбалтывали в течение 30 минут при -78°C и затем оставляли для нагревания до -55°C. Кетон в растворе ТГФ (49 мл) добавляли по капле при -78°C. Реакционную смесь взбалтывали в течение 1 часа при -78°C и затем оставляли для нагревания до -30°C и перемешивали в течение 3 часов. Реакцию гасили осторожным добавлением водной насыщенной NH4Cl (72 мл) при -78°C. После нагревания при комнатной температуре смесь разводили этилацетатом, промывали дважды насыщенным солевым раствором, высушивали (Na2SO4) и выпаривали досуха. Сырой материал очищали колоночной хроматографией с элюированием 1,5% MeOH в дихлорметане, чтобы получить названное соединение (8,59 г, 34%, 2 этапа). Пена бледно-желтого цвета. Молекулярная формула C31H55N5O6Si3. 1H ЯМР (ДМСО-d6, 250 МГц) δ (м.д.) 12,10 (1c, 1H, NH), 11,69 (1c, 1H, NH), 7,91 (с, 1H, H-8), 6,69 (с, 1H, OH), 5,94 (с, 1H, H-1'), 4,29 (д, 1H, H-3', J3'-4'=5,5 Гц), 3,85-3,95 (м, 3H, H-4', H-5' и H-5"), 2,46 (м, 1H, CH(CH3)2), 0,90-1,08 (м, 30H, iPr и CH(CH3)3), 0,00 (с, 9H, Si(CH3)2). LC/MS: (M+H+) 692,4 (24,96 мин). УФ λмакс1 254 нм, λмакс2 281 нм. Rf=0,34 (MeOH/CH2Cl, 15/85, об/об).
5b: N
2
-изобутирил-9-[(2R)-2-дезокси-2-фтор-3,5-O-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритропентофуранозил]гуанин
4b (2,89 ммоль) растворяли в безводном ДХМ (60 мл) в атмосфере аргона и добавляли пиридин (18,06 ммоль). Реакционную смесь охлаждали до -20°C, и по капле добавляли DAST (31,35 ммоль). После завершения добавления удаляли охлаждающую ванну. Перемешивание продолжали в течение 1 ч 15 мин, и смесь растворяли этилацетатом, вливали в насыщенный NaHCO3 и взбалтывали в течение 5 мин. Органический слой промывали насыщенным солевым раствором, высушивали (Na2SO4), концентрировали и очищали хроматографией на силикагеле, с элюированием этилацетатом в ДХМ (2%), чтобы получить названное соединение (1,41 г, 70%). Масло желтого цвета. Молекулярная формула C31H54FN5O5Si3. 1H ЯМР (ДМСО-d6, 250 МГц) δ (м.д.) 12,22 (с, 1H, NH), 8,09 (с, 1H, H-8), 6,21 (д, 1H, H-1', J1-'F=15,6 Гц), 4,54 (дд, 1H, H-3', J3'-F=23,6 Гц, J3'-4',=9,8 Гц), 4,33 (м, 1H, H-5', 2J5'-5"=13,1 Гц), 4,16 (м, 1H, H-5"), 2,81 (м, 1H, CH(CH3)2), 1,13-1,03 (м, 34H, iPr и CH(CH3)2), 0,08 (с, 9H, Si(CH3)3, 3JH-H=6,9 Гц). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) - 160,26 (дд, JF-1'=16,1 Гц, JF-3'=23,3 Гц). LC/MS: (M+H+) 694,7 (24,02 мин). LRFAB-MS (GT): 694 (M+H)+, 692 (M-H)-, УФ λмакс 256 нм, Rf=0,46 (MeOH/CH2Cl, 05/95, об/об).
6b: N
2
-изобутирил-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
5b (1,89 ммоль) растворяли в метаноле (13,8 мл) и добавляли фторид аммония (24,54 ммоль). Полученный раствор перемешивали и нагревали с обратным холодильником в течение 1 часа, и выпаривали досуха. Сырой материал очищали хроматографией на силикагеле с элюированием метанолом в ДХМ с поэтапным градиентом 6-10%, чтобы получить названное соединение (344 мг, 48%). Масло бледно-желтого цвета. Молекулярная формула C16H20FN5O4Si3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 12,18 (1c, 1H, NH), 11,77 (1c, 1H, NH), 8,34 (с, 1H, H-8), 6,29 (д, 1H, OH-3', JOH-3'=7,5 Гц), 6,20 (д, 1H, H-1', J1'-F=16,2 Гц), 5,39 (т, 1H, OH-5', JOH-5'=5,1 Гц), 4,52 (дт, 1H, H-3', J3'-F=22,9 Гц), 3,98 (м, 1H, H-4'), 3,90-3,85 (м, 2H, H-5' и этинил), 3,72 (м, 1H, H-5"), 2,52 (м, 1H, CH(CH3)2), 1,14 (д, 6H, CH(CH3)2, 3JH-H=6,9 Гц). 13C ЯМР (ДМСО-d6, 100 МГц) δ (м.д.) 180,7 (C-6), 155,3 (C-2), 148,9 (C-4), 137,3 (C-8), 120,4 (C-5), 95,8 (д, C-2', 1J1'-F=182,1 Гц), 87,7 (д, C-1', 2J1'-F=39,2 Гц), 83,4 (д, CCH, 3JC-F=9,1 Гц), 82,6 (C-4'), 75,9 (д, CCH, 2JC-F=31,2 Гц), 72,9 (д, C-3', 2J3'-F=19,1 Гц), 59,3 (C-5'). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) - 158,9 (м), LC/MS: (M+H+) 380,3 (8,34 мин), УФ λмакс1 256 нм, Rf=0,40 (MeOH/CH2Cl, 15/85, об/об).
7j: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
6b (1,33 ммоль) растворяли в насыщенном метанольном аммиаке (62 мл) и перемешивали при комнатной температуре в течение 20 часов. После этого реакционную смесь выпаривали досуха при пониженном давлении. Остаток растворяли в воде и промывали дважды этилацетатом. Водный слой выпаривали и очищали обращенно-фазовой колоночной хроматографией (C 18) с элюированием ацетонитрилом в воде с градиентом 2-15%. Полученный остаток затем растворяли в горячем этилацетате, фильтровали и высушивали, чтобы получить названное соединение (134 мг, 33%). Твердое вещество желтого цвета. Молекулярная формула C12H12FN5O4. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 10,70 (1c, 1H, NH), 7,98 (с, 1H, H-8), 6,60 (1c, 2H, NH2), 6,21 (д, 1H, OH-3', JOH-3'=7,6 Гц), 5,83 (д, 1H, H-1', J1'-F=16,9 Гц), 5,29 (т, 1H, OH-5', JOH-5'=5,2 Гц), 4,50 (тд, 1H, H-3', J3'-F=22,8 Гц, J3'-4'=9,2 Гц), 3,93-3,81 (м, 3H, H-4', H-5' и этинил), 3,70 (м, 1H, H-5"). 13C ЯМР (ДМСО-d6, 100 МГц) δ (м.д.) 157,2 (C-6), 154,3 (C-2), 151,05 (C-4), 135,1 (C-8), 116,7 (C-5), 96,4 (д, C-2', 1JC-F=182,1 Гц), 87,4 (д, C-1', 2JC-F=39,2 Гц), 83,1 (д, CCH, JC-F=9,1 Гц), 82,4 (C-4'), 76,2 (д, CCH, 2JC-F=31,2 Гц), 73,2 (д, C-3', 2JC-F=20,1 Гц), 59,5 (C-5'). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -158,5 (м). LC/MS (A): (M+H+) 310,1 (5,55 мин). LRFAB-MS (GT): 619 (2M+H)+, 310 (M+H)+, 152 (B+H)+, 617 (2M-H)-, 308 (M-H)-. УФ λмакс 253 нм.
3c: 1-[2-оксо-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-β-D-эритрофуранозил]урацил
3c синтезировали из 1-[3,5-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)рибофуранозил]урацила, согласно описанию 3a. Пена бледно-желтого цвета. Молекулярная формула C21H36N2O7Si2. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 11,58 (1c, 1H, NH), 7,74 (д, 1H, H-6, J6-5=8,0 Гц), 5,68 (д, 1H, H-5, J5-6=8,0 Гц), 5,45 (с, 1H, H-1'), 4,97 (д, 1H, H-3', J3'-4'=9,2 Гц), 4,06-3,90 (м, 3H, H-4', H-5'), 1,14-0,87 (м, 28H, iPr). LR LC/MS: (M+H+) 485,1 (M-H-) 483,1 (5,53 мин). УФ λмакс 262 нм, Rf=0,40 (MeOH/CH2Cl, 05/95, об/об).
4c: 1-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-арабинофуранозил]урацил Yoshimura, Y.; Iino, T.; Matsuda, A. Tetrahedron Lett. 1991, 32, 6003-6006.
Молекулярная формула C26H46N2O7Si3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 11,35 (1c, 1H, NH), 7,44 (д, 1H, H-6, J6-5=8,0 Гц), 6,54 (с, 1H, OH), 6,02 (с, 1H, H-1'), 5,54 (д, 1H, H-5, J6-5=8,0 Гц), 4,13-3,93 (м, 3H, H-3', H-5'), 3,75 (м, 1H, H-4'), 1,03-0,96 (м, 28H, iPr), 0,00 (с, 9H, Si(CH3)3). 13C ЯМР (ДМСО-d6, 100 МГц) δ (м.д.) 163,4 (C-4), 150,8 (C-2), 141,6 (C-6), 103,6 (CCSi), 101,2 (C-5), 92,5 (CCSi), 87,3 (C-1'), 80,9 (C-4'), 77,9 (C-2'), 75,9 (C-3'), 61,8 (C-5'), 17,7-17,1 (8C, 4 SiC(CH3)2), 13,3-12,6 (4C, 4SiC(CH3)2), 0,2 (3C, Si(CH3)3). LR LC/MS: (M+H+) 583,2 (M-H-) 581,2 (6,72 мин). УФ λmaK 261 нм. Rf=0,27 (этилацетат/CH2Cl, 10/90, об/об).
5c: 1-[(2R)-2-дезокси-2-фтор-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритропентофуранозил]урацил
5c синтезировали из 4c, согласно описанию 5a. Масло желтого цвета. Молекулярная формула C27H49FN2O6Si3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 11,62 (c1, 1H, NH), 7,43 (д1, 1H, H-6, J6-5=8,0 Гц), 6,12 (д, 1H, H-1', J1'-F=16,8 Гц), 5,68 (д, 1H, H-5, J5-6=8,0 Гц), 4,22-3,85 (м, 4H, H-3', H-4', H-5'), 1,16-1,00 (м, 28H, iPr), 0,00 (с, 9H, Si(CH3)3). 19F ЯМР (ДМСО-d6, 376 МГц) δ (м.д.) -159,7. LR LC/MS: (M+H+) 585,2 (M-H-) 583,3 (6,47 мин). УФ λмакс 261 нм. Rf=0,52 (этилацетат/CH2Cl, 15/85, об/об).
6c: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]урацил
Смесь 5c (0,56 ммоль) и фторида аммония (7,31 ммоль) растворяли в метаноле (10 мл), перемешивали и нагревали с обратным холодильником в течение 1 часа, и выпаривали досуха. Полученный остаток очищали колоночной флэш-хроматографией на силикагеле, с элюированием метанолом в ДХМ с градиентом 0-20%, после чего обращенно-фазовой колоночной хроматографией, с элюированием ацетонитрилом в воде с градиентом 0-100%, чтобы получить желаемый продукт, который лиофилизировали из воды (47 мг, 31%). Лиофилизированный порошок белого цвета. Молекулярная формула C11H11FN2O5. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 11,49 (c1, 1H, NH), 7,87 (д, 1H, H-6, J6-5=8,0 Гц), 6,18 (д, 1H, OH-3', JOH-3'=7,2 Гц), 6,10 (д, 1H, H-1', J1'-F=18,0 Гц), 5,69 (д, 1H, H-5, J5-6=8,0 Гц), 5,32 (м, 1H, OH-5'), 4,19-4,10 (м, 2H, H-3' и этинил), 3,85-3,75 (м, 2H, H-4' H-5'), 3,60 (м, 1H, H-5"). 13C ЯМР (ДМСО-d6, 100 МГц) δ (м.д.) 163,3 (C-4), 150,6 (C-2), 140,1 (C-6), 102,5 (C-5), 95,5 (д, C-2', J2'-F=186,1 Гц), 87,1 (д, C-1', 2J1'-F=40,2 Гц), 83,2 (д, CCH, 2JC-F=8,0 Гц), 82,1 (C-4'), 76,5 (д, CCH, 4JC-F=30,1 Гц), 73,3 (д, C-3', 2JC-F=19,1 Гц), 58,7 (C-5'). 19F ЯМР (ДМСО-d6, 376 МГц) δ (м.д.) -158,2. LR LC/MS: (M+H+) 271,1 (M-H-) 269,2 (1,12 мин). HRFAB-MS C11H12O5N2F, (M+H+) Рассчитано 271,0730, Найдено 271,0739. УФ λмакс 261 нм, Rf=0,33 (MeOH/CH2Cl, 20/80, об/об).
2d: 1-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-β-D-рибофуранозил]тимин
1-(β-D-рибофуранозил)тимин (40,9 ммоль) растворяли в пиридине (435 мл), и указанную смесь охлаждали до 0°C в ледяной ванне в течение 25 минут. Затем добавляли TIPSCl2 (16,2 мл) и после завершения добавления смесь оставляли для нагревания до комнатной температуры. Реакционную смесь взбалтывали при комнатной температуре в течение 3 часов, разводили дихлорметаном и водой, промывали насыщенным водным раствором NaHCO3. Органические фазы объединяли, высушивали Na2SO4, фильтровали и выпаривали. Остаток выпаривали совместно с толуолом для удаления пиридина. Полученный остаток очищали колоночной флэш-хроматографией с элюированием метанолом в дихлорметане с градиентом 0-2%, чтобы получить названное соединение. Порошок беловатого цвета. Молекулярная формула C22H40N2O7 Si2. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,94-1,04 (м, 28H), 1,73 (с, 3H), 3,86-3,96 (м, 1H), 4,06-4,13 (м, 2H), 4,14-4,20 (м, 1H), 5,44-5,48 (м, 1H), 5,53 (ушир.с, 1H), 5,77 (ушир.с, 1H) 7,42 (с, 1H), 11,35 (ушир.с, 1H). УФ λмакс 212 нм, 266 нм.
3d: 1-[2-оксо-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-β-D-рибофуранозил]тимин
К суспензии CrO3 (60 ммоль) в дихлорметане (200 мл) при 0°C добавляли уксусный ангидрид (59 ммоль) и безводный пиридин (120 ммоль). По капле добавляли 2d (20 ммоль) в растворе ДХМ. Удаляли охлаждающую ванну, и полученный раствор взбалтывали при комнатной температуре в течение 3 часов. Реакционную смесь вливали в холодный этилацетат, проводили гельфильтрацию на силикагеле и целите, концентрировали досуха и дважды выпаривали совместно с толуолом, чтобы получить названное соединение. Бесцветное масло. Молекулярная формула C22H38N2O7Si2.
4d: 1-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-арабинофуранозил]тимин
4d синтезировали из 3d и триметилацетилена, как описано для соединения 4a. Твердое вещество коричневого цвета. Молекулярная формула C27H48N2O7Si3. Сканирование ES+ 597 (M+H)+, УФ λмакс 265 нм.
5d: 1-[(2R)-2-дезокси-2-фтор-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритропентофуранозил]тимин
5d синтезировали из 4a согласно описанию 5a. Твердое вещество коричневого цвета. Молекулярная формула C27H47FN2O6Si3. 1H ЯМР (CDCl3-d6, 400 МГц) δ (м.д.) 0,1 (с, 9H), 1,05-1,14 (м, 28H), 1,92 (с, 3H), 3,99-4,13 (м, 1H), 4,44-4,9 (м, 3H), 6,35 (д, 1H, J=16,44 Гц), 7,2 (с, 1H), 8,86 (с, 1H). Сканирование ES+ 599 (M+H)+.
6d: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]тимин
6d синтезировали из 5d согласно описанию 7i. Молекулярная формула C12H13FN2O5. 1H ЯМР (CDCl3-d6, 400 МГц) δ (м.д.) 1,75 (с, 3H), 3,6-3,65 (м, 1H), 3,82-3,84 (м, 2H), 4,07 (д, 1H, J=5,27 Гц), 4,19 (м, 1H), 5,4 (ушир.с, 1H), 6,08 (д, 1H, J=17,8 Гц), 6,17 (ушир.с, 1H), 7,8 (с, 1H), 11,46 (ушир.с, 1H). Сканирование ES+ 285 (M+H)+. УФ λмакс 266 нм.
4e: N
4
-бензоил-1-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-арабинофуранозил]цитозин
4e синтезировали из 3e и триметилацетилена, как описано для соединения 4a. Твердое вещество коричневого цвета. Молекулярная формула C33H51N3O7Si3. Сканирование ES+ 686 (M+H)+, УФ λмакс 260 нм, 310 нм.
5e: N
4
-бензоил-1-[(2R)-2-дезокси-2-фтор-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритропентофуранозил]цитозин
5e синтезировали из N4-бензоил-1-[3,5-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-D-арабинофуранозил]цитозина согласно описанию 5a. Твердое вещество желтого цвета. Молекулярная формула C30H42FN3O6Si2. Сканирование ES+ 688 (M+H)+, УФ λмакс 260 нм, 310 нм.
6e: N
4
-бензоил-1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]цитозин
6e синтезировали из 5e согласно описанию 7i. Порошок белого цвета. Молекулярная формула C18H16FN3O5. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 3,63-3,69 (м, 1H), 3,82-,393 (м, 2H), 4 (д, 1H, J=5,27 Гц), 4,13-4,24 (м, 1H), 5,38 (ушир.с, 1H), 6,23-6,28 (м, 2H), 7,32-7,36 (м, 1H), 7,49-7,53 (м, 2H), 7,6-7,64 (м, 1H), 7,99-8,01 (м, 2H), 8,34 (д, 1H, J=7,32 Гц), 11,30 (ушир.с, 1H). Сканирование ES+ 374 (M+H)+, УФ λмакс 262 нм, 303 нм.
7k: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]цитозин
Молекулярная формула C11H12FN3O4. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 3,57-3,62 (м, 1H), 3,77-3,80 (м, 2H), 3,95 (д, 1H, J=5,53 Гц), 4,03-4,16 (м, 1H), 5,2 (ушир.с, 1H), 5,73 (д, 1H, J=7,19 Гц), 6,06 (д, 1H, J=7,19 Гц), 6,14-6,25 (м, 1H), 7,17-7,3 (2 ушир.с, 2H), 7,74 (д, 1H, J=7,74 Гц). Сканирование ES+ 270 (M+H)+. УФ λмакс 271 нм.
8k: N
4
-диметокситритил-1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]цитозин
К перемешиваемому раствору 7k (2,34 ммоль) в пиридине (7,2 мл) при комнатной температуре добавляли триметилсилилхлорид (9,36 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 2 часов. Затем добавляли 4-диметиламинопиридин (1,17 ммоль) и диметокситритилхлорид (3,51 ммоль). Смесь взбалтывали при комнатной температуре в течение 16 часов. Реакционную смесь разводили ДХМ и насыщенным раствором NaHCO3. Органическую фазу дважды промывали насыщенным раствором NaHCO3, высушивали Na2SO4, фильтровали и выпаривали. Сырой материал растворяли в растворе NH4OH/диоксан (2:1) и перемешивали в течение 4 часов. Растворитель выпаривали и остаток очищали хроматографией на силикагеле (ДХМ/EtOH), чтобы получить названное соединение. Пена белого цвета. Молекулярная формула C32H30FN3O6. Сканирование ES- 570 (M+H)-, УФ λмакс 277 нм.
9k: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]-4-N-диметокситритилцитозин-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
К перемешиваемому раствору 8k (0,35 ммоль) в растворе ангидрида ТГФ/тетразола (1,05 ммоль) при 0°C добавляли бис(S-пивалоил-2-тиоэтил)N,N-диизопропилфосфорамидит (0,42 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 3 часов. Реакционную смесь охлаждали до 0°C и добавляли трет-бутилгидропероксид (0,7 мл/ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 2 часов. Смесь разводили ДХМ, нейтрализовали насыщенным раствором Na2S2O3. Органическую фазу дважды промывали H2O, экстрагировали, высушивали Na2SO4, фильтровали и выпаривали. Сырой материал очищали хроматографией на силикагеле (ДХМ/EtOH) для получения названного соединения. Стекловидное соединение. Молекулярная формула C47H59FN3O11PS2. Сканирование ES+ 938 (M+H)+, УФ λмакс 277 нм.
11k: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]цитозин-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат
9k (34 ммоль) перемешивали в растворе AcOH/MeOH/H2O (3/6/1) в течение 2 часов и при 50°C в течение 4 часов. После этого реакционную смесь выпаривали и очищали хроматографией на силикагеле (ДХМ/EtOH) для получения названного соединения. Лиофилизированный порошок белого цвета. Молекулярная формула C25H37FN3O9PS2. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,17 (с, 18H), 3,08-3,11 (т, J=6,07 Гц, 4H), 3,99-4,08 (м, 7H), 4,22-4,28 (м, 2H), 5,73-5,75 (д, J=7,30 Гц, 1H), 6,30 (ушир.с, 2H), 7,26-7,31 (д, J=17,30 Гц, 2H), 7,47-7,48 (д, J=7,30 Гц, 1H). 19F ЯМР (ДМСО-d6, 376 МГц) δ (м.д.) -156,48 (с, 1F). 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) -1,96 (с, 1P), сканирование ES+ 638 (M+H)+, УФ λмакс 271 нм.
12: N
2
-метокситритил-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
К перемешиваемому раствору 7j (5,18 ммоль) в пиридине (7 мл/ммоль) при комнатной температуре добавляли триметилсилилхлорид. Реакционную смесь взбалтывали при комнатной температуре в течение 6 часов. Затем добавляли метокситритилхлорид (6,21 ммоль), и реакционную смесь взбалтывали при комнатной температуре в течение 16 часов и 2 часа с NH4OH (4 мл/ммоль). Смесь разводили этилацетатом, промывали H2O, насыщенным раствором NaHCO3 и насыщенным раствором NaCl, высушивали Na2SO4, фильтровали и выпаривали. Сырой материал очищали хроматографией на силикагеле (ДХМ/MeOH) для получения названного соединения. Масло желтоватого цвета. Молекулярная формула C32H28FN5O5.
13: N
2
-метокситритил-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-5-O-трет-бутилдиметилсилил-β-D-эритропентофуранозил]гуанин
К перемешиваемому раствору 12 (2,29 ммоль) в пиридине (5 мл) при 0°C добавляли трет-бутилдиметилсилилхлорид (2,75 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 24 часов. После этого ее разводили ДХМ и дважды промывали H2O. Органическую фазу экстрагировали, высушивали Na2SO4, фильтровали и выпаривали. Сырой материал очищали хроматографией на силикагеле (ДХМ/MeOH) для получения названного соединения. Масло желтоватого цвета. Молекулярная формула C38H44FN5O5Si. Сканирование ES+ 696 (M+H)+, λмакс 260 нм. Сканирование ES- 694 (M+H)-, УФ λмакс 260 нм.
14: N
2
-метокситритил-9-[(2R)-2,3-дидезокси-2-C-этинил-2-фтор-5-О-трет-бутилдиметилсилил-β-D-глицеропентофуранозил]гуанин
К перемешиваемому раствору 13 (0,14 ммоль) в ацетонитриле (47 мл/ммоль) при комнатной температуре добавляли 4-диметиламинопиридин (0,56 ммоль) и фенилхлортионоформиат (0,43 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 16 часов и выпаривали при пониженном давлении. Полученный остаток растворяли в ДХМ, органическую фазу промывали H2O, HCl (1Н), высушивали Na2SO4, фильтровали, выпаривали и выпаривали совместно с толуолом.
Сырой материал растворяли в толуоле (12 мл/ммоль), азо-бис-изобутиронитриле (0,02 ммоль) и при комнатной температуре добавляли трибутилстаннан (0,24 ммоль). Реакционную смесь взбалтывали при 125°C в течение 2 часов и концентрировали при пониженном давлении. Сырой материал очищали хроматографией на силикагеле (ДХМ/MeOH) для получения названного соединения. Масло желтоватого цвета. Молекулярная формула C38H44FN5O4Si. Сканирование ES+ 680 (M+H)+, УФ λмакс 260 нм.
15: N
2
-Метокситритил-9-[(2R)-2,3-дидезокси-2-C-этинил-2-фтор-β-D-глицеропентофуранозил]гуанин
14 (0,35 ммоль) растворяли в MeOH (20 мл/ммоль). Затем при комнатной температуре добавляли фторид аммония (3,55 ммоль), и реакционную смесь взбалтывали при 70°C в течение 2 часов. После выпаривания при пониженном давлении сырой материал очищали хроматографией на силикагеле (ДХМ/MeOH) для получения названного соединения. Пена бежевого цвета. Молекулярная формула C32H30FN5O4. 1H ЯМР (CDCl3-d6, 400 МГц) δ (м.д.) 2,38-2,45 (м, 2H), 2,75 (ушир.с, 2H), 3,64-3,67 (д, J=12,20 Гц, 2H), 3,77 (с, 4H), 4,20-4,23 (д, J=11,7 Гц, 1H), 4,41-4,42 (д, J=8,4 Гц, 1H), 5,83-5,87 (д, J=16,24 Гц, 1H), 6,80-6,82 (д, J=8,12 Гц, 4H), 7,26-7,31 (м, 11H), 7,84 (ушир.с, 1H), 9,26 (ушир.с, 1H).
16: 9-[(2R)-23-дидезокси-2-C-этинил-2-фтор-β-D-глицеропентофуранозил]гуанин
Соединение 15 (0,09 ммоль) перемешивали в растворе AcOH/ТГФ/H2O (3/6/1) при 50°C в течение 1 дня. После этого реакционную смесь выпаривали при пониженном давлении и очищали хроматографией на силикагеле, С18 (H2O/ACN). Лиофилизированный порошок бежевого цвета. Молекулярная формула C12H12FN5O5. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 2,57-2,74 (м, 2H), 3,56 (с, 1H), 3,61-3,64 (д, J=12,10 Гц, 1H), 3,79-3,82 (д, J=12,10 Гц, 1H), 3,91-3,93 (д, J=5,40 Гц, 1H), 4,32-4,35 (м, 1H), 5,25 (с, 1H), 6,06-6,10 (д, J=18,20 Гц, 1H), 6,64 (с, 1H), 8,01 (с, 1H), 10,82 (с, 1H). 19F ЯМР (ДМСО-d6, 376 МГц) δ (м.д.) -138,4 (с, 1F), Сканирование ES- 292 (M+H)-, сканирование ES+ 316 (M+Na)+, УФ λмакс 251 нм.
17: 9-[(2R)-2,3-дидезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
Соединение 17 синтезировали из 14 (0,35 ммоль) согласно описанию 9. После этого сырой материал смешивали с раствором AcOH/ТГФ/H2O (4/2/1) при 50°C в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на силикагеле (ДХМ/MeOH) для получения названного соединения. Твердое вещество бежевого цвета. Молекулярная формула C26H37FN5O8PSi2. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 2,48 (с, 18H), 2,65-2,68 (м, 2H), 3,06-3,10 (кв, J=3,71 Гц, и J=6,02 Гц, 4H), 3,97-4,04 (м, 5H), 4,31-4,35 (м, 2H), 4,50-4,52 (м, 1H), 6,13-6,18 (д, J=17,60 Гц, 1H), 6,63 (с, 2H), 7,82 (с, 1H), 10,85 (с, 1H). 19F ЯМР (ДМСО-d6, 376 МГц) δ (м.д.) -139,2 (с, 1F). Сканирование ES+ 662 (M+H)+, УФ λмакс 254 нм. ВЭЖХ (0-100% ACN в течение 8 минут). tR=5,65 минут.
18: N
2
-метокситритил-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-5-О-трет-бутилдиметилсилил-3-О-тетрагидропиранил-β-D-эритропентофуранозил]гуанин
К перемешиваемому раствору 13 (0,8 ммоль) в безводном ТГФ (20 мл/ммоль) при комнатной температуре добавляли п-толуолсульфоновую кислоту (0,12 ммоль) и дигидрофуран (2 мл/ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 3 дней и нейтрализовали ТЕА. Смесь разводили ДХМ, дважды промывали H2O. Органическую фазу высушивали Na2SO4, фильтровали и выпаривали. Сырой материал очищали хроматографией на силикагеле (ДХМ/MeOH) для получения названного соединения. Молекулярная формула C43H52FN5O6Si. Сканирование ES+ 780 (M+H)+.
19: N
2
-метокситритил-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-3-О-тетрагидропиранил-β-D-эритропентофуранозил]гуанин
Соединение 19 синтезировали из 18, согласно описанию для 15. Молекулярная формула C37H38FN5O6. Сканирование ES+ 666 (M+H)+.
21: N
2
-Тетрагидропиранил-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-5-О-трет-бутилдиметилсилил-3-О-тетрагидропиранил-β-D-эритропентофуранозил]гуанин
21 получали путем очистки 18. Молекулярная формула C28H42FN5O6Si. Сканирование ES+ 592 (M+H)+, УФ λмакс 273 нм.
22: N
2
-Тетрагидропиранил-9-[(2R)-2-дезокси-2-C-этинил-2-фтор-3-О-тетрагидропиранил-β-D-эритропентофуранозил]гуанин
Соединение 22 синтезировали из 21 (0,46 ммоль) согласно описанию 15. Молекулярная формула C22H28FN5O6.
20: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
Соединение 20 синтезировали из 19 (0,09 ммоль), согласно описанию для 9k. Затем неочищенный материал перемешивали при комнатной температуре в растворе AcOH/ТГФ/H2O (4/2/1) в течение ночи. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на силикагеле (ДХМ/MeOH) для получения названного соединения. Лиофилизированный порошок белого цвета. Молекулярная формула C26H37FN5O9PS2. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,16 (с, 20H), 3,06-3,09 (м, 4H), 3,90-3,91 (д, J=5,40 Гц, 1H), 3,99-4,10 (кв, J=6,70 Гц и J=7,00 Гц, 4H), 4,32-4,38 (м, 2H), 4,63 (м, 1H), 6,10-6,14 (д, J=16,93 Гц, 1H), 6,69 (с, 2H), 7,79 (с, 1H), 10,96 (с, 1H). 31P ЯМР (ДМСО-d6, 162 МГц) δ (м.д.) -1,91 (с, 1P). 19F ЯМР (ДМСО-d6, 376 МГц) δ (м.д.) -156,82 (с, 1F). Сканирование ES+ 678 (M+H)+.
25: 1-[(2R)-2-дезокси-2-фтор-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритропентофуранозил]-4-тиоурацил
Соединение 5c (820 мг, 1,40 ммоль) растворяли в безводном 1,2-дихлорэтане (35 мл) и обрабатывали реагентом Лавессона (Lawesson) (1,13 г, 2,80 ммоль). Реакционную смесь взбалтывали при нагревании с обратным холодильником в течение ночи и выпаривали досуха. Полученный остаток подвергали гельфильтрации на силикагеле, элюировали этилацетатом в дихлорметане с градиентом 0-5% для получения названного соединения. Масло желтого цвета. Молекулярная формула C26H45FN2O5SSi3. LR-LC/MS: (M+H+) 601,3 (M-H-) 599,3 (7,03 минут). УФ λмакс 332 нм. Rf=0,71 (этилацетат/CH2Cl, 7/93, об/об).
26: 1-[(2R)-2-дезокси-2-фтор-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-этинил-β-D-эритропентофуранозил]цитозин
Неочищенное соединение 25 растворяли в насыщенном аммонийном метаноле (9 мл). Полученный раствор нагревали посредством микроволн при 120°C в течение 20 минут и выпаривали при пониженном давлении для получения названного соединения. Маслянистый остаток. Молекулярная формула C26H46FN3O5Si3 LR-LC/MS (B): (M+H+) 512,3 (М-Н-) 510,3 (5,33 мин). УФ λмакс1 242 нм, λмакс2 273 нм.
27i: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]аденин 5'-трифосфата натриевая соль
К раствору 7i (0,286 ммоль) в триэтилфосфате (750 мкл), добавляли при 0°C фосфорилхлорид (75 мкл, 0,807 ммоль). Указанную реакционную смесь A взбалтывали в течение ночи при 5°C. Трибутиламмония пирофосфат (PPi/Bu3N 1/1,5, 1 г, 2,19 ммоль) растворяли в безводном ДМФ (2 мл). Трибутиламин (420 мкл, 1,76 ммоль) добавляли к PPi, и полученную смесь перемешивали в течение 15 минут при 0°C. К реакционной смеси A добавляли 2,4 мл указанного раствора. Реакционную смесь взбалтывали при 0°C в течение 1 мин. Реакционную смесь аккуратно гасили 1М TEAB (уровень pH=7,5, 10 мл), взбалтывали 20 минут при 0°C, после чего разводили этилацетатом и водой. Водную фазу концентрировали при пониженном давлении. Неочищенный материал подвергали хроматографии на колонке DEAE-Sephadex, с элюированием с градиентом 10-3-1М TEAB). Желаемые фракции объединяли, выпаривали при пониженном давлении, концентрировали совместно со смесью воды/метанола, и в конце выпаривали совместно с водой. Полученный остаток очищали полупрепаративной ВЭЖХ. Фракции, содержащие ожидаемый продукт, концентрировали при пониженном давлении, выпаривали совместно со смесью воды/метанола и лиофилизировали из воды. Соль трифосфат триэтиламмония элюировали трижды водой на колонке со смолой Dowex Na+, чтобы после лиофилизации из воды получить натриевую соль.
Молекулярная формула C12H11FN5O12P3 3Na. 1H ЯМР (D2O, 300 МГц) δ (м.д.) 8,31 (с, 1H, H-8), 8,14 (с, 1H, H-2), 6,28 (д, 1H, H-1', 3J1'-F=15,6 Гц), 4,64 (м, 1H, H-3'), 4,42 (м, 1H, H-5'), 4,35-4,25 (м, 2H, H-4' и H-5"), 2,82 (д, 1H, этинил, 4JH-F=5,5 Гц). 31P ЯМР (D2O, 121 МГц) δ (м.д.) -10,27 (д, 1P, Pγ, JPγ-Pβ=19,4 Гц), -11,03 (д, 1P, Pα, JPα-Pβ=19,4 Гц), -22,38 (т, 1P, Pβ, JPβ-Pγ=JPβ-Pα=19,4 Гц). 19F ЯМР (D2O, 282 МГц) δ (м.д.) - 160,0 (м). LRFAB-MS (GT): 600 (M+H)+, 578 (M-Na+2H)+, 556 (M-2Na+3H), 598 (M-H)-, 576 (M-Na)-, 554 (M-2Na+H)-, 532 (M-3Na+2H)-.
27i: 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин 5'-трифосфата натриевая соль
Соединение 27j синтезировали из 7j согласно описанию 27. Молекулярная формула C12H11FN5O13P3 3Na. 1H ЯМР (D2O, 400 МГц) δ (м.д.) 7,97 (с, 1H, H-8), 6,19 (д, 1H, H-1', 3J1'-F=16,0 Гц), 4,70 (м, 1H в H2O, H-3'), 4,39 (м, 1H, H-5'), 4,29-4,22 (м, 2H, H-4' и H-5"), 2,98 (д, 1H, этинил, 4JH-F=5,0 Гц). 31P ЯМР (D2O, 162 МГц): -10,50 (д, 1P, Pγ, JPγ-Pβ=19,4 Гц), -11,03 (д, 1P, Pα, JPα-Pβ=19,4 Гц), -22,38 (т, 1P, Pβ, JPβ-Pγ=JPβ-Pα=19,4 Гц). 19F ЯМР (ДМСО-d6, 376 МГц) δ (м.д.) -159,1 (м), LRFAB-MS (GT): 638 (M+Na)+, 616 (M+H)+, 594 (M-Na+2H)+, 572 (M-2Na+3H)+, 550 (M-3Na+4H)+, 592 (M-Na)-, 570 (M-2Na+H)-, 548 (M-3Na+2H)-.
2g: 4-хлор-7-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-β-D-рибофуранозил]пирроло[2,3-d]пиримидин
2g синтезировали из 9-[β-D-рибофуранозил]-7-деаза-6-хлорпурина, как описано для промежуточного продукта 12. Масло желтого цвета. Молекулярная формула C23H38ClN3O5Si2. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,96-1,04 (м, 28H), 3,92-3,95 (м, 3H), 4,41-4,58 (м, 2H), 5,65 (с, 1H), 6,08 (с, 1H), 6,71 (с, 1H), 7,83 (с, 1H), 8,62 (с, 1H).
3g: 4-хлор-7-[2-оксо-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-β-D-эритропентофуранозил]пирроло[2,3-d]пиримидин
3g синтезировали из 2g, согласно описанию 3d. Твердое вещество коричневого цвета. Молекулярная формула C23H36ClN3O5Si2. Сканирование ES+ (M+H)+ 528, УФ λмакс 271 нм.
4g: 4-хлор-[3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-арабинофуранозил]пирроло[2,3-d]пиримидин
4g синтезировали из 3g согласно описанию 4a. Твердое вещество бежевого цвета. Молекулярная формула: C28H46ClN3O5Si3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.), 0,12 (с, 9H),0,95-1,09 (м, 28H), 3,90-3,94 (м, 1H), 4,02-4,03 (м, 2H), 4,37-4,39 (д, J=6,74 Гц, 1H), 6,43 (с, 1H), 6,44 (с, 1H), 6,68 (д, J=3,71 Гц, 1H), 7,71-7,72 (д, J=3,84 Гц, 1H), 8,66 (с, 1H).
5g: 4-хлор-7-[(2R)-2-дезокси-2-фтор-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритропентофуранозил]пирроло[2,3-d]пиримидин
5g синтезировали из 4g, как описано для 5a. Масло желтого цвета. Молекулярная формула C28H45ClFN3O5Si3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,33 (с, 9H), 1,02-1,13 (м, 28H), 4,0-4,03 (д, J=13,42 Гц, 1H), 4,12-4,14 (д, J=9,43 Гц, 1H), 4,27-4,31 (д, J=14,00 Гц, 1H), 4,71 (ушир.с, 1H), 6,58-6,62 (д, J=17,07 Гц, 1H), 6,82-6,83 (д, J=3,80 Гц, 1H), 7,72 (д, J=3,80 Гц, 1H), 8,69 (с, 1H). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -159,6 (с, 1F).
6g: 4-хлор-7-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]пирроло[2,3-d]пиримидин
Соединение 6g синтезировали из 5g согласно описанию 6a. Масло желтого цвета. Молекулярная формула C13H11ClFN3O3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 3,60-3,65 (д, J=5,44 Гц, 1H), 3,68-3,71 (д, J=12,35 Гц, 1H), 3,85-3,88 (д, J=12,35 Гц, 1H), 3,95-3,97 (д, J=8,90 Гц, 1H), 4,46-4,54 (дд, J=23,23 Гц и J=9,39 Гц, 1H), 5,38 (с, 1H), 6,28 (с, 1H), 6,57-6,61 (д, J=16,47 Гц, 1H), 6,79 (д, J=3,82 Гц, 1H), 8,04 (д, J=3,78 Гц, 1H), 8,70 (с, 1H). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -158,30 (с, 1F). Сканирование ES+ 312 (M+H)+, сканирование ES- 356 (M+HCO2)-.
7l: 4-амино-7-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]пирроло[2,3-d]пиримидин
7l синтезировали из 6g, как описано для 6a. Лиофилизированный порошок белого цвета. Молекулярная формула C13H13FN4O3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 3,61 (д, J=5,52 Гц, 1H), 3,63-3,67 (м, 1H), 3,80-3,83 (д, J=12,14 Гц, 1H), 3,86-3,88 (д, J=9,38 Гц, 1H), 4,46-4,54 (дд, J=23,23 Гц и J=9,39 Гц, 1H), 5,30 (ушир.с, 1H), 6,1 (ушир.с, 1H), 6,41-6,47 (д, J=16,47 Гц, 1H), 6,57-6,61 (д, J=16,47 Гц, 1H), 7,04 (с, 2H), 7,37-7,38 (д, J=3,65 Гц, 1H), 8,05 (с, 1H). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -157,15 (с, 1F). Сканирование ES+ 293 (M+H)+, УФ λмакс 275 нм.
23: 9-[(2R)-2-дезокси-3,5-ди-О-изобутирил-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
Раствор 9-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритрофуранозил]гуанина (0,16 ммоль), 4-диметиламинопиридина (0,01 ммоль), триэтиламина (0,48 ммоль) и изобутирильного ангидрида (0,48 ммоль) в ацетонитриле (1 мл) перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь гидролизировали насыщенным раствором NaHCO3. Добавляли этилацетат. Органическую фазу отделяли, промывали насыщенным раствором NaCl, высушивали Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой материал очищали колоночной флэш-хроматографией (ДХМ/EtOH) для получения названного соединения в виде порошка белого цвета. Молекулярная формула C20H24FN5O6. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,02-1,22 (м, 12H), 2,53-2,59 (м, 1H), 2,65-2,70 (м, 1H), 4,04 (д, J=4,77 Гц, 1H), 4,35-4,40 (м, 3H), 5,88-5,94 (дд, J=9,39 Гц и J=8,21 Гц, 1H), 6,21-6,25 (д, J=17,28 Гц, 1H), 6,58 (с, 2H), 7,09 (с, 1H), 10,82 (с, 1H). Сканирование ES+ 450,0 (M+H)+, УФ λмакс 251 нм.
24: N-2-изобутирил-9-[(2R)-2-дезокси-3,5-ди-O-изобутирил-2-C-этинил-2-фтор-β-D-эритропентофуранозил]гуанин
Соединение 24 получали путем очистки 23. Порошок белого цвета. Молекулярная формула C24H30FN5O7. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 1,02-1,22 (м, 18H), 2,53-2,59 (м, 1H), 2,65-2,70 (м, 1H), 2,74-2,80 (м, 1H), 4,04 (д, J=4,90 Гц, 1H), 4,35-4,40 (м, 3H), 5,73-5,80 (дд, J=10,14 Гц и J=7,80 Гц, 1H), 6,29-6,34 (д, J=17,36 Гц, 1H), 8,23 (с, 1H), 11,80 (ушир.с, 1H), 12,3 (ушир.с, 1H). Сканирование ES+ 520 (M+H)+, УФ λмакс 257 нм.
4f: 5-фтор-1-[3,5-O-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-арабинофуранозил]урацил
4f синтезировали из 3f согласно описанию 4a. Твердое вещество оранжевого цвета. Молекулярная формула C26H45FN2O7Si3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,13 (с, 9H), 0,94-1,06 (м, 28 H), 3,75 (м, 1H), 3,96-4,09 (м, 3H), 5,99 (д, J=1,53 Гц, 1H), 6,53 (с, 1H), 7,58-7,60 (д, J=6,76 Гц, 1H), 11,8 (ушир.с, 1H). Сканирование ES- 599 (M-H)-, УФ λмакс 271 нм.
5f: 5-фтор-1-[(2R)-2-дезокси-2-фтор-3,5-О-(1,3-диил-1,1,3,3-тетраизопропилдисилоксан)-2-C-триметилсилилэтинил-β-D-эритропентофуранозил]урацил
5f синтезировали из 4f, как описано для 5a. Твердое вещество белого цвета. Молекулярная формула C26H44F2N2O6Si3. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 0,13 (с, 9H), 0,94-1,06 (м, 28 H), 3,92-3,95 (д, J=12,47 Гц, 1H), 3,96-4,09 (м, 1H), 4,21 (д, J=12,22 Гц, 1H), 5,20 (ушир.с, 1H), 6,10-6,15 (д, J=16,53 Гц, 1H), 7,56 (с, 1H), 12,23 (ушир.с, 1H). 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -160,06 (с, 1F), -165,94 (с, 1F). Сканирование ES+ 603 (M-H)+, УФ λмакс 272 нм.
6f: 5-фтор-1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]урацил
6f синтезировали из 5f, как описано для 6a. Твердое вещество белого цвета. Молекулярная формула C11H10F2N2O5. 1H ЯМР (ДМСО-d6, 400 МГц) δ (м.д.) 3,61-3,64 (д, J=12,55 Гц, 1H), 3,81-3,85 (м, 2H), 4,11 (д, J=4,91 Гц, 1H), 4,14-4,20 (м, 1H), 5,50 (с, 1H), 6,04-6,09 (д, J=16,91 Гц, 1H), 6,20 (д, J=7,64 Гц, 1H), 8,29 (д, J=7,09 Гц, 1H), 12,05 (с, 1H) 19F ЯМР (ДМСО-d6, 235 МГц) δ (м.д.) -158,74 (с, 1F), -166,27 (с, 1F). Сканирование ES+ 289,0 (M-H)+, УФ λмакс 270 нм.
11f: 1-[(2R)-2-дезокси-2-C-этинил-2-фтор-β-D-эритропентофуранозил]-5-фтороурацил-5'-ил-бис(S-пивалоил-2-тиоэтилфосфат)
11f синтезировали из 6f согласно описанию 9k. Твердое вещество белого цвета. Молекулярная формула C25H35F2N2O10PS2. 1H ЯМР (ДМСО-d6+D2O, 400 МГц) δ (м.д.) 1,15-1,17 (м, 18H), 3,10 (т, J=6,40 Гц, 4H), 4-4,08 (м, 5H), 4,19 (д, J=5,39 Гц, 1H), 4,24-4,39 (м, 3H), 6,12 (д, J=16,82 Гц, 1H), 6,39 (д, J=6,07 Гц, 1H), 7,86 (ушир.с, 1H), 12,12 (ушир.с, 1H).
28: 9-[(2R)-2,3-дидезокси-2-C-этинил-2-фтор-β-D-глицеропентофуранозил]гуанин 5'-трифосфат натрия
Соединение 28 синтезировали из 16 согласно описанию 27i. Порошок белого цвета. Молекулярная формула C12H12FN5Na3O12P3. 1H ЯМР (D2O, 400 МГц) δ (м.д.) 2,61-2,72 (м, 2H), 2,95-2,96 (м, 1H), 4,16-4,22 (м, 1H), 4,35-4,40 (м, 1H), 4,6-4,7 (м, 1H), 6,17 (д, J=16 Гц, 1H), 8,02 (с, 1H). 19F ЯМР (D2O, 235 МГц) δ (м.д.) (-138,95)-(-138,74) (м, 1F). 31P ЯМР (D2O, 162 МГц) δ (м.д.) -10,66 (д, J=19,44 Гц, 1P), -11,14 (д, J=19,44 Гц, 1P), -22,82 (т, J=19,44 Гц, 1P). Сканирование ES+ 599,6 (M-3Na)3+, УФ λмакс 253 нм.
Все публикации, патенты и патентные заявки, цитируемые в настоящем описании, включены в изобретение в качестве ссылки, как если присутствует конкретное и отдельное указание в виде ссылки на каждую включенную в изобретение отдельную публикацию или патентную заявку. Поскольку описание заявленного объекта изобретения приведено в плане разных вариантов осуществления, специалист в данной области техники будет подразумевать, что можно осуществлять различные модификации, замены, упущения и изменения, не отступая от сути изобретения. Соответственно, предполагается, что область объекта изобретения ограничена исключительно объемом приведенной ниже формулы изобретения, включающий в себя ее эквиваленты.
Claims (13)
1. Соединение формулы
или его фармацевтически приемлемая соль, в котором;
Ry представляет собой гидрокси(C1-C10)алкил;
Ra и Rb выбирают следующим образом:
i) Ra и Rb каждый независимо представляют собой водород, C1-С10 алкил, замещенный C1-C10 алкил, арил, арил(C1-C10)алкил или (C1-C10)циклоалкил; или
ii) Ra и Rb вместе с атомом азота, на который они замещены, образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов; и
R1 выбран из группы, состоящей из рибавирина, вирамидина, валопицитабина, PSI-6130, МК-0608, резиквимода, целгосивира, ламивудина, энтекавира, телбивудина, рацивира, эмтрицитабина, клевудина, амдоксовира и валторцитабина;
где “алкил” является незамещенным или замещенным одной или более группами, выбранными из галогена, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, и
и где “арил” является незамещенным или замещенным одной или более группами, выбранными из галогена, алкила, галоалкила, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, и где каждый арил представляет собой фенил, бифенил и нафтил.
или его фармацевтически приемлемая соль, в котором;
Ry представляет собой гидрокси(C1-C10)алкил;
Ra и Rb выбирают следующим образом:
i) Ra и Rb каждый независимо представляют собой водород, C1-С10 алкил, замещенный C1-C10 алкил, арил, арил(C1-C10)алкил или (C1-C10)циклоалкил; или
ii) Ra и Rb вместе с атомом азота, на который они замещены, образуют гетероциклическое или гетероарильное кольцо, имеющее от 3 до 7 членов; и
R1 выбран из группы, состоящей из рибавирина, вирамидина, валопицитабина, PSI-6130, МК-0608, резиквимода, целгосивира, ламивудина, энтекавира, телбивудина, рацивира, эмтрицитабина, клевудина, амдоксовира и валторцитабина;
где “алкил” является незамещенным или замещенным одной или более группами, выбранными из галогена, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, и
и где “арил” является незамещенным или замещенным одной или более группами, выбранными из галогена, алкила, галоалкила, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, и где каждый арил представляет собой фенил, бифенил и нафтил.
2. Соединение по п.1, имеющее формулу:

или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
3. Соединение по п.1, имеющее формулу:

или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
4. Соединение по п.1, имеющее формулу:

или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
5. Соединение по п.1, имеющее формулу:


 или
или
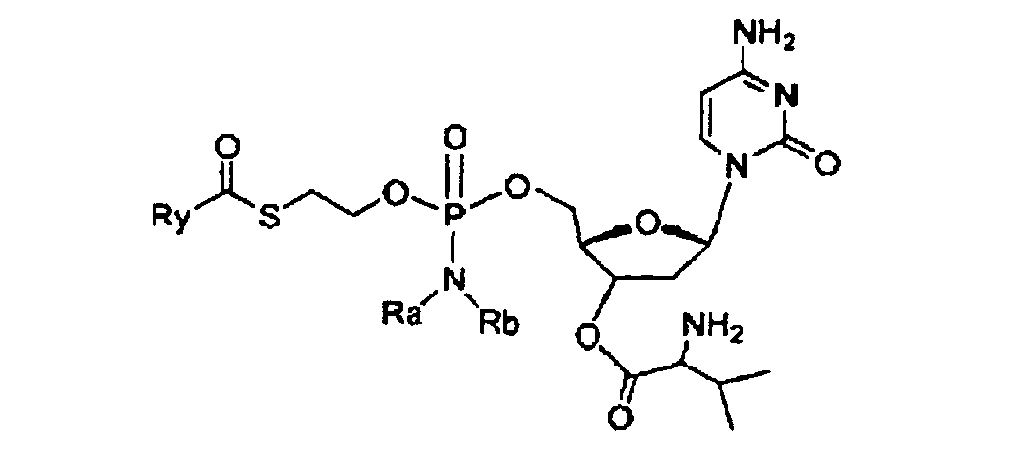
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
6. Соединение по п.1, в котором Ra и Rb независимо представляют собой водород, бензил или замещенный (C1-C10)алкил.
7. Соединение формулы:
или
8. Фармацевтическая композиция для лечения организма-хозяина, инфицированного вирусом гепатита B или C, содержащая соединение согласно любому из пп.1-7 и фармацевтически приемлемый наполнитель, носитель или разбавитель.
9. Композиция по п.8, в котором композиция представляет собой рецептуру для перорального приема.
10. Применение соединения или фармацевтической композиции согласно любому из пп.1-7, для лечения организма-хозяина, инфицированного вирусом гепатита B или вирусом гепатита C.
11. Применение по п.10, в котором указанное соединение или его фармацевтически приемлемую соль или стереоизомер вводят в комбинации или чередовании со вторым противовирусным средством, необязательно выбираемым из группы, состоящей из интерферона, рибавирина, интерлейкина, ингибитора протеазы NS3, ингибитора цистеин-протеазы, фенантренхинона, производного тиазолидина, тиазолидина, бензанилида, ингибитора геликазы, ингибитора полимеразы, нуклеотидного аналога, глиотоксина, церуленина, антисмыслового олигодезоксинуклеотида фосфоротиоата, ингибитора IRES-зависимой трансляции и рибозима; или в котором соединение или его фармацевтически приемлемую соль или стереоизомер вводят в комбинации или чередовании со вторым противовирусным средством, необязательно выбираемым из группы, состоящей из интерферона, рибавирина, интерлейкина, ингибитора протеазы NS3, ингибитора цистеин-протеазы, фенантренхинона, производного тиазолидина, тиазолидина, бензанилида, ингибитора геликазы, ингибитора полимеразы, нуклеотидного аналога, глиотоксина, церуленина, антисмыслового олигодезоксинуклеотида фосфоротиоата, ингибитора IRES-зависимой трансляции и рибозима, причем необязательно второе средство выбирают из группы, состоящей из пегилированного интерферона-альфа 2а, интерферона альфакон-1, интерферона природного происхождения, альбуферона, интерферона-бета 1а, интерферона омега, интерферона альфа, интерферона гамма, интерферона тау, интерферона дельта и интерферона γ-1b.
12. Применение по п.11, в котором организмом-хозяином является человек.
13. Применение по п.12, в котором при указанном введении существенное количество указанного соединения или фармацевтической композиции направленно поступает в печень указанного организма-хозяина.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US87794406P | 2006-12-28 | 2006-12-28 | |
| US60/877,944 | 2006-12-28 | ||
| US93629007P | 2007-06-18 | 2007-06-18 | |
| US60/936,290 | 2007-06-18 | ||
| US98589107P | 2007-11-06 | 2007-11-06 | |
| US60/985,891 | 2007-11-06 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2009128978/04A Division RU2466729C2 (ru) | 2006-12-28 | 2007-12-28 | Соединения и фармацевтические композиции для лечения вирусных инфекций |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2012132218A RU2012132218A (ru) | 2014-02-10 |
| RU2525392C2 true RU2525392C2 (ru) | 2014-08-10 |
Family
ID=39589128
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2012132218/04A RU2525392C2 (ru) | 2006-12-28 | 2007-12-28 | Соединения и фармацевтические композиции для лечения вирусных инфекций |
Country Status (18)
| Country | Link |
|---|---|
| US (5) | US7951789B2 (ru) |
| EP (2) | EP2120566B1 (ru) |
| JP (3) | JP2010514769A (ru) |
| KR (1) | KR101508018B1 (ru) |
| AU (1) | AU2007339226B2 (ru) |
| BR (1) | BRPI0720637A2 (ru) |
| CA (2) | CA2673722A1 (ru) |
| IL (1) | IL200116A0 (ru) |
| ME (1) | ME00816B (ru) |
| MX (1) | MX2009006851A (ru) |
| MY (1) | MY169568A (ru) |
| NO (1) | NO20092585L (ru) |
| NZ (1) | NZ577999A (ru) |
| RU (1) | RU2525392C2 (ru) |
| SG (1) | SG177923A1 (ru) |
| TW (2) | TWI412368B (ru) |
| UA (1) | UA96975C2 (ru) |
| WO (2) | WO2008082601A2 (ru) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2632431C2 (ru) * | 2016-06-29 | 2017-10-04 | Федеральное государственное бюджетное учреждение "Национальный медицинский исследовательский радиологический центр" Министерства здравоохранения Российской Федерации (ФГБУ "НМИРЦ" Минздрава России) | Гидрогель для получения композиционных материалов с антибактериальной активностью для замещения костно-хрящевых дефектов методом 3d печати |
| WO2018016999A1 (ru) * | 2016-07-20 | 2018-01-25 | Общество С Ограниченной Ответственностью "Остерос Биомедика" | Препарат для лечения костных поражений, вызванных злокачественными новообразованиями |
| US11219603B2 (en) | 2015-12-01 | 2022-01-11 | Maxwell Biotech Group Ltd. | Stable dosage form of etidronate-cytarabine conjugate, and use thereof |
Families Citing this family (172)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY164523A (en) | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| EA007867B1 (ru) | 2000-05-26 | 2007-02-27 | Айденикс (Кайман) Лимитед | Композиции для лечения флавивирусных и пестивирусных инфекций и способы их применения |
| KR20050088079A (ko) | 2002-11-15 | 2005-09-01 | 이데닉스 (케이만) 리미티드 | 2'-분지형 뉴클레오시드 및 플라비비리다에 돌연변이 |
| US8222257B2 (en) | 2005-04-01 | 2012-07-17 | The Regents Of The University Of California | Phosphono-pent-2-en-1-yl nucleosides and analogs |
| RU2008152171A (ru) | 2006-07-05 | 2010-08-10 | Интермьюн, Инк. (Us) | Новые ингибиторы вирусной репликации гепатита с |
| JP5452022B2 (ja) | 2006-12-11 | 2014-03-26 | 独立行政法人科学技術振興機構 | 植物生長調整剤及びその利用 |
| US7951789B2 (en) * | 2006-12-28 | 2011-05-31 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| US7964580B2 (en) | 2007-03-30 | 2011-06-21 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| CL2008001381A1 (es) | 2007-05-10 | 2008-11-03 | Intermune Inc Y Array Biopharma Inc | Compuestos derivados de tripeptidos que contienen heterociclos nitrogenados; composicion farmaceutica que comprende a dichos compuestos; y uso para tratar una infeccion de hepatitis c o vih. |
| GB0709791D0 (en) * | 2007-05-22 | 2007-06-27 | Angeletti P Ist Richerche Bio | Antiviral agents |
| CA2718396C (en) | 2008-03-14 | 2016-01-05 | Toyota Jidosha Kabushiki Kaisha | Gene for increasing the production of plant biomass and/or seeds and method for use thereof |
| JP2011519364A (ja) | 2008-04-15 | 2011-07-07 | インターミューン・インコーポレーテッド | C型肝炎ウイルス複製の新規大環状阻害剤 |
| EA019883B1 (ru) | 2008-04-23 | 2014-07-30 | Джилид Сайэнс, Инк. | Карбануклеозидные аналоги для противовирусной терапии |
| US8173621B2 (en) | 2008-06-11 | 2012-05-08 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| ES2458358T3 (es) * | 2008-07-02 | 2014-05-05 | Idenix Pharmaceuticals, Inc. | Compuestos y composiciones farmacéuticas para el tratamiento de infecciones víricas |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| KR20110104074A (ko) | 2008-12-23 | 2011-09-21 | 파마셋 인코포레이티드 | 퓨린 뉴클레오시드의 합성 |
| MX2011006890A (es) | 2008-12-23 | 2011-07-20 | Pharmasset Inc | Analogos de nucleosidos. |
| PE20120013A1 (es) | 2009-01-09 | 2012-02-02 | Univ Cardiff | Fosforamidato derivados de guanosina compuestos nucleosidos para tratamiento de infecciones virales |
| PT2396340E (pt) | 2009-02-10 | 2014-03-26 | Gilead Sciences Inc | Análogos de carba-nucleosídeo para tratamento antiviral |
| AR075584A1 (es) | 2009-02-27 | 2011-04-20 | Intermune Inc | COMPOSICIONES TERAPEUTICAS QUE COMPRENDEN beta-D-2'-DESOXI-2'-FLUORO-2'-C-METILCITIDINA Y UN DERIVADO DE ACIDO ISOINDOL CARBOXILICO Y SUS USOS. COMPUESTO. |
| JP5604657B2 (ja) | 2009-03-12 | 2014-10-08 | トヨタ自動車株式会社 | 植物のバイオマス量及び/又は種子量を増産させる遺伝子及びその利用方法 |
| TWI598358B (zh) | 2009-05-20 | 2017-09-11 | 基利法瑪席特有限責任公司 | 核苷磷醯胺 |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| JP5250807B2 (ja) | 2009-09-11 | 2013-07-31 | トヨタ自動車株式会社 | 植物のバイオマス量及び/又は種子量を増産させる方法、バイオマス量及び/又は種子量を増産できる植物の製造方法 |
| US7973013B2 (en) | 2009-09-21 | 2011-07-05 | Gilead Sciences, Inc. | 2'-fluoro substituted carba-nucleoside analogs for antiviral treatment |
| SI2480559T1 (sl) | 2009-09-21 | 2013-10-30 | Gilead Sciences, Inc. | Postopki in intermediati za pripravo 1'-ciano-karbanukleozidnih analogov |
| AP2012006189A0 (en) * | 2009-09-21 | 2012-04-30 | Gilead Sciences Inc | 2'-fluoro substituted carbanucleoside analogs for antiviral treatment. |
| WO2011063076A1 (en) | 2009-11-19 | 2011-05-26 | Itherx Pharmaceuticals, Inc. | Methods of treating hepatitis c virus with oxoacetamide compounds |
| WO2011072370A1 (en) | 2009-12-18 | 2011-06-23 | Boehringer Ingelheim International Gmbh | Hcv combination therapy |
| CN104017020B (zh) * | 2010-03-31 | 2017-04-12 | 吉利德制药有限责任公司 | 核苷氨基磷酸酯 |
| WO2011123586A1 (en) * | 2010-04-01 | 2011-10-06 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| SG185545A1 (en) * | 2010-05-18 | 2012-12-28 | Bukwang Pharm Co Ltd | Composition for treating chronic hepatitis b, containing clevudine and adefovir dipivoxil |
| AU2011282241B2 (en) | 2010-07-19 | 2015-07-30 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| MX2013000744A (es) | 2010-07-22 | 2013-03-07 | Gilead Sciences Inc | Metodos y compuestos para tratar infecciones virales por paramyxoviridae. |
| US9394313B2 (en) | 2010-09-14 | 2016-07-19 | Infinity Pharmaceuticals, Inc. | Transfer hydrogenation of cyclopamine analogs |
| BR112013008017A2 (pt) | 2010-09-20 | 2016-06-14 | Gilead Sciences Inc | análogos de carba-nucleosídeo 2-flúor substituídos para tratamento antiviral |
| US8871737B2 (en) | 2010-09-22 | 2014-10-28 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| WO2012048013A2 (en) * | 2010-10-06 | 2012-04-12 | Inhibitex, Inc. | Phosphorodiamidate derivatives of guanosine nucleoside compounds for treatment of viral injections |
| ES2564358T3 (es) * | 2010-10-15 | 2016-03-22 | The Trustees Of Columbia University In The City Of New York | Genes relacionados con la obesidad y sus proteínas y usos de los mismos |
| CA2818853A1 (en) | 2010-11-30 | 2012-06-07 | Gilead Pharmasset Llc | 2'-spirocyclo-nucleosides for use in therapy of hcv or dengue virus |
| CN102584756B (zh) * | 2010-12-23 | 2015-01-21 | 财团法人食品工业发展研究所 | 新颖紫红曲酮、其制备方法及紫红曲酮的用途 |
| US9351989B2 (en) | 2010-12-29 | 2016-05-31 | Inhibitex, Inc. | Substituted purine nucleosides, phosphoroamidate and phosphorodiamidate derivatives for treatment of viral infections |
| CN103403014B (zh) | 2011-01-03 | 2016-07-06 | 河南美泰宝生物制药有限公司 | O-(经取代的苯甲基)氨基磷酸酯化合物及其治疗用途 |
| US9156874B2 (en) | 2011-01-03 | 2015-10-13 | Nanjing Molecular Research, Inc. | Double-liver-targeting phosphoramidate and phosphonoamidate prodrugs |
| EP2680859B1 (en) | 2011-03-02 | 2022-04-27 | Jerome Schentag | Compositions, methods of treatment and diagnostics for treatment of hepatic steatosis alone or in combination with a hepatitis c virus infection |
| US20120252721A1 (en) | 2011-03-31 | 2012-10-04 | Idenix Pharmaceuticals, Inc. | Methods for treating drug-resistant hepatitis c virus infection with a 5,5-fused arylene or heteroarylene hepatitis c virus inhibitor |
| CA2843324A1 (en) | 2011-03-31 | 2012-11-15 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| ES2744587T3 (es) * | 2011-04-13 | 2020-02-25 | Gilead Sciences Inc | Análogos de N-nucleósido de pirimidina 1-sustituidos para un tratamiento antiviral |
| SG194485A1 (en) | 2011-04-13 | 2013-12-30 | Merck Sharp & Dohme | 2'-substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| WO2012142075A1 (en) | 2011-04-13 | 2012-10-18 | Merck Sharp & Dohme Corp. | 2'-azido substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| WO2013039920A1 (en) | 2011-09-12 | 2013-03-21 | Idenix Pharmaceuticals, Inc. | Substituted carbonyloxymethylphosphoramidate compounds and pharmaceutical compositions for the treatment of viral infections |
| CA2847892A1 (en) * | 2011-09-12 | 2013-03-21 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| ME03009B (me) | 2011-09-16 | 2018-10-20 | Gilead Pharmasset Llc | Metode za lecenje hcv-a |
| TW201331221A (zh) | 2011-10-14 | 2013-08-01 | Idenix Pharmaceuticals Inc | 嘌呤核苷酸化合物類之經取代的3’,5’-環磷酸酯及用於治療病毒感染之醫藥組成物 |
| US8466159B2 (en) | 2011-10-21 | 2013-06-18 | Abbvie Inc. | Methods for treating HCV |
| EA201490837A1 (ru) | 2011-10-21 | 2014-11-28 | Эббви Инк. | Способы лечения hcv, включающие по меньшей мере два противовирусных агента прямого действия, рибавирин, но не интерферон |
| US8492386B2 (en) | 2011-10-21 | 2013-07-23 | Abbvie Inc. | Methods for treating HCV |
| DE112012003457T5 (de) | 2011-10-21 | 2015-03-12 | Abbvie Inc. | Kombinationsbehandlung (z.B. mit ABT-072 oder ABT-333 von DAAs zur Verwendung in der Behandlung von HCV) |
| WO2013066991A1 (en) * | 2011-10-31 | 2013-05-10 | Inhibitex, Inc. | Crystalline solvates of nucleoside phosphoroamidates, their stereoselective preparation, novel intermediates thereof, and their use in the treatment of viral disease |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| WO2013084165A1 (en) * | 2011-12-05 | 2013-06-13 | Medivir Ab | Hcv polymerase inhibitors |
| JP5967780B2 (ja) | 2011-12-12 | 2016-08-10 | 岡山県 | 植物のアミノ酸含量を高めるための化合物およびその利用 |
| WO2013096680A1 (en) | 2011-12-22 | 2013-06-27 | Alios Biopharma, Inc. | Substituted phosphorothioate nucleotide analogs |
| US20130217644A1 (en) | 2012-02-13 | 2013-08-22 | Idenix Pharmaceuticals, Inc. | Pharmaceutical Compositions of 2'-C-Methyl-Guanosine, 5'-[2[(3-Hydroxy-2,2-Dimethyl-1-Oxopropyl)Thio]Ethyl N-(Phenylmethyl)Phosphoramidate] |
| BR112014019897A8 (pt) | 2012-02-14 | 2017-07-11 | Univ Georgia | Espiro[2.4]heptanos para tratamento de infecções por flaviviridae |
| CA2864469C (en) * | 2012-02-17 | 2020-07-07 | Celsion Corporation | Thermosensitive nanoparticle formulations and method of making the same |
| EP2828277A1 (en) | 2012-03-21 | 2015-01-28 | Vertex Pharmaceuticals Incorporated | Solid forms of a thiophosphoramidate nucleotide prodrug |
| EP2827876A4 (en) | 2012-03-22 | 2015-10-28 | Alios Biopharma Inc | PHARMACEUTICAL COMBINATIONS WITH A THIONUCLEOTIDE ANALOG |
| US9296778B2 (en) | 2012-05-22 | 2016-03-29 | Idenix Pharmaceuticals, Inc. | 3′,5′-cyclic phosphate prodrugs for HCV infection |
| WO2013177188A1 (en) | 2012-05-22 | 2013-11-28 | Idenix Pharmaceuticals, Inc. | 3',5'-cyclic phosphoramidate prodrugs for hcv infection |
| CA2873315A1 (en) | 2012-05-22 | 2013-11-28 | Idenix Pharamaceuticals, Inc. | D-amino acid compounds for liver disease |
| EA027929B1 (ru) | 2012-05-25 | 2017-09-29 | Янссен Сайенсиз Айрлэнд Юси | Нуклеозиды на основе урацила и спирооксетана |
| CN104640444B (zh) * | 2012-06-16 | 2016-12-14 | 河南美泰宝生物制药有限公司 | 双肝脏靶向氨基磷酸酯和氨基膦酸酯前药 |
| CN107312039B (zh) | 2012-08-30 | 2019-06-25 | 江苏豪森药业集团有限公司 | 一种替诺福韦前药的制备方法 |
| EP2900682A1 (en) | 2012-09-27 | 2015-08-05 | IDENIX Pharmaceuticals, Inc. | Esters and malonates of sate prodrugs |
| MD20150036A2 (ru) | 2012-10-08 | 2015-08-31 | Idenix Pharmaceuticals, Inc. | Аналоги 2'-хлор-нуклеозидов для лечения вирусного гепатита С |
| WO2014058729A1 (en) * | 2012-10-09 | 2014-04-17 | California Institute Of Technology | In vivo and in vitro carbene insertion and nitrene transfer reactions catalyzed by heme enzymes |
| JP2015534464A (ja) | 2012-10-09 | 2015-12-03 | カリフォルニア インスティチュート オブ テクノロジー | ヘム酵素によって触媒されるインビボおよびインビトロオレフィンシクロプロパン化 |
| AR092959A1 (es) | 2012-10-17 | 2015-05-06 | Merck Sharp & Dohme | Derivados de nucleosidos 2-metil sustituidos y metodos de uso de los mismos para el tratamiento de enfermedades virales |
| US9457039B2 (en) | 2012-10-17 | 2016-10-04 | Merck Sharp & Dohme Corp. | 2′-disubstituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| WO2014059901A1 (en) | 2012-10-17 | 2014-04-24 | Merck Sharp & Dohme Corp. | 2'-cyano substituted nucleoside derivatives and methods of use thereof for treatment of viral diseases |
| WO2014066239A1 (en) | 2012-10-22 | 2014-05-01 | Idenix Pharmaceuticals, Inc. | 2',4'-bridged nucleosides for hcv infection |
| KR20150086325A (ko) | 2012-11-19 | 2015-07-27 | 머크 샤프 앤드 돔 코포레이션 | 바이러스성 질환을 치료하기 위한 2''-알키닐 치환된 뉴클레오시드 유도체 |
| WO2014099941A1 (en) | 2012-12-19 | 2014-06-26 | Idenix Pharmaceuticals, Inc. | 4'-fluoro nucleosides for the treatment of hcv |
| CN103012520B (zh) * | 2012-12-28 | 2015-12-02 | 中国科学院上海有机化学研究所 | 抗乙肝病毒活性化合物菲糖苷类衍生物 |
| US20140212355A1 (en) * | 2013-01-28 | 2014-07-31 | Abbott Cardiovascular Systems Inc. | Trans-arterial drug delivery |
| HUE047777T2 (hu) | 2013-01-31 | 2020-05-28 | Gilead Pharmasset Llc | Két vírusellenes vegyület kombinációs készítménye |
| US9309275B2 (en) | 2013-03-04 | 2016-04-12 | Idenix Pharmaceuticals Llc | 3′-deoxy nucleosides for the treatment of HCV |
| WO2014137930A1 (en) | 2013-03-04 | 2014-09-12 | Idenix Pharmaceuticals, Inc. | Thiophosphate nucleosides for the treatment of hcv |
| US20140271547A1 (en) | 2013-03-13 | 2014-09-18 | Idenix Pharmaceuticals, Inc. | Amino acid phosphoramidate pronucleotides of 2'-cyano, azido and amino nucleosides for the treatment of hcv |
| WO2014165542A1 (en) | 2013-04-01 | 2014-10-09 | Idenix Pharmaceuticals, Inc. | 2',4'-fluoro nucleosides for the treatment of hcv |
| EP2984098A2 (en) | 2013-04-12 | 2016-02-17 | Achillion Pharmaceuticals, Inc. | Deuterated nucleoside prodrugs useful for treating hcv |
| WO2014197578A1 (en) * | 2013-06-05 | 2014-12-11 | Idenix Pharmaceuticals, Inc. | 1',4'-thio nucleosides for the treatment of hcv |
| US9765107B2 (en) | 2013-06-18 | 2017-09-19 | Merck Sharp & Dohme Corp. | Cyclic phosphonate substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| ES2952714T3 (es) | 2013-06-26 | 2023-11-03 | Janssen Pharmaceuticals Inc | Nucleósidos sustituidos, nucleótidos y análogos de los mismos |
| DK3013843T3 (en) | 2013-06-26 | 2018-09-03 | Alios Biopharma Inc | SUBSTITUTED NUCLEOSIDES, NUCLEOTIDES AND ANALOGS THEREOF |
| US20150037282A1 (en) | 2013-08-01 | 2015-02-05 | Idenix Pharmaceuticals, Inc. | D-amino acid phosphoramidate pronucleotides of halogeno pyrimidine compounds for liver disease |
| JP2016529293A (ja) | 2013-08-27 | 2016-09-23 | ギリアド ファーマセット エルエルシー | 2つの抗ウイルス化合物の組合せ製剤 |
| UA117375C2 (uk) | 2013-09-04 | 2018-07-25 | Медівір Аб | Інгібітори полімерази hcv |
| ES2927955T3 (es) | 2013-09-11 | 2022-11-14 | Univ Emory | Composiciones de nucleótidos y nucleósidos y sus usos |
| WO2015042375A1 (en) | 2013-09-20 | 2015-03-26 | Idenix Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| US9862743B2 (en) | 2013-10-11 | 2018-01-09 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US20160271160A1 (en) | 2013-10-17 | 2016-09-22 | Medivir Ab | Hcv polymerase inhibitors |
| EP3092255A4 (en) | 2014-01-10 | 2017-09-20 | Birdie Biopharmaceuticals Inc. | Compounds and compositions for treating egfr expressing tumors |
| US9399762B2 (en) | 2014-02-18 | 2016-07-26 | California Institute Of Technology | Methods and systems for sulfimidation or sulfoximidation of organic molecules |
| WO2015134560A1 (en) | 2014-03-05 | 2015-09-11 | Idenix Pharmaceuticals, Inc. | Solid forms of a flaviviridae virus inhibitor compound and salts thereof |
| WO2015143712A1 (en) | 2014-03-28 | 2015-10-01 | Merck Sharp & Dohme Corp. | 4'-substituted nucleoside reverse transcriptase inhibitors |
| WO2015161137A1 (en) | 2014-04-16 | 2015-10-22 | Idenix Pharmaceuticals, Inc. | 3'-substituted methyl or alkynyl nucleosides for the treatment of hcv |
| US11918728B2 (en) | 2014-04-17 | 2024-03-05 | ImMutriX Therapeutics, Inc. | Therapeutic compositions for viral-associated disease states and methods of making and using same |
| US9669151B2 (en) | 2014-04-17 | 2017-06-06 | ImMutriX Therapeutics, Inc. | Therapeutic compositions for viral-associated disease states and methods of making and using same |
| WO2015181624A2 (en) * | 2014-05-28 | 2015-12-03 | Idenix Pharmaceuticals, Inc | Nucleoside derivatives for the treatment of cancer |
| SG11201610599TA (en) | 2014-06-24 | 2017-01-27 | Alios Biopharma Inc | Substituted nucleosides, nucleotides and analogs thereof |
| CN105440135A (zh) | 2014-09-01 | 2016-03-30 | 博笛生物科技有限公司 | 用于治疗肿瘤的抗-pd-l1结合物 |
| WO2016004876A1 (en) | 2014-07-09 | 2016-01-14 | Shanghai Birdie Biotech, Inc. | Anti-pd-l1 combinations for treating tumors |
| RU2660438C1 (ru) | 2014-07-18 | 2018-07-06 | Джей ДаблЮ ФАРМАСЬЮТИКАЛ КОРПОРЭЙШН | Новая соль тенофовира дизопроксила |
| TWI767201B (zh) | 2014-10-29 | 2022-06-11 | 美商基利科學股份有限公司 | 絲狀病毒科病毒感染之治療 |
| KR20220025914A (ko) | 2015-03-06 | 2022-03-03 | 아테아 파마슈티컬즈, 인크. | HCV 치료를 위한 β-D-2'-데옥시-2'-α-플루오로-2'-β-C-치환된-2-변형된-N6-치환된 퓨린 뉴클레오티드 |
| JP6526904B2 (ja) * | 2015-04-26 | 2019-06-05 | マッカイ メモリアル ホスピタルMackay Memorial Hospital | 腎疾患を有する患者の腎臓及び/又は心臓機能の改善方法 |
| HRP20220740T1 (hr) | 2015-09-16 | 2022-11-11 | Gilead Sciences, Inc. | Postupci za liječenje virusnih infekcija arenaviridae |
| US10953029B2 (en) | 2015-09-23 | 2021-03-23 | Merck Sharp & Dohme Corp. | 4′-Substituted nucleoside reverse transcriptase inhibitors and preparations thereof |
| CN115554406A (zh) | 2016-01-07 | 2023-01-03 | 博笛生物科技有限公司 | 用于治疗肿瘤的抗-cd20组合 |
| CN115252792A (zh) | 2016-01-07 | 2022-11-01 | 博笛生物科技有限公司 | 用于治疗肿瘤的抗-egfr组合 |
| EP3448392A4 (en) | 2016-04-28 | 2020-01-15 | Emory University | ALCYNE-CONTAINING NUCLEOTIDES AND NUCLEOSIDES THERAPEUTIC COMPOSITIONS AND USES THEREOF |
| AU2017259960A1 (en) * | 2016-05-02 | 2018-12-06 | Paratek Pharmaceuticals, Inc. | 9-aminomethyl minocycline compounds and methods of use thereof in urinary tract infection (UTI) treatment |
| CN109641874A (zh) | 2016-05-10 | 2019-04-16 | C4医药公司 | 用于靶蛋白降解的c3-碳连接的戊二酰亚胺降解决定子体 |
| EP3454862B1 (en) | 2016-05-10 | 2024-09-11 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| CN109562107A (zh) | 2016-05-10 | 2019-04-02 | C4医药公司 | 用于靶蛋白降解的杂环降解决定子体 |
| US10899788B2 (en) | 2016-06-20 | 2021-01-26 | Merck Sharp & Dohme Corp. | Cyclic phosphate substituted nucleoside compounds and methods of use thereof for the treatment of viral diseases |
| US11174279B2 (en) | 2016-06-21 | 2021-11-16 | National Institute Of Advanced Industrial Science And Technology | Method for synthesizing ribonucleic acid H-phosphonate monomer, and oligonucleotide synthesis in which said monomer is used |
| US10202412B2 (en) | 2016-07-08 | 2019-02-12 | Atea Pharmaceuticals, Inc. | β-D-2′-deoxy-2′-substituted-4′-substituted-2-substituted-N6-substituted-6-aminopurinenucleotides for the treatment of paramyxovirus and orthomyxovirus infections |
| US10711029B2 (en) | 2016-07-14 | 2020-07-14 | Atea Pharmaceuticals, Inc. | Beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-c-substituted-4′fluoro-n6-substituted-6-amino-2-substituted purine nucleotides for the treatment of hepatitis c virus infection |
| KR20220146668A (ko) | 2016-09-07 | 2022-11-01 | 아테아 파마슈티컬즈, 인크. | Rna 바이러스 치료를 위한 2'-치환된-n6-치환된 퓨린 뉴클레오티드 |
| WO2018071814A1 (en) | 2016-10-14 | 2018-04-19 | The Trustees Of Columbia University In The City Of New York | Methods of treating alcohol abuse disorder |
| JP7545803B2 (ja) | 2016-11-01 | 2024-09-05 | パラテック ファーマシューティカルズ,インコーポレイテッド | 9-アミノメチルミノサイクリン化合物及び市中感染型細菌性肺炎(cabp)の治療におけるその使用 |
| EP3534903B1 (en) | 2016-11-07 | 2022-08-03 | Arbutus Biopharma Corporation | Substituted pyridinone-containing tricyclic compounds, and methods using same |
| EP3541825A1 (en) | 2016-11-21 | 2019-09-25 | Idenix Pharmaceuticals LLC. | Cyclic phosphate substituted nucleoside derivatives for the treatment of liver diseases |
| CN108276463A (zh) * | 2017-01-06 | 2018-07-13 | 米文君 | 一类新的化合物及其用途 |
| KR102592899B1 (ko) | 2017-02-01 | 2023-10-24 | 아테아 파마슈티컬즈, 인크. | C형 간염 바이러스를 치료하기 위한 뉴클레오티드 헤미-술페이트 염 |
| AU2018235754B2 (en) | 2017-03-14 | 2021-04-08 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| WO2018172852A1 (en) | 2017-03-21 | 2018-09-27 | Arbutus Biopharma Corporation | Substituted dihydroindene-4-carboxamides and analogs thereof, and methods using same |
| CN108794467A (zh) | 2017-04-27 | 2018-11-13 | 博笛生物科技有限公司 | 2-氨基-喹啉衍生物 |
| EP4219513A1 (en) | 2017-05-01 | 2023-08-02 | Gilead Sciences, Inc. | Crystalline form of (s)-2-ethylbutyl 2-(((s)-(((2r,3s,4r,5r)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate |
| CN110996966A (zh) * | 2017-06-05 | 2020-04-10 | 维京治疗公司 | 用于治疗纤维化的组合物 |
| GB201709471D0 (en) * | 2017-06-14 | 2017-07-26 | Nucana Biomed Ltd | Diastereoselective synthesis of hosphate derivatives |
| IL312120A (en) | 2017-06-23 | 2024-06-01 | Birdie Biopharmaceuticals Inc | medical preparation |
| CA3077489A1 (en) | 2017-07-11 | 2019-01-17 | Gilead Sciences, Inc. | Compositions comprising an rna polymerase inhibitor and cyclodextrin for treating viral infections |
| US10688112B2 (en) | 2017-07-13 | 2020-06-23 | Emory University | Lipid disulfide prodrugs and uses related thereto |
| US11040975B2 (en) | 2017-12-08 | 2021-06-22 | Merck Sharp & Dohme Corp. | Carbocyclic nucleoside reverse transcriptase inhibitors |
| WO2019120084A1 (zh) * | 2017-12-21 | 2019-06-27 | 深圳市塔吉瑞生物医药有限公司 | 用于抗病毒的核苷类逆转录酶抑制剂 |
| EA202091979A1 (ru) | 2018-03-22 | 2021-06-22 | Вайкинг Терапьютикс, Инк. | Кристаллические формы и способы получения кристаллических форм соединения |
| WO2019200005A1 (en) | 2018-04-10 | 2019-10-17 | Atea Pharmaceuticals, Inc. | Treatment of hcv infected patients with cirrhosis |
| TWI826492B (zh) | 2018-07-27 | 2023-12-21 | 加拿大商愛彼特生物製藥公司 | 經取代四氫環戊[c]吡咯、經取代二氫吡咯,其類似物及使用其之方法 |
| WO2020117962A1 (en) | 2018-12-05 | 2020-06-11 | Viking Therapeutics, Inc. | Compositions for the treatment of fibrosis and inflammation |
| TW202415643A (zh) | 2018-12-12 | 2024-04-16 | 加拿大商愛彼特生物製藥公司 | 經取代之芳基甲基脲類及雜芳基甲基脲類、其類似物及其使用方法 |
| JP2022546310A (ja) | 2019-08-22 | 2022-11-04 | エモリー ユニバーシティー | ヌクレオシドプロドラッグ及びそれに関する使用 |
| CN111330557B (zh) * | 2020-01-17 | 2022-11-18 | 中山职业技术学院 | 一种手性固定相及其制备和其在手性药物拆分应用 |
| TW202313067A (zh) | 2020-01-27 | 2023-04-01 | 美商基利科學股份有限公司 | 治療sars cov-2感染之方法 |
| TW202245800A (zh) | 2020-02-18 | 2022-12-01 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| TWI794742B (zh) | 2020-02-18 | 2023-03-01 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| CA3171648A1 (en) | 2020-02-18 | 2021-08-26 | Gilead Sciences, Inc. | Antiviral compounds |
| US10874687B1 (en) | 2020-02-27 | 2020-12-29 | Atea Pharmaceuticals, Inc. | Highly active compounds against COVID-19 |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| CA3172483A1 (en) | 2020-04-06 | 2021-10-14 | Scott Ellis | Inhalation formulations of 1'-cyano substituted carbanucleoside analogs |
| KR20230018473A (ko) | 2020-05-29 | 2023-02-07 | 길리애드 사이언시즈, 인코포레이티드 | 렘데시비르 치료 방법 |
| IL299202A (en) | 2020-06-24 | 2023-02-01 | Gilead Sciences Inc | 1'-cyano nucleoside analogs and uses thereof |
| BR112023000626A2 (pt) | 2020-07-16 | 2023-01-31 | Merck Sharp & Dohme Llc | Derivados de cianoenona cíclicos como moduladores de keap1 |
| FI4204421T3 (fi) | 2020-08-27 | 2024-06-25 | Gilead Sciences Inc | Koostumuksia ja menetelmiä virusinfektioiden hoitoon |
| JP2024512771A (ja) | 2021-04-16 | 2024-03-19 | ギリアード サイエンシーズ, インコーポレイテッド | アミドを使用してカルバヌクレオシドを調製する方法 |
| US12116380B2 (en) | 2021-08-18 | 2024-10-15 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| US20230295172A1 (en) | 2022-03-02 | 2023-09-21 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023250015A1 (en) * | 2022-06-21 | 2023-12-28 | The Children's Medical Center Corporation | Pyrimidine nucleoside treatments |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996040164A1 (en) * | 1995-06-07 | 1996-12-19 | Emory University | Nucleosides with anti-hepatitis b virus activity |
| WO2002057287A2 (en) * | 2001-01-22 | 2002-07-25 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2003072757A2 (en) * | 2002-02-28 | 2003-09-04 | Biota, Inc. | Nucleotide mimics and their prodrugs |
Family Cites Families (147)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3480613A (en) | 1967-07-03 | 1969-11-25 | Merck & Co Inc | 2-c or 3-c-alkylribofuranosyl - 1-substituted compounds and the nucleosides thereof |
| US4849513A (en) | 1983-12-20 | 1989-07-18 | California Institute Of Technology | Deoxyribonucleoside phosphoramidites in which an aliphatic amino group is attached to the sugar ring and their use for the preparation of oligonucleotides containing aliphatic amino groups |
| US5401726A (en) * | 1988-08-25 | 1995-03-28 | Yoshitomi Pharaceutical Industries, Ltd. | 2'methylidenepyrimidine nucleoside compounds, their use and method for production thereof |
| DE4207363A1 (de) | 1992-03-04 | 1993-09-09 | Max Delbrueck Centrum | Antivirale nucleosidanaloga, ihre herstellung und ihre pharmazeutische verwendung |
| US6469158B1 (en) | 1992-05-14 | 2002-10-22 | Ribozyme Pharmaceuticals, Incorporated | Synthesis, deprotection, analysis and purification of RNA and ribozymes |
| FR2692265B1 (fr) | 1992-05-25 | 1996-11-08 | Centre Nat Rech Scient | Composes biologiquement actifs de type phosphotriesters. |
| US6057305A (en) * | 1992-08-05 | 2000-05-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Antiretroviral enantiomeric nucleotide analogs |
| GB9217998D0 (en) * | 1992-08-24 | 1992-10-07 | Wellcome Found | Methods of treating cancer |
| US6423489B1 (en) | 1992-09-10 | 2002-07-23 | Isis Pharmaceuticals, Inc. | Compositions and methods for treatment of Hepatitis C virus-associated diseases |
| US6391542B1 (en) | 1992-09-10 | 2002-05-21 | Isis Pharmaceuticals, Inc. | Compositions and methods for treatment of Hepatitis C virus-associated diseases |
| CA2143678A1 (en) | 1992-09-10 | 1994-03-17 | Kevin P. Anderson | Compositions and methods for treatment of hepatitis c virus-associated diseases |
| US6995146B2 (en) | 1992-09-10 | 2006-02-07 | Isis Pharmaceuticals, Inc. | Compositions and methods for treatment of hepatitis C virus-associated diseases |
| US6174868B1 (en) | 1992-09-10 | 2001-01-16 | Isis Pharmaceuticals, Inc. | Compositions and methods for treatment of hepatitis C virus-associated diseases |
