EP0000057B1 - Selektiv geschützte 4,6-Di-O-(aminoglycosyl)-1,3-diaminocyclitole - Google Patents
Selektiv geschützte 4,6-Di-O-(aminoglycosyl)-1,3-diaminocyclitole Download PDFInfo
- Publication number
- EP0000057B1 EP0000057B1 EP78100099A EP78100099A EP0000057B1 EP 0000057 B1 EP0000057 B1 EP 0000057B1 EP 78100099 A EP78100099 A EP 78100099A EP 78100099 A EP78100099 A EP 78100099A EP 0000057 B1 EP0000057 B1 EP 0000057B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- sisomicin
- tetra
- methanol
- phenyl
- sisomycin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/20—Carbocyclic rings
- C07H15/22—Cyclohexane rings, substituted by nitrogen atoms
- C07H15/222—Cyclohexane rings substituted by at least two nitrogen atoms
- C07H15/226—Cyclohexane rings substituted by at least two nitrogen atoms with at least two saccharide radicals directly attached to the cyclohexane rings
- C07H15/234—Cyclohexane rings substituted by at least two nitrogen atoms with at least two saccharide radicals directly attached to the cyclohexane rings attached to non-adjacent ring carbon atoms of the cyclohexane rings, e.g. kanamycins, tobramycin, nebramycin, gentamicin A2
- C07H15/236—Cyclohexane rings substituted by at least two nitrogen atoms with at least two saccharide radicals directly attached to the cyclohexane rings attached to non-adjacent ring carbon atoms of the cyclohexane rings, e.g. kanamycins, tobramycin, nebramycin, gentamicin A2 a saccharide radical being substituted by an alkylamino radical in position 3 and by two substituents different from hydrogen in position 4, e.g. gentamicin complex, sisomicin, verdamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Definitions
- the invention relates to new, selectively acylated or sulfenylated 4,6-di-O- (aminoglycosyl) -1,3-diamino-cyclitols, which can serve as intermediates for the production of valuable new and known antibiotics.
- radicals R ' are the o-nitrophenylsulfenyl radical and the 2,4,5-trichlorophenylsulfenyl radical.
- FR-A-2 240 015 and 2 263 251 describe aminoglycosides which have protective groups in the 2'- and / or 6'-position. However, there is no reference whatsoever to derivatives with four protective groups and the person skilled in the art is also shown no way in which he could arrive at aminoglycoside derivatives in which an amino group is selectively unprotected.
- Preferred acylation reagents are those in which G 'denotes chlorine or the radical ⁇ O ⁇ CO ⁇ A, with the meaning given for A above.
- Examples include: acetic anhydride, acetyl chloride and di-t-butylpyrocarbonate, the use of dialkylpyrocarbonates as a blocking reagent being generally particularly preferred for the preparation of N-alkyloxycarbonyl derivatives of sisomicin.
- Suitable diluents for the reaction with sulfenoic acid halides are both inert organic solvents such as chloroform and toluene, but preferably water-miscible solvents such as dioxane, DMF and dimethoxyethane, and mixtures thereof with water.
- the reactions with activated esters of the above sulfenic acids are preferably carried out in inert organic solvents such as CHCl 3 , DMF, pyridine or mixtures of such solvents with alcohols, preferably methanol or ethanol.
- inert organic solvents such as CHCl 3 , DMF, pyridine or mixtures of such solvents with alcohols, preferably methanol or ethanol.
- the acyl compounds according to the invention are prepared in any inert organic solvents or in mixtures of organic solvents with water, mixtures of methanol, ethanol or acetone with water being preferred.
- All basic compounds common in organic chemistry such as, for. B. triethylamine, pyridine, diazabicyclonones can be used; but are preferably alkali metal hydroxides or carbonates such.
- the sulfenylations or acylations are carried out at temperatures between about -30 ° C and + 50 ° C, preferably between about 0 ° C and + 25 ° C.
- the reactions can be carried out both under normal pressure and under elevated pressure. Generally one works at normal pressure.
- the acid addition salts from the 4,6-di-O- (aminoglycosyl) -1,3-diaminocyclitols with inorganic and organic acids, preferably the chlorides and sulfates, can also be used.
- the amount of auxiliary base used must be varied according to the number of amino groups present in salt form.
- the derivatives according to the invention are valuable intermediates in the preparation of derivatives of 4,6-di-O- (aminoglycosyl) -1,3-diaminocyclitole gentamicin C ia , gentamicin C 2 , sisomicin and verdamicin.
- antibiotics are valuable substances for the effective fight against bacterial infections.
- their high effectiveness is often associated with relatively high nephro- and ototoxicity; in addition there is the development of resistance of the controlled germs
- it is desirable to produce derivatives of the aminoglycoside antibiotics with improved properties which, with reduced toxicity, may also make it possible to control resistant germs.
- Compounds such as 1-N-acetylsisomicin and 1-N-ethylsisomicin have become known as substances with such improved properties (DE-OS 2 437 160).
- the sulfenyl protecting groups can be used with nucleophiles such as e.g. B. H 2 S, thiophenol, etc. as well as by weak acids, that is, under conditions under which the newly introduced acyl or alkyl radicals are stable.
- nucleophiles such as e.g. B. H 2 S, thiophenol, etc.
- weak acids that is, under conditions under which the newly introduced acyl or alkyl radicals are stable.
- deblocking can be carried out with aqueous alkali metal or alkaline earth metal hydroxide or with acids such as trifluoroacetic acid, perchloric acid or boron trifluoride etherate in organic solvents or mixtures of organic solvents with water.
- acids such as trifluoroacetic acid, perchloric acid or boron trifluoride etherate in organic solvents or mixtures of organic solvents with water.
- acyl protective groups and cleavage reagents newly introduced alky
- the selective cleavage of the protective groups can take place both with equivalent amounts of cleaving reagent and with an excess, but preferably with a 20-50-fold excess.
- the optimal reaction time is determined by thin layer chromatography.
- 1-N-formylsisomicin is particularly advantageous.
- 2 ', 3.3 “, 6'-tetra-N- (o-nitro-phenylsulfenyl) sisomicin which is accessible by sulfenylation of sisomicin or its acid addition salts, is split with about 1 to 5 mol of formylating agent processes the o-nitrophenylsulfenyl groups with sulfur-containing, nucleophilic reagents such as H 2 S or thiophenol, works up the reaction set in the usual way for the free 1-N-formyl derivative and, if necessary, converts it into the pharmaceutically acceptable salts
- Sulfenyl derivatives are preferably carried out in pyridine as a diluent using p-nitrophenyl formate as a formylating agent.
- sulfenyl derivatives which can be obtained in an analogous manner by reacting sisomicin with corresponding sulfonic acid chlorides or the sulfonic acid p-nitrophenyl esters can also be used with great success.
- examples include: tritylsulfenyl, pentachlorophenylsulfenyl and 2,4-dinitrophenylsulfenylsisomicin derivatives.
- formic acid-acetic anhydride is also particularly suitable for the formylation of the sulfenyl derivative.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Oncology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Communicable Diseases (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
- Die Erfindung betrifft neue, selektiv acylierte oder sulfenylierte 4,6-Di-O-(aminoglykosyl)-1,3-diami- nocyclitole, die als Zwischenprodukte zur Herstellung wertvoller neuer und bekannter Antibiotika dienen können.
- Bei den neuen Aryl- oder Sulfenylderivaten der 4,6-Di-O-(aminoglykosyl)-1,3-diaminocyclitole handelt es sich um an 4 der 5 Aminogruppen durch Reste -SR' oder -CO-A blockierte Aminoglykoside aus der Gruppe Sisomicin, Verdamicin, Gentamicin C1a und Gentamicin C2, wobei R' Phenyl, durch 1 - Substituenten aus der Reihe Nitro, C1-C4-Alkyl, C1-C4-Alkoxy, C1- C4-Alkylcarbonyl oder Phenyl oder durch 1 -5 Halogenatome (vorzugsweise Chlor) substituiertes Phenyl, Di- oder Triphenylmethyl und A einen der Reste

- B und D für Wasserstoff, Phenyl oder durch einen oder zwei Substituenten aus der Reihe Nitro, C1-C4-Alkyl, Ci-C4-Alkoxy, Phenyl oder Halogen (vorzugsweise Chlor) substituiertes Phenyl und
- n, n1, n2
- und n3 unabhängig für eine Zahl von 0-5 stehen,
- Als Beispiele für Reste R' seien der o-Nitrophenylsulfenylrest und der 2,4,5-Trichlorphenylsulfenylrest genannt.
- Aus US-PS 3 997 524 sind zwar bereits an vier der fünf Aminogruppen durch Arylreste geschützte Derivate des Sisomicins und Verdamicins bekannt. Die Schutzgruppen aufweisenden Stickstoffatome sind jedoch jene in den 1,3,2'- und 3"-Positionen, während die Aminogruppe in der 6'-Position nicht blockiert ist. Bei den erfindungsgemäßen Verbindungen ist jedoch gerade die 6'-Position zwingend mit einer Schutzgruppe versehen. Gemäß US-PS 3997524 werden die tetra-blockierten Aminoglykoside hergestellt, indem man zunächst die reaktivste 6'-Aminogruppe mit einer leicht abspaltbaren Schutzgruppe versieht, danach die restlichen vier Aminogruppen acyliert und die Schutzgruppen in der 6'-Position wieder abspaltet. Es wird somit keinerlei Anregung gegeben, Derivate herzustellen, bei denen eine der Aminogruppen in der 1,3,2'- oder 3"-Position nicht geschützt ist.
- In FR-A-2 240 015 und 2 263 251 werden Aminoglykoside beschrieben, welche in der 2'- und/oder 6'-Position Schutzgruppen aufweisen. Es findet sich jedoch keinerlei Hinweis auf Derivate mit vier Schutzgruppen und dem Fachmann wird auch kein Weg gewiesen, auf welchem er zu Aminoglykosid-Derivaten gelangen könnte, bei denen selektiv eine Aminogruppe ungeschützt ist.
- Erfindungsgemäß bevorzugt sind Derivate von Sisomicin der Formel (1)

- R1 für einen Rest -SR' oder -CO-A steht, und
- R2, R3, R4
- und R5 Wasserstoff, einen Rest -SR' oder -CO-A darstellen, mit der Maßgabe, daß einer und nur einer der Reste R2―R5 für Wasserstoff steht, und
- R' und A die oben angegebene Bedeutung haben.
- Beispiele für erfindungsgemäß bevorzugte Verbindungen sind
- 2',3,3",6'-Tetra-N-(o-nitrophenylsulfenyl)-sisomicin,
- 1,2',3,6'-Tetra-N-acetyl-sisomicin,
- 1,3,3",6'-Tetra-N-(t-butoxycarbonyl)-sisomicin,
- 1,2',3,6'-Tetra-N-(ethoxycarbonyl)-sisomicin,
- 1,2',3",6'-Tetra-N-(o-nitrophenyisulfenyl)-sisomicin und
- 1,3,3",6'-Tetra-N-(o-nitrophenylsulfenyl)-sisomicin.
- Wie sich aus US-PS 3 997 524 ergibt, ist der Erfindungsgedanke aber auch auf Verdamicin übertragbar: Der Literaturstelle ist zu entnehmen, daß auch beim Verdamicin die Aminogruppe in der 6'-Position die reaktivste ist. Ähnliches gilt für Gentamicin C1a und C2, wie die Beispiele 9 und 10 der europäischen Patentanmeldung 80 108 216.5 zeigen.
