WO2012145888A1 - 一种罗库溴铵的纯化方法 - Google Patents
一种罗库溴铵的纯化方法 Download PDFInfo
- Publication number
- WO2012145888A1 WO2012145888A1 PCT/CN2011/073256 CN2011073256W WO2012145888A1 WO 2012145888 A1 WO2012145888 A1 WO 2012145888A1 CN 2011073256 W CN2011073256 W CN 2011073256W WO 2012145888 A1 WO2012145888 A1 WO 2012145888A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- rocuronium bromide
- rocuronium
- temperature
- impurity
- bromide
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
Definitions
- the invention relates to the field of medicine, in particular to a method for purifying a drug rocuronium bromide. Background technique
- Rocuronium Bromide is a non-depolarizing muscle relaxant developed by Organon in the Netherlands. It was first introduced in the United States in 1994. It is currently the most widely used antiseptic in the world, in North America and large. Most European countries use the first dose of muscle relaxants.
- Rocuronium bromide is a novel monoquaternary ammonium muscle relaxant. It is used as an anesthesia aid for tracheal intubation during anesthesia and muscle relaxation during surgery. It is the fastest-acting non-depolarizing muscle relaxant used clinically. medicine. It is characterized by rapid onset, rapid recovery, weak inhibition of the cardiovascular system, and no histamine release.
- the chemical structural formula of rocuronium bromide is:
- European Patent EP 0 287 150 first discloses the preparation of rocuronium bromide, 2 ⁇ -(4-morpholino) -16 ⁇ -(1-pyrrolidinyl) -5 ⁇ -androst-3 ⁇ -ol, 17 ⁇ - Acetate (hereinafter referred to as Roque VIII) and 3-bromopropene are prepared by quaternization.
- the reaction equation is as follows:
- ester group at position 17 in the rocuronium structure is hydrolyzed to form a hydrolyzed impurity (impurity C in the USP Pharmacopoeia), which is the raw material of the raw material Rocco, which is unreacted in the previous step and is reacted with 3-bromopropene.
- impurity C in the USP Pharmacopoeia is the raw material of the raw material Rocco, which is unreacted in the previous step and is reacted with 3-bromopropene.
- the post-treatment process of rocuronium bromide or the hydrolysis reaction with the water contained in itself is the main impurity to be controlled in the preparation product. Its chemical structure is:
- the raw material impurities (USP Pharmacopoeia impurity A) decomposed during the post-treatment and storage of unreacted Roque VIII and rocuronium are also the main impurities to be controlled in the preparation product. Its chemical structure is:
- U.S. Patent No. 2,005, 558, 275 discloses a freeze drying method of rocuronium bromide in which a buffer solution of acetic acid and sodium acetate is added during lyophilization. Since the water and acetic acid in the buffer system are gradually reduced as the lyophilization progresses during lyophilization, the pH value during lyophilization The change is unstable, and finally the product is seriously decomposed, and the single pure rocuronium bromide product is not obtained.
- British Patent GB2445746 mentions a method for lyophilization of a solution of rocuronium bromide: dissolving rocuronium bromide in an aqueous solution having a pH below 4-5 (pH is adjusted with carbon dioxide), and then adjusting the pH with carbon dioxide to After 8 or less, it was concentrated, and the amount of residual solvent was measured, followed by lyophilization.
- the process is cumbersome and the resulting 3-bromopropyl residue is more than 100 ppm. Therefore, it is of great significance to develop a preparation method of high-purity rocuronium bromide. Summary of the invention
- Rocuronium bromide is a water-soluble drug that is highly susceptible to hydrolysis, its stability is extremely poor, it is easy to produce unknown impurities when heated, and it is easy to form solvates with many solvents to make the residual solvent difficult to remove. Since rocuronium is dissolved in water, the bond and the organic solvent are released, which is easy to remove, and rocuronium is relatively stable at low temperatures. In view of this, the method of using freeze drying is very suitable for purifying the product. However, in the normal freeze-drying process, the residual solvent in the product is still not well controlled.
- rocuronium bromide is mixed with water to a mass percentage of 5% to 40% aqueous solution, and its pH range is between 8 and 9.5, and the higher the concentration of rocuronium, the lower the pH of the solution.
- the rocuronium bromide aqueous solution is slowed down at a low temperature, pH of 8 to 9.5, and good nitrogen protection.
- the inventors have found that the stability of the products treated by the method of the present invention is greatly improved.
- the inventors found through research that when the moisture in rocuronium is more than 4%, at room temperature After being left for about ten days, the hydrolyzed impurities exceeded the standard and a large amount of unknown impurities appeared.
- the water was controlled to be 4% or less, preferably 0.5% or less, after the purification by the method of the present invention, the impurities were left unchanged at room temperature for one month.
- the method for purifying crude rocuronium bromide provided by the invention adopts the following scheme:
- the crude rocuronium bromide to be purified is formulated into an aqueous solution having a mass percentage of 5 to 40% and distilled under reduced pressure at 20 ⁇ 5 mbar, and then added with activated carbon or silica gel having a mass of 1 to 5% of rocuronium bromide, and filtered. The filtrate was quickly frozen into ice and lyophilized to obtain rocuronium bromide.
- the aqueous solution of rocuronium has a mass percentage of 15 to 35%.
- the vacuum distillation is carried out under the protection of nitrogen.
- the vacuum distillation has a temperature of 0 to 15 ° C and a distillation time of not more than 5 hours, preferably 2 to 5 hours.
- the fast freezing ice-forming material has a temperature of -80 to -20 ° C, preferably -40 to -20 ° C.
- the temperature of the primary drying material at the beginning of the freeze-drying is -80 ⁇ -10 ° C, preferably -40 ⁇ -10 ° C,
- the primary drying takes 0 to 30 hours, more preferably 15 hours or less.
- the analytical drying temperature of the freeze-drying is 15 to 40 ° C, preferably 25 °
- the analysis drying time is 0 to 20 hours, preferably 3 to 15 hours.
- the purification method of the invention can effectively control the 3-bromopropene residue and the hydrolysis impurity C growth, and the moisture and the residual solvent are all in compliance with the USP33 standard, and the process is clean and the operation tube is cleaned. , easy to control, good product quality.
- the HPLC purity of the obtained rocuronium bromide is over 99%, and the HPLC area of impurity C is less than 0.20%, 3-bromopropene residue
- the residual amount is less than 10 ppm, the water content is less than 4.0%, and the residual solvent meets the requirements of the pharmacopoeia. It has improved and optimized the existing technology, which is very conducive to industrial production, and has four strong market competitiveness.
- Figure 1 is an X-ray diffraction pattern of lyophilized rocuronium bromide powder. detailed description
- Step 1 The crude rocuronium bromide to be purified is formulated into an aqueous solution of 5-40% by mass and distilled under reduced pressure at 20 ⁇ 5 mbar, and then added with activated carbon or silica gel of 1 to 5% by mass of rocuronium bromide. .
- the aqueous solution of rocuronium bromide preferably has a mass concentration of 15 to 35%.
- the temperature range for vacuum distillation is 0 to 15 ° C, preferably 0 to 10 ° C.
- the distillation time is from 0 to 5 hours, preferably from 2 to 5 hours.
- the vacuum distillation is carried out under the protection of nitrogen.
- Step 2 The aqueous rocuronium bromide solution was filtered, and the filtrate was taken for lyophilization.
- the mixture was filtered through an activated carbon or silica gel-treated aqueous solution, and the filtrate was dispensed into a freeze-drying tray of a freeze dryer.
- the thickness of the liquid should not be too thick.
- Step 3 The filtrate is snap frozen and then lyophilized.
- the temperature is rapidly frozen until the temperature of the material is -80 to -20 ° C, preferably -40 to -30 ° C.
- the freeze-drying drying setting temperature ranges from -40 to -10 ° C, preferably from -25 to -10 ° C. It takes 0 to 30 hours, and more preferably within 15 hours.
- Analytical drying set temperature range is 15 ⁇ 40 °C, preferably 25 ⁇ 40 °C. It takes 0 to 20 hours, and more preferably 3 to 15 hours.
- the X-ray diffraction pattern of the crystalline rocuronium bromide powder is shown in Fig. 1, from which it can be seen that the obtained powder is amorphous.
- the purification method of the invention can effectively control the residual amount of 3-bromopropene and the growth of hydrolyzed impurities C, and make the moisture and residual solvent conform to the USP33 standard of the United States Pharmacopoeia, the process is clean, the cylinder is easy to control, and the product quality is good. .
- the HPLC purity of the obtained rocuronium bromide is over 99%, the HPLC area of impurity C is less than 0.20%, the residual amount of 3-bromopropene is less than 10 ppm, the water content is less than 4.0%, and the total amount of residual solvent meets the requirements of the pharmacopoeia.
- Example 1 Purification of crude rocuronium bromide by the method of the present invention
- the crude rocuronium bromide was prepared by the method of EP 0287150.
- the residual solvent in the crude product was tested: 3-bromopropene 379 ppm, dichloromethane 3000 ppm, and diethyl ether 15%.
- the HPLC of impurity A was 0.08%, and impurity C was not detected.
- the above crude rocuronium bromide lO.Og was dissolved in 30.0 g of deionized water, cooled to 5 ° C, first replaced with nitrogen and then subjected to vacuum distillation.
- the distillation pressure was controlled at a positive pressure of 20 ⁇ 5 mbar.
- 0.4 g of silica gel was added and the mixture was stirred for 30 minutes.
- the filtrate was collected in a tray and rapidly frozen into ice at -40 °C. Freeze and dry under controlled vacuum of 0 ⁇ 5Pa.
- the temperature was raised to 35 ° C and vacuum-dried for 10 hours to obtain 8.2 g of rocuronium bromide.
- the residual solvent in rocuronium bromide after lyophilization was 3-bromopropene: 6.0 ppm, diethyl ether: 200 ppm.
- the water was divided into 0.50%, the HPLC area of the impurity A was 0.02%, and the HPLC area of the impurity C was 0.09%.
- Example 2 Comparison of the method of the present invention with conventional purification methods
- Preparation of crude rocuronium 200.0 g of 2 ⁇ -(4-morpholinyl)-16 ⁇ -(1-pyrrolidinyl)-5a-androst-3 ⁇ -ol, 17 ⁇ -acetate It was mechanically stirred with 140 ml of 3-bromopropene in a 1000 mL bottle under a nitrogen atmosphere at a constant temperature of 25 ° C, and the reaction was completed for 1.25 hours. The mixture was dissolved by stirring with 500 ml of acetonitrile, suction filtered, and the filtrate was dried.
- the above crude rocuronium bromide was tested, and the residual solvent was 550 ppm of 3-bromopropene, 9.0% of methyl tert-butyl ether, 2000 ppm of acetonitrile, and 0.5% of dichloromethane.
- the HPLC area of rocuronium was greater than 99.5%, and the HPLC area of impurity A was 0.02%. No impurity C was detected.
- the crude rocuronium bromide was purified by three methods, as detailed below:
- the above crude rocuronium lO.Og was ground to a fine powder, and dried in a vacuum drying oven at 40 ° C for one day to obtain 8.8 g of rocuronium bromide.
- the rocuronium bromide obtained by vacuum drying was tested as follows: residual solvent 3-bromopropene residue 60 ppm, methyl tert-butyl ether 400 ppm, acetonitrile 150 ppm, dichloromethane 60 ppm; water fraction 3.50%; HPLC area of impurity C
- the HPLC area of 0.11%, impurity A was 0.26%, and two unknown impurities appeared, which were 1.1 and 1.3 times of the main peak time, respectively, and the HPLC area was 0.05%.
- Method 2 the method of the present invention does not add activated carbon and silica gel to purify rocuronium bromide crude 10.0g of the above crude rocuronium bromide dissolved in 50.0g of deionized water, cooled to 10 ° C, first replaced with nitrogen Vacuum distillation. The distillation pressure was controlled at a positive pressure of about 20 ⁇ 5 mbar. After 3.5 hours of treatment, the raw material liquid was collected in a tray and rapidly frozen into ice at -40 °C. Freeze drying was carried out under controlled vacuum of 0 to 5 Pa. Finally, the temperature was raised to 35 ° C and vacuum-dried for 3 hours to obtain 8.9 g of rocuronium bromide.
- the residual solvent in rocuronium bromide after lyophilization was 3-bromopropene: 17.9 ppm, methyl tert-butyl ether: ND, acetonitrile: 78 ppm.
- the water was divided into 2.60%, the HPLC area of the impurity A was 0.10%, and the HPLC area of the impurity C was 0.30%.
- the above-mentioned crude rocuronium bromide lO.Og was dissolved in 50.0 g of deionized water, cooled to 10 ° C, first replaced with nitrogen, and then subjected to vacuum distillation.
- the distillation pressure was controlled at a positive pressure of 20 ⁇ 5 mbar.
- 0.4 g of activated carbon was added and stirred for 30 minutes.
- the filtrate was collected in a tray and rapidly frozen into ice at -40 °C. Freeze and dry under controlled vacuum of 0 ⁇ 5Pa.
- the temperature was raised to 35 ° C and vacuum-dried for 6 hours to obtain 8.7 g of rocuronium bromide.
- the residual solvent in rocuronium bromide after lyophilization was 3-bromopropene: 3.0 ppm, methyl tert-butyl ether: N.D, acetonitrile: 78 ppm.
- the water was divided into 2.60%, the HPLC area of impurity A was 0.02%, and the HPLC area of impurity C was 0.10%.
- Example 3 Moisture content and stability test of crude rocuronium bromide
- the inventors found through research that when the moisture in rocuronium is more than 4%, the hydrolyzed impurities are left at room temperature for about ten days, which exceeds the USP33 standard, and a large number of unknown impurities appear.
- the data is shown in Table 2; when the moisture is controlled at about 0.5% When the temperature is left for one month at room temperature, there is almost no change in impurities.
- Table 3 Moisture content and stability test of crude rocuronium bromide
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Saccharide Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
种罗库淡铵的纯化方法 技术领域
本发明涉及医药领域, 具体涉及药物罗库溴铵的纯化方法。 背景技术
罗库溴铵( Rocuronium Bromide )是荷兰 Organon公司开发的甾体类 非去极化肌肉松弛药, 1994年在美国首次上市, 该药是目前国际上应用 最广泛的几松药,在北美及大多数欧洲国家用量居于肌松药的第一位。 罗 库溴铵是新型单季铵类肌松药,作为麻醉辅助用药,用于麻醉时的气管插 管和手术中的肌肉松弛,是临床上使用的起效最快的非去极化肌松药。其 特点为起效快, 恢复迅速, 对心血管系统抑制作用弱, 无组胺释放作用。 罗库溴铵化学结构式为:

罗库溴铵
欧洲专利 EP0287150首先公开了罗库溴铵的制备方法, 2 β -(4-吗啉 基) -16 β -(1-吡咯烷基) -5 α -雄甾 -3 α -醇, 17 β -乙酸酯 (以下筒称罗库八) 与 3-溴丙烯经季铵化制得, 其反应方程式如下:

另外, 罗库溴铵结构中 17位的酯基会水解, 形成水解杂质 (USP 药典中的杂质 C ), 该杂质是原料罗库八上一步未反应完全的原料引入与 3-溴丙烯反应得到,同时罗库溴铵后处理过程或者与本身含有的水分反应 水解产生的, 是制剂产品中需要控制的主要杂质。 其化学结构式为:

杂质 C
而未反应完全的罗库八及罗库溴铵后处理及存储过程中分解生成的 原料杂质(USP药典中的杂质 A ),也是制剂产品中需要控制的主要杂质。 其化学结构式为:
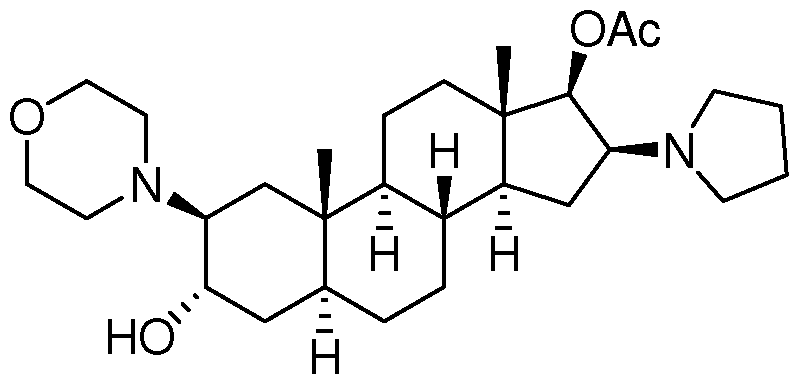
杂质 A
另夕卜,有研究报道低级 代烃有基因毒性,一般来说它们的毒性按溴 代烃、 氯代烃、 氟代烃依次降低。 因此合成罗库溴铵中使用的原料 3-溴 丙稀, 也是潜在的基因毒性物质。
美国专利 US2006058275公开了一种罗库溴铵的冷冻干燥方法,在冷 冻干燥的过程中,加入了乙酸和乙酸钠的緩沖溶液。由于在冻干的过程中, 緩沖体系中的水分和乙酸随着冻干的进行会逐渐减少,冻干过程中 PH值
变化不稳定,最后得到产品分解严重, 而且得到的不是单一纯的罗库溴铵 产品。 英国专利 GB2445746中提及了一种罗库溴铵水溶液冻干的方法: 将罗库溴铵溶于 PH低于 4 ~ 5的水溶液中 (用二氧化碳调节 PH值), 然 后再用二氧化碳调节 PH至 8以下, 然后浓缩, 检测残留溶剂的量, 再进 行冻干。该工艺操作繁瑣,而且得到产品的 3-溴丙稀残留量超过 100ppm。 因此开发一种高纯度罗库溴铵的制备方法具有重要意义。 发明内容
本发明的目的是提供一种高纯度罗库溴铵的制备方法。
罗库溴铵是一种水溶性药物, 该药物极易发生水解, 其稳定性极差, 受热易产生未知杂质,又容易与许多溶剂形成溶剂化物使得残留溶剂 4艮难 除去。 因为罗库溴铵溶于水后,键和的有机溶剂便游离出来, 这时就便于 除去, 罗库溴铵在低温时又比较稳定。鉴于此, 使用冷冻干燥的方法对于 纯化该产品是非常适合的。但是在正常的冷冻干燥处理工艺中,产品中的 残留溶剂依然不能得到良好的控制。
本发明人通过研究发现:
首先, 罗库溴铵与水配成质量百分比 5% ~ 40%水溶液, 其 PH值范 围在 8 ~ 9.5之间, 且罗库溴铵浓度越大溶液的 PH值越低。 罗库溴铵水 溶液在低温、 PH值为 8 ~ 9.5、 良好的氮气保护中, 水解速度减慢。
其次,使用活性炭或者硅胶对罗库溴铵去除杂质效果非常明显,尤其 是对 3-溴丙烯的吸附。 本发明人经过研究发现使用同样的工艺, 不加入 活性炭或硅胶处理得到的罗库溴铵中 3-溴丙烯的残留量超过 lOOppm, 而 加入后可以控制在 1 Oppm以下。
更欣喜的是发明人发现,用本发明所述方法处理后的产品,稳定性也 大大提高。 发明人通过研究发现当罗库溴铵中水分在 4%以上时, 室温下
放置十天左右水解杂质即超标并出现大量未知杂质;当经本发明所述方法 纯化后, 水分控制在 4%以下优选为 0.5%以下时, 室温下放置一个月杂质 基本无变化。
本发明提供的纯化罗库溴铵粗品的方法, 采用如下方案:
待纯化的罗库溴铵粗品配成质量百分比为 5~40%的水溶液在 20 ± 5 mbar减压蒸馏后, 加入质量为罗库溴铵粗品 1 ~ 5%的活性炭或者硅胶吸 附, 过滤后取滤液快速冻结成冰, 再经过冷冻干燥得到罗库溴铵。
作为优选, 罗库溴铵的水溶液质量百分比含量为 15~35%。
更优选地, 所述减压蒸馏在氮气保护下进行。
更优选地,所述减压蒸馏的温度为 0~ 15°C,蒸馏时间不超过 5小时, 优选为 2~5小时。
作为优选,所述快速冻结成冰物料温度为 -80~-20°C,优选的是 -40 ~ -20°C。
作为优选, 所述冷冻干燥开始时一次干燥物料温度在 -80 ~-10°C, 优 选的是 -40~-10°C,
作为优选, 所述一次干燥耗时 0 ~ 30小时, 更优选为 15小时以内。 作为优选,所述冷冻干燥的解析干燥温度为 15 ~ 40°C ,优选的是 25 ~
40°C。
作为优选, 所述解析干燥时间 0~ 20小时, 优选的是 3~15小时。 与现有技术相比, 本发明所述方法纯化能有效的控制 3-溴丙烯残留 量和水解杂质 C增长, 并使得水分及残留溶剂均符合美国药典 USP33标 准, 其工艺筒洁, 操作筒便, 易于控制, 产品质量好。 所得罗库溴铵的 HPLC纯度可达 99%以上, 杂质 C的 HPLC面积小于 0.20%, 3-溴丙烯残
留量小于 lOppm, 水分小于 4.0%, 残留溶剂符合药典要求。 实现了对现 有技术的改进和优化, 非常利于工业生产, 有 4艮强的市场竟争力。 附图说明
图 1为冻干的罗库溴铵粉末的 X射线衍射图谱。 具体实施方式
为了进一步了解本发明,下面结合实施例对本发明优选实施方案进行 描述, 但是应当理解, 这些描述只是为进一步说明本发明的特征和优点, 而不是对本发明权利要求的限制。
本发明提供的纯化罗库溴铵的方法, 具体实施方法为:
步骤 1:将待纯化的罗库溴铵粗品配成质量百分比为 5~40%的水溶液 在 20 ± 5 mbar减压蒸馏后, 加入质量为罗库溴铵粗品 1 ~ 5%的活性炭或 硅胶吸附。
其中罗库溴铵水溶液优选质量浓度为 15 ~ 35%。 减压蒸馏的温度范 围为 0~15°C, 优选的是 0~10°C。 蒸馏时间为 0~5小时, 优选为 2~5 小时。
作为优选, 所述减压蒸馏在氮气保护下进行。
步骤 2: 将罗库溴铵水溶液过滤, 取滤液进行冻干。
经过活性炭或者硅胶处理的水溶液过滤,滤液分装在冷冻干燥机的冻 干托盘中。 为保证冻干效果, 料液厚度不宜太厚。
步骤 3: 将滤液快速冷冻, 然后开始冻干。
其中快速冷冻至物料温度为 -80~-20°C, 优选的是 -40~-30°C。 冷冻 干燥一次干燥设置温度范围在 -40~-10°C,优选的是 -25~-10°C。耗时 0~ 30小时, 更为优选的是 15小时以内。 解析干燥设置温度范围在 15 ~ 40
°C , 优选的是 25 ~ 40°C。 耗时 0 ~ 20小时, 更为优选的是 3 ~ 15小时。 得到罗库溴铵晶状粉末的 X射线衍射图谱见图 1 ,从中可以看出得到的粉 末为无定形。
本发明所述纯化方法能有效的控制 3-溴丙烯残留量和水解杂质 C增 长, 并使得水分及残留溶剂均符合美国药典 USP33标准, 其工艺筒洁, 操作筒便, 易于控制,产品质量好。所得罗库溴铵的 HPLC纯度可达 99% 以上, 杂质 C的 HPLC面积小于 0.20%, 3-溴丙烯残留量小于 lOppm, 水 分小于 4.0%, 残留溶剂总量符合药典要求。
以下以具体实施例说明本发明的效果,但本发明的保护范围不受以下 实施例的限制。 实施例 1: 本发明所述方法纯化罗库溴铵粗品
参考 EP0287150方法, 制备罗库溴铵粗品。 经检测, 粗品中残留溶 剂: 3-溴丙稀 379ppm、 二氯甲烷 3000ppm、 乙醚 15%。 杂质 A的 HPLC 为 0.08%, 未检出杂质 C。
取以上罗库溴铵粗品 lO.Og溶于 30.0g的去离子水中, 冷却至 5°C , 先用氮气进行置换后进行减压蒸馏。 蒸馏压力控制在正压 20 ± 5 mbar左 右, 处理 3.5小时后, 加入硅胶 0.4g, 保温搅拌 30分钟。 过滤, 滤液收 集在托盘中, -40°C下快速冻结成冰。 控制真空度 0 ~ 5Pa下进行冷冻干 燥。 最后升温至 35°C保温真空干燥 10小时, 得到罗库溴铵 8.2g。
冻干后罗库溴铵中的残留溶剂为 3-溴丙烯: 6.0ppm、 乙醚: 200ppm。 水分为 0.50%, 杂质 A的 HPLC面积为 0.02%, 杂质 C的 HPLC面积为 0.09%。 实施例 2: 本发明所述方法与常规纯化方法比较
罗库溴铵粗品的制备:将 200.0克 2 β -(4-吗啉基 )-16 β -(1-吡咯烷基) -5 a -雄甾 -3 α -醇, 17 β -乙酸酯与 140毫升 3-溴丙稀于 lOOOmL瓶中氮气保 护恒温 25 °C机械搅拌, 反应 1.25小时结束。 加入 500ml乙腈搅拌溶解, 抽滤, 滤液旋干。 再用 800ml二氯甲烷溶解緩慢滴加到剧烈搅拌的 48L 冰甲基叔丁基醚中。 滴加完毕后冰浴搅拌 1小时。 过滤收集滤饼, 真空室 温干燥 2天得到罗库溴铵粗品 260.5克。
对以上罗库溴铵粗品进行检测, 残留溶剂: 3-溴丙烯残留为 550ppm、 甲基叔丁基醚为 9.0%、乙腈为 2000ppm、二氯甲烷 0.5%。罗库溴铵 HPLC 面积大于 99.5%, 杂质 A的 HPLC面积为 0.02%, 未检出杂质 C。
对该罗库溴铵粗品分别用三种方法进行纯化, 详述如下:
方法 1、 常规真空干燥
取上述罗库溴铵粗品 lO.Og研磨成细粉,在真空干燥箱中 40°C真空干 燥一天, 得到罗库溴铵 8.8g。
对真空干燥得到的罗库溴铵进行检测, 结果如下: 残留溶剂 3-溴丙 烯残留 60ppm、 甲基叔丁基醚 400ppm、 乙腈 150ppm、 二氯甲烷 60ppm; 水分为 3.50%; 杂质 C的 HPLC面积为 0.11%、 杂质 A的 HPLC面积为 0.26%、 并出现两个未知杂质, 分别在主峰时间 1.1和 1.3倍, HPLC面积 均为 0.05%。 方法 2、 本发明所述方法不加活性炭和硅胶纯化罗库溴铵粗品 取上述罗库溴铵粗品 10.0g溶于 50.0g的去离子水中, 冷却至 10°C , 先用氮气进行置换后进行减压蒸馏。 蒸馏压力控制在正压 20 ± 5 mbar左 右, 处理 3.5小时后, 将原料液收集在托盘中, -40°C下快速冻结成冰。 控制真空度 0 ~ 5Pa下进行冷冻干燥。最后升温至 35°C保温真空干燥 3小 时, 得到罗库溴铵 8.9g。
冻干后罗库溴铵中的残留溶剂为 3-溴丙烯: 17.9ppm、甲基叔丁基醚: N.D、 乙腈: 78ppm。 水分为 2.60%, 杂质 A的 HPLC面积为 0.10%, 杂 质 C的 HPLC面积为 0.30%。
方法 3、 本发明所述方法纯化罗库溴铵粗品
取上述罗库溴铵粗品 lO.Og溶于 50.0g的去离子水中, 冷却至 10°C , 先用氮气进行置换后进行减压蒸馏。 蒸馏压力控制在正压 20 ± 5 mbar左 右, 处理 3.5小时后, 加入活性炭 0.4g, 保温搅拌 30分钟。 过滤, 滤液 收集在托盘中, -40°C下快速冻结成冰。控制真空度 0 ~ 5Pa下进行冷冻干 燥。 最后升温至 35°C保温真空干燥 6小时, 得到罗库溴铵 8.7g。
冻干后罗库溴铵中的残留溶剂为 3-溴丙烯: 3.0ppm、 甲基叔丁基醚: N.D、 乙腈: 78ppm。 水分为 2.60%, 杂质 A的 HPLC面积为 0.02%, 杂 质 C的 HPLC面积为 0.10%。
详见表 1。
表 1.本发明所述纯化罗库溴铵粗品方法与常规方法比较
3-溴丙 甲基叔 杂质 杂质
乙腈 水分 未知杂质 烯 丁基醚 A c 方法 1 60ppm 400ppm 150ppm 3.5% 0.26% 0.11% 2个, 0.05% 方法 2 17.9ppm N.D 78ppm 2.6% 0.10% 0.30% N.D 方法 3 3.0ppm N.D 78ppm 1.7% 0.02% 0.10% N.D
实施例 3: 罗库溴铵粗品的水分含量与稳定性试验
对于罗库溴铵本身稳定性而言, 残留的水分越少, 产品越稳定。发明 人通过研究发现当罗库溴铵中水分在 4%以上时, 室温下放置十天左右水 解杂质即超过 USP33的标准, 并出现大量未知杂质, 数据见表 2; 当水 分控制在 0.5%左右时, 室温下放置一个月杂质基本无变化, 数据见表 3。
表 2 水分 4.5%的罗库溴铵样品室温存放稳定性 有关物质 (HPLC )
杂质 A 杂质 C 单个未知杂质 总杂质
0天 0.02% 0.06% N.D 0.08%
10天 0.25% 0.36% 0.12% 0.73%
20天 0.30% 0.40% 0.20% 0.90%
30天 0.35% 0.50% 0.20% 1.05% 表 3 水分 0.5%的罗库溴铵样品室温存放稳定性 有关物质 (HPLC )
杂质 A 杂质 C 单个未知杂质 总杂质
0天 0.02% 0.06% N.D 0.08%
10天 0.02% 0.06% N.D 0.08%
20天 0.03% 0.06% N.D 0.09%
30天 0.03% 0.06% N.D 0.09%
本发明提出的一种纯化罗库溴铵的方法已通过实施例进行了描述,相 关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的纯化 罗库溴铵的方法进行改动或适当变更与组合,来实现本发明技术。特别需 要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见 的, 它们都被视为包括在本发明的精神、 范围和内容中。
Claims
1、 一种纯化罗库溴铵粗品的方法, 其特征在于, 将待纯化的罗库溴 铵粗品配成质量百分比为 5~40%的水溶液在减压蒸馏后, 加入质量为罗 库溴铵粗品 1 ~ 5%的活性炭或者硅胶吸附, 过滤后取滤液快速冻结成冰, 再经过冷冻干燥得到罗库溴铵。
2、 根据权利要求 1所述的方法, 其特征在于, 罗库溴铵的水溶液质 量百分比含量为 15 ~ 35%。
3、 根据权利要求 1所述的方法, 其特征在于, 所述减压蒸馏在氮气 保护下进行。
4、 根据权利要求 1所述的方法, 其特征在于, 所述减压蒸馏的温度 为 0 ~ 15 °C。
5、 根据权利要求 1所述的方法, 其特征在于, 所述减压蒸馏时间不 超过 5小时, 优选为 2-5小时。
6、 根据权利要求 1所述的方法, 其特征在于, 所述快速冻结成冰物 料温度为 -80 ~ -20°C。
7、 根据权利要求 6所述的方法, 其特征在于, 所述快速冻结成冰物 料温度是 -40 ~ -20°C。
8、 根据权利要求 1所述的方法, 其特征在于, 所述冷冻干燥开始时 一次干燥物料温度在 -80 ~ -10°C。
9、 根据权利要求 8所述的方法, 其特征在于, 所述冷冻干燥开始时 一次干燥物料温度是 -40 ~ -10°C。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11864391.5A EP2703408B1 (en) | 2011-04-25 | 2011-04-25 | Method for purifying rocuronium bromide |
| PCT/CN2011/073256 WO2012145888A1 (zh) | 2011-04-25 | 2011-04-25 | 一种罗库溴铵的纯化方法 |
| US14/122,679 US9024013B2 (en) | 2011-04-25 | 2011-04-25 | Method for purifying rocuronium bromide |
| ES11864391.5T ES2553639T3 (es) | 2011-04-25 | 2011-04-25 | Método para purificar bromuro de rocuronio |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2011/073256 WO2012145888A1 (zh) | 2011-04-25 | 2011-04-25 | 一种罗库溴铵的纯化方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012145888A1 true WO2012145888A1 (zh) | 2012-11-01 |
Family
ID=47071543
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2011/073256 WO2012145888A1 (zh) | 2011-04-25 | 2011-04-25 | 一种罗库溴铵的纯化方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9024013B2 (zh) |
| EP (1) | EP2703408B1 (zh) |
| ES (1) | ES2553639T3 (zh) |
| WO (1) | WO2012145888A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103435674A (zh) * | 2013-09-09 | 2013-12-11 | 山东新华制药股份有限公司 | 高纯度、高稳定性罗库溴铵的制备方法 |
| WO2019242039A1 (zh) * | 2018-06-21 | 2019-12-26 | 上药东英(江苏)药业有限公司 | 一种低杂质水平的罗库溴铵注射液的制备方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10170072B2 (en) | 2015-09-21 | 2019-01-01 | Apple Inc. | Gate line layout configuration |
| CN106831926B (zh) * | 2017-01-24 | 2020-01-21 | 山东轻工职业学院 | 一种注射用罗库溴铵原料药的干燥方法 |
| CN110734468B (zh) * | 2018-07-20 | 2021-04-16 | 济南高德医药科技有限公司 | 罗库溴铵粗品的精制方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0287150A1 (en) | 1987-04-14 | 1988-10-19 | Akzo N.V. | Novel 2 beta-morpholino-androstane derivatives and processes for their preparation |
| US20060058275A1 (en) | 2004-07-15 | 2006-03-16 | Oded Friedman | Processes for preparing stabilized, highly pure rocuronium bromide |
| GB2445746A (en) | 2007-01-17 | 2008-07-23 | Texcontor Ets | Use of carbonated water as a solvent for freeze drying, and method of purification comprising dissolution of material in carbonated water and freeze drying |
| CN101993470A (zh) * | 2009-08-27 | 2011-03-30 | 济南诺和诺泰生物制药有限公司 | 一种制备高纯度罗库溴铵的工艺 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2008000933A (es) * | 2005-09-13 | 2008-03-26 | Sicor Inc | Bromuro de roncuronio puro. |
| CN101397329B (zh) * | 2007-09-25 | 2011-04-27 | 重庆人本药物研究院 | 罗库溴铵结晶水合物的制备方法 |
| CN101653412B (zh) * | 2009-09-15 | 2014-06-25 | 尹双保 | 一种稳定的罗库溴铵注射液 |
-
2011
- 2011-04-25 WO PCT/CN2011/073256 patent/WO2012145888A1/zh active Application Filing
- 2011-04-25 EP EP11864391.5A patent/EP2703408B1/en active Active
- 2011-04-25 ES ES11864391.5T patent/ES2553639T3/es active Active
- 2011-04-25 US US14/122,679 patent/US9024013B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0287150A1 (en) | 1987-04-14 | 1988-10-19 | Akzo N.V. | Novel 2 beta-morpholino-androstane derivatives and processes for their preparation |
| US20060058275A1 (en) | 2004-07-15 | 2006-03-16 | Oded Friedman | Processes for preparing stabilized, highly pure rocuronium bromide |
| GB2445746A (en) | 2007-01-17 | 2008-07-23 | Texcontor Ets | Use of carbonated water as a solvent for freeze drying, and method of purification comprising dissolution of material in carbonated water and freeze drying |
| CN101993470A (zh) * | 2009-08-27 | 2011-03-30 | 济南诺和诺泰生物制药有限公司 | 一种制备高纯度罗库溴铵的工艺 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2703408A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103435674A (zh) * | 2013-09-09 | 2013-12-11 | 山东新华制药股份有限公司 | 高纯度、高稳定性罗库溴铵的制备方法 |
| WO2019242039A1 (zh) * | 2018-06-21 | 2019-12-26 | 上药东英(江苏)药业有限公司 | 一种低杂质水平的罗库溴铵注射液的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2703408A4 (en) | 2014-10-08 |
| ES2553639T3 (es) | 2015-12-10 |
| EP2703408B1 (en) | 2015-08-26 |
| EP2703408A1 (en) | 2014-03-05 |
| US20140200340A1 (en) | 2014-07-17 |
| US9024013B2 (en) | 2015-05-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6692941B2 (ja) | スガマデックスの製造及び純化方法 | |
| WO2012145888A1 (zh) | 一种罗库溴铵的纯化方法 | |
| US8481780B2 (en) | Polymorphs of bromfenac sodium and methods for preparing bromfenac sodium polymorphs | |
| JP4530664B2 (ja) | 2−メチル−4−(4−メチル−1−ピペラジニル)−10H−チエノ[2,3−b][1,5]ベンゾジアゼピンの結晶形I、この結晶形Iの製造方法、及び医薬組成物 | |
| CN111655739A (zh) | 一种去除舒更葡糖钠中气相杂质和制备其无定型物的方法 | |
| US20130303754A1 (en) | Refining process of cefamandole sodium | |
| WO2013029293A1 (zh) | 一种制备厄他培南钠的方法 | |
| RU2009119411A (ru) | Новые кристаллические формы | |
| EP2311794B1 (en) | Polymorphs of bromfenac sodium and methods for preparing bromfenec sodium polymorphs | |
| US8927707B2 (en) | Purification method of aztreonam | |
| US20060276463A1 (en) | Pure levofloxacin hemihydrate and processes for preparation thereof | |
| WO2013121279A2 (en) | Process to prepare ertapenem | |
| EP2209787A1 (en) | Process for the preparation of carbapenem antibiotic | |
| CN104610411B (zh) | 一种化合物的纯化方法 | |
| DK170645B1 (da) | Krystallinsk vandfrit amoxycillin, fremgangsmåde til fremstilling heraf, samt præparater indeholdende dette | |
| WO2013132314A1 (en) | Tenofovir phosphate, processes for the preparation and pharmaceutical composition thereof | |
| WO2018032506A1 (zh) | 一种硫酸多粘菌素b结晶及其制备方法 | |
| WO2000075143A1 (fr) | Cristaux de derives de carbapenem et preparations pharmaceutiques pour injection | |
| JP3558684B2 (ja) | ピロリジルチオカルバペネム誘導体の乾燥方法 | |
| KR102595725B1 (ko) | 아트로핀의 제조방법 | |
| KR20110111494A (ko) | 티게사이클린의 단리방법 | |
| CN111057122B (zh) | 一种罗库溴铵的制备方法 | |
| CN113527098B (zh) | 一种氟比洛芬酯晶型及其制备方法 | |
| CA2840814A1 (en) | Solid dispersions of sitagliptin and processes for their preparation | |
| JP6198269B2 (ja) | オルメサルタンメドキソミルの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11864391 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011864391 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14122679 Country of ref document: US |