-
Die
vorliegende Erfindung betrifft neuartige Indolylpiperidinverbindungen
und pharmakologisch annehmbare Salze derselben, die antihistaminische
und antiallergische Aktivität
besitzen und als Arzneimittel für die
Behandlung von Bronchialasthma, allergischer Rhinitis, Konjunktivitis,
Dermatose, Urticaria und dergleichen nützlich sind.
-
Die
vorliegende Erfindung betrifft auch ein Verfahren zur Herstellung
der Indolylpiperidinverbindungen und zur Behandlung von allergischen
Erkrankungen und Bronchialasthma nützliche pharmazeutische Zusammensetzungen,
die eine wirksame Menge der Indolylpiperidinverbindung umfassen.
-
Mehrere
antihistaminische und antiallergische Mittel, die den Indolylpiperidinkern
enthalten, sind bekannt. Beispiele für Indolylpiperidinverbindungen,
dargestellt durch die folgende Formel, in der R = H, OH, OR' und n = 2–6, sind
in Arch. Pharm. 1996, 329 (1), 3–10 beschrieben.
-
-
Weiterhin
offenbart
EP 224919 ,
als zur Behandlung von allergischen Erkrankungen nützliche
Verbindungen, Beispiele, die durch die folgende Formel dargestellt
sind:
(in der R
1 =
fakultativ substituiertes Amino; R
2 = H,
Niederalkyl oder Aryl; R
3 = H, NO
2, fakultativ substituiertes Amino, OH oder
Niederalkoxy; A = Niederalkylen; Q = H oder Halogen).
-
Die
meisten dieser Verbindungen sind als antiallergische Mittel charakterisiert,
die zur Behandlung von allergischem Asthma, Rhinitis, Konjunktivitis
und Urticaria nützlich
sind.
-
Gegenwärtige Antihistaminika
können
unter einem Sicherheitsgesichtspunkt nicht als vollständig befriedigend
angesehen werden, und es verbleiben Probleme im Hinblick auf Nebenwirkungen,
wie Schläfrigkeit, Sedierung,
Hydrodipsie, Mydriasis, Palpitation und Arrhythmie, vermittelt durch
deren unerwünschtes
Eindringen in das zentrale Nervensystem, antiacetylcholinerge Aktivität, Aktivität gegen
das kardiovaskuläre
System und dergleichen. Folglich besteht das klinische Bedürfnis nach
Antihistaminika und antiallergischen Mitteln, die weitgehend frei
von sedativen und kardiovaskulären
Nebenwirkungen sind.
-
Die
vorliegende Erfindung stellt neuartige Indolylpiperidinverbindungen
mit verbesserter antihistaminischer und antiallergischer Aktivität bereit.
-
Die
vorliegende Erfindung stellt auch neuartige Indolylpiperidinverbindungen
bereit, die aufgrund ihres Mangels an lipophilen Eigenschaften fast
vollständig
unfähig
sind, in das Gehirn einzudringen, und denen daher sedative Sekundärwirkungen
fehlen. Man kann auch verstehen, daß die Verbindungen der vorliegenden Erfindung
verringerte kardiovaskuläre
Nebenwirkungen besitzen.
-
Eine
weitere Aufgabe der vorliegenden Erfindung ist, ein Verfahren zur
Herstellung besagter Verbindungen bereitzustellen.
-
Noch
eine weitere Aufgabe ist, eine pharmazeutische Zusammensetzung bereitzustellen,
die eine wirksame Menge besagter Verbindungen umfaßt.
-
Gemäß der vorliegenden
Erfindung werden Indolylpiperidinverbindungen bereitgestellt, die
dargestellt sind durch die Formel (I):
worin:
A
1 eine
Alkylen-, Alkylenoxy-, Alkylenthio-, Alkanoylen- oder Hydroxyalkylengruppe
darstellt;
A
2 eine Alkylen-, Alkylenoxy-,
Alkylenthio-, Alkanoylen- oder eine Alkylenoxyalkylengruppe darstellt;
W
1 eine Phenylen-, Furanylen- oder Pyridinylengruppe
darstellt, die unsubstituiert ist oder durch ein oder mehrere Halogenatome,
eine oder mehrere Alkoxygruppen und/oder Alkylgruppen substituiert
ist;
W
2 eine 3- bis 10-gliedrige monocyclische
oder bicyclische Gruppe darstellt, die 1 bis 3 Heteroatome enthält, wobei
die genannte Gruppe unsubstituiert ist oder durch ein oder mehrere
Halogenatome, eine oder mehrere Alkylgruppen, Alkoxygruppen und/oder
Oxogruppen substituiert ist;
R
1 ein
Wasserstoff- oder Halogenatom oder eine Alkyl-, Alkoxy- oder Methylaminogruppe
darstellt; und
R
2 eine Carboxylgruppe
darstellt;
und pharmazeutisch annehmbare Salze davon.
-
In
der obigen Formel (I) können
die Alkyl-, Alkylen-, Alkylenoxy-, Alkylenthio-, Alkanoylen-, Hydroxyalkylen-
und Alkoxygruppen, die in bezug auf die Gruppen A1,
A2, W1, W2 und R1 in den Verbindungen
der Erfindung erwähnt
sind, verzweigt oder gerade sein und vorzugsweise bis zu 7 und insbesondere
bis zu 5 Kohlenstoffatome enthalten.
-
In
der obigen Formel (I) kann die 3- bis 10-gliedrige monocyclische
oder bicyclische Gruppe, die von 1 bis 3 Heteroatome enthält, erwähnt im Zusammenhang
mit der Gruppe W2, gesättigt oder ungesättigt sein, einschließlich aromatisch.
Man wird verstehen, daß in
den monocyclischen oder bicyclischen Gruppen, die im Zusammenhang
mit der Gruppe W2 erwähnt sind, die 1, 2 oder 3 Heteroatome
innerhalb der cyclischen Struktur enthalten sind. In bevorzugten
Gruppen W2 sind die 1, 2 oder 3 Heteroatome
ausgewählt
aus der Gruppe, die aus Sauerstoff, Schwefel und Stickstoff besteht.
In den bevorzugteren Gruppen W2 besitzt
die monocyclische oder bicyclische Gruppe von 5 bis 9 Glieder, insbesondere
ist die monocyclische oder bicyclische Gruppe eine monocyclische
Gruppe mit 5 oder 6 Gliedern oder eine bicyclische Gruppe mit 9
Gliedern.
-
In
der obigen Formel (I) bedeutet der Ausdruck „ein oder mehrere", der die Anzahl
fakultativer Substituenten definiert, die in den Gruppen W1 und W2 vorhanden
sind, von einem bis zu der Anzahl substituierbarer Positionen auf
der chemischen Einheit, die substituiert werden soll. Vorzugsweise
haben die Gruppen, in Verbindungen der Erfindung, in denen die W1- und/oder W2-Gruppen
Substituenten enthalten, von 1–3
Substituenten. Man sollte verstehen, daß, in den Verbindungen der
Erfindung, die Substituenten, die im Zusammenhang mit den Gruppen
W1 und W2 erwähnt sind,
sich an jeder substituierbaren Position oder Kombination von substituierbaren
Positionen auf der chemischen Einheit, die substituiert werden soll,
befinden können.
Man sollte verstehen, daß die
Phenylen-, Furanylen- oder Pyridinylengruppe W1 durch
A1 und R2 an jeder
Kombination substituierbarer Ringpositionen relativ zueinander substituiert
sein kann, zum Beispiel 1,2; 1,3; oder 1,4. In Verbindungen der
Erfindung, in denen die Phenylen-, Furanylen- oder Pyrilidengruppe
W1 weiter substituiert ist, können die
weiteren Substituenten an jede der restlichen Positionen gebunden
sein.
-
Man
sollte verstehen, daß,
in der obigen Formel (I), der Substituent R1 an
die 4-, 5-, 6- oder 7-Position des
Indolylkerns gebunden sein kann. In bevorzugten Verbindungen der
Erfindung ist R1 an die 5- oder 6-Position
des Indolylkerns gebunden.
-
Weitere
Merkmale und Vorteile der vorliegenden Erfindung werden aus der
Beschreibung der bevorzugten Verbindungen deutlich werden, die folgt,
wenn sie im Lichte der beigefügten
Beispiele gelesen wird.
-
In
bevorzugten Verbindungen der Erfindung steht A1 für eine Alkylen-
oder eine Alkylenoxygruppe, bevorzugter eine C1-3-Alkylen-,
wie etwa eine Methylen-, Ethylen- oder Propylengruppe, oder eine
C1-5-Alkylenoxygruppe, wie etwa eine Methylenoxy-,
Ethylenoxy-, Propylenoxy-, Butylenoxy- oder Pentylenoxygruppe.
-
In
bevorzugten Verbindungen der Erfindung steht A2 für eine C1-5-Alkylen-, C1-5-Alkanoylen-,
C2-5-Alkylenoxy-, C2-5-Alkylenthio-
oder C2-5-Alkylenoxy-C1-5-alkylengruppe.
In bevorzugteren Verbindungen der Erfindung steht A2 für eine Methylen-,
Ethylen-, Propylen-, Butylen-, Ethanoylen-, Propanoylen-, Butanoylen-,
Ethylenoxy-, Propylenoxy-, Butylenoxy-, Ethylenthio-, Propylenthio-,
Butylenthio-, Ethylenoxyethylen- oder Ethylenoxymethylengruppe.
-
In
bevorzugten Verbindungen der Erfindung steht W1 für eine Phenylen-,
Furanylen- oder Pyridinylengruppe, die unsubstituiert oder mit einem
oder mehreren, vorzugsweise einem oder zwei, Substituenten substituiert
ist, ausgewählt
aus Fluor-, Chlor- oder Brom-Atomen und Methylen- und Methoxygruppen.
Bevorzugter steht W1 für eine unsubstituierte Phenylen-,
Furanylen- oder Pyridinylengruppe oder eine Phenylengruppe, die
mit einem Fluor-Atom,
Brom-Atom oder einer Methoxygruppe substituiert ist. Am bevorzugtesten
steht W1 für eine unsubstituierte Phenylen-
oder eine Phenylengruppe, die mit einer Methoxygruppe substituiert
ist.
-
In
den bevorzugten Verbindungen der Erfindung ist die fakultativ substituierte
3- bis 10-gliedrige
monocyclische oder bicyclische Gruppe, die von 1 bis 3 Heteroatome
enthält,
spezifiziert in der Definition als W2, eine
Dioxolanyl-, Dioxanyl-, Pyrazolidinyl-, Isoindolinyl-, Benzodioxolanyl-,
Tetrahydropyranyl-, Tetrahydrofuranyl-, Oxetanyl-, Furanyl-, Thienyl-,
Pyrrolyl-, Pyridinyl-, Imidazolyl-, Dihydrothiazolyl-, Benzothiazolyl-,
Pyrrolidinyl-, Benzooxazolyl-, Benzothienyl-, Pyranyl-, Benzofuranyl-,
Isobenzylfuranyl-, Chromenyl-, Pyrazolyl-, Oxazolyl-, Isooxazolyl-,
Furazanyl-, Isochromanyl-, Pyrrolinyl-, Imidazolidinyl-, Imidazolinyl-,
Pyrazolinyl-, Piperidyl-, Piperazinyl-, Indolinyl-, Morpholinyl-,
Pyrazinyl-, Pyrimidinyl-, Pyridazinyl-, Indolizinyl-, Isoindolyl-,
Indolyl-, Indazolyl-, Chinazolinyl-, Isochinazolinyl-, Chinolyl-,
Phthalazinyl-, Naphthyridinyl-, Chinoxalinyl-, Chinazolinyl- oder
Cinnolinylgruppe. Bevorzugter ist die fakultativ substituierte 3-
bis 10-gliedrige monocyclische oder bicyclische Gruppe, die von
1 bis 3 Heteroatome enthält,
eine Dioxolanyl-, Dioxanyl-, Pyrazolidinyl-, Isoindolinyl-, Benzodioxolanyl-,
Tetrahydropyranol-, Tetrahydrofuranyl-, Oxetanyl-, Furanyl-, Thienyl-,
Pyrrolyl-, Pyridinyl-, Imidazolyl-, Dihydrothiazolyl-, Benzothiazolyl-,
Pyrrolidinyl- oder eine Benzooxazolylgruppe. Bevorzugter ist die
fakultativ substituierte 3- bis 10-gliedrige monocyclische oder
bicyclische Gruppe, die von 1 bis 3 Heteroatome enthält, eine
Dioxolanyl-, Dioxanyl-, Pyrazolidinyl-, Benzodioxolanyl-, Tetrahydropyranyl-,
Tetrahydrofuranyl-, Oxetanyl-, Furanyl-, Thienyl-, Pyrrolyl-, Pyridinyl-,
Pyrrolidinyl- oder eine Benzooxazolylgruppe.
-
In
Verbindungen der Erfindung, in denen die 3- bis 10-gliedrige monocyclische
oder bicyclische Gruppe, die von 1 bis 3 Heteroatome enthält, spezifiziert
in der Definition als W2, substituiert ist,
sind der eine oder die mehreren Substituenten vorzugsweise unabhängig ausgewählt aus
Fluoratomen, Chloratomen, Bromatomen, C1-7-Alkylgruppen,
C1-7-Alkoxygruppen
und Oxogruppen. Am bevorzugtesten sind die Substituenten ausgewählt aus
Chloratomen, C1-4-Alkylgruppen, Methoxygruppen
und Oxogruppen.
-
In
besonders bevorzugten Verbindungen der Erfindung ist die fakultativ
substituierte 3- bis 10-gliedrige monocyclische oder bicyclische
Gruppe, die von 1 bis 3 Heteroatome enthält, spezifiziert in der Definition
als W2, ein 5-gliedriger Ring, der 1 oder
2 Heteroatome enthält,
und der Ring ist entweder unsubstituiert oder mit einer C1-7-Alkylgruppe oder einem Chloratom substituiert.
-
In
bevorzugten Verbindungen der Erfindung steht R1 für ein Wasserstoff-,
Fluor-, Chlor- oder Bromatom oder eine Methyl-, Methoxy- oder Methylaminogruppe.
Am bevorzugtesten steht R1 für ein Wasserstoff, ein
Floratom oder eine Methoxygruppe.
-
Bevorzugtere
Verbindungen von Formel (I) sind diejenigen, in denen A1 für eine Methylen-,
Ethylen- oder Ethylenoxygruppe steht; A2 für eine Methylen-,
Etyhlen-, Propylen-, Butylen-, Ethylenoxy-, Propylenoxy-, Ethylenoxyethylen-,
Ethylenoxymethylen-, Ethanoylen-, Butanoylen- oder eine Propylenthio-(Propylsulfanylen-)gruppe
steht; W1 für eine unsubstituierte Phenylen-,
Furanylen- oder Pyridinylengruppe oder eine Phenylengruppe, die
mit einem oder mehreren Fluor-, Brom- oder Methoxygruppen substituiert
ist, steht; W2 für eine (1,3)-Dioxolanyl-, (1,3)-Dioxanyl-,
2,5,5-Trimethyl-[1,3]-dioxan-2-yl, Isoindolyl-, 1,3-Dioxo-1,3-dihydroisoindolinyl-,
(1,3)-Benzodioxolanyl-, Tetrahydropyranyl-, Tetrahydropuranyl-,
Oxetanyl-, Furanyl-, Thienyl-, 5-Chlorthienyl-, Pyrrolyl-, Pyridinyl-,
Imidazolyl-, Methylimidazolyl-, Dihydrothiazolyl-, Benzothiazolyl-,
Pyrrolidinyl-, Pyrrolidinonyl-, Benzoxazolonyl-, Phthalimidoyl-,
Benzooxazolyl-, 2-Oxobenzooxazolyl- oder 5-Methyl-2-oxobenzooxazolylgruppe
steht; R1 für ein Wasserstoff, ein Fluoratom
oder eine Methoxygruppe steht, zum Beispiel ein Wasserstoffatom
oder ein Fluoratom; und R2 für eine Carboxylgruppe
steht.
-
Bevorzugtere
Verbindungen von Formel (I) sind diejenigen, in denen A1 für eine Methylen-,
Ethylen- oder Ethylenoxygruppe steht; A2 für eine Methylen-,
Ethylen-, Propylen-, Butylen- oder
eine Ethylenoxygruppe steht; W1 für eine unsubstituierte
Phenylen-, Furanylen- oder Pyridinylengruppe oder eine Phenylengruppe,
die mit einer oder mehreren Fluor-, Brom- oder Methoxygruppen substituiert
ist, steht; W2 für eine (1,3)-Dioxolanyl-, (1,3)-Dioxanyl-,
Tetrahydropyranyl-, Tetrahydropyranyl-, Oxetanyl-, Furanyl-, Thienyl-,
5-Chlorthienyl-, Pyrrolyl- oder eine Pyridinylgruppe steht; R1 für
ein Wasserstoff, ein Fluoratom oder eine Methoxygruppe steht; und
R2 für
eine Carboxylgruppe steht.
-
Die
pharmakologisch annehmbaren Salze der Verbindungen der vorliegenden
Erfindung, dargestellt durch Formel (I), können Säureadditionssalze oder Alkaliadditionssalze
sein. Beispiele für
die Säureadditionssalze
schließen
Mineralsäureadditionssalze,
wie etwa zum Beispiel Hydrochlorid, Hydrobromid, Hydroiodid, Sulfat,
Nitrat, Phosphat, und organische Säureadditionssalze, wie etwa
zum Beispiel Acetat, Maleat, Fumarat, Citrat, Oxalat, Succinat,
Tartrat, Malat, Mandelat, Methansulfonat und p-Toluolsulfonat, ein.
Beispiele für
die Alkaliadditionssalze schließen
anorganische Salze, wie etwa zum Beispiel Natrium-, Kalium-, Calcium-
und Ammoniumsalze, und organische Alkalisalze, wie etwa zum Beispiel
Ethylendiamin-, Ethanolamin-, N,N-Dialkylenethanolamin-, Triethanolamin-
und basische Aminosäuresalze,
ein.
-
Die
Verbindungen der vorliegenden Erfindung, dargestellt durch die oben
beschriebene Formel (I) können
in Abhängigkeit
von ihrer Asymmetrie Enantiomere oder Diastereoisomere einschließen. Die
einzelnen Isomere und Mischungen der Isomere fallen in den Schutzumfang
der vorliegenden Erfindung.
-
Die
bevorzugten Indolylpiperidinverbindungen der vorliegenden Erfindung
schließen
die folgenden Verbindungen ein:
- 1. 2-{2-[4-(1-[1,3]Dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 2. 2-(2-{4-[1-(Tetrahydro-pyran-2-ylmethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 3. 2-{2-[4-(1-Pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 4. 2-(2-{4-[1-(3-Pyrrol-1-yl-propyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 5. 2-(2-(4-[1-(3-Thiophen-2-yl-propyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 6. 2-[2-(4-(1-[3-(1-Methyl-1H-imidazol-2-ylsulfanyl)-propyl]-1H-indol-3-yl)-piperidin-1-yl)-ethoxy]-benzoesäure
- 7. 2-[2-(4-{1-[2-(2,5,5-Trimethyl-[1,3]dioxan-2-yl)-ethyl]-1H-indol-3-yl}-piperidin-1-yl)-ethoxy]-benzoesäure
- 8. 2-{2-[4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 9. 2-{2-(4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 10. 2-(2-{4-[1-(2-Oxo-2-pyrrolidin-1-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 11. 2-{2-[4-(1-Thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 12. 2-(2-{4-[1-(2-Thiophen-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 13. 2-(2-{4-[1-(2-Thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 14. 2-[2-(4-{1-[3-(Tetrahydro-furan-2-yl)-propyl]-1H-indol-3-yl}-piperidin-1-yl)-ethoxy]-benzoesäure
- 15. 2-(2-{4-[1-(4-[1,3]Dioxolan-2-yl-butyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 16. 2-[2-(4-{1-[3-(Benzo[1,3]dioxol-5-yloxy)propyl]-1H-indol-3-yl}piperidin-1-yl)ethoxy]-benzoesäure
- 17. 2-[2-{4-{1-[3-{1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-propyl]-1H-indol-3-yl}-piperidin-1-yl)-ethoxy]-benzoesäure
- 18. 2-{2-[4-(1-Benzo[1,3]dioxol-5-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 19. 2-(2-{4-[1-(5-Chlor-thiophen-2-ylmethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 20. 2-[2-(4-{1-[4-(5-Methyl-2-oxo-benzooxazol-3-yl)-butyl]-1H-indol-3-yl}-piperidin-1-yl)-ethoxy]-benzoesäure
- 21. 2-(2-{4-[1-(3-[1,3]Dioxolan-2-yl-propyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 22. 2-{2-[4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 23. 2-(2-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 24. 2-(2-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 25. 3-[4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 26. 3-[4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 27. 3-{4-[1-(5-Chlor-thiophen-2-ylmethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 28. 3-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 29. 3-{4-[1-(3-[1,3]Dioxolan-2-yl-propyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 30. 3-{4-(1-Pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 31. 3-(4-{1-[3-(Benzo[1,3]dioxol-5-yloxy)-propyl]-1H-indol-3-yl}-piperidin-1-ylmethyl)-benzoesäure
- 32. 3-[4-(1-(1,3]Dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 33. 3-[4-(1-Pyridin-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 34. 2-Methoxy-5-[4-(1-pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 35. 5-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxy-benzoesäure
- 36. 5-{4-(1-(3-[1,3]Dioxolan-2-yl-propyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxy-benzoesäure
- 37. 2-Methoxy-5-[4-(1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 38. 2-Methoxy-5-[4-(1-pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 39. 4-Brom-3-[4-(1-[1,3]dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 40. 4-Brom-3-[4-(1-pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 41. 4-Brom-3-{4-[1-(2-[1,3]dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 42. 4-Brom-3-{4-[1-(3-[1,3]dioxolan-2-yl-propyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 43. 4-Brom-3-[4-(1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 44. 2-{4-[5-Methoxy-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 45. 3-(4-{1-[3-(Benzo[1,3]dioxol-5-yloxy)-propyl]-1H-indol-3-yl}-piperidin-1-ylmethyl)-4-brombenzoesäure
- 46. 2-Fluor-5-[4-(1-pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 47. 5-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-fluor-benzoesäure
- 48. 5-{4-[1-(3-[1,3]Dioxolan-2-yl-propyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-fluor-benzoesäure
- 49. 2-Fluor-5-[4-(1-pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 50. 5-(4-{1-[3-(Benzo[1,3]dioxol-5-yloxy)-propyl]-1H-indol-3-yl}-piperidin-1-ylmethyl)-2-fluor-benzoesäure
- 51. 5-[4-(1-[1,3]Dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-fluor-benzoesäure
- 52. 2-Fluor-5-[4-(1-pyridin-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 53. 2-(2-{4-[1-(Tetrahydrofuran-3-ylmethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 54. 2-(2-{4-[1-(2-Morpholin-4-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 55. 2-(2-{4-[1-(3-Methyl-oxetan-3-ylmethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 56. 2-{2-[4-(1-Furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 57. 2-(2-{4-[1-(2-Pyridin-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 58. 3-{4-[1-(Tetrahydro-furan-3-ylmethyl)-1H-indol-3-yl}-piperidin-1-ylmethyl}-benzoesäure
- 59. 3-{4-[1-(3-Methyl-oxetan-3-ylmethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 60. 3-{4-[1-(2-Thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 61. 3-[4-(1-Furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 62. 2-[4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-nicotinsäure
- 63. 2-{4-[1-(2-Morpholin-4-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-nicotinsäure
- 64. 2-[4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-nicotinsäure
- 65. 3-{4-(1-(5-Chlor-thiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 66. 3-{4-[6-Fluor-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 67. 3-{4-[6-Fluor-1-(2-thiophen-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 68. 2-Methoxy-5-{4-[1-(2-thiophen-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 69. 5-{4-(6-Fluor-1-(2-thiophen-2-yl-ethyl)-1H-indol-3-yl}-piperidin-1-ylmethyl}-2-methoxybenzoesäure
- 70. 5-{4-(6-Fluor-1-(2-morpholin-4-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxy-benzoesäure
- 71. 5-{4-[1-(2-[1,4]Dioxan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxy-benzoesäure
- 72. 3-{4-[1-(2-(1,4]Dioxan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 73. 2-Methoxy-5-[4-(1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 74. 4-Brom-3-[4-(1-pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 75. 2-Methoxy-5-{4-[1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 76. 3-{4-[1-(2-Morpholin-4-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 77. 2-[2-(4-{1-[2-(Benzo(1,3]dioxol-5-yloxy)-ethyl]-1H-indol-3-yl}-piperidin-1-yl)-ethoxy]-benzoesäure
- 78. 5-{4-[6-Fluor-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxy-benzoesäure
- 79. 5-{4-[1-(5-Chlor-thiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxy-benzoesäure
- 80. 5-[4-(6-Fluor-1-furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxy-benzoesäure
- 81. 3-{4-[1-(2-Pyridin-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 82. 5-[4-(6-Fluor-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxy-benzoesäure
- 83. 3-[4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 84. 2-(2-{4-[1-(2-[1,4]Dioxan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 85. 5-[4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxy-benzoesäure
- 86. 5-[4-(1-Furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxy-benzoesäure
- 87. 3-{4-[5-Methoxy-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 88. 2-(2-{4-[5-Methoxy-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 89. 2-{2-[4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 90. 3-[4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 91. 2-Methoxy-5-{4-[5-methoxy-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 92. 2-{2-[4-(1-Furan-3-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 93. 3-[4-(1-Furan-3-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 94. 2-[4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 95. 2-[4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 96. 3-[4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 97. 5-[4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxy-benzoesäure
- 98. 4-Methoxy-2-[4-(5-methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 99. 2-[4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 100. 2-Methoxy-5-[4-(5-methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 101. 2-[4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-ylmethyl]-4-methoxy-benzoesäure
- 102. 3-[4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 103. 2-[4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 104. 5-(4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxy-benzoesäure
- 105. 2-{2-[4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 106. 4-Methoxy-2-{4-[5-methoxy-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 107. 2-{2-[4-(6-Fluor-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 108. 5-[4-(6-Fluor-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxy-benzoesäure
- 109. 2-{2-[4-(6-Fluor-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 110. 2-(2-{4-[6-Fluor-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 111. 2-(2-{4-[1-(5-Chlor-thiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 112. 2-{2-[4-(6-Fluor-1-furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 113. 2-{2-[4-(5-Methoxy-1-thiophen-3-ylmethyl-1H-indol-4-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
- 114. 3-[4-(5-Methoxy-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 115. 2-(2-{4-[1-(5-Chlor-thiophen-2-ylmethyl)-5-methoxy-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
- 116. 3-{4-[1-(5-Chlor-thiophen-2-ylmethyl)-5-methoxy-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 117. 2-Methoxy-5-[4-(1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 118. 3-[4-(1-Thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
- 119. 5-{4-[1-(5-Chlor-thiophen-2-ylmethyl)-5-methoxy-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxy-benzoesäure
- 120. 3-{4-[5-Methoxy-1-(2-thiophen-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
- 121. 2-Methoxy-5-{4-[5-methoxy-1-(2-thiophen-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
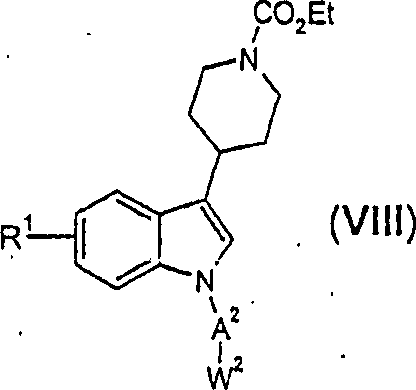
Gemäß einer
weiteren Ausführungsform
der vorliegenden Erfindung stellt sie ein Verfahren zur Herstellung
einer Verbindung, dargestellt durch Formel (I), bereit, welches
die Hydrolyse einer Verbindung von Formel (VI) umfaßt
worin A
1,
A
2, W
1, W
2 und R
1 sind, wie
oben definiert, und R
3 eine -COOR
4-Gruppe ist, worin R
4 für eine C
1-C
4-Alkylgruppe
steht.
-
Die
neuartigen Indolylpiperidinverbindungen der vorliegenden Erfindung,
dargestellt durch die Formel (I), können und werden vorzugsweise
gemäß Schema
1 hergestellt.
-
-
Das
Piperidinderivat der allgemeinen Struktur (II), worin R1 ist,
wie oben definiert, wird mit einem reaktiven Zwischenprodukt der
allgemeinen Formel (III) alkyliert: X-A1-W1-R3 (III), worin
A1 und W1 sind,
wie oben definiert, R3 eine -COOR4-Gruppe ist, worin R4 eine
C1-C4-Alkylgruppe ist,
und X eine Abgangsgruppe ist, wie etwa ein Chlor- oder Bromatom
oder eine Methansulfonat-, p-Toluolsulfonat- oder Benzolsulfonatgruppe.
-
Die
Reaktion wird vorzugsweise in einem inerten organischen Lösemittel,
wie etwa Toluol, Dioxan oder Methylisobutylketon, bei einer Temperatur
zwischen 80°C
und 140°C
und in Gegenwart einer anorganischen Base, wie etwa einem Alkalimetallcarbonat
oder -bicarbonat, durchgeführt.
-
In
der Reaktion wird das entsprechende Alkylierungsprodukt der allgemeinen
Formel (IV) gebildet:
-
-
Verbindung
(IV) wird am Indol-Stickstoff mit einem reaktiven Zwischenprodukt
der allgemeinen Formel (V) alkyliert: X-A2-W2 (V),worin X
eine Abgangsgruppe ist, wie etwa ein Chlor- oder Bromatom oder eine
Methansulfonat-, p-Toluolsulfonat- oder Benzolsulfonatgruppe, und
A2 und W2 sind,
wie oben definiert.
-
Die
Reaktion wird vorzugsweise in einem inerten organischen Lösemittel,
wie etwa Dimethylformamid, Tetrahydrofuran oder Ethylether, bei
einer Temperatur zwischen 0°C
und 80°C
in Gegenwart einer anorganischen Base, wie etwa Natriumhydrid oder
Natriumamid, durchgeführt.
In der Reaktion wird das entsprechende Alkylierungsprodukt der allgemeinen
Formel (VI) gebildet, worin R1, R3, W2, A1 und
A2 sind, wie oben definiert.
-
-
Üblicherweise
wird ein Überschuß der Reagentien
in beiden Alkylierungen eingesetzt, um vollständige Reaktion sicherzustellen.
In solchen Fällen
kann ein Polymer, wie etwa Methylisocyanatpolystyrol oder/und 3-(3-Mercaptophenyl)-propanamidomethylpolystyrol,
geeigneterweise zugesetzt werden, um mit dem überschüssigen Reagens zu reagieren.
Isolierung der Produkte aus Reaktionen, in denen ein Polymer-gebundenes Reagens
verwendet worden ist, ist in großem Maße vereinfacht, wobei nur Filtration
unter verringertem Druck erforderlich ist. Das Produkt aus diesen
Reaktionen kann durch Festphasenextraktion unter Verwendung eines geeigneten
Sorptionsmittels, wie etwa Varian SCX oder Varian C18, gereinigt
werden.
-
Wenn
man einem unterschiedlichen Weg folgt (siehe Schema 1), wird das
Piperidin von Verbindung (II) an seinem reaktiven Piperidin-Stickstoffatom
durch eine geeignete Schutzgruppe geschützt, wie etwa durch Bildung
einer Carbamateinheit (das Ethylcarbamat ist als Beispiel dargestellt),
um Verbindungen der allgemeinen Struktur (VII) zu ergeben, worin
R1 ist, wie oben definiert. Diese Reaktion
wird vorzugsweise in Methylenchlorid oder Chloroform als einem Lösemittel
in Gegenwart von Triethylamin und Ethylchlorformiat bei einer Temperatur
zwischen –20°C und 30°c durchgeführt.
-
-
Verbindung
(VII) wird am Indol mit einem reaktiven Zwischenprodukt der allgemeinen
Formel (V) alkyliert: X-A2-W2 (V),worin X
eine Abgangsgruppe ist, wie etwa ein Chlor- oder Bromatom oder eine
Methansulfonat-, p-Toluolsulfonat- oder Benzolsulfonatgruppe, und
A2 und W2 sind,
wie oben definiert.
-
Diese
Reaktion wird vorzugsweise in einem inerten organischen Lösemittel,
wie etwa Dimethylformamid, Tetrahydrofuran oder Ethylether, bei
einer Temperatur zwischen 0°C
und 80°C
in Gegenwart einer anorganischen Base, wie etwa Natriumhydrid oder
Natriumamid, durchgeführt.
In der Reaktion wird das entsprechende Alkylierungsprodukt der allgemeinen
Formel (VIII) gebildet, worin R1, W2 und A2 sind, wie
oben definiert.
-
-
Von
Verbindung (VIII) wird in der geeigneten Art und Weise für die im
vorhergehenden Schritt ausgewählte
Schutzgruppe diese abgespalten. Für die beispielhaft dargestellte
Carbamatgruppe kann dies durch Kochen in Gegenwart eines Überschusses
Natrium- oder Kaliumhydroxid in einem alkoholischen Lösemittel, wie
Ethanol, Isopropanol oder n-Butanol, bei einer Temperatur zwischen
80°C und
180°C erfolgen.
Weitere Neutralisation mit einer anorganischen Säure, wie etwa Salz- oder Schwefelsäure, führt zu einer
Verbindung der allgemeinen Struktur (IX), in der R1,
A2 und W2 sind,
wie oben definiert.
-
-
Weitere
Alkylierung von Verbindung (IX) mit einem reaktiven Zwischenprodukt
der allgemeinen Formel (III) ergibt eine Verbindung der allgemeinen
Struktur (VI) X-A1-W1-R3 (III),wobei
R1, A1, A2, W1 und W2 sind wie oben definiert, R3 eine
-COOR4-Gruppe ist, worin R4 eine
C1-C4-Alkylgruppe
ist, und X eine Abgangsgruppe ist, wie etwa ein Chlor- oder Bromatom
oder eine Methansulfonat-, p-Toluolsulfonat- oder Benzolsulfonatgruppe.
Die Reaktion wird vorzugsweise in einem inerten organischen Lösemittel,
wie etwa Toluol, Dioxan oder Methylisobutylketon, bei einer Temperatur
zwischen 80°C
und 140°C
in Gegenwart einer anorganischen Base, wie etwa einem Alkalimetallcarbonat
oder -bicarbonat, durchgeführt.
-
Verbindungen
der allgemeinen Formel (VI), worin R3 für einen
Alkylester steht, werden mit Natrium- oder Kaliumhydroxid behandelt
und weitere Neutralisation mit einer anorganischen Säure, wie
etwa Salz- oder Schwefelsäure,
liefert das entsprechende Indolderivat von Formel (I), worin R2 eine Carbonsäure ist. Die Reaktion wird
vorzugsweise in einem Lösemittel,
wie etwa Methanol, Ethanol, Tetrahydrofuran oder einer wäßrigen Mischung
eines der obengenannten Lösemittel,
bei dessen Siedepunkt durchgeführt.
-
Gelegentlich
werden die Verbindungen der vorliegenden Erfindung durch präparative
HPLC-MS gereinigt.
In diesen Fällen
wird eine Gilson-Termoquest-HPLC-MS mit präparativen C-18-Säulen
(5 μm, 1 × 5 cm, Waters)
und unter Verwendung von Wasser/Ameisensäure 0,1% als mobiler Phase
verwendet.
-
Die
Piperidinderviate von Formel (II) können aus dem 4-Piperidon hergestellt
werden, wie offenbart in der Literatur (J. Med. Chem. 1992, 35,
4813–4822).
Die reaktiven Zwischenprodukte der allgemeinen Formel (III) sind
entweder kommerziell erhältlich,
oder sie können
hergestellt werden, wie offenbart in der Literatur, oder ihre Herstellung
ist in der vorliegenden Erfindung eingeschlossen.
-
Ebenfalls
eingeschlossen im Schutzumfang der vorliegenden Erfindung sind pharmazeutische
Zusammensetzungen, die, als den aktiven Inhaltsstoff, wenigstens
ein Indolylpiperidinderivat der allgemeinen Formel (I) oder ein
pharmakologisch annehmbares Salz desselben, zusammen mit einem pharmazeutisch
annehmbaren Trägerstoff
oder Verdünnungsmittel
umfassen. Vorzugsweise wird die Zusammensetzung in einer für orale
oder parenterale Verabreichung geeigneten Form hergestellt.
-
Die
pharmazeutisch annehmbaren Trägerstoffe
oder Verdünnungsmittel,
die mit der aktiven Verbindung oder den aktiven Verbindungen, oder
Salzen derselben, vermischt werden, um die Zusammensetzung dieser
Erfindung zu bilden, sind „per
se" gut bekannt,
und die tatsächlich
verwendeten Hilfsstoffe hängen
unter anderem von der beabsichtigten Verabreichungsmethode der Zusammensetzungen
ab. Zusammensetzungen dieser Erfindung sind vorzugsweise für orale
Verabreichung angepaßt.
In diesem Fall kann die Zusammensetzung für orale Verabreichung die Form
von Tabletten, Kapseln oder Brausegranülen oder flüssigen Präparaten, wie etwa Elixieren,
Sirupen oder Suspensionen, annehmen, die alle eine oder mehrere
Verbindungen der Erfindung enthalten; solche Präparate können mit im Stand der Technik
gut bekannten Methoden hergestellt werden.
-
Die
Verdünnungsmittel,
die bei den Herstellungen der Zusammensetzungen verwendet werden
können,
schließen
diejenigen flüssigen
und festen Verdünnungsmittel
ein, mit denen der aktive Inhaltsstoff zusammen mit Färbemitteln
oder Geschmacksstoffen, falls gewünscht, vermischt wird. Tabletten
oder Kapseln können
geeigneterweise zwischen 0,2 und 500 mg, vorzugsweise von 0,5 bis
100 mg, aktiven Inhaltsstoff oder die äquivalente Menge eines pharmakologisch
annehmbaren Salzes desselben enthalten. Die Verbindungen können in
Pellets eingearbeitet werden, die mit einem geeigneten natürlichen
oder synthetischen Polymer, von dem im Stand der Technik bekannt
ist, daß es
Eigenschaften verzögerter
Freisetzung erzeugt, oder mit Polymeren in Tablettenform eingearbeitet
werden, um dieselben Eigenschaften zu erzeugen.
-
Die
flüssige
Zusammensetzung, die für
orale Verwendung angepaßt
ist, kann in Form einer Lösung oder
Suspension vorliegen. Die Lösung
kann eine wäßrige Lösung eines
Säureadditionssalzes
des Indolylpiperidinderivats zusammen mit zum Beispiel Saccharose
oder Sorbitol, um einen Sirup zu bilden, sein. Die Suspension kann
eine unlösliche
oder mikroverkapselte Form einer aktiven Verbindung der Erfindung
zusammen mit Wasser oder einem anderen pharmazeutisch annehmbaren
flüssigen
Medium zusammen mit einem Suspendiermittel oder Geschmacksstoff
umfassen.
-
Eine
Zusammensetzung für
parenterale Injektion kann aus löslichen
Salzen des Indolylpiperidinderivats hergestellt werden, die gefriergetrocknet
sein können
oder nicht und die in Wasser oder einer geeigneten parenteral injizierbaren
Flüssigkeit
gelöst
sein können.
-
In
der Humantherapie hängen
die Dosen der Verbindung der allgemeinen Formel (I) von der gewünschten
Wirkung und Behandlungsdauer ab; Dosen für Erwachsene liegen im allgemeinen
zwischen 0,2 mg und 500 mg pro Tag und vorzugsweise zwischen 0,5
mg und 100 mg pro Tag. Im allgemeinen wird der Arzt das Dosierungsregime
unter Berücksichtigung
des Alters und des Gewichts des zu behandelnden Patienten entscheiden.
-
Pharmakologische Wirkung
-
Die
folgenden Beispiele zeigen die hervorragenden pharmakologischen
Aktivitäten
der Verbindungen der vorliegenden Erfindung. Die Ergebnisse (1)
des Histamin-H1-Rezeptorbindungstestes, (2) der Histamin-induzierten
Hautgefäßpermeabilität in Ratten
mit Überwachung
antiallergischer Aktivität,
(3) der H1-ex-vivo-Bindungsstudien in Mäusen mit
der Überwachung
des Grades des Eindringens ins Gehirn und (4) der Messung des Blutdrucks
und der Herzrate in bewußten
unbehinderten Ratten mit Bluthochdruck mit Überwachung der kardiovaskulären Wirkungen,
wurden erhalten, wie unten beschrieben.
-
(1) Histamin-H1-Rezeptorbindungstest
-
Bindung
an die Histamin-H1-Rezeptoren wurde in Meerschweinchen-Cerebellum-Membranen
durchgeführt,
wie zuvor beschrieben (Chang et al., 1979). Kurz gesagt wurden die
Membransuspensionen (160 μg/ml)
bei 30°C
mit 0,7 nM [3H]-Mepyramin und verschiedenen
Konzentrationen der Testverbindungen in einem Endvolumen von 250 μl inkubiert.
Bindungsreaktionen wurden durch Filtration nach 30 min Inkubation
abgebrochen und die gebundene Radioaktivität wurde bestimmt. Die spezifische
Bindung wurde in Gegenwart von 10 μM Promethazin gemessen. Die
Affinität
jeder Testverbindung zum Rezeptor wurde durch Verwendung von wenigstens
sechs unterschiedlichen Konzentrationen, die man doppelt laufen
ließ,
bestimmt. IC50-Werte wurden durch nicht-lineare
Regression durch Verwendung von SAS auf einem DEC-AXP-Computer erhalten.
-
Tabelle
1. Histamin-H
1-Rezeptorbindungstest
-
Unsere
Ergebnisse zeigen, daß die
Verbindungen der vorliegenden Erfindung Affinitäten für die H1 Rezeptoren
besitzen, die sehr ähnlich
sind zu den Referenzverbindungen.
-
(2) Histamin-induzierte
Hautgefäßpermeabilität in Ratten
-
Männliche
Wistar-Ratten (180–210
g) wurden oral mit der Testverbindung oder Vehikel behandelt. 1, 4,
8 und 24 Stunden später
wurden die Ratten leicht mit Ether anästhesiert. Die Hautreaktion
wurde durch zwei intradermale Injektionen von 50 μl Histamin
(100 μg/ml)
auf den Rücken,
gefolgt von einer intravenösen
Injektion von 3 ml/kg Evans-Blau (5 mg/ml), beide in physiologischer
Kochsalzlösung
gelöst,
induziert. 60 Minuten später
wurden die Ratten durch Cervixdislokation getötet und die Rückenhaut
freiseziert. Der Durchmesser (in Millimetern) der Papula wurde in
zwei Richtungen gemessen und die Fläche wurde berechnet. Ergebnisse
sind als die % Hemmung bei einer gegebenen Dosis, verglichen mit
der mit Vehikel behandelten Gruppe, angegeben.
-
Die
in den Beispielen 22, 23, 24, 73, 75, 78, 79, 80, 82, 85 und 86
offenbarten Verbindungen zeigen eine Hemmung >50% der Histamin-induzierten Hautgefäßpermeabilität bei der
Dosis von 1 mg/kg 4 Stunden nach Verabreichung. Unter denselben
experimentellen Bedingungen zeigte Cetirizin eine Hemmung von 7%, wohingegen
Fexofenadin eine vernachlässigbare
Hemmung zeigt.
-
(3) H1-ex-vivo-Bindungsstudien
in Mäusen
-
Der
Test wurde im wesentlichen durchgeführt, wie von Leysen et al.
beschrieben, mit den folgenden Modifikationen. Über Nacht ausgehungerte männliche
Swiss-Albinomäuse
(21 ± 2
g) wurden oral mit unterschiedlichen Dosen der Testverbindungen
(10 ml/kg, p.o.) behandelt und wurden 90 Minuten später getötet. Das
gesamte Gehirn wurde herausseziert und in 10 ml eiskaltem 0,05 M
Na+/K+-Phosphatpuffer
(pH 7,4) homogenisiert. Ein 1 ml-Aliquot des Homogenats wurde, dreifach,
mit 0,1 ml [3H]-Mepyramin (2 nM Endkonzentration,
27 Ci/mmol, Amersham) während
40 Minuten bei 30°C
inkubiert. Das an die Membranen gebundene [3H]-Mepyramin
wurde durch sofortige Filtration der Homogenate unter Vakuum auf
die Glasfaserfilter (Whatman GF/B), gefolgt von drei schnellen Spülungen mit
5 ml kaltem Puffer, der 10 μM
kaltes Mepyramin enthielt, bestimmt. Die in den Filtern gebundene
Radioaktivität
wurde durch Flüssigszintillationsspektrometrie
bestimmt. Die nicht-spezifische Bindung wurde durch Behandeln der
Tiere mit 30 mg/kg p.o. D-Chlorpheniraminmaleat bestimmt. Mäuse, die
mit Vehikel (Methylcellulose 0,5% und Tween 0,1%) behandelt wurden,
wurden verwendet, um die Gesamtbindung zu bestimmen. Ergebnisse
sind ausgedrückt
als die % spezifische Bindung bei einer gegebenen Dosis der Testverbindung.
-
Die
Verbindungen der vorliegenden Erfindung zeigen geringe oder keine
Durchdringung durch die Blut-Hirn-Schranke.
-
(4) Messung des Blutdruckes
und der Herzrate in bewußten
unbeschränkten
Ratten mit Bluthochdruck
-
Erwachsene
männliche
Ratten mit spontanem Bluthochdruck (SAR) wurden operiert, um Blutdrucksensoren
in der Bauchaorta unmittelbar oberhalb der Darmbeinbifurkation zu
implantieren. Nach Aufwachen aus der Anästhesie wurden die Ratten einzeln
in Käfigen
untergebracht, die auf Radiofrequenzempfänger gestellt wurden. Amoxcillin
(15 mg/kg, i.m., nach chirurgischem Eingriff) wurde verabreicht,
um Infektion zu verhindern. Man ließ die Ratten sich für wenigstens
2 Wochen nach der Transmitterimplantation erholen. Arterieller Blutdruck
und Herzrate wurden aufgezeichnet und mit dem Dataquest-V-System
(Data Science, St. Paul, MN) analysiert. Die Tiere wurden in einem
Hell-Dunkel-Zyklus von 12:12 Stunden während des gesamten Aufzeichnungszeitraums
gehalten. Nach 18 Stunden Fasten mit Wasser „ad libitum" erhielten die Tiere
oral Arzneistoffe und wurden dann gefüttert. Hämodynamische Aufzeichnungen
wurden alle 15 Minuten vorgenommen, beginnend 4 Stunden nach Arzneistoffverabreichung
und sich bis zu 24 Stunden danach fortsetzend. Jede Aufzeichnung
dauerte 10 Sekunden, und die hämodynamischen
Werte aller Zyklen innerhalb dieses Zeitraums wurden gemittelt.
Alle Tiere erhielten alle Behandlungen, zwischen Verabreichungen
in derselben Ratte gab es einen siebentägigen Auswaschzeitraum und
eine vollständige
Erholung bis zu Basislinienwerten wurde sichergestellt. Die Wirkungen
der Behandlungen auf dem mittleren arteriellen Blutdruck und die
Herzrate wurden mit Einwegvarianzanalyse (ANOVA) bestimmt. Ein P-Wert <0,05 wurde als statistisch
signifikant angesehen.
-
Die
Verbindungen der vorliegenden Erfindung haben geringe oder keine
Wirkungen auf den Blutdruck und die Herzrate bei Dosen von 3 bis
30 mg/kg.
-
Aus
den oben beschriebenen Ergebnissen wird man verstehen, daß die Verbindungen
der vorliegenden Erfindung hervorragende antihistaminische und antiallergische
Aktivitäten
besitzen. Verbindungen der vorliegenden Erfindung haben verringerte
kardiovaskuläre
Nebenwirkungen und Nebenwirkungen auf das zentrale Nervensystem
und sind somit für
die Behandlung verschiedener allergischer Störungen nützlich, zum Beispiel Bronchialasthma,
Rhinitis, Konjunktivitis, Dermatitis und Urticaria. Die Erfindung
stellt somit ein Verfahren zur Behandlung einer allergischen Störung, zum
Beispiel Bronchialasthma, Rhinitis, Konjunktivitis, Dermatitis und
Urticaria, bereit, das den Schritt der Verabreichung einer wirksamen
Menge einer Verbindung von Formel (I) an einen menschlichen oder
tierischen Patienten, der einer solchen Behandlung bedarf, umfaßt. Die Erfindung
stellt auch die Verwendung der Verbindungen von Formel (I) bei der
Herstellung eines Arzneimittels zur Behandlung einer allergischen
Störung,
zum Beispiel Bronchialasthma, Rhinitis, Konjuntivitis, Dermatitis und
Urticaria, bereit.
-
Die
vorliegende Erfindung wird durch die folgenden Beispiele weiter
veranschaulicht werden. Diese Beispiele sind nur zu Veranschaulichungszwecken
angegeben und sollen nicht als beschränkend angesehen werden.
-
Tabelle
2. Liste von Beispielen
-
-
-
-
-
-
-
-
-
-
-
- Das Zeichen (*) in den Strukturen zeigt nur den Bindungspunkt,
es symbolisiert aber kein Kohlenstoffatom.
-
Beispiel 1
-
Darstellung von 2-{2-[4-(1-[1,3]Dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
A. Darstellung von 3-(1,2,3,6-Tetrahydro-pyridin-4-yl)-1H-indol.
-
30
g (0,26 mol) Indol wurden in einer Lösung von Kaliumhydroxid (77,6
g, 1,38 mol) in Methanol (692 ml) gelöst. 4-Piperidon-Monohydrat-Hydrochlorid
(102,3 g, 0,66 mol) wurde in einer Portion zugegeben und die Mischung
wurde für
5 h auf Rückfluß erhitzt.
Kaliumchlorid fiel bei Abkühlung
auf Raumtemperatur aus und wurde abfiltriert. Die flüssige Phase
wurde konzentriert, bis nur noch ein Drittel der Flüssigkeit
im Rundkolben zurückblieb.
Der Feststoff, der sich während
der Konzentration der flüssigen
Phase bildete, wurde abfiltriert und gründlich mit Ethanol und schließlich mit
Ethylether gewaschen. 31,9 g (63% Ausbeute) des Endproduktes wurden
erhalten.
Schmelzpunkt = 183–185°C.
-
B. Darstellung von 3-Piperidin-4-yl-1H-indol
-
19,03
g (0,096 mol) 3-(1,2,3,6-Tetrahydro-pyridin-4-yl)-1H-indol wurden
in einem Parr-Apparat über 18 h
bei 40 psi mit 2,2 g Pd/C 10% in 600 ml Methanol hydriert. Nach
Standardaufarbeitung wurden 16,76 g (87% Ausbeute) des gewünschten
Produktes erhalten.
Schmelzpunkt = 210–212°C.
-
C. Darstellung von 2-(2-Chlor-ethoxy)-benzoesäuremethylester
-
34
g (0,25 mol) Kaliumcarbonat wurden zu einer Lösung von 25 g (0,16 mol) Methylsalicylat
in 250 ml Methylethylketon zugegeben. Diese Mischung wurde für 1 h unter
Rückfluß gekocht,
dann wurden 27,3 ml (0,35 mol) 1-Brom-2-chlorethan zugegeben und
die Mischung wurde erneut auf Rückfluß gebracht.
Vier Stunden später
wurden weitere 34 g (0,25 mol) Kaliumcarbonat und weitere 16,3 ml
(0,2 mol) 1-Brom-2-chlorethan zugegeben. Dieser Vorgang wurde wiederholt,
bis die Reaktion beendet war. Die anorganischen Salze wurden abfiltriert
und die flüssige
Phase wurde mit demselben Volumen Hexan verdünnt. Diese organische Phase
wurde dreimal mit Wasser gewaschen und wie üblich aufgearbeitet. Die Ausbeute
in diesem Schritt war quantitativ und das Produkt war für den nächsten Syntheseschritt
rein genug.
NMR (300 MHz, CDCl3) δ 3,86–3,90 (m,
5H), 4,28–4,33
(t, 2H), 6,96–7,09
(m, 2H), 7,43–7,51
(m, 1H), 7,78–7,83
(m, 1H).
-
D. Darstellung von 2-{2-[4-(1H-Indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäuremethylester
-
0,14
g (0,65 mmol) 2-(2-Chlor-ethoxy)-benzoesäuremethylester wurden zu einer
Mischung von 0,1 g (0,5 mmol) 3-Piperidin-4-yl-1H-indol, 0,10 g
(0,75 mmol) Kaliumcarbonat und 0,06 g (0,37 mmol) Kaliumiodid in
1,5 ml Isobutylmethylketon unter Stickstoffatmosphäre zugegeben
und die Reaktionsmischung wurde für 18 Stunden unter Rückfluß gekocht.
Nach Abkühlung
auf Raumtemperatur wurden 1,5 ml Dichlormethan und 0,08 g (0,1 mmol)
Polystyrolmethylisocyanat zugegeben und die Mischung wurde bei dieser
Temperatur für
3 Stunden gerührt.
Nach Filtrieren wurde die Lösung
direkt auf eine 500 mg-Varian-SCX-Ionenaustauschsäule gegeben. Die Säulen wurden
mit 5 ml Methanol gewaschen und das Produkt wurde mit 5 ml Methanol/Ammoniak 20:1
eluiert, was, nach Entfernung des Lösemittels bei verringertem
Druck, 0,113 g (60% Ausbeute) 2-{2-[4-(1H-Indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäuremethylester
als ein gelbes Öl
lieferte.
ESI/MS m/e = 379 [(M + 1)+,
C23 H26 N2 03]
-
E. Darstellung von 2-{2-[4-(1-[1,3]Dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
0,02
g (0,42 mmol) einer Dispersion von 60% NaH in Mineralöl wurden
zu einer Lösung
von 0,06 g (0,16 mmol) 2-{2-[4-(1H-Indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäuremethylester,
hergestellt in Schritt D, in 1 ml wasserfreiem DMF unter einer inerten
Atmosphäre
zugegeben. Nach Rühren
für 30
Minuten bei Raumtemperatur wurden 0,04 mg (0,24 mmol) 2-Brommethyl-[1,3]-dioxolan
zugegeben und die Mischung wurde für 18 Stunden gerührt. Das
Lösemittel
wurde unter verringertem Druck abgezogen und die rohe Mischung wurde in
1 ml Ethanol gelöst.
0,1 ml 2N NaOH wurden zugegeben und die Mischung wurde bei 60°C für 3 Stunden gerührt. 0,1
ml 2N HCl wurden zugegeben und die Reaktionsmischung wurde bei Raumtemperatur
für 1 Stunde
gerührt.
Das Lösemittel
wurde unter verringertem Druck abgezogen und die rohe Mischung wurde
unter Verwendung einer 500 mg-Varian-C18-Chromatographiesäule gereinigt, was 0,040 g
(56% Ausbeute) 2-{2-[4-(1-[1,3]Dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure lieferte.
Schmelzpunkt
139–141°C.
NMR
(300 MHz, DMSO) δ =
1,90–2,10
(m, 4H), 2,58–2,72
(m, 2H), 2,90–2,98
(m, 3H), 3,20–3,24
(m, 2H), 3,76–3,80
(m, 4H), 4,25–4,27
(m, 2H), 4,41–4,45
(m, 2H), 5,09–5,13
(m, 1H), 7,00–7,12
(m, 2H), 7,12 (s, 2H), 7,38–7,54
(m, 4H), 7,63–7,65
(d, 1H).
-
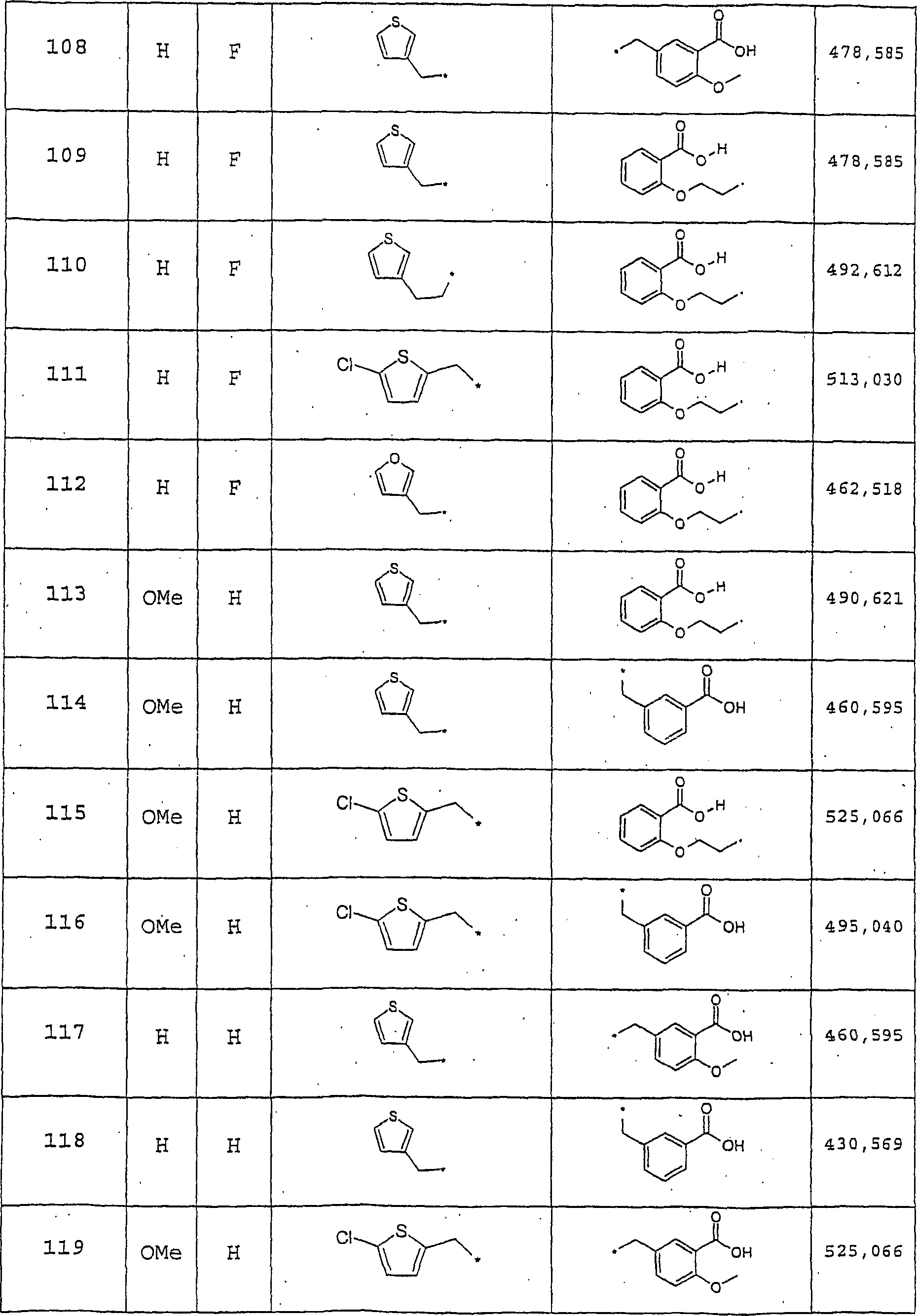
Beispiele 2–10, 17
und 20
-
Diese
Beispiele wurden unter Befolgung des in Beispiel 1 (Schritte D und
E) beschriebenen Verfahrens hergestellt. Die ESI/MS-Daten und Ausbeuten
sind in Tabelle 3 zusammengefaßt.
-
Tabelle
3. Beispiele 2–10,
17 und 20
-
Beispiel 13
-
Darstellung von 2-(2-{4-[1-(2-Thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
A. Darstellung von 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
-
Zu
einer Suspension von 30 g (0,15 mol) 3-Piperidin-4-yl-1H-indol und
28 ml (0,2 mol) in 185 ml wasserfreiem Dichlormethan wurden 17 ml
(0,18 mol) Ethylchlorformiat tropfenweise zugegeben, wobei die Temperatur
der Reaktion unter 20°C
gehalten wurde. Nach 2 h bei Raumtemperatur wurde die rohe Mischung
in 100 ml Wasser gegossen. Die organische Schicht wurde abgetrennt
und mit Natriumsulfat getrocknet. Nach Filtration wurde das Lösemittel
unter verringertem Druck abgezogen, was 39 g (95% Ausbeute) des
erwarteten Produktes lieferte.
ESI/MS m/e = 272 [(M + 1)+, C16 H20 N2 O2]
NMR (300 MHz, DMSO) δ = 1,16–1,23 (t,
2H), 1,41–1,65
(m, 2H), 1,92–1,99
(m, 2H), 2,90–23,10
(m, 3H), 3,99–4,10
(m, 4H), 6,95–7,10
(m, 3H), 7,31–7,34
(d, 1H), 7,53-7,57 (d, 1H), 10,81 (s, 1H).
-
B. Darstellung von 4-[1-(2-Thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Unter
einer inerten Atmosphäre
wurde eine Lösung
von 6,9 g (0,025 mol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester in 25 ml wasserfreiem
DMF tropfenweise zu einer Suspension zugegeben, die 1,2 g (0,030
mol) Natriumhydrid (60% in Mineralöl) in 70 ml wasserfreiem DMF
enthielt. Nach Rühren
bei Raumtemperatur für
1 Stunde wurde eine Lösung
von 6,2 g (0,03 mol) 2-Thiophen-3-yl-ethylmethansulfat in 15 ml wasserfreiem
DMF zugegeben. Die Reaktionsmischung wurde bei Raumtemperatur für 30 Minuten
gerührt und
dann bei 60°C
für 3 Stunden
erhitzt. Die rohe Mischung wurde in Wasser gegossen und mit Dichlormethan extrahiert.
Nach Trocknung wurde das Lösemittel
unter verringertem Druck abgezogen und 10,3 g eines rohen Öls wurden
erhalten. Die rohe Mischung wurde durch Flashchromatographie über Silicagel
gereinigt, was 8,3 g (86% Ausbeute) des erwarteten Produktes lieferte.
-
C. Darstellung von 3-Piperidin-4-yl-1-(2-thiophen-3-yl-ethyl)-1H-indol
-
Zu
einer Lösung
von 12,7 g (0,033 mol) 4-[1-(2-Thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
in 10 ml Isopropanol wurde eine Lösung von 22 g Kaliumhydroxid
in 220 ml Isopropanol zugegeben. Die rohe Mischung wurde für 16 Stunden
unter Rückfluß gekocht.
Nach Abkühlung
auf Raumtemperatur wurde das Lösemittel
bei verringertem Druck abgezogen und die rohe Mischung wurde zwischen
Toluol und Wasser extrahiert. Die organische Schicht wurde mit Natriumsulfat
getrocknet und das Lösemittel
wurde nach Filtration unter verringertem Druck abgezogen, was 9,3
g (90% Ausbeute) eines Öls
lieferte, das dem erwarteten Produkt entspricht.
-
D. Darstellung von 2-{2-(4-[1-(2-Thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
Eine
Lösung
von 1,5 g (0,007 mol) 2-(2-Chlorethoxy)-benzoesäuremethylester (hergestellt
in Beispiel 1, Teil C) in 5 ml Methylisobutylketon wurde zu einer
Suspension von 2 g (0,065 mol) 3-Piperidin-4-yl-1-(2-thiophen-3-yl-ethyl)-1H-indol
und 1,8 g (0,013 mol) Kaliumcarbonat in 45 ml Methylisobutylketon
zugegeben. Die Reaktionsmischung wurde für 18 h unter Rückfluß gekocht.
Die rohe Mischung wurde filtriert, um anorganische Salze zu entfernen,
und das Lösemittel
wurde unter verringertem Druck abgezogen, was 3,3 g eines rohen Öls lieferte.
Die rohe Mischung wurde durch Flashchromatographie über Silicagel
gereinigt, was 1,5 g (48% Ausbeute) 2-{2-(4-[1-(2-Thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäuremethylester
lieferte. Dieser Ester wurde in einer Mischung aus 25 ml Methanol/THF
3:2 gelöst
und mit 2N NaOH bei Raumtemperatur für 16 Stunden hydrolysiert.
Die rohe Mischung wurde mit einer wäßrigen 2N HCl-Lösung neutralisiert und
das Lösemittel
wurde unter verringertem Druck abgezogen. Der rohe Rückstand
wurde mit Dichlormethan ausgefällt
und dann mit Methanol umkristallisiert, was 1,3 g der erwarteten
Säure lieferte.
Schmelzpunkt
165–167°C
NMR
(300 MHz, DMSO) δ =
1,75–2,07
(m, 4H), 2,54–2,65
(m, 2H), 2,77–3,00
(m, 3H), 3,00–3,13
(t, 2H), 3,14–3,30
(m, 2H), 4,25–4,39
(t, 2H), 4,39–4,55
(m, 2H), 5,20–5,40
(m, 1H), 6,93–7,29
(m, 7H), 7,33–7,59
(m, 4H), 7,59–7,67
(d, 1H).
-
Beispiele 11, 12,14, 15
und 18
-
Diese
Beispiele wurden unter Befolgung des in Beispiel 13 beschriebenen
Verfahrens unter Verwendung eines geeigneten Methansulfonats oder
Bromids in Teil B hergestellt. Die ESI/MS-Daten und Ausbeuten sind
in Tabelle 4 zusammengefaßt.
-
Tabelle
4. Beispiele 11, 12, 14, 15 und 18
-
Beispiel 16
-
Darstellung von 2[2-(4-(1-[3-(Benzo[1,3]dioxol-5-yloxy)propyl]-1H-indol-3-yl}piperidin-1-yl)ethoxy]benzoesäure
-
A. Darstellung von 4-{1-[3-(Benzo[1,3]dioxol-5-yloxy)-propyl]-1H-indol-3-yl-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 2,2 g (8,1 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 2,68 g (10 mmol) 5-(3-Brompropoxy)-benzo[1,3]dioxol.
Nach Standardaufarbeitung wurden 3,8 g (100% Ausbeute) des erwarteten
Produktes erhalten.
-
B. Darstellung von 1-[3-(Benzo[1,3]dioxol-5-yloxy)-propyl]-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 2,68 g (8,1 mmol) 4-{1-[3-(Benzo[1,3]dioxol-5-yloxy)-propyl]-1H-indol-3-yl-piperidin-1-carbonsäureethylester.
-
C. Darstellung von 2-[2-(4-(1-[3-(Benzo[1,3]dioxol-5-yloxy)propyl]-1H-indol-3-yl)piperidin-1-yl)ethoxy]benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 8,1 mmol 1-[3-(Benzo[1,3]dioxol-5-yloxy)-propyl]-3-piperidin-4-yl-1H-indol
und 2,3 g (11 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Nach der Standardaufarbeitung
wurden 2,68 g der entsprechenden Säure erhalten. Die rohe Mischung
wurde durch Flashchromatographie über Silicagel gereinigt, was
1,15 g (26% Ausbeute) der erwarteten Säure lieferte.
Schmelzpunkt
147–152°C
NMR
(300 MHz, DMSO) δ =
1,70–2,00
(m, 4H), 2,07–2,16
(m, 2H), 2,60–2,68
(m, 2H), 2,81–2,97
(m, 3H), 3,16–3,24
(m, 2H), 3,76–3,82
(m, 2H), 4,25–4,30
(t, 2H), 4,31–4,35
(m, 2H), 4,30–4,70
(m, 1H), 5,94 (s, 2H), 6,32–6,36
(dd, 1H), 6,62–6,63
(m, 1H), 6,78–6,80
(d, 1H), 6,96–7,13
(m, 4H), 7,21–7,24
(m, 1H), 7,36–7,40
(m, 2H), 7,51–7,54
(m, 1H), 7,63–7,66
(d, 1H)
-
Beispiel 19
-
Darstellung von 2-(2-{4-[1-(5-Chlorthiophen-2-ylmethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
A. Darstellung von 4-[1-(5-Chlorthiophen-2-ylmethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung von des in Beispiel 13 (Teil B)
beschriebenen Verfahrens bei Raumtemperatur für 15 Stunden hergestellt, ausgehend
von 3,5 g (13 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 1,9 ml (16 mmol) 2-Chlor-5-chlormethylthiophen.
Nach Standardaufarbeitung wurden 5,2 g (99% Ausbeute) des erwarteten
Produktes erhalten.
-
B. Darstellung von 1-(5-Chlorthiophen-2-ylmethyl)-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 5,21 g (13 mmol) 4-[1-(5-Chlorthiophen-2-ylmethyl)-1H-indol-3-yl]-piperidin-1-carbonäureethylester.
Nach Standardaufarbeitung wurden 4,19 g (97% Ausbeute) des erwarteten
Produktes erhalten.
-
C. Darstellung von 2-(2-{4-[1-(5-Chlorthiophen-2-ylmethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 4,21 g (13 mmol) 1-(5-Chlorthiophen-2-ylmethyl)-3-piperidin-4-yl-1H-indol
und 3,6 g (17 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Nach der Standardaufarbeitung
wurden 2,47 g der entsprechenden Säure erhalten. Die rohe Mischung
wurde durch Flashchromatographie über Silicagel gereinigt, was
1,2 g (17% Ausbeute) der reinen Säure lieferte.
Schmelzpunkt
178–179°C
NMR
(300 MHz, DMSO) δ =
1,86–2,05
(m, 4H), 2,58–2,69
(m, 2H), 2,87–2,98
(m, 3H), 3,17–3,23
(m, 2H), 4,41–4,45
(m, 2H), 5,50 (s, 2H), 5,40–5,80
(m, 1H), 6,95–7,05
(m, 4H), 7,10–7,16
(m, 1H), 7,21–7,24
(m, 2H), 7,36–7,41
(m, 1H), 7,47–7,55
(m, 2H), 7,64–7,68
(d, 1H)
-
Beispiel 21
-
Darstellung von 2-(2-{4-[1-(3-[1,3]Dioxolan-2-yl-propyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
Eine
Lösung
von 2,75 g (7 mmol) 2-{2-[4-(1H-Indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäuremethylester
(hergestellt wie in Beispiel 1, Teil D) in 10 ml wasserfreiem DMF
wurde zu einer Suspension von 0,36 g (9,1 mmol) NaH (60% Dispersion
in Mineralöl)
in 5 ml wasserfreiem DMF unter einer inerten Atmosphäre zugegeben.
Nach Rühren
für 30
Minuten bei Raumtemperatur wurde eine Lösung von 1,1 ml (8,4 mmol)
2-(3-Chlorpropyl)-[1,3]dioxolan
in 3 ml DMF zugegeben. Die rohe Mischung wurde bei Raumtemperatur
für 16
Stunden gerührt
und das Lösemittel
wurde unter verringertem Druck abgezogen. Der erhaltene Rückstand
wurde mit 150 ml Ethanol aufgelöst
und 6 ml einer wäßrigen 2N
NaOH-Lösung wurden
zugegeben. Nach 12 h bei Raumtemperatur wurde das Lösemittel
unter verringertem Druck abgezogen. Die rohe Mischung wurde mit
50 ml Wasser aufgelöst
und mit einer wäßrigen 2N
HCl-Lösung
neutralisiert. Die rohe Mischung wurde mit Flashchromatographie über Silicagel
gereinigt, was 0,83 g (29% Ausbeute) des erwarteten Produktes lieferte.
Schmelzpunkt
147–149°C
NMR
(300 MHz, DMSO) δ =
1,50–1,56
(m, 2H), 1,75–1,86
(m, 2H), 1,89–1,97
(m, 4H), 2,61–2,69
(m, 2H), 2,79–2,99
(m, 3H), 3,21–3,24
(d, 2H), 3,70–3,75
(m, 2H), 3,82–3,87
(m, 2H), 4,13–4,17
(m, 2H), 4,42–4,46
(m, 2H), 4,76–4,80
(m, 1H), 5,00–5,40
(bs, 1H), 6,99–7,02
(m, 2H), 7,10–7,24
(m, 3H), 7,37–7,43
(m, 2H), 7,52–7,54
(d, 1H), 7,64–7,66
(d, 1H).
-
Beispiel 22
-
Darstellung von 2-{2-[4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
A. Darstellung von 6-Fluor-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teile A und
B) beschriebenen Verfahrens hergestellt, ausgehend von 1 g (7,4
mmol) 6-Fluorindol und 2,84 g (18,5 mmol) 4-Piperidon-Monohydrat-Hydrochlorid. In
diesem Fall lief der Hydrierungsschritt für 1 Stunde bei 30 psi ab und
der verwendete Katalysator war Platin(IV)-oxid. 0,640 g (51% Ausbeute)
6-Fluor-3-piperidin-4-yl-1H-indol wurden erhalten.
ESI/MS m/e
= 219 [(M + 1)+, C13 H15 F N2]
-
B. Darstellung von 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil A) beschriebenen
Verfahrens hergestellt, ausgehend von 4,4 g (20 mmol) 6-Fluor-3-piperidin-4-yl-1H-indol.
Nach Standardaufarbeitung wurden 5,2 g (90% Ausbeute) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
C. Darstellung von 4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil A) beschriebenen
Verfahrens bei Raumtemperatur für
5 Stunden erhalten, ausgehend von 5 g (17,2 mmol) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
und 3,2 g (20 mmol) 2-Brommethylfuran.
Nach Standardaufarbeitung wurden 6,4 g (99% Ausbeute) 4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
D. Darstellung von 6-Fluor-1-furan-2-ylmethyl-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 6,4 g (17,2 mmol) 4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäurethylester.
Nach Standardaufarbeitung wurden 4,4 g (86% Ausbeute) 6-Fluor-1-furan-2-ylmethyl-3-piperidin-4-yl-1H-indol
erhalten.
-
E. Darstellung von 2-{2-[4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 2 g (6,5 mmol) 6-Fluor-1-furan-2-ylmethyl-3-piperidin-4-yl-1H-indol
und 1,5 g (7,1 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Nach Standardaufarbeitung
und Reinigung durch Flashchromatographie über Silicagel wurden 0,9 g
(30% Ausbeute) der erwarteten Säure
erhalten.
Schmelzpunkt 174–175°C
NMR
(300 MHz, DMSO) δ =
1,83–1,95
(m, 4H), 2,58–2,66
(m, 2H), 2,79–2,94
(m, 3H), 3,16–3,22
(d, 2H), 4,00–4,40
(bs, 1H), 4,33–4,39
(m, 2H), 5,35 (s, 2H), 6,40 (s, 1H), 6,45–6,47 (m, 1H), 6,97–7,66 (m,
10H).
-
Beispiel 23
-
Darstellung von 2-(2-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
A. Darstellung von 4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 4 g (0,015 mol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 2,07 ml (0,018 mol) 2-(2-Bromethyl)-[1,3]-dioxolan. Nach Standardaufarbeitung
wurden 5,3 g 4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
erhalten.
ESI/MS m/e = 373 [(M + 1)+,
C21 H28 N2 O4]
NMR (300 MHz, CDCl3) δ = 1,25–1,28 (t,
3H), 1,64–1,70
(m, 4H), 2,01–2,17
(m, 4H), 2,88–3,00
(m, 3H), 3,82–4,05
(m, 4H), 4,18–4,27
(m, 4H), 4,81–4,86
(t, 1H), 6,86 (s, 1H), 7,05–7,26
(m, 2H), 7,34–7,38
(d, 1H), 7,59–7,63
(d, 1H).
-
B. Darstellung von 1-(2-[1,3]Dioxolan-2-yl-ethyl)-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens erhalten, ausgehend von 5,3 g (0,015 mol) 4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 4 g (89% Ausbeute) 1-(2-[1,3]Dioxolan-2-yl-ethyl)-3-piperidin-4-yl-1H-indol
erhalten.
ESI/MS m/e = 301 [(M + 1)+,
C18 H24 N2 O2]
NMR (300 MHz, CDCl3) δ = 1,61–1,76 (m,
2H), 2,01–2,21
(m, 5H), 2,74–3,02
(m, 3H), 3,16–3,22
(m, 2H), 3,82–4,04
(m, 4H), 4,20–4,27
(t, 2H), 4,81–4,86
(t, 1H), 6,87 (s, 1H), 7,07–7,25
(m, 2H), 7,32–7,36
(d, 1H), 7,61–7,65
(d, 1H).
-
C. Darstellung von 2-(2-(4-[1-(2-[1,3]-Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl)-ethoxy)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 3 g (0,01 mol) 1-(2-[1,3]Dioxolan-2-yl-ethyl)-3-piperidin-4-yl-1H-indol
und 2,8 g (0,013 mol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Die rohe Mischung
wurde durch Flashchromatographie über Silicagel gereinigt, was
1,86 g (40% Ausbeute) 2-(2-(4-[1-(2-[1,3]-Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-yl)-ethoxy)-benzoesäure lieferte.
Schmelzpunkt
118–120°C
NMR
(300 MHz, CDCl3) δ = 2,18–2,28 (m, 4H), 2,47–2,56 (m,
2H), 3,00–3,15
(m, 3H), 2,52–3,56
(m, 2H), 3,77–3,90
(m, 4H), 4,00–4,05
(m, 2H), 4,20–4,22
(t, 2H), 4,64–4,68
(m, 2H), 4,85–4,89
(m, 1H), 7,01–7,12
(m, 4H), 7,20–7,25
(t, 1H), 7,36–7,39
(d, 1H), 7,49–7,54
(t, 1H), 7,61–7,63
(d, 1H), 7,90–7,93
(d, 1H).
-
Beispiel 24
-
Darstellung von 2-(2-{4-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
A. Darstellung von 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil A) beschriebenen
Verfahrens hergestellt, ausgehend von 0,4 g (1,83 mmol) 6-Fluor-3-piperidin-4-yl-1H-indol, 0,2
ml (2,13 mmol) Ethylchlorformiat und 0,32 ml (2,13 mmol) Triethylamin.
0,32 g (60% Ausbeute) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
wurden erhalten.
-
B. Darstellung von 4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 0,1 g (0,37 mmol) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
und 0,081 g (0,45 mmol) 2-(2-Bromethyl)-[1,3]doxolan.
0,170 g (quantitative Ausbeute) 4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
wurden erhalten.
ESI/MS m/e = 391 [(M + 1)+,
C21 H27 F N2 O4]
-
C. Darstellung von 1-(2-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 170 g (0,448 mmol) 4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
0,04 g (28% Ausbeute) 1-(2-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-3-piperidin-4-yl-1H-indol
wurden erhalten.
ESI/MS m/e = 319 [(M + 1)+,
C18 H23 F N2 O2]
NMR (300 MHz, CDCl3) δ = 1,60–1,74 (m,
2H), 1,99–2,18
(m, 5H), 2,73–2,95
(m, 3H), 3,16–3,22
(m, 2H), 3,82–4,04
(m, 4H), 4,12–4,20
(t, 2H), 4,80–4,85
(t, 1H), 6,86 (s, 1H), 6,99–7,05
(dd, 1H), 7,15–7,25
(m, 1H), 7,48–7,55
(dd, 1H).
-
D. Darstellung von 2-(2-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,04 g (0,11 mmol) 1-(2-[1,3]Dioxolan-2-yl-ethyl)-6-fluor-3-piperidin-4-yl-1H-indol
und 0,03 g (0,15 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Nach Reinigung über einer
Varian-C18-Säule wurden
0,012 g (22% Ausbeute) erhalten.
Schmelzpunkt 150–151°C
NMR
(300 MHz, CDCl3) δ = 1,94–2,04 (m, 6H), 2,61–2,64 (m,
2H), 2,89–2,98
(m, 3H), 3,20–3,23
(d, 2H), 3,78–3,95
(m, 4H), 4,15–4,19
(t, 2H), 4,41–4,44
(m, 2H), 4,77–4,80
(t, 1H), 5,47 (bs, 1H), 6,82–6,88
(t, 1H), 6,99–7,04
(t, 1H), 7,16–7,27
(m, 3H), 7,36–7,41
(t, 1H), 7,52–7,55
(d, 1H), 7,64–7,69
(t, 1H).
-
Beispiel 25
-
Darstellung von 3-[4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
A. Darstellung von 4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 4 g (15 mmol) 4-(1H-Tndol-3-yl)-piperidin-1-carbonsäureethylester
und 30 ml einer frisch hergestellten 0,61M 2-Brommethylthiophen-Lösung in
wasserfreiem Ethylether. Nach Standardaufarbeitung wurden 5,6 g
(100% Ausbeute) des erwarteten Produktes erhalten.
-
B. Darstellung von 3-Piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 5,6 g (15 mmol) 4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 4,35 g (98% Ausbeute) des erwarteten
Produktes erhalten.
-
C. Darstellung von 3-[4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 4,35 g (15 mmol) 3-Piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
und 4,58 g (20 mmo) 3-Brommethylbenzoesäuremethylester. Nach der Standardaufarbeitung
wurden 3,3 g der entsprechenden Säure erhalten. Die rohe Mischung
wurde durch Flashchromatographie über Silicagel gereinigt, was
0,64 g (10% Ausbeute) der reinen Säure lieferte.
Schmelzpunkt
228–229°C
NMR
(300 MHz, DMSO) δ =
1,55–1,79
(m, 2H), 1,87–1,97
(m, 2H), 2,10–2,22
(t, 2H), 2,73–2,81
(t, 1H), 2,90–2,94
(m, 2H), 3,59 (s, 2H), 5,52 (s, 2H), 6,92–7,01 (m, 2H), 7,07–7,13 (m,
2H), 7,24 (s, 1H), 7,36–7,57 (m,
5H), 7,83–7,86
(d, 1H), 7,92–7,94
(m, 1H).
-
Beispiel 26
-
Darstellung von 3-[4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
A. Darstellung von 4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
16 Stunden hergestellt, ausgehend von 11 g (40 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 7,2 g (44 mol) 3-Chlormethylpyridin-Hydrochlorid. Nach Standardaufarbeitung
wurden 13 g eines rohen Öls
erhalten. Dieses Rohmaterial wurde mit Ethylether ausgefällt, was
10,8 g (90% Ausbeute) eines weißen
Feststoffes lieferte.
-
B. Darstellung von 3-Piperidin-4-yl-1-pyridin-3-ylmethyl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 10,8 g (30 mmol) 4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 9,3 g eines rohen Öls erhalten.
Das entsprechende Fumarat-Derivat wurde in Ethanol hergestellt,
was 9,8 g eines weißen
Feststoffes lieferte. Nach Behandlung mit wäßriger NaOH und Extraktion
mit Ethylacetat wurden 5,3 g (62% Ausbeute) reines 3-Piperidin-4-yl-1-pyridin-3-ylmethyl-1H-indol erhalten.
-
C. 3-[4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Eine
Lösung,
die 1,2 g (52 mmol) 3-Brommethylbenzoesäuremethylester in 10 ml wasserfreiem
Dichlormethan enthielt, wurde tropfenweise über einer Lösung von 1,5 g (5 mmol) 3-Piperidin-4-yl-1-pyridin-3-ylmethyl-1H-indol
und 0,8 ml (55 mmol) Triethylamin in 35 ml wasserfreiem Dichlormethan
zugegeben. Nach Rühren
bei Raumtemperatur für
5 Stunden wurde die rohe Mischung mit Wasser, gesättigter
Natriumhydrogencarbonatlösung
und Wasser gewaschen. Die organische Phase wurde getrocknet und
das Lösemittel
wurde unter verringertem Druck abgezogen. Die rohe Mischung wurde
durch Flashchromatographie über
Silicagel gereinigt, was 0,95 g (43% Ausbeute) 3-[4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester
lieferte. Dieser Ester wurde in 15 ml Methanol gelöst und mit
1N NaOH bei Raumtemperatur für
12 Stunden hydrolysiert. Die rohe Mischung wurde mit 1N HCl neutralisiert
und dann wurde das Lösemittel unter
verringertem Druck abgezogen. Der feste Rückstand wurde mit Wasser und
Dichlormethan gewaschen und die entsprechende Säure (0,6 g, 77% Ausbeute) wurde
durch Filtration isoliert.
Schmelzpunkt 190–192°C
NMR (300 MHz, DMSO) δ = 1,92–2,21 (m,
4H), 2,98–3,20
(m, 2H), 3,32–3,43
(m, 3H), 4,38 (s, 2H), 5,42 (s, 2H), 6,98–7,03 (t, 1H), 7,08–7,14 (t,
1H), 7,29–7,37
(m, 2H), 7,46–7,49
(d, 1H), 7,56–7,64
(m, 2H), 7,68–7,72 (d,
1H), 7,94–7,97
(d, 1H), 8,00–8,03
(d, 1H), 8,20 (s, 1H), 8,20 (s, 1H), 8,43–8,47 (d, 1H), 8,50 (s, 1H).
-
Beispiel 27
-
Darstellung von 3-{4-[1-(5-Chlorthiophen-2-ylmethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 19 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 1,48 g (4,48 mmol) 1-(5-Chlorthiophen-2-ylmethyl)-3-piperidin-4-yl-1H-indol
und 1,0 g (8,8 mmol) 3-Brommethylbenzoesäuremethylester. Die rohe Mischung
wurde durch Flashchromatographie über Silicagel gereinigt, was
0,29 g (14% Ausbeute der reinen Säure lieferte.
Schmelzpunkt
232–234°C
NMr
(300 MHz, DMSO) δ =
1,91–2,09
(m, 4H), 2,88–3,20
(m, 2H), 3,22–3,36
(m, 3H), 4,34 (s, 2H), 5,48 (s, 2H), 6,93–7,03 (m, 3H), 7,09–7,12 (m,
1H), 7,25 (s, 1H), 7,50–7,66
(m, 3H), 7,84–7,86
(m, 1H), 7,99–8,01
(d, 1H), 8,15 (s, 1H).
-
Beispiel 28
-
Darstellung von 3-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
A. Darstellung von 3-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,2 g (1 mmol) 3-Piperidin-4-yl-1H-indol
und 0,29 g (1,3 mmol) 3-Brommethyl-benzoesäuremethylester. Nach Ionenaustauschreinigung
wurden 0,276 g (79% Ausbeute) 3-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester
erhalten.
-
B. Darstellung von 3-{4-[1-(2-[1,3]Dioxolan-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,046 g (0,13 mmol) 3-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester und
0,019 ml (0,16 mmol) 2-(2-Bromethyl)-[1,3]dioxolan. Nach der beschriebenen
Reinigung wurden 0,023 g (40% Ausbeute) der erwarteten Säure erhalten.
NMR
(300 MHz, DMSO) δ =
1,64–1,75
(m, 2H), 1,90–1,95
(m, 2H), 1,99–2,06
(m, 2H), 2,12–2,19
(m, 2H), 2,72–2,80
(m, 1H), 2,90–2,94
(m, 2H), 3,59 (s, 2H), 3,72–3,80
(m, 2H), 3,87–3,93
(m, 2H), 4,11–4,21
(t, 2H), 4,75–4,78
(t, 2H), 6,99–7,00
(t, 1H), 7,08–7,14
(m, 2H), 7,35–7,46
(m, 2H), 7,54–7,56
(m, 2H), 7,83–7,85
(m, 2H), 7,93 (s, 2H).
-
Beispiele 29–33
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 28 beschriebenen
Verfahrens hergestellt. Die ESI/MS-Daten, Ausbeuten und Reinheit
sind in Tabelle 5 zusammengefaßt.
-
Tabelle
5. Beispiele 29–33
-
Beispiel 34
-
Darstellung von 2-Methoxy-5-[4-(1-pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
A. Darstellung von 5-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,2 g (1 mmol) 3-Piperidin-4-yl-1H-indol
und 0,34 g (1,3 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester.
Nach Ionenaustauschreinigung wurden 0,273 g (70% Ausbeute) 5-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäureethylestter
erhalten.
-
B. Darstellung von 2-Methoxy-5-[4-(1-pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,054 g (0,13 mmol) 5-[4-(1H-Indol-3-yl)-piperidin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester
und 0,029 mg (0,16 mmol) 3-Chlormethylpyridin-Hydrochlorid.
Nach der beschriebenen Reinigung wurden 0,007 g (11% Ausbeute) der
erwarteten Säure
erhalten.
NMR (300 MHz, DMSO) δ = 1,61–1,72 (m, 2H), 1,87–1,95 (m,
2H), 2,04–2,11
(m, 2H), 2,70–2,78
(m, 1H), 2,88–2,92
(d, 2H), 3,71 (s, 3H), 5,39 (s, 2H), 6,86–6,89 (d, 1H), 6,96–7,33 (m,
6H), 7,43–7,46
(d, 1H), 7,55–7,58 (d,
1H), 8,43–8,45
(d, 1H), 8,51 (s, 1H).
-
Beispiele 35–39
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 34 beschriebenen
Verfahrens hergestellt. Die ESI/MS-Daten, Ausbeuten und Reinheit
sind in Tabelle 6 zusammengefaßt.
-
Tabelle
6. Beispiel 35–38
-
Beispiel 39
-
Darstellung von 4-Brom-3-[4-(1-[1,3]dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
A. Darstellung von 4-Brom-3-[4-(1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,2 g (1 mmol) 3-Piperidin-4-yl-1-H-indol
und 0,39 g (1,3 mmol) 4-Brom-3-brommethylbenzoesäuremethylester. Nach Ionenaustauschreinigung
wurden 0,196 g (46% Ausbeute) 4-Brom-3-[4-(1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester
erhalten.
-
B. Darstellung von 4-Brom-3-[4-(1-[1,3]Dioxolan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,055 g (0,133 mol) 4-Brom-3-[4-(1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester
und 0,034 mg (0,16 mmol) 2-Brommethyl-[1,3]dioxolan. Nach der beschriebenen
Reinigung wurden 0,021 g (32% Ausbeute) der erwarteten Säure erhalten.
NMR
(300 MHz, DMSO) δ =
1,62–1,74
(m, 2H), 1,90–1,95
(m, 2H), 2,19–2,23
(m, 2H), 2,74–2,82
(m, 1H), 2,93–3,00
(m, 2H), 3,58 (s, 2H), 4,23–4,25
(m, 2H), 5,08–5,12
(t, 1H), 6,95–7,00
(t, 1H), 7,07–7,15
(m, 2H), 7,41–7,69
(m, 4H), 7,98–8,00
(m, 1H).
-
Beispiele 40–45
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 39 beschriebenen
Verfahrens hergestellt. Die ESI/MS-Daten, Ausbeuten und Reinheit
sind in Tabelle 7 zusammengefaßt.
-
Tabelle
7. Beispiele 40–45
-
Beispiel 46
-
Darstellung von 2-Fluor-5-[4-(1-pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
A. Darstellung von 2-Fluor-5-[4-(1H-indol-3-yl)piperidin-1-ylmethyl]-benzoesäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,2 g (1 mmol) 3-Piperidin-4-yl-1H-indol
und 0,33 g (1,3 mmol) 5-Brommethyl-2-fluorbenzoesäureethylester.
Nach Ionenaustauschreinigung wurden 0,30 g (79% Ausbeute) 2-Fluor-5-[4-(1H-indol-3-yl)piperidin-1-ylmethyl]-benzoesäureethylester
erhalten.
-
B. Darstellung von 2-Fluor-5-(4-(1-pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,63 g (0,17 mmol) 2-Fluor-5-[4-(1H-indol-3-yl)piperidin-1-ylmethyl]-benzoesäureethylester
und 0,035 mg (0,21 mmol) 3-Chlormethylpyridin-Hydrochlorid. Nach der beschriebenen
Reinigung wurden 0,053 g (71% Ausbeute) der erwarteten Säure erhalten.
NMR
(300 MHz, DMSO) δ =
1,67–1,77
(m, 2H), 1,90–1,95
(m, 2H), 2,09–2,17
(t, 2H), 2,73–2,81
(m, 1H), 2,88–2,93
(d, 2H), 3,49 (s, 2H), 5,38 (s, 2H), 6,95–7,10 (m, 3H), 7,24–7,35 (m,
2H), 7,43–7,46
(d, 1H), 7,54–7,61
(m, 4H), 8,42–8,45
(dd, 1H), 8,50–8,52
(m, 1H).
-
Beispiele 47–52
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 46 beschriebenen
Verfahrens hergestellt. Die ESI/MS-Daten, Ausbeuten und Reinheit
sind in Tabelle 8 zusammengefaßt.
-
Tabelle
8. Beispiele 47–52
-
Beispiel 53
-
Darstellung von 2-(2-{4-[1-(Tetrahydrofuran-3-ylmethyl)-1H-indol-3-yl)-piperidin-1-yl}-ethoxy)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,1 g (0,26 mmol) 2-{2-[4-(1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäuremethylester und
0,067 g (0,37 mmol) frisch hergestelltem Tetrahydrofuran-3-yl-methyl-Methansulfonat.
Nach Standardaufarbeitung wurden 0,045 g (99% Ausbeute) der erwarteten
Säure erhalten.
NMR
(300 MHz, DMSO) δ =
1,82–1,98
(m, 4H), 2,44–2,56
(m, 5H), 2,67–2,78
(m, 1H), 2,81–2,93
(m, 2H), 3,15–3,20
(m, 2H), 3,57–3,66
(m, 2H), 3,78–3,86
(m, 2H), 4,08–4,11
(m, 2H), 4,29–4,33
(m, 2H), 6,89–7,02
(m, 2H), 7,09–7,18
(m, 3H), 7,27–7,49
(m, 3H), 7,62–7,64
(d, 1H).
-
Beispiele 54–57
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 53 beschriebenen
Verfahrens hergestellt, ausgehend von dem geeigneten Methansulfonat
oder Halogenid. Die ESI/MS-Daten,
Ausbeuten und Reinheit sind in Tabelle 9 zusammengefaßt.
-
Tabelle
9. Beispiele 54–57
-
Beispiel 58
-
Darstellung von 3-(4-[1-(Tetrahydrofuran-3-ylmethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,1 g (0,28 mmol) 3-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-benzoesäuremethylester (Beispiel
28, Teil A) und 0,72 g (0,4 mmol) (Tetrahydrofuran-3-yl)-Methansulfonat. Nach
Standardreinigung wurden 0,04 g (34% Ausbeute) der erwarteten Säure erhalten.
NMR
(300 MHz, DMSO) δ =
1,53–1,94
(m, 5H), 2,07–2,14
(t, 2H), 2,61–2,79
(m, 2H), 2,89–2,94
(m, 3H), 3,40–3,71
(m, 4H), 3,77–3,85
(m, 2H), 4,05–4,09
(d, 2H), 6,95–7,00
(t, 1H), 7,07–7,13
(m, 1H), 7,25–7,34
(m, 3H), 7,42–7,45
(d, 1H), 7,53–7,56
(d, 1H), 7,74–7,77
(d, 1H), 7,83–7,87
(m, 1H).
-
Beispiele 59–61
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 58 beschriebenen
Verfahrens hergestellt, ausgehend von dem geeigneten Methansulfonat
oder Halogenid. Die ESI/MS-Daten,
Ausbeuten und Reinheit sind in Tabelle 10 zusammengefaßt.
-
Tabelle
10. Beispiele 59–61
-
Beispiel 62
-
Darstellung von 2-[4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-nicotinsäure
-
A. Darstellung von 2-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-nicotinsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,5 g (2,5 mmol) 3-Piperidin-4-yl-1H-indol
und 0,65 g (3,25 mmol) 2-Brommethylnicotinsäureethylester. Nach der Standardreinigung
wurden 0,84 g (92% Ausbeute) 2-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-nicotinsäureethylester
erhalten.
-
B. Darstellung von 2-[4-(1-Pyridin-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-nicotinsäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,76 g (0,21 mmol) 2-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-nicotinsäureethylester
und 0,04 g (0,25 mmol) 3-Chlormethylpyridin-Hydrochlorid. Nach Standardreinigung
wurden 0,040 g (45% Ausbeute) der erwarteten Säure erhalten.
NMR (300
MHz, DMSO) δ =
1,65–1,79
(m, 2H), 1,99–2,10
(m, 2H), 2,55–2,76
(m, 2H), 2,89–2,96
(t, 1H), 3,08–3,12
(d, 2H), 4,24 (s, 2H), 5,40 (s, 2H), 6,98–7,12 (dt, 2H), 7,29–7,39 (m,
3H), 7,44–7,47
(d, 1H), 7,55–7,61
(m, 2H), 8,06–8,08
(d, 1H), 8,43–8,45
(m, 1H), 8,49–8,51
(m, 1H).
-
Beispiele 63–64
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 62 beschriebenen
Verfahrens hergestellt, ausgehend von dem geeigneten Methansulfonat
oder Halogenid. Die ESI/MS-Daten,
Ausbeuten und Reinheit sind in Tabelle 11 zusammengefaßt.
-
Tabelle
11. Beispiele 63–64
-
Beispiel 65
-
Darstellung von 3-{4-[1-(5-Chlor-thiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
A. Darstellung von 3-[4-(6-Fluor-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzosäuremethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,5 g (2,3 mmol) 6-Fluor-3-piperidin-4-yl-1H-indol
und 0,7 g (3 mmol) 3-Brommethylbenzoesäure. Nach der Standardreinigung
wurden 0,842 g (93% Ausbeute) 3-[4-(6-Fluor-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzosäuremethylester
erhalten.
-
B. Darstellung von 3-{4-[1-(5-Chlor-thiophen-2-ylmethyl}-6-fluor-1H-indol-3yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,07 g (0,19 mmol) 3-[4-(6-Fluor-1H-indol-3-yl)-piperidin-1- ylmethyl]-benzosäuremethylester
und 0,05 g (0,29 mmol) 2-Chlor-5-chlormethylthiophen. Nach Standardreinigung
wurden 0,01 g (10% Ausbeute) der erwarteten Säure erhalten.
NMR (300
MHz, DMSO) δ =
2,14–2,29
(m, 4H), 2,76–2,85
(m, 2H), 2,94–3,01
(m, 1H), 3,47–3,54
(m, 2H), 4,17 (s, 2H), 5,25 (s, 2H), 6,71–6,79 (m, 2H), 6,83–6,90 (dt,
1H), 6,97–7,03
(m, 2H), 7,48–7,56
(m, 2H), 7,75–7,78 (d,
1H), 8,09–8,12
(m, 2H), 8,15–8,19
(m, 1H).
-
Beispiele 66–67
-
Verbindungen
66 und 67 wurden unter Befolgung des in Beispiel 65 beschriebenen
Verfahrens hergestellt, ausgehend von dem geeigneten Methansulfonat
oder Halogenid. Die ESI/MS-Daten, Ausbeuten und Reinheit sind in
Tabelle 12 zusammengefaßt.
-
Tabelle
12. Beispiele 66–67
-
Beispiel 68
-
Darstellung von 2-Methoxy-5-{4-[1-(2-thiophen-2-ylethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
A. Darstellung von 5-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,5 g (2,5 mmol) 6-Fluor-3-piperidin-4-yl-1H-indol
und 0,88 g (3,2 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester.
Nach Standardreinigung wurden 0,83 g (91% Ausbeute) 5-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäureethylester
erhalten.
-
B. Darstellung von 2-Methoxy-5-{4-[1-(2-thiophen-2-ylethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,07 g (0,18 mmol) 5-[4-(1H-Indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäureethylester
und 0,05 g (0,25 mmol) 2-Thiophen-2-ylethyl-Methansulfonat. Nach der Standardreinigung
wurden 0,009 g (10% Ausbeute) der erwarteten Säure erhalten.
-
Beispiele 69 und 70
-
A. Darstellung von 5-[4-(6-Fluor-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,05 (2,2 mmol) 6-Fluor-3-piperidin-yl-1H-indol
und 0,8 g (2,9 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester.
Nach Standardreinigung wurden 0,91 g (100% Ausbeute) 5-[4-(6-Fluor-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäureethylester
erhalten.
-
B. Darstellung von 5-{4-[6-Fluor-1-(2-thiophen-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxy-benzoesäure und
5-{4-[6-Fluor-1-(2-morpholin-4-ylethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxybenzoesäure
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 1 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 0,07 g (0,17 mmol) 5-[4-(6-Fluor-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäureethylester.
-
-
Beispiel 71
-
Darstellung von 5-{4-[1-(2-[1,4]Dioxan-2-yl-thyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxybenzoesäure
-
A. Darstellung von 4-[1-(2-[1,4]Dioxan-2-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 0,47 g (1,2 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 0,62 g (2,9 mmol) 2-[1,4]Dioxan-2-ylethyl-methansulfonat. Die
Reaktionsmischung wurde bei 40°C
für 24
Stunden gerührt.
Nach Standardaufarbeitung und -reinigung wurden 0,47 g (51% Ausbeute)
4-[1-(2-[1,4]Dioxan-2-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester erhalten.
-
B. Darstellung von 1-(2-[1,4]Dioxan-2-ylethyl)-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 0,47 g (1,2 mmol) 4-[1-(2-[1,4]Dioxan-2-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 0,2 g (53% Ausbeute) 1-(2-[1,4]Dioxan-2-ylethyl)-3-piperidin-4-yl-1H-indol
erhalten.
-
C. Darstellung von 5-{4-[1-(2-[1,4]Dioxan-2-ylethyl)-1H-indol-3-yl]piperidin-1-ylmethyl}-2-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,06 g (0,19 mmol) 1-(2-[1,4]Dioxan-2-ylethyl)-3-piperidin-4-yl-1H-indol
und 0,071 g (0,26 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester. Nach Standardreinigung
wurden 0,019 g (65% Ausbeute) erhalten.
NMR (300 MHz, CDCl3) δ =
1,73–1,80
(m, 2H), 2,15–2,30
(m, 4H), 2,55–2,80
(m, 2H), 2,99–3,10
(m, 1H), 3,20–3,45
(m, 3H), 3,52–3,67
(m, 3H), 3,52–3,67
(m, 5H), 3,78–3,82
(m, 1H), 3,98 (s, 3H), 4,03–4,10
(m, 2H), 4,18–4,23
(t, 2H), 6,91 (s, 1H), 6,99–7,10
(m, 2H), 7,17–7,22
(t, 1H), 7,30–7,35
(m, 1H), 7,56–7,58
(m, 3H), 8,03–8,08
(m, 1H).
-
Beispiel 72
-
Darstellung von 3-{4-[1-(2-[1,4]Dioxan-2-yl-ethyl)-1H-indol-3-yl]piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 71 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 0,06 g (0,19 mmol) 1-(2-[1,4]Dioxan-2-yl-ethyl)-3-piperidin-4-yl-1H-indol
und 0,060 g (0,26 mmol) 3-Brommethylbenzoesäuremethylester. Nach Standardreinigung
wurden 0,037 g (75% Ausbeute) erhalten.
NMR (300 MHz, CDCl3) δ =
1,65–1,80
(m, 2H), 2,10–2,24
(m, 2H), 2,35–2,52
(m, 2H), 2,81–3,09
(m, 3H), 3,18–3,33
(m, 3H), 3,51–3,66
(m, 5H), 3,77–3,80
(m, 1H), 4,15–4,27
(m, 4H), 6,93 (s, 1H), 7,02–7,07
(t, 1H), 7,15–7,20
(t, 1H), 7,25–7,33
(m, 1H), 7,40–7,56
(m, 2H), 7,62–7,85
(m, 1H), 8,08–8,10
(d, 1H), 8,34 (s, 1H).
-
Beispiel 73
-
Darstellung von 2-Methoxy-5-[4-(1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 25 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 1,90 g (0,065 mol) 3-Piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
und 1,92 g (0,071 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester.
Nach Standardreinigung und Umkristallisation mit Ethanol 1,2 g (40%
Ausbeute) der erwarteten Säure.
Schmelzpunkt
242–243°C
NMR
(300 MHz, DMSO) δ =
1,6–1,73
(m, 2H), 1,91–1,95
(d, 2H), 2,09–2,17
(t, 2H), 2,32–2,82
(m, 1H), 2,88–2,92
(d, 2H), 3,49 (s, 2H), 3,80 (s, 3H), 5,52 (s, 2H), 6,92–6,96 (m,
1H), 6,93–7,00
(m, 1H), 7,06–7,12
(m, 3H), 7,23 (s, 1H), 7,36–7,38
(dd, 1H), 7,42–7,46
(dd, 1H), 7,46–7,48
(m, 1H), 7,49–7,51
(m, 1H), 7,53–7,55
(m, 1H), 7,56–7,59
(m, 2H).
-
Beispiel 74
-
Darstellung von 4-Brom-3-[4-(1-pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
A. Darstellung von 4-(1-Pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 11 g (40 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 8 g (48 mmol) 4-Chlormethylpyridin-Hydrochlorid, unter Rühren bei
Raumtemperatur für
18 Stunden. Nach Standardaufarbeitung wurden 11,8 g (81% Ausbeute)
4-(1-Pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 3-Piperidin-4-yl-1-pyridin-4-ylmethyl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 11,8 g (0,032 mmol) 4-(1-Pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Reinigung über
das Fumarat-Derivat, wie beschrieben in Beispiel 26 (Teil B), wurden
6 g (64% Ausbeute) 3-Piperidin-4-yl-1-pyridin-4-ylmethyl-1H-indol erhalten.
-
C. Darstellung von 4-Brom-3-[4-(1-piperidin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 1,5 g (50 mmol) 3-Piperidin-4-yl-1-pyridin-4-ylmethyl-1H-indol
und 1,7 g (55 mmol) 4-Brom-3-brommethylbenzoesäuremethylester. Nach Standardaufarbeitung
und Umkristallisation mit Ethylether wurden 1,7 g (68% Ausbeute)
der erwarteten Säure
erhalten.
Schmelzpunkt 167–168°C
NMR
(300 MHz, DMSO) δ =
1,66–1,77
(m, 2H), 1,91–2,02
(m, 2H), 2,25–2,33
(t, 2H), 2,80–2,97
(m, 3H), 3,65 (s, 2H), 5,42 (s, 2H), 6,98–7,10 (m, 4H), 7,31–7,33 (m,
2H), 7,59–7,62
(d, 1H), 7,68–7,81
(m, 2H), 8,09 (s, 1H), 8,45–8,48
(m, 2H).
-
Beispiel 75
-
Darstellung von 2-Methoxy-5-{4-[1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 1 g (3,2 mmol) 3-Piperidin-4-yl-1-(2-thiophen-3-ylethyl)-1H-indol
und 1,15 g (4,2 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester.
Nach Standardaufarbeitung und -reinigung wurden 1,16 g (76% Ausbeute)
der erwarteten Säure
erhalten.
Schmelzpunkt 219–220°C
NMR
(300 MHz, DMSO) δ =
1,58–1,70
(m, 2H), 1,89–1,93
(d, 2H), 2,11–2,19
(t, 2H), 2,70–2,78
(m, 2H), 2,89–2,93
(d, 1H), 3,02–3,07
(m, 2H), 3,50 (s, 2H), 3,80 (s, 3H), 4,30–4,35 (m, 2H), 6,95–7,12 (m,
5H), 7,18 (s, 1H), 7,43–7,46
(m, 3H), 7,53–7,55
(d, 1H), 7,59 (s, 1H).
-
Beispiel 76
-
Darstellung von 3-{4-[1-(2-Morpholin-4-ylethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
A. Darstellung von 4-[1-(2-Morpholin-4-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 11 g (40 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 9 g (48 mmol) 4-(2-Chlorethyl)-morpholin-Hydrochlorid. Die Reaktionsmischung
wurde bei Raumtemperatur für
18 Stunden gerührt.
Nach Standardaufarbeitung und -reinigung wurden 13,5 g (88% Ausbeute)
4-[1-(2-Morpholin-4-ylethyl)-1H-indoL-3-yl]-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 1-(2-Morpholin-4-ylethyl)-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 13,5 g (35 mmol) 4-[1-(2-Morpholin-4-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 9,5 g (87% Ausbeute) 1-(2-Morpholin-4-ylethyl)-3-piperidin-4-yl-1H-indol
erhalten.
-
C. Darstellung von 3-{4-[1-(2-Morpholin-4-ylethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,4 g (7,5 mmol) 1-(2-Morpholin-4-ylethyl)-3-piperidin-4-yl-1H-indol
und 1,8 g (7,8 mmol) 3-Brommethylbenzoesäuremethylester. Nach Standardaufarbeitung
und -reinigung wurden 0,75 g (22% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
186–191 °C
NMR
(300 MHz, DMSO) δ =
1,91–2,10
(m, 4H), 2,42–2,51
(m, 4H), 2,66–2,82
(m, 4H), 3,16–3,26
(m, 3H), 3,54–3,58
(m, 4H), 4,15–4,26
(m, 4H), 6,96–7,00
(t, 1H), 7,09–7,14
(t, 1H), 7,18 (s, 1H), 7,42–7,45
(m, 2H), 7,54–7,64
(m, 2H), 7,84–7,86
(m, 1H), 7,95–7,98
(d, 1H), 8,11 (s, 1H).
-
Beispiel 77
-
Darstellung von 2-[2-(4-{1-[2-(Benzo[1,3]dioxol-5-yloxy)-ethyl]-1H-indol-3-yl}-piperidin-1-yl)-ethoxy]-benzoesäure
-
A. Darstellung von 4-{1-[2-(Benzo[1,3]dioxol-5-yloxy)-ethyl]-1H-indol-3-yl}-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahren hergestellt, ausgehend von 11 g (40 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 9,6 g (48 mmol) 5-(2-Chlorethoxy)-benzo[1,3]dioxol. Die Mischung
wurde bei Raumtemperatur für
18 Stunden gerührt.
Nach Standardaufarbeitung und -reinigung wurden 10,5 g (60% Ausbeute)
4-{1-[2-(Benzo[1,3]dioxol-5-yloxy)-ethyl]-1H-indol-3-yl}-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 1-[2-(Benzo[1,3]dioxol-5-yloxy)-ethyl]-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 12,5 g (0,028 mmol) 4-{1-[2-(Benzo[1,3]dioxol-5-yloxy)-ethyl]-1H-indol-3-yl}-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 9 g (87% Ausbeute) 1-[2-(Benzo[1,3]dioxol-5-yloxy)-ethyl]-3-piperidin-4-yl-1H-indol erhalten.
-
C. Darstellung von 2-[2-(4-(1-[2-(Benzo[1,3]dioxol-5-yloxy)-ethyl)-1H-indol-3-yl}-piperidin-1-yl)-ethoxyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,3 g (6,2 mmol) 1-[2-(Benzo[1,3]dioxol-5-yloxy)-ethyl]-3-piperidin-4-yl-1H-indol
und 1,5 g (7,1 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Nach Standardaufarbeitung
und Umkristallisation mit Methanol wurden 1,6 g (49% Ausbeute) der
erwarteten Säure
erhalten.
Schmelzpunkt 123–125°C
NMR
(300 MHz, DMSO) δ =
1,85–2,06
(m, 4H), 2,61–2,69
(m, 2H), 2,89–2,98
(m, 3H), 3,16–3,24
(m, 2H), 4,17–4,21
(m, 2H), 4,42–4,49
(m, 4H), 5,93 (s, 2H), 6,30–6,33
(dd, 1H), 6,56–6,57
(m, 1H), 6,76–6,78
(d, 1H), 6,97–7,04
(m, 2H), 7,11–7,16
(t, 1H), 7,20–7,24
(m, 2H), 7,36–7,41
(m, 1H), 7,48–7,55
(m, 2H), 7,64–7,66
(d, 1H).
-
Beispiel 78
-
Darstellung von 5-{4-[6-Fluor-1-(2-thiophen-3-yl-ethyl}-1H-indol-3-yl]-piperidin-1-ylmethyl)-2-methoxy-benzoesäure
-
A. Darstellung von 4-[6-Fluor-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 4 g (13,8 mmol) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
(Beispiel 24, Teil A) und 3,3 g (16 mmol) 2-Thiophen-3-ylethyl-Methansulfonat. Die
Reaktionsmischung wurde bei 60°C
für 3 Stunden
gerührt.
Nach Standardaufarbeitung wurden 5,6 g (100% Ausbeute) 4-[6-Fluor-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 6-Fluor-3-piperidin-4-yl-1-(2-thiophen-3-yl-ethyl)-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 5,6 g (13,8 mmol) 4-[6-Fluor-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 4,5 g (99% Ausbeute) 6-Fluor-3-piperidin-4-yl-1-(2-thiophen-3-yl-ethyl)-1H-indol
erhalten.
-
C. Darstellung von 5-{4-[6-Fluor-1-(2-thiophen-3-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,3 g (6,9 mmol) 6-Fluor-3-piperidin-4-yl-1-(2- thiophen-3-yl-ethyl)-1H-indol
und 2 g (7,5 mmol) 3-Brommethyl-4-methoxybenzoesäureethylester. Nach Standardaufarbeitung
und Umkristallisation mit Methanol wurde 1 g (29% Ausbeute) der
erwarteten Säure
erhalten.
Schmelzpunkt 228–229°C
NMR
(300 MHz, DMSO) δ =
1,56–1,67
(m, 2H), 1,87–1,91
(m, 2H), 2,10–2,17
(t, 2H), 2,68–2,76
(m, 1H), 2,88–2,92
(d, 2H), 2,99–3,05
(t, 2H), 3,50 (s, 2H), 3,80 (s, 3H), 4,29–4,31 (m, 2H), 6,78–6,85 (m,
1H), 6,99–7,02 (dd,
1H), 7,07–7,10
(m, 2H), 7,16–7,19
(m, 1H), 7,28–7,32
(m, 2H), 7,38–7,49
(m, 2H), 7,51–7,54
(m, 1H), 7,58–7,59
(m, 1H).
-
Beispiel 79
-
Darstellung von 5-{4-[1-(5-Chlorthiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxybenzoesäure
-
A. Darstellung 4-[1-(5-Chlorthiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 2,2 g (1,5 mmol) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
(Beispiel 24, Teil A) und 1,1 ml (8,8 mmol) 2-Chlor-5-chlormethylthiophen.
Die Reaktionsmischung wurde bei Raumtemperatur für 18 Stunden gerührt. Nach
Standardaufarbeitung wurden 2,2 g (68% Ausbeute) 4-[1-(5-Chlorthiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 1-(5-Chlor-thiophen-2-ylmethyl)-6-fluor-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 4,4 g (10,4 mmol) 4-[1-(5-Chlorthiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 2,4 g (67% Ausbeute) 1-(5-Chlor-thiophen-2-ylmethyl)-6-fluor-3-piperidin-4-yl-1H-indol
erhalten.
-
C. Darstellung von 5-(4-[1-(5-Chlor-thiophen-2-ylmethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,4 g (6,9 mmol) 1-(5-Chlor-thiophen-2-ylmethyl)-6-fluor-3-piperidin-4-yl-1H-indol
und 2 g (7,5 mmol) 3-Brommethyl-4-methoxybenzoesäureethylester. Nach Standardaufarbeitung
wurden 0,7 g (20% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
232–236°C
NMR
(300 MHz, DMSO) δ =
1,65–1,73
(m, 2H), 1,90–1,94
(d, 2H), 2,15–2,22
(t, 2H), 2,70–2,78
(m, 1H), 2,91–2,95
(m, 2H), 3,53 (s, 2H), 3,80 (s, 3H), 5,45 (s, 2H), 6,83–6,89 (t,
1H), 6,95–7,08
(m, 3H), 7,23 (s, 1H), 7,40–7,44
(m, 2H), 7,54–7,59
(m, 2H).
-
Beispiel 80
-
Darstellung von 5-[4-(6-Fluor-2-furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäure
-
A. Darstellung von 4-(6-Fluor-1-furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 4 g (13,8 mmol) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
(Beispiel 24, Teil A) und 17 ml (16 mmol) einer frisch hergestellten
0,94M Lösung
von 3-Brommethylfuran in Ethylether. Die Reaktionsmischung wurde
bei Raumtemperatur für
18 Stunden gerührt.
Nach Standardaufarbeitung wurden 5,3 g (99% Ausbeute) 4-(6-Fluor-1-furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 6-Fluor-1-furan-3-ylmethyl-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 5,3 g (13,7 mmol) 4-(6-Fluor-1-furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 3,5 g (86% Ausbeute) 6-Fluor-1-furan-3-ylmethyl-3-piperidin-4-yl-1H-indol
erhalten.
-
C. Darstellung von 5-[4-(6-Fluor-1-furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,1 g (6,9 mmol) 6-Fluor-1-furan-3-ylmethyl-3-piperidin-4-yl-1H-indol
und 2 g (7,5 mmol) 3-Brommethyl-4-methoxybenzoesäureethylester. Nach Standardaufarbeitung
wurden 0,9 g (28% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
228–229°C
NMR
(300 MHz, DMSO) δ =
1,56–1,73
(m, 2H), 1,76–1,89
(m, 2H), 2,11–2,18
(m, 2H), 2,62–2,82
(m, 1H), 2,90–2,93
(m, 2H), 3,51 (s, 2H), 5,11 (s, 2H), 6,40 (s, 1H), 6,76–6,92 (m,
1H), 7,07–7,10
(d, 1H), 7,22 (s, 1H), 7,38–7,43
(m, 2H), 7,46–7,59
(m, 3H), 7,72 (s, 1H).
-
Beispiel 81
-
Darstellung von 3-{4-[1-(2-Pyridin-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
A. Darstellung von 4-[1-(2-Pyridin-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 11 g (40 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 8,6 g (48 mmol) eines frisch hergestellten 2-Pyridin-2-yl-ethyl-Methansulfonats.
Die Reaktionsmischung wurde bei 60°C für 18 Stunden gerührt. Nach
Standardaufarbeitung wurden 3,2 g (21% Ausbeute) 4-[1-(2-Pyridin-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 3-Piperidin-4-yl-1-(2-pyridin-2-yl-ethyl)-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 8,8 g (12,9 mmol) 4-[1-(2-Pyridin-2-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 3,4 g (87% Ausbeute) 3-Piperidin-4-yl-1-(2-pyridin-2-yl-ethyl)-1H-indol
erhalten.
-
C. Darstellung von 3-{4-[1-(2-Pyridin-2-yl-ethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 3,4 g (11 mmol) 3-Piperidin-4-yl-1-(2-pyridin-2-yl-ethyl)-1H-indol und
2,7 g (11,5 mmol) 3-Brommethylbenzoesäuremethylester. Nach Standardaufarbeitung
und Umkristallisation mit Dichlormethan/Methanol wurden 1,4 g (29%
Ausbeute) der erwarteten Säure
erhalten.
Schmelzpunkt 141–142°C
NMR(300
MHz, DMSO) δ =
1,55–1,72
(m, 2H), 1,86–1,90
(m, 2H), 2,11–2,19
(t, 2H), 2,69–2,74
(m, 1H), 2,88–2,92
(m, 2H), 3,15–3,20
(t, 2H), 3,59 (s, 2H), 4,45–4,50
(t, 2H), 6,94–7,24
(m, 5H), 7,37–7,67
(m, 5H), 7,83–7,86
(m, 1H), 7,94 (s, 1H), 8,51–8,54
(m, 1H).
-
Beispiel 82
-
Darstellung von 5-[4-(6-Fluor-2-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxy-benzoesäure
-
A. Darstellung von 4-(6-Fluor-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 4 g (13,8 mmol) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
(Beispiel 24, Teil A) und 16 ml (16 mmol) einer frisch hergestellten
1M Lösung
von 2-Brommethylthiophen in Ethylether. Die Reaktionsmischung wurde
bei Raumtemperatur für
18 Stunden gerührt.
Nach Standardaufarbeitung wurden 5,42 g (100% Ausbeute) 4-(6-Fluor-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 6-Fluor-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 9,4 g (13,8 mmol) 4-(6-Fluor-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 2,9 g (69% Ausbeute) 6-Fluor-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
erhalten.
-
C. 5-[4-(6-Fluor-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäure
-
Dieser
Verbindung wurde Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,9 g (9,2 mmol) 6-Fluor-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol und 2,7
g (11,5 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester. Nach Standardaufarbeitung
wurden 1,2 g (27% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
245–246°C
NMR
(300 MHz, DMSO) δ =
1,60–1,68
(m, 2H), 1,89–1,93
(m, 2H), 2,10–2,18
(t, 2H), 2,65–2,80
(m, 1H), 2,89–2,93
(d, 2H), 3,50 (s, 2H), 3,80 (s, 3H), 5,50 (s, 2H), 6,81–6,87 (m,
1H), 6,94–6,96
(m, 1H), 7,09–7,13
(m, 2H), 7,23 (s, 1H), 7,36–7,44
(m, 3H), 7,52–7,58
(m, 2H).
-
Beispiel 83
-
Darstellung von 3-[4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
A. Darstellung von 4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Dieser
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 9,4 g (34,4 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäureethylester
und 40 ml (40 mmol) einer frisch hergestellten 1M Lösung von
2-Brommethylfuran
in Ethylether. Die Reaktionsmischung wurde bei Raumtemperatur für 18 Stunden
gerührt.
Nach Standardaufarbeitung wurden 13,2 g (100% Ausbeute) 4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 1-Furan-2-ylmethyl-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 13,2 g (37 mmol) 4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurde 10,2 g (98% Ausbeute) 1-Furan-2-ylmethyl-3-piperidin-4-yl-1H-indol
erhalten.
-
C. 3-[4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Dieser
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,8 g (10 mmol) 1-Furan-2-ylmethyl-3-piperidin-4-yl- 1H-indol und 2,5
g (11 mmol) 3-Brommethylbenzoesäuremethylester.
Nach Standardaufarbeitung wurden 1,5 g (36% Ausbeute) der erwarteten
Säure erhalten.
Schmelzpunkt
154–155°C
NMR
(300 MHz, DMSO) = 1,61–1,76
(m, 2H), 1,90–1,95
(m, 2H), 2,12–2,20
(t, 2H), 2,72–2,80
(m, 1H), 2,89–2,92
(m, 2H), 3,59 (s, 2H), 5,33 (s, 2H), 6,37–6,44 (m, 2H), 6,96–7,01 (m,
1H), 7,08–7,13
(m, 2H), 7,44–7,57
(m, 5H), 7,83–7,85
(m, 1H), 7,93 (s, 1H).
-
Beispiel 84
-
Darstellung von 2-(2-{4-[1-(2-[1,4]Dioxan-2-ylethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,58 g (1,84 mmol) 1-(2-[1,4]Dioxan-2-ylethyll-3-piperidin-4-yl-1H-indol
(Beispiel 71, Teil B) und 0,51 g (2,30 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester.
Nach Standardaufarbeitung und Reinigung durch Flashchromatographie über Silicagel
wurden 0,18 g (20% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
139–140°C
NMR
(300 MHz, DMSO) = 1,70–1,82
(m, 2H), 1,91–2,08
(m, 4H), 2,66–2,73
(m, 2H), 2,93–3,10
(m, 3H), 3,11–3,27
(m, 4H), 3,44–3,64
(m, 4H), 3,76–3,79
(m, 1H), 4,18–4,22
(m, 2H), 4,42–4,46
(m, 2H), 6,97–7,04
(m, 2H), 7,12–7,15
(m, 2H), 7,22–7,25
(m, 1H), 7,37–7,41
(m, 2H), 7,52–7,54
(d, 1H), 7,64–7,66
(d, 1H).
-
Beispiel 85
-
Darstellung von 5-[4-(-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 1,9 g (6,5 mmol) 1-Furan-2-ylmethyl-3-piperidin-4-yl-1H-indol (Beispiel
83, Teil B) und 1,9 g (7,1 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester. Nach – Standaradaufarbeitung
und Umkristallisation mit Ethanol wurden 0,5 g (16% Ausbeute) der
erwarteten Säure
erhalten.
Schmelzpunkt 237–238°C
NMR
(300 MHz, DMSO) = 1,65–1,75
(m, 2H), 1,90–1,95
(m, 2H), 2,11–2,18
(t, 2H), 2,68–2,83
(m, 1H), 2,89–2,93
(m, 2H), 3,50 (s, 2H), 3,81 (s, 3H), 5,33 (s, 2H), 6,37–6,44 (m,
2H), 6,96–7,17
(m, 4H), 7,42–7,59 (m,
5H).
-
Beispiel 86
-
Darstellung von 5-[4-{1-Furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäure
-
A. Darstellung von 4-(1-Furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 4 g (13,7 mmol) 4-(1H-Indol-3-yl)-piperidin-1- carbonsäureethylester
und 16 ml (16 mmol) einer frisch hergestellten 1M Lösung von
3-Brommethylfuran
in Ethylether. Die Reaktionsmischung wurde bei Raumtemperatur für 18 Stunden
gerührt.
Nach Standardaufarbeitung wurden 5,3 g (99% Ausbeute) 4-(1-Furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 1-Furan-3-ylmethyl-3-piperidin-4-yl-1H-indol
-
Dieser
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 7,3 g (20 mmol) 4-(1-Furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 5,6 g (99% Ausbeute) 1-Furan-3-ylmethyl-3-piperidin-4-yl-1H-indol
erhalten.
-
C. 5-[4-{1-Furan-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 1,9 g (6,5 mmol) 1-Furan-3-ylmethyl-3-piperidin-4-yl-1H-indol und 1,9
g (7,1 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester. Nach Standardaufarbeitung
wurden 1,2 g (42% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
253–255°C
NMR
(300 MHz, DMSO) = 1,61–1,78
(m, 2H), 1,91–1,95
(m, 2H), 2,08–2,12
(m, 2H), 2,72–2,82
(m, 1H), 2,91–2,94
(m, 2H), 3,52–3,62
(m, 2H), 3,81 (s, 3H), 5,14 (s, 2H), 6,38 (s, 1H), 6,95–7,00 (t,
1H), 7,08–7,11
(m, 2H), 7,21 (s, 1H), 7,44–7,60
(m, 5H), 7,69 (s, 1H).
-
Beispiel 87
-
Darstellung von 3-{4-[5-Methoxy-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
A. Darstellung von 5-Methoxy-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 1 (Teile A und
B) beschriebenen Verfahrens hergestellt, ausgehend von 5,9 g (40
mmol) 5-Methoxyindol und 15,5 g (100 mmol) 4-Piperidon. In diesem
Fall lief die Hydrierung für
24 Stunden bei 30 psi ab und der verwendete Katalysator war Platin(IV)-oxid.
6,8 g (7% Ausbeute) 5-Methoxy-3-piperidin-4-yl-1H-indol wurden erhalten.
-
B. Darstellung von 4-(5-Methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil A) beschriebenen
Verfahrens hergestellt, ausgehend von 5,8 g (25 mmol) 5-Methoxy-3-piperidin-4-yl-1H-indol.
Nach Standardaufarbeitung wurden 6,9 g (91% Ausbeute) 4-(5-Methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
C. Darstellung von 4-[5-Methoxy-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 8,7 g (28,6 mmol) 4-(5-Methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
und 6,9 g (33,4 mmol) 2-Thiophen-3-ylethyl-Methansulfonat. Die Reaktionsmischung
wurde bei Raumtemperatur für
18 Stunden gerührt.
-
Nach
Standardaufarbeitung wurden 6,7 g (57% Ausbeute) 4-[5-Methoxy-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
erhalten.
-
D. Darstellung von 5-Methoxy-3-piperidin-4-yl-1-(2-thiophen-3-ylethyl)-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 6,6 g (16 mmol) 4-[5-Methoxy-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 5,3 g (97% Ausbeute) 5-Methoxy-3-piperidin-4-yl-1-(2-thiophen-3-ylethyl)-1H-indol
erhalten.
-
E. Darstellung von 3-{4-[5-Methoxy-1-(2-thiaphen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 1,7 g (5 mmol) 5-Methoxy-3-piperidin-4-yl-1-(2-thiophen-3-ylethyl)-1H-indol
und 1,3 g (5,5 mmol) 3-Brommethylbenzoesäuremethylester. Nach Standardaufarbeitung
wurden 0,8 g (34% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
217–218°C
NMR
(300 MHz, DMSO) = 1,60–1,67
(m, 2H), 1,88–1,91
(m, 2H), 2,12–2,20
(t, 2H), 2,64–2,72
(m, 1H), 2,88–2,92
(m, 2H), 2,99–3,04
(m, 2H), 3,59 (s, 2H), 3,75 (s, 3H), 4,25–4,30 (m, 2H), 6,71–6,75 (m,
1H), 6,96–7,02
(m, 3H), 7,14–7,16
(m, 1H), 7,31–7,34
(d, 1H), 7,42–7,48
(m, 2H), 7,56–7,58
(d, 1H), 7,83–7,85
(d, 1H), 7,93 (s, 1H).
-
Beispiel 88
-
Darstellung von 2-(2-{4-[5-Methoxy-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
Dieser
Verbindung wurde unter Befolgung des in Beispiel 87 (Teil E) beschriebenen
Verfahrens hergestellt, ausgehend von 1,7 g (5 mmol) 5-Methoxy-3-piperidin-4-yl-1-(2-thiophen-3-ylethyl)-1H-indol
und 1,2 g (5,5 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Nach Standardaufarbeitung
und Reinigung durch Flashchromatographie wurden 0,6 g (24% Ausbeute)
der erwarteten Säure
erhalten.
Schmelzpunkt 145–148°C
NMR
(300 MHz, DMSO) = 1,94–2,03
(m, 4H), 2,64–2,67
(m, 2H), 2,82–2,87
(m, 1H), 2,98–3,05
(m, 4H), 3,21–3,25
(m, 2H), 3,80 (s, 3H), 4,27–4,32
(m, 2H), 4,46 (s, 2H), 6,73–6,77
(dd, 1H), 6,99–7,04
(m, 3H), 7,13 (s, 1H), 7,19–7,22
(m, 2H), 7,33–7,37
(m, 2H), 7,40–7,51
(m, 1H), 7,533–7,58
(m, 1H)
-
Beispiel 89
-
Darstellung von 2-{2-[4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
A. Darstellung von 4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 9,1 g (30 mmol) 4-(5-Methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
(Beispiel 87, Teil B) und 50 ml (50 mmol) einer frisch hergestellten
1M Lösung
von 2-Brommethylthiophen in Ethylether. Die Reaktionsmischung wurde
bei Raumtemperatur für
18 Stunden gerührt.
Nach Standardaufarbeitung wurden 7,7 g (65% Ausbeute) 4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 5-Methoxy-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 7,7 g (19 mmol) 4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 5 g (81% Ausbeute) 5-Methoxy-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
erhalten.
-
C. Darstellung von 2-{2-[4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,4 g (7,4 mmol) 5-Methoxy-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
und 1,8 g (8 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Nach Standardaufarbeitung
wurden 1,3 g (36% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
150–151 °C
NMR
(300 MHz, DMSO) = 1,94–2,10
(m, 4H), 2,63–2,70
(m, 2H), 2,86–2,98
(m, 3H), 3,22–3,26
(m, 2H), 3,79 (s, 3H), 4,44–4,47
(m, 2H), 4,80–5,25
(m, 1H), 5,50 (s, 2H), 6,74–6,77
(dd, 1H), 6,93–7,24
(m, 6H), 7,35–7,41 (m,
2H), 7,50–7,53
(dd, 1H).
-
Beispiel 90
-
Darstellung von 3-[4-(5-Methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 2,4 g (7,4 mmol) 5-Methoxy-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
(Beispiel 89, Teil C) und 1,9 g (8 mmol) 3-Brommethylbenzoesäuremethylester. Nach Standardaufarbeitung
wurden 1,4 g (41% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
185–186°C
NMR
(300 MHz, DMSO) = 1,60–1,76
(m, 2H), 1,91–1,95
(m, 2H), 2,16–2,23
(m, 2H), 2,70–2,78
(m, 1H), 2,91–2,94
(m, 2H), 3,56 (s, 2H), 3,74 (s, 3H), 5,47 (s, 2H), 6,72–6,76 (dd,
1H), 6,92–6,95
(m, 1H), 7,01–7,02 (m,
1H), 7,06–7,07
(m, 1H), 7,20 (s, 1H), 7,33–7,60
(m, 4H), 7,84–7,86
(d, 1H), 7,94 (s, 1H).
-
Beispiel 91
-
Darstellung von 2-Methoxy-5-{4-[5-methoxy-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 1,95 g (5,7 mmol) 5-Methoxy-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
(Beispiel 87, Teil E) und 1,7 g (6,2 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester.
Nach Standardaufarbeitung wurde 1 g (35% Ausbeute) der erwarteten
Säure erhalten.
Schmelzpunkt
229–230°C
NMR
(300 MHz, DMSO) = 1,55–1,66
(m, 2H), 1,87–1,92
(m, 2H), 2,08–2,16
(m, 2H), 2,53–2,74
(m, 1H), 2,87–2,91
(m, 2H), 2,99–3,04
(m, 2H), 3,48 (s, 2H), 3,75 (s, 3H), 3,81 (s, 3H), 4,25–4,30 (t,
2H), 6,72–6,75
(d, 1H), 6,96–7,01
(m, 3H), 7,07–7,10
(d, 1H), 7,16 (s, 1H), 7,32–7,34
(d, 1H), 7,42–7,45
(m, 3H), 7,58 (s, 1H).
-
Beispiel 92
-
Darstellung von 2-{2-[4-(1-Furan-3-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
A. Darstellung von 4-(1-Furan-3-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens hergestellt, ausgehend von 8 g (26,4 mmol) 4-(5-Methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
(Beispiel 87, Teil B) und 30 ml (30 mmol) einer frisch hergestellten
1M Lösung
von 3-Brommethylfuran in Ethylether. Die Reaktionsmischung wurde
bei Raumtemperatur für
18 Stunden gerührt.
Nach Standardaufarbeitung wurden 9,9 g (99% Ausbeute) 4-(1-Furan-3-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 1-Furan-3-ylmethyl-5-methoxy-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 9,9 g (25,8 mmol) 4-(1-Furan-3-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 7,5 g (94% Ausbeute) 1-Furan-3-ylmethyl-5-methoxy-3-piperidin-4-yl-1H-indol
erhalten.
-
C. Darstellung von 2-{2-[4-(1-Furan-3-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 3,7 g (11,9 mmol) 1-Furan-3-ylmethyl-5-methoxy-3-piperidin-4-yl-1H-indol
und 3 g (13,9 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester. Nach Standardaufarbeitung
wurden 3,2 g (57% Ausbeute) der erwarteten Säure erhalten.
Schmelzpunkt
153–154°C
NMR
(300 MHz, DMSO) = 1,86–2,02
(m, 4H), 2,63–2,69
(m 2H), 2,79–2,99
(m, 3H), 3,21–3,26
(m, 2H), 3,79 (s, 3H), 4,43–4,47
(m, 2H), 5,12 (s, 2H), 6,38 (s, 1H), 6,73–6,76 (dd, 1H), 6,99–7,04 (t,
1H), 7,15–7,16
(m, 2H), 7,22–7,25
(d, 1H), 7,35–7,40
(m, 2H), 7,50–7,52
(d, 1H), 7,56 (s, 1H), 7,69 (s, 1H).
-
Beispiel 93
-
Darstellung von 3-[4-(1-Furan-3-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 3,7 g (11,9 mmol) 1-Furan-3-ylmethyl-5-methoxy-3-piperidin-4-yl-1H-indol
(Beispiel 92, Teil B) und 3 g (13 mmol) 3-Brommethylbenzoesäuremethylester. Nach Standardaufarbeitung
wurden 2,4 g (45% Ausbeute) der erwarteten Säure erhalten. In diesem Falle
wurde ein p-Toluolsulfonatderivatsalz
hergestellt, was 2,9 g eines weißen Feststoffes lieferte.
Schmelzpunkt
214–215°C
NMR
(300 MHz, DMSO) = 1,78–1,91
(m, 2H), 2,13–2,18
(m, 2H), 2,28 (s, 3H), 2,94–3,12
(m, 3H), 3,46–3,49 (d,
2H), 3,75 (s, 3H), 4,45 (s, 2H), 5,12 (s, 2H), 6,34 (s, 1H), 6,76–6,79 (dd,
1H), 7,10–7,18
(s, 4H), 7,39–7,66 (m,
6H), 7,79–7,81
(d, 1H), 8,04–8,06
(d, 1H), 8,20 (s, 1H).
-
Beispiel 94
-
Darstellung von 2-[4-(1-Furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,05 g (0,18 mmol) 1-Furan-2-ylmethyl-3-piperidin-4-yl-1H-indol (Beispiel
83, Teil B) und 0,056 g (0,23 mmol) 2-Brommethylbenzoesäureethylester. Nach Standardaufarbeitung
und Reinigung unter Verwendung einer C18-Chromatographiesäule wurden
0,014 g (19% Ausbeute) der erwarteten Säure erhalten.
NMR (300
MHz, CDCl3) = 1,91–2,03 (m, 2H), 2,13–2,18 (m,
2H), 2,55–2,68
(m, 2H), 2,95–3,30
(m, 1H), 3,26–3,30
(m, 2H), 3,98 (s, 2H), 5,18 (m, 2H), 6,24–6,25 (d, 1H), 6,30–6,31 (m,
1H), 6,90 (s, 1H), 7,08–7,13
(t, 1H), 7,20–7,50
(m, 6H), 7,54–7,56
(d, 1H), 8,19–8,22
(dd, 1H).
-
Beispiel 95
-
Darstellung von 2-[4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 0,3 g (1,0 mmol) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
und 2,7 ml (1,6 mmol) einer frisch hergestellten 0,61M Lösung von
2-Brommethylfuran in Ethylether. Nach Standardaufarbeitung wurden
0,38 g (100% Ausbeute) 4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 6-Fluor-1-furan-2-ylmethyl-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 0,38 g (1,1 mmol) 4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 0,27 g (89% Ausbeute) 6-Fluor-1-furan-2-ylmethyl-3-piperidin-4-yl-1H-indol.
-
C. Darstellung von 2-[4-(6-Fluor-1-furan-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,05 g (0,17 mmol) und 0,054
g (0,22 mmol) 2-Brommethylbenzoesäureethylester.
Nach Standardaufarbeitung und Reinigung unter Verwendung einer C18-Chromatographiesäule wurden
0,021 g (29% Ausbeute) der erwarteten Säure erhalten.
NMR (300
MHz, CDCl3) = 1,95–2,04 (m, 2H), 2,18–2,22 (m,
2H), 2,72–2,88
(m, 3H), 3,37–3,41
(m, 2H), 4,10 (s, 2H), 5,14 (s, 2H), 6,27–6,28 (d, 1H), 6,31–6,33 (dd,
1H), 6,83–6,90
(td, 1H), 6,93 (s, 1H), 7,07–7,11
(dd, 1H), 7,23–7,26
(d, 1H), 7,36–7,53
(m, 4H), 8,11–8,14
(dd, 1H).
-
Beispiele 96 und 97
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 95 beschriebenen
Verfahrens hergestellt. Die ESI/MS-Daten, Ausbeuten und Reinheit
sind in Tabelle 13 zusammengefaßt.
-
Tabelle
13. Beispiele 96–97
-
Beispiel 98
-
Darstellung von 4-Methoxy-2-[4-(5-methoxy-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,06 g (0,19 mmol) 5-Methoxy-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
(Beispiel 89, Teil B) und 0,064 g (0,23 mmol) 2-Brommethyl-4-methoxybenzoesäuremethylester.
Nach Standardaufarbeitung und Reinigung durch Chromatographie unter
Verwendung einer C18-Säule
wurden 0,018 g (19% Ausbeute) der erwarteten Säure erhalten.
NMR (300
MHz, DMSO) = 1,58–1,72
(m, 2H), 2,07–2,11
(m, 2H), 2,69–2,77
(t, 2H), 2,82–3,11
(m, 3H), 3,76 (s, 3H), 3,81 (s, 3H), 4,02 (s, 2H), 5,49 (s, 2H),
6,76–6,79
(dd, 1H), 6,92–6,99
(m, 3H), 7,06 (s, 1H), 7,35–7,05
(m, 2H), 7,88–7,91
(d, 1H).
-
Beispiele 99–100
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 98 beschriebenen
Verfahrens hergestellt. Die ESI/MS-Daten, Ausbeuten und Reinheit
sind in Tabelle 14 zusammengefaßt.
-
Tabelle
14. Beispiele 99–100
-
Beispiel 101
-
Darstellung von 2-[4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-ylmethyl]-4-methoxybenzoesäure
-
A. Darstellung von 4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 0,3 (1,0 mmol) 4-(5-Methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
und 2,11 ml (1,3 mmol) einer frisch hergestellten 0,6 1M Lösung von
2-Brommethylfuran in Ethylether. Nach Standardaufarbeitung wurden
0,38 g (100% Ausbeute) 4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 1-Furan-2-ylmethyl-5-methoxy-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 0,38 g (1,1 mmol) 4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 0,27 g (86% Ausbeute) 1-Furan-2-ylmethyl-5-methoxy-3-piperidin-4-yl-1H-indol
erhalten.
-
C. Darstellung von 2-[4-(1-Furan-2-ylmethyl-5-methoxy-1H-indol-3-yl)-piperidin-1-ylmethyl]-4-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,05 g (0,17 mmol) 1-Furan-2-ylmethyl-5-methoxy-3-piperidin-4-yl-1H-indol
und 0,057 g (0,22 mmol) 2-Brommethyl-4-methoxybenzoesäuremethylester. Nach Standardaufarbeitung
und Reinigung durch Chromatographie unter Verwendung einer C18-Säule wurden
0,029 g (36% Ausbeute) der erwarteten Säure erhalten.
NMR (300
MHz, DMSO) = 1,64–1,75
(m, 2H), 2,07–2,12
(m, 2H), 2,75–2,83
(m, 2H), 2,88– 3,00
(m, 1H), 3,12–3,16
(d, 2H), 3,76-(s, 3H), 3,82 (s, 3H), 4,08 (s, 2H), 5,29 (s, 2H),
6,37–6,43
(m, 2H), 6,76–6,80
(dd, 1H), 6,99–7,07
(m, 2H), 7,18 (s, 1H), 7,40–7,43
(d, 1H), 7,55 (s, 1H), 7,90–7,93
(s, 1H).
-
Beispiele 102–105
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 101 beschriebenen
Verfahrens hergestellt. Die ESI/MS-Daten, Ausbeuten und Reinheiten
sind in Tabelle 15 zusammengefaßt.
-
Tabelle
15. Beispiele 102–105
-
Beispiel 106
-
Darstellung von 4-Methoxy-2-{4-[4-methoxy-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]piperidin-1-ylmethyl}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,05 g (0,16 mmol) 5-Methoxy-3-piperidin-4-yl-1-(2-thiophen-3-ylethyl)-1H-indol
(Beispiel 87, Teil D) und 0,054 g (0,21 mmol) 2-Brommethyl-4-methoxybenzoesäuremethylester.
Nach Standardaufarbeitung und Reinigung durch Chromatographie unter
Verwendung einer C18-Säule
wurden 0,019 g (24% Ausbeute) der erwarteten Säure erhalten.
NMR (300
MHz, DMSO) = 1,59–1,71
(m, 2H), 2,04–2,08
(m, 2H), 2,69–2,77
(m, 2H), 2,89–3,10
(m, 5H), 3,77 (s, 3H), 3,81 (s, 3H), 4,02 (s, 2H), 4,26–4,31 (t,
2H), 6,74–6,77
(dd, 1H), 6,97–7,01
(m, 3H), 7,04–7,05
(d, 1H), 7,08 (s, 1H), 7,15–7,18
(m, 1H), 7,34–7,37
(d, 1H), 7,43–7,45
(dd, 1H), 7,88–7,91
(d, 1H).
-
Beispiel 107
-
Darstellung von 2-{2-[4-(6-Fluor-1-thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,1 g (0,33 mmol) 6-Fluor-3-piperidin-4-yl-1-thiophen-2-ylmethyl-1H-indol
(Beispiel 82, Teil B) und 0,092 g (0,42 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester.
Die rohe Mischung wurde durch HPLC-MS unter Verwendung einer C-18-Säule gereinigt.
NMR
(300 MHz, DMSO) = 1,86–2,10
(m, 4H), 2,73–2,80
(m, 2H), 2,90–2,99
(m, 1H), 3,05–3,12
(m, 2H), 3,30–3,34
(m, 2H), 4,40–4,48
(m, 2H), 5,53 (s, 2H), 6,83–6,90
(td, 1H), 6,95–6,98
(td, 1H), 7,00–7,05
(t, 1H), 7,14–7,15
(m, 1H), 7,21–7,26
(m, 2H), 7,39–7,44
(m, 3H), 7,55–7,58
(m, 1H), 7,64–7,69
(dd, 1H).
-
Beispiel 108
-
Darstellung von 5-[4-(6-Fluor-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäure
-
Darstellung von 4-(6-Fluor-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 0,01 g (0,34 mmol) 4-(6-Fluor-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
und 0,77 ml (0,45 mmol) einer frisch hergestellten 0,58M Lösung von
3-Brommethylthiophen in Ethylether. Nach Standardaufarbeitung wurden
0,13 g (100% Ausbeute) 4-(6-Fluor-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 6-Fluor-3-piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 0,13 g (0,34 mmol) 4-(6-Fluor-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 0,12 g (99% Ausbeute) 6-Fluor-3-piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
erhalten.
-
C. 5-[4-(6-Fluor-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-2-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,12 g (0,34 mmol) 6-Fluor-3-piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
und 0,11 g (0,44 mmol) 5-Brommethyl-2-methoxybenzoesäureethlester. Die rohe Mischung
wurde durch HPLC-MS unter Verwendung einer C-18-Säule gereinigt.
NMR
(300 MHz, DMSO) = 1,03–1,15
(m, 2H), 1,25–1,31
(m, 2H), 1,78–2,10
(m, 2H), 2,69–2,81
(m, 1H), 3,00–3,16
(m, 2H), 3,79–3,83
(m, 5H), 5,29 (s, 2H), 6,79–6,86
(t, 1H), 6,99–7,01
(d, 1H), 7,09–7,15
(m, 2H), 7,38–7,46
(m, 3H), 7,59–7,64
(m, 2H), 7,72 (s, 1H).
-
Beispiel 109
-
Darstellung von 2-{2-[4-(6-Fluor-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,98 g (0,31 mmol) 6-Fluor-3-piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
(Beispiel 108, Teil B) und 0,09 g (0,40 mmol) 2-(2-Chlorethoxy)-benzoesäuremethylester.
Die rohe Mischung wurde durch HPLC-MS unter Verwendung einer C18-Säule gereinigt.
NMR
(300 MHz, DMSO) = 1,90–2,10
(m, 4H), 2,62–2,71
(m, 2H), 2,78–3,10
(m, 3H), 3,22–3,26
(d, 2H), 4,34–4,39
(m, 2H), 5,30 (s, 2H), 6,82–6,88
(t, 1H), 6,99–7,04
(m, 2H), 7,22–7,28
(m, 2H), 7,37–7,47
(m, 4H), 7,53–7,55
(d, 1H), 7,64–7,69
(dd, 1H).
-
Beispiel 110
-
Darstellung von 2-(2-{4-[6-Fluor-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,1 g (0,31 mmol) 6-Fluor-3-piperidin-4-yl-1-(2-thiophen-3-ylethyl)-1H-indol
(Beispiel 78, Teil B) und 0,09 g (0,42 mmol) 2-(2- Chlorethoxy)-benzoesäureethylester.
Nach Reinigung durch HPLC-MS unter Verwendung einer C-18-Säule wurden
0,01 g (99% Reinheit) 2-(2-{4-[6-Fluor-1-(2-thiophen-3-ylethyl)-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure erhalten.
NMR
(300 MHz, DMSO) = 1,84–2,02
(m, 4H), 2,77–2,87
(m, 3H), 3,01–3,06
(t, 2H), 3,10–3,18
(m, 2H), 4,29–4,34
(t, 2H), 4,42–4,46
(m, 2H), 6,79–6,88
(td, 1H), 7,01–7,07
(m, 2H), 7,10 (s, 1H), 7,20–7,25
(m, 2H), 7,32–7,35
(dd, 1H), 7,41–7,47
(m, 2H), 7,57–7,65
(m, 2H).
-
Beispiele 111–112
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 110 beschriebenen
Verfahrens unter Verwendung der geeigneten Halogenide hergestellt.
Die ESI/MS-Daten und Reinheit sind in Tabelle 16 zusammengefaßt.
-
Tabelle
16. Beispiele 111–112
-
Beispiel 113
-
Darstellung von 2-{2-[4-(5-Methoxy-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure
-
A. Darstellung von 4-(5-Methoxy-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des im Beispiel 3 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 4 g (13,2 mmol) 4-(5-Methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
und 2,65 g (15 mmol) 3-Brommethylthiophen.
Nach Standardaufarbeitung wurden 4,5 g (87% Ausbeute) 4-(5-Methoxy-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 5-Methoxy-3-piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 4,5 g (11,2 mmol) 4-(5-Methoxy-1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 3,4 g (93% Ausbeute) 5-Methoxy-3-piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
erhalten.
-
C. Darstellung von 2-{2-[4-(5-methoxy-1-thiophen-3-ylmethyl-1H-indol-3-yl-piperidin-1-yl]-ethoxy}-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 3,3 g (10 mmol) 5-Methoxy-3-piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
und 2,6 g (12 mmol) 2-(2-Chlorethoxy)-benzoesäureethylester. Nach Standardaufarbeitung
und Umkristallisation mit Ethanol wurden 1,8 g (37% Ausbeute) der
erwarteten Säure
erhalten.
NMR (300 MHz, DMSO) = 1,82–2,02 (m, 4H), 2,62–2,69 (t,
2H), 2,79–2,98
(m, 3H), 3,21–3,25
(d, 2H), 3,79 (s, 3H), 4,43–4,47
(s, 2H), 5,38 (s, 2H), 6,72–6,75
(dd, 1H), 6,97–7,04
(m, 2H), 7,15 (s, 1H), 7,22–7,25
(m, 2H), 7,37–7,38
(m, 3H), 7,40–7,46
(m, 1H), 7,50–7,53
(dd, 1H).
-
Beispiele 114–116
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 113 beschriebenen
Verfahrens unter Verwendung von 0,3 mmol der geeigneten Indole und
Halogenide hergestellt. Die rohen Mischungen wurden mit HPLC-MS
unter Verwendung einer C-18-Säule
gereinigt. Die ESI/MS-Daten und Reinheit sind in Tabelle 17 zusammengefaßt.
-
Tabelle
17. Beispiele 114–116
-
Beispiel 117
-
Darstellung von 2-Methoxy-5-[4-(1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
A. Darstellung von 4-(1-Thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 0,2 g (0,73 mmol) 4-(1H-Indol-3-yl)-piperidin-1-carbonsäurethylester
und 1,64 ml (0,95 mmol) einer frisch hergestellten 0,6M Lösung von
3-Brommethylthiophen in Ethylether. Nach Standardaufarbeitung wurden
0,27 g (100% Ausbeute) 4-(1-Thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 3-Piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 0,27 g (0,73 mmol) 4-(1-Thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 0,22 g (100% Ausbeute) 3-Piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
erhalten.
-
C. Darstellung von 2-Methoxy-5-[4-(1-thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,1 g (0,38 mmol) 3-Piperidin-4-yl-1-thiophen-3-ylmethyl-1H-indol
und 0,13 g (0,48 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester.
Die rohe Mischung wurde mit HPLC-MS unter Verwendung einer C-18-Säule gereinigt
und 0,002 g (94% Reinheit) der erwarteten Säure wurden isoliert.
NMR
(300 MHz, DMSO) = 1,06–1,31
(m, 4H), 1,98–2,18
(m, 2H), 2,60–2,78
(m, 1H), 2,85–2,99
(m, 2H), 3,84 (S, 3H), 3,89–4,05
(m, 2H), 5,32 (s, 2H), 6,96–7,00
(m, 2H), 7,05–7,18
(m, 3H), 7,37 (s, 1H), 7,43–7,49
(m, 2H), 7,63–7,78
(m, 3H).
-
Beispiel 118
-
Darstellung von 3-[4-(1-Thiophen-3-ylmethyl-1H-indol-3-yl)-piperidin-1-ylmethyl]-benzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 117 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 0,1 g (0,38 mmol) 3-Piperidin-4-yl-thiophen-3- ylmethyl-1H-indol
und 0,1 g (0,48 mmol) 3-Brommethylbenzoesäuremethylester. Die rohe Mischung
wurde mit HPLC-MS unter Verwendung einer C-18-Säule gereinigt und 0,005 g (98%
Reinheit) der erwarteten Säure
wurden isoliert.
-
Beispiel 119
-
Darstellung von 5-{4-[1-(5-Chlorthiophen-2-ylmethyl)-5-methoxy-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxybenzoesäure
-
A. Darstellung von 4-[1-(5-Chlorthiophen-2-ylmethyl)-5-methoxy-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil B) beschriebenen
Verfahrens bei Raumtemperatur für
15 Stunden hergestellt, ausgehend von 0,1 g (0,33 mmol) 4-(5-Methoxy-1H-indol-3-yl)-piperidin-1-carbonsäureethylester
und 0,052 ml (0,43 mmol) 2-Chlor-5-chlormethylthiophen.
Nach Standardaufarbeitung wurden 0,06 g (44% Ausbeute) 4-[1-(5-Chlorthiophen-2-ylmethyl)-5-methoxy-1H-indol-3-yl]-piperidin-1-carbonsäureethylester
erhalten.
-
B. Darstellung von 1-(5-Chlorthiophen-2-ylmethyl)-5-methoxy-3-piperidin-4-yl-1H-indol
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil C) beschriebenen
Verfahrens hergestellt, ausgehend von 0,06 g (0,15 mmol) 4-[1-(5-Chlorthiophen-2-ylmethyl)-5-methoxy-1H-indol-3-yl]-piperidin-1-carbonsäureethylester.
Nach Standardaufarbeitung wurden 0,05 g (89% Ausbeute) 1-(5-Chlorthiophen-2-ylmethyl)-5-methoxy-3-piperidin-4-yl-1H-indol
erhalten.
-
C. Darstellung von 5-{4-[1-(5-Chlorthiophen-2-ylmethyl)-5-methoxy-1H-indol-3-yl]-piperidin-1-ylmethyl}-2-methoxybenzoesäure
-
Diese
Verbindung wurde unter Befolgung des in Beispiel 13 (Teil D) beschriebenen
Verfahrens hergestellt, ausgehend von 0,05 g (0,13 mmol) 1-(5-Chlorthiophen-2-ylmethyl)-5-methoxy-3-piperidin-4-yl-1H-indol und
0,05 g (0,18 mmol) 5-Brommethyl-2-methoxybenzoesäureethylester. Die rohe Mischung
wurde mit HPLC-MS unter Verwendung einer C-18-Säule gereinigt und 0,002 g (99%
Reinheit) der erwarteten Säure
wurden isoliert.
NMR (300 MHz, DMSO) = 1,78–1,85 (m, 2H), 1,92–1,99 (m,
2H), 2,31–2,48
(m, 2H), 2,72– 2,85
(m, 1H), 3,03–3,06
(m, 2H), 3,70–3,74
(m, 5H), 3,81 (s, 3H), 5,44 (s, 2H), 6,67–6,77 (m, 1H), 6,93–6,96 (m,
1H), 7,05–7,11
(m, 3H), 7,20 (s, 1H), 7,37–7,40
(d, 1H), 7,51–7,54
(d, 1H), 7,64 (s, 1H).
-
Beispiele 120–121
-
Diese
Verbindungen wurden unter Befolgung des in Beispiel 119 beschriebenen
Verfahrens mit den entsprechenden Halogeniden hergestellt. Die rohen
Mischungen wurden mit Flashchromatographie unter Verwendung einer
C-18-Säule
gereinigt. Die ESI/MS-Daten und Reinheit sind in Tabelle 18 zusammengefaßt.
-
Tabelle
18. Beispiele 120–121
-
Beispiel 122
-
Herstellung einer pharmazeutischen
Zusammensetzung: Sirup
-
1000
Flaschen (150 ml Volumen), die jeweils eine Lösung von 750 mg 2-(2-{4-[1-(2-[1,3]Dioxolan-2-ylethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure enthielten,
wurden wie folgt hergestellt:
| 2-(2-{4-[1-(2-[1,3]Dioxolan-2-ylethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure | 750
g |
| Glycerin | 15.000
g |
| hydriertes
Riziniusöl-Ethylenoxid | 1.500
g |
| Natriummethyl-p-hydroxybenzoat | 240
g |
| Natriumpropyl-p-hydroxybenzoat | 60
g |
| Natriumsaccharin | 300
g |
| Geschmacksstoff | q.s. |
| Natriumhydroxid
q.s. | pH
= 4 |
| entmineralisiertes
Wasser q.s. | 150
Liter |
-
Verfahren:
-
Zu
einer Lösung
der Natriummethyl(und -propyl-)-p-hydroxybenzoate und des Natriumsaccharins
in 30 Litern entmineralisiertem Wasser wurde eine wäßrige Glycerinlösung und
hydriertes Rizinusöl-Ethylenoxid zugegeben.
Nach Rühren
wurde die 2-(2-{4-[1-(2-[1,3]Dioxolan-2-ylethyl)-6-fluor-1H-indol-3-yl]-piperidin-1-yl}-ethoxy)-benzoesäure zugegeben
und homogenisiert, um vollständige
Auflösung
zu erreichen. Danach wurde der Geschmacksstoff in die Lösung unter
kräftigem
Rühren
eingemischt, und die Mischung wurde mit entmineralisiertem Wasser
auf das Endvolumen aufgefüllt.
-
Die
resultierende Lösung
wurde in 150 ml-Flaschen unter Verwendung einer geeigneten Füllmaschine abgefüllt.
-
Beispiel 123
-
Herstellung einer pharmazeutischen
Zusammensetzung: Kapseln
-
50.000
Kapseln, die jeweils 50 mg 2-{2-[4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure enthielten,
wurden aus der folgenden Formulierung hergestellt:
| 2-{2-[4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure | 2.500
g |
| Magnesiumstearat | 225
g |
| sprühgetrocknete
Lactose | 18.350
g |
| quervernetzte
Natriumcarboxymethylcellulose | 900
g |
| Natriumlaurylsulfat | 450
g |
-
Verfahren:
-
Die
2-{2-[4-(1-Thiophen-2-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure, Natriumlaurylsulfat,
Lactose und quervernetzte Natriumcarboxymethylcellulose wurden miteinander
vermischt und durch ein Sieb mit einer Öffnung von 0,6 mm hindurchgegeben.
-
Das
Magnesiumstearat wurde zugegeben und die Mischung in Gelatinekapselen
geeigneter Größe verkapselt.
-
Beispiel 124
-
Herstellung einer pharmazeutischen
Zusammensetzung: Tabletten
-
100.000
Tabletten, die jeweils 25 mg 2-{2-[4-(1-Pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure enthielten,
wurden aus der folgenden Formulierung hergestellt:
| 2-{2-[4-(1-Pyridin-4-ylmethyl-1H-indol-3-yl)-piperidin-1-yl]-ethoxy}-benzoesäure | 2.500
g |
| mikrokristalline
Cellulose | 1.650
g |
| sprühgetrocknete
Lactose | 9.620
g |
| Carboxymethylstärke | 570
g |
| Natriumstearylfumarat | 80
g |
| kolloidales
Siliciumdioxid | 80
g |
-
Verfahren:
-
Alle
Pulver wurden durch ein Sieb mit Öffnungen von 0,6 mm hindurchgegeben.
Sie wurden dann alle in einem geeigneten Mischer für 30 Minuten
vermischt und zu 145 mg-Tabletten unter Verwendung von 6 mm-Scheiben
und flach abgeschrägten
Stempeln verpreßt.
Die Desintegrationszeit der Tabletten betrug etwa 60 Sekunden.