| US6433159B1 (en) | 1992-09-10 | 2002-08-13 | Isis Pharmaceuticals, Inc. | Compositions and methods for treatment of Hepatitis C virus associated diseases |
| US5955591A (en) * | 1993-05-12 | 1999-09-21 | Imbach; Jean-Louis | Phosphotriester oligonucleotides, amidites and method of preparation |
| ES2186690T3 (es) | 1993-09-02 | 2003-05-16 | Ribozyme Pharm Inc | Acido nucleico enzimatico que contiene no-nucleotidos. |
| US6146886A (en) | 1994-08-19 | 2000-11-14 | Ribozyme Pharmaceuticals, Inc. | RNA polymerase III-based expression of therapeutic RNAs |
| US5627185A (en) * | 1994-11-23 | 1997-05-06 | Gosselin; Gilles | Acyclovir derivatives as antiviral agents |
| GB9505025D0 (en) | 1995-03-13 | 1995-05-03 | Medical Res Council | Chemical compounds |
| US7034009B2 (en) | 1995-10-26 | 2006-04-25 | Sirna Therapeutics, Inc. | Enzymatic nucleic acid-mediated treatment of ocular diseases or conditions related to levels of vascular endothelial growth factor receptor (VEGF-R) |
| GB9708611D0 (en) | 1997-04-28 | 1997-06-18 | Univ Cardiff | Chemical compounds |
| EP0998280A4 (en) | 1997-05-29 | 2002-11-06 | Incara Pharmaceuticals Corp | CARBOHYDRATE COMPOUNDS WITH SCAFFOLDING STRUCTURE AND BANKS CONTAINING SAME |
| CN100349913C (zh) | 1998-02-25 | 2007-11-21 | 埃莫里大学 | 2′-氟代核苷 |
| US6833361B2 (en) | 1998-05-26 | 2004-12-21 | Ribapharm, Inc. | Nucleosides having bicyclic sugar moiety |
| US6274725B1 (en) * | 1998-06-02 | 2001-08-14 | Isis Pharmaceuticals, Inc. | Activators for oligonucleotide synthesis |
| GB9821058D0 (en) | 1998-09-28 | 1998-11-18 | Univ Cardiff | Chemical compound |
| US6752981B1 (en) | 1999-09-08 | 2004-06-22 | Metabasis Therapeutics, Inc. | Prodrugs for liver specific drug delivery |
| WO2001032153A2 (en) | 1999-11-04 | 2001-05-10 | Shire Biochem Inc. | Method for the treatment or prevention of flaviviridae viral infection using nucleoside analogues |
| WO2002081628A2 (en) | 2001-04-05 | 2002-10-17 | Ribozyme Pharmaceuticals, Incorporated | Modulation of gene expression associated with inflammation proliferation and neurite outgrowth, using nucleic acid based technologies |
| JP2003523978A (ja) | 2000-02-18 | 2003-08-12 | シャイアー・バイオケム・インコーポレイテッド | ヌクレオシドアナログを用いるflavivirus感染の処置もしくは予防するための方法 |
| DE60136620D1 (de) | 2000-04-13 | 2009-01-02 | Pharmasset Inc | 3 oder 2 hydroxymethyl substituierte nucleoside derivate und ihre verwendung zur behandlung von virusinfektionen |
| GB0011203D0 (en) | 2000-05-09 | 2000-06-28 | Univ Cardiff | Chemical compounds |
| MY164523A (en) | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| EA007867B1 (ru) | 2000-05-26 | 2007-02-27 | Айденикс (Кайман) Лимитед | Композиции для лечения флавивирусных и пестивирусных инфекций и способы их применения |
| US6787526B1 (en) * | 2000-05-26 | 2004-09-07 | Idenix Pharmaceuticals, Inc. | Methods of treating hepatitis delta virus infection with β-L-2′-deoxy-nucleosides |
| UA72612C2 (en) * | 2000-07-06 | 2005-03-15 | Pyrido[2.3-d]pyrimidine and pyrimido[4.5-d]pyrimidine nucleoside analogues, prodrugs and method for inhibiting growth of neoplastic cells | |
| US7045309B2 (en) * | 2000-07-07 | 2006-05-16 | The Research Foundation Of State University Of New York | 9-substituted adenine derivatives as prodrug regulators of cell and tissue function |
| ES2627903T3 (es) * | 2000-07-21 | 2017-08-01 | Gilead Sciences, Inc. | Profármacos de análogos de nucleótidos de fosfonato y métodos para seleccionar y preparar los mismos |
| US20030008841A1 (en) | 2000-08-30 | 2003-01-09 | Rene Devos | Anti-HCV nucleoside derivatives |
| EP1361847A2 (en) * | 2000-10-06 | 2003-11-19 | Xenoport, Inc. | Bile-acid conjugates for providing sustained systemic concentrations of drugs |
| WO2002044324A2 (en) | 2000-10-06 | 2002-06-06 | Xenoport, Inc. | Bile-acid derived compounds for enhancing oral absorption and systemic bioavailability of drugs |
| KR20040028657A (ko) | 2000-10-18 | 2004-04-03 | 파마셋, 리미티드 | 바이러스 감염 및 비정상적인 세포 증식의 치료를 위한변형된 뉴클레오시드 |
| CN1527836A (zh) | 2000-12-15 | 2004-09-08 | 用于治疗黄病毒科病毒感染的抗病毒剂 | |
| US7105499B2 (en) | 2001-01-22 | 2006-09-12 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase |
| KR20030092006A (ko) | 2001-03-01 | 2003-12-03 | 파마셋, 리미티드 | 2',3'-디데옥시-2',3'-디데하이드로뉴클레오시드 합성 방법 |
| US7109165B2 (en) | 2001-05-18 | 2006-09-19 | Sirna Therapeutics, Inc. | Conjugates and compositions for cellular delivery |
| US20050148530A1 (en) | 2002-02-20 | 2005-07-07 | Sirna Therapeutics, Inc. | RNA interference mediated inhibition of vascular endothelial growth factor and vascular endothelial growth factor receptor gene expression using short interfering nucleic acid (siNA) |
| US20040209831A1 (en) | 2002-02-20 | 2004-10-21 | Mcswiggen James | RNA interference mediated inhibition of hepatitis C virus (HCV) gene expression using short interfering nucleic acid (siNA) |
| WO2003070750A2 (en) | 2002-02-20 | 2003-08-28 | Sirna Therapeutics, Inc | Rna interference mediated inhibition of hepatitis c virus |
| GB0112617D0 (en) | 2001-05-23 | 2001-07-18 | Hoffmann La Roche | Antiviral nucleoside derivatives |
| GB0114286D0 (en) | 2001-06-12 | 2001-08-01 | Hoffmann La Roche | Nucleoside Derivatives |
| JP2004534830A (ja) | 2001-06-21 | 2004-11-18 | グラクソ グループ リミテッド | Hcvにおけるヌクレオシド化合物 |
| JP2005536440A (ja) | 2001-09-28 | 2005-12-02 | イデニクス(ケイマン)リミテツド | 4’位が修飾されたヌクレオシドを使用するフラビウイルスおよびペスチウイルスの治療のための方法および組成物 |
| US7138376B2 (en) | 2001-09-28 | 2006-11-21 | Idenix Pharmaceuticals, Inc. | Methods and compositions for treating hepatitis C virus using 4'-modified nucleosides |
| WO2003062256A1 (en) | 2002-01-17 | 2003-07-31 | Ribapharm Inc. | 2'-beta-modified-6-substituted adenosine analogs and their use as antiviral agents |
| US7323453B2 (en) | 2002-02-13 | 2008-01-29 | Merck & Co., Inc. | Methods of inhibiting orthopoxvirus replication with nucleoside compounds |
| EP1485396A2 (en) * | 2002-02-28 | 2004-12-15 | Biota, Inc. | Nucleoside 5'-monophosphate mimics and their prodrugs |
| BR0309581A (pt) * | 2002-05-06 | 2005-03-29 | Genelabs Tech Inc | Derivados de nucleosìdeo para tratamento de infecção por vìrus de hepatite c |
| AU2003241621A1 (en) | 2002-05-24 | 2003-12-12 | Isis Pharmaceuticals, Inc. | Oligonucleotides having modified nucleoside units |
| JP2005533777A (ja) * | 2002-06-17 | 2005-11-10 | メルク エンド カムパニー インコーポレーテッド | Rna抗ウイルス剤としての炭素環ヌクレオシドアナログ |
| US20070004669A1 (en) | 2002-06-21 | 2007-01-04 | Carroll Steven S | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| US20060234962A1 (en) * | 2002-06-27 | 2006-10-19 | Olsen David B | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| US7608600B2 (en) | 2002-06-28 | 2009-10-27 | Idenix Pharmaceuticals, Inc. | Modified 2′ and 3′-nucleoside prodrugs for treating Flaviviridae infections |
| BR0312278A (pt) | 2002-06-28 | 2007-06-19 | Idenix Cayman Ltd | éster 2'-c-metil-3'-o-l-valina de ribofuranosil citidina para tratamento de infecções por flaviviridae |
| CN100348607C (zh) | 2002-06-28 | 2007-11-14 | 埃迪尼克斯(开曼)有限公司 | 用于治疗黄病毒科病毒感染的2’和3’-核苷前药 |
| NZ537662A (en) | 2002-06-28 | 2007-10-26 | Idenix Cayman Ltd | 2'-C-methyl-3'-O-L-valine ester ribofuranosyl cytidine for treatment of flaviviridae infections |
| KR20050055630A (ko) | 2002-06-28 | 2005-06-13 | 이데닉스 (케이만) 리미티드 | 플라비비리다에 감염 치료를 위한 1'-, 2'- 및 3'-변형된뉴클레오시드 유도체 |
| US20060264389A1 (en) | 2002-07-16 | 2006-11-23 | Balkrishen Bhat | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2004009020A2 (en) | 2002-07-24 | 2004-01-29 | Merck & Co., Inc. | Pyrrolopyrimidine thionucleoside analogs as antivirals |
| EP1529172A1 (de) | 2002-08-15 | 2005-05-11 | DOW CORNING GmbH | Zusammensetzung und vorrichtung zum dämpfen mechanischer bewegung |
| AU2003259381A1 (en) | 2002-08-29 | 2004-03-19 | University Of Southampton | Use of apoptosis inducing agents in the preparation of a medicament for the treatment of liver diseases |
| CA2499889A1 (en) | 2002-09-26 | 2004-04-08 | Lg Life Sciences Ltd. | (+)-trans-isomers of (1-phosphonomethoxy-2-alkylcyclopropyl) methyl nucleoside derivatives, process for the preparation of stereoisomers thereof, and use of antiviral agents thereof |
| US20040229840A1 (en) * | 2002-10-29 | 2004-11-18 | Balkrishen Bhat | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase |
| KR20050088079A (ko) | 2002-11-15 | 2005-09-01 | 이데닉스 (케이만) 리미티드 | 2'-분지형 뉴클레오시드 및 플라비비리다에 돌연변이 |
| TWI332507B (en) | 2002-11-19 | 2010-11-01 | Hoffmann La Roche | Antiviral nucleoside derivatives |
| WO2004052899A2 (en) | 2002-12-12 | 2004-06-24 | Idenix (Cayman) Limited | Process for the production of 2'-branched nucleosides |
| AR043006A1 (es) | 2003-02-12 | 2005-07-13 | Merck & Co Inc | Proceso para preparar ribonucleosidos ramificados |
| US7741065B2 (en) * | 2003-03-13 | 2010-06-22 | Ramot At Tel Aviv University Ltd. | Non-invasive marker for liver function and disease |
| WO2004100960A2 (en) | 2003-04-25 | 2004-11-25 | Gilead Sciences, Inc. | Anti-inflammatory phosphonate compounds |
| US7427636B2 (en) | 2003-04-25 | 2008-09-23 | Gilead Sciences, Inc. | Inosine monophosphate dehydrogenase inhibitory phosphonate compounds |
| WO2004096237A2 (en) | 2003-04-25 | 2004-11-11 | Gilead Sciences, Inc. | Phosphonate analogs for treating metabolic diseases |
| WO2004096233A2 (en) | 2003-04-25 | 2004-11-11 | Gilead Sciences, Inc. | Nucleoside phosphonate conjugates |
| US7429565B2 (en) | 2003-04-25 | 2008-09-30 | Gilead Sciences, Inc. | Antiviral phosphonate analogs |
| US7300924B2 (en) | 2003-04-25 | 2007-11-27 | Gilead Sciences, Inc. | Anti-infective phosphonate analogs |
| CA2522845A1 (en) | 2003-04-25 | 2004-11-11 | Gilead Sciences, Inc. | Kinase inhibitor phosphonate conjugates |
| WO2005020884A2 (en) | 2003-05-14 | 2005-03-10 | Idenix (Cayman) Limited | Nucleosides for treatment of infection by corona viruses, toga viruses and picorna viruses |
| US20040229839A1 (en) * | 2003-05-14 | 2004-11-18 | Biocryst Pharmaceuticals, Inc. | Substituted nucleosides, preparation thereof and use as inhibitors of RNA viral polymerases |
| WO2005020885A2 (en) | 2003-05-21 | 2005-03-10 | Isis Pharmaceuticals, Inc. | Compositions and methods for the treatment of severe acute respiratory syndrome (sars) |
| EP4032897A1 (en) | 2003-05-30 | 2022-07-27 | Gilead Pharmasset LLC | Modified fluorinated nucleoside analogues |
| KR20060026426A (ko) | 2003-06-19 | 2006-03-23 | 에프. 호프만-라 로슈 아게 | 4'-아지도 뉴클레오사이드 유도체의 제조 방법 |
| US20050043266A1 (en) | 2003-07-25 | 2005-02-24 | Sumedha Jayasena | Short interfering RNA as an antiviral agent for hepatitis C |
| US7713941B2 (en) | 2003-08-27 | 2010-05-11 | Biota Scientific Management Pty Ltd | Tricyclic nucleosides or nucleotides as therapeutic agents |
| WO2005044279A1 (en) | 2003-10-24 | 2005-05-19 | Gilead Sciences, Inc. | Purine nucleoside phosphonate conjugates |
| WO2005044308A1 (en) | 2003-10-24 | 2005-05-19 | Gilead Sciences, Inc. | Phosphonate analogs of antimetabolites |
| CA2543116A1 (en) | 2003-10-27 | 2005-05-19 | Genelabs Technologies, Inc. | Methods for preparing 7-(2'-substituted-.szlig.-d-ribofuranosyl)-4-(nr2r3)-5-(substituted ethyn-1-yl)-pyrrolo[2,3-d]pyrimidine derivatives |
| EP2428516A1 (en) | 2003-11-19 | 2012-03-14 | Metabasis Therapeutics, Inc. | Novel phosphorus-containing thyromimetics |
| US20050182252A1 (en) | 2004-02-13 | 2005-08-18 | Reddy K. R. | Novel 2'-C-methyl nucleoside derivatives |
| US20050190302A1 (en) * | 2004-02-26 | 2005-09-01 | Goedde Kirby R. | Automation of field service color temperature alignment |
| WO2005087788A2 (en) | 2004-03-04 | 2005-09-22 | The Regents Of The University Of California | Methods for preparation of nucleoside phosphonate esters |
| EP1912643A2 (en) | 2004-06-23 | 2008-04-23 | Idenix (Cayman) Limited | 5-aza-7-deazapurine derivatives for treating infections with flaviviridae |
| CA2571079A1 (en) | 2004-06-24 | 2006-02-02 | Merck & Co., Inc. | Nucleoside aryl phosphoramidates for the treatment of rna-dependent rna viral infection |
| KR101235548B1 (ko) | 2004-07-21 | 2013-02-21 | 길리어드 파마셋 엘엘씨 | 2'-데옥시-2'-플루오로-2'-C-메틸-β-D-리보퓨라노실 뉴클레오사이드의 제조 방법 |
| JP2008510748A (ja) | 2004-08-23 | 2008-04-10 | エフ.ホフマン−ラ ロシュ アーゲー | 抗ウイルス4’−アジド−ヌクレオシド |
| US20080280842A1 (en) | 2004-10-21 | 2008-11-13 | Merck & Co., Inc. | Fluorinated Pyrrolo[2,3-D]Pyrimidine Nucleosides for the Treatment of Rna-Dependent Rna Viral Infection |
| US8399428B2 (en) | 2004-12-09 | 2013-03-19 | Regents Of The University Of Minnesota | Nucleosides with antiviral and anticancer activity |
| AP2007004128A0 (en) | 2005-02-28 | 2007-08-31 | Genelabs Tech Inc | Tricyclic-nucleoside compounds for treating viral infections |
| US8802840B2 (en) | 2005-03-08 | 2014-08-12 | Biota Scientific Management Pty Ltd. | Bicyclic nucleosides and nucleotides as therapeutic agents |
| US20100279974A1 (en) | 2005-03-09 | 2010-11-04 | Claire Pierra | Nucleosides With Non-Natural Bases as Anti-Viral Agents |
| WO2006116557A1 (en) | 2005-04-25 | 2006-11-02 | Genelabs Technologies, Inc. | Nucleoside compounds for treating viral infections |
| CA2609342A1 (en) | 2005-04-26 | 2006-11-02 | Andrei L. Gartel | Nucleoside compounds and methods of use thereof |
| CA3021449C (en) * | 2005-05-05 | 2021-12-14 | Drexel University | Diagnosis of liver pathology through assessment of protein glycosylation |
| WO2006122207A1 (en) * | 2005-05-10 | 2006-11-16 | Valeant Research & Development | 6-hydrazinopurine 2'-methyl ribonucleosides and nucleotides for treatment of hcv |
| WO2007027248A2 (en) | 2005-05-16 | 2007-03-08 | Valeant Research & Development | 3', 5' - cyclic nucleoside analogues for treatment of hcv |
| CA2618335C (en) | 2005-08-15 | 2015-03-31 | F.Hoffmann-La Roche Ag | Antiviral phosphoramidates of 4'-substituted pronucleotides |
| US7781576B2 (en) | 2005-12-23 | 2010-08-24 | Idenix Pharmaceuticals, Inc. | Process for preparing a synthetic intermediate for preparation of branched nucleosides |
| WO2007095269A2 (en) | 2006-02-14 | 2007-08-23 | Merck & Co., Inc. | Nucleoside aryl phosphoramidates for the treatment of rna-dependent rna viral infection |
| GB0623493D0 (en) | 2006-11-24 | 2007-01-03 | Univ Cardiff | Chemical compounds |
| US7951789B2 (en) | 2006-12-28 | 2011-05-31 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| CN101333235A (zh) | 2007-06-29 | 2008-12-31 | 天津大学 | 一种糖基部分具有乙炔基及氟原子取代的核苷衍生物 |
| GB0718575D0 (en) | 2007-09-24 | 2007-10-31 | Angeletti P Ist Richerche Bio | Nucleoside derivatives as inhibitors of viral polymerases |
| WO2011123586A1 (en) | 2010-04-01 | 2011-10-06 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| CA2843324A1 (en) | 2011-03-31 | 2012-11-15 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| WO2012142075A1 (en) | 2011-04-13 | 2012-10-18 | Merck Sharp & Dohme Corp. | 2'-azido substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| CA2832449A1 (en) | 2011-04-13 | 2012-10-18 | Vinay GIRIJAVALLABHAN | 2'-cyano substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| SG194485A1 (en) | 2011-04-13 | 2013-12-30 | Merck Sharp & Dohme | 2'-substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| WO2013039920A1 (en) | 2011-09-12 | 2013-03-21 | Idenix Pharmaceuticals, Inc. | Substituted carbonyloxymethylphosphoramidate compounds and pharmaceutical compositions for the treatment of viral infections |
| TW201331221A (zh) | 2011-10-14 | 2013-08-01 | Idenix Pharmaceuticals Inc | 嘌呤核苷酸化合物類之經取代的3’,5’-環磷酸酯及用於治療病毒感染之醫藥組成物 |
| US9296778B2 (en) | 2012-05-22 | 2016-03-29 | Idenix Pharmaceuticals, Inc. | 3′,5′-cyclic phosphate prodrugs for HCV infection |
| WO2013177188A1 (en) | 2012-05-22 | 2013-11-28 | Idenix Pharmaceuticals, Inc. | 3',5'-cyclic phosphoramidate prodrugs for hcv infection |
| CA2873315A1 (en) | 2012-05-22 | 2013-11-28 | Idenix Pharamaceuticals, Inc. | D-amino acid compounds for liver disease |
| EP2900682A1 (en) | 2012-09-27 | 2015-08-05 | IDENIX Pharmaceuticals, Inc. | Esters and malonates of sate prodrugs |
| MD20150036A2 (ru) | 2012-10-08 | 2015-08-31 | Idenix Pharmaceuticals, Inc. | Аналоги 2'-хлор-нуклеозидов для лечения вирусного гепатита С |
| AR092959A1 (es) | 2012-10-17 | 2015-05-06 | Merck Sharp & Dohme | Derivados de nucleosidos 2-metil sustituidos y metodos de uso de los mismos para el tratamiento de enfermedades virales |
| US9457039B2 (en) | 2012-10-17 | 2016-10-04 | Merck Sharp & Dohme Corp. | 2′-disubstituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| WO2014059901A1 (en) | 2012-10-17 | 2014-04-24 | Merck Sharp & Dohme Corp. | 2'-cyano substituted nucleoside derivatives and methods of use thereof for treatment of viral diseases |
| WO2014063019A1 (en) | 2012-10-19 | 2014-04-24 | Idenix Pharmaceuticals, Inc. | Dinucleotide compounds for hcv infection |
| WO2014066239A1 (en) | 2012-10-22 | 2014-05-01 | Idenix Pharmaceuticals, Inc. | 2',4'-bridged nucleosides for hcv infection |
| US20140140952A1 (en) | 2012-11-14 | 2014-05-22 | Idenix Pharmaceuticals, Inc. | D-Alanine Ester of Sp-Nucleoside Analog |
| US20140140951A1 (en) | 2012-11-14 | 2014-05-22 | Idenix Pharmaceuticals, Inc. | D-Alanine Ester of Rp-Nucleoside Analog |
| WO2014099941A1 (en) | 2012-12-19 | 2014-06-26 | Idenix Pharmaceuticals, Inc. | 4'-fluoro nucleosides for the treatment of hcv |
| WO2014137930A1 (en) | 2013-03-04 | 2014-09-12 | Idenix Pharmaceuticals, Inc. | Thiophosphate nucleosides for the treatment of hcv |
| US9309275B2 (en) | 2013-03-04 | 2016-04-12 | Idenix Pharmaceuticals Llc | 3′-deoxy nucleosides for the treatment of HCV |
| US20140271547A1 (en) | 2013-03-13 | 2014-09-18 | Idenix Pharmaceuticals, Inc. | Amino acid phosphoramidate pronucleotides of 2'-cyano, azido and amino nucleosides for the treatment of hcv |
| WO2014165542A1 (en) | 2013-04-01 | 2014-10-09 | Idenix Pharmaceuticals, Inc. | 2',4'-fluoro nucleosides for the treatment of hcv |
| WO2014197578A1 (en) | 2013-06-05 | 2014-12-11 | Idenix Pharmaceuticals, Inc. | 1',4'-thio nucleosides for the treatment of hcv |
| US9765107B2 (en) | 2013-06-18 | 2017-09-19 | Merck Sharp & Dohme Corp. | Cyclic phosphonate substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| US20150037282A1 (en) | 2013-08-01 | 2015-02-05 | Idenix Pharmaceuticals, Inc. | D-amino acid phosphoramidate pronucleotides of halogeno pyrimidine compounds for liver disease |
-
2007
- 2007-12-27 US US12/005,937 patent/US7951789B2/en active Active
- 2007-12-27 US US12/005,938 patent/US20080261913A1/en not_active Abandoned
- 2007-12-28 EP EP07872645.2A patent/EP2120566B1/en active Active
- 2007-12-28 TW TW096150890A patent/TWI412368B/zh not_active IP Right Cessation
- 2007-12-28 JP JP2009544101A patent/JP2010514769A/ja active Pending
- 2007-12-28 SG SG2011096476A patent/SG177923A1/en unknown
- 2007-12-28 TW TW096150931A patent/TW200844108A/zh unknown
- 2007-12-28 UA UAA200907835A patent/UA96975C2/ru unknown
- 2007-12-28 ME MEP-2009-232A patent/ME00816B/me unknown
- 2007-12-28 EP EP07868086A patent/EP2107909A4/en not_active Withdrawn
- 2007-12-28 AU AU2007339226A patent/AU2007339226B2/en not_active Ceased
- 2007-12-28 CA CA002673722A patent/CA2673722A1/en not_active Abandoned
- 2007-12-28 WO PCT/US2007/026408 patent/WO2008082601A2/en active Application Filing
- 2007-12-28 CA CA002673776A patent/CA2673776A1/en not_active Abandoned
- 2007-12-28 MX MX2009006851A patent/MX2009006851A/es active IP Right Grant
- 2007-12-28 JP JP2009544100A patent/JP5578853B2/ja not_active Expired - Fee Related
- 2007-12-28 WO PCT/US2007/026409 patent/WO2008082602A2/en active Application Filing
- 2007-12-28 MY MYPI20092752A patent/MY169568A/en unknown
- 2007-12-28 RU RU2012132218/04A patent/RU2525392C2/ru not_active IP Right Cessation
- 2007-12-28 KR KR1020097015742A patent/KR101508018B1/ko not_active IP Right Cessation
- 2007-12-28 BR BRPI0720637-2A2A patent/BRPI0720637A2/pt active Search and Examination
- 2007-12-28 NZ NZ577999A patent/NZ577999A/en not_active IP Right Cessation
-
2008
- 2008-04-25 US US12/150,327 patent/US7902202B2/en active Active
-
2009
- 2009-07-08 NO NO20092585A patent/NO20092585L/no not_active Application Discontinuation
- 2009-07-28 IL IL200116A patent/IL200116A0/en unknown
-
2011
- 2011-05-26 US US13/116,890 patent/US8691788B2/en active Active
-
2014
- 2014-03-04 US US14/197,078 patent/US9249173B2/en active Active
- 2014-03-24 JP JP2014060859A patent/JP2014139218A/ja active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996040164A1 (en) * | 1995-06-07 | 1996-12-19 | Emory University | Nucleosides with anti-hepatitis b virus activity |
| WO2002057287A2 (en) * | 2001-01-22 | 2002-07-25 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2003072757A2 (en) * | 2002-02-28 | 2003-09-04 | Biota, Inc. | Nucleotide mimics and their prodrugs |
Non-Patent Citations (2)
| Title |
|---|
| M.Д. МАШКОВСКИЙ, "Лекарственные средства, пособие для врачей", 1993, часть 1, Москва, "Медицина" * |
| THIERRY BELTRAN ET AL, BIOORGANIC&MEDICINAL CHEMISTRY LETTERS, 2001, 11(13), 1775-1777, * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11219603B2 (en) | 2015-12-01 | 2022-01-11 | Maxwell Biotech Group Ltd. | Stable dosage form of etidronate-cytarabine conjugate, and use thereof |
| RU2632431C2 (ru) * | 2016-06-29 | 2017-10-04 | Федеральное государственное бюджетное учреждение "Национальный медицинский исследовательский радиологический центр" Министерства здравоохранения Российской Федерации (ФГБУ "НМИРЦ" Минздрава России) | Гидрогель для получения композиционных материалов с антибактериальной активностью для замещения костно-хрящевых дефектов методом 3d печати |
| WO2018016999A1 (ru) * | 2016-07-20 | 2018-01-25 | Общество С Ограниченной Ответственностью "Остерос Биомедика" | Препарат для лечения костных поражений, вызванных злокачественными новообразованиями |
| EA030671B1 (ru) * | 2016-07-20 | 2018-09-28 | Общество С Ограниченной Ответственностью "Остерос Биомедика" | Препарат для лечения костных поражений, вызванных злокачественными новообразованиями |
| AU2017297989B2 (en) * | 2016-07-20 | 2019-10-03 | Maxwell Biotech Group Ltd. | Pharmaceutical preparation for treating bone lesions caused by malignant neoplasms |
| US11045487B2 (en) | 2016-07-20 | 2021-06-29 | Maxwell Biotech Group Ltd. | Pharmaceutical preparation treating bone lesions caused by malignant neoplasms |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2525392C2 (ru) | Соединения и фармацевтические композиции для лечения вирусных инфекций | |
| JP6069410B2 (ja) | フラビウィルス科ウィルス感染治療用の修飾2’および3’−ヌクレオシドプロドラッグ | |
| JP5087211B2 (ja) | フラビウィルス感染治療のための2′および3′−ヌクレオシドプロドラッグ | |
| US20070042990A1 (en) | 2' and 3'-nucleoside prodrugs for treating Flaviviridae infections | |
| AU2013329521A1 (en) | 2'-chloro nucleoside analogs for HCV infection | |
| KR20060020649A (ko) | 변형 불소화 뉴클레오시드 유사체 | |
| CN101626683B (zh) | 用于治疗病毒感染的化合物和药物组合物 | |
| RU2466729C2 (ru) | Соединения и фармацевтические композиции для лечения вирусных инфекций | |
| AU2013200925B2 (en) | Compounds and pharmaceutical compositions for the treatment of viral infections |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20161229 |