CN1016778B - 螺-取代的戊二酸单酰胺 - Google Patents
螺-取代的戊二酸单酰胺Info
- Publication number
- CN1016778B CN1016778B CN87107371A CN87107371A CN1016778B CN 1016778 B CN1016778 B CN 1016778B CN 87107371 A CN87107371 A CN 87107371A CN 87107371 A CN87107371 A CN 87107371A CN 1016778 B CN1016778 B CN 1016778B
- Authority
- CN
- China
- Prior art keywords
- group
- milliliters
- alkyl
- ethyl
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
- GTFMAONWNTUZEW-UHFFFAOYSA-N glutaramic acid Chemical class NC(=O)CCCC(O)=O GTFMAONWNTUZEW-UHFFFAOYSA-N 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 117
- 150000003839 salts Chemical class 0.000 claims abstract description 19
- -1 heterocyclic radical Chemical class 0.000 claims description 194
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 80
- 238000000034 method Methods 0.000 claims description 77
- 150000002148 esters Chemical class 0.000 claims description 75
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 69
- 239000002253 acid Substances 0.000 claims description 37
- 238000002360 preparation method Methods 0.000 claims description 37
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 36
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 33
- 238000006243 chemical reaction Methods 0.000 claims description 33
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 30
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Natural products CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 30
- 238000005984 hydrogenation reaction Methods 0.000 claims description 26
- 125000003118 aryl group Chemical group 0.000 claims description 21
- 229910052739 hydrogen Inorganic materials 0.000 claims description 21
- 229910052799 carbon Inorganic materials 0.000 claims description 18
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 18
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 18
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 16
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 15
- 230000032050 esterification Effects 0.000 claims description 13
- 238000005886 esterification reaction Methods 0.000 claims description 13
- XNMQEEKYCVKGBD-UHFFFAOYSA-N 2-butyne Chemical compound CC#CC XNMQEEKYCVKGBD-UHFFFAOYSA-N 0.000 claims description 12
- 150000001408 amides Chemical class 0.000 claims description 11
- 125000003368 amide group Chemical group 0.000 claims description 10
- 239000003795 chemical substances by application Substances 0.000 claims description 9
- 230000007062 hydrolysis Effects 0.000 claims description 9
- 238000006460 hydrolysis reaction Methods 0.000 claims description 9
- 125000000217 alkyl group Chemical group 0.000 claims description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 8
- 229920006395 saturated elastomer Polymers 0.000 claims description 8
- 230000015572 biosynthetic process Effects 0.000 claims description 7
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 7
- 229910052757 nitrogen Inorganic materials 0.000 claims description 7
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical group CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 claims description 6
- DGYIJVNZSDYBOE-UHFFFAOYSA-N [CH2]C1=CC=NC=C1 Chemical group [CH2]C1=CC=NC=C1 DGYIJVNZSDYBOE-UHFFFAOYSA-N 0.000 claims description 6
- 238000009903 catalytic hydrogenation reaction Methods 0.000 claims description 6
- 229910052736 halogen Inorganic materials 0.000 claims description 6
- 150000002367 halogens Chemical class 0.000 claims description 6
- 125000000623 heterocyclic group Chemical group 0.000 claims description 6
- 230000000903 blocking effect Effects 0.000 claims description 5
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 5
- 239000001257 hydrogen Substances 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 4
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 claims description 4
- PFUQSACCWFVIBW-UHFFFAOYSA-N [C].C1=CC=CC=C1 Chemical compound [C].C1=CC=CC=C1 PFUQSACCWFVIBW-UHFFFAOYSA-N 0.000 claims description 4
- 125000004104 aryloxy group Chemical group 0.000 claims description 4
- 125000002837 carbocyclic group Chemical group 0.000 claims description 4
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 claims description 4
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 claims description 4
- RAABOESOVLLHRU-UHFFFAOYSA-N diazene Chemical compound N=N RAABOESOVLLHRU-UHFFFAOYSA-N 0.000 claims description 4
- 229910000071 diazene Inorganic materials 0.000 claims description 4
- 125000004185 ester group Chemical group 0.000 claims description 4
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 claims description 4
- 125000004760 (C1-C4) alkylsulfonylamino group Chemical group 0.000 claims description 3
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 claims description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 239000003960 organic solvent Substances 0.000 claims description 3
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 claims description 3
- 125000006730 (C2-C5) alkynyl group Chemical group 0.000 claims description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 2
- IVSZLXZYQVIEFR-UHFFFAOYSA-N 1,3-Dimethylbenzene Natural products CC1=CC=CC(C)=C1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 claims description 2
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 claims description 2
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 claims description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 2
- 125000001539 acetonyl group Chemical group [H]C([H])([H])C(=O)C([H])([H])* 0.000 claims description 2
- 125000000043 benzamido group Chemical group [H]N([*])C(=O)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 2
- 239000003153 chemical reaction reagent Substances 0.000 claims description 2
- 125000005113 hydroxyalkoxy group Chemical group 0.000 claims description 2
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 claims 1
- 238000007327 hydrogenolysis reaction Methods 0.000 claims 1
- PQNCGXPHRHYJJP-UHFFFAOYSA-N n-phenylpropane-1-sulfonamide Chemical compound CCCS(=O)(=O)NC1=CC=CC=C1 PQNCGXPHRHYJJP-UHFFFAOYSA-N 0.000 claims 1
- 238000011282 treatment Methods 0.000 abstract description 9
- 206010019280 Heart failures Diseases 0.000 abstract description 8
- 206010020772 Hypertension Diseases 0.000 abstract description 7
- 239000002934 diuretic Substances 0.000 abstract description 7
- 208000001647 Renal Insufficiency Diseases 0.000 abstract description 6
- 201000006370 kidney failure Diseases 0.000 abstract description 6
- 229940030606 diuretics Drugs 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 208
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 162
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 140
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 128
- 239000000243 solution Substances 0.000 description 126
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 110
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 100
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 77
- 239000000047 product Substances 0.000 description 75
- 239000003921 oil Substances 0.000 description 73
- 238000003756 stirring Methods 0.000 description 67
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 63
- 235000019260 propionic acid Nutrition 0.000 description 63
- 239000000741 silica gel Substances 0.000 description 61
- 229910002027 silica gel Inorganic materials 0.000 description 61
- 229960001866 silicon dioxide Drugs 0.000 description 61
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 60
- 239000002904 solvent Substances 0.000 description 59
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 52
- 239000011259 mixed solution Substances 0.000 description 47
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 45
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 42
- 239000000203 mixture Substances 0.000 description 40
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 38
- 238000007738 vacuum evaporation Methods 0.000 description 35
- 238000005406 washing Methods 0.000 description 34
- 239000000460 chlorine Substances 0.000 description 29
- 239000007864 aqueous solution Substances 0.000 description 28
- 239000007787 solid Substances 0.000 description 27
- 238000001704 evaporation Methods 0.000 description 26
- 239000011734 sodium Substances 0.000 description 26
- 150000001412 amines Chemical class 0.000 description 25
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 24
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 24
- 239000006260 foam Substances 0.000 description 23
- 239000011541 reaction mixture Substances 0.000 description 23
- 238000001035 drying Methods 0.000 description 22
- 239000002585 base Substances 0.000 description 20
- 238000000605 extraction Methods 0.000 description 19
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 19
- 238000010898 silica gel chromatography Methods 0.000 description 19
- 235000002639 sodium chloride Nutrition 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- 239000002994 raw material Substances 0.000 description 18
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 17
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 17
- 229910001873 dinitrogen Inorganic materials 0.000 description 16
- 239000007788 liquid Substances 0.000 description 16
- QGLVWTFUWVTDEQ-UHFFFAOYSA-N 2-chloro-3-methoxyphenol Chemical compound COC1=CC=CC(O)=C1Cl QGLVWTFUWVTDEQ-UHFFFAOYSA-N 0.000 description 15
- JBDSSBMEKXHSJF-UHFFFAOYSA-N cyclopentanecarboxylic acid Chemical compound OC(=O)C1CCCC1 JBDSSBMEKXHSJF-UHFFFAOYSA-N 0.000 description 15
- 229960004756 ethanol Drugs 0.000 description 15
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 14
- 238000005859 coupling reaction Methods 0.000 description 14
- 239000000284 extract Substances 0.000 description 14
- 238000006722 reduction reaction Methods 0.000 description 14
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 13
- 238000013375 chromatographic separation Methods 0.000 description 13
- 230000008878 coupling Effects 0.000 description 13
- 238000010168 coupling process Methods 0.000 description 13
- 230000008020 evaporation Effects 0.000 description 13
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 13
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 13
- 230000009467 reduction Effects 0.000 description 13
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 11
- 239000012044 organic layer Substances 0.000 description 11
- 239000012074 organic phase Substances 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- 238000009834 vaporization Methods 0.000 description 10
- 230000008016 vaporization Effects 0.000 description 10
- 102000003729 Neprilysin Human genes 0.000 description 9
- 108090000028 Neprilysin Proteins 0.000 description 9
- 230000008030 elimination Effects 0.000 description 9
- 238000003379 elimination reaction Methods 0.000 description 9
- 238000010828 elution Methods 0.000 description 9
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 9
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 9
- 235000017557 sodium bicarbonate Nutrition 0.000 description 9
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 9
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 9
- 238000010025 steaming Methods 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 8
- 239000003054 catalyst Substances 0.000 description 8
- 239000012043 crude product Substances 0.000 description 8
- 238000002425 crystallisation Methods 0.000 description 8
- 230000008025 crystallization Effects 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 229910052708 sodium Inorganic materials 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 7
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 7
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 7
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 7
- 229910052801 chlorine Inorganic materials 0.000 description 7
- 238000011097 chromatography purification Methods 0.000 description 7
- 125000004494 ethyl ester group Chemical group 0.000 description 7
- UFWIBTONFRDIAS-UHFFFAOYSA-N naphthalene-acid Natural products C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 7
- 239000001301 oxygen Substances 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- JAELLLITIZHOGQ-UHFFFAOYSA-N tert-butyl propanoate Chemical compound CCC(=O)OC(C)(C)C JAELLLITIZHOGQ-UHFFFAOYSA-N 0.000 description 7
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 6
- NZNMSOFKMUBTKW-UHFFFAOYSA-N cyclohexanecarboxylic acid Chemical class OC(=O)C1CCCCC1 NZNMSOFKMUBTKW-UHFFFAOYSA-N 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- 238000001953 recrystallisation Methods 0.000 description 6
- 235000017550 sodium carbonate Nutrition 0.000 description 6
- 229910000029 sodium carbonate Inorganic materials 0.000 description 6
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 5
- 241000675108 Citrus tangerina Species 0.000 description 5
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 5
- 208000004880 Polyuria Diseases 0.000 description 5
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- GCTPMLUUWLLESL-UHFFFAOYSA-N benzyl prop-2-enoate Chemical compound C=CC(=O)OCC1=CC=CC=C1 GCTPMLUUWLLESL-UHFFFAOYSA-N 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 229910052794 bromium Inorganic materials 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 239000012230 colorless oil Substances 0.000 description 5
- 238000010790 dilution Methods 0.000 description 5
- 239000012895 dilution Substances 0.000 description 5
- 230000035619 diuresis Effects 0.000 description 5
- FKRCODPIKNYEAC-UHFFFAOYSA-N ethyl propionate Chemical compound CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 5
- 239000012362 glacial acetic acid Substances 0.000 description 5
- 150000002310 glutaric acid derivatives Chemical class 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 5
- 239000008108 microcrystalline cellulose Substances 0.000 description 5
- 229940016286 microcrystalline cellulose Drugs 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 5
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 4
- JIRULJUIQOAJPM-UHFFFAOYSA-N 3-methoxy-2-methylpropanoic acid Chemical compound COCC(C)C(O)=O JIRULJUIQOAJPM-UHFFFAOYSA-N 0.000 description 4
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- YNWXVXXRJDFANW-UHFFFAOYSA-N C[SiH](C)C.I(=O)(=O)O Chemical compound C[SiH](C)C.I(=O)(=O)O YNWXVXXRJDFANW-UHFFFAOYSA-N 0.000 description 4
- 206010007559 Cardiac failure congestive Diseases 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 4
- 235000019445 benzyl alcohol Nutrition 0.000 description 4
- 229960004217 benzyl alcohol Drugs 0.000 description 4
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 229960000935 dehydrated alcohol Drugs 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 4
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 239000011630 iodine Substances 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 4
- 150000003457 sulfones Chemical class 0.000 description 4
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- YVZVHNPTRCHYEY-UHFFFAOYSA-N 2-methylhex-4-ynoic acid Chemical compound CC#CCC(C)C(O)=O YVZVHNPTRCHYEY-UHFFFAOYSA-N 0.000 description 3
- OVBFMEVBMNZIBR-UHFFFAOYSA-N 2-methylvaleric acid Chemical compound CCCC(C)C(O)=O OVBFMEVBMNZIBR-UHFFFAOYSA-N 0.000 description 3
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 3
- AJZOYSGAEPJFKH-UHFFFAOYSA-N 4-methoxy-2-methylbutanoic acid Chemical compound COCCC(C)C(O)=O AJZOYSGAEPJFKH-UHFFFAOYSA-N 0.000 description 3
- 239000005541 ACE inhibitor Substances 0.000 description 3
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 3
- VHOMAPWVLKRQAZ-UHFFFAOYSA-N Benzyl propionate Chemical class CCC(=O)OCC1=CC=CC=C1 VHOMAPWVLKRQAZ-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- 150000001263 acyl chlorides Chemical class 0.000 description 3
- 239000003513 alkali Substances 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 3
- NDKBVBUGCNGSJJ-UHFFFAOYSA-M benzyltrimethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)CC1=CC=CC=C1 NDKBVBUGCNGSJJ-UHFFFAOYSA-M 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 238000003810 ethyl acetate extraction Methods 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- 210000002837 heart atrium Anatomy 0.000 description 3
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 229910052744 lithium Inorganic materials 0.000 description 3
- 239000006210 lotion Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 3
- 230000001452 natriuretic effect Effects 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000000813 peptide hormone Substances 0.000 description 3
- XCRBXWCUXJNEFX-UHFFFAOYSA-N peroxybenzoic acid Chemical compound OOC(=O)C1=CC=CC=C1 XCRBXWCUXJNEFX-UHFFFAOYSA-N 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 3
- 238000007614 solvation Methods 0.000 description 3
- 229940124530 sulfonamide Drugs 0.000 description 3
- ISXSCDLOGDJUNJ-UHFFFAOYSA-N tert-butyl prop-2-enoate Chemical compound CC(C)(C)OC(=O)C=C ISXSCDLOGDJUNJ-UHFFFAOYSA-N 0.000 description 3
- 210000002700 urine Anatomy 0.000 description 3
- 238000001291 vacuum drying Methods 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- MJUVRTYWUMPBTR-MRXNPFEDSA-N 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-n-[1-[(2r)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)indol-5-yl]cyclopropane-1-carboxamide Chemical compound FC=1C=C2N(C[C@@H](O)CO)C(C(C)(CO)C)=CC2=CC=1NC(=O)C1(C=2C=C3OC(F)(F)OC3=CC=2)CC1 MJUVRTYWUMPBTR-MRXNPFEDSA-N 0.000 description 2
- SRBTZBKETQNYPZ-UHFFFAOYSA-N 1-(2-carboxyethyl)cyclopentane-1-carboxylic acid Chemical compound OC(=O)CCC1(C(O)=O)CCCC1 SRBTZBKETQNYPZ-UHFFFAOYSA-N 0.000 description 2
- WUEPMEXUMQVEGN-UHFFFAOYSA-N 2,2,2-trifluoroacetic acid;2,2,2-trifluoro-n-[2-[2-[(2,2,2-trifluoroacetyl)amino]ethylamino]ethyl]acetamide Chemical compound OC(=O)C(F)(F)F.FC(F)(F)C(=O)NCCNCCNC(=O)C(F)(F)F WUEPMEXUMQVEGN-UHFFFAOYSA-N 0.000 description 2
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 2
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- MKACBEDLFQZKTI-UHFFFAOYSA-N 5-chloro-2-methylpentanoic acid Chemical compound OC(=O)C(C)CCCCl MKACBEDLFQZKTI-UHFFFAOYSA-N 0.000 description 2
- WDYVUKGVKRZQNM-UHFFFAOYSA-N 6-phosphonohexylphosphonic acid Chemical compound OP(O)(=O)CCCCCCP(O)(O)=O WDYVUKGVKRZQNM-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- 206010002383 Angina Pectoris Diseases 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 101150065749 Churc1 gene Proteins 0.000 description 2
- 241000257465 Echinoidea Species 0.000 description 2
- 108010059378 Endopeptidases Proteins 0.000 description 2
- 102000005593 Endopeptidases Human genes 0.000 description 2
- 208000010412 Glaucoma Diseases 0.000 description 2
- 206010020571 Hyperaldosteronism Diseases 0.000 description 2
- 206010020590 Hypercalciuria Diseases 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 206010030113 Oedema Diseases 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- 102100038239 Protein Churchill Human genes 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 239000005864 Sulphur Substances 0.000 description 2
- UGAPHEBNTGUMBB-UHFFFAOYSA-N acetic acid;ethyl acetate Chemical compound CC(O)=O.CCOC(C)=O UGAPHEBNTGUMBB-UHFFFAOYSA-N 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N anhydrous n-heptane Natural products CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 125000000440 benzylamino group Chemical group [H]N(*)C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 230000036772 blood pressure Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- DVECBJCOGJRVPX-UHFFFAOYSA-N butyryl chloride Chemical compound CCCC(Cl)=O DVECBJCOGJRVPX-UHFFFAOYSA-N 0.000 description 2
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical class [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 239000011575 calcium Chemical class 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 125000006244 carboxylic acid protecting group Chemical group 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 150000001924 cycloalkanes Chemical class 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 230000001882 diuretic effect Effects 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 239000002024 ethyl acetate extract Substances 0.000 description 2
- 238000000227 grinding Methods 0.000 description 2
- FUKUFMFMCZIRNT-UHFFFAOYSA-N hydron;methanol;chloride Chemical compound Cl.OC FUKUFMFMCZIRNT-UHFFFAOYSA-N 0.000 description 2
- PEHSSTUGJUBZBI-UHFFFAOYSA-N indan-5-ol Chemical compound OC1=CC=C2CCCC2=C1 PEHSSTUGJUBZBI-UHFFFAOYSA-N 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- OVEHNNQXLPJPPL-UHFFFAOYSA-N lithium;n-propan-2-ylpropan-2-amine Chemical compound [Li].CC(C)NC(C)C OVEHNNQXLPJPPL-UHFFFAOYSA-N 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 238000006386 neutralization reaction Methods 0.000 description 2
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 2
- 229960001597 nifedipine Drugs 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- IENZQIKPVFGBNW-UHFFFAOYSA-N prazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 IENZQIKPVFGBNW-UHFFFAOYSA-N 0.000 description 2
- 229960001289 prazosin Drugs 0.000 description 2
- 201000009395 primary hyperaldosteronism Diseases 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000012266 salt solution Substances 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- 229910001415 sodium ion Inorganic materials 0.000 description 2
- 229960002317 succinimide Drugs 0.000 description 2
- FDDDEECHVMSUSB-UHFFFAOYSA-N sulfanilamide Chemical compound NC1=CC=C(S(N)(=O)=O)C=C1 FDDDEECHVMSUSB-UHFFFAOYSA-N 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 125000005951 trifluoromethanesulfonyloxy group Chemical group 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- HKEJPRYQQADSPP-UHFFFAOYSA-N (2-methyloxolan-2-yl) trifluoromethanesulfonate Chemical compound FC(S(=O)(=O)OC1(OCCC1)C)(F)F HKEJPRYQQADSPP-UHFFFAOYSA-N 0.000 description 1
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 1
- ZMMRLWFGWQVVJD-UHFFFAOYSA-O (n,n-dimethylcarbamimidoyl)-dimethylazanium;azide Chemical compound [N-]=[N+]=[N-].CN(C)C(N)=[N+](C)C ZMMRLWFGWQVVJD-UHFFFAOYSA-O 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- QWUWMCYKGHVNAV-UHFFFAOYSA-N 1,2-dihydrostilbene Chemical group C=1C=CC=CC=1CCC1=CC=CC=C1 QWUWMCYKGHVNAV-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- YZUPZGFPHUVJKC-UHFFFAOYSA-N 1-bromo-2-methoxyethane Chemical compound COCCBr YZUPZGFPHUVJKC-UHFFFAOYSA-N 0.000 description 1
- MYFKLQFBFSHBPA-UHFFFAOYSA-N 1-chloro-2-methylsulfanylethane Chemical compound CSCCCl MYFKLQFBFSHBPA-UHFFFAOYSA-N 0.000 description 1
- SFOYQZYQTQDRIY-UHFFFAOYSA-N 1-chloro-3-iodopropane Chemical compound ClCCCI SFOYQZYQTQDRIY-UHFFFAOYSA-N 0.000 description 1
- QYQQTZFOFMPYCA-UHFFFAOYSA-N 1-iodoprop-1-ene Chemical group CC=CI QYQQTZFOFMPYCA-UHFFFAOYSA-N 0.000 description 1
- XKXXXODAXXAFNP-UHFFFAOYSA-N 1-o-benzyl 3-o-tert-butyl propanedioate Chemical compound CC(C)(C)OC(=O)CC(=O)OCC1=CC=CC=C1 XKXXXODAXXAFNP-UHFFFAOYSA-N 0.000 description 1
- JIXHYWCLUOGIMM-UHFFFAOYSA-N 2-(2-methoxyethoxy)propanoic acid Chemical compound COCCOC(C)C(O)=O JIXHYWCLUOGIMM-UHFFFAOYSA-N 0.000 description 1
- VPJRVTFFZXVLRP-UHFFFAOYSA-N 2-(2-methoxyethyl)propanedioic acid Chemical compound COCCC(C(O)=O)C(O)=O VPJRVTFFZXVLRP-UHFFFAOYSA-N 0.000 description 1
- HVRZYSHVZOELOH-UHFFFAOYSA-N 2-Methyl-4-pentenoic acid Chemical compound OC(=O)C(C)CC=C HVRZYSHVZOELOH-UHFFFAOYSA-N 0.000 description 1
- VRLUSLNMNQAPOH-UHFFFAOYSA-N 2-cyclohexylpropanoic acid Chemical compound OC(=O)C(C)C1CCCCC1 VRLUSLNMNQAPOH-UHFFFAOYSA-N 0.000 description 1
- YGGWENAMQGWVGL-UHFFFAOYSA-N 2-cyclopentyl-3-(2-methoxyethoxy)-2-methylpropanoic acid Chemical compound C1(CCCC1)C(C(=O)O)(C)COCCOC YGGWENAMQGWVGL-UHFFFAOYSA-N 0.000 description 1
- ASUDFOJKTJLAIK-UHFFFAOYSA-N 2-methoxyethanamine Chemical compound COCCN ASUDFOJKTJLAIK-UHFFFAOYSA-N 0.000 description 1
- ANUFAWHRSIJTHW-UHFFFAOYSA-N 2-methylheptanedioic acid Chemical class OC(=O)C(C)CCCCC(O)=O ANUFAWHRSIJTHW-UHFFFAOYSA-N 0.000 description 1
- KGIGUEBEKRSTEW-UHFFFAOYSA-N 2-vinylpyridine Chemical compound C=CC1=CC=CC=N1 KGIGUEBEKRSTEW-UHFFFAOYSA-N 0.000 description 1
- YFCIFWOJYYFDQP-PTWZRHHISA-N 4-[3-amino-6-[(1S,3S,4S)-3-fluoro-4-hydroxycyclohexyl]pyrazin-2-yl]-N-[(1S)-1-(3-bromo-5-fluorophenyl)-2-(methylamino)ethyl]-2-fluorobenzamide Chemical compound CNC[C@@H](NC(=O)c1ccc(cc1F)-c1nc(cnc1N)[C@H]1CC[C@H](O)[C@@H](F)C1)c1cc(F)cc(Br)c1 YFCIFWOJYYFDQP-PTWZRHHISA-N 0.000 description 1
- DOTGZROJTAUYFQ-UHFFFAOYSA-N 4-bromobut-2-enoic acid Chemical compound OC(=O)C=CCBr DOTGZROJTAUYFQ-UHFFFAOYSA-N 0.000 description 1
- WZNXBMDGFRTUOZ-UHFFFAOYSA-N 6-hydroxy-2-methylhexanoic acid Chemical compound OC(=O)C(C)CCCCO WZNXBMDGFRTUOZ-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 1
- 102400001282 Atrial natriuretic peptide Human genes 0.000 description 1
- 101800001890 Atrial natriuretic peptide Proteins 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- MXJWWBZRSZLYCM-UHFFFAOYSA-N C1(CCCC1)C(=O)O.[Li] Chemical class C1(CCCC1)C(=O)O.[Li] MXJWWBZRSZLYCM-UHFFFAOYSA-N 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 240000001879 Digitalis lutea Species 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- JIGUQPWFLRLWPJ-UHFFFAOYSA-N Ethyl acrylate Chemical compound CCOC(=O)C=C JIGUQPWFLRLWPJ-UHFFFAOYSA-N 0.000 description 1
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 1
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 206010017807 Gastric mucosal hypertrophy Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 208000000239 Hypertrophic Gastritis Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 208000027530 Meniere disease Diseases 0.000 description 1
- 238000006845 Michael addition reaction Methods 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- RKOTXQYWCBGZLP-UHFFFAOYSA-N N-[(2,4-difluorophenyl)methyl]-2-ethyl-9-hydroxy-3-methoxy-1,8-dioxospiro[3H-pyrido[1,2-a]pyrazine-4,3'-oxolane]-7-carboxamide Chemical compound CCN1C(OC)C2(CCOC2)N2C=C(C(=O)NCC3=C(F)C=C(F)C=C3)C(=O)C(O)=C2C1=O RKOTXQYWCBGZLP-UHFFFAOYSA-N 0.000 description 1
- IDGQXGPQOGUGIX-VIFPVBQESA-N O-BENZYL-l-SERINE Chemical compound OC(=O)[C@@H](N)COCC1=CC=CC=C1 IDGQXGPQOGUGIX-VIFPVBQESA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 206010036618 Premenstrual syndrome Diseases 0.000 description 1
- 108090000783 Renin Proteins 0.000 description 1
- 102100028255 Renin Human genes 0.000 description 1
- XPPULAHOJKTSAQ-SECBINFHSA-N SC[C@@]1(N(CCC1)C(CC)=O)C(=O)O Chemical compound SC[C@@]1(N(CCC1)C(CC)=O)C(=O)O XPPULAHOJKTSAQ-SECBINFHSA-N 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- FKNQFGJONOIPTF-UHFFFAOYSA-N Sodium cation Chemical compound [Na+] FKNQFGJONOIPTF-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical group O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 1
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 1
- 229920004482 WACKER® Polymers 0.000 description 1
- KPZIJOVUZXUKLF-UHFFFAOYSA-N [Br].C(CCC)C=CC Chemical group [Br].C(CCC)C=CC KPZIJOVUZXUKLF-UHFFFAOYSA-N 0.000 description 1
- PFRUBEOIWWEFOL-UHFFFAOYSA-N [N].[S] Chemical compound [N].[S] PFRUBEOIWWEFOL-UHFFFAOYSA-N 0.000 description 1
- DPDMMXDBJGCCQC-UHFFFAOYSA-N [Na].[Cl] Chemical compound [Na].[Cl] DPDMMXDBJGCCQC-UHFFFAOYSA-N 0.000 description 1
- JLCHNBRGUPQWKF-UHFFFAOYSA-J [OH-].[C+4].[OH-].[OH-].[OH-] Chemical compound [OH-].[C+4].[OH-].[OH-].[OH-] JLCHNBRGUPQWKF-UHFFFAOYSA-J 0.000 description 1
- WJGAPUXHSQQWQF-UHFFFAOYSA-N acetic acid;hydrochloride Chemical compound Cl.CC(O)=O WJGAPUXHSQQWQF-UHFFFAOYSA-N 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000005041 acyloxyalkyl group Chemical group 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 238000005576 amination reaction Methods 0.000 description 1
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 229940127088 antihypertensive drug Drugs 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 150000007860 aryl ester derivatives Chemical class 0.000 description 1
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 1
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 125000006278 bromobenzyl group Chemical group 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- NHZZUJIRMHDTKR-UHFFFAOYSA-N carbonic acid;toluene Chemical compound OC(O)=O.CC1=CC=CC=C1 NHZZUJIRMHDTKR-UHFFFAOYSA-N 0.000 description 1
- 238000003763 carbonization Methods 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000002327 cardiovascular agent Substances 0.000 description 1
- 229940125692 cardiovascular agent Drugs 0.000 description 1
- 238000005277 cation exchange chromatography Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000003518 caustics Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- HRYZWHHZPQKTII-UHFFFAOYSA-N chloroethane Chemical compound CCCl HRYZWHHZPQKTII-UHFFFAOYSA-N 0.000 description 1
- GGRHYQCXXYLUTL-UHFFFAOYSA-N chloromethyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OCCl GGRHYQCXXYLUTL-UHFFFAOYSA-N 0.000 description 1
- 229940061627 chloromethyl methyl ether Drugs 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- 235000009508 confectionery Nutrition 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- VZFUCHSFHOYXIS-UHFFFAOYSA-N cycloheptane carboxylic acid Natural products OC(=O)C1CCCCCC1 VZFUCHSFHOYXIS-UHFFFAOYSA-N 0.000 description 1
- 230000006837 decompression Effects 0.000 description 1
- 239000000645 desinfectant Substances 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- SPWVRYZQLGQKGK-UHFFFAOYSA-N dichloromethane;hexane Chemical compound ClCCl.CCCCCC SPWVRYZQLGQKGK-UHFFFAOYSA-N 0.000 description 1
- BOROJSBBTJGBRP-UHFFFAOYSA-N dichloromethylcyclopentane Chemical compound ClC(Cl)C1CCCC1 BOROJSBBTJGBRP-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 150000007520 diprotic acids Chemical class 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- AFOSIXZFDONLBT-UHFFFAOYSA-N divinyl sulfone Chemical compound C=CS(=O)(=O)C=C AFOSIXZFDONLBT-UHFFFAOYSA-N 0.000 description 1
- 235000015177 dried meat Nutrition 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000008451 emotion Effects 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- UJTPZISIAWDGFF-UHFFFAOYSA-N ethenylsulfonylbenzene Chemical compound C=CS(=O)(=O)C1=CC=CC=C1 UJTPZISIAWDGFF-UHFFFAOYSA-N 0.000 description 1
- ZKQFHRVKCYFVCN-UHFFFAOYSA-N ethoxyethane;hexane Chemical compound CCOCC.CCCCCC ZKQFHRVKCYFVCN-UHFFFAOYSA-N 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- HCPOCMMGKBZWSJ-UHFFFAOYSA-N ethyl 3-hydrazinyl-3-oxopropanoate Chemical compound CCOC(=O)CC(=O)NN HCPOCMMGKBZWSJ-UHFFFAOYSA-N 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 235000011194 food seasoning agent Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000027119 gastric acid secretion Effects 0.000 description 1
- 230000007160 gastrointestinal dysfunction Effects 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- NEXSMEBSBIABKL-UHFFFAOYSA-N hexamethyldisilane Chemical compound C[Si](C)(C)[Si](C)(C)C NEXSMEBSBIABKL-UHFFFAOYSA-N 0.000 description 1
- UKJFVOWPUXSBOM-UHFFFAOYSA-N hexane;oxolane Chemical compound C1CCOC1.CCCCCC UKJFVOWPUXSBOM-UHFFFAOYSA-N 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- DCPMPXBYPZGNDC-UHFFFAOYSA-N hydron;methanediimine;chloride Chemical compound Cl.N=C=N DCPMPXBYPZGNDC-UHFFFAOYSA-N 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- YIAPLDFPUUJILH-UHFFFAOYSA-N indan-1-ol Chemical compound C1=CC=C2C(O)CCC2=C1 YIAPLDFPUUJILH-UHFFFAOYSA-N 0.000 description 1
- SNWQUNCRDLUDEX-UHFFFAOYSA-N inden-1-one Chemical compound C1=CC=C2C(=O)C=CC2=C1 SNWQUNCRDLUDEX-UHFFFAOYSA-N 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical class CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 230000005906 menstruation Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical class [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 150000002780 morpholines Chemical class 0.000 description 1
- BXGTVNLGPMZLAZ-UHFFFAOYSA-N n'-ethylmethanediimine;hydrochloride Chemical class Cl.CCN=C=N BXGTVNLGPMZLAZ-UHFFFAOYSA-N 0.000 description 1
- SFMJNHNUOVADRW-UHFFFAOYSA-N n-[5-[9-[4-(methanesulfonamido)phenyl]-2-oxobenzo[h][1,6]naphthyridin-1-yl]-2-methylphenyl]prop-2-enamide Chemical compound C1=C(NC(=O)C=C)C(C)=CC=C1N1C(=O)C=CC2=C1C1=CC(C=3C=CC(NS(C)(=O)=O)=CC=3)=CC=C1N=C2 SFMJNHNUOVADRW-UHFFFAOYSA-N 0.000 description 1
- PVWOIHVRPOBWPI-UHFFFAOYSA-N n-propyl iodide Chemical compound CCCI PVWOIHVRPOBWPI-UHFFFAOYSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- QWVGKYWNOKOFNN-UHFFFAOYSA-N o-cresol Chemical compound CC1=CC=CC=C1O QWVGKYWNOKOFNN-UHFFFAOYSA-N 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 239000004533 oil dispersion Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000004817 pentamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- XGISHOFUAFNYQF-UHFFFAOYSA-N pentanoyl chloride Chemical compound CCCCC(Cl)=O XGISHOFUAFNYQF-UHFFFAOYSA-N 0.000 description 1
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- LIGACIXOYTUXAW-UHFFFAOYSA-N phenacyl bromide Chemical compound BrCC(=O)C1=CC=CC=C1 LIGACIXOYTUXAW-UHFFFAOYSA-N 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000007715 potassium iodide Nutrition 0.000 description 1
- 229960004839 potassium iodide Drugs 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 229960001153 serine Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- PODWXQQNRWNDGD-UHFFFAOYSA-L sodium thiosulfate pentahydrate Chemical compound O.O.O.O.O.[Na+].[Na+].[O-]S([S-])(=O)=O PODWXQQNRWNDGD-UHFFFAOYSA-L 0.000 description 1
- 235000013599 spices Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000005728 strengthening Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000006633 tert-butoxycarbonylamino group Chemical group 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- JMXKSZRRTHPKDL-UHFFFAOYSA-N titanium ethoxide Chemical compound [Ti+4].CC[O-].CC[O-].CC[O-].CC[O-] JMXKSZRRTHPKDL-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 238000003809 water extraction Methods 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/12—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/57—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C233/61—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by doubly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/44—Iso-indoles; Hydrogenated iso-indoles
- C07D209/46—Iso-indoles; Hydrogenated iso-indoles with an oxygen atom in position 1
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/56—Amides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/10—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/36—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/46—Acylated amino or imino radicals by carboxylic acids, or sulfur or nitrogen analogues thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/10—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/16—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/56—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D321/00—Heterocyclic compounds containing rings having two oxygen atoms as the only ring hetero atoms, not provided for by groups C07D317/00 - C07D319/00
- C07D321/02—Seven-membered rings
- C07D321/04—Seven-membered rings not condensed with other rings
- C07D321/08—1,4-Dioxepines; Hydrogenated 1,4-dioxepines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/34—Sulfur atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
具有通式I的化合物及其可作药用的盐及其生物前体都是利尿剂,可用于高血压,心衰,肾机能不全和其它方面疾病的治疗。
Description
本发明是关于一系列螺-取代的戊二酸单酰胺衍生物,它们是利尿药,可用于多种治疗范围,包括对各种心血管疾病如高血压及心衰等的治疗。
这些化合物是锌-依赖的、中性肽链内切酶E.C.3.4.24.11.的抑制剂。这个酶与几种肽激素的断裂有关,肽激素包括心房尿钠排泄因子(ANF),它是由心脏分泌的,具有强的血管扩张、利尿及尿钠排泄作用。这样,本发明的化合物由于抑制中性肽链内切酶E.C.3.4.24.11,能加强ANF的生物效应。因而,尤其是,这些化合物作为利尿剂,应用于许多疾病的治疗,包括高血压,心衰,心绞痛,肾机能不全,月经前综合征,周期水肿,梅尼埃尔氏病,醛甾酮过多症(初期及二期)及高钙尿。此外,由于它们有加强ANF效应的能力这些化合物可用于治疗青光眼。它们对中性肽链内切酶E.C.3.4.24.11,抑制能力的另一个结果是本发明的化合物在其他治疗领域中亦有作用,例如治疗气喘,炎症,疼痛,癫痫,情感紊乱,痴呆及老年性混乱,肥胖及胃肠紊乱(尤其腹泻及过敏的肠综合征),调整胃酸分泌及治疗血肾素过多。
化合物为下列通式:
(通式见下页)
及其可作药用的盐及生物前体。
其中A完成一个4至7员碳环,它们可以是饱和的或单一不饱和的,
它们可随意地与另外的饱和或不饱和5或6员碳环稠合;B是

(CH2)m,其中m是1至3的整数;R及R4分别为H,C1-C6烷基,苄基或是形成生物不稳定的酯的基团;
R1是H或C1-C4烷基;
R2及R3分别是H,OH,C1-C4烷基或C1-C4烷氧基;
及R5是C1-C6烷基,C2-C6链烯基,C2-C6炔基,芳基(C2-C6炔基),C3-C7环烷基,C3-C7环烯基,C1-C6烷氧基,-NR6R7,-NR8COR9,-NR8SO2R9或是一个饱和杂环基团;或被一个或多个取代基所取代的C1-C6烷基,取代基选自卤素,羟基,C1-C6烷氧基,C2-C6羟基烷氧基,C1-C6烷氧基,C1-C6烷氧基,C3-C7环烷基,C3-C7环烯基,芳基,芳氧基,芳氧基(C1-C4烷氧基),杂环基,杂环氧基,-NR6R7,-NR8COR9,-NR8SO2R9,-CONR6R7,-SH,-S(O)pR10,-COR11或-CO2R12;
其中R6及R7分别为H,C1-C4烷基,C3-C7环烷基(任意被羟基或C1-C4烷氧基取代),芳基,芳基(C1-C4烷基),C2-C6烷氧烷基,或杂环基;或R6及R7两个基团与氮连在一起形成一个吡咯烷基,哌啶子基,吗啉代,哌嗪基或N-(C1-C4烷基),-哌嗪基;
R8是H或C1-C4烷基;
R9是C1-C4烷基,CF3,芳基,芳基(C1-C4烷基),芳基(C1-C4烷氧基),杂环基,C1-C4烷氧基或NR6R7其中R6及R7定义如前;
R10是C1-C4烷基,芳基,杂环基或NR6R7其中R6及R7定义如前;
R11是C1-C4烷基,C3-C7环烷基,芳基或杂环基;
R12是H或C1-C4烷基;
及P是0,1或2;
在上述定义中,除非有其他所指,烷基有三个
或更多的碳原子,可以是直链的或支链的。在这里所用术语芳基意指一个芳香碳氢基团如苯基或萘基,它们可以任意地取代,例如被一个或多个OH,CN,CF3,C1-C4烷基,C1-C4烷氧基,卤素,氨基甲酰基,氨基磺酰基,氨基,单或双(C1-C4烷基)氨基或(C1-C4烷酰基)氨基基团取代。卤素为氟,氯,溴或碘。
术语杂环基意指一个包含氮,氧或硫的5或6员杂环基,除非有其他规定,可以是饱和的或不饱和的,并可任意地在环内再包含一个氧或一至三个氮原子,它可以任意地与苯稠合或取代例如被一个或多个卤素,C1-C4烷基,羟基,氨基甲酰基,苄基,氧代,氨基或单或双-(C1-C4烷基)氨基或(C1-C4烷酰基)氨基基团取代。杂环的具体例子包含吡啶基,吡嗪基,嘧啶基,哒嗪基,吡咯基,咪啶基,吡唑基,三唑基,四唑基,呋喃基,四氢呋喃基,四氢吡喃基,二噁烷基,噻吩基,噁唑基,异噁唑基,噻唑基,吲哚基,异吲哚基,喹啉基,喹喔啉基,喹唑啉基及苯并咪唑基,如上面所述每一个可任意取代。
通式(Ⅰ)化合物可以含有几个不对称中心,因而他们能作为对映体及非对映异构体存在。本发明包括分离的单独异构体和异构体混合物两者。
含有一个酸中心的通式(Ⅰ)化合物的可作药用的盐是与碱形成的无毒性的盐。实例包括碱金属盐(如钠,钾)或钙盐或与胺如二乙胺成的盐。具有碱中心的化合物也能与医药上可用的酸形成酸加成盐。实例包括盐酸盐,溴氢酸盐,硫酸盐或硫酸氢盐,磷酸盐或磷酸氢盐,乙酸盐,柠檬酸盐,富马酸盐,葡糖酸盐,乳酸盐,顺丁烯二酸盐,琥珀酸盐或酒石酸盐。
在上面定义中的术语生物前体意为通式(Ⅰ)化合物的可作为药用的生物可降解的衍生物,在对动物或人给药后它在体内转变产生通式(Ⅰ)的化合物。
通式(Ⅰ)较好的一组化合物是:其中A是(CH2)n,n是3至6的整数,及在其中R及R4分别是H,C1-C6烷基或苄基。
最好的一组通式(Ⅰ)化合物是:其中A是(CH2)4,R1是H及B是(CH2)2,即为通式(Ⅱ)化合物,其中R,R2,R3,R4及R5定义如前面通式(Ⅰ):
较好的还有通式(Ⅰ)及(Ⅱ)化合物,其中R及R4都是H(二酸),和其生物不稳定的单及双-酯衍生物,其中R及R4一个或两者是形成生物不稳定酯的基团。
术语形成生物不稳定酯的基团在技术上是很好理解的,意为能提供酯的基团,该酯能在体内断裂释出通式(Ⅰ)的相应二酸,其中R及R4均是H。许多这种酯是众所周知的,例如在盘尼西林范围内或在抗高血压药物ACE抑制剂例子中。
对通式(Ⅰ)及(Ⅱ)化合物来说,这种生物不稳定的前药酯在提供通式(Ⅰ)化合物作为口服给药时特别有利。任何个别的形成酯的基团的适合性能用常规的动物或体外酶水解研究来评价。这样,对于最佳作用,则需要酯仅在吸收后才水解。因此酯在吸收前应能抵抗消化酶的水解,但应能迅速地被例如肝脏酶水解。就这样有活性的二酸随着口服吸收被释放进入血流。
除了低级烷酯(特别乙基)及苄酯外,合适的生物不稳定酯包括烷酰氧基烷基酯,包括它们的烷基,环烷基及芳基取代的衍生物,及芳香酰氧烷基酯,芳基酯,芳烷基酯,及卤代烷基酯其中所说的烷酰基或烷基基团具有1至8个碳原子,是支链或直链的。所说的芳基是苯基,萘基或茚基,可任意被一个或多个C1-C4烷基或C1-C4烷氧基或卤原子所取代。
当R及R4是形成生物不稳定酯的基团时,除乙基、苄基外,还包括下列例子:
1-(2,2-二乙基丁酰氧基)乙基
2-乙基丙酰氧基甲基
1-(2-乙基丙酰氧基)乙基
1-(2,4-二甲基苯酰氧基)乙基
2-苯酰氧基苄基
1-(苯酰氧基)乙基
2-甲基-1-丙酰氧基-1-丙基
2,4,6-三甲基苯酰氧基甲基
1-(2,4,6-三甲基苯酰氧基)乙基
新戊酰氧基甲基
苯乙基
苯丙基
2,2,2-三氟乙基
1-或2-萘基
2,4-二甲基苯基
4-叔丁基苯基
及5-二氢茚基。
其中一个最好的形成生物不稳定酯的基团是5-二氢茚基。
通式(Ⅰ)及(Ⅱ)化合物,其中R及R4中一个或二个是C1-C6烷基,尤其是乙基或苄基,也是有活性的,由于它们在体内水解,另外,它们为制备二酸的有价值的中间体,二酸的R及R4均是H。尤其是发现单苄酯及单乙基酯在体内迅速水解成二酸。
通式(Ⅱ)的另一组较好化合物中,R是H,R2是H,R3是CH3或C2H5及R4是H。特别好的是:其中R,R2,R3及R4分别是H和羧基基团CO2R4是连接在环己烷环的3-或4-位上,最好是那些与酰胺基成顺式-立体化学关系的化合物。
由于它们良好的口服效果而有特殊兴趣的是式(Ⅱ)的单乙基酯,其中R,R2和R3各为H,R4是乙基,并且乙氧羰基是连接在环己烷环的3位,并与酰胺基处于顺式-立体化学关系。
基团R5较好为C2-C4烷基,C2-C4烯基,C2-C5炔基,C5-C6环烷基,C5-C6环烯基,C1-C4烷基磺酰氨基,或四氢呋喃基或其中R5是被下列基团所取代的C1-C3烷基,C1-C3烷氧基,C1-C6烷氧基(C2-C4烷氧基),C3-C6环烷基,4-吡啶基,2-咪唑基,C2-C4烷酰基,C2-C4烷氧羰基氨基,C1-C4烷基磺酰基,C1-C4烷基磺酰氨基,芳基磺酰氨基,杂芳磺酰氨基或苯甲酰氨基。
这样在一个特殊的及较好的方面,本发明提供了通式(Ⅱ)的二羧酸,其中R及R4都是H,及其中R2及R3都是H,CO2R4基团连接在环己烷环的4-位并与酰胺基团成顺式-立体化学关系,及其中R5是正丙基,甲氧基乙基,2-甲氧基乙氧基甲基,2-丁炔基,环己烯基,四氢呋喃基,4-吡啶基甲基,2-咪唑基甲基,丙酮基,乙基磺酰甲基,苯磺酰氨基甲基,正丙基磺酰氨基或1-甲氧基羰基氨基乙基。
特别好的个别化合物包括3-{1-[(顺-4-羰基环己基)氨基甲酰基]环戊基}-2-(正丙基)-丙酸。
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙基)丙酸。
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙氧基甲基)丙酸。
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-丁炔基)丙酸。
3-{1-[(顺-4-羧基环己基)氨基甲酰氨]环戊基}-2-(3-四氢呋喃基)丙酸。
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(n-丙基磺酰氨基)丙酸。
在另一个特殊的及较好方面,本发明提供了通式(Ⅱ)的二羧酸,其中R及R4都是H,R2及R3都是H,CO2R4基团链在环己烷环的3位并与酰胺基团成顺式-立体化学关系,及其中R5是正丙基,2-甲氧基乙氧基甲基,2-丁炔基,2-丙烯基,2-丁烯基,环戊基,环己基,环己烯基,环丙基甲基,四氢呋喃基,4-吡啶基甲基,n-丙基磺酰氨基,苯磺酰氨基甲基,或苯甲酰氨基甲基。
特别好的个别化合物包括3-{1-[(顺-3-羰基环己基)氨基甲酰基]环戊基}-2-(正丙基)-丙酸。
3-{1-[(顺-3-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙氧基甲基)丙酸。
3-{1-[(顺-3-羧基环己基)氨基甲酰基]环戊基}-2-(2-丁炔基)丙酸。及
3-{1-[(顺-3-羧基环己基)氨基甲酰基]环戊基}-2-(正丙基磺酰氨基)丙酸。
在另一特殊及较好的方面,本发明提供了通式(Ⅱ)的单乙基酯,其中R是H及R4是乙基及其中R2及R3都是H,乙氧羰基连在环己烷环的3-位并有顺式-立体化学及R5是2-甲氧基乙氧基甲基,正丙基,2-丁炔基,2-丙烯基,环己烯基,环己基,环戊基,环丙基甲基,四氢呋喃基,4-吡啶基甲基,苯磺酰氨基甲基,苯甲酰氨基甲基,或正丙基磺酰氨基。
特别好的个别化合物包括3-{1-[(顺-3-乙氧羰基环己基)氨基甲酰基]环戊基}-2-(2-甲氧基
乙氧基甲基)丙酸。
3-{1-[(顺-3-乙氧羰基环己基)氨基甲酰基]环戊基}-2-(正丙基)丙酸。
3-{1-[(顺-3-乙氧羰基环己基)氨基甲酰基]环戊基}-2-(2-丁炔基)丙酸。及
3-{1-[(顺-3-乙氧羰基环己基)氨基甲酰基]环戊基}-2-(n-丙基磺酰氨基)丙酸。
但在更为特殊及比较好的方面,本发明提供了通式(Ⅱ)的生物不稳定酯的衍生物,其中R及R4中之一或二者都是5-二氢茚基。
特别好的个别化合物包括:
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙氧基甲基)丙酸5-二氢茚酯,及
3-{1-[(顺-4-{5-二氢茚氧基羰基}环己基)氨基甲酰基]-环戊基}-2-(2-甲氧基乙氧基甲基)丙酸。
通式(Ⅰ)化合物由若干不同的方法来制备。基本方法包括合成一个部分保护的螺-取代的戊二酸衍生物,它与一个胺偶合得所需的戊二酸单酰胺。在胺中,如果羧酸为游离的或在R5中有任何活性基团则在偶合一步时需要保护,在该方法的最后阶段除去保护基团。
合成路线在方案1中说明,其中A,B,R1,R2及R3在前面已定义,R5′的定义如同具有被护的活性基团的R5,R13和R14定义如R和R4,但H除外,或它们是常规的羧酸保护基团:
(方案1见下页)
用常规的胺偶合技术完成通式(Ⅲ)及(Ⅳ)化合物的反应。这样,在一种方法中,为完成反应,反应物要溶于有机溶剂中如二氯甲烷,用一种二亚胺作缩合剂,例如1-乙基-3-(二甲基氨基丙基)-碳化

二亚胺,或N,N′-二环己基碳化二亚胺在1-羟基苯并三唑及一种有机碱如N-甲基吗啉存在下为有利。在室温12小时至24小时后反应通常完全,产物用常规方法分离出,如用水洗涤或过滤除去副产物尿素,然后蒸发溶剂。产物用结晶法再纯制,需要时用色谱法。通式(Ⅴ)化合物包括通式(Ⅰ)化合物,其中R及R4是C1-C6烷基或苄基。
在一些情况中偶合产物,为保护形式时,可以经受常规的化学变换反应,以制备通式(Ⅴ)的其他化合物。例如通式(Ⅴ)化合物其中R5′含有一个酯基,可以被水解或氢化产生羧酸,它可以再反应,例如与胺,生成酰胺衍生物。
同样地,其中R5′含有一个取代的或保护的氨基基团(例如苄氨基,双苄氨基,苄氧羰基氨基或叔丁氧基羰基氨基)的化合物可适当地用氢化或水解转成游离胺,产生的胺可以再反应,例如与磺酰氯反应生成相应的磺酰胺,与酰氯或酸酐酰化反应生成相应的酰胺,与异氰酸酯反应生成尿素衍生物及与氯甲酸酯或N-(芳氧基羰基)琥珀酰亚胺反应分别生成烷氧羰基氨基及芳氧基羰基氨基产物。其他的反应包括,例如,硫化物氧化生成相应的亚砜或砜的衍生物;顶端烯的华格(Wacker)氧化产生相应的甲基酮,它可以再反应,例如用还原胺化生成相应的胺;将化合物所含的苄氧基氢化得醇,叠氮基团还原得胺,或环烯还原成环烷化合物。所有这些转变完全是常规性的,为实行反应所要的合适条件及试剂以及其它变化及可能性对熟练技术人员都是熟知的。
通式(Ⅴ)的双酯可以进一步反应产生式(Ⅰ)的单酯或二酸衍生物,其中R及R4中之一或二者都是H。所用反应条件依通式(Ⅴ)的化合物中基团R13及R14的确切性质而定,但可能有一些改变。例如,当R13和R14两者都是苄基时,产物的氢化将生成通式(Ⅰ)的二酸,其中R和R4都是H。另外如果R13和R14中的一个是苄基而另一个是烷基,氢化将得到单酯产物。如果需要,产物能被水解,再生成二酸产物。当R13和R14中一个是叔丁基时,用三氟乙酸处理通式(Ⅴ)化合物产生相应的酸。双酯产物其中R13和R14是苄基或低级烷基时也能用碘化三甲基硅烷处理产生二
羧酸产物。如果R13或R14,是一些别的羧酸保护基时,则显然需要在最后一步用合适的条件将它除去以得通式(Ⅰ)的酯或二酸产物。在环A或取代基R5是不饱和的时候,必须用非还原方法去保护基,由此,例如若R或R4是苄基时,可用碘化三甲基硅烷处理除去。
在除去在R5′中所存在的保护基时,同样有一些化学转换反应可能发生在最后单酯或二酸产物,如同以前所描述的。在各种情况中得到产物可以是游离羧酸或可用合适的碱中和,以盐的形式分离。
通式(Ⅲ)的原料螺-取代的戊二酸单酯可以用一些不同方法来制备,如下列反应方案所阐明的:

通式(Ⅵ)的丙烯酸酯也是已知化合物,它们可以买到,或者可按文献用常规方法来制备(例如将一个合适的取代丙二酸单酯与多聚甲醛反应同时伴随以脱羧反应)。
丙烯酸酯直接与双阴离子反应,该双阴离子是从合适的环烷或环烯羧酸用强碱(如二异丙基氨基锂)处理得到的,生成通式(Ⅲ)的戊二酸单酯。另外,该酸与3-溴代丙酸酯反应产生相应的酯(Ⅷ)。然后与强碱(如二异丙基氨基锂)在低温下进行烷化反应产生双阴离子,随着加入合适的式R5′-X化合物(其中X是离去基团,如三氟甲烷磺酰氧基或卤基,最好为溴),或一个迈克尔加成接受体(如乙烯基砜)再产生通式(Ⅲ)的戊二酸单酯。
通式(Ⅳ)的胺一般都是已知的化合物,它们可得到其商品或按照文献用常规方法来制备(例如,在B为(CH2)2时,用相应的苯甲酸还原而得)。
通式(Ⅰ)化合物(其中R和R4中之一或二者都是形成生物不稳定酯的基团)是用上面描述过的相似方法制备。
由此,在方案1所概述方法的一种变法中,通式(Ⅲ)化合物(其中R13是一个形成生物不稳定酯的基团)与通式(Ⅳ)的合适的化合物(其中R14是苄基)进行偶合,产品被氢化,得到通式(Ⅰ)化合物,其中R是形成生物不稳定酯的基团及R4是H。
通式(Ⅲ)的戊二酸单酯(其中R13是一个形成生物不稳定酯的基团)是由相应的通式(Ⅲ)化合物(其中R13是一个常规的可选择性除去的羧酸保护基)来制备,例如叔丁基,首先保护游离羧基,例如它的苯甲酰甲基酯;用适合于所用的特定保护基团的常规方法除去R13;与所需的形成生物不稳定酯的基团形成一个酯,例如与式R17X′的卤化物反应或与式R17OH的醇和二亚胺缩合剂反应,其中R17是一个形成生物不稳定酯的基团及X′是氯,溴,或碘,较好的为氯;最后用常规方法,例如用锌及冰醋酸反应,除去苯甲酰甲基保护基。
然后应用以前描述过的偶合技术将产物与通式(Ⅳ)胺反应,并用常规的催化氢化最后除去苄基R14得到通式(Ⅰ)产物,其中R是一个形成生物不稳定酯的基团及R4是H。
在这方法的另一变法中,通式(Ⅳ)的胺(其中R14是一个形成生物不稳定酯的基团与通式(Ⅲ)化合物偶合,其中R13是一个常规的可选择性除去的保护基,例如苄基。然后偶合产物去保护基;在R13是苄基时,产物被氢化得到通式(Ⅰ)化合物,其中R是H及R4是一个形成生物不稳定酯的基团。
在另一方法中,通式(Ⅰ)的化合物(其中R及R4中的一个是形成生物不稳定酯的基团)是由通式(Ⅴ)的适宜的化合物制备的,其中R13及R14两者都是可选择性除去的保护基团,去保护除去R13或R14中的一个,随即酯化,例如与式R17X′卤化物反应,其中R17及X′定义如前,最后除去另一保护基得到单酯产物。
在这个方法的另一变法中,通式(Ⅴ)化合物(其中R13是叔丁基,R14为苄基)用三氟乙酸去保护得到通式(Ⅰ)化合物(其中R是H及R4是苄基)。完成酯化反应,例如,首先将单一苄酯用碳酸铯中和以转成它的铯盐,随即在惰性有机溶
剂如二甲基甲酰胺中与式R17X卤化物搅拌反应过夜。用常规的催化氢化除去苄基R4,得通式(Ⅰ)化合物,其中R4是一个形成生物不稳定酯的基团及R4是H。
在该方法的另一种变法中,R14从通式(Ⅴ)化合物中除去,经酯化及去保护得到通式(Ⅰ)化合物,其中R是H及R4是一个形成生物不稳定酯的基团。
最后,在另一方法中,化合物(其中R和R4都是形成生物不稳定酯的基团)可以从相应的通式(Ⅰ)的二酸,其中R和R4都是H,用简单的酯化一步就可制备,例如与一个以前描述过的式R17X卤化物反应或与一个醇在偶合剂碳化二亚胺存在下反应。
对上述步骤的所有适宜的偶合反应及保护方法及种种变化及操作方法对技术熟练人员来说经参照标准教科书和后面给出的实例将是熟知的。
如以前所提到的,本发明化合物是中性肽链内切酶(E.C.3.4.24.11.)的强抑制剂。此酶与一些肽激素断裂有关,并且,特别是我们发现它与心房尿钠排泄因子(ANF)断裂有关。这种激素的组成是属于心脏分泌的相关的尿钠排泄肽类一族,在人体内它主要的循环形式已知为28个氨基酸肽,可归于α-hANP(参看,例如G.A.Sagnella及G.A.Mac Greggor,Nature,1984,309,666及S.A.Atlas等,Nature,1984,309 717-725)。这样本发明化合物因阻止肽链内切酶E.C.3.4.24.11降解ANF,能增强其生物效应,化合物因而是利尿及排钠剂,可用在一些如前所述的疾病中。
用于评价对抗中性肽链内切酶E.C.3.4.24.11的活性的方法,是基于J.T.Gafford,R.A.Skidgel,E.G.Erdos及L.B.Hersh,Biochemistry,1983,32,3265-3271中所叙述的测定法。这方法包括测定所需化合物的浓度,该浓度能使应用大鼠肾脏的中性肽链内切酶制剂时从马尿酰-L-苯丙氨酰-L-精氨酸中释放同位素标记马尿酸的释放率降低50%。
化合物作为利尿剂的活性测定,是用测量它们使灌过盐水的清醒小鼠增加尿排出及钠离子排泄的能力。在此实验中,雄性小鼠(charles Rivel CD1,22-28克)在代谢碗中使适应并禁食过夜。测试的化合物溶于相当小鼠体重2.5%的盐水中,通过尾静脉对小鼠静脉给药。每小时将尿样收集在预先称过重量的管中共2小时,分析电解质的浓度。测试动物的尿量与钠离子浓度,与仅给盐水的对照组相比较。
对人给药,在高血压,充血性心力衰竭或肾机能不全的治疗或预防治疗方面,化合物用于成年病人(70千克)的口服剂量一般平均每日在10-1500毫克之间。因此为典型成年病人服用,每一片或每一胶囊中有2至3000毫克活性化合物,含在一种适宜的医药上可用的赋形剂或载体中,可为单次剂量,或分成数次量,一天一次或几次。静脉给药的剂量,典型地是根据需要每一单剂量在5至500毫克之间。在实践中,医生将决定个别病人的最适合的实际剂量,剂量将随年龄,体重及特殊病人的反应而改变。上述剂量为一般情况的范例,当然,对个别情况说来,剂量高一些或低一些可能更好,并且这些也在本发明的范围之内。
对人的应用,通式(Ⅰ)化合物能单独给药,但一般与药用载体成混合物给药,载体的选择与所打算的给药途径及标准医药实践有关。例如,他们可以口服给药,制成片剂形式,含有淀粉或乳糖等赋形剂,或制成胶束或胶丸,可单独或与赋形剂成混合物,或成含有香料或色素的酏剂或悬浮剂。他们可以肠胃外注射,例如,静脉注射,肌肉注射或皮下注射。对于肠胃外给药,他们最好以消毒的水溶液形式,该溶液可以含有其他物质,例如足够的盐或葡萄糖使溶液与血液成等渗。
化合物可以单独给药,也可以合用,如当医生对个别病人要致力于控制血压或治疗充血性心脏衰竭,肾机能不全或其他疾病时,则可按照已确定的医疗惯例与其他药物同时使用。这些化合物能与多种心血管药同时使用,例如在治疗高血压时与ACE抑制剂如甲巯丙脯酸(Captopril)或甘脯肽(enalapril)共用促进血压的控制,或与洋地黄或其他心脏刺激剂或与ACE抑制剂来治疗充血性心力衰竭。其他可能性包括和钙拮抗剂(如硝苯吡啶(nifedipine)或硫氮罩酮(diltiazem),一种β-阻断剂(如氨酰心安atenoeoe)或一种α-阻断剂(如哌唑嗪Prazosin)同时给药,这应由医生决定,以适应特殊病人或有关情况。
除上述外,化合物也可以和外原性ANF,或其衍生物或相关的肽或具有利尿/排钠活性的肽片
断或和其他ANF基因有关的肽联合给药(如D.L.Vesely等在,Biochem.Biophys.Res.Comm.,1987,143,186中所描述的)。
因此在本发明的另一方面是提供了包括通式(Ⅰ)或(Ⅱ)的一个化合物,或其可作药用的盐或其生物前体,以及医药上可用的稀释剂或载体的药物组合物。
本发明也包括通式(Ⅰ)或(Ⅱ)的化合物或其可作药用的盐或生物前体,用作药物,特别是用来治疗人的高血压,充血性心力衰竭或肾机能不全。
最后,基于我们的发现,中性肽链内切酶E.C.3.4.24.11与心房尿钠排泄因子(ANF)的断裂有关,并且本化合物是中性肽链内切酶E.C.3.4.24.11的抑制剂,可用来阻止肽链内切酶E.C.3.4.24.11对ANF的降解,因而增强了它的利尿及尿钠排泄活性;本发明包括用有抑制中性肽链内切酶E.C.3.4.24.11能力的化合物,以制造药物来防止ANF的降解,从而在治疗高血压,心力衰竭,心绞痛,肾机能不全,月经前综合症,周期性水肿,梅尼埃氏病,醛甾酮过多症(初期,二期),高钙尿,或青光眼时,可增强它的利尿及排钠作用。
本发明化合物以及在制备过程中所用中间体的制备,用以下实例来说明,其中实例1-14描述通式(Ⅵ)的某些原料的制备,实例15-69描述通式(Ⅲ)的戊二酸衍生物的制备,实例70-80描述通式(Ⅳ)的某些胺原料的制备,实例81-216描述通式(Ⅴ)的双酯的制备,实例217-415及439-440描述通式(Ⅰ)的单和二羧酸的制备,其中R及R4中之一或二者都是H,实例416-438描述各种前药酯的制备,其中R和R4中之一或二者都是形成生物不稳定酯的基团。
实例1
2-(2-甲氧基乙基)丙烯酸苄酯
在氮气流下,将丙二酸双苄酯(28.43克,0.1摩尔)在一小时内滴加入搅拌着的氢钠(3.15克,80%在油中分散剂;0.105摩尔)在干燥四氢呋喃(100毫升)的悬浮液中,温度可上升至40℃。将2-甲氧基乙基溴(13.9克,0.1摩尔)加入到生成的澄清溶液中,室温下搅拌2小时,然后回流过夜。加水,用二氯甲烷提取混合液。该有机提取液用水洗,干燥(MgSO4),真空蒸发得粗液(30.94克)。在硅胶(700克)柱上用色谱法分离,用乙醚和己烷混合液(2∶8体积比)洗脱,得到2-甲氧基乙基丙二酸双苄酯(15.6克)的无色液体。将其溶解于二噁烷(150毫升)中,在0℃搅拌下,将氢氧化钾(2.55克,45.44毫摩尔)的水溶液(40毫升)加入。混合物在室温搅拌过夜,真空蒸发除去溶剂。加水,用乙醚提取混合液以除去未反应的双酯。然后用2N盐酸(50毫升)酸化此水相,并用乙醚提取。有机提取液用水洗,干燥(MgSO4),真空蒸发,得单酯(8.95克,78%),该单酯为无色油状物。
将多聚甲醛(1.6克,53.34毫摩尔)加入到搅拌着的含粗单酯(8.95克,35.48毫摩尔)和六氢吡啶(502毫克,5.9毫摩尔)的吡啶(70毫升)溶液中。在60℃搅拌2.5小时以后,将该混合液冷却,倒入冰中,用浓盐酸酸化,用乙醚提取。有机提取液相继用水、饱和碳酸氢钠水溶液和水水洗,干燥(MgSO4),在真空中蒸发,得到液体(7.42克),在硅胶柱(300克)上用色谱分离。用乙醚及己烷(2∶8体积比)混合液洗脱,得到需要的丙烯酸苄酯(7.13克,92%),该酯为无色液体,C13H16O3实验值C,70.69;H,7.42%理论值C,70.89,H,7.32%
实例2-4
下列化合物是用实例1的通法制备的,分别用丙基碘,2-乙烯吡啶或叔丁基丙烯溴作为原料以代替2-甲氧基乙基溴(见表1)。
实例5
2-(2-甲基硫乙基)丙烯酸苄酯
在氮气流下将氢钠(0.96克,50%悬浮在矿物油中)加入到搅拌着的含有丙二酸叔丁基苄基酯(5克)的干燥二甲基甲酰胺(50毫升)溶液中,温度保持在0℃。搅拌15分钟后,将含有2-氯乙基-甲基硫(2.21克)的二甲基甲酰胺(10毫升)滴下,反应温度保持在10℃以下。把水小心加入时,该反应可升至室温并搅拌15小时。用乙酸乙酯(2×100毫升)提取该反应混合物,用水(4次)洗乙酸乙酯提取液,干燥(Na2SO4)及蒸发得到4-甲基硫代-2-叔丁氧基羰基-丁酸苄酯,该酯为油状物(6.4克)。在氮气流下,在0℃并剧烈搅拌。将此产物溶于三氟乙酸(50毫升)中,搅
拌45分钟后,在35℃以下减压蒸发三氟乙酸。最后的微量三氟乙酸是同四氯化羰(3×20毫升)共沸除去的,留下油状残留物,该残留物被溶于吡啶中(20毫升)。将六氢吡啶(0.44毫升)及多聚甲醛(1.47克)加入这溶液中,在氮气流下,该混合物在60℃加热2小时。将该反应混合物小心地倒入冰水,用浓硫酸调节pH至1。用乙醚(2×100毫升)提取该混合物,乙醚提取液经干燥(Na2SO4),减压蒸发,得到粗品(5.4克),该产物为油状。在硅胶柱上用色谱分离,用己烷及乙酸乙酯混合液洗脱,得到列于小标题的酯,该酯为无色油状物(1.22克)。C13H16O2O(0.25H2O)实验值:C,64.69;H,6.55,理论值C,64.85;H,6.91%
实例6
2-(2-苯乙基)丙烯酸苄酯
在氮气流下将经苄醇(50毫升)冲洗后的四乙氧基钛(7.72克,338毫摩尔)加入含有2-(2-苯乙基)丙烯酸乙酯(20.23克,99毫摩尔)的苄醇(400毫升)中。在氮气流下,100℃,搅拌所得到的溶液达18小时,冷却到室温,用1N盐酸(140毫升)酸化。该混合物用乙醚及己烷(1∶1体积比)混合液提取。用饱和碳酸氢钠水溶液洗涤有机提取液,使水相中形成一层厚的沉淀物,该水相经分开后,再用乙醚/己烷提取。合并的有机提取液用饱和氯化钠溶液洗,干燥(MgSO4),并在真空下蒸发该溶剂。蒸馏多余的苄醇(63℃,2托),得到粗产物,该产物为棕色油状物。在硅胶(600克)柱上用色谱分离,用含有逐步增加二氯甲烷比例的乙烷(2∶8至4∶6体积比)洗脱,得到需要的酯(19.04克,72%),使用时不需再纯化。
实例7
2-[2(1-氧代异二氢氮(杂)茚基)甲基]丙烯酸叔-丁酯。
在氮气流和室温下,将在二甲基甲酰胺(20毫升)中的异二氢氮(杂)茚酮(2.13克,16毫摩尔)加入到搅拌着的含80%氢钠(0.53克,17.6毫摩尔)的干燥二甲基甲酰胺(20毫升)的悬浮液中。2小时后该桔红色的悬浮液被冷却到0℃,将在干燥二甲基甲酰胺(5毫升)中的2-溴甲基丙烯酸叔丁酯(3.52克,16毫摩尔)溶液慢慢加入。在0.5小时后在0℃,将该反应混合物倒入乙醚中,然后该溶液用水(4次),稀盐酸(2次)及稀碳酸氢钠水溶液(2次)洗涤。干燥(Na2SO4)后,减压蒸发该溶剂,得到黄色油状物(3.0克)。在硅胶柱上用色谱法分离,用二氯甲烷/己烷及二氯甲烷/乙醚混合溶剂洗脱,得到纯的列于小标题的丙烯酯,该酯为无色油状物(2.06克,47%)。C16H19NO3实验值C,70.12;H,7.10;N,5.03。理论值C,70.31;H,7.01;N,5.12%
实例8-9
下列化合物按实例7的方法制备,用适宜的胺原料及用碳酸钾作碱及乙腈作溶剂,分别代替氢钠及二甲基甲酰胺(见表2)。
实例12
2-(苄氧羰基甲基)丙烯酸叔丁酯
在-78℃,将凝聚的异丁烯(50毫升)及浓硫酸(1毫升)加入搅拌着的含有2-(苄氧羰基甲基)丙烯酸(25.0克,114.0毫摩尔)的二氯甲烷溶液中。允许该混合物升至室温并保持72小时。此后,用10%碳酸钠液(3×200毫升)洗溶液,用硫酸镁干燥并蒸发溶剂,得到需要的酯,该酯为浅黄色油状物(28.4克,90%)。C16H20O4实验值:C,69.60;H,7.35。理论值:C,69.55;H,7.30%。
实例13
2-(叔-丁氧羰基氨基)丙烯酸苄酯
在室温,将1,1′羰基双咪唑(8.10克,50毫摩尔)分小部分加入到搅拌着的含叔-丁氧基羰基丝氨酸苄酯(14.75克、50毫摩尔)及三乙胺(5.05克,50毫摩尔)的干燥四氢呋喃(100毫升)溶液中。在室温搅拌16小时后,将该反应混合物倒入乙醚中,相继用稀盐酸,水及碳酸钠水溶液洗有机相,用硫酸钠干燥,在真空下蒸发,得到油(14克)。在硅胶柱上用色谱法分离,用己烷及二氯甲烷混合液洗脱,得到列于小标题的丙烯酯,该酯为黄色油状物(10.98克,79%)。Rf0.5(硅胶;二氯甲烷,己烷1∶1)
实例14
2-(苄氧羰基氨基)丙烯酸叔丁酯
列于小标题的化合物是经由N-苄氧羰基-O-苄基-L-丝氢酸叔丁酯制备的,后者是用与文献
(Recl.Trav.Chim.Pay3-Bas,1964,83,99)所描叙的不同路制备的。在室温,搅拌下将碳酸钠(7.46克,70毫摩尔)加入到含有O-苄基-L-丝氨酸(25.0克,12.8毫摩尔)的水(200毫升)及二噁烷(100毫升)的溶液中。将含有碳酸双苄酯(36.1克,126毫摩尔)的二噁烷(100毫升)溶液滴加下去,该混合物搅拌18小时。二噁烷真空蒸发,用乙醚提取水性残留物。用Na2SO4干燥的提取液在真空下蒸发,得到白色固体,用水洗涤,得到粗的N-苄氧羰基-O-苄基-L-丝氨酸(39.57克,95%)。该物质用在二氯甲烷中的异丁烯(360毫升)及浓硫酸(2毫升)处理。将该反应混合物放在一个压力瓶内,在室温振摇3天。用稀碳酸氢钠洗该混合物,并蒸发二氯甲烷。将该残留物溶在乙醚中,用稀碳酸氢钠洗,干燥(Na2SO4)及在真空下蒸发。用硅胶柱色谱法纯化,用乙醚-二氯甲烷混合液洗脱,得到N-苄氧羰基-O-苄基-L-丝氨酸叔丁酯(36.66克,79%),该酯为油状物。C22H27NO5实验值:C,68.90;H,7.06;N,3.46。理论值C,68.55;H,7.06;N,3.63%。
在氮气流下,室温,将上述化合物(36.06克,94毫摩尔)溶于干燥的叔丁醇(500毫升)中,用叔丁醇钾(12.59克,112毫摩尔)处理。2小时后将该反应混合物倒入2N盐酸(50毫升)及水(350毫升)中用乙醚提取。用盐水洗提取液,干燥(Na2SO4),在真空下蒸发溶剂。残留物用硅胶柱色谱法纯化,用二氯甲烷-己烷混合液洗脱,得列于小标题的化合物。该物为油状物(22.50克,80%)。Rf.0.65(硅胶;50%乙醚-己烷)。由于在室温聚合,不可能做元素分析,产品要储存在0℃以下
实例15
3-(1-羧基环戊基)-2-(2-甲氧基乙基)丙酸苄酯
在氮气流下,将正-丁基锂(2.5摩尔在己烷中,18.16毫升,45.4毫摩尔)滴加入搅拌着的含有二异丙基胺(4.59,45.4毫摩尔)的干燥四氢呋喃(20毫升)溶液中,保持温度-40℃至-20℃。在-20℃继续搅拌半小时,将含有环戊烷羧酸(2.59克,22.7毫摩尔)的干燥四氢呋喃(10毫升)在5分钟内加入,保持温度在-20℃。在1个半小时间允许该混合物达到室温,再搅拌一小时,然后冷却到-73℃。滴加含2-(2-甲氧基乙基)丙烯酸苄酯(5.0克,22.7毫摩尔)的干燥四氢呋喃(10毫升)液,温度保持在-70℃以下,在-77℃两小时后,该混合物很快温热至0℃,用5N盐酸酸化,用己烷提取。用水及饱和碳酸氢钠水溶液(1∶1体积比)的混合液洗该己烷提取液(7次)以除去未反应的环戊烷羧酸。用水洗该提取液,干燥(MgSO4),在真空下蒸发,得到淡黄色油(6.3克),用硅胶(600克)柱色谱法层离。梯度洗脱开始用乙酸乙酯及己烷混合液(3∶7体积比),再改换成净乙酸乙酯,得到需要的产物,该物为无色油(4.0克,53%)。C19H26O5实验值:C,68.39;H,7.99。理论值:C,68.24;H,7.84%。长期放置后,物质固化,用己烷重结晶得到白色固体,熔点41-42℃。
实例16-28
下列化合物是用实例15的方法制备的,用实例2至14的适宜的丙烯酸酯作原料。除实例16及17外,得到的产物都为油。实例24及25以盐酸盐分离。在实例20及28制备时用了2摩尔当量的二锂环戊烷羧酸二阴离子(见表3)
实例29-34
下列化合物是按实例15的方法制备的,但用适宜的环烷或环烯烷羧酸作原料代替环戊烷羧酸,将该阴离子与适宜的2-丙基丙烯酸苄酯或2-(2-甲氧基乙基)丙烯酸苄酯反应(见表4)。
实例35
3-(1-羧基环戊基)丙酸叔-丁酯
在氮气流下-20℃将环戊烷羧酸(22.7克;0.20摩尔)加入至搅拌的二异丙基锂胺(0.43摩尔)在干燥四氢呋喃(300毫升)的溶液中。该溶液可温热至室温并在2小时后冷至-10℃,用套管加到搅拌的3-溴代丙酸叔-丁酯(44.4克,0.21摩尔)在四氢呋喃(100毫升)的溶液中。得到的溶液可温热至室温保持过夜。小心地加入盐酸(3N,250毫升),接着加乙醚(500毫升)并使分层。水层用乙醚(300毫升)洗涤,合并醚层,用水(300毫升)洗,硫酸镁干燥并蒸发得油状物。将该油状物溶入乙醚(300毫升)用饱和碳酸氢钠溶液(3×100毫升)洗涤直至不再剩留环戊烷羧酸。然后用10%碳酸钠溶液(4×150毫升)提
取乙醚液,分出水相,用2N盐酸酸化,用乙醚提取(3×200毫升)。分出乙醚层,水洗(300毫升),硫酸镁干燥并蒸发得很容易结晶的油状物。由戊烷重结晶得无色固体(10.4克,21%),熔点78-81℃(由戊烷)。C13H22O4实验值:C,64.70;H,9.18。理论值:C,64.44;H,9.15%。
实例36
3-(1-羧基环戊基)丙酸乙酯是用3-溴代丙酸乙酯为原料按实例35方法制备。C11H18O4实验值:C,61.91;H,8.53。理论值:C,61.66;H,8.47%。
实例37
3-(1-羧基环戊基)丙酸苄酯是由3-溴代丙酸苄酯按实例35的方法制备,得到的苄酯为油状物。C15H20O4实验值:C,69.76;H,7.18。理论值:C,69.55;H,7.29%。
实例38
3-(1-羧基环戊基)-2-(甲氧基甲基)丙酸叔-丁酯
在氮气流下-78℃将3-(1-羧基环戊基)丙酸叔-丁酯(1.0克,4.13毫摩尔)的干燥四氢呋喃溶液加至搅拌的二异丙基锂胺(9.29毫摩尔)在干燥四氢呋喃(50毫升)的溶液中。0.5小时后,加入氯甲基甲基醚(0.53克,6.58毫摩尔)并在16小时内使该混合物温热至室温。将该溶液倒入水中,用2N盐酸酸化至pH3,用乙酸乙酯提取(3×50毫升)。分出有机层,用硫酸镁干燥并蒸发得无色油,该油用硅胶色谱分离,经乙醚和二氯甲烷(体积比为1∶9-1∶4)混合液洗脱。蒸发适当的部分得标题化合物,该物为无色油状物(0.78克,66%)。C15H26O5实验值:C,62.75;H,8.94。理论值:C,62.91;H,9.15%。
实例39-69
下列化合物系用实例35,36或37的适当的丙酸酯和适当的式R5′X的氯、溴、碘或三氟甲烷磺酰氧基衍生物为起始原料按实例38方法制备。实例50得到的是固体(熔点94-6℃),其它的产物是油(见表5)。
实例70
顺-5-氨基-顺2-乙基-r-1-环己烷羧酸甲酯盐酸盐
5-氨基-2-乙基苯甲酸(7.0克,42.37毫摩尔)溶于热的乙醇(120毫升)和水(200毫升)中,在45℃50磅/平方英寸(3.45巴)压力下,用铂(由PtO2,1.0克)催化氢化两天。在7小时和24小时后再次加入催化剂(1.0克)。通过微晶纤维素过滤并真空蒸发得白色固体。将它溶于水中并吸附在阳离子交换树脂(DOW AG 50W-X8)上。用0.5%吡啶水溶液洗脱,真空蒸发得氨基酸粗品为白色固体(5.81克)。用热丙酮研磨以除去少量起始原料,接着用丙酮和水的混合液重结晶,再用乙腈和水的混合液重结晶,得顺-5-氨基-顺-2-乙基-r-1-环己烷羧酸(3.4克)为一高熔点的白色固体。C9H17NO2实验值:C,63.02;H,10.28;N,8.13。理论值:C,63.13;H,10.00;N,8.18%。
将上述酸(3.35克,19.56毫摩尔)在甲醇(100毫升)中的冰冷的悬浮液搅拌并用氯化氢饱和,得到的溶液室温放置过夜。真空蒸去溶剂,用乙醚研磨残留物,并从乙醚和甲醇的混合液重结晶,得所需的甲酯盐酸盐为白色细结晶(3.57克,82%)。熔点200-201℃。C10H19NO2·HCl实验值:C,53.86;H,9.05;N,6.14。理论值:C,54.17;H,9.09;N,6.32%。
实例71
顺-3-氨基-顺-5-甲基-r-1-环己烷羧酸甲酯盐酸盐
将3-氨基-5-甲基苯甲酸(7.2克,47.6毫摩尔)按实例70中所述的方法氢化与后处理,得到该环乙烷羧酸(3.30克,44%)为一高熔点的白色固体。C8H15NO2实验值:C,60.81;H,9.67;N,8.80%。理论值C,61.12;H,9.62;N,6.91%。将该酸(3.17克,20.16毫摩尔),如先前所描述的用氯化氢甲醇酯化,得到其甲酯盐酸盐为白色固体(3.35克,80%)。熔点172.5-173.5℃。C9H17NO2·HCl实验值:C,52.36;H,8.63;N,6.62。理论值:C,52.04;H,8.73;N,6.74%。
实例72
顺-3-氨基-顺-5-甲基-r-1-环己烷羧酸乙酯盐酸盐
用乙醇和氯化氢酯化顺-3-氨基-顺-5-甲基-r-1-环己烷羧酸(2.0克)得到其乙酯盐酸盐
(1.4克,75%)。C10H20NO2Cl实验值:C,51.23;H,8.52;N,7.13。理论值:C,54.17;H,9.09;N,6.32%。
实例73
顺-3-氨基-顺-4-乙基-r-1-环己烷羧酸甲酯盐酸盐
将3-氨基-4-乙基苯甲酸(14.0克,84.7毫摩尔)如实例70中所述的方法氢化和后处理,得到的该环己烷羧酸(6.36克,43%)为一轻的纤维状白色固体。熔点大于250℃C9H17NO2·O·2H2O实验值:C,61.83;H,9.80;N,8.13。理论值:C,61.82;H,10.03;N,8.01%。上述酸(6.0克,35.04毫摩尔)用实例70中所述方法酯化,得其甲酯盐酸盐,该盐为白色固体(7.02克,90%)。熔点161.5-162℃。C10H19NO2·HCl实验值:C,53.79;H,9.13;N,6.33。理论值:C,54.17;H,9.09;N,6.32%。
实例74
顺-3-氨基-顺-4-乙基-r-1-环己烷羧酸乙酯盐酸盐
用乙醇和氯化氢酯化顺-3-氨基-顺-4-乙基-r-1-环己烷羧酸(2.5克),得到其乙酸盐酸盐(2.0克,75%)。C11H22NO2Cl实验值:C,54.31;H,9.53;N,5.92。理论值:C,56.04;H,9.41;N5.94%。
实例75
顺-3-氨基-顺-4-甲基-r-1-环己烷羧酸甲酯盐酸盐
将3-氨基-4-甲基苯甲酸(3.2克,21.2毫摩尔)如实例42中所述的方法氢化和后处理,但没有用阳离子交换色谱法分离,得到的该环己烷羧酸(1.31克,39%),为白色固体,熔点258-261℃(分解)。C8H15NO2实验值:C,61.03;H,9.89;N,8.90。理论值:C,61.11;H,9.62;N,8.91%。用实例42中所述方法酯化,得其甲酯盐酸盐为一吸潮的泡沫状物(1.31克,87%)。C9H17NO2·HCl实验值:C,51.84;H,8.99;N,6.73。理论值:C,52.03;H,8.73;N,6.74%。
实例76
顺-3-氨基环己烷羧酸乙酯盐酸盐
顺-3-氨基-环己烷羧酸盐酸盐(5.25克,36.7毫摩尔),如先前所述的用乙醇和氯化氢酯化,得其乙酯盐酸盐。该盐为白色固体(6.62克,87%),熔点163-4℃。C9H17NO2HCl实验值:C,51.94;H,8.73;N,6.47。理论值:C,52.03;H,8.73;N,6.74%。
实例77
顺-3-氨基环己烷羧酸苄酯对-甲苯磺酸盐
用迪安-斯达克分水器将顺-3-氨基环己烷羧酸盐酸盐(8.78克,49毫摩尔)与苄醇(26.4克,0.24摩尔)和对-甲苯磺酸单水合物(11.17克,0.59摩尔)在甲苯(150毫升)中回流24小时。冷却并加入乙醚,结晶出的所需苄酯为白色固体(18.68克,94%),熔点148-150℃。C21H27NO5S实验值:C,62.21;H,6.75;N,3.34。理论值:C,62.20;H,6.71;N,3.45%。
实例78
顺-4-氨基环己烷羧酸苄酯对-甲苯磺酸盐
该酯由顺-4-氨基环己烷羧酸盐酸盐按实例77中所述方法制备,得到白色固体,产率为97%,熔点172-4℃。C21H27NO5S实验值:C,62.21;H,6.61;N,3.33。理论值:C,62.20;H,6.71;N,3.45%。
实例79
反-3-氨基-1-丙基-1-环戊烷羧酸甲酯
(a)3-环戊烷羧酸(7.5克,66.9毫摩尔)在干燥四氢呋喃(15毫升)中的溶液在-60℃、搅拌、氮气流下滴加至由二异丙基胺(19.7毫升,0.14摩尔)和2.5M的正丁基锂的己烷液(56.2毫升0.14摩尔)制备的二异丙基锂胺的干燥四氢呋喃(150毫升)液中。产生的悬浮液可温热至室温然后再搅拌一小时,此时得一澄清溶液。在-60℃滴加入碘代丙烯(7.18毫升,73.6毫摩尔),并将该混合物再将温热至室温然后搅拌过夜。将该溶液冷至15℃,加水(10毫升)接着用2N盐酸调至pH3。分出有机层,水相用二氯甲烷提取两次。该合并的有机提取液用饱和食盐溶液洗涤、干燥(MgSO4)并真空蒸发得到粗产物,用硅胶(200克)色谱分离,乙酸乙酯和己烷混合液洗脱,得1-丙基-3-环戊烯-羧酸(7.96克,77%),该羧酸为一吸湿性固体。
(b)在6-10℃将含溴(7.28克,45毫摩尔)
的四氯化碳(20毫升)溶液滴加入搅拌着的上述产物(6.39克,41毫摩尔)的四氯化碳(30毫升)溶液中。半小时后真空蒸发溶剂,残留的二溴衍生物与碳酸钾(6.8克,49毫摩尔)在甲乙酮(500毫升)中回流1小时15分钟。将该混合物过滤并蒸发至小体积,残留物溶入乙醚中。用水洗、干燥(MgSO4)、蒸发得油以硅胶(120克)色谱分离。用乙酸乙酯在己烷中比例逐渐增长的混合液(体积比由35∶75至60∶40)梯度洗脱得6-内-溴-4-丙基-2-氧杂双环[2,2,1]庚-3-酮,该物为浅桔色油(8.79克,91%)。
(c)由(b)步得到的溴代-内酯(4.0克,17.2毫摩尔)于无水乙醇(35毫升)中在50磅/平方英寸(3.45巴)压力下,在氧化镁(6.92克,0.172摩尔)和10%钯-碳(800毫克)上氢化。该还原反应持续四十小时;在7小时和24小时后分别添加催化剂(1.0克)。通过微晶纤维过滤该混合物,真空蒸发溶剂,残留物用硅胶(150克)色谱分离,以乙醚在己烷中比例逐渐增长的混合液洗脱,得到的4-丙基-2-氧杂双环[2,2,1]庚-3-酮为无色油(1.13克;42%)。
(d)由(c)步得到的内酯(1.13克,7.28毫摩尔)在含有浓硫酸(0.15毫升)的甲醇(30毫升)中回流1小时15分钟。蒸发该混合物至小体积,将残留物分配于醚和水之间。有机层用水洗,硫酸镁干燥,减压蒸发,得到的顺-3-羟基-1-丙基-r-1-环戊烷羧酸甲酯为无色油(0.94克,70%)。
(e)将对-甲苯磺酰氯(1.43克,7.5毫摩尔)分批加入由(d)步得到的羟基酯(930毫克,5毫摩尔)在吡啶(10毫升)的冰冷的溶液中。让该混合物逐渐升至室温过夜,18小时后倒入冰水中。该混合物用乙醚提取,有机层相继用1N盐酸,水,饱和碳酸氢钠液和水洗。干燥(MgSO4),并真空蒸发得油状物,用硅胶(80克)色谱分离,乙酸乙酯和己烷的混合液洗脱得所需的3-对-甲苯磺酰基衍生物(1.5克,89%)。此化合物(1.45克,4.3毫摩尔)与叠氮化钠在甲醇(20毫升)和水(10毫升)中回流18小时。真空蒸发除去大部分甲醇,残留物用二氯甲烷提取。有机提取液经水洗、干燥(MgSO4)和真空蒸发,得到油状物,用硅胶(40克)色谱分离。以乙酸乙酯和己烷的混合液梯度洗脱,得到的反-3-叠氮基-1-丙基-r-1-环戊烷羧酸甲酯为无色油(0.79克,89%)。
(f)由(e)步得到的叠氮化物(730毫克,3.4毫摩尔),于甲醇(25毫升)中在50磅/平方英寸氢气压下10%钯-碳(80毫克)上氢化四小时。该混合物通过一短微晶纤维柱过滤并真空蒸发得所需标题胺(619毫克,97%),该胺为浅黄色吸湿性油。C10H19NO2·2H2O实验值:C,54.93;H,9.59;N,5.79%。理论值:C,54.27;H,10.47;N,6.33%。
实例80
反-3-氨基-顺-4-羟基-1-丙基-r-1-环戊烷羧酸甲酯
(a)由实例48(b)得到的6-内-溴-4-丙烯-2-氧杂双环[2,2,1]庚-3-酮(2.0克,8.6毫摩尔)在含有浓硫酸(0.15毫升)的甲醇(30毫升)中回流1小时15分钟。该混合物蒸发至小体积,将残留物分配于醚和水之间。该有机层用水洗、干燥(MgSO4)并减压蒸发,得顺-3-溴-顺-4-羟基-1-丙基-r-1-环戊烷羧酸甲酯,该酯为油(2.18克,98%)。
(b)由(a)步得到的产物(2.14克,8.1毫摩尔)与叠氮化钠(2.10克,32.3毫摩尔)在甲醇(25毫升)和水(15毫升)中回流。48小时后再加入叠氮化钠(1.0克),反应持续四天。真空蒸发除去大部分甲醇,残留物用二氯甲烷提取。提取液用水洗、干燥(MgSO4)并真空蒸发得到油状物,用硅胶(100克)色谱分离。以乙酸乙酯在己烷中逐渐增长比例的混合液梯度洗脱得反-3-叠氮基-顺-4-羟基-1-丙基-r-1-环戊烷羧酸甲酯,该酯为无色油(1.24克,68%)。
(c)由(b)步得到的叠氮化物用在实例79(f)中描述的方法还原,得到的所需氨基酯为油状物(1.06克,98%)。C10H19NO3,0.25H2O实验值:C,58.14;H,9.32;N,6.31。理论值:C,58.37;H,9.55;N,6.81%。
实例81
3-{1-[顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙基)丙酸苄酯
将1-乙基-3-(3-二甲氨基丙基)碳化二亚胺盐酸盐(1.15克,6毫摩尔),加入到3-(1-羧
基环戊基)-2-(2-甲氧基乙基)丙酸苄酯(1.0克,3毫摩尔)、顺-4-氨基-环己烷羧酸苄酯对-甲苯磺酸盐(1.21克,3毫摩尔)、1-羟基苯并三唑(405毫克,3毫摩尔)和N-甲基吗啉(910毫克,9毫摩尔)在干燥二氯甲烷中的冰冷的搅拌着的混合物中。半小时后让该混合物达到室温并搅拌18小时。真空蒸去溶剂并将残留物分配于醚和水间。该有机提取液相继用水、2N盐酸、水、饱和碳酸氢钠水溶液及水洗涤。干燥(MgSO4),真空蒸发得油状物(1.6克),用硅胶(110克)色谱分离,以醚和己烷(体积比1∶1)的混合液洗脱,得到的所需双酯为无色油(1.12克,67%)。C33H43NO6实验值:C,72.07;H,8.00;N,2.52。理论值:C,72.10;H,7.89;N,2.55%。
实例82-161
下列式(Ⅴ)化合物,其中R13和R14各为C1-C6烷基或苄基,是用由实例15-69得到的适当的式(Ⅲ)戊二酸衍生物按实例81的通法与适当的式(Ⅳ)的胺偶合制备。得到的产物为树胶状物或油。环戊烷取代基R2和CO2R4的立体化学是对照1-氨基甲酰取代基定的(见表6)。
实例160
3-{1-[顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-乙基硫代甲基丙酸叔-丁酯
(a)环戊烷羧酸二锂二阴离子(由环戊烷羧酸(2.58克)用实例15中描述的方法制备)在四氢呋喃(100毫升)中,先在室温用氯化锌(1.85克)处理30分钟,再在-78℃与2-溴代甲基丙烯酸叔-丁酯反应。该反应混合物经后处理并用先前所描述的方法纯化该产物得3-(1-羧基环戊基)-2-亚甲基丙酸叔-丁酯,该酯为油状物(5.1克,89%)。C14H22O4实验值:C,66.16;H,8.89。理论值:C,66.11;H,8.72%。
(b)上述酸(4.96克)与顺-4-氨基-环己烷羧酸乙酯(4.06克)按实例81的方法偶合得3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-亚甲基丙酸叔-丁酯,该酯为油状物(6.2克,78%)。C23H37NO5实验值:C,67.94;H,9.35;N,3.54。理论值:C,67.78;H,9.15;N,3.44%。
(c)上述双酯(1.38克,3.4毫摩尔)用纯乙硫醇(0.375毫升,5.1毫摩尔)和N-苄基三甲基-氢氧化铵(7滴)处理。5天后在氮气流下将该混合物溶于乙醚中,用稀盐酸、稀碳酸氢钠和水洗,干燥(Na2SO4),蒸除溶剂得黄色油(1.09克)。用硅胶色谱分离,以乙醚与二氯甲烷混合液洗脱得标题产物,该产物为油状物(0.51克,32%)。C25H43NO5S实验值:C,64.23;H,9.14;N,2.98。理论值:C,63.93;H,9.23;N,2.98%。
实例161
3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-乙基磺酰基甲基丙酸叔-丁酯
将间-氯过苯甲酸(0.36克,2.09毫摩尔)在0℃加至搅拌着的3-{1-[(顺-4-乙氧羰基环己基)-氨基甲酰基]环戊基}-2-乙基硫代甲基丙酸叔-丁酯(0.49克,1.04毫摩尔)的二氯甲烷(25毫升)溶液中。该溶液在室温搅拌16小时,再添加间-氯过苯甲酸(0.36克),该溶液再搅拌24小时。该反应混合物用二氯甲烷(25毫升)稀释,用稀碳酸氢钠、水和盐水洗涤,干燥(Na2SO4)并蒸去溶剂。该残留物用硅胶色谱分离,以乙醚与二氯甲烷的混合液洗脱得标题砜,该砜为白色晶状固体(0.38克,73%)。C25H43NO7S实验值:C,59.49;H,8.73;N,2.64。理论值:C,59.85;H,8.64;N,2.79%。
实例162
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(2-羧基乙基)丙酸苄酯
将3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]-环戊基}-2-[2-(叔-丁氧羰基)乙基]丙酸苄酯(3.72克,6毫摩尔)溶于三氟醋酸(35毫升)中在0℃贮放24小时。该反应混合物倒入冰水中用二氯甲烷提取。该有机相用水洗,干燥(Na2SO4)并真空蒸发。残留物用乙醚和己烷的混合液结晶得白色固体(1.93克),再用乙醚重结晶得标题酸,该酸为白色固体(1.43克,42%),熔点103-104℃。C33H41NO7实验值:C,70.16;H,7.47;N,2.45。理论值:C,70.31;H,7.33;N,2.49%。
实例163
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(2-异丙基氨甲酰基乙基)丙酸苄
酯
3-{1-[(顺-4-芳氧羰基-环己基)氨基甲酰基]-环戊基}-2-(2-羧基乙基)丙酸苄酯(0.41克,0.73毫摩尔)溶于干燥二氯甲烷(20毫升)中,在室温相继用1-(3-二甲氨基丙基)-3-乙基碳化二亚胺盐酸盐(0.28克,1.46毫摩尔)、N-甲基吗啉(0.22克,2.18毫摩尔)和1-羟基-苯并三唑(0.10克,0.74毫摩尔)处理。搅拌10分钟后,加入异丙胺(0.065克,94毫摩尔)。该混合物在室温氮气流下搅拌18小时。用二氯甲烷稀释该反应混合物,该有机相依次用水、稀盐酸和碳酸氢钠水溶液洗涤,干燥(Na2SO4),真空蒸发得油状物(0.46克)。硅胶柱色谱分离以二氯甲烷和乙醚-二氯甲烷混合液洗脱得标题丙酸衍生物,该衍生物为油状物(0.09克,20%)。Rf0.65(硅胶;甲醇,二氯甲烷1∶9)。
实例164-169
用适当的胺按实例163的方法制备下列化合物。分离得到的产物为树胶状物。(见表7)。
实例170
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(2-氨甲酰基乙基)丙酸苄酯
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]-环戊基}-2-{2-[(2,4-二甲氧基苯基)甲基氨基甲酰基]乙基}-丙酸苄酯(实例164)(0.37克,0.52毫摩尔)溶于三氟醋酸(10毫升)中。在室温搅拌18小时后将该深粉红色反应混合物倒入冰水中,用碳酸氢钠碱化,并用二氯甲烷提取。合并的提取液干燥(Na2SO4)并真空蒸发。残留物用硅胶柱色谱法纯化,以二氯甲烷-乙醚和甲醇-二氯甲烷混合液洗脱得标题酯(110毫克,38%)。Rf0.25(硅胶;乙酸乙酯)。
实例171
3-{1-[(顺-4-苄氧羰基-环己基-)-氨基甲酰基]环戊基}-2-(丁酰氨基)丙酸苄酯
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(叔-丁氧羰基氨基)丙酸苄酯(0.71克,1.2毫摩尔)的干燥乙醚(50毫升)溶液,在0℃用氯化氢气体饱和。在室温2小时后,真空蒸发该溶剂,得到的残留物溶于干燥二氯甲烷(30毫升)中。在0℃加入三乙胺(0.16毫升,5.9毫摩尔)接着加入丁酰氯(0.18毫升,1.8毫摩尔)。在室温搅拌16小时后再添加丁酰氯(0.18毫升,1.8毫摩尔)并继续搅拌0.75小时。该反应混合物依次用稀碳酸氢钠水溶液、1N氢氧化钠溶液和水洗,干燥(Na2SO4),真空蒸发溶剂。粗产物用硅胶柱色谱法纯化,以乙醚-二氯甲烷混合液洗脱,接着用制备薄层色谱(用10%甲醇-二氯甲烷展开)纯化得标题酰胺(0.15克,22%)。Rf0.7(硅胶;甲醇,二氯甲烷1∶9)。
实例172
3-{1-[(顺-4-乙氧羰基环己基)氨基甲酰基]环戊基}-2-(2-甲磺酰基乙基)丙酸苄酯
在室温将间-氯过苯甲酸(0.17克,80%)加入2-(2-甲基硫代乙基)-3-{1-[(顺-4-苄氧羰基环己基)氨基甲酰基]环戊基}丙酸苄酯(0.2克)的二氯甲烷(5毫升)溶液中,将该反应混合物搅拌三天。将该混合物减压蒸乾并将残留物分配于乙醚和碳酸氢钠水溶液(5%)间。该有机相经干燥(Na2SO4)、减压蒸发,得油状粗产物(0.2克)。硅胶色谱分离,用己烷与乙酸乙酯(1∶3)混合液洗脱,得到的标题砜,为油状物(0.15克)。C29H41NO6S实验值:C,60.20;H,7.60;N,2.48。理论值:C,60.74;H,7.83;N,2.53%。
实例173
2-(羧基甲基)-3-{1-[(顺-4-乙氧羰基-环己基)-氨基甲酰基]环戊基}丙酸叔-丁酯
2-(苄氧羰基甲基)-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯(3.50克,6.45毫摩尔)溶于四氢呋喃(100毫升)中。在室温50磅/平方英寸(3.5巴)氢气压下与5%钯-碳(200毫克)催化剂一起搅拌。36小时后滤去该催化剂。蒸发溶剂得产物,为无色油状物(2.8克,96%)。C24H39NO7实验值:C,63.28;H,8.70;N,3.20。理论值:C,63.55;H,8.67;N,3.09%。
实例174
3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-(吗啉代羰基甲基)丙酸叔-丁酯
在室温将2-(羧基甲基)-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯(1.0克,2.21毫摩尔)加入到含有吗啉(230毫克,2.65毫摩尔)、1-(3-二甲氨基丙基)-3-乙
基碳化二亚胺盐酸盐(845毫克,4.41毫摩尔)、1-羟基苯并三唑水合物(675毫克,4.41毫摩尔)和三乙胺(893毫克,8.84毫摩尔)的干燥二氯甲烷(50毫升)的溶液中。四天后蒸发该溶剂,将该残留物溶入乙酸乙酯(50毫升)中,用1N盐酸(50毫升)、水(50毫升)和饱和碳酸氢钠溶液(50毫升)洗涤。分出该有机层,用硫酸镁干燥,蒸发后得桔色油。硅胶色谱分离先用乙醚再用乙酸乙酯洗脱得标题产物,该产物为黄色泡沫状物(790毫克,68%)。C28H46N2O7实验值:C,64.63;H,9.09;N,5.23。理论值:C,64.34;H,8.87;N,5.36%。
实例175
3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-[(2-甲氧基乙基)氨甲酰基甲基]丙酸叔-丁酯
用2-甲氧基乙胺替代吗啉按实例174的方法得到标题产物,该产物为浅黄色油(740毫克,66%)。C27H46N2O7实验值:C,63.77;H,9.32;N,5.28。理论值:C,63.50;H,9.08;N,5.49%。
实例176
2-(2-丙酮基)-3-{1-[(顺-4-苄氧羰基-环己基)-氨基甲酰基]环戊基}丙酸叔-丁酯
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(2-丙烯基)丙酸叔-丁酯(400毫克,0.80毫摩尔)溶于水(0.7毫升)和二甲基甲酰胺(5毫升)的混合液中,在60℃与二氯化钯(15毫克)和氯化酮(25毫克)一起搅拌18小时,同时该溶液中通入空气。冷却该溶液,加入乙醚(20毫升)和1N盐酸(20毫升),分层。该水相用乙醚(20毫升)提取,合并有机层,用硫酸镁干燥,蒸发,残留物用硅胶柱色谱分离,以乙醚和戊烷(体积比2∶1)混合液洗脱。合并适当的部分,蒸发得标题产物,该产物为浅黄色油(210毫克,51%)。Rf0.2(硅胶:乙醚,己烷2∶1)。
实例177
2-(2-丙酮基)-3-{1-[(顺-4-乙氧羰基-环己基)-氨基甲酰基]环戊基}丙酸叔-丁酯
3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-(2-丙烯基)丙酸按实例176中所述方法氧化得标题产物。C25H41NO6实验值:C,66.00;H,9.16;N,3.15。理论值:C,66.49;H,9.15;N,3.10%。
实例178
2-(2-N-苄基氨基丙基)-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯
非对映体A和B
将2-(2-丙酮基)-3-{1-[(顺-4-乙氧羰基环己基)氨基甲酰基]环戊基}丙酸叔-丁酯(1.00克,2.2毫摩尔)溶于甲醇(20毫升)中,并相继用苄按(1.5毫升,13.8毫摩尔)、5N盐酸甲醇(0.8毫升)和氰基硼氢化钠(0.14克,2.2毫摩尔)处理并加入3A分子筛(10克)。该黑色溶液在室温搅拌16小时。再加入氰基硼氢化钠(0.23克,3.7毫摩尔),用5N盐酸甲醇和苄胺将该溶液调至pH7.16小时后真空除去溶剂,将该残留物分配于乙酸乙酯和1M碳酸钠水溶液间。该水相用乙酸乙酯(4×50毫升)提取并用氯化钠消散乳状液。该有机相干燥(MgSO4)、蒸发得桔色油(4克),用硅胶柱色谱法纯化,以乙酸乙酯-己烷混合液洗脱得标题的两个非对映体。非对映体A(490毫克,41%)Rf.0.41(硅胶;甲醇,乙酸乙酯,5∶95)。C32H50N2O5实验值:C,70.52;H,9.52;N,5.62。理论值:C,70.81;H,9.29;N,5.16%。非对映体B(650毫克,54%)Rf.0.27(硅胶;甲醇,乙酸乙酯,5∶95)。C32H50N2O5实验值:C,70.75;H,9.22;N,5.49。理论值:C,70.81;H,9.92;N,5.16%。
实例179
2-(1-N-苄基氨基丙基)-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯
标题化合物系由2-丙酰基-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯用实例178的方法制备。得到的是油。C32H50N2O5实验值:C,70.96;H,9.47;N,5.22。理论值:C,70.81;H,9.29;N,5.16%。
实例180
2-乙氨基甲基-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯
2-(N-苄基-N-乙氨基甲基)-3-{1-[(顺
-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯(3.38克,6.2毫摩尔)溶于乙醇(70毫升)中。在室温30磅/平方英寸(2巴)压力下,20%氢氧化钯-碳上,氢化16小时,通过海胆晶(Arbacell)垫板滤去该催化剂,该滤液真空蒸发。该残留物经硅胶色谱分离用甲醇-二氯甲烷混合液洗脱得标题化合物,该物为油状物(2.35克,83%)。C25H44N2O5·O·1CH2Cl2实验值:C,65.51;H,9.57;N,6.14。理论值:C,65.37;H,9.66;N,6.08%。
实例181-185
下列实例是按实例180的通法制备的,实例181-183用适当的N-苄胺为原料,实例184和185用适当的N,N-二苄胺(见表8)
实例186
2-氨基-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯
2-苄氧羰基氨基-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔丁酯(8.11克,15毫摩尔)在10%乙醇水溶液(320毫升)中,在30磅/平方英寸(2巴)用10%钯-碳氢化四小时。滤去该催化剂,该滤液真空蒸发。该残留物与二氯甲烷(3×50毫升)共沸,干燥,得粗的标题化合物,该物为树胶状物(6.86克)。Rf.0.6(硅胶,甲醇,二氯甲烷5∶95)。
实例187
2-氨基-3-{1-[(顺-3-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯
按上述的方法氢化2-苄氧羰基氨基-3-{1-[(顺-3-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯,得标题的胺,该胺为树胶状物。C22H38N2O5实验值:C,64.75;H,9.35;N,6.36。理论值:C,64.36;H,9.33;N,6.82%。
实例188
2-苯磺酰氨基-3-{1-[(顺-4-乙氧羰基-环己基)-氨基甲酰基]环戊基}丙酸叔-丁酯
2-氨基-3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯(0.605克,1.48毫摩尔)溶于干燥二氯甲烷(25毫升)中,在0℃用三乙胺(0.45克,4.4毫摩尔)和苯磺酰氯(0.24毫升,1.84毫摩尔)处理。在氮气流下室温搅拌16小时后,真空除去二氯甲烷,该残留物溶入乙醚(25毫升)中,用稀盐酸、稀碳酸氢钠和水洗,干燥(Na2SO4),真空蒸发该溶剂得粗产物。用硅胶柱色谱法纯化,用乙醚-二氯甲烷混合洗脱,得纯的标题化合物,该物为白色泡沫状物(0.60克,74%)。C28H42N2O7S实验值:C,61.38;H,7.85;N,5.06理论值:C,61.06;H,7.69;N,5.09%。
实例189-213
下列化合物是按实例188的通法制备的,从适当的胺出发,与适当的磺酰氯反应,生成磺酰胺,或分别与酰氯、异氰酸酯、氯代甲酸酯或N-(芳氧基羰基氧代)-琥珀酰亚胺反应生成酰胺、脲、烷氧基羰基氨基或芳烷氧基羰基氨基产物。产物为顺-3-及顺-4-环己烷羧酸酯(见表9)。
实例214
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(4-羟基丁基)丙酸叔-丁酯
2-(4-苄氧丁基)-3-(1-羧基环戊基)丙酸叔-丁酯(1克,2.48毫摩尔)溶于乙醇(35毫升)中,在室温、50磅/平方英寸(3.45巴)氢气压下与5%钯-碳催化剂(250毫克)一起搅拌。3小时后滤去该催化剂并蒸发去该溶剂。该残留物溶于二氯甲烷(40毫升)中,并加入顺-4-氨基-环己烷羧酸苄酯对-甲苯磺酸盐(1.5克,3.70毫摩尔)、1-(3-二甲氨基丙基)-3-乙基碳化二亚胺盐酸盐(1.22克,6.37毫摩尔)、1-羟基苯并三唑水合物(0.97克,6.34毫摩尔)和三乙胺(1.28克,12.65毫摩尔)。室温放置3天后,蒸发该溶剂,将该残留物溶于乙酸乙酯中(50毫升)。该溶液用水(50毫升)、1N盐酸(50毫升)、水(50毫升)和饱和碳酸氢钠溶液(50毫升)洗,硫酸镁干燥,蒸发该溶剂。该残留物用硅胶色谱分离以乙酸乙酯和戊烷(体积比1∶1)的混合液洗脱,合并适当部分,蒸发得标题产物,该产物为浅桔色油(912毫克,61%)。C31H47NO6实验值:C,70.29;H,8.94;N,2.64。理论值:C,70.05;H,9.08;N,2.59%。
实例215
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(4-戊酰氧基丁基)丙酸叔-丁酯
将戊酰氯(0.2毫升,1.98毫摩尔)滴加到搅
拌着的3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(4-羟基丁基丙酸叔-丁酯(0.7克,1.32毫摩尔)在干燥吡啶(5毫升)中的冰冷溶液中。该混合物室温放置过夜,加入冰,用2N盐酸调节pH至3,该混合物以乙醚提取。该有机提取液经干燥(Na2SO4)后蒸发得树胶状物,用硅胶色谱分离,以含有不断增长比例乙醚的己烷液洗脱,得到的标题产物为树胶状物。C38H55NO7实验值:C,70.06;H,8.91;N,2.58。理论值:C,70.44;H,9.03;N,2.28%
实例216
2-(4-叠氮基丁基)-3-{1-[(顺-4-苄氧羰基-环己基)-氨基甲酰基]环戊基}丙酸叔-丁酯
将2-(4-溴代丁基)-3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯(3.1克,5.24毫摩尔)加到叠氮化四甲基鈲盐(1.4克,8.86毫摩尔)和碘化钾(60毫克)在氯仿(60毫升)的溶液中。该溶液加热回流3天,然后用水(2×50毫升)洗,硫酸镁干燥,蒸发溶剂得黄色油状产物(2.9克,100%)。C31H48N4O5实验值:C,66.86;H,8.40;N,9.85。理论值:C,67.12;H,8.36;N,10.10%。
实例217
3-{1-[(顺-4-羧基-环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙基)丙酸
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙基)丙酸苄酯(1.08克)在无水乙醇(30毫升)和水(20毫升)中,在室温、30磅/平方英寸压力下用5%钯-碳为催化剂,氢化2小时。该反应混合物通过微晶纤维短柱过滤,减压蒸去溶剂。该残留物溶入1N氢氧化钠(4毫升)中用醚洗。该水层用2N盐酸酸化,用二氯甲烷提取。该有机提取液用水洗,干燥(MgSO4),减压蒸发得所需的二元酸,该酸为白色泡沫状物(620毫克,83%)。C19H31NO6·O·0.5CH2Cl2·0.5H2O实验值:C,60.07;H,7.64;N,4.07。理论值:C,59.89;H,8.46;N,3.67%。
实例218-234
下列化合物用适当的双苄酯为原料按实例217的方法制备(见表10)
实例235-240
下列化合物用适当的单或双苄酯为原料按实例217的方法制备(见表11)
实例241
3-{1-[(顺-4-乙氧羰基环己基)氨基甲酰基]环戊基}-2-[3-(N-甲基氨甲酰基)苯甲基]丙酸叔-丁酯
3-{1-[(顺-4-乙氧羰基环己基)氨基甲酰基]环戊基}-2-[3-(苄氧羰基)苯甲基]丙酸叔-丁酯(370毫克,0.60毫摩尔)溶于四氢呋喃(30毫升)中,在50磅/平方英寸(3.45巴)氢气压下与5%钯-碳(50毫克)为催化剂一起搅拌5小时。再加入该催化剂(50毫克)并继续氢化48小时。滤去该催化剂,蒸发该溶剂得黄色油。用硅胶色谱分离,以甲醇和乙酸乙酯(体积比1∶4至1∶1)混合液洗脱得无色油。将此油(240毫克)加至含1-乙基-3-(3-二甲氨基丙基)碳化二亚胺盐酸盐(170毫克,0.91毫摩尔)、1-羟基苯并三唑(140毫克,0.91毫摩尔)、三乙胺(182毫克,1.80毫摩尔)和甲胺盐酸盐(31毫克,0.45毫摩尔)的干燥二氯甲烷(20毫升)溶液中,该混合物在室温搅拌六天。减压蒸去该溶剂并将该残留物分配于乙酸乙酯与水间。分出水层用2N盐酸酸化,并以乙酸乙酯提取三次。合并该有机部分并依次用饱和碳酸氢钠水溶液、水和盐水洗。干燥(MgSO4),减压蒸发得黄色油。用硅胶色谱分离,以乙酸乙酯和戊烷(体积比1∶1)混合液洗脱。与二氯甲烷共沸得所需酰胺,该胺为无色油(200毫克,80%)。C31H48N2O6·1/8CH2Cl2实验值:C,67.58;H,8.80;N,5.22。理论值:C,67.57;H,8.42;N,5.06%。
实例242
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(甲氧基甲基)丙酸
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(甲氧基甲基)丙酸叔-丁酯(483毫克,0.96毫摩尔)在0℃溶于干燥三氟醋酸(5毫升)中。在18小时后,蒸发去溶剂,得到的油与二氯甲烷(4×20毫升)共沸。将该油溶入二氯甲烷(50毫升),用水(7×50毫升)洗涤直至洗涤液呈中性。该有机溶液用硫酸镁干燥,蒸去该溶剂得无色油(415毫克,97%)。C25H35NO6·0.75H2O实验值:C,65.07;H,
7.66;N,3.05。理论值:C,65.41;H,8.01;N,3.05%。
实例243-314
下列化合物以R13为叔丁基的式(Ⅴ)适当的酯为原料按实例242的方法制备。该类化合物为顺-3-和顺-4-环己烷-羧酸酯(见表12)
实例315
3-{1-[(顺-4-羧基-环己基)氨基甲酰基]环戊基}-2-(甲氧基甲基)丙酸
3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]-环戊基}-2-(甲氧基甲基)丙酸(390毫克,0.88毫摩尔)溶于四氢呋喃(25毫升)和水(10毫升)中,在50磅/平方英寸(3.45巴)氢气压下与催化剂5%钯-碳(100毫克)一起搅拌3.5小时。滤去该催化剂,蒸发溶剂得黄色树胶状物。将该残留物溶入乙酸乙酯(50毫升),并用饱和碳酸氢钠溶液(3×20毫升)提取。分出水层,用2N盐酸酸化并用乙酸乙酯(2×50毫升)提取。分出有机层,硫酸镁干燥,蒸发溶剂,将该残留物与二氯甲烷共沸得标题产物,该物为无色泡沫状物(264毫克,85%)。C18H29NO6·0.15CH2Cl2实验值:C,59.02;H,7.89;N,3.73。理论值:C,59.21;H,8.02;N,3.80%。
实例316-321
下列实例是用适当的酯为原料按实例315的方法制备的。产物为顺-3-及顺-4-环己烷羧酸(见表13)
实例322
3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-(环己基)丙酸
3-{1-[(顺-4-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-(环己烯-2-基)丙酸(185毫克,0.441毫摩尔)溶于绝对无水乙醇(50毫升)中,在50磅/平方英寸(3.45巴)氢气压下以5%钯-碳(10毫克)催化氢化。溶液过滤,蒸发溶剂得标题化合物为无色树胶状物(180毫克,97%)。C24H39NO5·H2O实验值:C,65.56;H,9.05;N,3.31。理论值:C,65.57;H,9.39;N,3.18%。
实例323
3-{1-[(顺-4-羧基-环己基)氨基甲酰基]环戊基}-2-(2-乙氧基乙基)丙酸
将3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}-2-(2-乙氧基乙基)丙酸(225毫克,0.476毫摩尔)溶于1,4-二噁烷(20毫升)、乙醇(4毫升)和水(6毫升)中并用1N氢氧化钠溶液(2.4毫升,2.4毫摩尔)处理。在室温放置20小时后,该溶液体积蒸发至一半,用水(20毫升)稀释,用乙醚洗涤。分出水层,用2N盐酸酸化、乙酸乙酯(3×20毫升)提取。分出乙酸乙酯层,硫酸镁干燥,蒸发去该溶剂,得标题产物为无色泡沫状物(161毫克,88%)。C20H33NO6·0.2H2O实验值:C,61.89;H,8.29;N,3.62。理论值:C,62.06;H,8.69;N,3.62%。
实例324-391
下列实例用适当的酯为原料按实例323的方法制备(见表14)
实例392
3-{1-[(顺-3-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-(4-羧基丁基)丙酸,钙盐
3-{1-[(顺-3-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-(4-苄氧丁基)丙酸(742毫克,1.48毫摩尔)溶于含有催化剂10%钯-碳(74毫克)的绝对无水乙醇(10毫升)中,该混合物在室温50磅/平方英寸(3.45巴)压力下氢化20小时。再添加该催化剂(70毫克),在同样条件下该混合物再氢化4.5小时。过滤该溶液,真空蒸去溶剂,该残留物与二氯甲烷共沸六次。将该粗产物溶入乙酸乙酯(50毫升)并提取入饱和碳酸氢钠溶液(2×50毫升)中。该水提取液用2N盐酸酸化至pH1,用乙酸乙酯(3×50毫升)提取。该合并的有机提取液经干燥(MgSO4)和蒸除溶剂,得标题酸为一无色泡沫状物(556毫克,92%)。将此产物溶于乙醇(15毫升)中,并向其中加入氢氧化钙(71毫克)的水(3毫升)悬浮液。该混合物在30℃搅拌45分钟,然后过滤、蒸发。该残留物溶于二氯甲烷中,MgSO4干燥,过滤,蒸除溶剂。该残留物用乙醚研磨在70℃真空干燥2天得到白色固体钙盐,熔点116-120℃。C44H72N2O12Ca实验值:C,61.30;H,8.32;N,3.61。理论值:C,61.37;H,8.43;N,3.25%。
实例393
3-{[1-(顺-5-羧基-顺-2-甲基-环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙基)丙酸
3-{1-[(顺-5-甲氧羰基-顺-2-甲基-环己基)-氨基甲酰基]环戊基}-2-(甲氧基乙基)丙酸苄酯(630毫克,1.29毫摩尔)在乙醇(20毫升)和水(15毫升)中,室温50磅/平方英寸(3.45巴)压力下,在5%钯-碳(200毫克)催化剂上氢化三小时,该悬浮液通过一微晶纤维短柱过滤,减压蒸除溶剂。得到的单酯(490毫克)为树胶状物,把它溶于1N氢氧化钠(5毫升)中,该溶液用醚洗,该醚洗涤液用水提取,该合并的水溶液在40℃减压蒸发至5毫升,然后室温放置过夜。
得到的澄清溶液用2N盐酸酸化,二氯甲烷提取。该有机提取液用水洗,干燥(MgSO4),蒸发得所需的二酸为泡沫状物(411毫克,83%)。C20H33NO6·0.25H2O实验值:C,61.95;H,8.58;N,3.69。理论值:C,61.91;H,8.70;N,3.61%。
实例394-403
下列实例系用R13为苄基、R14为甲基或乙基的式(Ⅴ)适当的酯为原料,按实例393的方法制备(见表15)。
实例404
2-(4-氨基丁基)-3-{1-[(顺-4-羧基-环己基)氨基甲酰基]-环戊基}丙酸叔-丁酯
2-(4-叠氮基丁基)-3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}丙酸叔-丁酯(2.73克,5.11毫摩尔)在四氢呋喃(150毫升)和水(75毫升)中在室温及50磅/平方英寸(3.45巴)氢气压下与5%钯-碳催化剂一起搅拌2.5小时。其后滤去该催化剂,蒸去溶剂,该残留用乙醚(3×50毫升)研磨,得标题产物为无色固体(1.45克,65%)熔点189-90℃(分解)。C24H42N2O5·0.33H2O实验值:C,64.80;H,9.69;N,5.92。理论值:C,64.83;H,9.67;N,6.30%。
实例405
2-(4-乙酰氨基丁基)-3-{1-[(顺-4-羧基环己基)氨基甲酰基]-环戊基}丙酸叔-丁酯
2-(4-氨基丁基)-3-{1-[(顺-4-羧基-环己基)-氨基甲酰基]环戊基}丙酸叔-丁酯(280毫克,0.64毫摩尔)溶于二氯甲烷(30毫升)中,用三乙胺(152毫克,1.51毫摩尔)和乙酸酐(78毫克0.76毫摩尔)处理。室温20分钟后蒸发溶剂,将该残留物溶于乙酸乙酯(50毫升)用1N盐酸(50毫升)和水(2×50毫升)洗。分出有机层,硫酸镁干燥,蒸发得无色泡沫状物。硅胶色谱分离,甲醇和二氯甲烷(体积比1∶19-1∶4)洗脱,适当部分蒸发得标题产物为无色泡沫状物(273毫克,89%)。C26H44N2O5·0.6CH2Cl2实验值:C,60.09;H,8.68;N,4.99。理论值:C,60.10;H,8.57;N,5.27%
实例406
2-(4-氨基丁基)-3-{1-[(顺-4-羧基-环己基)氨基甲酰基]-环戊基}丙酸
2-(4-氨基丁基)-3-{1-[(顺-4-羧基-环己基)-氨基甲酰基]-环戊基}丙酸叔-丁酯(1.0克,2.28毫摩尔)在0℃溶于三氟醋酸(8毫升)中。在该温度三天后,蒸发溶剂,残留物与二氯甲烷(3×50毫升)共沸。得到的棕色油,经通过离子交换树脂(Bio-Rad AG 50W-X8,30毫升)纯化,用吡啶和水(体积比3∶100)洗脱,适当部分蒸发,得标题产物为无色泡沫状物(700毫克,80%)。C20H34N2O5·0.1C5H5N·0.3H2O实验值:C,61.97;H,9.08;N,7.69。理论值:C,62.21;H,8.94;N,7.43%。
实例407
2-(4-乙酰氨基丁基)-3-{1-[(顺-4-羧基-环己基)氨基甲酰基]-环戊基}丙酸
2-(4-乙酰氨基丁基)-3-{1-[(顺-4-羧基-环己基)-氨基甲酰基]环戊基}丙酸叔-丁酯(270毫克,0.58毫摩尔)在0℃溶于三氟醋酸中。20小时后蒸去溶剂,得到的油与二氯甲烷(3×20毫升)共沸。将该油溶于乙酸乙酯(20毫升)中,用水(7×20毫升)洗直至洗液呈中性。分出有机层,硫酸镁干燥,蒸发。该残留物溶于饱和碳酸氢钠溶剂(20毫升)中,用乙酸乙酯(2×20毫升)洗,2N盐酸酸化,然后用乙酸乙酯(3×20毫升)提取。分出有机层,硫酸镁干燥,蒸发。该残留物与四氢呋喃共沸,得标题产物为无色泡沫状物(110毫克,45%)。C22H36N2O5·C4H8O实验值:C,62.58;H,8.71;N,5.27。理论值:C,62.88;H,8.93;N,5.64%。
实例408
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(咪唑-2-基甲基)丙酸
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(1-苄基-咪唑-2-基-甲基)丙酸(实例147)(800毫克,1.5毫摩尔)在含水乙醇(80毫升),体积比1∶1)中,30磅/平方英寸(2巴)压力下用5%钯-碳(400毫克)催化氢化七小时。滤去该催化剂,减压蒸去溶剂。产物溶于1N氢氧化钠(6毫升)中,并吸附在阳离子树脂(DOW,AG 50W-X8)上。用吡啶水溶液洗脱,该吡啶水溶液的浓度自0至5%逐渐增长,蒸去洗脱液,得标题二酸为白色泡沫状物(270毫克,34%)。C20H29N3O5·H2O实验值:C,58.76;H,7.50;N,10.18。理论值C,58.66;H,7.63;N,10.26%。
实例409
3-{1-[(顺-4-羧基环己基-氨基甲酰基]环戊基}-2-[2-(2-噻唑基氨基甲酰基)乙基]丙酸
将3-{1-[(顺-4-苄氧羰基-环己基)-氨基甲酰基]-环戊基}-2-[2-(2-噻唑基氨基甲酰基)乙基]丙酸苄酸(120毫克,0.18毫摩尔)和苯甲醚(121毫克,1.1毫摩尔)溶于二氯甲烷(2.5毫升)中。在氮气下0℃加入含氯化铝(149毫克,1.1毫摩尔)的硝基甲烷(2.5毫升)溶液。室温搅拌16小时后,将混合物倒入饱和碳酸氢钠水溶液中,过滤并用稀盐酸酸化该含水滤液。该溶液用固体氯化钠饱和,产物提取到乙酸乙酯中,干燥(Na2SO4),真空蒸去溶剂,得标题化合物(50毫克,58%)。Rf0.4(硅胶:CH2Cl2,CH3OH,CH3CO2H,90∶10∶1)。
实例410
2-氨基-3-{1-[(顺-4-羧基-环己基)氨基甲酰基]-环戊基}-丙酸
将2-苄氧羰基氨基-3-{1-[(顺-4-羧基-环己基)-氨基甲酰基]环戊基}-丙酸(260毫克,0.56毫摩尔)溶于乙醇(20毫升)和水(2毫升)的混合液中,在30磅/平方英寸(2巴)气压下,室温氢化三小时。通过海胆晶(Arbacell)过滤,蒸发溶剂得粗品,将其溶于甲醇中,过滤和蒸发滤液。该残留物用二氯甲烷洗涤并真空干燥,得标题化合物为膏状固体(33毫克,18%)。C16H26N2O5·0.2CH3OH·1.0H2O实验值:C,55.72;H,8.00;N,7.65。理论值:C,55.46;H,8.27;N,7.99%。
实例411
3-{1-[(顺-4-羧基-环己基)氨基甲酰基]-环戊基}-2-[2-(羧基)乙氧甲基]丙酸
3-{1-[(顺-4-羧基-环己基)氨基甲酰基]-环戊基}-2-[2-(甲氧基)乙氧甲基]丙酸(0.80克,2毫摩尔)溶于干燥二氯甲烷(15毫升)中,在0℃氮气流下用碘化三甲基硅烷(1.4毫升,10毫摩尔)处理。在0℃反应六小时后,将该反应物倒入冰冷的稀碳酸氢钠中,并用二氯甲烷洗涤。水相用浓盐酸酸化,用乙酸乙酯(3×50毫升)提取。第一份提取液中含有起始原料,合并第二份和第三份提取液,用稀硫代硫酸钠液洗涤,干燥(Na2SO4)和蒸发得桔色油。该残留物和二氯甲烷共沸,用二氯甲烷洗涤,得标题化合物为玻璃状泡沫物(0.22克,29%)。C19H31NO7·0.1CH2Cl20.25H2O实验值:C,57.24;H,8.07;N,3.42%。理论值:C,57.58;H,8.02;N,3.52%。
实例412
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊-3-烯基}-2-丙基丙酸
六甲基二硅烷(1.78克,12.19毫摩尔)和碘(2.8克,11.05毫摩尔)在氮气中65℃加热搅拌一小时。冷却该混合物,先加入环己烷(30毫升,300毫摩尔),接着加3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊-3-烯基}-2-丙基丙酸苄酯(1.78克,3.35毫摩尔)在四氯化碳(30毫升)的溶液,在65-70℃搅拌该混合物,用薄板色谱跟踪该反应的进程。18小时后加入碘化三甲基硅烷(2.0克,10.05毫摩尔),24小时后再加入一次(4.0克,20毫摩尔)。再过七小时后,冷却该混合物并倒入二氯甲烷和水的混合液中。有机相用饱和盐水洗涤并用0.1N氢氧化钠液提取。该碱性提取液用乙醚洗涤,用浓盐酸酸化至pH1-2并用二氯甲烷提取。该有机提取液用饱和盐水洗涤,干燥(Na2SO4),真空下蒸去溶剂,得标题二酸为黄色泡沫状物(1.05克)。在硅胶上色谱分离,用含有增加比例甲醇(1到5%体积比)的二氯甲烷洗脱,得纯品为浅黄色泡沫状物(650毫克,55%)
C19H29NO5·0.6H2O实验值:C,62.69;H,8.11;N,3.99。理论值:C,62.99;H,8.40;N,3.87%。
实例413-415
下列化合物是按实例412的方法用相应的二苄酯制备的(见表16)。
实例416
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧基乙氧基甲基)丙酸1-(2,2-二乙基丁酰氧基)乙酯
(a)3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]环戊基}2-[(2-甲氧基乙氧基甲基]丙酸(0.5克,1.02毫摩尔)溶于乙腈(20毫升)和水(10毫升)的混合液中。在该溶液中滴加入碳酸铯(1.0克)在水(10毫升)中的溶液,直至该溶液pH约为8。所得铯盐溶液在室温搅拌15分钟并真空蒸干。该残留物先后用甲苯(3次)乙腈(4次)共沸蒸馏以除去水。所得浅黄泡沫状物溶于干燥二甲基甲酰胺(40毫升)中,并向其中加入含1-(2,2-二乙基丁酰氧基)氯乙烷(232毫克,1.12毫摩尔)的二甲基甲酰胺(1毫升)溶液,室温搅拌该混合物过夜。减压蒸除溶剂并与甲苯共沸,以除去痕迹量二甲基甲酰胺。该残留物溶于乙酸乙酯(20毫升)中,并用2M盐酸(3×10毫升)洗涤。该有机相用硫酸镁干燥,减压蒸除溶剂,该残留物在硅胶上色谱分离。用乙醚和二氯甲烷(9∶1)混合液洗脱,得所要双酯为无色树胶状物(355毫克,53%)。C37H57NO9实验值:C,67.53;H,8.87;N,2.23。理论值:C,67.35;H,8.71;N,2.12%。(b)自上述(a)得到的产物(318毫克,0.482毫摩尔)溶于乙醇(27毫升)和水(3毫升)混合液中,在50磅/平方英寸(3.45巴)压力、10%钯碳(30毫克)催化剂上氢化4小时。过滤该反应混合物,真空蒸去溶剂。该残留物与二氯甲烷(4×50毫升)共沸,得所要的化合物为白色泡沫状物(275毫克,100%)。C30H51NO9实验值:C,63.60;H,9.22;N,2.46。理论值:C,63.24;H,9.02;N,2.46%。
实例417-428
下列化合物是按实例416的方法用铯盐和相应的氯化物反应制备的。这些产物都呈泡沫状或树胶状(见表17)
实例429
3-{1-[(顺-4-羧基(环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸1-萘酯半水合物
(a)2-(2-甲氧乙氧基甲基)-3-[(1-苯甲酰甲氧基羰基)-环戊基]丙酸叔丁酯
将无水碳酸钾(18.0克,0.130摩尔)加到搅拌着的含苯甲酰甲基溴(13.0克,0.0653摩尔)和3-(1-羧基环戊基)-2-(2-甲氧乙氧基甲基)丙酸叔丁酯(21.34克,0.0645摩尔)的干燥二甲基甲酰胺(100毫升)溶液中。所得的悬浮物在室温搅拌18小时,然后真空蒸除大部分溶剂,该残留物在乙酸乙酯(150毫升)和水(100毫升)中分配。分出有机相,相继用水(50毫升)、1N盐酸(3×50毫升)和饱和碳酸氢钠水溶液(50毫升)洗涤,然后用无水硫酸镁干燥。真空除去溶剂得油状物,在硅胶上用色谱法纯化,用己烷-乙酸乙酯梯度洗脱。合并适当部分,真空蒸去溶剂,得标题化合物无色油(16.0克,55%),Rf(硅胶)0.71(乙酸乙酯/甲苯,1∶1);0.91(乙醚)。
(b)2-(2-甲氧乙氧基甲基)-3-[(1-苯甲酰甲氧基羰基)环戊基]丙酸
在0℃20分钟内将干燥的三氟醋酸(40毫升)滴加到搅拌着的先前产物(8.0克,0.0178摩尔)在干燥二氯甲烷(50毫升)的溶液中。然后移去冷却浴并继续搅拌3小时,在这时间内该反应混合液的颜色变得很深。真空蒸发得一深色油,与二氯甲烷(3×50毫升)共沸,并溶于饱和碳酸氢钠水溶液(100毫升)中,用乙醚(2×50毫升)洗涤所得溶液,用浓盐酸酸化至pH3,并用乙酸乙酯(2×50毫升)提取。合并有机提取液并用饱和盐水(2×50毫升)洗涤,无水硫酸镁干燥,过滤。真空蒸发该滤液,得标题化合物为一浅黄色油(6.98克,100%),Rf(硅胶)0.89(二氯甲烷/甲醇/氨,80∶20∶1)。
(c)2-(2-甲氧乙氧基甲基)-3-[(1-苯甲酰甲氧基羰基)-环戊基]丙酸1-萘酯
在搅拌着的、冰冷的含先前产物(1.0克,2.55毫摩尔)的干燥二氯甲烷(30毫升)溶液中,先后加入1-羟基苯并三唑(0.38克,2.80毫
摩尔)、1-萘醇(1.83克,12毫摩尔)、N-甲基吗啉(0.33克,3.31毫摩尔)和1-乙基-3-(3-二甲氨基丙基)碳化二亚胺盐酸盐(0.64克,3.31毫摩尔)。10分钟后,该溶液浓缩至油状,在室温搅拌18小时,然后溶于二氯甲烷(200毫升)中。该溶液用水(50毫升)、2N盐酸(2×25毫升)、饱和碳酸氢钠水溶液(2×25毫升)和饱和盐水(2×25毫升)洗涤,然后用无水硫酸镁干燥并过滤。真空蒸去溶剂得油,该油在硅胶上用色谱法纯化,用己烷-乙酸乙酯梯度洗脱。合并适当部分,真空蒸去溶剂,得标题双酯为油状物(0.9克,68%),Rf(硅胶)0.57(乙酸乙酯/己烷1∶1)。
(d)3-[(1-羧基环戊基]-2-(2-甲氧乙氧基甲基)-丙酸1-萘酯
将活化了的锌粉(0.9克,13毫摩尔)在室温加到搅拌着的上述双酯(0.9克,1.74毫摩尔)的冰醋酸(10毫升)溶液中。18小时后,过滤该反应混合物,并先后用冰醋酸(2×10毫升)和二氯甲烷(2×20毫升)洗过滤的垫板。真空蒸发合并的母液和洗液,所得的油先用甲苯(3×20毫升),后用二氯甲烷(3×20毫升)共沸,然后溶于乙醚(50毫升)中。该乙醚液用饱和碳酸氢钠水溶液(3×20毫升)洗涤,无水硫酸镁干燥,并过滤。真空蒸发滤液得油状物,在硅胶上用色谱法纯化,用氯仿至含5%甲醇的氯仿梯度洗脱。蒸发适当部分,得标题化合物为油(0.63克,91%),Rf(硅胶),0.18-0.36(乙酸乙酯/己烷,1∶1)。
(e)3-{1-[(顺-4-苄氧羰基-环己基)氨基甲酰基]-环戊基}-2-(2-甲氧乙氧基甲基)丙酸1-萘酯
往一搅拌着的冰冷的含先前产物(0.77克,1.92毫摩尔)的干燥二氯甲烷(20毫升)溶液中,先后加入1-羟基苯并三唑(0.29克,2.11毫摩尔)、顺-4-氨基-环己烷-羧基苄酯对甲苯碳酸盐(0.78克,1.92毫摩尔)、N-甲基吗啉(0.45克4.42毫摩尔)和1-乙基-3-(3-二甲氨基丙基)-碳化二亚胺盐酸盐(0.48克,2.50毫摩尔)。10分钟后,所得溶液浓缩成油,然后室温搅拌18小时。该产物溶于乙酸乙酯(200毫升)中,用水(2×25毫升)、1N盐酸(2×25毫升)、饱和碳酸氢钠水溶液(2×25毫升)和饱和盐水(2×25毫升)洗涤。然后用无水硫酸镁干燥并过滤。真空蒸发该滤液得油,该油在硅胶上用色谱法纯化,用己烷-乙酸乙酯(3∶2)洗脱,合并适当部分并真空蒸去溶剂,得标题化合物为油(0.94克,79%),Rf(硅胶)0.20(乙酸乙酯/己烷,2∶3),0.71(乙酸乙酯)。
(f)3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸1-萘酯半水合物
搅拌着的上述产物(0.80克,1.3毫摩尔)的5%乙醇水溶液,在10%钯-碳催化剂(80毫克)中、60磅/平方英寸(4.1巴)压力下,室温氢化4.5小时。滤去该催化剂,真空蒸去滤液。所得树胶状物和二氯甲烷(3×20毫升)共沸,得标题酯为粘性白色泡沫(0.64克,92%)。C30H39NO7·0.5H2O实验值:C,67.81;H,7.62;N,2.68。理论值:C,67.39;H,7.54;N,2.62%。
实例430-433
下列化合物是按实例429的方法,在429(c)酯化一步中用相应的芳香醇制备的(见表18)。
实例434
3-{1-[(顺-4-{5-二氢化茚氧基羰基}环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基)丙酸
(a)顺-4-苄氧羰基氨基环己烷羧酸的制备
将碳酸钠(4.03克,38毫摩尔)分小批加到搅拌着的顺-4-氨基环己烷羧酸(10.0克,69毫摩尔)在二噁烷(80毫升)和水(40毫升)混合的溶液中。15分钟后,滴加二苄基二碳酸盐(19.47克,68毫摩尔)的二噁烷(40毫升)溶液,接着加入水(40毫升)。室温搅拌该混合物18小时,真空蒸去溶剂,该残留物在乙醚和水中分配。有机相用稀盐酸和水洗涤,无水硫酸钠干燥并真空蒸发,得顺-4-苄氧羰基氨基环己烷羧酸为油(14.5克,75%)。C15H19NO4实验值:C,64.88;H,6.98;N,5.13。理论值:C,64.97;H,6.91;H,5.05%。
(b)顺-4-苄氧羰基氨基-环己烷羧酸5-二氢化茚酯
往一搅拌着的冰冷的含(a)产物(1.70克,
6.13毫摩尔)的干燥二氯甲烷(30毫升)溶液中,依次加入1-羟基苯并三唑(0.92克,6.77毫摩尔)、5-二氯化茚醇(2.0克,15毫摩尔)、N-甲基吗啉(0.80克,7.92毫摩尔)和1-乙基-3-(3-二甲氨基丙基)碳化二亚胺盐酸盐(1.53克,7.98毫摩尔)。10分钟后将所得溶液浓缩成油,室温搅拌18小时,溶于二氯甲烷(150毫升)中。后者的溶液先后用水(2×50毫升)、饱和碳酸氢钠水溶液(2×25毫升)、2N盐酸(2×25毫升)和饱和盐水(2×25毫升)洗涤,然后用无水硫酸镁干燥,过滤。真空蒸除溶剂得油状物。该油在硅胶上用色谱法纯化,用己烷-乙酸乙酯(4∶1)洗脱。合并适当部分并真空蒸去溶剂,得标题化合物为白色固体(2.20克,91%),可用乙醚和正戊烷混合液结晶,熔点,68-69℃。C24H27NO4实验值:C,73.30;H,7.04;N,3.43。理论值:C,73.26;H,6.92;N,3.56%。
(c)顺-4-氨基环己烷羧酸5-二氢化茚酯盐酸盐1/4水合物
搅拌着的上述产物(1.10克,2.8毫摩尔)的乙醇(50毫升)和浓盐酸(3毫升)溶液在10%钯-碳催化剂(110毫克)上,压力为60磅/平方英寸(4.1巴),室温氢化2小时。过滤除去该催化剂,滤液真空蒸发,得标题化合物为膏状固体(0.77克,91%),用己烷-四氢呋喃结晶,熔点163-164℃。C16H21NO2;HCl;0.25H2O实验值:C,63.86;H,7.52;N,4.70。理论值:C,63.98;H,7.54;N,4.66%。
(d)2-(2-甲氧乙氧基甲基)-3-[(1-苯甲酰甲氧基羰基)环戊基]丙酸苄酯
将无水碳酸钾(10.55克,0.076摩尔)加到搅拌着的含2-(2-甲氧乙氧基甲基)-3-[(1-苯甲酰甲氧基羰基)环戊基]丙酸(实例14(b),15.0克,0.038摩尔)和溴苄(6.53克,0.038摩尔)的干燥二甲基甲酰胺(100毫升)溶液中。所得悬浮液在室温下搅拌18小时,然后真空蒸去大部分溶剂,残留物在乙酸乙酯(150毫升)和水(100毫升)中分配。分出有机相,相继用水(50毫升)、1N盐酸(50毫升)和饱和碳酸氢钠水溶液(50毫升)洗涤,然后无水硫酸镁干燥。过滤、接着真空蒸去溶剂,得标题苄酯为油(16.2克,88%),Rf(硅胶)0.69(乙酸乙酯/甲苯,1∶1),0.88(乙酸乙酯)。
(e)3-(1-羧基环戊基)-2-(2-甲氧乙氧基甲基)丙酸苄酯
在室温将活化的锌末(5.0克,0.076摩尔)加到搅拌着的、上述产物(8.0克,0.0165摩尔)的冰醋酸(50毫升)溶液中。三小时后,过滤该反应混合物并用冰醋酸洗过滤垫板。真空蒸发合并的母液和洗液,油状残留物先与甲苯(3×40毫升)共沸,然后溶于饱和碳酸氢钠水溶液(100毫升)中。该水溶液用正己烷(3×50毫升)洗涤,用2M浓盐酸酸化到pH3-4,并用乙醚(2×100毫升)提取。合并乙醚提取液,用无水硫酸镁干燥并真空蒸发。残留物与二氯甲烷(2×40毫升)共沸,得标题化合物为黄色油(5.15克,84%),Rf(硅胶)0.25-0.50(乙酸乙酯),0.85(二氯甲烷/甲醇/氨,80∶20∶1)。
(f)3-{1-[(顺-4-{5-二氢化茚氧基羰基}环己基)氨基甲酰基]-环戊基-}-2-(2-甲氧乙氧基甲基)丙酸苄酯
往一搅拌着的,冰冷的含上述产物(0.91克,2.52毫摩尔)的干燥二氯甲烷(30毫升)溶液中,依次加入1-羟基苯并三唑(0.38克,2.74毫摩尔)、自(c)得的顺-4-氨基环己烷羧酸5-二氢化茚酯盐酸盐1/4水合物(0.77克,2.52毫摩尔)的干燥二氯甲烷(20毫升)溶液,N-甲基吗啉(0.58克,5.74毫摩尔)和1-乙基-3-(3-二乙基氨基丙基)碳化二亚胺盐酸盐(0.63克,3.25毫摩尔)。十分钟后,浓缩所得溶液成油并在室温搅拌72小时,然后用二氯甲烷(250毫升)稀释。这溶液先后用水(2×50毫升)、2N盐酸(2×50毫升)、饱和碳酸氢钠水溶液(2×50毫升)和饱和盐水(2×50毫升)洗涤,然后用无水硫酸镁干燥并过滤。真空蒸去溶剂,得油,该油在硅胶上用色谱法纯化,用己烷和乙酸乙酯(2∶1)混合液洗脱。合并适当部分,真空蒸去溶剂,得标题的双酯为油(1.40克,91%),Rf(硅胶)0.33(乙酸乙酯/己烷,1∶1)。
(g)3-{1-[(顺-4-{5-二氢化茚氧基羰基}环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸
将一搅拌着的上述产物(1.4克,2.3毫摩尔)在乙醇(50毫升)中的溶液在10%钯碳催化
剂(140毫克)中,60磅/平方英寸(4.1巴)压力下室温氢化18小时。过滤除去该催化剂,真空蒸发滤液得一油,该油与二氯甲烷(3×40毫升)共沸,得标题化合物为油(0.95克,81%),Rf(硅胶)0.90(二氯甲烷/甲醇/氨,80∶20∶1)。C29H41NO7·0.2CH2Cl2,实验值:C,66.25;H,7.70;N,2.72。理论值:C,65.84;H,7.83;N,2.63%。
实例435
3-{1-[(顺-4-乙氧羰基环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸半水合物
(a)将草酰氯(0.26克,2.1毫摩尔)加到搅拌着的、冰冷的3-(1-羧基环戊基)-2-(2-甲氧乙氧基甲基)丙酸苄酯(0.50克,1.37毫摩尔)在干燥二氯甲烷(10毫升)的溶液中,接着加一滴二甲基甲酰胺。所得溶液在室温搅拌3.5小时,真空蒸去溶剂,该残留物和二氯甲烷(3×25毫升)共沸。所得的净酰氯用冰冷的顺-4-氨基环己烷羧酸乙酯盐酸盐(0.28g,1.37毫摩尔)在干燥二氯甲烷(5毫升)的溶液处理,接着在搅拌下再滴加三乙胺(0.42克,4.11毫摩尔)的干燥二氯甲烷(5毫升)溶液。该反应混合物在室温搅拌18小时,然后用二氯甲烷(200毫升)稀释,依次用2N盐酸(2×50毫升)、饱和碳酸氢钠水溶液(2×50毫升)和饱和盐水(2×50毫升)洗涤,并用无水硫酸镁干燥。真空发去溶剂得油,用色谱法在硅胶上纯化,用己烷和乙酸乙酯(3∶2)混合液洗脱。合并适当部分,真空蒸去溶剂,得3-{1-[(顺-4-乙氧羰基环己基)氨基甲酰基]-环戊基}-2-(2-甲氧乙氧基甲基)丙酸苄酯为油(0.51克,72%),Rf(硅胶)0.4(乙酸乙酯/甲苯,1∶1)。
(b)搅拌着的得自(a)的产物(0.50克,0.965毫摩尔)的5%乙醇水溶液(40毫升)在10%钯-碳催化剂(50毫克)上,在60磅/平方英寸(4.1巴)压力时,室温氢化18小时。过滤除去该催化剂,滤液真空蒸发,得树胶状物,该物与二氯甲烷(3×40毫升)共沸并在高真空下干燥,得所要的乙酯为树胶状物(0.41克。100%)C22H37NO7·0.5H2O,实验值:C,60.48;H,8.79;N,3.30。
理论值:C,60.52;H,8.77;N,3.21%。
实例436
3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸苄酯
(a)顺-4-氨基-环己烷羧酸叔丁酯盐酸盐
顺-4-苄氧羰基氨基-环己烷羧酸(实例191a)(14.5克,52毫摩尔)溶于二氯甲烷(100毫升)中并冷至-78℃。往其中加入液态异丁烯(100毫升)和浓硫酸(1.0毫升),该反应混合物密封于压力瓶中,使温热至室温并振摇过夜。排出过量异丁烯,加入稀碳酸氢钠溶液,真空下除去溶剂。该残留物在乙醚和稀碳酸氢钠水溶液间分配,有机相用无水碳酸钠干燥并蒸发,得油状物(35克)。用柱色谱法在硅胶上纯化,用乙醚和二氯甲烷混合液洗脱,随后从正戊烷在0℃结晶,得顺-4-苄氧羰基氨基-环己烷羧酸叔丁酯为白色针晶(3.77克,22%)熔点73-74℃。该产物溶于乙醇(200毫升)中并在30磅/平方英寸(2.0巴)压力,用10%钯-碳催化剂室温氢化4小时。过滤该反应混合物,真空蒸去溶剂。该残留物溶于二氯甲烷中,过滤和蒸除溶剂得油,将该油溶于干燥乙醚中并用盐酸乙醚处理到pH3。收集所得沉淀并干燥,得标题酯盐酸盐为白色固体(2.34克,90%)熔点180℃(分解)。C11H21NO2·HCl实验值:C,55.95;H,9.35;N,5.68。理论值:C,56.04;H,9.41;N,5.94%。
(b)3-{1-顺-4-叔丁氧羰基环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸苄酯
得自(a)的胺盐酸盐(1.37克,576毫摩尔)和得自实例434(e)的3-(1-羧基环戊基)-2-(2-甲氧乙氧基甲基)丙酸苄酯(1.98克,5.4毫摩尔)按实例434(f)的方法偶合,得标题的双酯为油状物(2.23克,75%)C31H47NO7实验值:C,68.39;H,8.75;N,2.37。理论值:C,68.22;H,8.68;N,2.57%。
(c)3-{[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸苄酯
得自上述(b)的产物(2.23克,4.09毫摩尔)溶于干燥的三氟醋酸(20毫升)中,该反应混合物在0-4℃放置过夜。真空除去三氟醋酸,该残留物溶于碳酸氢钠水溶液(50毫升)中,用乙醚(3×100毫升)提取。水相用稀盐酸酸化并用乙酸乙酯提取。该乙酸乙酯提取液用无水硫酸钠干
燥并真空蒸发。该残留物与二氯甲烷共沸并真空干燥得油,放置成结晶,得标题苄酯为白色固体(1.4克,70%),熔点83-85℃,C27H39NO7实验值:C,65.97;H,8.17;N,2.87。
理论值:C,66.24;H,8.03;N,2.86%。
实例437
3-{1-[(顺-4-((2,2-二甲基丙酰氧基甲氧基羰基)环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸
(a)将碳酸钯(133毫克,0.41毫摩尔)的水(5毫升)溶液加到3-{1-[(顺-4-羧基环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氧基甲基)丙酸苄酯(400毫克,0.82毫摩尔)的乙腈(15毫升)溶液中,并减压蒸去溶剂。该残留物与乙腈(2次)共沸得绝盐为泡沫状物。将其悬浮于二甲基甲酰胺(2毫升)中,加入新戊酸氯甲酯(148毫克,0.98毫摩尔),该混合物室温搅拌过夜。加入乙醚(30毫升),该溶液用水洗涤,无水硫酸镁干燥并蒸除溶剂。该残留物在硅胶上色谱分离,用乙酸乙酯和己烷(1∶5)混合液洗脱,得3-{1-〔顺-4-(2,2-二甲基丙酰氧基甲氧基羰基)环己基)氨基甲酰基〕环戊基}-2-(2-甲氧乙氧基甲基)丙酸苄酯为无色树胶状物(510毫无,100%)。C33H49NO9实验值:C,65.45;H,8.17;N,2.49。理论值:C,65.65;H,8.18;N,2.3%。
(b)自上述(a)得到的双酯(450毫克,0.75毫摩尔)在甲醇(18毫升)和水(12毫升)中的溶液,在50磅/平方英寸(3.45巴)用5%钯-碳催化剂(50毫克)室温氢化5小时。过滤除去该催化剂,减压蒸发该滤液,得所要的新戊酰氧基甲酯为无色树胶状物(265毫克,69%)。C26H43NO9实验值:C,60.56;H,8.50;N,2.8。理论值:C,60.80;H8.44;N,2.73%。
实例438
3-{1-[(顺-4-{5-二氢化茚氧基羰基}环己基)氨基甲酰基]-环戊基}-2-(2-甲氧乙氨基甲基)丙酸5-二氢化茚酯
往一搅拌着的、冰冷的含3-{1-[(顺-4-羰基环己基)氨基甲酰基]环戊基}-2-(2-甲氧乙氨基甲基)-丙酸(0.50毫克,1.25毫摩尔)的干燥二氯甲烷(30毫升)溶液中,依次加入1-羟基苯并三唑(0.37克,2.76毫摩尔)、5-二氢化茚醇(0.67克,5.0毫摩尔)、N-甲基吗啉(0.33克,3.3毫摩尔)和1-乙基-3-(3-二甲氨基丙基)-碳化二亚胺盐酸盐(0.62克,3.2毫摩尔)。10分钟后,浓缩所得溶液得油,将该油在室温搅拌18小时。该反应混合物用二氯甲烷(120毫升)稀释,这溶液先后用水(2×20毫升)饱和碳酸氢钠水溶液(2×20毫升)和饱和盐水(2×20毫升)洗涤,无水硫酸镁干燥,过滤。该滤液在真空蒸发得油,在硅胶上用色谱法纯化,用己烷-乙酸乙酯梯度洗脱。合并适当的部分并真空蒸发,得标题双-5-二氢化茚酯为油(0.52克,65%),Rf(硅胶)0.50(乙酸乙酯)。C38H49NO7·0.1CH2Cl2,实验值:C,71.35;H,8.01;N,2.06。理论值:C,71.50;H,7.74;N,2.19%。
实例439
3-{1-[(顺-3-羰基环己基)氨基甲酰基]环戊基}-2-(3-氯丙基)丙酸
3-(1-羧基环戊基)-2-(3-氯丙基)丙酸叔丁酯是按实例38方法,用实例35的丙酸酯和1-氯-3-碘丙烷作原料制备的。分出的产物为油(71%),Rf0.38(硅胶;氯仿,己烷,2-丙醇,2-丙胺,200∶100∶20∶1)。
上述戊二酸衍生物和顺-3-氨基-环己烷羧酸乙酯按实例81方法偶合,得3-{1-[顺-3-乙氧羰基环己基)氨基甲酰基]环戊基}-2-(3-氯丙基)丙酸叔丁酯为油(77%)。C25H42ClNO3实验值:C,63.31;H,9.01;N,2.94。理论值:C,63.60;H,8.97;N,2.97%。
上述的双酯按实例242的方法用三氟醋酸处理得3-{1-[顺-3-乙氧羰基-环己基)氨基甲酰基]-环戊基}-2-(3-氯丙基)丙酸为油(76%)。C21H34ClNO5实验值:C,60.13;H,8.30;N,3.10。理论值:C,60.63;H,8.24;N,3.37%。
上述的单酯按实例323的方法水解,得标题二酸为树胶状物(80%)。C15H30ClNO5。实验值:C,58.77;H,7.79;N,3.33。理论值:C,58.83;H,7.80;N,3.61%。
实例440
3-{1-[(顺-3-羧基环己基)氨基甲酰基]环戊基}-2-[2-(苯磺酰基)乙基]丙酸
3-(1-羧基环戊基)-2-[2-(苯磺酰基)乙基]-丙酸叔丁酯是按实例38方法用实例35的丙酸酯和苯基乙烯基砜做为原料制备的。分离出的产物为油(15%)。C21H30O6S·0.13CH2Cl2。实验值:C,60.25;H,7.24。理论值:C,60.24;H,7.24%。
上述戊二酸衍生物和顺-3-氨基-环己烷羧酸乙酯按实例81的通法偶合,得3-{1-[顺-3-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-[2-(苯磺酰基)乙基]丙酸叔丁酯为泡沫状物(71%)。C30H45NO7S实验值:C,63.53;H,8.02;N,2.38。理论值:C,63.91;H,8.05;N,2.48%。上述双酯用三氟醋酸按实例242方法处理得3-{1-[(顺-3-乙氧羰基-环己基)氨基甲酰基]环戊基}-2-[2-(苯磺酰基)乙基]丙酸为泡沫状物(96%)。
C26H37NO7S实验值:C,61.90;H,7.54;N,2.86。理论值:C,61.51;H,7.35;N,2.76%。上述的单酯按实例323的通法水解,得标题二酸为泡沫状物。C24H33NO7S·0.5H2O·0.1CH3CO2C2H5实验值:C,59.00;H,6.83;N,2.51。理论值:C,58.92;H,7.04;N,2.81%。

表2(续)
分析%
实例 R5(括号内为理论值)或薄层色谱
C H N
10
 74.41 9.39 5.38
74.41 9.39 5.38
(74.14 9.15 5.09)
11 (C6H5CH2)2N-CH278.09 8.20 4.18
(78.30 8.06 4.15)
表3

实例 分析%
编号 R R5(括号内为理论值)
C H N
16 C6H5CH2- CH3(CH2)2- 71.93 8.09 -
(71.67 8.23) -
17 C6H5CH2- C6H5(CH2)2- 75.77 7.49 -
(75.76 7.42) -
18 C6H5CH2-
 Rf0.2
Rf0.2
(硅胶;CH3OH,CH2Cl2
CH3CO2H,10∶90∶1)
19 C6H5CH2- (CH3)3CO2C(CH2)2- Rf 0.7
(硅胶;Et2O)
20 C6H5CH2- (CH3)3CO2CNH- 64.40 7.67 3.55
(64.43 7.47 3.58)
表3(续)
实例 R R5分析%
编号 (括号内为理论值)
C H N
21 C6H5CH2- CH3S(CH2)2- 64.88 7.29
(65.12 7.48) -
22 (CH3)3C- C6H5CH2O2CCH2- 67.52 7.88
(67.67 7.74) -
23 (CH3)3C-
Rf 0.3
(硅胶;CH3OH,CH2Cl2,
1∶9)
CH3Rf 0.5
24 (CH3)3C- C6H5CH2NCH2- (硅胶;CH3OH,CH2Cl2,
1∶9)
25 (CH3)3C-
 Rf 0.5
Rf 0.5
(硅胶;CH3OH,CH2Cl2,
1∶9)
26 (CH3)3C-
 69.76 9.06 3.55
69.76 9.06 3.55
(69.70 8.91 3.52)(1)
27 (CH3)3C- (C6H5CH2)2NCH2- 74.38 8.41 2.91
(74.47 8.26 3.10)
28 (CH3)3C- C6H5CH2OCONH- 64.33 7.71 3.29
(64.43 7.47 3.58)
(1)用0.1CH2Cl2作溶剂。
表4

实例 R
 分析%
分析%
编号 (括号内为理论值)
C H
29 CH3(CH2)2-
72.27 7.76
(72.12 7.65)
30 CH3O(CH2)2-
 68.22 7.31
68.22 7.31
(68.65 7.28)
表4(续)
实例 R
分析%
(括号内为理论值)
编号 C H
31 CH3O(CH2)2-
69.10 8.44
(68.94 8.10)
32 CH3O(CH2)2-
 69.03 7.85
69.03 7.85
(69.34 7.57)
33 CH3O(CH2)2-
68.95 8.38
(68.94 8.10)
34 CH3O(CH2)2-
 71.48 6.75
71.48 6.75
(72.23 6.85)
表5

实例 R R5分析%
编号 (括号内为理论值)
C H N
39 (CH3)3C-
 62.38 7.40 3.47
62.38 7.40 3.47
(61.94 7.72 3.42)(1)
40 (CH3)3C- CH3O(CH2)3- Rf 0.45
(硅胶,Et2O,已烷,
2∶1)
41 (CH3)3C- C2H5O(CH2)2- 64.94 9.55 -
(64.94 9.62) -
42 (CH3)3C- CH3O(CH2)2OCH2- 61.86 9.15 -
(61.79 9.15) -
表5(续)
实例 R R5分析%
编号 (括号内为理论值)
C H N
43 (CH3)3C-
64.35 8.06 8.14
(64.65 7.84 8.38)
44 (CH3)3C- CH2=CH-CH2- 67.72 9.47 -
(68.05 9.28)
45 (CH3)3C-
 69.28 7.58 6.55
69.28 7.58 6.55
(69.87 7.82 6.79)
46 (CH3)3C-
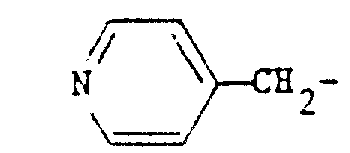 67.52 8.01 4.24
67.52 8.01 4.24
(67.53 8.20 4.15)(2)
47 (CH3)3C- CH3-C≡C-CH2- 68.70 8.78 -
(69.36 8.90) -
48 (CH3)3C- C6H5CH2O-(CH2)4- Rf 0.85
(硅胶;Et2O,CH2Cl2
1∶4)
49 (CH3)3C- Br-(CH2)4- 55.35 7.87 -
(55.07 8.27) -(3)
50 (CH3)3C-
 69.90 8.41 -
69.90 8.41 -
(69.59 8.34) -
51 (CH3)3C-
70.55 9.44 -
(70.77 9.38) -
52 (CH3)3C-
 Rf 0.4
Rf 0.4
(硅胶;Et2O,CH2Cl2
1∶2)
表5(续)
实例 R R5分析%
编号 (括号内为理论值)
C H N
53 C2H5- CH3OCH2CH2OCH2- Rf 0.15
(硅胶;Et2O,已烷
1∶1)
54 C2H5CH3(CH2)2- 65.40 9.51 -
(65.60 9.44) -
55 (CH3)3C- CH3CH2CO- 64.22 8.94 -
(64.41 8.78) -
56 C2H5- CH2=CHCH2- 65.91 8.60 -
(66.11 8.72) -
57 (CH3)3C- CH3CH=CHCH2- 67.70 9.44(4)
(67.85 9.54)(2)
58 (CH3)3C-
 Rf 0.91(硅胶;
Rf 0.91(硅胶;
乙酸乙酯,甲苯
1∶1)
59 (CH3)3C-
69.32 9.47
(69.64 9.74
60 (CH3)3C-
 65.42 8.49
65.42 8.49
(65.36 9.03)
61 (CH3)3C-
66.03 9.11
(65.36 9.03)
62 (CH3)3C-
 62.01 8.77
62.01 8.77
(62.05 8.87)(5)
63 (CH3)3C-
 62.83 9.42
62.83 9.42
(63.28 9.38)(6)
表5(续)
实例 R R5分析%
编号 (括号内为理论值)
C H N
64 (CH3)3C- C6H5CH2- 7.19 8.68
(7.26 8.41)
65 (CH3)3C-
 73.34 7.97
73.34 7.97
(73.57 7.83)(7)
66 (CH3)3C-
 Rf 0.61
Rf 0.61
(硅胶;Et2O,CH2Cl2,
1∶4)
67 C6H5CH2- CH3C≡C-CH2- 70.75 3.03
(70.85 3.30)(8)
68 CH3CH2- CH3C≡C-CH2- 66.69 8.79
(67.64 8.32)
69 CH3CH2-
69.28 9.05
(69.36 8.90)
(1)与CH3CO2H,0.2CH2Cl2溶剂化
(2)0.25H2O
(3)与0.5C4H10O溶剂化
(4)用2-三氟甲基磺酰氧基甲基-2,3,4,5-四氢呋喃
(5)0.33H2O
(6)与0.125CH2Cl2溶剂化
(7)0.125H2O
(8)半水合物
表6









表7(续)
实例 R16分析%
编号 (括号内为理论值)
C H N
164
 Rf 0.6
Rf 0.6
(硅胶;Et2O)
165 (CH3)2N- Rf 0.6
(硅胶;Et2O)
166 CH3NH- 70.65 7.66 4.83
(70.81 7.69 4.86)
167
72.17 8.10 4.61
(72.35 7.99 4.44)
168
 70.25 7.89 4.19
70.25 7.89 4.19
(70.88 7.93 4.24)
169
 66.67 6.86 6.31
66.67 6.86 6.31
(66.95 6.71 6.51)


































Claims (14)
1、一种制备具有通式(Ⅰ)化合物或其可作药用的盐的方法,

其中A完成一个5或6员碳环,它们可是饱和或单-不饱和的和它们
可任意地与另外一个饱和或不饱和的5或6员碳环稠合;
B是(CH2)m其中m是从1或2的整数;
R和R4为别为H,C1-C6烷基,苄基或是另一个形成生物不稳定酯的基团;
R1是氢或C1-C4烷基;
R2和R3分别是H,OH,C1-C4烷基或C1-C4烷氧基;
和R5是C1-C6烷基,C2-C6链烯基,C2-C6炔基,芳基(C2-C6炔基),C3-C7环烷基,C3-C7环烯基,C1-C6烷氧基,-NR6R7,-NR8COR9,-NR8SO2R9或是一个饱和杂环基团;
或C1-C6烷基被一个或多个取代基取代,取代基选自卤素,羟基,C1-C6烷氧基,C2-C6羟基烷氧基,C1-C6烷氧基(C1-C6烷氧基)C3-C7环烷基,C3-C7环烯基,芳基,芳氧基,芳氧基(C1-C4烷氧基),杂环基,杂环氧基,-NR6R7,-NR8COR0,-NR8SO2R0,-CONR6R9,-SH,-S(O)PR10,-COR11或-CO2R12;
其中R6和R7分别是H,C1-C4烷基,C3-C7环烷基(可任意被羟基或C1-C4烷氧基取代),芳基,芳基(C1-C4烷基),C2-C6烷氧烷基,或杂环基;或R6和R7两个基团与氮连在一起形成吡咯烷基,哌啶子基,吗啉代,哌嗪基或N-(C1-C4烷基),-哌嗪基;R8是H或C1-C4烷基;
R8是H或C1-C4烷基;
R9是C1-C4烷基,CF3,芳基,芳基(C1-C4烷基),芳基(C1-C4烷氧基),杂环基,C1-C4烷氧基或NR6R9其中R6和R7定义如前;
R10是C1-C4烷基,芳基,杂环基或NR6R9其中R6和R7定义如前;
R11是C1-C4烷基,C3-C7环烷基,芳基或杂环基;
R12是H或C1-C4烷基;
和P是0,1或2;
它包括的步骤有:
a)将式(Ⅲ)与(Ⅳ)化合物反应生成式(Ⅴ)化合物:

其中A、B、R1、R2和R3定义如前,R5′定义如R5在其中可有被任意保护的活性基团和R13和R14定义如R和R4,H除外,或它们是常规的羧酸保护基团;
b)当R13和R14中的一个或二者分别为C1-C6烷基或苄基时,除去所述基团中的一个或二者;或当R13和R14的一个或二者是常规的羧酸保护基团时,除去所述的羧酸保护基团;当R5′含有一个保护基团时,除去所述保护基团;得到式(Ⅰ)化合物其中R和R4二者都是H或R和R4中的一个是H而另一个是C1-C6烷基,苄基或一个形成生物不稳定酯的基团,和
c)可任意用一个形成生物不稳定酯的基团酯化产物(其中R13和R14另一个是C1-C6烷基,苄基或常规的羧酸保护基团而另一个是H),接着除去C1-C6烷基,苄基或羧酸保护基团而得到式(Ⅰ)化合物,其中R和R4中一个是形成生物不稳定酯的基团而中一个是H;或用一个形成生物不稳定酯的基团酯化式(Ⅰ)的产物(其中R和R4两者都是H),得到式(Ⅰ)产物,其中R和R4两者都是形成生物不稳定酯的基团,和
d)可任意地形成产物的可作药用的盐。
2、一种如权利要求1中的方法,其中式(Ⅲ)和(Ⅳ)化合物是在一种有机溶剂中,在一种二亚胺缩合剂存在下反应的,所述的试剂是1-乙基-3-(二甲基氨基丙基)碳化二亚胺或N,N′-二环己基碳化二亚胺。
3、一种如权利要求1中的方法,其中R13和R14两者都是苄基或其中一个是苄基和另一个是C1-C6烷基,或是另外的形成生物不稳定酯的基团,此时式(Ⅴ)的产物进行催化氢化得到式(Ⅰ)的化合物(其中R和R4两者都是H或其中一个是H而另一个是C1-C6烷基或另外的形成生物不稳定酯的基团),以及,在产物中,当R和R4中的一个是C1-C6烷基时,可任意水解得式(Ⅰ)化合物(其中R和R4都是H)。
4、一种如权利要求1中的方法,其中R13和R14中的一个是叔丁基和另一个是甲基,乙基或苄基,此时式(Ⅴ)产物可用三氟醋酸处理得式(Ⅰ)化合物,其中R和R4中一个是H而另一个是甲基、乙基或苄基,此时该产物可任意水解或氢化而得式(Ⅰ)化合物其中R和R4中一个是H和另一是甲基,乙基或苄基,并且产物也可水解或氢解得到式(Ⅰ)化合物(其中R和R4都是H)。
5、一种如权利要求1中的方法,其中A是(CH2)n,n是4或5的整数,和其中R和R4分别是H,C1-C6烷基或苄基。
6、一种如权利要求1中的方法,其中A是(CH2)4,R1是H和B是(CH2)2,则式(Ⅰ)化合物如下式:

其中R,R2,R3,R4和R5如权利要求1中的规定。
7、一种如权利要求1的方法,其中所述另外的形成生物不稳定酯的基团是
1-(2,2-二乙基丁酰氧基)乙基
2-乙基丙酰氧基甲基
1-(2-乙基丙酰氧基)乙基
1-(2,4-二甲基苯甲酰氧基)乙基
2-苯甲酰氧基苄基
1-(苯甲酰氧基)乙基
2-甲基-1-丙酰氧基-1-丙基
2,4,6-三甲基苯甲酰氧基甲基
1-(2,4,6-三甲基苯甲酰氧基)乙基
新戊酰氧基甲基
苯乙基
苯丙基
2,2,2-三氟乙基
1-或2-萘基
2,4-二甲基苯基
4-叔丁基苯基
5-二氢茚基。
8、一种如权利要求6中的方法,其中R,R2,R3和R4各自是H,其中羧基CO2R4是连接在环己烷环的3-或4-位上并与酰胺基成顺式-立体化学关系。
9、一种如权利要求6中的方法,其R,R2和R3各自是H,R4是C2G5,和其中乙氧羰基是连接在环己烷环3位上并与酰胺基成顺式-立体化学关系。
10、一种如前面任一项权利要求中的方法,其中R5是C2-C4烷基,C2-C4链烯基,C2-C5炔基,C5-C6环烷基,C5-C6环烯基,C1-C4烷基磺酰氨基,或四氢呋喃基或其中R5是被C1-C3烷氧基,C1-C6烷氧基(C2-C4烷氧基),C3-C6环烷基,4-吡啶基,2-咪唑基,C2-C4烷酰基,C2-C4烷氧羰基氨基,C1-C4烷磺酰基,C1-C4烷磺酰氨基,芳基磺酰氨基,杂芳基磺酰氨基或苯甲酰氨基取代的C1-C3烷基。
11、一种如权利要求6中的方法,其R和R4都是H,R2和R3都是H,其团CO2R4是连接在环己烷环4位上并与酰胺基成顺式-立体化学关系,以及其中R5是正丙基,甲氧基乙基,2-甲氧基乙氧基甲基,2-丁炔基,2-环己烯基,四氢呋喃基,4-吡啶基甲基,2-咪唑基甲基,丙酮基,乙基磺酰基甲基,苯磺酰氨基甲基,正丙基磺酰氨基或1-甲氧羰基氨基乙基。
12、一种如权利要求6中的方法,其中R和R4都是H,R2和R3都是H,基团CO2R4连接在环己烷环的3位上并与酰胺基成顺式-立体化学关系,其中R5是正丙基,2-甲氧基乙氧基甲基,2-丁炔基,2-丙烯基,2-丁烯基,环戊基,环己基,2-环己烯基,环丙基甲基,四氢呋喃基,4-吡啶基甲基,正丙基磺酰氨基,苯磺酰氨基甲基或苯甲酰氨基甲基。
13、一种如权利要求6中的方法,其中R是H,R4是乙基,和其中R2和R3都是H,乙氧羰基连接在环己烷环的3位上并有顺式立体化学和R5是2-甲氧基乙氧基甲基,正丙基,2-丁炔基,2-丙烯基,环己烯基,环己基,环戊基,环丙基甲基,四氢呋埚基,4-吡啶基甲基,苯磺酰氨基甲基,苯甲酰氨基甲基或正丙基磺酰氨基。
14、一种如权利要求6中的方法,其中R2和R3都是H,基团-CO2R4连接在环己烷环4位上并有顺式立体化学,R5是2-甲氧基乙氧基甲基和或在R是5-二氯茚基和R4是H或者R4是5-二氢茚基和R是H。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB868629663A GB8629663D0 (en) | 1986-12-11 | 1986-12-11 | Therapeutic agents |
| GB8629663 | 1986-12-11 | ||
| GB878715722A GB8715722D0 (en) | 1987-07-03 | 1987-07-03 | Therapeutic agents |
| GB8715722 | 1987-07-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN87107371A CN87107371A (zh) | 1988-07-06 |
| CN1016778B true CN1016778B (zh) | 1992-05-27 |
Family
ID=26291684
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN87107371A Expired CN1016778B (zh) | 1986-12-11 | 1987-12-11 | 螺-取代的戊二酸单酰胺 |
Country Status (25)
| Country | Link |
|---|---|
| US (1) | US5030654A (zh) |
| EP (1) | EP0274234B1 (zh) |
| KR (2) | KR880007441A (zh) |
| CN (1) | CN1016778B (zh) |
| AU (1) | AU595082B2 (zh) |
| CA (1) | CA1328264C (zh) |
| CY (1) | CY1723A (zh) |
| DE (1) | DE3772950D1 (zh) |
| DK (1) | DK174922B1 (zh) |
| EG (1) | EG18361A (zh) |
| ES (1) | ES2031523T3 (zh) |
| FI (1) | FI94336C (zh) |
| GR (1) | GR3002739T3 (zh) |
| HK (1) | HK65893A (zh) |
| HU (1) | HU202482B (zh) |
| IE (1) | IE60304B1 (zh) |
| IL (1) | IL84757A (zh) |
| MY (1) | MY100740A (zh) |
| NO (1) | NO174385C (zh) |
| NZ (1) | NZ222848A (zh) |
| PH (1) | PH25058A (zh) |
| PL (1) | PL150420B1 (zh) |
| PT (1) | PT86316B (zh) |
| SG (1) | SG50593G (zh) |
| YU (1) | YU47970B (zh) |
Families Citing this family (91)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1337400C (en) * | 1987-06-08 | 1995-10-24 | Norma G. Delaney | Inhibitors of neutral endopeptidase |
| GB8811873D0 (en) * | 1988-05-19 | 1988-06-22 | Pfizer Ltd | Therapeutic agents |
| GB8812597D0 (en) * | 1988-05-27 | 1988-06-29 | Pfizer Ltd | Therapeutic agents |
| GB8820844D0 (en) * | 1988-09-05 | 1988-10-05 | Pfizer Ltd | Therapeutic agents |
| GB8903740D0 (en) * | 1989-02-18 | 1989-04-05 | Pfizer Ltd | Therapeutic agents |
| GB8907704D0 (en) * | 1989-04-05 | 1989-05-17 | Pfizer Ltd | Preparation of glutaric acid derivatives |
| GB8925933D0 (en) * | 1989-11-16 | 1990-01-04 | Pfizer Ltd | Glutaric acid derivatives and preparation thereof |
| US5260467A (en) * | 1989-11-16 | 1993-11-09 | Pfizer Inc. | Glutaric acid derivatives and preparation thereof |
| US5157138A (en) * | 1989-11-16 | 1992-10-20 | Pfizer, Inc. | Glutaric acid derivatives and preparation thereof |
| GB8926063D0 (en) * | 1989-11-17 | 1990-01-10 | Pfizer Ltd | Therapeutic agents |
| WO1991007386A1 (en) * | 1989-11-21 | 1991-05-30 | Schering Corporation | Carboxyalkylcarbonyl aminoacid endopeptidase inhibitors |
| AU7168091A (en) * | 1989-12-22 | 1991-07-24 | Schering Corporation | Mercaptocycloacyl aminoacid endopeptidase inhibitors |
| GB9000725D0 (en) * | 1990-01-12 | 1990-03-14 | Pfizer Ltd | Therapeutic agents |
| GB9004260D0 (en) * | 1990-02-26 | 1990-04-18 | Pfizer Ltd | Therapeutic agents |
| US5075302A (en) * | 1990-03-09 | 1991-12-24 | Schering Corporation | Mercaptoacyl aminolactam endopeptidase inhibitors |
| NZ239846A (en) * | 1990-09-27 | 1994-11-25 | Merck & Co Inc | Sulphonamide derivatives and pharmaceutical compositions thereof |
| US5264420A (en) * | 1990-09-27 | 1993-11-23 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| IL99537A (en) * | 1990-09-27 | 1995-11-27 | Merck & Co Inc | Fibrinogen receptor antagonists and pharmaceutical preparations containing them |
| GB9103454D0 (en) * | 1991-02-19 | 1991-04-03 | Pfizer Ltd | Therapeutic agents |
| US5252601A (en) * | 1991-05-03 | 1993-10-12 | Merrell Dow Pharmaceuticals Inc. | 2-mercaptomethylene-tetrahydronaphthalene and indane-2-carboxamide derivatives as enkephalinase inhibitors |
| HUT68769A (en) * | 1991-05-07 | 1995-07-28 | Merck & Co Inc | FIBRINOGéN RECEPTOR ANTAGONIST COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS COMPRISING THEM AS EFFECTIVE SUBSTANCE |
| US5321034A (en) * | 1991-05-07 | 1994-06-14 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5389631A (en) * | 1991-10-29 | 1995-02-14 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5416099A (en) * | 1991-10-29 | 1995-05-16 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5272158A (en) * | 1991-10-29 | 1993-12-21 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5298492A (en) * | 1992-08-04 | 1994-03-29 | Schering Corporation | Diamino acid derivatives as antihypertensives |
| US5208236A (en) * | 1992-09-23 | 1993-05-04 | Schering Corporation | N-(acylaminomethyl)glutaryl amino acids and use |
| US5629343A (en) * | 1992-10-02 | 1997-05-13 | Merck & Co., Inc. | N-(mercaptoacyl) peptidyl derivatives as antidegenerative agents |
| WO1994008962A1 (en) * | 1992-10-14 | 1994-04-28 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5358956A (en) * | 1992-10-14 | 1994-10-25 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US6090785A (en) * | 1992-10-15 | 2000-07-18 | Merck & Co., Inc. | Substituted N-carboxyalkylpeptidyl derivatives as antidegenerative agents |
| US5672583A (en) * | 1992-11-25 | 1997-09-30 | Merck & Co., Inc. | Carboxy-peptidyl derivatives as antidegenerative active agents |
| JPH08504194A (ja) * | 1992-12-01 | 1996-05-07 | メルク エンド カンパニー インコーポレーテッド | フィブリノーゲンレセプターアンタゴニスト |
| JPH06199850A (ja) * | 1992-12-28 | 1994-07-19 | Tanabe Seiyaku Co Ltd | インドール含有ペプチド及びその製法 |
| US5441952A (en) * | 1993-04-05 | 1995-08-15 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5334596A (en) * | 1993-05-11 | 1994-08-02 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5397791A (en) * | 1993-08-09 | 1995-03-14 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5821241A (en) * | 1994-02-22 | 1998-10-13 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5719144A (en) * | 1995-02-22 | 1998-02-17 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5684152A (en) * | 1995-09-28 | 1997-11-04 | Merck & Co., Inc. | Preparation of carboxyalkyl derivatives as inhibitors of matrix metalloproteinases |
| US5852045A (en) * | 1995-10-19 | 1998-12-22 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5789421A (en) * | 1995-10-26 | 1998-08-04 | Merck & Co., Inc. | Fibrinogen receptor antagonist |
| US5780480A (en) * | 1996-02-28 | 1998-07-14 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5889023A (en) * | 1996-05-10 | 1999-03-30 | Merck & Co., Inc. | Fibrinogen receptor antagonist |
| US5981584A (en) * | 1997-02-06 | 1999-11-09 | Merck & Co., Inc. | Fibrinogen receptor antagonist prodrugs |
| US5939455A (en) * | 1997-03-11 | 1999-08-17 | Beacon Laboratories, Inc. | Therapeutic augmentation of oxyalkylene diesters and butyric acid derivatives |
| US6043389A (en) * | 1997-03-11 | 2000-03-28 | Mor Research Applications, Ltd. | Hydroxy and ether-containing oxyalkylene esters and uses thereof |
| EP1165088A4 (en) * | 1999-03-29 | 2002-09-04 | Bristol Myers Squibb Co | USE OF VASOPEPTIDASE INHIBITORS FOR THE TREATMENT OF ANGINA PECTORIS |
| IL139457A0 (en) * | 1999-11-08 | 2001-11-25 | Pfizer | Compounds for the treatment of female sexual dysfunction |
| US6660756B2 (en) | 2001-03-28 | 2003-12-09 | Pfizer Inc. | N-phenpropylcyclopentyl-substituted glutaramide derivatives as inhibitors of neutral endopeptidase |
| SK14102003A3 (sk) * | 2001-05-18 | 2004-08-03 | Solvay Pharmaceuticals Gmbh | Použitie zlúčenín s kombinovanou inhibičnou aktivitou k NEP/MP pri príprave liečiv |
| WO2003057200A2 (en) * | 2002-01-11 | 2003-07-17 | Novo Nordisk A/S | Compositions comprising inhibitors of dpp-iv and nep enzymes for the treatment of diabetes |
| US20070197552A1 (en) * | 2002-01-11 | 2007-08-23 | Novo Nordisk A/S | Method and composition for treatment of diabetes, hypertension, chronic heart failure and fluid retentive states |
| EP2316468A1 (en) | 2002-02-22 | 2011-05-04 | Shire LLC | Delivery system and methods for protecting and administering dextroamphetamine |
| NI200300043A (es) * | 2002-03-28 | 2003-11-05 | Warner Lambert Co | AMINOACIDOS CON AFINIDAD POR LA PROTEINA a2DELTA. |
| JO2479B1 (en) * | 2002-04-29 | 2009-01-20 | ميرك شارب اند دوم ليمتد | Rates for the activity of the chemokine receptor of tetrahydrobranyl cycloneptil tetrahydrobredo predine |
| US7045653B2 (en) | 2002-12-23 | 2006-05-16 | Pfizer, Inc. | Pharmaceuticals |
| TW200838501A (en) | 2007-02-02 | 2008-10-01 | Theravance Inc | Dual-acting antihypertensive agents |
| TWI448284B (zh) * | 2007-04-24 | 2014-08-11 | Theravance Inc | 雙效抗高血壓劑 |
| TWI406850B (zh) * | 2007-06-05 | 2013-09-01 | Theravance Inc | 雙效苯并咪唑抗高血壓劑 |
| EP2200975A1 (en) | 2007-09-07 | 2010-06-30 | Theravance, Inc. | Dual-acting antihypertensive agents |
| CN101896470B (zh) | 2007-12-11 | 2013-04-10 | 施万制药 | 双重作用的苯并咪唑衍生物和其作为抗高血压药剂的用途 |
| JP2011518884A (ja) | 2008-04-29 | 2011-06-30 | セラヴァンス, インコーポレーテッド | 二重活性抗高血圧剤 |
| WO2010011821A2 (en) | 2008-07-24 | 2010-01-28 | Theravance, Inc. | Dual-acting antihypertensive agents |
| WO2010132127A1 (en) | 2009-05-13 | 2010-11-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| RS53664B1 (en) * | 2009-05-28 | 2015-04-30 | Novartis Ag | SUBSTITUTED AMINOPROPIONIC ACID DERIVATIVES AS NEPRILIZINE INHIBITORS |
| EP2451782B1 (en) | 2009-07-07 | 2016-11-09 | Theravance Biopharma R&D IP, LLC | Dual-acting pyrazole antihypertensive agents |
| JP2012533626A (ja) | 2009-07-22 | 2012-12-27 | セラヴァンス, インコーポレーテッド | 二重作用オキサゾール降圧剤 |
| JP2013517295A (ja) | 2010-01-19 | 2013-05-16 | セラヴァンス, インコーポレーテッド | 二重に作用するチオフェン、ピロール、チアゾールおよびフラン抗高血圧症薬 |
| WO2012082853A1 (en) | 2010-12-15 | 2012-06-21 | Theravance, Inc. | Neprilysin inhibitors |
| WO2012082857A1 (en) | 2010-12-15 | 2012-06-21 | Theravance, Inc. | Neprilysin inhibitors |
| EP2675795B1 (en) | 2011-02-17 | 2016-04-20 | Theravance Biopharma R&D IP, LLC | Substituted aminobutyric derivatives as neprilysin inhibitors |
| EP2675792B1 (en) | 2011-02-17 | 2016-01-06 | Theravance Biopharma R&D IP, LLC | Substituted aminobutyric derivatives as neprilysin inhibitors |
| WO2012166387A1 (en) | 2011-05-31 | 2012-12-06 | Theravance, Inc. | Neprilysin inhibitors |
| WO2012166389A1 (en) | 2011-05-31 | 2012-12-06 | Theravance, Inc. | Neprilysin inhibitors |
| CA2835220A1 (en) | 2011-05-31 | 2012-12-06 | Theravance, Inc. | Neprilysin inhibitors |
| TWI560172B (en) | 2011-11-02 | 2016-12-01 | Theravance Biopharma R&D Ip Llc | Neprilysin inhibitors |
| EP2855464B1 (en) | 2012-05-31 | 2016-10-26 | Theravance Biopharma R&D IP, LLC | Nitric oxide donor neprilysin inhibitors |
| NZ702749A (en) | 2012-06-08 | 2017-01-27 | Theravance Biopharma R&D Ip Llc | Neprilysin inhibitors |
| US8871792B2 (en) | 2012-06-08 | 2014-10-28 | Theravance Biopharma R&D Ip, Llc | Neprilysin inhibitors |
| SI2882716T1 (sl) | 2012-08-08 | 2017-04-26 | Theravance Biopharma R&D Ip, Llc | Zaviralci neprilizina |
| WO2014127331A1 (en) | 2013-02-17 | 2014-08-21 | Intra-Cellular Therapies, Inc. | Novel uses |
| DK2964616T3 (en) | 2013-03-05 | 2017-08-28 | Theravance Biopharma R&D Ip Llc | Neprilysin inhibitors |
| CN105992763A (zh) | 2014-01-30 | 2016-10-05 | 施万生物制药研发Ip有限责任公司 | 脑啡肽酶抑制剂 |
| SG11201606057PA (en) | 2014-01-30 | 2016-08-30 | Theravance Biopharma R&D Ip Llc | 5-biphenyl-4-heteroarylcarbonylamino-pentanoic acid derivatives as neprilysin inhibitors |
| ES2745819T3 (es) | 2014-08-07 | 2020-03-03 | Intra Cellular Therapies Inc | Derivados de imidazo[1,2-a]-pirazolo[4,3-e]-pirimidin-4-ona con actividad inhibidora de la PDE1 |
| RU2726623C2 (ru) | 2015-02-11 | 2020-07-15 | ТЕРЕВАНС БАЙОФАРМА Ар энд Ди АйПи, ЭлЭлСи | (2s,4r)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановая кислота |
| HUE052732T2 (hu) | 2015-02-19 | 2021-05-28 | Theravance Biopharma R&D Ip Llc | (2R,4R)-5-(5'-klór-2'-fluorbifenil-4-il)-2-hidroxi-4-[(5-metiloxazol-2-karbonil)amino]pentánsav |
| AU2017229466C1 (en) | 2016-03-08 | 2021-02-11 | Theravance Biopharma R&D Ip, Llc | Crystalline(2S,4R)-5-(5'-chloro-2'-fluoro-[1,1'-biphenyl]-4-YL)-2-(ethoxymethyl)-4-(3-hydroxyisoxazole-5-carboxamido)-2-methylpentanoic acid and uses thereof |
| EP3436083A4 (en) | 2016-03-28 | 2019-11-27 | Intra-Cellular Therapies, Inc. | NEW COMPOSITIONS AND METHODS |
| WO2019152697A1 (en) | 2018-01-31 | 2019-08-08 | Intra-Cellular Therapies, Inc. | Novel uses |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3981915A (en) * | 1971-12-17 | 1976-09-21 | American Home Products Corporation | Amides of 1-aminocyclopentane carboxylic acid |
| FR2480747A1 (fr) * | 1980-04-17 | 1981-10-23 | Roques Bernard | Derives d'acides amines et leur application therapeutique |
| AU594645B2 (en) * | 1984-06-08 | 1990-03-15 | Ciba-Geigy Ag | N-substituted butyramide derivatives |
| GB2167748A (en) * | 1984-11-23 | 1986-06-04 | Squibb & Sons Inc | Acylamino hydroxyalkanoyl amino and imino acids and esters |
| EP0225292A3 (en) * | 1985-12-06 | 1988-11-30 | Ciba-Geigy Ag | Certain n-substituted butyramide derivatives |
| US4749688A (en) * | 1986-06-20 | 1988-06-07 | Schering Corporation | Use of neutral metalloendopeptidase inhibitors in the treatment of hypertension |
-
1986
- 1986-12-10 KR KR860014073A patent/KR880007441A/ko unknown
-
1987
- 1987-12-08 IL IL84757A patent/IL84757A/xx not_active IP Right Cessation
- 1987-12-08 DE DE8787310784T patent/DE3772950D1/de not_active Expired - Lifetime
- 1987-12-08 EP EP87310784A patent/EP0274234B1/en not_active Expired - Lifetime
- 1987-12-08 ES ES198787310784T patent/ES2031523T3/es not_active Expired - Lifetime
- 1987-12-09 PT PT86316A patent/PT86316B/pt not_active IP Right Cessation
- 1987-12-09 FI FI875413A patent/FI94336C/fi not_active IP Right Cessation
- 1987-12-09 NZ NZ222848A patent/NZ222848A/xx unknown
- 1987-12-09 CA CA000553863A patent/CA1328264C/en not_active Expired - Fee Related
- 1987-12-10 YU YU223987A patent/YU47970B/sh unknown
- 1987-12-10 PL PL1987269336A patent/PL150420B1/pl unknown
- 1987-12-10 EG EG714/87A patent/EG18361A/xx active
- 1987-12-10 HU HU875576A patent/HU202482B/hu not_active IP Right Cessation
- 1987-12-10 PH PH36203A patent/PH25058A/en unknown
- 1987-12-10 KR KR1019870014073A patent/KR910003335B1/ko not_active IP Right Cessation
- 1987-12-10 NO NO875169A patent/NO174385C/no unknown
- 1987-12-10 DK DK198706484A patent/DK174922B1/da not_active IP Right Cessation
- 1987-12-10 AU AU82407/87A patent/AU595082B2/en not_active Ceased
- 1987-12-10 IE IE335587A patent/IE60304B1/en not_active IP Right Cessation
- 1987-12-11 CN CN87107371A patent/CN1016778B/zh not_active Expired
- 1987-12-12 MY MYPI87003186A patent/MY100740A/en unknown
-
1989
- 1989-05-19 US US07/354,170 patent/US5030654A/en not_active Expired - Lifetime
-
1991
- 1991-09-16 GR GR91401354T patent/GR3002739T3/el unknown
-
1993
- 1993-04-20 SG SG50593A patent/SG50593G/en unknown
- 1993-07-08 HK HK658/93A patent/HK65893A/xx not_active IP Right Cessation
-
1994
- 1994-05-06 CY CY172394A patent/CY1723A/xx unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1016778B (zh) | 螺-取代的戊二酸单酰胺 | |
| CN1186332C (zh) | 新的甲状腺受体配位体及方法ⅱ | |
| CN1198839C (zh) | 作为凝血酶抑制剂的新的二肽脒类 | |
| CN1078889C (zh) | 非肽类速激肽受体拮抗剂 | |
| CN1243723C (zh) | 作为fsad的nep抑制剂的n-苯丙基环戊基取代的戊二酰胺衍生物 | |
| CN1259307C (zh) | 酰化的二氢化茚基胺及其作为药物的用途 | |
| CN1030251C (zh) | 取代的4-(喹啉-2-基-甲氧基)苯乙酸衍生物的制备方法 | |
| CN1107502C (zh) | 含氢化吡啶衍生物作为其活性成分的药用组合物的制备方法 | |
| CN1218943C (zh) | 4-嘧啶基-n-酰基-l-苯基丙氨酸类化合物 | |
| CN1040986A (zh) | 环烷基取代的戊二酰胺衍生物的制备 | |
| CN1310872C (zh) | 单和双取代的3-丙基-γ-氨基丁酸 | |
| CN1087902A (zh) | 吗啉和硫代吗啉速激肽受体拮抗药 | |
| CN1617852A (zh) | α-(N-磺酰氨基)乙酰胺衍生物作为β-淀粉样蛋白抑制剂 | |
| CN1880304A (zh) | 三酰胺取代的吲哚、苯并呋喃及苯并噻吩 | |
| CN1640877A (zh) | 新的螺环化合物 | |
| CN1043501A (zh) | 制备取代2-吡啶酮和吡啶-2-硫酮的方法 | |
| CN1032786A (zh) | N-酰基氨基酸衍生物及其用途 | |
| CN1479715A (zh) | 用于抑制由白介素8诱导的中性粒细胞趋化作用的酰胺 | |
| CN1761657A (zh) | Ep4受体拮抗剂 | |
| CN1138037A (zh) | 粘连受体拮抗剂 | |
| CN1501934A (zh) | 新型的3-c(o)r取代的10-环己基苯甲酰基吡咯并苯并二氮杂�;保胎催产素受体拮抗剂 | |
| CN1058019C (zh) | 环状氨基酸衍生物 | |
| CN1745065A (zh) | 4-氧代-3-(1-氧代-1h-异喹啉-2-基乙酰氨基)-戊酸酯和酰胺衍生物及其作为天冬氨酸特异性半胱氨酸蛋白酶抑制剂的用途 | |
| CN1204889C (zh) | 粘附受体拮抗剂 | |
| CN1139580C (zh) | 取代的苄胺及它们用于治疗抑郁症的用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C13 | Decision | ||
| GR02 | Examined patent application | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C53 | Correction of patent of invention or patent application | ||
| COR | Change of bibliographic data |
Free format text: CORRECT: APPLICANT; FROM: AMERICAN TELEPHONE + TELEGRAPH CO. TO: AT + T CORP. |
|
| C15 | Extension of patent right duration from 15 to 20 years for appl. with date before 31.12.1992 and still valid on 11.12.2001 (patent law change 1993) | ||
| OR01 | Other related matters | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |