WO2014167537A1 - Synthesis of sialylated/fucosylated oligosaccharides - Google Patents
Synthesis of sialylated/fucosylated oligosaccharides Download PDFInfo
- Publication number
- WO2014167537A1 WO2014167537A1 PCT/IB2014/060647 IB2014060647W WO2014167537A1 WO 2014167537 A1 WO2014167537 A1 WO 2014167537A1 IB 2014060647 W IB2014060647 W IB 2014060647W WO 2014167537 A1 WO2014167537 A1 WO 2014167537A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- moiety
- hydrogenolysis
- group
- Prior art date
Links
- 0 CC(C(C(C1O)OC)=O)O[C@]1OC(C1O)[C@@](OC(C([C@](**)OC2CO*)NC(C)=O)C2=*)OC2[C@@]1OC2O Chemical compound CC(C(C(C1O)OC)=O)O[C@]1OC(C1O)[C@@](OC(C([C@](**)OC2CO*)NC(C)=O)C2=*)OC2[C@@]1OC2O 0.000 description 9
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/18—Acyclic radicals, substituted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/04—Polysaccharides, i.e. compounds containing more than five saccharide radicals attached to each other by glycosidic bonds
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/18—Preparation of compounds containing saccharide radicals produced by the action of a glycosyl transferase, e.g. alpha-, beta- or gamma-cyclodextrins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/26—Preparation of nitrogen-containing carbohydrates
Definitions
- This invention relates to a chemo-enzymatic synthesis of oligosaccharides having important biological activities and significant commercial value for the pharmaceutical and food industry.
- HMOs oligosaccharides
- mucin oligosaccharides oligosaccharides
- Lewis type oligosaccharides have gained much interest and have become important commercial targets for nutrition and therapeutic applications.
- the synthesis of these oligosaccharides has increased significantly due to their role in numerous biological processes occurring in humans.
- oligosaccharides contain sialosides, mainly N-acetyl-neuraminic acid, that are most frequently found at the terminal end of oligosaccharides.
- the linkages of sialic acids, in which they are bound to galactose, N-acetyl-galactosamine and N-acetyl-glucosamine, are most commonly a- 2,3- and a-2,6-ketosidic bonds.
- Their terminally exposed positions allow sialoconjugates to be recognized by receptors of cells, viruses and bacteria, thus to be involved in a wide variety of biological processes, such as tumour metastasis, cell differentiation, cell-cell interactions, etc.
- N- Acetyl-glucosamine or -galactosamine to which a galactose is bound with ⁇ 1-3 interglycosidic linkage (that is lacto-N-biose and Gaip(l-3)-GalNAc, respectively), can be substituted by a sialic acid at its 6-position.
- These trisaccharide motifs often substituted with further sialic acid and/or fucosyl residue(s), can be found naturally in various glycoconjugates having diverse biological roles.
- Aminopropyl glycoside of Neu5Aca(2-3)-Gaip(l-3)-[Neu5Aca(2-6)-]GalNAc has been shown to bind to Sclerotium rolfsii lectin (SRL) (Chachadi et al. Glycoconj. J. 28, 49 (2011)).
- the pentasaccharide 6" '-0-sialyl-LNT (LST b, Galp(l-3)-[Neu5Aca(2-6)-]GlcNAcp(l-3)-Galp(l- 4)-Glc) is a human milk oligosaccharide and represents a common structural element in some other sialylated human milk oligosaccharides (Urashima et al. : Milk Oligosaccharides, Nova Medical Books, NY, 2011); they are listed in Table 1.
- DS-LNT has recently been found to be preventive against necrotising enterocolitis (NEC) in rat model (Jantscher-Krenn et al. Gut 61, 1417 (2012), WO 2012/106665).
- the first aspect of the invention relates to a method for making a sialylated or fucosylated 3-0- galactosyl-GlcNAc or -GalNAc derivative of formula 1 and salts thereof
- R is selected from -OH, -N 3 and -OR 5 wherein 5 is selected from: allyl optionally substituted by one or more methyl, propargyl optionally
- n is an integer of 1 to 10, preferably 2 or 3
- Ri is selected from sialyl moiety, -SO 3 H and -CH(R 5 )-COOH wherein R 5 is selected from H, alkyl and benzyl,
- R 2 is selected from H and fucosyl
- R 3 is selected from H and sialyl
- R 4 is selected from H and fucosyl, provided that one but only one of R 3 and R 4 is H, and
- A is selected from a bond and a divalent carbohydrate linker, comprising the steps of: a) sialylation, sulfation or carboxymethylation of a compound of formula 2
- R' is selected from -N 3 and -OR' 6 wherein R' 6 is selected from allyl optionally substituted by one or more methyl, propargyl optionally
- n is an integer of 1 to 10
- R 7 is independently acyl
- Y is selected from -NHAc, haloalkanoylamido, -NAc 2 , haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and -N 3 , and
- B is selected from a bond and a divalent carbohydrate linker in protected form, b) optional fucosylation of the sialylated, sulfated or carboxymethylated compound of formula 2 obtained in step a), c) de-O-acylation and/or basic hydrolysis, optional mild acidic hydrolysis and optional transformation of Y to -NHAc of the compound obtained in step a) or step b), d) sialylation or fucosylation of the compound obtained in step c), and e) optional catalytic hydrogenolysis and/or anomeric deprotection of the compound
- the second aspect of the invention provides a compound of formula 11 and salts thereof
- R' is selected from -N 3 and -OR' 6 which R' 6 is selected from allyl optionally substituted by one ore more methyl, propargyl optionally substituted by one ore more methyl, a group removable by hydrogenolysis, 2-trimethylsilyl-ethyl and -(CH 2 ) n -N3 wherein integer n is 1 to 10, preferably 2 or 3,
- A' is a divalent lactosyl moiety having the R' group on its C-l (anomeric) carbon atom and attached via the 3'-OH group to the lacto-N-biosyl residue of the compound of formula 11, A' being optionally substituted by a fucosyl moiety on its 3-OH or by a N-acetyllactosaminyl moiety on its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety on its 6-OH, or by a fucosyl on its 3 -OH or 2' -OH,
- Ri is selected from sialyl moiety, -SO 3 H and -CH(R 5 )-COOH wherein R 5 is selected from H, alkyl and benzyl,
- R 3 is selected from H and sialyl moiety
- R 4 is selected from H and fucosyl moiety, provided that at least one of R 3 and R 4 is H, and Ri 6 is selected from H and moiety C, preferably H,
- Rn and Ris independently, are selected from H and a group removable by hydrogenolysis, provided that the disodium salt of the following compound is excluded:
- R' is 2-trimethylsilyl- ethyloxy
- A' is unsubstituted lactosyl
- Ri and R 3 are sialyl moieties
- R 4 and Ri 6 are H.
- the alkyl and aryl residues can be unsubstituted or substituted one or several times, preferably 1-5 times, more preferably 1-3 times.
- the substituents can preferably be alkyl (for aromatic acyl), hydroxy, alkoxy, carboxy, oxo (for alkyl, forming a keto or aldehyde function), alkoxycarbonyl, alkylcarbonyl, formyl, aryl, aryloxycarbonyl, aryloxy, arylamino, arylcarbonyl, amino, mono- and dialkylamino, carbamoyl, mono- and dialkyl-aminocarbonyl, alkylcarbonylamino, cyano, alkanoyloxy, nitro, alkylthio and/or halogen (F, CI, Br, I).
- aryl and alkyl moieties of acyl groups can modify the general chemical characteristics of the acyl group, and thereby the characteristics, such as stability, solubility and the ability to form crystals, of a molecule as a whole.
- group removable by hydrogenolysis preferably means a protecting group whose C-0 bond can be cleaved by hydrogen in the presence of a catalytic amount of palladium, Raney nickel or any other conventional hydrogenolysis catalyst to regenerate the protected -OH group.
- Such protecting groups are described in Wuts and Greene: Protective Groups in Organic Synthesis, John Wiley & Sons, 2007, and include benzyl, diphenylmethyl (benzhydryl), 1-naphthylmethyl, 2- naphthylmethyl and triphenylmethyl (trityl) groups, each of which can be optionally substituted by one or more of the following groups: alkyl, alkoxy, phenyl, amino, acylamino, alkylamino, dialkylamino, nitro, carboxyl, alkoxycarbonyl, carbamoyl, N-alkylcarbamoyl, N,N- dialkylcarbamoyl, azido, halogenalkyl or halogen.
- a preferred protecting group is benzyl optionally substituted with one or more of the following groups: phenyl, alkyl and halogen, particularly unsubstituted benzyl, 4-chlorobenzyl, 3-phenylbenzyl and 4-methylbenzyl groups.
- phenyl, alkyl and halogen particularly unsubstituted benzyl, 4-chlorobenzyl, 3-phenylbenzyl and 4-methylbenzyl groups.
- These preferred and particularly preferred protecting groups have the advantage that the by-products of their hydrogenolysis are exclusively toluene or substituted toluene. Such by-products can easily be removed, even in multi-ton quantities, from water-soluble oligosaccharide products via evaporation and/or extraction processes.
- a-sialyl or “sialyl” preferably means - in accordance with Chen and Varki ACS Chem. Biol. 5, 163 (2010) - a glycosyl moiety of any naturally occurring or modified neuraminic acid or sialic acid derivative or an analogue thereof having an a-glycosidic linkage.
- Preferred neuraminic acid derivatives are N-acetyl- (Neu5Ac), N-glycolyl- (Neu5Gc) and deamino- neuraminic acid (3-deoxy-D-glycero-D-galacto-nonulosonic acid, KDN), as well as Neu5Ac, Neu5Gc and KDN derivatives that are derivatized with linkers, reactive functional groups, detectable labels or targeting moieties, and/or substituted at C-4, C-7-, C-8 and/or C-9, especially at C-9, with acyloxy, alkoxy, halogen or azido.
- the preferred sialyl moiety is the glycosyl residue of Neu5Ac (see Scheme 1). It should be noted that the term "sialyl" in the generally accepted trivial names of human milk oligosaccharides and Lewis type oligosaccharides always refers to Neu5Ac.
- succosyl preferably means a L-fucopyranosyl group (see Scheme 2) attached to a core oligosaccharide with a-interglycosidic linkage.
- salt in connection with sialylated, sulfated and carboxymethylated compounds described in the application preferably means an associated ion pair consisting of the negatively charged acid residue and one or more cations in any stoichiometric proportion.
- Cations which are atoms or molecules with a positive charge, can be inorganic or organic.
- Preferred inorganic cations are ammonium ion, alkali metal, alkali earth metal and transition metal ions, more preferably Na + , K + , Ca 2+ , Mg 2+ , Ba 2+ , Fe 2+ , Zn 2+ , Mn 2+ and Cu 2+ , more preferably K + , Ca 2+ , Mg 2+ , Ba 2+ , Fe 2+ and Zn 2+ .
- Preferred basic organic compounds in positively charged form are diethyl amine, triethyl amine, diisopropyl ethyl amine, ethanolamine, diethanolamine, triethanolamine, imidazole, piperidine, piperazine, morpholine, benzyl amine, ethylene diamine, meglumin, pyrrolidine, choline, tris-(hydroxymethyl)-methyl amine, N-(2-hydroxyethyl)-pyrrolidine, N-(2- hydroxyethyl)-piperidine, N-(2-hydroxyethyl)-piperazine, N-(2-hydroxyethyl)-morpholine, L- arginine, L-lysine, oligopeptides having L-arginine or L-lysine unit or oligopeptides having free amino group on N-terminal, etc., all in protonated form.
- Such salts can be used in a conventional manner to modify the characteristics of the compounds of this invention, such as their stability
- alkyl preferably means a linear or branched hydrocarbon group with 1-6 carbon atoms, such as methyl, ethyl, ⁇ -propyl, / ' -propyl, «-butyl, / ' -butyl, s-butyl, t-butyl, etc.;
- haloalkanoylamido preferably means a halogen substituted Ci-C 6 -alkanoylamido such as chloroacetamido, trichloroacetamido, trifluoroacetamido, etc.;
- haloalkoxycarbonylamino preferably means a Ci-C 6 -alkyloxycarbonyl- H-group substituted by one or more halogen atoms such as 2,2,2-trichloroethoxycarbonylamino, etc.;
- optionally substituted phenyl and "optionally substituted benzyl
- the present invention provides an efficient approach to a diverse group of sialylated and/or fucosylated 3'-0-galactosyl-6-0-sialyl-GlcNAc or -GalNAc derivatives and analogs thereof based on the unique combination of chemical and enzymatic glycosylation steps.
- the claimed method is attractive for scale-up developments and therefore may imply a potential industrial process.
- the first aspect of the invention relates to a method for making a sialylated or fucosylated 3-0- galactosyl-GlcNAc or -GalNAc derivative of formula 1 and salts thereof
- R is selected from -OH, -N 3 and -OR 6 wherein R 6 is selected from: allyl optionally substituted by one or more methyl, propargyl optionally
- n is an integer of 1 to 10, preferably 2 or 3
- Ri is selected from sialyl moiety, -SO 3 H and -CH(R 5 )-COOH wherein R 5 is selected from H, alkyl and benzyl,
- R 2 is selected from H and fucosyl
- R 3 is selected from H and sialyl
- R 4 is selected from H and fucosyl, provided that one but only one of R 3 and R 4 is H, and
- A is selected from a bond and a divalent carbohydrate linker, comprising the steps of: a) sialylation, sulfation or carbox methylation of a compound of formula 2
- R' is selected from -N 3 and -OR' 6 wherein R' 6 is selected from allyl optionally substituted by one or more methyl, propargyl optionally
- n is an integer of 1 to 10
- R 7 is independently acyl
- Y is selected from -NHAc, haloalkanoylamido, -NAc 2 , haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and -N 3 , and
- B is selected from a bond and a divalent carbohydrate linker in protected form, b) optional fucosylation of the sialylated, sulfated or carboxymethylated compound of formula 2 obtained in step a), c) de-O-acylation and/or basic hydrolysis, optional mild acidic hydrolysis and optional transformation of Y to -NHAc of the compound obtained in step a) or step b), d) sialylation or fucosylation of the compound obtained in step c), and e) optional catalytic hydrogenolysis and/or anomeric deprotection of the compound
- a compound of formula 2 is sialylated, sulfated, or carboxymethylated.
- B is a bond
- a compound of formula 2 is a partially protected disaccharide of formula 2A
- the secondary OH-group of the deoxy-aminohexose moiety of the disaccharide of formula 2A can be in equatorial orientation giving rise to a partially protected lacto-N-biosyl derivative of formula
- the disaccharide of formula 2 A is a GalpPl- 4GalNAc derivative of formula 2C
- the compound of formula 2 is a compound of formula 2B wherein Y is - HAc and R 7 groups are identical and are acetyl or benzoyl.
- Compounds of formula 2A can be made in a conventional manner by standard carbohydrate chemistry, for example by reacting a protected glucos- or galactosamine acceptor having a free 3- OH with a tetraacyl galactosyl donor followed by selective deprotection of the 4- and 6-OH groups (see e.g. Hendel at al. Carbohydr. Res. 343, 2914 (2008), Jain et al. Carbohydr. Res. 275, 231 (1995), WO 95/29927, Bao et al. Bioorg. Med. Chem. 18, 3760 (2010), Rana et al. Carbohydr. Res.
- B is a divalent carbohydrate linker in protected form.
- the term "carbohydrate linker in protected form” preferably means any mono-, di- or oligosaccharide glycosyl residue which is attached to the R' group by the C-l (aldoses) or C-2 (ketoses) anomeric carbon, and is also attached, via one of its non-glycosidic OH-groups, to the galactosylated aminodeoxy-hexose residue of the compound of formula 2.
- this divalent glycosyl residue B may represent a linear or branched structure consisting of monosaccharide units that are linked to each other by interglycosidic linkages.
- the monosaccharide unit(s) of the divalent glycosyl residue B can be selected from any 5- 9 carbon atom containing sugars consisting of aldoses (e.g. D-glucose, D-galactose, D-mannose, D- ribose, D-arabinose, L-arabinose, D-xylose, etc.), ketoses (e.g.
- D-fructose, D-sorbose, D-tagatose, etc. deoxysugars (e.g. L-rhamnose, L-fucose, etc.), deoxy-amino sugars (e.g. N-acetylglucosamine, N-acetylmannosamine, N-acetylgalactosamine, etc.), uronic acids, ketoaldonic acids (e.g. sialic acid) and like.
- the divalent glycosyl moiety B is a lactosyl moiety or an oligosaccharide moiety that consists of a lactosyl moiety and at least one monosaccharide unit selected from the group consisting of galactose, N-acetylglucosamine, fucose and N-acetyl neuraminic acid.
- the divalent lactosyl moiety is attached to the R' group by its C-1 anomeric carbon atom, and it is also attached, via one of its non-glycosidic OH-groups, preferably via its 3'-OH group to the
- the divalent lactosyl moiety defined above can be optionally substituted by other glycosyl residues, preferably on its 3- OH group by a fucosyl moiety, or on its 6' -OH group by a N-acetyllactosaminyl moiety, which N- acetyllactosaminyl moiety can optionally be further substituted by an N-acetyl neuraminyl moiety on its 6-OH, or by a fucosyl on its 3-OH or 2'-OH.
- These oligosaccharide substructures can be found in human milk oligosaccharides.
- the functional groups of the divalent glycosyl residue B are protected, preferably the free OH groups are acylated (e.g. acetylated, benzoylated) and the carboxy group of the optional N-acetyl neuraminyl moiety is blocked in ester form (e.g. methyl, ethyl or benzyl ester).
- ester form e.g. methyl, ethyl or benzyl ester
- the R' group has ⁇ -orientation.
- the moiety B is attached to the parent carbohydrate backbone by ⁇ -linkage.
- step a) the primary free OH group of a compound of formula 2 is regioselectively sialylated, sulfated or carboxymethylated, without affecting the neighbouring secondary OH-group, to give a compound of formula 4
- R', R 7 , B and Y are as defined above, and
- P is selected from protected sialyl moiety, -SO 3 H and -CH(R 5 )-COORio wherein R 5 is selected from H, alkyl and benzyl, R 10 is selected from alkyl and benzyl.
- sialyl donors For the chemical sialylation suitably protected and activated sialyl donors are used.
- functional group protection of the sialyl donor an array of protecting groups, mainly esters, ethers and acetals, are available to the skilled person.
- optionally substituted acyls such as acetyl, benzoyl, chloroacetyl or chlorobenzoyl, and ether-type groups such as benzyl are of synthetic usefulness;
- the carboxyl group can be protected by an ester, typically by a methyl, ethyl or benzyl ester;
- the amino function can be masked by diacetyl, trifluoroacetyl, trichloroacetyl, Troc or Fmoc group, or as a cyclic carbamate with the adjacent 4-OH.
- the anomeric centre activation can be varied among halo, alkyl- or arylthio, dialkyl, dibenzyl or diaryl phosphite, or trihaloacetimidate, each of which is commonly used in sialoglycosidation methods.
- the protective group introduction and anomeric centre activations mentioned above can be carried out by known processes (see e.g. Ress et al. Curr. Org. Synth. 1, 31 (2004), Chen et al. ACS Chem. Biol. 5, 163 (2010) and references cited therein).
- the preferred sialyl donors are those disclosed in WO 2011/100979 and WO 2012/113404, among which the N-acetyl neuraminyl phosphite donors of formula 3
- R 8 is acyl, preferably acetyl
- Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl, and
- R9 is optionally substituted phenyl or benzyl, are especially preferred.
- the coupling reaction runs in aprotic solvent, preferably in
- the sialylated product can be characterized by the following formula 4 A
- R', R 7 , R 8 , B, Q and Y are as defined above.
- step a) is a sulfation step
- a compound of formula 2 defined above is treated with SCVpyridine complex in a non-protic solvent to form a compound of formula 4B or a salt thereof
- a compound of formula 2 can be carboxymethylated to give rise to a compound of formula 4C
- R 5 is selected from H, alkyl and benzyl
- R 10 is selected from alkyl and benzyl
- R', R 7 , B and Y are as defined above.
- step b) the sialylated, sulfated or carboxymethylated compound obtained in step a) is optionally fucosylated with a suitably protected and activated fucosyl donor.
- the fucosyl moiety is introduced at the remaining free secondary OH-group (4-OH) of the compound of formula 4.
- X is selected from a halogen, -O-pentenyl, -OAc, -OBz and -SRi 3 , in which Ri 3 is alkyl or optionally substituted phenyl,
- Rii is selected from acyl and a group removable by hydrogenolysis
- Ri 2 is selected from a group removable by hydrogenolysis, acyl or two R 12 groups together form a moiety R-14 C R 15j wherein R14 and R15 independently are alkyl or phenyl, or wherein R14 and R15 together with the carbon atom, to which they are attached, form cycloalkylidene, can be used in this reaction to give a compound of formula 6
- R', R 7 , Rn, Ri 2 , B, P and Y are as defined above.
- the fucosylation can be carried out in a conventional manner in an aprotic solvent or in a mixture of aprotic solvents in the presence of an activator, see Demchenko (Ed.): Handbook of Chemical Glycosylation Wiley (2008).
- the fucosylation reaction is generally promoted by heavy metal ions, mainly mercury or silver, and Lewis acids such as trimethylsilyl triflate or BF 3 -etherate.
- a fucosyl halide i.e., X is F, CI, Br or I
- X is F, CI, Br or I
- anomeric halides follow the reactivity order F ⁇ Cl ⁇ Br ⁇ I for nucleophilic displacement.
- Glycosyl fluorides can be prepared by treating the appropriate precursors such as hemiacetals, glycosyl halides, glycosyl esters and ⁇ -glycosides with fluorinating reagents such as HF, AgF, AgBF 4 , tetrabutyl ammonium fluoride, diethyl amino sulfur trifluoride, 2- fluoro-l-methylpyridinium tosylate, Selectfluor, Deoxo-Fluor or 4-methyl(difluoroiodo)-benzene.
- fluorinating reagents such as HF, AgF, AgBF 4 , tetrabutyl ammonium fluoride, diethyl amino sulfur trifluoride, 2- fluoro-l-methylpyridinium tosylate, Selectfluor, Deoxo-Fluor or 4-methyl(difluoroiodo)-benzene.
- the resulting glycosyl donor can be activated by a catalytic amount of a Lewis acid, such as trimethylsilyl triflate or BF 3 -etherate, for the glycosylation reaction.
- Fucosyl acetates or benzoates are preferably first subjected to electrophilic activation to provide a reactive intermediate and then treated with a nucleophilic OH-acceptor.
- Typical activators of choice are Bronsted acids (e.g., /?-TsOH, HC10 4 or sulfamic acid), Lewis acids (e.g., ZnCl 2 , SnCl 4 , triflate salts, BF 3 -etherate, trityl perchlorate, A1C1 3 or triflic anhydride) or a mixture thereof.
- Pentenyl fucosides can be transglycosylated with appropriate glycosyl acceptors in the presence of a promoter such as NBS and NIS.
- a promoter such as NBS and NIS.
- Protic or Lewis acids triflic acid, Ag-triflate, etc. can enhance the reaction.
- the pentenyl glycosides can be prepared with the aid of «-pentenol by standard Fischer glycosylation of hemiacetals under acidic condition, by silver(I) salt promoted coupling of glycosyl bromides (Koenigs-Knorr method), or by glycosylation of 1 -acetyl glycosides in the presence of tin(IV) chloride.
- Thiofucosides i.e., X is alkylthio- or optionally substituted phenylthio-group
- thiophilic promoters such as mercury(II) salts, Br 2 , 1 2 , NBS, NIS, triflic acid, triflate salts, BF 3 - etherate, trimethylsilyl triflate, dimethyl-methlythio sulphonium triflate, phenylselenyl triflate, iodonium dicollidine perchlorate, tetrabutylammonium iodide or mixtures thereof, preferably by Br 2 , NBS, NIS or triflic acid.
- thiophilic promoters such as mercury(II) salts, Br 2 , 1 2 , NBS, NIS, triflic acid, triflate salts, BF 3 - etherate, trimethylsilyl triflate, dimethyl-methlythio sulphonium tri
- Aprotic solvents such as toluene, THF, methyl-THF, DCM, chloroform, dioxane, acetonitrile, chlorobenzene, ethylene di chloride, DMSO, DMF or N-methylpyrrolidone or mixtures thereof, preferably DMF, toluene, DCM or mixtures thereof, more preferably toluene or DMF-DCM mixture can be used in this glycosylation reaction at -20 to 20 °C, preferably at -10 to 5 °C, with reaction time of 5 min to 2 hours.
- Br 2 , NBS or NIS can be used, optionally in the presence of triflic acid or a triflate derivative.
- a slight excess of donor 1.1-1.2 eq.
- water or a Ci-C 6 alcohol is generally used, preferably an aqueous or alcoholic solution of a base like sodium carbonate, sodium bicarbonate, ammonia or triethyl amine, more preferably an aqueous Na 2 S 2 0 3 /NaHC0 3 solution.
- the fucosyl donor is a compound of formula 5 wherein Rn is as defined above, R 12 is acyl or a group removable by hydrogenolysis, and X is phenylthio optionally substituted with one or more alkyl. More preferably Rn is benzyl, 4-methylbenzyl, naphthylmethyl, 4-phenylbenzyl, 4- chlorobenzyl, 4-methoxybenzyl, 3,4-dimethoxybenzyl, 2,4,6-trimethylbenzyl or 2,3,4,5,6- pentamethylbenzyl, R 12 is benzyl, 4-methylbenzyl, naphthylmethyl, 4-phenylbenzyl, 4- chlorobenzyl, 4-methoxybenzyl, 3,4-dimethoxybenzyl, 2,4,6-trimethylbenzyl, 2,3,4,5,6- pentamethylbenzyl or benzoyl optionally substituted by one or more halogens and X is
- Rn is benzyl or 4-methylbenzyl and R 12 is benzoyl or 4-chlorobenzoyl.
- step b) a compound of formula 4A, 4B or 4C defined above is reacted with a fucosyl donor of formula 5 giving rise to a compound of formula 6A, 6B or 6C, respectively, and salts thereof
- R', R 5 , R 7 , R 8 , Rio, Rn, R 12 , B, Q and Y are as defined above.
- step c) the protecting groups of a compound obtained in step a) or step b) are removed except for the anomeric protection R', and optionally Y is converted into a - HAc group.
- protecting groups are O-acyls (R 7 , R 8 , the OH-protections in moiety B, and optionally Rn and R 12 ).
- Acyl protective groups can be removed by a conventional base catalysed
- transesterification deprotection reaction wherein the acyl groups are removed in an alcohol solvent such as methanol, ethanol, propanol or t-butanol in the presence of an alcoholate such as NaOMe, NaOEt or KO*Bu at 20-100 °C.
- an alcohol solvent such as methanol, ethanol, propanol or t-butanol
- an alcoholate such as NaOMe, NaOEt or KO*Bu at 20-100 °C.
- a co- solvent such as toluene or xylene can be beneficial in order to control particle size of the product and to avoid gel formation.
- a catalytic amount of NaOMe is used in methanol (Zemplen de-O-acylation). Under this condition only O-acyls can be deprotected.
- Y is -NAc 2
- one of the acetyl groups can also be removed to make the Y group -NHAc.
- Y is: haloalkanoylamido, haloalkoxycarbonylamino, 2,3-diphenylmaleimido, 2,3-dimethylmaleimido or -N 3 , and ester groups (see group -COOQ or -COORio) remain intact under these conditions.
- acyloxy groups to OH, esters to carboxyl see group -COOQ or -COORio
- Y-groups to -NH 2 haloalkanoylamido, halocarboxylamino, 2,3-diphenylmaleimido and 2,3-dimethylmaleimido
- basic hydrolysis which is a base catalysed hydrolysis in water, alcohol or water-organic solvent mixtures, in homogeneous or heterogeneous reaction conditions at 0-100 °C.
- a strong base such as Li OH, NaOH, KOH, Ba(OH) 2 , K 2 C0 3 , a basic ion exchange resin or tetraalkylammonium hydroxide, is used, but the base can also be in an aqueous solution as well.
- the base is NaOH and the solvent is methanol. If Y is -NAc 2 , one of the acetyl groups can also be removed to make the Y group -NHAc. Azido Y group is not affected in this reaction.
- R' is not azido or R' 6 is not -(CH 2 ) n -N 3 ).
- the free amino group obtained by one of the deprotective methods can then be acetylated without acetylating the free OH-groups.
- Selective N-acetylation in the presence of one or more hydroxyls can be carried out in a conventional manner with a slight excess of acetic anhydride or acetyl chloride (-1.5-3 equiv.) at about 0-35 °C with or without added base. Any resulting overacetylated by-product(s) can be readily transformed into the desired compounds with e.g. NaOH/MeOH or NaOMe/MeOH treatment.
- the deprotected compound having a free amino group can be peracetylated (that is the amino group and all available OH groups are acetylated) followed by base catalysed transesterification (see above).
- trichloroacetyl amide Y-group can be transformed in one step to -NHAc with tributyltin hydride.
- step c) an acetal/ketal group optionally present on the compounds of formulas 6A, 6B and
- 6C (i.e. when two Ri 2 groups together form a moiety ⁇ 14 C R 15 ) ca n be selectively deprotected by acid catalysed mild hydrolysis, i.e. by reacting the compounds with water or an alcohol in the presence of acid at pH>l-2 to produce OH-groups on the compounds.
- Acyl protective groups will not be affected because they can be deprotected only by extremely strong acidic hydrolysis (pH ⁇ l).
- the interglycosidic linkage and anomeric protecting groups of the compounds of formulas 6A, 6B and 6C can also be sensitive to acids, they can be split in the compounds of formulas 6A, 6B and 6C only by acidic hydrolysis at pH ⁇ l-2.
- Water which is a reagent, can also serve as solvent or co-solvent.
- Organic protic or aprotic solvents which are stable under acidic conditions and fully or partially miscible with water, such as Ci-C 6 alcohols, acetone, THF, dioxane, ethyl acetate or MeCN, can be also used in a mixture with water.
- the acid used is generally a protic acid, such as acetic acid, trifluoroacetic acid, HC1, formic acid, sulphuric acid, perchloric acid, oxalic acid, ⁇ -toluenesulfonic acid,
- benzenesulfonic acid or a cationic exchange resin can be present in from a catalytic amount to a large excess.
- the hydrolysis can be carried out at between 20 °C and reflux until completion of the reaction which can take from about 2 hours to 3 days depending on temperature, concentration and pH.
- an organic acid such as acetic acid, formic acid, chloroacetic acid or oxalic acid
- a Ci-C 6 alcohol-acetonitrile or Ci-C 6 alcohol-water mixture is used in the presence of HC1 or a sulfonic acid such as ⁇ -toluenesulfonic acid or camphorsulfonic acid.
- an anhydrous Ci-C 6 alcohol such as methanol, ethanol, propanol and butanol
- an anhydrous Ci-C 6 alcohol such as methanol, ethanol, propanol and butanol
- Catalytic amount of hydrogen chloride, sulphuric acid, perchloric acid, ⁇ -toluenesulfonic acid, acetic acid, oxalic acid, camphorsulfonic acid or a strong acidic ion-exchange resin can be used at temperatures of 20 °C to reflux.
- any compound of formula 6A, 6B and 6C, wherein at least one of the Rn and R 12 substituents is a group removable by hydrogenolysis can be subjected to catalytic hydrogenolysis in order to remove
- R' is as defined above, moiety A is a bond or is a divalent carbohydrate linker in deprotected form,
- Ri is selected from sialyl, -SO 3 H and -CH(R 5 )-COOH, wherein R 5 is as defined above,
- Ri6 is selected from H and a moiety of formula C
- Rn and Ris independently, are selected from H and a group removable by hydrogenolysis.
- divalent carbohydrate linker in deprotected form preferably means any mono-, di- or oligosaccharide glycosyl residue which is attached to the R' group by the C-l (aldoses) or C-2 (ketoses) anomeric carbon, and in the same time it is also attached, via one of its non-glycosidic OH-groups, to the galactosylated aminodeoxy-hexose residue of the compound of formula 7, as defined for moiety B above, in deprotected from, which means that the OH-protective groups and the ester protective group of the N-acetyl neuraminyl moiety (if present) in the divalent
- the divalent glycosyl moiety A is a lactosyl moiety or an oligosaccharide moiety that consists of a lactosyl moiety and at least one monosaccharide unit selected from the group consisting of galactose, N-acetylglucosamine, fucose and N-acetyl neuraminic acid.
- the divalent lactosyl moiety is attached to the R' group by its C-l anomeric carbon atom, and at the same time it is also attached, via one of its non-glycosidic OH-groups, preferably via its 3'-OH group to the galactosylated aminodeoxy-hexose residue of the compound of formula 7.
- the divalent lactosyl moiety defined above can be optionally substituted by other glycosyl residues, preferably on its 3 -OH group by a fucosyl moiety, or on its 6' -OH group by a N-acetyllactosaminyl moiety, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetyl neuraminyl moiety on its 6-OH, or by a fucosyl on its 3 -OH or 2' -OH.
- step d) a compound obtained in step c), that is a compound of formula 7 defined above, is brought into a glycosidase mediated transsialylation or transfucosylation reaction and a compound of formula 10 and salts thereof can be obtained wherein R', Ri, Ri 6 and A are as defined above, R 3 is selected from H and a sialyl, and
- R 4 is selected from H and a fucosyl moiety, provided that one but only one of R 3 and R 4 is H.
- a compound of formula 7 is reacted with a sialyl donor, preferably a N-acetyl neuraminyl donor, under the catalysis of an enzyme having a2-3- transsialidase activity.
- Typical natural sialyl donors can be selected from, but are not limited to, 3'- O-sialyl-lactose, fetuin, gangliosides, O- or N-linked glycopeptides, all of which contain a sialic acid a-2,3-linked to a terminal ⁇ -galactoside residue, or polysialic acid with a-2,8-linkage.
- the sialyl donor used in step d) can be characterized by formula 8 and salts thereof,
- Z is selected from the group consisting of azide, fluoro, optionally substituted phenoxy, optionally substituted pyridinyloxy, group D, group E, group F and group G
- alkylhydrazino dialkylhydrazino or trialkylhydrazino.
- sialyl donors 4- methylumbelliferyl and optionally substituted phenyl N-acetyl-a-neuraminosides and 3'-0-sialyl- lactose, more commonly /?-nitrophenyl N-acetyl-a-neuraminoside and 3'-0-sialyl-lactose, are of high preference.
- Enzymes having a2-3-transsialidase activity are preferably selected from a sialidase or
- transsialidase as described in the following, e.g. from sialidases (EC 3.2.1.18) and transsialidases (EC 2.4.1.-) as classified according to the the GH33 family. They are retaining enzymes. Sialidases and transsialidases are widely distributed in nature. They are found particularly in diverse virus families and bacteria, and also in protozoa, some invertebrates and mammals. These enzymes differ in their biochemical properties, e.g., kinetics, binding affinity or substrate preference. Nevertheless, they possess conserved domains and structural similarities.
- Transsialidases differ from sialidases since they can transfer sialic acids, preferably a-2, 3 -bonded sialic acids, from a donor molecule to an acceptor derivative, which is preferably a terminal galactose moiety with a ⁇ -interglycosidic linkage. As a result of this transfer, an a-glycosidic bond is formed between the sialic acid and the acceptor. However, if there is no suitable acceptor, the transsialidase hydrolyses the sialic acid.
- TcTS transsialidase
- transsialidases have been detected in several other trypanosome types such as Trypanosoma brucei gambiense, Trypanosoma brucei rhodesiense, Trypanosoma brucei brucei and Trypanosoma congolense. Moreover, the existence of transsialidases has been shown in Endotrypanum types, in Corynebacterium diphtheriae and even in human plasma.
- Sialidases can be classified into two different subgroups, endo- and exo-sialidases.
- the endo- sialidases hydrolyse sialic acid linkages internal to macromolecules, while the exo-sialidases attack terminal sialic acid linkages, and desialylate glycoproteins, glycopeptides, gangliosides,
- sialidases from Bifidobacterium bifidum and Bifidobacterium longum subsp. infantis have been identified, cloned and characterized. These sialidases can cleave and so recognize both a-2, 3- and a-2,6-linked sialosides.
- Sialidases from Bifidobacterium longum subsp. infantis have a consistent preference for a-2,6-linkage whereas sialidases from Bifidobacterium bifidum have a consistent preference for a-2, 3 -linkage.
- Sialidases which may be employed in the context of the present invention, may also comprise engineered sialidases. Based on sequence and structure comparisons, sialidase from Trypanosoma rangeli may be mutated at six positions, wherein the resulting mutant is able to display a significant level of transsialidase activity (see Paris et al. J. Mol. Biol. 345, 923 (2005)).
- the enzyme having a sialidase and/or transsialidase activity may be selected from sialidases or transsialidases derived from Bifidobacterium longum subsp. infantis ATCC 15697, Bifidobacterium bifidum JCM1254, Bifidobacterium bifidum S17, Bifidobacterium bifidum PRL2010, Bifidobacterium bifidum NCIMB 41171, Trypanosoma cruzi, etc.
- enzyme having a sialidase and/or transsialidase activity may be selected from sialidases or transsialidases as defined according to the following deposit numbers: gi
- siab2 (Bifidobacterium bifidum JCM1254), further sialidases or transsialidases from Bifidobacterium bifidum JCM1254, gi
- PRL2010 gi
- sialidases with sialidase/transsialidase activity are listed in the following Table 2:
- R 3 is a sialyl, preferably N-acetyl-neuraminyl moiety.
- a compound of formula 7 is reacted with a fucosyl donor under the catalysis of an enzyme having al-2-transfucosidase activity.
- Typical fucosyl donors can be selected from, but are not limited to, 2'-0-fucosyl-lactose, difucosyl-lactose, and fucose donors of formula 9 wherein Z is selected from the group consisting of azide, fluoro, optionally substituted phenoxy, optionally substituted pyridinyloxy, group D, group E, group F and group G
- the enzyme exhibiting al-2-transfucosidase activity is preferably selected from fucosidases, transfucosidases and fucosynthases as classified according to EC 3.2.1.38 and 3.2.1.51.
- a-L- Fucosidases are widely spread in living organisms such as mammals, plants, fungi and bacteria. These enzymes belong to the families 29 and 95 of the glycoside hydrolases (GH29 and GH95) as defined by the CAZY nomenclature (https://www.cazy.org).
