CN86103220A - 制备阿朴长春蔓胺醇衍生物的新方法 - Google Patents
制备阿朴长春蔓胺醇衍生物的新方法 Download PDFInfo
- Publication number
- CN86103220A CN86103220A CN86103220.9A CN86103220A CN86103220A CN 86103220 A CN86103220 A CN 86103220A CN 86103220 A CN86103220 A CN 86103220A CN 86103220 A CN86103220 A CN 86103220A
- Authority
- CN
- China
- Prior art keywords
- structural formula
- dehydrogenation
- acid
- acidylate
- vincamine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D461/00—Heterocyclic compounds containing indolo [3,2,1-d,e] pyrido [3,2,1,j] [1,5]-naphthyridine ring systems, e.g. vincamine
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明涉及制备结构式为(I)的17,18-脱氢-阿朴长春蔓胺醇的三甲氧基苯甲酸酯的新方法。以结构式为(IIa)的17,18-脱氢-长春蔓胺和/或结构式为(IIb)的17,18-脱氢-表长春蔓胺为原料,先用复合金属氢化物还原;再用3,4,5-三甲氧基苯甲酸或其有酰化作用的衍生物,选择酰化所得的羟基长春蔓胺醇衍生物;最后,在酰氯存在下,用甲酸处理此酰化的羟基长春蔓胺醇衍生物,得到结构式为(I)的化合物。如果需要,可把得到的结构式为(I)的化合物转化为其酸加成盐。
Description
本发明是关于制备阿朴长春蔓胺醇(Apovincaminol)衍生物的新方法。特别是,本发明是关于制备结构式为(Ⅰ)的17,18-脱氢-阿朴长春蔓胺醇的3′,4′,5′-三甲氧基-苯甲酸酯及其酸式加成盐的新方法。
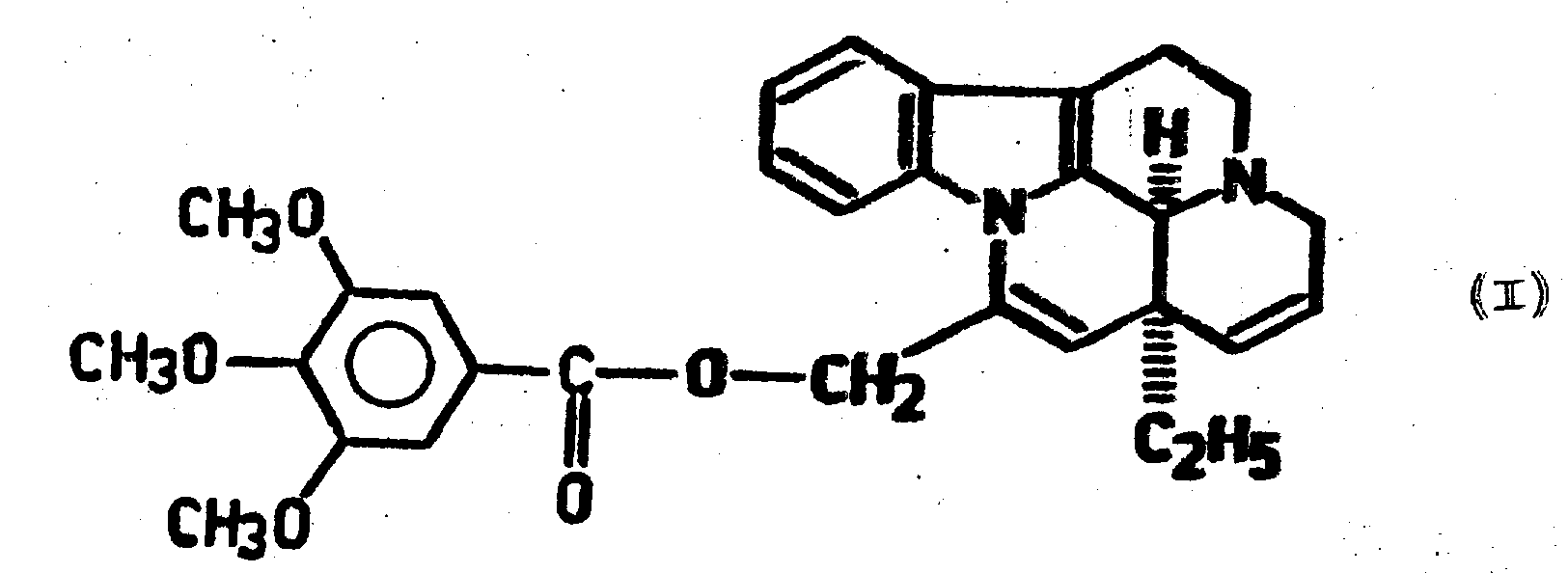
在结构式(Ⅰ)和所有其它结构式中,虚线表示此取代基是在α-位置上,箭头表示此取代基是在β-位置上,波纹线表示此取代的立体化学位置不定。
从例如英国专利说明书2094297中已知,结构式为(Ⅰ)的17,18-脱氢-阿朴长春蔓胺醇的3′,4′,5′-三甲氧基-苯甲酸酯抑制磷酸二脂酶的活性,因此对牛皮癣的治疗是满意的。按照引证的英国专利说明
书,制备这种化合物是从以下化合物起始,即从结构式为(Ⅱa)的17,18-脱氢-长春蔓胺或结构式为(Ⅱb)的17,18-脱氢-表长春蔓胺或者是它们的混合物起始。

用适当的脱水剂处理结构式如(Ⅱa)或(Ⅱb)的已知起始化合物或者是它们的混合物,得到17,18-脱氢-阿朴长春蔓胺。然后用选择的还原剂处理,把17,18-脱氢-阿朴长春蔓胺转化成17,18-脱氢-阿朴长春蔓胺醇。用3,4,5-三甲氧基-苯甲酸或者其具有可酰化作用的衍生物,酰化所得到的产物,最后得到结构式(Ⅰ)的化合物。
按照实例脱水作用是在甲酸和乙酰氯存在下在氯甲酸溶液中进行,还原作用是在乙醚中用氢化铝锂进行,而酰化作用是在苯溶液中用三甲氧基-苯甲酰氯进行。
在实验中我们发现,上述方法实际上有以下缺点:
1)当按照英国专利说明书2,094,297的实例Ⅰ或在类似化合物脱水工艺中已知的其它条件下,制备17,18-脱氢-阿朴长春蔓胺酸时,不能安全地消除结构式(Ⅱa)和(Ⅱb)化合物的羟基,起始的羟基化合物污染了反应所得到的产品。
2)用大过量的酰化剂进行酰化作用,从反应混合物中除掉这种酰化剂是困难的。过量的酰化剂可能与晶形产品一起分出,因此增加了产品的杂质含量。
3)当从结构式(Ⅱa)和(Ⅱb)的化合物起始进行多步合成时,需要分离两种中间体。经多次分离就降低了最后产品的产率。因为这些中间体是易结晶的化合物,如果留在反应混合物中,它们会与最后产品一起结晶,也就是会在最后产品中混进更多的杂质。分离两种中间化合物的另一个缺点是由于对两种中间体进行干燥需要时间,从而增加了制备时间。
本发明的目的是提供一个简单快速的方法,这种方法避免了上述缺点。
按照本发明,从结构式(Ⅱa)和/或(Ⅱb)通过新的中间体来制备结构式(Ⅰ)的化合物。更可取的是没有分离中间化合物步骤,并具有好的产率和高的纯度。
因此,本发明是关于制备结构式为(Ⅰ)的17,18-脱氢-阿朴长春蔓胺醇-3′,4′,5′-三甲氧基-苯甲酸酯的新方法,这种方法包括:
a)结构式为(Ⅱa)的17,18-脱氢-长春蔓胺和/或结构为(Ⅱb)的17,18-

脱氢-表长春蔓胺,与复合金属氢化物进行反应。

所得的结构式为(Ⅲ)的羟基长春蔓胺醇衍生物,选用3,4,5-三甲氧基-苯甲酸或其可酰化的衍生物,在任选的催化剂和/或酸结合剂存在下酰化。
在某种酰氯存在下,用甲酸处理所得的结构式为(Ⅳ)的酰化羟基衍生物,分离出结构式为(Ⅰ)的产品。如果要求,也可把它转化成酸式加成盐;或者
b)在任选的催化剂和/或酸结合剂存在下,用3,4,5-三甲氧基苯甲酸或它的可进行酰化的衍生物,选择酰化结构式为(Ⅲ)的羟基-长春蔓胺醇衍生物,然后在酰氯存在下,用甲酸处理上面得到的结构式为(Ⅳ)的酰化羟基-衍生物,分得结构式为(Ⅰ)的产品。如果需要,同样可转化成其酸式加成盐;或
C)在酰氯存在下,用甲酸处理结构式为(Ⅳ)的酰化羟基-衍生物,分得结构式为(Ⅰ)的产品。如果需要,同样可转化成其酸式加成盐。
按照本发明的方法,类似于英国专利说明书2,094,297,结构式(Ⅱa)或(Ⅱb)的化合物或它们的混合物作为起始材料。
作为复合金属氢化物一般是碱金属氢化物,最好用氢化铝锂。还原作用在溶剂中进行,最好在环状醚或甲苯中,或者是在它们的混合物中。作为可酰化的3,4,5-三甲氧基苯甲酸的衍生物,可用这样的酸本身或相应的酰氯或酸酐。典型的催化剂包括4-二甲氨基-吡啶,4-吡咯烷基-吡啶和其它的吡啶衍生物,而作为酸结合剂,例如可以用三乙胺,吡啶或其它碱性物质。
按照本发明的最佳实施例,在甲苯和四氢呋喃的混合物中,用氢化铝锂把结构式为(Ⅱa)或(Ⅱb)的化合物或者它们的混合物还原为结构式为(Ⅲ)的新化合物。然后,在催化剂存在下,最好是4-二甲基氨基-吡啶,用当量的酰化剂(例如,三甲氧基苯甲酰氯),选择性地酰化上述化合物中的羟甲基的伯羟基,得到结构式为(Ⅳ)的新的化合物。选择性酰化作用是基于叔醇和伯醇的不同反应活性。然后这种新化合物通过甲酸和乙酰氯,除去1克分子的水,转化成结构式为(Ⅰ)的17,18-脱氢-阿朴长春蔓胺醇的三甲氧基-苯甲酸酯。
如果需要,可以把得到的这种产品转化成酸的加成盐。无机酸加成盐的最好代表性化合物是例如盐酸盐,硫酸盐和磷酸盐。最好的有机酸加成盐包括例如酒石酸氢盐,琥珀酸盐,柠檬酸盐,抗坏血酸盐。通过把含酸成分的醇、醚或丙酮溶液加到结构式为(Ⅰ)的产品中,制备这类盐。这类盐的制备在PH3-6之间进行。
本发明方法的主要好处是在下一步反应之前,不需要分离中间体,只用萃取或过滤除掉过量的反应物以避免分离产品时,由于热处理而发生分解。在合成的最后,易从少量可结晶的付产物中分离出产品,该产品产率高和纯度好。
本发明的方法可由以下实例进一步说明,但并不限定本发明的范围。
实例1
A)把100.0克17,18-脱氢-长春蔓胺和17,18-脱氢-表长春蔓胺悬浮在435.0克(500毫升)甲苯中,将此混合物冷却到0℃。然后把15.0克氢化铝锂加到反应混合物中,接着在10分钟内加入88.7克(100毫升)无水四氢呋喃。加完后,把此反应混合物加热到60-70℃。0.5-1小时反应终止〔用3∶1的苯和甲醇混合物,在硅胶上经薄层色谱(t.l.G)监测反应的进展〕。然后把反应混合物冷却到0℃,并在激烈搅拌下于20分钟内加入50.0克(50毫升)水。接着把1320克(1000毫升)二氯甲烷和75.0克干燥的硫酸钠加入所得混合物中。把沉淀的氢氧化铝和硫酸钠滤出,用两份100毫升(132克)二氯甲烷洗涤。得到的二氯甲烷溶液含有95%的17,18-脱氢-14-羟基-长春蔓胺醇。
B)把1.0克二甲氨基-吡啶和27.2克(36毫升)三乙胺加到在步骤A)中得到的溶液中,接着,滴加溶于924.0克(700毫升)二氯甲烷中的1克分子当量的三甲氧基-苯甲酰氯溶液。加入时间为1小时。
用3∶1的苯和甲醇混合物,以硅胶薄层色谱监测反应的进展。此反应在2.5小时内完成。
然后用两份1000毫升2%的氢氧化钠水溶液洗涤此反应混合物,接着用两份1000毫升的水洗涤,有机相用硫酸钠干燥。滤出硫酸钠,二氯甲烷溶液中含有83.5%的17,18-脱氢-14-羟基长春蔓胺醇的3′,4′,5′-三甲氧基-苯甲酸酯。
C)在室温下,把183.0克(150毫升)干燥的甲酸和347克(250毫升)乙酰氯加到步骤B)所得的溶液中。搅拌4小时后,进一步把69.4克(50毫升)乙酰氯加到此反应混合物中。
通过薄层色谱(硅胶,3∶1的苯和甲醇混合物)监测反应的进展。反应终止后(总共约6小时),把500毫升水加入到反应混合物中,随后在连续搅拌和冷却下,用浓氢氧化铵调整至PH8。分离有机相。水相用660克(500毫升)二氯甲烷萃取。合并有机相用无水硫酸钠干燥,滤出硫酸钠,在最高60℃的温度下,真空蒸发滤液。随后,当蒸馏时,用干燥乙醇交换溶剂。最后体积约为150到200毫升。得到的悬浮液在0到5℃放置12小时。过滤出结晶,用两份100毫升冷乙醇洗涤并干燥(在避光下,最高温度在50℃)。得到114.1克17,18-脱氢-阿朴长春蔓胺醇的3′,4′,5′-三甲氧基-苯甲酸酯。
产率:80.3%
熔点:142-143℃
〔α〕D=+26.5°(C=1,氯仿)
紫外光谱(EtoH)λmax:261,303,314nm
红外光谱(KBr)νmax(cm-1):
1722(>C=0)
1650(>C=C<)

1215(Ar-O-C)
1128(C-O-C)
865〔Ar(1H)〕
743〔Ar(4H)〕
1H-核磁共振谱(CDCl3)δ(ppm):
1.04(t) H-21
1.78(q) H-20
2.45-3.60(m) H-5,6,19
3.73(s) O-CH3(3′,5′)
3.85(s) O-CH3(4′)
4.31(s) H-3
5.20(s) H-15
5.34(d) H-17
5.37;5.48(d) O-CH2-
5.53(m) H-18
7.00-7.65(m) H-9,10,11,12
7.21(s) H-2′,6′
中间体的物理数据:
A.)17,18-脱氢-14-羟基-长春蔓胺醇
紫外光谱(乙醇)λ最大(nm):230,282,290
红外光谱(溴化钾)ν最大(厘米-1):3380(OH),1653(C=C),1105〔C-O(H)〕,1038〔C-O(H)〕,741〔Ar(4H)〕
1H-核磁共振(CDCl3)δ(ppm)
1.01(t) H-21
1.64;1.91(m) H-20
2.23(d) H-15(ax)
2.42(d) H-15(eq)
2.45-3.50(m) H-5,6,19
2.75(br*) O-H
3.98(s) H-3
5.60(m) H-18
5.72(d) H-17
7.0-7.63(m) H-9,10,11,12
B)17,18-脱氢-14-羟基-长春蔓胺醇的3′,4′,5′-三甲氧基-苯甲酸酯
紫外光谱(乙醇)λ最大(nm):273
红外光谱(溴化钾)ν最大(厘米-1):
3524 OH
1718 (>C=O)
1653 (>C=C<)
1221 Ar-O-C
1128 C-O-C
1107 C-O(H)
865 Ar(1H)
741 Ar(4H)
1H-核磁共振(CDCl3)δ(ppm):
1.01(t) H-21
1.6(br*) OH
1.65;1.93(m) H-20
2.31(d) H-15(ax)
2.41(d) H-15(eq)
2.45-3.5(m) H-5,6,19
3.75(s) O-CH3(3′,5′)
3.85(s) O-CH3(4′)
3.99(s) H-3
4.9;5.0(d) O-CH2
5.62(m) H-18
5.75(d) H-17
7.01(s) H-2′,6′
7.0-7.7(m) H-9,10,11,12
br*:宽频带,可由重水交换
实例2
重复实例1的方法,但在步骤C)中得到的17,18-脱氢-阿朴长春蔓胺醇的3′,4′,5′-三甲氧基苯甲酸酯是被溶解在250毫升乙醇中,并且把D-酒石酸加到此溶液中,直至酒石酸盐沉淀完全。
熔点:110-112℃(分解)
元素分析 C34H38O11N2(650):
计算值:N=4.3%;
实验值:N=4.24%。
紫外光谱:λ最大(nm):209,251,304,315。
Claims (4)
1、结构式为(Ⅰ)的17,18-脱氢-阿朴长春蔓胺醇的3′,4′,5′-三甲氧基-苯甲酸酯及其酸加成盐的制备方法,

其特征是,
a)结构式为(Ⅱa)的17,18-脱氢-长春蔓胺和/或结构式为(Ⅱb)的17,18-脱氢-表长春蔓胺与一种复合金属氢化物反应,

用3,4,5-三甲氧基-苯甲酸或其有酰化作用的衍生物,任意地在催化剂和/或酸结合剂存在下,选择酰化得到的结构为(Ⅲ)的羟基长春蔓胺醇衍生物,

在一种酰氯存在下,用甲酸处理得到的结构式为(Ⅳ)的酰化羟基衍生物,或者

b)任意在催化剂和/或酸结合剂存在下,用3,4,5-三甲氧基

苯甲酸或其有酰化作用的衍生物选择酰化结构式(Ⅲ)的羟基-长春蔓胺醇衍生物,在一种酰氯存在下,用甲酸处理得到的结构式为(Ⅳ)的酰化羟基一衍生物,
c)在一种酰氯存在下,用甲酸处理结构式为(Ⅳ)的酰化羟基-衍生物,分离出由a),b)或c)制得的结构式为(Ⅰ)的产品,如果需要,可把这些产品转化成它的酸加成盐。
2、权利要求1的方法,其中方法a)包括用氢化铝锂作复合金属氢化物并在甲苯和四氢呋喃的混合物中进行反应。
3、权利要求1的方法,其中方法b)包括用二甲氨基-吡啶和/或三乙基胺作催化剂和/或酸结合剂。
4、权利要求1的方法,其中方法c)包括用乙酰氯作所说的酰氯。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HU1758/85 | 1985-05-10 | ||
| HU851758A HU192932B (en) | 1985-05-10 | 1985-05-10 | Process for producing apovincaminol derivative |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN86103220A true CN86103220A (zh) | 1987-11-18 |
| CN1014520B CN1014520B (zh) | 1991-10-30 |
Family
ID=10956041
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN86103220A Expired CN1014520B (zh) | 1985-05-10 | 1986-05-09 | 制备阿朴长春蔓胺醇衍生物的新方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US4758666A (zh) |
| KR (1) | KR860009013A (zh) |
| CN (1) | CN1014520B (zh) |
| AT (1) | AT394720B (zh) |
| CS (1) | CS257792B2 (zh) |
| DK (1) | DK215386A (zh) |
| ES (1) | ES8900139A1 (zh) |
| FI (1) | FI85375C (zh) |
| HU (1) | HU192932B (zh) |
| PL (1) | PL146325B1 (zh) |
| PT (1) | PT82559B (zh) |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HU183323B (en) * | 1981-02-11 | 1984-04-28 | Richter Gedeon Vegyeszet | Process for producing 17,18-dihydro-apovincaminol-3-comma down comma aaove-4-comma down comma above-5-comma down comma aaove-trimethoxy-benzoate and acid additional salts thereof |
-
1985
- 1985-05-10 HU HU851758A patent/HU192932B/hu not_active IP Right Cessation
-
1986
- 1986-05-08 CS CS863390A patent/CS257792B2/cs unknown
- 1986-05-09 CN CN86103220A patent/CN1014520B/zh not_active Expired
- 1986-05-09 ES ES554806A patent/ES8900139A1/es not_active Expired
- 1986-05-09 PL PL1986259414A patent/PL146325B1/pl unknown
- 1986-05-09 FI FI861952A patent/FI85375C/fi not_active IP Right Cessation
- 1986-05-09 PT PT82559A patent/PT82559B/pt not_active IP Right Cessation
- 1986-05-09 DK DK215386A patent/DK215386A/da not_active Application Discontinuation
- 1986-05-09 AT AT0124386A patent/AT394720B/de not_active IP Right Cessation
- 1986-05-09 KR KR1019860003632A patent/KR860009013A/ko not_active Application Discontinuation
- 1986-05-09 US US06/861,550 patent/US4758666A/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| FI861952A0 (fi) | 1986-05-09 |
| FI861952A (fi) | 1986-11-11 |
| CN1014520B (zh) | 1991-10-30 |
| PL146325B1 (en) | 1989-01-31 |
| DK215386A (da) | 1986-11-11 |
| FI85375C (fi) | 1992-04-10 |
| PT82559B (pt) | 1988-03-03 |
| HUT40122A (en) | 1986-11-28 |
| ES8900139A1 (es) | 1989-02-01 |
| AT394720B (de) | 1992-06-10 |
| CS339086A2 (en) | 1987-11-12 |
| PT82559A (en) | 1986-06-01 |
| FI85375B (fi) | 1991-12-31 |
| DK215386D0 (da) | 1986-05-09 |
| ES554806A0 (es) | 1989-02-01 |
| CS257792B2 (en) | 1988-06-15 |
| ATA124386A (de) | 1991-11-15 |
| HU192932B (en) | 1987-08-28 |
| US4758666A (en) | 1988-07-19 |
| KR860009013A (ko) | 1986-12-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1024545C (zh) | 4-脱甲氧-4-氨基正定霉素酮的制备方法 | |
| CN1229366C (zh) | 甲基磺酰胺基-苯并呋喃衍生物,它的制备方法与它作为合成中间体的应用 | |
| CN1107153A (zh) | 取代的苯并吡喃衍生物的制备方法 | |
| CN101058598A (zh) | 2α,3α-环氧-16α-溴-5α-雄甾-17-酮的合成方法 | |
| CN1178942C (zh) | 结晶青霉素和它的制备方法 | |
| CN86103220A (zh) | 制备阿朴长春蔓胺醇衍生物的新方法 | |
| CN108516952B (zh) | 一种3-酰基六元含氮杂环类化合物的合成方法 | |
| CN112457175B (zh) | 一种制备1,3-二苄氧基-2-丙酮的方法 | |
| CN1033583C (zh) | 制备福司可林的6-(取代氨基丙酰基)衍生物的方法 | |
| CN1175943A (zh) | 氨基四氢萘酮衍生物及其制备方法 | |
| CN114773378A (zh) | 一种培南类药物中间体的非对映异构体的合成方法 | |
| CN1291987C (zh) | 制备吉米沙星酸式盐的方法 | |
| CN1105658A (zh) | d-8-酰氧基别二氢葛缕酮类化合物及其合成方法 | |
| CN1863807A (zh) | 青霉素晶体及其制备方法 | |
| CN1016869B (zh) | 制备6,7-二酰基-7-脱乙酰福斯克林衍生物的新方法 | |
| CN108976198B (zh) | 一种3-(4-吡啶)吲哚类化合物的合成方法 | |
| CN101035801A (zh) | 合成季铵甾族化合物的环境友好的方法 | |
| CN108409561B (zh) | 一种5-氨基酮戊酸盐酸盐及中间体的制备方法 | |
| CN1152038C (zh) | D-和l-脱氧核糖的合成方法 | |
| CN1179957C (zh) | 制备苯并呋喃酮肟的方法 | |
| CN1492861A (zh) | 西酞普兰的制备方法 | |
| CN111072625A (zh) | 一种3,4-亚甲二氧苯乙酮的制备方法 | |
| JP6153209B2 (ja) | エゼチミブ及びその中間体の製造方法 | |
| CN1644583A (zh) | 一种头孢烯类鎓盐化合物及其制备方法和用它制备头孢吡肟的应用 | |
| CN1116294C (zh) | 3-甲氧基甲酰基-4,5二甲基噻吩的合成 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C13 | Decision | ||
| C14 | Grant of patent or utility model | ||
| C19 | Lapse of patent right due to non-payment of the annual fee |