- Die erfindungsgemäßen Derivate werden erhalten, indem man Sisomicin, Verdamicin, Gentamicin Cia, Gentamicin C2 oder deren Säureadditionssalze, mit etwa 4 Äquivalenten einer Verbindung der Formel

- R' die oben angegebene Bedeutung besitzt und
- G Halogen oder eine bei Sulfenylierungsreaktionen gebräuchliche Abgangsgruppe, vorzugsweise einen aktivierenden Esterrest, bezeichnet,
oder mit etwa 4 Äquivalenten einer Verbindung der Formel
- A die oben genannte Bedeutung besitzt und
- G' Halogen oder eine bei Acylierungsreaktionen gebräuchliche Abgangsgruppe, vorzugsweise einen aktivierenden Esterrest, oder eine Gruppe -O-CO-A mit der obigen Bedeutung für A bezeichnet,
- Verwendet man Sisomicin und o-Nitrophenylsulfensäure-p-nitrophenylester als Ausgangsstoffe, so kann der Reaktionsablauf durch das folgende Schema wiedergegeben werden:
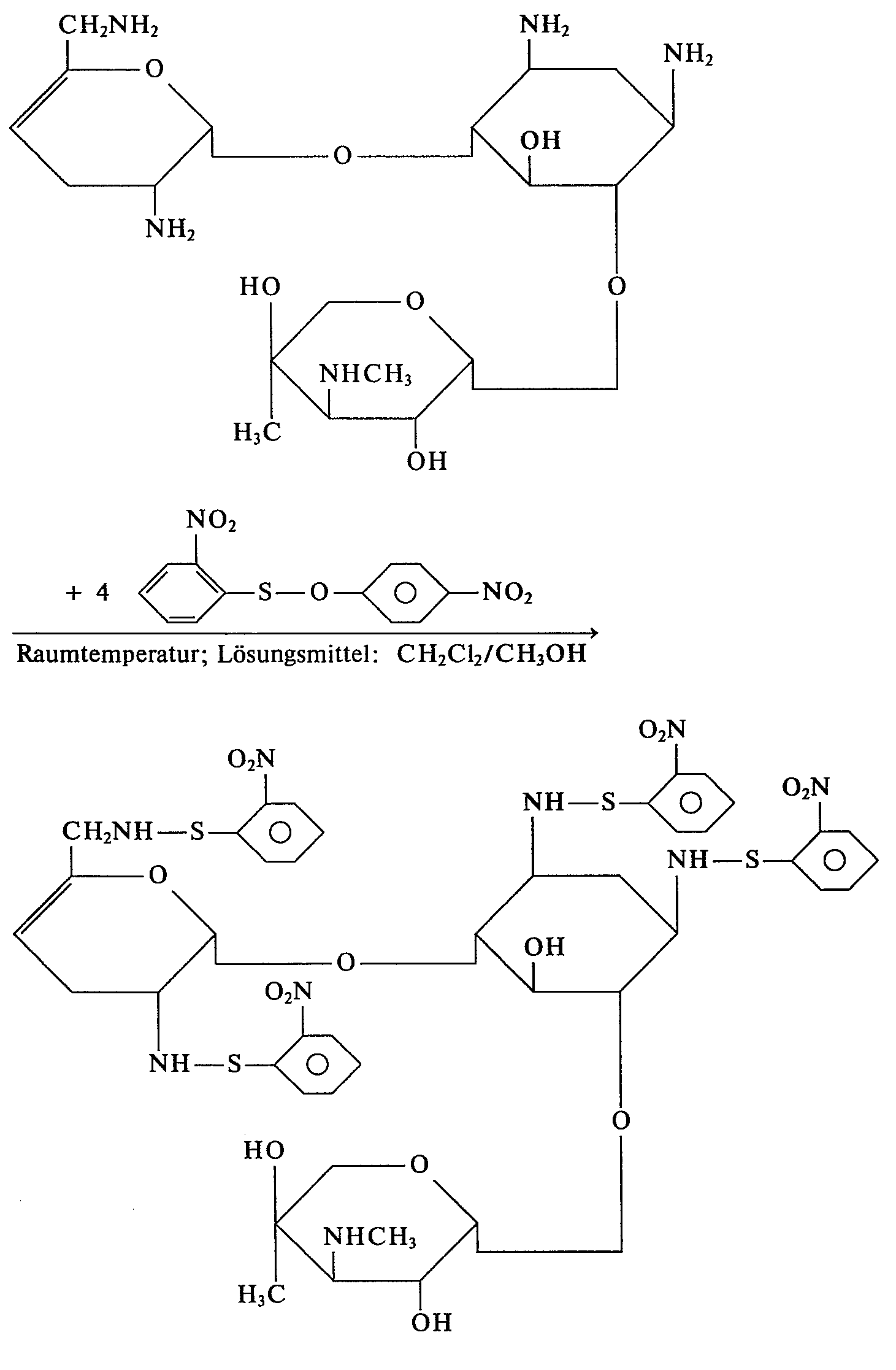
- Für die Darstellung der erfindungsgemäßen Verbindungen auf dem oben geschilderten Wege durch Umsetzung der ungeschützten 4,6-Di-O-(aminoglykosyl)-1,3-diaminocyclitole mit einem Sulfenylierungsreagenz finden vorzugsweise Sulfenylierungsreagenzien Verwendung, in denen G Chlor oder den p-Nitrophenyloxyrest bezeichnet. Als Beispiele seien genannt:
- Tritylsulfenylchlorid, o-Nitrophenylsulfenylchlorid,
- 2,4-Dinitrophenylsulfenylchlorid, 2,4,5-Trichlorphenylsulfenylchlorid,
- Pentachlorphenylsulfenylchlorid und o-Nitrophenylsulfensäure-p-nitrophenylester,
- 2,4-Dinitrophenylsulfenyl-p-nitrophenylester,
- 2,4,5-Trichlorphenylsulfensäure-p-nitrophenylester und
- Pentachlorphenylsulfensäure-p-nitrophenylester.
- Diese reaktiven Sulfensäurederivate sind entweder bereits bekannt (siehe z. B. Houben-Weyl, Methoden der Organischen Chemie, Band XV, 1, Seiten 203-222, Georg Thieme Verlag, Stuttgart, 1974' oder können nach analogen Verfahren wie die bereits bekannten Verbindungen hergestellt werden.
- Bevorzugte Acylierungsreagenzien sind solche, bei denen G' Chlor oder den Rest ―O―CO―A, mit der oben angegebenen Bedeutung für A, bezeichnet. Beispielhaft seien genannt: Acetanhydrid, Acetylchlorid und Di-t-Butylpyrocarbonat, wobei zur Darstellung von N-Alkyl-oxycarbonyl-derivaten des Sisomicins die Verwendung von Dialkylpyrocarbonaten als Blockierungsreagenz im allgemeinen besonders zu bevorzugen ist.
- Als Verdünnungsmittel für die Umsetzung mit Sulfensäurehalogeniden kommen sowohl inerte organische Lösungsmittel wie Chloroform und Toluol, vorzugsweise aber mit Wasser mischbare Lösungsmittel wie Dioxan, DMF und Dimethoxyethan sowie deren Gemische mit Wasser in Frage.
- Die Umsetzungen mit aktivierten Estern der obengenannten Sulfensäuren erfolgen vorzugsweise in inerten organischen Lösungsmitteln wie CHCI3, DMF, Pyridin oder Gemischen von solchen Lösungsmitteln mit Alkoholen, vorzugsweise Methanol oder Ethanol. Die Darstellung der erfindungsgemäßen Acylverbindungen erfolgt in beliebigen inerten organischen Lösungsmitteln oder in Gemischen organischer Lösungsmittel mit Wasser, wobei Gemische von Methanol, Ethanol oder Aceton mit Wasser zu bevorzugen sind.
- Als Basen können alle in der organischen Chemie üblichen basischen Verbindungen wie z. B. Triethylamin, Pyridin, Diazabicyclononen eingesetzt werden; vorzugsweise werden aber Alkali-hydroxide bzw. -carbonate wie z. B. Natronlauge oder Natriumcarbonat verwendet.
- Die Sulfenylierungen bzw. Acylierungen werden bei Temperaturen zwischen etwa -30°C und + 50° C, vorzugsweise zwischen etwa 0° C und + 25° C, durchgeführt.
- Die Umsetzungen können sowohl bei Normaldruck, aber auch bei erhöhtem Druck durchgeführt werden. Im allgemeinen arbeitet man bei Normaldruck.
- Bei der Sulfenylierung bzw. Acylierung der Aminoglycoside können auch die Säureadditionssalze aus den 4,6-Di-O-(aminoglykosyl)-1,3-Diaminocyclitolen mit anorganischen und organischen Säuren, vorzugsweise die Chloride und Sulfate, eingesetzt werden.
- In diesem Falle muß die Menge der verwendeten Hilfsbase entsprechend der Zahl der in Salzform vorliegenden Aminogruppen variiert werden.
- Die erfindungsgemäßen Derivate sind wertvolle Zwischenprodukte bei der Darstellung von Derivaten der4,6-Di-O-(aminoglykosyl)-1,3-diaminocyclitole Gentamicin Cia, Gentamicin C2, Sisomicin und Verdamicin.
- Diese Antibiotika sind wertvolle Substanzen zur wirkungsvollen Bekämpfung bakterieller Infektionen. Ihre hohe Wirksamkeit ist jedoch häufig verbunden mit relativ großer Nephro- und Ototoxizität; hinzu kommt Resistenzbildung der bekämpften Keime. Aus diesen Gründen ist es wünschenswert, Derivate der Aminoglykosid-Antibiotika mit verbesserten Eigenschaften herzustellen, die bei verringerter Toxizität eventuell auch die Bekämpfung resistenter Keime ermöglichen. Als Substanzen mit derartig verbesserten Eigenschaften sind Verbindungen wie 1-N-Acetylsisomicin und 1-N-Ethylsisomicin bekanntgeworden (DE-OS 2 437 160).
- Die Darstellung mono-N-substituierter Sisomicin-Derivate erweist sich jedoch ausgehend von ungeschütztem Sisomicin als schwierig, da im Sisomicin-Molekül fünf Aminogruppen vergleichbarer Reaktivität vorhanden sind. Man gelangt daher immer zu Reaktionsgemischen, was aufwendige chromatographische Verfahren zur Isolierung der gewünschten Verbindung notwendig macht. Erfindungsgemäß werden nunmehr Derivate von Aminoglysid-Antibiotika mit einer selektiv ungeschützten Aminogruppe zur Verfügung gestellt, wobei die Natur der Schutzgruppen so beschaffen ist, daß sowohl Alkylierungen als auch Acylierungen (in glatter Reaktion und mit hohen Ausbeuten) an den freien Stickstoffatomen möglich sind und eine anschließende einfache und schonende Entblockierung durchgeführt werden kann, ohne daß aufwendige-chromatographische Reinigungsoperationen erforderlich werden.
- Die Sulfenyl-Schutzgruppen können sowohl mit Nucleophilen wie z. B. H2S, Thiophenol usw. als auch durch schwache Säuren abgespalten werden, d. h. unter Bedingungen, unter denen die neu eingeführten Acyl- oder Alkylreste stabil sind. Werden Acylreste, wie z. B. Acetyl-, Ethyloxycarbonyl-oder t-Butyloxycarbonylgruppen als Schutzgruppen verwendet, kann die Deblockierung mit wäßrigem Alkali- oder Erdalkalihydroxid oder mit Säuren wie Trifluoressigsäure, Perchlorsäure oder Bortrifluoridetherat in organischen Lösungsmitteln bzw. Mischungen von organischen Lösungsmitteln mit Wasser erfolgen. Auch hier werden bei entsprechender Wahl von Acyl-Schutzgruppen und Abspaltungsreagenzien neu eingeführte Alkyl- bzw. Acylreste bei der Deblockierung nicht angegriffen.
- Die selektive Abspaltung der Schutzgruppen kann sowohl mit äquivalenten Mengen Abspaltungsreagens als auch mit einem Überschuß erfolgen, vorzugsweise aber mit einem 20-50fachen Überschuß. Dabei wird die optimale Reaktionszeit dünnschichtchromatographisch ermittelt.
- Zur Herstellung von 1-N-Acetyl-sisomicin unter Verwendung eines der erfindungsgemäßen Zwischenprodukte geht man z. B. so vor, daß man ein geschütztes Sisomicinderivat der Formel (I), in der Ri, R2, R3 und R5 den o-Nitrophenylsulfenylrest bezeichnen, mit 1 Äquivalent Essigsäure-p-nitrophenylester in Pyridin umsetzt, die o-Nitrophenylsulfenylschutzgruppen mit überschüssigem Schwefelwasserstoff in Alkohol bei pH 1 -3 abspaltet und 1-N-Acetylsisomicin durch Behandlung mit einem basischen lonenaustauscher isoliert.
- Zur Darstellung von 2'-N-Acetylsisomicin setzt man z. B. 1 Mol einer Verbindung der Formel (I), in der RI, R3, R4 und Rs tert.-Butyloxycarbonyl-Reste bezeichnen, mit 1 Äquivalent Essigsäureanhydrid in Methanol um und spaltet nach Beendigung der Acylierungsreaktion die Schutzgruppen mit überschüssigem Bortrifluorid-Etherat in Aceton ab und isoliert das entstandene 2'-N-Acetylsisomicin durch Behandlung mit einem basischen lonenaustauscher.
- Besonders vorteilhaft gelingt die Herstellung von 1-N-Formylsisomicin. Dazu setzt man 2',3,3",6'-Tetra-N-(o-nitro-phenylsulfenyl)-sisomicin, das durch Sulfenylierung von Sisomicin bzw. dessen Säureadditionssalzen zugänglich ist, mit etwa 1 bis 5 Mol Formylierungsmittel um, spaltet die o-Nitrophenylsulfenylgruppen mit schwefelhaltigen, nucleophilen Reagentien wie z. B. H2S oder Thiophenol ab, arbeitet den Reaktionssatz in üblicher Weise auf das freie 1-N-Formylderivat hin auf und überführt dieses gegebenenfalls in die pharmazeutisch verwendbaren Salze. Die Formylierung des Sulfenylderivates erfolgt vorzugsweise in Pyridin als Verdünnungsmittel unter Verwendung von Ameisensäure-p-nitrophenylester als Formylierungsmittel.
- Anstelle der o-Nitrophenylsulfenyl-Derivate können mit gutem Erfolg auch andere Sulfenylderivate, die in analoger Weise durch Umsetzung von Sisomicin mit entsprechenden Sulfonsäurechloriden bzw. den Sulfonsäuren-p-nitrophenylestern erhältlich sind, eingesetzt werden. Als Beispiele seien genannt: Tritylsulfenyl-, Pentachlorphenylsulfenyl- und 2,4-Dinitrophenylsulfenylsisomicin-Derivate.
- Neben Ameisensäure-p-nitrophenylester eignet sich auch Ameisensäure-essigsäure-anhydrid besonders gut zur Formylierung des Sulfenylderivates.
- 450 mg Sisomicin werden in 10 ml Wasser gelöst. Nach Zugabe von 10 ml Methanol versetzt man unter gutem Rühren mit 870 mg Pyrokohlensäure-di-t-butylester. Nach 1,3 Stunden Rühren bei Raumtemperatur versetzt man mit 5 ml Wasser, filtriert und dampft das Filtrat im Vakuum zur Trockene ein. Man löst den Rückstand in Methanol und fällt das gewünschte Produkt durch Zugabe von Ether und Petrolether. Ausbeute = 600 mg.
- 13C-NMR (CD2OD):
- δ=66,01 (C-3"); 52,23 (C-1); 51,67 (C-3); 48,23 (C-2'); 43,74 (C-6'); 157,69 ( c=0) ppm.
- 1,1 g Sisomicin werden in 50 ml Ethanol und 70 ml Wasser gelöst. Nach Abkühlen auf -10°C tropft man unter gutem Rühren 1,35 ml Pyrokohlensäurediethylester dazu. Nach weiteren 21/2 Stunden bei -10°C wird mit 100 ml Wasser versetzt. Man extrahiert anschließend mit 150 ml Petrolether und dampft die wäßrige Phase im Vakuum zur Trockene ein. Der Rückstand wird in Methanol gelöst. Durch Zugabe von überschüssigem Ether und Petrolether wird das gewünschte Produkt ausgefällt. Ausbeute = 1,5 g (91%).
- 13C-NMR (CD3OD/CDCl3):
- δ=50,86 (C-1); 49,90 (C-2); 46,33 (C-2'); 42,87 (C-6'); 157,94,157,73,157,29 und 157,22 ( c=0) ppm.
- 1,1 g Sisomicin werden in 120 ml Wasser gelöst. Nach Zusatz von 60 ml Methanol tropft man unter Rühren 2,5 ml Acetanhydrid dazu. Nach 15 Minuten wird im Vakuum zur Trockene eingedampft. Den Rückstand löst man in 10 ml Methanol und tropft diese Lösung in eine Mischung von 30 ml Ether und 30 ml Petrolether, wobei das gewünschte Produkt ausfällt. Ausbeute = 1,43 g, MS: m/e=615.
- 13C-NMR (CD30D):
- δ=50,14 (C-1); 49,20 (C-3); 48,88 (C-2'); 42,26 (C-6'); 173,24,173,13,172,63 ( c=0) ppm.
- 1,3 g des Produktes von Beispiel 3 in 20 ml Dimethylformamid werden mit 2,5 g Silberoxid und 2,2 g p-Nitrobenzylbromid für 1 Stunde bei Raumtemperatur gerührt. Anschließend verdünnt man mit 50 ml Chloroform, rührt kurz mit wenig Holzkohle und filtriert. Das Filtrat wird im Vakuum zur Trockene eingedampft und der Rückstand aus Chloroform mit Petrolether umgefällt. Ausbeute = 1,1 g.
- Zur Reduktion der Nitrogruppe löst man 200 mg des oben erhaltenen Produktes in 2 ml Wasser und gibt nach Zusatz von 0,06 ml Essigsäure 400 mg Eisenpulver dazu. Man erhitzt für 10 Minuten auf 70°C und läßt anschließend noch 1 Stunde bei Raumtemperatur reagieren. Zur Aufarbeitung wird filtriert, das Filtrat eingedampft und der Rückstand in 5 ml Methanol und 5 ml Wasser mit Holzkohle entfärbt. Nach Filtrieren der Lösung wird im Vakuum eingedampft. Zur Abspaltung der N-Acetylgruppen erhitzt man den so erhaltenen Rückstand in 2 ml Wasser mit 1 g Bariumhydroxidoctahydrat für 8 Stunden am Rückfluß. Man läßt erkalten, fällt die Bariumsalze mit Kohlendioxid und filtriert. Eindampfen des Filtrats und Fällen des erhaltenen Rückstands aus Methanol mit Ether liefert die Titelverbindung. Ausbeute = 95 mg.
- 175 mg 1,3,3",6'-Tetra-N-t-butoxycarbonyl-sisomicin werden in 7 ml Methanol gelöst. Diese Lösung versetzt man nacheinander mit 7 ml Aceton sowie 2 ml Acetanhydrid. Nach 2 Stunden wird im Vakuum vom Lösungsmittel befreit und der dabei erhaltene Rückstand in Methanol gelöst. Man fällt das gewünschte Produkt durch Zugabe von überschüssigem Ether und Petrolether. Ausbeute = 170 mg.
- 13C-NMR (CD3OD/CDCl3):
- δ=56,72 (C-2'); 23,14 und 175,80 (Acetyl) ppm.
- Zur Abspaltung der t-Butoxycarbonylschutzgruppen löst man 150 mg des nach a) erhaltenen Produktes in 3 ml Aceton und versetzt mit 0,6 ml Bortrifluorid-Diethylether-Komplex. Nach 1 Stunde gießt man in eine Mischung von 15 ml Ether und 5 ml Petrolether, wobei das gewünschte Produkt ausfällt. Man entionisiert durch Rühren mit einem basischen lonenaustauscher (OHe-Form) in wäßriger Lösung. Das Harz wird abfiltriert und das Filtrat im Vakuum vom-Lösungsmittel befreit. Der Rückstand wird in wenig Methanol gelöst und durch Eingießen in überschüssigen Ether gefällt. Ausbeute = 60 mg.
- 13C-NMR (D2O/Dioxan):
- 6=56,67 (C-2'); 21,62 und 177,01 (Acetyl) ppm.
- 265 mg (0,25 mMol) 1,3,3",6'-Tetra-N-NPS-sisomicin (Beispiel 9) und 47 mg Essigsäure-p-nitrophenylester werden in 1,25 ml absol. Pyridin 1 Stunde bei Raumtemperatur gerührt, das Lösungsmittel abgedampft, der Rückstand in 5 ml Dichlormethan aufgenommen und 5 ml einer methanolischen Schwefelwasserstofflösung (bei 0°C gesätt.) sowie 0,7 ml einer gesätt. (20°C) Lösung von HCI in Methanol 1 Minute bei Raumtemperatur geschüttelt, das Reaktionsgemisch eingedampft und der Rückstand mit 20 ml Wasser digeriert. Das Digerat wird filtriert, das Filtrat zweimal mit Ether ausgeschüttelt, die wäßrige Phase über 10 ml basischem Ionenaustauscher (OHe-Form) filtriert und eingedampft. Der farblose Eindampfrückstand besteht aus dem reinen 2'-Acetylsisomicin.
