WO2015037243A1 - Method for producing optically active hydantoin compound - Google Patents
Method for producing optically active hydantoin compound Download PDFInfo
- Publication number
- WO2015037243A1 WO2015037243A1 PCT/JP2014/004702 JP2014004702W WO2015037243A1 WO 2015037243 A1 WO2015037243 A1 WO 2015037243A1 JP 2014004702 W JP2014004702 W JP 2014004702W WO 2015037243 A1 WO2015037243 A1 WO 2015037243A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- acid
- potassium
- sodium
- formula
- Prior art date
Links
- 0 CC*1cc(OC(C)C)ccc1I Chemical compound CC*1cc(OC(C)C)ccc1I 0.000 description 2
- AYTGARGOCPEHGL-UHFFFAOYSA-N Cc1ccc2OCCOc2c1 Chemical compound Cc1ccc2OCCOc2c1 AYTGARGOCPEHGL-UHFFFAOYSA-N 0.000 description 2
- YJMADHMYUJFMQE-UHFFFAOYSA-O [NH3+]c1ccc2OCCc2c1 Chemical compound [NH3+]c1ccc2OCCc2c1 YJMADHMYUJFMQE-UHFFFAOYSA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B53/00—Asymmetric syntheses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
- C07D233/76—Two oxygen atoms, e.g. hydantoin with substituted hydrocarbon radicals attached to the third ring carbon atom
- C07D233/78—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
Definitions
- the present invention relates to a method for producing an optically active hydantoin compound having an LXR ⁇ activation action and a production intermediate thereof.
- the method of this document does not mention the production of optically active (a3), and does not describe the production method of the optical isomers of the compounds (A) to (J). Therefore, in the above production method, two diastereomers with different stereochemistry of the asymmetric carbon atom of the 4,4-disubstituted imidazolidine-2,5-dione moiety are produced for the compounds (A) to (J). The problem that it was not possible was left.
- Patent Documents 2 and 3 As a method for optical resolution of 4,4-disubstituted imidazolidine-2,5-dione derivatives, a preferential crystallization method using optically active benzylamine (Patent Documents 2 and 3) and a method using lipase (non-patent document) Although literature 7) has been reported, it has been found that all of them have a narrow substrate application range and are not suitable for the production of the compounds (A) to (J).
- Patent Document 5 Furthermore, a method for producing an optically active 1,4,4-trisubstituted imidazolidine-2,5-dione derivative represented by the following formula has also been reported (Patent Document 5).
- compound (c2) By reacting 2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl produced by the method described in the pamphlet, compound (c2) is obtained, and this is subjected to Wittig reaction to give compound (c3 to), an unsaturated bond and a compound by the hydrogenation reaction (c4), which compounds by hydrolyzing the (c5), by reacting it with the compound (c6), ribs with tmsCF 3
- Compound (c7) is obtained by a nolation reaction, and the optically active 2- [4- (2,5-dimethylpiperazi is obtained by deprotecting the piperazine compound.
- R 1 , R 2 and R 3 may be the same or different and each represents a C 1-3 alkyl group, * represents an asymmetric carbon atom, and R 4 represents
- the compound represented by the formula (1) is (S) -3- (2- ⁇ (2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro- 2-Hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazin-1-yl ⁇ -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methyl
- the method of the present invention can produce the target compound (1) in a higher yield than the conventional method, as shown in the Examples below.
- the asymmetric carbon atom of the imidazolidine-2,4-dione (3) can be constructed without racemization by following this method. Therefore, by using the method of the present invention, compound (1) useful as an LXR ⁇ selective agonist can be produced with high yield and high optical purity.
- the C 1-3 alkyl group means a linear or branched alkyl group having 1 to 3 carbon atoms, and examples thereof include a methyl group, an ethyl group, a propyl group, and an isopropyl group.
- the C 1-3 alkyl group for R 1 is preferably an ethyl group or a propyl group, and more preferably a propyl group.
- the C 1-3 alkyl group for R 2 is preferably a methyl group.
- the C 1-3 alkyl group for R 3 is preferably a methyl group.
- the C 1-3 alkyl group for R 6 is preferably a methyl group or an ethyl group.
- (1b) is preferable.
- the steric structure is (1b), R 1 is a propyl group, R 2 and R 3 are methyl groups, and R 4 is 4- (1- Mention may be made of compounds that are methylethoxy) phenyl groups.
- reaction process diagrams are shown, and the reaction of each process will be described in detail.
- the base is not particularly limited.
- alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide
- Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate
- metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium
- lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide
- n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium; triethylamine, N, N-diiso
- the reaction conditions are ⁇ 80 to 150 ° C., preferably 0 to 100 ° C., and 1 minute to 48 hours, preferably 1 hour to 24 hours.
- Step 2 In this step, compound (3) and compound (4) are reacted in a solvent in the presence of a condensing agent, in the presence or absence of a reaction accelerator, and in the presence or absence of a base.
- a condensing agent in the presence or absence of a reaction accelerator
- a base in the presence or absence of a base.
- the solvent is not particularly limited.
- dimethyl sulfoxide, acetonitrile, and methylene chloride are preferable.
- the condensing agent is not particularly limited.
- carbodiimide reagents such as dicyclohexylcarbodiimide (DCC), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC), diisopropylcarbodiimide (DIPCDI), (1H- Benzotriazol-1-yloxy) tris (dimethylamino) phosphonium hexafluorophosphate (BOP), (1H-benzotriazol-1-yloxy) tris (pyrrolidino) phosphonium hexafluorophosphate (PyBOP), 1- [bis (dimethyl) Amino) methylene] -1H-1,2,3-triazolo (4,5-b) pyridium-3-oxodohexafluorophosphate (HATU),
- the reaction accelerator is not particularly limited.
- 1-hydroxybenzotriazole (HOBt) 6-chloro-1-hydroxybenzotriazole (6-Cl-HOBt), 3,4-dihydro-3-hydroxy-4 -Oxo-1,2,3-benzotriazine (HOOBt), 1-hydroxy-7-azabenzotriazole (HOAt) and the like can be used.
- the base is not particularly limited.
- alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide
- Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate
- metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium
- lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide
- n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium; triethylamine, N, N-diisopropylethylamine, N
- phosphine reagents include trialkylphosphine such as trimethylphosphine, triethylphosphine, tripropylphosphine, triisopropylphosphine, tributylphosphine, triisobutylphosphine, and tricyclohexylphosphine, and triarylphosphine such as diphenylphosphinophenylpolystyrene Etc. can be used.
- trialkylphosphine such as trimethylphosphine, triethylphosphine, tripropylphosphine, triisopropylphosphine, tributylphosphine, triisobutylphosphine, and tricyclohexylphosphine
- triarylphosphine such as diphenylphosphinophenylpolystyrene Etc.
- Examples of the azo reagent or ethylenedicarboxylic acid reagent include diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD), 1,1′-azobis (N, N-dimethylformamide) (TMAD), 1,1′- (Azodicarbonyl) dipiperidine (ADDP), 1,1′-azobis (N, N-diisopropylformamide) (TIPA), 1,6-dimethyl-1,5,7-hexahydro-1,4,6,7- Tetrazocine-2,5-dione (DHTD) or the like can be used.
- the reaction conditions are ⁇ 80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
- compound (3) is reacted with an acid halogenating agent in a solvent to produce an acid halide derivative, and this is then combined with compound (4) in a solvent in the presence or absence of a base.
- the hydantoin derivative (1) can also be produced by reacting.
- Examples of the acid halogenating agent include N, N-diethylaminosulfur trifluoride (DAST), selenium tetrafluoride or its pyridine adduct, thionyl chloride, oxalyl chloride, pyrocatekylphosphotrichloride, dichlorotriphenylphosphorane, Thionyl bromide, dibromotriphenylphosphorane, 1-dimethyl-1-iodo-2-methylpropene and the like can be used.
- DAST N-diethylaminosulfur trifluoride
- selenium tetrafluoride or its pyridine adduct thionyl chloride
- oxalyl chloride oxalyl chloride
- pyrocatekylphosphotrichloride dichlorotriphenylphosphorane
- Thionyl bromide Thionyl bromide
- dibromotriphenylphosphorane 1-dimethyl-1-i
- alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide
- Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate
- metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium
- lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide
- n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium; triethylamine, N, N-diisopropylethylamine, N
- Step 3 is a step for producing a compound (7) by reacting the compound (5) with a compound (6) in a solvent in the presence or absence of a base.
- a compound (6) a commercially available compound can be used, and for example, a compound produced according to the method described in WO2010 / 12581 can be used, but the compound (6) is particularly limited to this. is not.
- the solvent is not particularly limited, but for example, tetrahydrofuran, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, methyl ethyl ketone, water, etc.
- alkali metal hydrides such as lithium hydride, sodium hydride, potassium hydride, Alkali metal hydroxides, such as lithium hydroxide, sodium hydroxide, potassium hydroxide; Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate; metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium; lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide; n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium;
- Step 4 is a step for producing compound (8) by reacting compound (7) with a Wittig reagent or Horner-Wadsworth-Emmons (HWE) reagent in a solvent in the presence or absence of a base.
- the solvent is not particularly limited, and for example, N, N-dimethylformamide, tetrahydrofuran, dioxane, acetonitrile, nitromethane, acetone, ethyl acetate, benzene, chlorobenzene, toluene, chloroform, methylene chloride and the like can be used.
- Tetrahydrofuran, ethyl acetate, toluene, chloroform and methylene chloride are preferred.
- phosphonium salts such as stable ylide and unstable ylide (methyltriphenylphosphonium bromide, ethyltriphenylphosphonium bromide) can be used.
- HWE reagent a phosphonic acid ester can be used.
- the base is not particularly limited.
- alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide
- Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate, metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium
- lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide
- n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium
- This process is a process of manufacturing a compound (10) by making a compound (9) react in a solvent in presence of a base or an acid.
- the solvent is not particularly limited.
- the base is not particularly limited.
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide
- alkali carbonates such as lithium carbonate, sodium carbonate, potassium carbonate and cesium carbonate
- an acid For example, hydrochloric acid, a sulfuric acid, an acetic acid, a tosylic acid etc. can be used.
- the reaction conditions are ⁇ 80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
- Step 7 is a step of removing the compound (10) in a solvent or in the absence of a solvent, in the presence or absence of a condensing agent, in the presence or absence of a reaction accelerator, in the presence or absence of an acid or base.
- compound (11) is produced by the reaction below.
- the substituent represented by R 9 can be converted into an acid halide, an acid anhydride or an ester with reference to, for example, literature (Comprehensive Organic Transformations Second Edition, John Wiley & Sons, Inc.).
- acid halides include acid fluorides and acid chlorides.
- Examples of the acid anhydride include an acid anhydride with an aliphatic carboxylic acid such as acetic acid, an acid anhydride with an aromatic carboxylic acid such as benzoic acid, and the like.
- Examples of the ester include an ester with an aliphatic alcohol such as methanol, an ester with an aromatic alcohol such as pentafluorophenol, and the like.
- R 9 is a pentafluorophenoxy group
- the solvent is not particularly limited.
- tetrahydrofuran, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, Methyl ethyl ketone, ethyl acetate and the like can be used alone or in combination.
- the condensing agent is not particularly limited, and examples thereof include carbodiimide reagents such as dicyclohexylcarbodiimide (DCC), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC), and diisopropylcarbodiimide (DIPCDI). Of these, dicyclohexylcarbodiimide and 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide are preferable.
- DEC dicyclohexylcarbodiimide
- EDC 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide
- DIPCDI diisopropylcarbodiimide
- Step 8 is a step for producing the compound (11) into the hexafluorocarbinol compound (12) using a trifluoromethylating reagent in a solvent in the presence of a base.
- the solvent is not particularly limited, and examples thereof include dimethoxyethane, tetrahydrofuran, toluene, dioxane, ethylene glycol dimethyl ether, N, N-dimethylformamide, N-methylpyrrolidone, tetramethylurea, dimethyl sulfoxide, acetonitrile, propionitrile and the like. They can be used alone or in combination, and ethylene glycol dimethyl ether is particularly preferable.
- Trifluoromethylating reagents include (trifluoromethyl) trimethylsilane, triethyl (trifluoromethyl) silane, triisopropyl (trifluoromethyl) silane, methyldiphenyl (trifluoromethyl) silane, dimethyl (diphenyl) trifluoromethylsilane Etc.
- the reaction conditions are ⁇ 80 to 150 ° C., preferably ⁇ 30 to 50 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
- compound (4) can be produced by reacting compound (12) with an acid in a solvent or without a solvent.
- the solvent is not particularly limited.
- N, N-dimethylformamide, tetrahydrofuran, dioxane, acetonitrile, nitromethane, acetone, ethyl acetate, benzene, chlorobenzene, toluene, chloroform, methylene chloride, water, methanol, ethanol, 1- Propanol, 2-propanol and the like can be used alone or in combination.
- the production method of the compound (2) used in the present invention is not particularly limited, but can be synthesized, for example, by the method shown below.
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide
- alkali carbonates such as lithium carbonate, sodium carbonate, potassium carbonate and cesium carbonate
- an acid For example, hydrochloric acid, a sulfuric acid, an acetic acid, a tosylic acid etc. can be used.
- the reaction conditions are ⁇ 80 to 200 ° C., preferably 0 to 150 ° C., for 1 minute to 7 days, preferably 1 hour to 2 days.
- Step 12 is a step for producing a compound (16) by reacting the compound (15) in a solvent or in the absence of a solvent in the presence of an optical resolution agent.
- the solvent is not particularly limited.
- esters such as isobutyl acetate, alcohols such as water, methanol, ethanol, 1-propanol, and 2-propanol can be used alone or in combination.
- the optical resolution agent is not particularly limited.
- (L)-(+)-mandelic acid, (D)-( ⁇ )-mandelic acid, malic acid, tartaric acid, lactic acid, camphorsulfonic acid, (1S) — (+)-Ketopic acid or commercially available optically pure compounds can also be used.
- the reaction conditions are -80 to 150 ° C, preferably -10 to 100 ° C, 1 minute to 48 hours, preferably 1 hour to 24 hours.
- the base is not particularly limited.
- alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide
- Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate
- metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium
- Specific examples of the salt allowed in the optically active compound represented by the general formula (3) of the present invention include acid addition salts with inorganic acids and organic acids, or base addition salts with inorganic bases and organic bases. Etc.
- solvate of the optically active compound represented by the general formula (3) of the present invention and its acceptable salt include hydrates and various solvates.
- N, N-dimethylformamide 50 mL of 2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (14.2 g, 61.26 mmol) produced according to the pamphlet of WO2010 / 12581
- the solution and 4-fluoro-3-formylbenzoic acid methyl ester 11.15 g, 61.26 mmol
- toluene and water were added to the reaction solution at 0 ° C.
- Step II 4- [4- (Methoxycarbonyl) -2- (prop-1-en-1-yl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl ( c3)
- Ethyltriphenylphosphonium bromide 25.8 g, 69.46 mmol
- potassium methoxide 4.87 g, 69.46 mmol
- trans-c3 4- ⁇ 4- (methoxycarbonyl) -2-[(E) -prop-1-en-1-yl] phenyl ⁇ -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl
- Step III Preparation of 4- [4- (methoxycarbonyl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (c4): 4- [4- (Methoxycarbonyl) -2- (prop-1-en-1-yl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (4.76 g) , 12.25 mmol) was dissolved in methanol (123 mL), and palladium on carbon was added under an argon atmosphere. Thereafter, the mixture was stirred at room temperature for 24 hours under a hydrogen atmosphere.
- Step IV Preparation of 4-[(2R, 5S) -4- (tert-butoxycarbonyl) -2,5-dimethylpiperazin-1-yl] -3-propylbenzoic acid (c5): 4- [4- (Methoxycarbonyl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (4.72 g, 12.09 mmol) in methanol (121 mL) 4N-aqueous sodium hydroxide solution (18.1 mL, 72.52 mmol) was added at 0 ° C., and the mixture was stirred at 60 ° C. for 2 hours.
- Step V Preparation of 2,5-dimethyl-4- ⁇ 4-[(perfluorophenoxy) carbonyl] -2-propylphenyl ⁇ piperazine-1-carboxylic acid (2S, 5R) -tert-butyl (c6): 4-[(2R, 5S) -4- (tert-butoxycarbonyl) -2,5-dimethylpiperazin-1-yl] -3-propylbenzoic acid (1.0 g, 2.656 mmol) was added to ethyl acetate (18 mL).
- Step VI 4- [4- (1,1,1,3,3,3-Hexafluoro-2-hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxyl
- 2S, 5R 2,5-dimethyl-4- ⁇ 4-[(perfluorophenoxy) carbonyl] -2-propylphenyl ⁇ piperazine-1-carboxylic acid (2S, 5R) -tert-butyl (5.75 g, 10.6 mmol) under reduced pressure After drying at 100 ° C.
- Step VII 2- ⁇ 4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl ⁇ -1,1,1,3,3,3-hexafluoropropane-2 -Manufacture of all (c8): 4- [4- (1,1,1,3,3,3-Hexafluoro-2-hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S , 5R) -tert-butyl (4.53 g, 9.08 mmol) was dissolved in methanol (14 mL), 2N hydrochloric acid methanol solution (41 mL, 81.7 mmol) was added at 0 ° C., and 5 hours at 35 ° C.
- Step IX Preparation of methyl 2-amino-2- [4- (1-methylethoxy) phenyl] propanoate (c11): 2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid hydrochloride (70.0 g, 270 mmol) was dissolved in methanol (225 mL), concentrated sulfuric acid (70 mL) was sequentially added at room temperature, and 24 Heated to reflux for hours. After confirming the completion of the reaction, the reaction solution was brought to room temperature and concentrated under reduced pressure to distill off methanol. The obtained residue was dissolved in dichloromethane and extracted with dichloromethane. The organic layer was washed successively with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the title compound (44.8 g, yield 70%) as a colorless oil.
- Step XI Preparation of 2- [3- (2-ethoxy-2-oxoethyl) ureido] -2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl ((S) -c12): 2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl (53.0 mg, 0.223 mmol) was dissolved in dichloromethane (740 ⁇ L) and ethyl isocyanatoacetate (0 ° C.) 29 mg, 0.223 mmol) was added, and the mixture was stirred at 0 ° C. for 1 hour. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain 91.5 mg (yield> 99%) of the title compound as a colorless oil.
- Steps XI & XII (S) -2- ⁇ 4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl ⁇ acetic acid ((S) -c13) Manufacturing: 2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl (2.90 g, 12.2 mmol) was dissolved in dimethyl sulfoxide (15 mL) and ethyl isocyanatoacetate at 15 ° C. (1.74 g, 13.4 mmol) was added, and the mixture was stirred at 15 ° C. for 1 hour.
- Oxoimidazolidin-1-yl ⁇ acetic acid (169 mg, 0.552 mmol) was added. Thereafter, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (HBTU) (228 mg, 0.602 mmol), diisopropylethylamine ( 156 mg, 1.20 mmol). The reaction solution was stirred at 0 ° C. for 10 minutes, and then stirred at room temperature for 20 hours.
- HBTU hexafluorophosphate
- Step XIII-2 (S) -3- (2- ⁇ (2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl) ) -2-Propylphenyl] -2,5-dimethylpiperazin-1-yl ⁇ -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione Production of ((S) -B): (S) -2- ⁇ 4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl ⁇ acetic acid was dissolved in toluene (2.0 mL), N, N-dimethylformamide (2.6 mg) and thionyl chloride (77.7 mg) were sequentially added, and the mixture was stirred at 120 ° C.
- Step XIV Preparation of 2-amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (R) -methyl ((R) -c11): (D)-( ⁇ )-Mandelic acid was used for the reaction and treatment in the same manner as in Step X to obtain the title compound as a colorless oil (99.4% ee).
- Step XV (R) -2- ⁇ 4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl ⁇ acetic acid ((R) -c13) Manufacturing: 2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (R) -methyl (1.00 g, 4.21 mmol) was dissolved in ethanol (5.0 mL), and Ethyl isocyanatoacetate (599 mg, 4.64 mmol) was added, and the mixture was stirred at 20 ° C. for 1 hr 30 min.
- Step XVI (R) -3- (2- ⁇ (2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)- 2-propylphenyl] -2,5-dimethylpiperazin-1-yl ⁇ -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione (( Production of R) -B): 2- ⁇ 4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl ⁇ -1,1,1,3,3,3-hexafluoropropan-2-ol ( 43.8 mg, 0.112 mmol) is dissolved in dimethyl sulfide (500 ⁇ L) and (R) -2- ⁇ 4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-
- the present invention provides a production method applicable to mass synthesis of the optically active hydantoin compound (1) represented by the general formula (1).
- Compound (1) has an LXR ⁇ agonistic action, and atherosclerosis such as atherosclerosis, arteriosclerosis, and diabetes caused by diabetes; dyslipidemia; hypercholesterolemia; lipid-related disease; inflammation Inflammatory diseases caused by sex cytokines; skin diseases such as allergic skin diseases; diabetes; or Alzheimer's disease preventive and / or therapeutic agents, etc. Has the above applicability.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
The present invention provides a method for producing an optically active hydantoin compound having an LXRβ-activating effect and a production intermediate thereof. In the present invention, an optically active carbinol compound represented by formula (1) is produced by reacting a compound represented by formula (3) and a compound represented by formula (4). In formulas (1), (3), and (4), R1, R2, and R3 show the same or different C1-3 alkyl groups, * shows an asymmetric carbon atom, and R4 shows any one structure selected from A-E.
Description
本発明は、LXRβ活性化作用を有する光学活性ヒダントイン化合物の製造方法およびその製造中間体に関する。
The present invention relates to a method for producing an optically active hydantoin compound having an LXRβ activation action and a production intermediate thereof.
肝臓X受容体(LXR)は、22-R-ヒドロキシコレステロールをはじめとするオキシステロール類の一部がリガンドとして作用する核内受容体である(非特許文献1~3)。哺乳類では二種のLXR遺伝子(α及びβ)の存在が知られている。LXRαは肝臓、小腸、脂肪組織などのコレステロール代謝に関わる組織に特異的に発現し、LXRβは調べられたほぼ全ての組織で普遍的に発現しているが、この受容体は共に、DNA上の同様の配列を認識し、付近の標的遺伝子の転写を活性化する(非特許文献4、5)。LXRの標的遺伝子として同定された遺伝子群のうちの多くは、ABCトランスポーター(ABCA1,ABCG1,ABCG5,ABCG8)をはじめとするコレステロール逆輸送(RCT)に関わる遺伝子(ApoE,CETP,及びLPL)である。このため、LXRの活性化はこれらの遺伝子発現上昇およびコレステロール逆輸送系経路を活性化して末梢からのコレステロール流出を増加させ、HDLコレステロールを増加させることにより、動脈硬化病変部位のコレステロール含量を減少させるものと期待されている(非特許文献6)。
Liver X receptor (LXR) is a nuclear receptor in which a part of oxysterols including 22-R-hydroxycholesterol acts as a ligand (Non-Patent Documents 1 to 3). In mammals, the presence of two LXR genes (α and β) is known. LXRα is specifically expressed in tissues involved in cholesterol metabolism such as liver, small intestine, adipose tissue, and LXRβ is ubiquitously expressed in almost all tissues examined. It recognizes similar sequences and activates transcription of nearby target genes (Non-Patent Documents 4 and 5). Many of the genes identified as LXR target genes are genes involved in reverse cholesterol transport (RCT) (ApoE, CETP, and LPL) including ABC transporters (ABCA1, ABCG1, ABCG5, ABCG8). is there. Thus, activation of LXR increases the expression of these genes and activates the reverse cholesterol transport pathway to increase peripheral cholesterol efflux and increase HDL cholesterol, thereby reducing the cholesterol content at the site of atherosclerotic lesions. It is expected (Non-Patent Document 6).
斯かる状況の下、本発明者らはLXRβ選択的活性化作用を有する化合物として、下記式:
Under such circumstances, the present inventors have expressed the following formula as a compound having an LXRβ selective activation action:

で示される化学構造を有する化合物(A)~(J)を見出し、特許出願している(特許文献1)。これらの化合物は、不斉炭素原子を有する4,4-二置換イミダゾリジン-2,5-ジオン構造を有するという点で共通している。
The compounds (A) to (J) having the chemical structure represented by the above have been found and a patent application has been filed (Patent Document 1). These compounds are common in that they have a 4,4-disubstituted imidazolidine-2,5-dione structure having an asymmetric carbon atom.
上記化合物(A)~(J)の製造法としては、例えば、以下の反応工程図に示すようなラセミ体の4,4-二置換イミダゾリジン-2,5-ジオン誘導体(a3)の製造を経由した方法が報告されている。すなわち、1-(4-ヒドロキシフェニル)エタノン(a1)の水酸基をアルキル化することによって、化合物(a2)を製造し、Bucherer-Bergs反応をすることによって、共通中間体となる4,4-二置換イミダゾリジン-2,5-ジオン誘導体(a3)を製造した後に、(a4)と反応させて、(B)を得る方法が知られている。
Examples of the method for producing the above compounds (A) to (J) include the production of a racemic 4,4-disubstituted imidazolidine-2,5-dione derivative (a3) as shown in the following reaction process diagram. The way through is reported. That is, the compound (a2) is produced by alkylating the hydroxyl group of 1- (4-hydroxyphenyl) ethanone (a1), and undergoes the Bucherer-Bergs reaction, thereby forming a 4,4-diphenyl which becomes a common intermediate. A method is known in which a substituted imidazolidine-2,5-dione derivative (a3) is produced and then reacted with (a4) to obtain (B).

しかしながら、当該文献の方法では光学活性な(a3)の製造には言及しておらず、上記化合物(A)~(J)の光学異性体の製造法については、記載されていない。そのため、上記製造法では(A)~(J)の化合物について、4,4-二置換イミダゾリジン-2,5-ジオン部位の不斉炭素原子の立体化学が異なる2つのジアステレオマーの作り分けができない、といった課題が残されていた。
However, the method of this document does not mention the production of optically active (a3), and does not describe the production method of the optical isomers of the compounds (A) to (J). Therefore, in the above production method, two diastereomers with different stereochemistry of the asymmetric carbon atom of the 4,4-disubstituted imidazolidine-2,5-dione moiety are produced for the compounds (A) to (J). The problem that it was not possible was left.
ラセミ体の一般的な光学分割法として、光学活性カラムクロマトグラフィーを用いる方法が知られているが、一方で、作業効率およびコストの観点から大量合成には適していない。
Although a method using optically active column chromatography is known as a general optical resolution method for racemates, it is not suitable for mass synthesis from the viewpoint of work efficiency and cost.
4,4-二置換イミダゾリジン-2,5-ジオン誘導体の光学分割の方法として、光学活性なベンジルアミンを用いた優先晶析法(特許文献2、3)やリパーゼを用いた方法(非特許文献7)が報告されているが、いずれも基質適用範囲が狭く、上記化合物(A)~(J)の製造には適さないことが分かった。
As a method for optical resolution of 4,4-disubstituted imidazolidine-2,5-dione derivatives, a preferential crystallization method using optically active benzylamine (Patent Documents 2 and 3) and a method using lipase (non-patent document) Although literature 7) has been reported, it has been found that all of them have a narrow substrate application range and are not suitable for the production of the compounds (A) to (J).
また、光学活性な1,4,4-三置換イミダゾリジン-2,5-ジオン誘導体の製造方法として、1,4,4-三置換イミダゾリジン-2,5-ジオン誘導体のラセミ体に対して、光学活性な化合物を用いた優先晶析法(特許文献4)が報告されているが、基質適用範囲が狭く、上記化合物(A)~(J)の製造には適さないことが分かった。
Further, as a method for producing an optically active 1,4,4-trisubstituted imidazolidine-2,5-dione derivative, a racemate of a 1,4,4-trisubstituted imidazolidine-2,5-dione derivative is used. Although a preferential crystallization method using an optically active compound (Patent Document 4) has been reported, it has been found that the substrate application range is narrow and is not suitable for the production of the compounds (A) to (J).
さらに、下式に示される光学活性な1,4,4-三置換イミダゾリジン-2,5-ジオン誘導体の製造方法も報告されている(特許文献5)。
Furthermore, a method for producing an optically active 1,4,4-trisubstituted imidazolidine-2,5-dione derivative represented by the following formula has also been reported (Patent Document 5).

しかしながら、当該文献に記載の方法を上記化合物(A)~(J)の製造に適用すると、上記vi)の工程において基質が分解してしまい、1,4,4-三置換イミダゾリジン-2,5-ジオン環が得られないことが分かった。
However, when the method described in this document is applied to the production of the compounds (A) to (J), the substrate is decomposed in the step vi), and 1,4,4-trisubstituted imidazolidine-2, It was found that a 5-dione ring could not be obtained.
本発明は、光学活性な1,4,4-三置換イミダゾリジン-2,5-ジオン環を経由して、LXRβ選択的活性化作用を有する光学活性ヒダントイン化合物の大量合成に適用できる製造法を提供することを目的とする。また、本発明は、光学活性ヒダントイン化合物の製造中間体として有用な光学活性2-(4-アリール-4-メチル-2,5-ジオキソイミダゾリジン-1-イル)酢酸誘導体化合物を提供することを目的とする。
The present invention provides a production method applicable to a large-scale synthesis of an optically active hydantoin compound having an LXRβ selective activation action via an optically active 1,4,4-trisubstituted imidazolidine-2,5-dione ring. The purpose is to provide. The present invention also provides an optically active 2- (4-aryl-4-methyl-2,5-dioxoimidazolidin-1-yl) acetic acid derivative compound useful as an intermediate for the production of an optically active hydantoin compound. With the goal.
前記の通り、光学活性ヒダントイン化合物を効率的かつ光学純度よく製造することは、医薬品、特にLXRβ選択的活性化剤の製造において非常に有用であると考えられる。そこで本発明者らは、前記化合物(B)で示される光学活性ヒダントイン体の有用な製造法について鋭意研究を行った結果、下図に示すように、安息香酸エステル体(c1)とWO2010/125811号パンフレットに記載の方法で製造した2,5-ジメチルピペラジン-1-カルボン酸(2S,5R)-tert-ブチルを反応させることにより、化合物(c2)とし、これをWittig反応することにより化合物(c3)にし、不飽和結合を水素添加反応することにより化合物(c4)とし、これを加水分解することにより化合物(c5)とし、これを反応することにより化合物(c6)にし、TMSCF3を用いてカルビノール化反応することにより化合物(c7)とし、ピペラジン体の脱保護を行うことで光学活性な2-[4-(2,5-ジメチルピペラジン-1-イル)-3-プロピルフェニル]-1,1,1,3,3,3-ヘキサフルオロプロパン-2-オール化合物(c8)とした。次にWO2010/125811号パンフレットに記載の方法で製造したヒダントイン体(c9)を加水分解させることにより化合物(c10)とし、これをエステル化反応することにより化合物(c11)とし、これを(L)-(+)-マンデル酸により優先晶析することにより光学活性なアミノ酸体((S)-c11)とし、これをイソシアナト酢酸エステルと反応することにより化合物((S)-c12)とし、これを塩基存在下、閉環反応することにより光学活性な化合物(S)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸化合物((S)-c13)とし、これと化合物(c8)とを縮合させることで、光学純度を損なうことなく光学活性ヒダントイン体((S)-B)を得られることを見出した。また、(c11)に対し、(D)-(-)-マンデル酸を用いて優先晶析することにより光学活性なアミノ酸体((R)-c11)を製造し、光学活性ヒダントイン体((R)-B)が同様に得られた。本発明は、この知見に基づいて完成されたものである。
As described above, it is considered that production of an optically active hydantoin compound efficiently and with high optical purity is very useful in the production of pharmaceuticals, particularly LXRβ selective activators. Therefore, as a result of intensive studies on a useful method for producing an optically active hydantoin compound represented by the compound (B), the present inventors have found that a benzoate ester compound (c1) and WO2010 / 12581 are shown in the figure below. By reacting 2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl produced by the method described in the pamphlet, compound (c2) is obtained, and this is subjected to Wittig reaction to give compound (c3 to), an unsaturated bond and a compound by the hydrogenation reaction (c4), which compounds by hydrolyzing the (c5), by reacting it with the compound (c6), ribs with tmsCF 3 Compound (c7) is obtained by a nolation reaction, and the optically active 2- [4- (2,5-dimethylpiperazi is obtained by deprotecting the piperazine compound. N-1-yl) -3-propylphenyl] -1,1,1,3,3,3-hexafluoropropan-2-ol compound (c8). Next, the hydantoin body (c9) produced by the method described in the pamphlet of WO2010 / 12581 is hydrolyzed to obtain a compound (c10), which is esterified to obtain a compound (c11). An optically active amino acid form ((S) -c11) is obtained by preferential crystallization with — (+)-mandelic acid, and this is reacted with isocyanatoacetate to give compound ((S) -c12). Optically active compound (S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl by ring-closing reaction in the presence of a base } Acetic acid compound ((S) -c13) and this compound (c8) are condensed to form an optically active hydantoin compound (( ) -B) it was found that the resulting. In addition, optically active amino acid form ((R) -c11) is produced by preferential crystallization of (c11) using (D)-(−)-mandelic acid, and optically active hydantoin form ((R ) -B) was obtained analogously. The present invention has been completed based on this finding.

すなわち本発明は、以下の発明に関する。
[1]式(1): That is, the present invention relates to the following inventions.
[1] Formula (1):
[1]式(1): That is, the present invention relates to the following inventions.
[1] Formula (1):

[式中、R1、R2、R3は同一又は異なってもよく、C1-3アルキル基を示し、*は不斉炭素原子を示し、R4は、
[Wherein R 1 , R 2 and R 3 may be the same or different and each represents a C 1-3 alkyl group, * represents an asymmetric carbon atom, and R 4 represents

から選択される構造のうちいずれかひとつを示す]で表される光学活性化合物の製造方法であって、
i) 式(2): A method for producing an optically active compound represented by any one of the structures selected from:
i) Formula (2):
i) 式(2): A method for producing an optically active compound represented by any one of the structures selected from:
i) Formula (2):

[式中、R4、*は前記と同じ定義であり、R5、R6はそれぞれ同一又は異なってもよく、C1-3アルキル基を示す]で表される化合物を塩基存在下、閉環反応させることにより、式(3):
[Wherein R 4 and * are as defined above, and R 5 and R 6 may be the same or different and each represents a C 1-3 alkyl group] By reacting, the formula (3):

[式中、R4、*は前記と同じ定義である]で表される化合物を製造し、
ii) 式(3)で表される化合物と、式(4): [Wherein R 4 and * are as defined above]
ii) a compound represented by formula (3) and formula (4):
ii) 式(3)で表される化合物と、式(4): [Wherein R 4 and * are as defined above]
ii) a compound represented by formula (3) and formula (4):

[式中、R1、R2、R3、*は前記と同じ定義である]で表される化合物を反応させることにより、式(1)で表される化合物を製造することを特徴とする方法。
[Wherein R 1 , R 2 , R 3 , * are as defined above] are reacted to produce a compound represented by the formula (1). Method.
[2]式(1)のR1がプロピル基、R2、R3がメチル基である、前記[1]に記載の製造方法。
[2] The production method according to [1], wherein R 1 in formula (1) is a propyl group, and R 2 and R 3 are methyl groups.
[3]式(1)で表される化合物が(S)-3-(2-{(2S,5R)-4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-イル}-2-オキソエチル)-5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオンである前記[1]又は[2]に記載の製造方法。
[3] The compound represented by the formula (1) is (S) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro- 2-Hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methyl The production method according to the above [1] or [2], which is imidazolidine-2,4-dione. *
[4]式(3):
[4] Formula (3):

[式中、*は不斉炭素原子を示し、R4は
[Wherein, * represents an asymmetric carbon atom and R 4 represents

から選択される構造のうちいずれかひとつを示す]で表される光学活性化合物若しくはその塩、又はそれらの溶媒和物。
An optically active compound represented by the formula: or a salt thereof, or a solvate thereof.
本発明の方法は、後記実施例に示すとおり、従来の方法に比べて高収率で目的の化合物(1)を製造することができる。また、イミダゾリジン-2,4-ジオン体(3)の不斉炭素原子は、本方法に従う事によりラセミ化が起こる事無く構築することができる。従って、本発明の方法を用いることにより、LXRβ選択的アゴニストとして有用な化合物(1)を高収率且つ高光学純度で製造できる。
The method of the present invention can produce the target compound (1) in a higher yield than the conventional method, as shown in the Examples below. In addition, the asymmetric carbon atom of the imidazolidine-2,4-dione (3) can be constructed without racemization by following this method. Therefore, by using the method of the present invention, compound (1) useful as an LXRβ selective agonist can be produced with high yield and high optical purity.
本発明の用語の定義は以下の通りである。
The definitions of the terms of the present invention are as follows.
本発明において、C1-3アルキル基とは、炭素数が1~3の直鎖又は分岐鎖のアルキル基を意味し、例えば、メチル基、エチル基、プロピル基、イソプロピル基が挙げられる。
In the present invention, the C 1-3 alkyl group means a linear or branched alkyl group having 1 to 3 carbon atoms, and examples thereof include a methyl group, an ethyl group, a propyl group, and an isopropyl group.
一般式(1)、(4)中、R1のC1-3アルキル基としては、エチル基、プロピル基が好ましく、プロピル基がより好ましい。
In general formulas (1) and (4), the C 1-3 alkyl group for R 1 is preferably an ethyl group or a propyl group, and more preferably a propyl group.
一般式(1)、(4)中、R2のC1-3アルキル基としては、メチル基が好ましい。
In general formulas (1) and (4), the C 1-3 alkyl group for R 2 is preferably a methyl group.
一般式(1)、(4)中、R3のC1-3アルキル基としては、メチル基が好ましい。
In general formulas (1) and (4), the C 1-3 alkyl group for R 3 is preferably a methyl group.
一般式(1)~(3)中、R4としては、
In general formulas (1) to (3), as R 4 ,

一般式(2)中、R5のC1-3アルキル基としては、メチル基又はエチル基が好ましい。
In general formula (2), the C 1-3 alkyl group for R 5 is preferably a methyl group or an ethyl group.
一般式(2)中、R6のC1-3アルキル基としては、メチル基又はエチル基が好ましい。
In general formula (2), the C 1-3 alkyl group for R 6 is preferably a methyl group or an ethyl group.
本発明において、*は不斉炭素原子を示す。本発明の一般式(1)は、下記(1a)~(1h):
In the present invention, * represents an asymmetric carbon atom. The general formula (1) of the present invention includes the following (1a) to (1h):

に示す8つの構造を包含している。
8 structures are included.
本発明の一般式(1)の立体構造としては、(1b)が好ましい。
As the three-dimensional structure of the general formula (1) of the present invention, (1b) is preferable.
本発明の一般式(1)の特に好ましい構造として、立体構造は(1b)であり、R1がプロピル基であり、R2およびR3がメチル基であり、R4が4-(1-メチルエトキシ)フェニル基である化合物を挙げることができる。
As a particularly preferred structure of the general formula (1) of the present invention, the steric structure is (1b), R 1 is a propyl group, R 2 and R 3 are methyl groups, and R 4 is 4- (1- Mention may be made of compounds that are methylethoxy) phenyl groups.
以下、本発明製造法について、反応工程図を示し、各工程の反応について詳細に説明する。
Hereinafter, with respect to the production method of the present invention, reaction process diagrams are shown, and the reaction of each process will be described in detail.

(式中、R1、R2、R3、R4、R5、R6および*は、前記と同じものを示す)
(Wherein R 1 , R 2 , R 3 , R 4 , R 5 , R 6 and * are the same as described above)
工程1:本工程は、化合物(2)を溶媒中又は無溶媒中、塩基の存在下、閉環反応させることにより化合物(3)を製造する工程である。溶媒としては、特に制限は無いが、例えばテトラヒドロフラン、エチレングリコールジメチルエーテル、トルエン、ジオキサン、N,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、アセトニトリル、プロピオニトリル、アセトン、メチルエチルケトン;酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸イソブチル等のエステル類;メタノール、エタノール、1-プロパノール、2-プロパノール等のアルコール類等を単独又は組み合わせて使用することができ、中でもエタノール、エタノール/水、エチレングリコールジメチルエーテル、エチレングリコールジメチルエーテル/水、ジメチルスルホキシド、ジメチルスルホキシド/水が好ましい。塩基としては、特に制限はないが、例えば水素化リチウム、水素化ナトリウム、水素化カリウム等の水素化アルカリ金属類;水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類;ナトリウムメトキシド、カリウムメトキシド、ナトリウムエトキシド、カリウムエトキシド、tert-ブトキシナトリウム、tert-ブトキシカリウム等のアルコールの金属塩類;リチウムジイソプロピルアミド、ナトリウムジイソプロピルアミド、カリウムジイソプロピルアミド、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド等の金属アミド類;n-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム等の有機金属化合物類;トリエチルアミン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、3,4-ルチジン、2,6-ルチジン、2,4,6-コリジン等を使用することができ、中でも水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウムが好ましい。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~48時間、好ましくは1時間~24時間である。
Step 1: This step is a step for producing a compound (3) by subjecting the compound (2) to a ring-closing reaction in a solvent or in the absence of a solvent in the presence of a base. The solvent is not particularly limited. For example, tetrahydrofuran, ethylene glycol dimethyl ether, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, methyl ethyl ketone; ethyl acetate, acetic acid Esters such as propyl, isopropyl acetate, and isobutyl acetate; alcohols such as methanol, ethanol, 1-propanol, and 2-propanol can be used alone or in combination, among which ethanol, ethanol / water, ethylene glycol dimethyl ether, Ethylene glycol dimethyl ether / water, dimethyl sulfoxide, and dimethyl sulfoxide / water are preferred. The base is not particularly limited. For example, alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride; alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide; Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate; metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium; lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide; n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium; triethylamine, N, N-diisopropylethylamine, N-methylmorpholine, pyridine, 3,4-lutidine, 2,6-lutidine, 2,4,6-collidine, etc. Among them, sodium hydroxide, potassium hydroxide, sodium carbonate, and potassium carbonate are preferable. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., and 1 minute to 48 hours, preferably 1 hour to 24 hours.
工程2:本工程は、化合物(3)と化合物(4)を溶媒中、縮合剤の存在下、反応促進剤の存在下又は非存在下、塩基の存在下又は非存在下で反応することにより、ヒダントイン誘導体(1)を製造する工程である。溶媒としては、特に制限は無いが、例えばN,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、アセトニトリル、ニトロメタン、アセトン、酢酸エチル、ベンゼン、クロロベンゼン、トルエン、クロロホルム、塩化メチレン等を用いることができ、中でもジメチルスルホキシド、アセトニトリル、塩化メチレンが好ましい。縮合剤としては、特に制限は無いが、例えばジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(EDC)、ジイソプロピルカルボジイミド(DIPCDI)等のカルボジイミド系試薬、(1H-ベンゾトリアゾール-1-イルオキシ)トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスファート(BOP)、(1H-ベンゾトリアゾール-1-イルオキシ)トリス(ピロリジノ)ホスホニウムヘキサフルオロホスファート(PyBOP)、1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ(4,5-b)ピリジウム-3-オキソドヘキサフルオロホスファート(HATU)、1-[ビス(ジメチルアミノ)メチレン]-1H-ベンゾトリアゾリウム-3-オキシドヘキサフルオロホスファート(HBTU)、1-[ビス(ジメチルアミノ)メチレン]-1H-ベンゾトリアゾリウム-3-オキシドテトラフルオロボラート(TBTU)、1-[ビス(ジメチルアミノ)メチレン]-5-クロロ-1H-ベンゾトリアゾリウム-3-オキシドヘキサフルオロホスファート(HCTU)、1-[ビス(ジメチルアミノ)メチレン]-5-クロロ-1H-ベンゾトリアゾリウム-3-オキシドテトラフルオロボラート(TCTU)等のホスホニウム塩型あるいはグアニジウム塩型試薬が挙げられ、このうち1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(EDC)、(1H-ベンゾトリアゾール-1-イルオキシ)トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスファート(BOP)、(1H-ベンゾトリアゾール-1-イルオキシ)トリス(ピロリジノ)ホスホニウムヘキサフルオロホスファート(PyBOP)、1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ(4,5-b)ピリジウム-3-オキソドヘキサフルオロホスファート(HATU)、1-[ビス(ジメチルアミノ)メチレン]-1H-ベンゾトリアゾリウム-3-オキシドヘキサフルオロホスファート(HBTU)が好ましい。反応促進剤としては、特に制限は無いが、例えば1-ヒドロキシベンゾトリアゾール(HOBt)、6-クロロ-1-ヒドロキシベンゾトリアゾール(6-Cl-HOBt)、3,4-ジヒドロ-3-ヒドロキシ-4-オキソ―1,2,3―ベンゾトリアジン(HOOBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)等を使用することができる。塩基としては、特に制限はないが、例えば水素化リチウム、水素化ナトリウム、水素化カリウム等の水素化アルカリ金属類;水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類;ナトリウムメトキシド、カリウムメトキシド、ナトリウムエトキシド、カリウムエトキシド、tert-ブトキシナトリウム、tert-ブトキシカリウム等のアルコールの金属塩類;リチウムジイソプロピルアミド、ナトリウムジイソプロピルアミド、カリウムジイソプロピルアミド、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド等の金属アミド類;n-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム等の有機金属化合物類;トリエチルアミン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、3,4-ルチジン、2,6-ルチジン、2,4,6-コリジン等を使用することができる。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~5日間、好ましくは1時間~3日間である。
Step 2: In this step, compound (3) and compound (4) are reacted in a solvent in the presence of a condensing agent, in the presence or absence of a reaction accelerator, and in the presence or absence of a base. This is a process for producing a hydantoin derivative (1). The solvent is not particularly limited. For example, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, tetrahydrofuran, dioxane, acetonitrile, nitromethane, acetone, ethyl acetate, benzene, chlorobenzene, toluene, chloroform, methylene chloride, etc. Among them, dimethyl sulfoxide, acetonitrile, and methylene chloride are preferable. The condensing agent is not particularly limited. For example, carbodiimide reagents such as dicyclohexylcarbodiimide (DCC), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC), diisopropylcarbodiimide (DIPCDI), (1H- Benzotriazol-1-yloxy) tris (dimethylamino) phosphonium hexafluorophosphate (BOP), (1H-benzotriazol-1-yloxy) tris (pyrrolidino) phosphonium hexafluorophosphate (PyBOP), 1- [bis (dimethyl) Amino) methylene] -1H-1,2,3-triazolo (4,5-b) pyridium-3-oxodohexafluorophosphate (HATU), 1- [bis (dimethylamino) methylene] -1H-benzotriazoly 3-oxide hexafluorophosphate (HBTU), 1- [bis (dimethylamino) methylene] -1H-benzotriazolium-3-oxide tetrafluoroborate (TBTU), 1- [bis (dimethylamino) Methylene] -5-chloro-1H-benzotriazolium-3-oxide hexafluorophosphate (HCTU), 1- [bis (dimethylamino) methylene] -5-chloro-1H-benzotriazolium-3-oxide Examples thereof include phosphonium salt or guanidinium salt type reagents such as tetrafluoroborate (TCTU), among which 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC), (1H-benzotriazol-1-yloxy) ) Tris (dimethylamino) phosphonium hexafluorophor Sulfate (BOP), (1H-benzotriazol-1-yloxy) tris (pyrrolidino) phosphonium hexafluorophosphate (PyBOP), 1- [bis (dimethylamino) methylene] -1H-1,2,3-triazolo (4 , 5-b) pyridium-3-oxodohexafluorophosphate (HATU), 1- [bis (dimethylamino) methylene] -1H-benzotriazolium-3-oxide hexafluorophosphate (HBTU). The reaction accelerator is not particularly limited. For example, 1-hydroxybenzotriazole (HOBt), 6-chloro-1-hydroxybenzotriazole (6-Cl-HOBt), 3,4-dihydro-3-hydroxy-4 -Oxo-1,2,3-benzotriazine (HOOBt), 1-hydroxy-7-azabenzotriazole (HOAt) and the like can be used. The base is not particularly limited. For example, alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride; alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide; Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate; metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium; lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide; n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium; triethylamine, N, N-diisopropylethylamine, N-methylmorpholine, pyridine, 3,4-lutidine, 2,6-lutidine, 2,4,6-collidine, etc. Can be used. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
また本工程は、化合物(3)と化合物(4)を溶媒中、ホスフィン試薬の存在下、アゾ系試薬又はエチレンジカルボン酸試薬で反応する光延反応を適応することによっても、ヒダントイン誘導体(1)を製造することができる。溶媒としては、特に制限は無いが、例えばN,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、アセトニトリル、ニトロメタン、アセトン、酢酸エチル、ベンゼン、クロロベンゼン、トルエン、クロロホルム、塩化メチレン等を用いることができる。ホスフィン試薬としては、トリメチルホスフィン、トリエチルホスフィン、トリプロピルホスフィン、トリイソプロピルホスフィン、トリブチルホスフィン、トリイソブチルホスフィン、トリシクロへキシルホスフィン等のトリアルキルホスフィン及びトリフェニルホスフィン、ジフェニルホスフィノフェニルポリスチレン等のトリアリールホスフィン等を用いることができる。アゾ系試薬又はエチレンジカルボン酸試薬としては、アゾジカルボン酸ジエチル(DEAD)、アゾジカルボン酸ジイソプロピル(DIAD)、1,1’-アゾビス(N,N-ジメチルホルムアミド)(TMAD)、1,1’-(アゾジカルボニル)ジピペリジン(ADDP)、1,1’-アゾビス(N,N-ジイソプロピルホルムアミド)(TIPA)、1,6-ジメチル-1,5,7-ヘキサヒドロ-1,4,6,7-テトラゾシン-2,5-ジオン(DHTD)等を用いることができる。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~5日間、好ましくは1時間~3日間である。
This step can also be carried out by adapting Mitsunobu reaction in which compound (3) and compound (4) are reacted with an azo reagent or ethylenedicarboxylic acid reagent in a solvent in the presence of a phosphine reagent. Can be manufactured. The solvent is not particularly limited. For example, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, tetrahydrofuran, dioxane, acetonitrile, nitromethane, acetone, ethyl acetate, benzene, chlorobenzene, toluene, chloroform, methylene chloride, etc. Can be used. Examples of phosphine reagents include trialkylphosphine such as trimethylphosphine, triethylphosphine, tripropylphosphine, triisopropylphosphine, tributylphosphine, triisobutylphosphine, and tricyclohexylphosphine, and triarylphosphine such as diphenylphosphinophenylpolystyrene Etc. can be used. Examples of the azo reagent or ethylenedicarboxylic acid reagent include diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD), 1,1′-azobis (N, N-dimethylformamide) (TMAD), 1,1′- (Azodicarbonyl) dipiperidine (ADDP), 1,1′-azobis (N, N-diisopropylformamide) (TIPA), 1,6-dimethyl-1,5,7-hexahydro-1,4,6,7- Tetrazocine-2,5-dione (DHTD) or the like can be used. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
また本工程は、化合物(3)を溶媒中、酸ハロゲン化剤と反応して酸ハライド誘導体を製造し、その後、これと化合物(4)とを溶媒中、塩基の存在下又は非存在下で反応させることによっても、ヒダントイン誘導体(1)を製造することができる。酸ハロゲン化剤としては、三フッ化N,N-ジエチルアミノ硫黄(DAST)、四フッ化セレン又はそのピリジン付加物、塩化チオニル、塩化オキサリル、ピロカテキルホスホ三塩化物、ジクロロトリフェニルホスホラン、臭化チオニル、ジブロモトリフェニルホスホラン、1-ジメチル-1-ヨード-2-メチルプロペン等を使用することができる。溶媒としては、特に制限は無いが、例えばトルエン、N,N-ジメチルホルムアミド、N-メチルピロリドン、アセトニトリル、ジクロロメタン、1,2-ジクロロエタン等を単独又は組み合わせて使用することができる。塩基としては、特に制限はないが、例えば水素化リチウム、水素化ナトリウム、水素化カリウム等の水素化アルカリ金属類;水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類;ナトリウムメトキシド、カリウムメトキシド、ナトリウムエトキシド、カリウムエトキシド、tert-ブトキシナトリウム、tert-ブトキシカリウム等のアルコールの金属塩類;リチウムジイソプロピルアミド、ナトリウムジイソプロピルアミド、カリウムジイソプロピルアミド、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド等の金属アミド類;n-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム等の有機金属化合物類;トリエチルアミン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、3,4-ルチジン、2,6-ルチジン、2,4,6-コリジン等を使用することができる。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~5日間、好ましくは1時間~3日間である。
In this step, compound (3) is reacted with an acid halogenating agent in a solvent to produce an acid halide derivative, and this is then combined with compound (4) in a solvent in the presence or absence of a base. The hydantoin derivative (1) can also be produced by reacting. Examples of the acid halogenating agent include N, N-diethylaminosulfur trifluoride (DAST), selenium tetrafluoride or its pyridine adduct, thionyl chloride, oxalyl chloride, pyrocatekylphosphotrichloride, dichlorotriphenylphosphorane, Thionyl bromide, dibromotriphenylphosphorane, 1-dimethyl-1-iodo-2-methylpropene and the like can be used. The solvent is not particularly limited, and for example, toluene, N, N-dimethylformamide, N-methylpyrrolidone, acetonitrile, dichloromethane, 1,2-dichloroethane and the like can be used alone or in combination. The base is not particularly limited. For example, alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride; alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide; Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate; metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium; lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide; n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium; triethylamine, N, N-diisopropylethylamine, N-methylmorpholine, pyridine, 3,4-lutidine, 2,6-lutidine, 2,4,6-collidine, etc. Can be used. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
本発明に用いる化合物(4)は、その製造方法は特に限定しないが、例えば、下記に示した方法で合成することができる。
The production method of the compound (4) used in the present invention is not particularly limited, but can be synthesized, for example, by the method shown below.

(式中、R1、R2、R3および*は、前記と同じものを示し、P1は、アミンの保護基を示し、R7は、C1-3アルキル基を示し、R8は、水素原子又はメチル基を示し、R9は、ハロゲン原子又はペンタフルオロフェノキシ基等の脱離基を示し、Xはハロゲン原子を示し、波線は単結合であって、それが結合している二重結合についての立体配置が、それぞれ独立して、E配置若しくはZ配置、又はそれらの混合であることを示す)
(Wherein R 1 , R 2 , R 3 and * represent the same as above, P 1 represents an amine protecting group, R 7 represents a C 1-3 alkyl group, and R 8 represents Represents a hydrogen atom or a methyl group, R 9 represents a leaving group such as a halogen atom or a pentafluorophenoxy group, X represents a halogen atom, the wavy line is a single bond, and (The configuration for a heavy bond is independently an E configuration or a Z configuration, or a mixture thereof)
工程3:本工程は、化合物(5)を溶媒中、塩基の存在下又は非存在下、化合物(6)と反応させることにより化合物(7)を製造する工程である。化合物(6)は、市販されているものを使用することができるほか、例えば、WO2010/125811号パンフレットに記載の方法に従って製造されたものを使用することができるが、特にこれに限定されるものではない。溶媒としては、特に制限は無いが、例えばテトラヒドロフラン、トルエン、ジオキサン、N,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、アセトニトリル、プロピオニトリル、アセトン、メチルエチルケトン、水等を単独又は組み合わせて使用することができる。塩基としては、特に制限はないが、例えば水素化リチウム、水素化ナトリウム、水素化カリウム等の水素化アルカリ金属類、水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類;ナトリウムメトキシド、カリウムメトキシド、ナトリウムエトキシド、カリウムエトキシド、tert-ブトキシナトリウム、tert-ブトキシカリウム等のアルコールの金属塩類;リチウムジイソプロピルアミド、ナトリウムジイソプロピルアミド、カリウムジイソプロピルアミド、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド等の金属アミド類;n-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム等の有機金属化合物類;フッ化リチウム、塩化リチウム、臭化リチウム、ヨウ化リチウム、フッ化ナトリウム、塩化ナトリウム、臭化ナトリウム、ヨウ化ナトリウム、フッ化カリウム、塩化カリウム、臭化カリウム、ヨウ化カリウム、フッ化セシウム、塩化セシウム、臭化セシウム、ヨウ化セシウム等のハロゲン化アルカリ金属類;トリエチルアミン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、3,4-ルチジン、2,6-ルチジン、2,4,6-コリジン等の有機アミン類、中でもトリエチルアミン、N,N-ジイソプロピルエチルアミンが好ましい。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~5日間、好ましくは1時間~3日間である。
Step 3: This step is a step for producing a compound (7) by reacting the compound (5) with a compound (6) in a solvent in the presence or absence of a base. As the compound (6), a commercially available compound can be used, and for example, a compound produced according to the method described in WO2010 / 12581 can be used, but the compound (6) is particularly limited to this. is not. The solvent is not particularly limited, but for example, tetrahydrofuran, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, methyl ethyl ketone, water, etc. are used alone or in combination. can do. Although there is no restriction | limiting in particular as a base, For example, alkali metal hydrides, such as lithium hydride, sodium hydride, potassium hydride, Alkali metal hydroxides, such as lithium hydroxide, sodium hydroxide, potassium hydroxide; Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate; metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium; lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide; n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium; lithium fluoride, lithium chloride, lithium bromide, lithium iodide, sodium fluoride, sodium chloride, sodium bromide, sodium iodide, potassium fluoride, potassium chloride Alkali metal halides such as potassium bromide, potassium iodide, cesium fluoride, cesium chloride, cesium bromide, cesium iodide; triethylamine, N, N-diisopropylethylamine, N-methylmorpholine, pyridine, 3,4 -Organic amines such as lutidine, 2,6-lutidine, 2,4,6-collidine, among them, triethylamine and N, N-diisopropylethylamine are preferred. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
工程4:本工程は、化合物(7)を溶媒中、塩基の存在下又は非存在下、Wittig試薬又はHorner-Wadsworth-Emmons(HWE)試薬を反応させることにより化合物(8)を製造する工程である。溶媒としては、特に制限は無いが、例えばN,N-ジメチルホルムアミド、テトラヒドロフラン、ジオキサン、アセトニトリル、ニトロメタン、アセトン、酢酸エチル、ベンゼン、クロロベンゼン、トルエン、クロロホルム、塩化メチレン等を用いることができ、中でも、テトラヒドロフラン、酢酸エチル、トルエン、クロロホルム、塩化メチレンが好ましい。Wittig試薬としては、安定イリド、不安定イリド(臭化メチルトリフェニルホスホニウム、臭化エチルトリフェニルホスホニウム)等のホスホニウム塩を用いることができる。またHWE試薬としては、ホスホン酸エステルを用いることができる。塩基としては、特に制限はないが、例えば水素化リチウム、水素化ナトリウム、水素化カリウム等の水素化アルカリ金属類;水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類、ナトリウムメトキシド、カリウムメトキシド、ナトリウムエトキシド、カリウムエトキシド、tert-ブトキシナトリウム、tert-ブトキシカリウム等のアルコールの金属塩類;リチウムジイソプロピルアミド、ナトリウムジイソプロピルアミド、カリウムジイソプロピルアミド、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド等の金属アミド類;n-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム等の有機金属化合物類;塩化リチウム、臭化リチウム、ヨウ化リチウム、フッ化ナトリウム、塩化ナトリウム、臭化ナトリウム、ヨウ化ナトリウム、塩化カリウム、臭化カリウム、ヨウ化カリウム、塩化セシウム、臭化セシウム、ヨウ化セシウム等のハロゲン化アルカリ金属類等を使用することができる。反応条件としては、-80℃~150℃、好ましくは0℃~100℃にて、1分~5日間、好ましくは1時間~3日間である。
Step 4: This step is a step for producing compound (8) by reacting compound (7) with a Wittig reagent or Horner-Wadsworth-Emmons (HWE) reagent in a solvent in the presence or absence of a base. is there. The solvent is not particularly limited, and for example, N, N-dimethylformamide, tetrahydrofuran, dioxane, acetonitrile, nitromethane, acetone, ethyl acetate, benzene, chlorobenzene, toluene, chloroform, methylene chloride and the like can be used. Tetrahydrofuran, ethyl acetate, toluene, chloroform and methylene chloride are preferred. As the Wittig reagent, phosphonium salts such as stable ylide and unstable ylide (methyltriphenylphosphonium bromide, ethyltriphenylphosphonium bromide) can be used. As the HWE reagent, a phosphonic acid ester can be used. The base is not particularly limited. For example, alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride; alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide; Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate, metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium; lithium Metal amides such as diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide; n-butyllithium, sec- Organometallic compounds such as til lithium and tert-butyl lithium; lithium chloride, lithium bromide, lithium iodide, sodium fluoride, sodium chloride, sodium bromide, sodium iodide, potassium chloride, potassium bromide, potassium iodide Alkali metal halides such as cesium chloride, cesium bromide, and cesium iodide can be used. The reaction conditions are −80 ° C. to 150 ° C., preferably 0 ° C. to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
工程5:本工程は、化合物(8)を溶媒中、金属炭素存在下、水素雰囲気下に反応させることにより化合物(9)を製造する工程である。溶媒としては、特に制限は無いが、トルエン、酢酸メチルや酢酸エチル等のエステル類等、メタノール、エタノール、1-プロパノール、2-プロパノール等のアルコール類等を単独又は組み合わせて使用することができる。金属炭素としては、例えば、パラジウム炭素、白金炭素、ロジウム炭素、ルテニウム炭素等が挙げられる。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~5日間、好ましくは1時間~3日間である。
Step 5: This step is a step for producing the compound (9) by reacting the compound (8) in a solvent in the presence of metallic carbon under a hydrogen atmosphere. The solvent is not particularly limited, and may be used alone or in combination with toluene, esters such as methyl acetate and ethyl acetate, and alcohols such as methanol, ethanol, 1-propanol and 2-propanol. Examples of the metal carbon include palladium carbon, platinum carbon, rhodium carbon, ruthenium carbon and the like. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
工程6:本工程は、化合物(9)を溶媒中、塩基又は酸の存在下、反応させることにより化合物(10)を製造する工程である。溶媒としては、特に制限は無いが、例えばテトラヒドロフラン、トルエン、ジオキサン、N,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、アセトニトリル、プロピオニトリル、アセトン、メチルエチルケトン、水やメタノール、エタノール、1-プロパノール、2-プロパノール等のアルコール類等を単独又は組み合わせて使用することができる。塩基としては、特に制限はないが、例えば水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類等を使用することができる。酸としては、特に制限はないが、例えば塩酸、硫酸、酢酸、トシル酸等を使用することができる。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~5日間、好ましくは1時間~3日間である。
Process 6: This process is a process of manufacturing a compound (10) by making a compound (9) react in a solvent in presence of a base or an acid. The solvent is not particularly limited. For example, tetrahydrofuran, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, methyl ethyl ketone, water, methanol, ethanol, 1- Alcohols such as propanol and 2-propanol can be used alone or in combination. The base is not particularly limited. For example, alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide, alkali carbonates such as lithium carbonate, sodium carbonate, potassium carbonate and cesium carbonate are used. can do. Although there is no restriction | limiting in particular as an acid, For example, hydrochloric acid, a sulfuric acid, an acetic acid, a tosylic acid etc. can be used. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
工程7:本工程は、化合物(10)を溶媒中又は無溶媒中で、縮合剤の存在下又は非存在下、反応促進剤の存在下又は非存在下、酸又は塩基の存在下又は非存在下に反応して、化合物(11)を製造する工程である。R9で示される置換基としては、例えば文献(Comprehensive Organic Transformations Second Edition, John Wiley & Sons, Inc.)を参考に、酸ハロゲン化物、酸無水物又はエステルに変換する事ができる。酸ハロゲン化物としては、酸フッ化物、酸塩化物等が挙げられる。酸無水物としては、酢酸等の脂肪族カルボン酸との酸無水物、安息香酸等の芳香族カルボン酸との酸無水物等が挙げられる。エステルとしては、メタノール等の脂肪族アルコールとのエステル、ペンタフルオロフェノール等の芳香族アルコールとのエステル等が挙げられる。
Step 7: This step is a step of removing the compound (10) in a solvent or in the absence of a solvent, in the presence or absence of a condensing agent, in the presence or absence of a reaction accelerator, in the presence or absence of an acid or base. In this step, compound (11) is produced by the reaction below. The substituent represented by R 9 can be converted into an acid halide, an acid anhydride or an ester with reference to, for example, literature (Comprehensive Organic Transformations Second Edition, John Wiley & Sons, Inc.). Examples of acid halides include acid fluorides and acid chlorides. Examples of the acid anhydride include an acid anhydride with an aliphatic carboxylic acid such as acetic acid, an acid anhydride with an aromatic carboxylic acid such as benzoic acid, and the like. Examples of the ester include an ester with an aliphatic alcohol such as methanol, an ester with an aromatic alcohol such as pentafluorophenol, and the like.
R9がペンタフルオロフェノキシ基の場合、溶媒としては、特に制限は無いが、例えばテトラヒドロフラン、トルエン、ジオキサン、N,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、アセトニトリル、プロピオニトリル、アセトン、メチルエチルケトン、酢酸エチル等を単独又は組み合わせて使用することができる。縮合剤としては、特に制限は無いが、例えばジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(EDC)、ジイソプロピルカルボジイミド(DIPCDI)等のカルボジイミド系試薬が挙げられ、中でも、ジシクロヘキシルカルボジイミド、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミドが好ましい。塩基としては、特に制限はないが、例えば水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類;トリエチルアミン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、3,4-ルチジン、2,6-ルチジン、2,4,6-コリジン等の有機アミン類等を使用することができる。酸としては、特に制限はないが、例えば塩酸、硫酸、酢酸、トシル酸等を使用することができる。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~5日間、好ましくは1時間~3日間である。
When R 9 is a pentafluorophenoxy group, the solvent is not particularly limited. For example, tetrahydrofuran, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, Methyl ethyl ketone, ethyl acetate and the like can be used alone or in combination. The condensing agent is not particularly limited, and examples thereof include carbodiimide reagents such as dicyclohexylcarbodiimide (DCC), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC), and diisopropylcarbodiimide (DIPCDI). Of these, dicyclohexylcarbodiimide and 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide are preferable. The base is not particularly limited, and examples thereof include alkali metal hydroxides such as lithium hydroxide, sodium hydroxide, and potassium hydroxide; alkali carbonate metals such as lithium carbonate, sodium carbonate, potassium carbonate, and cesium carbonate; triethylamine, Organic amines such as N, N-diisopropylethylamine, N-methylmorpholine, pyridine, 3,4-lutidine, 2,6-lutidine, 2,4,6-collidine and the like can be used. Although there is no restriction | limiting in particular as an acid, For example, hydrochloric acid, a sulfuric acid, an acetic acid, a tosylic acid etc. can be used. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
工程8:本工程は、化合物(11)を溶媒中、塩基の存在下、トリフルオロメチル化試薬を用いて、ヘキサフルオロカルビノール化合物(12)へと製造する工程である。溶媒としては、特に制限は無いが、例えばジメトキシエタン、テトラヒドロフラン、トルエン、ジオキサン、エチレングリコールジメチルエーテル、N,N-ジメチルホルムアミド、N-メチルピロリドン、テトラメチルウレア、ジメチルスルホキシド、アセトニトリル、プロピオニトリル等を単独又は組み合わせて使用することができ、中でもエチレングリコールジメチルエーテルが好ましい。塩基としては、テトラメチルアンモニウムフルオリド、テトラエチルアンモニウムフルオリド、テトラブチルアンモニウムフルオリド等のテトラアルキルアンモニウム塩や、フッ化リチウム、フッ化ナトリウム、フッ化カリウム、フッ化セシウム等のフッ化アルカリ金属塩等が挙げられる。トリフルオロメチル化試薬としては、(トリフルオロメチル)トリメチルシラン、トリエチル(トリフルオロメチル)シラン、トリイソプロピル(トリフルオロメチル)シラン、メチルジフェニル(トリフルオロメチル)シラン、ジメチル(ジフェニル)トリフルオロメチルシラン等が挙げられる。反応条件としては、-80~150℃、好ましくは-30~50℃にて、1分~5日間、好ましくは1時間~3日間である。
Step 8: This step is a step for producing the compound (11) into the hexafluorocarbinol compound (12) using a trifluoromethylating reagent in a solvent in the presence of a base. The solvent is not particularly limited, and examples thereof include dimethoxyethane, tetrahydrofuran, toluene, dioxane, ethylene glycol dimethyl ether, N, N-dimethylformamide, N-methylpyrrolidone, tetramethylurea, dimethyl sulfoxide, acetonitrile, propionitrile and the like. They can be used alone or in combination, and ethylene glycol dimethyl ether is particularly preferable. Examples of the base include tetraalkylammonium salts such as tetramethylammonium fluoride, tetraethylammonium fluoride and tetrabutylammonium fluoride, and alkali metal fluoride salts such as lithium fluoride, sodium fluoride, potassium fluoride and cesium fluoride. Etc. Trifluoromethylating reagents include (trifluoromethyl) trimethylsilane, triethyl (trifluoromethyl) silane, triisopropyl (trifluoromethyl) silane, methyldiphenyl (trifluoromethyl) silane, dimethyl (diphenyl) trifluoromethylsilane Etc. The reaction conditions are −80 to 150 ° C., preferably −30 to 50 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
工程9:本工程は、化合物(12)のアミノ基を脱保護して化合物(4)を製造する工程である。P1で示される保護基は、例えば文献(Protective Groups in Organic Synthesis Third Edition, John Wiley & Sons, Inc.)を参考にして脱保護を行うことができる。
Process 9: This process is a process of manufacturing a compound (4) by deprotecting the amino group of a compound (12). The protecting group represented by P 1 can be deprotected with reference to literature (Protective Groups in Organic Synthesis Third Edition, John Wiley & Sons, Inc.), for example.
P1がtert-ブトキシカルボニル基の場合は、化合物(12)を溶媒中又は無溶媒で、酸と反応することにより化合物(4)を製造することができる。溶媒としては、特に制限はないが、例えばN,N-ジメチルホルムアミド、テトラヒドロフラン、ジオキサン、アセトニトリル、ニトロメタン、アセトン、酢酸エチル、ベンゼン、クロロベンゼン、トルエン、クロロホルム、塩化メチレン、水やメタノール、エタノール、1-プロパノール、2-プロパノール等を単独又は組み合わせて用いることができる。酸としては、特に制限はないが、塩酸、塩酸/酢酸エチル溶液、塩酸/ジオキサン溶液、塩酸/メタノール溶液、臭化水素酸、硫酸、硝酸等を使用することができ、中でも塩酸/酢酸エチル溶液、塩酸/メタノール溶液が好ましい。反応条件としては、-20℃~100℃、好ましくは0℃~50℃で、1分~24時間、好ましくは5分~12時間である。
When P 1 is a tert-butoxycarbonyl group, compound (4) can be produced by reacting compound (12) with an acid in a solvent or without a solvent. The solvent is not particularly limited. For example, N, N-dimethylformamide, tetrahydrofuran, dioxane, acetonitrile, nitromethane, acetone, ethyl acetate, benzene, chlorobenzene, toluene, chloroform, methylene chloride, water, methanol, ethanol, 1- Propanol, 2-propanol and the like can be used alone or in combination. The acid is not particularly limited, but hydrochloric acid, hydrochloric acid / ethyl acetate solution, hydrochloric acid / dioxane solution, hydrochloric acid / methanol solution, hydrobromic acid, sulfuric acid, nitric acid, etc. can be used. Among them, hydrochloric acid / ethyl acetate solution A hydrochloric acid / methanol solution is preferred. The reaction conditions are −20 ° C. to 100 ° C., preferably 0 ° C. to 50 ° C., for 1 minute to 24 hours, preferably 5 minutes to 12 hours.
本発明に用いる化合物(2)は、その製造方法は特に限定しないが、例えば、下記に示した方法で合成することができる。
The production method of the compound (2) used in the present invention is not particularly limited, but can be synthesized, for example, by the method shown below.

(式中、R4、R5、R6および*は、前記と同じものを示す)
(In the formula, R 4 , R 5 , R 6 and * are the same as described above.)
工程10:本工程は、化合物(13)を溶媒中、水並びに塩基又は酸の存在下、加水分解反応させることにより化合物(14)を製造する工程である。溶媒としては、特に制限は無いが、例えばテトラヒドロフラン、トルエン、ジオキサン、N,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、アセトニトリル、プロピオニトリル、アセトン、メチルエチルケトン、水やメタノール、エタノール、1-プロパノール、2-プロパノール等のアルコール類等を単独又は組み合わせて使用することができる。塩基としては、特に制限はないが、例えば水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類等を使用することができる。酸としては、特に制限はないが、例えば塩酸、硫酸、酢酸、トシル酸等を使用することができる。反応条件としては、-80~200℃、好ましくは0~150℃にて、1分~7日間、好ましくは1時間~3日間である。
Step 10: This step is a step for producing the compound (14) by subjecting the compound (13) to a hydrolysis reaction in a solvent in the presence of water and a base or acid. The solvent is not particularly limited. For example, tetrahydrofuran, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, methyl ethyl ketone, water, methanol, ethanol, 1- Alcohols such as propanol and 2-propanol can be used alone or in combination. The base is not particularly limited. For example, alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide, alkali carbonates such as lithium carbonate, sodium carbonate, potassium carbonate and cesium carbonate are used. can do. Although there is no restriction | limiting in particular as an acid, For example, hydrochloric acid, a sulfuric acid, an acetic acid, a tosylic acid etc. can be used. The reaction conditions are −80 to 200 ° C., preferably 0 to 150 ° C., for 1 minute to 7 days, preferably 1 hour to 3 days.
工程11:本工程は、化合物(14)をアルコール(R5-OH)存在下、その他の溶媒中又は無溶媒中にて、縮合剤の存在下又は非存在下、反応促進剤の存在下又は非存在下、酸又は塩基の存在下又は非存在下に反応して、化合物(15)を製造する工程である。例えば文献(Comprehensive Organic Transformations Second Edition, John Wiley & Sons, Inc.)を参考に、エステルに変換する事ができる。アルコールとしては、メタノール、エタノール等の脂肪族アルコール等が挙げられる。塩基としては、特に制限はないが、例えば水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類等を使用することができる。酸としては、特に制限はないが、例えば塩酸、硫酸、酢酸、トシル酸等を使用することができる。反応条件としては、-80~200℃、好ましくは0~150℃にて、1分~7日間、好ましくは1時間~2日間である。
Step 11: In this step, the compound (14) is added in the presence of an alcohol (R 5 —OH), in another solvent or in the absence of a solvent, in the presence or absence of a condensing agent, in the presence of a reaction accelerator or In this step, the compound (15) is produced by reacting in the absence or presence of an acid or a base. For example, it can be converted into an ester with reference to literature (Comprehensive Organic Transformations Second Edition, John Wiley & Sons, Inc.). Examples of the alcohol include aliphatic alcohols such as methanol and ethanol. The base is not particularly limited. For example, alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide, alkali carbonates such as lithium carbonate, sodium carbonate, potassium carbonate and cesium carbonate are used. can do. Although there is no restriction | limiting in particular as an acid, For example, hydrochloric acid, a sulfuric acid, an acetic acid, a tosylic acid etc. can be used. The reaction conditions are −80 to 200 ° C., preferably 0 to 150 ° C., for 1 minute to 7 days, preferably 1 hour to 2 days.
工程12:本工程は、化合物(15)を溶媒中又は無溶媒中、光学分割剤の存在下にて反応して、化合物(16)を製造する工程である。溶媒としては、特に制限は無いが、例えばテトラヒドロフラン、トルエン、ジオキサン、N,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、アセトニトリル、プロピオニトリル、アセトン、メチルエチルケトン、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸イソブチル等のエステル類、水やメタノール、エタノール、1-プロパノール、2-プロパノール等のアルコール類等を単独又は組み合わせて使用することができる。光学分割剤としては、特に制限は無いが、例えば(L)-(+)-マンデル酸、(D)-(-)-マンデル酸、リンゴ酸、酒石酸、乳酸、カンファースルホン酸、(1S)-(+)-ケトピン酸、又は商業的に入手可能な光学的に純粋な化合物等も使用することができる。反応条件としては、-80~150℃、好ましくは-10~100℃にて、1分~48時間、好ましくは1時間~24時間である。
Step 12: This step is a step for producing a compound (16) by reacting the compound (15) in a solvent or in the absence of a solvent in the presence of an optical resolution agent. The solvent is not particularly limited. For example, tetrahydrofuran, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, methyl ethyl ketone, ethyl acetate, propyl acetate, isopropyl acetate In addition, esters such as isobutyl acetate, alcohols such as water, methanol, ethanol, 1-propanol, and 2-propanol can be used alone or in combination. The optical resolution agent is not particularly limited. For example, (L)-(+)-mandelic acid, (D)-(−)-mandelic acid, malic acid, tartaric acid, lactic acid, camphorsulfonic acid, (1S) — (+)-Ketopic acid or commercially available optically pure compounds can also be used. The reaction conditions are -80 to 150 ° C, preferably -10 to 100 ° C, 1 minute to 48 hours, preferably 1 hour to 24 hours.
工程13:本工程は、化合物(16)を溶媒中又は無溶媒中、塩基の存在下又は非存在下にてイソシアネート誘導体(17)と反応して、化合物(2)を製造する工程である。溶媒としては、特に制限は無いが、例えばテトラヒドロフラン、エチレングリコールジメチルエーテル、トルエン、ジオキサン、N,N-ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド、アセトニトリル、プロピオニトリル、アセトン、メチルエチルケトン、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸イソブチル等のエステル類、メタノール、エタノール、1-プロパノール、2-プロパノール等のアルコール類等を単独又は組み合わせて使用することができる。塩基としては、特に制限はないが、例えば水素化リチウム、水素化ナトリウム、水素化カリウム等の水素化アルカリ金属類;水酸化リチウム、水酸化ナトリウム、水酸化カリウム等の水酸化アルカリ金属類;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸アルカリ金属類;ナトリウムメトキシド、カリウムメトキシド、ナトリウムエトキシド、カリウムエトキシド、tert-ブトキシナトリウム、tert-ブトキシカリウム等のアルコールの金属塩類;リチウムジイソプロピルアミド、ナトリウムジイソプロピルアミド、カリウムジイソプロピルアミド、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド、n-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム、トリエチルアミン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、3,4-ルチジン、2,6-ルチジン、2,4,6-コリジン等を使用することができる。反応条件としては、-80~150℃、好ましくは0~100℃にて、1分~5日間、好ましくは1時間~3日間である。
Step 13: This step is a step for producing a compound (2) by reacting the compound (16) with an isocyanate derivative (17) in a solvent or without a solvent in the presence or absence of a base. The solvent is not particularly limited, but for example, tetrahydrofuran, ethylene glycol dimethyl ether, toluene, dioxane, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, acetonitrile, propionitrile, acetone, methyl ethyl ketone, ethyl acetate, acetic acid Esters such as propyl, isopropyl acetate and isobutyl acetate, alcohols such as methanol, ethanol, 1-propanol and 2-propanol can be used alone or in combination. The base is not particularly limited. For example, alkali metal hydrides such as lithium hydride, sodium hydride and potassium hydride; alkali metal hydroxides such as lithium hydroxide, sodium hydroxide and potassium hydroxide; Alkali metal carbonates such as lithium, sodium carbonate, potassium carbonate, cesium carbonate; metal salts of alcohols such as sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, tert-butoxy sodium, tert-butoxy potassium; lithium Diisopropylamide, sodium diisopropylamide, potassium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide, n-butyllithium, sec-butyllithium, ert- butyl lithium, triethylamine, N, N- diisopropylethylamine, N- methylmorpholine, pyridine, 3,4-lutidine, 2,6-lutidine, can be used 2,4,6-collidine. The reaction conditions are −80 to 150 ° C., preferably 0 to 100 ° C., for 1 minute to 5 days, preferably 1 hour to 3 days.
また、工程13で得られる化合物(2)は、単離精製することなく、継続して工程1の反応を行い、化合物(3)を得ることもできる。
Further, the compound (2) obtained in the step 13 can be continuously subjected to the reaction in the step 1 without isolation and purification to obtain the compound (3).
本発明の一般式(3)で表される光学活性化合物において許容される塩としては、具体的には、無機酸や有機酸との酸付加塩、又は無機塩基や有機塩基との塩基付加塩等が挙げられる。
Specific examples of the salt allowed in the optically active compound represented by the general formula (3) of the present invention include acid addition salts with inorganic acids and organic acids, or base addition salts with inorganic bases and organic bases. Etc.
本発明の一般式(3)で表される光学活性化合物、及びその許容される塩の溶媒和物としては、具体的には水和物や各種の溶媒和物が挙げられる。
Specific examples of the solvate of the optically active compound represented by the general formula (3) of the present invention and its acceptable salt include hydrates and various solvates.
以下に、実施例を挙げて本発明を更に詳細に説明する。
実施例1:(S)-3-(2-{(2S,5R)-4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-イル}-2-オキソエチル)-5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオン((S)-B)の製造 Hereinafter, the present invention will be described in more detail with reference to examples.
Example 1: (S) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione ( Production of (S) -B)
実施例1:(S)-3-(2-{(2S,5R)-4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-イル}-2-オキソエチル)-5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオン((S)-B)の製造 Hereinafter, the present invention will be described in more detail with reference to examples.
Example 1: (S) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione ( Production of (S) -B)

工程I:4-[2-ホルミル-4-(メトキシカルボニル)フェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(c2)の製造:
フッ化セシウム(14.0g,91.89mmol)を減圧下120℃にて2時間乾燥後、アルゴン雰囲気下、室温にてN,N-ジメチルホルムアミド(52mL)を加えた。その後、WO2010/125811号パンフレットに従い製造される2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(14.2g,61.26mmol)のN,N-ジメチルホルムアミド(50mL)溶液、4-フルオロ-3-ホルミル安息香酸メチルエステル(11.15g,61.26mmol)の順に加え、100℃にて18時間攪拌した。反応終結を確認後、0℃にて、反応液にトルエン、水を加えた。その後、トルエンで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物19.57g(収率85%)を黄色アモルファスとして得た。 Step I: Preparation of 4- [2-formyl-4- (methoxycarbonyl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (c2):
Cesium fluoride (14.0 g, 91.89 mmol) was dried under reduced pressure at 120 ° C. for 2 hours, and N, N-dimethylformamide (52 mL) was added at room temperature under an argon atmosphere. Thereafter, N, N-dimethylformamide (50 mL) of 2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (14.2 g, 61.26 mmol) produced according to the pamphlet of WO2010 / 12581 The solution and 4-fluoro-3-formylbenzoic acid methyl ester (11.15 g, 61.26 mmol) were added in this order, and the mixture was stirred at 100 ° C. for 18 hours. After confirming the completion of the reaction, toluene and water were added to the reaction solution at 0 ° C. Thereafter, extraction with toluene was performed, and the organic layer was washed with saturated brine, dried using anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 19.57 g (yield 85%) of the title compound as a yellow amorphous.
フッ化セシウム(14.0g,91.89mmol)を減圧下120℃にて2時間乾燥後、アルゴン雰囲気下、室温にてN,N-ジメチルホルムアミド(52mL)を加えた。その後、WO2010/125811号パンフレットに従い製造される2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(14.2g,61.26mmol)のN,N-ジメチルホルムアミド(50mL)溶液、4-フルオロ-3-ホルミル安息香酸メチルエステル(11.15g,61.26mmol)の順に加え、100℃にて18時間攪拌した。反応終結を確認後、0℃にて、反応液にトルエン、水を加えた。その後、トルエンで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物19.57g(収率85%)を黄色アモルファスとして得た。 Step I: Preparation of 4- [2-formyl-4- (methoxycarbonyl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (c2):
Cesium fluoride (14.0 g, 91.89 mmol) was dried under reduced pressure at 120 ° C. for 2 hours, and N, N-dimethylformamide (52 mL) was added at room temperature under an argon atmosphere. Thereafter, N, N-dimethylformamide (50 mL) of 2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (14.2 g, 61.26 mmol) produced according to the pamphlet of WO2010 / 12581 The solution and 4-fluoro-3-formylbenzoic acid methyl ester (11.15 g, 61.26 mmol) were added in this order, and the mixture was stirred at 100 ° C. for 18 hours. After confirming the completion of the reaction, toluene and water were added to the reaction solution at 0 ° C. Thereafter, extraction with toluene was performed, and the organic layer was washed with saturated brine, dried using anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 19.57 g (yield 85%) of the title compound as a yellow amorphous.
1H-NMR (CDCl3) δ:1.00 (3H, d, J = 6.0 Hz), 1.28 (3H, d, J = 6.4 Hz), 1.49 (9H, s), 2.86 (1H, d, J = 11.6 Hz), 3.63-3.70 (3H, m), 3.77 (1H, d, J = 12.6 Hz), 3.91 (3H, s), 4.44-4.49 (1H, m), 6.99 (1H, d, J = 8.8 Hz), 8.13 (1H, dd, J = 2.4, 8.8 Hz), 8.45 (1H, d, J = 2.4 Hz), 10.12 (1H, s).
1 H-NMR (CDCl 3 ) δ: 1.00 (3H, d, J = 6.0 Hz), 1.28 (3H, d, J = 6.4 Hz), 1.49 (9H, s), 2.86 (1H, d, J = 11.6 Hz), 3.63-3.70 (3H, m), 3.77 (1H, d, J = 12.6 Hz), 3.91 (3H, s), 4.44-4.49 (1H, m), 6.99 (1H, d, J = 8.8 Hz ), 8.13 (1H, dd, J = 2.4, 8.8 Hz), 8.45 (1H, d, J = 2.4 Hz), 10.12 (1H, s).
工程II:4-[4-(メトキシカルボニル)-2-(プロパ-1-エン-1-イル)フェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(c3)の製造:
臭化エチルトリフェニルホスホニウム(25.8g,69.46mmol)をテトラヒドロフラン(140mL)に溶解させ、アルゴン雰囲気下、0℃にてカリウムメトキシド(4.87g,69.46mmol)を加えた。0℃にて1時間30分攪拌後、4-[2-ホルミル-4-(メトキシカルボニル)フェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(5.23g,13.89mmol)のテトラヒドロフラン(140mL)溶液を加え、室温にて2時間30分攪拌した。反応終結を確認後、0℃にて反応液に水を加えた。その後、トルエンで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物4.76g(収率88%、cis/trans=0.7/1)を黄色アモルファスとして得た。 Step II: 4- [4- (Methoxycarbonyl) -2- (prop-1-en-1-yl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl ( c3) Production:
Ethyltriphenylphosphonium bromide (25.8 g, 69.46 mmol) was dissolved in tetrahydrofuran (140 mL), and potassium methoxide (4.87 g, 69.46 mmol) was added at 0 ° C. under an argon atmosphere. After stirring at 0 ° C. for 1 hour and 30 minutes, 4- [2-formyl-4- (methoxycarbonyl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (5. (23 g, 13.89 mmol) in tetrahydrofuran (140 mL) was added, and the mixture was stirred at room temperature for 2 hours 30 minutes. After confirming the completion of the reaction, water was added to the reaction solution at 0 ° C. Thereafter, extraction with toluene was performed, and the organic layer was washed with saturated brine, dried using anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain 4.76 g (yield 88%, cis / trans = 0.7 / 1) of the title compound as a yellow amorphous.
臭化エチルトリフェニルホスホニウム(25.8g,69.46mmol)をテトラヒドロフラン(140mL)に溶解させ、アルゴン雰囲気下、0℃にてカリウムメトキシド(4.87g,69.46mmol)を加えた。0℃にて1時間30分攪拌後、4-[2-ホルミル-4-(メトキシカルボニル)フェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(5.23g,13.89mmol)のテトラヒドロフラン(140mL)溶液を加え、室温にて2時間30分攪拌した。反応終結を確認後、0℃にて反応液に水を加えた。その後、トルエンで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物4.76g(収率88%、cis/trans=0.7/1)を黄色アモルファスとして得た。 Step II: 4- [4- (Methoxycarbonyl) -2- (prop-1-en-1-yl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl ( c3) Production:
Ethyltriphenylphosphonium bromide (25.8 g, 69.46 mmol) was dissolved in tetrahydrofuran (140 mL), and potassium methoxide (4.87 g, 69.46 mmol) was added at 0 ° C. under an argon atmosphere. After stirring at 0 ° C. for 1 hour and 30 minutes, 4- [2-formyl-4- (methoxycarbonyl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (5. (23 g, 13.89 mmol) in tetrahydrofuran (140 mL) was added, and the mixture was stirred at room temperature for 2 hours 30 minutes. After confirming the completion of the reaction, water was added to the reaction solution at 0 ° C. Thereafter, extraction with toluene was performed, and the organic layer was washed with saturated brine, dried using anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain 4.76 g (yield 88%, cis / trans = 0.7 / 1) of the title compound as a yellow amorphous.
cis-c3: 4-{4-(メトキシカルボニル)-2-[(Z)-プロパ-1-エン-1-イル]フェニル}-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル

cis-c3: 4- {4- (methoxycarbonyl) -2-[(Z) -prop-1-en-1-yl] phenyl} -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl
1H-NMR (CDCl3) δ:0.87 (3H, d, J = 6.8 Hz), 1.30 (3H, d, J = 6.8 Hz), 1.48 (9H,s), 1.90-1.92 (3H, m), 2.71 (1H, d, J = 11.6 Hz), 3.41-3.44 (1H, m), 3.53-3.58 (1H, m), 3.69-3.72 (2H, m), 3.86 (3H, s), 4.41-4.46 (1H, m), 5.81 (1H, dd, J = 6.8, 11.4 Hz), 6.42 (1H, dd, J = 1.6, 11.4 Hz), 6.84 (1H, d, J = 8.6 Hz), 7.84 (1H, dd, J = 2.0, 8.6 Hz), 7.90 (1H, d, J = 1.6 Hz).
1 H-NMR (CDCl 3 ) δ: 0.87 (3H, d, J = 6.8 Hz), 1.30 (3H, d, J = 6.8 Hz), 1.48 (9H, s), 1.90-1.92 (3H, m), 2.71 (1H, d, J = 11.6 Hz), 3.41-3.44 (1H, m), 3.53-3.58 (1H, m), 3.69-3.72 (2H, m), 3.86 (3H, s), 4.41-4.46 ( 1H, m), 5.81 (1H, dd, J = 6.8, 11.4 Hz), 6.42 (1H, dd, J = 1.6, 11.4 Hz), 6.84 (1H, d, J = 8.6 Hz), 7.84 (1H, dd , J = 2.0, 8.6 Hz), 7.90 (1H, d, J = 1.6 Hz).
trans-c3: 4-{4-(メトキシカルボニル)-2-[(E)-プロパ-1-エン-1-イル]フェニル}-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル

trans-c3: 4- {4- (methoxycarbonyl) -2-[(E) -prop-1-en-1-yl] phenyl} -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl
1H-NMR (CDCl3) δ:0.85 (3H, d, J = 6.8 Hz), 1.26 (3H, d, J = 6.8 Hz), 1.46 (9H,s), 1.87-1.89 (3H, m), 2.67 (1H, d, J = 12.0 Hz), 3.39-3.59 (3H, m), 3.67-3.73 (1H, m), 3.86 (3H, s), 4.41 (1H, brs), 6.25 (1H, dd, J = 6.4, 15.6 Hz), 6.54 (1H, dd, J = 1.6, 15.6 Hz), 6.80 (1H, d, J = 8.8 Hz), 7.80 (1H, dd, J = 1.6, 8.8 Hz), 8.03 (1H, d, J = 1.6 Hz).
1 H-NMR (CDCl 3 ) δ: 0.85 (3H, d, J = 6.8 Hz), 1.26 (3H, d, J = 6.8 Hz), 1.46 (9H, s), 1.87-1.89 (3H, m), 2.67 (1H, d, J = 12.0 Hz), 3.39-3.59 (3H, m), 3.67-3.73 (1H, m), 3.86 (3H, s), 4.41 (1H, brs), 6.25 (1H, dd, J = 6.4, 15.6 Hz), 6.54 (1H, dd, J = 1.6, 15.6 Hz), 6.80 (1H, d, J = 8.8 Hz), 7.80 (1H, dd, J = 1.6, 8.8 Hz), 8.03 ( (1H, d, J = 1.6 Hz).
工程III:4-[4-(メトキシカルボニル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(c4)の製造:
4-[4-(メトキシカルボニル) -2-(プロパ-1-エン-1-イル)フェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(4.76g,12.25mmol)をメタノール(123mL)に溶解させ、アルゴン雰囲気下、パラジウム炭素を加えた。その後、水素雰囲気下、室温にて24時間攪拌した。反応終結を確認後、セライトろ過し、酢酸エチルで洗浄し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物4.72g(収率99%)を無色油状物として得た。 Step III: Preparation of 4- [4- (methoxycarbonyl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (c4):
4- [4- (Methoxycarbonyl) -2- (prop-1-en-1-yl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (4.76 g) , 12.25 mmol) was dissolved in methanol (123 mL), and palladium on carbon was added under an argon atmosphere. Thereafter, the mixture was stirred at room temperature for 24 hours under a hydrogen atmosphere. After confirming the completion of the reaction, the mixture was filtered through Celite, washed with ethyl acetate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (4.72 g, yield 99%) as a colorless oil.
4-[4-(メトキシカルボニル) -2-(プロパ-1-エン-1-イル)フェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(4.76g,12.25mmol)をメタノール(123mL)に溶解させ、アルゴン雰囲気下、パラジウム炭素を加えた。その後、水素雰囲気下、室温にて24時間攪拌した。反応終結を確認後、セライトろ過し、酢酸エチルで洗浄し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物4.72g(収率99%)を無色油状物として得た。 Step III: Preparation of 4- [4- (methoxycarbonyl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (c4):
4- [4- (Methoxycarbonyl) -2- (prop-1-en-1-yl) phenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (4.76 g) , 12.25 mmol) was dissolved in methanol (123 mL), and palladium on carbon was added under an argon atmosphere. Thereafter, the mixture was stirred at room temperature for 24 hours under a hydrogen atmosphere. After confirming the completion of the reaction, the mixture was filtered through Celite, washed with ethyl acetate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (4.72 g, yield 99%) as a colorless oil.
1H-NMR (CDCl3) δ:0.91 (3H, d, J = 6.4 Hz), 0.97 (3H, t, J = 7.2 Hz), 1.30 (3H, d, J = 6.4 Hz), 1.49 (9H, s), 1.70 (2H, tq, J = 7.2, 7.6 Hz), 2.49-2.56 (2H, m), 2.79 (1H, td, J = 7.6, 14.4 Hz), 3.35-3.40 (1H, m), 3.51 (1H, dd, J = 3.2, 13.2 Hz), 3.61 (1H, dd, J = 4.0, 11.4 Hz), 3.74 (1H, dd, J = 1.6, 13.2 Hz), 3.88 (3H, s), 4.38-4.44 (1H, m), 6.90 (1H, d, J = 8.0 Hz), 7.80 (1H, dd, J = 2.0, 8.0 Hz), 7.89 (1H, d, J = 2.0 Hz).
1 H-NMR (CDCl 3 ) δ: 0.91 (3H, d, J = 6.4 Hz), 0.97 (3H, t, J = 7.2 Hz), 1.30 (3H, d, J = 6.4 Hz), 1.49 (9H, s), 1.70 (2H, tq, J = 7.2, 7.6 Hz), 2.49-2.56 (2H, m), 2.79 (1H, td, J = 7.6, 14.4 Hz), 3.35-3.40 (1H, m), 3.51 (1H, dd, J = 3.2, 13.2 Hz), 3.61 (1H, dd, J = 4.0, 11.4 Hz), 3.74 (1H, dd, J = 1.6, 13.2 Hz), 3.88 (3H, s), 4.38- 4.44 (1H, m), 6.90 (1H, d, J = 8.0 Hz), 7.80 (1H, dd, J = 2.0, 8.0 Hz), 7.89 (1H, d, J = 2.0 Hz).
工程IV:4-[(2R,5S)-4-(tert-ブトキシカルボニル)-2,5-ジメチルピペラジン-1-イル]-3-プロピル安息香酸(c5)の製造:
4-[4-(メトキシカルボニル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(4.72g,12.09mmol)をメタノール(121mL)に溶解させ、0℃にて4N-水酸化ナトリウム水溶液(18.1mL,72.52mmol)を加え、60℃にて2時間攪拌した。反応終結を確認後、0℃にて反応液に塩化アンモニウム水溶液を用いて中和した(pH=7)。その後、クロロホルムで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮し、表題化合物4.48g(収率99%)を無色アモルファスとして得た。 Step IV: Preparation of 4-[(2R, 5S) -4- (tert-butoxycarbonyl) -2,5-dimethylpiperazin-1-yl] -3-propylbenzoic acid (c5):
4- [4- (Methoxycarbonyl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (4.72 g, 12.09 mmol) in methanol (121 mL) 4N-aqueous sodium hydroxide solution (18.1 mL, 72.52 mmol) was added at 0 ° C., and the mixture was stirred at 60 ° C. for 2 hours. After confirming the completion of the reaction, the reaction solution was neutralized with an aqueous ammonium chloride solution at 0 ° C. (pH = 7). Thereafter, the mixture was extracted with chloroform, and the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain 4.48 g (yield 99%) of the title compound as a colorless amorphous.
4-[4-(メトキシカルボニル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(4.72g,12.09mmol)をメタノール(121mL)に溶解させ、0℃にて4N-水酸化ナトリウム水溶液(18.1mL,72.52mmol)を加え、60℃にて2時間攪拌した。反応終結を確認後、0℃にて反応液に塩化アンモニウム水溶液を用いて中和した(pH=7)。その後、クロロホルムで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮し、表題化合物4.48g(収率99%)を無色アモルファスとして得た。 Step IV: Preparation of 4-[(2R, 5S) -4- (tert-butoxycarbonyl) -2,5-dimethylpiperazin-1-yl] -3-propylbenzoic acid (c5):
4- [4- (Methoxycarbonyl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S, 5R) -tert-butyl (4.72 g, 12.09 mmol) in methanol (121 mL) 4N-aqueous sodium hydroxide solution (18.1 mL, 72.52 mmol) was added at 0 ° C., and the mixture was stirred at 60 ° C. for 2 hours. After confirming the completion of the reaction, the reaction solution was neutralized with an aqueous ammonium chloride solution at 0 ° C. (pH = 7). Thereafter, the mixture was extracted with chloroform, and the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain 4.48 g (yield 99%) of the title compound as a colorless amorphous.
1H-NMR (CDCl3) δ: 0.92 (3H, d, J = 6.8 Hz), 0.97 (3H, t, J = 7.2 Hz), 1.30 (3H, d, J = 6.8 Hz), 1.49 (9H, s), 1.71 (2H, tq, J = 7.2, 7.6 Hz), 2.50-2.58 (2H, m), 2.79 (1H, td, J = 7.6, 14.0 Hz), 3.37-3.43 (1H, m), 3.52 (1H, dd, J = 3.6, 12.8 Hz), 3.61 (1H, dd, J = 4.0, 11.4 Hz), 3.75 (1H, dd, J = 1.6, 11.6 Hz), 4.39-4.44 (1H, m), 6.91 (1H, d, J = 8.0 Hz), 7.87 (1H, dd, J = 2.0, 8.0 Hz), 7.94 (1H, d, J = 2.0 Hz).
1 H-NMR (CDCl 3 ) δ: 0.92 (3H, d, J = 6.8 Hz), 0.97 (3H, t, J = 7.2 Hz), 1.30 (3H, d, J = 6.8 Hz), 1.49 (9H, s), 1.71 (2H, tq, J = 7.2, 7.6 Hz), 2.50-2.58 (2H, m), 2.79 (1H, td, J = 7.6, 14.0 Hz), 3.37-3.43 (1H, m), 3.52 (1H, dd, J = 3.6, 12.8 Hz), 3.61 (1H, dd, J = 4.0, 11.4 Hz), 3.75 (1H, dd, J = 1.6, 11.6 Hz), 4.39-4.44 (1H, m), 6.91 (1H, d, J = 8.0 Hz), 7.87 (1H, dd, J = 2.0, 8.0 Hz), 7.94 (1H, d, J = 2.0 Hz).
工程V:2,5-ジメチル-4-{4-[(ペルフルオロフェノキシ)カルボニル]-2-プロピルフェニル}ピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(c6)の製造:
4-[(2R,5S)-4-(tert-ブトキシカルボニル)-2,5-ジメチルピペラジン-1-イル]-3-プロピル安息香酸(1.0g,2.656mmol)を酢酸エチル(18mL)に溶解させ、室温にて2,3,4,5,6-ペンタフルオロフェノール(513mg,2.789mmol)、N, N’-ジシクロヘキシルカルボジイミド(575mg,2.789mmol)を順に加えた。室温にて18時間攪拌した。反応終結を確認後、0℃にて反応液に水を加えた。その後、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物1.43g(収率>99%)を橙色油状物として得た。 Step V: Preparation of 2,5-dimethyl-4- {4-[(perfluorophenoxy) carbonyl] -2-propylphenyl} piperazine-1-carboxylic acid (2S, 5R) -tert-butyl (c6):
4-[(2R, 5S) -4- (tert-butoxycarbonyl) -2,5-dimethylpiperazin-1-yl] -3-propylbenzoic acid (1.0 g, 2.656 mmol) was added to ethyl acetate (18 mL). Then, 2,3,4,5,6-pentafluorophenol (513 mg, 2.789 mmol) and N, N′-dicyclohexylcarbodiimide (575 mg, 2.789 mmol) were sequentially added at room temperature. Stir at room temperature for 18 hours. After confirming the completion of the reaction, water was added to the reaction solution at 0 ° C. Thereafter, the mixture was extracted with ethyl acetate, and the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 1.43 g (yield> 99%) of the title compound as an orange oil.
4-[(2R,5S)-4-(tert-ブトキシカルボニル)-2,5-ジメチルピペラジン-1-イル]-3-プロピル安息香酸(1.0g,2.656mmol)を酢酸エチル(18mL)に溶解させ、室温にて2,3,4,5,6-ペンタフルオロフェノール(513mg,2.789mmol)、N, N’-ジシクロヘキシルカルボジイミド(575mg,2.789mmol)を順に加えた。室温にて18時間攪拌した。反応終結を確認後、0℃にて反応液に水を加えた。その後、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物1.43g(収率>99%)を橙色油状物として得た。 Step V: Preparation of 2,5-dimethyl-4- {4-[(perfluorophenoxy) carbonyl] -2-propylphenyl} piperazine-1-carboxylic acid (2S, 5R) -tert-butyl (c6):
4-[(2R, 5S) -4- (tert-butoxycarbonyl) -2,5-dimethylpiperazin-1-yl] -3-propylbenzoic acid (1.0 g, 2.656 mmol) was added to ethyl acetate (18 mL). Then, 2,3,4,5,6-pentafluorophenol (513 mg, 2.789 mmol) and N, N′-dicyclohexylcarbodiimide (575 mg, 2.789 mmol) were sequentially added at room temperature. Stir at room temperature for 18 hours. After confirming the completion of the reaction, water was added to the reaction solution at 0 ° C. Thereafter, the mixture was extracted with ethyl acetate, and the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 1.43 g (yield> 99%) of the title compound as an orange oil.
1H-NMR (CDCl3) δ: 0.96 (3H, d, J = 6.6 Hz), 1.00 (3H, t, J = 7.3 Hz), 1.31 (3H, d, J = 6.8 Hz), 1.50 (9H, s), 1.74 (2H, tq, J = 7.2, 8.2 Hz), 2.54-2.63 (2H, m), 2.81 (1H, td, J = 8.2, 14.1 Hz), 3.45-3.51 (1H, m), 3.54 (1H, dd, J = 3.6, 12.9 Hz), 3.65 (1H, dd, J = 4.1, 11.7 Hz), 3.77 (1H, dd, J = 1.4, 12.8 Hz), 4.41-4.47 (1H, m), 6.98 (1H, d, J = 8.5 Hz), 7.98 (1H, dd, J = 2.2, 8.5 Hz), 8.03 (1H, d, J = 2.2 Hz).
1 H-NMR (CDCl 3 ) δ: 0.96 (3H, d, J = 6.6 Hz), 1.00 (3H, t, J = 7.3 Hz), 1.31 (3H, d, J = 6.8 Hz), 1.50 (9H, s), 1.74 (2H, tq, J = 7.2, 8.2 Hz), 2.54-2.63 (2H, m), 2.81 (1H, td, J = 8.2, 14.1 Hz), 3.45-3.51 (1H, m), 3.54 (1H, dd, J = 3.6, 12.9 Hz), 3.65 (1H, dd, J = 4.1, 11.7 Hz), 3.77 (1H, dd, J = 1.4, 12.8 Hz), 4.41-4.47 (1H, m), 6.98 (1H, d, J = 8.5 Hz), 7.98 (1H, dd, J = 2.2, 8.5 Hz), 8.03 (1H, d, J = 2.2 Hz).
工程VI: 4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(c7)の製造:
2,5-ジメチル-4-{4-[(ペルフルオロフェノキシ)カルボニル]-2-プロピルフェニル}ピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(5.75g,10.6mmol)を減圧下100℃にて1時間乾燥後、アルゴン雰囲気下、室温にてエチレングリコールジメチルエーテル(110mL)を加えた。その後、-15℃にてトリフルオロメチルトリメチルシラン(3.32g,23.3mmol)、フッ化セシウム(7.08g,46.6mmol)の順に加えた。その後、-15℃にて1時間攪拌後、室温にて3時間30分攪拌した。反応終結を確認後、0℃にて反応液に1N-塩酸水溶液、トルエンを加えた。その後、トルエンで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物4.98g(収率90%)を褐色結晶性固体として得た。 Step VI: 4- [4- (1,1,1,3,3,3-Hexafluoro-2-hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxyl Preparation of acid (2S, 5R) -tert-butyl (c7):
2,5-dimethyl-4- {4-[(perfluorophenoxy) carbonyl] -2-propylphenyl} piperazine-1-carboxylic acid (2S, 5R) -tert-butyl (5.75 g, 10.6 mmol) under reduced pressure After drying at 100 ° C. for 1 hour, ethylene glycol dimethyl ether (110 mL) was added at room temperature under an argon atmosphere. Thereafter, trifluoromethyltrimethylsilane (3.32 g, 23.3 mmol) and cesium fluoride (7.08 g, 46.6 mmol) were added in this order at −15 ° C. Thereafter, the mixture was stirred at −15 ° C. for 1 hour and then at room temperature for 3 hours and 30 minutes. After confirming the completion of the reaction, a 1N hydrochloric acid aqueous solution and toluene were added to the reaction solution at 0 ° C. Thereafter, extraction with toluene was performed, and the organic layer was washed with saturated brine, dried using anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 4.98 g (yield 90%) of the title compound as a brown crystalline solid.
2,5-ジメチル-4-{4-[(ペルフルオロフェノキシ)カルボニル]-2-プロピルフェニル}ピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(5.75g,10.6mmol)を減圧下100℃にて1時間乾燥後、アルゴン雰囲気下、室温にてエチレングリコールジメチルエーテル(110mL)を加えた。その後、-15℃にてトリフルオロメチルトリメチルシラン(3.32g,23.3mmol)、フッ化セシウム(7.08g,46.6mmol)の順に加えた。その後、-15℃にて1時間攪拌後、室温にて3時間30分攪拌した。反応終結を確認後、0℃にて反応液に1N-塩酸水溶液、トルエンを加えた。その後、トルエンで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物4.98g(収率90%)を褐色結晶性固体として得た。 Step VI: 4- [4- (1,1,1,3,3,3-Hexafluoro-2-hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxyl Preparation of acid (2S, 5R) -tert-butyl (c7):
2,5-dimethyl-4- {4-[(perfluorophenoxy) carbonyl] -2-propylphenyl} piperazine-1-carboxylic acid (2S, 5R) -tert-butyl (5.75 g, 10.6 mmol) under reduced pressure After drying at 100 ° C. for 1 hour, ethylene glycol dimethyl ether (110 mL) was added at room temperature under an argon atmosphere. Thereafter, trifluoromethyltrimethylsilane (3.32 g, 23.3 mmol) and cesium fluoride (7.08 g, 46.6 mmol) were added in this order at −15 ° C. Thereafter, the mixture was stirred at −15 ° C. for 1 hour and then at room temperature for 3 hours and 30 minutes. After confirming the completion of the reaction, a 1N hydrochloric acid aqueous solution and toluene were added to the reaction solution at 0 ° C. Thereafter, extraction with toluene was performed, and the organic layer was washed with saturated brine, dried using anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 4.98 g (yield 90%) of the title compound as a brown crystalline solid.
1H-NMR (CDCl3) δ: 0.92 (3H, d, J = 6.8 Hz), 0.97 (3H, t, J = 7.2 Hz), 1.30 (3H, d, J = 6.8 Hz), 1.48 (9H, s), 1.67 (2H, tq, J = 7.2, 8.0 Hz), 2.47-2.56 (2H, m), 2.82 (1H, td, J = 8.0, 14.0 Hz), 3.28-3.34 (1H, m), 3.50 (1H, dd, J = 3.2, 13.6 Hz), 3.53 (1H, s), 3.59 (1H, dd, J = 4.0, 11.6 Hz), 3.74 (1H, dd, J = 1.6, 12.4 Hz), 4.37-4.43 (1H, m), 6.93 (1H, d, J = 8.4 Hz), 7.44 (1H, dd, J = 2.0, 8.4 Hz), 7.51 (1H, d, J = 2.0 Hz). Mp:170-172℃.
1 H-NMR (CDCl 3 ) δ: 0.92 (3H, d, J = 6.8 Hz), 0.97 (3H, t, J = 7.2 Hz), 1.30 (3H, d, J = 6.8 Hz), 1.48 (9H, s), 1.67 (2H, tq, J = 7.2, 8.0 Hz), 2.47-2.56 (2H, m), 2.82 (1H, td, J = 8.0, 14.0 Hz), 3.28-3.34 (1H, m), 3.50 (1H, dd, J = 3.2, 13.6 Hz), 3.53 (1H, s), 3.59 (1H, dd, J = 4.0, 11.6 Hz), 3.74 (1H, dd, J = 1.6, 12.4 Hz), 4.37- 4.43 (1H, m), 6.93 (1H, d, J = 8.4 Hz), 7.44 (1H, dd, J = 2.0, 8.4 Hz), 7.51 (1H, d, J = 2.0 Hz). Mp: 170-172 ° C.
工程VII:2-{4-[(2R,5S)-2,5-ジメチルピペラジン-1-イル]-3-プロピルフェニル}-1,1,1,3,3,3-ヘキサフルオロプロパン-2-オール(c8)の製造:
4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(4.53g,9.08mmol)をメタノール(14mL)に溶解させ、0℃にて2N-塩酸メタノール溶液(41mL,81.7mmol)を加え、35℃にて5時間攪拌した。反応終結を確認後、反応溶液を減圧濃縮した。残渣に水を加え、水酸化ナトリウム水溶液を用いてpH=8~9にし、ジクロロメタンにて抽出し、有機層を飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた残渣をヘプタンにて洗浄し、乾燥することにより表題化合物3.43g(収率95%)を淡黄色結晶性固体して得た。 Step VII: 2- {4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl} -1,1,1,3,3,3-hexafluoropropane-2 -Manufacture of all (c8):
4- [4- (1,1,1,3,3,3-Hexafluoro-2-hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S , 5R) -tert-butyl (4.53 g, 9.08 mmol) was dissolved in methanol (14 mL), 2N hydrochloric acid methanol solution (41 mL, 81.7 mmol) was added at 0 ° C., and 5 hours at 35 ° C. Stir. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure. Water was added to the residue, the pH was adjusted to 8-9 using an aqueous sodium hydroxide solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over sodium sulfate, and then decompressed. Concentrated. The obtained residue was washed with heptane and dried to give 3.43 g (yield 95%) of the title compound as a pale yellow crystalline solid.
4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-カルボン酸 (2S,5R)-tert-ブチル(4.53g,9.08mmol)をメタノール(14mL)に溶解させ、0℃にて2N-塩酸メタノール溶液(41mL,81.7mmol)を加え、35℃にて5時間攪拌した。反応終結を確認後、反応溶液を減圧濃縮した。残渣に水を加え、水酸化ナトリウム水溶液を用いてpH=8~9にし、ジクロロメタンにて抽出し、有機層を飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた残渣をヘプタンにて洗浄し、乾燥することにより表題化合物3.43g(収率95%)を淡黄色結晶性固体して得た。 Step VII: 2- {4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl} -1,1,1,3,3,3-hexafluoropropane-2 -Manufacture of all (c8):
4- [4- (1,1,1,3,3,3-Hexafluoro-2-hydroxypropan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazine-1-carboxylic acid (2S , 5R) -tert-butyl (4.53 g, 9.08 mmol) was dissolved in methanol (14 mL), 2N hydrochloric acid methanol solution (41 mL, 81.7 mmol) was added at 0 ° C., and 5 hours at 35 ° C. Stir. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure. Water was added to the residue, the pH was adjusted to 8-9 using an aqueous sodium hydroxide solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over sodium sulfate, and then decompressed. Concentrated. The obtained residue was washed with heptane and dried to give 3.43 g (yield 95%) of the title compound as a pale yellow crystalline solid.
1H-NMR (CDCl3) δ:0.76 (3H, d, J = 6.0 Hz), 0.95 (3H, t, J = 7.2 Hz), 1.04 (3H, d, J = 6.4 Hz), 1.63 (2H, qt, J = 7.2, 7.6 Hz), 2.17 (2H, brs), 2.30 (1H, d, J = 10.4 Hz), 2.33 (1H, d, J = 10.4 Hz), 2.63-2.77 (3H, m), 2.81 (1H, dd, J = 2.8, 11.8 Hz), 2.97-3.05 (2H, m), 7.22 (1H, d, J = 9.2 Hz), 7.52-7.54 (2H, m).Mp:165-167℃.
1 H-NMR (CDCl 3 ) δ: 0.76 (3H, d, J = 6.0 Hz), 0.95 (3H, t, J = 7.2 Hz), 1.04 (3H, d, J = 6.4 Hz), 1.63 (2H, qt, J = 7.2, 7.6 Hz), 2.17 (2H, brs), 2.30 (1H, d, J = 10.4 Hz), 2.33 (1H, d, J = 10.4 Hz), 2.63-2.77 (3H, m), 2.81 (1H, dd, J = 2.8, 11.8 Hz), 2.97-3.05 (2H, m), 7.22 (1H, d, J = 9.2 Hz), 7.52-7.54 (2H, m). Mp: 165-167 ° C .
工程VIII:2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸塩酸塩(c10)の製造:
WO2009/144961号パンフレットに従い製造される5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオン(110g,443mmol)を水(700mL)に溶解させ、室温にて水酸化ナトリウム(90g,2.25mol)を加え、48時間加熱還流した。反応終結を確認後、反応溶液を0℃にし、pH<6になるまで濃塩酸を加えた。その後、水を減圧濃縮し得られた粗体を次の工程に使用した(>99%)。 Step VIII: Preparation of 2-amino-2- [4- (1-methylethoxy) phenyl] propanoic acid hydrochloride (c10):
5- [4- (1-Methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione (110 g, 443 mmol) produced according to the pamphlet of WO2009 / 144961 is dissolved in water (700 mL) and brought to room temperature. Sodium hydroxide (90 g, 2.25 mol) was added, and the mixture was heated to reflux for 48 hours. After confirming the completion of the reaction, the reaction solution was brought to 0 ° C. and concentrated hydrochloric acid was added until pH <6. Thereafter, the crude product obtained by concentrating water under reduced pressure was used in the next step (> 99%).
WO2009/144961号パンフレットに従い製造される5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオン(110g,443mmol)を水(700mL)に溶解させ、室温にて水酸化ナトリウム(90g,2.25mol)を加え、48時間加熱還流した。反応終結を確認後、反応溶液を0℃にし、pH<6になるまで濃塩酸を加えた。その後、水を減圧濃縮し得られた粗体を次の工程に使用した(>99%)。 Step VIII: Preparation of 2-amino-2- [4- (1-methylethoxy) phenyl] propanoic acid hydrochloride (c10):
5- [4- (1-Methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione (110 g, 443 mmol) produced according to the pamphlet of WO2009 / 144961 is dissolved in water (700 mL) and brought to room temperature. Sodium hydroxide (90 g, 2.25 mol) was added, and the mixture was heated to reflux for 48 hours. After confirming the completion of the reaction, the reaction solution was brought to 0 ° C. and concentrated hydrochloric acid was added until pH <6. Thereafter, the crude product obtained by concentrating water under reduced pressure was used in the next step (> 99%).
1H-NMR (DMSO) δ: 1.22 (6H, d, J = 6.1 Hz), 1.62 (3H, s), 4.57 (1H, sept, J = 6.1 Hz), 6.68 (2H, brs), 6.84 (2H, d, J = 8.8 Hz), 7.37 (2H, d, J = 8.8 Hz).
1 H-NMR (DMSO) δ: 1.22 (6H, d, J = 6.1 Hz), 1.62 (3H, s), 4.57 (1H, sept, J = 6.1 Hz), 6.68 (2H, brs), 6.84 (2H , d, J = 8.8 Hz), 7.37 (2H, d, J = 8.8 Hz).
工程IX:2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸メチル(c11)の製造:
2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸塩酸塩(70.0g,270mmol)をメタノール(225mL)に溶解させ、室温にて濃硫酸(70mL)を順に加え、24時間加熱還流した。反応終結を確認後、反応溶液を室温にし、減圧濃縮してメタノールを留去した。得られた残渣をジクロロメタンに溶解させ、ジクロロメタンにて抽出した。有機層を飽和炭酸水素ナトリウム水溶液、飽和食塩水で順に洗浄し、無水硫酸ナトリウムにて乾燥し、減圧濃縮することにより表題化合物44.8g(収率70%)を無色油状物として得た。 Step IX: Preparation of methyl 2-amino-2- [4- (1-methylethoxy) phenyl] propanoate (c11):
2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid hydrochloride (70.0 g, 270 mmol) was dissolved in methanol (225 mL), concentrated sulfuric acid (70 mL) was sequentially added at room temperature, and 24 Heated to reflux for hours. After confirming the completion of the reaction, the reaction solution was brought to room temperature and concentrated under reduced pressure to distill off methanol. The obtained residue was dissolved in dichloromethane and extracted with dichloromethane. The organic layer was washed successively with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the title compound (44.8 g, yield 70%) as a colorless oil.
2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸塩酸塩(70.0g,270mmol)をメタノール(225mL)に溶解させ、室温にて濃硫酸(70mL)を順に加え、24時間加熱還流した。反応終結を確認後、反応溶液を室温にし、減圧濃縮してメタノールを留去した。得られた残渣をジクロロメタンに溶解させ、ジクロロメタンにて抽出した。有機層を飽和炭酸水素ナトリウム水溶液、飽和食塩水で順に洗浄し、無水硫酸ナトリウムにて乾燥し、減圧濃縮することにより表題化合物44.8g(収率70%)を無色油状物として得た。 Step IX: Preparation of methyl 2-amino-2- [4- (1-methylethoxy) phenyl] propanoate (c11):
2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid hydrochloride (70.0 g, 270 mmol) was dissolved in methanol (225 mL), concentrated sulfuric acid (70 mL) was sequentially added at room temperature, and 24 Heated to reflux for hours. After confirming the completion of the reaction, the reaction solution was brought to room temperature and concentrated under reduced pressure to distill off methanol. The obtained residue was dissolved in dichloromethane and extracted with dichloromethane. The organic layer was washed successively with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the title compound (44.8 g, yield 70%) as a colorless oil.
1H-NMR (acetone-d6) δ: 1.27 (6H, d, J = 5.8 Hz), 1.55 (3H, s), 3.63 (3H, s), 4.58 (1H, sept, J = 5.8 Hz), 6.84 (2H, d, J = 8.8 Hz), 7.39 (2H, d, J = 8.8 Hz).
1 H-NMR (acetone-d6) δ: 1.27 (6H, d, J = 5.8 Hz), 1.55 (3H, s), 3.63 (3H, s), 4.58 (1H, sept, J = 5.8 Hz), 6.84 (2H, d, J = 8.8 Hz), 7.39 (2H, d, J = 8.8 Hz).
工程X:2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (S)-メチル((S)-c11)の製造:
ラセミ体の2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸メチル(5.54g,23.3mmol)を酢酸エチル(52mL)に溶解させ、室温にて(L)-(+)-マンデル酸(3.55g,23.3mmol)を加え、エタノール(6mL)を加え、30分間加熱還流した。その後、徐々に温度を下げ、さらに0~5℃にて1時間放置した。得られた沈殿物をろ取し、ヘプタンにて洗浄、乾燥し、表題化合物の(L)-(+)-マンデル酸塩(3.35g,37%)を白色固体として得た。次に得られた表題化合物の(L)-(+)-マンデル酸塩を95%エタノールにて再結晶を行った。得られた表題化合物の(L)-(+)-マンデル酸塩をジクロロメタンに懸濁させ、pH=8になるまで炭酸ナトリウム水溶液を加えた。その後、ジクロロメタンにて抽出し、無水硫酸ナトリウムにより乾燥し、減圧濃縮することにより表題化合物1.12g(収率20%,99.4%ee)を無色油状物として得た。 Step X: Preparation of 2-amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl ((S) -c11):
Racemic methyl 2-amino-2- [4- (1-methylethoxy) phenyl] propanoate (5.54 g, 23.3 mmol) was dissolved in ethyl acetate (52 mL) and (L)-( +)-Mandelic acid (3.55 g, 23.3 mmol) was added, ethanol (6 mL) was added, and the mixture was heated to reflux for 30 min. Thereafter, the temperature was gradually lowered, and the mixture was further left at 0 to 5 ° C. for 1 hour. The resulting precipitate was collected by filtration, washed with heptane and dried to give the title compound (L)-(+)-mandelate (3.35 g, 37%) as a white solid. Next, the obtained (L)-(+)-mandelic acid salt of the title compound was recrystallized from 95% ethanol. The obtained title compound (L)-(+)-mandelate was suspended in dichloromethane, and an aqueous sodium carbonate solution was added until pH = 8. Thereafter, the mixture was extracted with dichloromethane, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain 1.12 g (yield 20%, 99.4% ee) of the title compound as a colorless oil.
ラセミ体の2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸メチル(5.54g,23.3mmol)を酢酸エチル(52mL)に溶解させ、室温にて(L)-(+)-マンデル酸(3.55g,23.3mmol)を加え、エタノール(6mL)を加え、30分間加熱還流した。その後、徐々に温度を下げ、さらに0~5℃にて1時間放置した。得られた沈殿物をろ取し、ヘプタンにて洗浄、乾燥し、表題化合物の(L)-(+)-マンデル酸塩(3.35g,37%)を白色固体として得た。次に得られた表題化合物の(L)-(+)-マンデル酸塩を95%エタノールにて再結晶を行った。得られた表題化合物の(L)-(+)-マンデル酸塩をジクロロメタンに懸濁させ、pH=8になるまで炭酸ナトリウム水溶液を加えた。その後、ジクロロメタンにて抽出し、無水硫酸ナトリウムにより乾燥し、減圧濃縮することにより表題化合物1.12g(収率20%,99.4%ee)を無色油状物として得た。 Step X: Preparation of 2-amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl ((S) -c11):
Racemic methyl 2-amino-2- [4- (1-methylethoxy) phenyl] propanoate (5.54 g, 23.3 mmol) was dissolved in ethyl acetate (52 mL) and (L)-( +)-Mandelic acid (3.55 g, 23.3 mmol) was added, ethanol (6 mL) was added, and the mixture was heated to reflux for 30 min. Thereafter, the temperature was gradually lowered, and the mixture was further left at 0 to 5 ° C. for 1 hour. The resulting precipitate was collected by filtration, washed with heptane and dried to give the title compound (L)-(+)-mandelate (3.35 g, 37%) as a white solid. Next, the obtained (L)-(+)-mandelic acid salt of the title compound was recrystallized from 95% ethanol. The obtained title compound (L)-(+)-mandelate was suspended in dichloromethane, and an aqueous sodium carbonate solution was added until pH = 8. Thereafter, the mixture was extracted with dichloromethane, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain 1.12 g (yield 20%, 99.4% ee) of the title compound as a colorless oil.
1H-NMR (acetone-d6) δ: 1.27 (6H, d, J = 5.8 Hz), 1.55 (3H, s), 3.63 (3H, s), 4.58 (1H, sept, J = 5.8 Hz), 6.84 (2H, d, J = 8.8 Hz), 7.39 (2H, d, J = 8.8 Hz).
1 H-NMR (acetone-d6) δ: 1.27 (6H, d, J = 5.8 Hz), 1.55 (3H, s), 3.63 (3H, s), 4.58 (1H, sept, J = 5.8 Hz), 6.84 (2H, d, J = 8.8 Hz), 7.39 (2H, d, J = 8.8 Hz).
光学純度測定:
カラム: CHIRALPAK AS-H, 5μm, 0.46*250mm
移動相: ヘキサン:イソプロピルアルコール=92:8 (0.1% DEA)
流速:1mL/min
温度:30℃
波長:234nm
保持時間:化合物(S)-c11;7.36min(化合物(R)-c11;8.59min) Optical purity measurement:
Column: CHIRALPAK AS-H, 5μm, 0.46 * 250mm
Mobile phase: Hexane: Isopropyl alcohol = 92: 8 (0.1% DEA)
Flow rate: 1 mL / min
Temperature: 30 ° C
Wavelength: 234nm
Retention time: Compound (S) -c11; 7.36 min (Compound (R) -c11; 8.59 min)
カラム: CHIRALPAK AS-H, 5μm, 0.46*250mm
移動相: ヘキサン:イソプロピルアルコール=92:8 (0.1% DEA)
流速:1mL/min
温度:30℃
波長:234nm
保持時間:化合物(S)-c11;7.36min(化合物(R)-c11;8.59min) Optical purity measurement:
Column: CHIRALPAK AS-H, 5μm, 0.46 * 250mm
Mobile phase: Hexane: Isopropyl alcohol = 92: 8 (0.1% DEA)
Flow rate: 1 mL / min
Temperature: 30 ° C
Wavelength: 234nm
Retention time: Compound (S) -c11; 7.36 min (Compound (R) -c11; 8.59 min)
工程XI:2-[3-(2-エトキシ-2-オキソエチル)ウレイド]-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (S)-メチル((S)-c12)の製造:
2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (S)-メチル(53.0mg,0.223mmol)をジクロロメタン(740μL)に溶解させ、0℃にてイソシアナト酢酸エチル(29mg,0.223mmol)を加え、0℃にて1時間攪拌した。反応終結を確認後、反応溶液を減圧濃縮することにより、表題化合物91.5mg(収率>99%)を無色油状物として得た。 Step XI: Preparation of 2- [3- (2-ethoxy-2-oxoethyl) ureido] -2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl ((S) -c12):
2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl (53.0 mg, 0.223 mmol) was dissolved in dichloromethane (740 μL) and ethyl isocyanatoacetate (0 ° C.) 29 mg, 0.223 mmol) was added, and the mixture was stirred at 0 ° C. for 1 hour. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain 91.5 mg (yield> 99%) of the title compound as a colorless oil.
2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (S)-メチル(53.0mg,0.223mmol)をジクロロメタン(740μL)に溶解させ、0℃にてイソシアナト酢酸エチル(29mg,0.223mmol)を加え、0℃にて1時間攪拌した。反応終結を確認後、反応溶液を減圧濃縮することにより、表題化合物91.5mg(収率>99%)を無色油状物として得た。 Step XI: Preparation of 2- [3- (2-ethoxy-2-oxoethyl) ureido] -2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl ((S) -c12):
2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl (53.0 mg, 0.223 mmol) was dissolved in dichloromethane (740 μL) and ethyl isocyanatoacetate (0 ° C.) 29 mg, 0.223 mmol) was added, and the mixture was stirred at 0 ° C. for 1 hour. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain 91.5 mg (yield> 99%) of the title compound as a colorless oil.
1H-NMR; (CDCl3) δ: 1.25 (3H, t, J = 7.1 Hz), 1.32 (6H, d, J = 6.1 Hz), 2.00 (3H, s), 3.69 (3H, s), 3.89 (1H, dd, J = 5.1, 18.6 Hz), 3.93 (1H, dd, J = 5.6, 18.6 Hz), 4.16 (2H, q, J = 7.1 Hz), 4.53 (1H, sept, J = 6.1 Hz), 4.94 (1H, brs), 5.82 (1H, brs), 6.84 (2H, d, J = 8.8 Hz). 7.36 (2H, d, J = 8.8 Hz).
1 H-NMR; (CDCl 3 ) δ: 1.25 (3H, t, J = 7.1 Hz), 1.32 (6H, d, J = 6.1 Hz), 2.00 (3H, s), 3.69 (3H, s), 3.89 (1H, dd, J = 5.1, 18.6 Hz), 3.93 (1H, dd, J = 5.6, 18.6 Hz), 4.16 (2H, q, J = 7.1 Hz), 4.53 (1H, sept, J = 6.1 Hz) , 4.94 (1H, brs), 5.82 (1H, brs), 6.84 (2H, d, J = 8.8 Hz). 7.36 (2H, d, J = 8.8 Hz).
工程XII:(S)- 2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸((S)-c13)の製造:
2-[3-(2-エトキシ-2-オキソエチル)ウレイド]-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (S)-メチル(49.6mg,0.135mmol)をエタノール(500μL)に溶解させ、室温にて炭酸水素ナトリウム(11.3mg,0.135mmol)を加え、室温にて7時間攪拌した。その後、反応溶液に室温にて2M炭酸カリウム水溶液(136μL,0.271mmol)を加え、70℃にて15時間攪拌した。反応終結を確認後、反応溶液に0℃にて1N塩酸を用いてpH=<2にして、酢酸エチルにて抽出した。有機層を飽和食塩水にて洗浄し、無水硫酸ナトリウムにて乾燥し、減圧濃縮することにより、表題化合物39.6mg(収率96%)を無色油状物として得た。 Step XII: (S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl} acetic acid ((S) -c13) Manufacturing:
2- [3- (2-Ethoxy-2-oxoethyl) ureido] -2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl (49.6 mg, 0.135 mmol) was added to ethanol (500 μL). ), Sodium hydrogen carbonate (11.3 mg, 0.135 mmol) was added at room temperature, and the mixture was stirred at room temperature for 7 hours. Then, 2M potassium carbonate aqueous solution (136 μL, 0.271 mmol) was added to the reaction solution at room temperature, and the mixture was stirred at 70 ° C. for 15 hours. After confirming the completion of the reaction, the reaction solution was adjusted to pH = <2 with 1N hydrochloric acid at 0 ° C. and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the title compound (39.6 mg, yield 96%) as a colorless oil.
2-[3-(2-エトキシ-2-オキソエチル)ウレイド]-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (S)-メチル(49.6mg,0.135mmol)をエタノール(500μL)に溶解させ、室温にて炭酸水素ナトリウム(11.3mg,0.135mmol)を加え、室温にて7時間攪拌した。その後、反応溶液に室温にて2M炭酸カリウム水溶液(136μL,0.271mmol)を加え、70℃にて15時間攪拌した。反応終結を確認後、反応溶液に0℃にて1N塩酸を用いてpH=<2にして、酢酸エチルにて抽出した。有機層を飽和食塩水にて洗浄し、無水硫酸ナトリウムにて乾燥し、減圧濃縮することにより、表題化合物39.6mg(収率96%)を無色油状物として得た。 Step XII: (S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl} acetic acid ((S) -c13) Manufacturing:
2- [3- (2-Ethoxy-2-oxoethyl) ureido] -2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl (49.6 mg, 0.135 mmol) was added to ethanol (500 μL). ), Sodium hydrogen carbonate (11.3 mg, 0.135 mmol) was added at room temperature, and the mixture was stirred at room temperature for 7 hours. Then, 2M potassium carbonate aqueous solution (136 μL, 0.271 mmol) was added to the reaction solution at room temperature, and the mixture was stirred at 70 ° C. for 15 hours. After confirming the completion of the reaction, the reaction solution was adjusted to pH = <2 with 1N hydrochloric acid at 0 ° C. and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the title compound (39.6 mg, yield 96%) as a colorless oil.
1H-NMR (CDCl3) δ: 1.32 (6H, d, J = 6.0 Hz), 1.84 (3H, s), 4.27 (1H, d, J = 18.0 Hz), 4.32 (1H, d, J = 18.0 Hz), 4.52 (1H, sept, J = 6.0 Hz), 6.50 (1H, brs), 6.87 (2H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz).
1 H-NMR (CDCl 3 ) δ: 1.32 (6H, d, J = 6.0 Hz), 1.84 (3H, s), 4.27 (1H, d, J = 18.0 Hz), 4.32 (1H, d, J = 18.0 Hz), 4.52 (1H, sept, J = 6.0 Hz), 6.50 (1H, brs), 6.87 (2H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz).
工程XI&XII:(S)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸((S)-c13)の製造:
2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (S)-メチル(2.90g,12.2mmol)をジメチルスルホキシド(15mL)に溶解させ、15℃にてイソシアナト酢酸エチル(1.74g,13.4mmol)を加え、15℃にて1時間攪拌した。その後、反応溶液に15℃にて1M炭酸カリウム水溶液(18.3mL,18.3mmol)を加え、70℃にて2時間攪拌した。反応終結を確認後、反応溶液に室温にて水、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルにて抽出し、有機層を飽和炭酸水素ナトリウム水溶液にて洗浄した。得られた水層を0℃にて4N塩酸を用いてpH=<2にして、酢酸エチルにて抽出し、有機層を飽和食塩水にて洗浄し、無水硫酸ナトリウムにて乾燥し、減圧濃縮することにより、表題化合物3.26g(収率87%)を無色油状物として得た。 Steps XI & XII: (S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl} acetic acid ((S) -c13) Manufacturing:
2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl (2.90 g, 12.2 mmol) was dissolved in dimethyl sulfoxide (15 mL) and ethyl isocyanatoacetate at 15 ° C. (1.74 g, 13.4 mmol) was added, and the mixture was stirred at 15 ° C. for 1 hour. Then, 1M potassium carbonate aqueous solution (18.3 mL, 18.3 mmol) was added to the reaction solution at 15 ° C., and the mixture was stirred at 70 ° C. for 2 hours. After confirming the completion of the reaction, water and a saturated aqueous sodium bicarbonate solution were added to the reaction solution at room temperature, followed by extraction with ethyl acetate, and the organic layer was washed with a saturated aqueous sodium bicarbonate solution. The obtained aqueous layer was adjusted to pH = <2 with 4N hydrochloric acid at 0 ° C., extracted with ethyl acetate, the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. As a result, 3.26 g (yield 87%) of the title compound was obtained as a colorless oil.
2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (S)-メチル(2.90g,12.2mmol)をジメチルスルホキシド(15mL)に溶解させ、15℃にてイソシアナト酢酸エチル(1.74g,13.4mmol)を加え、15℃にて1時間攪拌した。その後、反応溶液に15℃にて1M炭酸カリウム水溶液(18.3mL,18.3mmol)を加え、70℃にて2時間攪拌した。反応終結を確認後、反応溶液に室温にて水、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルにて抽出し、有機層を飽和炭酸水素ナトリウム水溶液にて洗浄した。得られた水層を0℃にて4N塩酸を用いてpH=<2にして、酢酸エチルにて抽出し、有機層を飽和食塩水にて洗浄し、無水硫酸ナトリウムにて乾燥し、減圧濃縮することにより、表題化合物3.26g(収率87%)を無色油状物として得た。 Steps XI & XII: (S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl} acetic acid ((S) -c13) Manufacturing:
2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (S) -methyl (2.90 g, 12.2 mmol) was dissolved in dimethyl sulfoxide (15 mL) and ethyl isocyanatoacetate at 15 ° C. (1.74 g, 13.4 mmol) was added, and the mixture was stirred at 15 ° C. for 1 hour. Then, 1M potassium carbonate aqueous solution (18.3 mL, 18.3 mmol) was added to the reaction solution at 15 ° C., and the mixture was stirred at 70 ° C. for 2 hours. After confirming the completion of the reaction, water and a saturated aqueous sodium bicarbonate solution were added to the reaction solution at room temperature, followed by extraction with ethyl acetate, and the organic layer was washed with a saturated aqueous sodium bicarbonate solution. The obtained aqueous layer was adjusted to pH = <2 with 4N hydrochloric acid at 0 ° C., extracted with ethyl acetate, the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. As a result, 3.26 g (yield 87%) of the title compound was obtained as a colorless oil.
工程XIII-1:(S)-3-(2-{(2S,5R)-4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-イル}-2-オキソエチル)-5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオン((S)-B)の製造:
2-{4-[(2R,5S)-2,5-ジメチルピペラジン-1-イル]-3-プロピルフェニル}-1,1,1,3,3,3-ヘキサフルオロプロパン-2-オール(200mg,0.502mmol)をジクロロメタン(2.0mL)に溶解させ、室温にて(S)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸(169mg,0.552mmol)を加えた。その後、0℃にてO-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロリン酸塩(HBTU)(228mg,0.602mmol)、ジイソプロピルエチルアミン(156mg,1.20mmol)の順に加えた。反応溶液を0℃にて10分間攪拌した後、室温にて20時間攪拌した。反応終結を確認後、減圧濃縮し、得られた残渣に水、メチル-tert-ブチルエーテルを加え、メチル-tert-ブチルエーテルで抽出した。有機層を20%リン酸カリウム水溶液、1N-塩酸水溶液、飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物294mg(収率85%)を淡黄色アモルファスとして得た。 Step XIII-1: (S) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl) ) -2-Propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione Production of ((S) -B):
2- {4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl} -1,1,1,3,3,3-hexafluoropropan-2-ol ( 200 mg, 0.502 mmol) was dissolved in dichloromethane (2.0 mL) and (S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-di- at room temperature. Oxoimidazolidin-1-yl} acetic acid (169 mg, 0.552 mmol) was added. Thereafter, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (HBTU) (228 mg, 0.602 mmol), diisopropylethylamine ( 156 mg, 1.20 mmol). The reaction solution was stirred at 0 ° C. for 10 minutes, and then stirred at room temperature for 20 hours. After confirming the completion of the reaction, the mixture was concentrated under reduced pressure, water and methyl-tert-butyl ether were added to the resulting residue, and the mixture was extracted with methyl-tert-butyl ether. The organic layer was washed with 20% aqueous potassium phosphate solution, 1N aqueous hydrochloric acid solution and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 294 mg (yield 85%) of the title compound as a pale yellow amorphous.
2-{4-[(2R,5S)-2,5-ジメチルピペラジン-1-イル]-3-プロピルフェニル}-1,1,1,3,3,3-ヘキサフルオロプロパン-2-オール(200mg,0.502mmol)をジクロロメタン(2.0mL)に溶解させ、室温にて(S)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸(169mg,0.552mmol)を加えた。その後、0℃にてO-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロリン酸塩(HBTU)(228mg,0.602mmol)、ジイソプロピルエチルアミン(156mg,1.20mmol)の順に加えた。反応溶液を0℃にて10分間攪拌した後、室温にて20時間攪拌した。反応終結を確認後、減圧濃縮し、得られた残渣に水、メチル-tert-ブチルエーテルを加え、メチル-tert-ブチルエーテルで抽出した。有機層を20%リン酸カリウム水溶液、1N-塩酸水溶液、飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物294mg(収率85%)を淡黄色アモルファスとして得た。 Step XIII-1: (S) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl) ) -2-Propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione Production of ((S) -B):
2- {4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl} -1,1,1,3,3,3-hexafluoropropan-2-ol ( 200 mg, 0.502 mmol) was dissolved in dichloromethane (2.0 mL) and (S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-di- at room temperature. Oxoimidazolidin-1-yl} acetic acid (169 mg, 0.552 mmol) was added. Thereafter, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (HBTU) (228 mg, 0.602 mmol), diisopropylethylamine ( 156 mg, 1.20 mmol). The reaction solution was stirred at 0 ° C. for 10 minutes, and then stirred at room temperature for 20 hours. After confirming the completion of the reaction, the mixture was concentrated under reduced pressure, water and methyl-tert-butyl ether were added to the resulting residue, and the mixture was extracted with methyl-tert-butyl ether. The organic layer was washed with 20% aqueous potassium phosphate solution, 1N aqueous hydrochloric acid solution and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 294 mg (yield 85%) of the title compound as a pale yellow amorphous.
工程XIII-2:(S)- 3-(2-{(2S,5R)-4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-イル}-2-オキソエチル)-5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオン((S)-B)の製造:
(S)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸をトルエン(2.0mL)に溶解させ、N,N-ジメチルホルムアミド(2.6mg)、塩化チオニル(77.7mg)を順に加え、120℃にて1時間攪拌した。反応終結を確認後、反応溶液を減圧濃縮して残渣を得た。次に、2-{4-[(2R,5S)-2,5-ジメチルピペラジン-1-イル]-3-プロピルフェニル}-1,1,1,3,3,3-ヘキサフルオロプロパン-2-オール(130mg,0.326mmol)をジクロロメタン(3.9mL)に溶解させ、室温にてトリエチルアミン(66.1mg,0.653mmol)を加えた。0℃にて先に得られた残渣を加え、0℃にて5分間攪拌した後、室温にて14時間攪拌した。反応終結を確認後、0℃にて水を加え、クロロホルムで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール)を用いて精製し、表題化合物176mg(収率78%)を無色アモルファスとして得た。 Step XIII-2: (S) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl) ) -2-Propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione Production of ((S) -B):
(S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl} acetic acid was dissolved in toluene (2.0 mL), N, N-dimethylformamide (2.6 mg) and thionyl chloride (77.7 mg) were sequentially added, and the mixture was stirred at 120 ° C. for 1 hour. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain a residue. Next, 2- {4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl} -1,1,1,3,3,3-hexafluoropropane-2 -Ol (130 mg, 0.326 mmol) was dissolved in dichloromethane (3.9 mL) and triethylamine (66.1 mg, 0.653 mmol) was added at room temperature. The residue obtained previously at 0 ° C. was added, stirred at 0 ° C. for 5 minutes, and then stirred at room temperature for 14 hours. After confirming the completion of the reaction, water was added at 0 ° C., and the mixture was extracted with chloroform. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (chloroform / methanol) to obtain 176 mg (yield 78%) of the title compound as a colorless amorphous substance.
(S)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸をトルエン(2.0mL)に溶解させ、N,N-ジメチルホルムアミド(2.6mg)、塩化チオニル(77.7mg)を順に加え、120℃にて1時間攪拌した。反応終結を確認後、反応溶液を減圧濃縮して残渣を得た。次に、2-{4-[(2R,5S)-2,5-ジメチルピペラジン-1-イル]-3-プロピルフェニル}-1,1,1,3,3,3-ヘキサフルオロプロパン-2-オール(130mg,0.326mmol)をジクロロメタン(3.9mL)に溶解させ、室温にてトリエチルアミン(66.1mg,0.653mmol)を加えた。0℃にて先に得られた残渣を加え、0℃にて5分間攪拌した後、室温にて14時間攪拌した。反応終結を確認後、0℃にて水を加え、クロロホルムで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール)を用いて精製し、表題化合物176mg(収率78%)を無色アモルファスとして得た。 Step XIII-2: (S) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl) ) -2-Propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione Production of ((S) -B):
(S) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl} acetic acid was dissolved in toluene (2.0 mL), N, N-dimethylformamide (2.6 mg) and thionyl chloride (77.7 mg) were sequentially added, and the mixture was stirred at 120 ° C. for 1 hour. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain a residue. Next, 2- {4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl} -1,1,1,3,3,3-hexafluoropropane-2 -Ol (130 mg, 0.326 mmol) was dissolved in dichloromethane (3.9 mL) and triethylamine (66.1 mg, 0.653 mmol) was added at room temperature. The residue obtained previously at 0 ° C. was added, stirred at 0 ° C. for 5 minutes, and then stirred at room temperature for 14 hours. After confirming the completion of the reaction, water was added at 0 ° C., and the mixture was extracted with chloroform. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (chloroform / methanol) to obtain 176 mg (yield 78%) of the title compound as a colorless amorphous substance.
1H-NMR (CDCl3) δ:0.96-0.99 (6H, m), 1.32-1.33 (9H, m), 1.68 (2H, qt, J = 7.0, 7.6 Hz), 1.90 (3H, s), 2.48-2.63 (2H, m), 2.79-2.87 (1H, m), 3.39-3.66 (4H, m), 4.29-4.33 (2H, m), 4.54 (1H, sept, J= 5.9 Hz), 4.84 (1H, s), 5.63 (1H, s), 6.90 (2H, d, J = 8.8 Hz), 6.93 (1H, d, J = 8.8 Hz), 7.45 (1H, d, J = 8.8 Hz), 7.46 (2H, d, J = 8.8 Hz), 7.52 (1H, d, J = 2.2 Hz).
1 H-NMR (CDCl 3 ) δ: 0.96-0.99 (6H, m), 1.32-1.33 (9H, m), 1.68 (2H, qt, J = 7.0, 7.6 Hz), 1.90 (3H, s), 2.48-2.63 (2H, m), 2.79-2.87 (1H, m), 3.39-3.66 (4H, m), 4.29-4.33 (2H, m), 4.54 (1H, sept, J = 5.9 Hz), 4.84 (1H, s), 5.63 (1H, s), 6.90 (2H, d, J = 8.8 Hz), 6.93 (1H, d, J = 8.8 Hz), 7.45 (1H, d, J = 8.8 Hz), 7.46 (2H, d, J = 8.8 Hz) , 7.52 (1H, d, J = 2.2 Hz).
光学純度:99% ee
測定条件:HPLC
カラム:CHIRALPAK AD-H
溶媒:ヘキサン/イソプロピルアルコール=25/75
流速:0.6 mL/min
保持時間:化合物(S)-B;23.4min
(異性体1;9.7min、異性体2;13.3min、異性体3;14.1min、異性体4;15.0min、異性体5;17.9min、異性体6;20.5min、異性体7;54.9min) Optical purity: 99% ee
Measurement conditions: HPLC
Column: CHIRALPAK AD-H
Solvent: hexane / isopropyl alcohol = 25/75
Flow rate: 0.6 mL / min
Retention time: Compound (S) -B; 23.4 min
(Isomer 1; 9.7 min, Isomer 2; 13.3 min, Isomer 3; 14.1 min, Isomer 4; 15.0 min, Isomer 5; 17.9 min, Isomer 6; 20.5 min, Isomer Body 7; 54.9 min)
測定条件:HPLC
カラム:CHIRALPAK AD-H
溶媒:ヘキサン/イソプロピルアルコール=25/75
流速:0.6 mL/min
保持時間:化合物(S)-B;23.4min
(異性体1;9.7min、異性体2;13.3min、異性体3;14.1min、異性体4;15.0min、異性体5;17.9min、異性体6;20.5min、異性体7;54.9min) Optical purity: 99% ee
Measurement conditions: HPLC
Column: CHIRALPAK AD-H
Solvent: hexane / isopropyl alcohol = 25/75
Flow rate: 0.6 mL / min
Retention time: Compound (S) -B; 23.4 min
(Isomer 1; 9.7 min, Isomer 2; 13.3 min, Isomer 3; 14.1 min, Isomer 4; 15.0 min, Isomer 5; 17.9 min, Isomer 6; 20.5 min, Isomer Body 7; 54.9 min)
実施例2(R)-3-(2-{(2S,5R)-4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-イル}-2-オキソエチル)-5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオン((R)-B)の製造
Example 2 (R) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)- 2-propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione (( Production of R) -B)

工程XIV:2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (R)-メチル((R)-c11)の製造:
(D)-(-)-マンデル酸を用いて、工程Xと同様に反応・処理し、表題化合物を無色油状物(99.4%ee)として得た。 Step XIV: Preparation of 2-amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (R) -methyl ((R) -c11):
(D)-(−)-Mandelic acid was used for the reaction and treatment in the same manner as in Step X to obtain the title compound as a colorless oil (99.4% ee).
(D)-(-)-マンデル酸を用いて、工程Xと同様に反応・処理し、表題化合物を無色油状物(99.4%ee)として得た。 Step XIV: Preparation of 2-amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (R) -methyl ((R) -c11):
(D)-(−)-Mandelic acid was used for the reaction and treatment in the same manner as in Step X to obtain the title compound as a colorless oil (99.4% ee).
工程XV:(R)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸((R)-c13)の製造:
2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (R)-メチル(1.00g,4.21mmol)をエタノール(5.0mL)に溶解させ、20℃にて2-イソシアナト酢酸エチル(599mg,4.64mmol)を加え、20℃にて1時間30分攪拌した。その後、反応溶液に20℃にて1M炭酸水素カリウム水溶液(4.64mL,4.64mmol)を加え、20℃にて2時間攪拌した。さらにその後、反応溶液に0℃にて2M炭酸カリウム水溶液(4.21mL,8.43mmol)を加え、70℃にて6時間攪拌した。反応終結を確認後、反応溶液に室温にて酢酸エチル、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルにて抽出し、有機層を飽和炭酸水素ナトリウム水溶液にて洗浄した。得られた水層を0℃にて4N塩酸を用いてpH=<2にして、酢酸エチルにて抽出した。得られた有機層をリン酸緩衝液(pH=5.8)、飽和食塩水の順にて洗浄し、無水硫酸ナトリウムにて乾燥し、減圧濃縮することにより、表題化合物1.12g(収率87%)を無色油状物として得た。 Step XV: (R) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl} acetic acid ((R) -c13) Manufacturing:
2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (R) -methyl (1.00 g, 4.21 mmol) was dissolved in ethanol (5.0 mL), and Ethyl isocyanatoacetate (599 mg, 4.64 mmol) was added, and the mixture was stirred at 20 ° C. for 1 hr 30 min. Then, 1M potassium hydrogen carbonate aqueous solution (4.64 mL, 4.64 mmol) was added to the reaction solution at 20 ° C., and the mixture was stirred at 20 ° C. for 2 hours. Further, 2M potassium carbonate aqueous solution (4.21 mL, 8.43 mmol) was added to the reaction solution at 0 ° C., and the mixture was stirred at 70 ° C. for 6 hours. After confirming the completion of the reaction, ethyl acetate and saturated aqueous sodium hydrogen carbonate solution were added to the reaction solution at room temperature, followed by extraction with ethyl acetate, and the organic layer was washed with saturated aqueous sodium hydrogen carbonate solution. The resulting aqueous layer was adjusted to pH = <2 with 4N hydrochloric acid at 0 ° C. and extracted with ethyl acetate. The obtained organic layer was washed successively with phosphate buffer (pH = 5.8) and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give 1.12 g (yield 87) of the title compound. %) As a colorless oil.
2-アミノ-2-[4-(1-メチルエトキシ)フェニル]プロパン酸 (R)-メチル(1.00g,4.21mmol)をエタノール(5.0mL)に溶解させ、20℃にて2-イソシアナト酢酸エチル(599mg,4.64mmol)を加え、20℃にて1時間30分攪拌した。その後、反応溶液に20℃にて1M炭酸水素カリウム水溶液(4.64mL,4.64mmol)を加え、20℃にて2時間攪拌した。さらにその後、反応溶液に0℃にて2M炭酸カリウム水溶液(4.21mL,8.43mmol)を加え、70℃にて6時間攪拌した。反応終結を確認後、反応溶液に室温にて酢酸エチル、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルにて抽出し、有機層を飽和炭酸水素ナトリウム水溶液にて洗浄した。得られた水層を0℃にて4N塩酸を用いてpH=<2にして、酢酸エチルにて抽出した。得られた有機層をリン酸緩衝液(pH=5.8)、飽和食塩水の順にて洗浄し、無水硫酸ナトリウムにて乾燥し、減圧濃縮することにより、表題化合物1.12g(収率87%)を無色油状物として得た。 Step XV: (R) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5-dioxoimidazolidin-1-yl} acetic acid ((R) -c13) Manufacturing:
2-Amino-2- [4- (1-methylethoxy) phenyl] propanoic acid (R) -methyl (1.00 g, 4.21 mmol) was dissolved in ethanol (5.0 mL), and Ethyl isocyanatoacetate (599 mg, 4.64 mmol) was added, and the mixture was stirred at 20 ° C. for 1 hr 30 min. Then, 1M potassium hydrogen carbonate aqueous solution (4.64 mL, 4.64 mmol) was added to the reaction solution at 20 ° C., and the mixture was stirred at 20 ° C. for 2 hours. Further, 2M potassium carbonate aqueous solution (4.21 mL, 8.43 mmol) was added to the reaction solution at 0 ° C., and the mixture was stirred at 70 ° C. for 6 hours. After confirming the completion of the reaction, ethyl acetate and saturated aqueous sodium hydrogen carbonate solution were added to the reaction solution at room temperature, followed by extraction with ethyl acetate, and the organic layer was washed with saturated aqueous sodium hydrogen carbonate solution. The resulting aqueous layer was adjusted to pH = <2 with 4N hydrochloric acid at 0 ° C. and extracted with ethyl acetate. The obtained organic layer was washed successively with phosphate buffer (pH = 5.8) and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give 1.12 g (yield 87) of the title compound. %) As a colorless oil.
1H-NMR (CDCl3) δ: 1.32 (6H, d, J = 6.0 Hz), 1.84 (3H, s), 4.27 (1H, d, J = 18.0 Hz), 4.32 (1H, d, J = 18.0 Hz), 4.52 (1H, sept, J = 6.0 Hz), 6.50 (1H, brs), 6.87 (2H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz).
1 H-NMR (CDCl 3 ) δ: 1.32 (6H, d, J = 6.0 Hz), 1.84 (3H, s), 4.27 (1H, d, J = 18.0 Hz), 4.32 (1H, d, J = 18.0 Hz), 4.52 (1H, sept, J = 6.0 Hz), 6.50 (1H, brs), 6.87 (2H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz).
工程XVI:(R)-3-(2-{(2S,5R)-4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-イル}-2-オキソエチル)-5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオン((R)-B)の製造:
2-{4-[(2R,5S)-2,5-ジメチルピペラジン-1-イル]-3-プロピルフェニル}-1,1,1,3,3,3-ヘキサフルオロプロパン-2-オール(43.8mg,0.112mmol)をジメチルスルフィド(500μL)に溶解させ、室温にて(R)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸(40.4mg,0.132mmol)を加えた。その後、室温にてO-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロリン酸塩(HBTU)(50.1mg,0.132mmol)、ジイソプロピルエチルアミン(17.1mg,0.132mmol)の順に加えた。反応溶液を室温にて3時間攪拌した。反応終結を確認後、メチル-tert-ブチルエーテル、20%リン酸カリウム水溶液を順に加え、メチル-tert-ブチルエーテルで抽出した。有機層を20%リン酸カリウム水溶液、1N-塩酸水溶液、飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物65.8mg(収率87%、99% ee)を淡黄色アモルファスとして得た。 Step XVI: (R) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)- 2-propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione (( Production of R) -B):
2- {4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl} -1,1,1,3,3,3-hexafluoropropan-2-ol ( 43.8 mg, 0.112 mmol) is dissolved in dimethyl sulfide (500 μL) and (R) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5- is dissolved at room temperature. Dioxoimidazolidin-1-yl} acetic acid (40.4 mg, 0.132 mmol) was added. Then, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (HBTU) (50.1 mg, 0.132 mmol), diisopropylethylamine at room temperature (17.1 mg, 0.132 mmol) was added in this order. The reaction solution was stirred at room temperature for 3 hours. After confirming the completion of the reaction, methyl-tert-butyl ether and 20% aqueous potassium phosphate solution were sequentially added, and the mixture was extracted with methyl-tert-butyl ether. The organic layer was washed with 20% aqueous potassium phosphate solution, 1N aqueous hydrochloric acid solution and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 65.8 mg (yield 87%, 99% ee) of the title compound as a pale yellow amorphous.
2-{4-[(2R,5S)-2,5-ジメチルピペラジン-1-イル]-3-プロピルフェニル}-1,1,1,3,3,3-ヘキサフルオロプロパン-2-オール(43.8mg,0.112mmol)をジメチルスルフィド(500μL)に溶解させ、室温にて(R)-2-{4-[4-(1-メチルエトキシ)フェニル]-4-メチル-2,5-ジオキソイミダゾリジン-1-イル}酢酸(40.4mg,0.132mmol)を加えた。その後、室温にてO-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロリン酸塩(HBTU)(50.1mg,0.132mmol)、ジイソプロピルエチルアミン(17.1mg,0.132mmol)の順に加えた。反応溶液を室温にて3時間攪拌した。反応終結を確認後、メチル-tert-ブチルエーテル、20%リン酸カリウム水溶液を順に加え、メチル-tert-ブチルエーテルで抽出した。有機層を20%リン酸カリウム水溶液、1N-塩酸水溶液、飽和食塩水で洗浄し、無水硫酸ナトリウムを用いて乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)を用いて精製し、表題化合物65.8mg(収率87%、99% ee)を淡黄色アモルファスとして得た。 Step XVI: (R) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)- 2-propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine-2,4-dione (( Production of R) -B):
2- {4-[(2R, 5S) -2,5-dimethylpiperazin-1-yl] -3-propylphenyl} -1,1,1,3,3,3-hexafluoropropan-2-ol ( 43.8 mg, 0.112 mmol) is dissolved in dimethyl sulfide (500 μL) and (R) -2- {4- [4- (1-methylethoxy) phenyl] -4-methyl-2,5- is dissolved at room temperature. Dioxoimidazolidin-1-yl} acetic acid (40.4 mg, 0.132 mmol) was added. Then, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (HBTU) (50.1 mg, 0.132 mmol), diisopropylethylamine at room temperature (17.1 mg, 0.132 mmol) was added in this order. The reaction solution was stirred at room temperature for 3 hours. After confirming the completion of the reaction, methyl-tert-butyl ether and 20% aqueous potassium phosphate solution were sequentially added, and the mixture was extracted with methyl-tert-butyl ether. The organic layer was washed with 20% aqueous potassium phosphate solution, 1N aqueous hydrochloric acid solution and saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (hexane / ethyl acetate) to obtain 65.8 mg (yield 87%, 99% ee) of the title compound as a pale yellow amorphous.
1H-NMR (CDCl3) δ:0.86-1.02 (3H, m), 0.96 (3H, t, J = 7.3 Hz), 1.25-1.45 (3H, m), 1.31 (6H, d, J = 5.9 Hz), 1.62-1.71 (2H, m), 1.90 (3H, s), 2.47-2.58 (2H, m), 2.77-2.85 (1H, m), 3.37-3.88 (4H, m), 4.06-4.85 (3H, m), 4.52 (1H, sept, J = 5.9 Hz), 5.85 (1H, brs), 6.88 (2H, d, J = 9.0 Hz), 6.91 (1H ,d , J = 8.6 Hz), 7.43 (2H, d, J = 9.0 Hz), 7.45 (1H, d, J = 8.6 Hz), 7.51 (1H, s).
1 H-NMR (CDCl 3 ) δ: 0.86-1.02 (3H, m), 0.96 (3H, t, J = 7.3 Hz), 1.25-1.45 (3H, m), 1.31 (6H, d, J = 5.9 Hz), 1.62-1.71 (2H, m), 1.90 (3H, s), 2.47-2.58 (2H, m), 2.77-2.85 (1H, m), 3.37-3.88 (4H, m), 4.06-4.85 (3H, m), 4.52 (1H, sept, J = 5.9 Hz), 5.85 (1H, brs), 6.88 (2H, d, J = 9.0 Hz), 6.91 (1H, d, J = 8.6 Hz), 7.43 (2H, d, J = 9.0 Hz) , 7.45 (1H, d, J = 8.6 Hz), 7.51 (1H, s).
比較例 ラセミ体との物性データ比較
WO2010/125811号パンフレットに記載されている化合物(B)について、化合物((S)-B)および化合物((R)-B)と物性データを比較した結果、NMRスペクトルにおいて有意な差が認められた。 Comparative Example Comparison of physical property data with racemic compound (B) described in WO 2010/12581 pamphlet As a result of comparing physical property data with compound ((S) -B) and compound ((R) -B), Significant differences were observed in the NMR spectra.
WO2010/125811号パンフレットに記載されている化合物(B)について、化合物((S)-B)および化合物((R)-B)と物性データを比較した結果、NMRスペクトルにおいて有意な差が認められた。 Comparative Example Comparison of physical property data with racemic compound (B) described in WO 2010/12581 pamphlet As a result of comparing physical property data with compound ((S) -B) and compound ((R) -B), Significant differences were observed in the NMR spectra.
本発明は、前記一般式(1)で表される光学活性ヒダントイン化合物(1)の大量合成に適用できる製造法を提供するものである。化合物(1)は、LXRβアゴニスト作用を有し、アテローム性動脈硬化症、動脈硬化症、糖尿病に起因する動脈硬化症等の動脈硬化症;脂質異常症;高コレステロール血症;脂質関連疾患;炎症性サイトカインにより引き起こされる炎症性疾患;アレルギー性皮膚疾患等の皮膚疾患;糖尿病;又は、アルツハイマー病の予防及び/又は治療剤等として有用なため、その高収率且つ高光学純度製造法は、産業上の利用可能性を有している。
The present invention provides a production method applicable to mass synthesis of the optically active hydantoin compound (1) represented by the general formula (1). Compound (1) has an LXRβ agonistic action, and atherosclerosis such as atherosclerosis, arteriosclerosis, and diabetes caused by diabetes; dyslipidemia; hypercholesterolemia; lipid-related disease; inflammation Inflammatory diseases caused by sex cytokines; skin diseases such as allergic skin diseases; diabetes; or Alzheimer's disease preventive and / or therapeutic agents, etc. Has the above applicability.
Claims (4)
- 式(1):


i)式(2):


ii)式(3)で表される化合物と、式(4):

i) Formula (2):
ii) a compound represented by formula (3) and formula (4):
- 式(1)のR1がプロピル基、R2、R3がメチル基である、請求項1に記載の製造方法。 The production method according to claim 1, wherein R 1 in the formula (1) is a propyl group, and R 2 and R 3 are methyl groups.
- 式(1)で表される化合物が(S)-3-(2-{(2S,5R)-4-[4-(1,1,1,3,3,3-ヘキサフルオロ-2-ヒドロキシプロパン-2-イル)-2-プロピルフェニル]-2,5-ジメチルピペラジン-1-イル}-2-オキソエチル)-5-[4-(1-メチルエトキシ)フェニル]-5-メチルイミダゾリジン-2,4-ジオンである請求項1又は2に記載の製造方法。 The compound represented by the formula (1) is (S) -3- (2-{(2S, 5R) -4- [4- (1,1,1,3,3,3-hexafluoro-2-hydroxy). Propan-2-yl) -2-propylphenyl] -2,5-dimethylpiperazin-1-yl} -2-oxoethyl) -5- [4- (1-methylethoxy) phenyl] -5-methylimidazolidine The production method according to claim 1 or 2, which is 2,4-dione.
- 式(3):

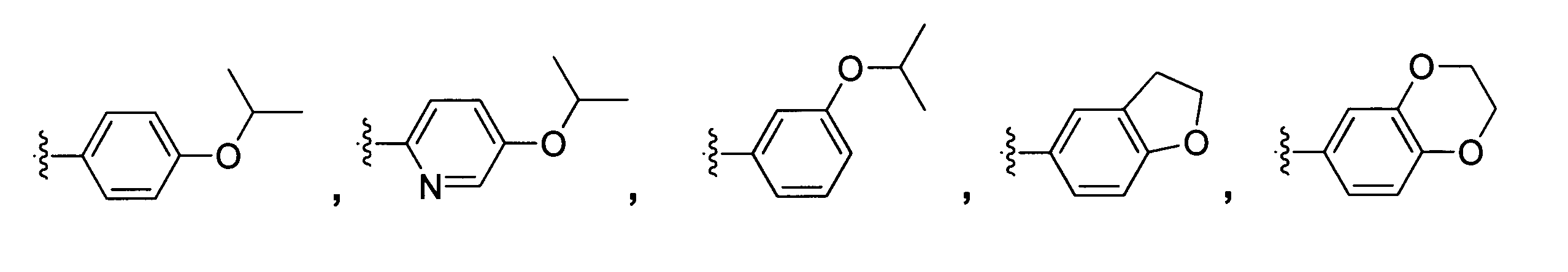
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015536452A JPWO2015037243A1 (en) | 2013-09-12 | 2014-09-11 | Method for producing optically active hydantoin compound |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-189691 | 2013-09-12 | ||
| JP2013189691 | 2013-09-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015037243A1 true WO2015037243A1 (en) | 2015-03-19 |
Family
ID=52665374
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/004702 WO2015037243A1 (en) | 2013-09-12 | 2014-09-11 | Method for producing optically active hydantoin compound |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2015037243A1 (en) |
| WO (1) | WO2015037243A1 (en) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE335993C (en) * | 1916-02-02 | 1921-04-20 | Chem Fab Von Heyden Aktien Ges | Process for the production of hydantoins |
| JPH05503718A (en) * | 1990-11-16 | 1993-06-17 | ジュヴェイナル エス アー | Method for dividing optically isomeric hydantoin |
| JPH07330737A (en) * | 1993-06-18 | 1995-12-19 | Rhone Poulenc Agrochim | Bactericidal optically active 2-imidazolin-5-one and 2-imidazoline-5-thion derivatives |
| JPH1059947A (en) * | 1996-06-20 | 1998-03-03 | Hoechst Ag | Production of chiral and non-racemic (4-aryl-2,5-dioxoimidazolidin-1-yl)acetic acid |
| JPH11504024A (en) * | 1995-04-28 | 1999-04-06 | ヘキスト・アクチエンゲゼルシヤフト | Hydantoin derivatives as intermediates of pharmaceutically active compounds |
| WO2010125811A1 (en) * | 2009-04-29 | 2010-11-04 | 興和株式会社 | Carbinol compound having heterocyclic linker |
-
2014
- 2014-09-11 WO PCT/JP2014/004702 patent/WO2015037243A1/en active Application Filing
- 2014-09-11 JP JP2015536452A patent/JPWO2015037243A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE335993C (en) * | 1916-02-02 | 1921-04-20 | Chem Fab Von Heyden Aktien Ges | Process for the production of hydantoins |
| JPH05503718A (en) * | 1990-11-16 | 1993-06-17 | ジュヴェイナル エス アー | Method for dividing optically isomeric hydantoin |
| JPH07330737A (en) * | 1993-06-18 | 1995-12-19 | Rhone Poulenc Agrochim | Bactericidal optically active 2-imidazolin-5-one and 2-imidazoline-5-thion derivatives |
| JPH11504024A (en) * | 1995-04-28 | 1999-04-06 | ヘキスト・アクチエンゲゼルシヤフト | Hydantoin derivatives as intermediates of pharmaceutically active compounds |
| JPH1059947A (en) * | 1996-06-20 | 1998-03-03 | Hoechst Ag | Production of chiral and non-racemic (4-aryl-2,5-dioxoimidazolidin-1-yl)acetic acid |
| WO2010125811A1 (en) * | 2009-04-29 | 2010-11-04 | 興和株式会社 | Carbinol compound having heterocyclic linker |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE; 17 April 2011 (2011-04-17), accession no. 281562-68-2 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE; 25 September 2008 (2008-09-25), accession no. 052555-32-4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2015037243A1 (en) | 2017-03-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011101861A1 (en) | Process for preparation of dpp-iv inhibitors | |
| EP3231793B1 (en) | Dihydropyrimidine-2-one compounds and medicinal uses thereof | |
| US8304576B2 (en) | Process for production of halogenated alpha-fluoroethers | |
| CA2295800A1 (en) | Resolution of amines | |
| KR20100059836A (en) | Novel process for preparing highly pure levocetirizine and salts thereof | |
| ES2602962T3 (en) | Substituted Phenylazole Derivative | |
| JP5585822B2 (en) | Method for producing optically active nipecotic acid derivative | |
| US5380866A (en) | Alkyl substituted nitroimidazole acetic acids | |
| WO2015037243A1 (en) | Method for producing optically active hydantoin compound | |
| US10683257B2 (en) | Methods and intermediates for synthesizing SK1-I | |
| JP2005179281A (en) | Biphenyl compound | |
| JP4763954B2 (en) | Process for the production of {2- [4- (α-phenyl-p-chlorobenzyl) piperazin-1-yl] ethoxy} acetic acid and novel intermediates therefor | |
| JP2015048324A (en) | Manufacturing method of optically active carbinol compound | |
| EP3442957B1 (en) | "process for preparing intermediates useful in the synthesis of antifungal drugs" | |
| WO2009139438A1 (en) | Process for production of optically active carboxylic acid | |
| US8119797B2 (en) | Production method | |
| WO2015029447A1 (en) | Method for manufacturing optically active carbinol compound | |
| CZ299711B6 (en) | Process for preparing 6-aryl-5,6-dioxohexanoic acid | |
| US5808085A (en) | Stereoselective preparation of 2-substituted succinic acid derivatives | |
| Burdzhiev et al. | Synthesis of piperidinones incorporating an amino acid moiety as potential SP antagonists | |
| JP3855686B2 (en) | 3,3-dialkoxy-2-hydroxyimino derivative and process for producing the same | |
| JP4561635B2 (en) | Process for producing 4-alkoxycarbonyltetrahydropyran or tetrahydropyranyl-4-carboxylic acid | |
| US8115019B2 (en) | Cis-2, 6-disubstituted tetrahydropyran derivatives and preparation method thereof | |
| FI107605B (en) | Optically Active 2- [1- (4-t-Butyl-phenyl) -2-pyrrolidon-4-yl] -methoxycarbonyl-benzoic acid alpha-phenylethylamide, Intermediate and Process for its Preparation | |
| CA1191856A (en) | Process for preparing optically active imidazolylpropanol compounds, and intermediate therein |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14843517 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015536452 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14843517 Country of ref document: EP Kind code of ref document: A1 |