WO2016079216A1 - Physical forms of ibrutinib, a bruton's kinase inhibitor - Google Patents
Physical forms of ibrutinib, a bruton's kinase inhibitor Download PDFInfo
- Publication number
- WO2016079216A1 WO2016079216A1 PCT/EP2015/077042 EP2015077042W WO2016079216A1 WO 2016079216 A1 WO2016079216 A1 WO 2016079216A1 EP 2015077042 W EP2015077042 W EP 2015077042W WO 2016079216 A1 WO2016079216 A1 WO 2016079216A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ibrutinib
- solvate
- amorphous
- temperature
- phenoxyphenyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention refers to amorphous and crystalline forms of the Bruton's tyrosine kinase (Btk) inhibitor l-((/?)-3-(4-amino-3-(4-phenoxyphenyl)-l/-/-pyrazolo[3 / 4-c ]pyrimidin-l- yl)piperidin-l-yl)prop-2-en-l-one (ibrutinib).
- Btk Bruton's tyrosine kinase
- the invention refers to new crystalline solvate forms of ibrutinib, a method of using these crystalline solvate forms for the preparation of essentially pure amorphous ibrutinib as well as pharmaceutical compositions that comprise essentially pure amorphous ibrutinib. Further, the invention relates to essentially pure amorphous ibrutinib for the treatment of cancer.
- Btk Bruton's tyrosine kinase
- BCR cell surface B-cell receptor
- l-((/?)-3-(4-amino-3-(4-phenoxyphenyl)-lW-pyrazolo[3,4-d]pyrimidin-l-yl)piperidin-l-yl)prop-2- en-l-one is also known by its lUPAC name as l- ⁇ (/?)-3-[4-amino-3-(4-phenoxyphenyl)-lH- pyrazolo [3,4-d]pyrimidin-l-yl]piperidin-l-yl ⁇ prop-2-en-l-one or 2-propen-l-one,l-[(3 ?)-3-[4- amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl]-l-piperidinyl]-, and has been given the USAN name "ibrutinib", which will be used further in the document and refers to the compound with the following structure:
- Ibrutinib is a selective, irreversible inhibitor of BTK first disclosed in WO 2008/039218, which has been shown to be highly clinically efficacious in relapsed/refractory CLL and mantle cell lymphoma (see e.g. Burger et. Leukemia & Lymphoma (2013), 54(11), 2385-91).
- Amorphous ibrutinib is moisture stable. It is therefore a good alternative to crystalline ibrutinib as amorphous material has a higher solubility, which is connected with better bioavailability, than corresponding crystalline forms.
- amorphous ibrutinib is the preferred physical form for the preparation of pharmaceutical compositions.
- CN 103121999 A discloses ibrutinib of undisclosed form with an HPLC purity of 98.6% and >98% ee by crystallization from toluene.
- CN 103923084 A discloses anhydrous, hydrous as well as solvate crystal forms of ibrutinib, the solvate forms of ibrutinib being solvates of oxolane and trichloromethane.
- solvates from methanol, methylisobutylketone (MIBK), and toluene of ibrutinib have been specifically disclosed in WO 2013/184572 including formulations containing them and their use.
- MIBK methylisobutylketone
- the toluene solvate has been reported as being unstable.
- solvates with methanol and MIBK suffer from low solubility of ibrutinib in those solvents (about 1 wt% or less), thereby limiting their use for purification and subsequent preparation of amorphous ibrutinib on industrial scale as this would require very large amounts of solvent.
- the solvates from methanol and methylisobutylketone are not suitable for the preparation of amorphous ibrutinib for use in the manufacture of a medicament.
- amorphous ibrutinib can be obtained from material prepared by fast evaporation of solvent from dichloromethane solution.
- material used for this process has been purified by chromatography on silica gel using dichloromethane/alcohol mixtures. This approach bears the disadvantage that the eluent in the final purification step contains unspecified amounts of silica gel leaking from the column. Because such impurities might affect chemical stability in an uncontrolled manner such a material is not appropriate for use in a medicament.
- amorphous ibrutinib material prepared by fast rotary evaporation as described in WO 2008/039218 features a solid state best described as honey-like, gum, or foam, depending on the amount of residual dichloromethane still present. Such a material is difficult or even impossible to handle and process on a large scale.
- amorphous form of ibrutinib prepared from these new solvates is essentially free from a ny chemical impurities, and is essentially free from any crystalline forms of ibrutinib, such as for example crystalline form A.
- the process of the present invention for the preparation of amorphous ibrutinib has the advantage over the prior art process that it relies not on any column chromatography, and therefore the resulting product is free from any residual silica. Fina lly, amorphous ibrutinib is obtained as a free fluent powder, appropriate for use in the preparation of a medicament on industrial scale.
- solvates obtained from e.g. anisole, chlorobenzene, 1,4-dioxane, and pyridine were found to be useful for the preparation of crystalline, anhydrous form of ibrutinib on industrial scale.
- the present invention refers to a solvate of l-[(3/?)-3-[4-amino-3- (4-phenoxyphenyl)pyrazolo[3 ; 4-d]pyrimidin-l-yl]piperidin-l-yl]prop-2-en-l-one (ibrutinib) and a solvent wherein the solubility of amorphous ibrutinib in the corresponding solvent is in the range of 20 to 900 mg/mL at 40°C, preferably is in the range of 25 to 800 mg/mL at 40°C, most preferably is in the range of 30 to 750 mg/mL.
- the invention refers to the use of a solvate of ibrutinib for preparing amorphous ibrutinib.
- the invention refers to a process for the preparation of a solvate of ibrutinib and an organic solvent selected from anisole, chlorobenzene, 1,4-dioxane, pyridine, and dichloromethane, the process comprising the steps of a) providing a solution of ibrutinib in the organic solvent,
- the invention refers to a process for the preparation of amorphous ibrutinib comprising the steps of: a) providing a crystalline solvate of ibrutinib characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
- the invention refers to the amorphous ibrutinib obtained by the process as described herein.
- the invention refers to the amorphous ibrutinib for use in the treatment of cancer.
- the invention refers to a process for the preparation of anhydrous crystalline form C of ibrutinib, the process comprising the steps of: a) providing a solvate of ibrutinib with an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
- Figure 1 illustrates an X-Ray powder diffraction (XRPD) pattern (diffractogram) of the anisole solvate of ibrutinib.
- XRPD X-Ray powder diffraction
- Figure 2 illustrates a TG/DTA thermogram of the anisole solvate of ibrutinib.
- Figure 3 illustrates an X-Ray powder diffraction (XRPD) pattern of the chlorobenzene solvate of ibrutinib.
- Figure 4 illustrates a TG/DTA thermogram of the chlorobenzene solvate of ibrutinib.
- Figure 5 illustrates an X-Ray powder diffraction (XRPD) pattern of the dichloromethane solvate of ibrutinib.
- Figure 6 illustrates a TG/DTA thermogram of the dichloromethane solvate of ibrutinib.
- Figure 7 illustrates an X-Ray powder diffraction (XRPD) pattern of the 1,4-dioxane solvate of ibrutinib.
- Figure 8 illustrates a TG/DTA thermogram of the 1,4-dioxane solvate of ibrutinib.
- Figure 9 illustrates an X-Ray powder diffraction (XRPD) pattern of the pyridine solvate of ibrutinib.
- Figure 10 illustrates a TG/DTA thermogram of the pyridine solvate of ibrutinib.
- Figure 11 illustrates an X-Ray powder diffraction (XRPD) pattern of amorphous ibrutinib containing substantial amount of crystalline form A prepared by evaporation of dichloromethane solution.
- XRPD X-Ray powder diffraction
- Figure 12 illustrates an X-Ray powder diffraction (XRPD) pattern of amorphous ibrutinib prepared by lyophilization from chlorobenzene solvate of ibrutinib.
- Figure 13 illustrates an X-Ray powder diffraction (XRPD) pattern of amorphous ibrutinib after storage at 40°C / 75% RH for 8 week.
- Figure 14 illustrates an X-Ray powder diffraction (XRPD) pattern of crystalline form C of ibrutinib prepared by thermal desolvation of chlorobenzene solvate of ibrutinib.
- XRPD X-Ray powder diffraction
- the present invention refers to new crystalline forms of ibrutinib.
- the present invention refers to new crystalline solvates of ibrutinib.
- These new solvate forms are preferably characterized by a solubility at 40°C of amorphous ibrutinib in the solvent used for the solvate formation in the range of 20 to 900 mg/mL, preferably in the range of 25 to 800 mg/mL, most preferably in the range of 30 to 750 mg/mL.
- Solubility can for example be determined by a solvent addition technique as described in the Example 1.
- the present invention relates to a solvate, wherein ibrutinib is solvated with an organic solvent selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane, and pyridine.
- an organic solvent selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane, and pyridine.
- the present invention relates to a solvate, wherein ibrutinib is solvated with anisole, chlorobenzene, 1,4-dioxane, or pyridine.
- the solvate of ibrutinib of the present invention may be characterized by a stoichiometric composition of the solvates, i.e. number of solvent molecules per ibrutinib molecule as follows:
- Anisole solvate 0.5 mol ( ⁇ 0.1 mol) of anisole
- Chlorobenzene solvate 1.0 mol ( ⁇ 0.1 mol) of chlorobenzene
- 1,4-Dioxane solvate 0.7 mol ( ⁇ 0.1 mol) of dioxane
- Dichloromethane solvate 0.9 mol ( ⁇ 0.1 mol) of dichloromethane.
- the present invention refers to an anisole solvate of ibrutinib.
- the solvate is typically characterized by having one or more of the following properties:
- thermogravimetric/differential thermal analysis TG/DTA thermogram substantially similar to the one set forth in Figure 2, when measured at a rate of 10°C/min from 25°C to 300°C.
- TG/DTA thermogravimetric/differential thermal analysis
- the present invention refers to a chlorobenzene solvate of ibrutinib.
- the solvate is typically characterized by having one or more of the following properties:
- thermogravimetric/differential thermal analysis TG/DTA thermogram substantially similar to the one set forth in Figure 4, when measured at a rate of 10°C/min from 25°C to 300°C.
- the present invention refers to a dichloromethane solvate of ibrutinib.
- the solvate is typically characterized by having one or more of the following properties:
- thermogravimetric/differential thermal analysis TG/DTA thermogram substantially similar to the one set forth in Figure 6, when measured at a rate of 10°C/min from 25°C to 300°C.
- the present invention refers to a 1,4-dioxane solvate of ibrutinib.
- the solvate is typically characterized by having one or more of the following properties: (a) an X-Ray powder diffraction (XRPD) pattern substantially the same as shown in Figure 7;
- thermogravimetric/differential thermal analysis TG/DTA thermogram substantially similar to the one set forth in Figure 8, when measured at a rate of 10°C/min from 25°C to 300°C.
- the present invention refers to a pyridine solvate of ibrutinib.
- the solvate is typically characterized by having one or more of the following properties:
- thermogravimetric/differential thermal analysis TG/DTA thermogram substantially similar to the one set forth in Figure 10, when measured at a rate of 10°C/min from 25°C to 300°C.
- the solvates of ibrutinib of the present invention may be characterized by the peaks listed in the following table, showing the main 6 peaks and the main 10-11 peaks in an XRPD diffractogram.
- the solvates of the present invention can be prepared by a process comprising the steps of ) providing a solution of ibrutinib in an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
- the organic solvent in step a) is preferably selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane and pyridine, most preferably anisole and chlorobenzene. Dissolution of ibrutinib can be obtained at temperatures in the range of from 4°C to 100°C depending on the solvent used.
- seed crystals may be added in step b).
- the seed crystals are prepared by the same solvent used in step a) of the process.
- the seed crystals are prepared by the same process steps as the solvates of the present invention.
- the seed crystals are typically added in an amount of 0.1 wt% to 10 wt%, preferably in an amount of 0.5 wt% to 7.0 wt%, most preferably 1.0 wt.% 5.0 wt%, on the basis of the total amount of the starting material, i.e. the solution obtained in step a).
- step c) the solution transforms into a slurry by performing one or more of stirring the solution, or submitting the solution to one or more of temperature cycling, crash cooling, evaporation, or anti-solvent addition.
- Stirring is performed at a temperature of from 10°C to 100°C, preferably between 20°C and 50°C, most preferably about room temperature, e.g. 22 to 25°C, typically for a period of time of from 2 to 20h.
- room temperature is understood to mean temperatures between 15 and 25 °C [see e.g. European Pharmacopoeia 8.2, 1.2 (2014)].
- Temperature cycling is typically performed at atmospheric pressure, preferably at 2-22°C or 22-50°C, but also other conditions may be used depending on the type of solvate to be produced. Temperature cycling is typically performed for a period of 16 to 24 hours by subjecting the solution or slurry to temperature cycles such as between 2°C and 22°C, wherein each cycle may have a length between 2 and 8 hours, preferably about 4 hours.
- a typical but not limiting temperature cycling protocol may be as follows: i) Cooling from 22 to 2°C (4 h)
- Steps i) to iv) are typically repeated from 2 to 6 times.
- Crash cooling is typically performed by directly and rapidly cooling the solution from a temperature of between 25-50°C to a temperature of 2°C or below, such as a temperature between 0°C and -18°C, depending on the type of solvate, and keeping the solution at this temperature for example for 4 h to 24 h, such as for example 16-20 h.
- Evaporation is typically performed at reduced pressure, but may also be achieved at atmospheric pressure.
- the solvent used for anti-solvent addition may typically be selected from water, acetonitrile, a C M dialkylether, preferably methyliertbutylether ( TBE), or an alkane,preferably is selected from an alkane, more preferably a C 5 alkane, a C 5 alkane, a C 7 alkane, a C a alkane, or a mixture of two or more thereof, more preferably a C 7 alkane, more preferably n-heptane.
- Additional solvent may be added in step c) in case of formation of thick slurries to allow easier stirring of the resulting slurry.
- isolation in step d) may be performed by using procedures known in the art, such as by filtration, centrifugation, or evaporation of solvent.
- the isolated crystals may optionally be dried, e.g. under reduced pressure, typically at room temperature, or heated up to a temperature between 25°C and 50°C or the crystals may directly be used in further processes, such as the preparation of amorphous ibrutinib or isolated crystals may be used as seed crystals for the preparation of a solvate of ibrutinib.
- the invention further relates to the use of the solvates as described herein for the manufacture of amorphous ibrutinib, which can be obtained by the process of the invention in pure amorphous form.
- amorphous ibrutinib can be prepared by using the solvates of the present invention. After desolvation of solvates of the present invention, amorphous ibrutinib with higher purity than the starting material can be obtained.
- the present invention refers to the use of the solvates of ibrutinib as described herein in a process for preparing amorphous ibrutinib, such as pure amorphous ibrutinib.
- the process for the preparation of amorphous ibrutinib comprises the steps of: a) providing a crystalline solvate of ibrutinib and a solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
- the organic solvent in step a) is preferably selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane, and pyridine, most preferably is selected from anisole and chlorobenzene.
- desolvating in step d) is typically performed by one or more of thermal desolvation, hot melt-extrusion, spray drying, or lyophilisation.
- Solvents in pure form or mixtures used for spray drying are preferably selected from Biopharmaceutics Classification System (BCS) Class 3 solvent(s), further preferably with a low boiling point, most preferably acetone and dichloromethane.
- Solvents used for lyophilisation are typically selected from dioxane, dimethylsulfoxide (DMSO), acetic acid and tert-butanol, preferably acetic acid and DMSO.
- the amount of the solvent is not particular restricted, preferably 10 to 100 fold amount with respect to the substrate.
- the time for lyophilization is not particularly restricted, preferably 1 to 240 hours.
- the process optionally includes the additional step of washing the solution with an acidic aqueous solution of pH 1-5, preferably pH 2-4; e.g. aqueous phosphoric acid.
- the product obtained in step d) may further optionally be dried, for example under reduced pressure, typically at room temperature, or heated up to a temperature between 25°C and 90°C.
- the present invention further relates to the amorphous ibrutinib obtained by the process described herewith. It has surprisingly been found in the present invention that amorphous ibrutinib can be prepared with the above process in substantially pure form, and in particular can be prepared in pure form.
- the terms "pure” or “substantially pure” in the context of the present invention refer to phase-purity and chemical purity of the amorphous ibrutinib.
- Phase purity of amorphous ibrutinib is characterized by the absence of any peaks in the XRPD pattern.
- Chemical purity of the amorphous ibrutinib can for example be determined by HPLC analysis.
- the amorphous ibrutinib obtained by the process described herewith thus preferably has a chemical purity of 99 wt.% or more, preferably of 99.5 wt.% or more, more preferably of 99.9 wt.%, based on the total weight of the amorphous ibrutinib.
- amorphous ibrutinib can be prepared according to the method of the present invention without using column chromatography. As a consequence, any possible product contamination with silica can be avoided.
- the amorphous ibrutinib thus preferably has a content of silica, which is less than 1.0 wt.%, preferably is less than 0.3 wt.%, more preferably is less than 0.1 wt.%, further more preferably is less than 0.03 wt.%, most preferably is less than 0.01 wt%, based on the total weight of the amorphous ibrutinib, which can be determined by sulphated ash analysis according to Ph. Eur. 6.0, 2.4.14.
- amorphous ibrutinib which is essentially free from any crystal form of ibrutinib, such as crystal form A of ibrutinib.
- "Essentially free" in the context of the present invention means that no crystalline form, such as crystalline form A can be detected by XRPD measurement, i.e. no peaks of a crystalline form can be observed in an XRPD measurement.
- the invention is directed to the amorphous form of ibrutinib having no noticeable peak in a powder X-ray diffraction.
- Amorphous ibrutinib was stable under these conditions, as determined by the absence of XRPD peaks, in particular the absence of characteristic XRPD peaks for form A of ibrutinib after 8 weeks at 40°C / 75% RH.
- the process of the present invention has the further advantage that the amorphous ibrutinib can be obtained in powder form, and therefore has beneficial properties making it suitable for use in the preparation of a medicament and preparing same on industrial scale.
- the amorphous ibrutinib obtained in powder form is free flowing, and its particle size and particle size distribution can be controlled by methods known in the art, such as by milling and/or sieving the amorphous ibrutinib in powder form.
- the present invention refers to a combination of the process for the preparation of the solvates of ibrutinib and the process for the preparation of pure amorphous ibrutinib, as both described herein.
- crude amorphous ibrutinib may be used as the starting material for the preparation of the solvates of ibrutinib of the present invention, which may contain any chemical impurities, such as residual amounts of silica, or may contain residual amounts of crystal forms of ibrutinib, such as the crystal form A of ibrutinib, it is possible with the combination of both processes to obtain amorphous ibrutinib with higher purity than in the starting material.
- the present invention refers to a process for the preparation of pure amorphous ibrutinib, comprising the steps of: a) providing a solution of crude ibrutinib in an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane and pyridine, b) optionally adding seed crystals, and
- Amorphous ibrutinib with higher purity than in the amorphous ibrutinib used as the starting material in step a) is obtained in step g) or optionally in step h).
- the amorphous ibrutinib obtained by the above process is substantially free from silica or from residual amounts of crystal forms of ibrutinib such as crystal form A of ibrutinib, which however may be present in the crude amorphous ibrutinib used as the starting material in step a) of the process.
- the preferred embodiments of the process are the same as described above for the process for the preparation of the solvates of ibrutinib of the present invention and the process for the preparation of pure amorphous ibrutinib and thus also refer to the combination of both processes.
- a process as described above for the purification of l-((R)-3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)piperidin-l-yl)prop-2-en-l-one of ⁇ 90% purity such as crude ibrutinib for obtaining a purified ibrutinib having a purity of >97 wt.%, preferably >98wt.%, more preferably > 99%wt.% and most preferably >99.5 wt.% is provided.
- the solvates of ibrutinib as described herein can be used for the preparation of the crystalline form C of ibrutinib.
- the present invention further refers to the use of the solvates of ibrutinib described herein in a method for preparing the crystalline form C of ibrutinib.
- the present invention relates to a process for the preparation of anhydrous crystalline form C of ibrutinib, the process comprising the steps of: providing a solvate of ibrutinib with an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
- step b) optionally drying the product obtained in step b) or optionally c).
- the organic solvent in step a) is preferably selected from anisole, chlorobenzene, 1,4-dioxane, and pyridine, most preferably is selected from anisole and chlorobenzene.
- Typical conditions for performing desolvation in step b) are 50-120°C, preferably at 60-100°C. Thus, the step may also be referred to as "thermal desolvation" .
- desolvation in step b) may be performed at reduced or atmospheric pressure, preferably at reduced pressure, such as between 1 and 100 mbar, preferably between 5 and 50 mbar.
- Step b) may further be performed with or without a solvent, preferably without, i.e. in the absence of, a solvent.
- desolvating of step b) is performed by heating the solvate to a temperature between 50-120°C, preferably at 60-100°C, while the pressure is between 1 and 100 mbar, preferably between 5 and 50 mbar.
- Desolvation is typically performed for 0.5 to 12 hours, preferably between 1 and 4 hours, depending on the temperature and pressure applied during desolvation.
- Desolvation may further be performed by changing the temperature and/or the pressure during desolvation, e.g. by firstly heating the solvate at a temperature between 50 and 90°C for a first period, e.g. between 0.5 and 2 hours, and then increasing the temperature in a second step, such as between 90 and 120°C for e.g.
- the process optionally includes the additional step of washing the solution with an acidic aqueous solution of pH 1-5, preferably pH 2-4; e.g. aqueous phosphoric acid.
- Form C of ibrutinib obtained by the above process can be characterized by the X PD peaks of 7.010.2, 14.010.2, 15.8+0.2, 18.310.2, 19.210.2, 19.610.2, 20.3+0.2, 22.110.2, and 23.010.2° 2- Theta.
- the present invention refers to a process for the preparation of the crystalline form C of ibrutinib, the process comprising the steps of: a) providing a solution of ibrutinib in an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, b) optionally adding seed crystals,
- step e) optionally drying the product obtained in step e) or optionally f).
- the preferred embodiments of the process are the same as described above for the process for the preparation of the solvates of ibrutinib of the present invention and the process for the preparation of crystalline form C of ibrutinib and thus also refer to the combination of both processes.
- the crystalline form C of ibrutinib can be obtained by the process as described herein in pure or substantially pure form.
- the term pure or substantially pure in the context of the present invention refers to phase-purity and chemical purity of the form C of ibrutinib.
- Phase purity of form C is characterized by the absence of any XRPD peaks of anhydrous crystalline forms A and B.
- Chemical purity of the form C of ibrutinib can for example be determined by HPLC analysis.
- the form C of ibrutinib obtained by the process described herewith thus preferably has a chemical purity, of 99 wt.% or more, preferably of 99.5 wt.% or more, more preferably of 99.9 wt.%, based on the total weight of the form C of ibrutinib.
- the invention is directed to the crystalline form C of l-[(3 ?)-3-[4- amino-3-(4-phenoxyphenyl)pyrazolo[3,4-c/]pyrimidin-l-yl]piperidin-l-yl]prop-2-en-l-one having no noticeable peak of form A and/or B in a X-ray powder diffraction, in particular no noticeable peak of crystalline form A.
- amorphous ibrutinib or the crystal form C of ibrutinib as described above, preferably the amorphous form of ibrutinib as obtained by the process of the present invention may be used for the manufacture of a pharmaceutical composition.
- the present invention relates to a pharmaceutical composition comprising the forms of ibrutinib as described herein.
- the pharmaceutical composition typically comprises 1.0 to 1000 mg of amorphous ibrutinib or the crystal form C of ibrutinib, preferably comprises 10 to 800mg of amorphous ibrutinib, most preferably comprises 50 to 550 mg of amorphous ibrutinib.
- the pharmaceutical composition may further comprise one or more pharmaceutically acceptable additives, such as binders, carriers, stabilizers, diluents, dispersing agents, suspending agents, thickening agents, and/or excipients.
- Suitable excipients include, for example, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatine, gum tragacanth, methylcellulose, micro crystalline cellulose, hydroxypropyl methylcellulose, sodium carboxymethylcellulose; or others such as: polyvinylpyrrolidone (PVP or povidone) or calcium phosphate.
- disintegrating agents may be added, such as the cross-linked croscarmellose sodium, polyvinylpyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
- the pharmaceutical composition facilitates administration of the compound to a mammal, preferably to a human.
- Ibrutinib can be used singly or in combination with one or more therapeutic agents as components of mixtures.
- the pharmaceutical composition is typically a solid oral dosage form. It may be administered to a subject by multiple administration routes, including but not limited to, oral, parenteral (e.g., intravenous, subcutaneous, intramuscular), intranasal, buccal, topical, rectal, or transdermal administration routes.
- the pharmaceutical formulations described herein include, but are not limited to, aqueous liquid dispersions, self-emulsifying dispersions, solid solutions, liposomal dispersions, aerosols, solid dosage forms, powders, immediate release formulations, controlled release formulations, fast melt formulations, tablets, capsules, pills, delayed release formulations, extended release formulations, pulsatile release formulations, multiparticulate formulations, and mixed immediate and controlled release formulations.
- the pharmaceutical dosage form is a tablet or capsule.
- compositions may be manufactured in a conventional manner, such as, by way of example only, by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or compression processes.
- cancer may be a B cell malignancy, preferably selected from chronic lymphocytic leukemia (CLL)/ small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), indolent non-Hodgkins's lymphoma, diffuse large B Cell lymphoma (DLBCL), multiple myeloma(MM), marginal zone lymphoma (NHL), hairy cell leukemia, acute lymphocyte leukemia (ALL), and breast cancer.
- CLL chronic lymphocytic leukemia
- SLL small lymphocytic lymphoma
- MCL mantle cell lymphoma
- MM diffuse large B Cell lymphoma
- NHL marginal zone lymphoma
- hairy cell leukemia acute lymphocyte leukemia
- ALL acute lymphocyte leukemia
- the present invention further refers to a pharmaceutical composition as described above and comprising amorphous ibrutinib or the crystal form C of ibrutinib for use in the treatment of cancer.
- XRPD diffractograms were obtained with an X'Pert PRO diffractometer (PANalytical, Almelo, The Netherlands) equipped with a theta/theta coupled goniometer in transmission geometry, programmable XYZ stage with well plate holder, Cu-Kalpha 1 2 radiation source (wavelength 0.15419 nm) with a focusing mirror, a 0.5° divergence slit, a 0.02° soller slit collimator and a 0.5° anti-scattering slit on the incident beam side, a 2 mm anti-scattering slit, a 0.02° soller slit collimator, a Ni-filter and a solid state PIXcel detector on the diffracted beam side.
- the diffractogram was recorded at room temperature at a tube voltage of 40 kV, tube current of 40 mA, applying a step size of 0.013° 2-theta with 40 sec per step in the angular range of 2 ° to 40 ° 2-theta.
- a typical precision of the 2-theta values is in the range of ⁇ 0.2° 2-theta.
- a diffraction peak that appears for example at 9.4° 2-theta can appear between 9.2 and 9.6° 2- theta on most X-ray diffractometers under standard conditions.
- HPLC measurements were performed with a Agilent 1100 Series (UV/Vis detector) and a reversed phase column YMC-Pack Pro C18 RS (150 x 4.60 mm, 3 ⁇ ), using the following method:
- UV wavelength 245 nm
- Comparative Example 1 Experiments for preparation of amorphous form of ibrutinib by evaporation of a dichloromethane solution
- Amorphous ibrutinib prepared according to Comparative Example 1 was used as the input material for the solubility screen. Solubility values were estimated by a solvent addition technique in order to provide approximate values for designing further crystallisation experiments. Approximately 10 mg or 15 mg of ibrutinib were weighed each into a vial. Each solvent was added to the appropriate vial in 10 aliquots of 50 ⁇ . or until material was fully dissolved. In between additions the sample was heated to 40°C. If 1000 ⁇ . of solvent was added without dissolution of the material, solubility was calculated to be below this point. Results obtained from the solvent solubility screen are detailed in Table 3.
- ibrutinib prepared according to comparative Example 1 were added 330 ⁇ of anisole, yielding a turbid solution.
- the solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of anisole solvate of ibrutinib (5 mg, prepared according to Example 2, Route A).
- the mixture was further stirred at room temperature for a few minutes, leading to rapid slurry formation.
- An aliquot of the slurry was taken out and centrifuged during 6 minutes at 12000 r.p.m..
- the solid residue was dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding anisole solvate of ibrutinib.
- the rest of the slurry was temperature cycled (4 hour cycles) between 22°C and 2°C for a period of time of 20 hours (the cooling/heating rate after the 4 hour period was 0.083°C/min) and filtered.
- the solid residue was dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding crystalline anisole solvate of ibrutinib
- the X PD pattern is shown in Figure 1 and the TG/TGA pattern in Figure 2.
- ibrutinib prepared according to comparative Example 1 To 1.00 g of ibrutinib prepared according to comparative Example 1 were added 3.3 mL of anisole under stirring conditions (ca. 300 r.p.m.), yielding a turbid solution. The solution was stirred at room temperature. After 5 minutes, solid had precipitated out of solution. 2.5 mL anisole were added to the thick slurry and the suspension was further stirred between 20 and 30°C for 6 hours and cooled to a temperature of from 2 to 5°C.
- the slurry was filtered and dried under vacuum at a pressure of from 20 to 30 mbar and a temperature of 40°C for 6 hours where-after heating was stopped and the temperature of the solid residue was allowed to decrease slowly to 25°C at a pressure of from 20 to 30 mbar for 20 h, yielding 0.90 g of crystalline anisole solvate of ibrutinib.
- Chlorobenzene was added to 10.2 mg of ibrutinib prepared according to comparative Example 1 in six aliquots of 50 ⁇ . until a total of 300 ⁇ had been added. In between additions the sample was heated to a temperature of from 40 to 50°C on a hotplate. After addition of the fourth aliquot, ie. 300 ⁇ in total and subsequent heating to 40°C, complete dissolution was observed. The solution was then left at room temperature for ca. 1 hour, without stirring. After 1 hour, solid had precipitated out producing chlorobenzene solvate of ibrutinib.
- ibrutinib prepared according to comparative Example 1 were added 650 ⁇ of chlorobenzene, yielding a turbid solution. The solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of ibrutinib chlorobenzene solvate (5 mg, prepared according to Example 3, Route A). The mixture was further stirred at room temperature for a few minutes, leading to rapid slurry formation. An aliquot of the slurry was taken out and centrifuged during 6 minutes at 12000 r.p.m.. The solid residue was dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding chlorobenzene solvate of ibrutinib.
- the rest of the slurry was temperature cycled (4 hour cycles) between 22°C and 2°C for a period of time of 20 hours (the cooling/heating rate after the 4 hour period was 0.083°C/min) and filtered.
- the solid residue was dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding chlorobenzene solvate of ibrutinib.
- the XRPD pattern is shown in Figure 3 and the TG/TGA pattern in Figure 4.
- ibrutinib prepared according to comparative Example 1 were added 6.6 mL of chlorobenzene under stirring conditions (ca. 300 rpm), yielding a turbid solution.
- the solution was stirred at room temperature. After ca. 5 minutes, solid had precipitated out of solution.
- the slurry was further stirred between 20 and 30°C for 6 hours and cooled to a temperature of from 2 to 5°C.
- the slurry was filtered and dried under vacuum at a pressure of from 20 to 30 mbar and a temperature of 40°C for 6 hours whereafter heating was stopped and the temperature of the solid residue was allowed to decrease slowly to 25°C at a pressure of from 20 to 30 mbar for 20 h, yielding 0.96 g of crystalline chlorobenzene solvate of ibrutinib with an HPLC purity of 99.2%.
- ibrutinib prepared according to comparative Example 1 100 mg were dissolved in 400 ⁇ of pre-heated chlorobenzene at 50°C in a 2 mL vial. The resultant was filtered and stirred (ca. 300 r.p.m.) at ambient temperature. After 15 minutes, solid had precipitated out of solution. The sample was then stirred for a further 15 hours at ambient temperature. The slurry was filtered, yielding chlorobenzene solvate of ibrutinib.
- ibrutinib prepared according to the comparative Example 1 were dissolved in 1.0 mL of dichloromethane. The solution was evaporated at room temperature. The solid obtained after evaporation overnight was found to be crystalline anhydrous ibrutinib (Form A, according to WO2013184572). A further 0.5 mL of dichloromethane was added to the crystalline material and the resulting slurry was stirred (ca. 300 r.p.m.) at room temperature for 3 days. The solid residue was filtrated and dried at room temperature for 24 hours, yielding dichloromethane solvate of ibrutinib. The XRPD pattern is shown in Figure 5 and the TG/TGA pattern in Figure 6.
- ibrutinib prepared according to the comparative Example 1 were added 200 ⁇ . of dichloromethane, yielding a turbid solution. The solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of ibrutinib dichloromethane solvate (5 mg, prepared according to Example 4, Route A). The mixture was temperature cycled (4 hour cycles) between 22°C and 2°C for a period of time of 20 hours (the cooling/heating rate after the 4 hour period was 0.083°C/min) to allow slurry formation. The solid residue was filtered and dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 17 hours, yielding dichloromethane solvate of ibrutinib.
- ibrutinib prepared according to the comparative Example 1 100 mg were dissolved in 250 ⁇ of 1,4-dioxane. The clear solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of ibrutinib 1,4-dioxane solvate (5 mg, prepared according to Example 5, Route A). The mixture was temperature cycled (4 hour cycles) between 22°C and 2°C for a period of time of 20 hours (the cooling/heating rate after the 4 hour period was 0.083°C/min) to allow slurry formation. The solid residue was filtered and dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 17 hours, yielding 1,4-dioxane solvate of ibrutinib.
- ibrutinib prepared according to the comparative Example 1 were added 1.0 mL of pyridine in 300-500 ⁇ aliquots to create a slurry.
- the slurry was stirred at a temperature of 25°C for 72 hours and filtered.
- the solid residue was dried at a pressure between 20 to 30 mbar and a temperature of 25°C, yielding pyridine solvate of ibrutinib.
- ibrutinib essentially pure amorphous form of ibrutinib was prepared from anisole, dichloromethane or chlorobenzene solvates by thermal desolvation, hot melt-extrusion, spay drying or lyophilization. b) Preparation of essentially pure amorphous by lyophilization
- ibrutinib Essentially pure amorphous form of ibrutinib was prepared according to the following protocol: 130 mg of chlorobenzene solvate of ibrutinib (prepared according to Example 3, Route C of this invention) were dissolved in 1.1 mL of 1,4-dioxane at a temperature of from 40 to 50°C. The solution was sonicated (2 to 5 min in a VWR Ultrasonic Cleaner apparatus) to allow faster solid dissolution.
- the homogeneous solution was rapidly frozen in a Christ Alpha 2- 4 LSC freeze dryer (plate temperature of -30°C) and lyophilized (plate temperature from -30 to +50°C, pressure from 0.150 to 0.020 mbar) during 48 hours, yielding essentially pure amorphous form of ibrutinib as a white powder, with a HPLC purity of 99.2%.
- the X PD pattern of the obtained amorphous ibrutinib is shown in Figure 12.
- ibrutinib essentially pure amorphous form of ibrutinib was prepared according to the following protocol: 1.0 g of e.g. dichloromethane or anisole solvate of ibrutinib was dissolved in 50 mL of dichloromethane at a temperature of 25°C. The solution is spray-dried through the nozzle of a Biichi Spray Dryer (inlet temperature: 43-55°C, outlet temperature: 33-40°C, spray rate of feed: 3-5 mL/min), yielding essentially pure amorphous form of ibrutinib as a white powder. d) Determination of the moisture stability of amorphous ibrutinib
- Example 8 a) Preparation of crystalline form C of ibrutinib from anisole, chlorobenzene, 1,4- dioxane and pyridine solvates
- Essentially pure form C of ibrutinib was prepared from anisole, chlorobenzene, 1,4-dioxane and pyridine solvates, e.g. by thermal desolvation. b) Preparation of essentially pure form C of ibrutinib by thermal desolvation
- Essentially pure form C of ibrutinib was prepared according to the following protocol: 100 mg of e.g. chlorobenzene solvate of ibrutinib was stored at 80°C for 1 hours and 100°C for 1 hour at a pressure of 20-30 mbar, yielding essentially pure form C of ibrutinib.
- the XRPD pattern is shown in Figure 14.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention refers to amorphous and crystalline forms of the Bruton's tyrosine kinase (Btk) inhibitor 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1- yl)piperidin-1-yl)prop-2-en-1-one (ibrutinib). In particular, the invention refers to new crystalline solvate forms of ibrutinib, a method of using these crystalline solvate forms for the preparation of essentially pure amorphous ibrutinib as well as pharmaceutical compositions that comprise essentially pure amorphous ibrutinib. Further, the invention relates to essentially pure amorphous ibrutinib for the treatment of cancer.
Description
PHYSICAL FORMS OF IBRUTINIB, A BRUTON'S KINASE INHIBITOR
FIELD OF THE INVENTION
The present invention refers to amorphous and crystalline forms of the Bruton's tyrosine kinase (Btk) inhibitor l-((/?)-3-(4-amino-3-(4-phenoxyphenyl)-l/-/-pyrazolo[3/4-c ]pyrimidin-l- yl)piperidin-l-yl)prop-2-en-l-one (ibrutinib). In particular, the invention refers to new crystalline solvate forms of ibrutinib, a method of using these crystalline solvate forms for the preparation of essentially pure amorphous ibrutinib as well as pharmaceutical compositions that comprise essentially pure amorphous ibrutinib. Further, the invention relates to essentially pure amorphous ibrutinib for the treatment of cancer.
BACKGROUND OF THE INVENTION
Bruton's tyrosine kinase (Btk), a member of the Tec family of non-receptor tyrosine kinases, is a key signaling enzyme expressed in all hematopoietic cells types except T lymphocytes and natural killer cells. Btk plays an essential role in the B-cell signaling pathway linking cell surface B-cell receptor (BCR) stimulation to downstream intracellular responses. l-((/?)-3-(4-amino-3-(4-phenoxyphenyl)-lW-pyrazolo[3,4-d]pyrimidin-l-yl)piperidin-l-yl)prop-2- en-l-one is also known by its lUPAC name as l-{(/?)-3-[4-amino-3-(4-phenoxyphenyl)-lH- pyrazolo [3,4-d]pyrimidin-l-yl]piperidin-l-yl}prop-2-en-l-one or 2-propen-l-one,l-[(3 ?)-3-[4- amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl]-l-piperidinyl]-, and has been given the USAN name "ibrutinib", which will be used further in the document and refers to the compound with the following structure:

CN 103923084 A discloses anhydrous, hydrous as well as solvate crystal forms of ibrutinib, the solvate forms of ibrutinib being solvates of oxolane and trichloromethane.
Three anhydrous polymorphs and solvates from methanol, methylisobutylketone (MIBK), and toluene of ibrutinib have been specifically disclosed in WO 2013/184572 including formulations containing them and their use. However, the toluene solvate has been reported as being unstable. Moreover, solvates with methanol and MIBK suffer from low solubility of ibrutinib in those solvents (about 1 wt% or less), thereby limiting their use for purification and subsequent preparation of amorphous ibrutinib on industrial scale as this would require very large amounts of solvent. Thus, the solvates from methanol and methylisobutylketone are not suitable for the preparation of amorphous ibrutinib for use in the manufacture of a medicament.
Furthermore, according to WO 2008/039218 amorphous ibrutinib can be obtained from material prepared by fast evaporation of solvent from dichloromethane solution. However, material used for this process has been purified by chromatography on silica gel using dichloromethane/alcohol mixtures. This approach bears the disadvantage that the eluent in the final purification step contains unspecified amounts of silica gel leaking from the column. Because such impurities might affect chemical stability in an uncontrolled manner such a material is not appropriate for use in a medicament. Moreover, amorphous ibrutinib material prepared by fast rotary evaporation as described in WO 2008/039218 features a solid state
best described as honey-like, gum, or foam, depending on the amount of residual dichloromethane still present. Such a material is difficult or even impossible to handle and process on a large scale. SUMMARY OF THE INVENTION
It was thus an object of the present invention to provide a physical form of ibrutinib and the provision of a process for the preparation of such physical forms of ibrutinib, which can overcome the disadvantages of the prior art processes. In particular, it was an object of the present invention to provide a process for the preparation of ibrutinib, in particular in its amorphous state, which can be used for the preparation of a medicament on industrial scale. Moreover, it was an object of the present invention to provide a process for the preparation of amorphous ibrutinib free from any chemical impurities such as byproducts or silica gel, which might affect stability of ibrutinib in an uncontrolled manner.
It has surprisingly been found in the present invention that the above objects can be solved by the provision of new crystalline forms of ibrutinib. In particular, it has surprisingly been found that new crystalline solvates can be provided, characterized by higher solubility of the amorphous form of ibrutinib in the corresponding solvents used for their preparation than in the solvents used for the preparation of the solvates described in WO 2013/184572.
Studies of solvates of ibrutinib obtained from solvents such as anisole, chlorobenzene, dichloromethane, dioxane, and pyridine, most prefera bly from anisole and chlorobenzene, surprisingly revealed that they are useful for the preparation of essentially pure amorphous ibrutinib on industrial scale.
Further, it has been surprisingly found that amorphous form of ibrutinib prepared from these new solvates is essentially free from a ny chemical impurities, and is essentially free from any crystalline forms of ibrutinib, such as for example crystalline form A. Moreover, the process of the present invention for the preparation of amorphous ibrutinib has the advantage over the prior art process that it relies not on any column chromatography, and therefore the resulting product is free from any residual silica. Fina lly, amorphous ibrutinib is obtained as a free fluent powder, appropriate for use in the preparation of a medicament on industrial scale.
Furthermore, solvates obtained from e.g. anisole, chlorobenzene, 1,4-dioxane, and pyridine were found to be useful for the preparation of crystalline, anhydrous form of ibrutinib on industrial scale.
Thus, in a first embodiment, the present invention refers to a solvate of l-[(3/?)-3-[4-amino-3- (4-phenoxyphenyl)pyrazolo[3;4-d]pyrimidin-l-yl]piperidin-l-yl]prop-2-en-l-one (ibrutinib) and a solvent wherein the solubility of amorphous ibrutinib in the corresponding solvent is in the range of 20 to 900 mg/mL at 40°C, preferably is in the range of 25 to 800 mg/mL at 40°C, most preferably is in the range of 30 to 750 mg/mL.
In a further embodiment, the invention refers to the use of a solvate of ibrutinib for preparing amorphous ibrutinib.
In a further embodiment, the invention refers to a process for the preparation of a solvate of ibrutinib and an organic solvent selected from anisole, chlorobenzene, 1,4-dioxane, pyridine, and dichloromethane, the process comprising the steps of a) providing a solution of ibrutinib in the organic solvent,
b) optionally adding seed crystals,
c) stirring the solution, or submitting the solution to one or more of temperature cycling, crash cooling, evaporation, or anti-solvent addition, to form a slurry,
d) optionally isolating the crystalline solvate in solid form, and
e) optionally drying the crystalline solvate.
In a further embodiment, the invention refers to a process for the preparation of amorphous ibrutinib comprising the steps of: a) providing a crystalline solvate of ibrutinib characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
b) optionally dissolving the solvate in the solvent,
c) optionally washing with an acidic aqueous solution,
d) desolvating the solvate, and
e) optionally drying the product.
In a further embodiment, the invention refers to the amorphous ibrutinib obtained by the process as described herein.
In a further embodiment, the invention refers to the amorphous ibrutinib for use in the treatment of cancer.
In a further embodiment, the invention refers to a process for the preparation of anhydrous crystalline form C of ibrutinib, the process comprising the steps of: a) providing a solvate of ibrutinib with an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
b) desolvating the solvate,
c) optionally washing with an acidic aqueous solution, and
d) optionally drying the product.
BRIEF DESCRIPTION OF DRAWINGS
Figure 1 illustrates an X-Ray powder diffraction (XRPD) pattern (diffractogram) of the anisole solvate of ibrutinib.
Figure 2 illustrates a TG/DTA thermogram of the anisole solvate of ibrutinib.
Figure 3 illustrates an X-Ray powder diffraction (XRPD) pattern of the chlorobenzene solvate of ibrutinib.
Figure 4 illustrates a TG/DTA thermogram of the chlorobenzene solvate of ibrutinib.
Figure 5 illustrates an X-Ray powder diffraction (XRPD) pattern of the dichloromethane solvate of ibrutinib.
Figure 6 illustrates a TG/DTA thermogram of the dichloromethane solvate of ibrutinib.
Figure 7 illustrates an X-Ray powder diffraction (XRPD) pattern of the 1,4-dioxane solvate of ibrutinib.
Figure 8 illustrates a TG/DTA thermogram of the 1,4-dioxane solvate of ibrutinib.
Figure 9 illustrates an X-Ray powder diffraction (XRPD) pattern of the pyridine solvate of ibrutinib.
Figure 10 illustrates a TG/DTA thermogram of the pyridine solvate of ibrutinib.
Figure 11 illustrates an X-Ray powder diffraction (XRPD) pattern of amorphous ibrutinib containing substantial amount of crystalline form A prepared by evaporation of dichloromethane solution.
Figure 12 illustrates an X-Ray powder diffraction (XRPD) pattern of amorphous ibrutinib prepared by lyophilization from chlorobenzene solvate of ibrutinib. Figure 13 illustrates an X-Ray powder diffraction (XRPD) pattern of amorphous ibrutinib after storage at 40°C / 75% RH for 8 week.
Figure 14 illustrates an X-Ray powder diffraction (XRPD) pattern of crystalline form C of ibrutinib prepared by thermal desolvation of chlorobenzene solvate of ibrutinib.
DETAILED DESCRIPTION OF THE INVENTION
Solvates of Ibrutinib In a first embodiment, the present invention refers to new crystalline forms of ibrutinib. In particular, the present invention refers to new crystalline solvates of ibrutinib. These new solvate forms are preferably characterized by a solubility at 40°C of amorphous ibrutinib in the solvent used for the solvate formation in the range of 20 to 900 mg/mL, preferably in the
range of 25 to 800 mg/mL, most preferably in the range of 30 to 750 mg/mL. Solubility can for example be determined by a solvent addition technique as described in the Example 1.
In one embodiment, the present invention relates to a solvate, wherein ibrutinib is solvated with an organic solvent selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane, and pyridine. In a preferred embodiment the present invention relates to a solvate, wherein ibrutinib is solvated with anisole, chlorobenzene, 1,4-dioxane, or pyridine.
In another preferred embodiment, the solvate of ibrutinib of the present invention may be characterized by a stoichiometric composition of the solvates, i.e. number of solvent molecules per ibrutinib molecule as follows:
Anisole solvate: 0.5 mol (±0.1 mol) of anisole,
Chlorobenzene solvate: 1.0 mol (±0.1 mol) of chlorobenzene,
1,4-Dioxane solvate: 0.7 mol (±0.1 mol) of dioxane,
Pyridine solvate: 0.7 mol (±0.1 mol) of pyridine,
Dichloromethane solvate: 0.9 mol (±0.1 mol) of dichloromethane.
In one aspect described herein, the present invention refers to an anisole solvate of ibrutinib. The solvate is typically characterized by having one or more of the following properties:
(a) an X-Ray powder diffraction (XRPD) pattern substantially the same as shown in Figure 1;
(b) an X-ray powder diffraction (XRPD) pattern with characteristic peaks at 6.5±0.2, 9.7±0.2, 10.5±0.2, 18.3±0.2, 20.1±0.2, and 21.1±0.2° 2-Theta, when measured at a temperature in the range of from 15 to 25°C with Cu-Kalpha^ radiation having a wavelength of 0.15419 nm; and
(c) a thermogravimetric/differential thermal analysis (TG/DTA) thermogram substantially similar to the one set forth in Figure 2, when measured at a rate of 10°C/min from 25°C to 300°C.
In one aspect described herein, the present invention refers to a chlorobenzene solvate of ibrutinib. The solvate is typically characterized by having one or more of the following properties:
(a) an X-Ray powder diffraction (XRPD) pattern substantially the same as shown in Figure 3;
(b) an X-ray powder diffraction (XRPD) pattern with characteristic peaks at 7.7±0.2, 8.9±0.2, 10.610.2, 17.810.2, 18.510.2, and 19.4+0.2° 2-Theta, when measured at a temperature in the range of from 15 to 25°C with Cu-Kalpha12 radiation having a wavelength of 0.15419 nm; and
(c) a thermogravimetric/differential thermal analysis (TG/DTA) thermogram substantially similar to the one set forth in Figure 4, when measured at a rate of 10°C/min from 25°C to 300°C.
In one aspect described herein, the present invention refers to a dichloromethane solvate of ibrutinib. The solvate is typically characterized by having one or more of the following properties:
(a) an X-Ray powder diffraction (XRPD) pattern substantially the same as shown in Figure 5;
(b) an X-ray powder diffraction (XRPD) pattern with characteristic peaks at 6.810.2, 11.5+0.2, 15.310.2, 18.410.2, 18.8+0.2, and 19.210.2° 2-Theta, when measured at a temperature in the range of from 15 to 25°C with Cu-Kalpha12 radiation having a wavelength of 0.15419 nm; and
(c) a thermogravimetric/differential thermal analysis (TG/DTA) thermogram substantially similar to the one set forth in Figure 6, when measured at a rate of 10°C/min from 25°C to 300°C.
In one aspect described herein, the present invention refers to a 1,4-dioxane solvate of ibrutinib. The solvate is typically characterized by having one or more of the following properties:
(a) an X-Ray powder diffraction (XRPD) pattern substantially the same as shown in Figure 7;
(b) an X-ray powder diffraction (XRPD) pattern with characteristic peaks at 6.2±0.2, 9.7±0.2, 12.410.2, 15.1+0.2, 16.810.2 and 20.210.2° 2-Theta, when measured at a temperature in the range of from 15 to 25°C with Cu-Kalpha^ radiation having a wavelength of 0.15419 nm; and
(c) a thermogravimetric/differential thermal analysis (TG/DTA) thermogram substantially similar to the one set forth in Figure 8, when measured at a rate of 10°C/min from 25°C to 300°C.
In one aspect described herein, the present invention refers to a pyridine solvate of ibrutinib. The solvate is typically characterized by having one or more of the following properties:
(a) an X-Ray powder diffraction (XRPD) pattern substantially the same as shown in Figure 9;
(b) an X-ray powder diffraction (XRPD) pattern with characteristic peaks at 6.310.2, 9.7+0.2, 12.6+0.2, 15.7+0.2, 16.610.2, and 21.2+0.2° 2-Theta, when measured at a temperature in the range of from 15 to 25°C with Cu-Kalpha1;2 radiation having a wavelength of 0.15419 nm; and
(c) a thermogravimetric/differential thermal analysis (TG/DTA) thermogram substantially similar to the one set forth in Figure 10, when measured at a rate of 10°C/min from 25°C to 300°C.
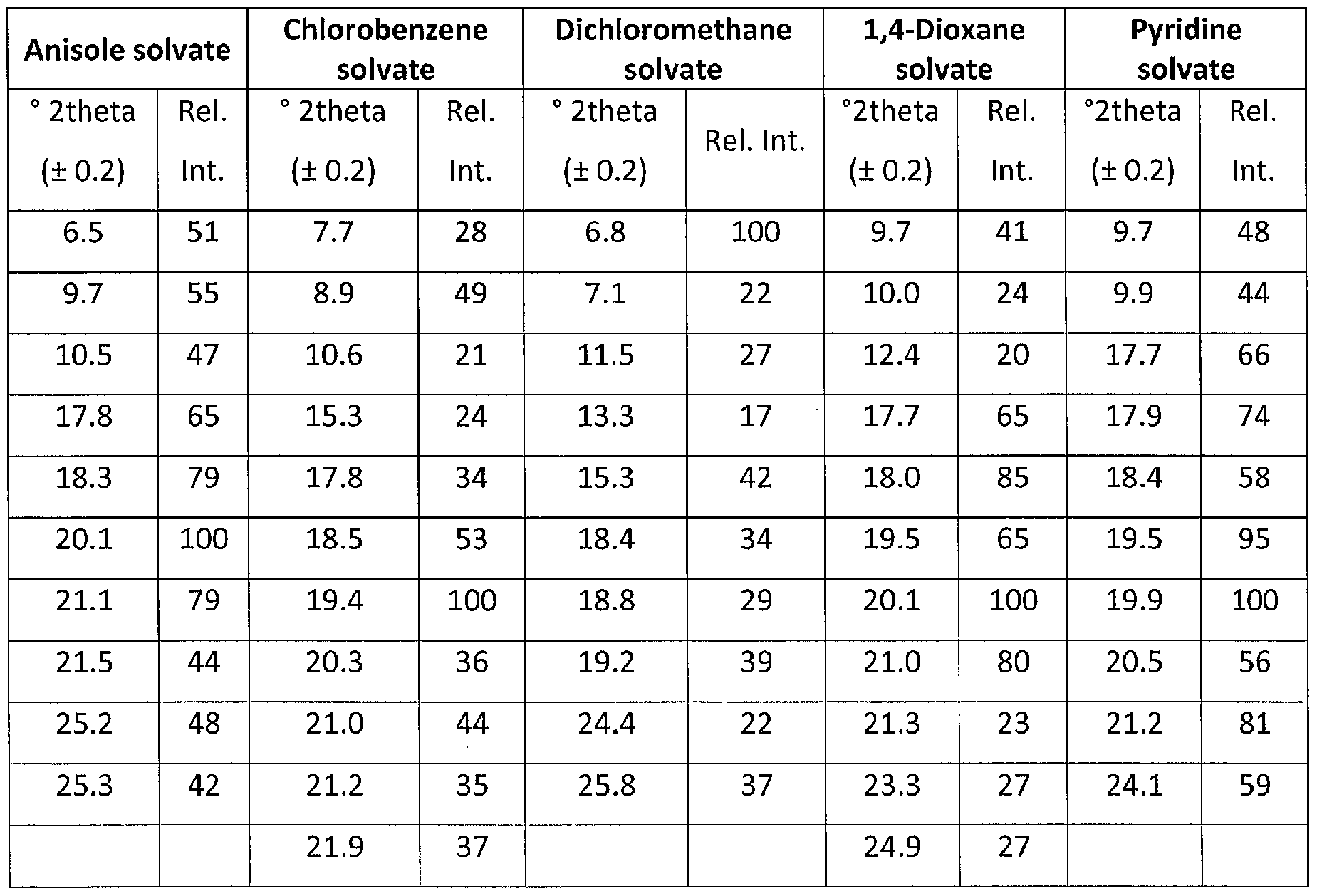
In particular, the solvates of ibrutinib of the present invention may be characterized by the peaks listed in the following table, showing the main 6 peaks and the main 10-11 peaks in an XRPD diffractogram.
Table 1 - Peak selection (6 peaks)

Table 2 - Peak selection (10-11 peaks)
Preparation of the Solvates of Ibrutinib
The solvates of the present invention can be prepared by a process comprising the steps of
) providing a solution of ibrutinib in an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
) optionally adding seed crystals, and
) stirring the solution, or performing one or more of temperature cycling, crash cooling, evaporation, or anti-solvent addition, to form a slurry,
) isolating the crystalline solvate in solid form, and
) optionally drying the crystalline solvate.
The organic solvent in step a) is preferably selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane and pyridine, most preferably anisole and chlorobenzene. Dissolution of ibrutinib can be obtained at temperatures in the range of from 4°C to 100°C depending on the solvent used.
Optionally, seed crystals may be added in step b). The seed crystals are prepared by the same solvent used in step a) of the process. In particular, the seed crystals are prepared by the same process steps as the solvates of the present invention.
The seed crystals are typically added in an amount of 0.1 wt% to 10 wt%, preferably in an amount of 0.5 wt% to 7.0 wt%, most preferably 1.0 wt.% 5.0 wt%, on the basis of the total amount of the starting material, i.e. the solution obtained in step a).
In step c) the solution transforms into a slurry by performing one or more of stirring the solution, or submitting the solution to one or more of temperature cycling, crash cooling, evaporation, or anti-solvent addition.
Stirring is performed at a temperature of from 10°C to 100°C, preferably between 20°C and 50°C, most preferably about room temperature, e.g. 22 to 25°C, typically for a period of time of from 2 to 20h. Usually, "room temperature" is understood to mean temperatures between 15 and 25 °C [see e.g. European Pharmacopoeia 8.2, 1.2 (2014)].
Temperature cycling is typically performed at atmospheric pressure, preferably at 2-22°C or 22-50°C, but also other conditions may be used depending on the type of solvate to be
produced. Temperature cycling is typically performed for a period of 16 to 24 hours by subjecting the solution or slurry to temperature cycles such as between 2°C and 22°C, wherein each cycle may have a length between 2 and 8 hours, preferably about 4 hours. A typical but not limiting temperature cycling protocol may be as follows: i) Cooling from 22 to 2°C (4 h)
ii) 2°C (4 h)
iii) Warming up from 2 to 22°C (4 h)
iv) 22° C (0.5 h).
Steps i) to iv) are typically repeated from 2 to 6 times.
Crash cooling is typically performed by directly and rapidly cooling the solution from a temperature of between 25-50°C to a temperature of 2°C or below, such as a temperature between 0°C and -18°C, depending on the type of solvate, and keeping the solution at this temperature for example for 4 h to 24 h, such as for example 16-20 h.
Evaporation is typically performed at reduced pressure, but may also be achieved at atmospheric pressure. The solvent used for anti-solvent addition may typically be selected from water, acetonitrile, a CM dialkylether, preferably methyliertbutylether ( TBE), or an alkane,preferably is selected from an alkane, more preferably a C5 alkane, a C5 alkane, a C7 alkane, a Ca alkane, or a mixture of two or more thereof, more preferably a C7 alkane, more preferably n-heptane. Additional solvent may be added in step c) in case of formation of thick slurries to allow easier stirring of the resulting slurry.
Optionally, isolation in step d) may be performed by using procedures known in the art, such as by filtration, centrifugation, or evaporation of solvent. Moreover, the isolated crystals may optionally be dried, e.g. under reduced pressure, typically at room temperature, or heated up to a temperature between 25°C and 50°C or the crystals may directly be used in further processes, such as the preparation of amorphous ibrutinib or isolated crystals may be used as seed crystals for the preparation of a solvate of ibrutinib.
Preparation of Amorphous Ibrutinib
The invention further relates to the use of the solvates as described herein for the manufacture of amorphous ibrutinib, which can be obtained by the process of the invention in pure amorphous form. In particular, it has surprisingly been found in the present invention that amorphous ibrutinib can be prepared by using the solvates of the present invention. After desolvation of solvates of the present invention, amorphous ibrutinib with higher purity than the starting material can be obtained. Thus, it is possible with the process of the present invention to obtain amorphous ibrutinib with a purity which could not be obtained with the conventional processes described in the prior art.
Thus, the present invention refers to the use of the solvates of ibrutinib as described herein in a process for preparing amorphous ibrutinib, such as pure amorphous ibrutinib. In particular, the process for the preparation of amorphous ibrutinib comprises the steps of: a) providing a crystalline solvate of ibrutinib and a solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
b) optionally dissolve the solvate in a solvent,
c) optionally washing with an acidic aqueous solution,
d) desolvating the solvate of step a), and
e) optionally drying the product.
The organic solvent in step a) is preferably selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane, and pyridine, most preferably is selected from anisole and chlorobenzene.
Further, desolvating in step d) is typically performed by one or more of thermal desolvation, hot melt-extrusion, spray drying, or lyophilisation. Solvents in pure form or mixtures used for spray drying are preferably selected from Biopharmaceutics Classification System (BCS) Class 3 solvent(s), further preferably with a low boiling point, most preferably acetone and dichloromethane. Solvents used for lyophilisation are typically selected from dioxane, dimethylsulfoxide (DMSO), acetic acid and tert-butanol, preferably acetic acid and DMSO.
Categories of solvents are defined in, for example, the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), "Impurities: Guidelines for Residual Solvents", Q3C(R3), (February 2009). The amount of the solvent is not particular restricted, preferably 10 to 100 fold amount with respect to the substrate. The time for lyophilization is not particularly restricted, preferably 1 to 240 hours.
In case the solvent is a base, such as pyridine, the process optionally includes the additional step of washing the solution with an acidic aqueous solution of pH 1-5, preferably pH 2-4; e.g. aqueous phosphoric acid.
Moreover, the product obtained in step d) may further optionally be dried, for example under reduced pressure, typically at room temperature, or heated up to a temperature between 25°C and 90°C. Thus, the present invention further relates to the amorphous ibrutinib obtained by the process described herewith. It has surprisingly been found in the present invention that amorphous ibrutinib can be prepared with the above process in substantially pure form, and in particular can be prepared in pure form. The terms "pure" or "substantially pure" in the context of the present invention refer to phase-purity and chemical purity of the amorphous ibrutinib. Phase purity of amorphous ibrutinib is characterized by the absence of any peaks in the XRPD pattern. Chemical purity of the amorphous ibrutinib can for example be determined by HPLC analysis. The amorphous ibrutinib obtained by the process described herewith thus preferably has a chemical purity of 99 wt.% or more, preferably of 99.5 wt.% or more, more preferably of 99.9 wt.%, based on the total weight of the amorphous ibrutinib.
In particular, amorphous ibrutinib can be prepared according to the method of the present invention without using column chromatography. As a consequence, any possible product contamination with silica can be avoided. The amorphous ibrutinib thus preferably has a content of silica, which is less than 1.0 wt.%, preferably is less than 0.3 wt.%, more preferably is less than 0.1 wt.%, further more preferably is less than 0.03 wt.%, most preferably is less than 0.01 wt%, based on the total weight of the amorphous ibrutinib, which can be determined by sulphated ash analysis according to Ph. Eur. 6.0, 2.4.14.
Further, it is possible with the method of the present invention to prepare amorphous ibrutinib which is essentially free from any crystal form of ibrutinib, such as crystal form A of ibrutinib. "Essentially free" in the context of the present invention means that no crystalline form, such as crystalline form A can be detected by XRPD measurement, i.e. no peaks of a crystalline form can be observed in an XRPD measurement. Thus, in a further embodiment, the invention is directed to the amorphous form of ibrutinib having no noticeable peak in a powder X-ray diffraction.
Stability of amorphous ibrutinib as obtained by the process described herewith at 40°C/75% relative humidity has been investigated over 8 weeks. Amorphous ibrutinib was stable under these conditions, as determined by the absence of XRPD peaks, in particular the absence of characteristic XRPD peaks for form A of ibrutinib after 8 weeks at 40°C / 75% RH.
The process of the present invention has the further advantage that the amorphous ibrutinib can be obtained in powder form, and therefore has beneficial properties making it suitable for use in the preparation of a medicament and preparing same on industrial scale. In particular, the amorphous ibrutinib obtained in powder form is free flowing, and its particle size and particle size distribution can be controlled by methods known in the art, such as by milling and/or sieving the amorphous ibrutinib in powder form.
Purification of Crude Ibrutinib
In another embodiment, the present invention refers to a combination of the process for the preparation of the solvates of ibrutinib and the process for the preparation of pure amorphous ibrutinib, as both described herein. As crude amorphous ibrutinib may be used as the starting material for the preparation of the solvates of ibrutinib of the present invention, which may contain any chemical impurities, such as residual amounts of silica, or may contain residual amounts of crystal forms of ibrutinib, such as the crystal form A of ibrutinib, it is possible with the combination of both processes to obtain amorphous ibrutinib with higher purity than in the starting material. Thus, the combination of both processes may be used as a process for purification of amorphous ibrutinib, such as crude amorphous ibrutinib.
Thus, in another embodiment, the present invention refers to a process for the preparation of pure amorphous ibrutinib, comprising the steps of: a) providing a solution of crude ibrutinib in an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably selected from anisole, chlorobenzene, dichloromethane, 1,4-dioxane and pyridine, b) optionally adding seed crystals, and
c) stirring the solution, or performing one or more of temperature cycling, crash cooling, evaporation, or anti-solvent addition, to form a slurry,
d) isolating the solvate in solid form from the slurry,
e) optionally dissolve the solvate in a solvent,
f) optionally washing with an acidic aqueous solution,
g) desolvating the solvate, and
h) optionally drying the product.
Amorphous ibrutinib with higher purity than in the amorphous ibrutinib used as the starting material in step a) is obtained in step g) or optionally in step h). In particular, the amorphous ibrutinib obtained by the above process is substantially free from silica or from residual amounts of crystal forms of ibrutinib such as crystal form A of ibrutinib, which however may be present in the crude amorphous ibrutinib used as the starting material in step a) of the process.
The preferred embodiments of the process are the same as described above for the process for the preparation of the solvates of ibrutinib of the present invention and the process for the preparation of pure amorphous ibrutinib and thus also refer to the combination of both processes.
Hence, in another preferred embodiment, the use of a process as described above for the purification of l-((R)-3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)piperidin-l-yl)prop-2-en-l-one of <90% purity, such as crude ibrutinib for obtaining a purified ibrutinib having a purity of >97 wt.%, preferably >98wt.%, more preferably > 99%wt.% and most preferably >99.5 wt.% is provided.
Preparation of pure Form C of Ibrutinib
It has further been surprisingly found that the solvates of ibrutinib as described herein can be used for the preparation of the crystalline form C of ibrutinib. Thus, the present invention further refers to the use of the solvates of ibrutinib described herein in a method for preparing the crystalline form C of ibrutinib. In particular, the present invention relates to a process for the preparation of anhydrous crystalline form C of ibrutinib, the process comprising the steps of: providing a solvate of ibrutinib with an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, preferably of 25 to 800 mg/mL, most preferably of 30 to 750 mg/mL,
desolvating the solvate of step a),
optionally washing with an acidic aqueous solution, and
optionally drying the product obtained in step b) or optionally c).
The organic solvent in step a) is preferably selected from anisole, chlorobenzene, 1,4-dioxane, and pyridine, most preferably is selected from anisole and chlorobenzene. Typical conditions for performing desolvation in step b) are 50-120°C, preferably at 60-100°C. Thus, the step may also be referred to as "thermal desolvation" . Moreover, desolvation in step b) may be performed at reduced or atmospheric pressure, preferably at reduced pressure, such as between 1 and 100 mbar, preferably between 5 and 50 mbar. Step b) may further be performed with or without a solvent, preferably without, i.e. in the absence of, a solvent. Therefore, in a preferred embodiment, desolvating of step b) is performed by heating the solvate to a temperature between 50-120°C, preferably at 60-100°C, while the pressure is between 1 and 100 mbar, preferably between 5 and 50 mbar. Desolvation is typically performed for 0.5 to 12 hours, preferably between 1 and 4 hours, depending on the temperature and pressure applied during desolvation. Desolvation may further be performed by changing the temperature and/or the pressure during desolvation, e.g. by firstly heating the solvate at a temperature between 50 and 90°C for a first period, e.g. between 0.5 and 2 hours, and then increasing the temperature in a second step, such as between 90 and 120°C for e.g. another 0.5 to 2 hours.
In case the solvent is a base, such as pyridine, the process optionally includes the additional step of washing the solution with an acidic aqueous solution of pH 1-5, preferably pH 2-4; e.g. aqueous phosphoric acid.
Form C of ibrutinib obtained by the above process can be characterized by the X PD peaks of 7.010.2, 14.010.2, 15.8+0.2, 18.310.2, 19.210.2, 19.610.2, 20.3+0.2, 22.110.2, and 23.010.2° 2- Theta.
In particular, the solvate provided in step a) may be obtained by the process described herein. Thus, in a further preferred embodiment, the present invention refers to a process for the preparation of the crystalline form C of ibrutinib, the process comprising the steps of: a) providing a solution of ibrutinib in an organic solvent characterized by a solubility of amorphous ibrutinib in the corresponding solvent at 40°C of 20 to 900 mg/mL, b) optionally adding seed crystals,
c) stirring the solution, or performing one or more of temperature cycling, crash cooling, evaporation, or anti-solvent addition, to form a slurry,
d) isolating the solvate in solid form from the slurry,
e) desolvating the solvate,
f) optionally washing with an acidic aqueous solution, and
g) optionally drying the product obtained in step e) or optionally f).
The preferred embodiments of the process are the same as described above for the process for the preparation of the solvates of ibrutinib of the present invention and the process for the preparation of crystalline form C of ibrutinib and thus also refer to the combination of both processes.
In particular, the crystalline form C of ibrutinib can be obtained by the process as described herein in pure or substantially pure form. The term pure or substantially pure in the context of the present invention refers to phase-purity and chemical purity of the form C of ibrutinib. Phase purity of form C is characterized by the absence of any XRPD peaks of anhydrous crystalline forms A and B. Chemical purity of the form C of ibrutinib can for example be determined by HPLC analysis. The form C of ibrutinib obtained by the process described
herewith thus preferably has a chemical purity, of 99 wt.% or more, preferably of 99.5 wt.% or more, more preferably of 99.9 wt.%, based on the total weight of the form C of ibrutinib. Thus, in another embodiment, the invention is directed to the crystalline form C of l-[(3 ?)-3-[4- amino-3-(4-phenoxyphenyl)pyrazolo[3,4-c/]pyrimidin-l-yl]piperidin-l-yl]prop-2-en-l-one having no noticeable peak of form A and/or B in a X-ray powder diffraction, in particular no noticeable peak of crystalline form A.
Dosage Forms The amorphous ibrutinib or the crystal form C of ibrutinib as described above, preferably the amorphous form of ibrutinib as obtained by the process of the present invention may be used for the manufacture of a pharmaceutical composition. Thus, in a further embodiment, the present invention relates to a pharmaceutical composition comprising the forms of ibrutinib as described herein.
The pharmaceutical composition typically comprises 1.0 to 1000 mg of amorphous ibrutinib or the crystal form C of ibrutinib, preferably comprises 10 to 800mg of amorphous ibrutinib, most preferably comprises 50 to 550 mg of amorphous ibrutinib. The pharmaceutical composition may further comprise one or more pharmaceutically acceptable additives, such as binders, carriers, stabilizers, diluents, dispersing agents, suspending agents, thickening agents, and/or excipients. Suitable excipients include, for example, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatine, gum tragacanth, methylcellulose, micro crystalline cellulose, hydroxypropyl methylcellulose, sodium carboxymethylcellulose; or others such as: polyvinylpyrrolidone (PVP or povidone) or calcium phosphate. If desired, disintegrating agents may be added, such as the cross-linked croscarmellose sodium, polyvinylpyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
The pharmaceutical composition facilitates administration of the compound to a mammal, preferably to a human. Ibrutinib can be used singly or in combination with one or more therapeutic agents as components of mixtures.
The pharmaceutical composition is typically a solid oral dosage form. It may be administered to a subject by multiple administration routes, including but not limited to, oral, parenteral (e.g., intravenous, subcutaneous, intramuscular), intranasal, buccal, topical, rectal, or transdermal administration routes. The pharmaceutical formulations described herein include, but are not limited to, aqueous liquid dispersions, self-emulsifying dispersions, solid solutions, liposomal dispersions, aerosols, solid dosage forms, powders, immediate release formulations, controlled release formulations, fast melt formulations, tablets, capsules, pills, delayed release formulations, extended release formulations, pulsatile release formulations, multiparticulate formulations, and mixed immediate and controlled release formulations. Preferably, the pharmaceutical dosage form is a tablet or capsule.
The pharmaceutical compositions may be manufactured in a conventional manner, such as, by way of example only, by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or compression processes.
In another embodiment, the amorphous ibrutinib or the crystal form C of ibrutinib prepared by the process as described herewith is used in the treatment of cancer. In particular, cancer may be a B cell malignancy, preferably selected from chronic lymphocytic leukemia (CLL)/ small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), indolent non-Hodgkins's lymphoma, diffuse large B Cell lymphoma (DLBCL), multiple myeloma(MM), marginal zone lymphoma (NHL), hairy cell leukemia, acute lymphocyte leukemia (ALL), and breast cancer.
Thus, the present invention further refers to a pharmaceutical composition as described above and comprising amorphous ibrutinib or the crystal form C of ibrutinib for use in the treatment of cancer.
EXAMPLES
In the following, the present invention will further be described by the way of non-limiting examples.
Methods of Measurement and Analyses
X-Ray Powder Diffraction (XRPD)
XRPD diffractograms were obtained with an X'Pert PRO diffractometer (PANalytical, Almelo, The Netherlands) equipped with a theta/theta coupled goniometer in transmission geometry, programmable XYZ stage with well plate holder, Cu-Kalpha1 2 radiation source (wavelength 0.15419 nm) with a focusing mirror, a 0.5° divergence slit, a 0.02° soller slit collimator and a 0.5° anti-scattering slit on the incident beam side, a 2 mm anti-scattering slit, a 0.02° soller slit collimator, a Ni-filter and a solid state PIXcel detector on the diffracted beam side. The diffractogram was recorded at room temperature at a tube voltage of 40 kV, tube current of 40 mA, applying a step size of 0.013° 2-theta with 40 sec per step in the angular range of 2 ° to 40 ° 2-theta. A typical precision of the 2-theta values is in the range of ± 0.2° 2-theta. Thus, a diffraction peak that appears for example at 9.4° 2-theta can appear between 9.2 and 9.6° 2- theta on most X-ray diffractometers under standard conditions.
Thermogravimetric/Differential Thermal Analysis (TG/DTA)
Approximately, 5mg of material was weighed into an open aluminium pan and loaded into a simultaneous thermogravimetric/differential thermal analyzer (TG/DTA) and held at room temperature. The sample was then heated at a rate of 10°C/min from 25°C to 300°C during which time the change in sample weight was recorded along with any differential thermal events (DTA). Nitrogen was used as the purge gas, at a flow rate of 100 cm3/nnin. Software: Muse Measurement v. 5.4 U (Build 263). High Performance Liquid Chromatography (HPLC)
HPLC measurements were performed with a Agilent 1100 Series (UV/Vis detector) and a reversed phase column YMC-Pack Pro C18 RS (150 x 4.60 mm, 3 μιη), using the following method:
Oven Temperature: 30°C
UV wavelength: 245 nm
Injection Volume: 3 μί
Flow Rate: 1.0 mL/min
Run time: 16 minutes
Post time: 2 minutes
Mobile Phase A: 0.39 g of sulfamic acid, 996 g of H20
Mobile Phase B: 0.39 g of sulfamic acid, 300 g of H20, 587 g of acetonitrile
Diluent: watenacetonitrile (1:1)
Sample preparation: 2 mg / 2.5 mL diluent
HPLC solvent gradient:

Comparative Example 1: Experiments for preparation of amorphous form of ibrutinib by evaporation of a dichloromethane solution
To 2.00 g of ibrutinib were added 60 mL of dichloromethane. The mixture was gently heated on a hotplate at 35-40°C for 15 minutes to encourage dissolution, forming a slightly cloudy solution. The solution was rapidly evaporated at a pressure between 600 and 30 mbar and a temperature of 40°C for 3-6 hours, following by drying in vacuum at a pressure of 20-30 mbar and a temperature of 40°C for 16-24 hours, yielding ibrutinib as a yellowish foam. This process frequently led to amorphous form of ibrutinib containing substantial amounts of crystal form A of ibrutinib, as can be seen by the sharp peaks present in the XRPD diffractogram of Figure 11. In this figure characteristic peaks of form A are presents at 5.7±0.2, 13.6±0.2, 16.1±0.2, 18.9±0.2, 21.3±0.2, and 21.6±0.2° 2-Theta. Example 1: Solubility Screen:
Amorphous ibrutinib prepared according to Comparative Example 1 was used as the input material for the solubility screen. Solubility values were estimated by a solvent addition
technique in order to provide approximate values for designing further crystallisation experiments. Approximately 10 mg or 15 mg of ibrutinib were weighed each into a vial. Each solvent was added to the appropriate vial in 10 aliquots of 50 μί. or until material was fully dissolved. In between additions the sample was heated to 40°C. If 1000 μί. of solvent was added without dissolution of the material, solubility was calculated to be below this point. Results obtained from the solvent solubility screen are detailed in Table 3.
Table 3 - Solubility Screen Results

Preparation of ibrutinib solvates
Example 2: Preparation of anisole solvates of ibrutinib Route A:
Anisole was added to 9.9 mg of ibrutinib prepared according to comparative Example 1 in four aliquots of 50 μί until a total of 200 μί had been added. In between additions the sample was heated to a temperature of from 40 to 50°C on a hotplate. After the addition of the fourth aliquots, i.e. 200 μί. in total, and subsequent heating to 40°C, complete dissolution was observed. The solution was then left at room temperature for ca. 1 hour, without stirring. After 1 hour, solid had precipitated out producing anisole solvate of ibrutinib.
Route B:
To 100 mg of ibrutinib prepared according to comparative Example 1 were added 330 μί of anisole, yielding a turbid solution. The solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of anisole solvate of ibrutinib (5 mg, prepared according to Example 2, Route A). The mixture was further stirred at room temperature for a few minutes, leading to rapid slurry formation. An aliquot of the slurry was taken out and centrifuged during 6 minutes at 12000 r.p.m.. The solid residue was dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding anisole solvate of ibrutinib.
The rest of the slurry was temperature cycled (4 hour cycles) between 22°C and 2°C for a period of time of 20 hours (the cooling/heating rate after the 4 hour period was 0.083°C/min) and filtered. The solid residue was dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding crystalline anisole solvate of ibrutinib The X PD pattern is shown in Figure 1 and the TG/TGA pattern in Figure 2.
Route C:
To 1.00 g of ibrutinib prepared according to comparative Example 1 were added 3.3 mL of anisole under stirring conditions (ca. 300 r.p.m.), yielding a turbid solution. The solution was stirred at room temperature. After 5 minutes, solid had precipitated out of solution. 2.5 mL anisole were added to the thick slurry and the suspension was further stirred between 20 and 30°C for 6 hours and cooled to a temperature of from 2 to 5°C. After 3 hours at this temperature, the slurry was filtered and dried under vacuum at a pressure of from 20 to 30 mbar and a temperature of 40°C for 6 hours where-after heating was stopped and the temperature of the solid residue was allowed to decrease slowly to 25°C at a pressure of from 20 to 30 mbar for 20 h, yielding 0.90 g of crystalline anisole solvate of ibrutinib.
Route D:
100 mg of ibrutinib prepared according to comparative Example 1 were dissolved in 500 μί of pre-heated anisole at 50°C in a 2 mL vial. The resultant solution was filtered and then stirred at ambient temperature (18-22°C, ca. 300 r.p.m.). After 15 minutes, solid had precipitated out of solution. The sample was then stirred for a further 15 hours at ambient temperature. The slurry was filtered yielding anisole solvate of ibrutinib.
Route E:
500 μί of anisole were added to 500 mg of ibrutinib prepared according to comparative Example 1 at ambient temperature (18 to 22°C) in a 2ml vial. After 20 minutes of stirring [ca. 300 r.p.m.) at ambient, the material was found to have dissolved. After ca. 1 hour of stirring, solid precipitated, resulting in a very thick immobile precipitate. To obtain stirring, an additional 1.5 mL of anisole was added and left stirring for 15 hours at ambient temperature (18-22°C). The slurry was still very thick but more mobile after the additional solvent had been added. The slurry was filtered yielding anisole solvate of ibrutinib. Route F:
500 mg of crude ibrutinib (88.2 % HPLC purity) were dissolved in 1.2 mL of anisole at room temperature. The solution was stirred at room temperature. After 2.5 hours, solid had precipitated out of solution. The suspension was further stirred at room temperature for 16 hours, followed by addition of 0.6 mL of anisole before filtration. The solid residue was washed with heptane and dried under vacuum (20 mbar) at room temperature for 16 hours, yielding anisole solvate of ibrutinib with a HPLC purity of 97.8%.
The peak list corresponding to the XRPD pattern of crystalline anisole solvate of ibrutinib shown in Figure 1 is given in the following table:
Pos. [°2Th.] Rel. Int. [%]
6.5 51
9.7 55
10.0 10
10.5 47
12.5 17
13.1 34
14.4 30
16.9 20
17.2 18
17.6 35
17.8 65
Pos. [°2Th.] Rel. Int. [ ]
18.3 79
19.4 28
20.1 100
20.5 14
21.1 79
21.5 44
21.6 33
22.0 15
22.2 39
23.4 21
24.0 20
25.2 48
25.3 42
26.1 24
27.6 16
28.5 14
29.0 18
Example 3: Preparation of chlorobenzene solvate of ibrutinib Route A:
Chlorobenzene was added to 10.2 mg of ibrutinib prepared according to comparative Example 1 in six aliquots of 50 μί. until a total of 300 μί had been added. In between additions the sample was heated to a temperature of from 40 to 50°C on a hotplate. After addition of the fourth aliquot, ie. 300 μί in total and subsequent heating to 40°C, complete dissolution was observed. The solution was then left at room temperature for ca. 1 hour, without stirring. After 1 hour, solid had precipitated out producing chlorobenzene solvate of ibrutinib.
Route B:
To 100 mg of ibrutinib prepared according to comparative Example 1 were added 650 μί of chlorobenzene, yielding a turbid solution. The solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of ibrutinib chlorobenzene solvate (5 mg, prepared according to Example 3, Route A). The mixture was further stirred at room
temperature for a few minutes, leading to rapid slurry formation. An aliquot of the slurry was taken out and centrifuged during 6 minutes at 12000 r.p.m.. The solid residue was dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding chlorobenzene solvate of ibrutinib.
The rest of the slurry was temperature cycled (4 hour cycles) between 22°C and 2°C for a period of time of 20 hours (the cooling/heating rate after the 4 hour period was 0.083°C/min) and filtered. The solid residue was dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding chlorobenzene solvate of ibrutinib. The XRPD pattern is shown in Figure 3 and the TG/TGA pattern in Figure 4.
Route C:
To 1.00 g of ibrutinib prepared according to comparative Example 1 were added 6.6 mL of chlorobenzene under stirring conditions (ca. 300 rpm), yielding a turbid solution. The solution was stirred at room temperature. After ca. 5 minutes, solid had precipitated out of solution. The slurry was further stirred between 20 and 30°C for 6 hours and cooled to a temperature of from 2 to 5°C. After 3 hours at this temperature, the slurry was filtered and dried under vacuum at a pressure of from 20 to 30 mbar and a temperature of 40°C for 6 hours whereafter heating was stopped and the temperature of the solid residue was allowed to decrease slowly to 25°C at a pressure of from 20 to 30 mbar for 20 h, yielding 0.96 g of crystalline chlorobenzene solvate of ibrutinib with an HPLC purity of 99.2%.
Route D:
100 mg of ibrutinib prepared according to comparative Example 1 were dissolved in 400 μί of pre-heated chlorobenzene at 50°C in a 2 mL vial. The resultant was filtered and stirred (ca. 300 r.p.m.) at ambient temperature. After 15 minutes, solid had precipitated out of solution. The sample was then stirred for a further 15 hours at ambient temperature. The slurry was filtered, yielding chlorobenzene solvate of ibrutinib.
Route E:
500 μί of chlorobenzene were added to 500 mg of ibrutinib prepared according to comparative Example 1 in a 2 mL vial and stirred (ca. 300 r.p.m.) at ambient temperature (18 to 22°C). After 20 minutes, the material was found to have dissolved. After ca. 1 hour of stirring, solid had precipitated out of solution resulting in a very thick, immobile slurry. To
obtain stirring, an additional 750 μΐ of chlorobenzene was added and the sample was left stirring for a further 15 hours at ambient temperature. The slurry was still very thick but more mobile after the additional solvent had been added. The slurry was filtered, yielding chlorobenzene solvate of ibrutinib.
Route F:
500 mg of crude ibrutinib (88.2 % HPLC purity) were dissolved in 2.2 mL of chlorobenzene at room tem perature. The solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of ibrutinib chlorobenzene solvate (25 mg, prepared according to Example 3, Route A) leading to direct solid precipitation. The suspension was further stirred at room temperature for 16 hours and filtered. The solid residue was washed with heptane and dried under vacuum (20 mbar) at room temperature for 16 hours, yielding chlorobenzene solvate of ibrutinib with a H PLC purity of 98.5%. The peak list corresponding to the XRPD pattern of crystalline chlorobenzene solvate of ibrutinib shown in Figure 3 is given in the following table:

22.7 23
24.1 16
24.9 17
25.1 30
Example 4: Preparation of dichloromethane solvate of ibrutinib Route A:
700-750 mg of ibrutinib prepared according to the comparative Example 1 were dissolved in 1.0 mL of dichloromethane. The solution was evaporated at room temperature. The solid obtained after evaporation overnight was found to be crystalline anhydrous ibrutinib (Form A, according to WO2013184572). A further 0.5 mL of dichloromethane was added to the crystalline material and the resulting slurry was stirred (ca. 300 r.p.m.) at room temperature for 3 days. The solid residue was filtrated and dried at room temperature for 24 hours, yielding dichloromethane solvate of ibrutinib. The XRPD pattern is shown in Figure 5 and the TG/TGA pattern in Figure 6.
Route B:
To 100 mg of ibrutinib prepared according to the comparative Example 1 were added 200 μί. of dichloromethane, yielding a turbid solution. The solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of ibrutinib dichloromethane solvate (5 mg, prepared according to Example 4, Route A). The mixture was temperature cycled (4 hour cycles) between 22°C and 2°C for a period of time of 20 hours (the cooling/heating rate after the 4 hour period was 0.083°C/min) to allow slurry formation. The solid residue was filtered and dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 17 hours, yielding dichloromethane solvate of ibrutinib.
The peak list corresponding to the XRPD pattern of crystalline dichloromethane solvate of ibrutinib shown in Figure 5 is given in the following table:
Pos. [°2Th.] Rel. Int. [%]
6.8 100
7.1 22
11.5 27
13.2 15
13.3 17
15.0 13
15.3 42
16.5 15
18.4 34
18.8 29
19.2 39
20.2 16
23.5 16
24.4 22
25.6 12
25.8 37
26.6 14
26.8 15
27.8 11
30.9 10
Example 5: Preparation of 1,4-dioxane solvate of ibrutinib Route A:
400 μΙ_ of 1,4-dioxane were added to 500 mg of ibrutinib prepared according to the comparative Example 1 at ambient temperature (18 to 22°C) in a 2 mL vial. After ca. 10 minutes, the material was found to have dissolved. The solution was stirred {ca. 300 r.p.m.) at ambient temperature for 15 hours. After 15 hours, solid was found to have precipitated resulting in a very thick, immobile slurry. The slurry was filtered yielding 1,4-dioxane solvate of ibrutinib. The XRPD pattern is shown in Figure 7 and the TG/TGA pattern in Figure 8.
Route B:
1.0 mL of 1,4-dioxane was added to 396.2 mg of ibrutinib prepared according to the comparative Example 1 to create a slurry. The sample was placed on temperature cycling (4 hour cycles) between 50°C and 22°C (the cooling/heating rate after the 4 hour period was ca. l°C/min). After approximately 1 hour, thick, viscous slurry was observed. A further 2.0 mL of 1,4-dioxane was then added in order to create a more mobile slurry for further temperature cycling. The sample was then temperature cycled for ca. 72 hours in total.
Approximately 200 μί of saturated solution obtained after filtration was transferred into a 2.0 mL vials, which was left uncapped to allow for open air evaporation, leading to crystallization. The solid residue was filtered and dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 16 hours, yielding 1,4-dioxane solvate of ibrutinib.
Route C:
100 mg of ibrutinib prepared according to the comparative Example 1 were dissolved in 250 μί of 1,4-dioxane. The clear solution was stirred at room temperature for 5 minutes, followed by addition of seed crystals of ibrutinib 1,4-dioxane solvate (5 mg, prepared according to Example 5, Route A). The mixture was temperature cycled (4 hour cycles) between 22°C and 2°C for a period of time of 20 hours (the cooling/heating rate after the 4 hour period was 0.083°C/min) to allow slurry formation. The solid residue was filtered and dried at a pressure between 20 to 30 mbar and a temperature of from 25 to 40°C for 17 hours, yielding 1,4-dioxane solvate of ibrutinib.
The peak list corresponding to the XRPD pattern of crystalline 1,4-dioxane solvate of ibrutinib shown in Figure 7 is given in the following table:

16.8 29
17.5 15
17.7 50
17.9 65
18.0 81
18.1 18
18.6 10
18.9 22
19.5 74
20.2 100
20.8 11
21.0 68
21.3 28
22.2 22
22.5 20
23.2 28
24.1 18
24.9 29
25.3 14
25.8 19
26.2 12
26.4 11
27.7 14
30.4 16
Example 6: Preparation of pyridine solvate Route A:
500 μί. of pyridine were added to 500 mg of ibrutinib prepared according to the comparative Example 1 at ambient temperature (18 to 22°C) in a 2 mL vial. After 20 minutes, the material was found to have dissolved. After ca. 1 hour of stirring {ca. 300 r.p.m.), solid had precipitated out of solution resulting in a very thick, immobile slurry. To obtain stirring, an additional 1.0 mL
of pyridine was added and the sample left stirring for 15 hours. The slurry was filtered, yielding pyridine solvate of ibrutinib. The XRPD pattern is shown in Figure 9 and the TG/TGA pattern in Figure 10.
Route B:
To 301 mg of ibrutinib prepared according to the comparative Example 1 were added 1.0 mL of pyridine in 300-500 μί aliquots to create a slurry. The slurry was stirred at a temperature of 25°C for 72 hours and filtered. The solid residue was dried at a pressure between 20 to 30 mbar and a temperature of 25°C, yielding pyridine solvate of ibrutinib.
The peak list corresponding to the XRPD pattern of crystalline pyridine solvate of ibrutinib shown in Figure 9 is given in the following table:
Table 6:
Pos. [°2Th.] Rel. Int. [%]
6.3 46
9.4 16
9.7 51
9.9 38
10.7 19
12.6 21
15.7 13
16.6 26
17.5 29
17.7 98
18.0 81
18.1 16
18.3 18
18.4 50
18.8 18
18.9 21
19.5 92
19.9 70
Pos. [°2Th.] Rel. Int. [%]
20.5 46
21.0 20
21.2 100
21.4 31
21.8 11
23.1 40
23.8 30
24.1 45
24.3 41
25.6 27
25.7 20
26.4 16
27.6 22
27.8 12
28.1 18
28.8 14
Preparation of amorphous ibrutinib Example 7: a) Preparation of pure amorphous ibrutinib from chlorobenzene solvates
Essentially pure amorphous form of ibrutinib was prepared from anisole, dichloromethane or chlorobenzene solvates by thermal desolvation, hot melt-extrusion, spay drying or lyophilization. b) Preparation of essentially pure amorphous by lyophilization
Essentially pure amorphous form of ibrutinib was prepared according to the following protocol: 130 mg of chlorobenzene solvate of ibrutinib (prepared according to Example 3, Route C of this invention) were dissolved in 1.1 mL of 1,4-dioxane at a temperature of from 40 to 50°C. The solution was sonicated (2 to 5 min in a VWR Ultrasonic Cleaner apparatus) to allow faster solid dissolution. The homogeneous solution was rapidly frozen in a Christ Alpha 2-
4 LSC freeze dryer (plate temperature of -30°C) and lyophilized (plate temperature from -30 to +50°C, pressure from 0.150 to 0.020 mbar) during 48 hours, yielding essentially pure amorphous form of ibrutinib as a white powder, with a HPLC purity of 99.2%. The X PD pattern of the obtained amorphous ibrutinib is shown in Figure 12. c) Preparation of essentially pure amorphous ibrutinib by spray-drying
Essentially pure amorphous form of ibrutinib was prepared according to the following protocol: 1.0 g of e.g. dichloromethane or anisole solvate of ibrutinib was dissolved in 50 mL of dichloromethane at a temperature of 25°C. The solution is spray-dried through the nozzle of a Biichi Spray Dryer (inlet temperature: 43-55°C, outlet temperature: 33-40°C, spray rate of feed: 3-5 mL/min), yielding essentially pure amorphous form of ibrutinib as a white powder. d) Determination of the moisture stability of amorphous ibrutinib
25-30 mg of amorphous form of ibrutinib prepared according to Example 7 were exposed to an atmosphere having a relative humidity of 75 % and a temperature of 40°C for a period of time of 8 weeks and analyzed via XRPD with respect to the amorphousness. The XRPD pattern is shown in Figure 13. The absence of peaks on the X-ray powder diffractogram suggests that the material is substantially amorphous. Example 8: a) Preparation of crystalline form C of ibrutinib from anisole, chlorobenzene, 1,4- dioxane and pyridine solvates
Essentially pure form C of ibrutinib was prepared from anisole, chlorobenzene, 1,4-dioxane and pyridine solvates, e.g. by thermal desolvation. b) Preparation of essentially pure form C of ibrutinib by thermal desolvation
Essentially pure form C of ibrutinib was prepared according to the following protocol: 100 mg of e.g. chlorobenzene solvate of ibrutinib was stored at 80°C for 1 hours and 100°C for 1 hour at a pressure of 20-30 mbar, yielding essentially pure form C of ibrutinib. The XRPD pattern is shown in Figure 14.
Claims
Claims
A solvate of l-(( ?)-3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)piperidin-l-yl)prop-2-en-l-one and a solvent, wherein the solubility of amorphous 1- (( ?)-3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3;4-d]pynmidin-l-yl)piperidin-l- yl)prop-2-en-l-one in the corresponding solvent at 40°C is in the range of from 20 to 900 mg/mL.
The solvate of claim 1, wherein the organic solvent is selected from anisole, chlorobenzene, 1,4-dioxane, and pyridine.
The solvate of claim 1, wherein the organic solvent is anisole, and which has an X-ray powder diffraction pattern with characteristic peaks at 6.5±0.2, 9.7±0.2, 10.5±0.2, 18.3±0.2, 20.110.2, and 21.1 +0.2° 2-Theta, when measured at a temperature in the range of from 15 to 25 °C with Cu-Kalpha^ radiation having a wavelength of 0.15419 nm.
The solvate of claim 1, wherein the organic solvent is chlorobenzene, and which has an X-ray powder diffraction pattern with peaks at 7.7±0.2, 8.9±0.2, 10.6±0.2, 17.8±0.2, 18.5±0.2, and 19.4+0.2° 2-Theta, when measured at a temperature in the range of from 15 to 25 °C with Cu-Kalpha1;2 radiation having a wavelength of 0.15419 nm.
The solvate of claim 1, wherein the organic solvent is 1,4-dioxane, and which has an X- ray powder diffraction pattern with peaks at 6.210.2, 9.7+0.2, 12.410.2, 15.110.2, 16.810.2 and 20.2+0.2° 2-Theta, when measured at a temperature in the range of from 15 to 25 °C with Cu-Kalpha1/2 radiation having a wavelength of 0.15419 nm.
The solvate of claim 1, wherein the organic solvent is pyridine, and which has an X-ray powder diffraction pattern with peaks at 6.310.2, 9.7+0.2, 12.6+0.2, 15.710.2, 16.610.2, and 21.2+0.2° 2-Theta, when measured at a temperature in the range of from 15 to 25 °C with Cu-Kalphai2 radiation having a wavelength of 0.15419 nm.
7. Use of the solvate of any one of claims 1 to 6 for preparing amorphous l-((/?)-3-(4- amino-3-(4-phenoxyphenyl)-lW-pyrazolo[3,4-c/]pyrimidin-l-yl)piperidin-l-yl)prop-2-en- 1-one.
8. A process for the preparation of the solvate of any one of claims 1 to 6, the process comprising the steps of:
a) providing a solution of l-((/?)-3-(4-amino-3-(4-phenoxyphenyl)-lH- pyrazolo[3,4-d]pyrimidin-l-yl)piperidin-l-yl)prop-2-en-l-one in the corresponding organic solvent,
b) optionally adding seed crystals,
c) stirring the solution, or performing one or more of temperature cycling, crash cooling, evaporation, or anti-solvent addition, to form a slurry, d) optionally isolating the solvate in solid form, and
e) optionally drying the crystalline solvate.
9. A process for the preparation of amorphous l-(( ?)-3-(4-amino-3-(4-phenoxyphenyl)- lH-pyrazolotS^-cyipyrimidin-l-ylipiperidin-l-yljprop^-en-l-one comprising the steps of:
a) providing a solvate of any one of claims 1 to 6,
b) dissolving the solvate in a solvent,
c) optionally washing with an acidic aqueous solution, and
d) desolvating the solvate of step a) by lyophilisation or by spray-drying.
10. The process according to claim 9, wherein the organic solvent is selected from anisole, chlorobenzene, 1,4-dioxane, and pyridine.
11. Amorphous l-((R)-3-(4-amino-3-(4-phenoxyphenyl)-l/-/-pyrazolo[3,4-c]pyrimidin-l- yl)piperidin-l-yl)prop-2-en-l-one obtained by the process of claim 9 or 10.
12. Amorphous l-(( ?)-3-(4-amino-3-(4-phenoxyphenyl)-l/-/-pyrazolo[3,4-c ]pyrimidin-l- yl)piperidin-l-yl)prop-2-en-l-one wherein the content of silica gel is less than 1.0 wt.%, based on the total weight of the amorphous l-((R)-3-(4-amino-3-(4-phenoxyphenyl)- l/-/-pyrazolo[3,4-c ]pyrimidin-l-yl)piperidin-l-yl)prop-2-en-l-one.
13. The amorphous l-((/?)-3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)piperidin-l-yl)prop-2-en-l-one claim 11 or 12 for use in the treatment of cancer.
14. A process for the preparation of anhydrous crystalline form C of l-(( ?)-3-(4-amino-3- (4-phenoxyphenyl)-lH-pyrazolo[3,4-c ]pyrimidin-l-yl)piperidin-l-yl)prop-2-en-l-one which has an X-ray powder diffraction pattern with peaks at 7.0+0.2, 14.0±0.2, 15.8±0.2, 18.3±0.2, 19.2±0.2, 19.6±0.2, 20.3±0.2, 22.1±0.2 and 23.0+0.2° 2-Theta, when measured at a temperature in the range of from 15 to 25 °C with Cu-Kalpha1 2 radiation having a wavelength of 0.15419 nm, the process comprising the steps of:
a) providing the solvate of claim 1,
b) desolvating the solvate of step a) by thermal desolvation at a temperature of 50°C to 120°C,
c) optionally washing with an acidic aqueous solution, and
d) optionally drying.
15. A pharmaceutical composition comprising amorphous l-((R)-3-(4-amino-3-(4- phenoxyphenyl)-lH-pyrazolo[3,4-ci]pyrimidin-l-yl)piperidin-l-yl)prop-2-en-l-one according to claim 11 or 12.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14194018 | 2014-11-20 | ||
| EP14194018.9 | 2014-11-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016079216A1 true WO2016079216A1 (en) | 2016-05-26 |
Family
ID=51987006
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2015/077042 WO2016079216A1 (en) | 2014-11-20 | 2015-11-19 | Physical forms of ibrutinib, a bruton's kinase inhibitor |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2016079216A1 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013184572A1 (en) | 2012-06-04 | 2013-12-12 | Pharmacyclics, Inc. | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| CN106008529A (en) * | 2016-08-08 | 2016-10-12 | 上海工程技术大学 | Ibrutinib solvate and preparation method thereof |
| CN106831788A (en) * | 2017-01-22 | 2017-06-13 | 鲁南制药集团股份有限公司 | Yi Bu replaces Buddhist nun's process for purification |
| WO2017137446A1 (en) * | 2016-02-09 | 2017-08-17 | Azad Pharmaceutical Ingredients Ag | Process for the synthesis of stable amorphous ibrutinib |
| US20180028537A1 (en) | 2014-08-07 | 2018-02-01 | Pharmacyclics Llc | Novel Formulations of a Bruton's Tyrosine Kinase Inhibitor |
| US10010507B1 (en) | 2015-03-03 | 2018-07-03 | Pharmacyclics Llc | Pharmaceutical formulations of a bruton's tyrosine kinase inhibitor |
| US10183024B2 (en) | 2016-12-02 | 2019-01-22 | Apotex Inc. | Crystalline forms of ibrutinib |
| WO2019070698A1 (en) * | 2017-10-02 | 2019-04-11 | Johnson Matthey Public Limited Company | Novel forms of ibrutinib |
| EP3501609A1 (en) | 2017-12-08 | 2019-06-26 | Zentiva K.S. | Pharmaceutical compositions comprising ibrutinib |
| EP3575300A1 (en) | 2018-05-31 | 2019-12-04 | Apotex Inc. | Novel crystalline forms of ibrutinib |
| RU2711106C2 (en) * | 2018-06-06 | 2020-01-15 | Общество с ограниченной ответственностью "АКСЕЛЬФАРМ" | Crystalline μ-modification of 1-[(3r)-3-[4-amino-3-(4-phenoxy-phenyl)-1h-pyrazolo[3,4-d]pyrimidin-1-yl]-1-piperidyl]-2-propenyl-1-one, method for production thereof and pharmaceutical composition based thereon |
| US10688050B1 (en) | 2018-12-21 | 2020-06-23 | Synthon B.V. | Pharmaceutical composition comprising ibrutinib |
| EP3669867A1 (en) | 2018-12-21 | 2020-06-24 | Synthon B.V. | Pharmaceutical composition comprising ibrutinib |
| WO2021097213A1 (en) * | 2019-11-14 | 2021-05-20 | Quogue Ip Holdings Llc | Combination of a btk inhibitor and an mdm2 inhibitor for cancer treatment |
| CN113135917A (en) * | 2020-01-16 | 2021-07-20 | 北京赛思源生物医药技术有限公司 | Amorphous substance of ibrutinib and medicinal composition thereof |
| WO2023242384A1 (en) | 2022-06-17 | 2023-12-21 | Krka, D.D., Novo Mesto | Crystalline form of ibrutinib |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008039218A2 (en) | 2006-09-22 | 2008-04-03 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| CN103121999A (en) | 2012-08-29 | 2013-05-29 | 苏州迪飞医药科技有限公司 | Method for synthesizing tyrosine kinase inhibitor PCI-32765 |
| WO2013184572A1 (en) | 2012-06-04 | 2013-12-12 | Pharmacyclics, Inc. | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| CN103923084A (en) | 2014-01-29 | 2014-07-16 | 苏州晶云药物科技有限公司 | Several new crystal forms and preparation methods thereof |
| WO2015145415A2 (en) * | 2014-03-27 | 2015-10-01 | Perrigo Api Ltd. | Ibrutinib solid forms and production process therefor |
-
2015
- 2015-11-19 WO PCT/EP2015/077042 patent/WO2016079216A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008039218A2 (en) | 2006-09-22 | 2008-04-03 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2013184572A1 (en) | 2012-06-04 | 2013-12-12 | Pharmacyclics, Inc. | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| CN103121999A (en) | 2012-08-29 | 2013-05-29 | 苏州迪飞医药科技有限公司 | Method for synthesizing tyrosine kinase inhibitor PCI-32765 |
| CN103923084A (en) | 2014-01-29 | 2014-07-16 | 苏州晶云药物科技有限公司 | Several new crystal forms and preparation methods thereof |
| WO2015145415A2 (en) * | 2014-03-27 | 2015-10-01 | Perrigo Api Ltd. | Ibrutinib solid forms and production process therefor |
Non-Patent Citations (3)
| Title |
|---|
| "International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH", IMPURITIES: GUIDELINES FOR RESIDUAL SOLVENTS, February 2009 (2009-02-01) |
| "Preparation of (R)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one, its salt and intermediates thereof", IP.COM JOURNAL, IP.COM INC., WEST HENRIETTA, NY, US, 23 September 2014 (2014-09-23), XP013164909, ISSN: 1533-0001 * |
| BURGER, LEUKEMIA & LYMPHOMA, vol. 54, no. 11, 2013, pages 2385 - 91 |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10106548B2 (en) | 2012-06-04 | 2018-10-23 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US10065968B2 (en) | 2012-06-04 | 2018-09-04 | Pharmacyclics Llc | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| US10294231B2 (en) | 2012-06-04 | 2019-05-21 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US10752634B2 (en) | 2012-06-04 | 2020-08-25 | Pharmacyclics Llc | Crystalline forms of a brutons tyrosine kinase inhibitor |
| US9828383B1 (en) | 2012-06-04 | 2017-11-28 | Pharmacyclic s LLC | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| US10294232B2 (en) | 2012-06-04 | 2019-05-21 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US10961251B1 (en) | 2012-06-04 | 2021-03-30 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| WO2013184572A1 (en) | 2012-06-04 | 2013-12-12 | Pharmacyclics, Inc. | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| US10125140B1 (en) | 2012-06-04 | 2018-11-13 | Pharmacyclics Llc | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| US10266540B2 (en) | 2012-06-04 | 2019-04-23 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US20180028537A1 (en) | 2014-08-07 | 2018-02-01 | Pharmacyclics Llc | Novel Formulations of a Bruton's Tyrosine Kinase Inhibitor |
| US10010507B1 (en) | 2015-03-03 | 2018-07-03 | Pharmacyclics Llc | Pharmaceutical formulations of a bruton's tyrosine kinase inhibitor |
| US10213386B2 (en) | 2015-03-03 | 2019-02-26 | Pharmacyclics Llc | Pharmaceutical formulations of a Bruton's tyrosine kinase inhibitor |
| US10828259B2 (en) | 2015-03-03 | 2020-11-10 | Pharmacyclics Llc | Pharmaceutical formulations of a Bruton's tyrosine kinase inhibitor |
| GB2558514A (en) * | 2016-02-09 | 2018-07-18 | Azad Pharmaceutical Ingredients Ag | Process for the synthesis of stable amorphous ibrutinib |
| WO2017137446A1 (en) * | 2016-02-09 | 2017-08-17 | Azad Pharmaceutical Ingredients Ag | Process for the synthesis of stable amorphous ibrutinib |
| CN106008529A (en) * | 2016-08-08 | 2016-10-12 | 上海工程技术大学 | Ibrutinib solvate and preparation method thereof |
| US10183024B2 (en) | 2016-12-02 | 2019-01-22 | Apotex Inc. | Crystalline forms of ibrutinib |
| CN106831788A (en) * | 2017-01-22 | 2017-06-13 | 鲁南制药集团股份有限公司 | Yi Bu replaces Buddhist nun's process for purification |
| CN106831788B (en) * | 2017-01-22 | 2020-10-30 | 鲁南制药集团股份有限公司 | Ibutotinib refining method |
| WO2019070698A1 (en) * | 2017-10-02 | 2019-04-11 | Johnson Matthey Public Limited Company | Novel forms of ibrutinib |
| EP3501609A1 (en) | 2017-12-08 | 2019-06-26 | Zentiva K.S. | Pharmaceutical compositions comprising ibrutinib |
| EP3575300A1 (en) | 2018-05-31 | 2019-12-04 | Apotex Inc. | Novel crystalline forms of ibrutinib |
| RU2711106C2 (en) * | 2018-06-06 | 2020-01-15 | Общество с ограниченной ответственностью "АКСЕЛЬФАРМ" | Crystalline μ-modification of 1-[(3r)-3-[4-amino-3-(4-phenoxy-phenyl)-1h-pyrazolo[3,4-d]pyrimidin-1-yl]-1-piperidyl]-2-propenyl-1-one, method for production thereof and pharmaceutical composition based thereon |
| WO2020127912A1 (en) | 2018-12-21 | 2020-06-25 | Synthon B.V. | Pharmaceutical composition comprising ibrutinib |
| EP3669867A1 (en) | 2018-12-21 | 2020-06-24 | Synthon B.V. | Pharmaceutical composition comprising ibrutinib |
| US10688050B1 (en) | 2018-12-21 | 2020-06-23 | Synthon B.V. | Pharmaceutical composition comprising ibrutinib |
| WO2021097213A1 (en) * | 2019-11-14 | 2021-05-20 | Quogue Ip Holdings Llc | Combination of a btk inhibitor and an mdm2 inhibitor for cancer treatment |
| CN113135917A (en) * | 2020-01-16 | 2021-07-20 | 北京赛思源生物医药技术有限公司 | Amorphous substance of ibrutinib and medicinal composition thereof |
| WO2023242384A1 (en) | 2022-06-17 | 2023-12-21 | Krka, D.D., Novo Mesto | Crystalline form of ibrutinib |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016079216A1 (en) | Physical forms of ibrutinib, a bruton's kinase inhibitor | |
| JP6916285B2 (en) | Cystic fibrosis membrane conductance regulator regulators, pharmaceutical compositions, therapeutic methods, and process of making regulators | |
| AU2011317040B2 (en) | Crystalline (8S,9R)-5-fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-8,9-dihydro-2H-pyrido[4,3,2-de]phthalazin-3(7H)-one tosylate salt | |
| CN103626803B (en) | Solid of tenofovir dipivoxil and its production and use | |
| CN110650963B (en) | Polymorphs and solid forms of GDC-0077 and process for preparing the same | |
| CN108602827A (en) | The Imidazopyrazines inhibitor of Bruton tyrosine kinase | |
| KR101669707B1 (en) | Purified pyrroloquinolinyl-pyrrolidine-2,5-dione compositions and methods for preparing and using same | |
| KR20120113285A (en) | Polymorphs of dasatinib, preparation methods and pharmaceutical compositions thereof | |
| KR102274755B1 (en) | NOVEL N-(2,3-DIHYDRO-1H-PYRROLO[2,3-b]PYRIDIN-5-YL)-4-QUINAZOLINAMINE AND N-(2,3-DIHYDRO-1H-INDOL-5-YL)-4-QUINAZOLINAMINE DERIVATIVES AS PERK INHIBITORS | |
| CA2963581C (en) | Crystal form of bisulfate of jak inhibitor and preparation method therefor | |
| WO2018045993A1 (en) | Crystal form, salt type of substituted 2-hydro-pyrazole derivative and preparation method therefor | |
| WO2016207172A1 (en) | Preparation of pure amorphous ibrutinib | |
| CN111777595A (en) | Novel crystal form of cyclohexane carboxamide compound and preparation method thereof | |
| KR102522895B1 (en) | Crystal Form of JAK Kinase inhibitor Bisulfate and a preparation method therefor | |
| JP2024038072A (en) | Voruciclib poly morphs and methods of making and using the same | |
| JP2023522012A (en) | crystal RET inhibitor | |
| CN113811539A (en) | Selective inhibitors of protein arginine methyltransferase 5(PRMT5) | |
| WO2020244348A1 (en) | Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereof | |
| EP3971175A1 (en) | Crystal form of quinazolinone compound and preparation method therefor | |
| EP3292115B1 (en) | Crystalline form of fused pyridine derivative's maleate and uses thereof | |
| CN112074517A (en) | Crystal forms of a compound | |
| EP3004102A1 (en) | Crystalline form of n,n-dicyclopropyl-4-(1,5-dimethyl-1 h-pyrazol-3-ylamino)-6-ethyl-1 -methyl-1,6-dihydroimidazo[4,5- d]fy rrolo[2,3-b]pyridine-7-carboxamide for the treatment of myeloproliferative disorders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15797102 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15797102 Country of ref document: EP Kind code of ref document: A1 |