CN110937985A - 一种姜酮酚的合成方法 - Google Patents
一种姜酮酚的合成方法 Download PDFInfo
- Publication number
- CN110937985A CN110937985A CN201911202624.0A CN201911202624A CN110937985A CN 110937985 A CN110937985 A CN 110937985A CN 201911202624 A CN201911202624 A CN 201911202624A CN 110937985 A CN110937985 A CN 110937985A
- Authority
- CN
- China
- Prior art keywords
- compound
- reaction
- organic solvent
- water
- gingerol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- CZNLTCTYLMYLHL-UHFFFAOYSA-N [6]-Paradol Chemical compound CCCCCCCC(=O)CCC1=CC=C(O)C(OC)=C1 CZNLTCTYLMYLHL-UHFFFAOYSA-N 0.000 title claims abstract description 22
- 238000010189 synthetic method Methods 0.000 title claims description 5
- 239000002994 raw material Substances 0.000 claims abstract description 24
- 238000000034 method Methods 0.000 claims abstract description 22
- -1 paradol Natural products 0.000 claims abstract description 5
- 150000001875 compounds Chemical class 0.000 claims description 63
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 57
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 53
- 238000006243 chemical reaction Methods 0.000 claims description 53
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 50
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 45
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 42
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 39
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 37
- 238000002360 preparation method Methods 0.000 claims description 34
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 32
- 239000003960 organic solvent Substances 0.000 claims description 31
- NLDDIKRKFXEWBK-AWEZNQCLSA-N gingerol Chemical compound CCCCC[C@H](O)CC(=O)CCC1=CC=C(O)C(OC)=C1 NLDDIKRKFXEWBK-AWEZNQCLSA-N 0.000 claims description 28
- JZLXEKNVCWMYHI-UHFFFAOYSA-N gingerol Natural products CCCCC(O)CC(=O)CCC1=CC=C(O)C(OC)=C1 JZLXEKNVCWMYHI-UHFFFAOYSA-N 0.000 claims description 28
- 235000002780 gingerol Nutrition 0.000 claims description 28
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 27
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 23
- 238000001514 detection method Methods 0.000 claims description 21
- 239000002904 solvent Substances 0.000 claims description 20
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 19
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 18
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 15
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 15
- 230000035484 reaction time Effects 0.000 claims description 15
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 14
- 230000009471 action Effects 0.000 claims description 14
- 238000001035 drying Methods 0.000 claims description 13
- 239000003208 petroleum Substances 0.000 claims description 13
- 238000001953 recrystallisation Methods 0.000 claims description 13
- MWOOGOJBHIARFG-UHFFFAOYSA-N vanillin Chemical compound COC1=CC(C=O)=CC=C1O MWOOGOJBHIARFG-UHFFFAOYSA-N 0.000 claims description 13
- FGQOOHJZONJGDT-UHFFFAOYSA-N vanillin Natural products COC1=CC(O)=CC(C=O)=C1 FGQOOHJZONJGDT-UHFFFAOYSA-N 0.000 claims description 13
- 235000012141 vanillin Nutrition 0.000 claims description 13
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 claims description 12
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 12
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 claims description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 12
- 239000002253 acid Substances 0.000 claims description 12
- 239000011230 binding agent Substances 0.000 claims description 12
- 229940126214 compound 3 Drugs 0.000 claims description 12
- 239000000047 product Substances 0.000 claims description 11
- 235000019441 ethanol Nutrition 0.000 claims description 10
- 238000010438 heat treatment Methods 0.000 claims description 10
- 229910000104 sodium hydride Inorganic materials 0.000 claims description 10
- 238000003756 stirring Methods 0.000 claims description 10
- 238000005406 washing Methods 0.000 claims description 10
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 claims description 8
- 238000001914 filtration Methods 0.000 claims description 8
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 8
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 claims description 8
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 8
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 8
- 239000012312 sodium hydride Substances 0.000 claims description 8
- 239000007818 Grignard reagent Substances 0.000 claims description 7
- 239000012043 crude product Substances 0.000 claims description 7
- 238000000967 suction filtration Methods 0.000 claims description 7
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 claims description 6
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 claims description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 6
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 6
- RHVNHKWMQOUSLJ-UHFFFAOYSA-N methoxymethanamine;hydrochloride Chemical compound Cl.COCN RHVNHKWMQOUSLJ-UHFFFAOYSA-N 0.000 claims description 6
- 239000012046 mixed solvent Substances 0.000 claims description 6
- 230000009467 reduction Effects 0.000 claims description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 claims description 6
- 235000017557 sodium bicarbonate Nutrition 0.000 claims description 6
- 239000000126 substance Substances 0.000 claims description 6
- 230000002194 synthesizing effect Effects 0.000 claims description 6
- BDZBKCUKTQZUTL-UHFFFAOYSA-N triethyl phosphite Chemical compound CCOP(OCC)OCC BDZBKCUKTQZUTL-UHFFFAOYSA-N 0.000 claims description 6
- 238000005804 alkylation reaction Methods 0.000 claims description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 5
- 235000011181 potassium carbonates Nutrition 0.000 claims description 5
- LSXKDWGTSHCFPP-UHFFFAOYSA-N 1-bromoheptane Chemical compound CCCCCCCBr LSXKDWGTSHCFPP-UHFFFAOYSA-N 0.000 claims description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 4
- 238000006052 Horner reaction Methods 0.000 claims description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 4
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 4
- 239000003513 alkali Substances 0.000 claims description 4
- 150000001408 amides Chemical group 0.000 claims description 4
- 239000003054 catalyst Substances 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 239000011777 magnesium Substances 0.000 claims description 4
- 229910052749 magnesium Inorganic materials 0.000 claims description 4
- 239000011736 potassium bicarbonate Substances 0.000 claims description 4
- 229910000028 potassium bicarbonate Inorganic materials 0.000 claims description 4
- 235000015497 potassium bicarbonate Nutrition 0.000 claims description 4
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 claims description 4
- 229940086066 potassium hydrogencarbonate Drugs 0.000 claims description 4
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 claims description 4
- 235000017550 sodium carbonate Nutrition 0.000 claims description 4
- 238000003747 Grignard reaction Methods 0.000 claims description 3
- 230000015572 biosynthetic process Effects 0.000 claims description 3
- 125000006278 bromobenzyl group Chemical group 0.000 claims description 3
- 239000006227 byproduct Substances 0.000 claims description 3
- 238000001704 evaporation Methods 0.000 claims description 3
- 150000004795 grignard reagents Chemical class 0.000 claims description 3
- 238000003786 synthesis reaction Methods 0.000 claims description 3
- DZMDPHNGKBEVRE-UHFFFAOYSA-N 1-chloroheptane Chemical compound CCCCCCCCl DZMDPHNGKBEVRE-UHFFFAOYSA-N 0.000 claims description 2
- LMHCYRULPLGEEZ-UHFFFAOYSA-N 1-iodoheptane Chemical compound CCCCCCCI LMHCYRULPLGEEZ-UHFFFAOYSA-N 0.000 claims description 2
- 239000002585 base Substances 0.000 claims description 2
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 claims description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 claims description 2
- DQWPFSLDHJDLRL-UHFFFAOYSA-N triethyl phosphate Chemical compound CCOP(=O)(OCC)OCC DQWPFSLDHJDLRL-UHFFFAOYSA-N 0.000 claims description 2
- 238000004809 thin layer chromatography Methods 0.000 claims 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims 3
- 150000007529 inorganic bases Chemical class 0.000 claims 1
- 238000002156 mixing Methods 0.000 claims 1
- 238000009776 industrial production Methods 0.000 abstract description 6
- 239000012467 final product Substances 0.000 abstract description 3
- 229930014626 natural product Natural products 0.000 abstract 1
- 238000006257 total synthesis reaction Methods 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 19
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 5
- 239000008346 aqueous phase Substances 0.000 description 5
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 5
- 239000012295 chemical reaction liquid Substances 0.000 description 5
- 238000006722 reduction reaction Methods 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 238000005882 aldol condensation reaction Methods 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 229940125797 compound 12 Drugs 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 3
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 3
- 229940126543 compound 14 Drugs 0.000 description 3
- 229940126086 compound 21 Drugs 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 229910001385 heavy metal Inorganic materials 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000000171 quenching effect Effects 0.000 description 3
- VOLGAXAGEUPBDM-UHFFFAOYSA-N $l^{1}-oxidanylethane Chemical compound CC[O] VOLGAXAGEUPBDM-UHFFFAOYSA-N 0.000 description 2
- PLGXEPHZCXBYLP-UHFFFAOYSA-N (-)-munitagine Chemical compound C1C2=CC=C(OC)C(O)=C2C2CC(C=C(C(=C3)O)OC)=C3C1N2C PLGXEPHZCXBYLP-UHFFFAOYSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- ASEQIFOGFYYURN-UHFFFAOYSA-N 6-bromo-6-methoxycyclohexa-2,4-dien-1-ol Chemical compound COC1(Br)C=CC=CC1O ASEQIFOGFYYURN-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- WWULHQLTPGKDAM-UHFFFAOYSA-N gamma-eudesmol Natural products CC(C)C1CC(O)C2(C)CCCC(=C2C1)C WWULHQLTPGKDAM-UHFFFAOYSA-N 0.000 description 2
- JARKCYVAAOWBJS-UHFFFAOYSA-N hexanal Chemical compound CCCCCC=O JARKCYVAAOWBJS-UHFFFAOYSA-N 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- VKCYHJWLYTUGCC-UHFFFAOYSA-N nonan-2-one Chemical compound CCCCCCCC(C)=O VKCYHJWLYTUGCC-UHFFFAOYSA-N 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- QQZOPKMRPOGIEB-UHFFFAOYSA-N 2-Oxohexane Chemical compound CCCCC(C)=O QQZOPKMRPOGIEB-UHFFFAOYSA-N 0.000 description 1
- 244000227206 Aframomum melegueta Species 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- 238000007341 Heck reaction Methods 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- 229930182821 L-proline Natural products 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- 244000131415 Zanthoxylum piperitum Species 0.000 description 1
- 235000008853 Zanthoxylum piperitum Nutrition 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- JMFRWRFFLBVWSI-UHFFFAOYSA-N cis-coniferyl alcohol Natural products COC1=CC(C=CCO)=CC=C1O JMFRWRFFLBVWSI-UHFFFAOYSA-N 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000002778 food additive Substances 0.000 description 1
- 235000013376 functional food Nutrition 0.000 description 1
- 238000007210 heterogeneous catalysis Methods 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- RDIMTXDFGHNINN-UHFFFAOYSA-N panaxytriol Chemical compound CCCCCCCC(O)C(O)CC#CC#CC(O)C=C RDIMTXDFGHNINN-UHFFFAOYSA-N 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 229960002429 proline Drugs 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- ZENOXNGFMSCLLL-UHFFFAOYSA-N vanillyl alcohol Chemical compound COC1=CC(CO)=CC=C1O ZENOXNGFMSCLLL-UHFFFAOYSA-N 0.000 description 1
- OJYLAHXKWMRDGS-UHFFFAOYSA-N zingerone Chemical compound COC1=CC(CCC(C)=O)=CC=C1O OJYLAHXKWMRDGS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/004—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reaction with organometalhalides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/04—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids
- C07C259/06—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids having carbon atoms of hydroxamic groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/41—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by hydrogenolysis or reduction of carboxylic groups or functional derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/64—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by introduction of functional groups containing oxygen only in singly bound form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4003—Esters thereof the acid moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4006—Esters of acyclic acids which can have further substituents on alkyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4071—Esters thereof the ester moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4075—Esters with hydroxyalkyl compounds
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明公开了一种天然产物姜酮酚的全合成方法,其路线如下所示:
Description
技术领域
本发明涉及一种姜酮酚(6-Paradol)的合成方法,属于药物合成技术及保健食品领域。
背景技术
姜酮酚,化学名: 1-(4-羟基-3-甲氧基苯基)-3-癸酮,是一类存在于非洲豆蔻类植物天堂椒里的化学物质,其化学结构和姜辣素十分类似。目前研究表明,姜酮酚一方面具有显著降低人体血糖和血脂的功效,同时可以促进人体新陈代谢,具有较好的减肥效果;另一方面,该化合物的毒性和刺激性小且目前尚无任何副作用的相关报道;因此,姜酮酚作为保健品和功能性食品添加剂具有较为广阔的开发前景。
目前,文献报道的关于姜酮酚的制备,主要有以下几种方法:
路线1

Eur. J. Org. Chem., 2017(48), 7295-7299
路线2

J. Med. Chem. 2017(60), 9821−9837.
路线3

Int. J. Mol. Sci. 2014(15), 3926-3951.
路线4

CN 103553889A
路线5

PLoS One, 2015, 10(3): 1-17.
路线1 是以4-羟基-3-甲氧基苄醇为起始原料和溴苄经烷基化反应得到化合物10,化合物10在磷酸钾的作用下通过Ir/chitin非均相催化生成化合物11,化合物11最后经Pd/C催化氢化制得目标产物。该路线较短、操作简单,但反应中需要用到价格昂贵的重金属铱作为催化剂,且中间体及终产品需通过硅胶柱层析进行纯化,因此该路线不适合工业化生产。
路线2是以香兰素为起始原料在L-脯氨酸和三乙胺的作用下与2-壬酮进行Aldol缩合得到化合物12,化合物12再通过Pd/C催化经氢化还原得到目标化合物姜酮酚,该路线较短,操作简单,但在Aldol缩合时收率较低且每步反应均需采用硅胶柱层析进行纯化,因此该路线不适用于工业化生产。
路线3是以香兰素为起始原料在氢氧化钠作用下和丙酮通过Aldol缩合得到化合物13,化合物13再在二(三甲基硅基)氨基锂的作用下和己酮进行Aldol缩合得到化合物14,化合物14最后经催化氢化得到目标化合物。该路线操作简单但收率较低,在化合物14的制备时收率仅为19%,因此该路线不适合工业化生产。
路线4是以2-溴2-甲氧基苯酚为起始原料在醋酸钯的催化下通过Heck偶联反应制备得到化合物12,化合物12再通过催化氢化反应得到目标化合物。该路线较短,但Heck反应需要在绝对无氧的条件下进行,因此对设备及操作要求较高。同时原料2-溴-2-甲氧基苯酚相比于路线2中报道的香兰素价格更为昂贵,且反应中多次用到重金属Pd催化剂,可能给终产品带来重金属残留的问题。
路线5是以香兰素为起始原料在咪唑的作用下和叔丁基二甲基氯硅烷经烷基化反应得到化合物16,化合物16再依次通过Aldol缩合反应以及催化氢化反应得到化合物18,化合物18在二异丙基氨基锂作用下和正己醛进行缩合得到化合物19,化合物19再依次通过四丁基氟化铵脱保护以及酸催化的分子内脱水反应得到化合物21,化合物21最后通过催化氢化反应制备得到目标化合物。该路线较长,成本较高,同时制备化合物21反应时间较长,不适合工业化生产。
发明内容
本发明要解决的技术问题是提供一种新的姜酮酚的制备方法。
本发明要解决的另一技术问题是提供一种可以适用于工业化生产的姜酮酚的合成方法。
本发明要解决的又一技术问题是提供一种可以得到高纯度姜酮酚的制备方法。
本发明的方法具有原料易得,操作简单,收率较高,成本低廉,姜酮酚终产品纯度较高等优点。
为解决本发明的技术问题,本发明提供如下合成姜酮酚的技术方案:

该技术方案包括如下步骤:
(1) 化合物3的制备
以氯乙酰氯为原料和甲氧基甲基胺盐酸盐在缚酸剂的作用下于有机溶剂中缩合得到具有酰胺结构的化合物3.
其中上述缚酸剂可独立选自:氢氧化钠、氢氧化钾、无水碳酸钾、无水碳酸钠、碳酸氢钠、碳酸氢钾、三乙胺、咪唑、1,8-二氮双环[5.4.0]十一-7-烯、吡啶、氢化钠、叔丁醇钾、碳酸铯,优选无水碳酸钾、无水碳酸钠、吡啶;
其中有机溶剂可独立选自:甲醇、乙腈、四氢呋喃、丙酮、乙酸乙酯、氯仿、二氯甲烷;
优选于乙腈、二氯甲烷、四氢呋喃;
具体步骤如下:
以氯乙酰氯为原料,在缚酸剂的作用下与一定量的甲氧基甲基胺盐酸盐于有机溶剂中0-100oC下反应1-24 h,得到化合物3;

其中,反应温度优选0-25oC,反应时间优选6-10 h;
另外,步骤 (1) 中,可在TLC检测反应完毕后,过滤,水洗,干燥,浓缩得化合物3。
(2) 化合物4的制备
上述合成得到的中间体3和一定量的亚磷酸三乙酯于50-120 oC下反应10-30 h,得到化合物4。

其中上述磷酸三乙酯的用量为1-5 eq.,优选1-1.5 eq.;
其中反应温度优选80-110 oC,反应时间优选 10-20 h;
另外,步骤 (2) 中,可在TLC检测反应完毕后,减压蒸除副产物,得中间体4。
(3) 化合物6的制备
以简单易得的香兰素5 为原料,在缚酸剂的作用下与溴苄在有机溶剂中经烃化反应得到化合物6。
其中上述缚酸剂可独立选自:氢氧化钠、氢氧化钾、无水碳酸钾、无水碳酸钠、碳酸氢钠、碳酸氢钾、三乙胺、咪唑、1,8-二氮双环[5.4.0]十一-7-烯、吡啶、氢化钠、叔丁醇钾、碳酸铯;
优选于无水碳酸钾、无水碳酸钠、吡啶;
其中有机溶剂可独立选自:甲醇、乙腈、四氢呋喃、丙酮、乙酸乙酯、氯仿、二氯甲烷;
优选于丙酮、四氢呋喃、乙腈;
具体步骤如下:
以香兰素为原料,在缚酸剂的作用下和一定量的溴苄在有机溶剂中于0-100 oC下反应1-24 h,重结晶得到化合物6。

其中溴苄的用量为1-5 eq.,优选1-1.2 eq.;
其中反应温度优选50-100 oC,反应时间优选6-12 h;
其中重结晶溶剂可独立选自甲醇、乙醇、乙酸乙酯、丙酮、叔丁醇、石油醚或它们之间任意两种溶剂的混合溶剂,优选甲醇、乙醇、丙酮;
另外,步骤 (3) 中,可在TLC检测反应完毕后抽滤,减压回收溶剂,水洗,浓缩,重结晶,得中间体6。
(4) 化合物7的制备
以上述制备得到的化合物6为原料,在碱性条件下和上述制备得到的中间体4经Witing-Horner反应得到化合物7。
其中上述所用的碱可独立选自:叔丁醇钾、氢化钠、甲醇钠、乙醇钠、碳酸铯、二(三甲基硅基)氨基钠、二(三甲基硅基)氨基钾、1,8-二氮双环[5.4.0]十一-7-烯;
优选于叔丁醇钾、氢化钠、二(三甲基硅基)氨基钠;
具体步骤如下:
将中间体4溶于一定量的有机溶剂中,加入一定量的碱,加毕0-50 oC下反应1-4 h,反应毕,于-78--10 oC下加入一定浓度的化合物6的有机溶液,加毕,搅拌1-3 h后升温至25-100 oC继续反应4-20 h,得到化合物7。

其中化合物4和碱的反应温度优选0-25 oC,反应时间优选1-2 h;
其中加入化合物6时的反应温度优选控制在-78--40 oC,加料后的反应温度优选25-40oC,反应时间优选4-10 h。
另外,步骤 (4) 中,可在TLC检测反应完毕后,减压回收溶剂,水洗,干燥,浓缩,得化合物7。
(5) 化合物8的制备
以上述制备得到的化合物7为原料,经Pd/C催化的氢气还原,制备得化合物8。
其中上述有机溶剂可独立选自:甲醇、乙醇、二氯甲烷、乙酸乙酯、乙醚;
具体步骤如下:
以上述化合物7为原料,在有机溶剂中以Pd/C为催化剂,常压下经氢气还原1-10 h,制备得到化合物8。

其中反应时间优选2-4h;
另外,步骤 (5) 中,可在TLC检测反应完毕后,抽滤,水洗,浓缩,得化合物8。
(6) 姜酮酚1的制备
以上述制备得到的化合物8为原料,与庚基格式试剂经Grignard 反应于有机溶剂中室温反应1-10 h,制备得到目标产物姜酮酚1。
其中上述有机溶剂可独立选自四氢呋喃、乙醚、甲苯或它们任意两个的混合溶剂;
其中上述卤代庚烷可独立选自:溴代正庚烷、氯代正庚烷、碘代正庚烷;
具体步骤如下:
以卤代庚烷为原料,在有机溶剂中和镁条加热制备得到庚基格式试剂,再在0-50 oC下向所得格式试剂中分批加入化合物7的四氢呋喃溶液,加毕, 0-80 oC下反应1-10 h,经重结晶制备得到目标产物姜酮酚 1。

其中反应温度优选40-80 oC,反应时间优选2-4 h;
其中重结晶溶剂可独立选自甲醇、乙醇、异丙醇、丙酮、正己烷、环己烷、石油醚、乙酸乙酯或任意两种溶剂的混合溶剂;优选正己烷、环己烷、石油醚、乙醇;
另外,步骤 (6) 中,可在TLC检测反应完毕后减压回收溶剂,水洗,干燥,浓缩,经石油醚重结晶得终产品姜酮酚。
本发明采用简单易得的香兰素为起始原料,依次经烷基化反应、Witing-horner反应、Pd/H2还原反应以及格式反应制备得到粗品姜酮酚,再经过石油醚重结晶得到高纯度姜酮酚。
本发明采用操作简单且原料廉价的Witing-horner反应来进行香兰素和烷基侧链的连接,从而制备得到纯度较高的化合物7,该方法克服了常规Adol缩合反应中副产物较多,需要柱层析的缺点,同时也避免了Heck反应中的绝对无氧条件,使大规模生产的可操作性大为提高。除此之外,本发明在制备化合物8时利用weineb酰胺结构较为稳定的特点,在Pd/H2还原双键时最大程度减少了不饱和羰基因过度还原的而产生的副产物,因而产品纯度和收率大幅度提高,使大规模生产高纯度姜酮酚成为可能。
其中,在步骤 (1)、步骤 (3) 和步骤 (5) 中,反应易控,溶剂可以回收利用,反应收率较高;在步骤 (2) 中,无溶剂反应,收率高,绿色环保,后处理操作简单;在步骤 (4)中,操作简单,产品纯度较高,后处理操作简单(反应中产生的磷酸配体可以通过水洗除去,无需经过柱层析纯化),有利于后续产品的纯化和成本降低;在步骤 (6) 中,通过格式试剂和weineb酰胺反应制备得到姜酮酚粗品,再通过石油醚重结晶,因此无需柱层析进行纯化,所得姜酮酚外观好、纯度高适合工业化生产。
具体实施方式
以下实施例中所涉及的试剂未特别说明,则为商业化产品,纯度为分析级。为了更加清楚的解释本发明所解决的技术问题及技术方案,以下所述的具体实施例对本发明进行了进一步详细说明。此处所述的具体实施列仅用于解释本发明,并不用于限定本发明。
实施例1
化合物3的制备

向500 mL 单口瓶中依次加入甲氧基甲基胺盐酸盐(20.0 g, 211 mmol, 1.0 eq.)、二氯甲烷 (100 mL)、氯乙酰氯水溶液(ca. 2.1 M H2O solution, 100 mL, 211 mmol, 1.0eq.),冰浴下冷却至0ºС,分批加入无水碳酸钾 (138.21 g, 253 mmol, 1.2 eq.)。加毕,升至室温继续反应 12 h,TLC检测,反应毕,将反应液倒入200 mL水中,水相用二氯甲烷(50 mL×3) 萃取。合并有机相,无水硫酸钠干燥,过滤收集滤液,旋干得无色透明油状物32.3 g,收率 93%。1H NMR (400 MHz, CDCl3) δ 4.25 (s, 2H, CH2), 3.76 (s, 3H,NCH3), 3.24 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ 167.62 (C=O), 61.77,40.81, 32.58; HRMS (ESI): calcd. for C4H8ClNO2 [M+H]+, 138.0244; found138.0318。
化合物4 的制备

向500 mL单口瓶中依次加入化合物3 (31.9 g, 230 mmol, 1.0 e.q)和亚磷酸三乙酯(38.6 g, 230 mmol, 1.0 eq.),加毕,升温至100 ºС 继续搅拌18 h,TLC检测,反应毕,减压蒸除多余的亚磷酸三乙酯,得浅黄色油状物 47.7g,收率 86%。1H NMR (400 MHz,CDCl3) δ 4.06 (m, 4H, CH2×2 of CH3CH2O), 3.66 (s, 3H, NCH3), 3.09 (s, 3H,OCH3), 3.04 (t, J = 21.9, 2H, CH2P=O), 1.21 (t, J = 7.2 Hz, 6H, CH3×2 ofCH3CH2O); 13C NMR (100 MHz, CDCl3) δ 166.13 (C=O), 62.52, 61.42, 32.08,30.69, 16.32, 16.29; HRMS (ESI): calcd. for C8H18NO5P [M+H]+, 240.0923; found240.0992。
化合物6的制备

向500 mL单口瓶中依次加入化合物5 (20.0 g, 131 mmol, 1.0 eq.)、乙腈 (200mL)、溴苄 (16.4 mL, 131 mmol, 1.0 eq.) 以及无水碳酸钾 (27.2 g, 197 mmol, 1.5eq.)。加毕,升温至100 ºС,继续搅拌8 h,TLC检测,反应毕,将上述反应液过滤,收集滤液。所得滤液经减压蒸馏回收溶剂得黄色油状残留物,将所得残留物溶于200 mL乙酸乙酯中,依次用水 (100 mL×3)、饱和氯化钠 (100 mL) 洗涤,所得有机相依次经无水硫酸钠干燥、过滤、浓缩得粗品,再经过无水乙醇 (W/V = 1/2) 重结晶得白色结晶状固体30.1 g,收率94%。1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H, CHO), 7.54 (dd, J = 1.8, 8.2 Hz,1H, Ar-H), 7.48 (m, 2H, Ar-H), 7.40 (m, 4H, Ar-H), 7.27 (d, J = 8.2 Hz, 1H,Ar-H), 5.22 (s, 2H, CH2), 3.33 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ191.90 (C=O), 153.69, 149.93, 136.84, 130.34, 129.03, 128.62, 128.50, 126.41,113.16, 110.27, 70.54, 56.10; HRMS (ESI): calcd. for C15H14O3 [M+H]+,243.0943; found 243.1012。
化合物7的制备

向100 mL单口瓶中分别加入化合物4 (2.0 g, 8.36 mmol, 1.2 eq.)、干燥四氢呋喃(30 mL)。将上述反应液冰浴下冷却至0 ºС,分批加入60% NaH (0.33 g, 8.36 mmol, 1.2eq.),加毕,冷却至-78 ºС,缓慢滴加化合物6的四氢呋喃溶液 (ca. 0.5 M THFsolution, 14 mL, 6.96 mmol, 1.0 eq.),加毕,-78 ºС下搅拌1 h后升至室温继续搅拌12 h,TLC检测,反应毕,加入水 (5 mL) 淬灭。将所得反应液倒入100 mL水中,水相用乙酸乙酯 (50 mL×3) 萃取,合并有机相,有机相依次用饱和碳酸氢钠水溶液 (30 mL)、水 (30mL×3)、饱和氯化钠 (30 mL) 洗涤,再分别经无水硫酸钠干燥、过滤、旋干得白色固体粉末2.28 g,收率 99%。1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 15.7 Hz, 1H, Ar-H),7.37 (m, 5H, Ar-H), 7.09 (m, 2H, Ar-H), 6.88 (m, 2H, CH=CH), 5.19 (s, 2H,CH2), 3.93 (s, 3H, NCH3), 3.76 (s, 3H, OCH3), 3.31 (s, 3H, OCH3); HRMS (ESI):calcd. for C19H21NO4 [M+H]+, 328.1471; found 328.1541。
化合物8的制备

向100 mL单口瓶中依次加入化合物7 (2.0 g, 6.1 mmol)、Pd/C (0.1 g)、甲醇(20mL)。加毕,室温常压下于氢气中搅拌3 h,TLC检测,反应毕,抽滤,收集滤液,旋干得浅黄色油状物 1.4 g, 收率 98%。1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 7.9 Hz, 1H, Ar-H), 6.72 (m, 2H, Ar-H), 5.73 (br, 1H, OH), 3.86 (s, 3H, NCH3), 3.60 (s, 3H,OCH3), 3.18 (s, 3H, OCH3), 2.89 (t, J = 7.4 Hz, 2H, CH2), 2.71 (t, J = 7.4Hz, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 207.53 (C=O), 146.55, 144.05,133.30, 120.93, 114.45, 111.33, 110.00, 61.32, 55.92, 34.19, 30.51; HRMS(ESI): calcd. for C12H17NO4 [M+H]+, 240.1158; found 240.1217。
姜酮酚 1的制备

向100 mL三口瓶中依次加入1-溴庚烷 (1.25 g, 7.0 mmol, 1.0 e.q)、镁条(0.18 g,7.6 mmol, 1.1 eq.)、干燥四氢呋喃(20 mL),加毕,氩气保护下40 ºС下反应1h,反应毕,将反应液降至0ºС,缓慢加入化合物8的四氢呋喃溶液(ca. 0.5 M THF solution, 14 mL,7.0 mmol, 1.0 eq.),加毕,升至室温继续反应1h,TLC检测,反应毕,反应液用饱和氯化铵水溶液(5 mL)淬灭,再用1 M 稀盐酸溶液调pH至1。将所得溶液倒入水 (40 mL) 中,水相用乙酸乙酯 (30 mL×3) 萃取,合并有机相,所得有机相依次经无水硫酸钠干燥、过滤、旋干,得黄色油状物粗品,所得粗品经石油醚重结晶得灰白色蜡状固体1.5 g, 收率79 %。1H NMR(400 MHz, CDCl3) δ 6.81 (d, J = 8.0 Hz, 1H, Ar-H), 6.66 (m, 2H, Ar-H), 5.54(br, 1H, OH), 3.86 (s, 3H, OCH3), 2.82 (t, J = 7.4 Hz, 2H, CH2), 2.68 (t, J =7.6 Hz, 2H, CH2), 2.36 (t, J = 7.4 Hz, 2H, CH2), 1.54 (m, 2H, CH2), 1.25 (m,8H, CH2×4), 0.87 (t, J = 6.7 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 210.75(C=O), 146.51, 143.99, 133.15, 120.83, 114.42, 111.15, 55.92, 44.68, 43.21,31.74, 29.62, 29.25, 29.13, 23.88, 22.67, 14.14; HRMS (ESI): calcd. forC17H26O3 [M+H]+, 279.1182; found 279.1921。
实施例2
化合物3的制备

向10 L 反应釜中依次加入甲氧基甲基胺盐酸盐(1.0 kg, 10.2 mol, 1.0 eq.)、二氯甲烷 (4 L)、氯乙酰氯水溶液(ca. 3 M H2O solution, 3.4 L, 10.2 mol, 1.0 eq.),冰浴下冷却至 -5 ℃,分批加入无水碳酸钾 (1.7 kg, 12.2 mol, 1.2 eq.)。加毕,升至室温继续反应 12 h,TLC检测,反应毕,将反应液倒入水(5 L)中,水相用二氯甲烷 (2 L×3) 萃取。合并有机相,无水硫酸钠干燥,过滤收集滤液,旋干得无色透明油状物 1.3 kg,收率94%。HRMS (ESI): calcd. for C4H8ClNO2 [M+H]+, 138.0244; found 138.0278。
化合物4 的制备

向10 L反应釜中依次加入化合物3 (1.1 kg, 8.0 mol, 1.0 eq.)和亚磷酸三乙酯(1.3 kg, 8.0 mol, 1.0 eq.),加毕,升温至110 ℃ 继续搅拌16 h,TLC检测,反应毕,减压蒸除多余的亚磷酸三乙酯,得浅黄色油状物 1.6 kg,收率 88%。HRMS (ESI): calcd. forC8H18NO5P [M+H]+, 240.0923; found 240.1002。
化合物6的制备

向5 L反应釜中依次加入化合物5 (1.0 kg, 6.6 mol, 1.0 eq.)、乙腈 (3 L)、溴苄(1.1 kg, 6.6 mol, 1.0 eq.) 以及无水碳酸钾 (1.4 kg, 9.9 mol, 1.5 eq.)。加毕,升温至100 ºС,继续搅拌8 h,TLC检测,反应毕,将上述反应液过滤,收集滤液。所得滤液经减压蒸馏回收溶剂得黄色油状残留物,将所得残留物溶于乙酸乙酯 (3 L) 中,依次用水 (2L×3)、饱和氯化钠 (1 L) 洗涤,所得有机相依次经无水硫酸钠干燥、过滤、浓缩得粗品,再经过无水乙醇 (W/V = 1/2) 重结晶得白色结晶状固体1.5 kg,收率 94%。HRMS (ESI):calcd. for C15H14O3 [M+H]+, 243.0943; found 243.1012。
化合物7的制备

向10 L反应釜中分别加入化合物4 (2.0 kg, 8.36 mol, 1.2 eq.)、干燥四氢呋喃 (3L)。将上述反应液冰浴下冷却至 -10 ºС,分批加入60% NaH (330 g, 8.36 mol, 1.2eq.),加毕,再冷却至 -78 ºС,缓慢滴加化合物6的四氢呋喃溶液(ca. 2 M THFsolution, 3.5 L, 6.96 mol, 1.0 eq.),加毕,-78 ºС下搅拌1 h后升至室温继续搅拌10h,TLC检测,反应毕,加入水 (300 mL) 淬灭。减压回收溶剂,将所得残留液倒入水 (3 L)中,水相用乙酸乙酯 (2 L×3) 萃取,合并有机相,有机相依次用饱和碳酸氢钠水溶液 (2L)、水 (2 L×3)、饱和氯化钠 (2 L)洗涤,再分别经无水硫酸钠干燥、过滤、旋干得白色固体粉末 2.7 kg,收率 98%。HRMS (ESI): calcd. for C19H21NO4 [M+H]+, 328.1471;found 328.1521。
化合物8的制备

向10 L氢化釜中依次加入化合物7 (2.0 kg, 6.1 mol)、Pd/C (10 g)、甲醇 (4 L)。加毕,室温常压下于氢气中搅拌5 h,TLC检测,反应毕,抽滤,收集滤液,旋干得浅黄色油状物1.4 kg, 收率 98%。HRMS (ESI): calcd. for C12H17NO4 [M+H]+, 240.1158; found240.1217。
姜酮酚1的制备

向10 L反应釜中依次加入1-溴庚烷 (770 g, 4.3 mol, 1.0 eq.)、镁条 (113 g,4.7mol, 1.1 eq.)、干燥四氢呋喃 (3 L),加毕,氩气保护下40 ºС下反应1h,反应毕,将反应液降至0 ºС,缓慢加入化合物8的四氢呋喃溶液 (ca. 1 M THF solution, 4.3 L, 4.3mol, 1.0 eq.),加毕,升至室温继续反应1h,TLC检测,反应毕,反应液用饱和氯化铵水溶液(300 mL) 淬灭,再用1 M 稀盐酸溶液调pH至1,减压回收溶剂,将所得残留液倒入水 (3 L)中,水相用乙酸乙酯 (2 L×3) 萃取,合并有机相,所得有机相依次经无水硫酸钠干燥、过滤、旋干,得黄色油状物粗品,所得粗品经石油醚 (W/V = 1/1.5) 重结晶得灰白色蜡状固体969.6 g, 收率81%。HRMS (ESI): calcd. for C17H26O3 [M+H]+, 279.1182; found279.1421。
随机抽取本发明合成得到的姜酮酚样品进行HPLC纯度检测。
检测条件:
仪器:Agilent 1100 series
色谱柱:YMC-C18 4.6×250 mm, 5μm
柱温:25 ºС
流速:1.0 mL/min
检测波长:210 nm
进样体积:5 μL
流动相:乙腈:0.1% CF3COOH水溶液 = 60:40 (v/v)
运行时间:30 min
检测结果:
本发明合成所得姜酮酚的液相分析结果如表1所示。
表1:本发明合成所得姜酮酚样品HPLC检测结果。

从表1可以看出本发明合成所得姜酮酚样品纯度较高,达到99.35%。
以上所述仅为本发明优选实施例,并不用于限制本发明,凡在本发明的原则和精神之内所作的任何改动,等同替换和改进等,均包含在本发明的保护范围之内。
Claims (6)
1.一种姜酮酚的合成方法,其特征在于,包括步骤:
(1)化合物3的制备:

以氯乙酰氯为原料和甲氧基甲基胺盐酸盐在缚酸剂的作用下于有机溶剂中缩合得到具有酰胺结构的化合物3;
其中上述缚酸剂可独立选自:氢氧化钠、氢氧化钾、无水碳酸钾、无水碳酸钠、碳酸氢钠、碳酸氢钾、三乙胺、咪唑、1,8-二氮杂双环[5.4.0]十一碳-7-烯、吡啶、氢化钠、叔丁醇钾、碳酸铯,优选无水碳酸钾、无水碳酸钠、吡啶;
其中有机溶剂可独立选自:甲醇、乙腈、四氢呋喃、丙酮、乙酸乙酯、氯仿、二氯甲烷;优选于乙腈、二氯甲烷、四氢呋喃;
(2)化合物4的制备:

上述合成得到的中间体3和一定量的亚磷酸三乙酯于50-120 oC下反应10-30 h,得到化合物4;
其中上述磷酸三乙酯的用量为1-5 eq.,优选1-1.5 eq.;
其中反应温度优选80-110 oC,反应时间优选 10-20 h;
(3)化合物6的制备:

以简单易得的香兰素5为原料,在缚酸剂的作用下与溴苄在有机溶剂中经烃化反应得到化合物6;
其中上述缚酸剂可独立选自:氢氧化钠、氢氧化钾、无水碳酸钾、无水碳酸钠、碳酸氢钠、碳酸氢钾、三乙胺、咪唑、1,8-二氮杂双环[5.4.0]十一碳-7-烯、吡啶、氢化钠、叔丁醇钾、碳酸铯;优选于无水碳酸钾、无水碳酸钠、吡啶;
其中有机溶剂可独立选自:甲醇、乙腈、四氢呋喃、丙酮、乙酸乙酯、氯仿、二氯甲烷;优选于丙酮、四氢呋喃、乙腈;
(4)化合物7的制备:
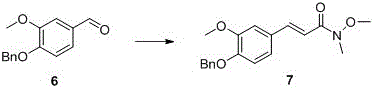
以上述制备得到的化合物6为原料,在碱性条件下和上述制备得到的中间体4经Witing-Horner反应得到化合物7;
其中上述所用的碱可独立选自:叔丁醇钾、氢化钠、甲醇钠、乙醇钠、碳酸铯、二(三甲基硅基)氨基钠、二(三甲基硅基)氨基钾、1,8-二氮杂双环[5.4.0]十一碳-7-烯;优选于叔丁醇钾、氢化钠、二(三甲基硅基)氨基钠;
(5)化合物8的制备:

以上述制备得到的化合物7为原料,经Pd/C催化的氢气还原,制备得到中间体8;
其中上述有机溶剂可独立选自:甲醇、乙醇、二氯甲烷、乙酸乙酯、乙醚;
(6)姜酮酚1的制备:

以上述制备得到的化合物8为原料,与庚基格式试剂经Grignard 反应于有机溶剂中室温反应1-10 h,制备得到目标产物姜酮酚1;
其中上述有机溶剂可独立选自四氢呋喃、乙醚、甲苯或它们任意两个的混合溶剂;
其中上述卤代庚烷可独立选自:溴代正庚烷、氯代正庚烷、碘代正庚烷。
2.如权利要求1所述的一种姜酮酚的合成方法,其特征在于:
所述步骤(1)中,以氯乙酰氯为原料,在缚酸剂的作用下与一定量的甲氧基甲基胺盐酸盐于有机溶剂中0-100oC下反应1-24 h,得到化合物3;
其中,反应温度优选0-25oC,反应时间优选6-10 h;
步骤(3)中,以香兰素为原料,在缚酸剂的作用下和一定量的溴苄在有机溶剂中于0-100 oC下反应1-24 h,重结晶得到化合物6;
其中溴苄的用量为1-5 eq.,优选1-1.2 eq;
其中反应温度优选50-100 oC,反应时间优选6-12 h;
其中重结晶溶剂可独立选自甲醇、乙醇、乙酸乙酯、丙酮、叔丁醇、石油醚或它们之间任意两种溶剂的混合溶剂,优选甲醇、乙醇、丙酮;
步骤(4)中,将中间体4溶于一定量的有机溶剂中,加入一定量的碱,加毕0-50 oC下反应1-4 h,反应毕,于-78--10 oC下加入一定浓度的化合物6的有机溶液,加毕,搅拌1-3 h后升温至25-100 oC继续反应4-20 h,得到化合物7;
其中中间体4和碱的反应温度优选0-25 oC,反应时间优选1-2 h;
其中加入化合物6时的反应温度优选控制在-78--40 oC,加料后的反应温度优选25-40oC,反应时间优选4-10 h;
步骤(5)中,以上述化合物7为原料,在有机溶剂中以Pd/C为催化剂,常压下经氢气还原1-10 h,制备得到中间体8;
其中反应时间优选2-4h;
步骤(6)中,以卤代庚烷为原料,在有机溶剂中和镁条加热制备得到庚基格式试剂,再在0-50 oC下向所得格式试剂中分批加入化合物7的四氢呋喃溶液,加毕,0-80oC下反应1-10 h,经重结晶制备得到目标产物姜醇酚1;
其中反应温度优选40-80 oC,反应时间优选2-4 h;
其中重结晶溶剂可独立选自甲醇、乙醇、异丙醇、丙酮、正己烷、环己烷、石油醚、乙酸乙酯或任意两种溶剂的混合溶剂;优选正己烷、环己烷、石油醚、乙醇。
3.如权利要求1所述的一种姜酮酚的合成方法,其特征在于:
所述步骤(1)中,以薄层层析检测反应完毕后,过滤,水洗,干燥,浓缩得化合物3;
步骤(2)中,以薄层层析检测反应完毕后,减压蒸除未反应的原料及副产物,得化合物4;
步骤(3)中,以薄层层析检测反应完毕后,减压回收乙腈,所得油状物用非水溶性有机溶剂溶解,水洗,干燥,抽滤,浓缩,重结晶,得化合物6;其中非水溶性有机溶剂包括:乙酸乙酯、二氯甲烷、甲基叔丁醚、氯仿、乙醚中的一种或多种;
步骤(4)中,以薄层层析检测反应完毕后,减压回收四氢呋喃,所得油状物用非水溶性有机溶剂溶解,水洗,干燥,抽滤,浓缩,重结晶,得中间体7;其中非水溶性有机溶剂包括:乙酸乙酯、二氯甲烷、甲基叔丁醚、氯仿、乙醚中的一种或多种;
步骤(5)中,以薄层层析检测反应完毕后,抽滤,水洗,干燥,过滤,浓缩,得中间体8;
步骤(6)中,以薄层层析检测反应完毕后,减压回收乙腈,所得油状物用非水溶性有机溶剂溶解,水洗,干燥,抽滤,浓缩,重结晶,得姜酮酚;其中非水溶性有机溶剂包括:乙酸乙酯、二氯甲烷、甲基叔丁醚、氯仿、乙醚中的一种或多种。
4.如权利要求1所述的一种姜酮酚的合成方法,其特征在于:所述步骤(1)中,水和非水溶性有机溶剂的混合体积比例为1:1;
无机碱为无水碳酸钾。
5.如权利要求1所述的一种姜酮酚的合成方法,其特征在于:所述步骤(3)中,重结晶溶剂为无水乙醇,用量为粗品/无水乙醇(W/V = 1/2)。
6.如权利要求1所述的一种姜酮酚的合成方法,其特征在于:所述步骤(6)中,重结晶溶剂为石油醚,用量为粗品/石油醚(W/V = 1/1.5)。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911202624.0A CN110937985B (zh) | 2019-11-29 | 2019-11-29 | 一种姜酮酚的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911202624.0A CN110937985B (zh) | 2019-11-29 | 2019-11-29 | 一种姜酮酚的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN110937985A true CN110937985A (zh) | 2020-03-31 |
| CN110937985B CN110937985B (zh) | 2022-10-14 |
Family
ID=69909439
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201911202624.0A Active CN110937985B (zh) | 2019-11-29 | 2019-11-29 | 一种姜酮酚的合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN110937985B (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112457192A (zh) * | 2020-11-03 | 2021-03-09 | 桂林理工大学 | 一种姜酮酚的合成方法 |
| CN113264850A (zh) * | 2021-05-27 | 2021-08-17 | 安徽农业大学 | 一种益智酮甲的合成方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103553889A (zh) * | 2013-10-31 | 2014-02-05 | 苏州永健生物医药有限公司 | 一种姜酮酚的合成方法 |
| CN106866393A (zh) * | 2016-12-29 | 2017-06-20 | 陕西嘉禾药业有限公司 | 一种姜酮酚的制备方法 |
-
2019
- 2019-11-29 CN CN201911202624.0A patent/CN110937985B/zh active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103553889A (zh) * | 2013-10-31 | 2014-02-05 | 苏州永健生物医药有限公司 | 一种姜酮酚的合成方法 |
| CN106866393A (zh) * | 2016-12-29 | 2017-06-20 | 陕西嘉禾药业有限公司 | 一种姜酮酚的制备方法 |
Non-Patent Citations (3)
| Title |
|---|
| CARINA GLAS等: "General Method for the Preparation of Indole-2-Weinreb Amides", 《SYNTHESIS》 * |
| XIAOYU LIU等: "Design, synthesis and biological evaluation of substituted (+)-SG-1 derivatives as novel anti-HIV agents", 《BIOORGANIC & MEDICINAL CHEMISTRY LETTERS》 * |
| 杨礼寿等: "一种酚羟基苄基保护基的脱除方法", 《化学试剂》 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112457192A (zh) * | 2020-11-03 | 2021-03-09 | 桂林理工大学 | 一种姜酮酚的合成方法 |
| CN112457192B (zh) * | 2020-11-03 | 2023-03-14 | 桂林理工大学 | 一种姜酮酚的合成方法 |
| CN113264850A (zh) * | 2021-05-27 | 2021-08-17 | 安徽农业大学 | 一种益智酮甲的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110937985B (zh) | 2022-10-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3260441B1 (en) | Method for preparing formamide compound | |
| KR20240135777A (ko) | 고순도 식물 유래 콜레스테롤의 합성방법 | |
| CN110937985B (zh) | 一种姜酮酚的合成方法 | |
| Lalic et al. | Enantioselective rhodium (I)-triethylamine catalyzed addition of potassium isopropenyl trifluoroborate to enones | |
| KR20240135778A (ko) | 고순도 콜레스테롤의 합성방법 | |
| Pietruszka et al. | Diastereo‐and Enantiomerically Pure Allylboronates: Their Synthesis and Scope | |
| CN101560191B (zh) | α-萘甲基取代的螺环双噁唑啉配体、合成方法及其在合成吡唑烷衍生物中的应用 | |
| CN110615811B (zh) | 一种手性亚磺酰胺单膦配体的大量制备方法 | |
| CN109776295B (zh) | 一种邻位含二氟亚甲基的芳基碘化合物及制备方法 | |
| CN109535120B (zh) | 7-取代-3,4,4,7-四氢环丁烷并香豆素-5-酮的制备方法 | |
| CN106966889A (zh) | 一种(E)‑β,γ‑烯基羧酸衍生物及其制备方法 | |
| CN101863954A (zh) | 一种N-叔丁基-4-氮杂-5α-雄甾-3-酮-17β-甲酰胺的制备方法 | |
| CN112250567B (zh) | 一种AMG837及手性γ-甲基苯戊醇的合成方法 | |
| Wen et al. | Perfectly green organocatalysis: quaternary ammonium base triggered cyanosilylation of aldehydes | |
| CN109265385B (zh) | 一种手性催化剂的合成工艺 | |
| CN110437277B (zh) | 一种磷酸烯基酯类化合物的合成方法 | |
| CN113264850A (zh) | 一种益智酮甲的合成方法 | |
| CN108727179B (zh) | 一种α-烯丙基取代的α,β-不饱和酮、酯或腈化合物的合成方法 | |
| CN109705014B (zh) | 一种新型手性氧化胺配体及其制备方法 | |
| CN108948055B (zh) | 一种8-甲基喹啉偕二硼化合物及其制备方法 | |
| CN104230880A (zh) | 2-((4r, 6r)-6-氨乙基-2,2-二甲基-1,3-二氧六环-4-基)乙酸酯的简便制备方法 | |
| EP2876108B1 (en) | Compounds of chiral aromatic spiroketal diphosphine ligands, preparation methods and uses thereof | |
| CN106748725B (zh) | 一种4-氯-2-氟-苯丙酸的制备方法 | |
| CN110627718B (zh) | 一种(E)-β-单氟烷基-β,γ-不饱和酰胺的合成方法 | |
| US20060281949A1 (en) | Method for production $g(a),$g(b)-unsaturated amide compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |