WO2022203466A1 - Novel phenylaminopyrimidine compound having inhibitory activity against lrrk2 and use thereof - Google Patents
Novel phenylaminopyrimidine compound having inhibitory activity against lrrk2 and use thereof Download PDFInfo
- Publication number
- WO2022203466A1 WO2022203466A1 PCT/KR2022/004259 KR2022004259W WO2022203466A1 WO 2022203466 A1 WO2022203466 A1 WO 2022203466A1 KR 2022004259 W KR2022004259 W KR 2022004259W WO 2022203466 A1 WO2022203466 A1 WO 2022203466A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- alkyl
- ring
- compound
- halogen
- Prior art date
Links
- -1 phenylaminopyrimidine compound Chemical class 0.000 title claims abstract description 144
- 230000002401 inhibitory effect Effects 0.000 title abstract description 10
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 51
- 201000010099 disease Diseases 0.000 claims abstract description 34
- 238000000034 method Methods 0.000 claims abstract description 25
- 230000001404 mediated effect Effects 0.000 claims abstract description 23
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 23
- 208000035475 disorder Diseases 0.000 claims abstract description 17
- 208000018737 Parkinson disease Diseases 0.000 claims abstract description 10
- 150000001875 compounds Chemical class 0.000 claims description 131
- 125000000217 alkyl group Chemical group 0.000 claims description 78
- 229910052736 halogen Inorganic materials 0.000 claims description 63
- 150000002367 halogens Chemical class 0.000 claims description 56
- 102100032693 Leucine-rich repeat serine/threonine-protein kinase 2 Human genes 0.000 claims description 53
- 101000941879 Homo sapiens Leucine-rich repeat serine/threonine-protein kinase 2 Proteins 0.000 claims description 51
- 125000000623 heterocyclic group Chemical group 0.000 claims description 42
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 42
- 150000003839 salts Chemical class 0.000 claims description 37
- 229910052757 nitrogen Inorganic materials 0.000 claims description 35
- 239000012453 solvate Substances 0.000 claims description 30
- 125000003545 alkoxy group Chemical group 0.000 claims description 27
- 125000004452 carbocyclyl group Chemical group 0.000 claims description 25
- 125000001424 substituent group Chemical group 0.000 claims description 25
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 23
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 18
- 229920006395 saturated elastomer Polymers 0.000 claims description 18
- 125000005843 halogen group Chemical group 0.000 claims description 15
- 229910052799 carbon Inorganic materials 0.000 claims description 13
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 11
- 238000011282 treatment Methods 0.000 claims description 11
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 10
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 9
- 125000001072 heteroaryl group Chemical group 0.000 claims description 9
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 9
- 208000014644 Brain disease Diseases 0.000 claims description 8
- 230000003412 degenerative effect Effects 0.000 claims description 8
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 7
- 229910052739 hydrogen Inorganic materials 0.000 claims description 7
- 230000002265 prevention Effects 0.000 claims description 7
- 125000003003 spiro group Chemical group 0.000 claims description 7
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims description 6
- 125000004432 carbon atom Chemical group C* 0.000 claims description 6
- 125000002950 monocyclic group Chemical group 0.000 claims description 6
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 6
- 125000003386 piperidinyl group Chemical group 0.000 claims description 6
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 6
- 125000006619 (C1-C6) dialkylamino group Chemical group 0.000 claims description 5
- 208000024827 Alzheimer disease Diseases 0.000 claims description 5
- 125000003282 alkyl amino group Chemical group 0.000 claims description 5
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 5
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 5
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 4
- 201000011240 Frontotemporal dementia Diseases 0.000 claims description 4
- 125000002618 bicyclic heterocycle group Chemical group 0.000 claims description 4
- 239000003814 drug Substances 0.000 claims description 4
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 3
- 125000003725 azepanyl group Chemical group 0.000 claims description 3
- 125000002393 azetidinyl group Chemical group 0.000 claims description 3
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 3
- 150000003951 lactams Chemical group 0.000 claims description 3
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 2
- 206010003591 Ataxia Diseases 0.000 claims description 2
- 208000014094 Dystonic disease Diseases 0.000 claims description 2
- 208000023105 Huntington disease Diseases 0.000 claims description 2
- 208000009829 Lewy Body Disease Diseases 0.000 claims description 2
- 201000002832 Lewy body dementia Diseases 0.000 claims description 2
- 208000002569 Machado-Joseph Disease Diseases 0.000 claims description 2
- 208000001089 Multiple system atrophy Diseases 0.000 claims description 2
- 208000000609 Pick Disease of the Brain Diseases 0.000 claims description 2
- 206010039966 Senile dementia Diseases 0.000 claims description 2
- 208000009415 Spinocerebellar Ataxias Diseases 0.000 claims description 2
- 208000036834 Spinocerebellar ataxia type 3 Diseases 0.000 claims description 2
- 208000034799 Tauopathies Diseases 0.000 claims description 2
- 208000000323 Tourette Syndrome Diseases 0.000 claims description 2
- 208000016620 Tourette disease Diseases 0.000 claims description 2
- 206010002022 amyloidosis Diseases 0.000 claims description 2
- 208000010877 cognitive disease Diseases 0.000 claims description 2
- 208000010118 dystonia Diseases 0.000 claims description 2
- 208000027061 mild cognitive impairment Diseases 0.000 claims description 2
- 201000006417 multiple sclerosis Diseases 0.000 claims description 2
- 201000002212 progressive supranuclear palsy Diseases 0.000 claims description 2
- 208000011580 syndromic disease Diseases 0.000 claims description 2
- 238000006467 substitution reaction Methods 0.000 claims 3
- 238000004519 manufacturing process Methods 0.000 claims 1
- 238000011321 prophylaxis Methods 0.000 claims 1
- 108010020246 Leucine-Rich Repeat Serine-Threonine Protein Kinase-2 Proteins 0.000 abstract description 5
- 239000000126 substance Substances 0.000 abstract description 2
- 102000009784 Leucine-Rich Repeat Serine-Threonine Protein Kinase-2 Human genes 0.000 abstract 3
- 239000000203 mixture Substances 0.000 description 90
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 66
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 53
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 42
- 239000007787 solid Substances 0.000 description 39
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 34
- 238000002360 preparation method Methods 0.000 description 33
- 238000005160 1H NMR spectroscopy Methods 0.000 description 29
- 239000012299 nitrogen atmosphere Substances 0.000 description 26
- 238000003786 synthesis reaction Methods 0.000 description 25
- 230000015572 biosynthetic process Effects 0.000 description 23
- 238000006243 chemical reaction Methods 0.000 description 23
- 235000019439 ethyl acetate Nutrition 0.000 description 23
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 22
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- 238000010898 silica gel chromatography Methods 0.000 description 18
- 239000004698 Polyethylene Substances 0.000 description 15
- 239000012043 crude product Substances 0.000 description 15
- 239000012071 phase Substances 0.000 description 14
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 13
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 13
- 229910001868 water Inorganic materials 0.000 description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 11
- 238000003756 stirring Methods 0.000 description 11
- 230000003287 optical effect Effects 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 238000002953 preparative HPLC Methods 0.000 description 9
- 239000011734 sodium Substances 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 8
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 239000003960 organic solvent Substances 0.000 description 8
- 102200092160 rs34637584 Human genes 0.000 description 8
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- 229910052731 fluorine Inorganic materials 0.000 description 7
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 7
- GDHHAOFBLGZCMI-UHFFFAOYSA-N 2,4-dichloro-6,7-dihydro-5h-cyclopenta[d]pyrimidine Chemical compound ClC1=NC(Cl)=NC2=C1CCC2 GDHHAOFBLGZCMI-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 6
- 125000003118 aryl group Chemical group 0.000 description 6
- JBDSSBMEKXHSJF-UHFFFAOYSA-N cyclopentanecarboxylic acid Chemical compound OC(=O)C1CCCC1 JBDSSBMEKXHSJF-UHFFFAOYSA-N 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 125000005842 heteroatom Chemical group 0.000 description 6
- BVCRERJDOOBZOH-UHFFFAOYSA-N bicyclo[2.2.1]heptanyl Chemical group C1C[C+]2CC[C-]1C2 BVCRERJDOOBZOH-UHFFFAOYSA-N 0.000 description 5
- 229910052794 bromium Inorganic materials 0.000 description 5
- 229910052801 chlorine Inorganic materials 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000003112 inhibitor Substances 0.000 description 5
- 229910052740 iodine Inorganic materials 0.000 description 5
- BIWOSRSKDCZIFM-UHFFFAOYSA-N piperidin-3-ol Chemical compound OC1CCCNC1 BIWOSRSKDCZIFM-UHFFFAOYSA-N 0.000 description 5
- ZMANZCXQSJIPKH-UHFFFAOYSA-O triethylammonium ion Chemical compound CC[NH+](CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-O 0.000 description 5
- GFPPPMAWVWTROZ-UHFFFAOYSA-N 1-(4-aminophenyl)piperidin-3-ol Chemical compound C1=CC(N)=CC=C1N1CC(O)CCC1 GFPPPMAWVWTROZ-UHFFFAOYSA-N 0.000 description 4
- JOHFJTBDUSVGQB-UHFFFAOYSA-N 3-(trifluoromethyl)piperidine Chemical compound FC(F)(F)C1CCCNC1 JOHFJTBDUSVGQB-UHFFFAOYSA-N 0.000 description 4
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N N-phenyl amine Natural products NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- 229910004298 SiO 2 Inorganic materials 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- JDUYOMPVOXGHIR-MMALYQPHSA-N (3s,4s)-3,4-difluoropyrrolidine;hydrochloride Chemical compound Cl.F[C@H]1CNC[C@@H]1F JDUYOMPVOXGHIR-MMALYQPHSA-N 0.000 description 3
- WXAAQKMTSQDMII-UHFFFAOYSA-N 1-(4-nitrophenyl)azepane Chemical compound C1=CC([N+](=O)[O-])=CC=C1N1CCCCCC1 WXAAQKMTSQDMII-UHFFFAOYSA-N 0.000 description 3
- RMFWVOLULURGJI-UHFFFAOYSA-N 2,6-dichloro-7h-purine Chemical compound ClC1=NC(Cl)=C2NC=NC2=N1 RMFWVOLULURGJI-UHFFFAOYSA-N 0.000 description 3
- RDUONPQXZRHVKQ-UHFFFAOYSA-N 4-(azepan-1-yl)aniline Chemical compound C1=CC(N)=CC=C1N1CCCCCC1 RDUONPQXZRHVKQ-UHFFFAOYSA-N 0.000 description 3
- QPJOJKMHNWAGPS-UHFFFAOYSA-N 4-[3-(trifluoromethyl)piperidin-1-yl]aniline Chemical compound C1=CC(N)=CC=C1N1CC(C(F)(F)F)CCC1 QPJOJKMHNWAGPS-UHFFFAOYSA-N 0.000 description 3
- XZAIBMLMZLSXBH-UHFFFAOYSA-N 4-bromo-2-chloro-6-iodopyrimidine Chemical compound ClC1=NC(Br)=CC(I)=N1 XZAIBMLMZLSXBH-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 102000010909 Monoamine Oxidase Human genes 0.000 description 3
- 108010062431 Monoamine oxidase Proteins 0.000 description 3
- 229940099433 NMDA receptor antagonist Drugs 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- ZSIQJIWKELUFRJ-UHFFFAOYSA-N azepane Chemical compound C1CCCNCC1 ZSIQJIWKELUFRJ-UHFFFAOYSA-N 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 229940125898 compound 5 Drugs 0.000 description 3
- 125000004093 cyano group Chemical group *C#N 0.000 description 3
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 239000012669 liquid formulation Substances 0.000 description 3
- 239000003703 n methyl dextro aspartic acid receptor blocking agent Substances 0.000 description 3
- 230000004770 neurodegeneration Effects 0.000 description 3
- 208000015122 neurodegenerative disease Diseases 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 125000003226 pyrazolyl group Chemical group 0.000 description 3
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 3
- 125000000168 pyrrolyl group Chemical group 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- GOMXWUJOKDHSBV-UHFFFAOYSA-N 1-(4-nitrophenyl)-3-(trifluoromethyl)piperidine Chemical compound C1=CC([N+](=O)[O-])=CC=C1N1CC(C(F)(F)F)CCC1 GOMXWUJOKDHSBV-UHFFFAOYSA-N 0.000 description 2
- WFQDTOYDVUWQMS-UHFFFAOYSA-N 1-fluoro-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C=C1 WFQDTOYDVUWQMS-UHFFFAOYSA-N 0.000 description 2
- IDRUEHMBFUJKAK-UHFFFAOYSA-N 2,4-dichloro-5-(trifluoromethyl)pyrimidine Chemical compound FC(F)(F)C1=CN=C(Cl)N=C1Cl IDRUEHMBFUJKAK-UHFFFAOYSA-N 0.000 description 2
- RTMVNQUZFLMAMR-UHFFFAOYSA-N 2-chloro-4-cyclopropyl-5-(trifluoromethyl)pyrimidine Chemical compound ClC1=NC=C(C(=N1)C1CC1)C(F)(F)F RTMVNQUZFLMAMR-UHFFFAOYSA-N 0.000 description 2
- LOPQDJJEZFCJMU-UHFFFAOYSA-N 2-chloro-4-iodopyrimidine Chemical compound ClC1=NC=CC(I)=N1 LOPQDJJEZFCJMU-UHFFFAOYSA-N 0.000 description 2
- DCUZDHOWLBKKDZ-UHFFFAOYSA-N 2-chloro-4-pyrrolidin-1-yl-5-(trifluoromethyl)pyrimidine Chemical compound FC(F)(F)C1=CN=C(Cl)N=C1N1CCCC1 DCUZDHOWLBKKDZ-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- HWHYTOJFRBCMNS-UHFFFAOYSA-N 4-(1,4-dioxa-7-azaspiro[4.4]nonan-7-yl)aniline Chemical compound C1=CC(N)=CC=C1N1CC2(OCCO2)CC1 HWHYTOJFRBCMNS-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 229940100578 Acetylcholinesterase inhibitor Drugs 0.000 description 2
- 101710134784 Agnoprotein Proteins 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 102000006378 Catechol O-methyltransferase Human genes 0.000 description 2
- 108020002739 Catechol O-methyltransferase Proteins 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- 208000012661 Dyskinesia Diseases 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 2
- HNYOPLTXPVRDBG-UHFFFAOYSA-N barbituric acid Chemical compound O=C1CC(=O)NC(=O)N1 HNYOPLTXPVRDBG-UHFFFAOYSA-N 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 208000015114 central nervous system disease Diseases 0.000 description 2
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 2
- 239000000544 cholinesterase inhibitor Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- QFVRLYRYKCLRMD-UHFFFAOYSA-N cyclodecane-1,6-diol Chemical compound OC1CCCCC(O)CCCC1 QFVRLYRYKCLRMD-UHFFFAOYSA-N 0.000 description 2
- NZNMSOFKMUBTKW-UHFFFAOYSA-N cyclohexanecarboxylic acid Chemical compound OC(=O)C1CCCCC1 NZNMSOFKMUBTKW-UHFFFAOYSA-N 0.000 description 2
- 230000005786 degenerative changes Effects 0.000 description 2
- 230000006806 disease prevention Effects 0.000 description 2
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 239000003136 dopamine receptor stimulating agent Substances 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 238000001361 intraarterial administration Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- ULKSWZAXQDJMJT-UHFFFAOYSA-M magnesium;2,2,6,6-tetramethylpiperidin-1-ide;chloride Chemical compound [Cl-].CC1(C)CCCC(C)(C)N1[Mg+] ULKSWZAXQDJMJT-UHFFFAOYSA-M 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- GFPPPMAWVWTROZ-LLVKDONJSA-N (3r)-1-(4-aminophenyl)piperidin-3-ol Chemical compound C1=CC(N)=CC=C1N1C[C@H](O)CCC1 GFPPPMAWVWTROZ-LLVKDONJSA-N 0.000 description 1
- BIWOSRSKDCZIFM-RXMQYKEDSA-N (3r)-piperidin-3-ol Chemical compound O[C@@H]1CCCNC1 BIWOSRSKDCZIFM-RXMQYKEDSA-N 0.000 description 1
- GFPPPMAWVWTROZ-NSHDSACASA-N (3s)-1-(4-aminophenyl)piperidin-3-ol Chemical compound C1=CC(N)=CC=C1N1C[C@@H](O)CCC1 GFPPPMAWVWTROZ-NSHDSACASA-N 0.000 description 1
- BIWOSRSKDCZIFM-YFKPBYRVSA-N (3s)-piperidin-3-ol Chemical compound O[C@H]1CCCNC1 BIWOSRSKDCZIFM-YFKPBYRVSA-N 0.000 description 1
- PHFTVEADDMICLE-IMJSIDKUSA-N (3s,4s)-3,4-difluoropyrrolidine Chemical compound F[C@H]1CNC[C@@H]1F PHFTVEADDMICLE-IMJSIDKUSA-N 0.000 description 1
- HVGGGVAREUUJQV-CHHVJCJISA-N (4z)-4-[3-(2,5-dichloro-4,6-dimethyl-1-oxidopyridin-1-ium-3-yl)-2h-1,2,4-oxadiazol-5-ylidene]-2-hydroxy-6-nitrocyclohexa-2,5-dien-1-one Chemical compound CC1=C(Cl)C(C)=[N+]([O-])C(Cl)=C1C(NO1)=N\C1=C\1C=C([N+]([O-])=O)C(=O)C(O)=C/1 HVGGGVAREUUJQV-CHHVJCJISA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 1
- WJUKOGPNGRUXMG-UHFFFAOYSA-N 1,2-dibromo-1,1,2,2-tetrachloroethane Chemical compound ClC(Cl)(Br)C(Cl)(Cl)Br WJUKOGPNGRUXMG-UHFFFAOYSA-N 0.000 description 1
- JTOUWLZSXWMCSH-UHFFFAOYSA-N 1,4-dioxa-7-azaspiro[4.4]nonane Chemical compound C1NCCC21OCCO2 JTOUWLZSXWMCSH-UHFFFAOYSA-N 0.000 description 1
- DPVIABCMTHHTGB-UHFFFAOYSA-N 2,4,6-trichloropyrimidine Chemical compound ClC1=CC(Cl)=NC(Cl)=N1 DPVIABCMTHHTGB-UHFFFAOYSA-N 0.000 description 1
- RGJNPJRAXMSHKN-UHFFFAOYSA-N 2,4-dichloro-5-iodopyrimidine Chemical compound ClC1=NC=C(I)C(Cl)=N1 RGJNPJRAXMSHKN-UHFFFAOYSA-N 0.000 description 1
- GHXBPCSSQOKKGB-UHFFFAOYSA-N 2,4-dichloro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=NC(Cl)=C2C=CNC2=N1 GHXBPCSSQOKKGB-UHFFFAOYSA-N 0.000 description 1
- BTTNYQZNBZNDOR-UHFFFAOYSA-N 2,4-dichloropyrimidine Chemical compound ClC1=CC=NC(Cl)=N1 BTTNYQZNBZNDOR-UHFFFAOYSA-N 0.000 description 1
- OQDPVLVUJFGPGQ-UHFFFAOYSA-N 2-[4-(1,3-benzodioxol-5-ylmethyl)-1-piperazinyl]pyrimidine Chemical compound C=1C=C2OCOC2=CC=1CN(CC1)CCN1C1=NC=CC=N1 OQDPVLVUJFGPGQ-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- TYCYTQLXAIDJNF-UHFFFAOYSA-N 2-chloro-5-(trifluoromethyl)pyrimidine Chemical compound FC(F)(F)C1=CN=C(Cl)N=C1 TYCYTQLXAIDJNF-UHFFFAOYSA-N 0.000 description 1
- UNCQVRBWJWWJBF-UHFFFAOYSA-N 2-chloropyrimidine Chemical compound ClC1=NC=CC=N1 UNCQVRBWJWWJBF-UHFFFAOYSA-N 0.000 description 1
- JHMBTUMIVBSJFS-UHFFFAOYSA-N 2-methyl-3h-isoindol-1-one Chemical compound C1=CC=C2C(=O)N(C)CC2=C1 JHMBTUMIVBSJFS-UHFFFAOYSA-N 0.000 description 1
- WDBQJSCPCGTAFG-QHCPKHFHSA-N 4,4-difluoro-N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclohexane-1-carboxamide Chemical compound FC1(CCC(CC1)C(=O)N[C@@H](CCN1CCC(CC1)N1C(=NN=C1C)C(C)C)C=1C=NC=CC=1)F WDBQJSCPCGTAFG-QHCPKHFHSA-N 0.000 description 1
- BWGRDBSNKQABCB-UHFFFAOYSA-N 4,4-difluoro-N-[3-[3-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl]-1-thiophen-2-ylpropyl]cyclohexane-1-carboxamide Chemical compound CC(C)C1=NN=C(C)N1C1CC2CCC(C1)N2CCC(NC(=O)C1CCC(F)(F)CC1)C1=CC=CS1 BWGRDBSNKQABCB-UHFFFAOYSA-N 0.000 description 1
- CTYPROOLWJDUTA-UHFFFAOYSA-N 4,6-dichloro-1h-pyrazolo[3,4-d]pyrimidine Chemical compound ClC1=NC(Cl)=C2C=NNC2=N1 CTYPROOLWJDUTA-UHFFFAOYSA-N 0.000 description 1
- JLNMBIKJQAKQBH-UHFFFAOYSA-N 4-cyclohexylaniline Chemical compound C1=CC(N)=CC=C1C1CCCCC1 JLNMBIKJQAKQBH-UHFFFAOYSA-N 0.000 description 1
- UBXDNWVNEZBDBN-UHFFFAOYSA-N 4-cyclopropylaniline Chemical compound C1=CC(N)=CC=C1C1CC1 UBXDNWVNEZBDBN-UHFFFAOYSA-N 0.000 description 1
- TVOSOIXYPHKEAR-UHFFFAOYSA-N 4-piperidin-1-ylaniline Chemical compound C1=CC(N)=CC=C1N1CCCCC1 TVOSOIXYPHKEAR-UHFFFAOYSA-N 0.000 description 1
- OUVVCECMELPNNT-UHFFFAOYSA-N 5-(trifluoromethyl)pyrimidin-2-amine Chemical compound NC1=NC=C(C(F)(F)F)C=N1 OUVVCECMELPNNT-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- KORNTPPJEAJQIU-KJXAQDMKSA-N Cabaser Chemical compound C1=CC([C@H]2C[C@H](CN(CC=C)[C@@H]2C2)C(=O)N(CCCN(C)C)C(=O)NCC)=C3C2=CNC3=C1 KORNTPPJEAJQIU-KJXAQDMKSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical group [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 description 1
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 229940124786 LRRK2 inhibitor Drugs 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 206010024229 Leprosy Diseases 0.000 description 1
- 108010006444 Leucine-Rich Repeat Proteins Proteins 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229940121948 Muscarinic receptor antagonist Drugs 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 206010028570 Myelopathy Diseases 0.000 description 1
- LFZAGIJXANFPFN-UHFFFAOYSA-N N-[3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-thiophen-2-ylpropyl]acetamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CCC(C=1SC=CC=1)NC(C)=O)C LFZAGIJXANFPFN-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- BKRGVLQUQGGVSM-KBXCAEBGSA-N Revanil Chemical compound C1=CC(C=2[C@H](N(C)C[C@H](C=2)NC(=O)N(CC)CC)C2)=C3C2=CNC3=C1 BKRGVLQUQGGVSM-KBXCAEBGSA-N 0.000 description 1
- XSVMFMHYUFZWBK-NSHDSACASA-N Rivastigmine Chemical compound CCN(C)C(=O)OC1=CC=CC([C@H](C)N(C)C)=C1 XSVMFMHYUFZWBK-NSHDSACASA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 244000299461 Theobroma cacao Species 0.000 description 1
- 235000005764 Theobroma cacao ssp. cacao Nutrition 0.000 description 1
- 235000005767 Theobroma cacao ssp. sphaerocarpum Nutrition 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- PMZXXNPJQYDFJX-UHFFFAOYSA-N acetonitrile;2,2,2-trifluoroacetic acid Chemical compound CC#N.OC(=O)C(F)(F)F PMZXXNPJQYDFJX-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- AEMOLEFTQBMNLQ-BKBMJHBISA-N alpha-D-galacturonic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-BKBMJHBISA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 150000001413 amino acids Chemical group 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 1
- 150000001448 anilines Chemical group 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 239000003006 anti-agglomeration agent Substances 0.000 description 1
- 229960004046 apomorphine Drugs 0.000 description 1
- VMWNQDUVQKEIOC-CYBMUJFWSA-N apomorphine Chemical compound C([C@H]1N(C)CC2)C3=CC=C(O)C(O)=C3C3=C1C2=CC=C3 VMWNQDUVQKEIOC-CYBMUJFWSA-N 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000012098 association analyses Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- VUFQYRAKSQTZEB-UHFFFAOYSA-N bicyclo[2.2.1]heptan-4-amine Chemical compound C1CC2CCC1(N)C2 VUFQYRAKSQTZEB-UHFFFAOYSA-N 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- SXDBWCPKPHAZSM-UHFFFAOYSA-M bromate Inorganic materials [O-]Br(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-M 0.000 description 1
- SXDBWCPKPHAZSM-UHFFFAOYSA-N bromic acid Chemical compound OBr(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229960002802 bromocriptine Drugs 0.000 description 1
- OZVBMTJYIDMWIL-AYFBDAFISA-N bromocriptine Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 OZVBMTJYIDMWIL-AYFBDAFISA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 235000014121 butter Nutrition 0.000 description 1
- 229960004596 cabergoline Drugs 0.000 description 1
- 235000001046 cacaotero Nutrition 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 125000003739 carbamimidoyl group Chemical group C(N)(=N)* 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 239000003543 catechol methyltransferase inhibitor Substances 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 239000007809 chemical reaction catalyst Substances 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- VZFUCHSFHOYXIS-UHFFFAOYSA-N cycloheptane carboxylic acid Natural products OC(=O)C1CCCCCC1 VZFUCHSFHOYXIS-UHFFFAOYSA-N 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 229960004132 diethyl ether Drugs 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- VFNGKCDDZUSWLR-UHFFFAOYSA-L disulfate(2-) Chemical compound [O-]S(=O)(=O)OS([O-])(=O)=O VFNGKCDDZUSWLR-UHFFFAOYSA-L 0.000 description 1
- 229960003530 donepezil Drugs 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 229940052760 dopamine agonists Drugs 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 229950005627 embonate Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- JRURYQJSLYLRLN-BJMVGYQFSA-N entacapone Chemical compound CCN(CC)C(=O)C(\C#N)=C\C1=CC(O)=C(O)C([N+]([O-])=O)=C1 JRURYQJSLYLRLN-BJMVGYQFSA-N 0.000 description 1
- 229960003337 entacapone Drugs 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229960003980 galantamine Drugs 0.000 description 1
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 125000004404 heteroalkyl group Chemical group 0.000 description 1
- 125000005326 heteroaryloxy alkyl group Chemical group 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 229910001867 inorganic solvent Inorganic materials 0.000 description 1
- 239000003049 inorganic solvent Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- VMPHSYLJUKZBJJ-UHFFFAOYSA-N lauric acid triglyceride Natural products CCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCC)COC(=O)CCCCCCCCCCC VMPHSYLJUKZBJJ-UHFFFAOYSA-N 0.000 description 1
- 210000004901 leucine-rich repeat Anatomy 0.000 description 1
- 229960004502 levodopa Drugs 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 229960003587 lisuride Drugs 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 229960003511 macrogol Drugs 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000000845 maltitol Substances 0.000 description 1
- 235000010449 maltitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-WUJBLJFYSA-N maltitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-WUJBLJFYSA-N 0.000 description 1
- 229940035436 maltitol Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- BUGYDGFZZOZRHP-UHFFFAOYSA-N memantine Chemical compound C1C(C2)CC3(C)CC1(C)CC2(N)C3 BUGYDGFZZOZRHP-UHFFFAOYSA-N 0.000 description 1
- 229960004640 memantine Drugs 0.000 description 1
- 210000001259 mesencephalon Anatomy 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 229940050176 methyl chloride Drugs 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 229950001673 opicapone Drugs 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 229940039748 oxalate Drugs 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 239000006201 parenteral dosage form Substances 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 229960004851 pergolide Drugs 0.000 description 1
- YEHCICAEULNIGD-MZMPZRCHSA-N pergolide Chemical compound C1=CC([C@H]2C[C@@H](CSC)CN([C@@H]2C2)CCC)=C3C2=CNC3=C1 YEHCICAEULNIGD-MZMPZRCHSA-N 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229960004310 piribedil Drugs 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229960003089 pramipexole Drugs 0.000 description 1
- FASDKYOPVNHBLU-ZETCQYMHSA-N pramipexole Chemical compound C1[C@@H](NCCC)CCC2=C1SC(N)=N2 FASDKYOPVNHBLU-ZETCQYMHSA-N 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- YAAWASYJIRZXSZ-UHFFFAOYSA-N pyrimidine-2,4-diamine Chemical compound NC1=CC=NC(N)=N1 YAAWASYJIRZXSZ-UHFFFAOYSA-N 0.000 description 1
- RUOKEQAAGRXIBM-GFCCVEGCSA-N rasagiline Chemical compound C1=CC=C2[C@H](NCC#C)CCC2=C1 RUOKEQAAGRXIBM-GFCCVEGCSA-N 0.000 description 1
- 229960000245 rasagiline Drugs 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 229960004136 rivastigmine Drugs 0.000 description 1
- 229960001879 ropinirole Drugs 0.000 description 1
- UHSKFQJFRQCDBE-UHFFFAOYSA-N ropinirole Chemical compound CCCN(CCC)CCC1=CC=CC2=C1CC(=O)N2 UHSKFQJFRQCDBE-UHFFFAOYSA-N 0.000 description 1
- NEMGRZFTLSKBAP-LBPRGKRZSA-N safinamide Chemical compound C1=CC(CN[C@@H](C)C(N)=O)=CC=C1OCC1=CC=CC(F)=C1 NEMGRZFTLSKBAP-LBPRGKRZSA-N 0.000 description 1
- 229950002652 safinamide Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- MEZLKOACVSPNER-GFCCVEGCSA-N selegiline Chemical compound C#CCN(C)[C@H](C)CC1=CC=CC=C1 MEZLKOACVSPNER-GFCCVEGCSA-N 0.000 description 1
- 229960003946 selegiline Drugs 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 159000000008 strontium salts Chemical class 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229960001685 tacrine Drugs 0.000 description 1
- YLJREFDVOIBQDA-UHFFFAOYSA-N tacrine Chemical compound C1=CC=C2C(N)=C(CCCC3)C3=NC2=C1 YLJREFDVOIBQDA-UHFFFAOYSA-N 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- MIQPIUSUKVNLNT-UHFFFAOYSA-N tolcapone Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC(O)=C(O)C([N+]([O-])=O)=C1 MIQPIUSUKVNLNT-UHFFFAOYSA-N 0.000 description 1
- 229960004603 tolcapone Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- 125000004953 trihalomethyl group Chemical group 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to a phenylaminopyrimidine compound of Formula 1, a leucine-rich repeat kinase 2 (leucine-rich repeat kinase 2: LRRK2)-mediated or related disease or disorder comprising the same, a pharmaceutical composition for preventing or treating, and It relates to a method for treating and preventing a disease or disorder using the same.
- Parkinson's disease affect millions of people. Parkinson's disease is caused by progressive deficits in midbrain dopamine neurons, impairing the patient's ability to direct and control movement.
- Leucine-rich repeat kinase 2 (leucine-rich repeat kinase 2: LRRK2) is implicated in hereditary Parkinson's disease.
- LRRK2 Gly2019Ser mutation causes an increase in kinase activity, resulting in hereditary Parkinson's disease.
- LRRK2 is also associated with Crohn's disease through genomic association analysis (Teri A. Manolio, N Engl J Med 2010;363:166-176).
- modulators or inhibitors of LRRK2 are being developed (eg, Patent Publication No. 2020-0085779 (July 15, 2020)).
- a phenylaminopyrimidine compound of Formula 1 that exhibits excellent inhibitory activity against LRRK2, and uses the same to treat and prevent diseases mediated by or related to LRRK2, for example, neurodegenerative diseases. to achieve high efficacy.
- Another object of the present invention is to provide a pharmaceutical composition for preventing or treating a disease or disorder mediated by or related to LRRK2, which includes a phenylaminopyrimidine compound of Formula 1.
- Another object of the present invention is to provide a method for preventing or treating a disease or disorder mediated by or related to LRRK2 using an LRRK2 inhibitor including a phenylaminopyrimidine compound of Formula 1.
- One aspect of the present invention provides a compound represented by the following formula (1), a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof:
- R 1 and R 2 are each independently H, halogen, OH, CN, amino, nitro, C 1-6 alkyl, halogen-substituted C 1-6 alkyl, C 1-6 alkoxy, or halogen It may be C 1-6 alkoxy substituted with .
- R 1 and R 2 can each independently be H, halogen, C 1-4 alkyl, or C 1-4 alkyl substituted with halogen.
- one of R 1 and R 2 may be H, and the other may be halogen, C 1-4 alkyl, or C 1-4 alkyl substituted with halogen.
- R 1 is H and R 2 can be halogen, C 1-6 alkyl, or C 1-6 alkyl substituted with a halogen atom.
- R 1 may be H and R 2 may be C 1-4 alkyl or C 1-4 alkyl substituted with halogen.
- R 2 is H and R 1 can be halogen, C 1-6 alkyl, or C 1-6 alkyl substituted with a halogen atom.
- R 2 can be H and R 1 can be halogen or C 1-4 alkyl.
- halogen may be F, Cl, Br or I.
- C 1-6 alkyl substituted with halogen or C 1-4 alkyl substituted with halogen may be substituted with 1, 2, 3 or more halogens (eg, F, Cl, Br, I), For example, it may be substituted with 2 or 3 or more halogens.
- halogen-substituted C 1-6 alkyl may be C 1-4 perhaloalkyl containing a trihalomethyl group (-CX′ 3 ; X′ is halogen).
- the halogen-substituted C 1-6 alkyl or halogen-substituted C 1-4 alkyl may be one substituted with fluorine, for example, a plurality of fluorine-substituted C 1-4 alkyl, tri It may include C 1-4 alkyl or C 1-4 perfluoroalkyl including a fluoromethyl group (—CF 3 ).
- R 1 and R 2 together are a substituted or unsubstituted 4-7 membered saturated or partially unsaturated carbocyclyl ring, or a substituted or unsubstituted 4-7 membered carbocyclyl ring comprising 1 to 3 nitrogen atoms. It may form a membered heteroaryl or heterocyclyl ring.
- R 1 and R 2 together represent a 5 or 6 membered saturated or partially unsaturated carbocyclyl ring, or a 5 or 6 membered heteroaryl or heterocyclyl ring containing 1 or 2 nitrogen atoms. can be formed
- R 1 and R 2 together can form, but are not limited to, cyclopentenyl, pyrrolyl, imidazolyl or pyrazolyl.
- the carbocyclyl ring, heteroaryl or heterocyclyl ring formed by R 1 and R 2 is C 1 -substituted with halogen, oxo, OH, CN, amino, nitro, C 1-6 alkyl, halogen or hydroxy selected from the group consisting of 6 alkyl, C 1-6 alkoxy, halogen substituted C 1-6 alkoxy, C 2-6 alkoxyalkyl, C 1-6 alkylamino and di-(C 1-6 alkyl)-amino may be optionally substituted with one or more substituents.
- the carbocyclyl ring, heteroaryl or heterocyclyl ring formed by R 1 and R 2 is halogen, OH, CN, amino, C 1-4 alkyl, halogen-substituted C 1-4 alkyl or may be optionally substituted with a substituent selected from C 1-4 alkoxy.
- R 3 may be —NR 31 R 32 or substituted or unsubstituted C 3-8 cycloalkyl.
- R 3 can be substituted or unsubstituted C 3-6 cycloalkyl.
- R 3 can be cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl.
- R 3 is —NR 31 R 32 , any one of R 31 and R 32 is H, the other is optionally substituted or unsubstituted 3-12 membered containing 1 nitrogen or oxygen atom It may be a saturated or partially unsaturated carbocyclyl or heterocyclyl ring.
- any one of R 31 and R 32 is H and the other is a substituted or unsubstituted 5-8 membered saturated or partially unsaturated carbocyclyl or heterocyclyl ringyl optionally containing 1 oxygen atom.
- the carbocyclyl or heterocyclyl ring may optionally be bridged or form a spiro ring.
- any one of R 31 and R 32 is H, and the other one is cyclohexyl, cycloheptyl, cyclooctyl, oxacyclohexyl, oxacycloheptyl, oxacyclooctyl, bicyclohexanyl, bicycloheptanyl.
- bicyclooctanyl oxabicyclohexanyl, oxabicycloheptanyl, oxabicyclooctanyl, spirohexanyl, spiroheptanyl, spirooctanyl, oxaspirohexanyl, oxaspiroheptanyl and oxaspirooctanyl It can be selected from the group consisting of.
- any one of R 31 and R 32 is H and the other is bicyclo[2.1.1]hexanyl, bicyclo[3.1.1]heptanyl, bicyclo[2.2.1]heptanyl, bicyclo[2.1.1]heptanyl, bicyclo[2.1.1]heptanyl Cyclo[4.1.1]octanyl, bicyclo[3.2.1]octanyl, bicyclo[2.2.2]octanyl, oxabicyclo[2.1.1]hexanyl, oxabicyclo[2.2.1]heptanyl , oxabicyclo[3.1.1]heptanyl, oxabicyclo[4.1.1]octanyl, oxabicyclo[3.2.1]octanyl, oxabicyclo[2.2.2]octanyl, bicyclo[3.1.
- R 3 is —NR 31 R 32 and R 31 and R 32 together with the nitrogen atom to which they are attached are substituted or unsubstituted 3-10 membered monocyclic comprising 1 or 2 nitrogen atoms or a bicyclic heterocyclyl ring.
- R 31 and R 32 together with the nitrogen atom to which they are attached may form a substituted or unsubstituted 3-8 membered heterocyclyl ring containing 1 or 2 nitrogen atoms.
- R 31 and R 32 together with the nitrogen atom to which they are attached may form a substituted or unsubstituted 4-7 membered heterocyclyl ring containing 1 nitrogen atom.
- R 31 and R 32 together can form, but are not limited to, azetidinyl, pyrrolidinyl, piperidinyl or azepanyl.
- a substituted C 3-8 cycloalkyl or C 3-6 cycloalkyl, a substituted carbocyclyl ring or a substituted heterocyclyl ring is halogen, oxo, OH, CN , amino, nitro, C 1-6 alkyl, halogen or hydroxy substituted C 1-6 alkyl, C 1-6 alkoxy, halogen substituted C 1-6 alkoxy, C 2-6 alkoxyalkyl, C 1 may be optionally substituted with one or more substituents selected from the group consisting of -6 alkylamino and di-(C 1-6 alkyl)-amino.
- the substituted C 3-8 cycloalkyl or C 3-6 cycloalkyl, substituted carbocyclyl ring or substituted heterocyclyl ring is each independently halogen, OH, CN, amino, C 1-4 may be substituted with one or more substituents selected from the group consisting of alkyl, C 1-4 alkoxy, C 1-4 alkylamino and di-(C 1-4 alkyl)amino.
- the substituted C 3-8 cycloalkyl or C 3-6 cycloalkyl, substituted carbocyclyl ring or substituted heterocyclyl ring is each independently halogen (eg, F, Cl, Br, I), OH, amino, C 1-4 alkoxy (eg methoxy, ethoxy) and C 1-4 alkylamino (eg methylamino, ethylamino) or It may be substituted with two substituents.
- R 4 may be ring X and R 5 may be H.
- ring X may be -NR X1 R X2 or substituted or unsubstituted C 3-8 cycloalkyl.
- ring X can be substituted or unsubstituted C 3-6 cycloalkyl.
- ring X can be cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl.
- ring X is —NR X1 R X2 , R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted 3-10 membered monocytic containing 1 or 2 nitrogen atoms It may form a cyclic or bicyclic heterocyclyl ring.
- R X1 and R X2 may form a substituted or unsubstituted 3- to 7-membered heterocyclyl ring containing one or two nitrogen atoms together with the nitrogen atom to which they are attached.
- R X1 and R X2 may form a substituted or unsubstituted 4- to 7-membered heterocyclyl ring including one nitrogen atom together with the nitrogen atom to which they are attached.
- R X1 and R X2 together may form, but are not limited to, azetidinyl, pyrrolidinyl, piperidinyl or azepanyl.
- ring X is halogen, oxo, OH, CN, amino, nitro, C 1-6 alkyl, halogen or hydroxy substituted C 1-6 alkyl, C 1-6 alkoxy, halogen substituted C 1-6 alkoxy , C 2-6 alkoxyalkyl, C 1-6 alkylamino and di-(C 1-6 alkyl)-amino may be optionally substituted with one or more substituents selected from the group consisting of.
- ring X is substituted with halogen, oxo, OH, C 1-4 alkyl, halogen substituted C 1-4 alkyl, hydroxy substituted C 1-4 alkyl, C 1-4 alkoxy and halogen It may be optionally substituted with one or more substituents selected from the group consisting of C 1-4 alkoxy.
- ring X can be halogen (eg F, Cl, Br, I), oxo, OH, C 1-4 alkyl (eg methyl, ethyl) and halogen substituted C 1-4 alkyl (eg, CHF 2 , CH 2 F, CF 3 , CH 2 CF 3 ) may be optionally substituted with one or two substituents selected from the group consisting of.
- halogen eg F, Cl, Br, I
- oxo OH
- C 1-4 alkyl eg methyl, ethyl
- halogen substituted C 1-4 alkyl eg, CHF 2 , CH 2 F, CF 3 , CH 2 CF 3
- substituents selected from the group consisting of.
- R 4 and R 5 may be joined together with the carbon atom of the benzene ring to which they are attached to a *—CO—NR 6 —CH 2 — group to form a five membered lactam ring.
- * represents the position at which R 4 is bonded to the benzene ring.
- R 6 may be H, C 1-6 alkyl, or C 1-6 alkyl substituted with a halogen atom.
- R 6 can be H or C 1-4 alkyl.
- R 3 is -NR 31 R 32
- R 31 and R 32 form a substituted or unsubstituted 3- to 10-membered heterocyclyl ring
- R 4 is Ring X
- Ring X is When -NR X1 R X2 , if either of R 1 and R 2 is H, then the other of R 1 and R 2 is not C 1-6 alkyl substituted with halogen.
- R 1 and R 2 together are a substituted or unsubstituted 5- or 6-membered saturated or partially unsaturated carbocyclyl ring, or a substituted or unsubstituted 4-membered ring containing 1 nitrogen atom.
- R 31 and R 32 together with the nitrogen atom to which they are attached form a substituted or unsubstituted 5- or 6-membered heterocyclyl ring containing 1 or 2 nitrogen atoms;
- R 4 is ring X,
- R 5 is H, wherein ring X is -NR X1 R X2 and R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted 4 members containing one nitrogen atom to 7 membered heterocyclyl rings.
- the substituted rings may each independently be optionally substituted with a substituent selected from halogen, OH, C 1-6 alkyl, or C 1-6 alkyl substituted with halogen or hydroxy.
- R 1 and R 2 together form cyclopentenyl, pyrrolyl, imidazolyl or pyrazolyl;
- R 31 and R 32 together with the nitrogen atom to which they are attached form pyrrolidinyl or piperidinyl optionally substituted with halogen, OH, or C 1-4 alkyl substituted with halogen;
- ring X is —NR X1 R X2 ,
- R 5 is H, and R X1 and R X2 taken together form halogen, C 1-4 alkyl substituted with halogen, or pyrrolidinyl or piperidinyl optionally substituted with OH can do.
- R 1 and R 2 are each independently H, halogen or C 1-6 alkyl; R 31 and R 32 together with the nitrogen atom to which they are attached form a substituted or unsubstituted 4-7 membered heterocyclyl ring containing 1 nitrogen atom; R 4 is ring X, R 5 is H, wherein ring X is -NR X1 R X2 and R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted 4 members containing one nitrogen atom to 7 membered heterocyclyl rings.
- the aforementioned R 31 , R 32 , R X1 and R X2 , And specific examples of substituents thereof may be equally applied to the present embodiment, if applicable.
- R One and R 2 are each independently H, halogen or C 1-6 alkyl, for example R One and R 2 one is H and the other is halogen or C 1-4 alkyl; R 31 and R 32 any one is H and the other is a substituted or unsubstituted 5-8 membered saturated or partially unsaturated carbocyclyl or heterocyclyl ring optionally containing 1 oxygen atom, wherein the carbocyclyl or heterocyclyl may optionally be bridged or form a spiro ring, for example R 31 and R 32 one is H and the other is oxabicyclo[2.2.1]heptanyl or bicyclo[2.2.1]heptanyl; R 4 is ring X and R 5 is H and ring X is -NR X1 R X2 and R X1 and R X2 may be a substituted or unsubstituted 4-7 membered heterocyclyl ring containing 1 nitrogen atom together with the nitrogen atom
- any one of R 1 and R 2 is H, and the other is C 1-6 alkyl substituted with halogen (eg, C 1-4 alkyl substituted with fluoro, such as For example, when CF 3 ), R 3 is —NR 31 R 32 or substituted or unsubstituted C 3-6 cycloalkyl, either of R 31 and R 32 is H and the other is optionally 1 oxygen.
- R 3 is —NR 31 R 32 or substituted or unsubstituted C 3-6 cycloalkyl, either of R 31 and R 32 is H and the other is optionally 1 oxygen.
- R 31 and R 32 are H and the other is oxabicyclo[2.2.1]heptanyl or bicyclo[2.2.1]heptanyl;
- R 4 is ring X
- R 5 is H, and
- ring X is —NR X1 R X2 or substituted or unsubstituted C 3-6 cycloalkyl, wherein R X1 and R X2 together with the nitrogen atom to which they are attached are 1
- a substituted or unsubstituted 4- to 7-membered heterocyclyl ring containing two nitrogen atoms may be formed.
- the aforementioned R 31 , R 32 , R X1 and R X2 , And specific examples of substituents thereof may be equally applied to the present embodiment, if applicable.
- any one of R 1 and R 2 is H, and the other is C 1-6 alkyl substituted with halogen (eg, C 1-4 alkyl substituted with fluoro, such as For example, in the case of CF 3 ), R 3 is —NR 31 R 32 , and R 31 and R 32 are substituted or unsubstituted 4-7 membered heterocycle containing 1 nitrogen atom together with the nitrogen atom to which they are attached. forming a reel ring; R 4 is ring X, R 5 is H, and ring X can be substituted or unsubstituted C 3-6 cycloalkyl.
- the aforementioned R 31 , R 32 , R X1 and R X2 , And specific examples of substituents thereof may be equally applied to the present embodiment, if applicable.
- the reaction for preparing the compound of the present invention can be carried out in a suitable solvent which can be appropriately selected by a person skilled in the art of organic synthesis.
- suitable solvents are those that are substantially unreactive with the starting materials (reactants), intermediates or objects at the temperature at which the reaction occurs.
- a person skilled in the art will be able to properly select a suitable solvent for a particular reaction step.
- the compound of the present invention can be synthesized according to the synthesis procedure described in the following examples, and based on this, reactants and reaction conditions (eg, reaction solvent, reaction temperature, reaction time, reaction catalyst, etc.) according to the structure of the target compound, etc. With appropriate modifications, the target compound may be prepared.
- reactants and reaction conditions eg, reaction solvent, reaction temperature, reaction time, reaction catalyst, etc.
- each reaction can be monitored by an appropriate method known in the art.
- the synthesis of the target compound may be monitored by spectroscopic means, such as NMR (eg, 1 H or 13 C), mass spectroscopy, or chromatography (HPLC or TLC).
- R 1 and R 5 are each H, and R 3 is -NHR 31
- a compound of Formula 1 may be prepared according to Scheme A below.
- step 1 the aniline compound and the dichloropyrimidine compound are mixed in a suitable organic solvent such as dichloroethane (DCE), t-butanol (tBuOH), etc. optionally in the presence of a suitable catalyst such as ZnCl 2 , triethylammonium (TEA), etc. can be reacted.
- a suitable organic solvent such as dichloroethane (DCE), t-butanol (tBuOH), etc.
- a suitable catalyst such as ZnCl 2 , triethylammonium (TEA), etc.
- the reaction may be carried out at about -10 °C to about 25 °C, about -5 °C to about 10 °C, or about 0 °C for about 1 to 5 hours, or about 2 to 3 hours.
- step 2 the compound obtained in step 1 may be mixed with an appropriate amine compound (NH 2 R 31 ) and an appropriate organic solvent, such as N,N-diisopropylethylamine (DIEA), acetonitrile (ACN), or the like.
- an appropriate amine compound such as N,N-diisopropylethylamine (DIEA), acetonitrile (ACN), or the like.
- the reaction may be carried out at about 50 °C to about 1500 °C, about 80 °C to about 120 °C, or about 100 °C.
- R 1 and R 5 are each H and R 3 is C 3-8 cycloalkyl may be prepared according to Scheme B below.
- step 1 the chloropyrimidine compound and the carboxylic acid compound (R 3 COOH) are optionally mixed with a suitable catalyst such as (NH 4 ) 2 S 2 O 8 , AgNO 3 in a suitable solvent such as a mixed solvent of ACN and H 2 O. It can be reacted by mixing in the presence of such.
- the reaction is at about 50 °C to about 120 °C, about 70 °C to about 100 °C, or about 80 °C for about 10 hours to about 20 hours, about 12 hours to about 18 hours, or about 16 hours. can be done
- step 2 the compound obtained in step 1 is mixed with an appropriate aniline compound and a suitable solvent such as butanol (BuOH), ACN, DIEA, optionally by dropwise addition of 1 drop of concentrated hydrochloric acid, to obtain the compound of formula 1.
- a suitable solvent such as butanol (BuOH), ACN, DIEA, optionally by dropwise addition of 1 drop of concentrated hydrochloric acid, to obtain the compound of formula 1.
- the reaction is at about 50 °C to about 120 °C, about 70 °C to about 100 °C, or about 80 °C for about 30 minutes to about 15 hours, about 30 minutes to about 10 hours, about 1 hour to about This can be done for 12 hours.
- the compound of Formula 1 wherein R 3 is —NR 31 R 32 , R 4 is Ring X, and R 5 is H may be prepared according to Scheme C below.
- step 1 the dichloropyrimidine compound and the amine compound (NHR 31 R 32 ) are mixed with an appropriate organic solvent, such as DIEA (diisopropylethylamine), DCM (dichloromethane), DMF (dimethylformamide) and ACN (aceto).
- an appropriate organic solvent such as DIEA (diisopropylethylamine), DCM (dichloromethane), DMF (dimethylformamide) and ACN (aceto
- the reaction may be carried out in one or more organic solvents selected from the group consisting of nitrile).
- a temperature of about -80 ° C. to about 80 ° C. for example, about 10 minutes to about 10 hours, preferably about 15 minutes to about 6 hours may be stirred and mixed.
- step 2 the compound obtained in step 1 may be reacted with aniline substituted with ring X by adding 1 drop of HCl optionally in a suitable solvent such as nBuOH.
- a suitable solvent such as nBuOH.
- the reaction of step 2 for example, at a temperature of about 50° C. to about 150° C., preferably about 80° C. to about 120° C., for example, about 30 minutes to about 10 hours, preferably about 1 hour to about 6 hours. It can be stirred and mixed.
- step 1 the dichloropyridine compound may be stirred and mixed in an appropriate organic solvent, such as dichloroethane (DCE) and t-BuOH, and ZnCl 2 may be added thereto, followed by stirring and mixing with the ring X-substituted aniline compound.
- an appropriate organic solvent such as dichloroethane (DCE) and t-BuOH
- ZnCl 2 may be added thereto, followed by stirring and mixing with the ring X-substituted aniline compound.
- step 2 the compound obtained in step 1 is mixed with an appropriate amine compound (NHR 31 R 32 ) with an appropriate organic solvent such as DIEA (diisopropylethylamine), DCM (dichloromethane), DMF (dimethylformamide) and
- an appropriate organic solvent such as DIEA (diisopropylethylamine), DCM (dichloromethane), DMF (dimethylformamide) and
- the reaction may be carried out in one or more organic solvents selected from the group consisting of ACN (acetonitrile).
- ACN acetonitrile
- the preparation method of Chemical Formula 1 described above is exemplary, and may be modified by selecting an appropriate solvent, catalyst, reaction conditions, etc. depending on the type of starting material for obtaining the final compound, and these solvents, catalysts, reaction conditions, etc. It is well known to those skilled in the art.
- the compound of Formula 1 may be selected from the group consisting of:
- alkyl refers to a fully saturated branched or unbranched (or straight-chain or linear) hydrocarbon.
- the alkyl may be a substituted or unsubstituted alkyl group.
- the C 1-6 alkyl may be a C 1 to C 5 , C 1 to C 4 , C 1 to C 3 , or C 1 to C 2 alkyl group.
- Non-limiting examples of the alkyl may be methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, n-pentyl, isopentyl, neopentyl, iso-amyl, or n-hexyl. have.
- halogen refers to an atom belonging to group 17 of the periodic table. Halogen atoms include fluorine, chlorine, bromine, and iodine.
- cycloalkyl refers to a saturated monocyclic hydrocarbon group.
- the cycloalkyl may contain 3 to 12, such as 3 to 10, 3 to 8, or 3 to 6 carbon atoms.
- C 3-8 cycloalkyl can be cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl or cyclooctyl.
- carbocyclyl refers to a saturated or partially unsaturated monocyclic or polycyclic hydrocarbon group. As used herein, the carbocyclyl is intended as a term inclusive of the cycloalkyl.
- carbocyclyl refers to a saturated or partially unsaturated carbocyclic ring having 3 to 20, 3 to 10, 3 to 8, 4 to 7, 5 to 8, 5 or 6 carbon atoms. can refer to The term carbocyclyl includes mono-, bi-, tri-, fused, bridged and spiro-ring systems and combinations thereof. In one embodiment, the bicyclic carbocyclyl may contain 6 to 12, 6 to 10, or 6 to 8 carbon atoms.
- the spiro carbocyclyl may contain 5 to 12, 6 to 10, or 6 to 8 carbon atoms.
- Representative examples of monocyclic carbocyclyl may include cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, and the like.
- Bicyclic carbocyclyl having 6 to 12 ring atoms is [4,3], [4,4], [4,5], [5,5], [5,6] or [6,6] ] ring systems, for example, bridged bicyclic rings such as bicyclo[2.2.1]heptanyl, bicyclo[2.2.2]octanyl, and bicyclo[3.2.2]nonanyl.
- Spiro carbocyclyls may include spiro[2.2]pentanyl, spiro[2.3]hexanyl, spiro[2.4]heptanyl, spiro[2.5]octanyl, spiro[4.5]decanyl, and the like.
- heterocyclyl refers to a saturated or partially unsaturated cyclic hydrocarbon containing at least one heteroatom.
- the heterocyclyl ring group may be a single ring group, a two ring group, or a three ring group.
- the two ring groups may be a spiro-ring group, a bridged-ring group, and a fused-ring group.
- Heterocyclyl ring groups are 3 to 20, 3 to 12, 3 to 10, 3 to 8, 3 to 7, 4 to 7, 5 to 7, 3 to 6, It may contain 4 to 6, or 5 to 6 ring atoms.
- the heteroatom may be any one or more selected from the group consisting of N, O and S.
- the heteroatom can be 1 to 3 N, or 1 or 2 N, 1 N, or 1 O.
- the heteroatom may be one or two N.
- monocyclic heterocyclyl rings include aziridine, azetidine, pyrrolidine, piperidine, azepane, and the like.
- Non-limiting examples of bridged or spiro heterocyclic rings include oxabicyclohexanyl, oxabicycloheptanyl, oxabicyclooctanyl, oxaspirohexanyl, oxaspiroheptanyl, oxaspirooctanyl, azabicyclohexane yl, azabicycloheptanyl, azabicyclooctanyl, azaspirohexanyl, azaspiroheptanyl, azaspirooctanyl, and the like.
- aryl also includes groups in which an aromatic ring is fused to one or more carbon rings.
- the C 6 -C 30 aryl group may be, for example, a C 6 to C 15 , or a C 6 to C 10 aryl group.
- Non-limiting examples of aryl include phenyl, naphthyl, or tetrahydronaphthyl.
- heteroaryl refers to a monocyclic or bicyclic aromatic compound containing one or more heteroatoms selected from the group consisting of N, O, P and S, the remaining ring atoms being carbon.
- the heteroaryl group may contain, for example, 1 to 3, 1 or 2 heteroatoms, and 4 to 10, 5 to 10, 4 to 7, 5 or 6 ring atoms. may include.
- the S or N may be oxidized to have several oxidation states.
- Non-limiting examples of "heteroaryl” include pyrrolyl, imidazolyl, pyrazolyl, thienyl, furyl, thiazolyl, isothiazolyl, or 1,2,3-oxadiazolyl, and the like.
- hydroxy refers to a -OH functional group (hydroxyl group).
- alkoxy refers to an alkyl bound to an oxygen atom.
- the C 1 to C 20 alkoxy group may be, for example, C 2 to C 15 , C 2 to C 10 , or C 2 to C 5 alkoxy group.
- C 1-6 alkoxy may include methoxy, ethoxy, propoxy, and the like.
- amino refers to a —NH 2 group.
- nitro refers to —NO 2 .
- cyano refers to a functional group consisting of a triple bond between a carbon atom and a nitrogen atom as -CN.
- substituted of “substituted or unsubstituted” refers to introduced instead of a hydrogen atom when a derivative is formed by substituting one or more hydrogen atoms in an organic compound with another atomic group, and “substituent” refers to an introduced atomic group .
- the substituent is, for example, a halogen atom, a C 1 to C 20 alkyl group substituted with a halogen atom (eg, CCF 3 , CHCF 2 , CH 2 F, CCl 3 etc.), C 1 to C 20 alkoxy, C 2 to C 20 alkoxyalkyl, hydroxy group, nitro group, cyano group, amino group, amidino group, hydrazine, hydrazone, carboxyl group or its Salt, sulfonyl group, sulfamoyl group, sulfonic acid group or a salt thereof, phosphoric acid or a salt thereof, or C 1 to C 20 alkyl group, C 2 to C 20 alkenyl group, C 2 to C 20 alkynyl group, C 1 to C 20 heteroalkyl group, C 6 to C 20 aryl group, C 6 to C 20 a halogen atom (eg, CCF 3 , CHCF 2 , CH 2 F, CCl 3
- the substituent may be halogen, oxo, OH, CN, amino, nitro, C 1-6 alkyl, halogen or hydroxy substituted C 1-6 alkyl, C 1-6 alkoxy, halogen substituted C 1 - and one or more substituents selected from the group consisting of 6 alkoxy, C 2-6 alkoxyalkyl, C 1-6 alkylamino and di-(C 1-6 alkyl)-amino.
- isomer of the term “stereoisomer” refers to a compound that has the same molecular formula but does not have the same spatial arrangement or connection mode of constituent atoms in the molecule.
- Isomers include, for example, structural isomers, and stereoisomers.
- the stereoisomer may be a diastereomer or an enantiomer.
- Enantiomers refer to isomers that do not overlap their mirror images, such as the relationship between the left and right hands, and are also called optical isomers. Enantiomers are divided into R (Rectus: clockwise) and S (Sinister: counterclockwise) when 4 or more substituents differ from each other at the chiral central carbon.
- Diastereomers refer to stereoisomers that are not in a mirror image relationship, and are isomers caused by different spatial arrangement of atoms.
- the diastereomers may be divided into cis-trans isomers and conformational isomers or conformers.
- solvate refers to a compound solvated in an organic or inorganic solvent.
- the solvate is, for example, a hydrate.
- salt refers to inorganic and organic acid addition salts of compounds.
- the pharmaceutically acceptable salt may be a salt that does not cause serious irritation to the organism to which the compound is administered and does not impair the biological activity and properties of the compound.
- the inorganic acid salt may be hydrochloride, bromate, phosphate, sulfate, or disulfate.
- the organic acid salt is formate, acetate, propionate, lactate, oxalate, tartrate, malate, maleate, citrate, fumarate, besylate, camsylate, edicyl salt, trichloroacetic acid, trifluoroacetate , benzoate, gluconate, methanesulfonate, glycolate, succinate, 4-toluenesulfonate, galacturonate, embonate, glutamate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, or aspartate It may be an acid.
- the metal salt may be a calcium salt, a sodium salt, a magnesium salt, a strontium salt, or a potassium salt.
- the compound of Formula 1 may be a Leucine-rich repeat kinase 2 (LRRK2) inhibitor.
- the LRRK2 may be a protein belonging to a leucine-rich repeat kinase family.
- the LRRK2 may also be referred to as AURA17, DARDARIN, PARK8, RIPK7, or ROCO2.
- the LRRK2 is Uniprot No. It may be a protein comprising the amino acid sequence of Q5S007.
- the LRRK2 may include a Gly2019Ser mutation.
- Another aspect of the present invention is to treat a disease or condition mediated by or related to LRRK2, including the compound of Formula 1, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof according to an aspect
- a pharmaceutical composition for preventing or treating is provided.
- the disease or disorder mediated by or related to LRRK2 may be a neurodegenerative disease.
- neurodegenerative disease refers to any disease associated with degenerative changes in the nervous system, and specifically may refer to degenerative brain diseases associated with degenerative changes in the brain.
- the degenerative brain disease is Parkinson's disease, Alzheimer's disease, Huntington's disease, mild cognitive impairment, amyloidosis, multiple system atrophy, multiple Multiple sclerosis, tauopathies, Pick's disease, senile dementia, amyotrophic lateral sclerosis, Spinocerebellar Atrophy, Tourette's Syndrome, Friedrich's Syndrome, Friedrich's Ataxia, Machado-Joseph's disease, Lewy Body Dementia, Dystonia, Progressive Supranuclear Palsy and Frontotemporal Dementia It can be selected from the group consisting of.
- diseases or disorders mediated by or associated with LRRK2 include dyskinesia; central nervous system disorders; cancer such as kidney cancer, breast cancer, prostate cancer, blood cancer, lung cancer, acute myeloid leukemia or multiple myelopathy; inflammatory diseases such as rheumatoid arthritis, ankylosing spondylitis, Crohn's disease, amyotrophic lateral sclerosis, leprosy, Inflammatory Bowel Disease, tuberculosis, and the like.
- the pharmaceutical composition may further include a known active ingredient having degenerative brain disease prevention, treatment, or amelioration activity.
- Known active ingredients having degenerative brain disease prevention, treatment, or amelioration activity are levodopa, catechol-O-methyltransferase (COMT) inhibitor, dopamine agonist, MAO-B (monoamine oxidase) B) an inhibitor, amantadine, an anticholinergic agent, an acetylcholinesterase inhibitor, or an NMDA receptor antagonist (N-Methyl-D-aspartate receptor antagonist).
- COMT inhibitors may be opicapone, entacapone and tolcapone.
- Dopamine agonists include bromocriptine, pergolide, pramipexole, ropinirole, piribedil, cabergoline, and apomorphine. and lisuride.
- MAO-B inhibitors are safinamide, selegiline, and rasagiline.
- the acetylcholinesterase inhibitor may be tacrine, rivastigmine, galantamine and donepezil.
- the NMDA receptor antagonist may be memantine.
- the compound represented by Formula 1, a derivative, stereoisomer, solvate, or pharmaceutically acceptable salt thereof and the known active ingredient may be a single or separate composition for simultaneous or sequential administration.
- prevention refers to any action of inhibiting the onset of or delaying the onset of a disease or disorder mediated by or related to LRRK2 by administration of the pharmaceutical composition.
- treatment refers to any action in which the symptoms of a disease or disorder mediated by or related to LRRK2 are ameliorated or beneficially altered by administration of the pharmaceutical composition.
- the pharmaceutical composition may include a pharmaceutically acceptable carrier.
- the carrier is used in the sense of including excipients, diluents or adjuvants.
- the carrier may be, for example, lactose, dextrose, sucrose, sorbitol, mannitol, xylitol, erythritol, maltitol, starch, gum acacia, alginate, gelatin, calcium phosphate, calcium silicate, cellulose, methyl cellulose, polyvinyl pi It may be selected from the group consisting of rolidone, water, physiological saline, buffers such as PBS, methyl hydroxy benzoate, propyl hydroxy benzoate, talc, magnesium stearate, and mineral oil.
- the composition may include a filler, an anti-agglomeration agent, a lubricant, a wetting agent, a flavoring agent, an emulsifying agent, a preservative, or a combination thereof
- the pharmaceutical composition may be prepared in any formulation according to a conventional method.
- the composition may be formulated, for example, as an oral dosage form (eg, a powder, tablet, capsule, syrup, pill, or granule), or a parenteral dosage form (eg, an injection).
- the composition may be prepared as a systemic formulation, or as a topical formulation.
- the solid preparation for oral administration may be a tablet, pill, powder, granule, or capsule.
- the solid formulation may further include an excipient.
- the excipient may be, for example, starch, calcium carbonate, sucrose, lactose, or gelatin.
- the solid formulation may further include a lubricant such as magnesium stearate or talc.
- the oral liquid formulation may be a suspension, an internal solution, an emulsion, or a syrup.
- the liquid formulation may contain water or liquid paraffin.
- the liquid formulation may contain excipients, for example, wetting agents, sweetening agents, perfuming agents, or preservatives.
- the preparation for parenteral administration may be a sterile aqueous solution, non-aqueous solution, suspension, emulsion, freeze-dried or suppository.
- Non-aqueous solvents or suspending agents may include vegetable oils or esters.
- the vegetable oil may be, for example, propylene glycol, polyethylene glycol, or olive oil.
- the ester may be, for example, ethyl oleate.
- the base of the suppository may be witepsol, macrogol, tween 61, cacao butter, laurin, or glycerogelatin.
- the pharmaceutical composition includes the compound according to an aspect, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof as an active ingredient of the pharmaceutical composition.
- Active ingredient refers to a physiologically active substance used to achieve pharmacological activity (eg, treatment of degenerative brain disease).
- the pharmaceutical composition may include the compound according to an aspect, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof in an effective amount.
- effective amount refers to an amount sufficient to effect the prevention or treatment of a disease or disorder when administered to a subject in need thereof.
- the effective amount can be appropriately selected by those skilled in the art depending on the cell or individual to be selected.
- the preferred dosage of the pharmaceutical composition varies depending on the condition and weight of the individual, the degree of disease or disease, the drug form, the route and duration of administration, but may be appropriately selected by those skilled in the art.
- the compound, stereoisomer, solvate, or pharmaceutically acceptable salt thereof may be, for example, in an amount from about 0.0001 mg/kg to about 100 mg/kg, or from about 0.001 mg/kg to about 100 mg/kg. may be administered in divided doses 1 to 24 times a day, 1 to 7 times per 2 days to 1 week, or 1 to 24 times in 1 month to 12 months.
- the compound, stereoisomer, solvate, or pharmaceutically acceptable salt thereof is present in an amount of from about 0.0001% to about 10% by weight, or from about 0.001% to about 1% by weight, based on the total weight of the composition. may be included.
- the administration method may be oral or parenteral administration.
- the method of administration can be, for example, oral, transdermal, subcutaneous, rectal, intravenous, intraarterial, intraperitoneal, intramuscular, intrasternal, topical, intranasal, intratracheal, or intradermal routes.
- the composition may be administered systemically or locally, alone or in combination with other pharmaceutically active compounds.
- Another aspect of the present invention is to prevent a disease or disorder mediated by or related to LRRK2, comprising administering to an individual a compound of Formula 1, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof according to an aspect or a method of treatment.
- the compounds of Formula 1, stereoisomers, solvates, pharmaceutically acceptable salts, and diseases mediated by or related to LRRK2, prevention, and treatment are as described above.
- the subject may be a mammal, such as a human, mouse, rat, cow, horse, pig, dog, monkey, sheep, goat, ape, or cat.
- the subject may be suffering from, or likely to suffer from, a condition associated with a disease mediated by or associated with LRRK2.
- the method may further comprise administering to the subject a known active ingredient having an effect of preventing or treating a disease or disorder mediated by or related to LRRK2.
- the known active ingredient may be administered to the subject simultaneously, separately, or sequentially with the compound according to an aspect, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof.
- the administration method may be oral or parenteral administration.
- the method of administration can be, for example, oral, transdermal, subcutaneous, rectal, intravenous, intraarterial, intraperitoneal, intramuscular, intrasternal, topical, intranasal, intratracheal, or intradermal routes.
- the pharmaceutical composition may be administered systemically or locally, alone or in combination with other pharmaceutically active compounds.
- the preferred dosage of the pharmaceutical composition varies depending on the condition and weight of the patient, the severity of the disease or disease, the drug form, the route and duration of administration, but may be appropriately selected by those skilled in the art.
- the dosage is, for example, in the range of about 0.001 mg/kg to about 100 mg/kg, about 0.01 mg/kg to about 10 mg/kg, or about 0.1 mg/kg to about 1 mg/kg, on an adult basis.
- the administration may be administered once a day, multiple times a day, or once a week, once every two weeks, once every three weeks, or once every four weeks to once a year.
- Another aspect of the present invention is the use of a compound of Formula 1, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof according to one aspect, for use in the prevention or treatment of a disease or condition mediated by or related to LRRK2. to provide.
- Another aspect of the present invention is a compound of Formula 1, a stereoisomer, a solvate, or a pharmaceutically acceptable compound according to one aspect, for preparing a medicament for preventing or treating a disease or condition mediated by or related to LRRK2. Uses of salts are provided.
- the disease or disorder mediated by or related to LRRK2, prevention, treatment, compound of Formula 1, stereoisomer, solvate, and pharmaceutically acceptable salt are as described above.
- the pyrimidine compound of Formula 1 according to the present invention has excellent LRRK2 inhibitory activity, and thus a pharmaceutical composition for preventing or treating a disease mediated by or related to LRRK2 (eg, Parkinson's disease), and treatment and prevention of a disease or disorder using the same method can be used effectively.
- LRRK2 eg, Parkinson's disease
- step 1 The compound obtained in step 1 (1.20 g, 4.376 mmol, 1.00 equiv) and Pd/C (300 mg, 10%) were mixed in MeOH (10 mL) under a hydrogen atmosphere (3 atm) at 25° C. by stirring for 1 hour. did. The solid was filtered off and the filtrate was concentrated under reduced pressure to obtain 4-[3-(trifluoromethyl)piperidin-1-yl]aniline (1 g, 84.21%) as a brown solid. MS m/z: 245 [M+H] + .
- 2-Chloro-pyrimidine (2.00 g, 17.46 mmol, 1.00 equiv) was mixed by stirring in THF (15 mL) and TMPMgCl.LiCl (1.1M in THF) (17.50 mL, 19.21 mmol, 1.10 equiv) was placed in a nitrogen atmosphere. It was added dropwise at -60°C under The resulting mixture was stirred under a nitrogen atmosphere at -60°C for 2 h, and ZnCl 2 (1.0 M in THF) (19.21 mL, 19.21 mmol, 1.10 equiv) was added dropwise at -60°C.
- step 1 The compound obtained in step 1 (900 mg, 3.74 mmol, 1.00 equiv) was mixed by stirring in THF (10 mL), and TMPMgCl.LiCl (1.1M in THF) (5.0 mL, 5.49 mmol, 1.47 equiv) was placed in a nitrogen atmosphere. It was added dropwise at -60 °C under The resulting mixture was stirred at -60°C under a nitrogen atmosphere for 1 hour, and then 1,2-dibromo-1,1,2,2-tetrachloroethane (1.82 g, 5.61 mmol, 1.50 eq.) in the resulting mixture.
- the optical isomer mixture (1.1 g, 3.59 mmol, 1 equiv) obtained in step 2 above was mixed in THF (15 mL) and NaH (215.38 mg, 5.39 mmol, 60% purity, 1.5 equiv) was added at 0 °C and 20 min. stirred for a while. Then MeI (2.32 g, 16.35 mmol, 1.02 mL, 4.55 equiv) was added to the mixture at 0° C., then warmed to 20° C. and stirred for 2 h. The reaction was quenched with anhydrous NH 4 Cl and the aqueous phase was extracted with ethyl acetate (50 mL ⁇ 3).
- the optical isomer mixture obtained in step 3 above (500 mg, 1.56 mmol, 1 equiv) was mixed in DCM (6 mL) and TFA (2.31 g, 20.29 mmol, 1.50 mL, 13 equiv) was added in one portion at 0°C. The mixture was stirred at 0° C. for 3 min, then warmed to 20° C. and stirred for 2 h. The reaction mixture was concentrated under reduced pressure to remove the solvent and compound 5 (520 mg, 99.67%, TFA salt) was obtained as a mixture of optical isomers in the form of a yellow oil.
- Step 1 Synthesis of N-[4-(azepan-1-yl)phenyl]-4-chloro-5-(trifluoromethyl)pyrimidin-2-amine
- Step 2 N2-[4-(azepan-1-yl)phenyl]-N4-[(1R,4R)-bicyclo[2.2.1]heptan-1-yl]-5-(trifluoromethyl) Synthesis of pyrimidine-2,4-diamine
- N-[4-(azepan-1-yl)phenyl]-4-chloro-5-(trifluoromethyl)pyrimidin-2-amine 100 mg, 0.27 mmol, 1.00 equiv) in ACN (3.00 mL) and bicyclo[2.2.1]heptan-1-amine (32 mg, 0.29 mmol, 1.10 equiv) were stirred mixed and DIEA (139 mg, 1.07 mmol, 4.00 equiv) was added. The resulting mixture was stirred at 100° C. for 16 h, and the mixture was cooled to room temperature. The mixture was purified by silica gel column chromatography, eluting with PE/EtOAc (5:1).
- the crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30 ⁇ 150 mm, 5 um, n; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (80% Phase B, up to 90% in 8 min); Detector, UV 254 nm.
- the collected fractions were lyophilized to N2-[4-(azepan-1-yl)phenyl]-N4-[(1R,4R)-bicyclo[2.2.1]heptan-1-yl]-5-(tri Fluoromethyl)pyrimidine-2,4-diamine (compound of Example 1; 11 mg, 9.56%) was obtained as a white solid.
- the crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30 ⁇ 150 mm, 5 um; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (80% Phase B, up to 90% in 10 min); Detector, UV 254 nm.
- the collected fractions were lyophilized to obtain N-(4-cyclohexylphenyl)-4-cyclopentyl-5-(trifluoromethyl)pyrimidin-2-amine (compound of Example 2; 9.7 mg, 6.22%). It was obtained as a white solid.
- the crude product was purified by Prep-HPLC under the following conditions: column, YMC-Actus Triart C18 ExRS, 30 ⁇ 150 mm, 5 um; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (70% Phase B, up to 90% in 10 min); Detector, UV 254/220 nm.
- the collected fractions were lyophilized to obtain 4-cyclopropyl-N-(4-cyclopropylphenyl)-5-(trifluoromethyl)pyrimidin-2-amine (compound of Example 3; 8.4 mg, 1.77%). It was obtained as a white solid.
- Step 1 4-Cyclohexyl-N-(4-[1,4-dioxa-7-azaspiro[4.4]nonan-7-yl]phenyl)-5-(trifluoromethyl)pyrimidine-2- Synthesis of amines
- the compound of preparation 5 (200 mg, 0.75 mmol, 1.00 equiv) was stirred mixed in DCE (8 mL) and t-BuOH (8 mL) and ZnCl 2 in Et 2 O (2.26 mL, 2.26 mmol, 3.00 equiv) (1M) was added at 0° C. under a nitrogen atmosphere. The resulting mixture was stirred at 0° C. for 30 min under a nitrogen atmosphere. To the reaction was added the compound of Preparation 2 (183 mg, 0.83 mmol, 1.10 equiv) and TEA (84 mg, 0.83 mmol, 1.10 equiv) under a nitrogen atmosphere at 0°C. The resulting mixture was stirred at 25° C.
- Step 2 Synthesis of 1-(4- ⁇ [4-cyclohexyl-5-(trifluoromethyl)pyrimidin-2-yl]amino ⁇ phenyl)pyrrolidin-3-one
- the crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30 ⁇ 150 mm, 5 um, n; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (Phase B 65%, up to 85% in 7 min); Detector, UV 254/220 nm.
- the product fractions were lyophilized to obtain 1-(4- ⁇ [4-cyclohexyl-5-(trifluoromethyl)pyrimidin-2-yl]amino ⁇ phenyl)pyrrolidin-3-one (compound of Example 4) 8.5 mg, 9.41%) was obtained as a white solid.
- Step 2 1-[4-([4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-5H,6H,7H-cyclopenta[d]pyrimidine-2 Synthesis of -yl]amino)phenyl]piperidin-3-ol
- the crude product was purified by Prep-HPLC under the following conditions: column, X Bridge Prep phenyl OBD column, 19 x 250 mm, 5um; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (up to 70% in 40% Phase B in 7 min); Detector, UV 254 & 220 nm.
- step 1 of Example 7 2,4-dichloro-7H-pyrrolo[2,3-d]pyrimidine was used instead of 2,4-dichloro-5H,6H,7H-cyclopenta[d]pyrimidine. Except in the same manner as in Example 7, 1-[4-( ⁇ 4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-7H-pyrrolo[2,3 -d]pyrimidin-2-yl ⁇ amino)phenyl]piperidin-3-ol (compound of Example 8; 6.0%) was obtained as a white solid.
- step 1 of Example 7 2,6-dichloro-9H-purine was used instead of 2,4-dichloro-5H,6H,7H-cyclopenta[d]pyrimidine, and in step 2 of Example 7, 1- Instead of (4-aminophenyl) piperidin-3-ol, (S)-3-hydroxypiperidine was used instead of 3- (trifluoromethyl) piperidine in Step 1 of Preparation Example 7 and (3S)-1-[4] in the same manner as in Example 7, except that (S)-1-(4-aminophenyl)piperidin-3-ol prepared in the same manner as in Preparation Example 7 was used.
- step 1 of Example 7 2,6-dichloro-9H-purine was used instead of 2,4-dichloro-5H,6H,7H-cyclopenta[d]pyrimidine, and in step 2 of Example 7, 1-
- (R) -3-hydroxypiperidine was used instead of 3- (trifluoromethyl) piperidine in Step 1 of Preparation Example 7 (3R)-1-[4 in the same manner as in Example 7, except that (R)-1-(4-aminophenyl)piperidin-3-ol prepared in the same manner as in Preparation Example 7 was used.
- step 1 The compound obtained in step 1 (100 mg, 0.28 mmol, 1.00 equiv) and concentrated hydrochloric acid (1 drop) were mixed with stirring in n-BuOH (3 mL), and the compound of Preparation 7 (90 mg, 0.36 mmol, 1.30 equiv.) ) was added. The resulting mixture was stirred at 80° C. for 4 h under a nitrogen atmosphere and cooled to room temperature. The resulting mixture was concentrated in vacuo and the residue was purified by silica gel column chromatography, eluting with DCM/MeOH (5:1).
- the crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30 x 150mm 5um; mobile phase, water (10 mmol/L NH4HCO3+0.1% NH3.H2O) and ACN (36% PhaseB, up to 48% in 11 min); Detector, UV 254/220 nm.
- 2,4,6-trichloro-pyrimidine (1.00 g, 5.452 mmol, 1.00 equiv) was stirred mixed in DCE (10 mL) and t-BuOH (10 mL) and ZnCl 2 (1M) in ether (10.85 mL) , 10.85 mmol, 1.99 equiv) were added dropwise at 0 °C.
- the resulting mixture was stirred at 0 °C for 1 h, and 1-(4-aminophenyl)piperidin-3-ol (1.04 g, 5.40 mmol, 0.99 equiv) was added dropwise at 0 °C.
- the resulting mixture was stirred under a hydrogen atmosphere at 0° C. for 0.5 h.
- Step 2 1-[4-([4-chloro-6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino)phenyl]pi Synthesis of peridin-3-ol
- step 1 Compound obtained in step 1 (80 mg, 0.23 mmol, 1.00 equiv) and (3S,4S)-3,4-difluoropyrrolidine hydrochloride (40 mg, 0.28 mmol, 1.20 equiv) were mixed with ACN (2mL) Stir to mix and DIEA (89 mg, 0.70 mmol, 3.00 equiv) was added dropwise. The resulting mixture was stirred at 80° C. under a nitrogen atmosphere for 6 hours. The mixture was cooled to room temperature.
- the crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30 x 150mm 5um, n; mobile phase, water (10 mmol/L NH4HCO3 + 0.1% NH 3 . H 2 O) and ACN (30% PhaseB, up to 60% in 8 min); Detector, UV 254&220nm.
- the collected fractions were freeze-dried to 1-[4-([4-chloro-6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino )phenyl]piperidin-3-ol (compound of Example 12; 8.3 mg, 8.5%) was obtained as a white solid.
- Examples 13 and 14 1-[4-( ⁇ 4-bromo-6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl ⁇ amino )phenyl]piperidin-3-ol and 1-[4-( ⁇ 4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-6-iodopyrimidine- 2-yl ⁇ amino)phenyl]piperidin-3-ol
- Step 2 1-[4-([4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino)phenyl]piperidin-3 -Synthesis of ol
- step 1 Compound obtained in step 1 (50 mg, 0.20 mmol, 1.00 equiv) and 1-(4-aminophenyl)piperidin-3-ol (43 mg, 0.20 mmol, 1.00 equiv) were mixed with n-BuOH (2.00mL) Stir to mix and add HCl (1 drop). The resulting mixture was stirred at 120° C. under a nitrogen atmosphere for 1 hour. The mixture was cooled to room temperature and the crude product was purified by Prep-HPLC under the following conditions: column, XBridge Prep phenyl OBD column, 19 x 250 mm, 5um; Mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .
- Example 20 5-((4-((3R,4R)-3-methoxy-4-(methylamino)pyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidine-2- yl)amino)-2-methylisoindolin-1-one
- Step 2 5-((4-((3R,4R)-3-(benzyl(methyl)amino)-4-methoxypyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidine-2 Synthesis of -yl)amino)-2-methylisoindolin-1-one (Compound 8-1)
- optical isomer mixture obtained in step 1 above (160.00 mg, 399.18 umol, 1 equiv) and compound 2A (97.11 mg, 598.77 ⁇ mol, 1.5 equiv) were mixed in dioxane (8 mL), XPhos (38.06 mg, 79.84 ⁇ mol) , 0.2 eq), Cs 2 CO 3 (260.12 mg, 798.35 ⁇ mol, 2 eq), and Pd 2 (dba) 3 (36.55 mg, 39.92 ⁇ mol, 0.1 eq) were added in one portion at 20° C. under N 2 . The mixture was warmed to 110° C. and stirred under N 2 for 12 h. The mixture was cooled to 20° C.
- Step 3 5-((4-((3R,4R)-3-methoxy-4-(methylamino)pyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl Synthesis of )amino)-2-methylisoindolin-1-one
- Step 1 Synthesis of 5-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-2-methylisoindolin-1-one
- step 1 The compound obtained in step 1 and (3S,4S)-3,4-difluoropyrrolidine were reacted in the same manner as in step 1 of Example 20 to 5-((4-((3S,4S)-3 ,4-Difluoropyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-2-methylisoindolin-1-one (compound of Example 21; 25.6 mg, 0.06 mmol, 29%) were obtained as a white powder.
- LRRK2 enzyme inhibitory activity was determined by Nat Biotechnol. 2011 Oct 30;29(11):1039-45.
- a stock solution was prepared by dissolving the compound in 100% DMSO.
- 50% inhibitory concentration half maximal inhibitory concentration: IC 50
- the compound was prepared at a maximum concentration of 10 ⁇ M and diluted 3 times to 10 concentrations.
- LRRK2 G2019S enzyme Invitrogen, PR8764C
- LRRKtide SynignalChem, L10-58
- reaction buffer 20 mM Hepes (pH 7.5), 10 mM MgCl 2 , 1 mM EGTA, 0.01% (v/v) Brij35, 0.02 mg/mL BSA) , 0.1 mM Na 3 VO 4 , 2 mM DTT, and 1% (w/v) DMSO
- LRRKtide 20 ⁇ M LRRKtide was added to fresh reaction buffer, 30 nM LRRK2 (G2019S) enzyme was added, and then gently mixed.
- the prepared compound (in 100% DMSO) was added to the LRRK2 (G2019S) mixture using Acoustic technology (Echo550; nano level).
- 33 P-ATP was added to the reaction mixture to initiate the reaction, followed by incubation at room temperature for 2 hours.
- the IC 50 for LRRK2 (G2019S) was calculated by the P81 filter-binding method, and the results were evaluated based on the criteria in Table 1 below, and are shown in Table 2.
- LRRK2 antagonist activity (IC 50 , nM) compound of example 1 ++ compound of example 2 - compound of example 3 ++ compound of example 4 ++ compound of example 6 + compound of example 7 + compound of example 8 +++ compound of example 9 +++ Compound of Example 9(S) +++ compound of example 9(R) +++ compound of example 10 +++ compound of example 11 - compound of example 12 - compound of example 13 + compound of example 14 + compound of example 15 + compound of example 16 ++ compound of example 17 ++ compound of example 18 - compound of example 19 + compound of example 20 ++ compound of example 21 +++
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Neurology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides a pyrimidine compound of chemical formula 1, a pharmaceutical composition containing same for preventing or treating a LRRK2-mediated or related disease (for example, Parkinson's disease), and a method for treating and preventing disorders or diseases by using same. Having inhibitory activity against LRRK2, the pyrimidine compound according to the present invention can be advantageously used for preventing or treating LRRK2-related diseases (for example, Parkinson's disease).
Description
본 발명은 화학식 1의 페닐아미노피리미딘 화합물, 이를 포함하는 류신-풍부 반복 키나제 2(leucine-rich repeat kinase 2: LRRK2)에 의해 매개되거나 이와 관련된 질병 또는 질환의 예방 또는 치료용 약학적 조성물, 및 이를 이용한 질병 또는 질환의 치료 및 예방 방법에 관한 것이다.The present invention relates to a phenylaminopyrimidine compound of Formula 1, a leucine-rich repeat kinase 2 (leucine-rich repeat kinase 2: LRRK2)-mediated or related disease or disorder comprising the same, a pharmaceutical composition for preventing or treating, and It relates to a method for treating and preventing a disease or disorder using the same.
퇴행성 뇌질환, 예컨대 파킨슨병은 수백만명에게 발병되고 있다. 파킨슨병은 중뇌 도파민 뉴런의 진행성 결손에 의해 유발되어, 환자의 움직임을 지시하고 제어하는 능력이 손상된다.Degenerative brain diseases, such as Parkinson's disease, affect millions of people. Parkinson's disease is caused by progressive deficits in midbrain dopamine neurons, impairing the patient's ability to direct and control movement.
류신-풍부 반복 키나제 2(leucine-rich repeat kinase 2: LRRK2)는 유전성 파킨슨병에 관련되어 있다. 예를 들어, LRRK2 Gly2019Ser 돌연변이는 키나제 활성의 증가를 야기하는 것으로서, 유전성 파킨슨병을 야기한다. 파킨슨병 외에도, 알츠하이머병, 운동이상증, 중추신경계 장애, 암, 류마티스 관절염, 강직성 척추염과 관련이 있는 것으로 보고되었다. 또한, 유전체 연관 분석을 통해 LRRK2가 크론병과도 관련이 있는 것으로 알려졌다(Teri A. Manolio, N Engl J Med 2010;363:166-176). LRRK2와 질병들간의 관련성이 알려지면서, LRRK2의 조절제 또는 억제제들이 개발되고 있다(예컨대, 공개특허 제2020-0085779호(2020.07.15)). Leucine-rich repeat kinase 2 (leucine-rich repeat kinase 2: LRRK2) is implicated in hereditary Parkinson's disease. For example, the LRRK2 Gly2019Ser mutation causes an increase in kinase activity, resulting in hereditary Parkinson's disease. In addition to Parkinson's disease, it has been reported to be associated with Alzheimer's disease, dyskinesia, central nervous system disorders, cancer, rheumatoid arthritis, and ankylosing spondylitis. In addition, it was known that LRRK2 is also associated with Crohn's disease through genomic association analysis (Teri A. Manolio, N Engl J Med 2010;363:166-176). As the relationship between LRRK2 and diseases is known, modulators or inhibitors of LRRK2 are being developed (eg, Patent Publication No. 2020-0085779 (July 15, 2020)).
따라서, 본 발명의 목적은 LRRK2에 대해 우수한 저해 활성을 나타내는 화학식 1의 페닐아미노피리미딘 화합물을 제공하고, 이를 이용하여 LRRK2에 의해 매개되거나 이와 관련된 질병, 예를 들어 퇴행성 신경질환의 치료 및 예방에 높은 효능을 달성하는 것이다.Accordingly, it is an object of the present invention to provide a phenylaminopyrimidine compound of Formula 1 that exhibits excellent inhibitory activity against LRRK2, and uses the same to treat and prevent diseases mediated by or related to LRRK2, for example, neurodegenerative diseases. to achieve high efficacy.
본 발명의 목적은 화학식 1의 페닐아미노피리미딘 화합물을 제공하는 것이다.It is an object of the present invention to provide a phenylaminopyrimidine compound of formula (1).
본 발명의 또다른 목적은 화학식 1의 페닐아미노피리미딘 화합물을 포함하는 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료하기 위한 약학적 조성물을 제공하는 것이다.Another object of the present invention is to provide a pharmaceutical composition for preventing or treating a disease or disorder mediated by or related to LRRK2, which includes a phenylaminopyrimidine compound of Formula 1.
본 발명의 또다른 목적은 화학식 1의 페닐아미노피리미딘 화합물을 포함하는 LRRK2 억제제를 이용하여 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료하는 방법을 제공하는 것이다.Another object of the present invention is to provide a method for preventing or treating a disease or disorder mediated by or related to LRRK2 using an LRRK2 inhibitor including a phenylaminopyrimidine compound of Formula 1.
본 출원에서 개시된 각각의 설명 및 실시형태는 각각의 다른 설명 및 실시 형태에도 적용될 수 있다. 즉, 본 출원에서 개시된 다양한 요소들의 모든 조합이 본 출원의 범주에 속한다. 또한, 하기 기술된 구체적인 서술에 의하여 본 출원의 범주가 제한된다고 볼 수 없다.Each description and embodiment disclosed in this application may also be applied to each other description and embodiment. That is, all combinations of the various elements disclosed in this application fall within the scope of this application. In addition, it cannot be seen that the scope of the present application is limited by the detailed description described below.
본 발명의 일 양상은 하기 화학식 1로 표시되는 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염을 제공한다:One aspect of the present invention provides a compound represented by the following formula (1), a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof:
[화학식 1][Formula 1]

