JP7162897B2 - 融合タンパク質 - Google Patents
融合タンパク質 Download PDFInfo
- Publication number
- JP7162897B2 JP7162897B2 JP2019526938A JP2019526938A JP7162897B2 JP 7162897 B2 JP7162897 B2 JP 7162897B2 JP 2019526938 A JP2019526938 A JP 2019526938A JP 2019526938 A JP2019526938 A JP 2019526938A JP 7162897 B2 JP7162897 B2 JP 7162897B2
- Authority
- JP
- Japan
- Prior art keywords
- scfv
- amino acids
- peptide
- intracellular
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 108020001507 fusion proteins Proteins 0.000 title claims description 72
- 102000037865 fusion proteins Human genes 0.000 title claims description 70
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 208
- 150000001413 amino acids Chemical class 0.000 claims description 181
- 230000003834 intracellular effect Effects 0.000 claims description 149
- 230000000087 stabilizing effect Effects 0.000 claims description 107
- 230000027455 binding Effects 0.000 claims description 75
- 239000000427 antigen Substances 0.000 claims description 70
- 108091007433 antigens Proteins 0.000 claims description 68
- 102000036639 antigens Human genes 0.000 claims description 68
- 230000002378 acidificating effect Effects 0.000 claims description 41
- 108091033319 polynucleotide Proteins 0.000 claims description 34
- 239000002157 polynucleotide Substances 0.000 claims description 34
- 102000040430 polynucleotide Human genes 0.000 claims description 34
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 27
- 239000013604 expression vector Substances 0.000 claims description 19
- 238000004519 manufacturing process Methods 0.000 claims description 4
- 229940024606 amino acid Drugs 0.000 description 177
- 235000001014 amino acid Nutrition 0.000 description 177
- 210000004027 cell Anatomy 0.000 description 136
- 108090000623 proteins and genes Proteins 0.000 description 63
- 102000004169 proteins and genes Human genes 0.000 description 53
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 50
- 235000018102 proteins Nutrition 0.000 description 50
- 239000013598 vector Substances 0.000 description 42
- 241000699670 Mus sp. Species 0.000 description 41
- 102000004196 processed proteins & peptides Human genes 0.000 description 39
- 230000014509 gene expression Effects 0.000 description 37
- 229960003638 dopamine Drugs 0.000 description 25
- 230000006870 function Effects 0.000 description 25
- 241000699666 Mus <mouse, genus> Species 0.000 description 24
- 238000000034 method Methods 0.000 description 23
- 239000000872 buffer Substances 0.000 description 22
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 20
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 17
- 201000010099 disease Diseases 0.000 description 16
- 230000002209 hydrophobic effect Effects 0.000 description 15
- 210000002569 neuron Anatomy 0.000 description 15
- 108020004414 DNA Proteins 0.000 description 14
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 14
- 229960002429 proline Drugs 0.000 description 14
- HVLSXIKZNLPZJJ-TXZCQADKSA-N HA peptide Chemical compound C([C@@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 HVLSXIKZNLPZJJ-TXZCQADKSA-N 0.000 description 13
- 210000004556 brain Anatomy 0.000 description 13
- 210000003523 substantia nigra Anatomy 0.000 description 13
- 239000013607 AAV vector Substances 0.000 description 12
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 12
- 230000002776 aggregation Effects 0.000 description 12
- 238000004220 aggregation Methods 0.000 description 12
- 238000001690 micro-dialysis Methods 0.000 description 12
- 230000007935 neutral effect Effects 0.000 description 12
- 239000008194 pharmaceutical composition Substances 0.000 description 12
- 239000013603 viral vector Substances 0.000 description 12
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 11
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 11
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 11
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 11
- 229960001230 asparagine Drugs 0.000 description 11
- 235000009582 asparagine Nutrition 0.000 description 11
- 238000001514 detection method Methods 0.000 description 11
- 239000012634 fragment Substances 0.000 description 11
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 11
- 229960002743 glutamine Drugs 0.000 description 11
- 239000007924 injection Substances 0.000 description 11
- 238000002347 injection Methods 0.000 description 11
- 229960004441 tyrosine Drugs 0.000 description 11
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 11
- 238000002965 ELISA Methods 0.000 description 10
- 238000002474 experimental method Methods 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- 230000001965 increasing effect Effects 0.000 description 10
- 239000011780 sodium chloride Substances 0.000 description 10
- 239000006228 supernatant Substances 0.000 description 10
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 9
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 9
- 102000003802 alpha-Synuclein Human genes 0.000 description 9
- 108090000185 alpha-Synuclein Proteins 0.000 description 9
- 210000005064 dopaminergic neuron Anatomy 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 230000004927 fusion Effects 0.000 description 9
- 235000014304 histidine Nutrition 0.000 description 9
- 210000001577 neostriatum Anatomy 0.000 description 9
- 238000011160 research Methods 0.000 description 9
- 229960004295 valine Drugs 0.000 description 9
- 239000004474 valine Substances 0.000 description 9
- 239000004471 Glycine Substances 0.000 description 8
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 8
- 206010028980 Neoplasm Diseases 0.000 description 8
- 229920004890 Triton X-100 Polymers 0.000 description 8
- 239000013504 Triton X-100 Substances 0.000 description 8
- 230000003247 decreasing effect Effects 0.000 description 8
- 229960002449 glycine Drugs 0.000 description 8
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 8
- 208000015181 infectious disease Diseases 0.000 description 8
- 210000004962 mammalian cell Anatomy 0.000 description 8
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 8
- 239000002953 phosphate buffered saline Substances 0.000 description 8
- 238000010825 rotarod performance test Methods 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- 241000588724 Escherichia coli Species 0.000 description 7
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 7
- 241000700605 Viruses Species 0.000 description 7
- 239000011324 bead Substances 0.000 description 7
- 210000004899 c-terminal region Anatomy 0.000 description 7
- 239000011575 calcium Substances 0.000 description 7
- 239000002299 complementary DNA Substances 0.000 description 7
- 230000001086 cytosolic effect Effects 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- 238000001262 western blot Methods 0.000 description 7
- 201000009030 Carcinoma Diseases 0.000 description 6
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 6
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 6
- 239000002033 PVDF binder Substances 0.000 description 6
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 235000003704 aspartic acid Nutrition 0.000 description 6
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 6
- 238000005119 centrifugation Methods 0.000 description 6
- 210000004748 cultured cell Anatomy 0.000 description 6
- 230000001419 dependent effect Effects 0.000 description 6
- 108010048367 enhanced green fluorescent protein Proteins 0.000 description 6
- 235000013922 glutamic acid Nutrition 0.000 description 6
- 239000004220 glutamic acid Substances 0.000 description 6
- 238000012405 in silico analysis Methods 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 239000013612 plasmid Substances 0.000 description 6
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 6
- GUUBJKMBDULZTE-UHFFFAOYSA-M potassium;2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid;hydroxide Chemical compound [OH-].[K+].OCCN1CCN(CCS(O)(=O)=O)CC1 GUUBJKMBDULZTE-UHFFFAOYSA-M 0.000 description 6
- 239000000523 sample Substances 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 5
- 102000005720 Glutathione transferase Human genes 0.000 description 5
- 108010070675 Glutathione transferase Proteins 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- 108091028043 Nucleic acid sequence Proteins 0.000 description 5
- 230000000259 anti-tumor effect Effects 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 238000010276 construction Methods 0.000 description 5
- 238000010586 diagram Methods 0.000 description 5
- 239000005090 green fluorescent protein Substances 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 230000007326 intracellular aggregation Effects 0.000 description 5