- Fucosidases from GH29 are retaining enzymes (3D structure: ( ⁇ / ⁇ ) 8 ) whereas fucosidases from GH95 are inverting enzymes (3D structure: ( ⁇ / ⁇ ) 6 ).
- the substrate specificity of the GH29 family is broad whereas that of the GH95 family is strict to al,2-linked fucosyl residues.
- a-L-Fucosidases generally hydrolyse the terminal fucosyl residue from glycans.
- Fucosidases which may be employed in the context of the present invention, may also comprise engineered fucosidases.
- Such engineered fucosidases preferably comprise engineered a-L- fucosidases, preferably engineered fucosidases derived from fucosidases as described above, e.g. an engineered a-l,2-L-fucosynthase from Bifidobacterium bifidum, a-L-fucosynthases from Sulfolobus solfataricus and Thermotoga maritime, etc.
- Such engineered fucosidases show an acceptor dependent regioselectivity and are devoid of product hydrolysis activity.
- engineered fucosidases preferably comprise a-L-fucosidase from Thermotoga maritima, which has also been recently converted into an efficient a-L-transfucosidase by directed evolution (see Osanjo et al. Biochemistry 46, 1022 (2007)).
- the enzyme having a fucosidase and/or trans-fucosidase and/or fucosynthase activity may be selected from a-L-fucosidases derived from Thermotoga maritima MSB 8,
- Bifidobacterium dentium Bdl or Lactobacillus casei BL23, etc.
- the enzyme having a fucosidase and/or trans-fucosidase and/or fucosynthase activity may be selected from following a-L-fucosidases as defined according to the following deposit numbers gi
- 4980806 Thermotoga maritima MSB8
- 13816464 Sulfolobus solfataricus P2
- 34451973 Bifidobacterium bifidum JCM 1254
- 310867039 (Bifidobacterium bifidum PRL2010), gi
- step d) provides a compound of formula 10B and salts thereof
- R', Ri, Ri 6 and A are as defined above.
- the enzymes comprising transsialidase or transfucosidase activity as defined above may also comprise engineered enzymes comprising transsialidase or transfucosidase activity. It is particularly envisaged that wild type or mutated glycosidases displaying a-transsialidase or a- transfucosidase activity can be used in the present invention to produce the target oligosaccharides. Preparation of such enzymes is preferably carried out via site directed mutagenesis approaches or directed evolution.
- mutants are created via site directed mutagenesis approaches, preferably by introduction of point mutations. This technique generally requires reliance on the static 3D protein structure.
- the mutations generally affect the active site of the enzymes such that they lose their ability to degrade their transglycosylation products but remain capable of synthesis.
- a preferred strategy consists of the replacement of the catalytic nucleophile by a non-nucleophilic residue. This modification results in the formation of an inactive mutant or an altered enzyme with reduced transglycosylation activity due the lack of appropriate environment for the formation of the reactive host-guest complex for transglycosylation.
- a more active glycosyl donor e.g.
- glycosynthase e.g. fucosynthase
- their development represents one of the major advances in the use of glycosidases for synthetic purposes.
- the glycosynthase concept can be applied to all GH specificities and offer a large panel of enzymes potentially able to synthesize various oligosaccharides with very high yields, up to 95%.
- the second preferred technique is called directed evolution.
- This strategy comprises random mutagenesis applied to the gene of the selected glycosidase and generates thus a library of genetically diverse genes expressing glycosidase. Generation of sequence diversity can be performed using well-known methodologies, the most preferable being the error prone polymerase chain reaction (epCR) method.
- epCR error prone polymerase chain reaction
- This gene library may be inserted into suitable microorganisms such as E. coli or S. cerevisiae for producing recombinant variants with slightly altered properties.
- Clones expressing improved enzymes are then identified with a fast and reliable screening method, selected and brought into a next round of mutation process.
- the recursive cycles of mutation, recombination and selection are continued as far as mutant(s) with the desired activity and/or specificity is/are evolved.
- different high-throughput screening methodologies for glycosidases including glycosynthases have been developed. Applying these approaches, effective engineered transglycosidases, including new and more efficient glycosynthases can and have been created and isolated.
- An a-L-fucosidase from Thermotoga maritima has been recently converted into an efficient a-L-transfucosidase by directed evolution.
- the transferase/hydrolysis ratio of the evolved enzyme was 30 times higher than the native enzyme (see Osanjo et al. Biochemistry 46, 1022 (2007)).
- Proteins comprising a transglycosidase and/or a glycosynthase activity as defined above may also comprise fragments or variants of those protein sequences.
- Such fragments or variants may typically comprise a sequence having a sequence identity with one of the above mentioned proteins sequences of at least 70%, more preferably at least 80%, equally more preferably at least 85%, even more preferably at least 90% and most preferably at least 95% or even 97%, 98% or 99% as compared to the entire wild type sequence on amino acid level.
- “Fragments” of proteins or peptides in the context of the present invention may also comprise a sequence of a protein or peptide as defined herein, which is, with regard to its amino acid sequence N-terminally, C-terminally and/or intrasequentially truncated compared to the amino acid sequence of the original (native) protein. Such truncation may thus occur either on the amino acid level or correspondingly on the nucleic acid level.
- a sequence identity with respect to such a fragment as defined herein may therefore preferably refer to the entire protein or peptide as defined herein or to the entire (coding) nucleic acid molecule of such a protein or peptide.
- fragments of nucleic acids in the context of the present invention may comprise a sequence of a nucleic acid as defined herein, which is, with regard to its nucleic acid molecule 5'-, 3'- and/or intrasequentially truncated compared to the nucleic acid molecule of the original (native) nucleic acid molecule.
- a sequence identity with respect to such a fragment as defined herein may therefore preferably refer to the entire nucleic acid as defined herein.
- “Variants” of proteins or peptides as defined in the context of the present invention may be encoded by the nucleic acid molecule of a polymeric carrier cargo complex.
- a protein or peptide may be generated, having an amino acid sequence which differs from the original sequence in one or more mutation(s), such as one or more substituted, inserted and/or deleted amino acid(s).
- these fragments and/or variants have the same biological function or specific activity compared to the full-length native protein, e.g. its specific antigenic property.
- “Variants” of proteins or peptides as defined in the context of the present invention may also comprise conservative amino acid substitution(s) compared to their native, i.e. non- mutated physiological, sequence. Those amino acid sequences as well as their encoding nucleotide sequences in particular fall under the term variants as defined herein. Substitutions in which amino acids, which originate from the same class, are exchanged for one another are called conservative substitutions. In particular, these are amino acids having aliphatic side chains, positively or negatively charged side chains, aromatic groups in the side chains or amino acids, the side chains of which can enter into hydrogen bridges, e.g. side chains which have a hydroxyl function.
- an amino acid having a polar side chain is replaced by another amino acid having a likewise polar side chain, or, for example, an amino acid characterized by a hydrophobic side chain is substituted by another amino acid having a likewise hydrophobic side chain (e.g. serine (threonine) by threonine (serine) or leucine (isoleucine) by isoleucine (leucine)).
- Insertions and substitutions are possible, in particular, at those sequence positions which cause no modification to the three-dimensional structure or do not affect the binding region. Modifications to a three-dimensional structure by insertion(s) or deletion(s) can easily be determined e.g.
- CD spectra circular dichroism spectra
- variants of proteins or peptides as defined herein may also comprise those sequences, wherein nucleotides of the nucleic acid are exchanged according to the degeneration of the genetic code, without leading to an alteration of the respective amino acid sequence of the protein or peptide, i.e. the amino acid sequence or at least part thereof may not differ from the original sequence in one or more mutation(s) within the above meaning.
- nucleic acid sequences or amino acid sequences as defined herein preferably the amino acid sequences encoded by a nucleic acid sequence of the polymeric carrier as defined herein or the amino acid sequences themselves
- the sequences can be aligned in order to be subsequently compared to one another. Therefore, e.g. a position of a first sequence may be compared with the corresponding position of the second sequence. If a position in the first sequence is occupied by the same component as is the case at a position in the second sequence, the two sequences are identical at this position. If this is not the case, the sequences differ at this position.
- the percentage to which two sequences are identical is then a function of the number of identical positions divided by the total number of positions including those positions which are only occupied in one sequence.
- the percentage to which two sequences are identical can be determined using a mathematical algorithm.
- a preferred, but not limiting, example of a mathematical algorithm which can be used is the algorithm of Karlin et al. Proc. Natl. Acad. Sci. USA 90, 5873 (1993) or Altschul et al. Nucleic Acids Res. 25, 3389 (1997). Such an algorithm is integrated in the BLAST program. Sequences which are identical to the sequences of the present invention to a certain extent can be identified by this program.
- the enzymes used in step d) may be provided in a free form or alternatively be bound to or are immobilized onto a surface. Binding to or immobilization onto a surface may be carried out e.g. via electrostatic bonds, van der Waals-bonds, covalent bonds, etc. Binding to or immobilization onto a surface may be furthermore carried out, using a covalent linker or a crosslinker, or a tag, as known to a skilled person for purification of proteins. Such tags comprise, inter alia, e.g. affinity tags or chromatography tags. Affinity tags may include e.g.
- CBP chitin binding protein
- MBP maltose binding protein
- GST glutathione-S-transferase
- Strep-Tag Strep-Tag
- the poly(His) tag is a widely- used protein tag, that binds to metal matrices. Chromatography tags are used to alter
- the surface may be the surface of a bioreactor, or any suitable reaction chamber.
- the enzymatic reaction can be carried out as described in WO 2012/007588, WO 2012/127410 or WO 2012/156897, preferably occurs with a concentration of respective enzyme in a concentration of 1 mU/1 to 1000 U/l, preferably 10 mU/1 to 100 U/l, when the activity capable of forming 1 ⁇ of specific product for a defined protein starting from a defined educt is defined as 1 unit (U), e.g. for a glycotransferase the production of a glycose-containing complex carbohydrate at 37 °C in 1 minute.
- the activity of each enzyme as defined herein may be assessed with respect to its naturally occurring or engineered substrate.
- the incubation may be carried out in a reaction medium, preferably an aqueous medium, comprising the compound obtained according to step c) and optionally water; a buffer such as a phosphate buffer, a carbonate buffer, an acetate buffer, a borate buffer, a citrate buffer and a tris buffer, or combinations thereof; alcohol, such as methanol and ethanol; ester such as ethyl acetate; ketone such as acetone; amide such as acetamide; and the like. Furthermore, the incubation may be carried out in a reaction medium as defined above, wherein optionally a surfactant or an organic solvent may be added, if necessary.
- a surfactant or an organic solvent may be added, if necessary.
- any surfactant capable of accelerating the formation of a complex carbohydrate as defined according to the present invention as a possible product of the invention can be used as the surfactant.
- examples include non-ionic surfactants such as poly oxy ethylene octadecylamine (e.g., Nymeen S-215, manufactured by Nippon Oil & Fats); cationic surfactants, such as cetyltrimethylammonium bromide and alkyldimethyl benzylammoniumchloride (e.g., Cation F2-40E, manufactured by Nippon Oil & Fats); anionic surfactants such as lauroyl sarcosinate; tertiary amines such as alkyldimethylamine (e.g., Tertiary Amine FB, manufactured by Nippon Oil & Fats); and the like, which are used alone or as a mixture of two or more.
- non-ionic surfactants such as poly oxy ethylene octade
- the surfactant may be used generally in a concentration of 0.1 to 50 g/1.
- the organic solvent may include xylene, toluene, fatty acid alcohol, acetone, ethyl acetate, and the like, which may be used in a concentration of generally 0.1 to 50 ml/1.
- the incubation may be furthermore carried out in a reaction medium as defined above, preferably having a pH 3 to 10, pH 5 to 10, preferably pH 6 to 8.
- the incubation may be furthermore carried out at a temperature of about 0 °C to about 100 °C, preferably at a temperature of about 10 to about 50 °C, e.g. at a temperature of about 20 °C to about 50 °C.
- inorganic salts such as MnCl 2 and MgCl 2 , may be added, if necessary.
- the incubation according to step d) of the method of the present invention may be carried out in a bioreactor.
- the bioreactor is preferably suitable for either a continuous mode or a discontinuous mode. If carried out in a continuous mode, the method preferably provides for a continuous flow of compounds and/or enzymes as necessary, preferably by continuously providing educts of the reaction to the reaction mixture and continuously removing products from the reaction mixture, while maintaining the concentration of all components, including enzymes at a predetermined level.
- the enzymes used in a continuous mode may be added either in free form or as bound or immobilized to a surface.
- step e) of the present invention a compound obtained in step d) is subjected to catalytic hydrogenolysis and/or anomeric deprotection.
- the catalytic hydrogenolysis is performed when, in a compound of formula 10 obtained in step d), R' is -N 3 or -OR' 6 wherein R' 6 is a group removable by hydrogenolysis or - (CH 2 ) n -N3, or when Ri 6 is a moiety C wherein at least one of the Rn and Ris groups is a group removable by hydrogenolysis and R' is 2-trimethylsilyl-ethyloxy.
- Such catalytic hydrogenolysis typically takes place in a protic solvent or in a mixture of protic solvents.
- a protic solvent may be selected from the group consisting of water, acetic acid or Ci-C 6 alcohols.
- a mixture of one or more protic solvents with one or more suitable aprotic organic solvents partially or fully miscible with the protic solvent(s) may also be used.
- suitable aprotic organic solvents partially or fully miscible with the protic solvent(s) (such as THF, dioxane, ethyl acetate or acetone) may also be used.
- Water, one or more Ci-C 6 alcohols or a mixture of water and one or more Ci-C 6 alcohols are preferably used as the solvent system. Solutions containing the carbohydrate derivatives in any concentration or suspensions of the carbohydrate derivatives in the solvent(s) used are also applicable.
- the reaction mixture is stirred at a temperature in the range of 10-100 °C, preferably between 20-50 °C, in a hydrogen atmosphere of 1-50 bar absolute (100 to 5000 kPa) in the presence of a catalyst such as palladium, Raney nickel or any other appropriate metal catalyst, preferably palladium on charcoal or palladium black, until reaching the completion of the reaction.
- a catalyst such as palladium, Raney nickel or any other appropriate metal catalyst, preferably palladium on charcoal or palladium black
- Transfer hydrogenolysis may also be performed, when the hydrogen is generated in situ from cyclohexene, cyclohexadiene, formic acid or ammonium formate.
- Addition of organic or inorganic bases or acids and/or basic and/or acidic ion exchange resins can also be used to improve the kinetics of the hydrogenolysis.
- anomeric azide group (R' is -N 3 ) in a the above-mentioned compound of formula 10 is reduced to amino under the conditions disclosed above, and the thus formed glycosyl amine easily undergoes hydrolysis to give the anomerically unprotected compound of formula 1 (R is OH).
- R' -0-(CH 2 ) n -N3 in a compound of formula 10 is converted to -0-(CH 2 ) n - H 2 under these conditions.
- R' group being -N 3 or -OR' 6 wherein R' 6 is selected from allyl optionally substituted by one or more methyl or 2-trimethylsilyl-ethyl in a compound of formula 10 obtained in step d) - provided that in moiety C, if it is present in a compound of formula 10, both Rn and Ri 8 groups are H - can be converted to OH in an anomeric deprotection reaction.
- the R' group being -N 3 in a compound of formula 10 can be reduced by complex metal hydrides like NaBH 4 , or by PPh 3 or Cu/Zn.
- 2- Trimethylsilyl-ethyl glycosides (R' is -OR' 6 wherein R' 6 is 2-trimethylsilyl-ethyl) can be deprotected with fluoride ion (from BF " ), an anion of a strong acid (e.g. trifluoroacetate) or a Lewis-acid (BF 3 -etherate, ZnCl 2 , SnCl 4 , FeCl 3 ) followed by careful hydrolysis of the resulting glycosyloxy derivative substituted with the remains of the reagent that is used (see Jansson et al. J. Org. Chem. 53, 5629 (1988)).
- fluoride ion from BF "
- an anion of a strong acid e.g. trifluoroacetate
- a Lewis-acid BF 3 -etherate, ZnCl 2 , SnCl 4 , FeCl 3
- R' is -N 3 or -OR' 6 wherein R' 6 is selected from allyl optionally substituted by one or more methyl or 2-trimethylsilyl-ethyl, and in moiety C at least one of the Rn and Ri 8 groups is a group removable by hydrogenolysis
- the anomeric deprotection reaction is followed by catalytic hydrogenolysis to give compounds of formula 1 wherein R is -OH and R 2 is fucosyl moiety.
- R is selected from -OH, -N 3 and -OR 5 wherein 5 is selected from allyl optionally substituted by one or more methyl, propargyl optionally substituted by one or more methyl, 2-trimethylsilyl-ethyl, -(CH 2 ) n -NH 2 and -(CH 2 ) n -N3 wherein integer n is i to 10, preferably 2 or 3,
- Ri is selected from sialyl moiety, -SO 3 H and -CH(R 5 )-COOH wherein R 5 is selected from H, alkyl and benzyl,
- R 2 is selected from H and fucosyl moiety
- R 3 is selected from H and sialyl moiety
- R 4 is selected from H and fucosyl moiety, provided that one but only one of R 3 and R 4 is H, and
- A is selected from a bond and a divalent carbohydrate linker, can be effectively synthesized.
- Both the anomerically deprotected derivatives (R is -OH), and the glycosides listed above have synthetic usefulness.
- the unsaturated bond of glycosides having -N 3 , -0-(CH 2 ) n -N 3 , -O-allyl or -O-propargyl aglycon or the -0-(CH 2 ) n -NH 2 group can be functionalized by a wide variety of selective and mild water-compatible chemical reactions. Thus e.g.
- the azido group of a compound of formula 1 can be brought into "click chemistry" with an alkyne to form bioconjugates (and so can the propargyl derivatives of formula 1 with an azido reagent).
- the allyl functionality can be converted to other functional groups by addition reactions to the double bond or by ozonolysis.
- the amino function can be used to make an amide or urea linkage to bind a compound of formula 1 to (bio)macromolecules/polymers.
- the present method readily provides the synthesis of complex sialylated and/or fucosylated 3'-0- galactosyl-6-O-sialyl-GlcNAc or -GalNAc derivatives, glycosides and analogs thereof, by a unique combination of chemical and enzymatic glycosylation steps in the reaction sequence.
- the process opens the possibility, via the permutation of the mandatory and optional glycosylation steps, to obtain a number of compounds being substituted by sialyl and/or fucosyl moiety/moieties in various positions.
- sialyl residue at position 6 can be replaced by simpler charged residues like carboxymethyl or sulfate while maintaining the biological effect of the parent sialylated compounds (see e.g. Chachadi et al. Glycoconj. J. 28, 49 (2011), WO 2011/130332, Liao et al. J. Am. Chem. Soc. 132, 14849 (2010), Wang et al. Proc. Nat. Acad. Sci. USA 106, 18137 (2009), Schwardt et al. J. Med. Chem. 52, 989 (2009)).
- the method disclosed above gives the chance for the synthesis of these kinds of analogs as well when the chemical sialylation in step a) is replaced by a chemical carboxymethylation or sulfation.
- the present method is advantageous in that these various groups of compounds can be obtained in a simpler manner and/or in fewer steps.
- Ando et al. ⁇ Carbohydr. Res. 338, 503 (2003)) taught that 6-O-sialylation of "bulky" acceptors where the OH-group to be sialylated belonged to an internal GlcNAc residue was not favourable because only low yields could be achieved. Therefore it was proposed to sialylate acceptors having a terminal GlcNAc moiety.
- the present inventors found that compounds of formula 2 having internal GlcNAc can be sialylated effectively.
- all the prior art methods (Ando et al. J. Carbohydr. Chem. 20, 425 (2001) and
- the present method avoids the fabrication and use of this type of difficult to obtain disaccharide donor, and instead introduces a direct enzymatic sialylation of intermediates of formula 7 obtained in step c) of the present method that takes place in good regio- and stereoselectivity.
- the intermediate of formula 7 can be a substrate for enzymatic fucosylation as well, and therefore the method is able to provide a diverse group of variously sialylated and/or fucosylated 3'-0-galactosyl-6-0-sialyl-GlcNAc or -GalNAc derivatives and analogs thereof.
- compounds of formula 1 wherein A is a divalent carbohydrate linker are preferred. More preferably, compounds of formula 1 are human milk oligosaccharides listed in Table 1 above and derivatives, even more preferably F-LST b, DS-LNT or FDS-LNT I and derivatives. Accordingly, a compound of formula 2D falling into the scope of compounds of formula 2 wherein Y and R 7 are as defined above,
- R' is as defined above, preferably R' is OR" wherein R" is a group removable by
- B' is a divalent lactosyl moiety having the R' group on its C-l anomeric carbon atom and attached via its 3'-OH group to the lacto-N-biose residue of the compound of formula 2D, optionally substituted by a fucosyl moiety on its 3-OH or by a N-acetyllactosaminyl moiety on its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety on its 6-OH, or by a fucosyl on its 3-OH or 2' -OH, and the functional groups of the divalent lactosyl residue B' are protected, preferably the free OH groups are acylated (e.g.
- R', B', Y and R 7 are as defined above, and Ri9 is selected from -S0 3 H, -CH(R 5 )-COORi 0 and moiety H
- R20 is independently acyl
- R21 is selected from H and acyl, more preferably a compound of formula 2E wherein Y is selected from - HAc and
- R 7 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl
- R20 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl
- R21 is selected from H, acetyl, benzoyl or 4-chlorobenzoyl, is reacted with a sialyl donor of formula 3 defined above to give a compound of formula 4E
- Y is selected from -NHAc, haloalkanoylamido, -NAc 2 , haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and azido, preferably -NHAc and
- R 7 is independently acyl, preferably R 7 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
- R 8 is independently acyl, preferably Rs groups are acetyl,
- R 2 o is independently acyl, preferably R 2 o groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
- R 21 is selected from H and acyl, preferably R 2 i is selected from H, acetyl, benzoyl or 4- chlorobenzoyl, and
- Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl.
- a compound of formula 4D preferably of formula 4E, is optionally fucosylated with a fucosyl donor of formula 5A
- XA is selected from alkylthio and optionally substituted phenylthio, preferably -SPh,
- R 22 and R 23 are, independently, selected from a group removable by hydrogenolysis and acyl, preferably acetyl, pivaloyl, benzoyl and 4-chlorobenzoyl, to give rise to a compound of formula 6D wherein R', B', Y, R 7 and R19 are as defined above, and R24 is a moiety I
- R" is a group removable by hydrogenolysis
- Y is selected from -NHAc, haloalkanoylamido, -NAc 2 , haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and azido, preferably -NHAc and
- R 7 is independently acyl, preferably R 7 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
- R 8 is independently acyl, preferably Rs groups are acetyl,
- R 2 o is independently acyl, preferably R 20 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
- R 2 i is selected from H and acyl, preferably R 2 i is selected from H, acetyl, benzoyl or 4- chlorobenzoyl, and Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl, and
- R-24 is moiety I as defined above.
- step c) a compound of formula 4D obtained in step a) or a compound of formula 6D obtained in step b) is subjected to deprotective treatment and, where necessary, transformation of Y to
- moiety A' is a divalent lactosyl moiety having the R' group on its C-1 anomeric carbon atom and attached via its 3'-OH group to the lacto-N-biose residue of the compound of formula 7A, optionally substituted by a fucosyl moiety on its 3-OH or by a N-acetyllactosaminyl moiety on its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety on its 6-OH, or by a fucosyl on its 3-OH or 2' -OH,
- Ri is selected from sialyl, -SO 3 H and -CH(R 5 )-COOH, wherein R 5 is as defined above, and
- Ri6 is selected from H and a moiety of formula C
- Rn and Ris independently, are selected from H and a group removable by hydrogenolysis, comprising the steps of i) base catalysed transesterification to remove O-acyl groups,
- step c) of the method a compound of formula 4E or a compound of formula 6E defined above is converted into a compound of formula 7B
- R" is a group removable by hydrogenolysis
- Ri6 is selected from H and moiety C defined above, preferably H comprising: i) base catalysed transesterification deprotection, preferably NaOMe/MeOH treatment, ii) basic hydrolysis, preferably NaOH/MeOH treatment, and
- haloalkoxycarbonylamino, 2,3-diphenylmaleimido, 2,3-dimethylmaleimido and azido, preferably trichloroacetamido, is deprotected to amino under the conditions used in step ii) or by other azido-amino transformations disclosed above.
- a compound of formula 7A is sialylated or fucosylated under the action of a sialidase or fucosidase having transsialidase or transfucosidase/fucosynthase activity, respectively, to give a compound of formula IOC
- R', A', Ri, R 3 , R 4 and Ri 6 are as defined above, provided that one but only one of the R 3 and R 4 is H.
- a compound according to formula 7B is reacted with a sialyl donor, preferably a N- acetyl neuraminyl donor selected from 3'-0-sialyllactose or /?-nitrophenyl N-acetyl-a- neuraminoside in the presence of transsialidase, preferably a transsialidase listed in Table 2 above, to make a compound of formula 10D
- a compound of formula 7A is subjected to enzymatic fucosylation under the action of a fucosidase having transfucosidase/fucosynthase activity, preferably those listed in Table 3, in the presence of a fucosyl donor selected from 2-O-fucosyllactose or fucosyl fluoride to give a compound of formula 10E
- R" and Ri 6 are as defined above.
- step e if it is desired, a compound of formula IOC is subjected to catalytic
- the hydrogenolysis of a compound of formula 10D provides a compound of formula IB
- R 2 is selected from H and fucosyl, that is DS-LNT and FDS-LNT I, respectively.
- R 2 is selected from H and fucosyl, that is DS-LNT and FDS-LNT I, respectively.
- R 2 is as defined above, is easily accessible.
- R 2 is H in a compound of formula 1C that corresponds to F-LST b.
- the method according to the first aspect of the invention involves useful novel synthetic intermediates for the synthesis of sialylated/fucosylated oligosaccharides.
- the second aspect of the invention provides a compound of formula 11 and salts thereof
- R' is selected from -N 3 and -OR' 6 which R' 6 is selected from allyl optionally substituted by one ore more methyl, propargyl optionally substituted by one ore more methyl, a group removable by hydrogenolysis, 2-trimethylsilyl-ethyl and -(CH 2 ) n -N3 wherein integer n is 1 to 10, preferably 2 or 3,
- A' is a divalent lactosyl moiety having the R' group on its C-l anomeric carbon atom and attached via its 3'-OH group to the lacto-N-biose residue of the compound of formula 11, A' being optionally substituted by a fucosyl moiety on its 3-OH or by a N-acetyllactosaminyl moiety on its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety on its 6-OH, or by a fucosyl on its 3-OH or 2' -OH,
- Ri is selected from sialyl moiety, -SO 3 H and -CH(R 5 )-COOH wherein R 5 is selected from H, alkyl and benzyl,
- R 3 is selected from H and sialyl moiety
- R 4 is selected from H and fucosyl moiety, provided that at least one of R 3 and R 4 is H, and Ri 6 is selected from H and moiety C, preferably H,
- Rn and Ris independently, are selected from H and a group removable by hydrogenolysis provided that the di sodium salt of the following compound is excluded: wherein R' is 2- trimethylsilyl-ethyl, A' is unsubstituted lactosyl, Ri and R 3 are sialyl moieties, and R 4 and Ri 6 are H.
- R' is -OR" wherein R" is a group removable by hydrogenolysis.
- a compound of formula 11 can be either an a- or ⁇ -anomer or an anomeric mixture of a- and ⁇ - anomers. It can be crystalline solid, oil, syrup, precipitated amorphous material or spray dried product. If crystalline, a compound of formula 11 can exist either in an anhydrous or hydrated crystalline form by incorporating one or several molecules of water into its crystal structure.
- a compound of formula 11 can exist as a crystalline substance, incorporating ligands such as organic molecules and/or ions into its crystal structure.
- a preferred compound of formula 11 is a compound of formula IOC being a precursor for DS-LNT, FDS-LNT I and F-LST b and analogs thereof
- R', A', Ri, R 3 , R 4 and Ri 6 are as defined above, provided that one but only one of R 3 and R 4 is H, particularly a compound of formula 10D
- Ri6 is selected from H and moiety C defined above, preferably H, and also particularly a compound of formula 10E wherein R" is a group removable by hydrogenolysis, and Ri 6 is selected from H and moiety C defined above, preferably H.
- a compound of formula 11 is characterized by formula 7A
- a preferred compound of formula 7A is a compound of formula 7B
- R" is a group removable by hydrogenolysis
- Ri 6 is selected from H and moiety C defined above, preferably H.
- Step 1 A solution of the compound from example 1 (6.57 g) in dry MeOH (50 ml) was treated with 25% (w/w) methanolic MeONa (0.5 ml) and stirred at r.t. for 21 hours, quenched with AcOH and evaporated in vacuo. The obtained residue (6.14 g) was sonicated with Et 2 0 and filtered (5x40 ml). Alternatively, methyl benzoates could be extracted by partitioning between water and organic solvent such as ether or hexane. The compound was dried in vacuum oven to give 3.90 g of a crude material.
- Step 2 The obtained solid from step 1 was taken up in water (30 ml) containing NaOH (0.514 g) and stirred at r.t. for 3 days and then at 60 °C for 2 hrs. The mixture was neutralized with AcOH and extracted with ether. The aqueous phase gave 5.52 g of solid after evaporation.
- LC-HRMS calc. for [C 4 2H 66 N 2 028-H] " 1045.3729, found 1045.3732.
- Step 3 The product from step 2 was taken up in pyridine and Ac 2 0 mixture (50 ml each) and stirred at 50 °C for 30 min then at r.t. for 18 hours. Extra amount of acetic anhydride (20 ml) was added and the mixture was stirred at 40 °C for 2 hours. The volatiles were removed in vacuo and the residue was coevaporated with toluene then partitioned between CH 2 C1 2 (200 ml) and 0.5 M HC1 (50 ml). The aqueous phase was re-extracted with CH 2 C1 2 (3x20 ml). Combined organic solution was dried and evaporated to give 5.0 g of crude product as white solid. It was chromatographed on silica to give 3.89 g of the title compound (80.5 %).
- reaction mixtures were stirred at 20 °C for 24 hours and the conversion of the acceptor to the desired product was determined by HPLC. Conversion yield was estimated to be from 70 to 80 % when using 3'-0-sialyllactose or from 40 to 50 % when using /?-nitrophenyl N-acetyl-a-neuraminoside.
- Reaction mixtures were loaded on a Dowex 1 (HC0 3 " -form) column. After washing with distilled water, the mono-acidic compounds (3'-0-sialyllactose or sialic acid and the remaining acceptor) were eluted with a 60 mM NaHC0 3 solution. The product was then eluted with a 125 mM NaHC0 3 solution. The eluted fractions containing acidic oligosaccharides were analysed by HPLC and pooled. The NaHC0 3 was removed by treating with Amberlite IR120 (H + form) until pH 3.0 was reached. The pH was then adjusted to 6.0 with NaOH. The mono-acidic fractions were freeze-dried and reused with another enzymatic run. The fractions containing the products were freeze-dried.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Polymers & Plastics (AREA)
- Medicinal Chemistry (AREA)
- Materials Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Saccharide Compounds (AREA)
Abstract
This invention relates to a chemo-enzymatic synthesis of oligosaccharidesof formula 1 wherein R is selected from -OH, -N3 and -OR6 wherein R6 is selected from allyl optionally substituted by one or more methyl, propargyl optionally substituted by one or more methyl, 2-trimethylsilyl-ethyl, -(CH2)n -NH2 and -(CH2)n -N3 wherein integer n is to 10, preferably 2 or 3, R1 is selected from sialyl moiety, -SO3H and -CH(R5)-COOH wherein R5 is selected from H, alkyl and benzyl, R2 is selected from H and fucosyl, R3 is selected from H and sialyl, R4 is selected from H and fucosyl, provided that one but only one of R3 and R4 is H, and A is selected from a bond and a divalent carbohydrate linker, having important biological activities and significant commercial value for the pharmaceutical and food industry.
Description
SYNTHESIS OF SIALYLATED FUCOSYLATED OLIGOSACCHARIDES
FIELD OF THE INVENTION
This invention relates to a chemo-enzymatic synthesis of oligosaccharides having important biological activities and significant commercial value for the pharmaceutical and food industry.
BACKGROUND OF THE INVENTION
In recent years, the manufacture and commercialization of complex carbohydrates, particularly secreted oligosaccharides, have increased significantly due to their roles in numerous biological processes occurring in living organisms. Secreted oligosaccharides such as human milk
oligosaccharides (HMOs), mucin oligosaccharides and Lewis type oligosaccharides have gained much interest and have become important commercial targets for nutrition and therapeutic applications. In particular, the synthesis of these oligosaccharides has increased significantly due to their role in numerous biological processes occurring in humans.
Many secreted oligosaccharides contain sialosides, mainly N-acetyl-neuraminic acid, that are most frequently found at the terminal end of oligosaccharides. The linkages of sialic acids, in which they are bound to galactose, N-acetyl-galactosamine and N-acetyl-glucosamine, are most commonly a- 2,3- and a-2,6-ketosidic bonds. Their terminally exposed positions allow sialoconjugates to be recognized by receptors of cells, viruses and bacteria, thus to be involved in a wide variety of biological processes, such as tumour metastasis, cell differentiation, cell-cell interactions, etc.
N- Acetyl-glucosamine or -galactosamine, to which a galactose is bound with β1-3 interglycosidic linkage (that is lacto-N-biose and Gaip(l-3)-GalNAc, respectively), can be substituted by a sialic acid at its 6-position. These trisaccharide motifs, often substituted with further sialic acid and/or fucosyl residue(s), can be found naturally in various glycoconjugates having diverse biological roles.
Thus the tetrasaccharide Neu5Aca(2-3)-Galp(l-3)-[Neu5Aca(2-6)-]GlcNAc (disialyl Lewis C) has been identified as a motif in cancer specific antigens (EP-A-264911, WO 94/03629). Its 3- aminopropyl glycoside and sulfated analogue of this glycoside bound to gold particle have been tested for virus binding (WO 2011/130332).
Aminopropyl glycoside of Neu5Aca(2-3)-Gaip(l-3)-[Neu5Aca(2-6)-]GalNAc has been shown to bind to Sclerotium rolfsii lectin (SRL) (Chachadi et al. Glycoconj. J. 28, 49 (2011)).
The pentasaccharide 6" '-0-sialyl-LNT (LST b, Galp(l-3)-[Neu5Aca(2-6)-]GlcNAcp(l-3)-Galp(l- 4)-Glc) is a human milk oligosaccharide and represents a common structural element in some other sialylated human milk oligosaccharides (Urashima et al. : Milk Oligosaccharides, Nova Medical Books, NY, 2011); they are listed in Table 1.

Table 1. Selected sialylated HMOs
DS-LNT has recently been found to be preventive against necrotising enterocolitis (NEC) in rat model (Jantscher-Krenn et al. Gut 61, 1417 (2012), WO 2012/106665).
More thorough research studies of cell-cell interactions, mechanism of cancer malignancy and/or determination of the bioactive conformation of compounds that are involved in protein-glycan recognition processes have required greater quantities of such oligosaccharides, but present enzymatic and/or microbial methods have not been able to provide with them in sufficient amounts and/or purities. Synthetic chemical methods, adapted to provide them in sufficient purity, have required many expensive reaction steps and the extensive use of protection-deprotection sequences during the synthesis, and afford compounds only in mg quantities, as demonstrated by the synthesis of an a(2-3)/a(2-6)-disialyl lactotetraosyl ceramide and a disialyl Lewis A ganglioside (Ando et al. J. Carbohydr. Chem. 20, 425 (2001), Carbohydr. Res. 338, 503 (2003)) or truncated analogs thereof (Bhunia et al. ChemBioChem 9, 2941 (2008), Hsu et al. Chem. Eur. J. 16, 1754 (2010)).
There has been a need, therefore, for an efficient method of making a diverse group of sialylated and/or fucosylated 3'-0-galactosyl-6-0-sialyl-GlcNAc or -GalNAc derivatives and their analogs. There has been a particular need for a method that can be readily scaled-up for potential industrial use.
SUMMARY OF THE INVENTION
The first aspect of the invention relates to a method for making a sialylated or fucosylated 3-0- galactosyl-GlcNAc or -GalNAc derivative of formula 1 and salts thereof

1 wherein R is selected from -OH, -N3 and -OR5 wherein 5 is selected from: allyl optionally substituted by one or more methyl, propargyl optionally
substituted by one or more methyl, 2-trimethylsilyl-ethyl, -(CH2)n-NH2 and - (CH2)n-N3 wherein n is an integer of 1 to 10, preferably 2 or 3,
Ri is selected from sialyl moiety, -SO3H and -CH(R5)-COOH wherein R5 is selected from H, alkyl and benzyl,
R2 is selected from H and fucosyl,
R3 is selected from H and sialyl,
R4 is selected from H and fucosyl, provided that one but only one of R3 and R4 is H, and
A is selected from a bond and a divalent carbohydrate linker, comprising the steps of: a) sialylation, sulfation or carboxymethylation of a compound of formula 2

2
wherein R' is selected from -N3 and -OR'6 wherein R'6 is selected from allyl optionally substituted by one or more methyl, propargyl optionally
substituted by one or more methyl, a group removable by hydrogenolysis, 2- trimethylsilyl-ethyl and -(CH2)n-N3 wherein n is an integer of 1 to 10,
preferably 2 or 3,
R7 is independently acyl,
Y is selected from -NHAc, haloalkanoylamido, -NAc2, haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and -N3, and
B is selected from a bond and a divalent carbohydrate linker in protected form, b) optional fucosylation of the sialylated, sulfated or carboxymethylated compound of formula 2 obtained in step a), c) de-O-acylation and/or basic hydrolysis, optional mild acidic hydrolysis and optional transformation of Y to -NHAc of the compound obtained in step a) or step b), d) sialylation or fucosylation of the compound obtained in step c), and e) optional catalytic hydrogenolysis and/or anomeric deprotection of the compound
obtained in step d).
The second aspect of the invention provides a compound of formula 11 and salts thereof

11 wherein R' is selected from -N3 and -OR'6 which R'6 is selected from allyl optionally substituted by one ore more methyl, propargyl optionally substituted by one ore more methyl, a group removable by hydrogenolysis, 2-trimethylsilyl-ethyl and -(CH2)n-N3 wherein integer n is 1 to 10, preferably 2 or 3,
A' is a divalent lactosyl moiety having the R' group on its C-l (anomeric) carbon atom and attached via the 3'-OH group to the lacto-N-biosyl residue of the compound of formula 11, A' being optionally substituted by a fucosyl moiety on its 3-OH or by a N-acetyllactosaminyl
moiety on its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety on its 6-OH, or by a fucosyl on its 3 -OH or 2' -OH,
Ri is selected from sialyl moiety, -SO3H and -CH(R5)-COOH wherein R5 is selected from H, alkyl and benzyl,
R3 is selected from H and sialyl moiety,
R4 is selected from H and fucosyl moiety, provided that at least one of R3 and R4 is H, and Ri6 is selected from H and moiety C, preferably H,

C wherein Rn and Ris, independently, are selected from H and a group removable by hydrogenolysis, provided that the disodium salt of the following compound is excluded: R' is 2-trimethylsilyl- ethyloxy, A' is unsubstituted lactosyl, Ri and R3 are sialyl moieties, and R4 and Ri6 are H.
DETAILED DESCRIPTION OF THE INVENTION
In accordance with this invention, the term "acyl" preferably means an R*-C(=0)- group, wherein R* can be H, linear or branched alkyl with 1-6 carbon atoms or aryl including phenyl and naphthyl, preferably phenyl, such as formyl, acetyl, propionyl, butyryl, pivaloyl, benzoyl, etc. The alkyl and aryl residues can be unsubstituted or substituted one or several times, preferably 1-5 times, more preferably 1-3 times. The substituents can preferably be alkyl (for aromatic acyl), hydroxy, alkoxy, carboxy, oxo (for alkyl, forming a keto or aldehyde function), alkoxycarbonyl, alkylcarbonyl, formyl, aryl, aryloxycarbonyl, aryloxy, arylamino, arylcarbonyl, amino, mono- and dialkylamino, carbamoyl, mono- and dialkyl-aminocarbonyl, alkylcarbonylamino, cyano, alkanoyloxy, nitro, alkylthio and/or halogen (F, CI, Br, I). The substituents on aryl and alkyl moieties of acyl groups can modify the general chemical characteristics of the acyl group, and thereby the characteristics, such as stability, solubility and the ability to form crystals, of a molecule as a whole.
Herein, the term "group removable by hydrogenolysis" preferably means a protecting group whose C-0 bond can be cleaved by hydrogen in the presence of a catalytic amount of palladium, Raney nickel or any other conventional hydrogenolysis catalyst to regenerate the protected -OH group.
Such protecting groups are described in Wuts and Greene: Protective Groups in Organic Synthesis, John Wiley & Sons, 2007, and include benzyl, diphenylmethyl (benzhydryl), 1-naphthylmethyl, 2- naphthylmethyl and triphenylmethyl (trityl) groups, each of which can be optionally substituted by one or more of the following groups: alkyl, alkoxy, phenyl, amino, acylamino, alkylamino, dialkylamino, nitro, carboxyl, alkoxycarbonyl, carbamoyl, N-alkylcarbamoyl, N,N- dialkylcarbamoyl, azido, halogenalkyl or halogen. Preferably, such substitution, if present, is on the aromatic ring(s). A preferred protecting group is benzyl optionally substituted with one or more of the following groups: phenyl, alkyl and halogen, particularly unsubstituted benzyl, 4-chlorobenzyl, 3-phenylbenzyl and 4-methylbenzyl groups. These preferred and particularly preferred protecting groups have the advantage that the by-products of their hydrogenolysis are exclusively toluene or substituted toluene. Such by-products can easily be removed, even in multi-ton quantities, from water-soluble oligosaccharide products via evaporation and/or extraction processes.
Also herein, the term "a-sialyl" or "sialyl" preferably means - in accordance with Chen and Varki ACS Chem. Biol. 5, 163 (2010) - a glycosyl moiety of any naturally occurring or modified neuraminic acid or sialic acid derivative or an analogue thereof having an a-glycosidic linkage. Preferred neuraminic acid derivatives are N-acetyl- (Neu5Ac), N-glycolyl- (Neu5Gc) and deamino- neuraminic acid (3-deoxy-D-glycero-D-galacto-nonulosonic acid, KDN), as well as Neu5Ac, Neu5Gc and KDN derivatives that are derivatized with linkers, reactive functional groups, detectable labels or targeting moieties, and/or substituted at C-4, C-7-, C-8 and/or C-9, especially at C-9, with acyloxy, alkoxy, halogen or azido. The preferred sialyl moiety is the glycosyl residue of Neu5Ac (see Scheme 1). It should be noted that the term "sialyl" in the generally accepted trivial names of human milk oligosaccharides and Lewis type oligosaccharides always refers to Neu5Ac.

Scheme 1
Further herein, the term "fucosyl" preferably means a L-fucopyranosyl group (see Scheme 2) attached to a core oligosaccharide with a-interglycosidic linkage.

Scheme 2
Still further herein, the term "salt" in connection with sialylated, sulfated and carboxymethylated compounds described in the application preferably means an associated ion pair consisting of the negatively charged acid residue and one or more cations in any stoichiometric proportion. Cations, which are atoms or molecules with a positive charge, can be inorganic or organic. Preferred inorganic cations are ammonium ion, alkali metal, alkali earth metal and transition metal ions, more preferably Na+, K+, Ca2+, Mg2+, Ba2+, Fe2+, Zn2+, Mn2+ and Cu2+, more preferably K+, Ca2+, Mg2+, Ba2+, Fe2+ and Zn2+. Preferred basic organic compounds in positively charged form are diethyl amine, triethyl amine, diisopropyl ethyl amine, ethanolamine, diethanolamine, triethanolamine, imidazole, piperidine, piperazine, morpholine, benzyl amine, ethylene diamine, meglumin, pyrrolidine, choline, tris-(hydroxymethyl)-methyl amine, N-(2-hydroxyethyl)-pyrrolidine, N-(2- hydroxyethyl)-piperidine, N-(2-hydroxyethyl)-piperazine, N-(2-hydroxyethyl)-morpholine, L- arginine, L-lysine, oligopeptides having L-arginine or L-lysine unit or oligopeptides having free amino group on N-terminal, etc., all in protonated form. Such salts can be used in a conventional manner to modify the characteristics of the compounds of this invention, such as their stability, compatibility to excipients, solubility and ability to form crystals.
Yet further herein, the term "alkyl" preferably means a linear or branched hydrocarbon group with 1-6 carbon atoms, such as methyl, ethyl, ^-propyl, /'-propyl, «-butyl, /'-butyl, s-butyl, t-butyl, etc.; the term "haloalkanoylamido" preferably means a halogen substituted Ci-C6-alkanoylamido such as chloroacetamido, trichloroacetamido, trifluoroacetamido, etc.; the term "haloalkoxycarbonylamino" preferably means a Ci-C6-alkyloxycarbonyl- H-group substituted by one or more halogen atoms such as 2,2,2-trichloroethoxycarbonylamino, etc.; the term "optionally substituted phenyl" and "optionally substituted benzyl" preferably mean a phenyl or benzyl group, respectively, that is not substituted or substituted by 1 to 5, preferably 1 to 3, more preferably 1 or 2 substituents selected from the group consisting of alkyl, halogen, alkoxy, phenyl, nitro, haloalkyl, haloalkoxy, amino, alkylamino, dialkylamino, acylamino, carbamoyl, N-alkylcarbamoyl and Ν,Ν-dialkyl carbamoyl; the term "cycloalkylidene" preferably means a Cs-Cs-cycloalkylidene group optionally substituted
with alkyl(s) wherein the cycloalkyl group with the optional substituent(s) is of 3-8 carbon atoms, such as cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl or 4,4-dimethyl-cyclohexyl.
The present invention provides an efficient approach to a diverse group of sialylated and/or fucosylated 3'-0-galactosyl-6-0-sialyl-GlcNAc or -GalNAc derivatives and analogs thereof based on the unique combination of chemical and enzymatic glycosylation steps. The claimed method is attractive for scale-up developments and therefore may imply a potential industrial process.
The first aspect of the invention relates to a method for making a sialylated or fucosylated 3-0- galactosyl-GlcNAc or -GalNAc derivative of formula 1 and salts thereof

1 wherein R is selected from -OH, -N3 and -OR6 wherein R6 is selected from: allyl optionally substituted by one or more methyl, propargyl optionally
substituted by one or more methyl, 2-trimethylsilyl-ethyl, -(CH2)n-NH2, and -(CH2)n-N3 wherein n is an integer of 1 to 10, preferably 2 or 3,
Ri is selected from sialyl moiety, -SO3H and -CH(R5)-COOH wherein R5 is selected from H, alkyl and benzyl,
R2 is selected from H and fucosyl,
R3 is selected from H and sialyl,
R4 is selected from H and fucosyl, provided that one but only one of R3 and R4 is H, and
A is selected from a bond and a divalent carbohydrate linker, comprising the steps of: a) sialylation, sulfation or carbox methylation of a compound of formula 2

2
wherein R' is selected from -N3 and -OR'6 wherein R'6 is selected from allyl optionally substituted by one or more methyl, propargyl optionally
substituted by one or more methyl, a group removable by hydrogenolysis, 2- trimethylsilyl-ethyl and -(CH2)n-N3 wherein n is an integer of 1 to 10,
preferably 2 or 3,
R7 is independently acyl,
Y is selected from -NHAc, haloalkanoylamido, -NAc2, haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and -N3, and
B is selected from a bond and a divalent carbohydrate linker in protected form, b) optional fucosylation of the sialylated, sulfated or carboxymethylated compound of formula 2 obtained in step a), c) de-O-acylation and/or basic hydrolysis, optional mild acidic hydrolysis and optional transformation of Y to -NHAc of the compound obtained in step a) or step b), d) sialylation or fucosylation of the compound obtained in step c), and e) optional catalytic hydrogenolysis and/or anomeric deprotection of the compound
obtained in step d).
In step a), a compound of formula 2 is sialylated, sulfated, or carboxymethylated. When B is a bond, a compound of formula 2 is a partially protected disaccharide of formula 2A

2A wherein R', R7 and Y are as defined above.
The secondary OH-group of the deoxy-aminohexose moiety of the disaccharide of formula 2A can be in equatorial orientation giving rise to a partially protected lacto-N-biosyl derivative of formula
2B

2B wherein R', R7 and Y are as defined above.
When the secondary OH-group is in axial orientation, the disaccharide of formula 2 A is a GalpPl- 4GalNAc derivative of formula 2C

2C wherein R', R7 and Y are as defined above.
Preferably, the compound of formula 2 is a compound of formula 2B wherein Y is - HAc and R7 groups are identical and are acetyl or benzoyl.
Compounds of formula 2A can be made in a conventional manner by standard carbohydrate chemistry, for example by reacting a protected glucos- or galactosamine acceptor having a free 3- OH with a tetraacyl galactosyl donor followed by selective deprotection of the 4- and 6-OH groups (see e.g. Hendel at al. Carbohydr. Res. 343, 2914 (2008), Jain et al. Carbohydr. Res. 275, 231 (1995), WO 95/29927, Bao et al. Bioorg. Med. Chem. 18, 3760 (2010), Rana et al. Carbohydr. Res.
96, 231 (1981), Aly et al. Eur. J. Org. Chem. 2305 (1998), Bedini et al. Carbohydr. Res. 349, 24
(2012)).
According to another embodiment of compounds of formula 2, B is a divalent carbohydrate linker in protected form. In this invention, the term "carbohydrate linker in protected form" preferably means any mono-, di- or oligosaccharide glycosyl residue which is attached to the R' group by the C-l (aldoses) or C-2 (ketoses) anomeric carbon, and is also attached, via one of its non-glycosidic OH-groups, to the galactosylated aminodeoxy-hexose residue of the compound of formula 2. If this divalent glycosyl residue B differs from a monosaccharide, it may represent a linear or branched structure consisting of monosaccharide units that are linked to each other by interglycosidic linkages. The monosaccharide unit(s) of the divalent glycosyl residue B can be selected from any 5- 9 carbon atom containing sugars consisting of aldoses (e.g. D-glucose, D-galactose, D-mannose, D-
ribose, D-arabinose, L-arabinose, D-xylose, etc.), ketoses (e.g. D-fructose, D-sorbose, D-tagatose, etc.), deoxysugars (e.g. L-rhamnose, L-fucose, etc.), deoxy-amino sugars (e.g. N-acetylglucosamine, N-acetylmannosamine, N-acetylgalactosamine, etc.), uronic acids, ketoaldonic acids (e.g. sialic acid) and like. Preferably, the divalent glycosyl moiety B is a lactosyl moiety or an oligosaccharide moiety that consists of a lactosyl moiety and at least one monosaccharide unit selected from the group consisting of galactose, N-acetylglucosamine, fucose and N-acetyl neuraminic acid. The divalent lactosyl moiety is attached to the R' group by its C-1 anomeric carbon atom, and it is also attached, via one of its non-glycosidic OH-groups, preferably via its 3'-OH group to the
galactosylated aminodeoxy-hexose residue of the compound of formula 2. The divalent lactosyl moiety defined above can be optionally substituted by other glycosyl residues, preferably on its 3- OH group by a fucosyl moiety, or on its 6' -OH group by a N-acetyllactosaminyl moiety, which N- acetyllactosaminyl moiety can optionally be further substituted by an N-acetyl neuraminyl moiety on its 6-OH, or by a fucosyl on its 3-OH or 2'-OH. These oligosaccharide substructures can be found in human milk oligosaccharides. The functional groups of the divalent glycosyl residue B are protected, preferably the free OH groups are acylated (e.g. acetylated, benzoylated) and the carboxy group of the optional N-acetyl neuraminyl moiety is blocked in ester form (e.g. methyl, ethyl or benzyl ester). In the divalent lactosyl moiety defined above the axial OH-group of any galactose may not be protected.
In a further preferred embodiment of the compounds of formula 2, the R' group has β-orientation. Also preferably, the moiety B is attached to the parent carbohydrate backbone by β-linkage.
Compounds of formula 2 wherein B is a divalent carbohydrate linker in protected form can be synthesized by using multistep chemical glycosylation procedures as described in or by analogy of e.g. Xia et al. Bioorg. Med. Chem. Lett. 9, 2941 (1999), Pozsgay et al. Tetrahedron 48, 10249 (1992), Aly et al. Carbohydr. Res. 316, 121 (1999) and WO 2012/155916.
In carrying out step a) the primary free OH group of a compound of formula 2 is regioselectively sialylated, sulfated or carboxymethylated, without affecting the neighbouring secondary OH-group, to give a compound of formula 4

P is selected from protected sialyl moiety, -SO3H and -CH(R5)-COORio wherein R5 is selected from H, alkyl and benzyl, R10 is selected from alkyl and benzyl.
For the chemical sialylation suitably protected and activated sialyl donors are used. For the functional group protection of the sialyl donor an array of protecting groups, mainly esters, ethers and acetals, are available to the skilled person. Among OH-protection possibilities, optionally substituted acyls, such as acetyl, benzoyl, chloroacetyl or chlorobenzoyl, and ether-type groups such as benzyl are of synthetic usefulness; the carboxyl group can be protected by an ester, typically by a methyl, ethyl or benzyl ester; and the amino function can be masked by diacetyl, trifluoroacetyl, trichloroacetyl, Troc or Fmoc group, or as a cyclic carbamate with the adjacent 4-OH. The anomeric centre activation can be varied among halo, alkyl- or arylthio, dialkyl, dibenzyl or diaryl phosphite, or trihaloacetimidate, each of which is commonly used in sialoglycosidation methods. The protective group introduction and anomeric centre activations mentioned above can be carried out by known processes (see e.g. Ress et al. Curr. Org. Synth. 1, 31 (2004), Chen et al. ACS Chem. Biol. 5, 163 (2010) and references cited therein). The preferred sialyl donors are those disclosed in WO 2011/100979 and WO 2012/113404, among which the N-acetyl neuraminyl phosphite donors of formula 3

3 wherein R8 is acyl, preferably acetyl,
Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl, and
R9 is optionally substituted phenyl or benzyl, are especially preferred. The coupling reaction runs in aprotic solvent, preferably in
dichloromethane, TFIF, methyl-THF, toluene, acetonitrile or in mixtures thereof, at temperatures between -78 - 0 °C, in the presence of promoter like BS, NIS, TMSOTf, TfOH, Tf20, ZnCl2, BF3 OEt2, L1CIO4, DTBPI, Bu4 I, AgC104, LiC104, Sn(OTf)2> iodine, montmorillonite, Tf2 H or mixtures thereof, preferably TMSOTf. The sialylated product can be characterized by the following formula 4 A

4A wherein R', R7, R8, B, Q and Y are as defined above.
If step a) is a sulfation step, a compound of formula 2 defined above is treated with SCVpyridine complex in a non-protic solvent to form a compound of formula 4B or a salt thereof

4B wherein R', R7, B and Y are as defined above. Also in step a), a compound of formula 2 can be carboxymethylated to give rise to a compound of formula 4C

4C wherein R5 is selected from H, alkyl and benzyl, R10 is selected from alkyl and benzyl, and R', R7, B and Y are as defined above.
This reaction can be carried out by the analogous process disclosed by Schwardt et al. J. Med. Chem. 52, 989 (2009). Thus the dihydroxy derivative of formula 2 is reacted with dibutyltin oxide to form a stannylidene acetal which reacts in a nucleophilic substitution reaction with the acetic acid derivative of formula L-CH(R5)-COORio wherein L is a good leaving group such as halogen or sulfonate ester (e.g. mesyloxy, tosyloxy, trifluoromethanesulfonyloxy, etc.), and R5 and Rio are as defined above. In the reaction only the primary OH-group is alkylated, and, if the reagent acetic acid derivative is chiral, the substitution takes place with the inversion of configuration.
In step b), the sialylated, sulfated or carboxymethylated compound obtained in step a) is optionally fucosylated with a suitably protected and activated fucosyl donor. The fucosyl moiety is introduced at the remaining free secondary OH-group (4-OH) of the compound of formula 4. Thus, a fucosyl donor of formula 5

5 wherein X is selected from a halogen,
 -O-pentenyl, -OAc, -OBz and -SRi3, in which Ri3 is alkyl or optionally substituted phenyl,
-O-pentenyl, -OAc, -OBz and -SRi3, in which Ri3 is alkyl or optionally substituted phenyl,
Rii is selected from acyl and a group removable by hydrogenolysis, and
Ri2 is selected from a group removable by hydrogenolysis, acyl or two R12 groups together form a moiety R-14 C R15j wherein R14 and R15 independently are alkyl or phenyl, or wherein R14 and R15 together with the carbon atom, to which they are attached, form cycloalkylidene, can be used in this reaction to give a compound of formula 6

The fucosylation can be carried out in a conventional manner in an aprotic solvent or in a mixture of aprotic solvents in the presence of an activator, see Demchenko (Ed.): Handbook of Chemical Glycosylation Wiley (2008). The fucosylation reaction is generally promoted by heavy metal ions, mainly mercury or silver, and Lewis acids such as trimethylsilyl triflate or BF3-etherate.
Preferably, a fucosyl halide (i.e., X is F, CI, Br or I) is used because of its easy accessibility and satisfactory reactivity. Typically, anomeric halides follow the reactivity order F<Cl<Br<I for nucleophilic displacement. Glycosyl fluorides can be prepared by treating the appropriate
precursors such as hemiacetals, glycosyl halides, glycosyl esters and ^-glycosides with fluorinating reagents such as HF, AgF, AgBF4, tetrabutyl ammonium fluoride, diethyl amino sulfur trifluoride, 2- fluoro-l-methylpyridinium tosylate, Selectfluor, Deoxo-Fluor or 4-methyl(difluoroiodo)-benzene.
A fucosyl trichloroacetimidate (i.e., X is -OC(=NH)CCl3) can be prepared by adding a sugar with a free anomeric OH to trichloroacetonitrile under inorganic or organic base catalysis. The resulting glycosyl donor can be activated by a catalytic amount of a Lewis acid, such as trimethylsilyl triflate or BF3-etherate, for the glycosylation reaction.
Fucosyl acetates or benzoates (i.e., X is -OAc or -OBz) are preferably first subjected to electrophilic activation to provide a reactive intermediate and then treated with a nucleophilic OH-acceptor. Typical activators of choice are Bronsted acids (e.g., /?-TsOH, HC104 or sulfamic acid), Lewis acids (e.g., ZnCl2, SnCl4, triflate salts, BF3-etherate, trityl perchlorate, A1C13 or triflic anhydride) or a mixture thereof.
Pentenyl fucosides (i.e., X is -O-(CH2)3-CH=CH2) can be transglycosylated with appropriate glycosyl acceptors in the presence of a promoter such as NBS and NIS. Protic or Lewis acids (triflic acid, Ag-triflate, etc.) can enhance the reaction. The pentenyl glycosides can be prepared with the aid of «-pentenol by standard Fischer glycosylation of hemiacetals under acidic condition, by silver(I) salt promoted coupling of glycosyl bromides (Koenigs-Knorr method), or by glycosylation of 1 -acetyl glycosides in the presence of tin(IV) chloride.
Thiofucosides (i.e., X is alkylthio- or optionally substituted phenylthio-group) can be activated by thiophilic promoters such as mercury(II) salts, Br2, 12, NBS, NIS, triflic acid, triflate salts, BF3- etherate, trimethylsilyl triflate, dimethyl-methlythio sulphonium triflate, phenylselenyl triflate, iodonium dicollidine perchlorate, tetrabutylammonium iodide or mixtures thereof, preferably by Br2, NBS, NIS or triflic acid.
Aprotic solvents such as toluene, THF, methyl-THF, DCM, chloroform, dioxane, acetonitrile, chlorobenzene, ethylene di chloride, DMSO, DMF or N-methylpyrrolidone or mixtures thereof, preferably DMF, toluene, DCM or mixtures thereof, more preferably toluene or DMF-DCM mixture can be used in this glycosylation reaction at -20 to 20 °C, preferably at -10 to 5 °C, with reaction time of 5 min to 2 hours. For thiophilic activation, Br2, NBS or NIS can be used, optionally in the presence of triflic acid or a triflate derivative. Usually a slight excess of donor (1.1-1.2 eq.) is used compared to the acceptor. For quenching the reaction, water or a Ci-C6 alcohol is generally
used, preferably an aqueous or alcoholic solution of a base like sodium carbonate, sodium bicarbonate, ammonia or triethyl amine, more preferably an aqueous Na2S203/NaHC03 solution.
More preferably, the fucosyl donor is a compound of formula 5 wherein Rn is as defined above, R12 is acyl or a group removable by hydrogenolysis, and X is phenylthio optionally substituted with one or more alkyl. More preferably Rn is benzyl, 4-methylbenzyl, naphthylmethyl, 4-phenylbenzyl, 4- chlorobenzyl, 4-methoxybenzyl, 3,4-dimethoxybenzyl, 2,4,6-trimethylbenzyl or 2,3,4,5,6- pentamethylbenzyl, R12 is benzyl, 4-methylbenzyl, naphthylmethyl, 4-phenylbenzyl, 4- chlorobenzyl, 4-methoxybenzyl, 3,4-dimethoxybenzyl, 2,4,6-trimethylbenzyl, 2,3,4,5,6- pentamethylbenzyl or benzoyl optionally substituted by one or more halogens and X is
unsubstituted phenylthio. Even more preferably, Rn is benzyl or 4-methylbenzyl and R12 is benzoyl or 4-chlorobenzoyl.
According to a preferred embodiment of step b), a compound of formula 4A, 4B or 4C defined above is reacted with a fucosyl donor of formula 5 giving rise to a compound of formula 6A, 6B or 6C, respectively, and salts thereof

6A 6B 6C wherein R', R5, R7, R8, Rio, Rn, R12, B, Q and Y are as defined above.
In step c), the protecting groups of a compound obtained in step a) or step b) are removed except for the anomeric protection R', and optionally Y is converted into a - HAc group.
Most of the protecting groups are O-acyls (R7, R8, the OH-protections in moiety B, and optionally Rn and R12). Acyl protective groups can be removed by a conventional base catalysed
transesterification deprotection reaction wherein the acyl groups are removed in an alcohol solvent such as methanol, ethanol, propanol or t-butanol in the presence of an alcoholate such as NaOMe, NaOEt or KO*Bu at 20-100 °C. The alcohol solvent and the alcoholate should be matched. A co- solvent such as toluene or xylene can be beneficial in order to control particle size of the product
and to avoid gel formation. Preferably, a catalytic amount of NaOMe is used in methanol (Zemplen de-O-acylation). Under this condition only O-acyls can be deprotected. If Y is -NAc2, one of the acetyl groups can also be removed to make the Y group -NHAc. When Y is: haloalkanoylamido, haloalkoxycarbonylamino, 2,3-diphenylmaleimido, 2,3-dimethylmaleimido or -N3, and ester groups (see group -COOQ or -COORio) remain intact under these conditions.
Deprotection of acyloxy groups to OH, esters to carboxyl (see group -COOQ or -COORio) and the following Y-groups to -NH2: haloalkanoylamido, halocarboxylamino, 2,3-diphenylmaleimido and 2,3-dimethylmaleimido can be carried out by basic hydrolysis, which is a base catalysed hydrolysis in water, alcohol or water-organic solvent mixtures, in homogeneous or heterogeneous reaction conditions at 0-100 °C. Preferably, a strong base, such as Li OH, NaOH, KOH, Ba(OH)2, K2C03, a basic ion exchange resin or tetraalkylammonium hydroxide, is used, but the base can also be in an aqueous solution as well. Preferably, the base is NaOH and the solvent is methanol. If Y is -NAc2, one of the acetyl groups can also be removed to make the Y group -NHAc. Azido Y group is not affected in this reaction.
Also in step c), a 2,2,2-trichloroethoxycarbonylamino Y-group can be converted into free amino groups by means of Zn/HCl, and Y= -N3 can be reduced to amino using e.g. PPh3 or Cu/Zn
(provided that R' is not azido or R'6 is not -(CH2)n-N3).
The free amino group obtained by one of the deprotective methods can then be acetylated without acetylating the free OH-groups. Selective N-acetylation in the presence of one or more hydroxyls can be carried out in a conventional manner with a slight excess of acetic anhydride or acetyl chloride (-1.5-3 equiv.) at about 0-35 °C with or without added base. Any resulting overacetylated by-product(s) can be readily transformed into the desired compounds with e.g. NaOH/MeOH or NaOMe/MeOH treatment. Alternatively, the deprotected compound having a free amino group can be peracetylated (that is the amino group and all available OH groups are acetylated) followed by base catalysed transesterification (see above).
Also in step c), trichloroacetyl amide Y-group can be transformed in one step to -NHAc with tributyltin hydride.
Also in step c), an acetal/ketal group optionally present on the compounds of formulas 6A, 6B and
_\ /_
6C (i.e. when two Ri2 groups together form a moiety ^14 C R15) can be selectively deprotected by acid catalysed mild hydrolysis, i.e. by reacting the compounds with water or an alcohol in the
presence of acid at pH>l-2 to produce OH-groups on the compounds. Acyl protective groups will not be affected because they can be deprotected only by extremely strong acidic hydrolysis (pH<l). Although the interglycosidic linkage and anomeric protecting groups of the compounds of formulas 6A, 6B and 6C can also be sensitive to acids, they can be split in the compounds of formulas 6A, 6B and 6C only by acidic hydrolysis at pH<l-2. The skilled person is able to distinguish which deprotective condition affects the acetal group while the acyl groups and the interglycosidic bonds remain intact. Water, which is a reagent, can also serve as solvent or co-solvent. Organic protic or aprotic solvents which are stable under acidic conditions and fully or partially miscible with water, such as Ci-C6 alcohols, acetone, THF, dioxane, ethyl acetate or MeCN, can be also used in a mixture with water. The acid used is generally a protic acid, such as acetic acid, trifluoroacetic acid, HC1, formic acid, sulphuric acid, perchloric acid, oxalic acid, ^-toluenesulfonic acid,
benzenesulfonic acid or a cationic exchange resin, and can be present in from a catalytic amount to a large excess. The hydrolysis can be carried out at between 20 °C and reflux until completion of the reaction which can take from about 2 hours to 3 days depending on temperature, concentration and pH. Preferably, an organic acid, such as acetic acid, formic acid, chloroacetic acid or oxalic acid, is used. Preferably, a Ci-C6 alcohol-acetonitrile or Ci-C6 alcohol-water mixture is used in the presence of HC1 or a sulfonic acid such as ^-toluenesulfonic acid or camphorsulfonic acid.
Alternatively, an anhydrous Ci-C6 alcohol, such as methanol, ethanol, propanol and butanol, can be used for the cleavage of acetal via acid catalysed trans-acetalization/trans-ketalization processes. Catalytic amount of hydrogen chloride, sulphuric acid, perchloric acid, ^-toluenesulfonic acid, acetic acid, oxalic acid, camphorsulfonic acid or a strong acidic ion-exchange resin can be used at temperatures of 20 °C to reflux.
Also in step c), any compound of formula 6A, 6B and 6C, wherein at least one of the Rn and R12 substituents is a group removable by hydrogenolysis (provided that R' is 2-trimethylsilylethyloxy and Y is not azido), can be subjected to catalytic hydrogenolysis in order to remove
benzyl/substituted benzyl group(s).
The above mentioned deprotective steps can be carried out in any order. As a result, a compound of formula 7 and salts thereof can be obtained

7
wherein R' is as defined above, moiety A is a bond or is a divalent carbohydrate linker in deprotected form,
Ri is selected from sialyl, -SO3H and -CH(R5)-COOH, wherein R5 is as defined above,
Ri6 is selected from H and a moiety of formula C

c wherein Rn and Ris, independently, are selected from H and a group removable by hydrogenolysis.
The term "divalent carbohydrate linker in deprotected form" above preferably means any mono-, di- or oligosaccharide glycosyl residue which is attached to the R' group by the C-l (aldoses) or C-2 (ketoses) anomeric carbon, and in the same time it is also attached, via one of its non-glycosidic OH-groups, to the galactosylated aminodeoxy-hexose residue of the compound of formula 7, as defined for moiety B above, in deprotected from, which means that the OH-protective groups and the ester protective group of the N-acetyl neuraminyl moiety (if present) in the divalent
carbohydrate linker of moiety B are removed when carrying out step c), thus transforming moiety B into moiety A. Preferably, the divalent glycosyl moiety A is a lactosyl moiety or an oligosaccharide moiety that consists of a lactosyl moiety and at least one monosaccharide unit selected from the group consisting of galactose, N-acetylglucosamine, fucose and N-acetyl neuraminic acid. The divalent lactosyl moiety is attached to the R' group by its C-l anomeric carbon atom, and at the same time it is also attached, via one of its non-glycosidic OH-groups, preferably via its 3'-OH group to the galactosylated aminodeoxy-hexose residue of the compound of formula 7. The divalent lactosyl moiety defined above can be optionally substituted by other glycosyl residues, preferably on its 3 -OH group by a fucosyl moiety, or on its 6' -OH group by a N-acetyllactosaminyl moiety, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetyl neuraminyl moiety on its 6-OH, or by a fucosyl on its 3 -OH or 2' -OH.
In step d) a compound obtained in step c), that is a compound of formula 7 defined above, is brought into a glycosidase mediated transsialylation or transfucosylation reaction and a compound of formula 10 and salts thereof can be obtained
 wherein R', Ri, Ri6 and A are as defined above, R3 is selected from H and a sialyl, and
wherein R', Ri, Ri6 and A are as defined above, R3 is selected from H and a sialyl, and
R4 is selected from H and a fucosyl moiety, provided that one but only one of R3 and R4 is H.
With regard to the transsialylation reaction of step d), a compound of formula 7 is reacted with a sialyl donor, preferably a N-acetyl neuraminyl donor, under the catalysis of an enzyme having a2-3- transsialidase activity. Typical natural sialyl donors can be selected from, but are not limited to, 3'- O-sialyl-lactose, fetuin, gangliosides, O- or N-linked glycopeptides, all of which contain a sialic acid a-2,3-linked to a terminal β-galactoside residue, or polysialic acid with a-2,8-linkage. Also preferably, the sialyl donor used in step d) can be characterized by formula 8 and salts thereof,

8 wherein Z is selected from the group consisting of azide, fluoro, optionally substituted phenoxy, optionally substituted pyridinyloxy, group D, group E, group F and group G

D E F G wherein Ra is independently H or alkyl, or two vicinal Ra groups represent a =C(Rb)2 group, wherein R is independently H or alkyl, Rc is independently selected from the group consisting of alkoxy, amino, alkylamino and dialkylamino, Rd is selected from the group consisting of H, alkyl and -C(=0)Re, wherein Re is OH, alkoxy, amino, alkylamino, dialkylamino, hydrazino,
alkylhydrazino, dialkylhydrazino or trialkylhydrazino. Among the sialyl donors 4- methylumbelliferyl and optionally substituted phenyl N-acetyl-a-neuraminosides and 3'-0-sialyl-
lactose, more commonly /?-nitrophenyl N-acetyl-a-neuraminoside and 3'-0-sialyl-lactose, are of high preference.
Enzymes having a2-3-transsialidase activity are preferably selected from a sialidase or
transsialidase as described in the following, e.g. from sialidases (EC 3.2.1.18) and transsialidases (EC 2.4.1.-) as classified according to the the GH33 family. They are retaining enzymes. Sialidases and transsialidases are widely distributed in nature. They are found particularly in diverse virus families and bacteria, and also in protozoa, some invertebrates and mammals. These enzymes differ in their biochemical properties, e.g., kinetics, binding affinity or substrate preference. Nevertheless, they possess conserved domains and structural similarities. Transsialidases differ from sialidases since they can transfer sialic acids, preferably a-2, 3 -bonded sialic acids, from a donor molecule to an acceptor derivative, which is preferably a terminal galactose moiety with a β-interglycosidic linkage. As a result of this transfer, an a-glycosidic bond is formed between the sialic acid and the acceptor. However, if there is no suitable acceptor, the transsialidase hydrolyses the sialic acid.
The first transsialidase enzyme described was found in Trypanosoma cruzi, a protozoa which causes Chagas disease. This transsialidase (TcTS) has been extensively studied. Since that time
transsialidases have been detected in several other trypanosome types such as Trypanosoma brucei gambiense, Trypanosoma brucei rhodesiense, Trypanosoma brucei brucei and Trypanosoma congolense. Moreover, the existence of transsialidases has been shown in Endotrypanum types, in Corynebacterium diphtheriae and even in human plasma.
Sialidases can be classified into two different subgroups, endo- and exo-sialidases. The endo- sialidases hydrolyse sialic acid linkages internal to macromolecules, while the exo-sialidases attack terminal sialic acid linkages, and desialylate glycoproteins, glycopeptides, gangliosides,
oligosaccharides and polysaccharides. Recently, sialidases from Bifidobacterium bifidum and Bifidobacterium longum subsp. infantis have been identified, cloned and characterized. These sialidases can cleave and so recognize both a-2, 3- and a-2,6-linked sialosides. Sialidases from Bifidobacterium longum subsp. infantis have a consistent preference for a-2,6-linkage whereas sialidases from Bifidobacterium bifidum have a consistent preference for a-2, 3 -linkage. These enzymes are also capable of acting as catalysts for sialylation reactions due to their transsialidase activity and thus may be used in the context of the method of the present invention, preferably under kinetically controlled conditions.
Sialidases, which may be employed in the context of the present invention, may also comprise engineered sialidases. Based on sequence and structure comparisons, sialidase from Trypanosoma rangeli may be mutated at six positions, wherein the resulting mutant is able to display a significant level of transsialidase activity (see Paris et al. J. Mol. Biol. 345, 923 (2005)).
Even more preferably, the enzyme having a sialidase and/or transsialidase activity may be selected from sialidases or transsialidases derived from Bifidobacterium longum subsp. infantis ATCC 15697, Bifidobacterium bifidum JCM1254, Bifidobacterium bifidum S17, Bifidobacterium bifidum PRL2010, Bifidobacterium bifidum NCIMB 41171, Trypanosoma cruzi, etc.
Even more preferably enzyme having a sialidase and/or transsialidase activity may be selected from sialidases or transsialidases as defined according to the following deposit numbers: gi|213524659 {Bifidobacterium longum subsp. infantis ATCC 15697), gi|213523006 Bifidobacterium longum subsp. infantis ATCC 15697), siab2 (Bifidobacterium bifidum JCM1254), further sialidases or transsialidases from Bifidobacterium bifidum JCM1254, gi|309252191 (Bifidobacterium bifidum S17), gi|309252190 (Bifidobacterium bifidum S17), gi|310867437 (Bifidobacterium bifidum
PRL2010), gi|310867438 (Bifidobacterium bifidum PRL2010), gi|224283484 (Bifidobacterium bifidum NCIMB 41171), gi|313140638 (Bifidobacterium bifidum NCIMB 41171), gi|47252690 (Trypanosoma cruzi), gi|432485 (Trypanosoma cruzi), gi|343957998 (Trypanosoma congolense), gi|343958004 (Trypanosoma congolense) etc., or a sequence exhibiting a sequence identity with the sequence of one of the above mentioned enzymes having a sialidase and/or transsialidase activity of at least 70%, more preferably at least 80%>, equally more preferably at least 85%>, even more preferably at least 90% and most preferably at least 95% or even 97%, 98% or 99% as compared to the entire wild type sequence on amino acid level.
Particularly preferred sialidases with sialidase/transsialidase activity are listed in the following Table 2:

gi 47252690 Trypanosoma cruzi
gi 432485 Trypanosoma cruzi
gi 343957998 Trypanosoma congolense
gi 343958004 Trypanosoma congolense
Table 2: Preferred sialidases/transsialidases
In the trans si alylation reaction according to step d) a compound of formula 10A and salts thereof can be made

10A wherein R', Ri, Ri6 and A are as defined above, and
R3 is a sialyl, preferably N-acetyl-neuraminyl moiety.
With regard to the transfucosylation reaction of step d), a compound of formula 7 is reacted with a fucosyl donor under the catalysis of an enzyme having al-2-transfucosidase activity. Typical fucosyl donors can be selected from, but are not limited to, 2'-0-fucosyl-lactose, difucosyl-lactose, and fucose donors of formula 9
 wherein Z is selected from the group consisting of azide, fluoro, optionally substituted phenoxy, optionally substituted pyridinyloxy, group D, group E, group F and group G
wherein Z is selected from the group consisting of azide, fluoro, optionally substituted phenoxy, optionally substituted pyridinyloxy, group D, group E, group F and group G

D E F G wherein Ra is independently H or alkyl, or two vicinal Ra groups represent a =C(Rb)2 group, wherein R is independently H or alkyl, Rc is independently selected from the group consisting of alkoxy, amino, alkylamino and dialkylamino, Rd is selected from the group consisting of H, alkyl
and -C(=0)Re, wherein Re is OH, alkoxy, amino, alkylamino, dialkylamino, hydrazino, alkylhydrazino, dialkylhydrazino or trialkylhydrazino.
The enzyme exhibiting al-2-transfucosidase activity is preferably selected from fucosidases, transfucosidases and fucosynthases as classified according to EC 3.2.1.38 and 3.2.1.51. a-L- Fucosidases are widely spread in living organisms such as mammals, plants, fungi and bacteria. These enzymes belong to the families 29 and 95 of the glycoside hydrolases (GH29 and GH95) as defined by the CAZY nomenclature (https://www.cazy.org). Fucosidases from GH29 are retaining enzymes (3D structure: (β/α)8) whereas fucosidases from GH95 are inverting enzymes (3D structure: (α/α)6 ). The substrate specificity of the GH29 family is broad whereas that of the GH95 family is strict to al,2-linked fucosyl residues. a-L-Fucosidases generally hydrolyse the terminal fucosyl residue from glycans. These enzymes are also capable of acting as catalysts for fucosylation reactions due to their transfucosylation activity and thus may be used in the context of the method of the present invention, preferably under kinetically controlled conditions.
Fucosidases, which may be employed in the context of the present invention, may also comprise engineered fucosidases. Such engineered fucosidases preferably comprise engineered a-L- fucosidases, preferably engineered fucosidases derived from fucosidases as described above, e.g. an engineered a-l,2-L-fucosynthase from Bifidobacterium bifidum, a-L-fucosynthases from Sulfolobus solfataricus and Thermotoga maritime, etc. Such engineered fucosidases show an acceptor dependent regioselectivity and are devoid of product hydrolysis activity. Furthermore, engineered fucosidases preferably comprise a-L-fucosidase from Thermotoga maritima, which has also been recently converted into an efficient a-L-transfucosidase by directed evolution (see Osanjo et al. Biochemistry 46, 1022 (2007)).
Even more preferably, the enzyme having a fucosidase and/or trans-fucosidase and/or fucosynthase activity may be selected from a-L-fucosidases derived from Thermotoga maritima MSB 8,
Sulfolobus solfataricus P2, Bifidobacterium bifidum JCM 1254, Bifidobacterium bifidum JCM 1254, Bifidobacterium longum subsp. infantis ATCC 15697, Bifidobacterium longum subsp.
infantis ATCC 15697, Bifidobacterium longum subsp. Infantis JCM 1222, Bifidobacterium bifidum PRL2010, Bifidobacterium bifidum SI 7, Bifidobacterium longum subsp longum JDM 301,
Bifidobacterium dentium Bdl, or Lactobacillus casei BL23, etc.
Even more preferably the enzyme having a fucosidase and/or trans-fucosidase and/or fucosynthase activity may be selected from following a-L-fucosidases as defined according to the following
deposit numbers gi|4980806 (Thermotoga maritima MSB8), gi| 13816464 (Sulfolobus solfataricus P2), gi|34451973 (Bifidobacterium bifidum JCM 1254), gi|242345155 (Bifidobacterium bifidum, JCM 1254), gi|213524647 (Bifidobacterium longum subsp. infantis, ATCC 15697), gi|213522629 (Bifidobacterium longum subsp. infantis ATCC 15697), gi|213522799 (Bifidobacterium longum subsp. infantis ATCC 15697), gi|213524646 (Bifidobacterium longum subsp. infantis ATCC 15697), gi|320457227 (Bifidobacterium longum subsp. infantis JCM 1222), gi|320457408
(Bifidobacterium longum subsp. infantis JCM 1222), gi|320459369 (Bifidobacterium longum subsp. infantis JCM 1222), gi|320459368 (Bifidobacterium longum subsp. infantis JCM 1222),
gi|310867039 (Bifidobacterium bifidum PRL2010), gi|310865953 (Bifidobacterium bifidum
PRL2010), gi|309250672 (Bifidobacterium bifidum S17), gi|309251774 (Bifidobacterium bifidum SI 7), gi 1296182927 (Bifidobacterium longum subsp longum JDM 301), gi (296182928
(Bifidobacterium longum subsp longum JDM 301), gi|283103603 (Bifidobacterium dentium Bdl), gi| 190713109 (Lactobacillus casei BL23), gi| 190713871 (Lactobacillus casei BL23), gi| 190713978 (Lactobacillus casei BL23), etc., or a sequence exhibiting a sequence identity with the sequences of one of the above mentioned enzymes having a fucosidase and/or trans-fucosidase activity of at least 70%, more preferably at least 80%>, equally more preferably at least 85%>, even more preferably at least 90%) and most preferably at least 95% or even 97%, 98%> or 99% as compared to the entire wild type sequence on amino acid level.
Particularly preferred a-L-fucosidases with fucosidase/trans-fucosidase/fucosynthase activity are listed in the following Table 3 :

gi 309250672 Bifidobacterium bifidum S17
gi 309251774 Bifidobacterium bifidum S17
gi 296182927 Bifidobacterium longum subsp longum JDM 301 gi 296182928 Bifidobacterium longum subsp longum JDM 301 gi 283103603 Bifidobacterium dentium Bdl
gi 190713109 Lactobacillus casei BL23
gi| 190713871 Lactobacillus casei BL23
gi| 190713978 Lactobacillus casei BL23
Table 3 : Preferred a-L-fucosidases
The enzymatic fucosylation reaction according to step d) provides a compound of formula 10B and salts thereof

In step d) the enzymes comprising transsialidase or transfucosidase activity as defined above may also comprise engineered enzymes comprising transsialidase or transfucosidase activity. It is particularly envisaged that wild type or mutated glycosidases displaying a-transsialidase or a- transfucosidase activity can be used in the present invention to produce the target oligosaccharides. Preparation of such enzymes is preferably carried out via site directed mutagenesis approaches or directed evolution.
In rational engineering novel altered enzymes (mutants) are created via site directed mutagenesis approaches, preferably by introduction of point mutations. This technique generally requires reliance on the static 3D protein structure. The mutations generally affect the active site of the enzymes such that they lose their ability to degrade their transglycosylation products but remain capable of synthesis. A preferred strategy consists of the replacement of the catalytic nucleophile by a non-nucleophilic residue. This modification results in the formation of an inactive mutant or an altered enzyme with reduced transglycosylation activity due the lack of appropriate environment for the formation of the reactive host-guest complex for transglycosylation. However, in the presence of a more active glycosyl donor (e.g. fucosyl fluoride) that mimics the glycosyl enzyme
intermediate, the mutated enzyme is able to transfer efficiently the glycosyl moiety to a suitable acceptor generating a glycoside with inverted anomeric stereochemistry. Such a mutant glycosidase is termed as glycosynthase (e.g. fucosynthase) and their development represents one of the major advances in the use of glycosidases for synthetic purposes. In principle, the glycosynthase concept can be applied to all GH specificities and offer a large panel of enzymes potentially able to synthesize various oligosaccharides with very high yields, up to 95%.
The second preferred technique is called directed evolution. This strategy comprises random mutagenesis applied to the gene of the selected glycosidase and generates thus a library of genetically diverse genes expressing glycosidase. Generation of sequence diversity can be performed using well-known methodologies, the most preferable being the error prone polymerase chain reaction (epCR) method. This gene library may be inserted into suitable microorganisms such as E. coli or S. cerevisiae for producing recombinant variants with slightly altered properties.
Clones expressing improved enzymes are then identified with a fast and reliable screening method, selected and brought into a next round of mutation process. The recursive cycles of mutation, recombination and selection are continued as far as mutant(s) with the desired activity and/or specificity is/are evolved. To date, different high-throughput screening methodologies for glycosidases including glycosynthases have been developed. Applying these approaches, effective engineered transglycosidases, including new and more efficient glycosynthases can and have been created and isolated. An a-L-fucosidase from Thermotoga maritima has been recently converted into an efficient a-L-transfucosidase by directed evolution. The transferase/hydrolysis ratio of the evolved enzyme was 30 times higher than the native enzyme (see Osanjo et al. Biochemistry 46, 1022 (2007)).
Proteins comprising a transglycosidase and/or a glycosynthase activity as defined above may also comprise fragments or variants of those protein sequences. Such fragments or variants may typically comprise a sequence having a sequence identity with one of the above mentioned proteins sequences of at least 70%, more preferably at least 80%, equally more preferably at least 85%, even more preferably at least 90% and most preferably at least 95% or even 97%, 98% or 99% as compared to the entire wild type sequence on amino acid level.
"Fragments" of proteins or peptides in the context of the present invention may also comprise a sequence of a protein or peptide as defined herein, which is, with regard to its amino acid sequence N-terminally, C-terminally and/or intrasequentially truncated compared to the amino acid sequence
of the original (native) protein. Such truncation may thus occur either on the amino acid level or correspondingly on the nucleic acid level. A sequence identity with respect to such a fragment as defined herein may therefore preferably refer to the entire protein or peptide as defined herein or to the entire (coding) nucleic acid molecule of such a protein or peptide. Likewise, "fragments" of nucleic acids in the context of the present invention may comprise a sequence of a nucleic acid as defined herein, which is, with regard to its nucleic acid molecule 5'-, 3'- and/or intrasequentially truncated compared to the nucleic acid molecule of the original (native) nucleic acid molecule. A sequence identity with respect to such a fragment as defined herein may therefore preferably refer to the entire nucleic acid as defined herein.
"Variants" of proteins or peptides as defined in the context of the present invention (e.g. as encoded by a nucleic acid as defined herein) may be encoded by the nucleic acid molecule of a polymeric carrier cargo complex. Thereby, a protein or peptide may be generated, having an amino acid sequence which differs from the original sequence in one or more mutation(s), such as one or more substituted, inserted and/or deleted amino acid(s). Preferably, these fragments and/or variants have the same biological function or specific activity compared to the full-length native protein, e.g. its specific antigenic property.
"Variants" of proteins or peptides as defined in the context of the present invention (e.g. as encoded by a nucleic acid as defined herein) may also comprise conservative amino acid substitution(s) compared to their native, i.e. non- mutated physiological, sequence. Those amino acid sequences as well as their encoding nucleotide sequences in particular fall under the term variants as defined herein. Substitutions in which amino acids, which originate from the same class, are exchanged for one another are called conservative substitutions. In particular, these are amino acids having aliphatic side chains, positively or negatively charged side chains, aromatic groups in the side chains or amino acids, the side chains of which can enter into hydrogen bridges, e.g. side chains which have a hydroxyl function. This means that e.g. an amino acid having a polar side chain is replaced by another amino acid having a likewise polar side chain, or, for example, an amino acid characterized by a hydrophobic side chain is substituted by another amino acid having a likewise hydrophobic side chain (e.g. serine (threonine) by threonine (serine) or leucine (isoleucine) by isoleucine (leucine)). Insertions and substitutions are possible, in particular, at those sequence positions which cause no modification to the three-dimensional structure or do not affect the binding region. Modifications to a three-dimensional structure by insertion(s) or deletion(s) can easily be determined e.g. using CD spectra (circular dichroism spectra) (Urry, 1985, Absorption,
Circular Dichroism and ORD of Polypeptides, in: Modern Physical Methods in Biochemistry, Neuberger et al. (ed.), Elsevier, Amsterdam).
Furthermore, variants of proteins or peptides as defined herein may also comprise those sequences, wherein nucleotides of the nucleic acid are exchanged according to the degeneration of the genetic code, without leading to an alteration of the respective amino acid sequence of the protein or peptide, i.e. the amino acid sequence or at least part thereof may not differ from the original sequence in one or more mutation(s) within the above meaning.
In order to determine the percentage to which two sequences are identical, e.g. nucleic acid sequences or amino acid sequences as defined herein, preferably the amino acid sequences encoded by a nucleic acid sequence of the polymeric carrier as defined herein or the amino acid sequences themselves, the sequences can be aligned in order to be subsequently compared to one another. Therefore, e.g. a position of a first sequence may be compared with the corresponding position of the second sequence. If a position in the first sequence is occupied by the same component as is the case at a position in the second sequence, the two sequences are identical at this position. If this is not the case, the sequences differ at this position. If insertions occur in the second sequence in comparison to the first sequence, gaps can be inserted into the first sequence to allow a further alignment. If deletions occur in the second sequence in comparison to the first sequence, gaps can be inserted into the second sequence to allow a further alignment. The percentage to which two sequences are identical is then a function of the number of identical positions divided by the total number of positions including those positions which are only occupied in one sequence. The percentage to which two sequences are identical can be determined using a mathematical algorithm. A preferred, but not limiting, example of a mathematical algorithm which can be used is the algorithm of Karlin et al. Proc. Natl. Acad. Sci. USA 90, 5873 (1993) or Altschul et al. Nucleic Acids Res. 25, 3389 (1997). Such an algorithm is integrated in the BLAST program. Sequences which are identical to the sequences of the present invention to a certain extent can be identified by this program.
The enzymes used in step d) may be provided in a free form or alternatively be bound to or are immobilized onto a surface. Binding to or immobilization onto a surface may be carried out e.g. via electrostatic bonds, van der Waals-bonds, covalent bonds, etc. Binding to or immobilization onto a surface may be furthermore carried out, using a covalent linker or a crosslinker, or a tag, as known to a skilled person for purification of proteins. Such tags comprise, inter alia, e.g. affinity tags or
chromatography tags. Affinity tags may include e.g. chitin binding protein (CBP), maltose binding protein (MBP), glutathione-S-transferase (GST), or the Strep-Tag. The poly(His) tag is a widely- used protein tag, that binds to metal matrices. Chromatography tags are used to alter
chromatographic properties of the protein to afford different resolution across a particular separation technique, and include e.g. polyanionic amino acids based tags, such as the FLAG-tag. The surface may be the surface of a bioreactor, or any suitable reaction chamber.
The enzymatic reaction can be carried out as described in WO 2012/007588, WO 2012/127410 or WO 2012/156897, preferably occurs with a concentration of respective enzyme in a concentration of 1 mU/1 to 1000 U/l, preferably 10 mU/1 to 100 U/l, when the activity capable of forming 1 μπιοΐ of specific product for a defined protein starting from a defined educt is defined as 1 unit (U), e.g. for a glycotransferase the production of a glycose-containing complex carbohydrate at 37 °C in 1 minute. The activity of each enzyme as defined herein may be assessed with respect to its naturally occurring or engineered substrate. The incubation may be carried out in a reaction medium, preferably an aqueous medium, comprising the compound obtained according to step c) and optionally water; a buffer such as a phosphate buffer, a carbonate buffer, an acetate buffer, a borate buffer, a citrate buffer and a tris buffer, or combinations thereof; alcohol, such as methanol and ethanol; ester such as ethyl acetate; ketone such as acetone; amide such as acetamide; and the like. Furthermore, the incubation may be carried out in a reaction medium as defined above, wherein optionally a surfactant or an organic solvent may be added, if necessary. Any surfactant capable of accelerating the formation of a complex carbohydrate as defined according to the present invention as a possible product of the invention can be used as the surfactant. Examples include non-ionic surfactants such as poly oxy ethylene octadecylamine (e.g., Nymeen S-215, manufactured by Nippon Oil & Fats); cationic surfactants, such as cetyltrimethylammonium bromide and alkyldimethyl benzylammoniumchloride (e.g., Cation F2-40E, manufactured by Nippon Oil & Fats); anionic surfactants such as lauroyl sarcosinate; tertiary amines such as alkyldimethylamine (e.g., Tertiary Amine FB, manufactured by Nippon Oil & Fats); and the like, which are used alone or as a mixture of two or more. The surfactant may be used generally in a concentration of 0.1 to 50 g/1. The organic solvent may include xylene, toluene, fatty acid alcohol, acetone, ethyl acetate, and the like, which may be used in a concentration of generally 0.1 to 50 ml/1.
The incubation may be furthermore carried out in a reaction medium as defined above, preferably having a pH 3 to 10, pH 5 to 10, preferably pH 6 to 8.
The incubation may be furthermore carried out at a temperature of about 0 °C to about 100 °C, preferably at a temperature of about 10 to about 50 °C, e.g. at a temperature of about 20 °C to about 50 °C. In the reaction medium, inorganic salts, such as MnCl2 and MgCl2, may be added, if necessary.
The incubation according to step d) of the method of the present invention may be carried out in a bioreactor. The bioreactor is preferably suitable for either a continuous mode or a discontinuous mode. If carried out in a continuous mode, the method preferably provides for a continuous flow of compounds and/or enzymes as necessary, preferably by continuously providing educts of the reaction to the reaction mixture and continuously removing products from the reaction mixture, while maintaining the concentration of all components, including enzymes at a predetermined level. The enzymes used in a continuous mode may be added either in free form or as bound or immobilized to a surface.
In the optional step e) of the present invention a compound obtained in step d) is subjected to catalytic hydrogenolysis and/or anomeric deprotection.
Accordingly, the catalytic hydrogenolysis is performed when, in a compound of formula 10 obtained in step d), R' is -N3 or -OR'6 wherein R'6 is a group removable by hydrogenolysis or - (CH2)n-N3, or when Ri6 is a moiety C wherein at least one of the Rn and Ris groups is a group removable by hydrogenolysis and R' is 2-trimethylsilyl-ethyloxy. Such catalytic hydrogenolysis typically takes place in a protic solvent or in a mixture of protic solvents. A protic solvent may be selected from the group consisting of water, acetic acid or Ci-C6 alcohols. A mixture of one or more protic solvents with one or more suitable aprotic organic solvents partially or fully miscible with the protic solvent(s) (such as THF, dioxane, ethyl acetate or acetone) may also be used. Water, one or more Ci-C6 alcohols or a mixture of water and one or more Ci-C6 alcohols are preferably used as the solvent system. Solutions containing the carbohydrate derivatives in any concentration or suspensions of the carbohydrate derivatives in the solvent(s) used are also applicable. The reaction mixture is stirred at a temperature in the range of 10-100 °C, preferably between 20-50 °C, in a hydrogen atmosphere of 1-50 bar absolute (100 to 5000 kPa) in the presence of a catalyst such as palladium, Raney nickel or any other appropriate metal catalyst, preferably palladium on charcoal or palladium black, until reaching the completion of the reaction. Transfer hydrogenolysis may also be performed, when the hydrogen is generated in situ from cyclohexene, cyclohexadiene, formic acid or ammonium formate. Addition of organic or inorganic bases or acids and/or basic and/or
acidic ion exchange resins can also be used to improve the kinetics of the hydrogenolysis. The conditions proposed above allow simple, convenient and delicate removal of benzyl-like protective groups giving rise to a group of compound of formula 1 wherein R is OH (from compounds of formula 10 wherein R' is -N3 or -OR'6 wherein R'6 is a group removable by hydrogenolysis), or R is -0-(CH2)n- H2 (from compounds of formula 10 wherein R' is -(CH2)n-N3), orto another group of compounds of formula 1 wherein R is 2-trimethylsilyl-ethyloxy and R2 is fucosyl moiety (from compounds of formula 10 wherein R' is 2-trimethylsilyl-ethyloxy and Ri6 is a moiety C wherein at least one of the Rn and Ri8 groups is a group removable by hydrogenolysis). It should be noted that the anomeric azide group (R' is -N3) in a the above-mentioned compound of formula 10 is reduced to amino under the conditions disclosed above, and the thus formed glycosyl amine easily undergoes hydrolysis to give the anomerically unprotected compound of formula 1 (R is OH). Similarly, R'= -0-(CH2)n-N3 in a compound of formula 10 is converted to -0-(CH2)n- H2 under these conditions.
Also optionally in step e), R' group being -N3 or -OR'6 wherein R'6 is selected from allyl optionally substituted by one or more methyl or 2-trimethylsilyl-ethyl in a compound of formula 10 obtained in step d) - provided that in moiety C, if it is present in a compound of formula 10, both Rn and Ri8 groups are H - can be converted to OH in an anomeric deprotection reaction. In an embodiment, the R' group being -N3 in a compound of formula 10 can be reduced by complex metal hydrides like NaBH4, or by PPh3 or Cu/Zn. Both types of reactions yield amine functionality at the anomeric position, the hydrolysis of which under neutral or slightly acidic pH (pH ~ 4-7) readily provides the fully deprotected oligosaccharides of formula 1 wherein R is -OH. In other embodiment, allyl glycosides (R' is -OR'6 wherein R'6 is allyl optionally substituted by one ore more methyl) can be removed in a delicate way by a) isomerization with Pd-, Rh- or Ir-complex catalyst followed by mild hydrolysis of the resulting 1-propenyl glycoside, or b) using Pd(0) or Ni(0) catalyst. 2- Trimethylsilyl-ethyl glycosides (R' is -OR'6 wherein R'6 is 2-trimethylsilyl-ethyl) can be deprotected with fluoride ion (from BF "), an anion of a strong acid (e.g. trifluoroacetate) or a Lewis-acid (BF3-etherate, ZnCl2, SnCl4, FeCl3) followed by careful hydrolysis of the resulting glycosyloxy derivative substituted with the remains of the reagent that is used (see Jansson et al. J. Org. Chem. 53, 5629 (1988)). In compounds of formula 10 wherein R' is -N3 or -OR'6 wherein R'6 is selected from allyl optionally substituted by one or more methyl or 2-trimethylsilyl-ethyl, and in moiety C at least one of the Rn and Ri8 groups is a group removable by hydrogenolysis, the
anomeric deprotection reaction is followed by catalytic hydrogenolysis to give compounds of formula 1 wherein R is -OH and R2 is fucosyl moiety.
Thus, in accordance with the present invention, complex sialylated or fucosylated 3-O-galactosyl- GlcNAc or -GalNAc derivatives of formula 1 and salts thereof

1 wherein R is selected from -OH, -N3 and -OR5 wherein 5 is selected from allyl optionally substituted by one or more methyl, propargyl optionally substituted by one or more methyl, 2-trimethylsilyl-ethyl, -(CH2)n-NH2 and -(CH2)n-N3 wherein integer n is i to 10, preferably 2 or 3,
Ri is selected from sialyl moiety, -SO3H and -CH(R5)-COOH wherein R5 is selected from H, alkyl and benzyl,
R2 is selected from H and fucosyl moiety,
R3 is selected from H and sialyl moiety,
R4 is selected from H and fucosyl moiety, provided that one but only one of R3 and R4 is H, and
A is selected from a bond and a divalent carbohydrate linker, can be effectively synthesized. Both the anomerically deprotected derivatives (R is -OH), and the glycosides listed above have synthetic usefulness. The unsaturated bond of glycosides having -N3, -0-(CH2)n-N3, -O-allyl or -O-propargyl aglycon or the -0-(CH2)n-NH2 group can be functionalized by a wide variety of selective and mild water-compatible chemical reactions. Thus e.g. the azido group of a compound of formula 1 can be brought into "click chemistry" with an alkyne to form bioconjugates (and so can the propargyl derivatives of formula 1 with an azido reagent). The allyl functionality can be converted to other functional groups by addition reactions to the double bond or by ozonolysis. The amino function can be used to make an amide or urea linkage to bind a compound of formula 1 to (bio)macromolecules/polymers.
The present method readily provides the synthesis of complex sialylated and/or fucosylated 3'-0- galactosyl-6-O-sialyl-GlcNAc or -GalNAc derivatives, glycosides and analogs thereof, by a unique
combination of chemical and enzymatic glycosylation steps in the reaction sequence. The process opens the possibility, via the permutation of the mandatory and optional glycosylation steps, to obtain a number of compounds being substituted by sialyl and/or fucosyl moiety/moieties in various positions. These possible substitution patterns are summarized in Table 4 below.

Table 4.
In addition, it has shown that the sialyl residue at position 6 can be replaced by simpler charged residues like carboxymethyl or sulfate while maintaining the the biological effect of the parent sialylated compounds (see e.g. Chachadi et al. Glycoconj. J. 28, 49 (2011), WO 2011/130332, Liao et al. J. Am. Chem. Soc. 132, 14849 (2010), Wang et al. Proc. Nat. Acad. Sci. USA 106, 18137 (2009), Schwardt et al. J. Med. Chem. 52, 989 (2009)). The method disclosed above gives the chance for the synthesis of these kinds of analogs as well when the chemical sialylation in step a) is replaced by a chemical carboxymethylation or sulfation.
Moreover, the present method is advantageous in that these various groups of compounds can be obtained in a simpler manner and/or in fewer steps. Ando et al. {Carbohydr. Res. 338, 503 (2003)) taught that 6-O-sialylation of "bulky" acceptors where the OH-group to be sialylated belonged to an internal GlcNAc residue was not favourable because only low yields could be achieved. Therefore it was proposed to sialylate acceptors having a terminal GlcNAc moiety. However the present inventors found that compounds of formula 2 having internal GlcNAc can be sialylated effectively. In addition, all the prior art methods (Ando et al. J. Carbohydr. Chem. 20, 425 (2001) and
Carbohydr. Res. 338, 503 (2003), Pazynina et al. Mendeleev Commun. 13, 245 (2003), Hsu et al. Chem. Eur. J. 16, 1754 (2010), Bhunia et al. ChemBioChem 9, 2941 (2008)) reported the introduction of the 3'-0-sialyl moiety in an indirect way, that is by sialylation of a galactose monosaccharide derivative first to obtain a sialyl-galactose disaccharide that - after transforming it to a disaccharide donor - was used to glycosylate the GlcNAc acceptor, often with unsatisfactory
regioselectivity. The present method however avoids the fabrication and use of this type of difficult to obtain disaccharide donor, and instead introduces a direct enzymatic sialylation of intermediates of formula 7 obtained in step c) of the present method that takes place in good regio- and stereoselectivity. Additionally, the intermediate of formula 7 can be a substrate for enzymatic fucosylation as well, and therefore the method is able to provide a diverse group of variously sialylated and/or fucosylated 3'-0-galactosyl-6-0-sialyl-GlcNAc or -GalNAc derivatives and analogs thereof.
Within the first aspect of the invention the synthesis of compounds of formula 1 wherein A is a divalent carbohydrate linker is preferred. More preferably, compounds of formula 1 are human milk oligosaccharides listed in Table 1 above and derivatives, even more preferably F-LST b, DS-LNT or FDS-LNT I and derivatives. Accordingly, a compound of formula 2D falling into the scope of compounds of formula 2
 wherein Y and R7 are as defined above,
wherein Y and R7 are as defined above,
R' is as defined above, preferably R' is OR" wherein R" is a group removable by
hydrogenolysis, and
B' is a divalent lactosyl moiety having the R' group on its C-l anomeric carbon atom and attached via its 3'-OH group to the lacto-N-biose residue of the compound of formula 2D, optionally substituted by a fucosyl moiety on its 3-OH or by a N-acetyllactosaminyl moiety on its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety on its 6-OH, or by a fucosyl on its 3-OH or 2' -OH, and the functional groups of the divalent lactosyl residue B' are protected, preferably the free OH groups are acylated (e.g. acetylated, benzoylated) and the carboxy group of the optional N- acetylneuraminyl moiety is blocked in ester form (e.g. methyl, ethyl or benzyl ester), except for the axial 4-OH-group of any galactosyl moiety in group B' that is optionally protected, is sialylated, sulfated or carboxymethylated in step a) giving a compound of formula 4D

4D wherein R', B', Y and R7 are as defined above, and Ri9 is selected from -S03H, -CH(R5)-COORi0 and moiety H

H wherein R5, R8 and Q are as defined above. Preferably a compound of formula 2E

2E wherein R", Y and R7 are as defined above,
R20 is independently acyl, and
R21 is selected from H and acyl, more preferably a compound of formula 2E wherein Y is selected from - HAc and
trichloroacetamido, R7 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl, R20 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl, and R21 is selected from H, acetyl, benzoyl or 4-chlorobenzoyl, is reacted with a sialyl donor of formula 3 defined above to give a compound of formula 4E

4E wherein R" is a group removable by hydrogenolysis,
Y is selected from -NHAc, haloalkanoylamido, -NAc2, haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and azido, preferably -NHAc and
trichloroacetamido,
R7 is independently acyl, preferably R7 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
R8 is independently acyl, preferably Rs groups are acetyl,
R2o is independently acyl, preferably R2o groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
R21 is selected from H and acyl, preferably R2i is selected from H, acetyl, benzoyl or 4- chlorobenzoyl, and
Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl.
In the subsequent step b) a compound of formula 4D, preferably of formula 4E, is optionally fucosylated with a fucosyl donor of formula 5A

5A wherein XA is selected from alkylthio and optionally substituted phenylthio, preferably -SPh,
R22 and R23 are, independently, selected from a group removable by hydrogenolysis and acyl, preferably acetyl, pivaloyl, benzoyl and 4-chlorobenzoyl, to give rise to a compound of formula 6D
 wherein R', B', Y, R7 and R19 are as defined above, and R24 is a moiety I
wherein R', B', Y, R7 and R19 are as defined above, and R24 is a moiety I

I wherein R22 and R23 are as defined above, Preferably, a compound of formula 6D is represented by the formula 6E

Y is selected from -NHAc, haloalkanoylamido, -NAc2, haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and azido, preferably -NHAc and
trichloroacetamido,
R7 is independently acyl, preferably R7 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
R8 is independently acyl, preferably Rs groups are acetyl,
R2o is independently acyl, preferably R20 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
R2i is selected from H and acyl, preferably R2i is selected from H, acetyl, benzoyl or 4- chlorobenzoyl, and
Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl, and
R-24 is moiety I as defined above.
In the subsequent step c) a compound of formula 4D obtained in step a) or a compound of formula 6D obtained in step b) is subjected to deprotective treatment and, where necessary, transformation of Y to
NHAc to result in the formation of a compound of formula 7A

7A wherein R' is as defined above, moiety A' is a divalent lactosyl moiety having the R' group on its C-1 anomeric carbon atom and attached via its 3'-OH group to the lacto-N-biose residue of the compound of formula 7A, optionally substituted by a fucosyl moiety on its 3-OH or by a N-acetyllactosaminyl moiety on its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety on its 6-OH, or by a fucosyl on its 3-OH or 2' -OH,
Ri is selected from sialyl, -SO3H and -CH(R5)-COOH, wherein R5 is as defined above, and
Ri6 is selected from H and a moiety of formula C

C wherein Rn and Ris, independently, are selected from H and a group removable by hydrogenolysis, comprising the steps of i) base catalysed transesterification to remove O-acyl groups,
ii) basic hydrolysis,
iii) and optional transformation of Y to -NHAc.
Alternatively, the base catalysed transestenfication deprotection can be avoided and the O-acyl groups can be removed by basic hydrolysis.
Preferably, in this step c) of the method a compound of formula 4E or a compound of formula 6E defined above is converted into a compound of formula 7B

7B wherein R" is a group removable by hydrogenolysis, and Ri6 is selected from H and moiety C defined above, preferably H comprising: i) base catalysed transesterification deprotection, preferably NaOMe/MeOH treatment, ii) basic hydrolysis, preferably NaOH/MeOH treatment, and
iii) optionally selective N-acetylation or peracetylation/de-O-acetylation if group Y in a
compound of formula 4E or 6E selected from haloalkanoylamido,
haloalkoxycarbonylamino, 2,3-diphenylmaleimido, 2,3-dimethylmaleimido and azido, preferably trichloroacetamido, is deprotected to amino under the conditions used in step ii) or by other azido-amino transformations disclosed above.
Still within the preferred embodiment of the method, in step d), a compound of formula 7A is sialylated or fucosylated under the action of a sialidase or fucosidase having transsialidase or transfucosidase/fucosynthase activity, respectively, to give a compound of formula IOC


10D wherein R" and Ri6 are as defined above.
With regard to enzymatic fucosylation in this step d) of the claimed method, a compound of formula 7A, preferably of formula 7B, is subjected to enzymatic fucosylation under the action of a fucosidase having transfucosidase/fucosynthase activity, preferably those listed in Table 3, in the presence of a fucosyl donor selected from 2-O-fucosyllactose or fucosyl fluoride to give a compound of formula 10E

Finally in step e), if it is desired, a compound of formula IOC is subjected to catalytic
hydrogenation to remove benzyl/substituted benzyl protective group(s) and/or to anomeric deprotection method described above to give complex oligosaccharides of formula 1A and analogs thereof, preferably human milk oligosaccharides listed in Table 1 above or analogs

1A wherein A', R, Ri, R2, R3 and R4 are as defined above, provided that one but only one of the R3 and R4 is H.
Preferably, the hydrogenolysis of a compound of formula 10D provides a compound of formula IB

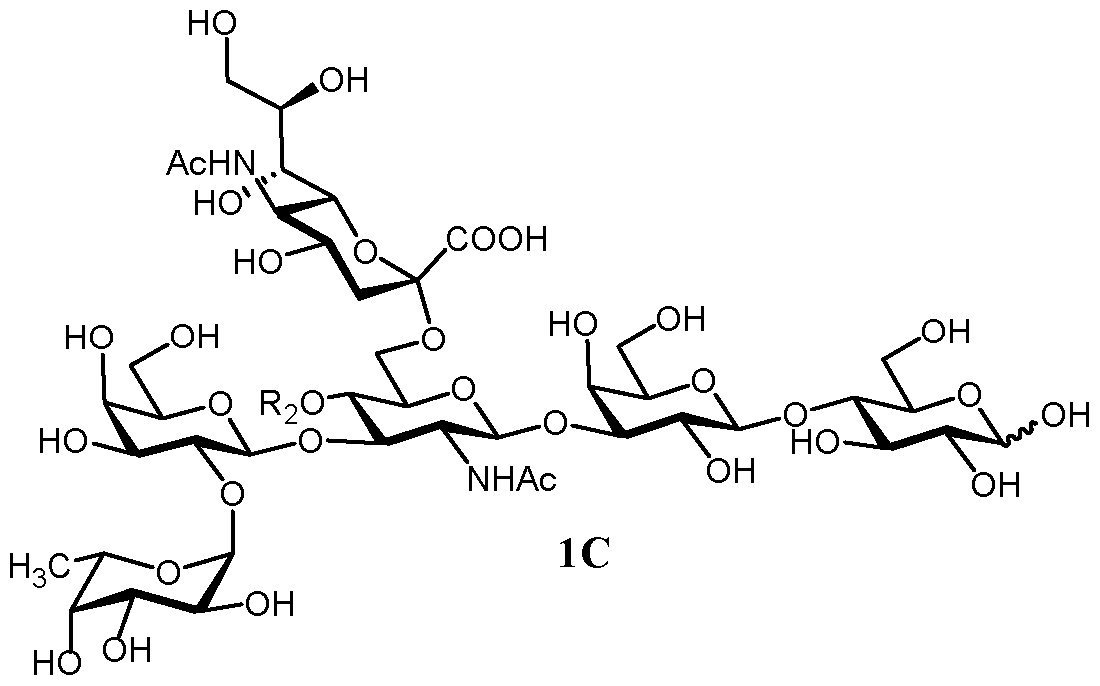

11 wherein R' is selected from -N3 and -OR'6 which R'6 is selected from allyl optionally substituted by one ore more methyl, propargyl optionally substituted by one ore more methyl, a group removable by hydrogenolysis, 2-trimethylsilyl-ethyl and -(CH2)n-N3 wherein integer n is 1 to 10, preferably 2 or 3,
A' is a divalent lactosyl moiety having the R' group on its C-l anomeric carbon atom and attached via its 3'-OH group to the lacto-N-biose residue of the compound of formula 11, A' being optionally substituted by a fucosyl moiety on its 3-OH or by a N-acetyllactosaminyl moiety on its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety on its 6-OH, or by a fucosyl on its 3-OH or 2' -OH,
Ri is selected from sialyl moiety, -SO3H and -CH(R5)-COOH wherein R5 is selected from H, alkyl and benzyl,
R3 is selected from H and sialyl moiety,
R4 is selected from H and fucosyl moiety, provided that at least one of R3 and R4 is H, and Ri6 is selected from H and moiety C, preferably H,

c wherein Rn and Ris, independently, are selected from H and a group removable by hydrogenolysis provided that the di sodium salt of the following compound is excluded: wherein R' is 2- trimethylsilyl-ethyl, A' is unsubstituted lactosyl, Ri and R3 are sialyl moieties, and R4 and Ri6 are H.
In a preferred embodiment R' is -OR" wherein R" is a group removable by hydrogenolysis.
A compound of formula 11 can be either an a- or β-anomer or an anomeric mixture of a- and β- anomers. It can be crystalline solid, oil, syrup, precipitated amorphous material or spray dried product. If crystalline, a compound of formula 11 can exist either in an anhydrous or hydrated crystalline form by incorporating one or several molecules of water into its crystal structure.
Likewise, a compound of formula 11 can exist as a crystalline substance, incorporating ligands such as organic molecules and/or ions into its crystal structure.
A preferred compound of formula 11 is a compound of formula IOC being a precursor for DS-LNT, FDS-LNT I and F-LST b and analogs thereof


10D wherein R" is a group removable by hydrogenolysis, and
Ri6 is selected from H and moiety C defined above, preferably H, and also particularly a compound of formula 10E
 wherein R" is a group removable by hydrogenolysis, and Ri6 is selected from H and moiety C defined above, preferably H. In another preferred embodiment a compound of formula 11 is characterized by formula 7A
wherein R" is a group removable by hydrogenolysis, and Ri6 is selected from H and moiety C defined above, preferably H. In another preferred embodiment a compound of formula 11 is characterized by formula 7A

7A wherein R', A', Ri and Ri6 are as defined above.
Compounds of formula 7A are direct precursors of compounds of formula IOC defined above. A preferred compound of formula 7A is a compound of formula 7B

7B wherein R" is a group removable by hydrogenolysis, and
Ri6 is selected from H and moiety C defined above, preferably H.
Other features of the invention will become apparent in view of the following exemplary embodiments which are illustrative but not limiting of the invention.
EXAMPLES Example 1

To a solution of the tetrasaccharide diol acceptor (10.0 g, ca. 93% purity, prepared as described in WO 2012/155916) and sialic acid phosphite donor (4.45 g, prepared as described in WO
2012/1 13404) in propionitrile (30 ml) was added trimethylsilyl triflate (0.4 ml) dropwise at -30 °C in the presence of 4A molecular sieves and the reaction mixture was allowed to warm up slowly to -20 °C and stirred at this temperature for 60 min. Extra amounts of the donor were added periodically (2.4 g + 2.4 g + 1.2 g) each followed by addition of TMSOTf (100 μΐ + 100 μΐ + 50 μΐ) every 60 min at -30 °C followed by stirring at -20 °C. After 7 hours the mixture was quenched with EtOAc (50 ml) and sat. aq. NaHC03 (20 ml), stirred for 10 min at 0 °C and it was partitioned between EtOAc (150 ml total) and NaHC03 (100 ml), washed with 5% NaHC03 and brine. The combined aqueous solution was re-extracted with EtOAc (50 ml) and washed as above. The combined organic solution was dried and evaporated to give a white foam (20.4 g). The title product was isolated by flash chromatography on silica gel yielding 7.9 g of the title pentasaccharide (62 %). LC-HRMS calculated for [Ci03Hi02Cl8N2O43 +H]+ 2340.3412, found m/z 2340.3371 ( rel.
intensity 100%).
1H MR (CDC13, 300 MHz, CHC13=7.26) δ: 8.00-7.06 (m, 28H), 6.91-6.85 (m, 2H), 6.62 (d, 1H, J = 7.1 Hz, H), 5.64 (d, 1H, J= 3.1 Hz), 5.56 (pt, 1H, J= 9.7 Hz, J= 9.1 Hz), 5.49-5.27 (m, 5H), 5.16 (bd, 1H, J= 9.5 Hz), 5.09 (dd, 1H, J= 8.0 Hz, J= 10.6 Hz), 4.99-4.89 (m, 2H), 4.81-4.74 (m,
2H), 4.64 (d, 2H, J= 8.0 Hz), 4.54 (d, 1H, 12.5 Hz), 4.40-4.33 (m, 3H), 4.31 (ddpo, 1H, J= 2.6 Hz, J= 12.6 Hz), 4.24 (dd, 1H, J= 3.4 Hz, J= 10.3 Hz), 4.16-3.94 (m, 8H), 3.88-3.79 (m, 3H), 3.76 (s0, 3H, C02Me), 3.77-3.71 (m0, 1H), 3.66 (pdt, 1H, J= 3.6 Hz, J= 10.6 Hz), 3.40-3.28 (m, 3H), 3.04 (pdt, 1H, J= 7.7 Hz, J= 9.8 Hz, H-2ghlcN), 2.58 (dd, 1H, J= 4.8 Hz, J= 13.2 Hz, H-3asiai), 2.12 (s, 3H), 2.11 (s, 6H), 2.02 (s0, 3H), 2.01 (s0, 3H), 1.99 (pt0, 1H, H-3bsiai) 1.98 (s0, 3H), 1.93 (s, 3H), 1.91 (s, 3H), 1.89 (s, 3H).
1 C MR (CDC13, 75 MHz) δ: 171.3, 171.1, 170.6, 170.5, 170.4 (3C), 170.3, 169.5, 168.5, 165.3, 165.2, 165.1, 164.8, 164.6, 164.0, 161.8, 140.5, 140.3 (2C), 140.2, 139.8, 138.2, 136.6, 133.8, 131.7-127.6 (33C), 125.6, 101.2, 100.8, 99.2, 99.0, 98.2, 92.0, 81.2, 76.0, 75.1 (2C), 73.3 (2C), 72.6, 72.4, 72.0 (2C), 71.3, 71.0, 70.9, 70.4, 69.4, 69.2, 68.8, 68.7, 67.5, 67.0, 64.4, 63.2, 62.7, 62.5, 61.3, 58.9, 53.1, 48.8, 37.3, 23.6, 21.8, 21.5, 21.2-20.8 (6C).
Example 2

A solution of the product obtained according to Example 1 (13.18 g) in dry MeOH (65 ml) was treated with 25% (w/w) methanolic MeONa (1.05 ml) at 65 °C. The mixture was stirred at this temperature for 3 h. The reaction mixture was neutralized by addition of AcOH and the obtained solution was concentrated to approx. 1/10 volume. The resulting solution was taken up in water and hexane. Layers were separated and the aqueous layer was washed twice with hexane. The water- MeOH phase was then concentrated to approx. 250 ml and treated with a 50% aqueous NaOH solution (2.6 ml) at 65 °C for 1.5 h. The reaction mixture was then evaporated in vacuo. The residue was taken up in dry pyridine (200 ml) and acetic anhydride (160 ml) and the mixture was stirred
under for 40 hours. Volatiles were evaporated and coevaporated twice with toluene. The residue was taken up in CH2CI2, washed with 5% aqueous HCl, then twice with brine. The organic phase was dried, filtered and concentrated. The residue was purified by flash chromatography to give the peracetylated compound (7.86 g, 81 %) as a beige solid. LC-HRMS m/z 860.2636 [M+2H]2+, 871.2552 [M+H+Na]2+, 879.2423 [M+H+K]2+.
1H NMR (DMSO-d6, 300 MHz) δ: 7.66 (d, 1H, J= 11.6 Hz, H), 7.58 (d, 1H, J= 8.9 Hz, H), 7.39-7.21 (m, 5H), 5.31-5.01 (m, 5H), 4.91-4.47 (m, 9H), 4,41-3.27 (m, 23H), 2.52 (dd0, 1H, J= 5.2 Hz, H-3asiai), 2.09 (s, 6H), 2.04 (s, 6H), 2.02 (s, 6H), 1.98 (s, 6H), 1.96 (s, 3H), 1.95 (s, 3H), 1.94 (s, 3H), 1.92 (s, 3H), 1.89 (s, 6H), 1.87 (s, 3H), 1.81 (s, 3H), 1.62 (s, 3H), 1.33 (pt, 1H, J= 11.8 Hz, J = 11.1 Hz, H-3bsiai).
Example 3

Step 1. A solution of the compound from example 1 (6.57 g) in dry MeOH (50 ml) was treated with 25% (w/w) methanolic MeONa (0.5 ml) and stirred at r.t. for 21 hours, quenched with AcOH and evaporated in vacuo. The obtained residue (6.14 g) was sonicated with Et20 and filtered (5x40 ml). Alternatively, methyl benzoates could be extracted by partitioning between water and organic solvent such as ether or hexane. The compound was dried in vacuum oven to give 3.90 g of a crude material.
Step 2. The obtained solid from step 1 was taken up in water (30 ml) containing NaOH (0.514 g) and stirred at r.t. for 3 days and then at 60 °C for 2 hrs. The mixture was neutralized with AcOH
and extracted with ether. The aqueous phase gave 5.52 g of solid after evaporation. LC-HRMS calc. for [C42H66N2028-H]" 1045.3729, found 1045.3732.
Step 3. The product from step 2 was taken up in pyridine and Ac20 mixture (50 ml each) and stirred at 50 °C for 30 min then at r.t. for 18 hours. Extra amount of acetic anhydride (20 ml) was added and the mixture was stirred at 40 °C for 2 hours. The volatiles were removed in vacuo and the residue was coevaporated with toluene then partitioned between CH2C12 (200 ml) and 0.5 M HC1 (50 ml). The aqueous phase was re-extracted with CH2C12 (3x20 ml). Combined organic solution was dried and evaporated to give 5.0 g of crude product as white solid. It was chromatographed on silica to give 3.89 g of the title compound (80.5 %).
Example 4

To a stirred solution of the compound according to Examples 2 or 3 (4.89 g) in dry MeOH (60 ml) was added 25 % (w/w) methanolic MeONa (2.02 ml). After 16 hours the reaction mixture was neutralized carefully by addition of Dowex 50W-X8 resin (H+-form) resin to pH=3. The suspension was filtered and the solvents were evaporated to give the title compound (3.05 g, 98 %) as a white solid. LC-HRMS calc. for [C44H68N2029-H]" 1087.3835, found 1087.3848.
1H MR (D20, 300 MHz) δ: 7.46-7.35 (m, 5H), 4.89 (d, 1H, J= 11.6 Hz, HBn), 4.71 (d, 1H, J = 11.6 Hz, HB„), 4.65 (d, 1H, J= 8.40 Hz), 4.51 (d, lH, J= 8.0 Hz), 4.39 (d, 1H, J= 7.6 Hz), 4.38 (d, 1H, J= 7.8 Hz), 4.12 (d, 1H, J= 3.1 Hz), 3.99-3.44 (m, 29H), 3.30 (pt, 1H, J= 8.5 Hz), 2.69 (dd, lH, J= 4.6 Hz, J= 12,7 Hz, H-3asiai), 1.99 (s, 3H), 1.98 (s, 3H), 1.68 (pt, 1H, J= 11.3 Hz, J= 12.3 Hz, H-3bsiai)
1JC MR (D20, 75 MHz) δ: 175.6, 175.5, 173.5, 137.1, 129.4 (2C), 129.3 (2C), 129.1, 104.1, 103.6, 103.3, 101.6, 100.4, 82.9, 82.5, 79.0, 75.9, 75.6, 75.4, 75.1, 74.3, 73.4, 73.3, 73.1, 72.2 (2C), 71.3, 71.1, 70.6, 69.2, 69.0, 68.9, 68.7, 63.4, 63.3, 61.7, 60.7, 55.4, 52.5, 49.5, 40.5, 22.9, 22.7.
Example 5

To a stirred solution of the compound according to Examples 2 or 3 (7.77 g) in dry MeOH (95 ml) was added 25 % (w/w) methanolic MeONa (2.12 ml). After 16 hours the reaction mixture was neutralized carefully by addition of Dowex 50W-X8 resin (H+-form) to pH=3. The suspension was filtered and 25 % aq. H3 (1.5 ml) was added to the filtrate. The volatiles were then evaporated to give the ammonium salt (5.0 g, 100 %) as a white solid.
Example 6
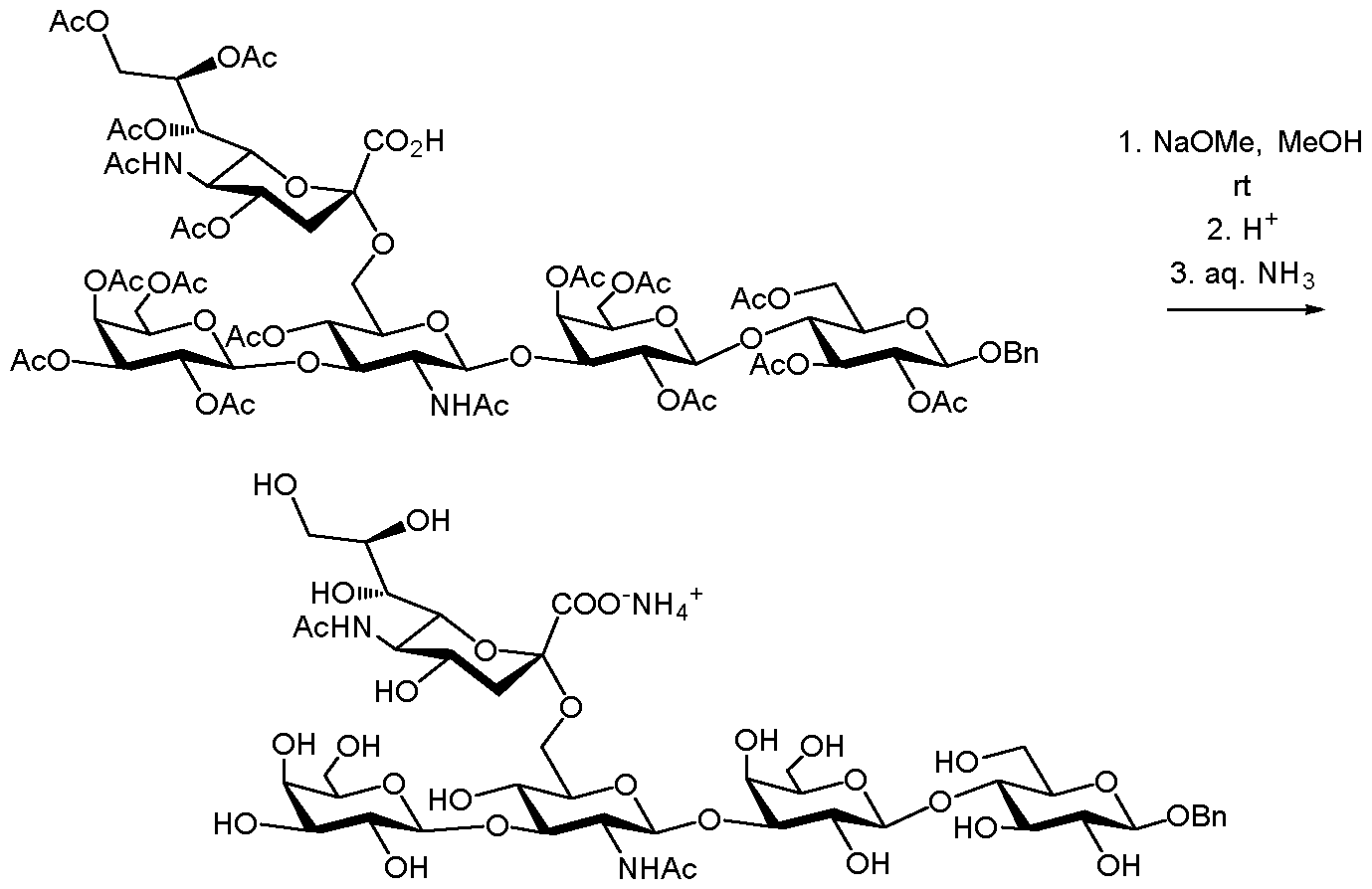
To a stirred solution of the compound according to Examples 2 or 3 (3.83 g) in dry MeOH (100 ml) was added 25 % (w/w) methanolic MeONa (0.5 ml) to give a neutral pH. Extra amount of MeONa added (0.5 ml) to give a strongly basic pH. After 13 hours at r.t. the reaction mixture was neutralized carefully by addition of Dowex 50W-X8 resin (H+-form) to pH=3. The suspension was filtered and 25 % aq. NH3 (1.0 ml) was added to the filtrate. The volatiles were then evaporated to give the ammonium salt (2.50 g, 100 %).
Example 7

Reaction mixtures were loaded on a Dowex 1 (HC03 " -form) column. After washing with distilled water, the mono-acidic compounds (3'-0-sialyllactose or sialic acid and the remaining acceptor) were eluted with a 60 mM NaHC03 solution. The product was then eluted with a 125 mM NaHC03 solution. The eluted fractions containing acidic oligosaccharides were analysed by HPLC and pooled. The NaHC03 was removed by treating with Amberlite IR120 (H+ form) until pH 3.0 was reached. The pH was then adjusted to 6.0 with NaOH. The mono-acidic fractions were freeze-dried and reused with another enzymatic run. The fractions containing the products were freeze-dried.
LC-HRMS: m/z calc. for [C55H85N3037 -H]" 1378.4789, found 1378.4807, calc. for [M-2H]2" 688.7358, found 688.7376 (main peak).
1H NMR (300 MHz, 100 mg in 0.7 ml D20, DHO=4.81 ppm) δ: 7.37-7.47 (m, 5H, Ph of Bn), 4.923 (d, J=11.6 Hz, IH, CH2 ofBn), 4.739 (d, J=11.5 Hz, IH, CH2 of Bn), 4.685 (d, J=8.4 Hz, IH), 4.534 (d, J=8.0 Hz, IH), 4.494 (d, J=7.8 Hz, IH), 4.419 (d, J=7.7 Hz, IH), 4.154 (d, J=3.1 Hz, IH, H-4'), 4.074 (dd, J=3.1 Hz, J=9.8 Hz, IH, H-3' "), 3.48-4.02 (overlapping m, 35 H), 3.921
(overlapping d, J=3.2 Hz, H-4'"), 3.335 (t, J=8.35 Hz, IH, H-2), 2.731 and 2.748 (two overlapping dd, J=12.5 Hz, J=4.9 Hz, 2H, two H-3" "eq(2,6) and (2,3)), 2.01 (three overlapping s, 9H, 3 AcNH), 1.775 (t, J=12.1 Hz, IH, H-3""^^)), and 1.679 (t, J=12.1 Hz, IH, H-3" "ax(2,6)).
1 C NMR (75 Hz, NaOAc = 182.02 & 23.95 ppm) δ: 175.80, 175.73, 175.65 (3 Ac), 174.70 (C- l " "(2-3)), 174.23 (C-l" "(2-6)), 137.29 (C-l of Ph), 129.58, 129.53 (C-2 & C-3 of Ph), 129.30 (C-4 ofPh), 104.19, 103.70, 103.37 (C-l' ", C-l ', C-l "), 101.77 (C-l), 100.96 (C-2""(2;6)), 100.41 (C- 2" "(2>3)), 83.07 (C-3"), 82.55 (C-3'), 79.17 (C-4), 76.37 (C-3' "), 75.87 (C-5' "), 75.75 (C-5'), 75.58 (C-5), 75.19 (C-3), 74.50 (C-5"), 73.57 (C-6" "(2-3)), 73.28 (C-6" "(2-6)), 72.62 (C-8" "(2-3)), 72.50 (C-8" "(2-6)), 72.30 (CH2 ofBn), 70.74 (C-2'), 69.89 (C-2' "), 69.15, 69.14, 69.11, 69.00, 68.80 (overlapping C-4', C-4", C-4""(2-6)&(2-3), C-7" "(2-6)&(2-3)), 68.07 (C-4' "), 63.60 (C-6"),
63.36 (C-9" "(2,3)), 63.21 (C-9" "(2>6)), 63.86 (overlapping C-6', C-6' "), 60.83 (C-6), 53.39 (C-2"), 52.63 (C-5" "(2>6)), 52.43 (C-5" "(2>3)), 40.86 (C-3""(2,6)), 40.53 (C-3" "(2>3)), 23.12 (Ac of
GlcNAc), 22.88 & 22.86 (2 overlapping Ac of NeuAc(2-6)&(2-3)).
Example 8: DS-LNT disodium salt
The compound obtained according to Example 7 (4.43 g) and 10 % Pd on carbon (0.4 g) were suspended in a mixture of deionized water (15 ml) and methanol (15 ml) and stirred under hydrogen at 5 bar pressure. After 24 hours extra amount of the catalyst (100 mg) was added and the hydrogenolysis continued at 5 bars for 3 days. The reaction mixture was filtered via short plug of Celite (5 g), which was further washed with 1 : 1 aqueous methanol and concentrated under reduced pressure to a small volume. The residue was treated with Amberlite IRC-86 (H+ form). The obtained solution was passed through Amberlite IRC-86 column (Na+ form). After all the product was eluted with water, it was freeze-dried to give the title disodium salt as a white foam (3.855 g, 93%). LC-MS: UV purity 97.2 % (205 nm); the retention time and MS/MS were identical with those of a reference sample purchased from Carbosynth. HRMS, m/z calculated for [C48H79N3O37 - 2H]2" 643.7123, found 643.7150. 1H MR (300 MHz, D20) was identical within 0.01 ppm with that reported in the literature (Sabesan et al. J. Am. Chem. Soc. 108, 2068 (1986)).
Claims
1. A method for making a sialylated or fucosylated 3-O-galactosyl-GlcNAc or -GalNAc
derivative of formula 1 and salts thereof

1
wherein R is selected from -OH, -N3 and -OR5 wherein 5 is selected from allyl optionally substituted by one or more methyl, propargyl optionally substituted by one or more methyl, 2-trimethylsilyl-ethyl, -(CH2)n- H2 and -(CH2)n-N3 wherein integer n is i to 10, preferably 2 or 3,
Ri is selected from sialyl moiety, -SO3H and -CH(R5)-COOH wherein R5 is selected from H, alkyl and benzyl,
R2 is selected from H and fucosyl,
R3 is selected from H and sialyl,
R4 is selected from H and fucosyl, provided that one but only one of R3 and R4 is H, and A is selected from a bond and a divalent carbohydrate linker,
lprising the steps:
a) sialylation, sulfation or carbox methylation of a compound of formula 2
 wherein R' is selected from -N3 and -OR'6 and wherein R'6 is selected from allyl optionally substituted by one ore more methyl, propargyl optionally substituted by one or more methyl, a group removable by hydrogenolysis, 2-trimethylsilyl-ethyl and -(CH2)n-N3 wherein integer n is 1 to 10, preferably 2 or 3,
wherein R' is selected from -N3 and -OR'6 and wherein R'6 is selected from allyl optionally substituted by one ore more methyl, propargyl optionally substituted by one or more methyl, a group removable by hydrogenolysis, 2-trimethylsilyl-ethyl and -(CH2)n-N3 wherein integer n is 1 to 10, preferably 2 or 3,
R7 is independently acyl,
Y is selected from -NHAc, haloalkanoylamido, -NAc2, haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and -N3, and
B is selected from a bond and a divalent carbohydrate linker in protected form, b) optional fucosylation of the compound obtained in step a),
c) de-O-acylation and/or basic hydrolysis, optional mild acidic hydrolysis and optional transformation of Y to - HAc of the compound obtained in step a) or step b), d) sialylation or fucosylation of the compound obtained in step c), and
e) optional catalytic hydrogenolysis and/or anomeric deprotection of the compound
obtained in step d).
2. The method according to claim 1, wherein sialylation, sulfation or carboxymethylation of a compound of formula 2 in step a) results in the formation of a compound of formula 4

4
wherein R', R7, B and Y are as defined in claim 1, and
P is selected from protected sialyl moiety, -SO3H and -CH(R5)-COORio wherein R5 is selected from H, alkyl and benzyl, Rio is selected from alkyl and benzyl.
3. The method according claim 2, wherein the sialylation in step a) is carried out by reacting a compound of formula 2 with a compound of formula 3

3
wherein R8 is acyl, preferably acetyl,
Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl, and
R9 is optionally substituted phenyl or benzyl,
to give a compound of formul 4A

4A
wherein R', R7, B and Y are as defined in claim 1, and
R8 and Q are as defined above.
4. The method according to claim 3, wherein a compound of formula 4A is represented by formula 4E

wherein R" is a group removable by hydrogenolysis,
Y is selected from -NHAc, haloalkanoylamido, -NAc2, haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and azido, preferably -NHAc and trichloroacetamido,
R7 is independently acyl, preferably R7 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
R8 is independently acyl, preferably Rs groups are acetyl,
R2o is independently acyl, preferably R2o groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
R21 is selected from H and acyl, preferably R2i is selected from H, acetyl, benzoyl or 4- chlorobenzoyl, and
Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl.
5. The method according to claim 1, wherein the optional fucosylation in step b) comprises the reaction of a compound of f rmula 4
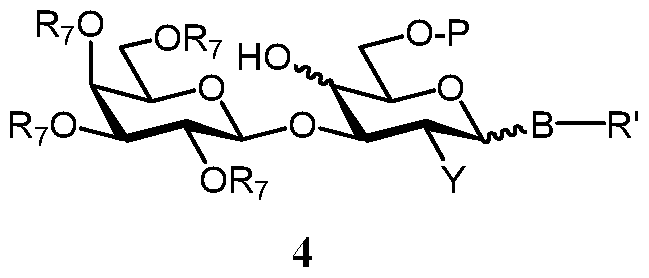
wherein R', R7, B and Y are as defined in claim 1, and
P is selected from protected sialyl moiety, -SO3H and -CH(R5)-COORio wherein R5 is selected from H, alkyl and benzyl, Rio is selected from alkyl and benzyl,
with a compound of formula 5

5
wherein X is selected from a halogen, -OC(= H)CCl3, -O-pentenyl, -OAc, -OBz and -SRi3, in which Ri3 is alkyl or optionally substituted phenyl,
Rii is selected from acyl and a group removable by hydrogenolysis, and
Ri2 is selected from a group removable by hydrogenolysis, acyl or two R12 groups together form a moiety
 , wherein R14 and R15 independently are alkyl or phenyl, or wherein R14 and R15 together with the carbon atom, to which they are attached, form a cycloalkylidene,
, wherein R14 and R15 independently are alkyl or phenyl, or wherein R14 and R15 together with the carbon atom, to which they are attached, form a cycloalkylidene,
to give a compound of form la 6
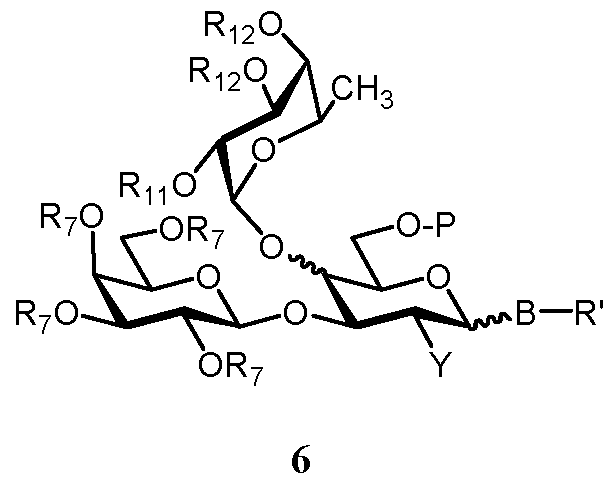
wherein R', R7, Rn, Ri2, B, P and Y are as defined above.
6. The method according to claim 5, wherein a compound of formula 4 is a compound of formula 4E defined in claim 4, a compound of formula 5 is a compound of formula 5A

5A wherein XA is selected from alkylthio and optionally substituted phenylthio, preferably -SPh,
R22 and R23 are, independently, selected from a group removable by hydrogenolysis and acyl, preferably acetyl, pivaloyl, benzoyl and 4-chlorobenzoyl,
and a compound formula 6 is a compound of formula 6E

6E
wherein R" is a group removable by hydrogenolysis,
Y is selected from -NHAc, haloalkanoylamido, -NAc2, haloalkoxycarbonylamino, 2,3- diphenylmaleimido, 2,3-dimethylmaleimido and azido, preferably -NHAc and trichloroacetamido,
R7 is independently acyl, preferably R7 groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
R8 is independently acyl, preferably Rs groups are acetyl,
R2o is independently acyl, preferably R2o groups are identical and selected from acetyl, benzoyl and 4-chlorobenzoyl,
R21 is selected from H and acyl, preferably R2i is selected from H, acetyl, benzoyl or 4- chlorobenzoyl, and
Q is selected from alkyl and benzyl, preferably methyl, ethyl and benzyl, and
R24 is moiety I

I
wherein R22 and R23 are as defined above.
7. The method according to claim 1, wherein a compound obtained in step a) or step b) is converted into a compound f formula 7

7
wherein R' and Ri are as defined in claim 1,
moiety A is bond or is a divalent carbohydrate linker in deprotected form, and
selected from H and a moiety of formula C

C
wherein Rn and Ris, independently, are selected from H and a group removable by hydrogenolysis,
comprising the steps of:
i) base catalysed transesterification and/or
ii) basic hydrolysis,
iii) optional mild acidic hydrolysis, and
iv) optional conversion to Y to - HAc.
8. The method according to claim 7, wherein a compound of formula 4E defined in claim 4 and obtained in step a) or a compound of formula 6E defined in claim 6 and obtained in step b) is subjected to deprotection and optional functional group transformation comprising: i) base catalysed transesterification deprotection, preferably NaOMe/MeOH treatment,
ii) basic hydrolysis, preferably NaOH/MeOH treatment, and
iii) where group Y in a compound of formula 4E or 6E is selected from haloalkanoylamido, haloalkoxycarbonylamino, 2,3-diphenylmaleimido, 2,3- dimethylmaleimido and azido, preferably trichloroacetamido, and is deprotected to amino under the conditions used in step ii) followed by selective N-acetylation, or peracetylation followed by de-O-acetylation,
to yield a compound of formula 7B

wherein R" is a group removable by hydrogenolysis, and
Ri6 is selected from H and moiety C defined in claim 7, preferably H.
9. The method according to claim 1, wherein in step d) a compound of formula 7 defined in claim 7 and obtained in step c) is reacted with a sialyl donor, preferably a N-acetyl neuraminyl donor, under the catalysis of an enzyme having a2-3-transsialidase activity or with a fucosyl donor under the catalysis of an enzyme having transfucosidase/fucosynthase activity to give a compound of formula 10

wherein R', Ri, Ri6 and A are as defined in claim 7,
R3 is selected from H and a sialyl, and
R4 is selected from H and fucosyl moiety, provided that one but only one of R3 and R4 is H.
10. The method according to claim 9, wherein a compound of formula 7 is a compound of
formula 7B defined in claim 8, the sialyl donor is a N-acetyl neuraminyl donor selected from 3'-0-sialyllactose or 2-O-(p-nitrophenyl) N-acetyl-a-neuraminoside, and a compound of formul 10 is a compound of formula 10D

10D
wherein R" and Ri6 are as defined in claim 8.
11. The method according to claim 9, wherein a compound of formula 7 is a compound of
formula 7B defined in claim 8, the fucosyl donor is selected from from 2-O-fucosyllactose or fucosyl fluoride, and a compound of formula 10 is a compound of formula 10E

wherein R" and Ri6 are as defined in claim 8.
12. The method according to claim 1, wherein a compound of formula 10 defined in claim 9 is subjected to catalytic hydrogenolysis and/or anomeric deprotection in the optional step e) to give a compound of formula 1 defined in claim 1.
13. The method according to claim 12, wherein a compound of formula 10 is a compound of formula 10D or 10E defined in claim 11, and is subjected to catalytic hydrogenolysis to give rise to DS-LNT, FDS-LNT or F-LST b.
14. A compound of formula 11 and salts thereof

11
wherein R' is selected from -N3 and -OR'6 which R'6 is selected from allyl optionally substituted by one or more methyl, propargyl optionally substituted by one or more methyl, a group removable by hydrogenolysis, 2-trimethylsilyl-ethyl and -(CH2)n-N3 wherein integer n is 1 to 10, preferably R' is -OR" wherein R" is a group removable by hydrogenolysis,
A' is a divalent lactosyl moiety having the R' group on its C-l anomeric carbon atom and attached via its 3'-OH group to the lacto-N-biose residue of the compound of formula 11, optionally substituted by a fucosyl moiety at its 3-OH or by a N- acetyllactosaminyl moiety at its 6'-OH, which N-acetyllactosaminyl moiety can optionally be further substituted by an N-acetylneuraminyl moiety at its 6-OH, or by a fucosyl at its 3-OH or 2' -OH,
Ri is selected from sialyl moiety, -SO3H and -CH(R5)-COOH wherein R5 is selected from H, alkyl and benzyl,
R3 is selected from H and sialyl moiety,
R4 is selected from H and fucosyl moiety, provided that at least one of R3 and R4 is H, and
Ri6 is selected from H and moiety C, preferably H,

C
wherein Rn and Ris, independently, are selected from H and a group removable by hydrogenolysis
provided that the di sodium salt of the following compound is excluded: R' is 2- trimethylsilyl-ethyl, A' is unsubstituted lactosyl, Ri and R3 are sialyl moieties, and R4 and
15. The compound according to claim 14, wherein one but only one of R3 and R4 is H.
16. The compound according to claim 15 characterized by formula 10D

10D
wherein R" is a group removable by hydrogenolysis, and
Ri6 is selected from H and moiety C defined in claim 14, preferably H.
17. The compound according to claim 15 characterized by formula 10E
wherein R" is a group removable by hydrogenolysis, and
Ri6 is selected from H and moiety C defined in claim 14, preferably H.
18. The compound according to claim 14, wherein R3 and R4 are H.
19. The compound according to claim 18 characterized by formula 7B

wherein R" is a group removable by hydrogenolysis, and
Ri6 is selected from H and moiety C defined above, preferably H.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1306687.3A GB201306687D0 (en) | 2013-04-12 | 2013-04-12 | Synthesis of sialylated/fucosylated oligosaccharides |
| GB1306687.3 | 2013-04-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014167537A1 true WO2014167537A1 (en) | 2014-10-16 |
Family
ID=48537173
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2014/060647 WO2014167537A1 (en) | 2013-04-12 | 2014-04-11 | Synthesis of sialylated/fucosylated oligosaccharides |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB201306687D0 (en) |
| WO (1) | WO2014167537A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021061991A1 (en) | 2019-09-24 | 2021-04-01 | Prolacta Bioscience, Inc. | Compositions and methods for treatment of inflammatory and immune diseases |
| EP3645146A4 (en) * | 2017-06-30 | 2021-05-05 | Glycom A/S | Synthesis of oligosaccharides |
| WO2022036225A1 (en) | 2020-08-14 | 2022-02-17 | Prolacta Bioscience, Inc. | Human milk oligosaccharide compositions for use with bacteriotherapies |
| WO2022155201A1 (en) | 2021-01-12 | 2022-07-21 | Prolacta Bioscience, Inc. | Synbiotic treatment regimens |
| WO2024130119A2 (en) | 2022-12-16 | 2024-06-20 | Prolacta Bioscience, Inc. | Synbiotic compositions for short chain fatty acid production |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0319253A2 (en) * | 1987-12-02 | 1989-06-07 | Alberta Research Council | Sialic acid glycosides, antigens, immunoadsorbents, and methods for their preparation |
| WO2003089574A2 (en) * | 2002-04-15 | 2003-10-30 | Biomira, Inc. | Synthetic glyco-lipo-peptides as vaccines |
| WO2005116088A2 (en) * | 2004-05-25 | 2005-12-08 | The Johns Hopkins University | Methods and compositions for treating diseases and disorders associated with siglec-8 expressing cells |
| EP1650226A1 (en) * | 2003-07-28 | 2006-04-26 | Yasuhiro Kajihara | Aminated complex-type sugar chain derivatives and process for the production thereof |
| WO2011100979A1 (en) * | 2010-02-19 | 2011-08-25 | Glycom A/S | Production of 6'-o-sialyllactose and intermediates |
| CN103555796A (en) * | 2013-10-24 | 2014-02-05 | 山东大学 | Synthetic method of double-sialylated tetrasaccharide |
-
2013
- 2013-04-12 GB GBGB1306687.3A patent/GB201306687D0/en not_active Ceased
-
2014
- 2014-04-11 WO PCT/IB2014/060647 patent/WO2014167537A1/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0319253A2 (en) * | 1987-12-02 | 1989-06-07 | Alberta Research Council | Sialic acid glycosides, antigens, immunoadsorbents, and methods for their preparation |
| WO2003089574A2 (en) * | 2002-04-15 | 2003-10-30 | Biomira, Inc. | Synthetic glyco-lipo-peptides as vaccines |
| EP1650226A1 (en) * | 2003-07-28 | 2006-04-26 | Yasuhiro Kajihara | Aminated complex-type sugar chain derivatives and process for the production thereof |
| WO2005116088A2 (en) * | 2004-05-25 | 2005-12-08 | The Johns Hopkins University | Methods and compositions for treating diseases and disorders associated with siglec-8 expressing cells |
| WO2011100979A1 (en) * | 2010-02-19 | 2011-08-25 | Glycom A/S | Production of 6'-o-sialyllactose and intermediates |
| CN103555796A (en) * | 2013-10-24 | 2014-02-05 | 山东大学 | Synthetic method of double-sialylated tetrasaccharide |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3645146A4 (en) * | 2017-06-30 | 2021-05-05 | Glycom A/S | Synthesis of oligosaccharides |
| US11214588B2 (en) | 2017-06-30 | 2022-01-04 | Glycom A/S | Synthesis of oligosaccharides |
| US11919919B2 (en) | 2017-06-30 | 2024-03-05 | Glycom A/S | Synthesis of oligosaccharides |
| WO2021061991A1 (en) | 2019-09-24 | 2021-04-01 | Prolacta Bioscience, Inc. | Compositions and methods for treatment of inflammatory and immune diseases |
| WO2022036225A1 (en) | 2020-08-14 | 2022-02-17 | Prolacta Bioscience, Inc. | Human milk oligosaccharide compositions for use with bacteriotherapies |
| WO2022155201A1 (en) | 2021-01-12 | 2022-07-21 | Prolacta Bioscience, Inc. | Synbiotic treatment regimens |
| WO2024130119A2 (en) | 2022-12-16 | 2024-06-20 | Prolacta Bioscience, Inc. | Synbiotic compositions for short chain fatty acid production |
Also Published As
| Publication number | Publication date |
|---|---|
| GB201306687D0 (en) | 2013-05-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2900829B1 (en) | Glycoconjugate synthesis | |
| AU2011278239B2 (en) | Synthesis of new sialooligosaccharide derivatives | |
| Gridley et al. | Recent advances in the construction of β-D-mannose and β-D-mannosamine linkages | |
| CN103562214B (en) | Human milk oligosaccharides (HMO) or the variation of its precursor | |
| EP2984096B1 (en) | Synthesis of sialylated and optionally fucosylated human milk oligosaccharides | |
| WO2014167537A1 (en) | Synthesis of sialylated/fucosylated oligosaccharides | |
| Shoda et al. | Green process in glycotechnology | |
| US20220106348A1 (en) | N-acetylated sialic acids and related sialosides | |
| Cao et al. | General consideration on sialic acid chemistry | |
| Wang et al. | Glycosylation of Nα-lauryl-O-(β-D-xylopyranosyl)-L-serinamide as a saccharide primer in cells | |
| US9187513B2 (en) | N-substituted mannosamine derivatives, process for their preparation and their use | |
| Shoda | Enzymatic glycosylation | |
| Aït-Mohand et al. | Efficient and stereocontrolled synthesis of chondroitin mono-and disaccharide linked to variously sulfated biotinylated trisaccharides of the linkage region of proteoglycans | |
| Huang et al. | Chemoenzymatic synthesis of α2–3-sialylated carbohydrate epitopes | |
| Jaiswal et al. | Sialic acid donors: stereoselective chemical and enzymatic O-glycosylations | |
| US20140303363A1 (en) | Preparation and Use of Isolactosamine and Intermediates therefor | |
| Guo | One-Pot Enzymatic Synthesis of UDP-GlcA and UDP-GalA and Chemoenzymatic Synthesis of a Library of Human Milk Oligosaccharides and Enzymatic Synthsis of O-antigen from P. aeruginosa serotype O11 | |
| CN118434851A (en) | Novel glycoside synthase | |
| Scheppokat | Efficient enzymatic syntheses of L-fucosylated and D-galactosylated oligosaccharides using glycosyltransferases from Helix pomatia albumen glands | |
| Feng | The Total Chemical Synthesis of Tumor-associated Forssman Antigen | |
| Chmielewski | ANOMERIC HYDROPEROXIDES; SYNTHESIS, ENANTIOSELECTIVE EPOXIDATION | |
| AU2012257396A1 (en) | Diversification of human milk oligosaccharides (HMOs) or precursors thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14783020 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14783020 Country of ref document: EP Kind code of ref document: A1 |