상기 화학식 1에서, R1 및 R2는 각각 독립적으로 H, 할로겐, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐으로 치환된 C1-6 알킬, C1-6 알콕시, 또는 할로겐으로 치환된 C1-6 알콕시일 수 있다. In Formula 1, R 1 and R 2 are each independently H, halogen, OH, CN, amino, nitro, C 1-6 alkyl, halogen-substituted C 1-6 alkyl, C 1-6 alkoxy, or halogen It may be C 1-6 alkoxy substituted with .
일 실시태양에서, R1 및 R2는 각각 독립적으로 H, 할로겐, C1-4 알킬, 또는 할로겐으로 치환된 C1-4 알킬일 수 있다. 일 실시태양에서, R1 및 R2 중 어느 하나는 H이고, 나머지 하나는 할로겐, C1-4 알킬, 또는 할로겐으로 치환된 C1-4 알킬일 수 있다. 일 실시태양에서, R1은 H이고, R2는 할로겐, C1-6 알킬, 또는 할로겐 원자로 치환된 C1-6 알킬일 수 있다. 예를 들어, R1은 H이고, R2는 C1-4 알킬 또는 할로겐으로 치환된 C1-4 알킬일 수 있다. 일 실시태양에서, R2는 H이고, R1은 할로겐, C1-6 알킬, 또는 할로겐 원자로 치환된 C1-6 알킬일 수 있다. 예를 들어, R2은 H이고, R1는 할로겐 또는 C1-4 알킬일 수 있다. In one embodiment, R 1 and R 2 can each independently be H, halogen, C 1-4 alkyl, or C 1-4 alkyl substituted with halogen. In one embodiment, one of R 1 and R 2 may be H, and the other may be halogen, C 1-4 alkyl, or C 1-4 alkyl substituted with halogen. In one embodiment, R 1 is H and R 2 can be halogen, C 1-6 alkyl, or C 1-6 alkyl substituted with a halogen atom. For example, R 1 may be H and R 2 may be C 1-4 alkyl or C 1-4 alkyl substituted with halogen. In one embodiment, R 2 is H and R 1 can be halogen, C 1-6 alkyl, or C 1-6 alkyl substituted with a halogen atom. For example, R 2 can be H and R 1 can be halogen or C 1-4 alkyl.
상기 R1 및 R2의 정의에서, 할로겐은 F, Cl, Br 또는 I일 수 있다. 할로겐으로 치환된 C1-6 알킬 또는 할로겐으로 치환된 C1-4 알킬은 1개, 2개, 3개 또는 그 이상의 할로겐(예컨대, F, Cl, Br, I)으로 치환된 것일 수 있으며, 예를 들어, 2개 또는 3개 이상의 할로겐으로 치환된 것일 수 있다. 예를 들어, 할로겐으로 치환된 C1-6 알킬은 트리할로메틸기(-CX'3; X'는 할로겐)를 포함하는 C1-4 퍼할로알킬일 수 있다. 예를 들어, 상기 할로겐으로 치환된 C1-6 알킬 또는 할로겐으로 치환된 C1-4 알킬은 불소로 치환된 것일 수 있으며, 예를 들어, 복수의 불소로 치환된 C1-4 알킬, 트리플루오로메틸기(-CF3)를 포함하는 C1-4 알킬 또는 C1-4 퍼플루오로알킬을 포함할 수 있다. In the above definitions of R 1 and R 2 , halogen may be F, Cl, Br or I. C 1-6 alkyl substituted with halogen or C 1-4 alkyl substituted with halogen may be substituted with 1, 2, 3 or more halogens (eg, F, Cl, Br, I), For example, it may be substituted with 2 or 3 or more halogens. For example, halogen-substituted C 1-6 alkyl may be C 1-4 perhaloalkyl containing a trihalomethyl group (-CX′ 3 ; X′ is halogen). For example, the halogen-substituted C 1-6 alkyl or halogen-substituted C 1-4 alkyl may be one substituted with fluorine, for example, a plurality of fluorine-substituted C 1-4 alkyl, tri It may include C 1-4 alkyl or C 1-4 perfluoroalkyl including a fluoromethyl group (—CF 3 ).
대안적으로, R1 및 R2는 함께 치환 또는 비치환된 4원 내지 7원 포화 또는 부분 불포화 카보사이클릴 고리, 또는 1개 내지 3개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로아릴 또는 헤테로사이클릴 고리를 형성할 수 있다. 일 실시태양에서, R1 및 R2는 함께 5원 또는 6원 포화 또는 부분 불포화 카보사이클릴 고리, 또는 1개 또는 2개의 질소 원자를 포함하는 5원 또는 6원 헤테로아릴 또는 헤테로사이클릴 고리를 형성할 수 있다. 예를 들어, R1 및 R2은 함께 사이클로펜텐일, 피롤릴, 이미다졸릴 또는 피라졸릴을 형성할 수 있지만, 이에 제한되지 않는다.Alternatively, R 1 and R 2 together are a substituted or unsubstituted 4-7 membered saturated or partially unsaturated carbocyclyl ring, or a substituted or unsubstituted 4-7 membered carbocyclyl ring comprising 1 to 3 nitrogen atoms. It may form a membered heteroaryl or heterocyclyl ring. In one embodiment, R 1 and R 2 together represent a 5 or 6 membered saturated or partially unsaturated carbocyclyl ring, or a 5 or 6 membered heteroaryl or heterocyclyl ring containing 1 or 2 nitrogen atoms. can be formed For example, R 1 and R 2 together can form, but are not limited to, cyclopentenyl, pyrrolyl, imidazolyl or pyrazolyl.
상기 R1 및 R2가 형성하는 카보사이클릴 고리, 헤테로아릴 또는 헤테로사이클릴 고리는 할로겐, 옥소, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐 또는 하이드록시로 치환된 C1-6 알킬, C1-6 알콕시, 할로겐으로 치환된 C1-6 알콕시, C2-6의 알콕시알킬, C1-6 알킬아미노 및 디-(C1-6 알킬)-아미노로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 임의로 치환될 수 있다. 예를 들어, 상기 R1 및 R2가 형성하는 카보사이클릴 고리, 헤테로아릴 또는 헤테로사이클릴 고리는 할로겐, OH, CN, 아미노, C1-4 알킬, 할로겐으로 치환된 C1-4 알킬 또는 C1-4 알콕시로부터 선택된 치환기로 임의로 치환될 수 있다.The carbocyclyl ring, heteroaryl or heterocyclyl ring formed by R 1 and R 2 is C 1 -substituted with halogen, oxo, OH, CN, amino, nitro, C 1-6 alkyl, halogen or hydroxy selected from the group consisting of 6 alkyl, C 1-6 alkoxy, halogen substituted C 1-6 alkoxy, C 2-6 alkoxyalkyl, C 1-6 alkylamino and di-(C 1-6 alkyl)-amino may be optionally substituted with one or more substituents. For example, the carbocyclyl ring, heteroaryl or heterocyclyl ring formed by R 1 and R 2 is halogen, OH, CN, amino, C 1-4 alkyl, halogen-substituted C 1-4 alkyl or may be optionally substituted with a substituent selected from C 1-4 alkoxy.
상기 화학식 1에서, R3은 -NR31R32 또는 치환 또는 비치환된 C3-8 사이클로알킬일 수 있다. In Formula 1, R 3 may be —NR 31 R 32 or substituted or unsubstituted C 3-8 cycloalkyl.
일 실시태양에서, R3는 치환 또는 비치환된 C3-6 사이클로알킬일 수 있다. 예를 들어, R3는 사이클로프로필, 사이클로부틸, 사이클로펜틸 또는 사이클로헥실일 수 있다. In one embodiment, R 3 can be substituted or unsubstituted C 3-6 cycloalkyl. For example, R 3 can be cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl.
일 실시태양에서, R3은 -NR31R32이고, R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 임의로 1개의 질소 또는 산소 원자를 포함하는 치환 또는 비치환된 3원 내지 12원 포화 또는 부분 불포화 카보사이클릴 또는 헤테로사이클릴 고리일 수 있다. 예를 들어, R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 임의로 1개의 산소 원자를 포함하는 치환 또는 비치환된 5원 내지 8원 포화 또는 부분 불포화 카보사이클릴 또는 헤테로사이클릴 고리일 수 있다. 상기 카보사이클릴 또는 헤테로사이클릴 고리는 임의로 가교되거나 스피로 고리를 형성할 수 있다. In one embodiment, R 3 is —NR 31 R 32 , any one of R 31 and R 32 is H, the other is optionally substituted or unsubstituted 3-12 membered containing 1 nitrogen or oxygen atom It may be a saturated or partially unsaturated carbocyclyl or heterocyclyl ring. For example, any one of R 31 and R 32 is H and the other is a substituted or unsubstituted 5-8 membered saturated or partially unsaturated carbocyclyl or heterocyclyl ringyl optionally containing 1 oxygen atom. can The carbocyclyl or heterocyclyl ring may optionally be bridged or form a spiro ring.
예를 들어, 상기 R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 사이클로헥실, 사이클로헵틸, 사이클로옥틸, 옥사사이클로헥실, 옥사사이클로헵틸, 옥사사이클로옥틸, 바이사이클로헥산일, 바이사이클로헵탄일, 바이사이클로옥탄일, 옥사바이사이클로헥산일, 옥사바이사이클로헵탄일, 옥사바이사이클로옥탄일, 스피로헥산일, 스피로헵탄일, 스피로옥탄일, 옥사스피로헥산일, 옥사스피로헵탄일 및 옥사스피로옥탄일로 이루어진 군에서 선택될 수 있다. 비제한적으로, R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 바이사이클로[2.1.1]헥산일, 바이사이클로[3.1.1]헵탄일, 바이사이클로[2.2.1]헵탄일, 바이사이클로[4.1.1]옥탄일, 바이사이클로[3.2.1]옥탄일, 바이사이클로[2.2.2]옥탄일, 옥사바이사이클로[2.1.1]헥산일, 옥사바이사이클로[2.2.1]헵탄일, 옥사바이사이클로[3.1.1]헵탄일, 옥사바이사이클로[4.1.1]옥탄일, 옥사바이사이클로[3.2.1]옥탄일, 옥사바이사이클로[2.2.2]옥탄일, 바이사이클로[3.1.0]헥산일, 바이사이클로[2.2.0]헥산일, 바이사이클로[4.1.0]헵탄일, 바이사이클로[3.2.0]헵탄일, 바이사이클로[5.1.0]옥탄일, 바이사이클로[4.2.0]옥탄일, 바이사이클로[3.3.0]옥탄일, 옥사바이사이클로[3.1.0]헥산일, 옥사바이사이클로[2.2.0]헥산일, 옥사바이사이클로[4.1.0]헵탄일, 옥사바이사이클로[3.2.0]헵탄일, 옥사바이사이클로[5.1.0]옥탄일, 옥사바이사이클로[4.2.0]옥탄일, 옥사바이사이클로[3.3.0]옥탄일, 스피로[2.3]헥산일, 스피로[2.4]헵탄일, 스피로[3.3]헵탄일, 스피로[2.5]옥탄일, 스피로[3.4]옥탄일, 옥사스피로[2.3]헥산일, 옥사스피로[2.4]헵탄일, 옥사스피로[3.3]헵탄일, 옥사스피로[2.5]옥탄일 및 옥사스피로[3.4]옥탄일로 이루어진 군에서 선택될 수 있다. 예를 들어, R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 바이사이클로[2.2.1]헵탄일 또는 옥사바이사이클로[2.2.1]헵탄일일 수 있다.For example, any one of R 31 and R 32 is H, and the other one is cyclohexyl, cycloheptyl, cyclooctyl, oxacyclohexyl, oxacycloheptyl, oxacyclooctyl, bicyclohexanyl, bicycloheptanyl. , bicyclooctanyl, oxabicyclohexanyl, oxabicycloheptanyl, oxabicyclooctanyl, spirohexanyl, spiroheptanyl, spirooctanyl, oxaspirohexanyl, oxaspiroheptanyl and oxaspirooctanyl It can be selected from the group consisting of. Without limitation, any one of R 31 and R 32 is H and the other is bicyclo[2.1.1]hexanyl, bicyclo[3.1.1]heptanyl, bicyclo[2.2.1]heptanyl, bicyclo[2.1.1]heptanyl, bicyclo[2.1.1]heptanyl Cyclo[4.1.1]octanyl, bicyclo[3.2.1]octanyl, bicyclo[2.2.2]octanyl, oxabicyclo[2.1.1]hexanyl, oxabicyclo[2.2.1]heptanyl , oxabicyclo[3.1.1]heptanyl, oxabicyclo[4.1.1]octanyl, oxabicyclo[3.2.1]octanyl, oxabicyclo[2.2.2]octanyl, bicyclo[3.1. 0]hexanyl, bicyclo[2.2.0]hexanyl, bicyclo[4.1.0]heptanyl, bicyclo[3.2.0]heptanyl, bicyclo[5.1.0]octanyl, bicyclo[4.2. 0] octanyl, bicyclo [3.3.0] octanyl, oxabicyclo [3.1.0] hexanyl, oxabicyclo [2.2.0] hexanyl, oxabicyclo [4.1.0] heptanyl, oxabi Cyclo[3.2.0]heptanyl, oxabicyclo[5.1.0]octanyl, oxabicyclo[4.2.0]octanyl, oxabicyclo[3.3.0]octanyl, spiro[2.3]hexanyl, spiro [2.4]heptanyl, spiro[3.3]heptanyl, spiro[2.5]octanyl, spiro[3.4]octanyl, oxaspiro[2.3]hexanyl, oxaspiro[2.4]heptanyl, oxaspiro[3.3]heptanyl , may be selected from the group consisting of oxaspiro [2.5] octanyl and oxaspiro [3.4] octanyl. For example, one of R 31 and R 32 may be H and the other may be bicyclo[2.2.1]heptanyl or oxabicyclo[2.2.1]heptanyl.
대안적으로, R3은 -NR31R32이고, R31 및 R32는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 3원 내지 10원 모노사이클릭 또는 바이사이클릭 헤테로사이클릴 고리를 형성할 수 있다. 예를 들어, R31 및 R32는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 3원 내지 8원 헤테로사이클릴 고리를 형성할 수 있다. 예를 들어, R31 및 R32는 이들이 결합한 질소 원자와 함께 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성할 수 있다. 예를 들어, R31 및 R32는 함께 아제티딘일, 피롤리딘일, 피페리딘일 또는 아제판일을 형성할 수 있지만 이에 제한되지 않는다.Alternatively, R 3 is —NR 31 R 32 and R 31 and R 32 together with the nitrogen atom to which they are attached are substituted or unsubstituted 3-10 membered monocyclic comprising 1 or 2 nitrogen atoms or a bicyclic heterocyclyl ring. For example, R 31 and R 32 together with the nitrogen atom to which they are attached may form a substituted or unsubstituted 3-8 membered heterocyclyl ring containing 1 or 2 nitrogen atoms. For example, R 31 and R 32 together with the nitrogen atom to which they are attached may form a substituted or unsubstituted 4-7 membered heterocyclyl ring containing 1 nitrogen atom. For example, R 31 and R 32 together can form, but are not limited to, azetidinyl, pyrrolidinyl, piperidinyl or azepanyl.
상기 R3, R31 및 R32의 정의에서, 치환된 C3-8 사이클로알킬 또는 C3-6 사이클로알킬, 치환된 카보사이클릴 고리 또는 치환된 헤테로사이클릴 고리는 할로겐, 옥소, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐 또는 하이드록시로 치환된 C1-6 알킬, C1-6 알콕시, 할로겐으로 치환된 C1-6 알콕시, C2-6의 알콕시알킬, C1-6 알킬아미노 및 디-(C1-6 알킬)-아미노로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 임의로 치환될 수 있다. 예를 들어, 상기 치환된 C3-8 사이클로알킬 또는 C3-6 사이클로알킬, 치환된 카보사이클릴 고리 또는 치환된 헤테로사이클릴 고리는 각각 독립적으로 할로겐, OH, CN, 아미노, C1-4 알킬, C1-4 알콕시, C1-4 알킬아미노 및 디-(C1-4 알킬)아미노로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 치환될 수 있다. 예를 들어, 상기 치환된 C3-8 사이클로알킬 또는 C3-6 사이클로알킬, 치환된 카보사이클릴 고리 또는 치환된 헤테로사이클릴 고리는 각각 독립적으로 할로겐(예를 들어, F, Cl, Br, I), OH, 아미노, C1-4 알콕시(예를 들어, 메톡시, 에톡시) 및 C1-4 알킬아미노(예를 들어, 메틸아미노, 에틸아미노)로 이루어진 군으로부터 선택되는 1개 또는 2개의 치환기로 치환될 수 있다.In the above definitions of R 3 , R 31 and R 32 , a substituted C 3-8 cycloalkyl or C 3-6 cycloalkyl, a substituted carbocyclyl ring or a substituted heterocyclyl ring is halogen, oxo, OH, CN , amino, nitro, C 1-6 alkyl, halogen or hydroxy substituted C 1-6 alkyl, C 1-6 alkoxy, halogen substituted C 1-6 alkoxy, C 2-6 alkoxyalkyl, C 1 may be optionally substituted with one or more substituents selected from the group consisting of -6 alkylamino and di-(C 1-6 alkyl)-amino. For example, the substituted C 3-8 cycloalkyl or C 3-6 cycloalkyl, substituted carbocyclyl ring or substituted heterocyclyl ring is each independently halogen, OH, CN, amino, C 1-4 may be substituted with one or more substituents selected from the group consisting of alkyl, C 1-4 alkoxy, C 1-4 alkylamino and di-(C 1-4 alkyl)amino. For example, the substituted C 3-8 cycloalkyl or C 3-6 cycloalkyl, substituted carbocyclyl ring or substituted heterocyclyl ring is each independently halogen (eg, F, Cl, Br, I), OH, amino, C 1-4 alkoxy (eg methoxy, ethoxy) and C 1-4 alkylamino (eg methylamino, ethylamino) or It may be substituted with two substituents.
상기 화학식 1에서, R4는 고리 X이고 R5는 H일 수 있다. 이 경우, 고리 X는 -NRX1RX2 또는 치환 또는 비치환된 C3-8 사이클로알킬일 수 있다. In Formula 1, R 4 may be ring X and R 5 may be H. In this case, ring X may be -NR X1 R X2 or substituted or unsubstituted C 3-8 cycloalkyl.
일 실시태양에서 고리 X는 치환 또는 비치환된 C3-6 사이클로알킬일 수 있다. 예를 들어, 고리 X는 사이클로프로필, 사이클로부틸, 사이클로펜틸 또는 사이클로헥실일 수 있다. In one embodiment, ring X can be substituted or unsubstituted C 3-6 cycloalkyl. For example, ring X can be cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl.
일 실시태양에서, 고리 X는 -NRX1RX2이고, RX1 및 RX2는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 3원 내지 10원 모노사이클릭 또는 바이사이클릭 헤테로사이클릴 고리를 형성할 수 있다. 예를 들어, 상기 RX1 및 RX2는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 3원 내지 7원 헤테로사이클릴 고리를 형성할 수 있다. 예를 들어, 상기 RX1 및 RX2는 이들이 결합한 질소 원자와 함께 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성할 수 있다. 예를 들어, 상기 RX1 및 RX2는 함께 아제티딘일, 피롤리딘일, 피페리딘일 또는 아제판일을 형성할 수 있지만 이에 제한되지 않는다.In one embodiment, ring X is —NR X1 R X2 , R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted 3-10 membered monocytic containing 1 or 2 nitrogen atoms It may form a cyclic or bicyclic heterocyclyl ring. For example, R X1 and R X2 may form a substituted or unsubstituted 3- to 7-membered heterocyclyl ring containing one or two nitrogen atoms together with the nitrogen atom to which they are attached. For example, R X1 and R X2 may form a substituted or unsubstituted 4- to 7-membered heterocyclyl ring including one nitrogen atom together with the nitrogen atom to which they are attached. For example, R X1 and R X2 together may form, but are not limited to, azetidinyl, pyrrolidinyl, piperidinyl or azepanyl.
상기 고리 X는 할로겐, 옥소, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐 또는 하이드록시로 치환된 C1-6 알킬, C1-6 알콕시, 할로겐으로 치환된 C1-6 알콕시, C2-6의 알콕시알킬, C1-6 알킬아미노 및 디-(C1-6 알킬)-아미노로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 임의로 치환될 수 있다. 일 실시태양에서, 고리 X는 할로겐, 옥소, OH, C1-4 알킬, 할로겐으로 치환된 C1-4 알킬, 하이드록시로 치환된 C1-4 알킬, C1-4 알콕시 및 할로겐으로 치환된 C1-4 알콕시로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 임의로 치환될 수 있다. 예를 들어, 고리 X는 할로겐(예를 들어, F, Cl, Br, I), 옥소, OH, C1-4 알킬 (예를 들어, 메틸, 에틸) 및 할로겐으로 치환된 C1-4 알킬(예를 들어, CHF2, CH2F, CF3, CH2CF3)로 이루어진 군으로부터 선택되는 1개 또는 2개의 치환기로 임의로 치환될 수 있다. wherein ring X is halogen, oxo, OH, CN, amino, nitro, C 1-6 alkyl, halogen or hydroxy substituted C 1-6 alkyl, C 1-6 alkoxy, halogen substituted C 1-6 alkoxy , C 2-6 alkoxyalkyl, C 1-6 alkylamino and di-(C 1-6 alkyl)-amino may be optionally substituted with one or more substituents selected from the group consisting of. In one embodiment, ring X is substituted with halogen, oxo, OH, C 1-4 alkyl, halogen substituted C 1-4 alkyl, hydroxy substituted C 1-4 alkyl, C 1-4 alkoxy and halogen It may be optionally substituted with one or more substituents selected from the group consisting of C 1-4 alkoxy. For example, ring X can be halogen (eg F, Cl, Br, I), oxo, OH, C 1-4 alkyl (eg methyl, ethyl) and halogen substituted C 1-4 alkyl (eg, CHF 2 , CH 2 F, CF 3 , CH 2 CF 3 ) may be optionally substituted with one or two substituents selected from the group consisting of.
대안적으로, R4 및 R5는 이들이 결합한 벤젠 고리의 탄소 원자와 함께 *-CO-NR6-CH2-기로 연결되어 5원 락탐 고리를 형성할 수 있다. 이 경우, *은 R4가 벤젠 고리에 결합된 위치를 나타낸다. 이 경우, R6은 H, C1-6 알킬, 또는 할로겐 원자로 치환된 C1-6 알킬일 수 있다. 예를 들어, R6은 H 또는 C1-4 알킬일 수 있다.Alternatively, R 4 and R 5 may be joined together with the carbon atom of the benzene ring to which they are attached to a *—CO—NR 6 —CH 2 — group to form a five membered lactam ring. In this case, * represents the position at which R 4 is bonded to the benzene ring. In this case, R 6 may be H, C 1-6 alkyl, or C 1-6 alkyl substituted with a halogen atom. For example, R 6 can be H or C 1-4 alkyl.
다만, 상기 화학식 1에서 R3이 -NR31R32이고, R31 및 R32가 치환 또는 비치환된 3원 내지 10원 헤테로사이클릴 고리를 형성하고, R4가 고리 X이고, 고리 X가 -NRX1RX2인 경우, R1 및 R2 중 어느 하나가 H이면, R1 및 R2 중 나머지 하나는 할로겐으로 치환된 C1-6 알킬이 아니다.However, in Formula 1, R 3 is -NR 31 R 32 , R 31 and R 32 form a substituted or unsubstituted 3- to 10-membered heterocyclyl ring, R 4 is Ring X, and Ring X is When -NR X1 R X2 , if either of R 1 and R 2 is H, then the other of R 1 and R 2 is not C 1-6 alkyl substituted with halogen.
본 발명의 일 실시태양에 따르면, R1 및 R2가 함께 치환 또는 비치환된 5원 또는 6원 포화 또는 부분 불포화 카보사이클릴 고리, 또는 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로아릴 또는 헤테로사이클릴 고리를 형성하고; R31 및 R32는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 5원 또는 6원 헤테로사이클릴 고리를 형성하고; R4는 고리 X이고, R5는 H이되, 고리 X는 -NRX1RX2이고, RX1 및 RX2는 이들이 결합한 질소 원자와 함께, 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성할 수 있다. 이 경우, 상기 치환된 고리는 각각 독립적으로 할로겐, OH, C1-6 알킬, 또는 할로겐 또는 하이드록시로 치환된 C1-6 알킬로부터 선택된 치환기로 임의로 치환될 수 있다.According to an embodiment of the present invention, R 1 and R 2 together are a substituted or unsubstituted 5- or 6-membered saturated or partially unsaturated carbocyclyl ring, or a substituted or unsubstituted 4-membered ring containing 1 nitrogen atom. to 7 membered heteroaryl or heterocyclyl ring; R 31 and R 32 together with the nitrogen atom to which they are attached form a substituted or unsubstituted 5- or 6-membered heterocyclyl ring containing 1 or 2 nitrogen atoms; R 4 is ring X, R 5 is H, wherein ring X is -NR X1 R X2 and R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted 4 members containing one nitrogen atom to 7 membered heterocyclyl rings. In this case, the substituted rings may each independently be optionally substituted with a substituent selected from halogen, OH, C 1-6 alkyl, or C 1-6 alkyl substituted with halogen or hydroxy.
예를 들어, R1 및 R2은 함께 사이클로펜텐일, 피롤릴, 이미다졸릴 또는 피라졸릴을 형성하고; R31 및 R32는 이들이 결합한 질소 원자와 함께, 할로겐, OH, 또는 할로겐으로 치환된 C1-4 알킬로 임의로 치환된 피롤리딘일 또는 피페리딘일을 형성하고; 고리 X는 -NRX1RX2이고, R5는 H이며, RX1 및 RX2는 함께 할로겐, 할로겐으로 치환된 C1-4 알킬, 또는 OH로 임의로 치환된 피롤리딘일 또는 피페리딘일을 형성할 수 있다.For example, R 1 and R 2 together form cyclopentenyl, pyrrolyl, imidazolyl or pyrazolyl; R 31 and R 32 together with the nitrogen atom to which they are attached form pyrrolidinyl or piperidinyl optionally substituted with halogen, OH, or C 1-4 alkyl substituted with halogen; ring X is —NR X1 R X2 , R 5 is H, and R X1 and R X2 taken together form halogen, C 1-4 alkyl substituted with halogen, or pyrrolidinyl or piperidinyl optionally substituted with OH can do.
본 발명의 일 실시태양에 따르면, R1 및 R2는 각각 독립적으로 H, 할로겐 또는 C1-6 알킬이고; R31 및 R32는 이들이 결합한 질소 원자와 함께, 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성하고; R4는 고리 X이고, R5는 H이되, 고리 X는 -NRX1RX2이고, RX1 및 RX2는 이들이 결합한 질소 원자와 함께, 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성할 수 있다. 전술한 R31, R32, RX1 및 RX2
,
및 이들의 치환기의 구체적인 예는 적용가능한 경우 본 실시태양에 동일하게 적용될 수 있다.According to an embodiment of the present invention, R 1 and R 2 are each independently H, halogen or C 1-6 alkyl; R 31 and R 32 together with the nitrogen atom to which they are attached form a substituted or unsubstituted 4-7 membered heterocyclyl ring containing 1 nitrogen atom; R 4 is ring X, R 5 is H, wherein ring X is -NR X1 R X2 and R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted 4 members containing one nitrogen atom to 7 membered heterocyclyl rings. The aforementioned R 31 , R 32 , R X1 and R X2 , And specific examples of substituents thereof may be equally applied to the present embodiment, if applicable.
본 발명의 일 실시태양에 따르면, R1 및 R2는 각각 독립적으로 H, 할로겐 또는 C1-6 알킬이고, 예를 들어, R1 및 R2 중 어느 하나는 H이고, 나머지 하나는 할로겐 또는 C1-4 알킬이고; R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 임의로 1개의 산소 원자를 포함하는 치환 또는 비치환된 5원 내지 8원 포화 또는 부분 불포화 카보사이클릴 또는 헤테로사이클릴고리이되, 상기 카보사이클릴 또는 헤테로사이클릴은 임의로 가교되거나 스피로 고리를 형성할 수 있고, 예를 들어, R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 옥사바이사이클로[2.2.1]헵탄일 또는 바이사이클로[2.2.1]헵탄일이고; R4는 고리 X이고, R5는 H이며, 고리 X는 -NRX1RX2이고, RX1 및 RX2는 이들이 결합한 질소 원자와 함께 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리일 수 있다. 전술한 R31, R32, RX1 및 RX2
,
및 이들의 치환기의 구체적인 예는 적용가능한 경우 본 실시태양에 동일하게 적용될 수 있다.According to one embodiment of the present invention, ROne and R2are each independently H, halogen or C1-6 alkyl, for example ROne and R2 one is H and the other is halogen or C1-4 alkyl; R31 and R32 any one is H and the other is a substituted or unsubstituted 5-8 membered saturated or partially unsaturated carbocyclyl or heterocyclyl ring optionally containing 1 oxygen atom, wherein the carbocyclyl or heterocyclyl may optionally be bridged or form a spiro ring, for example R31 and R32 one is H and the other is oxabicyclo[2.2.1]heptanyl or bicyclo[2.2.1]heptanyl; R4is ring X and R5is H and ring X is -NRX1RX2and RX1 and RX2may be a substituted or unsubstituted 4-7 membered heterocyclyl ring containing 1 nitrogen atom together with the nitrogen atom to which they are attached. above R.31, R32, RX1 and RX2
,
and specific examples of substituents thereof may be equally applied to the present embodiment, if applicable.
본 발명의 일 실시태양에 따르면, R1 및 R2 중 어느 하나가 H이고, 나머지 하나가 할로겐으로 치환된 C1-6 알킬(예를 들어, 플루오로로 치환된 C1-4 알킬, 예를 들어, CF3)인 경우, R3은 -NR31R32 또는 치환 또는 비치환된 C3-6 사이클로알킬이고, R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 임의로 1개의 산소 원자를 포함하는 치환 또는 비치환된 5원 내지 8원 포화 또는 부분 불포화 카보사이클릴 또는 헤테로사이클릴고리이되, 상기 카보사이클릴 또는 헤테로사이클릴 고리는 임의로 가교되거나 스피로 고리를 형성할 수 있고, 예를 들어, R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 옥사바이사이클로[2.2.1]헵탄일 또는 바이사이클로[2.2.1]헵탄일이고; R4는 고리 X이고, R5는 H이며, 고리 X는 -NRX1RX2 또는 치환 또는 비치환된 C3-6 사이클로알킬이되, RX1 및 RX2는 이들이 결합한 질소 원자와 함께, 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성할 수 있다. 전술한 R31, R32, RX1 및 RX2
,
및 이들의 치환기의 구체적인 예는 적용가능한 경우 본 실시태양에 동일하게 적용될 수 있다.According to an embodiment of the present invention, any one of R 1 and R 2 is H, and the other is C 1-6 alkyl substituted with halogen (eg, C 1-4 alkyl substituted with fluoro, such as For example, when CF 3 ), R 3 is —NR 31 R 32 or substituted or unsubstituted C 3-6 cycloalkyl, either of R 31 and R 32 is H and the other is optionally 1 oxygen. A substituted or unsubstituted 5- to 8-membered saturated or partially unsaturated carbocyclyl or heterocyclyl ring containing atoms, wherein the carbocyclyl or heterocyclyl ring may optionally be bridged or form a spiro ring, e.g. For example, one of R 31 and R 32 is H and the other is oxabicyclo[2.2.1]heptanyl or bicyclo[2.2.1]heptanyl; R 4 is ring X, R 5 is H, and ring X is —NR X1 R X2 or substituted or unsubstituted C 3-6 cycloalkyl, wherein R X1 and R X2 together with the nitrogen atom to which they are attached are 1 A substituted or unsubstituted 4- to 7-membered heterocyclyl ring containing two nitrogen atoms may be formed. The aforementioned R 31 , R 32 , R X1 and R X2 , And specific examples of substituents thereof may be equally applied to the present embodiment, if applicable.
본 발명의 일 실시태양에 따르면, R1 및 R2 중 어느 하나가 H이고, 나머지 하나가 할로겐으로 치환된 C1-6 알킬(예를 들어, 플루오로로 치환된 C1-4 알킬, 예를 들어, CF3)인 경우, R3은 -NR31R32이고, R31 및 R32가 이들이 결합한 질소 원자와 함께 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성하고; R4는 고리 X이고, R5는 H이며, 고리 X는 치환 또는 비치환된 C3-6 사이클로알킬일 수 있다. 전술한 R31, R32, RX1 및 RX2
,
및 이들의 치환기의 구체적인 예는 적용가능한 경우 본 실시태양에 동일하게 적용될 수 있다.According to an embodiment of the present invention, any one of R 1 and R 2 is H, and the other is C 1-6 alkyl substituted with halogen (eg, C 1-4 alkyl substituted with fluoro, such as For example, in the case of CF 3 ), R 3 is —NR 31 R 32 , and R 31 and R 32 are substituted or unsubstituted 4-7 membered heterocycle containing 1 nitrogen atom together with the nitrogen atom to which they are attached. forming a reel ring; R 4 is ring X, R 5 is H, and ring X can be substituted or unsubstituted C 3-6 cycloalkyl. The aforementioned R 31 , R 32 , R X1 and R X2 , And specific examples of substituents thereof may be equally applied to the present embodiment, if applicable.
본 발명의 화합물을 제조하기 위한 반응은 유기 합성 분야의 당업자가 적절히 선택할 수 있는 적합한 용매 중에서 수행될 수 있다. 적합한 용매는 반응이 일어나는 온도에서 출발물질(반응물), 중간체 또는 목적물과 실질적으로 비반응성인 것들이다. 당업자는 특정 반응 단계별로 적합한 용매를 적절하게 선택할 수 있을 것이다.The reaction for preparing the compound of the present invention can be carried out in a suitable solvent which can be appropriately selected by a person skilled in the art of organic synthesis. Suitable solvents are those that are substantially unreactive with the starting materials (reactants), intermediates or objects at the temperature at which the reaction occurs. A person skilled in the art will be able to properly select a suitable solvent for a particular reaction step.
본 발명의 화합물은 하기 실시예에 기재된 합성 과정에 따라서 합성될 수 있으며, 이를 기초로 목적 화합물의 구조에 따라서 반응물 및 반응 조건(예컨대, 반응용매, 반응온도, 반응시간, 반응촉매 등) 등을 적절히 변경하여 목적 화합물을 제조할 수 있을 것이다. The compound of the present invention can be synthesized according to the synthesis procedure described in the following examples, and based on this, reactants and reaction conditions (eg, reaction solvent, reaction temperature, reaction time, reaction catalyst, etc.) according to the structure of the target compound, etc. With appropriate modifications, the target compound may be prepared.
각 반응은 관련 분야에 공지된 적절한 방법에 의해 모니터링될 수 있다. 예컨대, 목적 화합물의 합성은 분광학적 수단, 예컨대, NMR (예: 1H 또는 13C), 질량 분석(mass spectroscopy), 또는 크로마토그래피(HPLC 또는 TLC) 등에 의해 모니터링될 수 있다.Each reaction can be monitored by an appropriate method known in the art. For example, the synthesis of the target compound may be monitored by spectroscopic means, such as NMR (eg, 1 H or 13 C), mass spectroscopy, or chromatography (HPLC or TLC).
일 실시태양에서, 화학식 1의 화합물 중 R1 및 R5가 각각 H이고, R3이 -NHR31인 화합물은 하기 반응식 A에 따라서 제조할 수 있다.In one embodiment, in the compound of Formula 1, R 1 and R 5 are each H, and R 3 is -NHR 31 A compound of Formula 1 may be prepared according to Scheme A below.
[반응식 A][Scheme A]

상기 단계 1에서, 아닐린 화합물 및 디클로로피리미딘 화합물을 적절한 유기 용매, 예컨대 디클로로에탄(DCE), t-부탄올(tBuOH) 등에서 임의로 적절한 촉매, 예컨대 ZnCl2, 트리에틸암모늄(TEA) 등의 존재 하에 혼합하여 반응시킬 수 있다. 일 실시태양에서, 상기 반응은 약 -10℃ 내지 약 25℃, 약 -5℃ 내지 약 10℃ 또는 약 0℃에서 약 1~5시간, 또는 약 2~3시간 동안 수행할 수 있다.In step 1, the aniline compound and the dichloropyrimidine compound are mixed in a suitable organic solvent such as dichloroethane (DCE), t-butanol (tBuOH), etc. optionally in the presence of a suitable catalyst such as ZnCl 2 , triethylammonium (TEA), etc. can be reacted. In one embodiment, the reaction may be carried out at about -10 °C to about 25 °C, about -5 °C to about 10 °C, or about 0 °C for about 1 to 5 hours, or about 2 to 3 hours.
상기 단계 2에서, 단계 1에서 수득한 화합물을 적절한 아민 화합물(NH2R31)과 적절한 유기 용매, 예컨대 N,N-디이소프로필에틸아민(DIEA), 아세토니트릴(ACN) 등에서 혼합할 수 있다. 일 실시태양에서, 상기 반응은 약 50℃ 내지 약 1500℃, 약 80℃ 내지 약 120℃, 또는 약 100℃에서 수행할 수 있다.In step 2, the compound obtained in step 1 may be mixed with an appropriate amine compound (NH 2 R 31 ) and an appropriate organic solvent, such as N,N-diisopropylethylamine (DIEA), acetonitrile (ACN), or the like. . In one embodiment, the reaction may be carried out at about 50 °C to about 1500 °C, about 80 °C to about 120 °C, or about 100 °C.
일 실시태양에서, 화학식 1의 화합물 중 R1 및 R5가 각각 H이고, R3이 C3-8 사이클로알킬인 화합물은 하기 반응식 B에 따라서 제조할 수 있다.In one embodiment, in the compound of Formula 1, R 1 and R 5 are each H and R 3 is C 3-8 cycloalkyl may be prepared according to Scheme B below.
[반응식 B][Scheme B]

상기 단계 1에서, 클로로피리미딘 화합물 및 카르복실산 화합물(R3COOH)을 적절한 용매, 예컨대 ACN 및 H2O의 혼합 용매 중에서 임의로 적절한 촉매, 예컨대 (NH4)2S2O8, AgNO3 등의 존재 하에 혼합하여 반응시킬 수 있다. 일 실시태양에서, 상기 반응은 약 50℃ 내지 약 120℃, 약 70℃ 내지 약 100℃, 또는 약 80℃에서 약 10시간 내지 약 20시간, 약 12시간 내지 약 18시간, 또는 약 16시간 동안 수행할 수 있다. In step 1, the chloropyrimidine compound and the carboxylic acid compound (R 3 COOH) are optionally mixed with a suitable catalyst such as (NH 4 ) 2 S 2 O 8 , AgNO 3 in a suitable solvent such as a mixed solvent of ACN and H 2 O. It can be reacted by mixing in the presence of such. In one embodiment, the reaction is at about 50 °C to about 120 °C, about 70 °C to about 100 °C, or about 80 °C for about 10 hours to about 20 hours, about 12 hours to about 18 hours, or about 16 hours. can be done
상기 단계 2에서, 단계 1에서 수득한 화합물을 적절한 아닐린 화합물과 적절한 용매, 예컨대 부탄올(BuOH), ACN, DIEA, 중에서 임의로 농염산 1방울을 적가하여 혼합시켜서 화학식 1의 화합물을 수득할 수 있다. 일 실시태양에서, 상기 반응은 약 50℃ 내지 약 120℃, 약 70℃ 내지 약 100℃, 또는 약 80℃에서 약 30분 내지 약 15시간, 약 30분 내지 약 10시간, 약 1시간 내지 약 12시간 동안 수행할 수 있다. In step 2, the compound obtained in step 1 is mixed with an appropriate aniline compound and a suitable solvent such as butanol (BuOH), ACN, DIEA, optionally by dropwise addition of 1 drop of concentrated hydrochloric acid, to obtain the compound of formula 1. In one embodiment, the reaction is at about 50 °C to about 120 °C, about 70 °C to about 100 °C, or about 80 °C for about 30 minutes to about 15 hours, about 30 minutes to about 10 hours, about 1 hour to about This can be done for 12 hours.
일 실시태양에서, 화학식 1의 화합물 중 R3가 -NR31R32이고, R4가 고리 X이고, R5가 H인 화합물은 하기 반응식 C에 따라서 제조될 수 있다.In one embodiment, the compound of Formula 1 wherein R 3 is —NR 31 R 32 , R 4 is Ring X, and R 5 is H may be prepared according to Scheme C below.
[반응식 C][Scheme C]

상기 단계 1에서, 디클로로피리미딘 화합물과 아민 화합물(NHR31R32)을 적절한 유기 용매, 예컨대, DIEA (디이소프로필에틸아민), DCM (디클로로메탄), DMF(디메틸포름아미드) 및 ACN(아세토니트릴)으로부터 이루어진 군으로부터 선택된 하나 이상의 유기용매 중에서 반응시킬 수 있다. 단계 1의 반응을 위하여 예컨대, 약 -80℃ 내지 약 80℃의 온도에서 예컨대, 약 10분 내지 약 10시간, 바람직하게는 약 15분 내지 약 6시간 동안 교반 혼합할 수 있다.In step 1, the dichloropyrimidine compound and the amine compound (NHR 31 R 32 ) are mixed with an appropriate organic solvent, such as DIEA (diisopropylethylamine), DCM (dichloromethane), DMF (dimethylformamide) and ACN (aceto The reaction may be carried out in one or more organic solvents selected from the group consisting of nitrile). For the reaction of step 1, for example, at a temperature of about -80 ° C. to about 80 ° C., for example, about 10 minutes to about 10 hours, preferably about 15 minutes to about 6 hours may be stirred and mixed.
상기 단계 2에서, 단계 1에서 수득된 화합물을 고리 X로 치환된 아닐린과 적절한 용매, 예컨대, nBuOH 중에서 임의로 HCl 1방울을 첨가하여 반응시킬 수 있다. 단계 2의 반응을 위하여 예컨대, 약 50℃ 내지 약 150℃, 바람직하게는 약 80℃ 내지 약 120℃의 온도에서 예컨대, 약 30분 내지 약 10시간, 바람직하게는 약 1시간 내지 약 6시간 동안 교반 혼합할 수 있다. In step 2 above, the compound obtained in step 1 may be reacted with aniline substituted with ring X by adding 1 drop of HCl optionally in a suitable solvent such as nBuOH. For the reaction of step 2, for example, at a temperature of about 50° C. to about 150° C., preferably about 80° C. to about 120° C., for example, about 30 minutes to about 10 hours, preferably about 1 hour to about 6 hours. It can be stirred and mixed.
대안으로, 화학식 1의 화합물 중 R3가 -NR31R32이고, R4가 고리 X이고, R5가 H인 화합물은 하기 반응식 D에 따라서 제조될 수 있다.Alternatively, a compound of Formula 1 wherein R 3 is —NR 31 R 32 , R 4 is Ring X and R 5 is H may be prepared according to Scheme D below.
[반응식 D][Scheme D]

상기 단계 1에서, 디클로로피리딘 화합물을 적절한 유기 용매, 예컨대 디클로로에탄(DCE) 및 t-BuOH에 교반 혼합하고, ZnCl2를 첨가한 후, 고리 X로 치환된 아닐린 화합물과 교반 혼합할 수 있다. In step 1, the dichloropyridine compound may be stirred and mixed in an appropriate organic solvent, such as dichloroethane (DCE) and t-BuOH, and ZnCl 2 may be added thereto, followed by stirring and mixing with the ring X-substituted aniline compound.
상기 단계 2에서, 단계 1에서 수득된 화합물을 적절한 아민 화합물(NHR31R32)과 적절한 유기 용매, 예컨대, DIEA (디이소프로필에틸아민), DCM (디클로로메탄), DMF(디메틸포름아미드) 및 ACN(아세토니트릴)으로부터 이루어진 군으로부터 선택된 하나 이상의 유기용매 중에서 반응시킬 수 있다. 단계 2의 반응을 위하여 예컨대, 약 -80℃ 내지 약 80℃의 온도에서 예컨대, 약 10분 내지 약 10시간, 바람직하게는 약 15분 내지 약 6시간 동안 교반 혼합할 수 있다.In step 2, the compound obtained in step 1 is mixed with an appropriate amine compound (NHR 31 R 32 ) with an appropriate organic solvent such as DIEA (diisopropylethylamine), DCM (dichloromethane), DMF (dimethylformamide) and The reaction may be carried out in one or more organic solvents selected from the group consisting of ACN (acetonitrile). For the reaction of step 2, for example, at a temperature of about -80 ° C. to about 80 ° C., for example, about 10 minutes to about 10 hours, preferably about 15 minutes to about 6 hours may be stirred and mixed.
상기 기술된 화학식 1의 제조방법은 예시적인 것으로서, 최종 화합물을 수득하기 위한 출발 물질의 종류에 따라서 적절한 용매, 촉매, 반응 조건 등을 선택하여 변형할 수 있으며, 이러한 용매, 촉매, 반응 조건 등은 당업자의 기술자에게 널리 알려져 있다. The preparation method of Chemical Formula 1 described above is exemplary, and may be modified by selecting an appropriate solvent, catalyst, reaction conditions, etc. depending on the type of starting material for obtaining the final compound, and these solvents, catalysts, reaction conditions, etc. It is well known to those skilled in the art.
일 실시태양에서, 상기 화학식 1의 화합물은 하기 화합물로 이루어진 군으로부터 선택될 수 있다:In one embodiment, the compound of Formula 1 may be selected from the group consisting of:





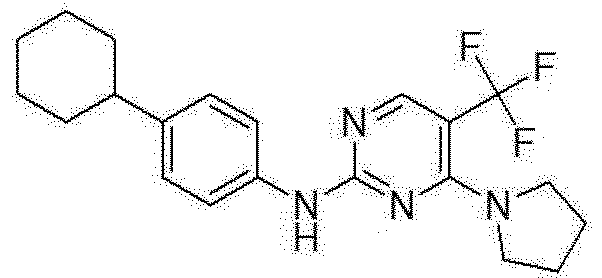

















본 명세서에서 사용된 용어 "알킬"은 완전 포화된 분지형 또는 비분지형 (또는, 직쇄 또는 선형) 탄화수소를 말한다. 상기 알킬은 치환 또는 비치환된 알킬기일 수 있다. 상기 C1-6 알킬은 C1 내지 C5, C1 내지 C4, C1 내지 C3, 또는 C1 내지 C2인 알킬기일 수 있다. 상기 알킬의 비제한적인 예로는 메틸, 에틸, n-프로필, 이소프로필, n-부틸, 이소부틸, sec-부틸, n-펜틸, 이소펜틸, 네오펜틸, iso-아밀, 또는 n-헥실일 수 있다.As used herein, the term “alkyl” refers to a fully saturated branched or unbranched (or straight-chain or linear) hydrocarbon. The alkyl may be a substituted or unsubstituted alkyl group. The C 1-6 alkyl may be a C 1 to C 5 , C 1 to C 4 , C 1 to C 3 , or C 1 to C 2 alkyl group. Non-limiting examples of the alkyl may be methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, n-pentyl, isopentyl, neopentyl, iso-amyl, or n-hexyl. have.
용어 "할로겐" 원자는 주기율표의 17족에 속하는 원자를 말한다. 할로겐 원자는 불소, 염소, 브롬, 및 요오드 등을 포함한다.The term "halogen" atom refers to an atom belonging to group 17 of the periodic table. Halogen atoms include fluorine, chlorine, bromine, and iodine.
용어 "사이클로알킬(cycloalkyl)"은 포화된 모노사이클릭(monocyclic) 탄화수소기를 지칭한다. 상기 사이클로알킬은 3 내지 12, 예컨대 3 내지 10, 3 내지 8, 또는 3 내지 6개의 탄소 원자를 포함할 수 있다. 예를 들어, C3-8 사이클로알킬은 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 사이클로헵틸 또는 사이클로옥틸일 수 있다.The term “cycloalkyl” refers to a saturated monocyclic hydrocarbon group. The cycloalkyl may contain 3 to 12, such as 3 to 10, 3 to 8, or 3 to 6 carbon atoms. For example, C 3-8 cycloalkyl can be cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl or cyclooctyl.
용어 "카보사이클릴"은 포화 또는 부분 불포화된 모노사이클릭 또는 폴리사이클릭(polycyclic) 탄화수소기를 지칭한다. 본원에서, 상기 카보사이클릴은 상기 사이클로알킬을 포함하는 용어로서 의도된다. 본원에서 용어 "카보사이클릴"은 3 내지 20개, 3 내지 10개, 3 내지 8개, 4 내지 7개, 5 내지 8개, 5개 또는 6개의 탄소 원자를 갖는 포화 또는 부분 불포화된 탄소 고리를 지칭할 수 있다. 용어 카보사이클릴은 모노-, 바이-, 트리-, 융합된, 브릿지(bridged) 및 스피로(spiro)-고리 시스템 및 이들의 조합을 포함한다. 일 실시태양에서, 바이사이클릭 카보사이클릴은 6개 내지 12개, 6개 내지 10개, 또는 6개 내지 8개의 탄소 원자를 포함할 수 있다. 다른 양태에서, 스피로 카보사이클릴은 5개 내지 12개, 6개 내지 10개, 또는 6개 내지 8개의 탄소 원자를 포함할 수 있다. 모노사이클릭 카보사이클릴의 대표적인 예시는 사이클로프로필, 사이클로부틸, 사이클로부텐일, 사이클로펜틸, 사이클로펜텐일, 사이클로헥실, 사이클로헥센일 등을 포함할 수 있다. 6개 내지 12개의 고리 원자를 갖는 바이사이클릭 카보사이클릴은 [4,3], [4,4], [4,5], [5,5], [5,6] 또는 [6,6] 고리 시스템, 예를 들면 바이사이클로[2.2.1]헵탄일, 바이사이클로[2.2.2]옥탄일, 및 바이사이클로[3.2.2]노난일과 같은 가교된 바이사이클릭 고리를 포함한다. 스피로 카보사이클릴은 스피로[2.2]펜탄일, 스피로[2.3]헥산일, 스피로[2.4]헵탄일, 스피로[2.5]옥탄일, 스피로[4.5]데칸일 등을 포함할 수 있다.The term "carbocyclyl" refers to a saturated or partially unsaturated monocyclic or polycyclic hydrocarbon group. As used herein, the carbocyclyl is intended as a term inclusive of the cycloalkyl. The term "carbocyclyl" as used herein refers to a saturated or partially unsaturated carbocyclic ring having 3 to 20, 3 to 10, 3 to 8, 4 to 7, 5 to 8, 5 or 6 carbon atoms. can refer to The term carbocyclyl includes mono-, bi-, tri-, fused, bridged and spiro-ring systems and combinations thereof. In one embodiment, the bicyclic carbocyclyl may contain 6 to 12, 6 to 10, or 6 to 8 carbon atoms. In other embodiments, the spiro carbocyclyl may contain 5 to 12, 6 to 10, or 6 to 8 carbon atoms. Representative examples of monocyclic carbocyclyl may include cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, and the like. Bicyclic carbocyclyl having 6 to 12 ring atoms is [4,3], [4,4], [4,5], [5,5], [5,6] or [6,6] ] ring systems, for example, bridged bicyclic rings such as bicyclo[2.2.1]heptanyl, bicyclo[2.2.2]octanyl, and bicyclo[3.2.2]nonanyl. Spiro carbocyclyls may include spiro[2.2]pentanyl, spiro[2.3]hexanyl, spiro[2.4]heptanyl, spiro[2.5]octanyl, spiro[4.5]decanyl, and the like.
용어 "헤테로사이클릴", "헤테로사이클릭" 또는 "헤테로고리"는 적어도 하나의 헤테로원자를 포함하는 포화 또는 부분 불포화 고리식 탄화수소를 말한다. 헤테로사이클릴 고리기는 하나의 고리기, 두개의 고리기, 또는 세개의 고리기일 수 있다. 상기 두개의 고리기는 스피로 고리기(spiro-ring), 가교된 고리기(bridged-ring), 및 융합 고리기(fused-ring)일 수 있다. 헤테로사이클릴 고리기는 3 내지 20개, 3 내지 12개, 3 내지 10개, 3개 내지 8개, 3개 내지 7개, 4개 내지 7개, 5개 내지 7개, 3개 내지 6개, 4개 내지 6개, 또는 5개 내지 6개의 고리 원자를 함유할 수 있다. 상기 헤테로원자는 N, O 및 S로 이루어진 군으로부터 선택된 어느 하나 이상일 수 있다. 예를 들어, 상기 헤테로원자는 1개 내지 3개의 N, 또는 1개 또는 2개의 N, 1개의 N일 수 있거나, 또는 1개의 O일 수 있다. 상기 헤테로원자는 1개 또는 2개의 N일 수 있다. 모노사이클릭 헤테로사이클릴 고리의 비제한적인 예로는, 아지리딘, 아제티딘, 피롤리딘, 피페리딘, 아제판 등을 들 수 있다. 가교 또는 스피로 헤테로사이클릭 고리의 비제한적인 예로는 옥사바이사이클로헥산일, 옥사바이사이클로헵탄일, 옥사바이사이클로옥탄일, 옥사스피로헥산일, 옥사스피로헵탄일, 옥사스피로옥탄일, 아자바이사이클로헥산일, 아자바이사이클로헵탄일, 아자바이사이클로옥탄일, 아자스피로헥산일, 아자스피로헵탄일, 아자스피로옥탄일 등을 들 수 있다. The term "heterocyclyl", "heterocyclic" or "heterocycle" refers to a saturated or partially unsaturated cyclic hydrocarbon containing at least one heteroatom. The heterocyclyl ring group may be a single ring group, a two ring group, or a three ring group. The two ring groups may be a spiro-ring group, a bridged-ring group, and a fused-ring group. Heterocyclyl ring groups are 3 to 20, 3 to 12, 3 to 10, 3 to 8, 3 to 7, 4 to 7, 5 to 7, 3 to 6, It may contain 4 to 6, or 5 to 6 ring atoms. The heteroatom may be any one or more selected from the group consisting of N, O and S. For example, the heteroatom can be 1 to 3 N, or 1 or 2 N, 1 N, or 1 O. The heteroatom may be one or two N. Non-limiting examples of monocyclic heterocyclyl rings include aziridine, azetidine, pyrrolidine, piperidine, azepane, and the like. Non-limiting examples of bridged or spiro heterocyclic rings include oxabicyclohexanyl, oxabicycloheptanyl, oxabicyclooctanyl, oxaspirohexanyl, oxaspiroheptanyl, oxaspirooctanyl, azabicyclohexane yl, azabicycloheptanyl, azabicyclooctanyl, azaspirohexanyl, azaspiroheptanyl, azaspirooctanyl, and the like.
용어 "아릴"은 방향족 고리가 하나 이상의 탄소 고리에 융합된 그룹도 포함한다. 상기 C6-C30의 아릴기는 예를 들면, C6 내지 C15, 또는 C6 내지 C10인 아릴기일 수 있다. 아릴의 비제한적인 예로는, 페닐, 나프틸, 또는 테트라하이드로나프틸 등을 들 수 있다.The term "aryl" also includes groups in which an aromatic ring is fused to one or more carbon rings. The C 6 -C 30 aryl group may be, for example, a C 6 to C 15 , or a C 6 to C 10 aryl group. Non-limiting examples of aryl include phenyl, naphthyl, or tetrahydronaphthyl.
용어 "헤테로아릴"은 N, O, P 및 S로 이루어진 군으로부터 선택된 하나 이상의 헤테로원자를 포함하고, 나머지 고리 원자가 탄소인 모노사이클릭 또는 바이사이클릭 방향족 화합물을 의미한다. 상기 헤테로아릴기는 예를 들어 1 내지 3개, 1개 또는 2개의 헤테로원자를 포함할 수 있고, 4개 내지 10개, 5개 내지 10개, 4개 내지 7개, 5개 또는 6개의 고리 원자를 포함할 수 있다. 상기 S 또는 N은 산화되어 여러 산화 상태를 가질 수 있다. "헤테로아릴"의 비제한적인 예로는, 피롤릴, 이미다졸릴, 피라졸릴, 티에닐, 퓨릴, 티아졸릴, 이소티아졸릴, 또는 1,2,3-옥사디아졸릴 등을 포함한다.The term “heteroaryl” refers to a monocyclic or bicyclic aromatic compound containing one or more heteroatoms selected from the group consisting of N, O, P and S, the remaining ring atoms being carbon. The heteroaryl group may contain, for example, 1 to 3, 1 or 2 heteroatoms, and 4 to 10, 5 to 10, 4 to 7, 5 or 6 ring atoms. may include. The S or N may be oxidized to have several oxidation states. Non-limiting examples of "heteroaryl" include pyrrolyl, imidazolyl, pyrazolyl, thienyl, furyl, thiazolyl, isothiazolyl, or 1,2,3-oxadiazolyl, and the like.
용어 "하이드록시(hydroxy)"는 -OH 기능기(수산기)를 말한다.The term "hydroxy" refers to a -OH functional group (hydroxyl group).
용어 "알콕시(alkoxy)"는 산소 원자에 결합된 알킬을 말한다. 예를 들어, C1 내지 C20의 알콕시기는 예를 들면, C2 내지 C15, C2 내지 C10, 또는 C2 내지 C5인 알콕시기일 수 있다. C1-6 알콕시는 메톡시, 에톡시, 프로폭시 등을 포함할 수 있다.The term “alkoxy” refers to an alkyl bound to an oxygen atom. For example, the C 1 to C 20 alkoxy group may be, for example, C 2 to C 15 , C 2 to C 10 , or C 2 to C 5 alkoxy group. C 1-6 alkoxy may include methoxy, ethoxy, propoxy, and the like.
용어 "아미노(amino)"기는 -NH2기를 말한다. The term “amino” group refers to a —NH 2 group.
용어 "니트로(nitro)"는 -NO2를 말한다.The term “nitro” refers to —NO 2 .
용어 "시아노(cyano)"는 -CN으로서, 탄소 원자와 질소 원자 사이에 삼중결합으로 이루어진 작용기를 말한다.The term "cyano" refers to a functional group consisting of a triple bond between a carbon atom and a nitrogen atom as -CN.
용어 "옥소(oxo)"는 =O를 지칭하며, "옥소로 치환된"은 탄소 원자가 -C(=O)-의 형태로 =O 치환기를 갖는 것을 의미한다.The term "oxo" refers to =O, and "substituted with oxo" means that the carbon atom has an =O substituent in the form -C(=O)-.
상기 "치환 또는 비치환된"의 용어 "치환"은 유기 화합물 중의 하나 이상의 수소 원자를 다른 원자단으로 치환하여 유도체를 형성한 경우 수소 원자 대신에 도입되는 것을 말하고, "치환기"는 도입된 원자단을 말한다. 본원에서 치환기의 한정이 없이 용어 “치환” 또는 “치환된”이 사용되는 경우, 치환기는 예를 들면, 할로겐 원자, 할로겐 원자로 치환된 C1 내지 C20의 알킬기(예: CCF3, CHCF2, CH2F, CCl3 등), C1 내지 C20의 알콕시, C2 내지 C20의 알콕시알킬, 하이드록시기, 니트로기, 시아노기, 아미노기, 아미디노기, 히드라진, 히드라존, 카르복실기나 그의 염, 술포닐기, 술파모일(sulfamoyl)기, 술폰산기나 그의 염, 인산이나 그의 염, 또는 C1 내지 C20 알킬기, C2 내지 C20 알케닐기, C2 내지 C20 알키닐기, C1 내지 C20 헤테로알킬기, C6 내지 C20 아릴기, C6 내지 C20 아릴알킬기, C6 내지 C20 헤테로아릴기, C7 내지 C20헤테로아릴알킬기, C6 내지 C20 헤테로아릴옥시기, 및 C6 내지 C20 헤테로아릴옥시알킬기 또는 C6 내지 C20 헤테로아릴알킬기일 수 있다.The term “substituted” of “substituted or unsubstituted” refers to introduced instead of a hydrogen atom when a derivative is formed by substituting one or more hydrogen atoms in an organic compound with another atomic group, and “substituent” refers to an introduced atomic group . When the term “substituted” or “substituted” is used herein without limitation of the substituent, the substituent is, for example, a halogen atom, a C 1 to C 20 alkyl group substituted with a halogen atom (eg, CCF 3 , CHCF 2 , CH 2 F, CCl 3 etc.), C 1 to C 20 alkoxy, C 2 to C 20 alkoxyalkyl, hydroxy group, nitro group, cyano group, amino group, amidino group, hydrazine, hydrazone, carboxyl group or its Salt, sulfonyl group, sulfamoyl group, sulfonic acid group or a salt thereof, phosphoric acid or a salt thereof, or C 1 to C 20 alkyl group, C 2 to C 20 alkenyl group, C 2 to C 20 alkynyl group, C 1 to C 20 heteroalkyl group, C 6 to C 20 aryl group, C 6 to C 20 arylalkyl group, C 6 to C 20 heteroaryl group, C 7 to C 20 heteroarylalkyl group, C 6 to C 20 heteroaryloxy group, and C It may be a 6 to C 20 heteroaryloxyalkyl group or a C 6 to C 20 heteroarylalkyl group.
예를 들면, 치환기는 할로겐, 옥소, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐 또는 하이드록시로 치환된 C1-6 알킬, C1-6 알콕시, 할로겐으로 치환된 C1-6 알콕시, C2-6 알콕시알킬, C1-6 알킬아미노 및 디-(C1-6 알킬)-아미노로 이루어진 군으로부터 선택되는 하나 이상의 치환기일 수 있다. For example, the substituent may be halogen, oxo, OH, CN, amino, nitro, C 1-6 alkyl, halogen or hydroxy substituted C 1-6 alkyl, C 1-6 alkoxy, halogen substituted C 1 - and one or more substituents selected from the group consisting of 6 alkoxy, C 2-6 alkoxyalkyl, C 1-6 alkylamino and di-(C 1-6 alkyl)-amino.
용어 "입체이성질체"의 "이성질체(isomer)"는 분자식은 같지만 분자 내에 있는 구성 원자의 연결 방식이나 공간 배열이 동일하지 않은 화합물을 말한다. 이성질체는 예를 들면, 구조 이성질체(structural isomers), 및 입체이성질체(stereoisomer)를 포함한다. 상기 입체이성질체는 부분입체 이성질체(diastereomer) 또는 거울상 이성질체(enantiomer)일 수 있다. 거울상이성질체는 왼손과 오른손의 관계처럼 그 거울상과 겹쳐지지 않는 이성질체를 말하고, 광학 이성질체(optical isomer)라고도 한다. 거울상 이성질체는 키랄 중심 탄소에 4개 이상의 치환기가 서로 다른 경우 R(Rectus: 시계방향) 및 S(Sinister: 반시계 방향)로 구분한다. 부분입체이성질체는 거울상 관계가 아닌 입체 이성질체를 말하고, 원자의 공간 배열이 달라 생기는 이성질체이다. 상기 부분입체이성질체는 시스(cis)-트랜스(trans) 이성질체 및 형태이성질체(conformational isomer 또는 conformer) 로 나뉠 수 있다.The term "isomer" of the term "stereoisomer" refers to a compound that has the same molecular formula but does not have the same spatial arrangement or connection mode of constituent atoms in the molecule. Isomers include, for example, structural isomers, and stereoisomers. The stereoisomer may be a diastereomer or an enantiomer. Enantiomers refer to isomers that do not overlap their mirror images, such as the relationship between the left and right hands, and are also called optical isomers. Enantiomers are divided into R (Rectus: clockwise) and S (Sinister: counterclockwise) when 4 or more substituents differ from each other at the chiral central carbon. Diastereomers refer to stereoisomers that are not in a mirror image relationship, and are isomers caused by different spatial arrangement of atoms. The diastereomers may be divided into cis-trans isomers and conformational isomers or conformers.
용어 "용매화물(solvate)"은 유기 또는 무기 용매에 용매화된 화합물을 말한다. 상기 용매화물은 예를 들어 수화물이다.The term “solvate” refers to a compound solvated in an organic or inorganic solvent. The solvate is, for example, a hydrate.
용어 "염(salt)"은 화합물의 무기 및 유기산 부가염을 말한다. 상기 약학적으로 허용가능한 염은 화합물이 투여되는 유기체에 심각한 자극을 유발하지 않고 화합물의 생물학적 활성과 물성들을 손상시키지 않는 염일 수 있다. 상기 무기산염은 염산염, 브롬산염, 인산염, 황산염, 또는 이황산염일 수 있다. 상기 유기산염은 포름산염, 아세트산염, 프로피온산염, 젖산염, 옥살산염, 주석산염, 말산염, 말레인산염, 구연산염, 푸마르산염, 베실산염, 캠실산염, 에디실염, 트리클로로아세트산, 트리플루오로아세트산염, 벤조산염, 글루콘산염, 메탄술폰산염, 글리콜산염, 숙신산염, 4-톨루엔술폰산염, 갈룩투론산염, 엠본산염, 글루탐산염, 에탄술폰산염, 벤젠술폰산염, p-톨루엔술폰산염, 또는 아스파르트산염일 수 있다. 상기 금속염은 칼슘염, 나트륨염, 마그네슘염, 스트론튬염, 또는 칼륨염일 수 있다.The term "salt" refers to inorganic and organic acid addition salts of compounds. The pharmaceutically acceptable salt may be a salt that does not cause serious irritation to the organism to which the compound is administered and does not impair the biological activity and properties of the compound. The inorganic acid salt may be hydrochloride, bromate, phosphate, sulfate, or disulfate. The organic acid salt is formate, acetate, propionate, lactate, oxalate, tartrate, malate, maleate, citrate, fumarate, besylate, camsylate, edicyl salt, trichloroacetic acid, trifluoroacetate , benzoate, gluconate, methanesulfonate, glycolate, succinate, 4-toluenesulfonate, galacturonate, embonate, glutamate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, or aspartate It may be an acid. The metal salt may be a calcium salt, a sodium salt, a magnesium salt, a strontium salt, or a potassium salt.
상기 화학식 1의 화합물은 LRRK2(Leucine-rich repeat kinase 2) 저해제일 수 있다. 상기 LRRK2는 류신-풍부 반복 키나제 패밀리(leucine-rich repeat kinase family)에 속하는 단백질일 수 있다. 상기 LRRK2는 AURA17, DARDARIN, PARK8, RIPK7, 또는 ROCO2로도 불릴 수 있다. 상기 LRRK2는 Uniprot No. Q5S007의 아미노산 서열을 포함하는 단백질일 수 있다. 상기 LRRK2는 Gly2019Ser 돌연변이를 포함할 수 있다.The compound of Formula 1 may be a Leucine-rich repeat kinase 2 (LRRK2) inhibitor. The LRRK2 may be a protein belonging to a leucine-rich repeat kinase family. The LRRK2 may also be referred to as AURA17, DARDARIN, PARK8, RIPK7, or ROCO2. The LRRK2 is Uniprot No. It may be a protein comprising the amino acid sequence of Q5S007. The LRRK2 may include a Gly2019Ser mutation.
본 발명의 다른 양상은 일 양상에 따른 화학식 1의 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염을 포함하는, LRRK2에 의해 매개되거나 이와 관련된 질병(disease) 또는 질환(condition)을 예방 또는 치료하기 위한 약학적 조성물을 제공한다.Another aspect of the present invention is to treat a disease or condition mediated by or related to LRRK2, including the compound of Formula 1, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof according to an aspect A pharmaceutical composition for preventing or treating is provided.
상기 화학식 1의 화합물, 입체이성질체, 용매화물, 약학적으로 허용가능한 염, 및 LRRK2는 전술한 바와 같다.The compound of Formula 1, stereoisomer, solvate, pharmaceutically acceptable salt, and LRRK2 are the same as described above.
LRRK2 에 의해 매개되거나 이와 관련된 질병 또는 질환은 퇴행성 신경질환일 수 있다. 용어 "퇴행성 신경질환(neurodegenerative disease)"은 신경계의 퇴행성 변화와 관련된 모든 질환을 지칭하며, 구체적으로 뇌의 퇴행성 변화와 관련된 퇴행성 뇌질환을 지칭할 수 있다. 상기 퇴행성 뇌질환은 파킨슨병(Parkinson's disease), 알츠하이머병(Alzheimer's disease), 헌팅턴병(Huntington's disease), 경도인지장애(mild cognitive impairment), 아밀로이드증(amyloidosis), 다계통위측증(Multiple system atrophy), 다발성경화증(multiple sclerosis), 타우병증(tauopathies), 픽병(Pick's disease), 노인성 치매, 근위축성 측삭 경화증(amyotrophic lateral sclerosis), 척수소뇌성 운동실조증(Spinocerebellar Atrophy), 뚜렛 증후군(Tourette's Syndrome), 프리드리히 보행실조(Friedrich's Ataxia), 마차도-조셉 병(Machado-Joseph's disease), 루이 소체 치매(Lewy Body Dementia), 근육긴장이상(Dystonia), 진행성 핵상 마비(Progressive Supranuclear Palsy) 및 전두측두엽 치매(Frontotemporal Dementia)로 이루어진 군에서 선택될 수 있다.The disease or disorder mediated by or related to LRRK2 may be a neurodegenerative disease. The term "neurodegenerative disease" refers to any disease associated with degenerative changes in the nervous system, and specifically may refer to degenerative brain diseases associated with degenerative changes in the brain. The degenerative brain disease is Parkinson's disease, Alzheimer's disease, Huntington's disease, mild cognitive impairment, amyloidosis, multiple system atrophy, multiple Multiple sclerosis, tauopathies, Pick's disease, senile dementia, amyotrophic lateral sclerosis, Spinocerebellar Atrophy, Tourette's Syndrome, Friedrich's Syndrome, Friedrich's Ataxia, Machado-Joseph's disease, Lewy Body Dementia, Dystonia, Progressive Supranuclear Palsy and Frontotemporal Dementia It can be selected from the group consisting of.
또한, LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환은 운동이상증; 중추신경계 장애; 암, 예를 들어, 신장암, 유방암, 전립선암, 혈액암, 폐암, 급성 골수성 백혈병 또는 다발성 골수증; 염증성 질환, 예를 들어, 류마티스 관절염, 강직성 척추염, 크론병, 근위축성 측삭 경화증, 나병, 염증성 장질환(Inflammatory Bowel Disease), 결핵 등을 포함할 수 있다.In addition, diseases or disorders mediated by or associated with LRRK2 include dyskinesia; central nervous system disorders; cancer such as kidney cancer, breast cancer, prostate cancer, blood cancer, lung cancer, acute myeloid leukemia or multiple myelopathy; inflammatory diseases such as rheumatoid arthritis, ankylosing spondylitis, Crohn's disease, amyotrophic lateral sclerosis, leprosy, Inflammatory Bowel Disease, tuberculosis, and the like.
상기 약학적 조성물은 퇴행성 뇌질환 예방, 치료, 또는 개선 활성을 갖는 공지의 유효 성분을 더 포함할 수 있다. 퇴행성 뇌질환 예방, 치료, 또는 개선 활성을 갖는 공지의 유효 성분은 레보도파(levodopa), 카테콜-O-메틸트랜스퍼라제(Catechol-O-methyltransferase: COMT) 저해제, 도파민 작용제, MAO-B(monoamine oxidase B) 저해제, 아만타딘, 항콜린제, 아세틸콜린에스터라제(acetylcholinesterase) 저해제 또는 NMDA 수용체 길항제(N-Methyl-D-aspartate receptor antagonist)일 수 있다. COMT 저해제는 오피카폰(opicapone), 엔타카폰(entacapone) 및 톨카폰(tolcapone)일 수 있다. 도파민 작용제는 브로모크립틴(bromocriptine), 페르골리드(pergolide), 프라미펙솔(pramipexole), 로피니롤(ropinirole), 피리베딜(piribedil), 카베르골린(cabergoline), 아포모르핀(apomorphine) 및 리수리드(lisuride)일 수 있다. MAO-B 저해제는 사핀아미드(safinamide), 세레길린(selegiline), 및 라사길린(rasagiline)이다. 아세틸콜린에스터라제 저해제는 타크린(tacrine), 리바스티그민(rivastigmine), 갈란타민(galantamine) 및 도네페질(donepezil)일 수 있다. NMDA 수용체 길항제는 메만틴(memantine)일 수 있다. 상기 화학식 1로 표시되는 화합물, 이의 유도체, 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염과 상기 공지의 유효 성분은 동시 또는 순차 투여를 위한 단일 또는 개별 조성물일 수 있다.The pharmaceutical composition may further include a known active ingredient having degenerative brain disease prevention, treatment, or amelioration activity. Known active ingredients having degenerative brain disease prevention, treatment, or amelioration activity are levodopa, catechol-O-methyltransferase (COMT) inhibitor, dopamine agonist, MAO-B (monoamine oxidase) B) an inhibitor, amantadine, an anticholinergic agent, an acetylcholinesterase inhibitor, or an NMDA receptor antagonist (N-Methyl-D-aspartate receptor antagonist). COMT inhibitors may be opicapone, entacapone and tolcapone. Dopamine agonists include bromocriptine, pergolide, pramipexole, ropinirole, piribedil, cabergoline, and apomorphine. and lisuride. MAO-B inhibitors are safinamide, selegiline, and rasagiline. The acetylcholinesterase inhibitor may be tacrine, rivastigmine, galantamine and donepezil. The NMDA receptor antagonist may be memantine. The compound represented by Formula 1, a derivative, stereoisomer, solvate, or pharmaceutically acceptable salt thereof and the known active ingredient may be a single or separate composition for simultaneous or sequential administration.
용어 "예방"은 상기 약학적 조성물의 투여에 의해 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환의 발생을 억제하거나 그의 발병을 지연시키는 모든 행위를 말한다. 용어 "치료"는 상기 약학적 조성물의 투여에 의해 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환의 증세가 호전되거나 이롭게 변경하는 모든 행위를 말한다.The term "prevention" refers to any action of inhibiting the onset of or delaying the onset of a disease or disorder mediated by or related to LRRK2 by administration of the pharmaceutical composition. The term “treatment” refers to any action in which the symptoms of a disease or disorder mediated by or related to LRRK2 are ameliorated or beneficially altered by administration of the pharmaceutical composition.
상기 약학적 조성물은 약학적으로 허용가능한 담체를 포함할 수 있다. 상기 담체는 부형제, 희석제 또는 보조제를 포함하는 의미로 사용된다. 상기 담체는 예를 들면, 락토스, 덱스트로스, 수크로스, 소르비톨, 만니톨, 자일리톨, 에리트리톨, 말티톨, 전분, 아카시아 고무, 알기네이트, 젤라틴, 칼슘 포스페이트, 칼슘 실리케이트, 셀룰로스, 메틸 셀룰로스, 폴리비닐 피롤리돈, 물, 생리식염수, PBS와 같은 완충액, 메틸하이드록시 벤조에이트, 프로필하이드록시 벤조에이트, 탈크, 마그네슘 스테아레이트, 및 미네랄 오일로 이루어진 군으로부터 선택된 것일 수 있다. 상기 조성물은 충진제, 항응집제, 윤활제, 습윤제, 풍미제, 유화제, 보존제, 또는 이들의 조합을 포함할 수 있다.The pharmaceutical composition may include a pharmaceutically acceptable carrier. The carrier is used in the sense of including excipients, diluents or adjuvants. The carrier may be, for example, lactose, dextrose, sucrose, sorbitol, mannitol, xylitol, erythritol, maltitol, starch, gum acacia, alginate, gelatin, calcium phosphate, calcium silicate, cellulose, methyl cellulose, polyvinyl pi It may be selected from the group consisting of rolidone, water, physiological saline, buffers such as PBS, methyl hydroxy benzoate, propyl hydroxy benzoate, talc, magnesium stearate, and mineral oil. The composition may include a filler, an anti-agglomeration agent, a lubricant, a wetting agent, a flavoring agent, an emulsifying agent, a preservative, or a combination thereof.
상기 약학적 조성물은 통상의 방법에 따라 임의의 제형으로 준비될 수 있다. 상기 조성물은 예를 들면, 경구 투여 제형(예를 들면, 분말, 정제, 캡슐, 시럽, 알약, 또는 과립), 또는 비경구 제형(예를 들면, 주사제)으로 제형화될 수 있다. 또한, 상기 조성물은 전신 제형, 또는 국부 제형으로 제조될 수 있다.The pharmaceutical composition may be prepared in any formulation according to a conventional method. The composition may be formulated, for example, as an oral dosage form (eg, a powder, tablet, capsule, syrup, pill, or granule), or a parenteral dosage form (eg, an injection). In addition, the composition may be prepared as a systemic formulation, or as a topical formulation.
상기 약학적 조성물에 있어서, 경구 투여를 위한 고형 제제는 정제, 환제, 산제, 과립제, 또는 캡슐제일 수 있다. 상기 고형 제제는 부형제를 더 포함할 수 있다. 부형제는 예를 들면, 전분, 칼슘 카보네이트(calcium carbonate), 수크로스(sucrose), 락토오스(lactose), 또는 젤라틴일 수 있다. 또한, 상기 고형 제제는 마그네슘 스테아레이트, 또는 탈크와 같은 윤활제를 더 포함할 수 있다. 상기 약학적 조성물에 있어서, 경구를 위한 액상 제제는 현탁제, 내용액제, 유제, 또는 시럽제일 수 있다. 상기 액상 제제는 물, 또는 리퀴드 파라핀을 포함할 수 있다. 상기 액상 제제는 부형제, 예를 들면 습윤제, 감미제, 방향제, 또는 보존제를 포함할 수 있다. 상기 약학적 조성물에 있어서, 비경구 투여를 위한 제제는 멸균된 수용액, 비수성용제, 현탁제, 유제, 동결건조 또는 및 좌제일 수 있다. 비수성용제 또는 현탁제는 식물성 기름 또는 에스테르를 포함할 수 있다. 식물성 기름은 예를 들면, 프로필렌글리콜(propylene glycol), 폴리에틸렌 글리콜, 또는 올리브 오일일 수 있다. 에스테르는 예를 들면 에틸올레이트일 수 있다. 좌제의 기제는 위텝솔(witepsol), 마크로골, 트윈(tween) 61, 카카오지, 라우린지, 또는 글리세로젤라틴일 수 있다.In the pharmaceutical composition, the solid preparation for oral administration may be a tablet, pill, powder, granule, or capsule. The solid formulation may further include an excipient. The excipient may be, for example, starch, calcium carbonate, sucrose, lactose, or gelatin. In addition, the solid formulation may further include a lubricant such as magnesium stearate or talc. In the pharmaceutical composition, the oral liquid formulation may be a suspension, an internal solution, an emulsion, or a syrup. The liquid formulation may contain water or liquid paraffin. The liquid formulation may contain excipients, for example, wetting agents, sweetening agents, perfuming agents, or preservatives. In the pharmaceutical composition, the preparation for parenteral administration may be a sterile aqueous solution, non-aqueous solution, suspension, emulsion, freeze-dried or suppository. Non-aqueous solvents or suspending agents may include vegetable oils or esters. The vegetable oil may be, for example, propylene glycol, polyethylene glycol, or olive oil. The ester may be, for example, ethyl oleate. The base of the suppository may be witepsol, macrogol, tween 61, cacao butter, laurin, or glycerogelatin.
상기 약학적 조성물은 일 양상에 따른 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염을 상기 약학적 조성물의 유효 성분으로 포함한다. "유효 성분"은 약리학적 활성(예를 들면, 퇴행성 뇌질환 치료)을 달성하기 위해 사용되는 생리활성 물질을 말한다.The pharmaceutical composition includes the compound according to an aspect, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof as an active ingredient of the pharmaceutical composition. "Active ingredient" refers to a physiologically active substance used to achieve pharmacological activity (eg, treatment of degenerative brain disease).
상기 약학적 조성물은 일 양상에 따른 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염을 유효한 양으로 포함할 수 있다. 용어 "유효한 양"은 예방 또는 치료를 필요로 하는 개체에게 투여되는 경우 질병 또는 질환의 예방 또는 치료의 효과를 나타내기에 충분한 양을 말한다. 상기 유효한 양은 당업자가 선택되는 세포 또는 개체에 따라 적절하게 선택할 수 있다. 상기 약학적 조성물의 바람직한 투여량은 개체의 상태 및 체중, 질병 또는 질환의 정도, 약물 형태, 투여 경로 및 기간에 따라 다르지만, 당업자에 의해 적절하게 선택될 수 있다. 그러나, 상기 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염은 예를 들면, 약 0.0001 ㎎/㎏ 내지 약 100 ㎎/㎏, 또는 약 0.001 ㎎/㎏ 내지 약 100 ㎎/㎏의 양을 1일 1회 내지 24회, 2일 내지 1주에 1 내지 7회, 또는 1개월 내지 12개월에 1 내지 24회로 나누어 투여할 수 있다. 상기 약학적 조성물에서 상기 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염은 전체 조성물 총 중량에 대하여 약 0.0001 중량% 내지 약 10 중량%, 또는 약 0.001 중량% 내지 약 1 중량%로 포함될 수 있다.The pharmaceutical composition may include the compound according to an aspect, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof in an effective amount. The term "effective amount" refers to an amount sufficient to effect the prevention or treatment of a disease or disorder when administered to a subject in need thereof. The effective amount can be appropriately selected by those skilled in the art depending on the cell or individual to be selected. The preferred dosage of the pharmaceutical composition varies depending on the condition and weight of the individual, the degree of disease or disease, the drug form, the route and duration of administration, but may be appropriately selected by those skilled in the art. However, the compound, stereoisomer, solvate, or pharmaceutically acceptable salt thereof may be, for example, in an amount from about 0.0001 mg/kg to about 100 mg/kg, or from about 0.001 mg/kg to about 100 mg/kg. may be administered in divided doses 1 to 24 times a day, 1 to 7 times per 2 days to 1 week, or 1 to 24 times in 1 month to 12 months. In the pharmaceutical composition, the compound, stereoisomer, solvate, or pharmaceutically acceptable salt thereof is present in an amount of from about 0.0001% to about 10% by weight, or from about 0.001% to about 1% by weight, based on the total weight of the composition. may be included.
투여 방법은 경구, 또는 비경구 투여일 수 있다. 투여 방법은 예를 들어, 경구, 경피, 피하, 직장, 정맥내, 동맥내, 복강내, 근육내, 흉골내, 국소, 코안(intranasal), 기관내(intratracheal), 또는 피내 경로일 수 있다. 상기 조성물은 전신적으로 또는 국부적으로 투여될 수 있고, 단독으로 또는 다른 약학적 활성 화합물과 함께 투여될 수 있다.The administration method may be oral or parenteral administration. The method of administration can be, for example, oral, transdermal, subcutaneous, rectal, intravenous, intraarterial, intraperitoneal, intramuscular, intrasternal, topical, intranasal, intratracheal, or intradermal routes. The composition may be administered systemically or locally, alone or in combination with other pharmaceutically active compounds.
본 발명의 다른 양상은 일 양상에 따른 화학식 1의 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염을 개체에게 투여하는 단계를 포함하는 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료하는 방법을 제공한다.Another aspect of the present invention is to prevent a disease or disorder mediated by or related to LRRK2, comprising administering to an individual a compound of Formula 1, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof according to an aspect or a method of treatment.
상기 화학식 1의 화합물, 입체이성질체, 용매화물, 약학적으로 허용가능한 염, LRRK2에 의해 매개되거나 이와 관련된 질병, 예방, 및 치료는 전술한 바와 같다.The compounds of Formula 1, stereoisomers, solvates, pharmaceutically acceptable salts, and diseases mediated by or related to LRRK2, prevention, and treatment are as described above.
상기 개체는 포유동물, 예를 들면, 인간, 마우스, 래트, 소, 말, 돼지, 개, 원숭이, 양, 염소, 유인원, 또는 고양이일 수 있다. 상기 개체는 LRRK2에 의해 매개되거나 이와 관련된 질병과 연관된 증상을 앓고 있거나, 앓을 가능성이 큰 개체일 수 있다.The subject may be a mammal, such as a human, mouse, rat, cow, horse, pig, dog, monkey, sheep, goat, ape, or cat. The subject may be suffering from, or likely to suffer from, a condition associated with a disease mediated by or associated with LRRK2.
상기 방법은 상기 개체에 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료하는 효과의 공지의 유효 성분을 투여하는 단계를 더 포함할 수 있다. 상기 공지의 유효 성분은 일 양상에 따른 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염과 동시, 개별, 또는 순차로 상기 개체에 투여될 수 있다.The method may further comprise administering to the subject a known active ingredient having an effect of preventing or treating a disease or disorder mediated by or related to LRRK2. The known active ingredient may be administered to the subject simultaneously, separately, or sequentially with the compound according to an aspect, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof.
투여 방법은 경구, 또는 비경구 투여일 수 있다. 투여 방법은 예를 들어, 경구, 경피, 피하, 직장, 정맥내, 동맥내, 복강내, 근육내, 흉골내, 국소, 코안(intranasal), 기관내(intratracheal), 또는 피내 경로일 수 있다. 상기 약학적 조성물은 전신적으로 또는 국부적으로 투여될 수 있고, 단독으로 또는 다른 약학적 활성 화합물과 함께 투여될 수 있다.The administration method may be oral or parenteral administration. The method of administration can be, for example, oral, transdermal, subcutaneous, rectal, intravenous, intraarterial, intraperitoneal, intramuscular, intrasternal, topical, intranasal, intratracheal, or intradermal routes. The pharmaceutical composition may be administered systemically or locally, alone or in combination with other pharmaceutically active compounds.
상기 약학적 조성물의 바람직한 투여량은 환자의 상태 및 체중, 질병 또는 질환의 정도, 약물 형태, 투여 경로 및 기간에 따라 다르지만, 당업자에 의해 적절하게 선택될 수 있다. 상기 투여량은 예를 들어, 성인 기준으로 약 0.001 ㎎/kg 내지 약 100 ㎎/kg, 약 0.01 ㎎/kg 내지 약 10 ㎎/kg, 또는 약 0.1 ㎎/kg 내지 약 1 ㎎/kg의 범위 내 일 수 있다. 상기 투여는 1일 1회, 1일 다회, 또는 1주일에 1회, 2주일에 1회, 3주일에 1회, 또는 4주일에 1회 내지 1년에 1회 투여될 수 있다.The preferred dosage of the pharmaceutical composition varies depending on the condition and weight of the patient, the severity of the disease or disease, the drug form, the route and duration of administration, but may be appropriately selected by those skilled in the art. The dosage is, for example, in the range of about 0.001 mg/kg to about 100 mg/kg, about 0.01 mg/kg to about 10 mg/kg, or about 0.1 mg/kg to about 1 mg/kg, on an adult basis. can be The administration may be administered once a day, multiple times a day, or once a week, once every two weeks, once every three weeks, or once every four weeks to once a year.
본 발명의 다른 양상은 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료에 사용하기 위한, 일 양상에 따른 화학식 1의 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염의 용도를 제공한다.Another aspect of the present invention is the use of a compound of Formula 1, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof according to one aspect, for use in the prevention or treatment of a disease or condition mediated by or related to LRRK2. to provide.
본 발명의 다른 양상은 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료하기 위한 의약을 제조하기 위한, 일 양상에 따른 화학식 1의 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염의 용도를 제공한다.Another aspect of the present invention is a compound of Formula 1, a stereoisomer, a solvate, or a pharmaceutically acceptable compound according to one aspect, for preparing a medicament for preventing or treating a disease or condition mediated by or related to LRRK2. Uses of salts are provided.
상기 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환, 예방, 치료, 화학식 1의 화합물, 입체이성질체, 용매화물, 및 약학적으로 허용가능한 염은 전술한 바와 같다.The disease or disorder mediated by or related to LRRK2, prevention, treatment, compound of Formula 1, stereoisomer, solvate, and pharmaceutically acceptable salt are as described above.
본 발명에 따른 화학식 1의 피리미딘 화합물은 LRRK2 저해 활성이 우수하여 LRRK2에 의해 매개되거나 이와 관련된 질병(예컨대, 파킨슨병)의 예방 또는 치료용 약학적 조성물, 및 이를 이용한 질병 또는 질환의 치료 및 예방 방법에 효과적으로 사용될 수 있다. The pyrimidine compound of Formula 1 according to the present invention has excellent LRRK2 inhibitory activity, and thus a pharmaceutical composition for preventing or treating a disease mediated by or related to LRRK2 (eg, Parkinson's disease), and treatment and prevention of a disease or disorder using the same method can be used effectively.
이하 실시예를 통하여 보다 상세하게 설명한다. 그러나, 이들 실시예는 예시적으로 설명하기 위한 것으로 본 발명의 범위가 이들 실시예에 한정되는 것은 아니다.Hereinafter, it will be described in more detail through examples. However, these examples are for illustrative purposes only, and the scope of the present invention is not limited to these examples.
제조예 1: 4-(아제판-1-일)아닐린Preparation Example 1: 4-(azepan-1-yl)aniline

단계 1: 1-(4-니트로페닐)아제판의 합성Step 1: Synthesis of 1-(4-nitrophenyl)azepane
ACN (10 mL) 중에 4-플루오로니트로벤젠 (1.00g, 7.08 mmol, 1.00당량) 및 헥사메틸렌이민 (1.00g, 10.63 mmol, 1.50당량)을 교반 혼합하고, DIEA (2.70g, 21.26 mmol, 3.00당량)를 첨가하였다. 생성된 혼합물을 80℃에서 2시간 동안 교반하고, 혼합물을 실온으로 냉각시켰다. 생성된 혼합물을 감압 농축시키고, 잔사를 PE/EtOAc (10:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 1-(4-니트로페닐)아제판 (1g, 57.65%)을 황색 고체로 수득하였다. 1H-NMR (300 MHz, CDCl3) δ (ppm): 8.11 (d, J = 12.0 Hz, 2H), 6.63 (d, J = 12.0 Hz, 2H), 3.62-3.53 (m, 4H), 1.91-1.76 (m, 4H), 1.67-1.51 (m, 4H); MS m/z: 221 [M+H]+.4-fluoronitrobenzene (1.00 g, 7.08 mmol, 1.00 equiv) and hexamethyleneimine (1.00 g, 10.63 mmol, 1.50 equiv) in ACN (10 mL) were stirred mixed and DIEA (2.70 g, 21.26 mmol, 3.00 equiv) equivalent) was added. The resulting mixture was stirred at 80° C. for 2 h, and the mixture was cooled to room temperature. The resulting mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with PE/EtOAc (10:1) to give 1-(4-nitrophenyl)azepane (1 g, 57.65%) as a yellow solid. . 1 H-NMR (300 MHz, CDCl 3 ) δ (ppm): 8.11 (d, J = 12.0 Hz, 2H), 6.63 (d, J = 12.0 Hz, 2H), 3.62-3.53 (m, 4H), 1.91 -1.76 (m, 4H), 1.67-1.51 (m, 4H); MS m/z: 221 [M+H] + .
단계 2: 4-(아제판-1-일)아닐린의 합성Step 2: Synthesis of 4-(azepan-1-yl)aniline
MeOH (15 mL) 중에 1-(4-니트로페닐)아제판 (1.00g, 4.540 mmol, 1.00당량) 및 Pd/C (300 mg, 10%)를 교반 혼합하고, 수소 대기하에 25℃에서 1시간 동안 교반하였다. 생성된 혼합물을 여과하고, 필터 케이크를 MeOH (3×30 mL)로 세척하였다. 여액을 감압 농축시키고, 잔사를 PE/EtOAc (1:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 4-(아제판-1-일)아닐린 (700 mg, 72.93%)을 흑색 오일로 수득하였다. MS m/z: 191 [M+H]+.Stir-mix 1-(4-nitrophenyl)azepane (1.00 g, 4.540 mmol, 1.00 equiv) and Pd/C (300 mg, 10%) in MeOH (15 mL) under hydrogen atmosphere at 25° C. for 1 h stirred for a while. The resulting mixture was filtered and the filter cake was washed with MeOH (3×30 mL). The filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with PE/EtOAc (1:1) to give 4-(azepan-1-yl)aniline (700 mg, 72.93%) as a black oil. . MS m/z: 191 [M+H] + .
제조예 2: 4-[1,4-디옥사-7-아자스피로[4.4]노난-7-일]아닐린Preparation 2: 4- [1,4-dioxa-7-azaspiro [4.4] nonan-7-yl] aniline

제조예 1의 단계 1에서 헥사메틸렌이민 대신에 1,4-디옥사-7-아자스피로[4.4]노난을 사용한 것을 제외하고, 제조예 1과 동일한 방법으로 4-[1,4-디옥사-7-아자스피로[4.4]노난-7-일]아닐린 (77.52%)을 갈색 고체로 수득하였다. MS m/z: 221 [M+H]+.In the same manner as in Preparation Example 1, except that 1,4-dioxa-7-azaspiro[4.4]nonane was used instead of hexamethyleneimine in step 1 of Preparation Example 1, 4-[1,4-dioxa- 7-azaspiro[4.4]nonan-7-yl]aniline (77.52%) was obtained as a brown solid. MS m/z: 221 [M+H] + .
제조예 3: 2-클로로-4-사이클로펜틸-5-(트리플루오로메틸)피리미딘Preparation 3: 2-chloro-4-cyclopentyl-5- (trifluoromethyl) pyrimidine

ACN (7 mL) 및 H2O (7 mL) 중에 2-클로로-5-(트리플루오로메틸)피리미딘 (1.00g, 5.47 mmol, 1.00당량) 및 사이클로펜탄카르복실산 (750 mg, 6.57 mmol, 1.20당량)을 교반 혼합하고, (NH4)2S2O8 (1.00g, 4.38 mmol, 0.80당량) 및 AgNO3 (148 mg, 0.87 mmol, 0.16당량)을 첨가하였다. 생성된 혼합물을 질소 대기하에 80℃에서 16시간 동안 교반하였다. 혼합물을 실온으로 냉각시키고, 부분적으로 감압 농축하였다. 혼합물을 CH2Cl2 (3×300 mL)로 추출하여 무수 Na2SO4 상에서 건조시키고, 감압 농축하였다. 잔사를 PE/EtOAc (10:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 2-클로로-4-사이클로펜틸-5-(트리플루오로메틸)피리미딘 (600 mg, 37.58%)을 황백색(off-white) 오일로 수득하였다. 1H-NMR (300 MHz, CDCl3) δ (ppm): 8.75 (s, 1H), 3.53-3.36 (m, 1H), 2.14-2.01 (m, 2H), 2.00-1.85 (m, 4H), 1.84-1.67 (m, 2H); MS m/z: 251 [M+H]+.2-chloro-5-(trifluoromethyl)pyrimidine (1.00 g, 5.47 mmol, 1.00 equiv) and cyclopentanecarboxylic acid (750 mg, 6.57 mmol) in ACN (7 mL) and H 2 O (7 mL) , 1.20 equiv) were stirred and mixed, (NH 4 ) 2 S 2 O 8 (1.00 g, 4.38 mmol, 0.80 equiv) and AgNO 3 (148 mg, 0.87 mmol, 0.16 equiv) were added. The resulting mixture was stirred at 80° C. under a nitrogen atmosphere for 16 hours. The mixture was cooled to room temperature and partially concentrated under reduced pressure. The mixture was extracted with CH 2 Cl 2 (3×300 mL), dried over anhydrous Na 2 SO 4 , and concentrated under reduced pressure. The residue was eluted with PE/EtOAc (10:1) and purified by silica gel column chromatography to give 2-chloro-4-cyclopentyl-5-(trifluoromethyl)pyrimidine (600 mg, 37.58%) off-white (off). -white) as an oil. 1 H-NMR (300 MHz, CDCl 3 ) δ (ppm): 8.75 (s, 1H), 3.53-3.36 (m, 1H), 2.14-2.01 (m, 2H), 2.00-1.85 (m, 4H), 1.84-1.67 (m, 2H); MS m/z: 251 [M+H] + .
제조예 4: 2-클로로-4-사이클로프로필-5-(트리플루오로메틸)피리미딘Preparation 4: 2-chloro-4-cyclopropyl-5- (trifluoromethyl) pyrimidine

제조예 3에서 사이클로펜탄카르복실산 대신에 사이클로프로판카르복실산을 사용한 것을 제외하고, 제조예 3과 동일한 방법으로 2-클로로-4-사이클로프로필-5-(트리플루오로메틸)피리미딘 (16.40%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 8.95 (s, 1H), 2.33-2.18 (m, 1H), 1.35-1.30 (m, 2H), 1.30-1.20 (m, 2H); MS m/z: 223 [M+H]+.2-chloro-4-cyclopropyl-5- (trifluoromethyl) pyrimidine (16.40 in the same manner as in Preparation Example 3, except that cyclopropanecarboxylic acid was used instead of cyclopentanecarboxylic acid in Preparation Example 3) %) as a white solid. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 8.95 (s, 1H), 2.33-2.18 (m, 1H), 1.35-1.30 (m, 2H), 1.30-1.20 (m, 2H); MS m/z: 223 [M+H] + .
제조예 5: 2-클로로-4-사이클로헥실-5-(트리플루오로메틸)피리미딘Preparation 5: 2-chloro-4-cyclohexyl-5- (trifluoromethyl) pyrimidine

제조예 3에서 사이클로펜탄카르복실산 대신에 사이클로헥산카르복실산을 사용한 것을 제외하고, 제조예 3과 동일한 방법으로 2-클로로-4-사이클로헥실-5-(트리플루오로메틸)피리미딘 (47.58%)을 무색 오일로 수득하였다. MS m/z: 265/267 [M+H]+.2-chloro-4-cyclohexyl-5-(trifluoromethyl)pyrimidine (47.58) in the same manner as in Preparation Example 3, except that cyclohexanecarboxylic acid was used instead of cyclopentanecarboxylic acid in Preparation Example 3 %) as a colorless oil. MS m/z: 265/267 [M+H] + .
제조예 6: 2-클로로-4-(피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘Preparation 6: 2-chloro-4- (pyrrolidin-1-yl) -5- (trifluoromethyl) pyrimidine

DCM (5 mM) 중에 2,4-디클로로-5-(트리플루오로메틸)피리미딘 (300 mg, 1.38 mmol, 1.00당량)을 교반 혼합하고, DCM (5 mM) 중의 피롤리딘 (98 mg, 1.38 mmol, 1.00당량) 및 DIEA (536 mg, 4.14 mmol, 3.00당량)를 질소 대기하에 -70℃에서 적가하였다. 생성된 혼합물을 질소 대기하에 -70℃에서 0.5시간 동안 교반하였다. 생성된 혼합물을 실온으로 가온한 다음 진공 하에 농축시켰다. 잔사를 EtOAc/PE (1/2)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 2-클로로-4-(피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘 (150 mg, 41%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 8.51 (s, 1H), 3.57 (s, 4H), 1.95-1.88 (m, 4H); MS m/z: 252/254 [M+H]+.2,4-dichloro-5-(trifluoromethyl)pyrimidine (300 mg, 1.38 mmol, 1.00 equiv) in DCM (5 mM) was stirred mixed and pyrrolidine (98 mg, 1.38 mmol, 1.00 equiv) and DIEA (536 mg, 4.14 mmol, 3.00 equiv) were added dropwise at -70°C under a nitrogen atmosphere. The resulting mixture was stirred at -70°C for 0.5 h under a nitrogen atmosphere. The resulting mixture was allowed to warm to room temperature and then concentrated in vacuo. The residue was purified by silica gel column chromatography, eluting with EtOAc/PE (1/2), to 2-chloro-4-(pyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidine (150 mg, 41%) as a white solid. 1 H-NMR (DMSO-d 6 , 300 MHz) δ (ppm): 8.51 (s, 1H), 3.57 (s, 4H), 1.95-1.88 (m, 4H); MS m/z: 252/254 [M+H] + .
제조예 7: 4-[3-(트리플루오로메틸)피페리딘-1-일]아닐린Preparation 7: 4-[3-(trifluoromethyl)piperidin-1-yl]aniline

단계 1: 1-(4-니트로페닐)-3-(트리플루오로메틸)피페리딘Step 1: 1-(4-nitrophenyl)-3-(trifluoromethyl)piperidine
ACN (10 mL) 중에 3-(트리플루오로메틸)피페리딘; 메틸 클로라이드 (1.00g, 4.91 mmol, 1.00당량) 및 4-플루오로니트로벤젠 (1.10g, 7.79 mmol, 1.59당량)을 교반하여 혼합하고 DIEA (2.0g, 15.47 mmol, 3.15당량)을 첨가하였다. 생성된 혼합물을 질소 대기하에 80℃에서 3시간 동안 교반하고 진공하에 농축시켰다. 잔사를 PE/EtOAc (1:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 1-(4-니트로페닐)-3-(트리플루오로메틸)피페리딘 (1.2g, 82.86%)을 연황색 고체로 수득하였다. MS m/z: 275 [M+H]+. 3-(trifluoromethyl)piperidine in ACN (10 mL); Methyl chloride (1.00 g, 4.91 mmol, 1.00 equiv) and 4-fluoronitrobenzene (1.10 g, 7.79 mmol, 1.59 equiv) were stirred to mix and DIEA (2.0 g, 15.47 mmol, 3.15 equiv) was added. The resulting mixture was stirred at 80° C. for 3 h under a nitrogen atmosphere and concentrated in vacuo. The residue was purified by silica gel column chromatography eluting with PE/EtOAc (1:1) to give 1-(4-nitrophenyl)-3-(trifluoromethyl)piperidine (1.2 g, 82.86%) light yellow. obtained as a solid. MS m/z: 275 [M+H] + .
단계 2: 4-[3-(트리플루오로메틸)피페리딘-1-일]아닐린Step 2: 4-[3-(trifluoromethyl)piperidin-1-yl]aniline
단계 1에서 수득된 화합물 (1.20g, 4.376 mmol, 1.00당량) 및 Pd/C (300 mg, 10%)을 MeOH (10mL) 중에서 수소 대기하(3기압)에 25℃에서 1시간 동안 교반하여 혼합하였다. 고체를 여과해 내고 여액을 감압 농축하여 4-[3-(트리플루오로메틸) 피페리딘-1-일]아닐린 (1g, 84.21%)을 갈색 고체로 수득하였다. MS m/z: 245[M+H]+.The compound obtained in step 1 (1.20 g, 4.376 mmol, 1.00 equiv) and Pd/C (300 mg, 10%) were mixed in MeOH (10 mL) under a hydrogen atmosphere (3 atm) at 25° C. by stirring for 1 hour. did. The solid was filtered off and the filtrate was concentrated under reduced pressure to obtain 4-[3-(trifluoromethyl)piperidin-1-yl]aniline (1 g, 84.21%) as a brown solid. MS m/z: 245 [M+H] + .
제조예 8: 4-브로모-2-클로로-6-요오도피리미딘Preparation 8: 4-bromo-2-chloro-6-iodopyrimidine

단계 1: 2-클로로-4-요오도피리미딘의 합성Step 1: Synthesis of 2-chloro-4-iodopyrimidine
2-클로로-피리미딘 (2.00 g, 17.46 mmol, 1.00당량)을 THF (15 mL) 중에 교반하여 혼합하고, TMPMgCl.LiCl (THF 중의 1.1M)(17.50 mL, 19.21 mmol, 1.10당량)을 질소 대기하에 -60℃에서 적가하였다. 생성된 혼합물을 질소 대기하에 -60℃에서 2시간 동안 교반하고, ZnCl2 (THF 중의 1.0 M)(19.21 mL, 19.21 mmol, 1.10당량)를 -60℃에서 적가하였다. 혼합물을 실온으로 가온시킨 다음, 생성된 혼합물에 건조 THF (15 mL)에 용해된 I2 (6.65g, 26.19 mmol, 1.50당량)를 적가하고, 생성된 혼합물을 25℃에서 1시간 동안 교반하였다. 반응을 NH4Cl 포화 수용액 (100 mL) 및 Na2S2O3 포화 수용액 (60 mL)로 실온에서 퀀칭(quenching)시키고, 혼합물을 물에 붓고 에테르 (5 x 100 mL)로 추출하였다. 유기층을 모아서 염수 (100 mL)로 세척하고, 무수 황산나트륨상에서 건조시키고 감압 농축시켰다. 조 생성물을 PE/EtOAc (10:1)로 용출시키고 실리카겔 컬럼 크로마토그래피로 정제하여 2-클로로-4-요오도피리미딘 (1.0g, 19.1%)을 황색 오일로서 수득하였다; MS m/z: 241/243[M+H]+.2-Chloro-pyrimidine (2.00 g, 17.46 mmol, 1.00 equiv) was mixed by stirring in THF (15 mL) and TMPMgCl.LiCl (1.1M in THF) (17.50 mL, 19.21 mmol, 1.10 equiv) was placed in a nitrogen atmosphere. It was added dropwise at -60°C under The resulting mixture was stirred under a nitrogen atmosphere at -60°C for 2 h, and ZnCl 2 (1.0 M in THF) (19.21 mL, 19.21 mmol, 1.10 equiv) was added dropwise at -60°C. The mixture was allowed to warm to room temperature, then to the resulting mixture was added dropwise I 2 (6.65 g, 26.19 mmol, 1.50 equiv) dissolved in dry THF (15 mL), and the resulting mixture was stirred at 25° C. for 1 h. The reaction was quenched with saturated aqueous NH 4 Cl (100 mL) and saturated aqueous Na 2 S 2 O 3 (60 mL) at room temperature, the mixture was poured into water and extracted with ether (5×100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was eluted with PE/EtOAc (10:1) and purified by silica gel column chromatography to give 2-chloro-4-iodopyrimidine (1.0 g, 19.1%) as a yellow oil; MS m/z: 241/243[M+H] + .
단계 2: 4-브로모-2-클로로-6-요오도피리미딘의 합성Step 2: Synthesis of 4-bromo-2-chloro-6-iodopyrimidine
단계 1에서 수득된 화합물 (900 mg, 3.74 mmol, 1.00당량)을 THF (10 mL) 중에 교반하여 혼합하고, TMPMgCl.LiCl (THF 중의 1.1M)(5.0 mL, 5.49 mmol, 1.47당량)을 질소 대기하에 -60 ℃에서 적가하였다. 생성된 혼합물을 질소 대기하에 -60℃에서 1시간 동안 교반한 다음, 생성된 혼합물에 1,2-디브로모-1,1,2,2-테트라클로로에탄 (1.82g, 5.61 mmol, 1.50당량)을 -78℃에서 첨가하고 생성된 혼합물을 -65℃로 가온하고 질소 대기하에 -65℃에서 3시간 동안 교반하였다. 생성된 혼합물을 감압 농축시켰다. 조 생성물을 PE/EtOAc (5:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 4-브로모-2-클로로-6-요오도피리미딘 (900 mg, 60.2%)을 황색 고체로 수득하였다. MS m/z: 319/321/323[M+H]+.The compound obtained in step 1 (900 mg, 3.74 mmol, 1.00 equiv) was mixed by stirring in THF (10 mL), and TMPMgCl.LiCl (1.1M in THF) (5.0 mL, 5.49 mmol, 1.47 equiv) was placed in a nitrogen atmosphere. It was added dropwise at -60 °C under The resulting mixture was stirred at -60°C under a nitrogen atmosphere for 1 hour, and then 1,2-dibromo-1,1,2,2-tetrachloroethane (1.82 g, 5.61 mmol, 1.50 eq.) in the resulting mixture. ) was added at -78°C and the resulting mixture was warmed to -65°C and stirred at -65°C for 3 hours under a nitrogen atmosphere. The resulting mixture was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography, eluting with PE/EtOAc (5:1) to give 4-bromo-2-chloro-6-iodopyrimidine (900 mg, 60.2%) as a yellow solid. MS m/z: 319/321/323[M+H] + .
제조예 9:(3S,4S)-N-벤질-4-메톡시-N-메틸피롤리딘-3-아민(화합물 5)Preparation 9: (3S, 4S) -N-benzyl-4-methoxy-N-methylpyrrolidin-3-amine (Compound 5)

단계 1: 화합물 2의 합성Step 1: Synthesis of compound 2
DCM(50 mL) 중 화합물 1(2 g, 11.82 mmol, 1 당량)의 혼합물에 m-CPBA(3.60 g, 17.73 mmol, 85% 순도, 1.5 당량)을 20℃에서 첨가하였다. 혼합물을 20℃에서 24 시간 동안 교반하였다. 혼합물에 Na2S2O3 포화 수용액 (30 mL)을 첨가하고 20℃에서 10분 동안 교반하였다. 반응은 요오드화 칼륨-녹말 시험 종이로 확인하여 m-CPBA가 분해되었는지 확인하였다. 그렇지 않은 경우, 시험 종이가 파란색으로 변하지 않을 때까지 Na2S2O3 수용액을 첨가하는 과정을 반복하고 교반하였다. 혼합물을 에틸 아세테이트(50 mLx3)로 추출하였다. 유기층을 모아서 염수로 세척(50mLx3)하고, 무수 Na2SO4로 건조시킨 후 여과하였다. 여과물은 진공에서 농축시켜 잔사를 얻고, 컬럼 크로마토그래피(SiO2, 석유 에테르/에틸 아세테이트=10/1 내지 5/1)로 정제하여 화합물 2(1.85 g, 84.5%)을 황색 오일로서 수득하였다. 1H NMR(400 MHz, CDCl3) δ ppm 3.77 - 3.63 (m, 2 H), 3.59 (d, J = 3.6 Hz, 2 H), 3.24 (dd, J = 12.8, 4.8 Hz, 2 H), 1.37 (s, 9 H).To a mixture of compound 1 (2 g, 11.82 mmol, 1 equiv) in DCM (50 mL) was added m-CPBA (3.60 g, 17.73 mmol, 85% purity, 1.5 equiv) at 20°C. The mixture was stirred at 20° C. for 24 h. To the mixture was added a saturated aqueous solution of Na 2 S 2 O 3 (30 mL) and stirred at 20° C. for 10 minutes. The reaction was confirmed with potassium iodide-starch test paper to determine whether m-CPBA was decomposed. Otherwise, the process of adding Na 2 S 2 O 3 aqueous solution was repeated and stirred until the test paper did not turn blue. The mixture was extracted with ethyl acetate (50 mLx3). The combined organic layers were washed with brine (50mLx3), dried over anhydrous Na 2 SO 4 and filtered. The filtrate was concentrated in vacuo to give a residue, which was purified by column chromatography (SiO 2 , petroleum ether/ethyl acetate=10/1 to 5/1) to give compound 2 (1.85 g, 84.5%) as a yellow oil. . 1 H NMR (400 MHz, CDCl 3 ) δ ppm 3.77 - 3.63 (m, 2 H), 3.59 (d, J = 3.6 Hz, 2 H), 3.24 (dd, J = 12.8, 4.8 Hz, 2 H), 1.37 (s, 9 H).
단계 2: 화합물 3의 합성Step 2: Synthesis of compound 3
화합물 2(1.8 g, 9.72 mmol, 1 당량)에 EtOH(25 mL) 중 화합물 6A(1.77 g, 14.58 mmol, 1.88 mL, 1.5 당량)을 첨가하고, 혼합물을 95℃로 가온하고 16시간 동안 교반하였다. 혼합물을 진공에서 농축하였다. 잔사를 컬럼 크로마토그래피(SiO2, 석유 에테르/에틸 아세테이트=10/1 내지 1/1)로 정제하여 화합물 3(1.3 g, 43.7% 수율)을 광학 이성질체의 혼합물로서 황색 오일의 형태로 수득하였다. 1H NMR(400 MHz, CDCl3) δ ppm 7.28 - 7.37 (m, 5 H), 4.33 (d, J = 6.4 Hz, 1 H), 3.73 (s, 1 H), 3.66 - 3.52 (m, 3 H), 3.34 (br s, 1 H), 3.25 - 3.13 (m, 1 H), 3.09 (br s, 1 H), 2.25 (s, 3 H), 1.46 (s, 9 H); LCMS: Rt = 0.977 분, MS (ESI) m/z = 307.2 [M+H]+.To compound 2 (1.8 g, 9.72 mmol, 1 equiv) was added compound 6A (1.77 g, 14.58 mmol, 1.88 mL, 1.5 equiv) in EtOH (25 mL), the mixture was warmed to 95° C. and stirred for 16 h. . The mixture was concentrated in vacuo. The residue was purified by column chromatography (SiO 2 , petroleum ether/ethyl acetate=10/1 to 1/1) to give compound 3 (1.3 g, 43.7% yield) as a mixture of optical isomers in the form of a yellow oil. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 7.28 - 7.37 (m, 5 H), 4.33 (d, J = 6.4 Hz, 1 H), 3.73 (s, 1 H), 3.66 - 3.52 (m, 3 H), 3.34 (br s, 1 H), 3.25 - 3.13 (m, 1 H), 3.09 (br s, 1 H), 2.25 (s, 3 H), 1.46 (s, 9 H); LCMS: Rt = 0.977 min, MS (ESI) m/z = 307.2 [M+H] + .
단계 3: 화합물 4의 합성Step 3: Synthesis of compound 4
상기 단계 2에서 수득된 광학 이성질체 혼합물(1.1 g, 3.59 mmol, 1 당량)을 THF(15 mL) 중에 혼합하고 NaH(215.38 mg, 5.39 mmol, 60% 순도, 1.5 당량)를 0℃에서 가하고 20분 동안 교반하였다. 그 후 MeI(2.32 g, 16.35 mmol, 1.02 mL, 4.55 당량)을 0℃에서 혼합물에 첨가한 후 20℃로 가온하고 2 시간 동안 교반하였다. 반응은 무수 NH4Cl로 퀀칭하고, 수성 상(aqueous phase)을 에틸 아세테이트(50 mLx3)로 추출하였다. 유기상을 모아서 염수(50 mL)로 세척하고, 무수 Na2SO4로 건조하고, 진공에서 여과 및 농축시켜 화합물 4(700 mg, 60.9%)를 광학 이성질체의 혼합물로서 황색 오일의 형태로 수득하였다. 1H NMR(400 MHz, CDCl3)δ ppm 7.16 (br d, J = 4.0 Hz, 3 H), 7.12 - 7.07 (m, 2 H), 3.96 - 3.57 (m, 2 H), 3.23 - 3.56 (m, 4 H), 3.19 (s, 3 H), 3.15- 2.87 (m, 2 H), 2.05 (br s, 3 H), 1.30 (s, 9 H); LCMS: RT = 0.773 min, MS (ESI) m/z = 321.3 [M+H]+.The optical isomer mixture (1.1 g, 3.59 mmol, 1 equiv) obtained in step 2 above was mixed in THF (15 mL) and NaH (215.38 mg, 5.39 mmol, 60% purity, 1.5 equiv) was added at 0 °C and 20 min. stirred for a while. Then MeI (2.32 g, 16.35 mmol, 1.02 mL, 4.55 equiv) was added to the mixture at 0° C., then warmed to 20° C. and stirred for 2 h. The reaction was quenched with anhydrous NH 4 Cl and the aqueous phase was extracted with ethyl acetate (50 mL×3). The combined organic phases were washed with brine (50 mL), dried over anhydrous Na 2 SO 4 , filtered and concentrated in vacuo to give compound 4 (700 mg, 60.9%) as a mixture of optical isomers in the form of a yellow oil. 1 H NMR(400 MHz, CDCl 3 )δ ppm 7.16 (br d, J = 4.0 Hz, 3 H), 7.12 - 7.07 (m, 2 H), 3.96 - 3.57 (m, 2 H), 3.23 - 3.56 ( m, 4 H), 3.19 (s, 3 H), 3.15- 2.87 (m, 2 H), 2.05 (br s, 3 H), 1.30 (s, 9 H); LCMS: RT = 0.773 min, MS (ESI) m/z = 321.3 [M+H] + .
단계 4: 화합물 5((3R,4R)-N-벤질-4-메톡시-N-메틸피롤리딘-3-아민)의 합성Step 4: Synthesis of compound 5 ((3R,4R)-N-benzyl-4-methoxy-N-methylpyrrolidin-3-amine)
상기 단계 3에서 수득된 광학 이성질체 혼합물(500 mg, 1.56 mmol, 1당량)을 DCM(6 mL) 중에 혼합하고 TFA(2.31 g, 20.29 mmol, 1.50 mL, 13 당량)을 0℃에서 한번에 가하였다. 혼합물을 0℃에서 3분 동안 교반한 후, 20℃로 가온하고 2시간 동안 교반하였다. 반응 혼합물은 감압하에서 농축시켜 용매를 제거하고 화합물 5(520 mg, 99.67%, TFA 염)를 광학 이성질체의 혼합물로서 황색 오일의 형태로 수득하였다. LCMS: RT = 0.597 min, MS (ESI) m/z = 221.2 [M+H]+.The optical isomer mixture obtained in step 3 above (500 mg, 1.56 mmol, 1 equiv) was mixed in DCM (6 mL) and TFA (2.31 g, 20.29 mmol, 1.50 mL, 13 equiv) was added in one portion at 0°C. The mixture was stirred at 0° C. for 3 min, then warmed to 20° C. and stirred for 2 h. The reaction mixture was concentrated under reduced pressure to remove the solvent and compound 5 (520 mg, 99.67%, TFA salt) was obtained as a mixture of optical isomers in the form of a yellow oil. LCMS: RT = 0.597 min, MS (ESI) m/z = 221.2 [M+H] + .
실시예 1: N2-[4-(아제판-1-일)페닐]-N4-[(1R,4R)-바이사이클로[2.2.1]헵탄-1-일]-5-(트리플루오로메틸)피리미딘-2,4-디아민Example 1: N2-[4-(azepan-1-yl)phenyl]-N4-[(1R,4R)-bicyclo[2.2.1]heptan-1-yl]-5-(trifluoromethyl ) pyrimidine-2,4-diamine

실시예 1의 화합물을 하기 반응식 1에 따라서 합성하였다.The compound of Example 1 was synthesized according to Scheme 1 below.
[반응식 1][Scheme 1]

단계 1: N-[4-(아제판-1-일)페닐]-4-클로로-5-(트리플루오로메틸)피리미딘-2-아민의 합성Step 1: Synthesis of N-[4-(azepan-1-yl)phenyl]-4-chloro-5-(trifluoromethyl)pyrimidin-2-amine
DCE : tBuOH (1:1) (3.00 mL) 중에 2,4-디클로로-5-(트리플루오로메틸)피리미딘 (100 mg, 0.46 mmol, 1.00당량)을 교반 혼합하고, 디에틸에테르 (1 mL, 1.00 mmol, 2.17당량) 중의 ZnCl2 (1M)를 질소 대기하에 0℃에서 적가하였다. 1시간 동안 교반한 후, 제조예 1의 화합물 (96.47 mg, 0.507 mmol, 1.10당량)을 첨가한 다음 DCE : tBuOH (1:1) (3.00 mL) 중의 TEA (51 mg, 0.50 mmol, 1.10당량) 용액을 적가하였다. 생성된 혼합물을 질소 대기하에 0℃에서 1.5시간 동안 교반하였다. 혼합물을 감압 농축시키고, 잔사를 PE/EtOAc (10:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 N-[4-(아제판-1-일)페닐]-4-클로로-5-(트리플루오로메틸)피리미딘-2-아민 (100 mg, 52.66%)을 황색 고체로 수득하였다. MS m/z: 371/373 [M+H]+.DCE: Stir mixed 2,4-dichloro-5-(trifluoromethyl)pyrimidine (100 mg, 0.46 mmol, 1.00 equiv) in tBuOH (1:1) (3.00 mL), diethylether (1 mL) , 1.00 mmol, 2.17 eq . ) was added dropwise at 0° C. under nitrogen atmosphere. After stirring for 1 h, the compound of Preparation 1 (96.47 mg, 0.507 mmol, 1.10 equiv) was added followed by DCE: TEA (51 mg, 0.50 mmol, 1.10 equiv) in tBuOH (1:1) (3.00 mL). The solution was added dropwise. The resulting mixture was stirred at 0° C. under a nitrogen atmosphere for 1.5 hours. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with PE/EtOAc (10:1) to N-[4-(azepan-1-yl)phenyl]-4-chloro-5-(tri Fluoromethyl)pyrimidin-2-amine (100 mg, 52.66%) was obtained as a yellow solid. MS m/z: 371/373 [M+H] + .
단계 2: N2-[4-(아제판-1-일)페닐]-N4-[(1R,4R)-바이사이클로[2.2.1]헵탄-1-일]-5-(트리플루오로메틸)피리미딘-2,4-디아민의 합성Step 2: N2-[4-(azepan-1-yl)phenyl]-N4-[(1R,4R)-bicyclo[2.2.1]heptan-1-yl]-5-(trifluoromethyl) Synthesis of pyrimidine-2,4-diamine
ACN (3.00 mL) 중에 N-[4-(아제판-1-일)페닐]-4-클로로-5-(트리플루오로메틸)피리미딘-2-아민 (100 mg, 0.27 mmol, 1.00당량) 및 바이사이클로[2.2.1]헵탄-1-아민 (32 mg, 0.29 mmol, 1.10당량)을 교반 혼합하고, DIEA (139 mg, 1.07 mmol, 4.00당량)를 첨가하였다. 생성된 혼합물을 100℃에서 16시간 동안 교반하고, 혼합물을 실온으로 냉각시켰다. 혼합물을 PE/EtOAc (5:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하였다. 조 생성물을 하기 조건으로 Prep-HPLC로 정제하였다: 컬럼, Xselect CSH OBD 컬럼 30×150 mm, 5 um, n; 이동상, 물 (10 mmol/L NH4HCO3 + 0.1% NH3·H2O) 및 ACN (80% Phase B, 8분에 90%까지); 검출기, UV 254 nm. 수집된 분획을 동결건조하여 N2-[4-(아제판-1-일)페닐]-N4-[(1R,4R)-바이사이클로[2.2.1]헵탄-1-일]-5-(트리플루오로메틸)피리미딘-2,4-디아민 (실시예 1의 화합물; 11 mg, 9.56%)을 백색 고체로 수득하였다. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 9.05 (s, 1H), 8.09 (s, 1H), 7.19 (br s, 2H), 6.60 (d, J = 9.2 Hz, 2H), 5.92 (m, 1H), 3.44-3.41 (m, 4H), 2.03-1.98 (m, 1H), 1.82-1.55 (m, 12H), 1.50-1.44 (m, 4H), 1.38-1.27 (m, 2H); MS m/z: 446 [M+H]+. N-[4-(azepan-1-yl)phenyl]-4-chloro-5-(trifluoromethyl)pyrimidin-2-amine (100 mg, 0.27 mmol, 1.00 equiv) in ACN (3.00 mL) and bicyclo[2.2.1]heptan-1-amine (32 mg, 0.29 mmol, 1.10 equiv) were stirred mixed and DIEA (139 mg, 1.07 mmol, 4.00 equiv) was added. The resulting mixture was stirred at 100° C. for 16 h, and the mixture was cooled to room temperature. The mixture was purified by silica gel column chromatography, eluting with PE/EtOAc (5:1). The crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30×150 mm, 5 um, n; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (80% Phase B, up to 90% in 8 min); Detector, UV 254 nm. The collected fractions were lyophilized to N2-[4-(azepan-1-yl)phenyl]-N4-[(1R,4R)-bicyclo[2.2.1]heptan-1-yl]-5-(tri Fluoromethyl)pyrimidine-2,4-diamine (compound of Example 1; 11 mg, 9.56%) was obtained as a white solid. 1 H-NMR (400 MHz, DMSO-d 6 ) δ (ppm): 9.05 (s, 1H), 8.09 (s, 1H), 7.19 (br s, 2H), 6.60 (d, J = 9.2 Hz, 2H) ), 5.92 (m, 1H), 3.44-3.41 (m, 4H), 2.03-1.98 (m, 1H), 1.82-1.55 (m, 12H), 1.50-1.44 (m, 4H), 1.38-1.27 (m) , 2H); MS m/z: 446 [M+H] + .
실시예 2: N-(4-사이클로헥실페닐)-4-사이클로펜틸-5-(트리플루오로메틸)피리미딘-2-아민Example 2: N-(4-cyclohexylphenyl)-4-cyclopentyl-5-(trifluoromethyl)pyrimidin-2-amine

BuOH (2 mL) 및 농염산 (1 방울) 중에 제조예 3의 화합물 (100 mg, 0.39 mmol, 1.00당량) 및 4-사이클로헥실-벤젠아민 (69 mg, 0.39 mmol, 1.00당량)을 80℃에서 1시간 동안 교반 혼합하였다. 혼합물을 실온으로 냉각시켰다. 조 생성물을 PE/EtOAc (10:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하였다. 조 생성물을 하기 조건으로 Prep-HPLC로 정제하였다: 컬럼, Xselect CSH OBD 컬럼 30×150 mm, 5 um; 이동상, 물 (10 mmol/L NH4HCO3 + 0.1% NH3·H2O) 및 ACN (80% Phase B, 10분에 90%까지); 검출기, UV 254 nm. 수집된 분획을 동결건조하여 N-(4-사이클로헥실페닐)-4-사이클로펜틸-5-(트리플루오로메틸)피리미딘-2-아민 (실시예 2의 화합물; 9.7 mg, 6.22%)을 백색 고체로 수득하였다. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.03 (s, 1H), 8.61 (s, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.8 Hz, 2H), 3.30-3.26 (m, 1H), 2.49-2.42 (m, 1H), 2.07-1.67 (m, 13H), 1.55-1.15 (m, 5H); MS m/z: 390 [M+H]+. Compound of Preparation 3 (100 mg, 0.39 mmol, 1.00 equiv) and 4-cyclohexyl-benzeneamine (69 mg, 0.39 mmol, 1.00 equiv) in BuOH (2 mL) and concentrated hydrochloric acid (1 drop) at 80 °C Stir and mix for 1 hour. The mixture was cooled to room temperature. The crude product was purified by silica gel column chromatography, eluting with PE/EtOAc (10:1). The crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30×150 mm, 5 um; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (80% Phase B, up to 90% in 10 min); Detector, UV 254 nm. The collected fractions were lyophilized to obtain N-(4-cyclohexylphenyl)-4-cyclopentyl-5-(trifluoromethyl)pyrimidin-2-amine (compound of Example 2; 9.7 mg, 6.22%). It was obtained as a white solid. 1 H-NMR (400 MHz, DMSO-d 6 ) δ (ppm): 10.03 (s, 1H), 8.61 (s, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.8 Hz, 2H), 3.30-3.26 (m, 1H), 2.49-2.42 (m, 1H), 2.07-1.67 (m, 13H), 1.55-1.15 (m, 5H); MS m/z: 390 [M+H] + .
실시예 3: 4-사이클로프로필-N-(4-사이클로프로필페닐)-5-(트리플루오로메틸)피리미딘-2-아민Example 3: 4-cyclopropyl-N-(4-cyclopropylphenyl)-5-(trifluoromethyl)pyrimidin-2-amine

ACN (5 mL) 중에 제조예 4의 화합물 (330 mg, 1.48 mmol, 1.00당량) 및 4-사이클로프로필아닐린 (237 mg, 1.78 mmol, 1.20당량)을 교반 혼합하고, DIEA (574 mg, 4.44 mmol, 3.00당량)를 적가하였다. 생성된 혼합물을 질소 대기하에 80℃에서 12시간 동안 교반하고, 혼합물을 실온으로 냉각시켰다. 혼합물을 DCM/MeOH (10:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 조 생성물을 수득하였다. 조 생성물을 하기 조건으로 Prep-HPLC로 정제하였다: 컬럼, YMC-Actus Triart C18 ExRS, 30×150 mm, 5 um; 이동상, 물 (10 mmol/L NH4HCO3 + 0.1% NH3·H2O) 및 ACN (70% Phase B, 10분에 90%까지); 검출기, UV 254/220 nm. 수집된 분획을 동결건조하여 4-사이클로프로필-N-(4-사이클로프로필페닐)-5-(트리플루오로메틸)피리미딘-2-아민 (실시예 3의 화합물; 8.4 mg, 1.77%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 9.91 (s, 1H), 8.55 (s, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.1 Hz, 2H), 2.20-2.10 (m, 1H), 1.90-1.78 (m, 1H), 1.25-0.98 (m, 4H), 0.98-0.75 (m, 2H), 0.70-0.50 (m, 2H); MS m/z: 320 [M+H]+.The compound of preparation 4 (330 mg, 1.48 mmol, 1.00 equiv) and 4-cyclopropylaniline (237 mg, 1.78 mmol, 1.20 equiv) in ACN (5 mL) were stirred and mixed, followed by DIEA (574 mg, 4.44 mmol, 3.00 equiv) was added dropwise. The resulting mixture was stirred at 80° C. for 12 h under a nitrogen atmosphere, and the mixture was cooled to room temperature. The mixture was purified by silica gel column chromatography, eluting with DCM/MeOH (10:1) to give the crude product. The crude product was purified by Prep-HPLC under the following conditions: column, YMC-Actus Triart C18 ExRS, 30×150 mm, 5 um; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (70% Phase B, up to 90% in 10 min); Detector, UV 254/220 nm. The collected fractions were lyophilized to obtain 4-cyclopropyl-N-(4-cyclopropylphenyl)-5-(trifluoromethyl)pyrimidin-2-amine (compound of Example 3; 8.4 mg, 1.77%). It was obtained as a white solid. 1 H-NMR (DMSO-d6, 300 MHz) δ (ppm): 9.91 (s, 1H), 8.55 (s, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.1) Hz, 2H), 2.20-2.10 (m, 1H), 1.90-1.78 (m, 1H), 1.25-0.98 (m, 4H), 0.98-0.75 (m, 2H), 0.70-0.50 (m, 2H); MS m/z: 320 [M+H] + .
실시예 4: 1-(4-{[4-사이클로헥실-5-(트리플루오로메틸)피리미딘-2-일]아미노}페닐)피롤리딘-3-온Example 4: 1-(4-{[4-cyclohexyl-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)pyrrolidin-3-one

실시예 4의 화합물을 하기 반응식 2에 따라서 합성하였다.The compound of Example 4 was synthesized according to Scheme 2 below.
[반응식 2][Scheme 2]

단계 1: 4-사이클로헥실-N-(4-[1,4-디옥사-7-아자스피로[4.4]노난-7-일]페닐)-5-(트리플루오로메틸)피리미딘-2-아민의 합성Step 1: 4-Cyclohexyl-N-(4-[1,4-dioxa-7-azaspiro[4.4]nonan-7-yl]phenyl)-5-(trifluoromethyl)pyrimidine-2- Synthesis of amines
DCE (8 mL) 및 t-BuOH (8 mL) 중에 제조예 5의 화합물 (200 mg, 0.75 mmol, 1.00당량)을 교반 혼합하고, Et2O (2.26 mL, 2.26 mmol, 3.00당량) 중의 ZnCl2 (1M)를 질소 대기하에 0℃에서 첨가하였다. 생성된 혼합물을 질소 대기하에 0℃에서 30분 동안 교반하였다. 반응물에 제조예 2의 화합물 (183 mg, 0.83 mmol, 1.10당량) 및 TEA (84 mg, 0.83 mmol, 1.10당량)를 질소 대기하에 0℃에서 첨가하였다. 생성된 혼합물을 질소 대기하에 25℃에서 16시간 동안 교반하였다. 생성된 혼합물을 진공 하에 농축시키고, 잔사를 PE/EtOAc (2:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 4-사이클로헥실-N-(4-[1,4-디옥사-7-아자스피로[4.4]노난-7-일]페닐)-5-(트리플루오로메틸)피리미딘-2-아민 (120 mg, 32.93%)을 담황색 고체로 수득하였다. MS m/z: 449 [M+H]+.The compound of preparation 5 (200 mg, 0.75 mmol, 1.00 equiv) was stirred mixed in DCE (8 mL) and t-BuOH (8 mL) and ZnCl 2 in Et 2 O (2.26 mL, 2.26 mmol, 3.00 equiv) (1M) was added at 0° C. under a nitrogen atmosphere. The resulting mixture was stirred at 0° C. for 30 min under a nitrogen atmosphere. To the reaction was added the compound of Preparation 2 (183 mg, 0.83 mmol, 1.10 equiv) and TEA (84 mg, 0.83 mmol, 1.10 equiv) under a nitrogen atmosphere at 0°C. The resulting mixture was stirred at 25° C. under a nitrogen atmosphere for 16 hours. The resulting mixture was concentrated in vacuo and the residue was purified by silica gel column chromatography eluting with PE/EtOAc (2:1) to 4-cyclohexyl-N-(4-[1,4-dioxa-7-aza) Spiro[4.4]nonan-7-yl]phenyl)-5-(trifluoromethyl)pyrimidin-2-amine (120 mg, 32.93%) was obtained as a pale yellow solid. MS m/z: 449 [M+H] + .
단계 2: 1-(4-{[4-사이클로헥실-5-(트리플루오로메틸)피리미딘-2-일]아미노}페닐)피롤리딘-3-온의 합성Step 2: Synthesis of 1-(4-{[4-cyclohexyl-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)pyrrolidin-3-one
HCl (1 mL) 및 1,4-디옥산 (3 mL) 중에 4-사이클로헥실-N-(4-[1,4-디옥사-7-아자스피로[4.4]노난-7-일]페닐)-5-(트리플루오로메틸)피리미딘-2-아민 (100 mg, 0.22 mmol, 1.00당량) 용액을 질소 대기하에 60℃에서 1시간 동안 교반하였다. 생성된 혼합물을 진공 하에 농축시켰다. 조 생성물을 하기 조건으로 Prep-HPLC로 정제하였다: 컬럼, Xselect CSH OBD 컬럼 30×150 mm, 5 um, n; 이동상, 물 (10 mmol/L NH4HCO3 + 0.1% NH3·H2O) 및 ACN (Phase B 65%, 7분에 85%까지); 검출기, UV 254/220 nm. 생성물 분획을 동결건조하여 1-(4-{[4-사이클로헥실-5-(트리플루오로메틸)피리미딘-2-일]아미노}페닐)피롤리딘-3-온 (실시예 4의 화합물; 8.5 mg, 9.41%)을 백색 고체로 수득하였다. 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.86 (s, 1H), 8.56 (s, 1H), 7.59 (d, J = 7.5 Hz, 2H), 6.70 (d, J = 9.0 Hz, 2H), 3.65-3.56 (m, 3H), 2.77-2.64 (m, 3H), 1.84-1.64 (m, 7H), 1.36-1.1.25 (m, 3H); MS m/z: 405 [M+H]+.4-cyclohexyl-N-(4-[1,4-dioxa-7-azaspiro[4.4]nonan-7-yl]phenyl) in HCl (1 mL) and 1,4-dioxane (3 mL) A solution of-5-(trifluoromethyl)pyrimidin-2-amine (100 mg, 0.22 mmol, 1.00 equiv) was stirred under nitrogen atmosphere at 60° C. for 1 h. The resulting mixture was concentrated in vacuo. The crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30×150 mm, 5 um, n; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (Phase B 65%, up to 85% in 7 min); Detector, UV 254/220 nm. The product fractions were lyophilized to obtain 1-(4-{[4-cyclohexyl-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)pyrrolidin-3-one (compound of Example 4) 8.5 mg, 9.41%) was obtained as a white solid. 1 H-NMR (300 MHz, DMSO-d 6 ) δ (ppm): 9.86 (s, 1H), 8.56 (s, 1H), 7.59 (d, J = 7.5 Hz, 2H), 6.70 (d, J = 9.0 Hz, 2H), 3.65-3.56 (m, 3H), 2.77-2.64 (m, 3H), 1.84-1.64 (m, 7H), 1.36-1.1.25 (m, 3H); MS m/z: 405 [M+H] + .
실시예 5: 1-(4-{[4-사이클로헥실-5-(트리플루오로메틸)피리미딘-2-일]아미노}페닐)피페리딘-3-올Example 5: 1-(4-{[4-cyclohexyl-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)piperidin-3-ol

제조예 5의 화합물 및 1-(4-아미노페닐)피페리딘-3-올을 출발물질로 하여 실시예 2와 동일한 방법으로 1-(4-{[4-사이클로헥실-5-(트리플루오로메틸)피리미딘-2-일]아미노}페닐)피페리딘-3-올 (실시예 5의 화합물; 5.2 mg, 8.12%)을 황백색 고체로 수득하였다.1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.90 (s, 1H), 8.57 (s, 1H), 7.57 (d, J = 9.0 Hz, 2H), 6.89 (d, J = 9.0 Hz, 2H), 4.80 (d, J = 4.8 Hz, 1H), 3.64-3.52 (m, 2H), 3.43-3.39 (m, 1H), 2.81-2.73 (m, 1H), 2.62-2.54 (m, 1H), 2.47-2.41 (m, 1H), 1.89-1.50 (m, 10 H), 1.47-1.18 (m, 4H); MS m/z: 421 [M+H]+. 1-(4-{[4-cyclohexyl-5-(trifluoro Rhomethyl)pyrimidin-2-yl]amino}phenyl)piperidin-3-ol (compound of Example 5; 5.2 mg, 8.12%) was obtained as an off-white solid. 1 H-NMR (300 MHz, DMSO-d 6 ) δ (ppm): 9.90 (s, 1H), 8.57 (s, 1H), 7.57 (d, J = 9.0 Hz, 2H), 6.89 (d, J = 9.0 Hz, 2H), 4.80 (d, J = 4.8 Hz, 1H), 3.64-3.52 (m, 2H), 3.43-3.39 (m, 1H), 2.81-2.73 (m, 1H), 2.62-2.54 (m) , 1H), 2.47-2.41 (m, 1H), 1.89-1.50 (m, 10 H), 1.47-1.18 (m, 4H); MS m/z: 421 [M+H] + .
실시예 6: N-(4-사이클로헥실페닐)-4-(피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-아민Example 6: N-(4-cyclohexylphenyl)-4-(pyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidin-2-amine

실시예 2에서 제조예 3의 화합물 대신에 제조예 6의 화합물을 사용한 것을 제외하고, 실시예 2와 동일한 방법으로 N-(4-사이클로헥실페닐)-4-(피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-아민 (실시예 6의 화합물; 7%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 9.52 (s, 1H), 8.30 (s, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.8 Hz, 2H), 3.59 (s, 4H), 2.44-2.39 (m, 1H), 1.94-1.92 (m, 4H), 1.90-1.67 (m, 5H), 1.41-1.33 (m, 4H), 1.29-1.25 (m, 1H); MS m/z: 391 [M+H]+.N-(4-cyclohexylphenyl)-4-(pyrrolidin-1-yl) in the same manner as in Example 2, except that the compound of Preparation Example 6 was used instead of the compound of Preparation Example 3 in Example 2 5-(trifluoromethyl)pyrimidin-2-amine (compound of Example 6; 7%) was obtained as a white solid. 1 H-NMR (DMSO-d 6 , 400 MHz) δ (ppm): 9.52 (s, 1H), 8.30 (s, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.8 Hz, 2H), 3.59 (s, 4H), 2.44-2.39 (m, 1H), 1.94-1.92 (m, 4H), 1.90-1.67 (m, 5H), 1.41-1.33 (m, 4H), 1.29 -1.25 (m, 1H); MS m/z: 391 [M+H] + .
실시예 7: 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-5H,6H,7H-사이클로펜타[d]피리미딘-2-일}아미노)페닐]피페리딘-3-올Example 7: 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-5H,6H,7H-cyclopenta[d]pyrimidine- 2-yl}amino)phenyl]piperidin-3-ol

실시예 7의 화합물을 하기 반응식 3에 따라서 제조하였다.The compound of Example 7 was prepared according to Scheme 3 below.
[반응식 3][Scheme 3]

단계 1: (3S,4S)-1-[2-클로로-5H,6H,7H-사이클로펜타[d]피리미딘-4-일]-3,4-디플루오로피롤리딘의 합성Step 1: Synthesis of (3S,4S)-1-[2-chloro-5H,6H,7H-cyclopenta[d]pyrimidin-4-yl]-3,4-difluoropyrrolidine
DCM (5 mL) 중에 (3S, 4S)-3,4-디플루오로피롤리딘 하이드로클로라이드 (60 mg, 0.42 mmol, 1.00당량)을 교반하여 혼합하고, DCM (2 mL) 중의 2,4-디클로로-5H, 6H, 7H-사이클로펜타[d]피리미딘 (94 mg, 0.502 mmol, 1.20당량) 및 DCM (1 mL) 중 DIEA (162 mg, 1.25 mmol, 3.00당량)를 질소 대기하에 -78 ℃에서 적가하였다. 생성된 혼합물을 질소 대기하에 -78℃에서 15분 동안 교반하였다. 혼합물을 실온으로 가온시키고, 생성된 혼합물을 감압 농축시켰다. 조 생성물을 PE/EtOAc (1:10)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 (3S,4S)-1-[2-클로로-5H,6H,7H-사이클로펜타[d]피리미딘-4-일]-3,4-디플루오로피롤리딘 (70 mg, 64.50%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 6.53 (s, 1H), 6.36 (s, 1H), 4.25-3.75 (m, 4H), 3.25-3.10 (m, 2H), 2.80-2.60 (m, 2H), 2.10-1.80 (m, 2H); MS m/z: 260/262 [M+H]+.(3S, 4S)-3,4-difluoropyrrolidine hydrochloride (60 mg, 0.42 mmol, 1.00 equiv) in DCM (5 mL) was mixed with stirring and 2,4- in DCM (2 mL) Dichloro-5H, 6H, 7H-cyclopenta[d]pyrimidine (94 mg, 0.502 mmol, 1.20 equiv) and DIEA (162 mg, 1.25 mmol, 3.00 equiv) in DCM (1 mL) under nitrogen atmosphere at -78 °C was added dropwise. The resulting mixture was stirred under nitrogen atmosphere at -78 °C for 15 min. The mixture was warmed to room temperature, and the resulting mixture was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography eluting with PE/EtOAc (1:10) to (3S,4S)-1-[2-chloro-5H,6H,7H-cyclopenta[d]pyrimidine-4- yl]-3,4-difluoropyrrolidine (70 mg, 64.50%) was obtained as a white solid. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 6.53 (s, 1H), 6.36 (s, 1H), 4.25-3.75 (m, 4H), 3.25-3.10 (m, 2H), 2.80- 2.60 (m, 2H), 2.10-1.80 (m, 2H); MS m/z: 260/262 [M+H] + .
단계 2: 1-[4-([4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-5H,6H,7H-사이클로펜타[d]피리미딘-2-일]아미노)페닐]피페리딘-3-올의 합성Step 2: 1-[4-([4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-5H,6H,7H-cyclopenta[d]pyrimidine-2 Synthesis of -yl]amino)phenyl]piperidin-3-ol
제조예 7의 단계 1에서 3-(트리플루오로메틸)피페리딘 대신에 3-하이드록시피페리딘을 사용하여 제조예 7과 동일한 방법으로 제조한 1-(4-아미노페닐)피페리딘-3-올 (62 mg, 0.32 mmol, 1.20당량)과 단계 1에서 수득된 화합물 (70 mg, 0.27 mmol, 1.00 당량)을 n-BuOH (2mL) 중에서 교반 혼합하고 HCl (1방울)을 첨가하였다. 생성된 혼합물을 질소 대기하에 80℃에서 6시간 동안 교반하였다. 혼합물을 실온으로 냉각시키고, DCM/MeOH (10:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 조 생성물을 수득하였다. 조 생성물을 하기 조건으로 Prep-HPLC로 정제하였다: 컬럼, X Bridge Prep 페닐 OBD 컬럼, 19 x 250 mm, 5um; 이동상, 물 (10mmol/L NH4HCO3 + 0.1% NH3.H2O) 및 ACN (7분에 40% Phase B에서 최대 70%); 검출기, UV 254 & 220 nm. 수집된 분획을 동결 건조하여 1-[4-([4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-5H,6H,7H-사이클로펜타[d]피리미딘-2-일]아미노)페닐]피페리딘-3-올 (실시예 7의 화합물; 9.8 mg, 8.57%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 8.72 (s, 1H), 7.57 (d, J = 9.0 Hz, 2H), 6.82 (d, J = 9.0 Hz, 2H), 5.58-5.30 (m, 2H), 4.76 (d, J = 4.8 Hz, 1H), 4.12-3.75 (m, 4H), 4.65-3.50 (m, 1H), 3.50-3.39 (m, 1H), 3.38-3.33 (m, 1H), 3.15-2.90 (m, 2H), 2.70-2.55 (m, 2H), 2.50-2.45 (m, 1H), 2.40-2.30 (m, 1H), 2.00-1.78 (m, 3H), 1.80-1.69 (m, 1H), 1.65-1.45 (m, 1H), 1.28-1.10 (m, 1H); MS m/z: 416[M+H]+.1-(4-aminophenyl)piperidine prepared in the same manner as in Preparation Example 7 using 3-hydroxypiperidine instead of 3-(trifluoromethyl)piperidine in Step 1 of Preparation Example 7 -3-ol (62 mg, 0.32 mmol, 1.20 equiv) and the compound obtained in step 1 (70 mg, 0.27 mmol, 1.00 equiv) were stirred and mixed in n-BuOH (2mL) and HCl (1 drop) was added. . The resulting mixture was stirred at 80° C. under a nitrogen atmosphere for 6 hours. The mixture was cooled to room temperature and purified by silica gel column chromatography eluting with DCM/MeOH (10:1) to give the crude product. The crude product was purified by Prep-HPLC under the following conditions: column, X Bridge Prep phenyl OBD column, 19 x 250 mm, 5um; mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 .H 2 O) and ACN (up to 70% in 40% Phase B in 7 min); Detector, UV 254 & 220 nm. The collected fractions were freeze-dried to 1-[4-([4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-5H,6H,7H-cyclopenta[d] Pyrimidin-2-yl]amino)phenyl]piperidin-3-ol (compound of Example 7; 9.8 mg, 8.57%) was obtained as a white solid. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 8.72 (s, 1H), 7.57 (d, J = 9.0 Hz, 2H), 6.82 (d, J = 9.0 Hz, 2H), 5.58-5.30 (m, 2H), 4.76 (d, J = 4.8 Hz, 1H), 4.12-3.75 (m, 4H), 4.65-3.50 (m, 1H), 3.50-3.39 (m, 1H), 3.38-3.33 (m) , 1H), 3.15-2.90 (m, 2H), 2.70-2.55 (m, 2H), 2.50-2.45 (m, 1H), 2.40-2.30 (m, 1H), 2.00-1.78 (m, 3H), 1.80 -1.69 (m, 1H), 1.65-1.45 (m, 1H), 1.28-1.10 (m, 1H); MS m/z: 416 [M+H] + .
실시예 8: 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-7H-피롤로[2,3-d]피리미딘-2-일}아미노)페닐]피페리딘-3-올Example 8: 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-7H-pyrrolo[2,3-d]pyrimidine- 2-yl}amino)phenyl]piperidin-3-ol

실시예 7의 단계 1에서 2,4-디클로로-5H,6H,7H-사이클로펜타[d]피리미딘 대신에 2,4-디클로로-7H-피롤로[2,3-d]피리미딘을 사용한 것을 제외하고, 실시예 7과 동일한 방법으로 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-7H-피롤로[2,3-d]피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 8의 화합물; 6.0%)을 백색 고체로서 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 11.12 (s, 1H), 8.43 (s, 1H), 7.63 (d, J = 9.0 Hz, 2H), 6.85-6.75 (m, 3H), 6.50-6.40 (m, 1H), 5.58-5.30 (m, 2H), 4.76 (d, J = 4.8 Hz, 1H), 4.20-3.99 (m, 3H), 3.99-3.85 (m, 1H), 3.65-3.57 (m, 1H), 3.48-3.44 (m, 1H), 3.32-3.29 (m, 1H), 2.45-2.30 (m, 2H), 1.90-1.80 (m, 1H), 1.80-1.65 (m, 1H), 1.65-1.45 (m, 1H), 1.28-1.10 (m, 1H); LCMS (ES, m/z): 415[M+H]+.In step 1 of Example 7, 2,4-dichloro-7H-pyrrolo[2,3-d]pyrimidine was used instead of 2,4-dichloro-5H,6H,7H-cyclopenta[d]pyrimidine. Except in the same manner as in Example 7, 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-7H-pyrrolo[2,3 -d]pyrimidin-2-yl}amino)phenyl]piperidin-3-ol (compound of Example 8; 6.0%) was obtained as a white solid. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 11.12 (s, 1H), 8.43 (s, 1H), 7.63 (d, J = 9.0 Hz, 2H), 6.85-6.75 (m, 3H) , 6.50-6.40 (m, 1H), 5.58-5.30 (m, 2H), 4.76 (d, J = 4.8 Hz, 1H), 4.20-3.99 (m, 3H), 3.99-3.85 (m, 1H), 3.65 -3.57 (m, 1H), 3.48-3.44 (m, 1H), 3.32-3.29 (m, 1H), 2.45-2.30 (m, 2H), 1.90-1.80 (m, 1H), 1.80-1.65 (m, 1H), 1.65-1.45 (m, 1H), 1.28-1.10 (m, 1H); LCMS (ES, m/z): 415 [M+H] + .
실시예 9: 1-[4-({6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-9H-퓨린-2-일}아미노)페닐]피페리딘-3-올Example 9: 1-[4-({6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-9H-purin-2-yl}amino)phenyl]piperi din-3-ol

실시예 7의 단계 1에서 2,4-디클로로-5H,6H,7H-사이클로펜타[d]피리미딘 대신에 2,6-디클로로-9H-퓨린을 사용한 것을 제외하고, 실시예 7과 동일한 방법으로 1-[4-({6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-9H-퓨린-2-일}아미노)페닐]피페리딘-3-올(실시예 9의 화합물; 3.8%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 12.46 (s, 1H), 8.64 (s, 1H), 7.81 (s, 1H), 7.61 (d, J = 9.0 Hz, 2H), 6.83 (d, J = 9.0 Hz, 2H), 5.60-5.43 (m, 2H), 4.76 (d, J = 4.8 Hz, 1H), 4.70-3.75 (m, 4H), 3.70-3.52 (m, 1H), 3.52-3.40 (m, 1H), 3.40-3.35 (m, 1H), 2.60-2.55 (m, 1H), 2.45-2.35 (m, 1H), 1.95-1.80 (m, 1H), 1.80-1.65 (m, 1H), 1.64-1.45 (m, 1H), 1.30-1.10 (m, 1H); MS m/z: 416[M+H]+.In the same manner as in Example 7, except that 2,6-dichloro-9H-purine was used instead of 2,4-dichloro-5H,6H,7H-cyclopenta[d]pyrimidine in step 1 of Example 7 1-[4-({6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-9H-purin-2-yl}amino)phenyl]piperidin-3- All (compound of Example 9; 3.8%) was obtained as a white solid. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 12.46 (s, 1H), 8.64 (s, 1H), 7.81 (s, 1H), 7.61 (d, J = 9.0 Hz, 2H), 6.83 (d, J = 9.0 Hz, 2H), 5.60-5.43 (m, 2H), 4.76 (d, J = 4.8 Hz, 1H), 4.70-3.75 (m, 4H), 3.70-3.52 (m, 1H), 3.52-3.40 (m, 1H), 3.40-3.35 (m, 1H), 2.60-2.55 (m, 1H), 2.45-2.35 (m, 1H), 1.95-1.80 (m, 1H), 1.80-1.65 (m) , 1H), 1.64-1.45 (m, 1H), 1.30-1.10 (m, 1H); MS m/z: 416 [M+H] + .
실시예 9(S): (3S)-1-[4-({6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-9H-퓨린-2-일}아미노)페닐]피페리딘-3-올Example 9(S): (3S)-1-[4-({6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-9H-purin-2-yl }amino)phenyl]piperidin-3-ol

실시예 7의 단계 1에서 2,4-디클로로-5H,6H,7H-사이클로펜타[d]피리미딘 대신에 2,6-디클로로-9H-퓨린을 사용하고, 실시예 7의 단계 2에서 1-(4-아미노페닐)피페리딘-3-올을 대신하여, 제조예 7의 단계 1에서 3-(트리플루오로메틸)피페리딘 대신에 (S)-3-하이드록시피페리딘을 사용하여 제조예 7과 동일한 방법으로 제조한 (S)-1-(4-아미노페닐)피페리딘-3-올을 사용한 것을 제외하고, 실시예 7과 동일한 방법으로 (3S)-1-[4-({6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-9H-퓨린-2-일}아미노)페닐]피페리딘-3-올(실시예 9(S)의 화합물)을 갈색 고체로 수득하였다. 1H-NMR (400 MHz, DMSO- d6); δ 12.47 (brs, 1H), 8.62 (s, 1H), 7.82 (s, 1H), 7.62 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 5.58 (brs, 1H), 5.45 (brs, 1H), 4.75 (d, J = 4.4 Hz, 1H), 4.12-4.04 (m, 4H), 3.61-3.59 (m, 1H), 3.49-3.46 (m, 1H), 3.31 (m, 1H), 2.57 (m, 1H), 2.40 (t, J = 10.0 Hz, 1H), 1.90-1.87 (m, 1H), 1.77-1.73 (m, 1H), 1.57-1.54 (m, 1H), 1.27-1.24 (m, 1H; MS m/z: 416 [M+H]+.In step 1 of Example 7, 2,6-dichloro-9H-purine was used instead of 2,4-dichloro-5H,6H,7H-cyclopenta[d]pyrimidine, and in step 2 of Example 7, 1- Instead of (4-aminophenyl) piperidin-3-ol, (S)-3-hydroxypiperidine was used instead of 3- (trifluoromethyl) piperidine in Step 1 of Preparation Example 7 and (3S)-1-[4] in the same manner as in Example 7, except that (S)-1-(4-aminophenyl)piperidin-3-ol prepared in the same manner as in Preparation Example 7 was used. -({6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-9H-purin-2-yl}amino)phenyl]piperidin-3-ol (Example 9(S)) was obtained as a brown solid. 1 H-NMR (400 MHz, DMSO-d 6 ); δ 12.47 (brs, 1H), 8.62 (s, 1H), 7.82 (s, 1H), 7.62 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 5.58 (brs, 1H), 5.45 (brs, 1H), 4.75 (d, J = 4.4 Hz, 1H), 4.12-4.04 (m, 4H), 3.61-3.59 (m, 1H), 3.49-3.46 (m, 1H), 3.31 (m, 1H), 2.57 (m, 1H), 2.40 (t, J = 10.0 Hz, 1H), 1.90-1.87 (m, 1H), 1.77-1.73 (m, 1H), 1.57-1.54 (m, 1H) ), 1.27-1.24 (m, 1H; MS m/z: 416 [M+H] + .
실시예 9(R): (3R)-1-[4-({6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-9H-퓨린-2-일}아미노)페닐]피페리딘-3-올Example 9(R): (3R)-1-[4-({6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-9H-purin-2-yl }amino)phenyl]piperidin-3-ol

실시예 7의 단계 1에서 2,4-디클로로-5H,6H,7H-사이클로펜타[d]피리미딘 대신에 2,6-디클로로-9H-퓨린을 사용하고, 실시예 7의 단계 2에서 1-(4-아미노페닐)피페리딘-3-올을 대신하여, 제조예 7의 단계 1에서 3-(트리플루오로메틸)피페리딘 대신에 (R)-3-하이드록시피페리딘을 사용하여 제조예 7과 동일한 방법으로 제조한 (R)-1-(4-아미노페닐)피페리딘-3-올을 사용한 것을 제외하고, 실시예 7과 동일한 방법으로 (3R)-1-[4-({6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-9H-퓨린-2-일}아미노)페닐]피페리딘-3-올(실시예 9(R)의 화합물)을 갈색 고체로 수득하였다. 1H-NMR (400 MHz, DMSO-d6); δ 12.47 (brs, 1H), 8.62 (s, 1H), 7.82 (s, 1H), 7.62 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 5.58 (brs, 1H), 5.45 (brs, 1H), 4.75 (d, J = 4.4 Hz, 1H), 4.10-3.97 (m, 4H), 3.61-3.60 (m, 1H), 3.49-3.46 (m, 1H), 3.31 (m, 1H), 2.57-2.50 (m, 1H), 2.40 (t, J = 9.6 Hz, 1H), 1.90-1.87 (m, 1H), 1.77-1.73 (m, 1H), 1.59-1.51 (m, 1H), 1.22-1.18 (m, 1H); MS m/z: 416 [M+H]+.In step 1 of Example 7, 2,6-dichloro-9H-purine was used instead of 2,4-dichloro-5H,6H,7H-cyclopenta[d]pyrimidine, and in step 2 of Example 7, 1- In place of (4-aminophenyl) piperidin-3-ol, (R) -3-hydroxypiperidine was used instead of 3- (trifluoromethyl) piperidine in Step 1 of Preparation Example 7 (3R)-1-[4 in the same manner as in Example 7, except that (R)-1-(4-aminophenyl)piperidin-3-ol prepared in the same manner as in Preparation Example 7 was used. -({6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-9H-purin-2-yl}amino)phenyl]piperidin-3-ol (Example 9(R)) was obtained as a brown solid. 1 H-NMR (400 MHz, DMSO-d 6 ); δ 12.47 (brs, 1H), 8.62 (s, 1H), 7.82 (s, 1H), 7.62 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 5.58 (brs, 1H), 5.45 (brs, 1H), 4.75 (d, J = 4.4 Hz, 1H), 4.10-3.97 (m, 4H), 3.61-3.60 (m, 1H), 3.49-3.46 (m, 1H), 3.31 (m, 1H), 2.57-2.50 (m, 1H), 2.40 (t, J = 9.6 Hz, 1H), 1.90-1.87 (m, 1H), 1.77-1.73 (m, 1H), 1.59-1.51 (m) , 1H), 1.22-1.18 (m, 1H); MS m/z: 416 [M+H] + .
실시예 10: 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-1H-피라졸로[3,4-d]피리미딘-6-일}아미노)페닐]피페리딘-3-올Example 10: 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-1H-pyrazolo[3,4-d]pyrimidine- 6-yl}amino)phenyl]piperidin-3-ol