- 238000002955 isolation Methods 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 5
- 229920001184 polypeptide Polymers 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 230000001105 regulatory effect Effects 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- 238000001890 transfection Methods 0.000 description 5
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 4
- DLZKEQQWXODGGZ-KCJUWKMLSA-N 2-[[(2r)-2-[[(2s)-2-amino-3-(4-hydroxyphenyl)propanoyl]amino]propanoyl]amino]acetic acid Chemical compound OC(=O)CNC(=O)[C@@H](C)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 DLZKEQQWXODGGZ-KCJUWKMLSA-N 0.000 description 4
- 241001655883 Adeno-associated virus - 1 Species 0.000 description 4
- 241000701022 Cytomegalovirus Species 0.000 description 4
- 101150105104 Kras gene Proteins 0.000 description 4
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 4
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 4
- 102100036407 Thioredoxin Human genes 0.000 description 4
- 238000010171 animal model Methods 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 210000000805 cytoplasm Anatomy 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 238000002513 implantation Methods 0.000 description 4
- 230000001939 inductive effect Effects 0.000 description 4
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 4
- 230000013016 learning Effects 0.000 description 4
- 239000003550 marker Substances 0.000 description 4
- 206010061289 metastatic neoplasm Diseases 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 108060008226 thioredoxin Proteins 0.000 description 4
- 229920000936 Agarose Polymers 0.000 description 3
- 239000012110 Alexa Fluor 594 Substances 0.000 description 3
- 239000012099 Alexa Fluor family Substances 0.000 description 3
- 206010002091 Anaesthesia Diseases 0.000 description 3
- 241000283690 Bos taurus Species 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- 108020004705 Codon Proteins 0.000 description 3
- ZGTMUACCHSMWAC-UHFFFAOYSA-L EDTA disodium salt (anhydrous) Chemical compound [Na+].[Na+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O ZGTMUACCHSMWAC-UHFFFAOYSA-L 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 241000672609 Escherichia coli BL21 Species 0.000 description 3
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 3
- 206010025323 Lymphomas Diseases 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 208000018737 Parkinson disease Diseases 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 102000019355 Synuclein Human genes 0.000 description 3
- 108050006783 Synuclein Proteins 0.000 description 3
- 208000009956 adenocarcinoma Diseases 0.000 description 3
- 230000037005 anaesthesia Effects 0.000 description 3
- 238000000540 analysis of variance Methods 0.000 description 3
- 230000003542 behavioural effect Effects 0.000 description 3
- 238000001574 biopsy Methods 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 239000004202 carbamide Substances 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 238000001378 electrochemiluminescence detection Methods 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 108091006047 fluorescent proteins Proteins 0.000 description 3
- 102000034287 fluorescent proteins Human genes 0.000 description 3
- 238000010230 functional analysis Methods 0.000 description 3
- 238000003364 immunohistochemistry Methods 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 229960002725 isoflurane Drugs 0.000 description 3
- 230000004807 localization Effects 0.000 description 3
- 238000004020 luminiscence type Methods 0.000 description 3
- 239000012139 lysis buffer Substances 0.000 description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 3
- 238000000691 measurement method Methods 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 230000028161 membrane depolarization Effects 0.000 description 3
- 230000001394 metastastic effect Effects 0.000 description 3
- 239000013642 negative control Substances 0.000 description 3
- 230000001537 neural effect Effects 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 description 3
- 230000001717 pathogenic effect Effects 0.000 description 3
- 230000006798 recombination Effects 0.000 description 3
- 108091008146 restriction endonucleases Proteins 0.000 description 3
- ZNJHFNUEQDVFCJ-UHFFFAOYSA-M sodium;2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid;hydroxide Chemical compound [OH-].[Na+].OCCN1CCN(CCS(O)(=O)=O)CC1 ZNJHFNUEQDVFCJ-UHFFFAOYSA-M 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 238000012549 training Methods 0.000 description 3
- 230000010474 transient expression Effects 0.000 description 3
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 3
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- 241000251468 Actinopterygii Species 0.000 description 2
- 206010000871 Acute monocytic leukaemia Diseases 0.000 description 2
- 241000202702 Adeno-associated virus - 3 Species 0.000 description 2
- 241000271566 Aves Species 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 241000700198 Cavia Species 0.000 description 2
- 206010008025 Cerebellar ataxia Diseases 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- 206010052804 Drug tolerance Diseases 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 241001198387 Escherichia coli BL21(DE3) Species 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 208000032612 Glial tumor Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 101000834898 Homo sapiens Alpha-synuclein Proteins 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- 102000016252 Huntingtin Human genes 0.000 description 2
- 108050004784 Huntingtin Proteins 0.000 description 2
- 208000027747 Kennedy disease Diseases 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- 239000012097 Lipofectamine 2000 Substances 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- 108010076504 Protein Sorting Signals Proteins 0.000 description 2
- 239000012722 SDS sample buffer Substances 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- 241000700584 Simplexvirus Species 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- 241000282887 Suidae Species 0.000 description 2
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 238000000862 absorption spectrum Methods 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 239000002390 adhesive tape Substances 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 230000006399 behavior Effects 0.000 description 2
- 238000009227 behaviour therapy Methods 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000005013 brain tissue Anatomy 0.000 description 2
- UDSAIICHUKSCKT-UHFFFAOYSA-N bromophenol blue Chemical compound C1=C(Br)C(O)=C(Br)C=C1C1(C=2C=C(Br)C(O)=C(Br)C=2)C2=CC=CC=C2S(=O)(=O)O1 UDSAIICHUKSCKT-UHFFFAOYSA-N 0.000 description 2
- AIYUHDOJVYHVIT-UHFFFAOYSA-M caesium chloride Chemical compound [Cl-].[Cs+] AIYUHDOJVYHVIT-UHFFFAOYSA-M 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 230000005907 cancer growth Effects 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- 230000009087 cell motility Effects 0.000 description 2
- 208000015114 central nervous system disease Diseases 0.000 description 2
- 230000002490 cerebral effect Effects 0.000 description 2
- 210000003792 cranial nerve Anatomy 0.000 description 2
- 239000003479 dental cement Substances 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 210000002257 embryonic structure Anatomy 0.000 description 2
- 210000001163 endosome Anatomy 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 238000002875 fluorescence polarization Methods 0.000 description 2
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 2
- 108010021843 fluorescent protein 583 Proteins 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 230000005714 functional activity Effects 0.000 description 2
- 206010017758 gastric cancer Diseases 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 230000026781 habituation Effects 0.000 description 2
- 230000000984 immunochemical effect Effects 0.000 description 2
- 238000003365 immunocytochemistry Methods 0.000 description 2
- 238000003125 immunofluorescent labeling Methods 0.000 description 2
- 230000016784 immunoglobulin production Effects 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 229960003136 leucine Drugs 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 description 2
- 210000001259 mesencephalon Anatomy 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 229960004452 methionine Drugs 0.000 description 2
- 230000004660 morphological change Effects 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 208000004019 papillary adenocarcinoma Diseases 0.000 description 2
- 239000013600 plasmid vector Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 210000000063 presynaptic terminal Anatomy 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 238000005215 recombination Methods 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 229960001153 serine Drugs 0.000 description 2
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 229940094937 thioredoxin Drugs 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 208000010576 undifferentiated carcinoma Diseases 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 238000012447 xenograft mouse model Methods 0.000 description 2
- YMHOBZXQZVXHBM-UHFFFAOYSA-N 2,5-dimethoxy-4-bromophenethylamine Chemical compound COC1=CC(CCN)=C(OC)C=C1Br YMHOBZXQZVXHBM-UHFFFAOYSA-N 0.000 description 1
- FMYBFLOWKQRBST-UHFFFAOYSA-N 2-[bis(carboxymethyl)amino]acetic acid;nickel Chemical compound [Ni].OC(=O)CN(CC(O)=O)CC(O)=O FMYBFLOWKQRBST-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 108020005065 3' Flanking Region Proteins 0.000 description 1
- 108020005345 3' Untranslated Regions Proteins 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 108020005029 5' Flanking Region Proteins 0.000 description 1
- HLXHCNWEVQNNKA-UHFFFAOYSA-N 5-methoxy-2,3-dihydro-1h-inden-2-amine Chemical compound COC1=CC=C2CC(N)CC2=C1 HLXHCNWEVQNNKA-UHFFFAOYSA-N 0.000 description 1
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 1
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 1
- 108010022752 Acetylcholinesterase Proteins 0.000 description 1
- 102000012440 Acetylcholinesterase Human genes 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 241000321096 Adenoides Species 0.000 description 1
- 208000009746 Adult T-Cell Leukemia-Lymphoma Diseases 0.000 description 1
- 208000016683 Adult T-cell leukemia/lymphoma Diseases 0.000 description 1
- 102000007698 Alcohol dehydrogenase Human genes 0.000 description 1
- 108010021809 Alcohol dehydrogenase Proteins 0.000 description 1
- 239000012103 Alexa Fluor 488 Substances 0.000 description 1
- 101000935845 Aliivibrio fischeri Blue fluorescence protein Proteins 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 101000760456 Anguilla japonica Bilirubin-inducible fluorescent protein UnaG Proteins 0.000 description 1
- 235000002198 Annona diversifolia Nutrition 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000228212 Aspergillus Species 0.000 description 1
- 102000014461 Ataxins Human genes 0.000 description 1
- 108010078286 Ataxins Proteins 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 206010003840 Autonomic nervous system imbalance Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 208000020925 Bipolar disease Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 208000019337 Bowen disease of the skin Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 201000011057 Breast sarcoma Diseases 0.000 description 1
- 208000003170 Bronchiolo-Alveolar Adenocarcinoma Diseases 0.000 description 1
- 208000029402 Bulbospinal muscular atrophy Diseases 0.000 description 1
- 206010068597 Bulbospinal muscular atrophy congenital Diseases 0.000 description 1
- 241000282832 Camelidae Species 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 108090000397 Caspase 3 Proteins 0.000 description 1
- 102100029855 Caspase-3 Human genes 0.000 description 1
- 102100035882 Catalase Human genes 0.000 description 1
- 108010053835 Catalase Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 241000606161 Chlamydia Species 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 241001573498 Compacta Species 0.000 description 1
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 201000008163 Dentatorubral pallidoluysian atrophy Diseases 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 206010061825 Duodenal neoplasm Diseases 0.000 description 1
- 102100037024 E3 ubiquitin-protein ligase XIAP Human genes 0.000 description 1
- 108091005941 EBFP Proteins 0.000 description 1
- 108091005942 ECFP Proteins 0.000 description 1
- 241001115402 Ebolavirus Species 0.000 description 1
- 101000935842 Escherichia coli O127:H6 (strain E2348/69 / EPEC) Major structural subunit of bundle-forming pilus Proteins 0.000 description 1
- 241001524679 Escherichia virus M13 Species 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 208000010368 Extramammary Paget Disease Diseases 0.000 description 1
- 101710163305 Fibril protein Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 208000004463 Follicular Adenocarcinoma Diseases 0.000 description 1
- 201000011240 Frontotemporal dementia Diseases 0.000 description 1
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 206010067807 Gingival cancer Diseases 0.000 description 1
- 239000004366 Glucose oxidase Substances 0.000 description 1
- 108010015776 Glucose oxidase Proteins 0.000 description 1
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 208000005331 Hepatitis D Diseases 0.000 description 1
- 108010093488 His-His-His-His-His-His Proteins 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000588302 Homo sapiens Nuclear factor erythroid 2-related factor 2 Proteins 0.000 description 1
- 101000741544 Homo sapiens Properdin Proteins 0.000 description 1
- 101001079872 Homo sapiens RING finger protein 112 Proteins 0.000 description 1
- 101000821100 Homo sapiens Synapsin-1 Proteins 0.000 description 1
- 206010020460 Human T-cell lymphotropic virus type I infection Diseases 0.000 description 1
- 241000714260 Human T-lymphotropic virus 1 Species 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- 241000282838 Lama Species 0.000 description 1
- 206010023825 Laryngeal cancer Diseases 0.000 description 1
- 208000020358 Learning disease Diseases 0.000 description 1
- 241000589248 Legionella Species 0.000 description 1
- 208000007764 Legionnaires' Disease Diseases 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- 208000000265 Lobular Carcinoma Diseases 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 1
- 101150056129 M4 gene Proteins 0.000 description 1
- 102000013460 Malate Dehydrogenase Human genes 0.000 description 1
- 108010026217 Malate Dehydrogenase Proteins 0.000 description 1
- 208000007054 Medullary Carcinoma Diseases 0.000 description 1
- 208000037196 Medullary thyroid carcinoma Diseases 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 241000713333 Mouse mammary tumor virus Species 0.000 description 1
- 208000016285 Movement disease Diseases 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 208000001089 Multiple system atrophy Diseases 0.000 description 1
- 241000711408 Murine respirovirus Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 206010028289 Muscle atrophy Diseases 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 1
- 206010028729 Nasal cavity cancer Diseases 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 206010060860 Neurological symptom Diseases 0.000 description 1
- 206010029333 Neurosis Diseases 0.000 description 1
- 208000010505 Nose Neoplasms Diseases 0.000 description 1
- 102100031701 Nuclear factor erythroid 2-related factor 2 Human genes 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 108700026244 Open Reading Frames Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 108010087702 Penicillinase Proteins 0.000 description 1
- 108010079855 Peptide Aptamers Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 208000009565 Pharyngeal Neoplasms Diseases 0.000 description 1
- 206010034811 Pharyngeal cancer Diseases 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 206010035603 Pleural mesothelioma Diseases 0.000 description 1
- 208000024777 Prion disease Diseases 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 208000035755 Psychosomatic disease Diseases 0.000 description 1
- 108010010469 Qa-SNARE Proteins Proteins 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 101000839071 Rattus norvegicus Heterogeneous nuclear ribonucleoprotein M Proteins 0.000 description 1
- 101100480719 Rattus norvegicus Mapt gene Proteins 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 241000208422 Rhododendron Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 108091058545 Secretory proteins Proteins 0.000 description 1
- 102000040739 Secretory proteins Human genes 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 1
- 206010054184 Small intestine carcinoma Diseases 0.000 description 1
- 208000009415 Spinocerebellar Ataxias Diseases 0.000 description 1
- 208000010112 Spinocerebellar Degenerations Diseases 0.000 description 1
- 208000034254 Squamous cell carcinoma of the cervix uteri Diseases 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- 238000003639 Student–Newman–Keuls (SNK) method Methods 0.000 description 1
- 102000050389 Syntaxin Human genes 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 208000028530 T-cell lymphoblastic leukemia/lymphoma Diseases 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 206010062129 Tongue neoplasm Diseases 0.000 description 1
- 206010044565 Tremor Diseases 0.000 description 1
- 241000223104 Trypanosoma Species 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108010046334 Urease Proteins 0.000 description 1
- 206010046770 Uterine carcinoma in situ Diseases 0.000 description 1
- 241000545067 Venus Species 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 241000710886 West Nile virus Species 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- 241001492404 Woodchuck hepatitis virus Species 0.000 description 1
- 208000006269 X-Linked Bulbo-Spinal Atrophy Diseases 0.000 description 1
- 108700031544 X-Linked Inhibitor of Apoptosis Proteins 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 229940022698 acetylcholinesterase Drugs 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 208000036676 acute undifferentiated leukemia Diseases 0.000 description 1
- 101150063416 add gene Proteins 0.000 description 1
- 210000002534 adenoid Anatomy 0.000 description 1
- 201000008395 adenosquamous carcinoma Diseases 0.000 description 1
- 201000006966 adult T-cell leukemia Diseases 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 230000009833 antibody interaction Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000004900 autophagic degradation Effects 0.000 description 1
- 201000004562 autosomal dominant cerebellar ataxia Diseases 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 244000052616 bacterial pathogen Species 0.000 description 1
- 102000005936 beta-Galactosidase Human genes 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 208000028683 bipolar I disease Diseases 0.000 description 1
- 206010005084 bladder transitional cell carcinoma Diseases 0.000 description 1
- 201000001528 bladder urothelial carcinoma Diseases 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 201000003714 breast lobular carcinoma Diseases 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 208000002458 carcinoid tumor Diseases 0.000 description 1
- 208000011892 carcinosarcoma of the corpus uteri Diseases 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 208000025434 cerebellar degeneration Diseases 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 201000006612 cervical squamous cell carcinoma Diseases 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 235000013330 chicken meat Nutrition 0.000 description 1
- 201000002797 childhood leukemia Diseases 0.000 description 1
- 229960005091 chloramphenicol Drugs 0.000 description 1
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 239000013599 cloning vector Substances 0.000 description 1
- 238000000749 co-immunoprecipitation Methods 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 239000013601 cosmid vector Substances 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 239000000385 dialysis solution Substances 0.000 description 1
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 230000003291 dopaminomimetic effect Effects 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 206010013781 dry mouth Diseases 0.000 description 1
- 201000000312 duodenum cancer Diseases 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 238000000835 electrochemical detection Methods 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 208000037828 epithelial carcinoma Diseases 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 239000013613 expression plasmid Substances 0.000 description 1
- 210000001723 extracellular space Anatomy 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 201000010175 gallbladder cancer Diseases 0.000 description 1
- 229940116332 glucose oxidase Drugs 0.000 description 1
- 235000019420 glucose oxidase Nutrition 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 208000005252 hepatitis A Diseases 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 201000010284 hepatitis E Diseases 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 210000004295 hippocampal neuron Anatomy 0.000 description 1
- 150000002411 histidines Chemical class 0.000 description 1
- 239000011539 homogenization buffer Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 102000056115 human SYN1 Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 238000002991 immunohistochemical analysis Methods 0.000 description 1
- 238000011532 immunohistochemical staining Methods 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 238000012744 immunostaining Methods 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000004068 intracellular signaling Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 206010073096 invasive lobular breast carcinoma Diseases 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 206010023841 laryngeal neoplasm Diseases 0.000 description 1
- 201000003723 learning disability Diseases 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 238000001638 lipofection Methods 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 208000025036 lymphosarcoma Diseases 0.000 description 1
- 201000000564 macroglobulinemia Diseases 0.000 description 1
- 201000004792 malaria Diseases 0.000 description 1
- 208000024407 malignant pericardial mesothelioma Diseases 0.000 description 1
- 201000009023 maxillary cancer Diseases 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 206010027191 meningioma Diseases 0.000 description 1
- 230000006371 metabolic abnormality Effects 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- 201000010879 mucinous adenocarcinoma Diseases 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 230000020763 muscle atrophy Effects 0.000 description 1
- 201000000585 muscular atrophy Diseases 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 208000001611 myxosarcoma Diseases 0.000 description 1
- 208000037830 nasal cancer Diseases 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 208000025189 neoplasm of testis Diseases 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 208000015238 neurotic disease Diseases 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 238000012346 open field test Methods 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 208000021090 palsy Diseases 0.000 description 1
- 201000010198 papillary carcinoma Diseases 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 229950009506 penicillinase Drugs 0.000 description 1
- 201000004266 pericardial mesothelioma Diseases 0.000 description 1
- 208000027232 peripheral nervous system disease Diseases 0.000 description 1
- 201000002513 peritoneal mesothelioma Diseases 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 230000001144 postural effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000004853 protein function Effects 0.000 description 1
- 238000010379 pull-down assay Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000000384 rearing effect Effects 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 108010054624 red fluorescent protein Proteins 0.000 description 1
- 238000002165 resonance energy transfer Methods 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 201000006845 reticulosarcoma Diseases 0.000 description 1
- 208000029922 reticulum cell sarcoma Diseases 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 208000020352 skin basal cell carcinoma Diseases 0.000 description 1
- 201000010106 skin squamous cell carcinoma Diseases 0.000 description 1
- 210000003625 skull Anatomy 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 125000002298 terpene group Chemical group 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 208000030901 thyroid gland follicular carcinoma Diseases 0.000 description 1
- 208000013818 thyroid gland medullary carcinoma Diseases 0.000 description 1
- 201000006134 tongue cancer Diseases 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 108010072106 tumstatin (74-98) Proteins 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 238000011870 unpaired t-test Methods 0.000 description 1
- 201000000334 ureter transitional cell carcinoma Diseases 0.000 description 1
- 201000007554 urethra adenocarcinoma Diseases 0.000 description 1
- 201000005645 urethra squamous cell carcinoma Diseases 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 201000005290 uterine carcinosarcoma Diseases 0.000 description 1
- 208000037965 uterine sarcoma Diseases 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Images


















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K19/00—Hybrid peptides, i.e. peptides covalently bound to nucleic acids, or non-covalently bound protein-protein complexes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/62—DNA sequences coding for fusion proteins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/40—Fusion polypeptide containing a tag for immunodetection, or an epitope for immunisation
- C07K2319/43—Fusion polypeptide containing a tag for immunodetection, or an epitope for immunisation containing a FLAG-tag
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biomedical Technology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physics & Mathematics (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Peptides Or Proteins (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Description
1)細胞内安定化ペプチドと、抗原結合性ペプチドとを含む融合タンパク質であって、
上記細胞内安定化ペプチドは、10~39個のアミノ酸からなり、かつ、当該アミノ酸の少なくとも45%が酸性アミノ酸であり、
上記抗原結合性ペプチドは、重鎖CDR1、重鎖CDR2、重鎖CDR3、軽鎖CDR1、軽鎖CDR2、および軽鎖CDR3のうちの少なくとも1つを含む、
融合タンパク質。
2)上記細胞内安定化ペプチドは、上記アミノ酸の28%以下が塩基性アミノ酸であるか、塩基性アミノ酸を含まない、1)に記載の融合タンパク質。
3)上記細胞内安定化ペプチドは、上記塩基性アミノ酸を含まない、2)に記載の融合タンパク質。
4)上記細胞内安定化ペプチドは、上記塩基性アミノ酸として少なくとも1個のヒスチジンを含む、2)に記載の融合タンパク質。
5)上記細胞内安定化ペプチドは、14~25個のアミノ酸からなり、当該アミノ酸の少なくとも50%が酸性アミノ酸であり、
上記細胞内安定化ペプチドにおいて、上記酸性アミノ酸が8個以上連続した部分、および、酸性アミノ酸以外のアミノ酸が4個以上連続した部分が存在しない、1)~4)の何れか1つに記載の融合タンパク質。
6)上記細胞内安定化ペプチドにおいて、上記酸性アミノ酸が3個以上連続した部分、および、酸性アミノ酸以外のアミノ酸が3個以上連続した部分が存在しない、5)に記載の融合タンパク質。
7)上記細胞内安定化ペプチドは、上記酸性アミノ酸をXA、酸性アミノ酸以外のアミノ酸をXnとしたときに、
以下のアミノ酸配列(1)または(2):
-Xn-XA-XA-Xn-XA-(XAまたはXn)-Xn-XA-(XAまたはXn)-Xn-(XAまたはXn)-XA- ・・・・・・(1)、
-Xn-XA-Xn-XA-Xn-XA-XA-Xn-XA-Xn-Xn-XA-XA-Xn-Xn-XA- ・・・・・・(2)、
を含んでなる、1)~6)の何れか1つに記載の融合タンパク質。
8)XAはアスパラギン酸またはグルタミン酸であり、Xnはアスパラギン、グルタミン、プロリン、チロシンおよびバリンからなる群より選択される何れかのアミノ酸である、7)に記載の融合タンパク質。
9)上記1)~8)の何れか1つに記載の融合タンパク質をコードするポリヌクレオチド。
10)上記9)に記載のポリヌクレオチドを含む発現ベクター。
11)細胞内において抗原結合性ペプチドを発現させる細胞を製造する方法であって、9)に記載のポリヌクレオチドまたは10)に記載の発現ベクターを細胞に導入する工程を含む、製造方法。
12)上記1)~8)の何れか1つに記載の融合タンパク質を発現する細胞。
(ペプチド)
本明細書において、「ペプチド」は、「ポリペプチド」または「タンパク質」とも換言し得る。「ペプチド」は、アミノ酸がペプチド結合してなる構造を含むが、さらに、例えば、糖鎖、またはイソプレノイド基などの構造を含んでいてもよい。「ペプチド」は、特に明記しない場合は、天然に存在するアミノ酸と同様に機能することができる、天然に存在するアミノ酸の既知の類似体を含有するペプチドを包含する。
本明細書において、「酸性アミノ酸」とはその等電点が3.99以下のアミノ酸を指す。アミノ酸は天然に存在するものであっても、天然に存在するアミノ酸の類似体であってもよい。天然に存在する酸性アミノ酸としては、アスパラギン酸とグルタミン酸とが挙げられる。
本明細書において、「塩基性アミノ酸」とはその等電点が7.40以上のアミノ酸を指す。アミノ酸は天然に存在するものであっても、天然に存在するアミノ酸の類似体であってもよい。天然に存在する塩基性アミノ酸としては、ヒスチジン、リジン、およびアルギニンが挙げられる。
本明細書において、アミノ酸自体のpHに着目した場合に、上記の酸性アミノ酸の定義にも、塩基性アミノ酸の定義にも当てはまらないアミノ酸を、「酸性アミノ酸および塩基性アミノ酸以外のアミノ酸」または「略中性アミノ酸」と総称する。すなわち、これらのアミノ酸の等電点は3.99を超え7.40未満である。アミノ酸は天然に存在するものであっても、天然に存在するアミノ酸の類似体であってもよい。
本明細書において、「Aおよび/またはB」は、AおよびBとAまたはBとの双方を含む概念であり、「AおよびBの少なくとも一方」とも換言できる。
本発明の融合タンパク質は、細胞内安定化ペプチドと、抗原結合性ペプチドとを含む融合タンパク質であって、
上記細胞内安定化ペプチドは、10~39個のアミノ酸からなり、かつ、当該アミノ酸の少なくとも45%が酸性アミノ酸であり、
上記抗原結合性ペプチドは、重鎖CDR1、重鎖CDR2、重鎖CDR3、軽鎖CDR1、軽鎖CDR2、および軽鎖CDR3のうちの少なくとも1つを含む。
(ペプチドの長さと酸性アミノ酸の含有)
細胞内安定化ペプチドは、構成されるアミノ酸数は特に限定されないが、好ましくは50個までのアミノ酸からなるペプチドであり、より好ましくは10~39個のアミノ酸からなるペプチドである。ここで、細胞内安定化ペプチドを構成する全アミノ酸の少なくとも45%が、酸性アミノ酸である。
細胞内安定化ペプチドを構成する全アミノ酸に占める塩基性アミノ酸の割合は特に限定されないが、28%以下であることが好ましく、27%以下または26.5%以下であることがより好ましく、25%以下、20%以下、15%以下、または10%以下であることがさらに好ましく、9%以下、8%以下、7%以下、6%以下、5%以下、4%以下または3%以下であることが特に好ましい。特に好ましい一形態では、細胞内安定化ペプチドは、塩基性アミノ酸を含まない。
細胞内安定化ペプチドは、上記の酸性アミノ酸および塩基性アミノ酸以外のアミノ酸(「略中性アミノ酸」と総称する)を含んでいてもよい。
細胞内安定化ペプチドの等電点は、当該細胞内安定化ペプチドを構成するアミノ酸の種類から算出することができる。細胞内安定化ペプチドの等電点の算出は、例えば、Protein Caluculator(https://protcalc.sourceforge.net)等の記載に従って行うことができる。
以下、細胞内安定化ペプチドのより具体的な例示について説明を行う。なお、以下の説明において、XAとは酸性アミノ酸を指し、Xnとは酸性アミノ酸でないアミノ酸、すなわち、上記した塩基性アミノ酸と略中性アミノ酸とを指す。
以下に箇条書きをした条件を全て満たす細胞内安定化ペプチド。
・細胞内安定化ペプチドは、10~39個のアミノ酸からなり、14~25個のアミノ酸からなることが好ましい。
・細胞内安定化ペプチドを構成するアミノ酸の少なくとも45%が酸性アミノ酸であり、少なくとも50%が酸性アミノ酸であることが好ましい。
・細胞内安定化ペプチドにおいて、XAが8個以上連続した部分、および、Xnが4個以上連続した部分が存在しない。すなわち、細胞内安定化ペプチドにおいて、XAの連続は7個までであり、Xnの連続は3個までである。
上記の第一の態様の条件を満たし、かつ、以下に箇条書きをした条件を満たす細胞内安定化ペプチド。
・細胞内安定化ペプチドにおいて、XAが6個以上連続した部分、および、Xnが4個以上連続した部分が存在しない。すなわち、細胞内安定化ペプチドにおいて、XAの連続は5個までであり、Xnの連続は3個までである。
上記の第一の態様の条件を満たし、かつ、以下に箇条書きをした条件を満たす細胞内安定化ペプチド。
・細胞内安定化ペプチドにおいて、XAが6個以上連続した部分、および、Xnが3個以上連続した部分が存在しない。すなわち、細胞内安定化ペプチドにおいて、XAの連続は5個までであり、Xnの連続は2個までである。
上記の第一の態様の条件を満たし、かつ、以下に箇条書きをした条件を満たす細胞内安定化ペプチド。
・細胞内安定化ペプチドにおいて、XAが3個以上連続した部分、および、Xnが3個以上連続した部分が存在しない。すなわち、細胞内安定化ペプチドにおいて、XAの連続は2個までであり、Xnの連続は2個までである。
以下に箇条書きをした条件を全て満たす細胞内安定化ペプチド。なお、第五の態様は、第一~第四の態様の何れかに当てはまるものであっても、当てはまらないものであってもよい。
・細胞内安定化ペプチドは、10~39個のアミノ酸からなり、14~25個のアミノ酸からなることが好ましい。
・細胞内安定化ペプチドを構成するアミノ酸の少なくとも45%が酸性アミノ酸であり、少なくとも50%が酸性アミノ酸であることが好ましい。
・以下に示すアミノ酸配列:
-Xn-XA-XA-Xn-XA-(XAまたはXn)-Xn-XA-(XAまたはXn)-Xn-(XAまたはXn)-XA-・・・・・(1)
を含んでいる。
上記の第五の態様の条件を満たし、第五の態様で示したアミノ酸配列(1)の左側にさらに3個~7個のXAまたはXnを有し、右側にさらに0個~6個のXAまたはXnを有する。
以下に箇条書きをした条件を全て満たす細胞内安定化ペプチド。なお、第七の態様は、第一~第四の態様の何れかに当てはまるものであっても、当てはまらないものであってもよい。
・細胞内安定化ペプチドは、10~39個のアミノ酸からなり、14~25個のアミノ酸からなることが好ましい。
・細胞内安定化ペプチドを構成するアミノ酸の少なくとも45%が酸性アミノ酸であり、少なくとも50%が酸性アミノ酸であることが好ましい。
・以下に示すアミノ酸配列:
-Xn-XA-Xn-XA-Xn-XA-XA-Xn-XA-Xn-Xn-XA-XA-Xn-Xn-XA-・・・・・・・(2)
を含んでいる。
上記の第七の態様の条件を満たし、第七の態様で示したアミノ酸配列(2)の左側にさらに0個~9個のXAまたはXnを有し、右側にさらに0個~9個のXAまたはXnを有する。但し、アミノ酸配列(2)の左側と右側とにあるアミノ酸の個数の合計は10個以下であることが好ましく、9個以下であることがより好ましく、6個または5個以下であることがさらに好ましい場合がある。
上記第一~第八の態様の何れかに当てはまるものであって、XAはアスパラギン酸、またはグルタミン酸であり、Xnはアスパラギン、グルタミン、プロリン、チロシン、およびバリンからなる群より選択される何れかのアミノ酸である。
上記第五~第六の態様の何れかに当てはまるものであって、XAはアスパラギン酸、またはグルタミン酸であり、Xnのうちアミノ酸配列(1)の左側から2~6番目(好ましくは2~4番目、より好ましくは2~3番目)のXnの一つまたはそれ以上がプロリンであり、プロリンでないXnは、アスパラギン、グルタミン、チロシン、およびバリンからなる群より選択される何れかのアミノ酸である。例えば、アミノ酸配列(1)の左側から2~3番目のXnの一つがプロリンであり、アミノ酸配列(1)に含まれるその他のXnが、アスパラギン、グルタミン、バリン、およびチロシンからなる群より選択される何れかのアミノ酸であるもの。さらには例えば、アミノ酸配列(1)の左側から2~3番目のXnの一つがプロリンであり、アミノ酸配列(1)に含まれるその他のXnが、アスパラギン、グルタミン、およびチロシンからなる群より選択される何れかのアミノ酸であるもの。
上記第七~第八の態様の何れかに当てはまるものであって、XAはアスパラギン酸、またはグルタミン酸であり、Xnのうちアミノ酸配列(2)の左側から3~6番目(好ましくは3~5番目、より好ましくは4~5番目)のXnの一つまたはそれ以上がプロリンであり、プロリンでないXnは、アスパラギン、グルタミン、チロシン、およびバリンからなる群より選択される何れかのアミノ酸である。例えば、アミノ酸配列(2)の左側から4~5番目のXnの一つがプロリン、Xnの他の一つがバリンであり、アミノ酸配列(2)に含まれるその他のXnが、アスパラギン、グルタミン、およびチロシンからなる群より選択される何れかのアミノ酸であるもの。
細胞内安定化ペプチドは、以下のアミノ酸配列の何れかからなる。
EEDQDDEDDEDQDD(配列番号2);
NDEYEDPDEQDDEND(配列番号3);
QDEVDEPEDEEDNDD(配列番号4);
QDEVDEPEDEDENDD(配列番号5);
QDEVDEPEDEDENQD(配列番号6);
QDNVDEPEDNDENQD(配列番号7);
QDNYDEPEDNDENQD(配列番号8);
EDNYDEPEDNDENQD(配列番号9);
DNNYDEQDENEQPED(配列番号10);
QENDYDEPEVNDENQD(配列番号11);
DEQENDYDEPEVNDENQD(配列番号12);
DEQENDYDEPEVNDENQDYDE(配列番号13);。
以下に箇条書きをした条件を全て満たす細胞内安定化ペプチド。
・細胞内安定化ペプチドは、10~39個のアミノ酸からなり、14~25個のアミノ酸からなることが好ましい。
・細胞内安定化ペプチドを構成するアミノ酸のすべてが酸性アミノ酸である。
(構造)
典型的な抗体構築物ユニットは、テトラマーを含むことが知られている。各々のテトラマーは、同一の2対のポリペプチド鎖から構成され、各々の対が1本の軽鎖(一例では、約25kDa)および1本の重鎖(一例では、約50~70kDa)を有する。各々の鎖のアミノ末端部分は約100アミノ酸以上の可変領域を有し、該可変領域は主に抗原(標的分子)認識の役割を担う。各々の鎖のカルボキシル末端部分は、主にエフェクター機能の役割を担う定常領域を規定する。軽鎖は、カッパまたはラムダの何れかに分類される。重鎖は、ガンマ、ミュー、アルファ、デルタまたはエプシロンとして分類され、抗体のアイソタイプをそれぞれIgG、IgM、IgA、IgD、およびIgEを規定する。軽鎖および重鎖内で、可変領域および可変領域が約12アミノ酸以上のJ領域によって連結されており、重鎖はまた、約10アミノ酸以上のD領域を含む。各々の軽鎖/重鎖対の可変領域は、抗体結合部位を形成する。これらの鎖はすべて、3つの超可変領域(相補性決定領域またはCDRとも呼ばれる)によって連結される相対的に保存されたフレームワーク領域(FR)の同一の一般構造を示す。各対の2本の鎖由来のCDRをフレームワーク領域によって並べることで、特定のエピトープへの結合を可能とする。
本発明の融合タンパク質は、細胞内安定化ペプチドおよび抗原結合性ペプチド以外の機能ペプチドを1つ以上さらに含んでいてもよい。そのような機能ペプチドとしては、精製タグペプチド、検出用ペプチド、分解促進ペプチド(例えばHSC70結合ペプチド、XIAP RINGドメインペプチド)、Neh2ドメインペプチド(Nrf2由来のペプチド)、酸素依存性分解ドメイン(Hif1α由来のペプチド)等のストレス応答性分解ペプチド、及び、薬剤結合性ペプチド等が挙げられる。
「融合タンパク質」とは、共有結合によって、直接またはリンカーを介して連結された少なくとも2種の異種ペプチドを有するタンパク質を指す。リンカーは、特に限定されないが、好ましくは、柔軟性を与えるために、例えば、グリシン、アラニン、およびセリンなどの低分子側鎖を有するアミノ酸を主に含有する。好ましくは、リンカー配列の80%、90%、またはそれ以上がグリシン残基、アラニン残基、またはセリン残基を含み、特にグリシン残基またはセリン残基を含有する。
本発明の融合タンパク質は、一例において、pH7.4における正味の電荷が負であることが好ましく、pH6.6における正味の電荷が負であることがより好ましく、pH6.0における正味の電荷が負であることがより好ましく、pH5.0における正味の電荷が負であることがさらに好ましい。
本発明のポリヌクレオチドは、本発明の融合タンパク質をコードする。「ポリヌクレオチド」は、DNA分子、RNA分子、およびDNAとRNAとのハイブリッド分子の何れであってもよい。また、「ポリヌクレオチド」は、一本鎖であってもよいし、二本鎖であってもよい。
〔薬学的用途〕
(薬学的組成物)
本発明はまた、上述の融合タンパク質、ポリヌクレオチドまたはベクターを含む薬学的組成物を提供する。
上述の薬学的組成物は、例えば、抗原が関連する疾患を治療および/または予防するために用いることができる。
(研究用試薬)
本発明はまた、上述のポリヌクレオチドまたはベクターを含む研究用試薬を提供する。研究用試薬は、上述のポリヌクレオチドまたはベクター以外の他の成分をさらに含んでいてもよい。当該他の成分は、上述の(薬学的組成物)で説明したものを参照可能である。
本発明の融合タンパク質は、細胞内における抗原(例えば、タンパク質またはポリペプチド等)の機能解析に用いることができる。一例において、機能解析の対象となる抗原に対する抗原結合性ペプチドを本発明の融合タンパク質として細胞内で発現させ、抗原の機能を制御する(好ましくは阻害する)ことにより、抗原の機能解析を行うことができる。
本発明はまた、細胞内において抗原結合性ペプチドを発現させる方法を提供する。当該発現方法は、上述のポリヌクレオチド(すなわち、本発明の融合タンパク質をコードするポリヌクレオチド)を細胞内において発現させる工程を含む。
本発明はまた、上述の細胞内安定化ペプチドと抗原結合性ペプチドとを含む融合タンパク質をコードするポリヌクレオチドを作製するためのキットであって、当該細胞内安定化ペプチドをコードするポリヌクレオチドを含む、キットを提供する。
理化学研究所の動物実験委員会のガイドラインに従って、すべての実験手順を行った。使用マウスを、12時間毎の明暗サイクルに置き、暗サイクルは20:00から8:00に起こるようにした。
この研究で用いた抗体を表S1に載せた。
組換えファージ抗体システム(RPAS)(GE Healthcare)を用いてscFv遺伝子を単離した。scFv-A36のDNA配列は、日本DNAデータバンク(DDBJ)にアクセッション番号AB472376で登録されている。
特定のプライマーを用いたPCRを基にして構築した発現ベクターは、下記に記載した。
ScFv-GFPA36およびscFv-GFPM4をAAV1血清型を用いて作製した;s3Flag-scFv-A36-HAおよびs3Flag-scFv-M4-HAは、AAV9血清型を用いて作製した。AAVベクタープラスミド、およびその作製の詳細は、下記に記載した。
オスの野生型B6マウス、もしくはオスのDAT-cre(+/-)マウス(8週齢)に麻酔器(MK-A110;Muromachi)を用いてイソフルレン(Escain; Mylan)で麻酔をかけ、定位フレーム(Stereotaxic Just for Mouse,Muromachi)に配置した。AAVベクターは、右半球の黒質に注入した(ブレグマに対するミリメートル単位で表された座標、AP;-3.08,LR;-1.25,DV;-4.5)。
組換え抗体scFv-GFPA36をE. coli系統BL21で発現し、変性もしくは非変性の条件下で、Ni-NTAアガロースクロマトグラフィーを用いて精製した。外来タンパク質の可溶な状態での発現を向上するため(Yasukawa et al.,1995)、チオレドキシン(Trx)を発現しているpT-Trxベクターによって共形質転換されたE. coli系統BL21から、非変性の条件下で、抗-Flag(M2)抗体結合アフィニティービーズ(Sigma)を用いて、組換え抗体s3Flag-scFv-A36-HAを精製した。
免疫染色を標準的な手法で行った。免疫蛍光シグナルは、Alexa Fluor 488もしくはAlexa Fluor 594でラベルされた二次抗体(Invitrogen)により視覚化した。蛍光ラベルされた試料は、Fluoview FV1000共焦点顕微鏡(Olympus)もしくはBZ-9000蛍光顕微鏡(Keyence)により画像表示した。
GST-Syt I-C2Aの抗体結合アフィニティーを、下記に記載された通りにELISAにより計測した。
以前に報告されたロータロッド試験(Shiotsuki et al.,2010)を改変し、改変ロータロッド試験を実施した。本研究では、マウス用のロータロッド(MK-610A,Muromachi)に、滑り止めテープ(Nitoflon粘着テープ,No.903UL,0.13mm厚,Nitto denko)で覆われた大きなロッド(直径9cm)を装備した。
scFv-A36とアラインメントしたscFvタンパク質のアクセッション番号およびアミノ酸残基を、表S2に載せた。
Dat+/IRES-cre マウス(Backman et al.,2006)はJackson laboratoryより購入した。scFv遺伝子を発現しているAAVウイルスベクターを、DAT-Creマウス系統とともに、黒質のドーパミン神経において抗体遺伝子を選択的に発現させるために用いた。マイクロダイアリシスおよび行動テストで用いた、ウイルスベクターを有するすべてのマウスは、一対のヘテロ接合体から生まれたオスの同腹子であった。Charles Liver Japanより購入したBalb/c、Balb/c-nuおよび野生型のC57B6/Jマウスは、抗体産生、もしくはレンチウイルスベクターおよびAAVベクターを用いた実験、ならびに初代ドーパミン神経培養に使用した。
scFvにおいて、上記抗体の軽鎖および重鎖の可変部は、ひとつの遺伝子にコードされたグリシンリンカーによって融合している。RPAS(GE Healthcare)により、scFvの多様なレパートリーをM13ファージの表面上で提示することが可能になる。GST-Syt II-C2Aにより免疫性を与えられたマウス(Balb/c)脾臓からトータルRNAを単離した(Fukuda et al.,1994; Fukuda et al.,1999)。上記軽鎖および重鎖の可変部(それぞれVHおよびVL)を、縮重プライマー(GE Healthcare)を用いて、2つの独立した反応により増幅した。結果得られたPCR産物を、フレキシブルな(Gly4-Ser)3の15アミノ酸鎖をコードするリンカーと結合した。VH-グリシンリンカー-VL複合体(scFv)を、pCANTAB 5Eベクター(GE Healthcare)のSfi IおよびNot I部位にサブクローニングした。E. coli TG-1細胞をscFv cDNAを含むプラスミドベクターおよびM13-KO7ヘルパーファージの感染によって形質転換することにより、組換えファージ抗体を産生した。抗体反応性ファージの単離は、製造元の使用説明書に従ってバイオパニングすることにより行った。対数期TG-1細胞を抗体反応性ファージに感染させた。別個の抗体を提示するファージを、マイクロタイターウェルに結合した組換えGST-Syt II-C2Aを用いたELISAにより、ファージライブラリーからスクリーニングした。抗体反応性ファージを、セイヨウワサビペルオシキダーゼ(HRP)結合抗M13抗体を用いて視覚化した。抗体反応性scFvクローン(scFv-A36と名付けた)の配列決定は、自動DNAシークエンサーを用いて行った。
scFv-A36のcDNA配列に基づいて、2つのリンカープライマーをPCR増幅に合わせて設計し、それにより、コザック配列、T7ペプチド、およびBamHI制限酵素認識部位をA36の5’フランキング領域に導入し、MunI部位、ヘキサヒスチジン残基、およびNot I部位をA36の3’フランキング領域に導入した(表1)。



初代ドーパミン神経培養を、胚日13~14日のオスおよびメスのマウス胚の腹側中脳から準備した。簡潔に言えば、腹側中脳をトリプシン処理により解離し(0.25% 20分、37℃)、DNase Iを含む10%FBSを添加したneurobasal培地中で、火で仕上げたパスツールピペットで粉砕した。解離した細胞(6×104)を、24ウェルプレート(Iwaki)中で、ポリ-L-リシン(1μg/mL)コートカバーガラス(Fisherbrand,直径;12mm)上にまき、それからB27(Invitrogen)を添加したneurobasal培地500μL中で培養した。それぞれのウェル内の7日目のin vitroの細胞(DIV)を、AAVベクター(タイター:1×1011マイクロg/mL)5μLで感染させ、GFP-タグ付きscFvタンパク質を発現させ、感染から7日後に、免疫細胞化学もしくはドーパミン放出アッセイを行った。理研バイオリソースセンターセルバンク(Tsukuba,Japan)から入手した293TおよびCOS-7細胞を、10%FBSを添加したDMEMで培養した。これらの細胞を、Lipofectamine2000を製造元の使用説明書(Invitrogen)に従って用いて、発現ベクターによりトランスフェクションし、トランスフェクションから1日後に、免疫化学的分析に用いた。
E. coli系統BL21(DE3)(Novagen)を、scFv-GFPA36の発現に用いた。scFv-GFPA36の発現を、1mMのイロプロピル-1-チオ-β-D-ガラクトピラノシド(IPTG)により、30℃で3時間誘導した。ScFv-GFPA36タンパク質を、バッファー(8M 尿素,0.1M NaH2PO4,10mM Tris-HCl[pH8.0])で可溶化し、Ni-NTAアガロースクロマトグラフィー(Qiagen)で製造元の推奨に従って精製した。変性条件下で、4mLのバッファー(8M 尿素,0.1M NaH2PO4,10mM Tris-HCl[pH6.3])でカラムを洗浄した。その後ScFv-GFPA36を、1mLのバッファー(8M 尿素,0.1M NaH2PO4,0.01M Tris-HCl[pH 4.5])でカラムから溶出した。溶出したscFv-GFPA36タンパク質を、バッファー(10mM HEPES-KOH;pH7.2)で透析した。非変性条件下でscFv-GFPA36を精製するために、scFv-GFPA36を発現している細胞を、プロテアーゼインヒビターカクテルを含むPBSバッファーに再度懸濁し、氷上でソニケートし、Triton X-100(1%)を加えて4℃で1時間可溶化した。遠心分離の後、上清をNi-NTA(ニッケル-ニトリロ三酢酸、GE Healthcare)クロマトグラフィーにかけた。発現させたscFv-GFPA36を、5mMヒスチジンを含むバッファー(10mM HEPES-KOH;pH7.2)を用いてカラムから溶出し、その後バッファー(10mM HEPES-KOH;pH7.2,150mM NaCl)で透析した。
バッファー中の精製されたGST-Syt I-C2A(0.25pmol)を、室温で16時間96ウェルプレートにコートした。それぞれのウェルを、5%スキムミルクを含むPBSバッファーで2時間室温でブロッキングし、0.22~36nMの精製されたs3Flag-scFv-A36-HAとともにインキュベートした。マウスの抗体Flag(M2)一次抗体、およびHRP結合抗マウス二次抗体とともにインキュベートすることにより、結合を定量した。特異的な結合は、0.25pmolのGSTでコートしたウェルに対するs3Flag-scFv-A36-HAの結合を差し引くことにより計算した。s3Flag-scFv-A36-HA結合データの非線形回帰は、ヒル-ラングミュアの式により行った。

免疫細胞化学および免疫組織化学は、下記の標準的な方法で実施した。細胞を4% PFAで室温、2分間固定し、0.3% Triton X-100を含むPBSで室温、2分間透過処理した。細胞をすぐにブロッキング溶液(1% BSAおよび0.1% Triton X-100を含むPBS)で3回洗浄し、ブロッキング溶液とともに室温で1時間インキュベートし、その後2時間室温で一次抗体とともにインキュベートした。免疫蛍光シグナルは、Alexa Fluor 488もしくはAlexa Fluor 594でラベルされた二次抗体(Invitrogen)とともにインキュベートすることにより視覚化した。Alexa Fluor色素と結合した一次抗体および二次抗体は、表S1に載せた。細胞内抗体により形成された凝集体を有する細胞の割合を定量する実験において、各実験で1つのディッシュ内の少なくとも100個の細胞を、凝集体を有する細胞の割合を定量するために数えた。データは3回の独立した実験により得た。免疫組織化学において、マウスは4%パラホルムアルデヒドで固定した。マウス脳の低温切片(16μm厚)を、0.3% Triton X-100を含むPBS中で室温で2時間透過処理し、一次抗体を含むブロッキング溶液(1% BSA,0.1% Triton X-100)とともに4℃で16時間インキュベートした。免疫蛍光シグナルを、Alexa Fluor 488もしくはAlexa Fluor 594でラベルされた二次抗体(Invitrogen)を用いて視覚化した。蛍光ラベルした試料は、Fluoview FV1000共焦点顕微鏡(Olympus)もしくはBZ-9000蛍光顕微鏡(Keyence)のもとで画像化した。
Flagタグつき全長マウスSyt I-XIを、以前に記載されたように調製した(Fukuda et al.,1999)。COS-7細胞の中で一過性発現させたFlagタグつきSyt I-XIの全ホモジネートを、10%ドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動(SDS-PAGE)にかけ、ポリフッ化ビニリデン(PVDF)膜に転写した。精製したscFv-GFPA36(1.6μg/mL)を一次抗体として用いた。HRPラベル抗T7モノクローナル抗体(1/5000希釈, Merck Millipore)を二次抗体として用いた。免疫反応性のあるバンドを、高感度ケミルミネセンス(ECL)検出システム(GE Healthcare)を用いて視覚化した。等量のFlagタグつきSytsがロードされたことを保証するため、ブロットを抗Flag抗体でリプローブした。精製したs3Flag-A36-HAが、脳でマウスSyt I/IIを認識したかを調べるために、マウス脳組織を単離し、ホモジナイゼーションバッファー(20mM HEPES-KOH,pH7.2,150mM NaCl,0.5% TrintonX-100,コンプリートプロテアーゼインヒビターカクテル;Roche)でホモジナイズし、4℃で1時間アジテートした。全ホモジネートの2.5μgのタンパク質を、精製したs3Flag-A36-HAを一次抗体として用いてウエスタンブロットした。免疫反応性のあるバンドを、抗Flag抗体(二次抗体)およびHRPラベル抗マウス抗体を用いたECLにより視覚化した。AAVベクターを注入したマウス脳を用いた実験では、マウス脳組織を単離し、バッファー(PBS,0.5% NP40,1mM 2ME,2mM EDTA,およびコンプリートプロテアーゼインヒビターカクテル)内でホモジナイズし、27Gニードルシリンジでせん断し、4℃で1時間アジテートした。全ホモジネートを、抗TH、抗チューブリン、抗Syt I抗体を一次抗体として用いてウエスタンブロットした。
COS-7細胞を、pIRES-scFv-GFPA36もしくはpIRES-scFv-GFPM4、およびpEF-BOS-Flag-Syt Iもしくはコントロールベクター(pEF-BOS)で共トランスフェクションした。プラスミドDNAのトランスフェクションは、LipofectAMINE(Invitrogen)により製造元の推奨に従って行った。トランスフェクションの2日後、細胞をばらばらにし、バッファー(10mM HEPES-KOH[pH7.2],100mM NaCl,1mM β-メルカプトエタノール)中でホモジナイズした。ホモジネートを、1,200×g、4℃で5分間遠心分離し、上清を溶解バッファー(10mM HEPES-KOH[pH7.2],0.1% Triton X-100,100mM NaCl,1mM β-メルカプトエタノール)で4℃で1時間可溶化した。20,400×g、4℃で15分間遠心分離した後、上清を新しいチューブに移し、Flag-Syt IもしくはIIを、抗Flag抗体(Sigma)およびプロテインAセファロース(GE Healthcare)により免疫沈降した。溶解バッファーでビーズを洗浄した後、ビーズを1×SDSサンプルバッファー(62.5mM Tris-HCl[pH6.8],2% SDS,2% β-メルカプトエタノール,0.001% ブロモフェノールブルー[BPB],10% グリセロール)中で煮沸した。13,000×gで5分間遠心分離した後、上清をウエスタンブロット分析に用いた。共免疫沈降したscFv-GFPA36を、はじめにHRPラベル抗T7抗体により検出した。Flag-Syt Iが沈降したことを保証するため、抗Syt I抗体(Stressgen)によりブロットをリプローブした。Syt Iと細胞内s3Flag-scFv-A36-HAとの相互作用を調べるため、293T細胞を、Lipofectamine2000を用いてpEF-BOS-Syt IおよびpEF-BOS-s3Flag-scFv-A36-HAもしくはpEF-BOS-s3Flag-scFv-M4-HAベクターにより共トランスフェクションした。トランスフェクションの28時間後、細胞をばらばらにし、バッファー(20mM HEPES-NaOH[pH7.4],150mM NaCl,0.5% Triton X-100,プロテアーゼインヒビター(EGTA-free,Roche))中で1時間溶解し、その後、27ゲージニードルシリンジでせん断した。ホモジネートを1,5000rpm、4℃で10分間遠心分離し、上清をCaCl2(500μM)を含む新しいチューブに移し、抗HAアガロース(Sigma)により免疫沈降した。500μM CaCl2を含む溶解バッファーでビーズを洗浄した後、ビーズを1×SDSサンプルバッファー中で3分間煮沸した。12,000rpmで5分間遠心分離した後、上清をウエスタンブロット分析に用いた。共免疫沈降したSyt Iを、一次抗Syt I(Stressgen)抗体および二次HRPラベル抗マウスIgG軽鎖特異的抗体で検出した。scFvタンパク質の沈降を保証するために、抗Flag(M2)一次抗体(Sigma)および抗HRPラベル抗マウスIgG軽鎖特異的二次抗体でブロットをプローブした。
AAV9/3ベクタープラスミドは、ヒトシナプシンIプロモーター(hSynIp)を含み、その後ろに、pAAV-DIO-eNpHR-YFPベクター(addgene: #26966)に由来するダブルloxP配列(loxP-lox2722)の間に目的の逆向きcDNAを含み、ラットのtau 3’-UTR(Brun et al.,2003)に由来するヌクレオチド2519-2760を含む241bp断片(FrgH)、ウッドチャック肝炎ウイルス転写後調節エレメント(WPRE)、およびシミアンウイルス40ポリアデニレーションシグナル配列(SV40pA)を、AAV3ゲノムの逆方向末端反復の間に含む。変異を有するAAV9 vp cDNA(T466F)(Gao et al.,2004)を、以前に記載された方法(Petrs-Silva et al.,2011)で合成した。組換えAAV9/3ベクターを、ベクタープラスミド、AAV3 repおよびAAV9 vp発現プラスミド、ならびにアデノウイルスヘルパープラスミドpHelper(Agilent Technologies)を用いたHEK293細胞の一過性トランスフェクションにより、以前に記述された方法で作製し(Li et al.,2006)、ベクターAAV9/3-hSynIp-DIO-s3Flag-scFv-A36もしくは-M4-HAを得た。組換えウイルスを2つの一連の連続的なCsCl勾配からの単離により精製し、ウイルスタイターをqRT-PCRにより決定した。
オスの野生型マウスもしくはメスのDAT-cre(+/-)マウス(8週齢)をイソフルレン(Escain;Mylan)により麻酔した。注入部位の頭蓋骨は、ドリル(Model 1474 Stereotaxic drill,Muromachi)でカットした。AAVベクターを右半球の黒質(ブレグマに対するミリメートル座標, AP;-3.08,LR;-1.25,DV;-4.5)に、注入ポンプ(Legato 130,Muromachi)により制御される33ゲージニードルおよび5μLシリンジ(Model 75 RN SYR,Hamilton)を用いて、分速0.2μLで2μL注入した。ニードルは、注入後、10分間その位置に放置した。注入の少なくとも33日後のマウスを、マイクロダイアリシス、行動テスト、もしくは免疫化学分析に用いた。
AAVベクターの注入7日後の初代ドーパミン神経を、500μLの予熱したPSS-LKバッファー(20mM HEPES-NaOH,pH7.4,140mM NaCl,4.7mM KCl,2.5mM CaCl2,1.2 mM MgSO4,1.2mM KH2PO4,11mM グルコース)で2回洗浄し、細胞を200μLのPSS-LKバッファー中で、37℃で5分間インキュベートした。PSS-LKバッファーを回収した後、細胞を200μLのPSS-HKバッファー(20mM HEPES-NaOH,pH7.4,85mM NaCl,60mM KCl,2.5mM CaCl2,1.2mM MgSO4,1.2mM KH2PO4,11mM グルコース)で37℃、5分間刺激した。回収された上清を、すぐに50μM EDTA-2Naを含む10μLの0.1M過塩素酸(PCA)と混合し、30秒ソニケートし、その後20,000G、0℃で15分間遠心分離した。上清(190μL)を新しいチューブに移し、pHを3.0あたりに調整するために1.7μLの5M CH3COONaと混ぜた。PSS-HKバッファーを回収した後、細胞を2.5μM EDTA-2Naを含む200μLの5 mM PCAに溶解した。サンプル(200μL)をソニケートし、2.4μLの5M CH3COONaと混合した。すべてのサンプルを、0.45μmポリフッ化ビニリデン(PVDF)マイクロポアフィルター(Millipore)で濾過し、濾過物を、電気化学検出システム(ECD300,Eicom)と連結した高圧液体クロマトグラフィー(HPLC)で、以前の報告(Kabayama et al.,2013)に従って分析した。高濃度KClにさらされた初代培養ドーパミン神経から放出されたドーパミンの量を、細胞に含まれた全ドーパミンの割合で表す。
マウスをイソフルレン(Escain;Mylan)で小動物麻酔器(MK-A110;Muromachi)を用いて麻酔し、定位フレーム(Stereotaxic Just for Mouse,Muromachi)に配置した。スチールガイドカニューレ(AG-4;Eicom)の移植のために、右半球の線条体上(ブレグマに対する座標,AP;+0.6 mm,LR;-2.0mm,DV;-2.0mm)に穴をあけ、固定するためのネジをしっかりと固定するためにもう一つの穴を設けた。ガイドカニューレおよび固定するためのネジを、歯科用セメント(Unifast3;GC)によって固定した。歯科用セメントが完全に乾いた後、マイクロダイアリシスプローブ(A-I-4-02;Eicom)をガイドに沿って線条体に挿入した。マイクロダイアリシスプローブの移植は、マウスの麻酔の開始後、3.5時間から4.5時間の間に終了した。マイクロダイアリシスプローブを挿入したマウスを、水と飼料を備えた透明のマイクロダイアリシスケージ(20cm長さ×20cm幅×21cm高さ)内のペーパータオル(キムタオル,Nippon Paper Crecia)の上に配置した。移植手術の次の日、マウスを、水と飼料を備えていない透明のマイクロダイアリシスケージ内の新しいペーパータオル(Comfort200,Nippon Paper Crecia)上に配置した。基底ドーパミン放出を測定するため、透析液の回収を、プローブ移植手術の次の日の10:00から11:00の間に行った。マイクロダイアリシスプローブは、147mM NaCl、4.02mM KCl、2.25mM CaCl2を含むリンガー溶液によって、シリンジポンプ(ESP-32,Eicom)を用いて分速1μLで連続的に潅流された。透析液は、冷蔵したフラクションコレクター(Refrigerated collector 820,Univentor)を用いて4℃で10分の間隔で自動的に、事前に5μLの0.1M CH3COOH/250μM EDTA-2Naをいれたプラスチックのマイクロチューブに回収した。透析液の回収は、マイクロダイアリシスプローブの移植後16.5時間から17.5時間後に行った。すべての透析液サンプルを4℃で保存し、次の日HPLCで分析した。
脳を骨頭切除の直後に回収し、50μm厚の凍結王冠切片を調製した。12ピースの黒質切片、および8ピースの線条体切片(4ピースの前部切片が以下の座標から得られた,AP;+0.6mm,および4ピースの後部切片が以下の座標から得られた,AP;+0.6mm)から、直径1.5mmのディスポーザブルバイオプシーニードル(Biopsy Punch;Kai Medical)を用いて、円形の組織パンチを回収した。サンプルを0.1mM EDTAを含む0.1M過塩素酸内でホモジナイズし、20,000×g、4℃で15分間遠心分離した。上清を0.22μmポリフッ化ビニリデン(PVDF)フィルター(GV Durapore,Millipore)に通して濾過し、濾過したサンプルをHPLCにより分析した。
初代培養神経に由来するサンプルおよび脳切片のバイオプシーを、HPLC(ECD300,Eicom)により、以前に報告されたプロトコル(Kabayama et al.,2013)に従って分析した。マイクロダイアリシスサンプルを、製造元の文書に従ってHPLC(ECD500,Eicom)により分析した。
ロッド(直径3cm)を用いたロータロッド試験が、一般的にマウスの運動技能学習のために用いられてきた。以前の研究におけるマウスの改変ロータロッド試験では、大きなドラムおよび遅い回転(直径9cmおよび10rpm)を組み合わせたロータロッド試験において、急勾配の学習曲線が得られることが実証された(Shiotsuki et al.,2010)。本研究では、マウス用ロータロッド(MK-610A,Muromachi)に、滑り止めテープ(Nitoflon粘着テープ,No.903UL,0.13mm厚,Nitto denko)で覆われた大きなロッド(直径9cm)を装備した。訓練セッションの前に、1日目にマウスを定位ドラムの上に3分間留まらせることにより馴化を行った。馴化は、毎日1分間、セッションの直前に繰り返した。回転は、遅い速度(5rpm,表面速度2.8m/min)に設定した。マウスは、落下後すぐに、一回のセッションにつき5回までドラムの上に戻した。テストは、1日1セッションで5日間繰り返した。落下による待ち時間を手動で記録し、訓練の(Day1)の前、および(Day5)の後のロッド上での合計の待ち時間を、二元反復測定ANOVAで分析した。
等電点および細胞内pH(7.4,7.03,6.60)における正味の電荷を、ペプチドタグを含むそれぞれの細胞内抗体のアミノ酸配列から、Chris Putnam ant the Scrips Research Institute(http://protcalc.sourceforge.net/)により開発されたProtein calculator v3.4を用いて計算した。
すべてのデータは、少なくとも3回の独立した実験の代表値である。結果は、平均±標準誤差で表している。データはGraphPad Prism 4.0 program(GraphPad Software)を用いて、両側独立t-検定、両側非反復ANOVAの後にニューマン・コイルス事後多重比較検定を行って、もしくは二元反復測定ANOVAの後にボンフェローニ事後多重比較検定を行って分析した。P<0.05は、統計学的に有意であるとみなした。
<実施例1:Syt I/IIのC2Aドメインに対する抗体特異的結合の単離>
まず、ファージディスプレイシステムを用いて、Syt I/IIのC2Aドメイン(Syt I/II-C2A)に対する特異的抗体をコードするScFv遺伝子を単離した。単離されたScFvを提示するファージの特異性を調べるために、Syt I/IIのC2Bドメイン(Syt I/II-C2B)をネガティブコントロールとして用いた。酵素結合免疫吸着アッセイ(ELISA)により、単離されたScFv提示ファージのうちの一つ(scFv-A36と呼ぶ)がグルタチオンS-トランスフェラーゼ(GST)-Syt II-C2Aに結合し、GST-Syt II-C2BもしくはGSTには結合しなかった(図1のA)。cDNA配列から、重鎖および軽鎖が互いにグリシンリンカーで融合された、723ヌクレオチドを含むオープンリーディングフレーム(361アミノ酸;アクセッションno.AB472376)が明らかになった(図1のB、上パネル)。発現したscFv-A36を視覚化および精製するために、T7、EGFP、およびHisタグを、A36のアミノ末端、もしくはカルボキシ末端に融合した(scFv-GFPA36と呼ぶ;図1のB、下パネル)。ScFv-GFPA36をE. coliで発現し、Ni-NTAカラムに通して精製した。ウエスタンブロット分析により、精製されたScFv-GFPA36タンパク質が全長Syt I/IIのみを認識し、COS-7細胞において外来的に発現させた他の9つのアイソフォームは認識しないことが確認された(図1のC)。これらの結果から、cFv-GFPA36がSyt I/II-C2Aドメインに対する特異的抗体であることが示された。scFv-A36の軽鎖の可変領域の3つのCDRのアミノ酸配列は、CRA5をコードしているscFvのそれらと同一であり、CRA5は免疫グロブリンE(IgE)受容体(アクセッションno.BAB82458、データは示されていない)に対する抗体であったことから、scFv-A36の軽鎖のCDRがSyt I/IIとの結合特異性の決定において重要な役割を持たないことが示唆された。一方、重鎖CDR3は抗体中で最も超可変な領域であり、ヒトにおいて約1014の多様性を生み出す可能性を有し(Sanz,1991)、それにより抗体の相互作用に貢献している(Chothia and Lesk,1987)ことが知られている。実際、scFv-A36の重鎖CDR3のアミノ酸配列と、国立生物工学情報センター(NCBI)DNAデータベースに蓄積されている他の4つのscFvの重鎖CDR3のアミノ酸配列(図2)との間に大きな多様性があることが見出され、scFv-A36の重鎖CDR3が結合特異性に重要であることが示唆された。次に、PCRを用いた突然変異誘発により、scFv-GFPA36からネガティブコントロールの抗体の構築を試みた。A36の重鎖CDR1(8アミノ酸)およびCDR3(8アミノ酸)の計16アミノ酸が縮重プライマーにより変異した。ELISAにおいて、単離された変異型ScFvファージクローン(scFv-M4と呼ぶ)がGST-Syt II-C2Aタンパク質に結合しなかったことを確認し(図1のD)、scFv-M4のCDR1およびCDR3のヌクレオチド配列を決定した(図2)。以上より、scFv-GFPA36のさらなる機能分析においてM4遺伝子をネガティブコントロール(scFv-GFPM4と呼ぶ)として用いることにした。次に、scFv-GFPA36を哺乳類細胞の細胞内で発現することができるか、また細胞内抗体と結合することができるのかを調べた。ScFv-GFPA36は、scFv-GFPA36とFlag-Syt Iの両方がCOS-7細胞内で共発現されたときのみ、抗Flag抗体とともに共免疫沈降したが、scFv-M4は抗Flag抗体とともに共免疫沈降しなかったことから(図1のE)、細胞内で発現したscFv-GFPA36が、発現したSyt Iと結合できることが示された。
<実施例2:インビトロおよびインビボにおける細胞内scFv-A36の特徴付け>
Syt I/IIが、脳における神経伝達物質の速い放出に対する主要なCa2+センサーとして働くことから、次にドーパミン放出におけるscFv-A36の阻害効果を調べた。scFv-GFPA36およびscFv-GFPM4を発現するためにAAVベクターを構築した(それぞれ、AAV-scFv-GFPA36およびAAV-scFv-GFPM4と呼ぶ)。マウス胚の黒質から調製したDIV7における初代培養ドーパミン神経に、AAVベクターを感染させた。AAVによる感染から7日後に細胞を固定し、分析した。scFv-GFPA36およびscFv-GFPM4の両方が、神経の細胞内において凝集なしに広がって発現した(図3,A~C;scFv-GFPA36,D~F;scFv-GFPM4)。ScFv発現はまた、ウエスタンブロット分析により確認した(図3のG,上;GFP,中;Syt I,下;アクチン)。次に脱分極により誘導されたドーパミン放出を比較した。初代培養細胞において、脱分極により誘導されたドーパミン放出は、細胞外Ca2+に依存していた(図3のH、右列)。このCa2+依存性は、以前の報告(Rouge-Pont et al.,1999)と矛盾しなかった。scFv-GFPA36を発現している細胞において、脱分極により誘導されたドーパミン放出は、scFv-GFPM4を発現している細胞、もしくは感染されていないコントロール細胞に比べて、有意に減少した(図3のH)。これらの結果から、細胞内抗体が初代培養神経の細胞内で発現することができ、scFv-GFPA36がSyt I/IIのC2Aドメインの機能を阻害することが示された。次に、これらの細胞内抗体が、マウス脳神経の細胞内において安定に発現されているのかを調べた。成体マウス(P56)の黒質に対し、片側だけにAAV-scFv-GFPA36(1.6×108 vg/mouse)を注入した。注入の4週間後、ほとんどすべてのドーパミン神経において、scFv-GFPA36の細胞内での発現が観察された。しかしながら、黒質緻密部(SNc)のいくつかの神経において、激しい凝集が観察された(図4のA~C,矢印)。これらの蛍光シグナルは、AAVを注入しなかった左半球のSNcでは観察されなかった(図4のD~F)。これらの結果から、インビトロでの培養細胞の細胞内において安定に発現する細胞内抗体が、インビボで常に安定に発現するわけではないことが示された。
<実施例3:強い負電荷を有する3xFlagペプチドタグ、および中程度の負電荷を有するHAペプチドタグの付加による細胞内抗体の安定性の向上>
細胞質内pH7.4における細胞内抗体の正味の負電荷と、哺乳類細胞の細胞質内の細胞内抗体の安定性との間には正の相関がある(Kvam et al.,2010)。例えば、pH7.4において高い正味の負電荷(-5.0)を有するVHH-H7細胞内抗体が、哺乳類の細胞質内であまり凝集することなく、うまく発現する(Kvam et al.,2010)。これは、pH7.4における正味の負電荷値が-5.0よりも小さいと、細胞内抗体の細胞内における安定な発現を予測できることを示唆している。事実、pH7.4において同じ正味の負電荷(-5.0)を有するscFv-GFPA36およびscFv-GFPM4は、初代培養ドーパミン神経の細胞内において7日間安定に発現された(図3)。しかしながら、scFv-GFPA36およびscFv-GFPM4は、マウス脳のドーパミン神経において細胞内凝集物を形成する(図4)。これらの状況より、インビボにおける安定な細胞内抗体を設計するためにインシリコ解析を行った。海馬神経の非興奮時の細胞内pHは~7.03-7.35であることが報告されており(Ruffin et al.,2014)、エンドソームを標的としたpH感受性蛍光タンパク質pHluorinを用いた以前の研究から、エンドソームの局所的な細胞質側表面pHが6.73±0.08であることが明らかになっている(Mitsui et al.,2011)。そこで、細胞質内pH6.60、7.03、および7.40それぞれにおけるscFv-A36およびscFv-M4のpIおよび正味の電荷を、他のペプチドタグと比較した(図5のA)。scFv-A36もしくはscFv-M4へのT7タグ、EGFPタグ、およびHisタグの融合により、pH7.4における正味の負電荷が増加した。しかしながら、scFv-GFPA36およびscFvGFPM4正味の負電荷は、より低いpH7.03およびpH6.6において劇的に減少した。これは、細胞質内pH7.03および6.6において、scFv-GFPA36およびsFv-GFPM4の安定性が減少することを示唆していた。細胞内抗体の正味の電荷をより低いpHにおいても向上させるため、2つの短いペプチドタグ、3×Flagタグ(MDYKDHDGDYKDHDIDYKDDDDK(配列番号1);s3Flagと呼ぶ)およびHAタグを検証した。これらのタグ自身はそれぞれ強い正味の負電荷(s3Flag;-7.0)および中程度の正味の負電荷(HA;-2.2)を有する。in silicoの解析の結果、scFv-A36/M4タンパク質へのs3FlagタグおよびHAタグの融合(s3Flag-scFv-HAと呼ぶ)により、その正味の負電荷が、pH6.6においても劇的に増加することが実証された(図5のA)。次に、scFv-A36タンパク質配列を用いたNCBI blast検索により得られた他の94種類のscFvタンパク質配列(表S2)を用いて、s3FlagおよびHAペプチドタグの付加による低pH環境下での細胞内抗体の正味の負電荷の増加効果の一般性について調べた(図6)。異なるペプチドタグと融合した94種類のscFvタンパク質のpH7.4(図6のA,上パネル)、およびpH6.6(図6のA,下パネル)における正味の電荷を比較した。T7タグ、EGFPタグ、およびHisタグと融合したscFvタンパク質の正味の負電荷の平均は、ペプチドタグを持たないscFvタンパク質と比較して、pH7.4において統計学的に増加した(上パネル)が、pH6.6においては増加しなかった(下パネル)。反対に、pH7.4とpH6.6の両方において、s3FlagおよびHAタグと融合したscFvタンパク質の正味の負電荷の平均は、他のペプチドタグを付加した、もしくはペプチドタグを付加しなかったscFvタンパク質の正味の負電荷の平均と比較して統計学的に増加した。タンパク質は、そのpIより低いpH下で正の正味の電荷を持ち、そのpIより高いpH下で負の正味の電荷を有する。s3FlagタグおよびHAタグと融合したscFvタンパク質の推定されたpIの平均は、T7タグ、EGFPタグ、およびHisタグと融合したscFvタンパク質、もしくはタグを付加しなかったscFvタンパク質の推定されたpIの平均と比べて有意に低かった(図5のA,図6のB)。これらのことから、s3FlagタグおよびHAタグの付加によるpH6.6でのscFvタンパク質の正味の負電荷を増加効果は、scFvタンパク質のpIを減少させることによりもたらされたことが示唆された。また、scFvタンパク質のscFv-A36とのアミノ酸同一性と、s3FlagタグおよびHAタグと融合したscFvタンパク質のpH7.4における正味の電荷との間の相関を調べたところ(図6のC)、それらの間に統計学的相関がないことが明らかになった。このことは、scFvタンパク質のscFv-A36に対する構造的な同一性そのものは、s3FlagタグおよびHAタグとの融合による他のscFvタンパク質の正味の負電荷を増加する効果に影響しないことを示している。以上より、これらの結果は、scFvタンパク質にs3FlagペプチドタグもしくはHAペプチドタグを融合することで、これらのタグの強い負電荷により、およびscFvタンパク質のpIが低下することにより、細胞質内pH6.6~7.4におけるscFvタンパク質の正味の負電荷が一般的に増加することを示している。
<実施例4:s3Flag-scFv-A36-HAはSyt I-C2Aドメインと相互作用し、Syt IとSyntaxin1-SNAREドメインとの間のCa2+依存的な相互作用を抑制する>
次に、s3Flag-scFvA-36-HA構築物の生化学的な性質を調べた。精製されたs3Flag-scFv-A36-HAのGST-Syt I-C2Aとの結合アフィニティーを計測した。s3Flag-scFv-A36-HAを、抗Flag抗体結合アフィニティービーズを用いてE.coliから精製した。ELISA実験により、精製されたs3Flag-scFv-A36-HAはSyt I-C2Aと相互作用するが、Syt I-C2Bとは相互作用しないことが明らかになった(図7のA)。このs3Flag-scFv-A36-HAの特異性は、図1に示されたscFv-GFPA36の結果と矛盾しなかった。異なる濃度のs3Flag-scFv-A36-HAを用いたELISA実験により、s3Flag-scFv-A36-HAのSytI-C2Aに対するKd値が、約11.97nMであることが明らかになった(図7のB)。加えて、ウエスタンブロット分析により、s3Flag-scFv-A36-HAがマウス脳のSyt I/IIを特異的に認識することが明らかになった(図7のC)。これらの結果は、s3Flag-scFv-A36-HAが高いアフィニティーを有する高度に特異的な細胞内抗体であることを示している。次に、細胞内s3Flag-scFv-A36-HAが細胞内においてSyt Iと結合できるかを293T細胞を用いて調べた。Syt Iはs3Flag-scFv-A36-HAと共免疫沈降したが、s3Flag-scFv-M4-HAとは共免疫沈降しなかった(図7のD)。このことは、哺乳類細胞の細胞内において、細胞内s3Flag-scFv-A36-HAがSyt Iと結合できることを示す。これらのことから、s3Flag-scFv-A36-HAがその相互作用を阻害する可能性が考えられた。このことを調べるため、図7のDで示された細胞溶解物を用いてGSTプルダウンアッセイを行った。精製されたGST-syntaxin1-SNAREを用いて500μM Ca2+の存在下で沈降を行い、沈降物をウエスタンブロットにより分析した。s3Flag-scFv-A36-HAを発現している細胞内におけるSyt IおよびGST-syntaxin1-SNAREの共沈降は、s3Flag-scFv-M4-HAを発現している細胞と比較して約46.5%減少した(図7のE,F)。このことは、細胞内s3Flag-scFv-A36-HAが、Syt IのC2Aドメインとの相互作用によって、細胞内におけるSyt IとSyntaxin Iとの間の相互作用に干渉することを示す。
<実施例5:インビボにおけるs3Flag-scFv-HAの安定した細胞内発現>
次に、s3Flag-scFv-HAが、マウス脳の神経の細胞内において安定に発現するかを調べた。AAV9/3-hSynIp-DIO-s3Flag-scFv-A36(もしくはM4)-HAを、DAT-creマウスの右半球の黒質に注入した。免疫組織化学的解析により、AAV9/3ベクターの注入から33日後、右半球のSNcおよびVTAのドーパミン神経の細胞内において、s3Flag-scFv-A36-HAおよびs3Flag-scFv-M4-HAが、安定に発現することが明らかになった(図8のA~L,矢印)。これらの発現パターンは、黒質領域のほとんどすべての広範囲(~768μm)において観察された(図9のA~J)。また、ウエスタンブロット分析を用いて、s3Flag-scFv-A36-HAおよびs3Flag-scFv-M4-HAの両方のSNにおける発現を確認した(図9のK,上パネル)。線条体ニューロンが中脳ドーパミン神経から長い求心的な入力を受け取る線条体では、s3Flag-scFv-A36-HAの発現レベルは、s3Flag-scFv-M4-HAの発現レベルより高かった(図9のK,上パネル)。このことは、s3Flag-scFv-A36-HAが内生的なSyt Iと結合し、線条体においてSyt Iとともに細胞体から軸索末端に向かって輸送されることを示唆している。加えて、AAV9/3注入の6か月後に、s3Flag-scFv-A36-HAおよびs3Flag-scFv-M4-HA両方が、ドーパミン神経を傷つけることなく安定して発現することが観察された(図10)。SNのドーパミン神経において、in vivoでs3Flag-scFv-A36-HAがSyt Iに対し機能活性を有することを確認するため、AAV9/3注入から33日後の覚醒時のマウスの線条体における基底ドーパミン放出を、マイクロダイアリシスにより測定した。線条体におけるドーパミン放出は、s3Flag-scFv-A36-HAを発現しているマウス、s3Flag-scFv-M4-HAを発現しているマウス、およびAAVを注入していないマウスの3つのグループで比較した。s3Flag-scFv-A36-HAを発現しているマウスの線条体からのドーパミン放出は、s3Flag-scFv-M4-HAを発現しているマウスと比べて55.3%、およびAAV9/3を注入されていないマウスに比べて52.7%と、有意に減少することがわかった(図11のB、C)。しかしながら、線条体および黒質におけるドーパミンの総量は、すべての3つのグループの間で統計学的に変わらなかった(図11のD)。以上より、これらの結果は、s3Flag-scFv-A36-HAが神経活性とドーパミン神経の軸索末端からの細胞外Ca2+とに依存的なドーパミン放出を阻害することを示された。
<実施例6:ドーパミン神経におけるs3Flag-scFv-A36-HAの細胞内発現による 運動学習の阻害>
パーキンソン病(PD)は、神経変性疾患であり、震え、硬直、動きの遅さ、および姿勢の不安定を含む運動欠損を伴う。患者の最も特徴的な点は、主にSNcにおけるドーパミン神経の喪失および線条体におけるドーパミン濃度の減少である。そこで、線条体のドーパミン放出の運動行動における直接的な役割を調べるために、オープンフィールドテストおよび改変ロータロッド試験を行った。AAV9/3-hSynIp-DIO-s3Flag-scFv-A36(もしくはM4)-HAをDAT-creマウスの黒質の両側に注入した。AAV注入の4週間後、マウスをオープンフィールドテストに用い、5または6日後に、改変ロータロッド試験に用いた。s3Flag-scFv-A36-HAを発現しているマウスとs3Flag-scFv-M4-HAを発現しているマウスとの間で、合計の移動距離、動きの速さ、center of time、および立ち上がりの合計数に違いはなかった(図12のA~D)。次に、以前に報告されたロータロッド試験(Shiotsuki et al.,2010)を改変し、運動学習を調べた(実験手順の欄を参照)。訓練の5日後、s3Flag-scFv-M4-HAを発現しているマウスは、s3Flag-scFv-A36-HAを発現しているマウスに比べて、ロッドの上に長くとどまった(図12のE,P=0.0132)。運動行動テストの後、免疫組織化学によりマウス脳を分析した。黒質のDA神経における3Flag-scFv-M4-HAおよびs3Flag-scFv-A36-HAタンパク質の両側における発現を確認することができた(図12のF,矢印)。
<実施例7:抗腫瘍活性の評価>
次に、抗Krasモノクローナル抗体(Y13-259;配列番号22)について、前述の実施例と同様にs3FlagおよびHAを融合させたs3Flag-scFv-Y13-259-HA(STAND-Y13-259;配列番号23)を作製し、抗腫瘍活性の有無を評価することにした。なお、Y13-259のDNA配列は、データベースに登録されていなかったため、アミノ酸配列に基づき、マウスのコドンに最適化した配列を設計した。STAND-Y13-259のDNA配列を配列番号24に示す。myc-Y13-259のDNA配列を配列番号25に示す。DNA合成はGenescript社に委託した。
STAND-Y13-259の設計にあたり、In silicoの解析を行った結果、s3FlagタグおよびHAタグの融合により、scFv-Y13-259の正味の負電荷が、pH6.6においても劇的に増加することが実証された(図13のA)。
次に、STAND-Y13-259とGST-Krasとの結合アフィニティーを、前述の実施例の通りにELISAにより計測した。その結果、STAND-Y13-259もKrasとの高い結合活性が維持されていることが確認された(図13のB)。続いて、レンチウイルスにSTAND-Y13-259、myc-Y13-259(配列番号26)またはscFv-Y13-259のみを組み込んだベクターをMIA PaCa2細胞に感染させ、免疫細胞染色を行った。その結果、従来のmyc-Y13-259は不安定で発現しないか、発現しても細胞内で凝集するのに対し、STAND-Y13-259が安定して発現していることが確認された(図13のC)。さらに、細胞内在性のKras細胞内相互作用を確認するために、抗HA抗体を用いて共免疫沈降を行ったところ、STAND-Y13-259を感染させたMIA PaCa2細胞由来の試料において、内在性KrasとSTAND-Y13-259との結合が確認された(図13のD)。
次に、前述と同様にレンチウイルスベクターを用いて感染させたMIA PaCa2細胞の生存率を、感染から4日後にMTS解析によって測定した。その結果、STAND-Y13-259の発現によって生存率が顕著に低下することが示された(図13のE)。続いて、膵臓癌細胞を皮下に移植したマウス異種移植モデルにおいて、STAND-Y13-259の細胞内発現によって癌の成長が抑制されるかを評価した。ヌードマウスの皮下で形成された腫瘍部分に対し、週に一度、4週に亘って前述のSTAND-Y13-259を発現するレンチウイルスベクターを投与した結果、癌を強力に阻害することが示された(図13のF、G)。最初の感染から25日後に摘出された腫瘍においてSTAND-Y13-259が発現していることも確認された(図13のH)。
<実施例8:他のペプチドタグの評価>
他のペプチドタグの性能評価を行うためDE2.0タグ及びDE5.0タグを作製し、その安定化の効果を検討した。実施例7と同様に、DE2.0タグ(DEQENDYDEPEVNDENQDYDE(配列番号13))を用いた融合タンパク質としては、抗Krasモノクローナル抗体由来のペプチド(Y13-259;配列番号22)のN末端側にDE2.0タグを融合させたDE2.0-Y13-259(配列番号27);DE2.0-Y13-259のC末端側にさらにHAペプチドを融合させたDE2.0-Y13-259-HA(配列番号28);DE2.0-Y13-259のC末端側にさらにPAペプチドを融合させたDE2.0-Y13-259-PA(配列番号29);並びに、凝集繊維化したhuman α‐synucleinに特異的に結合する抗体(Barkhordarian et al., 2006:Protein Engineering, Design and Selection, Volume 19, Issue 11, 1 November 2006, Pages 497-502)のscFV配列(scFv‐6E)のN、C両末端にDE2.0タグを融合させたタンパク質STAND-6E-LYS;を作製した。STAND-6E-LYSには、さらに機能ペプチドとしてHeat shock cognate protein 70 (HSC70)結合ペプチド(MARVKKDQAEPLHRKFERQPPG(配列番号31))(Bauer et al., Nature Biotech (2010):Nature Biotechnology volume 28, pages 256-263 (2010))をC末端側のDE2.0タグの下流に融合させ、さらにHSC70結合ペプチドの下流にはHAペプチドも付した。作成したSTAND-6E-LYSのアミノ酸配列の全長は、配列番号32に示す。


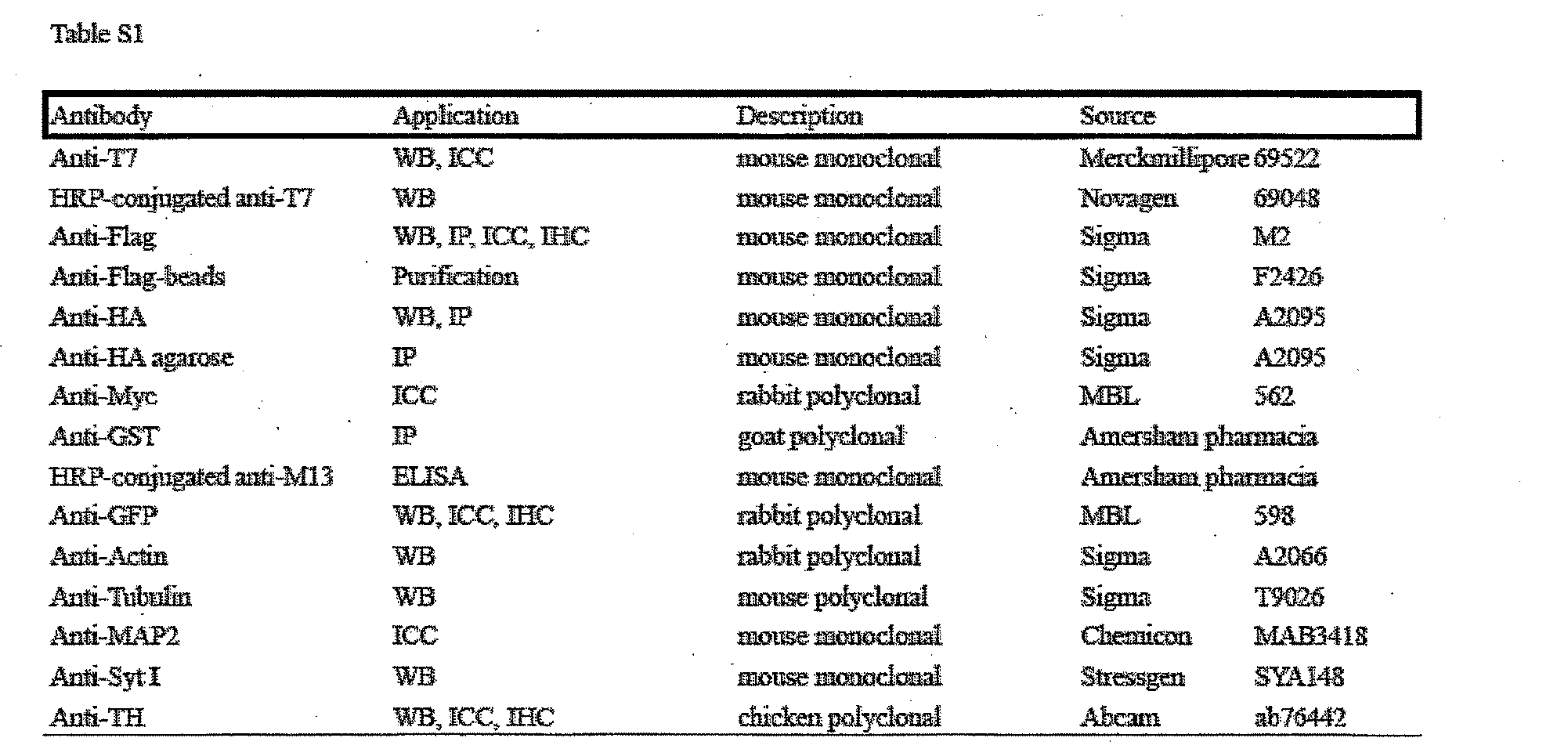

Claims (6)
- 細胞内安定化ペプチドと、抗原結合性ペプチドとを含む融合タンパク質であって、
上記細胞内安定化ペプチドは、39個以下のアミノ酸からなり、かつ、当該アミノ酸の少なくとも45%が酸性アミノ酸であり、
上記抗原結合性ペプチドは、重鎖CDR3を含み、
前記細胞内安定化ペプチドは、配列番号1、13または33によって示されるアミノ酸配列を含んでいる、
融合タンパク質。 - 上記細胞内安定化ペプチドは、配列番号13または33によって示されるアミノ酸配列を含んでおり、塩基性アミノ酸を含まない、請求項1に記載の融合タンパク質。
- 請求項1又は2に記載の融合タンパク質をコードするポリヌクレオチド。
- 請求項3に記載のポリヌクレオチドを含む発現ベクター。
- 細胞内において抗原結合性ペプチドを発現させる細胞を製造する方法であって、
請求項3に記載のポリヌクレオチドまたは請求項4に記載の発現ベクターを細胞に導入する工程を含む、製造方法。 - 請求項1又は2に記載の融合タンパク質を発現する細胞。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017124596 | 2017-06-26 | ||
| JP2017124596 | 2017-06-26 | ||
| PCT/JP2018/024197 WO2019004213A1 (ja) | 2017-06-26 | 2018-06-26 | 融合タンパク質 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2019004213A1 JPWO2019004213A1 (ja) | 2020-05-21 |
| JP7162897B2 true JP7162897B2 (ja) | 2022-10-31 |
Family
ID=64742311
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019526938A Active JP7162897B2 (ja) | 2017-06-26 | 2018-06-26 | 融合タンパク質 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20200157210A1 (ja) |
| EP (1) | EP3647426A4 (ja) |
| JP (1) | JP7162897B2 (ja) |
| CN (1) | CN111032873A (ja) |
| AU (1) | AU2018292846A1 (ja) |
| CA (1) | CA3070936A1 (ja) |
| WO (1) | WO2019004213A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2023008415A1 (ja) | 2021-07-27 | 2023-02-02 | ||
| CN118146376B (zh) * | 2024-05-09 | 2024-07-05 | 成都微芯新域生物技术有限公司 | Hla-g抗体及其制备方法和用途 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010536341A (ja) | 2007-08-15 | 2010-12-02 | アムニクス, インコーポレイテッド | 生物学的に活性なポリペプチドの特性を改変するための組成物および方法 |
| JP2016500511A (ja) | 2012-09-14 | 2016-01-14 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | 少なくとも2つの異なる実体を含む分子を作製および選択するための方法ならびにその使用 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2456121A1 (en) | 2001-08-03 | 2003-02-20 | Medical Research Council | Intracellular antibodies |
-
2018
- 2018-06-26 JP JP2019526938A patent/JP7162897B2/ja active Active
- 2018-06-26 CA CA3070936A patent/CA3070936A1/en active Pending
- 2018-06-26 CN CN201880055237.6A patent/CN111032873A/zh active Pending
- 2018-06-26 WO PCT/JP2018/024197 patent/WO2019004213A1/ja unknown
- 2018-06-26 EP EP18825349.6A patent/EP3647426A4/en not_active Withdrawn
- 2018-06-26 US US16/625,278 patent/US20200157210A1/en not_active Abandoned
- 2018-06-26 AU AU2018292846A patent/AU2018292846A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010536341A (ja) | 2007-08-15 | 2010-12-02 | アムニクス, インコーポレイテッド | 生物学的に活性なポリペプチドの特性を改変するための組成物および方法 |
| JP2016500511A (ja) | 2012-09-14 | 2016-01-14 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | 少なくとも2つの異なる実体を含む分子を作製および選択するための方法ならびにその使用 |
Non-Patent Citations (1)
| Title |
|---|
| Protein Engineering, Design & Selection,2010年,Vol.23, No.6,p.489-498 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019004213A1 (ja) | 2019-01-03 |
| JPWO2019004213A1 (ja) | 2020-05-21 |
| CA3070936A1 (en) | 2019-01-03 |
| CN111032873A (zh) | 2020-04-17 |
| EP3647426A1 (en) | 2020-05-06 |
| US20200157210A1 (en) | 2020-05-21 |
| EP3647426A4 (en) | 2021-04-14 |
| AU2018292846A1 (en) | 2020-02-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7095019B2 (ja) | 抗rho gtpaseコンホーメーションシングルドメイン抗体及びその使用 | |
| US10882902B2 (en) | Human islet amyloid polypeptide (hIAPP) specific antibodies and uses thereof | |
| US8404233B2 (en) | Neutralizing monoclonal antibody against human DLL4 | |
| US20220332808A1 (en) | Human-derived anti-huntingtin (htt) antibodies and uses thereof | |
| JP6952827B2 (ja) | 血液脳関門輸送分子およびそれらの使用 | |
| CN109310756A (zh) | 新型血管生成素2,vegf双特异性拮抗剂 | |
| JP2020023517A (ja) | Frizzledタンパク質に対する抗体及びその使用方法 | |
| JP2010530735A (ja) | 細胞内のprl−1ポリペプチドまたはprl−3ポリペプチドに結合する抗体 | |
| Kabayama et al. | An ultra-stable cytoplasmic antibody engineered for in vivo applications | |
| JP2017531426A (ja) | Lox1特異的結合タンパク質及びその使用 | |
| CN113631573A (zh) | 通过靶向新Tau物种治疗Tau蛋白病的方法 | |
| Bartl et al. | Inhibiting cellular uptake of mutant huntingtin using a monoclonal antibody: Implications for the treatment of Huntington's disease | |
| JP7162897B2 (ja) | 融合タンパク質 | |
| KR20220131233A (ko) | Semg2 항체 및 이의 용도 | |
| EP4269588A1 (en) | Anti-epha4 antibody | |
| JP2024509973A (ja) | Tdp-43に対する抗体およびその使用方法 | |
| US20130216546A1 (en) | Methods of Treating Glucose Metabolism Disorders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20201214 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20211214 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220210 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220621 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220722 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20220913 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20221012 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7162897 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |