WO2015110999A1 - Ezh2 inhibitors and uses thereof - Google Patents
Ezh2 inhibitors and uses thereof Download PDFInfo
- Publication number
- WO2015110999A1 WO2015110999A1 PCT/IB2015/050520 IB2015050520W WO2015110999A1 WO 2015110999 A1 WO2015110999 A1 WO 2015110999A1 IB 2015050520 W IB2015050520 W IB 2015050520W WO 2015110999 A1 WO2015110999 A1 WO 2015110999A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- oxo
- tetrahydro
- pyran
- ethyl
- Prior art date
Links
- 239000003112 inhibitor Substances 0.000 title description 12
- 150000001875 compounds Chemical class 0.000 claims abstract description 570
- 150000003839 salts Chemical class 0.000 claims abstract description 97
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 68
- 239000012453 solvate Substances 0.000 claims abstract description 54
- 229940002612 prodrug Drugs 0.000 claims abstract description 49
- 239000000651 prodrug Substances 0.000 claims abstract description 49
- 102000058017 Enhancer of Zeste Homolog 2 Human genes 0.000 claims abstract description 45
- 101710196274 Histone-lysine N-methyltransferase EZH2 Proteins 0.000 claims abstract description 45
- RCCPEORTSYDPMB-UHFFFAOYSA-N hydroxy benzenecarboximidothioate Chemical compound OSC(=N)C1=CC=CC=C1 RCCPEORTSYDPMB-UHFFFAOYSA-N 0.000 claims abstract description 45
- 230000000155 isotopic effect Effects 0.000 claims abstract description 45
- 150000001204 N-oxides Chemical class 0.000 claims abstract description 44
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 39
- 201000010099 disease Diseases 0.000 claims abstract description 39
- 201000011510 cancer Diseases 0.000 claims abstract description 34
- 208000035475 disorder Diseases 0.000 claims abstract description 29
- 230000001404 mediated effect Effects 0.000 claims abstract description 21
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 14
- -1 (CrCe)-alkyl Chemical group 0.000 claims description 163
- 229910003827 NRaRb Inorganic materials 0.000 claims description 154
- 125000000623 heterocyclic group Chemical group 0.000 claims description 87
- 125000005843 halogen group Chemical group 0.000 claims description 84
- 125000001072 heteroaryl group Chemical group 0.000 claims description 79
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 claims description 78
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 52
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims description 50
- 229910052736 halogen Inorganic materials 0.000 claims description 41
- 150000002367 halogens Chemical group 0.000 claims description 41
- 229910052739 hydrogen Inorganic materials 0.000 claims description 38
- 239000001257 hydrogen Substances 0.000 claims description 38
- 125000004043 oxo group Chemical group O=* 0.000 claims description 38
- 229910052757 nitrogen Inorganic materials 0.000 claims description 35
- 229910052717 sulfur Inorganic materials 0.000 claims description 34
- 229910003813 NRa Inorganic materials 0.000 claims description 28
- 238000000034 method Methods 0.000 claims description 25
- 229910052760 oxygen Inorganic materials 0.000 claims description 25
- 125000000217 alkyl group Chemical group 0.000 claims description 24
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 23
- 125000005842 heteroatom Chemical group 0.000 claims description 23
- 229910052698 phosphorus Inorganic materials 0.000 claims description 23
- 125000000081 (C5-C8) cycloalkenyl group Chemical group 0.000 claims description 22
- 229910052701 rubidium Inorganic materials 0.000 claims description 20
- 125000004648 C2-C8 alkenyl group Chemical group 0.000 claims description 19
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 17
- 125000002950 monocyclic group Chemical group 0.000 claims description 17
- 150000002431 hydrogen Chemical group 0.000 claims description 14
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 13
- 125000006652 (C3-C12) cycloalkyl group Chemical group 0.000 claims description 11
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 claims description 10
- 229920006395 saturated elastomer Polymers 0.000 claims description 10
- 208000009329 Graft vs Host Disease Diseases 0.000 claims description 9
- 241000725303 Human immunodeficiency virus Species 0.000 claims description 9
- 125000003118 aryl group Chemical group 0.000 claims description 9
- 125000005915 C6-C14 aryl group Chemical group 0.000 claims description 8
- 235000010290 biphenyl Nutrition 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 6
- 238000004519 manufacturing process Methods 0.000 claims description 6
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- 201000004681 Psoriasis Diseases 0.000 claims description 5
- 201000003790 Weaver syndrome Diseases 0.000 claims description 5
- 239000004305 biphenyl Substances 0.000 claims description 5
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 5
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 210000004185 liver Anatomy 0.000 claims description 5
- 206010028537 myelofibrosis Diseases 0.000 claims description 5
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 5
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 claims description 4
- YZPQQHXDCGVCFT-UHFFFAOYSA-N 2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-[4-(morpholin-4-ylmethyl)phenyl]-3-(2-oxoazepan-1-yl)benzamide Chemical compound CC1=C(C=C(C=C1N1C(CCCCC1)=O)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC(=C2CCCCC12)C)=O YZPQQHXDCGVCFT-UHFFFAOYSA-N 0.000 claims description 3
- DHHBDHJWKPUJHZ-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]-N-[[3-oxo-1-(trifluoromethyl)-2,5,6,7,8,9-hexahydrocyclohepta[c]pyridin-4-yl]methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC2=C3C(=C(NC2=O)C(F)(F)F)CCCCC3)C=C(C1)C=1C=NC(=CC1)N1CCN(CC1)C)C)C1CCOCC1 DHHBDHJWKPUJHZ-UHFFFAOYSA-N 0.000 claims description 3
- YJPWTRWOXOVHCD-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-2,5,6,7-tetrahydrocyclopenta[c]pyridin-4-yl)methyl]-5-[6-(morpholin-4-ylmethyl)pyridin-3-yl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC2=C3C(=C(NC2=O)C)CCC3)C=C(C1)C=1C=NC(=CC1)CN1CCOCC1)C)C1CCOCC1 YJPWTRWOXOVHCD-UHFFFAOYSA-N 0.000 claims description 3
- OSQBKPPJOOIYLC-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-(1,3,5-trimethylpyrazol-4-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C=C(C1)C=1C(=NN(C1C)C)C)C)C1CCOCC1 OSQBKPPJOOIYLC-UHFFFAOYSA-N 0.000 claims description 3
- JBJFHMLJDUTEFW-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(3-hydroxyoxetan-3-yl)-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C=C(C1)C1(COC1)O)C)C1CCOCC1 JBJFHMLJDUTEFW-UHFFFAOYSA-N 0.000 claims description 3
- KWVYQBWJLXQARS-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(3-hydroxyoxetan-3-yl)-2-methyl-N-[(1-methyl-3-oxo-7-propan-2-yl-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CN(CCC23)C(C)C)C)=O)C=C(C1)C1(COC1)O)C)C1CCOCC1 KWVYQBWJLXQARS-UHFFFAOYSA-N 0.000 claims description 3
- IODWEUPCDFFHJR-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(6-fluoro-5-methylpyridin-3-yl)-2-methyl-N-[(1-methyl-3-oxo-2,5,6,7-tetrahydrocyclopenta[c]pyridin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC2=C3C(=C(NC2=O)C)CCC3)C=C(C1)C=1C=NC(=C(C1)C)F)C)C1CCOCC1 IODWEUPCDFFHJR-UHFFFAOYSA-N 0.000 claims description 3
- JWNVSRAFFFFOJK-UHFFFAOYSA-N 5-[3-[(dimethylamino)methyl]phenyl]-3-[ethyl(oxan-4-yl)amino]-2-fluoro-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound CN(C)CC=1C=C(C=CC1)C1=CC(=C(C(=C1)N(C1CCOCC1)CC)F)C(=O)NCC=1C(NC(=C2CCCCC12)C)=O JWNVSRAFFFFOJK-UHFFFAOYSA-N 0.000 claims description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 3
- 125000006553 (C3-C8) cycloalkenyl group Chemical group 0.000 claims description 2
- LEWRCHGYSIWALT-UHFFFAOYSA-N 3-[2-(dimethylamino)pyrimidin-5-yl]-5-[ethyl(oxan-4-yl)amino]-N-[(1-methyl-3-oxo-2,5,6,7-tetrahydrocyclopenta[c]pyridin-4-yl)methyl]benzamide Chemical compound CN(C1=NC=C(C=N1)C=1C=C(C(=O)NCC2=C3C(=C(NC2=O)C)CCC3)C=C(C1)N(C1CCOCC1)CC)C LEWRCHGYSIWALT-UHFFFAOYSA-N 0.000 claims description 2
- ALCHMNYMCZWANQ-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-fluoro-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-[4-(1-morpholin-4-ylethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)C(C)N1CCOCC1)C(=O)NCC=1C(NC(=C2CCCCC12)C)=O)F)C1CCOCC1 ALCHMNYMCZWANQ-UHFFFAOYSA-N 0.000 claims description 2
- DWTVYUMSCWBXLJ-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]-N-[(3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC=C2CCCCC12)=O)C)C1CCOCC1 DWTVYUMSCWBXLJ-UHFFFAOYSA-N 0.000 claims description 2
- PGRRQBAKLANJDI-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]-N-[[3-oxo-1-(trifluoromethyl)-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl]methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C(F)(F)F)=O)C=C(C1)C=1C=NC(=CC1)N1CCN(CC1)C)C)C1CCOCC1 PGRRQBAKLANJDI-UHFFFAOYSA-N 0.000 claims description 2
- MUNYMJFUSNXKSS-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-2,5,6,7-tetrahydrocyclopenta[c]pyridin-4-yl)methyl]-5-quinolin-3-ylbenzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC2=C3C(=C(NC2=O)C)CCC3)C=C(C1)C=1C=NC3=CC=CC=C3C1)C)C1CCOCC1 MUNYMJFUSNXKSS-UHFFFAOYSA-N 0.000 claims description 2
- PPZIRERPKGPEJP-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-(4-morpholin-4-ylphenyl)benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)N1CCOCC1)C(=O)NCC=1C(NC(=C2CCCCC12)C)=O)C)C1CCOCC1 PPZIRERPKGPEJP-UHFFFAOYSA-N 0.000 claims description 2
- LBLDWWWKRZPURB-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-(6-methylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C=C(C1)C=1C=NC(=CC1)C)C)C1CCOCC1 LBLDWWWKRZPURB-UHFFFAOYSA-N 0.000 claims description 2
- WVVWPZJCYMGUMD-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 WVVWPZJCYMGUMD-UHFFFAOYSA-N 0.000 claims description 2
- YCGXQTKOFCEANA-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-[4-(2-piperidin-1-ylpropan-2-yl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)C(C)(C)N1CCCCC1)C(=O)NCC=1C(NC(=C2CCCCC12)C)=O)C)C1CCOCC1 YCGXQTKOFCEANA-UHFFFAOYSA-N 0.000 claims description 2
- BCGPNNFJEQFPIY-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-[6-(trifluoromethyl)pyridin-3-yl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C=C(C1)C=1C=NC(=CC1)C(F)(F)F)C)C1CCOCC1 BCGPNNFJEQFPIY-UHFFFAOYSA-N 0.000 claims description 2
- RINRKEZYQGIINW-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-7-propan-2-yl-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CN(CCC23)C(C)C)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 RINRKEZYQGIINW-UHFFFAOYSA-N 0.000 claims description 2
- MONNWAIPHCSYNX-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-7-propyl-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CN(CCC23)CCC)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 MONNWAIPHCSYNX-UHFFFAOYSA-N 0.000 claims description 2
- QFSJRDQTXKAZBQ-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-7-propyl-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CN(CCC23)CCC)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCN(CC1)C)C)C1CCOCC1 QFSJRDQTXKAZBQ-UHFFFAOYSA-N 0.000 claims description 2
- GEBNPPLAILUKMY-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(8-methyl-6-oxo-1,3,4,7-tetrahydropyrano[3,4-c]pyridin-5-yl)methyl]-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC2=C3C(=C(NC2=O)C)COCC3)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 GEBNPPLAILUKMY-UHFFFAOYSA-N 0.000 claims description 2
- YWGANRQZANWWSL-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(8-methyl-6-oxo-1,3,4,7-tetrahydropyrano[3,4-c]pyridin-5-yl)methyl]-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC2=C3C(=C(NC2=O)C)COCC3)C=C(C1)C=1C=NC(=CC1)N1CCN(CC1)C)C)C1CCOCC1 YWGANRQZANWWSL-UHFFFAOYSA-N 0.000 claims description 2
- KROUFTQZLRHZGF-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[[1-methyl-3-oxo-7-(2,2,2-trifluoroethyl)-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl]methyl]-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CN(CCC23)CC(F)(F)F)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 KROUFTQZLRHZGF-UHFFFAOYSA-N 0.000 claims description 2
- MIDZOMPLOWPROX-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[[1-methyl-7-(2-methylpropyl)-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl]methyl]-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CN(CCC23)CC(C)C)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 MIDZOMPLOWPROX-UHFFFAOYSA-N 0.000 claims description 2
- MDPKVZQKXHYIME-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(3-methoxyoxetan-3-yl)-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C=C(C1)C1(COC1)OC)C)C1CCOCC1 MDPKVZQKXHYIME-UHFFFAOYSA-N 0.000 claims description 2
- QEDIZHVLRQMABI-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(5-fluoro-6-morpholin-4-ylpyridin-3-yl)-2-methyl-N-[(1-methyl-3-oxo-2,5,6,7-tetrahydrocyclopenta[c]pyridin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC2=C3C(=C(NC2=O)C)CCC3)C=C(C1)C=1C=NC(=C(C1)F)N1CCOCC1)C)C1CCOCC1 QEDIZHVLRQMABI-UHFFFAOYSA-N 0.000 claims description 2
- GKLTZWCMMAKQSV-UHFFFAOYSA-N N-[(1,7-dimethyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound CC=1NC(C(=C2CCN(CC12)C)CNC(=O)C=1C=C(C=C(C1C)N(C1CCOCC1)CC)C1=CC=C(C=C1)CN1CCOCC1)=O GKLTZWCMMAKQSV-UHFFFAOYSA-N 0.000 claims description 2
- KYZSLLHBNXTVQM-UHFFFAOYSA-N N-[(7-butyl-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(CCC)N1CCC2=C(C(NC(=C2C1)C)=O)CNC(C1=C(C(=CC(=C1)C=1C=NC(=CC1)N1CCOCC1)N(C1CCOCC1)CC)C)=O KYZSLLHBNXTVQM-UHFFFAOYSA-N 0.000 claims description 2
- OQFCWMNINLFNIE-UHFFFAOYSA-N N-[(7-butyl-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzamide Chemical compound C(CCC)N1CCC2=C(C(NC(=C2C1)C)=O)CNC(C1=C(C(=CC(=C1)C=1C=NC(=CC1)N1CCN(CC1)C)N(C1CCOCC1)CC)C)=O OQFCWMNINLFNIE-UHFFFAOYSA-N 0.000 claims description 2
- SMTRJUFEQXFETB-UHFFFAOYSA-N N-[(7-ethyl-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC(=C2CN(CCC12)CC)C)=O)C)C1CCOCC1 SMTRJUFEQXFETB-UHFFFAOYSA-N 0.000 claims description 2
- PVWWAPULDLMEHV-UHFFFAOYSA-N N-[(7-ethyl-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CN(CCC23)CC)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCN(CC1)C)C)C1CCOCC1 PVWWAPULDLMEHV-UHFFFAOYSA-N 0.000 claims description 2
- 125000002619 bicyclic group Chemical group 0.000 claims description 2
- ZHXTWWCDMUWMDI-UHFFFAOYSA-N dihydroxyboron Chemical group O[B]O ZHXTWWCDMUWMDI-UHFFFAOYSA-N 0.000 claims description 2
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 claims description 2
- 125000006299 oxetan-3-yl group Chemical group [H]C1([H])OC([H])([H])C1([H])* 0.000 claims description 2
- DSKXJOQZIKYBJB-UHFFFAOYSA-N 5-diethoxyphosphoryl-3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C=C(C=C(C1C)C(NCC=1C(NC(=C2CCCCC12)C)=O)=O)P(OCC)(OCC)=O)C1CCOCC1 DSKXJOQZIKYBJB-UHFFFAOYSA-N 0.000 claims 1
- 238000002360 preparation method Methods 0.000 abstract description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 437
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 308
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 286
- 239000011541 reaction mixture Substances 0.000 description 265
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 190
- 239000000243 solution Substances 0.000 description 171
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 164
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 162
- 235000019439 ethyl acetate Nutrition 0.000 description 146
- 229940093499 ethyl acetate Drugs 0.000 description 145
- 238000005481 NMR spectroscopy Methods 0.000 description 137
- 238000004128 high performance liquid chromatography Methods 0.000 description 134
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 124
- 239000000203 mixture Substances 0.000 description 109
- YMWUJEATGCHHMB-UHFFFAOYSA-N dichloromethane Substances ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 108
- 239000000741 silica gel Substances 0.000 description 108
- 229910002027 silica gel Inorganic materials 0.000 description 108
- 229960001866 silicon dioxide Drugs 0.000 description 108
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 99
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 94
- 238000004440 column chromatography Methods 0.000 description 94
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 89
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 84
- 238000006243 chemical reaction Methods 0.000 description 77
- 239000013058 crude material Substances 0.000 description 62
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 54
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 54
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 52
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 51
- 229910000029 sodium carbonate Inorganic materials 0.000 description 51
- 229910052938 sodium sulfate Inorganic materials 0.000 description 47
- 235000011152 sodium sulphate Nutrition 0.000 description 47
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 46
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 45
- 239000007787 solid Substances 0.000 description 45
- 239000000284 extract Substances 0.000 description 44
- 239000007821 HATU Substances 0.000 description 43
- 239000007810 chemical reaction solvent Substances 0.000 description 34
- 239000012267 brine Substances 0.000 description 33
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 33
- 239000000706 filtrate Substances 0.000 description 32
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 32
- 239000002904 solvent Substances 0.000 description 30
- 239000010410 layer Substances 0.000 description 27
- 229910052786 argon Inorganic materials 0.000 description 26
- 239000012044 organic layer Substances 0.000 description 25
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 24
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 24
- 239000012298 atmosphere Substances 0.000 description 23
- 239000003208 petroleum Substances 0.000 description 22
- 238000005160 1H NMR spectroscopy Methods 0.000 description 21
- 239000012299 nitrogen atmosphere Substances 0.000 description 21
- 125000001424 substituent group Chemical group 0.000 description 21
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 19
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 19
- 239000001632 sodium acetate Substances 0.000 description 19
- 235000017281 sodium acetate Nutrition 0.000 description 19
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 18
- 238000003818 flash chromatography Methods 0.000 description 13
- 239000000377 silicon dioxide Substances 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 230000008569 process Effects 0.000 description 11
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 11
- DGJMPUGMZIKDRO-UHFFFAOYSA-N cyanoacetamide Chemical compound NC(=O)CC#N DGJMPUGMZIKDRO-UHFFFAOYSA-N 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- 229910052805 deuterium Inorganic materials 0.000 description 9
- JOIXYIWXEYXHHG-UHFFFAOYSA-N 4-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]morpholine Chemical compound O1C(C)(C)C(C)(C)OB1C(C=C1)=CC=C1CN1CCOCC1 JOIXYIWXEYXHHG-UHFFFAOYSA-N 0.000 description 8
- 201000009030 Carcinoma Diseases 0.000 description 8
- 206010039491 Sarcoma Diseases 0.000 description 8
- 208000009956 adenocarcinoma Diseases 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- 230000014509 gene expression Effects 0.000 description 8
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 7
- 239000001301 oxygen Substances 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- KVQXFNGVIIPCSX-UHFFFAOYSA-N 1-methyl-4-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl]piperazine Chemical compound C1CN(C)CCN1C1=CC=C(B2OC(C)(C)C(C)(C)O2)C=N1 KVQXFNGVIIPCSX-UHFFFAOYSA-N 0.000 description 6
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 206010025323 Lymphomas Diseases 0.000 description 6
- 241000124008 Mammalia Species 0.000 description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- WXIONIWNXBAHRU-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethylazanium Chemical compound C1=CN=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 WXIONIWNXBAHRU-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 206010041823 squamous cell carcinoma Diseases 0.000 description 6
- ZGDLVKWIZHHWIR-UHFFFAOYSA-N 4-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl]morpholine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N2CCOCC2)N=C1 ZGDLVKWIZHHWIR-UHFFFAOYSA-N 0.000 description 5
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- 206010014759 Endometrial neoplasm Diseases 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 108010033040 Histones Proteins 0.000 description 5
- 239000013543 active substance Substances 0.000 description 5
- 125000003342 alkenyl group Chemical group 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000003937 drug carrier Substances 0.000 description 5
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 238000010926 purge Methods 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 4
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 4
- 206010006187 Breast cancer Diseases 0.000 description 4
- 208000026310 Breast neoplasm Diseases 0.000 description 4
- 206010014733 Endometrial cancer Diseases 0.000 description 4
- 102000006947 Histones Human genes 0.000 description 4
- 241000282414 Homo sapiens Species 0.000 description 4
- 102000002272 Polycomb Repressive Complex 2 Human genes 0.000 description 4
- 108010000597 Polycomb Repressive Complex 2 Proteins 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 229910052796 boron Inorganic materials 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 230000008025 crystallization Effects 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 201000001441 melanoma Diseases 0.000 description 4
- 230000007935 neutral effect Effects 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 125000000547 substituted alkyl group Chemical group 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- VGBLRIBCUYWCDA-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzamide Chemical compound C=1C(C=2C=NC(=CC=2)N2CCN(C)CC2)=CC(C(N)=O)=C(C)C=1N(CC)C1CCOCC1 VGBLRIBCUYWCDA-UHFFFAOYSA-N 0.000 description 3
- WKEPYHBAAIKXKF-UHFFFAOYSA-N 7-(cyclopropylmethyl)-1-methyl-2,5,6,8-tetrahydro-2,7-naphthyridin-3-one Chemical compound C1(CC1)CN1CCC2=CC(NC(=C2C1)C)=O WKEPYHBAAIKXKF-UHFFFAOYSA-N 0.000 description 3
- 208000010667 Carcinoma of liver and intrahepatic biliary tract Diseases 0.000 description 3
- 201000008808 Fibrosarcoma Diseases 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 201000003741 Gastrointestinal carcinoma Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 206010073069 Hepatic cancer Diseases 0.000 description 3
- 208000007766 Kaposi sarcoma Diseases 0.000 description 3
- 206010024612 Lipoma Diseases 0.000 description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 3
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 3
- 208000003445 Mouth Neoplasms Diseases 0.000 description 3
- 208000000453 Skin Neoplasms Diseases 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- 206010043276 Teratoma Diseases 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 239000012300 argon atmosphere Substances 0.000 description 3
- 125000004104 aryloxy group Chemical group 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 206010006007 bone sarcoma Diseases 0.000 description 3
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 208000018554 digestive system carcinoma Diseases 0.000 description 3
- 230000004049 epigenetic modification Effects 0.000 description 3
- 206010016629 fibroma Diseases 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 201000002655 heart sarcoma Diseases 0.000 description 3
- 230000002489 hematologic effect Effects 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 description 3
- 201000002250 liver carcinoma Diseases 0.000 description 3
- 201000005296 lung carcinoma Diseases 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 210000000653 nervous system Anatomy 0.000 description 3
- JMJRYTGVHCAYCT-UHFFFAOYSA-N oxan-4-one Chemical compound O=C1CCOCC1 JMJRYTGVHCAYCT-UHFFFAOYSA-N 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 201000000849 skin cancer Diseases 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- WHDIUBHAKZDSJL-UHFFFAOYSA-N (4-morpholin-4-ylphenyl)boronic acid Chemical compound C1=CC(B(O)O)=CC=C1N1CCOCC1 WHDIUBHAKZDSJL-UHFFFAOYSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- IAEOYUUPFYJXHN-UHFFFAOYSA-N 1,5-diiodopentane Chemical compound ICCCCCI IAEOYUUPFYJXHN-UHFFFAOYSA-N 0.000 description 2
- WAHDCHNIVMRNLX-UHFFFAOYSA-N 1,7-dimethyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridine-4-carbonitrile Chemical compound C1N(C)CCC=2C1=C(C)NC(=O)C=2C#N WAHDCHNIVMRNLX-UHFFFAOYSA-N 0.000 description 2
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 2
- BDISBPIAIZBDKU-LLVKDONJSA-N 1-[(1R)-1-(4-bromophenyl)ethyl]piperidine Chemical compound BrC1=CC=C(C=C1)[C@@H](C)N1CCCCC1 BDISBPIAIZBDKU-LLVKDONJSA-N 0.000 description 2
- UELWLLSJFNZKKZ-UHFFFAOYSA-N 1-methyl-3-oxo-5,6,7,8-tetrahydro-2h-isoquinoline-4-carbonitrile Chemical compound C1CCCC2=C(C)NC(=O)C(C#N)=C21 UELWLLSJFNZKKZ-UHFFFAOYSA-N 0.000 description 2
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 2
- WNWPMWRKNLBJQO-UHFFFAOYSA-N 2-(3-dimethoxyphosphorylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound COP(=O)(OC)C1=CC=CC(B2OC(C)(C)C(C)(C)O2)=C1 WNWPMWRKNLBJQO-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 2
- DETQETQTBXHTLD-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]-N-[(3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC=C3CCCCC23)=O)C=C(C1)C=1C=NC(=CC1)N1CCN(CC1)C)C)C1CCOCC1 DETQETQTBXHTLD-UHFFFAOYSA-N 0.000 description 2
- YVGSCPPJZPRZMJ-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-(6-pyrrolidin-1-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCCC1)C)C1CCOCC1 YVGSCPPJZPRZMJ-UHFFFAOYSA-N 0.000 description 2
- UEVPBXKCXXRMGO-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-[3-(1-pyrrolidin-1-ylethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC(=CC=C1)C(C)N1CCCC1)C(=O)NCC=1C(NC(=C2CCCCC12)C)=O)C)C1CCOCC1 UEVPBXKCXXRMGO-UHFFFAOYSA-N 0.000 description 2
- FPCWSZFJIREHOF-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-[4-(2-morpholin-4-ylethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CCN1CCOCC1)C(=O)NCC=1C(NC(=C2CCCCC12)C)=O)C)C1CCOCC1 FPCWSZFJIREHOF-UHFFFAOYSA-N 0.000 description 2
- XHXCMSSUGKWKFF-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(8-methyl-6-oxo-1,3,4,7-tetrahydropyrano[3,4-c]pyridin-5-yl)methyl]-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC1=C2C(=C(NC1=O)C)COCC2)C)C2CCOCC2 XHXCMSSUGKWKFF-UHFFFAOYSA-N 0.000 description 2
- LJDWPGXZNNEFNY-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[[1-methyl-3-oxo-7-(trifluoromethyl)-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl]methyl]-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC(=C2CC(CCC12)C(F)(F)F)C)=O)C)C1CCOCC1 LJDWPGXZNNEFNY-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- ROADCYAOHVSOLQ-UHFFFAOYSA-N 3-oxetanone Chemical compound O=C1COC1 ROADCYAOHVSOLQ-UHFFFAOYSA-N 0.000 description 2
- IYZIARNSXPGYSC-UHFFFAOYSA-N 3-phenylbenzamide Chemical compound NC(=O)C1=CC=CC(C=2C=CC=CC=2)=C1 IYZIARNSXPGYSC-UHFFFAOYSA-N 0.000 description 2
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 2
- OZOUWOZKSMVEHE-UHFFFAOYSA-N 5-dimethylphosphoryl-3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound CP(=O)(C)C=1C=C(C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C1)C)N(C1CCOCC1)CC OZOUWOZKSMVEHE-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- 201000003076 Angiosarcoma Diseases 0.000 description 2
- 208000003950 B-cell lymphoma Diseases 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 208000032612 Glial tumor Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 208000001258 Hemangiosarcoma Diseases 0.000 description 2
- 102000011787 Histone Methyltransferases Human genes 0.000 description 2
- 108010036115 Histone Methyltransferases Proteins 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- AMIMRNSIRUDHCM-UHFFFAOYSA-N Isopropylaldehyde Chemical compound CC(C)C=O AMIMRNSIRUDHCM-UHFFFAOYSA-N 0.000 description 2
- CPLXHLVBOLITMK-UHFFFAOYSA-N Magnesium oxide Chemical compound [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 208000034578 Multiple myelomas Diseases 0.000 description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 201000004404 Neurofibroma Diseases 0.000 description 2
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 2
- 229910002666 PdCl2 Inorganic materials 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 description 2
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 239000005864 Sulphur Substances 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 208000033781 Thyroid carcinoma Diseases 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- BNTIPMNMTIAWIW-UHFFFAOYSA-N [6-(trifluoromethyl)pyridin-3-yl]boronic acid Chemical compound OB(O)C1=CC=C(C(F)(F)F)N=C1 BNTIPMNMTIAWIW-UHFFFAOYSA-N 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- QLDHWVVRQCGZLE-UHFFFAOYSA-N acetyl cyanide Chemical compound CC(=O)C#N QLDHWVVRQCGZLE-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 208000026563 adrenal gland neuroblastoma Diseases 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 230000003140 astrocytic effect Effects 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 229960002412 cediranib Drugs 0.000 description 2
- 208000019065 cervical carcinoma Diseases 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 201000003444 follicular lymphoma Diseases 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- QBKSWRVVCFFDOT-UHFFFAOYSA-N gossypol Chemical compound CC(C)C1=C(O)C(O)=C(C=O)C2=C(O)C(C=3C(O)=C4C(C=O)=C(O)C(O)=C(C4=CC=3C)C(C)C)=C(C)C=C21 QBKSWRVVCFFDOT-UHFFFAOYSA-N 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 201000010536 head and neck cancer Diseases 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 125000005956 isoquinolyl group Chemical group 0.000 description 2
- 206010027191 meningioma Diseases 0.000 description 2
- NMLOSXSDLWFBKT-UHFFFAOYSA-N methyl 3-amino-5-bromo-2-methylbenzoate Chemical compound COC(=O)C1=CC(Br)=CC(N)=C1C NMLOSXSDLWFBKT-UHFFFAOYSA-N 0.000 description 2
- 230000011987 methylation Effects 0.000 description 2
- 238000007069 methylation reaction Methods 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- WOOWBQQQJXZGIE-UHFFFAOYSA-N n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound CCN(C(C)C)C(C)C.CCN(C(C)C)C(C)C WOOWBQQQJXZGIE-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 201000008968 osteosarcoma Diseases 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- XSCHRSMBECNVNS-UHFFFAOYSA-N quinoxaline Chemical compound N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 2
- 229910052702 rhenium Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 238000004611 spectroscopical analysis Methods 0.000 description 2
- 210000000130 stem cell Anatomy 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 201000002510 thyroid cancer Diseases 0.000 description 2
- 208000013077 thyroid gland carcinoma Diseases 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 208000010570 urinary bladder carcinoma Diseases 0.000 description 2
- 229960000241 vandetanib Drugs 0.000 description 2
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- SOZMSEPDYJGBEK-ZCFIWIBFSA-N (1r)-1-(4-bromophenyl)ethanamine Chemical compound C[C@@H](N)C1=CC=C(Br)C=C1 SOZMSEPDYJGBEK-ZCFIWIBFSA-N 0.000 description 1
- SOZMSEPDYJGBEK-LURJTMIESA-N (1s)-1-(4-bromophenyl)ethanamine Chemical compound C[C@H](N)C1=CC=C(Br)C=C1 SOZMSEPDYJGBEK-LURJTMIESA-N 0.000 description 1
- YPWAJLGHACDYQS-UHFFFAOYSA-N (2-methoxypyrimidin-5-yl)boronic acid Chemical compound COC1=NC=C(B(O)O)C=N1 YPWAJLGHACDYQS-UHFFFAOYSA-N 0.000 description 1
- CVCLJVVBHYOXDC-IAZSKANUSA-N (2z)-2-[(5z)-5-[(3,5-dimethyl-1h-pyrrol-2-yl)methylidene]-4-methoxypyrrol-2-ylidene]indole Chemical compound COC1=C\C(=C/2N=C3C=CC=CC3=C\2)N\C1=C/C=1NC(C)=CC=1C CVCLJVVBHYOXDC-IAZSKANUSA-N 0.000 description 1
- AFSSVCNPDKKSRR-UHFFFAOYSA-N (3-bromophenyl)boronic acid Chemical compound OB(O)C1=CC=CC(Br)=C1 AFSSVCNPDKKSRR-UHFFFAOYSA-N 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- VXWBQOJISHAKKM-UHFFFAOYSA-N (4-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=C(C=O)C=C1 VXWBQOJISHAKKM-UHFFFAOYSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- QFCMBRXRVQRSSF-UHFFFAOYSA-N 1,2,3,3a,4,5,6,6a-octahydropyrrolo[3,4-c]pyrrole Chemical compound C1NCC2CNCC21 QFCMBRXRVQRSSF-UHFFFAOYSA-N 0.000 description 1
- ABIPLJQFLRXYES-UHFFFAOYSA-N 1,2,3,3a-tetrahydropentalene Chemical compound C1=CC=C2CCCC21 ABIPLJQFLRXYES-UHFFFAOYSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- IZNGYNMIIVJWSO-UHFFFAOYSA-N 1,3,5-trimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound CC1=NN(C)C(C)=C1B1OC(C)(C)C(C)(C)O1 IZNGYNMIIVJWSO-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- BGEOMACGJZGFEC-OAHLLOKOSA-N 1-[(1R)-1-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethyl]piperidine Chemical compound CC1(OB(OC1(C)C)C1=CC=C(C=C1)[C@@H](C)N1CCCCC1)C BGEOMACGJZGFEC-OAHLLOKOSA-N 0.000 description 1
- BDISBPIAIZBDKU-NSHDSACASA-N 1-[(1S)-1-(4-bromophenyl)ethyl]piperidine Chemical compound BrC1=CC=C(C=C1)[C@H](C)N1CCCCC1 BDISBPIAIZBDKU-NSHDSACASA-N 0.000 description 1
- BGEOMACGJZGFEC-HNNXBMFYSA-N 1-[(1S)-1-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethyl]piperidine Chemical compound CC1(OB(OC1(C)C)C1=CC=C(C=C1)[C@H](C)N1CCCCC1)C BGEOMACGJZGFEC-HNNXBMFYSA-N 0.000 description 1
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 1
- VDBUUILSIZWDAW-UHFFFAOYSA-N 1-[2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]propan-2-yl]piperidine Chemical compound CC1(OB(OC1(C)C)C1=CC=C(C=C1)C(C)(C)N1CCCCC1)C VDBUUILSIZWDAW-UHFFFAOYSA-N 0.000 description 1
- LLAPDLPYIYKTGQ-UHFFFAOYSA-N 1-aminoethyl Chemical group C[CH]N LLAPDLPYIYKTGQ-UHFFFAOYSA-N 0.000 description 1
- LHILJAUUTFEGCV-UHFFFAOYSA-N 1-ethoxy-3-oxo-5,6,7,8-tetrahydro-2h-isoquinoline-4-carbonitrile Chemical compound C1CCCC2=C(OCC)NC(=O)C(C#N)=C21 LHILJAUUTFEGCV-UHFFFAOYSA-N 0.000 description 1
- JONNLPPUAHXEMS-UHFFFAOYSA-N 1-methyl-3-oxo-2,5,6,7,8,9-hexahydrocyclohepta[c]pyridine-4-carbonitrile Chemical compound C1CCCCC2=C(C)NC(=O)C(C#N)=C21 JONNLPPUAHXEMS-UHFFFAOYSA-N 0.000 description 1
- KPKMQKOATGLSHP-UHFFFAOYSA-N 1-methyl-3-oxo-2,5,6,7-tetrahydrocyclopenta[c]pyridine-4-carbonitrile Chemical compound N1C(=O)C(C#N)=C2CCCC2=C1C KPKMQKOATGLSHP-UHFFFAOYSA-N 0.000 description 1
- JQUBPRMKFQPKQN-UHFFFAOYSA-N 1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-2,7-naphthyridine-4-carbonitrile Chemical compound CC=1NC(C(=C2CCNCC12)C#N)=O JQUBPRMKFQPKQN-UHFFFAOYSA-N 0.000 description 1
- YYIVKEBKLAZGOU-UHFFFAOYSA-N 1-methyl-3-oxo-7-(2,2,2-trifluoroethyl)-2,5,6,8-tetrahydro-2,7-naphthyridine-4-carbonitrile Chemical compound CC=1NC(C(=C2CCN(CC12)CC(F)(F)F)C#N)=O YYIVKEBKLAZGOU-UHFFFAOYSA-N 0.000 description 1
- ASWLKLSOBFPDEV-UHFFFAOYSA-N 1-methyl-3-oxo-7-(trifluoromethyl)-5,6,7,8-tetrahydro-2H-isoquinoline-4-carbonitrile Chemical compound CC=1NC(C(=C2CCC(CC12)C(F)(F)F)C#N)=O ASWLKLSOBFPDEV-UHFFFAOYSA-N 0.000 description 1
- FLWNBODUCACHHU-UHFFFAOYSA-N 1-methyl-3-oxo-7-propan-2-yl-2,5,6,8-tetrahydro-2,7-naphthyridine-4-carbonitrile Chemical compound C(C)(C)N1CCC2=C(C(NC(=C2C1)C)=O)C#N FLWNBODUCACHHU-UHFFFAOYSA-N 0.000 description 1
- CYUSWSJMPWOAJR-UHFFFAOYSA-N 1-methyl-3-oxo-7-propyl-2,5,6,8-tetrahydro-2,7-naphthyridine-4-carbonitrile Chemical compound CC=1NC(C(=C2CCN(CC12)CCC)C#N)=O CYUSWSJMPWOAJR-UHFFFAOYSA-N 0.000 description 1
- ZSDDJVUYAQJBOO-UHFFFAOYSA-N 1-methyl-7-(oxetan-3-yl)-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridine-4-carbonitrile Chemical compound CC=1NC(C(=C2CCN(CC12)C1COC1)C#N)=O ZSDDJVUYAQJBOO-UHFFFAOYSA-N 0.000 description 1
- HUUPVABNAQUEJW-UHFFFAOYSA-N 1-methylpiperidin-4-one Chemical compound CN1CCC(=O)CC1 HUUPVABNAQUEJW-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- WMDLGLZRZCXLSM-UHFFFAOYSA-N 2-(2,2,2-trifluoroacetyl)cycloheptan-1-one Chemical compound FC(F)(F)C(=O)C1CCCCCC1=O WMDLGLZRZCXLSM-UHFFFAOYSA-N 0.000 description 1
- YDFLGIXUFCIELL-UHFFFAOYSA-N 2-(2,2,2-trifluoroacetyl)cyclohexan-1-one Chemical compound FC(F)(F)C(=O)C1CCCCC1=O YDFLGIXUFCIELL-UHFFFAOYSA-N 0.000 description 1
- UWMPYUXPRBDDNF-UHFFFAOYSA-N 2-(2,2,2-trifluoroacetyl)cyclopentan-1-one Chemical compound FC(F)(F)C(=O)C1CCCC1=O UWMPYUXPRBDDNF-UHFFFAOYSA-N 0.000 description 1
- FHCWGLQDAMYXJS-UHFFFAOYSA-N 2-(3-bromophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC(Br)=C1 FHCWGLQDAMYXJS-UHFFFAOYSA-N 0.000 description 1
- DSOYNVOQUXAPQL-UHFFFAOYSA-N 2-Acetylcycloheptanone Chemical compound CC(=O)C1CCCCCC1=O DSOYNVOQUXAPQL-UHFFFAOYSA-N 0.000 description 1
- BBCSKDZLEVADMO-UHFFFAOYSA-N 2-[ethyl(oxan-4-yl)amino]-6-methyl-3-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C1CCOCC1)C1=C(C=CC(=C1C(=O)N)C)C1=CC=C(C=C1)CN1CCOCC1 BBCSKDZLEVADMO-UHFFFAOYSA-N 0.000 description 1
- VSBJILDTCQTQAJ-UHFFFAOYSA-N 2-acetyl-4-(trifluoromethyl)cyclohexan-1-one Chemical compound C(C)(=O)C1C(CCC(C1)C(F)(F)F)=O VSBJILDTCQTQAJ-UHFFFAOYSA-N 0.000 description 1
- OEKATORRSPXJHE-UHFFFAOYSA-N 2-acetylcyclohexan-1-one Chemical compound CC(=O)C1CCCCC1=O OEKATORRSPXJHE-UHFFFAOYSA-N 0.000 description 1
- OSWDNIFICGLKEE-UHFFFAOYSA-N 2-acetylcyclopentan-1-one Chemical compound CC(=O)C1CCCC1=O OSWDNIFICGLKEE-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- IBODJKKYTBNWTD-UHFFFAOYSA-N 2-azaspiro[3.5]nonane Chemical compound C1NCC11CCCCC1 IBODJKKYTBNWTD-UHFFFAOYSA-N 0.000 description 1
- JEECGGXGHJUYMN-UHFFFAOYSA-N 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound C1=NC(C)=CC=C1B1OC(C)(C)C(C)(C)O1 JEECGGXGHJUYMN-UHFFFAOYSA-N 0.000 description 1
- ZHAIMJRKJKQNQI-UHFFFAOYSA-N 2-oxa-7-azaspiro[3.4]octane Chemical compound C1OCC11CNCC1 ZHAIMJRKJKQNQI-UHFFFAOYSA-N 0.000 description 1
- UGQAXUDFJWQPHM-UHFFFAOYSA-N 2-oxa-7-azaspiro[4.4]nonane Chemical compound C1NCCC11COCC1 UGQAXUDFJWQPHM-UHFFFAOYSA-N 0.000 description 1
- PNLURCRXZLVRJR-UHFFFAOYSA-N 2-oxaspiro[3.5]nonane Chemical compound C1OCC11CCCCC1 PNLURCRXZLVRJR-UHFFFAOYSA-N 0.000 description 1
- GWJJTLUNSYVYHO-UHFFFAOYSA-N 2-pyrrolidin-1-yl-5-(4,4,5,5-tetramethyl-1,3-dioxolan-2-yl)pyridine Chemical compound N1(CCCC1)C1=NC=C(C=C1)C1OC(C(O1)(C)C)(C)C GWJJTLUNSYVYHO-UHFFFAOYSA-N 0.000 description 1
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 1
- ZZVDXRCAGGQFAK-UHFFFAOYSA-N 2h-oxazaphosphinine Chemical class N1OC=CC=P1 ZZVDXRCAGGQFAK-UHFFFAOYSA-N 0.000 description 1
- 125000003762 3,4-dimethoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C(OC([H])([H])[H])C([H])=C1* 0.000 description 1
- 125000004211 3,5-difluorophenyl group Chemical group [H]C1=C(F)C([H])=C(*)C([H])=C1F 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 1
- DRHDVPPLSDJQLK-UHFFFAOYSA-N 3-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound NC(=O)C1=CC=CC(C=2C=NC(=CC=2)N2CCOCC2)=C1 DRHDVPPLSDJQLK-UHFFFAOYSA-N 0.000 description 1
- SPNAYGRJPAYMLE-UHFFFAOYSA-N 3-(aminomethyl)-6-methyl-1h-pyridin-2-one;hydrochloride Chemical compound Cl.CC1=CC=C(CN)C(=O)N1 SPNAYGRJPAYMLE-UHFFFAOYSA-N 0.000 description 1
- FULGYVDDZHOGSA-UHFFFAOYSA-N 3-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzamide Chemical compound CN1CCN(CC1)C1=CC=C(C=N1)C=1C=CC=C(C(=O)N)C=1 FULGYVDDZHOGSA-UHFFFAOYSA-N 0.000 description 1
- JBNHPLSONVVRGC-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-(4-methyl-3-oxo-1,4-benzoxazin-6-yl)-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C=C(C1)C1=CC2=C(OCC(N2C)=O)C=C1)C)C1CCOCC1 JBNHPLSONVVRGC-UHFFFAOYSA-N 0.000 description 1
- SDQHCYXDLFCUJH-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-(4-morpholin-4-ylphenyl)-N-[(3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)N1CCOCC1)C(=O)NCC=1C(NC=C2CCCCC12)=O)C)C1CCOCC1 SDQHCYXDLFCUJH-UHFFFAOYSA-N 0.000 description 1
- AVCDNWPILFAYOH-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-(6-morpholin-4-ylpyridin-3-yl)-N-[[3-oxo-1-(trifluoromethyl)-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl]methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C(F)(F)F)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 AVCDNWPILFAYOH-UHFFFAOYSA-N 0.000 description 1
- MLMUHXYRSGRQIH-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C1CCOCC1)C=1C(=C(C(=O)N)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C MLMUHXYRSGRQIH-UHFFFAOYSA-N 0.000 description 1
- NBELDDPCFCEAAC-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-(6-morpholin-4-ylpyridin-3-yl)benzoic acid Chemical compound C(C)N(C=1C(=C(C(=O)O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 NBELDDPCFCEAAC-UHFFFAOYSA-N 0.000 description 1
- RUQDOKXCJIPILL-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]-N-[[3-oxo-1-(trifluoromethyl)-2,5,6,7,8,9-hexahydrocyclohepta[c]pyridin-4-yl]methyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC1=C2C(=C(NC1=O)C(F)(F)F)CCCCC2)C)C2CCOCC2 RUQDOKXCJIPILL-UHFFFAOYSA-N 0.000 description 1
- ILSWVVZGWSKSQA-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]-N-[[3-oxo-1-(trifluoromethyl)-2,5,6,7-tetrahydrocyclopenta[c]pyridin-4-yl]methyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC1=C2C(=C(NC1=O)C(F)(F)F)CCC2)C)C2CCOCC2 ILSWVVZGWSKSQA-UHFFFAOYSA-N 0.000 description 1
- KLYFLPQLTNAFGR-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]benzoic acid Chemical compound C=1C(C=2C=CC(CN3CCOCC3)=CC=2)=CC(C(O)=O)=C(C)C=1N(CC)C1CCOCC1 KLYFLPQLTNAFGR-UHFFFAOYSA-N 0.000 description 1
- FSFIMTOPQUYCHV-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzoic acid Chemical compound C(C)N(C=1C(=C(C(=O)O)C=C(C1)C=1C=NC(=CC1)N1CCN(CC1)C)C)C1CCOCC1 FSFIMTOPQUYCHV-UHFFFAOYSA-N 0.000 description 1
- AKKBISHSFVSXRC-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-2,5,6,7-tetrahydrocyclopenta[c]pyridin-4-yl)methyl]-5-[4-(oxan-4-yl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)C1CCOCC1)C(=O)NCC1=C2C(=C(NC1=O)C)CCC2)C)C2CCOCC2 AKKBISHSFVSXRC-UHFFFAOYSA-N 0.000 description 1
- FSIROVNCORSVQK-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-5-[4-(1-piperidin-1-ylethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)C(C)N1CCCCC1)C(=O)NCC=1C(NC(=C2CCCCC12)C)=O)C)C1CCOCC1 FSIROVNCORSVQK-UHFFFAOYSA-N 0.000 description 1
- YWZDWBFSLHVMFD-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-7-propan-2-yl-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC(=C2CN(CCC12)C(C)C)C)=O)C)C1CCOCC1 YWZDWBFSLHVMFD-UHFFFAOYSA-N 0.000 description 1
- ZYIVGCAANSPYEA-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-7-propyl-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC(=C2CN(CCC12)CCC)C)=O)C)C1CCOCC1 ZYIVGCAANSPYEA-UHFFFAOYSA-N 0.000 description 1
- DCOFNPMTSLWMIT-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(6-methyl-2-oxo-1H-pyridin-3-yl)methyl]-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=CC2)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 DCOFNPMTSLWMIT-UHFFFAOYSA-N 0.000 description 1
- HCJSBLXRKPSAFE-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(6-methyl-2-oxo-1H-pyridin-3-yl)methyl]-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC(=CC1)C)=O)C)C1CCOCC1 HCJSBLXRKPSAFE-UHFFFAOYSA-N 0.000 description 1
- PQBWQOABZORSPH-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(6-methyl-2-oxo-1H-pyridin-3-yl)methyl]-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=CC=2)C)=O)C=C(C=1)C=1C=NC(=CC=1)N1CCN(CC1)C)C)C1CCOCC1 PQBWQOABZORSPH-UHFFFAOYSA-N 0.000 description 1
- RJYZMYOMFQWJJG-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[[1-methyl-3-oxo-7-(trifluoromethyl)-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl]methyl]-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CC(CCC23)C(F)(F)F)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 RJYZMYOMFQWJJG-UHFFFAOYSA-N 0.000 description 1
- QKXZHVABQDFTND-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[[1-methyl-7-(2-methylpropyl)-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl]methyl]-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC(=C2CN(CCC12)CC(C)C)C)=O)C)C1CCOCC1 QKXZHVABQDFTND-UHFFFAOYSA-N 0.000 description 1
- IMVIAINDDSWEJP-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[[1-methyl-7-(oxetan-3-yl)-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl]methyl]-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC=C(C=C1)CN1CCOCC1)C(=O)NCC=1C(NC(=C2CN(CCC12)C1COC1)C)=O)C)C1CCOCC1 IMVIAINDDSWEJP-UHFFFAOYSA-N 0.000 description 1
- QCKUUNBCDCYXNF-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(2-methoxypyrimidin-5-yl)-2-methyl-N-[(1-methyl-3-oxo-2,5,6,7,8,9-hexahydrocyclohepta[c]pyridin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC2=C3C(=C(NC2=O)C)CCCCC3)C=C(C1)C=1C=NC(=NC1)OC)C)C1CCOCC1 QCKUUNBCDCYXNF-UHFFFAOYSA-N 0.000 description 1
- JUMSRUJAEOHVMA-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(3-fluoro-4-morpholin-4-ylphenyl)-2-methyl-N-[(1-methyl-3-oxo-2,5,6,7-tetrahydrocyclopenta[c]pyridin-4-yl)methyl]benzamide Chemical compound C(C)N(C=1C(=C(C=C(C1)C1=CC(=C(C=C1)N1CCOCC1)F)C(=O)NCC1=C2C(=C(NC1=O)C)CCC2)C)C2CCOCC2 JUMSRUJAEOHVMA-UHFFFAOYSA-N 0.000 description 1
- VCVUGZIUYDDYKG-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(3-hydroxyoxetan-3-yl)-2-methylbenzoic acid Chemical compound C(C)N(C=1C(=C(C(=O)O)C=C(C1)C1(COC1)O)C)C1CCOCC1 VCVUGZIUYDDYKG-UHFFFAOYSA-N 0.000 description 1
- HEXGBHZDRVYOMP-UHFFFAOYSA-N 3-[ethyl(oxan-4-yl)amino]-5-(3-methoxyoxetan-3-yl)-2-methylbenzoic acid Chemical compound C(C)N(C=1C(=C(C(=O)O)C=C(C1)C1(COC1)OC)C)C1CCOCC1 HEXGBHZDRVYOMP-UHFFFAOYSA-N 0.000 description 1
- BGKOEZJCYPBPPW-UHFFFAOYSA-N 3-acetyloxan-4-one Chemical compound CC(=O)C1COCCC1=O BGKOEZJCYPBPPW-UHFFFAOYSA-N 0.000 description 1
- LKEABTHVRADLLJ-UHFFFAOYSA-N 3-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound C1=C(F)C(C)=NC=C1B1OC(C)(C)C(C)(C)O1 LKEABTHVRADLLJ-UHFFFAOYSA-N 0.000 description 1
- FYJSFVVUSKOPRK-UHFFFAOYSA-N 3-oxo-1-(trifluoromethyl)-2,5,6,7,8,9-hexahydrocyclohepta[c]pyridine-4-carbonitrile Chemical compound O=C1C(=C2C(=C(N1)C(F)(F)F)CCCCC2)C#N FYJSFVVUSKOPRK-UHFFFAOYSA-N 0.000 description 1
- KPKZUELBFKHRPC-UHFFFAOYSA-N 3-oxo-1-(trifluoromethyl)-2,5,6,7-tetrahydrocyclopenta[c]pyridine-4-carbonitrile Chemical compound O=C1C(=C2C(=C(N1)C(F)(F)F)CCC2)C#N KPKZUELBFKHRPC-UHFFFAOYSA-N 0.000 description 1
- UAWYPSWCSFLTLP-UHFFFAOYSA-N 3-oxo-1-(trifluoromethyl)-5,6,7,8-tetrahydro-2H-isoquinoline-4-carbonitrile Chemical compound O=C1NC(=C2CCCCC2=C1C#N)C(F)(F)F UAWYPSWCSFLTLP-UHFFFAOYSA-N 0.000 description 1
- IGNWFQOCRCXUJK-UHFFFAOYSA-N 3-oxo-5,6,7,8-tetrahydro-2h-isoquinoline-4-carbonitrile Chemical compound C1CCCC2=C(C#N)C(O)=NC=C21 IGNWFQOCRCXUJK-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- DMLXOYKSKYTGKQ-UHFFFAOYSA-N 4,4,5,5-tetramethyl-2-[4-(oxan-4-yl)phenyl]-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(C2CCOCC2)C=C1 DMLXOYKSKYTGKQ-UHFFFAOYSA-N 0.000 description 1
- QDCIXBBEUHMLDN-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-indole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC2=C1C=CN2 QDCIXBBEUHMLDN-UHFFFAOYSA-N 0.000 description 1
- CQXDFDDHJWNGGF-UHFFFAOYSA-N 4-(aminomethyl)-1-(trifluoromethyl)-2,5,6,7,8,9-hexahydrocyclohepta[c]pyridin-3-one hydrochloride Chemical compound Cl.NCC1=C2C(=C(NC1=O)C(F)(F)F)CCCCC2 CQXDFDDHJWNGGF-UHFFFAOYSA-N 0.000 description 1
- PMIKDGCEXDEEQP-UHFFFAOYSA-N 4-(aminomethyl)-1-(trifluoromethyl)-2,5,6,7-tetrahydrocyclopenta[c]pyridin-3-one hydrochloride Chemical compound Cl.NCC1=C2C(=C(NC1=O)C(F)(F)F)CCC2 PMIKDGCEXDEEQP-UHFFFAOYSA-N 0.000 description 1
- UEMKQIANKPMPNV-UHFFFAOYSA-N 4-(aminomethyl)-1-ethoxy-5,6,7,8-tetrahydro-2H-isoquinolin-3-one Chemical compound NCC=1C(NC(=C2CCCCC12)OCC)=O UEMKQIANKPMPNV-UHFFFAOYSA-N 0.000 description 1
- BLGGSRMUSBMTSK-UHFFFAOYSA-N 4-(aminomethyl)-1-methyl-2,5,6,7-tetrahydrocyclopenta[c]pyridin-3-one hydrochloride Chemical compound Cl.NCC1=C2C(=C(NC1=O)C)CCC2 BLGGSRMUSBMTSK-UHFFFAOYSA-N 0.000 description 1
- IMQJERUFPGBLTJ-UHFFFAOYSA-N 4-(aminomethyl)-1-methyl-5,6,7,8-tetrahydro-2H-isoquinolin-3-one Chemical compound NCC=1C(NC(=C2CCCCC12)C)=O IMQJERUFPGBLTJ-UHFFFAOYSA-N 0.000 description 1
- BOPVXDGDIQPEFP-UHFFFAOYSA-N 4-(aminomethyl)-1-methyl-7-(2,2,2-trifluoroethyl)-2,5,6,8-tetrahydro-2,7-naphthyridin-3-one Chemical compound NCC=1C(NC(=C2CN(CCC12)CC(F)(F)F)C)=O BOPVXDGDIQPEFP-UHFFFAOYSA-N 0.000 description 1
- FOFHLPYGZLPQMA-UHFFFAOYSA-N 4-(aminomethyl)-1-methyl-7-(2-methylpropyl)-2,5,6,8-tetrahydro-2,7-naphthyridin-3-one hydrochloride Chemical compound Cl.NCC=1C(NC(=C2CN(CCC12)CC(C)C)C)=O FOFHLPYGZLPQMA-UHFFFAOYSA-N 0.000 description 1
- BICNTRLUYLOQOD-UHFFFAOYSA-N 4-(aminomethyl)-1-methyl-7-(oxetan-3-yl)-2,5,6,8-tetrahydro-2,7-naphthyridin-3-one hydrochloride Chemical compound Cl.NCC=1C(NC(=C2CN(CCC12)C1COC1)C)=O BICNTRLUYLOQOD-UHFFFAOYSA-N 0.000 description 1
- KRLXLJFMRWUOFC-UHFFFAOYSA-N 4-(aminomethyl)-1-methyl-7-(trifluoromethyl)-5,6,7,8-tetrahydro-2H-isoquinolin-3-one Chemical compound NCC=1C(NC(=C2CC(CCC12)C(F)(F)F)C)=O KRLXLJFMRWUOFC-UHFFFAOYSA-N 0.000 description 1
- QXRIEFBSUUHPHJ-UHFFFAOYSA-N 4-(aminomethyl)-1-methyl-7-propan-2-yl-2,5,6,8-tetrahydro-2,7-naphthyridin-3-one hydrochloride Chemical compound Cl.NCC=1C(NC(=C2CN(CCC12)C(C)C)C)=O QXRIEFBSUUHPHJ-UHFFFAOYSA-N 0.000 description 1
- MSEWAHNDPKEOLO-UHFFFAOYSA-N 4-(aminomethyl)-1-methyl-7-propyl-2,5,6,8-tetrahydro-2,7-naphthyridin-3-one hydrochloride Chemical compound Cl.NCC=1C(NC(=C2CN(CCC12)CCC)C)=O MSEWAHNDPKEOLO-UHFFFAOYSA-N 0.000 description 1
- XVZSHSRFIXZSKG-UHFFFAOYSA-N 4-(aminomethyl)-5,6,7,8-tetrahydro-2H-isoquinolin-3-one hydrochloride Chemical compound Cl.NCC=1C(NC=C2CCCCC12)=O XVZSHSRFIXZSKG-UHFFFAOYSA-N 0.000 description 1
- BJOMSSSCJOMVRF-UHFFFAOYSA-N 4-(aminomethyl)-7-(cyclopropylmethyl)-1-methyl-2,5,6,8-tetrahydro-2,7-naphthyridin-3-one hydrochloride Chemical compound Cl.NCC=1C(NC(=C2CN(CCC12)CC1CC1)C)=O BJOMSSSCJOMVRF-UHFFFAOYSA-N 0.000 description 1
- RIQMSWOLENZXFK-UHFFFAOYSA-N 4-[1-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethyl]morpholine Chemical compound C=1C=C(B2OC(C)(C)C(C)(C)O2)C=CC=1C(C)N1CCOCC1 RIQMSWOLENZXFK-UHFFFAOYSA-N 0.000 description 1
- OOGUDBLEQZQAGC-UHFFFAOYSA-N 4-[2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethyl]morpholine Chemical compound O1C(C)(C)C(C)(C)OB1C(C=C1)=CC=C1CCN1CCOCC1 OOGUDBLEQZQAGC-UHFFFAOYSA-N 0.000 description 1
- YOJYRVSDQHWBGS-UHFFFAOYSA-N 4-[2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]morpholine Chemical compound O1C(C)(C)C(C)(C)OB1C(C=C1F)=CC=C1N1CCOCC1 YOJYRVSDQHWBGS-UHFFFAOYSA-N 0.000 description 1
- MWMQQWANJUZVDR-UHFFFAOYSA-N 4-[3-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl]morpholine Chemical compound FC=1C(=NC=C(C1)B1OC(C(O1)(C)C)(C)C)N1CCOCC1 MWMQQWANJUZVDR-UHFFFAOYSA-N 0.000 description 1
- DDIPWDTXYFXZET-UHFFFAOYSA-N 4-[[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl]methyl]morpholine Chemical compound O1C(C)(C)C(C)(C)OB1C(C=N1)=CC=C1CN1CCOCC1 DDIPWDTXYFXZET-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- 125000004801 4-cyanophenyl group Chemical group [H]C1=C([H])C(C#N)=C([H])C([H])=C1* 0.000 description 1
- ZBZAZWWQFQFIAN-UHFFFAOYSA-N 4-methyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,4-benzoxazin-3-one Chemical compound C1=C2N(C)C(=O)COC2=CC=C1B1OC(C)(C)C(C)(C)O1 ZBZAZWWQFQFIAN-UHFFFAOYSA-N 0.000 description 1
- VRJHQPZVIGNGMX-UHFFFAOYSA-N 4-piperidinone Chemical compound O=C1CCNCC1 VRJHQPZVIGNGMX-UHFFFAOYSA-N 0.000 description 1
- FATPQDPUKVVCLT-UHFFFAOYSA-N 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-indole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(NC=C2)C2=C1 FATPQDPUKVVCLT-UHFFFAOYSA-N 0.000 description 1
- CKFXPBNBNPHZIG-UHFFFAOYSA-N 5-(aminomethyl)-8-methyl-1,3,4,7-tetrahydropyrano[3,4-c]pyridin-6-one hydrochloride Chemical compound Cl.NCC1=C2C(=C(NC1=O)C)COCC2 CKFXPBNBNPHZIG-UHFFFAOYSA-N 0.000 description 1
- IRPVABHDSJVBNZ-RTHVDDQRSA-N 5-[1-(cyclopropylmethyl)-5-[(1R,5S)-3-(oxetan-3-yl)-3-azabicyclo[3.1.0]hexan-6-yl]pyrazol-3-yl]-3-(trifluoromethyl)pyridin-2-amine Chemical compound C1=C(C(F)(F)F)C(N)=NC=C1C1=NN(CC2CC2)C(C2[C@@H]3CN(C[C@@H]32)C2COC2)=C1 IRPVABHDSJVBNZ-RTHVDDQRSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- XVUZJDSVZMDZBY-UHFFFAOYSA-N 5-bromo-2-methyl-3-(2-oxoazepan-1-yl)benzoic acid Chemical compound BrC=1C=C(C(=C(C(=O)O)C1)C)N1C(CCCCC1)=O XVUZJDSVZMDZBY-UHFFFAOYSA-N 0.000 description 1
- DXUUJILVHXQDCV-UHFFFAOYSA-N 5-bromo-2-methyl-3-nitrobenzoic acid Chemical compound CC1=C(C(O)=O)C=C(Br)C=C1[N+]([O-])=O DXUUJILVHXQDCV-UHFFFAOYSA-N 0.000 description 1
- TZWCNNXGMHLOFP-UHFFFAOYSA-N 5-bromo-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]-3-(2-oxoazepan-1-yl)benzamide Chemical compound BrC=1C=C(C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C1)C)N1C(CCCCC1)=O TZWCNNXGMHLOFP-UHFFFAOYSA-N 0.000 description 1
- JCBDKQUYDVUSCB-UHFFFAOYSA-N 5-bromo-3-[ethyl(oxan-4-yl)amino]-2-fluoro-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound BrC=1C=C(C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C1)F)N(C1CCOCC1)CC JCBDKQUYDVUSCB-UHFFFAOYSA-N 0.000 description 1
- GOUHEIQPTYPLPN-UHFFFAOYSA-N 5-bromo-3-[ethyl(oxan-4-yl)amino]-2-fluorobenzoic acid Chemical compound BrC=1C=C(C(=C(C(=O)O)C1)F)N(C1CCOCC1)CC GOUHEIQPTYPLPN-UHFFFAOYSA-N 0.000 description 1
- RLAJAQYAZGCECQ-UHFFFAOYSA-N 5-bromo-3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound BrC=1C=C(C(=C(C(=O)NCC=2C(NC(=C3CCCCC23)C)=O)C1)C)N(C1CCOCC1)CC RLAJAQYAZGCECQ-UHFFFAOYSA-N 0.000 description 1
- RHXNIHLWHDXUPP-UHFFFAOYSA-N 5-bromo-3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methyl]benzamide Chemical compound BrC=1C=C(C(=C(C(=O)NCC=2C(NC=C3CCCCC23)=O)C1)C)N(C1CCOCC1)CC RHXNIHLWHDXUPP-UHFFFAOYSA-N 0.000 description 1
- IQBUXDNMMHNFDS-UHFFFAOYSA-N 5-bromo-3-[ethyl(oxan-4-yl)amino]-2-methyl-N-[(6-methyl-2-oxo-1H-pyridin-3-yl)methyl]benzamide Chemical compound BrC=1C=C(C(=C(C(=O)NCC=2C(NC(=CC2)C)=O)C1)C)N(C1CCOCC1)CC IQBUXDNMMHNFDS-UHFFFAOYSA-N 0.000 description 1
- IPITXYVHPYUULP-UHFFFAOYSA-N 5-bromo-3-[ethyl(oxan-4-yl)amino]-2-methylbenzoic acid Chemical compound C=1C(Br)=CC(C(O)=O)=C(C)C=1N(CC)C1CCOCC1 IPITXYVHPYUULP-UHFFFAOYSA-N 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- NVRVNSHHLPQGCU-UHFFFAOYSA-N 6-bromohexanoic acid Chemical compound OC(=O)CCCCCBr NVRVNSHHLPQGCU-UHFFFAOYSA-N 0.000 description 1
- FIMGYEKEYXUTGD-UHFFFAOYSA-N 6-methyl-2-oxo-1h-pyridine-3-carbonitrile Chemical compound CC1=CC=C(C#N)C(O)=N1 FIMGYEKEYXUTGD-UHFFFAOYSA-N 0.000 description 1
- HMGRRRXXYQYVAO-UHFFFAOYSA-N 7-(cyclopropylmethyl)-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridine-4-carbonitrile Chemical compound C1(CC1)CN1CCC2=C(C(NC(=C2C1)C)=O)C#N HMGRRRXXYQYVAO-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- NFTKYRKVHAJCBX-UHFFFAOYSA-N 7-butyl-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridine-4-carbonitrile Chemical compound C(CCC)N1CCC2=C(C(NC(=C2C1)C)=O)C#N NFTKYRKVHAJCBX-UHFFFAOYSA-N 0.000 description 1
- VTNQATDUHKYASG-UHFFFAOYSA-N 7-ethyl-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridine-4-carbonitrile Chemical compound C(C)N1CCC2=C(C(NC(=C2C1)C)=O)C#N VTNQATDUHKYASG-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- SAZZPRNPIWGGMH-UHFFFAOYSA-N 8-methyl-6-oxo-1,3,4,7-tetrahydropyrano[3,4-c]pyridine-5-carbonitrile Chemical compound C1COCC2=C(C)NC(=O)C(C#N)=C21 SAZZPRNPIWGGMH-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 206010001233 Adenoma benign Diseases 0.000 description 1
- 102400000068 Angiostatin Human genes 0.000 description 1
- 108010079709 Angiostatins Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 201000008271 Atypical teratoid rhabdoid tumor Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000028564 B-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 239000012664 BCL-2-inhibitor Substances 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 229940123711 Bcl2 inhibitor Drugs 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000003170 Bronchiolo-Alveolar Adenocarcinoma Diseases 0.000 description 1
- GSVHDOOBWFHZMT-UHFFFAOYSA-N C(#N)C1=C2CCN(CC2=C(NC1=O)C)C(=O)OC(C)(C)C Chemical compound C(#N)C1=C2CCN(CC2=C(NC1=O)C)C(=O)OC(C)(C)C GSVHDOOBWFHZMT-UHFFFAOYSA-N 0.000 description 1
- GRMSPKGOZLXHQS-UHFFFAOYSA-N CC1(OB(OC1(C)C)C=1C(NC2=CC=CC=C2C1)=O)C Chemical compound CC1(OB(OC1(C)C)C=1C(NC2=CC=CC=C2C1)=O)C GRMSPKGOZLXHQS-UHFFFAOYSA-N 0.000 description 1
- GUBYHGBHMCORDG-UHFFFAOYSA-N CC1=C(C=C(C=C1N1C(CCCCC1)=O)C1=CC=C(C=C1)P(OC)(OC)=O)C(NCC=1C(NC(=C2CCCCC12)C)=O)=O Chemical compound CC1=C(C=C(C=C1N1C(CCCCC1)=O)C1=CC=C(C=C1)P(OC)(OC)=O)C(NCC=1C(NC(=C2CCCCC12)C)=O)=O GUBYHGBHMCORDG-UHFFFAOYSA-N 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282461 Canis lupus Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 208000009458 Carcinoma in Situ Diseases 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 241000282994 Cervidae Species 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 201000005262 Chondroma Diseases 0.000 description 1
- 108010077544 Chromatin Proteins 0.000 description 1
- WMIUPLULYBKZDK-UHFFFAOYSA-N Cl.NCC=1C(NC(=C2CN(CCC12)CC)C)=O Chemical compound Cl.NCC=1C(NC(=C2CN(CCC12)CC)C)=O WMIUPLULYBKZDK-UHFFFAOYSA-N 0.000 description 1
- BYZJWOCDLJHFQU-UHFFFAOYSA-N Cl.NCC=1C(NC(=C2CN(CCC12)CCCC)C)=O Chemical compound Cl.NCC=1C(NC(=C2CN(CCC12)CCCC)C)=O BYZJWOCDLJHFQU-UHFFFAOYSA-N 0.000 description 1
- 102100031162 Collagen alpha-1(XVIII) chain Human genes 0.000 description 1
- 206010048832 Colon adenoma Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 1
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- 201000009051 Embryonal Carcinoma Diseases 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 208000007659 Fibroadenoma Diseases 0.000 description 1
- 206010053717 Fibrous histiocytoma Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 208000007569 Giant Cell Tumors Diseases 0.000 description 1
- 201000005409 Gliomatosis cerebri Diseases 0.000 description 1
- 206010018404 Glucagonoma Diseases 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 206010019629 Hepatic adenoma Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000005045 Interdigitating dendritic cell sarcoma Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 1
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 1
- 239000002177 L01XE27 - Ibrutinib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 1
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000002404 Liver Cell Adenoma Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- HPYDJCRRYSTNSD-UHFFFAOYSA-N N-[(7-butyl-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C(CCC)N1CCC2=C(C(NC(=C2C1)C)=O)CNC(=O)C=1C=C(C=C(C1C)N(C1CCOCC1)CC)C1=CC=C(C=C1)CN1CCOCC1 HPYDJCRRYSTNSD-UHFFFAOYSA-N 0.000 description 1
- RYZYMGSCDMQWPF-UHFFFAOYSA-N N-[(7-ethyl-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-(6-morpholin-4-ylpyridin-3-yl)benzamide Chemical compound C(C)N(C=1C(=C(C(=O)NCC=2C(NC(=C3CN(CCC23)CC)C)=O)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 RYZYMGSCDMQWPF-UHFFFAOYSA-N 0.000 description 1
- VCUFZILGIRCDQQ-KRWDZBQOSA-N N-[[(5S)-2-oxo-3-(2-oxo-3H-1,3-benzoxazol-6-yl)-1,3-oxazolidin-5-yl]methyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C1O[C@H](CN1C1=CC2=C(NC(O2)=O)C=C1)CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F VCUFZILGIRCDQQ-KRWDZBQOSA-N 0.000 description 1
- GGEGVRNOMLNMKA-UHFFFAOYSA-N N-[[7-(cyclopropylmethyl)-1-methyl-3-oxo-2,5,6,8-tetrahydro-2,7-naphthyridin-4-yl]methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide Chemical compound C1(CC1)CN1CCC2=C(C(NC(=C2C1)C)=O)CNC(=O)C=1C=C(C=C(C1C)N(C1CCOCC1)CC)C1=CC=C(C=C1)CN1CCOCC1 GGEGVRNOMLNMKA-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- IDRGFNPZDVBSSE-UHFFFAOYSA-N OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F Chemical compound OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F IDRGFNPZDVBSSE-UHFFFAOYSA-N 0.000 description 1
- 239000012425 OXONE® Substances 0.000 description 1
- 201000010133 Oligodendroglioma Diseases 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 239000012828 PI3K inhibitor Substances 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 208000005678 Rhabdomyoma Diseases 0.000 description 1
- 201000010208 Seminoma Diseases 0.000 description 1
- 206010054184 Small intestine carcinoma Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric Acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- QHOPXUFELLHKAS-UHFFFAOYSA-N Thespesin Natural products CC(C)c1c(O)c(O)c2C(O)Oc3c(c(C)cc1c23)-c1c2OC(O)c3c(O)c(O)c(C(C)C)c(cc1C)c23 QHOPXUFELLHKAS-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 206010062129 Tongue neoplasm Diseases 0.000 description 1
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 1
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 241000282458 Ursus sp. Species 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 201000003761 Vaginal carcinoma Diseases 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 241000282485 Vulpes vulpes Species 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- 206010048214 Xanthoma Diseases 0.000 description 1
- 206010048215 Xanthomatosis Diseases 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- OFYQEYPITRRCBP-UHFFFAOYSA-N [3-[(dimethylamino)methyl]phenyl]boronic acid Chemical compound CN(C)CC1=CC=CC(B(O)O)=C1 OFYQEYPITRRCBP-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000002001 anti-metastasis Effects 0.000 description 1
- 229940044684 anti-microtubule agent Drugs 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940034982 antineoplastic agent Drugs 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 208000001119 benign fibrous histiocytoma Diseases 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 201000001531 bladder carcinoma Diseases 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 201000008211 brain sarcoma Diseases 0.000 description 1
- 201000003149 breast fibroadenoma Diseases 0.000 description 1
- 201000002143 bronchus adenoma Diseases 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 208000022033 carcinoma of urethra Diseases 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 201000005217 chondroblastoma Diseases 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 208000009060 clear cell adenocarcinoma Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 201000010989 colorectal carcinoma Diseases 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 201000010305 cutaneous fibrous histiocytoma Diseases 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- CGZZMOTZOONQIA-UHFFFAOYSA-N cycloheptanone Chemical compound O=C1CCCCCC1 CGZZMOTZOONQIA-UHFFFAOYSA-N 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- JMYVMOUINOAAPA-UHFFFAOYSA-N cyclopropanecarbaldehyde Chemical compound O=CC1CC1 JMYVMOUINOAAPA-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 150000001975 deuterium Chemical group 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000006003 dichloroethyl group Chemical group 0.000 description 1
- 125000004774 dichlorofluoromethyl group Chemical group FC(Cl)(Cl)* 0.000 description 1
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- LXCYSACZTOKNNS-UHFFFAOYSA-N diethoxy(oxo)phosphanium Chemical compound CCO[P+](=O)OCC LXCYSACZTOKNNS-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 description 1
- 125000006001 difluoroethyl group Chemical group 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- JMRYOSQOYJBDOI-UHFFFAOYSA-N dilithium;di(propan-2-yl)azanide Chemical compound [Li+].CC(C)[N-]C(C)C.CC(C)N([Li])C(C)C JMRYOSQOYJBDOI-UHFFFAOYSA-N 0.000 description 1
- WQAWEUZTDVWTDB-UHFFFAOYSA-N dimethyl(oxo)phosphanium Chemical compound C[P+](C)=O WQAWEUZTDVWTDB-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 150000004141 diterpene derivatives Chemical class 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000003968 dna methyltransferase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 201000009409 embryonal rhabdomyosarcoma Diseases 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 230000001973 epigenetic effect Effects 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- LJCDYNICHZCUGR-UHFFFAOYSA-N ethyl 2-[3-[4-[3-[ethyl(oxan-4-yl)amino]-4-methyl-5-[(1-methyl-3-oxo-5,6,7,8-tetrahydro-2H-isoquinolin-4-yl)methylcarbamoyl]phenyl]phenyl]oxetan-3-yl]acetate Chemical compound C(C)N(C=1C=C(C=C(C1C)C(NCC=1C(NC(=C2CCCCC12)C)=O)=O)C1=CC=C(C=C1)C1(COC1)CC(=O)OCC)C1CCOCC1 LJCDYNICHZCUGR-UHFFFAOYSA-N 0.000 description 1
- FGSGHBPKHFDJOP-UHFFFAOYSA-N ethyl 2-oxocyclohexane-1-carboxylate Chemical compound CCOC(=O)C1CCCCC1=O FGSGHBPKHFDJOP-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 1
- 201000001343 fallopian tube carcinoma Diseases 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 208000010749 gastric carcinoma Diseases 0.000 description 1
- 201000011587 gastric lymphoma Diseases 0.000 description 1
- 208000015419 gastrin-producing neuroendocrine tumor Diseases 0.000 description 1
- 201000000052 gastrinoma Diseases 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 230000030279 gene silencing Effects 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 229930000755 gossypol Natural products 0.000 description 1
- 229950005277 gossypol Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229960004198 guanidine Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 201000011066 hemangioma Diseases 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- 201000002735 hepatocellular adenoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 125000006343 heptafluoro propyl group Chemical group 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- MHLPKAGDPWUOOT-UHFFFAOYSA-N housane Chemical compound C1CC2CC21 MHLPKAGDPWUOOT-UHFFFAOYSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical class I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- XYFPWWZEPKGCCK-GOSISDBHSA-N ibrutinib Chemical compound C1=2C(N)=NC=NC=2N([C@H]2CN(CCC2)C(=O)C=C)N=C1C(C=C1)=CC=C1OC1=CC=CC=C1 XYFPWWZEPKGCCK-GOSISDBHSA-N 0.000 description 1
- 229960001507 ibrutinib Drugs 0.000 description 1
- YKLIKGKUANLGSB-HNNXBMFYSA-N idelalisib Chemical compound C1([C@@H](NC=2[C]3N=CN=C3N=CN=2)CC)=NC2=CC=CC(F)=C2C(=O)N1C1=CC=CC=C1 YKLIKGKUANLGSB-HNNXBMFYSA-N 0.000 description 1
- 229960003445 idelalisib Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 201000004933 in situ carcinoma Diseases 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 150000002475 indoles Chemical class 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 206010022498 insulinoma Diseases 0.000 description 1
- 210000002570 interstitial cell Anatomy 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 201000010985 invasive ductal carcinoma Diseases 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000004552 isoquinolin-4-yl group Chemical group C1=NC=C(C2=CC=CC=C12)* 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 208000022013 kidney Wilms tumor Diseases 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- 208000003849 large cell carcinoma Diseases 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- CUONGYYJJVDODC-UHFFFAOYSA-N malononitrile Chemical compound N#CCC#N CUONGYYJJVDODC-UHFFFAOYSA-N 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 210000002418 meninge Anatomy 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- FQPCDFQEIJXUGX-UHFFFAOYSA-N methyl 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-(6-morpholin-4-ylpyridin-3-yl)benzoate Chemical compound C(C)N(C=1C(=C(C(=O)OC)C=C(C1)C=1C=NC(=CC1)N1CCOCC1)C)C1CCOCC1 FQPCDFQEIJXUGX-UHFFFAOYSA-N 0.000 description 1
- SBEPWSRSEAKYPL-UHFFFAOYSA-N methyl 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]benzoate Chemical compound C=1C(C=2C=CC(CN3CCOCC3)=CC=2)=CC(C(=O)OC)=C(C)C=1N(CC)C1CCOCC1 SBEPWSRSEAKYPL-UHFFFAOYSA-N 0.000 description 1
- HADYIIRSJPWVPH-UHFFFAOYSA-N methyl 3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[6-(4-methylpiperazin-1-yl)pyridin-3-yl]benzoate Chemical compound C(C)N(C=1C(=C(C(=O)OC)C=C(C1)C=1C=NC(=CC1)N1CCN(CC1)C)C)C1CCOCC1 HADYIIRSJPWVPH-UHFFFAOYSA-N 0.000 description 1
- IYFKZFOVIFHVSD-UHFFFAOYSA-N methyl 3-[ethyl(oxan-4-yl)amino]-5-(3-methoxyoxetan-3-yl)-2-methylbenzoate Chemical compound C(C)N(C=1C(=C(C(=O)OC)C=C(C1)C1(COC1)OC)C)C1CCOCC1 IYFKZFOVIFHVSD-UHFFFAOYSA-N 0.000 description 1
- OPHPFMGGUDZNOU-UHFFFAOYSA-N methyl 3-amino-5-bromo-2-fluorobenzoate Chemical compound COC(=O)C1=CC(Br)=CC(N)=C1F OPHPFMGGUDZNOU-UHFFFAOYSA-N 0.000 description 1
- WVAMJHFEJPKDCD-UHFFFAOYSA-N methyl 5-bromo-2-fluoro-3-(oxan-4-ylamino)benzoate Chemical compound BrC=1C=C(C(=C(C(=O)OC)C1)F)NC1CCOCC1 WVAMJHFEJPKDCD-UHFFFAOYSA-N 0.000 description 1
- ODDWYOBMSSXDDU-UHFFFAOYSA-N methyl 5-bromo-2-fluoro-3-nitrobenzoate Chemical compound COC(=O)C1=CC(Br)=CC([N+]([O-])=O)=C1F ODDWYOBMSSXDDU-UHFFFAOYSA-N 0.000 description 1
- PMZVIXJYPPZPPJ-UHFFFAOYSA-N methyl 5-bromo-2-methyl-3-(2-oxoazepan-1-yl)benzoate Chemical compound BrC=1C=C(C(=C(C(=O)OC)C1)C)N1C(CCCCC1)=O PMZVIXJYPPZPPJ-UHFFFAOYSA-N 0.000 description 1
- SGCILHRPCZDQGD-UHFFFAOYSA-N methyl 5-bromo-2-methyl-3-(oxan-4-ylamino)benzoate Chemical compound COC(=O)C1=CC(Br)=CC(NC2CCOCC2)=C1C SGCILHRPCZDQGD-UHFFFAOYSA-N 0.000 description 1
- UPBDNPCJIJAMPI-UHFFFAOYSA-N methyl 5-bromo-2-methyl-3-nitrobenzoate Chemical compound COC(=O)C1=CC(Br)=CC([N+]([O-])=O)=C1C UPBDNPCJIJAMPI-UHFFFAOYSA-N 0.000 description 1
- SIYWDEGKWJEBES-UHFFFAOYSA-N methyl 5-bromo-3-(6-bromohexanoylamino)-2-methylbenzoate Chemical compound BrC=1C=C(C(=C(C(=O)OC)C1)C)NC(CCCCCBr)=O SIYWDEGKWJEBES-UHFFFAOYSA-N 0.000 description 1
- QRWYCWGGFYOMCF-UHFFFAOYSA-N methyl 5-bromo-3-[ethyl(oxan-4-yl)amino]-2-fluorobenzoate Chemical compound BrC=1C=C(C(=C(C(=O)OC)C1)F)N(C1CCOCC1)CC QRWYCWGGFYOMCF-UHFFFAOYSA-N 0.000 description 1
- SLSYENZIHLMJNW-UHFFFAOYSA-N methyl 5-bromo-3-[ethyl(oxan-4-yl)amino]-2-methylbenzoate Chemical compound C=1C(Br)=CC(C(=O)OC)=C(C)C=1N(CC)C1CCOCC1 SLSYENZIHLMJNW-UHFFFAOYSA-N 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 210000000258 minor salivary gland Anatomy 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000002200 mouth mucosa Anatomy 0.000 description 1
- RMPRVJCNMPFBCX-UHFFFAOYSA-N n,n-dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidin-2-amine Chemical compound C1=NC(N(C)C)=NC=C1B1OC(C)(C)C(C)(C)O1 RMPRVJCNMPFBCX-UHFFFAOYSA-N 0.000 description 1
- KWQWWUXRGIIBAS-UHFFFAOYSA-N n-[2-(4-hydroxyanilino)pyridin-3-yl]-4-methoxybenzenesulfonamide;hydrochloride Chemical compound Cl.C1=CC(OC)=CC=C1S(=O)(=O)NC1=CC=CN=C1NC1=CC=C(O)C=C1 KWQWWUXRGIIBAS-UHFFFAOYSA-N 0.000 description 1
- SFMJNHNUOVADRW-UHFFFAOYSA-N n-[5-[9-[4-(methanesulfonamido)phenyl]-2-oxobenzo[h][1,6]naphthyridin-1-yl]-2-methylphenyl]prop-2-enamide Chemical compound C1=C(NC(=O)C=C)C(C)=CC=C1N1C(=O)C=CC2=C1C1=CC(C=3C=CC(NS(C)(=O)=O)=CC=3)=CC=C1N=C2 SFMJNHNUOVADRW-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229950004847 navitoclax Drugs 0.000 description 1
- JLYAXFNOILIKPP-KXQOOQHDSA-N navitoclax Chemical compound C([C@@H](NC1=CC=C(C=C1S(=O)(=O)C(F)(F)F)S(=O)(=O)NC(=O)C1=CC=C(C=C1)N1CCN(CC1)CC1=C(CCC(C1)(C)C)C=1C=CC(Cl)=CC=1)CSC=1C=CC=CC=1)CN1CCOCC1 JLYAXFNOILIKPP-KXQOOQHDSA-N 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 102000037979 non-receptor tyrosine kinases Human genes 0.000 description 1
- 108091008046 non-receptor tyrosine kinases Proteins 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- JFNLZVQOOSMTJK-KNVOCYPGSA-N norbornene Chemical compound C1[C@@H]2CC[C@H]1C=C2 JFNLZVQOOSMTJK-KNVOCYPGSA-N 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 229950006584 obatoclax Drugs 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 208000003388 osteoid osteoma Diseases 0.000 description 1
- 208000008798 osteoma Diseases 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 150000002940 palladium Chemical class 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 208000021255 pancreatic insulinoma Diseases 0.000 description 1
- 229960000639 pazopanib Drugs 0.000 description 1
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- OKBMCNHOEMXPTM-UHFFFAOYSA-M potassium peroxymonosulfate Chemical compound [K+].OOS([O-])(=O)=O OKBMCNHOEMXPTM-UHFFFAOYSA-M 0.000 description 1
- 238000000634 powder X-ray diffraction Methods 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 201000001514 prostate carcinoma Diseases 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 210000001147 pulmonary artery Anatomy 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 201000010174 renal carcinoma Diseases 0.000 description 1
- 230000001718 repressive effect Effects 0.000 description 1
- 208000029922 reticulum cell sarcoma Diseases 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 208000013860 rhabdoid tumor of the kidney Diseases 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000011935 selective methylation Methods 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 210000003625 skull Anatomy 0.000 description 1
- 238000010583 slow cooling Methods 0.000 description 1
- 208000000649 small cell carcinoma Diseases 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- WGRULTCAYDOGQK-UHFFFAOYSA-M sodium;sodium;hydroxide Chemical compound [OH-].[Na].[Na+] WGRULTCAYDOGQK-UHFFFAOYSA-M 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 201000011099 spinal cord sarcoma Diseases 0.000 description 1
- LBJQKYPPYSCCBH-UHFFFAOYSA-N spiro[3.3]heptane Chemical compound C1CCC21CCC2 LBJQKYPPYSCCBH-UHFFFAOYSA-N 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 201000000498 stomach carcinoma Diseases 0.000 description 1
- 125000005415 substituted alkoxy group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 208000001608 teratocarcinoma Diseases 0.000 description 1
- ROUYFJUVMYHXFJ-UHFFFAOYSA-N tert-butyl 4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)CC1 ROUYFJUVMYHXFJ-UHFFFAOYSA-N 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 201000006134 tongue cancer Diseases 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000759 toxicological effect Toxicity 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 150000004654 triazenes Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- CYTQBVOFDCPGCX-UHFFFAOYSA-N trimethyl phosphite Chemical compound COP(OC)OC CYTQBVOFDCPGCX-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 208000022271 tubular adenoma Diseases 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 208000012991 uterine carcinoma Diseases 0.000 description 1
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N valeric aldehyde Natural products CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 1
- YCOYDOIWSSHVCK-UHFFFAOYSA-N vatalanib Chemical compound C1=CC(Cl)=CC=C1NC(C1=CC=CC=C11)=NN=C1CC1=CC=NC=C1 YCOYDOIWSSHVCK-UHFFFAOYSA-N 0.000 description 1
- 229950000578 vatalanib Drugs 0.000 description 1
- 229950011257 veliparib Drugs 0.000 description 1
- JNAHVYVRKWKWKQ-CYBMUJFWSA-N veliparib Chemical compound N=1C2=CC=CC(C(N)=O)=C2NC=1[C@@]1(C)CCCN1 JNAHVYVRKWKWKQ-CYBMUJFWSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 208000013013 vulvar carcinoma Diseases 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/052—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4003—Esters thereof the acid moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4021—Esters of aromatic acids (P-C aromatic linkage)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65586—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system at least one of the hetero rings does not contain nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6581—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and nitrogen atoms with or without oxygen or sulfur atoms, as ring hetero atoms
- C07F9/6584—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and nitrogen atoms with or without oxygen or sulfur atoms, as ring hetero atoms having one phosphorus atom as ring hetero atom
Definitions
- the present invention relates to heterocyclic compounds (referred to herein as the compounds of formula 1 ), processes for their preparation, pharmaceutical compositions comprising the compounds of formula 1 , and their use as EZH2 (enhancer of zeste homolog 2) inhibitors and methods of using said compounds in the treatment of diseases mediated by EZH2.
- EZH2 enhanced of zeste homolog 2
- Histone methyl transferases are a family of enzymes, which control selective methylation at specific amino acid sites on histones. Covalent modification of histones such as methylation control changes of chromatin structure in eukaryotic cell DNA, which lead to heritable alteration in gene expression. These modifications are referred to as epigenetic modifications. Polycomb genes are an illustration of epigenetic effectors structured in multimeric repressive complexes. Aberrant expression and/or activity of enzymes responsible for histone modification result in disease states such as cancer. Therefore, epigenetic modifications can be reversed and consequently, treatment of diseases such as cancer may be effected through selective inhibition of the enzymes involved.
- Histone-lysine N-methyl transferase EZH2 (enhancer of zeste homolog 2) is the catalytic subunit of Polycomb repressive complex 2 (PRC2), which methylates Lys27 of a specific histone H3 (H3K27) and is essential for the self- renewal of cancer stem cells.
- PRC2 Polycomb repressive complex 2
- H3K27 histone H3
- EZH2 is capable of silencing several anti-metastatic genes, favoring cell-invasion and uncontrolled cell-growth. For instance, somatic mutations of tyrosine 641 of EZH2 are reported to be associated with follicular lymphoma and diffused B-cell lymphoma (Nature Genet., 2010, 42, 2, 181 -185).
- Increased levels of trimethylated H3K27 resulting due to an increased expression of EZH2 contribute to cancer aggressiveness, metastasis, shorter disease free survival and increased death rate in many tumor types, for instance, in melanoma, prostate cancer, breast cancer and cancer of the endometrium (Bachmann et al, Journal of Clinical Oncology, 2006, 24, 4, 268-73). This is further substantiated in Oncogene, 2012, 31 , 3827-44, wherein it is reported that EZH2 expression is deregulated in prostate cancer, breast cancer and endometrial cancer, B-cell non-Hodgkin's lymphoma and bladder carcinoma.
- Increased expression of EZH2 also induces pulmonary artery smooth muscle proliferation (PLoS ONE, 2012, 7, 5, e37712). Increased expression of EZH2 has been further reported to have implication in myelofibrosis (Expert Review of Hematology, 2012, 5, 3, 313-324), HIV (PCT publication WO2012051492 A2), graft versus host diseases (GVHD) (Blood, 2012, 1 19, 5, 1274-1282), Weaver Syndrome (American Journal of Human Genetics, 2012, 90, 1 , 1 10-1 18), psoriasis vulgaris (European Journal of Dermatology, 201 1 , 21 , 4, 552-557) and liver fibrogenesis (PCT publication WO2010090723A2).
- PCT patent publication WO201 1 140324A1 discloses indoles as inhibitors of EZH2 and use of said compounds for the treatment of cancer.
- PCT patent publication WO20121 18812A2 discloses bicyclic heterocyclic compounds as EZH2 inhibitors and their use in the treatment of cancer.
- EZH2 activity would effectively decrease cellular proliferation and invasion and thereby provide therapeutic benefit in the treatment of diseases or disorders mediated by EZH2.
- the compounds of the current invention function as inhibitors of EZH2 and therefore, provide a therapeutic solution for the treatment of disorders mediated by EZH2, such as cancers.
- the present invention relates to a compound of formula 1 (as described herein), or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a polymorph, a prodrug, S-oxide or N- oxide thereof.
- the present invention relates to processes for the preparation of the compound of formula 1 or a pharmaceutically acceptable salt thereof.
- the present invention relates to pharmaceutical composition
- pharmaceutical composition comprising the compound of formula 1 or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof; and at least one pharmaceutically acceptable carrier or excipient.
- the present invention relates to a compound of formula 1 or a pharmaceutically acceptable salt thereof; for use as an EZH2 (enhancer of zeste homolog 2) inhibitor.
- the present invention relates to a compound of formula 1 or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate thereof; for use in the treatment of a disease or a disorder mediated by EZH2.
- the present invention relates to a method for the treatment of a disease or disorder mediated by EZH2; comprising administering to a subject in need thereof, a therapeutically effective amount of the compound of formula 1 or a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof.
- the present invention relates to a use of a compound of formula 1 or a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof; in combination with at least one further therapeutically active agent; for the treatment of a disease or disorder mediated by EZH2.
- the present invention relates to a use of a compound of formula 1 or a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof; for the manufacture of a medicament for the treatment of a disease or disorder mediated by EZH2.
- the present invention relates to a compound of formula 1 ,
- Formula 1 or an isotopic form, a stereoisomer ⁇ tautomer,a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof;
- Ri and R 2 are independently selected from hydrogen, halogen, hydroxy, cyano, (Ci-Ce)-alkyl, halo(Ci-C 8 )-alkyl,(C 2 -C 8 )-alkenyl, (Ci-C 8 )-alkoxy, C(0)R a ,C(0)OR a , C(0)NR a R b , OC(0)NR a R b , NR a R b , NR a C(0)R b ,S(0) q (C C 8 )-alkyl and S(0) q NR a R b ;or
- Ri and R 2 together can form a 3 to 10 membered monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N,S and P; wherein the ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(CrC 8 )-alkyl, (CrC 8 )-alkoxy, halo(CrC 8 )-alkoxy, (C 3 -Ci 2 )-cycloalkyl-(R c ) 0 -2, (C 6 -Ci 4 )-aryl-(R c ) 0 - 2 ,C(O)R a , C(0)OR a , C(0)NR a R b ,NR a R b , NR a C(0)R b , S(0) q (C C 8
- X and X 2 are independently selected from CR 6 and N;
- R 3 , R 4 and R 6 are independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (CrC 8 )-alkyl, halo(CrC 8 )-alkyl, halo(CrC 8 )-alkyl-OH, (Ci-C 8 )-alkoxy, halo(C C 8 )-alkoxy, (C 2 -C 8 )-alkenyl, (C 3 -Ci 2 )-cycloalkyl, (C 5 -C 8 )- cycloalkenyl, (C 6 -Ci 4 )-aryl, (C 6 -Ci 4 )ar-(CrC 8 )-alkyl, heterocyclyl, heteroaryl, C(0)R a , C(0)OR a , C(0)NR a R b , S(0) q (C C 8 )-alkyl, S(0) q NR a R b , NR
- R 4a is hydrogen, hydroxy, cyano, (CrC 8 )-alkyl, halo(CrC 8 )-alkyl, (CrC 8 )-alkoxy, halo(CrC 8 )-alkoxy, (C 2 -C 8 )-alkenyl, (C 3 -Ci 2 )-cycloalkyl,(C 5 -C 8 )-cycloalkenyl,(C 6 - Ci 4 )-aryl, (C 6 -Ci 4 )ar-(Ci-C 8 )-alkyl, heterocyclyl, heteroaryl, C(0)R a , C(0)NR a R b , S(0) q (Ci-C 8 )-alkyl or S(0) q NR a R b ; or
- R 3 and R 4 or R 3 and R 4a on A can join together to form a 3 to 10 membered saturated orunsaturated monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; wherein said ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (C C 8 )-alkyl, halo(C C 8 )-alkyl, (C C 8 )-alkoxy, halo(C C 8 )-alkoxy, (C 3 - Ci2)-cycloalkyl-(R c )o-2, 0-(C 3 -Ci 2 )-cycloalkyl-(R c )o-2, (C 6 -Ci 4 )-aryl-(R c ) 0 -2, 0-(C 6 - Ci 4 )-aryl-(R c
- R 5 is hydrogen, halogen, hydroxy, cyano, B(OH) 2 , P(0)(OR a )(OR b ), P(0)R a R b , (Ci-C 8 )-alkyl, halo(C C 8 )-alkyl, halo(C C 8 )-alkyl-OH, (C C 8 )-alkoxy, halo(C C 8 )- alkoxy, (C2-C 8 )-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C 8 )-cycloalkenyl, (C6-Ci 4 )-aryl, (C 6 -Ci 4 )ar-(CrC 8 )-alkyl, (C 6 -Ci 4 )-aryl-P(0)R a R b , (C 6 -Ci 4 )-aryl-P(0)(OR a )(OR b ), (C 6 -Ci
- L is --C(0)NR 7 --, --NR 7 C(0)--, -NR 7 C(0)NR 7 -, --NR 7 S(0) r --, --S(0) r NR 7 -, - CH 2 NR 7 --, -NR 7 CH 2 -, --CH(halo)NR 7 -, --NR 7 CH(halo)--, --C(halo) 2 NR 7 -, -- NR 7 C(halo) 2 -, --CH(halo-(C C 8 )-alkyl)NR 7 --, --NR 7 CH(halo-(C C 8 )-alkyl)-, - , -NR 7 C(halo-(Ci-C 8 )-alkyl) 2 - wherein the dotted line (--) indicates the point of attachment of L to the group p and ' " respectively; or
- L is a saturated or unsaturated 5 or 6 membered monocyclic ringoptionally containing one to three heteroatoms selected from the group consisting of O, N, S and P;
- R 7 is hydrogen, cyano, (CrC 8 )-alkyl, (C2-C 8 )-alkenyl, halo(Ci-C 8 )alkyl,C(0)R a , C(0)OR a ,C(0)NR a R b , S(0) q (CrC 8 )-alkyl or S(0) q NR a R b ;
- Q is:
- B is a 3 to 10 membered monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; wherein said ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano,(Ci -C e )-alkyl, halo(Ci-C 8 )-alkyl, (Ci-C 8 )-alkoxy, halo(Ci-C 8 )-alkoxy, (C 3 - Ci 2 )-cycloalkyl-(R c )o-2, (C 6 -Ci 4 )-aryl-(R c ) 0 -2, heterocyclyl-(R c ) 0 -2, heteroaryl-(R c ) 0 - 2 , C(0)R a , C(0)OR a , C(0)NR a R b ,NR
- R 8 and R 9 are independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (CrC 8 )-alkyl, halo(CrC 8 )alkyl, (CrC 8 )-alkoxy, halo(C C 8 )-alkoxy, (C2-C 8 )-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C 8 )-cycloalkenyl, (C6-Ci 4 )-aryl, (C 6 -Ci 4 )ar-(Ci -Ce)-alkyl, C(0)R a , C(0)OR a ,C(0)NR a R b , S(0) q (C C 8 )-alkyl, S(0) q NR a R b , NR a R b , NR a C(0)R b , NR a C(0)NR a R b and NR a C(0)OR b
- R a and R b are independently selected from the group consisting of hydrogen, (C C 8 )-alkyl, (C 2 -C 8 )-alkenyl, (C 3 -Ci 2 )-cycloalkyl, (C 5 -C 8 )-cycloalkenyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl; or R a and R b together with the nitrogen atom can form a 3 to 10 membered ring, optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P;
- R c at each occurrence is independently selected from the group consisting ofhydrogen, halogen, hydroxy, cyano, (Ci-C8)-alkyl, halo(Ci-C8)alkyl, (C3-C12)- cycloalkyl, (C 6 -Ci 4 )-aryl, (C 6 -Ci 4 )-ar-(Ci-C 8 )-alkyl, heteroaryl, heterocyclyl, COR a , C(0)OR a , C(0)NR a R b , C(0)NR a NR a R b , S(0) q (C C 8 )-alkyl, S(0) q NR a R b , NR a R b , NR a C(0)R b , NR a C(0)NR a R b , NR a C(0)OR b and NR a S(0) q NR a R b ;
- p and n are integers independently selected from 0, 1 , 2 and 3;
- q 1 or 2;
- r is 1 or 2;
- each of the (CrC 8 )-alkyl or (CrC 8 )-alkoxy can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkoxy, halo(CrC 8 )-alkoxy, (C 3 -Ci 2 )-cycloalkyl-(R c )o-2, O-(C 3 -Ci 2 )-cycloalkyl-(R c ) 0 -2, (C 6 -Ci 4 )-aryl-(R c ) 0 - 2 , O- (C 6 -Ci 4 )-aryl-(R c )o-2, heterocyclyl-(R c ) 0 -2, O-heterocyclyl-(R c ) 0 -2, heteroaryl-(R c ) 0 - 2 —
- each of the (C2-C 8 )-alkenyl, (C 3 -Ci2)-cycloalkyl, (C 3 -C 8 )-cycloalkenyl and (C 6 -Ci 4 )- aryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-CeJ-alkyl, halo(C C 8 )-alkyl, (C C 8 )-alkoxy, halo(C C 8 )-alkoxy, (C 3 -Ci 2 )-cycloalkyl-(R c ) 0 -2, 0-(C 3 -Ci2)-cycloalkyl-(R c )o-2, (C 6 -Ci 4 )-aryl-(R c ) 0 -2, O-(C 6 -Ci 4 )-aryl-(R c )
- the heterocyclyl is a 3 to 10 membered saturated or partially unsaturated, monocyclic or bicyclic ring system containing one to four heteroatoms independently selected from the group consisting of O, N, S and P; wherein the heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (C C 8 )-alkyl, halo(C C 8 )-alkyl, (C C 8 )-alkoxy, halo(C C 8 )-alkoxy, (C 3 - Ci 2 )-cycloalkyl-(R c )o-2, O-(C 3 -Ci 2 )-cycloalkyl-(R c ) 0 -2, (C 6 -Ci 4 )-aryl-(R c ) 0 -2, 0-(C 6 - Ci 4 )-aryl-(R c )o
- the heteroaryl is a 5 to 10 membered monocyclic or bicyclic aromatic ring system containing one to four heteroatoms independently selected from the group consisting of O, N, S and P, wherein the heteroaryl can be unsubstituted or substituted with one or more groups independently selected fromthe group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(CrC 8 )-alkyl, (C C 8 )-alkoxy, halo(CrC 8 )-alkoxy, (C 3 -Ci2)-cycloalkyl-(R c ) 0 -2, 0-(C 3 -Ci2)-cycloalkyl- (R c )o-2, (C 6 -Ci 4 )-aryl-(R c ) 0 -2, O-(C 6 -Ci 4 )-aryl-(R c ) 0 -2, heterocycly
- substitution means that one or more hydrogens of the specified moiety are replaced with a suitable substituent and includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and results in a stable compound.
- alkyl or "(CrC 8 )-alkyl” whether used alone or as part of a substituent group, refers to the radical of saturated aliphatic groups, including straight or branched-chain alkyl groups. If the number of carbon atoms is not specified, “alkyl” refers to alkyl group having 1 to 8 (both inclusive) carbon atoms. Accordingly, a straight-chain or branched chain alkyl has eight or fewer carbon atoms in its backbone, for instance, Ci-C 8 for straight- chain and C3-C8 for branched chain.
- alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, iso-butyl, sec-butyl, n- pentyl, isopentyl, 2-pentyl, 3-pentyl, neo-pentyl, n-hexyl, isohexyl, 2-hexyl, 3-hexyl and the like.
- alkyl groups can be unsubstituted or substituted with one or more substituents.
- a substituted alkyl refers to a (CrC 8 )-alkyl substituted preferably with 1 -7 groups, more preferably 1 - 3 groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkoxy, halo(CrC 8 )-alkoxy, (C 3 -Ci2)-cycloalkyl-(R c ) 0 -2, 0-(C 3 - Ci 2 )-cycloalkyl-(R c )o-2, (C 6 -Ci 4 )-aryl-(R c ) 0 -2, O-(C 6 -Ci 4 )-aryl-(R c ) 0 -2, heterocyclyl- (R c )o-2, 0-heterocyclyl-(R c )o-2,
- R c at each occurrence is independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (CrC 8 )-alkyl, halo(CrC 8 )alkyl, (C3-Ci2)-cycloalkyl, (C6-Ci 4 )-aryl, (C6-Ci 4 )-ar-(Ci-C 8 )-alkyl, heteroaryl, heterocyclyl, C(0)R a , C(0)OR a , C(0)NR a R b , CONR a NR a R b , S(0) q (C C 8 )-alkyl, S(0) q NR a R b , NR a R b , NR a C(0)R b , NR a C(0)NR a R b , NR a C(0)OR b and NR a S(0) q NR a R b ; q is 1 or 2; R a and
- substituted alkyls include, but are not limited to, trifluoromethyl, hydroxymethyl, hydroxyethyl, 1 -aminoethyl, benzyl, N- morpholino methyl, N-indolomethyl, and N-piperidinylmethyl.
- halogen refers to a fluorine, chlorine, bromine, or iodine atom.
- halo(CrC 8 )alkyl When the alkyl group is substituted with one or more halogens, it is specifically referred to as "halo(CrC 8 )alkyl" or "haloalkyl".
- a monohalo(Ci-C 8 )alkyl radical for example, can have a chlorine, bromine, iodine or fluorine atom.
- Dihalo and polyhalo(Ci-C8)alkyl radicals may have two or more of the same or different halogen atoms.
- halo(CrC 8 )-alkyl include, but are not limited to, chloromethyl, dichloromethyl, trichloromethyl, dichloroethyl, dichloropropyl, fluoromethyl, difluoromethyl, trifluoromethyl, pentafluoroethyl, heptafluoropropyl, difluorochloromethyl, dichlorofluoromethyl, difluoroethyl, difluoropropyl or the like groups.
- the haloalkyl group can be unsubstituted or substituted with one or more groupsas described in the definition of 'substituted alkyl' herein above.
- alkenyl or "(C 2 -C 8 )-alkenyl", as used herein; alone or as part of a substituent group, refers to a straight or branched chain hydrocarbon radical containing the indicated number of carbon atoms and at least one carbon-carbon double bond (two adjacent sp 2 carbon atoms).
- (C2-C8)-alkenyl refers to an alkenyl group having 2 to 8 (both inclusive) carbon atoms.
- the geometry of the double bond may be
- E Delta
- Z rocking
- Representative examples of alkenyl include, but are not limited to, vinyl, allyl and 2-propenyl.
- the alkenyl group can be unsubstituted or substituted with 1 -7 groups, preferably 1 -3 groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(Ci-C 8 )- alkyl, (C C 8 )-alkoxy, halo(C C 8 )-alkoxy, (C 3 -Ci 2 )-cycloalkyl-(R c ) 0 -2, 0-(C 3 -Ci 2 )- cycloalkyl-(R c )o-2,(C 6 -Ci 4 )-aryl-(R c ) 0 -2, O-(C 6 -Ci 4 )-aryl-(R c ) 0 - 2 , heterocyclyl-(R c ) 0 - 2 , heterocyclyl-(R c ) 0 - 2 ,
- alkoxy or "(CrC 8 )-alkoxy” refers to a (CrC 8 )- alkyl having an oxygen radical attached thereto.
- Representative examples of alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, isobutoxy and t-butoxy.
- An alkoxy group may be unsubstituted or substituted with one or more substituents.
- a substituted alkoxy refers to an (CrC 8 )-alkoxy group in which the alkyl is substituted with one or more groups as explained in the definition of 'substituted alkyl' herein above.
- Representative examples of substituted (Ci-C 8 )- alkoxy include, but are not limited to, chloromethoxy, 2-cyanoethoxy, trifluoromethoxy and benzyloxy group.
- a benzyloxy group refers to a benzyl having an oxygen radical attached thereto.
- cycloalkyi or "(C 3 -Ci2)-cycloalkyl” whether used alone or as part of a substituent group, refers to a saturated or partially unsaturated cyclic hydrocarbon radical including 1 , 2 or 3 rings and including a total of 3 to 12 carbon atoms forming the rings.
- the term cycloalkyi includes bridged, fused and spiro ring systems.
- (C 3 -Ci2)-cycloalkyl refers to a cycloalkyi group having 3 to 12 (both inclusive) carbon atoms.
- cycloalkyi include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, adamantyl, norbornyl, bicyclo[2.1 .0]pentane, bicyclo[2.2.1 ]heptyl, bicyclo[2.2.1 ]hept-2-ene, spiro[3.3]heptane and 1 ,2,3,3a- tetrahydropentalene.
- cycloalkenyl or "(C 5 -C 8 )-cycloalkenyl” refers to a non-aromatic monocyclic carboxycyclic ring having the specified number of carbon atoms and up to 3 carbon-carbon double bonds.
- Representative examples of cycloalkenyl include, but are not limited to, cyclopentenyl and cyclohexenyl.
- the "cycloalkyi” and "(C 5 -C 8 )-cycloalkenyl” can be unsubstituted or substituted with 1 -7, preferably 1 -3groups independently selected from halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(CrC 8 )-alkyl, (C C 8 )-alkoxy, halo(C C 8 )-alkoxy, (C 3 -C 12 )-cycloalkyl-(R c )o-2, 0-(C 3 -C 12 )-cycloalkyl- (R c )o-2, (C 6 -Ci 4 )-aryl-(R c ) 0 -2, 0-(C 6 -Ci 4 )-aryl-(R c ) 0 -2,heterocyclyl-(R c )o-2,0- heterocyclyl-(R c )o
- aryl or "(C 6 -Ci 4 )-aryl” as used herein refers to monocyclic or polycyclic hydrocarbon groups having 6 to 14 ring carbon atoms in which the carbocydic ring(s) present have a conjugated pi electron system.
- Representative examples of (C 6 -Ci 4 )-aryl residues include, but are not limited to, phenyl, naphthyl, fluorenyl or anthracenyl.
- Aryl groups can be unsubstituted or substituted with one or more, for example 1 -5 substituents independently selected from halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(CrC 8 )-alkyl, (CrC 8 )-alkoxy, halo(CrC 8 )- alkoxy, (C 3 -Ci 2 )-cycloalkyl-(R c )o-2, 0-(C 3 -Ci 2 )-cycloalkyl-(R c )o-2, (C 6 -Ci 4 )-aryl-(R c ) 0 - 2, O-(C 6 -Ci 4 )-aryl-(R c ) 0 -2, heterocyclyl-(R c ) 0 -2, O-heterocyclyl-(R c ) 0 -2, heteroaryl- (R c )o-2
- the substituent can be located in the 2-position, the 3-position or the 4-position. If the phenyl carries two substituents, they can be located in 2, 3-position, 2, 4-position, 2, 5-position, 2, 6- position, 3, 4-position or 3, 5-position.
- Representative examples of monosubstituted phenyl groups include, but are not limited to, 3- trifluoromethylphenyl, 4-chlorophenyl and 4-cyanophenyl.
- disubstituted phenyl groups include, but are not limited to, 3, 5-difluorophenyl and 3, 4-dimethoxyphenyl.
- aryloxy or "0-(C6-Ci 4 )-aryl” refers to an "(Ce- Ci 4 )-aryl” group having an oxygen radical attached thereto.
- the aryl of aryloxy group may be unsubstituted or substituted as indicated in the definition of (C 6 - Ci 4 )-aryl herein above.
- Representative examples of aryloxy groups include, but not limited to, phenoxy, 4-chlorophenoxy and 3, 4-dimethoxyphenoxy.
- aralkyl or "(C 6 -Ci 4 )ar-(Ci -C 8 )-alkyl” refers to an alkyl group substituted with an (C 6 -Ci 4 )-aryl group, wherein the terms alkyl and aryl are as defined above.
- Representative examples of aralkyl groups include (CH 2 ) p -phenyl, wherein p is an integer selected from 1 to 6, such as benzyl wherein p is 1 .
- the aryl of the (C 6 -C 14 )-aralkyl group can be unsubstituted or substituted as indicated in the definition of aryl herein above.
- heteroatom as used herein, includes nitrogen (N), oxygen (O), sulfur (S) and phosphorus (P). Any heteroatom with unsatisfied valency is assumed to have a hydrogen atom to satisfy the valency.
- heterocyclyl or “heterocyclic” whether used alone or as part of a substituent group, refers to a saturated, partially unsaturated, monocyclic, polycyclic, bridged cyclic or spirocyclic ring system containing 3 to 10 carbon atoms and 1 to 4 identical or different heteroatoms selected from oxygen, nitrogen, sulfurand phosphorus.
- heterocyclyls include, but are not limited to, oxetanyl, pyrrolyl, pyrrolidinyl, pyrazolyl, imidazolyl, pyrazinyl, piperazinyl, oxazolyl, oxadiazolyl, isoxazolyl, triazolyl, thiazolyl, tetrazolyl, furyl, thienyl, purinyl, pyridinyl, pyridazinyl, pyrimidinyl, piperidyl, benzoxazolyl, benzothiazolyl, benzofuranyl, purinyl, benzimidazolyl, benzoxazolyl, indolyl, indazolyl, isoindolyl, isothiazolyl, isoquinolyl, isoquinolyl, morpholinyl, thiomorpholinyl, thiomorpholinyl-1 , 1 -dioxid
- heteroaryl refers to a 5- to 10-membered aromatic monocyclic or bicyclic ring system containing one to four heteroatoms independently selected from: nitrogen, sulphur, oxygen and phosporus.
- heteroaryls include, but are not limited to, pyrrole, pyrazole, imidazole, pyrazine, furan, thiophene, oxazole, thiazole, benzimidazole, benzoxazole, benzothiazole, benzofuran, indole, indazole, isoindole, isoquinoline, isooxazole, triazine, purine, pyridine, quinoline, oxadiazole, thiene, pyridazine, pyrimidine, isothiazole, quinoxaline (benzopyrine) and tetrazole.
- the nitrogen or sulfur atom of the heterocyclyl can be optionally oxidized to the corresponding N- oxide, S-oxide or S, S-dioxide.
- a heterocyclyl or heteroaryl group can be unsubstituted or substituted with one or more groups independently selected from halogen, hydroxy, oxo, cyano, (CrCe)-alkyl, halo(C C 8 )-alkyl, (C C 8 )-alkoxy, halo(C C 8 )-alkoxy, (C3-C12)- cycloalkyl-(R c )o-2, O-(C 3 -Ci 2 )-cycloalkyl-(R c ) 0 -2, (C 6 -Ci 4 )-aryl-(R c ) 0 - 2 , 0-(C 6 -Ci 4 )- aryl-(R c )o-2, heterocyclyl-(R c ) 0 -2, O-heterocyclyl-(R c ) 0 -2, heteroaryl-(R c ) 0 - 2
- isotopic forms or “isotopically labeled forms” is a general term used for isotopic forms of the compounds of formula 1 , wherein one or more atoms of the compounds of formula 1 are replaced by their respective isotopes. All isotopes of any particular atom or element as specified are contemplated within the scope of the compounds of the present invention.
- isotopes that may be incorporated into the compounds disclosed herein include, but are not limited to, isotopes of hydrogen such as 2 H (deuterium or D) and 3 H (tritium or T), carbon such as 1 1 C, 13 C and 14 C, nitrogen such as 13 N and 15 N, oxygen such as 15 0, 17 0 and 18 0, chlorine such as 36 CI, fluorine such as 18 F and sulphur such as 35 S.
- isotopes of hydrogen such as 2 H (deuterium or D) and 3 H (tritium or T)
- carbon such as 1 1 C, 13 C and 14 C
- nitrogen such as 13 N and 15 N
- oxygen such as 15 0, 17 0 and 18 0, chlorine such as 36 CI
- fluorine such as 18 F
- sulphur such as 35 S.
- Substitution with heavier isotopes, for example, replacing one or more key carbon-hydrogen bonds with carbon-deuterium bond may show certain therapeutic advantages, resulting from longer metabolism cycles (e.g., increased in
- isotopic forms of the compounds of formula 1 can include, without limitation, deuterated compounds of formula 1 .
- deuterated as used herein, by itself or used to modify a compound or group, refers to replacement of one or more hydrogen atom(s), which is attached to carbon(s), with a deuterium atom.
- the compounds of formula 1 can include in the definitions of one or more of its various variables, wherever applicable, deuterium, deuterated-alkyl, deuterated-alkoxy, deuterated-cycloalkyl, deuterated-heterocyclyl, deuterated-aryl, deuterated-heteroaryl and the like.
- deuterated-alkyl refers to an (CrC 8 )-alkyl group as defined herein, wherein at least one hydrogen atom bound to carbon is replaced by a deuterium. That is, in a deuterated alkyl group, at least one carbon atom is bound to a deuterium. In a deuterated alkyl group, it is possible for a carbon atom to be bound to more than one deuterium; it is also possible that more than one carbon atom in the alkyl group is bound to a deuterium.
- deuterated and the terms deuterated-heterocyclyl, deuterated-heteroaryl, deuterated-cycloalkyl, deuterated-aryl, deuterated-alkoxy each refer to the corresponding chemical moiety wherein at least one carbon is bound to a deuterium.
- solvate refers to an aggregate of a molecule (in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof) with one or more solvent molecules.
- solvents for the purpose of the invention may not interfere with the biological activity of the molecule.
- the solvent used is a pharmaceutically acceptable solvent.
- suitable pharmaceutically acceptable solvents include, without limitation, water, ethanol and acetic acid.
- the solvent used is water and the solvates obtained are referred to as hydrates.
- suitable solvates are the mono- or di-hydrates or alcoholates of the compounds of the present invention.
- stereoisomer'Or “stereoisomeric form” is a general term used for all isomers of individual compounds (in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof) that differ only in the orientation of their atoms in space.
- stereoisomer includes mirror image isomers (enantiomers), mixtures of mirror image isomers (racemates, racemic mixtures), geometric (cis/trans or E/Z) isomers, and isomers of compounds with more than one chiral center that are not mirror images of one another (diastereoisomers).
- tautomer refers to the coexistence of two (or more) compounds that differ from each other only in the position of one (or more) mobile atoms and in electron distribution, for example, keto-enol tautomers.
- the term "pharmaceutically acceptable” means that the carrier, diluent, excipients and/or salt must be compatible with the other ingredients of the formulation (composition), and not deleterious to the recipient thereof.
- pharmaceutically acceptable salts or “pharmaceutically acceptable salfas used herein includes salts of the active compoundi.e.the compound of formula 1 , which retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects; and are prepared with suitable acids or bases, depending on the particular substituents found on the compounds described herein.
- polymorph or “pharmaceutically acceptable polymorph” or “polymorphic form” refers to crystals of the same compound (in the present invention, a compound of formula 1 )that differs only in the arrangement and/or conformation of the molecule in the crystal lattice.
- a prodrug or “prodrugs” refers to a compound, whichisa derivative of a parent compound(in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof), which following administration, releasesthe parent compoundin vivo via a chemical or physiological process, e.g., a prodrug on being brought to the physiological pHor through enzyme action is converted to the parent compound.
- N-oxide refers to an oxide of the nitrogen atom of a nitrogen-containing heteroaryl or heterocycle. N-oxide can be formed in the presence of an oxidizing agent such as m-chloro-perbenzoic acid or hydrogen peroxide. N-oxide refers to an amine oxide, also known as amine-N-oxide, and is a chemical compound that contains N-»0 bond.
- S-oxide refers to the oxide of the sulfur atom (S-oxide) or dioxide of the sulfur atom (S, S- dioxide) of a sulfur-containing heteroaryl or heterocycle.
- S-oxide and S, S-dioxides can be formed in the presence of an oxidizing agent such as m-chloro-perbenzoic acid or oxone (potassium peroxymonosulfate).
- the terms “compound of formula 1 ", “compounds of formula 1 ", and “compounds of the present invention” includeall the isotopic forms, stereoisomeric and tautomeric forms and mixtures thereof in all ratios, and the pharmaceutically acceptable salts, pharmaceutically acceptable solvates, pharmaceutically acceptable prodrugs, pharmaceutically acceptable polymorphs, N-oxides and S-oxides thereof.
- the compound(s) of the present invention can also be referred to herein as "the active compound” or "the active ingredient”.
- the term "pharmaceutically acceptable carrier” refers to a material that is non-toxic, inert, solid, semi-solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type which is compatible with a subject, preferably a mammal, more preferably a human, and is suitable for delivering an active agent (in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof or any other form recited herein),to the target site without adversely affecting activity of the agent.
- an active agent in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof or any other form recited herein
- subject refers to an animal, preferably a mammal, and most preferably a human.
- mammal refers to warm-blooded vertebrate animals of the class Mammalia, including humans, characterized by a covering of hair on the skin and, in the female, milk-producing mammary glands for nourishing the young.
- mammal includes animals such as cat, dog, rabbit, bear, fox, wolf, monkey, deer, mouse, pig as well as human.
- subject may be used interchangeably with the term patient.ln the context of the present invention the phrase "a subject in need thereof” means a subject in need of the treatment for the disease or disorder that is mediated by EZH2. Alternatively, the phrase "a subject in need thereof” means a subject (patient) diagnosed having a disease or disorder that is mediated by EZH2.
- therapeutically effective amount means an amount of the compound of formula 1 or a pharmaceutically acceptable salt thereof or a composition comprising a compound of formula 1 , effective in producing the desired therapeutic response in a particular patient (subject) suffering from a disease or disorder to be treated.
- therapeutically effective amount includes the amount of the compound of the present invention, when administered, that induces a positive modification in the condition (a disease or a disorder) to be treatedor is sufficient to prevent development of, or alleviate to some extent, one or more of the symptoms of the disease or disorder being treated in a patient (subject).
- the amount of the compound used for the treatment of a subject is low enough to avoid undue or severe side effects, within the scope of sound medical judgment.
- the therapeutically effective amount of the compound or composition will vary with the particular condition being treated, the age and physical condition of the patient (subject in need of the treatment), the severity of the condition being treated/prevented, the duration of the treatment, the nature of concurrent therapy, the specific compound or composition employed and the particular pharmaceutically acceptable carrier utilized.
- treatment is intended to mean to alleviate, slow the progression, attenuation or cure of existing disease or condition (e.g., cancer). Treatment also includes treating the symptoms of the disease or condition.
- Prevent or “Prevention”, as used herein, refers to delaying, slowing, inhibiting, reducing or ameliorating the onset of the disease or disorder e.g. cancer.
- EZH2 inhibitor refers to an agent (in the present invention, a compound of formula 1 ) which is capable of inhibiting the increased expression of Histone-lysine N-methyl transferase EZH2 (enhancer of zeste homolog 2), which is a catalytic subunit of polycomb repressive complex 2 (PRC2), responsible for methylation of Lys27 of a specific histone H3 (H3K27) and essential for the self-renewal of cancer stem cells.
- Histone-lysine N-methyl transferase EZH2 encodehancer of zeste homolog 2
- PRC2 polycomb repressive complex 2
- disease or disorder mediated by EZH2 refers to an abnormal condition in a subject due to aberrant expression of the enzyme 'Histone-lysine N- methyltransferase EZH2' leading to abnormal epigenetic modifications.
- the diseases or disorders mediated by EZH2 are selected from cancer, pulmonary arterial hypertension, myelofibrosis, human immunodeficiency virus (HIV) disease, graft versus host diseases, Weaver Syndrome, psoriasis vulgaris or liver fibrogenesis.
- HIV human immunodeficiency virus
- the cancers mediated by EZH2 include, but are not limited to, thyroid carcinoma, cardiac sarcoma, lung carcinoma, gastrointestinal carcinoma, genitourinary tract carcinoma, liver carcinoma, mantle cell lymphoma, bone sarcoma, sarcoma of the nervous system, gynaecological carcinoma, haematological cancer, adrenal gland neuroblastoma, skin cancer, astrocytic cancer, breast cancer, colorectal cancer, endometrial cancer, head and neck cancer and oral cancer.
- the present invention relates to a compound of formula
- Xi and X 2 are independently selected from CR 6 and N;
- R 3 , R 4 and R 5 are as defined in the first aspect
- the present invention relates to a compound of formula 1 , wherein,
- Xi and X 2 are independently selected from CR 6 and N;
- R3 and R 4 join together to form a 5 to 7 membered saturated or unsaturated monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; and
- R 5 is as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula
- A is:
- Xi and X 2 are CR 6 ;
- R 3 , R 4 , R 5 and R 6 are as defined in the first aspect; or
- the present invention relates to a compound of formula 1 , wherein,
- A is:
- Xi and X 2 are independently selected from CR 6 and N;
- R 3 , R 4a , R 5 and Re are as defined in the first aspect
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X2 are independently selected from CR 6 and N;
- R 3 and R 4a join together to form a 5 to 7 membered saturated or unsaturated monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; and
- R 5 and R 6 are as defined in the first aspect
- the present invention provides a compound of formula 1 ,
- L is -C(0)NR7--,-NR 7 C(0)--,--NR 7 C(0)NR7--, -NR 7 S(0) r - or -S(0) r NR 7 -; wherein the dotted line (--) indicates the point of attachment of L to the group
- the present invention relates to a compound of formula
- L is --CH 2 NR 7 -,-NR 7 CH2--,-CH(halo-(Ci-C 8 )-alkyl)NR 7 -, -NR 7 CH(halo-(Ci-C 8 )- alkyl)-, -C(halo-(CrC 8 )-alkyl) 2 NR 7 -, -NR 7 C(halo-(CrC 8 )-alkyl) 2 -, - CH(halo)NR 7 -, -NR 7 CH(halo)--, -C(halo) 2 NR 7 - or -NR 7 C(halo) 2 --;wherein the dotted line (-) indicates the point of attachment of L to the group
- A, R 1 ; R 2 , R 7 , Q, n and p are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula
- L is ; line (--) indicates the point o f attachment of L to the group respectively; where A, Ri , R 2 , R 7 , Q, n and p are as defined in the first aspect;
- the present invention relates to a compound of formula 1 ,
- R 8 and B are as defined in the first aspect
- the present invention relates to a compound of formula 1 ,
- R 8 , R9 and B are as defined in the first aspect
- the present invention relates to a compound of formula 1 ,
- the present invention relates to a compound of formula
- Xi and X 2 are independently selected from CR 6 and N;
- R 5 is P(0)(OR a )(OR b ), P(0)R a R b , (C 6 -Ci 4 )-aryl-P(0)R a R b ,(C 6 -Ci 4 )-aryl- P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)R a R b , (C 6 -Ci 4 )-aryl-heterocyclyl-R c , (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -heterocyclyl, (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ), (C 6 -Ci 4
- the present invention relates to a compound of formula
- A is:
- Xi and X2 are independently selected from CR6 and N;
- R 5 is P(0)(OR a )(OR b ), P(0)R a R b , (C 6 -Ci 4 )-aryl-P(0)R a R b , (C 6 -Ci 4 )-aryl- P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)R a R b , (C 6 -Ci 4 )-aryl-heterocyclyl-R c , (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -heterocyclyl, (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ), (C 6 -Ci 4
- R a , Rb, Rc, R3, R 4a and R 6 are as defined in the first aspect; or
- the present invention relates to a compound of formula
- A is:
- Xi and X 2 are independently selected from CR6 and N;
- L is -C(0)NR 7 - or -NR 7 C( -; wherein the dotted line (-) indicates the point of attachment of L to the group respectively;
- Ri, R2, R3, R 4 , R5, R6, R7, Q, n and p are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula
- A is:
- Xi and X 2 are independently selected from CR 6 and N;
- L is -C(0)NR 7 - or -NR 7 C(0)-; wherein the dotted line (-) indicates the point of attachment of L to the group respectively;
- Ri , R2, R3, R 4a , R5, R6, R7, Q, n and p are as defined in the first aspect;
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X 2 are independently selected from CR 6 and N;
- R 4 is NR a R b or NR a C(0)R b ;
- R 5 is P(0)(OR a )(OR b ), P(0)R a Rb, (C 6 -Ci 4 )-aryl-P(0)R a Rb, (C 6 -d 4 )-aryl- P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)R a R b , (C 6 -Ci 4 )-aryl-heterocyclyl-R c , (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -heterocyclyl, (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl
- n 0;
- each of the (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(CrC 8 )- alkyl, (d-CeJ-alkoxy, (C3-Ci 2)-cycloalkyl, (C6-Ci 4 )-aryl, heterocyclyl and heteroaryl;and
- R a , Rb, Rc, Ri , R2, R3, R6, R7, Re, B, p and r are as defined in the first aspect;
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X 2 are independently selected from CR 6 and N ;
- R 4 is NR a R b or NR a C(0)R b ;
- R 5 is P(0)(OR a )(OR b ), P(0) R a R b , (C 6 -Ci 4 )-aryl-P(0) R a R b ,(C 6 -Ci 4 )-aryl- P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0) R a R b , (C 6 -Ci 4 )-aryl-heterocyclyl-R c , (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ) or (C 6 -Ci 4 )-aryl-C(R c ) 2 -heterocyclyl; L is ⁇ C(0)NR 7 ⁇ , -NR 7 C(0) ⁇ , -NR 7 C(0)NR 7 -, -NR 7 S(0) r - or
- n 0;
- each of the (C 6 -Ci 4 )-aryl and heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(CrC 8 )-alkyl, (C Cs)-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci 4 )-aryl, heterocyclyl and heteroaryl;and R a , Rb, Rc, Ri , R2, R3, R6, R7, Re, Rc, P, r and B are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula 1 ,
- A is:
- X ⁇ and X 2 are independently selected from CR 6 and N ;
- R 4 is NR a R b or NR a C(0)R b ;
- R a and R b are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C 8 )-cycloalkenyl,(C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl;
- R 5 is (C 6 -Ci 4 )-aryl, (C 6 -Ci 4 )ar-(Ci -C 8 )-alkyl, heterocyclyl or heteroaryl;
- L is ⁇ C(0)NR 7 -, -NR 7 C(0)-, -NR 7 C(0)NR 7 -, -NR 7 S(0) r - or ⁇ S(0) r NR 7 ⁇ ;wherein the dotted line (--) indicates the point of attachment of L to the group
- n 0;
- each of the (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(CrC 8 )- alkyl, (d-CeJ-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci 4 )-aryl, heterocyclyl and heteroaryl;
- R2, R3, R6, R7, Re, P and B are as defined in the first aspect
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X 2 are independently selected from CR 6 and N;
- R 4 is NR a R b or NR a C(0)R b ;
- R 5 is P(0)(OR a )(OR b ), P(0)R a R b , (C 6 -Ci 4 )-aryl-P(0)R a R b , (C 6 -Ci 4 )-aryl- P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)R a R b , (C 6 -Ci 4 )-aryl-heterocyclyl-R c , (C 6 -Ci 4 )-aryl-C(R c )2-P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(Rc) 2 -heterocyclyl, (C 6 -Ci 4 )- aryl, (C 6 -Ci 4 )ar-(Ci-C 8 )-alkyl, heterocyclyl or heteroaryl;
- L is -C(0)NR 7 -, -N RTC(0) ⁇ , ⁇ NR 7 C(0)N R 7 ⁇ , ⁇ NR 7 S(0) R ⁇ or ⁇ S(0) R NR 7 ⁇ ; wherein the dotted line (--) indicates the point of attachment of L to the group
- each of the (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-CeJ-alkyl, halo(Ci-Ce)- alkyl, (CrC 8 )-alkoxy, (C 3 -C 12 )-cycloalkyl, (C 6 -C 14 )-aryl, heterocyclyl and heteroaryl; and
- Ra, Rb, Rc, Ri, R2, R3, R6, R7, Re, R9, Rc, P, r and B are as defined in the first aspect; an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X 2 are independently selected from CR 6 and N;
- R 4 is NR a R b or NR a C(0)R b ;
- R 5 is P(0)(ORa)(OR b ), P(0)R a R b , (C 6 -Ci 4 )-aryl-P(0)R a R b , (C 6 -Ci 4 )-aryl- P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(R c )2-P(0)R a R b , (C 6 -Ci 4 )-aryl-heterocyclyl-R c , (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ) or (C 6 -Ci 4 )-aryl-C(Rc) 2 -heterocyclyl;
- L is ⁇ C(0)NR 7 -, -NR 7 C(0)-, -NR 7 C(0)NR 7 -, -NR 7 S(0) r - or ⁇ S(0) r NR 7 ⁇ ;wherein the dotted line (--) indicates the point of attachment of L to the group
- n 0;
- each of the (C 6 -Ci 4 )-aryl and heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-C 8 )-alkyl, halo(CrC 8 )-alkyl, (C C 8 )-alkoxy, (C 3 -Ci2)-cycloalkyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl; and R a , Rb, Rc, Ri , R2, R3, R6, R7, Re, R9, P, and B are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt,a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X 2 are independently selected from CR 6 and N;
- R 4 is NR a R b or NR a C(0)R b ;
- R a and R b are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C 5 -C 8 )-cycloalkenyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl;
- R 5 is (C 6 -Ci 4 )-aryl, (C 6 -Ci 4 )ar-(Ci -C 8 )-alkyl, heterocyclyl or heteroaryl;
- L is ⁇ C(0)NR 7 -, -NR 7 C(0)-, -NR 7 C(0)NR 7 -, -NR 7 S(0) r - or ⁇ S(0) r NR 7 ⁇ ;wherein the dotted line (--) indicates the point of attachment of L to the group x 'p and respectively;
- n 0;
- each of the (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-C 8 )-alkyl, halo(C C 8 )- alkyl, (CrC 8 )-alkoxy, (C 3 -Ci2)-cycloalkyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl; and
- Ri , R 2 , R3, R6, R7, Re, R9, P and B are as defined in the first aspect; or
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X 2 are independently selected from CR 6 and N;
- R 4 is NR a R b or NR a C(0)R b ;
- R a and R b are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C 8 )-cycloalkenyl,(C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl;
- R 5 is P(0)(ORa)(OR b ), P(0)R a R b , (C 6 -Ci 4 )-aryl-P(0)R a R b , (C 6 -Ci 4 )-aryl- P(0)(ORa)(OR b ), (C 6 -Ci 4 )-aryl-C(R c )2-P(0) R a R b , (C 6 -Ci 4 )-aryl-heterocyclyl-R c , (C 6 -Ci 4 )-aryl-C(Rc) 2 -P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(Rc) 2 -heterocyclyl, (C 6 -Ci 4 )- aryl, (C 6 -Ci 4 )-ar-(Ci -C 8 )-alkyl, heterocyclyl or heteroaryl;
- L is ⁇ C(0)NR 7 ⁇ , -N RTC(0) ⁇ , -NR 7 C(0)NR 7 -, ⁇ NR 7 S(0) r ⁇ or ⁇ S(0) r NR 7 ⁇ ; wherein the dotted line (--) indicates the point of attachment of L to the group
- n 0;
- each of the (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(Ci -C 8 )- alkyl, (CrC 8 )-alkoxy, (C 3 -Ci 2)-cycloalkyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl; and
- R a , Rb, Rc, Ri , R2, R3, R6, R7, r and p are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X2 are independently selected from CR6 and N;
- R 4 is NR a R b or NR a C(0)R b ;
- R a and R b are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C 8 )-cycloalkenyl,(C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl;
- R5 IS (C6-Ci 4 )-aryl, (C6-Ci 4 )ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl;
- L is -C(0)NR 7 -,-NR 7 C(0)-, ⁇ NR 7 C(0)NR 7 ⁇ , -NR 7 S(0) r - or -S(0) r NR 7 ⁇ ; wherein the dotted line (--) indicates the point of attachment of L to the group
- n O
- each of the (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-CeJ-alkyl, halo(Ci-Ce)- alkyl, (CrC 8 )-alkoxy, (C 3 -C 12 )-cycloalkyl, (C 6 -C 14 )-aryl, heterocyclyl and heteroaryl; and
- Ri, R2, R3, R6, R? and p are as defined in the first aspect; or
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X2 are independently selected from CR6 and N;
- R 4a is NR a R b or NR a C(0)R b ;
- R 5 is P(0)(ORa)(OR b ), P(0)R a R b , (C 6 -Ci 4 )-aryl-P(0)R a R b , (C 6 -Ci 4 )-aryl- P(0)(ORa)(OR b ), (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)R a R b , (C 6 -Ci 4 )-aryl-heterocyclyl-R c , (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(Rc) 2 -heterocyclyl, (C 6 -Ci 4 )-aryl, (C 6 -Ci 4 )-aryl-C(Rc) 2 -P(0)(OR a )(OR b ), (C
- L is ⁇ C(0)N R 7 -, -NR 7 C(0)-, -NR 7 C(0)NR 7 -, -NR 7 S(0) r - or ⁇ S(0) r NR 7 ⁇ ;wherein the dotted line (--) indicates the point of attachment of L to the group
- n 0;
- each of the (C 6 -C 14 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(Ci-C 8 )- alkyl, (CrC 8 )-alkoxy, (C 3 -Ci 2)-cycloalkyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl; and
- R a , Rb, Rc, Ri , R2, R3, R6, R7, Re, p, r and B are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X 2 are independently selected from CR 6 and N ;
- R 4a is NR a R b or NR a C(0)R b ;
- R a and R b are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C 8 )-cycloalkenyl,(C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl;
- R 5 is (C 6 -Ci 4 )-aryl, (C 6 -Ci 4 )-ar-(Ci -C 8 )-alkyl, heterocyclyl or heteroaryl;
- L is -C(0)NR7-,-N RTC(0) ⁇ , -N R 7 C(0)NR 7 -, ⁇ NR 7 S(0) R - or -S(0) r NR 7 -
- n 0;
- each of the (C 6 -C 14 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(Ci -C 8 )- alkyl, (CrC 8 )-alkoxy, (C 3 -Ci 2)-cycloalkyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl; and
- Ri , R2, R3, R6, R7, Re, p and B are as defined in the first aspect; or
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X 2 are independently selected from CR 6 and N ;
- R 4a is NR a R b or NR a C(0)R b ;
- R a and R b are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C 5 -C 8 )-cycloalkenyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl;
- R 5 is the group consisiting of P(0)(OR a )(OR b ), P(0)R a R b , (C 6 -Ci 4 )-aryl- P(0)R a R b ,(C 6 -Ci 4 )-aryl-P(0)(OR a )(OR b ), (C 6 -Ci 4 )-aryl-C(Rc) 2 -P(0)R a R b , (C 6 -Ci 4 )- aryl-heterocyclyl-Rc, (C 6 -Ci 4 )-aryl-C(R c ) 2 -P(0)(OR a )(OR b ),
- n 0;
- each of the (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC 8 )-alkyl, halo(Ci -C 8 )- alkyl, (CrC 8 )-alkoxy, (C 3 -Ci 2 )-cycloalkyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl; and
- R a , Rb, Rc, Ri , R2, R3, R6, R7, Rs, R9, P, r and B are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
- the present invention relates to a compound of formula 1 ,
- A is:
- Xi and X2 are independently selected from CR6 and N;
- R 4a is NR a R b or NR a C(0)R b ;
- R a and R b are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C 8 )-cycloalkenyl,(C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl;
- R 5 is (C 6 -Ci 4 )-aryl, (C 6 -Ci 4 )-ar-(Ci-C 8 )-alkyl, heterocyclyl or heteroaryl;
- L is -C(0)NR 7 -, -NR 7 C(0)-, -NR 7 C(0)NR 7 -, -NR 7 S(0) r - or ⁇ S(0) r NR 7 ⁇
- n 0;
- each ofthe (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C 8 )-alkyl,halo(Ci-C 8 )-alkyl, (CrC 8 )- alkoxy, (C 3 -Ci2)-cycloalkyl, (C 6 -Ci 4 )-aryl, heterocyclyl and heteroaryl; and
- R 1 5 R 2 , R3, R6, R 7 , R 8 , R9, p, r and B are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
- Representative compounds of the present invention include:
- processes for the preparation of the compounds of formula 1 can be prepared using various methods including the methods well known to a person skilled in the art. Representative processes to prepare the compounds of the present invention are described below, and are particularly illustrated in Scheme 1 , but are not limited thereto. It will be appreciated by persons skilled in the art that within certain of the processes described herein, the order of the reaction steps employed can be varied and will depend inter alia on factors such as the nature of functional groups present in a particular substrate (starting compound or an intermediate) and the protecting group strategy (if any) to be adopted. Clearly, such factors will also influence the choice of reagent to be used in the reaction steps.
- Scheme 1 depicts a process for the preparation of the compound of formula 1 ,whereinl_ is -C(0)NR 7 -, n is 0, A is:
- Xi , X2, Ri , R2, R3, R 4 , R5, p and Q are as defined in the first aspect of the present invention.
- Xi , X 2 , Ri , R2, R3, R 4 , R5, P and Q are as defined in the first aspect.
- R 5 is as defined for the compounds of formula 1 , in the first aspect of the invention
- a base such as sodium carbonate
- a solvent or a mixture of solvents such as a mixture of dioxane and water.
- the reaction mixture is purged with argon for ten minutes to fifteen minutes followed by addition of 1 , 1 '- bis(diphenylphosphino)ferrocene palladium-dichloromethane adduct, which is followed by purging with argon again for another 15 minutes to 20 minutes.
- the reaction mass obtained is heated at a temperature ranging from 90 ° C to 120 ° C for 5 h to 9 h, which results in the formation of the compound of formula 1 , wherein L is -C(0)NR 7 -; Q, p, Ri , R 2 , R3, R 4 , Rs and R 7 are as defined in the first aspect of the present invention.
- pharmaceutically acceptable salt(s) means those salts of compounds of the formula 1 which retain the efficacy and the biological properties of the free bases or of the free acids and are safe for use in mammals.
- pharmaceutically acceptable salts include organic and inorganic salts of the compounds of the invention (the compounds of formula 1 ) depending on the particular group (acidic or basic group) present in the said compounds. When compounds of the present invention contain relatively basic groups, acid addition salts can be obtained.
- the pharmaceutically acceptable acid addition salts include those derived from inorganic acids, which are not limited to, hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, mono-hydrogensulfuric or hydroiodic acids and the like; as well as the salts derived from organic acids such as acetic, ascorbic, benzenesulfonic,benzoic, citric, ethanesulfonic, formic, fumaric, galacturonic, gluconic, glucuronic, glutamic, isobutyric, isonicotinic, lactic, maleic, malonic, mandelic, methanesulfonic, 4-methylbenzenesulfonic, nicotinic, oxalic, pantothenic, phthalic, propionic, saccharic, succinic, suberic, p-tolylsulfonic, tartaric acids and
- base addition salts can be obtained.
- examples of pharmaceutically acceptable organic base addition salts of the compounds of the present invention include those derived from organic bases selected from lysine, arginine, guanidine, diethanolamine, choline, tromethamine, metformin and the like.
- examples of pharmaceutically acceptable base addition salts of the compounds of the present invention include their alkali metal salts. Suitable alkali metal salts include, but are not limited to, aluminum, calcium, lithium, magnesium, potassium, sodium or zinc salts.
- the pharmaceutically acceptable salts of the present invention can be synthesized from the compound of formula 1 , which contains a basic or an acidic group, by using conventional chemical methods.
- the salts are prepared by treating the compound of formula 1 , which may be a free base or an acid with a suitable salt-forming inorganic or organic acid or a base in a suitable solvent or dispersant or from another salt by cation or anion exchange.
- suitable solvents that can be used for the preparation of pharmaceutically acceptable salts include, but are not limited to, ethyl acetate, diethyl ether, methanol, ethanol, acetone, tetrahydrofuran, dioxane or mixtures of these solvents.
- the compounds of formula 1 can be regenerated by contacting ⁇ pharmaceutically acceptable salt with a base or an acid depending on the type of saltand isolating the parent compound in the conventional manner.
- the present invention also encompasses within its scope the solvates of the compounds of formula 1 .
- Certain compounds of the present invention can exist in unsolvated forms as well as solvated forms, including hydrated forms. Certain compounds of the present invention can exist in multiple crystalline or amorphous forms. In general, all physical forms are suitable for the uses contemplated by the present invention and are intended to be within the scope of the present invention.
- polymorphs of the compound of formula 1 can be prepared by crystallization of the compound under different conditions.
- the different conditions are, for example, using different solvents or their mixtures for carrying out crystallization; crystallization at different temperatures; various modes of cooling, ranging from very fast to very slow cooling during crystallization or by heating or melting the compound followed by gradual or fast cooling.
- the presence of polymorphs can be determined by IR (Infra-red) spectroscopy, solid probe NMR (Nuclear Magnetic Resonance) spectroscopy, differential scanning calorimetry, powder X-ray diffraction or such other techniques.
- the present invention includes all possible stereoisomers and geometric isomers of the compound of formula 1 and includes not only racemic compounds but also the optically active isomers as well.
- a compound of formula 1 When a compound of formula 1 is desired as a single enantiomer, it may be obtained either by resolution of the final product or by stereospecific synthesis from either isomerically pure starting material or an appropriate intermediate. Resolution of the final product, an intermediate or a starting material may be effected by any suitable method known in the art, for example, Chiral reagents for asymmetric synthesis by Leo A. Paquette; John Wiley & Sons Ltd (2003).
- the present invention is intended to include all tautomeric forms of the compounds.
- prodrugs of the compound of formula 1 are those compounds that are converted to their parent compound intracellular ⁇ , where the cellular converting location is the site of therapeutic action.
- the prodrugs of the compounds of the present invention are derivatives, particularly simple derivatives of the said compounds which upon administration to a subject in need thereof undergo conversion by metabolic or chemical processes to release the parent drug in vivo from which the prodrug is derived.
- the preferred prodrugs are pharmaceutically acceptable ester derivatives e.g., alkyl esters, cycloalkyl esters, alkenyl esters, benzyl esters, mono- or di-substituted alkyl esters that are convertible by solvolysis under physiological conditions to the parent carboxylic acid (e.g. the compound of formula 1 containing the carboxylic acid group), and those conventionally known in the art.
- ester derivatives e.g., alkyl esters, cycloalkyl esters, alkenyl esters, benzyl esters, mono- or di-substituted alkyl esters that are convertible by solvolysis under physiological conditions to the parent carboxylic acid (e.g. the compound of formula 1 containing the carboxylic acid group), and those conventionally known in the art.
- the present invention furthermore relates to pharmaceutical compositions containing a therapeutically effective amount of at least one compound of formula 1 or a pharmaceutically acceptable salt thereof; in addition to a customary pharmaceutically acceptable carrier or excipient.
- the present invention also relatesto a process for the production of a pharmaceutical composition, which includes bringing at least one compound of formula 1 , into a suitable administration form using a pharmaceutically acceptable excipient and, if appropriate, further suitable additives or auxiliaries.
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising at least one compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable excipient.
- composition(s) of the present invention can be administered orally, for example in the form of pills, tablets, coated tablets, capsules, granules or elixirs. Administration, however, can also be carried out rectally, for example in the form of suppositories, or parenterally, for example intravenously, intramuscularly or subcutaneously, in the form of injectable sterile solutions or suspensions, or topically, for example in the form of ointments or creams or transdermal ⁇ , in the form of patches, or in other ways, for example in the form of aerosols or nasal sprays.
- composition(s) according to the invention are prepared in a manner known and familiar to one skilled in the art.
- Pharmaceutically acceptable inert inorganic and/or organic carriers and/or additives can be used in addition to the compound of formula 1 , or a stereoisomer, a tautomer or a pharmaceutically acceptable saltfor the production of oral dosage forms of compounds of formula 1 such as pills, tablets, coated tablets and hard gelatin capsules. It is possible to use, for example, lactose, corn starch or derivatives thereof, gum arabica, magnesia or glucose, etc. Carriers for soft gelatin capsules and suppositories are, for example, fats, waxes, natural or hardened oils, etc.
- Suitable carriers for the production of solutions for example injection solutions, or of emulsions or syrups are, for example, water, physiological sodium chloride solution or alcohols, for example, ethanol, propanol or glycerol, sugar solutions, such as glucose solutions or mannitol solutions, or a mixture of the said solvents.
- the pharmaceutical compositions normally contain about 1 % to 99 %, for example, about 5 % to 70 %, or from about 10 % to about 30 % by weight of the compound of formula 1 or its pharmaceutically acceptable salt.
- the amount of the compound of formula 1 or its pharmaceutically acceptable salt in the pharmaceutical compositions normally can be from about 5 mg to 500 mg or can be lower than or higher than the lower and the upper limit respectively.
- the dose of the compound of formula 1 which is to be administered, can cover a wide range depending on the type of disease or disorder to be treated. The dose to be administered daily is to be selected to suit the desired effect.
- a suitable dosage can be about 0.01 mg/kg to 100 mg/kg of the compound of formula 1 or its pharmaceutically acceptable salt depending on the body weight of the recipient (subject) per day, for example, about 0.1 mg/kg/day to 50 mg/kg/day of a compound of formula 1 or itspharmaceutically acceptable salt. If required, higher or lower daily doses can also be administered.
- the selected dosage level will depend upon a variety of factors including the activity of a compound of the present invention, or its salt employed, the route of administration, the time of administration, the rate of excretion of the particular compound being administered, the duration of the treatment, other concurrently administered drugs, compounds and/or materials, the age, sex, weight, condition, general health and prior medical history of the patient (subject) being treated, and like factors well known in the medical arts.
- the pharmaceutical compositions of the present invention can contain additives such as, for example, fillers, antioxidants, dispersants, emulsifiers, defoamers, flavors, preservatives, solubilizers or colorants.
- the pharmaceutical compositions can also contain more than one compound of formula 1 or their pharmaceutically acceptable salts.
- the pharmaceutical compositions can also contain one or more other therapeutically or prophylactically active agents.
- the present invention also encompasses within its scope the use of a compound of formula 1 or its pharmaceutically acceptable salt in combination, with other therapeutically active agents; wherein the compound of formula 1 and the further therapeutic agent are administered either simultaneously or sequentially.
- the therapeutically active agents used in combination with one or more compounds of formula 1 or its pharmaceutically acceptable salt can be selected from: anti-neoplastic agents or chemotherapeutic compounds such as anti- microtubule agents (diterpenoids (paclitaxel, docetaxel) and vinca alkaloids (vinblastine, vincristine, vinorelbine)); platinum coordination complexes (cisplatin, carboplatin), alkylating agents (nitrogen mustards (oxazaphosphorines, cyclophosphamide, melphalan, chlorambucil)); alkyl sulfonates (busulfan); nitrosoureas (carmustine); triazenes (dacarbazine); topoisomerase I inhibitors (camptothecins (irinotecan, topotecan)); topoisomerase II inhibitors (epipodophyllotoxins (etoposide, teniposide)); antimetabolite neoplastic agents (flu
- the compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof is an EZH2 inhibitor.
- the compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof is provided for use the treatment of a disease or a disorder mediated by EZH2.
- the present invention relates to a method for the treatment of a disease or a disorder mediated by EZH2, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof.
- the present invention relates to use of a compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof; for the treatment of a disease or a disorder mediated by EZH2.
- the present invention relates to use of a compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof; for the manufacture of a medicament for the treatment of a disease or a disorder mediated by EZH2.
- the disease or disorder mediated by EZH2 is selected from cancer, pulmonary arterial hypertension, myelofibrosis, human immunodeficiency virus (HIV) disease, graft versus host diseases (GVHD), Weaver Syndrome, psoriasis vulgaris or liver fibrogenesis.
- HIV human immunodeficiency virus
- GVHD graft versus host diseases
- Weaver Syndrome psoriasis vulgaris or liver fibrogenesis.
- the disease or disorder mediated by EZH2 is cancer.
- Cancers also include metastatic or malignant tumors.
- cancers that can be treated by the compound of formula 1 of the invention or pharmaceutical compositions containing the said compounds are selected from: thyroid carcinoma, cardiac sarcoma, lung carcinoma, gastrointestinal carcinoma, genitourinary tract carcinoma, liver carcinoma, mantle cell lymphoma, bone sarcoma, sarcoma of the nervous system, gynaecological carcinoma, haematological cancer, adrenal gland neuroblastoma, skin cancer, astrocytic cancer, breast cancer, colorectal cancer, endometrial cancer, head and neck cancer or oral cancer.
- the cancer is cardiac sarcoma selected from angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma, rhabdomyoma, fibroma, lipoma or teratoma.
- the cancer is lung carcinoma selected from squamous cell carcinoma, undifferentiated small or large cell carcinoma, adenocarcinoma, bronchiolar carcinoma, bronchial adenoma, bronchial sarcoma or bronchial lymphoma.
- the cancer is gastrointestinal carcinoma selected from stomach carcinoma, stomach lymphoma, pancreatic carcinoma (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma), small bowel carcinoma (adenocarcinoma, lymphoma, Kaposi's sarcoma, hemangioma, lipoma, neurofibroma, fibroma) or large bowel carcinoma (adenocarcinoma, tubular adenoma).
- pancreatic carcinoma ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma
- small bowel carcinoma adenocarcinoma, lymphoma, Kaposi's sarcoma, hemangioma, lipoma, neurofibroma, fibroma
- large bowel carcinoma adenocarcinoma, tubular adenoma
- the cancer is genitourinary tract carcinoma selected from carcinoma of kidney (adenocarcinoma, nephroblastoma, lymphoma, leukemia), malignant rhabdoid tumor of kidney, carcinoma of bladder and urethra (squamous cell carcinoma, adenocarcinoma), carcinoma of prostate (adenocarcinoma, sarcoma), or carcinoma of testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, lipoma).
- the cancer is liver carcinoma selected from hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma, angiosarcoma or hepatocellular adenoma.
- the cancer is bone sarcoma selected from osteogenic sarcoma (osteosarcoma), fibrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma or giant cell tumors.
- osteogenic sarcoma osteosarcoma
- fibrosarcoma fibrosarcoma
- Ewing's sarcoma malignant lymphoma (reticulum cell sarcoma)
- multiple myeloma benign chondroma
- chondroblastoma chondromyxofibroma
- osteoid osteoma giant cell tumors.
- the cancer is an oral cancer selected from tongue cancer, squamous cell carcinoma, Kaposi's sarcoma, teratoma, adenocarcinoma derived from a major or minor salivary gland, lymphoma from tonsillar or other lymphoid tissue, or melanoma from the pigment- producing cells of the oral mucosa.
- the cancer is sarcoma of the nervous system selected from sarcoma of skull (osteoma, granuloma, xanthoma), meninges (meningioma, meningiosarcoma, gliomatosis), sarcoma of brain (astrocytoma, medulloblastoma, glioma, glioblastoma multiform, oligodendroglioma, retinoblastoma, congenital tumors), sarcoma of spinal cord (neurofibroma, meningioma, glioma, sarcoma) or malignant rhabdoid tumor of brain.
- skull ovaloma, granuloma, xanthoma
- meninges meningioma, meningiosarcoma, gliomatosis
- sarcoma of brain astrocytoma, medulloblastoma, glio
- the cancer is carcinoma of gynaecological organs selected from carcinoma of uterus (endometrial carcinoma), carcinoma of cervix (cervical carcinoma, ovary carcinoma), carcinoma of vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), carcinoma of vagina (clear cell carcinoma, squamous cell carcinoma, embryonal rhabdomyosarcoma) or carcinoma of fallopian tubes.
- carcinoma of gynaecological organs selected from carcinoma of uterus (endometrial carcinoma), carcinoma of cervix (cervical carcinoma, ovary carcinoma), carcinoma of vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), carcinoma of vagina (clear cell carcinoma, squamous cell carcinoma, embryonal rhabdomyosarcoma) or carcinoma of fallopian tubes
- the cancer is haematological cancer selected from blood cancer (acute myeloid leukemia (AML), chronic myeloid leukemia (CML), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplastic syndrome, B-cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, Hodgkin's lymphoma (Hodgkin's disease), non-Hodgkin's lymphoma (malignant lymphoma) or mantle cell lymphoma.
- blood cancer acute myeloid leukemia (AML), chronic myeloid leukemia (CML), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplastic syndrome, B-cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, Ho
- the cancer is a skin cancer selected from malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Kaposi's sarcoma, angioma or dermatofibroma.
- reaction mixture was cooled, diluted with water and the residue was extracted with ethyl acetate.
- residue was washed with water and brine; and dried over anhydrous sodium sulphate to obtain a crude mixture, which was purified by column chromatography(silica gel, 10-20 % methanol in chloroform) to yield the title compound.
- reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate (2x25 mL) and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude mixture, which was purified by using column chromatograhy (silica gel, 0-15 % MeOH/CHCI 3 ) to yield the title compound.
- the reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate.
- the organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHC ) to yield the title compound.
- the reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate.
- the organic layers were concentrated to obtain a crude mixture, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHC ) to yield the title compound.
- the reaction mixture was stirred at 80 °C for 2 h under nitrogen atmosphere.
- the reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with water, brine and dried over anhydrous sodium sulphate.
- the organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI 3 ) to yield the title compound.
- the reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate.
- the organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0- 15% MeOH/CHC ) to yield the title compound.
- the reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate.
- the organic layers were concentrated to obtain a crude mixture, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHC ) to yield the title compound.
- reaction mixture was then cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate.
- the organic layers were concentrated to obtain a crude mixture, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI 3 ) to yield the title compound.
- reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI 3 ) to yield the title compound.
- the compound of example 40 (100 mg, 0.199 mmol) was added to a stirred solution of 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)piperazine (91 mg, 0.299 mmol), PdCI 2 (dppf)-CH 2 Cl2 adduct (16.25 mg, 0.020 mmol) and Na 2 C0 3 (63.3 mg, 0.597 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere.
- the reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with water and brine, and dried over anhydrous sodium sulphate.
- the organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHCI 3 ) to yield the title compound.
- the compound of example 46 (150 mg, 0.274 mmol) was added to a stirred solution of 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (125 mg, 0.412 mmol), PdCI 2 (dppf)-CH 2 CI 2 adduct (22.41 mg, 0.027 mmol) and Na 2 C0 3 (87 mg, 0.823 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3 h under nitrogen atmosphere.
- reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate was washed with waterand brine and dried over sodium sulphate to obtain a crude mixture, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
- the compound of example 46 (150 mg, 0.274 mmol) was added to a stirred solution of 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)piperazine (125 mg, 0.412 mmol), PdCI 2 (dppf)-CH 2 Cl2 adduct (22.41 mg, 0.027 mmol) and Na 2 C0 3 (87 mg, 0.823 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 ml_). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere.
- reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with waterand brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
- reaction solvent was distilled under vacuum; the residue obtained was extracted using ethyl acetate and washed with water (3x25 mL). The combined organic extracts were distilled off to obtain a crude mixture, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI 3 ) to yield the title compound.
- Example 51 5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((6-methyl-2-oxo-1 ,2- dihydropyridin-3-yl)methyl)-4'-(morpholinomethyl)-[1 ,1 '-biphenyl]-3- carboxamide
- reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
- the ethyl acetate layer was washed with water and brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
- the compound 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pyridin-2-yl)piperazine (197 mg, 0.649 mmol) was added to a stirred solution of the compound of example 50 (200 mg, 0.433 mmol), PdCI 2 (dppf)-CH 2 CI 2 adduct (35.3 mg, 0.043 mmol) and Na 2 C0 3 (138 mg, 1 .298 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere.
- reaction mixture was diluted with (250ml_) water, the phases were separated and the aqueous solution was washed with brine (100ml_), dried over anhydrous sodium sulphate and evaporated to obtain a solid, which was further treated with silica to obtain a waxy white solid, which was filtered and dried to yield the title compound.
- Example 76 3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7- propyl-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6-(4- methylpiperazin-1-yl)pyridin-3-yl)benzamide
- Example 88 4- (Aminomethyl)-7-isopropyl-1 -methyl-5,6,7,8-tetrahydro-2,7-naphthyridin- 3(2H)-one hydrochloride
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Virology (AREA)
- Physical Education & Sports Medicine (AREA)
- Tropical Medicine & Parasitology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- AIDS & HIV (AREA)
- Transplantation (AREA)
- Cardiology (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Heart & Thoracic Surgery (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides compound of formula 1, or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a polymorph, a prodrug, S-oxide or N-oxide thereof. The invention also relates toprocesses for their preparation, to pharmaceutical compositions containing them and use of the compound of formula 1, in the treatment of diseases or disorders mediated by EZH2 (enhancer of zeste homolog 2), particularly cancer.
Description
EZH2 INHIBITORS AND USES THEREOF
FIELD OF THE INVENTION
The present invention relates to heterocyclic compounds (referred to herein as the compounds of formula 1 ), processes for their preparation, pharmaceutical compositions comprising the compounds of formula 1 , and their use as EZH2 (enhancer of zeste homolog 2) inhibitors and methods of using said compounds in the treatment of diseases mediated by EZH2. BACKGROUND OF THE INVENTION
Histone methyl transferases (HMTs) are a family of enzymes, which control selective methylation at specific amino acid sites on histones. Covalent modification of histones such as methylation control changes of chromatin structure in eukaryotic cell DNA, which lead to heritable alteration in gene expression. These modifications are referred to as epigenetic modifications. Polycomb genes are an illustration of epigenetic effectors structured in multimeric repressive complexes. Aberrant expression and/or activity of enzymes responsible for histone modification result in disease states such as cancer. Therefore, epigenetic modifications can be reversed and consequently, treatment of diseases such as cancer may be effected through selective inhibition of the enzymes involved.
Histone-lysine N-methyl transferase EZH2 (enhancer of zeste homolog 2) is the catalytic subunit of Polycomb repressive complex 2 (PRC2), which methylates Lys27 of a specific histone H3 (H3K27) and is essential for the self- renewal of cancer stem cells. EZH2 is capable of silencing several anti-metastatic genes, favoring cell-invasion and uncontrolled cell-growth. For instance, somatic mutations of tyrosine 641 of EZH2 are reported to be associated with follicular lymphoma and diffused B-cell lymphoma (Nature Genet., 2010, 42, 2, 181 -185).
Increased levels of trimethylated H3K27 resulting due to an increased expression of EZH2 contribute to cancer aggressiveness, metastasis, shorter disease free survival and increased death rate in many tumor types, for instance, in melanoma, prostate cancer, breast cancer and cancer of the endometrium (Bachmann et al, Journal of Clinical Oncology, 2006, 24, 4, 268-73). This is further
substantiated in Oncogene, 2012, 31 , 3827-44, wherein it is reported that EZH2 expression is deregulated in prostate cancer, breast cancer and endometrial cancer, B-cell non-Hodgkin's lymphoma and bladder carcinoma. Increased expression of EZH2 also induces pulmonary artery smooth muscle proliferation (PLoS ONE, 2012, 7, 5, e37712). Increased expression of EZH2 has been further reported to have implication in myelofibrosis (Expert Review of Hematology, 2012, 5, 3, 313-324), HIV (PCT publication WO2012051492 A2), graft versus host diseases (GVHD) (Blood, 2012, 1 19, 5, 1274-1282), Weaver Syndrome (American Journal of Human Genetics, 2012, 90, 1 , 1 10-1 18), psoriasis vulgaris (European Journal of Dermatology, 201 1 , 21 , 4, 552-557) and liver fibrogenesis (PCT publication WO2010090723A2).
PCT patent publication WO201 1 140324A1 discloses indoles as inhibitors of EZH2 and use of said compounds for the treatment of cancer. PCT patent publication WO20121 18812A2 discloses bicyclic heterocyclic compounds as EZH2 inhibitors and their use in the treatment of cancer.
Thus, it is understood from the above discussion that the inhibition of EZH2 activity would effectively decrease cellular proliferation and invasion and thereby provide therapeutic benefit in the treatment of diseases or disorders mediated by EZH2. The compounds of the current invention function as inhibitors of EZH2 and therefore, provide a therapeutic solution for the treatment of disorders mediated by EZH2, such as cancers.
SUMMARY OF THE INVENTION
According to one aspect, the present invention relates to a compound of formula 1 (as described herein), or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a polymorph, a prodrug, S-oxide or N- oxide thereof.
According to another aspect, the present invention relates to processes for the preparation of the compound of formula 1 or a pharmaceutically acceptable salt thereof.
According to a further aspect, the present invention relates to pharmaceutical composition comprising the compound of formula 1 or an isotopic
form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof; and at least one pharmaceutically acceptable carrier or excipient.
According to a further aspect, the present invention relates to a compound of formula 1 or a pharmaceutically acceptable salt thereof; for use as an EZH2 (enhancer of zeste homolog 2) inhibitor.
According to yet another aspect, the present invention relates to a compound of formula 1 or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate thereof; for use in the treatment of a disease or a disorder mediated by EZH2.
According to a further aspect, the present invention relates to a method for the treatment of a disease or disorder mediated by EZH2; comprising administering to a subject in need thereof, a therapeutically effective amount of the compound of formula 1 or a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof.
In a further aspect, the present invention relates to a use of a compound of formula 1 or a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof; in combination with at least one further therapeutically active agent; for the treatment of a disease or disorder mediated by EZH2.
According to yet another aspect, the present invention relates to a use of a compound of formula 1 or a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof; for the manufacture of a medicament for the treatment of a disease or disorder mediated by EZH2.
One or more further aspects of the present inventions are discussed in detail herein below. These and other objectives and advantages of the present invention will be apparent to those skilled in the art from the following description.
DETAILED DESCRIPTION OF THE INVENTION
In a first aspect, the present invention relates to a compound of formula 1 ,

Formula 1
or an isotopic form, a stereoisomer^ tautomer,a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof;
wherein,
Ri and R2 are independently selected from hydrogen, halogen, hydroxy, cyano, (Ci-Ce)-alkyl, halo(Ci-C8)-alkyl,(C2-C8)-alkenyl, (Ci-C8)-alkoxy, C(0)Ra,C(0)ORa, C(0)NRaRb, OC(0)NRaRb, NRaRb, NRaC(0)Rb,S(0)q(C C8)-alkyl and S(0)qNRaRb;or
Ri and R2 together can form a 3 to 10 membered monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N,S and P; wherein the ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (CrC8)-alkoxy, halo(CrC8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)0-2, (C6-Ci4)-aryl-(Rc)0-2,C(O)Ra, C(0)ORa, C(0)NRaRb,NRaRb, NRaC(0)Rb, S(0)q(C C8)-alkyl and S(0)qNRaRb; A is:

Wherein X and X2 are independently selected from CR6 and N;
R3, R4 and R6 are independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, halo(CrC8)-alkyl-OH, (Ci-C8)-alkoxy, halo(C C8)-alkoxy, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)- cycloalkenyl, (C6-Ci4)-aryl, (C6-Ci4)ar-(CrC8)-alkyl, heterocyclyl, heteroaryl, C(0)Ra, C(0)ORa, C(0)NRaRb, S(0)q(C C8)-alkyl, S(0)qNRaRb, NRaRb, NRaC(0)Rb and NRaS(0)q(C C8)-alkyl;
R4a is hydrogen, hydroxy, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (CrC8)-alkoxy, halo(CrC8)-alkoxy, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl,(C5-C8)-cycloalkenyl,(C6- Ci4)-aryl, (C6-Ci4)ar-(Ci-C8)-alkyl, heterocyclyl, heteroaryl, C(0)Ra, C(0)NRaRb, S(0)q(Ci-C8)-alkyl or S(0)qNRaRb; or
R3 and R4 or R3 and R4a on A can join together to form a 3 to 10 membered saturated orunsaturated monocyclic ring optionally containing one to three
heteroatoms independently selected from the group consisting of O, N, S and P; wherein said ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (C C8)-alkyl, halo(C C8)-alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3- Ci2)-cycloalkyl-(Rc)o-2, 0-(C3-Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, 0-(C6- Ci4)-aryl-(Rc)o-2, heterocyclyl-(Rc)0-2, O-heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, S(0)q(C C8)-alkyl and S(0)qNRaRb;
R5 is hydrogen, halogen, hydroxy, cyano, B(OH)2, P(0)(ORa)(ORb), P(0)RaRb, (Ci-C8)-alkyl, halo(C C8)-alkyl, halo(C C8)-alkyl-OH, (C C8)-alkoxy, halo(C C8)- alkoxy, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)-cycloalkenyl, (C6-Ci4)-aryl, (C6-Ci4)ar-(CrC8)-alkyl, (C6-Ci4)-aryl-P(0)RaRb, (C6-Ci4)-aryl-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)- aryl-heterocyclyl-Rc,(C6-Ci4)-aryl-C(Rc)2-heterocyclyl, heterocyclyl, heteroaryl, C(0)Ra,C(0)ORa, C(0)NRaRb, C(0)NRaNRaRb,S(0)q(C C8)alkyl, S(0)qNRaRb, NRaS(0)q(CrC8)-alkyl, NRaS(0)q(C3-Ci2)-cycloalkyl, NRaS(0)q(C6-Ci4)-aryl, NRaS(0)qheterocyclyl, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, NRaC(0)ORb, NRaS(0)qNRaRb, NRaNRaRb,NRaNRaC(0)Rb, NRaNRaC(0)NRaRb or NRaNRaC(0)ORa;
L is --C(0)NR7--, --NR7C(0)--, -NR7C(0)NR7-, --NR7S(0)r--, --S(0)rNR7-, - CH2NR7--, -NR7CH2-, --CH(halo)NR7-, --NR7CH(halo)--, --C(halo)2NR7-, -- NR7C(halo)2-, --CH(halo-(C C8)-alkyl)NR7--, --NR7CH(halo-(C C8)-alkyl)-, - , -NR7C(halo-(Ci-C8)-alkyl)2-
 wherein the dotted line (--) indicates the point of attachment of L to the group p and' " respectively; or
wherein the dotted line (--) indicates the point of attachment of L to the group p and' " respectively; or
L is a saturated or unsaturated 5 or 6 membered monocyclic ringoptionally containing one to three heteroatoms selected from the group consisting of O, N, S and P;
R7 is hydrogen, cyano, (CrC8)-alkyl, (C2-C8)-alkenyl, halo(Ci-C8)alkyl,C(0)Ra, C(0)ORa,C(0)NRaRb, S(0)q(CrC8)-alkyl or S(0)qNRaRb;
Q is:

B is a 3 to 10 membered monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; wherein said ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano,(Ci -Ce)-alkyl, halo(Ci-C8)-alkyl, (Ci-C8)-alkoxy, halo(Ci-C8)-alkoxy, (C3- Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, C(0)ORa, C(0)NRaRb,NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(C C8)- alkyl and S(0)qNRaRb;
R8 and R9 are independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (CrC8)-alkyl, halo(CrC8)alkyl, (CrC8)-alkoxy, halo(C C8)-alkoxy, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)-cycloalkenyl, (C6-Ci4)-aryl, (C6-Ci4)ar-(Ci -Ce)-alkyl, C(0)Ra, C(0)ORa,C(0)NRaRb, S(0)q(C C8)-alkyl, S(0)qNRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb and NRaC(0)ORb;
Ra and Rb are independently selected from the group consisting of hydrogen, (C C8)-alkyl, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)-cycloalkenyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; or
Ra and Rb together with the nitrogen atom can form a 3 to 10 membered ring, optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P;
Rc at each occurrence is independently selected from the group consisting ofhydrogen, halogen, hydroxy, cyano, (Ci-C8)-alkyl, halo(Ci-C8)alkyl, (C3-C12)- cycloalkyl, (C6-Ci4)-aryl, (C6-Ci4)-ar-(Ci-C8)-alkyl, heteroaryl, heterocyclyl, CORa, C(0)ORa, C(0)NRaRb, C(0)NRaNRaRb, S(0)q(C C8)-alkyl, S(0)qNRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, NRaC(0)ORb and NRaS(0)qNRaRb;
p and n are integers independently selected from 0, 1 , 2 and 3;
q is 1 or 2; or
r is 1 or 2;
wherein each of the (CrC8)-alkyl or (CrC8)-alkoxy can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkoxy, halo(CrC8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)o-2, O-(C3-Ci2)-cycloalkyl-(Rc)0-2, (C6-Ci4)-aryl-(Rc)0-2, O- (C6-Ci4)-aryl-(Rc)o-2, heterocyclyl-(Rc)0-2, O-heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(Ci-Ce)-alkyl and S(0)qNRaRb;
each of the (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C3-C8)-cycloalkenyl and (C6-Ci4)- aryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-CeJ-alkyl, halo(C C8)-alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)0-2, 0-(C3-Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, O-(C6-Ci4)-aryl-(Rc)0-2, heterocyclyl-(Rc)o-2, O-heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(C C8)-alkyl and S(0)qNRaRb;
the heterocyclyl is a 3 to 10 membered saturated or partially unsaturated, monocyclic or bicyclic ring system containing one to four heteroatoms independently selected from the group consisting of O, N, S and P; wherein the heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (C C8)-alkyl, halo(C C8)-alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3- Ci2)-cycloalkyl-(Rc)o-2, O-(C3-Ci2)-cycloalkyl-(Rc)0-2, (C6-Ci4)-aryl-(Rc)0-2, 0-(C6-
Ci4)-aryl-(Rc)o-2, heterocyclyl-(Rc)0-2, O-heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(Ci-Ce)-alkyl and S(0)qNRaRb;and
the heteroaryl is a 5 to 10 membered monocyclic or bicyclic aromatic ring system containing one to four heteroatoms independently selected from the group consisting of O, N, S and P, wherein the heteroaryl can be unsubstituted or substituted with one or more groups independently selected fromthe group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (C C8)-alkoxy, halo(CrC8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)0-2, 0-(C3-Ci2)-cycloalkyl- (Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, O-(C6-Ci4)-aryl-(Rc)0-2, heterocyclyl-(Rc)0-2, O- heterocyclyl-(Rc)o-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(C C8)-alkyl and S(0)qNRaRb.
Definitions
Unless otherwise indicated, the following definitions are set forth to illustrate and define the meaning and scope of the various terms used to describe the invention herein and the appended claims. These definitions should not be interpreted in the literal sense as they are not general definitions and are relevant only for this application.
It will be understood that "substitution," "substituted" or "substituted with" means that one or more hydrogens of the specified moiety are replaced with a suitable substituent and includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and results in a stable compound.
The terms "a", "an" and "the" refers to "one or more" when used in the subject specification, including the claims. Thus, for example, reference to "a compound" may include a plurality of such compounds, or reference to "a disease" or "a condition" includes a plurality of diseases or disorders.
It should also be noted that the term "or" is generally employed in its sense including "and/or" unless the content clearly dictates otherwise.
The term "independently" when used in the context of selection of substituents for a variable, it means that where more than one substituent is
selected from a number of possible substituents, those substituents may be the same or different.
Also, use of "(s)" as part of a term, includes reference to the term singly or in plurality, e.g. the term compound(s) may indicate a single compound or more compounds.
As used herein, the term "alkyl" or "(CrC8)-alkyl" whether used alone or as part of a substituent group, refers to the radical of saturated aliphatic groups, including straight or branched-chain alkyl groups. If the number of carbon atoms is not specified, "alkyl" refers to alkyl group having 1 to 8 (both inclusive) carbon atoms. Accordingly, a straight-chain or branched chain alkyl has eight or fewer carbon atoms in its backbone, for instance, Ci-C8 for straight- chain and C3-C8 for branched chain. Representative examples of alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, iso-butyl, sec-butyl, n- pentyl, isopentyl, 2-pentyl, 3-pentyl, neo-pentyl, n-hexyl, isohexyl, 2-hexyl, 3-hexyl and the like.
Furthermore, unless stated otherwise, the alkyl groups can be unsubstituted or substituted with one or more substituents. A substituted alkyl refers to a (CrC8)-alkyl substituted preferably with 1 -7 groups, more preferably 1 - 3 groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkoxy, halo(CrC8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)0-2, 0-(C3- Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, O-(C6-Ci4)-aryl-(Rc)0-2, heterocyclyl- (Rc)o-2, 0-heterocyclyl-(Rc)o-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(Ci-C8)-alkyl or S(0)qNRaRb;
wherein Rc at each occurrence is independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (CrC8)-alkyl, halo(CrC8)alkyl, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, (C6-Ci4)-ar-(Ci-C8)-alkyl, heteroaryl, heterocyclyl, C(0)Ra, C(0)ORa, C(0)NRaRb, CONRaNRaRb, S(0)q(C C8)-alkyl, S(0)qNRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, NRaC(0)ORb and NRaS(0)qNRaRb; q is 1 or 2; Ra and Rb are independently selected from the group consisting of hydrogen, (CrC8)-alkyl, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)- cycloalkenyl,(C6-C14)-aryl, heterocyclyl and heteroaryl; or Ra and Rb together with the nitrogen atom can form 3 to 10 membered saturated or unsaturated ring,
optionally containing one to three heteroatoms independently selected from the group consisting of O, N,S and P.
Representative examples of substituted alkyls include, but are not limited to, trifluoromethyl, hydroxymethyl, hydroxyethyl, 1 -aminoethyl, benzyl, N- morpholino methyl, N-indolomethyl, and N-piperidinylmethyl.
The term "halogen" or "halo" refers to a fluorine, chlorine, bromine, or iodine atom.
When the alkyl group is substituted with one or more halogens, it is specifically referred to as "halo(CrC8)alkyl" or "haloalkyl". A monohalo(Ci-C8)alkyl radical, for example, can have a chlorine, bromine, iodine or fluorine atom. Dihalo and polyhalo(Ci-C8)alkyl radicals may have two or more of the same or different halogen atoms. Representative examples of halo(CrC8)-alkyl include, but are not limited to, chloromethyl, dichloromethyl, trichloromethyl, dichloroethyl, dichloropropyl, fluoromethyl, difluoromethyl, trifluoromethyl, pentafluoroethyl, heptafluoropropyl, difluorochloromethyl, dichlorofluoromethyl, difluoroethyl, difluoropropyl or the like groups. Unless stated otherwise, the haloalkyl group can be unsubstituted or substituted with one or more groupsas described in the definition of 'substituted alkyl' herein above.
As used herein, the term "alkenyl" or "(C2-C8)-alkenyl", as used herein; alone or as part of a substituent group, refers to a straight or branched chain hydrocarbon radical containing the indicated number of carbon atoms and at least one carbon-carbon double bond (two adjacent sp2 carbon atoms). For example, (C2-C8)-alkenyl refers to an alkenyl group having 2 to 8 (both inclusive) carbon atoms. Depending on the placement of double bond and substituents if any, the geometry of the double bond may be entgegen (E), or zusammen (Z), cis or trans. Representative examples of alkenyl include, but are not limited to, vinyl, allyl and 2-propenyl.
Unless stated otherwise, the alkenyl group can be unsubstituted or substituted with 1 -7 groups, preferably 1 -3 groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(Ci-C8)- alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)0-2, 0-(C3-Ci2)- cycloalkyl-(Rc)o-2,(C6-Ci4)-aryl-(Rc)0-2, O-(C6-Ci4)-aryl-(Rc)0-2, heterocyclyl-(Rc)0-2, 0-heterocyclyl-(Rc)o-2,heteroaryl-(Rc)o-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb,
NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(C C8)-alkyl or S(0)qNRaRb; wherein Ra, Rb, Rc and q are as defined herein above.
As used herein, the term "alkoxy" or "(CrC8)-alkoxy" refers to a (CrC8)- alkyl having an oxygen radical attached thereto. The terms "(C C8)-alkoxy" or O- (Ci-C6)-alkyl wherever used in this specification have the same meaning. Representative examples of alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, isobutoxy and t-butoxy.
An alkoxy group may be unsubstituted or substituted with one or more substituents. A substituted alkoxy refers to an (CrC8)-alkoxy group in which the alkyl is substituted with one or more groups as explained in the definition of 'substituted alkyl' herein above. Representative examples of substituted (Ci-C8)- alkoxy include, but are not limited to, chloromethoxy, 2-cyanoethoxy, trifluoromethoxy and benzyloxy group. A benzyloxy group refers to a benzyl having an oxygen radical attached thereto.
As used herein, the term "cycloalkyi" or "(C3-Ci2)-cycloalkyl" whether used alone or as part of a substituent group, refers to a saturated or partially unsaturated cyclic hydrocarbon radical including 1 , 2 or 3 rings and including a total of 3 to 12 carbon atoms forming the rings. The term cycloalkyi includes bridged, fused and spiro ring systems. As used herein, (C3-Ci2)-cycloalkyl refers to a cycloalkyi group having 3 to 12 (both inclusive) carbon atoms. Representative examples of cycloalkyi include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, adamantyl, norbornyl, bicyclo[2.1 .0]pentane, bicyclo[2.2.1 ]heptyl, bicyclo[2.2.1 ]hept-2-ene, spiro[3.3]heptane and 1 ,2,3,3a- tetrahydropentalene.
The term "cycloalkenyl" or "(C5-C8)-cycloalkenyl" refers to a non-aromatic monocyclic carboxycyclic ring having the specified number of carbon atoms and up to 3 carbon-carbon double bonds. Representative examples of cycloalkenyl include, but are not limited to, cyclopentenyl and cyclohexenyl.
Unless stated otherwise, the "cycloalkyi" and "(C5-C8)-cycloalkenyl" can be unsubstituted or substituted with 1 -7, preferably 1 -3groups independently selected from halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3-C12)-cycloalkyl-(Rc)o-2, 0-(C3-C12)-cycloalkyl- (Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, 0-(C6-Ci4)-aryl-(Rc)0-2,heterocyclyl-(Rc)o-2,0-
heterocyclyl-(Rc)o-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(C C8)-alkyl and S(0)qNRaRb; wherein Ra, Rb, Rc and q are as defined herein above.
The term "aryl" or "(C6-Ci4)-aryl" as used herein refers to monocyclic or polycyclic hydrocarbon groups having 6 to 14 ring carbon atoms in which the carbocydic ring(s) present have a conjugated pi electron system. Representative examples of (C6-Ci4)-aryl residues include, but are not limited to, phenyl, naphthyl, fluorenyl or anthracenyl. Aryl groups can be unsubstituted or substituted with one or more, for example 1 -5 substituents independently selected from halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (CrC8)-alkoxy, halo(CrC8)- alkoxy, (C3-Ci2)-cycloalkyl-(Rc)o-2, 0-(C3-Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0- 2, O-(C6-Ci4)-aryl-(Rc)0-2, heterocyclyl-(Rc)0-2, O-heterocyclyl-(Rc)0-2, heteroaryl- (Rc)o-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(Ci-C8)-alkyl and S(0)qNRaRb; wherein Ra, Rb, Rc and q are as defined herein above. In monosubstituted phenyl, the substituent can be located in the 2-position, the 3-position or the 4-position. If the phenyl carries two substituents, they can be located in 2, 3-position, 2, 4-position, 2, 5-position, 2, 6- position, 3, 4-position or 3, 5-position. Representative examples of monosubstituted phenyl groups include, but are not limited to, 3- trifluoromethylphenyl, 4-chlorophenyl and 4-cyanophenyl.
Representativeexamples of disubstituted phenyl groups include, but are not limited to, 3, 5-difluorophenyl and 3, 4-dimethoxyphenyl.
As used herein, the term "aryloxy" or "0-(C6-Ci4)-aryl" refers to an "(Ce- Ci4)-aryl" group having an oxygen radical attached thereto. The aryl of aryloxy group may be unsubstituted or substituted as indicated in the definition of (C6- Ci4)-aryl herein above. Representative examples of aryloxy groups include, but not limited to, phenoxy, 4-chlorophenoxy and 3, 4-dimethoxyphenoxy.
As used herein, the term "aralkyl" or "(C6-Ci 4)ar-(Ci -C8)-alkyl" refers to an alkyl group substituted with an (C6-Ci4)-aryl group, wherein the terms alkyl and aryl are as defined above. Representative examples of aralkyl groups include (CH2)p-phenyl, wherein p is an integer selected from 1 to 6, such as benzyl wherein p is 1 . The aryl of the (C6-C14)-aralkyl group can be unsubstituted or substituted as indicated in the definition of aryl herein above.
The term "heteroatom" as used herein, includes nitrogen (N), oxygen (O), sulfur (S) and phosphorus (P). Any heteroatom with unsatisfied valency is assumed to have a hydrogen atom to satisfy the valency.
As used herein, the terms "heterocyclyl" or "heterocyclic" whether used alone or as part of a substituent group, refers to a saturated, partially unsaturated, monocyclic, polycyclic, bridged cyclic or spirocyclic ring system containing 3 to 10 carbon atoms and 1 to 4 identical or different heteroatoms selected from oxygen, nitrogen, sulfurand phosphorus. Representative examples of heterocyclyls include, but are not limited to, oxetanyl, pyrrolyl, pyrrolidinyl, pyrazolyl, imidazolyl, pyrazinyl, piperazinyl, oxazolyl, oxadiazolyl, isoxazolyl, triazolyl, thiazolyl, tetrazolyl, furyl, thienyl, purinyl, pyridinyl, pyridazinyl, pyrimidinyl, piperidyl, benzoxazolyl, benzothiazolyl, benzofuranyl, purinyl, benzimidazolyl, benzoxazolyl, indolyl, indazolyl, isoindolyl, isothiazolyl, isoquinolyl, isoquinolyl, morpholinyl, thiomorpholinyl, thiomorpholinyl-1 , 1 -dioxide, quinoxalinyl, quinolinyl,thiophenyl, 1 ,4-azaphosphinanyl, 1 ,4-azaphosphinanyl-4-oxide, octahydropyrrolo[3,4- c]pyrrole, 2-oxa-7-azaspiro[4.4]nonane, 2-oxa-6-azaspiro[3.4]octane, 2- oxaspiro[3.5]nonane and 2-azaspiro[3.5]nonane. The nitrogen or sulfur atom of the heterocyclyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S, S-dioxide.
Heterocyclyl having an aromatic ring containing heteroatoms are herein referred to by the customary term "heteroaryl". Within the context of the present invention and as used herein, the term "heteroaryl" refers to a 5- to 10-membered aromatic monocyclic or bicyclic ring system containing one to four heteroatoms independently selected from: nitrogen, sulphur, oxygen and phosporus. Representative examples of heteroaryls include, but are not limited to, pyrrole, pyrazole, imidazole, pyrazine, furan, thiophene, oxazole, thiazole, benzimidazole, benzoxazole, benzothiazole, benzofuran, indole, indazole, isoindole, isoquinoline, isooxazole, triazine, purine, pyridine, quinoline, oxadiazole, thiene, pyridazine, pyrimidine, isothiazole, quinoxaline (benzopyrine) and tetrazole. The nitrogen or sulfur atom of the heterocyclyl can be optionally oxidized to the corresponding N- oxide, S-oxide or S, S-dioxide.
A heterocyclyl or heteroaryl group can be unsubstituted or substituted with one or more groups independently selected from halogen, hydroxy, oxo, cyano,
(CrCe)-alkyl, halo(C C8)-alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3-C12)- cycloalkyl-(Rc)o-2, O-(C3-Ci2)-cycloalkyl-(Rc)0-2, (C6-Ci4)-aryl-(Rc)0-2, 0-(C6-Ci4)- aryl-(Rc)o-2, heterocyclyl-(Rc)0-2, O-heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(C Cs)-alkyl and S(0)qNRaRb; wherein Ra, Rb, Rc and q are as defined herein above. The substituents can be present on either the ring carbon or the ring nitrogen atom(s). The substituents can be present at one or more positions provided that a stable molecule results.
As used herein, the term "oxo" whether used alone or as part of a substituent group, refers to a group of formula (=0).
Within the context of this present invention and as used herein the term "isotopic forms" or "isotopically labeled forms" is a general term used for isotopic forms of the compounds of formula 1 , wherein one or more atoms of the compounds of formula 1 are replaced by their respective isotopes. All isotopes of any particular atom or element as specified are contemplated within the scope of the compounds of the present invention. Representative examples of isotopes that may be incorporated into the compounds disclosed herein include, but are not limited to, isotopes of hydrogen such as 2H (deuterium or D) and 3H (tritium or T), carbon such as 1 1C, 13C and 14C, nitrogen such as 13N and 15N, oxygen such as 150, 170 and 180, chlorine such as 36CI, fluorine such as 18F and sulphur such as 35S. Substitution with heavier isotopes, for example, replacing one or more key carbon-hydrogen bonds with carbon-deuterium bond may show certain therapeutic advantages, resulting from longer metabolism cycles (e.g., increased in vivo half life or reduced dosage requirements), improved safety or greater effectiveness and hence, may be preferred in certain circumstances.
Representative examples of isotopic forms of the compounds of formula 1 can include, without limitation, deuterated compounds of formula 1 . The term "deuterated" as used herein, by itself or used to modify a compound or group, refers to replacement of one or more hydrogen atom(s), which is attached to carbon(s), with a deuterium atom. For example, the compounds of formula 1 can include in the definitions of one or more of its various variables, wherever applicable, deuterium, deuterated-alkyl, deuterated-alkoxy, deuterated-cycloalkyl, deuterated-heterocyclyl, deuterated-aryl, deuterated-heteroaryl and the like.
The term "deuterated-alkyl" refers to an (CrC8)-alkyl group as defined herein, wherein at least one hydrogen atom bound to carbon is replaced by a deuterium. That is, in a deuterated alkyl group, at least one carbon atom is bound to a deuterium. In a deuterated alkyl group, it is possible for a carbon atom to be bound to more than one deuterium; it is also possible that more than one carbon atom in the alkyl group is bound to a deuterium. Analogously, the term "deuterated" and the terms deuterated-heterocyclyl, deuterated-heteroaryl, deuterated-cycloalkyl, deuterated-aryl, deuterated-alkoxy each refer to the corresponding chemical moiety wherein at least one carbon is bound to a deuterium.
The term "pharmaceutically acceptable solvate" or "solvates" as used herein refers to an aggregate of a molecule (in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof) with one or more solvent molecules. Such solvents for the purpose of the invention may not interfere with the biological activity of the molecule. Preferably, the solvent used is a pharmaceutically acceptable solvent. Examples of suitable pharmaceutically acceptable solvents include, without limitation, water, ethanol and acetic acid. Most preferably, the solvent used is water and the solvates obtained are referred to as hydrates. Examples for suitable solvates are the mono- or di-hydrates or alcoholates of the compounds of the present invention.
Within the context of the present invention and as used herein, the term "stereoisomer'Or "stereoisomeric form"is a general term used for all isomers of individual compounds (in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof) that differ only in the orientation of their atoms in space. The term stereoisomer includes mirror image isomers (enantiomers), mixtures of mirror image isomers (racemates, racemic mixtures), geometric (cis/trans or E/Z) isomers, and isomers of compounds with more than one chiral center that are not mirror images of one another (diastereoisomers).
The term "tautomer" refers to the coexistence of two (or more) compounds that differ from each other only in the position of one (or more) mobile atoms and in electron distribution, for example, keto-enol tautomers.
As used herein, the term "pharmaceutically acceptable" means that the carrier, diluent, excipients and/or salt must be compatible with the other
ingredients of the formulation (composition), and not deleterious to the recipient thereof.
The term "pharmaceutically acceptable salts" or "pharmaceutically acceptable salfas used herein includes salts of the active compoundi.e.the compound of formula 1 , which retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects; and are prepared with suitable acids or bases, depending on the particular substituents found on the compounds described herein.
Within the context of the present invention and as used herein the term "polymorph" or "pharmaceutically acceptable polymorph" or "polymorphic form" refers to crystals of the same compound (in the present invention, a compound of formula 1 )that differs only in the arrangement and/or conformation of the molecule in the crystal lattice.
Within the context of the present invention and as used herein, "a prodrug" or "prodrugs" refers toa compound, whichisa derivative of a parent compound(in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof), which following administration, releasesthe parent compoundin vivo via a chemical or physiological process, e.g., a prodrug on being brought to the physiological pHor through enzyme action is converted to the parent compound.
Within the context of the present invention and as used herein, "N-oxide" refers to an oxide of the nitrogen atom of a nitrogen-containing heteroaryl or heterocycle. N-oxide can be formed in the presence of an oxidizing agent such as m-chloro-perbenzoic acid or hydrogen peroxide. N-oxide refers to an amine oxide, also known as amine-N-oxide, and is a chemical compound that contains N-»0 bond.
Within the context of the present invention and as used herein "S-oxide" refers to the oxide of the sulfur atom (S-oxide) or dioxide of the sulfur atom (S, S- dioxide) of a sulfur-containing heteroaryl or heterocycle. S-oxide and S, S-dioxides can be formed in the presence of an oxidizing agent such as m-chloro-perbenzoic acid or oxone (potassium peroxymonosulfate).
Within the context of the present invention and as used herein interchangeably throughout this application, the terms "compound of formula 1 ", "compounds of formula 1 ", and "compounds of the present invention" includeall
the isotopic forms, stereoisomeric and tautomeric forms and mixtures thereof in all ratios, and the pharmaceutically acceptable salts, pharmaceutically acceptable solvates, pharmaceutically acceptable prodrugs, pharmaceutically acceptable polymorphs, N-oxides and S-oxides thereof.The compound(s) of the present invention can also be referred to herein as "the active compound" or "the active ingredient".
As used herein, the term "pharmaceutically acceptable carrier" refers to a material that is non-toxic, inert, solid, semi-solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type which is compatible with a subject, preferably a mammal, more preferably a human, and is suitable for delivering an active agent (in the present invention, a compound of formula 1 or a pharmaceutically acceptable salt thereof or any other form recited herein),to the target site without adversely affecting activity of the agent.
The term "subject" as used herein refers to an animal, preferably a mammal, and most preferably a human.The term "mammal" as used herein refers to warm-blooded vertebrate animals of the class Mammalia, including humans, characterized by a covering of hair on the skin and, in the female, milk-producing mammary glands for nourishing the young. The term mammal includes animals such as cat, dog, rabbit, bear, fox, wolf, monkey, deer, mouse, pig as well as human.The term "subject" may be used interchangeably with the term patient.ln the context of the present invention the phrase "a subject in need thereof" means a subject in need of the treatment for the disease or disorder that is mediated by EZH2. Alternatively, the phrase "a subject in need thereof" means a subject (patient) diagnosed having a disease or disorder that is mediated by EZH2.
The term, "therapeutically effective amount" as used herein means an amount of the compound of formula 1 or a pharmaceutically acceptable salt thereof or a composition comprising a compound of formula 1 , effective in producing the desired therapeutic response in a particular patient (subject) suffering from a disease or disorder to be treated. Particularly, the term "therapeutically effective amount" includes the amount of the compound of the present invention, when administered, that induces a positive modification in the condition (a disease or a disorder) to be treatedor is sufficient to prevent development of, or alleviate to some extent, one or more of the symptoms of the
disease or disorder being treated in a patient (subject). In respect of the therapeutic amount of the compound, consideration is also given that the amount of the compound used for the treatment of a subject is low enough to avoid undue or severe side effects, within the scope of sound medical judgment.The therapeutically effective amount of the compound or composition will vary with the particular condition being treated, the age and physical condition of the patient (subject in need of the treatment), the severity of the condition being treated/prevented, the duration of the treatment, the nature of concurrent therapy, the specific compound or composition employed and the particular pharmaceutically acceptable carrier utilized.
Through use of the terms "treatment", "treat" and "therapy" in the context of the present invention, it is intended to mean to alleviate, slow the progression, attenuation or cure of existing disease or condition (e.g., cancer). Treatment also includes treating the symptoms of the disease or condition. "Prevent or "Prevention", as used herein, refers to delaying, slowing, inhibiting, reducing or ameliorating the onset of the disease or disorder e.g. cancer.
As used herein, the term "EZH2 inhibitor" refers to an agent (in the present invention, a compound of formula 1 ) which is capable of inhibiting the increased expression of Histone-lysine N-methyl transferase EZH2 (enhancer of zeste homolog 2), which is a catalytic subunit of polycomb repressive complex 2 (PRC2), responsible for methylation of Lys27 of a specific histone H3 (H3K27) and essential for the self-renewal of cancer stem cells.
The term "disease or disorder mediated by EZH2" refers to an abnormal condition in a subject due to aberrant expression of the enzyme 'Histone-lysine N- methyltransferase EZH2' leading to abnormal epigenetic modifications.
The diseases or disorders mediated by EZH2 are selected from cancer, pulmonary arterial hypertension, myelofibrosis, human immunodeficiency virus (HIV) disease, graft versus host diseases, Weaver Syndrome, psoriasis vulgaris or liver fibrogenesis. The cancers mediated by EZH2 include, but are not limited to, thyroid carcinoma, cardiac sarcoma, lung carcinoma, gastrointestinal carcinoma, genitourinary tract carcinoma, liver carcinoma, mantle cell lymphoma, bone sarcoma, sarcoma of the nervous system, gynaecological carcinoma, haematological cancer, adrenal gland neuroblastoma, skin cancer, astrocytic
cancer, breast cancer, colorectal cancer, endometrial cancer, head and neck cancer and oral cancer.
Embodiments
The invention encompasses all the compounds described by the formula 1 without limitation, however, for the purposes of further illustrations, preferred aspects and elements of the invention are discussed herein in the form of the following embodiments.
In an embodiment, the present invention relates to a compound of formula
1 ,
wherein,
A

Xi and X2 are independently selected from CR6 and N; and
R3, R4 and R5 are as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates to a compound of formula 1 , wherein,

Xi and X2 are independently selected from CR6 and N;
R3 and R4 join together to form a 5 to 7 membered saturated or unsaturated monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; and
R5 is as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention relates to a compound of formula
1 ,
wherein,
A is:

Xi and X2 are CR6;
R3, R4, R5 and R6 are as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention relates to a compound of formula 1 , wherein,
A is:

Xi and X2 are independently selected from CR6 and N; and
R3, R4a, R5 and Re are as defined in the first aspect;
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates to a compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R3 and R4a join together to form a 5 to 7 membered saturated or unsaturated monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; and
R5 and R6 are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention provides a compound of formula 1 ,
wherein,
L is -C(0)NR7--,-NR7C(0)--,--NR7C(0)NR7--, -NR7S(0)r- or -S(0)rNR7-; wherein the dotted line (--) indicates the point of attachment of L to the group

where A, Ri, R2, R7, Q, n, r and p are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention relates to a compound of formula
1 ,
wherein,
L is --CH2NR7-,-NR7CH2--,-CH(halo-(Ci-C8)-alkyl)NR7-, -NR7CH(halo-(Ci-C8)- alkyl)-, -C(halo-(CrC8)-alkyl)2NR7-, -NR7C(halo-(CrC8)-alkyl)2-, - CH(halo)NR7-, -NR7CH(halo)--, -C(halo)2NR7- or -NR7C(halo)2--;wherein the dotted line (-) indicates the point of attachment of L to the group

where A, R1 ; R2, R7, Q, n and p are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention relates to a compound of formula
1 ,
wherein,
L is ; line (--) indicates the point o
 f attachment of L to the group respectively; where A, Ri , R2, R7, Q, n and p are as defined in the first aspect;
f attachment of L to the group respectively; where A, Ri , R2, R7, Q, n and p are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates to a compound of formula 1 ,
wherein,
Q is:

Wherein R8 and B are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates to a compound of formula 1 ,
wherein,
Q is:

wherein R8, R9 and B are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates to a compound of formula 1 ,
wherein,
Q is:

or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention relates to a compound of formula
1 ,
wherein,

Xi and X2 are independently selected from CR6 and N;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb,(C6-Ci4)-aryl- P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)-aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)- aryl, (C6-C14)ar-(CrC8)-alkyl, heterocyclyl or heteroaryl; and
Ra, Rb, Rc, R3, R4 and Reare as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention relates to a compound of formula
1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb, (C6-Ci4)-aryl- P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)-aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)- aryl, (C6-Ci4)-ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl ; and
Ra, Rb, Rc, R3, R4a and R6 are as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention relates to a compound of formula
1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
L is -C(0)NR7- or -NR7C( -; wherein the dotted line (-) indicates the point of attachment of L to the group
 respectively;
respectively;
Ri, R2, R3, R4, R5, R6, R7, Q, n and p are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In an embodiment, the present invention relates to a compound of formula
1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
L is -C(0)NR7- or -NR7C(0)-; wherein the dotted line (-) indicates the point of attachment of L to the group
 respectively;
respectively;
Ri , R2, R3, R4a, R5, R6, R7, Q, n and p are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb, (C6-d4)-aryl- P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)-aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)- aryl, (C6-Ci4)-ar-(Ci -C8)-alkyl, heterocyclyl or heteroaryl ;
L is ~C(0)NR7 ~, -NR7C(0)~, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~ ;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)- alkyl, (d-CeJ-alkoxy, (C3-Ci 2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;and
Ra, Rb, Rc, Ri , R2, R3, R6, R7, Re, B, p and r are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N ;
R4 is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0) RaRb, (C6-Ci4)-aryl-P(0) RaRb,(C6-Ci4)-aryl- P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0) RaRb, (C6-Ci4)-aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb) or (C6-Ci4)-aryl-C(Rc)2-heterocyclyl;
L is ~C(0)NR7 ~, -NR7C(0)~, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~ ;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:
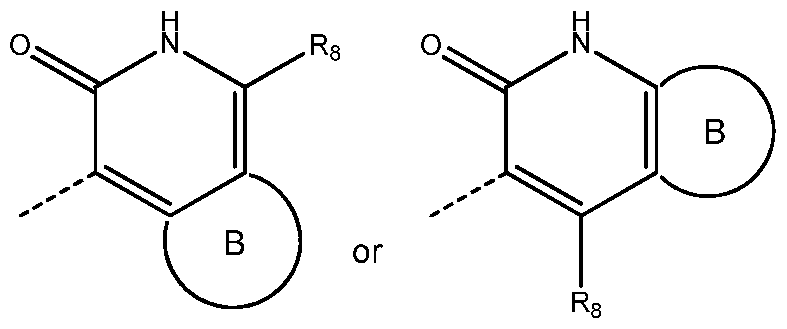
wherein each of the (C6-Ci4)-aryl and heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (C Cs)-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;and Ra, Rb, Rc, Ri , R2, R3, R6, R7, Re, Rc, P, r and B are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

X^ and X2 are independently selected from CR6 and N ;
R4 is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C8)-cycloalkenyl,(C6-Ci4)-aryl, heterocyclyl and heteroaryl;
R5 is (C6-Ci4)-aryl, (C6-Ci4)ar-(Ci -C8)-alkyl, heterocyclyl or heteroaryl;
L is ~C(0)NR7-, -NR7C(0)-, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~ ;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)- alkyl, (d-CeJ-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;
andRi , R2, R3, R6, R7, Re, P and B are as defined in the first aspect;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates to a compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb, (C6-Ci4)-aryl- P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)-aryl-heterocyclyl-Rc,
(C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)- aryl, (C6-Ci4)ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl;
L is -C(0)NR7-, -N RTC(0)~, ~NR7C(0)N R7 ~, ~NR7S(0)R ~ or ~S(0)RNR7 ~; wherein the dotted line (--) indicates the point of attachment of L to the group

wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-CeJ-alkyl, halo(Ci-Ce)- alkyl, (CrC8)-alkoxy, (C3-C12)-cycloalkyl, (C6-C14)-aryl, heterocyclyl and heteroaryl; and
Ra, Rb, Rc, Ri, R2, R3, R6, R7, Re, R9, Rc, P, r and B are as defined in the first aspect; an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb, (C6-Ci4)-aryl- P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)-aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb) or (C6-Ci4)-aryl-C(Rc)2-heterocyclyl;
L is ~C(0)NR7-, -NR7C(0)-, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~ ;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-Ci4)-aryl and heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-C8)-alkyl, halo(CrC8)-alkyl, (C C8)-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; and Ra, Rb, Rc, Ri , R2, R3, R6, R7, Re, R9, P, and B are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt,a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C8)-cycloalkenyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;
R5 is (C6-Ci4)-aryl, (C6-Ci4)ar-(Ci -C8)-alkyl, heterocyclyl or heteroaryl;
L is ~C(0)NR7-, -NR7C(0)-, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~ ;wherein the dotted line (--) indicates the point of attachment of L to the group x 'p and respectively;
n is 0;
Q is:

wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-C8)-alkyl, halo(C C8)- alkyl, (CrC8)-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; and
Ri , R2, R3, R6, R7, Re, R9, P and B are as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C8)-cycloalkenyl,(C6-Ci4)-aryl, heterocyclyl and heteroaryl;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb, (C6-Ci4)-aryl- P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0) RaRb, (C6-Ci4)-aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)- aryl, (C6-Ci4)-ar-(Ci -C8)-alkyl, heterocyclyl or heteroaryl;
L is ~C(0)NR7 ~, -N RTC(0)~, -NR7C(0)NR7-, ~NR7S(0)r ~ or ~S(0)rNR7 ~; wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(Ci -C8)- alkyl, (CrC8)-alkoxy, (C3-Ci 2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; and
Ra, Rb, Rc, Ri , R2, R3, R6, R7, r and p are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C8)-cycloalkenyl,(C6-Ci4)-aryl, heterocyclyl and heteroaryl;
R5 IS (C6-Ci4)-aryl, (C6-Ci4)ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl;
L is -C(0)NR7-,-NR7C(0)-, ~NR7C(0)NR7~, -NR7S(0)r- or -S(0)rNR7~; wherein the dotted line (--) indicates the point of attachment of L to the group

n is O;
Q is:

wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (d-CeJ-alkyl, halo(Ci-Ce)- alkyl, (CrC8)-alkoxy, (C3-C12)-cycloalkyl, (C6-C14)-aryl, heterocyclyl and heteroaryl; and
Ri, R2, R3, R6, R? and p are as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4a is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb, (C6-Ci4)-aryl- P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)-aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)- aryl, (C6-Ci4)-ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl;
L is ~C(0)N R7-, -NR7C(0)-, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7~ ;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-C14)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(Ci-C8)- alkyl, (CrC8)-alkoxy, (C3-Ci 2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; and
Ra, Rb, Rc, Ri , R2, R3, R6, R7, Re, p, r and B are as defined in the first aspect; or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N ;
R4a is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C8)-cycloalkenyl,(C6-Ci4)-aryl, heterocyclyl and heteroaryl;
R5 is (C6-Ci4)-aryl, (C6-Ci4)-ar-(Ci -C8)-alkyl, heterocyclyl or heteroaryl;
L is -C(0)NR7-,-N RTC(0)~, -N R7C(0)NR7-, ~NR7S(0)R- or -S(0)rNR7-
;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-C14)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(Ci -C8)- alkyl, (CrC8)-alkoxy, (C3-Ci 2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; and
Ri , R2, R3, R6, R7, Re, p and B are as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates toa compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N ;
R4a is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C8)-cycloalkenyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; R5 is the group consisiting of P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl- P(0)RaRb,(C6-Ci4)-aryl-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)- aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2- heterocyclyl, (C6-Ci4)-aryl, (C6-Ci4)-ar-(Ci -C8)-alkyl, heterocyclyl and heteroaryl; L is ~C(0)N R7-, -NR7C(0)-, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~ ;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:
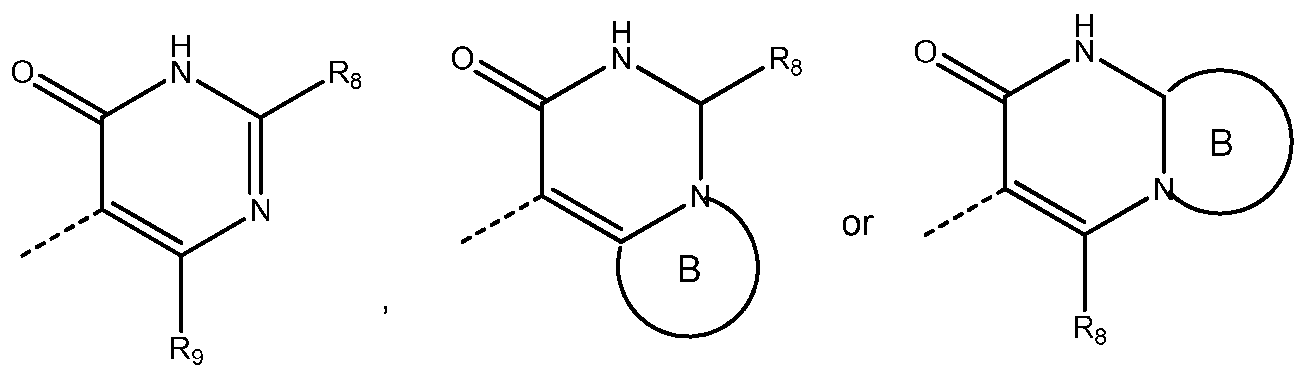
wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(Ci -C8)- alkyl, (CrC8)-alkoxy, (C3-Ci 2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; and
Ra, Rb, Rc, Ri , R2, R3, R6, R7, Rs, R9, P, r and B are as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
In another embodiment, the present invention relates to a compound of formula 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4a is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C8)-cycloalkenyl,(C6-Ci4)-aryl, heterocyclyl and heteroaryl;
R5 is (C6-Ci4)-aryl, (C6-Ci4)-ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl;
L is -C(0)NR7-, -NR7C(0)-, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~
;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:
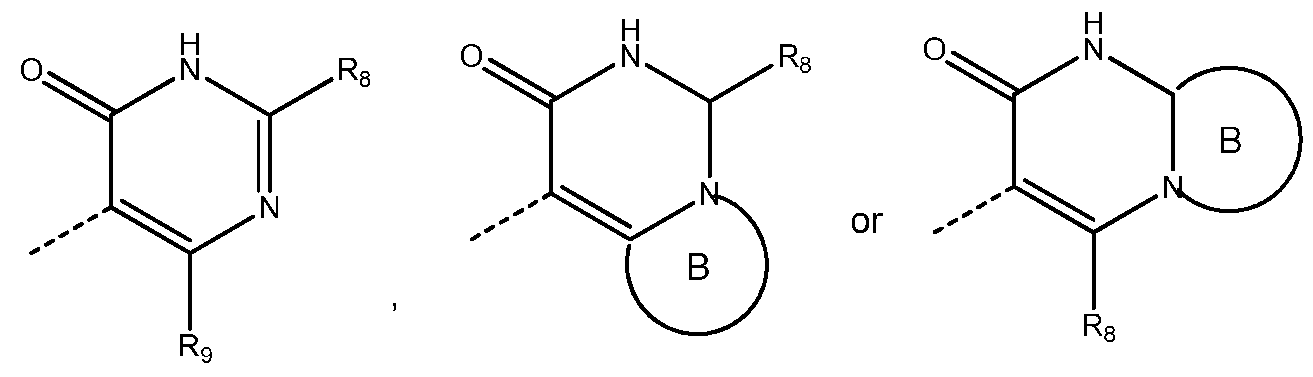
wherein each ofthe (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C8)-alkyl,halo(Ci-C8)-alkyl, (CrC8)- alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; and
R1 5 R2, R3, R6, R7, R8, R9, p, r and B are as defined in the first aspect; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
Representative compounds of the present invention include:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-fluoro-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8- hexahydroisoquinolin-4-yl)methyl)-4'-(1 -morpholinoethyl)-[1 , 1 '-biphenyl]-3- carboxamide;
3'-((Dimethylamino)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-fluoro-N- ((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3- carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((4-methyl-2-oxo-3, 5,6,7,8,9- hexahydro-2H-cyclohepta[c]pyridin-1 -yl)methyl)-4'-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo-3, 5,6,7,8,9- hexahydro-2H-cyclohepta[c]pyridin-1 -yl)methyl)-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo-3, 5,6,7,8,9- hexahydro-2H-cyclohepta[c]pyridin-1 -yl)methyl)-5-(6-morpholino pyridin-3- yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo-3, 5,6,7,8,9- hexahydro-2H-cyclohepta[c]pyridin-1 -yl)methyl)-5-(6-(trifluoro methyl)pyridin-3- yl)benzamide;
3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(2-methoxypyrimidin-5-yl)-2-methyl-N-
((1 -methyl-3-oxo-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-4- yl)methyl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((8-methyl-6-oxo-3,4,6,7- tetrahydro-1 H-pyrano[3,4-c]pyridin-5-yl)methyl)-4'-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((8-methyl-6-oxo-3,4,6,7- tetrahydro-1 H-pyrano[3,4-c]pyridin-5-yl)methyl)-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((8-methyl-6-oxo-3,4,6,7- tetrahydro-1H-pyrano[3,4-c]pyridin-5-yl)methyl)-5-(6-morpholinopyridin-3- yl)benzamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-N-((3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1'-biphenyl]-3-carboxamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-morpholino-N-((3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1'-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1- yl)pyridin-3-yl)-N-((3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide;
N-((1-ethoxy-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-
(et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-[1 ,1'- biphenyl]-3-carboxamide;
N-((1-ethoxy-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-3-
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1- yl)pyridin-3-yl)benzamide;
N-((1, 7-dimethyl-3-oxo-2, 3,5,6, 7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5- (et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-[1 ,1'- biphenyl]-3-carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1 -methyl-3-oxo-2,3, 5,6,7,8- hexa ydro-2,7-nap t yridin-4-yl)met yl)-4-met yl-4'-(morpholinomet yl)-[1 ,1'- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1-methyl-3-oxo-2,3, 5,6,7,8- exa ydro-2,7-nap t yridin-4-yl)met yl)-2-met yl-5-(6-(4-met ylpiperazin-1- yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1-methyl-3-oxo-2,3, 5,6,7,8- exa ydro-2,7-nap t yridin-4-yl)met yl)-2-met yl-5-(6-morpholinopyridin-3- yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-7-propyl- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4,-(morpholinomethyl)-[1,1'- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1-methyl-3-oxo-7-propyl-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6-morpholinopyridin-3- yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-propyl-
2,3,5,67 -hexahydro-27-naphthyridin-4-yl)methyl)-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
N-((7-butyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5 (et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-[1 ,1 '- biphenyl]-3-carboxamide;
N-((7-butyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-3
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
N-((7-butyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-3
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-morpholinopyridin-3- yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1 -methyl-3-oxo- 2,3,5,6,7,8-hexa ydro-2 ,7-nap t yridin-4-yl)met yl)-4-met yl-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-(4-methyl piperazin-1 -yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1 -methyl-3-oxo-
2,3,5,67 -hexahydro-27-naphthyridin-4-yl)methyl)-2-methyl-5-(6-morpholino pyridin-3-yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo- 2,3,5,6,7,8-hexa ydro-2 ,7-nap t yridin-4-yl)met yl)-4-met yl-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-(4- methylpiperazin-1 -yl)pyridin-3-yl)benzamide;
3- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6- morpholinopyridin-3-yl)benzamide;
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin
4- yl)met yl)-5-(et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide;
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin
4-yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4- methylpiperazin-1 -yl)pyridin-3-yl)benzamide;
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin
4- yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6- morpholinopyridin-3-yl)benzamide;
5- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-7-(oxetan-3-yl)-3 oxo-2,3,5,6,7,8-hexa ydro-2 ,7-nap t yridin-4-yl)met yl)-4'-(morpholinomet yl)- [1 ,1 '-biphenyl]-3-carboxamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-N-((3- oxo-1 -(trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)-N-((3-oxo-1 -(trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4- yl)methyl)benzamide;
3-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-2-met yl-5-(6-morpholinopyridin-3-yl)-N ((3-0X0-1 -(trifluoromethyl)-2, 3, 5,6,7,8-hexahydroisoquinolin-4- yl)methyl)benzamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-N-((3- oxo-1 -(trifluoromethyl)-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-4- yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)-N-((3-oxo-1 -(trifluoromethyl)-3,5,6,7,8,9-hexahydro-2H- cyclohepta[c]pyridin-4-yl)methyl)benzamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-N-((3- oxo-1 -(trifluoromethyl)-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-
[1 ,1 '-biphenyl]-3-carboxamide;
4- Methyl-N-((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'- (morpholinomethyl)-5-(2-oxoazepan-1 -yl)-[1 ,1 '-biphenyl]-3-carboxamide;
Dimethyl (4'-methyl-3'-(((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4- yl)methyl)carbamoyl)-5'-(2-oxoazepan-1 -yl)-[1 ,1 ,-biphenyl]-4-yl)phosphonate;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(5 luoro-6-morpholinopyridin-3-yl)-2- methyl-N-((1 -methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-3'-fluoro-4-met yl-N-((1 -rTiet yl-3-oxo- 3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-4'-morpholino-[1 ,1 '- biphenyl]-3-carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-3, 5,6,7- tetrahydro^H-cyclopentalclpyridin^-y methy ^'-^etrahydro^H-pyran^-y -II J '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(1 H-indol-4-yl)-2-methyl-N-((1 -methyl- 3-oxo-3,5, 6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl) benzamide;
3-(2-(Dimet ylamino)pyrimidin-5-yl)-5-(et yl(tetra ydro-2H-pyran-4-yl)amino)-N- ((1 -methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(6-fluoro-5-methylpyridin-3-yl)-2- methyl-N-((1 -methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(1 H-indol-5-yl)-2-methyl-N-((1 -methyl- 3-oxo-3,5, 6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl) benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-3, 5,6,7- tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-5-(quinolin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-3, 5,6,7- tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-5-(6-(morpholinomethyl) pyridin-3- yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-7- (trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-
(trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(6-morpholino pyridin-3-yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-7-(2,2,2- trifluoroethyl)-2, 3,5,6, 7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-(2,2,2- trifluoroethyl)-2, 3,5,6, 7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6-(4- methylpiperazin-1 -yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-(2,2,2- trifluoroethyl)-2, 3,5,6, 7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6- morpholinopyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(6-morpholinopyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(6-methylpyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(6-(trifluoromethyl)pyridin-3-yl)benzamide; 5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-4'-morpholino-[1 ,1 '-biphenyl]-3-carboxamide; Dimethyl (3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4,-methyl-5'-(((1 -methyl-3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 , 1 '-biphenyl]-3- yl)phosphonate;
(S)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(1 -(piperidin-1 -yl)ethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
(R)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(1 -(piperidin-1 -yl)ethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-4'-(1 -(piperidin-1 -yl)ethyl)-[1 ,1 '-biphenyl]-3- carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-4'-(2-morpholinoethyl)-[1 , 1 '-biphenyl]-3- carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-3'-(1 -(pyrrolidin-1 -yl)ethyl)-[1 ,1 '-biphenyl]-3- carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(4-methyl-3-oxo-3,4-dihydro-2H- benzo[b][1 ,4]oxazin-6-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(1 ,3,5-trimethyl-1 H-pyrazol-4-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(6-(pyrrolidin-1 -yl)pyridin-3-yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-4'-(2-(piperidin-1 -yl)propan-2-yl)-[1 ,1 '-biphenyl]
3-carboxamide;
Ethyl 2-(3-(3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4,-methyl-5'-(((1 -methyl-3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 , 1 '-biphenyl]-4- yl)oxetan-3-yl)acetate;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-hydroxyoxetan-3-yl)-2-methyl-N-((1 methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-methoxyoxetan-3-yl)-2-methyl-N-
((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro isoquinolin-4-yl)methyl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-hydroxyoxetan-3-yl)-N-((7- isopropyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2- methylbenzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4,-formyl-4-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
Diethyl (3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-5-(((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)phenyl)phosphonate;
5-(Dimethylphosphoryl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 - methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide;
Diethyl (((4-chlorophenyl)amino)(3,-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4'- methyl-5'-(((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl) carbamoyl)-[1 ,1 '-biphenyl]-4-yl)methyl)phosphonate;
Diethyl ((3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4,-methyl-5'-(((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 ,1 '-biphenyl]-4- yl)(morpholino)methyl)phosphonate and;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2,3,5,^ hexahydroisoquinolin-4-yl)metriyl)-4,-((4-metriyl-4-oxido-1 ,4-azaphosphinan-1 -yl) methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
or a stereoisomer, a tautomer, or a pharmaceutically acceptable salt thereof.
According to another aspect of the present invention, there are provided processes for the preparation of the compounds of formula 1 . The compounds of formula 1 can be prepared using various methods including the methods well known to a person skilled in the art. Representative processes to prepare the compounds of the present invention are described below, and are particularly illustrated in Scheme 1 , but are not limited thereto. It will be appreciated by persons skilled in the art that within certain of the processes described herein, the order of the reaction steps employed can be varied and will depend inter alia on factors such as the nature of functional groups present in a particular substrate (starting compound or an intermediate) and the protecting group strategy (if any) to be adopted. Clearly, such factors will also influence the choice of reagent to be used in the reaction steps.
The reagents, reactants and intermediates used in the following processes are either commercially available or can be prepared according to standard procedures known in the art, for instance those reported in the literature references. In the following schemes and the description of the processes for the synthesis of the compounds of formula 1 , the starting compounds and the intermediates that are used for the synthesis of the compounds of the present invention, are designated as compounds 2 and 3 for ease of reference. Unless stated otherwise, throughout the process description, the corresponding substituent groups in the various formulae representing starting compounds and intermediates have the same meaning as that for the compound(s) of formula 1 , unless stated otherwise.
Processes for the preparation of the compounds of formula 1 of the present invention are depicted in scheme 1 presented below. For ease of reference, the reaction steps shownin the Scheme 1 , are referred to as "step a" and "step b" respectively.
Scheme 1 depicts a process for the preparation of the compound of formula 1 ,whereinl_ is -C(0)NR7-, n is 0, A is:

Xi , X2, Ri , R2, R3, R4, R5, p and Q are as defined in the first aspect of the present invention.
Scheme 1


(compound of formula 1 ) wherein, L is -C(0)NR7-;n is 0;

Xi , X2, Ri , R2, R3, R4, R5, P and Q are as defined in the first aspect.
Reaction step a:
Preparation of compound of formula 3:
Ν,Ν-Diisopropylethylamine and 2-(7-aza-1 H-benzotriazole-1 -yl)-1 , 1 ,3,3- tetramethyl uronium hexafluorophosphate (HATU) are added to a solution of compound of formula 2 (wherein R3 and R4 are as defined for the compounds of formula 1 in the first aspect of the invention) in a solvent such as N, N-
dimethylformamide (DMF). The reaction mixture is stirred for 1 h at room temperature.To this reaction mixture, compound of formula E,
 HCI
HCI
E
(wherein Q, p, R1 5 R2 and R7 are as defined for the compounds of formula 1 in the first aspect of the invention) is added and the resulting mixture is stirred for 1 2 h at room temperature, which results in the formation of the compound of formula 3 (wherein L is -C(0)NR7-; Q, p, R1 ; R2, R3 and R4 are as defined for the compounds of formula 1 in the first aspect of the invention).
Reaction step b:
Preparation of compound of formula 1 :
Compound of formula 3 (as obtained in reaction step a) is reacted with compound of formula F,

F
(wherein R5 is as defined for the compounds of formula 1 , in the first aspect of the invention) in the presence of a base such as sodium carbonate in a solvent or a mixture of solvents such as a mixture of dioxane and water. The reaction mixture is purged with argon for ten minutes to fifteen minutes followed by addition of 1 , 1 '- bis(diphenylphosphino)ferrocene palladium-dichloromethane adduct, which is followed by purging with argon again for another 15 minutes to 20 minutes. The reaction mass obtained is heated at a temperature ranging from 90 °C to 120 °C for 5 h to 9 h, which results in the formation of the compound of formula 1 , wherein L is -C(0)NR7-; Q, p, Ri , R2, R3, R4, Rs and R7 are as defined in the first aspect of the present invention.
The compound of formula 1 , wherein A is:

can be prepared by a method analogous to the reaction steps a and b depicted in scheme 1 above, starting with compound of formula 2A,

2A
wherein X-\ , X2, R3, R4a and R5 are as defined in the first aspect.
The compounds of formula 1 , as obtained in Scheme 1 can be optionally converted into their corresponding pharmaceutically acceptable salts.
The term "pharmaceutically acceptable salt(s)", as used herein, means those salts of compounds of the formula 1 which retain the efficacy and the biological properties of the free bases or of the free acids and are safe for use in mammals. In the context of the present invention, the pharmaceutically acceptable salts include organic and inorganic salts of the compounds of the invention (the compounds of formula 1 ) depending on the particular group (acidic or basic group) present in the said compounds. When compounds of the present invention contain relatively basic groups, acid addition salts can be obtained. The pharmaceutically acceptable acid addition salts include those derived from inorganic acids, which are not limited to, hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, mono-hydrogensulfuric or hydroiodic acids and the like; as well as the salts derived from organic acids such as acetic, ascorbic, benzenesulfonic,benzoic, citric, ethanesulfonic, formic, fumaric, galacturonic, gluconic, glucuronic, glutamic, isobutyric, isonicotinic, lactic, maleic, malonic, mandelic, methanesulfonic, 4-methylbenzenesulfonic, nicotinic, oxalic, pantothenic, phthalic, propionic, saccharic, succinic, suberic, p-tolylsulfonic, tartaric acids and the like. When compounds of the present invention contain
relatively acidic groups, base addition salts can be obtained. Examples of pharmaceutically acceptable organic base addition salts of the compounds of the present invention include those derived from organic bases selected from lysine, arginine, guanidine, diethanolamine, choline, tromethamine, metformin and the like. Examples of pharmaceutically acceptable base addition salts of the compounds of the present invention include their alkali metal salts. Suitable alkali metal salts include, but are not limited to, aluminum, calcium, lithium, magnesium, potassium, sodium or zinc salts.
The pharmaceutically acceptable salts of the present invention can be synthesized from the compound of formula 1 , which contains a basic or an acidic group, by using conventional chemical methods. Generally, the salts are prepared by treating the compound of formula 1 , which may be a free base or an acid with a suitable salt-forming inorganic or organic acid or a base in a suitable solvent or dispersant or from another salt by cation or anion exchange. Suitable solvents that can be used for the preparation of pharmaceutically acceptable salts include, but are not limited to, ethyl acetate, diethyl ether, methanol, ethanol, acetone, tetrahydrofuran, dioxane or mixtures of these solvents.
The compounds of formula 1 can be regenerated by contacting ^pharmaceutically acceptable salt with a base or an acid depending on the type of saltand isolating the parent compound in the conventional manner.
The present invention also encompasses within its scope the solvates of the compounds of formula 1 .
Certain compounds of the present invention can exist in unsolvated forms as well as solvated forms, including hydrated forms. Certain compounds of the present invention can exist in multiple crystalline or amorphous forms. In general, all physical forms are suitable for the uses contemplated by the present invention and are intended to be within the scope of the present invention.
Various polymorphs of the compound of formula 1 can be prepared by crystallization of the compound under different conditions. The different conditions are, for example, using different solvents or their mixtures for carrying out crystallization; crystallization at different temperatures; various modes of cooling, ranging from very fast to very slow cooling during crystallization or by heating or melting the compound followed by gradual or fast cooling. The presence of
polymorphs can be determined by IR (Infra-red) spectroscopy, solid probe NMR (Nuclear Magnetic Resonance) spectroscopy, differential scanning calorimetry, powder X-ray diffraction or such other techniques.
Those skilled in the art will recognize that stereocentres exist in the compounds of formula 1 . Accordingly, the present invention includes all possible stereoisomers and geometric isomers of the compound of formula 1 and includes not only racemic compounds but also the optically active isomers as well. When a compound of formula 1 is desired as a single enantiomer, it may be obtained either by resolution of the final product or by stereospecific synthesis from either isomerically pure starting material or an appropriate intermediate. Resolution of the final product, an intermediate or a starting material may be effected by any suitable method known in the art, for example, Chiral reagents for asymmetric synthesis by Leo A. Paquette; John Wiley & Sons Ltd (2003).
Additionally, in situations wherein tautomers of the compounds of formula 1 are possible, the present invention is intended to include all tautomeric forms of the compounds.
The present invention also encompasses within its scope prodrugs of the compound of formula 1 . Preferably prodrugs are those compounds that are converted to their parent compound intracellular^, where the cellular converting location is the site of therapeutic action. The prodrugs of the compounds of the present invention are derivatives, particularly simple derivatives of the said compounds which upon administration to a subject in need thereof undergo conversion by metabolic or chemical processes to release the parent drug in vivo from which the prodrug is derived. The preferred prodrugs are pharmaceutically acceptable ester derivatives e.g., alkyl esters, cycloalkyl esters, alkenyl esters, benzyl esters, mono- or di-substituted alkyl esters that are convertible by solvolysis under physiological conditions to the parent carboxylic acid (e.g. the compound of formula 1 containing the carboxylic acid group), and those conventionally known in the art.
In another aspect, the present invention furthermore relates to pharmaceutical compositions containing a therapeutically effective amount of at least one compound of formula 1 or a pharmaceutically acceptable salt thereof; in addition to a customary pharmaceutically acceptable carrier or excipient. The
present invention also relatesto a process for the production of a pharmaceutical composition, which includes bringing at least one compound of formula 1 , into a suitable administration form using a pharmaceutically acceptable excipient and, if appropriate, further suitable additives or auxiliaries.
Accordingly, in an aspect, the present invention relates to a pharmaceutical composition comprising at least one compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable excipient.
The pharmaceutical composition(s) of the present invention can be administered orally, for example in the form of pills, tablets, coated tablets, capsules, granules or elixirs. Administration, however, can also be carried out rectally, for example in the form of suppositories, or parenterally, for example intravenously, intramuscularly or subcutaneously, in the form of injectable sterile solutions or suspensions, or topically, for example in the form of ointments or creams or transdermal^, in the form of patches, or in other ways, for example in the form of aerosols or nasal sprays.
The pharmaceutical composition(s) according to the invention are prepared in a manner known and familiar to one skilled in the art. Pharmaceutically acceptable inert inorganic and/or organic carriers and/or additives can be used in addition to the compound of formula 1 , or a stereoisomer, a tautomer or a pharmaceutically acceptable saltfor the production of oral dosage forms of compounds of formula 1 such as pills, tablets, coated tablets and hard gelatin capsules. It is possible to use, for example, lactose, corn starch or derivatives thereof, gum arabica, magnesia or glucose, etc. Carriers for soft gelatin capsules and suppositories are, for example, fats, waxes, natural or hardened oils, etc. Suitable carriers for the production of solutions, for example injection solutions, or of emulsions or syrups are, for example, water, physiological sodium chloride solution or alcohols, for example, ethanol, propanol or glycerol, sugar solutions, such as glucose solutions or mannitol solutions, or a mixture of the said solvents.
The pharmaceutical compositions normally contain about 1 % to 99 %, for example, about 5 % to 70 %, or from about 10 % to about 30 % by weight of the compound of formula 1 or its pharmaceutically acceptable salt. The amount of the compound of formula 1 or its pharmaceutically acceptable salt in the
pharmaceutical compositions normally can be from about 5 mg to 500 mg or can be lower than or higher than the lower and the upper limit respectively. The dose of the compound of formula 1 , which is to be administered, can cover a wide range depending on the type of disease or disorder to be treated. The dose to be administered daily is to be selected to suit the desired effect. A suitable dosage can be about 0.01 mg/kg to 100 mg/kg of the compound of formula 1 or its pharmaceutically acceptable salt depending on the body weight of the recipient (subject) per day, for example, about 0.1 mg/kg/day to 50 mg/kg/day of a compound of formula 1 or itspharmaceutically acceptable salt. If required, higher or lower daily doses can also be administered.
The selected dosage level will depend upon a variety of factors including the activity of a compound of the present invention, or its salt employed, the route of administration, the time of administration, the rate of excretion of the particular compound being administered, the duration of the treatment, other concurrently administered drugs, compounds and/or materials, the age, sex, weight, condition, general health and prior medical history of the patient (subject) being treated, and like factors well known in the medical arts.
In addition to the compound of formula 1 or its pharmaceutically acceptable salt and the pharmaceutically acceptable carrier substances, the pharmaceutical compositions of the present invention can contain additives such as, for example, fillers, antioxidants, dispersants, emulsifiers, defoamers, flavors, preservatives, solubilizers or colorants. The pharmaceutical compositionscan also contain more than one compound of formula 1 or their pharmaceutically acceptable salts. Furthermore, in addition to at least one compound of formula 1 or its pharmaceutically acceptable salt, the pharmaceutical compositions can also contain one or more other therapeutically or prophylactically active agents.
The present invention also encompasses within its scope the use of a compound of formula 1 or its pharmaceutically acceptable salt in combination, with other therapeutically active agents; wherein the compound of formula 1 and the further therapeutic agent are administered either simultaneously or sequentially.
The therapeutically active agents used in combination with one or more compounds of formula 1 or its pharmaceutically acceptable salt can be selected
from: anti-neoplastic agents or chemotherapeutic compounds such as anti- microtubule agents (diterpenoids (paclitaxel, docetaxel) and vinca alkaloids (vinblastine, vincristine, vinorelbine)); platinum coordination complexes (cisplatin, carboplatin), alkylating agents (nitrogen mustards (oxazaphosphorines, cyclophosphamide, melphalan, chlorambucil)); alkyl sulfonates (busulfan); nitrosoureas (carmustine); triazenes (dacarbazine); topoisomerase I inhibitors (camptothecins (irinotecan, topotecan)); topoisomerase II inhibitors (epipodophyllotoxins (etoposide, teniposide)); antimetabolite neoplastic agents (fluorouracil, methotrexate, cytarabine, mecaptopurine, thioguanine, gemcitabine); hormones and hormonal analogues (retinoids, histone deacetylase inhibitors); DNA methyltransferase inhibitors (azacitidine, decitabine); non-receptor tyrosine kinase angiogenesis inhibitors (endostatin, angiostatin); antibiotics (daunorubicin, doxorubicin, bleomycin); inhibitors of growth factor receptors (VEGFR inhibitors (pazopanib, ZD6474 (vandetanib, AstraZeneca), AZD2171 (cediranib, Astrazeneca), vatalanib, sunitinib and sorafenib), trastuzumab, cetuximab, bevacizumab, lapatinib, erlotinib, gefitinib, imatinib mesylate, ibrutinib, dasatinib),cell cycle signaling inhibitors (CDK inhibitors (ABT-751 (Eisai), veliparib)), PI3K inhibitors (Idelalisib (phase III) or Bcl-2-inhibitors (navitoclax (phase II), gossypol (phase II), obatoclax (phase III)).
In one aspect, the compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof; is an EZH2 inhibitor. In another aspect, the compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof; is provided for use the treatment of a disease or a disorder mediated by EZH2.
In one aspect, the present invention relates to a method for the treatment of a disease or a disorder mediated by EZH2, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof.
In another aspect, the present invention relates to use of a compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof; for the treatment of a disease or a disorder mediated by EZH2.
In yet another aspect, the present invention relates to use of a compound of formula 1 or a stereoisomer, a tautomer or a pharmaceutically acceptable salt thereof; for the manufacture of a medicament for the treatment of a disease or a disorder mediated by EZH2.
In an embodiment, the disease or disorder mediated by EZH2 is selected from cancer, pulmonary arterial hypertension, myelofibrosis, human immunodeficiency virus (HIV) disease, graft versus host diseases (GVHD), Weaver Syndrome, psoriasis vulgaris or liver fibrogenesis.
In an embodiment of the present invention, the disease or disorder mediated by EZH2 is cancer. Cancers also include metastatic or malignant tumors.
In an embodiment, cancers that can be treated by the compound of formula 1 of the invention or pharmaceutical compositions containing the said compounds; are selected from: thyroid carcinoma, cardiac sarcoma, lung carcinoma, gastrointestinal carcinoma, genitourinary tract carcinoma, liver carcinoma, mantle cell lymphoma, bone sarcoma, sarcoma of the nervous system, gynaecological carcinoma, haematological cancer, adrenal gland neuroblastoma, skin cancer, astrocytic cancer, breast cancer, colorectal cancer, endometrial cancer, head and neck cancer or oral cancer.
According to an embodiment of the present invention, the cancer is cardiac sarcoma selected from angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma, rhabdomyoma, fibroma, lipoma or teratoma.
According to another embodiment of the present invention, the cancer is lung carcinoma selected from squamous cell carcinoma, undifferentiated small or large cell carcinoma, adenocarcinoma, bronchiolar carcinoma, bronchial adenoma, bronchial sarcoma or bronchial lymphoma.
According to yet another embodiment of the present invention, the cancer is gastrointestinal carcinoma selected from stomach carcinoma, stomach lymphoma, pancreatic carcinoma (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma), small bowel carcinoma (adenocarcinoma, lymphoma, Kaposi's sarcoma, hemangioma, lipoma, neurofibroma, fibroma) or large bowel carcinoma (adenocarcinoma, tubular adenoma).
According to another embodiment of the present invention, the cancer is genitourinary tract carcinoma selected from carcinoma of kidney (adenocarcinoma, nephroblastoma, lymphoma, leukemia), malignant rhabdoid tumor of kidney, carcinoma of bladder and urethra (squamous cell carcinoma, adenocarcinoma), carcinoma of prostate (adenocarcinoma, sarcoma), or carcinoma of testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, lipoma).
According to yet another embodiment of the present invention, the cancer is liver carcinoma selected from hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma, angiosarcoma or hepatocellular adenoma.
According to yet another embodiment of the present invention, the cancer is bone sarcoma selected from osteogenic sarcoma (osteosarcoma), fibrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma or giant cell tumors.
According to another embodiment of the present invention, the cancer is an oral cancer selected from tongue cancer, squamous cell carcinoma, Kaposi's sarcoma, teratoma, adenocarcinoma derived from a major or minor salivary gland, lymphoma from tonsillar or other lymphoid tissue, or melanoma from the pigment- producing cells of the oral mucosa.
According to another embodiment of the present invention, the cancer is sarcoma of the nervous system selected from sarcoma of skull (osteoma, granuloma, xanthoma), meninges (meningioma, meningiosarcoma, gliomatosis), sarcoma of brain (astrocytoma, medulloblastoma, glioma, glioblastoma multiform, oligodendroglioma, retinoblastoma, congenital tumors), sarcoma of spinal cord (neurofibroma, meningioma, glioma, sarcoma) or malignant rhabdoid tumor of brain.
According to yet another further embodiment of the present invention, the cancer is carcinoma of gynaecological organs selected from carcinoma of uterus (endometrial carcinoma), carcinoma of cervix (cervical carcinoma, ovary carcinoma), carcinoma of vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), carcinoma of vagina
(clear cell carcinoma, squamous cell carcinoma, embryonal rhabdomyosarcoma) or carcinoma of fallopian tubes.
According to another embodiment of the present invention, the cancer is haematological cancer selected from blood cancer (acute myeloid leukemia (AML), chronic myeloid leukemia (CML), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplastic syndrome, B-cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, Hodgkin's lymphoma (Hodgkin's disease), non-Hodgkin's lymphoma (malignant lymphoma) or mantle cell lymphoma.
According to another embodiment of the present invention, the cancer is a skin cancer selected from malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Kaposi's sarcoma, angioma or dermatofibroma.
It is understood that modifications that do not substantially affect the activity of the various aspects of this invention are included. Accordingly, the following examples are intended to illustrate but not to limit the present invention.
EXAMPLES
The following abbreviations or terms are used herein:
AIBN Azobisisobutyronitrile
CHCI3 Chloroform
CDC Deuteriated chloroform
BSA Bovine serum albumin
DCE Dichloroethane
DCM Dichloromethane
DIEA N,N-Diisopropylethylamine
DIPEA N,N-Diisopropylethylamine
DMF N, N-dimethylformamide
DMSO Dimethylsulfoxide
DTT Dithiothreitol
EtOH Ethanol
EtOAc Ethyl acetate
Fe Iron
g Gram
h Hour
HATU : 2-(7-Aza- lH-benzotriazole- 1 -yl)- 1,1,3,3- tetramethyluronium
hexafluorophosphate)
HCI Hydrochloric acid
LDA Lithium diisopropylamide
L Litre
M Molar
mg Milligram
min Minute(s)
ml_ r/li Mi litre
μΙ- Microlitre
μΜ Micromolar
mmol Millimolar
MeOH Methanol
nM Nanomolar
ng Nanogram
Na2C03 Sodium carbonate
NaHC03 Sodium bicarbonate
NaOH Sodium hydroxide
NaCI Sodium chloride
NH4CI Ammonium chloride
°C Degree Centigrade
Pd-C Palladium-carbon catalyst
psi Pounds per square inch
RT Room temperature (20-35 °C)
TFA Trifluoroacetic acid
THF Tetrahydrofuran
Tris-HCI 2-Amino-2-hydroxymethyl-1 ,3-propanediol hydrochloride
PdCI2(dppf)-CH2CI2 [1 ,1 '-Bis(diphenylphosphino)ferrocene]dichloro palladium(ll),complex with dichloromethane
Example 1 :
1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinoline-4-carbonitrile
To a solution of 2-acetylcyclohexanone (20 g, 143 mmol) in 150 mL of ethanol were added 2-cyanoacetamide (12.00 g, 143 mmol) and piperidine (70.6 mL, 713 mmol) and the reaction mixture was heated for 12 h at 70 °C. After the reaction was complete, the reaction mixture was cooled to room temperature and filtered, followed by an ethanol wash to obtain the title compound.
Yield: 16 g (59.6 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.1 1 (s, 1 H), 2.71 (s, 2H), 2.36 (s, 2H), 2.20 (s, 3H) 1 .66 (br s, 4H); MS (ESI+) m/z 189.1 (M+H).
Example 2:
4-(Aminomethyl)-1-methyl-5,6,7,8-tetrahydroisoquinolin-3(2H)-one
hydrochloride
To a solution of 200 mLof acetic acid was added Pd-C (1 .696 g, 15.94 mmol) into a Parr bottle. To this were added 1 -methyl-3-oxo-2, 3, 5,6,7,8- hexahydroisoquinoline-4-carbonitrile (10 g, 53.1 mmol), sodium acetate (8.72 g, 106 mmol), and platinum oxide (362 mg, 1 .594 mmol). The bottle was capped and placed on a parr shaker under an atmosphere of 90 psi to 95 psi hydrogen for 2 days. The reaction mixture was filtered. The solvent was removed to give a residue, which was treated with 50 mL of concentrated HCI, and the resulting solids were filtered. The yellow filtrate was concentrated. To the crude compound was added 10 mL of concentrated HCI and 50 mL EtOH, the contents were cooled to 0 °C, and stirred at 0 °C for 2h. The formed solids were filtered, washed with cold ethanol and ether; and dried to obtain the title compound.
Yield: 6 g (58.7 %); 1 H NMR (DMSO-d6, 500 MHz): δ 1 1 .86 (s, 1 H), 7.95 (s,
2H), 3.85 (d, J =5.5Hz, 2H), 2.71 (s, 2H), 2.39 (s, 2H), 2.15 (s, 3H), 1 .64 (s, 4H); MS (ESI+) m/z 193.1 (M+H).
Example 3:
Methyl 3-amino-5-bromo-2-fluorobenzoate
To a stirred solution of methyl 5-bromo-2-fluoro-3-nitrobenzoate (4 g, 14.39 mmol) in ethanol (5 mL), was added ammonium chloride (3.85 g, 71 .9 mmol) in 5 mL water, followed by iron (6.43 g, 1 15 mmol). The resulting reaction mixture was
stirred at 90 °C for 1 h. On completion of the reaction, the reaction mixture was filtered and the filtrate was concentrated to dryness to obtain a solid which was dissolved in saturated sodium bicarbonate solution and extracted with ethyl acetate (3X50 mL). The combined organic layers were dried over anhydrous sodium sulphate and concentrated to yield the title compound.
Yield: 2.5 g (70.1 %).
Example 4:
Methyl 5-bromo-2-fluoro-3-((tetrahydro-2H-pyran-4-yl)amino)benzoate
To a solution of the compound of example 3 (3.0 g, 12 mmol) in 1 ,2- dichloroethane (48 mL) under nitrogen, was added oxan-4-one (2.3 ml, 25 mmol) followed by acetic acid (4.2 mL, 74 mmol). The reaction mixture was stirred for 5 min before the addition of sodium triacetoxyborohydride (7.8 g, 37 mmol). After stirring for 64 h, deionized water (100 mL) was added and the mixture was neutralized with solid NaHC03. The phases were separated and the aqueous layer was extracted with ethyl acetate (4 x 50 mL). The combined organic extracts were dried over magnesium sulphate, filtered and concentrated in-vacuo. The residue was purified by flash column chromatography (silica gel, 10-80 % ethyl acetate in petroleum ether) to yield the title compound.
1H NMR (DMSO-de, 300 MHz): δ 1.457-1 .537 (m, 2H), 1 .790-1 .812 (m,
2H), 3.415-3.437 (m, 2H), 3.570-3.58 (m, 1 H), 3.832 (s, 3H), 3.847-3.871 (s, 1 H), 5.873-5.887 (d 1 H), 7.010-7.024 (m, 1 H), 7.184-7.202 (m, 1 H); MS (ESI+): m/z 333.1 [M+H]+; HPLC Purity: 98.43 %. Example 5:
Methyl 5-bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-fluorobenzoate
To a stirred solution of the compound of example 4 (1 .5 g, 4.52 mmol) and acetaldehyde (0.510 mL, 9.03 mmol) in dichloroethane (15 mL), acetic acid (1 .551 mL, 27.1 mmol) was added and the reaction mixture was stirred at RT for 20 min. Sodium triacetoxyborohydride (2.87 g, 13.55 mmol) was added at 0 °C and the reaction mixture was stirred at room temperature for 2 h. The solvent was removed and water was added to the residue. The mixture was extracted with DCM.
The combined extracts were dried over sodium sulphate and concentrated to yield the crude material, which was purified by column chromatography (silica gel, 10- 80 % ethyl acetate in petroleum ether ) to yield the title compound.
Yield: 400 mg (24.59 %); 1 H NMR (DMSO-d6, 300 MHz): δ 0.3-0.9 (m, 3H), 1 .609-1 .658 (m, 4H), 3.163-3.204 (m, 2H), 3.287-3.340 (m, 3H), 3.833-3.874 (m, 5H), 7.498-7.529 (m, 2H); MS (ESI+): m/z 361 .1 [M+H]+; HPLC Purity: 95.92 %.
Example 6:
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-fluorobenzoic acid
To a stirred solution of the compound of example 5 (1 .4 g, 3.89 mmol) in ethanol (10 mL) was added aqueous NaOH (0.233 g, 5.83 mmol) and the resulting mixture was stirred at 60 °C for 1 h. Upon completion of the reaction, the solvent was removed and the residue obtained was acidified with IN HCI until a pH of 7 was obtained and then aqueous citric acid solution was added until a pH 5-6 was obtained. The aqueous layer was extracted with 10 % MeOH in DCM (200 mL X 3), the combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated to yield the title compound.
Yield: 350 mg (26.0 %); 1 H NMR (DMSO-d6, 300 MHz): δ 0.892-0.918 (t, 3H, J = 7.8 Hz), 1 .585-1 .636 (m, 4H), 3.167-3.343 (m, 4H), 3.855-3.876 (d, 2H), 7.473-7.483 (s, 2H), 13.501 (s, 1 H, COOH); MS (ESI+): m/z 347.1 [M+H]+; HPLC Purity: 97.37 %.
Example 7:
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-fluoro-N-((1 -methyl-3- oxo-2,3, 5, 6,7, 8-hexahydroisoquinolin-4-yl)methyl)benzamide
The compound of example 6 (185 mg, 0.534 mmol), the compound of example 2 (123 mg, 0.641 mmol) and HATU (305 mg, 0.802 mmol) were added in the DMF (10 mL), the reaction was cooled and Hunig's Base (280 μΐ, 1 .603 mmol) was added dropwise. The reaction mixture was stirred for 16 h, water was added and the resulting mixture was filtered to yield the title compound.
Yield: 150 mg (53.9 %); 1 H NMR (DMSO-d6, 300 MHz): δ 0.6-0.9 (t, 3H, J = 6.0 Hz), 1 .2-1.4 (m, 3H), 1 .600-1 .641 (m, 8H), 2.1 13 (s, 3H),2.382 (m, 2H), 2.38 (m, 2H), 2.695-2.709 (m, 2H), 3.127-3.168 (m, 2H), 3.275-3.297 (m, 2H), 3.849-
3.870 (m, 2H), 4.295-4.305(d, 2H, J = 3.0 Hz), 7.196-7.210 (m, 1 H), 7.313-7.332 (m, 1 H), 8.372 (s, 1 H), 1 1 .560 (s, 1 H); MS (ESI+): m/z 521 .2 [M+H]+; HPLC Purity: 96.30 %. Example 8:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-fluoro-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(1-morpholinoethyl)-[1 ,1 '- biphenyl]-3-carboxamide
The compound of example 7 (90 mg, 0.173 mmol), Na2C03 (55.0 mg, 0.519 mmol) and 4-(1 -(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)phenyl)ethyl)morpholine (65.8 mg, 0.208 mmol) was added in dioxane and 1 % water under an atmosphere of argon purging for 20 min, followed by triphenylphosphine and palladium salt (50.0 mg, 0.173 mmol) and continued purging for 10 min. The reaction mixture was heated to 200 °C for 16 h. Water was added and the resulting mixture was extracted with ethylacetate (20 mL x 2). The combined extracts were concentrated to yield a crude material, which was purified by flash column chromatography to yield the title compound.
Yield: 45 mg (41 .3 %); 1H NMR (DMSO-d6, 300 MHz): δ 0.882-0.927 (t, 3H, J = 6.6 Hz), 1 .261 -1.283 (m, 3H), 1 .528-1 .620 (m, 8H), 2.087 (s, 3H), 2.276-2.360 (m, 6H), 2.717 (m, 2H), 3.159-3.315 (m, 5H), 3.535 (m, 4H), 3.819- 3.854 (m, 2H), 4.307-4.321 (m,2 H), 7.341 -7.568 (m, 6H),8.306 (s, 1 H), 1 1 .534 (s, 1 H); MS (ESI+): m/z 631 .5 [M+H]+; HPLC Purity: 96.1 1 %.
Example 9:
3'-((Dimethylamino)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4- fluoro-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '- biphenyl]-3-carboxamide
The compound of example 7 (80 mg, 0.154 mmol), (3- ((dimethylamino)methyl)phenyl)boronic acid (33.0 mg, 0.184 mmol), Na2C03 (48.9 mg, 0.461 mmol) was dissolved and stirred in dioxane with argon purging for 20 min, followed by addition of PdCI2(dppf)CH2CI2 adduct (12.55 mg, 0.015 mmol) and continued purging of argon for 10 min. The reaction mixture was heated at 100 °C for 16 h. After completion of the reaction, the crude material was absorbed
on silica (100-200 mesh) and purified using column chromatography (silica gel, 10-15% methanol in chloroform) to yield the title compound.
Yield: 41 mg (46.5 %); 1 H NMR (DMSO-d6, 300 MHz): δ 0.907-0.953 (t, 3H), 1 .640 (m, 7H), 2.108 (s, 3H), 2.379 (s, 6H), 2.741 (m, 2H), 3.188 (m, 2H), 3.21 1 (m, 2H), 3.848 (m, 3H),4.331 (m, 2H), 7.382-7.651 (m, 5H), 8.319 (s, 1 H), 8.330 (bs, 1 H), 1 1 .540 (bs, 1 H); MS (ESI+): m/z 575.4 [M+H]+; HPLC Purity: 89.28 %.
Example 10:
1-Methyl-3-oxo-7-(trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinoline-4- carbonitrile
To a solution of 2-acetyl-4-(trifluoromethyl)cyclohexanone (1 .2g, 5.76 mmol) in ethanol (15 mL), were added 2-cyanoacetamide (0.485 g, 5.76 mmol) and piperidine (0.285 ml, 2.88 mmol), and the reaction mixture was stirred at 75 °C for 7 h. After completion of the reaction, the precipitated solid was filtered, washed with ethanol and dried to yield the title compound.
Yield: 1 .45 g (98 %); MS (ESI-): m/z 255 [M-H]\
Example 11 :
4-(Aminomethyl)-1-methyl-7-(trifluoromethyl)-5,6,7,8-tetrahydroisoquinolin- 3(2H)-one
To a solution of the compound of example 10 (0.6 g, 2.342 mmol) in acetic acid (50 mL) were added HCI (0.071 mL, 2.342 mmol), platinum (IV) oxide (0.013 g, 0.059 mmol) and Pd/C (0.062 g, 0.585 mmol). The flask was shaked under an atmosphere of hydrogen at 80 psi for 8 h. After completion of the reaction, the reaction mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 4.37 mL concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.9 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2 h. The solid was filtered, washed with ether and dried to yield the title compound.
Yield: 400 mg (57.6 %).
Example 12:
1- Methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridine-4-carbonitrile
To ethanol (200 mL, 3425 mmol), were added 2-acetylcyclopentanone (9.62 mL, 79 mmol), 2-cyanoacetamide (6.66 g, 79 mmol) and piperidine (7.85 mL, 79 mmol) The reaction mixture was heated to 75 °C for 16 h. The reaction mixture was cooled and filtered to obtain the title compound.
Yield: 1 1 .0 g (80 %).1 H NMR (DMSO-d6, 300 MHz): δ 12.13 (s, 1 H), 2.91 - 2.82 (m, 4H), 2.50 (s, 3H), 2.01 -2.05 (m, 2H); MS (ESI+): m/z 175.2 [M+H]+.
Example 13:
4-(aminomethyl)-1-methyl-6,7-dihydro-2H-cyclopenta[c]pyridin-3(5H)-one hydrochloride
To a solution of the compound of example 12 (2.0 g, 1 1 .48 mmol) in acetic acid (10 mL) were added Pd/C (0.305 g, 2.87 mmol), sodium acetate (1 .884 g, 22.96 mmol) and platinum(IV) oxide (0.065 g, 0.287 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the reaction mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 1 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The filtered solid was washed with ether and dried to yield the title compound.
Yield 1 .35 g (54.8 %). 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .89 - 1 1 .64 (m, 1 H), 8.06 - 8.05 (m, 2H), 3.80 - 3.71 (m, 2H), 2.86 - 2.73 (m, 2H), 2.63 - 2.58 (m, 2H), 2.16 - 1 .13 (m, 3H), 2.02 - 1 .91 (m, 2H); MS (ESI+): m/z 215.6 [M+H]+. Example 14:
2- (3-Bromophenyl)-4, 4, 5, 5-tetramethyl-1 , 3, 2-dioxaborolane
To a stirred suspension of (3-bromophenyl) boronic acid (3.0 g, 14.94 mmol) and acetonitrile (10 mL) was added pinacol (1 .765 g, 14.94 mmol) and the reaction mixture was purged using argon gas. The resulting mixture was then stirred for 6 hours at RT. After completion of the reaction, the acetonitrile was evaporated to yield the title compound.
Yield: 2.4 g (56.8 %).1 HNMR (DMSO, 300 MHz): δ 7.66 (m, 4H), 1 .3 (s,
12H).
Example 15:
Dimethyl (3-(4, 4, 5, 5-tetramethyl-1 , 3, 2-dioxaborolan-2-yl) phenyl) phosphonate
To a stirred solution of the compound of example 13 (1 .0 g, 3.53 mmol) in toluene (15 mL) was added trimethyl phosphite (1.535 g, 12.37 mmol) and the reaction mixture was heated to 105 °C. To the reaction mixture was added AIBN (0.093 g, 0.565 mmol) and tri-N-butyl tin hydride (1 .227 mL, 4.59 mmol) in toluene (10 mL) dropwise and the reaction mixture was stirred for 16 h at 105 °C. The solvent was removed to get the crude compound which was purified using column chromatography (silica gel, 9:1 petroleum ether and ethyl acetate) to yield the title compound.
Yield: 652 mg (59.1 %); 1 H NMR (DMSO,300 MHz): δ 7.97 (m, 1 H), 7.86 (m, 1 H),7.79 (m, 1 H), 7.57 (m, 1 H), 3.68 (s, 6H), 1 .31 (s, 12H).
Example 16:
Methyl 5-bromo-2-methyl-3-nitrobenzoate
To a stirred solution of 5-bromo-2-methyl-3-nitrobenzoic acid (25 g, 96 mmol) in DMF (300 mL), sodium carbonate (40.8 g, 385 mmol) and iodomethane (24.05 mL, 385 mmol) were added. Resulting reaction mass was heated at 60 °C for 16 h. On completion of reaction, solvent was evaporated to dryness. Ice cold water was added to the reaction mixture. The solid precipitated was filtered, and dried to yield the title compound.
Yield: 25.2 g (90 %); 1 H NMR (DMSO-d6, 300 MHz): δ 8.32 (d, J= 1 .8 Hz, 1 H), 8.14 (d, J= 2.1 Hz, 1 H), 3.87 (s, 3H), 2.40 (s, 3H); MS (ESI+): m/z 275.1 [M+H]+; HPLC Purity: 93.99 %.
Example 17:
Methyl 3-amino-5-bromo-2-methylbenzoate
To a solution of the compound of example 16 (25 g, 91 mmol) in ethanol
(250 ml) were added iron (12.74 g, 228 mmol) and ammonium chloride (12.20 g, 228 mmol) in water (50 mL) and stirred at 70 °C for 15 h. After completion of the reaction, the reaction mixture was filtered through celite and the filtrate was
distilled under vacuum. To the residue, water was added and extracted with ethyl acetate. The compound was adsorbed on silica and purified by column chromatography (silica gel, 10-50 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 20.2 g (84 %); 1 H NMR (DMSO-d6, 300 MHz): δ 6.96 (s, 2H), 5.43 (s,
2H), 3.78 (s, 3H), 2.1 1 (s, 3H); MS (ESI+): m/z 245.1 [M+H]+;HPLC Purity: 92.13 %.
Example 18:
Methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl)amino)benzoate
To a solution of the compound of example 17 (15 g, 61 .5 mmol) in DCE (100 ml) were added dihydro-2H-pyran-4(3H)-one (1 1 .35 ml_, 123 mmol) and acetic acid (21 .1 1 ml_, 369 mmol) and stirred at RT for 2h. The reaction mass was cooled to 0 °C and sodium triacetoxyborohydride (39.1 g, 184 mmol), was added lot wise in 1 h which was left to stir at RT for 16h. After completion of the reaction, water was added and extracted with ethyl acetate. The compound was adsorbed on silica and purified by column chromatography (silica gel, 10-80 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 15.1 g (73.6 %); 1 H NMR (DMSO-d6, 300 MHz): δ 6.97-6.93 (m, 2H), 3.87-3.85 (m, 2H), 3.80 (s, 3H), 3.59-3.55 (m, 1 H), 3.46-3.41 (m, 2H), 2.14 (s, 3H), 1 .84-1 .82 (m, 2H), 1 .56-1 .53 (m, 2H);MS (ESI+): m/z 329.1 [M+H]+; HPLC Purity: 98.3 %.
Example 19:
Methyl 5-bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methylbenzoate
To a solution of the compound of example 18 (15 g, 45.7 mmol) in DCE (50 mL) were added acetaldehyde (5.16 ml_, 91 mmol) and acetic acid (15.70 ml_, 274 mmol); and the reaction mixture was stirred at RT for 2 h. The reaction mixture was cooled to 0 °C and sodium triacetoxyborohydride (29.1 g, 137 mmol) was added in portions over a period of 1 h.The reaction mixturewas stirred at RT for 16 h. After completion of the reaction, water was added and the resulting mixture wasextracted with ethyl acetate. The compound was adsorbed on silica and
purified by column chromatography (silica gel, 10-70 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 14.2 g (86 %); 1 H NMR (DMSO-d6, 300 MHz): δ 7.60-7.53 (m, 2H), 3.82 (s, 3H), 3.81 -3.80 (m, 2H), 3.25 (t, J= 6.6 Hz, 2H), 3.03 (q, J= 4.2 Hz, 2H), 2.99-2.94 (m, 1 H), 2.35 (s, 3H), 1 .62-1 .59 (m, 2H), 1 .53-1.45 (m, 2H), 0.78(t, J= 4.2 Hz, 3H); MS (ESI+): m/z 357.1 [M+H]+; HPLC Purity: 99.02 %.
Example 20:
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methylbenzoic acid
A solution of NaOH (1 .572 g, 39.3 mmol) in water (2 mL) was added to a solution of the compound of example 19 (14 g, 39.3 mmol) in methanol (2ml_) and THF (25 mL). The reaction mixture was stirred at 60° C for 3 h.The methanol was removed and the residue was acidified to neutral pH. The precipitated solid was filtered, washed with water and dried to yield the title compound.
Yield: 10.0 g (72.9 %); 1 H NMR (DMSO-d6, 300 MHz): δ 13.18 (s, 1 H),
7.58-7.48 (m, 2H), 3.83-3.80 (m, 2H), 3.24 (t, J= 1 1 .4 Hz, 2H), 3.04-2.95 (m, 3H), 2.37 (s, 3H), 1 .62-1 .46 (m, 4H), 0.78 (t, J= 6.3 Hz, 3H); MS (ESI+): m/z 343.1 [M+H]+; HPLC Purity: 98.0 %. Example 21 :
2-Acetylcycloheptanone
To a solution of cycloheptanone (1 .052 mL, 8.92 mmol) in THF (45 mL) at - 78 °C was added a solution of LDA (Lithium diisopropylamide) solution (5.35 mL, 10.70 mmol) in THF drop wise via a syringe. After 1 h of stirring, acetyl cyanide (0.759 mL, 10.70 mmol) was added drop wise over a 10 min period. After 30 min, the reaction mixture was quenched with few drops of water. The solvent was evaporated and the residue was purified by column chromatography (silica gel, 20 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 0.659 g (46.9 %); 1H NMR (DMSO-d6, 300 MHz): δ 2.61 -2.35 (m, 5H), 2.1 1 (s, 3H), 1 .75-1 .40 (m, 6H); MS (ESI+): m/z 155.1 [M+H]+; HPLC Purity: 97.90 %.
Example 22:
1-Methyl-3-oxo-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridine-4- carbonitrile
To a solution of the compound of example 21 (0.6 g, 3.89 mmol) in ethanol (5 mL) were added 2-cyanoacetamide (0.327 g, 3.89 mmol) and piperidine (0.193 ml, 1 .945 mmol); and the reaction mixture was stirred at 75 °C for 7 h. After completion of the reaction, the precipitated solid was filtered, washed with ethanol and dried to yield the title compound.
Yield: 0.580 g (64.8%); 1 H NMR (DMSO-d6, 300 MHz): δ 12.2(s, 1 H), 2.75- 2.71 (m, 2H), 2.56-2.53 (m, 2H), 2.33 (s, 3H), 1 .73-1 .71 (m, 2H), 1.56-1 .46 (m, 4H); MS (ESI+): m/z 203.1 [M+H]+; HPLC Purity: 87.91 %.
Example 23:
4- (Aminomethyl)-1-methyl-6 ,8,9-tetrahydro-2H-cyclohepta[c]pyridin-3(5H)- one hydrochloride
To a solution of the compound of example 22 (550 mg, 2.72 mmol) in acetic acid (10 mL) were added Pd/C (72.3 mg, 0.680 mmol), sodium acetate (446 mg, 5.44 mmol) and platinum(IV) oxide (15.44 mg, 0.068 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 16 h. After completion of the reaction, the resulting mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 2.5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.5 mL of concentrated HCI and 2 mL of EtOH at 5-10 °C for 2 h. The filtered solid was washed with ether and dried to yield the title compound.
Yield: 0.539 g (59.7 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .76 (s, 1 H), 3.86-3.84 (m, 2H), 2.72-2.69 (m, 2H), 2.56-2.55 (m, 2H), 2.18 (s, 3H), 1.74-1 .44 (m, 6H); MS (ESI+): m/z 243.1 [M+H]+;HPLC Purity: 98.79 %.
Example 24:
5- Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2- oxo-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-1-yl)methyl)benzamide
To a solution of the compound of example 20 (2.8 g, 8.18 mmol) in DMF (20 mL) was added HATU (4.67 g, 12.27 mmol) and Hunig's base (4.29 mL, 24.55 mmol) and the resulting mixture was stirred at RT for 1 h. To the resulting mixture
was added the compound of example 23 (2.383 g, 9.82 mmol) and further stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate (2x25 mL) and washed with water (2x25 ml_).The combined organic extracts were distilled to yield a crude material. The crude material was purified by using column chromatography (silica gel, 0-15 % MeOH/CHC ) to yield the title compound.
Yield: 2.55 g (58.1 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .4 (s, 1 H), 8.23 (s, 1 H), 7.30 (s, 1 H), 7.08 (s, 1 H), 4.33 (d, J= 4.5 Hz, 2H),3.23 (t, J= 1 1 .4 Hz, 2H), 3.01 -2.93 (m, 3H), 2.62-2.63 (m, 2H), 2.48-2.49 (m, 2H), 2.16 (s, 3H), 2.14 (s, 3H), 1 .73-1 .46 (m,10H), 0.77 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 531 .2 [M+H]+; HPLC Purity: 98.89 %.
Example 25:
Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholino methyl) -[1 ,1 '-biphenyl]-3-carboxylate
The compound of example 19 (300 mg, 0.842 mmol) was added to a stirred solution of 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (383 mg, 1 .263 mmol), PdCI2(dppf)-CH2CI2 adduct (68.8 mg, 0.084 mmol) and Na2C03 (268 mg, 2.53 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 2 h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and the residue was extracted with ethyl acetate. The residue was washed with water and brine; and dried over anhydrous sodium sulphate to obtain a crude mixture, which was purified by column chromatography(silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.210 g (53.1 %); 1 H NMR (DMSO-d6, 300 MHz): δ 7.71 (s, 1 H), 7.59 (s, 1 H), 7.06 (d, J= 8.4 Hz, 2H), 6.68 (d, J= 8.1 Hz, 2H), 3.83 (s, 3H), 3.57-3.48 (m, 10H), 3.30-3.03 (m, 7H), 2.43 (s, 3H), 1 .67-1 .48 (m, 4H), 0.82 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 453.2 [M+H]+; HPLC Purity: 96.37 %.
Example 26:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)- [1 ,1 '-biphenyl]-3-carboxylic acid
A solution of NaOH (17.68 mg, 0.442 mmol) in water (0.5 mL) was added to a solution of the compound of example 25 (200 mg, 0.442 mmol) in methanol (0.500 mL) and THF (2 mL). The reaction mixture was stirred at 60 °C for 3 h. The methanol was removed and the residue was acidified to neutral pH. The precipitated solid was filtered, washed with water and dried to yield the title compound.
Yield: 0.077 g (39%); 1 H NMR (DMSO-d6, 300 MHz): δ 7.71 (s, 1 H), 7.60 (s, 1 H), 7.41 -7.43 (m, 2H), 7.1 1 -7.13 (m, 1 H), 6.72-6.73 (m, 1 H), 3.83-3.59 (m, 10H), 3.25-3.03 (m, 5H), 2.49 (s, 3H), 2.45 (s, 2H), 1.67-1 .48 (m, 4H), 0.82 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 439.5 [M+H]+;HPLC Purity: 98.13 %.
Example 27:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((4-methyl-2-oxo- 3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-1-yl)methyl)-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (70 mg, 0.160 mmol) in DMF (5 mL) was added HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and the reaction mixture was stirred at RT for 1 hour. To it was added the compound of example 23 (46.5 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate (2x25 mL) and washed with water (2x25 mL).The combined organic extracts were distilled off to obtain a crude mixture, which was purified by using column chromatograhy (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.031 g (28.6%); 1 H NMR (DMSO-d6, 300 MHz): δ 7.58-7.55 (m, 2H), 7.38-7.35 (m, 3H), 7.20 (s, 1 H), 4.37 (d, J= 4.2 Hz, 2H), 3.84-3.80(m, 2H), 3.56- 3.47(m, 6H), 3.24 (t, J= 1 1.4 Hz, 2H), 3.09-3.07 (m, 3H), 2.72-2.63 (m, 4H), 2.32- 2.35 (m, 4H), 2.25 (s, 3H), 2.14 (s, 3H), 1 .73-1 .39 (m, 10H), 0.82 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 627.4 [M+H]+; HPLC Purity: 92.21 %.
Example 28:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo-
3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-1-yl)methyl)-5-(6-(4-methyl piperazin-1-yl)pyridin-3-yl)benzamide
The compound of example 24 (175 mg, 0.329 mmol) was added to a stirred solution of 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)piperazine (150 mg, 0.494 mmol), PdCI2(dppf)-CH2CI2 adduct (26.9 mg, 0.033 mmol) and Na2C03 (105 mg, 0.988 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 ml_). The reaction mixture was stirred at 80 °C for 2 h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHC ) to yield the title compound.
Yield: 0.041 g (19.41 %); 1H NMR (DMSO-d6, 300 MHz): δ 8.39 (s, 1 H), 7.82-7.79 (m, 1 H), 7.34 (s, 1 H), 7.16 (s, 1 H), 6.90-6.87 (m, 1 H), 4.37 (d, J= 4.2 Hz, 2H), 3.83-3.80 (m, 2H), 3.52-3.48 (m, 4H), 3.24 (t, J= 1 1 .4 Hz, 2H), 3.08-3.05 (m, 3H), 2.64-2.63 (m, 2H), 2.36-2.39 (m, 6H), 2.28 (s, 3H), 2.21 (s, 6H), 1 .73- 1 .48 (m, 10H), 0.81 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 627.4 [M+H]+; HPLC Purity: 97.73 %.
Example 29:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo- 3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-1-yl)methyl)-5-(6-morpholino pyridi n-3-yl)benzamide
The compound of example 24 (175 mg, 0.329 mmol) was added to a stirred solution of 4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)morpholine (143 mg, 0.494 mmol), PdCI2(dppf)-CH2CI2 adduct (26.9 mg, 0.033 mmol) and Na2C03 (105 mg, 0.988 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 2 h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a
crude mixture, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHC ) to yield the title compound.
Yield: 0.085 g (42 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .41 (s, NH), 8.41 (s, 1 H), 8.17 (s, 1 H), 7.85 (s, NH), 7.36 (s, 1 H), 7.17 (s, 1 H), 6.89 (d, J= 9.0 Hz, 1 H), 4.37 (d, J= 4.2 Hz, 2H), 3.83-3.70(m, 6H), 3.42-3.46(m, 4H), 3.24 (t, J= 1 1 .4 Hz, 2H), 3.08-3.06 (m, 3H), 2.62-2.49 (m, 4H), 2.23 (s, 3H), 2.18 (s, 3H), 1 .73- 1 .48 (m, 10H), 0.81 (t, J= 6.0 Hz, 3H); MS (ESI+): m/z 614.2 [M+H]+; HPLC Purity: 99.83 %. Example 30:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo-
3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-1-yl)methyl)-5-(6-(trifluoro methyl)pyridin-3-yl)benzamide
The compound of example 24 (175 mg, 0.329 mmol) was added to a stirred solution of (6-(trifluoromethyl)pyridin-3-yl)boronic acid (94 mg, 0.494 mmol), PdCI2(dppf)-CH2CI2 adduct (26.9 mg, 0.033 mmol) and Na2C03 (105 mg, 0.988 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 ml_). The reaction mixture was stirred at 80 °C for 2 h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.091 g, 44.8%; 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .42 (s, NH), 9.06 (s, 1 H), 8.34 (d, J= 8.1 Hz, 1 H), 8.24 (s, NH), 7.95 (d, J= 8.1 Hz, 1 H), 7.57 (s, 1 H), 7.38 (s, 1 H), 4.37 (d, J= 4.2 Hz, 2H), 3.84-3.81 (m, 2H), 3.24 (t, J= 1 1 .4 Hz, 2H), 3.12-3.06 (m, 3H), 2.62-2.49 (m, 4H), 2.28 (s, 3H), 2.19 (s, 3H), 1 .68-1 .48 (m, 10H), 0.82 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 597.4 [M+H]+; HPLC Purity: 96.62 %.
Example 31 :
3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(2-methoxypyrimidin-5-yl)-2- methyl-N-((1-methyl-3-oxo-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-4- yl)methyl)benzamide
The compound of example 24 (125 mg, 0.235 mmol) was added to a stirred solution of (2-methoxypyrimidin-5-yl)boronic acid (36.2 mg, 0.235 mmol), PdCI2(dppf)-CH2CI2 adduct (19.21 mg, 0.024 mmol) and Na2C03 (74.8 mg, 0.706 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 ml_). The reaction mixture was stirred at 80 °C for 2 h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water, brine and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.031 g (23.46 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .40 (s, NH), 8.89 (s, 2H), 8.16 (s, NH), 7.47 (s, 1 H), 7.28 (s, 1 H), 4.37 (d, J= 4.5 Hz, 2H), 3.94 (s, 3H), 3.83-3.80 (m, 2H), 3.23 (t, J= 1 1 .1 Hz, 2H), 3.09-3.04 (m, 3H), 2.60-2.48 (m, 4H), 2.24 (s, 3H), 2.18 (s, 3H), 1 .72-1 .42 (m, 10H), 0.80 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 560.3 [M+H]+; HPLC Purity: 99.6 %.
Example 32:
3-Acetyldihydro-2H-pyran-4(3H)-one
Dihydro-2H-pyran-4(3H)-one (9.23 mL, 100 mmol) and pyrrolidine (16.52 ml, 200 mmol) were dissolved in toluene (50 ml); and the resulting mixture was refluxed for 3 h under nitrogen using a Dean and Stark apparatus, then cooled to RT and concentrated completely. The residue was dissolved in dioxane (30 mL) and acetic anhydride (20.73 mL, 220 mmol) was added and the resulting mixture was stirred under nitrogen for 16 h. Water (12 mL) was added and the resulting mixture was refluxed for 1 h, cooled to RT and concentrated. The residue was dissolved in water (40 mL) and the aqueous phase was extracted twice with ethyl acetate (30 mL).The combined organic extract was washed with 5 % w/w HCI solution (20 mL), dried over anhydrous sodium sulphate and concentrated to yield the title compound.
Yield: 10.2 g (71 .8%); 1 H NMR (DMSO-d6, 300 MHz): δ 3.75 (t, J= 5.7 Hz,
1 H), 3.36 (t, J= 6.6 Hz, 2H), 3.22 (t, J= 7.2 Hz, 2H), 2.38 (t, J= 6.0 Hz, 2H), 2.03 (s, 3H); MS (ESI+): m/z 143.2 [M+H]+; HPLC Purity: 88.41 %.
Example 33:
8-Methyl-6-oxo-3,4,6,7-tetrahydro-1 H-pyrano[3,4-c]pyridine-5-carbonitrile
To a solution of the compound of example 32 (10 g, 70.3 mmol) in ethanol (25 mL) were added 2-cyanoacetamide (5.91 g, 70.3 mmol) and diethyl amine (3.67 mL, 35.2 mmol); and the reaction mixture was stirred at 50 °C for 16 h. After completion of the reaction, the precipitated solid was filtered, washed with ethanol and dried to yield the title compound.
Yield: 2.02 g (14.7%); 1H NMR (DMSO-d6, 300 MHz): δ 12.43 (s, 1 H), 4.42 (s, 2H), 3.79 (t, J= 5.4 Hz, 2H), 2.76 (t, J= 5.7 Hz, 2H), 2.13 (s, 3H); MS (ESI+): m/z 191 .2 [M+H]+;HPLC Purity: 97.38 %.
Example 34:
5-(Aminomethyl)-8-methyl-3,4-dihydro-1 H-pyrano[3,4-c]pyridin-6(7H)-one hydrochloride
To a solution of the compound of example 33 (2 g, 10.52 mmol) in acetic acid (50 mL) were added Pd/C (0.280 g, 2.63 mmol), sodium acetate (1 .725 g, 21 .03 mmol) and platinum(IV) oxide (0.060 g, 0.263 mmol) and the flask was shaken under an atmosphere of hydrogen at 80 psi for 16h. After completion of the reaction, the resulting mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 4 mLof concentrated HCI and filtered. The filtrate was stirred in a mixture of 2 mL of concentrated HCI and 10 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.754 g (31 .1 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .94 (s, 1 H), 4.44 (s, 2H), 3.77-3.75 (m, 4H), 2.80 (t, J= 5.7 Hz, 2H), 2.08 (s, 3H); MS (ESI+): m/z 231 .1 [M+H]+; HPLC Purity: 69 %.
Example 35:
5-bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((5-methyl-7- oxo-2,4,6,7-tetrahydro-1 H-pyrano[3,4-c]pyridin-8-yl)methyl)benzamide
To the compound of example 20 (1 g, 2.92 mmol) in DMF (5 mL) were added HATU (1 .667 g, 4.38 mmol) and Hunig's Base (1 .531 mL, 8.77 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added
the compound of example 34 (0.741 g, 3.21 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue extracted using ethyl acetate and then washed with water (2x25 ml_). The combined organic extracts were distilled off to obtain a crude mixture, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHC ) to yield the title compound.
Yield: 0.795 g (52 %); 1H NMR (DMSO-d6, 300 MHz): δ 1 1 .59 (s, 1 H), 7.30 (s, 1 H), 7.08 (s, 1 H), 4.43 (s, 2H), 4.23 (d, J= 4.8 Hz, 2H),3.83-3.75 (m, 4H), 3.22 (d, J= 10.8 Hz, 2H), 3.03-2.93 (m, 3H), 2.81 -2.72 (m, 2H), 2.13 (s, 3H), 2.03 (s, 3H), 1 .60-1 .43 (m, 4H), 0.77 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 519.7 [M+H]+; HPLC Purity: 99.06 %.
Example 36:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((8-methyl-6-oxo- 3,4,6,7-tetrahydro-1 H-pyrano[3,4-c]pyridin-5-yl)methyl)-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide
The compound of example 35 (100 mg, 0.193 mmol) was added to a stirred solution of 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (88 mg, 0.289 mmol), PdCI2(dppf)-CH2CI2 adduct (15.75 mg, 0.019 mmol) and Na2C03 (61 .3 mg, 0.579 mmol) in 1 ,4-dioxane (5 mL) and water (1.667 ml_). The reaction mixture was stirred at 80 °C for 3 h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0- 15% MeOH/CHC ) to yield the title compound.
Yield: 0.010 g (7.36 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .58 (s, 1 H), 8.22 (s, 1 H), 7.58-7.37 (m, 6H), 4.44 (s, 2H), 4.28 (s, 4H), 3.84-3.79 (m, 4H), 3.56- 3.46 (m, 6H), 3.25 (t, J= 1 1 .4 Hz, 2H), 3.09-3.02 (m, 3H), 2.81 -2.83 (m, 2H), 2.36 (s, 3H), 2.24 (s, 3H), 2.21 (s, 2H), 1 .67-1 .52 (m, 4H), 0.83 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 615.2 [M+H]+; HPLC Purity: 87.23 %.
Example 37:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((8-methyl-6-oxo-
3,4,6,7-tetrahydro-1 H-pyrano[3,4-c]pyridin-5-yl)methyl)-5-(6-(4- methylpiperazin-1-yl)pyridin-3-yl)benzamide
The compound of example 35 (100 mg, 0.193 mmol) was added to a stirred solution of 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)piperazine (88 mg, 0.289 mmol), PdCI2(dppf)-CH2CI2adduct (15.75 mg, 0.019 mmol) and Na2C03 (61 .3 mg, 0.579 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 2h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a crude mixture, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHC ) to yield the title compound.
Yield: 0.030 g (24.43 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .57 (s, 1 H), 8.41 (s, 1 H), 8.19 (s, 1 H), 7.84 (d, J= 3.9 Hz, 1 H), 7.36 (s, 1 H), 7.18 (s, 1 H), 6.94 (d, J= 5.4 Hz, 1 H), 4.45 (s, 2H), 4.28 (s, 2H), 3.84-3.78 (m, 4H), 3.52-3.44 (m, 4H), 3.25 (t, J= 6.9 Hz, 2H), 3.09-3.02 (m, 3H), 2.81 -2.83 (m, 2H), 2.61 -2.65 (m, 4H), 2.41 (s, 3H), 2.22 (s, 3H), 2.04 (s, 3H), 1 .67-1 .36 (m, 4H), 0.82 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 615.6 [M+H]+; HPLC Purity: 96.55 %.
Example 38:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((8-methyl-6-oxo-
3,4,6,7-tetrahydro-1 H-pyrano[3,4-c]pyridin-5-yl)methyl)-5-(6- morpholinopyridin-3-yl)benzamide
The compound of example 35 (100 mg, 0.193 mmol) was added to a stirred solution of 4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)morpholine (67.2 mg, 0.231 mmol), PdCI2(dppf)-CH2CI2 adduct (15.75 mg, 0.019 mmol) and Na2C03 (61 .3 mg, 0.579 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The reaction mixture was then cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate. The organic layers were
concentrated to obtain a crude mixture, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.031 g (23.67 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .58 (s, 1 H), 8.41 (s, 1 H), 8.19 (s, 1 H), 7.85 (d, J= 3.9 Hz, 1 H), 7.36 (s, 1 H), 7.17 (s, 1 H), 6.91 (d, J= 5.4 Hz, 1 H), 4.43 (s, 2H), 4.26 (s, 2H), 3.83-3.70 (m, 8H), 3.42-3.46 (m, 4H), 3.24 (t, J= 6.9 Hz, 2H), 3.08-3.06 (m, 3H), 2.81 -2.83 (m, 2H), 2.21 (s, 3H), 2.03 (s, 3H), 1 .62-1 .46 (m, 4H), 0.81 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 602.7 [M+H]+; HPLC Purity: 88.64 %. Example 39:
4- (aminomethyl)-5,6,7,8-tetrahydroisoquinolin-3(2H)-one hydrochloride
To a solution of 3-oxo-2,3,5,6,7,8-hexahydroisoquinoline-4-carbonitrile (1 g, 5.74 mmol) in acetic acid (10 mL) were added Pd/C (0.153 g, 1 .435 mmol), sodium acetate (0.942 g, 1 1.48 mmol) and platinum (IV) oxide (0.033 g, 0.144 mmol) and the flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of reaction, the resulting mixturewas filtered through celite and concentrated. The resulting oily compound was stirred in 5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 1 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 1 .1 1 g (43.2 %); 1 H NMR (DMSO-d6, 300 MHz): δ 8.21 (s, 1 H), 7.38 (s, 1 H), 3.86 (s, 2H), 2.35-2.37 (m, 2H), 1 .62-1 .66 (m, 6H); MS (ESI+): m/z 215.5 [M+H]+; HPLC purity: 48 %. Example 40:
5- Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide
To a solution of the compound of example 20 (1 .2 g, 3.51 mmol) in DMF (20 mL) wereadded HATU (2.000 g, 5.26 mmol) and Hunig'sBase (1 .837 mL, 10.52 mmol) and the reaction mixture was stirred at RT for 1 hour. To it was added the compound of example 39 (0.903 g, 4.21 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL).The combined organic extracts
were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.753 g (41 .8 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .41 (s, 1 H), 8.58 (s, 1 H), 8.52 (s, 1 H), 7.52 (s, 1 H), 4.14 (d, J= 4.8 Hz, 2H),3.83-3.80 (m, 2H), 3.22 (d, J= 10.8 Hz, 2H), 3.05-2.95 (m, 3H), 2.35-2.37 (m, 4H), 2.18 (s, 3H), 1 .64- 1 .43 (m, 8H), 0.79 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 503.1 [M+H]+; HPLC Purity: 97.88 %.
Example 41 :
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-N- ((3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3- carboxamide
The compound of example 40 (100 mg, 0.199 mmol) was added to a stirred solution of 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (91 mg, 0.299 mmol), PdCI2(dppf)-CH2CI2 adduct (16.25 mg, 0.020 mmol) and
Na2C03 (63.3 mg, 0.597 mmol) in 1 ,4-dioxane (5 mL) and water (1.667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
The ethyl acetate layer was washed with water and brine; and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0-
15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.025 g (20.38 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .41 (s, 1 H),
8.57 (s, 1 H), 7.63-7.60 (m, 2H), 7.44-7.33 (m, 4H), 7.08 (s, 1 H), 4.17 (d, J= 5.1 Hz, 2H), 3.56-3.79 (m, 4H), 3.56-3.46 (m, 6H), 3.25 (t, J= 1 1 .4 Hz, 2H), 3.13-3.02
(m, 3H), 2.44-2.37 (m, 8H), 2.28 (s, 3H), 1 .62-1 .52 (m, 6H), 0.84 (t, J= 6.6 Hz,
3H); MS (ESI+): m/z 599.6 [M+H]+; HPLC Purity: 97.17 %.
Example 42:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-morpholino-N-((3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide
The compound of example 40 (100 mg, 0.199 mmol) was added to a stirred solution of (4-morpholinophenyl)boronic acid (61.8 mg, 0.299 mmol), PdCI2(dppf)-
CH2CI2 adduct (16.25 mg, 0.020 mmol) and Na2C03 (63.3 mg, 0.597 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 ml_). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with waterand brine; and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.016 g (13.61 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .40 (s, 1 H), 8.55 (s, 1 H), 7.53 (d, J= 8.1 Hz, 2H), 7.38 (s, 1 H), 7.28 (s, 1 H), 7.08 (s, 1 H), 7.02 (d, J= 7.8 Hz, 2H), 4.16 (d, J= 5.1 Hz, 2H), 3.84-3.73 (m, 6H), 3.25 (t, J= 1 1.4 Hz, 2H), 3.13-3.02 (m, 7H), 2.44-2.37 (m, 4H), 2.26 (s, 3H), 1.62-1 .51 (m, 8H), 0.84 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 585.3 [M+H]+; HPLC Purity: 99.0 %. Example 43:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1- yl)pyridin-3-yl)-N-((3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4- yl)methyl)benzamide
The compound of example 40 (100 mg, 0.199 mmol) was added to a stirred solution of 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)piperazine (91 mg, 0.299 mmol), PdCI2(dppf)-CH2Cl2 adduct (16.25 mg, 0.020 mmol) and Na2C03 (63.3 mg, 0.597 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine, and dried over anhydrous sodium sulphate. The organic layers were concentrated to obtain a crude material, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.016 g (12.73 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .39 (s, 1 H), 8.54 (s, 1 H), 8.42(s, 1 H), 7.85-7.82 (m, 1 H), 7.39 (s, 1 H), 7.29 (s, 1 H), 7.08 (s, 1 H), 6.90 (d, J= 8.7 Hz, 1 H), 4.16 (d, J= 5.1 Hz, 2H), 3.84-3.81 (m, 2H), 3.54-3.50 (m, 4H), 3.21 (t, J= 1 1 .4 Hz, 2H), 3.13-3.08 (m, 3H), 2.39-2.30 (m, 8H), 2.26 (s,
3H), 2.21 (s, 3H), 1 .68-1 .34 (m, 8H), 0.83 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 599.6 [M+H]+; HPLC Purity: 94.78 %.
Example 44:
1-Ethoxy-3-oxo-2,3,5,6,7,8-hexahydroisoquinoline-4-carbonitrile
A mixture of ethyl 2-oxocyclohexanecarboxylate (4.70 mL, 29.4 mmol), malononitrile (2.035 mL, 32.3 mmol), acetic acid (1 .682 mL, 29.4 mmol), ammonium acetate (250 mg) and benzene (20 mL) : ethanol (20.00 mL) (1 :1 ) were heated to reflux for 48 h. Water was removed from the reaction mixture continuously by allowing the condensed vapors to flow back through 3A molecular sieves into the reaction flask. Ateach interval of 4,8,18, and 24 h of the reaction time, portions of 200 mg ammonium acetate was added. Then the reaction mixture was evaporated to dryness under vacuum, the residue was taken up in boiling 1 N NaOH (60 mL).The resulting solution was then filtered and washed with cold 1 N NaOH. The solid was suspended in 75 mL water and acidified using concentrated HCI. The resulting precipitate was cooled in ice, filtered, washed with water, dried and recrystallised from benzene to yield the title compound.
Yield: 6.02 g (94 %); 1 H NMR (DMSO-d6, 300 MHz): δ 7.36 (s, 1 H), 4.33 (q, J= 4.2 Hz, 2H), 2.66-2.65 (m, 2H), 2.39-2.37 (m, 2H), 1 .69-1.60 (m, 4H), 1.30 (t, J= 4.2 Hz, 3H); MS (ESI+): m/z 219.1 [M+H]+; HPLC Purity: 99.66 %.
Example 45:
4-(Aminomethyl)-1-ethoxy-5,6,7,8-tetrahydroisoquinolin-3(2H)-one
hydrochloride
To a solution of the compound of example 44 (6 g, 27.5 mmol) in acetic acid (25 mL) were added Pd/C (0.731 g, 6.87 mmol), sodium acetate (4.51 g, 55.0 mmol) and platinum (IV) oxide (0.156 g, 0.687 mmol) The reaction flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the resulting mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 2.5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.5 mL concentrated HCI and 2 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether, and dried to yield the title compound.
Yield: 4.4 g (57.3 %); 1 H NMR (DMSO-d6, 300 MHz): δ 7.25 (s, 1 H), 4.28 (q, J= 4.2 Hz, 2H), 3.81 (s, 2H), 2.72-2.64 (m, 2H), 2.42-2.37 (m, 2H), 1.68-1 .52 (m, 4H), 1 .29 (t, J= 4.2 Hz, 3H); MS (ESI+): m/z 259.1 [M+H]+;HPLC Purity: 92.66 %.
Example 46:
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((5-methyl-7- oxo-2,4,6,7-tetrahydro-1 H-pyrano[3,4-c]pyridin-8-yl)methyl)benzamide
To the compound of example 20 (2 g, 5.84 mmol) in DMF (20 mL) were added HATU (3.33 g, 8.77 mmol) and Hunig's Base (3.06 mL, 17.53 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 45 (1 .815 g, 7.01 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum; the residue obtained was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 1 .4 g (42.3 %); 1 H NMR (DMSO-d6, 300 MHz): δ 10.20 (s, 1 H), 8.33 (s, 1 H), 7.32 (s, 1 H), 7.09 (s, 1 H), 4.29-4.25 (m, 4H), 3.82 (d, J= 6.6 Hz, 2H), 3.21 (t, J= 6.6 Hz, 2H),3.03-2.92 (m, 3H), 2.70-2.69 (m, 2H), 2.43-2.41 (m, 2H), 2.15 (s, 3H), 1 .66-1 .48 (m, 8H), 1 .26 (t, J= 6.9 Hz, 3H), 0.78 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 547.7 [M+H]+; HPLC Purity: 96.5 %.
Example 47:
N-((1-ethoxy-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide
The compound of example 46 (150 mg, 0.274 mmol) was added to a stirred solution of 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (125 mg, 0.412 mmol), PdCI2(dppf)-CH2CI2 adduct (22.41 mg, 0.027 mmol) and Na2C03 (87 mg, 0.823 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3 h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate.
The ethyl acetate was washed with waterand brine and dried over sodium sulphate to obtain a crude mixture, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.010 g (5.64 %); 1 H NMR (DMSO-d6, 300 MHz): δ 10.24 (s, 1 H), 8.37 (s, 1 H), 7.59-7.41 (m, 5H), 7.22 (s, 1 H), 4.32 (d, J= 3.0 Hz, 2H), 4.25 (q, J= 4.2 Hz, 2H), 3.83 (d, J= 6.0 Hz, 2H),3.58-3.49 (m, 6H), 3.25 (t, J= 6.6 Hz, 2H),3.10-3.02 (m, 3H), 2.73 (s, 2H), 2.42-2.36 (m, 6H), 2.25 (s, 3H), 1 .67-1 .52 (m, 8H), 1 .28 (t, J= 4.2 Hz, 3H), 0.83 (t, J= 3.9 Hz, 3H); MS (ESI+): m/z 643.5 [M+H]+; HPLC Purity: 99.6 %.
Example 48:
N-((1-ethoxy-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-3-
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1- yl)pyridin-3-yl)benzamide
The compound of example 46 (150 mg, 0.274 mmol) was added to a stirred solution of 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)piperazine (125 mg, 0.412 mmol), PdCI2(dppf)-CH2Cl2 adduct (22.41 mg, 0.027 mmol) and Na2C03 (87 mg, 0.823 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 ml_). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with waterand brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.008 g (4.10 %); 1 H NMR (DMSO-d6, 300 MHz): δ 10.25 (s, 1 H),
8.40 (s, 1 H), 8.38 (s, 1 H), 7.82 (d, J= 5.4 Hz, 1 H), 7.37 (s, 1 H), 7.17 (s, 1 H), 6.90 (d, J= 5.4 Hz, 1 H), 4.31 (d, J= 3.3 Hz, 2H), 4.26 (q, J= 4.8 Hz, 2H), 3.83 (d, J= 6.0 Hz, 2H),3.58-3.52 (m, 4H), 3.25 (t, J= 6.6 Hz, 2H), 3.09-2.98 (m, 3H), 2.73 (s, 2H), 2.42-2.36 (m, 6H), 2.23 (s, 6H), 1 .67-1.52 (m, 8H), 1 .28 (t, J= 4.2 Hz, 3H), 0.82 (t, J= 3.9 Hz, 3H); MS (ESI+): m/z 643.4 [M+H]+; HPLC Purity: 90.48 %.
Example 49:
3-(Aminomethyl)-6-methylpyridin-2(1 H)-one hydrochloride
To a solution of 6-methyl-2-oxo-1 ,2-dihydropyridine-3-carbonitrile (5 g, 37.3 mmol) in acetic acid (10 mL) were added Pd/C (0.992 g, 9.32 mmol), sodium acetate (6.12 g, 74.6 mmol) and platinum (IV) oxide (0.212 g, 0.932 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the resulting mixturewas filtered through celite and concentrated. The resulting oily compound was stirred in 25 mLof concentrated HCI and filtered. The filtrate was stirred in a mixture of 5 mL of concentrated HCI and 25 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 6.0 g (81 %); 1 H NMR (DMSO-d6, 300 MHz): δ 8.21 (s, 1 H), 7.40 (s,
1 H), 6.05 (s, 1 H), 3.87 (s, 2H), 2.18 (s, 3H); MS (ESI+): m/z175.5 [M+H]+; HPLC Purity: 87.81 %.
Example 50:
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((6-methyl-2- oxo-1 ,2-dihydropyridin-3-yl)methyl)benzamide
To the compound of example 20 (2 g, 5.84 mmol) in DMF (20 mL) wereadded HATU (3.33 g, 8.77 mmol) and Hunig's Base (3.06 mL, 17.53 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 49 (1 .225 g, 7.01 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum; the residue obtained was extracted using ethyl acetate and washed with water (3x25 mL).The combined organic extracts were distilled off to obtain a crude mixture, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 1 .5 g (34.7 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .65 (s, 1 H), 8.59 (s, 1 H), 7.35-7.06 (m, 3H), 5.99-5.92 (m, 1 H), 3.79-3.82 (m, 2H), 3.28-3.21 (m, 2H), 3.04-2.97 (m, 3H),2.15 (s, 3H), 1 .98 (s, 3H),1 .59-1.14 (m, 6H), 1 .85-0.73 (m, 3H); MS (ESI+): m/z 463.2 [M+H]+; HPLC Purity: 62.5 %.
Example 51 :
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((6-methyl-2-oxo-1 ,2- dihydropyridin-3-yl)methyl)-4'-(morpholinomethyl)-[1 ,1 '-biphenyl]-3- carboxamide
The compound of example 50 (200 mg, 0.433 mmol) was added to a stirred solution of 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (197 mg, 0.649 mmol), PdCI2(dppf)-CH2CI2adduct (35.3 mg, 0.043 mmol) and Na2C03 (138 mg, 1 .298 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 ml_). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The reaction mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.021 g (8.21 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .65 (s, 1 H), 8.59 (s, 1 H), 7.62 (d, J= 8.1 Hz, 2H), 7.43-7.34 (m, 4H), 7.26 (d, J= 6.9 Hz, 1 H), 5.99 (d, J= 6.9 Hz, 1 H), 4.17 (d, J= 5.4 Hz, 2H), 3.85-3.82 (m, 2H),3.59-3.49 (m, 6H), 3.31 -3.09 (m, 5H), 2.38-2.34 (m, 4H), 2.28 (s, 3H), 2.15 (s, 3H), 1 .65-1 .52 (m, 4H), 0.84 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 559.4 [M+H]+; HPLC Purity: 94.53 %. Example 52:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((6-methyl-2-oxo-1 ,2- di hydropyridi n-3-yl)methyl)-5-(6-(4-methyl pi perazi n-1 -yl)pyrid i n-3- yl)benzamide
The compound 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pyridin-2-yl)piperazine (197 mg, 0.649 mmol) was added to a stirred solution of the compound of example 50 (200 mg, 0.433 mmol), PdCI2(dppf)-CH2CI2 adduct (35.3 mg, 0.043 mmol) and Na2C03 (138 mg, 1 .298 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The resultingmixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-15 % methanol in chloroform ) to yield the title compound.
Yield: 0.017 g (6.66 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .66 (s, 1 H), 8.56 (s, 1 H), 8.49 (s, 1 H), 7.94 (d, J= 9.0 Hz, 1 H), 7.41 (s, 1 H), 7.32 (s, 1 H), 7.26 (d, J= 6.9 Hz, 1 H), 7.03 (d, J= 8.7 Hz, 1 H), 5.99 (d, J= 6.9 Hz, 1 H), 4.16 (s, 2H), 3.84-3.82 (m, 2H),3.25-3.15 (m, 4H), 3.18-3.02 (m, 6H),2.82-2.76 (m, 3H), 2.26 (s, 3H), 2.15 (s, 3H), 1 .65-1 .42 (m, 4H), 1 .17 (t, J= 7.2 Hz, 3H), 0.83 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 559.4 [M+H]+; HPLC Purity: 96.17 %.
Example 53:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((6-methyl-2-oxo-1 ,2- dihydropyridin-3-yl)methyl)-5-(6-morpholinopyridin-3-yl)benzamide
The compound of example 50 (200 mg, 0.433 mmol) was added to a stirred solution of 4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)morpholine (188 mg, 0.649 mmol), PdCI2(dppf)-CH2CI2 adduct (35.3 mg, 0.043 mmol) and Na2C03 (138 mg, 1 .298 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The resulting mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.060 g (24.41 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .64 (s, 1 H), 8.56 (s, 1 H), 8.46 (s, 1 H), 7.87 (d, J= 9.0 Hz, 1 H), 7.41 (s, 1 H), 7.31 (s, 1 H), 7.27 (d, J= 6.9 Hz, 1 H), 6.92 (d, J= 8.7 Hz, 1 H), 6.01 (d, J= 6.9 Hz, 1 H), 4.16 (s, 2H), 3.82-3.81 (m, 2H),3.74-3.70 (m, 4H), 3.48-3.44 (m, 4H), 3.28-3.24 (m, 2H),3.08- 3.02 (m, 3H), 2.26 (s, 3H), 2.15 (s, 3H), 1 .65-1 .42 (m, 4H), 0.84 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 546.4 [M+H]+; HPLC Purity: 96.03 %.
Example 54:
3- Acetyl -1 -methyl pi perid i n-4-one
To a solution of 1 -methylpiperidin-4-one (10 g, 88 mmol) in THF (45 mL) at
-78 °C was added a solution of LDA (53.0 mL, 106 mmol) in THF drop wise via a syringe. After 1 h of stirring, acetyl cyanide (7.52 mL, 106 mmol) was added drop wise over a 10 min period. After 30 min, the reaction mixture was quenched with
few drops of water. The solvent was evaporated and the residue was triturated to yield the title compound.
Yield: 13 g (85 %); 1 H NMR (DMSO-d6, 300 MHz): δ 3.58 (t, J= 6.3 Hz, 1 H), 2.61 -2.57 (m, 2H), 2.40-2.32 (m, 4H), 2.26 (s, 3H), 2.24 (s, 3H); MS (ESI+): m/z156.1 [M+H]+; HPLC Purity: 89.6 %.
Example 55:
1 ,7-Dimethyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4-carbonitrile
To a solution of the compound of example 54 (13 g, 84 mmol) in ethanol (25 mL) were added 2-cyanoacetamide (7.04 g, 84 mmol) and piperidine (4.15 mL, 41 .9 mmol) and the reaction mixture was stirred at 50 °C for 2 days. After completion of the reaction, the precipitated solid was filtered, washed with ethanol and dried to yield the title compound.
Yield: 0.410 g (0.024 %); 1 H NMR (DMSO-d6, 300 MHz):<5 3.14-3.12 (m, 2H), 2.58-2.54 (m, 2H), 2.45-2.41 (m, 2H), 2.29 (s, 3H), 2.01 (s, 3H); MS (ESI+): m/z 204.1 [M+H]+.
Example 56:
1 ,7-Dimethyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4-carbonitrile To a solution of the compound of example 55 (0.4 g, 1 .968 mmol) in acetic acid (10 mL) were added Pd/C (0.052 g, 0.492 mmol), sodium acetate (0.323 g, 3.94 mmol) and platinum (IV) oxide (0.01 1 g, 0.049 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the resulting mixturewas filtered through celite and concentrated. The resulting oily compound was stirred in 2.0 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.4 mL of concentrated HCI and 2 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.215 g (44.8 %); 1 H NMR (DMSO-d6, 300 ΜΗζ):δ 3.78 (s, 2H), 3.57 (S, 2H), 3.15-3.07 (m, 4H), 2.85 (s, 3H), 2.18 (s, 3H); MS (ESI+): m/z 244.5 [M+H]+.
Example 57:
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((5-methyl-7- oxo-2,4,6,7-tetrahydro-1 H-pyrano[3,4-c]pyridin-8-yl)methyl)benzamide
The compound of example 20 (50 mg, 0.094 mmol) was added to a stirred solution of 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (42.8 mg, 0.141 mmol), PdCI2(dppf)-CH2Cl2 adduct (7.68 mg, 9.41 μηιοΙ) and Na2C03 (29.9 mg, 0.282 mmol) in 1 ,4-dioxane (5 mL) and water (1.667 ml_). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The resultingmixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate was washed with water and brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography(silica gel, 10-15 % methanol in chloroform) to yield the title compound.
Yield: 0.103 g (26.3 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .61 (s, 1 H), 8.27 (s, 1 H), 7.30 (s, 1 H), 7.07 (s, 1 H), 4.23 (d, J= 4.5 Hz, 2H), 3.83-3.79 (m, 2H), 3.26-3.18 (m, 4H), 3.01 -2.89 (m, 5H), 2.78-2.67 (m, 2H),2.43 (s, 3H), 2.13 (s, 3H), 2.08 (s, 3H), 1 .60-1 .47 (m, 4H), 0.77 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 532.4 [M+H]+; HPLC Purity: 99.27 %.
Example 58:
N-((1 ,7-dimethyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5- (ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide
The compound of example 57 (50 mg, 0.094 mmol) was added to a stirred solution of 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (42.8 mg, 0.141 mmol), PdCI2(dppf)-CH2CI2 adduct (7.68 mg, 9.41 μηιοΙ) and Na2C03 (29.9 mg, 0.282 mmol) in 1 ,4-dioxane (5 mL) and water (1.667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The resultingmixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine and then dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.007 g (10.40 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .54 (s, 1 H), 8.24 (s, 1 H), 7.58-7.55 (m, 2H), 7.38-7.35 (s, 2H), 7.31 (s, 1 H), 7.07 (m, 1 H), 4.23 (d, J= 4.5 Hz, 2H), 3.83-3.79 (m, 4H),3.56-3.52 (m, 2H), 3.26-3.17 (m, 6H), 3.09- 2.72 (m, 7H), 2.35-2.31 (m, 2H), 2.30 (s, 3H),2.13 (s, 3H), 2.07 (s, 3H), 1.61 -1 .38 (m, 6H), 0.77 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 628.7 [M+H]+; HPLC Purity: 87.75 %.
Example 59:
Tert-butyl 4-oxopiperidine-1 -carboxylate
A solution of triethylamine (51 .4 ml_, 369 mmol) was added drop wise to a cold suspension of piperidin-4-one, HCI salt (50 g, 369 mmol) and BOC-anhydride (86 ml_, 369 mmol) in DCM (200 mL) under niotrogen atmosphere. The reaction mixture was stirred at RT for 16 h. The reaction mixture was diluted with (250ml_) water, the phases were separated and the aqueous solution was washed with brine (100ml_), dried over anhydrous sodium sulphate and evaporated to obtain a solid, which was further treated with silica to obtain a waxy white solid, which was filtered and dried to yield the title compound.
Yield: 53.6 g (73%); 1 H NMR (DMSO-d6, 300 MHz):<5 3.73 (t, J= 6.0 Hz, 4H), 2.45 (t, J= 6.0 Hz, 4H), 1 .51 (s, 9H);MS (ESI+): m/z 200.2 [M+H]+;HPLC Purity: 95.02 %.
Example 60:
Tert-butyl 3-acety l-4-oxop i perid i ne-1 -carboxyl ate
The compound of example 59 (50 g, 251 mmol) and pyrrolidine (41 .5 mL, 502 mmol) were dissolved in dioxane (50 mL) and the resulting mixture was refluxed for 3 hunder nitrogen using a Dean and Stark apparatus. The reaction mixture was then cooled to RT and concentrated. The residue was dissolved in toluene (250 mL), then acetic anhydride (52.1 mL, 552 mmol) was added and the resulting mixture was stirred under nitrogen. Water (60 mL) was added and the resulting mixture was refluxed for 1 h; then cooled to RT and concentrated. The residue was dissolved in water (100 mL) and the aqueous phase was extracted twice with ethyl acetate (100 mL).The combined organic extracts were washed
with 5 % w/w HCI solution (50 mL) and dried over anhydrous sodium sulphate. The organic extract was concentrated to yield the title compound.
Yield: 59.2 g (95 %); 1 H NMR (DMSO-d6, 300 MHz): δ 4.19 (s, 2H), 3.73 (t, J= 6.0 Hz, 1 H), 3.59 (t, J= 6.0 Hz, 2H), 2.45 (t, J= 6.0 Hz, 2H), 2.14 (s, 3H), 1 .49 (s, 9H); MS (ESI+): m/z 242.2 [M+H]+; HPLC Purity: 97.19 %.
Example 61 :
Tert-butyl 5-cyano-8-methyl-6-oxo-3,4,6,7-tetrahydro-2,7-naphthyridine- 2(1 H)-carboxylate
To a solution of the compound of example 60 (59 g, 245 mmol) in ethanol
(150 mL) were added 2-cyanoacetamide (20.56 g, 245 mmol) and piperidine (12.1 1 mL, 122 mmol) and the reaction mixture was stirred at 75 °C for 7 h. After completion of the reaction, the precipitated solid was filtered, washed with ethanol and dried to yield the title compound.
Yield: 27.5 g (38.9 %); 1 H NMR (DMSO-d6, 300 MHz): δ 4.20 (s, 2H), 3.52
(t, J= 5.7 Hz, 2H), 2.79 (t, J= 6.0 Hz, 2H), 2.23 (s, 3H), 1 .42 (s, 9H); MS (ESI+): m/z 290.1 [M+H]+.
Example 62:
1-Methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4-carbonitrile
To a stirred solution of the compound of example 61 (15 g, 51 .8 mmol) in dry DCM (100 mL), was added TFA (7.99 mL, 104 mmol) and the reaction mixture was stirred at RT for 16 h. The precipitated solid was filtered, washed with DCM and dried to yield the title compound.
Yield: 7.5 g (71 .6 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.75 (s, NH), 9.28
(s, NH), 4.01 (s, 2H), 3.33 (t, J= 6.0 Hz, 2H), 2.99 (t, J= 6.0 Hz, 2H), 2.62 (s, 3H); MS (ESI+): m/z 190.2 [M+H]+; HPLC Purity: 93.68 %.
Example 63:
7-Ethyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4- carbonitrile
To a solution of the compound of example 62 (1 .5 g, 7.93 mmol) in DCM (25 mL) and DMF (2.5 mL) were added acetaldehyde (0.895 mL, 15.86 mmol) and
acetic acid (2.72 mL, 47.6 mmol) and stirred at RT for 2h. The reaction mixture was cooled to 0 °C, sodium triacetoxyborohydride (5.04 g, 23.78 mmol) was added in portions over a period of 1 h and the reaction mixture was stirred at RT for 16h. After completion of the reaction, water was added and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-15% methanol in chloroform) to yield the title compound.
Yield: 0.850 g (45.5 %); 1H NMR (DMSO-d6, 300 MHz): δ 13.72 (s, NH), 3.03 (t, J= 6.0 Hz, 2H), 2.72-2.60 (m, 6H), 2.39 (s, 3H), 1 .20 (t, J= 6.0 Hz, 3H);MS (ESI+): m/z 218.2 [M+H]+; HPLC Purity: 92.26 %.
Example 64:
4-(aminomethyl)-7-ethyl-1 -methyl-5,6,7,8-tetrahydro-2,7-naphthyridin-3(2H)- one hydrochloride
To a solution of the compound of example 63 (800 mg, 3.68 mmol) in acetic acid (10 mL), were added Pd/C (98 mg, 0.921 mmol), sodium acetate (604 mg, 7.36 mmol) and platinum (IV) oxide (20.90 mg, 0.092 mmol). The flask was shaken under atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the resulting mixturewas filtered through celite and concentrated. The resulting oily compound was stirred in 4 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.8 mL concentrated HCI and 5 mL of EtOH at 5- 10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.51 1 g (53.8 %); 1H NMR (DMSO-d6, 300 MHz):<5 12.23 (s, NH),
8.07 (s, 2H), 4.29-3.75 (m, 6H), 3.22-3.17 (m, 4H), 2.22 (s, 3H), 1 .33 (t, J= 6.0 Hz, 3H);MS (ESI+): m/z 258.2 [M+H]+.
Example 65:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4-methyl-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution ofthe compound of example 26 (100 mg, 0.228 mmol) in DMF (5 mL) were added HATU (130 mg, 0.342 mmol) and Hunig's Base (0.1 19 ml_, 0.684 mmol) and the reaction mixture was stirred at RT for 1 hour. To it was added the compound of example 64 (70.5 mg, 0.274 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 ml_).The combined organic extracts were distilled off to obtain a crude material, which was purified by column chromatograhy(silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.090 g (57.8 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .65 (s, 1 H),
8.25 (s, 1 H), 7.59-7.56 (m, 2H), 7.39-7.36 (m, 3H), 7.21 (s, 1 H), 4.29 (s, 2H), 3.92- 3.81 (m, 2H), 3.68-3.50 (m, 8H), 3.25-3.07(m, 7H), 2.37-2.31 (m, 4H), 2.24 (s, 3H), 2.1 1 (s, 3H), 1 .64-1.23 (m, 8H), 1 .13(s, 3H), 0.83 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 642.7 [M+H]+; HPLC Purity: 94.01 %.
Example 66:
Methyl 3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methyl piperazin-1-yl)pyridin-3-yl)benzoate
The compound of example 19 (2 g, 5.61 mmol) was added to a stirred solution of 1 -methyl-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)piperazine (1 .702 g, 5.61 mmol), PdCI2(dppf)-CH2Cl2 adduct (0.458 g, 0.561 mmol) and Na2C03 (1.785 g, 16.84 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 mL). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The resulting mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 2.08 g (78 %); 1 H NMR (DMSO-d6, 300 MHz): δ 8.42 (s, 1 H), 7.85- 7.83 (m, 1 H), 7.65 (s, 1 H), 7.51 (s, 1 H), 6.90 (d, J= 9.0 Hz, 1 H), 3.84 (s, 3H), 3.83- 3.73 (m, 2H), 3.53-3.41 (m, 4H), 3.29-3.22 (m, 2H), 3.1 1 -3.01 (m, 3H), 2.49-2.42 (m, 4H), 2.41 (s, 3H), 2.23 (s, 3H), 1 .68-1 .13 (m, 4H), 0.81 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 453.3 [M+H]+; HPLC Purity: 95.67 %.
Example 67:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1- yl)pyridin-3-yl)benzoic acid
A solution of NaOH (0.177 g, 4.42 mmol) in water (2 mL) was added to a solution of the compound of example 66 (2 g, 4.42 mmol) in methanol (2 mL) and THF (25 mL). The reaction mixture was stirred at 60 °C for 3 h. Methanol was removed from the reaction mixture and the residue was acidified to neutral pH. The precipitated solid was filtered, washed with water and dried to yield the title compound.
Yield: 0.802 g (41 .2 %); 1 H NMR (DMSO-d6, 300 MHz): δ 8.41 (s, 1 H), 7.84-7.81 (m, 1 H), 7.62 (s, 1 H), 7.50 (s, 1 H), 6.90 (d, J= 9.0 Hz, 1 H), 3.84-3.80 (m, 2H), 3.53-3.49 (m, 4H), 3.25 (d, J= 1 1 .4 Hz, 2H), 3.10-3.02 (m, 3H), 2.49-2.44 (m, 4H), 2.42 (s, 3H), 2.24 (s, 3H), 1 .68-1 .48 (m, 4H), 0.82 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 439.3 [M+H]+; HPLC Purity: 97.35 %.
Example 68:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-(4- methylpiperazin-1-yl)pyridin-3-yl)benzamide
To the compound of example 67 (75 mg, 0.171 mmol) in DMF (5 mL) wereadded HATU (98 mg, 0.257 mmol) and Hunig's Base (0.090 mL, 0.513 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 64 (52.9 mg, 0.205 mmol) and stirredfor 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to give crude material which was purified using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.013 g (12.10 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .61 (s, 1 H),
8.41 (s, 1 H), 8.21 (s, 1 H), 7.83 (d, J= 6.6 Hz, 1 H), 7.36 (s, 1 H), 7.18 (s, 1 H), 6.92 (d, J= 6.6 Hz, 1 H), 4.28 (s, 2H), 3.84-3.81 (m, 2H), 3.59-3.44 (m, 6H), 3.33-3.09 (m, 4H), 3.06-2.71 (m, 5H), 2.64-2.52 (m, 6H), 2.38 (s, 3H), 2.22 (s, 3H), 2.1 1 (s,
3H), 1 .64-1 .48 (m, 4H), 1 .13(t, J= 6.0 Hz, 3H), 0.82 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 642.6 [M+H]+; HPLC Purity: 98.39 %.
Example 69:
Methyl 3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-morpholino pyridi n-3-yl)benzoate
The compound of example 19 (1 g, 2.81 mmol) was added to a stirred solution of 4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)morpholine (0.814 g, 2.81 mmol), PdCI2(dppf)-CH2CI2adduct (0.229 g, 0.281 mmol) and Na2C03 (0.893 g, 8.42 mmol) in 1 ,4-dioxane (5 mL) and water (1 .667 ml_). The reaction mixture was stirred at 80 °C for 3h under nitrogen atmosphere. The resulting mixture was cooled, diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine, and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-80 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 1 .1 g (89 %); 1 H NMR (DMSO-d6, 300 MHz): δ 8.45 (d, J= 2.4 Hz, 1 H), 7.89-7.86 (m, 1 H), 7.66 (s, 1 H), 7.58 (s, 1 H), 6.91 (d, J= 8.7 Hz, 1 H), 3.84 (s, 3H), 3.83-3.80 (m, 2H), 3.73-3.70 (m, 4H), 3.50-3.47 (m, 4H), 3.30-3.22 (m, 2H), 3.1 1 -3.09 (m, 3H), 2.41 (s, 3H), 1 .69-1.48 (m, 4H), 0.82 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 440.5 [M+H]+; HPLC Purity: 99.69 %.
Example 70:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-morpholinopyridin-3- yl)benzoic acid
A solution of NaOH (0.091 g, 2.275 mmol) in water (2 mL) was added to a solution of the compound of example 69 (1 g, 2.275 mmol) in methanol (2 mL) and THF (25 mL). The reaction mixture was stirred at 60 °C for 3 h. The methanol was removed and the residue was acidified to neutral pH. The precipitated solid was filtered, washed with water and dried to yield the title compound.
Yield: 0.420 g (30.6 %); 1 H NMR (DMSO-d6, 300 MHz): δ 13.08 (s, 1 H), 8.45 (s, 1 H), 7.87-7.84 (m, 1 H), 7.60 (s, 1 H), 7.49 (s, 1 H), 6.92-6.89 (m, 1 H), 3.84-3.80 (m, 2H), 3.71 -3.70 (m, 4H), 3.49-3.47 (m, 4H), 3.29-3.21 (m, 2H), 3.10-
3.02 (m, 3H), 2.41 (s, 3H), 1 .69-1 .45 (m, 4H), 0.82 (t, J= 6.0 Hz, 3H); MS (ESI+): m/z 426.2 [M+H]+; HPLC Purity: 70.5 %.
Example 71 :
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6- morpholinopyridin-3-yl)benzamide
To a solution of the compound of example 70 (75 mg, 0.176 mmol) in DMF (5 mL) wereadded HATU (101 mg, 0.264 mmol) and Hunig'sBase (0.092 mL, 0.529 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added compound of example 64 (54.5 mg, 0.212 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.015 g (13.46 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .57 (s, 1 H), 8.42 (s, 1 H), 8.23 (s, 1 H), 7.82 (s, 1 H), 7.37 (s, 1 H), 7.18 (s, 1 H), 6.91 (d, J= 8.4 Hz, 1 H), 4.28 (s, 2H), 3.84-3.71 (m, 6H), 3.48-3.44 (m, 4H), 3.28-3.21 (m, 4H), 3.09-2.87 (m, 5H), 2.87-2.84 (m, 2H), 2.22 (s, 3H), 2.10 (s, 3H), 1 .68-1 .48 (m, 6H), 1 .08 (t, J= 6.0 Hz, 3H), 0.81 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 629.3 [M+H]+; HPLC Purity: 99.43 %.
Example 72:
1-Methyl-3-oxo-7-propyl-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4- carbonitrile
To a solution of the compound of example 62 (1 .5 g, 7.93 mmol) in DMF (2.500 mL), DCM (25 mL) were added propionaldehyde (1 .151 mL, 15.86 mmol) and acetic acid (2.72 mL, 47.6 mmol) and the reaction mixture was stirred at RT for 2h. The reaction mixture was cooled to 0 °C and sodium triacetoxyborohydride (5.04 g, 23.78 mmol) was added in portions over a period of 1 h and stirred at RT for 16h. After completion of reaction, water was added and extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine; and dried over
anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-15 % methanol in chloroform) to yield the title compound.
Yield: 0.530 g (28.8 %); 1H NMR (DMSO-d6, 300 MHz): δ 12.38 (s, NH), 3.24 (s, 2H), 2.79 (t, J= 5.7 Hz, 2H), 2.58 (t, J= 5.7 Hz, 2H), 2.41 (t, J= 7.2 Hz, 2H), 2.20 (s, 3H), 1 .53-1 .47 (m, 2H), 0.87 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 232 [M+H]+; HPLC Purity: 99.68 %.
Example 73:
4-(Aminomethyl)-1-methyl-7-propyl-5,6,7,8-tetrahydro-2,7-naphthyridin- 3(2H)-one hydrochloride
To a solution of the compound of example 72 (500 mg, 2.162 mmol) in acetic acid (10 ml) were added Pd/C (57.5 mg, 0.540 mmol), sodium acetate (355 mg, 4.32 mmol) and platinum (IV) oxide (12.27 mg, 0.054 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the reaction mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 2.5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.5 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.495 g (84 %); 1H NMR (DMSO-d6, 300 MHz): δ 12.22 (s, NH), 4.27-4.23 (m, 2H), 3.97-3.90 (m, 2H), 3.84-3.81 (m, 2H), 3.20-3.17 (m, 2H), 3.1 1 - 3.02 (m, 2H), 2.21 (s, 3H), 1 .89-1 .71 (m, 2H), 0.92 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 272.5 [M+H]+.
Example 74:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-7- propyl-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (50 mg, 0.1 14 mmol) in DMF
(5 mL) were added HATU (65.0 mg, 0.171 mmol) and Hunig's Base (0.060 mL, 0.342 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 73 (37.2 mg, 0.137 mmol) and
stirred for 16 . The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.035 g (45.3 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .88 (s, 1 H), 9.71 (s, 1 H), 8.32 (s, 1 H), 7.51 -7.23 (m, 5H), 4.28 (s, 2H), 4.04-4.01 (m, 2H), 3.91 - 3.80 (m, 2 H), 3.71 -3.42 (m, 6H), 3.24-3.08 (m, 5H), 2.37-2.31 (m, 4H), 2.24 (s, 3H), 2.14 (s, 3H), 1.64-1 .48 (m, 6H), 1 .23-1 .51 (m, 6H), 0.91 (t, J= 7.2 Hz, 3H), 0.82 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 656.5 [M+H]+; HPLC Purity: 96.72 %.
Example 75:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7- propyl-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6- morpholinopyridin-3-yl)benzamide
To a solution of the compound of example 70 (50 mg, 0.1 18 mmol) in DMF (5 mL) were added HATU (67.0 mg, 0.176 mmol) and Hunig's Base (0.062 mL, 0.353 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 73 (38.3 mg, 0.141 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.035 g (45.3 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .82 (s, 1 H), 10.01 (s, 1 H), 8.42 (s, 1 H), 7.85-7.83 (m, 1 H), 7.37 (s, 1 H), 7.19 (s, 1 H), 6.91 -6.88 (s, 1 H), 4.28 (s, 2H), 3.84-3.81 (m, 2H), 3.71 -3.60 (m, 4H), 3.49-3.47 (m, 4H), 3.28-3.24 (m, 4H), ), 3.09-2.98 (m, 5H), 2.22 (s, 3H), 2.14 (s, 3H), 1 .71 -1 .42 (m, 6H), 1 .28-1 .17 (m, 4H), 0.90 (t, J= 7.2 Hz, 3H), 0.82 (t, J= 6.9 Hz, 3H);MS (ESI+): m/z 643.5 [M+H]+; HPLC Purity: 98.68 %.
Example 76:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7- propyl-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6-(4- methylpiperazin-1-yl)pyridin-3-yl)benzamide
To a solution ofthe compound of example 67 (100 mg, 0.228 mmol) in DMF (5 mL) wereadded HATU (130 mg, 0.342 mmol) and Hunig's Base (0.1 19 ml_, 0.684 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 73 (74.4 mg, 0.274 mmol) and stirred for 16 h. The reaction solvent was distilled, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.097 g (64.4 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .54 (s, 1 H),
8.39 (s, 1 H), 8.17 (s, 1 H), 7.82-7.79 (m, 1 H), 7.35 (s, 1 H), 7.16 (s, 1 H), 6.91 -6.88 (s, 1 H), 4.28 (s, 2H), 3.84-3.81 (m, 2H), 3.60-3.48 (m, 4H), 3.28-3.20 (m, 4H), 3.18-2.95 (m, 3H), 2.91 -2.81 (m, 2H), 2.58-2.35 (m, 8H), 2.26 (s, 3H), 2.22 (s, 3H), 2.09 (s, 3H), 1 .68-1 .41 (m, 6H), 0.87 (t, J= 7.2 Hz, 3H), 0.82 (t, J= 6.9 Hz, 3H); MS (ESI+): m/z 656.7 [M+H]+; HPLC Purity: 99.3 %.
Example 77:
7-Butyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4- carbonitrile
To a solution of the compound of example 62 (1 .5 g, 7.93 mmol) in a mixture of DCM (25 mL) and DMF (2.5 mL) were added butyraldehyde (1 .429 mL, 15.86 mmol) and acetic acid (2.72 mL, 47.6 mmol) and the reaction mixture was stirred at RT for 2h. The reaction mixture was cooled to 0 °C and sodium triacetoxyborohydride (5.04 g, 23.78 mmol) was added in portions over a period of 1 h and stirred at RT for 16h. After completion of the reaction, water was added and the resulting mixture was extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine, and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-15 % methanol in chloroform) to yield the title compound.
Yield: 0.475 g (21 .9 %); 1H NMR (DMSO-d6, 300 MHz): δ 12.38 (s, NH), 3.33 (s, 2H), 2.78 (t, J= 5.7 Hz, 2H), 2.58 (t, J= 5.7 Hz, 2H), 2.41 (t, J= 7.2 Hz, 2H), 2.20 (s, 3H), 1 .52-1 .43 (m, 2H), 1 .36-1 .24 (m, 2H), 0.89 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 246.1 [M+H]+; HPLC Purity: 89.67 %.
Example 78:
4-(Aminomethyl)-7-butyl-1-methyl-5,6,7,8-tetrahydro-2,7-naphthyridin-3(2H)- one hydrochloride
To a solution of the compound of example 77 (450 mg, 1 .834 mmol) in acetic acid (10 mL) were added Pd/C (48.8 mg, 0.459 mmol), sodium acetate (301 mg, 3.67 mmol) and platinum (IV) oxide (10.41 mg, 0.046 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the resulting mixturewas filtered through celite and concentrated. The resulting oily compound was stirred in 2.25 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.45 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.143 g (27.3 %); 1H NMR (DMSO-d6, 300 MHz): δ 12.21 (s, NH), 8.10 (s, 2H), 3.78 (s, 2H), 3.55 (s, 2H), 3.28-3.15 (m, 6H), 2.21 (s, 3H), 1.87-1 .78 (m, 2H), 1 .37-1 .30 (m, 2H), 0.92 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 286.5 [M+H]+.
Example 79:
N-((7-butyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4- yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (50 mg, 0.1 14 mmol) in DMF (5 mL) were added HATU (65.0 mg, 0.171 mmol) and Hunig's Base (0.060 mL, 0.342 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 78 (39.1 mg, 0.137 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by
using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.009 g (10.83 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .54 (s, 1 H), 8.20 (s, 1 H), 7.60-7.56 (m, 2H), 7.39-7.31 (m, 2H), 7.21 (s, 1 H), 4.29 (s, 2H), 3.87- 3.81 (m, 2H), 3.59-3.48 (m, 6H), 3.22-2.97 (m, 5H), 2.83-2.73 (m, 2H), 2.37-2.34 (m, 2H), 2.24 (s, 3H), 2.08 (s, 3H), 1 .68-1 .33 (m, 6H), 1 .34-1 .23 (m, 10H), 0.88- 0.83 (m, 6H); MS (ESI+): m/z670.6 [M+H]+; HPLC Purity: 91 .91 %.
Example 80:
N-((7-butyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4- yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4- methylpiperazin-1-yl)pyridin-3-yl)benzamide
To a solution of the compound of example 67 (50 mg, 0.1 14 mmol) in DMF (5 mL) wereadded HATU (65.0 mg, 0.171 mmol) and Hunig's Base (0.060 mL, 0.342 mmol) and the reaction mixture was stirred at RT for 1 hour. To it was added the compound of example 78 (39.1 mg, 0.137 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.01 1 g (12.9 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .58 (s, 1 H), 8.40 (s, 1 H), 8.17 (s, 1 H), 7.84-7.81 (m, 1 H), 7.36 (s, 1 H), 7.17 (s, 1 H), 6.92-6.90 (m, 1 H), 4.28 (s, 2H), 3.85-3.81 (m, 2H), 3.58-3.47 (m, 4H), 3.33-3.21 (m, 6H), 3.06-2.82 (m, 7H), 2.81 -2.68 (m, 2H), 2.33 (s, 3H), 2.22 (s, 3H), 2.09 (s, 3H), 1 .64-1 .23 (m, 10H), 0.91 -0.82 (m, 6H); MS (ESI+): m/z 670.6 [M+H]+; HPLC Purity: 89.57 %.
Example 81 :
N-((7-butyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4- yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6- morpholinopyridin-3-yl)benzamide
To a solution of the compound of example 70 (50 mg, 0.1 18 mmol) in DMF (5 mL) were added HATU (67.0 mg, 0.176 mmol) and Hunig's Base (0.062 ml_, 0.353 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 78 (40.3 mg, 0.141 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-15 % methanol in chloroform) to yield the title compound.
Yield: 0.013 g (14.62 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .58 (s, 1 H),
8.40 (s, 1 H), 8.17 (s, 1 H), 7.84-7.81 (m, 1 H), 7.36 (s, 1 H), 7.17 (s, 1 H), 6.92-6.90 (m, 1 H), 4.28 (s, 2H), 3.85-3.81 (m, 2H), 3.58-3.47 (m, 4H), 3.33-3.21 (m, 6H), 3.06-2.82 (m, 7H), 2.81 -2.68 (m, 2H), 2.33 (s, 3H), 2.22 (s, 3H), 2.09 (s, 3H), 1 .64-1 .23 (m, 10H), 0.91 -0.82 (m, 3H); MS (ESI+): m/z 657.6 [M+H]+; HPLC Purity: 86.83 %.
Example 82:
7-lsobutyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4- carbonitrile
To a solution of the compound of example 62 (1 g, 5.29 mmol) in DMF (2.500 ml), DCM (25 mL) were added isobutyraldehyde (0.965 ml, 10.57 mmol) and acetic acid (1 .815 mL, 31 .7 mmol) and the reaction mixture was stirred at RT for 2 h. The reaction mixture was cooled to 0 °C and sodium triacetoxyborohydride (3360 mg, 15.86 mmol) was added in portions over a period of 1 h and stirred at RT for 16 h. After completion of the reaction, water was added and the resulting mixture was extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine, and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-15 % methanol in chloroform) to yield the title compound.
Yield: 0.602 g (46.1 %); 1 H NMR (DMSO-d6, 300 MHz): δ 3.21 (s, 2H), 2.72-2.70 (m, 2H), 2.54 (t, J= 5.7 Hz, 2H), 2.20 (d, J= 7.5 Hz, 2H), 2.14 (s, 3H), 1 .88-1 .84 (m, 1 H), 0.87 (t, J= 6.3 Hz, 6H); MS (ESI+): m/z 246.2 [M+H]+; HPLC Purity: 99.2 %.
Example 83:
4- (Aminomethyl)-7-isobutyl-1-methyl-5,6,7,8-tetrahydro-2,7-naphthyridin- 3(2H)-one hydrochloride
To a solution of the compound of example 82 (500 mg, 2.038 mmol) in acetic acid (10 ml) were added Pd/C (54.2 mg, 0.510 mmol), sodium acetate (334 mg, 4.08 mmol) and platinum (IV) oxide (1 1 .57 mg, 0.051 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8 h. After completion of the reaction, the resulting mixturewas filtered through celite and concentrated. The resulting oily compound was stirred in 2.5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.5 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2 h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.371 g (63.7 %); 1H NMR (DMSO-d6, 300 MHz): δ 4.25-4.20 (m, 2H), 3.77 (s, 2H), 3.47-3.40 (m, 2H), 3.27-3.24 (m, 2H), 3.02-3.00 (m, 2H), 2.22 (s, 3H), 2.1 1 -2.01 (m, 1 H), 1 .05-1 .01 (m, 6H); MS (ESI+): m/z 286.5 [M+H]+.
Example 84:
5- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1-methyl-3-oxo- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4-methyl-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (70 mg, 0.160 mmol) in DMF
(5 mL) wereadded HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 83 (54.7 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.012 g (1 1 .1 1 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .55 (s, 1 H), 8.21 (s, 1 H), 7.58-7.56 (m, 2H), 7.40-7.36 (m, 3H), 7.22 (s, 1 H), 4.29 (s, 2H), 3.85-
3.81 (m, 2H), 3.58-3.48 (m, 6H), 3.28-3.21 (m, 4H), 3.10-3.02 (m, 5H), 2.87-2.84 (m, 2H), 2.36-2.31 (m, 4H), 2.25 (s, 3H), 2.17-2.15 (m, 2H), 2.08(s, 3H), 1 .88- 1 .86 (m, 1 H), 1 .68-1 .39 (m, 4H), 0.88-0.81 (m, 6H); MS (ESI+): m/z 670.6 [M+H]+; HPLC Purity: 98.98 %.
Example 85:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1-methyl-3-oxo-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-(4-methyl piperazin-1-yl)pyridin-3-yl)benzamide
To a solution of the compound of example 67 (70 mg, 0.160 mmol) in DMF (5 mL) were added HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 83 (54.7 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.073 g (67.2 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .54 (s, 1 H),
8.41 -8.40 (m, 1 H), 8.18 (s, 1 H), 7.85-7.83 (m, 1 H), 7.36 (s, 1 H), 7.17(s, 1 H), 6.95- 6.92 (m, 1 H), 4.29 (s, 2H), 3.84-3.81 (m, 2H), 3.81 -3.60 (m, 4H), 3.28-3.21 (m, 4H), 3.09-2.87 (m, 5H), 2.85-2.71 (m, 6H), 2.27 (s, 6H), 2.1 1 (s, 3H), 1 .87-1 .85 (m, 1 H), 1 .68-1 .50 (m, 4H), 1 .27-1 .17 (m, 2H), 0.88-0.79 (m, 9H); MS (ESI+): m/z 670.4 [M+H]+; HPLC Purity: 98.37 %.
Example 86:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1-methyl-3-oxo- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6- morpholino pyridin-3-yl)benzamide
To a solution of the compound of example 70 (75 mg, 0.176 mmol) in DMF (5 mL) were added HATU (101 mg, 0.264 mmol) and Hunig's Base (0.092 mL, 0.529 mmol) and the reaction mixture was stirred at RT for 1 h. To it was added
the compound of example 83 (60.5 mg, 0.212 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.029 g (24.97 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .52 (s, 1 H), 8.42 (s, 1 H), 8.17 (s, 1 H), 7.85-7.82 (m, 1 H), 7.37 (s, 1 H), 7.18 (s, 1 H), 6.91 -6.88 (m, 1 H), 4.29 (s, 2H), 3.84-3.81 (m, 2H), 3.71 -3.70 (m, 4H), 3.48-3.47 (m, 4H), 3.28-3.21 (m, 4H), 3.09-3.01 (m, 3H), 2.84-2.82 (m, 2H), 2.23 (s, 3H), 2.19-2.17 (m, 2H), 2.08 (s, 3H), 1 .99-1 .68 (m, 3H), 1 .68-1 .49 (m, 4H), 0.88-0.80 (m, 9H); MS (ESI+): m/z 657.8 [M+H]+; HPLC Purity: 99.69 %.
Example 87:
7-isopropyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4- carbonitrile
To a solution of the compound of example 62 (1 .5 g, 7.93 mmol) in DMF (2.5 mL) and DCM (25 mL) were added propan-2-one (1 .169 mL, 15.86 mmol) and acetic acid (2.72 mL, 47.6 mmol) and the reaction mixture was stirred at RT for 2 h. The reaction mixture was cooled to 0 °C and sodium triacetoxyborohydride (5.04 g, 23.78 mmol) was added in portions over a period of 1 h and stirred at RT for 16 h. After completion of the reaction, water was added and the residue was extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine, and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-15 % methanol in chloroform) to yield the title compound.
Yield: 0.780 g (32.5 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.36 (s, 1 H), 3.37 (s, 2H), 2.90-2.85 (m, 1 H), 2.76 (t, J= 6.0 Hz, 2H), 2.62 (d, J= 6.0 Hz, 2H), 2.20 (s, 3H), 1 .03 (t, J= 6.6 Hz, 6H); MS (ESI+): m/z 232.2 [M+H]+; HPLC Purity: 76.36 %.
Example 88:
4- (Aminomethyl)-7-isopropyl-1 -methyl-5,6,7,8-tetrahydro-2,7-naphthyridin- 3(2H)-one hydrochloride
To a solution of the compound of example 87 (700 mg, 3.03 mmol) in acetic acid (10 mL) were added Pd/C (81 mg, 0.757 mmol), sodium acetate (497 mg, 6.05 mmol) and platinum (IV) oxide (17.18 mg, 0.076 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8 h. After completion of the reaction, the reaction mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 3.5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.7 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2 h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.451 g (54.8 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.21 (s, 1 H),
8.12 (s, 2H), 4.12-3.96 (m, 2H), 3.80-3.78 (m, 2H), 3.59-3.49 (m, 2H), 3.21 -3.07 (m, 3H), 2.24 (s, 3H), 1 .35 (t, J= 6.6 Hz, 6H); MS (ESI+): m/z 272.5 [M+H]+.
Example 89:
5- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4-methyl-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (50 mg, 0.1 14 mmol) in DMF (5 mL) wereadded HATU (65.0 mg, 0.171 mmol) and Hunig's Base (0.060 mL, 0.342 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 88 (37.2 mg, 0.137 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.052 g (66.8 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .48 (s, 1 H), 8.18 (s, 1 H), 7.57-7.54 (m, 2H), 7.41 -7.37 (m, 3H), 7.22 (s, 1 H), 4.28 (s, 2H), 3.84- 3.82 (m, 2H), 3.58-3.50 (m, 4H), 3.49 (s, 2H), 3.27-3.23 (m, 2H), 3.15-3.02 (m, 5H), 2.88-2.83 (m, 1 H), 2.36 (s, 3H), 2.25 (s, 3H), 1 .67-1 .65 (m, 2H), 1.53-1 .51
(m, 2H), 1 .41 -1 .25 (m, 6H), 1 .04 (s, 3H), 0.84-0.83 (m, 6H); MS (ESI+): m/z 656.6 [M+H]+; HPLC Purity: 96.03 %.
Example 90:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-(4- methylpiperazin-1-yl)pyridin-3-yl)benzamide
To a solution of the compound of example 67 (75 mg, 0.171 mmol) in DMF (5 mL) were added HATU (98 mg, 0.257 mmol) and Hunig's Base (0.090 ml_, 0.513 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 88 (55.8 mg, 0.205 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.013 g (1 1.39%); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .80 (s, 1 H), 8.44 (s, 1 H), 8.32 (s, 1 H), 7.88-7.78 (m, 1 H), 7.38 (s, 1 H), 7.20 (s, 1 H), 7.00- 6.97(m, 1 H), 4.29 (s, 2H), 3.89-3.61 (m, 8H), 3.45-3.25 (m, 2H), 3.21 -2.98 (m, 9H), 2.87 (s, 2H), 2.65 (s, 3H), 2.22 (s, 3H), 2.17 (s, 3H), 1 .71 -1 .49 (m, 4H), 1 .38- 1 .21 (m, 9H); MS (ESI+): m/z 656.6 [M+H]+;HPLC Purity: 98.28 %.
Example 91 :
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6- morpholinopyridin-3-yl)benzamide
To a solution of the compound of example 70 in DMF (5 mL) were added HATU (87 mg, 0.229 mmol) and Hunig's Base (0.080 mL, 0.458 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 88 (49.8 mg, 0.183 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled to
obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.082 g (80 %); 1H NMR (DMSO-d6, 300 MHz): δ 1 1 .54 (s, 1 H), 8.42 (s, 1 H), 8.21 (s, 1 H), 7.85-7.83 (m, 1 H), 7.38 (s, 1 H), 7.19 (s, 1 H), 6.92-6.89 (m, 1 H), 4.28 (s, 2H), 3.84-3.81 (m, 2H), 3.71 -3.70 (m, 4H), 3.68-3.55 (m, 2H), 3.49- 3.47 (m, 4H), 3.28-3.21 (m, 4H), 3.12-3.01 (m, 5H), 2.87 (s, 1 H), 2.23 (s, 3H), 2.18 (s, 3H), 1 .71 -1 .49 (m, 4H), 1 .27-1 .24 (m, 9H); MS (ESI+): m/z 643.4 [M+H]+; HPLC Purity: 96.27 %. Example 92:
7-(Cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7- naphthyridine-4-carbonitrile
To a solution of the compound of example 62 (1 .5 g, 7.93 mmol) in DMF (2.5 mL) and DCM (25 mL) were added cyclopropanecarbaldehyde (1 .1 1 g, 15.86 mmol) and acetic acid (2.72 mL, 47.6 mmol) and the reaction mixture was stirred at RT for 2h. The reaction mixture was cooled to 0 "C and sodium triacetoxyborohydride (5.04 g, 23.78 mmol) was added in portions over a period of 1 h and stirred at RT for 16h. After completion of the reaction, water was added and the residue was extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine, and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-15 % methanol in chloroform) to yield the title compound.
Yield: 1 .01 g (44.6%); 1H NMR (DMSO-d6, 300 MHz): δ 12.39 (s, 1 H), 3.33 (s, 2H), 2.81 -2.79 (m, 2H), 2.69-2.68 (m, 2H), 2.37-2.36 (m, 2H), 2.21 (s, 3H), 0.94-0.89 (m, 1 H), 0.50-0.49 (m, 2H), 0.14-0.13 (m, 2H); MS (ESI+): m/z 244.1 [M+H]+; HPLC Purity: 85.23 %.
Example 93:
4-(Aminomethyl)-7-(cyclopropylmethyl)-1 -methyl-5,6,7,8-tetrahydro-2,7- naphthyridin-3(2H)-one hydrochloride
To a solution of the compound of example 92 (875 mg, 3.60 mmol) in acetic acid (10 mL) were added Pd/C (96 mg, 0.899 mmol), sodium acetate (590
mg, 7.19 mmol) and platinum (IV) oxide (20.42 mg, 0.090 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the resulting mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 4.37 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.9 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.757 g (74.2 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.37 (s, 1 H), 8.15 (s, 2H), 4.31 -4.26 (m, 2H), 3.89-3.79 (m, 2H), 3.64-3.52 (m, 2H), 3.24-3.22 (m, 2H), 3.09-3.02 (m, 2H), 2.21 (s, 3H), 1 .31 -1 .24 (m, 1 H), 0.67-0.64 (m, 2H), 0.46-0.44 (m, 2H); MS (ESI+): m/z 284.5 [M+H]+.
Example 94:
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7- naphthyridin-4-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl- 4'-(morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (70 mg, 0.160 mmol) in DMF (5 mL) were added HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 93 (54.4 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.016 g (14.89 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .54 (s, 1 H), 8.20 (s, 1 H), 7.58-7.56 (m, 2H), 7.38-7.36 (m, 3H), 7.21 (s, 1 H), 4.29 (s, 2H), 3.84- 3.81 (m, 2H), 3.57-3.48 (m, 6H), 3.31 -3.21 (m, 4H), 3.10-3.01 (m, 3H), 2.86-2.84 (m, 2H), 2.64-2.62 (m, 2H), 2.46-2.36 (m, 6H), 2.24 (s, 3H), 2.08 (s, 3H), 1 .68- 1 .51 (m, 4H), 0.94-0.86 (m, 1 H), 0.85-0.81 (m, 3H), 0.49-0.47 (m, 2H), 0.13-0.1 1 (m, 2H); MS (ESI+): m/z 668.4 [M+H]+; HPLC Purity: 99.2 %.
Example 95:
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7- naphthyridin-4-yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-
5-(6-(4-methylpiperazin-1 -yl)pyridin-3-yl)benzamide
To a solution of the compound of example 67 (70 mg, 0.160 mmol) in DMF (5 mL) were added HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 ml_, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 93 (54.4 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.072 g (65.4%); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .58 (s, 1 H),
8.40 (s, 1 H), 8.19 (s, 1 H), 7.83-7.81 (m, 1 H), 7.36 (m, 1 H), 7.17 (s, 1 H), 6.93-6.90 (m, 1 H), 4.27 (s, 2H), 3.84-3.81 (m, 2H), 3.55-3.48 (m, 6H), 3.34-3.21 (m, 1 H), 3.23-3.21 (m, 4H), 3.09-2.73 (m, 6H), 2.52-2.42 (m, 4H), 2.32 (s, 3H), 2.22 (s, 3H), 2.10 (s, 3H), 1.64-1 .52 (m, 4H), 0.94-0.92 (m, 1 H), 0.84-0.82 (m, 3H), 0.49- 0.46 (m, 2H), 0.14-0.12 (m, 2H); MS (ESI+): m/z 668.5 [M+H]+; HPLC Purity: 96.78 %.
Example 96:
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7- naphthyridin-4-yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-
5-(6-morpholinopyridin-3-yl)benzamide
To a solution of the compound of example 70 (75 mg, 0.176 mmol) in DMF (5 mL) wereadded HATU (101 mg, 0.264 mmol) and Hunig's Base (0.092 mL, 0.529 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 93 (60.0 mg, 0.212 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled to obtain a crude material, which was purified by
using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.025 g (21.58%); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .54 (s, 1 H), 8.42 (s, 1 H), 8.17 (s, 1 H), 7.86-7.83 (m, 1 H), 7.37 (m, 1 H), 7.18 (s, 1 H), 6.91 -6.88 (m, 1 H), 4.28 (s, 2H), 3.84-3.81 (m, 2H), 3.74-3.70 (m, 4H), 3.49-3.47 (m, 4H), 3.23-3.21 (m, 4H), 3.09-3.07 (m, 3H), 2.76-2.72 (m, 2H), 2.64-2.62 (m, 2H), 2.34- 2.32 (m, 2H), 2.23 (s, 3H), 2.09 (s, 3H), 1 .64-1 .49 (m, 4H), 0.93-0.91 (m, 1 H), 0.84-0.81 (m, 3H), 0.50-0.48 (m, 2H), 0.14-0.12 (m, 2H); MS (ESI+): m/z 655.3 [M+H]+; HPLC Purity: 99.62 %.
Example 97:
1-Methyl-7-(oxetan-3-yl)-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridine-4- carbonitrile
To a solution of the compound of example 62 (1 .5 g, 7.93 mmol) in DMF (2.5 mL) and DCM (25 ml_), were added oxetan-3-one (1.143 g, 15.86 mmol) and acetic acid (2.72 mL, 47.6 mmol) and the reaction mixture was stirred at RT for 16 h. The reaction mixture was cooled to 0 °C, sodium triacetoxyborohydride (5.04 g, 23.78 mmol) was added in portionsover a period of 1 h and stirred at RT for 5h. After completion of the reaction, water was added and the residue was extracted with ethyl acetate. The ethyl acetate layer was washed with water and brine, and dried over anhydrous sodium sulphate to obtain a crude material, which was purified by column chromatography (silica gel, 10-15 % methanol in chloroform) to yield the title compound.
Yield: 0.745 g (35.6%); 1 H NMR (DMSO-d6, 300 MHz): δ 12.45 (s, 1 H), 4.60 (t, J= 6.3 Hz, 2H), 4.50 (t, J= 6.0 Hz, 2H), 3.62-3.56 (s, 2H), 3.15 (s, 2H), 2.79-2.73 (m, 3H), 2.16 (m, 3H); MS (ESI+): m/z 246.1 [M+H]+; HPLC Purity: 93 %.
Example 98:
4-(Aminomethyl)-1-methyl-7-(oxetan-3-yl)-5,6,7,8-tetrahydro-2,7- naphthyridin-3(2H)-one hydrochloride
To a solution of the compound of example 97 (700 mg, 2.85 mmol) in acetic acid (10 ml) were added Pd/C (76 mg, 0.713 mmol), sodium acetate (468 mg, 5.71 mmol) and platinum (IV) oxide (16.20 mg, 0.071 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the reaction mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 3.5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.7 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.1 10 g (13.49 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.45 (s, 1 H),
8.17 (s, 2H), 4.60 (t, J= 6.3 Hz, 2H), 4.50 (t, J= 6.0 Hz, 2H), 3.34-3.32 (m, 2H), 3.60-3.55 (s, 2H), 3.15 (s, 2H), 2.78-2.73 (m, 3H), 2.15 (m, 3H); MS (ESI+): m/z 286.1 [M+H]+.
Example 99:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-7-(oxetan-3- yl)-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution ofthe compound of example 26 (70 mg, 0.160 mmol) in DMF
(5 mL) were added HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 98 (54.7 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain the crude material, which was purified by using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 0.005 g (3.58%); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .54 (s, 1 H), 8.20 (s, 1 H), 7.59-7.56 (m, 2H), 7.39-7.36 (m, 3H), 7.22 (s, 1 H), 4.62-4.60 (m, 2H), 4.51 -4.48 (m, 2H), 4.29 (s, 2H), 3.84-3.81 (m, 2H), 3.57-3.48 (m, 6H), 3.34- 3.12 (m, 6H), 2.86-2.84 (m, 3H), 2.72 (s, 1 H), 2.38-2.34 (m, 4H), 2.24 (s, 3H), 2.06 (s, 3H), 1 .70-1 .64 (m, 2H), 1 .53-1 .49 (m, 4H), 0.85-0.81 (m, 3H); MS (ESI+): m/z 670.5 [M+H]+; HPLC Purity: 76.65 %.
Example 100:
3- Oxo-l -(trifluoromethyl)-2, 3, 5,6,7, 8-hexahydroisoquinoline-4-carbonitrile
To a solution of 2-(2,2,2-trifluoroacetyl)cyclohexanone (2 g, 10.30 mmol) in ethanol (15 mL) were added 2-cyanoacetamide (0.866 g, 10.30 mmol) and piperidine (0.510 ml, 5.15 mmol). The reaction mixture was stirred at 75 °C for 7 h. After completion of the reaction, the precipitated solid was filtered, washed with ethanol and dried to yield the title compound.
Yield: 1 .45 g (57.9%); 1 H NMR (DMSO-d6, 300 MHz): δ 13.54 (s, 1 H), 2.90- 2.89 (m, 2H), 2.71 -2.69 (m, 2H), 1 .87-1 .84 (m, 4H); MS (ESI+): m/z 243.1 [M+H]+;HPLC Purity: 99.6 %.
Example 101 :
4- (Aminomethyl)-1-(trifluoromethyl)-5,6,7j8-tetrahydroisoquinolin-3(2H)-one hydrochloride
To a solution of the compound of example 100 (1 .4 g, 5.78 mmol) in acetic acid (10 mL) were added Pd/C (0.154 g, 1 .445 mmol), sodium acetate (0.948 g, 1 1 .56 mmol) and platinum (IV) oxide (0.033 g, 0.145 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the resulting mixture waswas filtered through celite and concentrated. The resulting oily compound was stirred in 7 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 1 .4 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.512 g (31 .2%); 1 H NMR (DMSO-d6, 300 MHz): δ 12.50 (s, 1 H), 8.28 (s, 2H), 3.92-2.89 (m, 2H), 3.32 (s, 2H), 2.64-2.61 (m, 2H), 1 .77-1 .67 (m, 4H); MS (ESI+): m/z 283.1 [M+H]+;HPLC Purity: 99.62 %.
Example 102:
5- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-N- ((3-OXO-1 -(trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-
[1 ,1 '-biphenyl]-3-carboxamide
no
To a solution of the compound of example 26 (70 mg, 0.160 mmol) in DMF (5 mL) wereadded HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 ml_, 0.479 mmol); and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 101 (54.1 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum and the residue was extracted using ethyl acetate. The ethyl acetate layer was washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15% MeOH/CHC ) to yield the title compound.
Yield: 0.017 g (15.76%); 1 H NMR (DMSO-d6, 300 MHz): δ 12.04 (s, 1 H),
8.32 (s, 1 H), 7.57-7.55 (m, 2H), 7.40-7.37 (m, 3H), 7.19 (s, 1 H), 4.45 (s, 2H), 3.85- 3.81 (m, 2H), 3.57-3.47 (m, 6H), 3.29-3.21 (m, 2H), 3.10-3.01 (m, 3H), 2.62-2.53 (m, 2H), 2.39-2.35 (m, 4H), 2.25 (s, 3H), 1 .65-1 .23 (m, 10H), 0.83 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 667.4 [M+H]+; HPLC Purity: 98.68 %.
Example 103:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1- yl)pyridin-3-yl)-N-((3-oxo-1 -(trifluoromethyl)-2,3,5,6,7,8-hexahydro
isoquinolin-4-yl)methyl)benzamide
To a solution of the compound of example 67 (70 mg, 0.160 mmol) in DMF (5 mL) were added HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 101 (54.1 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHC ) to yield the title compound.
Yield: 0.043 g (38.3%); 1 H NMR (DMSO-d6, 300 MHz): δ 12.04 (s, 1 H), 8.32 (s, 1 H), 8.29 (s, 1 H), 7.89-7.86 (m, 1 H), 7.38 (s, 1 H), 7.16 (s, 1 H), 7.03-7.00 (m, 1 H), 4.45 (s, 2H), 3.85-3.81 (m, 2H), 3.61 -3.60 (m, 2H), 3.34-3.21 (m, 2H), 3.15-3.02 (m, 5H), 2.87 (s, 3H), 2.73 (s, 3H), 2.62-2.50 (m, 2H), 2.23 (s, 2H), 1 .68-
1 .23 (m, 12H), 0.82 (t, J= 6.0 Hz, 3H); MS (ESI+): m/z 667.4 [M+H]+; HPLC Purity: 94.9 %.
Example 104:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-morpholinopyridin-3- yl)-N-((3-oxo-1-(trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4- yl)methyl)benzamide
To a solution of the compound of example 70 (60 mg, 0.141 mmol) in DMF (5 mL) wereadded HATU (80 mg, 0.212 mmol) and Hunig's Base (0.074 mL, 0.423 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 101 (47.8 mg, 0.169 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHC ) to yield the title compound.
Yield: 0.057 g (60.3 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.05 (s, 1 H), 8.40 (s, 1 H), 8.30 (s, 1 H), 7.83-7.81 (m, 1 H), 7.38 (s, 1 H), 7.81 (s, 1 H), 6.93-6.90 (m, 1 H), 4.45 (s, 2H), 3.85-3.81 (m, 2H), 3.79-3.68 (m, 4H), 3.52-3.48 (m, 4H), 3.28-3.21 (m, 2H), 3.09-2.98 (m, 5H), 2.62-2.53 (m, 2H), 2.23 (s, 3H), 1.65-1 .42 (m, 8H), 0.82 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 654.4 [M+H]+; HPLC Purity: 97.55 %.
Example 105:
3-Oxo-l -(trifluoromethyl)-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridine-4- carbonitrile
To a solution of 2-(2,2,2-trifluoroacetyl)cycloheptanone (2 g, 9.61 mmol) in ethanol (15 mL) were added 2-cyanoacetamide (0.808 g, 9.61 mmol) and piperidine (0.476 mL, 4.80 mmol) and the reaction mixture was heated at 75 °C for 7 h. After completion of the reaction, the precipitated solid was filtered, washed with ethanol and dried to yield the title compound.
Yield: 1 .71 g (69.4%); 1 H NMR (DMSO-d6, 300 MHz): δ 13.26 (s, 1 H), 2.90- 2.87 (m, 2H), 2.71 -2.64 (m, 2H), 1 .74-1 .73 (m, 2H), 1 .68-1 .59 (m, 2H), 1 .58-1 .49 (m, 2H); MS (ESI+): m/z 257.0 [M+H]+;HPLC Purity: 99.93 %.
Example 106:
4- (Aminomethyl)-1-(trifluoromethyl)-6,7,8,9-tetrahydro-2H-cyclohepta[c] pyridin-3(5H)-one hydrochloride
To a solution of the compound of example 105 (1 .5 g, 5.85 mmol) in acetic acid (10 mL) were added Pd/C (0.156 g, 1 .464 mmol), sodium acetate (0.961 g, 1 1 .71 mmol) and platinum (IV) oxide (0.033 g, 0.146 mmol). The flask was shaken under an atmosphere of hydroogen at 80 psi for 8h. After completion of the reaction, the resulting mixturewas filtered through celite and concentrated. The resulting oily compound was stirred in 7.5 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 1 .5 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered, washed with ether and dried to yield the title compound.
Yield: 0.247 g (14.22%); 1H NMR (DMSO-d6, 300 MHz): δ 13.26 (s, 1 H), 8.54 (s, 2H), 3.32 (s, 2H), 2.90-2.87 (m, 2H), 2.71 -2.64 (m, 2H), 1 .74-1 .73 (m, 2H), 1 .68-1 .59 (m, 2H), 1 .58-1 .49 (m, 2H); MS (ESI+): m/z 297.6 [M+H]+.
Example 107:
5- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-N- ((3-OXO-1 -(trifluoromethyl)-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-4- yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (70 mg, 0.160 mmol) in DMF
(5 mL) wereadded HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and to the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 106 (56.8 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by
using column chromatography (silica gel, 0-15% MeOH/CHCI3) to yield the title compound.
Yield: 0.042 g (36.9%); 1 H NMR (DMSO-d6, 300 MHz): δ 12.19 (s, 1 H), 8.34 (s, 1 H), 7.57-7.55 (m, 2H), 7.39-7.37 (m, 3H), 7.19 (s, 1 H), 4.47 (s, 2H), 3.84- 3.81 (m, 2H), 3.57-3.46 (m, 6H), 3.29-3.21 (m, 4H), 3.10-3.02 (m, 3H), 2.76-2.68 (m, 2H), 2.65-2.54 (m, 2H), 2.41 -2.35 (m, 4H), 2.25 (s, 3H), 1 .73-1 .23 (m, 8H), 0.83 (t, J= 6.0 Hz, 3H); MS (ESI+): m/z 681 .4 [M+H]+; HPLC Purity: 95.5 %.
Example 108:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1- yl)pyridin-3-yl)-N-((3-oxo-1 -(trifluoromethyl)-3,5,6,7,8,9-hexahydro-2H- cyclohepta[c]pyridin-4-yl)methyl)benzamide
To a solution ofthe compound of example 67 (70 mg, 0.160 mmol) in DMF (5 mL) were added HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 ml_, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture was added the compound of example 106 (56.8 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHCI3) to yield the title compound.
Yield: 0.018 g (15.34 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.19 (s, 1 H), 8.38 (s, 1 H), 8.36 (s, 1 H), 7.81 -7.74 (m, 1 H), 7.36 (s, 1 H), 7.14 (s, 1 H), 6.94-6.89 (s, 1 H), 4.46 (s, 2H), 3.84-3.81 (m, 2H), 3.57-3.48 (m, 4H), 3.29-3.23 (m, 2H), 3.09-3.02 (m, 3H), 2.80-2.77 (m, 2H), 2.68-2.59 (m, 2H), 2.43-2.36 (m, 4H), 2.24 (s, 3H), 2.23 (s, 3H), 1 .74-1 .48 (m, 10H), 0.82 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 681 .4 [M+H]+; HPLC Purity: 92.6 %.
Example 109:
3-Oxo-l -(trifluoromethyl)-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridine-4- carbonitrile
To a solution of 2-(2,2,2-trifluoroacetyl)cyclopentanone (2 g, 1 1 .10 mmol) in ethanol (15 mL) were added 2-cyanoacetamide (0.934 g, 1 1 .10 mmol) and piperidine (0.550 mL, 5.55 mmol) and the reaction mixture was stirred at 75 °C for 7 h. After completion of the reaction, the precipitated solid was filtered, washed with ethanol and dried to yield the title compound.
Yield: 0.640 g (35.2 %); 1 H NMR (DMSO-d6, 300 MHz): δ 13.57 (s, 1 H), 2.92-2.73 (m, 4H), 2.12-2.02 (m, 2H); MS (ESI+): m/z 229.2 [M+H]+; HPLC Purity: 99.9 %.
Example 110:
4- (Aminomethyl)-1-(trifluoromethyl)-6,7-dihydro-2H-cyclopenta[c]pyridin- 3(5H)-one hydrochloride
To a solution of the compound of example 109 (620 mg, 2.72 mmol) in acetic acid (10 mL) were added Pd/C (72.3 mg, 0.679 mmol), sodium acetate (446 mg, 5.43 mmol) and platinum (IV) oxide (15.43 mg, 0.068 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8h. After completion of the reaction, the resulting mixturewas filtered through celite and concentrated. The resulting oily compound was stirred in 3.1 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.62 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2h. The precipitated solid was filtered and washed with ether to yield the title compound.
Yield: 0.071 g (5.45%); 1 H NMR (DMSO-d6, 300 MHz): δ 13.04 (s, 1 H), 8.22 (s, 2H), 3.42 (s, 2H), 2.86-2.82 (m, 4H), 2.09-2.01 (m, 2H); MS (ESI+): m/z 269.2 [M+H]+.
Example 111 :
5- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-N- ((3-OXO-1 -(trifluoromethyl)-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (70 mg, 0.160 mmol) in DMF (5 mL) wereadded HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction
mixture was added the compound of example 1 10 (51 .5 mg, 0.192 mmol) and stirred for 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 ml_). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHC ) to yield the title compound.
Yield: 0.033 g (31 .2 %); 1 H NMR (DMSO-d6, 300 MHz): δ 12.45 (s, 1 H), 8.55 (s, 1 H), 7.57-7.54 (m, 2H), 7.40-7.36 (m, 3H), 7.19 (s, 1 H), 3.84-3.81 (m, 2H), 3.59-3.48(m, 6H), 3.29-3.21 (m, 4H), 3.10-3.01 (m, 3H), 2.79-2.67 (m, 4H), 2.31 -2.40 (m, 4H), 2.24 (s, 3H), 1 .99-1 .94 (m, 2H), 1 .68-1 .51 (m, 4H), 0.83 (t, J= 6.6 Hz, 3H); MS (ESI+): m/z 653.3 [M+H]+; HPLC Purity: 98.62 %.
Example 112:
Methyl 5-bromo-3-(6-bromohexanamido)-2-methylbenzoate
To a stirred solution of 6-bromohexanoic acid (3 g, 15.38 mmol) in DCM
(25 mL) at 0 °C was added drop-wise solution of oxalyl chloride (1 .481 ml_, 16.92 mmol) in DCM (10 mL). After complete addition, the reaction mixture was stirred at 0 °C and then at RT for 30 min. After completion of the reaction, solvents were removed, the resulting mixture was dried and was added drop-wise to the previously cooled solution of methyl 3-amino-5-bromo-2-methyl benzoate (3.75 g, 15.38 mmol) in DCM (30 mL). To the resulting mixture was added triethylamine (4.29 mL, 30.8 mmol) and stirred for 2 h. The reaction mixture was quenched with water, extracted with DCM and the organic layer was washed with brine solution. The organic layer was dried over anhydrous sodium sulphate and evaporated to obtain a brown residue. The residue was purified by column chromatography (silica gel, 2:8 ethyl acetate: petroleum ether) to yield the title compound.
Yield: 4.7 g (72 %); 1 H NMR (DMSO-d6, 300 MHz): δ 9.56 (s, 1 H), 7.78 (d, J = 1.8 Hz, 1 H), 7.68 (d, J = 1 .8 Hz, 1 H), 3.84 (s, 3H), 3.55 (t, J = 6.6 Hz, 2H), 2.37 (t, J = 7.2 Hz, 2H), 2.27 (s, 3H), 1 .89 (m, 2H), 1 .68 (m, 2H), 1 .49 (m, 2H); MS (ESI+): m/z 422.2 [M+H]+, HPLC Purity: 99.56 %.
Example 113:
Methyl 5-bromo-2-methyl-3-(2-oxoazepan-1 -yl)benzoate
To a stirred solution of the compound of example 1 12 (4.2 g, 9.97 mmol) in ethanol (10 mL) at RT was added sodium hydride (0.479 g, 1 1 .97 mmol) and the reaction mixture was heated to 65 °C for 3 h. After completion of the reaction, the solvent was removed to obtain a dark brown residue. To the residue, water was added and the reaction mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulphate and evaporated to obtain a brown residue. The residue was purified by column chromatography (silica gel, 10-70 % ethylacetate in pet ether using 3:7 ethyl acetate : petroleum ether) to yield the title compound.
Yield: 2.15 g (63 %); 1 H NMR (DMSO-d6, 300 MHz): δ 7.82 (d, J = 2.1 Hz,
1 H), 7.61 (d, J = 2.1 Hz, 1 H), 3.84 (s, 3H), 3.77 (m, 1 H), 3.44 (m, 1 H), 2.70 (m, 1 H), 2.21 (s, 3H), 1 .72 (bs, 7H); MS (ESI+): m/z 340 [M+H]+; HPLC Purity: 99.38 %. Example 114:
5-Bromo-2-methyl-3-(2-oxoazepan-1 -yl)benzoic acid
To a solution of the compound of example 1 13 (2 g, 5.88 mmol) in THF (4.00 mL) and MeOH (4 mL) was added 1 M NaOH solution (23.51 mL, 23.51 mmol) and the reaction mixture was heated to 65 °C for 1 h. The reaction mixture was cooled and the solvent was removed to obtain a residue. The residue was dissolved in water, acidified with dilute HCI solution (pH = 2) and the resulting solid was filtered and dried to yield the title compound.
Yield: 1 .6 g (83 %); 1 H NMR (DMSO-d6, 300 MHz): δ 13.32 (s, 1 H), 7.80 (s, 1 H), 7.57 (s, 1 H), 3.77 (m, 1 H), 3.47 (m, 1 H), 2.70 (m, 1 H), 2.23 (s, 3H), 1 .72 (bs, 7H); MS (ESI+): m/z 327 [M+H]+; HPLC Purity: 99.80 %.
Example 115:
5-Bromo-2-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4- yl)methyl)-3-(2-oxoazepan-1 -yl)benzamide
To a solution of the compound of example 1 14 (500 mg, 1.533 mmol) in
DMF (10 mL) was added HATU (874 mg, 2.299 mmol) and reaction mixture was stirred for 5 min at RT. To the reaction mixture was added the compound of example 2 (491 mg, 2.146 mmol) followed by DIPEA (0.803 ml, 4.60 mmol) and
the resulting mixture was stirred at 70 °C for 16 h. After completion of the reaction, water was added and solid was filtered and dried to yield the title compound.
Yield: 361 mg (47 %); 1H NMR (DMSO-d6, 300 MHz): δ 1 1 .50 (s, 1 H), 8.29 (s, 1 H), 7.38 (s, 1 H), 7.30 (s, 1 H), 4.28 (d, J = 4.2 Hz, 2H), 3.68 (m, 2H), 3.44 (m, 2H), 2.70 (m, 2H), 2.37 (s, 2H), 2.10 (s, 3H), 2.03 (s, 3H), 1 .70 (m, 10H); MS (ESI+): m/z 501 .1 [M+H]+; HPLC Purity: 92.51 %.
Example 116:
4-Methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)- 4'-(morpholinomethyl)-5-(2-oxoazepan-1 -yl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 1 15 (50 mg, 0.100 mmol) in dioxane were added 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)benzyl)morpholine (42.4 mg, 0.140 mmol) and PdCI2(dppf)-CH2CI2adduct (2.448 mg, 3.00 μηιοΙ) under nitrogen atmosphere and the reaction mixture was stirred at RT for 10 min. To this reaction mixture was added 2M Na2C03 solution (150 μΙ_, 0.300 mmol) and the reaction mixture was heated to 90 °C for 4 h. After completion of the reaction, the reaction mixture was cooled to RT, filtered through celite and filtrate was concentrated to obtain a dark brown residue.To the residue, water was added and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulphate, concentrated to obtain a crude material, which was purified by column chromatography (silica gel, 1 : 9 MeOH : CH2CI2) to yield the title compound.
Yield: 48 mg (81 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .50 (s, 1 H), 8.27 (s, 1 H), 7.63 (d, J = 7.8 Hz, 2H), 7.42 (m, 4H), 4.76 (s, 1 H), 4.33 (d, J = 4.2 Hz, 2H), 3.74 (m, 2H), 3.57 (s, 4H), 3.49 (s, 3H), 2.72 (m, 3H), 2.37 (s, 5H), 2.12 (s, 3H), 2.10 (s, 3H), 1 .74 (m, 4H), 1 .64 (s, 4H), 1 .23 (s, 2H); MS (ESI+): m/z 597.3 [M+H]+; HPLC Purity: 90.17 %.
Example 117:
Dimethyl (4'-methyl-3'-(((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4- yl)methyl)carbamoyl)-5'-(2-oxoazepan-1-yl)-[1 ,1 '-biphenyl]-4-yl)phosphonate
The title compound was synthesized by following the general procedure of compound of example 1 16 to yield the title compound.
Yield: 85 %; 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .50 (s, 1 H), 8.31 (s, 1 H), 7.85 (m, 4H), 7.49 (s, 2H), 4.34 (d, J = 4.5 Hz, 2H), 3.76 (m, 2H), 3.69 (s, 3H), 3.65 (s, 3H), 2.73 (m, 3H), 2.37 (s, 3H), 2.14 (s, 3H), 2.10 (s, 3H), 1 .75 (m, 5H), 1 .64 (s, 5H); MS (ESI+): m/z 606.2 [M+H]+; HPLC Purity: 98.33 %.
Example 118:
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2- oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-1-yl)methyl)benzamide
HATU((1 -[Bis(dimethylamino)methylene]-1 H-1 ,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate)) (15.00 g, 39.4 mmol) was added to a stirred solution of the compound of example 20 (9 g, 26.3 mmol) at 0 °C followed by addition of DIPEA (N,N-Diisopropylethylamine) (1 1 .48 mL, 65.7 mmol) and the compound of example 1 19 (6.09 g, 34.2 mmol). The reaction mixture was stirred for 18 h at RT. The reaction mixture was diluted with water and extracted with ethyl acetate, washed with brine and dried over anhydrous sodium sulphate to yield the title compound.
1 H NMR (DMSO-de, 300 MHz): δ 1 1 .68 (s, 1 H), 8.22 (s, 1 H), 7.31 (s, 1 H), 7.08 (s, 1 H), 4.29 (s, 2H), 3.83 (m, 2H), 3.24 (m, 2H), 2.98 (m, 3H), 2.69 (m, 2H), 2.60 (m, 2H), 2.17 (s, 6H), 1 .96 (m, 2H), 1 .53 (m, 4H), 0.74 (m, 3H); MS (ESI+):m/z 502.1 [M+H]+; HPLC Purity: 99.74 %.
Example 119:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(5-fluoro-6-morpholinopyridin-3- yl)-2-methyl-N-((1 -methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide
To a solution of the compound of example 1 18 (100 mg, 0.199 mmol) in 1 ,4-dioxane (10 mL), was added 4-(3-fluoro-5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridin-2-yl)morpholine (80 mg, 0.259 mmol) and the reaction mixture was stirred for 5 min, followed by addition of sodium carbonate (63.3 mg, 0.597 mmol) in water (2.0 mL) and stirred at 100 °C for 5h. After completion of the reaction, the resulting mixture was filtered through celite and the filtrate was distilled under vacuum. To the residue obtained, water was added and the resulting mixture was extracted with ethyl acetate. The compound was adsorbed
on silica and further purified by column chromatography (silica gel, 10-20 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 30 mg (24.97 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .62 - 1 1 .51 (m, 1 H), 8.19 - 8.01 (m, 1 H), 7.55 - 7.51 (m, 2H), 7.37 (s, 1 H), 7.04 - 7.01 (m, 1 H), 4.49 (s, 1 H), 4.37 - 4.21 (m, 3H), 3.88 - 3.70 (m, 6H), 3.33 - 3.13 (m, 3H), 3.1 1 - 2.94 (s, 3H), 2.92 - 2.88 (m, 1 H), 2.79 - 2.64 (m, 2H), 2.62 - 2.54 (m, 2H), 2.28 - 2.20 (m, 2H), 2.16 - 2.10 (m, 3H), 2.09 - 2.02 (m, 1 H), 2.01 - 1.90 (m, 2H), 1 .73 - 1 .41 (m, 4H), 0.85 - 0.78 (m, 3H); MS (ESI-): m/z 602.2 [M-H]"; HPLC Purity: 92.15 %.
Example 120:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-3'-fluoro-4-methyl-N-((1 -methyl-3- oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-4'-morpholino- [1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 1 18 (100 mg, 0.199 mmol) in
1 ,4-dioxane (10 mL) was added 4-(2-fluoro-4-(4,4,5,5- tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)morpholine (79 mg, 0.259 mmol). The reaction mixture was stirred for 5 min followed by addition of sodium carbonate (63.3 mg, 0.597 mmol) in water (2.0 mL) and stirring at 100 °C for 5 h. After completion of the reaction, the reaction mixture was filtered through celite, and the filtrate was distilled under vacuum.To the residue, water was added and the residue was extracted with ethyl acetate. The compound was adsorbed on silica and further purified using column chromatography (silica gel, 10-20 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 21 mg (17.51 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .67 (s, 1 H),
8.1 1 (s, 1 H), 7.61 - 7.39 (m, 3H), 7.21 - 7.1 1 (m, 1 H), 7.06 (t, 1 H), 4.33 - 4.23 (m, 2H), 3.84 - 3.76 (m, 6H), 3.29 - 3.25 (m, 2H), 3.21 - 2.86 (m, 6H), 2.84 - 2.73 (m, 1 H), 2.68 -2.60 (m, 4H), 2.23 - 2.18 (m, 6H), 2.09 - 2.00 (m, 2H), 1 .68 - 1 .53 (m, 4H), 0.82 (t, 3H); MS (ESI-): m/z 601 .2 [M-H]"; HPLC Purity: 96.34 %.
Example 121 :
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo- 3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-4'-(tetrahydro-2H- pyran-4-yl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 1 18 (100 mg, 0.199 mmol) in 1 ,4-dioxane (10 mL), was added 4,4,5,5-tetramethyl-2-(4-(tetrahydro-2H-pyran-4- yl)phenyl)-1 ,3,2-dioxaborolane (74.6 mg, 0.259 mmol). The reaction mixture was stirred for 5 min followed by addition of sodium carbonate (63.3 mg, 0.597 mmol) in water (2.0 mL). The reaction mixture was stirred at 100 °C for 5h. After completion of the reaction, the resulting mixture was filtered through celite and the filtrate was distilled under vacuum.To the residue, water was added and the resulting mixture was extracted with ethyl acetate. The compound was adsorbed on silica and purified by column chromatography (silica gel, 10-20 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 25 mg (21 .52 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .64 (s, 1 H), 8.17 (s, 1 H), 7.56 - 7.38 (m, 2H), 7.34 - 7.32 (m, 3H), 7.20 (s, 1 H), 4.33 - 4.32 (m, 2H), 3.98 - 3.94 (m, 2H), 3.84 - 3.81 (m, 2H), 3.49 - 3.42 (m, 2H), 3.33 - 3.28 (m, 2H), 3.25 - 3.21 (m, 3H), 2.86 - 2.80 (m, 2H), 2.73 - 2.62 (m, 4H), 2.24 - 2.20 (m, 5H), 2.18 (s, 1 H), 2.09 - 1 .93 (m, 2H), 1 .71 - 1 .65 (m, 5H), 1 .53 - 1 .51 (m, 2H), 0.83 (t, 3H); MS (ESI+): m/z 584.3 [M+H]+; HPLC Purity: 99.13 %.
Example 122:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(1 H-indol-4-yl)-2-methyl-N-((1- methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl) benzamide
To a solution of the compound of example 1 18 (100 mg, 0.199 mmol) in
1 ,4-dioxane (10 mL), was added 4-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)- 1 H-indole (62.9 mg, 0.259 mmol). The reaction mixture was stirred for 5 min, followed by addition of sodium carbonate (63.3 mg, 0.597 mmol) in water (2.0 mL) and stirring at 100 °C for 5 h. After completion of the reaction, the reaction mixture was filtered through celite, and the filtrate was distilled under vacuum.To the residue, water was added and the resulting mixture was extracted with ethyl acetate. The compound was adsorbed on silica and purified by column
chromatography (silica gel, 10-20 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 50 mg (46.7 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .66 (s, 1 H), 1 1 .27 (s, 1 H), 8.32 - 8.22 (m, 1 H), 7.44 - 7.41 (m, 3H), 7.38 - 7.23 (m, 1 H), 7.16 (t, 1 H), 7.07 - 7.05 (m, 1 H), 6.50 (s, 1 H), 4.41 - 4.29 (d, 2H), 3.92 - 3.78 (m, 2H), 3.33 - 3.24 (m, 2H), 3.19 - 2.96 (m, 3H), 2.75 - 2.67 (m, 2H), 2.65 - 2.56 (m, 2H), 2.30 (s, 2H), 2.15 (s, 2H), 2.13 - 1 .89 (m, 2H), 1 .75 - 1 .54 (m, 4H), 1 .08 (s, 2H), 0.83 (t, 3H); MS (ESI+): m/z 539.2 [M+H]+; HPLC Purity: 90.68 %. Example 123:
3-(2-(Dimethylamino)pyrimidin-5-yl)-5-(ethyl(tetrahydro-2H-pyran-4- yl)amino)-N-((1-methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide
To a solution of the compound of example 1 18 (100 mg, 0.199 mmol) in 1 ,4-dioxane (10 mL) were added PdCI2(dppf)- CH2CI2adduct (16.25 mg, 0.020 mmol) and N,N-dimethyl-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyrimidin- 2-amine (64.5 mg, 0.259 mmol). The reaction mixture was stirred for 5 min, a solution of sodium carbonate (63.3 mg, 0.597 mmol) in water (2.0 mL) was added and the mixture was stirred at 100 °C for 5h. After completion of the reaction, the reaction mixture was filtered through celite, and the filtrate was distilled under vacuum.To the residue, water was added and the resulting mixture was extracted with ethyl acetate. The compound was adsorbed on silica and purified by column chromatography (silica gel, 10-20 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 40 mg (37.0 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .27 (s, 1 H), 8.65
(s, 2H), 8.24 (s, 1 H), 7.39 (s, 1 H), 7.20 (s, 1 H), 4.23 - 4.21 (m, 1 H), 3.84 - 3.81 (m, 2H), 3.28 - 3.25 (m, 2H), 3.21 (s, 5H), 3.16 -3.06 (m, 3H), 2.87 - 2.86 (m, 2H), 2.73 - 2.72 (m, 1 H), 2.71 - 2,68 (m, 2H), 2.27 - 2.26 (m, 3H), 2.09 (s, 2H), 2.00 - 1 .91 (m, 2H), 1 .56 - 1 .45 (m, 2H), 0.81 (t, 3H); MS (ESI+): m/z 531 .3 [M+H]+; HPLC Purity: 97.97 %.
Example 124:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(6-fluoro-5-methylpyridin-3-yl)-2- methyl-N-((1-methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide
To a solution of the compound of example 1 18 (100 mg, 0.199 mmol) in 1 ,4-dioxane (10 mL), was added 3-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridine (47.2 mg, 0.199 mmol). The reaction mixture was stirred for 5 min followed by addition of a solution of sodium carbonate (63.3 mg, 0.597 mmol) in water (2.0 mL). The reaction mixture was stirred at 100 °C for 5h. After completion of the reaction, the resulting mixture was filtered through celite and the filtrate was distilled under vacuum.To the residue obtained, water was added and the resulting mixture was extracted with ethyl acetate. The compound was adsorbed on silica and purified by column chromatography (silica gel, 10-20 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 22 mg (20.75 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .68 (s, 1 H), 8.32 (s, 1 H), 8.18 - 8.16 (m, 2H), 7.55 - 7.47 (m, 1 H), 7.28 (s, 1 H), 4.50 (s, 1 H), 4.42 (d, 2H), 4.43 - 4.33 (m, 2H), 3.92 - 3.76 (m, 2H), 3.33 - 3.23 (m, 1 H), 3.21 - 2.98 (m, 2H), 2.86 - 2.78 (m, 2H), 2.77 - 2.56 (m, 3H), 2.31 (s, 3H), 2.54 (s, 2H), 2.18 (s, 1 H), 2.10 - 1 .90 (m, 2H), 1 .87 - 1 .78 (m, 2H), 1 .76 - 1 .58 (m, 2H), 0.83 (t, 3H); MS (ESI-): m/z 531 .1 [M-H]"; HPLC Purity: 91 .3 %.
Example 125:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(1 H-indol-5-yl)-2-methyl-N-((1- methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl) benzamide
To a solution of the compound of example 1 18 (100 mg, 0.199 mmol) in
1 ,4-dioxane (10 mL) was added 5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)- 1 H-indole (62.9 mg, 0.259 mmol). The reaction mixture was stirred for 5 min followed by addition of sodium carbonate (63.3 mg, 0.597 mmol) in water (2.0 mL). The reaction mixture was stirred at 100 °C for 5h. After completion of the reaction, the resulting mixture was filtered through celite and the filtrate was distilled under vacuum.To the residue, water was added and the resulting mixture was extracted with ethyl acetate. The compound was adsorbed on silica and
purified by column chromatography (silica gel, 10-20 % ethyl acetate in petroleum ether) to yield the title compound.
1 H NMR (DMSO-de, 300 MHz): δ 1 1 .67 (s, 1 H), 1 1 .27 (s, 1 H), 8.41 (s, 1 H), 7.83 (d, 1 H), 7.55 - 7.50 (m, 3H), 7.36 (s, 1 H), 7.19 - 7.18 (m, 1 H), 6.93 - 6.90 (m, 1 H), 4.34 - 4.14 (m, 2H), 3.91 - 3.75 (m, 2H), 3.56 - 3.48 (m, 2H), 3.45 - 3.35 (m, 3H), 3.33 - 3.15 (m, 2H), 3.13 - 2.97 (m, 2H), 2.84 - 2.72 (m, 1 H), 2.70 - 2.59 (m, 1 H), 2.24 - 2.12 (m, 3H), 2.10 (s, 1 H), 2.04 - 1.84 (m, 2H), 1 .73 - 1 .62 (m, 2H), 1.60 - 1 .56 (m, 2H), 0.83 (t, 3H);MS (ESI+): m/z 539.5 [M+H]+; HPLC Purity: 93.85 %.
Example 126:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-
3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-5-(quinolin-3- yl)benzamide
To a solution of the compound of example 1 18 (100 mg, 0.199 mmol) in
1 ,4-dioxane (10 ml_), were added PdCI2(dppf)-CH2CI2adduct (16.25 mg, 0.020 mmol) and 3-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)quinolone (50.8 mg, 0.199 mmol). The reaction mixture was stirred for 5 min, followed by addition of a solution of sodium carbonate (63.3 mg, 0.597 mmol) in water (2.0 ml_). The reaction mixture was stirred at 100 °C for 5h. After completion of the reaction, the reaction mixture was filtered through celite and the filtrate was distilled under vacuum. To the residue, water was added and the resulting mixture was extracted with ethyl acetate. The compound was adsorbed on silica and purified by column chromatography (silica gel, 10-20 % ethyl acetate in petroleum ether) to yield the title compound.
Yield: 19 mg (17.34 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .72 (s, 1 H), 9.23 (s, 1 H), 8.64 (s, 1 H), 8.24 (s, 1 H), 8.07 (t, 2H), 7.77 - 7.74 (m, 1 H), 7.65 (s, 2H), 7.62 - 7.46 (m, 1 H), 4.36 (s, 2H), 3.84 (d, 2H), 3.33 - 3.24 (m, 2H), 3.22 - 2.98 (m, 3H), 2.78 - 2.70 (m, 1 H), 2.69 - 2.53 (m, 2H), 2.29 (s, 3H), 2.20 (s, 4H), 2.04 - 1 .84 (m, 2H), 1 .82 - 1 .63 (m, 4H), 0.83 (t, 3H); MS (ESI+): m/z551 .2 [M+H]+; HPLC Purity: 98.21 %.
Example 127:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo- 3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-5-(6-(morpholino methyl) pyridin-3-yl)benzamide
To a solution of 10 mL of 1 ,4-dioxane in 2 mL of water, was added the compound of example 1 18 (150 mg, 0.299 mmol), 4-((5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridin-2-yl)methyl)morpholine (136 mg, 0.448 mmol), sodium carbonate (1 1 1 mg, 1 .045 mmol), [1 ,1 '-
Bis(diphenylphosphino)ferrocene]palladium(ll)dichloride (12.19 mg, 0.015 mmol). Argon gas was purged for 20 min and the reaction mixture was stirred for 16 h at 100 °C in a sealed tube. The reaction mixture was concentrated and 5 mL of water was added. The resulting mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulphate and concentrated to obtain a crude material, which was purified using DCM and methanol.
Yield: 67 mg (37.4 %);1H NMR (DMSO-d6, 300 MHz): δ 1 1 .67 (s, 1 H), 8.75 (s, 1 H), 8.19 (t, 1 H), 8.01 (d, 1 H), 7.50 - 7.44 (m, 2H), 7.25 (s, 1 H), 4.42 - 4.30 (m, 2H), 3.92 - 3.76 (m, 2H), 3.33 - 3.26 (m, 3H), 3.25 - 2.96 (m, 2H), 2.94 - 2.89 (m, 1 H), 2.88 - 2.76 (m, 4H), 2.74 - 2.56 (m, 6H), 2.48 (s, 3H), 2.41 (s, 2H), 2.24 (s, 1 H), 2.16 - 2.07 (m, 3H), 2.05 - 1 .89 (m, 2H), 1 .82 - 1 .73 (m, 2H), 1.70 - 1 .61 (m, 2H), 0.83 (t, 3H); MS (ESI+): m/z 600.2 [M+H]+; HPLC Purity: 94.57 %.
Example 128:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-7- (trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'- (morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide
HATU was added to a stirred solution of the compound of example 26 (0.1 g, 0228 mmol) at I CO followed by addition of Hunig's base. The reaction mixture was stirred for 10 min and the compound of example 1 1 (0.77 g, 0.296 mmol) was added. The reaction was stirred for 16 h at 25 °C. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and dried over anhydrous sodium sulphate. The resulting mixture was purified using flash column chromatography (silica-gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 80 mg (51 .5 %); 1 H NMR (DMSO-d6, 300 MHz): δ 8.29 - 8.09 (m, 1 H), 7.80 (s, 1 H), 7.62 (s, 2H), 7.42 - 7.38 (m, 3H), 7.32 (s, 1 H), 3.86 - 3.82 (m,
2H), 3.59 - 3.51 (m, 2H), 3.33 - 3.26 (m, 3H), 3.22 - 3.12 (m, 3H), 3.09 - 2.96 (m, 5H), 2.73 (s, 2H), 2.48 - 2.34 (m, 4H), 2.31 (s, 4H), 1 .67 - 1 .55 (m, 5H), 1 .52 - 1 .40 (m, 6H), 0.83 (t, 3H); MS (ESI-): m/z 681 .7 [M-H]"; HPLC Purity: 99.35 %. Example 129:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7- (trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(6- morpholino pyridin-3-yl)benzamide
HATU was added to a stirred solution of the compound of example 70 (0.1 g. 0.235 mmol) at 10 °C followed by addition of Hunig's base. The reaction mixture was stirred for 10 min and the compound of example 1 1 (0.08 g. O.306 mmol) was added. The reaction mixture was stirred for 16 h at 25 °C. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and dried over anhydrous sodium sulphate. The organic extract was concentrated and purified by flash column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 75 mg (47.8 %); 1 H NMR (DMSO-d6, 300 MHz): δ 8.45 (s, 1 H), 7.89 - 7.86 (m, 1 H), 7.85 (s, 1 H), 7.78 (s, 2H), 7.39 (s, 1 H), 6.92 (d, 1 H), 3.85 - 3.82 (m, 3H), 3.73 (m, 4H), 3.50 (s, 4H), 3.33 - 3.26 (m, 3H), 3.22 - 3.08 (m, 3H), 3.03 (s, 1 H), 2.96 - 2.81 (m, 1 H), 2.36 (s, 3H), 1 .96 - 1.34 (m, 10H), 1 .29 - 1 .14 (m, 2H), 0.83 (t, 3H); MS (ESI+): m/z 668.7 [M+H]+; HPLC Purity: 92.66 %.
Example 130:
1-methyl-3-oxo-7-(2,2,2-trifluoroethyl)-2,3,5,6,7,8-hexahydro-2,7- naphthyridine-4-carbonitrile
NaBH4 (2499 mg, 66.1 mmol) was added in portions to the compound of example 62 (250 mg, 1 .321 mmol) in TFA (10 mL) at RT over a period of 1 h. The reaction mixture was allowed to stir for 2 h, heated at 50 °C for 3 h and then stirred at RT for 16 h.The reaction mixture was poured onto ice, neutralised using 6M NaOH solution and extracted using ethyl acetate. The crude material was purified using column chromatography (silica gel, 10-20 % methanol in chloroform) to yield the title compound.
Yield: 89 mg (24.69 %); 1 H NMR (DMSO-d6, 300 MHz) δ 12.43 (s, NH), 3.54 (s, 2H), 3.43-3.36 (m, 2H), 2.88-2.73 (m, 4H), 2.18 (s, 3H); MS (ESI+): 272.1 [M+H]+; HPLC Purity: 99.43 %. Example 131 :
4-(Aminomethyl)-1-methyl-7-(2,2,2-trifluoroethyl)-5,6,7,8-tetrahydro-2,7- naphthyridin-3(2H)-one
To a solution of the compound of example 130 (800 mg, 2.95 mmol) in acetic acid (10 mL) were added Pd/C (78 mg, 0.737 mmol), sodium acetate (484 mg, 5.90 mmol) and platinum(IV) oxide (16.74 mg, 0.074 mmol). The flask was shaken under an atmosphere of hydrogen at 80 psi for 8 h. After completion of the reaction, the reaction mixture was filtered through celite and concentrated. The resulting oily compound was stirred in 4.0 mL of concentrated HCI and filtered. The filtrate was stirred in a mixture of 0.8 mL of concentrated HCI and 5 mL of EtOH at 5-10 °C for 2 h. The solid was filtered, washed with ether and dried to yield the title compound.
Yield: 750 mg (63.1 %); MS (ESI+): 312 [M+H]+; HPLC Purity: 77.3 %.
Example 132:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-7- (2,2,2-trifluoroethyl)-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 26 (70 mg, 0.160 mmol) in DMF (5 mL), were added HATU (91 mg, 0.239 mmol) and Hunig's Base (0.084 mL, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 131 (59.7 mg, 0.192 mmol) and the resulting mixture was stirred for 16 h.The reaction solvent was distilled under vacuum, the residue obtained was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0- 15 % MeOH/CHCI3) to yield the title compound.
Yield: 36 mg (22.76 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .57 (s, 1 H), 8.20 (s, 1 H), 7.58-7.56 (m, 2H), 7.40-7.36 (m, 3H), 7.21 (s, 1 H), 4.35-4.28 (s, 2H),
3.85-3.81 (m, 2H), 3.65-3.48 (m, 8H), 3.26-3.21 (m, 4H), 3.10-3.01 (m, 3H), 2.85- 2.73 (m, 4H), 2.38-2.31 (m, 4H), 2.24 (s, 3H), 2.07 (s, 3H), 1 .68-1.40 (m, 4H), 0.83 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 696.6 [M+H]+; HPLC Purity: 90.26 %. Example 133:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-
(2,2,2-trifluoroethyl)-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6-
(4-methylpiperazin-1-yl)pyridin-3-yl)benzamide
To a solution of the compound of example 67 (70 mg, 0.160 mmol) in DMF (5 mL) wereadded HATU (91 mg, 0.239 mmol) and Hunig'sBase (0.084 ml_, 0.479 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 131 (59.7 mg, 0.192 mmol) and the resulting mixture wasstirredfor 16 h. The reaction solvent was distilled under vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 mL). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHC ) to yield the title compound.
Yield: 65 mg (55.1 %); 1H NMR (DMSO-d6, 300 MHz): δ 1 1 .57 (s, 1 H), 8.39 (s, 1 H), 8.17 (s, 1 H), 7.80 (d, J= 6.6 Hz, 1 H), 7.36 (s, 1 H), 7.17 (s, 1 H), 6.91 (d, J= 6.6 Hz, 1 H), 4.34-4.27 (m, 2H), 3.84-3.81 (m, 2H), 3.62-3.51 (m, 8H), 3.28-3.21 (m, 4H), 3.09-3.01 (m, 5H), 2.87-2.71 (m, 4H), 2.27 (s, 3H), 2.22 (s, 3H), 2.07 (s, 3H), 1 .68-1 .49 (m, 4H), 0.82 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 696.3 [M+H]+; HPLC Purity: 94.1 1 %. Example 134:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-
(2,2,2-trifluoroethyl)-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6- morpholinopyridin-3-yl)benzamide
To a solution of the compound of example 70 (70 mg, 0.165 mmol) in DMF (5 mL), were added HATU (94 mg, 0.247 mmol) and Hunig's Base (0.086 mL, 0.494 mmol) and the reaction mixture was stirred at RT for 1 h. To the reaction mixture, was added the compound of example 131 (61 .5 mg, 0.197 mmol) and the resulting mixture was stirredfor 16 h.The reaction solvent was distilled under
vacuum, the residue was extracted using ethyl acetate and washed with water (2x25 ml_). The combined organic extracts were distilled off to obtain a crude material, which was purified by using column chromatography (silica gel, 0-15 % MeOH/CHC ) to yield the title compound.
Yield: 7 mg (5.1 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .58 (s, 1 H), 8.41
(s, 1 H), 8.18 (s, 1 H), 7.85 (s, 1 H), 7.37 (s, 1 H), 7.18 (s, 1 H), 6.92 (d, J= 8.4 Hz, 1 H), 4.28 (s, 2H), 3.84-3.71 (m, 6H), 3.51 -3.46 (m, 6H), 3.28-3.21 (m, 4H), 3.09- 3.01 (m, 3H), 2.87-2.81 (m, 4H), 2.22 (s, 3H), 2.07 (s, 3H), 1 .68-1 .40 (m, 4H), 0.82 (t, J= 7.2 Hz, 3H); MS (ESI+): m/z 683.3 [M+H]+; HPLC Purity: 81 .88 %.
Example 135:
5-Bromo-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3- oxo-2,3, 5, 6,7, 8-hexahydroisoquinolin-4-yl)methyl)benzamide
To a solution of the compound of example 20 (1 .33 g, 3.89 mmol) in 10 ml_ of DMF, were added DIEA (2.036 ml_, 1 1.66 mmol) and 2-(7-aza-1 H- benzotriazole-1 -yl)-1 ,1 ,3,3-tetramethyl uronium hexafluorophosphate (HATU) (2.217 g, 5.83 mmol). The reaction mixture was stirred for 1 h at room temperature. The compound of example 2 (1 .15 g, 5.05 mmol) was added to the reaction mixture and the reaction mixture was stirred for 12 h at room temperature. After the reaction was complete, the reaction mixture was concentrated and water was added to the same. The crude product obtained was extracted with ethyl acetate and purified by column chromatography (silica gel, ethyl acetate in petroleum ether) to obtain the title compound.
Yield: 1 .5 g (74.7%); 1 H NMR (DMSO-d6, 500 ΜΗζ):δ 1 1 .50 (s, 1 H), 8.19 (s, 1 H), 7.31 (s, 1 H), 7.08(s, 1 H), 4.24 (d, J = 4Hz, 2H), 3.83-3.81 (d, J = 10.5Hz, 2H), 3.26-3.21 (t, J = 6.9Hz, 2H), 3.02 (d, J = 4Hz, 2H), 2.94 (m, 1 H), 2.71 (s, 2H), 2.37 (s, 2H), 3.15 (s, 3H), 2.10 (s, 3H), 1 .64 (s, 2H), 1 .59 (s, 4H), 1 .50 (m, 2H), 0.79 (t, J = 6.5Hz, 3H); MS (ESI+) m/z 516 (M+H). Example 136:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(6-morpholinopyridin-3- yl)benzamide
To a stirred solution of the compound of example 135 (75 mg, 0.145 mmol) in dioxane (10 ml_), was added aqueous 2M sodium carbonate (254 μΙ_, 0.508 mmol) solution. To this mixture was added 4-(5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridin-2-yl)morpholine (63.2 mg, 0.218 mmol). The reaction mixture was purged with argon for 10 min. followed by addition of 1 ,1 '- bis(diphenylphosphino)ferrocene palladium-dichloromethane complex (20 mg, 0.024 mmol) and argon was purged again for 15 min. The reaction mixture was heated to 100 °C for 6 h to 8 h. On completion of the reaction, reaction mixture was diluted with water and extracted thrice with 10 % MeOH/DCM. The combined organic layer was dried over sodium sulphate. The solvent was removed under reduced pressure and further purified by column chromatography (silica gel, EtOAc in petroleum ether) to obtain the title compound.
Yield: 50 mg (57.4 %); 1 H NMR (DMSO-d6, 500 MHz): δ 1 1 .50 (s, 1 H), 8.42 (s, 1 H), 8.13 (s, 1 H), 7.85 (d, J = 10Hz, 1 H), 7.37 (s, 1 H), 7.17 (s, 1 H), 6.92 (d, J = 6Hz, 1 H), 4.31 (d, J = 4Hz, 2H), 3.84 (d, J = 9Hz, 2H), 3.71 (s, 4H), 3.47 (s, 4H), 3.2 (t, J = 6Hz, 2H), 3.08 (m , 3H), 2.7 (s, 2H), 2.37 (s, 2H), 2.23 (s, 3H), 2.21 (s, 3H), 1 .64 (m , 6H), 1 .52 (m, 2H), 0.82 (t, J = 3.9Hz, 3H); MS (ESI+): m/z 600.4 (M+H). Example 137:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-
2,3,5,6 ,8-hexahydroisoquinolin-4-yl)methyl)-5-(6-methylpyridin-3-yl) benzamide
To a stirred solution of the compound of example 135 (80 mg, 0.155 mmol) in dioxane (10 ml_), was added aqueous 2 M sodium carbonate (560 μΙ_, 0.560 mmol) solution. To this mixture was added 2-methyl-5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridine (32.9 mg, 0.240 mmol). The reaction mixture was purged with argon for 10 min, followed by addition of 1 ,1 '- bis(diphenylphosphino)ferrocene palladium-dichloromethane complex (13.07 mg, 0.016 mmol) and argon was purged again for 15 min. The reaction mixture was heated to 100 °C for 6 h to 8 h. On completion of the reaction, reaction mixture was diluted with water and extracted thrice with 10 % MeOH/DCM. The combined organic layer was dried over sodium sulphate. The solvent was removed under
reduced pressure and further purified by column chromatography (silica gel, DCM and methanol) to obtain the title compound.
Yield: 68 mg (40.2 %); 1 H NMR (CDCI3, 300 MHz): δ 8.58 (s, 1 H), 7.68 (d, J = 7.8Hz, 1 H), 7.41 (m, 1 H), 7.26 (s, 1 H), 7.15 (d, J = 7.8Hz, 1 H), 4.56 (s, 2H), 3.97 (d, J = 12Hz, 2H), 3.49 (d, J = 7.2Hz, 1 H), 3.32 (m, 3H), 3.1 1 (m, 2H), 2.96 (m, 4H), 2.5 (s, 3H), 2.36 (s, 5H), 2.0 (s, 3H), 1 .28-1 .25 (m, 5H), 0.91 (t, J = 6.9Hz, 3H); MS (ESI+) m/z 529.3 (M+H).
Example 138:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(6-(trifluoromethyl)pyridin-3- yl)benzamide
To a stirred solution of the compound of example 135 (100 mg, 0.194 mmol) in dioxane (10 ml_), was added aqueous 2 M sodium carbonate (339 μΙ_, 0.678 mmol) solution. To this mixture was added (6-(trifluoromethyl)pyridin-3- yl)boronic acid (55.4 mg, 0.290 mmol).The reaction mixture was purged with argon for 10 min. followed by addition of 1 ,1 '-Bis(diphenylphosphino)ferrocene palladium-dichloromethane complex (0.05 mg, 9.68 mmol) and argon was purged again for 15 min. The reaction mixture was heated to 100 °C for 6 h to 8 h. On completion of the reaction, the resulting mixture was diluted with water and extracted thrice with 10 % MeOH/DCM. The combined organic layer was dried over sodium sulphate. The solvent was removed under reduced pressure and further purified by column chromatography (silica gel, DCM and methanol) to obtain the title compound.
Yield: 67 mg (59.4 %); 1 H NMR (CDCI3, 300 MHz): δ 1 1 .07 (bs, 1 H), 8.85
(s, 1 H), 7.97 (d, J = 9Hz, 1 H), 7.71 (d, J = 8.1 Hz, 1 H), 7.38 (s, 1 H), 7.31 (s, 1 H),4.59 (d, J = 6Hz, 2H), 3.98 (d , J = 12Hz, 2H), 3.32 (m , 2H), 3.13 (m, 2H), 2.9 (m, 3H), 2.41 (m, 5H), 2.13 (s, 2H), 1 .71 (m, 7H), 1 .25 (s, 2H), 0.90 (t, J = 6.9Hz, 3H); MS (ESI+) m/z 583.2 (M+H).
Example 139:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-morpholino-[1 ,1 '-biphenyl]-
3-carboxamide
To a stirred solution of the compound of example 135 (150 mg, 0.290 mmol) in dioxane (10 ml_), was added aqueous sodium carbonate (508 μΙ_, 1 .017 mmol) solution. To this mixture was added (4-morpholinophenyl)boronic acid (84 mg, 0.407 mmol).The reaction mixture was purged with argon for 10 min. followed by addition of 1 ,1 '-bis(diphenylphosphino)ferrocene palladium-dichloromethane complex (1 1 .86 mg, 0.015 mmol) and argon was purged again for 15 min. The reaction mixture was heated to 100 °C for 6 h to 8 h. On completion of the reaction, the resulting mixture was diluted with water and extracted thrice with 10 % MeOH/DCM. The combined organic layer was dried over sodium sulphate. The solvent was removed under reduced pressure and further purified by column chromatography (silica gel, DCM and methanol) to obtain the title compound.
Yield: 90 mg (51 .8 %);1 H NMR (DMSO-d6, 300 ΜΗζ):δ 1 1 .489 (s, 1 H), 8.13
(s, 1 H), 7.50-7.47 (d, J = 8.7Hz, 2H), 7.33 (s, 1 H), 7.15 (s, 1 H), 7.01 (d, J = 8.7Hz, 2H), 4.31 (d, J = 6Hz, 2H), 3.84 (d, J = 12Hz, 2H), 3.74 (m,4H), 3.33 (m, 2H), 3.20 (m, 4H), 3.08 (m, 3H), 2.73 (s, 2H), 2.36 (s, 2H), 2.22 (s, 3H), 2.09 (s, 3H), 1 .63 (m, 6H), 1 .52 (m, 2H), 0.82(t, J = 6.6Hz, 3H); MS (ESI+) m/z 599.8 (M+H).
Example 140:
Dimethyl (3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4'-methyl-5'-(((1-methyl-
3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 ,1 '- biphenyl]-3-yl)phosphonate
To a stirred solution of the compound of example 135 (150 mg, 0.299 mmol) in 1 ,4-dioxane (10 mL) were added PdCI2(dppf)-CH2CI2adduct (24.38 mg, 0.030 mmol) and dimethyl (3-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)phenyl)phosphonate (93 mg, 0.299 mmol). The reaction mixture was then stirred for 5 min followed by addition of sodium carbonate (95 mg, 0.896 mmol) in water (2.0 ml_).The reaction mixture was stirred at 100 °C for 5 h. After completion of the reaction, the resulting mixture was filtered through celite and the filtrate was concentrated following which, water was added to it. The crude product was extracted using ethyl acetate. The compound obtained was purified by column
chromatography (silica gel, 20:80 ethyl acetate and petroleum ether) to yield the title compound.
Yield: 25 mg (13.47 %); 1 H NMR (DMSO,300 MHz): δ 1 1 .48 (s,1 H), 8.21 (s, 1 H), 7.92 (m, 2H), 7.89 (m, 2H), 7.52 (d, J=3.0 Hz, 1 H), 7.23 (d, J=3.0 Hz, 1 H), 4.33 (m, 2H), 3.85 (q, 2H),3.70 (s, 6H), 3.26 (m, 2H), 3.09 (m, 4H), 2.16 (m, 1 H), 2.38 (m, 2H), 2.27 (m, 2H), 2.10 (s, 3H), 1 .64 (m, 6H), 1 .55 (s, 3H), 0.86 (t, J=6.9Hz, 3H); MS (ESI+) m/z 622 (M+H).
Example 141 :
(S)-1 -(1-(4-bromophenyl)ethyl)piperidine
To a solution of (S)-1 -(4-bromophenyl)ethanamine (1 .439 ml_, 10.00 mmol) in DMF, were added potassium carbonate (4.14 g, 30.0 mmol) and 1 ,5-di- iodopentane (2.98 ml_, 19.99 mmol). The reaction mixture was heated at 90 °C in DMF for 16 h. The reaction mixture was concentrated, the product was extracted with ethyl acetate and purification was done using column chromatography (silica gel, 30 % CHCI3 in DCM) to yield the title compound.
Yield: 2 g (74.6%); 1H NMR (DMSO-d6, 300 MHz): δ 7.4 - 7.6 (d, 2H), 7.1 - 7.3 (d, 2H), 3.2 - 3.3 (m, 1 H), 2.1 - 2.4 (m, 4H), 1 .4 - 1 .5 (m, 4H), 1 .3 - 1 .4 (m, 2H), 1 .1 - 1 .2 (m, 3H); MS (ESI+): m/z 269 [M+H]+; HPLC Purity: 96.30 %.
Example 142:
(S)-1 -(1-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)ethyl) piperidine
To a solution of the compound of example 141 (1 .5 g, 5.59 mmol) and Bis(pinacolato)diboron (2.130 g, 8.39 mmol) in dioxane (15 ml_), was added potassium acetate (1 .372 g, 13.98 mmol), under the atmosphere of argon gas. PdCI2(dppf)-CH2CI2 adduct (0.228 g, 0.280 mmol) was added to the above reaction mixture and it was heated to 90 °C. The reaction mixture was concentrated and extracted with ethyl acetate. The purification was carried out using column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 1 g (56.7 %); 1 H NMR (DMSO-d6, 300 MHz): δ 7.62 (d, 2H), 7.29 (d, 2H), 3.2 - 3.3 (m, 1 H), 2.30 - 2.26 (m, 4H), 1 .45 - 1 .43 (m, 4H), 1 .24 (s, 2H),1 .20 (s, 3H), 1 .07 (s, 12H); MS (ESI+): m/z 316 [M+H]+; HPLC Purity: 87.88 %. Example 143:
(S)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(1-(piperidin-1 -yl)ethyl)-[1 ,1 '- biphenyl]-3-carboxamide
To a solution of the compound of example 135 (1 .5 g, 2.90 mmol) in dioxane were added the compound of example 142 (1 .099 g, 3.49 mmol), sodium carbonate (4.36 mL, 8.71 mmol) and PdCI2(dppf)-CH2Cl2 adduct (0.1 19 g, 0.145 mmol).Argon was purged and the reaction mixture was heated to 100 °C. The reaction mixture was concentrated, extracted with DCM and purification was done using 5 % MeOH in DCM to yield the title compound.
1 H NMR (DMSO-de, 300 MHz): δ 1 1 .48 (s, 1 H), 8.15 (s, 1 H), 7.54 (d, 2H),
7.39 - 7.33 (m, 3H), 7.21 (s, 1 H), 4.31 (d, 2H), 3.83 (d, 2H), 3.44 - 3.42 (m, 2H), 3.25 - 3.21 (m, 2H), 3.10 - 3.08 (m, 4H), 2.84 - 2.64 (m, 3H), 2.50 - 2.29 (m, 5H), 2.25 (s, 4H), 2.10 (s, 3H), 1 .74 - 1 .54 (m, 8H), 1 .30 - 1 .28 (m, 2H), 1 .26 - 1 .22 (m, 3H), 0.83 (t, 3H);MS (ESI+): m/z 625.4 [M+H]+; HPLC Purity: 98.5 %.
Example 144:
(R)-1-(1 -(4-bromophenyl)ethyl)piperidine
To a solution of (R)-1 -(4-bromophenyl)ethanamine (5 g, 24.99 mmol) in 70 mL of DMF, were added potassium carbonate (10.36 g, 75.0 mmol) and 1 ,5- diiodopentane (7.46 mL, 50.0 mmol) and the reaction mixture was heated to 90 °C. The reaction mixture was concentrated, the product was extracted with ethylacetate and purification was done by column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 2 g; 1 H NMR (DMSO-d6, 300 MHz): δ 7.47 - 7.50 (d, 2H), 7.23 - 7.26 (d, 2H), 4.12 - 4.22 (m, 1 H), 2.16 - 2.36 (m, 3H), 1 .62 - 1 .83 (m, 1 H), 1 .41 - 1 .58 (m, 4H),1 .20 - 1 .35 (m, 2H), 1 .1 1 - 1 .18 (m, 3H); MS (ESI+): m/z 269 [M+H]+; HPLC Purity: 98.72 %.
Example 145:
(R)-1 -(1 -(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)ethyl) piperidine
To a solution of (R)-1 -(1 -(4-bromophenyl)ethyl)piperidine (15.4 g, 57.4 mmol), Bis(pinacolato)diboron (21 .87 g, 86 mmol) in dioxane (15 mL) and potassium acetate (14.09 g, 144 mmol) was added. Argon gas was purged into the reaction mixture followed by addition of PdCI2(dppf)-CH2Cl2 adduct (2.34 g, 2.87 mmol). The reaction mixture was heated to 90 °C. The reaction mixture was concentrated and extracted with ethyl acetate. The purification was carried out using flash column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 1 g (56.7 %); 1 H NMR (DMSO-d6, 300 MHz): δ 7.61 (d, 2H), 7.29 (d, 2H), 2.50 - 2.34 (m, 4H), 1 .44 - 1 .34 (m, 2H), 1.26 (s, 8H), 1 .05 (s, 12H). Example 146:
(R)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1-methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(1-(piperidin-1 -yl)ethyl)-[1 ,1 '- biphenyl]-3-carboxamide
To a solution of 60 mL dioxane in 20 mL water, were added the compound of example 135 (6 g, 1 1 .62 mmol), the compound of example 145 (5.49 g, 17.43 mmol), PdCl2(dppf)-CH2Cl2 adduct (0.474 g, 0.581 mmol) and sodium carbonate (17.43 mL, 34.9 mmol).The reaction mixture was heated in argon atmosphere. The resulting mixture was concentrated and purified using flash column chromatography (silica gel, 5 % MeOH in DCM). The crude material was washed with petroleum ether, to yield the title compound.
Yield: 1 .7 g (23.42 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .48 (s, 1 H), 8.15 (s, 1 H), 7.54 (d, 2H), 7.39 - 7.33 (m, 3H), 7.21 (s, 1 H), 4.31 (d, 2H), 3.83 (d, 2H), 3.44 - 3.42 (m, 2H), 3.25 - 3.21 (m, 2H), 3.10 - 3.08 (m, 4H), 2.84 - 2.64 (m, 3H), 2.50 - 2.29 (m, 5H), 2.25 (s, 4H), 2.10 (s, 3H), 1 .74 - 1 .54 (m, 8H), 1 .30 - 1 .28 (m, 2H), 1 .26 - 1 .22 (m, 3H), 0.83 (t, 3H); MS (ESI+): m/z 625.4 [M+H]+; HPLC Purity: 98.76 %.
Example 147:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(1-(piperidin-1 -yl)ethyl)-[1 ,1 '- biphenyl]-3-carboxamide
To a solution of the compound of example 135 (6 g, 1 1 .62 mmol) in dioxane, the compound of example 145 (2.71 g, 1 1 .62 mmol), sodium carbonate (1 .231 g, 1 1 .62 mmol), PdCI2(dppf)-CH2CI2 adduct (9.49 g, 1 1 .62 mmol) were added. The reaction mixture was heated for 16 h at 100 °C, cooled to RT, concentrated and purified with flash column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 4 g (55.1 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .48 (s, 1 H), 8.15 (s,
1 H), 7.55 (d, 2H), 7.40 - 7.33 (m, 3H), 7.21 (s, 1 H), 4.32 (s, 2H), 3.90 - 3.80 (m, 2H), 3.52 - 3.45 (m, 1 H), 3.33 - 3.21 (m, 2H), 3.21 - 3.20 (m, 1 H), 3.19 - 2.93 (m, 3H), 2.82 - 2.78 (m, 2H), 2.40 - 2.26 (m, 5H), 2.25 (s, 3H), 2.10 (s, 3H), 1 .77 - 1 .58 (m, 6H), 1 .56 - 1 .48 (m, 6H), 1 .39 - 1 .14 (m, 5H), 0.83 (t, 3H); MS (ESI+): m/z 625.4 [M+H]+; HPLC Purity: 96.88 %.
Example 148:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(2-morpholinoethyl)-[1 ,1 '- biphenyl]-3-carboxamide
To a solution of the compound of example 135 (270mg, 0.523 mmol), in 20 mL of dioxane and 2 mL of water, were added 4-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenethyl)morpholine (249 mg, 0.784 mmol), sodium carbonate (222 mg, 2.091 mmol), PdCI2(dppf)-CH2CI2 adduct (21 .35 mg, 0.026 mmol). Argon gas was purged into the reaction mixture and the reaction mixture was heated for 16 h at 100 °C. The reaction mixture was concentrated and extracted with DCM. Purification was carried out using flash column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 100 mg (30.5 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .49 (s, 1 H), 8.13 (s, 1 H), 7.68 (d, 2H), 7.51 (d, 1 H), 7.33 (d, 2H), 7.27 (s, 1 H), 4.30 (d, 2H),
3.83 - 3.80 (m, 2H), 3.56 - 3.55 (m, 1 H), 3.33 - 3.16 (m, 2H), 3.14 - 2.94 (m, 4H),
2.84 - 2.66 (m, 4H), 2.62 - 2.56 (m, 3H), 2.44 - 2.36 (m, 7H), 2.32 - 2.22 (m, 3H),
2.14 - 2.01 (m, 3H), 1 .76 - 1 .54 (m, 6H), 1.43 - 1 .26 (m, 2H), 0.83 (t, 3H); MS (ESI+): m/z 627.5 [M+H]+; HPLC Purity: 96.32 %.
Example 149:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-3'-(1-(pyrrolidin-1 -yl)ethyl)- [1 ,1 '-biphenyl]-3-carboxamide
To a solution of 15 mL dioxane in 2 mL water, were added the compound of example 135 (300 mg, 0.581 mmol, (3-(1 -(pyrrolidin-1 -yl)et yl)phenyl)boronic acid (191 mg, 0.871 mmol), sodium carbonate (215 mg, 2.033 mmol), PdCI2(dppf)-CH2Cl2 adduct (23.72 mg, 0.029 mmol). Argon gas was purged into the reaction mixture and the reaction mixture was heated for 16 h at 100 °C. The reaction mixture was concentrated and purified using flash column chromatography (silica gel, 5% MeOH in DCM) to yield the title compound.
Yield: 150 mg (42.3 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .50 (s, 1 H),
8.17 (s, 1 H), 7.92 - 7.25 (m, 5H), 7.14 (s, 1 H), 4.31 (s, 2H), 3.80 (d, 2H), 3.33 - 3.26 (m, 4H), 3.24 - 2.98 (m, 2H), 2.94 - 2.88 (m, 2H), 2.82 - 2.74 (s, 2H), 2.50 - 2.32 (m, 4H), 2.30 - 2.14 (m, 4H), 2.06 - 1 .96 (m, 3H), 1 .86 - 1.49 (m, 9H), 1 .40 - 1 .06 (m, 5H), 0.83 (t, 3H); MS (ESI+): m/z 61 1 .4 [M+H]+; HPLC Purity: 97.33 %.
Example 150:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(4-methyl-3-oxo-3,4-dihydro-
2H-benzo[b][1 ,4]oxazin-6-yl)benzamide
To a mixture of 15 mL of dioxane and 3 mL of water, was added the compound of example 135 (300 mg, 0.581 mmol), 4-methyl-6-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-2H-benzo[b][1 ,4]oxazin-3(4H)-one (252 mg, 0.871 mmol), sodium carbonate (246 mg, 2.323 mmol) and PdCI2(dppf)-CH2CI2 adduct (23.72 mg, 0.029 mmol). Argon gas was purged into the reaction mixture and the reaction mixture was heated for 16 h at 100 °C. The reaction mixture was concentrated and purified using flash column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 130 mg (37.4 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .48 (s, 1 H), 8.14 (s, 1 H), 7.37 (s, 1 H), 7.29 (s, 1 H), 7.26 - 7.22 (m, 2H), 7.05 (d, 1 H), 4.66 (s, 2H), 4.30 (d, 2H), 3.94 (s, 1 H), 3.82 (d, 2H), 3.40 - 3.35 (m, 2H), 3.33 - 3.28 (m, 2H), 3.23 - 3.08 (m, 3H), 2.73 - 2.49 (m, 2H), 2.36 - 2.29 (m, 2H), 2.22 (s, 2H), 2.08 (s, 2H), 1 .70 - 1 .60 (m, 4H), 1 .56 - 1 .52 (m, 2H), 1.22 (s, 1 H), 1 .14 (s, 4H), 0.83 (t, 3H); MS (ESI+): m/z 599.4 [M+H]+; HPLC Purity: 94.46 %.
Example 151 :
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(1 ,3,5-trimethyl-1 H-pyrazol-4- yl)benzamide
To a mixture of 15 mL of dioxane and 3 mL of water, were added the compound of example 135 (200 mg, 0.387 mmol), 1 ,3,5-trimethyl-4-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (137 mg, 0.581 mmol), sodium carbonate (164 mg, 1 .549 mmol), PdCI2(dppf)-CH2CI2 adduct (15.81 mg, 0.019 mmol). Argon was purged into the reaction mixture for 1 h and the reaction mixture was heated for 16 h. The reaction mixture was concentrated and washed with water. The resulting mixture was extracted with DCM. The organic extract was concentrated and purified using flash column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 90 mg (42.6 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .48 (s, 1 H), 8.10 (s, 1 H), 6.99 (s, 1 H), 6.80 (s, 1 H), 4.29 (d, 2H), 3.84 - 3.80 (m, 2H), 3.67 (s, 3H), 3.33 - 3.23 (m, 2H), 3.20 - 3.02 (m, 3H), 2.81 - 2.61 (m, 2H), 2.46 - 2.29 (m, 2H), 2.21 - 2.14 (m, 6H), 2.09 (s, 2H), 1 .90 - 1 .63 (m, 8H), 0.83 (t, 6H); MS (ESI+): m/z 546.7 [M+H]+; HPLC Purity: 98.92 %.
Example 152:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(6-(pyrrolidin-1-yl)pyridin-3- yl)benzamide
To a solution of the compound of example 135 (150 mg, 0.290 mmol) in dioxane, was added 2-(pyrrolidin-1 -yl)-5-(4,4,5,5-tetramethyl-1 ,3-dioxolan-2- yl)pyridine (1 12 mg, 0.407 mmol), sodium carbonate (108 mg, 1 .017 mmol),
PdCl2(dppf)-CH2Cl2 adduct (1 1 .86 mg, 0.015 mmol). Argon was purged through the reaction mixture and the reaction mixture was heated at 100 °C for 16 h.The reaction mixture was concentrated and purified using flash column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 68.51 mg (40.3 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .48 (s, 1 H),
8.62 (s, 2H), 8.1 1 (t, 1 H), 7.36 (s, 1 H), 7.17 (s, 1 H), 4.29 (s, 2H), 3.89 - 3.70 (m, 2H), 3.50 (s, 4H), 3.33 - 3.24 (m, 3H), 3.08 - 2.93 (m, 4H), 2.73 (s, 2H), 2.46 - 2.26 (m, 2H), 2.21 (s, 3H), 2.12 (s, 3H), 2.09 (s, 4H), 1 .93 (s, 5H), 1 .63 - 1 .52 (m, 2H), 0.83 (t, 3H); MS (ESI+): m/z 584.7 [M+H]+; HPLC Purity: 96.21 %.
Example 153:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(2-(piperidin-1 -yl)propan-2- yl)-[1 ,1 '-biphenyl]-3-carboxamide
To a solution of the compound of example 135 (500 mg, 0.968 mmol) in dioxane, were added 1 -(2-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)phenyl)propan-2-yl)piperidine (351 mg, 1 .065 mmol), sodium carbonate (359 mg, 3.39 mmol) and PdCI2(dppf)-CH2CI2 adduct (39.5 mg, 0.048 mmol).The reaction mixture was heated at 100 °C for 16 h. The reaction mixture was concentrated and purified using flash column chromatography (silica gel, 5 % MeOH in DCM) to yield the title compound.
Yield: 230 mg (37.2 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .48 (s, 1 H), 8.14 (s, 1 H), 7.55 (s, 4H), 7.40 (s, 1 H), 7.21 (s, 1 H), 4.31 (s, 2H), 3.84 - 3.81 (m, 2H), 3.33 - 3.26 (m, 3H), 3.22 - 3.08 (m, 3H), 2.84 - 2.68 (m, 2H), 2.37 (s, 5H), 2.25 (s, 3H), 2.10 (s, 3H), 1 .64 (s, 5H), 1 .47 - 1 .40 (m, 5H), 1 .33 - 1 .29 (m, 2H), 1 .23 (s, 8H), 0.83 (t, 3H); MS (ESI+): m/z 639.4 [M+H]+; HPLC Purity: 98.50 %.
Example 154:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-hydroxyoxetan-3-yl)-2-methyl benzoic acid
The compound of example 20 (2 g, 5.84 mmol) was taken in dry THF under argon atmosphere and to the reaction mixture, was added N-butyl lithium (14.61 mL, 23.38 mmol) at -78 °C under argon atmosphere. The reaction mixture was
stirred for 30 min. To the reaction mixture was added, oxetan-3-one (0.547 g, 7.60 mmol) and the reaction mixture was gradually brought to RT. The resulting mixture was diluted with water and extracted with ethyl acetate after acidifing with citric acid. The crude material was purified through column chromatography.
Yield: 250 mg (12.45 %); 1 H NMR (CDCI3, 300 MHz): δ 12.83 (bs, 1 H),
7.74 (s, 1 H), 7.53 (s, 1 H), 6.42 (s, 1 H), 4.78 (d, 2H, J=6.3Hz), 4.64 (d, 2H, J=6.3 Hz), 3.84 (m, 2H), 3.28 (m, 2H), 3.06 (m, 3H), 2.43 (s, 3H), 1 .65 (m, 4H), 0.87 (t, 3H); MS (ESI+): m/z 336 (M+H)+, HPLC Purity: 97.64 %. Example 155:
Methyl 3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-methoxyoxetan-3-yl)-2- methylbenzoate
The compound of example 154 (200 mg, 0.596 mmol) was taken in DMF.
To the reaction mixture, NaH (47.7 mg, 1 .193 mmol) was added and the reaction mixture was stirred at RT followed by the addition of Mel (93 μΙ_, 1 .491 mmol).
The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was concentrated to obtain a crude compound, which was purified through column chromatography.
Yield: 150 mg (68.2 %); 1H NMR (CDCI3, 300 MHz): δ 7.52 (s, 1 H), 7.36 (s, 1 H), 4.78 (m, 4H), 3.83 (s, 3H), 3.80 (m, 2H), 3.31 (m, 1 H), 3.25 (m, 2H),
3.08 (m, 2H), 3.01 (s, 3H), 2.42 (s, 3H), 1 .66 (m, 4H), 0.81 (t, 3H, J=6.9 Hz) ; MS
(ESI+): m/z 364 (M+H)+; HPLC Purity: 98.51 %.
Example 156:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-methoxyoxetan-3-yl)-2-methyl benzoic acid
The compound of example 155 (150 mg, 0.413 mmol) and NaOH (24.76 mg, 0.619 mmol) were taken in a mixture of THF, methanol and water and heated at 60 °C for 16 h. The solvent was distilled off and pH was adjusted to 3. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was concentrated and the crude compound obtained was purified through precipitation.
Yield: 80 mg (54.5 %); 1 H NMR (CDCI3, 300 MHz): δ 12.92 (bs, 1 H), 7.52 (s, 1 H), 7.32 (s, 1 H), 4.78 (m, 4H), 3.84 (d , 2H, J=7.2Hz), 3.31 (m, 2H), 3.25 (m, 3H), 3.02 (s, 3H), 2.44 (s, 3H), 1 .65 (m, 4H), 0.87 (t, 3H); MS (ESI+): m/z 350 (M+H)+; HPLC Purity: 98.19 %.
Example 157:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-hydroxyoxetan-3-yl)-2-methyl- N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide
The compound of example 154 (50 mg, 0.149 mmol), 2-(3H- [1 ,2,3]triazolo[4,5-b]pyridin-3-yl)-1 , 1 ,3,3-tetramethylisouronium,
hexafluorophosphate(V) salt (85 mg, 0.224 mmol), DIPEA (39.1 μΙ_, 0.224 mmol) and the compound of example 2 (51 .1 mg, 0.224 mmol) were taken in DMF and the compound was stirred at 60 °C for 16 h. The reaction mixture was cooled to RT, diluted with water and extracted with ethyl acetate. The organic layer was concentrated to obtain a crude compound, which was purified using column chromatography (silica gel, methanol in chloroform).
Yield: 20 mg (26.0 %); 1 H NMR (CDCI3, 300 MHz): δ 1 1 .50 (bs, 1 H), 8.07 (bs, 1 H), 7.36 (s, 1 H), 7.20 (s, 1 H), 6.34 (s, 1 H), 4.75 (d, 2H, J=6.3Hz) 4.64 (d, 2H, J=6.3Hz), 4.31 (m, 2H), 3.84 (m, 2H), 3.33 (m, 2H), 3.04 (m, 3H), 2.73 (m, 2H), 2.23 (m, 2H), 2.21 (s, 3H), 2.10 (s, 3H), 1 .64 (m, 5H), 1 .48 (m, 3H), 0.81 (t, 3H); MS (ESI+): m/z 510 (M+H)+; HPLC Purity: 98.64 %.
Example 158:
Ethyl 2-(3-(3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4'-methyl-5'-(((1 -methyl- 3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 ,1 '- biphenyl]-4-yl)oxetan-3-yl)acetate
The compound of example 158 is prepared by a method analogous to the compound of example 157 by using appropriate reagents in a mixture of dioxane and water and heating at 100 °C for 16 h. The reaction mixture was concentrated and purified using DCM and methanol.
Yield: 33 %; 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .50 (s, 1 H), 8.17 (s, 1 H), 7.60 (d, 2H), 7.40 (s, 1 H), 7.33 (d, 2H), 7.22 (s, 1 H), 4.81 (s, 4H), 4.31 (d, 2H), 3.96 - 3.81 (m, 4H), 3.33 - 3.21 (m, 2H), 3.16 (s, 2H), 3.10 - 2.96 (m, 3H), 2.80 -
2.70 (m, 2H), 2.37 (m, 2H), 2.10 (s, 3H), 1 .64 - 1 .50 (m, 8H), 1 .23 (t, 3H), 0.86 (m, 3H); MS (ESI+) m/z 656.3 (M+H)+; HPLC: 96.45 %.
Example 159:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-methoxyoxetan-3-yl)-2-methyl- N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydro isoquinolin-4-yl)methyl)benzamide
The compound of example 156 (50 mg, 0.143 mmol),the compound of example 2 (49.1 mg, 0.215 mmol), N-ethyl-N-isopropylpropan-2-amine (27.7 mg, 0.215 mmol) and 2-(3H-[1 ,2,3]triazolo[4,5-b]pyridin-3-yl)-1 ,1 ,3,3- tetramethylisouronium, hexafluorophosphate(V) salt (82 mg, 0.215 mmol) were taken in DMF and the reaction mixture was stirred at 60 °C for 16 h. The reaction mixture was cooled to RT, water was added and the resulting mixture was extracted with ethyl acetate. The organic extract was dried over anhydrous sodium sulphate, filtered and concentrated to yield a crude mixture, which was purified using column chromatography (silica gel, methanol in chloroform) to yield the title compound.
Yield: 17 mg (22.25 %); 1 H NMR (CDCI3, 300 MHz): δ 1 1 .45 (bs, 1 H), 8.12 (s, 1 H), 7.14 (s, 1 H), 7.01 (s, 1 H), 4.91 (m, 4H), 4.31 (d, 2H, J=4.8Hz), 3.81 (m, 2H), 3.28 (m, 3H), 3.05 (m, 2H), 3.00 (s, 3H), 2.73 (m, 2H), 2.50 (m, 2H), 2.22 (s, 3H), 2.10 (s, 3H), 1 .64 (m, 4H), 1 .60 (m, 4H), 0.87 (t, 3H) ; MS (ESI+): m/z 524 (M+H)+; HPLC Purity: 98.07 %.
Example 160:
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-hydroxyoxetan-3-yl)-N-((7- isopropyl-1-methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4- yl)methyl)-2-methylbenzamide
The compound of example 154 (50 mg, 0.149 mmol), 2-(3H- [1 ,2,3]triazolo[4,5-b]pyridin-3-yl)-1 , 1 ,3,3-tetramethylisouronium
hexafluorophosphate (V) salt (85 mg, 0.224 mmol), DIPEA (39.1 μΙ, 0.224 mmol) and the compound of example 88 (60.8 mg, 0.224 mmol) were taken in DMF and the compound was stirred at 60 °C for 16 h. The reaction mixture was cooled to RT, water was added and the reaction mixture was extracted with ethyl acetate. The organic extract was dried over anhydrous sodium sulphate, filtered and
concentrated to yield a crude mixture, which was purified using column chromatography (silica gel, methanol in chloroform) to yield the title compound.
Yield: 10 mg (1 1 .91 %); 1 H NMR (CDCI3, 300 MHz): δ 1 1 .92 (bs, 1 H), 8.31 (bs, 1 H), 7.37 (s, 1 H), 7.20 (s, 1 H), 6.35 (s, 1 H), 4.76 (d, 2H, J=6.6 Hz) 4.64 (d, 2H, J=6.3Hz), 4.30 (m, 4H), 3.84 (m, 2H), 3.64 (m, 2H), 3.27 (m, 3H), 3.19 (m, 3H), 2.20 (s, 6H), 1 .60 (m, 6H), 1 .34 (d, 6H, J=6Hz), 0.87 (t, 3H) ; MS (ESI+): m/z 553 (M+H)+; HPLC Purity: 98.12 %.
Example 161 :
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4 ormyl-4-methyl-N-((1-methyl-3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3- carboxamide
To the compound of example 135 (3.00 g, 5.81 mmol) was added (4- formylphenyl)boronic acid (1 .045 g, 6.97 mmol) and a mixture of 1 ,4-dioxane (25 mL) and water (5 mL). Argon was bubbled through the reaction mixture for 10 min followed by the addition of sodium carbonate (2.155 g, 20.33 mmol) and PdCI2(dppf)-CH2Cl2 adduct (0.237 g, 0.290 mmol). The reaction mixture was stirred at 90 °C for 16 h and evaporated to dryness. DCM was added to the reaction mixture and washed with water. The organic layer was dried over anhydrous sodium sulphate and evaporated. The compound was purified by silica gel column chromatography to yield the title compound.
Yield: 2.40 g (76 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .07 (brs, 2H), 8.85 (s, 1 H), 7.95 (d, 1 H), 7.69 (d, 1 H), 7.38 - 7.29 (m, 5H), 4.58 (d, 2H), 3.96 (d, 2H), 3.53 - 3.46 (m, 2H), 3.13 - 3.1 1 (m, 2H), 3.08 - 2.9 (m, 2H), 2.41 - 2.36 (m, 4H), 2.13 (s, 2H), 1 .71 -1 .60 (m, 2H), 1 .25 (s, 7H), 0.93 - 0.90 (m, 2H), 0.86 (t, 3H); MS (ESI+): 583.6 [M+H]+; HPLC: 97.01 %.
Example 162:
Diethyl (3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-5-(((1-methyl-3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)phenyl) phosphonate
To the compound of example 135 (100 g, 0.194 mmol) in a sealed tube, was added acetonitrile (10 mL), diethyl phosphite (0.030 mL, 0.232 mmol) and
DIPEA (0.051 mL, 0.290 mmol). Argon was purged for 10 min and DPPF (1 ,1 '- Bis(diphenylphosphino)ferrocene)palladium(l l) dichloride) (1 .181 mg, 2.130 μηιοΙ) and Pd(OAc)2 (Palladium(ll) acetate) (0.435 mg, 1 .936 μιηοΙ) were added and the reaction mixture was stirred at 90 °C. The reaction mixture was adsorbed on silica gel and purified by column chromatography (silica gel, 6 % MeOH in DCM) to yield the title compound.
Yield: 70 mg (63.0 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .52 (s,1 H), 8.32 (s, 1 H), 7.39 (d, J=14.7Hz, 1 H), 7.24 (d,J=17.1 Hz,1 H), 4.30 (d, J=4.5Hz, 2H), 4.00 (m, 4H), 3.84 (d, J=10.2Hz, 2H), 3.24 (m, 2H), 3.06 (d, J=7.2Hz, 2H), 2.72 (s, 2H), 2.38 (bs, 2H), 2.25 (s, 3H), 2.10 (s,3H), 1 .64 (m,7H), 1 .27 (t, 6H), 0.87 (t, 3H); MS (ESI+): 574.3 [M+H]"; HPLC Purity: 99.05 %.
Example 163:
5-(Dimethylphosphoryl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl- N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide
To the compound of example 135 (0.09 g, 0.174 mmol) in a sealed tube, was added acetonitrile (10 mL), dimethylphosphine oxide (0.027 g, 0.349 mmol) and DIPEA (0.046 ml, 0.261 mmol). Argon was purged for 10 min and DPPF (1 .063 mg, 1 .917 μηιοΙ) and Pd(OAc)2 (0.391 mg, 1 .743 mol) (1 .181 mg, 2.130 mol) were added and the reaction mixture was stirred at 90 °C. The resulting mixture was adsorbed on silica gel and purified by column chromatography (silica gel, 6 % MeOH in DCM) to yield the title compound.
Yield: 60 mg (67.0 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .52 (s,1 H), 8.17 (s, 1 H), 7.51 (d, J=12.0Hz, 1 H), 7.31 (d,J=1 1 .1 Hz,1 H), 4.31 (d, J=4.5Hz, 2H), 3.85 (d, J=10.8Hz, 2H), 3.27 (t, J=10.2Hz, 2H), 2.98 (d, J=10.5Hz, 3H), 2.73 (bs,2H), 2.37 (s, 2H), 2.24 (s, 3H), 2.10 (s, 3H), 1 .63 (m,12H), 1 .24 (m, 3H), 0.87 (t, J=6.6Hz, 3H); MS (ESI+): 514.3 [M+H]+; HPLC Purity: 84.29 %.
Example 164:
Diethyl (((4-chlorophenyl)amino)(3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)- 4'-methyl-5'-(((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl) carbamoyl )-[1 ,1 '-biphenyl]-4-yl)methyl)phosphonate
To the compound of example 135 (0.10 g, 0.185 mmol) was added 4- chloroaniline (0.047 g, 0.369 mmol), tin (II) chloride (3.50 mg, 0.018 mmol) followed by addition of DCM to make a slurry. Diethyl phosphite (0.048 ml_, 0.369 mmol) was added and the reaction mixture was heated at 40 °C for 4 h. DCM was added to the reaction mixture, which was washed with water. The organic layer was dried over anhydrous sodium sulphate and evaporated. The compound was purified by silica gel column chromatography to yield the title compound.
Yield: 0.120 g (82 %); 1 H NMR (DMSO-d6, 300 MHz): δ 1 1 .52 (s,1 H), 8.15 (s, 1 H), 7.57 (m, 4H), 7.40 (s,1 H), 7.21 (s,1 H), 7.03 (d, J=9.0Hz, 2H), 6.82 (d, J=8.7Hz, 2H), 6.69 (d, J=8.7Hz, 1 H), 5.1 (d, J=9.9Hz, 1 H), 4.31 (d, J=4.2Hz, 2H), 4.10 (m, 2H), 4.05 (m,1 H), 3.82 (d, J=8.20Hz, 3H), 3.34 (m,2H), 3.20 (m,3H), 2.74 (bs,2H), 2.37 (bs, 2H), 2.23 (s,3H), 2.10 (s, 3H), 1 .52 (bs, 6H), 1 .49 (d,J=13.8Hz, 2H), 1 .23 (m, 3H), 1.1 1 (t, J=7.20Hz, 3H), 0.83 (t, J=6.9Hz, 3H); MS (ESI+): 789.4 [M+H]"; HPLC Purity: 99.54 %.
Example 165:
Diethyl ((3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4'-methyl-5'-(((1 -methyl-3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 ,1 '-biphenyl]- 4-yl)(morpholino)methyl)phosphonate
To the compound of example 161 (0.1 g, 0.185 mmol), was added morpholine (0.032 g, 0.369 mmol), tin (II) chloride (3.5 mg, 0.018 mmol) followed by addition of DCM to make a slurry and diethyl phosphonate (0.051 g, 0.369 mmol). The reaction mixture was heated at 45 °C for 4 h, DCM was added to the reaction mixture, which was washed with water. The organic layer was dried over anhydrous sodium sulphate and evaporated. The compound was purified by silica gel column chromatography to yield the title compound.
1 H NMR (DMSO-de, 300 MHz): δ 1 1 .52 (s,1 H), 8.32 (s,1 H), 7.67 (d,J=8.1 Hz, 2H), 7.54 (d, J=7.8Hz, 2H), 7.45 (s,1 H), 7.25 (s,1 H), 4.32 (d, J=4.5Hz, 2H), 4.19(m,3H), 3.82 (m,4H), 3.29 (d, J=12.0Hz, 2H), 3.1 1 (m, 3H), 2.75 (m,4H), 2.37 (m, 4H), 2.25 (s,3H), 2.10 (s, 3H), 1 .64 (m, 8H), 1.32 (t,J=6.9Hz, 3H), 1 .02 (t, J=6.9Hz, 3H), 0.86 (t, J=6.6Hz, 3H); MS (ESI+): 749.4 [M+H]+; HPLC Purity: 98.60 %.
Example 166:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-((4-methyl-4-oxido-1 ,4- azaphosphinan-1-yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide
To a stirred solution of the compound of example 135 (0.2 g, 0.387 mmol) in dioxane/water mixture (10 mL/2 mL) was added 4-methyl-1 -(4-(4, 4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzyl)-1 ,4-azaphosphinane 4-oxide (0.162 g, 0.465 mmol) followed by addition of Na2C03 (0.164 g, 1 .549 mmol). The solution was purged with argon for 15 min and then PdCI2(dppf)-CH2Cl2 adduct (0.032 g, 0.039 mmol) was added and the solution was again purged with argon for a further 10 min. The reaction mixture was heated at 100 °C for 4 h. After completion of the reaction, the resulting mixture was diluted with water and extracted with 10 % MeOH/DCM. The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated. The crude mixture was purified by column chromatography (silica gel, methanol in DCM) to yield the title compound.
1 H NMR (DMSO-de, 300 MHz): δ 1 1 .50 (s, 1 H), 8.16 (s, 1 H), 7.57 (d, 2H), 7.38 (d, 2H), 7.21 (s, 1 H), 5.76 (s, 1 H), 4.59 (d, 2H), 3.99 - 3.95 (d, 2H), 3.69 (s, 2H), 3.33 - 3.26 (m, 2H), 3.24 - 2.95 (m, 3H), 2.82 - 2.71 (m, 5H), 2.56 - 2.50 (m, 2H), 2.25 (s, 3H), 2.18 (s, 3H), 1 .90 - 1 .80 ( m, 4H), 1 .71 - 1 .66 (m, 6H), 1 .5 (s, 2H), 1.44 (s, 2H), 1 .05 - 1 .01 (m, 2H), 0.91 (t, 3H); MS (ESI+): m/z 659.3 [M+H]+; HPLC Purity: 98.8 %.
Biological Assays
Example 167:
In vitro EZH2 Assay
Representative compounds of formula 1 of the present invention (referred to as test compounds) were tested for their EZH2 inhibitory activity using the assays and the methods described below:
EZH2 biochemical activity assay was performed to measure the rate of trimethylation of Histone H3 at lysine 27. Serial dilutions of the test compounds ranging from 0.1 nM to 10 μΜ were pre-incubated for 10 minutes with 50 ng EZH2
protein complex and enzymatic reactions were initiated by the addition of 100 nM Histone H3 peptide (ATKAARKSAPATGGVKKPHRYRP-GG-K) substrate and 10 μΜ SAM S-(5'-Adenosyl)-L-methionine chloride. The assay buffer used in every reaction contained 0.5 % DMSO in assay buffer containing 50 mM Tris-HCI pH 9.0, 50 mM NaCI, 1 mM dithiothreitol (DTT), 0.01 % Tween-20 and 0.01 % bovine serum albumin (BSA). Detection mixture containing 500 ng of anti trimethyl Histone H3 Lysine 27 Acceptor Bead conjugate (Perkin Elmer) and 500 ng of Straptavidin donor beads (Perkin Elmer) were added to each reaction mixture and plate was incubated for one hour at room temperature, 25-35 °C. Alpha counts were detected using Multilable Plate Reader. The IC50 values for the test compounds were determined by a four-parameter sigmoidal curve fit (Sigma plot or Graph pad).
(Reference: Dillon SC, et al.; Genome Biology, 2005, 6, 227).
Table 1 : Symbols used to indicate IC50 range class

Table 2: IC50 values for the representative compounds of the present invention for their EZH2 inhibiting activity

150 +
151 +
152 +
165 +
166 +
Conclusion: The test compounds of the present invention were found to inhibit EZH2 activity in vitro.
All publications and patent applications in this specification are indicative of the level of ordinary skill in the art to which this invention pertains.
The invention has been described with reference to various specific and preferred embodiments and techniques. However, it should be understood that many variations and modifications may be made while remaining within the spirit and scope of the invention.
Claims
1 . A compound of formula 1 ,

Formula 1
wherein,
Ri and R2 are independently selected from hydrogen, halogen, hydroxy, cyano, (CrCe)-alkyl, halo(C C8)-alkyl, (C2-C8)-alkenyl, (C C8)-alkoxy, C(0)Ra,C(0)ORa, C(0)NRaRb, OC(0)NRaRb, NRaRb, NRaC(0)Rb, S(0)q(C C8)-alkyl and S(0)qNRaRb; or
Ri and R2 together can form a 3 to 10 membered monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; wherein the ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (CrC8)-alkoxy, halo(CrC8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)0-2, (C6-Ci4)-aryl-(Rc)0-2,C(O)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, S(0)q(C C8)-alkyl and S(0)qNRaRb; A is:

wherein X and X2 areindependently selected from CR6 and N;
R3, R4 and R6 are independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (C C8)-alkyl, halo(C C8)-alkyl, halo(C C8)-alkyl-OH, (Ci-C8)-alkoxy, halo(C C8)-alkoxy, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)- cycloalkenyl, (C6-Ci4)-aryl, (C6-Ci4)ar-(CrC8)-alkyl, heterocyclyl, heteroaryl, C(0)Ra,C(0)ORa, C(0)NRaRb, S(0)q(C C8)-alkyl, S(0)qNRaRb, NRaRb, NRaC(0)Rb and NRaS(0)q(C C8)-alkyl;
R4a is hydrogen, hydroxy, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (CrC8)-alkoxy, halo(CrC8)-alkoxy, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl,(C5-C8)-cycloalkenyl, (C6- Ci4)-aryl, (C6-Ci4)ar-(CrC8)-alkyl, heterocyclyl, heteroaryl, C(0)Ra, C(0)NRaRb, S(0)q(Ci-C8)-alkyl or S(0)qNRaRb; or
R3 and R4 or R3 and R4a on A can join together to form a 3 to 10 membered saturated or unsaturated monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; wherein said ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C8)-alkyl, halo(C C8)-alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3-C12)- cycloalkyl-(Rc)o-2, 0-(C3-Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, 0-(C6-d4)- aryl-(Rc)o-2, heterocyclyl-(Rc)0-2,O-heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, S(0)q(C C8)-alkyl and S(0)qNRaRb; R5 is hydrogen, halogen, hydroxy, cyano, B(OH)2, P(0)(ORa)(ORb), P(0)RaRb, (Ci-C8)-alkyl, halo(C C8)-alkyl, halo(C C8)-alkyl-OH, (C C8)-alkoxy, halo(C C8)- alkoxy, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)-cycloalkenyl, (C6-Ci4)-aryl, (C6-Ci4)ar-(Ci -C8)-alkyl, (C6-Ci4)-aryl-P(0)RaRb, (C6-Ci4)-aryl-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-P(0)RaRb, (C6-Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)- aryl-heterocyclyl-Rc, (C6-Ci4)-aryl-C(Rc)2-heterocyclyl, heterocyclyl, heteroaryl, C(0)Ra, C(0)ORa, C(0)NRaRb, C(0)NRaNRaRb, S(0)q(C C8)alkyl, S(0)qNRaRb, NRaS(0)q(Ci -C8)-alkyl, NRaS(0)q(C3-Ci2)-cycloalkyl, NRaS(0)q(C6-Ci4)-aryl, NRaS(0)qheterocyclyl, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, NRaC(0)ORb, NRaS(0)qNRaRb, NRaNRaRb, NRaNRaC(0)Rb, NRaNRaC(0)NRaRb or NRaNRaC(0)ORa;
L is --C(0)NR7--, --NR7C(0)--, -NR7C(0)NR7-, --NR7S(0)r--, --S(0)rNR7-, - CH2NR7--, -NR7CH2-, --CH(halo)NR7-, --NR7CH(halo)--, --C(halo)2NR7-, -- NR7C(halo)2-, --CH(halo-(Ci-C8)-alkyl)NR7--, --NR7CH(halo-(Ci-C8)-alkyl)-, - , -NR7C(halo-(C C8)-alkyl)2--, w ne (--) indicates the point of
 attachment of L to the group respectively; or
L is a saturated or unsaturated 5 or 6 membered monocyclic ring optionally containing one to three heteroatoms selected from the group consisting of O, N, S and P;
attachment of L to the group respectively; or
L is a saturated or unsaturated 5 or 6 membered monocyclic ring optionally containing one to three heteroatoms selected from the group consisting of O, N, S and P;
R7 is hydrogen, cyano, (C C8)-alkyl, (C2-C8)-alkenyl, halo(C C8)alkyl, C(0)Ra, C(0)ORa, C(0)NRaRb, S(0)q(Ci -C8)-alkyl or S(0)qNRaRb;
Q is:

B is a 3 to 10 membered monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; wherein said ring can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (C C8)-alkyl, halo(C C8)-alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3- Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, heterocyclyl-(Rc)o-2, heteroaryl-(Rc)0-2, C(0)Ra, C(0)ORa, C(0)NRaRb,NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(C C8)- alkyl and S(0)qNRaRb;
R8 and R9 are independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (CrC8)-alkyl, halo(CrC8)alkyl, (CrC8)-alkoxy, halo(C C8)-alkoxy, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)-cycloalkenyl, (C6-Ci4)-aryl,
(C6-Ci4)ar-(Ci-Ce)-alkyl, C(0)Ra, C(0)ORa, C(0)NRaRb, S(0)q(C C8)-alkyl, S(0)qNRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb and NRaC(0)ORb;
Ra and Rb are independently selected from the group consisting of hydrogen, (C C8)-alkyl, (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C5-C8)-cycloalkenyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; or
Ra and Rb together with the nitrogen atom can form a 3 to 10 membered ring, optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P;
Rc at each occurrence is independently selected from the group consisting of hydrogen, halogen, hydroxy, cyano, (CrC8)-alkyl, halo(CrC8)alkyl, (C3-Ci2)- cycloalkyl, (C6-Ci4)-aryl, (C6-Ci4)-ar-(Ci-C8)-alkyl, heteroaryl, heterocyclyl, CORa, C(0)ORa, C(0)NRaRb, C(0)NRaNRaRb, S(0)q(C C8)-alkyl, S(0)qNRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, NRaC(0)ORb and NRaS(0)qNRaRb;
p and n are integersindependently selected from 0, 1 , 2 and 3;
q is 1 or 2; or
r is 1 or 2;
wherein each of the (CrC8)-alkyl or (CrC8)-alkoxy can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkoxy, halo(CrC8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)o-2, O-(C3-Ci2)-cycloalkyl-(Rc)0-2, (C6-Ci4)-aryl-(Rc)0-2, O- (C6-Ci4)-aryl-(Rc)o-2, heterocyclyl-(Rc)o-2, 0-heterocyclyl-(Rc)o-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(Ci-C8)-alkyl and S(0)qNRaRb;
each of the (C2-C8)-alkenyl, (C3-Ci2)-cycloalkyl, (C3-C8)-cycloalkenyl and (C6-Ci4)- aryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(Ci-C8)-alkyl, (Ci-C8)-alkoxy, halo(CrC8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)0-2, 0-(C3-Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, O-(C6-Ci4)-aryl-(Rc)0-2, heterocyclyl-(Rc)o-2, O-heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(C C8)-alkyl and S(0)qNRaRb;
the heterocyclyl is a 3 to 10 membered saturated or partially unsaturated, monocyclic or bicyclic ring system containing one to four heteroatoms
independently selected from the group consisting of O, N, S and P; wherein the heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (C C8)-alkyl, halo(C C8)-alkyl, (C C8)-alkoxy, halo(C C8)-alkoxy, (C3- Ci2)-cycloalkyl-(Rc)o-2, 0-(C3-Ci2)-cycloalkyl-(Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, 0-(C6- Ci4)-aryl-(Rc)o-2, heterocyclyl-(Rc)0-2, O-heterocyclyl-(Rc)0-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(Ci-Ce)-alkyl and S(0)qNRaRb; and
the heteroaryl is a 5 to 10 membered monocyclic or bicyclic aromatic ring system containing one to four heteroatoms independently selected from the group consisting of O, N, S and P, wherein the heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting of halogen, hydroxy, oxo, cyano, (CrC8)-alkyl, halo(CrC8)-alkyl, (C C8)-alkoxy, halo(CrC8)-alkoxy, (C3-Ci2)-cycloalkyl-(Rc)0-2, 0-(C3-Ci2)-cycloalkyl- (Rc)o-2, (C6-Ci4)-aryl-(Rc)0-2, O-(C6-Ci4)-aryl-(Rc)0-2, heterocyclyl-(Rc)0-2, O- heterocyclyl-(Rc)o-2, heteroaryl-(Rc)0-2, C(0)Ra, OC(0)Ra, C(0)ORa, C(0)NRaRb, NRaRb, NRaC(0)Rb, NRaC(0)NRaRb, S(0)q(Ci-C8)-alkyl and S(0)qNRaRb;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
2. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
3. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R3 and R4 join together to form a 5 to 7 memberedsaturated or unsaturated monocyclic ring optionally containing one to three heteroatoms independently selected from the group consisting of O, N, S and P; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
4. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are CR6; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
5. The compound according to claim 1 ,
wherein,
L is -C(0)NR7-,-NR7C(0)-,-NR7C(0)NR7-, -NR7S(0)r- or -S(0)rNR7- ;wherein the dotted line (-) indicates the point of attachment of L to the group

or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
6. The compound according to claim 1 ,
wherein,
L is -CH2NR7~,-NR7CH2-,~CH(halo-(Ci-Ce)-alkyl)NR7-, -NR7CH(halo-(C C8)- alkyl)-, -C(halo-(CrC8)-alkyl)2NR7-, -NR7C(halo-(CrC8)-alkyl)2--, -- CH(halo)NR7-, --NR7CH(halo)--, --C(halo)2NR7-- or --NR7C(halo)2--; wherein the dotted line (--) indicates the point of attachment of L to the group

or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
7. The compound according to claim 1 ,
wherein,
L is
 ~ ; wherein the dotted line (-) indicates the point of attachment of L to the group
~ ; wherein the dotted line (-) indicates the point of attachment of L to the group
 respectively; where A, Ri, R2, R7, Q, n and p are as defined in the first aspect;
respectively; where A, Ri, R2, R7, Q, n and p are as defined in the first aspect;
oran isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
8. The compound according to claim 1 ,
wherein,
Q is:

or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
9. The compound according to claim 1 ,
wherein,
Q is:

or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
10. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb,(C6-Ci4)-aryl- P(0)(ORa)(ORb),(C6-Ci4)-aryl-C(Rc)2-P(0)RaRb,(C6-Ci4)-aryl-heterocyclyl-Rc, (C6- Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb),(C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)-aryl, (C6-Ci4)ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
1 1 . The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CP16 and N;
L is -C(0)NR7- or -NR7C( --; wherein the dotted line (--) indicates the point of attachment of L to the group
 respectively;
respectively;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
12. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-C14)-aryl-P(0)RaRb,(C6-C14)-aryl- P(0)(ORa)(ORb),(C6-Ci4)-aryl-C(Rc)2-P(0)RaRb,(C6-Ci4)-aryl-heterocyclyl-Rc, (C6- Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb),(C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)-aryl, (C6-Ci4)-ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl;
L is -C(0)NR7-, -NR7C(0)~, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~ ;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-C14)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C8)-alkyl,halo(Ci-C8)- alkyl, (CrC8)-alkoxy, (C3-Ci 2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
13. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb,(C6-Ci4)-aryl-
P(0)(ORa)(ORb),(C6-Ci4)-aryl-C(Rc)2-P(0)RaRb,(C6-Ci4)-aryl-heterocyclyl-Rc, (C6-
Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb)or(C6-Ci4)-aryl-C(Rc)2-heterocyclyl;
L is ~C(0)NR7 ~, -NR7C(0)~, -NR7C(0)NR7-, -NR7S(0)r- or ~S(0)rNR7 ~
;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-C14)-aryl and heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C8)-alkyl,halo(Ci-C8)-alkyl, (CrC8)- alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
14. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of(C3-Ci2)- cycloalkyl, (C5-C8)-cycloalkenyl,(C6-Ci4)-aryl, heterocyclyl and heteroaryl;
R5 is (C6-Ci4)-aryl, (C6-Ci4)ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl;
L is -C(0)NR7-, -NR7C(0)-, -NR7C(0)NR7-, -NR7S(0)r- or -S(0)rNR7-
;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-C14)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C8)-alkyl,halo(CrC8)- alkyl, (CrC8)-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
15. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb,(C6-Ci4)-aryl- P(0)(ORa)(ORb),(C6-Ci4)-aryl-C(Rc)2-P(0)RaRb,(C6-Ci4)-aryl-heterocyclyl-Rc, (C6- Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb), (C6-Ci4)-aryl-C(Rc)2-heterocyclyl, (C6-Ci4)-aryl, (C6-Ci4)ar-(Ci-C8)-alkyl, heterocyclyl or heteroaryl;
L is -C(0)NR7-, -NR7C(0)-, ~NR7C(0)NR7 ~, -NR7S(0)r- or ~S(0)rNR7 ~ ;wherein the dotted line (--) indicates the point of attachment of L to the group
A- respectively;
n is 0;
Q is:

wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C8)-alkyl,halo(CrC8)- alkyl, (CrC8)-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug, a polymorph, N-oxide or S-oxide thereof.
16. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
R5 is P(0)(ORa)(ORb), P(0)RaRb, (C6-Ci4)-aryl-P(0)RaRb,(C6-Ci4)-aryl-
P(0)(ORa)(ORb),(C6-Ci4)-aryl-C(Rc)2-P(0)RaRb,(C6-Ci4)-aryl-heterocyclyl-Rc, (C6-
Ci4)-aryl-C(Rc)2-P(0)(ORa)(ORb)or(C6-Ci4)-aryl-C(Rc)2-heterocyclyl;
L is --C(0)NR7--, --NR7C(0)--, -NR7C(0)NR7-, -NR7S(0)r- or -S(0)rNR7-
;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-Ci4)-aryl and heterocyclyl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C8)-alkyl,halo(Ci-C8)-alkyl, (Ci -Cs)- alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl;
or an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt,a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
17. The compound according to claim 1 ,
wherein,
A is:

Xi and X2 are independently selected from CR6 and N;
R4 is NRaRb or NRaC(0)Rb;
Ra and Rb are independently selected from the group consisting of (C3-C12)- cycloalkyl, (C5-C8)-cycloalkenyl,(C6-Ci4)-aryl, heterocyclyl and heteroaryl;
R5 IS (C6-Ci4)-aryl, (C6-Ci4)ar-(CrC8)-alkyl, heterocyclyl or heteroaryl;
L is -C(0)NR7-, -NR7C(0)-, -NR7C(0)NR7-, -NR7S(0)r- or -S(0)rNR7- ;wherein the dotted line (--) indicates the point of attachment of L to the group

n is 0;
Q is:

wherein each of the (C6-Ci4)-aryl, heterocyclyl and heteroaryl can be unsubstituted or substituted with one or more groups independently selected from the group consisting ofhalogen, hydroxy, oxo, cyano, (Ci-C8)-alkyl,halo(CrC8)- alkyl, (CrC8)-alkoxy, (C3-Ci2)-cycloalkyl, (C6-Ci4)-aryl, heterocyclyl and heteroaryl; or
an isotopic form, a stereoisomer, a tautomer, a pharmaceutically acceptable salt, a solvate, a prodrug,a polymorph, N-oxide or S-oxide thereof.
18. The compound according to claim 1 , selected from the group consisting of:
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-fluoro-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8- hexahydroisoquinolin-4-yl)methyl)-4'-(1 -morpholinoethyl)-[1 , 1 '-biphenyl]-3- carboxamide;
3'-((Dimethylamino)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-fluoro-N- ((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3- carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((4-methyl-2-oxo-3, 5,6,7,8,9- hexahydro-2H-cyclohepta[c]pyridin-1 -yl)methyl)-4'-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo-3, 5,6,7,8,9- hexahydro-2H-cyclohepta[c]pyridin-1 -yl)methyl)-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo-3, 5,6,7,8,9- hexahydro-2H-cyclohepta[c]pyridin-1 -yl)methyl)-5-(6-morpholino pyridin-3- yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((4-methyl-2-oxo-3, 5,6,7,8,9- hexahydro-2H-cyclohepta[c]pyridin-1 -yl)methyl)-5-(6-(trifluoro methyl)pyridin-3- yl)benzamide;
3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)
((1 -methyl-3-oxo-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-4- yl)methyl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((8-methyl-6-oxo-3,4,6,7- tetrahydro-I H-pyranoIS^-clpyridin-S-y methy ^'^morpholinomethy -II J '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((8-methyl-6-oxo-3,4,6,7- tetrahydro-1 H-pyrano[3,4-c]pyridin-5-yl)methyl)-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((8-methyl-6-oxo-3,4,6,7- tetrahydro-1 H-pyrano[3,4-c]pyridin-5-yl)methyl)-5-(6-morpholinopyridin-3- yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-N-((3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4,-morpholino-N-((3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)-N-((3-oxo-2,3,5,6J,8-hexahydroisoquinolin-4-yl)methyl)benzamide;
N-((1 -ethoxy-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
N-((1 -ethoxy-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-3-
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
N-((1 , 7-dimethyl-3-oxo-2, 3,5,6, 7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5- (ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4'-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1 -methyl-3-oxo-2,3, 5,6,7,8- hexahydro-2,7-naphthyridin-4-yl)methyl)-4-methyl-4'-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1 -methyl-3-oxo-2,3, 5,6,7,8- hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-ethyl-1 -methyl-3-oxo-2,3, 5,6,7,8- exa ydro-2,7-nap t yridin-4-yl)met yl)-2-met yl-5-(6-morpholinopyridin-3- yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-7-propyl- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4,-(morpholinomethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-propyl-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6-morpholinopyridin-3- yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-propyl-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
N-((7-butyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5 (et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-[1 ,1 '- biphenyl]-3-carboxamide;
N-((7-butyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-3
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)benzamide;
N-((7-butyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-3
(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-morpholinopyridin-3- yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1 -methyl-3-oxo- 2,3,5,6,7,8- exa ydro-2,7-nap t yridin-4-yl)met yl)-4-met yl-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-(4-methyl piperazin-1 -yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isobutyl-1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-morpholino pyridin-3-yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo- 2,3,5,6,7,8- exa ydro-2,7-nap t yridin-4-yl)met yl)-4-met yl-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6-(4- methylpiperazin-1 -yl)pyridin-3-yl)benzamide;
3- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-N-((7-isopropyl-1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2-methyl-5-(6- morpholinopyridin-3-yl)benzamide;
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin
4- yl)met yl)-5-(et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide;
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin
4-yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4- methylpiperazin-1 -yl)pyridin-3-yl)benzamide;
N-((7-(cyclopropylmethyl)-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin
4- yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6- morpholinopyridin-3-yl)benzamide;
5- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-7-(oxetan-3-yl)-3 oxo-2,3,5,6,7,8-hexa ydro-2 ,7-nap t yridin-4-yl)met yl)-4'-(morpholinomet yl)- [1 ,1 '-biphenyl]-3-carboxamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-N-((3- oxo-1 -(trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)-N-((3-oxo-1 -(trifluoromethyl)-2,3,5,6,7,8-hexahydroisoquinolin-4- yl)methyl)benzamide;
3-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-2-met yl-5-(6-morpholinopyridin-3-yl)-N ((3-0X0-1 -(trifluoromethyl)-2, 3, 5,6,7,8-hexahydroisoquinolin-4- yl)methyl)benzamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-N-((3- oxo-1 -(trifluoromethyl)-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-4- yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(6-(4-methylpiperazin-1 - yl)pyridin-3-yl)-N-((3-oxo-1 -(trifluoromethyl)-3,5,6,7,8,9-hexahydro-2H- cyclohepta[c]pyridin-4-yl)methyl)benzamide;
5-(Et yl(tetra ydro-2H-pyran-4-yl)amino)-4-met yl-4'-(morpholinomet yl)-N-((3- oxo-1 -(trifluoromethyl)-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)- [1 ,1 '-biphenyl]-3-carboxamide;
4- Methyl-N-((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'- (morpholinomethyl)-5-(2-oxoazepan-1 -yl)-[1 ,1 '-biphenyl]-3-carboxamide;
Dimethyl (4'-methyl-3'-(((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4- yl)methyl)carbamoyl)-5'-(2-oxoazepan-1 -yl)-[1 ,1 ,-biphenyl]-4-yl)phosphonate; 3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(5-fluoro-6-morpholinopyridin-3-yl)-2- methyl-N-((1 -methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide;
5- (Ethyl(tetrahydro-2H-pyran-4-yl)amino)-3'-fluoro-4-methyl-N-((1 -rTiethyl-3-oxo- 3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-4'-morpholino-[1 ,1 '- biphenyl]-3-carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-3, 5,6,7- tetrahydro^H-cyclopentalclpyridin^-y methy ^'-^etrahydro^H-pyran^-y -II J '- biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(1 H-indol-4-yl)-2-methyl-N-((1 -methyl- 3-oxo-3,5, 6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl) benzamide;
3-(2-(Dimethylamino)pyrimidin-5-yl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-N- ((1 -methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(6-fluoro-5-methylpyridin-3-yl)-2- methyl-N-((1 -methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4- yl)methyl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(1 H-indol-5-yl)-2-methyl-N-((1 -methyl- 3-oxo-3,5, 6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl) benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-3, 5,6,7- tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-5-(quinolin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-3, 5,6,7- tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-5-(6-(morpholinomethyl) pyridin-3- yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-7- (trifluoromethyl)-2,3,5,6J,8-hexahydroisoquinolin-4-yl)methyl)-4'-(morpholino methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-
(trifluoromethyl)-2,3,5,67,8-hexahydroisoquinolin-4-yl)methyl)-5-(6-morpholino pyridin-3-yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-7-(2,2,2- trifluoroethyl)-2, 3,5,6, 7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-4'- (morpholinomethyl)-[1 ,1 '-biphenyl]-3-carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-(2,2,2- trifluoroethyl)-2, 3,5,6, 7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6-(4- methylpiperazin-1 -yl)pyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-7-(2,2,2- trifluoroethyl)-2, 3,5,6, 7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-5-(6- morpholinopyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(6-morpholinopyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(6-methylpyridin-3-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(6-(trifluoromethyl)pyridin-3-yl)benzamide; 5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-4'-morpholino-[1 ,1 '-biphenyl]-3-carboxamide; Dimethyl (3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4,-methyl-5'-(((1 -methyl-3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 , 1 '-biphenyl]-3- yl)phosphonate;
(S)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(1 -(piperidin-1 -yl)ethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
(R)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-
2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(1 -(piperidin-1 -yl)ethyl)-[1 ,1 '- biphenyl]-3-carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexa ydroisoquinolin-4-yl)met yl)-4'-(1 -(piperidin-1 -yl)et yl)-[1 ,1 '-biphenyl]-3- carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 exa ydroisoquinolin-4-yl)met yl)-4'-(2-morpholinoet yl)-[1 , 1 '-biphenyl]-3- carboxamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexa ydroisoquinolin-4-yl)met yl)-3'-(1 -(pyrrolidin-1 -yl)et yl)-[1 ,1 '-biphenyl]-3- carboxamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 exa ydroisoquinolin-4-yl)met yl)-5-(4-met yl-3-oxo-3,4-di ydro-2H- benzo[b][1 ,4]oxazin-6-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-5-(1 ,3,5-trimethyl-1 H-pyrazol-4-yl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 exa ydroisoquinolin-4-yl)met yl)-5-(6-(pyrrolidin-1 -yl)pyridin-3-yl)benzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8 hexahydroisoquinolin-4-yl)methyl)-4'-(2-(piperidin-1 -yl)propan-2-yl)-[1 ,1 '-biphenyl]-
3-carboxamide;
Ethyl 2-(3-(3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4,-methyl-5'-(((1 -methyl-3- oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 , 1 '-biphenyl]-4- yl)oxetan-3-yl)acetate;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-hydroxyoxetan-3-yl)-2-methyl-N-((1 methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-methoxyoxetan-3-yl)-2-methyl-N-
((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro isoquinolin-4-yl)methyl)benzamide;
3-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-5-(3-hydroxyoxetan-3-yl)-N-((7- isopropyl-1 -methyl-3-oxo-2,3,5,6,7,8-hexahydro-2,7-naphthyridin-4-yl)methyl)-2- methylbenzamide;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4' ormyl-4-methyl-N-((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide; Diethyl (3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-5-(((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)phenyl)phosphonate;
5-(Dimethylphosphoryl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)am methyl-3-oxo-2,3,5,67,8-hexahydroisoquinolin-4-yl)methyl)benzamide;
Diethyl (((4-chlorophenyl)amino)(3,-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4'- methyl-5'-(((1 -methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl) carbamoyl)-[1 ,1 '-biphenyl]-4-yl)methyl)phosphonate;
Diethyl ((3'-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4,-methyl-5'-(((1 -methyl-3-oxo- 2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)carbamoyl)-[1 ,1 '-biphenyl]-4- yl)(morpholino)methyl)phosphonate and;
5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-N-((1 -methyl-3-oxo-2, 3, 5,6,7,8- hexahydroisoquinolin-4-yl)methyl)-4'-((4-methyl-4-oxido-1 ,4-azaphosphinan-1 - yl)methyl)-[1 ,1 '-biphenyl]-3-carboxamide;
or a stereoisomer, a tautomer, or a pharmaceutically acceptable salt thereof.
19. A pharmaceutical composition comprising a therapeutically effective amount of the compound of formula 1 , or an isotope, a stereoisomer, a tautomer, or a pharmaceutically acceptable salt, or a solvate thereof; along with at least one pharmaceutically acceptable excipient.
20. A method for the treatment of a disease or disorder mediated by EZH2 (enhancer of zeste homolog 2) comprising administering to a subject in need thereof a therapeutically effective amount of the compound of formula 1 , or a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof.
21 . The method according to claim 20, wherein the disease or disorder is selected from cancer, pulmonary arterial hypertension, myelofibrosis, human immunodeficiency virus (HIV) disease, graft versus host diseases (GVHD), Weaver Syndrome, psoriasis vulgaris or liver fibrogenesis.
22. The method according to claim 21 , wherein the disease or disorder is cancer.
23. Use of the the compound of formula 1 , or an isotope, a stereoisomer, a tautomer, a pharmaceutically acceptable salt or a solvate thereof; for the
manufacture of a medicament for the treatment of a disease or disorder mediated by EZH2.
24. The use according to claim 23, wherein the disease or disorder is selected from cancer, pulmonary arterial hypertension, myelofibrosis, human immunodeficiency virus (HIV) disease, graft versus host diseases (GVHD), Weaver Syndrome, psoriasis vulgaris or liver fibrogenesis.
25. The use according to claim 24, wherein the disease or disorder is cancer
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461931029P | 2014-01-24 | 2014-01-24 | |
| US61/931,029 | 2014-01-24 | ||
| US201462052649P | 2014-09-19 | 2014-09-19 | |
| US62/052,649 | 2014-09-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015110999A1 true WO2015110999A1 (en) | 2015-07-30 |
Family
ID=53680902
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2015/050520 WO2015110999A1 (en) | 2014-01-24 | 2015-01-23 | Ezh2 inhibitors and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015110999A1 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9683003B2 (en) | 2014-07-01 | 2017-06-20 | Millennium Pharmaceuticals, Inc. | Heteroaryl compounds useful as inhibitors of SUMO activating enzyme |
| US9695154B2 (en) | 2013-07-02 | 2017-07-04 | Millennium Pharmaceuticals, Inc. | Heteroaryl inhibitors of sumo activating enzyme |
| WO2018045971A1 (en) | 2016-09-07 | 2018-03-15 | 上海海和药物研究开发有限公司 | Pyrido five-element aromatic ring compound, preparation method therefor and use thereof |
| US9951069B1 (en) | 2017-01-11 | 2018-04-24 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| WO2018086592A1 (en) * | 2016-11-11 | 2018-05-17 | 上海海雁医药科技有限公司 | 4,5,6-tri-substituted indazoles derivatives, preparation thereof, and use thereof in medicines |
| CN109069508A (en) * | 2016-04-22 | 2018-12-21 | 达纳-法伯癌症研究所股份有限公司 | EZH2 inhibitor and application thereof |
| US10421756B2 (en) | 2015-07-06 | 2019-09-24 | Rodin Therapeutics, Inc. | Heterobicyclic N-aminophenyl-amides as inhibitors of histone deacetylase |
| WO2020011607A1 (en) | 2018-07-09 | 2020-01-16 | Fondation Asile Des Aveugles | Inhibition of prc2 subunits to treat eye disorders |
| WO2020192652A1 (en) * | 2019-03-25 | 2020-10-01 | 上海华汇拓医药科技有限公司 | Preparation method for amide compounds and use thereof in medical field |
| US10919902B2 (en) | 2015-07-06 | 2021-02-16 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| US10975056B2 (en) | 2016-06-13 | 2021-04-13 | Glaxosmithkline Intellectual Property Development Limited | Substituted pyridines as inhibitors of DNMT1 |
| US11225475B2 (en) | 2017-08-07 | 2022-01-18 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| US11236082B2 (en) | 2014-11-06 | 2022-02-01 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| WO2023025856A1 (en) | 2021-08-25 | 2023-03-02 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of ezh2 inhibitors for the treatment of aortic valve stenosis |
| US12018015B2 (en) | 2021-06-18 | 2024-06-25 | Aligos Therapeutics, Inc. | Methods and compositions for targeting PD-L1 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5387747A (en) * | 1992-02-24 | 1995-02-07 | Laboratoires Upsa | Triazolopyrimidine derivatives which are angiotensin II receptor antagonists, their methods of preparation and pharmaceutical compositions in which they are present |
| US5389632A (en) * | 1992-02-24 | 1995-02-14 | Laboratoires Upsa | Pyrazolopyrimidine derivatives which are angiotensin II receptor antagonists |
| US20060189617A1 (en) * | 2005-02-18 | 2006-08-24 | Wyeth | Imidazo[4,5-b]pyridine antagonists of gonadotropin releasing hormone receptor |
| WO2007076055A2 (en) * | 2005-12-22 | 2007-07-05 | Entremed, Inc. | Compositions and methods comprising proteinase activated receptor antagonists |
| WO2007136592A2 (en) * | 2006-05-18 | 2007-11-29 | Amphora Discovery Corporation | 2-0x0-l,2-dihydr0quin0line derivatives, compositions, and uses thereof as antiproliferative agents |
| WO2010017355A2 (en) * | 2008-08-08 | 2010-02-11 | New York Blood Center | Small molecule inhibitors of retroviral assembly and maturation |
| WO2013075083A1 (en) * | 2011-11-18 | 2013-05-23 | Constellation Pharmaceuticals | Modulators of methyl modifying enzymes, compositions and uses thereof |
| WO2014049488A1 (en) * | 2012-09-28 | 2014-04-03 | Pfizer Inc. | Benzamide and heterobenzamide compounds |
-
2015
- 2015-01-23 WO PCT/IB2015/050520 patent/WO2015110999A1/en active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5387747A (en) * | 1992-02-24 | 1995-02-07 | Laboratoires Upsa | Triazolopyrimidine derivatives which are angiotensin II receptor antagonists, their methods of preparation and pharmaceutical compositions in which they are present |
| US5389632A (en) * | 1992-02-24 | 1995-02-14 | Laboratoires Upsa | Pyrazolopyrimidine derivatives which are angiotensin II receptor antagonists |
| US20060189617A1 (en) * | 2005-02-18 | 2006-08-24 | Wyeth | Imidazo[4,5-b]pyridine antagonists of gonadotropin releasing hormone receptor |
| WO2007076055A2 (en) * | 2005-12-22 | 2007-07-05 | Entremed, Inc. | Compositions and methods comprising proteinase activated receptor antagonists |
| WO2007136592A2 (en) * | 2006-05-18 | 2007-11-29 | Amphora Discovery Corporation | 2-0x0-l,2-dihydr0quin0line derivatives, compositions, and uses thereof as antiproliferative agents |
| WO2010017355A2 (en) * | 2008-08-08 | 2010-02-11 | New York Blood Center | Small molecule inhibitors of retroviral assembly and maturation |
| WO2013075083A1 (en) * | 2011-11-18 | 2013-05-23 | Constellation Pharmaceuticals | Modulators of methyl modifying enzymes, compositions and uses thereof |
| WO2014049488A1 (en) * | 2012-09-28 | 2014-04-03 | Pfizer Inc. | Benzamide and heterobenzamide compounds |
Non-Patent Citations (5)
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9695154B2 (en) | 2013-07-02 | 2017-07-04 | Millennium Pharmaceuticals, Inc. | Heteroaryl inhibitors of sumo activating enzyme |
| US10335410B2 (en) | 2014-07-01 | 2019-07-02 | Millennium Pharmaceuticals, Inc. | Heteroaryl compounds useful as inhibitors of sumo activating enzyme |
| US9683003B2 (en) | 2014-07-01 | 2017-06-20 | Millennium Pharmaceuticals, Inc. | Heteroaryl compounds useful as inhibitors of SUMO activating enzyme |
| US9962386B2 (en) | 2014-07-01 | 2018-05-08 | Millennium Pharmaceuticals, Inc. | Heteroaryl compounds useful as inhibitors of SUMO activating enzyme |
| US10780090B2 (en) | 2014-07-01 | 2020-09-22 | Millennium Pharmaceuticals, Inc. | Heteroaryl compounds useful as inhibitors of SUMO activating enzyme |
| US11236082B2 (en) | 2014-11-06 | 2022-02-01 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| US10919902B2 (en) | 2015-07-06 | 2021-02-16 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| US10421756B2 (en) | 2015-07-06 | 2019-09-24 | Rodin Therapeutics, Inc. | Heterobicyclic N-aminophenyl-amides as inhibitors of histone deacetylase |
| US11858939B2 (en) | 2015-07-06 | 2024-01-02 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| EP3445365A4 (en) * | 2016-04-22 | 2019-10-16 | Dana-Farber Cancer Institute, Inc. | Ezh2 inhibitors and uses thereof |
| AU2017252460B2 (en) * | 2016-04-22 | 2021-11-18 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| US10633371B2 (en) * | 2016-04-22 | 2020-04-28 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| CN109069508A (en) * | 2016-04-22 | 2018-12-21 | 达纳-法伯癌症研究所股份有限公司 | EZH2 inhibitor and application thereof |
| US10975056B2 (en) | 2016-06-13 | 2021-04-13 | Glaxosmithkline Intellectual Property Development Limited | Substituted pyridines as inhibitors of DNMT1 |
| WO2018045971A1 (en) | 2016-09-07 | 2018-03-15 | 上海海和药物研究开发有限公司 | Pyrido five-element aromatic ring compound, preparation method therefor and use thereof |
| CN108699029A (en) * | 2016-11-11 | 2018-10-23 | 上海海雁医药科技有限公司 | Tri- substituted indazole analog derivatives of 4,5,6-, its preparation method and purposes pharmaceutically |
| US11104664B2 (en) | 2016-11-11 | 2021-08-31 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | 4,5,6-trisubstituted indazole derivatives, and preparation method and pharmaceutical use thereof |
| WO2018086592A1 (en) * | 2016-11-11 | 2018-05-17 | 上海海雁医药科技有限公司 | 4,5,6-tri-substituted indazoles derivatives, preparation thereof, and use thereof in medicines |
| CN108699029B (en) * | 2016-11-11 | 2021-07-30 | 上海海雁医药科技有限公司 | 4,5, 6-trisubstituted indazole derivatives, preparation method and medical application thereof |
| US10793567B2 (en) | 2017-01-11 | 2020-10-06 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US9951069B1 (en) | 2017-01-11 | 2018-04-24 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11987580B2 (en) | 2017-01-11 | 2024-05-21 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US10696673B2 (en) | 2017-01-11 | 2020-06-30 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11225479B2 (en) | 2017-01-11 | 2022-01-18 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US10519149B2 (en) | 2017-01-11 | 2019-12-31 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11286256B2 (en) | 2017-01-11 | 2022-03-29 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11225475B2 (en) | 2017-08-07 | 2022-01-18 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| US11912702B2 (en) | 2017-08-07 | 2024-02-27 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| WO2020011607A1 (en) | 2018-07-09 | 2020-01-16 | Fondation Asile Des Aveugles | Inhibition of prc2 subunits to treat eye disorders |
| CN113330008A (en) * | 2019-03-25 | 2021-08-31 | 上海华汇拓医药科技有限公司 | Preparation method of amide compound and application of amide compound in field of medicine |
| JP7396369B2 (en) | 2019-03-25 | 2023-12-12 | 上海華匯拓医薬科技有限公司 | Method for preparing amide compounds and their use in the medical field |
| JP7395807B2 (en) | 2019-03-25 | 2023-12-12 | 上海華匯拓医薬科技有限公司 | Method for preparing amide compounds and their use in the pharmaceutical field |
| JP2022528337A (en) * | 2019-03-25 | 2022-06-10 | 上海華匯拓医薬科技有限公司 | Method for preparing amide compounds and their use in the medical field |
| JP2022528042A (en) * | 2019-03-25 | 2022-06-08 | 上海華匯拓医薬科技有限公司 | Methods for preparing amide compounds and their use in the pharmaceutical field |
| CN113330008B (en) * | 2019-03-25 | 2024-05-07 | 上海华汇拓医药科技有限公司 | Preparation method of amide compound and application of amide compound in medicine field |
| WO2020192652A1 (en) * | 2019-03-25 | 2020-10-01 | 上海华汇拓医药科技有限公司 | Preparation method for amide compounds and use thereof in medical field |
| US12018015B2 (en) | 2021-06-18 | 2024-06-25 | Aligos Therapeutics, Inc. | Methods and compositions for targeting PD-L1 |
| WO2023025856A1 (en) | 2021-08-25 | 2023-03-02 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of ezh2 inhibitors for the treatment of aortic valve stenosis |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015110999A1 (en) | Ezh2 inhibitors and uses thereof | |
| CN106957314B (en) | Pyrimidine derivatives as RAF kinase inhibitors | |
| JP6726677B2 (en) | Substituted 2-H-pyrazole derivatives as anticancer agents | |
| AU2018391675A1 (en) | Sulphonyl urea derivatives as NLRP3 inflammasome modulators | |
| BR112019015707A2 (en) | COMPOUND, PHARMACEUTICAL COMPOSITION. USE OF A COMPOUND, COMBINATION OF A COMPOUND AND DEVICE | |
| IL259421A (en) | Modulators of ror-gamma | |
| AU2017263361A1 (en) | Cyclopropyl-amide compounds as dual LSD1/HDAC inhibitors | |
| WO2015104677A1 (en) | Heterocyclic compounds as ezh2 inhibitors | |
| WO2014137728A1 (en) | Compounds inhibiting leucine-rich repeat kinase enzyme activity | |
| WO2021127337A1 (en) | Trpml modulators | |
| KR20100099742A (en) | Bicyclic derivatives for use in the treatment of androgen receptor associated conditions | |
| WO2014155301A1 (en) | Substituted bicyclic compounds as inhibitors of ezh2 | |
| TWI793704B (en) | SOS1 inhibitors, pharmaceutical compositions comprising them and uses thereof | |
| US10301306B2 (en) | Substituted dihydro-1H-pyrrolo[3,2-c]pyridin-4(5H)-ones as RIPK3 inhibitors | |
| KR102667331B1 (en) | Aminopyridine derivatives and their use as selective alk-2 inhibitors | |
| CA3133751A1 (en) | Macrocyclic azolopyridine derivatives as eed and prc2 modulators | |
| JP2022521537A (en) | Imidazopyridinyl compounds and their use for the treatment of proliferative disorders | |
| CN110857293A (en) | Novel quinoline derivative inhibitor | |
| KR20200013718A (en) | Heteroaromatic Compounds as Banin Inhibitors | |
| AU2018360575A1 (en) | Alkene spirocyclic compounds as farnesoid X receptor modulators | |
| AU2022289869A1 (en) | Anticancer compounds | |
| CA3092770A1 (en) | Substituted imidazolidin-2-one derivatives as prmt5 inhibitors | |
| WO2011019060A1 (en) | Hedgehog signal inhibitor | |
| EP2632924A1 (en) | Diaza-spiro[5.5]undecanes useful as orexin receptor antagonists | |
| AU2017341999A1 (en) | Inhibitors of mutant isocitrate dehydrogenases and compositions and methods thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15740120 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15740120 Country of ref document: EP Kind code of ref document: A1 |