CN116217428A - D-泛酸钙的甲醇水合物晶体及d-泛酸钙的制备方法 - Google Patents
D-泛酸钙的甲醇水合物晶体及d-泛酸钙的制备方法 Download PDFInfo
- Publication number
- CN116217428A CN116217428A CN202211654801.0A CN202211654801A CN116217428A CN 116217428 A CN116217428 A CN 116217428A CN 202211654801 A CN202211654801 A CN 202211654801A CN 116217428 A CN116217428 A CN 116217428A
- Authority
- CN
- China
- Prior art keywords
- pantothenate
- calcium
- methanol
- calcium pantothenate
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- FAPWYRCQGJNNSJ-UBKPKTQASA-L calcium D-pantothenic acid Chemical compound [Ca+2].OCC(C)(C)[C@@H](O)C(=O)NCCC([O-])=O.OCC(C)(C)[C@@H](O)C(=O)NCCC([O-])=O FAPWYRCQGJNNSJ-UBKPKTQASA-L 0.000 title claims abstract description 204
- 239000013078 crystal Substances 0.000 title claims abstract description 80
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 title claims abstract description 45
- 238000000034 method Methods 0.000 title claims description 32
- 230000008569 process Effects 0.000 title claims description 20
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims abstract description 107
- 239000000243 solution Substances 0.000 claims abstract description 54
- 238000001816 cooling Methods 0.000 claims abstract description 46
- 238000004321 preservation Methods 0.000 claims abstract description 44
- 238000011282 treatment Methods 0.000 claims abstract description 43
- 239000011259 mixed solution Substances 0.000 claims abstract description 19
- 238000002360 preparation method Methods 0.000 claims abstract description 16
- 238000002156 mixing Methods 0.000 claims abstract description 4
- 238000000855 fermentation Methods 0.000 claims description 24
- 230000004151 fermentation Effects 0.000 claims description 24
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 claims description 21
- 239000011713 pantothenic acid Substances 0.000 claims description 17
- 235000019161 pantothenic acid Nutrition 0.000 claims description 17
- 229940014662 pantothenate Drugs 0.000 claims description 15
- 238000004519 manufacturing process Methods 0.000 claims description 14
- 239000012528 membrane Substances 0.000 claims description 13
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 claims description 10
- 239000003456 ion exchange resin Substances 0.000 claims description 7
- 229920003303 ion-exchange polymer Polymers 0.000 claims description 7
- 239000007788 liquid Substances 0.000 claims description 7
- 239000002994 raw material Substances 0.000 claims description 6
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 claims description 5
- 239000000920 calcium hydroxide Substances 0.000 claims description 5
- 229910001861 calcium hydroxide Inorganic materials 0.000 claims description 5
- 241000588724 Escherichia coli Species 0.000 claims description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 4
- 239000008103 glucose Substances 0.000 claims description 4
- 239000003729 cation exchange resin Substances 0.000 claims description 3
- 239000000919 ceramic Substances 0.000 claims description 3
- 230000003472 neutralizing effect Effects 0.000 claims description 3
- 229920000620 organic polymer Polymers 0.000 claims description 3
- 238000002425 crystallisation Methods 0.000 description 24
- 230000008025 crystallization Effects 0.000 description 23
- 230000000052 comparative effect Effects 0.000 description 14
- 239000012535 impurity Substances 0.000 description 12
- 150000004682 monohydrates Chemical class 0.000 description 10
- 239000007864 aqueous solution Substances 0.000 description 8
- 239000000047 product Substances 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 238000001035 drying Methods 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- GHOKWGTUZJEAQD-UHFFFAOYSA-N Chick antidermatitis factor Natural products OCC(C)(C)C(O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-UHFFFAOYSA-N 0.000 description 4
- -1 D-calcium pantothenate methanol compound Chemical class 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 239000007791 liquid phase Substances 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- DARDVQMPKFOPRT-UHFFFAOYSA-N 3-[(2-hydroxy-3-methylbutanoyl)amino]propanoic acid Chemical compound CC(C)C(O)C(=O)NCCC(O)=O DARDVQMPKFOPRT-UHFFFAOYSA-N 0.000 description 2
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 2
- 239000005516 coenzyme A Substances 0.000 description 2
- 229940093530 coenzyme a Drugs 0.000 description 2
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- LELOWRISYMNNSU-UHFFFAOYSA-N hydrogen cyanide Chemical compound N#C LELOWRISYMNNSU-UHFFFAOYSA-N 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 229940055726 pantothenic acid Drugs 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 206010012442 Dermatitis contact Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 206010033685 Pantothenic acid deficiency Diseases 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229960002079 calcium pantothenate Drugs 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 208000010247 contact dermatitis Diseases 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000003912 environmental pollution Methods 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 238000001000 micrograph Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000036284 oxygen consumption Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 238000010587 phase diagram Methods 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 238000011946 reduction process Methods 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/12—Preparation of carboxylic acid amides by reactions not involving the formation of carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/22—Separation; Purification; Stabilisation; Use of additives
- C07C231/24—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C235/12—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/74—Separation; Purification; Use of additives, e.g. for stabilisation
- C07C29/76—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment
- C07C29/78—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment by condensation or crystallisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C31/00—Saturated compounds having hydroxy or O-metal groups bound to acyclic carbon atoms
- C07C31/02—Monohydroxylic acyclic alcohols
- C07C31/04—Methanol
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/02—Amides, e.g. chloramphenicol or polyamides; Imides or polyimides; Urethanes, i.e. compounds comprising N-C=O structural element or polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/09—Geometrical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12R—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES C12C - C12Q, RELATING TO MICROORGANISMS
- C12R2001/00—Microorganisms ; Processes using microorganisms
- C12R2001/01—Bacteria or Actinomycetales ; using bacteria or Actinomycetales
- C12R2001/185—Escherichia
- C12R2001/19—Escherichia coli
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Crystallography & Structural Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
本申请涉及一种D‑泛酸钙的甲醇水合物晶体及D‑泛酸钙的制备方法,D‑泛酸钙的甲醇水合物晶体的制备方法包括以下步骤:于40℃~45℃下,将D‑泛酸钙溶液和甲醇混合溶解,得到D‑泛酸钙的混合液;将所述D‑泛酸钙的混合液进行第一次降温,以使所述D‑泛酸钙的混合液的温度在0.5h内降至25℃~15℃,加入晶种后进行第一次保温处理,然后进行第二次降温处理,以使所述D‑泛酸钙的混合液的温度在≥4h内降至5℃~0℃,再进行第二次保温处理,得到D‑泛酸钙的甲醇水合物晶体。
Description
技术领域
本申请涉及医药化合物技术领域,特别涉及到一种D-泛酸钙的甲醇水合物晶体及D-泛酸钙的制备方法。
背景技术
D-泛酸钙,化学名称D-N-(2,4-二羟基-3,3-二甲基丁酰基)-β-氨基丙酸钙,相对分子量:476.54,进入人体释放出钙元素和泛酸,而泛酸是辅酶A前体物质,进而转化为辅酶A而产生生理作用,参与碳水化合物、脂肪和蛋白质的代谢作用,是人体和动物维持正常生理机能不可缺少的微量物质,广泛用于泛酸缺乏症的预防和治疗、以及接触性皮炎、急慢性湿疹等的治疗。
D-泛酸钙的生产常采用微生物酶发酵法,微生物酶发酵法首先得到的是含D-泛酸钙的发酵液,其中含有大量不知名的杂质。EP1918383A1采用半连续发酵工艺,减少了泛酸相关代谢物的生成量,例如3-(2-羟基-3-甲基-丁酰氨基)-丙酸。而传统的纯化方法得到的D-泛酸钙晶体的纯度不高,且松密度较小,进而限制了D-泛酸钙的后续应用。
由此,现有技术仍有待改进。
发明内容
基于此,本申请提供了一种D-泛酸钙的甲醇水合物晶体及D-泛酸钙的制备方法,该D-泛酸钙的甲醇水合物晶体的制备方法可以制得块状的D-泛酸钙的甲醇水合物晶体,能提高制得的D-泛酸钙的甲醇水合物晶体的纯度,并提高其松密度。
本申请解决上述技术问题的技术方案如下:
本申请的一方面,提供一种D-泛酸钙的甲醇水合物晶体的制备方法,包括以下步骤:
于40℃~45℃下,将所述D-泛酸钙溶液和甲醇混合溶解,得到D-泛酸钙的混合液;
将所述D-泛酸钙的混合液进行第一次降温,以使所述D-泛酸钙的混合液的温度在0.5h内降至25℃~15℃,加入晶种后进行第一次保温处理,然后进行第二次降温处理,以使所述D-泛酸钙的混合液的温度在≥4h的时间内降至5℃~0℃,再进行第二次保温处理,得到D-泛酸钙的甲醇水合物晶体。
在其中一些实施例中,所述第二次降温处理步骤满足(a)~(b)中至少一个条件:
(a)所述第二次降温处理时,使所述D-泛酸钙的混合液的温度在4h~8h内降至5℃~0℃;
(b)所述第二次降温处理采用匀速降温处理。
在其中一些实施例中,所述D-泛酸钙的甲醇水合物晶体的制备方的保温步骤法满足(c)~(d)中至少一个条件:
(c)所述第一次保温处理的时间为1h~4h;
(d)所述第二次保温处理的时间为1h~4h。
在其中一些实施例中,所述D-泛酸钙溶液的制备包括如下步骤:
以葡萄糖作为原料,通过大肠杆菌发酵制备得到含D-泛酸铵的发酵液;
将含D-泛酸铵的发酵液预处理后,得到D-泛酸铵的预处理液,再将D-泛酸铵的预处理液用氢氧化钙中和,得到D-泛酸钙的预处理液;
将所述D-泛酸钙的预处理液进行浓缩处理,得到D-泛酸钙溶液。
在其中一些实施例中,所述预处理依次采用交换膜和离子交换树脂进行;
所述交换膜为陶瓷膜与有机高分子膜;及
所述离子交换树脂为阳离子交换树脂。
在其中一些实施例中,所述D-泛酸钙的预处理液中D-泛酸钙的折干纯度为60%~80%。
在其中一些实施例中,所述浓缩处理步骤满足(e)~(g)中至少一个条件:
(e)所述浓缩处理的温度为30℃~50℃;
(f)所述浓缩处理的压力为4000Pa~10000Pa;
(g)所述D-泛酸钙溶液中D-泛酸钙的含量为45wt%~55wt%。
在其中一些实施例中,所述D-泛酸钙溶液中的D-泛酸钙与所述甲醇的质量体积比为1g:(5~9)mL。
在其中一些实施例中,所述晶种为D-泛酸钙四甲醇类化合物,所述晶种的质量为所述D-泛酸钙溶液中D-泛酸钙含量的0.05%~1%。
本申请的另一方面,提供了一种D-泛酸钙的制备方法,采用上述的D-泛酸钙的甲醇水合物晶体的制备方法制得的D-泛酸钙的甲醇水合物晶体;采用所述D-泛酸钙的甲醇水合物晶体制备D-泛酸钙。
与传统技术相比较,本申请的D-泛酸钙的甲醇水合物晶体的制备方法具有如下有益效果:
上述D-泛酸钙的甲醇水合物晶体的制备方法中,将D-泛酸钙溶液在甲醇中进行重结晶,通过控制降温过程历经特定的两次降温处理,随温度的降低,D-泛酸钙的溶解度降低,溶液中的D-泛酸钙过饱和,容易结晶析出,且在晶种引发下,诱导晶核形成晶体,并控制每次降温处理的时间及温度,以控制其降温速率,从而得到非吸湿性的块状D-泛酸钙的甲醇水合物晶体,其松密度较高且纯度高,有利于后续应用。
附图说明
图1为实施例1中的D-泛酸钙四甲醇一水化合物晶体的显微图;
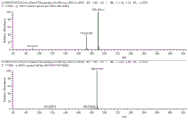
图2为实施例1中的D-泛酸钙溶液的液相检测图;
图3为实施例1中的D-泛酸钙溶液中杂质Y的质谱图谱。
具体实施方式
以下结合具体实施例对本申请的的方法作进一步详细的说明。本申请可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本申请公开内容理解更加透彻全面。
在本申请的描述中,需要理解的是,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。
除非另有定义,本文所使用的所有的技术和科学术语与属于本申请的技术领域的技术人员通常理解的含义相同。本文中在本申请的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本申请。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。在没有更多限制的情况下,由语句“包括一个……”限定的要素,并不排除在包括所述要素的过程、方法、物品或者设备中还存在另外的相同要素。本申请要素或组分前的不定冠词“一种”和“一个”对要素或组分的数量要求(即出现次数)无限制性。因此“一个”或“一种”应被解读为包括一个或至少一个,并且单数形式的要素或组分也包括复数形式,除非所述数量明显只指单数形式。“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。
需要说明的是,当本文中公开一个数值范围时,上述范围视为连续,且包括该范围的最小值及最大值,以及这种最小值与最大值之间的每一个值或任意两个值确定的范围。
本申请一实施方式提供了一种D-泛酸钙的甲醇水合物晶体的制备方法,包括步骤(1)~步骤(4)。
(1)将含D-泛酸铵的发酵液预处理后,得到D-泛酸铵的预处理液,再将D-泛酸铵的预处理液用氢氧化钙中和,得到D-泛酸钙的预处理液。
(2)将步骤(1)中的D-泛酸钙的预处理液进行浓缩处理,得到D-泛酸钙溶液。
(3)于40℃~45℃下,将步骤(2)中的D-泛酸钙溶液和甲醇混合溶解,得到D-泛酸钙的混合液。
(4)将步骤(3)中的D-泛酸钙的混合液进行第一次降温,以使D-泛酸钙的混合液的温度在0.5h内降至25℃~15℃,加入晶种后进行第一次保温处理,然后进行第二次降温处理,以使D-泛酸钙的混合液的温度在≥4h的时间内降至5℃~0℃,再进行第二次保温处理,得到D-泛酸钙的甲醇水合物晶体。
传统的D-泛酸或其盐的合成方法有三种:化学合成法、酶和生物发酵法,但化学合成过程有许多局限性,比如,氢氰酸和氰化钠造成严重的环境污染和人类危害,由此,本领域常采用酶和生物发酵法制备D-泛酸钙。然而,微生物酶发酵法首先得到的是含D-泛酸钙的发酵液,其中含有大量不知名的杂质,而传统的纯化方法得到的D-泛酸钙的纯度不高,且制得的D-泛酸钙的甲醇水合物晶体的松密度较小,进而限制了D-泛酸钙的后续应用。
由此,本申请的技术人员在长期的实际生产及研究中对酶和生物发酵法制备D-泛酸钙时的机理以及合成过程进行了大量分析研究,在掌握了酶和生物发酵法制备D-泛酸钙时的主要机理路线和杂质性质之后,创造性地提出了本申请中的D-泛酸钙的甲醇水合物晶体的制备方法:将D-泛酸钙溶液在甲醇中进行重结晶,通过控制降温过程历经特定的两次降温处理,随温度的降低,D-泛酸钙的甲醇水合物晶体的溶解度降低,溶液过饱和,容易结晶析出,且在晶种引发下,诱导晶核形成晶体,并控制每次降温处理的时间及温度,以控制其降温速率,从而得到非吸湿性的块状D-泛酸钙的甲醇水合物晶体,且松密度较高、纯度高,有利于后续应用。
在其中一些示例中,步骤(1)中含D-泛酸钙的发酵液通过以下方法制得:以葡萄糖作为原料,通过大肠杆菌发酵制备得到含D-泛酸铵的发酵液。
在其中一些实施例中,步骤(1)中含D-泛酸铵的发酵液与氢氧化钙的配比为:1000mL:(10.88~11.34)g。
在其中一些实施例中,步骤(1)中的预处理步骤依次采用交换膜和离子交换树脂进行。
具体地,交换膜为陶瓷膜与有机高分子膜,厂家为上海赛奥。
具体地,离子交换树脂为阳离子交换树脂,厂家为漂莱特。
将含D-泛酸钙的发酵液进行纯化预处理,可以筛除部分杂质,以利于后续步骤的进行。
在其中一些实施例中,步骤(1)中的D-泛酸钙的预处理液中D-泛酸钙的折干纯度为60%~80%。
具体地,步骤(1)中的D-泛酸钙的预处理液中D-泛酸钙的折干纯度可为60%、61%、62%、63%、64%、65%、66%、67%、68%、69%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%。
折干纯度是指扣除原料水分和残留溶剂后的固体原料中D-泛酸钙的质量含量。
在其中一些实施例中,步骤(2)中的浓缩步骤:在30℃~50℃,压力为4000Pa~10000Pa下浓缩至D-泛酸钙溶液中D-泛酸钙含量为45%~55%。
具体地,步骤(2)中的浓缩步骤的温度可为30℃、31℃、32℃、33℃、34℃、35℃、36℃、37℃、38℃、39℃、40℃、41℃、42℃、43℃、44℃、45℃、46℃、47℃、48℃、49℃、50℃。
具体地,步骤(2)中的浓缩步骤的压力可为4000Pa、4500Pa、5000Pa、5500Pa、6000Pa、6500Pa、7000Pa、7500Pa、8000Pa、8500Pa、9000Pa、9500Pa、10000Pa。
在其中一些实施例中,D-泛酸钙溶液中D-泛酸钙含量具体可为45%、46%、47%、48%、49%、50%、51%、52%、53%、54%、55%。
可理解,将D-泛酸钙的预处理液浓缩后,去除部分溶剂,可提高结晶效率。
在其中一些实施例中,步骤(3)中的D-泛酸钙溶液中的D-泛酸钙与甲醇的质量体积比为1g:(5~9)mL。
具体地,步骤(3)中的D-泛酸钙溶液中的D-泛酸钙与甲醇的质量体积比为可为1g:5mL、1g:6mL、1g:7mL、1g:8mL或1g:9mL。
在其中一些实施例中,步骤(3)中加入甲醇后,转移至结晶器中,进行搅拌。
在其中一些实施例中,步骤(3)中的搅拌处理速度为50rpm~200rpm。
上述结晶器可以采用本领域常用的结晶器进行,例如:冷却结晶器。
在其中一些实施例中,在进行第一次降温处理前,D-泛酸钙的混合液的温度为40℃~45℃。
可理解,步骤(3)中,于40℃~45℃下,将D-泛酸钙溶液和甲醇混合溶解,使得到的D-泛酸钙的混合液也达到了相应的温度。
在其中一些实施例中,第一次降温具体可降至25℃、24℃、23℃、22℃、21℃、20℃、19℃、18℃、17℃、16℃、15℃;第一次降温时间具体可为0.5h、0.4h、0.3h、0.2h、0.1h;第二次降温具体可降至5℃、4℃、3℃、2℃、1℃、0℃。
在结晶过程中,随温度的降低,D-泛酸钙的甲醇水合物晶体在甲醇中的溶解度降低,溶液过饱和,容易结晶析出,而主要杂质在甲醇中的溶解特性与D-泛酸钙的甲醇水合物晶体相差较大,由此就可以实现对生成D-泛酸钙的提纯。
其中若第二次降温处理的降温速度过快,则D-泛酸钙的晶体还未形成完整,无法析出或析出不完全结晶的D-泛酸钙,且夹杂的杂质Y多。
具体地,杂质Y即为3-(2-羟基-3-甲基-丁酰氨基)-丙酸。
在其中一些实施例中,步骤(4)中加入的晶种为D-泛酸钙四甲醇类化合物,且晶种的质量为D-泛酸钙溶液中D-泛酸钙含量的0.05%~1%。
具体地,晶种的质量为D-泛酸钙溶液中D-泛酸钙含量的0.05%~1%,具体可为0.05%、0.06%、0.07%、0.08%、0.09%、0.1%、0.2%、0.3%、0.4%、0.5%、0.6%、0.7%、0.8%、0.9%或1%。
通过加入晶种,是诱导晶核形成的有效手段,D-泛酸钙结晶化过程中具有高度的选择性,当加入与D-泛酸钙结构相似的同类分子时,在保温过程中有利于结晶进一步析出。
进一步地,通过调控在D-泛酸钙甲醇类化合物的添加量,使其在溶液中的浓度适,以析出纯度较高的D-泛酸钙的甲醇水合物晶体,避免因结晶析出的速度太快而导致的纯度较低的问题。
在其中一些示例中,步骤(4)中的第一次保温步骤:在15℃~25℃下保温处理1h~4h。
其中,第一次保温温度具体可为15℃、16℃、17℃、18℃、19℃、20℃、21℃、22℃、23℃、24℃、25℃;保温时间具体可为1h、1.5h、2h、2.5h、3h、3.5h、4h。
在其中一些示例中,步骤(4)中第二次保温步骤:在0℃~5℃下保温处理1h~4h。其中,第二次保温温度具体可为0℃、1℃、2℃、3℃、4℃、5℃;保温时间具体可为1h、1.5h、2h、2.5h、3h、3.5h、4h。
该D-泛酸钙的甲醇水合物晶体的制备方法能确保晶体充分析出的同时尽量不夹带杂质,得到了特定的形态的晶体:块状的非吸湿晶体,同时提高了产品的纯度及其松密度,且制备过程简单、效率高,有利于大规模生产。
本申请还提供一种D-泛酸钙的制备方法,包括如下步骤:
采用上述的D-泛酸钙的甲醇水合物晶体的制备方法制得D-泛酸钙的甲醇水合物晶体;采用D-泛酸钙的甲醇水合物晶体制备D-泛酸钙。
在其中一些实施例中,上述D-泛酸钙的甲醇水合物晶体经过溶解、脱甲醇、浓缩、喷雾干燥、包装得D-泛酸钙产品。
下面将结合具体的实施例对本申请进行了说明,但本申请并不局限于下述实施例,应当理解,所附权利要求概括了本申请的范围,在本申请构思的引导下本领域的技术人员应意识到,对本申请的各实施例所进行的一定的改变,都将被本申请的权利要求书的精神和范围所覆盖。
具体实施例
以下结合实施例对本申请作进一步阐述,但本申请的保护内容不限于实施例所述范围。
实施例1
1.D-泛酸钙溶液的制备
(1)预处理:将D-泛酸铵发酵液依次采用交换膜和离子交换树脂进行纯化预处理后,与氢氧化钙反应,其配比为1000mL:11.34g,获得到1500g D-泛酸钙水溶液的预处理液。
其中,D-泛酸铵发酵液是通过大肠杆菌以葡萄糖作为原料,经过耗氧发酵,发酵制备得到。
(2)浓缩:将D-泛酸钙的预处理液置于2L旋蒸瓶内蒸发,进行浓缩,浓缩得到质量浓度在50%左右的D-泛酸钙溶液,不同浓缩条件下得到的D-泛酸钙溶液的折干纯度不同,具体如表1所示。
表1

2.结晶
(1)溶解:将180.15g质量浓度为50.02wt%的D-泛酸钙溶液中加入720mL体积分数为99%的甲醇,得到748.95g D-泛酸钙甲醇水溶液,并转移至结晶器中,进行搅拌,搅拌速度为30rpm,D-泛酸钙甲醇水溶液的温度为40℃。
(2)结晶:将D-泛酸钙甲醇水溶液第一次15min降温至20℃,加入0.09g D-泛酸钙四甲醇一水化合物,进行第一次保温,保温时间为1h;保温结束后,进行第二次降温至2℃,降温时间为4.5h,降温结束后进行第二次保温,保温时间为3h;保温结束后,进行过滤和干燥,得到D-泛酸钙四甲醇一水化合物晶体。
实施例2
实施例2的D-泛酸钙产品的制备方法和实施例1基本一致,不同之处在于:结晶处理步骤不同,具体步骤如下:
2.结晶
(1)溶解:将180.15g质量浓度为50.02wt%的D-泛酸钙溶液中加入450mL体积分数为99%的甲醇,得到535.65g D-泛酸钙甲醇水溶液,并转移至结晶器中,进行搅拌,搅拌速度为30rpm,D-泛酸钙甲醇水溶液的温度为40℃。
(2)结晶:将D-泛酸钙甲醇水溶液第一次15min降温至15℃,加入0.045g D-泛酸钙四甲醇一水化合物,进行第一次保温,保温时间为1h;保温结束后,进行第二次降温至0℃,降温时间为4h,降温结束后进行第二次保温,保温时间为2h;保温结束后,进行过滤和干燥,得到D-泛酸钙的甲醇水合物晶体。
实施例3
实施例3的D-泛酸钙产品的制备方法和实施例1基本一致,不同之处在于:结晶处理步骤不同,具体步骤如下:
2.结晶
(1)溶解:将180.15g质量浓度为50.02wt%的D-泛酸钙溶液中加入540mL体积分数为99%的甲醇,得到606.75g D-泛酸钙甲醇水溶液,并转移至结晶器中,进行搅拌,搅拌速度为30rpm,D-泛酸钙甲醇水溶液的温度为40℃。
(2)结晶:将D-泛酸钙甲醇水溶液第一次15min降温至18℃,加入0.18g D-泛酸钙四甲醇一水化合物,进行第一次保温,保温时间为1.5h;保温结束后,进行第二次降温至1℃,降温时间为5h,降温结束后进行第二次保温,保温时间为2.5h;保温结束后,进行过滤和干燥,得到D-泛酸钙四甲醇一水化合物晶体。
实施例4
实施例4的D-泛酸钙产品的制备方法和实施例1基本一致,不同之处在于:结晶处理步骤不同,具体步骤如下:
2.结晶
(1)溶解:将180.15g质量浓度为50.02wt%的D-泛酸钙溶液中加入630mL体积分数为99%的甲醇,得到677.85g D-泛酸钙甲醇水溶液,并转移至结晶器中,进行搅拌,搅拌速度为30rpm,D-泛酸钙甲醇水溶液的温度为40℃。
(2)结晶:将D-泛酸钙甲醇水溶液第一次20min降温至21℃,加入0.36g D-泛酸钙四甲醇一水化合物,进行第一次保温,保温时间为2h;保温结束后,进行第二次降温至3℃,降温时间为6h,降温结束后进行第二次保温,保温时间为3h;保温结束后,进行过滤和干燥,得到D-泛酸钙四甲醇一水化合物晶体。
实施例5~6
实施例5~6与实施例1基本相同,不同之处仅在于:表2中的试验参数不同。
各实施例的具体试验参数设置如表2所示,其中:D-泛酸钙含量:有机溶剂体积(g:mL)记为M:V,晶种含量是指晶种的质量为D-泛酸钙的溶液中D-泛酸钙含量的百分占比。
表2


对比例1
对比例1与实施例1基本相同,不同点在于,D-泛酸钙溶液的制备中步骤(1)中不采用膜和树脂预处理。
其余步骤与实施例1相同。
对比例2
对比例2与实施例1基本相同,不同点在于,结晶处理步骤(2)中,加入晶种后,在3h降至0℃进行结晶。
其余步骤与实施例1相同。
2.结晶
(1)溶解:将180.15g质量浓度为50.02wt%的D-泛酸钙溶液中加入720mL体积分数为99%的甲醇,得到748.95g D-泛酸钙甲醇水溶液,并转移至结晶器中,进行搅拌,搅拌速度为30rpm,D-泛酸钙甲醇水溶液的温度为40℃。
(2)结晶:将D-泛酸钙甲醇水溶液第一次15min降温至20℃,加入0.09g D-泛酸钙四甲醇一水化合物,进行第一次保温,保温时间为1h;保温结束后,进行第二次降温至2℃,降温时间为3h,降温结束后进行第二次保温,保温时间为3h;保温结束后,进行过滤和干燥,得到D-泛酸钙四甲醇一水化合物晶体。
对比例3
对比例3与实施例1基本相同,不同点在于,结晶处理步骤(2)中,将D-泛酸钙甲醇水溶液在60min降温至20℃。
其余步骤与实施例1相同。
对比例4
对比例4与实施例1基本相同,不同点在于,结晶处理步骤(2)中,将D-泛酸钙甲醇水溶液第一次15min降温至30℃。
其余步骤与实施例1相同。
测试:
1、对实施例及对比例制得的D-泛酸钙四甲醇一水化合物晶体的进行观察。
2、对实施例及对比例中D-泛酸钙溶液(结晶原料)和制得的D-泛酸钙四甲醇一水化合物晶体的折干纯度进行测试,具体参照《GB/T 7299-2006D-泛酸钙》液相方法。
3、对实施例及对比例中D-泛酸钙溶液(结晶原料)和制得的D-泛酸钙四甲醇一水化合物晶体的杂质Y进行测试。液相色谱条件:流动相:1.36g磷酸二氢钾水溶液:乙腈=99:1(V/V);色谱柱:SBAQ C18 150*4.6mm,5μm;检测波长:200nm;温度:35℃;流速:1.2ml/min;运行时间:20min。。
4、对实施例及对比例制得的D-泛酸钙四甲醇一水化合物晶体的松密度进行测试,称取样品20g,缓慢倒入倾斜45度较的100ml量筒中,读取样品所占的体积V ml,20g/V ml即所测样品的松密度。
5、对实施例及对比例制得的D-泛酸钙四甲醇一水化合物晶体的吸湿性进行测试,对1g的结晶性泛酸钙进行精密第称量后,在称量瓶中展开,并且在40℃、75%RH的恒温恒湿机内静置24h,观察重量增加量。
具体测试结果请见表3。
表3

图1为D-泛酸钙四甲醇一水化合物晶体的显微图,图2为D-泛酸钙溶液的液相检测图,图3为D-泛酸钙溶液中杂质Y的质谱图谱,综合上述附图和表中测试结果可知,对比例的结晶产品的纯度、松密度均较实施例的低,且杂质Y含量均较实施例的高,由此可知,采用本申请的技术方案能制得非吸湿性的块状D-泛酸钙四甲醇一水化合物晶体,其松密度较高且纯度高。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本申请的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对申请专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干变形和改进,这些都属于本申请的保护范围。因此,本申请专利的保护范围应以所附权利要求为准。
Claims (10)
1.一种D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,包括以下步骤:
于40℃~45℃下,将D-泛酸钙溶液和甲醇混合溶解,得到D-泛酸钙的混合液;
将所述D-泛酸钙的混合液进行第一次降温,以使所述D-泛酸钙的混合液的温度在0.5h内降至25℃~15℃,加入晶种后进行第一次保温处理,然后进行第二次降温处理,以使所述D-泛酸钙的混合液的温度在≥4h的时间内降至5℃~0℃,再进行第二次保温处理,得到D-泛酸钙的甲醇水合物晶体。
2.如权利要求1所述的D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,所述第二次降温处理步骤满足(a)~(b)中至少一个条件:
(a)所述第二次降温处理时,使所述D-泛酸钙的混合液的温度在4h~8h内降至5℃~0℃;
(b)所述第二次降温处理采用匀速降温处理。
3.如权利要求1~2任一项所述的D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,所述D-泛酸钙的甲醇水合物晶体的制备方法的保温步骤满足(c)~(d)中至少一个条件:
(c)所述第一次保温处理的时间为1h~4h;
(d)所述第二次保温处理的时间为1h~4h。
4.如权利要求1~2任一项所述的D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,所述D-泛酸钙溶液的制备包括如下步骤:
以葡萄糖作为原料,通过大肠杆菌发酵制备得到含D-泛酸铵的发酵液;
将含D-泛酸铵的发酵液预处理后,得到D-泛酸铵的预处理液,再将D-泛酸铵的预处理液用氢氧化钙中和,得到D-泛酸钙的预处理液;
将所述D-泛酸钙的预处理液进行浓缩处理,得到所述D-泛酸钙溶液。
5.如权利要求4所述的D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,所述预处理依次采用交换膜和离子交换树脂进行;
所述交换膜为陶瓷膜与有机高分子膜;及
所述离子交换树脂为阳离子交换树脂。
6.如权利要求4所述的D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,所述D-泛酸钙的预处理液中D-泛酸钙的折干纯度为60%~80%。
7.如权利要求4所述的D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,所述浓缩处理步骤满足(e)~(g)中至少一个条件:
(e)所述浓缩处理的温度为30℃~50℃;
(f)所述浓缩处理的压力为4000Pa~10000Pa;
(g)所述D-泛酸钙溶液中D-泛酸钙的含量为45wt%~55wt%。
8.如权利要求1~2任一项所述的D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,所述D-泛酸钙溶液中的D-泛酸钙与所述甲醇的质量体积比为1g:(5~9)mL。
9.如权利要求4所述的D-泛酸钙的甲醇水合物晶体的制备方法,其特征在于,所述晶种为D-泛酸钙四甲醇类化合物,所述晶种的质量为所述D-泛酸钙溶液中D-泛酸钙含量的0.05%~1%。
10.一种D-泛酸钙的制备方法,其特征在于,包括如下步骤:
采用如权利要求1~9任一项所述的D-泛酸钙的甲醇水合物晶体的制备方法制得D-泛酸钙的甲醇水合物晶体;
采用所述D-泛酸钙的甲醇水合物晶体制备D-泛酸钙。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211654801.0A CN116217428B (zh) | 2022-12-22 | 2022-12-22 | D-泛酸钙的甲醇水合物晶体及d-泛酸钙的制备方法 |
| DE202023106581.3U DE202023106581U1 (de) | 2022-12-22 | 2023-11-10 | Kristallines Methanolhydrat von Calcium-D-Pantothenat und Calcium-D-Pantothenat |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211654801.0A CN116217428B (zh) | 2022-12-22 | 2022-12-22 | D-泛酸钙的甲醇水合物晶体及d-泛酸钙的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN116217428A true CN116217428A (zh) | 2023-06-06 |
| CN116217428B CN116217428B (zh) | 2024-04-02 |
Family
ID=86568759
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202211654801.0A Active CN116217428B (zh) | 2022-12-22 | 2022-12-22 | D-泛酸钙的甲醇水合物晶体及d-泛酸钙的制备方法 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN116217428B (zh) |
| DE (1) | DE202023106581U1 (zh) |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101948402A (zh) * | 2010-08-20 | 2011-01-19 | 新发药业有限公司 | 一种制备d-泛酸钙的方法 |
| CN108129345A (zh) * | 2018-01-10 | 2018-06-08 | 精晶药业股份有限公司 | 一种d-泛酸钙的制备方法 |
| CN108191688A (zh) * | 2017-12-28 | 2018-06-22 | 大连韦德生化科技有限公司 | 一种合成及结晶d-泛酸钙的方法 |
| CN108640852A (zh) * | 2018-06-11 | 2018-10-12 | 精晶药业股份有限公司 | 一种调节d-泛酸钙松密度和粒度的方法 |
| CN110845353A (zh) * | 2019-11-28 | 2020-02-28 | 安徽泰格生物科技有限公司 | 一种d-泛酸钙的结晶方法 |
| CN112209819A (zh) * | 2020-09-27 | 2021-01-12 | 安徽泰格生物科技有限公司 | 一种d-泛解酸钙的制备方法 |
| CN112457181A (zh) * | 2020-12-11 | 2021-03-09 | 黄冈美丰化工科技有限公司 | 一种d-泛酸钙的合成方法 |
| CN112592290A (zh) * | 2020-12-14 | 2021-04-02 | 广安摩珈生物科技有限公司 | 泛酸钙粗品纯化方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1918383A1 (en) | 2006-11-02 | 2008-05-07 | DSMIP Assets B.V. | Semi-continuous fermentation process for pantothenate production |
-
2022
- 2022-12-22 CN CN202211654801.0A patent/CN116217428B/zh active Active
-
2023
- 2023-11-10 DE DE202023106581.3U patent/DE202023106581U1/de active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101948402A (zh) * | 2010-08-20 | 2011-01-19 | 新发药业有限公司 | 一种制备d-泛酸钙的方法 |
| CN108191688A (zh) * | 2017-12-28 | 2018-06-22 | 大连韦德生化科技有限公司 | 一种合成及结晶d-泛酸钙的方法 |
| CN108129345A (zh) * | 2018-01-10 | 2018-06-08 | 精晶药业股份有限公司 | 一种d-泛酸钙的制备方法 |
| CN108640852A (zh) * | 2018-06-11 | 2018-10-12 | 精晶药业股份有限公司 | 一种调节d-泛酸钙松密度和粒度的方法 |
| CN110845353A (zh) * | 2019-11-28 | 2020-02-28 | 安徽泰格生物科技有限公司 | 一种d-泛酸钙的结晶方法 |
| CN112209819A (zh) * | 2020-09-27 | 2021-01-12 | 安徽泰格生物科技有限公司 | 一种d-泛解酸钙的制备方法 |
| CN112457181A (zh) * | 2020-12-11 | 2021-03-09 | 黄冈美丰化工科技有限公司 | 一种d-泛酸钙的合成方法 |
| CN112592290A (zh) * | 2020-12-14 | 2021-04-02 | 广安摩珈生物科技有限公司 | 泛酸钙粗品纯化方法 |
Non-Patent Citations (2)
| Title |
|---|
| MASAHIRO INAGAKI: "Crystal forms and optical resolution by preferential crystallization of calcium dl-pantothenate", 《CHEM.PHARM.BULL》, vol. 25, no. 5, 31 December 1977 (1977-12-31), pages 1001 - 1009 * |
| 韩丹丹: "有机小分子在溶液中的晶习调控", 《CNKI博士学位论文全文库》, no. 1, 15 January 2022 (2022-01-15) * |
Also Published As
| Publication number | Publication date |
|---|---|
| DE202023106581U1 (de) | 2023-12-07 |
| CN116217428B (zh) | 2024-04-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Li et al. | Tuning crystallization and stability of the metastable polymorph of DL-methionine by a structurally similar additive | |
| CN106187797B (zh) | 一种甘氨酸亚铁络合物的制备方法 | |
| CN102336647B (zh) | Ak糖结晶母液的用途及利用结晶母液制备钾盐的方法 | |
| CN116217428B (zh) | D-泛酸钙的甲醇水合物晶体及d-泛酸钙的制备方法 | |
| CN106187796B (zh) | 一种甘氨酸锌络合物的制备方法 | |
| KR101899015B1 (ko) | L-카르니틴 타르트레이트의 제조방법 | |
| CN109438268B (zh) | 一种酒石酸氢胆碱及其制备方法 | |
| CN107129446B (zh) | 一种2-丙烯酰胺基-2-甲基丙磺酸合成过程中降低硫酸根离子的工艺 | |
| EP3617191B9 (en) | Method for manufacturing diastereomer of citric acid derivative | |
| JP4453070B2 (ja) | 5’−グアニル酸ジナトリウム・5’−イノシン酸ジナトリウム混晶の製造法 | |
| Wu et al. | Molecular interaction transfer among solvents and solutes modulates the formation of linezolid crystals | |
| KR100343232B1 (ko) | 결정성 파미드론산 이나트륨염 수화물과 그의 제조방법 | |
| CN109694337A (zh) | 一种羟乙基磺酸钠椭球形晶体及其制备方法 | |
| CN115925569B (zh) | 一种无水l-苯丙氨酸晶型的制备方法 | |
| CN117166062B (zh) | 一种硫酸镁晶须制备方法 | |
| CN113474325B (zh) | 安赛蜜废液的处理方法 | |
| CN111187255B (zh) | 右旋艾普拉唑钾盐的制备方法和右旋艾普拉唑的制备方法 | |
| EA011409B1 (ru) | Способ получения морфологически чистой модификации "н" натеглинида | |
| CN109020826A (zh) | 一种l-天冬酰胺的制备方法 | |
| DE69024474T2 (de) | Herstellung von hydroxyaromatische Ketoacetale aus hydroxyaromatische Methylketone und Verfahren zur Herstellung von 5-(4'-Hydroxyphenyl)-hydantoin und D-p-Hydroxyphenylglycin aus 4-Hydroxyacetophenon | |
| CN117825627A (zh) | 一种测定溶解度的方法 | |
| SU1446108A1 (ru) | Способ получени кристаллического сульфата аммони | |
| CN116041246A (zh) | 一种格列齐特与2-甲基咪唑形成的共晶及其制备方法 | |
| CN115260087A (zh) | 一种盐酸尼卡地平杂质及其合成方法和应用 | |
| CN116239525A (zh) | 一种米力农-黄芩素共晶体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |