CN1345317A - (1-苯甲酰甲基-3-苯基-3-哌啶基乙基)哌啶衍生物,制备它们的方法以及包含它们的药物组合物 - Google Patents
(1-苯甲酰甲基-3-苯基-3-哌啶基乙基)哌啶衍生物,制备它们的方法以及包含它们的药物组合物 Download PDFInfo
- Publication number
- CN1345317A CN1345317A CN00805826A CN00805826A CN1345317A CN 1345317 A CN1345317 A CN 1345317A CN 00805826 A CN00805826 A CN 00805826A CN 00805826 A CN00805826 A CN 00805826A CN 1345317 A CN1345317 A CN 1345317A
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- structural formula
- salt
- obtains
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/34—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Pulmonology (AREA)
- Cardiology (AREA)
- Psychiatry (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Otolaryngology (AREA)
- Dermatology (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Hydrogenated Pyridines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本发明涉及一种结构式(I)的化合物,以及其与无机或有机酸的盐,其溶剂合物和/或水合物,所述物质对于P物质的人体NK1受体具有高的亲合力和高的选择性。另外,本发明还涉及所述物质的生产方法,在所述物质生产中使用的结构式(VII)的中间化合物,包含所述产物的药物组合物及其在生产药品中的用途,从而治疗涉及P物质和NK1人体受体的所有病症。
Description
本发明涉及一种新颖的哌啶衍生物,涉及所述衍生物的制备方法,以及包含这些物质作为活性成分的药物组合物。
更准确地说,本发明涉及用于治疗包括速激肽系统的病理现象的新颖哌啶衍生物,所述系统的非限定方式包括:(1)疼痛(L.Urban等人的TINS,1994,17,432-438;L.Seguin等人的《疼痛》(Pain),1995,61,325-343;S.H.Buck,1994,《速激肽受体》(TachykininReceptors),Humana出版社,Totowa,新泽西);(2)变应性变态反应和炎症(S.H.Buck,1994,《速激肽受体》,Humana出版社,Totowa,新泽西);(3)胃肠疾病(P.Holzer and U.Holzer-Petsche,Pharmacol.Ther.,1997,73,173-217和219-263);(4)呼吸疾病(J.Mizrahi等人,《药理学》(Pharma cology),1982,25,39-50;C.Advenier等人,Eur.Respir.J.,1997,10,1892-1906;C.Advenier和X.Emonds-Alt,《肺药理学》(PulmonaryPharmacol),1996,9,329-333);(5)泌尿疾病(S.H.Buck,1994,《速激肽受体》,Humana出版社,Totowa,新泽西;C.A.Maggi,《神经生物学的发展》(Progress in Neurobiology),1995,45,1-98);(6)神经病学上的疾病和神经精神病学上的疾病(C.A.Maggi等人,J.Autonomic Pharmacol.,1993,13,23-93;M.Otsuka和K.Yoshioka,《生物学综述》(Physiol.Rev.)1993,73.229-308)。
近年来,已对速激肽及其受体进行了许多研究。速激肽不仅分布在中枢神经系统中而且分布在外周神经系统中。速激肽受体已被识别并分成三类:NK1,NK2和NK3。物质P(SP)是NK1受体的内源配体,神经激肽A(NKA)是NK2受体的内源配体,而神经激肽B(NKB)是NK3受体的内源配体。
NK1,NK2和NK3受体由不同的种类表示。
C.A.Maggi等人的述评(J.Autonomic Pharmacol.,1993,13,23-93)和D.Regoli等人的述评(Pharmacol.Rev.,1994,46,551-559)讨论了速激肽受体及其拮抗剂,并披露了药理学研究以及在人体治疗方面的应用。
许多专利和专利申请描述了对于速激肽受体是活性的化合物。因此,欧洲专利申请EP 0 512 901涉及下式的化合物: 式中:
式中:
-Q′表示氧原子或两个氢原子,
-T′=-C(O)-或-CH2-,
-Y,Ar′,Z′,m′,n′,p′和q具有不同的值。
专利申请EP 0 714 891涉及下式的化合物: 式中:
式中:
-p″为1,2或3;
-m″和n″独立地从0-6;
-W,Ra,Rb,Rc,Rd和Re具有不同的值。
业已发现,新化合物对于物质P的人体NK1受体具有很强的亲合力和高的选择性,并且是所述受体的拮抗剂。
此外,当本发明的化合物进行口服给药时,它们具有良好的生物可利用率。
这些化合物能够用来制备药物,所述药物用于治疗:其中涉及物质P和NK1受体的病变,特别是治疗呼吸,胃肠,泌尿,免疫,心血管和中枢神经系统的病变,以及治疗疼痛,偏头痛,炎症,恶心,和呕吐,以及皮肤病。
因此,根据本发明的一方面,本发明的目的是下式的化合物,以及其与无机或有机酸可能的盐,和其溶剂合物和/或水合物: 式中:
式中:
-Ar表示被卤原子单取代或二取代的苯基;(C1-C3)烷基;-X表示
-R1表示氯原子,溴原子,(C1-C3)烷基或三氟甲基;
-R2表示-CR3R4CONR5R6;
-R3和R4表示相同的基团,所述基团选自:甲基,乙基,正丙基或正丁基;
-或者R3和R4与连接至其上的碳原子一起构成(C3-C6)环烷基;
-R5和R6各自独立地表示氢;(C1-C3)烷基;
-或者,R5和R6与连接至其上的氮原子一起构成杂环基团,所述基团选自:1-氮杂环丁烷基,1-吡咯烷基,1-哌啶基,4-吗啉基,4-硫代吗啉基或全氢化-1-吖庚因基。
根据本发明结构式(I)的化合物,不仅包括光学纯异构体,而且包括其任何比例的混合物。
能够形成结构式(I)化合物的盐。这些盐不仅包括与能使结构式(I)的化合物进行适当分离或结晶的无机或有机酸的盐,所述酸为如苦味酸或草酸或光学活性酸,例如杏仁酸或樟脑磺酸;而且包括形成药物上可接受盐的那些盐,如盐酸盐,氢溴酸盐,硫酸盐,硫酸氢盐,磷酸二氢盐,甲磺酸盐,甲基硫酸盐,草酸盐,马来酸盐,富马酸盐,丁二酸盐,2-萘磺酸盐,葡萄糖酸盐,柠檬酸盐,苯磺酸盐或对甲苯磺酸盐。
术语“卤素”意指:氯,溴,氟或碘原子。
在本说明书中,烷基基团是直链或支链的。
根据本发明,优选的化合物为如下结构式的化合物,或其与无机酸或有机酸的盐,以及其溶剂合物和/或水合物: 式中X和R1如结构式(I)化合物的定义。
式中X和R1如结构式(I)化合物的定义。
根据本发明,结构式(I)优选的化合物是:其中Ar表示3,4-二氯苯基,或3,4-二甲苯基的那些化合物。
根据本发明,结构式(I)优选的化合物是:其中取代基R1表示氯原子,甲基,乙基或三氟甲基的那些化合物。
根据本发明,结构式(I)优选的化合物是:其中X表示基团 的那些化合物,其中R2表示基团-CR3R4CONR5R6。
的那些化合物,其中R2表示基团-CR3R4CONR5R6。
特别是,优选的化合物是:其中R3和R4分别表示甲基,或与连接至其上的碳原子一起构成环己基的那些化合物。特别是,还优选的化合物是其中R5和R6分别表示氢或甲基的那些化合物。
根据本发明,结构式(I)优选的化合物是:其中X表示基团 的那些化合物,其中R2表示-CR3R4CONR5R6。
的那些化合物,其中R2表示-CR3R4CONR5R6。
特别是,优选的化合物是:其中R3和R4分别表示甲基,或与连接至其上的碳原子一起构成环丙基或环己基的那些化合物。特别是,还优选的化合物是其中R5和R6分别表示氢或甲基的那些化合物。
根据本发明,优选的化合物是下式的那些化合物,以及其与无机酸或有机酸的盐,和其溶剂合物和/或水合物:式中:
-R′1表示氯原子,甲基,乙基或三氟甲基;
-R′3和R′4各自表示甲基,或者与连接至其上的碳原子一起构成环己基;
-R′5和R′6各自表示氢或甲基。
根据本发明,优选的化合物是下式的那些化合物,以及其与无机酸或有机酸的盐,和其溶剂合物和/或水合物:式中:
-R′1表示氯原子,甲基,乙基或三氟甲基;
-R′3和R′4各自表示甲基,或者与连接至其上的碳原子一起构成环己基或环丙基;
-R′5和R′6各自表示氢或甲基。
根据本发明,优选的化合物是:光学纯形式的结构式(I),(I′)和(I″)的那些化合物。
更为优选的是下列化合物,及其盐,和其溶剂合物和/或水合物:
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌嗪基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-N,N-二甲基氨基甲酰基-1-甲基乙基)-1-哌嗪基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二(三氟甲基)苯基)乙酰基]哌啶,(+)异构体;
-3-[2-[4-(1-氨基甲酰基环己基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基环己基)-1-哌嗪基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基环己基)哌啶-1-基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)哌嗪-1-基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶,(+)异构体;
-3-[2-[4-(1-N,N-二甲基氨基甲酰基-1-甲基乙基)-1-哌嗪基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶,(+)异构体;
-3-[2-[4-(1-氨基甲酰基环己基)-1-哌嗪基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶,(+)异构体;
-3-[2-[4-(1-氨基甲酰基环己基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二(三氟甲基)-苯基]乙酰基]哌啶,(+)异构体;
-3-[2-[4-(1-氨基甲酰基环己基)-1-哌嗪基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二(三氟甲基)-苯基]乙酰基]哌啶,(+)异构体;
-3-[2-[4-(1-N,N-二甲基氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲基苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二乙基苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基环丙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲基苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基环丙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二(三氟甲基)-苯基]乙酰基]哌啶,(+)异构体;
-3-[2-[4-(1-氨基甲酰基环丙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶,(+)异构体;
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌嗪基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二乙基苯基)乙酰基]哌啶,(-)异构体;
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二甲苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶;
-3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌嗪基]乙基]-3-(3,4-二甲苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶。
根据本发明的另一方面,本发明涉及结构式(I)化合物,其盐以及其溶剂合物和或/和水合物的制备方法,其特征在于:
1a) 下式(II)的化合物用结构式(III)酸的官能衍生物进行处理,以得到结构式(IV)的化合物, 式中Ar如结构式(I)中所定义,E表示氢或氧(O-)保护基团,
式中Ar如结构式(I)中所定义,E表示氢或氧(O-)保护基团, 式中R1如结构式(I)化合物所定义;
式中R1如结构式(I)化合物所定义;
2a) 任选地,当E表示保护基团时,它通过酸或碱的作用而消除,以便得到下式的醇: (IV,E=H);
(IV,E=H);
3a) 步骤1a)或步骤2a)中得到的结构式(IV,E=H)的醇用下式
Y-SO2-Cl (V)的化合物进行处理,结构式(V)中,Y表示甲基,苯基,甲苯基或三氟甲基基团;以便得到下式(VI)的化合物:
4a) 结构式(VI)的化合物用下式(VII)的化合物进行处理: 式中X如结构式(I)化合物定义;
式中X如结构式(I)化合物定义;
5a) 任选地用无机或有机酸,将如此得到的化合物转化成其一种盐。
当E表示氧保护基团时,该基团选自:本领域技术人员熟知的常规氧保护基团,例如,2-四氢吡喃基,苯甲酰基或(C1-C4)烷基羰基。
在步骤1a)中,作为酸(III)的官能衍生物,使用酸本身,或者是与胺反应的官能衍生物之一,例如酸酐,混合酸酐,酰氯或活性酯,如对硝基苯基酯。
当使用结构式(III)本身的酸时,本发明方法在肽化学中使用的偶联剂,如1,3-二环己基碳二亚胺或苯并三唑-1-基氧代三(二甲氨基)六氟磷酸鏻的存在下,在碱如三乙胺或N,N-二异丙基乙基胺存在下,于惰性溶剂如二氯甲烷或N,N-二甲基甲酰胺中,在0℃和室温之间进行。
当使用酰氯时,反应在惰性溶剂如二氯甲烷或苯中,在碱如三乙基胺或N-甲基吗啉的存在下,以及在-60℃和室温之间进行。
根据本领域技术人员熟知的方法,在步骤2a)中,对如此得到的结构式(IV)的化合物进行非强制性的去保护。例如,当E表示2-四氢吡喃基时,通过利用于溶剂如乙醚,甲醇或其混合物中的盐酸,或利用在溶剂如甲醇中的对甲苯磺酸吡啶鎓,或利用于溶剂如甲醇中的Amberlyst的酸解而进行脱保护。反应在室温和溶剂回流温度之间进行。当E表示苯甲酰基或(C1-C4)烷基羰基时,通过在利用例如碱金属氢氧化物如氢氧化钠,氢氧化钾或氢氧化锂的碱介质中,于惰性溶剂如水,甲醇,乙醇,二噁烷或其混合物中,在0℃和溶剂的回流温度下的水解而进行脱保护。
在步骤3a)中,结构式(IV,E=H)醇与结构式(V)的磺酰氯的反应在碱如三乙胺,吡啶,N,N-二异丙基乙胺或N-甲基吗啉的存在下,于惰性溶剂如二氯甲烷,苯或甲苯中,在-20℃和溶剂回流温度之间的温度下进行。
如此得到的结构式(VI)的化合物以步骤4a)中与结构式(VII)的化合物进行反应。反应是在惰性溶剂如N,N-二甲基甲酰胺,乙腈,二氯甲烷,甲苯或异丙醇的存在下,以及存在或不存在碱下进行。当使用碱时,它选自:有机碱,如三乙胺,N,N-二异丙基乙基胺或N-甲基吗啉,和碱金属碳酸盐或碳酸氢盐,如碳酸钾,碳酸钠或碳酸氢钠。在没有碱的情况下,反应是利用过量的结构式(VII)化合物和在碱金属碘化物如碘化钾或碘化钠的存在下进行的。所述反应是在室温和100℃之间的温度下进行的。
根据该方法的一种变化形式为:
1b) 按照步骤1a)以及任选地按照步骤2a)进行;
2b) 使如此得到的结构式(IV,E=H)的化合物进行氧化,以便制备下式(VIII)的化合物:
3b) 在酸存在下,使结构式(VIII)的化合物与上面定义的结构式(VII)的化合物进行反应,然后,借助还原剂使所形成的中间亚胺鎓(imminium)盐进行还原;
4b) 利用无机或有机酸,将如此得到的化合物任选地转化成其一种盐。
根据该方法的变化形式,在步骤2b)中,使结构式(IV,E=H)的醇经过氧化,以便给出结构式(VIII)的醛。利用例如于溶剂如二氯甲烷中的乙酰氯,二甲基亚砜和三乙胺,并在-78℃和室温的温度下进行氧化反应。
接着,在步骤3b)中,在酸如乙酸的存在下,在惰性溶剂如甲醇或二氯甲烷中,使结构式(VII)的化合物与结构式(VIII)的醛反应,从而就地形成中间体胺,所述胺利用例如氰基硼氢化钠或三乙酰氧基硼氢化钠被化学还原,或者利用氢和催化剂如载于炭上的钯或Raney镍被催化还原。
最后,得到了根据本发明的结构式(I)的化合物。
根据常规的工艺,以游离碱的形式或以盐的形式分离如此得到的结构式(I)的化合物。
当以游离碱的形式得到结构式(I)的化合物时,借助在有机溶剂中所选的酸的处理而进行成盐作用。溶解于例如醚如二乙醚或醇如2-丙醇或丙酮或二氯甲烷,或醋酸乙酯中的游离碱,用上述溶剂之一中的所选酸溶液进行处理,将得到根据常规工艺被分离的相应盐。
因此,例如,制备盐酸盐,氢溴酸盐,硫酸盐,硫酸氢盐,磷酸二氢盐,甲磺酸盐,甲基硫酸盐,草酸盐,马来酸盐,丁二酸盐,富马酸盐,2-萘磺酸盐,苯磺酸盐,对甲苯磺酸盐或葡糖酸盐。
在反应结束时,结构式(I)的化合物能够以其一种盐的形式进行分离,例如盐酸盐或草酸盐;在这种情况下,如果需要,游离碱能够通过所述盐与无机或有机碱,或者与碱金属碳酸盐或碳酸氢盐的中和而制备,所述无机或有机碱如氢氧化钠或三乙胺,所述碱金属碳酸盐或碳酸氢盐如钠或钾的碳酸盐或碳酸氢盐。
结构式(II)的化合物通过已知的方法,特别是描述于EP-A-0512901,EP-A-0 591 040或EP-A-0 714 891中的方法来制备。
结构式(III)的化合物是市售产品,或根据已知的方法来制备。
因此,例如,根据如下方案1制备结构式(III)的化合物。方案1
根据描述于《美国化学会志》(J.Am.Chem.Soc.),(1941,63,3280-3282)中的方法,实施方案1的步骤a1和b1。
在步骤c1中,根据本领域技术人员已知的方法,由结构式(XI)的酸制备结构式(XII)的酯。
根据本领域技术人员已知的方法,在步骤d1中,将如此得到的酯(XII)还原成结构式(XIII)的醇。
根据《医药化学杂志》(J.Med.Chem.),(1973,16,684-687)中描述的方法,实施步骤e1和f1。
根据《有机化学》(J.Org.Chem.),(1968,33.4288)中或EP-A-0 714 891中描述的方法,在步骤g1中,将如此得到的结构式(XV)的苯基乙腈衍生物水解成结构式(III)的化合物。
结构式(IX)的溴代衍生物是已知的,或者根据已知的方法制备,所述方法如描述于《有机化学》,(1971,36(1),193-196),或描述于《美国化学会志》,(1941,63,3280-3282)中。
根据如下方案2,制备结构式(VII)的化合物,其中X表示基团 其中R2表示基团-CR3R4CONR5R6。方案2
其中R2表示基团-CR3R4CONR5R6。方案2
在方案2的步骤a2中,根据描述于《欧洲医药化学杂志》(Eur.J.Med.Chem.)(1990,25,609-615)中的方法,在2-羟基异丁腈的存在下,使化合物1与结构式(XVI)的酮反应。
根据本领域技术人员已知的方法,在步骤b2中,使如此得到的结构式(XVII)腈衍生物水解,以便得到结构式(XVIII)的酸衍生物。
根据肽偶联的常规方法,在步骤c2中,使酸(XVIII)与结构式(XIX)的胺进行反应,从而得到衍生物(XXI)。
另外,在步骤骤d2中,根据已知的方法,使结构式(XVII)的腈衍生物水解,从而得到结构式(XX)的羧酰胺(Carboxamide)衍生物,该衍生物可以在步骤e2中根据常规的方法任选地脱保护,从而得到其中R5=R6=H的化合物(VII)。
在步骤f2中,在强碱存在下,根据常规的烷基化方法,通过结构式(XX)的化合物,分别与(C1-C3)烷基卤化物,或顺序地与两种(C1-C3)烷基卤化物,或与结构式(Hal-R5-R6-Hal)的二卤化物进行反应,而制备结构式(XXI)的化合物,其中,R5表示(C1-C3)烷基,R6=H,或者R5和R6各自独立地表示(C1-C3)烷基,或者R5和R6与连接至其上的氮原子一起构成杂环。
根据已知的方法,在步骤g2中,使如此得到的化合物(XXI)脱保护,从而得到期望的化合物(VII)。
根据如下方案3,制备结构式(VII)的化合物,式中X表示基团 其中R2表示基团-CR3R4CONR5R6。方案3
其中R2表示基团-CR3R4CONR5R6。方案3
在方案3的步骤a3中,根据常规的烷基化方法,在强碱如氢化钠或氨基钠的存在下,在惰性溶剂如N,N-二甲基甲酰胺或二氯甲烷中以及在0℃和室温的温度下,化合物2分别与线性(C1-C4)烷基卤化物,或与式Hal(CH2)m-Hal的二卤化物(式中m=2-5,Hal表示卤原子)反应,可得到结构式(XXII)的化合物,式中R3和R4各自表示直链(C1-C4)烷基,或者与连接至其上的碳原子一起构成(C3-C6)环烷基。
根据本领域技术人员已知的方法,在步骤b3中,使如此得到的腈衍生物(XXII)水解,从而得到碳二亚胺衍生物(XXIII)。任选地,在步骤c3中,在催化剂如氧化铂的存在下,根据常规的方法,使吡啶环氢化,从而得到其中R5和R6=H的结构式(VII)的化合物。
在步骤d5中,根据前述常规的方法,使结构式(XXIII)的化合物进行烷基化反应,然后,借助常规的催化氢化使如此得到的化合物(XXIV)进行还原,结果得到其中R5和/或R6不等于H的结构式(VII)的化合物。
根据如下方案4,还能够获得其中X表示的结构式(VII)的化合物。方案4
在方案4的步骤a4中,根据描述于EP-A-0 625 509中的方法,使化合物3与适当的有机锂或有机镁衍生物反应,从而得到结构式(XXV)的醇;其中所述衍生物如甲基锂,乙基氯化镁,丙基氯化镁或戊烷-1,5-二(氯化镁)。
根据描述于Helvetica Chimica Acta(1972,55(7),2439)中的方法,在步骤b4中,使如此得到的醇(XXV)氧化成结构式(XXVI)的酸。
根据肽偶联的常规方法,在步骤c4中,使酸(XXVI)与结构式(XIX)的胺反应,从而得到化合物(XXVII)。
根据已知的方法,在步骤d4中,使化合物(XXVII)脱保护,从而得到期望的化合物(VII)。
根据常规的烷基化方法,在碱存在下,通过异哌啶甲酸乙酯与苄基溴的反应而制备化合物3。
结构式(VII)的化合物是新的并且将构成本发明的一部分。
因此,根据本发明的另一方面,本发明的目的是下式(VII)的化合物以及其与无机酸或有机酸的盐: 式中:-X表示基团
基团
式中:-X表示基团
基团

-R2表示基团-CR3R4CONR5R6;
-R3和R4表示相同的基团,它们选自甲基,乙基,正丙基或正丁基;
-或者R3和R4与连接至其上的碳原子一起构成(C3-C6)环烷基;
-R5和R6各自独立地表示氢;(C1-C3)烷基;
-或者R5和R6与连接至其上的氮原子一起构成杂环基团,所述杂环基团选自:1-氮杂环丁烷基,1-吡咯烷基,1-哌啶基,4-吗啉基,4-硫代吗啉基或全氢化-1-吖庚因基。
结构式(I)化合物的外消旋混合物的拆分使之能够分离下式(I*)的对映体,以及其与无机或有机酸可能的盐,和其溶剂合物和/或水合物: 式中“*”表示:如此标记的碳原子具有确定的(S)或(R)绝对构型;X,Ar和R1如结构式(I)化合物中的定义。
式中“*”表示:如此标记的碳原子具有确定的(S)或(R)绝对构型;X,Ar和R1如结构式(I)化合物中的定义。
然而,优选的是,由结构式(II,E=H)的中间化合物进行外消旋混合物的拆分,所述中间化合物是用于制备结构式(I)化合物,如下列专利申请所述:EP-A-0 512 901,EP-A-0 612 716和EP-A-0 591 040。
根据本发明的另一方面,本发明涉及:制备具有(S)构形的结构(I)化合物,其盐及其溶剂合物和/或水合物的立体有规方法,其特征在于:
1d)下式(II*,E=H)化合物的(S)异构体用下式酸(III)的官能衍生物进行处理: (II*,E=H)式中Ar如结构式(I)化合物的定义,式中R1如结构式(I)化合物中的定义,从而得到下式(IV*,E=H)的化合物:(IV*,E=H);
(II*,E=H)式中Ar如结构式(I)化合物的定义,式中R1如结构式(I)化合物中的定义,从而得到下式(IV*,E=H)的化合物:(IV*,E=H);
2d)将结构式(IV*)的化合物氧化,从而得到下式(VIII*)的化合物:
3d)在酸存在下,使结构式(VIII*)的化合物与下式(VII)化合物反应:式中X如结构式(I)化合物的定义,然后借助还原剂使所形成的中间亚胺鎓(imminium)盐进行还原;
4d)任选地,用无机或有机酸使如此得到的化合物转化成其一种盐。
上面结构式(I)的化合物也包含:其中一个或多个氢或碳原子已被其放射性同位素替换的那些化合物,所述同位素例如,氚或碳-14。所述的标记化合物可用于新陈代谢或药动学的研究,作为受体配体用于生化试验。
根据本发明的化合物进行生化试验。
借助利用放射配体的若干生化试验,在活体外,对所述化合物对于速激肽受体的亲合力进行评估:
1)[125I]BH-SP(利用Bolton-Hunter试剂,标记碘-125的物质P)与人体淋巴母细胞的NK1受体的结合(D.G.Payan等人,《免疫杂志》(J.Immunol.),1984,133,3260-3265)。
2)[125I]His-NKA与由CHO细胞表示的人体NK2克隆的受体的结合(Y.Takeda等人,J.Neurochem.,1992,59,740-745)。
3)[125I]His-[MePhe7]NKB与鼠大脑皮质的NK3受体、天竺鼠大脑皮质的NK3受体和沙土鼠大脑皮质的NK3受体,以及与由CHO细胞表示的人体NK3克隆的受体的结合(Buell等人,FEBS Letters,1992,299,90-95)。
根据X.Emonds-Alt等人的《欧洲药理学杂志》(Eur.J.Pharmacol.)(1993,250,403-413);《生命科学》(Life Sci.)(1995,56,PL 27-32),进行这些试验。
根据本发明的化合物可抑制物质P与人体IM9淋巴母细胞的NK1受体的结合。对于人体淋巴母细胞受体的抑制常数Ki约为10-11M。
对于人体NK2克隆受体的抑制常数Ki约为10-8M,而对于人体NK3克隆受体的抑制常数Ki大于10-7M。
对于人体NK1受体而言,结构式(I)的化合物是物质P强有力的且具有选择性的拮抗剂。
因此,针对动物模型,还在体内对结构式(I)的化合物进行评估。
在天竺鼠纹中,对NK1受体特异性的拮抗剂的局部应用,例如[Sar9,Met(O2)11]物质P,增加了乙酰胆碱的释放。通过口服或腹膜内施用本发明的化合物可抑制该释放。这一试验由R Steinberg等人的J.Neurochemistry 1995,65 2543-2548所述的方法一致。
这些结果证明式(I)化合物经口服是有效的,它可通过血脑屏障并且能够在枢神经系统中阻断对于NK1受体的特异性作用。
根据X.Emonds-Alt等人在《欧洲药理学杂志》(1993,250,403-413)中描述的方法,在天竺鼠支气管缩小的试验中,对结构式(I)的化合物进行评估。在这些试验条件下,静脉内给药的结构式(I)的化合物,将强烈地对抗由于肽(septide)静脉内给天竺鼠给药所产生的支气管缩小。
另外,还根据X.Emonds-Alt等人在《欧洲药理学杂志》(1993,250,403-413)中描述的方法,用狗的低血压模型,对结构式(I)化合物的体内药理学活性进行评估。在这些试验条件下,静脉内给药的结构式(I)的化合物,将强烈地抑制由于麻醉狗中[Sar9,Met(O2)11]物质P的静脉内给药所产生的低血压。
这些结果表明:结构式(I)的化合物将阻断与外周神经系统中NK1受体的特殊作用。
特别是,本发明的化合物是药物组合物的活性物质,其毒性与其用作药物产品相适合。
对于每公斤体重的待治疗的哺乳动物,上述结构式(I)的化合物能够以0.01-100mg的日剂量使用,优选的日剂量从0.1-50mg/kg。对于人类而言,取决于待治疗个体的年龄或治疗的类型:预防或治疗,所述剂量优选从0.1-4000mg/天,更优选从0.5-1000mg/天。
就其用作药物产品而言,结构式(I)的化合物通常以剂量单位给药。所述剂量单位优选用其中活性物质与一种或多种药物赋形剂混合的药物组合物来配制。
因此,根据本发明的另一方面,本发明涉及药物组合物,包含作为活性物质的结构式(I)化合物或其药物可接受盐,溶剂合物和/或水合物之一。
在用于口服,舌下,吸入,皮下,肌内,静脉内,经眼,局部,直肠给药的本发明的药物组合物中,与常规药物载体混合的活性物质能够以单位形式给动物和人类给药。合适的给药单位形式包括:口服路径形式,如片剂,凝胶,胶囊,粉剂,粒剂和口服液或口服悬浮液,舌下和口腔给药形式,气溶胶,局部给药形式,植入片,皮下,肌内,静脉内,鼻内或眼内给药形式,以及直肠给药形式。
当以片剂或胶囊形式制备固体组合物时,将由下列物质组成的药物赋形剂的混合物添加至微粒化或非微粒化的活性物质中,所述物质包括:稀释剂,如乳糖,微晶纤维素,淀粉,磷酸二钙;粘合剂,如聚乙烯吡咯烷酮,羟丙基甲基纤维素;崩解剂(délitant),如交联的聚乙烯吡咯烷酮,交联的羧甲基纤维素;流动剂,如硅石或滑石;以及润滑剂,如硬脂酸镁,硬脂酸,三榆树酸甘油酯或硬脂酰基富马酸钠。
另外,能够将润湿剂或表面活性剂,如月桂基硫酸钠,聚山梨酯80或泊咯沙姆188,添加至所述配方中。
可以通过各种工艺来制备片剂,所述工艺如直接压片,干法成粒,湿法成粒,热熔融。
片剂可以是裸露的或涂有糖(例如涂有蔗糖),或者涂有各种聚合物或其它合适的材料。
借助制备聚合物基体或借助使用特定的成膜聚合物,片剂可以有瞬间释放,迟延释放或持续释放的形式。
胶囊可以是软的或硬的,并且涂有膜等等,以便使之具有瞬间释放,迟延释放或持续释放的活性(例如通过肠的形式)。
它们不仅能够包含片剂用的如上配制的固体配方,而且能够包含液体或半固体的配方。
糖浆或酏剂形式的制剂可以包含活性物质以及甜味剂,优选是无卡路里甜味剂,作为抗茵剂的甲基对羟基苯甲酸酯和丙基对羟基苯甲酸酯,以及调味剂和合适的着色剂。
水可分散的粉末或颗粒可以包含活性物质,如与分散剂,润滑剂或悬浮剂,如聚乙烯吡咯烷酮,以及甜味剂或增味剂的混合物。
对于直肠投药,可以使用利用粘合剂制备的栓剂,所述粘合剂在直肠温度下将熔融,例如可可脂或聚乙二醇。
将水悬浮液,等渗盐溶液或无菌、可注射溶液用于肠胃外,鼻内或眼内给药,所述溶液包含:药理学相容的分散剂和/或加溶剂,例如丙二醇。
因此,为了制备能够进行静脉注射的水溶液,可以使用:助溶剂,如醇,例如乙醇或者乙二醇如聚乙二醇或丙二醇,以及亲水性表面活性剂,如聚山梨酯80或泊咯沙姆188。为制备肌内给药用的可注射的油性溶液,可以利用三甘油酯或甘油酯来溶解活性物质。
对于局部给药,可以使用乳膏,软膏,凝胶,滴眼剂和喷雾剂。
对于经眼的给药,可以使用多层或贮器形式的补片,其中活性成分可以在醇溶液中,和喷雾剂。
对于吸入给药,可以使用气溶胶,所述气溶胶包含:例如,三油酸脱水山梨糖醇酯或油酸以及三氯氟甲烷,二氯氟甲烷,二氯四氟乙烷,氟利昂取代物或其它生物相容的抛射气;另外也能够使用粉末形式的、只包含活性物质或与赋形剂复合的体系。
另外,活性物质也可以呈与环糊精,例如,α-,β-,或γ-环糊精或2-羟丙基-β-环糊精复合体的形式。
另外,也可以微胶囊或微球的形式配制活性物质,其中任选地含有一种或多种载体或添加剂。
在慢性治疗的情况下有用的持续释放的形式中,可以使用植入片。这些植入片能够以在等渗介质中微球的油性悬浮液形式或悬浮液形式来制备。
在各剂量单位中,结构式(I)的活性物质以与设计的每日剂量相当的量存在。通常,各剂量单位根据剂量和设计的给药方式进行适当调节,给药方式例如片剂,胶囊等,药袋,安瓿,糖浆等,或滴剂,以致使剂量单位包含0.1-1000mg的活性物质,优选的是0.5-250mg,所述剂量需要一天分1-4次进行给药。
尽管这些剂量是平均情况下的例子,但也存在着其中更高或更低剂量是适合的特定情况,并且所述剂量也将构成本发明的一部分。根据通常的实践,适合于各病人的剂量由医生根据给药方式,年龄,所述病人的体重和反应来确定。
根据本发明的另一方面,本发明涉及结构式(I)化合物,或其药物可接受盐,溶剂合物和/或水合物用于制备药品的用途,所述药品用于治疗其中涉及物质P和人体NK1受体的病变。
根据本发明的另一方面,本发明涉及结构式(I)化合物,或其药物可接受盐,溶剂合物和/或水合物用于制备药品的用途,所述药品用于治疗呼吸系统,胃肠系统,泌尿系统,免疫系统或心血管系统和中枢神经系统的病变,以及用于治疗疼痛,偏头痛,炎症,恶心和呕吐,以及皮肤病。
例如并且以非限定的方式,结构式(I)的化合物在下列各方面将是有用的:
-用作止痛剂,特别是治疗外伤性疼痛,如术后疼痛;臂丛神经痛;慢性疼痛,如由骨关节炎,类风湿性关节炎,牛皮癣性关节炎引起的关节炎疼痛;神经病性疼痛,如疱疹后神经痛,三叉神经的神经痛,节神经痛或肋间神经痛,纤肌痛,灼痛,周围神经病,糖尿病的神经病,由化学疗法引起的神经病,与爱滋病有关的神经病,枕骨神经痛,膝状神经痛或咽神经痛;切肢者因错觉产生的疼痛;各种头痛,如慢性或急性偏头痛,颞下颌疼痛,上颌窦疼痛,面部神经痛或牙痛;癌症患者所经受的疼痛;内脏起因疼痛;胃肠疼痛;由神经压缩引起的疼痛,由剧烈运动引起的疼痛;痛经;月经疼痛;由脑膜炎或蛛网膜炎引起的疼痛;肌与骨胳的疼痛;由脊柱狭窄引起的背部下方的疼痛,脱垂盘或坐骨神经痛;咽峡炎患者经受的疼痛;僵硬脊椎炎引起的疼痛;与痛风有关的疼痛;与灼伤,结瘢或痒疹皮肤病有关的疼痛;丘脑疼痛;
-抗炎症药,特别是用于治疗气喘,流感,慢性支气管炎(特别是阻塞性支气管炎和COPD(慢性阻塞性肺病)),咳嗽,变应性变态反应,支气管痉挛和风湿病样关节炎的炎症;胃肠系统的炎性疾病,例如,克罗恩氏病,溃疡性结膜炎,胰腺炎,胃炎,肠炎,由非类固醇抗火症药引起的疾病,由细茵感染引起的炎症和分泌作用,例如由难对付梭状芽胞杆菌引起的炎症和分泌作用;炎性皮肤病,例如疱疹和湿疹;炎性膀胱疾病,如膀胱炎和尿失禁;眼炎症,如结膜炎和玻璃体视网膜病变;牙炎症,如龈炎和牙周炎;
-用于治疗变应性疾病,特别是皮肤病,如荨麻疹,接触性皮炎,特异反应性皮炎和呼吸疾病,如鼻炎;
-用于治疗中枢神经系统的疾病,特别是精神病,如精神分裂症,躁狂和痴呆;认识障碍,如早老性痴呆,焦虑,与爱滋病有关的痴呆,糖尿病的神经病;抑郁症;帕金森氏病;药物依赖症;物质滥用;意识紊乱,睡眠障碍,近昼夜节律紊乱,心境紊乱和癫痫;Down氏综合征;慢性舞蹈病;压迫性躯体病症;神经变性病,如皮克氏病或Creutzfeldt-Jacob病;与恐慌,恐怖或紧张状态有关的病症;
-在中枢神经系统炎性和免疫处置期间,例如在与爱滋病有关的感染期间,用于治疗血脑障壁渗透性的改进;
-用于骨骼肌松弛药和镇痉剂;
-用于治疗急性或迟延且预期的恶心和呕吐,例如由药物,如在癌症的情况下在化疗中使用的制剂的引起的恶心和呕吐;在治疗癌症或类癌症(carcinoidose)时,在胸或腹照射期间,由放射疗法引起的恶心和呕吐;由于食入毒物所引起的恶心和呕吐;由新陈代谢或感染病症如胃炎造成的,或者在细菌或病毒性胃肠感染期间所产生的毒素所引起的恶心和呕吐;妊娠期间所引起的恶心和呕吐;在前庭病症期间如旅行病,眩晕或梅尼埃尔氏病所引起的恶心和呕吐;术后病症所引起的恶心和呕吐;由透析或前列腺素所引起的恶心和呕吐;由于胃肠阻塞所引起的恶心和呕吐;由于降低的胃肠能动力所引起的恶心和呕吐;由心肌梗塞或腹膜炎造成的内脏疼痛所引起的恶心和呕吐;偏头痛所引起的恶心和呕吐;高空病所引起的恶心和呕吐;食入鸦片止痛剂如吗啡所引起的恶心和呕吐;由胃食管回流所引起的恶心和呕吐;酸性消化不良或食物或饮料过份消耗,胃酸度或酸涩,反胃,和胃灼热,例如,插话式的或夜间的胃灼热,或由肉和消化不良造成的胃灼热所引起的恶心和呕吐;
-用于治疗胃肠系统的疾病,如过敏性肠综合征,胃和十二指肠溃疡,食管溃疡,腹泻,分泌过多,淋巴瘤,胃炎,胃食管回流,大便失禁,赫希施普龙病(Hirschsprung)和食物厌恶症;
-用于治疗皮肤疾病,如牛皮癣,搔痒症和灼伤,特别是晒伤;
-用于治疗心血管系统的疾病,如高血压,由偏头痛,水肿,血栓形成,心绞痛,血管痉挛,血管舒张造成的循环疾病,雷诺氏病,纤维变性,胶原疾病和动脉硬化症的血管方面;
-用于治疗小细胞(Petites cellules)肺癌;泌尿生殖系统的脑瘤和腺癌;
-用于治疗脱髓鞘疾病,如多硬化或肌萎缩外侧硬化;
-用于治疗与免疫细胞的抑制或刺激有关的免疫系统的疾病,例如,类风湿性关节炎,牛皮癣,克罗恩氏病,糖尿病,狼疮,移植术后的排异反应;
-用于治疗排尿紊乱,特别是频尿;
-用于治疗组织细胞性网状细胞增多,例如,淋巴组织增多;
-用于引起食欲缺乏的;
-用于治疗肺气肿,莱特尔氏病,和痔疮;
-用于治疗眼病,如青光眼,眼高血压,瞳孔缩小和过度泪分泌;
-用于治疗或防治由于血管的发作或闭合引起的癫痫发作,颅外伤,脊髓外伤,小脑的局部缺血损害;
-用于治疗心搏率和心节律紊乱,特别是由疼痛或紧张引起的心搏率和心节律紊乱;
-用于治疗过敏皮肤和用于防治皮肤或粘膜,皮屑,红斑或瘙痒的刺激;
-用于治疗神经学上皮肤病症,如苔癣,痒疹,痒疹的中毒性皮病和神经性严重瘙痒;
-用于治疗幽门螺杆菌或脲酶阳性革兰氏阴性的细菌引起的溃疡和所有疾病;
-用于治疗由血管生成或其中血管生成为症状所引起的疾病;
-用于治疗眼和/或眼睑(Palbébrales)和/或眼或眼睑的感觉迟钝;
-用作止汗剂。
另外,本发明还涉及在上述剂量下治疗所述病症的方法。
根据本发明的药物组合物还能够包含可用于治疗上述疾病或病症的其它活性物质,例如,支气管扩张药,镇咳剂,抗组胺剂,抗炎症药,抗吐剂和化疗剂。
在制备例和实施例中使用下列缩写:
DMF:二甲基甲酰胺
DMSO:二甲基亚砜
DCM:二氯甲烷
THF:四氢呋喃
醚:二乙醚
盐酸醚:在二乙醚中的盐酸饱和溶液
BOP:苯并三唑-1-基氧化三(二甲氨基)六氟磷酸鏻
F:熔点
Teb:沸点
TA:室温
二氧化硅H:由Merek(Darmstadt)出售的60H硅胶。
将DMSO-d6峰用作参考,在DMSO-d6峰中,于200MHz处记录质子核磁共振(RMN1H)光谱。化学位移δ以每百万的份数(ppm)表示。所观察到的信号表示如下:s:单峰;se:宽单峰;t:三重峰;qd:四重峰;m:多重峰。
制备例1.1
3-(3,4-二氯苯基)-3-(2-羟乙基)哌啶,(-)异构体,该化合物的制备描述于专利申请EP-A-0 591 040。制备例1.23-(3,4-二甲基苯基)-3-[2-(2-四氢吡喃氧基)-乙基]哌啶
A)2-(3,4-二甲基苯基)-4-(2-四氢吡喃氧基)-丁腈
于TA下,将6.6克于油中60%的氢化钠分批添加至于100ml无水THF中的20克3,4-二甲基苯基乙腈中,并在TA下对该混合物搅拌2小时。然后滴加29克1-溴-2-(2-四氢吡喃氧基)乙烷并在TA下对该混合物搅拌2天。将反应混合物倒入冰中并用AcOEt进行萃取,有机相用水和饱和氯化钠进行洗涤,并在硫酸钠上进行干燥,溶剂在真空下蒸发掉。残余物用硅胶进行色谱分析(用甲苯进行洗脱然后用梯度从99/1(v/v)至90/10(v/v)的甲苯/AcOEt混合物进行洗脱。结果得到17克期望的产物。
B)4-氰基-4-(3,4-二甲基苯基)-6-(2-四氢吡喃氧)己酸甲酯
将0.3ml于MeOH中的40%苄基三甲基氢氧化铵(TritonB)加至上面步骤中得到的化合物17克和于30ml二噁烷中的11ml丙烯酸甲酯的混合物中,并在TA下对该混合物搅拌48小时。在真空下使反应混合物浓缩,将残余物溶解于0.5N的盐酸水溶液中并用醚萃取,有机相用10%的碳酸钠水溶液进行洗涤并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。结果得到了23克期望的产物。
C)5-(3,4-二甲基苯基)-5-[2-(2-四氢吡喃氧)乙基]-2-哌啶酮。
将40ml氨水溶液加至上面步骤得到的23克化合物于250ml95%EtOH中的溶液中,然后添加Raney镍。再在40℃和16巴压力下使该混合物氢化24小时。在Célite上过滤掉催化剂并在真空下浓缩滤出液。结果得到22克期望的产物。
D)3-(3,4-二甲基苯基)-3-[2-(2-四氢吡喃氧)-乙基]哌啶。
将22克上面步骤得到的化合物添加至10克氢化锂铝于200mlTHF的悬浮液中,然后回流2小时。在冷却至TA之后,添加10ml水和80mlTHF,然后添加10ml 4N的氢氧化钠和30ml水。在Celite上过滤掉无机盐并在真空下使滤出液浓缩。结果得到15克期望的产物。
制备例2.1
3,5-二氯苯基乙酸。
(III):R1=Cl。
A)3,5-二氯苄基氯。
于TA,将12.5克亚硫酰氯于20ml氯仿中的溶液滴加至14.5克3,5-二氯苄醇于150ml氯仿中的溶液中,然后,在40-50℃加热8小时并于TA搅拌过夜。在真空下浓缩混合物,从而得到16克期望的产物,该产物就此使用。
B)3,5-二氯苯基乙腈。
将6.5克氰化钾于50ml水中的溶液添加至16克上面步骤得到的化合物于50ml EtOH中的溶液中,并使该混合物回流4小时。在真空下浓缩得到的混合物,将残余物溶于水中并用醚进行萃取,有机相用水洗涤并在硫酸钠上进行干燥,并在真空下蒸发掉溶剂。用二氧化硅H对残余物进行色谱分析(用庚烷/甲苯混合物(50/50;v/v)然后用甲苯进行洗脱)。结果得到了7克期望的产物,该产物就此使用。
C)3,5-二氯苯基乙酸。
将8.4克KOH于10ml水中的溶液添加至7克上面步骤得到的化合物于50ml EOH中的溶液中,然后加热回流5小时。在真空下浓缩该混合物,将残余物溶液于水中并用醚洗涤水相,通过添加浓盐酸酸化至pH=1,并在TA下搅拌过夜。将形成的结晶产物离心脱水,用水洗涤并在60℃真空下进行干燥。结果得到了7克期望的产物;F=112-114.5℃。
制备例2.2
3,5-二乙基苯基乙酸。
(III):R1=Et
A)3,5-二乙基溴苯。
将20克4-溴-2,6-二乙基苯胺,160ml乙酸,100ml浓盐酸溶液,30ml水和100ml EtOH的混合物冷却至-5℃,然后滴加6.6克亚硝酸钠于25ml水中的溶液,并在TA下对该混合物搅拌30分钟。将反应混合物倒入冷却至0℃的170ml 50%的H3PO2中,并在0℃搅拌2小时,然后在TA下搅拌48小时。反应混合物用醚进行萃取,有机相用水,用1N氢氧化钠溶液,再用水进行洗涤,并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。残余物用硅胶进行色谱分析(用环己烷洗脱)。结果得到了18克期望的产物。
B)3,5-二乙基苯甲腈。
使24.7克上面步骤得到的化合物与12克氰化亚铜于70ml DMF中溶液的混合物加热回流15小时。在冷却至TA之后,将反应混合物倒入50ml水中并在TA搅拌至形成胶质(gomme)。在冰浴中冷却该混合物,添加150ml乙二胺并在TA下对该混合物搅拌2小时。用AcOEt对该混合物进行萃取,有机相用水洗涤并在硫酸钠上进行干燥,并在真空下蒸发掉溶剂。用硅胶对残余物进行色谱分析(用环己烷/AcOEt混合物(95/5;v/v)进行洗脱)。结果得到了12克期望的产物。
C)3,5-二乙基苯甲酸。
将22克KOH于15ml水中的溶液添加至12克上面步骤得到的化合物于EtOH中的溶液中,然后加热回流24小时。在真空下浓缩反应混合物,残余物用水进行萃取,水相用醚进行洗涤并通过添加浓盐酸酸化至pH=2,所形成的沉淀物进行离心脱水,用水洗涤并在真空下进行干燥。结果得到了13克期望的产物。
D)3,5-二乙基苯甲酸甲酯
将13克上面步骤得到的化合物于90ml MeOH中的溶液和10滴硫酸的混合物加热回流48小时。在真空下浓缩反应混合物,残余物溶于水中,通过添加10%的碳酸氢钠溶液进行中和,并用醚进行萃取,有机相用10%碳酸氢钠溶液、水进行洗涤并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。结果得到了12克期望的产物。
E)3,5-二乙基苯甲醇。
将2.5克氢化锂铝于50ml THF中的悬浮液冷却至0℃,滴加12克上面步骤得到的化合物于50ml THF中的溶液,并对该混合物搅拌30分钟。通过添加2.5ml水,2.5ml 4N的氢氧化钠和7.5ml水而使反应混合物水解。过滤掉无机盐并在真空下浓缩滤出液。结果得到了10.9克期望的产物,该产物就此使用。
F)3,5-二乙基苄基甲磺酸酯。
在TA下,将8.4克甲磺酰氯于50ml DCM中的溶液滴加至10.9克上面步骤得到的化合物和7.4克三乙胺于100ml DCM中的溶液中,并对该混合物搅拌30分钟。在真空下浓缩反应混合物,残余物溶解于水中并用醚进行萃取,有机相用水进行洗涤并在硫酸钠上进行干燥,在真空下蒸发掉溶剂,结果得到了16克期望的产物,该产物就此使用。
G)3,5-二乙基苯基乙腈。
将5.15克氰化钾于20ml水中的溶液添加至16克上面步骤得到的化合物于100ml DMF中的溶液中,并在80℃对该混合物加热1小时。将反应混合物在真空下浓缩,残余物溶解于水中并用醚进行萃取,有机相用水进行洗涤并在硫酸钠上进行干燥,并在真空下蒸发掉溶剂。用硅胶H,对残余物进行色谱分析(用DCM进行洗脱)。结果得到了3克期望的产物。
H)3,5-二乙基苯基乙酸。
将7.8克KOH于10ml水中的溶液添加至3克上面步骤得到的化合物于50ml EtOH中的溶液中,然后加热回流5小时。在真空下对该混合物进行浓缩,残余物溶解于水中并用醚对水相进行洗涤,通过添加浓盐酸酸化至pH=1,并在TA下搅拌过夜。对所形成的结晶产物进行离心脱水,用水进行洗涤并在真空下进行干燥。结果得到了2.5克期望的产物。RMN1H:δ(ppm):1.1:t:6H;2.4:qd:4H;3.4:s:2H;6.8:m:3H;12.2:se:1H.制备例3.12-(4-哌啶基)异丁酰胺盐酸盐。
A)2-甲基-2-(4-吡啶基)丙腈。
向冷却至0℃的3克4-吡啶基乙腈盐酸盐于50ml DMF的混合物中,分小批加入2.6克于油中的60%氢化钠,并在TA下对该混合物搅拌2小时。在冰浴中对反应混合物进行冷却,滴加6克甲基碘并在TA下让该混合物搅拌过夜。将反应混合物倒入水/冰混合物中,并用醚进行萃取,有机相用饱和氯化钠溶液进行洗涤,在硫酸镁上进行干燥并进行过滤,在真空下蒸发掉溶剂。用硅胶H对残余物进行色谱分析(先用DCM后用DCM/MeOH混合物(98/2;v/v)进行洗脱)。结果得到了2.39克期望的产物,当结晶时呈油状。
B)2-(4-吡啶基)异丁酰胺盐酸盐。
在100℃,对2.39克上面步骤得到的化合物和10ml浓硫酸溶液的混合物加热15分钟。将反应混合物冷却至TA,添加50克冰,通过添加浓氢氧化钠溶液,将该混合物碱化至pH=14,过滤掉无机盐,用AcOEt然后用DCM对滤出液进行萃取,混合的有机相在硫酸镁上进行干燥并过滤,在真空下蒸发掉溶剂(F=134℃,碱)。将得到的产物溶解于丙酮中并通过添加盐酸醚酸化至pH=1,对形成的沉淀物进行离心脱水。结果得到了2.9克期望的产物。
C)2-(4-哌啶基)异丁酰胺盐酸盐。
将2.9克上面步骤得到的化合物,1克PtO2和50ml MeOH的混合物在60℃于60巴的压力下氢化3小时。在Celite上过滤掉催化剂并用MeOH进行洗涤,在真空下浓缩滤出液。残余物溶解于乙腈中并对形成的沉淀物进行离心脱水,再用乙腈然后用醚进行洗涤。结果得到了2.5克期望的产物;F>260℃。
制备例3.2
2-(1-哌嗪基)异丁酰胺二盐酸盐。
A)2-(4-苄基-1-哌嗪基)-2-甲基丙腈。
将4.5ml丙酮,20克无水硫酸镁,10克N,N-二甲基乙酰胺,10克1-苄基哌嗪和9.5ml 2-羟基异丁腈混合在一起并于45℃在剧烈搅拌下加热48小时。将反应混合物倒入冰中并搅拌30分钟。用醚对该混合物进行萃取,用水对有机相洗涤若干次并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。结果得到了13克期望的产物。
B)2-(4-苄基-1-哌嗪基)异丁酰胺二盐酸盐
将13克上面步骤得到的化合物和130ml 90%的硫酸溶液的混合物在110℃快速加热30分钟。在冷却至AT后,将反应混合物倒入冰中并通过添加浓氢氧化铵溶液碱化至pH=10,并对形成的结晶产物进行离心脱水。将该产物溶解于DCM中有机相在硫酸镁上进行干燥,在真空下蒸发掉溶剂。将该产物溶解于盐酸醚中,并对形成的沉淀物进行离心脱水。结果得到了9.5克期望的产物。
C)2-(1-哌嗪基)异丁酰胺二盐酸盐。
于AT和大气压下,对1.3克上面步骤得到的化合物和0.18克于10%钯/炭(于30ml 95%的EtOH中)的混合物氢化过夜。在Celite上过滤掉催化剂并在真空下使滤出液进行浓缩。结果得到了0.6克期望的产物。
制备例3.3
1-(1-哌嗪基)环己烷羧酰胺二盐酸盐
A)1-(-苄基-1-哌嗪基)环己腈。
将5.7克环己酮,20克无水硫酸镁,10克N,N-二甲基乙酰胺,10克1-苄基-哌嗪和9.5ml 2-羟基异丁腈混合在一起并在剧烈搅拌下于45℃加热48小时。将反应混合物倒入冰中并搅拌30分钟。用醚对该混合物进行萃取,用水对有机相洗涤若干次并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。结果得到了15克期望的产物。
B)1-(4-苄基-1-哌嗪基)环己烷羧酰胺二盐酸盐。
从15克上面步骤得到的化合物和50ml 90%的硫酸溶液为原料,根据制备例3.2的步骤B所述的操作方式得到该化合物。结果得到了5.5克期望的产物。
C)1-(1-哌嗪基)环己烷羧酰胺二盐酸盐。
从2.3克上面步骤得到的化合物和0.3克10%钯/炭(于30ml 95%的EtOH中)为原料,根据制备例3.2的步骤C所述的操作方式得到该化合物。结果得到了1.6克期望的产物。
制备例3.4
N,N-二甲基-2-(1-哌嗪基)异丁酰胺二甲酸酯。
A)N,N-二甲基-2-(4-苄基-1-哌嗪基)异丁酰胺。
将1.44克于油中60%的氢化钠分批加至2.6克制备例3.2的步骤B中得到的化合物(游离碱)于50ml无水THF中的混合物中。然后,滴加1.3ml甲基碘并于TA下对该混合物搅拌4小时。将反应混合物倒入水中并用醚进行萃取,有机相在硫酸镁上干燥,并在真空下蒸发掉溶剂。结果得到了1.8克期望的产物。
B)N,N-二甲基-2-(1-哌嗪基)异丁酰胺二甲酸酯。
将2克甲酸铵和0.5克5%的钯/炭添加至1.8克上面步骤中得到的化合物于30ml MeOH中的溶液中,并于TA下对该混合物搅拌4小时。在Celite上过滤掉催化剂并在真空下浓缩滤液。将残余物溶解于AcOEt中并对形成的沉淀物进行离心脱水,用AcOEt进行洗涤并干燥。结果得到了1.2克期望的产物。
制备例3.5
1-(4-哌啶基)环己烷羧酰胺盐酸盐。
A)1-(4-吡啶基)环己腈
将3克4-吡啶基乙腈盐酸盐于50ml DMF中的混合物冷却至0℃,分小批添加2.6克于油中60%的氢化钠,并在TA下对该混合物搅拌30分钟。在冰浴中对反应混合物冷却,滴加2.7ml 1,5-二溴戊烷并在TA下对该混合物搅拌48小时。将反应混合物倒入饱和氯化铵溶液中并用醚进行萃取,有机相用水洗涤三次并在硫酸镁上进行干燥,在真空下蒸发掉溶剂。用硅胶H对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(98/2;v/v)进行洗脱)。结果得到了2.5克期望的产物;F=79℃。
B)1-(4-吡啶基)环己烷羧酰胺盐酸盐。
在100℃对2.5克上面步骤得到的化合物和15ml浓硫酸溶液的混合物加热15分钟。将该反应混合物冷却至TA,倒入冰中并通过添加浓氢氧化钠溶液碱化至pH=14,并对形成的沉淀物进行离心脱水,用水洗涤并干燥。将得到的产物溶解于丙酮中,通过添加盐酸醚酸化至pH=1并在TA下搅拌30分钟,并对形成的沉淀物进行离心脱水。结果得到了3克期望的产物;F=224℃(déc.)。
C)1-(4-哌啶基)环己烷羧酰胺盐酸盐。
在60℃和80巴的压力下,使2.9克上面步骤得到的化合物、0.5克PtO2和50ml MeOH的混合物氢化3天。在Célite上过滤掉催化剂并在真空下浓缩滤出液。将残余物溶解于乙腈中并在TA下搅拌1小时,对形成的沉淀物进行离心脱水。结果得到了2.7克期望的产物;F=235℃。
制备例3.6
N,N-二甲基-2-(4-哌啶基)异丁酰胺盐酸盐。
A)1-苄基-4-哌啶羧酸乙酯。
将30克苄基溴滴加至25克六氢异烟酸乙酯和25克碳酸钾于125ml DMF中的混合物中,同时将反应混合物的温度保持在25和30℃之间,然后在TA下对该混合物搅拌1小时。将反应混合物倒至1升冰水中并用醚萃取两次,有机相用水洗涤并在硫酸镁上进行干燥,在真空下蒸发掉溶剂。得到的油在减压下蒸馏。结果得到29.2克期望的产物;在2.7Pa下,Teb=120-122℃。
B)2-(1-苄基-4-哌啶基)-2-丙醇。
在将介质的温度保持在25和30℃之间的同时,在氩气氛下,将24.73克上面步骤得到的化合物于100ml苯中的溶液滴加至200ml1.5M的甲基锂溶液中,作为在醚中与溴化锂的配合物,然后加热回流48小时。将反应混合物冷却至TA然后倒入400ml饱和氯化铵水溶液中,所述溶液事先已在冰浴中冷却。用醚对混合物萃取三次,混合有机相在硫酸镁上进行干燥,并在真空下浓缩溶剂。残余物溶解于丙酮中,冷却至10℃并通过添加盐酸醚而酸化至pH=1,并对形成的沉淀物进行离心脱水以及用丙酮/醚混合物(50/50;v/v)进行洗涤。结果得到24.5克盐酸盐形式的期望的产物;F=204℃。为使碱游离,将该盐酸盐溶解于浓氢氧化钠溶液中,用醚进行萃取并在硫酸镁上进行干燥,在真空下蒸发掉溶剂。结果得到21克期望的产物;F=66℃。
C)2-(1-苄基-4-哌啶基)-2-甲基丙酸。
将5.98克95%的硫酸和4.42克包含30%SO3的发烟硫酸的混合物冷却至3℃,并在温度保持在10℃以下的同时,滴加2克上面步骤得到的化合物于1.55克100%甲酸中的溶液。在3-5℃对该混合物搅拌2小时,然后使之返回至TA,并在TA下搁置过夜。将反应混合物倒入冰中,通过添加浓氢氧化钠溶液和添加浓氢氧化铵溶液将pH调节至6.5,并用DCM萃取三次,混合有机相在硫酸镁上进行干燥并在真空下浓缩溶剂。将残余物溶解于丙酮中并对沉淀物进行离心脱水和干燥。结果得到1.22克期望的产物;F=195℃。
D)N,N-二甲基-2-(1-苄基-4-哌啶基)异丁酰胺盐酸盐。
在TA下,对1.2克上面步骤得到的化合物,0.8ml三乙胺,2.8ml于THF中的2M二甲胺溶液和2.5克于20ml DCM中的BOP的混合物搅拌1小时。在真空下浓缩该反应混合物,残余物溶解于醚中,有机相用水,用1N氢氧化钠溶液,用饱和氯化钠溶液进行洗涤并在硫酸镁上进行干燥,在真空下浓缩溶剂。用硅胶H对残余物进行色谱分析(用DCM,然后用DCM/MeOH混合物(梯度(99/1;v/v至95/5;v/v))进行洗脱)。将得到的产物溶解于丙酮中并通过添加盐酸醚而酸化至pH=1,对形成的沉淀物进行离心脱水和干燥。结果得到0.8克期望的产物;F=229℃。
E)N,N-二甲基-2-(4-哌啶基)异丁酰胺盐酸盐。
在大气压和TA下,使0.8克上面步骤得到的化合物和0.2克在10%的钯/炭(于20ml MeOH中)氢化过夜。在Célite过滤掉催化剂并在真空下浓缩滤液。残余物溶解于乙腈中,添加醚并对形成的沉淀物进行离心脱水和干燥。结果得到0.51克期望的产物;F=258℃。
制备例3.7
1-(4-吡啶基)环丙烷羧酰胺盐酸盐。A)1-(4-吡啶基)环丙腈。
将3.5克4-吡啶基乙腈添加至2.5克于80ml DCM中的氨基钠的混合物中,然后,添加2.6ml 1,2-二溴乙烷,并在TA下对该混合物搅拌过夜。将反应混合物倒入水中并用AcOEt进行萃取,有机相用水洗涤并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。用硅胶对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1;v/v)至(95/5;v/v))进行洗脱)。结果得到2.5克期望的产物。
B)1-(4-吡啶基)环丙烷羧酰胺盐酸盐。
将2.5克上面步骤得到的化合物和20ml 96%的硫酸溶液的混合物迅速加热至100℃,并在100℃搅拌1小时。在冷却至TA之后,将反应混合物倒入冰中并通过添加20%的氢氧化铵溶液而中和至pH=7,对形成的沉淀物进行离心脱水,用水进行洗涤并干燥。沉淀物溶解于DCM中,通过添加盐酸醚而酸化至pH=1,并对形成的沉淀物进行离心脱水。结果得到1.8克期望的产物。
C)1-(4-吡啶基)环丙烷羧酰胺盐酸盐。
在80℃和100巴压力下,使1.8克上面步骤得到的化合物和0.6克于50ml MeOH中的PtO2氢化15小时。在Célite上过滤掉催化剂,在真空下使滤出液浓缩至5ml体积并添加乙腈,直至发生结晶为止。在离心脱水随后干燥之后,得到1.7克期望的产物。
制备例3.8
2-甲基-1-(4-吗啉基)-2-(4-哌啶基)-1-丙酮盐酸盐。
A)2-(1-苄基-4-哌啶基)-2-甲基-1-(4-吗啉基)-1-丙酮盐酸盐。
将1克制备例3.6的步骤C得到的化合物和1.2ml于20ml 1,2-二氯乙烷中的亚硫酰氯的混合物于80℃加热3小时。在真空下浓缩反应混合物,将如此得到的酰基氯溶解于20ml DCM中,将该溶液添加至0.7克吗啉和1.6ml于20ml事先冷却至0℃的DCM中的三乙胺的混合物中,并在TA下搅拌24小时。在真空下浓缩反应混合物,残余物用醚进行萃取,有机相用1N氢氧化钠溶液、水进行洗涤,并在硫酸镁上进行干燥,在真空下蒸发掉溶剂。将得到的产物溶解于丙酮中并通过添加盐酸醚而酸化至pH=1,并对形成的沉淀物进行离心脱水和干燥。结果得到0.7克期望的产物。
B)2-甲基-1-(4-吗啉基)-2-(4-哌啶基)-1-丙酮盐酸盐。
在TA下,对0.7克上面步骤得到的化合物,0.7克甲酸铵和0.2克10%的钯/炭(于10ml MeOH中)的混合物搅拌4小时。在Célite上过滤掉催化剂并在真空下浓缩滤出液。将残余物溶解于乙腈中,添加醚并对形成的沉淀物进行离心脱水和干燥。结果得到0.46克期望的产物;F=225℃。
实施例1
3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲基苯基)乙酰基]哌啶盐酸盐一水合物,(-)异构体。
A)3-(3,4-二氯苯基)-3-(2-羟乙基)-1-[2-(3,5-二甲基苯基)乙酰基]哌啶,单一异构体。
于TA下,将2.3ml三乙胺添加至2.0克3,5-二甲基苯基乙酸于100ml DCM中的混合物中,然后添加3克制备例1中得到的化合物和5.3克BOP,并在TA下对该混合物搅拌1小时。在真空下浓缩反应混合物,残余物用醚进行萃取,有机相用水,用2N盐酸溶液,用水,用10%氢氧化钠水溶液进行洗涤,在硫酸钠上进行干燥,并进行过滤,在真空下浓缩滤出液。用硅胶H对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(98/2;v/v)进行洗脱)。结果得到3.9克期望的产物,该产物就此在下列步骤中使用。
B)3-(3,4-二氯苯基)-3-(甲酰基甲基)-1-[2-(3,5-二甲基苯基)乙酰基]哌啶,单一异构体。
在氮气氛下,将0.25ml草酰氯于3ml DCM中的溶液冷却至-70℃,滴加0.35ml DMSO于3ml DCM中的溶液,然后添加0.5克上面步骤得到的化合物于5ml DCM中的溶液,并在-50℃下搅拌15分钟。然后添加0.9ml三乙胺并搅拌且同时回升至TA。反应混合物用水,用1N盐酸溶液和用10%的碳酸氢钠溶液进行洗涤,有机相在硫酸钠上进行干燥并过滤,并在真空下浓缩滤出液。结果得到0.5克期望的产物,该产物就此在下列步骤中使用。
C)3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲基苯基)乙酰基]哌啶盐酸盐一水合物,(-)异构体。
在TA和氮气氛下,将0.08ml乙酸添加至0.24克制备例3.1中得到的化合物(游离碱)于3ml MeOH中的溶液中,然后添加0.5克上面步骤中得到的化合物于5ml MeOH中的溶液。5分钟之后,添加0.08克氰基硼氢化钠并在TA下对该混合物搅拌过夜。将反应混合物倒入10%的碳酸氢钠水溶液中并用醚进行萃取,有机相用水进行洗涤,在硫酸钠上进行干燥并过滤,在真空下浓缩滤出液。用硅胶H对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1;v/v)至(90/10;v/v))进行洗脱),将该产物溶于DCM中,通过添加盐酸醚而酸化至pH=1并在真空下进行浓缩处理。在真空下由醚磨碎,离心脱水和干燥之后,得到0.5克期望的产物。 (c=1;MeOH).RMN1H:δ(ppm):0.7-1.2:se:6H;1.2-2.4:m:16H;2.5-4.8:m:12H;6.5-8.0:m:8H;10.2:se:1H.
(c=1;MeOH).RMN1H:δ(ppm):0.7-1.2:se:6H;1.2-2.4:m:16H;2.5-4.8:m:12H;6.5-8.0:m:8H;10.2:se:1H.
实施例2
3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌嗪基]乙基-3-(3,4-二氯苯基)-1-[2-(3,5-二甲基苯基)乙酰基]-哌啶二盐酸盐。2.7H2O,(-)异构体。
在TA和氮气氛下,将0.23克制备例3.2中得到的化合物(游离碱)添加至0.5克实施例1的步骤B中得到的化合物于20ml DCM中的溶液中,然后添加0.1ml乙酸,并在TA下搅拌30分钟。然后,添加0.55克三乙酰氧基硼氢化钠并在TA下搅拌过夜。向反应混合物中添加10%的碳酸钠水溶液并在TA下搅拌15分钟。反应混合物用DCM进行萃取,有机相用10%的碳酸钠水溶液进行洗涤,在硫酸钠上进行干燥并过滤,在真空下浓缩滤出液。用硅胶对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1;v/v)至(95/5;v/v))进行洗脱)。将得到的产物溶解于DCM中并通过添加盐酸醚而酸化至pH=1,对形成的沉淀物进行离心脱水,用醚进行洗涤并在真空下进行干燥。结果得到了0.4克期望的产物。(c=1;MeOH).RMN1H:δ(ppm):0.6-2.3:m:18H;2.3-4.7:m:16H;6.4-8.0:m:8H.
实施例3
3-[2-[4-(1-N,N-二甲基氨基甲酰基-1-甲基乙基)-1-哌嗪基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲基苯基)乙酰基]-哌啶二盐酸盐.1.25H2O,(-)异构体。
在TA下,将0.6克实施例1的步骤B中得到的化合物,0.3克制备例3.4中得到的化合物,0.1ml乙酸然后0.12克氰基硼氢化钠添加至20ml MeOH中,并在TA下搅拌过夜。将10%的碳酸钠水溶液添加至该反应混合物中并搅拌15分钟。混合物用AcOEt进行萃取,有机相用10%碳酸钠水溶液,用水,用饱和氯化钠溶液进行洗涤并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。用硅胶对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1;v/v)至(95/5;v/v))进行洗脱)。将得到的产物溶解于DCM中并通过添加盐酸醚而酸化至pH=1,并对形成的沉淀物进行离心脱水,用醚进行洗涤并在真空下进行干燥。结果得到了0.4克期望的产物。 (c=1;MeOH).RMN1H:δ(ppm):0.7-2.3:m:18H;2.35-4.7:m:22H;6.5-7.8:m:6H;10.3:s:1H.
(c=1;MeOH).RMN1H:δ(ppm):0.7-2.3:m:18H;2.35-4.7:m:22H;6.5-7.8:m:6H;10.3:s:1H.
实施例4
3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基-3-(3,4-二氯苯基)-1-[2-(3,5-二氯苯基)乙酰基]-哌啶盐酸倍半水合物,(-)异构体。
A)3-(3,4-二氯苯基)-3-(2-羟乙基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶,单一异构体。
在TA下,将4.75克制备例1中得到的化合物,3.55克制备例2.1中得到的化合物,3.6ml三乙胺然后8.4克BOP添加至150ml DCM中并在TA下对混合物搅拌2小时。在真空下对该混合物进行浓缩,残余物用AcOEt进行萃取,有机相用1N的盐酸溶液,用水,用1N氢氧化钠,用水,用饱和氯化钠溶液进行洗涤,在硫酸钠上进行干燥,并在真空下蒸发掉溶剂。结果得到了8克期望的产物,该产物就此使用。
B)3-(3,4-二氯苯基)-3-(甲酰基甲基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶,单一异构体。
从0.25ml于6ml DCM中的草酰氯,0.38ml于3ml DCM中的DMSO,1克在6ml DCM中的上面步骤中得到的化合物,以及1.5ml三乙胺为原料,根据实施例1步骤B中描述的操作方式制备该化合物。结果得到了1.0克期望的产物,该产物就此使用。
C)3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基-3-(3,4-二氯苯基)-1-[2-(3,5-二氯苯基)乙酰基]-哌啶盐酸倍半水合物,(-)异构体。
从0.25于3ml MeOH中的在制备例3.1中得到的化合物(游离碱),0.08ml乙酸,0.5克于5ml MeOH中的在上面步骤中得到的化合物以及0.08克氰基硼氢化钠为原料,根据实施例1的步骤C中描述的操作方式制备该化合物。结果得到0.52克期望的产物。(c=1;MeOH).
实施例5
3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-[3,5-二(三氟甲基)苯基]乙酰基]哌啶盐酸盐一水合物,(+)异构体。
A)3-(3,4-二氯苯基)-3-(2-羟乙基)-1-[2-[3,5-二(三氟甲基)苯基]乙酰基]哌啶,单一异构体。
在TA下,将1.2克制备例1.2中得到的化合物,1.2克3,5-二(三氟甲基)苯乙酸,1.7ml三乙胺,以及2.16克BOP添加至50ml DCM中并搅拌15分钟。反应混合物在真空下进行浓缩,残余物溶解于1N的HCl溶液中,用醚进行萃取,有机相用1N的HCl溶液,用水,用1N的氢氧化钠溶液,用水进行洗涤,并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。结果得到2.1克期望的产物,该产物就此使用。
B)3-(3,4-二氯苯基)-3-(甲酰基甲基)-1-[2-[3,5-二(三氟甲基)苯基]乙酰基]哌啶,单一异构体。
将20ml DCM冷却至-78℃,在氮气氛下添加1.5克上面步骤得到的化合物,0.45ml DMSO和0.3ml草酰氯,并在-78℃搅拌30分钟。然后添加2ml三乙胺并搅拌且同时回升至TA。将1N盐酸溶液添加至该反应混合物中,用DCM进行萃取,有机相用1N盐酸溶液,用水,用10%的碳酸钠水溶液进行洗涤,并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。结果得到1.5克期望的产物,该产物就此使用。
C)3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-[3,5-二(三氟甲基)苯基]乙酰基]哌啶盐酸盐一水合物,(+)异构体。
使0.35克制备例3.1得到的化合物和0.4克于10ml乙腈中的碳酸钾的混合物回流3小时。过滤掉不溶性物质,并在真空下浓缩滤液。将如此得到的游离碱形式的制备例3.1的产物溶解于3ml MeOH中,添加0.08ml乙酸,然后添加0.5克上面步骤得到的化合物于5ml MeOH中的溶液,并在TA下对该混合物搅拌5分钟。然后添加0.08克氰基硼氢化钠并在TA下搅拌过夜。将反应混合物倒入10%的碳酸氢钠水溶液中并用醚进行萃取,有机相用水进行洗涤并在硫酸钠上进行干燥,在真空下蒸发溶剂。用硅胶H对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1;v/v)至(90/10;v/v))进行洗脱)。将得到的产物溶解于DCM中并通过添加盐酸醚而酸化至pH=1,并对形成的沉淀物进行离心脱水。在真空干燥之后,得到了0.54克产物。 (c=1;MeOH).RMN1H:δ(ppm):0.6-2.2:m:16H;2.3-4.2:m:12H;6.6-8.0:m:8H;10.3:s:1H.
(c=1;MeOH).RMN1H:δ(ppm):0.6-2.2:m:16H;2.3-4.2:m:12H;6.6-8.0:m:8H;10.3:s:1H.
实施例6
3-[2-[4-(1-N,N-二甲基氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5二甲基苯基)乙酰基]哌啶盐酸盐半水合物,(-)异构体。
将0.35克制备例3.6中得到的化合物和0.4克于10ml乙腈中的碳酸钾的混合物回流3小时。过滤掉不溶性物质并在真空下浓缩滤液。将如此得到的游离碱形式的制备例3.6的产物溶解于3ml MeOH中,添加0.1ml乙酸,然后添加0.6克于5ml MeOH中的实施例1步骤B中得到的化合物,并在TA下搅拌5分钟。然后添加0.1克氰基硼氢化钠并在TA下搅拌过夜。将反应混合物倒入10%的碳酸氢钠水溶液中,用醚进行萃取,有机相用水进行洗涤,在硫酸镁上进行干燥,并在真空下蒸发掉溶剂。用硅胶H对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1;v/v)至(90/10;v/v))进行洗脱)。将得到的产物溶解于DCM中并通过添加盐酸醚而酸化至pH=1,在真空下浓缩溶剂。在由醚研碎,离心脱水和干燥之后,得到了0.68克期望的产物;F=202℃。(c=1;MeOH).RMN1H:δ(ppm):0.6-2.5:m:23H;2.5-4.6:m:18H;6.4-7.8:m:6H;10.1:s:1H.
实施例7
3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5二乙基苯基)乙酰基]哌啶盐酸盐半水合物,(-)异构体。
A)3-(3,4-二氯苯基)-3-(2-羟乙基)-1-[2-(3,5二乙基苯基)乙酰基]哌啶,单一异构体。
在TA下,将1.15克3,5-二乙基苯基乙酸添加至1.64克制备例1中得到的化合物于30ml DCM中的混合物中,然后添加3ml三乙胺和3.2克BOP,并在TA下对该混合物搅拌2小时。在真空下浓缩该反应混合物,将残余物溶解于1N的盐酸溶液中并用醚进行萃取,有机相用1N的盐酸溶液,用水,用1N的氢氧化钠溶液,用水,用饱和氯化钠溶液进行洗涤,并在硫酸钠上进行干燥,在真空下蒸发掉溶剂。用硅胶对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1;v/v)至(90/10;v/v))进行洗脱)。结果得到1.1克期望的产物,该产物就此使用。
B)3-(3,4-二氯苯基)-3-(甲酰基甲基)-1-[2-(3,5二乙基苯基)乙酰基]哌啶,单一异构体。
在氮气氛下,将0.5克上面步骤得到的化合物于10ml DCM中的溶液冷却至-78℃,添加0.23ml DMSO,然后添加0.16ml草酰氯,并在-78℃搅拌30分钟。然后,加入0.95ml三乙胺并搅拌且同时回升至TA。将1N的盐酸溶液添加至该反应混合物中,用DCM进行萃取,有机相用1N的盐酸溶液,用水,用10%的碳酸钠水溶液进行洗涤,在硫酸钠上进行干燥,并在真空下蒸发掉溶剂。结果得到0.5克期望的产物,该产物就此使用。
C)3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5二乙基苯基)乙酰基]哌啶盐酸盐半水合物,(-)异构体。
在TA和氮气氛下,将0.08ml乙酸添加至0.23克制备例3.1中得到的化合物(游离碱)于3ml MeOH中的溶液,然后添加0.5克上面步骤中得到的化合物于5ml MeOH中的溶液。5分钟之后,添加0.08克氰基硼氢化钠并在TA下搅拌过夜。将反应混合物倒入10%的碳酸氢钠水溶液中,用醚进行萃取,有机相用水进行洗涤并在硫酸镁上进行干燥,在真空下蒸发掉溶剂。用硅胶H对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1;v/v)至(93/7;v/v))进行洗脱)。将得到的产物溶解于DCM中并通过添加盐酸醚而酸化至pH=1,并在真空下浓缩溶剂。在由醚研碎,离心脱水和干燥之后,得到了0.51克期望的产物。(c=1;MeOH).RMN1H:δ(ppm):0.5-2.2:m:23H;2.2-4.65:m:16H;6.4-7.8:m:8H;9.85:s:1H.
借助上面实施例中描述的操作方式,制备下表I中排序的根据本发明的化合物。
表1



从实施例1步骤B中得到的化合物和呈游离碱形式的制备例3.5中得到的化合物为原料,根据实施例1步骤C中描述的操作方式,制备该化合物。
从实施例1步骤B中得到的化合物和呈游离碱形式的制备例3.3中得到的化合物为原料,根据实施例3中描述的操作方式,制备该化合物。
从实施例4步骤B中得到的化合物和呈游离碱形式的制备例3.5中得到的化合物为原料,根据实施例4步骤C中描述的操作方式,制备该化合物。
从实施例4步骤B中得到的化合物和呈游离碱形式的制备例3.2中得到的化合物为原料,根据实施例3中描述的操作方式,制备该化合物。
从实施例4步骤B中得到的化合物和制备例3.4中得到的化合物为原料,根据实施例3中描述的操作方式,制备该化合物。
从实施例4步骤B中得到的化合物和呈游离碱形式的制备例3.3中得到的化合物为原料,根据实施例3中描述的操作方式,制备该化合物。
从实施例5步骤B中得到的化合物和制备例3.5中得到的化合物为原料,根据实施例5步骤C中描述的操作方式,制备该化合物。
从实施例5步骤B中得到的化合物和呈游离碱形式的制备例3.3中得到的化合物为原料,根据实施例3中描述的操作方式,制备该化合物。
从实施例1步骤B中得到的化合物和呈游离碱形式的制备例3.7中得到的化合物为原料,根据实施例2中描述的操作方式,制备该化合物。
从实施例5步骤B中得到的化合物和制备例3.7中得到的化合物为原料,根据实施例5步骤C中描述的操作方式,制备该化合物。
从实施例4步骤B中得到的化合物和呈游离碱形式的制备例3.7中得到的化合物为原料,根据实施例4步骤C中描述的操作方式,制备该化合物。
从实施例7步骤B中得到的化合物和呈游离碱形式的制备例3.2中得到的化合物为原料,根据实施例7步骤C中描述的操作方式,制备该化合物。
从实施例1步骤B中得到的化合物和呈游离碱形式的制备例3.8中得到的化合物为原料,根据实施例1步骤C中描述的操作方式,制备该化合物。实施例8:RMN1H:δ(ppm):0.7-2.2:m:27H;2.3-4.6:m:14H;6.4-7.7:m:8H;10.1:S:1H.实施例 9:RMN1H:δ(ppm):0.6-2.35:m:22H;2.4-4.6:m:14H;6.4-8.2:m:8H.实施例 10:RMN1H:δ(ppm):0.7-2.25:m:21H;2.3-4.4:m:12H;6.7-7.8:m:8H;10.1:S:1H.实施例 11:RMN1H:δ(ppm):0.6-2.2:m:12H;2.3-4.4:m:16H;6.8-8.0:m:8H.实施例 12:RMN1H:δ(ppm):0.8-2.3:m:12H;2.35-4.4:m:22H;7.0-7.9:m:6H;10.6:s:1H.实施例 13:RMN1H:δ(ppm):0.9-2.3:m:16H;2.35-4.5:m:16H;7.0-7.9:m:8H.实施例 14:RMN1H:δ(ppm):0.9-2.3:m:21H;2.4-4.3:m:12H;6.8-8.1:m:8H;10.0:s:1H.实施例 15:RMN1H:δ(ppm):1.0-2.4:m:16H;2.5-4.5:m:16H;6.9-8.1:m:8H;11.0:se:1H.实施例 16:RMN1H:δ(ppm):0.4-2.3:m:20H;2.4-4.6:m:13H;6.5-7.7:m:8H;9.6:8:1H.实施例 17:RMN1H:δ(ppm):0.4-2.2:m:14H;2.3-4.4:m:13H;6.5-7.8:m:8H;9.9:s:1H.实施例 18:RMN1H:δ(ppm):0.4-2.2:m:14H;2.3-4.4:m:13H;6.6-7.8:m:8H;9.9:s:1H.实施例 19:RMN1H:δ(ppm):0.6-2.6:m:22H;2.6-4.8:m:16H;6.5-8.0:m:10H.实施例 20:RMN1H:δ(ppm):0.7-2,25:m:22H;2.3-4.6:m:21H;6.4-7.7:m:6H;10.4:s:1H.
实施例21
3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二甲基苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶盐酸盐。
A)1-[2-(3,5-二氯苯基)乙酰基]-3-(3,4-二甲基苯基)-3-[2-(2-四氢吡喃氧)乙基]哌啶。
在TA下,对3克制备例1.2中得到的化合物,1.3克制备例2.1中得到的化合物,3.2ml三乙胺和4.8克BOP于100ml DCM中的混合物搅拌2小时。在真空下浓缩该反应混合物,将残余物溶解于1N的盐酸溶液中并用AcOEt进行萃取,有机相用水,用1N的氢氧化钠溶液,用饱和的氯化钠溶液进行洗涤,在硫酸钠上进行干燥,并在真空下蒸发掉溶剂。结果得到4.5克期望的产物。
B)1-[2-(3,5-二氯苯基)乙酰基]-3-(3,4-二甲基苯基)-3-[2-羟乙基]哌啶。
在TA下,对4.5克上面步骤中得到的化合物和2ml浓盐酸溶液于10ml MeOH中的混合物搅拌2小时。在真空下浓缩该反应混合物,将残余物溶解于MeOH中并在真空下蒸发掉溶剂。用硅胶对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1,v/v)至(95/5;v/v))进行洗脱)。结果得到3克期望的产物。
C)1-[2-(3,5-二氯苯基)乙酰基]-3-(甲酰基)-3-(3,4-二甲基苯基)哌啶。
在氮气氛下,将10ml DCM冷却至-78℃,并添加0.5克上面步骤得到的化合物和0.18ml DMSO,然后添加0.13ml草酰氯,并在-78℃搅拌30分钟。然后添加0.75ml三乙胺并搅拌且同时回升至TA。将1N的盐酸溶液添加至该反应混合物中,得到的混合物用DCM进行萃取,有机相用水,用10%的碳酸钠溶液进行洗涤,在硫酸钠上进行干燥,并在真空下蒸发掉溶剂。结果得到0.5克期望的产物。
D)3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二甲基苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶盐酸盐。
在TA下,0.5克上面步骤得到的化合物,0.35克制备例3.1得到的化合物(游离碱),0.1ml乙酸和0.15克氰基硼氢化钠于30mlMeOH中的混合物在搅拌下过夜。将10%的碳酸钠水溶液添加至反应混合物,对得到的混合物搅拌15分钟,用AcOEt进行萃取,有机相用水,用饱和氯化钠溶液进行洗涤,在硫酸钠上进行干燥并在真空下蒸发掉溶剂。用硅胶对残余物进行色谱分析(用DCM然后用DCM/MeOH混合物(梯度(99/1,v/v)至(95/5;v/v))进行洗脱)。将得到的产物溶解于DCM中并通过添加盐酸醚而酸化至pH=1,并对形成的沉淀物进行离心脱水。结果得到0.35克期望的产物。RMN1H:δ(ppm):0.8-2.3:m:22H;2.3-4.0:m:13H;6.5-7.6:m:8H;9.5:s:1H.
实施例22
3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌嗪基]乙基]-3-(3,4-二甲基苯基)-1-[2-(3,5-二氯苯基)乙酰基]哌啶二盐酸盐一水合物。
从实施例21步骤C中得到的化合物和制备例3.2中得到的化合物(游离碱)开始,根据实施例21步骤D中描述的操作方式,制备该化合物。RMN1H:δ(ppm):1.4:1s:6H;2.2:2s:6H;1.3-4.0:m:26H;7.0-8.0:m:6H.
Claims (27)
1.一种下式(I)的化合物以及其与无机或有机酸的盐,其溶剂合物和/或水合物:式中:-X表示基团
 基团
基团

-Ar表示被卤原子单取代或二取代的苯基;(C1-C3)烷基;
-R1表示氯原子,溴原子,(C1-C3)烷基或三氟甲基;
-R2表示基团-CR3R4CONR5R6 ;
-R3和R4表示相同的基团,所述基团选自:甲基,乙基,正丙基或正丁基;
-或者R3和R4与连接至其上的碳原子一起构成(C3-C6)环烷基;
-R5和R6各自独立地表示氢;(C1-C3)烷基;
-或者,R5和R6与连接至其上的氮原子一起构成杂环基团,所述基团选自:1-氮杂环丁烷基,1-吡咯烷基,1-哌啶基,4-吗啉基,4-硫代吗啉基或全氢化-1-吖庚因基。
2.根据权利要求1的化合物,其中Ar表示3,4-二氯苯基或3,4-二甲基苯基。
3.根据权利要求1的化合物,其中,取代基Rx表示氯原子,甲基,乙基或三氟甲基。
4.根据权利要求1化合物,其中,X表示基团
 式中R2表示基团-CR3R4CONR5R6。
式中R2表示基团-CR3R4CONR5R6。
5.根据权利要求4的化合物,其中R3和R4表示甲基,或与连接至其上的碳原子一起构成环己基。
6.根据权利要求1的化合物,其中X表示
 ,式中R2表示-CR3R4CONR5R6。
,式中R2表示-CR3R4CONR5R6。
7.根据权利要求6的化合物,其中R3和R4表示甲基,或与连接至其上的碳原子一起构成环己基或环丙基。
8.根据权利要求4或6的化合物,其中R5和R6表示氢或甲基。
9.根据权利要求1的化合物,具有如下结构式(I′),以及其与无机或有机酸的盐,以及其溶剂合物和/或水合物: 式中:
式中:
-R′1表示氯原子,甲基,乙基或三氟甲基;
-R′3和R′4各自表示甲基,或者与连接至其上的碳原子一起构成环己基;
-R′5和R′6各自表示氢或甲基。
10.根据权利要求1的化合物,具有如下结构式(I″),以及其与无机或有机酸的盐,以及其溶剂合物和/或水合物: 式中:
式中:
-R′1表示氯原子,甲基,乙基或三氟甲基;
-R′3和R′4各自表示甲基,或者与连接至其上的碳原子一起构成环己基或环丙基;
-R′5和R′6各自表示氢或甲基。
11.根据权利要求1-10任一项的化合物,具有光学纯形式的结构式(I),(I′)或(I″)。
12. 3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲苯基)乙酰基]哌啶,(-)异构体,其盐以及其溶剂合物和/或水合物。
13. 3-[2-[4-(1-N,N-二甲基氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二甲苯基)乙酰基]哌啶,(-)异构体,其盐以及其溶剂合物和/或水合物。
14. 3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二乙基苯基)乙酰基]哌啶,(-)异构体,其盐以及其溶剂合物和/或水合物。
15. 3-[2-[4-(1-氨基甲酰基-1-甲基乙基)-1-哌啶基]乙基]-3-(3,4-二氯苯基)-1-[2-(3,5-二(三氟甲基)苯基]乙酰基]哌啶,(+)异构体,其盐以及其溶剂合物和/或水合物。
16.一种制备权利要求1的结构式(I)化合物,其盐以及其溶剂合物和/或水合物的方法,其特征在于:
1a) 下式(II)的化合物用结构式(III)酸的官能衍生物进行处理,以便得到结构式(IV)的化合物,式中Ar如权利要求1中结构式(I)化合物所定义,E表示氢或氧保护基团, 式中R1如权利要求1中结构式(I)化合物所定义;
式中R1如权利要求1中结构式(I)化合物所定义;
2a) 当E任选地表示保护基团时,它通过酸或碱的作用而消去,以便得到下式的醇: (IV,E=H);
(IV,E=H);
3a) 步骤1a)或步骤2a)中得到的结构式(IV,E=H)的醇用下式(V)化合物Y-SO2-Cl (V)进行处理,结构式(V)中,Y表示甲基,苯基,甲苯基或三氟甲基基团;以便得到下式(VI)的化合物:
4a) 结构式(VI)的化合物用下式(VII)的化合物反应:式中X如权利要求1中结构式(I)化合物所定义;
5a) 任选地用无机或有机酸,将如此得到的化合物转化成其盐的一种。
17.一种制备权利要求1的结构式(I)化合物,其盐以及其溶剂合物和/或水合物的方法,其特征在于:
1b)下式(II)的化合物用结构式(III)酸的官能衍生物进行处理,以便得到结构式(IV)的化合物,式中Ar如权利要求1中结构式(I)化合物所定义,E表示氢或氧保护基团,式中R1如权利要求1中结构式(I)化合物所定义;
当E表示或不表示保护基团时,它通过酸或碱的作用而消去,以便得到下式的醇: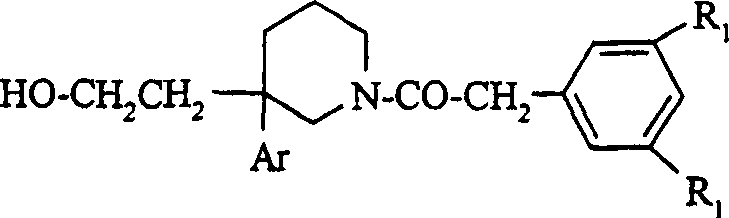 (IV,E=H);
(IV,E=H);
2b) 使如此得到的结构式(IV,E=H)的化合物进行氧化,以便制备下式(VIII)的化合物: 3b) 在酸存在下,使结构式(VIII)的化合物与结构式(VII)
3b) 在酸存在下,使结构式(VIII)的化合物与结构式(VII) 的化合物进行反应,式(VII)中X如权利要求1中结构(I)化合物所定义,然后,借助还原剂使所形成的中间亚胺鎓(imminium)盐进行还原;
的化合物进行反应,式(VII)中X如权利要求1中结构(I)化合物所定义,然后,借助还原剂使所形成的中间亚胺鎓(imminium)盐进行还原;
4b) 任选地用无机或有机酸,将如此得到的化合物转化成其盐的一种。
18.一种制备具有(S)构形的权利要求1中结构(I)化合物,其盐及其溶剂合物和/或水合物的立体有规方法,其特征在于:
1d)下式(II*,E=H)化合物的(S)异构体用下式酸(III)的官能衍生物进行处理:(II*,E=H)式中Ar如权利要求1中结构式(I)化合物所定义, 式中R1如权利要求1中结构式(I)化合物所定义,从而得到下式(IV*,E=H)的化合物:
式中R1如权利要求1中结构式(I)化合物所定义,从而得到下式(IV*,E=H)的化合物: (IV*,E=H);
(IV*,E=H);
2d)将结构式(IV*)的化合物氧化,从而得到下式(VIII*)的化合物:
3d)在酸存在下,使结构式(VIII*)的化合物与下式(VII)化合物反应: 式中X如权利要求1中结构式(I)化合物所定义,然后借助还原剂使所形成的中间亚胺鎓(imminium)盐进行还原;
式中X如权利要求1中结构式(I)化合物所定义,然后借助还原剂使所形成的中间亚胺鎓(imminium)盐进行还原;
4d)任选地,用无机或有机酸使如此得到的化合物转化成其盐的一种。
19.一种制备权利要求1的结构式(I)化合物,其盐以及其溶剂合物和/或水合物的制备方法,其特征在于:
将具有如下结构式(VI)的化合物与结构式(VII)的化合物进行反应,并且非强制性地,利用无机或有机酸,将如此得到的化合物转化成其盐的一种; 式中:
式中:
-Ar表示被卤原子单取代或二取代的苯基;(C1-C3)烷基;
-Y表示甲基,苯基,甲苯基或三氟甲基;
-R1表示氯原子,溴原子,(C1-C3)烷基或三氟甲基; 式中X如权利要求1中结构(I)化合物所定义。
式中X如权利要求1中结构(I)化合物所定义。
20.一种制备权利要求1的结构式(I)化合物,其盐以及其溶剂合物和/或水合物的方法,其特征在于:
在酸存在下,使如下结构式(VIII)的化合物与如下结构式(VII)的化合物进行反应,然后借助还原剂使所形成的中间亚胺鎓(imminium)盐进行还原;并任选地,利用无机或有机酸,将如此得到的化合物转化成其盐的一种; 式中Ar和R1如权利要求1中结构式(I)化合物所定义,式中X如权利要求1中结构式(I)化合物所定义。
式中Ar和R1如权利要求1中结构式(I)化合物所定义,式中X如权利要求1中结构式(I)化合物所定义。
21.一种制备具有(S)构形的结构(I)化合物,其盐及其溶剂合物和/或水合物的立体有规方法,其特征在于:
在酸存在下,将具有如下结构式(VIII*)的化合物与结构式(VII)的化合物进行反应,然后借助还原剂使形成的中间亚胺鎓盐(immimium)还原,并且任选地,利用无机或有机酸使如此得到的化合物转化成其盐的一种;式中:Ar和R1如权利要求1中结构式(I)化合物所定义;式中X如权利要求1中结构式(I)化合物所定义。
22.结构式(VII)的化合物以及其与无机或有机酸的盐: 式中:-X表示基团基团
式中:-X表示基团基团
-R2表示基团-CR3R4CONR5R6;
-R3和R4表示相同的基团,所述基团选自:甲基,乙基,正丙基或正丁基;
-或者R3和R4与连接至其上的碳原子一起构成(C3-C6)环烷基;
-R5和R6各自独立地表示氢;(C1-C3)烷基;
-或者,R5和R6与连接至其上的氮原子一起构成杂环基团,所述基团选自:1-氮杂环丁烷基,1-吡咯烷基,1-哌啶基,4-吗啉基,4-硫代吗啉基或全氢化-1-吖庚因基。
23.药物组合物,包含作为活性物质的根据权利要求1-15任一项的化合物,或其药物可接受的盐,溶剂合物和/或水合物。
24.根据权利要求23的药物组合物,以单位剂量形式,包含0.1至1000mg活性物质,其中将所述活性物质与至少一种药物赋形剂进行混合。
25.根据权利要求1-15任一项的化合物,或其药物可接受盐,溶剂合物和/或水合物用于制备药物的用途,所述药物用于治疗其中涉及物质P和人体NK1受体的各种病变。
26.根据权利要求25的用于制备药物的用途,所述药物用于治疗呼吸系统,胃肠系统,泌尿系统,免疫系统,心血管系统,中枢神经系统的病变,以及用于治疗疼痛,偏头痛,炎症,恶心和呕吐,以及皮肤病。
27.根据权利要求25的用于制备药物的用途,所述药物用于治疗慢性梗阻性支气管炎,气喘,尿失禁;过敏性肠综合征,克罗恩氏病,溃疡性结膜炎,抑郁症和焦虑症。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR99/01593 | 1999-02-10 | ||
| FR9901593A FR2789389B3 (fr) | 1999-02-10 | 1999-02-10 | Nouveaux derives de piperidine, procede pour leur obtention et compositions pharmaceutiques les contenant |
| FR9904429A FR2789390B3 (fr) | 1999-02-10 | 1999-04-07 | Nouveaux derives de piperidine, procede pour leur obtention et compositions pharmaceutiques les contenant |
| FR99/04429 | 1999-04-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1345317A true CN1345317A (zh) | 2002-04-17 |
| CN1273463C CN1273463C (zh) | 2006-09-06 |
Family
ID=26234812
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB008058261A Expired - Fee Related CN1273463C (zh) | 1999-02-10 | 2000-02-08 | (1-苯甲酰甲基-3-苯基-3-哌啶基乙基)哌啶衍生物,制备它们的方法以及包含它们的药物组合物 |
Country Status (37)
| Country | Link |
|---|---|
| US (2) | US6642233B1 (zh) |
| EP (1) | EP1150970B1 (zh) |
| JP (1) | JP4648544B2 (zh) |
| KR (1) | KR100611261B1 (zh) |
| CN (1) | CN1273463C (zh) |
| AR (1) | AR028813A1 (zh) |
| AT (1) | ATE370942T1 (zh) |
| AU (1) | AU763716B2 (zh) |
| BG (1) | BG65031B1 (zh) |
| BR (1) | BR0008067A (zh) |
| CA (1) | CA2360894C (zh) |
| CO (1) | CO4980896A1 (zh) |
| CZ (1) | CZ300765B6 (zh) |
| DE (1) | DE60036081T2 (zh) |
| DK (1) | DK1150970T3 (zh) |
| EE (1) | EE04437B1 (zh) |
| ES (1) | ES2291187T3 (zh) |
| FR (1) | FR2789390B3 (zh) |
| HK (1) | HK1038927A1 (zh) |
| HR (1) | HRP20010566A2 (zh) |
| HU (1) | HUP0201281A3 (zh) |
| ID (1) | ID29219A (zh) |
| IL (2) | IL144363A0 (zh) |
| ME (1) | MEP11908A (zh) |
| NO (2) | NO321149B1 (zh) |
| NZ (1) | NZ513053A (zh) |
| PE (1) | PE20001463A1 (zh) |
| PL (1) | PL198561B1 (zh) |
| PT (1) | PT1150970E (zh) |
| RS (1) | RS50210B (zh) |
| RU (1) | RU2220956C2 (zh) |
| SI (1) | SI1150970T1 (zh) |
| SK (1) | SK286401B6 (zh) |
| TR (1) | TR200102331T2 (zh) |
| TW (1) | TWI254712B (zh) |
| UA (1) | UA70986C2 (zh) |
| WO (1) | WO2000047572A1 (zh) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003070745A2 (en) * | 2002-02-20 | 2003-08-28 | Sepracor Inc. | Carbonate and carbamate modified forms of glucocorticoids |
| US20050009798A1 (en) | 2002-02-20 | 2005-01-13 | Sepracor Inc. | Carbonate and carbamate modified forms of glucocorticoids in combination with B2 adrenergic agonists |
| JP2007519702A (ja) * | 2004-01-30 | 2007-07-19 | ファイザー・プロダクツ・インク | ニューロキニン受容体拮抗薬及びシクロデキストリンからなる医薬組成物、ならびに注射部位の耐容性を改善するための方法 |
| FR2873373B1 (fr) * | 2004-07-23 | 2006-09-08 | Sanofi Synthelabo | Derives de 4-arylmorpholin-3-one, leur preparation et leur application en therapeutique |
| GB2452696B (en) * | 2007-08-02 | 2009-09-23 | Cambridge Entpr Ltd | 3-(2',2'-dimethylpropanoylamino)-tetrahydropyridin-2-one and its use in pharmaceutical compositions |
| DE102008015426A1 (de) * | 2008-03-20 | 2009-09-24 | Beiersdorf Ag | Kühlende kosmetische oder dermatologische Zubereitungen mit einem Gehalt an (1R,2S,5R)-2-Isopropyl-5-methyl-N-(2-(pyridyn-2-yl)ethyl-cyclohexancarboxamid und/oder (1R,2S,5R)-N-(4-(Cyanomethyl)-phenyl)-2-isopropyl-5-methylcyclohexancarboxamid zu Reduktion von Hautrötungen |
| DE102008015425A1 (de) * | 2008-03-20 | 2010-01-21 | Beiersdorf Ag | Kosmetische oder dermatologische Zubereitungen zur Verringerung von Juckreiz und anderen dermatologischen Missempfindungen, die insbesondere bei Altershaut auftreten können, mit einem Gehalt an (1R,2S,5R)-2-Isopropyl-5-methyl-N-(2-(pyridin-2-yl)ethyl-cyclohexancarboxamid und/oder (1R,2S,5R)-N-(4-(Cyanomethyl)-phenyl)-2-isopropyl-5-methylcyclohexancarboxamid |
| DE102008015428A1 (de) * | 2008-03-20 | 2009-09-24 | Beiersdorf Ag | Kühlende Zubereitungen für den humanen Haut- und/oder Schleimhautkontakt mit einem Gehalt an (1R,2S,5R)-2-Isopropyl-5-methyl-N-(2-(pyridin-2-yl)ethyl-cyclohexancarboxamid und/oder (1R,2S,5R)-N-(4-(Cyanomethyl)-phenyl)-2-isopropyl-5-methylcyclohexancarboxamid |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US512901A (en) * | 1894-01-16 | Folding bed | ||
| GB821012A (en) * | 1957-01-15 | 1959-09-30 | Rhone Poulenc Sa | New phenthiazine derivatives and processes for their preparation |
| FR2676055B1 (fr) | 1991-05-03 | 1993-09-03 | Sanofi Elf | Composes polycycliques amines et leurs enantiomeres, procede pour leur preparation et compositions pharmaceutiques les contenant. |
| AU693936B2 (en) | 1994-08-25 | 1998-07-09 | Merrell Pharmaceuticals Inc. | Novel substituted piperidines useful for the treatment of allergic diseases |
| EP0714891A1 (en) * | 1994-11-22 | 1996-06-05 | Eli Lilly And Company | Heterocyclic tachykinin receptor antagonists |
-
1999
- 1999-04-07 FR FR9904429A patent/FR2789390B3/fr not_active Expired - Lifetime
-
2000
- 2000-02-08 HU HU0201281A patent/HUP0201281A3/hu unknown
- 2000-02-08 BR BR0008067-5A patent/BR0008067A/pt not_active Application Discontinuation
- 2000-02-08 DE DE60036081T patent/DE60036081T2/de not_active Expired - Lifetime
- 2000-02-08 TR TR2001/02331T patent/TR200102331T2/xx unknown
- 2000-02-08 CA CA002360894A patent/CA2360894C/en not_active Expired - Fee Related
- 2000-02-08 KR KR1020017009983A patent/KR100611261B1/ko not_active IP Right Cessation
- 2000-02-08 NZ NZ513053A patent/NZ513053A/xx not_active IP Right Cessation
- 2000-02-08 IL IL14436300A patent/IL144363A0/xx not_active IP Right Cessation
- 2000-02-08 ID IDW00200101651A patent/ID29219A/id unknown
- 2000-02-08 CN CNB008058261A patent/CN1273463C/zh not_active Expired - Fee Related
- 2000-02-08 CZ CZ20012888A patent/CZ300765B6/cs not_active IP Right Cessation
- 2000-02-08 ES ES00903744T patent/ES2291187T3/es not_active Expired - Lifetime
- 2000-02-08 PL PL350243A patent/PL198561B1/pl not_active IP Right Cessation
- 2000-02-08 WO PCT/FR2000/000284 patent/WO2000047572A1/fr active IP Right Grant
- 2000-02-08 PT PT00903744T patent/PT1150970E/pt unknown
- 2000-02-08 JP JP2000598492A patent/JP4648544B2/ja not_active Expired - Fee Related
- 2000-02-08 DK DK00903744T patent/DK1150970T3/da active
- 2000-02-08 SK SK1139-2001A patent/SK286401B6/sk not_active IP Right Cessation
- 2000-02-08 US US09/913,106 patent/US6642233B1/en not_active Expired - Fee Related
- 2000-02-08 UA UA2001075184A patent/UA70986C2/uk unknown
- 2000-02-08 EE EEP200100417A patent/EE04437B1/xx not_active IP Right Cessation
- 2000-02-08 ME MEP-119/08A patent/MEP11908A/xx unknown
- 2000-02-08 AT AT00903744T patent/ATE370942T1/de not_active IP Right Cessation
- 2000-02-08 RU RU2001121989/04A patent/RU2220956C2/ru not_active IP Right Cessation
- 2000-02-08 AU AU25531/00A patent/AU763716B2/en not_active Ceased
- 2000-02-08 RS YUP-564/01A patent/RS50210B/sr unknown
- 2000-02-08 EP EP00903744A patent/EP1150970B1/fr not_active Expired - Lifetime
- 2000-02-08 SI SI200030968T patent/SI1150970T1/sl unknown
- 2000-02-09 AR ARP000100555A patent/AR028813A1/es active IP Right Grant
- 2000-02-09 TW TW089102106A patent/TWI254712B/zh active
- 2000-02-10 CO CO00008757A patent/CO4980896A1/es unknown
- 2000-02-10 PE PE2000000102A patent/PE20001463A1/es not_active Application Discontinuation
-
2001
- 2001-07-26 HR HR20010566A patent/HRP20010566A2/hr not_active Application Discontinuation
- 2001-08-08 NO NO20013878A patent/NO321149B1/no not_active IP Right Cessation
- 2001-08-08 BG BG105794A patent/BG65031B1/bg unknown
-
2002
- 2002-01-29 HK HK02100715A patent/HK1038927A1/xx not_active IP Right Cessation
-
2003
- 2003-09-16 US US10/663,124 patent/US6951940B2/en not_active Expired - Fee Related
-
2005
- 2005-04-21 NO NO20051958A patent/NO20051958L/no unknown
-
2007
- 2007-01-07 IL IL180579A patent/IL180579A0/en unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1142144C (zh) | 4-苯基-吡啶衍生物 | |
| CN1034934C (zh) | 血管紧张素ii拮抗性吡啶衍生物的制备方法 | |
| CN1202093C (zh) | 取代的杂环化合物 | |
| CN1142161C (zh) | 作为5-ht4受体拮抗剂的二氢苯并二噁烯羧酰胺与酮衍生物 | |
| CN1016778B (zh) | 螺-取代的戊二酸单酰胺 | |
| CN1113236A (zh) | 非肽基速激肽受体拮抗剂 | |
| CN1152030C (zh) | 新的杂环化合物 | |
| CN1058405A (zh) | 奎宁环衍生物 | |
| CN1055925A (zh) | 吡啶衍生物及其制备方法 | |
| CN1294577A (zh) | 钾通道抑制剂 | |
| CN1063430C (zh) | 乙醇胺衍生物 | |
| CN1922161A (zh) | 1-哌嗪-和1-高哌嗪-羧酸酯衍生物、制备和其治疗用途 | |
| CN1270585A (zh) | 取代的1,2,3,4-四氢化萘衍生物 | |
| CN1812980A (zh) | 哌啶基-和哌嗪基-烷基氨基甲酸酯衍生物,其制备及治疗用途 | |
| CN1787818A (zh) | 趋化因子受体活性的氨基环丁基酰胺调节剂 | |
| CN1092766A (zh) | 药物 | |
| CN1918160A (zh) | 用作趋化因子受体活性调节剂的新颖三环螺环衍生物 | |
| CN1045583A (zh) | 四氢苯并咪唑衍生物 | |
| CN1745065A (zh) | 4-氧代-3-(1-氧代-1h-异喹啉-2-基乙酰氨基)-戊酸酯和酰胺衍生物及其作为天冬氨酸特异性半胱氨酸蛋白酶抑制剂的用途 | |
| CN1020900C (zh) | 制备新的取代n-(3-羟基-4-哌啶基)苯甲酰胺的方法 | |
| CN1273463C (zh) | (1-苯甲酰甲基-3-苯基-3-哌啶基乙基)哌啶衍生物,制备它们的方法以及包含它们的药物组合物 | |
| CN1286683A (zh) | 四氢苯并吲哚衍生物 | |
| CN1178923C (zh) | 1,4-二氮杂环庚烷-2,5-二酮衍生物及其作为nk-1受体拮抗剂的应用 | |
| CN1189829A (zh) | 用作神经激肽拮抗药的哌嗪衍生物 | |
| CN1249059C (zh) | 新型哌啶羧酸酰胺衍生物、其制备方法以及含有它们的药物组合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20060906 Termination date: 20120208 |