WO2022256490A9 - Improved synthesis of phosphoramidates for the treatment of hepatitis b virus - Google Patents
Improved synthesis of phosphoramidates for the treatment of hepatitis b virus Download PDFInfo
- Publication number
- WO2022256490A9 WO2022256490A9 PCT/US2022/031904 US2022031904W WO2022256490A9 WO 2022256490 A9 WO2022256490 A9 WO 2022256490A9 US 2022031904 W US2022031904 W US 2022031904W WO 2022256490 A9 WO2022256490 A9 WO 2022256490A9
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- acid
- ati
- reaction
- Prior art date
Links
- 230000015572 biosynthetic process Effects 0.000 title claims abstract description 45
- 238000003786 synthesis reaction Methods 0.000 title abstract description 37
- 238000011282 treatment Methods 0.000 title abstract description 19
- 241000700721 Hepatitis B virus Species 0.000 title abstract description 18
- 150000008298 phosphoramidates Chemical class 0.000 title abstract description 8
- 238000006243 chemical reaction Methods 0.000 claims description 112
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 90
- 238000000034 method Methods 0.000 claims description 71
- 239000000203 mixture Substances 0.000 claims description 62
- FTMNZJASLMPPNW-MHFVGYLCSA-N propan-2-yl (2S)-2-[[[(2S,3S,4R,5S)-4-fluoro-3-hydroxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate Chemical compound CC(C)OC(=O)[C@H](C)N[P@](=O)(OC[C@@H]1O[C@@H]([C@H](F)[C@H]1O)n1cc(C)c(=O)[nH]c1=O)Oc1ccccc1 FTMNZJASLMPPNW-MHFVGYLCSA-N 0.000 claims description 57
- 229940125898 compound 5 Drugs 0.000 claims description 43
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 42
- 239000002904 solvent Substances 0.000 claims description 40
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 39
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 38
- 230000008569 process Effects 0.000 claims description 38
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims description 30
- 238000002360 preparation method Methods 0.000 claims description 30
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 claims description 26
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 24
- 239000002585 base Substances 0.000 claims description 23
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 20
- DCFKHNIGBAHNSS-UHFFFAOYSA-N chloro(triethyl)silane Chemical group CC[Si](Cl)(CC)CC DCFKHNIGBAHNSS-UHFFFAOYSA-N 0.000 claims description 18
- 229940125782 compound 2 Drugs 0.000 claims description 17
- -1 ethylsilyl Chemical group 0.000 claims description 17
- 239000003795 chemical substances by application Substances 0.000 claims description 16
- 239000012445 acidic reagent Substances 0.000 claims description 15
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 14
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 14
- 229910000042 hydrogen bromide Inorganic materials 0.000 claims description 14
- 239000005711 Benzoic acid Substances 0.000 claims description 13
- 235000010233 benzoic acid Nutrition 0.000 claims description 13
- 229940125904 compound 1 Drugs 0.000 claims description 13
- 229940126214 compound 3 Drugs 0.000 claims description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical group [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 12
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 claims description 12
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 claims description 12
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 10
- 238000010511 deprotection reaction Methods 0.000 claims description 10
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 claims description 9
- APOYTRAZFJURPB-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)-n-(trifluoro-$l^{4}-sulfanyl)ethanamine Chemical compound COCCN(S(F)(F)F)CCOC APOYTRAZFJURPB-UHFFFAOYSA-N 0.000 claims description 9
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 claims description 9
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 claims description 9
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 8
- 229920001429 chelating resin Polymers 0.000 claims description 8
- 229940125773 compound 10 Drugs 0.000 claims description 8
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 claims description 8
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 claims description 8
- 230000002378 acidificating effect Effects 0.000 claims description 7
- 239000003153 chemical reaction reagent Substances 0.000 claims description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 7
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 claims description 6
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 claims description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 claims description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 6
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 6
- 229910021529 ammonia Inorganic materials 0.000 claims description 6
- XMPZTFVPEKAKFH-UHFFFAOYSA-P ceric ammonium nitrate Chemical compound [NH4+].[NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O XMPZTFVPEKAKFH-UHFFFAOYSA-P 0.000 claims description 6
- 229920005989 resin Polymers 0.000 claims description 6
- 239000011347 resin Substances 0.000 claims description 6
- 238000006884 silylation reaction Methods 0.000 claims description 6
- NHGXDBSUJJNIRV-UHFFFAOYSA-M tetrabutylammonium chloride Chemical compound [Cl-].CCCC[N+](CCCC)(CCCC)CCCC NHGXDBSUJJNIRV-UHFFFAOYSA-M 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 claims description 5
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 claims description 5
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 claims description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 claims description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 claims description 4
- 229920002125 Sokalan® Polymers 0.000 claims description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 4
- ZMZINYUKVRMNTG-UHFFFAOYSA-N acetic acid;formic acid Chemical compound OC=O.CC(O)=O ZMZINYUKVRMNTG-UHFFFAOYSA-N 0.000 claims description 4
- 239000002253 acid Substances 0.000 claims description 4
- 239000012043 crude product Substances 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 235000019253 formic acid Nutrition 0.000 claims description 4
- 229920001467 poly(styrenesulfonates) Polymers 0.000 claims description 4
- 239000000376 reactant Substances 0.000 claims description 4
- 239000012312 sodium hydride Substances 0.000 claims description 4
- 229910000104 sodium hydride Inorganic materials 0.000 claims description 4
- QVCUKHQDEZNNOC-UHFFFAOYSA-N 1,2-diazabicyclo[2.2.2]octane Chemical compound C1CC2CCN1NC2 QVCUKHQDEZNNOC-UHFFFAOYSA-N 0.000 claims description 3
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 claims description 3
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 claims description 3
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 3
- UCKORWKZRPKRQE-UHFFFAOYSA-N bromo(triethyl)silane Chemical compound CC[Si](Br)(CC)CC UCKORWKZRPKRQE-UHFFFAOYSA-N 0.000 claims description 3
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 claims description 3
- 229940043279 diisopropylamine Drugs 0.000 claims description 3
- CQRPUKWAZPZXTO-UHFFFAOYSA-M magnesium;2-methylpropane;chloride Chemical group [Mg+2].[Cl-].C[C-](C)C CQRPUKWAZPZXTO-UHFFFAOYSA-M 0.000 claims description 3
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 claims description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 claims description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 claims description 3
- 239000000741 silica gel Substances 0.000 claims description 3
- 229910002027 silica gel Inorganic materials 0.000 claims description 3
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 claims description 3
- MTHYDHNQXLRHFM-UHFFFAOYSA-N triethyl(1-methoxyprop-1-enoxy)silane Chemical compound CC[Si](CC)(CC)OC(OC)=CC MTHYDHNQXLRHFM-UHFFFAOYSA-N 0.000 claims description 3
- PPLMQFARLJLZAO-UHFFFAOYSA-N triethyl(iodo)silane Chemical compound CC[Si](I)(CC)CC PPLMQFARLJLZAO-UHFFFAOYSA-N 0.000 claims description 3
- 229940086542 triethylamine Drugs 0.000 claims description 3
- MKMPBMJIGMMCPB-UHFFFAOYSA-N triethylsilylformonitrile Chemical compound CC[Si](CC)(CC)C#N MKMPBMJIGMMCPB-UHFFFAOYSA-N 0.000 claims description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 claims description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 2
- 230000008878 coupling Effects 0.000 claims description 2
- 238000010168 coupling process Methods 0.000 claims description 2
- 238000005859 coupling reaction Methods 0.000 claims description 2
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 claims description 2
- 229910000041 hydrogen chloride Inorganic materials 0.000 claims description 2
- 235000011007 phosphoric acid Nutrition 0.000 claims description 2
- 150000003138 primary alcohols Chemical class 0.000 claims description 2
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 claims description 2
- 239000012973 diazabicyclooctane Substances 0.000 claims 1
- MJSFTFFOEQQBLP-UHFFFAOYSA-N ethyl(1H-imidazol-2-yl)silane Chemical compound C(C)[SiH2]C=1NC=CN=1 MJSFTFFOEQQBLP-UHFFFAOYSA-N 0.000 claims 1
- NQBWPVUOYMLSRI-UHFFFAOYSA-N ethyl(trifluoromethyl)silane Chemical compound C(C)[SiH2]C(F)(F)F NQBWPVUOYMLSRI-UHFFFAOYSA-N 0.000 claims 1
- 208000036142 Viral infection Diseases 0.000 abstract description 5
- 230000009385 viral infection Effects 0.000 abstract description 5
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 abstract description 4
- 229940002612 prodrug Drugs 0.000 abstract description 3
- 239000000651 prodrug Substances 0.000 abstract description 3
- 239000000243 solution Substances 0.000 description 85
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 71
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 46
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 42
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 40
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 33
- 229960005338 clevudine Drugs 0.000 description 31
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 30
- 150000001875 compounds Chemical class 0.000 description 29
- 238000002474 experimental method Methods 0.000 description 29
- 239000012044 organic layer Substances 0.000 description 26
- 239000007787 solid Substances 0.000 description 26
- 238000003756 stirring Methods 0.000 description 26
- GBBJCSTXCAQSSJ-XQXXSGGOSA-N clevudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1[C@H](F)[C@@H](O)[C@H](CO)O1 GBBJCSTXCAQSSJ-XQXXSGGOSA-N 0.000 description 25
- 238000004128 high performance liquid chromatography Methods 0.000 description 25
- 230000002829 reductive effect Effects 0.000 description 24
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 23
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 20
- 235000017557 sodium bicarbonate Nutrition 0.000 description 20
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 20
- 229910052938 sodium sulfate Inorganic materials 0.000 description 20
- 235000011152 sodium sulphate Nutrition 0.000 description 20
- 239000000047 product Substances 0.000 description 19
- 235000019439 ethyl acetate Nutrition 0.000 description 18
- 239000010410 layer Substances 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 239000012535 impurity Substances 0.000 description 15
- 238000002425 crystallisation Methods 0.000 description 14
- 230000008025 crystallization Effects 0.000 description 14
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 13
- 208000015181 infectious disease Diseases 0.000 description 13
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 13
- 239000000463 material Substances 0.000 description 13
- 239000007858 starting material Substances 0.000 description 13
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 12
- 239000005695 Ammonium acetate Substances 0.000 description 12
- 229940043376 ammonium acetate Drugs 0.000 description 12
- 235000019257 ammonium acetate Nutrition 0.000 description 12
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 12
- 229940011051 isopropyl acetate Drugs 0.000 description 12
- 238000012369 In process control Methods 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 11
- 238000010965 in-process control Methods 0.000 description 11
- 239000000543 intermediate Substances 0.000 description 11
- 238000001228 spectrum Methods 0.000 description 11
- 239000007864 aqueous solution Substances 0.000 description 10
- 238000013341 scale-up Methods 0.000 description 10
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 9
- 239000013078 crystal Substances 0.000 description 9
- 238000001914 filtration Methods 0.000 description 9
- 150000003839 salts Chemical class 0.000 description 9
- 238000010791 quenching Methods 0.000 description 8
- 230000000171 quenching effect Effects 0.000 description 8
- 238000003556 assay Methods 0.000 description 7
- 238000005292 vacuum distillation Methods 0.000 description 7
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 239000012025 fluorinating agent Substances 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 238000010626 work up procedure Methods 0.000 description 6
- 239000003242 anti bacterial agent Substances 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 238000000926 separation method Methods 0.000 description 5
- 239000006227 byproduct Substances 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 238000000634 powder X-ray diffraction Methods 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 4
- WDBQJSCPCGTAFG-QHCPKHFHSA-N 4,4-difluoro-N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclohexane-1-carboxamide Chemical compound FC1(CCC(CC1)C(=O)N[C@@H](CCN1CCC(CC1)N1C(=NN=C1C)C(C)C)C=1C=NC=CC=1)F WDBQJSCPCGTAFG-QHCPKHFHSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 239000007848 Bronsted acid Substances 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 230000005526 G1 to G0 transition Effects 0.000 description 3
- 239000002841 Lewis acid Substances 0.000 description 3
- 208000021642 Muscular disease Diseases 0.000 description 3
- 201000009623 Myopathy Diseases 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000003115 biocidal effect Effects 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 230000002255 enzymatic effect Effects 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 150000007517 lewis acids Chemical class 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 238000005580 one pot reaction Methods 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 108020004638 Circular DNA Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- NUGPIZCTELGDOS-QHCPKHFHSA-N N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclopentanecarboxamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CC[C@@H](C=1C=NC=CC=1)NC(=O)C1CCCC1)C NUGPIZCTELGDOS-QHCPKHFHSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 239000011149 active material Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 230000008384 membrane barrier Effects 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- MEQHOJFTETWNFH-UHFFFAOYSA-N triethyl(1h-imidazol-2-yl)silane Chemical compound CC[Si](CC)(CC)C1=NC=CN1 MEQHOJFTETWNFH-UHFFFAOYSA-N 0.000 description 2
- ZHSKFONQCREGOG-UHFFFAOYSA-N triethyl(trifluoromethyl)silane Chemical compound CC[Si](CC)(CC)C(F)(F)F ZHSKFONQCREGOG-UHFFFAOYSA-N 0.000 description 2
- STMPXDBGVJZCEX-UHFFFAOYSA-N triethylsilyl trifluoromethanesulfonate Chemical compound CC[Si](CC)(CC)OS(=O)(=O)C(F)(F)F STMPXDBGVJZCEX-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- 238000004293 19F NMR spectroscopy Methods 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- 238000012573 2D experiment Methods 0.000 description 1
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 1
- 238000004679 31P NMR spectroscopy Methods 0.000 description 1
- TWGNOYAGHYUFFR-UHFFFAOYSA-N 5-methylpyrimidine Chemical compound CC1=CN=CN=C1 TWGNOYAGHYUFFR-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- QRAHVGYUKDBANU-OJJOVDNXSA-N CC(C)OC([C@H](C)N=P(OC(C(C(F)=C1F)F)(C(F)=C1F)OC1=CC=CC=C1)=O)=O Chemical compound CC(C)OC([C@H](C)N=P(OC(C(C(F)=C1F)F)(C(F)=C1F)OC1=CC=CC=C1)=O)=O QRAHVGYUKDBANU-OJJOVDNXSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 206010067125 Liver injury Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 238000012565 NMR experiment Methods 0.000 description 1
- GRZSFABZWSZOST-UHFFFAOYSA-N NP(O)(=O)Oc1ccc(cc1Cl)[N+]([O-])=O Chemical compound NP(O)(=O)Oc1ccc(cc1Cl)[N+]([O-])=O GRZSFABZWSZOST-UHFFFAOYSA-N 0.000 description 1
- XVEFAFPICKTHBB-UHFFFAOYSA-N N[SH3] Chemical compound N[SH3] XVEFAFPICKTHBB-UHFFFAOYSA-N 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 208000037581 Persistent Infection Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000009572 RNA Polymerase II Human genes 0.000 description 1
- 108010009460 RNA Polymerase II Proteins 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- RUKRVHYQIIURNV-XQXXSGGOSA-N [[(2s,3s,4r,5s)-4-fluoro-3-hydroxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1[C@H](F)[C@@H](O)[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 RUKRVHYQIIURNV-XQXXSGGOSA-N 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 229960001997 adefovir Drugs 0.000 description 1
- WOZSCQDILHKSGG-UHFFFAOYSA-N adefovir depivoxil Chemical compound N1=CN=C2N(CCOCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C)C=NC2=C1N WOZSCQDILHKSGG-UHFFFAOYSA-N 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- HUMHYXGDUOGHTG-HEZXSMHISA-N alpha-D-GalpNAc-(1->3)-[alpha-L-Fucp-(1->2)]-D-Galp Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](O[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](O)[C@@H](CO)OC1O HUMHYXGDUOGHTG-HEZXSMHISA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 239000012296 anti-solvent Substances 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000012223 aqueous fraction Substances 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000000084 colloidal system Substances 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000012045 crude solution Substances 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 230000003467 diminishing effect Effects 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 229960000980 entecavir Drugs 0.000 description 1
- YXPVEXCTPGULBZ-WQYNNSOESA-N entecavir hydrate Chemical compound O.C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)C1=C YXPVEXCTPGULBZ-WQYNNSOESA-N 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- 231100000234 hepatic damage Toxicity 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000001095 inductively coupled plasma mass spectrometry Methods 0.000 description 1
- 238000002354 inductively-coupled plasma atomic emission spectroscopy Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000013383 initial experiment Methods 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 229960001627 lamivudine Drugs 0.000 description 1
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 230000008818 liver damage Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- CCERQOYLJJULMD-UHFFFAOYSA-M magnesium;carbanide;chloride Chemical compound [CH3-].[Mg+2].[Cl-] CCERQOYLJJULMD-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004065 mitochondrial dysfunction Effects 0.000 description 1
- 150000004712 monophosphates Chemical class 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 1
- BTQLZQAVGBUMOG-UHFFFAOYSA-N n-silylacetamide Chemical compound CC(=O)N[SiH3] BTQLZQAVGBUMOG-UHFFFAOYSA-N 0.000 description 1
- WDFKBRQZGBFDAB-UHFFFAOYSA-N n-triethylsilylacetamide Chemical compound CC[Si](CC)(CC)NC(C)=O WDFKBRQZGBFDAB-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 229940042404 nucleoside and nucleotide reverse transcriptase inhibitor Drugs 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229940042443 other antivirals in atc Drugs 0.000 description 1
- LUYQYZLEHLTPBH-UHFFFAOYSA-N perfluorobutanesulfonyl fluoride Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)S(F)(=O)=O LUYQYZLEHLTPBH-UHFFFAOYSA-N 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- ZAEQTGTVGUJEFV-UHFFFAOYSA-N phenylmethanesulfonate;pyridin-1-ium Chemical compound C1=CC=[NH+]C=C1.[O-]S(=O)(=O)CC1=CC=CC=C1 ZAEQTGTVGUJEFV-UHFFFAOYSA-N 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 238000010935 polish filtration Methods 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- FCFXLXGZHDHJLB-UHFFFAOYSA-N pyridine-2-sulfonyl fluoride Chemical compound FS(=O)(=O)C1=CC=CC=N1 FCFXLXGZHDHJLB-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000013074 reference sample Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229960002063 sofosbuvir Drugs 0.000 description 1
- TTZHDVOVKQGIBA-IQWMDFIBSA-N sofosbuvir Chemical compound N1([C@@H]2O[C@@H]([C@H]([C@]2(F)C)O)CO[P@@](=O)(N[C@@H](C)C(=O)OC(C)C)OC=2C=CC=CC=2)C=CC(=O)NC1=O TTZHDVOVKQGIBA-IQWMDFIBSA-N 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- QHMQWEPBXSHHLH-UHFFFAOYSA-N sulfur tetrafluoride Chemical compound FS(F)(F)F QHMQWEPBXSHHLH-UHFFFAOYSA-N 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 229960005311 telbivudine Drugs 0.000 description 1
- IQFYYKKMVGJFEH-CSMHCCOUSA-N telbivudine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1O[C@@H](CO)[C@H](O)C1 IQFYYKKMVGJFEH-CSMHCCOUSA-N 0.000 description 1
- 229960004556 tenofovir Drugs 0.000 description 1
- VCMJCVGFSROFHV-WZGZYPNHSA-N tenofovir disoproxil fumarate Chemical compound OC(=O)\C=C\C(O)=O.N1=CN=C2N(C[C@@H](C)OCP(=O)(OCOC(=O)OC(C)C)OCOC(=O)OC(C)C)C=NC2=C1N VCMJCVGFSROFHV-WZGZYPNHSA-N 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- NYJIUCJXMLNPHU-UHFFFAOYSA-N thiophene-2-sulfonyl fluoride Chemical compound FS(=O)(=O)C1=CC=CS1 NYJIUCJXMLNPHU-UHFFFAOYSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- BZNAUEBFCXWMIV-UHFFFAOYSA-N triethyl(pent-2-en-3-yloxy)silane Chemical compound C(C)C(=CC)O[Si](CC)(CC)CC BZNAUEBFCXWMIV-UHFFFAOYSA-N 0.000 description 1
- SVGQCVJXVAMCPM-UHFFFAOYSA-N triethyl(prop-2-enyl)silane Chemical compound CC[Si](CC)(CC)CC=C SVGQCVJXVAMCPM-UHFFFAOYSA-N 0.000 description 1
- WILBTFWIBAOWLN-UHFFFAOYSA-N triethyl(triethylsilyloxy)silane Chemical compound CC[Si](CC)(CC)O[Si](CC)(CC)CC WILBTFWIBAOWLN-UHFFFAOYSA-N 0.000 description 1
- UFXIRMVZNARBDL-UHFFFAOYSA-N trifluoro(morpholin-4-yl)-$l^{4}-sulfane Chemical compound FS(F)(F)N1CCOCC1 UFXIRMVZNARBDL-UHFFFAOYSA-N 0.000 description 1
- SIOVKLKJSOKLIF-HJWRWDBZSA-N trimethylsilyl (1z)-n-trimethylsilylethanimidate Chemical compound C[Si](C)(C)OC(/C)=N\[Si](C)(C)C SIOVKLKJSOKLIF-HJWRWDBZSA-N 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
- C07H1/02—Phosphorylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H13/00—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids
- C07H13/02—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids
- C07H13/08—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids having the esterifying carboxyl radicals directly attached to carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon, or a metal, e.g. chelates, vitamin B12
Definitions
- Embodiments disclosed herein relate to the synthesis of phosphoramidate prodrugs useful in the treatment of viral infections. Specifically, embodiments relate to an improved synthesis of phosphoramidate nucleotides useful in the treatment of Hepatitis B virus.
- Hepatitis B virus is an infectious disease that targets the liver resulting in either an acute infection, with symptoms arising in 45 to 160 days, or a chronic infection, which 350 million people worldwide are affected by. Estimates indicate that 600,000 deaths occur each year as a result of consequences related to HBV infection.
- HBV possesses a 3.2- kb relaxed circular DNA (rcDNA) genome that is used to form covalently closed circular DNA (cccDNA) in a host cell.
- the cccDNA is then transcribed by RNA polymerase II, a host DNA-dependent RNA polymerase, to produce pregenomic RNA (pgRNA).
- pgRNA pregenomic RNA
- the pgRNA is then used by the virally encoded reverse transcriptase to form rcDNA.
- the goals of current treatments for chronic HBV infections are to reduce HBV replication and reduce liver damage.
- NRTIs nucleoside/nucleotide reverse transcriptase inhibitors
- Clevudine is an NRTI that is no longer being developed for the treatment of chronic HBV because of drug-related skeletal myopathy that was a result of mitochondrial dysfunction in patients.
- clevudine triphosphate has been shown to be a competitive nonsubstrate inhibitor of the HBV encoded polymerase, and due to its long intracellular half-life, is able to suppress HBV replication for an extended period of time after drug withdrawal.
- the discovery and synthesis of the (S,S) and (S,R) diastereomers of clevudine phosphoramidate has been previously reported. These studies were undertaken to address the myopathy concerns associated with clevudine. The phosphoramidate moiety was utilized to deliver clevudine, as its 5 '-monophosphate, to the liver reducing 1) systemic exposure to clevudine and 2) the possibility of skeletal myopathy. Both phosphoramidates showed anti-HBV activity similar to clevudine with the (S,S) diastereomer being slightly more potent.
- WO20 16099982 discloses compounds having the general formula: where R 1 is any one of several phosphoramidate groups. Specifically, they discovered the pro-drug compound referred to as EIDD-2173; also known as ATI-2173. See U.S. Patent No. 10,683,319.
- ATI-2173 is able to effectively and selectively deliver clevudine 5' monophosphate to the liver, bypassing the first systemic phosphorylation and thus reducing systemic exposure to clevudine allowing for the improved treatment of HBV.
- WO20 17223421 discloses that clevudine phosphoramidate compounds, such as
- ATI-2173 are additive or synergistic when combined with other antivirals such as lamivudine, adefovir, tenofovir, telbivudine, entecavir, or combinations thereof.
- the synthesis begins with commercially available clevudine, herein referred to as Compound-D, that is reacted with t-BuMgCl in THF solution in the presence of an excess of the pentafluoro compound SM-3, herein referred to as Compound-3.
- the reaction yields both ATI- 2173 and the clevudine 3', 5 '-bis phosphoramidate, as well as unreacted clevudine.
- Table 1 Summary of impurities from 1kg and 3kg batches [0017] Thus, what are needed are methods of more efficient, specific and selective synthesis for the clevudine phosphoramidates useful for the treatment of HBV.
- the present disclosure provides materials and methods of synthesizing clevudine phosphoramidates useful for the treatment of HBV infections. Specifically, disclosed are more efficient, specific and selective syntheses for the clevudine phosphoramidates that avoid the use of expensive and difficult to obtain starting materials, yield fewer impurities, provide greater stereoselectivity, are relatively stable under routine conditions allowing for the reaction to be performed at larger scales, and allow isolation by recrystallization.
- FIG. 1 is the X-Ray Powder Diffraction (XRPD) spectra of ATI-2173.
- FIG. 2 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with l.Oeq of TMSOTf.
- FIG. 3 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with 0.5eq of PhCOOH.
- FIG. 4 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with l.Oeq of PhCOOH.
- FIG. 5 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with 0.5eq of PhCOOH at 30g scale.
- FIG. 6 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with 0.5eq of PhCOOH at 30g scale, as the wet cake isolate.
- FIG. 7 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with 0.5eq of PhCOOH at 30g scale, recrystallized from EtOH.
- reduce or other forms of the word, such as “reducing” or “reduction,” is meant lowering of an event or characteristic (e.g., viral infection). It is understood that this is typically in relation to some standard or expected value, in other words it is relative, but that it is not always necessary for the standard or relative value to be referred to. For example, “reduces viral infection” means decreasing the amount of viral load relative to a standard or a control.
- an event or characteristic e.g., viral infection
- prevent or other forms of the word, such as “preventing” or “prevention,” is meant to stop a particular event or characteristic, to stabilize or delay the development or progression of a particular event or characteristic, or to minimize the chances that a particular event or characteristic will occur. Prevent does not require comparison to a control as it is typically more absolute than, for example, reduce. As used herein, something could be reduced but not prevented, but something that is reduced could also be prevented. Likewise, something could be prevented but not reduced, but something that is prevented could also be reduced. It is understood that where reduce or prevent are used, unless specifically indicated otherwise, the use of the other word is also expressly disclosed. [0039] As used herein, “treatment” refers to obtaining beneficial or desired clinical results.
- Beneficial or desired clinical results include, but are not limited to, any one or more of: alleviation of one or more symptoms (such as infection), diminishment of extent of infection, stabilized (i.e., not worsening) state of infection, preventing or delaying spread of the infection, preventing or delaying occurrence or recurrence of infection, and delay or slowing of infection progression.
- symptoms such as infection
- stabilized i.e., not worsening
- the term “patient” may refer to a human in need of treatment with an antibiotic or treatment for any purpose, such as a human in need of such a treatment to treat viral infection.
- the term “patient” can also refer to non-human animals, including but limited to, mammals such as dogs, cats, horses, cows, pigs, sheep and non-human primates, among others, that are in need of treatment with an antibiotic or treatment for any purpose, such as with an antiviral compound.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- References in the specification and concluding claims to parts by weight of a particular element or component in a composition denotes the weight relationship between the element or component and any other elements or components in the composition or article for which a part by weight is expressed.
- X and Y are present at a weight ratio of 2:5, and are present in such ratio regardless of whether additional components are contained in the mixture.
- a weight percent (wt.%) of a component unless specifically stated to the contrary, is based on the total weight of the formulation or composition in which the component is included.
- substantially pure means sufficiently homogeneous to appear free of readily detectable impurities as determined by standard methods of analysis, such as thin layer chromatography (TLC), nuclear magnetic resonance (NMR), gel electrophoresis, high performance liquid chromatography (HPLC) and mass spectrometry (MS), gas-chromatography mass spectrometry (GC-MS), and similar, used by those of skill in the art to assess such purity, or sufficiently pure such that further purification would not detectably alter the physical and chemical properties, such as enzymatic and biological activities, of the substance.
- TLC thin layer chromatography
- NMR nuclear magnetic resonance
- HPLC high performance liquid chromatography
- MS mass spectrometry
- GC-MS gas-chromatography mass spectrometry
- a “pharmaceutically acceptable” component is one that is suitable for use with humans and/or animals without undue adverse side effects (such as toxicity, irritation, and allergic response) commensurate with a reasonable benefit/risk ratio.
- “Pharmaceutically acceptable salt” refers to a salt that is pharmaceutically acceptable and has the desired pharmacological properties. Such salts include those that may be formed where acidic protons present in the compounds are capable of reacting with inorganic or organic bases. Suitable inorganic salts include those formed with the alkali metals, e.g., sodium, potassium, magnesium, calcium, and aluminum. Suitable organic salts include those formed with organic bases such as the amine bases, e.g., ethanolamine, diethanolamine, triethanolamine, tromethamine, N-methylglucamine, and the like.
- Such salts also include acid addition salts formed with inorganic acids (e.g., hydrochloric and hydrobromic acids) and organic acids (e.g., acetic acid, citric acid, maleic acid, and the alkane- and arene-sulfonic acids such as methanesulfonic acid and benzenesulfonic acid).
- inorganic acids e.g., hydrochloric and hydrobromic acids
- organic acids e.g., acetic acid, citric acid, maleic acid, and the alkane- and arene-sulfonic acids such as methanesulfonic acid and benzenesulfonic acid.
- acetic acid e.g., citric acid, maleic acid
- alkane- and arene-sulfonic acids such as methanesulfonic acid and benzenesulfonic acid.
- “Pharmaceutically acceptable excipient” refers to an excipient that is conventionally useful in preparing a pharmaceutical composition that is generally safe, non-toxic, and desirable, and includes excipients that are acceptable for veterinary use as well as for human pharmaceutical use. Such excipients can be solid, liquid, semisolid, or, in the case of an aerosol composition, gaseous.
- a “pharmaceutically acceptable carrier” is a carrier, such as a solvent, suspending agent or vehicle, for delivering the disclosed compounds to the patient.
- the carrier can be liquid or solid and is selected with the planned manner of administration in mind.
- Liposomes are also a pharmaceutical carrier.
- carrier includes any and all solvents, dispersion media, vehicles, coatings, diluents, antibacterial and antifungal agents, isotonic and absorption delaying agents, buffers, carrier solutions, suspensions, colloids, and the like. The use of such media and agents for pharmaceutical active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in the therapeutic compositions is contemplated.
- terapéuticaally effective amount means that amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- the present disclosure provides materials and methods of synthesizing clevudine phosphoramidates useful for the treatment of HBV infections.
- the methods disclosed herein are more efficient, specific and provide for the more selective synthesis for the clevudine phosphoramidates.
- the methods disclosed avoid the use of expensive commercially available starting materials, yield fewer impurities, provide greater stereoselectivity, are stable under routine conditions allowing for the reaction to be performed at larger scales, and allow isolation by recrystallization.
- Table 2 depicts the designated name used throughout the current specification for each of the respective compounds and intermediates. The table also shows the standard IUPAC (International Union of Pure and Applied Chemistry) name for each as well as the structure.
- IUPAC International Union of Pure and Applied Chemistry
- Certain embodiments disclosed herein provide a process for the preparation of ATI-
- reaction temperature is cooled to 25 °C or less
- the fluorinating agent of step (A-2) is selected from, but not limited to, trihydrofluorine trimethylamine, HF pyridine complex, bis(2-methoxy- ethyl)aminosulfur trifluoride (BAST, also called Deoxo-Fluor®), diethylaminosulfur trifluoride (DAST), sulfur tetrafluoride, morpholinosulfur trifluoride, (diethylamino)difluorosulfonium tetrafluoroborate, 2-pyridinesulfonyl fluoride, N-fluorobenzenesulfonimide, perfluoro- 1 -butanesulfonyl fluoride, 2-thiophenesulfonyl fluoride, l-chloromethyl-4-fluoro-l,4-diazoniabicyclo- [2.2.2]octane bis(tetrafluoroborate), or
- step (A-3) can be raised continuously up to 75 °C-85 °C or carried out incrementally in a step-wise fashion.
- the temperature of step (A-3) is first raised to 40 °C-50 °C, the raised 75 °C-85 °C.
- the reaction at step (A-3) is stirred at 75 °C-85 °C for 15-20 hours.
- the reaction at step (A-3) is stirred at 75 °C-85 °C until Compound-SM-1 is consumed by HPLC.
- the reaction in step (A-4) is cooled to 15 °C-25 °C.
- the NaHCCh aqueous solution of step (A-5) is a 7% NaHCOs aqueous solution.
- the NaHCOs of step (A-5) is added slowly.
- the NaHCOs solution of step (A-5) is added dropwise.
- the reaction mixture is first diluted with additional EtOAc and MeOH.
- the organic solvent fraction of step (A-6) may be washed one or more time. In embodiments, the organic solvent fraction of step (A-6) may be washed one, two, three, four, five, six, seven, eight, nine, or even ten times. In embodiments, the organic solvent fraction of step (A-6) may be washed more than ten times.
- the EtOAc of step (A-7) is reduced to about 2 volumes at a temperature of not more than 45 °C. In an embodiment the EtOAc of step (A-7) is removed entirely except for a trace quantity at a temperature of not more than 45 °C. In an embodiment the EtOAc of step (A-7) is removed entirely at a temperature of not more than 45 °C.
- step (A-8) before charging with EtOH and warming, the reaction is charged with an separate initial amount of EtOH. This initial EtOH may then be distilled off at a temperature of not more than 45 °C to about 2 volumes; then, the reaction mixture may be charged with the EtOH to be warmed. In an embodiment the reaction mixture of step (A-8) is warmed from about 70 °C to about 80 °C for 1-2 hours.
- step (A-9) the reaction is cooled to ⁇ 5 °C.
- the mixture of step (A-9) is stirred at the cooled temperature for 8-14 hours.
- the mixture of step (A-9) is stirred at the cooled temperature for 9 hours.
- the filtration of step (A-9) is carried out in a centrifuge.
- the filtrate of step (A- 10) is warmed to about 45 °C while drying under vacuum.
- Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-5, where Compound-5 is prepared from the reaction of Compound-4 in hydrogen bromide (HBr), acetic acid (AcOH), and dichloromethane (DCM).
- HBr hydrogen bromide
- AcOH acetic acid
- DCM dichloromethane
- reaction mixture is diluted with additional dichloromethane and quenched with sodium bicarbonate solution to a pH of about 6 to about 7;
- the temperature of step (B-l) is adjusted between about 10 °C and about 20 °C.
- the temperature of step (B-l) may be adjusted between about 11 °C and about 19 °C, between about 12 °C and about 18 °C, between about 13 °C and about 17 °C, or between about 14 °C and about 16 °C.
- step (B-3) is stirred for about 12 hours to about 20 hours. In another embodiment, the mixture of step (B-3) is stirred for about 15 hours. In an embodiment, step (B-3) is monitored for completeness by HPLC. In an embodiment, in step (B-3) additional portions of HBr/acetic acid can be added with additional time spent stirring until the starting material is consumed.
- the sodium bicarbonate solution of step (B-4) is a 7% sodium bicarbonate solution.
- the temperature during step (B-4) is maintained below 25 °C.
- the mixture of step (B-4) is cooled to between about 15 °C and about 20 °C with stirring.
- the sodium sulfate solution of step (B-5) is a 5% sodium sulfate solution.
- the organic layer of step (B-5) is washed more than once. In an embodiment, the organic layer of step (B-5) is washed twice.
- the filtrate of step (B-7) is reduced to about 1-2 volumes.
- Compound-B has a purity of greater than about 95% or greater based on the potency of the product solution.
- Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-6, where Compound-6 may be prepared by the reaction of Compound-5 with Compound-2 in the presence of A,O-bis(trimethylsilyl)acetamide (BSA), benzoic acid and 1,4-di oxane.
- BSA A,O-bis(trimethylsilyl)acetamide
- the preparation of Compound-6 may be carried out where: [0097] (C-l) Compound-2 is loaded in 1,4-dioxane with BSA;
- Compound-6 may be produced as a wet-cake, where in step C-10 the product is not dried rigorously. Compound-6 may be produced with a purity of greater than or equal to about 99%.
- HMDS hexamethyldisilazane
- the reaction in step (C-2) is stirred for up to 48 hours. In an embodiment, the reaction in step (C-2) is stirred for 2-8 hours. In an embodiment, the reaction in step (C-2) is stirred until the solution becomes clear.
- the Compound-5 added in step (C-3) is used as a crude solution from the production of Compound-5.
- di chloromethane is also added during step (C-3) before the addition of benzoic acid.
- the reaction at step (C-3) is maintained at about 70 °C to about 75 °C.
- the reaction at step (C-3) is first cooled to about 20 °C to about 30 °C before addition of Compound-5.
- the reaction at step (C-3) is cooled to about 20 °C to about 25 °C before addition of benzoic acid.
- the benzoic acid of step (C-3) is added in portions.
- the temperature of step (C-3) is maintained at about 20 °C to about 25 °C during addition of the benzoic acid. In an embodiment, if the reaction mixture of (C-3) is cooled, it is returned to about 70 °C to about 75 °C after addition of the benzoic acid. In an embodiment, the reaction mixture of (C-3) is stirred at about 70 °C to about 75 °C for about 42 hours to about 75 hours. In an embodiment, the reaction mixture of (C-3) is stirred at about 70 °C to about 75 °C for about 48 hr. [OHl] In an embodiment, the sodium bicarbonate solution used in quenching and or washing is a 7% sodium bicarbonate solution.
- reaction mixture of (C-4) is first cooled between about 20 °C to about 30 °C before quenching, and the temperature is maintained in this range during quenching.
- sodium bicarbonate solution of (C- 4) is added until the mixture has a pH of about 7 to about 8.
- the mixture of (C-5) can be filtered before separation of the aqueous and organic layers.
- the organic layer of step (C-5) can be washed with sodium bicarbonate solution two or more times.
- step (C-7) the addition and removal of EtOH of step (C-7) can be carried out two or more times.
- distillation of EtOH in step (C-7) is carried out at temperature of about 65 °C or less under reduced pressure.
- the EtOH mixture of step (C-8) is warmed to a temperature of about 80 °C to about 85 °C. In an embodiment, the EtOH mixture of step (C-8) is warmed for about 1 hour to about 5 hours until clear.
- step (C-9) is cooled to a temperature of about 20 °C to about 25 °C. In an embodiment, step (C-9) is cooled gradually over 5 hours to 6 hours. In an embodiment, step (C-9) is stirred at the cooled temperature for about 5 hours to about 18 hours. In an embodiment, step (C-9) can be repeated to obtain the desired purity.
- Step D Synthesis of Compound-7 (Clevudine)
- Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-7, where Compound-7 (Clevudine) is synthesized by reacting Compound-6 in the presence of sodium methoxide (NaOMe).
- the preparation of Compound-7 is carried out where: [0118] (D-l) Compound-6 wet cake is added with methanol;
- the resulting Compound-7 has a purity of about 99% or greater and about 80% to about 90% yield.
- the reaction mixture of step (D-2) is cooled to about 10 °C to about 15 °C before addition of sodium methoxide.
- the base is selected from sodium hydroxide, sodium methoxide, ammonia, pyridine, methylamine, potassium cyanide, or a combination thereof.
- the base is selected from sodium hydroxide or sodium methoxide.
- the base may be used in stoichiometric or catalytic amounts.
- there reaction mixture of step (D-2) is not allowed to rise above 20 °C.
- the reaction mixture of step (D-2) is warmed to a temperature of about 40 °C to about 45 °C. In an embodiment, the reaction mixture is stirred for about 12 hours to about 18 hours.
- step (D-3) is maintained below about 30 °C.
- step (D-4) is maintained below about 45 °C.
- step (D-5) is stirred for about 5 hours to about 12 hours.
- the wet cake of step (D-6) may be rinsed with additional IP Ac.
- the wet cake of step (D-7) is crystallized.
- crude Compound-7 can be crystallized from ethanol.
- Step E triethylsilylating selective agent deprotection
- Step E triethylsilylating selective agent deprotection
- the new and improved synthesis includes a selective deprotection of Compound-8 to afford Compound-9.
- Compound-7 (Clevudine) is first reacted in the presence of chlorotriethylsilane, imidazole, and methyl-tert butyl ether (MTBE) to yield the intermediate Compound-8, which is subsequently deprotected with a triethylsilylating agent to provide Compound-9.
- the isolated yield for the two steps, protection (Step E) and selective deprotection (Step F) may be at least about 70%, such as at least 75%, at least 80%, at least 85%, at least 90%, or even at least 95%.
- Compound-9 may be recrystallized to afford a crystalline, white solid with a purity of >99%.
- Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-8, where:
- (E-2) triethylsilylating agent is added at a rate to maintain the temperature at below about 5 °C;
- the resulting Compound-9 has a purity of greater than 99% in a solution of greater than 40% assay.
- Triethylsilylating agent refers to a reagent capable of converting an alcohol to a triethylsilyl ether.
- the triethylsilylating agent is selected from triethylsilyl chloride (TES), A-methyl-A-triethylsilyltrifluoroacetamide, -tri ethyl silyl acetamide, allyl tri ethyl si lane, A-triethylsilylacetamide, 1-methoxy-l-triethylsiloxypropene, l-methoxy-2-m ethyl - 1-triethylsiloxypropene, triethylsilyl triflate, triethylsilyl trifluoromethane, triethylsilyl imidazole, triethylsilyl bromide, triethylsilyl iodide, triethy
- a base is loaded along with Compound-7 to promote silylation.
- the base is selected from imidazole, pyridine, triethylamine, diisopropyl ethylamine, 2,6-lutidine, 1 -methylimidazole, ammonia, pyrrolidine, pyrrole, pyrimidine, piperidine, l,8-diazabicyclo[5.4.0]undec-7-ene, 4-methylmorpholine, 1,4- diazabicyclo[2.2.2]octane (DABCO), tetramethylethylenediamine (TMEDA), trimethylamine, diisopropylamine, sodium hydride, n-butyllithium, lithium diisopropylamide (LDA), potassium tert-butoxide or a combination thereof.
- a catalyst is loaded along with Compound-7 to promote silylation.
- the catalyst is selected from an ammonium halide, sodium halide, potassium halide, dimethylaminopyridine, 2-hydroxypyridine, sodium cyanide, or a combination thereof.
