JP2012509085A - マイクロキャリア上での多能性幹細胞の培養 - Google Patents
マイクロキャリア上での多能性幹細胞の培養 Download PDFInfo
- Publication number
- JP2012509085A JP2012509085A JP2011537603A JP2011537603A JP2012509085A JP 2012509085 A JP2012509085 A JP 2012509085A JP 2011537603 A JP2011537603 A JP 2011537603A JP 2011537603 A JP2011537603 A JP 2011537603A JP 2012509085 A JP2012509085 A JP 2012509085A
- Authority
- JP
- Japan
- Prior art keywords
- cells
- microcarriers
- cell
- stem cells
- pluripotent stem
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 210000001778 pluripotent stem cell Anatomy 0.000 title claims abstract description 83
- 238000000034 method Methods 0.000 claims abstract description 99
- 210000004027 cell Anatomy 0.000 claims description 703
- 210000001900 endoderm Anatomy 0.000 claims description 104
- 239000003550 marker Substances 0.000 claims description 40
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 39
- 230000009996 pancreatic endocrine effect Effects 0.000 claims description 31
- 239000003590 rho kinase inhibitor Substances 0.000 claims description 29
- 238000012258 culturing Methods 0.000 claims description 23
- 108090001061 Insulin Proteins 0.000 claims description 21
- 102000004877 Insulin Human genes 0.000 claims description 20
- 229940125396 insulin Drugs 0.000 claims description 20
- 210000000750 endocrine system Anatomy 0.000 claims description 16
- 229920002307 Dextran Polymers 0.000 claims description 7
- 230000002062 proliferating effect Effects 0.000 claims description 6
- 239000004793 Polystyrene Substances 0.000 claims description 5
- 229920002223 polystyrene Polymers 0.000 claims description 5
- 230000002255 enzymatic effect Effects 0.000 claims description 4
- 210000001671 embryonic stem cell Anatomy 0.000 description 93
- 239000002609 medium Substances 0.000 description 71
- 230000004069 differentiation Effects 0.000 description 58
- ILDBNQGLZFSHQZ-UTLKBRERSA-N 2-amino-1-[(3s)-3-methyl-4-(4-methylisoquinolin-5-yl)sulfonyl-1,4-diazepan-1-yl]ethanone;dihydrochloride Chemical compound Cl.Cl.C[C@H]1CN(C(=O)CN)CCCN1S(=O)(=O)C1=CC=CC2=CN=CC(C)=C12 ILDBNQGLZFSHQZ-UTLKBRERSA-N 0.000 description 38
- 230000014509 gene expression Effects 0.000 description 34
- 239000000758 substrate Substances 0.000 description 34
- 230000012010 growth Effects 0.000 description 31
- 108010076089 accutase Proteins 0.000 description 30
- 210000000130 stem cell Anatomy 0.000 description 28
- 238000010899 nucleation Methods 0.000 description 27
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 26
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 description 26
- 239000004033 plastic Substances 0.000 description 24
- 229920003023 plastic Polymers 0.000 description 24
- 210000001519 tissue Anatomy 0.000 description 22
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 21
- 108010082117 matrigel Proteins 0.000 description 21
- 230000010261 cell growth Effects 0.000 description 18
- 108010023082 activin A Proteins 0.000 description 17
- 210000003890 endocrine cell Anatomy 0.000 description 17
- 210000002950 fibroblast Anatomy 0.000 description 17
- 108060005980 Collagenase Proteins 0.000 description 16
- 102000029816 Collagenase Human genes 0.000 description 16
- 229960002424 collagenase Drugs 0.000 description 16
- 238000004113 cell culture Methods 0.000 description 13
- 238000005516 engineering process Methods 0.000 description 13
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 13
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 12
- 239000011324 bead Substances 0.000 description 12
- 230000035755 proliferation Effects 0.000 description 12
- 102000004190 Enzymes Human genes 0.000 description 11
- 108090000790 Enzymes Proteins 0.000 description 11
- 230000002354 daily effect Effects 0.000 description 11
- 229940088598 enzyme Drugs 0.000 description 11
- 238000002474 experimental method Methods 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 description 10
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 10
- 108020004999 messenger RNA Proteins 0.000 description 10
- 108090000623 proteins and genes Proteins 0.000 description 10
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 9
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 9
- -1 GATA-6 Proteins 0.000 description 9
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 9
- 239000003636 conditioned culture medium Substances 0.000 description 9
- 230000001419 dependent effect Effects 0.000 description 9
- 239000003112 inhibitor Substances 0.000 description 9
- WDHRPWOAMDJICD-FOAQWNCLSA-N n-[2-[(3'r,3'as,6's,6as,6bs,7'ar,9r,11as,11br)-3',6',10,11b-tetramethyl-3-oxospiro[1,2,4,6,6a,6b,7,8,11,11a-decahydrobenzo[a]fluorene-9,2'-3,3a,5,6,7,7a-hexahydrofuro[3,2-b]pyridine]-4'-yl]ethyl]-6-(3-phenylpropanoylamino)hexanamide Chemical compound C([C@@H](C)C[C@@H]1[C@@H]2[C@H]([C@]3(C(=C4C[C@@H]5[C@@]6(C)CCC(=O)CC6=CC[C@H]5[C@@H]4CC3)C)O1)C)N2CCNC(=O)CCCCCNC(=O)CCC1=CC=CC=C1 WDHRPWOAMDJICD-FOAQWNCLSA-N 0.000 description 9
- 102000051325 Glucagon Human genes 0.000 description 8
- 108060003199 Glucagon Proteins 0.000 description 8
- 102000052651 Pancreatic hormone Human genes 0.000 description 8
- 238000013019 agitation Methods 0.000 description 8
- 210000002744 extracellular matrix Anatomy 0.000 description 8
- 238000000684 flow cytometry Methods 0.000 description 8
- 229960004666 glucagon Drugs 0.000 description 8
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 8
- 230000006698 induction Effects 0.000 description 8
- 239000004025 pancreas hormone Substances 0.000 description 8
- 229940032957 pancreatic hormone Drugs 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- 210000002966 serum Anatomy 0.000 description 8
- 108010035532 Collagen Proteins 0.000 description 7
- 102000008186 Collagen Human genes 0.000 description 7
- 101800001268 Pancreatic hormone Proteins 0.000 description 7
- 101710183548 Pyridoxal 5'-phosphate synthase subunit PdxS Proteins 0.000 description 7
- 238000011529 RT qPCR Methods 0.000 description 7
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 7
- 108090000631 Trypsin Proteins 0.000 description 7
- 102000004142 Trypsin Human genes 0.000 description 7
- 230000001464 adherent effect Effects 0.000 description 7
- 239000001913 cellulose Substances 0.000 description 7
- 229920002678 cellulose Polymers 0.000 description 7
- 229920001436 collagen Polymers 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 239000012588 trypsin Substances 0.000 description 7
- 239000012583 B-27 Supplement Substances 0.000 description 6
- 108010011459 Exenatide Proteins 0.000 description 6
- 102100028071 Fibroblast growth factor 7 Human genes 0.000 description 6
- 101001060261 Homo sapiens Fibroblast growth factor 7 Proteins 0.000 description 6
- 101000578254 Homo sapiens Homeobox protein Nkx-6.1 Proteins 0.000 description 6
- 102100038553 Neurogenin-3 Human genes 0.000 description 6
- 102100041030 Pancreas/duodenum homeobox protein 1 Human genes 0.000 description 6
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 6
- 230000021164 cell adhesion Effects 0.000 description 6
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 6
- 239000000306 component Substances 0.000 description 6
- 210000004039 endoderm cell Anatomy 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 229960001519 exenatide Drugs 0.000 description 6
- 102000045246 noggin Human genes 0.000 description 6
- 108700007229 noggin Proteins 0.000 description 6
- 229930002330 retinoic acid Natural products 0.000 description 6
- 238000012546 transfer Methods 0.000 description 6
- 229960001727 tretinoin Drugs 0.000 description 6
- LBPKYPYHDKKRFS-UHFFFAOYSA-N 1,5-naphthyridine, 2-[3-(6-methyl-2-pyridinyl)-1h-pyrazol-4-yl]- Chemical compound CC1=CC=CC(C2=C(C=NN2)C=2N=C3C=CC=NC3=CC=2)=N1 LBPKYPYHDKKRFS-UHFFFAOYSA-N 0.000 description 5
- DWJXYEABWRJFSP-XOBRGWDASA-N DAPT Chemical compound N([C@@H](C)C(=O)N[C@H](C(=O)OC(C)(C)C)C=1C=CC=CC=1)C(=O)CC1=CC(F)=CC(F)=C1 DWJXYEABWRJFSP-XOBRGWDASA-N 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 5
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 241000508269 Psidium Species 0.000 description 5
- 238000003795 desorption Methods 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 238000010494 dissociation reaction Methods 0.000 description 5
- 230000005593 dissociations Effects 0.000 description 5
- 238000011977 dual antiplatelet therapy Methods 0.000 description 5
- 210000002889 endothelial cell Anatomy 0.000 description 5
- 210000002919 epithelial cell Anatomy 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 150000004665 fatty acids Chemical class 0.000 description 5
- 229940126864 fibroblast growth factor Drugs 0.000 description 5
- 210000001654 germ layer Anatomy 0.000 description 5
- 229920001184 polypeptide Polymers 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 108090000765 processed proteins & peptides Proteins 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 101710201246 Eomesodermin Proteins 0.000 description 4
- 102100030751 Eomesodermin homolog Human genes 0.000 description 4
- 101000578258 Homo sapiens Homeobox protein Nkx-6.2 Proteins 0.000 description 4
- 101000603702 Homo sapiens Neurogenin-3 Proteins 0.000 description 4
- 102100024392 Insulin gene enhancer protein ISL-1 Human genes 0.000 description 4
- 102100032063 Neurogenic differentiation factor 1 Human genes 0.000 description 4
- 230000005913 Notch signaling pathway Effects 0.000 description 4
- 239000012979 RPMI medium Substances 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 210000002257 embryonic structure Anatomy 0.000 description 4
- 230000003203 everyday effect Effects 0.000 description 4
- 239000003102 growth factor Substances 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 229940088597 hormone Drugs 0.000 description 4
- 239000005556 hormone Substances 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 210000002569 neuron Anatomy 0.000 description 4
- 210000000496 pancreas Anatomy 0.000 description 4
- 239000011148 porous material Substances 0.000 description 4
- 210000001811 primitive streak Anatomy 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 239000011435 rock Substances 0.000 description 4
- 230000003248 secreting effect Effects 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 230000035899 viability Effects 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 3
- 102100036008 CD48 antigen Human genes 0.000 description 3
- 102000024905 CD99 Human genes 0.000 description 3
- 108060001253 CD99 Proteins 0.000 description 3
- 102100037986 Dickkopf-related protein 4 Human genes 0.000 description 3
- 102100035308 Fibroblast growth factor 17 Human genes 0.000 description 3
- 102100028072 Fibroblast growth factor 4 Human genes 0.000 description 3
- 102100037680 Fibroblast growth factor 8 Human genes 0.000 description 3
- 102100023374 Forkhead box protein M1 Human genes 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 102100028096 Homeobox protein Nkx-6.2 Human genes 0.000 description 3
- 102100030634 Homeobox protein OTX2 Human genes 0.000 description 3
- 102100038146 Homeobox protein goosecoid Human genes 0.000 description 3
- 108010048671 Homeodomain Proteins Proteins 0.000 description 3
- 102000009331 Homeodomain Proteins Human genes 0.000 description 3
- 101000716130 Homo sapiens CD48 antigen Proteins 0.000 description 3
- 101000951340 Homo sapiens Dickkopf-related protein 4 Proteins 0.000 description 3
- 101000878124 Homo sapiens Fibroblast growth factor 17 Proteins 0.000 description 3
- 101001060274 Homo sapiens Fibroblast growth factor 4 Proteins 0.000 description 3
- 101001027382 Homo sapiens Fibroblast growth factor 8 Proteins 0.000 description 3
- 101000907578 Homo sapiens Forkhead box protein M1 Proteins 0.000 description 3
- 101000584400 Homo sapiens Homeobox protein OTX2 Proteins 0.000 description 3
- 101001053263 Homo sapiens Insulin gene enhancer protein ISL-1 Proteins 0.000 description 3
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 3
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 101100310648 Mus musculus Sox17 gene Proteins 0.000 description 3
- 101710144033 Pancreas/duodenum homeobox protein 1 Proteins 0.000 description 3
- 102000035195 Peptidases Human genes 0.000 description 3
- 108091005804 Peptidases Proteins 0.000 description 3
- 239000004365 Protease Substances 0.000 description 3
- 102000005157 Somatostatin Human genes 0.000 description 3
- 108010056088 Somatostatin Proteins 0.000 description 3
- 108091023040 Transcription factor Proteins 0.000 description 3
- 102000040945 Transcription factor Human genes 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 210000002459 blastocyst Anatomy 0.000 description 3
- 229940098773 bovine serum albumin Drugs 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 3
- 210000003494 hepatocyte Anatomy 0.000 description 3
- 230000033001 locomotion Effects 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 108010041788 rho-Associated Kinases Proteins 0.000 description 3
- 102000000568 rho-Associated Kinases Human genes 0.000 description 3
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 3
- 229960000553 somatostatin Drugs 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- 239000013589 supplement Substances 0.000 description 3
- 238000004114 suspension culture Methods 0.000 description 3
- HFDKKNHCYWNNNQ-YOGANYHLSA-N 75976-10-2 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)N)C(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 HFDKKNHCYWNNNQ-YOGANYHLSA-N 0.000 description 2
- 102100025745 Cerberus Human genes 0.000 description 2
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 2
- 108010087745 Hepatocyte Nuclear Factor 3-beta Proteins 0.000 description 2
- 102000009094 Hepatocyte Nuclear Factor 3-beta Human genes 0.000 description 2
- 108700014808 Homeobox Protein Nkx-2.2 Proteins 0.000 description 2
- 102100028098 Homeobox protein Nkx-6.1 Human genes 0.000 description 2
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 2
- 101000898034 Homo sapiens Hepatocyte growth factor Proteins 0.000 description 2
- 101001076408 Homo sapiens Interleukin-6 Proteins 0.000 description 2
- 101000613495 Homo sapiens Paired box protein Pax-4 Proteins 0.000 description 2
- 101000868152 Homo sapiens Son of sevenless homolog 1 Proteins 0.000 description 2
- 101000819074 Homo sapiens Transcription factor GATA-4 Proteins 0.000 description 2
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 2
- 101100013973 Mus musculus Gata4 gene Proteins 0.000 description 2
- GKWTZACONBQTNK-IVIZKJKWSA-N N-benzyl-7H-purin-6-amine (1R,2R,5R,8R,9S,10R,12S)-12-hydroxy-11-methyl-6-methylidene-16-oxo-15-oxapentacyclo[9.3.2.15,8.01,10.02,8]heptadecane-9-carboxylic acid (1R,2R,5R,8R,9S,10R,12S)-12-hydroxy-11-methyl-6-methylidene-16-oxo-15-oxapentacyclo[9.3.2.15,8.01,10.02,8]heptadec-13-ene-9-carboxylic acid Chemical compound C(Nc1ncnc2nc[nH]c12)c1ccccc1.CC12[C@H]3[C@H](C(O)=O)[C@@]45C[C@@H](CC[C@H]4[C@@]3(CC[C@@H]1O)OC2=O)C(=C)C5.CC12[C@H]3[C@H](C(O)=O)[C@@]45C[C@@H](CC[C@H]4[C@]3(OC1=O)C=C[C@@H]2O)C(=C)C5 GKWTZACONBQTNK-IVIZKJKWSA-N 0.000 description 2
- 101150079937 NEUROD1 gene Proteins 0.000 description 2
- 108050000588 Neurogenic differentiation factor 1 Proteins 0.000 description 2
- 101710096141 Neurogenin-3 Proteins 0.000 description 2
- 102100040909 Paired box protein Pax-4 Human genes 0.000 description 2
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 2
- 238000001190 Q-PCR Methods 0.000 description 2
- 101000983124 Sus scrofa Pancreatic prohormone precursor Proteins 0.000 description 2
- 102100021380 Transcription factor GATA-4 Human genes 0.000 description 2
- 239000012867 bioactive agent Substances 0.000 description 2
- 238000005266 casting Methods 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 239000002458 cell surface marker Substances 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 230000001143 conditioned effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 210000003981 ectoderm Anatomy 0.000 description 2
- 210000002242 embryoid body Anatomy 0.000 description 2
- 210000002308 embryonic cell Anatomy 0.000 description 2
- 210000003989 endothelium vascular Anatomy 0.000 description 2
- 238000005530 etching Methods 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 108010069764 helospectin I Proteins 0.000 description 2
- HTMVMVKJOPFRMK-OYZAELBCSA-N helospectin i Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=C(O)C=C1 HTMVMVKJOPFRMK-OYZAELBCSA-N 0.000 description 2
- 238000010191 image analysis Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- OGQSCIYDJSNCMY-UHFFFAOYSA-H iron(3+);methyl-dioxido-oxo-$l^{5}-arsane Chemical compound [Fe+3].[Fe+3].C[As]([O-])([O-])=O.C[As]([O-])([O-])=O.C[As]([O-])([O-])=O OGQSCIYDJSNCMY-UHFFFAOYSA-H 0.000 description 2
- 210000002510 keratinocyte Anatomy 0.000 description 2
- 238000002386 leaching Methods 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 210000003716 mesoderm Anatomy 0.000 description 2
- 239000011859 microparticle Substances 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- QNILTEGFHQSKFF-UHFFFAOYSA-N n-propan-2-ylprop-2-enamide Chemical compound CC(C)NC(=O)C=C QNILTEGFHQSKFF-UHFFFAOYSA-N 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 230000035935 pregnancy Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 238000003753 real-time PCR Methods 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 238000013341 scale-up Methods 0.000 description 2
- 238000009331 sowing Methods 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000003068 static effect Effects 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- LAQPKDLYOBZWBT-NYLDSJSYSA-N (2s,4s,5r,6r)-5-acetamido-2-{[(2s,3r,4s,5s,6r)-2-{[(2r,3r,4r,5r)-5-acetamido-1,2-dihydroxy-6-oxo-4-{[(2s,3s,4r,5s,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxy}hexan-3-yl]oxy}-3,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy}-4-hydroxy-6-[(1r,2r)-1,2,3-trihydrox Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]([C@@H](NC(C)=O)C=O)[C@@H]([C@H](O)CO)O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O)[C@@H](CO)O1 LAQPKDLYOBZWBT-NYLDSJSYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 108090000145 Bacillolysin Proteins 0.000 description 1
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 1
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- 101800001318 Capsid protein VP4 Proteins 0.000 description 1
- 241000202252 Cerberus Species 0.000 description 1
- 101710010675 Cerberus Proteins 0.000 description 1
- 206010010099 Combined immunodeficiency Diseases 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 101100518002 Danio rerio nkx2.2a gene Proteins 0.000 description 1
- 108010014258 Elastin Proteins 0.000 description 1
- 102000016942 Elastin Human genes 0.000 description 1
- 102100031785 Endothelial transcription factor GATA-2 Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 108090000394 Erythropoietin Proteins 0.000 description 1
- 102000003951 Erythropoietin Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 102100037060 Forkhead box protein D3 Human genes 0.000 description 1
- 108010086527 Hepatocyte Nuclear Factor 6 Proteins 0.000 description 1
- 102100029284 Hepatocyte nuclear factor 3-beta Human genes 0.000 description 1
- 102100029087 Hepatocyte nuclear factor 6 Human genes 0.000 description 1
- 102100027886 Homeobox protein Nkx-2.2 Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000914195 Homo sapiens Cerberus Proteins 0.000 description 1
- 101001066265 Homo sapiens Endothelial transcription factor GATA-2 Proteins 0.000 description 1
- 101001029308 Homo sapiens Forkhead box protein D3 Proteins 0.000 description 1
- 101001062347 Homo sapiens Hepatocyte nuclear factor 3-beta Proteins 0.000 description 1
- 101001032602 Homo sapiens Homeobox protein goosecoid Proteins 0.000 description 1
- 101000971533 Homo sapiens Killer cell lectin-like receptor subfamily G member 1 Proteins 0.000 description 1
- 101100460496 Homo sapiens NKX2-2 gene Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000819088 Homo sapiens Transcription factor GATA-6 Proteins 0.000 description 1
- 101000979205 Homo sapiens Transcription factor MafA Proteins 0.000 description 1
- 101000652324 Homo sapiens Transcription factor SOX-17 Proteins 0.000 description 1
- 101000687905 Homo sapiens Transcription factor SOX-2 Proteins 0.000 description 1
- 101000976622 Homo sapiens Zinc finger protein 42 homolog Proteins 0.000 description 1
- 108010085895 Laminin Proteins 0.000 description 1
- 102000007547 Laminin Human genes 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 101100351017 Mus musculus Pax4 gene Proteins 0.000 description 1
- 101100043062 Mus musculus Sox7 gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- GIYXAJPCNFJEHY-UHFFFAOYSA-N N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]-1-propanamine hydrochloride (1:1) Chemical compound Cl.C=1C=CC=CC=1C(CCNC)OC1=CC=C(C(F)(F)F)C=C1 GIYXAJPCNFJEHY-UHFFFAOYSA-N 0.000 description 1
- 102100034388 Netrin-4 Human genes 0.000 description 1
- 101710121532 Netrin-4 Proteins 0.000 description 1
- 102000035092 Neutral proteases Human genes 0.000 description 1
- 108091005507 Neutral proteases Proteins 0.000 description 1
- 108010032788 PAX6 Transcription Factor Proteins 0.000 description 1
- 102000007354 PAX6 Transcription Factor Human genes 0.000 description 1
- 101150081664 PAX6 gene Proteins 0.000 description 1
- 102100037506 Paired box protein Pax-6 Human genes 0.000 description 1
- 108700020479 Pancreatic hormone Proteins 0.000 description 1
- 101150075928 Pax4 gene Proteins 0.000 description 1
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 1
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 1
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 229920002385 Sodium hyaluronate Polymers 0.000 description 1
- 102000013275 Somatomedins Human genes 0.000 description 1
- 102000004874 Synaptophysin Human genes 0.000 description 1
- 108090001076 Synaptophysin Proteins 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- 102100021382 Transcription factor GATA-6 Human genes 0.000 description 1
- 102100030243 Transcription factor SOX-17 Human genes 0.000 description 1
- 102100024270 Transcription factor SOX-2 Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 208000037280 Trisomy Diseases 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 108010031318 Vitronectin Proteins 0.000 description 1
- 102100035140 Vitronectin Human genes 0.000 description 1
- 210000002593 Y chromosome Anatomy 0.000 description 1
- 102100023550 Zinc finger protein 42 homolog Human genes 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 210000001789 adipocyte Anatomy 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 210000002403 aortic endothelial cell Anatomy 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 238000006065 biodegradation reaction Methods 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000002798 bone marrow cell Anatomy 0.000 description 1
- 229940112869 bone morphogenetic protein Drugs 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 210000004413 cardiac myocyte Anatomy 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 239000002771 cell marker Substances 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 210000001612 chondrocyte Anatomy 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000011437 continuous method Methods 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 238000012136 culture method Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000032459 dedifferentiation Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000011143 downstream manufacturing Methods 0.000 description 1
- 238000007877 drug screening Methods 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 229920002549 elastin Polymers 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 229940105423 erythropoietin Drugs 0.000 description 1
- 210000000646 extraembryonic cell Anatomy 0.000 description 1
- 230000035558 fertility Effects 0.000 description 1
- 210000003754 fetus Anatomy 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 230000007045 gastrulation Effects 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 210000002064 heart cell Anatomy 0.000 description 1
- 238000010562 histological examination Methods 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 210000003917 human chromosome Anatomy 0.000 description 1
- 210000003090 iliac artery Anatomy 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 108010090448 insulin gene enhancer binding protein Isl-1 Proteins 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 210000002752 melanocyte Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 238000001682 microtransfer moulding Methods 0.000 description 1
- 210000004925 microvascular endothelial cell Anatomy 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 210000003098 myoblast Anatomy 0.000 description 1
- 210000000107 myocyte Anatomy 0.000 description 1
- 210000005155 neural progenitor cell Anatomy 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 210000000963 osteoblast Anatomy 0.000 description 1
- 239000013618 particulate matter Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 210000001147 pulmonary artery Anatomy 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 210000004116 schwann cell Anatomy 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 230000008410 smoothened signaling pathway Effects 0.000 description 1
- 229940010747 sodium hyaluronate Drugs 0.000 description 1
- YWIVKILSMZOHHF-QJZPQSOGSA-N sodium;(2s,3s,4s,5r,6r)-6-[(2s,3r,4r,5s,6r)-3-acetamido-2-[(2s,3s,4r,5r,6r)-6-[(2r,3r,4r,5s,6r)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2- Chemical compound [Na+].CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 YWIVKILSMZOHHF-QJZPQSOGSA-N 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 230000000920 spermatogeneic effect Effects 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 210000002948 striated muscle cell Anatomy 0.000 description 1
- 210000002536 stromal cell Anatomy 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 229920000208 temperature-responsive polymer Polymers 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 1
- 210000001644 umbilical artery Anatomy 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 230000036266 weeks of gestation Effects 0.000 description 1
Images















































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0603—Embryonic cells ; Embryoid bodies
- C12N5/0606—Pluripotent embryonic cells, e.g. embryonic stem cells [ES]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0068—General culture methods using substrates
- C12N5/0075—General culture methods using substrates using microcarriers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0676—Pancreatic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/25—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving enzymes not classifiable in groups C12Q1/26 - C12Q1/66
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/72—Transferases [EC 2.]
- C12N2501/727—Kinases (EC 2.7.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/999—Small molecules not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/02—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from embryonic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2509/00—Methods for the dissociation of cells, e.g. specific use of enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2531/00—Microcarriers
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Cell Biology (AREA)
- Developmental Biology & Embryology (AREA)
- Reproductive Health (AREA)
- Gynecology & Obstetrics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Transplantation (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
Abstract
Description
本発明は、多能性幹細胞をマイクロキャリア上で増殖、増量(expansion)及び分化させるための方法を目的とする。
a.多能性幹細胞集団を、第1容量のマイクロキャリアに付着させる工程と、
b.多能性幹細胞を、第1容量のマイクロキャリア上で培養する工程と、
c.多能性幹細胞を、第1容量のマイクロキャリアから取り外す工程と、
d.多能性幹細胞集団を、第2容量のマイクロキャリアへと付着させる工程と、を含む。
幹細胞は、単一の細胞レベルで自己複製し、分化して後代細胞を生成する、それら両方の能力で定義される未分化細胞であり、後代細胞には、自己複製前駆細胞、非再生前駆細胞、及び最終分化細胞が含まれる。同様に、幹細胞は、インビトロで複数の胚葉(内胚葉、中胚葉及び外胚葉)から様々な細胞系の機能細胞へと分化する能力によって、並びに移植後に複数の胚葉の組織を生じ、胚盤胞への注入後、全てではないとしても殆どの組織を提供する能力によっても特徴付けられる。
「マイクロキャリア」は、足場依存性の細胞を培養する際に、この細胞の付着及び増殖に有用な粒子、ビーズ又はペレットを指す。「マイクロキャリア」は以下の特性を有する:(a)(マイクロキャリア又は細胞に、有意なせん断ダメージを引き起こさないような撹拌速度での)懸濁培養で使用可能な程度に十分小さい;(b)中実である、あるいは表面に多孔性コーティングを備える中実コアを有している;及び(c)表面(多孔性キャリアの場合、外側表面と内側表面)が正又は負に帯電し得る。一態様では、マイクロキャリアは全般的に約150〜350μmの粒径を有し、かつ約0.8〜2.0meq/gの正電荷密度を有する。有用なマイクロキャリアとしては、限定するものではないが、Cytodex 1(登録商標)、Cytodex 2(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が挙げられる。
マイクロキャリア培養は、足場依存性の細胞(例えばヒト胚性幹細胞)の実用的な高収率培養を可能にする技術である。マイクロキャリアは、特に数ミリリットル〜1000Lを超える範囲の培養容量でヒト胚性幹細胞などの細胞を培養するために開発されてきた。マイクロキャリアは生物学的に不活性であり、撹拌式マイクロキャリア培養法に、丈夫であるが非剛性の基材を提供する。マイクロキャリアは透明であり得、付着した細胞の顕微鏡検査が可能である。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)は、架橋デキストランマトリックスに化学結合させた、変性コラーゲンの薄い層から構成される。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)の変性コラーゲン層は、トリプシン及びコラゲナーゼが挙げられる、様々なプロテアーゼにより消化されやすく、細胞の生存率、機能及び完全性を最大限維持しつつ、細胞をマイクロキャリアから取り外すという性能を提供する。
多能性幹細胞の特徴付け
多能性幹細胞は、ステージ特異的胚抗原(SSEA)3及び4の1種以上、並びにTra−1−60及びTra−1−81と呼ばれる抗体によって検出可能なマーカーを発現し得る(Thomsonら、Science 282:1145,1998)。インビトロで多能性幹細胞を分化させると、SSEA−4、Tra−1−60、及びTra−1−81の発現が消失し(存在する場合)、SSEA−1の発現が増大する。未分化の多能性幹細胞は、一般にアルカリホスファターゼ活性を有し、この活性は、製造業者(Vector Laboratories,Burlingame,Calif.)により説明されるように、細胞を4%パラホルムアルデヒドで固定した後、基質としてVector Redを使用して展開させることにより検出することができる。未分化の多能性幹細胞はまた、RT−PCRにより検出されるように、一般にOCT4及びTERTも発現する。
使用が可能な多能性幹細胞の種類としては、妊娠期間中の任意の時期(必ずしもではないが、通常は妊娠約10〜12週よりも前)に採取した前胚性組織(例えば胚盤胞など)、胚性組織、胎児組織などの、妊娠後に形成される組織に由来する多能性細胞の株化細胞系が挙げられる。非限定的な例は、例えばヒト胚幹細胞株H1、H7、及びH9(WiCell)などのヒト胚幹細胞又はヒト胚生殖細胞の確立株である。それらの細胞の最初の樹立又は安定化中に本開示の組成物を使用することも想定され、その場合、源となる細胞は、源となる組織から直接採取した一次多能性細胞であろう。フィーダー細胞の非存在下で既に培養された多能性幹細胞集団から採取した細胞も好適である。例えば、BG01v(BresaGen,Athens,GA)などの変異ヒト胚性幹細胞株も好適である。同様に、例えば成体体細胞などの非多能性細胞由来の多能性幹細胞も好適である。
多能性幹細胞は、マイクロキャリアに付着させる前に、当該技術分野の任意の方法により、平面状基材にて培養してもよい。例えば多能性幹細胞は、細胞外マトリックスタンパク質(例えばMATRIGEL)で処理した平面状基材にて培養することができる。別の方法としては、多能性幹細胞は、フィーダー細胞層を播種した平面状基材にて培養することもできる。
一実施形態では、多能性幹細胞は、胚体内胚葉系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。あるいは、多能性幹細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。あるいは、多能性幹細胞は、膵内分泌系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。
多能性幹細胞は、当該技術分野における任意の方法により、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
多能性幹細胞は、当該技術分野における任意の方法により、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
ヒト胚性幹細胞がマイクロキャリア上に付着しかつ増殖し得るか否かを判定するために、TrypLE(商標)Expressにより、継代数52のH9細胞をMATRIGEL(商標)(BD Biosciences,CA)コートプレートから放出させた。次いでそれらをマイクロキャリアとMEF−CMと共にインキュベートした。製造元からの取扱説明書に従い、ProNectinF(PN)、Plastic(P)、PlasticPlus(PP)、HILLEX(登録商標)II(H)、コラーゲン(Col)及びFACT III(SoloHill,MI)マイクロキャリアの懸濁液を調製した。毎日の観察結果に基づき、表1はH9細胞の、37℃にて2日経過後の、マイクロキャリアに対する付着と増殖を記載する。試験したほとんどのマイクロキャリアに対して、大部分の細胞は付着及び/又は増殖しなかった。H9細胞は、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対しては付着し増殖したが、画像で示される細胞−ビーズ凝集体は、2日間の静置培養後にはより少なくなった(図1B)。
マイクロキャリアに対するヒト胚性幹細胞の付着と増殖を最も支持するRhoキナーゼ阻害剤濃度を決定するため、以下の実験を実施した。
播種密度の改善は、必要とされる細胞の総数を減少させるための一手段である。適切な播種密度を決定するために、4x対物視野あたりのマイクロキャリア数を計数した。Cytodex 3マイクロキャリア(GE Healthcare Life Sciences,NJ)を含有している、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMの入った10cmプレートに、0.4×104個細胞/cm2(低密度)、1.2×104個細胞/cm2(中間密度)、又は3×104個細胞/cm2(高密度)の密度でH1細胞を播種した。次いでプレートを、37℃で6時間にわたり、45分毎に撹拌した。37℃にて30rpmのスピナーフラスコ(実施例5に記載)内の、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM 50mLに、細胞及びマイクロキャリアを移した。24時間後に、5μMのY27632(Sigma−Aldrich,MO)添加MEF−CM 25mLを添加した。24時間後、回転速度を40rpmに上昇させた。培養の3日目及び5日目に、75mLのうちの50mLを除去し、MEF−CMで交換した。6時間、3日、5日及び7日の時点で、スピナーフラスコからアリコートを取って画像を撮影した。細胞の付着したマイクロキャリアの百分率を、図9の画像の右下に記載する。播種後3日目では、細胞でコートされたマイクロキャリアの数は開始播種密度と対応したが、低密度で播種した細胞では、細胞でコートされたマイクロキャリアの数は5日目及び7日目でも増加しなかった。このことは、0.4×104個細胞/cm2が、細胞をマイクロキャリアへと組み込ませて凝集体にするのに十分な細胞数ではないことを意味する。3×104個細胞/cm2では、細胞が付着したマイクロキャリアの数は、1.2×104個細胞/cm2を播種した場合の5日目及び7日目と同様であった(図9)。細胞数を見た場合、高密度での細胞播種では、より多くの細胞がマイクロキャリアに付着していることは明らかである(図10)。開始播種細胞数と比較しての倍率変化(fold change)の解析では、高密度播種した培養物において、3日目及び5日目に、より多数の細胞が付着していたことを明らかになった(図11)。7日目には、対照と高密度播種培養物は、それらの開始播種密度から、細胞数について同様の倍率で変化した。これらのデータから、我々は、H1細胞をマイクロキャリア上に効果的に付着させ、増殖させるためには、1.2×104個細胞/cm2が最小の細胞数であると結論付けた。より高い播種密度へと移行させることは、細胞の増量に必要とされる日数を減少させる助けとなり得る。
増殖速度を判定するためには、マイクロキャリアから細胞を解離させる必要がある。Rhoキナーゼ阻害剤Y27632(Sigma−Aldrich,MO)を除去しても、H1細胞はマイクロキャリアから解離しなかった(実施例2、図12)。マイクロキャリアから細胞を解離する前に、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上のH9細胞を、倍率10x及び20xで画像解析した(それぞれ図13A,B)。HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上のH9細胞を酵素処理することで、生存細胞を脱着させた(図13C,D及び14)。37℃にて固定式プラットフォーム上で、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)を含有する6ウェルディッシュ中で、6日間にわたってH9細胞を増殖させた。マイクロキャリアに付着させた細胞を15mLコニカルチューブに設置し、マイクロキャリアを沈殿させた後に培地を吸引除去した。沈殿したマイクロキャリアを4mLのPBS(マグネシウムイオン及びカルシウムイオン不含)で3回洗浄し、重量沈降によりマイクロキャリアを沈殿させた。PBSを吸引除去し、1mLのPBSを加えた。細胞を有するマイクロキャリアを、12ウェルの組織培養用無処理プレートの単一ウェルに移した。プレートを、マイクロキャリアを沈殿させることができるような角度で静止させた。PBSを吸引除去し、1mLのTrypLE(商標)Express(Invitrogen,CA)又は0.05%トリプシン/EDTAをウェルに加えた。プレートを、37℃にて固定式プラットフォーム上に、10〜20分間にわたって設置した。プレートを取り外し、3mLのDMEM/F12又はMEF−CMをウェルに添加した。培地を激しくピペッティングし、細胞を放出させた(図13C,D)。顕微鏡下でのマイクロキャリアの観察は、細胞がマイクロキャリアから脱着したことを決定づけた。次いで細胞を、200×gで5分間にわたって遠心した。培地を吸引除去し、ペレットを1mLのDMEM/F12又はMEF−CM培地に再懸濁した。次いでViacount dyeを用いて、Guava PCA−96(Guava Technologies,Hayward,CA)で細胞を計数した。具体的には、培地で適切に希釈した200μL容量の細胞を、2μLのViacountと共に10分間にわたってインキュベートした。生存率と細胞数を決定した(図14)。TrypLE(商標)Express及びトリプシン/EDTAの両方が、細胞をマイクロキャリアから効率的に解離した。
マイクロキャリア上の細胞を増量させるためには、細胞を、マイクロキャリアから脱着又は酵素的に解離させ、かつ新しいマイクロキャリアへと再付着させることができなければならない。マイクロキャリア上での一般的な細胞増殖方法は、細胞の脱着及び再付着特性に依存する。以下の実験は、これがヒト胚性幹細胞の特徴ではなかったことを示す。具体的には、継代数43のH9細胞をHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上に播種し、125mLのスピナーフラスコ中でインキュベートした(以下を参照のこと)。フェノールレッドが培地中に存在し、かつHILLEX(登録商標)IIマイクロキャリアにより取り込まれた(Solohill,MI)。8日間の増殖後に、マイクロキャリア上の細胞のアリコート10mLを、フェノールレッド不含MEF−CMと、440mgのHILLEX(登録商標)IIマイクロキャリアと、5μMのY27632(Sigma−Aldrich,MO)とを含有している、新しいスピナーフラスコに設置した。37℃にて30rpm回転でのインキュベーションの5日目に、マイクロキャリアを取り出して画像を得た(図16)。黒っぽいマイクロキャリアは、フェノールレッドを含有している培地で増殖したH9細胞で覆われた、マイクロキャリアを示す。色味の明るいマイクロキャリアは、新しく加えたマイクロキャリアである。H9細胞が脱着し、新しいマイクロキャリアに再付着することが見込まれていたが、細胞は再付着する代わりに新しいマイクロキャリアと共に凝集体を形成した。色味の明るくないマイクロキャリアには細胞が付着しており、この細胞が黒っぽいマイクロキャリアとの凝集体を形成していないことは、細胞はマイクロキャリアから脱着しかつマイクロキャリアへと再付着することはできなかったことを示唆する。マイクロキャリア上の細胞の増殖を伝播させるためには、細胞をマイクロキャリアから酵素的に解離させる必要があった(実施例4を参照されたい)。
治療用製品を製造するためには、ヒト胚性幹細胞培養培地からいずれの動物成分も除外されていることが好ましい。現在、ヒト胚性幹細胞は、マウス胚性線維芽細胞を用いて馴化させた培地(MEF−CM)中の、MATRIGEL(商標)(BD Biosciences,CA)上で維持されている。MATRIGEL(商標)(BD Biosciences,CA)及びMEF−CMのいずれもがマウス細胞由来である。加えて、MEF−CMは高価であり、培地を生成するのに時間がかかる。ヒト胚性幹細胞が、制限培地を含むマイクロキャリア上で維持され得るかを判断するために、Stem Pro(Invitrogen,CA)、mTESR(StemCell Technologies,Vancouver,Canada)又はMEF−CM中の、Rhoキナーゼ阻害剤の10μMのY27632(Sigma−Aldrich,MO)又は2.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下で、H9細胞をCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)II(Solohill,MI)マイクロキャリア上に播種した。細胞を、37℃にて固定式プラットフォーム上の12ウェルディッシュ中に設置した。細胞を3、5及び7日目に計数した。継代数39のH9細胞はMEF−CM中で、どちらの種類のビーズ上でも増殖し、一般的な増量特性を示した(図24)。mTESR(StemCell Technologies,Vancouver,Canada)中で増殖させた同様の細胞は、10μMのY27632(Sigma−Aldrich,MO)の存在下で、Cytodex 3(登録商標)マイクロキャリア上で良好に増殖したが、HILLEX(登録商標)IIマイクロキャリア上では低い増殖率を呈した。20回にわたって継代することでStemPro培地に馴化された、継代数64のヒト胚性幹細胞株H9(継代数64のH9)細胞は、10μMのY27632(Sigma−Aldrich,MO)の存在下で、HILLEX(登録商標)II及びCytodex 3(登録商標)のいずれでも良好に増殖した。驚くべきことに、これらの細胞は、2.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下では、Cytodex 3(登録商標)マイクロキャリア上では良好には増殖しなかった。したがって、マイクロキャリアの種類、Rhoキナーゼ阻害剤、及び培地の全てが、ヒト胚性幹細胞の増殖能を決定する役割を果たす。
ヒト胚性幹細胞がマイクロキャリア上で増量可能であることから、これらの細胞の分化の可能性が決定されるべきである。継代数43のヒト胚性幹細胞株H9の細胞を、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代した。5回目の継代で、細胞をマイクロキャリア上で6日間にわたって増殖させた後、TrypLE(商標)Expressを用いてマイクロキャリアから解離させた(実施例4を参照されたい)。次いで細胞を、1:30 MATRIGEL(商標):DMEM/F12コートしたプレートに設置した。細胞がプレート上で80〜90%コンフルエントに達した後、細胞を分化誘導剤に曝露させた。100ng/mLのアクチビンA(PeproTech,NJ)と、20ng/mLのWnt3a(R&D Biosciences,MN)と、8ng/mL bFGF(PeproTech,NJ)とを添加した、2%ウシ血清アルブミンフラクションV(脂肪酸不含)(FAF BSA,MP Biomedicals,Ohio)含有RPMIで細胞を2日間にわたって処理することで、胚体内胚葉へのヒト胚性幹細胞の分化を実施した。細胞は、100ng/mLのアクチビンA(PeproTech,NJ)と、8ng/mLのbFGF(PeproTech,NJ)とを添加した、2% FAF BSA含有RPMIで更に2日にわたって処理した。培地は毎日交換した。胚体内胚葉の細胞表面マーカーのCXCR4についてFACS解析を実施したところ、87%の細胞がマーカータンパク質を発現していたことが示された(図27A)。継代数49のヒト胚性幹細胞株H1の細胞を、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代して増殖させて、同様の実験を実施したところ、マイクロキャリア上で分化した細胞の91%がCXCR4を発現していたことが明らかになった(図27B)。このことは、マイクロキャリア上で増殖させた細胞は、インスリン産生細胞に分化するための第1段階である、胚体内胚葉へと分化できるということを実証している。
H1ヒト胚性幹細胞を、実施例5に記載の方法に従ってマイクロキャリア上で培養した。細胞をマイクロキャリアから取り外し、3μMのGlycyl−H 1152ジヒドロクロリドを添加したMEFCM16と共に、Nunc4、Nunc13、CELLBIND(商標)、又はPRIMARIA(商標)組織培養ポリスチレン(TCPS)平面状表面に蒔いた。細胞は6ウェルプレートに100,000個細胞/cm2の密度で播種し、次いで各表面上で更に1継代培養した。次いで細胞を、TrypLEで浮かせてフローサイトメトリーにより多能性マーカーについて試験するか、mRNA精製及びqRT−PCRのためにウェル中でRLTに溶解させるか、あるいは胚体内胚葉へと分化させるかした。2% BSA、100ng/mLのアクチビンA、20ng/mLのWnt3a、8ng/mLのbFGF及び3μMのGlycyl−H 1152ジヒドロクロリドを添加したRPMI培地で24時間にわたって細胞を処理することで、分化を誘導した。次いで培地を、2% BSA、100ng/mLのアクチビンA、8ng/mLのbFGF、及び3μMのGlycyl−H 1152ジヒドロクロリドを添加したRPMI培地に交換し、更に48時間にわたって培地を毎日交換した。
ヒト胚性幹細胞株は、現在のところ、単独の細胞が増殖したコロニー、又は胚盤胞由来の少数の細胞のクラスターを発展させる方法に由来する。次いでこのコロニーを連続的に継代し、細胞株を構成するのに十分な細胞クラスター/コロニーが利用できるようになるまで増殖させる。ヒト胚性幹細胞の多能性と核型安定特性を維持することを目的として、一度細胞株が誘導されると、高度に再現性のあるヒト胚性幹細胞培養のための、当該技術分野の現行の標準方法では、有糸分裂不活性化線維芽細胞のフィーダー細胞層上の、ヒト胚性幹細胞クラスター/コロニーを維持し、及び手作業での撹乱(manual disruption)又はコラゲナーゼ若しくは中性プロテアーゼ若しくはこれらのブレンドでの穏やかな酵素的大容量継代を用いて、細胞を継代する。これらの継代法はヒト胚性幹細胞クラスターを維持し、かつヒト胚性幹細胞の、コロニー形式の増殖を促進する。安定したヒト胚性幹細胞株の確立後、この細胞をMATRIGEL(商標)などの細胞外マトリックス(ECM)基質へと移すことができる。しかしながら、細胞を線維芽細胞フィーダー上で増殖させるかあるいはECM基質上で増殖させるかのいずれにせよ、技術者に具体的に指示されるヒト胚性幹細胞に関して推奨される継代法は、ヒト胚性幹コロニーを十分に解離させはしない。
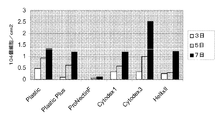
H1細胞を、PRIMARIA(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)組織培養プレート(実施例9の方法)で培養し、TrypLE(商標)Expressにより3〜5分処理することで放出させ、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM中にCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)又はHILLEX(登録商標)II(Solohill,MI)マイクロキャリアを含む6ウェルの組織培養用無処理プレートに播種した。対照として、MATRIGEL(BD Biosciences,CA)でコートしたプレートで増殖させ、かつコラゲナーゼ(1mg/mL)を用いて継代した、継代数46のH1細胞を放出させ、同様の方法でマイクロキャリアに播種した。プレートを、37℃で5時間にわたって、45分ごとに手で撹拌しながらインキュベートした。次いでプレートを37℃にて固定式プラットフォーム上に設置した。培地は、毎日10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMで交換した。画像解析は、3日目の時点で、マイクロキャリアに対する細胞の良好な付着を示す(図44)。7日後に細胞を放出させ(以下の実施例4に記載)、多能性マーカーのCD9、SSEA−4、SSEA−3、TRA−1−60、TRA−1−81に関してFACSで解析した(図45)。多能性マーカーのほとんどが、90〜100%の細胞で発現していた。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアで増殖させようが、HILLEX(登録商標)II(Solohill,MI)マイクロキャリアで増殖させようが、Accutase(商標)(Millipore,MA)で継代した細胞とTrypLE(商標)Express(Invitrogen,CA)で継代した細胞の間に明瞭な違いは存在しない。全般的に、細胞は、PRIMARIA(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)細胞培養用プラスチックからマイクロキャリアに移した場合に多能性を保持した。
米国特許出願第61/116,452号に開示の方法に従って、セルロース混合エステルからなる平面状基材で、H1細胞を12継代にわたって培養した。TrypLE(商標)Expressで3〜5分処理することで平面状基材から細胞を放出させ、10mMのY27632(Sigma−Aldrich,MO)添加MEF−CM中にCYTODEX 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア又はHILLEX(登録商標)II(Solohill,MI)マイクロキャリアを含む6ウェルの組織培養用無処理プレートに播種した。対照として、MATRIGEL(商標)(BD Biosciences,CA)でコートしたプレートで増殖させ、かつコラゲナーゼ(1mg/mL)を用いて継代した、継代数44のH1細胞を放出させ、同様の方法でマイクロキャリアに播種した。プレートを、37℃で5時間にわたって、45分ごとに手で撹拌しながらインキュベートした。次いでプレートを37℃にて固定式プラットフォーム上に設置した。培地は毎日交換した。7日後に細胞を放出させ(実施例4に記載)、多能性マーカーのCD9、SSEA−4、SSEA−3、TRA−1−60、TRA−1−81に関してFACSで解析した(図47)。ほとんどの多能性マーカーが、90%を超える細胞で発現していた。CYTODEX 3(登録商標)マイクロキャリアで増殖させた細胞と、HILLEX(登録商標)IIマイクロキャリアで増殖させた細胞との間に明瞭な違いは存在しなかった。継代数44のH1対照細胞は、多能性が他の実験により確認されていたことから、HILLEX(登録商標)IIマイクロキャリアで増殖させた後の多能性について試験しなかった(実施例5を参照されたい)。全般的に、細胞は、セルロース混合エステルからなる平面状基材からマイクロキャリアへと移した場合にも多能性を維持していた。

Claims (10)
- 多能性幹細胞の増殖方法であって、
a.多能性幹細胞集団を、第1容量のマイクロキャリアに付着させる工程と、
b.前記多能性幹細胞を、前記第1容量のマイクロキャリア上で培養する工程と、
c.前記多能性幹細胞を、前記第1容量のマイクロキャリアから取り外す工程と、
d.前記多能性幹細胞集団を、第2容量のマイクロキャリアに付着させる工程と、を含む、方法。 - 前記マイクロキャリア上の多能性幹細胞を培養する工程、取り外す工程及び付着させる工程が、連続容量のマイクロキャリアを用いて繰り返される、請求項1に記載の方法。
- 前記第1容量のマイクロキャリアが、デキストランマイクロキャリア及びポリスチレンマイクロキャリアからなる群から選択される、請求項1に記載の方法。
- 前記第2容量のマイクロキャリアが、デキストランマイクロキャリア及びポリスチレンマイクロキャリアからなる群から選択される、請求項1に記載の方法。
- 前記多能性幹細胞を、Rhoキナーゼ阻害剤を含有している培地中で、前記第1容量のマイクロキャリアに付着させる、請求項1に記載の方法。
- 前記多能性幹細胞を、Rhoキナーゼ阻害剤を含有している培地中で、前記第2容量のマイクロキャリアに付着させる、請求項1に記載の方法。
- 前記多能性幹細胞を、酵素処理により前記第1容量のマイクロキャリアから取り外す、請求項1に記載の方法。
- 前記多能性幹細胞を、酵素処理により前記第2容量のマイクロキャリアから取り外す、請求項1に記載の方法。
- 前記多能性幹細胞を前記第2容量のマイクロキャリアに付着させる前に、前記細胞から前記第1容量のマイクロキャリアを取り外す、請求項1に記載の方法。
- 多能性幹細胞を、インスリンを発現している細胞へとマイクロキャリア上で分化させる方法であって、
a.前記多能性幹細胞を、ある容量のマイクロキャリアに付着させる工程と、
b.前記多能性幹細胞を、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させる工程と、
c.前記胚体内胚葉系に特徴的なマーカーを発現している細胞を、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと分化させる工程と、
d.前記膵臓内胚葉系に特徴的なマーカーを発現している細胞を、膵内分泌系に特徴的なマーカーを発現している細胞へと分化させる工程と、
e.前記膵内分泌系に特徴的なマーカーを発現している細胞を、インスリンを発現している細胞へと分化させる工程と、を含む、方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11644708P | 2008-11-20 | 2008-11-20 | |
| US61/116,447 | 2008-11-20 | ||
| PCT/US2009/065062 WO2010059775A1 (en) | 2008-11-20 | 2009-11-19 | Pluripotent stem cell culture on micro-carriers |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012509085A true JP2012509085A (ja) | 2012-04-19 |
Family
ID=41820296
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011537603A Pending JP2012509085A (ja) | 2008-11-20 | 2009-11-19 | マイクロキャリア上での多能性幹細胞の培養 |
Country Status (15)
| Country | Link |
|---|---|
| US (3) | US20100124781A1 (ja) |
| EP (2) | EP3260534A1 (ja) |
| JP (1) | JP2012509085A (ja) |
| KR (2) | KR101774546B1 (ja) |
| CN (2) | CN107267442A (ja) |
| AU (2) | AU2009316580B2 (ja) |
| BR (1) | BRPI0920956A2 (ja) |
| CA (1) | CA2744234C (ja) |
| ES (1) | ES2642070T3 (ja) |
| HK (1) | HK1246356A1 (ja) |
| MX (2) | MX2011005288A (ja) |
| PL (1) | PL2366021T3 (ja) |
| RU (1) | RU2555538C2 (ja) |
| WO (1) | WO2010059775A1 (ja) |
| ZA (2) | ZA201104506B (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016502839A (ja) * | 2013-01-04 | 2016-02-01 | 国立大学法人京都大学 | 初期化幹細胞 |
| JP2016504037A (ja) * | 2012-12-31 | 2016-02-12 | ヤンセン バイオテツク,インコーポレーテツド | 膵内分泌細胞への分化のためのヒト多能性細胞の懸濁及びクラスタリング |
| JP2016508726A (ja) * | 2013-02-22 | 2016-03-24 | セルラー ダイナミクス インターナショナル, インコーポレイテッド | 組み合わせた遺伝的および化学的操作によるフォワードプログラミングを介した肝細胞の産生 |
| JP2016529920A (ja) * | 2013-09-19 | 2016-09-29 | ザ ボード オブ トラスティーズ オブ ザ レランド スタンフォード ジュニア ユニバーシティー | 肝細胞様細胞を産生するための方法および組成物 |
| WO2019102593A1 (ja) * | 2017-11-24 | 2019-05-31 | 株式会社Ihi | 細胞培養装置 |
Families Citing this family (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100381768B1 (ko) * | 2001-08-21 | 2003-04-26 | 김승환 | 무선 에이티엠망의 동적 자원할당 매체접속제어 프로토콜 |
| US9080145B2 (en) | 2007-07-01 | 2015-07-14 | Lifescan Corporation | Single pluripotent stem cell culture |
| BRPI0814894A2 (pt) | 2007-07-31 | 2014-10-21 | Lifescan Inc | Diferenciação de células-tronco embrionárias de ser humano. |
| WO2009070592A2 (en) | 2007-11-27 | 2009-06-04 | Lifescan, Inc. | Differentiation of human embryonic stem cells |
| US10066203B2 (en) | 2008-02-21 | 2018-09-04 | Janssen Biotech Inc. | Methods, surface modified plates and compositions for cell attachment, cultivation and detachment |
| EP2271747B1 (en) | 2008-03-17 | 2016-09-07 | Agency for Science, Technology And Research | Microcarriers for pluripotent stem cell culture |
| US8691569B2 (en) | 2008-03-17 | 2014-04-08 | Agency For Science, Technology And Research | Microcarriers for stem cell culture |
| US9458431B2 (en) | 2008-03-17 | 2016-10-04 | Agency For Science, Technology And Research | Microcarriers for stem cell culture |
| US8828720B2 (en) * | 2008-03-17 | 2014-09-09 | Agency For Science, Technology And Research | Microcarriers for stem cell culture |
| US20110143433A1 (en) * | 2008-03-17 | 2011-06-16 | Agency For Science, Technology And Research | Microcarriers for Stem Cell Culture |
| EP2942392B1 (en) | 2008-06-30 | 2018-10-03 | Janssen Biotech, Inc. | Differentiation of pluripotent stem cells |
| CN102333862B (zh) | 2008-10-31 | 2018-04-27 | 詹森生物科技公司 | 人胚胎干细胞向胰腺内分泌谱系的分化 |
| US9234178B2 (en) | 2008-10-31 | 2016-01-12 | Janssen Biotech, Inc. | Differentiation of human pluripotent stem cells |
| JP2012509085A (ja) | 2008-11-20 | 2012-04-19 | ヤンセン バイオテツク,インコーポレーテツド | マイクロキャリア上での多能性幹細胞の培養 |
| CN102257132B (zh) | 2008-11-20 | 2014-09-03 | 森托科尔奥索生物科技公司 | 用于在平面基底上进行细胞附着和培养的方法和组合物 |
| KR20110137805A (ko) | 2009-03-20 | 2011-12-23 | 에이전시 포 사이언스, 테크놀로지 앤드 리서치 | 마이크로캐리어 상의 다분화성 및 다능성 세포의 배양 방법 |
| GB2484873B (en) | 2009-07-20 | 2014-04-30 | Janssen Biotech Inc | Differentiation of pancreatic endoderm cells. |
| RU2610176C2 (ru) | 2009-12-23 | 2017-02-08 | Янссен Байотек, Инк. | Дифференцировка человеческих эмбриональных стволовых клеток |
| US9969981B2 (en) | 2010-03-01 | 2018-05-15 | Janssen Biotech, Inc. | Methods for purifying cells derived from pluripotent stem cells |
| ES2728900T3 (es) | 2010-05-12 | 2019-10-29 | Janssen Biotech Inc | Diferenciación de células madre embrionarias humanas |
| EP2582399A4 (en) * | 2010-06-16 | 2015-04-15 | Minerva Biotechnologies Corp | NEW PROGRAMMING OF CANCER CELLS |
| CN103221536B (zh) | 2010-08-31 | 2016-08-31 | 詹森生物科技公司 | 人胚胎干细胞的分化 |
| SG187944A1 (en) * | 2010-08-31 | 2013-04-30 | Janssen Biotech Inc | Differentiation of human embryonic stem cells |
| RU2599420C2 (ru) | 2010-08-31 | 2016-10-10 | Янссен Байотек, Инк. | Дифференцирование плюрипотентных стволовых клеток |
| EP2611452B1 (en) * | 2010-09-02 | 2018-10-24 | Life Technologies Corporation | Cell culture system for bioreactor scale-up of cells |
| EP2622065B1 (en) | 2010-10-01 | 2016-09-07 | The Government of the United States of America as represented by the Secretary of the Department of Health and Human Services | Manipulation of stem cell function by p53 isoforms |
| WO2012056997A1 (ja) | 2010-10-28 | 2012-05-03 | 国立大学法人熊本大学 | 多能性幹細胞の分化誘導効率を改善するための方法及び培地 |
| US9279106B2 (en) | 2010-11-12 | 2016-03-08 | Georgetown University | Immortalization of epithelial cells and methods of use |
| US9518249B2 (en) | 2010-12-16 | 2016-12-13 | General Electric Company | Cell carrier, associated methods for making cell carrier and culturing cells using the same |
| US9534206B2 (en) | 2010-12-16 | 2017-01-03 | General Electric Company | Cell carrier, associated methods for making cell carrier and culturing cells using the same |
| US9926523B2 (en) | 2010-12-16 | 2018-03-27 | General Electric Company | Cell carriers and methods for culturing cells |
| US9453197B2 (en) | 2010-12-16 | 2016-09-27 | General Electric Company | Methods of making cell carrier |
| US9453196B2 (en) | 2010-12-16 | 2016-09-27 | General Electric Company | Cell carrier, methods of making and use |
| WO2013095953A1 (en) | 2011-12-22 | 2013-06-27 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells into single hormonal insulin positive cells |
| EP2823037A4 (en) | 2012-03-07 | 2015-09-16 | Janssen Biotech Inc | DEFINED MEDIA FOR THE EXPANSION AND CARE OF PLURIPOTENTAL STEM CELLS |
| US20150133340A1 (en) * | 2012-03-07 | 2015-05-14 | Hoffmann-La Roche Inc. | Method of determining teratogenic risk |
| JP6469003B2 (ja) | 2012-06-08 | 2019-02-13 | ヤンセン バイオテツク,インコーポレーテツド | 膵内分泌細胞へのヒト胚性幹細胞の分化 |
| US9664671B2 (en) | 2012-07-24 | 2017-05-30 | Nissan Chemical Industries, Ltd. | Culture medium composition and method of culturing cell or tissue using thereof |
| JP5629893B2 (ja) | 2012-07-24 | 2014-11-26 | 日産化学工業株式会社 | 培地組成物及び当該組成物を用いた細胞又は組織の培養方法 |
| US10017805B2 (en) | 2012-08-23 | 2018-07-10 | Nissan Chemical Industries, Ltd. | Enhancing ingredients for protein production from various cells |
| EP2893000B1 (en) | 2012-09-03 | 2019-04-10 | Novo Nordisk A/S | Generation of pancreatic endoderm from pluripotent stem cells using small molecules |
| KR101942769B1 (ko) | 2012-12-31 | 2019-01-28 | 얀센 바이오테크 인코포레이티드 | Hb9 조절제를 사용하는 인간 배아 줄기세포의 췌장 내분비 세포로의 분화 |
| CN105008518B (zh) | 2012-12-31 | 2020-08-07 | 詹森生物科技公司 | 在空气-液体界面处培养人胚胎干细胞以用于分化成胰腺内分泌细胞 |
| US10370644B2 (en) | 2012-12-31 | 2019-08-06 | Janssen Biotech, Inc. | Method for making human pluripotent suspension cultures and cells derived therefrom |
| CN106414707A (zh) | 2014-01-23 | 2017-02-15 | 日产化学工业株式会社 | 培养基组合物的制造方法 |
| KR102411750B1 (ko) | 2014-01-23 | 2022-06-22 | 닛산 가가쿠 가부시키가이샤 | 배양 배지 조성물 |
| JP6588969B2 (ja) | 2014-05-16 | 2019-10-09 | ヤンセン バイオテツク,インコーポレーテツド | 膵内分泌細胞内のmafa発現を強化するための小分子の使用 |
| CN104450604A (zh) * | 2014-11-25 | 2015-03-25 | 成都威尔诺生物科技有限公司 | 用于制备疫苗工艺的微载体的清洁、灭菌的方法 |
| US9944894B2 (en) | 2015-01-16 | 2018-04-17 | General Electric Company | Pluripotent stem cell expansion and passage using a rocking platform bioreactor |
| EP3368657B1 (en) | 2015-10-30 | 2021-06-30 | Biolamina AB | Methods for producing hepatocytes |
| WO2017170849A1 (ja) * | 2016-03-30 | 2017-10-05 | 国立研究開発法人医薬基盤・健康・栄養研究所 | ナイーブ型多能性幹細胞培養用培地および多能性幹細胞の培養方法 |
| MA45479A (fr) | 2016-04-14 | 2019-02-20 | Janssen Biotech Inc | Différenciation de cellules souches pluripotentes en cellules de l'endoderme de l'intestin moyen |
| WO2018075940A1 (en) * | 2016-10-21 | 2018-04-26 | Georgia Tech Research Corporation | Methods and systems for t cell expansion |
| US11662296B2 (en) * | 2017-05-19 | 2023-05-30 | Thrive Bioscience, Inc. | Systems and methods for cell dissociation |
| CN108753681B (zh) * | 2018-04-28 | 2022-02-15 | 中山大学附属第一医院 | 一种鼻上皮干细胞培养方法及鼻上皮干细胞增殖培养基 |
| KR102115360B1 (ko) * | 2018-05-30 | 2020-05-26 | 주식회사 바이블리오테카 | 성체 줄기세포 배양액 및 그 제조방법 |
| CN110564675A (zh) * | 2019-09-30 | 2019-12-13 | 广东华夏健康生命科学有限公司 | 一种毛囊干细胞的分离提取方法 |
| CN111040983A (zh) * | 2019-12-25 | 2020-04-21 | 杭州原生生物科技有限公司 | 一种3d微载体细胞吸附培养的方法 |
| CN111424011A (zh) * | 2020-04-17 | 2020-07-17 | 深圳市旷逸生物科技有限公司 | 一种能够维持脐带间充质干细胞细胞形态的三维培养方法 |
| CN111206015B (zh) * | 2020-04-21 | 2020-07-31 | 广东省生物资源应用研究所 | 一种利用fact ⅲ微载体体外扩增精原干细胞的三维动态培养方法 |
| CN113637635B (zh) * | 2021-10-13 | 2022-01-25 | 北京华龛生物科技有限公司 | 微载体细胞培养体系中微载体聚团状态的解散方法 |
| CN115089614A (zh) * | 2022-06-28 | 2022-09-23 | 中国人民解放军军事科学院军事医学研究院 | 一种增强骨骼干细胞性能的方法及其在治疗骨关节炎中的应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007149182A2 (en) * | 2006-06-19 | 2007-12-27 | Geron Corporation | Differentiation and enrichment of islet-like cells from human pluripotent stem cells |
| JP2008099662A (ja) * | 2006-09-22 | 2008-05-01 | Institute Of Physical & Chemical Research | 幹細胞の培養方法 |
Family Cites Families (248)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US264713A (en) * | 1882-09-19 | Neck-strap buckle and rein-ring | ||
| US3209652A (en) | 1961-03-30 | 1965-10-05 | Burgsmueller Karl | Thread whirling method |
| AT326803B (de) | 1968-08-26 | 1975-12-29 | Binder Fa G | Maschenware sowie verfahren zur herstellung derselben |
| US3935067A (en) | 1974-11-22 | 1976-01-27 | Wyo-Ben Products, Inc. | Inorganic support for culture media |
| CA1201400A (en) | 1982-04-16 | 1986-03-04 | Joel L. Williams | Chemically specific surfaces for influencing cell activity during culture |
| US4499802A (en) | 1982-09-29 | 1985-02-19 | Container Graphics Corporation | Rotary cutting die with scrap ejection |
| US4537773A (en) | 1983-12-05 | 1985-08-27 | E. I. Du Pont De Nemours And Company | α-Aminoboronic acid derivatives |
| US4557264A (en) | 1984-04-09 | 1985-12-10 | Ethicon Inc. | Surgical filament from polypropylene blended with polyethylene |
| US5215893A (en) | 1985-10-03 | 1993-06-01 | Genentech, Inc. | Nucleic acid encoding the ba chain prodomains of inhibin and method for synthesizing polypeptides using such nucleic acid |
| US5089396A (en) | 1985-10-03 | 1992-02-18 | Genentech, Inc. | Nucleic acid encoding β chain prodomains of inhibin and method for synthesizing polypeptides using such nucleic acid |
| US4737578A (en) | 1986-02-10 | 1988-04-12 | The Salk Institute For Biological Studies | Human inhibin |
| US5863531A (en) | 1986-04-18 | 1999-01-26 | Advanced Tissue Sciences, Inc. | In vitro preparation of tubular tissue structures by stromal cell culture on a three-dimensional framework |
| CA1340581C (en) | 1986-11-20 | 1999-06-08 | Joseph P. Vacanti | Chimeric neomorphogenesis of organs by controlled cellular implantation using artificial matrices |
| US5567612A (en) | 1986-11-20 | 1996-10-22 | Massachusetts Institute Of Technology | Genitourinary cell-matrix structure for implantation into a human and a method of making |
| US5804178A (en) | 1986-11-20 | 1998-09-08 | Massachusetts Institute Of Technology | Implantation of cell-matrix structure adjacent mesentery, omentum or peritoneum tissue |
| NZ229354A (en) | 1988-07-01 | 1990-09-26 | Becton Dickinson Co | Treating polymer surfaces with a gas plasma and then applying a layer of endothelial cells to the surface |
| EP0363125A3 (en) | 1988-10-03 | 1990-08-16 | Hana Biologics Inc. | Proliferated pancreatic endocrine cell product and process |
| US5837539A (en) | 1990-11-16 | 1998-11-17 | Osiris Therapeutics, Inc. | Monoclonal antibodies for human mesenchymal stem cells |
| HU218140B (hu) | 1991-04-25 | 2000-06-28 | Chugai Seiyaku Kabushiki Kaisha | Humán interleukin-6-receptorral szembeni átalakított humán antitest |
| US5449383A (en) | 1992-03-18 | 1995-09-12 | Chatelier; Ronald C. | Cell growth substrates |
| GB9206861D0 (en) | 1992-03-28 | 1992-05-13 | Univ Manchester | Wound healing and treatment of fibrotic disorders |
| CA2114282A1 (en) | 1993-01-28 | 1994-07-29 | Lothar Schilder | Multi-layered implant |
| JP3525221B2 (ja) | 1993-02-17 | 2004-05-10 | 味の素株式会社 | 免疫抑制剤 |
| AU687386B2 (en) | 1993-04-08 | 1998-02-26 | Human Cell Cultures, Inc. | Cell culturing method and medium |
| US5523226A (en) * | 1993-05-14 | 1996-06-04 | Biotechnology Research And Development Corp. | Transgenic swine compositions and methods |
| GB9310557D0 (en) | 1993-05-21 | 1993-07-07 | Smithkline Beecham Plc | Novel process and apparatus |
| TW257671B (ja) | 1993-11-19 | 1995-09-21 | Ciba Geigy | |
| US5834308A (en) | 1994-04-28 | 1998-11-10 | University Of Florida Research Foundation, Inc. | In vitro growth of functional islets of Langerhans |
| US6703017B1 (en) | 1994-04-28 | 2004-03-09 | Ixion Biotechnology, Inc. | Reversal of insulin-dependent diabetes by islet-producing stem cells, islet progenitor cells and islet-like structures |
| US6001647A (en) | 1994-04-28 | 1999-12-14 | Ixion Biotechnology, Inc. | In vitro growth of functional islets of Langerhans and in vivo uses thereof |
| US6083903A (en) | 1994-10-28 | 2000-07-04 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| EP0800829B2 (en) | 1994-12-29 | 2012-07-25 | Chugai Seiyaku Kabushiki Kaisha | Use of a pm-1 antibody or of a mh 166 antibody for enhancing the anti-tumor effect of cisplatin or carboplatin |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5718922A (en) | 1995-05-31 | 1998-02-17 | Schepens Eye Research Institute, Inc. | Intravitreal microsphere drug delivery and method of preparation |
| US5908782A (en) | 1995-06-05 | 1999-06-01 | Osiris Therapeutics, Inc. | Chemically defined medium for human mesenchymal stem cells |
| US5681561A (en) | 1995-06-07 | 1997-10-28 | Life Medical Sciences, Inc. | Compositions and methods for improving autologous fat grafting |
| UA65572C2 (en) | 1997-04-24 | 2004-04-15 | Ortho Mcneil Pharm Inc | Substituted imidazoles, intermediate compounds for the preparation thereof, a method for the preparation of substituted imidazoles and a method for the treatment of inflammatory diseases |
| DK1028737T3 (da) | 1997-07-03 | 2007-08-13 | Osiris Therapeutics Inc | Humane mesenchymale stamceller fra perifert blod |
| DK1015576T3 (da) | 1997-09-16 | 2005-08-29 | Egea Biosciences Llc | Fremgangsmåde til fuldstændig kemisk syntese og aggregering af gener og genomer |
| US6670127B2 (en) | 1997-09-16 | 2003-12-30 | Egea Biosciences, Inc. | Method for assembly of a polynucleotide encoding a target polypeptide |
| AU729377B2 (en) | 1997-10-23 | 2001-02-01 | Asterias Biotherapeutics, Inc. | Methods and materials for the growth of primate-derived primordial stem cells in feeder-free culture |
| AR014195A1 (es) | 1997-12-29 | 2001-02-07 | Ortho Mcneil Pharm Inc | Compuestos de trifenilpropanamida utiles para el tratamiento de procesos inflamatorios, composiciones anti-inflamatorias que los comprenden, ymetodos para prepararlos |
| DK1066052T3 (da) | 1998-03-18 | 2006-06-12 | Osiris Therapeutics Inc | Mesenchymstamceller til forebyggelse og behandling af immunreaktioner ved transplantationer |
| MY132496A (en) | 1998-05-11 | 2007-10-31 | Vertex Pharma | Inhibitors of p38 |
| US6413773B1 (en) | 1998-06-01 | 2002-07-02 | The Regents Of The University Of California | Phosphatidylinositol 3-kinase inhibitors as stimulators of endocrine differentiation |
| US6667176B1 (en) | 2000-01-11 | 2003-12-23 | Geron Corporation | cDNA libraries reflecting gene expression during growth and differentiation of human pluripotent stem cells |
| US7410798B2 (en) | 2001-01-10 | 2008-08-12 | Geron Corporation | Culture system for rapid expansion of human embryonic stem cells |
| US6610540B1 (en) | 1998-11-18 | 2003-08-26 | California Institute Of Technology | Low oxygen culturing of central nervous system progenitor cells |
| US6413556B1 (en) | 1999-01-08 | 2002-07-02 | Sky High, Llc | Aqueous anti-apoptotic compositions |
| IL144359A0 (en) | 1999-01-21 | 2002-05-23 | Vitro Diagnostics Inc | Immortalized cell lines and methods of making the same |
| US6815203B1 (en) | 1999-06-23 | 2004-11-09 | Joslin Diabetes Center, Inc. | Methods of making pancreatic islet cells |
| US6333029B1 (en) | 1999-06-30 | 2001-12-25 | Ethicon, Inc. | Porous tissue scaffoldings for the repair of regeneration of tissue |
| US6306424B1 (en) | 1999-06-30 | 2001-10-23 | Ethicon, Inc. | Foam composite for the repair or regeneration of tissue |
| CA2385628A1 (en) | 1999-09-27 | 2001-04-05 | Ammon B. Peck | Reversal of insulin-dependent diabetes by islet-producing stem cells, islet progenitor cells and islet-like structures |
| US6685936B2 (en) | 1999-10-12 | 2004-02-03 | Osiris Therapeutics, Inc. | Suppressor cells induced by culture with mesenchymal stem cells for treatment of immune responses in transplantation |
| US20030082155A1 (en) | 1999-12-06 | 2003-05-01 | Habener Joel F. | Stem cells of the islets of langerhans and their use in treating diabetes mellitus |
| EP1240518A4 (en) | 1999-12-13 | 2006-05-17 | Scripps Research Inst | MARKERS FOR THE IDENTIFICATION AND INSULATION OF PRE-GENERIC CELLS OF A AND B PANCREAS ISOLATED CELLS |
| US7005252B1 (en) | 2000-03-09 | 2006-02-28 | Wisconsin Alumni Research Foundation | Serum free cultivation of primate embryonic stem cells |
| US7439064B2 (en) | 2000-03-09 | 2008-10-21 | Wicell Research Institute, Inc. | Cultivation of human embryonic stem cells in the absence of feeder cells or without conditioned medium |
| US6436704B1 (en) | 2000-04-10 | 2002-08-20 | Raven Biotechnologies, Inc. | Human pancreatic epithelial progenitor cells and methods of isolation and use thereof |
| US6458589B1 (en) | 2000-04-27 | 2002-10-01 | Geron Corporation | Hepatocyte lineage cells derived from pluripotent stem cells |
| AU2001274624A1 (en) | 2000-06-26 | 2002-01-08 | Renomedix Institute Inc. | Cell fraction containing cells capable of differentiating into nervous system cells |
| WO2002059083A2 (en) | 2000-10-23 | 2002-08-01 | Smithkline Beecham Corporation | Novel compounds |
| CA2431166A1 (en) | 2000-12-08 | 2002-06-13 | Ortho-Mcneil Pharmaceutical, Inc. | Indazolyl-substituted pyrroline compounds as kinase inhibitors |
| CA2431187A1 (en) | 2000-12-08 | 2002-06-13 | Ortho-Mcneil Pharmaceutical, Inc. | Macroheterocylic compounds useful as kinase inhibitors |
| US6599323B2 (en) | 2000-12-21 | 2003-07-29 | Ethicon, Inc. | Reinforced tissue implants and methods of manufacture and use |
| JP2005503759A (ja) | 2001-01-24 | 2005-02-10 | アメリカ合衆国 | 幹細胞の膵臓内分泌細胞への分化方法 |
| EP3078667B1 (en) | 2001-01-25 | 2018-11-21 | The United States of America, represented by the Secretary, Department of Health and Human Services | Formulation of boronic acid compounds |
| US6656488B2 (en) | 2001-04-11 | 2003-12-02 | Ethicon Endo-Surgery, Inc. | Bioabsorbable bag containing bioabsorbable materials of different bioabsorption rates for tissue engineering |
| DE10290025T1 (de) | 2001-04-19 | 2003-10-09 | Develogen Ag | Verfahren zur Differenzierung von Stammzellen in Insulin-produzierende Zellen |
| CN100516204C (zh) | 2001-04-24 | 2009-07-22 | 味之素株式会社 | 干细胞及其分离方法 |
| EP1393066A4 (en) | 2001-05-15 | 2006-01-25 | Rappaport Family Inst For Res | INSULIN-PRODUCING CELLS DERIVED FROM HUMAN EMBRYONAL STEM CELLS |
| US6626950B2 (en) | 2001-06-28 | 2003-09-30 | Ethicon, Inc. | Composite scaffold with post anchor for the repair and regeneration of tissue |
| KR100418195B1 (ko) | 2001-07-05 | 2004-02-11 | 주식회사 우리기술 | 전력케이블의 다중절연진단장치 및 그 방법 |
| GB0117583D0 (en) | 2001-07-19 | 2001-09-12 | Astrazeneca Ab | Novel compounds |
| WO2003014313A2 (en) | 2001-08-06 | 2003-02-20 | Bresagen, Ltd. | Alternative compositions and methods for the culture of stem cells |
| US6617152B2 (en) | 2001-09-04 | 2003-09-09 | Corning Inc | Method for creating a cell growth surface on a polymeric substrate |
| EP1298201A1 (en) | 2001-09-27 | 2003-04-02 | Cardion AG | Process for the production of cells exhibiting an islet-beta-cell-like state |
| EP1444345A4 (en) | 2001-10-18 | 2004-12-08 | Ixion Biotechnology Inc | TRANSFORMATION OF STEM CELLS AND LIVER PROGENITORS INTO FUNCTIONAL CELLS OF PANCREAS |
| DE60233248D1 (de) | 2001-11-15 | 2009-09-17 | Childrens Medical Center | Verfahren zur isolierung, expansion und differenzierung fötaler stammzellen aus chorionzotte, fruchtwasser und plazenta und therapeutische verwendungen davon |
| JP4653952B2 (ja) | 2001-12-07 | 2011-03-16 | サイトリ セラピューティクス インコーポレイテッド | 処理済み脂肪吸引細胞で患者を治療するためのシステムと方法 |
| EP2264146A1 (en) | 2001-12-07 | 2010-12-22 | Geron Corporation | Islet cells from human embryonic stem cells |
| AU2002218893A1 (en) | 2001-12-21 | 2003-07-09 | Thromb-X Nv | Compositions for the in vitro derivation and culture of embryonic stem (es) cell lines with germline transmission capability |
| US20050095703A1 (en) | 2001-12-28 | 2005-05-05 | Henrik Semb | Method for the establishment of a pluripotent human blastocyst - derived stem cell line |
| US20030162290A1 (en) | 2002-01-25 | 2003-08-28 | Kazutomo Inoue | Method for inducing differentiation of embryonic stem cells into functioning cells |
| US20050208029A1 (en) | 2002-04-17 | 2005-09-22 | Akihiro Umezawa | Method of forming pancreatic beta cells from mesenchymal cells |
| US20040161419A1 (en) | 2002-04-19 | 2004-08-19 | Strom Stephen C. | Placental stem cells and uses thereof |
| CA2485527A1 (en) | 2002-05-08 | 2003-11-20 | Janssen Pharmaceutica N.V. | Substituted pyrroline kinase inhibitors |
| US20060003446A1 (en) | 2002-05-17 | 2006-01-05 | Gordon Keller | Mesoderm and definitive endoderm cell populations |
| WO2003102134A2 (en) | 2002-05-28 | 2003-12-11 | Becton, Dickinson And Company | Pancreatic acinar cells into insulin producing cells |
| CN1671694A (zh) | 2002-06-05 | 2005-09-21 | 詹森药业有限公司 | 作为激酶抑制剂的二吲哚基-顺丁烯二酰亚胺衍生物 |
| GB0212976D0 (en) | 2002-06-06 | 2002-07-17 | Tonejet Corp Pty Ltd | Ejection method and apparatus |
| CN1171991C (zh) | 2002-07-08 | 2004-10-20 | 徐如祥 | 人神经干细胞的培养方法 |
| US6877147B2 (en) | 2002-07-22 | 2005-04-05 | Broadcom Corporation | Technique to assess timing delay by use of layout quality analyzer comparison |
| US7838290B2 (en) | 2002-07-25 | 2010-11-23 | The Scripps Research Institute | Hematopoietic stem cells and methods of treatment of neovascular eye diseases therewith |
| EP1539930A4 (en) | 2002-07-29 | 2006-08-09 | Es Cell Int Pte Ltd | METHOD IN MULTIPLE STAGES OF DIFFERENTIATION OF POSITIVE INSULIN-SENSITIVE CELLS, GLUCOSE |
| US20040063204A1 (en) | 2002-08-14 | 2004-04-01 | Lijun Yang | Bone marrow cell differentiation |
| US7371576B2 (en) | 2002-09-06 | 2008-05-13 | Reneuron, Inc. | CD56 positive human adult pancreatic endocrine progenitor cells |
| US9969977B2 (en) | 2002-09-20 | 2018-05-15 | Garnet Biotherapeutics | Cell populations which co-express CD49c and CD90 |
| US20040062753A1 (en) | 2002-09-27 | 2004-04-01 | Alireza Rezania | Composite scaffolds seeded with mammalian cells |
| AU2003285172A1 (en) | 2002-11-08 | 2004-06-03 | The Johns Hopkins University | Human embryonic stem cell cultures, and compositions and methods for growing same |
| US7144999B2 (en) | 2002-11-23 | 2006-12-05 | Isis Pharmaceuticals, Inc. | Modulation of hypoxia-inducible factor 1 alpha expression |
| AU2003302702B2 (en) | 2002-12-05 | 2008-08-07 | Technion Research & Development Foundation Ltd. | Cultured human pancreatic islets, and uses thereof |
| WO2004055155A2 (en) | 2002-12-16 | 2004-07-01 | Technion Research & Development Foundation Ltd. | Methods of preparing feeder cells-free, xeno-free human embryonic stem cells and stem cell cultures prepared using same |
| RU2359671C2 (ru) | 2003-01-29 | 2009-06-27 | Такеда Фармасьютикал Компани Лимитед | Способ получения препарата с покрытием |
| PL377403A1 (pl) | 2003-01-29 | 2006-02-06 | Takeda Pharmaceutical Company Limited | Sposób wytwarzania preparatu powlekanego |
| WO2005045001A2 (en) | 2003-02-14 | 2005-05-19 | The Board Of Trustees Of The Leland Stanford Junior University | Insulin-producing cells derived from stem cells |
| US20070155661A1 (en) | 2003-02-14 | 2007-07-05 | The Board Of Trustees Of The Leland Standord Junior University | Methods and compositions for modulating the development of stem cells |
| CA2520861A1 (en) | 2003-03-27 | 2004-10-14 | Ixion Biotechnology, Inc. | Method for transdifferentiation of non-pancreatic stem cells to the pancreatic pathway |
| WO2004090110A2 (en) | 2003-03-31 | 2004-10-21 | Bresagen Inc. | Compositions and methods for the control, differentiation and/or manipulation of pluripotent cells through a gamma-secretase signaling pathway |
| US20090203141A1 (en) | 2003-05-15 | 2009-08-13 | Shi-Lung Lin | Generation of tumor-free embryonic stem-like pluripotent cells using inducible recombinant RNA agents |
| CA2530533C (en) | 2003-06-27 | 2015-02-10 | Ethicon, Incorporated | Postpartum cells derived from umbilical cord tissue, and methods of making and using the same |
| IL161903A0 (en) | 2003-07-17 | 2005-11-20 | Gamida Cell Ltd | Ex vivo progenitor and stem cell expansion for usein the treatment of disease of endodermally- deri ved organs |
| ITRM20030395A1 (it) | 2003-08-12 | 2005-02-13 | Istituto Naz Per Le Malattie Infettive Lazz | Terreno di coltura per il mantenimento, la proliferazione e il differenziamento di cellule di mammifero. |
| US7569385B2 (en) | 2003-08-14 | 2009-08-04 | The Regents Of The University Of California | Multipotent amniotic fetal stem cells |
| US7157275B2 (en) | 2003-08-15 | 2007-01-02 | Becton, Dickinson And Company | Peptides for enhanced cell attachment and growth |
| EP1670900A4 (en) | 2003-08-27 | 2008-06-11 | Stemcells California Inc | ENHANCED PANCREATIC STEM CELL AND PRECURSOR CELL POPULATIONS AND METHOD OF IDENTIFYING, INSULATING AND ENRICHING SUCH POPULATIONS |
| EP1696899A1 (en) | 2003-12-17 | 2006-09-06 | Allergan, Inc. | Methods for treating retinoid responsive disorders using selective inhibitors of cyp26a and cyp26b |
| US20060030042A1 (en) | 2003-12-19 | 2006-02-09 | Ali Brivanlou | Maintenance of embryonic stem cells by the GSK-3 inhibitor 6-bromoindirubin-3'-oxime |
| US7625753B2 (en) | 2003-12-23 | 2009-12-01 | Cythera, Inc. | Expansion of definitive endoderm cells |
| AU2004309421B2 (en) | 2003-12-23 | 2011-04-21 | Viacyte, Inc. | Definitive endoderm |
| US20050266554A1 (en) | 2004-04-27 | 2005-12-01 | D Amour Kevin A | PDX1 expressing endoderm |
| CN112813019A (zh) | 2003-12-23 | 2021-05-18 | 维亚希特公司 | 定形内胚层 |
| TWI334443B (en) | 2003-12-31 | 2010-12-11 | Ind Tech Res Inst | Method of single cell culture of undifferentiated human embryonic stem cells |
| WO2005065354A2 (en) | 2003-12-31 | 2005-07-21 | The Burnham Institute | Defined media for pluripotent stem cell culture |
| US7794704B2 (en) | 2004-01-23 | 2010-09-14 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells for treatment of retinal degeneration |
| US20080241107A1 (en) | 2004-01-23 | 2008-10-02 | Copland Iii John A | Methods and Compositions For Preparing Pancreatic Insulin Secreting Cells |
| GB2441530B (en) | 2004-02-12 | 2009-09-23 | Univ Newcastle | Stem Cells |
| WO2005080598A1 (ja) | 2004-02-19 | 2005-09-01 | Dainippon Sumitomo Pharma Co., Ltd. | 体細胞核初期化物質のスクリーニング方法 |
| CA2581424A1 (en) | 2004-03-09 | 2005-09-22 | Gang Xu | Methods for generating insulin-producing cells |
| US20090155218A1 (en) | 2004-03-10 | 2009-06-18 | Alberto Hayek | Compositions and methods for growth of embryonic stem cells |
| RU2375448C2 (ru) | 2004-03-23 | 2009-12-10 | Асубио Фарма Ко., Лтд. | Способ выращивания плюрипотентных стволовых клеток |
| WO2005097980A2 (en) | 2004-03-26 | 2005-10-20 | Geron Corporation | New protocols for making hepatocytes from embryonic stem cells |
| GB2427874B (en) | 2004-04-01 | 2008-09-10 | Wisconsin Alumni Res Found | Differentiation of stem cells to endoderm and pancreatic lineage |
| EP1740612B1 (en) | 2004-04-27 | 2019-08-07 | Viacyte, Inc. | Pdx1 expressing endoderm |
| AU2005272078B2 (en) | 2004-07-09 | 2011-04-14 | Viacyte, Inc. | Methods for identifying factors for differentiating definitive endoderm |
| CA2576872C (en) | 2004-08-13 | 2013-11-12 | University Of Georgia Research Foundation, Inc. | Compositions and methods for self-renewal and differentiation in human embryonic stem cells |
| US20080268533A1 (en) | 2004-08-25 | 2008-10-30 | University Of Georgia Research Foundation, Inc. | Methods and Compositions Utilizing Myc and Gsk3Beta to Manipulate the Pluripotency of Embryonic Stem Cells |
| DE102004043256B4 (de) | 2004-09-07 | 2013-09-19 | Rheinische Friedrich-Wilhelms-Universität Bonn | Skalierbarer Prozess zur Kultivierung undifferenzierter Stammzellen in Suspension |
| MX2007002389A (es) | 2004-09-08 | 2009-02-12 | Wisconsin Alumni Res Found | Cultivo de celulas progenitoras embrionarias humanas. |
| WO2006029197A1 (en) | 2004-09-08 | 2006-03-16 | Wisconsin Alumni Research Foundation | Medium and culture of embryonic stem cells |
| AU2006208944A1 (en) | 2005-01-28 | 2006-08-03 | Imperial College Innovations Limited | Methods for embryonic stem cell culture |
| WO2006083782A2 (en) | 2005-01-31 | 2006-08-10 | Es Cell International Pte Ltd. | Directed differentiation of embryonic stem cells and uses thereof |
| WO2006088867A2 (en) | 2005-02-15 | 2006-08-24 | Medistem Laboratories, Incorporated | Method for expansion of stem cells |
| JP5265204B2 (ja) | 2005-03-04 | 2013-08-14 | ライフスキャン・インコーポレイテッド | 成人膵臓由来間質細胞 |
| GB0505970D0 (en) | 2005-03-23 | 2005-04-27 | Univ Edinburgh | Culture medium containing kinase inhibitor, and uses thereof |
| CN100425694C (zh) | 2005-04-15 | 2008-10-15 | 北京大学 | 诱导胚胎干细胞向胰腺细胞分化的方法 |
| WO2006113470A2 (en) | 2005-04-15 | 2006-10-26 | Geron Corporation | Cancer treatment by combined inhibition of proteasome and telomerase activities |
| EP1875234B1 (en) | 2005-04-26 | 2011-06-22 | Aarhus Universitet | Biosurface structure array |
| WO2006126574A1 (ja) | 2005-05-24 | 2006-11-30 | Kumamoto University | Es細胞の分化誘導方法 |
| AU2006202209B2 (en) | 2005-05-27 | 2011-04-14 | Lifescan, Inc. | Amniotic fluid derived cells |
| CA2610598A1 (en) | 2005-06-10 | 2006-12-21 | Irm Llc | Compounds that maintain pluripotency of embryonic stem cells |
| WO2006138433A2 (en) | 2005-06-14 | 2006-12-28 | The Regents Of The University Of California | Induction of cell differentiation by class i bhlh polypeptides |
| WO2006137787A1 (en) | 2005-06-21 | 2006-12-28 | Ge Healthcare Bio-Sciences Ab | Method for cell culture |
| EP1910516B1 (en) | 2005-06-22 | 2019-06-19 | Asterias Biotherapeutics, Inc. | Suspension culture of human embryonic stem cells |
| WO2007003525A2 (en) | 2005-06-30 | 2007-01-11 | Janssen Pharmaceutica N.V. | Cyclic anilino-pyridinotriazines as gsk-3 inhibitors |
| WO2007012144A1 (en) | 2005-07-29 | 2007-02-01 | Australian Stem Cell Centre Limited | Compositions and methods for growth of pluripotent cells |
| WO2007016485A2 (en) | 2005-07-29 | 2007-02-08 | Athersys, Inc. | Use of a gsk-3 inhibitor to maintain potency of cultured cells |
| WO2007025234A2 (en) | 2005-08-26 | 2007-03-01 | The Trustees Of Columbia University In The City Of New York | Generation of pancreatic endocrine cells from primary duct cell cultures and methods of use for treatment of diabetes |
| SG177946A1 (en) | 2005-08-29 | 2012-02-28 | Technion Res & Dev Foundation | Media for culturing stem cells |
| WO2007027156A1 (en) | 2005-09-02 | 2007-03-08 | Agency For Science, Technology And Research | Method of deriving mesenchymal stem cells |
| AU2006292021B2 (en) | 2005-09-12 | 2012-05-31 | Es Cell International Pte Ltd. | Cardiomyocyte production |
| SG169324A1 (en) | 2005-10-14 | 2011-03-30 | Univ Minnesota | Differentiation of non-embryonic stem cells to cells having a pancreatic phenotype |
| CN101351547A (zh) | 2005-10-27 | 2009-01-21 | 赛瑟拉公司 | 表达pdx1的背侧和腹侧前肠内胚层 |
| EA018039B1 (ru) | 2005-12-13 | 2013-05-30 | Киото Юниверсити | Ядерный фактор перепрограммирования |
| WO2007082963A1 (es) | 2006-01-18 | 2007-07-26 | Fundación Instituto Valenciano De Infertilidad | Líneas de células madre embrionarias humanas y métodos para usar las mismas |
| SG10201405107YA (en) | 2006-02-23 | 2014-10-30 | Viacyte Inc | Compositions and methods useful for culturing differentiable cells |
| US7695965B2 (en) | 2006-03-02 | 2010-04-13 | Cythera, Inc. | Methods of producing pancreatic hormones |
| DK1999253T3 (da) | 2006-03-02 | 2019-08-12 | Viacyte Inc | Endokrine precursorceller, pankreatiske hormon-udtrykkende celler og fremgangsmåder til fremstilling |
| EP2021462B1 (en) | 2006-04-28 | 2019-01-09 | Lifescan, Inc. | Differentiation of human embryonic stem cells |
| US8741643B2 (en) | 2006-04-28 | 2014-06-03 | Lifescan, Inc. | Differentiation of pluripotent stem cells to definitive endoderm lineage |
| SE534150C2 (sv) | 2006-05-02 | 2011-05-10 | Wisconsin Alumni Res Found | Förfarande för differentiering av stamceller till celler av den endodermala och pankreatiska utvecklingslinjen |
| US8685730B2 (en) | 2006-05-02 | 2014-04-01 | Wisconsin Alumni Research Foundation | Methods and devices for differentiating pluripotent stem cells into cells of the pancreatic lineage |
| WO2007139929A2 (en) | 2006-05-25 | 2007-12-06 | The Burnham Institute For Medical Research | Methods for culture and production of single cell populations of human embryonic stem cells |
| CN101541953A (zh) | 2006-06-02 | 2009-09-23 | 佐治亚大学研究基金会 | 通过从人胚胎干细胞获得的定形内胚层细胞的分化得到胰和肝内胚层细胞及组织 |
| US20090298169A1 (en) | 2006-06-02 | 2009-12-03 | The University Of Georgia Research Foundation | Pancreatic and Liver Endoderm Cells and Tissue by Differentiation of Definitive Endoderm Cells Obtained from Human Embryonic Stems |
| CN100494359C (zh) * | 2006-06-23 | 2009-06-03 | 中日友好医院 | 神经干细胞三维立体培养体外扩增的方法 |
| CA2893679C (en) | 2006-06-26 | 2018-10-30 | Lifescan, Inc. | Conditioned medium for pluripotent stem cell culture |
| US20080003676A1 (en) | 2006-06-26 | 2008-01-03 | Millipore Corporation | Growth of embryonic stem cells |
| WO2008004990A2 (en) * | 2006-07-06 | 2008-01-10 | Es Cell International Pte Ltd | Method for stem cell culture and cells derived therefrom |
| WO2008013664A2 (en) | 2006-07-26 | 2008-01-31 | Cythera, Inc. | Methods of producing pancreatic hormones |
| DK3441459T3 (da) | 2006-08-02 | 2021-06-07 | Technion Res & Dev Foundation | Fremgangsmåder til ekspansion af embryonale stamceller i en suspensionskultur |
| KR101331510B1 (ko) | 2006-08-30 | 2013-11-20 | 재단법인서울대학교산학협력재단 | 저농도의 포도당을 함유하는 인간 배아줄기세포용 배지조성물 및 이를 이용한 인간 배아 줄기세포로부터 인슐린생산 세포 또는 세포괴로 분화시키는 방법, 그리고그로부터 유도된 인슐린 생산 세포 또는 세포괴 |
| WO2008039521A2 (en) | 2006-09-26 | 2008-04-03 | Nmt Medical, Inc. | Method for modifying a medical implant surface for promoting tissue growth |
| WO2008048647A1 (en) | 2006-10-17 | 2008-04-24 | Cythera, Inc. | Modulation of the phosphatidylinositol-3-kinase pathway in the differentiation of human embryonic stem cells |
| EP2114895A4 (en) | 2006-10-17 | 2011-06-22 | Stiefel Laboratories | TALARAZOLMETABOLITEN |
| US8835163B2 (en) | 2006-10-18 | 2014-09-16 | The Board Of Trustees Of The University Of Illinois | Embryonic-like stem cells derived from adult human peripheral blood and methods of use |
| RU2323252C1 (ru) * | 2006-10-25 | 2008-04-27 | Антонина Ивановна Колесникова | Способ культивирования мезенхимальных стволовых клеток человека ex vivo |
| US20110151554A1 (en) | 2006-11-09 | 2011-06-23 | Akira Yuo | Method for culturing and subculturing primate embryonic stem cell, as well as method for inducing differentiation thereof |
| TW200836749A (en) | 2007-01-09 | 2008-09-16 | Vioquest Pharmaceuticals Inc | Compositions including triciribine and bortezomib and derivatives thereof and methods of use thereof |
| SE0950586L (sv) * | 2007-01-17 | 2009-08-13 | Wisconsin Alumni Res Found | Förbättrad odling av stamceller |
| WO2008094597A2 (en) | 2007-01-30 | 2008-08-07 | University Of Georgia Research Foundation, Inc. | Early mesoderm cells, a stable population of mesendoderm cells that has utility for generation of endoderm and mesoderm lineages and multipotent migratory cells (mmc) |
| GB0703188D0 (en) * | 2007-02-19 | 2007-03-28 | Roger Land Building | Large scale production of stem cells |
| US20090053182A1 (en) | 2007-05-25 | 2009-02-26 | Medistem Laboratories, Inc. | Endometrial stem cells and methods of making and using same |
| DK2173863T3 (en) | 2007-06-29 | 2019-01-21 | Fujifilm Cellular Dynamics Inc | Automated method and apparatus for embryonic stem cell culture |
| RU2465323C2 (ru) | 2007-07-18 | 2012-10-27 | Лайфскен, Инк. | Дифференцировка эмбриональных стволовых клеток человека |
| ES2665434T3 (es) | 2007-07-31 | 2018-04-25 | Lifescan, Inc. | Diferenciación de células madre pluripotentes usando células alimentadoras humanas |
| BRPI0814894A2 (pt) | 2007-07-31 | 2014-10-21 | Lifescan Inc | Diferenciação de células-tronco embrionárias de ser humano. |
| CA2696622C (en) | 2007-08-24 | 2016-07-19 | Stichting Het Nederlands Kanker Instituut | Compositions for the treatment of neoplastic diseases |
| US20110151447A1 (en) | 2007-11-06 | 2011-06-23 | Children's Medical Center Corporation | Method to produce induced pluripotent stem (ips) cells from non-embryonic human cells |
| WO2009070592A2 (en) | 2007-11-27 | 2009-06-04 | Lifescan, Inc. | Differentiation of human embryonic stem cells |
| SG154367A1 (en) | 2008-01-31 | 2009-08-28 | Es Cell Int Pte Ltd | Method of differentiating stem cells |
| WO2009096049A1 (ja) | 2008-02-01 | 2009-08-06 | Kyoto University | 人工多能性幹細胞由来分化細胞 |
| US20100330677A1 (en) | 2008-02-11 | 2010-12-30 | Cambridge Enterprise Limited | Improved Reprogramming of Mammalian Cells, and Cells Obtained |
| US10066203B2 (en) * | 2008-02-21 | 2018-09-04 | Janssen Biotech Inc. | Methods, surface modified plates and compositions for cell attachment, cultivation and detachment |
| JPWO2009110215A1 (ja) | 2008-03-03 | 2011-07-14 | 独立行政法人科学技術振興機構 | 繊毛細胞の分化誘導方法 |
| EP2271747B1 (en) | 2008-03-17 | 2016-09-07 | Agency for Science, Technology And Research | Microcarriers for pluripotent stem cell culture |
| DK2283117T3 (da) | 2008-04-21 | 2014-01-20 | Viacyte Inc | Fremgangsmåde til oprensning af pancreatiske endodermceller afledt fra humane embryoniske stamceller |
| US8338170B2 (en) | 2008-04-21 | 2012-12-25 | Viacyte, Inc. | Methods for purifying endoderm and pancreatic endoderm cells derived from human embryonic stem cells |
| WO2009132083A2 (en) | 2008-04-22 | 2009-10-29 | President And Fellows Of Harvard College | Compositions and methods for promoting the generation of pdx1+ pancreatic cells |
| US8623648B2 (en) | 2008-04-24 | 2014-01-07 | Janssen Biotech, Inc. | Treatment of pluripotent cells |
| US7939322B2 (en) | 2008-04-24 | 2011-05-10 | Centocor Ortho Biotech Inc. | Cells expressing pluripotency markers and expressing markers characteristic of the definitive endoderm |
| US20090298178A1 (en) | 2008-06-03 | 2009-12-03 | D Amour Kevin Allen | Growth factors for production of definitive endoderm |
| DK2297319T3 (en) | 2008-06-03 | 2015-10-19 | Viacyte Inc | GROWTH FACTORS FOR PREPARING DEFINITIVE ENDODERM |
| EP2942392B1 (en) | 2008-06-30 | 2018-10-03 | Janssen Biotech, Inc. | Differentiation of pluripotent stem cells |
| DE102008032236A1 (de) | 2008-06-30 | 2010-04-01 | Eberhard-Karls-Universität Tübingen | Isolierung und/oder Identifizierung von Stammzellen mit adipozytärem, chondrozytärem und pankreatischem Differenzierungspotential |
| US20100028307A1 (en) | 2008-07-31 | 2010-02-04 | O'neil John J | Pluripotent stem cell differentiation |
| US9683215B2 (en) | 2008-08-22 | 2017-06-20 | President And Fellows Of Harvard College | Methods of reprogramming cells |
| CN102333862B (zh) | 2008-10-31 | 2018-04-27 | 詹森生物科技公司 | 人胚胎干细胞向胰腺内分泌谱系的分化 |
| US9234178B2 (en) | 2008-10-31 | 2016-01-12 | Janssen Biotech, Inc. | Differentiation of human pluripotent stem cells |
| NZ592622A (en) | 2008-11-04 | 2012-10-26 | Viacyte Inc | Stem cell aggregate suspension compositions and methods for differentiation thereof |
| US8008075B2 (en) | 2008-11-04 | 2011-08-30 | Viacyte, Inc. | Stem cell aggregate suspension compositions and methods of differentiation thereof |
| CN105349517B (zh) | 2008-11-14 | 2021-05-04 | 维赛特公司 | 源于人多能干细胞的胰腺细胞的包封 |
| JP2012509085A (ja) | 2008-11-20 | 2012-04-19 | ヤンセン バイオテツク,インコーポレーテツド | マイクロキャリア上での多能性幹細胞の培養 |
| WO2010063848A1 (en) | 2008-12-05 | 2010-06-10 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and medium for neural differentiation of pluripotent cells |
| AR077766A1 (es) | 2009-07-20 | 2011-09-21 | Janssen Biotech Inc | Diferenciacion de celulas madre embrionarias humanas |
| GB2484873B (en) | 2009-07-20 | 2014-04-30 | Janssen Biotech Inc | Differentiation of pancreatic endoderm cells. |
| AU2010319921A1 (en) | 2009-10-29 | 2012-05-17 | Janssen Biotech Inc. | Pluripotent stem cells |
| FI20096288A0 (fi) | 2009-12-04 | 2009-12-04 | Kristiina Rajala | Formulations and methods for culturing stem cells |
| RU2610176C2 (ru) | 2009-12-23 | 2017-02-08 | Янссен Байотек, Инк. | Дифференцировка человеческих эмбриональных стволовых клеток |
| JP5882226B2 (ja) | 2009-12-23 | 2016-03-09 | ヤンセン バイオテツク,インコーポレーテツド | ヒト胚性幹細胞の分化 |
| CA2791846A1 (en) | 2010-03-02 | 2011-09-09 | National University Of Singapore | Culture additives to boost stem cell proliferation and differentiation response |
| CN102959076B (zh) | 2010-03-31 | 2015-09-16 | 斯克里普斯研究所 | 重编程细胞 |
| WO2011139628A1 (en) | 2010-04-25 | 2011-11-10 | Mount Sinai School Of Medicine | Generation of anterior foregut endoderm from pluripotent cells |
| ES2728900T3 (es) | 2010-05-12 | 2019-10-29 | Janssen Biotech Inc | Diferenciación de células madre embrionarias humanas |
| JP6043999B2 (ja) | 2010-08-05 | 2016-12-14 | ウィスコンシン アラムニ リサーチ ファンデーション | ヒト多能性細胞培養のための簡易基本培地 |
| RU2599420C2 (ru) | 2010-08-31 | 2016-10-10 | Янссен Байотек, Инк. | Дифференцирование плюрипотентных стволовых клеток |
| WO2012117333A1 (en) | 2011-02-28 | 2012-09-07 | Stempeutics Research Malaysia Sdn Bhd | Isolation and expansion of adult stem cells, their therapeutic composition and uses thereof |
| WO2013055834A2 (en) | 2011-10-11 | 2013-04-18 | The New York Stem Cell Foundation | Er stress relievers in beta cell protection |
| EP2766474B1 (en) | 2011-10-14 | 2020-10-07 | Children's Medical Center Corporation | Inhibition and enhancement of reprogramming by chromatin modifying enzymes |
| WO2013095953A1 (en) | 2011-12-22 | 2013-06-27 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells into single hormonal insulin positive cells |
| US10519422B2 (en) | 2012-02-29 | 2019-12-31 | Riken | Method of producing human retinal pigment epithelial cells |
| JP6469003B2 (ja) | 2012-06-08 | 2019-02-13 | ヤンセン バイオテツク,インコーポレーテツド | 膵内分泌細胞へのヒト胚性幹細胞の分化 |
| EP2893000B1 (en) | 2012-09-03 | 2019-04-10 | Novo Nordisk A/S | Generation of pancreatic endoderm from pluripotent stem cells using small molecules |
| KR101942769B1 (ko) | 2012-12-31 | 2019-01-28 | 얀센 바이오테크 인코포레이티드 | Hb9 조절제를 사용하는 인간 배아 줄기세포의 췌장 내분비 세포로의 분화 |
| US8859286B2 (en) | 2013-03-14 | 2014-10-14 | Viacyte, Inc. | In vitro differentiation of pluripotent stem cells to pancreatic endoderm cells (PEC) and endocrine cells |
| TW201522637A (zh) | 2013-03-15 | 2015-06-16 | Jackson Lab | 非胚胎幹細胞之單離及其用途 |
| US11645208B2 (en) | 2021-03-29 | 2023-05-09 | International Business Machines Corporation | Translation bandwidth optimized prefetching strategy through multiple translation lookaside buffers |
-
2009
- 2009-11-19 JP JP2011537603A patent/JP2012509085A/ja active Pending
- 2009-11-19 RU RU2011124900/10A patent/RU2555538C2/ru not_active IP Right Cessation
- 2009-11-19 WO PCT/US2009/065062 patent/WO2010059775A1/en active Application Filing
- 2009-11-19 AU AU2009316580A patent/AU2009316580B2/en not_active Ceased
- 2009-11-19 CN CN201710585295.7A patent/CN107267442A/zh active Pending
- 2009-11-19 EP EP17184333.7A patent/EP3260534A1/en not_active Withdrawn
- 2009-11-19 KR KR1020117013824A patent/KR101774546B1/ko active IP Right Grant
- 2009-11-19 BR BRPI0920956-5A patent/BRPI0920956A2/pt not_active Application Discontinuation
- 2009-11-19 ES ES09759851.0T patent/ES2642070T3/es active Active
- 2009-11-19 CA CA2744234A patent/CA2744234C/en not_active Expired - Fee Related
- 2009-11-19 PL PL09759851T patent/PL2366021T3/pl unknown
- 2009-11-19 KR KR1020177024255A patent/KR101837080B1/ko active IP Right Grant
- 2009-11-19 CN CN200980155382.2A patent/CN102307991B/zh not_active Expired - Fee Related
- 2009-11-19 MX MX2011005288A patent/MX2011005288A/es active IP Right Grant
- 2009-11-19 US US12/621,686 patent/US20100124781A1/en not_active Abandoned
- 2009-11-19 MX MX2014000085A patent/MX356756B/es unknown
- 2009-11-19 EP EP09759851.0A patent/EP2366021B1/en not_active Not-in-force
-
2011
- 2011-06-17 ZA ZA2011/04506A patent/ZA201104506B/en unknown
-
2012
- 2012-02-27 HK HK18105925.9A patent/HK1246356A1/zh unknown
-
2015
- 2015-02-25 US US14/631,019 patent/US9969972B2/en not_active Expired - Fee Related
-
2016
- 2016-02-26 AU AU2016201220A patent/AU2016201220B2/en not_active Ceased
- 2016-09-16 ZA ZA2016/06403A patent/ZA201606403B/en unknown
-
2018
- 2018-05-14 US US15/978,913 patent/US20180265842A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007149182A2 (en) * | 2006-06-19 | 2007-12-27 | Geron Corporation | Differentiation and enrichment of islet-like cells from human pluripotent stem cells |
| JP2008099662A (ja) * | 2006-09-22 | 2008-05-01 | Institute Of Physical & Chemical Research | 幹細胞の培養方法 |
Non-Patent Citations (2)
| Title |
|---|
| JPN5012004399; PHILLIPS B W: 'ATTACHMENT AND GROWTH OF HUMAN EMBRYONIC STEM CELLS ON MICROCARRIERS' JOURNAL OF BIOTECHNOLOGY V138 N1-2, 20081106, P24-32, ELSEVIER SCIENCE PUBLISHERS * |
| JPN5012004401; WATANABE KIICHI: 'A ROCK INHIBITOR PERMITS SURVIVAL OF DISSOCIATED HUMAN EMBRYONIC STEM CELLS' NATURE BIOTECHNOLOGY V25 N6, 20070601, P681-686, NATURE PUBLISHING GROUP * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016504037A (ja) * | 2012-12-31 | 2016-02-12 | ヤンセン バイオテツク,インコーポレーテツド | 膵内分泌細胞への分化のためのヒト多能性細胞の懸濁及びクラスタリング |
| JP2016502839A (ja) * | 2013-01-04 | 2016-02-01 | 国立大学法人京都大学 | 初期化幹細胞 |
| JP2016508726A (ja) * | 2013-02-22 | 2016-03-24 | セルラー ダイナミクス インターナショナル, インコーポレイテッド | 組み合わせた遺伝的および化学的操作によるフォワードプログラミングを介した肝細胞の産生 |
| JP2016529920A (ja) * | 2013-09-19 | 2016-09-29 | ザ ボード オブ トラスティーズ オブ ザ レランド スタンフォード ジュニア ユニバーシティー | 肝細胞様細胞を産生するための方法および組成物 |
| US11291691B2 (en) | 2013-09-19 | 2022-04-05 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions for producing hepatocyte-like cells |
| WO2019102593A1 (ja) * | 2017-11-24 | 2019-05-31 | 株式会社Ihi | 細胞培養装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2744234C (en) | 2018-11-06 |
| ES2642070T3 (es) | 2017-11-15 |
| US20150166950A1 (en) | 2015-06-18 |
| EP2366021B1 (en) | 2017-08-02 |
| US20180265842A1 (en) | 2018-09-20 |
| MX2011005288A (es) | 2011-06-01 |
| AU2016201220B2 (en) | 2017-09-28 |
| EP3260534A1 (en) | 2017-12-27 |
| AU2009316580B2 (en) | 2016-04-14 |
| HK1246356A1 (zh) | 2018-09-07 |
| EP2366021A1 (en) | 2011-09-21 |
| MX356756B (es) | 2018-06-11 |
| KR101837080B1 (ko) | 2018-03-09 |
| RU2555538C2 (ru) | 2015-07-10 |
| RU2011124900A (ru) | 2012-12-27 |
| CN107267442A (zh) | 2017-10-20 |
| CA2744234A1 (en) | 2010-05-27 |
| KR20170102075A (ko) | 2017-09-06 |
| ZA201104506B (en) | 2018-11-28 |
| BRPI0920956A2 (pt) | 2015-08-18 |
| ZA201606403B (en) | 2018-05-30 |
| US9969972B2 (en) | 2018-05-15 |
| PL2366021T3 (pl) | 2017-12-29 |
| CN102307991B (zh) | 2017-08-15 |
| KR101774546B1 (ko) | 2017-09-04 |
| KR20110091768A (ko) | 2011-08-12 |
| WO2010059775A1 (en) | 2010-05-27 |
| AU2009316580A1 (en) | 2010-05-27 |
| CN102307991A (zh) | 2012-01-04 |
| US20100124781A1 (en) | 2010-05-20 |
| AU2016201220A1 (en) | 2016-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2016201220B2 (en) | Pluripotent stem cell culture on micro-carriers | |
| CA2691975C (en) | Single pluripotent stem cell culture | |
| JP5719305B2 (ja) | 平面支持体上での細胞付着及び培養のための方法及び組成物 | |
| JP5922147B2 (ja) | 多能性幹細胞に由来するインスリン産生細胞 | |
| US9080145B2 (en) | Single pluripotent stem cell culture |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20121115 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140430 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140730 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140806 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140814 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20141203 |