WO2022003405A1 - One-pot process to obtain a pyrrolidine-2-carbonitrile intermediate compound and industrial scale telescopic process to prepare (2s)-1-[n-(3-hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (vildagliptin) using same - Google Patents
One-pot process to obtain a pyrrolidine-2-carbonitrile intermediate compound and industrial scale telescopic process to prepare (2s)-1-[n-(3-hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (vildagliptin) using same Download PDFInfo
- Publication number
- WO2022003405A1 WO2022003405A1 PCT/IB2020/056279 IB2020056279W WO2022003405A1 WO 2022003405 A1 WO2022003405 A1 WO 2022003405A1 IB 2020056279 W IB2020056279 W IB 2020056279W WO 2022003405 A1 WO2022003405 A1 WO 2022003405A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- vildagliptin
- formula
- solvent
- intermediate compound
- Prior art date
Links
- 0 *CC(N1[C@](*)CCC1)=O Chemical compound *CC(N1[C@](*)CCC1)=O 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
Definitions
- the high-boiling polar solvent is N,N-dimethylformamide, N,N- dimethylacetamide, or a compound having the following formula:
- R 3 is hydrogen, alkyl or aryl; R 1 and R 2 are alkyl, aryl, or R 1 and R 2 are linked together to form a ring.
- the reaction is quenched by the addition of water and a base selected from K 2 CO 3 , Na 2 CO 3 , KHCO 3 , NaHCO 3 , NH 3 , or a tertiary amine.
- a base selected from K 2 CO 3 , Na 2 CO 3 , KHCO 3 , NaHCO 3 , NH 3 , or a tertiary amine.
- the extraction solvent of the additional step is dichloromethane.
- the intermediate compound is (S)-1-(2-chloroacetyl)- pyrrolidine-2-carbonitrile.
- the extraction organic solvent of step (c) is dichloromethane.
- X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the second reagent is chloroacetyl chloride;
- the high-boiling polar solvent is N,N-dimethylformamide, N,N- dimethylacetamide, or a compound having the following formula: wherein R 3 is hydrogen, alkyl or aryl; R 1 and R 2 are alkyl, aryl, or R 1 and R 2 are linked together to form a ring.
- the extraction solvent of the additional step is dichloromethane.
- X and Y are, independently, a halogen selected from Cl, Br and
- the high-boiling polar solvent of step (a) is N,N-dimethylformamide, N,N-dimethylacetamide, or a compound having the following formula: wherein R 3 is hydrogen, alkyl or aryl; R 1 and R 2 are alkyl, aryl, or R 1 and R 2 are linked together to form a ring.
- the obtained solution is dosed on a suspension of 3-amino-1- adamantanol (16.1 g, 0.0964 mole), potassium carbonate (12.1 g, 0.0876 mole) and potassium iodide (0.55 g, 0.0031 mole) in 100 ml of tetrahydrofuran at 50°C for a period of about 0.5 hour. The resulting suspension is stirred at 50°C for 2 hours.
- the reaction mixture is cooled to 30°C, vacuum filtered to remove the salts, and the solvent is evaporated under reduced pressure.
- the obtained residue is re- dissolved in 50 ml of water, the pH is adjusted to 5-6 with glacial acetic add and washed with dichloromethane (2 x 30 ml).
- the aqueous phase is led to pH 9-10 with a 28% ammonium hydroxide solution and extracted with dichloromethane (4 x
- a suspension of L-prolinamide (10.0 g, 0.0876 mole) in 40 ml of N,N- dimethylformamide is dosed with chloroacetyl chloride (24.7 g, 0.219 mole) while keeping the temperature in the 0 - 5°C range and under a stream of nitrogen.
- the obtained suspension is stirred at 20°C for 26 hours.
- 15.3 ml of water are added and stirred for 0.5 hour at room temperature until complete dissolution of the salts.
- the aqueous solution is extracted with dichloromethane (4 x 30 ml), the organic phases are pooled, and the solvent is evaporated under reduced pressure.
- a viscous residue is obtained with (S)-1-(2-chloroacetyl)-pyrrolidine-2-carbonitrile as the main component, which is re-dissolved in 30 ml of tetrahydrofuran.
- 3-amino-1-adamantanol (16.1 g, 0.0964 mole)
- potassium carbonate (12.1 g, 0.0876 mole)
- potassium iodide (0.55 g, 0.0031 mole).
- the resulting suspension is stirred at 30°C for 2 hours.
- the reaction mixture is vacuum filtered to remove the salts, and the solvent is evaporated under reduced pressure.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
An industrial scale telescopic process for preparing (2S)-1-[N-(3-hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (Vildagliptin) of formula:, comprising: (a) reacting the compound of formula: with an excess of a compound of formula:, wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, in a high-boiling polar solvent, such that the reaction product is intermediate compound: wherein X is as defined above, and is obtained in a single step; (b) quenching the reaction by the addition of water and a base; (c) extracting the reaction product in a polar organic solvent and evaporating the extraction solvent thus obtaining a product which mostly comprises the intermediate compound; (d) dissolving the product comprising the intermediate compound by the addition of an organic solvent; and (e) reacting the intermediate compound with 3-amino-1-adamantanol, in the presence of potassium carbonate and potassium iodide to obtain crude Vildagliptin from the reaction mixture. Also disclosed is a one-pot process to obtain the intermediate compound (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile.
Description
ONE-POT PROCESS TO OBTAIN A PYRROLIDINE-2-CARBONITRILE
INTERMEDIATE COMPOUND AND INDUSTRIAL SCALE TELESCOPIC
PROCESS TO PREPARE (2S)-1-[N-
(3-HYDROXYADAMANTAN-1-YL)GLYCYL]-2-PYRROUDINECARBONITRILE
(VILDAGLIPTIN) USING SAME
Field of the Invention
The present invention is related to processes for preparing (2S)-1-[N-(3- hidroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (Vildagliptin) from intermediate compound (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile without separating the latter from the reaction mixture. More particularly, the present invention is directed to industrial scale processes for producing Vildagliptin; still more particularly, the present invention is directed to an industrial scale telescopic process for preparing Vildagliptin.
Background Art
Vildagliptin, lUPAC name (2S)-1-[N-(3-hydroxyadamantan-1-yl)glycyl]-2- pyrrolidinecarbonitrile (LAF237; CAS 274901-16-5), has the formula:
 and is a hypoglycemic drug used for the treatment of Type-2 diabetes mellitus.
and is a hypoglycemic drug used for the treatment of Type-2 diabetes mellitus.
Specifically, Vildagliptin is an anti-diabetic agent belonging to the DPP4 inhibitor class of drugs (Dipeptidyl peptidase-4).
Vildagliptin inhibits GLP-1 inactivation by DPP-4, allowing GLP-1 and GIP to enhance insulin secretion by beta cells in the pancreas, while suppressing glucagon release by alpha cells in the pancreatic islets of Langerhans.
This compound is employed in combination treatments with metformin, sulphonylureas or thiazolidinediones.
Several prior art documents may be found which are directed to Vildagliptin, pharmaceutically acceptable salts, crystalline forms and various synthetic methods thereof.
The PCT patent application published as W02000034241A1 by NOVARTIS-
ERFINDUNGEN, titled “N-substituted 2-Cyanopyrrolidines”, discloses the following synthetic method in Example 1 :

The PCT patent application published as W02004092127A1 by NOVARTIS
PHARMA GMBH, titled “Process for the preparation of N-substituted 2- cyanopyrrolidines”, provides the following synthetic method in Example 1 :

The PCT patent application published as W02006078593A2 by NOVARTIS
PHARMA GMBH, titled “Direct compression formulation and process”, wherein crystalline Form A of Vildagliptin is described by XRD (X-Ray Diffraction).
The PCT patent application published as W02007019255A2 by NOVARTIS
PHARMA GMBH, titled “New compounds”, refers to new saline forms of
Vildagliptin (LAF237). Clause 1 refers to a Vildagliptin salt of a pharmaceutically acceptable add in a 1:1 stoichiometric ratio. The second dause makes reference to possible Vildagliptin salts, namely: 4-acetam idobenzoate, acetate, adipate, alginate, aminosalydlate, ascorbate, aspartate, benzenesulfonate, benzoate, butirate, camphorate, camphorsulfonate, carbonate, cinnamate, citrate, cydamate, cydopentanepropionate, decanoate, 2,2-dichloroacetate, digluconate, dodecylsulfate, ethane-1 ,2-disulfonate, ethanesulfonate, formate, fumarate, galactarate, gentisate, glucoheptanoate, gluconate, glucuronate, glutamate,
glycerophosphate, glycolate, hemisulfate, heptanoate, hexanoate, hippurate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, isobutirate, lactate, lactobionate, laurate, malate, maleate, malonate, mandelate, methanesulfonate, naphthalene-1 ,5-disulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, octanoate, oleate, orotate, oxalate, 2-oxoglutarate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, pycrate, pidolate
(L-pyroglutamate), pivalate, propionate, salydlate, sebacate, hydrogen sebacate, stearate, sucdnate, sulfate, tannate, tartrate, hydrogen tartrate, thiocyanate, tosylate, or undecanoate. Particularly, hydrochloride, sulfate or dicarboxylate salts of Vildagliptin are herein daimed.
Furthermore, in the PCT patent application published as W02008084383A2 by MEDICHEM S.A., titled “Process for preparing Vildagliptin”, a process is described which comprises reacting a 2-cyanopyrrolidine derivative of formula (IV):
 wherein X is a halogen and Y is -CN, with 1-amino-3-adamantol of formula (III), wherein the hydroxyl group is protected:
wherein X is a halogen and Y is -CN, with 1-amino-3-adamantol of formula (III), wherein the hydroxyl group is protected:

Vildagliptin is obtained in good yield and with high optical purity. The process requires removal of the protecting group present in the OH moiety.
In another PCT patent application which was published under No.
WO201 0022690A2 by ZENTIVA KS, titled “A method of preparation of highly pure
Vildagliptin”, a process for preparing highly pure Vildagliptin is described which consists of preparing 1-haloacetyl-2-(S)pyrrolidinecarboxamide of formula (VI):
 wherein X is chloro or bromo, mixing it with trialkylamine hydrohalides, dehydrating the mixture with an appropriate agent, and isolating 1-haloacetyl2-(S) cyanopyrrolidine of general formula (IV):
wherein X is chloro or bromo, mixing it with trialkylamine hydrohalides, dehydrating the mixture with an appropriate agent, and isolating 1-haloacetyl2-(S) cyanopyrrolidine of general formula (IV):
 which is alkylated by reacting it with 3-amino-1-adamantanol of formula (V):
which is alkylated by reacting it with 3-amino-1-adamantanol of formula (V):
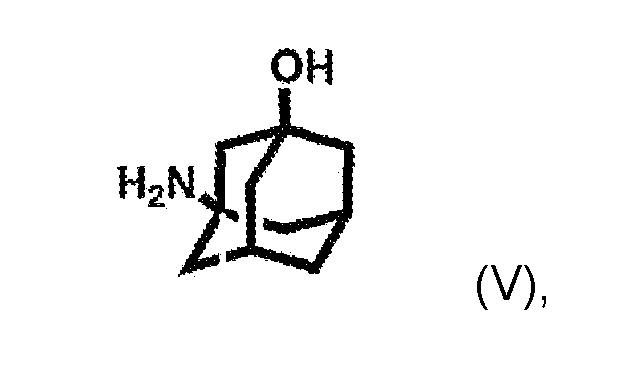 according to the conventional process for the synthesis of Vildagliptin.
according to the conventional process for the synthesis of Vildagliptin.
In the PCT patent application published under No. WO2011012322A9 by
KRKA, d.d., Novo mesto, titled “Synthesis and use of Vildagliptin for the preparation of pharmaceutical dosage forms”, a synthetic process is described employing a phase transference catalyst in the last step. This invention refers to a
method for preparing Vildagliptin which includes the step of reacting a 2-acetyl substituted (S)-1-(acetylpyrrolidine-2-cart)onitrile) having an LG leaving group of formula (II):
 preferably (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile, and 3-amino-1- adamantol of formula (III) in the presence of a phase transference catalyst. The reaction mixture may be made up of a first and a second phase, wherein the first phase determines the liquid phase and the second phase preferably determines the solid phase, optionally of different compounds, not dissolved in the solvent of the first phase. Optionally, a base is present.
preferably (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile, and 3-amino-1- adamantol of formula (III) in the presence of a phase transference catalyst. The reaction mixture may be made up of a first and a second phase, wherein the first phase determines the liquid phase and the second phase preferably determines the solid phase, optionally of different compounds, not dissolved in the solvent of the first phase. Optionally, a base is present.
In the PCT patent application published under No. W02011101861A1 on behalf of MSN LABORATORIES LIMITED, titled “Processes for preparation of
DPP-IV inhibitors”, alternative synthetic routes for Vildagliptin are described.
According to Scheme 1 described in this document:

In Scheme 2, the following process is proposed:

In the PCT patent application published under No. WO2012004210A1 on behalf of CHEMELECTIVA S.R.L., titled “Process and intermediates for preparation of an active ingredient", an alternative synthetic route for Vildagliptin is disclosed. The synthetic path disclosed in this document is based on new intermediates and is different from the synthetic strategies of the previously known processes, such that the process comprises the following steps: a) reacting 1-
amino-3-adamantanol (III) with glyoxylic add to afford the intermediate of formula
(VI):
 b) reacting intermediate (VI) with L-prolinamide to afford the intermediate of formula (VII):
b) reacting intermediate (VI) with L-prolinamide to afford the intermediate of formula (VII):
 c) dehydrating intermediate (VII) to afford the nitrile derivative corresponding to formula (VIII):
c) dehydrating intermediate (VII) to afford the nitrile derivative corresponding to formula (VIII):
 and d) deformylating intermediate (VIII) to obtain Vildagliptin.
and d) deformylating intermediate (VIII) to obtain Vildagliptin.
In the PCT case published as WO2012022994A1 on behalf of EGIS
GYOGYSZERGYAR NYILVANOSAN MUKODO RESZVENYTARASAG, titled
“Preparation process of Vildagliptin”, an alternative synthetic route for Vildagliptin is
disclosed. The process for preparing 1 -[2-(3-hydroxyadamant-1 -yl- amino)acetyl]pyrrolidine-(2S)-carbonitrile (Vildagliptin) from (±)-1-[2-(3- hydroxyadamant-1-ylamino)acetylpyrrolidine-2-carbonitrile of formula (III):
 which is a racemic mixture. Also, another process is disclosed for preparing
which is a racemic mixture. Also, another process is disclosed for preparing
Vildagliptin complexes, in particular the Vildagliptin trihydrate - calcium chloride complex of Formula (II):
 starting from the racemic base of Formula (III) and optionally converting the
starting from the racemic base of Formula (III) and optionally converting the
Vildagliptin complexes of Formula (IV) into Vildagliptin. This document also discloses diastereomeric salt Vildagliptin dihydrate - O,O'-di-p-toluen-(25',5S)- hydrogen tartrate of Formula (XV):
 solvates and hydrates thereof. Also disclosed is Vildagliptin - Ο,Ο'-di-p-benzoyl-
solvates and hydrates thereof. Also disclosed is Vildagliptin - Ο,Ο'-di-p-benzoyl-
(25',5S)-hydrogen tartrate of Formula (XVI):
 solvates and hydrates thereof.
solvates and hydrates thereof.
In the PCT application published under No. WO2013083326A1 on behalf of
CHEMELECTIVA S.R.L., titled “New process and intermediates for the synthesis of
Vildagliptin”, a synthetic process for Vildagliptin is claimed which comprises: a) salification of L-prolinamide with haloacetic acid (XCH2COOH) in an appropriate solvent to obtain the salt of formula (VI):
 b) condensation of the salt of formula (VI) by reaction with a condensation agent and subsequent reaction with 1 -amino-3-adamantol in an appropriate solvent to afford the intermediate of formula (VII), without isolating the condensation product:
b) condensation of the salt of formula (VI) by reaction with a condensation agent and subsequent reaction with 1 -amino-3-adamantol in an appropriate solvent to afford the intermediate of formula (VII), without isolating the condensation product:

In the PCT document published as W02013179300A2 on behalf of
MEGAFINE PHARMA (P) LTD., titled “A process for the preparation of Vildagliptin and its intermediate thereof, a process for preparing Vildagliptin of formula (I) is disdosed:
 which comprises: a) reacting 3-amino-1-adamantanol of formula (IV) with 1- chloroacetyl-(S)-2-cyanopyrrolidine of formula (V) in a solvent and a base to obtain crude Vildagliptin; and
which comprises: a) reacting 3-amino-1-adamantanol of formula (IV) with 1- chloroacetyl-(S)-2-cyanopyrrolidine of formula (V) in a solvent and a base to obtain crude Vildagliptin; and
 and b) isolating pure Vildagliptin of formula (I). Also, a process for isolating pure
and b) isolating pure Vildagliptin of formula (I). Also, a process for isolating pure
Vildagliptin is provided, which comprises: i) treating crude Vildagliptin with add and
water, ii) washing the contents of step i) with an organic solvent, iii) basifying the aqueous layer of step ii), iv) extracting Vildagliptin of step iii) using an organic solvent, v) separating the organic layer, repeating steps i) to iv), and concentrating the organic layer to obtain pure Vildagliptin; or concentrating the organic layer, repeating steps i) to iv), and concentrating the organic layer to obtain pure
Vildagliptin, and vi) treating the Vildagliptin obtained in step v) with an organic solvent, heating to obtain a dear solution, partially distilling off the solvent, cooling, filtering and drying to obtain pure Vildagliptin. Also provided is a process carried out in the same reactor for preparing 1-chloroacetyl-(S)-2-cyanopyrrolidine, a
Vildagliptin intermediate, comprising the steps of: A. reacting L-prolinamide of formula (II):
 with chloroacetyl chloride in a solvent, optionally in the presence of a first base; B. reacting the reaction mass of step (A) with a dehydrating agent optionally in the presence of a second base; and C. isolating pure 1-chloroacetyl-(S)-2- cyanopyrrolidine. Also disdosed is an amorphous co-predpitate of Vildagliptin or the add addition salt thereof with a pharmaceutically acceptable exdpient characterized by its X-ray diffraction (XRD) pattern, and a process for obtaining same.
The PCT document published under No. W02014013505A2 on behalf of
with chloroacetyl chloride in a solvent, optionally in the presence of a first base; B. reacting the reaction mass of step (A) with a dehydrating agent optionally in the presence of a second base; and C. isolating pure 1-chloroacetyl-(S)-2- cyanopyrrolidine. Also disdosed is an amorphous co-predpitate of Vildagliptin or the add addition salt thereof with a pharmaceutically acceptable exdpient characterized by its X-ray diffraction (XRD) pattern, and a process for obtaining same.
The PCT document published under No. W02014013505A2 on behalf of
HETERO RESEARCH FOUNDATION, titled “Amorphous Vildagliptin”, describes a novel amorphous form of Vildagliptin which is stable, reproducible and, therefore, suitable for pharmaceutical preparations. Also disclosed is a solid dispersion of
Vildagliptin in combination with a pharmaceutically acceptable carrier, which is stable, reproducible and, therefore, amenable for conducting a large scale preparation. Likewise, a process is provided for preparing the amorphous
Vildagliptin, comprising: a) dissolving Vildagliptin in water; and b) removing the water to obtain amorphous Vildagliptin.
The PCT document published under No. W02014102815A1 on behalf of
GLENMARK PHARMACEUTICALS LIMITED, titled “Improved process for preparation of Vildagliptin”, discloses a process for preparing Vildagliptin, comprising: a) reacting a compound of formula (VI): with a compound of formula (V):
 in the presence of a nitrile solvent to afford a compound of formula (IV):
in the presence of a nitrile solvent to afford a compound of formula (IV):


DMF and triphosgene, a mixture of diethylformamide (DBF) and POCI3, a mixture of DMF and thionyl chloride (SOCI), a mixture of DMF and oxalyl chloride, and the like, preferably Vilsmeier reagent; and c) reacting the compound of formula (III) with a compound of formula (II):
 to form Vildagliptin.
to form Vildagliptin.
The PCT document published under No. WO2015128718A1 on behalf of
HIKAL LIMITED, titled “Novel economic process for Vildagliptin”, discloses
alternative synthetic routes to prepare Vildagliptin. These processes are illustrated in the following general synthetic scheme (6):

The prior art does not provide a few-step strategy to obtain an intermediate for synthetizing Vildagliptin such as (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile.
A synthesis which allows facilitating to prepare this intermediate at an industrial scale would concomitantly improve the synthesis of Vildagliptin as the final product.
At the same time, it is beneficial to achieve the production of high purity crude
Vildagliptin from which a very pure product can be obtained with few purification steps. In this manner, the synthesis yields of the pharmaceutically pure product would be also improved.
Summary of the Invention
It is, therefore, an object of the present invention, a process for a one-pot preparation of a compound of the following formula:
 which is an intermediate in the synthesis of Vildagliptin, and wherein X is a halogen selected from Cl, Br and I, preferably Cl, which comprises reacting the compound of formula:
which is an intermediate in the synthesis of Vildagliptin, and wherein X is a halogen selected from Cl, Br and I, preferably Cl, which comprises reacting the compound of formula:
 with an excess of a compound of formula:
with an excess of a compound of formula:
 wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, and in a high-boiling polar solvent, such that the reaction product is obtained in a single step.
wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, and in a high-boiling polar solvent, such that the reaction product is obtained in a single step.
Preferably, the high-boiling polar solvent is N,N-dimethylformamide, N,N- dimethylacetamide, or a compound having the following formula:

Accessorily, the reaction is quenched by the addition of water and a base selected from K2CO3, Na2CO3, KHCO3, NaHCO3, NH3, or a tertiary amine.
Additionally, the intermediate compound of formula:
 is extracted in an extraction solvent and then the extraction solvent is evaporated to obtain a product mostly comprising said intermediate compound.
is extracted in an extraction solvent and then the extraction solvent is evaporated to obtain a product mostly comprising said intermediate compound.
Preferably, the extraction solvent of the additional step is dichloromethane.
It is a further object of the present invention, an industrial scale telescopic process for preparing (2S)-1 -[N-(3-hydroxyadamantan-1-yl)glycyl]-2- pyrrolidinecarbonitrile (Vildagliptin) of formula:
 which comprises:
which comprises:
(a) reacting the compound of formula:
 with an excess of a compound of formula:
with an excess of a compound of formula:
 wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, such that the reaction product is intermediate compound:
wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, such that the reaction product is intermediate compound:
 wherein X is as defined above, and is obtained in a single step;
wherein X is as defined above, and is obtained in a single step;
(b) quenching the reaction by the addition of water and a base;
(c) extracting the reaction product in an organic solvent and evaporating the extraction organic solvent, thus obtaining a product which mostly comprises the intermediate compound;
(d) dissolving the product comprising the intermediate compound by the addition of an organic solvent; and
(e) reacting the intermediate compound with 3-amino-l-adamantanol, in the presence of potassium carbonate and potassium iodide to obtain crude Vildagliptin from the reaction mixture.
Preferably, the high-boiling polar solvent of step (a) is N,N- dimethylformamide, N,N-dimethylacetamide, or a compound having the following formula:
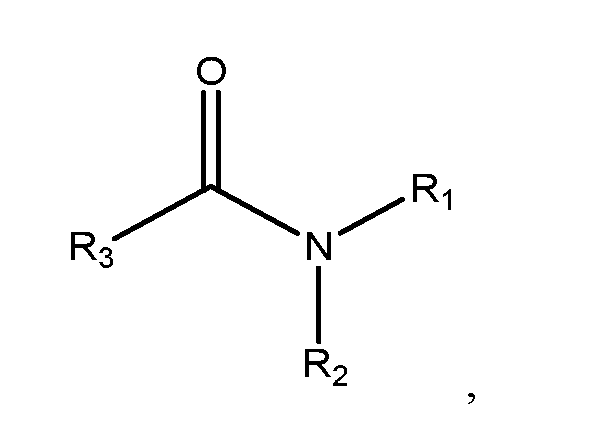 wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
Also preferably, the intermediate compound is (S)-1-(2-chloroacetyl)- pyrrolidine-2-carbonitrile.
More preferably, the base of step (b) is selected from K2CO3, Na2CO3, KHCO3, NaHCO3, NH3, or a tertiary amine.
Preferably, the extraction organic solvent of step (c) is dichloromethane.
Also preferably, the organic solvent of step (d) is tetrahydrofuran.
Additionally, the reaction mixture of step (e) is filtered, the solvent is evaporated, the residue is re-dissolved with water and extracted with an extraction solvent which is evaporated at reduced pressure to obtain a viscous residue.
Even more preferably, the extraction with the extraction solvent is carried out first at about pH 5 and then at about pH 9.
Still more preferably, crude Vildagliptin is precipitated from the viscous residue with an isopropanol/butanone (1:1) mixture.
More preferably, crude Vildagliptin is recrystallized from isopropanol.
Detailed Descrlption of the Invention
The processes known from the prior art teachings for the synthesis of
Vildagliptin comprise many synthetic stages, and the same is true for the synthetic process of intermediate (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile.
However, said intermediate has now been obtained in a single stage (a one- pot process) in good yield and with the necessary purity to address the industrial scale telescopic synthesis of Vildagliptin in an accessible manner and with good yields of the crude final product.
Indeed, as can be seen in the following general reaction scheme, said intermediate compound (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile can be obtained in a straighter manner and without the need for isolation of intermediate products:
 (i) Hiah-boilina polar solvent selected from N,N- dimethylformamide, N,N-dimethylacetamide, or a compound having the following formula:
(i) Hiah-boilina polar solvent selected from N,N- dimethylformamide, N,N-dimethylacetamide, or a compound having the following formula:
 wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring;
wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring;
X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the second reagent is chloroacetyl chloride;
(ii) Water and base, the base being selected from K2CO3, Na2CO3, KHCO3, NaHCO3 , NH3, or a tertiary amine / extraction with an extraction solvent, preferably dichloromethane.
Thus, it is an object of the present invention, a one-pot process for preparing the compound of the following formula:
 which is a synthesis intermediate of Vildagliptin, and wherein X is a halogen selected from Cl, Br and I, preferably Cl; which comprises reacting the compound of formula:
which is a synthesis intermediate of Vildagliptin, and wherein X is a halogen selected from Cl, Br and I, preferably Cl; which comprises reacting the compound of formula:


Preferably, the high-boiling polar solvent is N,N-dimethylformamide, N,N- dimethylacetamide, or a compound having the following formula:
 wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
Accessorily, the reaction is quenched by the addition of water and a base selected from K2CO3, Na2CO3, KHCO3, NaHCO3, NH3, or a tertiary amine.
Additionally, the obtained intermediate compound is extracted in an extraction solvent and then the extraction solvent is evaporated to obtain a product mostly comprising said intermediate compound.
Preferably, the extraction solvent of the additional step is dichloromethane.
In particular, X and Y are, independently, a halogen selected from Cl, Br and
I, preferably Cl, thereby obtaining intermediate compound (S)-1-(2-chloroacetyl)- pyrrolidine-2-carbonitrile, which is useful for the synthesis of Vildagliptin.
Therefore, it is another object of the present invention, an industrial scale telescopic process for preparing (2S)-1 -[N-(3-hydroxyadamantan-1 -yl)glycyl]-2- pyrrolidinecarbonitrile (Vildagliptin) of formula:
 which comprises:
which comprises:
(a) reacting the compound of formula:
 with an excess of a compound of formula:
with an excess of a compound of formula:
 wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, in a high-boiling polar solvent, such that the reaction product is intermediate compound:
wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, in a high-boiling polar solvent, such that the reaction product is intermediate compound:
 wherein X is as defined above, and is obtained in a single step;
wherein X is as defined above, and is obtained in a single step;
(b) quenching the reaction by the addition of water and a base;
(c) extracting the reaction product in a polar organic solvent and evaporating the extraction solvent thus obtaining a product mostly comprising the intermediate compound;
(d) dissolving the product comprising the intermediate compound by the addition of an organic solvent; and
(e) reacting the intermediate compound with 3-amino-1 -adamantanol, in the presence of potassium carbonate and potassium iodide to obtain crude Vildagliptin from the reaction mixture.
Wherein the high-boiling polar solvent of step (a) is N,N-dimethylformamide, N,N-dimethylacetamide, or a compound having the following formula:
 wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
Particularly, the base of step (b) is selected from K2CO3, Na2CO3, KHCO3, NaHCO3, NH3, or a tertiary amine.
Also particularly, the polar organic solvent of step (c) is dichloromethane.
In a preferred embodiment, the intermediate compound is (S)-1-(2- chloroacetyl)pyrrolidine-2-carbonitrile.
In a preferred embodiment, the organic solvent of step (d) is tetrahydrofuran.
Also, the reaction mixture of step (e) is filtered, the solvent is evaporated, the residue is redissolved with water and extracted with an extraction solvent which is evaporated at reduced pressure thus obtaining a viscous residue. In a preferred embodiment, the extraction solvent is dichloromethane, and the extraction with the extraction solvent is first carried out at about pH 5 and then at about pH 9.
Finally, from said viscous residue crude Vildagliptin is precipitated with an isopropanol/butanone mixture (1:1), and crude Vildagliptin is recrystallized from isopropanol until the necessary pharmacological purity is attained.
EXAMPLES
Preparation of (S)-1-(2- chloroacetyl)pyrrolidine -2-carbonitrile
EXAMPLE 1
L-prolinamide (10.0 g, 0.0867 mole) is mixed with N,N-dimethylformamide
(40 ml) and the resulting suspension is cooled to 0°C - 5°C. Chloroacetyl chloride
(29.7 g, 0.263 mole) is added dropwise to the cold reaction mixture and the
obtained yellow solution is stirred at 35°C for 4 hours. The reaction mass is cooled to 0-5°C and 18% ammonia (65 ml) is slowly added. The obtained aqueous solution is extracted with methylene chloride (4 x 30 ml) and the combined organic phases are concentrated at reduced pressure. The oily residue is dosed with n- heptane (20 ml) and isopropanol (20 ml), and the solvents are evaporated under vacuum. Isopropanol (40 ml) is added to the honey-like product and stirred for 1 hour at room temperature. The obtained solid is filtered off and washed with I PA.
Finally, it is vacuum dried (10-15 mbar, 50°C) to afford 3.84 g (yield: 25.4 %) of an off-white crystalline solid having a melting point of 64-66°C with 97% purity as determined by high performance liquid chromatography (HPLC).
EXAMPLE 2
L-prolinamide (10.0 g, 0.0867 mole) is mixed with N,N-dimethylformamide
(40 ml) and the resulting suspension is cooled to 0°C - 5°C. Chloroacetyl chloride
(39.6 g, 0.350 mole) is added dropwise to the cold reaction mixture and the obtained yellow solution is stirred at 40°C for 2 hours. The reaction mass is cooled to 0-5°C and a saturated potassium carbonate solution (100 ml) is slowly added.
The obtained solid is filtered off and washed with dichloromethane (DCM) (30 ml).
The layers are separated and the aqueous phase is re-extracted with methylene chloride (3 x 30 ml). To the combined organic phases activated carbon (2.0 g) is added and then vacuum filtration is performed. The filtrate is washed with a saturated solution of sodium chloride (40 ml) and then concentrated at reduced pressure. The oily residue is dosed with n-heptane (20 ml) and isopropanol (20 ml),
and the solvents are evaporated under vacuum. Isopropanol (40 ml) is added to the honey-like product and stirred for 1 hour at room temperature. The obtained solid is filtered off and washed with I PA. Finally, it is vacuum dried (10-15 mbar,
50°C) to afford 4.40 g (yield: 29.1 %) of a white crystalline solid having a melting point of 64-66°C with 96.4% purity (HPLC).
EXAMPLE 3
L-prolinamide (10.0 g, 0.0867 mole) is mixed with N,N-dimethylformamide
(40 ml) and the resulting suspension is cooled to 0°C - 5°C. Chloroacetyl chloride
(24.7 g, 0.219 mole) is added dropwise to the cold reaction mixture and the obtained yellow solution is stirred at 50°C for 2 hours. The reaction mass is cooled to 0-5°C, methylene chloride is added (50 ml) and the mass is washed with a saturated solution of sodium bicarbonate (4 x 60 ml). The organic phase is dried with anhydrous sodium sulfate and then concentrated at reduced pressure. The oily residue is dosed with n-heptane (20 ml) and isopropanol (20 ml), and the solvents are evaporated under vacuum. Isopropanol (40 ml) is added to the honey- like product and stirred for 1 hour at room temperature. The obtained solid is filtered off and washed with IPA. Finally, it is vacuum dried (10-15 mbar, 50°C) to afford 7.24 g (yield: 47.9 %) of a light beige crystalline solid having a melting point of 58-64°C with 90.4% purity (HPLC).
Pre Da ration of (2S)-1-[N-(3-hvdroxyadamantan-1-yl)glycyl]-2-pyrrolidine- carbonitrile (Vildagliptin)
EXAMPLE 4
A suspension of L-prolinamide (10.0 g, 0.0876 mole) in 40 ml of N,N- dimethylformamide is dosed with chloroacetyl chloride (39.6 g, 0.350 mole) while keeping the temperature in the 0 - 5°C range. The obtained solution is stirred at
37°C for 1.5 hours. The reaction mixture is led to pH 7-8 with 55 ml of a 28% ammonium hydroxide solution. To the resulting solution, 25 ml of water are added and the mixture is stirred for 0.5 hour at room temperature until complete dissolution of the salts. The aqueous solution is extracted with dichloromethane (4 x 30 ml), the organic phases are pooled and the solvent is evaporated under reduced pressure. A viscous residue is obtained with (S)-1-(2-chloroacetyl)- pyrrolidine-2-carbonitrile as the main component, which is re-dissolved in 30 ml of tetrahydrofuran. The obtained solution is dosed on a suspension of 3-amino-1- adamantanol (16.1 g, 0.0964 mole), potassium carbonate (12.1 g, 0.0876 mole) and potassium iodide (0.55 g, 0.0031 mole) in 100 ml of tetrahydrofuran at 50°C for a period of about 0.5 hour. The resulting suspension is stirred at 50°C for 2 hours.
The reaction mixture is cooled to 30°C, vacuum filtered to remove the salts, and the solvent is evaporated under reduced pressure. The obtained residue is re- dissolved in 50 ml of water, the pH is adjusted to 5-6 with glacial acetic add and washed with dichloromethane (2 x 30 ml). The aqueous phase is led to pH 9-10 with a 28% ammonium hydroxide solution and extracted with dichloromethane (4 x
30 ml). The organic phases are pooled, then dried with anhydrous sodium sulfate, and the solvent is evaporated under reduced pressure. A honey-like residue is obtained which crystallizes in 45 ml of an isopropanol/butanone (1:1) mixture. The
obtained suspension is stirred for 1 hour at 0°C, vacuum filtered, washed with 20 ml of cold precipitation solvent mixture, and vacuum dried (10-15 mbar, 50°C). 11.9 g (yield: 44.8%) are obtained of a white crystalline solid having a melting point of
147-149°C with 97.2% purity (HPLC).
EXAMPLE 5
A suspension of L-prolinamide (10.0 g, 0.0876 mole) in 40 ml of N,N- dimethylformamide is dosed with chloroacetyl chloride (24.7 g, 0.219 mole) while keeping the temperature in the 0 - 5°C range and under a stream of nitrogen. The obtained suspension is stirred at 20°C for 26 hours. The reaction mixture is cooled to 5-10°C with a water/ice bath and led to pH = 7-8 with 27.7 ml of a 28% ammonium hydroxide solution. To the resulting suspension, 15.3 ml of water are added and stirred for 0.5 hour at room temperature until complete dissolution of the salts. The aqueous solution is extracted with dichloromethane (4 x 30 ml), the organic phases are pooled, and the solvent is evaporated under reduced pressure.
A viscous residue is obtained with (S)-1-(2-chloroacetyl)-pyrrolidine-2-carbonitrile as the main component, which is re-dissolved in 30 ml of tetrahydrofuran. The obtained solution is dosed on a suspension of 3-amino-1-adamantanol (16.1 g,
0.0964 mole), potassium carbonate (12.1 g, 0.0876 mole) and potassium iodide
(0.55 g, 0.0031 mole) in 100 ml of tetrahydrofuran at 50°C for a period of about 0.5 hour. The resulting suspension is stirred at 50°C for 2 hours. The reaction mixture is cooled to 30°C, vacuum filtered to remove the salts, and the solvent is evaporated under reduced pressure. The obtained residue is re-dissolved in 50 ml
of water, the pH is adjusted to 5-6 with glacial acetic acid and washed with dichloromethane (2 x 30 ml). The aqueous phase is led to pH 9-10 with a 28% ammonium hydroxide solution and extracted with dichloromethane (4 x 30 ml). The organic phases are pooled, then dried with anhydrous sodium sulfate, and the solvent is evaporated under reduced pressure. A honey-like residue is obtained which crystallizes in 51 ml of an isopropanol/butanone (1:1) mixture. The obtained suspension is stirred for 1 hour at 0°C, vacuum filtered, washed with 13 ml of cold precipitation solvent mixture, and vacuum dried (10-15 mbar, 50°C). 10.7 g (yield:
40.2%) are obtained of a white crystalline solid having a melting point of 148-150°C with 99.1% purity (HPLC).
EXAMPLE 6
A suspension of L-prolinamide (10.0 g, 0.0876 mole) in 40 ml of N,N- dimethylformamide is dosed with chloroacetyl chloride (29.7 g, 0.263 mole) while keeping the temperature in the 0 - 5°C range. The obtained suspension is stirred at
30°C for 4 hours. The reaction mixture is led to pH = 7-8 with 40 ml of a 28% ammonium hydroxide solution. To the resulting suspension, 25 ml of water are added and stirred for 0.5 hour at room temperature until complete dissolution of the salts. The aqueous solution is extracted with dichloromethane (4 x 30 ml), the organic phases are pooled, and the solvent is evaporated under reduced pressure.
A viscous residue is obtained with (S)-1-(2-chloroacetyl)-pyrrolidine-2-carbonitrile as the main component, which is re-dissolved in 30 ml of tetrahydrofuran. To the obtained solution is added 3-amino-1-adamantanol (16.1 g, 0.0964 mole),
potassium carbonate (12.1 g, 0.0876 mole) and potassium iodide (0.55 g, 0.0031 mole). The resulting suspension is stirred at 30°C for 2 hours. The reaction mixture is vacuum filtered to remove the salts, and the solvent is evaporated under reduced pressure. The obtained residue is re-dissolved in 50 ml of water, the pH is adjusted to 5-6 with glacial acetic acid and washed with dichloromethane (2 x 30 ml). The aqueous phase is led to pH 9-10 with a 28% ammonium hydroxide solution and extracted with dichloromethane (4 x 30 ml). The organic phases are pooled, and the solvent is evaporated under reduced pressure. A honey-like residue is obtained which crystallizes in 45 ml of an isopropanol/butanone (1:1) mixture. The obtained suspension is stirred for 1 hour at 0°C, vacuum filtered, washed with 20 ml of cold predpitation solvent mixture, and vacuum dried (10-15 mbar, 50°C). 13.5 g (yield: 50.8%) are obtained of a white crystalline solid having a melting point of 145-148°C with 98.9% purity (HPLC).
Claims
1. A one-pot process for preparing a compound of the following formula:
 wherein X is a halogen selected from Cl, Br and I; comprising reacting a compound of formula:
wherein X is a halogen selected from Cl, Br and I; comprising reacting a compound of formula:
 with an excess of a compound of formula:
with an excess of a compound of formula:
 wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, in a high-boiling polar solvent, such that the reaction product is obtained in a single step.
wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, in a high-boiling polar solvent, such that the reaction product is obtained in a single step.
2. The process of claim 1 , wherein the high-boiling polar solvent is N,N- dimethylformamide, N,N-dimethylacetamide, or a compound having the following formula:
 wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
3. The process of claim 1 , wherein the reaction is quenched by the addition of water and a base selected from K2CO3, Na2CO3, KHCO3, NaHCO3, NH3, or a tertiary amine.
4. The process of claim 1 , wherein additionally the intermediate compound of formula:
 is extracted in an extraction solvent and then the extraction solvent is evaporated to obtain a product which mostly comprises said intermediate compound.
is extracted in an extraction solvent and then the extraction solvent is evaporated to obtain a product which mostly comprises said intermediate compound.
5. The process of claim 4, wherein the extraction solvent is dichloromethane.
6. An industrial scale telescopic process for preparing (2S)-1-[N-(3- hydroxyadamantan-1 -yl)glycyl]-2-pyrrolidinecarbonitrile (Vildagliptin) of formula:
 comprising:
comprising:
(a) reacting the compound of formula:
 with an excess of a compound of formula:
with an excess of a compound of formula:
 wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, in a high-boiling polar solvent, such that the reaction product is intermediate compound:
wherein X and Y are, independently, a halogen selected from Cl, Br and I, preferably Cl, thereby the compound is chloroacetyl chloride, in a high-boiling polar solvent, such that the reaction product is intermediate compound:
 wherein X is as defined above, and is obtained in a single step;
wherein X is as defined above, and is obtained in a single step;
(b) quenching the reaction by the addition of water and a base;
(c) extracting the reaction product in a polar extraction solvent, and evaporating the extraction solvent thus obtaining a product which mostly comprises the intermediate compound;
(d) dissolving the product comprising the intermediate compound by the addition of an organic solvent; and
(e) reacting the intermediate compound with 3-amino-1 -adamantanol, in the presence of potassium carbonate and potassium iodide to obtain crude
Vildagliptin from the reaction mixture.
7. The process of claim 6, wherein the high-boiling polar solvent of step (a) is N,N-dimethylformamide, N,N-dimethylacetamide, or is a compound having the following formula:
 wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
wherein R3 is hydrogen, alkyl or aryl; R1 and R2 are alkyl, aryl, or R1 and R2 are linked together to form a ring.
8. The process of claim 6, wherein the base of step (b) is selected from K2CO3, Na2CO3, KHCO3, NaHCO3, NH3, or a tertiary amine.
9. The process of claim 6, wherein the polar solvent of step (c) is dichloromethane.
10. The process of claim 6, wherein the intermediate compound is (S)-1-(2- chloroacetyl)pyrrolidine-2-carbonitrile.
11. The process of claim 6, wherein the organic solvent of step (d) is tetrahydrofuran.
12. The process of claim 6, wherein, additionally, the reaction mixture of step
(e) is filtered, the solvent is evaporated, the residue is re-dissolved with water and extracted in an extraction solvent, which is evaporated under reduced pressure to obtain a viscous residue.
13. The process of claim 12, wherein the extraction solvent is dichloromethane.
14. The process of claim 12, wherein the extraction with the extraction solvent is first carried out at about pH 5 and then at about pH 9.
15. The process of claim 12, wherein crude Vildagliptin is precipitated from the viscous residue with an isopropanol/butanone mixture (1 :1).
16. The process of claim 15, wherein crude Vildagliptin is recrystallized from isopropanol.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2020/056279 WO2022003405A1 (en) | 2020-07-03 | 2020-07-03 | One-pot process to obtain a pyrrolidine-2-carbonitrile intermediate compound and industrial scale telescopic process to prepare (2s)-1-[n-(3-hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (vildagliptin) using same |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2020/056279 WO2022003405A1 (en) | 2020-07-03 | 2020-07-03 | One-pot process to obtain a pyrrolidine-2-carbonitrile intermediate compound and industrial scale telescopic process to prepare (2s)-1-[n-(3-hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (vildagliptin) using same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022003405A1 true WO2022003405A1 (en) | 2022-01-06 |
Family
ID=71994665
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2020/056279 WO2022003405A1 (en) | 2020-07-03 | 2020-07-03 | One-pot process to obtain a pyrrolidine-2-carbonitrile intermediate compound and industrial scale telescopic process to prepare (2s)-1-[n-(3-hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (vildagliptin) using same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2022003405A1 (en) |
Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998019998A2 (en) * | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2000034241A1 (en) | 1998-12-10 | 2000-06-15 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2004092127A1 (en) | 2003-04-16 | 2004-10-28 | Novartis Ag | Process for the preparation of n-substituted 2-cyanopyrrolidines |
| WO2006078593A2 (en) | 2005-01-18 | 2006-07-27 | Novartis Ag | Direct compression formulation and process |
| WO2006116157A2 (en) * | 2005-04-22 | 2006-11-02 | Alantos Pharmaceuticals Holding, Inc. | Dipeptidyl peptidase-iv inhibitors |
| WO2007019255A2 (en) | 2005-08-04 | 2007-02-15 | Novartis Ag | Salts of vildagliptin |
| WO2008084383A2 (en) | 2007-01-10 | 2008-07-17 | Medichem, S.A. | Process for preparing vildagliptin |
| WO2010022690A2 (en) | 2008-08-26 | 2010-03-04 | Zentiva, K.S. | A method of preparation of highly pure vildagliptin |
| WO2011012322A2 (en) | 2009-07-31 | 2011-02-03 | Krka, D.D., Novo Mesto | Synthesis and use of vildagliptin for the preparation of pharmaceutical dosage forms |
| WO2011101861A1 (en) | 2010-01-29 | 2011-08-25 | Msn Laboratories Limited | Process for preparation of dpp-iv inhibitors |
| WO2012004210A1 (en) | 2010-07-06 | 2012-01-12 | Chemelectiva Srl | Process and intermediates for preparation of an active ingredient |
| WO2012022994A1 (en) | 2010-08-19 | 2012-02-23 | Egis Gyogyszergyar Nyilvanosan Mukodo Reszvenytarsasag | Preparation process of vildagliptin |
| WO2013083326A1 (en) | 2011-12-06 | 2013-06-13 | Chemelectiva S.R.L. | New process and intermediates for the synthesis of vildagliptin |
| WO2013179300A2 (en) | 2012-05-04 | 2013-12-05 | Megafine Pharma (P) Ltd. | A process for the preparation of vildagliptin and its intermediate thereof |
| WO2014013505A2 (en) | 2012-07-20 | 2014-01-23 | Hetero Research Foundation | Amorphous vildagliptin |
| WO2014102815A1 (en) | 2012-12-26 | 2014-07-03 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Improved process for preparation of vildagliptin |
| WO2015128718A1 (en) | 2014-02-28 | 2015-09-03 | Hikal Limited | Novel economic process for vildagliptin |
-
2020
- 2020-07-03 WO PCT/IB2020/056279 patent/WO2022003405A1/en active Application Filing
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998019998A2 (en) * | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2000034241A1 (en) | 1998-12-10 | 2000-06-15 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2004092127A1 (en) | 2003-04-16 | 2004-10-28 | Novartis Ag | Process for the preparation of n-substituted 2-cyanopyrrolidines |
| WO2006078593A2 (en) | 2005-01-18 | 2006-07-27 | Novartis Ag | Direct compression formulation and process |
| WO2006116157A2 (en) * | 2005-04-22 | 2006-11-02 | Alantos Pharmaceuticals Holding, Inc. | Dipeptidyl peptidase-iv inhibitors |
| WO2007019255A2 (en) | 2005-08-04 | 2007-02-15 | Novartis Ag | Salts of vildagliptin |
| WO2008084383A2 (en) | 2007-01-10 | 2008-07-17 | Medichem, S.A. | Process for preparing vildagliptin |
| WO2010022690A2 (en) | 2008-08-26 | 2010-03-04 | Zentiva, K.S. | A method of preparation of highly pure vildagliptin |
| WO2011012322A2 (en) | 2009-07-31 | 2011-02-03 | Krka, D.D., Novo Mesto | Synthesis and use of vildagliptin for the preparation of pharmaceutical dosage forms |
| WO2011101861A1 (en) | 2010-01-29 | 2011-08-25 | Msn Laboratories Limited | Process for preparation of dpp-iv inhibitors |
| WO2012004210A1 (en) | 2010-07-06 | 2012-01-12 | Chemelectiva Srl | Process and intermediates for preparation of an active ingredient |
| WO2012022994A1 (en) | 2010-08-19 | 2012-02-23 | Egis Gyogyszergyar Nyilvanosan Mukodo Reszvenytarsasag | Preparation process of vildagliptin |
| WO2013083326A1 (en) | 2011-12-06 | 2013-06-13 | Chemelectiva S.R.L. | New process and intermediates for the synthesis of vildagliptin |
| WO2013179300A2 (en) | 2012-05-04 | 2013-12-05 | Megafine Pharma (P) Ltd. | A process for the preparation of vildagliptin and its intermediate thereof |
| WO2014013505A2 (en) | 2012-07-20 | 2014-01-23 | Hetero Research Foundation | Amorphous vildagliptin |
| WO2014102815A1 (en) | 2012-12-26 | 2014-07-03 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Improved process for preparation of vildagliptin |
| WO2015128718A1 (en) | 2014-02-28 | 2015-09-03 | Hikal Limited | Novel economic process for vildagliptin |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7550479B2 (en) | Modified Pictet-Spengler reaction and products prepared therefrom | |
| KR910007882B1 (en) | Process for preparing 1 - benzyl - aminoalkyl - prrolidinones | |
| KR20200131241A (en) | 2 kinds of 4-v[(2S)-2-v4-[5-chloro-2-(1H-1,2,3-triazol-1-yl)phenyl]-5-methoxy-2-oxo Method for producing pyridin-1(2H)-yl}butanoyl]amino}-2-fluorobenzamide derivative | |
| RU2369598C2 (en) | Method of obtaining n-substituted 2-cyanopyrrolidines | |
| WO2012059941A1 (en) | Process for preparation of sunitinib malate and salts thereof | |
| SK14672001A3 (en) | Novel synthesis and crystallization of piperazine ring-containing compounds | |
| EP0154490B1 (en) | Process for the preparation of pyrrolidone derivatives | |
| WO2022003405A1 (en) | One-pot process to obtain a pyrrolidine-2-carbonitrile intermediate compound and industrial scale telescopic process to prepare (2s)-1-[n-(3-hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (vildagliptin) using same | |
| US20110172444A1 (en) | Method for purifying aminoacetylpyrrolidinecarbonitrile derivative and salt thereof | |
| SK15572000A3 (en) | Method for producing enantiomer-free n-methyl-n-[(1s)-1-phenyl- 2-((3s)-3-hydroxypyrrolidine-1-yl)ethyl]-2,2-diphenyl acetamide | |
| JP2020518573A (en) | Preparation of 2-([1,2,3]triazol-2-yl)-benzoic acid derivative | |
| JPS63303971A (en) | Benzazepine derivative | |
| JP2011524909A (en) | Process for the preparation of RHO-kinase inhibiting compounds | |
| TW200418850A (en) | Acyl derivatives of 5-(2-(4-(1,2 benzisothiazole-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one having neuroleptic activity | |
| JP3989832B2 (en) | Process for the preparation of N-protected-3-pyrrolidine-lactam substituted phosphonium salts | |
| NO128570B (en) | ||
| KR100850558B1 (en) | Process for preparing useful in synthesis of atorvastatin | |
| CA1133494A (en) | Substituted 4-aryl-2,5-dihydro-furane- 2-ones and 4-aryl-1,5-dihydro-2h-pyrrole-2-ones and method of their production | |
| KR102244415B1 (en) | Method for preparing chiral pyrrolidin-2-yl-methanol derivative | |
| RU2741389C1 (en) | Method for preparing intermediate compound for synthesis of medicinal agent | |
| IE922236A1 (en) | Process for the preparation of the dextrorotatory isomer of a 2-aminonaphthyridine derivative | |
| JPS6163677A (en) | Antibacterial-v | |
| AU2023206696A1 (en) | Method for preparing pyrrole compound and intermediate thereof | |
| JPH04230379A (en) | 5-isothiazolamine derivative | |
| FI68830B (en) | DL-ELLER D-TRANS-8-FLUORO-5- (P-FLUORPHENYL) -2,3,4,4A, 5,9B-HEXSAHYDRO-1H-PYRIDO (4,3-B) INDOL SOM ANVAENDS SOM MELLANPROTUKT VID FRAMSTAELLNING AV THERAPEUTIC ANVAENDBARA 2-SUBSTITUERADE DL- OCH D-TRANS-8-FLUOR-5- (P-FLUORPHENYL) -2,3,4,4A, 5,9B-HEXSAHYDRO-1H-PYRIDO (4,3-B) INDOLER |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20753419 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 32PN | Ep: public notification in the ep bulletin as address of the adressee cannot be established |
Free format text: NOTING OF LOSS OF RIGHTS PURSUANT TO RULE 112(1) EPC (EPO FORM 1205A DATED 04/05/2023) |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20753419 Country of ref document: EP Kind code of ref document: A1 |