- the solvent is a solvent or solvent system in which selective silylation is carried out successfully.
- the solvent is selected from methyl /-butyl ether (MTBE), tetrahydrofuran (THF), diethyl ether, 1,4-di oxane, hexanes, n-heptane, chloroform, dichloromethane (DCM), dimethylformamide (DMF), methanol, acetonitrile, ethyl acetate, isopropyl acetate, water, or a combination thereof.
- the solvent is MTBE.
- step (E-l) is adjusted to between about -5 °C and about 5 °C.
- step (E-2) the temperature of step (E-2) is maintained at between about -5 °C and about 5 °C during addition of the tri ethyl silylating agent. In an embodiment, during step (E-2) additional solvent can be added.
- the reaction in step (E-3) is stirred for about 5 hours to about 16 hours.
- additional base can be added and the reaction stirred for additional time.
- the amount of imidazole used may be regulated so as to prevent over de-protection in Step F.
- the amount of residual imidazole present may be less than 0.1 weight % (“wt%”), less than 0.09 wt%, less than 0.08 wt%, less than 0.07 wt%, less than 0.06 wt%, less than 0.05 wt%, less than 0.04 wt%, less than 0.03 wt%, less than 0.02 wt%, less than 0.01 wt%, or even less than 0.005 wt%.
- the amount of residual imidazole present may be about 0.015 wt%. In some embodiments, there may be no measurable quantity of residual imidazole.
- the ammonium acetate of step (E-4) is a 10% ammonium acetate solution.
- quenching of step (E-4) is carried out at a temperature below about 5 °C.
- the organic phase of (E-5) can be washed with ammonium acetate one or more time. In an embodiment, the organic phase of (E-5) can be washed with water one or more time. In an embodiment, the filtrate of (E-5) is concentrated to about 50% volume. In an embodiment, the filtrate of (E-5) is concentrated to provide MTBE solution of greater than about 70% assay.
- Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-9, where: [0154] (F-l) The MTBE solution containing Compound-8 is added with an additional solvent;
- Compound-8 of step (F-l) is carried forward as a dried solid after concentration of the previous step, a paste after concentration of the previous step, or in concentrated solution from the previous step; and added to the additional solvent of step (F-l).
- the additional solvent of step (F-l) is selected from methanol, ethanol, //-propanol, isopropanol, THF, water, dichloromethane or combinations thereof.
- Compound-8 of step (F-l) may be added with additional MTBE.
- the acidic reagent of step (F-2) is selected from pyridinium p- toluenesulfonate (PPTS); acetic acid; formic acid; proprionic acid; acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®; hydrochloric acid; trifluoroacetic acid; trichloroacetic acid; tetra-n-butyl ammonium fluoride (TBAF); tetrabutyl ammonium chloride; ceric ammonium nitrate immobilized on silica gel; ( ⁇ ) camphorsulfonic acid or combinations thereof.
- PPTS pyridinium p- toluenesulfonate
- acetic acid formic acid
- proprionic acid acidic resin
- acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®
- hydrochloric acid trifluoroacetic acid
- step (F-2) is cooled to abut -15 °C to about -5 °C and the temperature is maintained within this range during addition of the acidic reagent of step (F-2).
- step (F-2) is stirred for 5-28 hr.
- the reaction at step (F-4) can first be diluted with MTBE before quenching.
- the ammonium acetate solution of (F-4) is 10% ammonium acetate.
- the aqueous layer of step (F-4) can be further extracted with additional MTBE.
- the organic layer of step (F-4) can be washed with additional ammonium acetate solution or water.
- solvent is removed under pressure at a temperature of less than about 50 °C, such as less than about 45 °C.
- Compound-9 is obtained in greater than about 75%. In another embodiment, Compound-9 is obtained in greater than about 85%. In yet another embodiment,
- Compound-9 is obtained in greater than about 95%. In an embodiment, Compound-9 is obtained in greater than about 99%.
- Step G & Step H Synthesis of ATI-2173:
- Step H the crude reaction solution from Step G can be treated with 2% HC1 and stirred at ambient temperature to afford crude ATI-2173 (Step H).
- the crude material may be recrystallized from isopropyl acetate/n-heptane or MTBE/n-heptane to afford purified ATI-2173.
- ATI-2173 is produced in greater than about 70% yield with greater than about 90% purity.
- the solvent of step (G-l) is selected from tetrahydrofuran, diethyl ether, isopropyl ether, diglyme, 1,4-di oxane, dibutyle ether, or combinations thereof.
- the base of step (G-2) is selected from Zc/7-butylmagnesium chloride, Zc/7-butylmagnesium bromide, methylmagnesium chloride, methylmagnesium bromide, sodium hydride, or lithium aluminum hydride.
- the base of step (G-2) is Zc/7- butylmagnesiumchloride, 1.7 M, in THF solution.
- the reaction mixture of (G- 2) is cooled between about -10 °C and about -5 °C.
- the base of step (G-2) is added dropwise.
- the base of step (G-2) is added slowly enough such that the temperature is maintained below about -5 °C.
- the reaction mixture of (G-3) is first stirred at about -10 to about -5 °C for about 0.5 hour to about 1.0 hr. In an embodiment, the reaction mixture of (G-3) is adjusted to between about 20 °C to about 50 °C and stirred for the prescribed time.
- the acidic reagent of step (H-l) may be added neat or as a solution. In an embodiment, the acidic reagent of step (H-l) may be added as an aqueous solution. In an embodiment the acidic reagent of step (H-l) is selected from hydrochloric acid, pyridinium -toluenesulfonate; acetic acid; formic acid; proprionic acid; acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®; or combinations thereof. In an embodiment the acidic reagent of step (H-l) is a 2% solution of hydrochloric acid.
- the reaction mixture is maintained below about 25 °C during addition of the acidic reagent of step (H-l).
- the reaction mixture of (H-l) is stirred for about 12 hours to about 18 hours. In an embodiment, if there is not complete conversion during step (H-l), additional acidic reagent may be added and the reaction stirred for additional time.
- the sodium bicarbonate solution of step (H-2) is a 7% sodium bicarbonate solution.
- the work up of step (H-3) may also use THF in the organic layer.
- the organic layer of (H-3) may be distilled to a smaller volume before or between washing.
- the organic layer of (H-3) may also be washed using NaCl aqueous solution.
- the concentrated solution of step (H-4) may be diluted with IP Ac and reconcentrated.
- seed crystals are added to the mixture of step (H-5).
- the mixture of step (H-5) is stirred for about 12 hours to about 16 hours.
- the solid of step (H-6) is filtered by centrifuge.
- crude ATI-2173 can be crystallized from an isopropyl acetate/heptane mixture. In an embodiment, crude ATI-2173 may be crystallized from MTBE/n-heptane to afford a crystalline, white solid with a purity of >99%. In an embodiment, ATI-2173 is produced in greater than about 75% yield. In another embodiment, ATI-2173 is produced in greater than about 85% yield. In yet another embodiment, ATI-2173 is produced in greater than about 95% yield. In an embodiment, ATI-2173 is produced with greater than about 95% purity. In another embodiment, ATI-2173 is produced with greater than about 99% purity.
- a process for the preparation of ATI-2173 comprises silylating Compound-7 to form selectively silylated Compound-9; followed by coupling with Compound-3; and deprotection to provide ATI-2173.
- ATI-2173 is substantively free of Impurity- 1.
- a process for the formation of Compound-9 comprises reacting Compound-7 with a triethylsilylating agent to form intermediate Compound-8 then selectively deprotecting the primary alcohol to form Compound- 9.
- the triethylsilylating agent is selected from triethylsilyl chloride, A-methyl-A-triethylsilyltrifluoroacetamide, N- tri ethyl silyl acetamide, allyl tri ethylsilane, A-tri ethyl silyl acetamide, 1 -methoxy- 1- triethylsiloxypropene, l-methoxy-2-methyl-l-triethylsiloxypropene, tri ethylsilyl triflate, triethylsilyl trifluoromethane, triethylsilyl imidazole, triethylsilyl bromide, triethylsilyl iodide, triethylsilyl cyanide, or a combination thereof.
- the base selected from imidazole, pyridine, tri ethylamine, diisopropyl ethylamine, 2,6-lutidine, 1 -methylimidazole, ammonia, pyrrolidine, pyrrole, pyrimidine, piperidine, l,8-diazabicyclo[5.4.0]undec-7-ene, 4- methylmorpholine, l,4-diazabicyclo[2.2.2]octane (DABCO), tetramethylethylenediamine (TMEDA), trimethylamine, diisopropylamine, sodium hydride, n-butyllithium, lithium diisopropylamide (LDA), potassium tert-butoxide or a combination thereof.
- the base selected from imidazole, pyridine, tri ethylamine, diisopropyl ethylamine, 2,6-lutidine, 1 -methylimidazole, ammonia, pyrrolidine
- methyl /-butyl ether is used as a solvent.
- the selective deprotection is carried out in the presence of a reagent selected from pyridinium /?-toluenesulfonate (PPTS); acetic acid; formic acid; proprionic acid; acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®; hydrochloric acid; trifluoroacetic acid; trichloroacetic acid; tetra-n-butylammonium fluoride (TBAF); tetrabutyl ammonium chloride; ceric ammonium nitrate immobilized on silica gel; ( ⁇ ) camphorsulfonic acid or combinations thereof.
- a reagent selected from pyridinium /?-toluenesulfonate (PPTS); acetic acid; formic acid; proprionic acid; acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®; hydrochloric acid; tri
- the triethylsilylating agent is triethylsilyl chloride, where the selective silylation is carried out in the presence of imidazole in methyl /-butyl ether, and the selective deprotection is carried out in the presence of pyridinium p- toluenesulfonate.
- a process for the formation of ATI-2173 comprises reacting Compound-9 with Compound-3 forming the intermediate Compound- 10 where the silyl group is then removed to provide ATI-2173.
- the basic reactant is /c/7-butylmagnesiumchloride.
- the silyl group is removed in the presence of an acidic reagent, the acidic reagent selected from hydrogen chloride, acetic acid, acid resin, citric acid, sulfuric acid, sulfurous acid, phosphoric acid, methanoic acid, benzoic acid, formic acid, hydrogen bromide, or combinations thereof.
- an acidic reagent selected from hydrogen chloride, acetic acid, acid resin, citric acid, sulfuric acid, sulfurous acid, phosphoric acid, methanoic acid, benzoic acid, formic acid, hydrogen bromide, or combinations thereof.
- ATI-2173 is substantively free of Impurity- 1
- ATI-2173 is crystallized in a mixture of IP Ac and //-heptane.
- a process for the formation of Compound-4 comprises reacting Compound-1 in the presence of bis(2- methoxyethyl)aminosulfur trifluoride.
- a process for the formation of Compound-6 comprises reacting Compound-4 with HBr/acetic acid to form Compound-5 then reacting Compound-2 with BSA, benzoic acid, and Compound-4 to form Compound-5.
- Compound-4 may be purified before reaction with Compound-2.
- Compound-4 may be used as a crude product in reaction with Compound-2.
- Compound-6 is reacted with a base to form Compound-7, wherein the base is selected from sodium hydroxide, sodium methoxide, ammonia, pyridine, methylamine, potassium cyanide, or a combination thereof.
- the base is selected from sodium hydroxide, sodium methoxide, ammonia, pyridine, methylamine, potassium cyanide, or a combination thereof.
- Compound-4 was prepared from the reaction of Compound-1 and BAST in EtOAc. In order to avoid production of several unknown impurities caused by prolonging the reaction time, a modified purification process of Compound-4 was developed that utilized crystallization in EtOH. By crystallization in EtOH, the unknown impurities were purged and the purity of Compound-4 could be improved from 85-89% to >98%. The typical yield of this step in kilogram batches was in range of about 83-88%. [0218] Experimental Procedure 1
- Compound-5 was prepared from the reaction of Compound-4 and HBr (33% AcOH sol.). In initial experiments, it was found that conversion of Compound-4 with HBr in EtOAc at 20 °C gave incomplete conversion of the starting material. In order to improve the conversion rate, the solvent was changed from EtOAc to DCM and the charging amount of HBr was increased. In addition, it was found that Compound-5 was unstable under the original HPLC conditions. At the beginning, the HPLC purity of isolated Compound-5 was -70%, but the 'H-NMR spectrum was clean and Q-NMR assay was high. After optimization of the analytical HPLC method, the HPLC purity of Compound-5 in typical kilo-lab scale-up batches was about 94%.
- Compound-6 was prepared from the reaction of Compound-2 (5-methylpyrimidine-
- N,O-bis(trimethylsilyl)acetamide could replace HMDS to prepare the Compound-2 intermediate and avoid generation of the nucleophilic compound TMS-NH2 and avoid degradation of Compound-5.
- BZA N,O-bis(trimethylsilyl)acetamide
- the addition of BzOH in the reaction of the Compound-2 intermediate and Compound-5 could help to increase the diastereoselectivity.
- the typical IPC purity level of Compound-6 was 85-89%. After crystallization in EtOH, the diastereoisomer could be removed almost completely, and the purity of Compound-6 was >99%.
- the yield of this step was >70%. Further experiments determined that a possible solvent is THF/DCM (5V:5V).
- the impurity Compound- 11 was generated with 23.28%.
- THF and ACN were used as solvents, and after stirring for total 22 hr at 63 °C, Compound-5 was consumed completely and >73% of Compound- 6 was generated.
- the impurity Compound-11 was generated with 16.00%.
- 1,4-dioxane was used as the solvent, and after stirring for a total of 41 hr at 65-70 °C, 87.83% of Compound-6 was generated, the residual amount of Compound-5 was 3% and the impurity Compound-11 was 2.86%.
- 1,4- dioxane would be selected over the other solvents. However, it was found that upon scale up, 1,4- dioxane did not work well with an isolated yield of product Compound-6 of only -36%.
- the compound Compound-7 (Clevudine) was prepared from the reaction of compound Compound-6 and NaOMe in MeOH. Losing a charging amount of NaOMe 0.4-0.5 eq and trifluoroacetic acid (TFA) quenching and an isopropyl acetate (IP Ac) crystallization process, Compound-7 (Clevudine) could be obtained as white solid with >99% purity and >85% yield.
- TFA trifluoroacetic acid
- IP Ac isopropyl acetate
- IP Ac is charged to the mixtrure and concentrated under reduced pressure at ⁇ 35 °C (in-process check: residual THF (%, w/w) specification ⁇ 5%);
- ATI-2173 The final compound, ATI-2173, is crystalline based on extensive XRPD and compared to a reference standard. It has also been subjected to DSC and shows a consistent, sharp melting point. ATI-2173 has been fully characterized by TGA, residual solvents by GC, heavy metals by ICP-MS and/or ICP-OES, residue on ignition and water content. Purity was established using HPLC and chiral HPLC; extensive NMR experiments ( 1 H, 13 C, 19 F and 31 P NMR as well as 2D experiments) were performed to determine/verify the structure. FIG. 1 shows the XRPD spectrum of the crystalline ATI-2173
- Examples of methods to obtain optically active materials include at least the following, i) physical separation of crystals-a technique whereby macroscopic crystals of the individual enantiomers are manually separated. This technique can be used if crystals of the separate enantiomers exist, i.e., the material is a conglomerate, and the crystals are visually distinct; ii) simultaneous crystallization-a technique whereby the individual enantiomers are separately crystallized from a solution of the racemate, possible only if the latter is a conglomerate in the solid state; iii) enzymatic resolutions-a technique whereby partial or complete separation of a racemate by virtue of differing rates of reaction for the enantiomers with an enzyme; iv) enzymatic asymmetric synthesis-a synthetic technique whereby at least one step of the synthesis uses an enzymatic reaction to obtain an enantiomerically pure or enriched synthetic precursor of the desired enantiomer; v) chemical separation of crystals-a technique whereby mac
- the resulting diastereomers are then separated by chromatography or crystallization by virtue of their now more distinct structural differences and the chiral auxiliary later removed to obtain the desired enantiomer; vii) first- and second-order asymmetric transformations-a technique whereby diastereomers from the racemate equilibrate to yield a preponderance in solution of the diastereomer from the desired enantiomer or where preferential crystallization of the diastereomer from the desired enantiomer perturbs the equilibrium such that eventually in principle all the material is converted to the crystalline diastereomer from the desired enantiomer.
- kinetic resolutions-this technique refers to the achievement of partial or complete resolution of a racemate (or of a further resolution of a partially resolved compound) by virtue of unequal reaction rates of the enantiomers with a chiral, non-racemic reagent or catalyst under kinetic conditions; ix) enantiospecific synthesis from non-racemic precursors— a synthetic technique whereby the desired enantiomer is obtained from non-chiral starting materials and where the stereochemical integrity is not or is only minimally compromised over the course of the synthesis; x) chiral liquid chromatography— a technique whereby the enantiomers of a racemate are separated in a liquid mobile phase by virtue of their differing interactions with a stationary phase.
- the stationary phase can be made of chiral material or the mobile phase can contain an additional chiral material to provoke the differing interactions; xi) chiral gas chromatography-a technique whereby the racemate is volatilized and enantiomers are separated by virtue of their differing interactions in the gaseous mobile phase with a column containing a fixed non-racemic chiral adsorbent phase; xii) extraction with chiral solvents-a technique whereby the enantiomers are separated by virtue of preferential dissolution of one enantiomer into a particular chiral solvent; xiii) transport across chiral membranes-a technique whereby a racemate is placed in contact with a thin membrane barrier.
- the barrier typically separates two miscible fluids, one containing the racemate, and a driving force such as concentration or pressure differential causes preferential transport across the membrane barrier. Separation occurs as a result of the non-racemic chiral nature of the membrane that allows only one enantiomer of the racemate to pass through.
- Chiral chromatography including simulated moving bed chromatography, is used in one embodiment.
- a wide variety of chiral stationary phases are commercially available.
- some of the nucleosides described herein may exist as tautomers, such as, keto-enol tautomers. The individual tautomers as well as mixtures thereof are intended to be encompassed within the compounds of the present subject matter.
- compositions and methods disclosed and claimed herein can be made and executed without undue experimentation in light of the present disclosure. While the subject matter disclosed herein may be embodied in many different forms, there are described in detail herein specific embodiments. The present disclosure is an exemplification of the principles of the disclosed subject matter and is not intended to limit the subject matter to the particular embodiments illustrated.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
The synthesis of phosphoramidate prodrugs useful in the treatment of viral infections is disclosed. Specifically, an improved synthesis of phosphoramidate nucleotides useful in the treatment of Hepatitis B virus is disclosed.
Description
IMPROVED SYNTHESIS OF PHO SPHORAMID ATES FOR THE TREATMENT OF HEPATITIS B VIRUS
[0001] CROSS-REFERENCE TO RELATED APPLICATION
[0002] This application claims priority to U.S. Provisional Application No. 63/196,418, filed on June 3, 2021, the entire content of which is incorporated by reference herein.
[0003] FIELD
[0004] Embodiments disclosed herein relate to the synthesis of phosphoramidate prodrugs useful in the treatment of viral infections. Specifically, embodiments relate to an improved synthesis of phosphoramidate nucleotides useful in the treatment of Hepatitis B virus.
[0005] BACKGROUND
[0006] Hepatitis B virus (HBV) is an infectious disease that targets the liver resulting in either an acute infection, with symptoms arising in 45 to 160 days, or a chronic infection, which 350 million people worldwide are affected by. Estimates indicate that 600,000 deaths occur each year as a result of consequences related to HBV infection. HBV possesses a 3.2- kb relaxed circular DNA (rcDNA) genome that is used to form covalently closed circular DNA (cccDNA) in a host cell. The cccDNA is then transcribed by RNA polymerase II, a host DNA-dependent RNA polymerase, to produce pregenomic RNA (pgRNA). The pgRNA is then used by the virally encoded reverse transcriptase to form rcDNA. The goals of current treatments for chronic HBV infections are to reduce HBV replication and reduce liver damage.
[0007] Current treatments for chronic HBV infections include pegylated alpha interferon and nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs). NRTIs are converted to their corresponding 5 '-triphosphate, or diphosphate in the case of phosphonate containing NRTIs, and reduce viral replication by inhibiting the HBV encoded polymerase. Clevudine is an NRTI that is no longer being developed for the treatment of chronic HBV because of drug-related skeletal myopathy that was a result of mitochondrial dysfunction in patients.
[0008] Interestingly, clevudine triphosphate has been shown to be a competitive nonsubstrate inhibitor of the HBV encoded polymerase, and due to its long intracellular half-life, is able to suppress HBV replication for an extended period of time after drug withdrawal.
[0009] The discovery and synthesis of the (S,S) and (S,R) diastereomers of clevudine phosphoramidate has been previously reported. These studies were undertaken to address the myopathy concerns associated with clevudine. The phosphoramidate moiety was utilized to deliver clevudine, as its 5 '-monophosphate, to the liver reducing 1) systemic exposure to clevudine and 2) the possibility of skeletal myopathy. Both phosphoramidates showed anti-HBV activity similar to clevudine with the (S,S) diastereomer being slightly more potent.
[0010] WO20 16099982 discloses compounds having the general formula:
 where R1 is any one of several phosphoramidate groups. Specifically, they discovered the pro-drug compound referred to as EIDD-2173; also known as ATI-2173. See U.S. Patent No. 10,683,319.
where R1 is any one of several phosphoramidate groups. Specifically, they discovered the pro-drug compound referred to as EIDD-2173; also known as ATI-2173. See U.S. Patent No. 10,683,319.

ATI-2173
[0011] It was successfully demonstrated that ATI-2173 is able to effectively and selectively deliver clevudine 5' monophosphate to the liver, bypassing the first systemic phosphorylation and thus reducing systemic exposure to clevudine allowing for the improved treatment of HBV.
[0012] WO20 17223421 discloses that clevudine phosphoramidate compounds, such as
ATI-2173, are additive or synergistic when combined with other antivirals such as lamivudine, adefovir, tenofovir, telbivudine, entecavir, or combinations thereof.
[0013] Both publications disclose the synthesis of ATI-2173 by the following route:
 Compound-7
Compound-7
[0014] The synthesis begins with commercially available clevudine, herein referred to as Compound-D, that is reacted with t-BuMgCl in THF solution in the presence of an excess of the pentafluoro compound SM-3, herein referred to as Compound-3. The reaction yields both ATI- 2173 and the clevudine 3', 5 '-bis phosphoramidate, as well as unreacted clevudine.
[0015] However, there are several problems with this particular synthetic method, namely: i) poor selectivity for the 5' position leading to several by-products in the reaction including clevudine 3',5'-bisphosphoramidate (Impurity-1) as the major by-product at 27.8-32.4%; ii) chromatography was required to purify ATI-2173 in the final step; iii) yield of ATI-2173 was low ( <52.2%); and overall scalability issues due to the poor selectivity, chromatography and low yield. Example impurity levels can be found in Table 1.
[0016] Table 1. Summary of impurities from 1kg and 3kg batches
 [0017] Thus, what are needed are methods of more efficient, specific and selective synthesis for the clevudine phosphoramidates useful for the treatment of HBV.
[0017] Thus, what are needed are methods of more efficient, specific and selective synthesis for the clevudine phosphoramidates useful for the treatment of HBV.
[0018] SUMMARY
[0019] The present disclosure provides materials and methods of synthesizing clevudine phosphoramidates useful for the treatment of HBV infections. Specifically, disclosed are more efficient, specific and selective syntheses for the clevudine phosphoramidates that avoid the use of expensive and difficult to obtain starting materials, yield fewer impurities, provide greater stereoselectivity, are relatively stable under routine conditions allowing for the reaction to be performed at larger scales, and allow isolation by recrystallization.
[0020] These and other embodiments and features of the disclosure will become more apparent through reference to the following description, the accompanying figures, and the claims. Furthermore, it is to be understood that the features of the various embodiments described herein are not mutually exclusive and can exist in various combinations and permutations.
[0021] BRIEF DESCRIPTION OF THE FIGURES
[0022] Various other features and attendant advantages of the subject matter disclosed herein will be more fully appreciated from the following detailed description when considered in connection with the accompanying drawings in which like reference characters designate like or corresponding parts throughout the several views, and wherein:
[0023] FIG. 1 is the X-Ray Powder Diffraction (XRPD) spectra of ATI-2173.
[0024] FIG. 2 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with l.Oeq of TMSOTf.
[0025] FIG. 3 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with 0.5eq of PhCOOH.
[0026] FIG. 4 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with l.Oeq of PhCOOH.
[0027] FIG. 5 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with 0.5eq of PhCOOH at 30g scale.
[0028] FIG. 6 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with 0.5eq of PhCOOH at 30g scale, as the wet cake isolate.
[0029] FIG. 7 is the high performance liquid chromatography (HPLC) spectra for the preparation of Compound-6 with 0.5eq of PhCOOH at 30g scale, recrystallized from EtOH.
[0030] DETAILED DESCRIPTION
[0031] The materials, compounds, compositions, and methods described herein may be understood more readily by reference to the following detailed description of specific aspects of the disclosed subject matter, the Figures, and the Examples included therein. Before the present materials, compounds, compositions, and methods are disclosed and described, it is to be understood that the aspects described below are not limited to specific synthetic methods or specific reagents, as such may, of course, vary. It is also to be understood that the terminology used herein is for the purpose of describing particular aspects only and is not intended to be limiting.
[0032] Also, throughout this specification, various publications are referenced. The disclosures of these publications in their entireties are hereby incorporated by reference into this application in order to more fully describe the state of the art to which the disclosed matter pertains. The references disclosed are also individually and specifically incorporated by reference herein for the material contained in them that is discussed in the sentence in which the reference is relied upon.
[0033] General Definitions
[0034] In this specification and in the claims that follow, reference will be made to a number of terms, which shall be defined to have the following meanings:
[0035] Throughout the specification and claims the word “comprise” and other forms of the word, such as “comprising” and “comprises,” means including but not limited to, and is not intended to exclude, for example, other additives, components, integers, or steps. As used in the description and the appended claims, the singular forms “a,” “an,” and “the” include plural referents unless the context clearly dictates otherwise. Thus, for example, reference to “a composition” includes mixtures of two or more such compositions, reference to “an antibiotic” includes mixtures of two or more such antibiotics, reference to “the compound” includes mixtures of two or more such compounds, and the like. “Optional” or “optionally” means that the
subsequently described event or circumstance can or cannot occur, and that the description includes instances where the event or circumstance occurs and instances where it does not.
[0036] Notwithstanding that the numerical ranges and parameters setting forth the broad scope of the disclosure are approximations, the numerical values set forth in the specific examples are reported as precisely as possible. Any numerical value, however, inherently contain certain errors necessarily resulting from the standard deviation found in their respective testing measurements. Furthermore, when numerical ranges of varying scope are set forth herein, it is contemplated that any combination of these values inclusive of the recited values may be used. Further, ranges can be expressed herein as from “about” one particular value, and/or to “about” another particular value. When such a range is expressed, another aspect includes from the one particular value and/or to the other particular value. Similarly, when values are expressed as approximations, by use of the antecedent “about,” it will be understood that the particular value forms another aspect. It will be further understood that the endpoints of each of the ranges are significant both in relation to the other endpoint, and independently of the other endpoint. Unless stated otherwise, the term “about” means within 10% (e.g., within 8% or 5% or 2% or 1%) of the particular value modified by the term “about.”
[0037] By “reduce” or other forms of the word, such as “reducing” or “reduction,” is meant lowering of an event or characteristic (e.g., viral infection). It is understood that this is typically in relation to some standard or expected value, in other words it is relative, but that it is not always necessary for the standard or relative value to be referred to. For example, “reduces viral infection” means decreasing the amount of viral load relative to a standard or a control.
[0038] By “prevent” or other forms of the word, such as “preventing” or “prevention,” is meant to stop a particular event or characteristic, to stabilize or delay the development or progression of a particular event or characteristic, or to minimize the chances that a particular event or characteristic will occur. Prevent does not require comparison to a control as it is typically more absolute than, for example, reduce. As used herein, something could be reduced but not prevented, but something that is reduced could also be prevented. Likewise, something could be prevented but not reduced, but something that is prevented could also be reduced. It is understood that where reduce or prevent are used, unless specifically indicated otherwise, the use of the other word is also expressly disclosed.
[0039] As used herein, “treatment” refers to obtaining beneficial or desired clinical results. Beneficial or desired clinical results include, but are not limited to, any one or more of: alleviation of one or more symptoms (such as infection), diminishment of extent of infection, stabilized (i.e., not worsening) state of infection, preventing or delaying spread of the infection, preventing or delaying occurrence or recurrence of infection, and delay or slowing of infection progression.
[0040] The term “patient” may refer to a human in need of treatment with an antibiotic or treatment for any purpose, such as a human in need of such a treatment to treat viral infection. However, the term “patient” can also refer to non-human animals, including but limited to, mammals such as dogs, cats, horses, cows, pigs, sheep and non-human primates, among others, that are in need of treatment with an antibiotic or treatment for any purpose, such as with an antiviral compound.
[0041] It is understood that throughout this specification the identifiers “first” and “second” are used solely to aid in distinguishing the various components and steps of the disclosed subject matter. The identifiers “first” and “second” are not intended to imply any particular order, amount, preference, or importance to the components or steps modified by these terms.
[0042] Chemical Definitions
[0043] As used herein, the term “composition” is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts. References in the specification and concluding claims to parts by weight of a particular element or component in a composition denotes the weight relationship between the element or component and any other elements or components in the composition or article for which a part by weight is expressed. Thus, in a mixture containing 2 parts by weight of component X and 5 parts by weight component Y, X and Y are present at a weight ratio of 2:5, and are present in such ratio regardless of whether additional components are contained in the mixture. A weight percent (wt.%) of a component, unless specifically stated to the contrary, is based on the total weight of the formulation or composition in which the component is included.
[0044] As used herein, substantially pure means sufficiently homogeneous to appear free of readily detectable impurities as determined by standard methods of analysis, such as thin layer chromatography (TLC), nuclear magnetic resonance (NMR), gel electrophoresis, high performance liquid chromatography (HPLC) and mass spectrometry (MS), gas-chromatography
mass spectrometry (GC-MS), and similar, used by those of skill in the art to assess such purity, or sufficiently pure such that further purification would not detectably alter the physical and chemical properties, such as enzymatic and biological activities, of the substance. Both traditional and modem methods for purification of the compounds to produce substantially chemically pure compounds are known to those of skill in the art. A substantially chemically pure compound may, however, be a mixture of stereoisomers.
[0045] A “pharmaceutically acceptable” component is one that is suitable for use with humans and/or animals without undue adverse side effects (such as toxicity, irritation, and allergic response) commensurate with a reasonable benefit/risk ratio.
[0046] “Pharmaceutically acceptable salt” refers to a salt that is pharmaceutically acceptable and has the desired pharmacological properties. Such salts include those that may be formed where acidic protons present in the compounds are capable of reacting with inorganic or organic bases. Suitable inorganic salts include those formed with the alkali metals, e.g., sodium, potassium, magnesium, calcium, and aluminum. Suitable organic salts include those formed with organic bases such as the amine bases, e.g., ethanolamine, diethanolamine, triethanolamine, tromethamine, N-methylglucamine, and the like. Such salts also include acid addition salts formed with inorganic acids (e.g., hydrochloric and hydrobromic acids) and organic acids (e.g., acetic acid, citric acid, maleic acid, and the alkane- and arene-sulfonic acids such as methanesulfonic acid and benzenesulfonic acid). When two acidic groups are present, a pharmaceutically acceptable salt may be a mono-acid-mono-salt or a di-salt. Similarly, where there are more than two acidic groups present, some or all of such groups can be converted into salts.
[0047] “Pharmaceutically acceptable excipient” refers to an excipient that is conventionally useful in preparing a pharmaceutical composition that is generally safe, non-toxic, and desirable, and includes excipients that are acceptable for veterinary use as well as for human pharmaceutical use. Such excipients can be solid, liquid, semisolid, or, in the case of an aerosol composition, gaseous.
[0048] A “pharmaceutically acceptable carrier” is a carrier, such as a solvent, suspending agent or vehicle, for delivering the disclosed compounds to the patient. The carrier can be liquid or solid and is selected with the planned manner of administration in mind. Liposomes are also a pharmaceutical carrier. As used herein, “carrier” includes any and all solvents, dispersion media, vehicles, coatings, diluents, antibacterial and antifungal agents, isotonic and absorption delaying
agents, buffers, carrier solutions, suspensions, colloids, and the like. The use of such media and agents for pharmaceutical active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in the therapeutic compositions is contemplated.
[0049] The term “therapeutically effective amount” as used herein means that amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
[0050] Reference will now be made in detail to specific aspects of the disclosed materials, compounds, compositions, articles, and methods, examples of which are illustrated in the accompanying Examples and Figures.
[0051] Synthesis
[0052] The present disclosure provides materials and methods of synthesizing clevudine phosphoramidates useful for the treatment of HBV infections. The methods disclosed herein are more efficient, specific and provide for the more selective synthesis for the clevudine phosphoramidates. Specifically, the methods disclosed avoid the use of expensive commercially available starting materials, yield fewer impurities, provide greater stereoselectivity, are stable under routine conditions allowing for the reaction to be performed at larger scales, and allow isolation by recrystallization.
[0053] Compound and Intermediate Nomenclature:
[0054] Table 2 depicts the designated name used throughout the current specification for each of the respective compounds and intermediates. The table also shows the standard IUPAC (International Union of Pure and Applied Chemistry) name for each as well as the structure.
[0055] Table 2. Compound names.



[0056] Synthesis of Clevudine
[0057] REACTION SCHEME I
Step B
Step A OBz
HBr/AcOH, DCM fluorinating agent, 20 °C, 16 h
 EtOAc
EtOAc

 80 °C, 24 h
80 °C, 24 h
Compound-1 Compound-4 Compound-5
 clevudine
clevudine
[0058] Step A: Synthesis of Compound-4
OBz Step A OBz fluorinating agent,

HO'' OBz EtOAc
 80 °C, 24 h
80 °C, 24 h
Compound-1 Compound-4
[0059] Certain embodiments disclosed herein provide a process for the preparation of ATI-
2173 involving the preparation of Compound-4, where Compound-4 is prepared from the reaction of Compound- 1 and a fluorinating agent in ethyl acetate (EtOAc) or other suitable solvent. In embodiments, the preparation of Compound-4 may be carried out where:
[0060] (A-l) Compound-1 is added to a reaction vessel in EtOAc and the temperature adjusted to <5 °C with stirring;
[0061] (A-2) a fluorinating agent is added slowly and the temperature is maintained below 5 °C;
[0062] (A-3)) the temperature of the reaction mixture is warmed to 75 °C-85 °C and the reaction is stirred at 75 °C-85 °C for up to 24 hours;
[0063] (A-4), the reaction temperature is cooled to 25 °C or less;
[0064] (A-5) the reaction is quenched using a NaHCOs aqueous solution;
[0065] (A-6) the organic and aqueous fractions are separated, and the organic layer is washed with water, sodium sulfate aqueous solution, or both;
[0066] (A-7) the EtOAc is removed by distillation;
[0067] (A-8) EtOH is added and the mixture is warmed;
[0068] (A-9) the mixture is then cooled to <10 °C to allow for precipitation, and the precipitate is collected by filtration; and
[0069] (A-10) the wet precipitate is next washed with EtOH and dried under vacuum.
[0070] The resulting product Compound-4 has a purity of greater than about 99%.
[0071] In an embodiment the fluorinating agent of step (A-2) is selected from, but not limited to, trihydrofluorine trimethylamine, HF pyridine complex, bis(2-methoxy- ethyl)aminosulfur trifluoride (BAST, also called Deoxo-Fluor®), diethylaminosulfur trifluoride (DAST), sulfur tetrafluoride, morpholinosulfur trifluoride, (diethylamino)difluorosulfonium tetrafluoroborate, 2-pyridinesulfonyl fluoride, N-fluorobenzenesulfonimide, perfluoro- 1 -butanesulfonyl fluoride, 2-thiophenesulfonyl fluoride, l-chloromethyl-4-fluoro-l,4-diazoniabicyclo- [2.2.2]octane bis(tetrafluoroborate), or other aminosulfurane. In another embodiment the fluorinating agent of step (A-2) is BAST.
[0072] In an embodiment the temperature of step (A-3) can be raised continuously up to 75 °C-85 °C or carried out incrementally in a step-wise fashion. In an embodiment the temperature of step (A-3) is first raised to 40 °C-50 °C, the raised 75 °C-85 °C. In an embodiment, the reaction at step (A-3) is stirred at 75 °C-85 °C for 15-20 hours. In another embodiment, the reaction at step (A-3) is stirred at 75 °C-85 °C until Compound-SM-1 is consumed by HPLC. In an embodiment, the reaction in step (A-4) is cooled to 15 °C-25 °C.
[0073] In an embodiment the NaHCCh aqueous solution of step (A-5) is a 7% NaHCOs aqueous solution. In an embodiment, the NaHCOs of step (A-5) is added slowly. In an embodiment, the NaHCOs solution of step (A-5) is added dropwise. In another embodiment, before quenching with NaHCOs aqueous solution in step (A-5), the reaction mixture is first diluted with additional EtOAc and MeOH.
[0074] In an embodiment, the organic solvent fraction of step (A-6) may be washed one or more time. In embodiments, the organic solvent fraction of step (A-6) may be washed one, two, three, four, five, six, seven, eight, nine, or even ten times. In embodiments, the organic solvent fraction of step (A-6) may be washed more than ten times.
[0075] In an embodiment the EtOAc of step (A-7) is reduced to about 2 volumes at a temperature of not more than 45 °C. In an embodiment the EtOAc of step (A-7) is removed entirely except for a trace quantity at a temperature of not more than 45 °C. In an embodiment the EtOAc of step (A-7) is removed entirely at a temperature of not more than 45 °C.
[0076] In an embodiment, during step (A-8) before charging with EtOH and warming, the reaction is charged with an separate initial amount of EtOH. This initial EtOH may then be distilled off at a temperature of not more than 45 °C to about 2 volumes; then, the reaction mixture may be
charged with the EtOH to be warmed. In an embodiment the reaction mixture of step (A-8) is warmed from about 70 °C to about 80 °C for 1-2 hours.
[0077] In an embodiment, in step (A-9) the reaction is cooled to <5 °C. In an embodiment, the mixture of step (A-9) is stirred at the cooled temperature for 8-14 hours. In another embodiment the mixture of step (A-9) is stirred at the cooled temperature for 9 hours. In an embodiment the filtration of step (A-9) is carried out in a centrifuge.
[0078] In an embodiment, the filtrate of step (A- 10) is warmed to about 45 °C while drying under vacuum.
[0079] Step B: Synthesis of Compound-5

Compound-4 Compound-5
[0080] Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-5, where Compound-5 is prepared from the reaction of Compound-4 in hydrogen bromide (HBr), acetic acid (AcOH), and dichloromethane (DCM). The preparation of Compound-5 may be carried out where:
[0081] (B-l) Compound-4 is added to dichloromethane and the temperature is adjusted to about 20 °C or below;
[0082] (B-2) HBr/acetic acid is added while maintaining a temperature below
20 °C;
[0083] (B-3) the mixture is stirred at 15 °C-25 °C and the reaction is monitored for completeness;
[0084] (B-4) the reaction mixture is diluted with additional dichloromethane and quenched with sodium bicarbonate solution to a pH of about 6 to about 7;
[0085] (B-5) the layers are separated and the organic fraction is washed with sodium sulfate solution;
[0086] (B-6) the mixture is filtered through a sodium sulfate cake and washed with additional dichloromethane; and
[0087] (B-7) the filtrate is reduced by vacuum distillation at a temperature less than or equal to about 45 °C.
[0088] The resulting Compound-B is typically used directly in the next step. Compound- B has a purity of about 92% or greater, as estimated on the potency of the product solution.
[0089] In an embodiment, the temperature of step (B-l) is adjusted between about 10 °C and about 20 °C. For example, in embodiments the temperature of step (B-l) may be adjusted between about 11 °C and about 19 °C, between about 12 °C and about 18 °C, between about 13 °C and about 17 °C, or between about 14 °C and about 16 °C.
[0090] In an embodiment, the mixture of step (B-3) is stirred for about 12 hours to about 20 hours. In another embodiment, the mixture of step (B-3) is stirred for about 15 hours. In an embodiment, step (B-3) is monitored for completeness by HPLC. In an embodiment, in step (B-3) additional portions of HBr/acetic acid can be added with additional time spent stirring until the starting material is consumed.
[0091] In an embodiment, the sodium bicarbonate solution of step (B-4) is a 7% sodium bicarbonate solution. In an embodiment, the temperature during step (B-4) is maintained below 25 °C. In an embodiment, the mixture of step (B-4) is cooled to between about 15 °C and about 20 °C with stirring.
[0092] In an embodiment, the sodium sulfate solution of step (B-5) is a 5% sodium sulfate solution. In an embodiment, the organic layer of step (B-5) is washed more than once. In an embodiment, the organic layer of step (B-5) is washed twice.
[0093] In an embodiment, the filtrate of step (B-7) is reduced to about 1-2 volumes.
[0094] In an embodiment, Compound-B has a purity of greater than about 95% or greater based on the potency of the product solution.
[0095] Step C: Synthesis of Compound-6
Step C

[0096] Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-6, where Compound-6 may be prepared by the
reaction of Compound-5 with Compound-2 in the presence of A,O-bis(trimethylsilyl)acetamide (BSA), benzoic acid and 1,4-di oxane. The preparation of Compound-6 may be carried out where: [0097] (C-l) Compound-2 is loaded in 1,4-dioxane with BSA;
[0098] (C-2) the reaction is warmed to about 70 °C to about 75 °C with stirring;
[0099] (C-3) Compound-5 and benzoic acid are added to the reaction mixture with stirring;
[0100] (C-4) the reaction is quenched with sodium bicarbonate solution;
[0101] (C-5) the layers are separated and the organic layer is washed with additional sodium bicarbonate solution;
[0102] (C-6) the solvent is removed by vacuum distillation;
[0103] (C-7) EtOH is added and removed by vacuum distillation;
[0104] (C-8) EtOH is added, and the mixture is warmed with stirring;
[0105] (C-9) the reaction is cooled slowly and stirred; and
[0106] (C-10) the product is filtered and dried under vacuum.
[0107] Compound-6 may be produced as a wet-cake, where in step C-10 the product is not dried rigorously. Compound-6 may be produced with a purity of greater than or equal to about 99%.
[0108] In an embodiment, hexamethyldisilazane (HMDS) can be used instead of BSA.
[0109] In an embodiment, the reaction in step (C-2) is stirred for up to 48 hours. In an embodiment, the reaction in step (C-2) is stirred for 2-8 hours. In an embodiment, the reaction in step (C-2) is stirred until the solution becomes clear.
[0110] In an embodiment, the Compound-5 added in step (C-3) is used as a crude solution from the production of Compound-5. In an embodiment, di chloromethane is also added during step (C-3) before the addition of benzoic acid. In an embodiment, the reaction at step (C-3) is maintained at about 70 °C to about 75 °C. In an embodiment, the reaction at step (C-3) is first cooled to about 20 °C to about 30 °C before addition of Compound-5. In an embodiment, the reaction at step (C-3) is cooled to about 20 °C to about 25 °C before addition of benzoic acid. In an embodiment, the benzoic acid of step (C-3) is added in portions. In an embodiment, the temperature of step (C-3) is maintained at about 20 °C to about 25 °C during addition of the benzoic acid. In an embodiment, if the reaction mixture of (C-3) is cooled, it is returned to about 70 °C to about 75 °C after addition of the benzoic acid. In an embodiment, the reaction mixture of
(C-3) is stirred at about 70 °C to about 75 °C for about 42 hours to about 75 hours. In an embodiment, the reaction mixture of (C-3) is stirred at about 70 °C to about 75 °C for about 48 hr. [OHl] In an embodiment, the sodium bicarbonate solution used in quenching and or washing is a 7% sodium bicarbonate solution. In an embodiment, the reaction mixture of (C-4) is first cooled between about 20 °C to about 30 °C before quenching, and the temperature is maintained in this range during quenching. In an embodiment, sodium bicarbonate solution of (C- 4) is added until the mixture has a pH of about 7 to about 8.
[0112] In an embodiment, the mixture of (C-5) can be filtered before separation of the aqueous and organic layers. In an embodiment, the organic layer of step (C-5) can be washed with sodium bicarbonate solution two or more times.
[0113] In an embodiment, the addition and removal of EtOH of step (C-7) can be carried out two or more times. In an embodiment, distillation of EtOH in step (C-7) is carried out at temperature of about 65 °C or less under reduced pressure.
[0114] In an embodiment, the EtOH mixture of step (C-8) is warmed to a temperature of about 80 °C to about 85 °C. In an embodiment, the EtOH mixture of step (C-8) is warmed for about 1 hour to about 5 hours until clear.
[0115] In an embodiment, step (C-9) is cooled to a temperature of about 20 °C to about 25 °C. In an embodiment, step (C-9) is cooled gradually over 5 hours to 6 hours. In an embodiment, step (C-9) is stirred at the cooled temperature for about 5 hours to about 18 hours. In an embodiment, step (C-9) can be repeated to obtain the desired purity.
[0116] Step D: Synthesis of Compound-7 (Clevudine)

Compound-6 Compound-7 clevudine
[0117] Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-7, where Compound-7 (Clevudine) is synthesized by reacting Compound-6 in the presence of sodium methoxide (NaOMe). The preparation of Compound-7 is carried out where:
[0118] (D-l) Compound-6 wet cake is added with methanol;
[0119] (D-2) base is added and the reaction mixture is stirred;
[0120] (D-3) the temperature is adjusted from about 20 °C to about 30 °C, and the reaction is quenched with trifluoro acetic acid (TFA);
[0121] (D-4) the reaction mixture is concentrated under reduced pressure;
[0122] (D-5) isopropyl acetate (IP Ac) is added to form a slurry and the temperature is adjusted to about 15 °C to about 25 °C with stirring;
[0123] (D-6) the mixture is filtered to provide a wet cake; and
[0124] (D-7) the wet-cake is dried under reduced pressure to provide a white solid.
[0125] The resulting Compound-7 has a purity of about 99% or greater and about 80% to about 90% yield.
[0126] In an embodiment, the reaction mixture of step (D-2) is cooled to about 10 °C to about 15 °C before addition of sodium methoxide. In an embodiment, the base is selected from sodium hydroxide, sodium methoxide, ammonia, pyridine, methylamine, potassium cyanide, or a combination thereof. In an embodiment, the base is selected from sodium hydroxide or sodium methoxide. In an embodiment, the base may be used in stoichiometric or catalytic amounts. In another embodiment, there reaction mixture of step (D-2) is not allowed to rise above 20 °C. In an embodiment, the reaction mixture of step (D-2) is warmed to a temperature of about 40 °C to about 45 °C. In an embodiment, the reaction mixture is stirred for about 12 hours to about 18 hours.
[0127] In an embodiment, the temperature of step (D-3) is maintained below about 30 °C.
[0128] In an embodiment, the temperature of step (D-4) is maintained below about 45 °C.
[0129] In an embodiment, the mixture of step (D-5) is stirred for about 5 hours to about 12 hours.
[0130] In an embodiment, the wet cake of step (D-6) may be rinsed with additional IP Ac.
[0131] In an embodiment, the wet cake of step (D-7) is crystallized. In an embodiment, crude Compound-7 can be crystallized from ethanol.
[0132] Synthesis of ATI-2173:
[0133] The improved synthesis of ATI-2173 is depicted in Reaction Scheme II.
REACTION SCHEME II:
Step E Step F triethylsilylating selective agent deprotection



Compound-7 clevudine
Compound-8 Compound-9
Step G

Compound-10 ATI-2173
Step E & Step F: Synthesis of Compound-9:
Step E Step F triethylsilylating selective agent deprotection



Compound-7 clevudine
Compound-8 Compound-9
[0136] In embodiments, the new and improved synthesis includes a selective deprotection of Compound-8 to afford Compound-9. Compound-7 (Clevudine) is first reacted in the presence of chlorotriethylsilane, imidazole, and methyl-tert butyl ether (MTBE) to yield the intermediate Compound-8, which is subsequently deprotected with a triethylsilylating agent to provide Compound-9. The isolated yield for the two steps, protection (Step E) and selective deprotection (Step F), may be at least about 70%, such as at least 75%, at least 80%, at least 85%, at least 90%, or even at least 95%. Compound-9 may be recrystallized to afford a crystalline, white solid with a purity of >99%.
[0137] Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-8, where:
[0138] (E-l) Compound-7 is loaded in a solvent, optionally along with a base, and the temperature is adjusted to less than about 5 °C;
[0139] (E-2) triethylsilylating agent is added at a rate to maintain the temperature at below about 5 °C;
[0140] (E-3) the reaction is stirred at a temperature less than about 5 °C;
[0141] (E-4) the reaction is quenched with ammonium acetate and the resulting layers are separated;
[0142] (E-5) the organic layer is washed with additional ammonium acetate and water;
[0143] (E-6) the organic layer is filtered through sodium sulfate and the filtrate solution concentrated under reduced pressure.
[0144] The resulting Compound-9 has a purity of greater than 99% in a solution of greater than 40% assay.
[0145] Triethylsilylating agent refers to a reagent capable of converting an alcohol to a triethylsilyl ether. In an embodiment, the triethylsilylating agent is selected from triethylsilyl chloride (TES), A-methyl-A-triethylsilyltrifluoroacetamide, -tri ethyl silyl acetamide, allyl tri ethyl si lane, A-triethylsilylacetamide, 1-methoxy-l-triethylsiloxypropene, l-methoxy-2-m ethyl - 1-triethylsiloxypropene, triethylsilyl triflate, triethylsilyl trifluoromethane, triethylsilyl imidazole, triethylsilyl bromide, triethylsilyl iodide, triethylsilyl cyanide, or a combination thereof. In another embodiment, the triethylsilylating agent is tri ethylsilyl chloride.
[0146] In an embodiment, a base is loaded along with Compound-7 to promote silylation. In an embodiment, the base is selected from imidazole, pyridine, triethylamine, diisopropyl ethylamine, 2,6-lutidine, 1 -methylimidazole, ammonia, pyrrolidine, pyrrole, pyrimidine, piperidine, l,8-diazabicyclo[5.4.0]undec-7-ene, 4-methylmorpholine, 1,4- diazabicyclo[2.2.2]octane (DABCO), tetramethylethylenediamine (TMEDA), trimethylamine, diisopropylamine, sodium hydride, n-butyllithium, lithium diisopropylamide (LDA), potassium tert-butoxide or a combination thereof. In an embodiment, a catalyst is loaded along with Compound-7 to promote silylation. In an embodiment, the catalyst is selected from an ammonium
halide, sodium halide, potassium halide, dimethylaminopyridine, 2-hydroxypyridine, sodium cyanide, or a combination thereof.
[0147] In an embodiment, the solvent is a solvent or solvent system in which selective silylation is carried out successfully. In an embodiment, the solvent is selected from methyl /-butyl ether (MTBE), tetrahydrofuran (THF), diethyl ether, 1,4-di oxane, hexanes, n-heptane, chloroform, dichloromethane (DCM), dimethylformamide (DMF), methanol, acetonitrile, ethyl acetate, isopropyl acetate, water, or a combination thereof. In an embodiment, the solvent is MTBE.
[0148] In an embodiment, the temperature of step (E-l) is adjusted to between about -5 °C and about 5 °C.
[0149] In an embodiment, the temperature of step (E-2) is maintained at between about -5 °C and about 5 °C during addition of the tri ethyl silylating agent. In an embodiment, during step (E-2) additional solvent can be added.
[0150] In an embodiment, the reaction in step (E-3) is stirred for about 5 hours to about 16 hours. In an embodiment, if the reaction is not complete after step (E-3), additional base can be added and the reaction stirred for additional time. In embodiments in which imidazole is used as the base, the amount of imidazole used may be regulated so as to prevent over de-protection in Step F. For instance, the amount of residual imidazole present may be less than 0.1 weight % (“wt%”), less than 0.09 wt%, less than 0.08 wt%, less than 0.07 wt%, less than 0.06 wt%, less than 0.05 wt%, less than 0.04 wt%, less than 0.03 wt%, less than 0.02 wt%, less than 0.01 wt%, or even less than 0.005 wt%. In some embodiments, the amount of residual imidazole present may be about 0.015 wt%. In some embodiments, there may be no measurable quantity of residual imidazole.
[0151] In an embodiment, the ammonium acetate of step (E-4) is a 10% ammonium acetate solution. In an embodiment, quenching of step (E-4) is carried out at a temperature below about 5 °C.
[0152] In an embodiment, the organic phase of (E-5) can be washed with ammonium acetate one or more time. In an embodiment, the organic phase of (E-5) can be washed with water one or more time. In an embodiment, the filtrate of (E-5) is concentrated to about 50% volume. In an embodiment, the filtrate of (E-5) is concentrated to provide MTBE solution of greater than about 70% assay.
[0153] Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173 involving the preparation of Compound-9, where:
[0154] (F-l) The MTBE solution containing Compound-8 is added with an additional solvent;
[0155] (F-2) the reaction is cooled to less than about -5 °C and an acidic reagent is added while maintaining temperature below about -5 °C;
[0156] (F-3 ) the reaction is stirred at a temperature of about -15 °C to about -5 °C;
[0157] (F-4) the reaction mixture is quenched using ammonium acetate and the resulting layers are separated;
[0158] (F-5) the organic layer is then concentrated under reduced pressure;
[0159] (F-6) //-heptane is added, and the mixture is stirred at a temperature of about
15 °C to about 25 °C, then filtered; and
[0160] (F-7) the material is dried under reduced pressure.
[0161] Compound-9 is obtained in a greater than about 70% yield.
[0162] In an embodiment, Compound-8 of step (F-l) is carried forward as a dried solid after concentration of the previous step, a paste after concentration of the previous step, or in concentrated solution from the previous step; and added to the additional solvent of step (F-l). In an embodiment the additional solvent of step (F-l) is selected from methanol, ethanol, //-propanol, isopropanol, THF, water, dichloromethane or combinations thereof. In an embodiment, Compound-8 of step (F-l) may be added with additional MTBE.
[0163] In an embodiment the acidic reagent of step (F-2) is selected from pyridinium p- toluenesulfonate (PPTS); acetic acid; formic acid; proprionic acid; acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®; hydrochloric acid; trifluoroacetic acid; trichloroacetic acid; tetra-n-butyl ammonium fluoride (TBAF); tetrabutyl ammonium chloride; ceric ammonium nitrate immobilized on silica gel; (±) camphorsulfonic acid or combinations thereof. In an embodiment, the reaction at step (F-2) is cooled to abut -15 °C to about -5 °C and the temperature is maintained within this range during addition of the acidic reagent of step (F-2). In an embodiment, step (F-2) is stirred for 5-28 hr.
[0164] In an embodiment, the reaction at step (F-4) can first be diluted with MTBE before quenching. In an embodiment, the ammonium acetate solution of (F-4) is 10% ammonium acetate. In an embodiment, the aqueous layer of step (F-4) can be further extracted with additional MTBE. In an embodiment, the organic layer of step (F-4) can be washed with additional ammonium acetate solution or water.
[0165] In an embodiment, solvent is removed under pressure at a temperature of less than about 50 °C, such as less than about 45 °C.
[0166] In an embodiment, Compound-9 is obtained in greater than about 75%. In another embodiment, Compound-9 is obtained in greater than about 85%. In yet another embodiment,
Compound-9 is obtained in greater than about 95%. In an embodiment, Compound-9 is obtained in greater than about 99%.
[0167] Step G & Step H: Synthesis of ATI-2173:
Step G


ATI-2173
[0168] Another highlight of the synthesis is the facile cleavage of the triethylsilyl protecting group in the final step. Removal of ZcW-butyldimethyl silyl (TBS) protecting group requires much harsher conditions (fluoride source or strong acid) that will degrade the phosphoramidate moiety. In this synthesis, the crude reaction solution from Step G can be treated with 2% HC1 and stirred at ambient temperature to afford crude ATI-2173 (Step H). In embodiments, the crude material may be recrystallized from isopropyl acetate/n-heptane or MTBE/n-heptane to afford purified ATI-2173. US 2019/0062364 (WO2016/099982) discloses the use of clevudine and 2-chloro-4-nitrophenyl phosphoramidate to synthesize ATI-2173, albeit in a low yield. See also Ross, B. S., et al., J. Org. Chem. 2011, 76, pp. 8311-8319, in which sofosbuvir was synthesized using /?-nitrophenyl phosphoramidate. (See id. at scheme 5.)
[0169] Certain embodiments disclosed herein provide a process for the preparation of ATI- 2173, where:
[0170] (G-l) Compound-9 is added in a solvent with isopropyl (S)-2-(phenoxy-
2,3,4,5,6-pentafluoro-phenoxy-phosphorylamino)-propionate (Compound- 3);
[0171] (G-2) the reaction mixture is cooled to below -5 °C and base is added slowly;
[0172] (G-3) the reaction is stirred for about 12 hours to about 20 hr;
[0173] (H-l) the reaction mixture is adjusted to less than about 25 °C and acidic reagent is added to the reaction with stirring;
[0174] (H-2) the pH of the reaction mixture is adjusted between about 6 to about 7 using sodium bicarbonate solution and diluted with isopropyl acetate;
[0175] (H-3) the resulting layers are separated, and the organic layer is washed with sodium sulfate solution and water
[0176] (H-4) the organic phase is filtered through a pad of sodium sulfate and concentrated;
[0177] (H-5) the reaction mixture is cooled between about 10 °C to about 15 °C, after which //-heptane is slowly added and the resulting mixture is stirred;
[0178] (H-6) the solid is collected by filtration and rinsed with //-heptane; and
[0179] (H-7) the wetcake is dried at about 45 °C to about 50 °C under vacuum to a constant weight.
[0180] ATI-2173 is produced in greater than about 70% yield with greater than about 90% purity.
[0181] In an embodiment, the solvent of step (G-l) is selected from tetrahydrofuran, diethyl ether, isopropyl ether, diglyme, 1,4-di oxane, dibutyle ether, or combinations thereof.
[0182] In an embodiment, the base of step (G-2) is selected from Zc/7-butylmagnesium chloride, Zc/7-butylmagnesium bromide, methylmagnesium chloride, methylmagnesium bromide, sodium hydride, or lithium aluminum hydride. In an embodiment, the base of step (G-2) is Zc/7- butylmagnesiumchloride, 1.7 M, in THF solution. In an embodiment, the reaction mixture of (G- 2) is cooled between about -10 °C and about -5 °C. In an embodiment, the base of step (G-2) is
added dropwise. In an embodiment, the base of step (G-2) is added slowly enough such that the temperature is maintained below about -5 °C.
[0183] In an embodiment, the reaction mixture of (G-3) is first stirred at about -10 to about -5 °C for about 0.5 hour to about 1.0 hr. In an embodiment, the reaction mixture of (G-3) is adjusted to between about 20 °C to about 50 °C and stirred for the prescribed time.
[0184] In an embodiment, the acidic reagent of step (H-l) may be added neat or as a solution. In an embodiment, the acidic reagent of step (H-l) may be added as an aqueous solution. In an embodiment the acidic reagent of step (H-l) is selected from hydrochloric acid, pyridinium -toluenesulfonate; acetic acid; formic acid; proprionic acid; acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®; or combinations thereof. In an embodiment the acidic reagent of step (H-l) is a 2% solution of hydrochloric acid. In an embodiment, the reaction mixture is maintained below about 25 °C during addition of the acidic reagent of step (H-l). In an embodiment, the acidic reagent of (H-l) is added until the pH = about 4.5 to about 5.5. In an embodiment the reaction mixture of (H-l) is stirred for about 12 hours to about 18 hours. In an embodiment, if there is not complete conversion during step (H-l), additional acidic reagent may be added and the reaction stirred for additional time.
[0185] In an embodiment, the sodium bicarbonate solution of step (H-2) is a 7% sodium bicarbonate solution.
[0186] In an embodiment, the work up of step (H-3) may also use THF in the organic layer. In an embodiment, the organic layer of (H-3) may be distilled to a smaller volume before or between washing. In an embodiment, the organic layer of (H-3) may also be washed using NaCl aqueous solution.
[0187] In an embodiment, the concentrated solution of step (H-4) may be diluted with IP Ac and reconcentrated.
[0188] In an embodiment, seed crystals are added to the mixture of step (H-5). In an embodiment, the mixture of step (H-5) is stirred for about 12 hours to about 16 hours.
[0189] In an embodiment, the solid of step (H-6) is filtered by centrifuge.
[0190] In an embodiment, crude ATI-2173 can be crystallized from an isopropyl acetate/heptane mixture. In an embodiment, crude ATI-2173 may be crystallized from MTBE/n-heptane to afford a crystalline, white solid with a purity of >99%. In an embodiment, ATI-2173 is produced in greater than about 75% yield. In another embodiment, ATI-2173 is
produced in greater than about 85% yield. In yet another embodiment, ATI-2173 is produced in greater than about 95% yield. In an embodiment, ATI-2173 is produced with greater than about 95% purity. In another embodiment, ATI-2173 is produced with greater than about 99% purity.
[0191] In a first aspect, either alone or with any other aspect, a process for the preparation of ATI-2173 comprises silylating Compound-7 to form selectively silylated Compound-9; followed by coupling with Compound-3; and deprotection to provide ATI-2173.
[0192] In a second aspect, either alone or with any other aspect, ATI-2173 is substantively free of Impurity- 1.
[0193] In a third aspect, either alone or with any other aspect, a process for the formation of Compound-9 comprises reacting Compound-7 with a triethylsilylating agent to form intermediate Compound-8 then selectively deprotecting the primary alcohol to form Compound- 9.
[0194] In a fourth aspect, either alone or with any other aspect, the triethylsilylating agent is selected from triethylsilyl chloride, A-methyl-A-triethylsilyltrifluoroacetamide, N- tri ethyl silyl acetamide, allyl tri ethylsilane, A-tri ethyl silyl acetamide, 1 -methoxy- 1- triethylsiloxypropene, l-methoxy-2-methyl-l-triethylsiloxypropene, tri ethylsilyl triflate, triethylsilyl trifluoromethane, triethylsilyl imidazole, triethylsilyl bromide, triethylsilyl iodide, triethylsilyl cyanide, or a combination thereof.
[0195] In a fifth aspect, either alone or with any other aspect, the base selected from imidazole, pyridine, tri ethylamine, diisopropyl ethylamine, 2,6-lutidine, 1 -methylimidazole, ammonia, pyrrolidine, pyrrole, pyrimidine, piperidine, l,8-diazabicyclo[5.4.0]undec-7-ene, 4- methylmorpholine, l,4-diazabicyclo[2.2.2]octane (DABCO), tetramethylethylenediamine (TMEDA), trimethylamine, diisopropylamine, sodium hydride, n-butyllithium, lithium diisopropylamide (LDA), potassium tert-butoxide or a combination thereof.
[0196] In a sixth aspect, either alone or with any other aspect, methyl /-butyl ether is used as a solvent.
[0197] In a seventh aspect, either alone or with any other aspect, the selective deprotection is carried out in the presence of a reagent selected from pyridinium /?-toluenesulfonate (PPTS); acetic acid; formic acid; proprionic acid; acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®; hydrochloric acid; trifluoroacetic acid; trichloroacetic acid;
tetra-n-butylammonium fluoride (TBAF); tetrabutyl ammonium chloride; ceric ammonium nitrate immobilized on silica gel; (±) camphorsulfonic acid or combinations thereof.
[0198] In an eighth aspect, either alone or with any other aspect, the triethylsilylating agent is triethylsilyl chloride, where the selective silylation is carried out in the presence of imidazole in methyl /-butyl ether, and the selective deprotection is carried out in the presence of pyridinium p- toluenesulfonate.
[0199] In a ninth aspect, either alone or with any other aspect, a process for the formation of ATI-2173 comprises reacting Compound-9 with Compound-3 forming the intermediate Compound- 10 where the silyl group is then removed to provide ATI-2173.
[0200] In a tenth aspect, either alone or with any other aspect, Compound-9 and Compound-3 are reacted with addition of a basic reactant.
[0201] In an eleventh aspect, either alone or with any other aspect, the basic reactant is /c/7-butylmagnesiumchloride.
[0202] In a twelfth aspect, either alone or with any other aspect, the silyl group is removed in the presence of an acidic reagent, the acidic reagent selected from hydrogen chloride, acetic acid, acid resin, citric acid, sulfuric acid, sulfurous acid, phosphoric acid, methanoic acid, benzoic acid, formic acid, hydrogen bromide, or combinations thereof.
[0203] In a thirteenth aspect, either alone or with any other aspect, ATI-2173 is substantively free of Impurity- 1
[0204] In a fourteenth aspect, either alone or with any other aspect, ATI-2173 is crystallized in a mixture of IP Ac and //-heptane.
[0205] In a fifteenth aspect, either alone or with any other aspect, a process for the formation of Compound-4 comprises reacting Compound-1 in the presence of bis(2- methoxyethyl)aminosulfur trifluoride.
[0206] In a sixteenth aspect, either alone or with any other aspect, Compound-4 is crystallized in ethanol.
[0207] In a seventeenth aspect, either alone or with any other aspect, a process for the formation of Compound-6 comprises reacting Compound-4 with HBr/acetic acid to form Compound-5 then reacting Compound-2 with BSA, benzoic acid, and Compound-4 to form Compound-5.
[0208] In an eighteenth aspect, either alone or with any other aspect, Compound-4 may be purified before reaction with Compound-2.
[0209] In a nineteenth aspect, either alone or with any other aspect, Compound-4 may be used as a crude product in reaction with Compound-2.
[0210] In a twentieth aspect, either alone or with any other aspect, Compound-6 is reacted with a base to form Compound-7, wherein the base is selected from sodium hydroxide, sodium methoxide, ammonia, pyridine, methylamine, potassium cyanide, or a combination thereof.
[0211] Further reference is made to the following experimental examples.
[0212] EXAMPLES
[0213] The following examples are set forth below to illustrate the methods, compositions, and results according to the disclosed subject matter. These examples are not intended to be inclusive of all aspects of the subject matter disclosed herein, but rather to illustrate representative methods, compositions, and results. These examples are not intended to exclude equivalents and variations of the presently disclosed subject matter, which are apparent to one skilled in the art.
[0214] Efforts have been made to ensure accuracy with respect to numbers (e.g., amounts, temperature, etc.) but some errors and deviations should be accounted for. Unless indicated otherwise, parts are parts by weight, temperature is in °C or is at ambient temperature, and pressure is at or near atmospheric. There are numerous variations and combinations of reaction conditions, e.g., component concentrations, temperatures, pressures, and other reaction ranges and conditions that can be used to optimize the product purity and yield obtained from the described process. Only reasonable and routine experimentation will be required to optimize such process conditions.
[0215] EXAMPLE 1 :
[0216] Synthesis of Compound-4.
[0217] Compound-4 was prepared from the reaction of Compound-1 and BAST in EtOAc. In order to avoid production of several unknown impurities caused by prolonging the reaction time, a modified purification process of Compound-4 was developed that utilized crystallization in EtOH. By crystallization in EtOH, the unknown impurities were purged and the purity of Compound-4 could be improved from 85-89% to >98%. The typical yield of this step in kilogram batches was in range of about 83-88%.
[0218] Experimental Procedure 1
[0219] (A-l) A reactor was charged with 83.6 kg of Compound-1 (181 mol) and
378 kg of ethyl acetate (5V) under a nitrogen atmosphere, and the solution was cooled to <5 °C;
[0220] (A-2) 51.6 kg of BAST (233 mol, 1.29eq) was slowly added;
[0221] (A-3) the solution was warmed to 75 °C-85 °C with stirring until the
Compound- 1 was consumed, as shown by HPLC;
[0222] (A-4) the reaction was cooled to <25 °C;
[0223] (A-5) the reaction was diluted with an additional 2V of ethyl acetate
(150 kg), and methanol was added at a rate to maintain the temperature less than 20 °C, followed by 7% sodium bicarbonate;
[0224] (A-6) the layers were separated and the organic layer was washed with water or 5% sodium sulfate aqueous solution (2x);
[0225] (A-7) the reaction was distilled under reduced pressure at not more than
(NMT) 45 °C to approximately 2 volumes, ethanol (2V) was added to the reactor and the solution was distilled again under vacuum at NMT 45 °C to approximately 2 volumes;
[0226] (A-8) ethanol (4.5V) was added, the reactor was warmed to 75 °C-80 °C, and stirred for Ihr;
[0227] (A-9) the reaction was slowly cooled to <5 °C, stirred for 9hr and the precipitates were collected by filtration;
[0228] (A- 10) The precipitate was washed with 75 kg of ethanol (IV) and dried under vacuum at 45 °C to yield 70.74 kg of Compound-4 as a white solid (83.3% yield, 99.8% purity).
[0229] Additional scale-up batches can be carried out for preparation of Compound-4. Conversion of Compound-1 can be >96%. After workup and crystallization in EtOH, Compound- 4 was isolated as an off-white solid with 99.7-100% purity in the assay yield of 83-88%.
[0230] Table 3. Reaction Conditions for Conversion of Compound- 1 to Compound-4

[0231] Experimental Procedure 2
[0232] (A-l) A reactor was charged with 100 g of Compound-1 (1.0 eq.) and 450 g of ethyl acetate (4.5 - 5.5 V) under a nitrogen atmosphere, and the solution was cooled to -5 °C to 5 °C;
[0233] (A-2) 61 g of BAST (1.27 eq.) was slowly added;
[0234] (A-3A) the solution was warmed to 75 °C-85 °C with stirring for 15 hours to 20 hours;
[0235] (A-4A) the reaction was cooled to 15 °C - 25 °C;
[0236] (A-3B) the reaction was heated and stirred at 75 °C - 85 °C for 5 hours to
10 hours;
[0237] (A-4B) the reaction was cooled to 15 °C - 25 °C, and HPLC was used to confirm consumption of the starting material, Compund-1;
[0238] (A-5) the reaction was diluted with an additional 4.5V - 5.5V of ethyl acetate, and methanol was added at a rate to maintain the temperature less than 25 °C, followed by 7% sodium bicarbonate; the reaction was stirred for 0.5 hour to 1 hour at 15 °C to 25 °C and then allowed to stand for an additional 0.5 hour to 1 hour;
[0239] (A-6) the layers were separated and the organic layer was washed with water with stirring for 0.5 hour to 1 hour at 15 °C - 25 °C (3x; with the layers being allowed to separate for 0.5 hour to 1 hour between each separation);
[0240] (A-7) the reaction was distilled under reduced pressure at not more than
(NMT) 45 °C to 1.5 - 2.5 volumes, ethanol was added to the reactor and the
solution was distilled again under vacuum at NMT 45 °C to 1.5 - 2.5 volumes;
[0241] (A-8) ethanol (3.5 - 4.5 V) was added, the reactor was warmed to 70 °C-
80 °C, and stirred for 1 hour to 2 hours;
[0242] (A-9) the reaction was slowly cooled to 0 °C to 10 °C, stirred for 8 hours to
14 hours, and the precipitates were collected by filtration;
[0243] (A- 10) The precipitate was washed with ethanol and dried under vacuum at
45 °C - 55 °C until HPLC showed NMT 0.5% water content to yield 26 g of Compound-4 as a white solid (83.6% yield, 98.6% purity).
[0244] EXAMPLE 2:
[0245] Synthesis of Compound-5.
[0246] Compound-5 was prepared from the reaction of Compound-4 and HBr (33% AcOH sol.). In initial experiments, it was found that conversion of Compound-4 with HBr in EtOAc at 20 °C gave incomplete conversion of the starting material. In order to improve the conversion rate, the solvent was changed from EtOAc to DCM and the charging amount of HBr was increased. In addition, it was found that Compound-5 was unstable under the original HPLC conditions. At the beginning, the HPLC purity of isolated Compound-5 was -70%, but the 'H-NMR spectrum was clean and Q-NMR assay was high. After optimization of the analytical HPLC method, the HPLC purity of Compound-5 in typical kilo-lab scale-up batches was about 94%. The typical yield of this step was >90%. But for the kilo-lab scale-up batches, there was no actual yield at this step as the concentrated DCM solution of Compound-5 was immediately used in the next step for synthesis of Compound-6. The production yield of this step was >90% calculated by a reference sample.
[0247] Experimental Procedure 1 :
[0248] (B-l) A reactor was charged with 71 kg of Compound-4, 370 kg of di chloromethane and the reactor temperature was maintained below 20 °C; [0249] (B-2) 85 kg of HBr/acetic acid was added to the reactor while maintaining the temperature below 20 °C;
[0250] (B-3) the resulting solution was stirred at 15 °C-25 °C for 15 hr and monitored for reaction completeness; residual Compound-4 was present after 23 hr; an additional 40 kg of HBr/acetic acid was added to the reactor,
with stirring for 21 hr; an additional 10 kg of HBr/acetic acid was added to the reactor, with stirring for an additional 15 hr; after stirring for this 36 hr period, the starting material was consumed;
[0251] (B-4) the reaction was diluted with an additional 370 kg of di chloromethane and quenched with 7% sodium bicarbonate solution, pH = 6-7;
[0252] (B-5) the layers were separated and the organic layer was washed twice with a 5% sodium sulfate solution;
[0253] (B-6) the mixture was filtered through a sodium sulfate cake and washed with additional dichloromethane;
[0254] (B-7) the filtrate was reduced to ~l-2 volumes by vacuum distillation at
45 °C to yield 132 kg of di chloromethane solution of -92% purity Compound-5, which was used directly in the next step.
[0255] As the crude product was used directly in the next step as a dichloromethane solution and not isolated, the yields are an estimate based on the potency of the solution.
[0256] Experimental Procedure 2:
[0257] (B-l) A reactor was charged with 73.9 kg of Compound-4, 372 kg of di chloromethane and the temperature was maintained below 20 °C;
[0258] (B-2) 84.8 kg of HBr/acetic acid was added to the reactor followed by an additional 10 kg of DCM while maintaining the temperature below 20 °C;
[0259] (B-3) the resulting solution was stirred at 15 °C-25 °C for 12-20 hr and monitored for reaction completeness until the starting material was consumed;
[0260] (B-4) the reaction was diluted with an additional 572 kg of di chloromethane and quenched with 7% sodium bicarbonate solution, pH = 6-7, at or below 25 °C;
[0261] (B-5) the layers were separated, and the organic layer was washed twice with a 5% sodium sulfate solution;
[0262] (B-6) the mixture was filtered through a sodium sulfate cake and washed with additional dichloromethane;
[0263] (B-7) the filtrate was reduced to -1-2 volumes by vacuum distillation at
45 °C to yield 132 kg of di chloromethane solution of -95% purity Compound-5, which was used directly in the next step.
[0264] As the crude product was used directly in the next step as a dichloromethane solution and not isolated, the yields are an estimate based on the potency of the solution.
[0265] Table 4. Reaction conditions for the synthesis of Compound-5.

[0266] EXAMPLE S:
[0267] Synthesis of Compound-6.
[0268] Compound-6 was prepared from the reaction of Compound-2 (5-methylpyrimidine-
2,4(lH,3H)-dione), HMDS (1,1,1,3,3,3-Hexamethyldisilazane), (NH^SCU and Compound-5 in CHCh. Compound-6 could be isolated with >99% purity in -65% isolated yield. However, CHCL is highly volatile and carcinogenic, other alternative solvents were sought for improved scale up and production. It was discovered that 1,4-di oxane could gave better diastereoselectivity and in- process control (IPC) purity of Compound-6 than other solvents systems. However, in initial studies, the yield was only -36% in scale up synthesis, most likely due to the degradation of Compound-5 by the by-product TMS-NH2 from HMDS. To solve this problem, it was discovered that N,O-bis(trimethylsilyl)acetamide (BSA) could replace HMDS to prepare the Compound-2 intermediate and avoid generation of the nucleophilic compound TMS-NH2 and avoid degradation of Compound-5. In addition, it was further discovered that the addition of BzOH in the reaction of the Compound-2 intermediate and Compound-5 could help to increase the diastereoselectivity. Thus, the conversion of Compound-4 was then improved to >93% and diastereoisomer Compound-11 was below 8%. The typical IPC purity level of Compound-6 was 85-89%. After
crystallization in EtOH, the diastereoisomer could be removed almost completely, and the purity of Compound-6 was >99%. The yield of this step was >70%. Further experiments determined that a possible solvent is THF/DCM (5V:5V).

Compound-6 Compound-11
[0269] Experimental Procedure 1 :
[0270] (C-l) A reactor was charged with 21.7 kg of Compound-2 (172 mol, 1.09 eq.), 350L of 1,4-dioxane (5 volumes) and 79.5 kg of BSA (392 mol, 2.48 eq );
[0271] (C-2) the temperature of the reactor was adjusted to 70 °C-75 °C and stirred for 48 hr;
[0272] (C-3) the crude Compound-5 (67 kg, 158 mol, as 132 kg of a di chloromethane solution) from the previous step was added along with 9.6 kg of benzoic acid (79 mol, 0.5 eq) and the resulting solution was stirred at 70 °C-75 °C for 48 hr;
[0273] (C-4) the reaction was quenched with 7% sodium bicarbonate solution, diluted with additional dichloromethane;
[0274] (C-5) the layers were separated and the organic layer washed with 7% sodium bicarbonate, water (2x);
[0275] (C-6) the solution was concentrated by vacuum distillation;
[0276] (C-7) the solvent was switched to ethanol by adding 5V of ethanol and distilling to ~2V, repeated;
[0277] (C-8) 15V of ethanol were added and the mixture was stirred at 80 °C for
30 min;
[0278] (C-9) the mixture was slowly cooled to 25 °C and stirred at 25 °C for 18 hr; and
[0279] (C-10) the solid was filtered, then dried under a vacuum to yield 51.7 kg of
Compound-6 wet-cake (yield 75.6%, purity 99.8%).
[0280] The product in this reaction is readily isolable as a white solid but was not dried rigorously (hence ‘wet-cake’) due the methanol being used as the solvent in the next step.
[0281] Table 5. Reaction batches for the synthesis of Compound-6.

[0282] Experimental Procedure 2:
[0283] (C- 1 ) A reactor was charged with 33 g of Compound-2 (172 mol, 1.09 eq.),
1,4-dioxane (5 volumes) and 120 g of BSA (1.15 - 1.25X);
[0284] (C-2A) the temperature of the reactor was adjusted to 65 °C-75 °C and stirred for 2 hours to 8 hours until the solution became clear;
[0285] (C-2B) the temperature of the reactor was adjusted to 20 °C to 30 °C;
[0286] (C-3) the crude Compound-5 (100 g) and dichloromethane (1 - 3V) were added to the reactor under a N2 atmosphere; 15 g of benzoic acid was added to the reactor under an N2 atmosphere at 15 °C - 25 °C, and the resulting solution was stirred at 65 °C - 75 °C for 50 hours to 75 hours or until HPLC shows consumption of Compound-5;
[0287] (C-4) the reaction was quenched with 5% sodium bicarbonate solution, diluted with additional dichloromethane; the pH was adjusted to 7 to 8;
[0288] (C-5) the layers were separated and the organic layer washed with 7% sodium bicarbonate, water (2x);
[0289] (C-6) the solution was concentrated by vacuum distillation;
[0290] (C-7) the solvent was switched to ethanol by adding 5 - 8 V of ethanol and distilling to ~2 - 3 V, repeated;
[0291] (C-8) 15V of ethanol were added and the mixture was stirred at 75 °C -
85 °C for 1 hour to 5 hours;
[0292] (C-9) the mixture was slowly cooled to 15 °C - 25 °C and stirred at 25 °C for 5 hours to 6 hours; and
[0293] (C- 10) the solid was filtered, then dried under a vacuum to yield 51.7 kg of
Compound-6 wet-cake (yield 88%, purity 99.7%).
[0294] EXAMPLE 4:
[0295] Preparation of Compound-6 with a One-Pot Method
[0296] Experimental Procedure:
[0297] Several experiments were carried out to prepare Compound-6 with different solvents in one-pot. In the first, THF was used as solvent, and after stirring for 42 hr at 65 °C, 8.60% Compound-5 was residual and 78.30% Compound-6 was generated. The impurity Compound-11 was generated with 6.88%. In a second experiment, DMSO was used as solvent, and after stirring for 6 hr at 63 °C, 0.23% Compound-5 was residual and only 0.05% of Compound- 6 was generated. However, some new unknown impurities were found following HPLC. In a third experiment, ACN was used as solvent, and after stirring for 6 hr at 63 °C, Compound-5 was consumed completely and >70% of Compound-6 was generated. The impurity Compound- 11 was generated with 23.28%. In yet a fourth experiment, THF and ACN were used as solvents, and after stirring for total 22 hr at 63 °C, Compound-5 was consumed completely and >73% of Compound- 6 was generated. The impurity Compound-11 was generated with 16.00%. In another experiment, 1,4-dioxane was used as the solvent, and after stirring for a total of 41 hr at 65-70 °C, 87.83% of Compound-6 was generated, the residual amount of Compound-5 was 3% and the impurity Compound-11 was 2.86%. Thus, it was concluded that the reaction using a one-pot method in 1,4- dioxane would be selected over the other solvents. However, it was found that upon scale up, 1,4- dioxane did not work well with an isolated yield of product Compound-6 of only -36%.
[0298] EXAMPLE S:
[0299] Preparation of Compound-6 from Compound-5 and Compound-2 using a Lewis acid or a Bronsted Acid.
[0300] To improve yield, a series of experiments were conducted: i) in order to try to prepare the Compound-2 using BSA instead of HMDS; ii) to try to utilize a Lewis acid (TMSOTf) or a Bronsted acid (PhCOOH) to inhibit the formation of an impurity, which is suspected to be an isomer to Compound-6; and iii) and to use more PhCOOH to improve diastereoselectivity.
[0301] Three experiments were carried out to prepare Compound-6 from Compound-5 and Compound-2 using BSA as reagent in 1,4-dioxane. In the first experiment using TMSOTf as a catalyst, after adding the solution of Compound-5 in DCM and stirring for 23 hr at 70 °C,
Compound-6 and presumed isomer of Compound-6 were generated with >70% and -27% respectively. An impurity was also observed that is presumed to be the silylated base, which disappeared during the course of the reaction. In a second experiment in presence of 0.5 eq PhCOOH, after adding Compound-5 and stirring for 43 hr at 70 °C, Compound-6 was generated with >86% in 80% in-situ yield and the presumed isomer of Compound-6 was decreased to -4.8%. When the reaction temperature was further increased to 90 °C and stirred for another 3.0 hr, residual Compound-5 was decreased from to -4% and Compound-6 was increased to -90%. In a third experiment with 1.0 eq PhCOOH, after adding Compound-5 and stirring for 78 hr at 70 °C, >92% Compound-6 was generated with -5.1% of the presumed isomer of Compound-6, and the residual Compound-5 was only 1%. Thus, while using BSA worked well, PhCOOH as additive gave a better diastereoselectivity than TMSOTf. Increasing the amount of PhCOOH from 0.5 eq to 1.0 eq did not improve diastereoselectivity. The HPLC spectra for the reactions in Table 6 can be found in FIG. 2-5, along with the HPLC spectra for the wetcake and final crystalized form of Compound-6 found in FIG. 6 and 7, respectively.
[0302] Table 6. Effect of Bronsted or Lewis acid on the conversion of Compound-5 into
Compound-6.
 a) All reactions performed on a scale of 2g of Compound-5 using 1. leq of SM-3, 2.5eq of BSA and 10 volumes of 1,4-di oxane b) Reaction done on 30g scale of Compound-5 using l.leq of SM-3, 2.5eq BSA and 10 volumes of 1,4-di oxane
a) All reactions performed on a scale of 2g of Compound-5 using 1. leq of SM-3, 2.5eq of BSA and 10 volumes of 1,4-di oxane b) Reaction done on 30g scale of Compound-5 using l.leq of SM-3, 2.5eq BSA and 10 volumes of 1,4-di oxane
[0303] Scale up Production of Compound-6:
[0304] In initial scale up studies, after workup and crystallization in 8V EtOH, yield and purity of Compound-6 was improved with Compound-6 obtained as white solid with >95% HPLC purity. After further re-crystallization in 18V EtOH, the purity of Compound-6 was improved to 99.9% with a >84% yield. Further scale up on a 100 g scale, following work up and crystallization, purity was found to be 100% with HPLC purity with a 78.0% assay yield. Further scale up to
kilogram scales showed comparable yield of between 80-98% assay yield and greater than 99.9% purity.
[0305] EXAMPLE 6:
[0306] Synthesis of Compound-7 (Clevudine)
[0307] The compound Compound-7 (Clevudine) was prepared from the reaction of compound Compound-6 and NaOMe in MeOH. Losing a charging amount of NaOMe 0.4-0.5 eq and trifluoroacetic acid (TFA) quenching and an isopropyl acetate (IP Ac) crystallization process, Compound-7 (Clevudine) could be obtained as white solid with >99% purity and >85% yield.
[0308] Experimental Procedure 1 :
[0309] (D-l) A reactor was charged with 51.6 kg of Compound-6 wet-cake (110 mol) and 10V of methanol
[0310] (D-2) sodium methoxide (2.5 kg, 46.3 mol, 0.42 eq) was added and the resulting solution was stirred at 40-45 °C for 18 hr;
[0311] (D-3) the reaction was quenched with trifluoro acetic acid (TFA);
[0312] (D-4) the reaction mixture was concentrated under reduced pressure;
[0313] (D-5) after workup and crystallization from ethanol, 24.4 kg of the desired product (93.8 mol, 86.3% yield) was obtained as a white solid.
[0314] Table 7. Reaction batches for the synthesis of Compound-7.

[0315] In yet further studies, it was determined that after stirring the mixture of Compound-6 and 0.4 eq NaOMe for 20 hr at 40 °C, Compound-7 was generated with >99% purity but with 0.47% of the partially deprotected 3'-OBz analogue remaining. By adjusting the procedure and quenching the reacting mixture with TFA followed by IP Ac, and then slurried with IP Ac for 16 hr, the product could be obtained >99% purity and >80-90% yield.
[0316] Experimental Procedure 2:
[0317] (D-l) A reactor was charged with 100 g of Compound-6 wet-cake and 7.8
- 8 V of methanol under a N2 atmosphere; the temperature of the reactor was adjusted to 5 °C to 15 °C
[0318] (D-2) sodium methoxide (3.5 - 5.8 g, 0.3 - 0.5 eq.) was added and the resulting solution was stirred at 35-45 °C for 12 hours to 18 hours;
[0319] (D-3) the reaction was brought to a temperature of 20 °C to 30 °C and quenched with trifluoro acetic acid (TFA);
[0320] (D-4) the reaction mixture was concentrated under reduced pressure at
NMT 45 °C;
[0321] (D-5) after workup with isopropyl acetate washing with stirring at 15 °C to
25 °C for 5 hours to 12 hours, followed by filtering and drying under vacuum, the desired product was obtained as a white solid in 90.6 % yield and 99.8 % purity.
[0322] EXAMPLE 7:
[0323] Synthesis of Compound-8
[0324] Early attempts to synthesize ATI-2173 utilized reacting Compound-7 with
Compund-3 in the presence of LBuMgCl and THF. This yielded not only ATI-2173 but also unacceptable amounts of Impurity- 1 along with unreacted Compound-7:

[0325] The procedure was changed to first synthesize Compound-8 in an intermediate step.
[0326] Experimental Procedures:
[0327] (E-l) A reactor was charged with 240 L of methyl /-butyl ether (MTBE) (10V), 24.4 kg of Compound-7 (93.8 moles, 1 eq) and 22.4 kg of imidazole (328.3 moles, 3.5 eq), and the reaction mixture was cooled to <5 °C with stirring;
[0328] (E-2) 25.5 kg of triethylsilylchloride (234.5 moles, 2.5 eq) was added at a rate to maintain the temperature at <5 °C;
[0329] (E-3) the reaction was for 16 hr at -5 °C, at which point an IPC sample indicated incomplete conversion; an additional 7.1 kg of imidazole (104.1 moles, 1.11 eq) was charged to the reactor and stirred for another 5 hr at -5 °C; an IPC test result indicated 99.9% conversion to Compound-8;
[0330] (E-4) the reaction was quenched using 10% ammonium acetate and the layers were separated;
[0331] (E-5) the organic layer was washed with 10% ammonium acetate and water
(x2);
[0332] (E-6) the organic layer was filtered through a pad of sodium sulfate and the filtrate was concentrated under reduced pressure at 45 °C to -50% volume. Yield: 92.4 kg of MTBE solution of 43.7% assay and 99.5% purity, solution yield was 90%.
[0333] EXAMPLE 8:
[0334] Synthesis of Compound-9
[0335] Experimental Procedures:
[0336] (F-l) The MTBE solution containing Compound-8 was added with an additional solvent; a reactor was charged with the MTBE solution containing 40.3 kg (82.5 moles, 1.0 eq) of Compound-8 from the previous step, 240 L of MTBE (up to 6 volumes), 80 L of methanol (2 volumes);
[0337] (F-2) the reaction was cooled to <-5 °C, such as from -15 °C to -5 °C, and charged with 0.19 kg of pyridinium /?-toluenesulfonate (PPTS) (0.74 moles, 0.009eq) to the reactor;
[0338] (F-3) the reaction was stirred at <-5 °C for 21 hr or less, such as from 5 hours to 18 hours; IPC indicated 85.4% of the title compound present along with 5.5% clevudine (Compound-7) and 9% starting material;
[0339] (F-4) the reaction was diluted with MTBE, quenched with 10% ammonium acetate and the layers were separated;
[0340] (F-5 ) the aqueous layer was extracted with MTBE and the combined organic layers were washed with water, then concentrated under reduced pressure to ~3 volumes at NMT 50 °C;
[0341] (F-6) the solution was treated with seed crystals and n-heptane was slowly added to induce complete crystallization; the resulting mixture was stirred at 15-25 °C for less thanl6 hr, such as from 1 hour to 3 hours, and the solid was collected by filtration and washed with n-heptane;
[0342] (F-7) the solid was dried to a constant weight to yield 24.8 kg of the title compound as a white solid (66.2 moles, 78.3% yield); in some experiments, the yield was 77.2% and the purity was 99.6%.
[0343] EXAMPLE 9:
[0344] Synthesis of ATI-2173.
[0345] Experimental Procedure 1 :
[0346] (G-l) A reactor was charged with 24.55 kg of Compound-9 (65.6 moles),
240 L of THF (10 V) and isopropyl (S)-2-(phenoxy-2, 3,4,5, 6-pentafluoro- phenoxy-phosphorylamino)-propionate (Compound-3) (78.7 moles, 1.2 eq);
[0347] (G-2) the reaction was cooled to less than -5 °C, such as from -15 °C to
-5 °C or from -10 °C to -5 °C, and then /c/V-butylmagnesiumchloride (1.7 M in THF, 78.7 moles, 1.2 eq) was added;
[0348] (G-3) after stirring for 16 hr at 15 °C to 25 °C, an IPC showed complete consumption of starting material;
[0349] (H-l) the reaction temperature was adjusted to 15 °C to 25 °C, such as
20 °C, and 132 kg of 2% HC1 was added, with the resulting solution being stirred for 13 hr; an IPC showed incomplete conversion of the starting material; an additional 24 kg of 2% HC1 was added (pH 1.4) and the reaction was stirred for an additional great than or equal to 8 hours, such as from 12 hours to 18 hours; in some experiments, the pH was maintained between 4 and 6; an IPC showed complete conversion to desired product;
[0350] (H-2) the pH of reaction solution was adjusted to 6-7 using 7% sodium bicarbonate solution then diluted with 300 L of isopropyl acetate (13 V);
[0351] (H-3) the layers were separated and the organic layer was washed with sodium sulfate and water;
[0352] (H-4) the solution was filtered through a pad of sodium sulfate and concentrated to one-third volume under vacuum at NMT 35 °C; 5x isopropyl acetate was added and then concentrated to one-third volume; repeated;
[0353] (H-5) the solution was treated with seed crystals then cooled to 15 °C to
25 °C, such as 15 °C, and ^-heptane was slowly added; the resulting mixture was stirred for 16 hr;
[0354] (H-6) the solid was collected by filtration and rinsed with ^-heptane;
[0355] (H-7) the wetcake was dried at 45 °C-50 °C under vacuum to a constant weight (such as for 5 to 24 hours) to yield 25.2 kg of the title compound as white solid (47.6 moles, 72.5% yield); in some experiments, 83.7% yield and 99.6% purity were obtained.
[0356] Experimental Procedure 2:
[0357] (G-l) A reactor was charged with 26.5 kg of Compound-9 (70.5 moles),
234 kg of THF (9 V) and Compound-3 (38.2 kg, 84.3 moles, 1.2 eq);
[0358] (G-2) the reaction was cooled to -10 °C to -5 °C and then tertbutylmagnesiumchloride (1.7 M in THF, 85.6 moles, 2 eq) was added dropwise; additional THF was added dropwise below -5 °C, and the reaction was stirred for 0.5-1.0 hr at -10 °C -5 °C;
[0359] (G-3) the temperature was adjusted to 20-50 °C and stirred for 12-20 hr, until an IPC showed complete consumption of starting material;
[0360] (H-l) addition of 183 kg of 2% HC1 was carried out until the pH = 4.5-5.5 while keeping the temperature below 25 °C; the resulting solution was stirred for 12-18 hr at 20-25 °C;
[0361] (H-2) the pH of reaction solution was adjusted to 6-7 using 7% sodium bicarbonate solution then diluted with water (10 kg) and isopropyl acetate (341 kg, 13 V), then stirred for 20 min at 20-25 °C;
[0362] (H-3) the layers were separated and the organic layer was washed with sodium sulfate and water, along with additional IP Ac and THF; the organic
layer was distilled down to 4-7 V and washed again using additional IP Ac (10 V) and 5% sodium sulfate solution (5 V); the solution was charged with 25% NaCl aqueous solution (9 V), THF (3.5 V), and IP Ac (9 V); stirred and the phases were separated; and the organic phase was washed again with NaCl aqueous solution;
[0363] (H-4) the organic phase was filtered through a pad of sodium sulfate, and the filtrate was concentrated to approx, one-third volume and diluted with 5 volumes of isopropyl acetate; the solution was filtered through a pad of sodium sulfate and concentrated to 1-3 V;
[0364] (H-5) 5.7 V IP Ac was added and then treated with seed crystals, cooled to
10-15 °C and ^-heptane was slowly added; the resulting mixture was stirred for 12-16 hr;
[0365] (H-6) the solid was collected by centrifuge and rinsed with ^-heptane; an
IPC of the wetcake showed 99.8 w/w% ATI-2173;
[0366] (H-7) the wetcake was dried at 45-50 °C under reduced pressure to a constant weight to yield 34.1 kg of the title compound as white solid (91% yield).
[0367] EXAMPLE 10:
[0368] Preparation of Intermediate Compound- 10.
[0369] Experimental Procedures
[0370] (G-l) A mixture of Compound-9 (44.68 g, 1.0 eq) and Compound-3 (1.2 eq) was prepared in THF (10 V);
[0371] (G-2) the reaction mixture was cooled to below -5 °C and LBuMgCl was added slowly, maintaining the temperature below -5 °C. ;
[0372] (G-3) the reaction was stirred for 1 -2 hr, then warmed to 20-25 °C and stirred
16 hr; Compound- 10 was formed having a 99.63% conversion to product and a residual Compound-9 of 0.37%.
[0373] EXAMPLE 11 :
[0374] Preparation of intermediate ATI-2173 from Compound-10.
[0375] Experimental Procedure
[0376] (H-l) Crude Compound-10 in THF (see Example 10) was divided into three aliquots and stirred with 2% HC1 aq. solution at pH 5-6, 4-5, and 3-4; for 16 h at 20-25 °C; the aliquots were combined and stirred at 15-20 °C for 72 h, with no degradation of ATI-2173 observed over the second time period;
[0377] (H-2) the pH of the mixture was adjusted to 7 with 7% NaHCCh;
[0378] (H-3) phase separation was carried out using 2-MeTHF, the organic phase was washed with NA2SO4 aqueous soltion, then concentrated to 1-3 V, and MTBE (5 V) was added; this operation was repeated twice;
[0379] (H-5) ATI-2173 was precipitated gradually upon addition of seed crystal and addition of n-heptane (5 V);
[0380] (H-6) the product was filtered; and
[0381] (H-7) the wetcake was dried, resulting in ATI-2173 in 99.73a% purity and
84.4% yield.
[0382] EXAMPLE 12:
[0383] Crystallization of ATI-2173
[0384] Initial studies examined the use of single or mixed solvent systems to crystalize the amorphous product, ATI-2173. Several solvent conditions were screened, including single solvent and mixed solvent systems, in order to determine the potential for obtaining a crystalline material from the amorphous material. None of the solvents tested worked and all conditions produced an oil product. The results are shown below in Tables 8 and 9.
[0385] Table 8: Single Solvent Systems


[0386] Table 9: Multi- Solvent Screening


[0387] Successful Recrystallization Using Isopropyl Acetate/Heptane
[0388] The crude ATI-2173 was first dissolved in THF for polish filtration. After filtration, the clear filtrate was switched to IP Ac solution and 2% seed was added for crystallization. N- heptane was added as an anti-solvent to reduce yield loss in the mother liquor. 23.4 kg pure ATI- 2173 was obtained in 96.6% yield.
[0389] Crystallization via Precipitation:
[0390] (1-1) Crude ATI-2173 is charged to a reactor with THF and the temperature is adjusted to 20 °C -25 °C;
[0391] (1-2) the mixture is stirred until clear, filtered and concentrated under reduced pressure at < 35 °C;
[0392] (1-3) IP Ac is charged to the mixtrure and concentrated under reduced pressure at <35 °C (in-process check: residual THF (%, w/w) specification < 5%);
[0393] (1-4) the internal temperature of the reactor is adjusted to 15 °C-25°C and seed crystals of ATI-2173 are added;
[0394] (1-5) the mixture is stirred for 1 hr, then the temperature is lowered to 10 °C to 20 °C and n-heptane is added slowly;
[0395] (1-6) the resulting mixture is stirred for 12-16 hrs at 10 °C-20 °C;
[0396] (1-7) the mixture is filtered, and the resulting filter cake is washed with n- heptane and dried under vacuum at 45 °C-50 °C for 16-24hrs.
[0397] The final compound, ATI-2173, is crystalline based on extensive XRPD and compared to a reference standard. It has also been subjected to DSC and shows a consistent, sharp melting point. ATI-2173 has been fully characterized by TGA, residual solvents by GC, heavy metals by ICP-MS and/or ICP-OES, residue on ignition and water content. Purity was established using HPLC and chiral HPLC; extensive NMR experiments (1H, 13C, 19F and 31P NMR as well as 2D experiments) were performed to determine/verify the structure. FIG. 1 shows the XRPD spectrum of the crystalline ATI-2173
[0398] Purity:
[0399] To date, there is always observed a final purity >97% and the last 3 batches of material have been substantially better. The purity of earlier batches, using the old synthesis, was 98.7% by HPLC. The purity of the 2 batches made using the new and improved synthesis are 99.7% and 100.0% (due to rounding) for the cGMP batch. All specified impurities (starting materials, intermediates and known byproducts) have been identified and are reported by HPLC. [0400] Optically Active Materials:
[0401] Examples of methods to obtain optically active materials are known in the art, and include at least the following, i) physical separation of crystals-a technique whereby macroscopic crystals of the individual enantiomers are manually separated. This technique can be used if crystals of the separate enantiomers exist, i.e., the material is a conglomerate, and the crystals are visually distinct; ii) simultaneous crystallization-a technique whereby the individual enantiomers are separately crystallized from a solution of the racemate, possible only if the latter is a conglomerate in the solid state; iii) enzymatic resolutions-a technique whereby partial or complete separation of a racemate by virtue of differing rates of reaction for the enantiomers with an enzyme; iv) enzymatic asymmetric synthesis-a synthetic technique whereby at least one step of the synthesis uses an enzymatic reaction to obtain an enantiomerically pure or enriched synthetic precursor of the desired enantiomer; v) chemical asymmetric synthesis— a synthetic technique whereby the desired enantiomer is synthesized from an achiral precursor under conditions that
produce asymmetry (i.e., chirality) in the product, which may be achieved using chiral catalysts or chiral auxiliaries; vi) diastereomer separations-a technique whereby a racemic compound is reacted with an enantiomerically pure reagent (the chiral auxiliary) that converts the individual enantiomers to diastereomers. The resulting diastereomers are then separated by chromatography or crystallization by virtue of their now more distinct structural differences and the chiral auxiliary later removed to obtain the desired enantiomer; vii) first- and second-order asymmetric transformations-a technique whereby diastereomers from the racemate equilibrate to yield a preponderance in solution of the diastereomer from the desired enantiomer or where preferential crystallization of the diastereomer from the desired enantiomer perturbs the equilibrium such that eventually in principle all the material is converted to the crystalline diastereomer from the desired enantiomer. The desired enantiomer is then released from the diastereomer; viii) kinetic resolutions-this technique refers to the achievement of partial or complete resolution of a racemate (or of a further resolution of a partially resolved compound) by virtue of unequal reaction rates of the enantiomers with a chiral, non-racemic reagent or catalyst under kinetic conditions; ix) enantiospecific synthesis from non-racemic precursors— a synthetic technique whereby the desired enantiomer is obtained from non-chiral starting materials and where the stereochemical integrity is not or is only minimally compromised over the course of the synthesis; x) chiral liquid chromatography— a technique whereby the enantiomers of a racemate are separated in a liquid mobile phase by virtue of their differing interactions with a stationary phase. The stationary phase can be made of chiral material or the mobile phase can contain an additional chiral material to provoke the differing interactions; xi) chiral gas chromatography-a technique whereby the racemate is volatilized and enantiomers are separated by virtue of their differing interactions in the gaseous mobile phase with a column containing a fixed non-racemic chiral adsorbent phase; xii) extraction with chiral solvents-a technique whereby the enantiomers are separated by virtue of preferential dissolution of one enantiomer into a particular chiral solvent; xiii) transport across chiral membranes-a technique whereby a racemate is placed in contact with a thin membrane barrier. The barrier typically separates two miscible fluids, one containing the racemate, and a driving force such as concentration or pressure differential causes preferential transport across the membrane barrier. Separation occurs as a result of the non-racemic chiral nature of the membrane that allows only one enantiomer of the racemate to pass through. Chiral chromatography, including simulated moving bed chromatography, is used in one embodiment. A wide variety of chiral
stationary phases are commercially available. In addition, some of the nucleosides described herein, may exist as tautomers, such as, keto-enol tautomers. The individual tautomers as well as mixtures thereof are intended to be encompassed within the compounds of the present subject matter.
[0402] All of the compositions and methods disclosed and claimed herein can be made and executed without undue experimentation in light of the present disclosure. While the subject matter disclosed herein may be embodied in many different forms, there are described in detail herein specific embodiments. The present disclosure is an exemplification of the principles of the disclosed subject matter and is not intended to limit the subject matter to the particular embodiments illustrated.
[0403] Furthermore, the disclosed subject matter encompasses any and all possible combinations of some or all of the various embodiments described herein. It should also be understood that various changes and modifications to the embodiments presently described herein will be apparent to those skilled in the art. Such changes and modifications can be made without departing from the spirit and scope of the invention and without diminishing its intended advantages. It is therefore intended that such changes and modifications be covered by the appended claims.
Claims
1. A process for the preparation of ATI-2173 having the formula:

ATI-2173 the process comprising silylating Compound-7 having the formula:

Compound-7 to form selectively silylated Compound-9 having the formula:

Compound-9; followed by coupling with Compound-3 having the formula

Compound-3; and deprotection to provide ATI-2173 of formula:

ATI-2173.
2. The process of claim 1, wherein ATI-2173 is substantively free of Impurity-1, where Impurity- 1 has the formula:

Impurity- 1.
3. A process for the formation of Compound-9 having the formula:

Compound-9; wherein the process comprises reacting Compound-7 having the formula:

Compound-7 with a triethylsilylating agent to form intermediate Compound-8 having the formula:

Compound-8; then selectively deprotecting the primary alcohol to form Compound-9.
4. The process of claim 3, wherein the tri ethyl silyl ating agent is selected from triethylsilyl chloride, A-methyl-A-triethylsilyltrifluoroacetamide, A-triethylsilylacetamide, allyl tri ethyl si lane, A-triethylsilylacetamide, 1-methoxy-l-triethylsiloxypropene, l-methoxy-2- methyl-l-triethylsiloxypropene, tri ethylsilyl tritiate, tri ethylsilyl trifluoromethane, tri ethylsilyl imidazole, triethylsilyl bromide, triethylsilyl iodide, triethylsilyl cyanide, or a combination thereof.
5. The process of claim 3, further comprising a base during silylation, the base selected from imidazole, pyridine, tri ethylamine, diisopropyl ethylamine, 2,6-lutidine, 1- methylimidazole, ammonia, pyrrolidine, pyrrole, pyrimidine, piperidine, 1,8- diazabicyclo[5.4.0]undec-7-ene, 4-methylmorpholine, l,4-diazabicyclo[2.2.2]octane (DABCO), tetramethylethylenediamine (TMEDA), trimethylamine, diisopropylamine, sodium hydride, n- butyllithium, lithium diisopropylamide (LDA), potassium tert-butoxide or a combination thereof.
6. The process of claim 3, wherein methyl Lbutyl ether is used as a solvent.
7. The process of claim 3, wherein the selective deprotection is carried out in the presence of a reagent selected from pyridinium /?-toluenesulfonate (PPTS); acetic acid; formic acid; proprionic acid; acidic resin such as AMBERLITE®, DOWEX®, AMBERLYST® or DUOLITE®; hydrochloric acid; trifluoroacetic acid; trichloroacetic acid; tetra-n- butylammonium fluoride (TBAF); tetrabutyl ammonium chloride; ceric ammonium nitrate immobilized on silica gel; (±) camphorsulfonic acid or combinations thereof.
8. The process of claim 3, wherein the tri ethyl silyl ating agent is triethylsilyl chloride, where the selective silylation is carried out in the presence of imidazole in methyl t- butyl ether, and the selective deprotection is carried out in the presence of pyridinium p- toluenesulfonate.
9. A process for the formation of ATI-2173 having the formula:

ATI-2173 wherein the process comprises reacting Compound-9 having the formula:

Compound-9 with Compound-3 having the formula:

Compound-3 forming the intermediate Compound- 10 having the formula:

Compound- 10; where the silyl group is then removed to provide ATI-2173.
10. The process of claim 9, wherein Compound-9 and Compound-3 are reacted with addition of a basic reactant.
11. The process of claim 10, wherein the basic reactant is tertbutylmagnesiumchloride.
12. The process of claim 9, wherein the silyl group is removed in the presence of an acidic reagent, the acidic reagent selected from hydrogen chloride, acetic acid, acid resin, citric acid, sulfuric acid, sulfurous acid, phosphoric acid, methanoic acid, benzoic acid, formic acid, hydrogen bromide, or combinations thereof.
13. The process of claim 9, wherein ATI-2173 is substantively free of Impurity- 1, where Impurity- 1 has the formula:

Impurity- 1.
14. The process of claim 9, wherein ATI-2173 is crystallized in a mixture of IP Ac and ^-heptane.
15. A process for the formation of Compound-4 having the formula:

Compound-4 comprising reacting Compound- 1 having the formula:
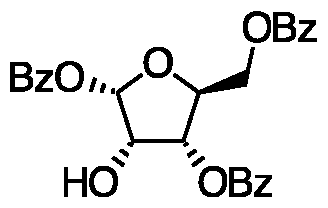
Compound- 1 in the presence of bis(2-methoxyethyl)aminosulfur trifluoride.
16. The process of claim 15, wherein Compound-4 is crystallized in ethanol.
17. A process for the formation of Compound-6 having the formula:

Compound-6 comprising reacting Compound-4 having the formula:

Compound-4 with HBr/acetic acid to form Compound-5 having the formula:
OBz

Compound-5 then reacting Compound-2 having the formula:

Compound-2 with BSA, benzoic acid, and Compound-4 to form Compound-6 having the formula:

Compound-6.
18. The process of claim 17, wherein Compound-4 may be purified before reaction with Compound-2.
19. The process of claim 17, wherein Compound-4 may be used as a crude product in reaction with Compound-2.
20. The process of claim 17, wherein Compound-6 is reacted with a base to form
Compound-7 having the formula:

Compound-7; wherein the base is selected from sodium hydroxide, sodium methoxide, ammonia, pyridine, methylamine, potassium cyanide, or a combination thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163196418P | 2021-06-03 | 2021-06-03 | |
| US63/196,418 | 2021-06-03 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2022256490A2 WO2022256490A2 (en) | 2022-12-08 |
| WO2022256490A3 WO2022256490A3 (en) | 2023-01-05 |
| WO2022256490A9 true WO2022256490A9 (en) | 2023-08-17 |
Family
ID=84323546
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2022/031904 WO2022256490A2 (en) | 2021-06-03 | 2022-06-02 | Improved synthesis of phosphoramidates for the treatment of hepatitis b virus |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2022256490A2 (en) |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5672697A (en) * | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| SG10202013032YA (en) * | 2014-12-15 | 2021-02-25 | Univ Emory | Phosphoramidates for the treatment of hepatitis b virus |
| EA201990059A1 (en) * | 2016-06-24 | 2019-07-31 | Эмори Юниверсити | PHOSPHORAMIDATES FOR THE TREATMENT OF HEPATITIS B VIRUS |
-
2022
- 2022-06-02 WO PCT/US2022/031904 patent/WO2022256490A2/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2022256490A2 (en) | 2022-12-08 |
| WO2022256490A3 (en) | 2023-01-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2021201474B2 (en) | Methods for the preparation of ribosides | |
| CN101541818B (en) | Process for preparation of 4'-azido cytidine derivatives | |
| US10118941B2 (en) | Methods for the preparation of diastereomerically pure phosphoramidate prodrugs | |
| US7582748B2 (en) | Methods of manufacture of 2′-deoxy-β-L-nucleosides | |
| US9908914B2 (en) | Methods of preparing substituted nucleoside analogs | |
| AU2018284131B2 (en) | Synthesis of substantially diastereomerically pure phosphate protides | |
| KR20130064064A (en) | Stereoselective synthesis of phosphorus containing actives | |
| WO2017218580A1 (en) | Synthetic methods for the preparation of nicotinamide riboside and related compounds | |
| IL224045A (en) | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs | |
| EP3684780B1 (en) | Floxuridine synthesis | |
| JP6975166B2 (en) | Synthesis of 2'-fluoro-6'-methylene-carbon ring adenosine (FMCA) and 2'-fluoro-6'-methylene-carbon ring guanosine (FMCG) | |
| EP0362967B1 (en) | Nucleoside derivatives | |
| WO2022256490A9 (en) | Improved synthesis of phosphoramidates for the treatment of hepatitis b virus | |
| US20170114087A1 (en) | Method of preparation of antiviral compounds and useful intermediates thereof | |
| EP4196131A1 (en) | Synthesis of fluorinated nucleotides | |
| US20170313735A1 (en) | Improved Fluorination Process | |
| KR20190002779A (en) | Regioselective and stereoselective preparation method of N-glycoside compounds using metal catalysts from alkoxyallene compounds | |
| US20190077797A1 (en) | Processes for preparing 2-dihalo ribolactones | |
| EP4308580A1 (en) | Chiral synthons for the synthesis of chiral phosphorothioates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22816827 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 22816827 Country of ref document: EP Kind code of ref document: A2 |