CN109897029B - 三联吡啶类衍生物及其制备方法、应用和器件 - Google Patents
三联吡啶类衍生物及其制备方法、应用和器件 Download PDFInfo
- Publication number
- CN109897029B CN109897029B CN201711293130.9A CN201711293130A CN109897029B CN 109897029 B CN109897029 B CN 109897029B CN 201711293130 A CN201711293130 A CN 201711293130A CN 109897029 B CN109897029 B CN 109897029B
- Authority
- CN
- China
- Prior art keywords
- terpyridyl
- carbazole
- added
- layer
- terpyridyl derivative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



Landscapes
- Indole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Electroluminescent Light Sources (AREA)
Abstract
本发明属于有机光电材料应用科技领域,具体涉及三联吡啶类衍生物及其制备方法、应用和器件。发明所提供的主要基于三联吡啶核心,外围采用咔唑类衍生物等来调控适当的给电子能力,通过D‑A结构设计变换不同的桥联基团,调控合适的最低占据轨道(HOMO)和最高非占据轨道(LUMO)的分布,主要应用于TADF客体材料,主体材料,以及载流子传输材料。
Description
技术领域
本发明属于有机光电材料应用科技领域,具体涉及三联吡啶类衍生物及其制备方法、应用和器件。
背景技术
有机发光二极管(OLED)具有轻薄,自发光,高对比度,易于大面积制造,可用于柔性和透明显示与照明领域等优点,被誉为“梦幻显示”和下一代固态照明技术。1963年,Pope教授以数百伏的偏压施加于蒽的晶体上发出微弱的蓝光,但是由于过高的电压和不佳的发光效率,并未引起人们重视。自从1987年,邓青云博士(C.W.Tang)以及Steve VanSlyke以真空蒸镀的方法制成多层的OLED器件,极大的降低了器件的驱动电压提高了发光效率,才使得OLED在照明和显示领域引起了广泛的关注。OLED的发展,大致经历了荧光→磷光→热致延迟荧光的三个发展阶段。第一代的纯荧光,第二代的磷光的材料发展均趋于完善,也是目前产业上使用较多的材料。为实现红绿蓝的三原色,产业上主要是采用蓝光荧光材料;绿光、红光的磷光材料。然而蓝光荧光在电致条件理论内量子效率只有25%,另有光波导影响,耦合出光常数在20%左右,故其理论最大外量子产率在5%左右。第二代,磷光蓝光材料可以通过外重原子效应,实现理论100%的内量子产率,但是由于含有贵金属会提高成本,且在器件电致发光过程中,重金属的发光材料易发生降解,导致器件稳定性很差。对于第三代的热致延迟荧光材料(TADF),是一种有潜力实现高效率的材料,但是其效率滚降问题非常严重,在蓝光上表现尤为明显。因此,对于OLED的蓝光材料,高效率和效率滚降问题都亟待解决。
对于实现蓝光TADF的D-A结构设计中,吸电子基团A与供电子基D的合理搭配以及D、A之间的桥联基团,分子实现弱的分子内电荷转移态,是实现蓝光的重要途径。三嗪,二苯甲酮,二苯硫砜,磷氧等,由于其强的吸电子能力,搭配弱电子基团,实现的蓝光大多属于天蓝光,而深蓝光高效发光材料是紧缺的。三联吡啶在金属配合物中应用广泛,但是由于其本身的量子产率比较低,在有机发光小分子中的应用有限。
发明内容
本发明所提供的主要基于三联吡啶核心,外围采用咔唑类衍生物等来调控适当的吸电子能力,通过D-A结构设计变换不同的桥联基团,调控合适的最低占据轨道(HOMO)和最高非占据轨道(LUMO)的分布,主要应用于TADF客体材料,主体材料,以及载流子传输材料。其目的在于通过采用简单可行的合成方案合成三联吡啶类衍生物并将其应用于电致发光材料,由此解决现有技术的蓝光材料稳定性差、效率低的技术问题。
本发明所提供的技术方案如下:
三联吡啶类衍生物,其特征在于,结构通式如下:

其中,桥联基团L选自以下四种任一:

R为碳原子数为6~60的烷基、取代或者未取代的芳香杂环基或者取代或者无取代的芳香环基,所述取代或者未取代的芳香杂环基的碳原子及杂原子数之合为6~60,所述取代或者无取代的芳香环基的碳原子数为6~60。
优选的,R为咔唑类衍生基团。
优选的,咔唑类衍生基团的通式为:A交B,其中,A为咔唑基,B为氮芴基、氧芴基、硫芴基、1、1-二甲基芴基、1、1-二苯基芴基或螺芴基,A与B交于苯环。
具体的,咔唑类衍生基团选自以下任一:



具体的,三联吡啶类衍生物选自以下任一:







本发明还提供了一种三联吡啶类衍生物的制备方法,包括以下步骤:R-H与Br-L-三联吡啶偶联,得到三联吡啶类衍生物 其中:
其中:
桥联基团L选自以下四种任一:

R为碳原子数为6~60的烷基、取代或者未取代的芳香杂环基或者取代或者无取代的芳香环基,所述取代或者未取代的芳香杂环基的碳原子及杂原子数之合为6~60,所述取代或者无取代的芳香环基的碳原子数为6~60。
优选的,R为咔唑类衍生基团。
咔唑类衍生基团和得到的三联吡啶类衍生物如上所述。
本发明还提供了本发明所提供的三联吡啶类衍生物的应用,作为有机电致发光器件中的电致发光层材料,所述电致发光层材料为荧光或热致延迟荧光材料、磷光主体材料或荧光主体材料。
本发明还提供了一种电致发光器件,所述电致发光器件至少包含一对电极和设置在该对电极之间的有机发光介质所述有机发光介质中至少含有一种本发明所提供的三联吡啶类衍生物。
三联吡啶基衍生物具有较弱的吸电子能力,搭配不同的供电基团可以实现从深蓝到天蓝的光色调控。由于在OLED器件中,有机材料的空穴迁移率一般要比电子的高一到两个数量级,而三联吡啶类衍生物通常可以利用分子内氢键作用提高电子迁移率,从而实现发光区域的载流子更平衡效率滚降小。该类三联吡啶基衍生物制备简单,且热稳定性好,适合通过热蒸镀作为有机发光功能层材料。
附图说明
图1是本发明所提供的化合物13、14和15的电致发光条件下的光谱图。
图2是本发明所提供的化合物13、14和15的光致条件下的寿命衰减曲线。
图3是本发明所提供的电致器件结构图。
具体实施方式
以下对本发明的原理和特征进行描述,所举实施例只用于解释本发明,并非用于限定本发明的范围。
实施例1

(5-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-12,12-二甲基-5,12-二氢茚并[1,2-c]咔)的制备:
(1)在干燥的500ml的双口烧瓶中,将3-咔唑硼酸频哪醇酯(15g,51mmol),邻二溴苯(14.5g,61.2mmol),甲苯(120mL),乙醇(60mL)和2mol/L碳酸钾溶液(60mL)加入,先超声5-10分钟,然后快速搅拌鼓氮气5分钟,迅速加入催化剂四(三苯基膦)化钯(1.8g,1.53mmol),大量通氮气10分钟。加热至100℃,搅拌回流12h。处理时,先萃取,旋干,用石油醚和二氯甲烷柱层析可以得到白色固体产物3-(2-溴苯基)-9H-咔唑,产率93%。
(2)将3-(2-溴苯基)-9H-咔唑(20.73g,64.4mmol),二碳酸二叔丁酯(19.67g,90.13mmol)和4-二甲氨基吡啶(0.787g,6.44mmol)置于500mL的单口瓶中,加入250mL的四氢呋喃溶解,室温搅拌4小时。旋干,用乙酸乙酯:石油醚=1:50的淋洗液进行柱层析,得到白色中间产物3-(2-溴苯基)-9H-咔唑-9-羧酸叔丁酯,产率95%。
(3)将烘干的两口的250mL圆底烧瓶,磁子和恒压滴液漏斗准备好,然后再加入步骤(2)获得的3-(2-溴苯基)-9H-咔唑-9-羧酸叔丁酯(8g,19mmol),把圆底烧瓶中抽成真空状态再通氮气,立即加入重蒸的四氢呋喃(80mL),搅拌溶解后,加液氮冷却,10分钟后逐滴加入正丁基锂(8.36mL,20.9mmol),30分钟加完。加完反应1h后,逐滴加入干燥的丙酮(1.2g,20.9mmol),反应2h以后撤掉低温锅,自然升温到室温。反应12h后加少量水猝灭。旋干以除去水和THF。用100mL单口瓶旋干是粘稠状固体。加乙酸(100mL)做溶剂,盐酸(25mL)关环。加热到120℃,回流搅拌12h。反应结束后加水析出固体,抽滤。用石油醚和乙酸乙酯柱层析得到白色产物12,12-二甲基-5,12-二氢茚并[1,2-c]咔,产率63%。
(4)在50mL的单口瓶中加入按照实施例1步骤(1)~(3)合成得到的12,12-二甲基-5,12-二氢茚并[1,2-c]咔(2g,7.06mmol),4'-(4-溴苯基)-2,2':6',2”-三联吡啶(2.73g,7.06mmol),碘化亚铜(0.8g,4.2mmol),K2CO3(5.83g,42.0mmol)和18-冠-6(1.08g,4.2mmol)溶解到5mLDMPU溶液中。在N2的保护下,升温至175℃,反应36h。待反应终止后,冷却至室温,萃取,旋干,柱层析。用石油醚和二氯甲烷柱层析得到淡黄色固体产物,产率78%。13C-NMR(CDCl3,100MHz):δ(ppm)155.9,155.3,152.0,149.2,147.8,145.4,141.0,139.1,137.3,137.2,134.0,133.9,130.9,129.7,129.6,128.0,126.7,126.6,123.6,123.2,121.6,121.4,119.8,118.0,117.3,114.1,109.5,109.1,104.4,42.9,31.2.高分辨质谱:590.568.
由上述核磁共振结果和质谱可知,该淡黄色固体产品结构正确,为式13所示的化合物(5-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-12,12-二甲基-5,12-二氢茚并[1,2-c]咔)。
实施例2

(5-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-12,12-二苯基-5,12-二氢茚并[1,2-c]咔)的制备
(1)在干燥的500ml的双口烧瓶中,将3-咔唑硼酸频哪醇酯(15g,51mmol),邻二溴苯(14.5g,61.2mmol),甲苯(120mL),乙醇(60mL)和2mol/L碳酸钾溶液(60mL)加入,先超声5-10分钟,然后快速搅拌鼓氮气5分钟,迅速加入催化剂四(三苯基膦)化钯(1.8g,1.53mmol),大量通氮气10分钟。加热至100℃,搅拌回流12h。处理时,先萃取,旋干,用石油醚和二氯甲烷柱层析可以得到白色固体产物3-(2-溴苯基)-9H-咔唑,产率93%。
(2)将3-(2-溴苯基)-9H-咔唑(20.73g,64.4mmol),二碳酸二叔丁酯(19.67g,90.13mmol)和4-二甲氨基吡啶(0.787g,6.44mmol)置于500mL的单口瓶中,加入250mL的四氢呋喃溶解,室温搅拌4小时。旋干,用乙酸乙酯:石油醚=1:50的淋洗液进行柱层析,得到白色中间产物3-(2-溴苯基)-9H-咔唑-9-羧酸叔丁酯,产率95%。
(3)将烘干的两口的250mL圆底烧瓶,磁子和恒压滴液漏斗准备好,然后再加入步骤(2)获得的3-(2-溴苯基)-9H-咔唑-9-羧酸叔丁酯(8g,19mmol),把圆底烧瓶中抽成真空状态再通氮气,立即加入重蒸的四氢呋喃(80mL),搅拌溶解后,加液氮冷却,10分钟后逐滴加入正丁基锂(8.36mL,20.9mmol),30分钟加完。加完反应1h后,逐滴加入二苯甲酮(3.8g,20.9mmol),反应2h以后撤掉低温锅,自然升温到室温。反应12h后加少量水猝灭。旋干以除去水和THF。用100mL单口瓶旋干是粘稠状固体。加乙酸(100mL)做溶剂,盐酸(25mL)关环。加热到120℃,回流搅拌12h。反应结束后加水析出固体,抽滤。用石油醚和乙酸乙酯柱层析得到白色产物12,12-二苯基-5,12-二氢茚并[1,2-c]咔,产率70%。
(4)在50mL的单口瓶中加入按照实施例2步骤(1)~(3)合成得到的12,12-二苯基-5,12-二氢茚并[1,2-c]咔(2g,4.9mmol),4'-(4-溴苯基)-2,2':6',2”-三联吡啶(1.9g,4.9mmol),碘化亚铜(0.55g,2.91mmol),K2CO3(4.03g,29.1mmol)和18-冠-6(0.75g,2.91mmol)溶解到5mLDMPU溶液中。在N2的保护下,升温至175℃,反应36h。待反应终止后,冷却至室温,萃取,旋干,柱层析。用石油醚和二氯甲烷柱层析得到淡黄色固体产物,产率80%。13C-NMR(CDCl3,100MHz):δ(ppm)155.9,155.3,152.0,149.2,147.7,145.4,141.9,141.0,139.1,137.3,137.2,134.0,133.9,131.9,130.9,129.7,129.2,128.7,128.1,128.0,126.7,126.6,126.2,123.6,121.4,119.8,118.0,117.3,114.1,109.5,109.1,104.4,
60.3.高分辨质谱:714.893.
由上述核磁共振结果和质谱可知,该淡黄色固体产品结构正确,为式14所示的化合物(5-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-12,12-二甲基-5,12-二氢茚并[1,2-c]咔)。
实施例3

(5'-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-5'H-螺[芴9,12'茚并[1,2-c〕咔唑])的制备
(1)在干燥的500ml的双口烧瓶中,将3-咔唑硼酸频哪醇酯(15g,51mmol),邻二溴苯(14.5g,61.2mmol),甲苯(120mL),乙醇(60mL)和2mol/L碳酸钾溶液(60mL)加入,先超声5-10分钟,然后快速搅拌鼓氮气5分钟,迅速加入催化剂四(三苯基膦)化钯(1.8g,1.53mmol),大量通氮气10分钟。加热至100℃,搅拌回流12h。处理时,先萃取,旋干,用石油醚和二氯甲烷柱层析可以得到白色固体产物3-(2-溴苯基)-9H-咔唑,产率93%。
(2)将3-(2-溴苯基)-9H-咔唑(20.73g,64.4mmol),二碳酸二叔丁酯(19.67g,90.13mmol)和4-二甲氨基吡啶(0.787g,6.44mmol)置于500mL的单口瓶中,加入250mL的四氢呋喃溶解,室温搅拌4小时。旋干,用乙酸乙酯:石油醚=1:50的淋洗液进行柱层析,得到白色中间产物3-(2-溴苯基)-9H-咔唑-9-羧酸叔丁酯,产率95%。
(3)将烘干的两口的250mL圆底烧瓶,磁子和恒压滴液漏斗准备好,然后再加入步骤(2)获得的3-(2-溴苯基)-9H-咔唑-9-羧酸叔丁酯(8g,19mmol),把圆底烧瓶中抽成真空状态再通氮气,立即加入重蒸的四氢呋喃(80mL),搅拌溶解后,加液氮冷却,10分钟后逐滴加入正丁基锂(8.36mL,20.9mmol),30分钟加完。加完反应1h后,逐滴加入9-芴酮(3.7g,20.9mmol),反应2h以后撤掉低温锅,自然升温到室温。反应12h后加少量水猝灭。旋干以除去水和THF。用100mL单口瓶旋干是粘稠状固体。加乙酸(100mL)做溶剂,盐酸(25mL)关环。加热到120℃,回流搅拌12h。反应结束后加水析出固体,抽滤。用石油醚和乙酸乙酯柱层析得到白色产物5'H-螺芴-9,8-茚咔唑,产率75%。
(4)在50mL的单口瓶中加入按照实施例3步骤(1)~(3)合成得到的5'H-螺芴-9,8-茚咔唑(1.98g,4.9mmol),4'-(4-溴苯基)-2,2':6',2”-三联吡啶(1.89g,4.9mmol),碘化亚铜(0.55g,2.91mmol),K2CO3(4.03g,29.1mmol)和18-冠-6(0.75g,2.91mmol)溶解到5mLDMPU溶液中。在N2的保护下,升温至175℃,反应36h。待反应终止后,冷却至室温,萃取,旋干,柱层析。用石油醚和二氯甲烷柱层析得到淡黄色固体产物,产率80%。13C-NMR(CDCl3,100MHz):δ(ppm)155.9,155.3,152.0,149.2,145.4,141.9,141.0,139.1,137.3,137.2,134.0,133.9,130.9,129.7,128.7,128.1,128.0,126.7,126.6,126.2,123.6,121.4,119.8,118.0,117.3,114.1,109.5,109.1,104.4,60.3.高分辨质谱:712.725.
由上述核磁共振结果和质谱可知,该淡黄色固体产品结构正确,为式15所示的化合物(5'-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-5'H-螺[芴9,12'茚并[1,2-c〕咔唑)。
实施例4

(5-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-5H-苯并呋喃并[3,2-c]咔)的制备
(1)50ml的三口瓶中,4-溴二苯并呋喃(7.4g,30mmol,1eq),加入干燥的四氢呋喃溶液,降温至-78℃,抽换N2,三次,保证体系的无氧环境,滴加正丁基锂(2.5M,13.2ml,33mmol,1.1eq)后,保持在-78℃反应1.5h,在滴加硼酸三异丙酯(ρ=0.815g/ml,7.6ml,33mmol,1.1eq),滴加完毕后升至室温,8h后滴加稀盐酸猝灭反应,减压蒸馏出去四氢呋喃,加二氯甲烷溶解,无水硫酸镁水洗,干燥,过滤,滤液进行重结晶,得白色固体产物4-硼酸二苯并呋喃6g(产率94%)。
(2)500ml的三口瓶中,6g的4-硼酸二苯并呋喃(28mmol,1.1eq)与5.05g邻溴硝基苯(25mmol),6.9g碳酸钾(50mmol,2eq),溶解在四氢呋喃:水=3:1(体积比)共200ml,抽排N2三次,保证体系的无氧环境,在N2的保护下加入催化剂四三苯基膦化钯0.72g(0.62mmol,2.5%eq),回流状态下反应16h,反应结束后,减压蒸馏除去四氢呋喃,用二氯甲烷溶解,水洗有机相,萃取水相三次,拌样过柱,用石油醚,二氯甲烷混合溶剂过柱,得亮黄色凝胶4-(2-硝基苯基)二苯并[b,d]呋喃7.0g(产率96%)。
(3)5-氢-苯并呋喃[3,2-c]咔唑的制备:
7.0g的4-(2-硝基苯基)二苯并[b,d]呋喃(24mmol,1eq),15.7g的三苯基膦(60mmol,2.5eq)加入500ml的三口瓶中,加入220ml的邻二氯苯,抽排N2三次,保证体系的无氧环境,回流48h,体系呈棕黑色,减压蒸馏出o-DCB,二氯甲烷溶解,水洗三次有机相,无水硫酸镁干燥过滤,拌样,过柱,得灰白色固体5-氢-苯并呋喃[3,2-c]咔唑3.9g(产率63%)。
(4)在50mL的单口瓶中加入按照实施例4步骤(1)~(3)合成得到的5-氢-苯并呋喃[3,2-c]咔唑(1.26g,4.9mmol),4'-(4-溴苯基)-2,2':6',2”-三联吡啶(1.89g,4.9mmol),碘化亚铜(0.55g,2.91mmol),K2CO3(4.03g,29.1mmol)和18-冠-6(0.75g,2.91mmol)溶解到5mLDMPU溶液中。在N2的保护下,升温至175℃,反应36h。待反应终止后,冷却至室温,萃取,旋干,柱层析。用石油醚和二氯甲烷柱层析得到淡黄色固体产物5-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-5H-苯并呋喃并[3,2-c]咔,产率78%。13C-NMR(CDCl3,100MHz):δ(ppm)156.5,155.9,155.3,152.0,149.2,145.4,141.2,139.1,137.3,137.2,134.0,130.9,128.0,126.6,124.7,124.6,123.6,123.3,123.2,121.4,120.9,119.8,118.0,114.1,111.8,111.5,109.5,106.4.高分辨质谱:564.812.
由上述核磁共振结果和质谱可知,该淡黄色固体产品结构正确,为式19所示的化合物(5-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-5H-苯并呋喃并[3,2-c]咔)。
实施例5
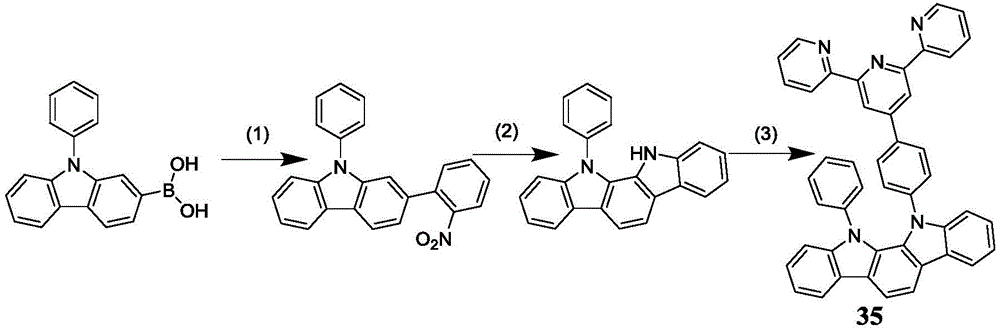
11-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-12-苯基-11,12-二氢吲哚并[2,3-a]咔唑的制备。
(1)将(9-苯基-9H-咔唑-2-基)硼酸(10g,34.84mmol),邻溴硝基苯(7.74g,38.32mmol),碳酸钾(13.8g,100mmol)和Pd(PPh3)4(0.4g,0.34mmol)加入500mL的三口瓶中,加入100mL甲苯,50mL乙醇和50mL水依次加入瓶中,排空气5min,在N2保护下搅拌加热到100度回流12小时。用二氯甲烷和石油醚进行柱层析,得到白色固体2-(2-硝基苯基)-9-苯基-9H-咔唑。产率92%。
(2)将2-(2-硝基苯基)-9-苯基-9H-咔唑(5g,14.75mmol)和三苯基膦(9.67g,36.87mmol)用150mL邻二氯苯溶解,N2保护条件下加热到180度,回流24小时。反应处理,先将邻二氯苯减压蒸馏除去,然后用二氯甲烷和石油醚进行柱层析提纯,得到白色固体11-苯基-11,12-二氢吲哚并[2,3-a]咔唑。产率55%。
(3)在50mL的单口瓶中加入按照实施例5步骤(1)~(2)合成得到的11-苯基-11,12-二氢吲哚并[2,3-a]咔唑(1.62g,4.9mmol),4'-(4-溴苯基)-2,2':6',2”-三联吡啶(1.89g,4.9mmol),碘化亚铜(0.55g,2.91mmol),K2CO3(4.03g,29.1mmol)和18-冠-6(0.75g,2.91mmol)溶解到5mLDMPU溶液中。在N2的保护下,升温至175℃,反应36h。待反应终止后,冷却至室温,萃取,旋干,柱层析。用石油醚和二氯甲烷柱层析得到淡黄色固体产物11-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-12-苯基-11,12-二氢吲哚并[2,3-a]咔唑,产率78%。13C-NMR(CDCl3,100MHz):δ(ppm)155.9,155.3,152.0,149.2,145.4,139.1,137.3,137.2,136.8,135.5,129.3,128.0,126.6,125.5,125.1,123.6,121.4,120.5,120.0,119.8,118.8,118.0,114.1,111.0,109.5,105.4.高分辨质谱:639.584.
由上述核磁共振结果和质谱可知,该淡黄色固体产品结构正确,为式35所示的化合物11-(4-([2,2':6',2”-三联吡啶]-4'-基)苯基)-12-苯基-11,12-二氢吲哚并[2,3-a]咔唑。
实施例6
将实施例1制备得到的三联吡啶类衍生物式13作为发光客体制备器件。这个实施例展示了13作为客体发光材料而制备的电致发光器件的性能验证。ITO(氧化铟锡)玻璃相继在清洗剂和去离子水中以超声波清洗30分钟。然后真空干燥2小时(105℃),再将ITO玻璃放入等离子反应器中进行5分钟的氧等离子处理,传送到真空室内制备有机膜和金属电极,接着通过真空蒸镀的方法制备一层10nm的空穴注入材料三氧化钼,接着蒸镀60nm厚的空穴传输材料4,4'-双[N-(1-萘基)-N-苯基氨基]联苯(NPB),电子阻挡层为10nm厚的1,3-二-9-咔唑基苯(mCP);然后蒸镀一层发光层30nm双[2-(二苯基膦基)苯基]醚氧化物(DPEPO):10%13;接着蒸镀一层10nm的DPEPO作为空穴阻挡层,然后蒸镀40nm厚的4,7-二苯基-1,10-菲咯啉(Bphen),最后再蒸镀一层1nm的LiF和100nm的Al。
铝作为器件的阴极,将直流电的正极加于ITO(氧化铟锡)层,将负极加于金属层,即可得到从ITO(氧化铟锡)层发出的明亮均匀的光,CIE色坐标为(0.155,0.108),启亮电压4.9V,最大外部量子效率15.4%,最大电流效率为12.4cd/A。本实验器件结构为:ITO(氧化铟锡)/MoO3(10nm)/NPB(60nm)/mCP(10nm)/DPEPO:10wt%13(30nm)/Bphen(40nm)/LiF(1nm)/Al(100nm)。
实施例7
将实施例2制备得到的三联吡啶类衍生物式14作为发光客体制备器件。这个实施例展示了14作为客体发光材料而制备的电致发光器件的性能验证。ITO(氧化铟锡)玻璃相继在清洗剂和去离子水中以超声波清洗30分钟。然后真空干燥2小时(105℃),再将ITO玻璃放入等离子反应器中进行5分钟的氧等离子处理,传送到真空室内制备有机膜和金属电极,接着通过真空蒸镀的方法制备一层10nm的空穴注入材料三氧化钼,接着蒸镀60nm厚的空穴传输材料4,4'-双[N-(1-萘基)-N-苯基氨基]联苯(NPB),电子阻挡层为10nm厚的1,3-二-9-咔唑基苯(mCP);然后蒸镀一层发光层30nm双[2-(二苯基膦基)苯基]醚氧化物(DPEPO):10%14;接着蒸镀一层10nm的DPEPO作为空穴阻挡层,然后蒸镀40nm厚的4,7-二苯基-1,10-菲咯啉(Bphen),最后再蒸镀一层1nm的LiF和100nm的Al。
铝作为器件的阴极,将直流电的正极加于ITO(氧化铟锡)层,将负极加于金属层,即可得到从ITO(氧化铟锡)层发出的明亮均匀的光,CIE色坐标为(0.154,0.109),启亮电压4.8V,最大外部量子效率17.8%,最大电流效率为14.9cd/A。本实验器件结构为:ITO(氧化铟锡)/MoO3(10nm)/NPB(60nm)/mCP(10nm)/DPEPO:10wt%14(30nm)/Bphen(40nm)/LiF(1nm)/Al(100nm)。
实施例8
将实施例3制备得到的三联吡啶类衍生物式15作为发光客体制备器件。这个实施例展示了15作为客体发光材料而制备的电致发光器件的性能验证。ITO(氧化铟锡)玻璃相继在清洗剂和去离子水中以超声波清洗30分钟。然后真空干燥2小时(105℃),再将ITO玻璃放入等离子反应器中进行5分钟的氧等离子处理,传送到真空室内制备有机膜和金属电极,接着通过真空蒸镀的方法制备一层10nm的空穴注入材料三氧化钼,接着蒸镀60nm厚的空穴传输材料4,4'-双[N-(1-萘基)-N-苯基氨基]联苯(NPB),电子阻挡层为10nm厚的1,3-二-9-咔唑基苯(mCP);然后蒸镀一层发光层30nm双[2-(二苯基膦基)苯基]醚氧化物(DPEPO):10%15;接着蒸镀一层10nm的DPEPO作为空穴阻挡层,然后蒸镀40nm厚的4,7-二苯基-1,10-菲咯啉(Bphen),最后再蒸镀一层1nm的LiF和100nm的Al。
铝作为器件的阴极,将直流电的正极加于ITO(氧化铟锡)层,将负极加于金属层,即可得到从ITO(氧化铟锡)层发出的明亮均匀的光,CIE色坐标为(0.158,0.157),启亮电压4.7V,最大外部量子效率8.35%,最大电流效率为10.3cd/A。本实验器件结构为:ITO(氧化铟锡)/MoO3(10nm)/NPB(60nm)/mCP(10nm)/DPEPO:10wt%15(30nm)/Bphen(40nm)/LiF(1nm)/Al(100nm)。
本发明以化合物13、14和15的相关图谱为例,说明本发明所提供的化合物材料的相关性能:
如图1所示,可以从图中的在电致发光器件中三个材料的发射分别在448nm,448nm和456nm可以看出,它们均可以实现深蓝光发射。
如图2所示,三个材料的瞬态延迟寿命非常明显,且均达到了上百微妙,表明这三种材料均有热致延迟荧光现象。
如图3所示,是该类深蓝光TADF材料电致发光的器件结构,由于在发光过程中三重态起到关键的作用,所以主体(Host)需要选用三重态比较高的,比如DPEPO,为了使得激子限制在发光层中,采用了电子阻挡层(EBL)和空穴阻挡层(HBL)。另外的电子传输层(ETL)和空穴传输层(HTL)需要和其他功能层能级搭配,且电子和空穴的传输速率相当。
其他实施中化合物的器件性能如下表所示,均具有良好表现:


以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
Claims (5)
1.三联吡啶类衍生物,其特征在于,结构通式如下:

桥联基团L选自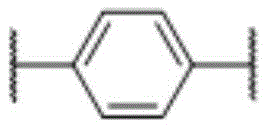 时,R选自以下任一:
时,R选自以下任一:

桥联基团L选自 时,R选自以下任一:
时,R选自以下任一:



2.根据权利要求1所述的三联吡啶类衍生物,其特征在于,选自以下任一:



3.一种根据权利要求1或2所述的三联吡啶类衍生物的制备方法,其特征在于,包括以下步骤:R-H与Br-L-三联吡啶偶联,得到三联吡啶类衍生物
4.一种根据权利要求1或2任一所述的三联吡啶类衍生物的应用,其特征在于:作为有机电致发光器件中的电致发光层材料,所述电致发光层材料为热致延迟荧光材料。
5.一种电致发光器件,所述电致发光器件至少包含一对电极和设置在该对电极之间的有机发光层,其特征在于:所述有机发光层中至少含有根据权利要求1或2任一所述的三联吡啶类衍生物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201711293130.9A CN109897029B (zh) | 2017-12-08 | 2017-12-08 | 三联吡啶类衍生物及其制备方法、应用和器件 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201711293130.9A CN109897029B (zh) | 2017-12-08 | 2017-12-08 | 三联吡啶类衍生物及其制备方法、应用和器件 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN109897029A CN109897029A (zh) | 2019-06-18 |
| CN109897029B true CN109897029B (zh) | 2021-03-30 |
Family
ID=66940170
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201711293130.9A Active CN109897029B (zh) | 2017-12-08 | 2017-12-08 | 三联吡啶类衍生物及其制备方法、应用和器件 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN109897029B (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110452227A (zh) * | 2019-08-05 | 2019-11-15 | 北京大学深圳研究生院 | 一种基于吲哚衍生物的有机蓝光荧光材料与蓝光器件 |
| CN113278018B (zh) * | 2020-02-20 | 2022-12-16 | 苏州大学 | 基于吡啶的热活化延迟荧光材料及其应用 |
| CN111689946A (zh) * | 2020-06-17 | 2020-09-22 | 深圳大学 | 一种咔唑并芳环热活化延迟荧光材料及其有机电致发光器件 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011108901A2 (ko) * | 2010-03-05 | 2011-09-09 | 덕산하이메탈(주) | 스파이로 골격을 포함하는 스파이로 카바졸 화합물 및 이를 이용한 유기전자소자, 그 단말 |
| CN106935712A (zh) * | 2015-12-29 | 2017-07-07 | 三星显示有限公司 | 有机发光器件 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101718887B1 (ko) * | 2013-07-01 | 2017-04-05 | 삼성디스플레이 주식회사 | 유기 발광 소자 |
| US20170170407A1 (en) * | 2014-06-12 | 2017-06-15 | Duk San Neolux Co., Ltd. | Compound for organic electronic element, organic electronic element using same, and electronic device thereof |
| CN107586299A (zh) * | 2017-09-29 | 2018-01-16 | 江苏三月光电科技有限公司 | 一种以氮杂苯为核心的有机化合物及其应用 |
-
2017
- 2017-12-08 CN CN201711293130.9A patent/CN109897029B/zh active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011108901A2 (ko) * | 2010-03-05 | 2011-09-09 | 덕산하이메탈(주) | 스파이로 골격을 포함하는 스파이로 카바졸 화합물 및 이를 이용한 유기전자소자, 그 단말 |
| CN106935712A (zh) * | 2015-12-29 | 2017-07-07 | 三星显示有限公司 | 有机发光器件 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN109897029A (zh) | 2019-06-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101427605B1 (ko) | 신규한 유기 발광 화합물 및 이를 채용하고 있는 유기 전계발광 소자 | |
| JP6182145B2 (ja) | 発光素子用のスピロビフルオレン化合物 | |
| CN108586188B (zh) | 䓛衍生物、包含该䓛衍生物的材料和有机电致发光器件 | |
| TWI454469B (zh) | 用於有機電激發光裝置之化合物及使用該化合物之有機電激發光裝置 | |
| JP7119204B2 (ja) | 有機発光化合物及びこれを用いた有機電界発光素子 | |
| KR20110132721A (ko) | 신규한 유기 발광 화합물 및 이를 채용하고 있는 유기 전계 발광 소자 | |
| CN110078755B (zh) | 化合物、显示面板以及显示装置 | |
| CN111777633B (zh) | 一种含硼化合物及含有其的有机电致发光器件 | |
| CN113336782A (zh) | 含咔唑骨架的绿光窄光谱三配位硼发光化合物、制备方法及其应用 | |
| KR20190049525A (ko) | 헤테로고리 화합물 및 이를 포함하는 유기 발광 소자 | |
| CN109053696B (zh) | 一种联吡嗪衍生物及其在有机光电器件中的应用 | |
| CN109897029B (zh) | 三联吡啶类衍生物及其制备方法、应用和器件 | |
| KR20130121516A (ko) | 신규한 아릴아민을 사용한 정공 수송 물질 및 이를 포함한 유기 전계 발광 소자 | |
| CN113563871A (zh) | 主体材料、有机光电器件及显示或照明装置 | |
| CN113980040B (zh) | 一种含硼的不对称螺环化合物及其在有机电致发光器件中的应用 | |
| CN113528123B (zh) | 主体材料和包含其的有机电致发光器件 | |
| KR101815653B1 (ko) | 아자보라디벤조크라이센 유도체 유기 발광 화합물, 잉크 조성물 및 유기 발광 소자 | |
| KR20130120855A (ko) | 티오펜를 사용한 정공 수송 물질 및 이를 포함한 유기 전계 발광 소자 | |
| CN111574505B (zh) | 一种以苯并[c]噌啉为受体的化合物及其应用 | |
| KR20120128386A (ko) | 유기전계 발광 재료용 아릴아민유도체와 이를 제조하는 방법 및 유기전계 발광 재료용 아릴아민유도체를 포함하는 유기전계 발광 재료 | |
| CN107344923B (zh) | 电场发光装置的有机化合物 | |
| KR101327301B1 (ko) | 아민계 정공수송 물질 및 이를 포함한 유기전계 발광소자 | |
| KR20110119282A (ko) | 신규한 유기 전자재료용 화합물 및 이를 채용하고 있는 유기 전계 발광 소자 | |
| CN113004262B (zh) | 一种有机材料及其应用 | |
| CN111635420B (zh) | 一种新型多杂环化合物及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |