CN101723883B - 一种羟尼酮的制备方法 - Google Patents
一种羟尼酮的制备方法 Download PDFInfo
- Publication number
- CN101723883B CN101723883B CN 200810201706 CN200810201706A CN101723883B CN 101723883 B CN101723883 B CN 101723883B CN 200810201706 CN200810201706 CN 200810201706 CN 200810201706 A CN200810201706 A CN 200810201706A CN 101723883 B CN101723883 B CN 101723883B
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- preparation
- reaction
- suc
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 20
- BRUQQQPBMZOVGD-XFKAJCMBSA-N Oxycodone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C BRUQQQPBMZOVGD-XFKAJCMBSA-N 0.000 title description 9
- 229960002085 oxycodone Drugs 0.000 title description 9
- 150000001875 compounds Chemical class 0.000 claims abstract description 90
- 239000003153 chemical reaction reagent Substances 0.000 claims abstract description 28
- 238000005906 dihydroxylation reaction Methods 0.000 claims abstract description 26
- SOHMZGMHXUQHGE-UHFFFAOYSA-N 5-methyl-1h-pyridin-2-one Chemical compound CC1=CC=C(O)N=C1 SOHMZGMHXUQHGE-UHFFFAOYSA-N 0.000 claims abstract description 15
- 125000001033 ether group Chemical group 0.000 claims abstract description 4
- 238000002156 mixing Methods 0.000 claims abstract description 3
- 238000006243 chemical reaction Methods 0.000 claims description 34
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 30
- 238000002360 preparation method Methods 0.000 claims description 29
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims description 24
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 19
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 16
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 15
- 229940125782 compound 2 Drugs 0.000 claims description 15
- GZFGOTFRPZRKDS-UHFFFAOYSA-N 4-bromophenol Chemical compound OC1=CC=C(Br)C=C1 GZFGOTFRPZRKDS-UHFFFAOYSA-N 0.000 claims description 12
- 239000000243 solution Substances 0.000 claims description 11
- 229940125898 compound 5 Drugs 0.000 claims description 10
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 claims description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 8
- 235000006408 oxalic acid Nutrition 0.000 claims description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 7
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 7
- 229910021595 Copper(I) iodide Inorganic materials 0.000 claims description 5
- 239000003223 protective agent Substances 0.000 claims description 5
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 claims description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 4
- 125000006278 bromobenzyl group Chemical group 0.000 claims description 4
- WZIYCIBURCPKAR-UHFFFAOYSA-N 4-(chloromethyl)pyridine Chemical compound ClCC1=CC=NC=C1 WZIYCIBURCPKAR-UHFFFAOYSA-N 0.000 claims description 3
- 239000007864 aqueous solution Substances 0.000 claims description 3
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 claims description 3
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 claims description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 3
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 claims description 2
- QARVLSVVCXYDNA-UHFFFAOYSA-N phenyl bromide Natural products BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- 208000019425 cirrhosis of liver Diseases 0.000 description 11
- 238000003756 stirring Methods 0.000 description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 239000007787 solid Substances 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 7
- HUHXLHLWASNVDB-UHFFFAOYSA-N 2-(oxan-2-yloxy)oxane Chemical class O1CCCCC1OC1OCCCC1 HUHXLHLWASNVDB-UHFFFAOYSA-N 0.000 description 6
- 206010019668 Hepatic fibrosis Diseases 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 125000001743 benzylic group Chemical group 0.000 description 6
- -1 bromobenzene tetrahydropyranyl ethers Chemical class 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 6
- 0 *c(cc1)ccc1Br Chemical compound *c(cc1)ccc1Br 0.000 description 4
- JTSSUEWTRDWHGY-UHFFFAOYSA-N 4-(pyridin-4-ylmethoxymethyl)pyridine Chemical compound C=1C=NC=CC=1COCC1=CC=NC=C1 JTSSUEWTRDWHGY-UHFFFAOYSA-N 0.000 description 4
- OCDXZFSOHJRGIL-UHFFFAOYSA-N cyclohexyloxycyclohexane Chemical compound C1CCCCC1OC1CCCCC1 OCDXZFSOHJRGIL-UHFFFAOYSA-N 0.000 description 4
- AQEFLFZSWDEAIP-UHFFFAOYSA-N di-tert-butyl ether Chemical compound CC(C)(C)OC(C)(C)C AQEFLFZSWDEAIP-UHFFFAOYSA-N 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- NETTXQJYJRFTFS-UHFFFAOYSA-N CC(C=C1)=CN(c(cc2)ccc2O)C1=O Chemical compound CC(C=C1)=CN(c(cc2)ccc2O)C1=O NETTXQJYJRFTFS-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical group CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 241000700721 Hepatitis B virus Species 0.000 description 2
- 206010019799 Hepatitis viral Diseases 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 208000002672 hepatitis B Diseases 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 125000005188 oxoalkyl group Chemical group 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 201000001862 viral hepatitis Diseases 0.000 description 2
- ITQTTZVARXURQS-UHFFFAOYSA-N 3-methylpyridine Chemical compound CC1=CC=CN=C1 ITQTTZVARXURQS-UHFFFAOYSA-N 0.000 description 1
- CMBSSVKZOPZBKW-UHFFFAOYSA-N 5-methylpyridin-2-amine Chemical class CC1=CC=C(N)N=C1 CMBSSVKZOPZBKW-UHFFFAOYSA-N 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- OUQSGILAXUXMGI-UHFFFAOYSA-N Brc(cc1)ccc1OCc1ccccc1 Chemical compound Brc(cc1)ccc1OCc1ccccc1 OUQSGILAXUXMGI-UHFFFAOYSA-N 0.000 description 1
- SSCLBSMWOMTJMY-UHFFFAOYSA-N CC(C)COc(cc1)ccc1N(C=C(C)C=C1)C1=O Chemical compound CC(C)COc(cc1)ccc1N(C=C(C)C=C1)C1=O SSCLBSMWOMTJMY-UHFFFAOYSA-N 0.000 description 1
- QWPYXLRGLVOZPL-UHFFFAOYSA-N CC(C=C1)=C[I]=C1OC1OCCCC1 Chemical compound CC(C=C1)=C[I]=C1OC1OCCCC1 QWPYXLRGLVOZPL-UHFFFAOYSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 206010008190 Cerebrovascular accident Diseases 0.000 description 1
- 206010008635 Cholestasis Diseases 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 208000000419 Chronic Hepatitis B Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 230000002300 anti-fibrosis Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 231100000359 cholestasis Toxicity 0.000 description 1
- 230000007870 cholestasis Effects 0.000 description 1
- 230000009693 chronic damage Effects 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000005868 electrolysis reaction Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N ferric oxide Chemical compound O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 230000007096 poisonous effect Effects 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000010865 sewage Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229960001866 silicon dioxide Drugs 0.000 description 1
- 235000002639 sodium chloride Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract



Description














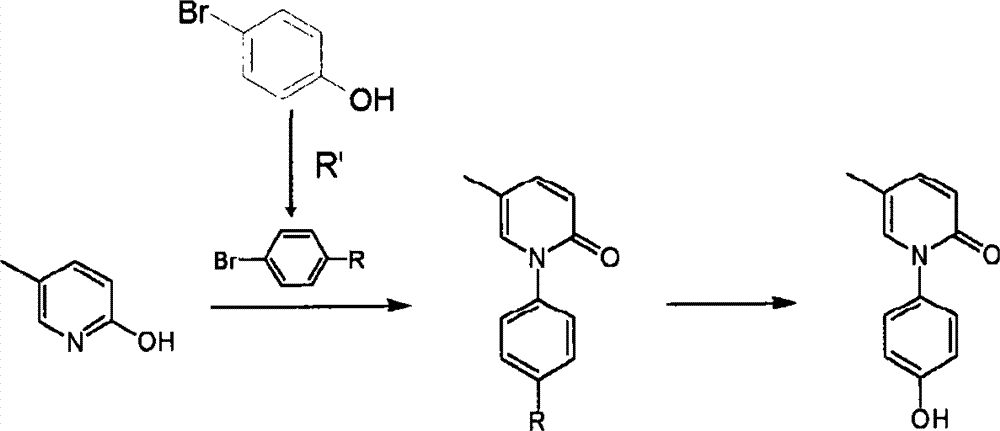




















Claims (10)




Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 200810201706 CN101723883B (zh) | 2008-10-24 | 2008-10-24 | 一种羟尼酮的制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 200810201706 CN101723883B (zh) | 2008-10-24 | 2008-10-24 | 一种羟尼酮的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101723883A CN101723883A (zh) | 2010-06-09 |
| CN101723883B true CN101723883B (zh) | 2013-06-05 |
Family
ID=42445511
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 200810201706 Active CN101723883B (zh) | 2008-10-24 | 2008-10-24 | 一种羟尼酮的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN101723883B (zh) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116082225A (zh) * | 2016-08-08 | 2023-05-09 | 北京康蒂尼药业股份有限公司 | 一种羟尼酮的制备方法 |
| CN116023327A (zh) * | 2016-08-08 | 2023-04-28 | 北京康蒂尼药业股份有限公司 | 一种羟尼酮的制备方法 |
| CN108285431B (zh) * | 2018-03-23 | 2021-04-09 | 北京康蒂尼药业股份有限公司 | 一种吡非尼酮有关物质及其制备方法和用途 |
| CN113173881B (zh) * | 2021-03-17 | 2022-12-20 | 北京康蒂尼药业股份有限公司 | 羟尼酮的晶型及其制备方法和用途 |
| EP4326265A1 (en) * | 2021-04-19 | 2024-02-28 | Gyre Therapeutics, Inc. | Pharmaceutical hydronidone formulations for diseases |
| CN113476445A (zh) * | 2021-05-14 | 2021-10-08 | 北京康蒂尼药业股份有限公司 | 羟尼酮在制备治疗或预防慢性乙肝伴肝纤维化药物中的应用 |
| CN116239452A (zh) * | 2022-12-20 | 2023-06-09 | 浙江圣效化学品有限公司 | 一种制备叔丁基苯醚类化合物的方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5310562A (en) * | 1989-11-22 | 1994-05-10 | Margolin Solomon B | Composition and method for reparation and prevention of fibrotic lesions |
| CN1386737A (zh) * | 2002-06-11 | 2002-12-25 | 中南大学湘雅医学院 | 抗纤维化吡啶酮药物及其生产工艺方法 |
-
2008
- 2008-10-24 CN CN 200810201706 patent/CN101723883B/zh active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5310562A (en) * | 1989-11-22 | 1994-05-10 | Margolin Solomon B | Composition and method for reparation and prevention of fibrotic lesions |
| CN1386737A (zh) * | 2002-06-11 | 2002-12-25 | 中南大学湘雅医学院 | 抗纤维化吡啶酮药物及其生产工艺方法 |
Non-Patent Citations (2)
| Title |
|---|
| 吡非尼酮的合成;马臻等;《中国医药工业杂志》;20061231;第37卷(第6期);372-373 * |
| 马臻等.吡非尼酮的合成.《中国医药工业杂志》.2006,第37卷(第6期),372-373. |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101723883A (zh) | 2010-06-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101723883B (zh) | 一种羟尼酮的制备方法 | |
| CN103265426A (zh) | 一种基于两相中自由基反应制备2-(4-溴甲基苯基)丙酸的绿色环保性方法 | |
| CN104447934B (zh) | 一种醋酸阿比特龙的纯化方法 | |
| CN102898422B (zh) | 一种苯醚甲环唑的制备方法 | |
| CN105968023A (zh) | 一种盐酸安非他酮的制备方法 | |
| CN104045669A (zh) | 一种适合工业化生产的化学合成红景天苷的分离方法 | |
| CN101648839B (zh) | 一种溴甲基联苯化合物的绿色合成方法 | |
| CN106883175A (zh) | 一种托伐普坦的制备方法 | |
| CN105646285B (zh) | 一种维兰特罗中间体及其制备方法和应用 | |
| CN102786404B (zh) | 安息香双甲醚的制备方法 | |
| CN105461525B (zh) | 1,3,5‑三醛基均苯三酚的制备及制备过程三氟乙酸的回用方法 | |
| CN103570668A (zh) | 4-氯甲基-5-甲基-1,3-二氧杂环戊烯-2-酮的合成方法 | |
| CN106749098B (zh) | 一种以氧气为氧化剂制备盐酸二氧丙嗪的制备方法 | |
| CN102850379A (zh) | 甲氧头孢中间体7-mac的合成方法 | |
| CN106674135A (zh) | 一种合成尿嘧啶的方法 | |
| CN102924255A (zh) | 一种液相氧化制备9-芴酮的方法 | |
| CN105418547A (zh) | 索非布韦关键中间体的制备 | |
| CN106977543A (zh) | 改进的索非布韦中间体的制备工艺 | |
| CN114315575A (zh) | 一种光引发剂中间体的制备方法及其应用 | |
| CN113336706B (zh) | 一种连续流制备奥美沙坦酯中间体的合成方法 | |
| CN109369492A (zh) | 一种(1S,4S)-2-Boc-2,5-二氮双环[2.2.1]庚烷的制备方法 | |
| CN102690211B (zh) | 托伐普坦中间体的制备方法 | |
| CN101671333B (zh) | 一种恩曲他滨的分离方法 | |
| CN113636938A (zh) | 一种5,5`-(全氟丙烷-2,2-二基)双(2-(烯丙氧基)苯胺)的制备方法 | |
| CN107698498A (zh) | 一种羟尼酮的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| TR01 | Transfer of patent right | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20201228 Address after: 101300 No.60, Shunkang Road, Linhe Industrial Development Zone, Shunyi District, Beijing Patentee after: BEIJING CONTINENT PHARMACEUTICAL Co.,Ltd. Address before: 201203 no.5-6, building 1, Lane 647, Songtao Road, Zhangjiang, Pudong, Shanghai Patentee before: SHANGHAI GENOMICS, Inc. |
|
| CP01 | Change in the name or title of a patent holder | ||
| CP01 | Change in the name or title of a patent holder |
Address after: 101300 No.60, Shunkang Road, Linhe Industrial Development Zone, Shunyi District, Beijing Patentee after: Beijing kangdini Pharmaceutical Co.,Ltd. Address before: 101300 No.60, Shunkang Road, Linhe Industrial Development Zone, Shunyi District, Beijing Patentee before: BEIJING CONTINENT PHARMACEUTICAL Co.,Ltd. |