WO2020125673A1 - 流感病毒复制抑制剂及其中间体和用途 - Google Patents
流感病毒复制抑制剂及其中间体和用途 Download PDFInfo
- Publication number
- WO2020125673A1 WO2020125673A1 PCT/CN2019/126277 CN2019126277W WO2020125673A1 WO 2020125673 A1 WO2020125673 A1 WO 2020125673A1 CN 2019126277 W CN2019126277 W CN 2019126277W WO 2020125673 A1 WO2020125673 A1 WO 2020125673A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- preparation
- pyrrolo
- amino
- influenza
- Prior art date
Links
- 0 C*1ccc(C(C(N=C(C2=CC=IN2)N[C@@](C2CCC3CC2)[C@@]3C(O)=O)=N)=*)c(N)n1 Chemical compound C*1ccc(C(C(N=C(C2=CC=IN2)N[C@@](C2CCC3CC2)[C@@]3C(O)=O)=N)=*)c(N)n1 0.000 description 8
- WCDWAPXAJLVNHO-WYXOVGLDSA-N C[C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2c[nH]c3c2cccn3)n[n]2c1ccc2 Chemical compound C[C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2c[nH]c3c2cccn3)n[n]2c1ccc2 WCDWAPXAJLVNHO-WYXOVGLDSA-N 0.000 description 2
- PPBFZTZQAPUGOK-ULCALQBOSA-N C[C@@H](C1CCC2CC1)[C@H]2Nc1nc(Cl)n[n](cc2)c1c2F Chemical compound C[C@@H](C1CCC2CC1)[C@H]2Nc1nc(Cl)n[n](cc2)c1c2F PPBFZTZQAPUGOK-ULCALQBOSA-N 0.000 description 2
- VEJLQVWTSIXAPJ-UHFFFAOYSA-O CC(C1=C(CC2)CCC2C1Nc1nc(C(c2c([NH3+])ncc(F)c2)=N)n[n](cc2)c1c2F)=O Chemical compound CC(C1=C(CC2)CCC2C1Nc1nc(C(c2c([NH3+])ncc(F)c2)=N)n[n](cc2)c1c2F)=O VEJLQVWTSIXAPJ-UHFFFAOYSA-O 0.000 description 1
- QWQCIAOSKJVYND-AONTZOIISA-N CC(C[C@@H](CC12)[C@]11Nc3nc(-c4n[nH]c(nc5)c4cc5F)n[n]4c3ccc4)[C@H]2[C@@H]1C(O)=O Chemical compound CC(C[C@@H](CC12)[C@]11Nc3nc(-c4n[nH]c(nc5)c4cc5F)n[n]4c3ccc4)[C@H]2[C@@H]1C(O)=O QWQCIAOSKJVYND-AONTZOIISA-N 0.000 description 1
- ILDFWQPYJXWKKP-YWTDHEFTSA-N CC(C[C@@H]1C(O)=O)C(CC2C3)C3[C@]12Nc1nc(-c2c[nH]c(nc3)c2cc3Cl)n[n]2c1ccc2 Chemical compound CC(C[C@@H]1C(O)=O)C(CC2C3)C3[C@]12Nc1nc(-c2c[nH]c(nc3)c2cc3Cl)n[n]2c1ccc2 ILDFWQPYJXWKKP-YWTDHEFTSA-N 0.000 description 1
- GPFNGAJDZLCRQR-MEKGUCOXSA-N CC([C@@H](C1CCC2CC1)[C@H]2N/C(/c1cc(Cl)c[nH]1)=N/C(C(CN1)c2c1ncc(F)c2)=N)O Chemical compound CC([C@@H](C1CCC2CC1)[C@H]2N/C(/c1cc(Cl)c[nH]1)=N/C(C(CN1)c2c1ncc(F)c2)=N)O GPFNGAJDZLCRQR-MEKGUCOXSA-N 0.000 description 1
- XAMBYRNHWOMBKE-IBYVOGSZSA-N CC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2c[nH]c(nc3)c2cc3Cl)n[n](cc2)c1c2F)=O Chemical compound CC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2c[nH]c(nc3)c2cc3Cl)n[n](cc2)c1c2F)=O XAMBYRNHWOMBKE-IBYVOGSZSA-N 0.000 description 1
- HBQJUHWBQZHSKY-WGCDDVKFSA-N CC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2n[nH]c(nc3)c2cc3Cl)n[n]2c1ccc2)O Chemical compound CC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2n[nH]c(nc3)c2cc3Cl)n[n]2c1ccc2)O HBQJUHWBQZHSKY-WGCDDVKFSA-N 0.000 description 1
- VXINTLFZOLFNGX-LGCIXGJRSA-N CCC(C1CC2[C@]34C(O)=O)C23[C@@]14Nc1nc(-c2c[nH]c(nc3)c2cc3F)n[n]2c1ccc2 Chemical compound CCC(C1CC2[C@]34C(O)=O)C23[C@@]14Nc1nc(-c2c[nH]c(nc3)c2cc3F)n[n]2c1ccc2 VXINTLFZOLFNGX-LGCIXGJRSA-N 0.000 description 1
- CFRDVZHAVFJIFL-QLEHZGMVSA-N CCOC([C@@H](C1CCC2CC1)[C@H]2N)=O Chemical compound CCOC([C@@H](C1CCC2CC1)[C@H]2N)=O CFRDVZHAVFJIFL-QLEHZGMVSA-N 0.000 description 1
- CGBFRCZRVQMEOY-PJODQICGSA-N CC[C@H]([C@@H]1C(O)=O)C=CC[C@@H]1Nc1nc(-c2n[nH]c(nc3)c2cc3F)n[n]2c1ccc2 Chemical compound CC[C@H]([C@@H]1C(O)=O)C=CC[C@@H]1Nc1nc(-c2n[nH]c(nc3)c2cc3F)n[n]2c1ccc2 CGBFRCZRVQMEOY-PJODQICGSA-N 0.000 description 1
- HDEBEXMMRFOWDY-BRHYBRFTSA-N C[C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c(c2c3)c[nH]c2ncc3Cl)n[n](cc2)c1c2F Chemical compound C[C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c(c2c3)c[nH]c2ncc3Cl)n[n](cc2)c1c2F HDEBEXMMRFOWDY-BRHYBRFTSA-N 0.000 description 1
- GAUQNRMZBRCTFV-PLHMTAAQSA-N C[C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c(c2c3nccc2)n[n]3[Tl])n[n](cc2)c1c2F Chemical compound C[C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c(c2c3nccc2)n[n]3[Tl])n[n](cc2)c1c2F GAUQNRMZBRCTFV-PLHMTAAQSA-N 0.000 description 1
- CVIVNKOXNULYBA-RDTNZQFSSA-N C[C@]1(C2C1CC1CC2)[C@H]1Nc1nc(Cl)n[n]2c1ccc2 Chemical compound C[C@]1(C2C1CC1CC2)[C@H]1Nc1nc(Cl)n[n]2c1ccc2 CVIVNKOXNULYBA-RDTNZQFSSA-N 0.000 description 1
- BSZGZNRUUJXKKQ-UHFFFAOYSA-N Clc1nc(Cl)n[n]2c1ccc2 Chemical compound Clc1nc(Cl)n[n]2c1ccc2 BSZGZNRUUJXKKQ-UHFFFAOYSA-N 0.000 description 1
- BTNFWFCWVZFQTG-UHFFFAOYSA-N Fc(cc1)c(c(Cl)n2)[n]1nc2Cl Chemical compound Fc(cc1)c(c(Cl)n2)[n]1nc2Cl BTNFWFCWVZFQTG-UHFFFAOYSA-N 0.000 description 1
- JHIYIGRBYXLHDA-IBYVOGSZSA-N N#Cc(cc12)c[n]1nc(-c1c[nH]c(nc3)c1cc3F)nc2N[C@@H](C1CCC2CC1)[C@H]2C(O)=O Chemical compound N#Cc(cc12)c[n]1nc(-c1c[nH]c(nc3)c1cc3F)nc2N[C@@H](C1CCC2CC1)[C@H]2C(O)=O JHIYIGRBYXLHDA-IBYVOGSZSA-N 0.000 description 1
- AOMRDQIIMQVAGM-UHBICSCXSA-N N#Cc1ccc2[n]1nc(-c1c[nH]c(nc3)c1cc3F)nc2N[C@@H]1[C@@H](C(O)O)C2CCC1CC2 Chemical compound N#Cc1ccc2[n]1nc(-c1c[nH]c(nc3)c1cc3F)nc2N[C@@H]1[C@@H](C(O)O)C2CCC1CC2 AOMRDQIIMQVAGM-UHBICSCXSA-N 0.000 description 1
- NXUIHPBVHDZWGB-JEBKPEFCSA-N NC(c1c(N)nccc1)c(nc1N[C@H]2C(CC3)CCC3[C@@H]2C/C=C/O)n[n]2c1ccc2 Chemical compound NC(c1c(N)nccc1)c(nc1N[C@H]2C(CC3)CCC3[C@@H]2C/C=C/O)n[n]2c1ccc2 NXUIHPBVHDZWGB-JEBKPEFCSA-N 0.000 description 1
- RJWQRVFVWBDRQC-GVSXRFJSSA-N Nc(nccc1)c1C(c(nc1N[C@@H](C2CCC3CC2)[C@H]3C(O)=O)n[n](cc2)c1c2F)=N Chemical compound Nc(nccc1)c1C(c(nc1N[C@@H](C2CCC3CC2)[C@H]3C(O)=O)n[n](cc2)c1c2F)=N RJWQRVFVWBDRQC-GVSXRFJSSA-N 0.000 description 1
- ZTYRINJUQJIQRH-WYBZEITLSA-N OC(C([C@H]1Nc2nc(-c3c[nH]c(nc4)c3cc4F)n[n]3c2ccc3)[C@H]2C=C[C@@H]1C=C2)=O Chemical compound OC(C([C@H]1Nc2nc(-c3c[nH]c(nc4)c3cc4F)n[n]3c2ccc3)[C@H]2C=C[C@@H]1C=C2)=O ZTYRINJUQJIQRH-WYBZEITLSA-N 0.000 description 1
- BGPLJGLQKVUPAV-KRWDZBQOSA-N OC(C1=C(CC2)CCC2[C@@H]1Nc1nc(-c2c[nH]c(nc3)c2cc3F)n[n](cc2)c1c2F)=O Chemical compound OC(C1=C(CC2)CCC2[C@@H]1Nc1nc(-c2c[nH]c(nc3)c2cc3F)n[n](cc2)c1c2F)=O BGPLJGLQKVUPAV-KRWDZBQOSA-N 0.000 description 1
- UEKDRRQBKZUDFV-BUZSGMRCSA-N OC([C@@H](C1CCC2CC1)/C2=N\c1nc(-c2n[nH]c(nc3)c2cc3Cl)n[n](cc2)c1c2F)=O Chemical compound OC([C@@H](C1CCC2CC1)/C2=N\c1nc(-c2n[nH]c(nc3)c2cc3Cl)n[n](cc2)c1c2F)=O UEKDRRQBKZUDFV-BUZSGMRCSA-N 0.000 description 1
- CRISJBHFEZHFON-DFYNNNJYSA-N OC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2c[nH]c(nc3)c2cc3F)n[n]2c1ccc2)=O Chemical compound OC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2c[nH]c(nc3)c2cc3F)n[n]2c1ccc2)=O CRISJBHFEZHFON-DFYNNNJYSA-N 0.000 description 1
- PPPUYCAZGHOCTL-YCWMGOOZSA-N OC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2c[nH]c3c2cccn3)n[n]2c1ccc2)=O Chemical compound OC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2c[nH]c3c2cccn3)n[n]2c1ccc2)=O PPPUYCAZGHOCTL-YCWMGOOZSA-N 0.000 description 1
- DNNYFDLJDXXFMA-HBGGEXNNSA-N OC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2n[nH]c3ncccc23)n[n](cc2)c1c2F)=O Chemical compound OC([C@@H](C1CCC2CC1)[C@H]2Nc1nc(-c2n[nH]c3ncccc23)n[n](cc2)c1c2F)=O DNNYFDLJDXXFMA-HBGGEXNNSA-N 0.000 description 1
- WHBFRQOVBLUOFP-UGGGCRDHSA-N OC([C@@H]([C@H]1C2CC3C1)[C@@]23Nc1nc(-c2n[nH]c(nc3)c2cc3F)n[n](cc2)c1c2F)=O Chemical compound OC([C@@H]([C@H]1C2CC3C1)[C@@]23Nc1nc(-c2n[nH]c(nc3)c2cc3F)n[n](cc2)c1c2F)=O WHBFRQOVBLUOFP-UGGGCRDHSA-N 0.000 description 1
- CMLZNAKIPJWUQK-VWLBSPQZSA-N OC([C@@]([C@@H]12)(C11C3C2=C[C@@H]32)[C@@]12Nc1nc(-c2n[nH]c3c2cccn3)n[n]2c1ccc2)=O Chemical compound OC([C@@]([C@@H]12)(C11C3C2=C[C@@H]32)[C@@]12Nc1nc(-c2n[nH]c3c2cccn3)n[n]2c1ccc2)=O CMLZNAKIPJWUQK-VWLBSPQZSA-N 0.000 description 1
- NZQMTKIKNZYQCQ-UHFFFAOYSA-O [NH3+]c(ncc(Cl)c1)c1C(c(nc1NC(C2CC3CC2)C3C(O)=O)n[n]2c1ccc2)=N Chemical compound [NH3+]c(ncc(Cl)c1)c1C(c(nc1NC(C2CC3CC2)C3C(O)=O)n[n]2c1ccc2)=N NZQMTKIKNZYQCQ-UHFFFAOYSA-O 0.000 description 1
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/06—Peri-condensed systems
Definitions
- This application belongs to the field of chemical medicine, and specifically relates to an influenza virus replication inhibitor and its intermediates and uses.
- Influenza is a respiratory disease caused by the Influenza virus.
- influenza viruses can be divided into three types: A (A), B (B), and C (C).
- A A
- B B
- C C
- influenza A is the most common and highly pathogenic. It is prone to large-scale epidemics and seriously threatens human life and health.
- vaccination and anti-influenza treatment are usually used: vaccination and anti-influenza treatment.
- Vaccination is an effective measure to prevent influenza.
- adults can achieve better preventive effects after vaccination, but infants, the elderly, and those with lower immunity are not ideal after vaccination.
- influenza viruses are constantly mutating, making it difficult for old vaccines to fight new viruses.
- Chemical drugs are another important means of treating influenza, but so far, the number of anti-influenza chemical drugs on the market is small, and the most used are M2 ion channel inhibitors, neuraminidase (NA) inhibitors and nucleoside antivirus drug.
- M2 ion channel inhibitors M2 ion channel inhibitors
- NA neuraminidase
- influenza RNA polymerase has received widespread attention.
- RdRp is a heterotrimer composed of three subunits of PA, PB1 and PB2, and plays an important role in the transcription and replication of influenza virus genome.
- Influenza virus RNA transcription has a special "cap” mechanism.
- the PB2 subunit is responsible for recognizing and binding the "cap structure" of the host precursor mRNA, and then the PA subunit cleaves the "cap” as a primer to initiate transcription process. Inhibiting "capping" can block the transcription process and achieve the effect of inhibiting the proliferation of influenza virus. Therefore, PB2 is regarded as a promising anti-influenza drug target, which has attracted great attention from pharmaceutical companies and academic research institutions.
- Pimodivir the world's first drug candidate for influenza virus polymerase complex PB2 subunit inhibitor is Pimodivir. Johnson & Johnson obtained global drug development rights from Vertex Pharmaceuticals in 2014. The drug is currently in Phase III clinical trials. Tests have shown that Pimodivir can significantly reduce the viral load of patients compared to the placebo group. Its structure is as follows:
- the present application discloses a class of compounds as inhibitors of influenza virus replication and their use in the preparation of drugs for the prevention or treatment of viral infectious diseases. Compared with existing compounds of the same type, the compounds of the present application show better resistance Viral activity.
- the application provides compounds of formula (I), formula (II) or formula (III):
- R 1 and R 2 are independently selected from hydrogen, C1-C6 alkyl, cyano, halogen, nitro or amino;
- X is selected from C or N.
- R 1 and R 2 are each independently selected from hydrogen, cyano, or halogen.
- R 1 is selected from hydrogen, cyano, fluorine, or chlorine.
- R 2 is selected from hydrogen, fluorine, or chlorine.
- the present application relates to the following compounds and pharmaceutically acceptable salts, esters, solvates, polymorphs, prodrugs, stereoisomers or tautomers, but not limited to these compounds :
- the present application provides the use of the compound for preparing a medicine for preventing or treating viral infectious diseases.
- the viral infection is an influenza virus infection.
- the viral infection is an influenza A virus infection.
- R 1 is selected from hydrogen, C1-C6 alkyl, cyano, halogen, nitro or amino;
- R 3 is selected from chlorine, bromine or iodine.
- R 1 is selected from hydrogen, cyano, fluorine, chlorine, or bromine; R 3 is chlorine.
- the compound of formula (IV), formula (V), or formula (VI) has the following structure:
- FIG. 1 Survival diagram of mice treated with H1N1 Puerto Rico/8/34 virus 2 hours before infection;
- FIG. 1 Survival map of mice treated with H1N1 Puerto Rico/8/34 virus 24 hours after infection.
- DIPEA N,N-diisopropylethylamine
- Pd(dppf)Cl 2 [1,1'-bis(diphenylphosphino)ferrocene]palladium dichloride;
- MgSO 4 magnesium sulfate
- Zn(CN) 2 zinc cyanide
- DPPF 1,1'-bis(diphenylphosphine)ferrocene
- Pd(PPh 3 ) 4 tetrakis(triphenylphosphine)palladium
- K 3 PO 4 potassium phosphate
- DMSO dimethyl sulfoxide
- DMAC N,N-dimethylacetamide.
- 2,4-Dichloropyrrolo[2,1-f][1,2,4]triazine (10.00g, 53.18mmol, 1.0eq.) and THF (100mL) were added to the reaction flask, and the temperature was lowered in an ice water bath.
- DIPEA 13.75g, 106.4mmol, 2.0eq.
- (2S, 3S)-3-aminobicyclo[2.2.2]octane-2-carboxylic acid was added to the above reaction system in portions Ethyl ester (12.59g, 63.82mmol, 1.2eq.), stirred at room temperature for 2 hours.
- Compound 4 was prepared according to a similar method for synthesizing Compound 3 in Example 3, using 6-bromo-2,4-dichloropyrrolo[2,1-f][1,2,4]triazine as a starting material.
- reaction mixture was concentrated, tetrahydrofuran and methanol were removed, and the residue was diluted with water.
- Compound 7 was prepared according to the preparation method similar to compound 1 in Example 1, with 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2-yl)- 1H-Pyrrolo[2,3-b]pyridine-1-carboxylic acid tert-butyl ester was prepared as the starting material.
- Compound 10 was prepared according to the preparation method similar to compound 7 in Example 7, with 5-chloro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2 -Yl)-1-p-toluenesulfonyl-1H-pyrrolo[2,3-b]pyridine as the starting material.
- Compound 14 was prepared according to the preparation method similar to compound 1 in Example 1, starting with (1S, 2S, 3S, 4R)-3-aminobicyclo[2.2.2]oct-5-ene-2-carboxylic acid ethyl ester Made from raw materials.
- Compound 15 was prepared according to the preparation method similar to compound 1 in Example 1, with 5-fluoro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2 -Yl)-1-trityl-1H-pyrazolo[3,4-b]pyridine as the starting material.
- Compound 16 was prepared similarly to compound 15 in Example 15, with (2S, 3S)-3-((2-chloro-5-fluoropyrrolo[2,1-f][1,2,4] Azin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid ethyl ester as the starting material.
- the reaction mixture was concentrated to remove tetrahydrofuran and methanol, and the residue was diluted with water.
- Test article Some compounds 1, 2, and 3 of this application.
- MDCK cells were cultured in MEM medium containing 10% fetal bovine serum. Virus-infected cells were cultured in MEM medium containing 0.42% bovine serum albumin and 5 ⁇ g/mL trypsin. The day before the virus inoculation, MDCK cells were seeded onto 96-well cell culture plates at a density of 3 ⁇ 10 4 /well. On the next day, 50 PFU of influenza virus (influenza A/CA/07/2009) was added to 100 ⁇ L of MEM medium containing bovine serum albumin to infect MDCK cells, and incubated at 37°C for 1 hour.
- influenza virus influenza A/CA/07/2009
- the virus-infected cells were diluted twice with 100 ⁇ L of MEM medium, and incubated in medium containing different concentrations of test compounds (0-10 ⁇ M), and three replicate wells were set for each concentration. Three MDCK cell wells not infected by virus were used as control group. 20 ⁇ L of 0.15 mg/mL resazurin solution was added to each well of cells infected with virus for 72 hours and incubated for 4 hours. Fluorescence quantification experiments were carried out under 560nm excitation/590nm emission conditions. The MDCK cell protection rate is calculated according to the following formula. The EC 50 of the test compound to suppress influenza virus is obtained by transformation of the cell protection rate.
- Test object some compounds of this application, the structural formula and preparation method are shown in the preparation examples of each compound.
- MDCK of canine kidney cells was purchased from ATCC, catalog number CCL-34. The cells were cultured using EMEM (Sigma) medium supplemented with 10% fetal bovine serum (Hyclone), 1% double antibody (Hyclone), 1% L-glutamine (Gibco), and 1% non-essential amino acids (Gibco). OptiPRO SFM (Gibco) culture medium supplemented with 1% double antibody, 1% L-glutamine and 1% non-essential amino acids was used as the experimental culture medium. The culture medium for the experiment to which pancreatin (Invitrogen) was added was the culture medium for virus infection.
- Influenza virus A/PR/8/34 (H1N1) strain was purchased from ATCC, catalog number VR-1469.
- MDCK cells were seeded into 384-well test plates at a density of 2,000 cells per well and cultured in a 5% CO 2 , 37°C incubator overnight. The next day, compounds (8 concentration points, double wells) and virus were added to 384-well cell culture plates. The final concentrations of DMSO and pancreatin in the culture medium were 0.5% and 2.5 ⁇ g/mL, respectively. The cells were cultured in a 5% CO 2 and 37°C incubator for 5 days until the cytopathy of the compound-free virus control wells reached 80-95%. Cell counting kit 8 kit (Shanghai Li Ji) was used to detect cell viability. The antiviral activity of the compound is represented by the inhibition rate (%) of the compound at different concentrations on the virus-induced cytopathic effect. Calculated as follows:
- Inhibition rate (%) (test well reading-average virus control value) / (average cell control value-average virus control value) x 100
- DMSO N,N-dimethyl sulfoxide
- Solutol HS15 Polyethylene glycol 15 hydroxystearate
- mice Female BALB/c mice aged 6-8 weeks were selected to be infected with influenza A H1N1A/PuertoRico/8/34 virus by nasal drip.
- the day of infection was set to the 0th day of the experimental cycle.
- the oral administration of the test compound was started 2 hours before infection (PI-2) and 24 hours after infection (PI24), and the anti-influenza A virus H1N1 in vivo of the compound was evaluated by observing the survival rate and body weight changes of mice Medicine effect. The administration was continued for 10 days, and the observation period was 20 days. After 20 days, all surviving mice were euthanized.
- test compound solutions were prepared in physiological saline containing 5% N,N-dimethyl sulfoxide (DMSO) and 10% polyethylene glycol 15 hydroxystearate (Solutol HS15).
- DMSO N,N-dimethyl sulfoxide
- Solutol HS15 polyethylene glycol 15 hydroxystearate
- mice were placed in the anesthesia box, they were anesthetized with oxygen containing 5% v/v isoflurane (2.5 L/min). Under the condition of maintaining anesthesia with oxygen containing 2-2.5% isoflurane, they were slowly instilled into the nasal cavity 50 microliters of LD90 dose of H1N1 Puerto Rico/8/34 virus was used for infection.
- oral administration was started two hours before infection and 24 hours after infection.
- the positive control Pimodivir was administered twice daily in two different infection time experiments.
- the test compound 17 was administered twice a day in the PI-2 experiment and once a day in the PI24 experiment.
- the administered dose is shown in the results in Figures 1 and 2. Observe the state of mice every day and record body weight and survival rate. When the weight loss of the tested mice exceeded the standards stipulated by the animal welfare organization, the health index reached 6, severe drowsiness, and paralysis and other serious pathological conditions, euthanasia was carried out. At the end of the experiment, all surviving mice were euthanized.
- mice All the three doses (1, 3, 10 mg/kg, BID) of compound 17 were used in the experiment under the condition of starting medication two hours before infection, and the survival rate of mice reached 100%.
- the control compound Pimodivir has a survival rate of 0% at low doses (1 mg/kg, BID), and 80% at medium doses (3 mg/kg, BID), only at the highest dose (10 mg/kg, BID) to reach full survival.
- the survival rate results, after all three doses of the compound 17 group were treated, there was no significant decrease in the body weight of the animals.
- the control compound Pimodivir the low-dose group began to lose weight from day 4 and began to lose more than 20% of mouse weight on day 8.
- the survival rate of compound 17 at low dose once-daily administration (1 mg/kg, QD) was 60%, medium-dose and high-dose once-daily administration (3,10 mg/kg, QD), the survival rate of mice reached 100%.
- the control compound Pimodivir has a survival rate of 40% when administered at a low dose twice daily (1 mg/kg, BID), and 80% at a medium dose (3 mg/kg, BID), only at the highest dose condition (10mg/kg, BID) to achieve complete survival.
- the currently approved oseltamivir has a survival rate of 80% at 10 mg/kg and a BID dose.
- Compound 17 showed significantly superior anti-influenza A efficacy compared to the control compound Pimodivir at different administration times. In the case of administration 24 hours after infection, Compound 17 showed a similar drug effect when the total daily dose (3 mg/kg/Day) was much lower than Pimodivir (20 mg/kg/Day).
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Virology (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pulmonology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Epidemiology (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本申请公开了一类可作为流感病毒复制抑制剂的化合物,制备所述化合物的中间体,以及所述化合物用于制备预防或治疗病毒感染性疾病药物,尤其是A型流感病毒感染药物中的用途。
Description
本申请属于化学医药领域,具体涉及一种流感病毒复制抑制剂及其中间体和用途。
流行性感冒(简称流感,Flu)是由流感病毒(Influenza virus)引起的呼吸系统疾病。根据病毒核蛋白和基质蛋白的抗原决定簇的差异,可将流感病毒分为甲(A)、乙(B)、丙(C)三种类型,其中A型流感最为常见,致病性强,易发生大规模流行,严重威胁人类生命与健康。预防和治疗流感,通常采用疫苗接种和抗流感药物治疗两种方法。疫苗接种是目前预防流感的一种有效措施。成年人接种后可以达到较好的预防效果,但是婴幼儿、老年人等免疫力较低者接种后效果并不理想。而且流感病毒不断地变异,旧疫苗难以对抗新病毒。化学药物是治疗流感的另一种重要手段,但是迄今为止,上市抗流感化学药物数量少,应用较多的是M2离子通道抑制剂、神经氨酸酶(NA)抑制剂以及核苷类抗病毒药物。
近年来,流感RNA聚合酶(RdRp)受到了广泛关注。RdRp是由PA、PB1和PB2三个亚基组成的异源三聚体,在流感病毒基因组转录和复制过程中发挥重要作用。流感病毒RNA的转录具有特殊的“夺帽”机制,在此过程中,PB2亚基负责识别和结合宿主前体mRNA的“帽子结构”,然后PA亚基剪切“帽子”作为引物,启动转录过程。抑制“夺帽”可以阻断转录过程,达到抑制流感病毒增殖的效果。因此,PB2被认为是很有前途的抗流感药物靶标,已引起了制药公司和学术研究机构的高度重视。
目前,全球首创的流感病毒聚合酶复合物PB2亚单位抑制剂候选药物是Pimodivir,由强生公司在2014年从Vertex pharmaceuticals获得了药物的全球开发权益,该药目前正处于III期临床。试验表明,相比安慰剂组,Pimodivir可以显著降低病患的病毒载量。其结构如下所示:

公开日期为2017年2月3日的国际专利公开文本WO2017133664A1中也公开了一类用于制备抗流感药物的化合物,其中一些化合物表现出了较好的抗病毒活性,如:

目前临床上可应用的抗流感药物的种类和数量都较少,且均存在耐药性、临床效果差等问题,因此基于该新作用机理的临床候选化合物仍迫切地需要进行开发。
发明内容
本申请公开了一类作为流感病毒复制抑制剂的化合物以及其在制备预防或治疗病毒感染性疾病药物中的用途,与现有的同类型化合物相比,本申请的化合物显示出更好的抗病毒活性。
一方面,本申请提供了如式(I)、式(II)或式(III)的化合物:

其中,R
1、R
2分别独立地选自氢、C1-C6烷基、氰基、卤素、硝基或氨基;
X选自C或N。
在一些实施方案中,R
1、R
2分别独立地选自氢、氰基或卤素。
在另一些实施方案中,R
1选自氢、氰基、氟或氯。
在另一些实施方案中,R
2选自氢、氟或氯。
在另一些实施方案中,本申请涉及到以下化合物及其药学上可接受的盐、酯、溶剂化物、多晶型、前药、立体异构体或互变异构体,但不限于这些化合物:


另一方面,本申请提供了所述化合物用于制备预防或治疗病毒感染性疾病药物中的用途。
在一些实施方案中,所述病毒感染为流感病毒感染。
在另一些实施方案中,所述病毒感染为A型流感病毒感染。
另一方面,本申请提供可作为制备前述具有抗病毒活性化合物中间体的式(IV)、式(V)或式(VI)的化合物:

其中,R
1选自氢、C1-C6烷基、氰基、卤素、硝基或氨基;
R
3选自氯、溴或碘。
在一些实施方案中,R
1选自氢、氰基、氟、氯或溴;R
3为氯。
在另一些实施方案中,所述式(IV)、式(V)或式(VI)的化合物具有如下结构:

图1.H1N1 Puerto Rico/8/34病毒感染前2小时用药小鼠生存图;
图2.H1N1 Puerto Rico/8/34病毒感染后24小时用药小鼠生存图。
下面简写词的使用贯穿本申请:
THF:四氢呋喃;
DIPEA:N,N-二异丙基乙胺;
Pd(dppf)Cl
2:[1,1'-双(二苯基膦基)二茂铁]二氯化钯;
MgSO
4:硫酸镁;
Zn(CN)
2:氰化锌;
DPPF:1,1'-双(二苯基膦)二茂铁;
Pd(PPh
3)
4:四(三苯基膦)钯;
DCM:二氯甲烷;
MeCN:乙腈;
K
3PO
4:磷酸钾;
DMF:N,N-二甲基甲酰胺;
DMSO:二甲基亚砜;
DMAC:N,N-二甲基乙酰胺。
实施例1 (2S,3S)-3-((2-(5-氟-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物1)

步骤1 中间体1.1的制备
向反应瓶中加入2,4-二氯吡咯并[2,1-f][1,2,4]三嗪(10.00g,53.18mmol,1.0eq.)和THF(100mL),冰水浴降温,搅拌下滴加DIPEA(13.75g,106.4mmol,2.0eq.),加毕,向上述反应体系中分批加入(2S,3S)-3-氨基双环[2.2.2]辛烷-2-羧酸乙酯(12.59g,63.82mmol,1.2eq.),室温搅拌反应2小时。浓缩反应液,向浓缩液中加入乙酸乙酯和水,分层分液,有机相用无水硫酸镁干燥,浓缩制砂,柱层析(石油醚∶乙酸乙酯=5∶1)得中间体1.1,为黄色油状物18.47g, 收率:100%。
1HNMR(400MHz,CDCl
3)δ(ppm)7.51(s,1H),6.60(m,2H),5.58(brs,1H),4.68(m,1H),4.22(q,J=7.2Hz,2H),2.45(d,J=4.8Hz,1H),1.45-2.05(m,10H),1.26(t,J=7.2Hz,3H)。
步骤2 中间体1.2的制备
向反应瓶中加入中间体1.1(1.15g,3.31mmol,1.2eq.)、5-氟-3-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1H-吡咯并[2,3-b]吡啶-1-羧酸叔丁酯(1.00g,2.76mmol,1.0eq.)和1,4-二氧六环(30mL),向其中加入碳酸钾(1.14g,8.28mmol,3.0eq.)的水(10mL)溶液,再向反应体系中加入催化量的Pd(dppf)Cl
2,氮气保护下加热至回流反应8小时,向反应液中加入水后用乙酸乙酯萃取,有机相用饱和食盐水洗涤,无水硫酸镁干燥,浓缩制砂,柱层析(石油醚∶乙酸乙酯=3∶1)得中间体1.2为类白色固体0.43g,收率:35%。
1HNMR(400MHz,CDCl
3)δ(ppm)9.52(s,1H),8.62(dd,J=2.8Hz,9.3Hz,1H),8.31(d,J=2.6Hz,1H),8.26(t,J=1.8Hz,1H),7.62(dd,J=1.6Hz,2.5Hz,1H),6.63(dd,J=2.6Hz,4.4Hz,1H),6.56(t,J=1.2Hz,1H),5.41(d,J=6.7Hz,1H),4.93(m,1H),4.04-4.24(m,2H),2.49-2.51(m,1H),2.07(m,2H),1.62-1.93(m,8H),1.15(t,J=7.2Hz,3H)。
步骤3 化合物1的制备
将中间体1.2(430mg,0.96mmol,1.0eq.)溶于四氢呋喃(30mL)置于反应瓶中,加入一水合氢氧化锂(80mg,1.92mmol,2.0eq.)的水(10mL)溶液,反应体系升温至回流反应16小时,浓缩反应液后加入水,用6N盐酸水溶液调节pH至5~6,用乙酸乙酯萃取,有机相用无水硫酸镁干燥,浓缩制砂,柱层析(二氯甲烷∶甲醇=30∶1)得化合物1为类白色固体200mg,收率:50%。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.31(brs,1H),12.18(brs,1H),8.52(d,J=8.6Hz,1H),8.28(s,1H),8.18(s,1H),7.88(s,1H),7.68(s,1H),6.97(s,1H),6.60(s,1H),4.81(s,1H),2.78(s,1H),1.42-2.03(m,10H);LC-MS(m/z):421[M+H]
+。
实施例2 (2S,3S)-3-((5-氟-2-(5-氟-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物2)

步骤1 中间体2.1的制备
将3-氟-1H-吡咯-2-羧酸乙酯(7.20g,45.8mmol,1.0eq.)溶于DMF置于反应瓶中,冰水浴条件下分批加入氢化钠(60%,2.40g,59.6mmol,1.3eq.),加毕,搅拌1小时。分批加入O-(2,4-二硝基苯基)羟胺(13.67g,68.7mmol,1.5eq.),加毕,室温搅拌17小时。将反应混合物倒入冷水中,并用乙酸乙酯萃取。将有机相用盐水洗涤,MgSO
4干燥并浓缩至干。柱层析(石油醚∶乙酸乙酯=30∶1)得中间体2.1,7.00g,收率89%,为黄色油状物。
步骤2 中间体2.2的制备
将中间体2.1(7.00g,40.7mmol,1.0eq.)的饱和氨甲醇溶液(200mL)在密封管中加热至150℃反应48小时。将反应混合物浓缩至干,柱层析(石油醚∶乙酸乙酯=30∶1)得中间体2.2,2.30g,收率40%,为灰白色固体。
步骤3 中间体2.3的制备
将化合物2.2(2.30g,16.0mmol,1.0eq.)悬浮于甲苯(20mL)中,加入草酰氯(3.4mL,40.2mmol,2.5eq.),反应混合物加热回流17小时。反应物浓缩至干,甲醇洗涤,过滤,收集滤饼即得中间体2.3,1.80g,收率67%,为灰白色固体。
步骤4 中间体2.4的制备
密闭试管中加入中间体2.3(1.00g,6.0mmol,1.0eq.),POCl
3(4.60g,30.0mmol,5.0eq.),DIPEA(2.33g,18.0mmol,3.0eq.),反应混合物加热至120℃反应12小时。将反应混合物倒入冷水中,并用饱和碳酸钠水溶液调节pH=7-8。用乙酸乙酯萃取混合物。有机相用盐水洗涤,MgSO
4干燥并浓缩至干。柱层析(石油醚∶乙酸乙酯=100∶1)得中间体2.4,0.97g,收率79%,为黄色油状物。
步骤5 化合物2的制备
根据实施例1中的合成方法,使用化合物2.4作为起始原料获得化合物2。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.24-12.27(m,2H),8.49(dd,J=2.8Hz,9.6Hz,1H),8.30(s,1H),8.18(d,J=2.8Hz,1H),7.61-7.63(m,1H),7.14(d,J=6.4Hz,1H),6.50(d,J=3.1Hz,1H),4.82(t,J=6.5Hz,1H),2.96(d,J=6.7Hz,1H),1.41-2.00(m,10H);LC-MS(m/z):439[M+H]
+。
实施例3 (2S,3S)-3-((7-氰基-2-(5-氟-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物3)

步骤1 中间体3.1的制备
中间体3.1以7-溴-2,4-二氯吡咯并[2,1-f][1,2,4]三嗪作为起始原料,使用与实施例1中的化合物1.1类似的方法合成。
步骤2 中间体3.2的制备
将中间体3.1(800mg,1.87mmol,1.0eq.)溶于DMAC(15mL)中,加入Zn(CN)
2(164mg,1.40mmol,0.75eq.),再加入催化量的DPPF和Pd(PPh
3)
4,将所得混合物在氮气条件下加热回流12小时。将反应混合物倒入冷水中,乙酸乙酯萃取混合物。有机相用盐水洗涤,MgSO
4干燥并浓缩至干。柱层析(石油醚∶乙酸乙酯=2∶1)得中间体3.2,300mg,收率43%,为淡黄色固体。
步骤3 化合物3的制备
化合物3根据实施例1中合成化合物1的类似方法,以中间体3.2为起始原料制得。
1HNMR (400MHz,DMSO-d
6)δ(ppm)12.39(br,2H),8.46(dd,J=2.6Hz,9.6Hz,1H),8.40(d,J=6.4Hz,1H),8.33(s,1H),8.28(s,1H),7.35(d,J=4.7Hz,1H),7.11(d,J=4.7Hz,1H),4.80(t,J=6.4Hz,1H),2.75(d,J=6.6Hz,1H),1.43-2.02(m,10H);LC-MS(m/z):446[M+H]
+。
实施例4 (2S,3S)-3-((6-氰基-2-(5-氟-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物4)

化合物4根据实施例3中合成化合物3的类似方法,以6-溴-2,4-二氯吡咯并[2,1-f][1,2,4]三嗪为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.34(br,2H),8.49(d,J=8.2Hz,1H),8.44(s,1H),8.25-8.31(m,3H),7.43(s,1H),4.80(m,1H),2.73(d,J=5.9Hz,1H),1.44-2.03(m,10H);LC-MS(m/z):446[M+H]
+。
实施例5 (1R,2S,4R,5S,6S,7S)-7-((2-(5-氟-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)三环[3.2.2.0
2,4]壬烷-6-羧酸(化合物5)

步骤1 中间体5.1的制备
将(1S,2S,3S,4R)-3(((苄氧基)羰基)氨基)双环[2.2.2]辛-5-烯-2-羧酸乙酯(40.00g,121.4mmol,1.0eq.)溶于DCM,在冰水浴温度和氮气保护的条件下滴加2M二乙基锌的正己烷溶液(121.4mL,242.8mmol,2.0eq.)。向体系中滴加CH
2I
2(130.1g,485.7mmol,4.0eq.),在冰水浴温度下再滴加2M二乙基锌的正己烷溶液(121.4mL,242.8mmol,2.0eq.)。反应混合物升至室温反应过夜,用饱和氯化铵水溶液淬灭反应,然后用DCM萃取。有机相用盐水洗涤,用无水MgSO
4干燥并浓缩至干。柱层析(石油醚∶乙酸乙酯=10∶1)得中间体5.1,21.00g,收率50%,为无色油状物。
步骤2 中间体5.2的制备
将中间体5.1(21.00g,61.1mmol,1.0eq.)溶于MeCN,冰水浴温度下滴加三甲基碘硅烷(26.91g,134.5mmol,2.2eq.),反应2小时后加入三乙胺(15.46g,152.8mmol,2.5eq.),搅拌15分钟。反应混合物浓缩至干,加入乙酸乙酯萃取。有机相用水洗涤,用无水MgSO
4干燥并浓缩至干,即得中间体5.2,9.50g,收率75%。
步骤3 中间体5.3的制备
将2,4-二氯吡咯并[2,1-f][1,2,4]三嗪(8.48g,45.4mmol,1.0eq.)溶于四氢呋喃(95mL),在冰水浴温度下滴加DIPEA(17.59g,136.17mmol,3.0eq.),再加入中间体5.2(9.50g,45.4mmol,1.0eq.),室温搅拌2小时。反应混合物浓缩至干,加入乙酸乙酯萃取。有机相用水洗涤,用无水MgSO
4干燥并浓缩至干。柱层析(石油醚∶乙酸乙酯=8∶1)得中间体5.3,12.00g,收率73%,为黄色固体。
步骤4 中间体5.4的制备
将5-氟-3-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-对甲苯磺酰基-1H-吡咯并[2,3-b]吡啶(28.04g,67.4mmol,2.2eq.)和中间体5.3(11.10g,30.6mmol,1.0eq.)溶于1,4-二氧六环(300mL),向体系中加入K
3PO
4(21.62g,101.8mmol,3.0eq.)的水溶液(60mL),再加入催化量的Pd(dppf)Cl
2,混合物在氮气条件下加热回流8小时。反应物用水稀释,加入乙酸乙酯萃取。有机相用盐水洗涤,用无水MgSO
4干燥并浓缩至干。柱层析(石油醚∶乙酸乙酯=10∶1)得中间体5.4,15.00g,收率71%,为白色固体。
步骤5 化合物5的制备
将中间体5.4(15.00g,21.3mmol,1.0eq.)溶于DCM(150mL),加入三氟乙酸(24.30g,213.1mmol,10.0eq.)和三乙基硅烷(12.38g,106.6mmol,5.0eq.),室温搅拌过夜。反应混合物浓缩至干后,用甲醇(80mL),水(15mL)和四氢呋喃(20mL)溶解,再加入一水合氢氧化锂(2.18g,52.0mmol,3.0eq.),将反应混合物在50℃下搅拌5小时。浓缩反应混合物,除去四氢呋喃和甲醇,并将残余物用水稀释。将得到的混合物用6N HCl调节pH=5-6,过滤收集固体,用乙酸乙酯(10mL)洗涤,得到化合物5,3.85g,收率51%,为白色固体。
1HNMR(400MHz,DMSO-d
6)δ(ppm)14.15(brs,1H),12.56(brs,1H),8.65(s,1H),8.55(d,J=8.6Hz,1H),7.79(s,1H),7.73(d,J=4.1Hz,1H),7.06(d,J=3.2Hz,1H),6.70(dd,J=2.6Hz,4.2Hz,1H),4.62(brs,1H),2.85(d,J=6.4Hz,1H),2.76(s,1H),1.51-1.69(m,4H),1.10-1.12(m,2H),0.75-0.85(m,1H),0.30-0.39(m,1H);LC-MS(m/z):434[M+H]
+;[α]=-26.6°(c=1.08g/100mL,MeOH)。
实施例6 (1R,2S,4R,5S,6S,7S)-7-((5-氟-2-(5-氟-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)三环[3.2.2.0
2,4]壬烷-6-羧酸(化合物6)

化合物6根据实施例5中合成化合物5类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.28(s,1H),12.23(d,1H),8.50-8.47(dd,1H),8.29(d,1H),8.23(d,1H),7.64(t,1H),6.63(d,1H),6.51(d,1H),4.53(brs,1H),2.72(d,1H),2.51(m,1H),2.35(d,1H),1.69-1.67(m,2H),1.59-1.54(m,2H),1.10(m,1H),0.91-0.82(m,2H),0.49-0.37(m,1H);LC-MS(m/z):451[M+H]
+。
实施例7 (2S,3S)-3-((2-(1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物7)

化合物7根据实施例1中合成化合物1类似的制备方法,以3-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1H-吡咯并[2,3-b]吡啶-1-羧酸叔丁酯为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.30(s,1H),11.98(s,1H),8.75(d,J=1.36Hz,1H),8.27(d,J=1.6Hz,1H),8.09(d,J=2.7Hz,1H),7.84(d,J=6.6Hz,1H),7.65(t,J=2.3Hz,1H),7.18(d,J=4.7Hz,1H),6.95(d,J=1.4Hz,1H),6.58(d,J=2.6Hz,1H),4.78(t,J=6.7Hz,1H),2.76(d,J=7.0Hz,1H),2.03(s,2H),1.39-1.80(m,7H);LC-MS(m/z):404[M+H]
+。
实施例8 (2S,3S)-3-((5-氟-2-(1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物8)

化合物8根据实施例7中合成化合物7类似的制备方法,以实施例2中的中间体2.4为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.25(brs,1H),12.07(brs,1H),8.73(d,1H),8.29(d,1H),8.10(s,1H),7.58(t,1H),7.20(q,1H),7.08(d,1H),6.49(d,1H),4.82(m,1H),2.96(d,1H),2.03-1.99(m,2H),1.78-1.60(m,3H),1.58-1.40(m,5H);LC-MS(m/z):422[M+H]
+。
实施例9 (1R,2S,4R,5S,6S,7S)-7-((2-(1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)三环[3.2.2.0
2,4]壬烷-6-羧酸(化合物9)

化合物9根据实施例7中合成化合物7类似的制备方法,以实施例5中的中间体5.2为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.37(s,1H),11.98(s,1H),8.74-8.76(d,J=7.9Hz,2H),8.29(s,1H),8.12(s,1H),7.64-7.66(d,J=8.4Hz,1H),7.20(s,1H),7.00(s,1H),6.58(s,1H),4.46(s,1H),2.68(s,2H),2.35(s,1H),1.71(s,1H),1.55(s,1H),1.01-1.06(d,J=18.1,2H),0.84(s,1H),0.36(s,1H);LC-MS(m/z):416[M+H]
+。
实施例10 (2S,3S)-3-((2-(5-氯-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物10)

化合物10根据实施例7中合成化合物7类似的制备方法,以5-氯-3-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-对甲苯磺酰基-1H-吡咯并[2,3-b]吡啶为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.32(s,1H),12.28(s,1H),8.78(d,J=2.4Hz,1H),8.29(d,J=2.4Hz,1H),8.17(d,J=2.7Hz,1H),7.90(d,J=6.7Hz,1H),7.69(t,J=2.2Hz,1H),6.97(q,1H),6.59(q,1H),4.80(t,1H),2.77(d,1H),2.03(s,2H),1.59-1.87(m,8H);LC-MS(m/z):438[M+H]
+。
实施例11 (2S,3S)-3-((2-(5-氯-1H-吡咯并[2,3-b]吡啶-3-基)-5-氟-吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物11)

化合物11根据实施例10中合成化合物10类似的制备方法,以实施例2中的中间体2.4为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.34(brs,1H),12.26(brs,1H),8.74(s,1H),8.30(s,1H),8.17(s,1H),7.63(m,1H),7.16(m,1H),6.50(s,1H),4.84(m,1H),2.98(d,1H),2.01(m,2H),1.79-1.91(m,3H),1.40-1.57(m,5H);LC-MS(m/z):456[M+H]
+。
实施例12 (1R,2S,4R,5S,6S,7S)-7-((2-(5-氯-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)三环[3.2.2.0
2,4]壬烷-6-羧酸(化合物12)

化合物12根据实施例10中合成化合物10类似的制备方法,以实施例5中的中间体5.2为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.38(s,1H),12.27(s,1H),8.76(s,1H),8.29(s,1H),8.20(s,1H),7.68-7.71(t,2H,J=14.0Hz),7.01-7.02(d,1H,J=2.6Hz),6.60(s,1H),4.47(s,1H),2.66-2.70(t,2H,J=16.0),2.40(s,1H),1.71-1.75(t,2H,J=15.8),1.55(s,2H),1.18-1.19(d,2H,J=6.8),0.82-0.96(d,2H),0.36(s,1H);LC-MS(m/z):450[M+H]
+。
实施例13 (1R,2S,3S,4R)-3-((6-氯-2-(5-氟-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物13)

化合物13根据实施例2中合成化合物2类似的制备方法,以4-氯-1H-吡咯-2-羧酸乙酯为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.34(s,1H),12.24(s,1H),8.48(d,J=2.8Hz,1H),8.29(d,J=1.4Hz,1H),8.19(d,J=2.8Hz,1H),7.93(d,J=6.7Hz,1H),7.88(d,J=1.8Hz,1H),7.04(d,J=1.8Hz,1H),4.76(t,J=6.1Hz,1H),2.67(m,1H),2.03(s,2H),1.62-1.81(m,7H);LC-MS(m/z):456[M+H]
+。
实施例14 (1S,2S,3S,4R)-3-((2-(5-氟-1H-吡咯并[2,3-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛-5-烯-2-羧酸(化合物14)

化合物14根据实施例1中合成化合物1类似的制备方法,以(1S,2S,3S,4R)-3-氨基双环[2.2.2]辛-5-烯-2-羧酸乙酯为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)12.45(s,1H),12.19(s,1H),8.52(d,J=2.8Hz,1H),8.29(d,J=1.5Hz,1H),8.28(d,J=1.4Hz,1H),8.23(d,J=2.7Hz,1H),7.68(t,J=1.7Hz,1H),7.58(d,J=6.3Hz,1H),6.94(d,J=1.5Hz,1H),6.52-6.57(m,2H),6.27(t,J=7.0Hz,1H),4.76(s,1H),3.08(d,J=3.0Hz,1H),2.91(d,J=3.52Hz,1H),1.61-1.78(m,2H),1.13-1.34(m,3H);LC-MS(m/z):420[M+H]
+。
实施例15 (2S,3S)-3-((2-(5-氟-1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物15)

化合物15根据实施例1中合成化合物1类似的制备方法,以5-氟-3-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-三苯甲基-1H-吡唑并[3,4-b]吡啶为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)14.13(brs,1H),12.41(brs,1H),8.64(s,1H),8.58(d,J=8.6Hz,1H),8.11(d,J=7.7Hz,1H),7.77(s,1H),7.04(d,J=3.8Hz,1H),6.69(t,J=2.4Hz,1H),4.92(m,1H),2.85(t,J=7.0Hz,1H),1.57-2.07(m,10H);LC-MS(m/z):422[M+H]
+;[α]=-22.6°(c=0.87g/100mL,DMSO)。
实施例16 (2S,3S)-3-((5-氟-2-(5-氟-1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物16)

化合物16根据实施例15中合成化合物15类似的制备方法,以(2S,3S)-3-((2-氯-5-氟吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸乙酯为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)14.16(brs,1H),12.31(brs,1H),8.64(s,1H),8.57(d,J=8.7Hz,1H),7.70(s,1H),7.43(d,J=6.9Hz,1H),6.60(s,1H),4.98(m,1H),3.11(d,J=6.3Hz,1H),1.46-2.04(m,10H);LC-MS(m/z):440[M+H]
+;[α]=-19.0°(c=0.89g/100mL,DMSO)。
实施例17 (1R,2S,4R,5S,6S,7S)-7-((2-(5-氟-1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)三环[3.2.2.0
2,4]壬烷-6-羧酸(化合物17)

将5-氟-3-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1-三苯甲基-1H-吡唑并[3,4-b]吡啶(28.04g,67.4mmol,2.2eq.)和中间体5.3(11.10g,30.6mmol,1.0eq.)溶于1,4-二氧六环(300mL),加入K
3PO
4(21.62g,101.8mmol,3.0eq.)的水(60mL)溶液,再加入催化量的Pd(dppf)Cl
2,将所得混合物在氮气下加热回流8小时。将反应混合物用水稀释,并用乙酸乙酯萃取。有机相用盐水洗涤,用无水MgSO
4干燥并浓缩至干。柱层析(石油醚∶乙酸乙酯=10∶1)得中间体17.1,15.00g,收率71%,为白色固体。
将中间体17.1(15.00g,21.3mmol,1.0eq.)溶于DCM(150mL),加入三氟乙酸(24.30g,213.1mmol,10.0eq.)和三乙基硅烷(12.38g,106.6mmol,5.0eq.),将反应混合物在室温下搅拌过夜。将反应混合物浓缩至干,并将获得的残余物用甲醇(80mL),水(15mL)和四氢呋喃(20mL)溶解,加入一水合氢氧化锂(2.18g,52.0mmol,3.0eq.)。将反应混合物在50℃下搅拌5小时。浓缩反应混合物以除去四氢呋喃和甲醇,并将残余物用水稀释。将得到的混合物用6N HCl调节pH=5-6,过滤收集固体,用乙酸乙酯(10ml)洗涤,得到化合物17,3.85g,收率51%,为白色固体。
1HNMR(400MHz,DMSO-d
6)δ(ppm)14.15(brs,1H),12.56(brs,1H),8.65(s,1H),8.55(d,J=8.6Hz,1H),7.79(s,1H),7.73(d,J=4.1Hz,1H),7.06(d,J=3.2Hz,1H),6.70(dd,J=2.6Hz,4.2Hz,1H),4.62(brs,1H),2.85(d,J=6.4Hz,1H),2.76(s,1H),1.51-1.69(m,4H),1.10-1.12(m,2H),0.75-0.85(m,1H),0.30-0.39(m,1H);LC-MS(m/z):434[M+H]
+;[α]=-26.6°(c=1.08g/100mL,MeOH)。
实施例18 (1R,2S,4R,5S,6S,7S)-7-((5-氟-2-(5-氟-1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)三环[3.2.2.0
2,4]壬烷-6-羧酸(化合物18)

化合物18根据实施例15中合成化合物15类似的制备方法,以2,4-二氯-5-氟-[2,1-f][1,2,4]三嗪和实施例5中的中间体5.2为起始原料制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)8.57(s,1H),8.49-8.47(d,1H),7.66(s,1H),4.77(s,1H),2.43(s,1H),2.35(s,1H),1.68-1.63(m,3H),1.41-1.39(m,1H),0.98(s,1H),0.79(s,2H),0.33-0.32(m,1H);LC-MS(m/z):452[M+H]
+。
实施例19 (1S,2S,3S,4R)-3-((2-(5-氟-1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛-5-烯-2-羧酸(化合物19)

化合物19根据实施例15中合成化合物15类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)8.70-8.60(m,2H),7.80-7.75(m,2H),7.03-7.02(d,1H),6.66-6.64(dd,1H),6.59-6.55(t,1H),6.24-6.21(t,1H),4.92(s,1H),3.14(s,1H),2.98(m,1H),2.63(s,1H),1.78-1.75(m,2H),1.62(m,1H),1.36-1.29(m,1H),1.16-1.14(m,1H);LC-MS(m/z):421[M+H]
+。
实施例20 (1R,2S,3S,4R)-3-((2-(1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物20)

化合物20根据实施例15中合成化合物15类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)13.91(s,1H),12.40(s,1H),8.84(d,J=1.5Hz,1H),8.57(d,J=1.5Hz,1H),8.06(d,J=7.0Hz,1H),7.74(d,J=1.8Hz,1H),7.30(d,J=4.4Hz,1H),7.03(d,J=1.4Hz,1H),6.68(d,J=2.6Hz,1H),4.89(t,J=6.8Hz,1H),2.84(d,J=7.0Hz,1H),2.06(s,1H),1.63-1.79(m,7H);LC-MS(m/z):405[M+H]
+。
实施例21 (1R,2S,3S,4R)-3-((5-氟-2-(1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物21)

化合物21根据实施例15中合成化合物15类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6) δ(ppm)13.98(brs,1H),12.31(brs,1H),8.84-8.82(d,1H),8.58(d,1H),7.68(t,1H),7.38(d,1H),7.32(q,1H),6.60(d,1H),4.96(m,1H),3.10(d,1H),2.04-1.99(m,2H),1.82-1.72(m,3H),1.61-1.38(m,5H);LC-MS(m/z):423[M+H]
+。
实施例22 (1R,2S,4R,5S,6S,7S)-7-((2-(1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)三环[3.2.2.02,4]壬烷-6-羧酸(化合物22)

化合物22根据实施例15中合成化合物15类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)8.82-8.85(m,1H),8.57-8.89(m,1H),7.74-7.76(d,2H),7.30-7.34(m,1H),7.07-7.08(m,1H),6.68-6.70(m,1H),4.61(s,1H),2.86-2.87(t,1H),2.77(s,1H),1.68(s,2H),1.50-1.52(d,2H),1.11-1.13(m,1H),1.08(m,1H),0.82(s,1H),0.33(d,1H);LC-MS(m/z):417[M+H]
+。
实施例23 (1S,2S,3S,4R)-3-((2-(1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-(基)氨基)双环[2.2.2]辛-5-烯-2-羧酸(化合物23)
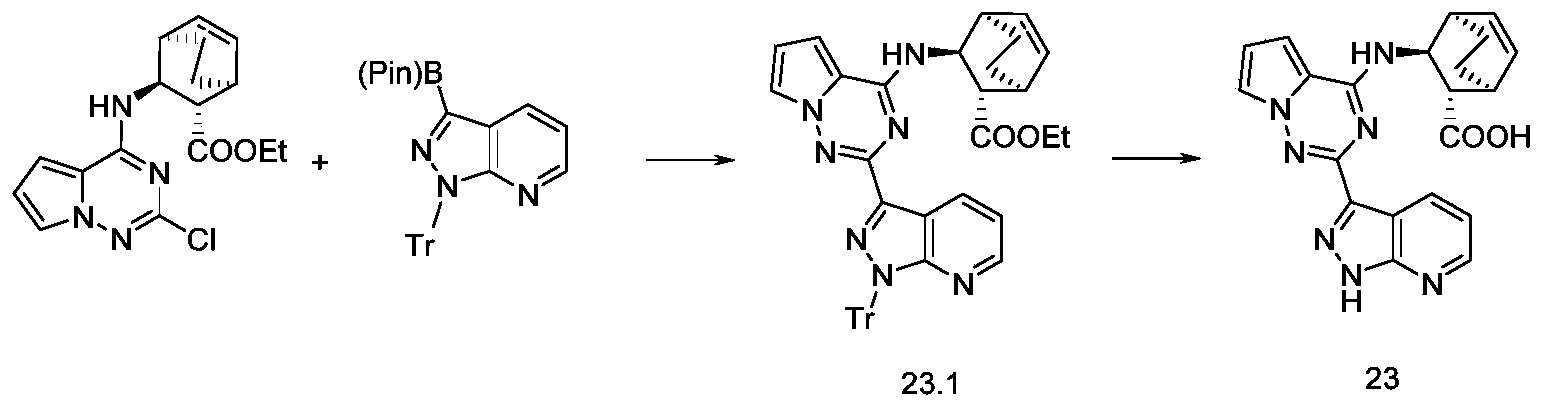
化合物23根据实施例15中合成化合物15类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)13.94(s,1H),12.60(s,1H),8.87-8.90(m,1H),8.58-8.59(m,1H),7.74-7.77(m,2H),7.31-7.34(m,1H),7.00-7.01(m,1H),6.65-6.67(m,1H),6.55-6.59(m,1H),6.22-6.25(m,1H),4.91(s,1H),2.97-3.17(m,2H),2.67(s,1H),1.59-1.63(m,2H);LC-MS(m/z):403[M+H]
+。
实施例24 (1R,2S,3S,4R)-3-((2-(5-氯-1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物24)

化合物24根据实施例15中合成化合物15类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)14.20(s,1H),12.40(s,1H),8.90(d,J=2.1Hz,1H),8.61(d,J=2.2Hz,1H),8.13(d,J=6.9Hz,1H),7.78(s,1H),7.05(d,J=3.2Hz,1H),6.69(q,J=2.7Hz,1H),4.91(t,J=6.4Hz,1H),2.85(d,J=6.8Hz,1H),2.04(d,J=13.9Hz,2H),1.78-1.87(m,3H),1.57-1.66(m,5H);LC-MS(m/z):439[M+H]
+。
实施例25 (1R,2S,3S,4R)-3-((2-(5-氯-1H-吡唑并[3,4-b]吡啶-3-基)-5-氟吡咯[2,1-f][1,2,4]三嗪-4-基)氨基)双环[2.2.2]辛烷-2-羧酸(化合物25)

化合物25根据实施例15中合成化合物15类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)14.25(brs,1H),12.32(brs,1H),8.88(d,1H),8.62(d,1H),7.71(t,1H),7.47(d,1H),6.61(d,1H),4.99(t,1H),3.11(d,1H),2.04-2.02(m,2H),1.83-1.62(m,3H),1.60-1.31(m,5H);LC-MS(m/z):457[M+H]
+。
实施例26 (1R,2S,4R,5S,6S,7S)-7-((2-(5-氯-1H-吡唑并[3,4-b]吡啶-3-基)吡咯并[2,1-f][1,2,4]三嗪-4-基)氨基)三环[3.2.2.0
2,4]壬烷-6-羧酸(化合物26)

化合物26根据实施例15中合成化合物15类似的制备方法制得。
1HNMR(400MHz,DMSO-d
6)δ(ppm)14.21(s,1H),12.53(s,1H),8.85(s,1H),8.61(s,1H),7.79(s,1H),7.74(s,1H),7.07(s,1H),6.70(s,1H),4.63(s,1H),2.85-2.86(d,J=5.20Hz,2H),2.75(s,1H),1.70-1.80(d,J=38.6Hz,2H),1.55-1.57(d,J=9.9Hz,2H),1.10-1.15(d,2H),0.85-0.89(m,2H),0.36(d,2H);LC-MS(m/z):451[M+H]
+。
实验例1 抑制流感病毒(influenza A/CA/07/2009)活性实验
测试物:本申请部分化合物1、2、3,其结构式和制备方法见各个化合物的制备实施例。
对照物:Pimodivir,其结构式见背景技术部分。
实验方法简述如下:
MDCK细胞在含有10%胎牛血清的MEM培养基中培养。病毒感染细胞在含有0.42%牛血清白蛋白和5μg/mL的胰蛋白酶的MEM培养基中培养。在接种病毒的前一天,将MDCK细胞按3×10
4/孔的密度接种到96孔的细胞培养板上。第二天,在100μL含有牛血清白蛋白的MEM培养基中加入50PFU的流感病毒(influenza A/CA/07/2009)感染MDCK细胞,置于37℃孵育1小时。移除病毒后,用100μL MEM培养基将病毒感染过的细胞进行两倍梯度稀释,分别在含有不同浓度测试化合物(0~10μM)的培养基中孵育,每个浓度设定三个复孔。三个未被病毒感染的MDCK细胞孔作为对照组。在被病毒感染72小时细胞的每孔中加入0.15mg/mL刃天青溶液20μL,孵育4小时。采用荧光剂在560nm激发/590nm发射条件下进行荧光定量实验。MDCK细胞保护率按照下列公式计算。测试化合物抑制流感病毒的EC
50通过细胞保护率转化得到。

刃天青溶液的制备:
1、用pH 7.4的DPBS溶液溶解刃天青至浓度为0.15mg/mL;
2、采用0.2μm过滤器过滤刃天青溶液至无菌避光试管中;
3、刃天青溶液避光保存在-20℃冰箱中。
实验结果:
表1 本申请部分化合物的流感病毒抑制活性

由表1可知,化合物1、2、3在细胞内显示出足够好的流感病毒抑制活性,且均优于对照物。
实验例2 抑制流感病毒(A/PR/8/34(H1N1))活性实验
测试物:本申请部分化合物,其结构式和制备方法见各个化合物的制备实施例。
对照物:Pimodivir,及WO2017133664A1中公开的化合物:

细胞:犬肾细胞MDCK购自ATCC,货号CCL-34。细胞使用添加了10%胎牛血清(Hyclone),1%双抗(Hyclone),1%L-谷氨酰胺(Gibco)和1%非必需氨基酸(Gibco)的EMEM(Sigma)培养液培养。添加了1%双抗,1%L-谷氨酰胺和1%非必需氨基酸的OptiPRO SFM(Gibco)培养液为实验用培养液。添加了胰酶(Invitrogen)的实验用培养液为病毒感染培养液。
病毒:流感病毒A/PR/8/34(H1N1)株购自ATCC,货号VR-1469。
实验步骤:
MDCK细胞以每孔2,000个细胞的密度接种到384孔测试板中并于5%CO
2、37℃培养箱中培养过夜。第二天,化合物(8个浓度点、双复孔)和病毒加入384孔细胞培养板。培养液中DMSO和胰酶的终浓度分别为0.5%和2.5μg/mL。细胞于5%CO
2、37℃培养箱中培养5天直至无化合物的病毒对照孔内细胞病变达80-95%。使用Cell counting kit 8试剂盒(上海李记)检测细胞活力。化合物的抗病毒活性由不同浓度下的化合物对病毒引起的细胞病变效应的抑制率(%)表示。计算公式如下:
抑制率(%)=(测试孔读值-病毒对照平均值)/(细胞对照平均值-病毒对照平均值)×100使用GraphPad Prism软件对化合物的抑制率和细胞活率进行非线性拟合分析,计算化合物的半数有效浓度(EC
50)值。
实验结果:
表2 本申请部分化合物的流感病毒抑制活性





可见本申请的化合物均具有良好的抗病毒活性,其中化合物15、16、17、19、22、24、25、26的抗病毒均显著优于对照物Pimodivir。
实验例3 体内药代动力学研究
将雄性CD-1小鼠(购自LC Laboratory Animal Co.LTD;22-23g;6-8周;n=18,每种给药途径9只,每个时间点3只动物)在测试化合物单一剂量条件下分别通过尾部静脉注射(5mg/kg)和口服饲喂(10mg/kg)进行给药处理,所用化合物溶液均在含5%N,N-二甲基亚砜(DMSO)和10%聚乙二醇15羟硬脂酸酯(Solutol HS15)的生理盐水中配制。实验动物在给药前一天禁食过夜并在给药之后4小时进食,自由饮水。所述研究符合实验动物管理评估和认可协会(国际AAALAC)和美国国立卫生研究院的指南和标准。在指定的取样时间点,使用异氟烷麻醉动物后,通过面部静脉或心脏穿刺,在异氟醚吸入麻醉状态下采用交错出血的方式采集大约110μL的血液样品到EDTA-2K管中存储。所采集血液样品保持在湿冰中并且在采样后15分钟内离心以获得血浆(2000g,4℃,5min)。将血浆样品储存在大约-70℃冷冻条件下,直至分析。在分析所采集血浆样品前,将20μL未稀释的血浆样品的等分试样加入200μL IS(含20ng/mL格列甲嗪的乙腈溶液)。将混合物在750rpm条件下涡旋10min并在5800rpm下离心10min。取2μL上清液的等分试样,采用UPLC-MS/MS-22(Triple QuadTM 6500)进行化合物浓度定量分析。通过分析含有格列甲嗪(20ng/mL)作为内参的3.0-3,000ng/mL测试化合物的一系列对照血浆等分式样来构建标准校准曲线。对于10倍稀释的血浆样品,将2μL血液样品的等分试样加入18μL空白稀释血浆,稀释因子为10。随后的操作与上述未稀释的血浆样品相同。
实验结果如表3所示。
表3

化合物15,16,17和对照化合物Pimodivir相比,均具有更低的体内清除率,更高的峰值浓度和总暴露量,呈现明显更为优良的成药性。
实验例4 甲型流感H1N1病毒小鼠感染模型体内药效研究
实验选用6-8周雌性BALB/c小鼠,经鼻滴感染甲型流感H1N1A/PuertoRico/8/34病毒。感染当天设定为实验周期第0天。分别在感染前2小时(PI-2)以及感染后24小时(PI24)开始进行待测化合物口服给药处理,通过观察小鼠存活率以及体重变化来评估化合物的抗甲型流感病毒H1N1的体内药效。连续给药10天,观察周期为20天。20天后所有存活小鼠实施安乐死。
待测化合物溶液均在含5%N,N-二甲基亚砜(DMSO)和10%聚乙二醇15羟硬脂酸酯(Solutol HS15)的生理盐水中配制。实验小鼠安置于麻醉箱后,采用含5%v/v异氟烷的氧气(2.5L/min)麻醉,在含2-2.5%异氟烷的氧气维持麻醉的状态下,缓慢鼻腔滴注50微升LD90剂量的H1N1 Puerto Rico/8/34病毒进行感染。根据实验要求分别在感染前两个小时以及感染后24小时开始口服给药。阳性对照物Pimodivir在两个不同感染时间实验中均采用每日两次给药方式。而受试化合物17在PI-2实验中采用每天两次给药方式,PI24实验给药方式为每天一次。给药剂量如图1,2结果所示。每天观察小鼠状态并记录体重及存活率。当受试小鼠出现体重下降超过动物福利组织所规定标准,健康指数到达6,严重嗜睡,以及瘫痪等严重病理状态时,实行安乐死。 实验结束,所有存活小鼠均实施安乐死。
实验结果如图1,2所示。
感染前两小时开始用药条件下,化合物17在实验所采用全部三个剂量(1,3,10mg/kg,BID),小鼠存活率均达到100%。而对照化合物Pimodivir在低剂量(1mg/kg,BID)时存活率为0%,在中剂量(3mg/kg,BID)时,存活率为80%,只有在最高剂量条件下(10mg/kg,BID)达到完全存活。与存活率结果相对应,化合物17组所有三个剂量用药处理后,动物体重未出现明显下降。而对照化合物Pimodivir,低剂量组从第4天开始出现体重下降,第8天开始出现小鼠体重下降超过20%现象,按照动物福利规范实施安乐死,直至第10天,所有动物体重下降超过20%。中剂量组从第6天开始出现明显体重下降。随着用药继续,从第9天开始体重逐渐恢复。其中在第10天,出现1例体重下降超过20%。
感染后24小时开始用药条件下,化合物17在低剂量每日一次给药(1mg/kg,QD)时存活率为60%,中剂量和高剂量每日一次给药(3,10mg/kg,QD)时,小鼠存活率均达到100%。而对照化合物Pimodivir在低剂量每日两次给药(1mg/kg,BID)时存活率为40%,在中剂量(3mg/kg,BID)时,存活率为80%,只有在最高剂量条件下(10mg/kg,BID)达到完全存活。目前已获批上市的奥司他韦在10mg/kg,BID剂量条件下,存活率为80%。
化合物17在不同给药时间都显示出和对照化合物Pimodivir相比明显优越抗甲型流感病毒的药效。感染后24小时后给药情况下,化合物17在每日总用药量(3mg/kg/Day)远低于Pimodivir的情况下(20mg/kg/Day),显示出类似的药效。
Claims (13)
- 式(I)、式(II)或式(III)的化合物:
 其中,R 1、R 2分别独立地选自氢、C1-C6烷基、氰基、卤素、硝基或氨基;X选自C或N。
其中,R 1、R 2分别独立地选自氢、C1-C6烷基、氰基、卤素、硝基或氨基;X选自C或N。 - 根据权利要求1所述的化合物,其中,R 1、R 2分别独立地选自氢、氰基或卤素。
- 根据权利要求1或2所述的化合物,其中,R 1选自氢、氰基、氟或氯。
- 根据权利要求1或2所述的化合物,其中,R 2选自氢、氟或氯。
- 根据权利要求1所述的化合物,其结构式为以下中的任意一种:


- 根据权利要求1-2和5中任一项的所述的化合物,其中,所述化合物以药学上可接受的盐、酯、溶剂化物、多晶型、前药、立体异构体或互变异构体的形式存在。
- 权利要求1-2和5中任一项所述的化合物用于制备预防或治疗病毒感染性疾病药物中的用途。
- 根据权利要求7所述的用途,其中,所述病毒感染为流感病毒感染。
- 根据权利要求8所述的用途,其中,所述病毒感染为A型流感病毒感染。
- 式(IV)、式(V)或式(VI)的化合物:
 其中,R 1选自氢、C1-C6烷基、氰基、卤素、硝基或氨基;R 3选自氯、溴或碘。
其中,R 1选自氢、C1-C6烷基、氰基、卤素、硝基或氨基;R 3选自氯、溴或碘。 - 根据权利要求10所述的化合物,其中,R 1选自氢、氰基、氟、氯或溴;R 3为氯。
- 根据权利要求10或11所述的化合物,其结构式为以下中的任意一种:

- 权利要求10所述化合物用于制备权利要求1-2和5中任一项所述的化合物的用途。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980062669.4A CN112771048B (zh) | 2018-12-19 | 2019-12-18 | 流感病毒复制抑制剂及其中间体和用途 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201811556309 | 2018-12-19 | ||
| CN201811556309.3 | 2018-12-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020125673A1 true WO2020125673A1 (zh) | 2020-06-25 |
Family
ID=71102499
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2019/126277 WO2020125673A1 (zh) | 2018-12-19 | 2019-12-18 | 流感病毒复制抑制剂及其中间体和用途 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN112771048B (zh) |
| WO (1) | WO2020125673A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022007966A1 (zh) * | 2020-07-10 | 2022-01-13 | 四川海思科制药有限公司 | Pb2抑制剂及其制备方法和用途 |
| CN115260105A (zh) * | 2021-04-30 | 2022-11-01 | 启东东岳药业有限公司 | 一种芳杂氨基甲酸类化合物及其制备方法和用途 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017133664A1 (en) * | 2016-02-05 | 2017-08-10 | Savira Pharmaceuticals Gmbh | Bicyclic pyridine and pyrimidine derivatives and their use in the treatment, amelioration or prevention of influenza |
-
2019
- 2019-12-18 WO PCT/CN2019/126277 patent/WO2020125673A1/zh active Application Filing
- 2019-12-18 CN CN201980062669.4A patent/CN112771048B/zh active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017133664A1 (en) * | 2016-02-05 | 2017-08-10 | Savira Pharmaceuticals Gmbh | Bicyclic pyridine and pyrimidine derivatives and their use in the treatment, amelioration or prevention of influenza |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022007966A1 (zh) * | 2020-07-10 | 2022-01-13 | 四川海思科制药有限公司 | Pb2抑制剂及其制备方法和用途 |
| CN115698011A (zh) * | 2020-07-10 | 2023-02-03 | 四川海思科制药有限公司 | Pb2抑制剂及其制备方法和用途 |
| CN115698011B (zh) * | 2020-07-10 | 2023-10-20 | 四川海思科制药有限公司 | Pb2抑制剂及其制备方法和用途 |
| CN115260105A (zh) * | 2021-04-30 | 2022-11-01 | 启东东岳药业有限公司 | 一种芳杂氨基甲酸类化合物及其制备方法和用途 |
| CN115260105B (zh) * | 2021-04-30 | 2024-01-16 | 启东东岳药业有限公司 | 一种芳杂氨基甲酸类化合物及其制备方法和用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN112771048A (zh) | 2021-05-07 |
| CN112771048B (zh) | 2022-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2023173708A1 (zh) | 三嗪类化合物或其可药用盐、异构体、药物组合物和用途 | |
| ES2684755T3 (es) | Métodos para preparar inhibidores de la replicación de virus de la gripe | |
| WO2022143473A1 (zh) | 一种核苷类化合物及其用途 | |
| JP2020128441A (ja) | インフルエンザウイルスの複製の阻害剤 | |
| WO2021151265A1 (zh) | 一种醛基类化合物的药物用途 | |
| AU2015298876A1 (en) | Indoles for use in influenza virus infection | |
| JP5665538B2 (ja) | (1s,2s,3s,4r)−3−[(1s)−1−アセチルアミノ−2−エチル−ブチル]−4−グアニジノ−2−ヒドロキシ−シクロペンチル−1−カルボン酸水和物及びその使用 | |
| WO2017198122A1 (zh) | 抗流感小分子化合物及其制备方法和用途 | |
| TWI844564B (zh) | 流感病毒複製之抑制劑 | |
| WO2020125673A1 (zh) | 流感病毒复制抑制剂及其中间体和用途 | |
| JP2021514967A (ja) | ピリジノイミダゾール系化合物の結晶型、塩型及びその製造方法 | |
| KR101902567B1 (ko) | 플루오로치환된 (3r,4r,5s)-5-구아니디노-4-아세트아미도-3-(펜탄-3-일옥시)사이클로헥센-1-카복실릭 엑시드, 이의 에스테르 및 이의 용도 | |
| AU2024204863A1 (en) | Antiviral 1,3-di-oxo-indene compounds | |
| CN115135646A (zh) | 取代的多环化合物及其药物组合物和用途 | |
| WO2020098710A1 (zh) | 一种取代的双芳香基酰胺化合物及其制备方法和应用 | |
| WO2022143424A1 (zh) | 一种多环吡啶酮衍生物和药物组合物及其应用 | |
| US11034655B1 (en) | Compounds for inhibiting the activity of SARS-COV-2 spike glycoprotein | |
| CN113603689B (zh) | 多环吡啶酮化合物及其药物组合物和用途 | |
| CA3106556A1 (en) | Pyrrolo[2,3-b]pyridin derivatives as inhibitors of influenza virus replication | |
| CN110590768B (zh) | 杂环化合物、其组合物及其作为抗流感病毒药物的应用 | |
| WO2024078618A1 (zh) | 一种含氰基取代的多肽类化合物的晶型及其制备方法 | |
| CN114057635A (zh) | 2-芳脲基-n-[2-(2-甲氧基苯氧基)乙基]烟酰胺类化合物及其应用 | |
| CN117800916A (zh) | 1-[(1h-吲唑-7-基)甲基]-3-芳基硫脲衍生物的制备及其应用 | |
| WO2018184392A1 (zh) | 一种含有肼基的吲哚胺2,3-双加氧化酶抑制剂 | |
| CN113979936A (zh) | 2-芳脲基-n-[3-(4-吗啉基)丙基]烟酰胺类化合物及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19899908 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19899908 Country of ref document: EP Kind code of ref document: A1 |