WO2013173382A1 - Benzothiophene sulfonamides and other compounds that interact with glucokinase regulatory protein - Google Patents
Benzothiophene sulfonamides and other compounds that interact with glucokinase regulatory protein Download PDFInfo
- Publication number
- WO2013173382A1 WO2013173382A1 PCT/US2013/041011 US2013041011W WO2013173382A1 WO 2013173382 A1 WO2013173382 A1 WO 2013173382A1 US 2013041011 W US2013041011 W US 2013041011W WO 2013173382 A1 WO2013173382 A1 WO 2013173382A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyridinyl
- methyl
- benzothiophen
- methylethyl
- cyclopropanesulfonamide
- Prior art date
Links
- 0 C1C2C1CCC*2 Chemical compound C1C2C1CCC*2 0.000 description 16
- PFWWSGFPICCWGU-UHFFFAOYSA-N O=S(C1CC1)(Cl)=O Chemical compound O=S(C1CC1)(Cl)=O PFWWSGFPICCWGU-UHFFFAOYSA-N 0.000 description 3
- WAILDPYIBNGEGR-UHFFFAOYSA-N CC(C)(C)OC(Nc(cc1)nc(Cc2cc3cccc(-c4cc(C5(C)OC(C)(C)OC5)ccn4)c3[s]2)c1Cl)=O Chemical compound CC(C)(C)OC(Nc(cc1)nc(Cc2cc3cccc(-c4cc(C5(C)OC(C)(C)OC5)ccn4)c3[s]2)c1Cl)=O WAILDPYIBNGEGR-UHFFFAOYSA-N 0.000 description 2
- CESUXLKAADQNTB-UHFFFAOYSA-N CC(C)(C)S(N)=O Chemical compound CC(C)(C)S(N)=O CESUXLKAADQNTB-UHFFFAOYSA-N 0.000 description 2
- ZRAYQPCAUZXCPZ-JEMSNHSXSA-N CC(C)(C)[S@](/N=C\c(cccc1)c1Cl)=O Chemical compound CC(C)(C)[S@](/N=C\c(cccc1)c1Cl)=O ZRAYQPCAUZXCPZ-JEMSNHSXSA-N 0.000 description 2
- DWSIXGXPMDAJFT-UHFFFAOYSA-N CC(CO)(c1ccnc(-c2c3[s]c(Cc(nc(cc4)N)c4Cl)cc3ccc2)c1)O Chemical compound CC(CO)(c1ccnc(-c2c3[s]c(Cc(nc(cc4)N)c4Cl)cc3ccc2)c1)O DWSIXGXPMDAJFT-UHFFFAOYSA-N 0.000 description 2
- NTALLVUYGBBGFE-BPARTEKVSA-N C[C@](CO)(c1ccnc(-c2c3[s]c(C(c(nc(cc4)N)c4Cl)N)cc3ccc2)c1)O Chemical compound C[C@](CO)(c1ccnc(-c2c3[s]c(C(c(nc(cc4)N)c4Cl)N)cc3ccc2)c1)O NTALLVUYGBBGFE-BPARTEKVSA-N 0.000 description 2
- WMSPXQIQBQAWLL-UHFFFAOYSA-N NS(C1CC1)(=O)=O Chemical compound NS(C1CC1)(=O)=O WMSPXQIQBQAWLL-UHFFFAOYSA-N 0.000 description 2
- GTPINKYWKTZDBU-UHFFFAOYSA-N NS(CC1CC1)(=O)=O Chemical compound NS(CC1CC1)(=O)=O GTPINKYWKTZDBU-UHFFFAOYSA-N 0.000 description 2
- XJPMSKSCGIBWPC-UHFFFAOYSA-N O=S(CC1CC1)(Cl)=O Chemical compound O=S(CC1CC1)(Cl)=O XJPMSKSCGIBWPC-UHFFFAOYSA-N 0.000 description 2
- SKVVBLQXOGSYSY-UHFFFAOYSA-N CC(C(C(F)(F)F)=O)c1ccnc(-c2c3[s]c(Cc4ccccc4[O]=S(c4ccccc4)(N)=O)cc3ccc2)c1 Chemical compound CC(C(C(F)(F)F)=O)c1ccnc(-c2c3[s]c(Cc4ccccc4[O]=S(c4ccccc4)(N)=O)cc3ccc2)c1 SKVVBLQXOGSYSY-UHFFFAOYSA-N 0.000 description 1
- POHVRPGQHISCFD-UHFFFAOYSA-N CC(C(F)(F)F)(c1ccnc(-c2c3[s]c(Cc(cccc4)c4Cl)cc3cc(Cl)c2)c1)O Chemical compound CC(C(F)(F)F)(c1ccnc(-c2c3[s]c(Cc(cccc4)c4Cl)cc3cc(Cl)c2)c1)O POHVRPGQHISCFD-UHFFFAOYSA-N 0.000 description 1
- CIFODHMEKBQUNJ-XQZUBTRRSA-N CC(C(F)(F)F)(c1ccnc(-c2c3[s]c([C@@H](c(cccc4)c4Cl)NS(C4CC4)(=O)=O)cc3cc(Cl)c2)c1)O Chemical compound CC(C(F)(F)F)(c1ccnc(-c2c3[s]c([C@@H](c(cccc4)c4Cl)NS(C4CC4)(=O)=O)cc3cc(Cl)c2)c1)O CIFODHMEKBQUNJ-XQZUBTRRSA-N 0.000 description 1
- XDCOTNHYFWIQFI-UHFFFAOYSA-N CC(C(F)(F)F)C1=CC(c2c3[s]c(Cc(cccc4)c4Cl)cc3ccn2)=NC(C)C1 Chemical compound CC(C(F)(F)F)C1=CC(c2c3[s]c(Cc(cccc4)c4Cl)cc3ccn2)=NC(C)C1 XDCOTNHYFWIQFI-UHFFFAOYSA-N 0.000 description 1
- WBPTXGLVQXKWML-PSRIUPCASA-N CC(C(F)(F)F)c1ccnc(-c2c3[s]c([C@@H](c(cccc4)c4Cl)NS(C4CC4)(=O)=O)cc3ccn2)c1 Chemical compound CC(C(F)(F)F)c1ccnc(-c2c3[s]c([C@@H](c(cccc4)c4Cl)NS(C4CC4)(=O)=O)cc3ccn2)c1 WBPTXGLVQXKWML-PSRIUPCASA-N 0.000 description 1
- DWPNRFUMTYEDRP-VCHYOVAHSA-N CC(C)(C)OC(N(C)c(cc1)nc(/C=N/S(C(C)(C)C)=O)c1Cl)=O Chemical compound CC(C)(C)OC(N(C)c(cc1)nc(/C=N/S(C(C)(C)C)=O)c1Cl)=O DWPNRFUMTYEDRP-VCHYOVAHSA-N 0.000 description 1
- QELHOZLOTUGMOU-UHFFFAOYSA-N CC(C)(C)OC(Nc(cc1)cc(CO)c1Cl)=O Chemical compound CC(C)(C)OC(Nc(cc1)cc(CO)c1Cl)=O QELHOZLOTUGMOU-UHFFFAOYSA-N 0.000 description 1
- SKPJBYJNNGSCNG-UHFFFAOYSA-N CC(C)(C)OC(Nc(cc1)nc(C(CNS(C2CC2)(=O)=O)c2cc3cccc(-c4cc(C5(COC5)O)ccn4)c3[s]2)c1Cl)=O Chemical compound CC(C)(C)OC(Nc(cc1)nc(C(CNS(C2CC2)(=O)=O)c2cc3cccc(-c4cc(C5(COC5)O)ccn4)c3[s]2)c1Cl)=O SKPJBYJNNGSCNG-UHFFFAOYSA-N 0.000 description 1
- WAILDPYIBNGEGR-SSEXGKCCSA-N CC(C)(C)OC(Nc(cc1)nc(Cc2cc3cccc(-c4cc([C@]5(C)OC(C)(C)OC5)ccn4)c3[s]2)c1Cl)=O Chemical compound CC(C)(C)OC(Nc(cc1)nc(Cc2cc3cccc(-c4cc([C@]5(C)OC(C)(C)OC5)ccn4)c3[s]2)c1Cl)=O WAILDPYIBNGEGR-SSEXGKCCSA-N 0.000 description 1
- PXPZXNKBZYLYIW-UHFFFAOYSA-N CC(C)(C)OC(Nc(cc1)nc(Cc2cc3cccc(B4OC(C)(C)C(C)(C)O4)c3[s]2)c1Cl)=O Chemical compound CC(C)(C)OC(Nc(cc1)nc(Cc2cc3cccc(B4OC(C)(C)C(C)(C)O4)c3[s]2)c1Cl)=O PXPZXNKBZYLYIW-UHFFFAOYSA-N 0.000 description 1
- JZFJREFQXPSLGX-UHFFFAOYSA-N CC(C)(C)OC(Nc(ccc(Cl)c1CO)c1F)=O Chemical compound CC(C)(C)OC(Nc(ccc(Cl)c1CO)c1F)=O JZFJREFQXPSLGX-UHFFFAOYSA-N 0.000 description 1
- ZZAVVAQGEWNMET-GHRIWEEISA-N CC(C)(C)S(/N=C/c(cccc1)c1C#C[Si](C)(C)C)=O Chemical compound CC(C)(C)S(/N=C/c(cccc1)c1C#C[Si](C)(C)C)=O ZZAVVAQGEWNMET-GHRIWEEISA-N 0.000 description 1
- ZRAYQPCAUZXCPZ-MDWZMJQESA-N CC(C)(C)S(/N=C/c1ccccc1Cl)=O Chemical compound CC(C)(C)S(/N=C/c1ccccc1Cl)=O ZRAYQPCAUZXCPZ-MDWZMJQESA-N 0.000 description 1
- LBTNVFVSDBAKCW-FZPQMKKPSA-N CC(C)(C)[S@](NC(c1cc2ccnc(-c3cc(C(C)(C(F)(F)F)O)ccn3)c2[s]1)c(cccc1)c1Cl)=O Chemical compound CC(C)(C)[S@](NC(c1cc2ccnc(-c3cc(C(C)(C(F)(F)F)O)ccn3)c2[s]1)c(cccc1)c1Cl)=O LBTNVFVSDBAKCW-FZPQMKKPSA-N 0.000 description 1
- NBBPTIZRGFHLNS-SSEXGKCCSA-N CC(C)(c1ccnc(-c(cccc23)c2nc([C@@H](c2ccccc2Cl)NS(C2CC2)(=O)=O)[n]3S(c2ccc(C)cc2)(=O)=O)c1)O Chemical compound CC(C)(c1ccnc(-c(cccc23)c2nc([C@@H](c2ccccc2Cl)NS(C2CC2)(=O)=O)[n]3S(c2ccc(C)cc2)(=O)=O)c1)O NBBPTIZRGFHLNS-SSEXGKCCSA-N 0.000 description 1
- NBBPTIZRGFHLNS-PMERELPUSA-N CC(C)(c1ccnc(-c(cccc23)c2nc([C@H](c2ccccc2Cl)NS(C2CC2)(=O)=O)[n]3S(c2ccc(C)cc2)(=O)=O)c1)O Chemical compound CC(C)(c1ccnc(-c(cccc23)c2nc([C@H](c2ccccc2Cl)NS(C2CC2)(=O)=O)[n]3S(c2ccc(C)cc2)(=O)=O)c1)O NBBPTIZRGFHLNS-PMERELPUSA-N 0.000 description 1
- JCLIXWNZEUICKG-UHFFFAOYSA-N CC(C)(c1ccnc(-c2c3[s]c(C(c(cccc4)c4Cl)NS(C(CC4)CS4(=O)=O)(=O)=O)cc3ccc2)c1)O Chemical compound CC(C)(c1ccnc(-c2c3[s]c(C(c(cccc4)c4Cl)NS(C(CC4)CS4(=O)=O)(=O)=O)cc3ccc2)c1)O JCLIXWNZEUICKG-UHFFFAOYSA-N 0.000 description 1
- LRGFIHGKRSYRTO-UHFFFAOYSA-N CC(C)(c1ccnc(-c2c3[s]c(C(c4ccccc4Cl)NS(c4c[nH]nc4)(=O)=O)cc3ccc2)c1)O Chemical compound CC(C)(c1ccnc(-c2c3[s]c(C(c4ccccc4Cl)NS(c4c[nH]nc4)(=O)=O)cc3ccc2)c1)O LRGFIHGKRSYRTO-UHFFFAOYSA-N 0.000 description 1
- KOENNEPWYDOHIV-UHFFFAOYSA-N CC(C)(c1ccnc(-c2c3[s]ccc3ccc2)c1)O Chemical compound CC(C)(c1ccnc(-c2c3[s]ccc3ccc2)c1)O KOENNEPWYDOHIV-UHFFFAOYSA-N 0.000 description 1
- RLXGFUYXIWWMGB-HSZRJFAPSA-N CC(C)(c1ccnc(-c2cccc3c2[nH]c([C@@H](c2ccccc2Cl)NS(C2CC2)(=O)=O)n3)c1)O Chemical compound CC(C)(c1ccnc(-c2cccc3c2[nH]c([C@@H](c2ccccc2Cl)NS(C2CC2)(=O)=O)n3)c1)O RLXGFUYXIWWMGB-HSZRJFAPSA-N 0.000 description 1
- RLXGFUYXIWWMGB-QHCPKHFHSA-N CC(C)(c1ccnc(-c2cccc3c2[nH]c([C@H](c2ccccc2Cl)NS(C2CC2)(=O)=O)n3)c1)O Chemical compound CC(C)(c1ccnc(-c2cccc3c2[nH]c([C@H](c2ccccc2Cl)NS(C2CC2)(=O)=O)n3)c1)O RLXGFUYXIWWMGB-QHCPKHFHSA-N 0.000 description 1
- UJYCQEQOWVMVCW-UHFFFAOYSA-N CC(C)(c1ccnc(Cl)c1)O Chemical compound CC(C)(c1ccnc(Cl)c1)O UJYCQEQOWVMVCW-UHFFFAOYSA-N 0.000 description 1
- XBGXJONBPQZJCI-UHFFFAOYSA-N CC(C1)(C1(C)O[B+]c1c2[s]ccc2cc(F)c1)O Chemical compound CC(C1)(C1(C)O[B+]c1c2[s]ccc2cc(F)c1)O XBGXJONBPQZJCI-UHFFFAOYSA-N 0.000 description 1
- IHIBDHNUWZWCQU-UHFFFAOYSA-N CC(CO)(c1ccnc(-c2c3[s]c(C(c(nc(cc4)N)c4Cl)NI)cc3cc(F)c2)c1)O Chemical compound CC(CO)(c1ccnc(-c2c3[s]c(C(c(nc(cc4)N)c4Cl)NI)cc3cc(F)c2)c1)O IHIBDHNUWZWCQU-UHFFFAOYSA-N 0.000 description 1
- AOKQNWTVCOBPQO-UHFFFAOYSA-N CC(CO)(c1ccnc(-c2c3[s]c(Cc(c(F)c(cc4)N)c4Cl)cc3ccc2)c1)O Chemical compound CC(CO)(c1ccnc(-c2c3[s]c(Cc(c(F)c(cc4)N)c4Cl)cc3ccc2)c1)O AOKQNWTVCOBPQO-UHFFFAOYSA-N 0.000 description 1
- VXGAVBVCVRPSKV-UHFFFAOYSA-N CC(COC(C)(C)O)C(C=CN1)=CC1Cl Chemical compound CC(COC(C)(C)O)C(C=CN1)=CC1Cl VXGAVBVCVRPSKV-UHFFFAOYSA-N 0.000 description 1
- KNYBXXWDMUGHEZ-UHFFFAOYSA-N CC(COC(C)(C)O)c1ccnc(-c2c3[s]ccc3cc(F)c2)c1 Chemical compound CC(COC(C)(C)O)c1ccnc(-c2c3[s]ccc3cc(F)c2)c1 KNYBXXWDMUGHEZ-UHFFFAOYSA-N 0.000 description 1
- QZUULRNNSNGSNB-UHFFFAOYSA-N CC(c1cc2cc(F)cc(-c3cc(C4(C)OC(C)(C)OC4)ccn3)c2[s]1)c(nc(cc1)NC(OC(C)(C)C)=O)c1Cl Chemical compound CC(c1cc2cc(F)cc(-c3cc(C4(C)OC(C)(C)OC4)ccn3)c2[s]1)c(nc(cc1)NC(OC(C)(C)C)=O)c1Cl QZUULRNNSNGSNB-UHFFFAOYSA-N 0.000 description 1
- KBPHUTKKSLBETB-UHFFFAOYSA-N CC(c1cc2cc(F)cc(-c3cc(C4(C)[O](C)C(C)(C)OC4)ccn3)c2[s]1)c(nc(cc1)NC(OC(C)(C)C)=O)c1Cl Chemical compound CC(c1cc2cc(F)cc(-c3cc(C4(C)[O](C)C(C)(C)OC4)ccn3)c2[s]1)c(nc(cc1)NC(OC(C)(C)C)=O)c1Cl KBPHUTKKSLBETB-UHFFFAOYSA-N 0.000 description 1
- XZUGZNGUTJJMDV-QQFDVULRSA-N CC(c1ccnc(-c2c3[s]c(C(c(cccc4)c4Cl)N[S@@](C(C)(C)C)=O)cc3ccc2)c1)O Chemical compound CC(c1ccnc(-c2c3[s]c(C(c(cccc4)c4Cl)N[S@@](C(C)(C)C)=O)cc3ccc2)c1)O XZUGZNGUTJJMDV-QQFDVULRSA-N 0.000 description 1
- DYGIVULIWAJGHE-UHFFFAOYSA-N CC(c1ccnc(-c2c3[s]c(Cc(c([O]=S(C4CC4)(N)=O)ccc4)c4Cl)cc3ccc2)c1)O Chemical compound CC(c1ccnc(-c2c3[s]c(Cc(c([O]=S(C4CC4)(N)=O)ccc4)c4Cl)cc3ccc2)c1)O DYGIVULIWAJGHE-UHFFFAOYSA-N 0.000 description 1
- TXDLXCNMGUZXDA-UHFFFAOYSA-N CC(c1ccnc(-c2c3[s]c(Cc(cccc4)c4Cl)cc3ccc2)c1)O Chemical compound CC(c1ccnc(-c2c3[s]c(Cc(cccc4)c4Cl)cc3ccc2)c1)O TXDLXCNMGUZXDA-UHFFFAOYSA-N 0.000 description 1
- PNPHOIZEFCAWEB-UHFFFAOYSA-N CC1(C)OB(c2c3[s]ccc3ccc2)OC1(C)C Chemical compound CC1(C)OB(c2c3[s]ccc3ccc2)OC1(C)C PNPHOIZEFCAWEB-UHFFFAOYSA-N 0.000 description 1
- BODHLVDFIURQNX-UHFFFAOYSA-N CCN(C=C(C=C1)S(C)(=O)=O)C1=O Chemical compound CCN(C=C(C=C1)S(C)(=O)=O)C1=O BODHLVDFIURQNX-UHFFFAOYSA-N 0.000 description 1
- XGNXNRSPNLBGJP-UHFFFAOYSA-N CCOC(c(c(Cl)ccc1N)c1F)=O Chemical compound CCOC(c(c(Cl)ccc1N)c1F)=O XGNXNRSPNLBGJP-UHFFFAOYSA-N 0.000 description 1
- XYVZEMDKQBWARC-UHFFFAOYSA-N CNS(C1CC1)(=O)=O Chemical compound CNS(C1CC1)(=O)=O XYVZEMDKQBWARC-UHFFFAOYSA-N 0.000 description 1
- UWKUPRIZGJVGHG-YCXDTOJBSA-N C[C@@](C(F)(F)F)(c1ccnc(-c2c3[s]c(Cc(c([O]=S(c(cc4)cc5c4[o]cc5)(N)=O)ccc4)c4Cl)cc3ccc2)c1)O Chemical compound C[C@@](C(F)(F)F)(c1ccnc(-c2c3[s]c(Cc(c([O]=S(c(cc4)cc5c4[o]cc5)(N)=O)ccc4)c4Cl)cc3ccc2)c1)O UWKUPRIZGJVGHG-YCXDTOJBSA-N 0.000 description 1
- CFBNBLCJHJGTAK-IERCKHOLSA-N C[C@@](CO)(c1ccnc(-c2c3[s]c(Cc(c([O]=S(C4CC4)(N)=O)c(cc4)N)c4Cl)cc3ccc2)c1)O Chemical compound C[C@@](CO)(c1ccnc(-c2c3[s]c(Cc(c([O]=S(C4CC4)(N)=O)c(cc4)N)c4Cl)cc3ccc2)c1)O CFBNBLCJHJGTAK-IERCKHOLSA-N 0.000 description 1
- DWSIXGXPMDAJFT-JOCHJYFZSA-N C[C@@](CO)(c1ccnc(-c2c3[s]c(Cc(nc(cc4)N)c4Cl)cc3ccc2)c1)O Chemical compound C[C@@](CO)(c1ccnc(-c2c3[s]c(Cc(nc(cc4)N)c4Cl)cc3ccc2)c1)O DWSIXGXPMDAJFT-JOCHJYFZSA-N 0.000 description 1
- ALYDOVLNTCMHJR-XSWBTSGESA-N C[C@@](CO)(c1ccnc(-c2cccc3c2[s]c(C(c(nc(cc2)N)c2Cl)NS(C2CC2)(=O)=O)c3)c1)O Chemical compound C[C@@](CO)(c1ccnc(-c2cccc3c2[s]c(C(c(nc(cc2)N)c2Cl)NS(C2CC2)(=O)=O)c3)c1)O ALYDOVLNTCMHJR-XSWBTSGESA-N 0.000 description 1
- IBFGLNWXUJEPFN-SVNIRARHSA-N C[C@](C(F)(F)F)(c1ccnc(-c2c3[s]c(C(c4ccccc4)NS(c4ccccc4)(=O)=O)cc3ccc2)c1)O Chemical compound C[C@](C(F)(F)F)(c1ccnc(-c2c3[s]c(C(c4ccccc4)NS(c4ccccc4)(=O)=O)cc3ccc2)c1)O IBFGLNWXUJEPFN-SVNIRARHSA-N 0.000 description 1
- PGJIGRHCTAFKNO-RRZXGEHQSA-N C[C@](C(F)(F)F)(c1ccnc(-c2c3[s]c(Cc4ccccc4[O]=S(c4ccccc4)(N)=O)cc3ccc2)c1)O Chemical compound C[C@](C(F)(F)F)(c1ccnc(-c2c3[s]c(Cc4ccccc4[O]=S(c4ccccc4)(N)=O)cc3ccc2)c1)O PGJIGRHCTAFKNO-RRZXGEHQSA-N 0.000 description 1
- OTNFQASUEIKHDF-OAHLLOKOSA-N C[C@](C(F)(F)F)(c1ccnc(-c2c3[s]ccc3ccc2)c1)O Chemical compound C[C@](C(F)(F)F)(c1ccnc(-c2c3[s]ccc3ccc2)c1)O OTNFQASUEIKHDF-OAHLLOKOSA-N 0.000 description 1
- ISQZFFYROHMOIV-ZCJYOONXSA-N C[C@](C(F)(F)F)(c1ccnc(-c2cccc3c2[s]c(C(c(cccc2)c2C#C)NS(C2CC2)(=O)=O)c3)c1)O Chemical compound C[C@](C(F)(F)F)(c1ccnc(-c2cccc3c2[s]c(C(c(cccc2)c2C#C)NS(C2CC2)(=O)=O)c3)c1)O ISQZFFYROHMOIV-ZCJYOONXSA-N 0.000 description 1
- OEVAOWRTTMWLAU-SSDOTTSWSA-N C[C@](C(F)(F)F)(c1ccnc(Cl)c1)O Chemical compound C[C@](C(F)(F)F)(c1ccnc(Cl)c1)O OEVAOWRTTMWLAU-SSDOTTSWSA-N 0.000 description 1
- IROGCMISRBINQX-UHFFFAOYSA-N C[Si](C)(C)C#Cc1c(C=O)cccc1 Chemical compound C[Si](C)(C)C#Cc1c(C=O)cccc1 IROGCMISRBINQX-UHFFFAOYSA-N 0.000 description 1
- RKJHSFXXXAWOLC-UHFFFAOYSA-N Cc(cc1)ccc1S([n](c(C(c(cccc1)c1Cl)NS(C1CC1)(=O)=O)cc1ccc2)c1c2Br)(=O)=O Chemical compound Cc(cc1)ccc1S([n](c(C(c(cccc1)c1Cl)NS(C1CC1)(=O)=O)cc1ccc2)c1c2Br)(=O)=O RKJHSFXXXAWOLC-UHFFFAOYSA-N 0.000 description 1
- ONHMWUXYIFULDO-UHFFFAOYSA-N Clc1cc(Br)ccn1 Chemical compound Clc1cc(Br)ccn1 ONHMWUXYIFULDO-UHFFFAOYSA-N 0.000 description 1
- KJKIPRQNFDUULB-UHFFFAOYSA-N Clc1nccc(I)c1 Chemical compound Clc1nccc(I)c1 KJKIPRQNFDUULB-UHFFFAOYSA-N 0.000 description 1
- RCOBEYPKYCHEAJ-UHFFFAOYSA-N FC(Cc1ccnc(-c2c3[s]ccc3ccc2)c1)(F)F Chemical compound FC(Cc1ccnc(-c2c3[s]ccc3ccc2)c1)(F)F RCOBEYPKYCHEAJ-UHFFFAOYSA-N 0.000 description 1
- FXICAYWEQJUXJH-UHFFFAOYSA-N Fc1cc(Cl)c2[s]ccc2c1 Chemical compound Fc1cc(Cl)c2[s]ccc2c1 FXICAYWEQJUXJH-UHFFFAOYSA-N 0.000 description 1
- OKFCHDGDLOXVKY-PWZPOPPBSA-N N=C/C=C/C=C(\S)/I Chemical compound N=C/C=C/C=C(\S)/I OKFCHDGDLOXVKY-PWZPOPPBSA-N 0.000 description 1
- CSFDTBRRIBJILD-UHFFFAOYSA-N Nc(c(F)c1)ccc1Cl Chemical compound Nc(c(F)c1)ccc1Cl CSFDTBRRIBJILD-UHFFFAOYSA-N 0.000 description 1
- GVCFFVPEOLCYNN-UHFFFAOYSA-N Nc(cc1)cc(C(O)=O)c1Cl Chemical compound Nc(cc1)cc(C(O)=O)c1Cl GVCFFVPEOLCYNN-UHFFFAOYSA-N 0.000 description 1
- QDLBAKWXWLAADL-UHFFFAOYSA-N Nc(cc1)nc(C(c2cc3cccc(-c4cc(C5(COC5)O)ccn4)c3[s]2)NS(C2CC2)(=O)=O)c1Cl Chemical compound Nc(cc1)nc(C(c2cc3cccc(-c4cc(C5(COC5)O)ccn4)c3[s]2)NS(C2CC2)(=O)=O)c1Cl QDLBAKWXWLAADL-UHFFFAOYSA-N 0.000 description 1
- FPYUJUBAXZAQNL-UHFFFAOYSA-N O=Cc1ccccc1Cl Chemical compound O=Cc1ccccc1Cl FPYUJUBAXZAQNL-UHFFFAOYSA-N 0.000 description 1
- KKYCMBXAWUPEBS-UHFFFAOYSA-N O=S(C1CC1)(NC(c1cc2cccc(Br)c2[nH]1)c1ccccc1Cl)=O Chemical compound O=S(C1CC1)(NC(c1cc2cccc(Br)c2[nH]1)c1ccccc1Cl)=O KKYCMBXAWUPEBS-UHFFFAOYSA-N 0.000 description 1
- JRZMYXYOFIUZKA-ONNFQVAWSA-N O=S/N=C/c(c(F)ccc1)c1Cl Chemical compound O=S/N=C/c(c(F)ccc1)c1Cl JRZMYXYOFIUZKA-ONNFQVAWSA-N 0.000 description 1
- UBYBTESBTLUPDX-YCRREMRBSA-N O=S/N=C/c(nc(cc1)Cl)c1Cl Chemical compound O=S/N=C/c(nc(cc1)Cl)c1Cl UBYBTESBTLUPDX-YCRREMRBSA-N 0.000 description 1
- KBUAFNFDYUSKAM-UITAMQMPSA-N O=[S-]/N=C\c(cccc1)c1Cl Chemical compound O=[S-]/N=C\c(cccc1)c1Cl KBUAFNFDYUSKAM-UITAMQMPSA-N 0.000 description 1
- VEFRDVVLKQVKJS-UHFFFAOYSA-N OC1(COC1)c1ccnc(Cl)c1 Chemical compound OC1(COC1)c1ccnc(Cl)c1 VEFRDVVLKQVKJS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention relates to benzothiophene sulfonamides and other compounds, or pharmaceutically acceptable salts thereof, as defined herein, that interact with glucokinase regulatory protein.
- the present invention relates to methods of treating type 2 diabetes, and other diseases and/or conditions where glucokinase regulatory protein is involved using the
- Glucokinase is a member of a family of four hexokinases that are critical in the cellular metabolism of glucose. Specifically GK, also known as hexokinase IV or hexokinase D, facilitates glucose induced insulin secretion from pancreatic ⁇ -cells as well as glucose conversion into glycogen in the liver. GK has a unique catalytic activity that enables the enzyme to be active within the physiological range of glucose (from 5mM glucose to lOmM glucose).
- mice lacking both copies of the GK gene die soon after birth from severe hyperglycemia, whereas mice lacking only one copy of the GK gene present with only mild diabetes. Mice that are made to overexpress the GK gene in their livers are hypoglycemic.
- GK activity in the liver is transiently regulated by glucokinase regulatory protein (GK P).
- GK catalytic activity is inhibited when GK is bound to GKRP. This interaction is antagonized by increasing concentrations of both glucose and fructose -1 -phosphate (F1P).
- the complex of the two proteins is localized primarily to the nuclear compartment of a cell. Post prandially as both glucose and fructose levels rise, GK released from GKRP translocates to the cytoplasm. Cytoplasmic GK is now free of the inhibitory effects of GKRP and able to kinetically respond to glucose. Evidence from the Zucker diabetic fatty rat (ZDF) indicates that their glucose intolerance may be a result of this mechanism failing to function properly.
- ZDF Zucker diabetic fatty rat
- a compound that acts directly on GKRP to disrupt its interaction with GK and hence elevate levels of cytoplasmic GK is a viable approach to modulate GK activity. Such an approach would avoid the unwanted hypoglycemic effects of over stimulation of GK catalytic activity, which has been seen in the
- GK activators A compound having such an effect would be useful in the treatment of diabetes and other diseases and/or conditions in which GKRP and/or GK plays a role.
- the present invention provides compounds that bind GKRP and disrupts its interaction with GK.
- the present invention provides compounds of Formula I, or pharmaceutically acceptable salts thereof, wherein:
- I * is a chiral center having the R configuration
- X 1 is N or CH
- X 2 is N or CH
- X' is N or CH
- X 4 is N or CH
- X 5 is N or CH
- heteroaryl group has from 1 to 4 heteroatoms independently selected from O, N or S, which cycloalkyl, aryl or heteroaryl group may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_ 6 alkyl, -OCi_ 6 haloalkyl,-OH, -NH 2 , Ci_ 6 alkyl or Ci_ 6 haloalkyl;
- R 2 is phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl, where the phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl group can be unsubstituted or substituted with from one to three substituents independently selected from Ci_ 6 alkyl, Ci_ 6 haloalkyl, C 2 _ 6 alkenyl, halo, -SCi_ 6 alkyl, -OCi_ 6 alkyl,
- -OCi_ 6 haloalkyl -OH, -NH 2 , -N0 2 or -N(Ci_ 6 alkyl) 2 ;
- n 0, 1, 2, 3 or 4;
- R 4 is hydrogen or Ci_ 6 alkyl
- R 5 is hydrogen, Ci_ 6 alkyl or Ci_ 6 haloalkyl.
- the invention provides compounds in accordance with embodiment 1 , or pharmaceutically acceptable salts thereof, wherein Y is
- the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X 1 is CH.
- the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X 1 is N.
- the invention provides compounds in accordance with any one of embodiments 1 to 4, or pharmaceutically acceptable salts thereof, wherein X 2 is CH.
- the invention provides compounds in accordance with any one of embodiments 1 to 5, or pharmaceutically acceptable salts thereof, wherein X 3 is CH.
- the invention provides compounds in accordance with any one of embodiments 1 to 6, or pharmaceutically acceptable salts thereof, wherein X 4 is CH.
- the invention provides compounds in accordance with any one of embodiments 1 to 6, or pharmaceutically acceptable salts thereof, wherein X 4 is N.
- the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X 1 , X 2 , X 3 , X 4 and X 5 are CH, and X 6 is S.
- the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X 1 , X 2 , X 3 and X 4 are CH, X 5 is N, and X 6 is S.
- the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X 1 is N, X 2 , X 3 and X 4 are CH, X 5 is CH, and X 6 is S.
- the invention provides compounds in accordance with any one of embodiments 1 to 11 , or pharmaceutically acceptable salts thereof, wherein R 4 is hydrogen.
- the invention provides compounds in accordance with any one of embodiments 1 to 12, or pharmaceutically acceptable salts thereof, wherein R 1 is Ci_ 6 alkyl, three to eight membered cycloalkyl, phenyl or pyridyl, which cycloalkyl, phenyl or pyridyl groups may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_ 6 alkyl, -NH 2 or Ci_ 6 alkyl.
- the invention provides compounds in accordance with any one of embodiments 1 to 12, or pharmaceutically acceptable salts thereof, wherein R 1 is three to eight membered cycloalkyl, substituted phenyl or substituted pyridyl.
- the invention provides compounds in accordance with any one of embodiments 1 to 12, or pharmaceutically acceptable salts thereof, wherein R 1 is cyclopropyl or substituted cyclopropyl.
- the invention provides compounds in accordance with any one of embodiments 1 to 15, or pharmaceutically acceptable salts thereof, wherein R 2 is phenyl or substituted phenyl.
- the invention provides compounds in accordance with any one of embodiments 1 to 15, or pharmaceutically acceptable salts thereof, wherein R 2 is pyridyl or substituted pyridyl.
- the invention provides compounds in accordance with any one of embodiments 1 to 15, or pharmaceutically acceptable salts thereof, wherein R 2 is
- the invention provides compounds in accordance with any one of embodiments 1 to 18, or pharmaceutically acceptable salts thereof, wherein R 3 is
- the invention provides compounds in accordance with any one of embodiments 1 to 18, or pharmaceutically acceptable salts thereof, wherein R 3 is
- the present invention also provides a compound, or a pharmaceutically acceptable salt thereof, selected from:
- the present invention provides methods of treating type 2 diabetes, hyperglycemia, impaired glucose tolerance, insulin resistance, retinopathy, nephropathy, neuropathy, cataracts, glaucoma, Syndrome X, or polycystic ovarian syndrome, the methods comprising administering to a patient in need thereof a therapeutically effective amount of a compound in accordance with any one of embodiments 1 to 20, or a pharmaceutically acceptable salt thereof.
- the present invention provides a method of embodiment 22 wherein the method of treatment is for type 2 diabetes.
- the present invention provides pharmaceutical compositions comprising a compound in accordance with any one of
- the present invention provides compounds of Formula
- I * is a chiral center having the R configuration, except when X 6 is N, then * is a chiral center having the S configuration;
- X 1 is N, CH or C-halo
- X 2 is N, CH or C-halo
- X 3 is N, CH or C-halo
- X 4 is N or CH
- X 5 is N or CH
- X b is S or NH
- R 1 is Ci_ 6 alkyl, -N(C 1-6 alkyl) 2 , -N(H)three to eight membered cycloalkyl,
- cycloalkyl six to ten membered aryl or five to ten membered heteroaryl, where the heteroaryl group has from 1 to 4 heteroatoms independently selected from O, N or S, which cycloalkyl, aryl or heteroaryl group may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_ 6 alkyl, -OCi_ 6 haloalkyl,-OH, -NH 2 , Ci_ 6 alkyl or Ci_ 6 haloalkyl;
- R is phenyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl or thiazolyl where the phenyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl or thiazolyl group can be unsubstituted or substituted with from one to three substituents independently selected
- n 0, 1, 2, 3 or 4
- m 0, 1 or 2;
- R 4 is hydrogen or Ci_6alkyl; hydrogen, Ci- 6 alkyl or Ci_ 6 haloalkyl;
- R 6 is hydrogen or halo, provided that R 1 is not dimethoxyphenyl.
- the present invention provides compounds in accordance with embodiment 1 A, or pharmaceutically acceptable salts thereof, wherein:
- X 1 is N or CH
- X 2 is N or CH
- X 3 is N or CH;
- R 1 is Ci_ 6 alkyl, -N(Ci_ 6 alkyl) 2 , three to eight membered cycloalkyl, six to ten membered aryl or five to ten membered heteroaryl, where the heteroaryl group has from 1 to 4 heteroatoms independently selected from O, N or S, which cycloalkyl, aryl or heteroaryl group may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_ 6 alkyl, -OCi_ 6 haloalkyl,-OH, -NH 2 , Ci_ 6 alkyl or Ci_ 6 haloalkyl;
- R 2 is phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl, where the phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl group can be unsubstituted or substituted with from one to three substituents independently selected from Ci_ 6 alkyl, Ci_ 6 haloalkyl, C 2 _ 6 alkenyl, halo, -SCi_ 6 alkyl, -OCi_ 6 alkyl,
- the present invention provides compounds in accordance with any one of embodiments 1 A or 2A, or pharmaceutically acceptable salts thereof, wherein Y is
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A, or pharmaceutically acceptable salts thereof, wherein X 1 is CH.
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A, or pharmaceutically acceptable salts thereof, wherein X 1 is N.
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 5 A, or pharmaceutically acceptable salts thereof, wherein X 2 is CH.
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 5 A, or pharmaceutically acceptable salts thereof, wherein X 2 is C-halo.
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 7 A, or pharmaceutically acceptable salts thereof, wherein X 3 is CH.
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 7 A, or pharmaceutically acceptable salts thereof, wherein X 3 is N.
- the present invention provides compounds in accordance with any one of embodiments 1 A to 9 A, or pharmaceutically acceptable salts thereof, wherein X 4 is CH.
- the present invention provides compounds in accordance with any one of embodiments 1 A to 9 A, or pharmaceutically acceptable salts thereof, wherein X 4 is N.
- the present invention provides compounds in accordance with embodiment 1 A, or pharmaceutically acceptable salts thereof, wherein X 1 , X 2 , X 3 , X 4 and X 5 are CH, and X 6 is S.
- the present invention provides compounds in accordance with embodiment 1 A, or pharmaceutically acceptable salts thereof, wherein X 1 , X 2 , X 3 and X 4 are CH, X 5 is N, and X 6 is S.
- the present invention provides compounds in accordance with embodiment 1 A, or pharmaceutically acceptable salts thereof, wherein X 1 is N, X 2 , X 3 and X 4 are CH, X 5 is CH, and X 6 is S.
- the present invention provides compounds in accordance with any one of embodiments 1 A to 14A, or pharmaceutically acceptable salts thereof, wherein R 4 is hydrogen.
- the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 15 A, or pharmaceutically acceptable salts thereof, wherein R 1 is Ci_ 6 alkyl, three to eight membered cycloalkyl, phenyl or pyridyl, which cycloalkyl, phenyl or pyridyl groups may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_ 6 alkyl, -NH 2 or Ci_ 6 alkyl.
- the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 15 A, or pharmaceutically acceptable salts thereof, wherein R 1 is three to eight membered cycloalkyl, substituted phenyl or substituted pyridyl.
- the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 15 A, or pharmaceutically acceptable salts thereof, wherein R 1 is cyclopropyl or substituted cyclopropyl.
- the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 18 A, or pharmaceutically acceptable salts thereof, wherein R 2 is phenyl or substituted phenyl.
- the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 18 A, or pharmaceutically acceptable salts thereof, wherein R 2 is pyridyl or substituted pyridyl.
- the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 18 A, or pharmaceutically acceptable salts thereof, wherein R 2 is
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 21 A, or pharmaceutically acceptable salts thereof, wherein R 3 is
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 21 A, or pharmaceutically acceptable salts thereof, wherein R 3 is
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 21 A, or pharmaceutically acceptable salts thereof, wherein R 3 is
- the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 21 A, or pharmaceutically acceptable salts thereof, wherein R 3 is
- the present invention provides compounds in accordance with any one of embodiments 1 A to 25 A, or pharmaceutically acceptable salts thereof, wherein m is 0.
- the present invention provides compounds in accordance with any one of embodiments 1 A to 25 A, or pharmaceutically acceptable salts thereof, wherein m is 1.
- the present invention provides compounds, or pharmaceutically acceptable salts thereof, selected from:
- the present invention provides compounds, or pharmaceutically acceptable salts thereof, selected from:
- the present invention provides methods of treating type 2 diabetes, hyperglycemia, impaired glucose tolerance, insulin resistance, retinopathy, nephropathy, neuropathy, cataracts, glaucoma, Syndrome X, or polycystic ovarian syndrome, the methods comprising administering to a patient in need thereof a therapeutically effective amount of a compound in accordance with any one of embodiments 1 A to 29 A, or a pharmaceutically acceptable salt thereof.
- the present invention provides methods of embodiment 30A wherein the treatment is for type 2 diabetes.
- the present invention provides pharmaceutical compositions comprising a compound in accordance with any one of
- the present invention provides benzothiophene sulfonamides and other compounds, as defined above, or pharmaceutically acceptable salts thereof.
- the present invention also provides pharmaceutical compositions comprising a compound of the present invention, or pharmaceutically acceptable salts thereof, and methods of treating diseases and/or conditions, such as diabetes, using compounds of the present invention, or pharmaceutically acceptable salts thereof.
- alkyl means a straight or branched chain hydrocarbon.
- alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, sec-butyl, pentyl and hexyl.
- Typical alkyl groups are alkyl groups having from 1 to 8 carbon atoms, which groups are commonly represented as Ci-galkyl.
- haloalkyl mean a straight or branched chain hydrocarbon wherein one or more hydrogen atom is replaced with a halogen.
- Typical haloalkyl groups are haloalkyl groups having from 1 to 8 carbon atoms, which groups are commonly represented as Ci-shaloalkyl.
- Common haloalkyl groups include -CF 3 , -CHF 2 or -CH 2 F.
- alkoxy means an alkyl group bonded to an oxygen atom.
- alkoxy groups include methoxy, ethoxy, tert-butoxy, propoxy and isobutoxy.
- Common alkoxy groups are Ci-galkoxy.
- halogen or halo means chlorine, fluorine, bromine or iodine.
- alkenyl means a branched or straight chain hydrocarbon having one or more carbon-carbon double bonds.
- Representative examples alkenyl groups include ethenyl, propenyl, allyl, butenyl and 4-methylbutenyl.
- Common alkenyl groups are C 2 - 8 alkenyl.
- alkynyl means a branched or straight chain hydrocarbon having one or more carbon-carbon triple bonds. Representative examples of alkynyl groups include ethynyl, propynyl (propargyl) and butynyl. Common alkynyl groups are C 2 -g alkynyl.
- cycloalkyl means a cyclic, nonaromatic hydrocarbon.
- cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- a cycloalkyl group can contain one or more double bond.
- Examples of cycloalkyl groups that contain double bonds include cyclopentenyl, cyclohexenyl, cyclohexadienyl and cyclobutadienyl.
- Common cycloalkyl groups are C3-8 cycloalkyl groups.
- perfluoroalkyl means an alkyl group in which all of the hydrogen atoms have been replaced with fluorine atoms.
- perfluoroalkyl groups are Ci-sperfluoroalkyl.
- An example of a common perfluoroalkyl group is CF 3 .
- acyl means a group derived from an organic acid by removal of the hydroxy group (-OH).
- aryl means a cyclic, aromatic hydrocarbon. Examples of aryl groups include phenyl and naphthyl. Common aryl groups are six to thirteen membered rings.
- heteroatom as used herein means an oxygen, nitrogen or sulfur atom.
- heteroaryl means a cyclic, aromatic hydrocarbon in which one or more carbon atoms of an aryl group have been replaced with a heteroatom. If the heteroaryl group contains more than one heteroatom, the heteroatoms may be the same or different.
- heteroaryl groups include pyridyl, pyrimidinyl, imidazolyl, thienyl, furyl, pyrazinyl, pyrrolyl, indolyl, triazolyl, pyridazinyl, indazolyl, purinyl, isoquinolyl, quinolyl, naphthyridinyl, quinoxalinyl, isothiazolyl and benzo[b]thienyl.
- Common heteroaryl groups are five to thirteen membered rings that contain from 1 to 4 heteroatoms. Heteroaryl groups that are five and six membered rings that contain 1 to 3 heteroatoms are particularly common.
- heterocycloalkyl means a cycloalkyl group in which one or more of the carbon atoms has been replaced with a heteroatom.
- heterocycloalkyl group contains more than one heteroatom, the heteroatoms may be the same or different.
- heterocycloalkyl groups include tetrahydrofuryl, morpholinyl, piperazinyl, piperidinyl and pyrrolidinyl. It is also possible for the heterocycloalkyl group to have one or more double bonds, but is not aromatic. Examples of heterocycloalkyl groups containing double bonds include dihydrofuran.
- Common heterocycloalkyl groups are three to ten membered rings containing from 1 to 4 heteroatoms. Heterocycloalkyl groups that are five and six membered rings that contain 1 to 2 heteroatoms are particularly common.
- cyclic ring groups i.e., aryl, heteroaryl, cycloalkyl, and heterocycloalkyl, can comprise more than one ring.
- the naphthyl group is a fused bicyclic ring system.
- the present invention include ring groups that have bridging atoms, or ring groups that have a spiro orientation.
- five to six membered aromatic rings are phenyl, furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, pyridinyl, pyridiazinyl, pyrimidinyl, and pyrazinyl.
- Representative examples of partially saturated, fully saturated or fully unsaturated five to eight membered rings, optionally having one to three heteroatoms, are cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and phenyl.
- Further exemplary five membered rings are furyl, thienyl, pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, pyrrolidinyl, 1,3-dioxolanyl, oxazolyl, thiazolyl, imidazolyl, 2H- imidazolyl, 2-imidazolinyl, imidazolidinyl, pyrazolyl, 2-pyrazolinyl,
- FIG. 1 For exemplary six membered rings are 2H-pyranyl, 4H-pyranyl, pyridinyl, piperidinyl, 1 ,2-dioxinyl, 1,3-dioxinyl, 1 ,4-dioxanyl, morpholinyl, 1,4- dithianyl, thiomorpholinyl, pyridazinyl, pyrimidinyl, pyrazinyl, piperazinyl, 1,3,5-triazinyl, 1,2,4-triazinyl, 1,2,3-triazinyl, 1,3,5-trithianyl, 4H-l,2-oxazinyl, 2H-l,3-oxazinyl, 6H-l,3-oxazinyl, 6H-l,2-oxazinyl, 1 ,4-oxazinyl, 2H-1,2- oxazinyl, 4H-l,4-oxazinyl
- exemplary seven membered rings are azepinyl, oxepinyl, thiepinyl and 1,2,4-triazepinyl.
- FIG. 1 Further exemplary eight membered rings are cyclooctyl, cyclooctenyl and cyclooctadienyl.
- Exemplary bicyclic rings consisting of two fused partially saturated, fully saturated or fully unsaturated five and/or six membered rings, optionally having one to four heteroatoms, are indolizinyl, indolyl, isoindolyl, indolinyl, cyclopenta(b)pyridinyl, pyrano(3,4-b)pyrrolyl, benzofuryl, isobenzofuryl, benzo(b)thienyl, benzo(c)thienyl, lH-indazolyl, indoxazinyl, benzoxazolyl, anthranilyl, benzimidazolyl, benzthiazolyl, purinyl, quinolinyl, isoquinolinyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 1,8-naphthyridinyl, pteridinyl, indenyl, is
- a cyclic ring group may be bonded to another group in more than one way. If no particular bonding arrangement is specified, then all possible arrangements are intended.
- pyridyl includes 2-, 3-, or 4- pyridyl
- thienyl includes 2-, or 3-thienyl.
- substituted means that a hydrogen atom on a molecule or group is replaced with a group or atom.
- R x groups may be joined together with the nitrogen atom to form a ring.
- a group or atom that replaces a hydrogen atom is also called a
- Any particular molecule or group can have one or more substituent depending on the number of hydrogen atoms that can be replaced.
- the symbol "-" represents a covalent bond and can also be used in a radical group to indicate the point of attachment to another group. In chemical structures, the symbol is commonly used to represent a methyl group in a molecule.
- terapéuticaally effective amount means an amount of a compound that ameliorates, attenuates or eliminates one or more symptom of a particular disease or condition, or prevents or delays the onset of one of more symptom of a particular disease or condition.
- patient means animals, such as dogs, cats, cows, horses, sheep and humans. Particular patients are mammals.
- patient includes males and females.
- pharmaceutically acceptable means that the referenced substance, such as a compound of the present invention or a formulation containing a compound of the present invention, or a particular excipient, are suitable for administration to a patient.
- treating include preventative (e.g., prophylactic) and palliative treatment.
- patient in need thereof means a patient who has or is at risk of having a GKRP/GK mediated disease or condition, such as type 2 diabetes.
- excipient means any pharmaceutically acceptable additive, carrier, diluent, adjuvant, or other ingredient, other than the active pharmaceutical ingredient (API), which is typically included for formulation and/or
- the compounds of the present invention are administered to a patient in a therapeutically effective amount.
- the compounds can be administered alone or as part of a pharmaceutically acceptable composition or formulation.
- the compounds or compositions can be administered all at once, as for example, by a bolus injection, multiple times, such as by a series of tablets, or delivered substantially uniformly over a period of time, as for example, using transdermal delivery. It is also noted that the dose of the compound can be varied over time.
- the compounds of the present invention can be administered alone, in combination with other compounds of the present invention, or with other pharmaceutically active compounds.
- the other pharmaceutically active compounds can be intended to treat the same disease or condition as the compounds of the present invention or a different disease or condition. If the patient is to receive or is receiving multiple pharmaceutically active compounds, the compounds can be administered simultaneously, or sequentially.
- the active compounds may be found in one tablet or in separate tablets, which can be administered at once or sequentially in any order.
- the compositions may be different forms. For example, one or more compound may be delivered via a tablet, while another is administered via injection or orally as a syrup. All combinations, delivery methods and administration sequences are contemplated.
- the compounds of the present invention may be used in the manufacture of a medicament for the treatment of a disease and/or condition mediated by GKRP/GK, such as type 2 diabetes.
- the compounds of the present invention may be used in combination with other pharmaceutically active compounds. It is noted that the term
- pharmaceutically active compounds can include biologies, such as proteins, antibodies and peptibodies.
- examples of other pharmaceutically active compounds include, but are not limited to: (a) dipeptidyl peptidase IV (DPP-IV) inhibitors such as Vildagliptin (Novartis), Sitagliptin (Merck&Co.), Saxagliptin (BMS) Alogliptin (Takeda); (b) insulin sensitizers including (i) PPARy agonists such as the glitazones (e.g., troglitazone, pioglitazone, edaglitazone,
- DPP-IV dipeptidyl peptidase IV
- glitazones e.g., troglitazone, pioglitazone, edaglitazone
- PPARa/ ⁇ dual agonists such as muraglitazar (BMS) and tesaglitazar (AstraZeneca), and PPARa agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (II) biguanides such as metformin and phenformin, and (iii) protein tyrosine phosphatase-lB (PTP-1B) inhibitors; (c) insulin or insulin mimetics; (d) incretin and incretin mimetics such as (i) Exenatide available from Amylin Pharmaceuticals, (i) amylin and amylin mimetics such as pramlintide acetate, available as Symlin ® , (iii) GLP-1, GLP-1 mimetics, and GLP-1
- dialkylaminoalkyl derivatives of a cross-linked dextran (iii) nicotinyl alcohol, nicotinic acid or a salt thereof, (iv) PPARa agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (v) PPARa/ ⁇ dual agonists such as muraglitazar (BMS) and tesaglitazar (AstraZeneca), (vi) inhibitors of cholesterol absorption, such as beta-sitosterol and ezetimibe, (vii) acyl CoAxholesterol acyltransferase inhibitors such as avasimibe, and (viii) antioxidants such as probucol; (j) PPAR5 agonists such as GW-501516 from GSK; (k) anti-obesity compounds such as fenfluramine, dexfenflur
- the compounds of the present invention may also be used in combination with GPR40 agonists.
- glucokinase activators that can be used in combination with the compounds of the present invention include those set forth in published PCT patent application no. WO 2009/042435, published April 2, 2009.
- ILl-Rl compounds set forth in U.S. patent no. 7,438,910.
- a particular disease that can be treated with the combination is type 2 diabetes.
- the compounds of the present invention can also be used in combination with FGF-21 compounds, and particularly for the treatment of type 2 diabetes.
- FGF-21 compounds are disclosed in U.S. patent no. 7,671,180; U.S. patent no. 7,667,008; U.S. patent no. 7,459,540; U.S. patent no. 7,696,172; PCT application publication no. WO 2010/042747; and PCT application publication no. WO 2009/149171.
- the compounds of the present invention can be also be used in combination with anakinra, particularly for the treatment of type 2 diabetes.
- the compounds of the present invention may be used in combination with metformin.
- the compounds of the present invention are used in the treatment diseases or symptoms mediated by GKRP and/or GK (GKRP/GK).
- diseases or symptoms mediated by GKRP/GK include, but are not limited to, Type II (type 2) diabetes and related disorders, such as hyperglycemia, low or impaired glucose tolerance, insulin resistance, obesity, lipid disorders such as
- dyslipidemia hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels, atherosclerosis, and vascular restenosis, irritable bowel syndrome, inflammatory bowel disease, including Crohn's disease and ulcerative colitis, other inflammatory conditions, pancreatitis, abdominal obesity, neurodegenerative disease, retinopathy, nephropathy, neuropathy, cataracts, glaucoma, glomerulosclerosis, foot ulcerations and unlcerative colitis, altered gastrointestinal motility, Syndrome X, ovarian hyperandrogenism, polycystic ovarian syndrome, premenstrual syndrome, other disorders where insulin resistance is a component.
- Syndrome X also known as Metabolic Syndrome
- obesity is thought to promote insulin resistance, diabetes, dyslipidemia, hypertension, and increased cardiovascular risk, growth hormone deficiency, neutropenia, neuronal disorders, tumor invasion and metastasis, benign prostatic hypertrophy, gingivitis, osteoporosis, frailty of aging, intestinal injury, benign prostatic hypertrophy (BPH), and sperm motility/male contraception.
- BPH benign prostatic hypertrophy
- cardiovascular diseases or damages e.g. cardiac hypertrophy, cardiac remodeling after myocardial infarction, pulmonary congestion and cardiac fibrosis in dilated or in hypertrophic cardiomyopathy, cardiomyopathy such as dilated cardiomyopathy or hypertrophic
- cardiomyopathy mesanglial hypertrophy, or diabetic cardiomyopathy, left or right ventricular hypertrophy, arrhythmia, cardiac dysrhythmia, syncopy, angina pectoris, cardiac bypass reocclusion, intermittent claudication, diastolic and/or systolic dysfunction, diabetic myopathy, stroke prevention in congestive heart failure, hypertrophic medial thickening in arteries and/or large vessels, mesenteric vasculature hypertrophy or atherosclerosis, preferably atherosclerosis in mammalian patients with hypertension of diabetes; (ii) renal diseases or damages like renal hyperfiltration such as after portal renal ablation, proteinuria in chronic renal disease, renal arteriopathy as a consequence of hypertension, nephrosclerosis, hypertensive nephrosclerosis or mesanglial hypertrophy; (iii) Heart Failure to be treated is secondary to idiopathic dilated cardiomyopathy and/or coronary ischemic disease.
- the compounds of the present invention can also be used for the prevention, the delay of the onset, the delay of progression or the treatment of neurodegenerative disorders, cognitive disorders and for improving memory (both short term and long term) and learning ability wherein the (i)
- neurodegenerative disorder is dementia, senile dementia, schizophrenia, mild cognitive impairment, Alzheimer related dementia, Huntington's chores, tardive dyskinesia, hyperkinesias, mania, Morbus Parkinson, Steel-Richard syndrome, Down's syndrome, myasthenia gravis, nerve and brain trauma, vascular amyloidosis, cerebral hamorrhage I with amyloidosis, brain inflammation, Friedrich ataxia, acute confusion disorders, acute confusion disorders with apoptotic necrocytosis, amyotrophic lateral sclerosis, glaucoma, and Alzheimer's disease; (ii) cognitive disorders like cognitive deficits associated with
- the compounds of the present invention can also be used for stimulating an immune response in a subject having or at risk of having cancer wherein the cancer is selected from the group consisting of basal cell carcinomas including cancers of the binary tract, bladder, urinary system, bone, brain, breast, cervical, endometrial, ovarian, uterine, choriocarcinoma, central nervous system, colon and rectal cancers, connective tissue cancer, cancer of the digestive system, esophageal, gastric, stomach, larynx, liver, pancreatic, colorectal, renal cancers; cancers of the urinary system; cancers of eye, head and neck, oral cavity, skin, prostate; cancers of biliary tract, testicular, thyroid; intra- epithelial neoplasm, leukemia, acute myeloid leukemia, acute lymphoid leukemia, chronic myeloid leukemia, chronic lymphoid leukemia; and other cancers of the respiratory system, lung, small cell lung, non-small cell lung; lymphom
- rhabdomyosarcoma and other cancers including neoplastic conditions, adipose cell tumors, adipose cell carcinomas, such as liposarcoma.
- the compounds of the present invention can also be used for the treatment or prophylaxis of chronic inflammatory diseases such as autoimmune disorders like rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, psoriasis, allergies or asthma.
- chronic inflammatory diseases such as autoimmune disorders like rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, psoriasis, allergies or asthma.
- the compounds of the present invention can also be used in the treatment of pain, neuropathic pain, rheumatoid pain, osteoarthritis pain, anesthesia adjunct in mammalian patients undergoing surgery, chronic pain in advanced cancer, treatment of refractory diarrhea, biliary pain caused by gallstones.
- the compounds of the present invention can also be used for the treatment of mammalian patients undergoing islet/pancreas transplantation, for the prevention or the delay of transplant rejection, or allograft rejection in transplantation, for improving pancreatic function by increasing the number and size of pancreatic beta-cells in the treatment of Type 1 diabetes patients, and for improving pancreatic function by increasing the number and size of pancreatic beta-cells in general.
- the compounds of the present invention can be used for the treatment of mammalian patients with acne, skin disorders (e.g. pigmentation disorders or psoriasis), scleroderma, mycoses; anxiety, anxiety neurosis, major depression disorder, drug abuse, alcohol addiction, insomnia, chronic fatigue, sleep apnea; anorexia nervosa; epilepsy; migraine; encephalomyelitis;
- osteoarthritis osteoporosis, calcitonin-induced osteoporosis; male and female sexual dysfunction, infertility; Type 1 diabetes; immunosuppression, HIV infection; hematopoiesis, anemia; and for weight reduction.
- the compounds of the present invention are useful for the prevention, delay of progression or treatment of (i) bacterial infections from Escherichia coli, Staphylococcus, Streptoococcus, Pseudomonas, Clostridium difficile infection, Legionella, Pneumococcus, Haemophilus, Klebsiella,
- cryptococcosis aspergillosis, chromomycosis, mycetoma infections
- Trichomonas vaginalis Trichomonas vaginalis, Taenia, Hymenolepsis, Echinococcus, Schistosomiasis, neurocysticerosis, Necator americanus, and Trichuris trichuria.
- kits comprises two separate pharmaceutical compositions: a compound of the present invention, and a second pharmaceutical compound.
- the kit comprises a container for containing the separate compositions such as a divided bottle or a divided foil packet.
- kits include syringes, boxes and bags.
- the kit comprises directions for the use of the separate components.
- the kit form is particularly advantageous when the separate components are preferably administered in different dosage forms (e.g., oral and parenteral), are
- Blister packs are well known in the packaging industry and are being widely used for the packaging of pharmaceutical unit dosage forms (tablets, capsules, and the like). Blister packs generally consist of a sheet of relatively stiff material covered with a foil of a preferably transparent plastic material. During the packaging process recesses are formed in the plastic foil. The recesses have the size and shape of the tablets or capsules to be packed. Next, the tablets or capsules are placed in the recesses and the sheet of relatively stiff material is sealed against the plastic foil at the face of the foil which is opposite from the direction in which the recesses were formed. As a result, the tablets or capsules are sealed in the recesses between the plastic foil and the sheet. Preferably the strength of the sheet is such that the tablets or capsules can be removed from the blister pack by manually applying pressure on the recesses whereby an opening is formed in the sheet at the place of the recess. The tablet or capsule can then be removed via said opening.
- a memory aid on the kit, e.g., in the form of numbers next to the tablets or capsules whereby the numbers correspond with the days of the regimen which the tablets or capsules so specified should be ingested.
- a memory aid is a calendar printed on the card, e.g., as follows "First Week, Monday, Tuesday, . . . etc . . . Second Week, Monday, Tuesday, . . . " etc.
- a “daily dose” can be a single tablet or capsule or several pills or capsules to be taken on a given day.
- a daily dose of a compound of the present invention can consist of one tablet or capsule, while a daily dose of the second compound can consist of several tablets or capsules and vice versa.
- the memory aid should reflect this and aid in correct administration of the active agents.
- a dispenser designed to dispense the daily doses one at a time in the order of their intended use.
- the dispenser is equipped with a memory-aid, so as to further facilitate compliance with the regimen.
- a memory-aid is a mechanical counter which indicates the number of daily doses that has been dispensed.
- a battery-powered microchip memory coupled with a liquid crystal readout, or audible reminder signal which, for example, reads out the date that the last daily dose has been taken and/or reminds one when the next dose is to be taken.
- the compounds of the present invention and other pharmaceutically active compounds can be administered to a patient either orally, rectally, parenterally, (for example, intravenously, intramuscularly, or
- compositions suitable for parenteral injection may comprise
- aqueous and nonaqueous carriers, diluents, solvents, or vehicles include water, ethanol, polyols (propylene glycol, polyethylene glycol, glycerol, and the like), suitable mixtures thereof, vegetable oils (such as olive oil) and injectable organic esters such as ethyl oleate.
- a coating such as lecithin
- surfactants for reconstitution into sterile injectable solutions or dispersions.
- compositions may also contain adjuvants such as preserving, wetting, emulsifying, and dispersing agents.
- adjuvants such as preserving, wetting, emulsifying, and dispersing agents.
- Microorganism contamination can be prevented by adding various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, and the like.
- isotonic agents for example, sugars, sodium chloride, and the like.
- Prolonged absorption of injectable pharmaceutical compositions can be brought about by the use of agents delaying absorption, for example, aluminum monostearate and gelatin.
- Solid dosage forms for oral administration include capsules, tablets, powders, and granules.
- the active compound is admixed with at least one inert customary excipient (or carrier) such as sodium citrate or dicalcium phosphate or
- fillers or extenders as for example, starches, lactose, sucrose, mannitol, and silicic acid;
- binders as for example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose, and acacia;
- humectants as for example, glycerol;
- disintegrating agents as for example, agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain complex silicates, and sodium carbonate;
- solution retarders as for example, paraffin;
- absorption accelerators as for example, quaternary ammonium compounds;
- wetting agents as for example, paraffin
- the dosage forms may also comprise buffering agents.
- Solid compositions of a similar type may also be used as fillers in soft and hard filled gelatin capsules using such excipients as lactose or milk sugar, as well as high molecular weight polyethylene glycols, and the like.
- Solid dosage forms such as tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells, such as enteric coatings and others well known in the art. They may also contain opacifying agents, and can also be of such composition that they release the active compound or compounds in a certain part of the intestinal tract in a delayed manner. Examples of embedding compositions that can be used are polymeric substances and waxes. The active compounds can also be in micro-encapsulated form, if appropriate, with one or more of the above-mentioned excipients.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs.
- the liquid dosage form may contain inert diluents commonly used in the art, such as water or other solvents, solubilizing agents and emulsifiers, as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethylformamide, oils, in particular, cottonseed oil, groundnut oil, corn germ oil, olive oil, castor oil, and sesame seed oil, glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, or mixtures of these substances, and the like.
- inert diluents commonly used in the art, such as water or other solvents, solubilizing
- the composition can also include adjuvants, such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- Suspensions in addition to the active compound, may contain suspending agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar, and tragacanth, or mixtures of these substances, and the like.
- compositions for rectal administration are preferable suppositories, which can be prepared by mixing the compounds of the present invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax, which are solid at ordinary room temperature, but liquid at body temperature, and therefore, melt in the rectum or vaginal cavity and release the active component.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax, which are solid at ordinary room temperature, but liquid at body temperature, and therefore, melt in the rectum or vaginal cavity and release the active component.
- Dosage forms for topical administration of a compound of the present invention include ointments, powders, sprays and inhalants.
- the active compound or fit compounds are admixed under sterile condition with a physiologically acceptable carrier, and any preservatives, buffers, or propellants that may be required.
- Opthalmic formulations, eye ointments, powders, and solutions are also contemplated as being within the scope of this invention.
- the compounds of the present invention can be administered to a patient at dosage levels in the range of about 0.1 to about 3,000 mg per day.
- dosage levels in the range of about 0.1 to about 3,000 mg per day.
- a dosage in the range of about 0.01 to about 100 mg per kilogram body weight is typically sufficient.
- the specific dosage and dosage range that can be used depends on a number of factors, including the requirements of the patient, the severity of the condition or disease being treated, and the pharmacological activity of the compound being administered. The determination of dosage ranges and optimal dosages for a particular patient is within the ordinary skill in the art.
- the compounds of the present invention can be administered as pharmaceutically acceptable salts, esters, amides or prodrugs.
- salts refers to inorganic and organic salts of compounds of the present invention.
- the salts can be prepared in situ during the final isolation and purification of a compound, or by separately reacting a purified compound in its free base or acid form with a suitable organic or inorganic base or acid and isolating the salt thus formed.
- Representative salts include the hydrobromide, hydrochloride, sulfate, bisulfate, nitrate, acetate, oxalate, palmitate, stearate, laurate, borate, benzoate, lactate, phosphate, tosylate, citrate, maleate, fumarate, succinate, tartrate, naphthylate, mesylate, glucoheptonate, lactobionate, and laurylsulphonate salts, and the like.
- the salts may include cations based on the alkali and alkaline earth metals, such as sodium, lithium, potassium, calcium, magnesium, and the like, as well as non-toxic ammonium, quaternary ammonium, and amine cations including, but not limited to, ammonium, tetramethylammonium,
- esters of the compounds of the present invention include Ci-Cg alkyl esters. Acceptable esters also include C5-C7 cycloalkyl esters, as well as arylalkyl esters such as benzyl. C 1 -C4 alkyl esters are commonly used. Esters of compounds of the present invention may be prepared according to methods that are well known in the art.
- Examples of pharmaceutically acceptable amides of the compounds of the present invention include amides derived from ammonia, primary Ci-Cs alkyl amines, and secondary Ci-Cg dialkyl amines. In the case of secondary amines, the amine may also be in the form of a 5 or 6 membered heterocycloalkyl group containing at least one nitrogen atom. Amides derived from ammonia, C 1 -C3 primary alkyl amines and C 1 -C 2 dialkyl secondary amines are commonly used. Amides of the compounds of the present invention may be prepared according to methods well known to those skilled in the art.
- prodrug means compounds that are transformed in vivo to yield a compound of the present invention. The transformation may occur by various mechanisms, such as through hydrolysis in blood.
- a discussion of the use of prodrugs is provided by T. Higuchi and W. Stella, "Pro-drugs as Novel Delivery Systems,” Vol. 14 of the A.C.S. Symposium Series, and in
- a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as (Ci-Cg alkyl, (C 2 - Cl 2 )alkanoyloxymethyl, l-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1 -methyl- l-(alkanoyloxy)ethyl having from 5 to 10 carbon atoms,
- alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1- (alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1 -methyl- 1- (alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N- (alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1-(N- (alkoxycarbonyl)aminomethyl having from 4 to 10 carbon atoms, 3-phthalidyl, 4- crotonolactonyl, gamma-butyrolacton-4-yl, di-N,N-(C i -C 2 )alkylamino(C 2 - C 3 )alkyl (such as ⁇ -dimethylaminoethyl), carbamoyl-(Ci-C 2 )alkyl, N,N-di(C 1 - C 2 )alkylcarbamoyl-(Ci-C 2
- a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as (Ci-C6)alkanoyloxymethyl, 1- ((Ci-C6)alkanoyloxy)ethyl, 1 -methyl- l-((Ci-C6)alkanoyloxy)ethyl, (Ci- C6)alkoxycarbonyloxymethyl, N-(C i -C6)alkoxycarbonylaminomethyl, succinoyl, (Ci-C 6 )alkanoyl, a-amino(Ci-C4)alkanoyl, arylacyl and a-aminoacyl, or a- aminoacyl-a-aminoacyl, where each a-aminoacyl group is independently selected from the naturally occurring L-amino acids, -P(0)(OH) 2 , -P(0)(0(Ci-C6)
- the compounds of the present invention may contain asymmetric or chiral centers, and therefore, exist in different stereoisomeric forms. It is contemplated that all stereoisomeric forms of the compounds as well as mixtures thereof, including racemic mixtures, form part of the present invention. In addition, the present invention contemplates all geometric and positional isomers. For example, if the compound contains a double bond, both the cis and trans forms (designated as S and E, respectively), as well as mixtures, are
- stereoisomers such as diastereomeric mixtures
- Enantiomers can also be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., an alcohol), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereomers to the corresponding pure enantiomers.
- an appropriate optically active compound e.g., an alcohol
- Atropisomers e.g., substituted biaryls
- the compounds of the present invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water (hydrate), ethanol, and the like.
- pharmaceutically acceptable solvents such as water (hydrate), ethanol, and the like.
- the present invention contemplates and encompasses both the solvated and unsolvated forms.
- compounds of the present invention may exist in different tautomeric forms. All tautomers of compounds of the present invention are contemplated. For example, all of the tautomeric forms of the tetrazole moiety are included in this invention. Also, for example, all keto-enol or imine- enamine forms of the compounds are included in this invention.
- the present invention encompass compounds that are synthesized in vitro using laboratory techniques, such as those well known to synthetic chemists; or synthesized using in vivo techniques, such as through metabolism, fermentation, digestion, and the like. It is also contemplated that the compounds of the present invention may be synthesized using a combination of in vitro and in vivo techniques.
- the present invention also includes isotopically-labeled compounds, which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2 H, 3 H, 13 C, 14 C, 15 N, 16 0, 17 0, 31 P, 32 P, 35 S, 18 F, and 36 C1.
- the compounds of the present invention contain one or more deuterium atoms (2H) in place of one or more hydrogen atoms.
- Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detection. Further, substitution with heavier isotopes such as deuterium, i.e., 2 H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some
- Isotopically labeled compounds of this invention can generally be prepared by substituting a readily available isotopically labeled reagent for a non- isotopically labeled reagent.
- the compounds of the present invention may exist in various solid states including crystalline states and as an amorphous state.
- crystalline states also called polymorphs, and the amorphous states of the present compounds are contemplated as part of this invention.
- LG generally refer to groups that are displaceable by a nucleophile.
- Such leaving groups are known in the art.
- Examples of leaving groups include, but are not limited to, halides (e.g., I, Br, F, CI), sulfonates (e.g., mesylate, tosylate), sulfides (e.g., SCH 3 ), N- hydroxsuccinimide, N-hydroxybenzotriazole, and the like.
- nucleophiles include, but are not limited to, amines, thiols, alcohols, Grignard reagents, anionic species (e.g., alkoxides, amides, carbanions) and the like.
- This assay is used to directly measure the formation of 13 C-glucose-6- phosphate from 13 C-glucose by liquid chromatography-mass spectrometry (LC MS/MS).
- CB Compound Buffer
- EB Enzyme Buffer
- EB 50mM Tris, pH 7.5 / 4mM MgCl 2 / 6% DMSO / fresh 0.1% BSA / fresh 0.01% Brij-35 (10% BSA and 1% Brij-35 stock).
- GK (Glucokinase) Working Stock (5X): Dilute human His-hepatic GK to 30nM in EB buffer.
- Substrate Working Stock (1.47X): Dilute 13C-D-glucose (Sigma-Aldrich, St. Louis, MO) to 7.35mM from 1M stock (1M 13 C-D-glucose 186.1 1 mg/ml in 50mM Tris pH 7.5, 4mM MgCl 2 ) and dilute ATP (EMD Chemical Inc., Gibbstown, NJ) to 0.3528mM from frozen lOOmM stock and dilute 20mM fructose-6-phosphate (F6P) (Sigma-Aldrich, St.
- GKRP Glucokinase Regulatory Protein
- G6P glucose-6-phosphate
- Assay format is the same as for GKRP LC MS/MS Biochemical Assay with the following exceptions.
- This assay is used to directly measure the interaction between glucokinase (GK) and glucokinase regulatory protein (GKRP).
- GK glucokinase
- GKRP glucokinase regulatory protein
- Assay Buffer 20mM Tris, pH 7.5 / 0.05% BSA / ImM DTT / ⁇ sorbitol-6-phosphate.
- Assay Procedure Dilute avi-tagged GKRP to 10.7 nM in assay buffer. Combine the following reagents in a white 96-well half area plate. Pipette 14 ⁇ 1 of the diluted avi-tagged GKRP into each well. Add ⁇ of compound to be tested and incubate at room temperature for 20 minutes.
- This assay is used to directly measure the formation of 13 C-glucose-6- phosphate from 13 C-glucose by LC MS/MS.
- CB Compound Buffer
- EB Enzyme Buffer
- EB 50mM Tris, pH 7.5 / 4mM MgCl 2 / 6% DMSO / fresh 0.1% BSA / fresh 0.01% Brij-35 (10% BSA and 1% Brij-35 stock).
- GK (Glucokinase) Working Stock (5X): Dilute human His-hepatic GK to 30nM in EB buffer.
- Substrate Working Stock (1.47X): Dilute 13 C-D-glucose (Sigma- Aldrich, St. Louis, MO) to 7.35mM from 1M stock and dilute ATP (EMD Chemical, Gibbstown, NJ) to 0.3528mM from frozen lOOmM stock in CB buffer (1M 13 C-D-glucose 186.1 1 mg/ml in water).
- Dilute 20mM fructose-6-phosphate (F6P) (Sigma- Aldrich, St. Louis, MO) to 441 ⁇ in the substrate working stock.
- GKRP Glucokinase Regulatory Protein
- Lincoln, NE e.g., RediSep ®
- Krackeler Scientific Albany, NY
- Teflon ® is polyfluoroethylene, DuPont, Wilmington, DE
- the cross coupling (Z a or Z b is a halogen or a metal salt of B, Zn, Mg, etc.) can be achieved using a transition metal catalyst (e.g., a salt of Pd, Ni, etc.) in either anhydrous (e.g., tetrahydrofuran, dioxane, etc.) or an aqueous (e.g., tetrahydrofuran-water, dioxane-water, etc.) solvent under an inert atmosphere (e.g., N 2 , Ar, etc.).
- a transition metal catalyst e.g., a salt of Pd, Ni, etc.
- anhydrous e.g., tetrahydrofuran, dioxane, etc.
- an aqueous e.g., tetrahydrofuran-water, dioxane-water, etc.
- solvent e.g., N 2 , Ar, etc.
- the coupling of the heterocycle with the sulfinylimine can be executed by deprotonation of the heterocycle with an strong base (e.g., alkyllithium, amide, etc.), followed by exposure to the sulfinylimine, in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.).
- an strong base e.g., alkyllithium, amide, etc.
- anhydrous solvent e.g., tetrahydrofuran, toluene, etc.
- the sulfmamide can be converted to the sulfonamide by deprotection of the sulfmamide with acid (e.g., HC1, TFA, etc.) in either anhydrous (e.g., methanol, diethyl ether, etc.) or aqueous (e.g., methanol-water, diethyl ether- water, etc.) solvent followed by sulfonylation with a sulfonyl chloride in the presence of an amine base (e.g., triethylamine, diisopropylethylamine, etc.) in an anhydrous solvent (e.g., N,N- dimethylformamide, dichloromethane, etc.).
- acid e.g., HC1, TFA, etc.
- anhydrous e.g., methanol, diethyl ether, etc.
- aqueous e.g., methanol-water, diethyl ether
- the coupling of the heterocycle with the sulfinylimine can be executed by deprotonation of the heterocycle with a strong base (e.g., alkyllithium, amide, etc.), followed by exposure to the sulfinylimine in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.).
- a strong base e.g., alkyllithium, amide, etc.
- an anhydrous solvent e.g., tetrahydrofuran, toluene, etc.
- the sulfinamide can be converted to the sulfonamide by deprotection of the sulfinamide with an acid (e.g., HC1, TFA, etc.) in either anhydrous (e.g., methanol, diethyl ether, etc.) or aqueous (e.g., methanol-water, diethyl ether-water, etc.) solvent followed by sulfonylation with a sulfonyl chloride in the presence of an amine base (e.g., triethylamine, diisopropylethylamine, etc.) in an anhydrous solvent (e.g., N,N- dimethylformamide, dichloromethane, etc.).
- an acid e.g., HC1, TFA, etc.
- anhydrous e.g., methanol, diethyl ether, etc.
- aqueous e.g., methanol-water, diethyl
- the cross coupling (Z a or Z b is a halogen or a metal salt of B, Zn, Mg, etc.) can be achieved using a transition metal catalyst (e.g., a salt of Pd, Ni, etc.) in either anhydrous (e.g.,
- tetrahydrofuran dioxane, etc.
- an aqueous e.g., tetrahydrofuran-water, dioxane-water, etc.
- an inert atmosphere e.g., N 2 , Ar, etc.
- the heterocycle can be formylated by by deprotonation of the heterocycle with a strong base (e.g., alkyllithium, amide, etc.), followed by exposure to a formylation reagent (e.g., ⁇ , ⁇ -dimethylformamide, etc.) in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.).
- a strong base e.g., alkyllithium, amide, etc.
- a formylation reagent e.g., ⁇ , ⁇ -dimethylformamide, etc.
- anhydrous solvent e.g., tetrahydrofuran, toluene, etc.
- the aldehyde can be converted to the sulfony limine by treatment with a sulfonamide and an acid (e.g., Montmorillonite K10 clay, p-toluenesulfonic acid, etc.) with removal of water (e.g., Dean-Stark trap, dessicant, etc.) in an anhydrous solvent (e.g., toluene, dichloromethane, etc.).
- a sulfonamide and an acid e.g., Montmorillonite K10 clay, p-toluenesulfonic acid, etc.
- water e.g., Dean-Stark trap, dessicant, etc.
- an anhydrous solvent e.g., toluene, dichloromethane, etc.
- R 2 deprotonation of R 2 with an strong base (e.g., alkyllithium, amide, etc.), followed by exposure to the sulfonylimine, in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.).
- anhydrous solvent e.g., tetrahydrofuran, toluene, etc.
- the cross coupling (Z a or Z b is a halogen or a metal salt of B, Zn, Mg, etc.) can be achieved using a transition metal catalyst (e.g., a salt of Pd, Ni, etc.) in either anhydrous (e.g., tetrahydrofuran, dioxane, etc.) or aqueous (e.g., tetrahydrofuran-water, dioxane-water, etc.) solvent under an inert atmosphere (e.g., N 2 , Ar, etc.).
- a transition metal catalyst e.g., a salt of Pd, Ni, etc.
- anhydrous e.g., tetrahydrofuran, dioxane, etc.
- aqueous e.g., tetrahydrofuran-water, dioxane-water, etc.
- solvent e.g., N 2 , Ar, etc.
- the coupling of the heterocycle with the sulfonylimine can be executed by deprotonation of the heterocycle with an strong base (e.g., alkyllithium, amide, etc.), followed by exposure to the sulfonylimine, in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.).
- an strong base e.g., alkyllithium, amide, etc.
- anhydrous solvent e.g., tetrahydrofuran, toluene, etc.
- n-Butyllithium (33 mL of a 2.5 M solution in toluene, 0.083 mol) was added to a stirring solution of 2-chloro-4-iodopyridine (20 g, 0.082 mol, Matrix Scientific, Columbia, SC) and tetrahydrofuran (200 mL) at -78 °C. After 15 min, 1,1,1-trifluoroacetone (28 g, 0.25 mol) was added. After 1 h, saturated aqueous ammonium chloride was added, the mixture was warmed to room temperature, partitioned between more saturated aqueous ammonium chloride and ethyl acetate.
- Biotools/BoMem ChirallR instrument Biotools, Inc., Jupiter, FL
- concentrations of about 30 mg/mL in CDCI 3 in a 100 ⁇ BaF2 cell over a 4 h acquisition time at 4 cm "1 resolution.
- Step 1 n-Butyllithium (8.44 mL of a 1.6 M solution in hexanes, 13.5 mmol) was added to a solution of (2R)-2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (1.82 g, 5.63 mmol, Intermediate X4) in THF (27 mL) at -78 °C and the mixture was stirred at -78 °C for 10 min. A solution of N- ((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide
- Step 2a Hydrogen chloride (8.16 mL of a 2 M solution in diethyl ether, 16.3 mmol) was added to a solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-((lR)- 2,2,2-trif uoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (1.85 g, 3.26 mmol) in Et 2 0 (24 mL) and the resulting suspension was stirred at room temperature for 18 h.
- Step 2b Hydrogen chloride (0.97 mL of a 4.0 M solution in 1,4-dioxane, 3.9 mmol) was added to a stirring solution of (S)-N-((S)-(2-chlorophenyl)(7-(4- (( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (0.22 g, 0.39 mmol) and methanol at room temperature.
- Step 2a Hydrogen chloride (2.90 mL of a 2 M solution in Et 2 0, 5.80 mmol) was added to a solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfinamide (658 mg, 1.16 mmol) in Et 2 0 (8 mL) and the resulting suspension was stirred at room temperature for 6 h.
- Step 2b Hydrogen chloride (261 of a 2 M solution in Et 2 0, 0.522 mmol) was added to a solution of (S)- N-((S)-(2-chlorophenyl)(7-(4-((lS)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfinamide (74 mg, 0.130 mmol) in Et 2 0 (1 mL) and the resulting suspension was stirred at room temperature for 6 h.
- Step 1 Trimethyl(trifiuoromethyl)silane (2.52 mL, 17.1 mmol) was added to a mixture of 2-chloro-4-pyridinecarbonyl chloride (1.00 g, 5.68 mmol, Sigma- Aldrich, St. Louis, MO) and tetramethylammonium fluoride (1.59 g, 17.1 mmol) in DME (15 mL) at -78 °C and the mixture was stirred for 17 h while it warmed to room temperature.
- Step 2 A mixture of 2-(2-chloro-4-pyridinyl)-l,l,l,3,3,3-hexafluoro-2- propanol (1.27 g, 4.55 mmol), 2-(l-benzothiophen-7-yl)-4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (2.01 g, 7.73 mmol, Intermediate Y2), dicyclohexyl(2',4',6'- tris(l-methylethyl)-2-biphenylyl)phosphane (108 mg, 0.227 mmol), chloro(2- propen-l-yl)palladium dimer (41.6 mg, 0.114 mmol), and sodium carbonate monohydrate (204 mg, 13.7 mmol) in 1 ,4-dioxane/water (4: 1, 10 mL) was stirred at 80 °C for 5 h.
- the mixture was allowed to cool to room temperature, diluted with MeOH/DCM, absorbed onto silica gel, and purified by flash chromatography (100 g of silica gel, 10% to 30%> EtOAc/hexanes) to deliver 2- (2-(l-benzothiophen-7-yl)-4-pyridinyl)-l,l,l,3,3,3-hexafluoro-2-propanol (1.50 g) as an off-white solid.
- Step 3 n-Butyllithium (3.32 mL of a 2.5M solution in toluene, 8.29 mmol) was added to a solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l,3,3,3-hexaf uoro-2-propanol (1.49 g, 3.95 mmol) in THF (20 mL) at -78 °C.
- Step 4 Hydrogen chloride (7.92 mL of a 1 M solution in Et 2 0, 7.92 mmol) was added to a solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (1.23 g, 1.98 mmol) in Et 2 0 (12 mL) and the resulting suspension was stirred at room temperature for 5 h.
- Step 1 To a 2-L three-necked round-bottomed flask containing a solution of 7-bromo-l-benzothiophene (50 g, 236 mmol, Intermediate XI 3) in THF (400 mL) at -78 °C was added LDA (236 mL of a 1.8 M solution in THF, 472 mmol, Sigma- Aldrich, India) and the mixture was stirred at -78 °C for 45 min.
- LDA 236 mL of a 1.8 M solution in THF, 472 mmol, Sigma- Aldrich, India
- the resulting product was purified by column chromatography (basic alumina, 0% to 10% EtOAc/hexane to deliver (S)-N-((R)-(7-bromo-l-benzothiophen-2- yl)(2-chlorophenyl)methyl)-2-methyl-2-propanesulfinamide (25 g) as a pale- yellow liquid.
- Step 2 To a 100-mLround-bottomed flask containing a solution of (S)-N- ((R)-(7-bromo-l-benzothiophen-2-yl)(2-chlorophenyl)methyl)-2-methyl-2- propanesulfmamide (5 g, 11.0 mmol) in MeOH (25 mL) at 0 °C was added saturated hydrogen chloride in 1 ,4-dioxane (20 mL) over the course of 5 min.
- Step 3 To a 100-mL round-bottomed flask charged with a solution of (R)- 1 -(7-bromo- 1 -benzothiophen-2-yl)- 1 -(2-chlorophenyl)methanamine hydrochloride (4 g, 10.3 mmol) in N-methyl-2-pyrrolidinone (30 mL) at 0 °C was added triethylamine (3.14 g, 30.9 mmol). After 5 min at 0 °C,
- cyclopropylsulfonyl chloride (4.34 g, 31.0 mmol) was added over the course of 5 min and the reaction was then stirred at room temperature for 16 h. The mixture was partitioned between EtOAc (100 mL) and water (50 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (50 mL). The combined organic layers were dried (Na 2 S0 4 ), filtered, and concentrated under reduced pressure.
- Step 1 Potassium carbonate (260 g, 1.9 mmol) was added to a stirring solution of 2-bromobenzenethiol (180 g, 0.96 mol, Matrix Scientific, Columbia, SC), 2-bromo-l,l-dimethoxy ethane (170 g, 1.0 mol, Spectrochem, India), and acetone (1.8 L) at room temperature, and then the reaction mixture was heated at 55 °C. After 4 h, the reaction mixture was cooled to room temperature, filtered, the filter cake was washed with acetone, and the combined filtrate was concentrated under a vacuum.
- 2-bromobenzenethiol 180 g, 0.96 mol, Matrix Scientific, Columbia, SC
- 2-bromo-l,l-dimethoxy ethane 170 g, 1.0 mol, Spectrochem, India
- acetone 1.8 L
- Step 2 Polyphosphoric acid (PPA) (100 g) was added to a stirring solution of l-bromo-2-((2,2-dimethoxyethyl)sulfanyl)benzene (220 g, 0.84 mol, from Step 1) and toluene (2.2 L) at room temperature, and then the reaction mixture was heated at 110 °C. After 12 h, the reaction mixture was cooled to room temperature and then partitioned between water and ethyl acetate. The layers were separated, the organic material was washed sequentially with water and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum. The residue was subjected to flash chromatography on silica gel (49: 1 hexane-ethyl acetate) to give 7-bromo-l-benzothiophene (82 g) as a colorless liquid.
- PPA Polyphosphoric acid
- the mixture was stirred at 90 °C for 12 h, allowed to cool to room temperature, and filtered through a pad of diatomaceous earth, which was washed with toluene (100 mL). The combined filtrates were concentrated under reduced pressure, hexane (100 mL) was added to the residue, and the mixture was stirred at room temperature for 1 h.
- Titanium(IV) ethoxide (220 mL, 1100 mmol) was added to a stirring solution of (S)-2-methyl-2-propanesulfinamide (26 g, 210 mmol, AK Scientific, Mountain View, CA), dichloromethane (430 mL), and 2-chlorobenzaldehyde (30 g, 210 mmol, Sigma- Aldrich, St. Louis, MO), at room temperature. After 22 h, the reaction mixture was added to water and dichloromethane. The mixture was agitated vigorously and then filtered through a pad of diatomaceous earth.
- the reaction mixture was cooled to room temperature, filtered through a pad of diatomaceous earth, and the filtrate was concentrated under a vacuum.
- the residue was partitioned between water and ethyl acetate, the layers were separated, and the aqueous material was washed with ethyl acetate (2x).
- the combined organic extracts were washed sequentially with water and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum.
- n-Butyllithium (8.4 mL of a 2.5 M solution in toluene, 0.021 mol) was added to a stirring solution of 2-chloro-4-iodopyridine (5.0 g, 0.021 mol, Matrix Scientific, Columbia, SC) and tetrahydrofuran (50 mL) at -78 °C. After 15 min, acetone (4.6 mL, 0.063 mol) was added, and the mixture was stirred at - 78 °C for an additional hour. Saturated aqueous ammonium chloride was added, the mixture was warmed to room temperature, partitioned between more saturated aqueous ammonium chloride and ethyl acetate.
- allylpalladium(II) chloride dimer (0.16 g, 0.44 mmol), 9,9-dimethyl-4,5- bisdiphenylphosphino)xanthene (0.51 g, 0.89 mmol), and cesium fluoride (2.0 g, 13 mol) was heated at 80 °C under a nitrogen atmosphere. After 24 h, the reaction mixture was heated at 100 °C. After an additional 13h, the reaction mixture was allowed to cool to room temperature and concentrated under a vacuum. The residue was partitioned between saturated aqueous sodium bicarbonate and ethyl acetate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine.
- Step 2 n-Butyllithium (1.4 mL of a 1.6 M solution in hexanes, 2.3 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (0.37 g, 1.1 mmol, Intermediate Y3) and
- Step 3 Hydrogen chloride (5.0 mL of a 1.0 M solution with diethyl ether, 5.0 mmol) was added to a stirring solution of 2-methyl-N-(phenyl(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-propanesulfinamide (0.15 g, 0.28 mmol, from Step 2) and ethyl acetate (10 mL) at room temperature.
- Step 1 Montmorillonite K10 (2.2 g) was added to a stirring solution of 2- (methylsulfanyl)benzaldehyde (2.2 g, 14 mmol), 2-methyl-2-propanesulfinamide (1.8 g, 14 mmol, AK Scientific, Mountain View, CA), and toluene (100 mL).
- the reaction vessel was fitted with a Dean-Stark trap and a reflux condenser, and then the reaction mixture was heated at reflux. After 3 h, the reaction mixture was allowed to cool to room temperature and filtered. Silica gel (2.0 g) was added to the filtrate, and the volatiles were removed under a vacuum.
- Step 2 n-Butyllithium (0.41 mL of a 1.6 M solution in hexanes, 0.65 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.10 g, 0.31 mmol, Intermediate Y3) and THF (3.0 mL) at -78 °C under a nitrogen atmosphere.
- Step 3 Hydrogen chloride (0.69 mL of a 1.0 M solution with diethyl ether, 0.69 mmol) was added to a stirring solution of 2-methyl-N-((2- (methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-propanesulfinamide (0.080 g, 0.14 mmol, from Step 2) and ethyl acetate (10 mL) at room temperature.
- Step 1 n-Butyllithium (2.0 mL of a 1.6 M solution with hexanes, 3.2 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.51 g, 1.6 mmol, Intermediate Y3) and THF (7.9 mL) at -78 °C under a nitrogen atmosphere.
- Step 2 Hydrogen chloride (2.56 mL of a 1.0 M solution with diethyl ether, 2.56 mmol) was added to a stirring solution of (S)-N-((R)-(2- chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.29 g, 0.451 mmol, from Step 1) and ethyl acetate (10 mL) at room temperature.
- Step 1 n-Butyllithium (3.9 mL of a 1.6 M solution in hexanes, 6.2 mmol) was added to a stirring solution of 4-bromo-2-chloropyridine (0.58 mL, 5.2 mmol, Alfa Aesar, Ward Hill, MA) and diethyl ether (26 mL) at -78 °C under a nitrogen atmosphere. After 15 min, acetaldehyde (1.5 mL, 26 mmol) was added. After an additional 10 min, water (5.0 mL) was added and the mixture was warmed to room temperature. The mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate.
- Step 2 A stirring mixture of allylpalladium(II) chloride dimer (0.093 g, 0.25 mmol), 2-(dicyclohexylphosphino)-2',4',6',-tri-isopropyl-l, -biphenyl (0.24 g, 0.51 mmol), 1,4-dioxane (10 mL), 2-(l-benzothiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (2.0 g, 7.6 mmol, Intermediate Y2), l-(2-chloro- 4-pyridinyl)ethanol (0.80 g, 5.1 mmol, from Step 1), water (3.0 mL), and sodium carbonate monohydrate (1.6 g, 15 mmol), was heated at 80 °C.
- Step 1 n-Butyllithium (7.8 mL of a 1.6 M solution with hexanes, 12 mmol) was added to a stirring solution of 4-bromo-2-chloropyridine (1.2 mL, 10 mmol, Alfa Aesar, Ward Hill, MA) and diethyl ether (52 mL) at -78 °C. After 30 min, cyclobutanone (3.9 mL, 52 mmol) and water (50 mL) were added sequentially, and then the reaction mixture was warmed to room temperature, partitioned between ethyl acetate and more water, and the layers were separated.
- 4-bromo-2-chloropyridine 1.2 mL, 10 mmol, Alfa Aesar, Ward Hill, MA
- diethyl ether 52 mL
- cyclobutanone 3.9 mL, 52 mmol
- water 50 mL
- reaction mixture was allowed to cool to room temperature, and partitioned between ethyl acetate and water. The layers were separated, the organic material was washed with brine, silica gel (1.0 g) was added, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (40 g RediSep ® normal phase column, gradient elution of 0% to 30% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to afford l-(2-(l- benzothiophen-7-yl)-4-pyridinyl)cyclobutanol (0.49 g) as a clear oil.
- silica gel 40 g RediSep ® normal phase column, gradient elution of 0% to 30% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE
- Step 1 n-Butyllithium (3.9 mL of a 1.6 M solution with hexanes, 6.2 mmol) was added to a stirring solution of 4-bromo-2-chloropyridine (0.63 mL, 5.7 mmol, Alfa Aesar, Ward Hill, MA) and diethyl ether (28 mL) at -78 °C under a nitrogen atmosphere. After 15 min, l-(tert-butyldimethylsilyloxy)-2-propanone (3.3 mL, 17 mmol, Sigma- Aldrich, St. Louis, MO) was added. After an additional 10 min, water (5.0 mL) was added, and then the reaction mixture was allowed to warm to room temperature and concentrated under a vacuum.
- 4-bromo-2-chloropyridine (0.63 mL, 5.7 mmol, Alfa Aesar, Ward Hill, MA
- diethyl ether 28 mL
- Step 2 A stirring mixture of allylpalladium(II) chloride dimer (0.049 g, 0.13 mmol), 2-(dicyclohexylphosphino)-2',4',6',-tri-isopropyl-l, -biphenyl (X- Phos) (0.13 mg, 0.27 mmol), 1,4-dioxane (13 mL), 2-(l-benzothiophen-7-yl)- 4,4,5,5-tetramethyl-l,3,2-dioxaborolane (1.0 g, 4.0 mmol, Intermediate Y2), 1- ((tert-butyl(dimethyl)silyl)oxy)-2-(2-chloro-4-pyridinyl)-2-propanol (0.80 g, 2.7 mmol, from Step 1), water (4.0 mL), and sodium carbonate monohydrate (0.84 g, 8.0 mmol) was heated at 80 °C.
- reaction mixture was cooled to room temperature, concentrated, and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, the organic material was washed with brine, dried (magnesium sulfate), filtered, silica gel (1.0 g) was added to the filtrate, and the volatiles were removed under a vacuum.
- Step 1 A stirring mixture of di-tert-butyl (6-bromo-2- pyridinyl)imidodicarbonate (5.0 g, 13 mmol, Sigma- Aldrich, St. Louis, MO), potassium trifluoro(vinyl)borate (2.0 g, 15 mmol), cesium carbonate (13 g, 40 mmol), 2-(dicyclohexylphosphino)-2',4',6',-tri-isopropyll, -biphenyl (X-Phos) (0.64 g, 1.3 mmol), palladium(II) acetate (0.15 g, 0.67 mmol), tetrahydrofuran (60 mL) and water (7.0 mL) was heated at 80 °C.
- Step 2 Sodium periodate (8.6 g, 40 mmol) was added to a stirring mixture of di-tert-butyl (6-ethenyl-2-pyridinyl)imidodicarbonate (4.3 g, 13 mmol, from Step 1), tetrahydrofuran (54 mL), water (14 mL), and osmium tetroxide (4.1 mL of a 4.0 wt% solution with water, 0.67 mmol) at room temperature. After 2 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried
- Step 3 Copper(II) sulfate (6.4 g, 40 mmol) was added to a stirring solution of di-tert-butyl (6-formyl-2-pyridinyl)imidodicarbonate (4.3 g, 13 mmol, from Step 2), dichloromethane (53 mL), and (R)-2-methyl-2-propanesulfinamide (1.9 g, 16 mmol, AK Scientific, Mountain View, CA) at room temperature. After 50 h, the reaction mixture was filtered through a 0.45 ⁇ Teflon®
- Step 4 n-Butyllithium (1.9 mL of a 2.5 M solution in toluene, 4.7 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (0.84 g, 2.6 mmol, Intermediate Y3) in tetrahydrofuran (20 mL) at -78 °C under a nitrogen atmosphere.
- Step 5 Hydrogen chloride (2.7 mL of a 4.0 M solution with 1,4-dioxane, 11 mmol) was added to a stirring solution of di-tert-butyl (6-((S)-(((R)-tert- butylsulfinyl)amino)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-pyridinyl)imidodicarbonate (0.82 g, containing about 7 mol% of tert-butyl (6-((S)-(((R)-tert-butylsulfmyl)amino)(7- (4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-
- Step 6 4-(Dimethylamino)pyridine (0.013 g, 0.11 mmol) was added to a stirring solution of 2-(2-(2-((S)-amino(6-amino-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (0.49 g, containing about 5 mol% of 2-(2-(2-((R)-amino(6-amino-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol, from Step 5), N,N- dimethylformamide (11 mL), N,N-diisopropylethylamine (0.96 mL, 5.5 mmol), and cyclopropanesulfonyl chloride (0.11 mL, 1.1 mmol
- reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, and the layers were separated. The organic layer was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine (2x), dried (sodium sulfate), filtered, the filtrate was concentrated. The residue was dissolved with
- Step 1 n-Butyllithium (6.6 mL of a 2.5 M solution in toluene, 16 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-2- propanol (2.4 g, 9.0 mmol, Intermediate Y5) and tetrahydrofuran (74 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of N-((S,E)-(2- chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (2.0 g, 8.2 mmol, Intermediate Yl) in tetrahydrofuran (8.0 mL) was added.
- Step 2 Hydrogen chloride (12 mL of a 4.0 M solution with 1,4-dioxane, 48 mmol) was added to a stirring solution of (S)-N-((R)-(2-chlorophenyl)(7-(4- ( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl- 2-propanesulfmamide (2.5 g, 4.9 mol, from Step 1) and methanol (49 mL) at room temperature.
- Step 3 4-(Dimethylamino)pyridine (0.060 g, 0.49 mmol) was added to a stirring solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l-benzothiophen- 7-yl)-4-pyridinyl)-2-propanol hydrochloride (2.0 g, 4.9 mmol, from Step 2), N,N- dimethylformamide (49 mL), N,N-diisopropylethylamine (8.5 mL, 49 mmol), and cyclopropanesulfonyl chloride (0.50 mL, 4.9 mmol, Matrix Scientific, Columbia, SC) at room temperature.
- 2-(2-(2-(R)-amino(2-chlorophenyl)methyl)-l-benzothiophen- 7-yl)-4-pyridinyl)-2-propanol hydrochloride 2.0 g, 4.9 mmol, from Step 2)
- Step 1 A stirring solution of 6-bromo-2-pyridinamine (15 g, 87 mmol, Sigma- Aldrich, St. Louis, MO), acetonitrile (170 mL), and N-chlorosuccinimide (14 g, 100 mmol) was heated at reflux under a nitrogen atmosphere. After 18 h, the reaction mixture was concentrated and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic layer was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), and filtered. Silica gel (45 g) was added to the filtrate, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (gradient elution; 6: 1 to 4: 1 hexane-ethyl acetate). The isolated material was dissolved with
- Step 2 Di-tert-butyl dicarbonate (19 g, 87 mmol) was added to a stirring mixture of 6-bromo-5-chloro-2-pyridinamine (8.6 g, 42 mmol, from Step 1), dichloromethane (83 mL), and N,N-diisopropylethylamine (22 mL, 120 mmol) at room temperature under a nitrogen atmosphere. After 14 h, 4- (dimethylamino)pyridine (0.51 g, 4.2 mmol) was added. After an additional 3 h, the reaction mixture was concentrated under a vacuum, and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate.
- the organic layer was isolated, and was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and concentrated under a vacuum.
- the residue was dissolved with dichloromethane, silica gel (40 g) was added to the solution, and the volatiles were removed under a vacuum.
- the residue was subjected to flash chromatography on silica gel (9: 1 hexane- ethyl acetate).
- the isolated material was dissolved with dichloromethane, silica gel (20 g) was added to the solution, and the volatiles were removed under a vacuum.
- Step 3 A stirring mixture of di-tert-butyl (6-bromo-5-chloro-2- pyridinyl)imidodicarbonate (3.0 g, 7.4 mmol, from Step 2), potassium trifluoro(vinyl)borate (1.1 g, 8.1 mmol), cesium carbonate (7.2 g, 22 mmol), 2- (dicyclohexylphosphino)-2',4',6',-tri-isopropyll, -biphenyl (0.35 g, 0.74 mmol), palladium(II) acetate (0.083 g, 0.37 mmol), tetrahydrofuran (33 mL) and water (4.0 mL) was heated at 80 °C.
- reaction mixture was allowed to cool to room temperature, concentrated under a vacuum, and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The organic layer was separated, and washed with brine, dried (sodium sulfate), filtered, then concentrated under a vacuum. The residue was dissolved with dichloromethane, silica gel (13 g) was added, and the volatiles were removed under a vacuum.
- Step 4 Sodium periodate (1.1 g, 5.2 mmol) was added to a stirring mixture of di-tert-butyl (5-chloro-6-ethenyl-2-pyridinyl)imidodicarbonate (0.61 g, containing about 35 mol% of di-tert-butyl (6-bromo-5-chloro-2- pyridinyl)imidodicarbonate, from Step 3), tetrahydrofuran (7.0 mL), water (1.6 mL), and osmium tetroxide (0.53 mL of a 4.0 wt% solution with water, 0.086 mmol) at room temperature.
- di-tert-butyl (5-chloro-6-ethenyl-2-pyridinyl)imidodicarbonate (0.61 g, containing about 35 mol% of di-tert-butyl (6-bromo-5-chloro-2- pyridinyl)imi
- Step 5 Copper(II) sulfate (0.64 g, 4.0 mmol) was added to a stirring solution of di-tert-butyl (5-chloro-6-formyl-2-pyridinyl)imidodicarbonate (0.48 g, from Step 4), dichloromethane (5.4 mL), and (S)-2-methyl-2-propanesulfinamide (0.20 g, 1.6 mmol, AK Scientific, Mountain View, CA) at room temperature. After 43 h, the reaction mixture was filtered through a 0.20 ⁇ Teflon®
- silica gel (3.0 g) was added to the filtrate, and the volatiles were removed under a vacuum. This material was subjected to flash chromatography on silica gel (9: 1 hexane-ethyl acetate) to give di-tert-butyl (6-((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5- chloro-2-pyridinyl)imidodicarbonate (0.37 g) as a clear pale yellow tar.
- Step 6 n-Butyllithium (0.62 mL of a 2.5 M solution in toluene, 1.6 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-2- propanol (0.23 g, 0.86 mmol, Intermediate Y5) and tetrahydrofuran (6.5 mL) at - 78 °C under a nitrogen atmosphere.
- Step 7 Hydrogen chloride (0.93 mL of a 4.0 M solution with 1,4-dioxane, 3.7 mmol) was added to a stirring solution of di-tert-butyl (6-((R)-(((S)-tert- butylsulfinyl)amino)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)imidodicarbonate (0.27 g, containing about 17 mol% of di-tert-butyl (6-((S)-(((S)-tert- butylsulfinyl)amino)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-5-chloro-2-pyridin
- Step 8 4-(Dimethylamino)pyridine (0.0046 g, 0.038 mmol) was added to a stirring solution of 2-(2-(2-((R and S)-amino(6-amino-3-chloro-2- pyridinyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.16 g, 0.38 mmol, from Step 7), N,N-dimethylformamide (3.8 mL), N,N- diisopropylethylamine (0.33 mL, 1.9 mmol), and cyclopropanesulfonyl chloride (0.038 mL, 0.38 mmol, Matrix Scientific, Columbia, SC) at room temperature.
- reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, and then
- Step 1 Montmorillonite K 10 (2.0 g) was added to a stirring mixture of 2- chloro-6-methoxybenzaldehyde (2.0 g, 12 mmol, Chem-Impex, Wood Dale, IL), cyclopropanesulfonamide (1.4 g, 12 mmol, Matrix, Columbia, SC), and toluene (59 mL).
- the reaction vessel was fitted with a Dean-Stark trap and a reflux condenser, and then the reaction mixture was heated at reflux. After 15 h, the reaction mixture was filtered and the filtrate was allowed to cool to room temperature. The filtrate was concentrated under a vacuum to a volume of about 20 mL, and then filtered again.
- Step 2 n-Butyllithium (0.73 mL of a 2.5 M solution in toluene, 1.8 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-2- propanol (0.27 g, 1.0 mmol, Intermediate Y5) and tetrahydrofuran (8.0 mL) at - 78 °C. After 10 min, a solution of N-((lE)-(2-chloro-6- methoxyphenyl)methylidene)cyclopropanesulfonamide (0.25 g, 0.91 mmol, from Step 1) in tetrahydrofuran (1.1 mL) was added.
- Step 1 Methylmagnesium chloride (4.8 mL of a 3.0 mL solution with tetrahydrofuran, 14 mmol) was added to a stirring solution of methyl 2-chloro-4- pyrimidinecarboxylate (1.0 g, 5.8 mmol, Maybridge, Tintagel, England) in tetrahydrofuran (58 mL) at 0 °C under a nitrogen atmosphere. After 10 min, saturated aqueous sodium bicarbonate was added and the reaction mixture was allowed to warm to room temperature. The organics were partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate.
- Step 2 A stirring mixture of 2-(2-chloro-4-pyrimidinyl)-2-propanol (1.0 g, 5.8 mol, from Step 1), 2-(l-benzothiophen-7-yl)-4,4,5,5-tetramethyl-l,3,2- dioxaborolane (1.8 g, 7.0 mol, Intermediate Y2), 1,4-dioxane (9.5 mL), water (2.5 mL), allylpalladium(II) chloride dimer (0.11 g, 0.29 mmol), 2- (dicyclohexylphosphino)-2',4',6',-tri-isopropyll, -biphenyl (X-Phos) (0.28 g, 0.58 mmol), and sodium carbonate (18 g, 17 mmol) was heated at 80 °C under a nitrogen atmosphere.
- reaction mixture was allowed to cool to room temperature, and partitioned between saturated aqueous sodium bicarbonate and ethyl acetate. The layers were separated and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, then concentrated under a vacuum. The residue was dissolved with dichloromethane, silica gel (10 g) was added to the solution, and the volatiles were removed under a vacuum.
- Step 3 Triethylsilyl trifluoromethanesulfonate (0.43 mL, 1.9 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyrimidinyl)-2- propanol (0.47 g, 1.7 mmol, from Step 2), dichloroethane (8.7 mL), and N,N- diisopropylethylamine (0.91 mL, 5.2 mmol) at room temperature. After 1 h, additional triethylsilyl trifluoromethanesulfonate (0.22 mL, 0.95 mmol) was added.
- Step 4 n-Butyllithium (0.23 mL of a 2.5 M solution with toluene, 0.57 mmol) was added to a stirring solution of 2,2,6,6-tetramethylpiperidine (0.097 mL, 57 mmol) and tetrahydrofuran (5.0 mL) at -78 °C under a nitrogen atmosphere. After 5 min, the reaction mixture was warmed to room temperature.
- tetrahydrofuran (0.50 mL) was added. After another hour, the reaction mixture was allowed to warm to room temperature. After 3 h at rt, tetrabutylammonium fluoride (0.69 mL of a 1.0 M solution with tetrahydrofuran, 0.69 mmol) was added. After 25 min, saturated aqueous sodium bicarbonate was added, and the mixture was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and then concentrated. The residue was dissolved with dichloromethane, silica gel (1.5 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash
- Step 5 Hydrogen chloride (0.32 mL of a 4.0 M solution with 1,4-dioxane, 1.3 mmol) was added to a stirring solution of (S)-N-((R)-(2-chlorophenyl)(7-(4- ( 1 -hydroxy- 1 -methylethyl)-2-pyrimidinyl)- 1 -benzothiophen-2-yl)methyl)-2- methyl-2-propanesulfmamide (0.065 g, 0.13 mmol, from Step 4) and methanol (1.3 mL) at room temperature. After 15 min, the reaction mixture was
- Step 6 4-(Dimethylamino)pyridine (1.5 mg, 12 ⁇ ) was added to a stirring solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l-benzothiophen- 7-yl)-4-pyrimidinyl)-2-propanol (0.049 g, 0.12 mmol, from Step 5), N,N- dimethylformamide (1.2 mL), N,N-diisopropylethylamine (0.10 mL, 0.60 mmol), and cyclopropanesulfonyl chloride (0.012 mL, 0.12 mmol, Matrix Scientific, Columbia, SC) at room temperature.
- Step 1 n-Butyllithium (0.23 mL of a 2.5 M solution in toluene, 0.57 mmol) was added to a stirring solution of 1,1,1 -trifluoro-2-(2-thieno[2,3- c]pyridin-7-yl-4-pyridinyl)-2-propanol (0.10 g, 0.32 mmol, Intermediate Y6) in tetrahydrofuran (2.0 mL) at -78 °C under a nitrogen atmosphere.
- Step 2 Hydrogen chloride (0.17 mL of a 4.0 M solution with 1,4-dioxane, 0.68 mmol) was added to a stirring solution of (S)-N-((R)-(2-chlorophenyl)(7-(4- (2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-2- yl)methyl)-2-methyl-2-propanesulfinamide (0.039 g, 0.069 mmol, from Step 1) and methanol (1.4 mL) at room temperature.
- Step 3 4-(Dimethylamino)pyridine (0.83 mg, 6.8 ⁇ ) was added to a stirring solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)thieno[2,3- c]pyridin-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol hydrochloride (0.034 g, 0.068 mmol, from Step 2), N,N-dimethylformamide (1.4 mL), N,N- diisopropylethylamine (0.071 mL, 0.41 mmol), and cyclopropanesulfonyl chloride (0.0069 mL, 0.068 mmol, Matrix Scientific, Columbia, SC) at room temperature.
- Step 1 n-Butyllithium (0.25 mL of a 2.5 M solution in toluene, 0.61 mmol) was added to a stirring solution of 1,1,1 -trifluoro-2-(2-thieno[2,3- c]pyridin-7-yl-4-pyridinyl)-2-propanol (0.11 g, 0.34 mmol, Intermediate Y6) in tetrahydrofuran (2.5 mL) at -78 °C under a nitrogen atmosphere.
- Step 3 4-(Dimethylamino)pyridine (1.3 mg, 11 ⁇ ) was added to a stirring solution of 2-(2-(2-((R)-amino(3-chloro-2-pyridinyl)methyl)thieno[2,3- c]pyridin-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol hydrochloride (0.050 g, 0.11 mmol, from Step 2), N,N-dimethylformamide (1.1 mL), N,N- diisopropylethylamine (0.19 mL, 1.1 mmol), and cyclopropanesulfonyl chloride (0.011 mL, 0.11 mmol, Matrix Scientific, Columbia, SC) at room temperature.
- Benzenesulfonyl chloride (0.019 mL, 0.15 mmol) was added to a stirring solution of 2-(2-(2-(amino(phenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol hydrochloride (0.035 g, 0.075 mmol, Intermediate Zl), N,N-diisopropylethylamine (0.065 mL, 0.38 mmol), and dichloromethane (5.0 mL) at room temperature. After 3 h, silica gel (1.0 g) was added and the volatiles were removed under a vacuum. The residue was subjected to flash
- Step 1 Titanium(IV) ethoxide (34 mL, 160 mmol) was added to a stirring solution of 2-(methylsulfanyl)benzaldehyde (5.0 g, 33 mmol), (S)-2-methyl-2- propanesulfinamide (4.0 g, 33 mmol, AK Scientific, Mountain View, CA), and dichloromethane (66 mL) at room temperature. After 24 h, water (10 mL) was added and the resulting suspension was filtered through a pad of diatomaceous earth. The two layers comprising the filtrate were separated, and the organic material was washed with brine, dried (magnesium sulfate), filtered, and concentrated.
- 2-(methylsulfanyl)benzaldehyde 5.0 g, 33 mmol
- (S)-2-methyl-2- propanesulfinamide 4.0 g, 33 mmol, AK Scientific, Mountain View, CA
- dichloromethane 66 mL
- Step 2 n-Butyllithium (0.74 mL of a 1.6 M solution in hexanes, 1.2 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.20 g, 0.62 mmol, Intermediate Y3) in tetrahydrofuran (5.0 mL) at -78 °C under a nitrogen atmosphere.
- Step 1 A solution of bromine (2.3 mL, 46 mmol) and chloroform (10 mL) was added to a stirring solution of l-methyl-2-pyridone (5.0 mL, 456 mmol) and chloroform (46 mL) at reflux. After 1 h, the reaction mixture was cooled to room temperature. After 72 h, the reaction mixture was filtered and the filter cake was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, silica gel (1.0 g) was added to the organic material, and the volatiles were removed under a vacuum.
- Step 2 n-Butyllithium (3.7 mL, 5.9 mmol) was added to a stirring solution of 5-bromo-l-methylpyridin-2(lH)-one (1.0 g, 5.3 mmol, from Step 1) in tetrahydrofuran (27 mL) at -78 °C. After 10 min, the reaction mixture was sparged with sulfur dioxide (0.27 mL, 5.3 mmol) for 2 min and then allowed to warm to room temperature. The mixture was concentrated under a vacuum and the residue was dissolved with dichloromethane (20 mL), and NCS (0.71 g, 5.3 mmol) was added.
- Step 3 l-Methyl-6-oxo-l,6-dihydro-3-pyridinesulfonyl chloride (0.050 g, 0.24 mmol, from Step 2) was added to a stirring solution of 2-(2-(2-((R)- amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol hydrochloride (0.040 g, 0.080 mmol, Intermediate Z3), N,N- diisopropylethylamine (0.14 mL, 0.80 mmol), and dichloromethane (3.0 mL) at room temperature.
- 6-Methoxy-3-pyridinesulfonyl chloride (0.025 mL, 0.12 mmol, Acros, Geel, Belgium) was added to a stirring solution of 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol hydrochloride (0.050 g, 0.10 mmol, Intermediate Z3), N,N- diisopropylethylamine (0.087 mL, 0.50 mmol), N,N-(dimethylamino)pyridine (0.12 mg, 1.0 ⁇ ), and dichloroethane (10 mL) at room temperature.
- 6-Aminopyridine-3-sulfonyl chloride (0.039 g, 0.20 mmol) was added to a stirring mixture of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol hydrochloride (0.10 g, 0.20 mmol, Intermediate Z3), dichloroethane (10 mL), N,N- diisopropylethylamine (0.17 mL, 1.0 mmol), and N,N-(dimethylamino)pyridine (0.024 mg, 0.20 ⁇ ) at room temperature.
- the isolated material was subjected to flash chromatography on silica gel (40 g RediSep ® normal phase column, elution of 5% methanol (with 2.0 M NH 3 ) in dichloromethane, Teledyne Isco, Lincoln, NE) to afford 6-amino-N-((R)-(2- chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-3-pyridinesulfonamide (0.0058 g) as a colorless solid (mixture of 2 diastereomers).
- Step 1 n-Butyllithium (3.8 mL of a 1.6 M solution in hexanes, 6.1 mmol) was added to a stirring solution of l-(2-(l-benzothiophen-7-yl)-4- pyridinyl)ethanol (0.77 g, 3.1 mmol, Intermediate Z4) and tetrahydrofuran (15 mL) at -78 °C under a nitrogen atmosphere. After 20 min, a solution of N-((S,E)- (2-chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (0.60 mg, 2.4 mmol, Intermediate Yl) in tetrahydrofuran (3.0 mL) was added.
- Step 2 Hydrogen chloride (2.4 mL of a 1.0 M solution with diethyl ether, 2.4 mmol) was added to a stirring solution of (S)-N-((R)-(2-chlorophenyl)(7-(4- ( 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (0.24 g, containing about 15 mol% of (S)-N-((S)-(2- chlorophenyl)(7-(4-( 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfinamide), from Step 1) and methanol (2.4 mL) at room temperature.
- Step 3 N,N-(Dimethylamino)pvridine (0.029 g, 0.24 mmol) was added to a stirring solution of l-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)ethanol hydrochloride (0.21 g, 0.48 mmol, from Step 2), dichloromethane (2.4 mL), N,N-diisopropylethylamine (0.25 mL, 1.4 mmol), and cyclopropanesulfonyl chloride (0.098 mL, 0.96 mmol, Matrix Scientific, Columbia, SC) at room temperature.
- Step 1 n-Butyllithium (2.2 mL of a 1.6 M solution with hexanes, 3.5 mmol) was added to a stirring solution of l-(2-(l-benzothiophen-7-yl)-4- pyridinyl)cyclobutanol (0.49 g, 1.7 mmol, Intermediate Z5) and tetrahydrofuran (17 mL) at -78 °C under a nitrogen atmosphere.
- Step 2 Hydrogen chloride (0.45 mL of a 1.0 M solution with water, 15 mmol) was added to a stirring solution of N-((R)-(2-chlorophenyl)(7-(4-(l- hydroxycyclobutyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (0.15 g, 0.29 mmol, from Step 1) and ethyl acetate (2.0 mL). After 1 h, the reaction mixture was concentrated under a vacuum. The residue was partitioned between saturated aqueous sodium bicarbonate and
- Step 3 N,N-(Dimethylamino)pyridine (0.0036 mg, 0.029 mmol) was added to a stirring solution of l-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)cyclobutanol (0.12 mg, 0.29 mmol, from Step 2), N,N-diisopropylethylamine (0.25 mL, 1.5 mmol), N,N-dimethylformamide (3.0 mL), and cyclopropanesulfonyl chloride (0.30 mL, 2.9 mmol, Matrix Scientific, Columbia, SC) at room temperature.
- reaction mixture was filtered and the filtrate was subjected to reversed-phase preparative HPLC (Phenomenex Gemini C 18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 ⁇ ) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 0% to 100% over 10 min).
- HPLC Henomenex Gemini C 18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 ⁇ ) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 0% to 100% over 10 min).
- the isolated material was subjected to flash chromatography on silica gel (40 g RediSep ® normal phase column, gradient elution of 0% to 100% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to provide N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy cyclobutyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.014 g) as a colorless solid.
- silica gel 40 g RediSep ® normal phase column, gradient elution of 0% to 100% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Endocrinology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The present invention relates to benzothiophene sulfonamides and other compounds that interact with glucokinase regulatory protein. In addition, the present invention relates to methods of treating type 2 diabetes, and other diseases and/or conditions where glucokinase regulatory protein is involved using the compounds, or pharmaceutically acceptable salts thereof, and pharmaceutical compositions that contain the compounds, or pharmaceutically acceptable salts thereof.
Description
BENZOTHIOPHENE SULFONAMIDES AND OTHER COMPOUNDS THAT INTERACT WITH GLUCOKINASE REGULATORY PROTEIN
FIELD OF THE INVENTION
The present invention relates to benzothiophene sulfonamides and other compounds, or pharmaceutically acceptable salts thereof, as defined herein, that interact with glucokinase regulatory protein. In addition, the present invention relates to methods of treating type 2 diabetes, and other diseases and/or conditions where glucokinase regulatory protein is involved using the
compounds, or the pharmaceutically acceptable salts thereof, and pharmaceutical compositions that contain the compounds, or pharmaceutically acceptable salts thereof.
BACKGROUND OF THE INVENTION
Glucokinase (GK) is a member of a family of four hexokinases that are critical in the cellular metabolism of glucose. Specifically GK, also known as hexokinase IV or hexokinase D, facilitates glucose induced insulin secretion from pancreatic β-cells as well as glucose conversion into glycogen in the liver. GK has a unique catalytic activity that enables the enzyme to be active within the physiological range of glucose (from 5mM glucose to lOmM glucose).
Genetically modified mouse models support the role of GK playing an important role in glucose homeostasis. Mice lacking both copies of the GK gene die soon after birth from severe hyperglycemia, whereas mice lacking only one copy of the GK gene present with only mild diabetes. Mice that are made to overexpress the GK gene in their livers are hypoglycemic.
Numerous human mutations in the GK gene have been identified, with the vast majority of them resulting in proteins with impaired or absent enzymatic activity. These loss-of- function mutations are thought to contribute to the hyperglycemia seen with maturity-onset diabetes of the young type II (MODY- 2). A small fraction of these mutations result in a GK with increased catalytic function. These individuals present with moderate to severe hypoglycemia.
GK activity in the liver is transiently regulated by glucokinase regulatory protein (GK P). GK catalytic activity is inhibited when GK is bound to GKRP. This interaction is antagonized by increasing concentrations of both glucose and fructose -1 -phosphate (F1P). The complex of the two proteins is localized primarily to the nuclear compartment of a cell. Post prandially as both glucose and fructose levels rise, GK released from GKRP translocates to the cytoplasm. Cytoplasmic GK is now free of the inhibitory effects of GKRP and able to kinetically respond to glucose. Evidence from the Zucker diabetic fatty rat (ZDF) indicates that their glucose intolerance may be a result of this mechanism failing to function properly.
A compound that acts directly on GKRP to disrupt its interaction with GK and hence elevate levels of cytoplasmic GK is a viable approach to modulate GK activity. Such an approach would avoid the unwanted hypoglycemic effects of over stimulation of GK catalytic activity, which has been seen in the
development of GK activators. A compound having such an effect would be useful in the treatment of diabetes and other diseases and/or conditions in which GKRP and/or GK plays a role. The present invention provides compounds that bind GKRP and disrupts its interaction with GK.
SUMMARY OF THE INVENTION
In embodiment 1, the present invention provides compounds of Formula I, or pharmaceutically acceptable salts thereof, wherein:

I
* is a chiral center having the R configuration;
Yis

X1 is N or CH;
X2 is N or CH;
X' is N or CH;
X4 is N or CH;
X5 is N or CH;
X6isSorNH;
-N(Ci_6alkyl)2

R2 is phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl, where the phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl group can be unsubstituted or substituted with from one to three substituents independently selected from Ci_6alkyl, Ci_6haloalkyl, C2_6alkenyl, halo, -SCi_6alkyl, -OCi_6alkyl,
-OCi_6haloalkyl -OH, -NH2, -N02 or -N(Ci_6alkyl)2;
3is

n is 0, 1, 2, 3 or 4;
R4 is hydrogen or Ci_6alkyl; and
R5 is hydrogen, Ci_6alkyl or Ci_6haloalkyl.
In embodiment 2, the invention provides compounds in accordance with embodiment 1 , or pharmaceutically acceptable salts thereof, wherein Y is

In embodiment 3, the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X1 is CH.
In embodiment 4, the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X1 is N.
In embodiment 5, the invention provides compounds in accordance with any one of embodiments 1 to 4, or pharmaceutically acceptable salts thereof, wherein X2 is CH.
In embodiment 6, the invention provides compounds in accordance with any one of embodiments 1 to 5, or pharmaceutically acceptable salts thereof, wherein X3 is CH.
In embodiment 7, the invention provides compounds in accordance with any one of embodiments 1 to 6, or pharmaceutically acceptable salts thereof, wherein X4 is CH.
In embodiment 8, the invention provides compounds in accordance with any one of embodiments 1 to 6, or pharmaceutically acceptable salts thereof, wherein X4 is N.
In embodiment 9, the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X1, X2, X3, X4 and X5 are CH, and X6 is S.
In embodiment 10, the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X1, X2, X3 and X4 are CH, X5 is N, and X6 is S.
In embodiment 11 , the invention provides compounds in accordance with any one of embodiments 1 or 2, or pharmaceutically acceptable salts thereof, wherein X1 is N, X2, X3 and X4 are CH, X5 is CH, and X6 is S.
In embodiment 12, the invention provides compounds in accordance with any one of embodiments 1 to 11 , or pharmaceutically acceptable salts thereof, wherein R4 is hydrogen.
In embodiment 13, the invention provides compounds in accordance with any one of embodiments 1 to 12, or pharmaceutically acceptable salts thereof, wherein R1 is Ci_6alkyl, three to eight membered cycloalkyl, phenyl or pyridyl, which cycloalkyl, phenyl or pyridyl groups may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_ 6alkyl, -NH2 or Ci_6alkyl.
In embodiment 14, the invention provides compounds in accordance with any one of embodiments 1 to 12, or pharmaceutically acceptable salts thereof, wherein R1 is three to eight membered cycloalkyl, substituted phenyl or substituted pyridyl.
In embodiment 15, the invention provides compounds in accordance with any one of embodiments 1 to 12, or pharmaceutically acceptable salts thereof, wherein R1 is cyclopropyl or substituted cyclopropyl.
In embodiment 16, the invention provides compounds in accordance with any one of embodiments 1 to 15, or pharmaceutically acceptable salts thereof, wherein R2 is phenyl or substituted phenyl.
In embodiment 17, the invention provides compounds in accordance with any one of embodiments 1 to 15, or pharmaceutically acceptable salts thereof, wherein R2 is pyridyl or substituted pyridyl.
In embodiment 18, the invention provides compounds in accordance with any one of embodiments 1 to 15, or pharmaceutically acceptable salts thereof, wherein R2 is

In embodiment 19, the invention provides compounds in accordance with any one of embodiments 1 to 18, or pharmaceutically acceptable salts thereof, wherein R3 is

In embodiment 20, the invention provides compounds in accordance with any one of embodiments 1 to 18, or pharmaceutically acceptable salts thereof, wherein R3 is

In embodiment 21, the present invention also provides a compound, or a pharmaceutically acceptable salt thereof, selected from:
N-((R)-(6-amino-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((2-chloro-6-methoxyphenyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyrimidinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide;
4-methoxy-N-(phenyl(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)benzenesulfonamide;
N-(phenyl(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)benzenesulfonamide;
4-methoxy-N-((2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)benzenesulfonamide; N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -methyl-6-oxo- 1 ,6-dihydro-3 - pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-methoxy-3 -pyridinesulfonamide; 6-amino-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-fluorobenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy cyclobutyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
6-chloro-N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-6-oxo-l,6-dihydro-3- pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-3-methoxybenzenesulfonamide;
N'-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-N,N-dimethylsulfamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)ethanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)methanesulfonamide;
N-((R)-(3-(dimethylamino)-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-chloro-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-methoxy-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-methyl-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-methoxyphenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-nitrophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-methylphenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
6-chloro-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-6-oxo-l,6-dihydro-3- pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
(lS,2S)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- ethylcyclopropanesulfonamide;
( 1 R,2R)-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- ethylcyclopropanesulfonamide;
(lS,2R)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-( 1 - methylethyl)cyclopropanesulfonamide;
(lR,2S)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-( 1 - methylethyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-3-methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-oxo- 1 ,6- dihydro-3 -pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -
(trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(5 -( 1 -hydroxy- 1 -methylethyl)-2-thiophenyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(4-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H- benzimidazol-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H-indol- 2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -benzofuran-5 -sulfonamide;
N-((R)-(2-(l -methylethenyl)phenyl)(7-(4-((lR)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-(l -methylethenyl)phenyl)(7-(4-((l S)-2,2,2-trifluoro-l -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(5 -( 1 -hydroxy- 1 -methylethyl)-6-oxo- 1 ,6-dihydro-3 - pyridazinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifiuoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-3- methylbenzofuran-5 -sulfonamide;
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifiuoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo [b]thiophen-2-yl)methyl)- 1 -ethy 1-6-oxo- 1 ,6-dihydropyridine-3 -sulfonamide; or
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifiuoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)benzofuran-5- sulfonamide.
In embodiment 22, the present invention provides methods of treating type 2 diabetes, hyperglycemia, impaired glucose tolerance, insulin resistance, retinopathy, nephropathy, neuropathy, cataracts, glaucoma, Syndrome X, or polycystic ovarian syndrome, the methods comprising administering to a patient in need thereof a therapeutically effective amount of a compound in accordance with any one of embodiments 1 to 20, or a pharmaceutically acceptable salt thereof.
In embodiment 23, the present invention provides a method of embodiment 22 wherein the method of treatment is for type 2 diabetes.
In embodiment 24, the present invention provides pharmaceutical compositions comprising a compound in accordance with any one of
embodiments 1 to 21, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. In embodiment 1A, the present invention provides compounds of Formula
I, or pharmaceutically acceptable salts thereof, wherein:

I * is a chiral center having the R configuration, except when X6 is N, then * is a chiral center having the S configuration;

X1 is N, CH or C-halo; X2 is N, CH or C-halo;
X3 is N, CH or C-halo;
X4 is N or CH;
X5 is N or CH;
Xb is S or NH;
R1 is Ci_6alkyl, -N(C1-6alkyl)2, -N(H)three to eight membered cycloalkyl,




-S(=0)Ci_6alkyl, -S02NH2 or -S02Ci_6alkyl; n is 0, 1, 2, 3 or 4; m is 0, 1 or 2;
R4 is hydrogen or Ci_6alkyl; hydrogen, Ci-6alkyl or Ci_6haloalkyl; and
R6 is hydrogen or halo, provided that R1 is not dimethoxyphenyl. In embodiment 2A, the present invention provides compounds in accordance with embodiment 1 A, or pharmaceutically acceptable salts thereof, wherein:
X1 is N or CH;
X2 is N or CH;
X3 is N or CH; R1 is Ci_6alkyl, -N(Ci_6alkyl)2,
 three to eight membered cycloalkyl, six to ten membered aryl or five to ten membered heteroaryl, where the heteroaryl group has from 1 to 4 heteroatoms independently selected from O, N or S, which cycloalkyl, aryl or heteroaryl group may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_6alkyl, -OCi_6haloalkyl,-OH, -NH2, Ci_ 6alkyl or Ci_6haloalkyl;
three to eight membered cycloalkyl, six to ten membered aryl or five to ten membered heteroaryl, where the heteroaryl group has from 1 to 4 heteroatoms independently selected from O, N or S, which cycloalkyl, aryl or heteroaryl group may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_6alkyl, -OCi_6haloalkyl,-OH, -NH2, Ci_ 6alkyl or Ci_6haloalkyl;
R2 is phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl, where the phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl group can be unsubstituted or substituted with from one to three substituents independently selected from Ci_6alkyl, Ci_6haloalkyl, C2_6alkenyl, halo, -SCi_6alkyl, -OCi_6alkyl,
-OCi_6haloalkyl -OH, -NH2, -N02 or -N(Ci_6alkyl)2; and
R3 is




In embodiment 3 A, the present invention provides compounds in accordance with any one of embodiments 1 A or 2A, or pharmaceutically acceptable salts thereof, wherein Y is

In embodiment 4A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A, or pharmaceutically acceptable salts thereof, wherein X1 is CH.
In embodiment 5 A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A, or pharmaceutically acceptable salts thereof, wherein X1 is N.
In embodiment 6A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 5 A, or pharmaceutically acceptable salts thereof, wherein X2 is CH.
In embodiment 7A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 5 A, or pharmaceutically acceptable salts thereof, wherein X2 is C-halo.
In embodiment 8A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 7 A, or pharmaceutically acceptable salts thereof, wherein X3 is CH.
In embodiment 9A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 7 A, or pharmaceutically acceptable salts thereof, wherein X3 is N.
In embodiment 10A, the present invention provides compounds in accordance with any one of embodiments 1 A to 9 A, or pharmaceutically acceptable salts thereof, wherein X4 is CH.
In embodiment 11 A, the present invention provides compounds in accordance with any one of embodiments 1 A to 9 A, or pharmaceutically acceptable salts thereof, wherein X4 is N.
In embodiment 12 A, the present invention provides compounds in accordance with embodiment 1 A, or pharmaceutically acceptable salts thereof, wherein X1, X2, X3, X4 and X5 are CH, and X6 is S.
In embodiment 13 A, the present invention provides compounds in accordance with embodiment 1 A, or pharmaceutically acceptable salts thereof, wherein X1, X2, X3 and X4 are CH, X5 is N, and X6 is S.
In embodiment 14 A, the present invention provides compounds in accordance with embodiment 1 A, or pharmaceutically acceptable salts thereof, wherein X1 is N, X2, X3 and X4 are CH, X5 is CH, and X6 is S.
In embodiment 15 A, the present invention provides compounds in accordance with any one of embodiments 1 A to 14A, or pharmaceutically acceptable salts thereof, wherein R4 is hydrogen.
In embodiment 16A, the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 15 A, or pharmaceutically acceptable salts thereof, wherein R1 is Ci_6alkyl, three to eight membered cycloalkyl, phenyl or pyridyl, which cycloalkyl, phenyl or pyridyl groups may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_6alkyl, -NH2 or Ci_6alkyl.
In embodiment 17A, the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 15 A, or pharmaceutically acceptable salts thereof, wherein R1 is three to eight membered cycloalkyl, substituted phenyl or substituted pyridyl. In embodiment 18A, the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 15 A, or pharmaceutically acceptable salts thereof, wherein R1 is cyclopropyl or substituted cyclopropyl.
In embodiment 19A, the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 18 A, or pharmaceutically acceptable salts thereof, wherein R2 is phenyl or substituted phenyl.
In embodiment 20A, the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 18 A, or pharmaceutically acceptable salts thereof, wherein R2 is pyridyl or substituted pyridyl.
In embodiment 21 A, the present invention provides compounds in accordance with any one of embodiments 1A or 3 A to 18 A, or pharmaceutically acceptable salts thereof, wherein R2 is

In embodiment 22A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 21 A, or pharmaceutically acceptable salts thereof, wherein R3 is

In embodiment 23 A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 21 A, or pharmaceutically acceptable salts thereof, wherein R3 is

In embodiment 24A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 21 A, or pharmaceutically acceptable salts thereof, wherein R3 is

In embodiment 25 A, the present invention provides compounds in accordance with any one of embodiments 1 A or 3 A to 21 A, or pharmaceutically acceptable salts thereof, wherein R3 is

In embodiment 26A, the present invention provides compounds in accordance with any one of embodiments 1 A to 25 A, or pharmaceutically acceptable salts thereof, wherein m is 0.
In embodiment 27A, the present invention provides compounds in accordance with any one of embodiments 1 A to 25 A, or pharmaceutically acceptable salts thereof, wherein m is 1.
In embodiment 28A, the present invention provides compounds, or pharmaceutically acceptable salts thereof, selected from:
N-((R)-(6-amino-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((2-chloro-6-methoxyphenyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyrimidinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide;
4-methoxy-N-(phenyl(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)benzenesulfonamide;
N-(phenyl(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)benzenesulfonamide;
4-methoxy-N-((2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)benzenesulfonamide; N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -methyl-6-oxo- 1 ,6-dihydro-3 - pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-methoxy-3 -pyridinesulfonamide; 6-amino-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-fluorobenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy cyclobutyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
6-chloro-N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-6-oxo-l,6-dihydro-3- pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-3-methoxybenzenesulfonamide;
N'-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-N,N-dimethylsulfamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)ethanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)methanesulfonamide;
N-((R)-(3-(dimethylamino)-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-chloro-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-methoxy-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-methyl-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-methoxyphenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-nitrophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-methylphenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
6-chloro-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-6-oxo-l,6-dihydro-3- pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
(lS,2S)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- ethylcyclopropanesulfonamide;
( 1 R,2R)-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- ethylcyclopropanesulfonamide;
(lS,2R)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-( 1 - methylethyl)cyclopropanesulfonamide;
(lR,2S)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-( 1 - methylethyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-3-methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-oxo- 1 ,6- dihydro-3 -pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -
(trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(5 -( 1 -hydroxy- 1 -methylethyl)-2-thiophenyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(4-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H- benzimidazol-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H-indol- 2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -benzofuran-5 -sulfonamide;
N-((R)-(2-(l -methylethenyl)phenyl)(7-(4-((lR)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-(l -methylethenyl)phenyl)(7-(4-((l S)-2,2,2-trifluoro-l -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(5 -( 1 -hydroxy- 1 -methylethyl)-6-oxo- 1 ,6-dihydro-3 - pyridazinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifiuoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-3- methylbenzofuran-5 -sulfonamide;
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifiuoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo [b]thiophen-2-yl)methyl)- 1 -ethy 1-6-oxo- 1 ,6-dihydropyridine-3 -sulfonamide; or
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifiuoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)benzofuran-5- sulfonamide.
In embodiment 29A, the present invention provides compounds, or pharmaceutically acceptable salts thereof, selected from:
N-((R)-(5-amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((5-Amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)thieno[3,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno [3 ,2- b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)tetrahydro-3 -thiophenesulfonamide 1 , 1 -dioxide hydrochloride;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(lR)-2,2,2-trifluoro-l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5-chloro-l,3-thiazol-4-yl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(5-amino-2-chlorophenyl)(7-(4-(l -hydroxy- l-methylethyl)-2 -pyridinyl)-
1- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((5-amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl) cyclopropanesulfonamide;
N-((S)-(2-amino-5 -chloro-4-pyrimidinyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5-chloro-4-pyrimidinyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5 -chloro-4-pyrimidinyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-
2- yl)(2-(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide;
N-((R)-(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methyl ethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)(2-(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - (hydroxymethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - (hydroxymethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)(2-
(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide;
N-((7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)(2-
(methylsulfonyl)phenyl)methyl)cyclopropanesulfonamide;
N-((2-amino-5-chloro-4-pyridinyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -cyclopropylmethanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -ethyl-6-oxo- 1 ,6-dihydro-3- pyridinesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-oxo- 1 -(2,2,2- trifluoroethyl)- 1 ,6-dihydro-3 -pyridinesulfonamide;
N-((R)-(3 -chloro-2-pyridiny l)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-chloro-2-pyridinyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro- 1 -ethyl-6-oxo- 1 ,6-dihydro-3 -pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 - (hydroxymethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -methyl- 1 -benzofuran-5 -sulfonamide; N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((S)-(2-chlorophenyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)-
1- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((S)-(2-chlorophenyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethy l)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-
2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methyl-3 -butyn- 1 -yl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2-cyano- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)-
1- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy-2-methoxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2-cyano- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(S-methylsulfonimidoyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(5-chloro-7-(4-(l,2-dihydroxy-l-methylethyl)-
2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((R)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((S)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((S)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((R)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((S)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((S)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-ethynylphenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
2-(2-((6-amino-3-chloro-2-pyridinyl)((cyclopropylsulfonyl)amino)methyl)-l- benzothiophen-7-yl)-4-pyridinesulfonamide;
N-((6-amino-3 -chloro-2-pyridinyl)(7-(4-(3 -hydroxytetrahydro-3 -furanyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylpropyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(methylsulfinyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(methylsulfonyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((2-chlorophenyl)(7-(4-( 1 -cyclopropyl- 1 -hydroxy ethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((3S)-3 -hydroxytetrahydro-3 -furanyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((3R)-3-hydroxytetrahydro-3-furanyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l -cyclopropyl- 1 -hydroxy ethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy cyclopropyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3 -chloro-2-pyridinyl)(7-(4-(2 -methyl- 1 H-imidazol- 1 -yl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((7-(4-acetyl-2-pyridinyl)-l-benzothiophen-2-yl)(6-amino-3-chloro-2- pyridinyl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(3-hydroxy-3-oxetanyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 H-indol-2-yl)methyl)cyclopropanesulfonamide;
R)-N-((2-chlorophenyl)(7-(4-(2-hydroxypropan-2-yl)pyridin-2-yl)-lH-indol-2- yl)methyl)cyclopropanesulfonamide;
2-(2-(2-(amino(3-amino-6-chloro-2-fluorophenyl)methyl)-l-benzothiophen-7- yl)-4-pyridinyl)- 1 ,2-propanediol;
N-((R)-(3 -amino-6-chloro-2-fluorophenyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3 -amino-6-chloro-2-fluorophenyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((3-amino-6-chloro-2-fluorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -cyclopropylmethanesulfonamide; N-((3-amino-6-chloro-2-fluorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -cyclopropylmethanesulfonamide; N-((R)-(5-amino-2-chloro-4-fluoro-3-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(5-amino-2-chloro-4-fluoro-3-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((5-amino-2-chloro-4-fluorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(2-hydroxypropan-2-yl)pyridin-2- yl)thieno [3 ,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l,2dihydroxypropan-2-yl)pyridin-2- yl)thieno [3 ,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l,2-dihydroxypropan-2-yl)pyridine-2- yl)-5-fluorobenzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((2-chloro-6-fluorophenyl)(7-(4-(l,2-dihydroxypropan-2-yl)pyridin-2- yl)thieno [3 ,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-((R)-l,2-dihydroxypropan-2-yl)pyridin- 2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-((S)-l,2-dihydroxypropan-2-yl)pyridin- 2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-morpholinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -azetidinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-4-morpholinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-N'-cyclopropylsulfamide;
N-((6-amino-3 -chloro-2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((6-amino-3 -chloro-2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -methyl- 1 H-pyrazole-4-sulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)- 2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)- 2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -methyl- 1 H-pyrazole-4-sulfonamide; N-((R)-(2-chlorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((2-chloro-6-fluorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l,3-benzothiazol-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
(1 S)-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)spiro [22]pentane- 1 - sulfonamide;
(lR)-N-((R)-(2-chlorophenyl)(7-(4-((l S)-2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)spiro [22]pentane- 1 - sulfonamide;
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-bromo-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-bromo-6-fluorophenyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-bromo-6-fluorophenyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-5-fluoro-2-pyridinyl)(7-(4-(l -hydroxy- 1-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(l -hydroxy- 1-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-5-fluoro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide; or
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide.
In embodiment 3 OA, the present invention provides methods of treating type 2 diabetes, hyperglycemia, impaired glucose tolerance, insulin resistance, retinopathy, nephropathy, neuropathy, cataracts, glaucoma, Syndrome X, or polycystic ovarian syndrome, the methods comprising administering to a patient in need thereof a therapeutically effective amount of a compound in accordance with any one of embodiments 1 A to 29 A, or a pharmaceutically acceptable salt thereof.
In embodiment 31 A, the present invention provides methods of embodiment 30A wherein the treatment is for type 2 diabetes.
In embodiment 32A, the present invention provides pharmaceutical compositions comprising a compound in accordance with any one of
embodiments 1 A to 29 A, or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable excipient.
DETAILED DESCRIPTION
The present invention provides benzothiophene sulfonamides and other compounds, as defined above, or pharmaceutically acceptable salts thereof. The present invention also provides pharmaceutical compositions comprising a compound of the present invention, or pharmaceutically acceptable salts thereof, and methods of treating diseases and/or conditions, such as diabetes, using compounds of the present invention, or pharmaceutically acceptable salts thereof.
The term "alkyl" means a straight or branched chain hydrocarbon.
Representative examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, sec-butyl, pentyl and hexyl. Typical alkyl groups are alkyl groups having from 1 to 8 carbon atoms, which groups are commonly represented as Ci-galkyl.
The term "haloalkyl" mean a straight or branched chain hydrocarbon wherein one or more hydrogen atom is replaced with a halogen. Typical haloalkyl groups are haloalkyl groups having from 1 to 8 carbon atoms, which groups are commonly represented as Ci-shaloalkyl. Common haloalkyl groups include -CF3, -CHF2 or -CH2F.
The term "alkoxy" means an alkyl group bonded to an oxygen atom.
Representative examples of alkoxy groups include methoxy, ethoxy, tert-butoxy, propoxy and isobutoxy. Common alkoxy groups are Ci-galkoxy.
The term "halogen" or "halo" means chlorine, fluorine, bromine or iodine.
The term "alkenyl" means a branched or straight chain hydrocarbon having one or more carbon-carbon double bonds. Representative examples alkenyl groups include ethenyl, propenyl, allyl, butenyl and 4-methylbutenyl. Common alkenyl groups are C2-8alkenyl.
The term "alkynyl" means a branched or straight chain hydrocarbon having one or more carbon-carbon triple bonds. Representative examples of alkynyl groups include ethynyl, propynyl (propargyl) and butynyl. Common alkynyl groups are C2-g alkynyl.
The term "cycloalkyl" means a cyclic, nonaromatic hydrocarbon.
Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl. A cycloalkyl group can contain one or more double bond. Examples of cycloalkyl groups that contain double bonds include cyclopentenyl, cyclohexenyl, cyclohexadienyl and cyclobutadienyl. Common cycloalkyl groups are C3-8 cycloalkyl groups.
The term "perfluoroalkyl" means an alkyl group in which all of the hydrogen atoms have been replaced with fluorine atoms. Common
perfluoroalkyl groups are Ci-sperfluoroalkyl. An example of a common perfluoroalkyl group is CF3.
The term "acyl" means a group derived from an organic acid by removal of the hydroxy group (-OH). For example, the acyl group CH3C(=0)- is formed by the removal of the hydroxy group from CH3C(=0)OH .
The term "aryl" means a cyclic, aromatic hydrocarbon. Examples of aryl groups include phenyl and naphthyl. Common aryl groups are six to thirteen membered rings.
The term "heteroatom" as used herein means an oxygen, nitrogen or sulfur atom.
The term "heteroaryl" means a cyclic, aromatic hydrocarbon in which one or more carbon atoms of an aryl group have been replaced with a heteroatom. If the heteroaryl group contains more than one heteroatom, the heteroatoms may be the same or different. Examples of heteroaryl groups include pyridyl, pyrimidinyl, imidazolyl, thienyl, furyl, pyrazinyl, pyrrolyl, indolyl, triazolyl, pyridazinyl, indazolyl, purinyl, isoquinolyl, quinolyl, naphthyridinyl, quinoxalinyl, isothiazolyl and benzo[b]thienyl. Common heteroaryl groups are five to thirteen membered rings that contain from 1 to 4 heteroatoms. Heteroaryl groups that are five and six membered rings that contain 1 to 3 heteroatoms are particularly common.
The term "heterocycloalkyl" means a cycloalkyl group in which one or more of the carbon atoms has been replaced with a heteroatom. If the
heterocycloalkyl group contains more than one heteroatom, the heteroatoms may
be the same or different. Examples of heterocycloalkyl groups include tetrahydrofuryl, morpholinyl, piperazinyl, piperidinyl and pyrrolidinyl. It is also possible for the heterocycloalkyl group to have one or more double bonds, but is not aromatic. Examples of heterocycloalkyl groups containing double bonds include dihydrofuran. Common heterocycloalkyl groups are three to ten membered rings containing from 1 to 4 heteroatoms. Heterocycloalkyl groups that are five and six membered rings that contain 1 to 2 heteroatoms are particularly common.
It is also noted that the cyclic ring groups, i.e., aryl, heteroaryl, cycloalkyl, and heterocycloalkyl, can comprise more than one ring. For example, the naphthyl group is a fused bicyclic ring system. It is also intended that the present invention include ring groups that have bridging atoms, or ring groups that have a spiro orientation.
Representative examples of five to six membered aromatic rings, optionally having one or two heteroatoms, are phenyl, furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, pyridinyl, pyridiazinyl, pyrimidinyl, and pyrazinyl.
Representative examples of partially saturated, fully saturated or fully unsaturated five to eight membered rings, optionally having one to three heteroatoms, are cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and phenyl. Further exemplary five membered rings are furyl, thienyl, pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, pyrrolidinyl, 1,3-dioxolanyl, oxazolyl, thiazolyl, imidazolyl, 2H- imidazolyl, 2-imidazolinyl, imidazolidinyl, pyrazolyl, 2-pyrazolinyl,
pyrazolidinyl, isoxazolyl, isothiazolyl, 1 ,2-dithiolyl, 1,3-dithiolyl, 3H-1,2- oxathiolyl, 1,2,3-oxadizaolyl, 1,2,4-oxadiazolyl, 1,2,5-oxadiazolyl,
l,3,4oxadiazolyl, 1,2,3-triazolyl, 1,2,4-trizaolyl, 1,3,4-thiadiazolyl, 3H-1,2,3- dioxazolyl, 1,2,4-dioxazolyl, 1,3,2-dioxazolyl, 1,3,4-dioxazolyl, 5H-1,2,5- oxathiazolyl, and 1,3-oxathiolyl.
Further exemplary six membered rings are 2H-pyranyl, 4H-pyranyl, pyridinyl, piperidinyl, 1 ,2-dioxinyl, 1,3-dioxinyl, 1 ,4-dioxanyl, morpholinyl, 1,4- dithianyl, thiomorpholinyl, pyridazinyl, pyrimidinyl, pyrazinyl, piperazinyl,
1,3,5-triazinyl, 1,2,4-triazinyl, 1,2,3-triazinyl, 1,3,5-trithianyl, 4H-l,2-oxazinyl, 2H-l,3-oxazinyl, 6H-l,3-oxazinyl, 6H-l,2-oxazinyl, 1 ,4-oxazinyl, 2H-1,2- oxazinyl, 4H-l,4-oxazinyl, 1,2,5-oxathiazinyl, 1 ,4-oxazinyl, o-isoxazinyl, p- isoxazinyl, 1,2,5-oxathiazinyl, 1,2,6-oxathiazinyl, and 1,4,2-oxadiazinyl.
Further exemplary seven membered rings are azepinyl, oxepinyl, thiepinyl and 1,2,4-triazepinyl.
Further exemplary eight membered rings are cyclooctyl, cyclooctenyl and cyclooctadienyl.
Exemplary bicyclic rings consisting of two fused partially saturated, fully saturated or fully unsaturated five and/or six membered rings, optionally having one to four heteroatoms, are indolizinyl, indolyl, isoindolyl, indolinyl, cyclopenta(b)pyridinyl, pyrano(3,4-b)pyrrolyl, benzofuryl, isobenzofuryl, benzo(b)thienyl, benzo(c)thienyl, lH-indazolyl, indoxazinyl, benzoxazolyl, anthranilyl, benzimidazolyl, benzthiazolyl, purinyl, quinolinyl, isoquinolinyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 1,8-naphthyridinyl, pteridinyl, indenyl, isoindenyl, naphthyl, tetralinyl, decalinyl, 2H-1- benzopyranyl, pyrido(3,4-b)pyridinyl, pyrido(3,2-b)pyridinyl, pyrido(4,3-b)- pyridinyl, 2H-l,3-benzoxazinyl, 2H-l,4-benzoxazinyl, lH-2,3-benzoxazinyl, 4H- 3,1-benzoxazinyl, 2H-l,2-benzoxazinyl and 4H-l,4-benzoxazinyl.
A cyclic ring group may be bonded to another group in more than one way. If no particular bonding arrangement is specified, then all possible arrangements are intended. For example, the term "pyridyl" includes 2-, 3-, or 4- pyridyl, and the term "thienyl" includes 2-, or 3-thienyl.
The term "substituted" means that a hydrogen atom on a molecule or group is replaced with a group or atom. Typical substituents include: halogen, Ci-galkyl, hydroxyl, Ci-galkoxy, -NRXRX, nitro, cyano, halo or perhaloCi-galkyl, C2-8alkenyl, C2-8alkynyl, -SRX, -S(=0)2Rx, -C(=0)ORx, -C(=0)Rx, wherein each Rx is independently hydrogen or Ci-Cs alkyl. It is noted that when the substituent is -NRXRX, the Rx groups may be joined together with the nitrogen atom to form a ring.
The term "oxo", when used as a substituent, means the =0 group, which is typically attached to a carbon atom.
A group or atom that replaces a hydrogen atom is also called a
substituent.
Any particular molecule or group can have one or more substituent depending on the number of hydrogen atoms that can be replaced.
The symbol "-" represents a covalent bond and can also be used in a radical group to indicate the point of attachment to another group. In chemical structures, the symbol is commonly used to represent a methyl group in a molecule.
The term "therapeutically effective amount" means an amount of a compound that ameliorates, attenuates or eliminates one or more symptom of a particular disease or condition, or prevents or delays the onset of one of more symptom of a particular disease or condition.
The term "patient" means animals, such as dogs, cats, cows, horses, sheep and humans. Particular patients are mammals. The term patient includes males and females.
The term "pharmaceutically acceptable" means that the referenced substance, such as a compound of the present invention or a formulation containing a compound of the present invention, or a particular excipient, are suitable for administration to a patient.
The terms "treating", "treat" or "treatment" and the like include preventative (e.g., prophylactic) and palliative treatment.
The term "patient in need thereof means a patient who has or is at risk of having a GKRP/GK mediated disease or condition, such as type 2 diabetes.
The term "excipient" means any pharmaceutically acceptable additive, carrier, diluent, adjuvant, or other ingredient, other than the active pharmaceutical ingredient (API), which is typically included for formulation and/or
administration to a patient.
The compounds of the present invention are administered to a patient in a therapeutically effective amount. The compounds can be administered alone or as
part of a pharmaceutically acceptable composition or formulation. In addition, the compounds or compositions can be administered all at once, as for example, by a bolus injection, multiple times, such as by a series of tablets, or delivered substantially uniformly over a period of time, as for example, using transdermal delivery. It is also noted that the dose of the compound can be varied over time.
In addition, the compounds of the present invention can be administered alone, in combination with other compounds of the present invention, or with other pharmaceutically active compounds. The other pharmaceutically active compounds can be intended to treat the same disease or condition as the compounds of the present invention or a different disease or condition. If the patient is to receive or is receiving multiple pharmaceutically active compounds, the compounds can be administered simultaneously, or sequentially. For example, in the case of tablets, the active compounds may be found in one tablet or in separate tablets, which can be administered at once or sequentially in any order. In addition, it should be recognized that the compositions may be different forms. For example, one or more compound may be delivered via a tablet, while another is administered via injection or orally as a syrup. All combinations, delivery methods and administration sequences are contemplated.
The compounds of the present invention may be used in the manufacture of a medicament for the treatment of a disease and/or condition mediated by GKRP/GK, such as type 2 diabetes.
The compounds of the present invention may be used in combination with other pharmaceutically active compounds. It is noted that the term
"pharmaceutically active compounds" can include biologies, such as proteins, antibodies and peptibodies. Examples of other pharmaceutically active compounds include, but are not limited to: (a) dipeptidyl peptidase IV (DPP-IV) inhibitors such as Vildagliptin (Novartis), Sitagliptin (Merck&Co.), Saxagliptin (BMS) Alogliptin (Takeda); (b) insulin sensitizers including (i) PPARy agonists such as the glitazones (e.g., troglitazone, pioglitazone, edaglitazone,
rosiglitazone, and the like) and other PPAR ligands, including PPARa/γ dual agonists such as muraglitazar (BMS) and tesaglitazar (AstraZeneca), and PPARa
agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (II) biguanides such as metformin and phenformin, and (iii) protein tyrosine phosphatase-lB (PTP-1B) inhibitors; (c) insulin or insulin mimetics; (d) incretin and incretin mimetics such as (i) Exenatide available from Amylin Pharmaceuticals, (i) amylin and amylin mimetics such as pramlintide acetate, available as Symlin®, (iii) GLP-1, GLP-1 mimetics, and GLP-1 receptor agonists, (iv) GIP, GIP mimetics and GIP receptor agonists; (e) sulfonylureas and other insulin secretagogues, such as tolbutamide, glyburide, gliclazide, glipizide, glimepiride, meglitinides, and repaglinide; (f) a-glucosidase inhibitors (such as acarbose and miglitol); (g) glucagon receptor antagonists; (h) PACAP, PACAP mimetics, and PACAP receptor agonists; (i) cholesterol lowering agents such as (i) HMG-CoA reductase inhibitors (lovastatin, simvastatin, pravastatin, cerivastatin, fluvastatin, atorvastatin, itavastatin, and rosuvastatin, and other statins), (ii) sequestrants such as cholestyramine, colestipol and
dialkylaminoalkyl derivatives of a cross-linked dextran, (iii) nicotinyl alcohol, nicotinic acid or a salt thereof, (iv) PPARa agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (v) PPARa/γ dual agonists such as muraglitazar (BMS) and tesaglitazar (AstraZeneca), (vi) inhibitors of cholesterol absorption, such as beta-sitosterol and ezetimibe, (vii) acyl CoAxholesterol acyltransferase inhibitors such as avasimibe, and (viii) antioxidants such as probucol; (j) PPAR5 agonists such as GW-501516 from GSK; (k) anti-obesity compounds such as fenfluramine, dexfenfluramine, phentermine, sibutramine, orlistat, neuropeptide Yl or Y5 antagonists, MTP inhibitors, squalene synthase inhibitor, lipoxygenase inhibitor, ACAT inhibitor,
Neuropeptide Cannabinoid CB-1 receptor antagonists, CB-1 receptor inverse agonists and antagonists, fatty acid oxidation inhibitors, appetite suppressants (1) adrenergic receptor agonists, melanocortin receptor agonists, in particular - melanocortin-4 receptor agonists, ghrelin antagonists, and melanin- concentrating hormone (MCH) receptor antagonists; (m) ileal bile acid transporter inhibitors; (n) agents intended for use in inflammatory conditions such as aspirin, non steroidal anti-inflammatory drugs, glucocorticoids, azalfidine, and selective
cyclooxygenase-2 inhibitors; (o) antihypertensive agents such as ACE inhibitors (enalapril, lisinopril, captopril, quinapril, fosinoprol, ramipril, spirapril, tandolapril), angiotensin-II (AT-1) receptor blockers (losartan, candesartan, irbesartan, valsartan, telmisartan, eprosartan), beta blockers and calcium channel blockers; and (p) glucokinase activators (GKAs); (q) agents which can be used for the prevention, delay of progression or treatment of neurodegenerative disorders, cognitive disorders or a drug for improving memory such as antiinflammatory drugs, antioxidants, neuroprotective agents, glutamate receptor antagonists, acetylcholine esterase inhibitors, butyrylcholinesterase inhibitors, MAO inhibitors, dopamine agonists or antagonists, inhibitors of gamma and beta secretases, inhibitors of amyloid aggregation, amyloid beta peptide, antibodies to amyloid beta peptide, inhibitors of acetylcholinesterase, glucokinase activators, agents directed at modulating GAB A, NMD A, cannabinoid, AMP A, kainate, phosphodiesterase (PDE), PKA, PKC, CREB or nootropic systems; ( r ) leukocyte growth promotors intended for the treatment and prevention of reduced bone marrow production, infectious diseases, hormone dependent disorders, inflammatory diseases, HIV, allergies, leukocytopenia, and rheumatism; (s) SGLT2 inhibitor; (t) glycogen phosphorylase inhibitor; (u) aP2 inhibitors; (v) aminopeptidase N inhibitor (w) vasopeptidase inhibitors like neprilysin inhibitors and/or ACE inhibitors or dual NEP/ACE inhibitor; (x) growth hormone secretagogue for enhancing growth hormone levels and for treating growth retardation / dwarfism or metabolic disorders or where the disorder is an injury, or a wound in need of healing, or a mammalian patient recovering from surgery; (y) 5-HT 3 or 5-HT 4 receptor modulators (tegaserod, cisapride, nor-cisapride, renzapride, zacopride, mosapride, prucalopride, buspirone, norcisapride, cilansetron, ramosetron, azasetron, ondansetron, etc.); (Za) aldose reductase inhibitors; (Zb) sorbitol dehydrogenase inhibitors; (Zc) AGE inhibitors; (Zd) erythropoietin agonist such as EPO, EPO mimetics, and EPO receptor agonists. The compounds of the present invention may also be used in combination with GPR40 agonists.
Examples of glucokinase activators that can be used in combination with the compounds of the present invention include those set forth in published PCT patent application no. WO 2009/042435, published April 2, 2009. Examples of specific compounds, or pharmaceutically acceptable salts thereof, disclosed in the published application that may be used in combination with the compounds of the present invention, or pharmaceutically acceptable salts thereof, include compounds selected from:
(S)-l-(5-(5-bromo-3-(2-methylpyridin-3-yloxy)pyridin-2-ylamino)-l,2,4- thiadiazol-3-yl)ethane- 1 ,2-diol;
(S)-l-(5 -(5 -trifluoromethyl-3 -(2-methylpyridin-3 -yloxy)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(S)-l-(5-(5 -phenylthio-3-(2-methylpyridin-3 -yloxy)pyridin-2-ylamino)- 1 ,2,4-thiadiazol-3-yl) ethane- 1 ,2-diol;
(S)-l-(5-(5-phenylthio-3-(pyridin-3-yloxy)pyridin-2-ylamino)-l,2,4- thiadiazol-3-yl)piperidin- 1 -yl)ethane- 1 ,2-diol;
(S)- 1 -(5 -(3 -(2-methylpyridin-3 -yloxy)-5 -(pyridin-2-ylthio)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(S)-l-(5 -(5 (2hydroxyethylthio)-3 -(2-methylpyridin-3 -yloxy)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(S)-l-(5-(4-fluorophenoxy)-5-pyridin-2-ylthio)pyridin-2-ylamino)-l,2,4- thiadiazol-3-yl) ethane- 1 ,2-diol;
(R)- 1 -(2-(5 -bromo-3 -(4-fluorophenoxy)pyridin-2-ylamino)thiazol-4- yl)ethane- 1 ,2-diol;
(S)-l-(2-(5 -bromo-3 -(4-fluorophenoxy)pyridin-2-ylamino)thiazol-4- yl)ethane- 1 ,2-diol;
(R)- 1 -(2-(3-(4-fluorophenoxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)thiazol-4-yl)-ethane- 1 ,2-diol;
( 1 S)- 1 -(5 -(5 -bromo-3 -(5 ,6,7, 8-tetrahydroquinolin-5 -yloxy )pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(S)-l -(5 -(5 -bromo-3 -(l-(2-hydroxy ethyl)- 1 H-pyrazol-4-yloxy )pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(R)- 1-(2-(5 -bromo-3 -(2-methylpyridin-3 -yloxy)pyridin-2- ylamino)thiazol-4-yl)-ethane- 1 ,2-diol;
(S)-l-(5 -(5 -(2-hydroxyethylthio)-3 -(pyridin-3 -yloxy)pyridin-2-ylamino)- 1 ,2,4-thiadiazol-3-yl)ethane- 1 ,2-diol;
(S)-l -(5 -(5 -bromo-3 -(1 -methyl- lH-pyrazol-4-yloxy)pyridin-2-ylamino)- 1 ,2,4-thiadiazol-3-yl)ethane- 1 ,2-diol;
(S)-l-(5-(3-(l-methyl-lH-pyrazol-4-yloxy)-5-(2-methylpyridin-3- ylthio)pyridin-2-ylamino)- 1 ,2,4-thiadiazol-3-yl)ethane- 1 ,2-diol;
(S)-l-(5-(5-(2-methylpyridin-3-ylthio)-3-(l,3,5-trimethyl-lH-pyrazol-4- yloxy)-pyridin-2-ylamino)-l,2,4-thiadiazol-3-yl)ethane-l,2-diol;


-47-

-48 -

-49-

-50-

-51 -

-52-



-55 -

-56-




-60-

-61 -


-63 -

(S)- 1 -(5 -(3 -(2-methylpyridin-3 -yloxy)-5 -(pyridin-2-ylthio)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l ,2-diol;
(S)- 1 -(5 -(3 -(2,6-dimethylpyridin-3 -yloxy)-5 -(pyridin-2-ylthio)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l ,2-diol;
(S)- 1 -(5-(5-(cyclopropylmethylthio)-3-(2-methylpyridin-3-yloxy)pyridin-
2-yl-amino)- 1 ,2,4-thiadiazol-3-yl)ethane- 1 ,2-diol;
(S)-l-(5-(3-(2-ethylpyridin-3-yloxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l ,2-diol;
(S)-l-(5-(5 -(3 -methoxypropylthio)-3 -(2-methylpyridin-3 -yloxy)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l ,2-diol;
(S)-l-(5-(3-(l-Ethyl-lH-pyrazol-5-yloxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l ,2-diol;
(S)- 1 -(5-(3-(l -ethyl- lH-pyrazol-5-yloxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)- 1 ,2,4-thiadiazol-3-yl)-2-methylpropane- 1 ,2-diol;
(S)- 1 -(5-(5-(3-methylpyridin-2-ylthio)-3-(2-methylpyridin-3- yloxy)pyridin-2-yl-amino)-l ,2,4-thiadiazol-3-yl)ethane-l ,2-diol;
(S)-l-(5-(3-(2,4-dimethylpyridin-3-yloxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(S)-2-methyl- 1 -(5-(3-(2-methylpyridin-3-yloxy)-5-(pyridin-2- ylthio)pyridin-2-ylamino)-l,2,4-thiadiazol-3-yl)propane-l,2-diol;
(S)-l-(5-(5 -(2-methoxyethylthio)-3 -(2-methylpyridin-3 -yloxy)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(lS,2S)-l-(5-(3-(2-ethylpyridin-3-yloxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)- 1 ,2,4-thiadiazol-3-yl)-3-methoxypropane- 1 ,2-diol;
(S)-2-methyl -(5-(5-(pyridin-2-ylthio)-3-(l,3,5-trimethyl-lH-pyrazol-4- yloxy)-pyridin-2-ylamino)- 1 ,2,4-thiadiazol-3-yl)propane- 1 ,2-diol;
(S)-l-(5-(5-(pyridin-2-ylthio)-3-(l,3,5-trimethyl-lH-pyrazol-4- yloxy)pyridin-2-yl-amino)-l,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(S)-l-(5-(5-(2-methoxyethylthio)-3-(l,3,5-trimethyl-lH-pyrazol-4- yloxy)pyridin-2-yl-amino)-l,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(R)-l-(5-(3-(2-methylpyridin-3-yloxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)-l ,2,4-thiadiazol-3-yl)ethane-l,2-diol;
(S)-2-(5-(3-(2-methylpyridin-3-yloxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)-l,2,4-thiadiazol-3-yl)propane-l,2-diol; or
(R)-2-(5-(3-(2-methylpyridin-3-yloxy)-5-(pyridin-2-ylthio)pyridin-2- ylamino)-l,2,4-thiadiazol-3-yl)propane-l,2-diol, or the pharmaceutically acceptable salts thereof.
Other compounds that may be used in combination with the compounds of the present invention include the ILl-Rl compounds set forth in U.S. patent no. 7,438,910. A particular disease that can be treated with the combination is type 2 diabetes.
The compounds of the present invention can also be used in combination with FGF-21 compounds, and particularly for the treatment of type 2 diabetes. Examples of FGF-21 compounds are disclosed in U.S. patent no. 7,671,180; U.S. patent no. 7,667,008; U.S. patent no. 7,459,540; U.S. patent no. 7,696,172; PCT application publication no. WO 2010/042747; and PCT application publication no. WO 2009/149171.
The compounds of the present invention can be also be used in combination with anakinra, particularly for the treatment of type 2 diabetes.
In one particular aspect, the compounds of the present invention may be used in combination with metformin.
The compounds of the present invention are used in the treatment diseases or symptoms mediated by GKRP and/or GK (GKRP/GK). Examples of diseases or symptoms mediated by GKRP/GK include, but are not limited to, Type II (type 2) diabetes and related disorders, such as hyperglycemia, low or impaired glucose tolerance, insulin resistance, obesity, lipid disorders such as
dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels, atherosclerosis, and vascular restenosis, irritable bowel syndrome, inflammatory bowel disease, including Crohn's disease and ulcerative colitis, other inflammatory conditions, pancreatitis, abdominal obesity, neurodegenerative disease, retinopathy, nephropathy, neuropathy, cataracts, glaucoma, glomerulosclerosis, foot ulcerations and unlcerative colitis, altered gastrointestinal motility, Syndrome X, ovarian hyperandrogenism, polycystic ovarian syndrome, premenstrual syndrome, other disorders where insulin resistance is a component. In Syndrome X, also known as Metabolic Syndrome, obesity is thought to promote insulin resistance, diabetes, dyslipidemia, hypertension, and increased cardiovascular risk, growth hormone deficiency, neutropenia, neuronal disorders, tumor invasion and metastasis, benign prostatic hypertrophy, gingivitis, osteoporosis, frailty of aging, intestinal injury, benign prostatic hypertrophy (BPH), and sperm motility/male contraception.
The compounds of the present invention are also useful for the prevention, delay of progression or the treatment of an early cardiac or early cardiovascular diseases or damages, renal diseases or damages, heart Failure, or heart Failure associated diseases like (i) cardiovascular diseases or damages e.g. cardiac hypertrophy, cardiac remodeling after myocardial infarction, pulmonary congestion and cardiac fibrosis in dilated or in hypertrophic cardiomyopathy, cardiomyopathy such as dilated cardiomyopathy or hypertrophic
cardiomyopathy, mesanglial hypertrophy, or diabetic cardiomyopathy, left or
right ventricular hypertrophy, arrhythmia, cardiac dysrhythmia, syncopy, angina pectoris, cardiac bypass reocclusion, intermittent claudication, diastolic and/or systolic dysfunction, diabetic myopathy, stroke prevention in congestive heart failure, hypertrophic medial thickening in arteries and/or large vessels, mesenteric vasculature hypertrophy or atherosclerosis, preferably atherosclerosis in mammalian patients with hypertension of diabetes; (ii) renal diseases or damages like renal hyperfiltration such as after portal renal ablation, proteinuria in chronic renal disease, renal arteriopathy as a consequence of hypertension, nephrosclerosis, hypertensive nephrosclerosis or mesanglial hypertrophy; (iii) Heart Failure to be treated is secondary to idiopathic dilated cardiomyopathy and/or coronary ischemic disease.
The compounds of the present invention can also be used for the prevention, the delay of the onset, the delay of progression or the treatment of neurodegenerative disorders, cognitive disorders and for improving memory (both short term and long term) and learning ability wherein the (i)
neurodegenerative disorder is dementia, senile dementia, schizophrenia, mild cognitive impairment, Alzheimer related dementia, Huntington's chores, tardive dyskinesia, hyperkinesias, mania, Morbus Parkinson, Steel-Richard syndrome, Down's syndrome, myasthenia gravis, nerve and brain trauma, vascular amyloidosis, cerebral hamorrhage I with amyloidosis, brain inflammation, Friedrich ataxia, acute confusion disorders, acute confusion disorders with apoptotic necrocytosis, amyotrophic lateral sclerosis, glaucoma, and Alzheimer's disease; (ii) cognitive disorders like cognitive deficits associated with
schizophrenia, age-induced memory impairment, cognitive deficits associated with psychosis, cognitive impairment associated with diabetes, cognitive deficits associated with post-stroke, memory defects associated hypoxia, cognitive and attention deficits associated with senile dementia, attention deficits disorders, memory problems associated with mild cognitive impairment, impaired cognitive function associated with vascular dementia, cognitive problems associated with brain tumors, Pick's disease, cognitive deficits due to autism, cognitive deficits post electroconvulsive therapy, cognitive deficits associated with traumatic brain
injury, amnesic disorders, deliriums, vitamin deficiency, dementias, impaired cognitive function associated with Parkinson's disease, attention-deficit disorders; (iii) prevention of memory impairment as a result of Alzheimer disease, Creutzfeld- Jakob disease, Pick disease, Huntington disease, AIDS, brain injury, brain aneurysm, epilepsy, stroke, toxicant exposure, mental retardation in children, Huntington's disease; (iv) to improve learning speed and potential in educational and rehabilitation contexts.
The compounds of the present invention can also be used for stimulating an immune response in a subject having or at risk of having cancer wherein the cancer is selected from the group consisting of basal cell carcinomas including cancers of the binary tract, bladder, urinary system, bone, brain, breast, cervical, endometrial, ovarian, uterine, choriocarcinoma, central nervous system, colon and rectal cancers, connective tissue cancer, cancer of the digestive system, esophageal, gastric, stomach, larynx, liver, pancreatic, colorectal, renal cancers; cancers of the urinary system; cancers of eye, head and neck, oral cavity, skin, prostate; cancers of biliary tract, testicular, thyroid; intra- epithelial neoplasm, leukemia, acute myeloid leukemia, acute lymphoid leukemia, chronic myeloid leukemia, chronic lymphoid leukemia; and other cancers of the respiratory system, lung, small cell lung, non-small cell lung; lymphoma, Hodgkin's lymphoma, Non-Hodgkin's lymphoma; melanoma, myeloma, neuroblastoma, retinoblastoma, fibrosarcoma (bone or connective tissue sarcoma),
rhabdomyosarcoma; and other cancers including neoplastic conditions, adipose cell tumors, adipose cell carcinomas, such as liposarcoma.
The compounds of the present invention can also be used for the treatment or prophylaxis of chronic inflammatory diseases such as autoimmune disorders like rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, psoriasis, allergies or asthma.
The compounds of the present invention can also be used in the treatment of pain, neuropathic pain, rheumatoid pain, osteoarthritis pain, anesthesia adjunct in mammalian patients undergoing surgery, chronic pain in advanced cancer, treatment of refractory diarrhea, biliary pain caused by gallstones.
The compounds of the present invention can also be used for the treatment of mammalian patients undergoing islet/pancreas transplantation, for the prevention or the delay of transplant rejection, or allograft rejection in transplantation, for improving pancreatic function by increasing the number and size of pancreatic beta-cells in the treatment of Type 1 diabetes patients, and for improving pancreatic function by increasing the number and size of pancreatic beta-cells in general.
Furthermore, the compounds of the present invention can be used for the treatment of mammalian patients with acne, skin disorders (e.g. pigmentation disorders or psoriasis), scleroderma, mycoses; anxiety, anxiety neurosis, major depression disorder, drug abuse, alcohol addiction, insomnia, chronic fatigue, sleep apnea; anorexia nervosa; epilepsy; migraine; encephalomyelitis;
osteoarthritis, osteoporosis, calcitonin-induced osteoporosis; male and female sexual dysfunction, infertility; Type 1 diabetes; immunosuppression, HIV infection; hematopoiesis, anemia; and for weight reduction.
Additionally, the compounds of the present invention are useful for the prevention, delay of progression or treatment of (i) bacterial infections from Escherichia coli, Staphylococcus, Streptoococcus, Pseudomonas, Clostridium difficile infection, Legionella, Pneumococcus, Haemophilus, Klebsiella,
Enterobacter, Citrobacter, Neisseria, Shigella, Salmonella, Listeria, Pasteurella, StreptobaciUus, Spirillum, Treponema, Actinomyces, Borrelia, Corynebacterium, Nocardia, Gardnerella, Campylobacter, Spirochaeta, Proteus, Bacteriodes, Helicobacter pylori, and anthrax infection; (ii) mycobacterial infection from tuberculosis and leprosy; (iii) viral infection from HIV, Herpes simplex virus 1 , Herpes simplex virus 2, Cytomegalovirus, hepatitis A virus, hepatitis B virus, hepatitis C virus, human papilloma virus, Epstein Barr virus, rotavirus, adenovirus, influenza A virus, respiratory syncytial virus, varicella-zoster virus, small pox, monkey pox and SARS; (iv) fungal infection from candidiasis, ringworm, histoplasmosis, blastomycosis, paracoccidioidomycosis,
cryptococcosis, aspergillosis, chromomycosis, mycetoma infections,
pseudallescheriasis, Tinea versicolor infection; (v) parasite infection from
amebiasis, Trypanosoma cruzi, Fascioliasis, Leishmaniasis, Plasmodium, Onchocerciasis, Paragonimiasis, Trypanosoma brucei, Pneumocystis,
Trichomonas vaginalis, Taenia, Hymenolepsis, Echinococcus, Schistosomiasis, neurocysticerosis, Necator americanus, and Trichuris trichuria.
Since one aspect of the present invention contemplates the treatment of the disease/conditions with a combination of pharmaceutically active compounds that may be administered separately, the invention further relates to combining separate pharmaceutical compositions in kit form. The kit comprises two separate pharmaceutical compositions: a compound of the present invention, and a second pharmaceutical compound. The kit comprises a container for containing the separate compositions such as a divided bottle or a divided foil packet.
Additional examples of containers include syringes, boxes and bags. Typically, the kit comprises directions for the use of the separate components. The kit form is particularly advantageous when the separate components are preferably administered in different dosage forms (e.g., oral and parenteral), are
administered at different dosage intervals, or when titration of the individual components of the combination is desired by the prescribing physician or veterinarian.
An example of such a kit is a so-called blister pack. Blister packs are well known in the packaging industry and are being widely used for the packaging of pharmaceutical unit dosage forms (tablets, capsules, and the like). Blister packs generally consist of a sheet of relatively stiff material covered with a foil of a preferably transparent plastic material. During the packaging process recesses are formed in the plastic foil. The recesses have the size and shape of the tablets or capsules to be packed. Next, the tablets or capsules are placed in the recesses and the sheet of relatively stiff material is sealed against the plastic foil at the face of the foil which is opposite from the direction in which the recesses were formed. As a result, the tablets or capsules are sealed in the recesses between the plastic foil and the sheet. Preferably the strength of the sheet is such that the tablets or capsules can be removed from the blister pack by manually applying pressure on
the recesses whereby an opening is formed in the sheet at the place of the recess. The tablet or capsule can then be removed via said opening.
It may be desirable to provide a memory aid on the kit, e.g., in the form of numbers next to the tablets or capsules whereby the numbers correspond with the days of the regimen which the tablets or capsules so specified should be ingested. Another example of such a memory aid is a calendar printed on the card, e.g., as follows "First Week, Monday, Tuesday, . . . etc . . . Second Week, Monday, Tuesday, . . . " etc. Other variations of memory aids will be readily apparent. A "daily dose" can be a single tablet or capsule or several pills or capsules to be taken on a given day. Also, a daily dose of a compound of the present invention can consist of one tablet or capsule, while a daily dose of the second compound can consist of several tablets or capsules and vice versa. The memory aid should reflect this and aid in correct administration of the active agents.
In another specific embodiment of the invention, a dispenser designed to dispense the daily doses one at a time in the order of their intended use is provided. Preferably, the dispenser is equipped with a memory-aid, so as to further facilitate compliance with the regimen. An example of such a memory-aid is a mechanical counter which indicates the number of daily doses that has been dispensed. Another example of such a memory-aid is a battery-powered microchip memory coupled with a liquid crystal readout, or audible reminder signal which, for example, reads out the date that the last daily dose has been taken and/or reminds one when the next dose is to be taken.
The compounds of the present invention and other pharmaceutically active compounds, if desired, can be administered to a patient either orally, rectally, parenterally, (for example, intravenously, intramuscularly, or
subcutaneously) intracisternally, intravaginally, intraperitoneally, intravesically, locally (for example, powders, ointments or drops), or as a buccal or nasal spray. All methods that are used by those skilled in the art to administer a
pharmaceutically active agent are contemplated.
Compositions suitable for parenteral injection may comprise
physiologically acceptable sterile aqueous or nonaqueous solutions, dispersions,
suspensions, or emulsions, and sterile powders for reconstitution into sterile injectable solutions or dispersions. Examples of suitable aqueous and nonaqueous carriers, diluents, solvents, or vehicles include water, ethanol, polyols (propylene glycol, polyethylene glycol, glycerol, and the like), suitable mixtures thereof, vegetable oils (such as olive oil) and injectable organic esters such as ethyl oleate. Proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
These compositions may also contain adjuvants such as preserving, wetting, emulsifying, and dispersing agents. Microorganism contamination can be prevented by adding various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for example, sugars, sodium chloride, and the like. Prolonged absorption of injectable pharmaceutical compositions can be brought about by the use of agents delaying absorption, for example, aluminum monostearate and gelatin.
Solid dosage forms for oral administration include capsules, tablets, powders, and granules. In such solid dosage forms, the active compound is admixed with at least one inert customary excipient (or carrier) such as sodium citrate or dicalcium phosphate or (a) fillers or extenders, as for example, starches, lactose, sucrose, mannitol, and silicic acid; (b) binders, as for example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose, and acacia; (c) humectants, as for example, glycerol; (d) disintegrating agents, as for example, agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain complex silicates, and sodium carbonate; (a) solution retarders, as for example, paraffin; (f) absorption accelerators, as for example, quaternary ammonium compounds; (g) wetting agents, as for example, cetyl alcohol and glycerol monostearate; (h) adsorbents, as for example, kaolin and bentonite; and (i) lubricants, as for example, talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, or mixtures thereof. In the case of capsules, and tablets, the dosage forms may also comprise buffering agents.
Solid compositions of a similar type may also be used as fillers in soft and hard filled gelatin capsules using such excipients as lactose or milk sugar, as well as high molecular weight polyethylene glycols, and the like.
Solid dosage forms such as tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells, such as enteric coatings and others well known in the art. They may also contain opacifying agents, and can also be of such composition that they release the active compound or compounds in a certain part of the intestinal tract in a delayed manner. Examples of embedding compositions that can be used are polymeric substances and waxes. The active compounds can also be in micro-encapsulated form, if appropriate, with one or more of the above-mentioned excipients.
Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs. In addition to the active compounds, the liquid dosage form may contain inert diluents commonly used in the art, such as water or other solvents, solubilizing agents and emulsifiers, as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethylformamide, oils, in particular, cottonseed oil, groundnut oil, corn germ oil, olive oil, castor oil, and sesame seed oil, glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, or mixtures of these substances, and the like.
Besides such inert diluents, the composition can also include adjuvants, such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents. Suspensions, in addition to the active compound, may contain suspending agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar, and tragacanth, or mixtures of these substances, and the like.
Compositions for rectal administration are preferable suppositories, which can be prepared by mixing the compounds of the present invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a
suppository wax, which are solid at ordinary room temperature, but liquid at body temperature, and therefore, melt in the rectum or vaginal cavity and release the active component.
Dosage forms for topical administration of a compound of the present invention include ointments, powders, sprays and inhalants. The active compound or fit compounds are admixed under sterile condition with a physiologically acceptable carrier, and any preservatives, buffers, or propellants that may be required. Opthalmic formulations, eye ointments, powders, and solutions are also contemplated as being within the scope of this invention.
The compounds of the present invention can be administered to a patient at dosage levels in the range of about 0.1 to about 3,000 mg per day. For a normal adult human having a body weight of about 70 kg, a dosage in the range of about 0.01 to about 100 mg per kilogram body weight is typically sufficient. The specific dosage and dosage range that can be used depends on a number of factors, including the requirements of the patient, the severity of the condition or disease being treated, and the pharmacological activity of the compound being administered. The determination of dosage ranges and optimal dosages for a particular patient is within the ordinary skill in the art.
The compounds of the present invention can be administered as pharmaceutically acceptable salts, esters, amides or prodrugs. The term "salts" refers to inorganic and organic salts of compounds of the present invention. The salts can be prepared in situ during the final isolation and purification of a compound, or by separately reacting a purified compound in its free base or acid form with a suitable organic or inorganic base or acid and isolating the salt thus formed. Representative salts include the hydrobromide, hydrochloride, sulfate, bisulfate, nitrate, acetate, oxalate, palmitate, stearate, laurate, borate, benzoate, lactate, phosphate, tosylate, citrate, maleate, fumarate, succinate, tartrate, naphthylate, mesylate, glucoheptonate, lactobionate, and laurylsulphonate salts, and the like. The salts may include cations based on the alkali and alkaline earth metals, such as sodium, lithium, potassium, calcium, magnesium, and the like, as well as non-toxic ammonium, quaternary ammonium, and amine cations
including, but not limited to, ammonium, tetramethylammonium,
tetraethylammonium, methylamine, dimethylamine, trimethylamine,
triethylamine, ethylamine, and the like. See, for example, S. M. Berge, et al., "Pharmaceutical Salts," J Pharm Sci, 66: 1-19 (1977).
Examples of pharmaceutically acceptable esters of the compounds of the present invention include Ci-Cg alkyl esters. Acceptable esters also include C5-C7 cycloalkyl esters, as well as arylalkyl esters such as benzyl. C1-C4 alkyl esters are commonly used. Esters of compounds of the present invention may be prepared according to methods that are well known in the art.
Examples of pharmaceutically acceptable amides of the compounds of the present invention include amides derived from ammonia, primary Ci-Cs alkyl amines, and secondary Ci-Cg dialkyl amines. In the case of secondary amines, the amine may also be in the form of a 5 or 6 membered heterocycloalkyl group containing at least one nitrogen atom. Amides derived from ammonia, C1-C3 primary alkyl amines and C1-C2 dialkyl secondary amines are commonly used. Amides of the compounds of the present invention may be prepared according to methods well known to those skilled in the art.
The term "prodrug" means compounds that are transformed in vivo to yield a compound of the present invention. The transformation may occur by various mechanisms, such as through hydrolysis in blood. A discussion of the use of prodrugs is provided by T. Higuchi and W. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A.C.S. Symposium Series, and in
Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American
Pharmaceutical Association and Pergamon Press, 1987.
To illustrate, if the compound of the invention contains a carboxylic acid functional group, a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as (Ci-Cg alkyl, (C2- Cl2)alkanoyloxymethyl, l-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1 -methyl- l-(alkanoyloxy)ethyl having from 5 to 10 carbon atoms,
alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1- (alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1 -methyl- 1-
(alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N- (alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1-(N- (alkoxycarbonyl)aminomethyl having from 4 to 10 carbon atoms, 3-phthalidyl, 4- crotonolactonyl, gamma-butyrolacton-4-yl, di-N,N-(C i -C2)alkylamino(C2- C3)alkyl (such as β-dimethylaminoethyl), carbamoyl-(Ci-C2)alkyl, N,N-di(C1- C2)alkylcarbamoyl-(Ci-C2)alkyl and piperidino-, pyrrolidino- or morpholino(C2- 3)alkyl.
Similarly, if a compound of the present invention comprises an alcohol functional group, a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as (Ci-C6)alkanoyloxymethyl, 1- ((Ci-C6)alkanoyloxy)ethyl, 1 -methyl- l-((Ci-C6)alkanoyloxy)ethyl, (Ci- C6)alkoxycarbonyloxymethyl, N-(C i -C6)alkoxycarbonylaminomethyl, succinoyl, (Ci-C6)alkanoyl, a-amino(Ci-C4)alkanoyl, arylacyl and a-aminoacyl, or a- aminoacyl-a-aminoacyl, where each a-aminoacyl group is independently selected from the naturally occurring L-amino acids, -P(0)(OH)2, -P(0)(0(Ci-C6)alkyl)2 or glycosyl (the radical resulting from the removal of a hydroxyl group of the hemiacetal form of a carbohydrate).
The compounds of the present invention may contain asymmetric or chiral centers, and therefore, exist in different stereoisomeric forms. It is contemplated that all stereoisomeric forms of the compounds as well as mixtures thereof, including racemic mixtures, form part of the present invention. In addition, the present invention contemplates all geometric and positional isomers. For example, if the compound contains a double bond, both the cis and trans forms (designated as S and E, respectively), as well as mixtures, are
contemplated.
Mixture of stereoisomers, such as diastereomeric mixtures, can be separated into their individual stereochemical components on the basis of their physical chemical differences by known methods such as chromatography and/or fractional crystallization. Enantiomers can also be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., an alcohol), separating the
diastereomers and converting (e.g., hydrolyzing) the individual diastereomers to the corresponding pure enantiomers. Also, some compounds may be
atropisomers (e.g., substituted biaryls).
The compounds of the present invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water (hydrate), ethanol, and the like. The present invention contemplates and encompasses both the solvated and unsolvated forms.
It is also possible that compounds of the present invention may exist in different tautomeric forms. All tautomers of compounds of the present invention are contemplated. For example, all of the tautomeric forms of the tetrazole moiety are included in this invention. Also, for example, all keto-enol or imine- enamine forms of the compounds are included in this invention.
Those skilled in the art will recognize that the compound names and structures contained herein may be based on a particular tautomer of a compound. While the name or structure for only a particular tautomer may be used, it is intended that all tautomers are encompassed by the present invention, unless stated otherwise.
It is also intended that the present invention encompass compounds that are synthesized in vitro using laboratory techniques, such as those well known to synthetic chemists; or synthesized using in vivo techniques, such as through metabolism, fermentation, digestion, and the like. It is also contemplated that the compounds of the present invention may be synthesized using a combination of in vitro and in vivo techniques.
The present invention also includes isotopically-labeled compounds, which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2H, 3H, 13C, 14C, 15N, 160, 170, 31P, 32P, 35S, 18F, and 36C1. In another aspect, the
compounds of the present invention contain one or more deuterium atoms (2H) in place of one or more hydrogen atoms.
Compounds of the present invention that contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of this invention. Certain isotopically-labeled compounds of the present invention, for example those into which radioactive isotopes such as 3H and 14C are
incorporated, are useful in drug and/or substrate tissue distribution assays.
Tritiated, i.e., 3H, and carbon-14, i.e., 14C, isotopes are particularly preferred for their ease of preparation and detection. Further, substitution with heavier isotopes such as deuterium, i.e., 2H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some
circumstances. Isotopically labeled compounds of this invention can generally be prepared by substituting a readily available isotopically labeled reagent for a non- isotopically labeled reagent.
The compounds of the present invention may exist in various solid states including crystalline states and as an amorphous state. The different crystalline states, also called polymorphs, and the amorphous states of the present compounds are contemplated as part of this invention.
In synthesizing compounds of the present invention, it may be desirable to use certain leaving groups. The term "leaving groups" ("LG") generally refer to groups that are displaceable by a nucleophile. Such leaving groups are known in the art. Examples of leaving groups include, but are not limited to, halides (e.g., I, Br, F, CI), sulfonates (e.g., mesylate, tosylate), sulfides (e.g., SCH3), N- hydroxsuccinimide, N-hydroxybenzotriazole, and the like. Examples of nucleophiles include, but are not limited to, amines, thiols, alcohols, Grignard reagents, anionic species (e.g., alkoxides, amides, carbanions) and the like.
All patents and other publications recited herein are hereby incorporated by reference in their entirety.
The examples presented below illustrate specific embodiments of the present invention. These examples are meant to be representative and are not intended to limit the scope of the claims in any manner.
EXAMPLES
BIOLOGICAL ASSAYS
GKRP LC MS/MS Biochemical Assay
This assay is used to directly measure the formation of 13C-glucose-6- phosphate from 13C-glucose by liquid chromatography-mass spectrometry (LC MS/MS). Begin by preparing the following solutions: Compound Buffer (CB): 50mM Tris, pH 7.5 / 4mM MgCl2 / 6% DMSO / fresh lOmM DTT from 1M frozen stock. Enzyme Buffer (EB): 50mM Tris, pH 7.5 / 4mM MgCl2 / 6% DMSO / fresh 0.1% BSA / fresh 0.01% Brij-35 (10% BSA and 1% Brij-35 stock). GK (Glucokinase) Working Stock (5X): Dilute human His-hepatic GK to 30nM in EB buffer. Substrate Working Stock (1.47X): Dilute 13C-D-glucose (Sigma-Aldrich, St. Louis, MO) to 7.35mM from 1M stock (1M 13C-D-glucose = 186.1 1 mg/ml in 50mM Tris pH 7.5, 4mM MgCl2) and dilute ATP (EMD Chemical Inc., Gibbstown, NJ) to 0.3528mM from frozen lOOmM stock and dilute 20mM fructose-6-phosphate (F6P) (Sigma-Aldrich, St. Louis, MO) to 441 μΜ in CB buffer. GKRP (Glucokinase Regulatory Protein) (1 OX): Dilute GKRP to Ι μΜ from 33.366mM stock in EB buffer. Combine the following reagents in a 96-well polypropylene plate: 34μ1 of Substrate Working Stock (1.47X), 5μ1 of Ι μΜ GKRP (10X), and Ι μΐ of compound or DMSO. Seal the plate and incubate for 30 minutes at room temperature while mixing. After 30 minutes add ΙΟμΙ of GK Working Stock (5X). Re-seal the plate and incubate for another 30 minutes at room temperature while mixing. After the second 30 minutes, stop the reaction by the addition of 50μ1 of 100% acetonitrile, seal, and mix for 5-10 minutes. Run ΙΟμΙ of this sample through the LC MS/MS (API 3200, Applied Biosystems Inc., Carlsbad, CA). Detection settings are for 265.2/78.8 atomic mass units.
GKRP NADPH Coupled Assay
This assay is used as an indirect measure of glucose-6-phosphate (G6P) formed from glucose due to the enzymatic activity of glucokinase. Assay format is the same as for GKRP LC MS/MS Biochemical Assay with the following exceptions. GK Working Stock (5X): Dilute human His-hepatic GK to 20nM in EB buffer. Stop & Detection Reagent (2X): Dilute β-nicotinamide adenine dinucleotide phosphate sodium salt (β-NADP) (Aldrich-Sigma, St. Louis, MO) to 2mM from lOOmM stock ( stock in lOmM Tris, pH 9.2) and dilute glucoses- phosphate dehydrogenase (Aldrich-Sigma, St. Louis, MO) to 0.04υηή7μ1 from lOUnit/μΙ ( stock in lOmM Tris pH 7.5 / 0.05% Brij-35) in 0.2 M Tris, pH 9.2 / 8% DMSO. After the initial 30 minute incubation add ΙΟμΙ of 20nM GK diluted in EB (5X). Re-seal the plate and incubate for another 1 hour at room
temperature while mixing. After 1 hour remove the seal and add 50μ1 of Stop & Detection Reagent and incubate for 5 minutes at room temperature while mixing. After 5 minutes read the plate using an Infinite M1000 (Tecan Systems Inc., San Jose CA) with the following detection settings: Mode: Fluorescence Top
Reading, Excitation Wavelength: 340nm, Emission Wavelength: 450nm, Excitation Bandwidth: 20nm, Emission Bandwidth: 20nm, Gain: 95, Number of Flashes: 10, Flash Frequency: 400Hz, Integration Time: 20μ8.
GK-GKRP Binding Assay Protocol
This assay is used to directly measure the interaction between glucokinase (GK) and glucokinase regulatory protein (GKRP). Begin by preparing the following solutions. Assay Buffer: 20mM Tris, pH 7.5 / 0.05% BSA / ImM DTT / ΙμΜ sorbitol-6-phosphate. Assay Procedure: Dilute avi-tagged GKRP to 10.7 nM in assay buffer. Combine the following reagents in a white 96-well half area plate. Pipette 14μ1 of the diluted avi-tagged GKRP into each well. Add Ιμΐ of compound to be tested and incubate at room temperature for 20 minutes. After 20 minutes, add 5μ1 of assay buffer containing 6nM GK-fluorescein. Add ΙΟμΙ
of AlphaScreen beads (Perkin Elmer, Waltham MA) that have been diluted 1 :333 in assay buffer. Incubate in a dark room for 2 hours at room temperature. After 2 hours read the plate using an Envision plate reader (Perkin Elmer, Waltham MA).
GKRP LC MS/MS-2 Biochemical Assay
This assay is used to directly measure the formation of 13C-glucose-6- phosphate from 13C-glucose by LC MS/MS. Begin by preparing the following solutions: Compound Buffer (CB): 50mM Tris, pH 7.5 / 4mM MgCl2 / 6% DMSO / fresh lOmM DTT from 1M frozen stock. Enzyme Buffer (EB): 50mM Tris, pH 7.5 / 4mM MgCl2 / 6% DMSO / fresh 0.1% BSA / fresh 0.01% Brij-35 (10% BSA and 1% Brij-35 stock). GK (Glucokinase) Working Stock (5X): Dilute human His-hepatic GK to 30nM in EB buffer. Substrate Working Stock (1.47X): Dilute 13C-D-glucose (Sigma- Aldrich, St. Louis, MO) to 7.35mM from 1M stock and dilute ATP (EMD Chemical, Gibbstown, NJ) to 0.3528mM from frozen lOOmM stock in CB buffer (1M 13C-D-glucose = 186.1 1 mg/ml in water). Dilute 20mM fructose-6-phosphate (F6P) (Sigma- Aldrich, St. Louis, MO) to 441 μΜ in the substrate working stock. GKRP (Glucokinase Regulatory Protein) (10X): Dilute GKRP to 280nM from 33.366mM stock in EB buffer. Combine the following reagents in a 96-well polypropylene plate: 34μΙ^ of Substrate Working Stock (1.47X), 5μ1 of 280nM GKRP (10X), and Ι μΐ of compound or DMSO. Seal the plate and incubate for 30 minutes at room temperature while mixing. After 30 minutes add ΙΟμΙ of GK Working Stock (5X). Re-seal the plate and incubate for another 30 minutes at room temperature while mixing. After the second 30 minutes, stop the reaction by the addition of 50μ1 of 100% acetonitrile, seal, and mix for 5-10 minutes. Run ΙΟμΙ of this sample through the LC MS/MS (API 3200, Applied Biosystems, Carlsbad, CA ). Detection settings are for 265.2/78.8 atomic mass units.
Results for compounds tested in these biological assays are set forth in the numbered examples below.
The following abbreviations may be used herein:
HATU [dimethylamino(triazolo [4,5 -b]pyridin-3 - yloxy)methylidene]-dimethylazanium
Hunig's base or DIPEA diisopropylethylamine
dppf Ι,Γ- bis(diphenylphosphanyl) ferrocene dba 1 ,5-diphenylpenta- 1 ,4-dien-3-one
LAH lithium aluminum hydride
TMS trimethylsilyl
EDC 3-(ethyliminomethyleneamino)-N,N-dimethyl- propan-1 -amine
HOBt 1 -hydroxybenzotriazole
TLC thin layer chromatography
MHz megahertz
br. broad
s singlet
d doublet
t triplet
dt doublet of triplet
dd doublet of doublet
quin quintuplet
q quartet
about
+ve or pos. ion positive ion
Δ heat
Ac acetyl
AcOH acetic acid
Ac20 acetic anhydride
ACN acetonitrile
A-phos, Am-Phos (bis[4-di-tert-butylphosphino)-N,N-dimethylaniline] palladium dichloride)
aq or aq. aqueous
ATP adenosine 5 '-triphosphate
BOC or Boc tert-butyloxycarbonyl
BOCOBOC or
BOC-O-BOC ditert-butyl dicarbonate
Bu butyl
Bn benzyl
B(pin)2 bis(pinacolato)diboron
Calcd or Calc'd calculated
cat or cat. catalytic
cone. concentrated
DCE 1 ,2-dichloroethane
DCM dichloromethane
DEA diethylamine
Dess-Martin Periodinane 1,1,1 -triacetoxy- 1 , 1 -dihydro-
(DMP) l,2-benziodoxol-3(lH)-one
DIBALH Diisobutylaluminum hydride
DIEA diisopropylethylamine
DMAP 4-dimethylaminopyridine
DME dimethoxyl ethyl ether
DMF N,N-dimethylformamide
DMSO dimethyl sulfoxide
DTT dithiothreitol
ESI or ES electrospray ionization
Et ethyl
Et20 diethyl ether
Et3N triethylamine
EtOAc ethyl acetate
EtOH ethyl alcohol
FBS fetal bovine serum
g grams
h hour
HC02H formic acid
Hex hexanes
HOAc acetic acid
HPLC high pressure liquid chromatography
IPA or iPrOH isopropyl alcohol
'PrMgCl Isopropyl magnesium chloride iPr2NEt N-ethyl diisopropylamine
KOAc potassium acetate
LC MS, LCMS, LC-MS or
LC/MS liquid chromatography mass spectroscopy
LDA lithium diisopropylamide
LHMDS or LiHMDS lithium hexamethyldisilazide
LiTMP lithium tetramethylpiperidide
m/z mass divided by charge
mCPBA or MCPBA m-chloroperoxybenzoic acid
Me methyl
MeCN acetonitrile
Mel iodomethane
MeOD deuterated methyl alcohol
MeOH methyl alcohol
mg milligrams
min minutes
mL milliliters
MS mass spectra
MsCl mesylchloride
NaBH4 sodium borohydride
NaHMDS sodium hexamethyldisilazide
NaOtBu sodium tert-butoxide
NBS N-bromosuccinimide
NIS N-iodosuccinimide
n-BuLi n-butyllithium
NMO N-methylmorpholine-N-oxide
NMP l-methyl-2-pyrrolidinone
NMR nuclear magnetic resonance
Pd2dba3 tris(dibenzylideneacetone)dipalladium(0)
PMB paramethoxybenzyl
PPA polyphosphoric acid
PTSA, p-TSA, TSOH para-toluenesulfonic acid; 4-methylbenzenesulfonic acid pyr pyridine
RT or rt room temperature
RuPhos 2-dicyclohexyl(2',6'-diisopropoxybiphenyl-2- yl)phosphine
RuPhos Palladacycle chloro(2-dicyclohexylphosphino-2',6'-di-i- propoxy- 1 , 1 '-biphenyl)[2-(2- aminoethylphenyl)]palladium(II), methyl-t- butylether adduct
Sat. or sat'd or satd saturated
SFC supercritical fluid chromatography
TESTf triethylsilyltrifluoromethane sulfonate
TFA trifluoro acetic acid
TfOH trifluoromethanesulfonic acid
THF tetrahydrofuran
Ti(0-iPr)4 titanium isopropoxide
TPAP tetrapropylammonium perruthenate
Tris tris(hydroxymethyl)aminomethane
Ts, Tos or TOS tosyl
xantphos
(9,9-dimethyl-9H-xanthene-4,5-
diyl)bis(diphenylphosphine)
X-Phos 2-Dicyclohexylphosphino-2 ' ,4 ' ,6 ' -triisopropyl- 1,1 '- biphenyl
X-Phos palladacycle chloro(2-dicyclohexylphosphino-2',4',6'-triisopropyl- 1 , - biphenyl)[2-(2-aminoethyl)phenyl)]palladium(II) Johnphos 2-biphenylyl(di-tert-butyl)phosphane
BINAP 1 , 1 '-binaphthalene-2,2'- diylbis(diphenylphosphane)
NCS 1 -chloro-2,5-pyrrolidinedione
Rochelle's salt potassium sodium tartrate
XtalFluor-E (diethylamino)difluorosulfonium
tetrafluoroborate
NIS 1 -iodo-2,5-pyrrolidinedione
It is noted that when a percent (%) is used with regard to a liquid, it is a percent by volume with respect to the solution. When used with a solid, it is the percent with regard to the solid composition. Throughout the Examples,
chromatography columns are used for separations and purifications. Below are some representative suppliers of columns: Phenomenex, Torrance,CA (e.g.,
Gemini); Diacel Inc., Fort Lee, NJ (e.g.,Chiralcel®, Chiralpak®); Teledyne Isco,
Lincoln, NE (e.g., RediSep®) and Krackeler Scientific, Albany, NY (e.g.,
AccuBOND). Also, Teflon® is polyfluoroethylene, DuPont, Wilmington, DE
General Synthetic Schemes Scheme 1

The cross coupling (Za or Zb is a halogen or a metal salt of B, Zn, Mg, etc.) can be achieved using a transition metal catalyst (e.g., a salt of Pd, Ni, etc.) in either anhydrous (e.g., tetrahydrofuran, dioxane, etc.) or an aqueous (e.g., tetrahydrofuran-water, dioxane-water, etc.) solvent under an inert atmosphere (e.g., N2, Ar, etc.). The coupling of the heterocycle with the sulfinylimine can be executed by deprotonation of the heterocycle with an strong base (e.g., alkyllithium, amide, etc.), followed by exposure to the sulfinylimine, in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.). The sulfmamide can be converted to the sulfonamide by deprotection of the sulfmamide with acid (e.g., HC1, TFA, etc.) in either anhydrous (e.g., methanol, diethyl ether, etc.) or aqueous (e.g., methanol-water, diethyl ether- water, etc.) solvent followed by sulfonylation with a sulfonyl chloride in the presence of an amine base (e.g., triethylamine, diisopropylethylamine, etc.) in an anhydrous solvent (e.g., N,N- dimethylformamide, dichloromethane, etc.).
Scheme 2

The coupling of the heterocycle with the sulfinylimine can be executed by deprotonation of the heterocycle with a strong base (e.g., alkyllithium, amide, etc.), followed by exposure to the sulfinylimine in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.). The sulfinamide can be converted to the sulfonamide by deprotection of the sulfinamide with an acid (e.g., HC1, TFA, etc.) in either anhydrous (e.g., methanol, diethyl ether, etc.) or aqueous (e.g., methanol-water, diethyl ether-water, etc.) solvent followed by sulfonylation with a sulfonyl chloride in the presence of an amine base (e.g., triethylamine, diisopropylethylamine, etc.) in an anhydrous solvent (e.g., N,N- dimethylformamide, dichloromethane, etc.). The cross coupling (Za or Zb is a halogen or a metal salt of B, Zn, Mg, etc.) can be achieved using a transition metal catalyst (e.g., a salt of Pd, Ni, etc.) in either anhydrous (e.g.,
tetrahydrofuran, dioxane, etc.) or an aqueous (e.g., tetrahydrofuran-water, dioxane-water, etc.) solvent under an inert atmosphere (e.g., N2, Ar, etc.).
Scheme 3

deprotonation of R2 with an strong base (e.g., alkyllithium, amide, etc.), followed by exposure to the sulfonylimine, in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.).
Scheme 4

The cross coupling (Za or Zb is a halogen or a metal salt of B, Zn, Mg, etc.) can be achieved using a transition metal catalyst (e.g., a salt of Pd, Ni, etc.) in either anhydrous (e.g., tetrahydrofuran, dioxane, etc.) or aqueous (e.g., tetrahydrofuran-water, dioxane-water, etc.) solvent under an inert atmosphere (e.g., N2, Ar, etc.). The coupling of the heterocycle with the sulfonylimine can be executed by deprotonation of the heterocycle with an strong base (e.g., alkyllithium, amide, etc.), followed by exposure to the sulfonylimine, in an anhydrous solvent (e.g., tetrahydrofuran, toluene, etc.).
Intermediate XI
2-(2-C hloro-4-pyridinyl)- 1, 1 ,l-trifluoro-2-pr opanol

A mixture of l-(2-chloro-4-pyridinyl)ethanone (14.8 g, 95.0 mmol, Maybridge, Tintagel, United Kingdom) and trimethyl(trifluoromethyl)silane (18.3 mL, 124 mmol) in DME (100 mL) was cooled until the inside temperature of the reaction mixture reached -78 °C. Cesium fluoride (0.867 g, 5.71 mmol) was added, and the reaction mixture was allowed to warm to room temperature. 10% Aqueous HCI (200 mL) was added and the mixture was stirred at room temperature for 20 h. Water (300 mL) was added, and the resulting mixture was extracted with Et20 (4 x 150 mL). The combined organic layers were washed with brine (200 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 25% EtOAc/hexanes) to deliver 2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (19.0 g, Intermediate XI) as a light-yellow solid.
Intermediate XI (Alternate Synthesis) 2-(2-Chloro-4-pyridinyl)-l,l,l-trifluoro-2-propanol

Intermediates X2 and X3
(2R)-2-(2-Chloro-4-pyridinyl)-l,l,l-trifluoro-2-propanol and (2S)-2-(2- chloro-4-pyridinyl)-l,l,l-trifluoro-2-propanol

Racemic 2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol
(Intermediate XI) was resolved using preparative SFC (Chiralcel® OJ-H column) (two 250 mm x 30 mm, 5 μιη columns in series) eluting with 90% liquid C02 in 10% mixture of hexanes/isopropanol (75:25 v/v) containing 20 mM ammonia in methanol at a flow rate of 120 mL/min) to give two products in greater than 98% enantiomeric excess.
The absolute configurations at the chiral carbinol center of these two enantiomeric products were determined via vibrational circular dichroism studies (See: Stephens, P.J.; Devlin, F.J.; Pan, J.-J. Chirality 2008, 20, 643-663).
1. Computational: The conformational ensemble of the enantiomer with the (S)- configuration was initially generated via stochastic search with the MMFF94 molecular mechanics force field as implemented in the MOE (2008.10) package (Chemical Computing Group, Montreal, CA). This was followed by geometry optimization, vibrational frequency, and VCD rotational strength determination of the structurally unique conformers using the B3PW91 density functional and the 6-31 lG(2d,2p) basis set as implemented in the Gaussian 03 quantum chemical program system (Gaussian, Inc., Wallingford, CT). The resultant VCD line spectra were convolved utilizing a Lorentzian lineshape function (4 cm"1
half-width), and summed over the conformational ensemble based on the relative free energies (and thus population) of each conformer.
2. Experimental: VCD spectra of each enantiomer were measured on a
Biotools/BoMem ChirallR instrument (Biotools, Inc., Jupiter, FL) at
concentrations of about 30 mg/mL in CDCI3 in a 100 μΜ BaF2 cell over a 4 h acquisition time at 4 cm"1 resolution.
Comparison of the predicted and experimental VCD spectra (from 1. and 2. above, respectively) afforded the absolute configuration assignment.
First eluting peak (peak #1): (2R)-2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (Intermediate X2).
Second eluting peak (peak #2): (2S)-2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (Intermediate X3).
Intermediate X4
-(l-Benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol

A mixture of (2R)-2-(2-chloro-4-pyridinyl)-l,l,l-trifluoro-2-propanol (3.00 g, 13.3 mmol, Intermediate X2), 2-(l-benzothiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (6.23 g, 23.9 mmol, Intermediate Y2), dicyclohexyl(2',4',6'-tris(l -methylethyl)-2-biphenylyl)phosphane (X-Phos) (0.697 g, 1.46 mmol), chloro(2-propen-l-yl)palladium dimer (0.122 g, 0.332 mmol), and sodium carbonate (4.23 g, 39.9 mmol) in 1 ,4-dioxane/water (4: 1, 40 mL) was stirred at 80 °C for 2 h. The mixture was allowed to cool to room temperature, H20 (300 mL) was added, and the resulting mixture was extracted with EtOAc (2 x 350 mL). The combined organic layers were washed with brine (200 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was
purified by flash chromatography (100 g of silica gel, 25% EtOAc/hexanes) and again by flash chromatography (150 g of silica gel, 10% EtOAc/hexanes) to afford (2R)-2-(2-( 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol (2.8 g, Intermediate X4) as a light-yellow solid. Intermediates X5 and X6
(2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)- 1 , 1 ,l-trifluoro-2-propanol and (2R)-2-(2-(2-((S)-amino(2- chlorophenyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2- propanol

Step 1. n-Butyllithium (8.44 mL of a 1.6 M solution in hexanes, 13.5 mmol) was added to a solution of (2R)-2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (1.82 g, 5.63 mmol, Intermediate X4) in THF (27 mL) at -78 °C and the mixture was stirred at -78 °C for 10 min. A solution of N- ((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide
(Intermediate Yl) (2.06 g, 8.44 mmol) in THF (8 mL) was added dropwise via a cannula and the resulting mixture was stirred at -78 °C for 2 h. Saturated aqueous
NH4CI (4 mL) was added and the mixture was allowed to warm to room temperature. The mixture was partitioned between aqueous NH4CI (25 mL of saturated NH4CI and 25 mL of H20) and EtOAc (50 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure. The resulting yellow oil was absorbed onto silica gel and purified by flash chromatography (100 g of silica gel, 10% to 60%>
EtOAc/hexanes) and again by flash chromatography (120 g of silica gel, 30% to 60% EtOAc/hexanes) to deliver (S)-N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfmamide (1.87 g) as a white foam. 280 mg of the minor isomer was further purified by flash chromatography (14 g of silica gel, 33% to 50% EtOAc/hexanes) to deliver (S)-N-((S)-(2-chlorophenyl)(7-(4-((lR)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfmamide (220 mg) as a colorless solid.
Step 2a. Hydrogen chloride (8.16 mL of a 2 M solution in diethyl ether, 16.3 mmol) was added to a solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-((lR)- 2,2,2-trif uoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (1.85 g, 3.26 mmol) in Et20 (24 mL) and the resulting suspension was stirred at room temperature for 18 h. The solid was collected by filtration and washed with Et20 (20 mL). The solid was dissolved in 2 M NH4 in MeOH/DCM, absorbed onto silica gel, and purified by flash chromatography (lOOg of silica gel, 30%> to 70%> EtOAc/hexanes) to deliver (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (1.34 g, Intermediate X5) as a white solid.
Step 2b. Hydrogen chloride (0.97 mL of a 4.0 M solution in 1,4-dioxane, 3.9 mmol) was added to a stirring solution of (S)-N-((S)-(2-chlorophenyl)(7-(4- (( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (0.22 g, 0.39 mmol) and methanol at
room temperature. After 10 min, the reaction mixture was concentrated under a vacuum, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate and the layers were separated. The organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), and filtered. The filtrate was concentrated to give (2R)-2-(2-(2- ((S)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol (0.16 g, Intermediate X6) as an off-white solid. This material was used without further purification.
Intermediate X7
(2 -2-(2-(l-Benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol

A mixture of (2S)-2-(2-chloro-4-pyridinyl)-l,l,l-trifluoro-2-propanol (1.07 g, 4.74 mmol, Intermediate X3), 2-(l-benzothiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (2.10 g, 8.06 mmol, Intermediate Y2), dicyclohexyl(2',4',6'-tris(l-methylethyl)-2-biphenylyl)phosphane (226 mg, 0.474 mmol), chloro(2-propen-l-yl)palladium dimer (87 mg, 0.237 mmol), and sodium carbonate monohydrate (1.76 g, 14.2 mmol) in 1 ,4-dioxane/water (4: 1, 10 mL) was stirred at 80 °C for 7 h and then at room temperature for 14 h. The mixture was concentrated under reduced pressure. The resulting residue was dissolved in MeOH/DCM, absorbed onto silica gel and purified by flash chromatography (100 g of silica gel, 10% to 30% EtOAc/hexanes) and again by flash chromatography (100 g of silica gel, 10% to 30% EtOAc/hexanes) to deliver (2S)-2-(2-(l- benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol (769 mg,
Intermediate X7) as a colorless oil.
Intermediates X8 and X9
(2S)-2-(2-(2-((R)-Amino(2-chlorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol and (2S)-2-(2-(2-((S)-amino(2- chlorophenyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2- propanol

n-Butyllithium (3.21 mL of a 1.6 M solution in hexanes, 5.14 mmol) was added to a solution of (2S)-2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 - trifluoro-2-propanol (755 mg, 2.34 mmol, Intermediate X7) in THF (2 mL) at - 78 °C and the mixture was stirred at -78 °C for 30 min. A solution of N-((S,E)- (2-chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (Intermediate Y 1 ) (740 mg, 3.04 mmol) in THF (1 mL) at -78 °C was added dropwise via a cannula and the resulting mixture was stirred at -78 °C for 3 h. Saturated aqueous NH4C1 (2 mL) was added, and the mixture was allowed to warm to room temperature. The mixture was partitioned between aqueous NH4C1 (10 mL of saturated NH4C1 and 10 mL of H20) and EtOAc (20 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure. The resulting yellow oil was purified by flash chromatography (80 g of silica gel, 10% to 60% EtOAc/hexanes) and again by flash chromatography (25 g of silica
gel, 30% to 60% EtOAc/hexanes) to deliver (S)-N-((R)-(2-chlorophenyl)(7-(4- (( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (674 mg) as a light-yellow foam and (S)- N-((S)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfinamide (85 mg) as a light-yellow foam.
Step 2a. Hydrogen chloride (2.90 mL of a 2 M solution in Et20, 5.80 mmol) was added to a solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfinamide (658 mg, 1.16 mmol) in Et20 (8 mL) and the resulting suspension was stirred at room temperature for 6 h. The solid was removed by filtration, washed with Et20 (10 mL), and dried under reduced pressure to deliver a yellow solid. The solid was dissolved in 2 M NH4 in MeOH/DCM, absorbed onto silica gel, and purified by flash chromatography (100 g of silica gel, 30% to 70% EtOAc/hexanes) to deliver (2S)-2-(2-(2-((R)- amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol (505 mg, Intermediate X8) as a white foam.
Step 2b. Hydrogen chloride (261 of a 2 M solution in Et20, 0.522 mmol) was added to a solution of (S)- N-((S)-(2-chlorophenyl)(7-(4-((lS)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfinamide (74 mg, 0.130 mmol) in Et20 (1 mL) and the resulting suspension was stirred at room temperature for 6 h. The solid was removed by filtration, washed with Et20 (10 mL) and dried under reduced pressure to deliver (2S)-2-(2-(2-((S)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol hydrochloride (58 mg, Intermediate X9) as a light-yellow solid.
Intermediate X10
(S)-N-((lE)-(3-Chloro-2-pyridinyl)methylidene)-2-methyl-2- propanesulflnamide

Diego, CA) was added to a mixture of (S)-2-methyl-2-propanesulfinamide (2.40 g, 19.8 mmol, AK Scientific, Mountain View, CA) and copper(II) sulfate (6.77 g, 42.4 mmol) in DCM (50 mL) and the mixture was stirred at room temperature for 2 h. The mixture was filtered, the filter residue was washed with DCM (30 mL), and the combined filtrate and washing were concentrated under reduced pressure. The residue was dissolved in EtOAc (100 mL) and filtered. The filter residue was washed with EtOAc (100 mL), and the combined filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 100% EtOAc) to afford (S)-N-((lE)-(3-chloro-2- pyridinyl)methylidene)-2-methyl-2-propanesulfinamide (2.5 g, Intermediate X10) as a light-yellow oil.
Intermediate XI 1
2-(2-(2-((R)-Amino(2-chlorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l,3,3,3-hexafluoro-2-propanol hydrochloride



Step 1. Trimethyl(trifiuoromethyl)silane (2.52 mL, 17.1 mmol) was added to a mixture of 2-chloro-4-pyridinecarbonyl chloride (1.00 g, 5.68 mmol, Sigma- Aldrich, St. Louis, MO) and tetramethylammonium fluoride (1.59 g, 17.1 mmol) in DME (15 mL) at -78 °C and the mixture was stirred for 17 h while it warmed to room temperature. The mixture was cooled to -78 °C, tetramethylammonium fluoride (0.529 g, 5.68 mmol) followed by trimethyl(trifiuoromethyl)silane (0.840 mL, 5.68 mmol) was added, and the mixture was stirred for 4 h while it warmed to room temperature. The mixture was cooled to 0 °C, saturated aqueous NaHC03 (20 mL) was added, and the mixture was extracted with DCM (3 x 30 mL). The combined organic layers were dried (MgS04), filtered, and
concentrated under reduced pressure. The resulting yellow oil was purified by flash chromatography (100 g of silica gel, 10% to 30% EtOAc/hexanes) to
deliver 2-(2-chloro-4-pyridinyl)-l, 1,1,3, 3,3-hexafluoro-2-propanol (1.29 g) as an off-white solid.
Step 2. A mixture of 2-(2-chloro-4-pyridinyl)-l,l,l,3,3,3-hexafluoro-2- propanol (1.27 g, 4.55 mmol), 2-(l-benzothiophen-7-yl)-4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (2.01 g, 7.73 mmol, Intermediate Y2), dicyclohexyl(2',4',6'- tris(l-methylethyl)-2-biphenylyl)phosphane (108 mg, 0.227 mmol), chloro(2- propen-l-yl)palladium dimer (41.6 mg, 0.114 mmol), and sodium carbonate monohydrate (204 mg, 13.7 mmol) in 1 ,4-dioxane/water (4: 1, 10 mL) was stirred at 80 °C for 5 h. Dicyclohexyl(2^4^6'-tris(l-methylethyl)-2- biphenylyl)phosphane (108 mg, 0.227 mmol) and chloro(2-propen-l- yl)palladium dimer (41.6 mg, 0.114 mmol) were added and the mixture was stirred at 80 °C for 2 h. The mixture was allowed to cool to room temperature, diluted with MeOH/DCM, absorbed onto silica gel, and purified by flash chromatography (100 g of silica gel, 10% to 30%> EtOAc/hexanes) to deliver 2- (2-(l-benzothiophen-7-yl)-4-pyridinyl)-l,l,l,3,3,3-hexafluoro-2-propanol (1.50 g) as an off-white solid.
Step 3. n-Butyllithium (3.32 mL of a 2.5M solution in toluene, 8.29 mmol) was added to a solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l,3,3,3-hexaf uoro-2-propanol (1.49 g, 3.95 mmol) in THF (20 mL) at -78 °C. After stirring at -78 °C for 0.5 h, a solution of N-((S,E)-(2- chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (1.25 mg, 5.13 mmol, Intermediate Yl) in THF (4 mL) at -78 °C was added dropwise via a cannula. After an additional 2.5 h at -78 °C, saturated aqueous NH4C1 (10 mL) was added, and the mixture was allowed to warm to room temperature. The mixture was partitioned between aqueous NH4C1 (15 mL of saturated NH4C1 and 15 mL of H20) and EtOAc (40 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 40 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure. The resulting yellow oil was purified by flash chromatography (100 g of silica gel, 30%> to 70%>
EtOAc/hexanes) and again by flash chromatography (80 g of silica gel, 30% to 70% EtOAc/hexanes) to deliver (S)-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (1.26 g) as a white solid.
Step 4. Hydrogen chloride (7.92 mL of a 1 M solution in Et20, 7.92 mmol) was added to a solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (1.23 g, 1.98 mmol) in Et20 (12 mL) and the resulting suspension was stirred at room temperature for 5 h. The solid was removed by filtration, washed with Et20 (10 mL) and dried under reduced pressure to deliver 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l-benzothiophen- 7-yl)-4-pyridinyl)- 1,1,1 ,3 ,3 ,3 -hexafluoro-2-propanol hydrochloride (Intermediate XI 1) (1.05 g) as a light-yellow solid.
Intermediate X12
N-((R)-(7-Bromo-l-benzothiophen-2-yl)(2- chlorophenyl)methyl)cyclopropanesulfonamide

Step 1. To a 2-L three-necked round-bottomed flask containing a solution of 7-bromo-l-benzothiophene (50 g, 236 mmol, Intermediate XI 3) in THF (400 mL) at -78 °C was added LDA (236 mL of a 1.8 M solution in THF, 472 mmol, Sigma- Aldrich, India) and the mixture was stirred at -78 °C for 45 min. A
solution of N-((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2- propanesulfinamide (57.3 g, 236 mmol, Intermediate Yl) in THF (100 mL) was added dropwise and the mixture was stirred at -78 °C for 16 h. Water (500 mL) was added, the mixture was allowed to warm to room temperature, and was extracted with EtOAc (2 x 500 mL and 2 x 200 mL). The combined organic layers were dried (Na2S04), filtered, and concentrated under reduced pressure. The resulting product was purified by column chromatography (basic alumina, 0% to 10% EtOAc/hexane to deliver (S)-N-((R)-(7-bromo-l-benzothiophen-2- yl)(2-chlorophenyl)methyl)-2-methyl-2-propanesulfinamide (25 g) as a pale- yellow liquid.
Step 2. To a 100-mLround-bottomed flask containing a solution of (S)-N- ((R)-(7-bromo-l-benzothiophen-2-yl)(2-chlorophenyl)methyl)-2-methyl-2- propanesulfmamide (5 g, 11.0 mmol) in MeOH (25 mL) at 0 °C was added saturated hydrogen chloride in 1 ,4-dioxane (20 mL) over the course of 5 min. After 2 h at room temperature, the mixture was concentrated under reduced pressure and the residue was triturated with Et20 (100 mL) to deliver (R)-l-(7- bromo- 1 -benzothiophen-2-yl)- 1 -(2-chlorophenyl)methanamine hydrochloride (4 g) as a brown solid.
Step 3. To a 100-mL round-bottomed flask charged with a solution of (R)- 1 -(7-bromo- 1 -benzothiophen-2-yl)- 1 -(2-chlorophenyl)methanamine hydrochloride (4 g, 10.3 mmol) in N-methyl-2-pyrrolidinone (30 mL) at 0 °C was added triethylamine (3.14 g, 30.9 mmol). After 5 min at 0 °C,
cyclopropylsulfonyl chloride (4.34 g, 31.0 mmol) was added over the course of 5 min and the reaction was then stirred at room temperature for 16 h. The mixture was partitioned between EtOAc (100 mL) and water (50 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (50 mL). The combined organic layers were dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 0% to 20% EtOAc/hexanes) to deliver N-((R)-(7-bromo-l-benzothiophen-2-
yl)(2-chlorophenyl)methyl)cyclopropanesulfonamide (3.5 g, Intermediate XI 2) as a brown solid.
Intermediate X13
7-Bromo-l-benzothiophene

Step 1. Potassium carbonate (260 g, 1.9 mmol) was added to a stirring solution of 2-bromobenzenethiol (180 g, 0.96 mol, Matrix Scientific, Columbia, SC), 2-bromo-l,l-dimethoxy ethane (170 g, 1.0 mol, Spectrochem, India), and acetone (1.8 L) at room temperature, and then the reaction mixture was heated at 55 °C. After 4 h, the reaction mixture was cooled to room temperature, filtered, the filter cake was washed with acetone, and the combined filtrate was concentrated under a vacuum. The residue was partitioned between ethyl acetate and water, the layers were separated, the organic material was washed with brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum to give l-bromo-2-((2,2-dimethoxyethyl)sulfanyl)benzene (240 g) as a pale yellow oil. The material was used in the next step of the synthesis without purification.
Step 2. Polyphosphoric acid (PPA) (100 g) was added to a stirring solution of l-bromo-2-((2,2-dimethoxyethyl)sulfanyl)benzene (220 g, 0.84 mol, from Step 1) and toluene (2.2 L) at room temperature, and then the reaction mixture was heated at 110 °C. After 12 h, the reaction mixture was cooled to room temperature and then partitioned between water and ethyl acetate. The layers were separated, the organic material was washed sequentially with water and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum. The residue was subjected to flash chromatography on silica gel (49: 1
hexane-ethyl acetate) to give 7-bromo-l-benzothiophene (82 g) as a colorless liquid.
Intermediate X14
N-((R)-(2-Chlorophenyl)(7-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l-

A 100-mL sealable tube containing a solution of N-((R)-(7-bromo-l- benzothiophen-2-yl)(2-chlorophenyl)methyl)cyclopropanesulfonamide (2.0 g, 4.40 mmol, Intermediate XI 2) in toluene (35 mL) was purged with argon for 15 min. Bis(pinacolato)diboron (B2Pin2) (5.58 g, 22.0 mmol, Sigma Aldrich, India), tris(dibenzylideneacetone)dipalladium(0) (2.00 g, 2.19 mmol, Sigma- Aldrich, India), 2-dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl (209 mg, 0.439 mmol, Sigma Aldrich, India), and potassium acetate (2.15 g, 21.9 mmol, Sigma- Aldrich, India) were added and the vessel was purged with argon for 15 min and sealed. The mixture was stirred at 90 °C for 12 h, allowed to cool to room temperature, and filtered through a pad of diatomaceous earth, which was washed with toluene (100 mL). The combined filtrates were concentrated under reduced pressure, hexane (100 mL) was added to the residue, and the mixture was stirred at room temperature for 1 h. The solvent was decanted and the resulting solid was dried under reduced pressure to deliver N-((R)-(2-chlorophenyl)(7-(4,4,5,5- tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (1.5 g, Intermediate XI 4) as an off-white solid.
Intermediate Yl
-(2-Chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide

Titanium(IV) ethoxide (220 mL, 1100 mmol) was added to a stirring solution of (S)-2-methyl-2-propanesulfinamide (26 g, 210 mmol, AK Scientific, Mountain View, CA), dichloromethane (430 mL), and 2-chlorobenzaldehyde (30 g, 210 mmol, Sigma- Aldrich, St. Louis, MO), at room temperature. After 22 h, the reaction mixture was added to water and dichloromethane. The mixture was agitated vigorously and then filtered through a pad of diatomaceous earth. The two layers comprising the filtrate were separated, the organic material was dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum. The residue was subjected to flash chromatography on silica gel (9 to 1 hexane-ethyl acetate) to afford N-((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2- propanesulfmamide (46 g, Intermediate Yl) as a clear yellow oil.
Intermediate Y2
2-(l-Benzothiophen-7-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane

Dichloro l,l '-bis(diphenylphosphino)ferrocene palladium(II) (14 g, 0.018 mol) was added to a mixture of 7-bromo-l-benzothiophene (Intermediate XI 3) (75 g, 0.35 mol, from Step 2), bis(pinacolato)diboron (110 g, 0.42 mol), potassium acetate (170 g, 1.8 mol), and toluene (750 mL) at room temperature under an argon atmosphere. The reaction mixture was then heated at 100 °C. After 12 h, the reaction mixture was cooled to room temperature, partitioned
between water and ethyl acetate, and the layers were separated. The organic material was washed sequentially with water and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum. The residue was subjected to flash chromatography on silica gel (99: 1 hexane-ethyl acetate) to give 2-(l-benzothiophen-7-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (60 g, Intermediate Y2) as a colorless solid.
Intermediate Y3 (racemate of Intermediates X4 and X7) 2-(2-(l-Benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol

A mixture of 2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol (7.4 g, 0.033 mol, Intermediate XI), 2-(l-benzothiophen-7-yl)-4,4,5,5-tetramethyl-l,3,2- dioxaborolane (11 g, 0.042 mol, Intermediate Y2), 1,4-dioxane (40 mL), water (10 mL), allylpalladium(II) chloride dimer (0.60 g, 1.6 mmol), 2- (dicyclohexylphosphino)-2',4',6',-tri-isopropyll, -biphenyl (1.5 g, 3.2 mmol), and sodium carbonate (10 g, 0.099 mol) was heated at 80 °C under an argon atmosphere. After 16 h, the reaction mixture was cooled to room temperature, filtered through a pad of diatomaceous earth, and the filtrate was concentrated under a vacuum. The residue was partitioned between water and ethyl acetate, the layers were separated, and the aqueous material was washed with ethyl acetate (2x). The combined organic extracts were washed sequentially with water and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum. The residue was subjected to flash chromatography on silica gel (9 to 1 hexane-ethyl acetate) to give 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -
trifluoro-2-propanol (5.0 g, Intermediate Y3) as a pale brown solid (racemic mixture).
Intermediate Y4
2-(2-Chloro-4-pyridinyl)-2-propanol

Intermediate Y5
2-(2-(l-Benzothiophen-7-yl)-4-pyridinyl)-2-propanol

chromatography on silica gel (9 tol hexane-ethyl acetate) to give 2-(2-(l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol (20 g, Intermediate Y5) as a pale yellow solid.
Intermediate Y6
1 ,1 , l-Trifluoro-2-(2-thieno [2,3-c] pyridin-7-yl-4-pyridinyl)-2-propanol

A stirring mixture of 2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol (1.0 g, 4.4 mol, Intermediate XI), 7-chlorothieno[2,3-c]pyridine (1.1 g, 6.7 mol, Ellanova, Inc., Hamden, CT), 1,4-dioxane (14 mL), water (3.5 mL),
allylpalladium(II) chloride dimer (0.16 g, 0.44 mmol), 9,9-dimethyl-4,5- bisdiphenylphosphino)xanthene (0.51 g, 0.89 mmol), and cesium fluoride (2.0 g,
13 mol) was heated at 80 °C under a nitrogen atmosphere. After 24 h, the reaction mixture was heated at 100 °C. After an additional 13h, the reaction mixture was allowed to cool to room temperature and concentrated under a vacuum. The residue was partitioned between saturated aqueous sodium bicarbonate and ethyl acetate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine. The combined extracts were dried (sodium sulfate), filtered, and concentrated under a vacuum. The residue was dissolved with dichloromethane, silica gel (10 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (4: 1 hexane-ethyl acetate) to give l,l,l-trifluoro-2-(2-thieno[2,3-c]pyridin-7-yl-4-pyridinyl)-2- propanol (0.25 g, Intermediate Y6) as an off-white solid (racemic mixture).
Intermediate Zl
2-(2-(2-(Amino(phenyl)methyl)- l-benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol hydrochloride

Step 1. Montmorillonite K10 (clay catalyst; Sigma-Aldrich, St. Louis, MO) (1.0 g) was added to a stirring solution of benzaldehyde (0.96 mL, 9.4 mmol), 2-methyl-2-propanesulfinamide (1.4 g, 11 mmol, AK Scientific, Mountain View, CA), and toluene (47 mL), the reaction vessel was fitted with a
Dean-Stark trap and a reflux condenser, and then the reaction mixture was heated at reflux. After 3 h, the reaction mixture was allowed to cool to room temperature and filtered. Silica gel (2.0 g) was added to the filtrate, and the volatiles were removed under a vacuum. The residue was purified by flash chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 0% to 20% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to provide 2-methyl-N-((lE)- phenylmethylidene)-2-propanesulfinamide (1.3 g) as pale yellow oil.
Step 2. n-Butyllithium (1.4 mL of a 1.6 M solution in hexanes, 2.3 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (0.37 g, 1.1 mmol, Intermediate Y3) and
tetrahydrofuran (6.0 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of 2-methyl-N-((lE)-phenylmethylidene)-2-propanesulfinamide (0.24 g, 1.1 mmol, from Step 1) and THF (3.0 mL) was added. After 10 min, the reaction mixture was allowed to warm to room temperature, where silica gel (2.0 g) was added and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 10% to 60% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to give 2-methyl-N-(phenyl(7-(4-(2,2,2-trifluoro-l -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-propanesulfinamide (0.15 g) as an off- white oil.
Step 3. Hydrogen chloride (5.0 mL of a 1.0 M solution with diethyl ether, 5.0 mmol) was added to a stirring solution of 2-methyl-N-(phenyl(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-propanesulfinamide (0.15 g, 0.28 mmol, from Step 2) and ethyl acetate (10 mL) at room temperature. After 1 min, the reaction mixture was concentrated to afford 2-(2-(2-(amino(phenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol hydrochloride (0.13 g, Intermediate Zl) as a colorless solid (mixture of 4 stereoisomers). This intermediate was used without purification.
- I l l -
Intermediate Z2
2-(2-(2-(Amino(2-(methylsulfanyl)phenyl)methyl)-l-benzothiophi
pyridinyl)- 1 , 1 ,l-trifluoro-2-propanol hydrochloride

Step 1. Montmorillonite K10 (2.2 g) was added to a stirring solution of 2- (methylsulfanyl)benzaldehyde (2.2 g, 14 mmol), 2-methyl-2-propanesulfinamide (1.8 g, 14 mmol, AK Scientific, Mountain View, CA), and toluene (100 mL). The reaction vessel was fitted with a Dean-Stark trap and a reflux condenser, and then the reaction mixture was heated at reflux. After 3 h, the reaction mixture was allowed to cool to room temperature and filtered. Silica gel (2.0 g) was added to the filtrate, and the volatiles were removed under a vacuum. The residue was purified by flash chromatography on silica gel (80 g RediSep® normal phase column, 10% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to provide 2- methyl-N-((lE)-(2-(methylsulfanyl)phenyl)methylidene)-2-propanesulfinamide (1.6 g) as a pale yellow oil.
Step 2. n-Butyllithium (0.41 mL of a 1.6 M solution in hexanes, 0.65 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.10 g, 0.31 mmol, Intermediate Y3) and THF (3.0 mL) at -78 °C under a nitrogen atmosphere. After 30 min, a solution of
2-methyl-N-((lE)-(2-(methylsulfanyl)phenyl)methylidene)-2-propanesulfinamide (0.079 g, 0.31 mmol, from Step 1) in THF (2.0 mL) was added. After 5 min, saturated aqueous sodium bicarbonate (2.0 mL) was added and the mixture was warmed to room temperature. Silica gel (1.0 g) was added and then the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (12 g RediSep® normal phase column, gradient elution of 10% to 50% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to give 2-methyl-N-((2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-propanesulfinamide (0.080 g).
Step 3. Hydrogen chloride (0.69 mL of a 1.0 M solution with diethyl ether, 0.69 mmol) was added to a stirring solution of 2-methyl-N-((2- (methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-propanesulfinamide (0.080 g, 0.14 mmol, from Step 2) and ethyl acetate (10 mL) at room temperature. After 1 h, the reaction mixture was concentrated to give 2-(2-(2-(amino(2- (methylsulfanyl)phenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol hydrochloride (0.071 g, Intermediate Z2, mixture of 4 stereoisomers). This intermediate was used without purification.
Intermediate Z3 (carbinol stereoisomer mixture of
Intermediates X5 and X8)
2-(2-(2-((R)-Amino(2-chlorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)- 1 , 1 ,l-trifluoro-2-propanol hydrochloride

Step 1. n-Butyllithium (2.0 mL of a 1.6 M solution with hexanes, 3.2 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.51 g, 1.6 mmol, Intermediate Y3) and THF (7.9 mL) at -78 °C under a nitrogen atmosphere. After 30 min, a solution of N-((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (0.46 g, 1.9 mmol, Intermediate Yl) in tetrahydrofuran (4.0 mL) was added. After 5 min, saturated aqueous sodium bicarbonate (5.0 mL) was added, and the mixture was warmed to room temperature. Silica gel (1.0 g) was added, and then the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (120 g RediSep® normal phase column, gradient elution of 35 to 40% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to give (S)-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.26 g).
Step 2. Hydrogen chloride (2.56 mL of a 1.0 M solution with diethyl ether, 2.56 mmol) was added to a stirring solution of (S)-N-((R)-(2- chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.29 g, 0.451 mmol, from Step 1) and ethyl acetate (10 mL) at room temperature. After 2 h, the
reaction mixture was concentrated under a vacuum to give 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol hydrochloride (0.25 g, Intermediate Z3) as a white solid (mixture of 2 diastereomers).
Intermediate Z4
l-(2-(l-Benzothiophen-7-yl)-4-pyridinyl)ethanol

Step 1. n-Butyllithium (3.9 mL of a 1.6 M solution in hexanes, 6.2 mmol) was added to a stirring solution of 4-bromo-2-chloropyridine (0.58 mL, 5.2 mmol, Alfa Aesar, Ward Hill, MA) and diethyl ether (26 mL) at -78 °C under a nitrogen atmosphere. After 15 min, acetaldehyde (1.5 mL, 26 mmol) was added. After an additional 10 min, water (5.0 mL) was added and the mixture was warmed to room temperature. The mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, the organic material was washed with brine, dried (magnesium sulfate), filtered, and the filtrate was concentrated to afford l-(2-chloro-4-pyridinyl)ethanol (0.80 g). The material was used in the next step of the synthesis without purification.
Step 2. A stirring mixture of allylpalladium(II) chloride dimer (0.093 g, 0.25 mmol), 2-(dicyclohexylphosphino)-2',4',6',-tri-isopropyl-l, -biphenyl (0.24 g, 0.51 mmol), 1,4-dioxane (10 mL), 2-(l-benzothiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (2.0 g, 7.6 mmol, Intermediate Y2), l-(2-chloro- 4-pyridinyl)ethanol (0.80 g, 5.1 mmol, from Step 1), water (3.0 mL), and sodium carbonate monohydrate (1.6 g, 15 mmol), was heated at 80 °C. After 3 h, the reaction mixture was allowed to cool to room temperature, concentrated under a
vacuum, and the residue was partitioned between water and ethyl acetate. The layers were separated, the aqueous material was washed with ethyl acetate (2x). The combined organic extracts were washed with brine, dried (magnesium sulfate) and filtered. Silica gel (1.0 g) was added to the filtrate, and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 0% to 100% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to afford l-(2-(l-benzothiophen-7-yl)-4-pyridinyl)ethanol (0.77 g, Intermediate Z4, racemic mixture).
Intermediate Z5
l-(2-(l-Benzothiophen-7-yl)-4-pyridinyl)cyclobutanol

Step 1. n-Butyllithium (7.8 mL of a 1.6 M solution with hexanes, 12 mmol) was added to a stirring solution of 4-bromo-2-chloropyridine (1.2 mL, 10 mmol, Alfa Aesar, Ward Hill, MA) and diethyl ether (52 mL) at -78 °C. After 30 min, cyclobutanone (3.9 mL, 52 mmol) and water (50 mL) were added sequentially, and then the reaction mixture was warmed to room temperature, partitioned between ethyl acetate and more water, and the layers were separated. The organic material was dried (magnesium sulfate), silica gel (2.0 g) was added, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 0% to 30% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to afford l-(2-chloro-4-pyridinyl)cyclobutanol (1.6 g).
Step 2. A stirring mixture of allylpalladium(II) chloride dimer (1.0 g, 2.7 mmol), l-(2-chloro-4-pyridinyl)cyclobutanol (0.50 g, 2.7 mmol, from Step 1), 2- (dicyclohexylphosphino)-2',4',6',-tri-isopropyl-l, -biphenyl (X-Phos)(1.3 g, 2.7 mmol), 2-(l-benzothiophen-7-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (0.71 g, 2.7 mmol, Intermediate Y2), sodium carbonate monohydrate (0.29 g, 2.7 mmol), 1,4-dioxane (9.1 mL), and water (4.5 mL) was sparged with nitrogen for 5 min, the reaction vessel was then sealed and heated at 80 °C. After 15 h, the reaction mixture was allowed to cool to room temperature, and partitioned between ethyl acetate and water. The layers were separated, the organic material was washed with brine, silica gel (1.0 g) was added, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 0% to 30% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to afford l-(2-(l- benzothiophen-7-yl)-4-pyridinyl)cyclobutanol (0.49 g) as a clear oil.
Intermediate Z6
2-(2-(l-Benzothiophen-7-yl)-4-pyridinyl)-l-((tert-butyl(dimethyl)silyl)oxy)-2- propanol

Step 1. n-Butyllithium (3.9 mL of a 1.6 M solution with hexanes, 6.2 mmol) was added to a stirring solution of 4-bromo-2-chloropyridine (0.63 mL, 5.7 mmol, Alfa Aesar, Ward Hill, MA) and diethyl ether (28 mL) at -78 °C under a nitrogen atmosphere. After 15 min, l-(tert-butyldimethylsilyloxy)-2-propanone (3.3 mL, 17 mmol, Sigma- Aldrich, St. Louis, MO) was added. After an additional 10 min, water (5.0 mL) was added, and then the reaction mixture was
allowed to warm to room temperature and concentrated under a vacuum. The residue was partitioned between ethyl acetate and water and the layers were separated. The organic material was washed with brine, dried (magnesium sulfate), filtered, silica gel (1.0 g) was added to the filtrate, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (80 g RediSep® normal phase column, gradient elution of 0% to 30% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to afford l-((tert- butyl(dimethyl)silyl)oxy)-2-(2-chloro-4-pyridinyl)-2-propanol (0.80 g).
Step 2. A stirring mixture of allylpalladium(II) chloride dimer (0.049 g, 0.13 mmol), 2-(dicyclohexylphosphino)-2',4',6',-tri-isopropyl-l, -biphenyl (X- Phos) (0.13 mg, 0.27 mmol), 1,4-dioxane (13 mL), 2-(l-benzothiophen-7-yl)- 4,4,5,5-tetramethyl-l,3,2-dioxaborolane (1.0 g, 4.0 mmol, Intermediate Y2), 1- ((tert-butyl(dimethyl)silyl)oxy)-2-(2-chloro-4-pyridinyl)-2-propanol (0.80 g, 2.7 mmol, from Step 1), water (4.0 mL), and sodium carbonate monohydrate (0.84 g, 8.0 mmol) was heated at 80 °C. After 3 h, the reaction mixture was cooled to room temperature, concentrated, and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, the organic material was washed with brine, dried (magnesium sulfate), filtered, silica gel (1.0 g) was added to the filtrate, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 0% to 100% ethyl acetate- hexane, Teledyne Isco, Lincoln, NE) to afford 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l-((tert-butyl(dimethyl)silyl)oxy)-2-propanol (0.76 g, racemic mixture).
Example 1
N-((R)-(6-Amino-2-pyridinyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide



Step 6
Step 1. A stirring mixture of di-tert-butyl (6-bromo-2- pyridinyl)imidodicarbonate (5.0 g, 13 mmol, Sigma- Aldrich, St. Louis, MO), potassium trifluoro(vinyl)borate (2.0 g, 15 mmol), cesium carbonate (13 g, 40 mmol), 2-(dicyclohexylphosphino)-2',4',6',-tri-isopropyll, -biphenyl (X-Phos) (0.64 g, 1.3 mmol), palladium(II) acetate (0.15 g, 0.67 mmol), tetrahydrofuran (60 mL) and water (7.0 mL) was heated at 80 °C. After 18 h, more potassium trifluoro(vinyl)borate (0.72 g, 5.4 mmol), 2-(dicyclohexylphosphino)-2',4',6',-tri- isopropyll,l'-biphenyl (0.64 g, 1.3 mmol), and palladium(II) acetate (0.15 g, 0.67 mmol) were added. After 4 h, the reaction mixture was allowed to cool to room temperature and then partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine. The organics extracts were dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum give di-tert-butyl (6-ethenyl-2-pyridinyl)imidodicarbonate (4.3
g) as a light brown tar. The material was used in the next step of the synthesis without purification.
Step 2. Sodium periodate (8.6 g, 40 mmol) was added to a stirring mixture of di-tert-butyl (6-ethenyl-2-pyridinyl)imidodicarbonate (4.3 g, 13 mmol, from Step 1), tetrahydrofuran (54 mL), water (14 mL), and osmium tetroxide (4.1 mL of a 4.0 wt% solution with water, 0.67 mmol) at room temperature. After 2 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried
(magnesium sulfate), filtered, and concentrated to afford di-tert-butyl (6-formyl- 2-pyridinyl)imidodicarbonate (4.3 g) as a dark tar. The material was used in the next step of the synthesis without purification.
Step 3. Copper(II) sulfate (6.4 g, 40 mmol) was added to a stirring solution of di-tert-butyl (6-formyl-2-pyridinyl)imidodicarbonate (4.3 g, 13 mmol, from Step 2), dichloromethane (53 mL), and (R)-2-methyl-2-propanesulfinamide (1.9 g, 16 mmol, AK Scientific, Mountain View, CA) at room temperature. After 50 h, the reaction mixture was filtered through a 0.45 μιη Teflon®
(polytetrafluoroethylene, DuPont, Wilmington, DE) filter. Silica gel (30 g) was added to the filtrate, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (gradient elution; 6: 1 to 4: 1 hexane-ethyl acetate) to give di-tert-butyl (6-((E)-(((R)-tert- butylsulfinyl)imino)methyl)-2-pyridinyl)imidodicarbonate di-tert-butyl (6-((E)- (((R)-tert-butylsulfinyl)imino)methyl)-2-pyridinyl)imidodicarbonate (2.8 g) as a pale yellow solid.
Step 4. n-Butyllithium (1.9 mL of a 2.5 M solution in toluene, 4.7 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (0.84 g, 2.6 mmol, Intermediate Y3) in tetrahydrofuran (20 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of di-
tert-butyl (6-((E)-(((R)-tert-butylsulfinyl)imino)methyl)-2- pyridinyl)imidodicarbonate (1.0 g, 2.4 mmol, from Step 3) in tetrahydrofuran (3.5 mL) was added. After an additional 10 min, saturated aqueous sodium bicarbonate was added. The mixture was allowed to warm to room temperature and then partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate. The layers were separated and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine. The organic extract was dried (sodium sulfate), and filtered. Silica gel (9.0 g) was added was added to the filtrate, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (1 : 1 hexane-ethyl acetate). The isolated material was dissolved with dichloromethane. Silica gel (5.0 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (gradient elution; 1 :4 to 1 :9 hexane-diethyl ether) to give di-tert-butyl (6-((S)-(((R)-tert- butylsulfinyl)amino)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-pyridinyl)imidodicarbonate (0.82 g, containing about 7 mol% of tert-butyl (6-((S)-(((R)-tert-butylsulfmyl)amino)(7- (4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-pyridinyl)carbamate and about 5 mol% of di-tert-butyl (6-((R)- (((R)-tert-butylsulfinyl)amino)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-pyridinyl)imidodicarbonate) as an off-white solid. The material was used in the next step of the synthesis without further purification.
Step 5. Hydrogen chloride (2.7 mL of a 4.0 M solution with 1,4-dioxane, 11 mmol) was added to a stirring solution of di-tert-butyl (6-((S)-(((R)-tert- butylsulfinyl)amino)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-pyridinyl)imidodicarbonate (0.82 g, containing about 7 mol% of tert-butyl (6-((S)-(((R)-tert-butylsulfmyl)amino)(7- (4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-pyridinyl)carbamate and about 5 mol% of di-tert-butyl (6-((R)-
(((R)-tert-butylsulfinyl)amino)(7-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-pyridinyl)imidodicarbonate, from Step 4) and methanol (11 mL) at room temperature. After 15 h, more hydrogen chloride (2.7 mL of a 4.0 M solution with 1,4-dioxane, 11 mmol) was added. After 24 h, the reaction mixture was concentrated under a vacuum, and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to afford 2-(2-(2-((S)-amino(6- amino-2-pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (0.49 g, containing about 5 mol% of 2-(2-(2-((R)-amino(6-amino-2- pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol) as an off-white solid. The material was used in the next step of the synthesis without purification.
Step 6. 4-(Dimethylamino)pyridine (0.013 g, 0.11 mmol) was added to a stirring solution of 2-(2-(2-((S)-amino(6-amino-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (0.49 g, containing about 5 mol% of 2-(2-(2-((R)-amino(6-amino-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol, from Step 5), N,N- dimethylformamide (11 mL), N,N-diisopropylethylamine (0.96 mL, 5.5 mmol), and cyclopropanesulfonyl chloride (0.11 mL, 1.1 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 5 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, and the layers were separated. The organic layer was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine (2x), dried (sodium sulfate), filtered, the filtrate was concentrated. The residue was dissolved with
dichloromethane, silica gel (3.0 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (1 : 1 hexane-ethyl acetate). The isolated material was subjected to preparative SFC (Chiralpak® AD-H column)(250 mm x 21 mm,
5 μηι) eluting with 65% liquid C02 in 35% methanol (with 40 mM NH3) at a flow rate of 75 mL/min) to afford N-((R)-(6-amino-2-pyridinyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.023 g) as an off-white solid (mixture of two diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.83 (d, J= 4.7 Hz, 1 H), 8.29 (br. s., 1 H), 8.25 (d, J= 8.8 Hz, 1 H), 8.07 (d, J= 7.2 Hz, 1 H), 7.90 (d, J= 7.4 Hz, 1 H), 7.60 (d, J= 4.1 Hz, 1 H), 7.52 (t, J= 7.7 Hz, 1 H), 7.42 (t, J= 7.7 Hz, 1 H), 7.28 (s, 1 H), 6.99 (s, 1 H), 6.79 (d, J= 7.2 Hz, 1 H), 6.38 (d, J= 8.0 Hz, 1 H), 6.01 (br. s., 2 H), 5.70 (d, J= 8.4 Hz, 1 H), 2.32 (m, 1 H), 1.79 (br. s., 3 H), 0.89 (m, 2 H), 0.73 (m, 2 H). m/z (ESI, pos. ion) 548.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.22 μΜ.
Example 2
N-((R)-(2-Chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 2. Hydrogen chloride (12 mL of a 4.0 M solution with 1,4-dioxane, 48 mmol) was added to a stirring solution of (S)-N-((R)-(2-chlorophenyl)(7-(4- ( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl- 2-propanesulfmamide (2.5 g, 4.9 mol, from Step 1) and methanol (49 mL) at room temperature. After 10 min, the reaction mixture was concentrated under a vacuum to give 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l-benzothiophen-7- yl)-4-pyridinyl)-2-propanol hydrochloride (2.0 g) as an off-white solid.
Step 3. 4-(Dimethylamino)pyridine (0.060 g, 0.49 mmol) was added to a stirring solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l-benzothiophen-
7-yl)-4-pyridinyl)-2-propanol hydrochloride (2.0 g, 4.9 mmol, from Step 2), N,N- dimethylformamide (49 mL), N,N-diisopropylethylamine (8.5 mL, 49 mmol), and cyclopropanesulfonyl chloride (0.50 mL, 4.9 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 24 h, more cyclopropanesulfonyl chloride (0.50 mL, 4.9 mmol) was added. After an additional 48h, saturated aqueous sodium bicarbonate was added, and the mixture was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine (2x), dried (sodium sulfate), filtered, and concentrated. The residue was dissolved in dichloromethane, silica gel (10 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (gradient elution of 3:1 to 2: 1 to 1 : 1 hexane-ethyl acetate). The isolated material was dissolved with dichloromethane, silica gel (5.0 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (2: 1 hexane-ethyl acetate). The isolated material was dissolved with dichloromethane, silica gel (5.0 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (49: 1 dichloromethane -methanol). The isolated material was subjected to preparative SFC (Chiralpak® AD-H
column)(15 cm x 21 mm, 5 μιη) eluting with 50% liquid C02 in 50% methanol (with 20 mM NH3) at a flow rate of 50 mL/min) to afford N-((R)-(2- chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.59 g) as a colorless solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.87 (br. s, 1 H), 8.71 (d, J= 5.1 Hz, 1 H), 8.21 (s, 1 H), 8.09 (d, J= 7.4 Hz, 1 H), 7.92 - 7.80 (m, 2 H), 7.58 - 7.45 (m, 4 H), 7.45 - 7.35 (m, 1 H), 6.99 (d, J= 1.0 Hz, 1 H), 6.28 (s, 1 H), 5.38 (s, 1 H), 2.27 - 2.13 (m, 1 H), 1.50 (s, 6 H), 0.94 - 0.83 (m, 1 H), 0.83 - 0.72 (m, 2 H), 0.67 - 0.54 (m, 1 H). m/z (ESI, pos. ion) 512.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.10 μΜ.
Example 3
N-((R)-(6-Amino-3-chloro-2-pyridinyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 8
Step 1. A stirring solution of 6-bromo-2-pyridinamine (15 g, 87 mmol, Sigma- Aldrich, St. Louis, MO), acetonitrile (170 mL), and N-chlorosuccinimide (14 g, 100 mmol) was heated at reflux under a nitrogen atmosphere. After 18 h, the reaction mixture was concentrated and the residue was partitioned between
ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic layer was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), and filtered. Silica gel (45 g) was added to the filtrate, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (gradient elution; 6: 1 to 4: 1 hexane-ethyl acetate). The isolated material was dissolved with
dichloromethane, silica gel (25 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (2: 1 dichloromethane-pentane) to give 6-bromo-5- chloro-2-pyridinamine (9.8 g) as a colorless solid.
Step 2. Di-tert-butyl dicarbonate (19 g, 87 mmol) was added to a stirring mixture of 6-bromo-5-chloro-2-pyridinamine (8.6 g, 42 mmol, from Step 1), dichloromethane (83 mL), and N,N-diisopropylethylamine (22 mL, 120 mmol) at room temperature under a nitrogen atmosphere. After 14 h, 4- (dimethylamino)pyridine (0.51 g, 4.2 mmol) was added. After an additional 3 h, the reaction mixture was concentrated under a vacuum, and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The organic layer was isolated, and was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and concentrated under a vacuum. The residue was dissolved with dichloromethane, silica gel (40 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (9: 1 hexane- ethyl acetate). The isolated material was dissolved with dichloromethane, silica gel (20 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (19: 1 hexane-ethyl acetate) to give di-tert-butyl (6-bromo-5-chloro-2- pyridinyl)imidodicarbonate (6.3 g) as a colorless solid.
Step 3. A stirring mixture of di-tert-butyl (6-bromo-5-chloro-2- pyridinyl)imidodicarbonate (3.0 g, 7.4 mmol, from Step 2), potassium
trifluoro(vinyl)borate (1.1 g, 8.1 mmol), cesium carbonate (7.2 g, 22 mmol), 2- (dicyclohexylphosphino)-2',4',6',-tri-isopropyll, -biphenyl (0.35 g, 0.74 mmol), palladium(II) acetate (0.083 g, 0.37 mmol), tetrahydrofuran (33 mL) and water (4.0 mL) was heated at 80 °C. After 22 h, the reaction mixture was allowed to cool to room temperature, concentrated under a vacuum, and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The organic layer was separated, and washed with brine, dried (sodium sulfate), filtered, then concentrated under a vacuum. The residue was dissolved with dichloromethane, silica gel (13 g) was added, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (19: 1 hexane-ethyl acetate, twice to give di-tert-butyl (5-chloro-6-ethenyl-2- pyridinyl)imidodicarbonate (0.61 g, containing about 35 mol% of di-tert-butyl (6- bromo-5-chloro-2-pyridinyl)imidodicarbonate) as a colorless oil. The material was used in the next step of the synthesis without further purification.
Step 4. Sodium periodate (1.1 g, 5.2 mmol) was added to a stirring mixture of di-tert-butyl (5-chloro-6-ethenyl-2-pyridinyl)imidodicarbonate (0.61 g, containing about 35 mol% of di-tert-butyl (6-bromo-5-chloro-2- pyridinyl)imidodicarbonate, from Step 3), tetrahydrofuran (7.0 mL), water (1.6 mL), and osmium tetroxide (0.53 mL of a 4.0 wt% solution with water, 0.086 mmol) at room temperature. After 17 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate and the layers were separated. The organic layer was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (magnesium sulfate), filtered, and the filtrate was concentrated to afford di-tert-butyl (5-chloro-6-formyl-2- pyridinyl)imidodicarbonate (0.48 g) as a dark tar. The material was used in the next step of the synthesis without purification.
Step 5. Copper(II) sulfate (0.64 g, 4.0 mmol) was added to a stirring solution of di-tert-butyl (5-chloro-6-formyl-2-pyridinyl)imidodicarbonate (0.48 g, from Step 4), dichloromethane (5.4 mL), and (S)-2-methyl-2-propanesulfinamide
(0.20 g, 1.6 mmol, AK Scientific, Mountain View, CA) at room temperature. After 43 h, the reaction mixture was filtered through a 0.20 μιη Teflon®
(polytetrafluoroethylene, DuPont, Wilmington, DE) filter, silica gel (3.0 g) was added to the filtrate, and the volatiles were removed under a vacuum. This material was subjected to flash chromatography on silica gel (9: 1 hexane-ethyl acetate) to give di-tert-butyl (6-((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5- chloro-2-pyridinyl)imidodicarbonate (0.37 g) as a clear pale yellow tar.
Step 6. n-Butyllithium (0.62 mL of a 2.5 M solution in toluene, 1.6 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-2- propanol (0.23 g, 0.86 mmol, Intermediate Y5) and tetrahydrofuran (6.5 mL) at - 78 °C under a nitrogen atmosphere. After 10 min, a solution of di-tert-butyl (6- ((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5-chloro-2- pyridinyl)imidodicarbonate (0.36 g, 0.78 mmol, from Step 5) and tetrahydrofuran (1.5 mL) was added. After 45 min, saturated aqueous sodium bicarbonate was added and the mixture was allowed to warm to room temperature. The mixture was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate. The organic layer was isolated and was washed sequentially with saturated aqueous sodium bicarbonate and brine. The combined extracts were dried (sodium sulfate), filtered, and then concentrated. The residue was dissolved with dichloromethane, silica gel (3.0 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (gradient elution; 2: 1 to 1 : 1 hexane-ethyl acetate) to give di-tert-butyl (6-((R)-(((S)-tert-butylsulfinyl)amino)(7-(4-(l -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-2- pyridinyl)imidodicarbonate (0.27 g, containing about 17 mol% of di-tert-butyl (6- ((S)-(((S)-tert-butylsulfinyl)amino)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)imidodicarbonate and about 25 mol% of tert-butyl (6-(((S)-tert-butylsulfmyl)amino)(7-(4-(l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-2-
pyridinyl)carbamate) as a clear pale-yellow tar. The material was used in the next step of the synthesis without further purification.
Step 7. Hydrogen chloride (0.93 mL of a 4.0 M solution with 1,4-dioxane, 3.7 mmol) was added to a stirring solution of di-tert-butyl (6-((R)-(((S)-tert- butylsulfinyl)amino)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)imidodicarbonate (0.27 g, containing about 17 mol% of di-tert-butyl (6-((S)-(((S)-tert- butylsulfinyl)amino)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)imidodicarbonate and about 25 mol% of tert-butyl (6-(((S)-tert-butylsulfinyl)amino)(7-(4-(l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-2- pyridinyl)carbamate), from Step 6, and methanol (3.7 mL) at room temperature. After 3 h, more hydrogen chloride (0.93 mL of a 4.0 M solution with 1,4- dioxane, 3.7 mmol) was added. After an additional 21 h, the reaction mixture was concentrated under a vacuum and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to afford 2-(2-(2-((R and S)-amino(6-amino-3-chloro-2- pyridinyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.16 g) as a clear pale yellow tar. The material was used in the next step of the synthesis without purification.
Step 8. 4-(Dimethylamino)pyridine (0.0046 g, 0.038 mmol) was added to a stirring solution of 2-(2-(2-((R and S)-amino(6-amino-3-chloro-2- pyridinyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.16 g, 0.38 mmol, from Step 7), N,N-dimethylformamide (3.8 mL), N,N- diisopropylethylamine (0.33 mL, 1.9 mmol), and cyclopropanesulfonyl chloride (0.038 mL, 0.38 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 3 h, the reaction mixture was partitioned between ethyl acetate and
saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, and then
concentrated. The residue was dissolved with dichloromethane, silica gel (2.0 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (1 : 1 hexane-ethyl acetate). The isolated material was subjected to preparative SFC (Chiralcel® AD- H column) (250 mm x 21 mm, 5 μιη) eluting with 55% liquid C02 in 45% methanol (with 40 mM NH3) at a flow rate of 70 mL/min) to afford N-((R)-(6- amino-3 -chloro-2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.10 g) as a colorless solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.70 (d, J= 5.3 Hz, 1 H), 8.19 (s, 1 H), 8.06 (d, J= 7.4 Hz, 1 H), 7.88 (d, J= 7.6 Hz, 1 H), 7.76 (d, J= 4.5 Hz, 1 H), 7.56 - 7.43 (m, 3 H), 7.24 (s, 1 H), 6.49 (d, J= 8.6 Hz, 1 H), 6.26 (s, 2 H), 6.12 (d, J = 4.5 Hz, 1 H), 5.35 (s, 1 H), 2.33 - 2.21 (m, 1 H), 1.50 (s, 6 H), 0.94 - 0.83 (m, 2 H), 0.82 - 0.72 (m, 1 H), 0.71 - 0.67 (m, 1 H). m/z (ESI, pos. ion) 528.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.0040 μΜ.
Example 4
N-((2-Chloro-6-methoxyphenyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 2. n-Butyllithium (0.73 mL of a 2.5 M solution in toluene, 1.8 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-2- propanol (0.27 g, 1.0 mmol, Intermediate Y5) and tetrahydrofuran (8.0 mL) at - 78 °C. After 10 min, a solution of N-((lE)-(2-chloro-6- methoxyphenyl)methylidene)cyclopropanesulfonamide (0.25 g, 0.91 mmol, from Step 1) in tetrahydrofuran (1.1 mL) was added. After 20 min, the reaction mixture was allowed to warm to room temperature, methanol (2.0 mL) and silica gel (2.5 g) were added sequentially and then the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (1 : 1 hexane-ethyl acetate). The isolated material was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (49: 1 dichloromethane -methanol) to give N-((2-chloro-6-methoxyphenyl)(7-(4- ( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.099 g) as a colorless solid (racemic mixture).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.68 (d, J= 5.1 Hz, 1 H), 8.19 (s, 1 H), 8.05 (d, J= 7.0 Hz, 1 H), 7.97 (d, J= 9.2 Hz, 1 H), 7.83 (d, J= 7.2 Hz, 1 H), 7.55
- 7.44 (m, 2 H), 7.43 - 7.35 (m, 1 H), 7.20 - 7.00 (m, 3 H), 6.48 (br. s., 1 H), 5.36 (s, 1 H), 3.79 (br. s., 3 H), 2.18 (m, 1 H), 1.50 (s, 6 H), 0.92 - 0.80 (m, 2 H), 0.67 (m, 2 H). m/z (ESI, pos. ion) 542.9 (M+H)+. GK-GKRP IC50 (Binding) = 1.9 μΜ.
Example 5
N-((R)-(2-Chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2-pyrimidinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. Methylmagnesium chloride (4.8 mL of a 3.0 mL solution with tetrahydrofuran, 14 mmol) was added to a stirring solution of methyl 2-chloro-4- pyrimidinecarboxylate (1.0 g, 5.8 mmol, Maybridge, Tintagel, England) in tetrahydrofuran (58 mL) at 0 °C under a nitrogen atmosphere. After 10 min, saturated aqueous sodium bicarbonate was added and the reaction mixture was allowed to warm to room temperature. The organics were partitioned between
ethyl acetate and more saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, then concentrated to give 2-(2-chloro-4-pyrimidinyl)-2-propanol (1.0 g) as a pale yellow oil. The material was used in the next step of the synthesis without purification.
Step 2. A stirring mixture of 2-(2-chloro-4-pyrimidinyl)-2-propanol (1.0 g, 5.8 mol, from Step 1), 2-(l-benzothiophen-7-yl)-4,4,5,5-tetramethyl-l,3,2- dioxaborolane (1.8 g, 7.0 mol, Intermediate Y2), 1,4-dioxane (9.5 mL), water (2.5 mL), allylpalladium(II) chloride dimer (0.11 g, 0.29 mmol), 2- (dicyclohexylphosphino)-2',4',6',-tri-isopropyll, -biphenyl (X-Phos) (0.28 g, 0.58 mmol), and sodium carbonate (18 g, 17 mmol) was heated at 80 °C under a nitrogen atmosphere. After 5 h, the reaction mixture was allowed to cool to room temperature, and partitioned between saturated aqueous sodium bicarbonate and ethyl acetate. The layers were separated and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, then concentrated under a vacuum. The residue was dissolved with dichloromethane, silica gel (10 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (4: 1 hexane-ethyl acetate) to give 2-(2-(l- benzothiophen-7-yl)-4-pyrimidinyl)-2-propanol (0.90 g) as a clear pale orange oil.
Step 3. Triethylsilyl trifluoromethanesulfonate (0.43 mL, 1.9 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyrimidinyl)-2- propanol (0.47 g, 1.7 mmol, from Step 2), dichloroethane (8.7 mL), and N,N- diisopropylethylamine (0.91 mL, 5.2 mmol) at room temperature. After 1 h, additional triethylsilyl trifluoromethanesulfonate (0.22 mL, 0.95 mmol) was added. After 10 min, silica gel (3.5 g) was added and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel
(49: 1 hexane-ethyl acetate) to give 2-(l-benzothiophen-7-yl)-4-(l -methyl- 1- ((triethylsilyl)oxy)ethyl)pyrimidine (0.63 g) as a clear colorless oil.
Step 4. n-Butyllithium (0.23 mL of a 2.5 M solution with toluene, 0.57 mmol) was added to a stirring solution of 2,2,6,6-tetramethylpiperidine (0.097 mL, 57 mmol) and tetrahydrofuran (5.0 mL) at -78 °C under a nitrogen atmosphere. After 5 min, the reaction mixture was warmed to room temperature. After 30 min, the reaction mixture was cooled to -78 °C, and a solution of 2-(l- benzothiophen-7-yl)-4-(l -methyl- l-((triethylsilyl)oxy)ethyl)pyrimidine (0.24 g, 0.63 mmol, from Step 3) in tetrahydrofuran (0.50 mL) was added. After an additional 55 min, a solution of N-((S,E)-(2-chlorophenyl)methylidene)-2- methyl-2-propanesulfinamide (0.14 g, 0.57 mmol, Intermediate Yl) in
tetrahydrofuran (0.50 mL) was added. After another hour, the reaction mixture was allowed to warm to room temperature. After 3 h at rt, tetrabutylammonium fluoride (0.69 mL of a 1.0 M solution with tetrahydrofuran, 0.69 mmol) was added. After 25 min, saturated aqueous sodium bicarbonate was added, and the mixture was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and then concentrated. The residue was dissolved with dichloromethane, silica gel (1.5 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (gradient elution; 4: 1 to 2: 1 to 1 : 1 hexane-ethyl acetate) to give (S)-N-((R)-(2-chlorophenyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2- pyrimidinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.070 g) as an off- white solid.
Step 5. Hydrogen chloride (0.32 mL of a 4.0 M solution with 1,4-dioxane, 1.3 mmol) was added to a stirring solution of (S)-N-((R)-(2-chlorophenyl)(7-(4- ( 1 -hydroxy- 1 -methylethyl)-2-pyrimidinyl)- 1 -benzothiophen-2-yl)methyl)-2- methyl-2-propanesulfmamide (0.065 g, 0.13 mmol, from Step 4) and methanol
(1.3 mL) at room temperature. After 15 min, the reaction mixture was
concentrated under a vacuum and the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to give 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyrimidinyl)-2-propanol (0.049 g) as a clear pale yellow tar. The material was used in the next step of the synthesis without purification.
Step 6. 4-(Dimethylamino)pyridine (1.5 mg, 12 μιηοΐ) was added to a stirring solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l-benzothiophen- 7-yl)-4-pyrimidinyl)-2-propanol (0.049 g, 0.12 mmol, from Step 5), N,N- dimethylformamide (1.2 mL), N,N-diisopropylethylamine (0.10 mL, 0.60 mmol), and cyclopropanesulfonyl chloride (0.012 mL, 0.12 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 19 h, more cyclopropanesulfonyl chloride (0.0060 mL, 0.060 mmol) was added. After an additional 7 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and then concentrated. The residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (2: 1 hexane-ethyl acetate). The isolated material was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (2: 1 hexane-ethyl acetate) to give N-((R)-(2- chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyrimidinyl)- 1 -benzothiophen- 2-yl)methyl)cyclopropanesulfonamide (0.034 g) as a colorless solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.97 (d, J= 5.1 Hz, 1 H), 8.90 (br. s., 1 H), 8.58 (d, J= 7.3 Hz, 1 H), 7.98 (d, J= 7.3 Hz, 1 H), 7.89 (dd, J= 1.5, 7.7 Hz,
1 H), 7.65 (d, J = 5.1 Hz, 1 H), 7.59 - 7.45 (m, 3 H), 7.45 - 7.36 (m, 1 H), 7.18 (s, 1 H), 6.31 (br. s., 1 H), 5.53 (s, 1 H), 2.36 - 2.22 (m, 1 H), 1.56 (s, 3 H), 1.54 (s, 3 H), 0.97 - 0.87 (m, 1 H), 0.87 - 0.76 (m, 2 H), 0.71 - 0.59 (m, 1 H). m/z (ESI, pos. ion) 513.8 (M+H)+. GK-GK P IC50 (Binding) = 1.6 μΜ.
Example 6
N-((R)-(2-Chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (0.23 mL of a 2.5 M solution in toluene, 0.57 mmol) was added to a stirring solution of 1,1,1 -trifluoro-2-(2-thieno[2,3- c]pyridin-7-yl-4-pyridinyl)-2-propanol (0.10 g, 0.32 mmol, Intermediate Y6) in tetrahydrofuran (2.0 mL) at -78 °C under a nitrogen atmosphere. After 5 min, a solution of N-((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2- propanesulfmamide (0.070 g, 0.29 mmol, Intermediate Yl) in tetrahydrofuran (1.0 mL) was added. After 15 min, the reaction mixture was allowed to warm to room temperature, methanol (1.0 mL) and silica gel (0.50 g) were added sequentially, and then the volatiles were removed under a vacuum. The residue
was subjected to flash chromatography on silica gel (1.5: 1 hexane-ethyl acetate). The isolated material was subjected to reversed-phase preparative HPLC
(Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 μιη) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 10%) to 90%) over 10 min). The isolated material was re-subjected to reversed- phase preparative HPLC (Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 μιη) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 10% to 90% over 10 min). The isolated material was again re-subjected to reversed-phase preparative HPLC (Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 50 mm, 10 μιη) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 10% to 95% over 16 min) to give (S)-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro-l- hydroxy-l-methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)-2-methyl- 2-propanesulfinamide (0.040 g) as a pale pink solid.
Step 2. Hydrogen chloride (0.17 mL of a 4.0 M solution with 1,4-dioxane, 0.68 mmol) was added to a stirring solution of (S)-N-((R)-(2-chlorophenyl)(7-(4- (2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-2- yl)methyl)-2-methyl-2-propanesulfinamide (0.039 g, 0.069 mmol, from Step 1) and methanol (1.4 mL) at room temperature. After 5 h, the reaction mixture was concentrated under a vacuum to give 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)thieno[2,3-c]pyridin-7-yl)-4-pyridinyl)-l , 1 , 1 -trifluoro-2- propanol hydrochloride (0.034 g) as a pale yellow solid. The material was used in the next step of the synthesis without purification.
Step 3. 4-(Dimethylamino)pyridine (0.83 mg, 6.8 μιηοΐ) was added to a stirring solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)thieno[2,3- c]pyridin-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol hydrochloride (0.034 g, 0.068 mmol, from Step 2), N,N-dimethylformamide (1.4 mL), N,N- diisopropylethylamine (0.071 mL, 0.41 mmol), and cyclopropanesulfonyl chloride (0.0069 mL, 0.068 mmol, Matrix Scientific, Columbia, SC) at room
temperature. After 16 h, more cyclopropanesulfonyl chloride (0.0069 mL, 0.068 mmol) was added. After another 24 h, saturated aqueous sodium bicarbonate was added, and the mixture was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and then concentrated. The residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (gradient elution; 3: 1 to 1 : 1 hexane-ethyl acetate). The isolated material was subjected to reversed-phase preparative HPLC
(Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 μιη) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 10% to 90% over 10 min) to afford N-((R)-(2-chlorophenyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-2- yl)methyl)cyclopropanesulfonamide (0.010 g) as a colorless tar (mixture of two diastereomers).
1H NMR (400 MHz, CDC13) δ ppm 8.90 (s, 1 H), 8.78 (d, J= 5.1 Hz, 1 H), 8.57 (d, J= 5.3 Hz, 1 H), 7.63 (d, J= 7.4 Hz, 1 H), 7.60 - 7.50 (m, 2 H), 7.50 - 7.30 (m, 3 H), 7.09 (s, 1 H), 6.42 (d, J= 8.4 Hz, 1 H), 5.67 (d, J= 7.8 Hz, 1 H), 3.24 (br. s., 1 H), 2.33 - 2.19 (m, 1 H), 1.85 (s, 3 H), 1.22 - 1.07 (m, 2 H), 0.95 - 0.70 (m, 2 H). m/z (ESI, pos. ion) 567.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.41 μΜ.
Example 7
N-((R)-(3-Chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)thieno [2,3-c] pyridin-2- yl)methyl)cyclopropanesulfonamide


Step 1. n-Butyllithium (0.25 mL of a 2.5 M solution in toluene, 0.61 mmol) was added to a stirring solution of 1,1,1 -trifluoro-2-(2-thieno[2,3- c]pyridin-7-yl-4-pyridinyl)-2-propanol (0.11 g, 0.34 mmol, Intermediate Y6) in tetrahydrofuran (2.5 mL) at -78 °C under a nitrogen atmosphere. After 5 min, a solution of (S)-N-((lE)-(3-chloro-2-pyridinyl)methylidene)-2-methyl-2- propanesulfmamide (0.075 g, 0.31 mmol, Intermediate XI 0) in tetrahydrofuran (0.50 mL) was added. After 10 min, the reaction mixture was allowed to warm to room temperature, saturated aqueous sodium bicarbonate was added, and the mixture was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and then concentrated. The residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (gradient elution; 1 : 1 to 1 :2 hexane-ethyl acetate) to give N-((R)-(3-chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)-2-methyl-2- propanesulfmamide (0.061 g) as a pale yellow tar.
Step 2. Hydrogen chloride (0.27 mL of a 4.0 M solution with 1,4-dioxane, 1.1 mmol) was added to a stirring solution of N-((R)-(3-chloro-2-pyridinyl)(7-(4- (2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-2- yl)methyl)-2-methyl-2-propanesulfinamide (0.061 g, 0.11 mmol, from Step 1) and methanol (1.1 mL) at room temperature. After 30 min, the reaction mixture was concentrated under a vacuum to give 2-(2-(2-((R)-amino(3-chloro-2- pyridinyl)methyl)thieno[2,3-c]pyridin-7-yl)-4-pyridinyl)-l , 1 , 1 -trifluoro-2- propanol hydrochloride (0.050 g) as a yellow solid. The material was used in the next step of the synthesis without purification.
Step 3. 4-(Dimethylamino)pyridine (1.3 mg, 11 μιηοΐ) was added to a stirring solution of 2-(2-(2-((R)-amino(3-chloro-2-pyridinyl)methyl)thieno[2,3- c]pyridin-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol hydrochloride (0.050 g, 0.11 mmol, from Step 2), N,N-dimethylformamide (1.1 mL), N,N- diisopropylethylamine (0.19 mL, 1.1 mmol), and cyclopropanesulfonyl chloride (0.011 mL, 0.11 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 16 h, more cyclopropanesulfonyl chloride (0.0069 mL, 0.068 mmol) was added. After 68 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, the filtrate was
concentrated. The residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (99: 1
dichloromethane-methanol) to afford N-((R)-(3-chloro-2-pyridinyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-2- yl)methyl)cyclopropanesulfonamide (0.022 g) as a pale yellow tar (mixture of two diastereomers).
1H NMR (400 MHz, CDC13) δ ppm 8.91 (br. s., 1 H), 8.86 - 8.75 (m, 1 H), 8.59 (br. s., 2 H), 7.75 (d, J= 8.0 Hz, 1 H), 7.64 (d, J= 4.5 Hz, 1 H), 7.54 (br. s., 1 H),
7.37 - 7.27 (m, 2 H), 6.71 (d, J= 8.5 Hz, 1 H), 6.54 (d, J= 8.5 Hz, 1 H), 3.69 (br. s., 1 H), 2.17 (m, 1 H), 1.80 (s, 3 H), 1.14 (m, 2 H), 0.72 (m, 2 H). m/z (ESI, pos. ion) 568.8 (M+H)+. GK-GK P IC50 (Binding) = 0.44 μΜ.
Example 8
4-Methoxy-N-(phenyl(7-(4-(2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)benzenesulfonamide

4-Methoxybenzenesulfonyl chloride (0.016 g, 0.075 mmol) was added to a stirring solution of 2-(2-(2-(amino(phenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol hydrochloride (0.035 g, 0.075 mmol, Intermediate Zl), N,N-diisopropylethylamine (0.013 mL, 0.075 mmol), and dichloromethane (5.0 mL) at room temperature. After 3 h, silica gel (1.0 g) was added, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (12 g, RediSep® normal phase column, gradient elution of 0% to 50% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to give 4-methoxy-N-(phenyl(7-(4-(2,2,2-trifluoro-l -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)benzenesulfonamide (0.015 g) as a colorless oil (mixture of 4 stereoisomers).
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.73 (d, J = 5.3 Hz, 1 H), 8.21 (s, 1 H), 7.98 (d, J = 7.6 Hz, 1 H), 7.75 (d, J = 7.9 Hz, 1 H), 7.70 - 7.60 (m, 2 H), 7.54 (d, J = 5.1 Hz, 1 H), 7.51 - 7.43 (m, 1 H), 7.34 - 7.23 (m, 5 H), 6.97 (s, 1 H), 6.88 - 6.78 (m, 2 H), 6.59 (d, J = 8.8 Hz, 1 H), 5.87 (d, J = 8.8 Hz, 1 H), 4.67 (s, 1 H), 3.74 (s, 3 H), 1.81 (s, 3 H). m/z (ESI, pos. ion) 598.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.73 μΜ.
Example 9
N-(Phenyl(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)benzenesulfonamide

Benzenesulfonyl chloride (0.019 mL, 0.15 mmol) was added to a stirring solution of 2-(2-(2-(amino(phenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol hydrochloride (0.035 g, 0.075 mmol, Intermediate Zl), N,N-diisopropylethylamine (0.065 mL, 0.38 mmol), and dichloromethane (5.0 mL) at room temperature. After 3 h, silica gel (1.0 g) was added and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (12 g RediSep® normal phase column, gradient elution of 0% to 50% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to give N-(phenyl(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -
benzothiophen-2-yl)methyl)benzenesulfonamide (0.0041 g) as a pale yellow oil (mixture of 4 stereoisomers).
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.74 (d, J= 5.3 Hz, 1 H), 8.20 (s, 1 H), 7.98 (d, J= 7.6 Hz, 1 H), 7.80 - 7.71 (m, 3 H), 7.58 - 7.35 (m, 5 H), 7.28 (s, 5 H), 7.02 - 6.94 (m, 1 H), 6.74 (d, J= 8.6 Hz, 1 H), 5.91 (d, J= 8.6 Hz, 1 H), 4.67 (s, 1 H), 1.81 (s, 3 H). m/z (ESI, pos. ion) 568.8 (M+H)+. GK-GKRP IC50 (Binding) = 2.4 μΜ.
Example 10
4-Methoxy-N-((2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)benzenesulfonamide
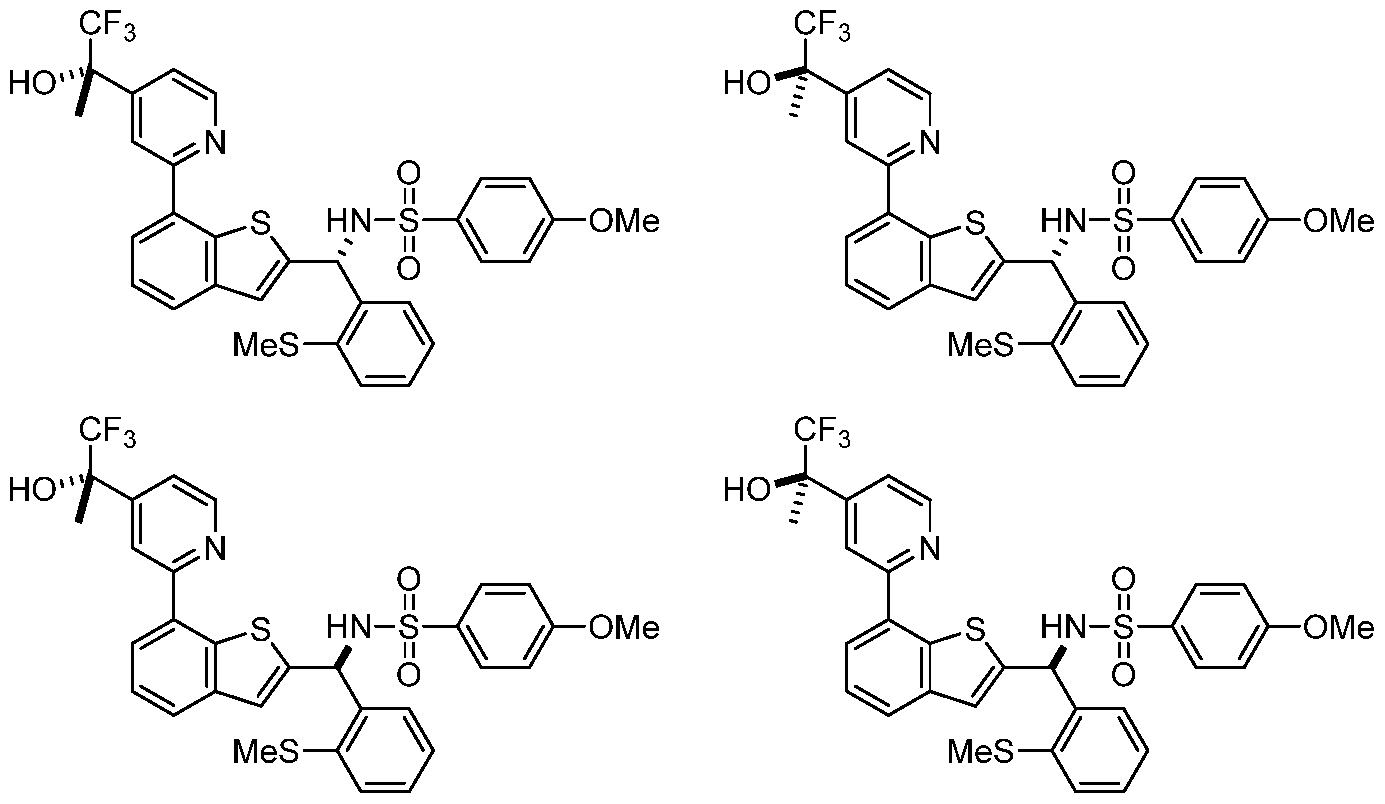
4-Methoxybenzenesulfonyl chloride (0.086 g, 0.42 mmol) was added to stirring solution of 2-(2-(2-(amino(phenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol hydrochloride (0.071 g, 0.14 mmol, Intermediate Z2), N,N-diisopropylethylamine (0.74 mL, 0.70 mmol), and dichloroethane (5.0 mL) at room temperature. After 48 h, the reaction mixture was allowed to cool to room temperature, silica gel (2.0 g) was added and the volatiles were removed under a vacuum. The residue was subjected to flash
chromatography on silica gel (12 g, RediSep normal phase column, gradient elution of 0% to 50% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to give 4-methoxy-N-((2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)benzenesulfonamide (0.0027 g) as a pale yellow oil (mixture of 4 stereoisomers).
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.75 (dd, J= 0.6, 5.3 Hz, 1 H), 8.21 (s, 1 H), 8.06 - 7.91 (m, 1 H), 7.76 - 7.63 (m, 3 H), 7.55 (d, J= 4.8 Hz, 1 H), 7.51 - 7.40 (m, 2 H), 7.36 - 7.24 (m, 2 H), 7.22 - 7.13 (m, 1 H), 6.95 - 6.86 (m, 2 H), 6.84 - 6.78 (m, 1 H), 6.70 (d, J= 8.5 Hz, 1 H), 6.28 (d, J= 8.0 Hz, 1 H), 4.70 (s, 1 H), 3.79 (s, 3 H), 2.41 (s, 3 H), 1.81 (s, 3 H). m/z (ESI, pos. ion) 644.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.012 μΜ.
Examples 11 and 12
N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((R)-(2-
(methylsulfanyl)phenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

EtN( -Pr)2, CICH2CH2CI
4. chiral chromatography
Step 2
Step 1. Titanium(IV) ethoxide (34 mL, 160 mmol) was added to a stirring solution of 2-(methylsulfanyl)benzaldehyde (5.0 g, 33 mmol), (S)-2-methyl-2- propanesulfinamide (4.0 g, 33 mmol, AK Scientific, Mountain View, CA), and
dichloromethane (66 mL) at room temperature. After 24 h, water (10 mL) was added and the resulting suspension was filtered through a pad of diatomaceous earth. The two layers comprising the filtrate were separated, and the organic material was washed with brine, dried (magnesium sulfate), filtered, and concentrated. The residue was subjected to flash chromatography on silica gel (120 g RediSep® normal phase column, gradient elution of 0% to 20% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to give (S)-2-methyl-N-((lE)-(2- (methylsulfanyl)phenyl)methylidene)-2-propanesulfinamide (5.8 g) .
Step 2. n-Butyllithium (0.74 mL of a 1.6 M solution in hexanes, 1.2 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.20 g, 0.62 mmol, Intermediate Y3) in tetrahydrofuran (5.0 mL) at -78 °C under a nitrogen atmosphere. After 1 h, a solution of (S)-2-methyl-N-((l E)-(2-(methylsulfanyl)phenyl)methylidene)-2- propanesulfmamide (0.17 g, 0.68 mmol, from Step 1) in tetrahydrofuran (3.0 mL) was added. After 10 min, saturated aqueous sodium bicarbonate (2.0 mL) was added and the mixture was allowed to warm to room temperature. Silica gel (2.0 g) was added, and then the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (120 g RediSep® normal phase column, gradient elution of 30% to 50% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE). The isolated material was dissolved with methanol then hydrogen chloride (1.0 M solution with diethyl ether) was added. The reaction mixture was stirred at room temperature for 3 h and then was concentrated. The residue was dissolved with dichloroethane and N,N-diisopropylethylamine, cyclopropanesulfonyl chloride (1.1 mL, 10 mmol, Matrix Scientific, Columbia, SC) was added at rt. The reaction was then heated to reflux. After 48 h, the reaction mixture was allowed to cool to room temperature and silica gel (1.0 g) was added. The volatiles were removed under a vacuum and the residue was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, 40% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE). The isolated material was resolved using preparative SFC (Chiralpak® AS-H column) (250
mm x 20 mm, 5 μηι) eluting with 80% liquid C02 in 20% methanol (with 20 mM NH3) at a flow rate of 70 mL/min) to give two products in greater than 99% enantiomeric excess.
N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-(( 1 S)-2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide Third eluting peak (peak #3); Example 11
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.78 (d, J= 5.4 Hz, 1 H), 8.23 (s, 1 H), 8.01 (d, J= 7.6 Hz, 1 H), 7.81 (d, J= 7.9 Hz, 1 H), 7.68 - 7.61 (m, 1 H), 7.58
- 7.28 (m, 5 H), 7.06 (d, J= 1.2 Hz, 1 H), 6.55 (d, J= 8.9 Hz, 1 H), 6.45 (d, J = 8.9 Hz, 1 H), 4.71 (s, 1 H), 2.48 (s, 3 H), 2.36 - 2.22 (m, 1 H), 1.81 (s, 3 H), 1.07
- 0.68 (m, 4 H). m/z (ESI, pos. ion) 578.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.0090 μΜ.
N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide Fourth eluting peak (peak #4); Example 12
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.82 - 8.74 (m, 1 H), 8.24 (s, 1 H), 8.06 - 7.97 (m, 1 H), 7.81 (dd, J = 0.9, 7.9 Hz, 1 H), 7.65 (dd, J= 1.5, 7.5 Hz, 1 H), 7.59 - 7.28 (m, 5 H), 7.06 (d, J= 1.2 Hz, 1 H), 6.55 (d, J= 8.9 Hz, 1 H), 6.45 (d, J= 8.8 Hz, 1 H), 4.70 (s, 1 H), 2.48 (s, 3 H), 2.37 - 2.22 (m, 1 H), 1.82 (s, 3 H), 1.07 - 0.68 (m, 4 H). m/z (ESI, pos. ion) 578.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.0050 μΜ.
Example 13
N-((R)-(2-Chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)- l-benzothiophen-2-yl)methyl)- l-methyl-6-oxo- 1 ,6-dihydro-3- pyridinesulfonamide

Step 1. A solution of bromine (2.3 mL, 46 mmol) and chloroform (10 mL) was added to a stirring solution of l-methyl-2-pyridone (5.0 mL, 456 mmol) and chloroform (46 mL) at reflux. After 1 h, the reaction mixture was cooled to room temperature. After 72 h, the reaction mixture was filtered and the filter cake was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, silica gel (1.0 g) was added to the organic material, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (120 g RediSep® normal phase column, gradient elution of 0 to 100% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to afford 5-bromo-l-methylpyridin-2(lH)-one (1.2 g).
Step 2. n-Butyllithium (3.7 mL, 5.9 mmol) was added to a stirring solution of 5-bromo-l-methylpyridin-2(lH)-one (1.0 g, 5.3 mmol, from Step 1) in tetrahydrofuran (27 mL) at -78 °C. After 10 min, the reaction mixture was sparged with sulfur dioxide (0.27 mL, 5.3 mmol) for 2 min and then allowed to warm to room temperature. The mixture was concentrated under a vacuum and the residue was dissolved with dichloromethane (20 mL), and NCS (0.71 g, 5.3 mmol) was added. After 90 min, the reaction mixture was filtered, silica gel (2.0 g) was added, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (40 g RediSep® normal phase
column, gradient elution of 0 to 100% ethyl acetate -hexane, Teledyne Isco, Lincoln, NE) to afford l-methyl-6-oxo-l,6-dihydro-3-pyridinesulfonyl chloride (0.050 g) as a yellow oil.
Step 3. l-Methyl-6-oxo-l,6-dihydro-3-pyridinesulfonyl chloride (0.050 g, 0.24 mmol, from Step 2) was added to a stirring solution of 2-(2-(2-((R)- amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol hydrochloride (0.040 g, 0.080 mmol, Intermediate Z3), N,N- diisopropylethylamine (0.14 mL, 0.80 mmol), and dichloromethane (3.0 mL) at room temperature. After 1 h, silica gel (1.0 g) was added and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (12 g RediSep® normal phase column, gradient elution of 30%> to 100% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to give N-((R)-(2- chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)- 1 -methyl-6-oxo- 1 ,6-dihydro-3 -pyridinesulfonamide (0.0055 g) as a colorless solid (mixture of 2 diastereomers).
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.78 (d, J= 5.3 Hz, 1 H), 8.23 (s, 1 H), 8.02 (d, J= 7.5 Hz, 1 H), 7.91 (d, J= 2.8 Hz, 1 H), 7.79 (d, J= 7.7 Hz, 1 H), 7.63 - 7.26 (m, 7 H), 7.00 (s, 1 H), 6.89 (d, J= 9.2 Hz, 1 H), 6.30 - 6.20 (m, 2 H), 4.71 (s, 1 H), 3.31 (s, 3 H), 1.82 (s, 3 H). m/z (ESI, pos. ion) 633.8 (M+H)+. GK- GKRP IC50 (Binding) = 0.055 μΜ.
Example 14
N-((R)-(2-Chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-6-methoxy-3-pyridinesulfonamide

6-Methoxy-3-pyridinesulfonyl chloride (0.025 mL, 0.12 mmol, Acros, Geel, Belgium) was added to a stirring solution of 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol hydrochloride (0.050 g, 0.10 mmol, Intermediate Z3), N,N- diisopropylethylamine (0.087 mL, 0.50 mmol), N,N-(dimethylamino)pyridine (0.12 mg, 1.0 μιηοΐ), and dichloroethane (10 mL) at room temperature. The mixture was heated at reflux for 24 h and then allowed to cool to room temperature. Silica gel (1.0 g) was added, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 20% to 50% ethyl acetate- hexane, Teledyne Isco, Lincoln, NE) to give N-((R)-(2-chlorophenyl)(7-(4- (2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-6-methoxy-3-pyridinesulfonamide (0.0080 g) as a yellow oil (mixture of 2 diastereomers).
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.76 (d, J= 5.3 Hz, 1 H), 8.48 (d, J = 2.5 Hz, 1 H), 8.23 (s, 1 H), 8.02 (d, J= 7.6 Hz, 1 H), 7.86 (dd, J= 2.6, 8.8 Hz, 1 H), 7.76 (d, J= 7.9 Hz, 1 H), 7.62 - 7.45 (m, 3 H), 7.43 - 7.33 (m, 1 H), 7.32 - 7.24 (m, 2 H), 6.92 (s, 2 H), 6.69 (d, J= 8.9 Hz, 1 H), 6.28 (d, J= 7.0 Hz, 1 H), 4.69 (s, 1 H), 3.87 (s, 3 H), 1.82 (s, 3 H). m/z (ESI, pos. ion) 633.8 (M+H)+. GK- GKRP IC50 (Binding) = 0.20 μΜ.
Example 15
6-Amino-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-3- pyridinesulfonamide

6-Aminopyridine-3-sulfonyl chloride (0.039 g, 0.20 mmol) was added to a stirring mixture of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol hydrochloride (0.10 g, 0.20 mmol, Intermediate Z3), dichloroethane (10 mL), N,N- diisopropylethylamine (0.17 mL, 1.0 mmol), and N,N-(dimethylamino)pyridine (0.024 mg, 0.20 μιηοΐ) at room temperature. The mixture was heated at reflux for 24 h and then allowed to cool to room temperature. Silica gel (1.0 g) was added, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 0% to 100% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE). The isolated material was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, elution of 5% methanol (with 2.0 M NH3) in dichloromethane, Teledyne Isco, Lincoln, NE) to afford 6-amino-N-((R)-(2- chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-3-pyridinesulfonamide (0.0058 g) as a colorless solid (mixture of 2 diastereomers).
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.79 (dd, J= 0.7, 5.2 Hz, 1 H), 8.33 (d, J= 2.0 Hz, 1 H), 8.22 (s, 1 H), 8.09 - 7.96 (m, 1 H), 7.76 (dd, J= 0.9, 7.9 Hz, 1 H), 7.67 - 7.44 (m, 4 H), 7.42 - 7.24 (m, 3 H), 6.94 - 6.83 (m, 1 H), 6.72 (br. s.,
1 H), 6.49 - 6.33 (m, 1 H), 6.20 (s, 1 H), 5.54 (br. s., 2 H), 4.73 (br. s., 1 H), 1.82 (s, 3 H). m/z (ESI, pos. ion) 618.8 (M+H)+. GK-GK P IC50 (Binding) = 0.58 μΜ.
Example 16
N-((R)-(2-Chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-fluorobenzenesulfonamide

diastereomers).
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.76 (d, J= 5.3 Hz, 1 H), 8.22 (s, 1 H), 8.01 (d, J= 7.5 Hz, 1 H), 7.85 - 7.71 (m, 3 H), 7.56 (d, J= 4.8 Hz, 1 H), 7.53 - 7.43 (m, 2 H), 7.39 - 7.24 (m, 3 H), 7.19 - 7.09 (m, 2 H), 6.88 (s, 2 H), 6.27 (br.
s., 1 H), 4.71 (s, 1 H), 1.82 (s, 3 H). m/z (ESI, pos. ion) 620.9 (M+H)+. GK- GKRP IC50 (Binding) = 0.65 μΜ.
Example 17
N-((R)-(2-Chlorophenyl)(7-(4-(l-hydroxyethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (3.8 mL of a 1.6 M solution in hexanes, 6.1 mmol) was added to a stirring solution of l-(2-(l-benzothiophen-7-yl)-4- pyridinyl)ethanol (0.77 g, 3.1 mmol, Intermediate Z4) and tetrahydrofuran (15 mL) at -78 °C under a nitrogen atmosphere. After 20 min, a solution of N-((S,E)- (2-chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (0.60 mg, 2.4 mmol, Intermediate Yl) in tetrahydrofuran (3.0 mL) was added. After 5 min, saturated aqueous sodium bicarbonate was added and the mixture was concentrated. Methanol (3.0 mL) was added, the mixture was filtered, and the filtrate was concentrated. The residue was subjected to reversed-phase preparative HPLC (Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 μπι) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 0% to 100% over 21 min) to afford (S)-N-((R)-(2-
chlorophenyl)(7-(4-( 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfinamide (0.24 g, containing about 15 mol% of (S)-N-((S)- (2-chlorophenyl)(7-(4-( 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide).
Step 2. Hydrogen chloride (2.4 mL of a 1.0 M solution with diethyl ether, 2.4 mmol) was added to a stirring solution of (S)-N-((R)-(2-chlorophenyl)(7-(4- ( 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (0.24 g, containing about 15 mol% of (S)-N-((S)-(2- chlorophenyl)(7-(4-( 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfinamide), from Step 1) and methanol (2.4 mL) at room temperature. After 2 h, the reaction mixture was concentrated under a vacuum to provide 1 -(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)ethanol hydrochloride (0.21 g).
Step 3. N,N-(Dimethylamino)pvridine (0.029 g, 0.24 mmol) was added to a stirring solution of l-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)ethanol hydrochloride (0.21 g, 0.48 mmol, from Step 2), dichloromethane (2.4 mL), N,N-diisopropylethylamine (0.25 mL, 1.4 mmol), and cyclopropanesulfonyl chloride (0.098 mL, 0.96 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 24 h, the reaction mixture was concentrated. The residue was subjected to reversed-phase preparative HPLC (Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 μιη) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 0% to 100% over 21 min) to provide N-((R)-(2-chlorophenyl)(7-(4-(l- hydroxyethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.025 g) as a white solid.
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.66 (d, J= 5.1 Hz, 1 H), 8.09 - 7.91 (m, 2 H), 7.75 (dd, J= 7.7, 17.2 Hz, 2 H), 7.56 - 7.22 (m, 5 H), 7.06 (d, J= 1.2 Hz, 1 H), 6.59 (d, J= 8.9 Hz, 1 H), 6.40 (d, J= 8.9 Hz, 1 H), 4.90 (q, J= 6.4 Hz,
1 H), 3.54 (br. s., 1 H), 2.44 - 2.25 (m, 1 H), 1.52 - 1.31 (m, 3 H), 1.07 - 0.67 (m, 4 H). m/z (ESI, pos. ion) 499.0 (M+H)+. GK-GK P IC50 (Binding) = 1.5 μΜ.
Example 18
N-((R)-(2-Chlorophenyl)(7-(4-(l-hydroxycyclobutyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (2.2 mL of a 1.6 M solution with hexanes, 3.5 mmol) was added to a stirring solution of l-(2-(l-benzothiophen-7-yl)-4- pyridinyl)cyclobutanol (0.49 g, 1.7 mmol, Intermediate Z5) and tetrahydrofuran (17 mL) at -78 °C under a nitrogen atmosphere. After 30 min, a solution of N- ((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (0.21 g, 0.87 mmol, Intermediate Yl) in tetrahydrofuran (3.0 mL) was added followed by saturated aqueous sodium bicarbonate (1.0 mL). The resulting mixture was allowed to warm to room temperature and then partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, silica gel (1.0 g) was added to the organic material, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (120 g RediSep® normal phase column, elution of 60% ethyl acetate-hexane, Teledyne
Isco, Lincoln, NE) to afford N-((R)-(2-chlorophenyl)(7-(4-( 1 - hydroxycyclobutyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfinamide (0.15 g) as a clear oil.
Step 2. Hydrogen chloride (0.45 mL of a 1.0 M solution with water, 15 mmol) was added to a stirring solution of N-((R)-(2-chlorophenyl)(7-(4-(l- hydroxycyclobutyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (0.15 g, 0.29 mmol, from Step 1) and ethyl acetate (2.0 mL). After 1 h, the reaction mixture was concentrated under a vacuum. The residue was partitioned between saturated aqueous sodium bicarbonate and
dichloromethane. The layers were separated, the aqueous material was washed with dichloromethane (2x), and the combined organic extract was washed with brine. The combined extracts were dried (magnesium sulfate), filtered, and concentrated under a vacuum to give l-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)cyclobutanol (0.12 g) as a clear oil. The material was used in the next step of the synthesis without purification.
Step 3. N,N-(Dimethylamino)pyridine (0.0036 mg, 0.029 mmol) was added to a stirring solution of l-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)cyclobutanol (0.12 mg, 0.29 mmol, from Step 2), N,N-diisopropylethylamine (0.25 mL, 1.5 mmol), N,N-dimethylformamide (3.0 mL), and cyclopropanesulfonyl chloride (0.30 mL, 2.9 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 15 h, the reaction mixture was filtered and the filtrate was subjected to reversed-phase preparative HPLC (Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 μιη) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 0% to 100% over 10 min). The isolated material was subjected to flash chromatography on silica gel (40 g RediSep® normal phase column, gradient elution of 0% to 100% ethyl acetate-hexane, Teledyne Isco, Lincoln, NE) to provide N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy cyclobutyl)-2-pyridinyl)- 1 -
benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.014 g) as a colorless solid.
1H NMR (300 MHz, methanol-d4) δ ppm 8.69 (d, J= 5.1 Hz, 1 H), 8.17 - 8.08 (m, 1 H), 7.92 - 7.83 (m, 1 H), 7.81 - 7.71 (m, 2 H), 7.57 - 7.30 (m, 5 H), 7.01 (d, J= 1.2 Hz, 1 H), 6.44 (s, 1 H), 2.64 - 2.51 (m, 2 H), 2.50 - 2.36 (m, 2 H), 2.30 (tt, J= 4.9, 7.9 Hz, 1 H), 2.20 - 2.03 (m, 1 H), 1.96 - 1.77 (m, 1 H), 1.10 - 0.91 (m, 2 H), 0.90 - 0.78 (m, 1 H), 0.77 - 0.64 (m, 1 H). m/z (ESI, pos. ion) 525.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.26 μΜ.
Example 19
N-((R)-(2-Chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (2.3 mL of a 1.6 M solution in hexanes, 3.7 mmol) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-l- ((tert-butyl(dimethyl)silyl)oxy)-2-propanol (0.68 g, 1.7 mmol, Intermediate Z6) and tetrahydrofuran (8.5 mL) at -78°C under a nitrogen atmosphere. After 20 min, a solution of N-((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2-
propanesulfinamide (0.31 g, 1.3 mmol, Intermediate Yl) in tetrahydrofuran (5.0 mL) was added. After 5 min, saturated aqueous sodium bicarbonate (3.0 mL) was added. The reaction mixture was concentrated under a vacuum, the residue was subjected to reversed-phase preparative HPLC (Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 μιη) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 0% to 100% over 21 min) to afford (S)-N-((R)-(7-(4-(2-((tert-butyl(dimethyl)silyl)oxy)- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)(2-chlorophenyl)methyl)-2- methyl-2-propanesulfinamide (0.13 ) as a white solid.
Step 2. Hydrogen chloride (2.0 mL of a 1.0 M solution with diethyl ether, 2.0 mmol) was added to a stirring solution of (S)-N-((R)-(7-(4-(2-((tert- butyl(dimethyl)silyl)oxy)- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)(2-chlorophenyl)methyl)-2-methyl-2 -propanesulfinamide (130 mg, 0.20 mmol, from Step 1) in methanol (2.0 mL) at room temperature . After 2 h, the reaction mixture was concentrated under a vacuum to provide 2-(2- (2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2- propanediol hydrochloride (0.093 g).
Step 3. N,N-(dimethylamino)pvridine (0.012 g, 0.10 mmol) was added to a stirring solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,2-propanediol hydrochloride (0.093 mg, 0.20 mmol, from Step 2), dichloromethane (1.0 mL), N,N-diisopropylethylamine (0.18 mL, 1.0 mmol), and cyclopropanesulfonyl chloride (0.021 mL, 0.20 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 24 h, the reaction mixture was concentrated. The residue was subjected to reverse-phase preparative HPLC (Phenomenex Gemini C18 column (Phenomenex, Inc., Torrance, CA)(150 x 30 mm, 10 μιη) eluting with 0.10% trifluoroacetic acid in acetonitrile-water, gradient of 0% to 100% over 21 min) to provide N-((R)-(2- chlorophenyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -
benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.0025 g) as a tan oil (mixture of 2 diastereomers).
1H NMR (300 MHz, acetonitrile-d3) δ ppm 8.73 - 8.63 (m, 1 H), 8.13 (s, 1 H), 8.07 - 7.97 (m, 1 H), 7.85 - 7.66 (m, 1 H), 7.77 (dd, J= 8.2, 18.4 Hz, 2 H), 7.56 - 7.33 (m, 5 H), 7.08 (d, J= 1.2 Hz, 1 H), 6.56 (d, J= 8.8 Hz, 1 H), 6.40 (d, J= 9.1 Hz, 1 H), 3.74 - 3.65 (m, 1 H), 3.64 - 3.55 (m, 1 H), 3.00 (t, J= 6.3 Hz, 1 H), 2.40 - 2.24 (m, 1 H), 1.49 (s, 3 H), 1.08 - 0.81 (m, 4 H). m/z (ESI, pos. ion) 528.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.058 μΜ.
Example 20
6-Chloro-N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-3- pyridinesulfonamide

4-Dimethylaminopyridine (3.43 mg, 0.028 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (130 mg, 0.281 mmol, Intermediate X5), 6- chloro-3-pyridinesulfonyl chloride (71.5 mg, 0.337 mmol, Beta Pharma, Branford, CT), and diisopropylethylamine (147 μί, 0.842 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 1 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under
reduced pressure to deliver 6-chloro-N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 3-pyridinesulfonamide (144 mg) as a white foam.
1H NMR (400 MHz, CDC13) δ ppm 8.81 (d, J= 5.3 Hz, 1 H), 8.72 (d, J= 2.3 Hz,
1 H), 8.15 (s, 1 H), 7.92 - 7.85 (m, 2 H), 7.69 (d, J= 7.8 Hz, 1 H), 7.50 - 7.43 (m,
2 H), 7.38 (dd, J= 2.1 , 7.1 Hz, 1 H), 7.34 - 7.17 (m, 4 H), 6.91 (d, J= 1.0 Hz, 1 H), 6.32 (d, J= 8.2 Hz, 1 H), 5.91 (d, J= 8.0 Hz, 1 H), 2.52 (br. s., 1 H), 1.85 (s,
3 H). m/z (ESI, pos. ion) 637.7 (M+H)+. GK-GKRP IC50 (Binding) = 2.51 μΜ.
Example 21
N-((R)-(2-Chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-6-oxo-l,6-dihydro-
3-pyridinesulfonamide

Hydrogen chloride (196 μΐ, of a 4 M solution in 1,4-dioxane, 0.783 mmol) was added to a solution of 6-chloro-N-((R)-(2-chlorophenyl)(7-(4-((lR)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-3-pyridinesulfonamide (125 mg, 0.196 mmol, Example 20) in 1,4- dioxane/water (1 : 1, 2 mL) and the mixture was stirred in a sealed tube at 120 °C for 24 h. The mixture was allowed to cool to room temperature, filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgSC^), filtered, and
concentrated under reduced pressure to deliver N-((R)-(2-chlorophenyl)(7-(4-
(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-6-oxo-l,6-dihydro-3-pyridinesulfonamide (71 mg) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 11.96 (br. s, 1 H), 9.24 (br. s, 1 H), 8.85 (d, J= 5.3 Hz, 1 H), 8.31 (s, 1 H), 8.12 (d, J= 7.6 Hz, 1 H), 7.86 (d, J= 7.6 Hz, 1 H), 7.69 - 7.59 (m, 3 H), 7.51 (t, J= 7.7 Hz, 1 H), 7.45 - 7.37 (m, 2 H), 7.37 - 7.28 (m, 2 H), 7.01 (s, 1 H), 6.90 (s, 1 H), 6.23 (d, J= 9.6 Hz, 1 H), 6.18 (br. s., 1 H), 1.80 (s, 3 H). m/z (ESI, pos. ion) 619.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.041 μΜ.
Example 22
N-((R)-(2-Chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

4-Dimethylaminopyridine (26.2 mg, 0.215 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (497 mg, 1.07 mmol, Intermediate X5), cyclopropanesulfonyl chloride (142 μΐ^, 1.40 mmol, Matrix Scientific, Columbia, SC), and diisopropylethylamine (560 μί, 3.22 mmol) in DMF (5 mL) and the mixture was stirred at room temperature for 20 h. The mixture was concentrated under reduced pressure, and the resulting yellow oil was dissolved in DCM, absorbed onto silica gel, and purified by flash chromatography (100 g of silica gel, 20% to 60% EtOAc/hexanes) and again by flash chromatography (100 g of silica gel, 30% to 60% EtOAc/hexanes) to deliver N-((R)-(2-chlorophenyl)(7-(4-
(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (487 mg) as a white foam.
1H NMR (400 MHz, CDC13) δ ppm 8.81 (d, J= 5.3 Hz, 1 H), 8.14 (s, 1 H), 7.88 (d, J= 7.6 Hz, 1 H), 7.74 (d, J= 7.8 Hz, 1 H), 7.64 (dd, J= 1.4, 7.6 Hz, 1 H), 7.50 - 7.41 (m, 3 H), 7.41 - 7.29 (m, 2 H), 7.07 (s, 1 H), 6.42 (d, J= 8.0 Hz, 1 H), 5.49 (d, J= 8.0 Hz, 1 H), 2.72 (br. s, 1 H), 2.33 - 2.22 (m, 1 H), 1.84 (s, 3 H), 1.22 - 1.06 (m, 2 H), 0.92 - 0.83 (m, 1 H), 0.81 - 0.71 (m, 1 H). m/z (ESI, pos. ion) 566.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.190 μΜ.
Example 23
N-((R)-(2-Chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- methoxybenzenesulfonamide

4-Dimethylaminopyridine (1.48 mg, 0.012 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (56 mg, 0.121 mmol, Intermediate X5), 4- methoxybenzenesulfonyl chloride (30.0 mg, 0.145 mmol, Sigma-Aldrich, St. Louis, MO), and diisopropylethylamine (63.1 μΐ,, 0.363 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 1.5 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The
combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure to deliver N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-4- methoxybenzenesulfonamide (70 mg) as a colorless film.
1H NMR (400 MHz, CDC13) δ ppm 8.79 (d, J= 5.1 Hz, 1 H), 8.12 (s, 1 H), 7.86 (d, J= 7.4 Hz, 1 H), 7.72 (d, J= 8.8 Hz, 2 H), 7.67 (d, J= 7.8 Hz, 1 H), 7.49 - 7.40 (m, 3 H), 7.33 - 7.28 (m, 1 H), 7.25 - 7.17 (m, 2 H), 6.89 (s, 1 H), 6.83 (d, J = 8.8 Hz, 2 H), 6.22 (d, J= 7.4 Hz, 1 H), 5.50 (d, J= 7.4 Hz, 1 H), 3.81 (s, 3 H), 2.29 (br. s., 1 H), 1.84 (s, 3 H). m/z (ESI, pos. ion) 632.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.282 μΜ.
Example 24
N-((R)-(2-Chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-3- methoxybenzenesulfonamide

4-Dimethylaminopyridine (1.48 mg, 0.012 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (56 mg, 0.121 mmol, Intermediate X5), 3- methoxybenzenesulfonyl chloride (20.6 μί, 0.145 mmol, Sigma- Aldrich, St. Louis, MO), and diisopropylethylamine (63.1 μΐ,, 0.363 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 1.5 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined,
basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgSC^), filtered, and concentrated under reduced pressure to deliver N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 - methoxybenzenesulfonamide (68 mg) as a white foam.
1H NMR (400 MHz, CDC13) δ ppm 8.80 (d, J= 5.3 Hz, 1 H), 8.13 (s, 1 H), 7.87 (d, J= 7.4 Hz, 1 H), 7.68 (d, J= 7.8 Hz, 1 H), 7.50 - 7.37 (m, 4 H), 7.32 - 7.26 (m, 3 H), 7.24 - 7.17 (m, 2 H), 7.00 (d, J= 7.4 Hz, 1 H), 6.89 (s, 1 H), 6.26 (d, J = 7.6 Hz, 1 H), 5.57 (d, J= 7.6 Hz, 1 H), 3.75 (s, 3 H), 1.85 (s, 3 H). m/z (ESI, pos. ion) 632.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.406 μΜ.
Example 25
N'-((R)-(2-Chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-N,N- dimethylsulfamide

4-Dimethylaminopyridine (2.64 mg, 0.022 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (50 mg, 0.108 mmol, Intermediate X5), dimethylsulfamoyl chloride (15.1 μΐ,, 0.140 mmol, Acros, Geel, Belgium), and diisopropylethylamine (56.4 μΐ^, 0.324 mmol) in DMF (1 mL) and the mixture was stirred at room temperature for 3 d. The mixture was diluted with MeOH, filtered, and purified by reverse phase HPLC (Phenomenex Gemini-NX 10μ CI 8 11 OA, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted
with DCM (2 x 20 mL). The combined organic layers were dried (MgSC^), filtered, and concentrated under reduced pressure to deliver N'-((R)-(2- chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifiuoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)-N,N-dimethylsulfamide (32 mg) as a white solid.
1H NMR (400 MHz, CDC13) δ ppm 8.83 (d, J= 5.3 Hz, 1 H), 8.15 (s, 1 H), 7.89 (d, J= 7.4 Hz, 1 H), 7.75 (d, J= 7.6 Hz, 1 H), 7.64 (d, J= 7.2 Hz, 1 H), 7.50 - 7.44 (m, 2 H), 7.44 - 7.37 (m, 2 H), 7.33 (dt, J= 1.0, 7.8 Hz, 1 H), 7.07 (s, 1 H), 6.31 (d, J= 7.6 Hz, 1 H), 5.29 (d, J= 7.8 Hz, 1 H), 2.74 (s, 6 H), 1.85 (s, 3 H). m/z (ESI, pos. ion) 669.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.641 μΜ.
Example 26
N-((R)-(2-Chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- -benzothiophen-2-yl)methyl)ethanesulfonamide

Ethanesulfonyl chloride (10.3 μΐ,, 0.109 mmol, Sigma-Aldrich, St. Louis, MO) was added to a mixture of (2R)-2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (42 mg, 0.091 mmol, Intermediate X5) and diisopropylethylamine (47.3 μί, 0.272 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 2 h. Ethanesulfonyl chloride (10.3 μΐ^, 0.109 mmol) was added and the reaction was stirred at room temperature for 1.5 h to give a mixture. The reaction was then repeated using DCM as the solvent. Ethanesulfonyl chloride (12.0 μΐ^, 0.127 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (49 mg, 0.106 mmol, Intermediate X5) and diisopropylethylamine (55.2
μί, 0.318 mmol) in DCM (0.7 mL) and the mixture was stirred at room temperature for 1 h. Ethanesulfonyl chloride (10.3 μί, 0.109 mmol) was added and the reaction was stirred at room temperature for 1.5 h to give a second mixture. The resulting reaction mixtures from both runs were combined, diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 1 ΙθΑ, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgSC^), filtered, and concentrated under reduced pressure to deliver N-((R)-(2- chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)ethanesulfonamide (12 mg) as a white foam.
1H NMR (400 MHz, CDC13) δ ppm 8.82 (d, J= 3.9 Hz, 1 H), 8.16 (s, 1 H), 7.89 (d, J= 7.4 Hz, 1 H), 7.76 (d, J= 7.6 Hz, 1 H), 7.61 (d, J= 6.7 Hz, 1 H), 7.52 - 7.41 (m, 3 H), 7.41 - 7.30 (m, 2 H), 7.12 (br s, 1 H), 6.40 (d, J= 7.8 Hz, 1 H), 5.36 (d, J= 7.6 Hz, 1 H), 3.12 - 2.87 (m, 2 H), 1.84 (s, 3 H), 1.33 (t, J = 6.8 Hz, 3 H). m/z (ESI, pos. ion) 554.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.755 μΜ.
Example 27
N-((R)-(2-Chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)methanesulfonamide

Methanesulfonyl chloride (8.42 μί, 0.109 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (42 mg, 0.091 mmol, Intermediate X5) and diisopropylethylamine (47.3 μΐ^, 0.272 mmol) in DMF (0.7 mL) and the mixture
was stirred at room temperature for 1 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 1 lOA, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure to deliver N-((R)-(2- chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)methanesulfonamide (44 mg) as a colorless film.
1H NMR (400 MHz, CDC13) δ ppm 8.82 (d, J= 5.3 Hz, 1 H), 8.16 (s, 1 H), 7.90 (d, J= 7.4 Hz, 1 H), 7.77 (d, J= 7.8 Hz, 1 H), 7.61 (dd, J= 1.6, 7.4 Hz, 1 H), 7.52 - 7.43 (m, 3 H), 7.42 - 7.31 (m, 2 H), 7.13 (s, 1 H), 6.44 (d, J= 7.8 Hz, 1 H), 5.43 (d, J= 7.8 Hz, 1 H), 2.90 (s, 3 H), 1.85 (s, 3 H). m/z (ESI, pos. ion) 540.8 (M+H)+. GK-GKRP ICso (Binding) = 1.41 μΜ.
Example 28
N-((R)-(3-(Dimethylamino)-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro-l- hydroxy-l-methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 2. Montmorillonite K10 (500 mg) was added to a mixture of 7-(4- (( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophene-2- carbaldehyde (400 mg, 1.14 mmol) and cyclopropanesulfonamide (152 mg, 1.25 mmol, Sigma- Aldrich, St. Louis, MO) in toluene (30 mL) and the mixture was heated at reflux for 18 h while water was removed using a Dean-Stark trap. The reaction was allowed to cool to room temperature and filtered. The filter residue was washed with DCM (100 mL). The combined filtrate and washings were concentrated under reduced pressure to afford N-((lE)-(7-(4-((lR)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methylidene)cyclopropanesulfonamide (401 mg) as a light-yellow solid.
Step 3. THF (3 mL) was added into a flame-dried round-bottomed flask charged with magnesium turnings (48.1 mg, 1.98 mmol). 2-Bromo-N,N- dimethylaniline (158 mg, 0.792 mmol, Sigma-Aldrich, St. Louis, MO) was added, and the mixture was stirred at room temperature for 20 min. Iodine (10.1
mg, 0.040 mmol) was added and the mixture was stirred at room temperature for 20 min and at 60 °C for 20 min. The mixture was cooled to -78 °C and a solution of N-(( 1 E)-(7-(4-(( 1 R)-2,2,2-trifiuoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methylidene)cyclopropanesulfonamide (60 mg, 0.132 mmol) in THF (1 mL) was added dropwise. The cooling bath was removed and the mixture was stirred at room temperature for 30 min. Saturated aqueous NH4C1 (75 mL) was added and the mixture was extracted with EtOAc (2 x 100 mL). The combined organic layers were washed with brine (200 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 25% EtO Ac/hex anes). The residue was subjected to preparative SFC (Chiralpak® AD-H column) (150 mm x 21 mm, 5 μιη) eluting with 70% liquid C02 in 30% ethanol (with 0.1% NH4OH) at a flow rate of 65 mL/min) to afford N-((R)-(3-(dimethylamino)-2-pyridinyl)(7-(4-((lR)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (16 mg) as an off-white solid.
1H NMR (400 MHz, methanol-d4) δ ppm 9.40 (d, J= 5.1 Hz, 1 H), 8.90 (s, 1 H), 8.52 (d, J= 7.4 Hz, 1 H), 8.41 (d, J= 7.8 Hz, 1 H), 8.28 (d, J= 7.6 Hz, 1 H), 8.23 (d, J= 4.7 Hz, 1 H), 8.12 (t, J= 7.7 Hz, 1 H), 8.06 - 7.94 (m, 2 H), 7.88 (t, J= 6.9 Hz, 1 H), 7.74 (s, 1 H), 7.25 (s, 1 H), 5.49 (s, 6 H), 2.91 - 2.80 (m, 1 H), 2.46 (s, 3 H), 1.69 - 1.57 (m, 2 H), 1.43 - 1.31 (m, J= 7.6 Hz, 2 H). m/z (ESI, pos. ion) 575.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.159 μΜ.
Example 29
N-((R)-(3-Chloro-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide


Step 1. n-Butyllithium (3.21 mL of a 1.6 M solution in hexanes, 5.13 mmol) was added to a solution of (2R)-2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (830 mg, 2.57 mmol, Intermediate X4) in THF (5 mL) at -78 °C and the mixture was stirred at -78 °C for 15 min. A solution of (S)-N- ((lE)-(3-chloro-2-pyridinyl)methylidene)-2-methyl-2-propanesulfinamide (628 mg, 2.57 mmol) in THF (3 mL) was added dropwise and the resulting mixture was allowed to warm slowly. Saturated aqueous NH4C1 (40 mL) was added and the mixture was allowed to warm to room temperature. The solution was partitioned between aqueous NH4C1 (25 mL of saturated NH4C1 and 25 mL of H20) and EtOAc (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic layers were washed with brine (100 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 20% to 100% EtOAc/hexanes) to afford (S)-N-((R)-(3-chloro-2-pyridinyl)(7-(4- (( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (601 mg) as a light-yellow solid.
Step 2. A mixture of (S)-N-((R)-(3-chloro-2-pyridinyl)(7-(4-((lR)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-
2-methyl-2-propanesulfinamide (580 mg, 1.02 mmol), hydrogen chloride (7.1 mL of a 4 M solution in 1,4-dioxane, 204 mmol), and MeOH (2 mL) was stirred at room temperature for 2 h. The mixture was concentrated under reduced pressure and the residue was dissolved in saturated aqueous NaHC03 (100 mL). The mixture was filtered and the filter residue was dissolved in MeOH (50 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 100% EtOAc) to afford (2R)-2-(2-(2-((R)-amino(3-chloro-2-pyridinyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (295 mg) as an off-white solid.
Step 3. 4-Dimethylaminopyridine (7.5 mg, 0.061 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(3-chloro-2-pyridinyl)methyl)- 1 - benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (285 mg, 0.614 mmol), cyclopropanesulfonyl chloride (125 μί, 1.23 mmol, Matrix Scientific, Columbia, SC), and diisopropylethylamine (534 μί, 3.07 mmol) in DMF (5 mL) and the mixture was stirred at room temperature for 16 h. The mixture was diluted with MeOH (8 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined, saturated aqueous NaHC03 (75 mL) was added, and the mixture was extracted with DCM (2 x 50 mL). The combined organic layers were dried (Na2S04), filtered, and concentrated under reduced pressure to deliver N-((R)-(3-Chloro-2- pyridinyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (245 mg) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.85 (d, J= 5.3 Hz, 1 H), 8.68 (dd, J = 1.1, 4.6 Hz, 1 H), 8.62 (d, J= 9.6 Hz, 1 H), 8.31 (s, 1 H), 8.11 (d, J = 7.4 Hz, 1 H), 8.05 (dd, J= 1.2, 8.2 Hz, 1 H), 7.91 (d, J= 7.6 Hz, 1 H), 7.62 (d, J= 5.1 Hz, 1 H), 7.53 (t, J= 7.7 Hz, 1 H), 7.49 (dd, J= 4.7, 8.2 Hz, 1 H), 7.24 (s, 1 H), 7.01 (s, 1 H), 6.41 (d, J= 9.4 Hz, 1 H), 2.26 - 2.16 (m, 1 H), 1.80 (s, 3 H), 0.92 - 0.83
(m, 1 H), 0.83 - 0.69 (m, 2 H), 0.59 - 0.50 (m, 1 H). m/z (ESI, pos. ion) 567.8 (M+H)+. GK-GK P ICso (Binding) = 0.213 μΜ.
Example 30
N-((R)-(3-Methoxy-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide


Step 1. 3-Methoxy-2-pyridinecarbaldehyde (1.50 g, 10.9 mmol, Chem- Impex, Wood Dale, IL) was added to a solution of (S)-2-methylpropane-2- sulfinamide (1.86 g, 15.3 mmol, AK Scientific, Mountain View, CA) and copper(II)sulfate (5.24 g, 32.8 mmol) in DCM (50 mL) and the mixture was stirred at room temperature for 2 h. The mixture was filtered, the filter residue was washed with DCM (30 mL), and the combined filtrate and washing were concentrated under reduced pressure. The residue was dissolved in EtOAc (100
mL) and filtered. The filter residue was washed with EtOAc (50 mL), and the combined filtrate and washing were concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 100% EtOAc) and again by flash chromatography (100 g of silica gel, 100% EtOAc) to afford (S)-N-((lE)-(3-methoxy-2-pyridinyl)methylidene)-2-methyl-2- propanesulfmamide contaminated with about 20% (S)-2-methylpropane-2- sulfmamide (1.50 g) as an off-white solid.
Step 2. n-Butyllithium (3.13 mL of a 1.6 M solution in hexanes, 5.01 mmol) was added to a solution of (2R)-2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-
1.1.1- trifluoro-2-propanol (810 mg, 2.51 mmol, Intermediate X4) in THF (5 mL) at -78 °C and the mixture was stirred at -78 °C for 15 min. A solution of (S)-N- (( 1 E)-(3 -methoxy-2-pyridinyl)methylidene)-2-methyl-2-propanesulfinamide contaminated with about20% (S)-2-methylpropane-2-sulfinamide (602 mg, 2.51 mmol) in THF (3 mL) was added dropwise and the resulting mixture was allowed to warm slowly. Saturated aqueous NH4C1 (40 mL) was added and the mixture was allowed to warm to room temperature. The solution was partitioned between aqueous NH4C1 (25 mL of saturated NH4C1 and 25 mL of H20) and EtOAc (50 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic layers were washed with brine (100 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 20%> to 100%
EtOAc/hexanes) to afford (S)-N-((R)-(3-methoxy-2-pyridinyl)(7-(4-((lR)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-methyl-2-propanesulfinamide (356 mg) as a light-yellow solid.
Step 3. A mixture of (S)-N-((R)-(3-methoxy-2-pyridinyl)(7-(4-((lR)-
2.2.2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (350 mg, 0.621 mmol), hydrogen chloride (6.47 mL of a 4 M solution in 1 ,4-dioxane, 186 mmol), and MeOH (2 mL) was stirred at room temperature for 2 h. The resulting solid was removed by
filtration, washed with MeOH (2 mL), and dried under reduced pressure to deliver (2R)-2-(2-(2-((R)-amino(3-methoxy-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol hydrochloride (135 mg) as an off-white solid.
Step 4. 4-Dimethylaminopyridine (3.82 mg, 0.031 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(3-methoxy-2-pyridinyl)methyl)- 1 - benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol hydrochloride (155 mg, 0.313 mmol), cyclopropanesulfonyl chloride (63.8 μΐ,, 0.625 mmol, Matrix Scientific, Columbia, SC), and diisopropylethylamine (272 μΐ,, 1.56 mmol) in DMF (4 mL) and the mixture was stirred at room temperature for 16 h. The mixture was diluted with MeOH (8 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 1 ΙθΑ, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1%) TFA). The product containing fractions were combined, saturated aqueous NaHCC>3 (75 mL) was added, and the mixture was extracted with DCM (2 x 50 mL). The combined organic layers were dried (Na2S04), filtered, and concentrated under reduced pressure to deliver N-((R)-(3-methoxy- 2-pyridinyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (78 mg) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.84 (d, J= 5.3 Hz, 1 H), 8.33 - 8.26 (m, 2 H), 8.23 (dd, J= 1.0, 4.7 Hz, 1 H), 8.07 (d, J= 7.2 Hz, 1 H), 7.88 (d, J= 7.4 Hz, 1 H), 7.61 (d, J= 5.3 Hz, 1 H), 7.57 - 7.48 (m, 2 H), 7.40 (dd, J= 4.6, 8.3 Hz, 1 H), 7.23 (d, J= 0.6 Hz, 1 H), 7.00 (s, 1 H), 6.31 (d, J= 9.6 Hz, 1 H), 3.91 (s, 3 H), 2.22 - 2.12 (m, 1 H), 1.80 (s, 3 H), 0.87 - 0.76 (m, 2 H), 0.71 - 0.63 (m, 1 H), 0.59 - 0.50 (m, 1 H). m/z (ESI, pos. ion) 653.7 (M+H)+. GK-GKRP IC50
(Binding) = 0.220 μΜ.
Example 31
N-((R)-(3-Methyl-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. 3-Methyl-2-pyridinecarbaldehyde (0.750 mL, 6.69 mmol, Sigma- Aldrich, St. Louis, MO) was added to a solution of (S)-2-methylpropane-2- sulfmamide (1.14 g, 9.36 mmol, AK Scientific, Mountain View, CA) and copper(II) sulfate (3.20 g, 20.1 mmol) in DCM (50 mL) and the mixture was stirred at room temperature for 2 h. The mixture was filtered, the filter residue was washed with DCM (30 mL), and the combined filtrate and washing were concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 33% EtOAc/hexanes) to afford (S)-N-((1E)- (3-methyl-2-pyridinyl)methylidene)-2-methyl-2-propanesulfinamide (980 mg) as a light-yellow solid.
Step 2. n-Butyllithium (1.80 mL of a 1.6 M solution in hexanes, 2.88 mmol) was added to a solution of (2R)-2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (310 mg, 0.959 mmol, Intermediate X4) in THF (5 mL) at -78 °C and the mixture was stirred at -78 °C for 10 min. A solution of (S,E)-2-methyl-N-((3-methoxypyridin-2-yl)methylene)propane-2-sulfinamide (323 mg, 1.44 mmol) in THF (3 mL) was added dropwise and the resulting mixture was stirred at -78 °C for 10 min. The cold bath was removed and the mixture was allowed to warm to room temperature. Saturated aqueous NH4CI (40 mL) was added and the mixture was partitioned between aqueous NH4C1 (25 mL of saturated NH4CI and 25 mL of H20) and EtOAc (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic layers were washed with brine (100 mL), dried (Na2SC"4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 20% to 100% EtOAc/hexanes). The material was dissolved in MeOH (6 mL), filtered, and purified further by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 l lOA, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, saturated aqueous NaHCOs (100 mL) was added, and the mixture was extracted with DCM (2 x 50 mL). The combined organic layers were dried (Na2SC"4), filtered, and concentrated under reduced pressure to deliver (S)- 2- methyl-N-((R)-(3-methyl-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-propanesulfinamide (145 mg) as an off- white solid.
Step 3. A mixture of (S)- 2-methyl-N-((R)-(3-methyl-2-pyridinyl)(7-(4- (( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-propanesulfinamide (140 mg, 0.256 mmol) and hydrogen chloride (7.67 mL of a 1 M solution in Et20, 7.67 mmol) was stirred at room temperature for 16 h. Saturated aqueous NaHCOs (40 mL) was added and the resulting mixture was extracted with EtOAc (2 x 50 mL). The combined organic layers
were washed with brine (100 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (100 g of silica gel, 5% MeOH/DCM). The material was dissolved in MeOH (6 mL), filtered, and purified further by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 1 lOA, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, saturated aqueous NaHC03 (100 mL) was added, and the mixture was extracted with DCM (2 x 50 mL). The combined organic layers were dried (Na2S04), filtered, and concentrated under reduced pressure to deliver (2R)-2-(2-(2-((R)-amino(3-methyl-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (101 mg) as an off- white solid.
Step 4. 4-Dimethylaminopyridine (2.75 mg, 0.023 mmol) was added to a mixture of (2R)-2-(2-(2-((R)-amino(3-methyl-2-pyridinyl)methyl)- 1 - benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (100 mg, 0.225 mmol), cyclopropanesulfonyl chloride (30.6 μί, 0.293 mmol, Matrix Scientific, Columbia, SC), and diisopropylethylamine (157 μί, 0.902 mmol) in DMF (1 mL) and the mixture was stirred at room temperature for 1.5 h. The mixture was diluted with MeOH (3 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined, saturated aqueous NaHC03 (100 mL) was added, and the mixture was extracted with DCM (2 x 50 mL). The combined organic layers were dried (Na2S04), filtered, and concentrated under reduced pressure to deliver N-((R)-(3-methyl-2- pyridinyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (59 mg) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.82 (d, J= 5.3 Hz, 1 H), 8.51 (d, J= 4.7 Hz, 1 H), 8.31 - 8.24 (m, 2 H), 8.08 (d, J= 7.2 Hz, 1 H), 7.89 (d, J= 7.4 Hz, 1 H), 7.68 (d, J= 7.0 Hz, 1 H), 7.60 (d, J= 5.1 Hz, 1 H), 7.52 (t, J= 7.6 Hz, 1 H), 7.32 (dd, J= 4.7, 7.6 Hz, 1 H), 7.26 (s, 1 H), 6.99 (s, 1 H), 6.16 (d, J= 9.0 Hz, 1
H), 2.40 (s, 3 H), 2.25 - 2.16 (m, 1 H), 1.79 (s, 3 H), 0.90 - 0.77 (m, 2 H), 0.72 - 0.63 (m, 1 H), 0.62 - 0.53 (m, 1 H). m/z (ESI, pos. ion) 547.8 (M+H)+. GK- GKRP IC50 (Binding) = 0.386 μΜ.
Example 32
N-((R)-(2-Methoxyphenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((S)-(2-methoxyphenyl)(7-(4-
((lR)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. Montmorillonite K10 (560 mg) was added to a mixture of 2- methoxybenzaldehyde (1.05 g, 7.70 mmol, Sigma- Aldrich, St. Louis, MO) and cyclopropanesulfonamide (777 mg, 6.41 mmol, Sigma- Aldrich, St. Louis, MO) in toluene (30 mL) and the mixture was heated at reflux for 2 h while water was removed using a Dean-Stark trap. The mixture was allowed to cool to room temperature, filtered, and the filtrate was concentrated under reduced pressure. The residue was triturated with hexanes (4 x 10 mL) and the resulting solid was
dried under reduced pressure to afford N-((lE)-(2- methoxyphenyl)methylidene)cyclopropanesulfonamide (1.00 g) as a white solid.
Step 2. n-Butyllithium (1.55 mL of a 1.6 M solution in hexanes, 2.47 mmol) was added dropwise to a solution of (2R)-2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (160 mg, 0.495 mmol, Intermediate X4) in THF (8 mL) at -78 °C and the mixture was stirred at -78 °C for 15 min. A solution of N-(( 1 E)-(2-methoxyphenyl)methylidene)cyclopropanesulfonamide (154 mg, 0.643 mmol) in THF (2 mL) was added and the mixture was stirred at - 78 °C for 15 min. The mixture was allowed to warm to room temperature, saturated aqueous NH4C1 (75 mL) was added, and the mixture was extracted with EtOAc (2 x 100 mL). The combined organic layers were washed with brine (200 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 25% EtOAc/hexanes) to afford a mixture of N-((R)-(2-methoxyphenyl)(7-(4-((lR)-2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((S)-(2-methoxyphenyl)(7-(4-((lR)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (154 mg) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.84 (d, J= 5.3 Hz, 1 H), 8.55 (d, J= 10.0 Hz, 1 H), 8.30 (s, 1 H), 8.07 (d, J= 7.4 Hz, 1 H), 7.87 (d, J= 7.8 Hz, 1 H), 7.64 (d, J= 7.6 Hz, 1 H), 7.61 (d, J= 5.1 Hz, 1 H), 7.51 (t, J= 7.7 Hz, 1 H), 7.38 - 7.32 (m, 1 H), 7.11 - 7.00 (m, 4 H), 6.26 (d, J= 9.8 Hz, 1 H), 3.84 (s, 3 H), 2.21 - 2.12 (m, 1 H), 1.80 (s, 3 H), 0.88 - 0.80 (m, 2 H), 0.71 (dd, J= 2.9, 10.8 Hz, 1 H), 0.67 - 0.59 (m, 1 H). m/z (ESI, pos. ion) 562.8 (M+H)+. GK-GKRP IC50
(Binding) = 0.443 μΜ.
Example 33
N-((R)-(2-Nitrophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide and N-
((S)-(2-nitrophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. Montmorillonite K10 (1.50 g) was added to a mixture of 2- nitrobenzaldehyde (2.72 mg, 18.0 mmol, Sigma- Aldrich, St. Louis, MO) and cyclopropanesulfonamide (2.18 g, 18.0 mmol, Sigma- Aldrich, St. Louis, MO) in toluene (30 mL) and the mixture was heated at reflux for 2 h while water was removed using a Dean-Stark trap. The mixture was cooled to 50 °C and filtered. The filtrate was cooled to room temperature and the resulting solid was removed by filtration, washed with hexanes, and dried under reduced pressure to afford N- ((lE)-(2-nitrophenyl)methylidene)cyclopropanesulfonamide (2.05 g) as a colorless solid.
Step 2. n-Butyllithium (1.35 mL of a 1.6 M solution in hexanes, 2.17 mmol) was added dropwise to a solution of (2R)-2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (140 mg, 0.433 mmol, Intermediate X4) in THF (4.3 mL) at -78 °C and the mixture was stirred at -78 °C for 15 min. A solution of N-((lE)-(2-nitrophenyl)methylidene)cyclopropanesulfonamide (275 mg, 1.08 mmol) in THF (2 mL) was added and the mixture was stirred at -78 °C
for 15 min. The mixture was allowed to warm to room temperature, saturated aqueous NH4C1 (75 mL) was added, and the mixture was extracted with EtOAc (2 x 100 mL). The combined organic layers were washed with brine (200 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was diluted with DMSO (7 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined, basified with saturated aqueous NaHC03 (100 mL), and extracted with DCM (2 x 80 mL). The combined organic layers were dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 33% EtOAc/hexanes) to afford a mixture of N-((R)- (2-nitrophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)cyclopropanesulfonamide and N-((S)- (2-nitrophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (89 mg) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.94 (d, J= 9.4 Hz, 1 H), 8.84 (d, J= 5.3 Hz, 1 H), 8.32 (s, 1 H), 8.13 (d, J= 7.6 Hz, 1 H), 8.09 - 8.03 (m, 2 H), 7.94 - 7.86 (m, 2 H), 7.67 (dt, J= 1.1, 7.8 Hz, 1 H), 7.61 (d, J= 5.3 Hz, 1 H), 7.53 (t, J= 7.7 Hz, 1 H), 7.05 (s, 1 H), 7.00 (s, 1 H), 6.53 (d, J= 9.0 Hz, 1 H), 2.34 - 2.27 (m, 1 H), 1.80 (s, 3 H), 0.94 - 0.73 (m, 3 H), 0.71 - 0.62 (m, 1 H). m/z (ESI, pos. ion) 577.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.778 μΜ.
Example 34
N-((R)-(2-Methylphenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((S)-(2-methylphenyl)(7-(4-((lR)-
2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide


Step 1. Montmorillonite K10 (500 mg) was added to a mixture of o- tolualdehyde (536 μΐ,, 4.62 mmol, Sigma-Aldrich, St. Louis, MO) and cyclopropanesulfonamide (466 mg, 3.85 mmol, Sigma-Aldrich, St. Louis, MO) in toluene (30 mL) and the mixture was heated at reflux for 3 h while water was removed using a Dean-Stark trap. The mixture was allowed to cool to room temperature, filtered, and the filtrate was concentrated under reduced pressure. The residue was triturated with hexanes (4 x 10 mL) and the resulting solid was dried under reduced pressure to afford N-((lE)-(2- methylphenyl)methylidene)cyclopropanesulfonamide (500 mg) as a white solid.
Step 2. n-Butyllithium (0.79 mL of a 1.6 M solution in hexanes, 1.27 mmol) was added dropwise to a solution of (2R)-2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (137 mg, 0.424 mmol, Intermediate X4) in THF (5 mL) at -78 °C and the mixture was stirred at -78 °C for 15 min. A solution of N-(( 1 E)-(2-methylphenyl)methylidene)cyclopropanesulfonamide (123 mg, 0.551 mmol) in THF (2 mL) was added and the mixture was stirred at -78 °C for an additional 15 min. The mixture was allowed to warm to room temperature, saturated aqueous NH4C1 (75 mL) was added, and the mixture was extracted with
EtOAc (2 x 100 mL). The combined organic layers were washed with brine (200 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 25% EtOAc/hexanes) to afford a mixture of N-((R)-(2-methylphenyl)(7-(4-((lR)-2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((S)-(2-methylphenyl)(7-(4-((lR)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (65 mg) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.84 (d, J= 5.1 Hz, 1 H), 8.64 (d, J= 9.4 Hz, 1 H), 8.32 (s, 1 H), 8.11 (d, J= 7.2 Hz, 1 H), 7.90 (d, J= 7.4 Hz, 1 H), 7.66 - 7.60 (m, 2 H), 7.54 (t, J= 7.6 Hz, 1 H), 7.33 - 7.25 (m, 3 H), 7.10 (s, 1 H), 7.01 (s, 1 H), 6.08 (d, J= 8.8 Hz, 1 H), 2.38 (s, 3 H), 2.25 - 2.17 (m, 1 H), 1.81 (s, 3 H), 0.95 - 0.86 (m, 1 H), 0.86 - 0.64 (m, 3 H). m/z (ESI, pos. ion) 546.8 (M+H)+. GK-GKRP IC50 (Binding) = 1.05 μΜ.
Example 35
6-Chloro-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-3- pyridinesulfonamide

4-Dimethylaminopyridine (1.74 mg, 0.014 mmol) was added to a mixture of (2S)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (66 mg, 0.143 mmol, Intermediate X8), 6- chloro-3-pyridinesulfonyl chloride (36.3 mg, 0.171 mmol, Beta Pharma,
Branford, CT), and diisopropylethylamine (74.4 μί, 0.428 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 1.5 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure to deliver 6-chloro-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 3-pyridinesulfonamide (65 mg) as a white solid.
1H NMR (400 MHz, CDC13) δ ppm 8.82 (d, J= 5.3 Hz, 1 H), 8.72 (d, J= 2.3 Hz, 1 H), 8.15 (s, 1 H), 7.89 (d, J= 7.8 Hz, 2 H), 7.70 (d, J= 7.8 Hz, 1 H), 7.51 - 7.43 (m, 2 H), 7.38 (dd, J= 2.0, 7.0 Hz, 1 H), 7.33 - 7.18 (m, 4 H), 6.93 (s, 1 H), 6.34 (d, J= 8.0 Hz, 1 H), 5.83 (d, J= 8.2 Hz, 1 H), 2.24 (br. s, 1 H), 1.85 (s, 3 H). m/z (ESI, pos. ion) 637.7 (M+H)+. GK-GKRP IC50 (Binding) = 2.28 μΜ.
Example 36
N-((R)-(2-Chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-6-oxo-l
3-pyridinesulfonamide

Hydrogen chloride (39.9 of a 4 M solution in 1,4-dioxane, 0.160 mmol) was added to a suspension of 6-chloro-N-((R)-(2-chlorophenyl)(7-(4- (( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-3-pyridinesulfonamide (51 mg, 0.080 mmol, Example 35) in 1,4-
dioxane/H20 (1 : 1, 1 mL) and the mixture was stirred at 120 °C for 66 h and at 140 °C for 24 h. The mixture was allowed to cool to room temperature, filtered, and concentrated. The material was purified by reversed-phase HPLC
(Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure to deliver N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-oxo- 1,6- dihydro-3-pyridinesulfonamide (22 mg) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 11.93 (br. s, 1 H), 9.25 (br. s, 1 H), 8.87 (d, J= 5.3 Hz, 1 H), 8.34 (s, 1 H), 8.14 (d, J= 7.4 Hz, 1 H), 7.88 (d, J= 7.6 Hz, 1 H), 7.71 - 7.60 (m, 3 H), 7.53 (t, J= 7.7 Hz, 1 H), 7.47 - 7.38 (m, 2 H), 7.38 - 7.28 (m, 2 H), 7.02 (s, 1 H), 6.92 (d, J= 1.0 Hz, 1 H), 6.24 (d, J= 9.6 Hz, 1 H), 6.20 (s, 1 H), 1.82 (s, 3 H). m/z (ESI, pos. ion) 619.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.023 μΜ.
Example 37
N-((R)-(2-Chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

4-Dimethylaminopyridine (1.29 mg, 10.6 μηιοΐ) was added to a mixture of (2S)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (49 mg, 0.106 mmol, Intermediate X8), cyclopropanesulfonyl chloride (12.9 μί, 0.127 mmol, Matrix Scientific,
Columbia, SC), and diisopropylethylamine (55.2 μί, 0.318 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 24 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95%
H20/MeCN, 0.1%) TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure to deliver N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (47 mg) as a white foam.
1H NMR (400 MHz, CDC13) δ ppm 8.83 (d, J= 5.3 Hz, 1 H), 8.15 (s, 1 H), 7.89 (d, J= 7.4 Hz, 1 H), 7.75 (d, J= 7.8 Hz, 1 H), 7.65 (dd, J= 1.4, 7.6 Hz, 1 H), 7.51 - 7.41 (m, 3 H), 7.41 - 7.30 (m, 2 H), 7.08 (s, 1 H), 6.42 (d, J= 8.0 Hz, 1 H), 5.44 (d, J= 8.0 Hz, 1 H), 2.31 - 2.24 (m, 1 H), 1.85 (s, 3 H), 1.22 - 1.07 (m, 2 H),
0.92 - 0.83 (m, 1 H), 0.82 - 0.72 (m, 1 H). m/z (ESI, pos. ion) 566.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.143 μΜ.
Examples 38 and 39
(lS,2S)-N-((R)-(2-Chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2- ethylcyclopropanesulfonamide and (lR,2R)-N-((R)-(2-chlorophenyl)(7-(4- ((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-2-ethylcyclopropanesulfonamide

Trans-2-ethylcyclopropane-l-sulfonyl chloride (52.5 μΐ,, 0.156 mmol, Chemizon, Longmont, CO) was added to a mixture of (2S)-2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (60 mg, 0.130 mmol, Intermediate X8), and diisopropylethylamine (67.6 μί, 0.389 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 8 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and
concentrated under reduced pressure. The residue was subjected to preparative SFC (Chiralpak® IC column) (250 mm x 21 mm, 5 μιη) eluting with 80% liquid C02 in 20% methanol (with 20 mM NH3) at a flow rate of 75 mL/min) to give two products in greater than 99%> purity.
First eluting peak (peak #1):
1H NMR (600 MHz, CDC13) δ ppm 8.80 (d, J= 3.2 Hz, 1 H), 8.15 (s, 1 H), 7.88 (d, J= 7.3 Hz, 1 H), 7.73 (d, J= 7.7 Hz, 1 H), 7.65 (d, J= 7.3 Hz, 1 H), 7.50 -
7.40 (m, 3 H), 7.38 (t, J= 7.3 Hz, 1 H), 7.32 (t, J= 7.5 Hz, 1 H), 7.05 (s, 1 H),
6.41 (d, J= 7.7 Hz, 1 H), 5.43 (d, J= 7.7 Hz, 1 H), 2.77 (br. s, 1 H), 2.06 (td, J = 4.3, 8.1 Hz, 1 H), 1.84 (s, 3 H), 1.54 - 1.45 (m, 1 H), 1.29 (td, J= 4.8, 9.5 Hz, 1 H), 1.23 (tt, J= 6.9, 13.9 Hz, 1 H), 1.02 (quind, J= 7.3, 14.2 Hz, 1 H), 0.86 (t, J = 7.4 Hz, 3 H), 0.73 - 0.68 (m, 1 H). m/z (ESI, pos. ion) 594.8 (M+H)+. GK- GKRP IC50 (Binding) = 0.280 μΜ.
Second eluting peak (peak #2):
1H NMR (600 MHz, CDC13) δ ppm 8.80 (d, J= 5.0 Hz, 1 H), 8.14 (s, 1 H), 7.88 (d, J= 7.5 Hz, 1 H), 7.73 (d, J= 7.9 Hz, 1 H), 7.64 (d, J = 7.1 Hz, 1 H), 7.49 - 7.41 (m, 3 H), 7.38 (t, J= 7.7 Hz, 1 H), 7.33 (dt, J= 0.8, 7.7 Hz, 1 H), 7.05 (s, 1 H), 6.40 (d, J= 7.9 Hz, 1 H), 5.48 (d, J= 7.9 Hz, 1 H), 2.81 (br. s, 1 H), 2.03 (td, J= 4.4, 8.3 Hz, 1 H), 1.83 (s, 3 H), 1.56 - 1.48 (m, 1 H), 1.35 (quind, J= 7.1, 13.9 Hz, 1 H), 1.21 (td, J = 5.0, 9.5 Hz, 1 H), 1.11 (quind, J= 7.3, 14.2 Hz, 1 H), 0.89 (t, J= 7.4 Hz, 3 H), 0.58 (td, J= 6.0, 7.9 Hz, 1 H). m/z (ESI, pos. ion) 594.8 (M+H)+. GK-GKRP ICso (Binding) = 0.042 μΜ.
Examples 40 and 41
(lS,2R)-N-((R)-(2-Chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-(l- methylethyl)cyclopropanesulfonamide and (lR,2S)-N-((R)-(2- chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-(l- methylethyl)cyclopropanesulfonamide

Trans-2-isopropylcyclopropane-l-sulfonyl chloride (31.8 μί, 0.174 mmol, Chemizon, Longmont, CO) was added to a mixture of (2S)-2-(2-(2-((R)- amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol (62 mg, 0.134 mmol, Intermediate X8) and
diisopropylethylamine (69.9 μΐ^, 0.402 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 18 h. Trans-2-isopropylcyclopropane-l- sulfonyl chloride (31.8 μί, 0.174 mmol) was added and the mixture was stirred at room temperature for 6 h. Additional trans-2-isopropylcyclopropane-l-sulfonyl chloride (31.8 μΐ,, 0.174 mmol) was added and the mixture was stirred at room temperature for a total of 78 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 1 lOA, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure. The residue was subjected to preparative SFC (Chiralpak® AS-H column) (250 mm x 21 mm, 5 μιη) eluting with 75% liquid C02 in 25% methanol (with 20 mM NH3) at a flow rate of 75 mL/min) to give two products in greater than 99% purity.
First eluting peak (peak #1):
1H NMR (400 MHz, CDC13) δ ppm 8.82 (d, J= 4.4 Hz, 1 H), 8.15 (s, 1 H), 7.88 (d, J= 7.3 Hz, 1 H), 7.74 (d, J= 7.7 Hz, 1 H), 7.65 (d, J= 7.3 Hz, 1 H), 7.51 - 7.41 (m, 3 H), 7.38 (t, J= 7.1 Hz, 1 H), 7.33 (t, J= 7.5 Hz, 1 H), 7.07 (s, 1 H), 6.41 (d, J= 7.7 Hz, 1 H), 5.42 (d, J= 7.3 Hz, 1 H), 2.72 (br. s, 1 H), 2.12 (td, J = 4.2, 8.1 Hz, 1 H), 1.84 (s, 3 H), 1.48 - 1.40 (m, 1 H), 1.14 (td, J= 4.9, 9.5 Hz, 1
H), 1.08 (qd, J= 6.8, 13.6 Hz, 1 H), 0.94 (d, J= 6.7 Hz, 3 H), 0.88 (d, J = 6.5 Hz, 3 H), 0.64 - 0.56 (m, 1 H). m/z (ESI, pos. ion) 608.8 (M+H)+. GK-GK P IC50 (Binding) = 0.580 μΜ.
Second eluting peak (peak #2):
1H NMR (600 MHz, CDC13) δ ppm 8.81 (d, J= 5.2 Hz, 1 H), 8.15 (s, 1 H), 7.89 (d, J= 7.5 Hz, 1 H), 7.75 (d, J= 7.7 Hz, 1 H), 7.65 (dd, J= 1.4, 7.7 Hz, 1 H), 7.51 - 7.40 (m, 3 H), 7.38 (dt, J= 1.1, 7.5 Hz, 1 H), 7.32 (dt, J= 1.4, 7.7 Hz, 1 H), 7.08 (s, 1 H), 6.41 (d, J= 7.9 Hz, 1 H), 5.40 (d, J= 8.1 Hz, 1 H), 2.73 (br. s, 1 H), 2.13 - 2.06 (m, 1 H), 1.84 (s, 3 H), 1.49 - 1.42 (m, 1 H), 1.27 - 1.21 (m, 1 H), 1.06 (tt, J = 6.9, 14.0 Hz, 1 H), 0.92 (d, J= 6.7 Hz, 3 H), 0.87 (d, J= 6.7 Hz, 3 H), 0.74 (td, J= 6.1, 7.9 Hz, 1 H). m/z (ESI, pos. ion) 608.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.989 μΜ.
Example 42
N-((R)-(2-Chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- methoxybenzenesulfonamide

4-Dimethylaminopyridine (1.32 mg, 0.011 mmol) was added to a mixture of (2S)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (50 mg, 0.108 mmol, Intermediate X8), 4- methoxybenzenesulfonyl chloride (26.8 mg, 0.130 mmol, Sigma-Aldrich, St. Louis, MO), and diisopropylethylamine (56.4 μΕ, 0.324 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 1 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex
Gemini-NX 10μ CI 8 1 ΙθΑ, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure to deliver N-((R)-(2- chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)-4-methoxybenzenesulfonamide (60 mg) as a white foam.
1H NMR (400 MHz, CDC13) δ ppm 8.79 (d, J= 5.1 Hz, 1 H), 8.12 (s, 1 H), 7.86 (d, J= 7.2 Hz, 1 H), 7.76 - 7.69 (m, 2 H), 7.67 (d, J= 7.4 Hz, 1 H), 7.48 - 7.42 (m, 3 H), 7.31 - 7.27 (m, 1 H), 7.25 - 7.19 (m, 2 H), 6.88 (d, J= 1.0 Hz, 1 H), 6.86 - 6.79 (m, 2 H), 6.22 (d, J= 7.4 Hz, 1 H), 5.49 (d, J= 7.6 Hz, 1 H), 3.81 (s, 3 H), 2.35 (br. s, 1 H), 1.84 (s, 3 H). m/z (ESI, pos. ion) 632.7 (M+H)+. GK- GKRP IC50 (Binding) = 0.102 μΜ.
Example 43
N-((R)-(2-Chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-3- methoxybenzenesulfonamide

4-Dimethylaminopyridine (1.32 mg, 10.8 μιηοΐ) was added to a mixture of (2S)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (50.0 mg, 0.108 mmol, Intermediate X8), 3- methoxybenzenesulfonyl chloride (18.4 μΐ^, 0.130 mmol), and 4- dimethylaminopyridine (1.32 mg, 10.8 μιηοΐ) in DMF (0.7 mL) and the mixture was stirred at room temperature for 1.5 h. The mixture was diluted with MeOH (1
mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 1 lOA, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure to deliver N-((R)-(2- chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)-3-methoxybenzenesulfonamide (47 mg) as a white foam.
1H NMR (400 MHz, CDC13) δ ppm 8.80 (d, J= 5.3 Hz, 1 H), 8.12 (s, 1 H), 7.86 (d, J = 7.4 Hz, 1 H), 7.67 (d, J = 7.6 Hz, 1 H), 7.48 - 7.37 (m, 4 H), 7.31 - 7.25 (m, 3 H), 7.24 - 7.17 (m, 2 H), 7.00 (ddd, J= 0.8, 2.6, 8.3 Hz, 1 H), 6.88 (d, J = 1.0 Hz, 1 H), 6.26 (d, J= 7.6 Hz, 1 H), 5.57 (d, J= 7.6 Hz, 1 H), 3.75 (s, 3 H), 2.36 (br. s, 1 H), 1.84 (s, 3 H). m/z (ESI, pos. ion) 632.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.283μΜ.
Example 44
N-((R)-(2-Chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-
(trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-6-oxo-l,6- dihydro-3-pyridinesulfonamide

Step 1. 4-Dimethylaminopyridine (2.83 mg, 0.023 mmol) was added to a mixture of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l,3,3,3-hexafluoro-2-propanol hydrochloride (128 mg, 0.231 mmol, Intermediate XI 1), 6-chloro-3-pyridinesulfonyl chloride (63.8 mg, 0.301 mmol, Beta Pharma, Branford, CT), and diisopropylethylamine (121 μΐ^, 0.694
mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 1.5 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 HOA, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure. The resulting colorless film was further purified by flash chromatography (50 g of silica gel, 20%> to 40%> EtOAc/hexanes) to deliver 6- chloro-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 - pyridinesulfonamide (109 mg) as a colorless film.
Step 2. Hydrogen chloride, (70.8 of a 4.0 M solution in 1,4-dioxane, 0.283 mmol) was added to 6-chloro-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-3 -pyridinesulfonamide (98 mg, 0.142 mmol) in 1 ,4-dioxane/water (1 : 1, 1 mL) and the mixture was stirred at room temperature for 2 h, at 60 °C for 1 h and then at 80 °C for 18 h. The mixture was transferred into a sealed vial and stirred at 100 °C for 3 h, at 120 °C for 21 h, and at 140 °C for 30 h. The mixture was allowed to cool to room temperature, filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ CI 8 1 ΙθΑ, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1%) TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure to deliver N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro-l- hydroxy- 1 -(trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6- oxo-l,6-dihydro-3-pyridinesulfonamide (29 mg) as a white solid.
1H NMR (300 MHz, DMSO-d6) δ ppm 11.95 (br. s, 1 H), 9.31 (br. s, 1 H), 9.25 (br. d, J= 8.3 Hz, 1 H), 8.99 (d, J= 5.4 Hz, 1 H), 8.37 (s, 1 H), 8.06 (d, J= 7.5 Hz, 1 H), 7.90 (d, J= 7.5 Hz, 1 H), 7.71 - 7.60 (m, 3 H), 7.55 (t, J= 7.7 Hz, 1 H),
7.46 - 7.30 (m, 4 H), 6.94 (d, J= 1.2 Hz, 1 H), 6.23 (d, J= 9.8 Hz, 1 H), 6.19 (br. d, J= 6.0 Hz, 1 H). m/z (ESI, pos. ion) 673.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.147 μΜ.
Example 45
N-((R)-(2-Chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l- (trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)i
methoxybenzenesulfonamide

4-Dimethylaminopyridine (1.10 mg, 9.04 μιηοΐ) was added to a mixture of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l,3,3,3-hexafluoro-2-propanol hydrochloride (50 mg, 0.090 mmol, Intermediate XI 1), 4-methoxybenzenesulfonyl chloride (24.3 mg, 0.117 mmol, Sigma- Aldrich, St. Louis, MO), and diisopropylethylamine (47.2 μί, 0.271 mmol) in DMF (0.7 mL) and the mixture was stirred at room temperature for 2 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed- phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1 %> TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and concentrated under reduced pressure. The resulting colorless film was further purified by flash chromatography (50g of silica gel, 20%> to 40%> EtOAc/hexanes) to deliver N- ((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)- 2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-4-methoxybenzenesulfonamide (47 mg) as a white foam.
1H NMR (300 MHz, CDC13) δ ppm 8.90 (d, J= 5.4 Hz, 1 H), 8.27 (s, 1 H), 7.89 (d, J= 7.3 Hz, 1 H), 7.79 - 7.68 (m, 3 H), 7.60 (d, J= 4.7 Hz, 1 H), 7.52 - 7.43 (m, 2 H), 7.33 - 7.30 (m, 1 H), 7.26 - 7.21 (m, 2 H), 6.92 (s, 1 H), 6.85 (d, J= 8.8 Hz, 2 H), 6.24 (d, J= 7.6 Hz, 1 H), 5.48 (d, J= 7.5 Hz, 1 H), 3.83 (s, 3 H). m/z (ESI, pos. ion) 686.6 (M+H)+. GK-GKRP IC50 (Binding) = 0.520 μΜ.
Example 46
N-((R)-(2-Chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-
(trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

4-Dimethylaminopyridine (1.14 mg, 9.40 μηιοΐ) was added to a mixture of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l,3,3,3-hexafluoro-2-propanol hydrochloride (52.0 mg, 0.094 mmol, Intermediate XI 1), cyclopropanesulfonyl chloride (12.5 μί, 0.122 mmol, Matrix Scientific, Columbia, SC), and diisopropylethylamine (49.0 μΐ,, 0.282 mmol) in DCM (0.7 mL) and the mixture was stirred at room temperature for 2 h. The solvent was removed by blowing a stream of nitrogen over the solution and the resulting residue was dissolved in DMF (0.7 mL) and stirred at room temperature for 20 h. The mixture was diluted with MeOH (1 mL), filtered, and purified by reversed-phase HPLC (Phenomenex Gemini-NX 10μ C18 1 ΙθΑ, 100 x 50 mm, 10% to 95% H20/MeCN, 0.1% TFA). The product containing fractions were combined, basified with solid NaHC03, and extracted with DCM (2 x 20 mL). The combined organic layers were dried (MgS04), filtered, and
concentrated under reduced pressure to deliver N-((R)-(2-Chlorophenyl)(7-(4- (2,2,2-trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)-2-pyridinyl)- 1 -
benzothiophen-2-yl)methyl)cyclopropanesulfonamide (34 mg) as a colorless film.
1H NMR (300 MHz, CDC1 ) δ ppm 8.91 (d, J= 5.1 Hz, 1 H), 8.28 (s, 1 H), 7.89 (d, J= 7.6 Hz, 1 H), 7.77 (d, J = 7.6 Hz, 1 H), 7.65 (d, J= 6.7 Hz, 1 H), 7.59 (d, J = 4.5 Hz, 1 H), 7.55 - 7.28 (m, 4 H), 7.10 (s, 1 H), 6.42 (d, J= 8.0 Hz, 1 H), 5.45 (d, J= 7.7 Hz, 1 H), 2.36 - 2.19 (m, 1 H), 1.25 - 1.05 (m, 2 H), 0.96 - 0.70 (m, 2 H). m/z (ESI, pos. ion) 620.8 (M+H)+. GK-GKRP IC50 (Binding) = 1.52 μΜ.
Example 47
N-((R)-(2-Chloropheny])(7-(5-(l-hydroxy-l-methylethyl)-2-thiophenyl)-l- b nzothiophen-2-y])methyl)cyclopropanesulfonamide

Step 1. To a 100-mL, single-necked, round-bottomed flask charged with a solution of thiophene-2-carboxylic acid (1.36 g, 10.6 mmol, Sigma-Aldrich,
India) in acetonitrile (40 mL) was added CsF-Diatomaceous earth (2.42 g, 15.9 mmol, See: Lee, J.C.; Choi, Y. Synth. Commun. 1998, 28, 2021-2026) and iodoethane (3.32 g, 21.3 mmol, Sigma-Aldrich, India) and the mixture was stirred at 80 °C for 12 h. The mixture was filtered, the filter residue was washed with acetonitrile, and the combined filtrates and washings were concentrated under reduced pressure. The residue was partitioned between EtOAc (25 mL) and H20 (25 mL), the layers were separated, and the organic layer was washed with 2 M aqueous Na2S203, dried (Na2S04), filtered, and concentrated under reduced pressure to afford ethyl 2-thiophenecarboxylate (1 g) as a colorless liquid. This intermediate was used in next step without further purification.
Step 2. A 50-mL sealable tube containing a solution of ethyl 2- thiophenecarboxylate (1 g, 6.41 mmol) in hexanes (20 mL) was purged with argon for 15 min. (l,5-cyclooctadiene)(methoxy)iridium(I) dimer (127 mg, 0.192 mmol, Sigma-Aldrich, India), 4,4'-di-tert-butyl-2,2'-bipyridine (51.0 mg, 0.192 mmol, Sigma-Aldrich, India), and 4,4,5,5-tetramethyl-l,3,2-dioxaborolane (2.0 mL, 12.8 mmol, Sigma-Aldrich, India) was added, the reaction vessel was sealed, and the mixture was stirred at room temperature for 12 h. The mixture was concentrated under reduced pressure and the residue was purified by flash chromatography (silica gel, 100% DCM) to obtain ethyl 5-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)-2-thiophenecarboxylate (1.50 g) as a brown liquid.
Step 3. A 100-mL sealable tube containing a solution of N-((R)-(7- bromo-l-benzothiophen-2-yl)(2-chlorophenyl)methyl)cyclopropanesulfonamide (100 mg, 0.022 mmol, Intermediate XI 2) in DME (20 mL) was purged with argon for 15 min. Ethyl 5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2- thiophenecarboxylate (88 mg, 0.032 mmol),
tetrakis(triphenylphosphine)palladium(0) (76 mg, 0.006 mmol, Sigma-Aldrich, India), and K3P04 (70 mg, 0.0329 mmol) was added and the reaction vessel was purged with argon for 15 min and sealed. The mixture was stirred at 90 °C for 12 h, allowed to cool to room temperature, and diluted with EtOAc (100 mL). The
mixture was washed with water (2 x 50 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, 30% to 35% EtOAc/hexanes) to deliver ethyl 5-(2- ((R)-(2-chlorophenyl)((cyclopropylsulfonyl)amino)methyl)-l-benzothiophen-7- yl)-2-thiophenecarboxylate (70 mg) as an off-white solid.
Step 4. To a 100-mL, single-necked, round-bottomed flask containing a solution of ethyl 5-(2-((R)-(2- chlorophenyl)((cyclopropylsulfonyl)amino)methyl)-l-benzothiophen-7-yl)-2- thiophenecarboxylate (400 mg, 0.753 mmol) in THF (50 mL) was added dropwise methylmagnesium bromide (1.50 mL of a 3 M solution in hexanes, 4.51 mmol) and the mixture was stirred at room temperature for 2 h. The mixture was cooled to 0 °C and partitioned between saturated aqueous NH4C1 (25 mL) and EtOAc (100 mL). The layers were separated and the organic layer was washed with water (2 x 50 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 40% to 42% EtOAc/hexanes) to deliver N-((R)-(2-chlorophenyl)(7-(5-(l -hydroxy- 1- methylethyl)-2-thioph 8enyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (120 mg) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ ppm 8.93 (d, J = 9.6 Hz, 1 H), 7.87 (d, J = 7.6 Hz, 1 H), 7.75 (d, J = 8.0 Hz, 1 H), 7.53 - 7.47 (m, 3 H), 7.43 - 7.38 (m, 3 H), 7.05 (s, 1 H), 7.03 - 6.99 (m, 1 H), 6.28 (d, J = 9.6 Hz, 1 H), 5.62 (d, J = 2.4 Hz, 1 H), 2.24 - 2. 23 (m, 1 H), 1.55 (s, 6 H), 0.87 - 0.61 (m, 4 H). m/z (APCI, neg. ion) 516 (M-H) . GK-GKRP IC50 (Binding) = 1.70 μΜ.
Example 48
N-((R)-(2-chlorophenyl)(4-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-lH- benzimidazol-2-yl)methyl)cyclopropanesulfonamide



Step 1. 4-bromo-lH-benzo[d]imidazole. To a stirring dark suspension of 3-bromo-l,2-benzenediamine (Combi-Blocks, Inc. 4.7 g, 25.1 mmol) and 4- methylbenzene sulfonic acid, monohydrate (0.048 g, 0.251 mmol) in THF (40 mL) at 20 °C under argon was added triethyl orthoformate (Sigma- Aldrich, St. Louis MO, 4.18 mL, 25.1 mmol) dropwise over a period of 5 min. The reaction was stirred for 10 min then heated to 60 °C for 20 min. The reaction was then cooled and concentrated under reduced pressure. The resulting dark solid was then triturated with diethyl ether (50 mL) and dried under reduced pressure to afford 4-bromo-lH-benzo[d]imidazole (4.9 g) as dark solid.

Step 2. tert-Butyl 4-bromo-lH-benzo[d]imidazole-l-carboxylate. To a stirring dark solution of 4-bromo-lH-benzo[d]imidazole (4.8 g, 24.36 mmol) and N,N-diisopropylethylamine (4.24 ml, 24.36 mmol) in THF (20 mL) was added di-tert-butyl dicarbonate (24.36 ml, 24.36 mmol) [1 M in THF] at 20 °C. After 45 min, DMAP (0.030 g, 0.244 mmol) was added. After 30 min, the reaction was then diluted with EtOAc (75 mL) and extracted with 5% NaHCCh (75 mL). The organic extracts were dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 0^30% EtO Ac/Hex to afford tert-butyl 4-bromo-lH- benzo[d]imidazole-l-carboxylate (5.60 g) as an amber oil.

Step 3 4-bromo-l-tosyl-lH-benzo[d]imidazole. A solution of tert-butyl 4- bromo-lH-benzo[d]imidazole-l-carboxylate (5.5 g, 18.51 mmol) in CH2CI2 (20 mL) and TFA (20 mL) was stirred for 1 h at 25 °C. The solvents were then removed under reduced pressure and the residue dissolved in CH2C12 (20 mL) then chilled to 0 °C. To this was added DIEA (32.3 mL, 185 mmol) and 4- toluenesulfonyl chloride (Sigma-Aldrich, St. Louis MO, 3.88 g, 20.36 mmol) followed by DMAP (5 mg). The solution was stirred for 2 h, then extracted with 1 M KH2P04 (150 mL). The organics were dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography
(120 g) eluting products with 0 to 40% EtOAc/Hexanes to afford 4-bromo-l- tosyl-lH-benzo[d]imidazole (5.0 g) as white solid.

Step 4. 4-(4.4.5.5-tetramethvl- 1.3 ,2-dioxaborolan-2-v0- 1 -tosvl- 1 H- benzo[d]imidazole. An appropriate vessel was charged with 4-bromo-l-tosyl-lH- benzo[d]imidazole (1620 mg, 4.61 mmol), bis(pinacolato)diboron (1288 mg, 5.07 mmol), (l, -bis(diphenylphosphino)ferrocene)dichloropalladium(II) (188 mg, 0.231 mmol), potassium acetate (679 mg, 6.92 mmol), 1,4-dioxane (15 mL) then sparged with argon for 30 seconds. The reaction was sealed and then heated to 120 °C with stirring for 4 h. The reaction was then partitioned between EtO Ac (50 mL) and sat. aqueous NaCl (10 mL). The organics were dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 10 to 40% of EtO Ac/Hex to afford 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l-tosyl-lH-benzo[d]imidazole (1720 mg) as white solid.

Step 5. 2-(2-(l-tosyl-lH-benzo[d]imidazol-4-yl)pyridin-4-yl)propan-2-ol. A suspension of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l-tosyl-lH- benzo[d]imidazole (1720 mg, 4.32 mmol), 2-(2-chloro-4-pyridinyl)-2-propanol (741 mg, 4.32 mmol, Intermediate Y4), tetrakis(triphenylphosphine)palladium(0)
(250 mg, 0.216 mmol), sodium carbonate (1831 mg, 17.27 mmol) in 1,4-dioxane (20 mL) and water (4 mL) then sparged for 1 min. The suspension was heated to 100 °C for 2 h with stirring. The reaction was then partitioned between EtOAc (75 mL) and 5% NaHC03 (25 mL). The organics were dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 30 to 90% EtOAc/Hexanes to afford 2-(2-(l-tosyl-lH-benzo[d]imidazol-4-yl)pyridin-4-yl)propan-2-ol (600 mg) as white solid, m/z (ESI, pos. ion) 408.0 (M+H)+.

Step 6. (S)-N-((S and R)-(2-chlorophenyl)(4-(4-(2-hydroxypropan-2- yl)pyridin-2-yl)-l-tosyl-lH-benzo[d]imidazol-2-yl)methyl)-2-methylpropane-2- sulfinamide. To a stirring solution of 2-(2-(l-tosyl-lH-benzo[d]imidazol-4- yl)pyridin-4-yl)propan-2-ol (275 mg, 0.675 mmol) in THF (3 mL) at -65 °C under argon was added n-butyllithium, (2.5m solution in hexanes,810 μί, 2.025 mmol). The resulting red suspension was stirred for 15 min then a solution of (S,E)-N-(2-chlorobenzylidene)-2-methylpropane-2-sulfinamide (329 mg, 1.350 mmol, Intermediate Yl) in dry THF (1.5 mL) was added at -60 °C. The reaction was stirred for 90 min at -65 °C and then quenched with sat. aqueous NH4C1 (2 mL) dropwise followed by dilution with EtOAc (20 mL). The organics were dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 50% MeCN/CH2Ci2 to afford (S)-N-((S)-(2-chlorophenyl)(4-(4-(2-hydroxypropan-2-yl)pyridin-2-yl)- l-tosyl-lH-benzo[d]imidazol-2-yl)methyl)-2-methylpropane-2-sulfinamide and (S)-N-((R)-(2-chlorophenyl)(4-(4-(2-hydroxypropan-2-yl)pyridin-2-yl)-l-tosyl- lH-benzo[d]imidazol-2-yl)methyl)-2-methylpropane-2-sulfinamide (240 mg) as white foam.

Step 7. (R and S)-2-(2-(2-(amino(2-chlorophenyl)methyl)-l-tosyl-lH- benzo[d]imidazol-4-yl)pyridin-4-yl)propan-2-ol. To a solution of (S)-N-((R and S)-(2-chlorophenyl)(4-(4-(2-hydroxypropan-2-yl)pyridin-2-yl)- 1 -tosyl- 1 H- benzo[d]imidazol-2-yl)methyl)-2-methylpropane-2-sulfinamide (190 mg, 0.292 mmol) in MeOH (10 mL) was added both 1.25 M HCl in MeOH (10 mL) and 5 M HCl in water (5 mL). The solution was stirred for 1 h. The solvents were then removed under reduced pressure. The residue was partitioned between 9: 1 CHCI3/IPA (30 mL) and 1 M NaOH (20 mL). The organics were dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 2 to 6% of 2 M NH3 in
MeOH/CH2Cl2 to afford a mixture of (R)-2-(2-(2-(amino(2- chlorophenyl)methyl)-l -tosyl- lH-benzo[d]imidazol-4-yl)pyridin-4-yl)propan-2- ol and (S)-2-(2-(2-(amino(2-chlorophenyl)methyl)- 1 -tosyl- 1 H-benzo[d]imidazol- 4-yl)pyridin-4-yl)propan-2-ol (140 mg) as white foam.

Step 8. (S and R)-N-((2-chlorophenyl)(4-(4-(2-hydroxypropan-2- yl)pyridin-2-yl)- 1 -tosyl- 1 H-benzo[d]imidazol-2- yl)methyl)cyclopropanesulfonamide. A solution of (S and R)-2-(2-(2-(amino(2- chlorophenyl)methyl)- 1 -tosyl- lH-benzo[d]imidazol-4-yl)pyridin-4-yl)propan-2- ol (130 mg, 0.238 mmol) cyclopropanesulfonyl chloride (Sigma- Aldrich, St.
Louis MO, 48.4 μΐ,, 0.475 mmol), DMAP (2.90 mg, 0.024 mmol), DIEA (62.3 μί, 0.356 mmol) in DMF (1 mL) was stirred for 18 h at 25 °C. The reaction was then partitioned between EtOAc (20 mL) and 5% NaHC03 (20 mL). The organics were dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 1.5 to 5% MeOH/CH2Cl2 to afford a mixture of (S)-N-((2-chlorophenyl)(4-(4-(2- hydroxypropan-2-yl)pyridin-2-yl)- 1 -tosyl- 1 H-benzo[d]imidazol-2- yl)methyl)cyclopropanesulfonamide and (R)-N-((2-chlorophenyl)(4-(4-(2- hydroxypropan-2-yl)pyridin-2-yl)- 1 -tosyl- 1 H-benzo[d]imidazol-2- yl)methyl)cyclopropanesulfonamide (127 mg) as yellow film, m/z (ESI, pos. ion) 651.0 (M+H)+.

Step 9. N-((R and S)-(2-chlorophenyl)(4-(4-(l -hydroxy- 1-methylethyl)- 2-pyridinyl)- 1 H-benzimidazol-2 -yl)methyl)cyclopropanesulfonamide. A suspension of (R and S)-N-((2-chlorophenyl)(4-(4-(2-hydroxypropan-2- yl)pyridin-2-yl)- 1 -tosyl- 1 H-benzo[d]imidazol-2- yl)methyl)cyclopropanesulfonamide (125 mg, 0.192 mmol) with lithium hydroxide monohydrate (40.3 mg, 0.960 mmol) in THF (1 mL), MeOH (0.2 mL) and water (0.2 mL) was combined and heated to 40 °C with stirring. After 3 h, the reaction was partitioned between 9: 1 CHC13/IPA (15 mL) and 5% NaHC03 (10 mL). The organics were then concentrated under reduced pressure followed by purification by silica gel chromatography (12 g) eluting products with 1.5 to 5% of 2 M NH3 in MeOH/CH2Cl2 to afford a mixture of N-((S)-(2- chlorophenyl)(4-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H-benzimidazol-2- yl)methyl)cyclopropanesulfonamide and N-((R)-(2-chlorophenyl)(4-(4-( 1 - hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H-benzimidazol-2 -
yl)methyl)cyclopropanesulfonamide (35 mg) as yellow film. The film was dissolved in 1 : 1 MeCN/water (1.6 mL), frozen, then lyophilized to afford a mixture of desired compounds as yellow fluffy solid.
1H NMR (400 MHz, CDC13) δ ppm 12.15 (br. s., 1 H) 8.64 (d, J=5.28 Hz, 1 H) 8.14 (s, 1 H) 7.85 (d, J=7.63 Hz, 1 H) 7.76 (d, J=8.02 Hz, 1 H) 7.68 (dd, J=7.43, 1.96 Hz, 1 H) 7.45 (dd, J=7.53, 1.66 Hz, 1 H) 7.21 - 7.37 (m, 5 H) 6.45 - 6.58 (m, 2 H) 2.24 - 2.36 (m, 1 H) 1.63 (s, 6 H) 1.13 - 1.23 (m, 1 H) 1.03 - 1.13 (m, 1 H) 0.79 - 0.89 (m, 1 H) 0.65 - 0.79 (m, 1 H). m/z (ESI, pos. ion) 497.0 (M+H)+. GK-GKRP IC50 (Binding) = 1.04 μΜ.
Example 49
N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-lH- indol-2-yl)methyl)cyclopropanesulfonamide


Step 1. To a stirred solution of 7-bromoindole (Sigma- Aldrich Corp, St. Louis, MO; 5.0 g, 25.5 mmol) in dry DMF (100 mL) at 0 °C under nitrogen was added sodium hydride, 60% dispersion in mineral oil (1.22 g, 30.6 mmol) in two portions. Gas evolution was evident. The suspension was stirred for 1 h at 0 °C. To this solution was added para -to luenesulfonyl chloride (3.26 mL, 25.5 mmol) in one portion and the reaction was stirred for 20 min then quenched with saturated NH4C1 (10 mL). The reaction was diluted with EtOAc (200 mL) and washed with water (600 mL). The separated aqueous layer was extracted with
EtOAc (100 mL). The organics layers were combined, dried over MgS04, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (120 g) with 5 to 20% EtO Ac/Hex as eluant to afford 7-bromo- 1-tosyl-lH-indole (6.24 g, 17.82 mmol, 69.9 % yield) as an off-white solid, m/z
+.

Step 2. To a stirred solution of diisopropylamine (2.76 mL, 19.70 mmol) in THF (20 mL) at -70 °C under nitrogen was added n-butyllithium (2.5M in hexanes) (5.25 mL, 13.13 mmol). The reaction was stirred for 20 min then a solution of 7-bromo-l-tosyl-lH-indole (4.60 g, 13.13 mmol) in THF (20 mL) was added. The cooling bath was removed and the red solution warmed to -15 °C. The reaction was then chilled back to -70 °C and a solution of N-((S,E)-(2- chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (3.44 g, 16.42 mmol, Intermediate Yl) in THF (20 mL) was added. The cooling bath removed and the reaction was allowed to warm to 20 °C over 2 h. The reaction was then quenched with saturated NH4C1 (20 mL) and diluted with EtO Ac (150 mL). The organic layer was dried over MgS04, filtered, and concentrated under reduced pressure.
The residue was purified by silica gel chromatography (330 g) eluting products with 30 to 70% EtOAc/Hex to afford (S)-N-((R)-(7-bromo-l-tosyl-lH-indol-2- yl)(2-chlorophenyl)methyl)-2-methylpropane-2-sulfinamide (6.50 g, 10.94 mmol, 83 % yield) as white foam, m/z (ESI, +ve ion) 593.0/595.0 (M+H)+.

Step 3. To a solution of (S)-N-((R)-(7-bromo-l-tosyl-lH-indol-2-yl)(2- chlorophenyl)methyl)-2-methylpropane-2-sulfinamide (1.65 g, 2.78 mmol) in MeOH (20 mL) was added 5 M HCI in water (5 mL). The solution was stirred for 18 h at 25 °C. The solvents were removed under reduced pressure. The residue was partitioned between 9: 1 CHC13/IPA (30 mL) and 5 M NaOH (15 mL). The organic layer was the dried over MgS04 and concentrated under reduced pressure to afford (R)-(7-bromo-l-tosyl-lH-indol-2-yl)(2- chlorophenyl)methanamine (1.45 g, 2.96 mmol, 107 % yield) as colorless oil. m/z (ESI, +ve ion) 510.9/512.9 (M+Na)+.

Step 4. To a stirred solution of (R)-(7-bromo-l-tosyl-lH-indol-2-yl)(2- chlorophenyl)methanamine (1.36 g, 2.78 mmol), DIEA (0.606 mL, 3.47 mmol), DMAP (0.017 g, 0.139 mmol) in DMF (5 mL) at 25 °C under argon was added cyclopropanesulfonyl chloride (0.354 mL, 3.47 mmol). The reaction was stirred for 22 h at 25 °C then partitioned between EtOAc (80 mL) and 5% NaHC03 (100 mL). The organic layer was dried over MgS04, filtered, and concentrated under
reduced pressure. The residue was purified by silica gel chromatography (80 g) eluting products with 25 to 50% EtO Ac/Hex to afford (R)-N-((7-bromo-l-tosyl- lH-indol-2-yl)(2-chlorophenyl)methyl)cyclopropanesulfonamide (0.86 g, 1.448 mmol, 52.1 % yield) as a colorless oil. m/z (ESI, +ve ion) 614.9/616.8 (M+Na)+.

Step 5. To a solution of (R)-N-((7-bromo-l-tosyl-lH-indol-2-yl)(2- chlorophenyl)methyl)cyclopropanesulfonamide (0.85 g, 1.431 mmol) in THF (6 mL) and MeOH (6 mL) was added a solution of lithium hydroxide monohydrate (0.300 g, 7.16 mmol) in water (3 mL). The reaction was heated to 80 °C in a capped vessel for 3 d. The reaction was then concentrated under reduced pressure and the residue partitioned between 9: 1 CHC13/IPA (30 mL) and 5%> NaHC03 (15 mL). The organic layer was then concentrated under reduced pressure and purified by silica gel chromatography (80 g) with 0 to 2.5%> of 2 M NH3 in MeOH/CH2Cl2 as eluant to afford (R)-N-((7-bromo-lH-indol-2-yl)(2- chlorophenyl)methyl)cyclopropanesulfonamide (0.39 g, 0.887 mmol, 62.0 %>
+.

Step 6. A suspension of (R)-N-((7-bromo-lH-indol-2-yl)(2- chlorophenyl)methyl)cyclopropanesulfonamide (360 mg, 0.819 mmol),
bis(pinacolato)diboron (270 mg, 1.064 mmol), [1,1- bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with
dichloromethane (33.4 mg, 0.041 mmol), potassium acetate (77 μΐ, 1.228 mmol) in 1,4-dioxane (4 mL) was sparged with argon for 1 min then heated at 120 °C for 20 min. The reaction was then partitioned between EtOAc (30 mL) and 5 NaHC03 (10 mL). The organic layer was dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 2% MeOH/CH2Cl2 to afford (R)-N-((2- chlorophenyl)(7-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-indol-2- yl)methyl)cyclopropanesulfonamide (180 mg, 0.370 mmol, 45.2 % yield) as colorless oil. +.

Step 7. A suspension of (R)-N-((2-chlorophenyl)(7-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)-lH-indol-2-yl)methyl)cyclopropanesulfonamide (300 mg, 0.616 mmol), 2-(2-chloro-4-pyridinyl)-2-propanol (127 mg, 0.739 mmol, Intermediate Y4), tetrakis(triphenylphosphine)palladium(0) (35.6 mg, 0.031 mmol), sodium carbonate (261 mg, 2.465 mmol) in 1,4-dioxane (3 mL) and water (2 mL) was sparged with argon for 1 min then heated at 100 °C for 30 min. The reaction was then partitioned between EtOAc (20 mL) and 5% NaHC03 (10 mL). The organic layer was dried over MgS04, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (40 g) with 0 to 3% of 2 M NH3 in MeOH/CH2Cl2 as eluant to afford 80% pure material. The material was then further purified by silica gel chromatography (40 g) with 10 to 20% MeCN/CH2Cl2 as eluant to afford
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H-indol- 2-yl)methyl)cyclopropanesulfonamide (109 mg, 0.220 mmol, 35.7 %> yield) as a
colorless film. The product was suspended 1 : 1 MeCN/water, frozen, then lyophilized to afford the desired compound as white fluffy powder.
1H NMR (400 MHz, CDC13) δ ppm 0.71 - 0.80 (m, 1 H) 0.81 - 0.89 (m, 1 H) 1.06 - 1.21 (m, 2 H) 1.63 (d, J=1.57 Hz, 6 H) 1.81 - 1.98 (br. s., 1 H) 2.26 (tt, J= 7.95, 4.87 Hz, 1 H) 5.45 (br s, 1 H) 6.16 (d, J= 1.76 Hz, 1 H) 6.39 (d, J= 8.22 Hz, 1 H) 7.16 (t, J= 7.73 Hz, 1 H) 7.23 - 7.30 (m, 1 H) 7.35 (quind, J= 7.58, 7.58, 7.58, 7.58, 1.76 Hz, 2 H) 7.43 - 7.49 (m, 1 H) 7.54 - 7.62 (m, 2 H) 7.78 (d, J = 7.43 Hz, 1 H) 8.12 (d, J= 0.78 Hz, 1 H) 8.61 (d, J= 5.28 Hz, 1 H) 11.52 (br s, 1 H). m/z (ESI, +ve ion) 496.1 (M+H)+. GK-GKRP IC50 (Binding) = 0.14 μΜ.
Example 50
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)-l-benzofuran-5-sulfonamide

To a solution of (2S)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (10 mg, 0.022 mmol, Intermediate X8) in DMF (1.0 ml) was added diisopropylethylamine (11.27 μΐ, 0.065 mmol, Sigma-Aldrich, St. Louis, MO), benzofuran-5-sulfonyl chloride (5.62 mg, 0.026 mmol, Astatech, Bristol, PA) and 4-dimethylaminopyridine (2.64 mg, 0.022 mmol, Sigma-Aldrich, St. Louis, MO). The reaction was stirred at room temperature for 1.5 h and then quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with water and brine. The solution was concentrated and the residue was purified by flash chromatography (4 g RediSep® Rf column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-
hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 - benzofuran-5 -sulfonamide (6.4 mg) as a colorless film.
1H NMR (400 MHz, CDC13) δ ppm 8.69 - 8.74 (m, 1 H), 8.08 - 8.11 (m, 1 H), 8.06 (d, J= 2.0 Hz, 1 H), 7.81 - 7.85 (m, 1 H), 7.73 (dd, J= 8.6, 2.0 Hz, 1 H), 7.68 (d, J= 2.3 Hz, 1 H), 7.60 - 7.64 (m, 1 H), 7.37 - 7.47 (m, 4 H), 7.17 - 7.23 (m, 1 H), 7.08 - 7.15 (m, 2 H), 6.85 (d, J= 1.4 Hz, 1 H), 6.75 (dd, J= 2.2, 1.0 Hz, 1 H), 6.24 - 6.28 (m, 1 H), 5.59 (d, J= 7.8 Hz, 1 H), 2.71 (s, 1 H), 1.83 (m, 3 H). m/z (ESI, pos. ion) 643.2 (M+H)+. GK-GKRP IC50 (Binding) = 0.058 μΜ.
Example 51
N-((R)-(2-(l-Methylethenyl)phenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. A solution of 2-bromobenzaldehyde (10.0 g, 54.34 mmol, Sigma- Aldrich, India), potassium isopropenyl trifluoroborate (9.64 g, 65.21 mmol, Frontier Scientific, USA), Cs2C03 (52.8 g, 163.04 mmol, Molekula Life
Sciences, India)) and triphenylphosphane (1.42 g, 5.43 mmol, Sigma- Aldrich, India) in THF:H20 (100 mL : 20 mL), was degassed with argon for 10 min. To the above reaction mixture, Pd(OAc)2 (1.22 g, 5.43 mmol Sigma-Aldrich, India,) was added and again degassed with argon for 10 min and the solution was slowly heated to 100 °C and maintained at 100 °C for 16 h. The reaction mixture was allowed to cool to ambient temperature and diluted with water (100 mL) and product was extracted into the ethyl acetate (100 mL). The aqueous layer was back extracted with ethyl acetate (100 mL). The organic layer were combined and dried over anhydrous Na2S04 and concentrated. The resulting product was purified by column chromatography using silica gel (100-200 mesh) eluting with
10 % ethyl acetate in hexane to deliver 4.6 g of 2-(l- methylethenyl)benzaldehyde.
Step 2. To a solution of 2-(l-methylethenyl)benzaldehyde (4.6 g, 31.29 mmol, 113289-74) and (S)-2-methylpropane-2-sulfinamide (3.78 g, 31.29 mmol, Combi Blocks, India) in dichloromethane (50 mL, Ranchem, India) was added Ti(OEt)4 (35.7 g, 156.46 mmol, Sigma- Aldrich, India) at ambient temperature and the solution was stirred for 16 h. The reaction mixture was quenched with water (100 mL) and product was extracted into the dichloromethane (100 mL). The aqueous layer was back extracted with dichloromethane (100 mL). The organic layers were combined and dried over anhydrous Na2S04 and
concentrated. The resulting product was purified by column chromatography using silica gel (100-200 mesh) eluting with 15% ethyl acetate in hexane to afford 5.8 g (S)-2-methyl-N-((lZ)-(2-(l-methylethenyl)phenyl)methylidene)-2- propanesulfmamide as a brown color oil.
Step 3. To a solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 - trifluoro-2-propanol (2.5 g, 7.73 mmol, Intermediate Y3) in THF (80 mL), was added n-butyllithium (2.5M solution in hexane, 1.48 g, 23.2 mmol, Acros, India) at -80 °C and stirred for 30 min. To the above reaction mixture, a solution of (S)- 2-methyl-N-(( 1 Z)-(2-( 1 -methylethenyl)phenyl)methylidene)-2- propanesulfmamide (1.9 g, 7.73 mmol) in THF (20 mL) was added slowly dropwise and the reaction mixture was stirred for 2 h at same temperature. The reaction was slowly quenched with water and product was extracted into the ethyl acetate (100 mL). The aqueous layer was back extracted with ethyl acetate (100 mL). The organic layers were combined and dried over anhydrous Na2S04 and concentrated. The resulting product was purified by column chromatography using silica gel (60-120) eluting with 40 % ethyl acetate in hexane to give 2.2 g of (S)-2-methyl-N-((R)-(2-(l-methylethenyl)phenyl)(7-(4-(2,2,2-trifiuoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- propanesulfmamide a brown solid.
Step 4. The diastereomers of (S)-2-methyl-N-((R)-(2-(l- methylethenyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-propanesulfinamide were separated using two sequential preparative chiral SFC columns. Column 1 : Chiralcel® AD- H column (250 mm, 21 mm, 5 um) eluting with 50% liquid CO2/50%> isopropanol containing 0.2% diethylamine at a flow rate of 50 mL/minute.
Column 2: Chiralcel® AD-H column (250 mm, 21 mm, 5 um) eluting with 70% liquid CO2/30% isopropanol containing 0.2% diethylamine at a flow rate of 60 mL/minute. The absolute stereochemistries of the two diastereomers were assigned arbitrarily.
First eluting peak: (S)- 2-methyl-N-((R)-(2-(l-methylethenyl)phenyl)(7- (4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)methyl)-2-propanesulfinamide. Second eluting peak: (S)- 2-methyl-N-((R)- (2-( 1 -methylethenyl)phenyl)(7-(4-(( 1 S)-2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-propanesulfinamide.
Step 5. (S)-2-Methyl-N-((R)-(2-( 1 -methylethenyl)phenyl)(7-(4-(( 1 R)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-propanesulfinamide (first eluting peak from Step 4) was dissolved in 10 mL of MeOH. Hydrogen chloride, 4.0M solution in 1,4-dioxane (2.62 ml, 10.48 mmol, Sigma Aldrich, St. Louis, MO) was added to the mixture. After 30 min, the solvent was removed under reduced pressure. The residue was partitioned between 20 mL of EtOAc and 20 mL of aq NaHCOs. The organic portion was washed with 10 mL of (2R)-2-(2-(2-((R)-amino(2-(l- methylethenyl)pheny l)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol (0.49 g, 1.046 mmol), which was carried on without further purification.
Step 6. (2R)-2-(2-(2-((R)- Amino(2-( 1 -methylethenyl)phenyl)methyl)- 1 - benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (0.49 g, 1.046
mmol), cyclopropanesulfonyl chloride (0.426 ml, 4.18 mmol, Matrix Scientific, Columbia, SC), and Hunig's base (0.728 ml, 4.18 mmol, Sigma Aldrich, St. Louis, MO) were dissolved in 10 mL of 1 ,2-dichloroethane and heated at 80 °C. After 24 h, the mixture was cooled to rt and quenched with 20 mL of aq
NaHC03. The mixture was extracted with DCM (15 mL x 2) and the combined organic extracts were dried over MgS04, and filtered. The solution was concentrated under reduced pressure, and the residue was purified by flash chromatography on silica gel (40 g RediSep® Rf column, eluting with 5% to 30% EtOAc/hexanes). Further purification by reverse phase HPLC (Phenomenox Gemini 5 micron C18 column, 100 mm X 30 mm, eluting with 10% to 95% MeCN/water gradient a 30 mL/min flow rate for 14 min) afforded N-((R)-(2-(l- methylethenyl)phenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.025g) as a white solid.
1H NMR (400MHz , CDC13) δ ppm 8.83 (d, J= 5.3 Hz, 1 H), 8.15 (s, 1 H), 7.88 (d, J = 7.6 Hz, 1 H), 7.73 (d, J = 7.6 Hz, 1 H), 7.66 - 7.54 (m, 1 H), 7.51 - 7.42 (m, 2 H), 7.39 - 7.31 (m, 2 H), 7.24 - 7.16 (m, 1 H), 7.03 (s, 1 H), 6.38 (d, J= 7.4 Hz, 1 H), 5.28 (s, 1 H), 5.20 (d, J= 7.4 Hz, 1 H), 4.88 (s, 1 H), 2.66 (s, 1 H), 2.28 - 2.16 (m, 1 H), 2.09 (s, 3 H), 1.85 (s, 3 H), 1.17 - 1.01 (m, 2 H), 0.88 - 0.70 (m, 2 H). m/z (ESI, pos. ion) 573.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.067 μΜ.
Example 52
N-((R)-(2-(l-Methylethenyl)phenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. (S)-2-Methyl-N-((R)-(2-( 1 -methylethenyl)phenyl)(7-(4-(( 1 S)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-propanesulfinamide (second eluting peak from example 51 , Step 4) was dissolved in 13 mL of MeOH. Hydrogen chloride, 4.0M solution in 1,4- dioxane (2.69 ml, 10.76 mmol, Sigma Aldrich, St. Louis, MO) was added to the mixture. After 1 h, the solvent was removed under reduced pressure. The residue was partitioned between 20 mL of DCM and 20 mL of aq NaHC03. The organic portion was washed with 10 mL of brine and dried over MgS04. The solution was filtered and concentrated under reduced pressure to afford (2S)-2-(2- (2-((R)-amino(2-( 1 -methyletheny l)phenyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.63 g, 1.345 mmol) as a clear oil. The product was carried on to the next step without additional purification.
Step 2. (2S)-2-(2-(2-((R)-amino(2-(l -methylethenyl)phenyl)methyl)- 1 - benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (0.63 g, 1.345 mmol) cyclopropanesulfonyl chloride (0.548 ml, 5.38 mmol, Matrix Scientific, Columbia, SC), and Hunig's base (0.936 ml, 5.38 mmol Sigma Aldrich, St. Louis, MO) were dissolved in 20 mL of 1,2 dichloroethane and heated to 90 °C. After 4 h, the mixture was cooled to room temperature and quenched with 20 mL of aq NaHC03. The mixture was extracted twice with 20 mL of DCM and the combined organic extracts were dried over MgS04. . The solution was filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on in silica gel (40 g RediSep® Rf column, eluting with 5% to 30% EtOAc/hexanes) afforded 400 mg of product. 50 mg of the product was further purified by reverse phase HPLC ((Phenomenox Gemini 5 micron C18
column, 100 mm X 30 mm, eluting with 30% to 95% MeCN/water gradient a 30 mL/min flow rate for 12 min) to afford N-((R)-(2-(l -methylethenyl)phenyl)(7-(4- (( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.025 g) as a white solid.
1H NMR (400MHz ,CDC13) δ ppm 8.83 (d, J= 5.1 Hz, 1 H), 8.14 (s, 1 H), 7.88 (d, J= 7.4 Hz, 1 H), 7.73 (d, J= 7.0 Hz, 1 H), 7.63 - 7.55 (m, 1 H), 7.46 (t, J = 7.6 Hz, 2 H), 7.34 (dquin, J= 1.8, 7.0 Hz, 2 H), 7.22 (dd, J = 2.2, 6.8 Hz, 1 H), 7.03 (s, 1 H), 6.38 (d, J= 7.4 Hz, 1 H), 5.27 (s, 1 H), 5.15 (d, J= 7.2 Hz, 1 H), 4.87 (s, 1 H), 2.52 (s, 1 H), 2.31 - 2.14 (m, 1 H), 2.08 (s, 3 H), 1.85 (s, 3 H), 1.16 - 0.97 (m, 2 H), 0.88 - 0.61 (m, 2 H). m/z (ESI, pos. ion) 573.0 (M+H)+. GK- GKRP IC50 (Binding) = 0.033 μΜ.
Example 53
N-((R)-(2-chlorophenyl)(7-(5-(l-hydroxy-l-methylethyl)-6-oxo-l,6-dihydro- 3-pyridazinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide


Step 1. To a 100-mL, single-necked, round-bottomed flask containing a solution of benzyl alcohol (560 mg, 5.18 mmol, Sigma- Aldrich, India) in DMF (40 mL) at 0 °C was added sodium hydride (260 mg, 10.9 mmol, prepared by washing a 60% dispersion of sodium hydride in mineral oil with n-pentane and decantation of the solvent two times followed by drying under reduced pressure, Sigma- Aldrich, India) and the mixture was stirred at room temperature for 30 min. A solution of 3,6-dichloropyridazine-4-carboxylic acid (1.0 g, 5.18 mmol, Frontier Scientific) in DMF (5 mL) was added slowly and the mixture was stirred at room temperature for 2 h. Saturated aqueous NH4C1 (30 mL) was added and the resulting solid was removed by filtration, washed with water, and dried under reduced pressure to deliver 3-(benzyloxy)-6-chloro-4-pyridazinecarboxylic acid (1.0 g) as a white solid.
Step 2. To a 100-mL, single-necked, round-bottomed flask containing a solution of 3-(benzyloxy)-6-chloro-4-pyridazinecarboxylic acid (1.2 g, 3.36 mmol) in MeOH (50 mL) at 0 °C was added (trimethylsilyl)diazomethane (5.0 mL of a 2.0 M solution in diethylether, 10.0 mmol, Sigma- Aldrich, India) and the mixture was stirred at 0 °C for 2 h. The mixture was sparged with nitrogen gas for 15 min and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 6% EtOAc/hexanes) to deliver methyl 3- (benzyloxy)-6-chloro-4-pyridazinecarboxylate (1.1 g) as white solid.
Step 3. To a 100-mL, two-necked, round-bottomed flask containing a solution of methyl 3-(benzyloxy)-6-chloro-4-pyridazinecarboxylate (400 mg, 1.43 mmol) in THF (40 mL) at -40 °C was added methylmagnesium bromide (1.43 mL of a 3.0 M solution in diethylether, 4.29 mmol, Sigma- Aldrich, India) and the mixture was stirred at -40 °C for 2 h. Saturated aqueous NH4C1 (30 mL) was added, the mixture was allowed to warm to room temperature, and extracted with EtOAc (2 x 100). The combined organic layers were washed with water, dried (Na2S04), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 20% EtOAc/hexanes) to deliver 3-(benzyloxy)-6-chloro-4-(l-methoxy-l-methylethyl)pyridazine (200 mg) as an off-white solid.
Step 4. A 20-mL microwave tube containing a solution of 3-(benzyloxy)- 6-chloro-4-(l-methoxy-l-methylethyl)pyridazine (200 mg, 0.72 mmol) in 1,4- dioxane/water (4: 1, 10 mL) was purged with argon for 15 min. N-((R)-(2- Chlorophenyl)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen- 2-yl)methyl)cyclopropanesulfonamide (400 mg, 0.790 mmol, Intermediate X14), [l, -bis(diphenylphosphino)ferrocene]dichloropalladium(II) (30 mg, 0.036 mmol, Sigma- Aldrich, India) and 2 M aqueous Na2C03 (0.5 mL) was added and the mixture was irradiated in a microwave reactor at 110 °C at 10 bar pressure for 30 min. The mixture was concentrated under reduced pressure and the residue was purified by flash chromatography (silica gel, 20% EtOAc/hexanes) to deliver
N-((R)-(7-(6-(benzyloxy)-5-(l -hydroxy- l-methylethyl)-3-pyridazinyl)-l- benzothiophen-2-yl)(2-chlorophenyl)methyl)cyclopropanesulfonamide (110 mg) as a light-yellow solid.
Step 5. A 50-mL round-bottomed flask containing a solution of N-((R)- (7-(6-(benzyloxy)-5 -( 1 -hydroxy- 1 -methylethyl)-3 -pyridazinyl)- 1 -benzothiophen- 2-yl)(2-chlorophenyl)methyl)cyclopropanesulfonamide (100 mg) in
MeOH/EtOAc (1 : 1, 30 mL) was purged with argon for 15 min. 20% Palladium hydroxide on carbon (10.0 mg, Sigma-Aldrich, India) was added and the mixture was stirred under 20 psi of hydrogen gas at ambient temperature for 1 h. The mixture was filtered through a pad of diatomaceous earth, which was washed with methanol. The combined filtrate and washing were concentrated under reduced pressure and the residue was subjected to preparative TLC (silica gel, 50%) EtOAc/hexane). The silica gel containing the desired product was removed from the plate, washed with 5% MeOH/CHCl3 (2 x 20 mL), and filtered through a pad of diatomaceous earth. The combined filtrates were concentrated under reduced pressure and the residue was triturated with n-pentane (10 ml) to deliver N-((R)-(2-chlorophenyl)(7-(5 -( 1 -hydroxy- 1 -methylethyl)-6-oxo- 1 ,6-dihydro-3 - pyridazinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (10 mg) as an off-white solid.
1H NMR (400 MHz, methanol-^): δ ppm 8.29 (s, 1 H), 7.85 (d, J= 7.2 Hz, 1 H), 7.96 - 7.74 (m, 2 H), 7.50 - 7.45 (m, 2 H), 7.43 - 7.37 (m, 2 H), 7.00 (s, 1 H), 6.42 (s, 1 H), 2.27 (m, 1 H), 1.63 (s, 6 H), 1.01 - 0.94 (m, 2 H), 0.81 - 0.70 (m, 2 H). m/z (APCI, pos. ion) 529.7 (M+l)+. GK-GKRP IC50 (Binding) = 0.108 μΜ.
Example 54
V-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)-l,l,l-trifluoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-3- methylbenzofuran-5-sulfonamide



Step 1. To a solution of 3,6-dichloro-pyridinecarboxaldehyde (40.0 g,
227 mmol) in CH2CI2 (400 mL) was added (S)-2-methylpropane-2-sulfinamide (33.0 g, 272 mmol) and Ti(OEt)4 (259.09 g, 1136.36 mmol). The reaction was stirred at ambient temperature for 18 h. After completion of reaction as monitored by TLC, the reaction mixture was cooled to 0 °C and was quenched with water (600 mL). The mixture was filtered through a pad of diatomaceous
earth. The aqueous layer was extracted with CH2CI2 (200 mL x 2). The combined organic layers were washed with brine (150 mL x 2), dried over anhydrous sodium sulfate, concentrated under reduced pressure and was purified by column chromatography (silica, gradient elution from petroleum ether to 10% ethyl acetate/petroleum ether) to provide (S,E)-N-((3,6-dichloropyridin-2- yl)methylene)-2-methylpropane-2-sulfinamide (49.0 g) as yellow solid.

Step 2. (S)-2-(2-(benzo[b]thiophen-7-yl)pyridin-4-yl)- 1,1,1- trifluoropropan-2-ol (3.32 g, 10.27 mmol, Intermediate X7) was placed in a flask and was azeotropically dried with 5 mL of toluene. THF (45 mL) was added to the flask and the resulting clear solution was cooled to -78 °C. To this solution was added n-butyllithium solution (2.5M in hexanes, 8.21 mL, 20.5 mmol, Sigma- Aldrich, St. Louis, MO), which resulted in a deeply reddish color. After 5 min, a solution of (S,E)-N-((3,6-dichloropyridin-2-yl)methylene)-2- methylpropane-2-sulfmamide (2.61 g, 9.33 mmol) in THF (30 mL) was added down the wall of the reaction flask. The resulting dark reaction solution was stirred at -78 °C for 1.5 h and was quenched with water (50 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, dried over MgS04, concentrated and purified by flash
chromatography (80 g RediSep® Rf column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide (S)-N-((R)-(3,6-dichloropyridin-2-yl)(7-(4-((R)- 1,1,1 -trifluoro-2-hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)-2-methylpropane-2-sulfinamide (1.62 g) as a pale yellow foam.

Step 3. To a sealed tube was added (S)-N-((R)-(3,6-dichloropyridin-2- yl)(7-(4-((S)- 1,1,1 -trifluoro-2-hydroxypropan-2-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)methyl)-2-methylpropane-2-sulfinamide (731 mg, 1.21 mmol), tert-butyl carbamate (171 mg, 1.46 mmol, Sigma- Aldrich, St. Louis, MO), cesium carbonate (593 mg, 1.820 mmol, Sigma-Aldrich, St. Louis, MO), tris(dibenzylideneacetone)dipalladium (0) (55.5 mg, 0.061 mmol, Strem
Chemicals, Newbury Port, MA), 2-(dicyclohexylphosphino)-2',4',6'-tri-i-propyl- Ι,Γ-biphenyl (57.8 mg, 0.121 mmol, Strem Chemicals, Newbury Port, MA) and 1,4-dioxane (10 mL). The mixture was purged with N2 for 5 min, and stirred at 100 °C for 8 h. The mixture was then cooled to room temperature and was quenched with water. The aqueous layer was extracted with EtOAc. The combined organic layers were concentrated and purified by flash chromatography (40 g RediSep® Rf column, gradient elution from hexanes to 40%
EtOAc/hexanes) to provide tert-butyl (6-((R)-(((S)-tert-butylsulfinyl)amino)(7- (4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (70 mg) as a pale yellow powder.

Step 5. To a solution of (S)-2-(2-(2-((R)-amino(6-amino-3-chloropyridin- 2-yl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)- 1,1,1 -trifluoropropan-2-ol hydrochloride (13.2 mg, 0.026 mmol) in DMF (2 mL) at room temperature was added N,N-diisopropylethylamine (22.3 μί, 0.128 mmol, Sigma- Aldrich, St. Louis, MO), 3-methylbenzofuran-5-sulfonyl chloride (Intermediate Ql below) (7.09 mg, 0.031 mmol), and 4-dimethylaminopyridine (3.13 mg, 0.026 mmol, Sigma- Aldrich, St. Louis, MO). The reaction was stirred at room temperature for 1.5 h and was quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, and concentrated. The residue was dried under high vacuum at 55 °C overnight to remove residual DMF. The resulting mixture was then purified by flash chromatography (4 g RediSep® Rf column, gradient elution from hexanes to 60% EtOAc/hexanes) to provide N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifluoro-2-hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)-3-methylbenzofuran-5 -sulfonamide (7.2 mg) as a pale yellow film.
1H NMR (300 MHz, chloroform-d) δ ppm 8.75 - 8.79 (m, 1 H), 8.04 - 8.08 (m, 1 H), 7.88 - 7.91 (m, 1 H), 7.76 - 7.80 (m, 1 H), 7.61 - 7.70 (m, 2 H), 7.31 - 7.44 (m, 3 H), 7.23 (s, 1 H), 7.03 - 7.06 (m, 2 H), 6.58 (d, J= 9.5 Hz, 1 H), 6.18 - 6.24 (m, 1 H), 6.11 (d, J = 8.6 Hz, 1 H), 4.43 (s, 2 H), 2.82 (s, 1 H), 2.07 (s, 3 H), 1.79 - 1.86 (m, 3 H). m/z (ESI, pos. ion) 673.1 (M+H)+. GK-GKRP IC50 (Binding) = 0.009 μΜ.
Intermediate Ql
3-Methylbenzofuran-5-sulfonyl chloride

To a solution of 5-bromo-3-methylbenzofuran (500 mg, 2.369 mmol, Milestone PharmTech, New Brunswick, NJ) in THF (20 mL) at -78 °C was added n-butyllithium solution, (2.5M in hexanes, 1137 μΐ,, 2.84 mmol, Sigma- Aldrich, St. Louis, MO) dropwise. The resulting dark solution was stirred at -78 °C for 15 min. A stream of sulfur dioxide (Sigma- Aldrich, St. Louis, MO) was bubbled through the solution for 10 min, resulting in a pale yellow slurry. The cold bath was removed and the reaction was slowly warmed to room temperature and stirred for 1 h. The solvent was removed under a vacuum. The residue was taken up in CH2CI2 (10 mL) and cooled to 0 °C. N-chlorosuccinimide (316 mg, 2.37 mmol, Alfa Aesar, Ward Hill, MA) was added in one portion. The reaction mixture was stirred at 0 °C for 1 h. The reaction was then quenched with saturated NaHC03 solution. The organic layer was washed with brine, concentrated and purified by flash chromatography (40 g RediSep® normal phase column, gradient elution from hexanes to 5% EtOAc/hexanes) to provide 3- methylbenzofuran-5-sulfonyl chloride (50 mg) as a white crystalline solid.
Intermediate Q2
l-Ethyl-6-oxo-l,6-dihydropyridine-3-sulfonyl chloride


Step 1. To a solution of 5-bromo-2(7H)-pyridone (5.97 g, 34.3 mmol, Sigma- Aldrich, St. Louis, MO) in DMF (40 mL) at 0 °C was added sodium hydride (60% dispersion in mineral oil (1.65 g, 41.2 mmol, Sigma-Aldrich, St. Louis, MO) in one portion. The resulting mixture was stirred at 0 °C for 40 min and was treated with iodoethane (3.32 mL, 41.2 mmol, Sigma-Aldrich, St. Louis, MO). The reaction mixture was stirred at room temperature for 15 h, and was quenched with saturated NaHC03 solution. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with water, brine, concentrated and purified by flash chromatography (80 g RediSep® Rf column, gradient elution from hexanes to 60% EtOAc/hexanes) to provide the desired product that was contaminated with some residual DMF. The mixture was dried under high vacuum at 75 °C for 12 h to provide 5-bromo-l-ethylpyridin-2(7H)- one (3.5 g) as a pale yellow oil.


Step 3. To a solution of methyl 3-((l-ethyl-6-oxo-l,6-dihydropyridin-3- yl)thio)propanoate (1.96 g, 8.12 mmol) in THF (30 mL) at 0 °C was added potassium tert-butoxide (1.276 g, 11.37 mmol, Sigma-Aldrich, St. Louis, MO) in one portion. The resulting yellowish slurry was stirred at 0 °C for 20 min. The reaction mixture was quenched with IN HC1 solution (25 mL). The aqueous layer was extracted with CH2C12 and the combined organic layers were concentrated and purified by flash chromatography (40 g RediSep® Rf, gradient elution from CH2C12 to 8% MeOH (containing 2 M NH3)/CH2C12) to provide l-ethyl-5- mercaptopyridin-2(lH)-one (1.236 g) as a pale yellow oil, which solidified upon standing at room temperature.

Step 4. To a solution of l-ethyl-5-mercaptopyridin-2(7H)-one (1.236 g, 7.96 mmol) in CH3CN (40 mL)/AcOH (1.5 mL)/H20 (1 mL) at 0 °C was added l ,3-dichloro-5,5-dimethylhydantoin (2.353 g, 1 1.94 mmol, Alfa Aesar, Ward Hill, MA). The reaction was stirred at 0 °C for 75 min and was slowly quenched with saturated NaHC03 solution. The aqueous layer was extracted with EtOAc and the combined organic layers were concentrated and purified by flash chromatography (40 g RediSep® Rf column, gradient elution from hexanes to 30% EtOAc/hexanes) to provide l-ethyl-6-oxo-l ,6-dihydropyridine-3-sulfonyl chloride (244 mg) as a thin white crystalline solid.
Example 55
V-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)-l,l,l-trifluoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-l oxo-l,6-dihydropyridine-3-sulfonamide

To a solution of (S)-2-(2-(2-((R)-amino(6-amino-3-chloropyridin-2- yl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)- 1 , 1 ,1 -trifluoropropan-2-ol hydrochloride (13.2 mg, 0.026 mmol) in DMF (2 mL) was added
diisopropylethylamine (22.28 μΐ, 0.128 mmol, Sigma-Aldrich, St. Louis, MO), 1- ethyl-6-oxo-l ,6-dihydropyridine-3-sulfonyl chloride (6.81 mg, 0.031 mmol) and 4-dimethylaminopyridine (3.13 mg, 0.026 mmol, Sigma-Aldrich, St. Louis, MO).
The reaction was stirred at room temperature for 1.5 h, and was quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were concentrated and dried under high vacuum at 50 °C overnight to remove residual DMF. The residue was purified by flash chromatography (4 g RediSep® Rf column, gradient elution from hexanes to EtOAc) to provide N- ((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifluoro-2-hydroxypropan- 2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-l-ethyl-6-oxo-l,6- dihydropyridine-3 -sulfonamide (7.3 mg) as an off- white powder.
1H NMR (300 MHz, chloroform-d) δ ppm 8.77 (d, J= 5.3 Hz, 1 H), 8.00 - 8.07 (m, 1 H), 7.71 - 7.80 (m, 2 H), 7.62 (d, J= 2.8 Hz, 1 H), 7.21 - 7.48 (m, 5 H), 6.72 (d, J= 8.5 Hz, 1 H), 6.35 (d, J= 8.6 Hz, 1 H), 6.19 (d, J= 8.2 Hz, 1 H), 6.09 (d, J= 9.6 Hz, 1 H), 4.61 (s, 2 H), 3.86 (s, 1 H), 3.58 - 3.71 (m, 1 H), 3.29 - 3.41 (m, 1 H), 1.82 - 1.86 (m, 3 H), 1.09 (t, J= 7.2 Hz, 3 H). m/z (ESI, pos. ion) 664.1 (M+H)+. GK-GKRP IC50 (Binding) = 0.002 μΜ.
Example 56
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)-l,l,l-trifluoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)benzofuran-
5-sulfonamide

To a solution of (S)-2-(2-(2-((R)-amino(6-amino-3-chloropyridin-2- yl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)- 1,1,1 -trifluoropropan-2-ol hydrochloride (27.8 mg, 0.047 mmol) in DMF (2 ml) at room temperature was added N,N-diisopropylethylamine (40.8 μί, 0.234 mmol, Sigma-Aldrich, St. Louis, MO), benzofuran-5-sulfonyl chloride (12.2 mg, 0.056 mmol, Astatech,
Bristol, PA), and 4-dimethylaminopyridine (5.7 mg, 0.047 mmol, Sigma- Aldrich, St. Louis, MO). The reaction was stirred at room temperature for 1 h and was quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, and concentrated. The residue was dried under high vacuum at 55 °C overnight to remove residual DMF. The resulting mixture was purified by flash chromatography (4 g RediSep® Rf column, gradient elution from hexanes to 60% EtOAc/hexanes) to provide N- ((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifluoro-2-hydroxypropan- 2-yl)pyridin-2-y l)benzo [b]thiophen-2-yl)methyl)benzofuran-5 -sulfonamide (21 mg) as a pale yellow foam.
1H NMR (300 MHz, chloroform-d) δ ppm 8.71 - 8.76 (m, 1 H), 7.99 - 8.07 (m, 1 H), 7.71 - 7.79 (m, 1 H), 7.58 - 7.71 (m, 2 H), 7.56 (d, J= 2.3 Hz, 1 H), 7.24 - 7.45 (m, 4 H), 7.02 - 7.09 (m, 2 H), 6.57 - 6.65 (m, 2 H), 6.16 - 6.22 (m, 1 H), 6.10 (d, J= 8.6 Hz, 1 H), 4.46 (s, 2 H), 3.12 (br. s., 1 H), 1.78 - 1.85 (m, 3 H). m/z (ESI, pos. ion) 659.1 (M+H)+. GK-GKRP IC50 (Binding) = 0.009 μΜ.
Intermediate AA1 (AA1-R and AA1-5) 2-(l-benzothiophen-7-yl)-4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine

Step 2
Step 1. To a solution of 7-bromo-l-benzothiophene (160 g, 0.755 mol, Combi-Blocks, San Diego, CA), bis(pinacolato)diboron (230 g, 0.905 mol) and potassium acetate (362.6 g, 3.780 mol), were dissolved in toluene (750 mL) at ambient temperature under an argon atmosphere. The solution was degassed by
purging argon gas for 15 min at ambient temperature. Pd(dppf)Cl2 (30.8 g, 0.038 mol) was added to the above solution under argon atmosphere. The resulting reaction mixture was again degassed by purging with argon gas (15 min). The reaction mixture was gradually heated to 100 °C and stirred at the same temperature for further 12 h under argon atmosphere. After completion of reaction (monitored by TLC, TLC eluent: 10 % EtOAc in hexane, UV active). The reaction mixture was diluted with water (1 L) and ethyl acetate (1 L). The organic layer was separated, washed with water, brine and dried over anhydrous Na2S04 and concentrated. The resulting product was purified by column chromatography using silica gel (60 to 120 mesh) eluting with 1% EtOAc- hexanes to afford 2-(l-benzothiophen-7-yl)-4,4,5,5-tetramethyl-l,3,2- dioxaborolane (140 g) as a white solid.
Step 2. A suspension of 2-chloro-4-(2,2,4-tetramethyl- 1 ,3-dioxolan-4- yl)pyridine (6.83 g, 30.0 mmol, Intermediate AA2), 2-(l-benzothiophen-7-yl)- 4,4,5,5-tetramethyl-l,3,2-dioxaborolane (8.01 g, 45 mmol, from Step 1), tetrakis(triphenylphosphine)palladium(0) (1.733 g, 1.5 mmol), sodium carbonate (9.54g, 90.0 mmol) in 1,4-dioxane (100 mL) and water (10 mL) was purged for 10 minutes. The suspension was heated to 80 °C overnight with stirring. The reaction mixture was then partitioned between EtOAc (250 mL) and 5% NaHC03 (75 mL). The organics were dried over sodium sulfate filtered, concentrated under reduced pressure. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (120 g), eluting with a gradient of 0% to 50% EtOAc in hexane to afford 2-(l-benzothiophen-7-yl)-4- (2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine (9.12g).
1H NMR (400 MHz, CDC13) δ 8.77 (d, J= 5.09 Hz, 1H), 8.01 (d, J= 0.59 Hz, 1H), 7.90 (d, J= 7.82 Hz, 2H), 7.56 (d, J= 5.48 Hz, 1H), 7.50 (t, J= 7.73 Hz, 1H), 7.42 (d, J= 5.67 Hz, 1H), 7.19 - 7.33 (m, lH), 4.16 (s, 2H), 1.64 (s, 3H), 1.57 (s, 3H), 1.44 (s, 3H).
The individual isomers were separated using preparative SFC (Chiralpak OJ-H column, 150 mm x 30 mm, 5 μΜ) eluting with 85 % liquid C02 in 15% MeOH. This method provided both compounds with enantiomeric excesses of > 95%. The first eluting peak was assigned as "S" with the second eluting peak assigned as "R" based on comparisons of the theoretical and experimental VCD spectra.
Intermediate AA2
2-chloro-4- 2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)pyridine

Step 1. In a 500 mL round bottom flask, a solution of l-hydroxy-2- propanone (24.3g, 0.32 mol, Sigma-Aldrich, India) in DMF (225 mL) maintained at rt, was treated with TBDMS-chloride (37. lg, 0.229 mol, Sigma-Aldrich, India), and imidazole (40.19g, 0.591 mol, Sigma-Aldrich, India). The solution was heated to 40°C for 2 hours under nitrogen atmosphere. Upon completion of reaction (TLC, 10% EtOAc-hexane, R 0.6), the reaction mixture was diluted with cold water (300 mL) and extracted with hexanes (250 mL x 4). The combined organic extract was washed with brine and concentrated under reduced pressure to give l-((tert-butyl(dimethyl)silyl)oxy)-2-propanone (35 g) as a colorless liquid.
Step 2. In 1.0 L three necked round bottom flask, 2-chloro-4-iodopyridine (95g, 0.396 mol, Apollo Sci. UK) was dissolved in dry THF (500 mL) at rt under nitrogen atmosphere. The resulting solution was cooled to -78°C and at this
temperature n-BuLi (1.6M, 297 mL, 0.476 mol, Acros, India) was added dropwise over a period of 45 minutes under nitrogen atmosphere. The reaction mixture was stirred at same temperature for 30 minutes under nitrogen atmosphere. After 30 minutes, l-((tert-butyl(dimethyl)silyl)oxy)-2-propanone (89.5g, 0.476 mol, from Step 1) was added dropwise over a period of 20 minutes at the same temperature The reaction mixture was stirred under nitrogen atmosphere for 2 h. Upon completion of reaction, the mixture was diluted with saturated NH4C1 solution and EtOAc. The organic layer was separated, washed with water, brine and dried over anhydrous Na2S04. The solution was concentrated under reduced pressure and the residue obtained was purified by silica gel (60 to 120 mesh) column chromatography (eluent, 8% EtOAc-hexanes) to give of l-((tert-butyl(dimethyl)silyl)oxy)-2-(2-chloro-4-pyridinyl)-2-propanol (62 g) as a colorless liquid.
Step 3. In a 1000 mL round bottom flask, a solution of l-((tert- butyl(dimethyl)silyl)oxy)-2-(2-chloro-4-pyridinyl)-2-propanol (62 g, 0.205 mol, from Step 2) was dissolved in THF (300 mL). The solution was cooled to 0°C and TBAF (1M in THF, 226 mL, 0.226 mol, Sigma-Aldrich, India ) was added drop wise over a period of 15 minutes. The reaction mixture was gradually warmed to rt and stirred at rt for 2 h (TLC, 30% EtOAc-hexanes, Rf 0.2). The reaction mixture was diluted with water (200 mL) and ethyl acetate (500 mL). The organic layer was separated, washed with water, brine and dried over anhydrous Na2S04. The solution was concentrated under reduced pressure to afford 2-(2-chloro-4-pyridinyl)-l ,2-propanediol (37 g) as a light brown liquid.
Step 4. In a 1000 mL round bottom flask, a solution of 2-(2-chloro-4- pyridinyl)-l ,2-propanediol (37 g, 0.197 mol, from Step 3) was dissolved in acetone (370 mL). The solution was cooled to 0°C and 2,2-dimethoxypropane (102.8 g, 0.98 mol, Sigma-Aldrich, India) was added followed by PTSA (4.5g, 0.023 mol, Sigma-Aldrich, India). The reaction mixture was gradually warmed to rt and stirred at rt for 12 h (TLC, 10% EtOAc-Hexanes, Rf 0.5). The reaction
mixture was diluted with saturated NaHC03 solution (50 mL) and acetone was distilled off under reduce pressure. The residue obtained was dissolved in water (200 mL) and ethyl acetate (500 mL). The organic layer was separated, washed with water, brine and dried over anhydrous Na2S04. The solution was concentrated under reduced pressure and the residue obtained was purified by silica gel (60 to 120 mesh) column chromatography (eluent, 5% EtOAc-hexanes) to give of 2-chloro-4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine (30 g) as a light brown liquid.
1H NMR (400 MHz, DMSO-d6) δ 8.39 (d, J= 5.2 Hz, 1H), 7.56 - 7.42 (m, 2H), 4.09 (q, J= 8.9 Hz, 2H), 1.47 (d, J= 24.5 Hz, 6H), 1.32 (s, 3H).
Intermediate AA3
2-(l-benzothiophen-7-yl)-4-(2,2-dimethyl-4-(trifluoromethyl)-l,3-dioxolan-4- yl)pyridine

Step 3
Step 1. A mixture of 4-bromo-2-chloropyridine (4.0 g, 20.79 mmol, Alfa Aesar, Ward Hill, MA), l-(trifluoromethyl)vinylboronic acid hexylene glycol ester (5.08 g, 22.86 mmol, Frontier Scientific, Inc. Logan, UT), sodium carbonate (6.61 g, 62.4 mmol), and bis(di-tert-butyl(4-
dimethylaminophenyl)phosphine)dichloropalladium(II) (0.368 g, 0.520 mmol) in 1 ,4-dioxane/water (18 ml/2 ml) under nitrogen atmosphere in microwave reaction vessel was irradiated in a microwave reactor at 130 °C for 45 min. The reaction mixture was partitioned between EtOAc and saturated aqueous NaHC03 solution. The organic layer was washed sequentially with aqueous NaHC03 solution and brine, dried (sodium sulfate), and filtered. The filtrate was concentrated and the residue was purified by flash chromatography (80 g silica gel, 5%
EtOAc/hexanes) to provide 2-chloro-4-(l-(trifluoromethyl)ethenyl)pyridine (2.49 g) as a clear colorless oil.
Step 2. To a stirred mixture of 2-chloro-4-(l- (trifluoromethyl)ethenyl)pyridine (2.8 g, 13.49 mmol, from Step 1) and 4- methylmorpholine 4-oxide (1.738 g, 14.84 mmol, Sigma Aldrich, St. Louis, MO) in acetone/H20 (1 :1 , 27 ml) was added osmium(VIII) oxide (4.0 wt% solution with H20, 4.29 g, 0.674 mmol, Sigma Aldrich, St. Louis, MO). The reaction mixture was stirred at ambient temperature for 3 h and was quenched with solid sodium sulfite (8.5 g, 67.4 mmol). The mixture was stirred for 10 min, filtered through a 0.45 μιη Teflon® filter, and the filtrate was concentrated under a vacuum to give 2-(2-chloro-4-pyridinyl)-3,3,3-trifluoro-l,2-propanediol (3.3 g) as a thick black tar. The alcohol was dissolved in acetone (34 mL) and was treated with 2,2-dimethoxypropane (28.5 g, 273 mmol, Fluka Chemie GmbH, Buchs, Switzerland) and 4-methylbenzene sulfonic acid, monohydrate (1.04 g, 5.46 mmol Sigma Aldrich, St. Louis, MO). After stirring at ambient temperature for 18 h, another portion of 4-methylbenzene sulfonic acid, monohydrate (0.52 g, 2.73 mmol, Sigma Aldrich, St. Louis, MO) was added. After stirring for additional 6 h, the reaction mixture was concentrated under a vacuum. The residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The organic layer was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and concentrated. The residue was purified by flash chromatography on silica gel (10%
EtOAc/hexanes) to provide 2-chloro-4-(2,2-dimethyl-4-(trifluoromethyl)-l,3- dioxolan-4-yl)pyridine (2.3 g) as a clear orange oil.
Step 3. A mixture of 2-chloro-4-(2,2-dimethyl-4-(trifluoromethyl)-l ,3- dioxolan-4-yl)pyridine (2.3 g, 8.17 mmol), 2-(l-benzothiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (2.55 g, 9.80 mmol, Intermediate Y2), sodium carbonate (2.60 g, 24.50 mmol), dicyclohexyl(2',4',6'-triisopropyl-[l, -biphenyl]- 2-yl)phosphine (0.389 g, 0.817 mmol, Strem Chemicals, Newbury Port, MA), and allylpalladium(II) chloride (0.149 g, 0.408 mmol, Sigma Aldrich, St. Louis, MO) in 1,4-dioxane (13 ml) and water (3.0 ml) was heated at 80 °C for 6 h. The reaction mixture was cooled to room temperature and was quenched with saturated aqueous sodium bicarbonate. The mixture was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate. The organic layer was washed sequentially with saturated aqueous sodium bicarbonate and brine (twice), dried (sodium sulfate), and concentrated. The residue was purified by flash chromatography on silica gel (19: 1 hexane-ethyl acetate) to provide 2-(l- benzothiophen-7-yl)-4-(2,2-dimethyl-4-(trifluoromethyl)-l,3-dioxolan-4- yl)pyridine (1.8 g) as a pale yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 8.95 - 8.86 (m, 1 H), 8.25 (s, 1 H), 8.16 (d, J = 7.4 Hz, 1 H), 8.07 - 7.99 (m, 1 H), 7.84 (d, J= 5.5 Hz, 1 H), 7.63 - 7.51 (m, 3 H), 4.91 (d, J= 10.3 Hz, 1 H), 4.47 (d, J= 10.3 Hz, 1 H), 1.57 (s, 3 H), 1.34 (s, 3 H).
Intermediate AA4
Di-tert-butyl (6-((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5-chloro-2- pyridinyl)imidodicarbonate

Step 3
Step 1. To a solution of methyl 6-amino-3-chloro-2-pyridinecarboxylate (50.0 g, 268 mmol, Combi-Blocks, san Diego, CA) in t-BuOH/acetone (500 ml/150 ml) was added 4-(dimethylamino)pyridine (0.466 g, 3.82 mmol), and di- tert-butyl dicarbonate (193 g, 884 mmol). The reaction was stirred at room temperature for 36 h. The mixture was concentrated and dissolved in EtOAc (1000 ml) and water (500 ml). The layers were separated and the organic layer was dried (MgS04), filtered and concentrated. The residue was passed through a silica gel plug rinsing with 10% EtOAc/hexanes to provide methyl 6-(bis(tert- butoxycarbonyl)amino)-3-chloro-2-pyridinecarboxylate (62.50 g) as a white solid.
Step 2. To a solution of methyl 6-(bis(tert-butoxycarbonyl)amino)-3- chloro-2-pyridinecarboxylate (62.50 g, 162 mmol, from Step 1) in MeOH (500 ml) at 0 °C was added sodium borohydride (30.6 g, 808 mmol) over 15 min. The reaction mixture was stirred at 0 °C for 1 h. MeOH was removed and the resulting oil was dissolved in EtOAc (500 ml) and washed with water (300 ml). The organic layer was dried (MgS04), and concentrated to provide a alcohol. The alcohol was dissolved in CH2CI2 (500 ml) and Dess-Martin periodinane (68.5 g, 162 mmol, Sigma Aldrich, St. Louis, MO) was added. After stirring at ambient temperature for 1 h, the mixture was cooled to 0 °C and quenched with saturated aq. Na2S203 solution (100 ml). The mixture was diluted with saturated NaHC03 solution and water. The organics were separated, dried (MgS04), and concentrated. The residue was purified by flash chromatography (1.5 kg silica gel
column, 10% EtOAc/hexanes) to provide a mixture of di-tert-butyl (5-chloro-6- formyl-2-pyridinyl)imidodicarbonate and tert-butyl (5-chloro-6-formyl-2- pyridinyl)carbamate (41.88 g) as a white solid.
Step 3. To a solution of di-tert-butyl (5-chloro-6-formyl-2- pyridinyl)imidodicarbonate and tert-butyl (5-chloro-6-formyl-2- pyridinyl)carbamate (41.88 g, 117 mmol) in CH2C12 (300 ml) was added (S)-2- methylpropane-2-sulfinamide (14.23 g, 117 mmol, AK Scientific, Mountain View, CA), and copper(II) sulfate (56.2 g, 352 mmol). After stirring at ambient temperature for 12 h, the mixture was filtered and the filtrate was concentrated. The residue was purified by flash chromatography (1.5 kg silica gel column, gradient elution from hexanes to 30% EtOAc/hexanes) to give a mixture of mono- and bis-Boc amines. This mixture was dissolved in in CH2C12 (250 mL) and di-tert-butyl dicarbonate (27.3 ml, 117 mmol) and DMAP (3.58 g, 29.3 mmol) were added. After stirring for 1 h, the mixture was diluted with water (500 ml). The organic layer was separated, dried (MgS04), and concentrated. The residue was purified by flash chromatography (1.5 kg silca gel column, gradient elution from hexanes to 40% EtOAc/hexanes) to provide di-tert-butyl (6- ((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5-chloro-2- pyridinyl)imidodicarbonate (34.5 g) as a tacky yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1 H), 8.19 (d, J= 8.6 Hz, 1 H), 7.68 (d, J= 8.6 Hz, 1 H), 1.41 (s, 18 H), 1.20 (s, 9 H).
Intermediate AA5
N-((R)-(2-Chlorophenyl)(7-(4-(2-methyl-2-oxiranyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a 20 mL vial, N-((R)-(2-chlorophenyl)(7-(4,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen-2-yl)methyl)cyclopropanesulfonamide (1.18 g, 2.34 mmol, Intermediate X14), 2-chloro-4-(2,2,4-trimethyl-l,3-dioxolan- 4-yl)pyridine (0.489 g, 2.15 mmol, Intermediate AA2), l,l-bis[(di-t-butyl-p- methylaminophenyl]palladium(II) chloride (AmPhos) (0.167 g, 0.236 mmol, Sigma- Aldrich, St. Louis, MO) and potassium carbonate (2 N, 3.50 mL, 7.01 mmol) were mixed into 1,4-dioxane (8 mL). The mixture was degassed by bubbling argon gas through the reaction mixture for 5 min. The vial was sealed and the mixture was stirred at 100 °C for 1 h. Water (100 mL) was added and the reaction mixture was extracted with EtOAc (2 x 50 mL). The combined organic phases were washed with saturated aqueous sodium chloride (100 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (100 g, EtOAc in hexanes 0 to 50%) followed by another silica gel column chromatography (50 g, acetone in heptanes 0 to 60 %) to afford N-((R)-(2-
chlorophenyl)(7-(4-(2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.374 g) as a white solid.
Step 2. To a 150-mL round-bottomed flask, N-((R)-(2-chlorophenyl)(7- (4-(2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.370 g, 0.650 mmol) and hydrogen chloride (4 N in 1,4-dioxane, 1.0 mL, 4.0 mmol, Sigma- Aldrich, St. Louis, MO) were dissolved into MeOH (6 mL). The mixture was stirred at rt for 18 h.
Additional hydrogen chloride (4 N in 1,4-dioxane, 0.5 mL, 2.0 mmol, Sigma- Aldrich, St. Louis, MO) was added and the mixture was stirred at rt for another 3.5 h. 1M K3PO4 (5 mL) was added followed by saturated aqueous sodium bicarbonate (40 mL). The aqueous phase was extracted with EtOAc (2 x 30 mL). The combined organic phases were washed with water (40 mL) and saturated aqueous sodium chloride (40 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (50 g, (3: 1 EtOAc/EtOH) in heptanes 10 to 60%) to afford N-((R)-(2-chlorophenyl)(7-(4-(l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.295 g) as a white solid.
Step 3. To a 150-mL round-bottomed flask, N-((R)-(2-chlorophenyl)(7- (4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.472 g, 0.891 mmol) and p- toluenesulfonyl chloride (0.255 g, 1.34 mmol, Sigma- Aldrich, St. Louis, MO) were added into pyridine (5 mL). The mixture was stirred at rt for 8 h then additional 4-methylbenzene-l-sulfonyl chloride (0.294 g, 1.54 mmol, Sigma- Aldrich, St. Louis, MO) was added and the mixture was stirred at rt for 18 h. Cesium carbonate (0.597 g, 1.83 mmol, Sigma-Aldrich, St. Louis, MO) was added and the mixture was stirred at rt for 1 h. The reaction mixture was concentrated under a vacuum and THF (10 mL) was added (making white suspension). The mixture was stirred at rt for 2 h. At this point, DMF (5 mL) was
added and the mixture was stirred at rt for 2 h. Water (50 mL) was added to the reaction mixture and the aqueous phase was extracted with EtOAc (2 x 50 mL). The combined organic phases were washed with water (100 mL) and saturated aqueous sodium chloride (100 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. This residue was taken into THF/2N NaOH (1 : 1, 5 mL each) and stirred at rt for 1 h. The reaction mixture was partitioned between saturated aqueous ammonium chloride (50 mL) and EtOAc (50 mL). The layers were separated and the aqueous phase was extracted with EtOAc (50 mL). The combined organic phases were washed with water (100 mL) and saturated aqueous sodium chloride (100 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (50 g, EtOAc in hexanes 10 to 60%) to afford N-((R)-(2-chlorophenyl)(7-(4-(2-methyl-2- oxiranyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.423 g) as a white foam.
1H NMR (400MHz, CDC13) δ= 8.77 (d, J= 5.1 Hz, 1 H), 7.91 (d, J= 8.0 Hz, 2 H), 7.75 (d, J= 7.8 Hz, 1 H), 7.65 (dd, J= 1.7, 7.5 Hz, 1 H), 7.52 - 7.28 (m, 5 H), 7.07 (s, 1 H), 6.41 (d, J= 7.8 Hz, 1 H), 5.41 (d, J= 7.8 Hz, 1 H), 3.08 (d, J= 5.3 Hz, 1 H), 2.85 (d, J= 5.3 Hz, 1 H), 2.28 (tt, J= 4.9, 8.0 Hz, 1 H), 1.81 (s, 3 H), 1.23 - 1.06 (m, 2 H), 0.93 - 0.71 (m, 2 H).
Intermediate AA6
2-methyl-N-((lE)-(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l- benzothiophen-2-yl)methylidene)-2-propanesulfinamide

Purification via silica gel chromatography (330 g of silica, 0 to 40% EA/Hex,) delivered 7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l- benzothiophene-2-carbaldehyde (5.76 g) as an oil that slowly solidified.
Step 2. A 250-mL round-bottomed flask was charged with 7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophene-2-carbaldehyde (5.67 g, 16.0 mmol), (R)-2-methylpropane-2-sulfinamide (2.24 g, 18.5 mmol), 50 mL CH2CI2, copper(II) sulfate (25.6 g, 160 mmol). The mixture was stirred at room temperature for 2.5 d and then another 5 equivalents of both sulfanamide and CuSC"4 were added and the mixture was heated at 55 °C. Heating was continued for an additional 3 d then the mixture was allowed to cool to room temperature and filtered. The filtrate was concentrated and purified via silica gel chromatography (330 g of silica, 0 to 40% EA/Hex,) to give 2-methyl-N-((lE)- (7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2- yl)methylidene)-2-propanesulfinamide (5.2 g) as a brittle foam.
Intermediate AA7
Tert-butyl (5-chloro-6-(((cyclopropylsulfonyl)amino)(7-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)-l-benzothiophen-2-yl)methyl)-2- pyridinyl)carbamate

Step 1. To a stirring solution of diisopropylamine (2.374 g, 23.46 mmol) in THF (12 mL) at -70 °C under nitrogen was added butyllithium (8.26 mL, 20.65 mmol, 2.5M solution in hexanes). After 15 minutes at -70 °C was added 7- bromo-l-benzothiophene (4.0 g, 18.77 mmol, Intermediate X13) in THF (10 mL). The cooling bath was removed and reaction warmed to -30 °C then returned to -70 °C. To the colorless solution was added di-tert-butyl (6-((E)- (((S)-tert-butylsulfinyl)imino)methyl)-5-chloro-2-pyridinyl)imidodicarbonate (8.63 g, 18.77 mmol, Intermediate AA4) in THF (15 mL) at a rate not to exceed - 60 °C. After 18 h of warming to 20 °C, the reaction was quenched with sat. NH4C1 (20 mL). The reaction was then partitioned between EtOAc (120 mL) and sat NH4C1 (50 mL). The organic was the dried over MgS04, concentrated onto dry silica (30 g) under reduced pressure, then purified by silica gel
chromatography (330 g) eluting products with 10 to 50% EtOAc in hexanes to afford tert-butyl (6-((7-bromo-l-benzothiophen-2-yl)(((S)-tert- butylsulfinyl)amino)methyl)-5-chloro-2-pyridinyl)carbamate (6.0 g) as a white solid.
Step 2. To a stirring suspension of tert-butyl (6-((7-bromo-l- benzothiophen-2-yl)(((S)-tert-butylsulfinyl)amino)methyl)-5-chloro-2- pyridinyl)carbamate (6.0 g, 10.47 mmol, from Step 1) in MeOH (100 mL) was added aqueous HC1 (21 mL, 5M solution in water) at rt. After 18 hrs the reaction was chilled to 0 °C, CH2C12 (300 mL) added, and the pH adjusted to 14 with 1 M NaOH (100 mL). The organic was then dried over MgSC^, filtered, then
concentrated under reduced pressure afford tert-butyl (6-(amino(7-bromo-l- benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (4.96 g) as an amber tar.
Step 3. To a stirring solution of tert-butyl (6-(amino(7-bromo-l- benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (4.9 g, 10.45 mmol, from Step 2) and DIEA (2.282 mL, 13.07 mmol) in CH2C12 (30 ml) at 25 °C was added cyclopropanesulfonyl chloride (5.32 mL, 52.3 mmol, Matrix Scientific, Columbia, SC). The reaction was stirred for 3 days at 25 °C then partitioned between CH2C12 (30 mL) and 5% NaHC03 (30 mL). The separated organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 15 to 30% EtOAc in hexanes to afford tert-butyl (6-((7-bromo-l-benzothiophen-2- yl)((cyclopropylsulfonyl)amino)methyl)-5-chloro-2-pyridinyl)carbamate (3.64 g) as white solid.
Step 4. A suspension of tert-butyl (6-((7-bromo-l-benzothiophen-2- yl)((cyclopropylsulfonyl)amino)methyl)-5-chloro-2-pyridinyl)carbamate (3.6 g, 6.28 mmol, from Step 3), bis(pinacolato) diboron (1.915 g, 7.54 mmol, Combi- Blocks, San Diego, CA), potassium acetate (1.233 g, 12.57 mmol), dichloro Ι ,Γ- bis(diphenylphosphino)ferrocene palladium (II) dichloromethane adduct (0.257 g, 0.314 mmol, Sigma Aldrich, St. Louis, MO) in 1,4-dioxane (30 mL) was sparged with argon for 1 minute then heated to 120 °C for 2 hrs in an
appropriately capped vessel. The reaction was then partitioned between EtOAc (10 mL) and 5% NaHC03 (5 mL). The separated organic was then dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 0 to 20% EtO Ac/Hex to afford tert-butyl (5-chloro-6-(((cyclopropylsulfonyl)amino)(7-(4,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (3.0 g) as a colorless tar.
1H NMR (400 MHz, CDC13) δ 7.97 - 7.88 (m, 1 H), 7.67 (d, J=8.80 Hz, 1 H), 7.76 (ddd, J=7.43, 6.16, 1.27 Hz, 2 H), 7.38 (s, 1 H), 7.11 (s, 1 H), 7.31 (dd, J=7.92, 7.14 Hz, 1 H), 6.38 - 6.32 (m, 1 H), 6.33 - 6.26 (m, 1 H), 2.17 - 2.07 (m, 1 H), 1.54 (s, 9 H), 1.39 (s, 12 H), 0.92 - 0.82 (m, 2 H), 0.80 - 0.66 (m, 2 H).
Intermediate AA8
2-(5-chloro-l-benzothiophen-7-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane

Step 1. In a 1 L round bottom flask, 2-bromo-4-chloroaniline (50. Og 245 mmol, Combi-blocks, USA) was treated with a solution of HCI (50 mL in 200 mL of water) at -10°C and treated with NaN02 solution (20.2 g in 100 mL of water 292.78 mmol, Sigma-Aldrich, India) and stirred at the same temperature for lh. The slightly orange diazonium solution was then added with a addition funnel to a stirred solution of potassium ethyl xanthate (66.34 g, in 120 ml. of water, 413.85 mmol, Sigma-Aldrich, India), at 80°C. Initial mild sputtering occurred. Heating was continued for 1 h, after completion of the addition, the red oil which separated was washed with aq.NaHC03 (3 X 150 mL) and then water (2 X 150 mL). The red oil was added in portions to a hot solution of potassium hydroxide (68.42 g, 1219.51 mmol, in 52 ml. of water and 280 ml. of ethanol. The reaction mixture was stirred at refluxing temperature for 21h. The mixture was cooled to 0°C, diluted with ice water (500 mL) and made acidic with hydrochloric acid (500 mL, pH about 2). The oil which was formed was extracted by diethyl ether (3 X 600 mL), and washed with water (2 X 300 mL), brine (2 X 300 mL) and dried over Na2S04, and the solution was concentrated under
reduced pressure, to afford 2-bromo-4-chlorobenzenethiol (46 g) as a brown liquid.
Step 2. In a 1 L round bottom flask, 2-bromo-4-chlorobenzenethiol (46 g, 207.20 mmol, from Step 1), and 2-bromo-l, 1-dimethoxyethane (34.80 g, 207.20 mmol, Sigma- Aldrich, India) were dissolved in DMF (300 mL) at ambient temperature, and was treated with K2CO3 (42.89 g, 310.81 mmol) at ambient temperature. The reaction mixture was gradually heated to 70°C and continued at the same temperature for further 3 h (TLC, 10% EtOAc-hexanes). The reaction mixture was cooled to ambient temperature, diluted with ice water (200 mL), diethyl ether (500 mL), separated the organic layer, and aq. layer was extracted by diethyl ether (2 X500 mL), The combined organic layer was washed with water (250 mL X 2), brine (150 mL X 2) and dried over Na2S04, and
concentrated under reduced pressure, the residue obtained was purified by silica gel (60 to 120 mesh) column chromatography (eluent 5% EtOAc-hexanes) to afford 2-bromo-4-chloro-l-((2,2-dimethoxyethyl)sulfanyl)benzene (30 g).
Step 3. In a 2 L round bottom flask 2-bromo-4-chloro-l-((2,2- dimethoxyethyl)sulfanyl)benzene (30 g, 96.77 mmol, from Step 2) and polyphosphoric acid (60 g, Sigma- Aldrich, India) were dissolved in
chlorobenzene (500 mL, Spectrochem, India) at rt under an nitrogen atmosphere. The resulting reaction mixture was heated to 131°C for 16 h. Upon completion of reaction (TLC 10% EtOAc-hexanes), the viscous material was decanted while hot and additional chlorobenzene (200 mL) was added to the flask and stirred at 120° C for 15 min., then decanted. The remaining viscous material in the flask was treated with toluene (200 mL), water (200 mL) and saturated aq.Na2C03 (150 mL). The resulting layers were separated and aq. phase was extracted by toluene (2 X 200 mL). The toluene and chlorobenzene layers were combined, washed with aq. Na2C03 (2 X 200 mL), brine (2 X 200 mL), and dried over anhydrous Na2S04. The solution was concentrated under reduced pressure and residue obtained was purified by silica gel (60 to 120mesh) column
chromatography (eluent 3% EtOAc-hexanes) to give 7-bromo-5-chloro-l- benzothiophene (18 g) as pale yellow solid.
Step 4. In a 1 L round bottom flask 7-bromo-5-chloro-l-benzothiophene (16 g, 65.06 mmol, from Step 3), bis(pinacolato)diborane (18.17 g, 71.54 mmol) and potassium acetate (31.23 g, 325.34 mmol), were dissolved in toluene (200 mL) at r.t under an argon atmosphere. The solution was degassed by purging with argon gas for 15 min at rt. Pd (dppf) Cl2 (26.54 g, 32.53 mmol, Sigma- Aldrich, India) was added to the above solution under argon atmosphere. The resulting reaction mixture was again degassed by purging with argon gas (15 min). The reaction mixture was gradually heated to 100°C and stirred at the same temperature for further 16 h under argon atmosphere. Upon completion of reaction (TLC 10% EtOAc-hexanes), the reaction mixture was diluted with water (150 mL) and ethyl acetate (200 mL). The organic layer was separated, washed with water (150 mL), brine (2 X 150 mL) and dried over anhydrous Na2S04. The solution was concentrated under reduced pressure and the resulting residue obtained was purified by silica gel (60 to 120 mesh) column chromatography (eluent 5% EtOAc-hexanes) to give 2-(5-chloro-l-benzothiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (17 g) as brown liquid.
1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J= 2.4 Hz, 1H), 7.88 (d, J
1H), 7.59 (d, J= 2.0 Hz, 1H), 7.45 (d, J= 5.6 Hz, 1H), 1.35 (s, 12H).
Intermediate AA9
2-(2-(2-(amino(2-chloro-6-fluorophenyl)methyl)-l-benzothiophi
pyridinyl)- 1 ,2-propanediol

Intermediate AA1
Step 1. To a solution of 2-(l-benzothiophen-7-yl)-4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)pyridine (2.18 g, 6.70 mmol, Intermediate AA1) (azeotropically dried with 4 mL of toluene) in THF (60 mL) at -78 °C was added butyllithium solution, 2.5M in hexanes (2.68 mL, 6.70 mmol). After 10 min, a solution of N- ((S, E)-(2-chloro-6-fluorophenyl)methylidene)-2-methyl-2-propanesulfinamide (1.461 g, 5.58 mmol, Intermediate AA12) in THF (12 mL) was added down the side of the reaction flask followed by 8 mL of rinse with THF. The reaction was stirred at -78 °C for 1 h. The reaction was quenched with water (50 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, dried (MgS04), concentrated and purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide N-(S)-((2-chloro-6-fiuorophenyl)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2- methyl-2-propanesulfinamide (2.83 g) as a colorless foam.
Step 2. To a solution of N-(S)-((2-chloro-6-fluorophenyl)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2- methyl-2-propanesulfinamide (3.32 g, 5.65 mmol, from Step 1) in MeOH (15 mL) was added hydrochloric acid (28.3 mL, 141 mmol, 5N solution in water). The reaction was stirred at room temperature for 16 h. The reaction was carefully quenched with saturated NaHC03 solution. The aqueous layer was extracted with
CH2CI2, and the combined organic layers were washed with brine, dried
(MgSC^), concentrated and purified by flash chromatography (40g silica gel column, gradient elution from CH2CI2 to 10 % MeOH (containing 2 M NH3) in CH2CI2) to provide 2-(2-(2-(amino(2-chloro-6-fluorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,2-propanediol (2.2 g) as a white foam.
1H NMR (300MHz ,CDC13) δ 8.77 (d, J= 5.7 Hz, 1 H), 8.03 (br. s, 1 H), 7.84 (d, J= 6.7 Hz, 1 H), 7.71 (dd, J= 0.8, 7.8 Hz, 1 H), 7.49 - 7.40 (m, 1 H), 7.31 (dd, J = 1.5, 5.2 Hz, 1 H), 7.26 - 7.22 (m, 2 H), 7.12 - 7.00 (m, 1 H), 6.98 (d, J= 1.3 Hz, 1 H), 6.00 (s, 1 H), 3.86 (d, J= 1.0 Hz, 1 H), 3.72 (d, J= 1.0 Hz, 1 H), 1.59 (s, 3 H).
Intermediate AA10
-(2-chlorophenyl)methylidene)cyclopropanesulfonamide

Dean-Stark
In a 1L 3 neck round bottom flask, 2-chlorobenzaldehyde (50 g, 357.14 mmol, Sigma- Aldrich, India), cyclopropanesulfonamide (47.4 g, 391.29 mmol, Matrix scientific, USA) and Montmorillonite K10 clay (20 g, Sigma- Aldrich, India ), were dissolved in toluene (300 mL) at ambient temperature. The reaction mixture was gradually heated to 110 °C with dean-stark and continued at the same temperature for further 12 h (TLC, 40% EtOAc-hexanes). The reaction mixture was cooled to rt, filtered through a diatomaceous earth pad and the residue was washed with toluene (100 mL). The combined filtrate was distilled off under reduced pressure. The residue obtained was re-crystallized with DCM- Hexane (1 : 1, 200mL) to give N-((lE)-(2- chlorophenyl)methylidene)cyclopropanesulfonamide (50 g) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.16 - 8.13 (dd, J=1.6, 8Hz, 1H), 7.77 - 7.68 (m, 2H), 7.56 (t, J= 8 Hz, 1H), 2.59 - 2.89 (m, 1H), 1.18 - 1.08 (m, 4H).
Intermediate AA11
2-(2-(2-(amino(6-amino-3-chloro-2-pyridinyl)methyl)-l-benzothiophi -pyridinyl)- 1 ,2-propanediol

Step 1. To a solution of 2-(l-benzothiophen-7-yl)-4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)pyridine (4.34 g, 13.34 mmol, Intermediate AA1) in THF (100 mL) at -78 °C was added butyllithium solution (5.33 mL, 13.34 mmol, 2.5M in hexanes). After 10 min, a solution of di-tert-butyl (6-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-5-chloro-2-pyridinyl)imidodicarbonate (5.11 g, 11.11 mmol, Intermediate AA4) in THF (20 mL) was added down the wall of the reaction flask followed by 20 mL of rinse with THF. The reaction was stirred at - 78 °C for 50 min then the reaction was quenched with water (50 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, concentrated and purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to 50% EtOAc in hexanes) to provide a mixture of di-tert-butyl (6-((((S)-tert-butylsulfinyl)amino)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5- chloro-2-pyridinyl)imidodicarbonate and tert-butyl (6-((((S)-tert- butylsulfinyl)amino)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-
benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (4.19 g) as a white foam.
Step 2. To a solution of mixture of di-tert-butyl (6-((((S)-tert- bu1ylsulfinyl)amino)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-5 -chloro-2-pyridinyl)imidodicarbonate and tert- butyl (6-((((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)- 2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (4.19 g, 5.70 mmol) in MeOH (20 mL) was added hydrochloric acid (20 mL, 100 mmol, 5N solution in water). The reaction was stirred at room temperature for 40 h, the reaction was then carefully quenched with saturated NaHC03 solution. The aqueous layer was extracted with CH2C12, and the combined organic layers were washed with brine, dried (MgS04), concentrated and purified by flash
chromatography (40g silca gel column, gradient elution from CH2C12 to 10 % MeOH (containing 2 M NH3) in CH2C12) to provide 2-(2-(2-(amino(6-amino-3- chloro-2-pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2-propanediol (1.63 g) as a white foam.
Intermediate AA12
(S)-N-((lE)-(2-chloro-6-fluorophenyl)methylidene)-2-methyl-2- propanesulflnami

Titanium tetraethoxide (110 g, 470 mmol, Sigma- Aldrich, India) was added to a stirring solution of 2-chloro-6-fluorobenzaldehyde (15 g, 95 mmol, Sigma- Aldrich, India), (S)-2-methyl-2-propanesulfinamide (14 g, 110 mmol, Combi-Blocks, San Diego, CA), and dichloromethane (150 mL) at room temperature. After stirring for 12 h, the reaction mixture was cooled to 0 °C,
quenched with ice water (100 mL), the mixture was filtered through a pad of diatomaceous earth, the layers of the filtrate were separated, the aqueous material was washed with dichloromethane (3x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (9: 1 hexane-ethyl acetate) to give (S)-N-((lE)-(2- chloro-6-fluorophenyl)methylidene)-2-methyl-2-propanesulfinamide (18 g) as a brown solid.
Example 57
N-((R)-(5-amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide



Step 1. 5-Amino-2-chlorobenzoic acid (15 g, 87 mmol, Sigma-Aldrich, St. Louis, MO) was taken up in 300 mL of EtOH and chilled to 0 °C. Thionyl chloride (14.03 mL, 192 mmol, Reidel-de Haen AG, Seelze, Germany) was added slowly and the mixture was warmed to rt and stirred for 1 h before heating to 80 °C for 2 h. The reaction was cooled to rt and poured into 1000 mL of aqueous NaHC03. The mixture was then extracted three times with 150 mL of DCM, and the organic extracts were dried over MgSC^. Filtration and concentration under reduced pressure afforded ethyl 5-amino-2-chlorobenzoate (14.3 g, 71.6 mmol) as a dark oil.
Step 2. Ethyl 5-amino-2-chlorobenzoate (14.3 g, 71.6 mmol, from Step 1) was taken up in 350 mL of THF and di-tert-butyl dicarbonate (39.1 g, 179 mmol), triethylamine (39.9 mL, 287 mmol), and DMAP (4.38 g, 35.8 mmol) were added. After 15 h, the mixture was diluted with 500 mL of EtOAc, washed with 300 mL of aqueous NH4C1, 300 mL of brine, and then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (330 g column, eluted with 5% to 30%
EtOAc/hexanes gradient) afforded ethyl 5-(bis(tert-butoxycarbonyl)amino)-2- chlorobenzoate (5.9 g, 14.75 mmol)) as a white solid. This material was taken up in 75 mL of ether and methanol (2.092 mL, 51.6 mmol) and lithium borohydride (25.8 mL, 51.6 mmol, 2.0M solution in THF) were added. After 3 h, the reaction was carefully quenched with 60 mL of aqueous NH4C1 and stirred for 30 min. The mixture was diluted with 60 mL of water and extracted twice with 100 mL of EtOAc, the combined organic extracts were washed with 100 mL of brine and dried over MgS04. Filtration and concentration under reduced pressure afforded tert-butyl (4-chloro-3-(hydroxymethyl)phenyl)carbamate (3.76g, 14.59 mmol) as a white solid.
Step 3. tert-Butyl (4-chloro-3-(hydroxymethyl)phenyl)carbamate (3.76 g, 14.59 mmol, from Step 2) was taken up in 100 mL of DCM and Dess-Martin periodinane (7.12 g, 16.78 mmol, Sigma-Aldrich, St. Louis, MO) was added. After 2 h the reaction was quenched with 50 mL of aqueous Na2S203 and 50 mL of aqueous NaHCOs, then stirred for 30 min. The mixture was partitioned, and the aqueous portion was extracted twice with 75 mL of DCM. The combined organic extracts were then dried over MgS04. Filtration and concentration under reduced pressure afforded tert-butyl (4-chloro-3-formylphenyl)carbamate (3.7 g, 14.47 mmol) as a yellow solid. This material was taken up in 75 mL of 5:1 lBuOH:acetone and di-tert-butyl dicarbonate (4.74 g, 21.71 mmol) and DMAP (0.354 g, 2.89 mmol) were added. The reaction was heated to 50 °C and after 15 h an additional 4 g of di-tert-butyl dicarbonate was added. After a further 2 h another 4 g of di-tert-butyl dicarbonate was added, and after stirring for an
additional 2 h the reaction was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (120 g column, eluted with 5% to 20% EtOAc/hexanes) to afford di-tert-butyl (4-chloro-3- formylphenyl)imidodicarbonate (4.64 g, 13.04 mmol) as a yellow oil.
Step 4. Di-tert-butyl (4-chloro-3-formylphenyl)imidodicarbonate (4.64 g, 13.04 mmol, from Step 3) was taken up in 15 mL of DCM and anhydrous copper(II) sulfate (6.24 g, 39.1 mmol, Fluka, St. Louis, MO) and (S)-2- methylpropane-2-sulfinamide (2.371 g, 19.56 mmol, AK Scientific, Inc. Union City, CA) were added. The mixture was heated to 30 °C for 5 days, and after this time the mixture was filtered through a plug of diatomaceous earth and the plug was washed with 150 mL of DCM. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (80 g column, eluted with 2.5% to 30% EtOAc/hexanes gradient) to afford di- tert-butyl (3 -((Z)-(((S)-tert-butylsulfmyl)imino)methy l)-4- chlorophenyl)imidodicarbonate (2.371 g, 19.56 mmol) as a sticky white solid.
Step 5. To a solution of dry 2-(l-benzothiophen-7-yl)-4-(2,4,4-trimethyl- l,3-dioxolan-2-yl)pyridine (2.340 g, 7.19 mmol, Intermediate AA1) in THF (40 mL) at -78 °C was added butyllithium solution (4.29 mL, 6.86 mmol, 1.6M in hexanes). The reaction was stirred at this temp for 20 min after which time a solution of di-tert-butyl (3-((Z)-(((S)-tert-butylsulfinyl)imino)methyl)-4- chlorophenyl)imidodicarbonate (2.340 g, 7.19 mmol, from Step 4) in THF (15 mL) was added, the reaction was stirred at the low temp for 15 min before warming to rt for lh. The reaction was then diluted with aqueous NH4C1 and EtOAc, and the organic layer was separated, dried over Na2S04, filtered and concentrated. Purified by flash chromatography on silica gel (330 g column, eluted with 0% to 60% EtOAc/hexanes) to afford the desired product (0.47g, 0.6 mmol) as a mixture of mono- and di-Boc, and a 3: 1 mix of diastereomers at the newly formed center, in favor of the (R) stereochemistry.
Step 6. A solution of di-tert-butyl (3-((R)-(((S)-tert- bu1ylsulfinyl)amino)(7-(4-(2,2,4 rimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-4-chlorophenyl)imidodicarbonate (2.6 g, 3.31 mmol, from Step 5) in MeOH (30 mL) and 4N hydrogen chloride (0.829 mL, 3.31 mmol) in dioxane was stirred at rt for lh. The reaction showed only 50% removal of the auxiliary so another equivalent of HC1 was added and after a further 20 min the reaction was complete. Then the mixture was diluted with aqueous NaHC03 and EtOAc, and the organic layer was separated, dried over Na2S04, filtered and concentrated. This material (2.25 g, 3.31 mmol) was used directly in the next reaction.
Step 7. To a solution of di-tert-butyl (3-((R)-amino(7-(4-(2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-4- chlorophenyl)imidodicarbonate (2.25 g, 3.31 mmol, from Step 6) and DMAP (0.404 g, 3.31 mmol) in Hunig's base (2.89 mL, 16.54 mmol) was added a solution of cyclopropanesulfonyl chloride (0.558 g, 3.97 mmol, Matrix Scientific, Albion, NY) in DMF (4 mL). The reaction was stirred overnight and then diluted with aqueous NaHC03 and EtOAc. The organic layer was separated, dried over Na2S04, filtered and concentrated and the resultant residue used directly in the next step. This material (2.6 g, 3.31 mmol) was taken up in THF (10 mL) and 4N HC1 (15 mL, 60.0 mmol) in dioxane, and was stirred at rt for 6 h. A solid precipitate began to form so 5 mL of MeOH was added and the reaction was heated to 70 °C for 3 h. The reaction was neutralized by the addition of 10 mL of triethylamine and diluted with EtOAc and water. The organic layer was separated, dried over Na2S04, filtered, concentrated onto silica and purified by flash chromatography on silica gel (eluted with 0% to 10% MeOH/DCM) to afford N-((5-amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (1.3g, 2.39 mmol) as a mixture of 4 isomers. The isolated material was subjected to preparative SFC (Chiralpak® AD-H column)(15 cm x 4.6 mm, 5 μιη) eluting with
50% liquid C02 in 50% methanol (with 20 mM NH3) at a flow rate of 4 mL/min to afford the two major products in greater than 99% enantiomeric excess.

N-((R)-(5-amino-2-chlorophenyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide; N-((R)-(5- amino-2-chlorophenyl)(7-(4-(( 1 S)-l ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide.
First Eluting Peak (Peak 1):
1H NMR (400MHz, CD3OD) δ = 8.66 (d, J= 5.3 Hz, 1 H), 8.18 - 8.11 (m, 1 H), 7.86 (d, J= 7.0 Hz, 1 H), 7.76 (d, J= 7.4 Hz, 1 H), 7.52 - 7.42 (m, 2 H), 7.15 (d, J= 8.4 Hz, 1 H), 7.09 - 6.97 (m, 2 H), 6.69 (dd, J= 2.7, 8.4 Hz, 1 H), 6.33 - 6.27 (m, 1 H), 3.77 - 3.59 (m, 2 H), 2.40 - 2.26 (m, 1 H), 1.57 (s, 3 H), 1.08 - 0.94 (m, 2 H), 0.92 - 0.82 (m, 1 H), 0.81 - 0.69 (m, 1 H). m/z (ESI, pos. ion) 544.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.010 μΜ.
Second Eluting Peak (Peak 2):
1H NMR (400MHz, CD3OD) δ = 8.66 (d, J = 5.1 Hz, 1 H), 8.15 - 8.10 (m, 1 H), 7.86 (d, J= 7.0 Hz, 1 H), 7.76 (d, J= 7.4 Hz, 1 H), 7.51 - 7.42 (m, 2 H), 7.15 (d, J= 8.4 Hz, 1 H), 7.07 - 7.00 (m, 2 H), 6.69 (dd, J= 2.7, 8.6 Hz, 1 H), 6.34 - 6.20 (m, 1 H), 3.76 - 3.62 (m, 2 H), 2.41 - 2.27 (m, 1 H), 1.57 (s, 3 H), 1.08 - 0.95 (m, 2 H), 0.91 - 0.82 (m, 1 H), 0.81 - 0.69 (m, 1 H). m/z (ESI, pos. ion) 544.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.039 μΜ.
Example 58
N-((5-Amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide

A solution of di-tert-butyl (3-((((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- chlorophenyl)imidodicarbonate (330 mg, 0.421 mmol, from Step 5 in above Example 57), tfa (0.156 mL, 2.103 mmol) and hydrogen chloride (2.103 mL, 8.41 mmol, 4N in dioxane) in THF (10 mL) was stirred at rt overnight. The reaction was not complete so was heated to 70 °C for 2h. The reaction was concentrated and slurried with EtOAc. The resultant yellow precipitate was collected and dried on the high vacuum for lh, then used directly in the next reaction. This HCI salt was taken up in DMF (4 mL) and lH-pyrazole-4-sulfonyl chloride (60.6 mg, 0.364 mmol, Matrix Scientific, Columbia, SC), N-ethyl-N- isopropylpropan-2-amine (0.318 mL, 1.820 mmol), and N,N-dimethylpyridin-4- amine (22.24 mg, 0.182 mmol) added. After 5 days complete bis-sulfonylation was observed so the reaction was diluted with EtOAc and aqueous NaHC03 and the organic layer separated, dried over Na2S04, filtered and concentrated. This residue was taken up in MeOH (4.00 mL) and potassium carbonate (252 mg, 1.820 mmol) was added. After 2h the reaction was diluted with water and extracted with EtOAc, the organic layer was separated, dried over Na2S04, filtered and concentrated. Purification by reverse-phase preparative HPLC using a Phemomenex Gemini-NX C18 110A column (Phenomenex, Torrance, CA) (150 x 30 mm), eluting with 0.1% TFA in CH3CN/H20 (20% to 60% over 15 min) to provide N-((5-amino-2-chlorophenyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide as a mixture of 4 isomers.

N-((R)-(5-amino-2-chlorophenyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide; N-((R)-(5- amino-2-chlorophenyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide; N-((S)-(5-amino-2- chlorophenyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methyl ethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide; N-((S)-(5-amino-2- chlorophenyl)(7-(4-(( 1 S)-l ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide.
1H NMR (400MHz, DMSO-d6) δ = 8.82 (d, J= 8.0 Hz, 1 H), 8.70 (d, J= 5.1 Hz, 1 H), 8.20 - 8.14 (m, 1 H), 8.01 (d, J= 7.6 Hz, 1 H), 7.80 (d, J= 7.2 Hz, 1 H), 7.74 - 7.56 (m, 1 H), 7.51 - 7.40 (m, 2 H), 7.00 (d, J= 8.6 Hz, 1 H), 6.96 - 6.89 (m, 1 H), 6.84 - 6.75 (m, 1 H), 6.56 - 6.45 (m, 1 H), 6.04 (d, J= 7.8 Hz, 1 H),
5.32 - 5.20 (m, 3 H), 4.86 - 4.77 (m, 1 H), 3.58 - 3.46 (m, 2 H), 1.46 (s, 3 H). m/z (ESI, pos. ion) 570.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.071 μΜ.
Example 59
N-((R)-(2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)thieno[3,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide

Step 1. A solution of 7-chlorothieno[3,2-b]pyridine (4.5 g, 26.5 mmol,
Accela ChemBio Inc., San Diego, CA), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi- 1,3,2-dioxaborolane (13.47 g, 53.1 mmol), l, -bis(diphenylphosphino)ferrocene palladium(II)dichloride dichloromethane adduct (0.650 g, 0.796 mmol), Ι,Γ- bis(diphenylphosphino)ferrocene (0.441 g, 0.796 mmol) and potassium acetate (7.81 g, 80 mmol) in 1,4-dioxane (30 mL) was heated in a sealed tube in the microwave at 140 °C for lh. The reaction was filtered through a pad of
diatomaceous earth and the filtrate was concentrated. This was loaded onto a plug of silica with DCM and flushed with hexanes. This removed the borane, and the desired material was then isolated after a flush with EtOAc and concentration of the filtrate. The material was used directly in the next reaction.
Step 2. A degassed solution of 7-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)thieno[3,2-b]pyridine (4.4 g, 16.85 mmol, from Step 1), 2-chloro-4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridine (3.6 g, 15.81 mmol, Intermediate AA2), tetrakis(triphenylphosphine)palladium(0) (0.914 g, 0.791 mmol) and sodium carbonate (5.03 g, 47.4 mmol) in 1,4-dioxane (100 mL)/water (10 mL) was heated to 80 °C overnight. The reaction was filtered and concentrated and purified by flash chromatography on silica gel (eluted with 0% to 40%
EtOAc/DCM) to afford the desired product (3.3g, 10.11 mmol).
Step 3. To a solution of 7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2- pyridinyl)thieno[3,2-b]pyridine (500 mg, 1.532 mmol, from Step 2) in THF (5 mL) at -78 °C was added butyllithium (0.613 mL, 1.532 mmol, 2.5M in hexanes). The reaction turned red and was stirred at this temp for 10 min before the addition of N-((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2- propanesulfmamide (411 mg, 1.685 mmol, Intermediate Yl) as a solution in THF (2 mL). After 15 min the reaction was diluted with aqueous NH4CI and EtOAc and the organic layer separated, dried over Na2SC"4, filtered and concentrated. The reaction mixture was purified by flash chromatography on silica gel (eluted with 0% to 100% EtOAc/hexanes) to afford the desired product (0.32g, 0.56 mmol) as an 8: 1 (5: 1 after reaction) mixture of isomers at the newly formed center.
Step 4. A solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-(2,2,4-trimethyl- l,3-dioxolan-4-yl)-2-pyridinyl)thieno[3,2-b]pyridin-2-yl)methyl)-2-methyl-2- propanesulfmamide (320 mg, 0.561 mmol, from Step 3) in MeOH (5 mL)/HCl (1.40 mL, 5.61 mmol, 4N in dioxane) was stirred overnight at rt. The reaction
was quenched with triethylamine and concentrated onto silica. The reaction mixture was purified by flash chromatography on silica gel (eluted with 0% to 10% (2M NH3 in MeOH)/DCM) to afford the desired product (0.24g, 0.56 mmol).
Step 5. A solution of 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)thieno [3 ,2-b]pyridin-7-yl)-4-pyridinyl)- 1 ,2-propanediol (240 mg, 0.563 mmol, from Step 4), cyclopropanesulfonyl chloride (158 mg, 1.127 mmol, Matrix Scientific, Columbia, SC), tea (0.393 mL, 2.82 mmol) and DMAP (68.8 mg, 0.563 mmol) in DMF (2 mL) was stirred at rt overnight. The reaction was quenched with aqueous NaHC03 and diluted with EtOAc, the organic layer was separated, dried over Na2S04, filtered and concentrated. The reaction was purified by flash chromatography on silica gel (eluted with 0% to 10% MeOH/DCM) to afford the desired product (0.060g, 0.11 mmol) as a mixture of four isomers, with a ratio of 8: 1 at the biaryl stereocenter, in favor of the (R) stereochemistry.

1H NMR (400MHz, CD3OD) δ = 8.78 (d, J= 5.1 Hz, 1 H), 8.69 (d, J = 5.1 Hz, 1 H), 8.38 - 8.32 (m, 1 H), 7.98 (d, J= 5.1 Hz, 1 H), 7.77 (dd, J= 1.8, 7.6 Hz, 1 H), 7.60 (dd, J= 1.6, 5.1 Hz, 1 H), 7.53 (dd, J= 1.5, 7.7 Hz, 1 H), 7.50 - 7.37 (m, 2 H), 7.17 - 7.08 (m, 1 H), 6.56 - 6.46 (m, 1 H), 3.80 - 3.64 (m, 2 H), 2.40 - 2.28 (m, 1 H), 1.58 (s, 3H), 1.10 - 0.96 (m, 2 H), 0.94 - 0.81 (m, 1 H), 0.80 - 0.68 (m, 1 H). m/z (ESI, pos. ion) 529.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.047 μΜ.
Example 60
N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)thieno[3,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide

Step 1. Performed as Step 1 in above Example 59.
Step 2. A degassed solution of 7-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)thieno[3,2-b]pyridine (5.5 g, 21.06 mmol, from Step 1), 2-(2-chloropyridin- 4-yl)propan-2-ol (3.61 g, 21.06 mmol, Intermediate Y4),
tetrakis(triphenylphosphine)palladium(0) (1.217 g, 1.053 mmol) and sodium carbonate (6.70 g, 63.2 mmol) in 1,4-dioxane (100 mL)/water (10.00 mL) was heated at 80 °C overnight. The reaction was filtered, concentrated and purified by flash chromatography on silica gel (e luted with 0% to 40% EtOAc/DCM
followed by 4-10% MeOH/DCM) to afford the desired product (1.4 g, 5.18 mmol).
Step 3. To a solution of dry 2-(2-(thieno[3,2-b]pyridin-7-yl)pyridin-4- yl)propan-2-ol (1.2 g, 4.44 mmol, from Step 2) in THF (25 mL) at -78 °C was added butyllithium (3.55 mL, 8.88 mmol). After 10 min a solution of N-((S,E)- (2-chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (1.082 g, 4.44 mmol, Intermediate Yl) in THF (10 mL) was added and after 15 min the reaction was quenched with aqueous NH4C1 and EtOAc and the organic layer separated, dried over Na2S04, filtered and concentrated. Purified by flash chromatography on silica gel (eluted with 0% to 100% EtOAc/hexanes) to afford the desired product (0.60 g, 1.17 mmol) which is now a 95:5 mix of stereoisomers at the newly formed center (5: 1 after reaction) in favor of the (R) stereochemistry.
Step 4. A solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-(l -hydroxy- 1- methylethyl)-2-pyridinyl)thieno[3,2-b]pyridin-2-yl)methyl)-2-methyl-2- propanesulfinamide (600 mg, 1.167 mmol, from Step 3) in MeOH (10 mL)/4N HCl (2.92 mL, 11.67 mmol) in dioxane was stirred at rt for 2h. The reaction was quenched with triethylamine and diluted with MeOH and concentrated onto silica. Purified by flash chromatography on silica gel (eluted with 0% to 10% (2M NH3 in MeOH)/DCM) to afford the desired product (0.48 g, 1.17 mmol) which was used directly in the next reaction.
Step 5. A solution of 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)thieno[3,2-b]pyridin-7-yl)-4-pyridinyl)-2-propanol (480 mg, 1.171 mmol), triethylamine (0.812 mL, 5.85 mmol), cyclopropanesulfonyl chloride (329 mg, 2.342 mmol, Matrix Scientific, Albion, NY) and N,N- dimethylpyridin-4-amine (143 mg, 1.171 mmol) in DMF (3 mL) was stirred at rt overnight. The reaction was quenched with aqueous NaHC03 and EtOAc and the organic layer separated, dried over Na2S04, filtered, concentrated onto silica and purified by flash chromatography on silica gel (eluted with 0%> to 10%>
MeOH/DCM) to afford the desired product (0.30 g, 0.58 mmol) as a mixture of two isomers, with a ratio of 95:5 in favor of the (R) stereochemistry.

N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno [3 ,2- b]pyridin-2-yl)methyl)cyclopropanesulfonamide; N-((S)-(2-chlorophenyl)(7-(4- ( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno [3 ,2-b]pyridin-2- yl)methyl)cyclopropanesulfonamide.
1H NMR (400MHz, CD3OD) δ = 8.76 (d, J= 5.1 Hz, 1 H), 8.69 (d, J = 5.1 Hz, 1 H), 8.37 - 8.29 (m, 1 H), 7.97 (d, J= 5.1 Hz, 1 H), 7.80 - 7.72 (m, 1 H), 7.61 - 7.56 (m, 1 H), 7.55 - 7.50 (m, 1 H), 7.49 - 7.37 (m, 2 H), 7.17 - 7.09 (m, 1 H), 6.55 - 6.46 (m, 1 H), 2.37 - 2.26 (m, 1 H), 1.61 (s, 6 H), 1.10 - 0.95 (m, 2 H), 0.92 - 0.81 (m, 1 H), 0.80 - 0.68 (m, 1 H). m z (ESI, pos. ion) 514.0 (M+H)+. GK- GKRP IC50 (Binding) = 0.136 μΜ.
Example 61
N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)tetrahydro-3-thiophenesulfonamide 1,1-dioxide hydrochloride


Step 1. To a 100 mL flame-dry 3-neck round bottomed flask was added 2- (2-(benzo[b]thiophen-7-yl)pyridin-4-yl)propan-2-ol (1.5 g, 5.57 mmol,
Intermediate Y5) and THF (15.0 mL). The reaction mixture was cooled to -78 °C followed by addition of butyllithium (5.57 mL, 13.92 mmol, 2.5 M solution in hexanes) dropwise via an addition funnel. The resulting mixture was stirred for 10 min before addition of N-((S,E)-(2-chlorophenyl)methylidene)-2-methyl-2- propanesulfmamide (2.1 g, 8.62 mmol, Intermediate Yl) in THF (10 mL) dropwise via the addition funnel. After 40 min the reaction was quenched with saturated NH4C1 (5 mL) and the resulting mixture was partitioned between EtOAc (70 mL) and water (40 mL). The aqueous layer was extracted with EtOAc (1 x 40 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. Purified twice by flash chromatography on silica gel (120 g column, eluted with 0% to 40% acetone/hexanes) to afford the desired product (2.00 g, 3.90 mmol) as a single diastereomer.
1H NMR (300MHz, CDC13) δ = 8.73 (dd, J= 0.5, 5.2 Hz, 1 H), 8.05 (d, J= 0.9 Hz, 1 H), 7.87 (dd, J= 1.0, 7.5 Hz, 1 H), 7.76 (dd, J= 0.9, 7.7 Hz, 1 H), 7.67 (dd, J= 1.8, 7.6 Hz, 1 H), 7.46 (t, J= 7.7 Hz, 1 H), 7.42 - 7.32 (m, 3 H), 7.32 - 7.28 (m, 1 H), 7.26 - 7.23 (m, 1 H), 6.38 (d, J= 5.3 Hz, 1 H), 4.13 (d, J= 5.3 Hz, 1 H), 1.86 (s, 1 H), 1.64 (s, 6 H), 1.32 (s, 9 H).
Step 2. To a solution of (S)-N-((R)-(2-chlorophenyl)(7-(4-(l -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (1.9 g, 3.70 mmol) in THF (5 mL) was added hydrogen chloride (9.26 mL, 37.0 mmol, 4.0M solution in 1,4-dioxane). A solid precipitate formed immediately and the reaction was diluted with EtOAc (20 mL) and stirred for lh and a free flowing solid was observed. The reaction mixture was filtered and the solid collected and dried to give the desired product as the HCl salt (1.7 g, 3.53 mmol) that was used directly in the next reaction.
Step 3. A solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol dihydrochloride (100 mg, 0.208 mmol), tetrahydrothiophene-3-sulfonyl chloride 1,1 -dioxide (91 mg, 0.415 mmol, Enamine Building Blocks, Kiev, Ukraine), N,N-dimethylpyridin-4-amine (25.4 mg, 0.208 mmol) and N-ethyl-N-isopropylpropan-2-amine (0.184 mL, 1.038 mmol) in DMF (2 mL) was stirred at rt overnight. The reaction was quenched with aqueous NaHC03 and diluted with EtOAc. The organic layer was separated, dried over Na2S04, filtered, concentrated and purified by flash chromatography on silica gel (eluted with 0% to 10% MeOH/DCM) followed by a second purification of flash chromatography on silica gel (eluted with 0% to 100% EtOAc/hexanes). This material was then taken up in EtOAc and 4N HCl in dioxane added to form the HCl salt. The precipitate that formed was collected and gave the desired material (10 mg, 0.016 mmol) as a mixture of two diastereomers.

(3R)-N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)tetrahydro-3 -thiophenesulfonamide 1 , 1 -dioxide hydrochloride; (3 S)-N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)tetrahydro-3 -thiophenesulfonamide 1,1- dioxide hydrochloride.
1H NMR (400MHz, CD3OD) δ = 8.75 (t, J= 5.3 Hz, 1 H), 8.38 - 8.29 (m, 1 H), 7.86 (dd, J= 7.7, 15.4 Hz, 2 H), 7.77 (d, J= 5.5 Hz, 1 H), 7.73 - 7.66 (m, 1 H), 7.59 - 7.38 (m, 5 H), 7.12 - 7.05 (m, 1 H), 6.50 - 6.45 (m, 1 H), 4.15 - 3.88 (m, 1 H), 3.50 - 3.34 (m, 1 H), 3.29 - 3.24 (m, 1 H), 3.18 - 3.08 (m, 1 H), 2.60 - 2.41 (m, 2 H), 1.63 (s, 6 H). m/z (ESI, pos. ion) 590.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.286 μΜ.
Examples 62 and 63
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((R)-(6-amino-3-chloro-2- pyridinyl)(7-(4-(lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide


Step 1 : To a solution of 3,6-dichloropicolinic acid (30 g, 1.6 mol, Sigma Aldrich, India) in THF (300 mL) at -60°C, was added DIBAL-H (44 g, 310 mmol, Sigma Aldrich, India) and the reaction mixture was stirred at same temperature for 3 h. After completion of reaction, the reaction mixture was quenched by saturated NH4C1 (100 mL), diluted by water (150 mL), EtOAc (300 mL), separated the organic layer, aqueous layer was extracted by EtOAc (150 mL x 2). The combined organic layer was dried on anhydrous sodium sulfate and
concentrated under reduced pressure afforded 15 g of 3,6-dichloro-2- pyridinecarbaldehyde as yellow solid.
Step 2: To a solution of 3,6-dichloro-2-pyridinecarbaldehyde (40.0 g, 227 mmol) in DCM (400 mL), was added (S)-2-methylpropane-2-sulfinamide (3) (33.0 g, 273 mmol, Sigma Aldrich, India ), Ti(OEt)4 (259 g, 1140 mmol, Sigma Aldrich, India) and stirred the reaction mixture at ambient temperature for 18 h. The reaction mixture was cooled to 0 0 C quenched with water (600 mL), filtered through a diatomaceous earth pad, and washed with DCM (200 mL x 2). The mixture was partitioned and the organic extracts were washed with saturated NaCl solution (150 mL x 2) and dried over Na2S04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (0 to 10% ethyl acetate in petroleum ether gradient) afforded 49 g of (S)-N-((lE)-(3,6- dichloro-2-pyridinyl)methylidene)-2-methyl-2-propanesulfinamide as yellow solid.
Step 3 : Lithium diisopropylamide (3.8g, 36 mmol, India) was added slowly to a solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro- 2-propanol (5.8 g, 18 mmol, intermediate Y3) in dry THF (150 mL) at -78 °C under argon atmosphere. After stirring for 30 min, a solution of (S)-N-((1E)- (3,6-dichloro-2-pyridinyl)methylidene)-2-methyl-2-propanesulfinamide (5.0 g, 18 mmol) in 100 mL of THF was added. The mixture was stirred for 2 h at -78 °C under a argon atmosphere. The reaction was quenched with 100 mL of aqueous Na2C03 and the mixture was allowed to warm to room temperature. The mixture was diluted with 15 mL of water (150 mL) and, extracted twice with 150 mL of EtOAc. The organic extracts were dried over Na2S04. Filtration and concentration under reduced pressure followed by flash chromatography on silica gel (0 to 50% EtOAc/hexanes gradient) afforded 4g of (S)-N-((3,6-dichloro-2- pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide as a yellow solid.
Step 4: In a 100 mL sealed tube, THF (30 mL) was degassed with argon gas for 15 min(S)-N-((3,6-dichloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro-l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (3.0 g, 5.0 mmol), K3P04 (1.3 g, 6.0 mmol, Sigma Aldrich, India), BocNH2 (0.70 g, 6.0 mmol, Sigma Aldrich, India),
tris(dibenzylideneacetone)dipalladium(0) (1.14 g, 1.2 mmol, Sigma Aldrich, India), 2-dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl (1.2g, 2.5 mmol, Sigma Aldrich, India) was added and degassed with argon for 15 min and sealed the tube and heated at 100 °C for 18 h. The reaction mixture was cooled to room temperature, diluted with 30 mL of water and 100 mL of EtOAc, and filtered through a Diatomaceous earthpad. The mixture was partitioned and the organic layer was washed twice with 25 mL of brine and dried over Na2S04. Filtration and concentrated under reduced pressure, followed by flash chromatography on silica gel (0 to 40% ethyl acetate in petroleum ether gradient) afforded 0.95 g of tert-butyl (6-((((S)-tert-butylsulfinyl)amino)(7-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-2- pyridinyl)carbamate as brown solid.
Step 5: Tert-butyl (6-((((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro- 2-pyridinyl)carbamate (3.75 g) was subjected to preparative SFC (Chiralpak® ASH column)(250 mm x 30 mm) eluting with 85% liquid C02 in 15% methanol (with 20 mM NH3) at a flow rate of 120 mL/min), affording 3.25 g of tert-butyl (6-((R)-(((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-2- pyridinyl)carbamate as the major eluting peak.
Step 6: Tert-butyl (6-((R)-(((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 5-chloro-2-pyridinyl)carbamate (3.15 g, 4.61 mmol) was taken up in 40 mL of MeOH. Hydrogen chloride, 4.0 M solution in 1,4-dioxane (9.2 mL, 37 mmol,
Sigma Aldrich, St. Louis, MO) was added to the mixture. After 3 h, an additional hydrogen chloride, 4.0 M solution in 1,4-dioxane (9.2 mL, 37 mmol, Sigma Aldrich, St. Louis, MO) was added to the mixture. The mixture was stirred for 18 h. The solvent was removed under reduced pressure and the residue was partitioned between 100 mL of 9: 1 chloroformTPA and 100 mL of aqueous NaHC03. The aqueous portion was extracted twice with 100 mL of 9: 1 chloroformTPA and the combined organic extracts were dried over MgS04. Filtration and concentration under reduced pressure afforded 2.2 g of 2-(2-(2- ((R)-amino(6-amino-3-chloro-2-pyridinyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol as a brown solid. The product was carried on without further purification. 2-(2-(2-((R)-amino(6-amino-3-chloro-2- pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol (2.2 g, 4.6 mmol), N,N-diisopropylethylamine (4.0 mL, 23 mmol, Sigma Aldrich, St. Louis, MO), N,N-dimethylaminopyridine (0.056 g, 0.46 mmol, Sigma Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (0.47 mL, 4.6 mmol, Matrix Scientific, Columbia, SC) were taken up in 20 mL of DMF and stirred. After 24 h, the reaction was quenched with 20 mL of aqueous NH4C1 and dilute with 50 mL of water. The mixture was extracted with 100 mL of EtOAc and the organic extracts were washed twice with 100 mL of water and once with 100 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (40 g column, eluted with 25% to 75% EtOAc/hexanes gradient) afforded 1.9 g of N-((R)-(6-amino-3- chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide as a tan solid.
1H NMR (400 MHz, CHLOROFORM-d) δ 8.84 (d, J= 5.28 Hz, 1H), 8.13 (s, 1H), 7.86 (d, J= 7.43 Hz, 1H), 7.78 (d, J= 7.83 Hz, 1H), 7.38 - 7.49 (m, 3H), 7.30 (s, 1H), 6.48 (d, J= 9.00 Hz, 1H), 6.42 (d, J= 8.61 Hz, 1H), 6.32 (d, J = 8.80 Hz, 1H), 4.58 (s, 2H), 2.65 (s, 1H), 2.10 - 2.21 (m, 1H), 1.84 (s, 3H), 1.14 (dd, J= 5.38, 7.53 Hz, 2H), 0.72 (d, J= 8.22 Hz, 2H). m/z (ESI, pos. ion) 583.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.008 μΜ.
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (1.9g) was subjected to preparative
SFC(Chiralpak® IA column)(250 mm x 30 mm, 5 μΜ) eluting with 50% liquid C02 in 50% 2-propanol (with 20 mM NH4OH) at a flow rate of 100 mL/min, affording two compounds.
First eluting peak: 0.69 g N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide
1H NMR (400 MHz, CHLOROFORM-d) δ 8.85 (d, J= 5.28 Hz, 1H), 8.14 (s, 1H), 7.84 - 7.91 (m, 1H), 7.71 - 7.82 (m, 1H), 7.39 - 7.51 (m, 3H), 7.31 (d, J = 0.78 Hz, 1H), 6.40 - 6.57 (m, 2H), 6.33 (d, J= 8.22 Hz, 1H), 4.59 (s, 2H), 2.61 (s, 1H), 2.07 - 2.24 (m, 1H), 1.85 (s, 3H), 1.08 - 1.18 (m, 2H), 0.63 - 0.81 (m, 2H). m/z (ESI, pos. ion) 583.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.006 μΜ.
Second eluting peak: 0.59 g N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide as an off-white solid.
1H NMR (400MHz ,CHLOROFORM-d) δ = 8.83 (dd, J = 0.8, 5.3 Hz, 1 H), 8.13 (s, 1 H), 7.85 (dd, J = 1.0, 7.6 Hz, 1 H), 7.77 (dd, J = 0.9, 7.9 Hz, 1 H), 7.53 - 7.36 (m, 3 H), 7.30 (d, J = 0.8 Hz, 1 H), 6.48 (d, J = 8.8 Hz, 1 H), 6.42 (d, J = 8.6 Hz, 1 H), 6.32 (dd, J = 0.8, 9.0 Hz, 1 H), 4.58 (s, 2 H), 2.65 (s, 1 H), 2.15 (td, J = 3.9, 8.0 Hz, 1 H), 1.83 (s, 3 H), 1.14 (dt, j = 2.2, 5.3 Hz, 2 H), 0.72 (dd, J = 1.8, 8.0 Hz, 2 H). m/z (ESI, pos. ion) 583.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.008 μΜ.
Example 64
N-((R)-(2-amino-5-chloro-l,3-thiazol-4-yl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide


Step 1 : Tert-butyl (4-bromo-l,3-thiazol-2-yl)carbamate (2.5 g, 8.9 mmol, WO07/084391) was taken up in 40 mL of DMF and chilled to 0 °C. Sodium hydride, 60% in mineral oil (0.39 g, 9.8 mmol, Sigma Aldrich, St. Louis, MO)
was added to the mixture. After 15 min, 2-(trimethylsilyl)-ethoxymethyl chloride (2.0 mL, 12 mmol, Sigma Aldrich, St. Louis, MO) was added. The mixture was stirred for 10 min and then warmed to room temperature. After 3 h, the reaction was quenched with 40 mL of aqueous NH4C1 and diluted with 40 mL of water. The mixture was extracted with 100 mL of EtOAc and the organic extracts were washed twice with 100 mL of water and once with 100 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (40 g column, eluted with 1% to 10%
EtOAc/hexanes gradient) afforded 2.7 g of tert-butyl (4-bromo-l,3-thiazol-2- yl)((2-(trimethylsilyl)ethoxy)methyl) carbamate as a clear oil.
Step 2: Tert-butyl (4-bromo-l,3-thiazol-2-yl)((2- (trimethylsilyl)ethoxy)methyl)carbamate (2.7 g, 6.5 mmol) was taken up in 100 mL of THF and chilled to -78 °C. Tert-butyllithium, 1.7 M solution in pentane (8.4 mL, 14 mmol, Sigma Aldrich, St. Louis, MO) was added rapidly to the mixture. After 5 min, dimethylformamide (2.5 mL, 33 mmol, Sigma Aldrich, St. Louis, MO) was added, and the mixture was warmed to room temperature and stirred for 30 min. The reaction was quenched with 25 mL of aqueous NH4C1 and diluted with 25 mL of water. The mixture was extracted with 100 mL of EtOAc and the combined organic extracts were washed with 100 mL of brine and dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (40 g column, eluted with 1% to 20% EtOAc/hexanes gradient) afforded 1.0 g of tert-butyl (4-formyl-l,3-thiazol-2- yl)((2-(trimethylsilyl)ethoxy)methyl)carbamate as a yellow oil.
Step 3: tert-Butyl (4-formyl-l,3-thiazol-2-yl)((2- (trimethylsilyl)ethoxy)methyl)carbamate (1.0 g, 2.8 mmol) was taken up in 15 mL of DMF. N-chlorosuccinimide (0.46 g, 3.4 mmol, Sigma Aldrich, St. Louis, MO) was added, and the mixture was heated to 60 °C. After 18 h, the mixture was cooled to rt. The mixture was diluted with 100 mL of EtOAc and washed with 50 mL of aqueous Na2S203, twice with 50 mL of water, and once with 50
mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure afforded 1.1 g of tert-butyl (5-chloro-4-formylthiazol-2-yl)((2- (trimethylsilyl)ethoxy)methyl)carbamate as an orange oil. The product was carried on without additional purification.
Step 4: tert-Butyl (5-chloro-4-formylthiazol-2-yl)((2- (trimethylsilyl)ethoxy)methyl)carbamate (1.1 g, 2.8 mmol) was taken up in 15 mL of DCM. (S)-2-Methylpropane-2-sulfinamide (0.40 g, 3.3 mmol, Sigma Aldrich, St. Louis, MO) and copper sulfate (1.5 g, 8.2 mmol, Fluka, Germany) were added. The mixture was stirred for 48 h. The mixture was then filtered through a plug of diatomaceous earth and washed with 30 mL of DCM. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (40 g column, eluted with 5% to 15%
EtOAc/hexanes gradient) affording 0.85 g of tert-butyl (4-((Z)-(((S)-tert- butylsulfinyl)imino)methyl)-5-chloro-l,3-thiazol-2-yl)((2- (trimethylsilyl)ethoxy)methyl) carbamate as a yellow oil.
Step 5 : 2-(2-( 1 -Benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (0.37 g, 1.1 mmol, intermediate Y3) was taken up in 11 mL of THF and chilled to -78 °C. Butyllithium solution, 2.5 M in hexanes (0.96 mL, 2.4 mmol, Sigma Aldrich, St. Louis, MO) was added dropwise to the mixture. After 15 min, (S,Z)-tert-butyl (4-(((tert-butylsulfinyl)imino)methyl)-5-chlorothiazol-2- yl)((2-(trimethylsilyl) ethoxy)methyl)carbamate (0.65 g, 1.3 mmol) was added dropwise to the mixture in 3 mL of THF. The mixture was stirred for 15 min, then warmed to room temperature and stirred for 1 hour. The reaction was quenched with 5 mL of aqueous NH4C1 and diluted with 10 mL of water. The mixture was extracted with 25 mL of EtOAc and the combined organic extracts were washed with 10 mL of brine and dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (12 g column, eluted with 5% to 50% EtOAc/hexanes gradient ) afforded 0.29 g of tert-butyl (4-((R)-(((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,2-
trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 5-chloro- 1 ,3-thiazol-2-yl)((2-(trimethylsilyl)ethoxy)methyl)carbamate as a yellow solid.
Step 6: tert-Butyl (5-chloro-4-((lR)-((S)-l ,l- dimethylethylsulfinamido)(7-(4-( 1 , 1 ,1 -trifluoro-2-hydroxypropan-2-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)methyl)thiazol-2-yl)((2-
(trimethylsilyl)ethoxy)methyl)carbamate (0.29 g, 0.35 mmol) was taken up in 1 mL of MeOH. Hydrogen chloride, 4.0 M in 1 ,4-dioxane (2.0 mL, 8.0 mmol, Sigma Aldrich, St. Louis, MO) was added. The mixture was stirred for 24 h. The solvent was removed under reduced pressure and the residue was partitioned between 25 mL of 9: 1 chloroformTPA and 25 mL of aqueous NaHC03. The mixture was partitioned and the aqueous portion was extracted twice with 15 mL of 9: 1 chloroformTPA. The combined organic extracts were dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (12 g column, eluted with 1% to 10% MeOH/DCM gradient) afforded 0.049 g of 2-(2-(2-((R)-amino(2-amino-5-chloro-l ,3-thiazol-4- yl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 , 1 ,1 -trifluoro-2-propanol as an orange solid.
Step 7: 2-(2-(2-((R)-Amino(2-amino-5-chloro- 1 ,3-thiazol-4-yl)methyl)- 1 - benzothiophen-7-yl)-4-pyridinyl)-l ,l , l-trifluoro-2-propanol (0.049 g, 0.10 mmol), cyclopropanesulfonyl chloride (0.01 1 mL, 0.1 1 mmol, Matrix Scientific, Columbia, SC), N,N-dimethylaminopyridine (1.2 mg, 10 μιηοΐ, Sigma Aldrich, St. Louis, MO), and diisopropylethylamine (0.053 mL, 0.30 mmol, Sigma Aldrich, St. Louis, MO) were taken up in 1 mL of DMF. The mixture was stirred for 24 h. The mixture was quenched with 5 mL of aqueous NH4C1 and extracted with 20 mL of EtOAc. The organic extracts were washed twice with 5 mL of water and once with 5 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (4 g column, eluted with 25% to 75 %> EtOAc/hexanes gradient)
afforded 0.015 g of N-((R)-(2-amino-5-chloro-l,3-thiazol-4-yl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide as a yellow solid.
1H NMR (400MHz ,CHLOROFORM-d) δ = 8.85 (d, J = 5.3 Hz, 1 H), 8.14 (s, 1 H), 7.87 (d, J = 7.4 Hz, 1 H), 7.79 (d, J = 7.0 Hz, 1 H), 7.52 - 7.39 (m, 2 H), 7.31 (d, J= 1.0 Hz, 1 H), 6.03 (dd, J= 1.0, 9.2 Hz, 1 H), 5.69 (d, J= 9.0 Hz, 1 H), 4.94 (s, 2 H), 2.71 - 2.61 (m, 1 H), 2.34 - 2.21 (m, 1 H), 1.84 (s, 3 H), 1.22 - 1.07 (m, 2 H), 0.90 - 0.77 (m, 2 H). m/z (ESI, pos. ion) 589.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.027 μΜ.
Example 65
N-((R)-(5-amino-2-chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide


Step 1 : 5-Amino-2-chlorobenzoic acid (5.0 g, 29 mmol, Sigma Aldrich, St. Louis, MO) was taken up in 100 mL of EtOH and chilled to 0 °C. Thionyl chloride (5.1 mL, 70 mmol, Riedel-de Haen, Germany)) was added dropwise to the mixture. The mixture was warmed to room temperature and stirred for 1 hour. The reaction was then heated to 80 °C and stirred for 2 h. The mixture was cooled to room temperature and poured into 300 mL of aqueous NaHC03. The mixture was then extracted three times with 150 mL of DCM and the organic extracts were dried over MgS04. Filtration and concentration under reduced pressure afforded 5.8 g of ethyl 5-amino-2-chlorobenzoate. The product was carried on without additional purification.
Step 2: Ethyl 5-amino-2-chlorobenzoate (5.8 g, 29 mmol) was taken up in 60 mL of DMF. Di-tert-butyl dicarbonate (14 g, 64 mmol, Sigma Aldrich, St.
Louis, MO) and N,N-dimethylaminopyridine (0.18 g, 1.4 mmol, Sigma Aldrich, St. Louis, MO) were added, and the mixture was warmed to 45 °C. The mixture was stirred for 15 h, and then cooled to room temperature. The reaction was diluted with 250 mL of EtOAc , washed three times with 200 mL of water and once with 200 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (120 g Rf column, eluted with 1% to 20% EtOAc/hexanes gradient) afforded 3.8 g of ethyl 5-(bis(tert-butoxycarbonyl)amino)-2-chlorobenzoate as a white solid.
Step 3: Ethyl 5-(bis(tert-butoxycarbonyl)amino)-2-chlorobenzoate (2.5 g, 6.2 mmol) was taken up in 31 mL of ether. Methanol (0.89 mL, 22 mmol) and lithium borohydride, 2.0 M solution in tetrahydrofuran (11 mL, 22 mmol, Sigma Aldrich, St. Louis, MO) were added. The mixture was stirred for 2 h. The reaction was carefully quenched with 20 mL of aqueous NH4C1 and diluted with 20 mL of water. The mixture was stirred for 45 min and diluted with 25 mL of EtOAc. The mixture was partitioned and the aqueous layer was extracted with 50 mL of EtOAc. The combined organic extracts were washed with 25 mL of brine and dried over MgS04. Filtration and concentration under reduced pressure afforded 1.6 g of tert-butyl (4-chloro-3-(hydroxymethyl)phenyl) carbamate as a white solid.
Step 4: tert- utyl (4-chloro-3-(hydroxymethyl)phenyl)carbamate (1.6 g, 6.2 mmol) was taken up in 60 mL of DCM. Dess-Martin periodinane (2.9 g, 6.8 mmol, Sigma Aldrich, St. Louis, MO) was added. After 30 min, the reaction was quenched by the addition of 20 mL of aqueous Na2S203 and 20 mL of aqueous NaHC03. The mixture was stirred for 20 min. The mixture was partitioned and the aqueous portion was extracted twice with 30 mL of DCM. The combined organic extracts were dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (40 g column, eluted with 1% to 10% EtOAc/hexanes gradient) afforded 1.4 g of tert-butyl (4- chloro-3-formylphenyl)carbamate as a white solid.
Step 5: tert-Butyl (4-chloro-3-formylphenyl)carbamate (1.4 g, 5.6 mmol) was taken up in 25 mL of t-BuOH and 5 mL of acetone. Di-tert-butyl dicarbonate (1.5 g, 6.7 mmol, Sigma Aldrich, St. Louis, MO) and N,N- dimethylaminopyridine (0.068 g, 0.56 mmol, Sigma Aldrich, St. Louis, MO) were added. The mixture was heated to 50 °C. After 15 h, an additional 1.0 g of di-tert-butyl dicarbonate was added. After 1 hour, an additional 1.0 g of di-tert- butyl dicarbonate was added. The mixture was stirred for 30 more min, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (40 g column, eluted with 1% to 10%
EtOAc/hexanes gradient), affording 1.8 g di-tert-butyl (4-chloro-3-formylphenyl) imidodicarbonate as a clear oil.
Step 6: di-tert-Butyl (4-chloro-3-formylphenyl) imidodicarbonate (1.8 g, 5.1 mmol) was taken up in 10 mL of DCM. (S)-2-Methylpropane-2-sulfinamide (0.92 g, 7.6 mmol, Sigma Aldrich, St. Louis, MO) and copper sulfate (2.7 g, 15 mmol, Fluka, Germany) were added. After 5 days, the mixture was filtered through a plug of diatomaceous earth and washed with 50 mL of DCM. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (40 g column, eluted with 1% to 15%
EtOAc/hexanes gradient) affording 1.1 g of di-tert-butyl (3-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-4-chlorophenyl)imidodicarbonate as sticky solid.
Step 7: 2-(2-(l-Benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.29 g, 1.1 mmol, intermediate Y5) was taken up in 11 mL of THF and chilled to -78 °C. Butyllithium solution, 2.5 M in hexanes (0.92 mL, 2.3 mmol, Sigma Aldrich, St. Louis, MO) was added to the mixture. After 10 min, di-tert-butyl (3-((E)-(((S)- tert-butylsulfinyl)imino)methyl)-4-chlorophenyl)imidodicarbonate (0.50 g, 1.1 mmol) was added to the mixture in 2 mL of THF. The mixture was stirred for 15 min, and then warmed to room temperature. After 3 h, the reaction was quenched with 5 mL of aqueous NH4C1 and diluted with 10 mL of water. The
mixture was extracted twice with 15 mL of EtOAc and the combined organic extracts were washed with 15 mL of brine and dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (12 g column, eluted with 5% to 60% EtOAc/hexanes gradient) afforded 0.31 g of di-tert-butyl (3-((((S)-tert-butylsulfmyl)amino)(7-(4-(l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-4- chlorophenyl)imidodicarbonate as white solid.
Step 8: di-tert-butyl (3-((((S)-tert-butylsulfmyl)amino)(7-(4-(l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-4- chlorophenyl)imidodicarbonate (0.30 g, 0.41 mmol) was taken up in 4 mL of MeOH. Hydrogen chloride, 4.0 M solution in 1,4-dioxane (0.11 mL, 0.45 mmol, Sigma Aldrich, St. Louis, MO) was added. After 1 hour, the solvent was removed under reduced pressure. The product was taken up in 3 mL of DMF. Hunig's base (0.22 mL, 1.2 mmol, Sigma Aldrich, St. Louis, MO),
cyclopropanesulfonyl chloride (0.042 mL, 0.41 mmol, Matrix Scientific, Columbia, SC), and N,N-dimethylaminopyridine (10 mg, 0.082 mmol, Sigma Aldrich, St. Louis, MO) were added. After 15 h, the mixture was diluted with 20 mL of EtOAc and washed with 15 mL of aqueous NH4CI, three times with 15 mL of water, and once with 15 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (12 g Isco RediSep Rf column, eluted with 10%> to 50%> EtOAc/hexanes gradient) afforded 0.18 g of di-tert-butyl (4-chloro-3-
(((cyclopropylsulfonyl)amino)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)phenyl)imidodicarbonate as a sticky yellow solid.
Step 9: di-tert-Butyl (4-chloro-3-(((cyclopropylsulfonyl)amino)(7-(4-(l - hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)phenyl)imidodicarbonate (0.18 g, 0.25 mmol) was taken up in 4 mL of 1 : 1 MeOH/THF. Hydrogen chloride, 4.0 M solution in 1,4-dioxane (0.25 mL, 1.0 mmol, Sigma Aldrich, St. Louis, MO) was added. After 3 h, an additional
0.75 mL of 4.0 N HCl in 1,4-dioxane was added. The mixture was stirred for 60 h. The mixture was diluted with 15 mL of EtOAc and washed with 10 mL of aqueous NaHC03 and 10 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure afforded 0.13 of of N-((5-amino-2- chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide as a white solid.
N-((5 -amino-2-chloropheny l)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.13g ) was subjected to two sequential preparative SFC purifications. First purification: (Chiralcel OJ column)(250 mm x 21 mm, 5 μΜ) eluting with 70% liquid C02 in 30% methanol (with 20 mM NH3) at a flow rate of 75 mL/min). Second purification: Chiralcel OJ column)(250 mm x 21 mm, 5 μΜ) eluting with 66% liquid C02 in 34% 2-propanol (with 20 mM NH3) at a flow rate of 70 mL/min). The major peak isolated from this purification was 0.57 g of N-((R)-(5-amino-2- chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide as a white solid.
1H NMR (400 MHz, CHLOROFORM-d) δ 8.75 (d, J= 5.28 Hz, 1H), 8.05 (s, 1H), 7.88 (d, J= 7.43 Hz, 1H), 7.73 (d, J= 7.04 Hz, 1H), 7.46 (t, J= 7.73 Hz, 1H), 7.35 (dd, J= 1.66, 5.18 Hz, 1H), 7.16 (d, J= 8.41 Hz, 1H), 7.11 (d, J= 1.17 Hz, 1H), 6.90 (d, J = 2.93 Hz, 1H), 6.62 (dd, J = 2.84, 8.51 Hz, 1H), 6.26 (d, J = 8.80 Hz, 1H), 5.38 (d, J= 8.22 Hz, 1H), 3.74 - 3.88 (m, 2H), 2.24 - 2.43 (m, 1H), 1.84 (s, 1H), 1.64 (s, 6H), 1.21 - 1.33 (m, 2H), 0.75 - 0.92 (m, 2H). m/z (ESI, pos. ion) 528.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.019 μΜ.
Example 66
N-((5-amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl) cyclopropanesulfonamide

Step 1 : 2-(l-Benzothiophen-7-yl)-4-(2,2,4-trimethyl-l,3-dioxolan-4- yl)pyridine (0.35 g, 1.1 mmol, Intermediate AA1) was taken up in 9 mL of THF and chilled to -78 °C. Butyllithium solution, 2.5 M in hexanes (0.46 mL, 1.1 mmol, Sigma Aldrich, St. Louis, MO) was added slowly to the mixture. After 20 min, di-tert-butyl (3 -((E)-(((S)-tert-butylsulfinyl)imino)methyl)-4- chlorophenyl)imidodicarbonate (0.55 g, 1.2 mmol, from Example 65, Step 6) was added to the mixture in 3 mL of THF. The mixture was stirred for 15 min, and then warmed to rt. The mixture was stirred for 2 h, then quenched with 5 mL of aqueous NH4CI and diluted with 5 mL of water. The mixture was extracted with 20 mL of EtOAc and the organic extracts were washed with 10 mL of brine and dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (12 g column, eluted with 5% to 60% EtOAc/hexanes gradient) afforded 0.42 g of di-tert-butyl (3-((((S)-tert- bu1ylsulfinyl)amino)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-4-chlorophenyl)imidodicarbonate as a white solid.
Step 2: Di-tert-butyl (3-((((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-4- chlorophenyl)imidodicarbonate (0.42 g, 0.54 mmol) was taken up in 5 mL of MeOH. Hydrogen chloride, 4.0 M solution in 1,4-dioxane (0.20 mL, 0.80 mmol, Sigma Aldrich, St. Louis, MO) was added. After 1 hour, the solvent was removed under reduced pressure. The residue was taken up in 5 mL of DMF. Hunig's base (0.37 mL, 2.1 mmol, Sigma Aldrich, St. Louis, MO),
cyclopropanesulfonyl chloride (0.065 mL, 0.64 mmol, Matrix Scientific,
Columbia, SC), and N,N-dimethylaminopyridine (6.5 mg, 0.053 mmol, Sigma Aldrich, St. Louis, MO) were added. The mixture was stirred for 18 h, and then diluted with 20 mL of EtOAc. The mixture was then washed three times with 10 mL of water and once with 10 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (12 g column, eluted with 5% to 60% EtOAc/hexanes gradient) afforded 0.12 g of di-tert-butyl (4-chloro-3-(((cyclopropylsulfonyl)amino)(7-(4- (2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)phenyl)imidodicarbonate as a white solid.
Step 3 : Di-tert-butyl (4-chloro-3-(((cyclopropylsulfonyl)amino)(7-(4- (2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)phenyl)imidodicarbonate (0.12 g, 0.15 mmol) was taken up in 5 mL of MeOH. Hydrochloric acid 5.0 normal solution (0.92 mL, 4.56 mmol) was added. After 18 h, the acetal had been cleaved, but one of the Boc groups remained. The solvent was removed under reduced pressure. The residue was taken up in 4 mL of THF. Hydrogen chloride, 4.0 M solution in 1,4-dioxane (0.46 mL, 1.8 mmol, Sigma Aldrich, St. Louis, MO) was added. After 1 hour, the mixture was diluted with 20 mL of EtOAc. The mixture was washed with 5 mL of aqueous NaHC03 and 5 mL of brine, and then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (4 g column, eluted with 20% to 90%> EtOAc/hexanes gradient) afforded 0.50 g of N-
((5-amino-2-chlorophenyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl) cyclopropanesulfonamide as a white solid.
1H NMR (400 MHz, CD3OD) δ 8.69 (d, J= 5.28 Hz, 1H), 8.16 (s, 1H), 7.89 (d, J = 7.04 Hz, 1H), 7.79 (d, J= 7.43 Hz, 1H), 7.41 - 7.58 (m, 2H), 7.18 (d, J= 8.61 Hz, 1H), 7.09 (d, J= 1.17 Hz, 1H), 7.05 (d, J= 2.74 Hz, 1H), 6.72 (dd, J= 2.64, 8.51 Hz, 1H), 6.33 (s, 1H), 3.59 - 3.86 (m, 2H), 2.27 - 2.45 (m, 1H), 1.60 (s, 3H), 0.97 - 1.12 (m, 2H), 0.86 - 0.96 (m, 1H), 0.72 - 0.83 (m, 1H). m/z (ESI, pos. ion) 544.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.012 μΜ.
Examples 67, 68, and 69
N-((S)-(2-amino-5-chloro-4-pyrimidinyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide, N-((R)-(2-amino-5-chloro-4- pyrimidinyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide, and N-((R)-(2-amino- 5-chloro-4-pyrimidinyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

-289 -
Step 1 : 1,3,5-Trichloroisocyanuric acid (37 g, 160 mmol, Sigma Aldrich, St. Louis, MO) and 4-chloropyrimidin-2-amine (42 g, 320 mmol, Waterstone Technology, Carmel, IN) were taken up in 375 mL of water and 42 mL of acetic acid and heated to 50 °C. After 15 h, the mixture was cooled to room
temperature. The mixture was poured into a 4 L flask and diluted with 1 L of ice. The mixture as basified with 10 M aqueous NaOH and stirred for 4 h. The resulting solid precipitate was collected by filtration and washed with 300 mL of water. The solid was suspended in 500 mL of 0.5 M aqueous NaOH and stirred for 2 h. The solid was then collected by filtration and washed with 500 mL of water. The yellow solid was then dried under high vac at 60 °C and was found to be 38 g of 4,5-dichloro-2-pyrimidinamine.
Step 2: 4,5-dichloro-2-pyrimidinamine (38g, 230 mmol) was taken up in 750 mL of THF. N,N,-dimethylaminopyridine (1.4 g, 12 mmol, Sigma Aldrich, St. Louis, MO) was added, followed by dropwise addition of di-tert-butyl dicarbonate (117 g, 536 mmol, Sigma Aldrich, St. Louis, MO) in 250 mL of THF. After 60 h, the reaction was diluted with 500 mL of EtOAc and transferred to a separatory funnel. The mixture was washed with 500 mL of aqueous NH4C1, 500 mL of water, and 500 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure afforded a sticky solid that was crystallized from ether/hexanes (200 mL of ether, followed by 1 L of hexanes) and, upon isolation, was found to be 62 g of di-tert-butyl (4,5-dichloro-2- pyrimidinyl)imidodicarbonate as a yellow solid.
Step 3 : Di-tert-butyl (4,5-dichloro-2-pyrimidinyl)imidodicarbonate (32 g, 87 mmol), l,l-bis[(di-t-butyl-p-methylaminophenyl]palladium(II) chloride (2.5 g, 3.50 mmol, Sigma Aldrich, St. Louis, MO), potassium vinyltrifluoroborate (14.6 g, 109 mmol, Sigma Aldrich, St. Louis, MO), and potassium acetate (25.7 g, 262 mmol, Sigma Aldrich, St. Louis, MO) were taken up in 400 mL of 3 : 1 MeCN: water. The mixture was purged with nitrogen for 5 min, then heated to 70 °C. After 3 h, the mixture was cooled to room temperature. The mixture was
diluted with 300 mL of EtOAc and washed with 250 mL of water, 250 mL of 1.0 N aqueous HC1, and 250 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (330 g column, eluted with 5% to 25% EtOAc/hexanes gradient) afforded 24 g of di-tert-butyl (5-chloro-4-ethenyl-2-pyrimidinyl)
imidodicarbonate as yellow solid.
Step 4: Di-tert-butyl (5-chloro-4-ethenyl-2-pyrimidinyl) imidodicarbonate (24 g, 67 mmol), sodium periodate (39.5 g, 185 mmol, Sigma Aldrich, St. Louis, MO), and potassium osmate dihydrate (0.099 g, 0.27 mmol, Strem Chemicals, Newburyport, MA) were taken up in 500 mL of 4: 1 THF: water. After 18 h, an additional 0.050 g of potassium osmate dihydrate was added. The mixture was stirred for 4 h, and then quenched with 30 g of solid Na2S03. The mixture was diluted with 400 mL of water and stirred for 45 min. The mixture was extracted twice with 250 mL of EtOAc and the combined organic extracts were washed with 250 mL of water and 250 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure afforded 24 g of di-tert-butyl (5-chloro- 4-formyl-2-pyrimidinyl) imidodicarbonate as a thick oil.
Step 5: Di-tert-butyl (5-chloro-4-formyl-2-pyrimidinyl) imidodicarbonate (24 g, 67.1 mmol), (s)-(-)-2-methyl-2-propane-sulfinamide (9.8 g, 80 mmol, Sigma Aldrich, St. Louis, MO), and copper(II) sulfate (32 g, 200 mmol, Fluka, Germany) were taken up in 200 mL of DCM. After 15 h, the mixture was filtered through a plug of diatomaceous earth and washed with 200 mL of DCM. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (330 g column, eluted with 5% to 30%
EtOAc/hexanes gradient), affording 18 g of di-tert-butyl (4-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-5-chloro-2-pyrimidinyl)imidodicarbonate as a sticky yellow solid.
Step 6: 2-(Benzo[b]thiophen-7-yl)-4-(2,2,4-trimethyl- 1 ,3-dioxolan-4- yl)pyridine (0.97 g, 3.0 mmol) was taken up in 30 mL of THF and chilled to -78 °C. Butyllithium,2.5 M in hexanes (1.2 mL, 3.1 mmol, Sigma Aldrich, St. Louis, MO) was added dropwise to the mixture. After 10 min, di-tert-butyl (4-((E)- (((S)-tert-butylsulfinyl)imino)methyl)-5-chloro-2-pyrimidinyl)imidodicarbonate (1.4 g, 3.0 mmol) was added dropwise to the mixture in 5 mL of THF. After 1.5 h, the reaction was quenched with 20 mL of aqueous NH4CI and warmed to room temperature. The mixture was diluted with 20 mL of water and extracted twice with 30 mL of EtOAc. The combined organic extracts were washed with 25 mL of brine and dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (40 g column, eluted with 2.5% to 60% EtOAc/hexanes gradient) afforded 0.65 g of di-tert-butyl (4- ((((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5-chloro-2- pyrimidinyl)imidodicarbonate as a white solid.
Step 7: Di-tert-butyl (4-((((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5- chloro-2-pyrimidinyl)imidodicarbonate (0.80 g, 1.0 mmol) was taken up in 10 mL of MeOH. Hydrogen chloride, 4.0 M in 1,4-dioxane (0.38 mL, 1.5 mmol, Sigma Aldrich, St. Louis, MO) was added. After 20 min, the reaction was quenched with 10 mL of aqueous NaHC03 and diluted with 10 mL of water. The mixture was extracted twice with 20 mL of 9: 1 chloroformTPA and the combined organic extracts were dried over MgS04. Filtration and concentration under reduced pressure afforded a sticky yellow solid. This solid was taken up in 5 mL of DMF. Diisopropylethylamine (0.44 mL, 2.5 mmol, Sigma Aldrich, St. Louis, MO), N,N-dimethylaminopyridine (0.012 g, 0.10 mmol, Sigma Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (0.16 mL, 1.5 mmol, Matrix Scientific, Columbia, SC) were added. After 18 h, the mixture was diluted with 30 mL of EtOAc and washed with 15 mL of aqueous NH4C1, twice with 15 mL of water, and once with 15 mL of brine, then dried over MgS04. Filtration and
concentration under reduced pressure, followed by flash chromatography on silica gel (12 g column, eluted with 10% to 75% EtOAc/hexanes gradient) afforded 0.30 g of di-tert-butyl (5-chloro-4-(((cyclopropylsulfonyl)amino)(7-(4- (2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-pyrimidinyl)imidodicarbonate as a yellow solid.
Step 8: Di-tert-butyl (5-chloro-4-(((cyclopropylsulfonyl)amino)(7-(4- (2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 2-pyrimidinyl)imidodicarbonate (0.30 g, 0.38 mmol) was taken up in 4 mL of MeOH. Hydrogen chloride, 4.0 M in 1,4-dioxane (1.9 mL, 7.6 mmol, Sigma Aldrich, St. Louis, MO) was added. After 18 h, the mixture was quenched with 10 mL of a NaHC03. The mixture was extracted twice with 15 mL of 9: 1 chloroformTPA and the combined organic extracts were dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (12 g column, eluted with 1% to 10% MeOH/DCM gradient) afforded 0.11 g of N-((2-amino-5-chloro-4-pyrimidinyl)(7-(4-(l,2- dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide as a yellow solid.
1H NMR (400MHz, CD3OD) δ = 8.69 (d, J= 5.3 Hz, 1 H), 8.27 (s, 1 H), 8.16 (s, 1 H), 7.93 (d, J= 7.0 Hz, 1 H), 7.83 (d, J= 7.6 Hz, 1 H), 7.61 - 7.43 (m, 2 H), 7.30 (s, 1 H), 6.24 (s, 1 H), 3.92 - 3.59 (m, 2 H), 2.32 (tt, J= 4.8, 8.0 Hz, 1 H), 1.58 (s, 3 H), 1.13 - 0.95 (m, 2 H), 0.90 - 0.81 (m, 1 H), 0.79 - 0.64 (m, 1 H). 546.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.024μΜ.
Step 9 : N-((2-amino-5 -chloro-4-pyrimidinyl)(7-(4-( 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.11 g) was subjected to two preparative SFC purifications. Purification 1 : Chiralpak® AD-H column (250 mm x 21 mm, 5 μΜ) eluting with 50% C02 in 50% 2-propanol (with 20 mM NH3) at a flow rate of 60 mL/min. Purification 2: Two AD-H column (250 mm x 21 mm, 5 μΜ)
in series, eluting with 60% C02, 40% MeOH (with 20 mM NH3) at a flow rate of 40 mL/min. The following products were isolated:
First eluting peak: 0.012 g, 1H NMR (400MHz, CD3OD) δ = 8.69 (d, J= 5.3 Hz, 1 H), 8.27 (s, 1 H), 8.17 (d, J= 0.8 Hz, 1 H), 7.93 (dd, J= 0.9, 7.5 Hz, 1 H), 7.87
- 7.74 (m, 1 H), 7.57 - 7.44 (m, 2 H), 7.30 (d, J= 0.8 Hz, 1 H), 6.24 (d, J= 0.8 Hz, 1 H), 3.82 - 3.57 (m, 2 H), 2.33 (tt, J= 4.8, 8.0 Hz, 1 H), 1.59 (s, 3 H), 1.09 - 0.95 (m, 2 H), 0.93 - 0.65 (m, 2 H). m/z (ESI, pos. ion) 546.0 (M+H)+. GK- GKRP IC50 (Binding) = 0.288 μΜ.
Third eluting peak: 0.022 g, 1H NMR (400MHz ,CD3OD) δ = 8.69 (d, J= 5.3 Hz, 1 H), 8.27 (s, 1 H), 8.17 (s, 1 H), 7.93 (d, J= 6.8 Hz, 1 H), 7.83 (d, J= 7.4 Hz, 1 H), 7.56 - 7.46 (m, 2 H), 7.30 (d, J= 0.6 Hz, 1 H), 6.24 (d, J= 0.8 Hz, 1 H), 3.77
- 3.66 (m, 2 H), 2.33 (tt, J= 4.8, 8.0 Hz, 1 H), 1.59 (s, 3 H), 1.09 - 0.96 (m, 2 H), 0.90 - 0.80 (m, 1 H), 0.79 - 0.66 (m, 1 H). m/z (ESI, pos. ion) 546.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.057μΜ.
Fourth eluting peak: 0.023 g, 1H NMR (400MHz, CD3OD) δ = 8.69 (d, J= 5.3 Hz, 1 H), 8.27 (s, 1 H), 8.17 (s, 1 H), 7.92 (s, 1 H), 7.82 (s, 1 H), 7.52 - 7.42 (m, 2 H), 7.30 (s, 1 H), 6.24 (s, 1 H), 3.80 - 3.64 (m, 2 H), 2.33 (tt, J= 4.8, 8.0 Hz, 1 H), 1.59 (s, 3 H), 1.12 - 0.95 (m, 2 H), 0.90 - 0.81 (m, 1 H), 0.80 - 0.66 (m, 1 H), m z (ESI, pos. ion) 546.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.006 μΜ.
Examples 70 and 71
N-((R)-(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)(2-
(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide
and N-((R)-(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)(2-
(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide


Step 1. To a solution of 2-(methylthio)benzaldehyde (5.0 g, 32.8 mmol) and (S)-2-methylpropane-2-sulfinamide (3.98 g, 32.8 mmol, AK Scientific, Mountain View, CA) in CH2CI2 (66 mL) was added tetraethoxytitanium (34.4 mL, 164 mmol). The reaction was stirred at room temperature for 18 h. The reaction was quenched with water (100 mL) and the resulting slurry was vigorously stirred at room temperature for 30 min. The aqueous layer was extracted with CH2CI2 (80 mL) three times. The combined organic layers were washed with water, brine, concentrated and purified by flash chromatography (80 g column, gradient elution from hexanes to 30% EtOAc/hexanes) to provide
(S,E)-2-methyl-N-(2-(methylthio)benzylidene)propane-2-sulfinamide (8.10 g, 31.7 mmol) as a pale yellow oil.
Step 2. To a solution of 2-(benzo[b]thiophen-7-yl)-4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)pyridine (1.01 g, 3.10 mmol, Intermediate AA1) (azeotropically dried with toluene) in THF (25 mL) at -78 °C was added n-butyllithium solution (2.5 M in hexanes, 1.241 mL, 3.10 mmol). After 10 min, a solution of (S,E)-2- methyl-N-(2-(methylthio)benzylidene)propane-2-sulfinamide (0.661 g, 2.59 mmol, from Step 1) in THF (15 mL) was added down the wall of the reaction flask. The reaction was stirred at -78 °C for 50 min. The reaction was quenched with water (20 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, dried (MgS04), concentrated and purified by flash chromatography (80 g column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide (S)-2-methyl-N-((2-(methylthio)phenyl)(7-(4- (2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)propane-2-sulfinamide (1.09 g, 1.877 mmol) as a white foam.
Step 3. To a solution of (S)-2-methyl-N-((2-(methylthio)phenyl)(7-(4- (2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)propane-2-sulfinamide (1.09 g, 1.877 mmol, from Step 2) in MeOH (10 mL) was added hydrochloric acid (5.0 M in H20, 9.38 mL, 46.9 mmol). The reaction was stirred at room temperature for 3.5 h. The reaction was then carefully quenched with saturated NaHC03 solution. The aqueous layer was extracted with CH2C12, and the combined organic layers were washed with water, brine, and dried (MgS04). The solvent was removed and the residue was purified by flash chromatography (40 g column, gradient elution from CH2C12 to 10 % MeOH (containing 2 M NH3) in CH2C12) to provide 2-(2-(2-(amino(2- (methylthio)phenyl)methyl)benzo [b]thiophen-7-yl)pyridin-4-yl)propane- 1 ,2-diol (0.687 g, 1.574 mmol) as a white form.
Step 4. To a solution of 2-(2-(2-(amino(2- (methylthio)phenyl)methyl)benzo [b]thiophen-7-yl)pyridin-4-yl)propane- 1 ,2-diol (687 mg, 1.574 mmol, from Step 3) in DMF (10 mL) at room temperature was added N,N-diisopropylethylamine (821 μΐ,, 4.72 mmol), 4- dimethylaminopyridine (96 mg, 0.787 mmol), and cyclopropanesulfonyl chloride (240 μί, 2.360 mmol, Matrix Scientific, Columbia, SC). The reaction was stirred at room temperature for 22 h and was quenched with water. The aqueous layer was extracted with CH2CI2, and the combined organic layers were washed with brine, dried (MgS04) and concentrated. The residue was purified by flash chromatography (80 g column, gradient elution from hexanes to 80%
EtOAc/hexanes) to provide N-((7-(4-(-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)(2-
(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide (650 mg, 1.202 mmol) as a white foam.
Step 5 : N-((7-(4-(l ,2-dihydroxypropan-2-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)(2-
(methylthio)phenyl)methyl)cyclopropanesulfonamide (650 mg, 1.202 mmol) as a mixture of diastereomers was subjected to preparative SFC (Chiralcel® AD-H column) (250 mm x 21 mm, 5 μιη) eluting with 40% liquid C02 in 60%> methanol (with 20 mM NH3) at a flow rate of 50 mL/min).
Peak 3 was collected as a white powder (297 mg, 0.549 mmol).
1H NMR (300 MHz, CDC13) δ ppm 8.69 - 8.74 (m, 1 H), 7.99 - 8.03 (m, 1 H), 7.84 (d, J= 6.4 Hz, 1 H), 7.70 (dd, J= 8.0, 0.9 Hz, 1 H), 7.54 (m, 1 H), 7.42 - 7.26 (m, 5 H), 7.02 (d, J= 1.5 Hz, 1 H), 6.43 (m, 1H), 5.80 (m, 1 H), 3.84 (m, 1H), 3.68 (m, 1H), 2.82 (br s, 1 H), 2.42 (s, 3H), 2.23 (m, 1 H), 1.99 (m, 1H), 1.57 (s, 3 H), 1.12 (m, 2 H), 0.70 - 0.85 (m, 2 H). m/z (ESI, pos. ion) 540 (M+H)+. GK-GKRP IC50 (Binding) = 0.001 μΜ.
Peak 4 was collected as a white powder (273 mg, 0.549 mmol).
1H NMR (300 MHz, CDC13) δ ppm 8.69 - 8.74 (m, 1 H), 7.99 - 8.03 (m, 1 H), 7.84 (d, J= 6.4 Hz, 1 H), 7.70 (dd, J= 8.0, 0.9 Hz, 1 H), 7.54 (m, 1 H), 7.42 - 7.26 (m, 5 H), 7.02 (d, J= 1.5 Hz, 1 H), 6.43 (m, 1H), 5.80 (m, 1 H), 3.84 (m, 1H), 3.68 (m, 1H), 2.82 (br s, 1 H), 2.42 (s, 3H), 2.23 (m, 1 H), 1.99 (m, 1H), 1.57 (s, 3 H), 1.12 (m, 2 H), 0.70 - 0.85 (m, 2 H). m/z (ESI, pos. ion) 540 (M+H)+. GK-GKRP IC50 (Binding) = 0.015 μΜ.
Examples 72 and 73
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy- l-(hydroxymethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((R)-(6-amino-3-chloro-2- pyridinyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l-(hydroxymethyl)ethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a solution of 2-(benzo[b]thiophen-7-yl)-4-(2,2-dimethyl-4- (trifluoromethyl)-l,3-dioxolan-4-yl)pyridine (1.14 g, 3.00 mmol, Intermediate AA3, azeotropically dried with 3 mL of toluene) in THF (50 mL) at -78 °C was added butyllithium solution (2.5M in hexanes, 1.202 mL, 3.00 mmol). After 10 min, a solution of the Intermediate AA4 (1.152 g, 2.504 mmol) in THF (30 mL) was added down the wall of the reaction flask. The reaction was stirred at -78 °C for 2 h. The reaction was quenched with water (50 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water,
brine, and were concentrated. The residue was purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide tert-butyl (5-chloro-6-((7-(4-(2,2-dimethyl-4-(trifluoromethyl)-l ,3- dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)((S)-l,l- dimethylethylsulfinamido)methyl)pyridin-2-yl)carbamate (1.29 g, 1.745 mmol) as a white foam.
Step 2. To a solution of tert-butyl (5-chloro-6-((7-(4-(2,2-dimethyl-4- (trifluoromethyl)- 1 ,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)((S)- 1,1- dimethylethylsulfinamido)methyl)pyridin-2-yl)carbamate (1.29 g, 1.745 mmol) in MeOH (15 mL) was added hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.309 mL, 5.23 mmol). After 1.5 h, the reaction was quenched with saturated NaHCC"3 solution. The aqueous layer was extracted with CH2CI2 and the combined organic layers were washed with brine, dried (MgSC^) and
concentrated to provide tert-butyl (6-(amino(7-(4-(2,2-dimethyl-4- (trifluoromethyl)- 1 ,3 -dioxolan-4-yl)pyridin-2-yl)benzo [b]thiophen-2-yl)methyl)- 5-chloropyridin-2-yl)carbamate, which was directly used in the next step.
Step 3. To a solution of tert-butyl (6-(amino(7-(4-(2,2-dimethyl-4- (trifluoromethyl)- 1 ,3 -dioxolan-4-yl)pyridin-2-yl)benzo [b]thiophen-2-yl)methyl)- 5-chloropyridin-2-yl)carbamate (1108 mg, 1.745 mmol) in DMF (15 mL) at room temperature was added N,N-diisopropylethylamine (1821 μί, 10.47 mmol), 4- dimethylaminopyridine (320 mg, 2.62 mmol), and cyclopropanesulfonyl chloride (889 μί, 8.73 mmol, Matrix Scientific, Columbia, SC). The reaction was stirred at room temperature for 25 h. The reaction was then quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, and concentrated. The residue was purified by flash chromatography (40 g silica gel column, gradient elution from hexanes to 40% EtOAc/hexanes) to provide a semi-pure product. The semi-pure product was further purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to 30% acetone/hexanes) to provide tert-butyl (5-chloro-6-
(cyclopropanesulfonamido(7-(4-(2,2-dimethyl-4-(trifluoromethyl)-l,3-dioxolan- 4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)pyridin-2-yl)carbamate (792 mg, 1.071 mmol) as a white powder.
Step 4. To a solution of tert-butyl (5-chloro-6- (cyclopropanesulfonamido(7-(4-(2,2-dimethyl-4-(trifluoromethyl)-l,3-dioxolan- 4-yl)pyridin-2-y l)benzo [b]thiophen-2-yl)methyl)pyridin-2-y l)carbamate (0.792 g, 1.071 mmol) in MeOH (15 mL) was added hydrochloric acid (5.0 M in H20, 13.29 mL, 66.4 mmol). The reaction was stirred at 50 °C for 36 h. The reaction was carefully quenched with saturated NaHC03 solution. The aqueous layer was extracted with CH2C12, and the combined organic layers were concentrated and purified by flash chromatography (40g silica gel column, gradient elution from CH2C12 to 7 % MeOH (containing 2 M NH3) in CH2C12) to provide N-((6-amino- 3-chloro-2-pyridinyl)(7-(4-2,2,2-trifluoro- 1 -hydroxy- 1 -(hydroxymethyl)ethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.536 g, 0.895 mmol) as a white powder.
1H NMR (300MHz, CDC13) δ 8.78 (dd, J= 2.0, 5.3 Hz, 1 H), 8.11 (s, 1 H), 7.84 (d, J= 7.6 Hz, 1 H), 7.77 (d, J= 7.7 Hz, 1 H), 7.48 - 7.35 (m, 3 H), 7.27 (s, 1 H), 6.50 (d, J= 8.8 Hz, 1 H), 6.42 (d, J= 8.6 Hz, 1 H), 6.32 (d, J= 8.8 Hz, 1 H), 4.61 (s, 2 H), 4.34 (dd, J= 5.1, 11.7 Hz, 1 H), 4.02 (s, 1 H), 3.89 (d, J= 6.0 Hz, 1 H), 2.42 (t, J= 6.2 Hz, 1 H), 2.22 - 2.10 (m, 1 H), 1.19 - 1.09 (m, 2 H), 0.73 (dd, J = 1.8, 7.9 Hz, 2 H). m/z (ESI, pos. ion) 599 (M+H)+. GK-GKRP IC50 (Binding) = 0.008 μΜ.
Step 5 : N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l , 1 , 1 -trifluoro-2,3- dihydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)cyclopropanesulfonamide (0.536 g, 0.895 mmol) as a mixture of diastereomers was subjected to preparative SFC (Chiralcel® AD-H column) (250 mm x 21 mm, 5 μιη) eluting with 58% liquid C02 in 42% methanol (with 20 mM NH3) at a flow rate of 70 mL/min).
Peak 3 was isolated as a white powder (257 mg, 0.429 mmol).
1H NMR (300MHz, CDC13) δ = 8.78 (dd, J= 2.0, 5.3 Hz, 1 H), 8.11 (s, 1 H), 7.84 (d, J= 7.6 Hz, 1 H), 7.77 (d, J= 7.7 Hz, 1 H), 7.48 - 7.35 (m, 3 H), 7.27 (s, 1 H), 6.50 (d, J= 8.8 Hz, 1 H), 6.42 (d, J= 8.6 Hz, 1 H), 6.32 (d, J= 8.8 Hz, 1 H), 4.61 (s, 2 H), 4.34 (dd, J= 5.1, 11.7 Hz, 1 H), 4.02 (s, 1 H), 3.89 (d, J= 6.0 Hz, 1 H), 2.42 (t, J= 6.2 Hz, 1 H), 2.22 - 2.10 (m, 1 H), 1.19 - 1.09 (m, 2 H), 0.73 (dd, J= 1.8, 7.9 Hz, 2 H). m/z (ESI, pos. ion) 599 (M+H)+. GK-GKRP IC50 (Binding) = 0.004 μΜ.
Peak 4 was collected as a white powder (198 mg, 0.331 mmol).
1H NMR (300MHz, CDC13) δ = 8.78 (dd, J= 2.0, 5.3 Hz, 1 H), 8.11 (s, 1 H), 7.84 (d, J= 7.6 Hz, 1 H), 7.77 (d, J= 7.7 Hz, 1 H), 7.48 - 7.35 (m, 3 H), 7.27 (s, 1 H), 6.50 (d, J= 8.8 Hz, 1 H), 6.42 (d, J= 8.6 Hz, 1 H), 6.32 (d, J= 8.8 Hz, 1 H), 4.61 (s, 2 H), 4.34 (dd, J= 5.1, 11.7 Hz, 1 H), 4.02 (s, 1 H), 3.89 (d, J= 6.0 Hz, 1 H), 2.42 (t, J= 6.2 Hz, 1 H), 2.22 - 2.10 (m, 1 H), 1.19 - 1.09 (m, 2 H), 0.73 (dd, J= 1.8, 7.9 Hz, 2 H). m/z (ESI, pos. ion) 599 (M+H)+. GK-GKRP IC50 (Binding) = 0.02 μΜ.
Example 74
N-((7-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)(2- (methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide

Step 1. To a solution of 2-(2-(benzo[b]thiophen-7-yl)pyridin-4-yl)propan- 2-ol (1.38 g, 5.14 mmol, intermediate Y5) in THF (40 mL) at -78 °C was added n-butyllithium solution (2.5M in hexanes) until the reaction turned yellowish. At this point, 2.1 mL of n-BuLi solution had been added. Another 2.1 mL of n-BuLi solution was added to the reaction. The color of the reaction mixture turned to dark brownish. After 10 min, a solution of (S,E)-2-methyl-N-(2- (methylthio)benzylidene)propane-2-sulfinamide (1.092 g, 4.28 mmol, see Examples 70 and 71, step 1) in THF (20 mL) was added down the side of the reaction flask. The reaction was stirred at -78 °C for 40 min. The reaction was quenched with saturated NaHC03 solution (40 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, concentrated and purified by flash chromatography (40 g silica gel column, gradient elution from hexanes to 60% EtOAc/hexanes) to provide (S)-N-((7-(4- (2-hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)(2- (methylthio)phenyl)methyl)-2-methylpropane-2-sulfinamide (1.51 g, 2.88 mmol, dr = 5.5: 1) as a white foam.
Step 2. To a solution of (S)-N-((7-(4-(2-hydroxypropan-2-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)(2-(methylthio)phenyl)methyl)-2-methylpropane-2- sulfinamide (1.51 g, 2.88 mmol) in MeOH (15 mL) was added hydrogen chloride
(4M in 1,4-dioxane, 1.727 mL, 8.63 mmol). After 40 min, the reaction was quenched with saturated NaHC03 slolution. The aqueous layer was extracted with CH2CI2 and the combined organic layers were washed with brine, dried (MgS04) and concentrated to provide 2-(2-(2-(amino(2- (methylthio)phenyl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)propan-2-ol, which was directly used in the next step.
Step 3. To a solution of 2-(2-(2-(amino(2- (methylthio)phenyl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)propan-2-ol (1.211 g, 2.88 mmol) in DMF (18 mL) at room temperature was added N,N- diisopropylethylamine (3.01 mL, 17.28 mmol), cyclopropanesulfonyl chloride (0.734 mL, 7.20 mmol, Matrix Scientific, Columbia, SC), and 4- dimethylaminopyridine (0.176 g, 1.440 mmol). The reaction was stirred at room temperature for 3.5 h. The reaction was then quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, and concentrated. The residue was purified by flash chromatography (40 g silica gel column, gradient elution from hexanes to 60% EtOAc/hexanes) to provide N-((7-(4-(l -hydroxy- l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)(2-(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide (1.04 g, 1.982 mmol) as a white foam.
1H NMR (300MHz, CDC13) δ = 8.73 (d, J= 5.1 Hz, 1 H), 8.03 (s, 1 H), 7.86 (d, J = 7.2 Hz, 1 H), 7.70 (d, J= 7.5 Hz, 1 H), 7.59 - 7.53 (m, 1 H), 7.47 - 7.26 (m, 5 H), 7.03 (d, J= 1.0 Hz, 1 H), 6.42 (d, J= 8.0 Hz, 1 H), 5.77 (d, J= 8.0 Hz, 1 H), 2.46 (s, 3 H), 2.30 - 2.18 (m, 1 H), 1.89 (s, 1 H), 1.63 (s, 6 H), 1.20 - 1.03 (m, 2 H), 0.87 - 0.70 (m, 2 H). m/z (ESI, pos. ion) 525 (M+H)+. GK-GKRP IC50 (Binding) = 0.004 μΜ.
Example 75
N-((7-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)(2- (methylsulfonyl)phenyl)methyl)cyclopropanesulfonamide

To a solution of N-((7-(4-(2-hydroxypropan-2-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)(2-
(methylthio)phenyl)methyl)cyclopropanesulfonamide (34 mg, 0.065 mmol) in THF/EtOH (4 mL, 1 : 1) at room temperature was added oxone, monopersulfate compound (80 mg, 0.130 mmol). The reaction mixture was stirred at room temperature for 66 h. The reaction mixture was quenched with saturated Na2S203 solution. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, and concentrated. The residue was purified by flash chromatography (4 g silica gel column, gradient elution from hexanes to 80% EtOAc/hexanes) to provide N-((7-(4-(l -hydroxy- l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)(2-
(methylsulfonyl)phenyl)methyl)cyclopropanesulfonamide (6.3 mg, 0.011 mmol) as a colorless oil.
1H NMR (300MHz, CDC13) δ 8.72 (d, J = 5.1 Hz, 1 H), 8.11 (dd, J= 1.2, 7.9 Hz, 1 H), 8.04 (d, J= 0.9 Hz, 1 H), 7.89 (ddd, J= 1.0, 3.4, 7.6 Hz, 2 H), 7.72 (d, J = 7.3 Hz, 2 H), 7.59 - 7.52 (m, 1 H), 7.49 - 7.42 (m, 1 H), 7.34 (dd, J= 1.7, 5.2 Hz, 1 H), 7.17 - 7.11 (m, 2 H), 5.76 (d, J= 7.9 Hz, 1 H), 3.02 (s, 3 H), 2.51 - 2.40 (m, 1 H), 1.93 (br. s., 1 H), 1.63 (s, 6 H), 1.20 - 0.81 (m, 4 H). m/z (ESI, pos. ion) 557 (M+H)+. GK-GKRP IC50 (Binding) = 0.133 μΜ.
Example 76
N-((2-amino-5-chloro-4-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a solution of methyl 2-amino-5-chloroisonicotinate (4.75 g, 25.5 mmol) in DMF (40 mL) was added 4-(dimethylamino)pyridine (0.466 g, 3.82 mmol), triethylamine (10.62 mL, 76 mmol), and di-tert-butyl dicarbonate (16.67 g, 76 mmol). The reaction was stirred at room temperature for 3 d. The reaction was quenched with water (40 mL). The aqueous layer was extracted with EtOAc (100 mL, three times), and the combined organic layers were washed with water, brine, dried (MgS04) and concentrated. The residue was purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to 15% EtOAc/hexanes) to provide bis-Boc aminopyridine (6.36 g, 16.44 mmol) as a white powder.
Step 2. To a solution of the bis-Boc aminopyridine (6.21 g, 16.05 mmol) in MeOH/THF/CH2Cl2 (80 mL, 60 mL/lOmL/lOmL) at 0 °C was added sodium borohydride (2.262 mL, 64.2 mmol) in portions. The reaction mixture was stirred at 0 °C for 1 h. The reaction was carefully quenched with saturated NaHCC"3 solution (30 mL). The aqueous layer was extracted with EtOAc (80 mL, three
times), and the combined organic layers were washed with water, brine, dried (MgS04) and concentrated to provide alcohol (5.19 g, 14.46 mmol) as a white powder, which was directly used in the next step without further purification.
Step 3. To a solution of alcohol (5.19 g, 14.46 mmol) in CH2C12 (100 mL) at 0 °C was added Dess-Martin periodinane (7.98 g, 18.80 mmol) in one portion. The reaction mixture was stirred at 0 °C for 1 h. The reaction was quenched with saturated Na2S203 solution (40 mL). The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, dried (MgS04) and concentrated. The residue was purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to 20% EtOAc/hexanes) to provide 5.28 g of aldehyde (5.16 g, 14.46 mmol). Without further purification, the semi-pure product was directly used in the next step.
Step 4. To a slurry of aldehyde (5.16 g, 14.46 mmol) and copper(II) sulfate (6.92 g, 43.4 mmol) in CH2C12 (100 mL) was added (s)-(-)-2-methyl-2- propane-sulfmamide (1.753 g, 14.46 mmol, AK Scientific, Mountain View, CA). The reaction mixture was stirred at room temperature for 2.5 d. The reaction mixture was filtered through a fritted funnel, rinsed with CH2C12 and the filtrated was concentrated and purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to 20% EtOAc/hexanes) to provide imine (1.71 g, 3.72 mmol) as a white powder.
Step 5. To a solution of 2-(benzo[b]thiophen-7-yl)-4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)pyridine (331 mg, 1.017 mmol, Intermediate AAl, azeotropically dried with toluene) in THF (15 mL) at -78 °C was added butyllithium solution (2.5 M in hexanes, 407 μί, 1.017 mmol). After 10 min, a solution of the bis-Boc aminopyridine (390 mg, 0.848 mmol) in THF (15 mL) was added down the wall of the reaction flask. The reaction was stirred at -78 °C for 50 min. The reaction was quenched with water (20 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, dried (MgS04),
concentrated and purified by flash chromatography (40 g silica gel column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide sulfmamide (301 mg, 0.383 mmol) as a pale yellow oil.
Step 6. To a solution of the sulfmamide (301 mg, 0.383 mmol) in MeOH (5 mL) was added hydrochloric acid (5.0 M in H20, 4 mL, 20.00 mmol). The reaction was stirred at room temperature for 42 h. The reaction was carefully quenched with saturated NaHC03 solution. The aqueous layer was extracted with CH2C12, and the combined organic layers were washed with brine, concentrated and purified by flash chromatography (12 g silica gel column, gradient elution from CH2C12 to 10 % MeOH (containing 10 M NH3) in CH2C12) to provide 2-(2- (2-(amino(2-amino-5-chloropyridin-4-yl)methyl)benzo[b]thiophen-7-yl)pyridin- 4-yl)propane-l,2-diol (93 mg, 0.211 mmol) as a colorless film.
Step 7. To a solution of 2-(2-(2-(amino(2-amino-5-chloropyridin-4- yl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)propane-l,2-diol (93 mg, 0.211 mmol) in DMF (3 mL) at room temperature was added N,N- diisopropylethylamine (110 μί, 0.633 mmol), 4-dimethylaminopyridine (12.88 mg, 0.105 mmol), and cyclopropanesulfonyl chloride (32.2 μΐ,, 0.316 mmol). The reaction was stirred at room temperature for 66 h. The reaction was then quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, dried (MgSC^) and concentrated. The residue was purified by flash chromatography (12 g silica gel column, gradient elution from hexanes to EtOAc) to provide N-((2-amino-5- chloro-4-pyridinyl)(7-(4-(( 1 S)-l ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (44 mg, 0.081 mmol) as a colorless film.
1H NMR (300MHz, CDC13) δ 8.65 (d, J= 5.3 Hz, 1 H), 8.01 (s, 1 H), 7.91 (s, 1 H), 7.83 (d, J= 7.6 Hz, 1 H), 7.72 (d, J= 7.9 Hz, 1 H), 7.48 - 7.40 (m, 1 H), 7.33 - 7.30 (m, 1 H), 7.09 (d, J= 0.9 Hz, 1 H), 6.87 (s, 1 H), 6.18 (s, 1 H), 3.80 - 3.62
(m, 2 H), 2.45 - 2.31 (m, 1 H), 1.53 (s, 3 H), 1.23 - 1.03 (m, 2 H), 1.01 - 0.78 (m, 2 H). m/z (ESI, pos. ion) 545 (M+H)+. GK-GKRP IC50 (Binding) = 0.006 μΜ.
Example 77
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-l-cyclopropylmethanesulfonamide


To a solution of 2-(2-(2-(amino(6-amino-3-chloropyridin-2- yl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)propan-2-ol (0.309 g, 0.727 mmol, Example 3, step 7) in DMF (3 mL) at room temperature was added N,N- diisopropylethylamine (0.379 mL, 2.181 mmol), 4-dimethylaminopyridine (0.044 g, 0.364 mmol) and cyclopropylmethanesulfonyl chloride (0.169 g, 1.091 mmol). The reaction was stirred at room temperature for 24 h. The reaction was then quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, and concentrated. The residue was purified by flash chromatography (40 g silica gel column, gradient elution from hexanes to 80% EtOAc/hexanes) to provide N-((6-amino-3-chloro-
2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-l -cyclopropylmethanesulfonamide (0.240 g, 0.442 mmol) as a white foam.
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(2-hydroxypropan-2-yl)pyridin- 2-yl)benzo[b]thiophen-2-yl)methyl)- 1 -cyclopropylmethanesulfonamide (240 mg, 0.442 mmol) as a mixture of enantiomers was subjected to preparative SFC (Chiralcel® AD-H column) (250 mm x 21 mm, 5 μιη) eluting with 65% liquid C02 in 35% methanol (with 20 mM NH3) at a flow rate of 75 niL/min). Peak 2 was collected to provide N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 - cyclopropylmethanesulfonamide (172 mg, 0.317 mmol) as a white solid.
1H NMR (300MHz, CDC13) δ 8.71 (d, J= 5.1 Hz, 1 H), 8.01 (d, J= 0.7 Hz, 1 H), 7.87 - 7.81 (m, 1 H), 7.75 (d, J= 7.9 Hz, 1 H), 7.43 (t, J= 7.7 Hz, 1 H), 7.37 - 7.29 (m, 3 H), 6.54 (d, J= 8.2 Hz, 1 H), 6.41 - 6.32 (m, 2 H), 4.64 (s, 2 H), 2.83 (dd, J= 3.3, 7.1 Hz, 2 H), 2.09 (br s, 1 H), 1.71 (br s, 1 H), 1.61 (s, 3 H), 1.17 - 1.02 (m, 1 H), 0.63 - 0.48 (m, 2 H), 0.24 (m, 2 H). m/z (ESI, pos. ion) 543 (M+H)+. GK-GKRP ICso (Binding) = 0.01 μΜ.
Example 78
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)- 2-pyridinyl)- l-benzothiophen-2-yl)methyl)- l-ethyl-6-oxo- 1 ,6-dihydr o-3- pyridinesulfonamide

1H NMR (300 MHz, CDC13) δ 8.80 (d, J= 5.3 Hz, 1 H), 8.11 (s, 1 H), 7.84 (d, J = 7.0 Hz, 1 H), 7.75 - 7.66 (m, 2 H), 7.53 - 7.39 (m, 4 H), 7.37 - 7.32 (m, 1 H), 7.31 - 7.24 (m, 2 H), 6.99 (d, J= 0.9 Hz, 1 H), 6.31 (d, J= 7.2 Hz, 1 H), 6.24 (d, J= 9.6 Hz, 1 H), 5.78 (d, J= 7.2 Hz, 1 H), 3.71 (q, J= 7.2 Hz, 2 H), 3.51 (s, 1 H), 1.86 (s, 3 H), 1.18 (t, J= 7.2 Hz, 3 H). m/z (ESI, pos. ion) 648 (M+H)+. GK- GKRP IC50 (Binding) = 0.015 μΜ.
Example 79
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy- l-methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-6-oxo-l-(2,2,2- trifluoroethyl)- 1 ,6-dihydro-3-pyridinesulfonamide

Step 5
Step 1. To a solution of 5-bromo-2(lH)-pyridone (1.96 g, 11.26 mmol) in DMF (30 mL) at 0 °C was added sodium hydride (60% dispersion in mineral oil, 0.541 g, 13.52 mmol) in one portion. The resulting mixture was stirred at 0 °C for 20 min and was treated with l-iodo-2,2,2-trifluoroethane (1.332 mL, 13.52 mmol). The reaction was stirred at 110 °C for 12 h. The reaction was quenched with saturated NaHCC"3 solution. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with water, brine, concentrated and purified by flash chromatography (40 g silica gel column, gradient elution from hexanes to 30% EtOAc/hexanes) to provide 5-bromo- 1 -(2,2,2- trifluoroethyl)pyridin-2(lH)-one (1.25 g, 4.88 mmol) as a pale yellow solid.
Step 2. To a sealed tube was added 5-bromo-l-(2,2,2- trifluoroethyl)pyridin-2(lH)-one (1.25 g, 4.88 mmol), methyl 3- mercaptopropionate (0.595 mL, 5.37 mmol), N,N-diisopropylethylamine (1.705 mL, 9.76 mmol), 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene (Xantphos) (0.283 g, 0.488 mmol), tris(dibenzylideneacetone)dipalladium(0) (0.224 g, 0.244 mmol) and 1,4-dioxane (25 mL). The mixture was purged with N2 for 2 min, the tube was sealed and the reaction mixture was heated at 90 °C for 12 h. The reaction mixture was cooled to room temperature, and was quenched with
saturated NaHC03 solution. The aqueous layer was extracted with EtOAc, and the combined organic layers were concentrated and purified by flash
chromatography (40 g silica gel column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide methyl 3-((6-oxo-l-(2,2,2-trifluoroethyl)-l,6- dihydropyridin-3-yl)thio)propanoate (1.43 g, 4.84 mmol) as a yellow crystalline solid.
Step 3. To a solution of methyl 3-((6-oxo-l-(2,2,2-trif uoroethyl)-l,6- dihydropyridin-3-yl)thio)propanoate (1.43 g, 4.84 mmol) in THF (30 mL) at 0 °C was added potassium tert-butoxide (0.761 g, 6.78 mmol) in one portion. The resulting yellowish slurry was stirred at 0 °C for 25 min. The reaction mixture was quenched with IN HC1 solution (25 mL). The aqueous layer was extracted with CH2CI2 and the combined organic layers were concentrated and purified by flash chromatography (40 g silica gel column, gradient elution from hexanes to EtOAc) to provide 5-mercapto-l-(2,2,2-trifluoroethyl)pyridin-2(lH)-one (776 mg, 3.71 mmol) as a pale yellow oil.
Step 4. To a solution of 5-mercapto-l-(2,2,2-trifluoroethyl)pyridin-2(lH)- one (776 mg, 3.71 mmol) in CH3CN (20 mL)/AcOH (0.75 mL)/H20 (0.5 mL) at 0 °C was added l,3-dichloro-5,5-dimethylhydantoin (877 mg, 4.45 mmol). The reaction was stirred at 0 °C for 25 min. The reaction was slowly quenched with saturated NaHC03 solution. The aqueous layer was extracted with EtOAc and the combined organic layers were concentrated and purified by flash chromatography (12 g silica gel column, gradient elution from hexanes to 30% EtOAc/hexanes) to provide 6-oxo-l-(2,2,2-trifluoroethyl)-l,6-dihydropyridine-3-sulfonyl chloride (284 mg, 1.030 mmol) as a white crystalline solid.
Step 5. To a solution of (S)-2-(2-(2-((R)-amino(6-amino-3-chloropyridin- 2-yl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)- 1,1,1 -trifluoropropan-2-ol hydrochloride (13.2 mg, 0.026 mmol, compound prepared as intermediate in Example 54) in DMF (2 mL) was added diisopropylethylamine (22.28 μί, 0.128
mmol), 6-oxo-l-(2,2,2-trifluoroethyl)-l,6-dihydropyridine-3-sulfonyl chloride (8.47 mg, 0.031 mmol) and 4-dimethylaminopyridine (3.13 mg, 0.026 mmol). The reaction was stirred at room temperature for 1.5 h, and was quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, concentrated and dried under high vacuum at 50 °C overnight (to remove residual DMF). The residue was purified by flash chromatography (4 g silica gel column, gradient elution from hexanes to 80% EtOAc/hexanes) to provide N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)- 2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-6-oxo-l-(2,2,2-trif uoroethyl)-l,6-dihydro-3-pyridinesulfonamide (7.3 mg, 10.17 μιηοΐ) as a pale yellow powder.
1H NMR (300 MHz, CDC13) δ 8.77 (d, J= 5.3 Hz, 1 H), 8.08 (s, 1 H), 8.01 (s, 1 H), 7.81 (d, J= 7.6 Hz, 1 H), 7.75 (d, J= 7.9 Hz, 1 H), 7.61 (s, 1 H), 7.51 - 7.38 (m, 2 H), 7.31 (d, J= 8.6 Hz, 1 H), 7.23 (s, 1 H), 6.86 (d, J= 7.9 Hz, 1 H), 6.37 (d, J= 8.6 Hz, 1 H), 6.24 - 6.11 (m, 2 H), 4.65 (s, 2 H), 4.28 (dd, J= 8.4, 14.8 Hz, 1 H), 3.78 (dd, J= 8.3, 14.8 Hz, 1 H), 3.64 (br. s., 1 H), 1.83 (s, 3 H). m/z (ESI, pos. ion) 717 (M+H)+. GK-GKRP IC50 (Binding) = 0.018 μΜ.
Examples 80 and 81
N-((R)-(3-chloro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide and N- ((R)-(3-chloro-2-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a solution of 2-(benzo[b]thiophen-7-yl)-4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)pyridine (1.92 g, 5.90 mmol, Intermediate AAl) (azeotropically dried with 4 mL of toluene) in THF (40 mL) at -78 °C was added butyllithium solution (2.5 M in hexanes, 2.36 mL, 5.90 mmol). After 10 min, a solution of (S,E)-N-((3-chloropyridin-2-yl)methylene)-2-methylpropane-2-sulfinamide (1.20 g, 4.92 mmol, intermediate X10) in THF (15 mL) was added down the wall of the reaction flask. The reaction was stirred at -78 °C for 1.5 h. The reaction was quenched with saturated NaHCOs solution (20 mL). The aqueous layer was extracted with EtOAc and the combined organic layers were washed with water, brine, concentrated and purified by flash chromatography (80 g of silica gel, gradient elution from hexanes to 70% EtOAc/hexanes) to provide (S)-N-((3- chloropyridin-2-yl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2-
yl)benzo[b]thiophen-2-yl)methyl)-2-methylpropane-2-sulfinamide (1.2 g, 2.11 mmol) as a white foam.
Step 2. To a solution of (S)-N-((3-chloropyridin-2-yl)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-2- methylpropane-2-sulfinamide (2.44 g, 4.28 mmol) in MeOH (20 mL) was added hydrogen chloride (4M in 1,4-dioxane, 2.57 mL, 12.8 mmol). The reaction was quenched with saturated NaHC03 solution. The aqueous layer was extracted with CH2CI2 and the combined organic layers were washed with brine, dried (MgS04), filtered, and concentrated to provide (3-chloropyridin-2-yl)(7-(4-(2,2,4-trimethyl- l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methanamine as a dark oil, which was directly used in the next step.
Step 3. To a solution of (3-chloropyridin-2-yl)(7-(4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methanamine (1.99 g, 4.28 mmol) in DMF (18 mL) at room temperature was added N,N- diisopropylethylamine (3.72 mL, 21.4 mmol), cyclopropanesulfonyl chloride (0.872 mL, 8.56 mmol), and 4-dimethylaminopyridine (0.261 g, 2.14 mmol). The reaction was stirred at room temperature for 3 h and was then quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, dried (MgS04), filtered, and concentrated. The residue was purified by flash chromatography (80 g of silica gel, gradient elution from hexanes to 70% EtOAc/hexanes) to provide N-((3-chloropyridin-2- yl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)cyclopropanesulfonamide (2.17 g, 3.81 mmol) as a white foam.
Step 4. To a solution of N-((3-chloropyridin-2-yl)(7-(4-(2,2,4-trimethyl- l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)cyclopropanesulfonamide (2.17 g, 3.81 mmol) in MeOH (10 mL) was added hydrochloric acid (5.0 M in H2O, 15.2 mL, 76 mmol). The reaction was stirred at room temperature for 22 h and then carefully quenched with saturated
NaHCC"3 solution. The aqueous layer was extracted with CH2CI2, and the combined organic layers were washed with brine, dried (MgSC^), filtered, and concentrated and purified by flash chromatography (80g silca gel column, gradient elution from hexanes to EtOAc) to provide N-((3-chloro-2-pyridinyl)(7- (4-1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (1.84 g, 3.47 mmol) as a white form.
1H NMR (300 MHz, CDC13) δ 8.76 (d, J = 5.1 Hz, 1 H), 8.57 (dd, J= 1.4, 4.6 Hz, 1 H), 8.02 (s, 1 H), 7.86 (d, J= 7.5 Hz, 1 H), 7.79 - 7.74 (m, 1 H), 7.72 (dd, J = 1.4, 8.1 Hz, 1 H), 7.49 - 7.41 (m, 1 H), 7.33 - 7.26 (m, 3 H), 6.65 - 6.58 (m, 1 H), 6.53 - 6.46 (m, 1 H), 3.90 - 3.81 (m, 1 H), 3.78 - 3.67 (m, 1 H), 2.76 (s, 1 H), 2.21 - 2.07 (m, 1 H), 1.88 (t, J= 6.1 Hz, 1 H), 1.58 (s, 3 H), 1.20 - 1.05 (m, 2 H), 0.80
- 0.62 (m, 2 H). m/z (ESI, pos. ion) 530 (M+H)+. GK-GKRP IC50 (Binding) = 0.055 μΜ.
Step 5 : N-((3-chloropyridin-2-yl)(7-(4-(l ,2-dihydroxypropan-2- yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide (1.84 g, 3.47 mmol) as a mixture of diastereomers was was subjected to preparative SFC (Chiralcel® AD-H column) (250 mm x 30 mm, 5 μιη) eluting with 55% liquid CO2 in 45% isopropanol (with 20 mM NH3) at a flow rate of 110 mL/min).
Peak 1 was collected as a white foam (706 mg, 1.332 mmol).
1H NMR (300 MHz, CDC13) δ 8.76 (d, J = 5.1 Hz, 1 H), 8.57 (dd, J= 1.4, 4.6 Hz, 1 H), 8.02 (s, 1 H), 7.86 (d, J= 7.5 Hz, 1 H), 7.79 - 7.74 (m, 1 H), 7.72 (dd, J = 1.4, 8.1 Hz, 1 H), 7.49 - 7.41 (m, 1 H), 7.33 - 7.26 (m, 3 H), 6.65 - 6.58 (m, 1 H), 6.53 - 6.46 (m, 1 H), 3.90 - 3.81 (m, 1 H), 3.78 - 3.67 (m, 1 H), 2.76 (s, 1 H), 2.21 - 2.07 (m, 1 H), 1.88 (t, J= 6.1 Hz, 1 H), 1.58 (s, 3 H), 1.20 - 1.05 (m, 2 H), 0.80
- 0.62 (m, 2 H). m/z (ESI, pos. ion) 530 (M+H)+. GK-GKRP IC50 (Binding) = 0.214 μΜ.
Peak 2 was collected as a white foam (803 mg, 1.515 mmol).
1H NMR (300 MHz, CDC13) δ 8.76 (d, J = 5.1 Hz, 1 H), 8.57 (dd, J= 1.4, 4.6 Hz, 1 H), 8.02 (s, 1 H), 7.86 (d, J= 7.5 Hz, 1 H), 7.79 - 7.74 (m, 1 H), 7.72 (dd, J = 1.4, 8.1 Hz, 1 H), 7.49 - 7.41 (m, 1 H), 7.33 - 7.26 (m, 3 H), 6.65 - 6.58 (m, 1 H), 6.53 - 6.46 (m, 1 H), 3.90 - 3.81 (m, 1 H), 3.78 - 3.67 (m, 1 H), 2.76 (s, 1 H), 2.21 - 2.07 (m, 1 H), 1.88 (t, J= 6.1 Hz, 1 H), 1.58 (s, 3 H), 1.20 - 1.05 (m, 2 H), 0.80 - 0.62 (m, 2 H). m/z (ESI, pos. ion) 530 (M+H)+. GK-GKRP IC50 (Binding) = 0.0215 μΜ.
Examples 82 and 83
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide and N- ((R)-(2-chloro-6-fluorophenyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a mixture of 2-chloro-6-fluorobenzaldehyde (34.50 g, 218 mmol) and copper(II) sulfate anhydrous (53.6 mL, 1088 mmol) in DCM (500 mL) was added (S)-2-methylpropane-2-sulfinamide (26.4 g, 218 mmol). This mixture was stirred for 3 d and then filtered through diatomaceous earth. The filtrate was concentrated and purified by flash chromatography (750 g silica gel column, gradient elution from hexanes to 40% EtOAc/hexanes) to provide (S,E)- N-(2-chloro-6-fluorobenzylidene)-2-methylpropane-2-sulfinamide (40.0 g, 153 mmol) as a yellow oil.
Step 2. To a solution of 2-(benzo[b]thiophen-7-yl)-4-(2,2,4-trimethyl- l,3-dioxolan-4-yl)pyridine (2.18 g, 6.70 mmol, Intermediate AA1, azeotropically dried with 4 mL of toluene) in THF (60 mL) at -78 °C was added butyllithium solution (2.5 M in hexanes, 2.68 mL, 6.70 mmol). After 10 min, a solution of (S,E)-N-(2-chloro-6-fluorobenzylidene)-2-methylpropane-2-sulfinamide (1.461 g, 5.58 mmol) in THF (20 mL) was added down the side of the reaction flask. The reaction was stirred at -78 °C for 1 h. The reaction was quenched with water (50 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, dried (MgS04), concentrated and purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide (S)-N-((2-chloro-6-fluorophenyl)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-2- methylpropane-2-sulfinamide (2.83 g, 4.82 mmol) as a colorless foam.
Step 3. To a solution of (S)-N-((2-chloro-6-fluorophenyl)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-2- methylpropane-2-sulfinamide (3.32 g, 5.65 mmol) in MeOH (15 mL) was added hydrochloric acid (5.0 M in H20, 28.3 mL, 141 mmol). The reaction was stirred at room temperature for 16 h. The reaction was carefully quenched with saturated NaHC03 solution. The aqueous layer was extracted with CH2C12, and the combined organic layers were washed with brine, dried (MgS04), concentrated and purified by flash chromatography (40g silica gel column, gradient elution from CH2C12 to 10 % MeOH (containing 2 M NH3) in CH2C12) to provide 2-(2- (2-(amino(2-chloro-6-fluorophenyl)methyl)benzo[b]thiophen-7-yl)pyridin-4- yl)propane-l,2-diol (2.2 g, 4.97 mmol) as a white form.
Step 4. To a solution of 2-(2-(2-(amino(2-chloro-6- fluorophenyl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)propane-l,2-diol (1.95 g, 4.40 mmol) in DMF (15 mL) at room temperature was added N,N- diisopropylethylamine (2.297 mL, 13.21 mmol), 4-dimethylaminopyridine (0.269 g, 2.201 mmol), and cyclopropanesulfonyl chloride (0.673 mL, 6.60 mmol,
Matrix Scientific, Columbia, SC). The reaction was stirred at room temperature for 19 h. The reaction was then quenched with water (15 mL). The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, dried (MgS04) and concentrated. The residue was purified by flash chromatography (80 g silica gel column, gradient elution from hexanes to EtOAc) to provide N-((2-chloro-6-fluorophenyl)(7-(4-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (1.935 g, 3.54 mmol) as a white foam.
1H NMR (300 MHz, CDC13) δ 8.77 (d, J= 5.3 Hz, 1 H), 8.05 (s, 1 H), 7.89 (d, J = 6.9 Hz, 1 H), 7.73 (d, J= 7.0 Hz, 1 H), 7.51 - 7.43 (m, 1 H), 7.39 - 7.28 (m, 3 H), 7.16 - 7.09 (m, 1 H), 7.09 - 7.06 (m, 1 H), 6.64 (d, J= 10.4 Hz, 1 H), 5.87 (d, J= 9.6 Hz, 1 H), 3.93 - 3.83 (m, 1 H), 3.74 (dd, J= 6.4, 10.8 Hz, 1 H), 2.77 (s, 1 H), 2.29 (tt, J = 4.8, 8.0 Hz, 1 H), 1.87 (t, J= 5.6 Hz, 1 H), 1.59 (s, 3 H), 1.25 - 1.14 (m, 2 H), 0.94 - 0.75 (m, 2 H). m/z (ESI, pos. ion) 547 (M+H)+. GK-GKRP IC50 (Binding) = 0.024 μΜ.
Step 5 : N-((2-chloro-6-fluorophenyl)(7-(4-(l ,2-dihydroxypropan-2- yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide (2.01 g, 3.67 mmol) as a mixture of diastereomers was subjected to preparative SFC (Chiralcel® IC-H column) (250 mm x 30 mm, 5 μιη) eluting with 65% liquid C02 in 35% MeOH (with 20 mM NH3) at a flow rate of 120 rnL/min).
Peak 4 was collected as a white foam (640 mg, 1.170 mmol).
1H NMR (300 MHz, CDC13) δ 8.77 (d, J= 5.3 Hz, 1 H), 8.05 (s, 1 H), 7.89 (d, J = 6.9 Hz, 1 H), 7.73 (d, J= 7.0 Hz, 1 H), 7.51 - 7.43 (m, 1 H), 7.39 - 7.28 (m, 3 H), 7.16 - 7.09 (m, 1 H), 7.09 - 7.06 (m, 1 H), 6.64 (d, J= 10.4 Hz, 1 H), 5.87 (d, J= 9.6 Hz, 1 H), 3.93 - 3.83 (m, 1 H), 3.74 (dd, J= 6.4, 10.8 Hz, 1 H), 2.77 (s, 1 H), 2.29 (tt, J = 4.8, 8.0 Hz, 1 H), 1.87 (t, J= 5.6 Hz, 1 H), 1.59 (s, 3 H), 1.25 -
1.14 (m, 2 H), 0.94 - 0.75 (m, 2 H). m/z (ESI, pos. ion) 547 (M+H)+. GK-GKRP IC50 (Binding) = 0.0357 μΜ.
Peak 3 was collected as a white foam (616 mg, 1.126 mmol). 1H NMR (300 MHz, CDC13) δ 8.77 (d, J= 5.3 Hz, 1 H), 8.05 (s, 1 H), 7.89 (d, J = 6.9 Hz, 1 H), 7.73 (d, J= 7.0 Hz, 1 H), 7.51 - 7.43 (m, 1 H), 7.39 - 7.28 (m, 3 H), 7.16 - 7.09 (m, 1 H), 7.09 - 7.06 (m, 1 H), 6.64 (d, J= 10.4 Hz, 1 H), 5.87 (d, J= 9.6 Hz, 1 H), 3.93 - 3.83 (m, 1 H), 3.74 (dd, J= 6.4, 10.8 Hz, 1 H), 2.77 (s, 1 H), 2.29 (tt, J = 4.8, 8.0 Hz, 1 H), 1.87 (t, J= 5.6 Hz, 1 H), 1.59 (s, 3 H), 1.25 - 1.14 (m, 2 H), 0.94 - 0.75 (m, 2 H). m/z (ESI, pos. ion) 547 (M+H)+. GK-GKRP IC50 (Binding) = 0.120 μΜ.
Example 84
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy- l-methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5-chloro-l-ethyl- -oxo- 1 ,6-dihydro-3-pyridinesulfonamide

Step 2
Step 1. To a solution of l-ethyl-5-mercaptopyridin-2(lH)-one (1.236 g, 7.96 mmol) in CH3CN (40 mL)/AcOH (1.5 mL)/H20 (1 mL) at 0 °C was added l,3-dichloro-5,5-dimethylhydantoin (2.353 g, 11.94 mmol). The reaction was stirred at 0 °C for 75 min. The reaction was slowly quenched with saturated NaHC03 solution. The aqueous layer was extracted with EtOAc and the combined organic layers were concentrated and purified by flash chromatography
(40 g silica gel column, gradient elution from hexanes to 30% EtOAc/hexanes) to provide 5-chloro-l-ethyl-6-oxo-l,6-dihydropyridine-3-sulfonyl chloride (375 mg, 1.464 mmol) as a white crystalline solid.
Step 2. To a solution of (S)-2-(2-(2-((R)-amino(6-amino-3-chloropyridin- 2-yl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)- 1,1,1 -trifluoropropan-2-ol hydrochloride (13.2 mg, 0.026 mmol, compound prepared as intermediate in Example 54) in DMF (1 mL) was added diisopropylethylamine (22.28 μί, 0.128 mmol), 5-chloro-l-ethyl-6-oxo-l,6-dihydropyridine-3-sulfonyl chloride (7.87 mg, 0.031 mmol) and 4-dimethylaminopyridine (3.13 mg, 0.026 mmol). The reaction was stirred at room temperature for 3 h and was quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were concentrated and dried under high vacuum at 50 °C overnight to remove residual DMF. The residue was purified by flash chromatography (4 g silica gel column, gradient elution from hexanes to 80% EtOAc/hexanes) to provide N- ((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro- 1 -ethyl-6-oxo- l,6-dihydro-3-pyridinesulfonamide (7.9 mg, 0.011 mmol) as a pale yellow powder.
1H NMR (300 MHz, CDC13) δ 8.76 (d, J= 5.3 Hz, 1 H), 8.06 (s, 1 H), 7.76 (dd, J = 2.9, 7.7 Hz, 2 H), 7.56 (d, J= 2.5 Hz, 1 H), 7.49 - 7.41 (m, 2 H), 7.38 (d, J = 4.7 Hz, 1 H), 7.34 - 7.28 (m, 2 H), 6.76 (d, J= 8.0 Hz, 1 H), 6.38 (d, J= 8.6 Hz, 1 H), 6.20 (d, J= 8.0 Hz, 1 H), 4.65 (s, 2 H), 3.74 - 3.56 (m, 2 H), 3.23 (dd, J= 7.2, 13.4 Hz, 1 H), 1.86 (s, 3 H), 1.08 (t, J= 7.2 Hz, 3 H). m/z (ESI, pos. ion) 698 (M+H)+. GK-GKRP IC50 (Binding) = 0.024 μΜ.
Example 85
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-
(hydroxymethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. To a solution of 2-(benzo[b]thiophen-7-yl)-4-(2,2-dimethyl-4- (trifluoromethyl)-l,3-dioxolan-4-yl)pyridine (0.514g, 1.354 mmol, Intermediate AA3) in THF (11 mL) at -78 °C was added butyllithium solution (2.5 M in hexanes, 0.492 mL, 1.231 mmol). After 10 min, a solution of (S,E)-N-(2- chlorobenzylidene)-2-methylpropane-2-sulfinamide (0.30 g, 1.231 mmol, intermediate Yl) in THF (2 mL) was added down the side of the reaction flask. The reaction was stirred at -78 °C for 70 min. The reaction was quenched with saturated NaHCOs solution. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, and dried (MgS04), The solution was concentrated and purified by flash chromatography (40 g silica gel column, gradient elution from 10% to 60% EtOAc/hexanes) to provide (S)-N- ((lR)-(2-chlorophenyl)(7-(4-(2,2-dimethyl-4-(trifluoromethyl)-l,3-dioxolan-4- yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-2-methylpropane-2-sulfinamide (0.36 g, 0.578 mmol).
Step 2. (S)-N-((l R)-(2-chlorophenyl)(7-(4-(2,2-dimethyl-4- (trifluoromethyl)- 1 ,3 -dioxolan-4-yl)pyridin-2-yl)benzo [b]thiophen-2-yl)methyl)- 2-methylpropane-2-sulfinamide (.36 g, 0.578 mmol) was taken up in 5 mL of MeOH. Hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.289 mL, 1.155 mmol) was added. The mixutre was stirred for 2 h. The reaction was quenched with 10 mL of aqueous NaHC03 and diluted with 10 mL of water. The mixture was then extracted twice with 15 mL of CH2CI2 and the combined organic extracts were dried over MgSC^. Filtration and concentration under reduced pressure afforded ( 1 R)-(2-chlorophenyl)(7-(4-(2,2-dimethyl-4-(trifluoromethyl)- 1 ,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methanamine (0.30 g, 0.578 mmol) as a yellow oil. The product was carried on without additional purification.
Step 3. (lR)-(2-chlorophenyl)(7-(4-(2,2-dimethyl-4-(trifluoromethyl)- 1 ,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methanamine (0.30 g, 0.578 mmol), diisopropylethylamine (0.302 mL, 1.734 mmol),
cyclopropanesulfonyl chloride (0.088 mL, 0.867 mmol), and 4- dimethylaminopyridine (7.06 mg, 0.058 mmol) were taken up in 3 mL of DMF. After 60 h, the reaction was diluted with 25 mL of EtOAc and washed with 10 mL of aqueous NH4C1, 10 mL of water, and 10 mL of brine, then dried over MgS04. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (12 g silica gel column, gradient elution from 5% to 60% EtOAc/hexanes) afforded N-((lR)-(2-chlorophenyl)(7-(4-(2,2-dimethyl-4- (trifluoromethyl)- 1 ,3 -dioxolan-4-yl)pyridin-2-yl)benzo [b]thiophen-2- yl)methyl)cyclopropanesulfonamide (0.18 g, 0.289 mmol) as an orange oil.
Step 4. To a solution of N-((2-chlorophenyl)(7-(4-(2,2-dimethyl-4- (trifluoromethyl)- 1 ,3 -dioxolan-4-yl)pyridin-2-yl)benzo [b]thiophen-2- yl)methyl)cyclopropanesulfonamide (100 mg, 0.160 mmol) in MeOH (2 mL) was added hydrochloric acid (5.0 M in H20, 2 mL, 10.00 mmol). The reaction was stirred at 50 °C for 6 h. The reaction was carefully quenched with saturated
NaHC03 solution. The aqueous layer was extracted with CH2CI2, and the combined organic layers were concentrated and purified by flash chromatography (4g silica gel column, gradient elution from CH2CI2 to 10% MeOH (containing 10 M NH3) in CH2C12) to provide N-((2-chlorophenyl)(7-(4-(l,l,l-trifiuoro-2,3- dihydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)cyclopropanesulfonamide (40 mg) as a mixture of diastereomers. The mixture was subjected to preparative SFC (Chiralcel® IA-H column) (250 mm x 21 mm, 5 μπι) eluting with 60% liquid C02 in 40% MeOH (with 20 mM NH3) at a flow rate of 60 mL/min). Peak 1 was as a pale yellow foam (16.1 mg, 0.028 mmol).
1H NMR (300 MHz, CDC13) δ = 8.76 (d, J= 5.1 Hz, 1 H), 8.04 (s, 1 H), 7.88 (d, J= 6.7 Hz, 1 H), 7.73 (d, J= 7.7 Hz, 1 H), 7.64 (dd, J= 1.8, 7.4 Hz, 1 H), 7.49 - 7.28 (m, 5 H), 7.06 (d, J= 1.2 Hz, 1 H), 6.41 (d, J= 7.7 Hz, 1 H), 5.40 (d, J= 7.9 Hz, 1 H), 3.90 - 3.82 (m, 1 H), 3.77 - 3.69 (m, 1 H), 2.74 (s, 1 H), 2.27 (m, 1 H), 1.82 (dd, J= 5.0, 7.2 Hz, 1 H), 1.13 (m, 2 H), 0.93 - 0.70 (m, 2 H). ). m/z (ESI, pos. ion) 583 (M+H)+. GK-GKRP IC50 (Binding) = 0.04 μΜ.
Example 86
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)-3-methyl-l-benzofuran-5- sulfonamide

Step 2
Step 1. To a solution of 5-bromo-3-methylbenzofuran (500 mg, 2.369 mmol, Milestone PharmTech, New Brunswick, NJ) in THF (20 mL) at -78 °C was dropwise added n-butyllithium solution (2.5M in hexanes, 1137 μΐ,, 2.84 mmol, Sigma- Aldrich, St. Louis, MO). The resulting dark solution was stirred at -78 °C for 15 min. A stream of sulfur dioxide (Sigma-Aldrich, St. Louis, MO) was bubbled through the solution for 10 min, resulting in a pale yellow slurry. The cold bath was removed and the reaction was slowly warmed to room temperature and stirred for 1 h. The solvent was removed under a vacuum. The residue was taken up in CH2CI2 (10 mL) and cooled to 0 °C. N-chlorosuccinimide (316 mg, 2.369 mmol, Alfa Aesar, Ward Hill, MA) was added in one portion. The reaction mixture was stirred at 0 °C for 1 h. The reaction was then quenched with saturated NaHC03 solution. The organic layer was washed with brine, concentrated and purified by flash chromatography (40 g silica gel column, gradient elution from hexanes to 5% EtOAc/hexanes) to provide 3- methylbenzofuran-5-sulfonyl chloride (50 mg, 0.217 mmol) as a white crystalline solid.
Step 2. To a solution of (S)-2-(2-(2-((R)-amino(2- chlorophenyl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)- 1,1,1 -trifluoropropan- 2-ol (16 mg, 0.035 mmol, intermediate X8) in DMF (1 mL) was added diisopropylethylamine (18.04 μί, 0.104 mmol), 3-methylbenzofuran-5-sulfonyl chloride (9.57 mg, 0.041 mmol) and 4-dimethylaminopyridine (4.22 mg, 0.035 mmol). The reaction was stirred at room temperature for 1 h, and was quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water, brine, and concentrated under a vacuum. The residue was dried under high vacuum at 50 °C overnight (to remove residual DMF) and purified by flash chromatography (4 g silica gel column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide N-((R)-(2- chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -methyl- 1 -benzofuran-5 -sulfonamide (8.9 mg, 0.014 mmol) as a colorless film.
1H NMR (300MHz, CDC13) δ = 8.72 (d, J= 5.3 Hz, 1 H), 8.09 (s, 1 H), 7.94 (d, J = 1.9 Hz, 1 H), 7.82 (dd, J= 0.8, 7.5 Hz, 1 H), 7.73 (dd, J= 1.9, 8.6 Hz, 1 H), 7.62 (dd, J= 0.8, 7.8 Hz, 1 H), 7.47 - 7.35 (m, 5 H), 7.18 (s, 1 H), 7.15 - 7.06 (m, 2 H), 6.86 (d, J= 1.0 Hz, 1 H), 6.28 (d, J= 6.9 Hz, 1 H), 5.58 (d, J= 7.7 Hz, 1 H), 2.73 (s, 1 H), 2.15 (d, J= 1.3 Hz, 3 H), 1.83 (s, 3 H). m/z (ESI, pos. ion) 656 (M+H)+. GK-GKRP IC50 (Binding) = 0.049 μΜ.
Examples 87, 88, 89, and 90
N-((R)-(2-chlorophenyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide; N-((R)- (2-chlorophenyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide; N-((S)-(2- chlorophenyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide; and
N-((S)-(2-chlorophenyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide



Step 1. To a solution of 2-(benzo[b]thiophen-7-yl)-4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)pyridine (3.94 g, 12.11 mmol, Intermediate AAl, azeotropically dried with 5 mL of toluene) in THF (50 mL) at -78 °C was added rc-BuLi (2.5 M in hexanes, 4.84 mL, 12.11 mmol). After 5 min, a solution of (S,E)-N-(2- chlorobenzylidene)-2-methylpropane-2-sulfinamide (2.46 g, 10.09 mmol, intermediate Yl) in THF (20 mL) was added down the wall of the reaction flask. The reaction was stirred at -78 °C for 50 min, and was quenched with water (50 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, brine, and dried (MgS04), and filtered. The solution was concentrated and purified by flash chromatography (80 g of silica gel, gradient elution from hexanes to 50% EtOAc/hexanes) to provide (S)-N-((2- chlorophenyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2-
yl)benzo[b]thiophen-2-yl)methyl)-2-methylpropane-2-sulfinamide (5.13 g, 9.01 mmol) as a white foam (diastereomeric ratio at the amino center is 6.5: 1 as determined by 1H NMR).
Step 2. To a solution of (S)-N-((2-chlorophenyl)(7-(4-(2,2,4-trimethyl- l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-2-methylpropane- 2-sulfinamide (5.13 g, 9.01 mmol) in MeOH (10 mL) was added hydrochloric acid (5.0 M in H20, 20 mL, 100 mmol). The reaction was stirred at room temperature for 4 h. The reaction was carefully quenched with saturated NaHC03 solution. The aqueous layer was extracted with CH2C12, and the combined organic layers were washed with brine, dried (MgS04), filtered, and concentrated and purified by flash chromatography (40 g of silica gel, gradient elution from CH2C12 to 10 % MeOH (containing 2 M NH3) in CH2C12) to provide 2-(2-(2- (amino(2-chlorophenyl)methyl)benzo [b]thiophen-7-yl)pyridin-4-yl)propane- 1,2- diol (3.65 g, 8.59 mmol) as a white foam.
Step 3. To a solution of 2-(2-(2-(amino(2- chlorophenyl)methyl)benzo[b]thiophen-7-yl)pyridin-4-yl)propane-l,2-diol (3.65 g, 8.59 mmol) in DMF (40 mL) at room temperature was added N,N- diisopropylethylamine (4.48 mL, 25.8 mmol), 4-dimethylaminopyridine (0.525 g, 4.29 mmol), and cyclopropanesulfonyl chloride (1.313 mL, 12.88 mmol, Matrix Scientific, Columbia, SC). After 22 h, the reaction was quenched with water. The aqueous layer was extracted with CH2C12, and the combined organic layers were washed with brine, dried (MgS04), filtered, and concentrated. The residue was purified by flash chromatography (80 g of silica gel, gradient elution from hexanes to 80% EtOAc) to provide N-((2-chlorophenyl)(7-(4-(l,2- dihydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)cyclopropanesulfonamide (4.2 g, 7.94 mmol) as a pale yellow oil.
Step_4. N-((2-chlorophenyl)(7-(4-( 1 ,2-dihydroxypropan-2-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide (1.1 g, 2.08 mmol) as
a mixture of stereoisomers was subjected to preparative SFC (Chiralcel AD-H column) (250 mm x 30 mm, 5 μιη) eluting with 65% liquid C02 in 35% MeOH (with 20 mM NH3) at a flow rate of 120 mL/min) to give 4 diastereomers.
Peak 1 : 1H NMR (300 MHz, CDC13) δ = 8.76 (d, J= 5.1 Hz, 1 H), 8.04 (s, 1 H), 7.88 (d, J= 6.7 Hz, 1 H), 7.73 (d, J= 7.7 Hz, 1 H), 7.64 (dd, J= 1.8, 7.4 Hz, 1 H), 7.49 - 7.28 (m, 5 H), 7.06 (d, J= 1.2 Hz, 1 H), 6.41 (d, J= 7.7 Hz, 1 H), 5.40 (d, J= 7.9 Hz, 1 H), 3.90 - 3.82 (m, 1 H), 3.77 - 3.69 (m, 1 H), 2.74 (s, 1 H), 2.27 (m, 1 H), 1.82 (dd, J= 5.0, 7.2 Hz, 1 H), 1.58 (s, 3 H), 1.13 (m, 2 H), 0.93 - 0.70 (m, 2 H). ). m/z (ESI, pos. ion) 529 (M+H)+. GK-GKRP IC50 (Binding) = 0.144 μΜ.
Peak 2: 1H NMR (300 MHz, CDC13) δ = 8.76 (d, J= 5.1 Hz, 1 H), 8.04 (s, 1 H), 7.88 (d, J= 6.7 Hz, 1 H), 7.73 (d, J= 7.7 Hz, 1 H), 7.64 (dd, J= 1.8, 7.4 Hz, 1 H), 7.49 - 7.28 (m, 5 H), 7.06 (d, J= 1.2 Hz, 1 H), 6.41 (d, J= 7.7 Hz, 1 H), 5.40 (d, J= 7.9 Hz, 1 H), 3.90 - 3.82 (m, 1 H), 3.77 - 3.69 (m, 1 H), 2.74 (s, 1 H), 2.27 (m, 1 H), 1.82 (dd, J= 5.0, 7.2 Hz, 1 H), 1.58 (s, 3 H), 1.13 (m, 2 H), 0.93 - 0.70 (m, 2 H). ). m/z (ESI, pos. ion) 529 (M+H)+. GK-GKRP IC50 (Binding) = 0.22 μΜ.
Peak 3: (351 mg, 0.663 mmol). 1H NMR (300 MHz, CDC13) δ = 8.76 (d, J= 5.1 Hz, 1 H), 8.04 (s, 1 H), 7.88 (d, J= 6.7 Hz, 1 H), 7.73 (d, J= 7.7 Hz, 1 H), 7.64 (dd, J= 1.8, 7.4 Hz, 1 H), 7.49 - 7.28 (m, 5 H), 7.06 (d, J= 1.2 Hz, 1 H), 6.41 (d, J= 7.7 Hz, 1 H), 5.40 (d, J= 7.9 Hz, 1 H), 3.90 - 3.82 (m, 1 H), 3.77 - 3.69 (m, 1 H), 2.74 (s, 1 H), 2.27 (m, 1 H), 1.82 (dd, J= 5.0, 7.2 Hz, 1 H), 1.58 (s, 3 H), 1.13 (m, 2 H), 0.93 - 0.70 (m, 2 H). ). m/z (ESI, pos. ion) 529 (M+H)+. GK- GKRP IC50 (Binding) = 0.021 μΜ.
Peak 4: 1H NMR (300 MHz, CDC13) δ = 8.76 (d, J= 5.1 Hz, 1 H), 8.04 (s, 1 H), 7.88 (d, J= 6.7 Hz, 1 H), 7.73 (d, J= 7.7 Hz, 1 H), 7.64 (dd, J= 1.8, 7.4 Hz, 1
H), 7.49 - 7.28 (m, 5 H), 7.06 (d, J= 1.2 Hz, 1 H), 6.41 (d, J= 7.7 Hz, 1 H), 5.40 (d, J= 7.9 Hz, 1 H), 3.90 - 3.82 (m, 1 H), 3.77 - 3.69 (m, 1 H), 2.74 (s, 1 H), 2.27 (m, 1 H), 1.82 (dd, J= 5.0, 7.2 Hz, 1 H), 1.58 (s, 3 H), 1.13 (m, 2 H), 0.93 - 0.70 (m, 2 H). ). m/z (ESI, pos. ion) 529 (M+H)+. GK-GKRP IC50 (Binding) = 0.256 μΜ.
Examples 91 and 92
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((R)-(6-amino-3-chloro-2- pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a solution of 2-(benzo[b]thiophen-7-yl)-4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)pyridine (1.01 g, 3.10 mmol, Intermediate AAl) (azeotropically dried with 2 mL toluene) in THF (40 mL) at -78 °C was added butyllithium solution (2.5 M in hexanes, 1.241 mL, 3.10 mmol). After 10 min, a solution of the imine (1.190 g, 2.59 mmol, Intermediate AA4) in THF (20 mL) was added
down the wall of the reaction flask. The reaction was stirred at -78 °C for 40 min. The reaction was quenched with water (50 mL). The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, and brine. The solution was concentrated and the residue was purified by flash
chromatography (80 g silica gel column, gradient elution from hexanes to 50% EtOAc/hexanes) to provide 1.34 g of a mixture of bis(Boc)-aminopyridine (893 mg, 1.137 mmol) and tert-butyl (5-chloro-6-(((S)-l,l- dimethylethylsulfinamido)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)methyl)pyridin-2-yl)carbamate (447 mg, 0.652 mmol) in about a 2: 1 ratio.
Step 2. To a solution of bis(Boc)-aminopyridine (0.893 g, 1.137 mmol) and tert-butyl (5-chloro-6-(((S)-l,l-dimethylethylsulfmamido)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)pyridin- 2-yl)carbamate (0.447 g, 0.652 mmol) in MeOH (15 mL) was added hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.853 mL, 3.41 mmol). After 1 h, the reaction was quenched with saturated NaHC03 slolution. The aqueous layer was extracted with CH2CI2 and the combined organic layers were washed with brine, dried (MgSC^) and concentrated to provide 1.59 g of a material containing about 2:1 ratio of bis-Boc and mono-Boc aminopyridines that was directly used in the next step.
Step 3. To a solution of bis-Boc aminopyridine (775 mg, 1.137 mmol) and tert-butyl (6-(amino(7-(4-(2,2,4-trimethyl-l ,3-dioxolan-4-yl)pyridin-2- yl)benzo [b]thiophen-2-yl)methyl)-5 -chloropyridin-2-yl)carbamate (330 mg, 0.569 mmol) in DMF (10 mL) at room temperature was added N,N- diisopropylethylamine (1187 μί, 6.82 mmol), cyclopropanesulfonyl chloride (290 μί, 2.84 mmol, Matrix Scientific, Columbia, SC), and 4- dimethylaminopyridine (208 mg, 1.706 mmol). The reaction was stirred at room temperature for 16 h. The reaction was then quenched with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed
with water, brine, and concentrated. The residue was purified by flash
chromatography (40 g silica gel column, gradient elution from hexanes to 40% EtOAc/hexanes) to provide a mixture of bis-Boc aminopyridine (766 mg, 0.975 mmol, 57.2 % yield) and tert-butyl (5-chloro-6-(cyclopropanesulfonamido(7-(4- (2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2- yl)methyl)pyridin-2-yl)carbamate (383 mg, 0.559 mmol) in about a 2: 1 ratio as a white foam.
Step 4. To a solution of bis-Boc aminopyridine (766 mg, 0.975 mmol) and tert-butyl (5-chloro-6-(cyclopropanesulfonamido(7-(4-(2,2,4-trimethyl- 1 ,3- dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)pyridin-2-yl)carbamate (383 mg, 0.559 mmol) in MeOH (10 mL) was added hydrochloric acid (5.0 M in H20, 1951 μί, 9.75 mmol). The reaction was stirred at room temperature for 3 d. The reaction was carefully quenched with saturated NaHC03 solution. The aqueous layer was extracted with CH2C12, and the combined organic layers were concentrated and purified by flash chromatography (40g silica gel column, gradient elution from CH2C12 to 7 % MeOH (containing 10 M NH3) in CH2C12) to provide N-((6-amino-3-chloro-2-pyridinyl)(7-(4- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (720 mg, 1.321 mmol) as a white foam.
1H NMR (300 MHz, CDC13) δ 8.75 (d, J= 4.8 Hz, 1H), 8.01 (s, 1H), 7.84 (dd, J = 0.8, 7.5 Hz, 1H), 7.76 (dd, J= 0.8, 7.8 Hz, 1H), 7.43 - 7.47 (m, 1H), 7.38 - 7.43 (m, 1H), 7.27 - 7.32 (m, 2H), 6.48 (d, J= 8.9 Hz, 1H), 6.42 (d, J= 8.6 Hz, 1H), 6.31 (d, J= 8.8 Hz, 1H), 4.60 (s, 2H), 3.82 - 3.89 (m, 1H), 3.68 - 3.76 (m, 1H), 3.49 (d, J= 5.6 Hz, 1H), 2.79 (s, 1H), 2.10 - 2.21 (m, 1H), 1.93 - 1.99 (m, 1H), 1.54 - 1.59 (m, 3H), 1.09 - 1.17 (m, 2H), 0.72 (m, 2H). m/z (ESI, pos. ion) 545 (M+H)+. GK-GKRP IC50 (Binding) = 0.005 μΜ.
Step 5 : N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l,2-dihydroxypropan-2- yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide (0.700
g, 1.284 mmol) as a mixture of diastereomers was subjected to preparative SFC (Chiralcel® AD-H column) (250 mm x 21 mm, 5 μιη) eluting with 40% liquid C02 in 60% isopropanol (with 20 mM NH3) at a flow rate of 50 mL/min) to provide two diastereomers
Peak 2 (237 mg, 0.435 mmol): 1H NMR (300 MHz, CDC13) δ 8.75 (d, J= 4.8 Hz, 1H), 8.01 (s, 1H), 7.84 (dd, J= 0.8, 7.5 Hz, 1H), 7.76 (dd, J= 0.8, 7.8 Hz, 1H), 7.43 - 7.47 (m, 1H), 7.38 - 7.43 (m, 1H), 7.27 - 7.32 (m, 2H), 6.48 (d, J = 8.9 Hz, 1H), 6.42 (d, J= 8.6 Hz, 1H), 6.31 (d, J= 8.8 Hz, 1H), 4.60 (s, 2H), 3.82 - 3.89 (m, 1H), 3.68 - 3.76 (m, 1H), 3.49 (d, J= 5.6 Hz, 1H), 2.79 (s, 1H), 2.10 - 2.21 (m, 1H), 1.93 - 1.99 (m, 1H), 1.54 - 1.59 (m, 3H), 1.09 - 1.17 (m, 2H), 0.72 (m, 2H). m/z (ESI, pos. ion) 545 (M+H)+. GK-GKRP IC50 (Binding) = 0.002 μΜ.
Peak 1 (218 mg, 0.400 mmol): 1H NMR (300 MHz, CDC13) δ 8.75 (d, J= 4.8 Hz, 1H), 8.01 (s, 1H), 7.84 (dd, J= 0.8, 7.5 Hz, 1H), 7.76 (dd, J= 0.8, 7.8 Hz, 1H), 7.43 - 7.47 (m, 1H), 7.38 - 7.43 (m, 1H), 7.27 - 7.32 (m, 2H), 6.48 (d, J= 8.9 Hz, 1H), 6.42 (d, J= 8.6 Hz, 1H), 6.31 (d, J= 8.8 Hz, 1H), 4.60 (s, 2H), 3.82 - 3.89 (m, 1H), 3.68 - 3.76 (m, 1H), 3.49 (d, J= 5.6 Hz, 1H), 2.79 (s, 1H), 2.10 - 2.21 (m, 1H), 1.93 - 1.99 (m, 1H), 1.54 - 1.59 (m, 3H), 1.09 - 1.17 (m, 2H), 0.72 (m, 2H). m/z (ESI, pos. ion) 545 (M+H)+. GK-GKRP IC50 (Binding) = 0.017 μΜ
Example 93
N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-l-methyl-3-butyn-l-yl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Potassium carbonate (2 N, 2.0 mL, 4.0 mmol) was added and the mixture was stirred at rt for 2 h. The reaction mixture was concentrated under a vacuum and the residue was partitioned between water (20 mL) and EtOAc (10 mL). The layers were separated and the aqueous phase was extracted with EtOAc (10 mL). The combined organic phases were washed with saturated aqueous sodium chloride (20 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (25 g, (3: 1 EtOAc/EtOH) in heptanes 0 to 60%). The corresponding fractions were concentrated and the residue was taken into about 1 mL of ACN and about 1 mL water to make slightly turbid solution, which was frozen in acetone dry-ice bath and lyophilized to afford N-((R)-(2- chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methyl-3 -butyn- 1 -yl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.0556 g) as a white solid (diastereomeric mixture).
1H NMR (400MHz, CDC13) δ ppm 8.77 (d, J= 5.3 Hz, 1 H), 8.08 (s, 1 H), 7.91 (d, J = 6.8 Hz, 1 H), 7.78 - 7.72 (m, 1 H), 7.66 (dd, J = 1.6, 7.6 Hz, 1 H), 7.51 - 7.30 (m, 5 H), 7.08 (s, 1 H), 6.42 (d, J= 8.0 Hz, 1 H), 5.45 (d, J= 7.8 Hz, 1 H), 2.87 - 2.72 (m, 2 H), 2.57 (br. s., 1 H), 2.28 (tt, J= 4.8, 8.0 Hz, 1 H), 2.11 (t, J =
2.6 Hz, 1 H), 1.69 (s, 3 H), 1.22 - 1.07 (m, 2 H), 0.92 - 0.83 (m, 1 H), 0.82 (m, 1 H). m/z (ESI, pos. ion) 536.9 (M+H)+. GK-GKRP IC50 (Binding) = 0 μΜ.
Example 94
N-((R)-(2-chlorophenyl)(7-(4-(2-cyano-l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

To a 5 mL vial, N-((lR)-(2-chlorophenyl)(7-(4-(2-methyloxiran-2- yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide (0.020 g, 0.039 mmol, Intermediate AA5) and potassium cyanide (0.0111 g, 0.170 mmol) were mixed into DMF (0.5 mL). The mixture was stirred at rt for 19 h. After that time the mixture was stirred at 40 °C for 2.5 h then at 50 °C for 4 h. Water (10 mL) was added to the reaction mixture and the aqueous phase was extracted with EtOAc (2 x 10 mL). The combined organic phases were washed with saturated aqueous sodium bicarbonate (20 mL), water (20 mL) and saturated aqueous sodium chloride (20 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (10 g, EtOAc in hexanes 30 to 80%) to afford N-((R)-(2-chlorophenyl)(7-(4-(2-cyano-l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.0130 g) as a white residue
(diastereomeric mixture).
1H NMR (300MHz, CD3OD) δ ppm 8.74 (dd, J= 0.6, 5.3 Hz, 1 H), 8.20 (d, J = 0.9 Hz, 1 H), 7.94 (dd, J= 1.0, 7.5 Hz, 1 H), 7.83 - 7.74 (m, 2 H), 7.58 - 7.33 (m,
5 H), 7.02 (t, J= 1.2 Hz, 1 H), 6.46 (d, J= 0.7 Hz, 1 H), 3.12 - 2.96 (m, 2 H), 2.32 (tt, J = 4.8, 7.9 Hz, 1 H), 1.74 (s, 3 H), 1.13 - 0.66 (m, 4 H). m/z (ESI, pos. ion) 537.9 (M+H)+. GK-GK P IC50 (Binding) = 0.057 μΜ.
Example 95
N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-2-methoxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

To a 5 mL vial, N-((lR)-(2-chlorophenyl)(7-(4-(2-methyloxiran-2- yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide (0.0538 g, 0.105 mmol, Intermediate AA5) and sodium methoxide (25 wt. % solution in methanol, 0.10 mL, 0.437 mmol, Sigma- Aldrich, St. Louis, MO) were dissolved into MeOH (1 mL). The vial was sealed and the reaction mixture was stirred at rt for 5 h. The reaction mixture was concentrated under a vacuum. The residue was partitioned between saturated aqueous ammonium chloride (2 mL) and EtOAc (2 mL). The layers were separated and the aqueous phase was extracted with EtOAc (2 x 2 mL). The combined organic phases were washed with water (6 mL) and saturated aqueous sodium chloride (6 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (10 g, EtOAc in hexanes 20 to 70%) to afford N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-2-methoxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.0478 g) as a white foam (diastereomeric mixture).
1H NMR (300MHz, CDC13) δ ppm 8.75 (d, J= 5.3 Hz, 1 H), 8.06 (s, 1 H), 7.91 (d, J= 7.5 Hz, 1 H), 7.73 (d, J= 7.5 Hz, 1 H), 7.65 (dd, J= 1.8, 7.5 Hz, 1 H), 7.52 - 7.28 (m, 5 H), 7.07 (s, 1 H), 6.42 (d, J= 7.6 Hz, 1 H), 5.44 (d, J= 7.9 Hz, 1 H), 3.70 - 3.61 (m, 1 H), 3.57 (s, 1 H), 3.40 (s, 3 H), 3.03 (s, 1 H), 2.35 - 2.21 (m, 1 H), 1.55 (s, 3 H), 1.24 - 1.05 (m, 2 H), 0.94 - 0.70 (m, 2 H). m/z (ESI, pos. ion) 542.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.089 μΜ.
Example 96
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2-cyano-l-hydroxy-l-methylethyl)- -pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a 250-mL round-bottomed flask, 2-chloro-4-(2,2,4-trimethyl- l,3-dioxolan-4-yl)pyridine (2.18 g, 9.59 mmol, Intermediate AA2) and hydrogen chloride (4.0 M in 1,4-dioxane, 12.0 mL, 48.0 mmol, Sigma- Aldrich, St. Louis, MO) were mixed into MeOH (50 mL). The mixture was stirred at rt for 2 h. The reaction mixture was concentrated and 1 M K3PO4 (70 mL) was added to the residue. The aqueous phase was extracted with EtOAc (2 x 70 mL) and 5% IPA in CHCI3 (3 x 50 mL). The combined organic phases were dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (50 g, (3: 1 EtOAc/EtOH) in heptanes 10 - 60%) to afford 2-(2-chloro-4-pyridinyl)-l,2-propanediol (1.71 g) as a clear viscous oil.
Step 2. To a 250-mL round-bottomed flask, 2-(2-chloro-4-pyridinyl)-l,2- propanediol (1.71 g, 9.11 mmol) and p-toluenesulfonyl chloride (5.26 g, 27.6
mmol, Sigma-Aldrich, St. Louis, MO) were dissolved into pyridine (50 mL). The solution was stirred at rt for 18 h. Water (100 mL) was added to the reaction mixture. The aqueous phase was extracted with EtOAc (2 x 60 mL). The combined organic phases were washed with water (100 mL) and saturated aqueous sodium chloride (100 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. To the residue, THF (50 mL) and sodium hydroxide solution (2 N, 50 mL, 100 mmol) were added and the mixture was stirred at rt for 10 min. Saturated aqueous ammonium chloride (300 mL) was added to the reaction mixture. The aqueous phase was extracted with EtOAc (2 x 150 mL). The combined organic phases were washed with saturated aqueous sodium chloride (300 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (50 g, EtOAc in hexanes 0 to 50%) to afford 2-chloro-4-(2-methyl-2-oxiranyl)pyridine in 2 batches: 1st batch (0.880 g) as a colorless oil, >99%> purity; 2nd batch (0.504 g) as a colorless oil, 95% purity.
Step 3. To a 5 mL vial, 2-chloro-4-(2-methyl-2-oxiranyl)pyridine (0.0519 g, 0.306 mmol), potassium cyanide (0.0625 g, 0.960 mmol, Sigma-Aldrich, St. Louis, MO) and water (0.20 mL, 1 1 mmol) were mixed into DMF (2 mL). The mixture was stirred at rt for 2.5 h. Water (20 mL) was added and the aqueous phase was extracted with EtOAc (2 x 20 mL). The combined organic phases were washed with water (2 x 20 mL) and saturated aqueous sodium chloride (20 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column
chromatography (10 g, EtOAc in hexanes 0 - 70%) to afford 3-(2-chloro-4- pyridinyl)-3-hydroxybutanenitrile (0.0342 g) as a clear viscous oil.
Step 4. To a 5 mL vial, 3-(2-chloro-4-pyridinyl)-3-hydroxybutanenitrile (0.034 g, 0.173 mmol), tert-butyl (5-chloro-6-(((cyclopropylsulfonyl)amino)(7- (4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen-2-yl)methyl)-2-
pyridinyl)carbamate (0.1098 g, 0.177 mmol, Intermediate AA7) Pd(PPli3)4 (0.0205 g, 0.018 mmol, Strem Chemical Inc, Newburyport, MA) and potassium carbonate (2 N, 0.259 mL, 0.519 mmol) were mixed into 1,4-dioxane (2 mL). The mixture was degassed by bubbling argon gas through the reaction mixture for 5 min. The vial was sealed and the reaction mixture was stirred at 100 °C for 1 h 15 min. Water (30 mL) was added to the reaction mixture and the aqueous phase was extracted with EtOAc (2 x 30 mL). The combined organic phases were washed with saturated aqueous sodium chloride (40 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (25 g, EtOAc in hexanes 30 - 80%) to afford tert-butyl (5-chloro-6-((7-(4-(2-cyano-l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)((cyclopropylsulfonyl)amino)methyl)-2-pyridinyl)carbamate (0.0822 g) as a light yellow foam.
Step 5. To a 20 mL scintillation vial, tert-butyl (5-chloro-6-((7-(4-(2- cyano- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)((cyclopropylsulfonyl)amino)methyl)-2-pyridinyl)carbamate (0.082 g, 0.13 mmol) and trifluoroacetic acid (0.50 mL, 6.5 mmol, Sigma-Aldrich, St. Louis, MO) were dissolved into DCM (1 mL). The mixture was stirred at rt for 35 min. K3PO4 solution (1 M, 5 mL) was added and the aqueous phase was extracted with DCM (2 x 10 mL). The combined organic phases were dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (10 g, EtOAc in hexanes 20 to 80%). The corresponding fractions were concentrated and ACN (0.5 mL) was added to the residue followed by 0.5 mL water to obtain slightly turbid solution, which was frozen and lyophilized to obtain N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2- cyano- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.0489 g) as an off white powder (mixture of 4 stereoisomers).
1H NMR (400MHz, CDC13) δ ppm 8.80 (d, J= 5.3 Hz, 1 H), 8.03 (s, 1 H), 7.84 (d, J= 7.2 Hz, 1 H), 7.76 (d, J= 7.2 Hz, 1 H), 7.50 - 7.38 (m, 2 H), 7.35 - 7.27 (m, J= 1.9, 1.9, 5.2 Hz, 2 H), 6.52 (br. s., 1 H), 6.43 (d, J= 8.6 Hz, 1 H), 6.31 (d, J= 8.8 Hz, 1 H), 5.17 - 4.30 (m, 2 H), 2.92 - 2.80 (m, 2 H), 2.50 (br. s., 1 H), 2.23 - 2.11 (m, 1 H), 1.81 (s, 3 H), 1.20 - 1.05 (m, 2 H), 0.79 - 0.67 (m, 2 H). m/z (ESI, pos. ion) 553.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.006 μΜ.
Example 97
N-((R)-(2-chlorophenyl)(7-(4-(S-methylsulfonimidoyl)-2-pyridinyl)-l- benzothio hen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a 20 mL vial, 2-chloro-4-fluoropyridine (1.26 mL, 9.55 mmol, Matrix Scientific, Columbia, SC) and sodium thiomethoxide (0.70 g, 10 mmol, Fluka Chemie GmbH, Switzerland) were mixed into DMF (10 mL). The slightly cloudy mixture was stirred at rt for 18 h. Additional sodium thiomethoxide (0.43 g, 6.2 mmol, Fluka Chemie GmbH, Switzerland) was added and the mixture was stirred for 6 h at rt. More sodium thiomethoxide (0.64 g, 9.1 mmol, Fluka Chemie GmbH, Switzerland) was added and the mixture was stirred at rt for 18 h. Water (80 mL) was added to the reaction mixture and the aqueous phase was extracted with EtOAc (2 x 50 mL). The combined organic phases were washed with water (2 x 100 mL) and saturated aqueous sodium chloride (100 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum.
The resulting product was purified by silica gel column chromatography (50 g, EtOAc in hexanes 0 to 30%) to afford 2-chloro-4-(methylsulfanyl)pyridine (1.43 g) as a clear oil.
Step 2. To a stirred solution of 2-chloro-4-(methylsulfanyl)pyridine (0.261 g, 1.64 mmol) in DCM (5 mL) in 250-mL round-bottomed flask, 3- chloroperoxybenzoic acid (77% max. 0.463 g, 2.06 mmol, Sigma-Aldrich, St. Louis, MO) was added in 3 potions at 0 °C. The mixture was stirred at that temperature for 30 min. The reaction was quenched by adding 1 M K3PO4 (40 mL). The aqueous phase was extracted with DCM (2 x 20 mL). The combined organic phases were washed with water (40 mL) and saturated aqueous sodium chloride (40 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (25 g, EtOAc in hexanes 50 to 100%) to afford 2- chloro-4-(methylsulfinyl)pyridine (0.193 g) as a clear oil.
Step 3. To a 20 mL vial, 2-chloro-4-(methylsulfinyl)pyridine (0.0975 g, 0.555 mmol) and sodium azide (0.0853 g, 1.31 mmol, Sigma-Aldrich, St. Louis, MO) were mixed into chloroform (0.6 mL). The mixture was cooled to 0 °C and sulfuric acid (0.28 mL, 5.3 mmol, Sigma-Aldrich, St. Louis, MO) was added drop-wise. The vial was sealed and the mixture was stirred at 45 °C for 40 h. The reaction was quenched with 2 N NaOH (about 4 mL) at 0 °C then diluted with water (20 mL) and EtOAc (20 mL). The layers were separated and the aqueous phase was extracted with EtOAc (20 mL) and 10% IPA/CHCI3 (2 x 20 mL). Each organics were washed with water and brine. The combined wash was extracted with 10% IPA/CHCI3 (2 x 20 mL). The combined organic phases were dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (25 g, EtOAc in hexanes 50 to 100%) to afford 2-chloro-4-(S-methylsulfonimidoyl)pyridine (0.056 g) as a clear oil.
Step 4. To a 5 niL vial, N-((R)-(2-chlorophenyl)(7-(4,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.353 g, 0.700 mmol, Intermediate X14), 2-chloro-4-(S- methylsulfonimidoyl)pyridine (0.055 g, 0.29 mmol), Pd(PPh3)4 (0.038 g, 0.033 mmol, Strem Chemical Inc, Newburyport, MA) and sodium carbonate (1 N, 0.90 mL, 0.90 mmol) were mixed into 1,4-dioxane (4 mL). The mixture was degassed by bubbling argon gas through the reaction mixture for 5 min. The vial was sealed and the reaction mixture was stirred at 100 °C for 2.5 h. The reaction mixture was partitioned between water (40 mL) and EtOAc (40 mL). The aqueous phase was extracted with EtOAc (40 mL). The combined organic phases were washed with saturated aqueous sodium chloride (60 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (50 g, EtOAc in hexanes 0 to 100%) to afford N-((R)-(2-chlorophenyl)(7-(4-(S- methylsulfonimidoyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.066 g) as a white solid (diastereomeric mixture).
1H NMR (300MHz, CDC13) δ ppm 9.03 - 8.95 (m, 1 H), 8.48 (s, 1 H), 7.96 (s, 1 H), 7.84 - 7.74 (m, 2 H), 7.67 (d, J= 7.3 Hz, 1 H), 7.56 - 7.31 (m, 4 H), 7.07 (s, 1 H), 6.43 (d, J= 8.2 Hz, 1 H), 5.74 - 5.63 (m, 1 H), 3.23 - 3.12 (m, 3 H), 2.32 - 2.19 (m, 1 H), 1.23 - 1.06 (m, 2 H), 0.93 - 0.69 (m, 2 H). NH was not observed, m/z (ESI, pos. ion) 553.8 (M+Na)+. GK-GKRP IC50 (Binding) = 0.182 μΜ.
Example 98
N-((6-amino-3-chloro-2-pyridinyl)(5-chloro-7-(4-(l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. To a 20 mL vial, 2-(5-chloro-l-benzothiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (0.698 g, 2.37 mmol, Intermediate AA8), 2- chloro-4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine (0.515 g, 2.26 mmol, Intermediate AA2), Pd(PPli3)4 (0.273 g, 0.236 mmol, Strem Chemical Inc, Newburyport, MA) and potassium carbonate (2 N, 2.30 mL, 4.60 mmol) were mixed into 1,4-dioxane (8 mL). The mixture was degassed by bubbling argon gas through the reaction mixture for 5 min. The vial was sealed and the mixture was stirred at 80 °C for 1 h. The reaction was quenched by adding water (80 mL). The aqueous phase was extracted with EtOAc (2 x 80 mL). The combined organic phases were washed with saturated aqueous sodium chloride (80 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (120 g, EtOAc in hexanes 0 to 20%) followed by another silica gel column chromatography (80 g, acetone in hexanes 0 - 15%) to afford 2-(5-chloro-l- benzothiophen-7-yl)-4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine (0.558 g) as a clear oil.
Step 2. Diisopropylamine (0.150 mL, 1.06 mmol, Sigma-Aldrich, St. Louis, MO) was dissolved into THF (5 mL) in a 20 mL scintillation vial and nBuLi (2.5 M, 0.450 mL, 1.13 mmol, Sigma-Aldrich, St. Louis, MO) was added at -78 °C. After 15 min, this solution was added to a solution of 2-(5-chloro-l- benzothiophen-7-yl)-4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine (0.550 g, 1.53 mmol) in THF (20 mL) in 150 mL round-bottomed flask at -78 °C. The cold bath was switched to another cold bath which was prepared to have the temperature of -45 °C and the mixture was stirred at -45 about -35 °C (bath temperature) for 20 min. The bath was switched back to -78°C. After 5 min of switching the bath, tert-butyl (6-(((tert-butylsulfinyl)imino)methyl)-5-chloro-2-pyridinyl)carbamate (0.411 g, 0.893 mmol, Intermediate AA4) in 5 mL of THF was added down the wall of the flask followed by THF rinse solution (5 mL). The mixture was stirred at -78 °C for 1 h. The reaction was quenched cold with water (20 mL) and NH4C1 (20 mL) and the mixture was allowed to warm up to rt. The aqueous phase was extracted with EtOAc (2 x 50 mL). The combined organic phases were washed with water (100 mL) and saturated aqueous sodium chloride (100 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (80 g, EtOAc in hexanes 0 to 60 %) to afford a mixture of di-tert-butyl (6-(((tert- butylsulfinyl)amino)(5-chloro-7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-2-pyridinyl)imidodicarbonate and tert-butyl (6-(((tert-butylsulfinyl)amino)(5-chloro-7-(4-(2,2,4-trimethyl- 1 ,3- dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5-chloro-2- pyridinyl)carbamate (0.239 g) as a mixture of diastereomers. This material was dissolved into MeOH (5 mL) in a 150-mL round-bottomed flask and HC1 (4 N in l,4-dioxane,1.00 mL, 4.00 mmol, Sigma-Aldrich, St. Louis, MO) was added. The mixture was stirred at rt for 2.5 h. The reaction mixture was concentrated under a vacuum and the residue was dissolved into DCM (5 mL) and TFA (2.5 mL). The reaction mixture was stirred at rt for 1 h 45 min. The reaction was quenched by slow addition of saturated aqueous sodium bicarbonate (40 mL). The aqueous phase was extracted with EtOAc (2 x 30 mL). The combined organic phases were
washed with saturated aqueous sodium bicarbonate (50 mL) and saturated aqueous sodium chloride (50 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (40 g, (2 M NH3 in MeOH) in DCM 0 to 10%) to afford 2-(2-(2-(amino(6-amino-3-chloro-2-pyridinyl)methyl)- 5-chloro-l-benzothiophen-7-yl)-4-pyridinyl)-l,2-propanediol (0.124 g) as a white solid.
Step 3. To a 150-mL round-bottomed flask, 2-(2-(2-(amino(6-amino-3- chloro-2-pyridinyl)methyl)-5-chloro- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2- propanediol (0.124 g, 0.261 mmol), DMAP (0.0204 g, 0.167 mmol, Sigma- Aldrich, St. Louis, MO), DIPEA (0.15 mL, 0.86 mmol, Sigma-Aldrich, St. Louis, MO) and cyclopropanesulfonyl chloride (0.040 mL, 0.39 mmol, Matrix
Scientific, Columbia, SC) were mixed into DMF (3 mL). The mixture was stirred at rt for 2 h 10 min. The reaction was quenched by adding water (40 mL). The aqueous phase was extracted with EtOAc (2 x 30 mL). The combined organic phases were washed with water (2 x 40 mL) and saturated aqueous sodium chloride (40 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (40 g, (2 M NH3 in MeOH) in DCM 0 to 10 %). The corresponding fractions were concentrated and the residue was taken into about 1 mL ACN. Water was added until the solution became slightly turbid. This mixture was frozen and lyophilized to obtain N-((6-amino-3-chloro-2- pyridinyl)(5 -chloro-7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.0947 g) as a white solid.
1H NMR (300MHz, CDC13) δ ppm 8.70 (dd, J= 3.1, 5.0 Hz, 1 H), 7.99 (s, 1 H), 7.80 - 7.74 (m, 1 H), 7.70 - 7.66 (m, J= 2.6 Hz, 1 H), 7.41 (d, J= 8.8 Hz, 1 H), 7.30 (ddd, J= 1.5, 3.3, 5.1 Hz, 1 H), 7.18 (d, J= 3.1 Hz, 1 H), 6.64 - 6.53 (m, 1 H), 6.43 (d, J= 8.6 Hz, 1 H), 6.30 (d, J= 8.6 Hz, 1 H), 4.69 (br. s., 2 H), 3.90 - 3.80 (m, 1 H), 3.77 - 3.67 (m, 1 H), 3.21 - 2.77 (m, 1 H), 2.22 - 2.11 (m, 1 H),
1.57 (s, 3 H), 1.22 - 1.06 (m, 2 H), 0.80 - 0.66 (m, 2 H). One exchangable proton was not observed, m/z (ESI, pos. ion) 578.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.003 μΜ.
Example 99, 100, 101, and 102
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((R)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide; N-((R)- (6-amino-3-chloro-2-pyridinyl)(7-(4-((S)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide; N-((S)- (6-amino-3-chloro-2-pyridinyl)(7-(4-((R)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide; and N- ((S)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((S)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide


Step 2. To a 20 mL scintillation vial, tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(S-methylsulfonimidoyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (0.0921 g, 0.142 mmol) and TFA (0.50 mL, 6.5 mmol, Sigma-Aldrich, St. Louis, MO) were dissolved into DCM (1 mL). The mixture was stirred at rt for 1.5 h. 1 M K3P04 (6 mL) was added and the aqueous phase was extracted with DCM (2 x 10 mL). The combined organic phases were dried over sodium sulfate, filtered and
concentrated under a vacuum. The resulting product was purified by silica gel column chromatography (25 g, EtOAc in hexanes 20 to 100%, then (3: 1
EtOAc/EtOH) in heptanes 50 to 100%) to afford 0.0729 g of white solid. The solid was taken into 1 mL of ACN and water was added until the mixture became
slightly turbid (about 1 mL). The mixture was frozen then lyophilized to give N- ((6-amino-3-chloro-2-pyridinyl)(7-(4-(S-methylsulfonimidoyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.0633 g) as a white solid (mixture of 4 diastereomers).
1H NMR (400MHz, CDC13) δ ppm 9.06 (d, J= 4.9 Hz, 1 H), 8.49 (s, 1 H), 7.96 (d, J= 7.6 Hz, 1 H), 7.87 - 7.78 (m, 2 H), 7.54 - 7.42 (m, 2 H), 7.32 (s, 1 H), 6.59 (d, J= 6.5 Hz, 1 H), 6.47 (d, J= 8.6 Hz, 1 H), 6.35 (d, J= 9.0 Hz, 1 H), 4.78 (br. s., 2 H), 3.18 (s, 3 H), 2.24 - 2.13 (m, 1 H), 1.23 - 1.11 (m, 2 H), 0.81 - 0.70 (m, 2 H). m/z (ESI, pos. ion) 569.8 (M+Na)+. GK-GKRP IC50 (Binding) = 0.015 μΜ.
Step 3 : The mixture was resolved using preparative SFC (Chiralpak® IC column) (250 mm x 21 mm, 5 μιη) eluting with 50% liquid C02 in 50% isopropanol (with 20 mM NH3) at a flow rate of 65 mL/min resulting in Peak 1 , 2, 3, and 4 with DE values of > 99%, 98.0%, 95.7%, and 95.9%, respectively.
Peak 1 : 1H NMR (400MHz, CDC13) δ = 9.07 (d, J= 5.1 Hz, 1 H), 8.49 (s, 1 H), 7.96 (d, J= 7.0 Hz, 1 H), 7.87 - 7.78 (m, 2 H), 7.54 - 7.42 (m, 2 H), 7.31 (s, 1 H), 6.55 (d, J= 8.4 Hz, 1 H), 6.47 (d, J= 8.8 Hz, 1 H), 6.35 (d, J= 9.0 Hz, 1 H), 4.71 (br. s., 2 H), 3.18 (s, 3 H), 2.24 - 2.12 (m, 1 H), 1.24 - 1.09 (m, 2 H), 0.81 - 0.68 (m, 2 H). One proton not observed, m/z (ESI, pos. ion) 547.8 (M+H)+. GK- GKRP IC50 (Binding) = 0.129 μΜ.
Peak 2: 1H NMR (400MHz, CDC13) δ = 9.06 (d, J= 5.1 Hz, 1 H), 8.48 (s, 1 H), 7.95 (d, J= 7.2 Hz, 1 H), 7.86 - 7.77 (m, 2 H), 7.54 - 7.40 (m, 2 H), 7.30 (s, 1 H), 6.52 (s, 1 H), 6.45 (d, J= 8.6 Hz, 1 H), 6.34 (d, J= 8.8 Hz, 1 H), 4.69 (br. s., 2 H), 3.17 (s, 3 H), 2.24 - 2.11 (m, J= 12.7 Hz, 1 H), 1.21 - 1.08 (m, 2 H), 0.79 - 0.67 (m, 2 H). One proton not observed, m/z (ESI, pos. ion) 547.8 (M+H)+. GK- GKRP IC50 (Binding) = 0.009 μΜ.
Peak 3: 1H NMR (400MHz, CDC13) δ = 9.07 (d, J= 5.5 Hz, 1 H), 8.50 (s, 1 H), 7.97 (d, J= 7.2 Hz, 1 H), 7.87 - 7.79 (m, 2 H), 7.54 - 7.45 (m, 2 H), 7.35 (s, 1 H), 6.64 (br. s., 1 H), 6.50 (d, J= 8.6 Hz, 1 H), 6.37 (d, J= 8.8 Hz, 1 H), 4.90 (br. s., 2 H), 3.19 (s, 3 H), 2.22 (br. s., 1 H), 1.20 - 1.14 (m, 2 H), 0.82 - 0.73 (m, 2 H). One proton not observed, m/z (ESI, pos. ion) 547.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.418 μΜ.
Peak 4: 1H NMR (400MHz, CDC13) δ = 9.06 (d, J= 4.7 Hz, 1 H), 8.49 (s, 1 H), 7.96 (d, J= 7.6 Hz, 1 H), 7.86 - 7.78 (m, 2 H), 7.53 - 7.43 (m, 2 H), 7.33 (s, 1 H), 6.60 (br. s., 1 H), 6.48 (d, J= 8.6 Hz, 1 H), 6.36 (d, J= 8.8 Hz, 1 H), 4.82 (br. s., 2 H), 3.17 (s, 3 H), 2.25 - 2.17 (m, 1 H), 1.19 - 1.12 (m, 2 H), 0.80 - 0.71 (m, 2 H). One proton not observed, m/z (ESI, pos. ion) 547.8 (M+H)+. GK-GKRP IC50 (Binding) = 1.31 μΜ.
Example 103
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide

Step 4
Step 1. To a stirring solution of 7-chlorothieno[2,3-c]pyridine (1.06 g, 6.25 mmol, see Examples 104 and 105, Step 3) in THF (10 mL) at -72 °C under nitrogen was added n-butyllithium, 2.5 M solution in hexanes (2.75 mL, 6.87 mmol) (on the side of round bottom flask) at a rate that did not exceed an internal
temp of -60 °C. To the reaction, after 5 min, was added a solution of di-tert-butyl (6-((E)-(((R)-tert-butylsulfinyl)imino)methyl)-5-chloro-2- pyridinyl)imidodicarbonate (2.87 g, 6.25 mmol, Intermediate AA4) in THF (1 mL). After 20 min the cooling bath was removed, the reaction warmed to -30 °C, then quenched with saturated NH4C1 (4 mL). The reaction was then partitioned between EtOAc (60 mL) and saturated NH4CI (30 mL). The organic was the dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 10 to 50% gradient of
EtO Ac/Hex to afford tert-butyl (6-((((R)-tert-butylsulfmyl)amino)(7- chlorothieno[2,3-c]pyridin-2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (1.27 g) as white solid (mixture of 2 diastereomers).
Step 2. A suspension of tert-butyl (6-((((R)-tert-butylsulfinyl)amino)(7- chlorothieno[2,3-c]pyridin-2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (1.25 g, 2.361 mmol) and hydrochloric acid, 5M (1.889 mL, 9.44 mmol) in MeOH (15 mL) was stirred for 5 h at 25 °C. The reaction was then diluted with CH2CI2 (50 mL), chilled to 0 °C, then basified to pH 14 with 1 M NaOH (14 mL). The separated organic was then dried over MgSC^, filtered, and concentrated under reduced pressure to afford tert-butyl (6-(amino(7-chlorothieno[2,3-c]pyridin-2- yl)methyl)-5-chloro-2-pyridinyl)carbamate (1.1 g) as colorless tar (mixture of enantiomers).
Step 3. A solution of tert-butyl (6-(amino(7-chlorothieno[2,3-c]pyridin- 2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (1.0 g, 2.351 mmol),
cyclopropanesulfonyl chloride (Matrix Scientific, 4.07 mL, 40.0 mmol), DIEA (0.821 mL, 4.70 mmol) in CH2C12 (5 mL) was stirred for 18 h at 25 °C. The reaction was then partitioned between CH2C12 (20 mL) and 5% NaHC03 (20 mL). The organic was then dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 15 to 40% gradient of EtO Ac/hex to afford tert-butyl (5-chloro-6-((7-
chlorothieno[2,3-c]pyridin-2-yl)(cyclopropanesulfonamido)methyl)pyridin-2- yl)carbamate (0.81 g) as yellow foam (mixture of enantiomers).
Step 4. A suspension of tert-butyl (5-chloro-6-((7-chlorothieno[2,3- c]pyridin-2-yl)((cyclopropylsulfonyl)amino)methyl)-2-pyridinyl)carbamate (800 mg, 1.51 1 mmol), 2-(2-chloro-4-pyridinyl)-2-propanol (389 mg, 2.266 mmol, Intermediate Y4), bis(pinacolato) diboron (576 mg, 2.266 mmol),
allylpalladium(II) chloride dimer (55.3 mg, 0.151 mmol), xantphos (175 mg, 0.302 mmol), cesium fluoride (689 mg, 4.53 mmol) in 1 ,4-dioxane (4 mL) and water (1 mL) was sparged with argon for 3 min then heated to 100 °C for 2.5 h. The reaction was then partitioned between EtOAc (50 mL) and 5% NaHC03 (20 mL). The organic was then dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 10 to 50% gradient of EtO Ac/Hex to afford tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)-2-pyridinyl)carbamate (250 mg) as colorless film (mixture of enantiomers).
Step 5. A solution of tert-butyl tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)-2-pyridinyl)carbamate (250 mg, 0.397 mmol) in TFA (10 mL) was stirred for 30 min at 25 °C. The solvent was then removed under reduced pressure and the residue dissolved in MeCN (3 mL) and purified by reverse phase HPLC eluting products with 0 to 100% gradient of MeCN/water (0.1% TFA). The culled fractions were then concentrated under reduced pressure. The resulting residue was then partitioned 9: 1 CHC13/IPA (10 mL) and 1 M K2HP04 (10 mL). The separated organic was then dried over MgS04 and concentrated under reduced pressure to afford N-((6-amino-3-chloro- 2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno [2,3 -c]pyridin-2- yl)methyl)cyclopropanesulfonamide (52 mg) as off white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.73 - 8.80 (m, 2 H) 8.62 (d, J=5.28 Hz, 1 H) 7.98 (d, J=9.00 Hz, 1 H) 7.88 (d, J=5.28 Hz, 1 H) 7.58 (dd, J=5.18, 1.86 Hz, 1 H) 7.55 (d, J=8.80 Hz, 1 H) 7.37 (d, J=0.98 Hz, 1 H) 6.53 (d, J=8.80 Hz, 1 H) 6.29 (s, 2 H) 6.21 (d, J=8.22 Hz, 1 H) 5.42 (s, 1 H) 2.28 - 2.39 (m, 1 H) 1.51 (s, 6 H) 0.85 - 0.96 (m, 2 H) 0.68 - 0.82 (m, 2 H). m/z (APCI, pos. ion) 530.0 (M+l)+. GK-GKRP ICso (Binding) = 0.017 μΜ (mixture of enantiomers).
Examples 104 and 105
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy- l-methylethyl)-2-pyridinyl)thieno [2,3-c] pyridin-3- yl)methyl)cyclopropanesulfonamide and N-((R)-(6-amino-3-chloro-2- pyridinyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-3-yl)methyl)cyclopropanesulfonamide


Step 1. A suspension of thiophene-2-carbaldehyde (Sigma- Aldrich, 30 g, 267 mmol), piperidine (2.67 mL), malonic acid (83.57g, 804 mmol), and pyridine (134 mL) was stirred at 100° C for 16h. The reaction mixture was cooled to 25 °C, slowly quenched with water (300 mL) then acidified with cone. HC1. The resulting precipitate was filtered and re-crystallized from ethanol: water (1 : 1; 100 mL) to afford (2E)-3-(2-thiophenyl)-2-propenoic acid as brown solid (30 g).
Step 2. In a 1 L round bottom flask as suspension of (2E)-3-(2- thiophenyl)-2-propenoic acid (30.0 g, 195 mmol) in CHCI3 (250 mL) was cooled to 0 °C followed by the addition of thionyl chloride (27.8 mL, 234 mmol) and DMF (3 mL, 42 mmol). The reaction was then refluxed for 16 hrs. After cooling, the reaction was concentrated under reduced pressure and the residue partitioned between NaHCC"3 (250 mL) and chloroform (150 mL). The organic layer was dried over Na2S04 and concentrated under reduced pressure to afford (2E)-3-(2- thiophenyl)-2-propenoyl chloride as brown liquid (40 g).
Step 3. To a stirring solution of sodium azide (33.25g, 512 mmol) in a mixture of 1 :1 dioxane/water (160 mL) at 5 °C was added dropwise a solution of (2E)-3-(2-thiophenyl)-2-propenoyl chloride (40.0 g, 256 mmol) in dioxane (150 mL). After 30 min, the organic layer was isolated, concentrated under reduced pressure, and dissolved in CH2C12 (250 mL). This solution was dried over Na2S04, filtered, and the filtrate was added dropwise to refluxing diphenyl ether (200 mL). The resulting reaction mixture was stirred at refluxing temperature for lhr, cooled to 20 °C, then concentrated under reduced pressure. The resulting was triturated with pentane and isolated by filtration to afford thieno[2,3- c]pyridin-7-ol as a brown solid (22 g).
Step 4. A suspension of thieno[2,3-c]pyridin-7-ol (22 g, 146 mmol) in phosphoryl trichloride (100 mL) was stirred at reflux for 2.5hr. The reaction mixture was cooled to 20 °C, poured into ice water (500 mL), stirred for 30 min, then extracted into CH2C12 (3 x 100 mL). The organic layer was dried over Na2S04, filtered, then concentrated under reduced pressure to afford 7- chlorothieno[2,3-c]pyridine as brown solid (14 g).
Step 5. In a 250 mL sealed tube 7-chlorothieno[2,3-c]pyridine (12. Og g, 71 mmol) and 2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol (24 g, 107 mmol) was dissolved in a 4: 1 mixture of 1 ,4-dioxane/water (30 mL) at 20 °C under an argon atmosphere. A stream of argon was bubbled through the solution
for 10 min followed by the addition of Pd2(allyl)2Cl2 (2.59 g, 7.1 mmol), X-Phos (8.21 g, 14.2 mmol) bis(pinacolato)diboron (27 g, 107 mmol) and cesium fluoride (32.3 g, 214 mmol). The tube was sealed and mixture gradually heated to 100 °C for 16 h with stirring. The reaction mixture was cooled and partitioned between water (100 mL) and EtOAc (300 mL). The organic layer was separated, washed with water (100 mL), brine (100 mL) and dried over anhydrous Na2S04. The organic was concentrated under reduced pressure then purified by silica gel chromatography eluting products with isocratic 3% EtOAc/ Hex to afford 1,1,1- trifluoro-2-(2-thieno[2,3-c]pyridin-7-yl-4-pyridinyl)-2-propanol as off white solid (1.8 g).
Step 6. To a solution of 3,6-dichloro-2-pyridinecarboxylic acid (10 g, 52 mmol, Matrix Scientific USA) in THF (100 mL) at -60 °C under nitrogen was added DIBAL-H (1M in THF, 110 mL, 110 mmol) and the reaction mixture was stirred for 3 h at -60 °C. The reaction mixture was quenched with saturated NH4C1 (35 mL), diluted with water (50 mL), and extracted with EtOAc (100 mL). The aqueous was further extracted with EtOAc (50 mL x 2). The combined organics were then dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford 3,6-dichloro-2-pyridinecarbaldehyde as yellow solid (5.0 g).
Step 7. A suspension of 3,6-dichloro-2-pyridinecarbaldehyde (5.0 g, 28 mmol), (S)-2-methylpropane-2-sulfinamide (4.13 g, 34 mmol, Combi-blocks, USA), titanium ethoxide (32.38 g, 142 mmol) in CH2C12 (50 mL) was stirred for 18 h at 20 °C. The reaction mixture was cooled to 0 °C then quenched with water (120 mL). The resulting solid was isolated by a diatomaceous earth pad and washed with CH2C12 (50 mL x 2). The organic was isolated by separation and extracted with saturated NaCl (50 mL x 2). The organic was then dried over anhydrous sodium sulfate, concentrated under reduced pressure, then purified by silica gel chromatography eluting products with 0 to 10% gradient of
EtOAc/Et20 to afford S-N-((3,6-dichloro-2-pyridinyl)methylidene)-2-methyl-2- propanesulfinamide as pale yellow solid.(5.0 g).
Step 8. To a stirring solution of 1 , 1 , 1 -trifluoro-2-(2-thieno[2,3-c]pyridin- 7-yl-4-pyridinyl)-2-propanol (1.55 g, 4.78 mmol) in THF (26 mL) at -70 °C was added n-butyllithium, 2.5M solution in hexanes (4.21 mL, 10.5 mmol). The resulting dark red suspension was removed from cooling bath and warmed to -35 °C. The suspension was then returned to -60 °C and a solution of S-N-((3,6- dichloro-2-pyridinyl)methylidene)-2-methyl-2-propanesulfinamide (1.601 g, 5.74 mmol) in THF (10 mL) added which warmed the internal temperature to -50 °C. After 20 min the reaction was then quenched with saturated NH4CI (15 mL). After warming to 5 °C the reaction was diluted with EtOAc (75 mL). The organic was isolated, dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 0 to 100% gradient of EtO Ac/Hex to afford a mixture of (S)-N-((3,6-dichloro-2- pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno[2,3- c]pyridin-3-yl)methyl)-2-methyl-2-propanesulfinamide (2.17 g) as yellow foam from diethylether (mixture of 4 diastereomers).
Step 9. A solution of (S)-N-((3,6-dichloro-2-pyridinyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-3- yl)methyl)-2-methyl-2-propanesulfinamide (1.59 g, 2.63 mmol) in MeOH (25 mL) was added 5 N HC1 (5 mL; 25 mmol). The suspension was stirred for 15 min to become a solution. After 90 min, the reaction was then chilled to 0 °C with external wet ice bath then basified with 1 N NaOH ( mL). To the suspension was added 9: 1 CHCI3/IPA (30 mL). The organic was separated, dried over MgS04, filtered, then concentrated under reduced pressure to afford 2-(2-(3- (amino(3,6-dichloro-2-pyridinyl)methyl)thieno[2,3-c]pyridin-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (1.33 g) as yellow foam (mixture of 4 diastereomers).
Step 10. To a 150 mL round-bottmed flask charged with 2-(2-(3- (amino(3,6-dichloro-2-pyridinyl)methyl)thieno[2,3-c]pyridin-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (1.03 g, 2.063 mmol) and stirring under nitrogen at 0 °C was added Boc-anhydride (10.31 mL, 10.31 mmol) [1 M in THF]. The cooling bath was removed and stirred for 45 min. The reaction mixture was purified directly, without aqueous workup, by silica gel chromatography (120 g) eluting products with 0 to 80% gradient of EtO Ac/Hex to afford tert-butyl ((3,6- dichloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-3-yl)methyl)carbamate (0.98 g) as amber oil (mixture of 4 diastereomers).
Step 11. A suspension of tert-butyl ((3,6-dichloro-2-pyridinyl)(7-(4- (2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-3- yl)methyl)carbamate (710 mg, 1.184 mmol), 4-methoxybenzylamine (615 μί, 4.74 mmol), (xphos) palladium(II) phenethylamine chloride (87 mg, 0.118 mmol), cesium carbonate (1554 mg, 4.74 mmol) in 1,4-dioxane (5 mL) was sparged with argon for 2 min then heated to 100 °C with stirring for 10 h. The reaction was then partitioned between EtO Ac (60 mL) and 5% NaHC03 (20 mL). The organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 30 to 70%> gradient of EtO Ac/Hex to afford tert-butyl ((3-chloro-6-((4- methoxybenzyl)amino)-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)thieno[2,3-c]pyridin-3-yl)methyl)carbamate (460 mg) as colorless oil (mixture of 4 diastereomers).
Step 12. A solution of tert-butyl ((3-chloro-6-((4-methoxybenzyl)amino)- 2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-3-yl)methyl)carbamate (460 mg, 0.657 mmol) in TFA (5 mL) was stirred for 2 h at 25 °C. The solution was then concentrated under reduced pressure and the residue partitioned between 9: 1 CHC13 (30 mL) and 1 M K2HP04 (50 mL). The organic was dried over MgS04, filtered, then
concentrated under reduced pressure to afford 2-(2-(3-(amino(6-amino-3-chloro- 2-pyridinyl)methyl)thieno[2,3-c]pyridin-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (286 mg) as red foam (mixture of 4 diastereomers).
Step 13. To a stirring suspension of 2-(2-(3-(amino(6-amino-3-chloro-2- pyridinyl)methyl)thieno[2,3-c]pyridin-7-yl)-4-pyridinyl)-l , 1 , 1 -trifluoro-2- propanol (286 mg, 0.596 mmol) and DIEA (114 μΐ,, 0.656 mmol) was added cyclopropanesulfonyl chloride (Matrix Scientific, 182 μΐ^, 1.788 mmol). After 18 h the suspension remained; IP A (2 mL) and DMF (1 mL) added to create a solution. After 3 h, the reaction was then partitioned between CH2CI2 (30 mL) and 5% NaHC03 (30 mL). The organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 50% gradient of MeCN/CH2C12 to afford N-((6-amino-3- chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide (175 mg) as colorless oil (mixture of 4 diastereomers).
The material above was subjected to two preparative SFC methods to separated the 4 diastereomers: Method #1 (Chiralpak® AD-H column; 25 cm x 21 mm, 5 μιη) eluting with 65% liquid C02 in 35% isopropanol (with 20 mM diethylamine) at a flow rate of 70 mL/min) and Method #2 (Chiralpak® IC column; 25 cm x 21 mm, 5 μιη) eluting with 67% liquid C02 in 33% isopropanol (with 20 mM diethylamine) at a flow rate of 70 mL/min) to afford two major components:
1H NMR (400 MHz, DMSO-d6) δ ppm 8.78 (d, J=5.28 Hz, 1 H) 8.61 (s, 1 H) 8.54 (d, J=5.48 Hz, 1 H) 8.48 (s, 1 H) 8.09 (d, J=5.48 Hz, 1 H) 7.95 (d, J=8.61 Hz, 1 H) 7.67 (d, J=4.89 Hz, 1 H) 7.51 (d, J=8.80 Hz, 1 H) 7.00 (s, 1 H) 6.49 (d, J=8.80 Hz, 1 H) 6.26 (br. s., 2 H) 6.22 (d, J=8.61 Hz, 1 H) 2.29 - 2.40 (m, 1 H) 1.75 (s, 3 H) 0.84 - 0.97 (m, 2 H) 0.69 - 0.83 (m, 2 H)
m z (APCI, pos. ion) 584.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.428 μΜ.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.78 (d, J=5.28 Hz, 1 H) 8.61 (s, 1 H) 8.54 (d, J=5.48 Hz, 1 H) 8.48 (s, 1 H) 8.09 (d, J=5.48 Hz, 1 H) 7.95 (d, J=8.61 Hz, 1 H) 7.67 (d, J=4.89 Hz, 1 H) 7.51 (d, J=8.80 Hz, 1 H) 7.00 (s, 1 H) 6.49 (d, J=8.80 Hz, 1 H) 6.26 (br. s., 2 H) 6.22 (d, J=8.61 Hz, 1 H) 2.29 - 2.40 (m, 1 H) 1.75 (s, 3 H) 0.84 - 0.97 (m, 2 H) 0.69 - 0.83 (m, 2 H)
m/z (APCI, pos. ion) 584.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.239 μΜ.
Example 106
N-((R)-(2-ethynylphenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. To a stirring solution of 2-((trimethylsilyl)ethynyl)benzaldehyde (Sigma-Aldrich, 23.0 g, 114 mmol) in CH2C12 (230 mL), was added (S)-2- methylpropane-2-sulfinamide (16.53 g, 137 mmol), Ti(OEt)4 (129.8 g, 569
mmol) and stirred at 20 °C for 18 h. The reaction mixture was then cooled to 0 °C and quenched with water (300 mL). The solids were isolated on a diatomaceous earth pad and washed with DCM (150 mL x 2).The organic layer was isolated and extracted with saturated NaCl (150 mL x 2). The organic layer was dried over sodium sulfate, concentrated under reduced pressure then purified by silica gel chromatography eluting products with a 0 to 7% gradient of EtOAc/Et20 to afford (S,E)-2-methyl-N-(2-((trimethylsilyl)ethynyl)benzylidene)propane-2- sulfmamide as off white solid (28.0 g).
Step 2. To a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (5.29 g, 16.4 mmol, Intermediate Y3) in THF (100 mL) at -78 °C under a nitrogen atmosphere n-butyllithium, 2.5 M solution in hexanes (13.1 mL, 32.8 mmol) was added slowly. After stirring for 10 min, a solution of (S,E)-2-methyl-N-(2-
((trimethylsilyl)ethynyl)benzylidene)propane-2-sulfinamide (5.0 g, 16.4 mmol) in THF (50 mL) was added and reaction stirred for 2 h at -78 °C. The reaction was quenched with sodium bicarbonate (50 mL) and reaction extracted with EtOAc (2 x 100 mL). The organics were then dried over anhydrous sodium sulfate, concentrated under reduced pressure. Then purified by silica gel chromatography eluting products with 0 to 40% gradient of EtOAc/Et20 to afford 2-methyl- N- (( 1 E)-(-((7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)(2-((trimethylsilyl)ethynyl)phenyl)methyl)-2- propanesulfmamide as off white solid.(3.2 g). Mixture of 4 diastereomers.
Step 3. 2-methyl- N-((lE)-(-((7-(4-(2,2,2-trifiuoro-l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)(2- ((trimethylsilyl)ethynyl)phenyl)methyl)-2-propanesulfinamide as off white solid.(3.2 g). Mixture of 4 diastereomers.
was subjected to preparative SFC (Chiralpak® ADH column; 25 cm x 30 mm, 5 μιη) eluting with 65% liquid C02 in 35%> isopropanol (with 20 mM NH3) at a flow rate of 120 mL/min) to afford 2-methyl-N-((R)-(7-(4-((lR)-2,2,2-trifiuoro-
1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)(2- ((trimethylsilyl)ethynyl)phenyl)methyl)-2-propanesulfinamide (1050 mg) as white solid (carbinol stereochemistry is arbitrarily assigned).
Step 4. To a stirring solution of (S)-2-methyl-N-((R)-(7-(4-((lR)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)(2- ((trimethylsilyl)ethynyl)phenyl)methyl)-2-propanesulfinamide (1050 mg, 1.670 mmol) in MeOH (5 mL) at 25 °C was added HC1 (5 N; 2 mL). After 18 h, the reaction was diluted with CH2CI2 (80 mL) and then extracted with NaOH (1 M; 20 mL). The separated organic was then concentrated under reduced pressure then purified by silica gel chromatography (80 g) eluting with 2 to 5% gradient of 2M NH3 in MeOH/CH2Cl2 to afford (2R)-2-(2-(2-((R)-amino(2- ((trimethylsilyl)ethynyl)phenyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifluoro-2-propanol (780 mg) as colorless oil.
Step 5. To a stirring solution of (2R)-2-(2-(2-((R)-amino(2- ((trimethylsilyl)ethynyl)phenyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)- l,l,l-trifiuoro-2-propanol (780 mg, 1.487 mmol) and DIEA (286 μί, 1.635 mmol) in CH2C12 (10 mL) at 25 °C was added cyclopropanesulfonyl chloride (Matrix Scientific, 606 μί, 5.95 mmol) followed by DMAP (18.16 mg, 0.149 mmol). After stirring for 3 days at 20 °C, the reaction was then diluted with CH2C12 (20 mL) and extracted with 5% NaHC03 (15 mL). The organic was dried over MgSC^, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 20% gradient of MeCN/CH2Cl2 to N-((R)-(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)(2-
((trimethylsilyl)ethynyl)phenyl)methyl)cyclopropanesulfonamide (692 mg) as amber oil.
Step 6. To a stirring solution of N-((R)-(7-(4-((lR)-2,2,2-trifhioro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)(2-
((trimethylsilyl)ethynyl)phenyl)methyl)cyclopropanesulfonamide (680 mg, 1.081 mmol) in THF (5 mL) at 0 °C under nitrogen was added tetrabutylammonium fluoride, 1.0M solution in tetrahydrofuran (1081 μί, 1.081 mmol). After 10 min, the reaction was diluted with EtOAc (20 mL) and extracted twice with 5% NaHCC"3 (2x30 mL). The organic was then dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 15% gradient of MeCN/CH2Cl2 to afford N- ((R)-(2-ethynylphenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (298 mg) as amber film.
1H NMR (400 MHz, CHLOROFORM-;/) δ ppm 8.81 (d, J=5.28 Hz, 1 H) 8.15 (s, 1 H) 7.87 (d, J=7.43 Hz, 1 H) 7.87 (d, J=7.43 Hz, 1 H) 7.58 (dd, J=7.24, 3.91 Hz, 2 H) 7.45 (t, J=7.63 Hz, 3 H) 7.32 - 7.40 (m, 1 H) 7.02 (d, J=0.98 Hz, 1 H) 6.39 (d, J=8.61 Hz, 1 H) 5.75 (d, J=8.61 Hz, 1 H) 3.36 (s, 1 H) 2.63 - 2.79 (m, 1 H) 2.12 - 2.27 (m, 1 H) 1.84 (s, 3 H) 1.11 (q, J=4.63 Hz, 2 H) 0.64 - 0.88 (m, 2 H). m/z (APCI, pos. ion) 557.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.050 μΜ.
Example 107
2-(2-((6-amino-3-chloro-2-pyridinyl)((cyclopropylsulfonyl)amino)methyl)-l- benzothiophen-7-yl)-4-pyridinesulfonamide

Step 1. A 250 mL round-bottomed flask was charged with 4-bromo-2- chloropyridine (1.07 g, 5.56 mmol), diethylether (20 mL), and magnetic stirrer, a Teflon® thermocouple, and equipped with a nitrogen line. After cooling to -70 °C, n-butyllithium, 2.5 M solution in hexanes (2.224 mL, 5.56 mmol) was added at a rate not to exceed -60 °C (a white precipitate formed). After 5 min at -70°C, sulfur dioxide (0.285 mL, 5.56 mmol) gas was bubbled through the solution for 2 min (exotherm noticed to -50 °C). The mixture was allowed to warm to 0 °C then the mixture was concentrated under reduced pressure. To the slurry was added CH2CI2 (20 mL) followed by n-chlorosuccinimide (0.742 g, 5.56 mmol). The suspension was stirred for 30 min at 25 °C; reaction suspension turned to a purple hue. The reaction suspension was then chilled to 0 °C and 4- methoxybenzylamine (2.89 mL, 22.24 mmol) added followed by DIEA (3.88 mL, 22.24 mmol). The cooling bath was removed and suspension stirred for 90 min warming to 25 °C. The suspension was then partitioned between CH2CI2 (30 mL) and 5% NaHC03 (50 mL). The organic was dried over MgS04,
concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 20 to 50% gradient of EtO Ac/Hex to afford 2-
chloro-N-(4-methoxybenzyl)-4-pyridinesulfonamide (1.16 g) as yellow solid (mixture of enantiomers).
Step 2. To a stirring solution of diisopropylamine (2.374 g, 23.46 mmol) in THF (12 mL) at -70 °C under nitrogen was added n-butyllithium, 2.5 M solution in hexanes (8.26 mL, 20.65 mmol). After 15 min at -70 °C was added 7- bromo-l-benzothiophene (Intermediate X13, 4.0 g, 18.77 mmol) in THF (10 mL). The cooling bath was removed and reaction warmed to -30 °C then returned to -70 °C. To the colorless solution was added (6-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-5-chloro-2-pyridinyl)imidodicarbonate (8.63 g, 18.77 mmol, Intermediate AA4) in THF (15 mL) at a rate not to exceed -60 °C. After 18 h warming to 20 °C, the reaction was then quenched with saturated NH4CI (20 mL). The reaction was then partitioned between EtO Ac (120 mL) and saturated NH4C1 (50 mL). The organic was the dried over MgS04, concentrated onto dry silica (30 g) under reduced pressure, then purified by silica gel chromatography (330 g) eluting products with 10 to 50% gradient of EtO Ac/Hex to afford tert-butyl (6-((7-bromo-l-benzothiophen-2-yl)(((S)-tert- butylsulfinyl)amino)methyl)-5-chloro-2-pyridinyl)carbamate as white solid (mixture of 2 diastereomers). m/z (APCI, pos. ion) 572.0/574.0 (M+l)+.
Step 3. A suspension of tert-butyl (6-((7-bromo-l-benzothiophen-2- yl)(((S)-tert-butylsulfinyl)amino)methyl)-5 -chloro-2-pyridinyl)carbamate (550 mg, 0.960 mmol), bis(pinacolato) diboron (293 mg, 1.152 mmol), [1 , 1- bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (78 mg, 0.096 mmol), potassium acetate (188 mg, 1.920 mmol) in 1 ,4-dioxane (5 mL) was sparged with argon for 1 min then placed in a 120 °C sand bath for 30 min. The reaction was then partitioned between EtO Ac (30 mL) and 5% NaHC03 (10 mL). The organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 50%> gradient of EtO Ac/Hex to afford tert-butyl (6-(((S-tert- butylsulfinyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -
benzothiophen-2-yl)methyl)-5-chloro-2-pyridinyl)carbamate (550 mg) as purple brittle foam (mixture of diastereomers).
Step 4. A suspension of (6-(((S-tert-butylsulfinyl)amino)(7-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-l-benzothiophen-2-yl)methyl)-5-chloro-2- pyridinyl)carbamate (150 mg, 0.242 mmol), 2-chloro-N-(4-methoxybenzyl)-4- pyridinesulfonamide (91 mg, 0.290 mmol),
tetrakis(triphenylphosphine)palladium(o) (28.0 mg, 0.024 mmol), sodium carbonate (103 mg, 0.968 mmol) in 1,4-dioxane (0.5 mL) and water (0.25 mL) was heated to 100 °C for 30 min. The reaction was then partitioned between EtOAc (15 mL) and 5% NaHC03 (5 mL). The organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 30 to 80% gradient of EtO Ac/Hex to afford tert- butyl (6-(((S-tert-butylsulfinyl)amino)(7-(4-((4-methoxybenzyl)sulfamoyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-2-pyridinyl)carbamate (158 mg) as white foam (mixture of 2 diastereomers).
Step 5. A suspension of tert-butyl (6-(((S-tert-butylsulfinyl)amino)(7-(4- ((4-methoxybenzyl)sulfamoyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 - chloro-2-pyridinyl)carbamate (150 mg, 0.195 mmol) was stirred in MeOH (10 mL) and aqueous HC1 (5 M; 1 mL) for 18 h at 25 °C. The reaction became a solution then chilled to 0 °C, CH2CI2 (10 mL) added, and pH adjusted to 13 with NaOH (1 M; 6 mL). The reaction was then diluted with more CH2CI2 (20 mL). The separated organic was then dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 50% gradient of MeCN/CH2Cl2 to afford tert-butyl (6-(amino(7-(4-((4- methoxybenzyl)sulfamoyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro- 2-pyridinyl)carbamate (85 mg) as colorless tar (mixture of enantiomers).
Step 6. To a stirring solution of tert-butyl (6-(amino(7-(4-((4- methoxybenzyl)sulfamoyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-
2-pyridinyl)carbamate (85 mg, 0.128 mmol) in CH2CI2 (1 mL) was added 1 M NaOH (1 mL) and cyclopropanesulfonyl chloride (Matrix Scientific, 130 μί, 1.276 mmol). The reaction was stirred for 48 h at 25 °C. The reaction was then diluted with CH2CI2 (10 mL) and 1 M NaOH (5 mL). The separated organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 20 to 60% EtO Ac/Hex to afford tert-butyl (5-chloro-6-(((cyclopropylsulfonyl)amino)(7-(4-((4- methoxybenzyl)sulfamoyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2- pyridinyl)carbamate (58 mg) as colorless film (mixture of enantiomers). m/z (APCI, pos. ion) 770.0 (M+l)+.
Step 7. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-((4-methoxybenzyl)sulfamoyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (55 mg, 0.071 mmol) in TFA (2 mL) was stirred for 4 days at 25 °C. The solvent was removed under reduced pressure and residue partitioned between CH2C12 (15 mL) and 1 M K2HPO4 (10 mL). The organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 1 to 4% gradient of MeOH/CH2Cl2 to afford 2-(2-((6-amino-3- chloro-2-pyridinyl)((cyclopropylsulfonyl)amino)methyl)-l-benzothiophen-7-yl)- 4-pyridinesulfonamide (25 mg) as white foam.
1H NMR (400 MHz, DMSO- 6) δ ppm 8.96 - 9.10 (m, 1 H) 8.53 (s, 1 H 8.11 (d, J=7.04 Hz, 1 H) 7.97 (d, J=7.24 Hz, 1 H) 7.89 (d, J=8.80 Hz, 1 H) 7.72 - 7.82 (m, 3 H) 7.48 - 7.63 (m, 2 H) 7.25 - 7.33 (m, 1 H) 6.44 - 6.56 (m, 1 H) 6.14 (dd, J=8.80, 0.78 Hz, 1 H) 2.24 - 2.37 (m, 1 H) 0.84 - 0.92 (m, 2 H) 0.74 - 0.82 (m, 1 H) 0.65 - 0.74 (m, 1 H). m/z (APCI, pos. ion) 550.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.078 μΜ (mixture of 2 enantiomers).
Example 108
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(3-hydroxytetrahydro-3-furanyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a stirring solution of 4-bromo-2-chloropyridine (1.154 mL, 10.39 mmol) in diethyl ether (40 mL) at -70 °C was added n-butyllithium, 2.5 M solution in hexanes (4.57 mL, 11.43 mmol) at a rate that did not exceed an internal temperature of -65 °C. After stirring the resulting suspension for 5 min a solution of 3 -oxotetrahydrofuran (CombiBlocks, Inc., 0.984 g, 11.43 mmol) in diethyl ether (4 mL) was added at a rate that did not exceed an internal temperature of 60 °C. The suspension was stirred for 15 min then warmed to -50 °C at which point it was quenched with saturated NH4C1 (20 mL). To the reaction was added water (20 mL). The separated organic was then dried onto silica gel (20 g) under reduced pressure. The product was then purified by silica gel chromatography (80 g) eluting products with 0 to 5% gradient of
MeOH/CH2Cl2 to afford 3-(2-chloro-4-pyridinyl)tetrahydro-3-furanol (1.0 g) as colorless oil (mixture of 2 enantiomers).
Step 2. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (250 mg, 0.403 mmol, Intermediate AA7), 3-(2-chloro-4-pyridinyl)tetrahydro-3-furanol (89 mg, 0.444 mmol), 1 , 1 '-bis(diphenylphosphino)ferrocene-palladium(II)dichloride
dichloromethane adduct (32.9 mg, 0.040 mmol), sodium carbonate (171 mg, 1.613 mmol) in 1,4-dioxane (1.5 mL) and water (1 mL) was sparged with argon for 1 min the heated to 100 °C with stirring. The reaction was then partitioned between EtOAc (15 mL) and 5% NaHC03 (15 mL). The separated organic was then dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 20 to 60% gradient of EtO Ac/Hex to afford tert-butyl (5-chloro-6-(((cyclopropylsulfonyl)amino)(7-(4- (3 -hydroxytetrahydro-3 -furanyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- pyridinyl)carbamate (155 mg) as colorless film (mixture of 4 diastereomers).
Step 3. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(3-hydroxytetrahydro-3-furanyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (150 mg, 0.228 mmol) in TFA (5 mL) was stirred for 30 min at 20 °C. The solvent was removed under reduced pressure and residue partitioned between CH2CI2 (15 mL) and 1 M K2HP04 (10 mL). The organic was dried over MgS04 and concentrated under reduced pressure to afford a colorless foam. This foam was suspended in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford N-((6-amino-3-chloro- 2-pyridinyl)(7-(4-(3 -hydroxytetrahydro-3 -furanyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)methyl)cyclopropanesulfonamide (105 mg) as white fluffy solid (mixture of 4 diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.74 (d, J=5.09 Hz, 1 H) 8.23 (s, 1 H) 8.08 (d, J=7.24 Hz, 1 H) 7.88 (d, J=7.24 Hz, 1 H) 7.47 - 7.55 (m, 3 H) 7.76 (d, J=9.00 Hz, 1 H) 7.25 (d, J=0.78 Hz, 1 H) 6.49 (d, J=8.80 Hz, 1 H) 6.26 (br. s., 2 H) 6.12 (dd, J=9.00, 0.78 Hz, 1 H) 5.75 (br. s., 1 H) 4.01 - 4.11 (m, 2 H) 3.81 - 3.91 (m, 2 H) 2.39 (dt, J=12.62, 8.95 Hz, 1 H) 2.23 - 2.33 (m, 1 H) 2.18 (dt, J=12.67, 4.91 Hz, 1 H) 0.83 - 0.92 (m, 2 H) 0.72 - 0.81 (m, 1 H) 0.63 - 0.72 (m, 1 H). m/z (APCI, pos. ion) 557.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.009 μΜ
Example 109
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l-hydroxy-l-methylpropyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a stirring solution l-(2-chloro-4-pyridinyl)ethanone (Sigma- Aldrich, 415 mg, 2.67 mmol) in THF (4 mL) at 0 °C under nitrogen was added ethylmagnesium bromide, 3.0 M solution in diethyl ether (978 μί, 2.93 mmol) over a 1 minute period. After 30 min the reaction was quenched with saturated NH4CI (2 mL). The reaction was then partitioned between 5% NaHC03 (5 mL) and EtOAc (10 mL). The organic was dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 40% gradient of EtO Ac/Hex to afford 2-(2-chloro-4- pyridinyl)-2-butanol (80 mg) as colorless oil (mixture of 2 enantiomers).
Step 2. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (200 mg, 0.323 mmol, Intermediate AA7), 2-(2-chloro-4-pyridinyl)-2-butanol (71.9 mg, 0.387 mmol), 1 , 1 '-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane adduct (26.3 mg, 0.032 mmol), sodium carbonate (137 mg, 1.290 mmol) in 1,4- dioxane (2 mL) and water (0.75 mL) was sparged with argon for 30 seconds then heated to 100 °C for 20 min. The reaction was then partitioned between EtOAc (15 mL) and extracted with 5% NaHC03 (5 mL). The separated organic was dried over MgSC^, concentrated under reduced pressure, then purified by silica
gel chromatography (40 g) eluting products with 0 to 50% gradient of
EtO Ac/Hex to afford tert-butyl (5-chloro-6-(((cyclopropylsulfonyl)amino)(7-(4- ( 1 -hydroxy- 1 -methylpropyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- pyridinyl)carbamate (86 mg) as white solid (mixture of 4 diastereomers).
Step 3. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-( 1 -hydroxy- 1 -methylpropyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (85 mg, 0.132 mmol) in TFA (5 mL) was stirred for 30 min at 25 °C. The solvents were then removed under reduced pressure and the residue partitioned between 9: 1 CHCI3/IPA (10 mL) and 1 M K2HP04 (10 mL). The separated organic was then dried over MgS04, filtered, and concentrated under reduced pressure to afford an amber film. This film was then dissolved in MeCN (1 mL) and water (1 mL) added. This suspension was then frozen and lyophilized to afford N-((6-amino-3-chloro-2- pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylpropyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (60 mg) as white fluffy powder (mixture of 4 diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.72 (d, J=5.28 Hz, 1 H) 8.17 (s, 1 H) 8.07 (d, J=6.85 Hz, 1 H) 7.90 (d, J=7.24 Hz, 1 H) 7.78 (d, J=9.00 Hz, 1 H) 7.49 - 7.56 (m, 2 H) 7.45 (dd, J=5.18, 1.47 Hz, 1 H) 7.27 (d, J=0.78 Hz, 1 H) 6.51 (d, J=8.80 Hz, 1 H) 6.28 (br. s., 2 H) 6.14 (dd, J=9.00, 0.78 Hz, 1 H) 5.17 (br. s., 1 H) 2.24 - 2.35 (m, 1 H) 1.71 - 1.90 (m, 2 H) 1.50 (s, 3 H) 0.84 - 0.94 (m, 2 H) 0.64 - 0.83 (m, 5 H). m/z (APCI, pos. ion) 543.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.023 μΜ.
Example 110
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(methylsulfinyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a stirring solution of 4-bromo-2-chloropyridine (Sigma- Aldrich, 1.442 mL, 12.99 mmol) in diethyl ether (40 mL) at -70 °C under nitrogen was added n-butyllithium, 2.5 M solution in hexanes (5.72 mL, 14.29 mmol) at a rate that did not exceed an internal temp of -50 °C. The resulting thick white suspension was stirred for 15 min at -70 °C then methyl disulfide (1.755 mL, 19.49 mmol) in diethyl ether (4 mL) was added. After 15 min the reaction was quenched with saturated NH4C1 (10 mL) and reaction warmed to 20 °C over 18 h. The organic was then dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 10 to 20% gradient of EtO Ac/Hex to afford 2-chloro-4- (methylsulfanyl)pyridine (1.72 g) as colorless oil
Step 2. To a stirring solution of 2-chloro-4-(methylsulfanyl)pyridine (410 mg, 2.57 mmol) in CH2CI2 (10 mL) at 25 °C was added 3-chloroperoxybenzoic acid, 77% max. (863 mg, 3.85 mmol) in one portion. After 10 min the reaction was diluted with CH2C12 (10 mL) and extracted with 1 M HC1 (10 mL). The organic was then dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 100%
gradient of MeCN/CH2Cl2 to afford 2-chloro-4-(methylsulfmyl)pyridine (150 mg) as colorless oil.
Step 3. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (100 mg, 0.161 mmol, Intermediate AA7), 2-chloro-4-(methylsulfinyl)pyridine (85 mg, 0.484 mmol), tetrakis(triphenylphosphine)palladium(o) (18.64 mg, 0.016 mmol), sodium carbonate (68.4 mg, 0.645 mmol) in 1,4-dioxane (1 mL) and water (0.5 mL) was sparged with argon for 30 seconds then heated to 100 °C for 10 min. The reaction was then diluted with EtO Ac (5 mL) and extracted with 1 M K2HP04 (10 mL). The separated organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 20 to 80% gradient of EtO Ac/Hex to afford tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(methylsulfinyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (86 mg) as colorless film (mixture of 2 enantiomers).
Step 4. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(methylsulfinyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (86 mg, 0.136 mmol) in TFA (5 mL) was stirred for 45 min at 25 °C. The solvent was then removed under reduced pressure and residue partitioned between CH2C12 (15 mL) and 1 M K2HP04 (10 mL). The organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 1 to 3% gradient of MeOH/CH2Cl2 to afford N-((6-amino-3- chloro-2-pyridinyl)(7-(4-(methylsulfinyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide (44 mg, 0.083 mmol, 60.8 % yield) as colorless film (mixture of 2 enantiomers). 1H NMR (400 MHz, DMSO- 6) δ ppm 8.92 (d, J=5.09 Hz, 1 H) 8.37 (s, 1 H) 8.10 (d, J=7.04 Hz, 1 H) 7.88 (d, J=7.24 Hz, 1 H) 7.79 (d, J=9.00 Hz, 1 H) 7.66 (dd, J=5.18, 1.47 Hz, 1 H) 7.41 -
7.51 (m, 2 H) 7.21 (d, J=0.78 Hz, 1 H) 6.42 (d, J=8.80 Hz, 1 H) 6.22 (br. s., 2 H) 6.07 (dd, J=9.00, 0.78 Hz, 1 H) 2.87 (s, 3 H) 2.17 - 2.25 (m, 1 H) 0.75 - 0.86 (m, 2 H) 0.66 - 0.74 (m, 1 H) 0.58 - 0.66 (m, 1 H). m/z (APCI, pos. ion) 533.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.219 μΜ Example 111
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(methylsulfonyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a stirring solution of 2-chloro-4-(methylsulfanyl)pyridine (1.25 g, 7.83 mmol) in CH2C12 (100 mL) at 25 °C was added 3- chloroperoxybenzoic acid, 77% max. (5.40 g, 31.3 mmol) in 4 portions over 2 min. The reaction was stirred for 45 min at 25 °C then filtered. The filtrate was then dried onto dry silica (20 g) by removing solvent under reduced pressure. The product was then purified by silica gel chromatography (120 g) eluting products with a 0 to 30% gradient of MeCN/CH2Cl2 to afford a white solid having some 3-chlorobenzoic acid. This material was then partitioned between CH2C12 (50 mL) and 1 M NaOH (50 mL). The organic portion was then dried over MgS04, filtered, and concentrated under reduced pressure to afford 2- chloro-4-(methylsulfonyl)pyridine (1.25 g) as white solid.
Step 2. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (100 mg, 0.161 mmol, Intermediate AA7), 2-chloro-4-(methylsulfonyl)pyridine (61.8 mg, 0.323 mmol), tetrakis(triphenylphosphine)palladium(o) (18.64 mg, 0.016 mmol), sodium carbonate (68.4 mg, 0.645 mmol) in 1,4-dioxane (1 mL) and water (0.5 mL) was sparged with argon for 15 seconds then heated to 100 °C for 15 min. The reaction was then diluted with EtOAc (5 mL) and extracted with 5% NaHC03 (5 mL). The separated organic was dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 0 to 10% gradient of MeCN/CH2Cl2 to afford tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(methylsulfonyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (78 mg) as white solid (mixture of 2 enantiomers).
Step 3. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(methylsulfonyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (78 mg, 0.120 mmol) in TFA (5 mL) was stirred for 15 min at 25 °C. The solvent was then removed under reduced pressure and residue partitioned between CH2C12 (15 mL) and 1 M K2HPC"4 (10 mL). The organic was dried over MgSC^ and concentrated under reduced pressure to afford N-((6-amino-3-chloro-2-pyridinyl)(7-(4- (methylsulfonyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (68 mg) as colorless film. The film was then suspended in MeCN/water (1 : 1; 2 mL), frozen, and lyophilized to afford desired compound as light fluffy powder (mixture of 2 enantiomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 9.13 (dd, J=5.09, 0.59 Hz, 1 H) 8.64 (s, 1 H) 0.63 - 0.72 (m, 1 H) 8.27 (d, J=7.04 Hz, 1 H) 7.98 (d, J=7.04 Hz, 1 H) 7.82 - 7.93 (m, 2 H) 7.47 - 7.62 (m, 2 H) 7.29 (d, J=0.98 Hz, 1 H) 6.49 (d, J=8.80 Hz, 1 H) 6.21 - 6.39 (m, 2 H) 6.13 (dd, J=9.00, 0.78 Hz, 1 H) 2.28 (s, 1 H) 3.45 (s, 3 H)
0.81 - 0.93 (m, 2 H) 0.72 - 0.81 (m, 1 H). m/z (APCI, pos. ion) 549.0 (M+l) GK-GK P IC50 (Binding) = 0.256 μΜ.
Example 112
N-((2-chlorophenyl)(7-(4-(l-cyclopropyl-l-hydroxyethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a stirring solution of 4-bromo-2-chloropyridine (2.0 g, 10.39 mmol) in diethyl ether (30 mL) at -70 °C under nitrogen was added n- butyllithium, 2.5 M solution in hexanes (4.57 mL, 11.43 mmol) at a rate not to exceed -65 °C. A white precipitate had formed. After 5 min a solution of cyclopropyl methyl ketone (Lancaster Synthesis, Ltd., 1.030 mL, 10.39 mmol) in diethyl ether (5 mL) was added to the suspension. After 5 min the cooling bath was removed and warmed to -15 °C. The suspension was then quenched with saturated NH4C1 (15 mL) and EtOAc (20 mL) added. The separated organic was then dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 10 to 30% gradient of
EtO Ac/Hex to afford l-(2-chloro-4-pyridinyl)-l-cyclopropylethanol (1.35 g, 6.83 mmol, 65.7 % yield) as amber oil (mixture of 2 enantiomers).
Step 2. To a stirring solution of diisopropylamine (0.594 g, 5.87 mmol) in THF (3 mL) at -70 °C under nitrogen was added n-butyllithium, 2.5 M solution in hexanes (2.065 mL, 5.16 mmol) at a rate not to exceed -60 °C. After 15 min a solution of 7-bromo-l-benzothiophene (Maybridge Chemical Co., Ltd., 1.0 g, 4.69 mmol) in THF (4 mL) was added dropwise at a rate that did not exceed an internal temperature of -65 °C. This solution was stirred for 1 h at -75 °C. To this was added a solution of N-((lE)-(2- chlorophenyl)methylidene)cyclopropanesulfonamide (1.144 g, 4.69 mmol, Intermediate AAIO) in THF (4 mL) at a rate that did not exceed an internal temperature of -65 °C. After 15 min, the reaction was warmed to -30 °C then quenched with saturated NH4C1 (20 mL). To the biphasic solution was added EtO Ac (20 mL). The isolated organic was then dried over MgS04, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 30% gradient of EtO Ac/Hex to afford N-((7- bromo-l-benzothiophen-2-yl)(2-chlorophenyl)methyl)cyclopropanesulfonamide (0.71 g, 1.554 mmol, 33.1 % yield) as colorless oil (mixture of 2 enantiomers).
Step 3. To a suspension of N-((7-bromo-l -benzothiophen-2-yl)(2- chlorophenyl)methyl)cyclopropanesulfonamide (0.71 g, 1.554 mmol), bis(pinacolato)diboron (0.474 g, 1.865 mmol), Ι,Γ- bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane adduct (0.095 g, 0.117 mmol), potassium acetate (0.194 mL, 3.11 mmol) in 1,4- dioxane (3 mL) was sparged with argon for 1 minute then heated to 100 °C with stirring for 45 min. The reaction was then partitioned between EtO Ac (20 mL) and 5% NaHC03 (10 mL). The separated organic was then dried over MgS04, filtered, concentrated under pressure, then purified by silica gel chromatography (80 g) eluting products with 15 to 40% gradient of EtO Ac/Hex to afford N-((2- chlorophenyl)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen-
2-yl)methyl)cyclopropanesulfonamide (0.57 g, 1.131 mmol, 72.8 % yield) as colorless oil (mixture of 2 enantiomers).
Step 4. A suspension of N-((2-chlorophenyl)(7-(4,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen-2-yl)methyl)cyclopropanesulfonamide (200 mg, 0.397 mmol), l-(2-chloro-4-pyridinyl)-l-cyclopropylethanol (86 mg, 0.437 mmol), tetrakis(triphenylphosphine)palladium(o) (45.9 mg, 0.040 mmol), sodium carbonate (168 mg, 1.588 mmol) in 1,4-dioxane (1 mL) and water (1 mL) was sparged with argon for 1 min then heated to 100 °C with stirring for 1 h. The reaction was then partitioned between EtOAc (20 mL) and 5% NaHC03 (10 mL). The separated organic was dried over MgSC^, filtered, concentrated under pressure, then purified by silica gel chromatography (40 g) eluting products with 15 to 50% gradient of EtO Ac/Hex to afford N-((2-chlorophenyl)(7-(4-(l- cyclopropyl- 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (150 mg, 0.278 mmol, 70.1 % yield) as colorless film. This film was then suspended in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford desired compound as white fluffy solid
(mixture of 4 diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.88 (d, J=9.59 Hz, 1 H) 8.71 (d, J=5.28 Hz, 1 H) 8.22 (s, 1 H) 8.08 (d, J=7.04 Hz, 1 H) 7.83 - 7.91 (m, 2 H) 7.46 - 7.58 (m, 4 H) 7.37 - 7.45 (m, 1 H) 6.99 (d, J=0.98 Hz, 1 H) 6.28 (d, J=9.39 Hz, 1 H) 5.05 (s, 1 H) 2.16 - 2.26 (m, 1 H) 1.50 (s, 3 H) 1.21 - 1.36 (m, 1 H) 0.88 (dd, J=6.75, 4.99 Hz, 1 H) 0.72 - 0.84 (m, 2 H) 0.50 - 0.66 (m, 2 H) 0.32 - 0.46 (m, 2 H) 0.16 - 0.28 (m, 1 H). m/z (APCI, pos. ion) 539.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.312 μΜ.
Examples 113 and 114
N-((R)-(2-chlorophenyl)(7-(4-((3S)-3-hydroxytetrahydro-3-furanyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide and N-
((R)-(2-chlorophenyl)(7-(4-((3R)-3-hydroxytetrahydro-3-furanyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a suspension of N-((2-chlorophenyl)(7-(4,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -benzothiophen-2-yl)methyl)cyclopropanesulfonamide (200 mg, 0.397 mmol, see Example 112, step 3), 3-(2-chloro-4- pyridinyl)tetrahydro-3-furanol (95 mg, 0.476 mmol, see Example 108, Step 1), tetrakis(triphenylphosphine)palladium(o) (34.4 mg, 0.030 mmol), sodium carbonate (168 mg, 1.588 mmol) in 1,4-dioxane (1 mL) and water (1 mL) was sparged 1 min with argon then heated to 100 °C with stirring for 1 h. The reaction was then partitioned between EtOAc (20 mL) and 5% NaHC03 (10 mL). The separated organic was dried over MgS04, filtered, concentrated under pressure, then purified by silica gel chromatography (40 g) eluting products with 20 to 70% gradient of EtO Ac/Hex to afford N-((2-chlorophenyl)(7-(4-(3- hydroxytetrahydro-3 -furanyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (160 mg, 0.296 mmol, 74.5 % yield) as colorless film. This film was then suspended in MeCN/water (1.5 mL), frozen, then lyophilized to afford desired compound as white fluffy solid (mixture of 4 diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.88 (d, J=9.59 Hz, 1 H) 8.75 (d, J=5.28 Hz, 1 H) 8.26 (s, 1 H) 8.12 (d, J=7.24 Hz, 1 H) 7.82 - 7.92 (m, 2 H) 7.46 - 7.56 (m, 4 H) 7.37 - 7.45 (m, 1 H) 6.99 (d, J=1.17 Hz, 1 H) 6.28 (d, J=9.19 Hz, 1 H) 5.77 (s, 1 H) 4.01 - 4.11 (m, 2 H) 3.81 - 3.91 (m, 2 H) 2.40 (dt, J=12.67, 9.12 Hz, 1 H) 2.13 - 2.26 (m, 2 H) 0.84 - 0.94 (m, 1 H) 0.72 - 0.84 (m, 2 H) 0.54 - 0.67 (m, 1 H). m/z (APCI, pos. ion) 541.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.089 μΜ.
The material above was subjected to preparative SFC (Chiralpak® AS-H column; 25 cm x 21 mm, 5 μιη) eluting with 75% liquid C02 in 25% methanol (with 20 mM NH3) at a flow rate of 70 mL/min) to afford two products:
Sample 1 : 1H NMR (400 MHz, DMSO-d6) δ ppm 8.88 (d, J=9.59 Hz, 1 H) 8.75 (d, J=5.28 Hz, 1 H) 8.26 (s, 1 H) 8.12 (d, J=7.24 Hz, 1 H) 7.82 - 7.92 (m, 2 H) 7.46 - 7.56 (m, 4 H) 7.37 - 7.45 (m, 1 H) 6.99 (d, J=1.17 Hz, 1 H) 6.28 (d, J=9.19 Hz, 1 H) 5.77 (s, 1 H) 4.01 - 4.11 (m, 2 H) 3.81 - 3.91 (m, 2 H) 2.40 (dt, J=12.67, 9.12 Hz, 1 H) 2.13 - 2.26 (m, 2 H) 0.84 - 0.94 (m, 1 H) 0.72 - 0.84 (m, 2 H) 0.54
- 0.67 (m, 1 H). m/z (APCI, pos. ion) 541.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.053 μΜ
Sample 2: 1H NMR (400 MHz, DMSO-d6) δ ppm 8.88 (d, J=9.59 Hz, 1 H) 8.75 (d, J=5.28 Hz, 1 H) 8.26 (s, 1 H) 8.12 (d, J=7.24 Hz, 1 H) 7.82 - 7.92 (m, 2 H) 7.46 - 7.56 (m, 4 H) 7.37 - 7.45 (m, 1 H) 6.99 (d, J=1.17 Hz, 1 H) 6.28 (d, J=9.19 Hz, 1 H) 5.77 (s, 1 H) 4.01 - 4.11 (m, 2 H) 3.81 - 3.91 (m, 2 H) 2.40 (dt, J=12.67, 9.12 Hz, 1 H) 2.13 - 2.26 (m, 2 H) 0.84 - 0.94 (m, 1 H) 0.72 - 0.84 (m, 2 H) 0.54
- 0.67 (m, 1 H). m/z (APCI, pos. ion) 541.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.118 μΜ
Example 115
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l-cyclopropyl-l-hydroxyethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (200 mg, 0.323 mmol, Intermediate AA7), l-(2-chloro-4-pyridinyl)-l-cyclopropylethanol (77 mg, 0.387 mmol, see Example 112, step 1), l, -bis(diphenylphosphino)ferrocene- palladium(II)dichloride dichloromethane adduct (26.3 mg, 0.032 mmol), sodium carbonate (137 mg, 1.290 mmol) in 1,4-dioxane (1.5 mL) and water (1 mL) was sparged with argon for 1 min then heated to 100 °C for 45 min. The reaction was then partitioned between EtOAc (15 mL) and extracted with 5% NaHC03 (5 mL). The separated organic was dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 15 to 50% gradient of EtO Ac/Hex to afford tert-butyl (5-chloro-6-((7-(4-(l- cyclopropyl- 1 -hydroxy ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)((cyclopropylsulfonyl)amino)methyl)-2-pyridinyl)carbamate (85 mg) as whtie solid (mixture of 4 diastereomers).
Step 2. A solution of tert-butyl (5-chloro-6-((7-(4-(l-cyclopropyl-l- hydroxyethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)((cyclopropylsulfonyl)amino)methyl)-2-pyridinyl)carbamate (85 mg, 0.130 mmol) was stirred in TFA (10 mL) for 1 h. The solvent was removed under reduced pressure and residue partitioned between CH2CI2 (15 mL) and 1 M K2HPO4 (10 mL). The organic was dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting
products with 1.5 to 4% gradient of MeOH/CH2Ci2 to afford a colorless film. This film was then suspended in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l-cyclopropyl-l- hydroxyethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (51 mg, 0.092 mmol, 70.8 % yield) as white fluffy solid (mixture of 4 diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.71 (d, J=5.28 Hz, 1 H) 8.20 (s, 1 H) 8.06 (d, J=7.04 Hz, 1 H) 7.88 (d, J=7.24 Hz, 1 H) 7.82 (d, J=8.41 Hz, 1 H) 7.46 - 7.56 (m, 3 H) 7.24 (d, J=0.98 Hz, 1 H) 6.49 (d, J=8.61 Hz, 1 H) 6.28 (s, 2 H) 6.12 (d, J=7.63 Hz, 1 H) 5.05 (s, 1 H) 2.21 - 2.31 (m, 1 H) 1.50 (s, 3 H) 1.21 - 1.34 (m, 1 H) 0.82 - 0.93 (m, 2 H) 0.72 - 0.81 (m, 1 H) 0.63 - 0.72 (m, 1 H) 0.50 - 0.60 (m, 1 H) 0.33 - 0.45 (m, 2 H) 0.16 - 0.28 (m, 1 H). m/z (APCI, pos. ion) 555.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.009 μΜ.
Example 116
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l-hydroxycyclopropyl)-2-pyridinyl)- -benzothiophen-2-yl)methyl)cyclopropanesulfonamide

trifluoromethanesulfonate (1299 μΐ,, 5.66 mmol) in CH2C12 (5 mL) dropwise (exothermic reaction) over a period of 2 min. After 20 min the reaction was extracted with 1 M KH2P04 (20 mL) and the separated organic dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 10% gradient of EtO Ac/Hex to afford 4-(l-((tert-butyl(dimethyl)silyl)oxy)ethenyl)-2-chloropyridine (585 mg) as colorless oil.
Step 2. To a stirring solution of 4-(l-((tert- butyl(dimethyl)silyl)oxy)ethenyl)-2-chloropyridine (550 mg, 2.038 mmol) in CH2C12 (5 mL) at 0 °C under nitrogen was added trimethylaluminum, 2.0M solution in toluene (6115 μί, 12.23 mmol) dropwise. After 5 min a solution of diiodomethane (1313 μΐ,, 16.31 mmol) in CH2C12 (1 mL) was added. The reaction slowly warmed to 20 °C over 10 days. The reaction was then chilled to 0 °C and slowly queched with dropwise addtion of 1 M HC1 (1 mL). Reaction then diluted with 1 M HC1 (20 mL) and CH2C12 (20 mL). The isolated organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 10% gradient of
EtO Ac/Hex to afford 4-(l-((tert-butyl(dimethyl)silyl)oxy)cyclopropyl)-2- chloropyridine (120 mg) as colorless oil.
Step 3. To a stirring solution of 4-(l-((tert- butyl(dimethyl)silyl)oxy)cyclopropyl)-2-chloropyridine (120 mg, 0.423 mmol) in MeCN (2 mL) at 20 °C was added hydrogen fluoride pyridine, 70% as hydrogen fluoride, 30% as pyridine (210 μΐ,, 1.691 mmol). The reaction was stirred for 1 h at 20 °C. The solvents were then removed under reduced pressure and the residue partitioned between K2HP04 (5 mL) and CH2C12 (10 mL). The aqueous was extracted further with CH2C12 (5 mL). The combined organics were then dried
over MgS04 and concentrated under reduced pressure to afford l-(2-chloro-4- pyridinyl)cyclopropanol (85 mg) as colorless oil.
Step 4. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (329 mg, 0.531 mmol, Intermediate AA7), l-(2-chloro-4-pyridinyl)cyclopropanol (90 mg, 0.531 mmol), tetrakis(triphenylphosphine)palladium(o) (61.3 mg, 0.053 mmol), sodium carbonate (225 mg, 2.123 mmol) in 1,4-dioxane (2 mL) and water (1 mL) was sparged with argon for 1 min then heated to 100 °C for 1.5 h. The reaction was then partitioned between EtOAc (20 mL) and 5% NaHC03 (10 mL). The organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 60% gradient of EtO Ac/Hex to afford tert-butyl (5-chloro-6-(((cyclopropylsulfonyl)amino)(7-(4- (1 -hydroxy cyclopropyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- pyridinyl)carbamate (113 mg) as colorless foam (mixture of 2 enantiomers).
Step 5. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-( 1 -hydroxy cyclopropyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (105 mg, 0.167 mmol) in TFA (5 mL) was stirred for 1 h at 25 °C. The solvent was then removed under reduced pressure and the residue partitioned between CH2CI2 (10 mL) and 1 M K2HP04 (10 mL). The organic was dried over MgS04, filtered, and concentrated under reduced pressure to afford desired compound as off white foam. This foam was then suspended in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy cyclopropyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (80 mg) as off white fluffy solid (mixture of 2 enantiomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.66 (d, J=5.28 Hz, 1 H) 8.06 (d, J=7.04 Hz, 1 H) 7.86 - 7.96 (m, 2 H) 7.78 (d, J=9.19 Hz, 1 H) 7.48 - 7.54 (m, 2 H) 7.22 -
7.27 (m, 2 H) 6.51 (d, J=8.80 Hz, 1 H) 6.28 (br. s., 2 H) 6.14 (dd, J=9.00, 0.98 Hz, 1 H) 3.24 - 3.57 (m, 1 H) 2.29 (s, 1 H) 1.28 (t, J=3.03 Hz, 2 H) 1.17 - 1.26 (m, 2 H) 0.82 - 0.96 (m, 2 H) 0.74 - 0.83 (m, 1 H) 0.66 - 0.74 (m, 1 H). m/z (APCI, pos. ion) 527.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.049 μΜ. Example 117
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2-methyl-lH-imidazol-l-yl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. A suspension of 2-methylimidazole (Alfa Aesar, 0.5 g, 6.09 mmol), 2-chloro-4-fluoropyridine (Alfa Aesar, 1.60 g, 12.18 mmol), cesium carbonate (4.17 g, 12.79 mmol) in DMF (5 mL) was heated to 80 °C for 18 h. The reaction was then partitioned between EtOAc (30 mL) and 5% NaHC03 (100 mL). The separated organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 10% gradient of MeOH/CH2C12 to afford 2-chloro-4-(2-methyl-lH- imidazol-l-yl)pyridine (0.87 g) as white solid.
Step 2. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (200 mg, 0.323 mmol,
Intermediate AA7), 2-chloro-4-(2-methyl-lH-imidazol-l-yl)pyridine (75.0 mg, 0.387 mmol), tetrakis(triphenylphosphine)palladium(o) (28.0 mg, 0.024 mmol), sodium carbonate (137 mg, 1.290 mmol) in 1,4-dioxane (1 mL) and water (1 mL) was sparged with argon for 30 seconds then heated to 100 °C for 0.5 h. The reaction was then partitioned between EtOAc (10 mL) and extracted with 5% NaHCC"3 (15 mL). The separated organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 70% gradient of MeCN/CH2Cl2 to afford tert-butyl (5-chloro- 6-(((cyclopropylsulfonyl)amino)(7-(4-(2 -methyl- 1 H-imidazol- 1 -yl)-2-pyridinyl)-
1- benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (140 mg) as white solid (mixture of 2 enantiomers).
Step 3. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(2 -methyl- 1 H-imidazol- 1 -yl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (140 mg, 0.215 mmol) in TFA (10 mL) was stirred for 0.5 h. The solvent was removed under reduced pressure and residue partitioned between CH2C12 (15 mL) and 1 M K2HP04 (10 mL). The organic was dried over MgS04, filtered, and concentrated under reduced pressure to afford a colorless film. The material was then purified by silica gel chromatography (40 g) eluting products with 1 to 5% gradient of MeOH/CH2Cl2 to afford a colorless film. This film was then suspended in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford N-((6-amino-3-chloro-
2- pyridinyl)(7-(4-(2 -methyl- 1 H-imidazol- 1 -yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (60 mg) as white fluffy solid (mixture of 2 enantiomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.86 (d, J=5.28 Hz, 1 H) 8.23 (d, J=1.56 Hz, 1 H) 8.15 (d, J=7.04 Hz, 1 H) 7.87 (d, J=7.24 Hz, 1 H) 7.79 (d, J=9.00 Hz, 1 H) 7.55 (d, J=1.56 Hz, 1 H) 7.52 (dd, J=5.48, 1.96 Hz, 1 H) 7.41 - 7.48 (m, 2 H) 7.20 (d, J=0.98 Hz, 1 H) 6.95 (d, J=1.56 Hz, 1 H) 6.43 (d, J=8.80 Hz, 1 H) 6.22 (s, 2 H) 6.07 (dd, J=9.10, 0.88 Hz, 1 H) 2.42 (s, 3 H) 2.21 (s, 1 H) 0.81 (t, J=3.42
Hz, 2 H) 0.66 - 0.75 (m, 1 H) 0.57 - 0.66 (m, 1 H). m/z (APCI, pos. ion) 551.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.399 μΜ.
Example 118
N-((7-(4-acetyl-2-pyridinyl)-l-benzothiophen-2-yl)(6-amino-3-chloro-2- pyridinyl)methyl)cyclopropanesulfonamide

Step 1. To a suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (200 mg, 0.323 mmol, Intermediate AA7), 4-acetyl-2-chloropyridine (Sigma-Aldrich, 55.2 mg, 0.355 mmol), 1 , 1 '-bis(diphenylphosphino)ferrocene-palladium(II)dichloride
dichloromethane adduct (26.3 mg, 0.032 mmol), sodium carbonate (137 mg, 1.290 mmol) in 1,4-dioxane (1.5 mL) and water (1 mL) was spaged argon for 1 min then heated to 100 °C with stirring for 1 h. The reaction was then partitioned between EtOAc (15 mL) and 5% NaHC03 (25 mL). The separated organic was then dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 20 to 50% gradient of EtO Ac/Hex to afford tert-butyl (6-((7-(4-acetyl-2-pyridinyl)-l-benzothiophen-2- yl)((cyclopropylsulfonyl)amino)methyl)-5 -chloro-2-pyridinyl)carbamate (95 mg, 0.155 mmol, 48.0 % yield) as colorless film (mixture of 2 enantiomers).
Step 2. A solution of tert-butyl (6-((7-(4-acetyl-2-pyridinyl)-l- benzothiophen-2-yl)((cyclopropylsulfonyl)amino)methyl)-5-chloro-2- pyridinyl)carbamate (90 mg, 0.147 mmol) in TFA (5 mL) was stirred for 30 min
at 25 °C. The solvent was removed under reduced pressure and residue partitioned between CH2CI2 (15 mL) and 1 M K2HPO4 (10 mL). The organic was dried over MgSC^ concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 1 to 3% gradient of
MeOH/CIH Cb to afford a colorless foam. This foam was suspended in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford N-((7-(4-acetyl-2- pyridinyl)- 1 -benzothiophen-2-yl)(6-amino-3-chloro-2- pyridinyl)methyl)cyclopropanesulfonamide (68 mg) as white fluffy solid
(mixture of 2 enantiomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 9.01 (d, J=5.09 Hz, 1 H) 8.56 (s, 1 H) 8.24 (d, J=7.04 Hz, 1 H) 7.94 (d, J=7.24 Hz, 1 H) 7.77 - 7.84 (m, 2 H) 7.49 - 7.59 (m, 2 H) 7.28 (d, J=0.78 Hz, 1 H) 6.50 (d, J=8.80 Hz, 1 H) 6.27 (br. s., 2 H) 6.14 (dd, J=9.00, 0.78 Hz, 1 H) 2.74 (s, 3 H) 2.21 - 2.34 (m, 1 H) 0.82 - 0.94 (m, 2 H) 0.73 - 0.81 (m, 1 H) 0.63 - 0.73 (m, 1 H). m/z (APCI, pos. ion) 513.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.49 μΜ.
Example 119
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-
(trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 2. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (125 mg, 0.202 mmol, Intermediate AA7), 2-(2-chloro-4-pyridinyl)- 1,1,1 ,3 ,3 ,3-hexafluoro-2-propanol (85 mg, 0.302 mmol), tetrakis(triphenylphosphine)palladium(o) (23.30 mg, 0.020 mmol), sodium carbonate (107 mg, 1.008 mmol), in 1,4-dioxane (1 mL) and water (0.5 mL) was sparged with argon for 30 seconds then heated to 100 °C for 1 h. The reaction was then diluted with EtO Ac (5 mL) and extracted with 5% NaHCC"3 (5 mL). The separated organic was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 0 to 50% gradient of EtO Ac/Hex to afford tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2- pyridinyl)carbamate (120 mg) as white solid (mixture of 2 enantiomers).
Step 3. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2- pyridinyl)carbamate (120 mg, 0.163 mmol) in TFA (5 mL) was stirred for 30 min
at 25 °C. The solvent was then removed under reduced pressure and the residue partitioned between 9: 1 CHCI3/IPA (15 mL) and 1 M K2HP04 (10 mL). The organic was dried over MgS04 and concentrated under reduced pressure to afford a white foam. This foam was then suspended in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2,2,2- trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (102 mg) as white fluffy solid (mixture of 2 enantiomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 9.33 (s, 1 H) 8.98 (d, J=5.48 Hz, 1 H) 8.35 (s, 1 H) 8.04 (d, J=7.04 Hz, 1 H) 7.96 (d, J=7.24 Hz, 1 H) 7.86 (d, J=9.00 Hz, 1 H) 7.67 (d, J=5.28 Hz, 1 H) 7.48 - 7.61 (m, 2 H) 7.29 (d, J=0.98 Hz, 1 H) 6.49 (d, J=8.80 Hz, 1 H) 6.29 (br. s., 2 H) 6.13 (dd, J=9.00, 0.78 Hz, 1 H) 2.20 - 2.35 (m, 1 H) 0.82 - 0.93 (m, 2 H) 0.72 - 0.80 (m, 1 H) 0.62 - 0.72 (m, 1 H). m/z (APCI, pos. ion) 637.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.088 μΜ.
Example 120
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(3-hydroxy-3-oxetanyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 2. A suspension of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (270 mg, 0.435 mmol, Intermediate AA7), 3-(2-chloro-4-pyridinyl)-3-oxetanol (89 mg, 0.479 mmol), 1 , 1 '-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane adduct (35.6 mg, 0.044 mmol), sodium carbonate (138 mg, 1.306 mmol) in 1,4- dioxane (1.5 mL) and water (1 mL) was sparged with argon for 1 minute then heated to 100 °C for 1 h. The reaction mixture was then partitioned between EtOAc (15 mL) and 5% NaHC03 (25 mL). The separated organic layer was then dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 20 to 60% gradient of
EtO Ac/Hex to afford tert-butyl (5-chloro-6-(((cyclopropylsulfonyl)amino)(7-(4- (3-hydroxy-3-oxetanyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2- pyridinyl)carbamate (120 mg, 0.187 mmol) as colorless film (mixture of 2 diastereomers).
Step 3. A solution of tert-butyl (5-chloro-6- (((cyclopropylsulfonyl)amino)(7-(4-(3 -hydroxy-3 -oxetanyl)-2-pyridinyl)- 1 -
benzothiophen-2-yl)methyl)-2-pyridinyl)carbamate (120 mg, 0.187 mmol) in TFA (5 mL) was stirred at 25 °C for 30 min. The solvent was removed under reduced pressure and residue partitioned between CH2CI2 (15 mL) and 1 M K2HP04 (10 mL). The organic was dried over MgS04 and concentrated under reduced pressure to afford a colorless film. This film was suspended in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford N-((6-amino-3-chloro- 2-pyridinyl)(7-(4-(3 -hydroxy-3 -oxetanyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (90 mg, 0.166 mmol, 89 % yield) as white fluffy solid (mixture of 2 enantiomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.82 (d, J=5.28 Hz, 1 H) 8.30 (s, 1 H) 8.08 (d, J=7.04 Hz, 1 H) 7.90 (d, J=7.24 Hz, 1 H) 7.77 (d, J=9.00 Hz, 1 H) 7.64 (dd, J=5.28, 1.57 Hz, 1 H) 7.47 - 7.56 (m, 2 H) 7.26 (d, J=0.98 Hz, 1 H) 6.70 (s, 1 H) 6.49 (d, J=8.80 Hz, 1 H) 6.26 (br. s., 2 H) 6.13 (dd, J=9.00, 0.78 Hz, 1 H) 4.81 - 4.87 (m, 2 H) 4.75 - 4.81 (m, 2 H) 2.22 - 2.34 (m, 1 H) 0.82 - 0.94 (m, 2 H) 0.73 - 0.81 (m, 1 H) 0.64 - 0.73 (m, 1 H). m/z (APCI, pos. ion) 543.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.055 μΜ.
Example 121
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)- 2-pyridinyl)-lH-indol-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a stirring solution of 7-bromoindole (5.0 g, 25.5 mmol) in dry DMF (100 mL) at 0 °C under nitrogen was added sodium hydride, 60% dispersion in mineral oil (1.22 g, 30.6 mmol) in two portions. The suspension was stirred for 1 h at 0 °C. To this was added para-toluenesulfonyl chloride (3.26 mL, 25.5 mmol) whole. The reaction mixture was stirred for 20 min then quenched with saturated NH4C1 (10 mL). The reaction was then diluted with EtO Ac (200 mL) and extracted with water (600 mL). The separated aqueous was then extracted with EtO Ac (100 mL). The organics were dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 5 to 20%> gradient of EtO Ac/Hex to afford 7- bromo-l-((4-methylphenyl)sulfonyl)-lH-indole (6.24 g) as off white solid.
Step 2. To a stirring solution of diisopropylamine (2.76 mL, 19.70 mmol) in THF (20 mL) at -70 °C under nitrogen was added n-butyllithium solution, 2.5 M in hexanes (5.25 mL, 13.13 mmol). The reaction was stirred for 20 min then a solution of 7-bromo-l-((4-methylphenyl)sulfonyl)-lH-indole (4.60 g, 13.13 mmol) in THF (20 mL) was added. The cooling bath was removed and the red solution warmed to -15 °C. The reaction was then returned to -70 °C and a separate solution of (S,E)-N-(2-chlorobenzylidene)-2-methylpropane-2- sulfmamide (3.44 g, 16.42 mmol, Intermediate Yl) in THF (20 mL) was added. The reaction was then warmed to 20 °C over 2 h. The reaction mixture was then quenched with saturated NH4C1 (20 mL) and diluted with EtOAc (150 mL). The organic was dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 30 to 70% gradient of EtO Ac/Hex to afford (S)-N-((R)-(7-bromo-l-((4- methylphenyl)sulfonyl)-lH-indol-2-yl)(2-chlorophenyl)methyl)-2-methyl-2- propanesulfmamide (6.50 g) as white foam.
Step 3. To a solution of (S)-N-((R)-(7-bromo-l-((4- methylphenyl)sulfonyl)-lH-indol-2-yl)(2-chlorophenyl)methyl)-2-methyl-2- propanesulfmamide (1.65 g, 2.78 mmol) in MeOH (20 mL) was added 5 M HC1 in water (5 mL). The solution was stirred for 18 h at 25 °C then solvents removed under reduced pressure. The residue was partitioned between 9: 1 CHCI3/IPA (30 mL) and 5 M NaOH (15 mL). The organic layer was dried over MgS04, then concentrated under reduced pressure to afford (R)-l-(7-bromo-l- ((4-methylphenyl)sulfonyl)- 1 H-indol-2-yl)- 1 -(2-chlorophenyl)methanamine (1.45 g) as colorless oil.
Step 4. To a stirring solution of (R)-l-(7-bromo-l-((4- methylphenyl)sulfonyl)-lH-indol-2-yl)-l-(2-chlorophenyl)methanamine (1.36 g, 2.78 mmol), DIEA (0.606 mL, 3.47 mmol), DMAP (0.017 g, 0.139 mmol) in DMF (5 mL) at 25 °C under argon was added cyclopropanesulfonyl chloride (Matrix Scientific, 0.354 mL, 3.47 mmol). The reaction mixture was stirred for
22 h at 25 °C then partitioned between EtOAc (80 mL) and 5% NaHC03 (100 mL). The organic was dried over MgSC^, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 25 to 50% gradient of EtO Ac/Hex to afford N-((R)-(7-bromo-l -((4- methylphenyl)sulfonyl)- 1 H-indol-2-yl)(2- chlorophenyl)methyl)cyclopropanesulfonamide (0.86 g) as colorless oil.
Step 5. To a solution of N-((R)-(7-bromo-l-((4-methylphenyl)sulfonyl)- lH-indol-2-yl)(2-chlorophenyl)methyl)cyclopropanesulfonamide (0.85 g, 1.431 mmol) in THF (6 mL) and MeOH (6 mL) was added a solution of lithium hydroxide monohydrate (0.300 g, 7.16 mmol) in water (3 mL). The reaction mixture was heated to 80 °C for 3 days in an appropiately capped vessel. The reaction mixture was then concentrated under reduced pressure and the residue partitioned between 9: 1 CHC13/IPA (30 mL) and 5% NaHC03 (15 mL). The organic was then concentrated under reduced pressure and purified by silica gel chromatography (80 g) eluting products with 0 to 2.5% gradient of 2M NH3 in MeOH/CH2Cl2 to afford N-((R)-(7-bromo-lH-indol-2-yl)(2- chlorophenyl)methyl)cyclopropanesulfonamide (0.39 g) as colorless oil.
Step 6. A suspension of N-((R)-(7-bromo-lH-indol-2-yl)(2- chlorophenyl)methyl)cyclopropanesulfonamide (360 mg, 0.819 mmol), bis(pinacolato)diboron (270 mg, 1.064 mmol), [1,1- bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane (33.4 mg, 0.041 mmol), potassium acetate (121 mg, 1.228 mmol) in 1 ,4-dioxane (4 mL) was sparged with argon for 1 minute then heated to 120 °C for 20 min. The reaction mixture was then partitioned between EtO Ac (30 mL) and 5 NaHC03 (10 mL). The organic was dried over MgSC^, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 2% gradient of MeOH/CH2Cl2 to afford N-((R)- (2-chlorophenyl)(7-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-indol-2- yl)methyl)cyclopropanesulfonamide (180 mg) as colorless oil.
Step 7. A suspension of N-((R)-(2-chlorophenyl)(7-(4,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H-indol-2-yl)methyl)cyclopropanesulfonamide (119 mg, 0.244 mmol), (2S)-2-(2-chloro-4-pyridinyl)-l,l,l-trifluoro-2-propanol (55.1 mg, 0.244 mmol, Intermediate X2), tetrakis(triphenylphosphine)palladium(o) (14.12 mg, 0.012 mmol), sodium carbonate (104 mg, 0.978 mmol) in 1,4-dioxane (1.5 mL) and water (1 mL) was sparged with argon for 1 minute then heated to 100 °C for 30 min. LCMS suggested clean conversion. The reaction mixture was then partitioned between EtOAc (20 mL) and 5% NaHC03 (10 mL). The organic layer was then dried over MgSC^, filtered, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 20% gradient of MeCN/CH2Cl2 to afford N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H-indol-2- yl)methyl)cyclopropanesulfonamide (100 mg) as colorless film. The product was then suspended 1 : 1 MeCN/water (6 mL), frozen, then lyophilized to afford desired compound as white fluffy powder.
1H NMR (400 MHz, CHLOROFORM- ) δ ppm 11.46 (br. s., 1 H) 8.70 (d, J=5.28 Hz, 1 H) 8.20 (s, 1 H) 7.78 (d, J=7.24 Hz, 1 H) 7.54 - 7.62 (m, 2 H) 7.43 - 7.49 (m, 1 H) 7.30 - 7.42 (m, 3 H) 7.17 (t, J=7.73 Hz, 1 H) 6.38 (d, J=8.22 Hz, 1 H) 6.16 (dd, J=2.45, 0.88 Hz, 1 H) 5.41 (d, J=7.82 Hz, 1 H) 2.42 - 2.77 (m, 1 H) 2.25 (tt, J=7.95, 4.87 Hz, 1 H) 1.83 (s, 3 H) 1.08 - 1.19 (m, 2 H) 0.70 - 0.90 (m, 2 H). m/z (APCI, pos. ion) 550.1 (M+l)+. GK-GKRP IC50 (Binding) = 0.216 μΜ.
Example 122
R)-N-((2-chlorophenyl)(7-(4-(2-hydroxypropan-2-yl)pyridin-2-yl)-lH-indol- 2-yl)methyl)cyclopropanesulfonamide

Step 1 : To a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4- pyridinyl)-2-propanol (30.0 g, 111 mmol) and DIEA (29.2 mL, 167 mmol) in THF (100 mL) at 0 °C under nitrogen was added a solution of triethylsilyl trifluoromethanesulfonate (31.5 ml, 139 mmol) in THF (50 mL) at a rate that did not exceed an internal temperature of 10 °C. After 3 hrs the reaction mixture was chilled to 5 °C, diethyl ether (250 mL) added and 1 M KH2P04 (200 ml) added at a rate that did not exceed an internal temp of 12 °C. The isolated organic layer was extracted with brine then dried over MgS04, concentrated under reduced pressure, and purified in two portions by silica gel chromatography (2 x 330 g) eluting products with a 0 to 10% gradient of EtO Ac/Hex to afford 2-(l- benzothiophen-7-yl)-4-(l -methyl- l-((triethylsilyl)oxy)ethyl)pyridine (36.5 g) as colorless oil.
Step 2: To a stirring colorless solution of 2-(l-benzothiophen-7-yl)-4-(l- methyl-l-((triethylsilyl)oxy)ethyl)pyridine (19.0 g, 49.5 mmol) in lithium chloride, 0.5 m in anhydrous tetrahydrofuran (Sigma- Aldrich, 198 mL, 99 mmol) at -73 °C under nitrogen in a 500-mL round bottom flask was added n-BuLi, (2.5M solution in hexanes (21.79 mL, 54.5 mmol) at a rate not to exceed -66 °C. This darkened solution was stirred for 30 minutes at -76 °C then added via canula to a stirring cold (-78 °C) solution of di-tert-butyl (4-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-5-chloro-2-pyrimidinyl)imidodicarbonate (22.83 g, 49.5 mmol) in THF (200 mL) in a different 1-L round-bottomed flask under nitrogen. The transfer lasted approximately 10 minutes. After an additional 20 minutes the reaction mixture was slowly quenched with sat. NH4C1 (50 mL) at - 65 °C. The reaction mixture was then partitioned between EtOAc (500 mL) and 5% NaHCC"3 (150 mL). The separated aqueous layer was further extracted with EtOAc (100 mL) and combined organic layers extracted with sat. NaCl (2 x50 mL). The separated organic layer was dried over MgSC^ and concentrated under reduced pressure to afford di-tert-butyl (4-((((S)-tert-butylsulfinyl)amino)(7-(4- ( 1 -methyl- 1 -((triethylsilyl)oxy)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-5-chloro-2-pyrimidinyl)imidodicarbonate (49.8 g) as amber oil (mixture of 2 diastereomers).
Step 3 : To a stirring suspension of di-tert-butyl (4-((((S)-tert- butylsulfinyl)amino)(7-(4-( 1 -methyl- 1 -((triethylsilyl)oxy)ethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-5-chloro-2-pyrimidinyl)imidodicarbonate (41.8 g, 49.5 mmol) in MeOH (200 mL) at 10 °C was added HC1 (4.0M in 1,4-dioxane (198 mL, 792 mmol). The reaction was stirred for 72 hrs at 20 °C to produce a suspension. To the stirring suspension was added diethyl ether (250 mL) dropwise over a 90 min period. The solid was then isolated by filtration and washed with diethyl ether (200 mL). The isolated solid was then air dried for 1 hr to afford 2-(2-(2-(amino(2-amino-5-chloro-4-pyrimidinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol trihydrochloride (17.3 g) as pale yellow dry solid (mixture of 2 enantiomers).
Step 4. To a stirring solution of 2-(2-(2-(amino(2-amino-5-chloro-4- pyrimidinyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol
trihydrochloride (17.3 g, 32.3 mmol), DMAP (0.395 g, 3.23 mmol), DIEA (22.58 mL, 129 mmol) in DMF (100 mL) at 13 °C was added cyclopropanesulfonyl chloride (Matrix Scientific, 4.94 ml, 48.5 mmol) over a 1 minute period. The ice bath was removed and amber solution stirred for 18 hrs at 20 °C. The reaction mixture was then partitioned between EtOAc (500 mL) and 5% NaHC03 (750 mL). The organic layer was then futher extracted with water (2 x 750 mL) and sat. NaCl (50 mL). The organic layer was dried over MgS04 and concentrated under reduced pressure to afford an amber tar. This tar was then suspended in 9: 1 CH2C12/IPA (200 mL) to afford as suspension. The solid was isolated by filtration, washed with diethyl ether (50 mL), then air dried to afford the desired product (2.1 g) as white solid. The filtrate was then concentrated under reduced pressure onto dry silica (25 g) and purified by silica gel chromatography (330 g) eluting products with a 2 to 5% gradient of 2M NH3 in MeOH/CH2Cl2 to afford 10.4 grams of N-((2-amino-5-chloro-4-pyrimidinyl)(7-(4-(l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (12.5 g) as dry tan foam.
The material above was subjected to preparative SFC (Chiralpak® ADH column; 25 cm x 30 mm, 5 μιη) eluting with 60% liquid C02 in 40%> isopropanol (with 20 mM NH3) at a flow rate of 120 mL/min) to afford two products with the peak assignments of:
First eluting peak: N-((S)-(2-amino-5-chloro-4-pyrimidinyl)(7-(4-(l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide
1H NMR (400 MHz, DMSO-d6) δ 8.71 (d, J=5.09 Hz, 1 H) 8.35 (s, 1 H) 8.22 (s, 1 H) 8.11 (d, J=8.61 Hz, 2 H) 7.90 (d, J=7.24 Hz, 1 H) 7.45 - 7.58 (m, 2 H) 7.33
(d, J=0.78 Hz, 1 H) 6.98 (br. s., 2 H) 6.03 - 6.14 (m, 1 H) 5.35 (s, 1 H) 2.35 - 2.45 (m, 1 H) 1.50 (s, 6 H) 0.79 - 0.97 (m, 3 H) 0.68 - 0.78 (m, 1 H) m/z (APCI, pos. ion) 530.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.232 μΜ.
Second eluting peak:N-((R)-(2-amino-5-chloro-4-pyrimidinyl)(7-(4-(l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide
1H NMR (400 MHz, DMSO-d6) δ ppm 8.71 (d, J=5.09 Hz, 1 H) 8.34 (s, 1 H) 8.21 (s, 1 H) 8.10 (d, J=8.61 Hz, 2 H) 7.85 - 7.94 (m, 1 H) 7.44 - 7.58 (m, 2 H) 7.33 (d, J=0.98 Hz, 1 H) 6.98 (br. s., 2 H) 6.09 (dd, J=8.90, 0.88 Hz, 1 H) 5.35 (br. s., 1 H) 2.33 - 2.45 (m, 1 H) 1.50 (s, 6 H) 0.79 - 0.97 (m, 3 H) 0.68 - 0.78 (m, 1 H). m/z (APCI, pos. ion) 530.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.014 μΜ.
Example 123
2-(2-(2-(amino(3-amino-6-chloro-2-fluorophenyl)methyl)-l-benzothiophen-7- yl)-4-pyridinyl)- 1 ,2-propanediol
BuLi


BuLi Step 7

Step 1. To a 2L 3 -neck round bottom flask equipped with an additional funnel and a Teflon® thermocouple was added 4-chloro-2-fluoroaniline (12 mL, 108 mmol) and 200 mL of THF. The solution was cooled to -76 °C and n- butyllithium solution, 2.5 M in hexanes (45 mL, 113 mmol) was slowly added while maintaining the reaction temperature below -70 °C. The reaction mixture was stirred at -72 °C for 30 min. A solution of 1 ,2-bis(chlorodimethylsilyl)ethane (24.8 g, 115 mmol) in THF (90 mL) was slowly added via the addition funnel while maintaining the reaction temperature below -70 °C. The reaction mixture was stirred at -76 °C for 1 h and n-butyllithium solution, 2.5 M in hexanes (45 mL, 113 mmol) was slowly added while maintaining the reaction temperature below -70 °C. The reaction mixture was stirred at -74 °C for 30 min and warmed up to 15 °C over 50 min. The reaction was re-cooled to -74 °C and n-BuLi solution (2.4 M in hexanes, 50 mL) was slowly added while maintaining the reaction temperature below -70 °C. The reaction was stirred at -70 °C for 1 h and
was slowly treated with ethyl chloro formate (13.4 mL, 140 mmol) while maintaining the reaction temperature below -70 °C. The reaction mixture was allowed to slowly warm to room temperature and stirred for 16 h. The reaction mixture was cooled to 0 °C and was quenched with 3N HC1 solution (200 mL). After stirring at room temperature for 1 h, the mixture was carefully basified with solid Na2C03. The aqueous layer was extracted with EtOAc (150 mL, twice) and the combined organic layers were washed with water (50 m), brine (50 mL), and concentrated. The residue was purified by flash chromatography (120 g silica gel column, gradient elution from hexanes to 30% EtOAc/hexanes) to afford ethyl 3- amino-6-chloro-2-fluorobenzoate (19.4 g) as a pale yellow oil.
Step 2. To a stirring solution of ethyl 3-amino-6-chloro-2-fluorobenzoate (13 g, 59.7 mmol), DMAP (0.365 g, 2.99 mmol), DIEA (31.3 mL, 179 mmol) in DMF (50 mL) at 20 °C was added di-tert-butyl dicarbonate (39.1 g, 179 mmol) in 5 gram portions. The reaction solution was stirred for 21 h at 20 °C then partitioned between EtOAc (250 mL) and 5% NaHC03 (750 mL). The organic layer was further extracted with water (750 mL) and the cloudy aqueous layer extracted with diethylether (100 mL). The combined organic layers were dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (330 g) eluting products with 0 to 15% gradient of EtO Ac/Hex to afford ethyl 3-(bis(tert-butoxycarbonyl)amino)-6-chloro-2-fluorobenzoate (14.3 g) as white solid.
Step 3. To stirring solution of 3-(bis(tert-butoxycarbonyl)amino)-6- chloro-2-fluorobenzoate (9.2 g, 33.4 mmol, 98 % yield) in diethyl ether (100 mL) and methyl alcohol, anhydrous (4.85 mL, 120 mmol) at 0 °C under nitrogen in a 500 mL round bottom flask was added lithium borohydride, 2.0 M solution in THF (59.9 mL, 120 mmol) dropwise. The reaction suspension was stirred for 18 h warming to 18 °C. The reaction was then chilled to 0 °C and slowly quenched with saturated NH4CI (25 mL). This produced a chalky aqueous layer and 1 M HC1 (25 mL) was added that created a clear aqueous layer. The organic layer
was diluted with EtOAc (50 mL) and separated from the aqueous layer. The organic alyer was dried over MgSC^ and concentrated under reduced pressure to afford tert-butyl (4-chloro-2-fluoro-3-(hydroxymethyl)phenyl)carbamate (9.2 g) as white solid.
Step 4. To a stirring solution of tert-butyl (4-chloro-2-fluoro-3- (hydroxymethyl)phenyl)carbamate (9.2 g, 33.4 mmol) in CH2CI2 (250 mL) at 0 °C under nitrogen was added Dess-Martin periodinane (15.57 g, 36.7 mmol) portionwise over 1 minute. After 2 h, the reaction was quenched by the addition of 200 mL of 1 M KH2PO4. The reaction mixture was stirred for 20 min and the organic layer isolated. The aqueous portion was extracted twice with 30 mL of DCM. The combined organic extracts were dried over MgSC^. concentrated under reduced pressure onto dry silica (15 g), then purified by silica gel chromatography (120 g) eluting products with 0 to 30% gradient of EtO Ac/Hex to afford tert-butyl (4-chloro-2-fluoro-3-formylphenyl)carbamate (4.1 g) as white solid.
Step 5. To a 500 mL round bottom flask was charged copper(II) sulfate (14.00 g, 88 mmol) and (s)-(-)-2-methyl-2-propanesulfmamide (7.09 g, 58.5 mmol) followed by a solution of tert-butyl (4-chloro-2-fluoro-3- formylphenyl)carbamate (4.0 g, 14.62 mmol) in CH2CI2 (80 mL). The suspension was stirred for 3 days at 20 °C under nitrogen followed by filtration. The filter cake was washed with CH2CI2 (25 mL). The filtrate was then purified by silica gel chromatography (120 g) eluting products with 0 to 10% gradient of EtOAc/CH2Cl2 to afford tert-butyl (3-((E)-(((S)-tert-butylsulfmyl)imino)methyl)- 4-chloro-2-fluorophenyl)carbamate (4.9 g) as white solid.
Step 6. To a stirring solution of tert-butyl (3-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-4-chloro-2-fluorophenyl)carbamate (4.9 g, 13.00 mmol) and DMAP (0.032 g, 0.260 mmol) in CH2C12 (25 mL) was added di-tert- butyl dicarbonate (6.24 g, 28.6 mmol) at 20 °C. After 1 h, the reaction was then
directly purified by silica gel chromatography (120 g) eluting products with 0 to 10% gradient of EtOAc/CH2Cl2 to afford di-tert-butyl (3-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-4-chloro-2-fluorophenyl)imidodicarbonate (5.64 g) as colorless tar (mixture of 4 diasteromers).
Step 7. To a stirring solution of 2-(l-benzothiophen-7-yl)-4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridine (1.70 g, 5.22 mmol, Intermediate AA1) in THF (15 mL) at -70 °C under nitrogen was added n-butyllithium, 2.5 M solution in hexanes (2.507 mL, 6.27 mmol) on the side of flask at a rate not to have internal temperature exceed -60 °C. After 15 min a solution of di-tert-butyl (3- ((E)-(((S)-tert-butylsulfinyl)imino)methyl)-4-chloro-2- fluorophenyl)imidodicarbonate (2.99 g, 6.27 mmol) in THF (12 mL) was added to the internal side of flask at a rate not to have internal temperature exceed -60 °C. The reaction mixture was stirred for 45 min at -70 °C, then the cooling bath removed. At 0 °C the reaction was slowly quenched with drop-wise addition of saturated NH4C1 (total 25 mL). The reaction was diluted with 5% NaHC03 (25 mL) and ethyl acetate (75 mL). The isolated organic layer was dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (120 g) eluting products with 0 to 50%> gradient of EtO Ac/Hex to afford tert-butyl (3-((((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,4-trimethyl-l ,3- dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4-chloro-2- fluorophenyl)carbamate (2.4 g) as colorless tar (mixture of 4 diastereomers).
Step 8. tert-Butyl (3-((((S)-tert-butylsulfmyl)amino)(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- chloro-2-fluorophenyl)carbamate (2.4 g, 3.42 mmol) was digested in 4 M HC1 in 1,4-dioxane (20 mL). After 10 min the reaction produced a solid. The reaction solvents were removed under reduced pressure and then the residue was partitioned between 9: 1 CHC13/IPA (75 mL) and 1 M K2HP04 (75 mL). The organic was dried over MgS04 then concentrated under reduced pressure to afford a colorless tar. This tar was dissolved and stirred in TFA (50 mL) for 30
min. The TFA was removed under reduced pressure and the reaction mixture was dissolved in MeOH (25 mL) and 4 M HC1 in 1,4-dioxane (20 mL) added. After 30 min the solvents were then removed under reduced pressure. The residue was then partitioned between 5% NaHC03 (25 mL) and 9: 1 CHC13/IPA (75 mL). The aqueous was pH adjusted to 7 with solid NaHC03. The aqueous was further extracted with 9: 1 CHC13/IPA (2x25 mL). The combined organic extracts were dried over MgSC^, concentrated under reduced pressure (1.27 g), then purified by silica gel chromatography (80 g) eluting products with 0 to 10% gradient of 2M NH3 in MeOH/CH2Cl2 to afford 2-(2-(2-(amino(3-amino-6- chloro-2-fluorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2- propanediol (1.08 g) as colorless tar (mixture of 4 diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.63 - 8.71 (m, 1 H) 8.11 - 8.19 (m, 1 H) 7.92 - 7.99 (m, 1 H) 7.77 - 7.85 (m, 1 H) 7.41 - 7.53 (m, 2 H) 7.05 - 7.11 (m, 1 H) 6.97 - 7.04 (m, 1 H) 6.68 - 6.78 (m, 1 H) 5.63 - 5.72 (m, 1 H) 5.29 (s, 2 H) 5.25 (s, 1 H) 4.78 - 4.85 (m, 1 H) 3.45 - 3.60 (m, 2 H) 2.65 - 2.80 (m, 2 H) 1.46 (s, 3 H). m/z (APCI, pos. ion) 458.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.129 μΜ.
Examples 124 and 125
N-((R)-(3-amino-6-chloro-2-fluorophenyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((R)-(3-amino-6-chloro-2- fluorophenyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide
Chr

Step 1. To a stirring solution of 2-(2-(2-(amino(3-amino-6-chloro-2- fluorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2-propanediol (115 mg, 0.251 mmol, Example 123), DIEA (132 μί, 0.753 mmol), DMAP (30.7 mg, 0.251 mmol) in DMF (1 mL) at 10 °C was added cyclopropanesulfonyl chloride (38.4 μΕ, 0.377 mmol). The reaction solution was stirred for 18 h at 20 °C, then partitioned between EtOAc (10 mL) and 5% NaHC03 (15 mL). The separated aqueous layer was further extracted with EtOAc (2x5 mL). The combined organic layers were dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 3 to 5% gradient of 2M NH3 in MeOH/CH2Cl2 to afford N-((3-amino-6-chloro-2- fluorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)methyl)cyclopropanesulfonamide (72 mg) as colorless film. This film was then dissolved in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford desired compound as white fluffy solid (mixture of 4 diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.68 (d, J=5.28 Hz, 1 H) 8.43 (d, J=9.19 Hz, 1 H) 8.18 (s, 1 H) 8.04 (d, J=7.04 Hz, 1 H) 7.87 (d, J=7.43 Hz, 1 H) 7.51 (t, J=7.63 Hz, 1 H) 7.46 (dd, J=5.09, 1.37 Hz, 1 H) 7.26 (d, J=1.17 Hz, 1 H) 7.05 (d,
J=8.61 Hz, 1 H) 6.80 (t, J=9.00 Hz, 1 H) 6.38 (d, J=9.19 Hz, 1 H) 5.38 (s, 2 H) 5.25 (s, 1 H) 4.81 (t, J=5.87 Hz, 1 H) 3.45 - 3.58 (m, 2 H) 2.29 - 2.39 (m, 1 H) 1.45 (s, 3 H) 0.91 (d, J=4.69 Hz, 2 H) 0.80 (td, J=8.12, 2.15 Hz, 2 H). m/z (APCI, pos. ion) 562.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.003 μΜ.
The material above was subjected to preparative SFC (Chiralpak® IC SFC column; 25 cm x 21 mm, 5 μιη) eluting with 60% liquid C02 in 40% methanol (with 20 mM NH3) at a flow rate of 60 mL/min) to afford two products:
Product 1 : 1H NMR (400 MHz, DMSO-d6) δ ppm 8.68 (d, J=5.28 Hz, 1 H) 8.49 (d, J=9.00 Hz, 1 H) 8.19 (s, 1 H) 8.05 (d, J=7.04 Hz, 1 H) 7.88 (d, J=7.24 Hz, 1 H) 7.52 (t, J=7.73 Hz, 1 H) 7.46 (dd, J=5.18, 1.47 Hz, 1 H) 7.26 (d, J=1.37 Hz, 1 H) 7.06 (d, J=8.80 Hz, 1 H) 6.79 (t, J=9.00 Hz, 1 H) 6.37 (d, J=9.19 Hz, 1 H) 5.41 (s, 2 H) 5.29 (s, 1 H) 4.85 (t, J=5.87 Hz, 1 H) 3.44 - 3.60 (m, 2 H) 2.29 - 2.40 (m, 1 H) 1.45 (s, 3 H) 0.85 - 0.96 (m, 2 H) 0.73 - 0.85 (m, 2 H). m/z (APCI, pos. ion) 562.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.008 μΜ.
Product 2: 1H NMR (400 MHz, DMSO-d6) δ ppm 8.65 - 8.71 (m, 1 H) 8.49 (d, J=8.80 Hz, 1 H) 8.19 (s, 1 H) 8.05 (d, J=7.04 Hz, 1 H) 7.88 (d, J=7.24 Hz, 1 H) 7.52 (t, J=7.73 Hz, 1 H) 7.46 (dd, J=5.09, 1.37 Hz, 1 H) 7.26 (d, J=1.37 Hz, 1 H) 7.06 (d, J=8.61 Hz, 1 H) 6.79 (t, J=9.00 Hz, 1 H) 6.37 (d, J=8.61 Hz, 1 H) 5.41 (s, 2 H) 5.29 (s, 1 H) 4.85 (t, J=5.87 Hz, 1 H) 3.44 - 3.62 (m, 2 H) 2.27 - 2.40 (m, 1 H) 2.27 - 2.40 (m, 1 H) 0.85 - 0.96 (m, 2 H) 0.71 - 0.85 (m, 2 H). m/z (APCI, pos. ion) 562.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.001 μΜ.
Example 126
N-((3-amino-6-chloro-2-fluorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide

Step 1. To a stirring solution of 2-(2-(2-(amino(3-amino-6-chloro-2- fluorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2-propanediol (200 mg, 0.437 mmol, Example 123), DIEA (152 μί, 0.873 mmol), DMAP (53.4 mg, 0.437 mmol) in DMF (2 mL) was added 4-chlorosulfonylpyrazole (Matrix Scientific, 218 mg, 1.310 mmol) at 20 °C for 20 h. The reaction mixture was then partitioned between EtOAc (10 mL) and 5% NaHC03 (15 mL). The separated aqueous layer was further extracted with EtOAc (2x5 mL). The combined organic layers were dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 2 to 10% gradient of 2M NH3 in MeOH/CH2Cl2 to afford a bis-sulfonated intermediate (with 25% desired product) as colorless film. This intermediate was dissolved in MeOH (3 mL) and sodium methoxide (283 mg, 1.310 mmol) added. This solution was stirred at 20 °C for 2 h. The reaction mixture was then partitioned between 9: 1 CHC13/IPA (15 mL) and 5% NaHC03 (10 mL). The separated aqueous was further extracted with 9: 1 CHC13/IPA (10 mL). The combined organic layers were dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 4 to 8% gradient of 2M NH3 in MeOH/CH2Cl2 to N-((3-amino-6-chloro-2- fluorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)methyl)-lH-pyrazole-4-sulfonamide (20 mg) as colorless film (mixture of 4 diastereomers). This film was dissolved in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford desired compound as white fluffy solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 13.39 (br. s., 1 H) 8.62 - 8.72 (m, 2 H) 8.17 (s, 1 H) 7.95 - 8.13 (m, 2 H) 7.82 (d, J=7.24 Hz, 1 H) 7.65 (br. s., 1 H) 7.43 - 7.53 (m, 2 H) 7.12 (d, J=1.17 Hz, 1 H) 6.94 (d, J=8.61 Hz, 1 H) 6.72 (t, J=9.00
Hz, 1 H) 6.29 (d, J=8.02 Hz, 1 H) 5.29 (s, 2 H) 5.25 (s, 1 H) 4.82 (t, J=5.87 Hz, 1 H) 3.45 - 3.59 (m, 2 H) 1.45 (s, 3 H). m/z (APCI, pos. ion) 587.9 (M+l)+. GK- GKRP IC50 (Binding) = 0.014 μΜ.
Example 127
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-l-cyclopropylmethanesulfonamide

Step 1. To a stirring solution of 2-(2-(2-(amino(3-amino-6-chloro-2- fluorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2-propanediol (200 mg, 0.454 mmol, Intermediate AA11), DIEA (158 μί, 0.907 mmol), DMAP (55.4 mg, 0.454 mmol) in DMF (2 mL) was added cyclopropylmethanesulfonyl chloride (Matrix Scientific, 105 mg, 0.680 mmol) at 20 °C. The reaction mixture was then partitioned between EtOAc (10 mL) and 5% NaHC03 (15 mL). The separated aqueous layer was further extracted with EtOAc (2x5 mL). The combined organic layers were dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 2 to 4% gradient of 2M NH3 in MeOH/CH2Cl2 to afford N-((6-amino-3-chloro-2- pyridinyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-l-cyclopropylmethanesulfonamide (35 mg) as colorless film (mixture of 4 diastereomers). This film was then dissolved in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford desired compound as white fluffy solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.68 (d, J=5.09 Hz, 1 H) 8.17 (s, 1 H) 8.00 - 8.06 (m, 1 H) 7.88 (d, J=7.24 Hz, 1 H) 7.65 (d, J=8.22 Hz, 1 H) 7.43 - 7.55 (m, 3 H) 7.29 (s, 1 H) 6.44 - 6.52 (m, 1 H) 6.24 - 6.33 (m, 2 H) 6.19 (d, J=8.02 Hz, 1 H) 5.25 (s, 1 H) 4.81 (t, J=5.87 Hz, 1 H) 3.44 - 3.60 (m, 2 H) 2.82 - 2.91 (m, 2 H) 1.45 (s, 3 H) 0.89 - 1.02 (m, 1 H) 0.42 - 0.52 (m, 2 H) 0.14 - 0.25 (m, 2 H). m/z (APCI, pos. ion) 559.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.006 μΜ.
Example 128
N-((3-amino-6-chloro-2-fluorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-l-cyclopropylmethanesulfonamide

Step 1. To a stirring solution of 2-(2-(2-(amino(3-amino-6-chloro-2- fluorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2-propanediol (200 mg, 0.437 mmol, Example 123), DIEA (152 μί, 0.873 mmol), DMAP (53.4 mg, 0.437 mmol) in DMF (2 mL) was added cyclopropylmethanesulfonyl chloride (Hande Science, 101 mg, 0.655 mmol) at 20 °C for 20 h. The reaction mixture was partitioned between EtOAc (10 mL) and 5% NaHC03 (15 mL). The separated aqueous layer was further extracted with EtOAc (2x5 mL). The combined organic layers were dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 2 to 4% gradient of 2M NH3 in MeOH/CH2Cl2 to afford N-((3-amino-6-chloro-2- fluorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)methyl)-l-cyclopropylmethanesulfonamide (13 mg) as colorless film
(mixture of 4 diastereomers). This film was then dissolved in 1 :1 MeCN/water
(1.5 mL), frozen, then lyophilized to afford desired compound as white fluffy solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.74 (d, J=5.09 Hz, 1 H) 8.21 (s, 1 H) 8.01 (d, J=7.04 Hz, 1 H) 7.83 (d, J=7.24 Hz, 1 H) 7.44 - 7.54 (m, 2 H) 7.09 (d, J=1.37 Hz, 1 H) 7.00 (dd, J=8.71, 0.88 Hz, 1 H) 6.73 (t, J=9.10 Hz, 1 H) 5.89 (s, 1 H) 5.67 (s, 1 H) 5.29 (s, 2 H) 4.38 - 4.44 (m, 1 H) 4.31 (d, J=9.98 Hz, 1 H) 3.23 (d, J=7.24 Hz, 2 H) 2.76 - 2.92 (m, 2 H) 1.55 (s, 3 H) 0.83 - 0.95 (m, 1 H) 0.41 - 0.52 (m, 2 H) 0.23 - 0.32 (m, 2 H). m/z (APCI, pos. ion) 576.0 (M+l)+. GK- GKRP IC50 (Binding) = 0.498 μΜ.
Examples 129 and 130
N-((R)-(5-amino-2-chloro-4-fluoro-3-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide and N-((R)-(5-amino-2-chloro-4-fluoro-
3-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. A solution of 5-amino-2-chloropyridine (Combi-Blocks Inc., 5.0 g, 38.9 mmol), di-tert-butyl dicarbonate (8.49 g, 38.9 mmol) in 1,4-dioxane (50 mL) was heated to 50 °C for 48 h under argon. The suspension was the partitioned between EtOAc (100 mL) and 1 M HC1 (100 mL). The isolated organic layer was then dried over MgSC^, filtered, then concentrated to an oil by reduced pressure. The oil was then dissolved in diethyl ether (75 mL), but after 1 hour no solid formed. The material was concentrated onto dry silica (20 g) then purified by silica gel chromatography (120 g) eluting products with 0 to 20%
gradient of EtO Ac/Hex to afford tert-butyl (6-chloro-3-pyridinyl)carbamate (5.4 g) as white solid.
Step 2. To a stirring solution of tert-butyl (6-chloro-3- pyridinyl)carbamate (5.4 g, 23.61 mmol) and Ν,Ν,Ν',Ν'-tetra-methyl- ethylenediamine (8.13 mL, 54.3 mmol) in diethyl ether (150 mL) at -70 °C under nitrogen was added n-butyllithium, 2.5 M solution in hexanes (13.12 mL, 32.8 mmol) at a rate not to exceed -60 °C. The resulting orange solution was stirred for 10 min then cooling bath removed. The solution was warmed to -20 °C, then returned to -70 °C, and a solution of n-fluorobenzenesulfonimide (11.17 g, 35.4 mmol) in THF (30 mL) was added at a rate not to exceed -60 °C. A precipitate was generated upon addition that became difficult to stir upon full addition. The slurry was stirred for 20 min at -70 °C then cooling bath removed. The suspension was quenched with saturated NH4C1 (50 mL total) at -25 °C. To the reaction was added EtO Ac (75 mL) and 1 M KH2P04 (25 mL). The separated aqueous layer was futher extracted with EtO Ac (25 mL). The combined organic layers were dried over MgS04, concentrated onto dry silica (20 g), then purified by silica gel chromatography (120 g) eluting products with 5 to 10% gradient of EtO Ac/Hex to afford tert-butyl (6-chloro-4-fluoro-3-pyridinyl)carbamate (3.25 g) as white crystalline solid.
Step 3. To a stirring solution of tert-butyl (6-chloro-4-fluoro-3- pyridinyl)carbamate (3.25 g, 13.18 mmol) in CH2C12 (5 mL) was added TFA (10 mL) and solution stirred for 1 h at 20 °C. The solvents were then removed under reduced pressure and the residue partitioned between 9: 1 CHC13/IPA (30 mL) and 1 M K2HP04 (30 mL; pH adjusted to 9 with 1 M NaOH). The separated aqueous layer was further extracted with 9: 1 CHCI3/IPA 3x25 mL). The combined organic layers were dried over MgS04, filtered, and concentrated under reduced pressure to afford tert-butyl (6-chloro-4-fluoropyridin-3- yl)carbamate (3.25 g) as yellow crystalline solid.
Step 4. To a stirring solution of 2-(l-benzothiophen-7-yl)-4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridine (1.1 g, 3.38 mmol, Intermediate AA1) in THF (10 mL) at -70 °C under nitrogen was added n-butyllithium, 2.5 M solution in hexanes (2.028 mL, 5.07 mmol) on side of flask at a rate not to exceed an internal temperature of -65 °C. This solution was stirred for 30 min at -75 °C then a solution of DMF (0.785 mL, 10.14 mmol) in THF (2 mL) was added onto the side of the flask at a rate that did not exceed -67 °C. After 15 min the cooling bath was removed, reaction solution warmed to -30 °C, then quenched with saturated NH4C1 (10 mL). The organic layer was dried over MgS04,
concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 10 to 30% gradient of EtO Ac/Hex to afford 7-(4- (2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophene-2- carbaldehyde (1.1 g) as white foam (racemic).
Step 5. A suspension of 7-(4-(2,2,4-trimethyl-l ,3-dioxolan-4-yl)-2- pyridinyl)-l-benzothiophene-2-carbaldehyde (1.1 g, 3.11 mmol), (R)-(+)-2- methyl-2-propanesulfinamide (1.509 g, 12.45 mmol), cupric sulfate anhydrous (1.106 g, 6.22 mmol) in CH2C12 (10 mL) was stirred for 48 h at 20 °C. To the suspension was added more cupric sulfate anhydrous (1.106 g, 6.22 mmol) and the reaction mixture heated to 45 °C for 24 h to form a suspension. The suspension was filtered, the filtrate concentrated onto dry silica (15 g) under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 10 to 40% gradient of EtO Ac/Hex to afford (R)-2-methyl-N-((7-(4- (2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methylidene)-2-propanesulfinamide (1.25 g) as white foam (mixture of 2 diastereomers).
Step 6. To a stirring solution of 6-chloro-4-fluoropyridin-3 -amine (385 mg, 2.63 mmol) in THF (3.0 mL) at -70 °C under nitrogen was added n- butyllithium, 2.5 M solution in hexanes (1104 μί, 2.76 mmol) on the side of the flask dropwise at rate not to exceed and internal temperature of -55 °C. The
resulting suspension was stirred for 10 min at -70 °C. To this suspension was added a solution of 1 ,2-bis(dimethylchlorosilyl)ethane (594 mg, 2.76 mmol) in THF (3 mL) at a rate not to exceed an internal temperature of -65 °C. The resulting solution was stirred for 10 min at -74 °C. To this was added n- butyllithium, 2.5 M solution in hexanes (1104 μΐ^, 2.76 mmol) at a rate not to exceed and internal temperature of -60 °C then the solution was stirred for an additional 25 min at -74 °C. To this solution was added n-butyllithium, 2.5 M solution in hexanes (1104 μΐ^, 2.76 mmol) on the side of the flask at a rate not to exceed an internal temperature of -65 °C. This was stirred for 2 min at <-70 °C then warmed to -30 °C with reaction turning a darker color. The dark solution was stirred at -30 °C for 15 min then chilled to -70 °C. To this was added a solution of (R)-2-methyl-N-((7-(4-(2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methylidene)-2-propanesulfinamide (600 mg, 1.314 mmol) in THF (4 mL) at a rate not to exceed an internal temperature of -60 °C. After 30 min the reaction was warmed to -20 °C, and slowly quenched with saturated NH4C1 (20 mL), then diluted with EtOAc (10 mL). The biphasic system was stirred for 15 min at 20 °C. The separated organic layer was extracted with 1 M HC1 (30 mL). The organic layer was isolated and aqueous further extracted with EtOAc (30 mL). The combined organic alyers were dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 0 to 100% gradient of EtO Ac/Hex to afford N-((5- amino-2-chloro-4-fluoro-3-pyridinyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-(R)-methyl-2-propanesulfinamide (240 mg) as colorless film (mixture of 4 diastereomers).
Step 7. To a stirring solution of N-((5-amino-2-chloro-4-fluoro-3- pyridinyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-2-(R)-methyl-2-propanesulfinamide (280 mg, 0.464 mmol) in MeOH (3 mL) at 20 °C was added hydrogen chloride, 4.0 M solution in 1,4-dioxane (232 μί, 0.928 mmol). The solution was stirred for 75 min at 20 °C then partitioned between 1 M K2HP04 (10 mL) and CHC13 (50 mL). The
separated organic alyer was dried over MgSC^, filtered, then concentrated under reduced pressure to afford 5-(amino(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-chloro-4-fluoro-3 -pyridinamine (235 mg) as tan oil (mixture of 4 diastereomers).
Step 8. To a stirring solution of 5-(amino(7-(4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-6-chloro-4-fluoro-3- pyridinamine (230 mg, 0.461 mmol), DIEA (161 μί, 0.922 mmol), DMAP (56.3 mg, 0.461 mmol) in DMF (2 mL) at 20 °C under nitrogen was added
cyclopropanesulfonyl chloride (Matrix Scientific, 56.3 μΐ,, 0.553 mmol) and stirred for 24 h at 20 °C. The reaction mixture was then partitioned between EtOAc (15 mL) and 5% NaHC03 (20 mL). The separated aqueous layer was further extracted with EtOAc (10 mL). The combined organic layers were dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (40 g) eluting products with 0 to 100% gradient of EtO Ac/Hex to afford N-((5-amino-2-chloro-4-fluoro-3-pyridinyl)(7-(4-(2,2,4-trimethyl- 1 ,3- dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (141 mg) as white foam (mixture of 4 diastereomers).
Step 9. A solution of N-((5-amino-2-chloro-4-fluoro-3-pyridinyl)(7-(4- (2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (141 mg, 0.234 mmol) in CH2CI2 (1 mL) and TFA (1 mL) was stirred for 90 min at 20 °C. The solvents were removed under reduced pressure then dissolved in 9: 1 CHC13/IPA (30 mL) and extracted with 1 M K2HPO4 (20 mL). The separated aqueous was further extracted with 9: 1 CHC13/IPA (2x10 mL). The combined organics were dried over MgSC^, concentrated under reduced pressure, then purified by silica gel chromatography (12 g) eluting products with 3 to 7% gradient of 2M NH3 in MeOH/CH2Cl2 to afford N-((5-amino-2-chloro-4-fluoro-3-pyridinyl)(7-(4-(l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-
yl)methyl)cyclopropanesulfonamide (85 mg) as colorless film. This film was then dissolved in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford desired compound as white fluffy powder (mixture of 4 diastereomers).
1H NMR (400 MHz, DMSO-d6) δ ppm 8.68 (d, J=5.09 Hz, 1 H) 8.64 (d, J=8.80 Hz, 1 H) 8.20 (s, 1 H) 8.07 (d, J=7.04 Hz, 1 H) 7.86 - 7.96 (m, 2 H) 7.53 (t, J=7.73 Hz, 1 H) 7.46 (dd, J=5.18, 1.47 Hz, 1 H) 7.31 (d, J=1.17 Hz, 1 H) 6.31 (d, J=8.61 Hz, 1 H) 5.72 (s, 2 H) 5.25 (s, 1 H) 4.82 (t, J=5.87 Hz, 1 H) 3.45 - 3.60 (m, 2 H) 2.37 - 2.46 (m, 1 H) 1.46 (s, 3 H) 0.75 - 1.02 (m, 4 H). m/z (APCI, pos. ion) 563.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.086 μΜ.
The material above was subjected to preparative SFC (Chiralpak® AD-H Sepax column; 15 cm x 21 mm, 5 μιη) eluting with 71% liquid C02 in 29% isopropanol (with 20 mM NH3) at a flow rate of 70 mL/min) to afford two products:
Product 1 : 1H NMR (400 MHz, DMSO-d6) δ ppm 8.68 (d, J=5.09 Hz, 1 H) 8.64 (d, J=8.80 Hz, 1 H) 8.20 (s, 1 H) 8.07 (d, J=7.04 Hz, 1 H) 7.86 - 7.96 (m, 2 H) 7.53 (t, J=7.73 Hz, 1 H) 7.46 (dd, J=5.18, 1.47 Hz, 1 H) 7.31 (d, J=1.17 Hz, 1 H) 6.31 (d, J=8.61 Hz, 1 H) 5.72 (s, 2 H) 5.25 (s, 1 H) 4.82 (t, J=5.87 Hz, 1 H) 3.45 - 3.60 (m, 2 H) 2.37 - 2.46 (m, 1 H) 1.46 (s, 3 H) 0.75 - 1.02 (m, 4 H). m/z (APCI, pos. ion) 563.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.257 μΜ.
Product 2: 1H NMR (400 MHz, DMSO-d6) δ ppm 8.68 (d, J=5.09 Hz, 1 H) 8.64 (d, J=8.80 Hz, 1 H) 8.20 (s, 1 H) 8.07 (d, J=7.04 Hz, 1 H) 7.86 - 7.96 (m, 2 H) 7.53 (t, J=7.73 Hz, 1 H) 7.46 (dd, J=5.18, 1.47 Hz, 1 H) 7.31 (d, J=1.17 Hz, 1 H) 6.31 (d, J=8.61 Hz, 1 H) 5.72 (s, 2 H) 5.25 (s, 1 H) 4.82 (t, J=5.87 Hz, 1 H) 3.45 - 3.60 (m, 2 H) 2.37 - 2.46 (m, 1 H) 1.46 (s, 3 H) 0.75 - 1.02 (m, 4 H). m/z (APCI, pos. ion) 563.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.033 μΜ.
Example 131
N-((5-amino-2-chloro-4-fluorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. To a stirring hazy suspension of 5-amino-2-chloro-4- fluorobenzoic acid (Combi-Blocks, 5.0 g, 26.4 mmol) and ethyl alcohol, anhydrous (101 mL, 1741 mmol) at 0 °C was added thionyl chloride (4.23 mL, 58.0 mmol) dropwise. After addition the ice bath was removed and after delay was then heated to 50 °C for 18 h then further to 85 °C for 2 h. The reaction was then partitioned between EtOAc (75 mL) and 1 M K2HP04 (50 mL). The separated aqueous layer was further extracted with EtOAc (25 mL). The combined organic layers were then dried over MgS04 and concentrated under reduced pressure to afford ethyl 5-amino-2-chloro-4-fluorobenzoate (4.7 g) as amber oil.
Step 2. A solution of ethyl 5-amino-2-chloro-4-fluorobenzoate (4.7 g, 21.60 mmol), di-tert-butyl dicarbonate (13.86 mL, 64.8 mmol), DMAP (0.079 g, 0.648 mmol), DIEA (11.32 mL, 64.8 mmol) in CH2C12 (15 mL) was stirred for 3 h at 20 °C. The reaction was then directly purified by silica gel chromatography (120 g) eluting products with 0 to 15% gradient of EtO Ac/Hex to afford ethyl 5- (bis(tert-butoxycarbonyl)amino)-2-chloro-4-fluorobenzoate as yellow oil: 2.62g.
Step 3. To a stirring solution of ethyl 5-(bis(tert-butoxycarbonyl)amino)- 2-chloro-4-fluorobenzoate (6.2 g, 14.84 mmol) in diethylether (100 mL) and methyl alcohol, anhydrous (2.104 mL, 51.9 mmol) at 0 °C under nitrogen was added lithium borohydride, 2.0 M solution in THF (26.0 mL, 51.9 mmol) at a rate that did not exceed an internal temp of 5 °C. After addition the reaction solution was stirred for 15 min at 0 °C then the cooling bath was removed. After 2 h the reaction was chilled to 0 °C and slowly quenched with saturated NH4C1 (5 mL). This produced a chalky aqueous layer and 1 M HC1 (25 mL) added that created a clear aqueous layer. The organic layer was diluted with EtO Ac (75 mL) and separated from the aqueous. The organic layer was dried over MgS04 and concentrated under reduced pressure to afford tert-butyl (4-chloro-2-fluoro-5- (hydroxymethyl)phenyl)carbamate (4.05 g) as colorless oil.
Step 4. To a stirring solution of tert-butyl (4-chloro-2-fluoro-5- (hydroxymethyl)phenyl)carbamate (4.0 g, 14.51 mmol) in CH2C12 (25 mL) at 0 °C under nitrogen was added Dess-Martin periodinane (7.38 g, 17.41 mmol) in 3 portions over one minute. After 5 min the ice bath was removed then reaction extracted with 1 M KH2P04 (50 mL). The organic was dried over MgS04, concentrated onto dry silica (20 g), then purified by silica gel chromatography (120 g) eluting products with 0 to 5% gradient of EtOAc/CH2Cl2 to afford tert- butyl (4-chloro-2-fluoro-5-formylphenyl)carbamate (3.4 g) as white solid.
Step 5. A suspension of tert-butyl (4-chloro-2-fluoro-5- formylphenyl)carbamate (3.4 g, 12.42 mmol), (s)-(-)-2-methyl-2-propane-
sulfinamide (3.01 g, 24.85 mmol), cupric sulfate anhydrous (4.41 g, 24.85 mmol) in CH2CI2 (15 mL) was stirred for 20 h at 20 °C. To the suspension was added (s)-(-)-2-methyl-2-propane-sulfinamide (3.01 g, 24.85 mmol) and cupric sulfate anhydrous (4.41 g, 24.85 mmol) then heated to 37 °C for 24 h. The suspension was then filtered and the cake washed with CH2CI2 (25 mL). The filtrate was then purified by silica gel chromatography (120 g) eluting products with 0 to 10% gradient of EtO Ac/Hex to afford tert-butyl (5-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-4-chloro-2-fluorophenyl)carbamate (4.55 g) as sticky foam.
Step 6. To a stirring solution of tert-butyl (5-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-4-chloro-2-fluorophenyl)carbamate (4.5 g, 11.94 mmol) and 4-(dimethylamino)pyridine (0.073 g, 0.597 mmol) in CH2CI2 (15 mL) at 20 °C under nitrogen was added di-tert-butyl dicarbonate (3.07 mL, 14.33 mmol). After 45 min at 20 °C, the reaction was then directly purified by silica gel chromatography (120 g) eluting products with 5 to 15% gradient of
EtO Ac/Hex to afford di-tert-butyl (5-((E)-(((S)-tert-butylsulfmyl)imino)methyl)- 4-chloro-2-fluorophenyl)imidodicarbonate (5.23 g) as white solid.
Step 7. To a stirring solution of 2-(l-benzothiophen-7-yl)-4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridine (0.59 g, 1.813 mmol, Intermediate AA1) in THF (8 mL) at -70 °C under nitrogen was added n-butyllithium, 2.5 M solution in hexanes (0.870 mL, 2.176 mmol) on the side of flask at a rate not to have internal temperature exceed -60 °C. After 15 min a solution of di-tert-butyl (5-((E)-(((S)- tert-butylsulfinyl)imino)methyl)-4-chloro-2-fluorophenyl)imidodicarbonate (1.038 g, 2.176 mmol) in THF (4 mL) was added to the internal side of flask at a rate not to have internal temperature exceed -65 °C. The reaction was stirred for 45 min at -70 °C then cooling bath removed. At 0 °C the reaction was slowly quenched with dropwise addition of saturated NH4C1 (total 25 mL). The reaction was diluted with 1 M KH2PO4 (25 mL) and ethyl acetate (75 mL). The isolated organic layer was dried over MgSC^, concentrated under reduced pressure, then
purified by silica gel chromatography (120 g) eluting products with 0 to 50% gradient of EtO Ac/Hex to afford tert-butyl (5-((((S)-tert-butylsulfinyl)amino)(7- (4-(2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-4-chloro-2-fluorophenyl)carbamate (0.76 g) as white foam (mixture of 4 diastereomers).
Step 8. To a stirring solution of tert-butyl (5-((((S)-tert- butylsulfinyl)amino)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-4-chloro-2-fluorophenyl)carbamate (0.76 g) in MeOH (5 mL) was added hydrogen chloride, 4.0 M solution in 1,4-dioxane (0.667 mL, 2.67 mmol) at 20 °C. After 90 min at 20 °C the solvents were then removed under reduced pressure to afford tert-butyl (5-(amino(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- chloro-2-fluorophenyl)carbamate as white foam (0.65 g) that was carried on without purification (mixture of 4 diastereomers).
Step 9. To a stirring solution of tert-butyl (5-(amino(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- chloro-2-fluorophenyl)carbamate (0.65 g, 1.087 mmol) and DIEA (0.949 mL, 5.43 mmol) in DMF (6 mL) at 20 °C under nitrogen was added DMAP (0.133 g, 1.087 mmol) followed by a solution of cyclopropanesulfonyl chloride (Matrix Scientific, 0.133 mL, 1.304 mmol) in DMF (1 mL). The solution was stirred for 18 h at 20 °C. To the reaction was added TFA (30 mL) and stirred for 45 min at 20 °C. The solvents were then removed under reduced pressure and the residue partitioned between 9: 1 CHC13/IPA (75 mL) and 1 M K2HP04 (75 mL). The aqueous layer was further extracted with 9: 1 CHCI3/IPA (25 mL). The combined organic layers were dried over MgS04, concentrated under reduced pressure, then purified by silica gel chromatography (80 g) eluting products with 3 to 5% gradient of 2M NH3 in MeOH/CH2Cl2 to afford N-((5-amino-2-chloro-4- fluorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)methyl)cyclopropanesulfonamide (160 mg) as colorless film. This film was
then dissolved in 1 : 1 MeCN/water (1.5 mL), frozen, then lyophilized to afford desired compound as white fluffy solid (mixture of 4 diastereomers). 1H NMR (400 MHz, DMSO-de) δ ppm 8.66 - 8.76 (m, 2 H) 8.19 (s, 1 H) 8.05 (d, J=7.24 Hz, 1 H) 7.87 (d, J=7.24 Hz, 1 H) 7.40 - 7.54 (m, 2 H) 7.23 (d, J=10.95 Hz, 1 H) 7.05 - 7.13 (m, 2 H) 6.12 (dd, J=9.00, 0.98 Hz, 1 H) 5.44 (s, 2 H) 5.29 (s, 1 H) 4.85 (t, J=5.77 Hz, 1 H) 3.44 - 3.59 (m, 2 H) 2.32 (t, J=6.16 Hz, 1 H) 1.45 (s, 3 H) 0.80 - 0.94 (m, 3 H) 0.70 - 0.79 (m, 1 H). m/z (APCI, pos. ion) 562.0 (M+l)+ GK-GKRP IC50 (Binding) = 0.016 μΜ
The material above was subjected to preparative SFC (Chiralpak® AD-H Sepax column; 25 cm x 21 mm, 5 μιη) eluting with 50% liquid C02 in 50% methanol (with 20 mM NH3) at a flow rate of 65 mL/min) to afford one diastereomer.
1H NMR (400 MHz, DMSO-d6) δ ppm 8.66 - 8.76 (m, 2 H) 8.19 (s, 1 H) 8.05 (d, J=7.24 Hz, 1 H) 7.87 (d, J=7.24 Hz, 1 H) 7.40 - 7.54 (m, 2 H) 7.23 (d, J=10.95 Hz, 1 H) 7.05 - 7.13 (m, 2 H) 6.12 (dd, J=9.00, 0.98 Hz, 1 H) 5.44 (s, 2 H) 5.29 (s, 1 H) 4.85 (t, J=5.77 Hz, 1 H) 3.44 - 3.59 (m, 2 H) 2.32 (t, J=6.16 Hz, 1 H) 1.45 (s, 3 H) 0.80 - 0.94 (m, 3 H) 0.70 - 0.79 (m, 1 H). m/z (APCI, pos. ion) 562.0 (M+l)+. GK-GKRP IC50 (Binding) = 0.005 μΜ
Example 132
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(2-hydroxypropan-2-yl)pyridin-2- yl)thieno[3,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide

Step 1. Dichloro-l, -bis(diphenylphosphino)ferrocene palladium(II) (0.074 g, 0.091 mmol) was added to a mixture of 7-chlorothieno[3,2-b]pyridine (Frontier) (0.515 g, 3.04 mmol), bis(pinacolato)diboron (1.542 g, 6.07 mmol), potassium acetate (0.894 g, 9.11 mmol), and 1,4-dioxane (100 mL) at room temperature under an argon atmosphere. The reaction mixture was then heated in a microwave reactor at 140 °C for 40 min. The resulting material was filtered through diatomaceous earth and the cake was washed with DCM/EtOAc and the filtrate was concentrated under a vacuum. The resulting material was basified with IN NaOH and water, and extracted with DCM. The aqueous layer was acidified with IN HCl and the product was extracted with DCM and dried over sodium sulfate, filtered and concentrated under a vacuum to give thieno[3,2- b]pyridin-7-ylboronic acid (0.310 g, 1.732 mmol, 57.0 % yield) as light yellow solid.
Step 2. A mixture of 2-(2-chloropyridin-4yl)propan-2-ol (0.374 g, 2.179 mmol, Intermediate Y4), thieno[3,2-b]pyridin-7-ylboronic acid (0.300 g, 1.676 mmol) 1,4-dioxane (5 mL), water (0.1 mL), bis(di-tert-butyl(4- dimethylaminophenyl)phosphine)-dichloropalladium(II) (0.059 g, 0.084 mmol, Aldrich) and potassium carbonate (0.695 g, 5.03 mmol). The reaction mixture
was stirred and heated in a microwave reactor at 140 °C for 60 min. The reaction mixture was filtered through a pad of diatomaceous earth, and the cake was washed with DCM/EtOAc and concentrated under a vacuum. The residue was partitioned between water and ethyl acetate, the layers were separated, and the aqueous material was washed with ethyl acetate (2x). The combined organic extracts were washed sequentially with water and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under a vacuum. The resulting product was adsorbed onto a plug of silica gel and then purified by silica gel
chromatographed through a column (40 g), eluting with a gradient 0% to 50% EtOAc/Hexanes to give 2-(2-(thieno[3,2-b]pyridin-7-yl)pyridin-4-yl)propan-2-ol (0.210 g, 0.777 mmol, 46.3 % yield), as a pale yellow solid.
Step 3. n-Butyllithium (0.710 mL of a 2.5 M solution in hexanes, 1.75 mmol) was added to a stirring solution of 2-(2-(thieno[3,2-b]pyridin-7-yl)pyridin- 4-yl)propan-2-ol (0.240 g, 0.888 mmol) and tetrahydrofuran (10.0 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of di-tert-butyl (6-((E)- (((S)-tert-butylsulfinyl)imino)methyl)-5-chloro-2-pyridinyl)imidodicarbonate (0.613 g, 1.332 mmol) and THF (3.0 mL) was added. After 10 min, the reaction mixture was allowed to warm to room temperature, where silica gel (2.0 g) was added and the volatiles were removed under a vacuum. The resulting product was adsorbed onto a plug of silica gel and then purified by silica gel chromatographed through a column (40 g), eluting with a gradient 0% to 50% EtOAc/Hexanes to give tert-butyl (5-chloro-6-(((S)-l,l-dimethylethylsulfinamido)(7-(4-(2- hydroxypropan-2-yl)pyridin-2-yl)thieno[3,2-b]pyridin-2-yl)methyl)pyridin-2- yl)carbamate as light yellow oil.
Step 4. Hydrogen chloride (5.0 mL of a 4.0 M solution with 1,4-dioxane, 5.0 mmol) and TFA (2 mL) was added to a stirring solution of tert-butyl (5- chloro-6-(((S)- 1 , 1 -dimethylethylsulfinamido)(7-(4-(2-hydroxypropan-2- yl)pyridin-2-yl)thieno [3 ,2-b]pyridin-2-yl)methyl)pyridin-2-yl)carbamate (0.190 g, 0.260 mmol) and methanol (10 mL) at room temperature and stirred for one
hour. The reaction mixture was concentrated under a vacuum and the residue was partitioned between DCM and saturated aqueous sodium bicarbonate. The organic layer was dried (sodium sulfate), filtered, and the filtrate was
concentrated to afford 2-(2-(2-(amino(6-amino-3-chloropyridin-2- yl)methyl)thieno[3,2-b]pyridin-4-yl)propan-ol (0.16 g) as a clear pale yellow tar. The material was used in the next step of the synthesis without purification.
Step 5. 4-(Dimethylamino)pyridine (0.013 g, 0.101 mmol) was added to a stirring solution of 2-(2-(2-(amino(6-amino-3-chloropyridin-2- yl)methyl)thieno[3,2-b]pyridin-4-yl)propan-ol (0.088 g, 0.207 mmol,
intermediate 4), N,N-dimethylformamide (3.8 mL), N,N-diisopropylethylamine (0.108 mL, 0.620 mmol), and cyclopropanesulfonyl chloride (0.032 mL, 0.310 mmol, Matrix Scientific, Columbia, SC) at room temperature. After 3 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried over sodium sulfate, filtered, and then concentrated. The residue was dissolved with dichloromethane, silica gel (2.0 g) was added to the solution, and the volatiles were removed under a vacuum. The residue was subjected to flash chromatography on silica gel (10% NH3 in MeOH/DCM 0 to 10%). The resulting product was re-purified by reverse-phase preparative HPLC using a Phenomenex Gemini column (Phenomenex, Torrance, CA), 10 micron, CI 8, 100 A, 150 x 30 mm, 0.1% TFA in CH3CN/H20, gradient 25% to 100% over 15 min. to afford N- ((6-amino-3-chloropyridin-2-yl)(7-(4-(2-hydroxypropan-2-yl)thieno[3,2- b]pyridin-2-yl)methylcyclopropanesulfonamide.
(0.022 g) as a colorless solid.
1H NMR (400 MHz, DMSO-d6) δ 8.78 (dd, J= 5.18, 8.51 Hz, 2H), 8.37 (s, 1H), 8.08 (d, J= 5.09 Hz, 1H), 7.97 (d, J= 8.80 Hz, 1H), 7.59 - 7.65 (m, 1H), 7.53 (d, J= 8.61 Hz, 1H), 7.38 (s, 1H), 6.51 (d, J= 8.80 Hz, 1H), 6.19 (d, J= 8.80 Hz, 1H), 2.34 (s, 1H), 1.52 (s, 6H), 1.24 (br. s., 2H), 0.90 (d, J= 4.89 Hz, 3H), 0.65 -
0.81 (m, 2H). m/z (ESI, pos. ion) 531.4 (M+H)+. GK-GK P IC50 (Binding) = 0.008 μΜ.
Example 133
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l,2dihydroxypropan-2-yl)pyridin-2- yl)thieno[3,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide

Step 1. Dichloro l,l '-bis(diphenylphosphino)ferrocene palladium(II) (0.074 g, 0.091 mmol) was added to a mixture of 7-chlorothieno[3,2-b]pyridine (Frontier) (0.515 g, 3.04 mmol), bis(pinacolato)diboron (1.542 g, 6.07 mmol), potassium acetate (0.894 g, 9.11 mmol), and dioxane (100 mL) at room temperature under an argon atmosphere. The reaction mixture was then heated in a microwave reactor at 140 °C for 40 min. The resulting material was filtered through diatomaceous earth and the cake was washed with DCM/EtOAc and the filtrate was concentrated under a vacuum. The resulting material was basified with IN NaOH and water, and extracted with DCM. The aqueous layer was acidified with IN HC1 and the product was extracted with DCM and dried over Na2S04, filtered and concentrated under a vacuum to give thieno[3,2-b]pyridin-7- ylboronic acid (0.310 g, 1.732 mmol, 57.0 % yield) as light yellow solid.
Step 2. A suspension of 2-chloro-4-(4,4,5,5-tetramethyl-l,3-dioxolan-4- yl)pyridine (0.267 g, 1.173 mmol), thieno[3,2-b]pyridin-7-ylboronic acid (0.210 g, 1.173 mmol), tetrakis(triphenylphosphine)palladium(0) (0.068 g, 0.059 mmol), sodium carbonate (0.373g, 3.52 mmol) in 1,4-dioxane (10 mL) and water (0.1 mL) were purged for 10 min and the suspension was heated to 80 °C overnight with stirring. The reaction mixture was then partitioned between EtOAc (75 mL) and 5% NaHC03 (25 mL). The organic layers were dried over sodium sulfate filtered and concentrated under reduced pressure. The resulting product was adsorbed onto a plug of silica gel and then purified by silica gel chromatographed through a column (40 g), eluting with a gradient 0% to 50% EtOAc/Hexanes to afford 7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)thieno[3,2-b]pyridine (0.310 g, 0.950 mmol, 81%).
Step 3. n-Butyllithium (0.441 mL of a 2.5 M solution in hexanes, 1.103 mmol) was added to a stirring solution of 7-(4-(2,2,4-trimethyl-l,3-dioxolan-4- yl)pyridin-2-yl)thieno[3,2-b]pyridine (0.180 g, 0.551 mmol) and tetrahydrofuran (10.0 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of di- tert-butyl (6-((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5-chloro-2- pyridinyl)imidodicarbonate (0.254 g, 0.551 mmol) and THF (3.0 mL) was added. After 10 min, the reaction mixture was quenched with saturated ammonium chloride solution and warmed to room temperature. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x) and dried over sodium sulfate and concentrated under vacuum. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a column (40 g), eluting with a gradient of 0% to 50% EtOAc in hexane, to provide tert-butyl(5- chloro-6-(((S)- 1 , 1 -dimetylethylsulfinamido)(7-(4-(2,2,4-trimethyl- 1 ,3-dioxolan- 4-yl)pyridin-2-yl)thieno [3 ,2-b]pyridin-2-yl)methyl)pyridin-2-yl)carbamate as light yellow oil.
Step 4. Hydrogen chloride (5.0 mL of a 4.0 M solution with 1,4-dioxane, 5.0 mmol) and TFA (2 mL) was added to a stirring solution of tert-butyl (5- chloro-6-(((S)-l,l-dimethylethylsulfinamido)(7-(4-(2,2,4-trimethyl-l ,3-dioxolan- 4-yl)pyridin-2-yl)thieno[3,2-b]pyridin-2-yl)methyl)pyridin-2-yl)carbamate (1.2 g, 1.749 mmol) and methanol (10 mL) at room temperature and stirred for one hour. The reaction mixture was concentrated under a vacuum and the residue was partitioned between DCM and saturated aqueous sodium bicarbonate. The organic layer was dried (sodium sulfate), filtered, and the filtrate was
concentrated to afford 2-(2-(2-(amino(6-amino-3-chloropyridin-2- yl)methyl)thieno[3,2-b]pyridin-7yl)pyridine -4-yl)propane-l ,2-diol(0.652 g, 1.475 mmol, 81%) as a clear pale yellow tar. The material was used in the next step of the synthesis without purification.
Step 5. 4-(Dimethylamino)pyridine (0.003 g, 0.045 mmol) was added to a stirring solution of 2-(2-(2-(amino(6-amino-3-chloropyridin-2- yl)methyl)thieno[3,2-b]pyridin-7yl)pyridine -4-yl)propane-l,2-diol (0.040 g, 0.091 mmol), Ν,Ν-dimethylformamide (1 mL), N,N-diisopropylethylamine (0.047 mL, 0.272 mmol), and cyclopropanesulfonyl chloride (0.014 mL, 0.136 mmol, Oakwood product Inc.) at room temperature. After 3 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried over sodium sulfate, filtered, and concentrated. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silca gel column (40 g), eluting with a gradient of 0% to 50% EtOAc in hexane. The resulting product was repurified by reverse-phase preparative HPLC using a Phenomenex Gemini column (Phenomenex, Torrance, CA), 10 micron, CI 8, 100 A, 150 x 30 mm, 0.1% TFA in CH3CN/H20, gradient 25% to 100% over 15 min. to afford N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l ,2-dihydroxypropan-2- yl)pyridine-2-yl)thieno[3,2-b]pyridin-2-yl)cyclopropanesulfonamide (0.015 g, 0.027 mmol, 30.3%>) as a colorless solid (a mixture of 4-isomers).
1H NMR (400 MHz, MeOH) δ 8.75 - 8.85 (m, 1H), 8.64 - 8.74 (m, 1H), 8.37 (s, 1H), 7.91 - 8.02 (m, 2H), 7.45 - 7.57 (m, 1H), 7.31 (s, 1H), 6.52 - 6.61 (m, 1H), 6.33 (s, 1H), 3.55 - 3.84 (m, 2H), 2.13 - 2.42 (m, 1H), 1.60 (s, 3H), 1.25 - 1.38 (m, 1H), 1.03 (d, J= 3.33 Hz, 2H), 0.63 - 0.86 (m, 2H). m/z (ESI, pos. ion) 547.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.007 μΜ.
Example 134
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l,2-dihydroxypropan-2-yl)pyridine- 2-yl)-5-fluorobenzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. Dichloro l,l '-bis(diphenylphosphino)ferrocene palladium(II) (0.074 g, 0.091 mmol) was added to a mixture of 7-chloro-5- fluorobenzo[b]thiophene (Ellanova) (0.515 g, 2.219 mmol),
bis(pinacolato)diboron (1.322 g, 4.46 mmol), potassium acetate (0.656 g, 6.69 mmol), and 1,4-dioxane (100 mL) at room temperature under an argon atmosphere. The reaction mixture was then heated in a microwave reactor at 140 °C for 40 min. The resulting material was filtered through diatomaceous earth and the cake was washed with DCM/EtOAc and the filtrate was concentrated under a vacuum. The resulting material was basified with IN NaOH and water, and extracted with DCM. The aqueous layer was acidified with IN HCl and the
product was extracted with DCM and dried over Na2S04, filtered and
concentrated under a vacuum to give 2-(5-fluorobenzo[b]thiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (0.385g 1.385 mmol, 62 % yield) as light yellow solid.
Step 2. A suspension of 2-chloro-4-(4,4,5,5-tetramethyl-l,3-dioxolan-4- yl)pyridine (0.311 g, 1.366 mmol), 2-(5-fluorobenzo[b]thiophen-7-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (0.380 g, 1.366 mmol),
tetrakis(triphenylphosphine)palladium(0) (0.068 g, 0.059 mmol), sodium carbonate (0.373g, 3.52 mmol) in 1,4-dioxane (10 mL) and water (0.1 mL) purged for 10 min. The suspension was heated to 80 °C overnight with stirring. The reaction mixture was then partitioned between EtOAc (75 mL) and 5% NaHCC"3 (25 mL). The organic layers were dried over sodium sulfate filtered, concentrated under reduced pressure. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (40 g), eluting with a gradient of 0% to 50% EtOAc in hexane to afford 2-(5- fluorobenzo[b]thiophen-7-yl)-4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine. (0.248 g, 0.722 mmol, 52.9%).
Step 3. n-Butyllithium (0.856 mL of a 2.5 M solution in hexanes, 2.14 mmol) was added to a stirring solution of 2-(5-fluorobenzo[b]thiophen-7-yl)-4- (2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine. (0.240 g, 0.888 mmol) and tetrahydrofuran (10.0 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of di-tert-butyl (6-((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5-chloro- 2-pyridinyl)imidodicarbonate (0.245 g, 0.713 mmol, Intermediate AA4) and THF (5.0 mL) was added. After 10 min, the reaction mixture was quenched with saturated ammonium chloride solution and warmed to room temperature. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x) and dried over sodium sulfate dried under vacuum. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (40 g), eluting with a gradient of 0%> to 50%> EtOAc in hexane, to provide
tert-butyl (5-chloro-6-(((S)- 1 , 1 -dimethylethylsulfmamido)(5-fluoro-7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)pyridin- 2-yl)carbamate (0.159 g, 0.226 mmol, 31.7 % yield), as tan solid.
Step 4. Hydrogen chloride (5.0 mL of a 4.0 M solution with 1,4-dioxane, 5.0 mmol) and TFA (2 mL) was added to a stirring solution of tert-butyl (5- chloro-6-(((S)-l,l-dimethylethylsulfinamido)(5-fluoro-7-(4-(2,2,4-trimethyl-l ,3- dioxolan-4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)pyridin-2- yl)carbamate) (0.150 g, 0.213 mmol) and methanol (2 mL) at room temperature and stirred for one hour. The reaction mixture was concentrated under a vacuum and the residue was partitioned between DCM and saturated aqueous sodium bicarbonate. The organic layer was dried (sodium sulfate), filtered, and the filtrate was concentrated to afford 2-(2-(2-(amino(6-amino-3-chloropyridin-2- yl)methyl-5-fluorobenzo[b]thiophen-7-yl)pyridin-4yl)propane-l ,2-diol (0.089 g, 0.194 mmol, 91%) as a clear pale yellow tar. The material was used in the next step of the synthesis without purification.
Step 5. 4-(Dimethylamino)pyridine (0.012 g, 0.100 mmol) was added to a stirring solution of 2-(2-(2-(amino(6-amino-3-chloropyridin-2-yl)methyl-5- fluorobenzo[b]thiophen-7-yl)pyridin-4yl)propane-l,2-diol yl) (0.092 g, 0.200 mmol), Ν,Ν-dimethylformamide (1 mL), N,N-diisopropylethylamine (0.105 mL, 0.621 mmol), and cyclopropanesulfonyl chloride (0.031 mL, 0.301 mmol at room temperature. After 1 hour, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried over sodium sulfate filtered, and then concentrated. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (12 g), eluting with a gradient of 0 % to 10% 2M NH3 MeOH in CH2C12, to provide N-((6-amino-3-chloropyridin-2- yl)(7-(4-(l,2-dihydroxypropan-2-yl)pyridin-2-yl)-5-fluorobenzo[b]thiophen-2-
yl)methyl)cyclopropanesulfonamide (0.043 g, 0.076 mmol), as off white solid (mixture of isomers).
1H NMR (400 MHz, DMSO-d6) 58.72 (d, J= 4.50 Hz, 1H), 8.21 (br. s., 1H), 7.97 (d, J= 9.98 Hz, 1H), 7.69 - 7.87 (m, 2H), 7.43 - 7.61 (m, 2H), 7.24 (s, 1H), 6.50 (d, J= 8.80 Hz, 1H), 6.27 (br. s., 2H), 6.13 (br. s., 1H), 5.25 (s, 1H), 4.81 (br. s., 1H), 3.55 (dd, J= 5.38, 14.77 Hz, 2H), 2.29 (br. s., 1H), 1.47 (br. s., 3H), 0.89 (br. s., 2H), 0.61 - 0.82 (m, 2H). ). m/z (ESI, pos. ion) 562.8 (M+H)+. GK-GKRP ICso (Binding) = 0.010 μΜ.
Example 135
N-((2-chloro-6-fluorophenyl)(7-(4-(l,2-dihydroxypropan-2-yl)pyridin-2- yl)thieno [3,2-b] pyridin-2-yl)methyl)cyclopropanesulfonamide.

(S)-N-((2-chlorophenyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2- yl)thieno[3,2-b]pyridin-2-yl)methyl)-2-methylpropane-2-sulfinamide
Step 1. n-Butyllithium (0.856 mL of a 2.5 M solution in hexanes, 2.14 mmol) was added to a stirring solution of 7-(4-(2,2,4-trimethyl-l,3-dioxolan-4- yl)pyridin-2-yl)thieno[3,2-b]pyridine (0.160 g, 0.490 mmol, see Example 132, step 2) and tetrahydrofuran (10.0 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of (S,E)-N-(2-chloro-6-fluorobenzylidene)-2- methylpropane-2-sulfinamide (0.119 g, 0.490 mmol, Intermediate AA12) and THF (5.0 mL) was added. After 10 min, the reaction mixture was quenched with saturated ammonium chloride solution and warmed to room temperature. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x) and dried over sodium sulfate concentrated under vacuum. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (40 g), eluting with a gradient of 0% to 50% EtOAc in hexane, to provide (S)-N-((2-chlorophenyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2- yl)thieno[3,2-b]pyridin-2-yl)methyl)-2-methylpropane-2-sulfinamide as tan solid.
2-(2-(2-(amino(2-chloro-6-fluorophenyl)methyl)thieno[3,2-b]pyridin-7- yl)pyridin-4-yl)propane- 1 ,2-diol
Step 2. Hydrogen chloride (5.0 mL of a 4.0 M solution with 1,4-dioxane, 5.0 mmol) and TFA (2 mL) was added to a stirring solution of (S)-N-((2- chlorophenyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2-yl)thieno[3,2- b]pyridin-2-yl)methyl)-2-methylpropane-2-sulfinamide (0.085 g, 0.146 mmol) and methanol (2 mL) at room temperature and stirred for one hour. The reaction mixture was concentrated under a vacuum and the residue was partitioned between DCM and saturated aqueous sodium bicarbonate. The organic layer was dried over sodium sulfate, filtered, and the filtrate was concentrated to afford 2- (2-(2-(amino(2-chloro-6-fluorophenyl)methyl)thieno[3,2-b]pyridin-4-yl)propane- 1,2-diol (0.034 g, 0.077 mmol, 52.6%) as a light yellow oil. The material was used in the next step of the synthesis without purification.
N-((2-chloro-6-fluorophenyl)(7-(4-(l,2-dihydroxypropan-2-yl)pyridin-2- yl)thieno [3,2-b] pyridin-2-yl)methyl)cyclopropanesulfonamide.
Step 3. 4-(Dimethylamino)pvridine (0.023 g, 0.191 mmol) was added to a stirring solution of 2-(2-(2-(amino(2-chloro-6-fluorophenyl)methyl)thieno[3,2- b]pyridin-4-yl)propane-l,2-diol (0.170 g, 0.383 mmol), N,N-dimethylformamide (5 mL), N,N-diisopropylethylamine (0.200 mL, 1.149 mmol), and
cyclopropanesulfonyl chloride (0.031 mL, 0.301 mmol.) at room temperature. After 1 hour, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, and then
concentrated. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (12 g), eluting with a gradient of 0 % to 10% 2M NH3 MeOH in DCM, to provide N-((2-chloro-6-fiuorophenyl)(7- (4-(l ,2-dihydroxypropan-2-yl)pyridin-2-yl)thieno[3,2-b]pyridin-2- yl)methyl)cyclopropanesulfonamide (0.061 g, 0.111 mmol, 29 % yield), as off white solid (mixture of isomers).
1H NMR (400 MHz, DMSO-d6) δ 8.77 - 8.81 (m, 1H), 8.74 - 8.77 (m, 1H), 8.67 - 8.73 (m, 1H), 8.33 - 8.38 (m, 1H), 8.04 - 8.09 (m, 1H), 7.57 - 7.61 (m, 1H), 7.44 - 7.54 (m, 2H), 7.39 - 7.43 (m, 1H), 7.30 - 7.39 (m, 1H), 6.50 - 6.57 (m, 1H), 4.80 - 4.87 (m, 1H), 3.54 - 3.61 (m, 1H), 3.47 - 3.54 (m, 1H), 2.38 - 2.47 (m, 1H), 1.47 (s, 1H), 1.19 - 1.30 (m, 1H), 1.07 - 1.15 (m, 3H), 0.89 - 0.98 (m, 1H), 0.76 - 0.88 (m, 2H). m/z (ESI, pos. ion) 549.2 (M+H)+. GK-GKRP IC50 (Binding) = 0.042 μΜ.
Example 136
N-((6-amino-3-chloropyridin-2-yl)(7-(4-((R)-l,2-dihydroxypropan-2- yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide


Step 1. n-Butyllithium (0.856 mL of a 2.5 M solution in hexanes, 2.14 mmol) was added to a stirring solution of (R)-2-(benzo[b]thiophen-7-yl)-4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridine (0.147 g, 0.452 mmol, Intermediate AAl-i?) and tetrahydrofuran (10.0 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of di-tert-butyl (6-((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5- chloro-2-pyridinyl)imidodicarbonate sulfinamide (0.208 g, 0.452 mmol,
Intermediate AA4) and THF (5.0 mL) was added. After 10 min, the reaction mixture was quenched with saturated ammonium chloride solution and warmed to room temperature.The organic layer was separated and the aqueous layer was extracted with EtOAc (3x) and dried over sodium sulfate concentrated under vacuum. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (40 g), eluting with a gradient of 0% to 50% EtOAc in hexane, to tert-butyl(R) (5-chloro-6-(((S)-l,l- dimethylethylsulfinamido)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)methyl)pyridin-2-yl)carbamate as pale yellow solid.
Step 2. Hydrogen chloride (5.0 mL of a 4.0 M solution with 1,4-dioxane, 5.0 mmol) and TFA (2 mL) was added to a stirring solution of tert-butyl(R) (5- chloro-6-(((S)-l,l-dimethylethylsulfinamido)(7-(4-(2,2,4-trimethyl-l ,3-dioxolan- 4-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)pyridin-2-yl)carbamate (0.235 g, 0.299 mmol) and methanol (2 mL) at room temperature and stirred for 2 h. The reaction mixture was concentrated under a vacuum and the residue was partitioned between DCM and saturated aqueous sodium bicarbonate. The organic layer was dried (sodium sulfate), filtered, and the filtrate was
concentrated to afford (2R)-2-(2-(amino(6-amino-3-chloropyridin-2- yl)benzo[b]thiophen-7-yl)pyridin-4-yl)propane-l,2-diol (0.115 g, 0.261 mmol, 87%) as a light yellow oil. The material was used in the next step of the synthesis without purification.
Step 3. 4-(Dimethylamino)pyridine (0.023 g, 0.191 mmol) was added to a stirring solution of (2R)-2-(2-(amino(6-amino-3-chloropyridin-2- yl)benzo[b]thiophen-7-yl)pyridin-4-yl)propane-l,2-diol (0.125 g, 0.283 mmol, intermediate 17), N,N-dimethylformamide (5 mL), N,N-diisopropylethylamine (0.148 mL, 0.850 mmol), and cyclopropanesulfonyl chloride (0.043 mL, 0.425 mmol, Oakwood product Inc.) at room temperature. After 4 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, and then concentrated. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (12 g), eluting with a gradient of 0 % to 10% 2M NH3 MeOH in DCM, to provide N-((2-chloro-6-fluorophenyl)(7-(4-(R)-(l ,2-dihydroxypropan-2- yl)pyridin-2-yl)thieno[3,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide (0.061 g, 0.111 mmol, 29 %> yield), as off white solid (mixture of isomers).
1H NMR (400 MHz, DMSO-d6) δ 8.71 (d, J= 5.28 Hz, 1H), 8.19 (s, 1H), 8.05 (d, J= 7.43 Hz, 1H), 7.77 - 7.92 (m, 2H), 7.41 - 7.56 (m, 3H), 7.25 (s, 1H), 6.50 (d, J
= 8.80 Hz, 1H), 6.28 (s, 2H), 6.13 (br. s., 1H), 4.85 (t, J= 5.67 Hz, 1H), 3.40 - 3.65 (m, 2H), 2.18 - 2.38 (m, 1H), 1.46 (s, 3H), 1.18 (t, J= 7.14 Hz, 1H), 0.88 (d, J= 3.72 Hz, 2H), 0.56 - 0.82 (m, 2H). m/z (ESI, pos. ion) 546.2 (M+H)+. GK- GKRP IC50 (Binding) = 0.004 μΜ. Example 137
N-((6-amino-3-chloropyridin-2-yl)(7-(4-((S)-l,2-dihydroxypropan-2- yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide.

Step 1. n-Butyllithium (0.856 mL of a 2.5 M solution in hexanes, 2.14 mmol) was added to a stirring solution of (S)-2-(benzo[b]thiophen-7-yl)-4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)pyridine (0.147 g, 0.452 mmol, Intermediate AA1-5) and tetrahydrofuran (10.0 mL) at -78 °C under a nitrogen atmosphere. After 10 min, a solution of di-tert-butyl (6-((E)-(((S)-tert-butylsulfinyl)imino)methyl)-5- chloro-2-pyridinyl)imidodicarbonate sulfinamide (0.208 g, 0.452 mmol,
Intermediate AA4) and THF (5.0 mL) was added. After 10 min, the reaction mixture was quenched with saturated ammonium chloride solution and warmed
to room temperature. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x) and dried over sodium sulfate concentrated under vacuum. The resulting product was adsorbed onto a plug of silica gel and chromatographed through a silica gel column (40 g), eluting with a gradient of 0% to 50% EtOAc in hexane, to tert-butyl(S) (5-chloro-6-(((S)-l,l- dimethylethylsulfinamido)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridin-2- yl)benzo[b]thiophen-2-yl)methyl)pyridin-2-yl)carbamate as pale yellow solid.
Step 2. Hydrogen chloride (5.0 mL of a 4.0 M solution with 1,4-dioxane, 5.0 mmol) and TFA (2 mL) was added to a stirring solution of tert-butyl(S) (5- chloro-6-(((S)-l,l-dimethylethylsulfinamido)(7-(4-(2,2,4-trimethyl-l ,3-dioxolan- 4-yl)pyridin-2-y l)benzo [b]thiophen-2-yl)methyl)pyridin-2-y l)carbamate (0.259 g, 0.330 mmol) and methanol (2 mL) at room temperature and stirred for 2 h. The reaction mixture was concentrated under a vacuum and the residue was partitioned between DCM and saturated aqueous sodium bicarbonate. The organic layer was dried (sodium sulfate), filtered, and the filtrate was
concentrated to afford (2R)-2-(2-(amino(6-amino-3-chloropyridin-2- yl)benzo[b]thiophen-7-yl)pyridin-4-yl)propane-l,2-diol (0.095 g, 0.261 mmol, 63%) as a light yellow oil. The material was used in the next step of the synthesis without purification.
Step 3. 4-(Dimethylamino)pyridine (0.023 g, 0.191 mmol) was added to a stirring solution of (2S)-2-(2-(amino(6-amino-3-chloropyridin-2- yl)benzo[b]thiophen-7-yl)pyridin-4-yl)propane-l,2-diol (0.095 g, 0.255 mmol), N,N-dimethylformamide (5 mL), N,N-diisopropylethylamine (0.148 mL, 0.850 mmol), and cyclopropanesulfonyl chloride (0.043 mL, 0.425 mmol, Oakwood product Inc.) at room temperature. After 4 h, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The layers were separated, and the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, and then concentrated. The resulting product was adsorbed onto a plug of silica gel
and chromatographed through a silica gel column (12 g), eluting with a gradient of 0 % to 10% 2M NH3 MeOH in DCM, to provide N-((2-chloro-6- fluorophenyl)(7-(4-(S)-(l,2-dihydroxypropan-2-yl)pyridin-2-yl)thieno[3,2- b]pyridin-2-yl)methyl)cyclopropanesulfonamide (0.061 g, 0.111 mmol, 29 % yield) as a off-white solid (mixture of isomers).
1H NMR (400 MHz, DMSO-d6) δ 8.71 (d, J= 4.89 Hz, 1H), 8.19 (br. s., 1H), 8.05 (d, J= 7.43 Hz, 1H), 7.77 - 7.94 (m, 2H), 7.42 - 7.60 (m, 3H), 7.25 (s, 1H), 6.50 (d, J= 8.80 Hz, 1H), 6.28 (br. s., 2H), 6.13 (d, J= 8.80 Hz, 1H), 4.74 - 4.96 (m, 1H), 4.03 (d, J= 6.85 Hz, 1H), 3.42 - 3.66 (m, 2H), 2.28 (br. s., 1H), 2.00 (s, 1H), 1.46 (s, 3H), 1.18 (t, J= 7.04 Hz, 1H), 0.88 (br. s., 2H), 0.63 - 0.81 (m, 2H), m/z (ESI, pos. ion) 546.2 (M+H)+. GK-GKRP IC50 (Binding) = 0.021 μΜ
Example 138
N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4-morpholinesulfonamide

To a solution of (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (0.100 g, 0.216 mmol, Intermediate X5) in pyridine (3.0 mL) was added 4- dimethylaminopyridine (0.013 g, 0.108 mmol, Sigma- Aldrich, St. Louis, MO) and morpholine-4-sulfonyl chloride (0.200 mL, 1.080 mmol, CamBridge Corp., San Diego, CA). The resulting mixture was heated at 90 °C under N2 for 20 h. The reaction mixture was cooled to rt and poured into the beaker which filled with EtOAc (30 mL) and hand stirred for 10 min. The organic layer was
separated from the black paste by decanting into a round-bottom flask. More EtOAc (20 mL) was added to the black paste, hand stirred for 10 min, and decanting into the same round-bottom flask. The solvent was concentrated. The resulting product was purified by column chromatography (24 g of silica, 10% to 40% acetone in hexanes) to afford N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2- trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 4-morpholinesulfonamide (0.200 g) as a light yellow solid.
1H NMR (300MHz ,CHLOROFORM-d) δ = 8.82 (d, J= 5.3 Hz, 1 H), 8.20 (s, 1 H), 7.91 (d, J= 7.3 Hz, 1 H), 7.74 (d, J= 7.6 Hz, 1 H), 7.63 (dd, J= 1.6, 7.5 Hz, 1 H), 7.53 - 7.29 (m, 5 H), 7.01 (d, J= 1.0 Hz, 1 H), 6.31 (d, J= 8.0 Hz, 1 H), 5.48 (d, J= 8.0 Hz, 1 H), 3.69 - 3.58 (m, 2 H), 3.57 - 3.46 (m, 2 H), 3.26 - 3.06 (m, 4 H), 1.84 (s, 3 H). m/z (ESI, +ve ion) 611.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.385 μΜ.
Example 139
N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-l- azetidinesulfonamide

To a solution of (2R)-2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (0.100 g, 0.216 mmol, Intermediate X5) in pyridine (3.0 mL) was added 4- dimethylaminopyridine (0.013 g, 0.108 mmol, Sigma- Aldrich, St. Louis, MO) and azetidine (0.040 mL, 0.562 mmol, Acros Organics, NJ). After stirred for 10
min, sulfuryl chloride (0.060 mL, 0.650 mmol, Sigma-Aldrich, St. Louis, MO) was added and the reaction mixture was continued to stir at rt for 20 h. The reaction mixture was cooled to rt and poured into the beaker which filled with EtOAc (30 mL) and hand stirred for 10 min. The organic layer was separated from the black paste by decanting into a round-bottom flask. More EtOAc (20 mL) was added to the black paste, hand stirred for 10 min, and decanting into the same round-bottom flask. The solvent was concentrated. The resulting product was purified by column chromatography (24 g of silica, 10% to 40% acetone in hexanes) to afford N-((R)-(2-chlorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 - azetidinesulfonamide (0.015 g) as a light yellow solid.
1H NMR (300MHz ,CHLOROFORM-d) δ = 8.81 (d, J= 5.3 Hz, 1 H), 8.19 (s, 1 H), 7.91 (d, J= 7.2 Hz, 1 H), 7.75 (d, J= 7.3 Hz, 1 H), 7.64 (dd, J = 1.7, 7.5 Hz, 1 H), 7.54 - 7.29 (m, 5 H), 7.04 (d, J= 1.2 Hz, 1 H), 6.33 (d, J= 8.2 Hz, 1 H), 5.36 (d, J= 8.3 Hz, 1 H), 3.98 - 3.70 (m, 4 H), 2.21 - 2.02 (m, 2 H), 1.85 (s, 3 H). m/z (ESI, +ve ion) 581.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.475 μΜ.
Example 140
N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide

To a solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.060 g, 0.147 mmol, see Example 2, step 2) in DMF (2.0 mL) was added 4-dimethylaminopyridine (5.40 mg, 0.044
mmol, Sigma- Aldrich, St. Louis, MO), diisopropylethylamine (0.100 mL, 0.575 mmol, Sigma-Aldrich, St. Louis, MO), and 4-chlorosulfonylpyrazole (0.040 g, 0.220 mmol, Matrix scientific, Columbia, SC). The resulting mixture was heated at 70 °C for 2 h. The reaction was cooled to rt and partitioned between EtOAc (50 mL) and water (30 mL). The organic layer was washed with water (2 x 30 mL), dried over MgS04, filtered, and concentrated. The resulting product was redissolved in MeOH (15 mL) and treated with 2 M K2C03 (5 mL, 10.00 mmol). The resulting mixture was stirred at rt for 2 h. Solvent was concentrated; the residue was partitioned between EtOAc (60 mL) and water (30 mL). The aqueous layer was extracted with EtOAc (2 x 30 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The resulting product was purified by column chromatography (24 g of silica, 2 to 8% MeOH in DCM) to afford N-((R)-(2-chlorophenyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide (0.047 g) as a white solid.
1H NMR (300MHz ,DMSO-d6) δ = 13.35 (br. s., 1 H), 9.03 (br. s., 1 H), 8.71 (d, J= 5.3 Hz, 1 H), 8.20 (s, 1 H), 8.07 (d, J= 7.0 Hz, 1 H), 7.81 (d, J= 7.3 Hz, 2 H), 7.70 - 7.59 (m, 1 H), 7.55 - 7.38 (m, 3 H), 7.37 - 7.22 (m, 2 H), 6.87 (d, J = 1.0 Hz, 1 H), 6.23 (s, 1 H), 5.35 (s, 1 H), 1.50 (s, 6 H). One exchangeable proton was not observed, m/z (ESI, +ve ion) 538.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.101 μΜ.
Example 141
N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-4-morpholinesulfonamide

1H NMR (300MHz ,CHLOROFORM-d) δ = 8.75 (dd, J= 0.6, 5.3 Hz, 1 H), 8.08 (s, 1 H), 7.95 - 7.87 (m, 1 H), 7.73 (dd, J= 0.9, 7.9 Hz, 1 H), 7.63 (dd, J= 1.8, 7.5 Hz, 1 H), 7.52 - 7.30 (m, 5 H), 7.01 (d, J= 1.2 Hz, 1 H), 6.36 - 6.26 (m, 1 H), 5.41 (d, J= 7.9 Hz, 1 H), 3.69 - 3.57 (m, 2 H), 3.57 - 3.43 (m, 2 H), 3.26 - 3.05 (m, 4 H), 1.89 (d, J= 18.9 Hz, 1 H), 1.65 (s, 6 H). m/z (ESI, +ve ion) 557.9 (M+H)+. GK-GKRP ICso (Binding) = 0.221 μΜ.
Example 142
N-((R)-(2-chlorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-N'-cyclopropylsulfamide

To a stirred suspension of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol hydrochloride (0.050 g, 0.112
mmol, see Example 2, step 2) in DCM (3 mL) was added diisopropylethylamine (0.200 mL, 1.123 mmol, Sigma-Aldrich, St. Louis, MO), 4- dimethylaminopyridine (7.10 mg, 0.056 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropylamine (0.160 mL, 2.24 mmol, Fluka, St. Louis, MO ). The resulting mixture was cooled to 0 °C and added sulfuryl chloride (0.050 mL, 0.555 mmol, Sigma-Aldrich, St. Louis, MO). After the addition, ice bath was removed. The reaction mixture was warmed to rt and continued to be stirred for 4 h. The reaction mixture was partitioned between water (30 mL) and DCM (60 mL). The aqueous layer was extracted with DCM (1 x 30 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The resulting product was purified by column chromatography (24 g of silica, 10 to 40% acetone in hexanes) to afford N-((R)-(2-chlorophenyl)(7-(4-(l -hydroxy- 1- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-N'- cyclopropylsulfamide (0.027 g) as yellow solid.
1H NMR (400MHz ,CHLOROFORM-d) δ = 8.75 (d, J= 5.3 Hz, 1 H), 8.09 (s, 1 H), 7.91 (d, J= 7.2 Hz, 1 H), 7.75 (d, J= 7.4 Hz, 1 H), 7.67 (dd, J= 1.7, 7.5 Hz, 1 H), 7.48 (t, J= 7.7 Hz, 1 H), 7.45 - 7.28 (m, 4 H), 7.12 (d, J = 1.0 Hz, 1 H), 6.33 (d, J= 7.4 Hz, 1 H), 5.50 (d, J= 7.4 Hz, 1 H), 4.72 (s, 1 H), 2.50 - 2.41 (m, 1 H), 1.65 (s, 6 H), 0.72 - 0.64 (m, 1 H), 0.64 - 0.53 (m, 3 H). (1 exchangeable proton was not observed), m/z (ESI, +ve ion) 527.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.290 μΜ.
Example 143
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide

To a solution of 2-(2-(2-(amino(6-amino-3-chloro-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.200 g, 0.471 mmol, see Example 3, step 7) in DMF (4.0 mL) was added 4-dimethylaminopyridine (0.029 g, 0.235 mmol, Sigma-Aldrich, St. Louis, MO), diisopropylethylamine (0.246 mL, 1.412 mmol, Sigma-Aldrich, St. Louis, MO), and 4-chlorosulfonylpyrazole (0.240 g, 1.41 mmol, Matrix scientific, Columbia, SC). The resulting mixture was heated at 70 °C under N2 for 20 h. The reaction was cooled to rt and partitioned between EtOAc (50 mL) and water (30 mL). The organic layer was washed with water (2 x 30 mL), dried over MgS04, filtered, and concentrated. The resulting product was redissolved in MeOH (15 mL) and added 2 M potassium carbonate (5.0 mL, 10.00 mmol). The resulting mixture was heated at 60 °C for 20 h. The reaction mixture was cooled to rt and partitioned between EtOAc (70 mL) and water (30 mL). The organic layer was washed with water (2 x 20 mL), dried over MgS04, filtered, and concentrated. The resuting product was purified by column chromatography (40 g of silica, 1 to 5% MeOH in DCM) to afford N-((6-amino- 3 -chloro-2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide (0.135 g) as an off- white solid.
1H NMR (300MHz ,DMSO-d6) δ = 13.36 (br. s., 1 H), 8.71 (d, J= 5.3 Hz, 1 H), 8.18 (s, 1 H), 8.12 (br. s., 1 H), 8.05 (d, J= 7.2 Hz, 1 H), 7.94 - 7.76 (m, 2 H), 7.58 (br. s., 1 H), 7.53 - 7.41 (m, 2 H), 7.35 (d, J= 8.8 Hz, 1 H), 7.12 (d, J= 0.7 Hz, 1 H), 6.38 (d, J= 8.8 Hz, 1 H), 6.22 (s, 2 H), 6.00 (d, J= 7.9 Hz, 1 H), 5.35 (s, 1 H), 1.50 (s, 6 H). m/z (ESI, +ve ion) 554.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.028 μΜ.
Example 144
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-l-methyl-lH-pyrazole-4- sulfonamide

To a solution of 2-(2-(2-(amino(6-amino-3-chloro-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.200 g, 0.471 mmol, see Example 3, step 7) in DMF (4.0 mL) was added 4-dimethylaminopyridine (0.029 g, 0.235 mmol, Sigma-Aldrich, St. Louis, MO), diisopropylethylamine (0.246 mL, 1.412 mmol, Sigma-Aldrich, St. Louis, MO), and 1 -methyl- lH-pyrazole-4-sulfonyl chloride (0.170 g, 0.941 mmol, ChemBridge, San Diego, CA). The resulting mixture was heated at 70 °C under N2 for 30 min. The reaction mixture was cooled to rt and partitioned between EtOAc (70 mL) and water (30 mL). The organic layer was washed with water (2 x 20 mL), dried over MgS04, filtered, and concentrated. The resulting product was purified by column chromatography (40 g of silica, 1 to 5% MeOH in DCM) to afford N-((6-amino-3-chloro-2- pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-l -methyl- lH-pyrazole-4-sulfonamide (0.210 g) as an off-white solid.
1H NMR (300MHz ,CHLOROFORM-d) δ = 8.74 (d, J= 5.3 Hz, 1 H), 8.04 (s, 1 H), 7.90 - 7.83 (m, 1 H), 7.77 - 7.69 (m, 1 H), 7.57 (s, 1 H), 7.51 - 7.40 (m, 2 H), 7.36 (dd, J= 1.6, 5.3 Hz, 1 H), 7.32 (d, J= 8.8 Hz, 1 H), 7.18 (s, 1 H), 6.68 (d, J = 8.8 Hz, 1 H), 6.37 (d, J= 8.6 Hz, 1 H), 6.23 (d, J= 8.9 Hz, 1 H), 4.64 (br. s., 2 H), 3.55 (s, 3 H), 2.05 (br. s., 1 H), 1.69 - 1.60 (m, 6 H). m/z (ESI, +ve ion) 568.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.136 μΜ.
Examples 145 and 146
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-lH-pyrazole-4- sulfonamide and N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)-l,2- dihydroxy-l-methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-lH- pyrazole-4-sulfonamide

To a solution of 2-(2-(2-(amino(6-amino-3-chloro-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,2-propanediol (0.200 g, 0.454 mmol, Intermediate AA11) in DMF (3.0 mL) was added 4-dimethylaminopyridine (0.028 g, 0.227 mmol, Sigma-Aldrich, St. Louis, MO), diisopropylethylamine (0.237 mL, 1.361 mmol, Sigma-Aldrich, St. Louis, MO), and 4- chlorosulfonylpyrazole (0.226 g, 1.36 mmol, Matrix scientific, Columbia, SC). The resulting mixture was heated at 70 °C under N2 for 20 h. The reaction was cooled to rt and partitioned between EtOAc (50 mL) and water (30 mL). The organic layer was washed with water (2 x 30 mL), dried over MgS04, filtered, and concentrated. The resulting product was redissolved in MeOH (15 mL) and added 2 M potassium carbonate (5.0 mL, 10.00 mmol). The resulting mixture was heated at 60 °C under N2 for 2 h. The reaction mixture was cooled to rt and
and partitioned between EtOAc (70 mL) and water (30 mL). The organic layer was washed with more water (2 x 20 mL), dried over MgSC^, filtered, and concentrated. The resulting product was purified by column chromatography (40 g of silica, 1 to 5% MeOH in DCM) to afford N-((6-amino-3-chloro-2- pyridinyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-lH-pyrazole-4-sulfonamide (0.152 g) as an off-white solid.
The racemic mixture was resolved using preparative SFC Chiralpak® AD- H (Sepax) (250 x 21 mm, 5 mm) columns coupled in tandem 55% i-PrOH with 0.2%) NH3 in supercritical fluid C02 diode ray detector (DAD) [following at 254nm]) to obtain 2 products.
First eluting peak (PEAK# 1)
1H NMR (300MHz ,DMSO-d6) δ = 13.47 - 13.24 (m, 1 H), 8.70 (d, J= 5.3 Hz, 1 H), 8.17 (s, 1 H), 8.03 (d, J= 6.9 Hz, 1 H), 7.84 (d, J= 7.3 Hz, 3 H), 7.55 - 7.42 (m, 2 H), 7.36 (d, J= 8.8 Hz, 1 H), 7.13 (d, J= 0.9 Hz, 1 H), 6.38 (d, J= 8.8 Hz, 1 H), 6.23 (s, 2 H), 6.00 (br. s., 1 H), 5.26 (s, 1 H), 4.83 (br. s., 1 H), 3.64 - 3.40 (m, 2 H), 1.91 (s, 1 H), 1.46 (s, 3 H). m/z (ESI, +ve ion) 570.8 (M+H)+. GK- GKRP IC50 (Binding) = 0.079 μΜ.
Second eluting peak (PEAK# 2)
1H NMR (300MHz ,DMSO-d6) δ = 13.36 (br. s., 1 H), 8.70 (d, J= 5.3 Hz, 1 H), 8.17 (s, 1 H), 8.02 (d, J= 7.2 Hz, 1 H), 7.88 (br. s., 1 H), 7.83 (d, J= 7.3 Hz, 1 H), 7.69 - 7.53 (m, 1 H), 7.53 - 7.42 (m, 2 H), 7.35 (d, J= 8.8 Hz, 1 H), 7.12 (d, J = 1.0 Hz, 1 H), 6.38 (d, J= 8.8 Hz, 1 H), 6.22 (s, 2 H), 6.00 (br. s., 1 H), 5.25 (s, 1 H), 4.82 (t, J= 5.8 Hz, 1 H), 3.62 - 3.39 (m, 2 H), 1.46 (s, 3 H). m/z (ESI, +ve ion) 570.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.009 μΜ.
Example 147
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-l-methyl-lH-pyrazole-4- sulfonamide

To a solution of 2-(2-(2-(amino(6-amino-3-chloro-2-pyridinyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,2-propanediol (0.200 g, 0.454 mmol, Intermediate AA11) in DMF (3.0 mL) was added 4-dimethylaminopyridine (0.028 g, 0.227 mmol, Sigma-Aldrich, St. Louis, MO), diisopropylethylamine (0.237 mL, 1.361 mmol, Sigma-Aldrich, St. Louis, MO), and 1-methyl-lH- pyrazole-4-sulfonyl chloride (0.123 g, 0.680 mmol, ChemBridge, San Diego, CA). The resulting mixture was stirred at rt under N2 for 20 min. The reaction mixture was partitioned between EtOAc (70 mL) and water (30 mL). The organic layer was washed with water (2 x 20 mL), dried over MgS04, filtered, and concentrated. The resulting product was purified by column chromatography (40 g of silica, 1 to 5% MeOH in DCM) to afford N-((6-amino-3-chloro-2- pyridinyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-l -methyl- lH-pyrazole-4-sulfonamide (0.210 g) as an off-white solid.
1H NMR (300MHz ,DMSO-d6) δ = 8.70 (d, J= 5.1 Hz, 1 H), 8.17 (s, 1 H), 8.08 - 7.98 (m, 2 H), 7.90 (d, J= 8.6 Hz, 1 H), 7.84 (d, J= 7.3 Hz, 1 H), 7.55 - 7.43 (m, 3 H), 7.36 (d, J= 8.8 Hz, 1 H), 7.12 (d, J= 0.9 Hz, 1 H), 6.39 (d, J= 8.8 Hz, 1 H), 6.20 (s, 2 H), 5.99 (d, J= 8.0 Hz, 1 H), 5.25 (s, 1 H), 4.82 (t, J= 5.8 Hz, 1 H), 3.68 (s, 3 H), 3.62 - 3.41 (m, 2 H), 1.46 (s, 3 H). m/z (ESI, +ve ion) 584.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.092 μΜ.
Example 148
N-((R)-(2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide

To a solution of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,2-propanediol (0.200 g, 0.471 mmol, see Example 19, step 2) in DMF (3.0 mL) was added 4-dimethylaminopyridine (0.029 g, 0.235 mmol, Sigma-Aldrich, St. Louis, MO), diisopropylethylamine (0.246 mL, 1.412 mmol, Sigma-Aldrich, St. Louis, MO), and 4- chlorosulfonylpyrazole (0.157 g, 0.941 mmol, Matrix scientific, Columbia, SC). The resulting mixture was heated at 75 °C under N2 for 5 h. The reaction mixture was cooled to rt and partitioned between EtOAc (50 mL) and water (30 mL). The organic layer was washed with water (2 x 30 mL), dried over MgS04, filtered, and concentrated. The resulting product was redissolved in MeOH (15 mL) and added 2M potassium carbonate (2.0 mL, 4.00 mmol). The resulting mixture was heated at 60 °C under N2 for 2 h. The reaction mixture was cooled to rt and concentrated. The residue was partitioned between EtOAc (70 mL) and water (30 mL). The organic layer was dried over MgS04, filtered, and concentrated. The resulting product was purified by column chromatography (40 g of silica, 1 to 5% MeOH in DCM) to afford N-((R)-(2-chlorophenyl)(7-(4-(l,2- dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H- pyrazole-4-sulfonamide (0.093 g) as a white solid.
1H NMR (300MHz ,DMSO-d6) δ = 13.36 (br. s., 1 H), 9.03 (br. s., 1 H), 8.71 (d, J= 5.1 Hz, 1 H), 8.19 (s, 1 H), 8.05 (d, J= 7.5 Hz, 1 H), 7.81 (d, J= 7.3 Hz, 3 H), 7.69 - 7.61 (m, 1 H), 7.53 - 7.45 (m, 2 H), 7.45 - 7.38 (m, 1 H), 7.37 - 7.25 (m, 2 H), 6.87 (d, J= 1.2 Hz, 1 H), 6.23 (s, 1 H), 5.26 (br. s., 1 H), 4.83 (br. s., 1 H), 3.64 - 3.44 (m, 2 H), 1.46 (s, 3 H). m/z (ESI, +ve ion) 554.8 (M+H)+. GK- GKRP IC50 (Binding) = 0.075 μΜ.
Example 149
N-((2-chloro-6-fluorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-lH-pyrazole-4-sulfonamide

To a solution of 2-(2-(2-(amino(2-chloro-6-fluorophenyl)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-l,2-propanediol (0.200 g, 0.452 mmol, Intermediate AA9) in DMF (3.0 mL) was added 4-dimethylaminopyridine (0.028 g, 0.226 mmol, Sigma-Aldrich, St. Louis, MO), diisopropylethylamine (0.236 mL, 1.355 mmol, Sigma-Aldrich, St. Louis, MO), and 4-chlorosulfonylpyrazole (0.225 g, 0.903 mmol, Matrix scientific, Columbia, SC). The resulting mixture was heated at 75 °C under N2 for 20 h. The reaction mixture was cooled to rt and partitioned between EtOAc (50 mL) and water (30 mL). The organic layer was washed with water (2 x 30 mL), dried over MgS04, filtered, and concentrated. The resulting product was redissolved in MeOH (15 mL) and added 2M potassium carbonate (2.0 mL, 4.00 mmol). The resulting mixture was heated at 60 °C under N2 for 2 h. The reaction mixture was cooled to rt and concentrated. The residue was partitioned between EtOAc (70 mL) and water (30 mL). The
organic layer was dried over MgSC^, filtered, and concentrated. The resulting product was purified by column chromatography (40 g of silica, 1 to 5% MeOH in DCM) to afford N-((2-chloro-6-fluorophenyl)(7-(4-(l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4- sulfonamide (0.082 g) as a light yellow solid.
1H NMR (300MHz ,DMSO-d6) δ = 13.38 (br. s., 1 H), 8.80 (d, J= 8.8 Hz, 1 H), 8.69 (d, J= 5.3 Hz, 1 H), 8.19 (s, 1 H), 8.11 - 8.00 (m, 2 H), 7.83 (d, J= 7.3 Hz, 1 H), 7.60 (d, J= 1.3 Hz, 1 H), 7.55 - 7.44 (m, 2 H), 7.43 - 7.35 (m, 1 H), 7.35 - 7.28 (m, 1 H), 7.23 - 7.13 (m, 1 H), 7.10 (d, J = 1.2 Hz, 1 H), 6.40 (d, J= 8.6 Hz, 1 H), 5.26 (s, 1 H), 4.82 (t, J= 5.8 Hz, 1 H), 3.67 - 3.40 (m, 2 H), 1.46 (s, 3 H). m/z (ESI, +ve ion) 572.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.172 μΜ.
Example 150
2-(2-(2-((6-amino-3-chloro-2-pyridinyl)(hydroxy)methyl)-l-benzothiophen-7- yl)-4-pyridinyl)-2-propanol

Step 1. In a 100 mL round-bottmed flask, a solution of 2-(2-(l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol (400 mg, 1.485 mmol, Intermediate Y5, azeotroped with toluene (2 x 5 mL)) in THF (8 mL) was cooled in a dry-ice acetone bath under N2. After 10 min, a solution of n-BuLi (2.5 M in hexane) (1.9 mL, 4.75 mmol) was added. The resulting dark brown mixture was stirred for 1 h. A solution of di-tert-butyl (5-chloro-6-formyl-2-pyridinyl)imidodicarbonate (500 mg, 1.401 mmol, see Intermediate AA4, step 2) in THF (4 mL) was added
via syringe followed by THF rinsing (about 2 mL total). After 40 min, saturated NH4C1 (5 mL) was added followed by HC1 (5 N, 1 mL). EtOAc (10 mL) was added. The mixture was allowed to warm to rt. The orange organic layer was dried over Na2S04, filtered, and concentrated. The residue was purified by chromatography on silica using EtOAc in hexane (10 to 50%) as eluent to give a white foam (440 mg, 50%). Both NMR and LCMS indicated this was a mixture (about 1 : 1) of mono-/bis-Boc derivatives. This material was used directly in the next step.
Step 2. 2-(2-(2-((6-amino-3-chloro-2-pyridinyl)(hydroxy)methyl)-l- benzothiophen-7-yl)-4-pyridinyl)-2-propanol. A solution of di-tert-butyl (5- chloro-6-(hydroxy(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-2-pyridinyl)imidodicarbonate (85 mg, 0.136 mmol) (1 : 1 mixture of mono-/bis Boc-derivative) in DCM (54 mL) was treated with TFA (0.6 mL). The mixture was stirred at rt. After the completion of the reaction (1.5 h), the solvent was removed and the residue was treated with MeOH (2 mL) and then with saturated NaHC03 (10 mL). The resulting slurry was stirred at rt for 2 days and filtered. The solid was dissolved in DCM and purified by chromatography on silica using MeOH-EtOAc (5%) in hexanes (0 to 40%) as eluent to give a light yellow powder (36 mg, 62%>).
1H NMR (400 MHz, CHLOROFORM- ) δ ppm 1.63 (s, 6 H), 1.85 (br. s., 1 H), 4.61 (br. s., 1 H), 6.24 (s, 1 H), 6.44 (d, J=8.6 Hz, 1 H), 7.26 (s, 2H, NH2), 7.34 (dd, J=5.2, 1.7 Hz, 1 H), 7.38 - 7.48 (m, 2 H), 7.72 - 7.79 (m, 1 H), 7.80 - 7.87 (m, 1 H), 8.03 (d, J=0.8 Hz, 1 H), 8.74 (d, J=4.9 Hz, 1 H). LCMS (ESI, pos.): calcd for C22H2oClN302S: 425.1; found: (M+l). GK-GKRP IC50 (Binding) = 0.27 μΜ.
Example 151
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l,3-benzothiazol-2-yl)methyl)cyclopropanesulfonamide (Diastereomer of Example 152)

One diastereomer (third peak off HPLC)

Step 2. A stirring mixture of 2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (3.0 g, 13 mmol, Intermediate XI), 7-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-l,3-benzothiazole (4.5 g, 17 mmol, from Step 1),
allylpalladium (II) chloride dimer (240 mg, 0.70 mmol, Sigma-Aldrich, India), 2- dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl (0.63 g, 1.3 mmol, Alfa Aesar, Waltham, MA), sodium carbonate (4.2 g, 40 mmol, Rankem, India), 1,4- dioxane (30 mL), and water (10 mL) was heated at 80 °C under an argon atmosphere. After stirring for 16 h, the reaction mixture was allowed to cool to room temperature, partitioned between water and ethyl acetate, the layers were separated, the aqueous layer was washed with ethyl acetate, and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (4: 1 hexane-ethyl acetate) to give 2-(2-(l,3-benzothiazol-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (1.3 g) as a colorless solid.
Step 3. Lithium diisopropylamide (34 mL of a 1.8 M solution with tetrahydrofuran, 61 mmol, Sigma-Aldrich, India) was added to a stirring solution
of 2-(2-(l ,3-benzothiazol-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol (6.6 g, 20 mmol, from Step 2) and tetrahydrofuran (100 mL) at -78 °C under a nitrogen atmosphere. After stirring for 45 min, a solution of (S)-N-((lE)-(2- chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (5.0 g, 20 mmol, Intermediate Yl) and tetrahydrofuran (50 mL) was added slowly dropwise. After stirring for 2 h, the reaction mixture was quenched with water, the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (70:30 hexane-ethyl acetate) to give (S)-N-((2- chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1,3- benzothiazol-2-yl)methyl)-2-methyl-2-propanesulfinamide (8.0 g) as a colorless solid.
Step 4. (S)-N-((2-chlorophenyl)(7-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 ,3 -benzothiazol-2-yl)methyl)-2-methyl-2- propanesulfmamide as a mixture of four stereoisomers (7.7 g, from Step 3) was subjected to preparative SFC (Chiralpak® AD-H column)(150 mm x 30 mm, 5 μΜ) eluting with 80% liquid C02 in 20% methanol (with 20 mM NH4OH) at a flow rate of 100 mL/min, affording one diastereomer of (S)-N-((R)-(2- chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1,3- benzothiazol-2-yl)methyl)-2-methyl-2-propanesulfinamide as the third eluting peak (3.4 g).
Step 5. Hydrogen chloride (15 mL of a 4.0 M solution with 1,4-dioxane, 60 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of (S)-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 ,3 -benzothiazol-2-yl)methyl)-2-methyl-2- propanesulfmamide (3.4 g, 6.0 mmol, from Step 4) and methanol (60 mL) at room temperature. After stirring for 90 min, the reaction mixture was concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the
organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to give one diastereomer of 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 ,3-benzothiazol-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (2.8 g) as a yellow solid.
Step 6. 4-(Dimethylamino)pyridine (0.074 g, 0.60 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of 2-(2-(2- ((R)-amino(2-chlorophenyl)methyl)- 1 ,3-benzothiazol-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol (2.8 g, 6.0 mmol, from Step 5), N,N-dimethylformamide (30 mL), diisopropylethylamine (5.3 mL, 30 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (0.62 mL, 6.0 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 17 h, saturated aqueous sodium
bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure, the residue was dissolved with
dichloromethane, silica gel (15 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 99: 1 to 49: 1 dichloromethane- methanol), the product-containing fractions were combined, the volatiles were removed under reduced pressure, the residue was dissolved with
dichloromethane, silica gel (2.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (4: 1 hexane-ethyl acetate) to give one diastereomer ofN-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifiuoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l,3-benzothiazol-2-yl)methyl)cyclopropanesulfonamide (0.27 g) as an orange solid.
1H NMR (400 MHz, DMSO-d6) δ 9.05 (d, J= 5.7 Hz, 1 H), 8.86 (d, J= 5.3 Hz, 1 H), 8.40 (s, 1 H), 8.29 (d, J= 7.6 Hz, 1 H), 8.05 (d, J= 7.6 Hz, 1 H), 7.80 (dd, J = 1.9, 7.5 Hz, 1 H), 7.73 - 7.61 (m, 2 H), 7.58 - 7.51 (m, 1 H), 7.49 - 7.35 (m, 2 H), 7.03 (s, 1 H), 6.44 (d, J= 6.3 Hz, 1 H), 2.43 - 2.29 (m, 1 H), 1.81 (s, 3 H), 0.96 - 0.83 (m, 2 H), 0.83 - 0.75 (m, 1 H), 0.75 - 0.65 (m, 1 H). m/z (ESI, pos. ion) 567.7 (M+H)+. GK-GKRP IC50 (Binding) = 0.32 μΜ.
Example 152
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l,3-benzothiazol-2-yl)methyl)cyclopropanesulfonamide
(Diastereomer to Example 151)

Step 1. (S)-N-((2-chlorophenyl)(7-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 ,3 -benzothiazol-2-yl)methyl)-2-methyl-2- propanesulfmamide as a mixture of four stereoisomers (7.7 g, Example 151, Step 3) was subjected to preparative SFC (Chiralpak® AD-H column)(150 mm x 30
mm, 5 μΜ) eluting with 80% liquid C02 in 20% methanol (with 20 mM NH4OH) at a flow rate of 100 mL/min, affording the one diastereomer of (S)-N-((R)-(2- chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1,3- benzothiazol-2-yl)methyl)-2-methyl-2-propanesulfinamide as the fourth eluting peak: (3.4 g).
Step 2. Hydrogen chloride (15 mL of a 4.0 M solution with 1,4-dioxane, 60 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of (S)-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 ,3 -benzothiazol-2-yl)methyl)-2-methyl-2- propanesulfmamide (3.4 g, 6.0 mmol, from Step 1) and methanol (60 mL) at room temperature. After stirring for 90 min, the reaction mixture was
concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to give one diastereomer of 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 ,3-benzothiazol-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (2.8 g) as a yellow solid.
Step 3. 4-(Dimethylamino)pyridine (0.074 g, 0.60 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of 2-(2-(2- ((R)-amino(2-chlorophenyl)methyl)- 1 ,3-benzothiazol-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol (2.8 g, 6.0 mmol, from Step 2), N,N-dimethylformamide (30 mL), diisopropylethylamine (5.3 mL, 30 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (0.62 mL, 6.0 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 17 h, saturated aqueous sodium
bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was
concentrated under reduced pressure, the residue was dissolved with
dichloromethane, silica gel (15 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 99: 1 to 49: 1 dichloromethane- methanol), the product-containing fractions were combined, the volatiles were removed under reduced pressure, the residue was dissolved with
dichloromethane, silica gel (2.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (4: 1 hexane-ethyl acetate) to give one diastereomer of N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l,3-benzothiazol-2-yl)methyl)cyclopropanesulfonamide (0.59 g) as an orange solid.
1H NMR (400 MHz, DMSO-d6) δ 9.04 (br. s., 1 H), 8.86 (d, J= 5.3 Hz, 1 H), 8.40 (s, 1 H), 8.29 (d, J= 7.8 Hz, 1 H), 8.09 - 8.01 (m, 1 H), 7.80 (dd, J= 1.8, 7.6 Hz, 1 H), 7.72 - 7.62 (m, 2 H), 7.54 (dd, J= 1.6, 7.6 Hz, 1 H), 7.49 - 7.36 (m, 2 H), 7.03 (s, 1 H), 6.43 (br. s., 1 H), 2.44 - 2.28 (m, 1 H), 1.81 (s, 3 H), 0.96 - 0.84 (m, 2 H), 0.84 - 0.75 (m, 1 H), 0.75 - 0.65 (m, 1 H). m/z (ESI, pos. ion) 567.8 (M+H)+. GK-GKRP ICso (Binding) = 0.29 μΜ.
Example 153
N-((R)-(2-chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide (Diastereomer to Example 154).


Step 1. A stirring mixture of 2-(2-chloro-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (8.0 g, 36 mmol, Intermediate XI), 2-(5-chloro-l-benzothiophen-7-yl)- 4,4,5,5-tetramethyl-l,3,2-dioxaborolane (10 g, 36 mmol, Intermediate AA8), allylpalladium (II) chloride dimer (0.65 g, 1.8 mmol, Sigma- Aldrich, India), 2- dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl (1.7 g, 3.6 mmol, Alfa Aesar, Waltham, MA), sodium carbonate (11 g, 110 mmol, Rankem, India), 1,4- dioxane (48 mL), and water (11 mL) was heated at 80 °C under an argon atmosphere. After stirring for 16 h, the reaction mixture was allowed to cool to room temperature, partitioned between water and ethyl acetate, the layers were
separated, the aqueous layer was washed with ethyl acetate (3x), the combined organic material was washed with brine (2x), dried (sodium sulfate), and concentrated. The residue was subjected to flash chromatography on silica gel (4: 1 hexane-ethyl acetate) to give 2-(2-(5-chloro-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (8.0 g) as a brown oil.
Step 2. Lithium diisopropylamide (31 mL of a 1.8 M solution with tetrahydrofuran, 56 mmol, Sigma- Aldrich, India) was added to a stirring solution of 2-(2-(5-chloro- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2-propanol (8.0 g, 23 mmol, from Step 1) and tetrahydrofuran (400 mL) at -78 °C under a nitrogen atmosphere. After stirring for 2 h, a solution of (S)-N-((lE)-(2- chlorophenyl)methylidene)-2-methyl-2-propanesulfinamide (5.5 g, 23 mmol, Intermediate Yl) and tetrahydrofuran (100 mL) was added slowly dropwise. After stirring for 2 h, the reaction mixture was quenched with saturated aqueous sodium bicarbonate, allowed to warm to room temperature, partitioned between water and ethyl acetate, the layers were separated, the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash
chromatography on silica gel (3:2 hexane-ethyl acetate) to give (S)-N-((2- chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (5.1 g) as an off-white solid.
Step 3. (S)-N-((2-chlorophenyl)(5-chloro-7-(4-(2,2,2-trifiuoro-l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide as a mixture of four stereoisomers (1.1 g, from Step 2) was subjected to preparative SFC (Chiralcel® OJ-H column)(250 mm x 21 mm, 5 μΜ) eluting with 80% liquid C02 in 20% methanol (with 20 mM NH4OH) at a flow rate of 70 mL/min, affording one diastereomer of (S)-N-((R)-(2- chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-
pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide as the first eluting peak: (0.43 g).
Step 4. Hydrogen chloride (1.8 mL of a 4.0 M solution with 1,4-dioxane, 7.2 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of (S)-N-((R)-(2-chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro- 1 - hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (0.43 g, 0.72 mmol, from Step 3) and methanol (7.2 mL) at room temperature. After stirring for 90 min, the reaction mixture was
concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to give one diastereomer of 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)-5 -chloro- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol (0.36 g) as an off-white solid.
Step 5. 4-(Dimethylamino)pyridine (0.0012 g, 0.010 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of 2-(2-(2-((R)-amino(2-chlorophenyl)methyl)-5 -chloro- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.050 g, 0.10 mmol, from Step 4), N,N- dimethylformamide (1.0 mL), diisopropylethylamine (0.088 mL, 0.50 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (10 μί, 0.10 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 19 h, more cyclopropanesulfonyl chloride (2.0 μί, 0.020 mmol, Matrix, Columbia, SC) was added. After stirring for 23 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was
added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (99: 1
dichloromethane -methanol) to give one diastereomer of N-((R)-(2- chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.040 g) as colorless solid.
1H NMR (400 MHz, chloroform-d) δ 8.78 (d, J= 5.3 Hz, 1 H), 8.10 (s, 1 H), 7.80 (d, J= 2.0 Hz, 1 H), 7.67 (d, J= 2.0 Hz, 1 H), 7.60 (dd, J= 1.8, 7.4 Hz, 1 H), 7.49 - 7.40 (m, 2 H), 7.40 - 7.29 (m, 2 H), 6.97 (d, J= 1.2 Hz, 1 H), 6.37 (d, J = 7.8 Hz, 1 H), 5.56 (d, J= 8.2 Hz, 1 H), 2.82 (br. s., 1 H), 2.24 (m, 1 H), 1.84 (s, 3 H), 1.19 - 1.03 (m, 2 H), 0.89 - 0.80 (m, 1 H), 0.80 - 0.70 (m, 1 H). m/z (ESI, pos. ion) 600.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.19 μΜ.
Example 154
N-((R)-(2-chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide (Diastereomer to Example 153)


Step 1. (S)-N-((2-chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro-l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfinamide as a mixture of four stereoisomers (1.1 g, Example 153, Step 2) was subjected to preparative SFC (Chiralcel® OJ-H column)(250 mm x 21 mm, 5 μΜ) eluting with 80% liquid C02 in 20% methanol (with 20 mM NH4OH) at a flow rate of 70 mL/min, affording one diastereomer of (S)-N-((R)-(2- chlorophenyl)(5-chloro-7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide as the second eluting peak (0.52 g).
Step 2. Hydrogen chloride (2.2 mL of a 4.0 M solution with 1,4-dioxane, 8.6 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of (S)-N-((R)-(2-chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro- 1 - hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (0.52 g, 0.86 mmol, from Step 1) and methanol (8.6 mL) at room temperature. After stirring for 90 min, the reaction mixture was
concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to give one diastereomer of 2-(2-(2-((R)-amino(2- chlorophenyl)methyl)-5 -chloro- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1- trifluoro-2-propanol (0.43 g) as an off-white solid.
Step 3. 4-(Dimethylamino)pyridine (0.0012 g, 0.010 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of 2- (2-(2-((R)-amino(2-chlorophenyl)methyl)-5 -chloro- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.050 g, 0.10 mmol, from Step 2), N,N- dimethylformamide (0.50 mL), diisopropylethylamine (0.088 mL, 0.50 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (10 μί, 0.10 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 19 h, more cyclopropanesulfonyl chloride (5.0 μί, 0.050 mmol, Matrix, Columbia, SC) was added. After stirring for 23 h, more cyclopropanesulfonyl chloride (5.0 μί, 0.050 mmol, Matrix, Columbia, SC) was added. After stirring for 22 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium
bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (99: 1 dichloromethane - methanol), the product-containing fractions were combined and concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (99: 1 dichloromethane-methanol) to give one diastereomer of N-((R)-(2-
chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.018 g) as a pale yellow tar.
1H NMR (400 MHz, chloroform-d) δ 8.79 (d, J= 5.1 Hz, 1 H), 8.10 (s, 1 H), 7.81 (d, J= 1.8 Hz, 1 H), 7.68 (d, J= 1.8 Hz, 1 H), 7.60 (dd, J= 1.8, 7.4 Hz, 1 H), 7.50 - 7.41 (m, 2 H), 7.41 - 7.29 (m, 2 H), 6.98 (s, 1 H), 6.38 (d, J= 8.2 Hz, 1 H), 5.53 (d, J= 8.2 Hz, 1 H), 2.79 (s, 1 H), 2.30 - 2.18 (m, 1 H), 1.84 (s, 3 H), 1.20 - 1.05 (m, 2 H), 0.90 - 0.80 (m, 1 H), 0.80 - 0.69 (m, 1 H). m/z (ESI, pos. ion) 600.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.12 μΜ.
Examples 155a and 155b
(lS)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)spiro[22]pentane-l- sulfonamide and (lR)-N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l- hydroxy-l-methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)spiro[22]pentane-l-sulfonamide

Step 1. A solution of l-bromospiro[22]pentane (11 g, 76 mmol, de Meijere, A.; Ernst, K.; Zuck, B.; Brandl, M.; Kozhushkov, S. I.; Tamm, M.; Yufit, D. S.; Howard, J. A. K.; Labahn, T. Eur. J. Org. Chem. 1999, 3105-3115) and tetrahydrofuran (30 mL) was added slowly dropwise to a stirring mixture of magnesium (3.6 g, 150 mmol, Sigma- Aldrich, India) and tetrahydrofuran (100 mL) at room temperature under an argon atmosphere. After stirring for 30 min, the reaction mixture was cooled to 0 °C and then sulfuryl chloride (20 g, 150 mmol, Sigma- Aldrich, India) was added. After stirring for 15 min, the reaction mixture was concentrated under reduced pressure, diethyl ether was added to the residue, the mixture filtered through a pad of diatomaceous earth, and the filtrate was dried (sodium sulfate) and concentrated under reduced pressure. The residue was subjected to flash chromatography on silica gel (49: 1 pentane- dichloromethane) to give spiro[22]pentane-l-sulfonyl chloride (4.0 g) as a yellow oil.
Step 2. Spiro[22]pentane-l-sulfonyl chloride (0.11 mL, 0.65 mmol, from Step 1) was added to a stirring mixture of (2S)-2-(2-(2-((R)-amino(2- chlorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (0.20 g, 0.43 mmol, Intermediate X8), dichloromethane (1.1 mL), and saturated aqueous sodium bicarbonate (1.1 mL) at room temperature. After stirring for 19 h, more spiro[22]pentane-l-sulfonyl chloride (0.11 mL, 0.65 mmol, from Step 1) was added. After stirring for 24 h, the reaction mixture was partitioned between more ethyl acetate and saturated aqueous sodium
bicarbonate, the layers were separated, the organic material was dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (4: 1 hexane-ethyl acetate) to give N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)spiro [22]pentane- 1 -sulfonamide (0.11 g) as an off-white solid.
Step 3. N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)spiro [22]pentane- 1 - sulfonamide as a mixture of two stereoisomers (0.11 g) was subjected to preparative SFC (Chiralpak® AD-H column)(250 mm x 20 mm, 5 μΜ) eluting with 70% liquid C02 in 30% 1 : 1 : 1 methanol:ethanol:2-propanol (with 20 mM NH4OH) at a flow rate of 60 mL/min, affording two major stereoisomers.
First eluting peak (0.045 g) as an off-white solid.
1H NMR (400 MHz, chloroform-d) δ 8.82 (d, J= 5.3 Hz, 1 H), 8.18 (s, 1 H), 7.90 (d, J= 6.8 Hz, 1 H), 7.74 (d, J= 7.2 Hz, 1 H), 7.59 (dd, J= 1.9, 7.5 Hz, 1 H), 7.52 - 7.41 (m, 3 H), 7.41 - 7.29 (m, 2 H), 7.05 (d, J= 1.4 Hz, 1 H), 6.38 (d, J = 7.4 Hz, 1 H), 5.47 (d, J= 8.4 Hz, 1 H), 2.68 (br. s., 1 H), 2.57 (dd, J= 4.1, 7.4 Hz, 1 H), 1.84 (s, 3 H), 1.57 (t, J= 4.6 Hz, 1 H), 1.24 - 1.18 (m, 1 H), 1.18 - 1.12
(m, 1 H), 1.10 - 1.02 (m, 1 H), 0.84 - 0.76 (m, 1 H), 0.76 - 0.67 (m, 1 H). m/z (ESI, pos. ion) 592.8 (M+H)+. GK-GKRP IC50 (Binding) = 1.5 μΜ.
Second eluting peak (0.044 g) as an off-white solid. 1H NMR (400 MHz, chloroform-d) δ 8.82 (d, J= 5.5 Hz, 1 H), 8.23 (s, 1 H), 7.93 (d, J= 7.6 Hz, 1 H), 7.75 (d, J= 7.8 Hz, 1 H), 7.65 (dd, J= 1.6, 7.6 Hz, 1 H), 7.56 - 7.45 (m, 2 H), 7.45 - 7.40 (m, 1 H), 7.40 - 7.29 (m, 2 H), 7.06 (d, J= 1.0 Hz, 1 H), 6.40 (d, J= 7.6 Hz, 1 H), 5.44 (d, J= 7.6 Hz, 1 H), 2.83 (br. s., 1 H), 2.70 (dd, J = 4.1 , 7.6 Hz, 1 H), 1.85 (s, 3 H), 1.63 (t, J = 4.5 Hz, 1 H), 1.34 (dd, J = 4.8, 7.5 Hz, 1 H), 1.20 - 1.12 (m, 1 H), 1.11 - 1.03 (m, 1 H), 0.89 - 0.77 (m, 2 H). m/z (ESI, pos. ion) 592.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.38 μΜ.
Example 156
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide (Diastereomer to Example 157)


Step 1. Lithium diisopropylamide (40 mL of a 1.8 M solution with tetrahydrofuran, 72 mmol, Sigma- Aldrich, India) was added to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-l,l,l-trifluoro-2-propanol (11 g, 72 mmol, Intermediate Y3) and tetrahydrofuran (150 mL) at -78 °C. After stirring for 30 min, a solution of (S)-N-((lE)-(2-chloro-6-fluorophenyl)methylidene)-2- methyl-2-propanesulfinamide (7.5 g, 29 mmol, Intermediate AA12) and tetrahydrofuran (100 mL) was added slowly dropwise. After stirring for an additional 2 h, the reaction mixture was quenched with saturated aqueous
ammonium chloride (100 mL), the mixture was allowed to warm to room temperature, diluted with water (100 mL), the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (70:30 hexane-ethyl acetate) to give (S)-N-((2-chloro-6-fluorophenyl)(7-(4- (2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)-2-methyl-2-propanesulfinamide (7.0 g) as an off-white solid.
Step 2. (S)-N-((2-chloro-6-fiuorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide as a mixture of four stereoisomers (2.5 g, from Step 2) was subjected to preparative SFC in two steps; Step 1 : (Chiralpak® AD-H
column)(150 mm x 20 mm, 5 μΜ) eluting with 77% liquid C02 in 23% 2- propanol (with 20 mM NH4OH) at a flow rate of 90 mL/min; Step 2: (Chiralcel® OJ-H column)(250 mm x 20 mm, 5 μΜ) eluting with 75% liquid C02 in 25% 2- propanol (with 20 mM NH4OH) at a flow rate of 60 mL/min, affording one diastereomer of (S)-N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide as third eluting peak (1.0 g) as a colorless solid.
Step 3. Hydrogen chloride (4.3 mL of a 4.0 M solution with 1,4-dioxane, 17 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of (S)-N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (1.0 g, 1.7 mmol, from Step 3) and methanol (17 mL) at room temperature. After stirring for 25 min, the reaction mixture was
concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to give one diastereomer of 2-(2-(2-((R)-amino(2-chloro-6-
fluorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (0.82 g) as an off-white solid.
Step 4. 4-(Dimethylamino)pyridine (0.051 g, 0.42 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of 2-(2-(2- ((R)-amino(2-chloro-6-fluorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.20 g, 0.42 mmol, from Step 4), N,N- dimethylformamide (2.1 mL), diisopropylethylamine (0.36 mL, 2.1 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (64 L, 0.62 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 15 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (3x) and brine (2x), dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure. The residue was subjected to flash chromatography on silica gel (99: 1 dichloromethane -methanol), the product-containing fractions were combined and concentrated under reduced pressure, the residue was dissolved with
dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (99: 1 dichloromethane-methanol) to give one diastereomer of N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.068 g) as a yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 8.82 (d, J= 5.3 Hz, 1 H), 8.59 (d, J= 7.2 Hz, 1 H), 8.31 (s, 1 H), 8.11 (d, J= 7.4 Hz, 1 H), 7.91 (d, J= 7.6 Hz, 1 H), 7.60 (d, J = 5.1 Hz, 1 H), 7.58 - 7.41 (m, 3 H), 7.33 (t, J= 9.0 Hz, 1 H), 7.26 (s, 1 H), 7.00 (s, 1 H), 6.49 (d, J = 5.7 Hz, 1 H), 2.41 - 2.30 (m, 1 H), 1.79 (s, 3 H), 0.98 - 0.69 (m, 4 H). m/z (ESI, pos. ion) 585.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.090 μΜ.
Example 157
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. (S)-N-((2-chloro-6-fiuorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide as a mixture of four stereoisomers (2.5 g, Example 156, Step 2) was subjected to preparative SFC in two steps; Step 1 : (Chiralpak® AD-H column)(150 mm x 20 mm, 5 μΜ) eluting with 77% liquid C02 in 23% 2- propanol (with 20 mM NH4OH) at a flow rate of 90 mL/min; Step 2: (Chiralcel® OJ-H column)(250 mm x 20 mm, 5 μΜ) eluting with 75% liquid C02 in 25% 2- propanol (with 20 mM NH4OH) at a flow rate of 60 mL/min; affording one diastereomer of (S)-N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide as the first eluting peak (0.83 g) as an off-white solid.
Step 2. Hydrogen chloride (3.6 mL of a 4.0 M solution with 1,4-dioxane, 14 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of (S)-N-((R)-(2-chloro-6-fluorophenyl)(7-(4-((2,2,2-trifluoro- 1 - hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (0.83 g, 1.4 mmol, from Step 1) and methanol (14 mL) at room temperature. After stirring for 25 min, the reaction mixture was
concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated to give one diastereomer of 2-(2-(2-((R)-amino(2-chloro-6- fluorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1,1,1 -trifluoro-2- propanol (0.60 g) as an off-white solid.
Step 3. 4-(Dimethylamino)pyridine (0.025 g, 0.21 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of one diastereomer of 2-(2-(2- ((R)-amino(2-chloro-6-fluorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,l,l-trifluoro-2-propanol (0.10 g, 0.21 mmol, from Step 2), N,N- dimethylformamide (1.0 mL), diisopropylethylamine (0.18 mL, 1.0 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (32 μί, 0.31 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 41 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (3x) and brine (2x), dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure. The residue was subjected to flash chromatography on silica gel (99: 1 dichloromethane -methanol), the product-containing fractions were combined and concentrated under reduced pressure, the residue was dissolved with
dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash
chromatography on silica gel (99: 1 dichloromethane-methanol) to give one diastereomer of N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(2,2,2-trifluoro-l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.031 g) as a yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 8.82 (d, J= 5.1 Hz, 1 H), 8.59 (d, J= 7.2 Hz, 1 H), 8.31 (s, 1 H), 8.11 (d, J= 7.4 Hz, 1 H), 7.91 (d, J= 7.8 Hz, 1 H), 7.60 (d, J = 5.3 Hz, 1 H), 7.58 - 7.41 (m, 3 H), 7.33 (t, J= 9.0 Hz, 1 H), 7.26 (s, 1 H), 7.00 (s, 1 H), 6.49 (d, J = 5.7 Hz, 1 H), 2.36 (m, 1 H), 1.79 (s, 3 H), 0.99 - 0.70 (m, 4 H). m/z (ESI, pos. ion) 584.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.15 μΜ.
Example 158
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (7.2 mL of a 1.6 M solution with hexane, 11 mmol, Sigma- Aldrich, India) was added to a stirring solution of 2-(2-(l-benzothiophen- 7-yl)-4-pyridinyl)-2-propanol (1.9 g, 6.9 mmol, Intermediate Y5) and
tetrahydrofuran (25 mL) at -78 °C. After stirring for 30 min, (S)-N-((lE)-(2- chloro-6-fluorophenyl)methylidene)-2-methyl-2-propanesulfinamide (1.5 g, 5.7 mmol, Intermediate AA12) was added. After stirring for 2 h, the reaction mixture was quenched with saturated aqueous ammonium chloride (30 mL), water (20 mL) was added, the aqueous material was washed with ethyl acetate (3x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (70:30 hexane-ethyl acetate) to give (S)-N-((2-chloro-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1-
methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfinamide (0.60 g) as a brown solid.
Step 2. (S)-N-((2-chloro-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide as a mixture of two stereoisomers (0.50 g, from Step 1) was subjected to preparative SFC (Chiralcel® OJ-H column)(250 mm x 20 mm, 5 μΜ) eluting with 85% liquid C02 in 15% methanol (with 0.10% NH4OH) at a flow rate of 60 mL/min, affording the desired diastereomer, the second eluting peak: (S)-N-((R)-(2-chloro-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.32 g) as an off-white solid.
Step 3. Hydrogen chloride (1.5 mL of a 4.0 M solution with 1,4-dioxane, 6.0 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of (S)- N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2-pyridinyl)-
1- benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.32 g, 0.60 mmol, from Step 2) and methanol (6.0 mL) at room temperature. After stirring for 30 min, the reaction mixture was concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduce pressure to give
2- (2-(2-((R)-amino(2-chloro-6-fluorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-2-propanol (0.26 g) as an off-white solid.
Step 4. 4-(Dimethylamino)pyridine (0.0074 g, 0.061 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of 2-(2-(2-((R)-amino(2- chloro-6-fluorophenyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.26 g, 0.61 mmol, from Step 3), N,N-dimethylformamide (3.0 mL), diisopropylethylamine (0.53 mL, 3.0 mmol, Sigma-Aldrich, St. Louis, MO), and
cyclopropanesulfonyl chloride (62 μΐ,, 0.61 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 68 h, more cyclopropanesulfonyl chloride (30 L, 0.30 mmol, Matrix, Columbia, SC) was added. After stirring for 23 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (3x) and brine (2x), dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure. The residue was subjected to flash chromatography on silica gel (99: 1 dichloromethane -methanol) to give N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.16 g) as a yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 8.68 (d, J= 5.3 Hz, 1 H), 8.58 (d, J= 8.8 Hz, 1 H), 8.21 (s, 1 H), 8.09 (d, J= 7.4 Hz, 1 H), 7.88 (d, J= 7.6 Hz, 1 H), 7.58 - 7.40 (m, 4 H), 7.33 (t, J= 9.0 Hz, 1 H), 7.24 (s, 1 H), 6.48 (d, J= 8.4 Hz, 1 H), 5.35 (s, 1 H), 2.41 - 2.29 (m, 1 H), 1.50 (s, 6 H), 0.99 - 0.69 (m, 4 H). m/z (ESI, pos. ion) 530.9 (M+H)+. GK-GKRP IC50 (Binding) = 0.035 μΜ.
Example 159
N-((R)-(2-bromo-6-fluorophenyl)(7-(4-(l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide

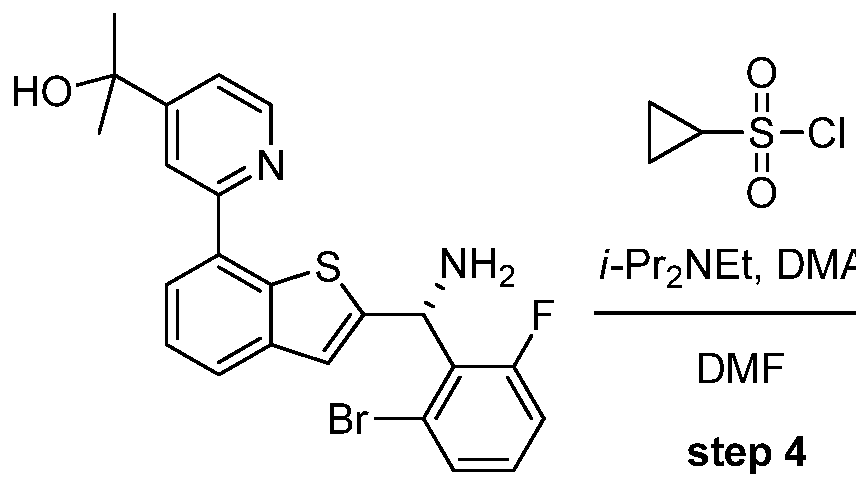
Step 1. Titanium tetraethoxide (26 mL, 120 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of 2-bromo-6-fluorobenzaldehyde (5.0 g, 25 mmol, Matrix, Columbia, SC), dichloromethane (49 mL), and (S)-2- methyl-2-propanesulfinamide (3.0 g, 25 mmol, AK Scientific, Mountain View, CA) at room temperature. After stirring for 5.5 h, more (S)-2-methyl-2- propanesulfmamide (0.30 g, 2.5 mmol, AK Scientific, Mountain View, CA) was added. After stirring for 1.5 h, the reaction mixture was added to a mixture of water (200 mL) and dichloromethane (200 mL), the mixture was stirred vigorously for 20 min, the layers were separated, the aqueous material was washed with dichloromethane (2x), the organic material was filtered through a 0.45 μιη Teflon® filter, the combined organic material was dried (sodium sulfate), filtered, the filtrate was concentrated, the residue was dissolved with
dichloromethane, silica gel (17 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (19: 1 hexane-ethyl acetate) to give (S)-N-((lE)-(2- bromo-6-fluorophenyl)methylidene)-2-methyl-2-propanesulfinamide (7.5 g) as a clear pale yellow oil.
Step 2. n-Butyllithium (0.65 mL of a 2.5 M solution with toluene, 1.6 mmol, Sigma- Aldrich, St. Louis, MO) was added slowly to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.24 g, 0.90 mmol, Intermediate Y5) and tetrahydrofuran (7.0 mL) at -78 °C under a nitrogen atmosphere. After stirring for 10 min, a solution of (S)-N-((lE)-(2-bromo-6- fluorophenyl)methylidene)-2-methyl-2-propanesulfinamide (0.25 g, 0.82 mmol, from Step 1) and tetrahydrofuran (1.1 mL) was added. After stirring for 80 min, saturated aqueous sodium bicarbonate was added, the mixture was allowed to warm to room temperature, partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed with brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (1.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 4: 1 to 2: 1 to 1 : 1 hexane-ethyl acetate) to give (S)-N-((2- bromo-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.27 g) as a colorless solid. (S)-N-((2-bromo-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide as a mixture of two stereoisomers (0.27 g) was subjected to preparative SFC (Chiralcel® OJ-H column)(250 mm x 21 mm, 5 μΜ) eluting with 75% liquid C02 in 25% methanol (with 20 mM NH4OH) at a flow rate of 70 mL/min, affording the desired diastereomer, the second eluting peak: (S)-N-((R)- (2-bromo-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -
benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.15 g) as a colorless solid.
Step 3. Hydrogen chloride (0.65 mL of a 4.0 M solution with 1,4-dioxane, 2.6 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of (S)- N-((R)-(2-bromo-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)-
1- benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.15 g, 0.26 mmol, from Step 2) and methanol (2.6 mL) at room temperature. After stirring for 70 min, the reaction mixture was concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduce pressure to give
2- (2-(2-((R)-amino(2-bromo-6-fluorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-2-propanol (0.12 g) as an off-white solid.
Step 4. 4-(Dimethylamino)pyridine (0.0030 g, 0.025 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of 2-(2-(2-((R)-amino(2- bromo-6-fluorophenyl)methyl)-l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.12 g, 0.26 mmol, from Step 3), N,N-dimethylformamide (1.3 mL),
diisopropylethylamine (0.22 mL, 1.3 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (26 μί, 0.26 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 17 h, more cyclopropanesulfonyl chloride (26 L, 0.26 mmol, Matrix, Columbia, SC) was added. After stirring for 67 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine (2x), dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure.
The residue was subjected to flash chromatography on silica gel (99: 1 dichloromethane -methanol) to give N-((R)-(2-bromo-6-fluorophenyl)(7-(4-(l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.061 g) as a yellow solid.
1H NMR (400 MHz, DMSO-d6) δ 8.69 (d, J= 5.1 Hz, 1 H), 8.57 (d, J= 8.6 Hz, 1 H), 8.21 (s, 1 H), 8.09 (d, J= 7.2 Hz, 1 H), 7.88 (d, J= 7.6 Hz, 1 H), 7.60 (d, J = 8.0 Hz, 1 H), 7.52 (t, J= 7.7 Hz, 1 H), 7.48 (dd, J= 1.4, 5.3 Hz, 1 H), 7.45 - 7.31 (m, 2 H), 7.22 (s, 1 H), 6.47 (d, J= 8.8 Hz, 1 H), 5.35 (s, 1 H), 2.41 - 2.25 (m, 1 H), 1.50 (s, 6 H), 0.98 - 0.68 (m, 4 H). m/z (ESI, pos. ion) 474.8, 476.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.024 μΜ.
Examples 160 and 161
N-((R)-(2-bromo-6-fluorophenyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide and N- ((R)-(2-bromo-6-fluorophenyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide


Step 1. n-Butyllithium (0.77 mL of a 2.5 M solution with toluene, 1.9 mmol, Sigma- Aldrich, St. Louis, MO) was added slowly to a stirring solution of 2-(l-benzothiophen-7-yl)-4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)pyridine (0.69 g, 2.1 mmol, Intermediate AA1) and tetrahydrofuran (18 mL) at -78 °C under a nitrogen atmosphere. After stirring for 10 min, a solution of (S)-N-((lE)-(2- bromo-6-fluorophenyl)methylidene)-2-methyl-2-propanesulfinamide (0.59 g, 1.9 mmol, Example 159, Step 1) and tetrahydrofuran (1.3 mL) was added. After stirring for 85 min, saturated aqueous sodium bicarbonate was added, the mixture was allowed to warm to room temperature, partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed with brine, dried (sodium sulfate), filtered, the
filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (3.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 3: 1 to 2: 1 hexane-ethyl acetate) to give (S)-N-((2-bromo-6-fluorophenyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.85 g) as a colorless solid.
Step 2. Hydrogen chloride (3.4 mL of a 4.0 M solution with 1,4-dioxane, 13 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of (S)- N-((2-bromo-6-fluorophenyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.85 g, 1.3 mmol, from Step 1) and methanol (13 mL) at room temperature. After stirring for 48 h, the reaction mixture was concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduce pressure to give 2-(2-(2-(amino(2-bromo-6-fluorophenyl)methyl)-l-benzothiophen-7-yl)-4- pyridinyl)-l,2-propanediol (0.66 g) as a purple solid.
Step 3. 4-(Dimethylamino)pyridine (0.017 g, 0.14 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of 2-(2-(2-(amino(2-bromo-6- fluorophenyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2-propanediol (0.66 g, 1.4 mmol, from Step 2), N,N-dimethylformamide (6.8 mL),
diisopropylethylamine (1.2 mL, 6.8 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (140 μί, 1.4 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 63 h, more cyclopropanesulfonyl chloride (69 μί, 0.69 mmol, Matrix, Columbia, SC) was added. After stirring for 10 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium
bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine (2x), dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (2.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 49: 1 to 19: 1 dichloromethane-methanol) to give N-((2-bromo-6- fluorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)methyl)cyclopropanesulfonamide (0.41 g) as a light tan solid. N-((2-bromo- 6-fluorophenyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide as a mixture of four stereoisomers (0.41 g) was subjected to preparative SFC in two steps; Step 1 : (Chiralpak® AD-H column)(150 mm x 21 mm, 5 μΜ) eluting with 57% liquid C02 in 43% 2-propanol (with 20 mM NH4OH) at a flow rate of 75 mL/min; Step 2: (Chiralpak® AD-H column)(150 mm x 21 mm, 5 μΜ) eluting with 65% liquid C02 in 35% 2-propanol (with 20 mM NH4OH) at a flow rate of 70 mL/min; affording two major stereoisomers of N-((R)-(2-bromo-6-fluorophenyl)(7-(4- (1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide.
Second eluting peak: (0.10 g) as an off- white solid.
1H NMR (400 MHz, DMSO-d6) δ 8.68 (d, J= 5.1 Hz, 1 H), 8.60 (br. s., 1 H), 8.19 (s, 1 H), 8.07 (d, J= 7.2 Hz, 1 H), 7.88 (d, J= 7.4 Hz, 1 H), 7.60 (d, J= 7.8 Hz, 1 H), 7.52 (t, J= 7.7 Hz, 1 H), 7.49 - 7.31 (m, 3 H), 7.25 - 7.17 (m, 1 H), 6.47 (br. s., 1 H), 5.28 (s, 1 H), 4.84 (t, J= 5.9 Hz, 1 H), 3.61 - 3.43 (m, 2 H), 2.39 - 2.23 (m, 1 H), 1.45 (s, 3 H), 0.97 - 0.67 (m, 4 H). m z (ESI, pos. ion) 590.8, 592.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.068 μΜ.
Third eluting peak: (0.11 g) as a colorless solid.
1H NMR (400 MHz, DMSO-d6) δ 8.68 (d, J= 5.3 Hz, 1 H), 8.60 (br. s., 1 H), 8.19 (s, 1 H), 8.07 (d, J= 7.2 Hz, 1 H), 7.88 (d, J= 7.4 Hz, 1 H), 7.60 (d, J= 7.6 Hz, 1 H), 7.52 (t, J= 7.6 Hz, 1 H), 7.49 - 7.30 (m, 3 H), 7.25 - 7.17 (m, 1 H), 6.47 (d, J= 4.7 Hz, 1 H), 5.28 (s, 1 H), 4.84 (t, J= 5.9 Hz, 1 H), 3.62 - 3.42 (m, 2 H), 2.40 - 2.25 (m, 1 H), 1.45 (s, 3 H), 0.96 - 0.67 (m, 4 H). m/z (ESI, pos. ion) 590.9, 592.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.0080 μΜ.
Example 162
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (0.88 mL of a 2.5 M solution with toluene, 2.2 mmol, Sigma- Aldrich, St. Louis, MO) was added slowly to a stirring solution of
5-chloro-3-fluoro-2-pyridinamine (0.18 g, 1.2 mmol, Combi-Blocks, San Diego, CA) and tetrahydrofuran (9.0 mL) at -78 °C under a nitrogen atmosphere. After stirring for 2 h, a solution of (R)-2-methyl-N-((lE)-(7-(4-(2,2,4-trimethyl-l,3- dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methylidene)-2- propanesulfmamide (0.50 g, 1.1 mmol, Intermediate AA6) and tetrahydrofuran (2.0 mL) was added. After stirring for 4 h, saturated aqueous sodium bicarbonate was added, the reaction mixture was allowed to warm to room temperature, partitioned between ethyl acetate and more saturated aqueous sodium
bicarbonate, the layers were separated, the organic material was washed with brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (3.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 49: 1 to 19: 1 dichloromethane-methanol), the product-containing fractions were combined, concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (1.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (1 :2 hexane-ethyl acetate) to give N-((R)-(2- amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan-4-yl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.11 g) as an off-white solid.
Step 2. Hydrogen chloride (0.10 mL of a 4.0 M solution with 1,4-dioxane, 0.40 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of N- ((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(2,2,4-trimethyl-l,3-dioxolan- 4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2-propanesulfinamide (0.11 g, 0.18 mmol, from Step 1) and methanol (1.8 mL) at 0 °C. After stirring for 1 h, more hydrogen chloride (0.10 mL of a 4.0 M solution with 1,4-dioxane, 0.40 mmol, Sigma-Aldrich, St. Louis, MO) was added. After stirring for 2.5 h, saturated aqueous sodium bicarbonate was added, the mixture was allowed to warm to room temperature, the volatiles were removed under reduced pressure,
the concentrate was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure to give 4-((R)-amino(7-(4-(2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-5-chloro-3-fluoro-2-pyridinamine (0.077 g) as an off-white solid.
Step 3. 4-(Dimethylamino)pyridine (1.9 mg, 0.015 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of 4-((R)-amino(7-(4-(2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5- chloro-3-fluoro-2-pyridinamine (0.077 g, 0.15 mmol, from Step 2), N,N- dimethylformamide (1.5 mL), diisopropylethylamine (0.13 mL, 0.77 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (16 μί, 0.15 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 15 h, more cyclopropanesulfonyl chloride (16 μί, 0.15 mmol, Matrix, Columbia, SC) was added. After stirring for 9 h, more triethylamine (0.13 mL, 0.77 mmol, Sigma- Aldrich, St. Louis, MO) and cyclopropanesulfonyl chloride (16 μί, 0.15 mmol, Matrix, Columbia, SC) were added. After stirring for 15 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was stirred for 1 h, partitioned between more ethyl acetate and saturated aqueous sodium
bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (3x) and brine (2x), dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (1 : 1 hexane-ethyl acetate) to give N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(2,2,4-trimethyl- 1 ,3- dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.041 g) as a clear yellow tar.
Step 4. Hydrogen chloride (0.10 mL of a 4.0 mL solution with 1,4- dioxane, 0.41 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(2,2,4-trimethyl- l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.041 g, 0.68 mmol, from Step 3) and methanol (0.68 mL) at room temperature. After stirring for 24 h, the reaction mixture was concentrated, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (19: 1 dichloromethane -methanol) to give N-((R)-(2-amino-5-chloro-3-fluoro-4- pyridinyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.033 g) as an off- white solid.
1H NMR (400 MHz, CDC13) δ 8.76 (d, J= 5.1 Hz, 1 H), 8.07 (s, 1 H), 7.96 (s, 1 H), 7.90 (d, J= 6.3 Hz, 1 H), 7.75 (d, J= 7.8 Hz, 1 H), 7.53 - 7.44 (m, 1 H), 7.34 (d, J= 5.1 Hz, 1 H), 7.14 (s, 1 H), 6.50 (d, J= 10.5 Hz, 1 H), 5.88 (d, J= 10.5 Hz, 1 H), 4.75 (br. s., 2 H), 3.87 (d, J= 9.4 Hz, 1 H), 3.74 (d, J= 10.8 Hz, 1 H), 2.84 (br. s., 1 H), 2.42 - 2.30 (m, 1 H), 1.58 (s, 3 H), 1.30 (dd, J= 7.0, 14.1 Hz, 1 H), 1.21 (m, 2 H), 0.99 - 0.80 (m, 2 H). m/z (ESI, pos. ion) 562.9 (M+H)+. GK- GKRP IC50 (Binding) = 0.0030 μΜ.
Example 163
N-((R)-(6-amino-3-chloro-5-fluoro-2-pyridinyl)(7-(4-(l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (92 mL of a 2.0 M solution with hexane, 0.18 mol, Sigma-Aldrich, India) was added to a stirring solution of 5-chloro-2,3- difluoropyridine (25 g, 0.17 mol, Combi-B locks, San Diego, CA) and
tetrahydrofuran (1.0 L) at -78 °C under a nitrogen atmosphere. After stirring for 30 min, a solution of trimethylsilylchloride (27 g, 0.25 mol) and tetrahydrofuran (200 mL) was added slowly dropwise. After stirring for an additional 2 h, water was added to the reaction mixture, the aqueous material was washed with ethyl acetate (4x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (hexane) to give (5-chloro-2,3-difluoro-4-(trimethylsilyl)pyridine (21 g) as a pale yellow oil.
Step 2. 2-(Dimethylamino)ethanol (20 g, 90 mmol, Sigma-Aldrich, India) was added slowly dropwise to a stirring solution of n-butyllithium (180 mL of a 2.0 M solution with hexane, 360 mmol, Sigma-Aldrich, India) and hexane (200 mL) at -78 °C under a nitrogen atmosphere. After stirring for 10 min, a solution of (5-chloro-2, 3-difluoro-4-(trimethylsilyl) pyridine (16.28 g, 180.0 mmol, from Step 1) and hexane (200 mL) was added slowly dropwise. After stirring for an additional 2 h, the reaction mixture was slowly poured over crushed dry ice, the mixture was allowed to slowly warm to ambient temperature, the solvent was removed under reduced pressure, the residue was washed with diethyl ether, dissolved with water, the solution was acidified with dilute HC1 (pH = 1.0), the resulting mixture was filtered, and the filter cake was collected to give 3-chloro- 5,6-difluoro-4-(trimethylsilyl)picolinic acid (15 g).
Step 3. Tetrabutylammonium fluoride (150 mL of a 1.0 M solution with tetrahydrofuran, 150 mmol) was added dropwise to a stirring solution of (3- chloro-5,6-difluoro-4-(trimethylsilyl)picolinic acid (15 g, 57 mmol, from Step 2) in tetrahydrofuran (150 mL) at room temperature. After stirring for 6 h, the reaction mixture was diluted with water, acidified with dilute HC1 (pH = 1.0), the aqueous material was extracted with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated to give 3-chloro-5,6- difluoropicolinic acid (8.0 g) as a color less oil.
Step 4. Concentrated sulfuric acid (2.0 mL) was added to a stirring solution of 3-chloro-5,6-difluoropicolinic acid (20 g, 100 mmol, from Step 3) and ethanol (200 mL) at room temperature, and then the reaction mixture was heated at 60 °C. After stirring for 12 h, the reaction mixture was cooled to room temperature, quenched with water and neutralized with saturated aqueous sodium bicarbonate, the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated to give ethyl 3-chloro-5, 6-difluoropicolinate (18 g) as a colorless oil.
Step 5. Sodium borohydride (12 g, 330 mmol, Sigma-Aldrich, India) was added to a stirring solution of ethyl 3 -chloro-5, 6-difluoropicolinate (18 g, 82 mmol, from Step 4), tetrahydrofuran (200 mL), and methanol (60 mL), and then the reaction mixture was heated at 90 °C. After stirring for 2 h, the reaction mixture was cooled to room temperature, the volatiles were removed under reduced pressure, the residue was partitioned between water and ethyl acetate, the layers were separated, the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (19: 1 hexane- ethyl acetate) to give (3 -chloro-5, 6-difluoropyridin-2-yl)methanol (14 g) as a colorless oil.
Step 6. A solution of (3 -chloro-5, 6-difluoropyridin-2-yl)methanol (1.0 g, 5.6 mmol, from Step 5) in aqueous ammonia (20 mL) was heated at 100 °C in a sealed tube. After heating for 24 h, the reaction mixture was cooled to room temperature, extracted with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (4: 1 hexene-ethyl acetate) to give (6-amino-3- chloro-5-fluoropyridin-2-yl) methanol (7.0 g) as a pale yellow oil.
Step 7. tert-Butyldimethylsilylchloride (6.6 g, 44 mmol, Sigma-Aldrich, India) was added to a stirring solution of (6-amino-3 -chloro-5 -fluoropyridin-2-yl)
methanol (7.0 g, 40 mmol, from Step 6), imidazole (8.1 g, 120 mmol), and dichloromethane (70 mL) at 0 °C, and then the reaction mixture was warmed to room temperature. After stirring for 2 h, the reaction mixture was diluted with water, the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography silica gel (19: 1 hexane-ethyl acetate) to give 6-(((tert-butyldimethylsilyl)oxy)methyl)-5-chloro-3- fluoropyridin-2-amine (7.0 g) colorless oil.
Step 8. 4-(Dimethylamino)pyridine (2.9 g, 24 mmol, Sigma- Aldrich, India) was added to a stirring mixture of 6-(((tert- butyldimethylsilyl)oxy)methyl)-5-chloro-3-fluoropyridin-2-amine (7.0 g, 24 mmol, from Step 7), tetrahydrofuran (500 mL), di-tert-butyl dicarbonate (64 g, 250 mmol, Leonide, India), and triethylamine (9.8 g, 97 mmol, Rankem, India) at room temperature. After stirring for 24 h, the reaction mixture was diluted with water and extracted with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (49: 1 hexane-ethyl acetate) to give di-tert-butyl (6- (((tert-butyl(dimethyl)silyl)oxy)methyl)-5 -chloro-3 -fluoro-2- pyridinyl)imidodicarbonate (10 g) as a colorless oil.
Step 9. Tetrabutylammonium fluoride (200 mL of a 1.0 M solution with tetrahydrofuran, 200 mmol) was added to a stirring solution of di-tert-butyl (6- (((tert-butyl(dimethyl)silyl)oxy)methyl)-5 -chloro-3 -fluoro-2- pyridinyl)imidodicarbonate (10 g, 57 mmol, from Step 8) and tetrahydrofuran (200 mL) at room temperature. After stirring for 4 h, the reaction mixture was diluted with water, the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (9: 1 hexane-ethyl acetate) to give di-tert-butyl (5-chloro-3-fluoro-6-(hydroxymethyl)-2- pyridinyl)imidodicarbonate (7.0 g) as a colorless oil.
Step 10. Manganese dioxide (14 g, 160 mmol, Sigma- Aldrich, India) was added to a stirring solution of di-tert-butyl (5-chloro-3-fluoro-6-(hydroxymethyl)- 2-pyridinyl)imidodicarbonate (7.0 g, 84 mmol, from Step 9) and tetrahydrofuran (250 mL) at room temperature. After stirring for 24 h, the reaction mixture was filtered through pad of diatomaceous earth, the filter cake was washed with tetrahydrofuran, and the filtrate was concentrated under reduced pressure to give di-tert-butyl (5-chloro-3-fluoro-6-formyl-2-pyridinyl)imidodicarbonate (7.0 g) as a colorless oil.
Step 11. Copper (II) sulfate (5.0 g, 32 mmol, Sigma-Aldrich, India) was added to a stirring solution of di-tert-butyl (5-chloro-3-fluoro-6-formyl-2- pyridinyl)imidodicarbonate (8.0 g, 21 mmol, from Step 10), (S)-2- methylpropane-2-sulfinamide (3.0 g, 25 mmol, Combi-b locks, USA), and dichloromethane (100 mL) at room temperature. After 24 h, the reaction mixture was filtered through a pad of diatomaceous earth and the filtrate was concentrated under reduced pressure. The residue was subjected to flash chromatography on silica gel (4: 1 hexane-ethyl acetate) to give di-tert-butyl (6-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-5-chloro-3-fluoro-2-pyridinyl)imidodicarbonate (1.3 g) as a colorless oil.
Step 12. n-Butyllithium (1.4 mL of a 2.5 M solution with toluene, 3.6 mmol, Sigma-Aldrich, St. Louis, MO) was added slowly to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol (0.53 g, 2.0 mmol,
Intermediate Y5) and tetrahydrofuran (15 mL) at -78 °C under a nitrogen atmosphere. After stirring for 10 min, a solution of di-tert-butyl (6-((E)-(((S)-tert- butylsulfinyl)imino)methyl)-5-chloro-3-fluoro-2-pyridinyl)imidodicarbonate (0.85 g, 1.8 mmol, from Step 11) and tetrahydrofuran (3.0 mL) was added. After stirring for 3.5 h, saturated aqueous sodium bicarbonate was added, the mixture was allowed to warm to room temperature, partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate, the layers were separated, the
organic material was washed with brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (3.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 2: 1 to 1 : 1 hexane-ethyl acetate) to give di-tert-butyl (6-((R)-(((S)-tert-butylsulfinyl)amino)(7-(4-(l -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5-chloro-3-fluoro-2- pyridinyl)imidodicarbonate (0.056 g) as an off-white solid.
Step 13. Hydrogen chloride (0.19 mL of a 4.0 M solution with 1,4- dioxane, 0.75 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of di-tert-butyl (6-((R)-(((S)-tert-butylsulfinyl)amino)(7-(4-(l-hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-3 -fluoro-2- pyridinyl)imidodicarbonate (0.056 g, 0.075 mmol) and methanol (0.75 mL) at room temperature. After stirring for 4 h, more hydrogen chloride (0.38 mL of a 4.0 M solution with 1,4-dioxane, 1.5 mmol, Sigma-Aldrich, St. Louis, MO) was added. After stirring for 23 h, the reaction mixture was concentrated, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure to give 2-(2-(2-((R)- amino(6-amino-3 -chloro-5 -fluoro-2-pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-2-propanol (0.033 g) as an off-white solid.
Step 14. 4-(Dimethylamino)pyridine (0.91 mg, 7.5 μιηοΐ, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of 2-(2-(2-((R)-amino(6-amino-3- chloro-5 -fluoro-2-pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)-2- propanol (0.033 g, 0.075 mmol, from Step 2), N,N-dimethylformamide (0.75 mL), diisopropylethylamine (0.13 mL, 0.75 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (7.6 μί, 0.075 mmol, Matrix,
Columbia, SC) at room temperature. After stirring for 23 h, saturated aqueous
sodium bicarbonate and ethyl acetate were added, the mixture was stirred for 1 h, partitioned between more ethyl acetate and saturated aqueous sodium
bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (3x) and brine (2x), dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (99: 1 dichloromethane- methanol) to give N-((R)-(6-amino-3-chloro-5-fluoro-2-pyridinyl)(7-(4-(l- hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.027 g) as a colorless solid.
1H NMR (400 MHz, chloroform-d) δ 8.75 (d, J= 5.1 Hz, 1 H), 8.03 (s, 1 H), 7.86 (d, J= 7.6 Hz, 1 H), 7.76 (d, J= 7.8 Hz, 1 H), 7.45 (t, J= 7.6 Hz, 1 H), 7.35 (d, J = 4.7 Hz, 1 H), 7.31 - 7.26 (m, 2 H), 6.31 (s, 2 H), 4.77 (br. s., 2 H), 2.23 - 2.10 (m, 1 H), 1.87 (br. s., 1 H), 1.63 (s, 6 H), 1.19 - 1.08 (m, 2 H), 0.78 - 0.71 (m, 2 H). m/z (ESI, pos. ion) 547.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.010 μΜ.
Example 164
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(l-hydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. Sodium hydride (21 g of a 60 wt% dispersion in mineral oil, 520 mmol, Sigma- Aldrich, India) was added slowly portionwise to a stirring solution of 2-(2-(l-benzothiophen-7-yl)-4-pyridinyl)-2-propanol (70 g, 260 mmol, Intermediate Y5) and tetrahydrofuran (700 mL) at 0 °C. After stirring for an additional 30 min, triethylsilylchloride (78 g, 520 mmol, Sigma-Aldrich, India) was added slowly dropwise, and then the reaction mixture was allowed to warm to room temperature. After stirring for 6 h, the reaction mixture was diluted with water, the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (19: 1 hexane-ethyl acetate) to give 2-(l-benzothiophen-7-yl)-4-(l -methyl- 1- ((triethylsilyl)oxy)ethyl)pyridine (70 g) as a yellow solid.
Step 2. n-Butyllithium (230 mL of a 2.0 M solution with hexane, 460 mmol, Sigma- Aldrich, India) was added to a stirring solution of 2-(l- benzothiophen-7-yl)-4-(l -methyl- l-((triethylsilyl)oxy)ethyl)pyridine (70 g, 180 mmol, from Step 1) and tetrahydrofuran (700 mL) at -78 °C. After stirring for an additional 30 min, N,N-dimethylformamide (80 mL, 1.1 mol) was added. After stirring for 2 h, saturated aqueous sodium bicarbonate was added, the aqueous material was washed with ethyl acetate (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (9: 1 hexane-ethyl acetate) to give 7-(4-( 1 -methyl- 1 - ((triethylsilyl)oxy)ethyl)-2-pyridinyl)-l-benzothiophene-2-carbaldehyde (40 g) as a pale yellow solid.
Step 3. Titanium tetraethoxide (89 g, 390 mmol, Sigma- Aldrich, India) was added to a stirring solution of 7-(4-(l -methyl- l-((triethy lsilyl)oxy)ethyl)-2- pyridinyl)-l-benzothiophene-2-carbaldehyde (40 g, 97 mmol, from Step 2), (R)- 2-methyl-2-propanesulfinamide (13 g, 110 mmol, Combi-Blocks, India), and dichloromethane (400 mL) at room temperature. After stirring for 16 h, water (500 mL) was added, the mixture was filtered through a pad of diatomaceous earth, the aqueous material was washed with dichloromethane (2x), and the combined organic material was dried (sodium sulfate) and concentrated. The residue was subjected to flash chromatography on silica gel (17:3 hexane-ethyl acetate) to give (R)-2-methyl-N-((lE)-(7-(4-(l -methyl- 1- ((triethylsilyl)oxy)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methylidene)-2- propanesulfmamide (25 g) as a pale yellow solid.
Step 4. n-Butyllithium (32 mL of a 2.5 M solution with toluene, 80 mmol, Sigma- Aldrich, St. Louis, MO) was added slowly to a stirring solution of 5- chloro-3-fluoro-2-pyridinamine (6.3 g, 43 mmol, Combi-Blocks, San Diego, CA) and tetrahydrofuran (80 mL) at -78 °C under a nitrogen atmosphere. After stirring for 2 h, a solution of (R)-2-methyl-N-((lE)-(7-(4-(l -methyl- 1- ((triethylsilyl)oxy)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methylidene)-2-
propanesulfinamide (10 g, 19 mmol, from Step 3) and tetrahydrofuran (17 mL) was slowly added. After stirring for 80 min, saturated aqueous sodium
bicarbonate was added, the reaction mixture was allowed to warm to room temperature, partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed with brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (39 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 5 : 1 to 3 : 1 hexane-acetone), the product-containing fractions were combined and concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (19 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (1 :1 hexane-ethyl acetate) to give N-((R)-(2- amino-5 -chloro-3 -fluoro-4-pyridinyl)(7-(4-( 1 -methyl- 1 -((triethy lsilyl)oxy)ethyl)- 2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-methyl-2 -propanesulfinamide (2.9 g) as a pale yellow solid.
Step 5. Hydrogen chloride (11 mL of a 4.0 M solution with 1,4-dioxane, 44 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of N- ((R)-(2-amino-5 -chloro-3 -fluoro-4-pyridinyl)(7-(4-(l -methyl- 1 - ((triethylsilyl)oxy)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (2.9 g, 4.4 mmol, from Step 4) and methanol (44 mL) at room temperature. After stirring for 20 min, the reaction mixture was
concentrated under reduced pressure, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure to give 2-(2-(2-((R)-amino(2-amino-5- chloro-3 -fluoro-4-pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)-2- propanol (1.9 g) as a pale yellow solid.
Step 6. 4-(Dimethylamino)pyridine (0.052 g, 0.43 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of 2-(2-(2-((R)-amino(2-amino-5- chloro-3 -fluoro-4-pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)-2- propanol (1.9 g, 4.3 mmol, from Step 5), N,N-dimethylformamide (4.3 mL), diisopropylethylamine (2.2 mL, 13 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (0.66 mL, 6.4 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 21 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was stirred for 20 min, partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (4x) and brine (2x), dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (8.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (49: 1 dichloromethane -methanol) to give N- ((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(l -hydroxy- l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide (2.0 g) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ 8.70 (d, J= 5.1 Hz, 1 H), 8.67 (d, J= 8.8 Hz, 1 H), 8.22 (s, 1 H), 8.11 (d, J= 7.2 Hz, 1 H), 7.92 (s, 1 H), 7.90 (d, J= 7.4 Hz, 1 H), 7.53 (t, J= 7.6 Hz, 1 H), 7.48 (dd, J= 1.5, 5.2 Hz, 1 H), 7.36 - 7.29 (m, 1 H), 6.56 (s, 2 H), 6.31 (d, J= 8.8 Hz, 1 H), 5.35 (s, 1 H), 2.48 - 2.39 (m, 1 H), 1.50 (s, 6 H), 1.02 - 0.76 (m, 4 H). m/z (ESI, pos. ion) 547.0 (M+H)+. GK-GKRP IC50 (Binding) = 0.0035 μΜ.
Example 165
N-((R)-(6-amino-3-chloro-5-fluoro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (1.2 mL of a 2.5 M solution with toluene, 3.0 mmol, Sigma- Aldrich, St. Louis, MO) was added slowly to a stirring solution of 2-( 1 -benzothiophen-7-yl)-4-((4R)-2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)pyridine (1.1 g, 3.3 mmol, Intermediate AAl-i?) and tetrahydrofuran (22 mL) at -78 °C under a nitrogen atmosphere. After stirring for 10 min, a solution of di-tert-butyl (6-((E)- (((S)-tert-butylsulfinyl)imino)methyl)-5-chloro-3-fluoro-2- pyridinyl)imidodicarbonate (1.3 g, 2.7 mmol, Example 163, Step 11) and tetrahydrofuran (5.2 mL) was added. After stirring for 2.0 h, saturated aqueous sodium bicarbonate was added, the mixture was allowed to warm to room temperature, partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed with brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (6.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 3: 1 to 2: 1 hexane-ethyl acetate) to give di-tert-butyl (6-((R)-
(((S)-tert-butylsulfmyl)amino)(7-(4-((4R)-2,2,4-trimethyl-l,3-dioxolan-4-yl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro-3 -fluoro-2- pyridinyl)imidodicarbonate (0.33 g) as a colorless solid.
Step 2. Hydrogen chloride (2.0 mL of a 4.0 M solution with 1,4-dioxane, 8.0 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of di- tert-butyl (6-((R)-(((S)-tert-butylsulfmyl)amino)(7-(4-((4R)-2,2,4-trimethyl- 1 ,3- dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5 -chloro-3 -fluoro-2- pyridinyl)imidodicarbonate (0.32 g, 0.40 mmol, from Step 1) and methanol (4.0 mL) at 0 °C, and then the reaction mixture was warmed to room temperature. After stirring for 19 h, more hydrogen chloride (2.0 mL of a 4.0 M solution with 1,4-dioxane, 8.0 mmol, Sigma-Aldrich, St. Louis, MO) was added. After stirring for 5 h, the reaction mixture was concentrated, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure to give (2R)-2-(2-(2-((R)-amino(6-amino-3- chloro-5-fluoro-2-pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4-pyridinyl)- 1 ,2- propanediol (0.18 g) as tan solid.
Step 3. 4-(Dimethylamino)pyridine (4.8 mg, 0.039 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of (2R)-2-(2-(2-((R)-amino(6- amino-3 -chloro-5 -fluoro-2-pyridinyl)methyl)- 1 -benzothiophen-7-yl)-4- pyridinyl)-l,2-propanediol (0.18 g, 0.39 mmol, from Step 2), N,N- dimethylformamide (1.3 mL), diisopropylethylamine (0.21 mL, 1.2 mmol, Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (60 μί, 0.59 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 18 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (3x) and brine (2x), dried
(sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (0.50 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (19: 1 chloroform- isopropanol) to give N-((R)-(6-amino-3-chloro-5-fluoro-2-pyridinyl)(7-(4-((lR)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.15 g) as an off- white solid.
1H NMR (400 MHz, DMSO-d6) δ 8.70 (d, J= 5.3 Hz, 1 H), 8.18 (s, 1 H), 8.04 (d, J= 7.4 Hz, 1 H), 7.87 (d, J= 7.8 Hz, 1 H), 7.81 (d, J= 8.8 Hz, 1 H), 7.71 (d, J = 10.4 Hz, 1 H), 7.51 (t, J= 7.7 Hz, 1 H), 7.48 - 7.43 (m, 1 H), 7.22 (s, 1 H), 6.54 (s, 2 H), 6.13 (d, J= 8.4 Hz, 1 H), 5.25 (s, 1 H), 4.82 (t, J= 5.9 Hz, 1 H), 3.66 - 3.43 (m, 2 H), 2.38 - 2.20 (m, 1 H), 1.46 (s, 3 H), 0.99 - 0.83 (m, 2 H), 0.83 - 0.74 (m, 1 H), 0.74 - 0.64 (m, 1 H). m/z (ESI, pos. ion) 562.8 (M+H)+. GK-GKRP ICso (Binding) = 0.0030 μΜ.
Example 166
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)-l-benzothiophen-2- yl)methyl)cyclopropanesulfonamide

Step 1. n-Butyllithium (7.4 mL of a 2.5 M solution with toluene, 18 mmol, Sigma- Aldrich, St. Louis, MO) was added to a stirring solution of 2-(l- benzothiophen-7-yl)-4-((4R)-2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)pyridine (6.0 g, 18 mmol, Intermediate AAl-i?) and tetrahydrofuran (62 mL) at -78 °C under a nitrogen atmosphere. After stirring for 10 min, N,N-dimethylformamide (7.1 mL, 92 mmol, Sigma-Aldrich, St. Louis, MO) was added. After stirring for 20 min, saturated aqueous sodium bicarbonate was added, the reaction vessel was removed from the cooling bath, the mixture was stirred for 10 min, partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure to give 7-(4-((4R)-2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophene-2-carbaldehyde (6.5 g) as a brown-yellow solid.
Step 2. Titanium tetraethoxide (19 mL, 92 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of 7-(4-((4R)-2,2,4-trimethyl-l,3- dioxolan-4-yl)-2-pyridinyl)-l-benzothiophene-2-carbaldehyde (6.5 g, 18 mmol, from Step 1), dichloromethane (37 mL), and (R)-2-methyl-2-propanesulfinamide (2.2 g, 18 mmol, AK Scientific, Mountain View, CA) at 0 °C under a nitrogen atmosphere, and then the reaction mixture was allowed to warm to room temperature. After stirring for 15 h, the reaction mixture was added to a rapidly stirring mixture of saturated aqueous sodium bicarbonate (125 mL), water (125 mL), and dichloromethane (250 mL), the mixture was stirred vigorously for 20 min, partitioned between more water (100 mL) and dichloromethane (100 mL), the layers were separated, the aqueous material was washed with
dichloromethane (3x), the organic material was filtered through a 0.45 μιη Teflon® filter, the combined organic material was dried (sodium sulfate), filtered, the filtrate was concentrated, the residue was dissolved with dichloromethane, silica gel (42 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (gradient elution; 4:1 to 2: 1 hexane-ethyl acetate) to give (R)-2-methyl-N- ((lE)-(7-(4-((4R)-2,2,4-trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l- benzothiophen-2-yl)methylidene)-2-propanesulfinamide (7.5 g) as an off-white solid.
Step 3. n-Butyllithium (8.6 mL of a 2.5 M solution with toluene, 22 mmol, Sigma-Aldrich, St. Louis, MO) was added slowly to a stirring solution of 5-chloro-3-fiuoro-2-pyridinamine (1.7 g, 12 mmol, Combi-Blocks, San Diego, CA) and tetrahydrofuran (20 mL) at -78 °C under a nitrogen atmosphere. After stirring for 2 h, a solution of (R)-2-methyl-N-((lE)-(7-(4-((4R)-2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methylidene)-2- propanesulfmamide (2.4 g, 5.3 mmol, from Step 2) and tetrahydrofuran (6.3 mL) was added. After stirring for 1 h, saturated aqueous sodium bicarbonate was added, the reaction mixture was allowed to warm to room temperature, partitioned between ethyl acetate and more saturated aqueous sodium
bicarbonate, the layers were separated, the organic material was washed with brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (13 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (1 :2 hexane- ethyl acetate) to give N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-((4R)- 2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- methyl-2-propanesulfinamide (1.1 g) as a yellow solid.
Step 4. Hydrogen chloride (1.4 mL of a 4.0 M solution with 1,4-dioxane, 5.5 mmol, Sigma- Aldrich, St. Louis, MO) was added slowly to a stirring solution ofN-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-((4R)-2,2,4-trimethyl- l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-2-methyl-2- propanesulfmamide (1.1 g, 1.8 mmol, from Step 3) and methanol at 0 °C. After stirring for 6 h at 0 °C, the reaction vessel was cooled to -20 °C. After standing for 15 h at -20 °C, the reaction mixture was warmed to 0 °C. After stirring for 8 h at 0 °C, saturated aqueous sodium bicarbonate was added, the reaction mixture was allowed to warm to room temperature, the volatiles were removed under reduced pressure, the concentrate was partitioned between ethyl acetate and more saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried (sodium sulfate), filtered, and the filtrate was concentrated under reduced pressure to give 4-((R)-amino(7-(4-((4R)-2,2,4-trimethyl-l,3-dioxolan-4- yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5-chloro-3-fluoro-2-pyridinamine (0.91 g) as a light tan solid.
Step 5. 4-(Dimethylamino)pyridine (0.022 g, 0.18 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of 4-((R)-amino(7-(4-((4R)-2,2,4- trimethyl-l,3-dioxolan-4-yl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-5- chloro-3-fluoro-2-pyridinamine (0.91 g, 1.8 mmol, from Step 4), N,N- dimethylformamide (1.8 mL), diisopropylethylamine (0.95 mL, 5.5 mmol,
Sigma-Aldrich, St. Louis, MO), and cyclopropanesulfonyl chloride (0.28 mL, 2.7 mmol, Matrix, Columbia, SC) at room temperature. After stirring for 18 h, saturated aqueous sodium bicarbonate and ethyl acetate were added, the mixture was stirred for 10 min, partitioned between more ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (3x) and brine (2x), dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (5.0 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (1 : 1 hexane- ethyl acetate) to give N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-((4R)- 2,2,4-trimethyl- 1 ,3-dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.75 g) as an off- white solid.
Step 6. Hydrogen chloride (1.9 mL of a 4.0 M solution with 1,4-dioxane, 7.5 mmol, Sigma-Aldrich, St. Louis, MO) was added to a stirring solution of N- ((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-((4R)-2,2,4-trimethyl-l,3- dioxolan-4-yl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide (0.75 g, 1.2 mmol, from Step 5) and methanol (12 mL) at room temperature. After stirring for 14 h, the reaction mixture was concentrated, the residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate, the layers were separated, the organic material was washed sequentially with saturated aqueous sodium bicarbonate (2x) and brine, dried (sodium sulfate), filtered, the filtrate was concentrated under reduced pressure, the residue was dissolved with dichloromethane, silica gel (3.5 g) was added to the solution, and the volatiles were removed under reduced pressure. The residue was subjected to flash chromatography on silica gel (19: 1 chloroform-isopropanol) to give N-((R)-(2-amino-5-chloro-3-fluoro-4- pyridinyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide (0.60 g) as a colorless solid.
1H NMR (400 MHz, DMSO-d6) δ 8.74 - 8.63 (m, 2 H), 8.20 (s, 1 H), 8.08 (d, J = 7.2 Hz, 1 H), 7.92 (s, 1 H), 7.90 (d, J= 7.6 Hz, 1 H), 7.53 (t, J= 7.6 Hz, 1 H), 7.47 (dd, J= 1.3, 5.2 Hz, 1 H), 7.36 - 7.28 (m, 1 H), 6.56 (s, 2 H), 6.31 (d, J= 8.6 Hz, 1 H), 5.26 (s, 1 H), 4.82 (t, J= 5.9 Hz, 1 H), 3.63 - 3.44 (m, 2 H), 2.48 - 2.39 (m, 1 H), 1.46 (s, 3 H), 1.03 - 0.74 (m, 4 H). m/z (ESI, pos. ion) 562.8 (M+H)+. GK-GKRP IC50 (Binding) = 0.0010 μΜ.
The stereochemistry of the imine addition using an organolithium species was assigned based on literature precedence. (Synthesis and Applications of tert- Butanesulfinamide, M. T. Robak; M. A. Herbage; J. A. Ellman, Chem. Rev. 2010, 110, 3600-3740.)
Claims
1. A compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein:

I
* is a chiral center having the R configuration, except when X6 is N, then * is a chiral center having the S configuration;
Y is

X1 is N, CH or C-halo;
X2 is N, CH or C-halo;
X3 is N, CH or C-halo; X4 is N or CH; X5 is N or CH; X6 is S or NH;
R1 is Ci_6alkyl, -N(Ci_6alkyl)2, -N(H)three to eight membered cycloalkyl,


R2 is phenyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl or thiazolyl where the phenyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl or thiazolyl group can be
unsubstituted or substituted with from one to three substituents independently selected from Ci_6alkyl, Ci_6haloalkyl, C2_6alkenyl, halo, -SCi_6alkyl, -OCi_6alkyl, -OCi_6haloalkyl -OH, -NH2, -N02 or -N(Ci_6alkyl)2, C2_6alkynyl, -S02Ci_6alkyl, or -C(=0)Ci_6alkyl; 3 is



-S(=0)Ci_6alkyl, -S02NH2 or -S02Ci_6alkyl; n is 0, 1, 2, 3 or 4; m is 0, 1 or 2;
R4 is hydrogen or Ci_6alkyl;
R5 is hydrogen,
 or Ci_6haloalkyl; and
or Ci_6haloalkyl; and
R6 is hydrogen or halo, provided that R1 is not dimethoxyphenyl.
2. A compound in accordance with claim 1, or a pharmaceutically acceptable salt thereof, wherein:
X1 is N or CH;
X2 is N or CH;
X3 is N or CH;
_6alkyl, -N(Ci_6alkyl)2

R2 is phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl, where the phenyl, pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl group can be unsubstituted or substituted with from one to three substituents independently selected from Ci_6alkyl, Ci_6haloalkyl, C2_6alkenyl, halo, -SCi_6alkyl, -OCi_6alkyl,
-OCi_6haloalkyl -OH, -NH2, -N02 or -N(Ci_6alkyl)2; and
R3 is




3. A compound in accordance with any one of claims 1 or 2, or a pharmaceutically acceptable salt thereof, wherein Y is

4. A compound in accordance with any one of claims 1 or 3, or a
pharmaceutically acceptable salt thereof, wherein X1 is CH.
5. A compound in accordance with any one of claims 1 or 3, or a
pharmaceutically acceptable salt thereof, wherein X1 is N.
6. A compound in accordance with any one of claims 1 or 3 to 5, or a pharmaceutically acceptable salt thereof, wherein X2 is CH.
7. A compound in accordance with any one of claims 1 or 3 to 5, or a pharmaceutically acceptable salt thereof, wherein X2 is C-halo.
8. A compound in accordance with any one of claims 1 or 3 to 7, or a pharmaceutically acceptable salt thereof, wherein X3 is CH.
9. A compound in accordance with any one of claims 1 or 3 to 7, or a pharmaceutically acceptable salt thereof, wherein X3 is N.
10. A compound in accordance with any one of claims 1 to 9, or a
pharmaceutically acceptable salt thereof, wherein X4 is CH.
11. A compound in accordance with any one of claims 1 to 9, or a
pharmaceutically acceptable salt thereof, wherein X4 is N.
12. A compound in accordance with claim 1, or a pharmaceutically acceptable salt thereof, wherein X1, X2, X3, X4 and X5 are CH, and X6 is S.
13. A compound in accordance with claim 1, or a pharmaceutically acceptable salt thereof, wherein X1, X2, X3 and X4 are CH, X5 is N, and X6 is S.
14. A compound in accordance with claim 1, or a pharmaceutically acceptable salt thereof, wherein X1 is N, X2, X3 and X4 are CH, X5 is CH, and X6 is S.
15. A compound in accordance with any one of claims 1 to 14, or a
pharmaceutically acceptable salt thereof, wherein R4 is hydrogen.
16. A compound in accordance with any one of claims 1 or 3 to 15, or a pharmaceutically acceptable salt thereof, wherein R1 is Ci_6alkyl, three to eight membered cycloalkyl, phenyl or pyridyl, which cycloalkyl, phenyl or pyridyl groups may be unsubstituted or substituted with from one to three substituents independently selected from halo, -OCi_6alkyl, -NH2 or Ci_6alkyl.
17. A compound in accordance with any one of claims 1 or 3 to 15, or a pharmaceutically acceptable salt thereof, wherein R1 is three to eight membered cycloalkyl, substituted phenyl or substituted pyridyl.
18. A compound in accordance with any one of claims 1 or 3 to 15, or a pharmaceutically acceptable salt thereof, wherein R1 is cyclopropyl or substituted cyclopropyl.
19. A compound in accordance with any one of claims 1 or 3 to 18, or a pharmaceutically acceptable salt thereof, wherein R2 is phenyl or substituted phenyl.
20. A compound in accordance with any one of claims 1 or 3 to 18, or a pharmaceutically acceptable salt thereof, wherein R2 is pyridyl or substituted pyridyl.
21. A compound in accordance with any one of claims 1 or 3 to 18, or a pharmaceutically acceptable salt thereof, wherein R2 is

22. A compound in accordance with any one of claims 1 or 3 to 21, or a pharmaceutically acceptable salt thereof, wherein R3 is

23. A compound in accordance with any one of claims 1 or 3 to 21, or a pharmaceutically acceptable salt thereof, wherein R3 is

24. A compound in accordance with any one of claims 1 or 3 to 21, or a pharmaceutically acceptable salt thereof, wherein R3 is

25. A compound in accordance with any one of claims 1 or 3 to 21, or a pharmaceutically acceptable salt thereof, wherein R3 is

26. A compound in accordance with any one of claims 1 to 25, or a
pharmaceutically acceptable salt thereof, wherein m is 0.
27. A compound in accordance with any one of claims 1 to 25, or a
pharmaceutically acceptable salt thereof, wherein m is 1.
28. The compound, or a pharmaceutically acceptable salt thereof, selected from: N-((R)-(6-amino-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((2-chloro-6-methoxyphenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyrimidinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(3 -chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide;
4-methoxy-N-(phenyl(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)benzenesulfonamide;
N-(phenyl(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)benzenesulfonamide;
4-methoxy-N-((2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)benzenesulfonamide; N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-(methylsulfanyl)phenyl)(7-(4-(( 1 R)-2,2,2-trifiuoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -methyl-6-oxo- 1 ,6-dihydro-3 - pyridinesulfonamide ;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-methoxy-3 -pyridinesulfonamide; 6-amino-N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-fluorobenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxyethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxycyclobutyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
6-chloro-N-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-oxo- 1 ,6-dihydro-3- pyridinesulfonamide ;
N-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -methoxybenzenesulfonamide;
N'-((R)-(2-chlorophenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-N,N-dimethylsulfamide;
N-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)ethanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)methanesulfonamide;
N-((R)-(3-(dimethylamino)-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-chloro-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-methoxy-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3 -methyl-2-pyridinyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-methoxyphenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-nitrophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-methylphenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
6-chloro-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -pyridinesulfonamide; N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-oxo- 1 ,6-dihydro-3- pyridinesulfonamide ;
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
( 1 S,2S)-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- ethylcyclopropanesulfonamide;
( 1 R,2R)-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2- ethylcyclopropanesulfonamide;
( 1 S ,2R)-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-( 1 - methylethyl)cyclopropanesulfonamide;
( 1 R,2S)-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-2-( 1 - methylethyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-oxo- 1,6- dihydro-3 -pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -
(trifluoromethyl)ethyl)-2-pyridinyl)-l-benzothiophen-2-yl)methyl)-4- methoxybenzenesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(5 -( 1 -hydroxy- 1 -methylethyl)-2-thiophenyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(4-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H- benzimidazol-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 H-indol- 2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -benzofuran-5-sulfonamide;
N-((R)-(2-( 1 -methylethenyl)phenyl)(7-(4-(( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-( 1 -methylethenyl)phenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(5-(l -hydroxy- 1 -methylethyl)-6-oxo- 1 ,6-dihydro-3- pyridazinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifluoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)-3- methylbenzofuran-5 -sulfonamide;
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifluoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo [b]thiophen-2-yl)methyl)- 1 -ethyl-6-oxo- 1 ,6-dihydropyridine-3-sulfonamide; or
N-((R)-(6-amino-3-chloropyridin-2-yl)(7-(4-((S)- 1,1,1 -trifluoro-2- hydroxypropan-2-yl)pyridin-2-yl)benzo[b]thiophen-2-yl)methyl)benzofuran-5- sulfonamide.
29. The compound, or a pharmaceutically acceptable salt thereof, selected from: N-((R)-(5-amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((5-Amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)thieno[3,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)thieno [3 ,2- b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)tetrahydro-3-thiophenesulfonamide 1 , 1 -dioxide hydrochloride;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-( 1 R)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5-chloro- 1 ,3-thiazol-4-yl)(7-(4-(2,2,2-trifluoro-l -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(5-amino-2-chlorophenyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2-pyridinyl)-
1- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((5-amino-2-chlorophenyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl) cyclopropanesulfonamide;
N-((S)-(2-amino-5 -chloro-4-pyrimidinyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5 -chloro-4-pyrimidinyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5 -chloro-4-pyrimidinyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(7-(4-(( 1 S)-l ,2-dihydroxy- 1 -methy lethyl)-2-pyridinyl)- 1 -benzothiophen-
2- yl)(2-(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide;
N-((R)-(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen- 2-yl)(2-(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- (hydroxymethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)-2,2,2-trifluoro-l -hydro xy-1 - (hydroxymethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)(2-
(methylsulfanyl)phenyl)methyl)cyclopropanesulfonamide;
N-((7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)(2-
(methylsulfonyl)phenyl)methyl)cyclopropanesulfonamide;
N-((2-amino-5 -chloro-4-pyridinyl)(7-(4-(( 1 S)-l ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy- l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -cyclopropylmethanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -ethyl-6-oxo- 1 ,6-dihydro-3- pyridinesulfonamide ;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-6-oxo- 1 -(2,2,2- trifluoroethyl)- 1 ,6-dihydro-3-pyridinesulfonamide;
N-((R)-(3-chloro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-chloro-2-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-((lS)-l ,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)-5 -chloro- 1 -ethyl-6-oxo- l,6-dihydro-3-pyridinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (hydroxymethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifluoro-l-hydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)-3 -methyl- 1 -benzofuran-5 -sulfonamide; N-((R)-(2-chlorophenyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((lS)-l,2-dihydroxy-l-methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((S)-(2-chlorophenyl)(7-(4-(( 1 R)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)-
1- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((S)-(2-chlorophenyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)-
2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3 -chloro-2-pyridinyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methyl-3 -butyn- 1 -yl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2-cyano- 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)-
1- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy-2-methoxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2-cyano-l -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(S-methylsulfonimidoyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(5-chloro-7-(4-(l ,2-dihydroxy- 1 -methylethyl)-
2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((R)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((S)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((S)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((R)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((S)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((S)-S-methylsulfonimidoyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy- l-methylethyl)-2- pyridinyl)thieno[2,3-c]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-ethynylphenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
2-(2-((6-amino-3-chloro-2-pyridinyl)((cyclopropylsulfonyl)amino)methyl)-l- benzothiophen-7-yl)-4-pyridinesulfonamide;
N-((6-amino-3 -chloro-2-pyridinyl)(7-(4-(3 -hydroxytetrahydro-3 -furanyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-( 1 -hydroxy- 1 -methylpropyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3 -chloro-2-pyridinyl)(7-(4-(methylsulfinyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(methylsulfonyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((2-chlorophenyl)(7-(4-( 1 -cyclopropyl- 1 -hydroxy ethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((3S)-3-hydroxytetrahydro-3-furanyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((3R)-3-hydroxytetrahydro-3-furanyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l -cyclopropyl- 1 -hydroxy ethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy cyclopropyl)-2 -pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2 -methyl- 1 H-imidazol- 1 -yl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((7-(4-acetyl-2-pyridinyl)-l-benzothiophen-2-yl)(6-amino-3-chloro-2- pyridinyl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - (trifluoromethyl)ethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(3-hydroxy-3-oxetanyl)-2-pyridinyl)-l- benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((lS)-2,2,2-trifiuoro-l-hydroxy-l-methylethyl)-2- pyridinyl)- 1 H-indol-2-yl)methyl)cyclopropanesulfonamide;
R)-N-((2-chlorophenyl)(7-(4-(2-hydroxypropan-2-yl)pyridin-2-yl)-lH-indol-2- yl)methyl)cyclopropanesulfonamide;
2-(2-(2-(amino(3-amino-6-chloro-2-fluorophenyl)methyl)-l-benzothiophen-7- yl)-4-pyridinyl)- 1 ,2-propanediol;
N-((R)-(3-amino-6-chloro-2-fiuorophenyl)(7-(4-((lR)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(3-amino-6-chloro-2-fiuorophenyl)(7-(4-((l S)-l ,2-dihydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((3-amino-6-chloro-2-fluorophenyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -cyclopropylmethanesulfonamide; N-((3-amino-6-chloro-2-fluorophenyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -cyclopropylmethanesulfonamide; N-((R)-(5-amino-2-chloro-4-fiuoro-3-pyridinyl)(7-(4-((lR)-l ,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(5-amino-2-chloro-4-fluoro-3-pyridinyl)(7-(4-((lS)-l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((5-amino-2-chloro-4-fluorophenyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(2-hydroxypropan-2-yl)pyridin-2- yl)thieno [3 ,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l,2dihydroxypropan-2-yl)pyridin-2- yl)thieno [3 ,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-(l,2-dihydroxypropan-2-yl)pyridine-2- yl)-5-fluorobenzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((2-chloro-6-fluorophenyl)(7-(4-(l,2-dihydroxypropan-2-yl)pyridin-2- yl)thieno [3 ,2-b]pyridin-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-((R)-l,2-dihydroxypropan-2-yl)pyridin- 2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((6-amino-3-chloropyridin-2-yl)(7-(4-((S)-l ,2-dihydroxypropan-2-yl)pyridin- 2-yl)benzo[b]thiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)-4-morpholinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-((l R)-2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -azetidinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methy 1)- 1 H-pyrazole-4-sulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-4-morpholinesulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)-N'-cyclopropylsulfamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy- l-methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l -hydroxy- l-methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -methyl- 1 H-pyrazole-4-sulfonamide;
N-((R)-(6-amino-3 -chloro-2-pyridinyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)- 2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((R)-(6-amino-3-chloro-2-pyridinyl)(7-(4-((lR)-l,2-dihydroxy-l-methylethyl)- 2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((6-amino-3-chloro-2-pyridinyl)(7-(4-(l,2-dihydroxy-l-methylethyl)-2- pyridinyl)- 1 -benzothiophen-2-yl)methyl)- 1 -methyl- 1 H-pyrazole-4-sulfonamide; N-((R)-(2-chlorophenyl)(7-(4-( 1 ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methyl)- 1 H-pyrazole-4-sulfonamide;
N-((2-chloro-6-fluorophenyl)(7-(4-(l ,2-dihydroxy- 1 -methylethyl)-2-pyridinyl)- 1 - benzothiophen-2-yl)methy 1)- 1 H-pyrazole-4-sulfonamide;
N-((R)-(2-chlorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -methylethyl)-2- pyridinyl)- 1 ,3 -benzothiazol-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chlorophenyl)(5-chloro-7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
( 1 S)-N-((R)-(2-chlorophenyl)(7-(4-((l S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)spiro [22]pentane- 1 - sulfonamide;
( 1 R)-N-((R)-(2-chlorophenyl)(7-(4-(( 1 S)-2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2-yl)methyl)spiro [22]pentane- 1 - sulfonamide;
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 - methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-chloro-6-fluorophenyl)(7-(4-( 1 -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-bromo-6-fluorophenyl)(7-(4-(l -hydroxy- 1 -methylethyl)-2-pyridinyl)- l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-bromo-6-fluorophenyl)(7-(4-(( 1 S)- 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-bromo-6-fluorophenyl)(7-(4-(( 1 R) - 1 ,2-dihydroxy- 1 -methylethyl)-2- pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(l,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-5-fluoro-2-pyridinyl)(7-(4-(l -hydroxy- 1-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-(l -hydroxy- 1-methylethyl)- 2-pyridinyl)-l-benzothiophen-2-yl)methyl)cyclopropanesulfonamide;
N-((R)-(6-amino-3-chloro-5-fluoro-2-pyridinyl)(7-(4-((lR)-l ,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide; or
N-((R)-(2-amino-5-chloro-3-fluoro-4-pyridinyl)(7-(4-((lR)-l ,2-dihydroxy-l- methylethyl)-2-pyridinyl)- 1 -benzothiophen-2- yl)methyl)cyclopropanesulfonamide.
30. A method of treating type 2 diabetes, hyperglycemia, impaired glucose tolerance, insulin resistance, retinopathy, nephropathy, neuropathy, cataracts, glaucoma, Syndrome X, or polycystic ovarian syndrome, the method comprising administering to a patient in need thereof a therapeutically effective amount of a compound in accordance with any one of claims 1 to 29, or a pharmaceutically acceptable salt thereof.
31. The method of claim 30 wherein the treatment is for type 2 diabetes.
32. A pharmaceutical composition comprising a compound in accordance with any one of claims 1 to 29, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261647336P | 2012-05-15 | 2012-05-15 | |
| US61/647,336 | 2012-05-15 | ||
| US201361775344P | 2013-03-08 | 2013-03-08 | |
| US61/775,344 | 2013-03-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013173382A1 true WO2013173382A1 (en) | 2013-11-21 |
Family
ID=48576524
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/041011 WO2013173382A1 (en) | 2012-05-15 | 2013-05-14 | Benzothiophene sulfonamides and other compounds that interact with glucokinase regulatory protein |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2013173382A1 (en) |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005095418A1 (en) * | 2004-04-02 | 2005-10-13 | Novartis Ag | Sulfonamide-thiazolpyridine derivatives as glucokinase activators useful the treatment of type 2 diabetes |
| WO2007084391A2 (en) | 2006-01-18 | 2007-07-26 | Amgen Inc. | Thiazole compounds as protein kinase b ( pkb) inhibitors |
| US7438910B2 (en) | 2002-09-06 | 2008-10-21 | Amgen Inc. | Therapeutic human anti-IL1-R1 monoclonal antibody |
| US7459540B1 (en) | 1999-09-07 | 2008-12-02 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| WO2009042435A1 (en) | 2007-09-21 | 2009-04-02 | Array Biopharma Inc. | Pyridin-2 -yl-amino-i, 2, 4 -thiadiazole derivatives as glucokinase activators for the treatment of diabetes mellitus |
| WO2009149171A2 (en) | 2008-06-04 | 2009-12-10 | Amgen Inc. | Fgf21 mutants and uses thereof |
| WO2010042747A2 (en) | 2008-10-10 | 2010-04-15 | Amgen Inc. | Fgf21 mutants and uses thereof |
| WO2012027261A1 (en) * | 2010-08-23 | 2012-03-01 | Amgen Inc. | Sulfonylpiperazine derivatives that interact with glucokinase regulatory protein for the treatment of diabetes |
| WO2012138776A1 (en) * | 2011-04-05 | 2012-10-11 | Amgen Inc. | Benzodioxepine and benzodioxine compounds that interact with glucokinase regulatory protein for the treatment of diabetes |
-
2013
- 2013-05-14 WO PCT/US2013/041011 patent/WO2013173382A1/en active Application Filing
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7459540B1 (en) | 1999-09-07 | 2008-12-02 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| US7667008B2 (en) | 1999-09-07 | 2010-02-23 | Amgen, Inc. | Fibroblast growth factor-like polypeptides and variants thereof |
| US7671180B2 (en) | 1999-09-07 | 2010-03-02 | Amgen, Inc. | Fibroblast growth factor-like polypeptides and variants thereof |
| US7696172B2 (en) | 1999-09-07 | 2010-04-13 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| US7438910B2 (en) | 2002-09-06 | 2008-10-21 | Amgen Inc. | Therapeutic human anti-IL1-R1 monoclonal antibody |
| WO2005095418A1 (en) * | 2004-04-02 | 2005-10-13 | Novartis Ag | Sulfonamide-thiazolpyridine derivatives as glucokinase activators useful the treatment of type 2 diabetes |
| WO2007084391A2 (en) | 2006-01-18 | 2007-07-26 | Amgen Inc. | Thiazole compounds as protein kinase b ( pkb) inhibitors |
| WO2009042435A1 (en) | 2007-09-21 | 2009-04-02 | Array Biopharma Inc. | Pyridin-2 -yl-amino-i, 2, 4 -thiadiazole derivatives as glucokinase activators for the treatment of diabetes mellitus |
| WO2009149171A2 (en) | 2008-06-04 | 2009-12-10 | Amgen Inc. | Fgf21 mutants and uses thereof |
| WO2010042747A2 (en) | 2008-10-10 | 2010-04-15 | Amgen Inc. | Fgf21 mutants and uses thereof |
| WO2012027261A1 (en) * | 2010-08-23 | 2012-03-01 | Amgen Inc. | Sulfonylpiperazine derivatives that interact with glucokinase regulatory protein for the treatment of diabetes |
| WO2012138776A1 (en) * | 2011-04-05 | 2012-10-11 | Amgen Inc. | Benzodioxepine and benzodioxine compounds that interact with glucokinase regulatory protein for the treatment of diabetes |
Non-Patent Citations (7)
| Title |
|---|
| "Bioreversible Carriers in Drug Design", 1987, AMERICAN PHARMACEUTIOAL ASSOCIATION AND PERGAMON PRESS |
| LEE, J.C.; CHOI, Y., SYNTH. COMMUN., vol. 28, 1998, pages 2021 - 2026 |
| PAUTSCH ET AL.: "Crystal Structure of Glucokinase Regulatory Protein", BIOCHEMISTRY, vol. 52, no. 20, 26 April 2013 (2013-04-26), pages 3523 - 3531, XP055072249, ISSN: 0006-2960, DOI: 10.1021/bi4000782 * |
| S. M. BERGE ET AL.: "Pharmaceutical Salts", J PHARM SCI, vol. 66, 1977, pages 1 - 19, XP002675560, DOI: doi:10.1002/jps.2600660104 |
| SARABU ET AL.: "Novel glucokinase activators: a patent review (2008 - 2010)", EXPERT OPIN. THER. PATENTS, vol. 21, no. 1, 1 January 2011 (2011-01-01), pages 13 - 33, XP009150497, ISSN: 1354-3776 * |
| STEPHENS, P.J.; DEVLIN, F.J.; PAN, J.-J., CHIRALITY, vol. 20, 2008, pages 643 - 663 |
| T. HIGUCHI; W. STELLA: "Pro-drugs as Novel Delivery Systems", vol. 14, A.C.S. SYMPOSIUM SERIES |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6734896B2 (en) | Bromodomain inhibitor | |
| JP6377123B2 (en) | Heteroaryl morpholinone compounds as MDM2 inhibitors for the treatment of cancer | |
| AU2016263564B2 (en) | Triazole agonists of the APJ receptor | |
| JP6545199B2 (en) | 3-Amino-1,5,6,7-tetrahydro-4H-indol-4-ones | |
| CA2758986C (en) | Inhibitors of pi3 kinase and/or mtor | |
| RU2688938C2 (en) | PYRROLO[3,2-B]PYRIDINE DERIVATIVE USEFUL AS A MUSCARINE ACETYLCHOLINE RECEPTOR MODULATOR (mAChR)M1 (mAChR M1) | |
| AU2015284135A1 (en) | Heteroaryl compounds useful as inhibitors of SUMO activating enzyme | |
| MX2014003642A (en) | Heterocyclic compounds as mdm2 inhibitors for the treatment of cancer. | |
| JP6559699B2 (en) | Substituted nitrogen-containing heterocyclic derivatives, pharmaceutical compositions containing them, and their antitumor applications | |
| TW201217362A (en) | Heteroaryls and uses thereof | |
| AU2011293584B2 (en) | Sulfonylpiperazine derivatives that interact with glucokinase regulatory protein for the treatment of diabetes | |
| TW202346288A (en) | Compounds for inhibiting kif18a | |
| AU2013303824B2 (en) | Bicyclic heteroaryl cycloalkyldiamine derivatives as inhibitors of spleen tyrosine kinases (SYK) | |
| CA2885167A1 (en) | Inhibitors of viral replication, their process of preparation and their therapeutical uses | |
| EP2694491A1 (en) | Benzodioxepine and benzodioxine compounds that interact with glucokinase regulatory protein for the treatment of diabetes | |
| WO2013123444A1 (en) | Sulfonyl compounds that interact with glucokinase regulatory protein | |
| WO2013173382A1 (en) | Benzothiophene sulfonamides and other compounds that interact with glucokinase regulatory protein | |
| CA3009485C (en) | Phenylimidazole compound | |
| WO2014035872A1 (en) | Tricyclic alkynes that interact with glucokinase regulatory protein |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13726930 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13726930 Country of ref document: EP Kind code of ref document: A1 |