실시예 7의 단계 1에서 2,4-디클로로-5H,6H,7H-사이클로펜타[d]피리미딘 대신에 4,6-디클로로-1H-피라졸로[3,4-d]피리미딘을 사용한 것을 제외하고, 실시예 7과 동일한 방법으로 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-1H-피라졸로[3,4-d]피리미딘-6-일}아미노)페닐]피페리딘-3-올(실시예 10의 화합물; 4.31%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 12.86 (s, 1H), 8.81 (s, 1H), 7.97 (s, 1H), 7.62 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 9.0 Hz, 2H), 5.63-5.48 (m, 2H), 4.77 (d, J = 4.8 Hz, 2H), 4.30-3.80 (m, 4H), 3.65-3.52 (m, 1H), 3.52-3.42 (m, 1H), 3.42-3.39 (m, 1H), 2.60-2.55 (m, 1H), 2.45-2.35 (m, 1H), 1.95-1.80 (m, 1H), 1.80-1.65 (m, 1H), 1.64-1.45 (m, 1H), 1.32-1.15 (m, 1H); MS m/z: 416[M+H]+.Using 4,6-dichloro-1H-pyrazolo[3,4-d]pyrimidine instead of 2,4-dichloro-5H,6H,7H-cyclopenta[d]pyrimidine in step 1 of Example 7 Except in the same manner as in Example 7, 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-1H-pyrazolo[3,4 -d]pyrimidin-6-yl}amino)phenyl]piperidin-3-ol (compound of Example 10; 4.31%) was obtained as a white solid. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 12.86 (s, 1H), 8.81 (s, 1H), 7.97 (s, 1H), 7.62 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 9.0 Hz, 2H), 5.63-5.48 (m, 2H), 4.77 (d, J = 4.8 Hz, 2H), 4.30-3.80 (m, 4H), 3.65-3.52 (m, 1H), 3.52-3.42 (m, 1H), 3.42-3.39 (m, 1H), 2.60-2.55 (m, 1H), 2.45-2.35 (m, 1H), 1.95-1.80 (m, 1H), 1.80-1.65 (m) , 1H), 1.64-1.45 (m, 1H), 1.32-1.15 (m, 1H); MS m/z: 416 [M+H] + .
실시예 11: 5-요오도-N4-[(1R,4R)-2-옥사바이사이클로[2.2.1]헵탄-4-일]-N2-{4-[3-(트리플루오로메틸)피페리딘-1-일]페닐}피리미딘-2,4-디아민Example 11: 5-iodo-N4-[(1R,4R)-2-oxabicyclo[2.2.1]heptan-4-yl]-N2-{4-[3-(trifluoromethyl)p Peridin-1-yl]phenyl}pyrimidine-2,4-diamine

실시예 11의 화합물을 하기 반응식 4에 따라서 제조하였다.The compound of Example 11 was prepared according to Scheme 4 below.
[반응식 4][Scheme 4]

단계 1: 2-클로로-5-요오도-N-[2-옥사바이사이클로[2.2.1]헵탄-4-일]피리미딘-4-아민의 합성Step 1: Synthesis of 2-chloro-5-iodo-N-[2-oxabicyclo[2.2.1]heptan-4-yl]pyrimidin-4-amine
2,4-디클로로-5-요오도피리미딘 (360 mg, 1.31 mmol, 1.00당량) 및 2-옥사 바이사이클로[2.2.1]헵탄-4-아민 (150 mg, 1.32 mmol, 1.01당량)을 ACN (5 mL) 중에서 교반 혼합하고, DIEA (513 mg, 3.96 mmol, 3.03당량)을 첨가하였다. 생성된 혼합물을 질소 대기하에 80℃에서 3시간 동안 교반하였다. 혼합물을 실온으로 냉각시키고, 진공하에 농축시켰다. 잔사를 PE/EtOAc (2:1)로 용출시킨 실리카겔 컬럼크로마토그래피로 정제하여 2-클로로-5-요오도-N-[2-옥사바이사이클로[2.2.1]헵탄-4-일]피리미딘-4-아민 (220 mg, 43.96%)을 황색 고체로 수득하였다. MS m/z: 352/354 [M+H]+. 2,4-dichloro-5-iodopyrimidine (360 mg, 1.31 mmol, 1.00 equiv) and 2-oxabicyclo[2.2.1]heptan-4-amine (150 mg, 1.32 mmol, 1.01 equiv) were combined with ACN (5 mL) was stirred and DIEA (513 mg, 3.96 mmol, 3.03 equiv) was added. The resulting mixture was stirred at 80° C. under a nitrogen atmosphere for 3 hours. The mixture was cooled to room temperature and concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with PE/EtOAc (2:1), and 2-chloro-5-iodo-N-[2-oxabicyclo[2.2.1]heptan-4-yl]pyrimidine -4-amine (220 mg, 43.96%) was obtained as a yellow solid. MS m/z: 352/354 [M+H] + .
단계 2: 5-요오도-N4-[(1R,4R)-2-옥사바이사이클로[2.2.1]헵탄-4-일]-N2-{4-[3-(트리플루오로메틸)피페리딘-1-일]페닐}피리미딘-2,4-디아민의 합성Step 2: 5-Iodo-N4-[(1R,4R)-2-oxabicyclo[2.2.1]heptan-4-yl]-N2-{4-[3-(trifluoromethyl)piperi Synthesis of diin-1-yl]phenyl}pyrimidine-2,4-diamine
단계 1에서 수득된 화합물 (100 mg, 0.28 mmol, 1.00당량) 및 농염산(1방울)을 n-BuOH (3 mL)에서 교반하여 혼합하고 제조예 7의 화합물 (90 mg, 0.36 mmol, 1.30당량)을 첨가하였다. 생성된 혼합물을 질소 대기하에 80℃에서 4시간 동안 교반하고, 실온으로 냉각시켰다. 생성된 혼합물을 진공하에 농축시키고, 잔사를 DCM/MeOH (5:1)로 용출시켜 실리카겔 컬럼 크로마토그래피로 정제하였다. 조 생성물을 하기 조건으로 Prep-HPLC로 정제하였다: 컬럼, Xselect CSH OBD 컬럼 30 x150mm 5um; 이동상, 물 (10mmol/L NH4HCO3 + 0.1% NH3.H2O) 및 ACN (36% PhaseB, 11 분에 48%까지); 검출기, UV 254/220 nm. 생성물 분획을 동결 건조하여 5-요오도-N4-[(1R,4R)-2-옥사바이사이클로[2.2.1]헵탄-4-일]-N2-{4-[3-(트리플루오로메틸)피페리딘-1-일]페닐}피리미딘-2,4-디아민 (실시예 11의 화합물; 9.4 mg, 5.90%)을 연황색 고체로 수득하였다. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.48 (s, 1H), 7.41 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 4.64 (s, 1H), 4.27-4.25 (m, 1H), 4.10-4.00 (m, 2H), 3.72-3.69 (m, 1H), 3.61-3.58 (m, 1H), 2.67-2.57 (m, 2H), 2.02-1.94 (m, 3H), 1.85-1.51 (m, 6H), 1.48-1.36 (m, 1H); MS m/z: 560 [M+H]+.The compound obtained in step 1 (100 mg, 0.28 mmol, 1.00 equiv) and concentrated hydrochloric acid (1 drop) were mixed with stirring in n-BuOH (3 mL), and the compound of Preparation 7 (90 mg, 0.36 mmol, 1.30 equiv.) ) was added. The resulting mixture was stirred at 80° C. for 4 h under a nitrogen atmosphere and cooled to room temperature. The resulting mixture was concentrated in vacuo and the residue was purified by silica gel column chromatography, eluting with DCM/MeOH (5:1). The crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30 x 150mm 5um; mobile phase, water (10 mmol/L NH4HCO3+0.1% NH3.H2O) and ACN (36% PhaseB, up to 48% in 11 min); Detector, UV 254/220 nm. The product fractions were lyophilized to 5-iodo-N4-[(1R,4R)-2-oxabicyclo[2.2.1]heptan-4-yl]-N2-{4-[3-(trifluoromethyl )piperidin-1-yl]phenyl}pyrimidine-2,4-diamine (compound of Example 11; 9.4 mg, 5.90%) was obtained as a light yellow solid. 1 H-NMR (400 MHz, DMSO-d 6 ) δ (ppm): 8.48 (s, 1H), 7.41 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 4.64 (s, 1H), 4.27-4.25 (m, 1H), 4.10-4.00 (m, 2H), 3.72-3.69 (m, 1H), 3.61-3.58 (m, 1H), 2.67 -2.57 (m, 2H), 2.02-1.94 (m, 3H), 1.85-1.51 (m, 6H), 1.48-1.36 (m, 1H); MS m/z: 560 [M+H] + .
실시예 12: 1-[4-([4-클로로-6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일]아미노)페닐]피페리딘-3-올Example 12: 1-[4-([4-chloro-6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino)phenyl] piperidin-3-ol

실시예 12의 화합물을 하기 반응식 5에 따라서 제조하였다.The compound of Example 12 was prepared according to Scheme 5 below.
[반응식 5][Scheme 5]

단계 1: 1-[4-[(4,6-디클로로피리미딘-2-일)아미노]페닐]피페리딘-3-올의 합성Step 1: Synthesis of 1-[4-[(4,6-dichloropyrimidin-2-yl)amino]phenyl]piperidin-3-ol
DCE (10 mL) 및 t-BuOH (10 mL) 중에서 2,4,6-트리클로로-피리미딘 (1.00 g, 5.452 mmol, 1.00당량)을 교반 혼합하고, 에테르 중의 ZnCl2 (1M)(10.85 mL, 10.85 mmol, 1.99당량)을 0℃에서 적가하였다. 생성된 혼합물을 0℃에서 1시간 동안 교반하고, 1-(4-아미노페닐)피페리딘-3-올 (1.04 g, 5.40 mmol, 0.99당량)을 0 ℃에서 적가하였다. 생성된 혼합물을 수소 대기하에 0℃에서 0.5시간 동안 교반하였다. 상기 혼합물에 TEA (606 mg, 5.98 mmol, 1.10당량)를 적가하고, 생성된 혼합물을 질소 대기하에 실온에서 12시간 동안 교반하고, 생성된 혼합물을 진공하에 농축시켰다. 조 생성물을 PE/EtOAc (1:5)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 1-[4-[(4,6-디클로로피리미딘-2-일)아미노]페닐]피페리딘-3-올 (80 mg, 3.5%)를 황색 오일로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 10.12(s, 1H), 7.55-7.30 (m, 2H), 7.15-6.75 (m, 3H), 3.75-3.50 (m, 2H), 3.50-3.00 (m, 1H), 2.75-2.55 (m, 1H), 2.50-2.35 (m, 1H), 2.00-1.82 (m, 1H), 1.82-1.65 (m, 1H), 1.65-1.35 (m, 1H), 1.35-1.15 (m, 1H); MS m/z: 339[M+H]+. 2,4,6-trichloro-pyrimidine (1.00 g, 5.452 mmol, 1.00 equiv) was stirred mixed in DCE (10 mL) and t-BuOH (10 mL) and ZnCl 2 (1M) in ether (10.85 mL) , 10.85 mmol, 1.99 equiv) were added dropwise at 0 °C. The resulting mixture was stirred at 0 °C for 1 h, and 1-(4-aminophenyl)piperidin-3-ol (1.04 g, 5.40 mmol, 0.99 equiv) was added dropwise at 0 °C. The resulting mixture was stirred under a hydrogen atmosphere at 0° C. for 0.5 h. To the mixture was added dropwise TEA (606 mg, 5.98 mmol, 1.10 equiv), the resulting mixture was stirred under nitrogen atmosphere at room temperature for 12 h, and the resulting mixture was concentrated in vacuo. The crude product was purified by silica gel column chromatography eluting with PE/EtOAc (1:5) to 1-[4-[(4,6-dichloropyrimidin-2-yl)amino]phenyl]piperidine-3- The ol (80 mg, 3.5%) was obtained as a yellow oil. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 10.12(s, 1H), 7.55-7.30 (m, 2H), 7.15-6.75 (m, 3H), 3.75-3.50 (m, 2H), 3.50-3.00 (m, 1H), 2.75-2.55 (m, 1H), 2.50-2.35 (m, 1H), 2.00-1.82 (m, 1H), 1.82-1.65 (m, 1H), 1.65-1.35 (m) , 1H), 1.35-1.15 (m, 1H); MS m/z: 339 [M+H] + .
단계 2: 1-[4-([4-클로로-6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일]아미노)페닐]피페리딘-3-올의 합성Step 2: 1-[4-([4-chloro-6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino)phenyl]pi Synthesis of peridin-3-ol
단계 1에서 수득된 화합물 (80 mg, 0.23 mmol, 1.00당량) 및 (3S,4S)-3,4-디플루오로피롤리딘 하이드로클로라이드 (40 mg, 0.28 mmol, 1.20당량)을 ACN (2mL) 중에서 교반하여 혼합하고, DIEA (89 mg, 0.70 mmol, 3.00당량)를 적가하였다. 생성된 혼합물을 질소 대기하에 80℃에서 6시간 동안 교반하였다. 혼합물을 실온으로 냉각시켰다. 조 생성물을 하기 조건으로 Prep-HPLC로 정제하였다: 컬럼, Xselect CSH OBD 컬럼 30 x 150mm 5um, n; 이동상, 물 (10mmol/L NH4HCO3 + 0.1% NH3
.H2O) 및 ACN (30% PhaseB, 8 분에 최대 60%); 검출기, UV 254&220nm. 수집된 분획을 동결 건조하여 1-[4-([4-클로로-6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일]아미노)페닐]피페리딘-3-올 (실시예 12의 화합물; 8.3 mg, 8.5%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 9.30 (s, 1H), 7.54 (d, J = 9.0 Hz, 2H), 6.85 (d, J = 9.0 Hz, 2H), 6.07 (s, 1H), 5.75-5.35 (m, 2H), 4.78 (s, 1H), 4.10-3.35 (m, 7H), 2.65-2.52 (m, 1H), 2.45-2.30 (m, 1H), 1.90-1.80 (m, 1H), 1.80-1.65 (m, 1H), 1.65-1.45 (m, 1H), 1.28-1.10 (m, 1H); MS m/z: 410[M+H]+.Compound obtained in step 1 (80 mg, 0.23 mmol, 1.00 equiv) and (3S,4S)-3,4-difluoropyrrolidine hydrochloride (40 mg, 0.28 mmol, 1.20 equiv) were mixed with ACN (2mL) Stir to mix and DIEA (89 mg, 0.70 mmol, 3.00 equiv) was added dropwise. The resulting mixture was stirred at 80° C. under a nitrogen atmosphere for 6 hours. The mixture was cooled to room temperature. The crude product was purified by Prep-HPLC under the following conditions: column, Xselect CSH OBD column 30 x 150mm 5um, n; mobile phase, water (10 mmol/L NH4HCO3 + 0.1% NH 3 . H 2 O) and ACN (30% PhaseB, up to 60% in 8 min); Detector, UV 254&220nm. The collected fractions were freeze-dried to 1-[4-([4-chloro-6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino )phenyl]piperidin-3-ol (compound of Example 12; 8.3 mg, 8.5%) was obtained as a white solid. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 9.30 (s, 1H), 7.54 (d, J = 9.0 Hz, 2H), 6.85 (d, J = 9.0 Hz, 2H), 6.07 (s) , 1H), 5.75-5.35 (m, 2H), 4.78 (s, 1H), 4.10-3.35 (m, 7H), 2.65-2.52 (m, 1H), 2.45-2.30 (m, 1H), 1.90-1.80 (m, 1H), 1.80-1.65 (m, 1H), 1.65-1.45 (m, 1H), 1.28-1.10 (m, 1H); MS m/z: 410 [M+H] + .
실시예 13 및 14: 1-[4-({4-브로모-6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일}아미노)페닐]피페리딘-3-올 및 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-6-요오도피리미딘-2-일}아미노)페닐]피페리딘-3-올Examples 13 and 14: 1-[4-({4-bromo-6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl}amino )phenyl]piperidin-3-ol and 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-6-iodopyrimidine- 2-yl}amino)phenyl]piperidin-3-ol


실시예 12의 단계 1에서 2,4,6-트리클로로피리미딘 대신에 제조예 8의 화합물을 사용한 것을 제외하고, 실시예 12와 동일한 방법으로 1-[4-({4-브로모-6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 13의 화합물; 6.43%) 및 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-6-요오도피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 14의 화합물; 14.0%)을 각각 백색 고체로 수득하였다.1-[4-({4-bromo-6 -[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl}amino)phenyl]piperidin-3-ol (compound of Example 13; 6.43% ) and 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-6-iodopyrimidin-2-yl}amino)phenyl]p Peridin-3-ol (compound of Example 14; 14.0%) was obtained as a white solid, respectively.
1-[4-({4-브로모-6-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 13의 화합물)1-[4-({4-bromo-6-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl}amino)phenyl]piperidine -3-ol (compound of Example 13)
1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 9.32 (s, 1H), 7.53 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 9.0 Hz, 2H), 6.21 (s, 1H), 5.65-5.30 (m, 2H), 4.77 (d, J = 4.5 Hz, 1H), 4.10-3.35 (m, 7H), 2.48-2.30 (m, 2H), 1.90-1.80 (m, 1H), 1.80-1.65 (m, 1H), 1.65-1.35 (m, 1H), 1.35-1.10 (m, 1H); MS m/z: 454 [M+H]+.1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 9.32 (s, 1H), 7.53 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 9.0 Hz, 2H), 6.21 (s) , 1H), 5.65-5.30 (m, 2H), 4.77 (d, J = 4.5 Hz, 1H), 4.10-3.35 (m, 7H), 2.48-2.30 (m, 2H), 1.90-1.80 (m, 1H) ), 1.80-1.65 (m, 1H), 1.65-1.35 (m, 1H), 1.35-1.10 (m, 1H); MS m/z: 454 [M+H] + .
1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-6-요오도피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 14의 화합물)1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-6-iodopyrimidin-2-yl}amino)phenyl]piperidine -3-ol (compound of Example 14)
1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 9.25 (s, 1H), 7.53 (d, J =9.0 Hz, 2H), 6.83 (d, J =9.0 Hz, 2H), 6.44 (s, 1H), 5.57-5.55 (m, 1H), 5.40-5.38 (m, 1H), 4.77 (d, J =4.8 Hz, 1H), 4.05-3.38 (m, 7H), 2.45-2.32 (m, 2H), 1.95-1.80 (m, 1H), 1.80-1.62 (m, 1H), 1.62-1.40 (m, 1H), 1.30-1.10 (m, 1H); 502[M+H]+.1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 9.25 (s, 1H), 7.53 (d, J =9.0 Hz, 2H), 6.83 (d, J =9.0 Hz, 2H), 6.44 (s) , 1H), 5.57-5.55 (m, 1H), 5.40-5.38 (m, 1H), 4.77 (d, J =4.8 Hz, 1H), 4.05-3.38 (m, 7H), 2.45-2.32 (m, 2H) ), 1.95-1.80 (m, 1H), 1.80-1.62 (m, 1H), 1.62-1.40 (m, 1H), 1.30-1.10 (m, 1H); 502[M+H] + .
실시예 15: 1-[4-([4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일]아미노)페닐]피페리딘-3-올Example 15: 1-[4-([4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino)phenyl]piperidine- 3-ol

실시예 15의 화합물을 하기 반응식 6에 따라서 제조하였다.The compound of Example 15 was prepared according to Scheme 6 below.
[반응식 6][Scheme 6]

단계 1: 2-클로로-4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘의 합성Step 1: Synthesis of 2-chloro-4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidine
DMF (2 mL) 중에서 2,4-디클로로피리미딘 (51 mg, 0.34 mmol, 1.00당량) 및 DIEA (230 mg, 1.03 mmol, 3.00당량)을 교반하여 혼합하고, (3S,4S)-3,4-디플루오로피롤리딘 하이드로클로라이드 (49 mg, 0.34 mmol, 1.00당량)을 첨가하였다. 생성된 혼합물을 질소 대기하에 25℃에서 0.5시간 동안 교반하였다. 실온에서 물 (10 mL)을 첨가하여 반응을 퀀칭시켰다. 생성된 혼합물을 EtOAc (3 x 20 mL)로 추출하였다. 유기층을 모아서 무수 Na2SO4로 건조시키고, 여과 후 여액을 감압 농축하였다. 잔사를 PE/EtOAc (3:1)로 용출시켜서 실리카겔 컬럼 크로마토그래피로 정제하여 2-클로로-4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘 (60 mg, 81.1%)을 백색 고체로 수득하였다. MS m/z: 220/222[M+H]+.Mix 2,4-dichloropyrimidine (51 mg, 0.34 mmol, 1.00 equiv) and DIEA (230 mg, 1.03 mmol, 3.00 equiv) in DMF (2 mL) by stirring, (3S,4S)-3,4 -difluoropyrrolidine hydrochloride (49 mg, 0.34 mmol, 1.00 equiv) was added. The resulting mixture was stirred at 25° C. under a nitrogen atmosphere for 0.5 h. The reaction was quenched by addition of water (10 mL) at room temperature. The resulting mixture was extracted with EtOAc (3 x 20 mL). The organic layers were collected, dried over anhydrous Na 2 SO 4 , filtered, and the filtrate was concentrated under reduced pressure. The residue was eluted with PE/EtOAc (3:1) and purified by silica gel column chromatography to 2-chloro-4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidine (60 mg, 81.1%) was obtained as a white solid. MS m/z: 220/222[M+H]+.
단계 2: 1-[4-([4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일]아미노)페닐]피페리딘-3-올의 합성Step 2: 1-[4-([4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino)phenyl]piperidin-3 -Synthesis of ol
단계 1에서 수득된 화합물 (50 mg, 0.20 mmol, 1.00 당량) 및 1-(4-아미노페닐)피페리딘-3-올 (43 mg, 0.20 mmol, 1.00당량)을 n-BuOH (2.00mL) 중에서 교반하여 혼합하고, HCl (1 방울)을 첨가하였다. 생성된 혼합물을 질소 대기하에 120℃에서 1시간 동안 교반하였다. 혼합물을 실온으로 냉각시키고, 조 생성물을 하기 조건으로 Prep-HPLC로 정제하였다: 컬럼, XBridge Prep 페닐 OBD 컬럼, 19 x 250 mm, 5um; 이동상, 물 (10mmol/L NH4HCO3 + 0.1% NH3
.H2O) 및 ACN (32% Phase B, 7 분에 62%까지). 수집된 분획을 동결 건조하여 1-[4-([4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]피리미딘-2-일]아미노)페닐]피페리딘-3-올 (실시예 15의 화합물; 5.1 mg, 6.3%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): δ 8.85 (s, 1H), 7.94 (d, J = 5.7 Hz, 1H), 7.58 (d, J = 9.0 Hz, 2H), 6.83 (m, J = 9.0 Hz, 2H), 5.98 (d, J = 5.8 Hz, 1H), 5.58-5.40 (m, 2H), 4.77 (d, J = 4.8 Hz, 1H), 4.08-3.41 (m, 6H), 3.36-3.34 (m, 1H), 2.602.56 (m, 1H), 2.42-2.35 (m, 1H), 1.97-1.80 (m, 1H), 1.77-1.74 (m, 1H), 1.60-1.50 (m, 1H), 1.28-1.18 (m, 1H); MS m/z: 376 [M+H]+.Compound obtained in step 1 (50 mg, 0.20 mmol, 1.00 equiv) and 1-(4-aminophenyl)piperidin-3-ol (43 mg, 0.20 mmol, 1.00 equiv) were mixed with n-BuOH (2.00mL) Stir to mix and add HCl (1 drop). The resulting mixture was stirred at 120° C. under a nitrogen atmosphere for 1 hour. The mixture was cooled to room temperature and the crude product was purified by Prep-HPLC under the following conditions: column, XBridge Prep phenyl OBD column, 19 x 250 mm, 5um; Mobile phase, water (10 mmol/L NH 4 HCO 3 + 0.1% NH 3 . H 2 O) and ACN (32% Phase B, up to 62% in 7 min). The collected fractions were freeze-dried to 1-[4-([4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]pyrimidin-2-yl]amino)phenyl]pyr Peridin-3-ol (compound of Example 15; 5.1 mg, 6.3%) was obtained as a white solid. 1 H-NMR (DMSO-d 6 , 300 MHz) δ (ppm): δ 8.85 (s, 1H), 7.94 (d, J = 5.7 Hz, 1H), 7.58 (d, J = 9.0 Hz, 2H), 6.83 (m, J = 9.0 Hz, 2H), 5.98 (d, J = 5.8 Hz, 1H), 5.58-5.40 (m, 2H), 4.77 (d, J = 4.8 Hz, 1H), 4.08-3.41 (m) , 6H), 3.36-3.34 (m, 1H), 2.602.56 (m, 1H), 2.42-2.35 (m, 1H), 1.97-1.80 (m, 1H), 1.77-1.74 (m, 1H), 1.60 -1.50 (m, 1H), 1.28-1.18 (m, 1H); MS m/z: 376 [M+H] + .
실시예 16: 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-5-메틸피리미딘-2-일}아미노)페닐]피페리딘-3-올Example 16: 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-5-methylpyrimidin-2-yl}amino)phenyl] piperidin-3-ol

실시예 15의 단계 1에서 2,4-디클로로피리미딘 대신에 2,4-디클로로-5-메틸피리미딘을 사용한 것을 제외하고, 실시예 15와 동일한 방법으로 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-5-메틸피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 16의 화합물; 11.78%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 8.66 (s, 1H), 7.74 (s, 1H), 7.54 (d, J = 8.9 Hz, 2H), 6.82 (d, J = 8.9 Hz, 2H), 5.52-5.33 (m, 2H), 4.73 (d, J = 4.8 Hz, 1H), 4.18-3.88 (m, 4H), 3.64-3.56 (m, 1H), 3.49-3.45 (m, H), 3.35-3.33 (m, 1H), 2.57-2.55 (m, 1H), 2.43-2.36 (m, 1H), 2.23 (s, 3H), 1.93-1.85 (m, 1H), 1.76-1.73 (m, 1H), 1.65-1.55 (m, 1H), 1.30-1.12 (m, 1H); MS m/z: 390 [M+H]+.In the same manner as in Example 15, 1-[4-({4-[ (3S,4S)-3,4-difluoropyrrolidin-1-yl]-5-methylpyrimidin-2-yl}amino)phenyl]piperidin-3-ol (compound of Example 16; 11.78%) as a white solid. 1 H-NMR (DMSO-d 6 , 300 MHz) δ (ppm): 8.66 (s, 1H), 7.74 (s, 1H), 7.54 (d, J = 8.9 Hz, 2H), 6.82 (d, J = 8.9 Hz, 2H), 5.52-5.33 (m, 2H), 4.73 (d, J = 4.8 Hz, 1H), 4.18-3.88 (m, 4H), 3.64-3.56 (m, 1H), 3.49-3.45 (m) , H), 3.35-3.33 (m, 1H), 2.57-2.55 (m, 1H), 2.43-2.36 (m, 1H), 2.23 (s, 3H), 1.93-1.85 (m, 1H), 1.76-1.73 (m, 1H), 1.65-1.55 (m, 1H), 1.30-1.12 (m, 1H); MS m/z: 390 [M+H] + .
실시예 17: 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-5-에틸피리미딘-2-일}아미노)페닐]피페리딘-3-올Example 17: 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-5-ethylpyrimidin-2-yl}amino)phenyl] piperidin-3-ol

실시예 15의 단계 1에서 2,4-디클로로피리미딘 대신에 2,4-디클로로-5-에틸피리미딘을 사용한 것을 제외하고, 실시예 15와 동일한 방법으로 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-5-에틸피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 17의 화합물; 4.48%)을 백색 고체로 수득하였다. 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 8.76 (s, 1H), 7.83 (s, 1H), 7.54 (d, J = 8.0 Hz, 2H), 6.82 (d, J = 8.0 Hz, 2H), 5.51-5.38 (m, 2H), 4.78 (d, J = 4.8 Hz, 1H), 4.12-3.85 (m, 4H), 3.79-3.67 (m, 2H), 2.69-2.57 (m, 2H), 2.40-2.35 (m, 1H), 1.95-1.86 (m, 1H), 1.81-1.72 (m, 1H),1.64-1.60 (m,1H) 1.25-1.15 (m, 1H), 1.10-1.00 (m, 3H); MS m/z: 404 [M+H]+.In the same manner as in Example 15, 1-[4-({4-[ (3S,4S)-3,4-difluoropyrrolidin-1-yl]-5-ethylpyrimidin-2-yl}amino)phenyl]piperidin-3-ol (compound of Example 17; 4.48%) as a white solid. 1 H-NMR (DMSO-d 6 , 400 MHz) δ (ppm): 8.76 (s, 1H), 7.83 (s, 1H), 7.54 (d, J = 8.0 Hz, 2H), 6.82 (d, J = 8.0 Hz, 2H), 5.51-5.38 (m, 2H), 4.78 (d, J = 4.8 Hz, 1H), 4.12-3.85 (m, 4H), 3.79-3.67 (m, 2H), 2.69-2.57 (m) , 2H), 2.40-2.35 (m, 1H), 1.95-1.86 (m, 1H), 1.81-1.72 (m, 1H), 1.64-1.60 (m, 1H) 1.25-1.15 (m, 1H), 1.10- 1.00 (m, 3H); MS m/z: 404 [M+H] + .
실시예 18: 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-6-메틸피리미딘-2-일}아미노)페닐]피페리딘-3-올Example 18: 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-6-methylpyrimidin-2-yl}amino)phenyl] piperidin-3-ol

실시예 15의 단계 1에서 2,4-디클로로피리미딘 대신에 2,4-디클로로-6-메틸피리미딘을 사용한 것을 제외하고, 실시예 15와 동일한 방법으로 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-6-메틸피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 18의 화합물; 8.48%)을 백색 고체로 수득하였다. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.83 (s, 1H), 7.60 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 5.86 (s, 1H), 5.57-5.54 (m, 1H), 5.48-5.41 (m, 1H), 4.76 (d, J = 4.8 Hz, 1H), 3.80-3.66 (m, 4H), 3.61-3.54 (m, 1H), 3.48-3.44 (m, 1H), 3.35-3.32 (m, 1H), 2.55-2.54 (m, 1H), 2.41-2.35 (m, 1H), 2.16 (s, 3H), 1.89-1.87 (m, 1H), 1.86-1.70 (m, 1H), 1.57-1.48 (m, 1H), 1.25-1.16 (m, 1H); MS m/z: 390 [M+H]+. In the same manner as in Example 15, 1-[4-({4-[ (3S,4S)-3,4-difluoropyrrolidin-1-yl]-6-methylpyrimidin-2-yl}amino)phenyl]piperidin-3-ol (compound of Example 18; 8.48%) as a white solid. 1 H-NMR (400 MHz, DMSO-d 6 ) δ (ppm): 8.83 (s, 1H), 7.60 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 5.86 (s, 1H), 5.57-5.54 (m, 1H), 5.48-5.41 (m, 1H), 4.76 (d, J = 4.8 Hz, 1H), 3.80-3.66 (m, 4H), 3.61-3.54 (m , 1H), 3.48-3.44 (m, 1H), 3.35-3.32 (m, 1H), 2.55-2.54 (m, 1H), 2.41-2.35 (m, 1H), 2.16 (s, 3H), 1.89-1.87 (m, 1H), 1.86-1.70 (m, 1H), 1.57-1.48 (m, 1H), 1.25-1.16 (m, 1H); MS m/z: 390 [M+H] + .
실시예 19: 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-6-에틸피리미딘-2-일}아미노)페닐]피페리딘-3-올Example 19: 1-[4-({4-[(3S,4S)-3,4-difluoropyrrolidin-1-yl]-6-ethylpyrimidin-2-yl}amino)phenyl] piperidin-3-ol

실시예 15의 단계 1에서 2,4-디클로로피리미딘 대신에 2,4-디클로로-6-에틸피리미딘을 사용한 것을 제외하고, 실시예 15와 동일한 방법으로 1-[4-({4-[(3S,4S)-3,4-디플루오로피롤리딘-1-일]-6-에틸피리미딘-2-일}아미노)페닐]피페리딘-3-올(실시예 19의 화합물; 12.24%)을 백색 고체로 수득하였다. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.81 (s, 1H), 7.63-7.59 (m, 2H), 6.84-6.80 (m, 2H), 5.86 (s, 1H), 5.57-5.56 (m, 1H), 5.48-5.44 (m, 1H), 4.76 (d, J = 4.8 Hz, 1H), 3.85-3.76 (m, 3H), 3.75-3.67 (m, 1H), 3.60-3.56 (m, 1H), 3.48-3.44 (m, 1H), 3.35-3.32 (m, 1H), 2.55-2.51 (m, 1H), 2.49-2.35 (m, 3H), 1.89-1.85 (m, 1H), 1.74-1.70 (m, 1H), 1.54-1.51 (m, 1H), 1.23-1.17 (m, 4H); 404 [M+H]+.In the same manner as in Example 15, 1-[4-({4-[ (3S,4S)-3,4-difluoropyrrolidin-1-yl]-6-ethylpyrimidin-2-yl}amino)phenyl]piperidin-3-ol (compound of Example 19; 12.24%) as a white solid. 1 H-NMR (400 MHz, DMSO-d 6 ) δ (ppm): 8.81 (s, 1H), 7.63-7.59 (m, 2H), 6.84-6.80 (m, 2H), 5.86 (s, 1H), 5.57-5.56 (m, 1H), 5.48-5.44 (m, 1H), 4.76 (d, J = 4.8 Hz, 1H), 3.85-3.76 (m, 3H), 3.75-3.67 (m, 1H), 3.60- 3.56 (m, 1H), 3.48-3.44 (m, 1H), 3.35-3.32 (m, 1H), 2.55-2.51 (m, 1H), 2.49-2.35 (m, 3H), 1.89-1.85 (m, 1H) ), 1.74-1.70 (m, 1H), 1.54-1.51 (m, 1H), 1.23-1.17 (m, 4H); 404 [M+H] + .
실시예 20: 5-((4-((3R,4R)-3-메톡시-4-(메틸아미노)피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온Example 20: 5-((4-((3R,4R)-3-methoxy-4-(methylamino)pyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidine-2- yl)amino)-2-methylisoindolin-1-one

실시예 20의 화합물을 하기 반응식 7에 따라서 제조하였다.The compound of Example 20 was prepared according to Scheme 7 below.
[반응식 7][Scheme 7]

단계 1: (3R,4R)-N-벤질-1-(2-클로로-5-(트리플루오로메틸)피리미딘-4-일)-4-메톡시-N-메틸피롤리딘-3-아민(화합물 7-1)의 합성Step 1: (3R,4R)-N-benzyl-1-(2-chloro-5-(trifluoromethyl)pyrimidin-4-yl)-4-methoxy-N-methylpyrrolidine-3- Synthesis of amines (Compound 7-1)
제조예 9에서 수득된 화합물 5의 광학 이성질체 혼합물(화합물 5-1; 520 mg, 1.56 mmol, 1당량, TFA) 및 화합물 6(337.47 mg, 1.56 mmol, 1 당량)을 DMF(5 mL) 중에 혼합하고 DIPEA(402.02 mg, 3.11 mmol, 541.81 uL, 2당량)를 첨가하였다. 혼합물을 20℃에서 12시간 동안 교반하였다. 혼합물에 에틸 아세테이트(50 mL)를 가하였다. 유기상을 모아서 염수(60 mLx3)로 세척하고, 무수 Na2SO4로 건조시키고 진공에서 여과 및 농축시켰다. 잔사는 prep-HPLC(방법: 물(0.1% TFA-ACN; 컬럼: phenomenex Luna C18 100^30 mm*5 ㎛)으로 정제하여 (3R,4R)-N-벤질-1-(2-클로로-5-(트리플루오로메틸)피리미딘-4-일)-4-메톡시-N-메틸피롤리딘-3-아민(화합물 7-1)(150 mg, 24.1%)을 광학 이성질체의 혼합물로서 황색 오일의 형태로 수득하였다. 1H NMR(400 MHz, CDCl3)δ ppm 8.53 (s, 1 H), 7.49 (br s, 3 H), 7.47 - 7.48 (m, 2 H), 4.71 (d, J = 6.4 Hz, 1 H) 4.38 - 4.51 (m, 2 H), 4.21 - 4.34 (m, 2 H), 4.06 - 4.20 (m, 1 H), 3.66 - 3.81 (m, 2 H), 3.45 - 3.49 (m, 3 H), 2.76 - 2.81 (m, 3 H); LCMS: RT = 0.823분, MS (ESI) m/z = 401.1 [M+H]+.Mixture of optical isomers of compound 5 obtained in Preparation 9 (compound 5-1; 520 mg, 1.56 mmol, 1 equiv, TFA) and compound 6 (337.47 mg, 1.56 mmol, 1 equiv) in DMF (5 mL) and DIPEA (402.02 mg, 3.11 mmol, 541.81 uL, 2 eq) was added. The mixture was stirred at 20° C. for 12 h. To the mixture was added ethyl acetate (50 mL). The combined organic phases were washed with brine (60 mLx3), dried over anhydrous Na 2 SO 4 , filtered and concentrated in vacuo. The residue was purified by prep-HPLC (method: water (0.1% TFA-ACN; column: phenomenex Luna C18 100^30 mm*5 µm)) (3R,4R)-N-benzyl-1-(2-chloro-5) -(trifluoromethyl)pyrimidin-4-yl)-4-methoxy-N-methylpyrrolidin-3-amine (compound 7-1) (150 mg, 24.1%) yellow as a mixture of optical isomers Obtained in the form of an oil: 1 H NMR (400 MHz, CDCl 3 )δ ppm 8.53 (s, 1 H), 7.49 (br s, 3 H), 7.47 - 7.48 (m, 2 H), 4.71 (d, J = 6.4 Hz, 1 H) 4.38 - 4.51 (m, 2 H), 4.21 - 4.34 (m, 2 H), 4.06 - 4.20 (m, 1 H), 3.66 - 3.81 (m, 2 H), 3.45 - 3.49 (m, 3 H), 2.76 - 2.81 (m, 3 H) LCMS: RT = 0.823 min, MS (ESI) m/z = 401.1 [M+H] + .
단계 2: 5-((4-((3R,4R)-3-(벤질(메틸)아미노)-4-메톡시피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온(화합물 8-1)의 합성Step 2: 5-((4-((3R,4R)-3-(benzyl(methyl)amino)-4-methoxypyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidine-2 Synthesis of -yl)amino)-2-methylisoindolin-1-one (Compound 8-1)
상기 단계 1에서 수득된 광학 이성질체 혼합물(160.00 mg, 399.18 umol, 1당량) 및 화합물 2A(97.11 mg, 598.77 μmol, 1.5당량)를 디옥산(8 mL) 중에 혼합하고, XPhos(38.06 mg, 79.84 μmol, 0.2당량), Cs2CO3(260.12 mg, 798.35 μmol, 2 당량), 및 Pd2(dba)3(36.55 mg, 39.92 μmol, 0.1 당량)을 한번에 20℃에 N2 하에서 가하였다. 혼합물을 110℃로 가온하고 12시간 동안 N2 하에서 교반하였다. 혼합물을 20℃로 냉각시킨 후 에틸 아세테이트로 희석하였다. 혼합물을 진공에서 여과 및 농축하고 잔사를 얻었다. 잔사는 prep-TLC(SiO2, 석유 에테르:에틸 아세테이트 = 1:1)을 정제하여 5-((4-((3R,4R)-3-(벤질(메틸)아미노)-4-메톡시피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온(화합물 8-1)(40 mg, 19.0%)을 광학 이성질체의 혼합물로서 황색 고체의 형태로 얻었다. 1H NMR(400 MHz, CDCl3) δ ppm 8.22 (s, 1 H), 7.87 - 7.69 (m, 2 H), 7.66 - 7.49 (m, 1 H), 7.26 - 7.15 (m, 4 H), 6.89 (br s, 1 H), 4.35 - 4.10 (m, 2 H), 4.07 - 3.86 (m, 2 H), 3.85 - 3.66 (m, 2 H), 3.64 - 3.45 (m, 3 H), 3.34 (s, 3 H), 3.30 - 3.21 (m, 1 H), 3.17 - 3.02 (m, 3 H), 2.19 (d, J = 4.0 8 Hz, 3 H); LCMS: RT = 1.280 min, MS (ESI) m/z = 527.2 [M+H]+.The optical isomer mixture obtained in step 1 above (160.00 mg, 399.18 umol, 1 equiv) and compound 2A (97.11 mg, 598.77 µmol, 1.5 equiv) were mixed in dioxane (8 mL), XPhos (38.06 mg, 79.84 µmol) , 0.2 eq), Cs 2 CO 3 (260.12 mg, 798.35 μmol, 2 eq), and Pd 2 (dba) 3 (36.55 mg, 39.92 μmol, 0.1 eq) were added in one portion at 20° C. under N 2 . The mixture was warmed to 110° C. and stirred under N 2 for 12 h. The mixture was cooled to 20° C. and then diluted with ethyl acetate. The mixture was filtered and concentrated in vacuo to give a residue. The residue was purified by prep-TLC (SiO 2 , petroleum ether:ethyl acetate = 1:1) to obtain 5-((4-((3R,4R)-3-(benzyl(methyl)amino)-4-methoxypyrroly) Din-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-2-methylisoindolin-1-one (compound 8-1) (40 mg, 19.0%) optically Obtained in the form of a yellow solid as a mixture of isomers. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 8.22 (s, 1 H), 7.87 - 7.69 (m, 2 H), 7.66 - 7.49 (m, 1 H), 7.26 - 7.15 (m, 4 H), 6.89 (br s, 1 H), 4.35 - 4.10 (m, 2 H), 4.07 - 3.86 (m, 2 H), 3.85 - 3.66 (m, 2 H), 3.64 - 3.45 (m, 3 H), 3.34 (s, 3 H), 3.30 - 3.21 (m, 1 H), 3.17 - 3.02 (m, 3 H), 2.19 (d, J = 4.0 8 Hz, 3 H); LCMS: RT = 1.280 min, MS (ESI) m/z = 527.2 [M+H] + .
단계 3: 5-((4-((3R,4R)-3-메톡시-4-(메틸아미노)피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온의 합성Step 3: 5-((4-((3R,4R)-3-methoxy-4-(methylamino)pyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl Synthesis of )amino)-2-methylisoindolin-1-one
상기 단계 2에서 수득된 광학 이성질체의 혼합물(20 mg, 37.98 μmol, 1당량)을 THF(2 mL) 중에 혼합하고 Pd(OH)2/C (10.67 mg, 7.60 μmol, 10% 순도, 0.2당량)를 가하였다. 현탁액을 진공 하에서 탈기하고 H2로 3회 퍼징하였다. 혼합물을 H2(15psi) 하에서 20℃에서 1 시간 동안 교반하였다. 반응 혼합물을 여과하고 여과물을 농축하였다. 잔사를 prep-TLC(SiO2, 디클로로메탄:메탄올 = 20:1)로 정제하여 5-((4-((3R,4R)-3-메톡시-4-(메틸아미노)피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온(실시예 20의 화합물; 5.4 mg, 31.0%)을 백색 고체로 수득하였다. 1H NMR(400 MHz, CDCl3)δ ppm 8.19 - 8.32 (m, 1 H), 7.72 - 7.86 (m, 2 H), 7.62 - 7.69 (m, 1 H), 6.91 (br s, 1 H), 4.28 - 4.42 (m, 2 H), 3.77 - 3.94 (m, 3 H), 3.56 - 3.72 (m, 2 H), 3.44 - 3.56 (m, 1 H), 3.40 (s, 3 H), 3.28 - 3.35 (m, 1 H), 3.14 - 3.23 (m, 3 H), 2.53 (d, J = 16.8 Hz, 3 H); LCMS: 생성물 RT = 0.716분, MS (ESI) m/z = 437.2 [M+H]+; HPLC: 순도 = 95.2% (면적 %); 키랄 SFC: ee% = 97.1% (면적 %).The mixture of optical isomers obtained in step 2 above (20 mg, 37.98 μmol, 1 eq) was mixed in THF (2 mL) and Pd(OH) 2 /C (10.67 mg, 7.60 μmol, 10% purity, 0.2 eq) was added. The suspension was degassed under vacuum and purged 3 times with H 2 . The mixture was stirred under H 2 (15 psi) at 20° C. for 1 h. The reaction mixture was filtered and the filtrate was concentrated. The residue was purified by prep-TLC (SiO 2 , dichloromethane:methanol = 20:1) to 5-((4-((3R,4R)-3-methoxy-4-(methylamino)pyrrolidine-1) Obtained -yl)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-2-methylisoindolin-1-one (compound of Example 20; 5.4 mg, 31.0%) as a white solid did. 1 H NMR (400 MHz, CDCl 3 )δ ppm 8.19 - 8.32 (m, 1 H), 7.72 - 7.86 (m, 2 H), 7.62 - 7.69 (m, 1 H), 6.91 (br s, 1 H) , 4.28 - 4.42 (m, 2 H), 3.77 - 3.94 (m, 3 H), 3.56 - 3.72 (m, 2 H), 3.44 - 3.56 (m, 1 H), 3.40 (s, 3 H), 3.28 - 3.35 (m, 1 H), 3.14 - 3.23 (m, 3 H), 2.53 (d, J = 16.8 Hz, 3 H); LCMS: product RT = 0.716 min, MS (ESI) m/z = 437.2 [M+H] + ; HPLC: purity = 95.2% (area %); Chiral SFC: ee% = 97.1% (area %).
실시예 21: 5-((4-((3S,4S)-3,4-디플루오로피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온Example 21: 5-((4-((3S,4S)-3,4-difluoropyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl)amino) -2-methylisoindolin-1-one

실시예 21의 화합물을 하기 반응식 8에 따라서 제조하였다.The compound of Example 21 was prepared according to Scheme 8 below.
[반응식 8][Scheme 8]

단계 1: 5-((4-클로로-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온의 합성Step 1: Synthesis of 5-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-2-methylisoindolin-1-one
5-아미노-2-메틸이소인돌린-1-온 및 5-트리플루오로메틸-2,4-디클로로피리미딘을 실시예 20의 단계 2와 동일한 방법으로 반응시켜서 5-((4-클로로-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온을 백색 고체 (78.9 mg, 0.20 mmol, 43 % 수율)로 수득하였다. 1H NMR (400 MHz, DMSO- d6); δ 8.87 (s, 1H), 8.00 (s, 1H), 7.72 (d, J = 8.4 Hz, 1H), 6.64 (d, J = 8.4 Hz, 1H), 4.45 (s, 2H), 3.05 (s, 3H).5-((4-chloro- 5-(trifluoromethyl)pyrimidin-2-yl)amino)-2-methylisoindolin-1-one was obtained as a white solid (78.9 mg, 0.20 mmol, 43 % yield). 1H NMR (400 MHz, DMSO-d 6 ); δ 8.87 (s, 1H), 8.00 (s, 1H), 7.72 (d, J = 8.4 Hz, 1H), 6.64 (d, J = 8.4 Hz, 1H), 4.45 (s, 2H), 3.05 (s, 3H).
단계 2: 5-((4-((3S,4S)-3,4-디플루오로피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온의 합성Step 2: 5-((4-((3S,4S)-3,4-difluoropyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- Synthesis of 2-methylisoindolin-1-one
단계 1에서 수득된 화합물 및 (3S,4S)-3,4-디플루오로피롤리딘을 실시예 20의 단계 1과 동일한 방법으로 반응시켜서 5-((4-((3S,4S)-3,4-디플루오로피롤리딘-1-일)-5-(트리플루오로메틸)피리미딘-2-일)아미노)-2-메틸이소인돌린-1-온(실시예 21의 화합물; 25.6 mg, 0.06 mmol, 29%)을 백색 분말로 수득하였다. 1H NMR (400 MHz, DMSO- d6); δ 10.10 (s, 1H), 8.47 (s, 1H), 8.03 (s, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 5.58 (brs, 1H), 5.44 (brs, 1H), 4.44 (s, 2H), 4.05-3.93 (m, 4H), 3.05 (s, 3H); MS m/z 414 [M+H]+
.
The compound obtained in step 1 and (3S,4S)-3,4-difluoropyrrolidine were reacted in the same manner as in step 1 of Example 20 to 5-((4-((3S,4S)-3 ,4-Difluoropyrrolidin-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-2-methylisoindolin-1-one (compound of Example 21; 25.6 mg, 0.06 mmol, 29%) were obtained as a white powder. 1H NMR (400 MHz, DMSO-d 6 ); δ 10.10 (s, 1H), 8.47 (s, 1H), 8.03 (s, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 5.58 (brs, 1H), 5.44 (brs, 1H), 4.44 (s, 2H), 4.05-3.93 (m, 4H), 3.05 (s, 3H); MS m/z 414 [M+H] + .
실험예 1. LRRK2 효소 억제 효과의 확인Experimental Example 1. Confirmation of LRRK2 enzyme inhibitory effect
실시예에서 제조된 화합물의 LRRK2 효소 저해 활성이 있는지 여부를 확인하였다. LRRK2 효소 저해 활성은 Nat Biotechnol. 2011 Oct 30;29(11):1039-45에 기재된 방법에 따라 수행하였다.It was confirmed whether the compound prepared in Examples had LRRK2 enzyme inhibitory activity. LRRK2 enzyme inhibitory activity was determined by Nat Biotechnol. 2011 Oct 30;29(11):1039-45.
화합물을 100% DMSO에 용해시켜 스톡 용액을 준비하였다. 50% 저해 농도(half maximal inhibitory concentration: IC50)을 결정하기 위해, 화합물을 최고 농도 10 μM로 하고, 3배씩 희석하여 10가지 농도로 준비하였다.A stock solution was prepared by dissolving the compound in 100% DMSO. In order to determine the 50% inhibitory concentration (half maximal inhibitory concentration: IC 50 ), the compound was prepared at a maximum concentration of 10 μM and diluted 3 times to 10 concentrations.
LRRK2 G2019S 효소(Invitrogen, PR8764C)와 기질로서 LRRKtide(SignalChem, L10-58)를 준비하였다. 30 nM LRRK2(G2019S), 20 μM LRRKtide, 10 μM ATP, 및 반응 완충액(20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.01%(v/v) Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, 및 1%(w/v) DMSO)을 준비하였다.LRRK2 G2019S enzyme (Invitrogen, PR8764C) and LRRKtide (SignalChem, L10-58) as a substrate were prepared. 30 nM LRRK2 (G2019S), 20 μM LRRKtide, 10 μM ATP, and reaction buffer (20 mM Hepes (pH 7.5), 10 mM MgCl 2 , 1 mM EGTA, 0.01% (v/v) Brij35, 0.02 mg/mL BSA) , 0.1 mM Na 3 VO 4 , 2 mM DTT, and 1% (w/v) DMSO) were prepared.
신선한 반응 완충액에 20 μM LRRKtide을 가하고, 30 nM LRRK2(G2019S) 효소를 가한 후 부드럽게 혼합하였다. 준비된 화합물(100% DMSO 중)을 Acoustic technology (Echo550; 나노 수준)를 사용하여 LRRK2(G2019S) 혼합물에 가하였다. 반응 혼합물에 33P-ATP를 가하여 반응을 개시한 후, 실온에서 2시간 동안 인큐베이션하였다. P81 필터-결합 방법으로 LRRK2(G2019S)에 대한 IC50를 산출하고, 그 결과를 하기 표 1의 기준으로 평가하여 표 2에 나타내었다.20 μM LRRKtide was added to fresh reaction buffer, 30 nM LRRK2 (G2019S) enzyme was added, and then gently mixed. The prepared compound (in 100% DMSO) was added to the LRRK2 (G2019S) mixture using Acoustic technology (Echo550; nano level). 33 P-ATP was added to the reaction mixture to initiate the reaction, followed by incubation at room temperature for 2 hours. The IC 50 for LRRK2 (G2019S) was calculated by the P81 filter-binding method, and the results were evaluated based on the criteria in Table 1 below, and are shown in Table 2.
| IC50(LRRK2 G2019S)IC 50 (LRRK2 G2019S) | |||
| 기준(IC50)Criterion (IC 50 ) | <50nM<50nM | 50-500nM50-500nM | >500nM>500nM |
| 표시값display value | ++++++ | ++++ | ++ |
| LRRK2 길항 활성(IC50, nM)LRRK2 antagonist activity (IC 50 , nM) | |
| 실시예 1의 화합물compound of example 1 | ++++ |
| 실시예 2의 화합물compound of example 2 | -- |
| 실시예 3의 화합물compound of example 3 | ++++ |
| 실시예 4의 화합물compound of example 4 | ++++ |
| 실시예 6의 화합물compound of example 6 | ++ |
| 실시예 7의 화합물compound of example 7 | ++ |
| 실시예 8의 화합물compound of example 8 | ++++++ |
| 실시예 9의 화합물compound of example 9 | ++++++ |
| 실시예 9(S)의 화합물Compound of Example 9(S) | ++++++ |
| 실시예 9(R)의 화합물compound of example 9(R) | ++++++ |
| 실시예 10의 화합물compound of example 10 | ++++++ |
| 실시예 11의 화합물compound of example 11 | -- |
| 실시예 12의 화합물compound of example 12 | -- |
| 실시예 13의 화합물compound of example 13 | ++ |
| 실시예 14의 화합물compound of example 14 | ++ |
| 실시예 15의 화합물compound of example 15 | ++ |
| 실시예 16의 화합물compound of example 16 | ++++ |
| 실시예 17의 화합물compound of example 17 | ++++ |
| 실시예 18의 화합물compound of example 18 | -- |
| 실시예 19의 화합물compound of example 19 | ++ |
| 실시예 20의 화합물compound of example 20 | ++++ |
| 실시예 21의 화합물compound of example 21 | ++++++ |
표 2의 실험 결과로부터 본원 화합물이 우수한 LRRK2 길항 활성을 나타냄을 확인할 수 있었다. From the experimental results in Table 2, it was confirmed that the present compound exhibited excellent LRRK2 antagonistic activity.
Claims (16)
- 하기 화학식 1로 표시되는 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염:A compound represented by the following formula (1), a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof:[화학식 1][Formula 1]
 상기 화학식 1에서,In Formula 1,R1 및 R2는 각각 독립적으로 H, 할로겐, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐으로 치환된 C1-6 알킬, C1-6 알콕시, 또는 할로겐으로 치환된 C1-6 알콕시이거나, 또는 R1 및 R2는 함께 치환 또는 비치환된 4원 내지 7원 포화 또는 부분 불포화 카보사이클릴 고리, 또는 1개 내지 3개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로아릴 또는 헤테로사이클릴 고리를 형성하고;R 1 and R 2 are each independently H, halogen, OH, CN, amino, nitro, C 1-6 alkyl, halogen substituted C 1-6 alkyl, C 1-6 alkoxy, or halogen substituted C 1 -6 alkoxy, or R 1 and R 2 together are substituted or unsubstituted 4-7 membered saturated or partially unsaturated carbocyclyl ring, or substituted or unsubstituted 4 membered containing 1 to 3 nitrogen atoms to 7 membered heteroaryl or heterocyclyl ring;R3은 -NR31R32 또는 치환 또는 비치환된 C3-8 사이클로알킬이고,R 3 is —NR 31 R 32 or substituted or unsubstituted C 3-8 cycloalkyl,R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 임의로 1개의 질소 또는 산소 원자를 포함하는 치환 또는 비치환된 3원 내지 12원 포화 또는 부분 불포화 카보사이클릴 또는 헤테로사이클릴 고리이되, 상기 카보사이클릴 또는 헤테로사이클릴 고리는 임의로 가교되거나 스피로 고리를 형성할 수 있거나, 또는 any one of R 31 and R 32 is H and the other is a substituted or unsubstituted 3- to 12-membered saturated or partially unsaturated carbocyclyl or heterocyclyl ring optionally containing 1 nitrogen or oxygen atom, wherein The carbocyclyl or heterocyclyl ring may optionally be bridged or form a spiro ring, orR31 및 R32는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 3원 내지 10원 모노사이클릭 또는 바이사이클릭 헤테로사이클릴 고리를 형성할 수 있고;R 31 and R 32 together with the nitrogen atom to which they are attached may form a substituted or unsubstituted 3- to 10-membered monocyclic or bicyclic heterocyclyl ring containing 1 or 2 nitrogen atoms;R4는 고리 X이고 R5는 H이거나, 또는R 4 is ring X and R 5 is H, orR4 및 R5는 이들이 결합한 벤젠 고리의 탄소 원자와 함께 *-CO-NR6-CH2-기로 연결되어 5원 락탐 고리를 형성할 수 있되, *은 R4가 벤젠 고리에 결합된 위치를 나타내며, R 4 and R 5 may be connected to a *-CO-NR 6 -CH 2 - group together with a carbon atom of the benzene ring to which they are attached to form a 5-membered lactam ring, wherein * represents the position at which R 4 is bonded to the benzene ring. indicate,고리 X는 -NRX1RX2 또는 치환 또는 비치환된 C3-8 사이클로알킬이되, RX1 및 RX2는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 3원 내지 10원 모노사이클릭 또는 바이사이클릭 헤테로사이클릴 고리를 형성하고,Ring X is -NR X1 R X2 or substituted or unsubstituted C 3-8 cycloalkyl, wherein R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted containing 1 or 2 nitrogen atoms to form a 3- to 10-membered monocyclic or bicyclic heterocyclyl ring,R6은 H, C1-6 알킬, 또는 할로겐 원자로 치환된 C1-6 알킬이고;R 6 is H, C 1-6 alkyl, or C 1-6 alkyl substituted with a halogen atom;상기 ”치환”은 각각 독립적으로 할로겐, 옥소, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐 또는 하이드록시로 치환된 C1-6 알킬, C1-6 알콕시, 할로겐으로 치환된 C1-6 알콕시, C2-6의 알콕시알킬, C1-6 알킬아미노 및 디-(C1-6 알킬)-아미노로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 치환됨을 의미하되;The "substitution" is each independently halogen, oxo, OH, CN, amino, nitro, C 1-6 alkyl, halogen or hydroxy substituted C 1-6 alkyl, C 1-6 alkoxy, halogen substituted C means substituted with one or more substituents selected from the group consisting of 1-6 alkoxy, C 2-6 alkoxyalkyl, C 1-6 alkylamino and di-(C 1-6 alkyl)-amino;다만, R3이 -NR31R32이고, R31 및 R32가 치환 또는 비치환된 3원 내지 10원 헤테로사이클릴 고리를 형성하고, R4가 고리 X이고, 고리 X가 -NRX1RX2인 경우, R1 및 R2 중 어느 하나가 H이면, R1 및 R2 중 나머지 하나는 할로겐으로 치환된 C1-6 알킬이 아니다.provided that R 3 is —NR 31 R 32 , R 31 and R 32 form a substituted or unsubstituted 3- to 10-membered heterocyclyl ring, R 4 is Ring X, and Ring X is —NR X1 R In case of X2 , if any one of R 1 and R 2 is H, the other of R 1 and R 2 is not C 1-6 alkyl substituted with halogen.
상기 화학식 1에서,In Formula 1,R1 및 R2는 각각 독립적으로 H, 할로겐, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐으로 치환된 C1-6 알킬, C1-6 알콕시, 또는 할로겐으로 치환된 C1-6 알콕시이거나, 또는 R1 및 R2는 함께 치환 또는 비치환된 4원 내지 7원 포화 또는 부분 불포화 카보사이클릴 고리, 또는 1개 내지 3개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로아릴 또는 헤테로사이클릴 고리를 형성하고;R 1 and R 2 are each independently H, halogen, OH, CN, amino, nitro, C 1-6 alkyl, halogen substituted C 1-6 alkyl, C 1-6 alkoxy, or halogen substituted C 1 -6 alkoxy, or R 1 and R 2 together are substituted or unsubstituted 4-7 membered saturated or partially unsaturated carbocyclyl ring, or substituted or unsubstituted 4 membered containing 1 to 3 nitrogen atoms to 7 membered heteroaryl or heterocyclyl ring;R3은 -NR31R32 또는 치환 또는 비치환된 C3-8 사이클로알킬이고,R 3 is —NR 31 R 32 or substituted or unsubstituted C 3-8 cycloalkyl,R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 임의로 1개의 질소 또는 산소 원자를 포함하는 치환 또는 비치환된 3원 내지 12원 포화 또는 부분 불포화 카보사이클릴 또는 헤테로사이클릴 고리이되, 상기 카보사이클릴 또는 헤테로사이클릴 고리는 임의로 가교되거나 스피로 고리를 형성할 수 있거나, 또는 any one of R 31 and R 32 is H and the other is a substituted or unsubstituted 3- to 12-membered saturated or partially unsaturated carbocyclyl or heterocyclyl ring optionally containing 1 nitrogen or oxygen atom, wherein The carbocyclyl or heterocyclyl ring may optionally be bridged or form a spiro ring, orR31 및 R32는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 3원 내지 10원 모노사이클릭 또는 바이사이클릭 헤테로사이클릴 고리를 형성할 수 있고;R 31 and R 32 together with the nitrogen atom to which they are attached may form a substituted or unsubstituted 3- to 10-membered monocyclic or bicyclic heterocyclyl ring containing 1 or 2 nitrogen atoms;R4는 고리 X이고 R5는 H이거나, 또는R 4 is ring X and R 5 is H, orR4 및 R5는 이들이 결합한 벤젠 고리의 탄소 원자와 함께 *-CO-NR6-CH2-기로 연결되어 5원 락탐 고리를 형성할 수 있되, *은 R4가 벤젠 고리에 결합된 위치를 나타내며, R 4 and R 5 may be connected to a *-CO-NR 6 -CH 2 - group together with a carbon atom of the benzene ring to which they are attached to form a 5-membered lactam ring, wherein * represents the position at which R 4 is bonded to the benzene ring. indicate,고리 X는 -NRX1RX2 또는 치환 또는 비치환된 C3-8 사이클로알킬이되, RX1 및 RX2는 이들이 결합한 질소 원자와 함께, 1개 또는 2개의 질소 원자를 포함하는 치환 또는 비치환된 3원 내지 10원 모노사이클릭 또는 바이사이클릭 헤테로사이클릴 고리를 형성하고,Ring X is -NR X1 R X2 or substituted or unsubstituted C 3-8 cycloalkyl, wherein R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted containing 1 or 2 nitrogen atoms to form a 3- to 10-membered monocyclic or bicyclic heterocyclyl ring,R6은 H, C1-6 알킬, 또는 할로겐 원자로 치환된 C1-6 알킬이고;R 6 is H, C 1-6 alkyl, or C 1-6 alkyl substituted with a halogen atom;상기 ”치환”은 각각 독립적으로 할로겐, 옥소, OH, CN, 아미노, 니트로, C1-6 알킬, 할로겐 또는 하이드록시로 치환된 C1-6 알킬, C1-6 알콕시, 할로겐으로 치환된 C1-6 알콕시, C2-6의 알콕시알킬, C1-6 알킬아미노 및 디-(C1-6 알킬)-아미노로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 치환됨을 의미하되;The "substitution" is each independently halogen, oxo, OH, CN, amino, nitro, C 1-6 alkyl, halogen or hydroxy substituted C 1-6 alkyl, C 1-6 alkoxy, halogen substituted C means substituted with one or more substituents selected from the group consisting of 1-6 alkoxy, C 2-6 alkoxyalkyl, C 1-6 alkylamino and di-(C 1-6 alkyl)-amino;다만, R3이 -NR31R32이고, R31 및 R32가 치환 또는 비치환된 3원 내지 10원 헤테로사이클릴 고리를 형성하고, R4가 고리 X이고, 고리 X가 -NRX1RX2인 경우, R1 및 R2 중 어느 하나가 H이면, R1 및 R2 중 나머지 하나는 할로겐으로 치환된 C1-6 알킬이 아니다.provided that R 3 is —NR 31 R 32 , R 31 and R 32 form a substituted or unsubstituted 3- to 10-membered heterocyclyl ring, R 4 is Ring X, and Ring X is —NR X1 R In case of X2 , if any one of R 1 and R 2 is H, the other of R 1 and R 2 is not C 1-6 alkyl substituted with halogen. - 제1항에 있어서,According to claim 1,R3은 -NR31R32 또는 치환 또는 비치환된 C3-6 사이클로알킬이고,R 3 is —NR 31 R 32 or substituted or unsubstituted C 3-6 cycloalkyl,R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 임의로 1개의 산소 원자를 포함하는 치환 또는 비치환된 5원 내지 8원 포화 또는 부분 불포화 카보사이클릴 또는 헤테로사이클릴 고리이되, 상기 카보사이클릴 또는 헤테로사이클릴 고리는 임의로 가교되거나 스피로 고리를 형성할 수 있거나, 또는 any one of R 31 and R 32 is H and the other is a substituted or unsubstituted 5-8 membered saturated or partially unsaturated carbocyclyl or heterocyclyl ring optionally containing 1 oxygen atom, wherein the carbocycle the ryl or heterocyclyl ring may optionally be bridged or form a spiro ring, orR31 및 R32는 이들이 결합한 질소 원자와 함께 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성할 수 있고;R 31 and R 32 together with the nitrogen atom to which they are attached may form a substituted or unsubstituted 4-7 membered heterocyclyl ring containing 1 nitrogen atom;상기 R3, R31 및 R32의 정의에서 “치환”은 각각 독립적으로 할로겐, OH, CN, 아미노, C1-4 알킬, C1-4 알콕시, C1-4 알킬아미노 및 디-(C1-4 알킬)아미노로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 치환됨을 의미하는, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염.In the above definitions of R 3 , R 31 and R 32 “substitution” is each independently halogen, OH, CN, amino, C 1-4 alkyl, C 1-4 alkoxy, C 1-4 alkylamino and di-(C A compound, a stereoisomer, a solvate or a pharmaceutically acceptable salt thereof, which means substituted with one or more substituents selected from the group consisting of 1-4 alkyl)amino.
- 제2항에 있어서,3. The method of claim 2,R31 및 R32 중 어느 하나는 H이고, 나머지 하나는 사이클로헥실, 사이클로헵틸, 사이클로옥틸, 옥사사이클로헥실, 옥사사이클로헵틸, 옥사사이클로옥틸, 바이사이클로헥산일, 바이사이클로헵탄일, 바이사이클로옥탄일, 옥사바이사이클로헥산일, 옥사바이사이클로헵탄일, 옥사바이사이클로옥탄일, 스피로헥산일, 스피로헵탄일, 스피로옥탄일, 옥사스피로헥산일, 옥사스피로헵탄일 및 옥사스피로옥탄일로 이루어진 군에서 선택되거나, 또는 any one of R 31 and R 32 is H, and the other is cyclohexyl, cycloheptyl, cyclooctyl, oxacyclohexyl, oxacycloheptyl, oxacyclooctyl, bicyclohexanyl, bicycloheptanyl, bicyclooctanyl , oxabicyclohexanyl, oxabicycloheptanyl, oxabicyclooctanyl, spirohexanyl, spiroheptanyl, spirooctanyl, oxaspirohexanyl, oxaspiroheptanyl and oxaspirooctanyl , orR31 및 R32는 이들이 결합한 질소 원자와 함께 아제티딘일, 피롤리딘일 또는 피페리딘일을 형성할 수 있고;R 31 and R 32 together with the nitrogen atom to which they are attached may form azetidinyl, pyrrolidinyl or piperidinyl;상기 R31 및 R32는 각각 독립적으로 할로겐, OH, 아미노, C1-4 알콕시 및 C1-4 알킬아미노로 이루어진 군으로부터 선택되는 1개 또는 2개의 치환기로 임의로 치환되는, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염.wherein R 31 and R 32 are each independently optionally substituted with 1 or 2 substituents selected from the group consisting of halogen, OH, amino, C 1-4 alkoxy and C 1-4 alkylamino, a compound, a stereoisomer thereof , a solvate or a pharmaceutically acceptable salt.
- 제1항 내지 제3항 중 어느 한 항에 있어서,4. The method according to any one of claims 1 to 3,R1 및 R2는 각각 독립적으로 H, 할로겐, C1-4 알킬, 또는 할로겐으로 치환된 C1-4 알킬인, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염.R 1 and R 2 are each independently H, halogen, C 1-4 alkyl, or halogen-substituted C 1-4 alkyl, a compound, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof.
- 제4항에 있어서,5. The method of claim 4,R1 및 R2 중 어느 하나는 H이고, 나머지 하나는 할로겐, C1-4 알킬, 또는 할로겐으로 치환된 C1-4 알킬인, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염.wherein any one of R 1 and R 2 is H and the other is halogen, C 1-4 alkyl, or halogen-substituted C 1-4 alkyl, a stereoisomer, solvate or pharmaceutically acceptable salt thereof. .
- 제1항 내지 제3항 중 어느 한 항에 있어서, 4. The method according to any one of claims 1 to 3,R1 및 R2는 함께 5원 또는 6원 포화 또는 부분 불포화 카보사이클릴 고리, 또는 1개 또는 2개의 질소 원자를 포함하는 5원 또는 6원 헤테로아릴 또는 헤테로사이클릴 고리를 형성하는, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염.R 1 and R 2 taken together form a 5 or 6 membered saturated or partially unsaturated carbocyclyl ring, or a 5 or 6 membered heteroaryl or heterocyclyl ring comprising 1 or 2 nitrogen atoms, A stereoisomer, solvate or pharmaceutically acceptable salt thereof.
- 제1항 내지 제6항 중 어느 한 항에 있어서,7. The method according to any one of claims 1 to 6,고리 X는 -NRX1RX2 또는 치환 또는 비치환된 C3-6 사이클로알킬이되, RX1 및 RX2는 이들이 결합한 질소 원자와 함께, 1개의 질소 원자를 포함하는 치환 또는 비치환된 4원 내지 7원 헤테로사이클릴 고리를 형성하고;Ring X is -NR X1 R X2 or substituted or unsubstituted C 3-6 cycloalkyl, wherein R X1 and R X2 together with the nitrogen atom to which they are attached are substituted or unsubstituted 4 membered containing one nitrogen atom to 7 membered heterocyclyl ring;상기 고리 X의 정의에서 “치환”은 할로겐, 옥소, OH, C1-4 알킬, 할로겐으로 치환된 C1-4 알킬, 하이드록시로 치환된 C1-4 알킬, C1-4 알콕시 및 할로겐으로 치환된 C1-4 알콕시로 이루어진 군으로부터 선택되는 하나 이상의 치환기로 치환됨을 의미하는, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염.“Substitution” in the above definition of Ring X means halogen, oxo, OH, C 1-4 alkyl, halogen substituted C 1-4 alkyl, hydroxy substituted C 1-4 alkyl, C 1-4 alkoxy and halogen A compound, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof, which means substituted with one or more substituents selected from the group consisting of C 1-4 alkoxy substituted with .
- 제7항에 있어서,8. The method of claim 7,고리 X는 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 피롤리딘일, 피페리딘일 및 아제판일로 이루어진 군으로부터 선택되고;ring X is selected from the group consisting of cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyrrolidinyl, piperidinyl and azepanyl;상기 고리 X는 할로겐, 옥소, OH, C1-4 알킬 및 할로겐으로 치환된 C1-4 알킬 로 이루어진 군으로부터 선택되는 1개 또는 2개의 치환기로 임의로 치환되는, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염.wherein ring X is optionally substituted with 1 or 2 substituents selected from the group consisting of halogen, oxo, OH, C 1-4 alkyl and halogen-substituted C 1-4 alkyl, compound, stereoisomer, solvate thereof or a pharmaceutically acceptable salt.
- 제1항 내지 제5항 중 어느 한 항에 있어서, 6. The method according to any one of claims 1 to 5,R4 및 R5는 이들이 결합한 벤젠 고리의 탄소 원자와 함께 *-CO-NR6-CH2-기로 연결되어 5원 락탐 고리를 형성하되, *은 R4가 벤젠 고리에 결합된 위치를 나타내고, R6은 H 또는 C1-4 알킬인, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염.R 4 and R 5 are connected with a carbon atom of the benzene ring to which they are attached to a *-CO-NR 6 -CH 2 - group to form a 5-membered lactam ring, wherein * represents the position at which R 4 is bonded to the benzene ring, R 6 is H or C 1-4 alkyl, a stereoisomer, solvate or pharmaceutically acceptable salt thereof.
- 제1항 내지 제9항 중 어느 한 항에 있어서,10. The method according to any one of claims 1 to 9,하기 화합물로부터 이루어진 군으로부터 선택되는, 화합물, 이의 입체이성질체, 용매화물 또는 약학적으로 허용가능한 염:A compound, a stereoisomer, a solvate or a pharmaceutically acceptable salt thereof, selected from the group consisting of:;
 ;
; ;;;
;;; ;
; ;;;
;;; ;
; ;;;
;;; ;
; ;;;
;;; ;
; ;;;
;;; ;
; ;;;
;;; ;
; ;;;
;;; ;
; ;;;
;;; ;
; ;;;
;;; ;
; ;;;
;;; ; 및
; 및 .
. ;; and.
;; and. - 제1항 내지 제10항 중 어느 한 항에 따른 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염을 포함하는 LRRK2(Leucine-rich repeat kinase 2)에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료하기 위한 약학적 조성물.A disease or disorder mediated by or related to Leucine-rich repeat kinase 2 (LRRK2) comprising the compound according to any one of claims 1 to 10, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof A pharmaceutical composition for preventing or treating.
- 제11항에 있어서,12. The method of claim 11,상기 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환은 퇴행성 뇌질환인 것인 약학적 조성물.The disease or disorder mediated by or related to the LRRK2 is a degenerative brain disease pharmaceutical composition.
- 제12항에 있어서,13. The method of claim 12,상기 퇴행성 뇌질환은 파킨슨병(Parkinson's disease), 알츠하이머병(Alzheimer's disease), 헌팅턴병(Huntington's disease), 경도인지장애(mild cognitive impairment), 아밀로이드증(amyloidosis), 다계통위측증(Multiple system atrophy), 다발성경화증(multiple sclerosis), 타우병증(tauopathies), 픽병(Pick's disease), 노인성 치매, 근위축성 측삭 경화증(amyotrophic lateral sclerosis), 척수소뇌성 운동실조증(Spinocerebellar Atrophy), 뚜렛 증후군(Tourette's Syndrome), 프리드리히 보행실조(Friedrich's Ataxia), 마차도-조셉 병(Machado-Joseph's disease), 루이 소체 치매(Lewy Body Dementia), 근육긴장이상(Dystonia), 진행성 핵상 마비(Progressive Supranuclear Palsy) 및 전두측두엽 치매(Frontotemporal Dementia)로 이루어진 군에서 선택된 것인 약학적 조성물.The degenerative brain disease is Parkinson's disease, Alzheimer's disease, Huntington's disease, mild cognitive impairment, amyloidosis, multiple system atrophy, multiple Multiple sclerosis, tauopathies, Pick's disease, senile dementia, amyotrophic lateral sclerosis, Spinocerebellar Atrophy, Tourette's Syndrome, Friedrich's Syndrome, Friedrich's Ataxia, Machado-Joseph's disease, Lewy Body Dementia, Dystonia, Progressive Supranuclear Palsy and Frontotemporal Dementia A pharmaceutical composition selected from the group consisting of.
- 제1항 내지 제10항 중 어느 한 항에 따른 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염을 개체에게 투여하는 단계를 포함하는 LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료하는 방법.Prevention of a disease or disorder mediated by or related to LRRK2, comprising administering to a subject a compound according to any one of claims 1 to 10, a stereoisomer, solvate, or pharmaceutically acceptable salt thereof or how to treat it.
- LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환의 예방 또는 치료에 사용하기 위한 제1항 내지 제10항 중 어느 한 항에 따른 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염의 용도.Use of a compound according to any one of claims 1 to 10, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof, for use in the prophylaxis or treatment of a disease or condition mediated by or associated with LRRK2.
- LRRK2에 의해 매개되거나 이와 관련된 질병 또는 질환을 예방 또는 치료하기 위한 의약을 제조하기 위한 제1 내지 제10항 중 어느 한 항에 따른 화합물, 이의 입체이성질체, 용매화물, 또는 약학적으로 허용가능한 염의 용도.Use of a compound according to any one of claims 1 to 10, a stereoisomer, a solvate, or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for preventing or treating a disease or condition mediated by or related to LRRK2 .

Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020237025047A KR102585194B1 (en) | 2021-03-26 | 2022-03-25 | Novel phenylaminopyrimidine compounds with inhibitory activity against LRRK2 and their uses |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2021-0039634 | 2021-03-26 | ||
| KR20210039634 | 2021-03-26 | ||
| KR20210040137 | 2021-03-29 | ||
| KR10-2021-0040137 | 2021-03-29 | ||
| KR10-2021-0042666 | 2021-04-01 | ||
| KR20210042666 | 2021-04-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022203466A1 true WO2022203466A1 (en) | 2022-09-29 |
Family
ID=83397670
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2022/004259 WO2022203466A1 (en) | 2021-03-26 | 2022-03-25 | Novel phenylaminopyrimidine compound having inhibitory activity against lrrk2 and use thereof |
Country Status (2)
| Country | Link |
|---|---|
| KR (1) | KR102585194B1 (en) |
| WO (1) | WO2022203466A1 (en) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070259904A1 (en) * | 2005-11-01 | 2007-11-08 | Targegen, Inc. | Bi-aryl meta-pyrimidine inhibitors of kinases |
| US20130029944A1 (en) * | 2008-04-22 | 2013-01-31 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| KR20180097162A (en) * | 2017-02-22 | 2018-08-30 | 국립암센터 | Pyrolo-pyrimidine derivatives, preparation method thereof, and pharmaceutical composition for use in preventing or treating protein kinase related disease as an active ingredient |
| KR20200110250A (en) * | 2019-03-13 | 2020-09-23 | 보로노이바이오 주식회사 | Heteroaryl derivatives, and pharmaceutical composition comprising the same |
| CN111732548A (en) * | 2020-06-11 | 2020-10-02 | 浙江大学 | N2-carbamyl aromatic ring-2-aminopyrimidine derivatives and medical application thereof |
-
2022
- 2022-03-25 WO PCT/KR2022/004259 patent/WO2022203466A1/en active Application Filing
- 2022-03-25 KR KR1020237025047A patent/KR102585194B1/en active IP Right Grant
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070259904A1 (en) * | 2005-11-01 | 2007-11-08 | Targegen, Inc. | Bi-aryl meta-pyrimidine inhibitors of kinases |
| US20130029944A1 (en) * | 2008-04-22 | 2013-01-31 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| KR20180097162A (en) * | 2017-02-22 | 2018-08-30 | 국립암센터 | Pyrolo-pyrimidine derivatives, preparation method thereof, and pharmaceutical composition for use in preventing or treating protein kinase related disease as an active ingredient |
| KR20200110250A (en) * | 2019-03-13 | 2020-09-23 | 보로노이바이오 주식회사 | Heteroaryl derivatives, and pharmaceutical composition comprising the same |
| CN111732548A (en) * | 2020-06-11 | 2020-10-02 | 浙江大学 | N2-carbamyl aromatic ring-2-aminopyrimidine derivatives and medical application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20230117756A (en) | 2023-08-09 |
| KR102585194B1 (en) | 2023-10-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2020377805B2 (en) | Melanocortin-4 receptor agonists | |
| WO2017026718A1 (en) | Novel 3-(isoxazol-3-yl)-pyrazolo[3,4-d]pyrimidin-4-amine compound, which is ret kinase inhibitor | |
| WO2022055181A1 (en) | Compounds for suppressing egfr mutant cancer and pharmaceutical use thereof | |
| WO2017142325A1 (en) | Novel 2,3,5-substituted thiophene compound as protein kinase inhibitor | |
| WO2023018155A1 (en) | Compound having shp2 protein degrading activity, and medical uses thereof | |
| WO2013081400A2 (en) | Novel benzamide derivative and use thereof | |
| WO2023017442A1 (en) | Novel plk1 degradation inducing compound | |
| EP3681877A1 (en) | Pyrazole derivative compound and use thereof | |
| WO2017034245A1 (en) | Janus kinase 1 selective inhibitor and pharmaceutical use thereof | |
| WO2018151562A2 (en) | Novel benzimidazole derivative having jnk inhibitory activity and use thereof | |
| AU2020360000B2 (en) | N-(1H-imidazol-2-yl)benzamide compound and pharmaceutical composition comprising the same as active ingredient | |
| WO2022216098A1 (en) | Novel lrrk2 inhibitor | |
| WO2022270987A1 (en) | Aurka selective degradation inducing compound | |
| WO2022203466A1 (en) | Novel phenylaminopyrimidine compound having inhibitory activity against lrrk2 and use thereof | |
| WO2019107943A1 (en) | Jak inhibitor compound and preparation method therefor | |
| WO2021172871A1 (en) | Novel imidazole derivative having protein kinase inhibitory activity, and use thereof | |
| WO2023027518A1 (en) | Substituted aminopyridine compounds as egfr inhibitors | |
| WO2022019597A1 (en) | Compound for androgen receptor degradation, and pharmaceutical use thereof | |
| WO2024162746A1 (en) | Indole compound decomposing ikzf2, and use thereof | |
| WO2024151112A1 (en) | Pyrazolopyrimidine derivative as cyclin-dependent kinase 9 inhibitor | |
| WO2023277583A1 (en) | Novel plx1 protein degradation-inducing compound | |
| WO2023121207A1 (en) | Pharmaceutical composition, which inhibits aak1, for preventing or treating viral diseases or brain diseases | |
| WO2023075479A1 (en) | Novel thieno[2,3-d]pyrimidine derivative, method for preparing same, and pharmaceutical composition for prevention or treatment of cancer or autoimmune disease comprising same as active ingredient | |
| WO2010074409A2 (en) | 3-aminopyrrolidine derivatives as ccr2 antagonists | |
| WO2023229378A1 (en) | Heterocyclic compound as diacylglycerol kinase inhibitor and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22776166 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20237025047 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 22776166 Country of ref document: EP Kind code of ref document: A1 |