- 4,5 g Sisomicin in 10 ml Methanol und 90 ml Dichlormethan werden mit 13 g o-Nitrophenylsulfensäure-p-nitrophenylester versetzt, das Reaktionsgemisch nach 1 Stunde auf 300 ml Methanol gegossen, der Niederschlag mit 50 ml Dichlormethan digeriert und das unlösliche Produkt (3,6 g) getrocknet. Filtrat und Digerat werden vereinigt und an einer 6 x 15 cm SiO2-Säule chromatographiert (Laufmittel: Dichlormethan mit steigendem Methanol-Zusatz, zuletzt 10% Methanol).
- Nach Abdampfen des Lösungsmittels werden aus dem Eluat 3,4g 1,2',3,6'-Tetra-NPS-sisomicin (Gesamtausbeute 7,0 g = 66%) und 2,5 g (21%) Penta-NPS-sisomicin isoliert.
- 13-C-NMR (δ-6-DMSO):
- 63,87 (C-3"), 146,07,147,28,145,67,145,11 (jeweils C-1 der arom. Ringe) ppm.
- 13,84 g (20 mMol) Sisomicinsulfat in 100 ml 1 n-NaOH und 450 ml frisch destilliertem Dioxan werden mit 38 g (0,20 Mol) o-Nitrophenylsulfenylchlorid in 200 ml Dioxan und mit 260 ml 1 n-NaOH versetzt, so daß der pH zwischen 12 und 14 liegt. Der Niederschlag wird abfiltriert, in CH2CI2/H20 gelöst, und die CH2Cl2-Phase mit Na2S04 getrocknet.
- Das Filtrat wird mit CH2Cl2 versetzt, die wäßrige Phase verworfen und die organische Phase über Na2S04 getrocknet. Die vereinigten organischen Phasen werden zur Trockne eingedampft und über 250 g Kieselgel (Säulendurchmesser 8 cm) zuerst mit CH2C12, dann mit CH2CI2/MeOH = 97,5/2,5 filtriert. Das Eluat liefert nach dem Abdampfen des Lösungsmittels 22 g (91%) Penta-N-(o-nitrophenylsulfenyl)-sisomicin als orangefarbenen Schaum.
- 13-C-NMR (CDCl3):
- δ=124-148 (arom. H); 102,30 (C-1"); 99,00 (C-1'); 97,92 (C-4'); 89,05 (C-6); 82,33 (C-4); 57,31 (C-1); 56,73 (C-3) ppm.
- 16,0 g (13,2 mMol) Penta-N-NPS-sisomicin in 80 ml absol. Pyridin werden mit 160 ml Thiophenol versetzt, nach 1 Stunde auf 500 ml Diethylether gegossen, der Niederschlag in Dichlormethan/Methanol = 8/2 aufgenommen und über Kieselgel filtriert (Säule 5,5 x 12 cm, Laufmittel Dichlormethan/Methanol = 8/2, steigender Zusatz des Laufmittelgemisches Methanol/Dichlormethan/20proz. Ammoniak =4/2/1); die rote Zone liefert nach dem Abdampfen des Lösungsmittels6,6 g(83%)3"-N-o-Nitrophenylsulfenyl-sisomicin als tiefroten Schaum.
- 13-C-NMR (CD30D):
- 33,59 (CH3N); 52,23 (C-1); 51,16 (C-3); 53 (C-2'); 43,84 (C-6') ppm.
- 3,0 g (5,0 Mol) 3"-N-NPS-sisomicin in 5 ml Methanol und 45 Mol Dichlormethan werden mit 4,4 g (15,0 Mol) o-Nitrophenylsulfensäure-p-nitrophenylester in 85 ml Dichlormethan versetzt, das Reaktionsgemisch sofort zur Trockne eingedampft, in Dichlormethan aufgenommen und an Kieselgel (Säule 5,5 x 30 cm) mit 200 ml Dichlormethan, dann mit Dichlormethan/Methanol = chromatographiert. Es werden 500 Fraktionen aufgefangen, wobei aus den vereinigten Fraktionen 150 bis 250 das 1,2',3",6'-Tetra-NPS-sisomicin und aus den Fraktionen 270 bis 500 das 2',3,3",6'-Tetra-NPS-Derivat jeweils als orangefarbener Schaum erhalten wird.
- 1,2',3",6'-Tetra-NPS-sisomicin:
- RF (CH2Cl2/CH3OH=9/1):0,62
- IR (KBr): 1501, 1360, 1300 (stark); 1587,1562,755 (mittel); 1442, 780, 890 (schwach)
- 2',3,3",6'-Tetra-NPS-sisomicin:
- RF(CH2Cl2/CH3OH=9/1):0,42
- IR (KBr): 1500,1358,1296 (stark); 1586,1560,753 (mittel), 1442,890,779 (schwach)
- 9 g Sisomicin in 20 ml Methanol/180 ml Dichlormethan werden mit 45 g o-Nitrophenylsulfensäure-p-nitrophenylester in 500 ml Dichlormethan versetzt, 10 Stunden bei Raumtemperatur gerührt, auf ein Volumen von 100 ml eingedampft und mit Cyclohexan im Überschuß versetzt. Der Niederschlag (37 g) wird abfiltriert, in 150 ml absol. Pyridin gelöst, das Gemisch mit 200 ml Thiophenol versetzt und nach 1 Stunde auf 500 ml Diethylether gegossen. Der Niederschlag wird in Dichlormethan/Methanol = 8/2 aufgenommen und über Kieselgel filtriert (Säule 5,5 x 12 cm, Laufmittel Dichlormethan/Methanol = 8/2, steigender Zusatz des Laufmittelgemisches Methanol/Dichlormethan/20proz. Ammoniak = 4/2/1). Die rote Zone wird eingedampft; 1800 mg des entstandenen Schaums werden in 3 ml Methanol und 27 ml Dichlormethan gelöst, mit 2,7 g o-Nitrophenylsulfensäure-p-nitrophenylester und 100 ml Methanol versetzt, eingedampft und der Rückstand an 100 g Kieselgel mit Dichlormethan/Methanol = 98/2 chromatographiert. Die gelbe Zone liefert nach dem Eindampfen 1,5 g 1,3,3",6'-Tetra-NPS-sisomicin.
- RF (CH2Cl2/CH3OH =9/1): 0,37
- 13-C-NMR(d-6-Dioxan): 124-147 (arom. C); 146,8 (C-5'); 103,85 (C-1"); 102,40 (C-1'); 88,89 (C-6); 76,48 (C-5) ppm.
- 265 g (0,25 mMol) 2',3,3",6'-Tetra-NPS-sisomicin (Beispiel 8c) und 58 mg Methoxyessigsäure-p-nitrophenylester werden in 1,25 ml absol. Pyridin 1 Stunde bei Raumtemperatur gerührt, das Lösungsmittel abgedampft, der Rückstand in 5 ml Dichlormethan aufgenommen und mit 5 ml einer methanolischen Schwefelwasserstofflösung (bei 0°C gesätt.) sowie 0,7 ml einer gesätt. (20° C) Lösung von HCI in Methanol 1 Minute bei Raumtemperatur geschüttelt, das Reaktionsgemisch mit einigen Tropfen wäßriger Ammoniaklösung neutralisiert, eingedampft und der Rückstand mit 20 ml Wasser digeriert. Das Digerat wird filtriert, das Filtrat zweimal mit Ether ausgeschüttelt, die wäßrige Phase über 10 ml basischem lonenaustauscher (OHe-Form) filtriert und eingedampft. Der farblose Eindampfrückstand besteht aus dem reinen 1-N-Methoxyacetyl-sisomicin.
- RF(CHCl3/CH3OH/20% NH4OH =2/4/1):0,53
- IR (KBr): 1650, 1105, 1048, 1000 (stark)
- 265 g (0,25 mMol) 2',3,3",6'-Tetra-NPS-sisomicin (Beispiel 8c) werden mit 47 mg Essigsäure-p-nitrophenylester wie für das 1-N-Methoxyacetylsisomicin beschrieben, umgesetzt. Nach Aufarbeitung des Reaktionsansatzes wird ganz analog das reine 1-N-Acetylsisomicin durch Eindampfen des wäßrigen lonenaustauscher-Eluats erhalten.
- 13-C-NMR (CD30D):
- 23,03 (CH3CO); 173,94 (C=0); 51,14 (C-3); 48,41 (C-2'); 150,62 (C-5); 44,21 (C-6'); 65,89 (C-3").
- 265 mg (0,25 mMol) 1,3,3",6'-Tetra-NPS-sisomicin (Beispiel 9) werden mit 47 mg Essigsäure-p-nitrophenylester wie für das 1-N-Acetyl-sisomicin beschrieben umgesetzt. Nach identischer Aufarbeitung des Ansatzes wird das reine 3-N-Acetyl-sisomicin als einziges Produkt isoliert.
- 13-C-NMR (CD30D):
- 23,22 (CH3CO); 173,13 (CO); 35,33 (C-2); 82,23 (C-4); 48,50 (C-2'); 148,42 (C-5'); 43,73 (C-6'); 66,01 (C-3").
- 265 mg (0,25 mMol) 1,2',3,6'-Tetra-NPS-sisomicin (Beispiel 8c) werden mit 47 mg Essigsäure-paranitrophenylester wie für das 1-N-Acetyl-sisomicin beschrieben umgesetzt. Nach identischer Aufarbeitung des Ansatzes wird das reine 3"-N-Acetylsisomicin als einziges Produkt isoliert.
- 13-C-NMR-(D20/d-6-Dioxan):
- Rotamerengemisch:
- 22,37/22,18 (CH3CO); 177,13/176,76 (CO); 65,67/62,06 (C-3"); 21,59/21,36 (C(OH)-CH3); 32,97/30,35 (CH3N).
- 265 mg des nach Beispiel 8c) erhaltenen 2',3,3",6'-Tetra-N-(o-nitro-phenylsulfenyl)-sisomicins und 44 g Ameisensäure-p-nitrophenylester werden in 1 ml Pyridin gelöst. Man läßt 1 Stunde bei Raumtemperatur stehen, dampft den Ansatz im Vakuum ein und nimmt in 2 ml Dichlormethan auf. Die Lösung wird mit 7,5 ml einer methanolischen, bei 0°C gesättigten H2S-Lösung sowie 0,75 ml einer bei 20°C gesättigten, methanolischen Chlorwasserstofflösung versetzt und nach 1 Minute mit konz. Ammoniaklösung neutralisiert. Man dampft den Ansatz im Vakuum ein, digeriert den Rückstand mit 10 ml Wasser und filtriert. Das Filtrat wird zweimal mit Ether gewaschen, die wäßrige Phase über 12 ml basischen lonenaustauscher in der OHe-Form filtriert und eingedampft.
- Ausbeute: 102 mg 1-N-Formylsisomicin
- IR (KBr-Preßling):

mit der Maßgabe, daß die Aminogruppe in 6'-Stellung eine der Schutzgruppen -SR' oder -CO-A aufweist.
in einem inerten Lösungsmittel, gegebenenfalls unter Zusatz von Wasser, bei Temperaturen zwischen etwa -30°C und +50°C, vorzugsweise zwischen etwa 0°C und +25°C, in Gegenwart einer Base umsetzt und das Reaktionsprodukt in üblicher Weise aufarbeitet.
Claims (8)

dadurch gekennzeichnet, daß die Aminogruppe in 6'-Stellung eine der Schutzgruppen -SR' oder ―CO―A aufweist.

Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE2726197 | 1977-06-10 | ||
| DE19772726208 DE2726208A1 (de) | 1977-06-10 | 1977-06-10 | 1-n-formylsisomicin, verfahren zu seiner herstellung sowie seine verwendung als arzneimittel |
| DE19772726197 DE2726197A1 (de) | 1977-06-10 | 1977-06-10 | Selektiv geschuetzte 4,6-di-o-(aminoglykosyl)-1,3-diaminocyclitole |
| DE2726208 | 1977-06-10 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0000057A1 EP0000057A1 (de) | 1978-12-20 |
| EP0000057B1 true EP0000057B1 (de) | 1982-07-14 |
Family
ID=25772130
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP78100099A Expired EP0000057B1 (de) | 1977-06-10 | 1978-06-06 | Selektiv geschützte 4,6-Di-O-(aminoglycosyl)-1,3-diaminocyclitole |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US4190722A (de) |
| EP (1) | EP0000057B1 (de) |
| JP (1) | JPS545946A (de) |
| CA (1) | CA1092101A (de) |
| DE (1) | DE2861947D1 (de) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2840907A1 (de) * | 1978-09-20 | 1980-04-03 | Bayer Ag | Selektiv geschuetzte 4,6-di-o-(aminoglykosyl)-1,3-diaminocyclitole |
| DE2924659A1 (de) * | 1979-06-19 | 1981-01-22 | Bayer Ag | Pseudotrisaccharide, ihre herstellung und verwendung als arzneimittel |
| DE3000841A1 (de) * | 1980-01-11 | 1981-07-16 | Bayer Ag, 5090 Leverkusen | Isolierung und reinigung von aminoglycosid-antibiotiaka |
| CN101868472B (zh) | 2007-11-21 | 2013-05-29 | 尔察祯有限公司 | 抗菌性氨基糖苷类似物 |
| WO2010132765A2 (en) | 2009-05-15 | 2010-11-18 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| WO2010132768A1 (en) | 2009-05-15 | 2010-11-18 | Achaogen, Inc. | Antibacterial derivatives of sisomicin |
| WO2010132760A1 (en) | 2009-05-15 | 2010-11-18 | Achaogen, Inc. | Antibacterial derivatives of tobramycin |
| WO2010132759A1 (en) | 2009-05-15 | 2010-11-18 | Achaogen, Inc. | Antibacterial derivatives of dibekacin |
| WO2010132757A2 (en) | 2009-05-15 | 2010-11-18 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2512587A1 (de) * | 1974-03-22 | 1975-09-25 | Microbial Chem Res Found | Kanamycinderivate, verfahren zu ihrer herstellung und sie enthaltende pharmazeutische mittel |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3929762A (en) * | 1972-08-23 | 1975-12-30 | Microbial Chem Res Found | 3{40 -Deoxy derivatives of neamine and its related aminoglycosidic antibiotics, and the production thereof |
| JPS5644076B2 (de) * | 1973-07-12 | 1981-10-16 | ||
| US4029882A (en) * | 1974-03-19 | 1977-06-14 | Schering Corporation | Selective acylation of the C-1 amino group of aminoglycoside antibiotics |
| CH606076A5 (de) * | 1973-08-06 | 1978-10-13 | Scherico Ltd | |
| JPS573679B2 (de) * | 1974-03-07 | 1982-01-22 | ||
| JPS5116642A (en) * | 1974-08-01 | 1976-02-10 | Meiji Seika Co | 3** deokishineamin oyobi sonokanrenkoseibutsushitsuno seizoho |
| US4048430A (en) * | 1974-10-29 | 1977-09-13 | Smithkline Corporation | Mercaptopseudotrisaccharides |
| US3997524A (en) * | 1975-05-02 | 1976-12-14 | Schering Corporation | Process for the manufacture of 6'-N-alkyl derivatives of sisomicin and verdamicin; novel intermediates useful therein, and novel 6'-N-alkylverdamicins prepared thereby |
-
1978
- 1978-06-06 EP EP78100099A patent/EP0000057B1/de not_active Expired
- 1978-06-06 DE DE7878100099T patent/DE2861947D1/de not_active Expired
- 1978-06-06 US US05/913,135 patent/US4190722A/en not_active Expired - Lifetime
- 1978-06-08 CA CA304,991A patent/CA1092101A/en not_active Expired
- 1978-06-08 JP JP6840178A patent/JPS545946A/ja active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2512587A1 (de) * | 1974-03-22 | 1975-09-25 | Microbial Chem Res Found | Kanamycinderivate, verfahren zu ihrer herstellung und sie enthaltende pharmazeutische mittel |
Also Published As
| Publication number | Publication date |
|---|---|
| CA1092101A (en) | 1980-12-23 |
| EP0000057A1 (de) | 1978-12-20 |
| JPS545946A (en) | 1979-01-17 |
| DE2861947D1 (en) | 1982-09-02 |
| US4190722A (en) | 1980-02-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0030295B1 (de) | Verfahren zur Herstellung von 4'-Epidaunorubicin, 3',4'-Diepidaunorubicin, hierbei entstehende Zwischenprodukte sowie deren Verwendung | |
| DE2618822B2 (de) | Anthracyclin-Glycoside und Verfahren zu deren Herstellung | |
| EP0048967B1 (de) | Verfahren zur Herstellung von 4'-Deoxydaunorubicin und 4'-Deoxydoxorubicin, sowie 4'-Epi-4'-trifluormethyl-sulfonyloxy-N-trifluoracetyldaunorubicin | |
| EP0032591B1 (de) | Verfahren zur Herstellung von gereinigten Aminoglycosid-Antibiotika | |
| DE2750812C2 (de) | 4-Alkoxy-4-desmethoxydaunomycine und Verfahren zu deren Herstellung | |
| DE69602834T2 (de) | Verfahren zur herstellung von anthracyclin antibiotica | |
| EP0000057B1 (de) | Selektiv geschützte 4,6-Di-O-(aminoglycosyl)-1,3-diaminocyclitole | |
| DE2708008C3 (de) | Verfahren zur Herstellung von 1-N-substituierten Kanamycinen, zur 1-N-Substitution eingesetzte cyclische Urethane und Verfahren zu deren Herstellung | |
| EP0021215B1 (de) | Pseudotrisaccharide, ihre Herstellung und Verwendung als Arzneimittel | |
| DE3026045A1 (de) | Verfahren zur herstellung von estern der polyen-makroliden und ihrer n-substituierten derivate | |
| DE3044970C2 (de) | ||
| DE2735455C3 (de) | Daunomycinanaloga, Verfahren zu deren Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| EP0286926B1 (de) | Semisynthetische Rhodomycine, Verfahren zu ihrer Herstellung und ihre Verwendung als Zytostatika | |
| DE3719377C2 (de) | Anthracyclinglykoside, Verfahren zu deren Herstellung und Arzneimittel, welche diese enthalten | |
| DE3028339A1 (de) | Neue zwischenprodukte fuer die herstellung von spectinomycin und seiner analoger sowie verfahren zur herstellung der betreffenden zwischenprodukte | |
| DE2366288B2 (de) | Verfahren zur Herstellung von 3' -Desoxykanamycin B und 3' -Desoxyneamin | |
| DE3308196A1 (de) | Verfahren zur herstellung von 6'-alkyl-spectinomycin sowie alkyl-spectinomycin-analogen | |
| DE69116423T2 (de) | 3'-Deamino-4'-deoxy-4'-amino-8-fluoranthracycline und Verfahren zu ihrer Herstellung | |
| DE2756057A1 (de) | Verfahren zur herstellung von 3',4'-dideoxykanamycin b | |
| DE69907101T2 (de) | Neue Hydroxyderivate von Tylosin und Verfahren zu deren Herstellung | |
| DE3004178C2 (de) | ||
| EP0009201B1 (de) | Selektiv geschützte 4,6-Di-O-(Aminoglykosyl)-1,3-diamino-cyclitole, Verfahren zu deren Herstellung und deren Verwendung als Zwischenprodukte | |
| DE69124700T4 (de) | 4-0-(aminoglycosyl)-oder 4,6-di-0-(aminoglycosyl)-2,5-dideoxy-5,5-difluorostreptaminderivat und seine herstellung | |
| CH661513A5 (de) | 14-de-(hydroxymethyl)-mycaminosyltylonolid-verbindungen. | |
| DE2726197A1 (de) | Selektiv geschuetzte 4,6-di-o-(aminoglykosyl)-1,3-diaminocyclitole |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): CH DE FR GB |
|
| 17P | Request for examination filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): CH DE FR GB |
|
| REF | Corresponds to: |
Ref document number: 2861947 Country of ref document: DE Date of ref document: 19820902 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19840526 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19840601 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19840924 Year of fee payment: 7 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19860630 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19870227 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19880301 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19881117 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |