CN109311812B - 作为c-MET抑制剂的吡啶酮类化合物 - Google Patents
作为c-MET抑制剂的吡啶酮类化合物 Download PDFInfo
- Publication number
- CN109311812B CN109311812B CN201780036464.XA CN201780036464A CN109311812B CN 109311812 B CN109311812 B CN 109311812B CN 201780036464 A CN201780036464 A CN 201780036464A CN 109311812 B CN109311812 B CN 109311812B
- Authority
- CN
- China
- Prior art keywords
- acid
- mmol
- compound
- pharmaceutically acceptable
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4412—Non condensed pyridines; Hydrogenated derivatives thereof having oxo groups directly attached to the heterocyclic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
Description















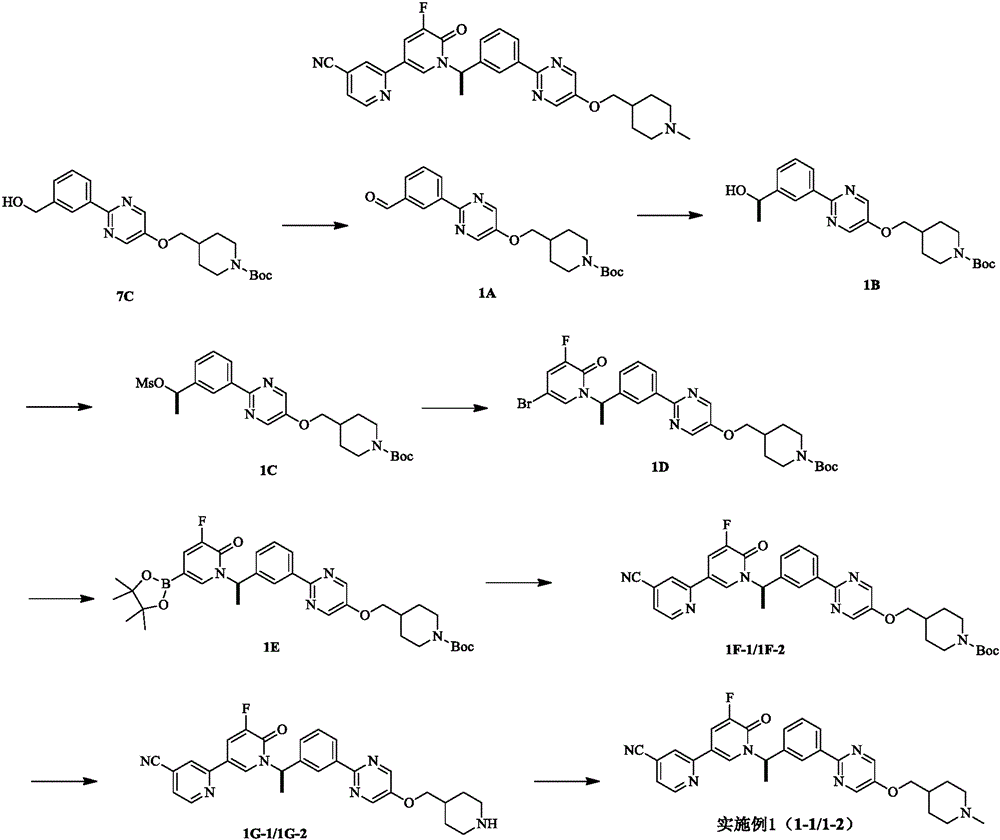









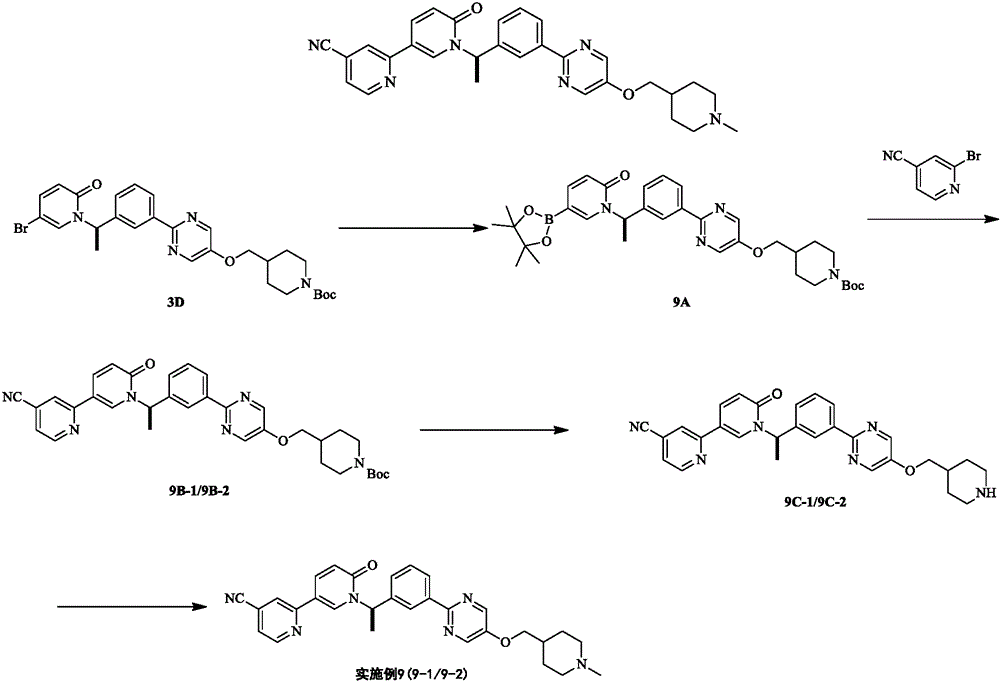




| 待测化合物 | c-MET IC<sub>50</sub>(nM) | 待测化合物 | c-MET IC<sub>50</sub>(nM) |
| 实施例1-2 | 1.09 | 实施例7 | 15.50 |
| 实施例2-2 | 9.33 | 实施例8-2 | 3.79 |
| 实施例4 | 6.16 | 实施例10 | 69.50 |
| 实施例5 | 2.90 | 实施例11 | 5.00 |
| 实施例6 | 4.37 |
| 待测化合物 | MHCC97H cell IC<sub>50</sub>(nM) | 待测化合物 | MHCC97H cell IC<sub>50</sub>(nM) |
| 实施例1-2 | 8.80 | 实施例7 | 22.30 |
| 实施例2-2 | 13.80 | 实施例9-2 | 22.10 |
| 实施例3-2 | 19.0 | 实施例10 | 166.00 |
| 实施例4 | 72.90 | 实施例11 | 93.80 |
| 实施例5 | 58.80 | 实施例12 | 51.40 |
| 实施例6 | 32.90 |

Claims (12)









Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610954377 | 2016-10-27 | ||
| CN201610954377X | 2016-10-27 | ||
| PCT/CN2017/107964 WO2018077227A1 (zh) | 2016-10-27 | 2017-10-27 | 作为c-MET抑制剂的吡啶酮类化合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN109311812A CN109311812A (zh) | 2019-02-05 |
| CN109311812B true CN109311812B (zh) | 2020-03-17 |
Family
ID=62024383
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201780036464.XA Active CN109311812B (zh) | 2016-10-27 | 2017-10-27 | 作为c-MET抑制剂的吡啶酮类化合物 |
Country Status (28)
| Country | Link |
|---|---|
| US (1) | US10501443B2 (zh) |
| EP (1) | EP3533787B1 (zh) |
| JP (1) | JP6719679B2 (zh) |
| KR (1) | KR102070748B1 (zh) |
| CN (1) | CN109311812B (zh) |
| AU (1) | AU2017348810B2 (zh) |
| BR (1) | BR112019008415B1 (zh) |
| CA (1) | CA3041164C (zh) |
| CO (1) | CO2019005165A2 (zh) |
| DK (1) | DK3533787T3 (zh) |
| EA (1) | EA038108B1 (zh) |
| ES (1) | ES2835301T3 (zh) |
| HR (1) | HRP20201985T1 (zh) |
| HU (1) | HUE051734T2 (zh) |
| IL (1) | IL266126B (zh) |
| LT (1) | LT3533787T (zh) |
| MX (1) | MX2019004626A (zh) |
| MY (1) | MY189557A (zh) |
| PE (1) | PE20190912A1 (zh) |
| PH (1) | PH12019500875A1 (zh) |
| PL (1) | PL3533787T3 (zh) |
| PT (1) | PT3533787T (zh) |
| RS (1) | RS61126B1 (zh) |
| SG (1) | SG11201903801YA (zh) |
| SI (1) | SI3533787T1 (zh) |
| UA (1) | UA122737C2 (zh) |
| WO (1) | WO2018077227A1 (zh) |
| ZA (1) | ZA201903074B (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019206268A1 (zh) * | 2018-04-26 | 2019-10-31 | 福建广生堂药业股份有限公司 | 一种c-MET抑制剂的晶型及其盐型和制备方法 |
| CN113365997B (zh) * | 2019-02-01 | 2022-06-07 | 南京明德新药研发有限公司 | 作为c-Met抑制剂的含嘧啶基团的三并环类化合物 |
| WO2022063869A2 (en) | 2020-09-24 | 2022-03-31 | Merck Patent Gmbh | Compounds for the treatment of viral infections |
| WO2022253935A1 (en) | 2021-06-04 | 2022-12-08 | Merck Patent Gmbh | Compounds for the treatment of glioblastoma |
| WO2024206858A1 (en) | 2023-03-30 | 2024-10-03 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101743241A (zh) * | 2007-07-12 | 2010-06-16 | 默克专利有限公司 | 哒嗪酮衍生物 |
| CN104144925A (zh) * | 2011-10-17 | 2014-11-12 | 拜耳知识产权有限责任公司 | 作为hif抑制剂的取代的噁二唑基吡啶酮和噁二唑基哒嗪酮 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2413241A1 (en) | 2000-06-29 | 2002-01-10 | Bristol-Myers Squibb Pharma Company | Thrombin or factor xa inhibitors |
| US7732456B2 (en) | 2004-03-05 | 2010-06-08 | Banyu Pharmaceutical Co., Ltd. | Pyridone derivative |
| WO2008103277A2 (en) | 2007-02-16 | 2008-08-28 | Amgen Inc. | Nitrogen-containing heterocyclyl ketones and their use as c-met inhibitors |
| DE102008062826A1 (de) * | 2008-12-23 | 2010-07-01 | Merck Patent Gmbh | Pyridazinonderivate |
| CN104203243A (zh) * | 2012-03-19 | 2014-12-10 | 默克专利股份公司 | 具有抗肿瘤活性的6-氧代-1,6-二氢-哒嗪衍生物与其他抗肿瘤化合物的组合 |
| RU2684407C2 (ru) * | 2012-10-11 | 2019-04-09 | Мерк Патент Гмбх | Комбинация производного 6-оксо-1,6-дигидро-пиридазина, имеющего противораковую активность, с мек ингибитором |
| BR112017010232A2 (pt) | 2014-12-11 | 2018-01-02 | Merck Patent Gmbh | Combinação de um derivado de 6-oxo-1,6-di-hidro- piridazina tendo atividade anticâncer com um derivado de quinazolina |
-
2017
- 2017-10-27 CN CN201780036464.XA patent/CN109311812B/zh active Active
- 2017-10-27 DK DK17864813.5T patent/DK3533787T3/da active
- 2017-10-27 KR KR1020197014236A patent/KR102070748B1/ko active IP Right Grant
- 2017-10-27 UA UAA201904417A patent/UA122737C2/uk unknown
- 2017-10-27 MY MYPI2019002142A patent/MY189557A/en unknown
- 2017-10-27 PE PE2019000855A patent/PE20190912A1/es unknown
- 2017-10-27 ES ES17864813T patent/ES2835301T3/es active Active
- 2017-10-27 RS RS20201448A patent/RS61126B1/sr unknown
- 2017-10-27 JP JP2019540379A patent/JP6719679B2/ja active Active
- 2017-10-27 WO PCT/CN2017/107964 patent/WO2018077227A1/zh active Application Filing
- 2017-10-27 US US16/343,387 patent/US10501443B2/en active Active
- 2017-10-27 SI SI201730558T patent/SI3533787T1/sl unknown
- 2017-10-27 AU AU2017348810A patent/AU2017348810B2/en active Active
- 2017-10-27 CA CA3041164A patent/CA3041164C/en active Active
- 2017-10-27 LT LTEP17864813.5T patent/LT3533787T/lt unknown
- 2017-10-27 PL PL17864813T patent/PL3533787T3/pl unknown
- 2017-10-27 SG SG11201903801YA patent/SG11201903801YA/en unknown
- 2017-10-27 MX MX2019004626A patent/MX2019004626A/es active IP Right Grant
- 2017-10-27 EP EP17864813.5A patent/EP3533787B1/en active Active
- 2017-10-27 EA EA201990952A patent/EA038108B1/ru unknown
- 2017-10-27 BR BR112019008415-0A patent/BR112019008415B1/pt active IP Right Grant
- 2017-10-27 PT PT178648135T patent/PT3533787T/pt unknown
- 2017-10-27 HU HUE17864813A patent/HUE051734T2/hu unknown
-
2019
- 2019-04-18 IL IL266126A patent/IL266126B/en active IP Right Grant
- 2019-04-22 PH PH12019500875A patent/PH12019500875A1/en unknown
- 2019-05-16 ZA ZA2019/03074A patent/ZA201903074B/en unknown
- 2019-05-21 CO CONC2019/0005165A patent/CO2019005165A2/es unknown
-
2020
- 2020-12-10 HR HRP20201985TT patent/HRP20201985T1/hr unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101743241A (zh) * | 2007-07-12 | 2010-06-16 | 默克专利有限公司 | 哒嗪酮衍生物 |
| CN104144925A (zh) * | 2011-10-17 | 2014-11-12 | 拜耳知识产权有限责任公司 | 作为hif抑制剂的取代的噁二唑基吡啶酮和噁二唑基哒嗪酮 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN109311812B (zh) | 作为c-MET抑制剂的吡啶酮类化合物 | |
| JP7394074B2 (ja) | 治療用化合物 | |
| ES2938899T3 (es) | Compuesto de aril-fósforo-oxígeno como inhibidor de la quinasa del EGFR | |
| JP6948659B1 (ja) | ピリダジニルチアアゾールカルボキシアミド化合物 | |
| WO2022061251A1 (en) | Compounds and methods for kras modulation and indications therefor | |
| US11623923B2 (en) | Uracil compound as c-MET/AXL inhibitor | |
| US9181264B2 (en) | Fused heteroaryls and their uses | |
| KR102359993B1 (ko) | 피리미도[5,4-b]인돌리진 또는 피리미도[5,4-b]피롤리진 화합물, 그의 제조방법 및 용도 | |
| EP4092024A1 (en) | Pyrimidine-4(3h)-ketone heterocyclic compound, preparation method therefor and use thereof in medicine and pharmacology | |
| JP7328263B2 (ja) | 免疫調節化合物 | |
| TWI628180B (zh) | 作為PI3K抑制劑的吡啶并[1,2-a]嘧啶酮類似物 | |
| CN117460737A (zh) | 杂芳环化合物、其制备方法及用途 | |
| CN112771049B (zh) | Fgfr4抑制剂及其应用 | |
| WO2023036156A1 (zh) | Dna-pk选择性抑制剂及其制备方法和用途 | |
| WO2023077259A1 (en) | Fused tetracyclic quinazoline derivatives as inhibitors of erbb2 | |
| CN113874379A (zh) | 作为Cdc7抑制剂的四并环类化合物 | |
| CN114907350B (zh) | 一类含氮稠环类化合物、制备方法和用途 | |
| BR112020008991A2 (pt) | compostos de piridopirimidina que atuam como inibidores duais de mtorc 1/2 | |
| NZ753020B2 (en) | Pyridone compound as c-met inhibitor | |
| WO2024044713A1 (en) | Naphthyridine compounds for inhibition of raf kinases | |
| TW202330519A (zh) | 作為tam抑制劑的吡唑并吡啶化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| CB02 | Change of applicant information | ||
| CB02 | Change of applicant information |
Address after: 355399 Fuyuan Industrial Zone, Dongyuan Township, Zherong County, Ningde City, Fujian Province Applicant after: Fujian Cosunter Pharmaceutical Co., Ltd. Address before: 350003 Block B, Building 10, Software Park B, 89 Software Avenue, Gulou District, Fuzhou City, Fujian Province Applicant before: Fujian Cosunter Pharmaceutical Co., Ltd. |
|
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 40002592 Country of ref document: HK |
|
| TR01 | Transfer of patent right | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20220127 Address after: 355300 building 1-7, Fuyuan Industrial Park, Zherong County, Ningde City, Fujian Province Patentee after: Fujian Guangsheng Zhonglin Biotechnology Co.,Ltd. Address before: 355399 Fuyuan Industrial Zone, Dongyuan Township, Zherong County, Ningde City, Fujian Province Patentee before: FUJIAN COSUNTER PHARMACEUTICAL Co.,Ltd. |