WO2008076043A1 - Novel 2-amino-5,5-diaryl-imidazol-4-ones - Google Patents
Novel 2-amino-5,5-diaryl-imidazol-4-ones Download PDFInfo
- Publication number
- WO2008076043A1 WO2008076043A1 PCT/SE2007/001116 SE2007001116W WO2008076043A1 WO 2008076043 A1 WO2008076043 A1 WO 2008076043A1 SE 2007001116 W SE2007001116 W SE 2007001116W WO 2008076043 A1 WO2008076043 A1 WO 2008076043A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dihydro
- methyl
- imidazol
- amino
- heterocyclyl
- Prior art date
Links
- SDJXSDIFSNKUIO-UHFFFAOYSA-N CC(C)(CC1)Oc2c1cc(C(C(N1C)=O)(c(cc3Br)ccc3F)NC1=S)cc2 Chemical compound CC(C)(CC1)Oc2c1cc(C(C(N1C)=O)(c(cc3Br)ccc3F)NC1=S)cc2 SDJXSDIFSNKUIO-UHFFFAOYSA-N 0.000 description 1
- IWBZWXLPXQEVMC-VOTSOKGWSA-N CN(C1=O)C(N)=NC1(c(cc1)cc2c1OCCC2)c(cc1)cc(/C=C/C2CC2)c1F Chemical compound CN(C1=O)C(N)=NC1(c(cc1)cc2c1OCCC2)c(cc1)cc(/C=C/C2CC2)c1F IWBZWXLPXQEVMC-VOTSOKGWSA-N 0.000 description 1
- QWXVFUOWERMKCG-UHFFFAOYSA-N CN(C1=O)C(N)=NC1(c1cccc(Br)c1)c(cc1)cc2c1OCCC2 Chemical compound CN(C1=O)C(N)=NC1(c1cccc(Br)c1)c(cc1)cc2c1OCCC2 QWXVFUOWERMKCG-UHFFFAOYSA-N 0.000 description 1
- GEYPSNLUSFJKJV-UHFFFAOYSA-N Fc(c(Br)c1)ccc1C#Cc(cc1)cc2c1OCCC2 Chemical compound Fc(c(Br)c1)ccc1C#Cc(cc1)cc2c1OCCC2 GEYPSNLUSFJKJV-UHFFFAOYSA-N 0.000 description 1
- SRFMSUYFBNWTJS-UHFFFAOYSA-N N#Cc1c2OCCc2cc(C2(C(c(cc3)cc(Br)c3F)O)SCCCS2)c1 Chemical compound N#Cc1c2OCCc2cc(C2(C(c(cc3)cc(Br)c3F)O)SCCCS2)c1 SRFMSUYFBNWTJS-UHFFFAOYSA-N 0.000 description 1
- VOXZRLBTJPPTQE-UHFFFAOYSA-N N#Cc1c2OCCc2cc(C2SCCCS2)c1 Chemical compound N#Cc1c2OCCc2cc(C2SCCCS2)c1 VOXZRLBTJPPTQE-UHFFFAOYSA-N 0.000 description 1
- PSZXPORAJUGOGF-UHFFFAOYSA-N OC(C1(c(cc2)cc3c2OCC3)SCCCS1)c(cc1Br)ccc1F Chemical compound OC(C1(c(cc2)cc3c2OCC3)SCCCS1)c(cc1Br)ccc1F PSZXPORAJUGOGF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
Definitions
- the present invention relates to novel compounds and their pharmaceutical compositions.
- the present invention relates to therapeutic methods for the treatment and/or
- a ⁇ -related pathologies such as Downs syndrome, ⁇ -amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such aso Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- MCI mimild cognitive impairment
- BACE has an optimum activity at pH 4.0-5.0 (Vassar et al, 1999) and is inhibited weakly by standard pepsin inhibitors such as pepstatin. It has been shown that the catalytic domain minus the transmembrane and cytoplasmic domain has activity against substrate peptides (Lin et al, 2000). BACE is a membrane bound type 1 protein that is synthesized as a partially active proenzyme, and is abundantly expressed in brain tissue. It is thought to represent the major ⁇ -secretase activity, and is considered to be the rate-limiting step in the production of amyloid- ⁇ -protein (A ⁇ ). It is thus of special interest in the pathology of Alzheimer's disease, and in the development of drugs as a treatment for Alzheimer's disease.
- a ⁇ or amyloid- ⁇ -protein is the major constituent of the brain plaques which are characteristic of Alzheimer's disease (De Strooper et al, 1999).
- a ⁇ is a 39-42 residue peptide formed by the specific cleavage of a class 1 transmembrane protein called APP, or amyloid precursor protein. Cleavage of APP by BACE generates the extracellular soluble APP ⁇ fragment and the membrane bound CTF ⁇ (C99) fragment that is subsequently cleaved by ⁇ -secretase to generate A ⁇ peptide.
- Alzheimer's disease is estimated to afflict more than 20 million people worldwide and is believed to be the most common form of dementia.
- Alzheimer's disease is a progressive dementia in which massive deposits of aggregated protein breakdown products - amyloid plaques and neurofibrillary tangles accumulate in the brain. The amyloid plaques are thought to be responsible for the mental decline seen in Alzheimer's patients.
- Alzheimer's disease increases with age, and as the aging population of the developed world increases, this disease becomes a greater and greater problem.
- this disease becomes a greater and greater problem.
- any individuals possessing the double mutation of APP known as the Swedish mutation (in which the mutated APP forms a considerably improved substrate for BACE) have a much higher risk of developing AD, and also of developing the disease at an early age ⁇ see also US 6,245,964 and US 5,877,399 pertaining to transgenic rodents comprising APP-Swedish). Consequently, there is also a strong need for developing a compound that can be used in a prophylactic fashion for these individuals.
- APP The gene encoding APP is found on chromosome 21, which is also the chromosome found as an extra copy in Down's syndrome.
- Down's syndrome patients tend to develop Alzheimer's disease at an early age, with almost all those over 40 years of age showing Alzheimer's-type pathology (Oyama et al., 1994). This is thought to be due to the extra copy of the APP gene found in these patients, which leads to overexpression of APP and therefore to increased levels of A ⁇ causing the high prevalence of Alzheimer's disease seen in this population.
- inhibitors of BACE could be useful in reducing Alzheimer's-type pathology in Down's syndrome patients.
- Drugs that reduce or block BACE activity should therefore reduce A ⁇ levels and levels of fragments of A ⁇ in the brain, or elsewhere where A ⁇ or fragments thereof deposit, and thus slow the formation of amyloid plaques and the progression of AD or other maladies involving deposition of A ⁇ or fragments thereof (Yankner, 1996; De Strooper and Konig, 1999).
- BACE is therefore an important candidate for the development of drugs as a treatment and/or prophylaxis of A ⁇ -related pathologies such as Downs syndrome, ⁇ - amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- a ⁇ -related pathologies such as Downs syndrome, ⁇ - amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage
- disorders associated with cognitive impairment such as but not limited to MCI (“mild cognitive impairment")
- Alzheimer Disease memory loss
- the compounds of the present invention show beneficial properties compared to the potential inhibitors known in the art, e.g. improved hERG selectivity.
- the present invention provides potent BACE inhibitors of formula I:
- R 1 is selected from phenyl, heteroaryl, d- ⁇ alkyl optionally substituted with a C 3- ⁇ cycloalkyl, C 2-6 alkenyl optionally substituted with a heterocyclyl, C 3- ecycloalkyl, Cs ⁇ cycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R 3 ;
- R 2 is independently selected from hydrogen, cyano or halo;
- A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R 4 ;
- R 3 is independently selected from halo, cyano, d ⁇ alkyl, trifluoromethyl, methoxy, trifluoromethoxy and acetyl;
- R 4 is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R 4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
- R 5 is Ci- ⁇ alkyl optionally substituted with halogen, cyano, NH 2 , OH, CO 2 H, OC ⁇ alkyl, SO 2 H, C(O)C 1-6 alkyl,' C(O)OC 1-6 alkyl, C(O)NH 2 , C(O)NHC 1-6 alkyl, C(O)N(Ci -6 alkyl) 2 , SO 2 C 1-6 alkyl, SO 2 NHC 1-6 alkyl, SO ⁇ d ⁇ U ⁇ , NH(d -6 alkyl), N(Ci -6 alkyl) 2 , NHC(O)C 1-6 alkyl, NC(O)(C 1-6 alkyl) 2 , aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl) 2 , S0 2 aryI, SO 2 NHaryl, SO 2 N(aryl) 2 , NH(aryl),
- the present invention further provides potent BACE inhibitors of formula I: wherein
- R 1 is selected from phenyl, heteroaryl, Chalky! optionally substituted with a C 3- ⁇ cycloalkyl, C 2-6 alkenyl optionally substituted with a C 3-6 cycloalkyl, heterocyclyl, C 3 . ⁇ cycloalkyl, Cs- ⁇ cycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R 3 ;
- R 2 is independently selected from hydrogen, cyano or halo
- A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R 4 ;
- R 3 is independently selected from halo, cyano, d- ⁇ alkyl, trifluoromethyl, methoxy, trifluoromethoxy and acetyl;
- R is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R 4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
- R 5 is optionally substituted with halogen, cyano, NH 2 , OH, CO 2 H, OCi- ⁇ alkyl, SO 2 H, C(O)Ci -6 alkyl, C(O)OC 1-6 alkyl, C(O)NH 2 , C(O)NHC 1-6 alkyl, C(O)N(C 1-6 alkyl) 2 , SO 2 C 1-6 alkyl, SO 2 NHC 1-6 alkyl, SO 2 N(C 1-6 alkyl) 2 , NH(Ci -6 alkyl), N(C 1-6 alkyl) 2 , NHC(O)C 1-6 alkyl, NC(O)(C 1 .
- the present invention further provides potent BACE inhibitors of formula I: wherein
- R 1 is selected from phenyl, heteroaryl, C 1-6 alkyl optionally substituted with a C 3- ⁇ cycloalkyl, C 2- 6alkenyl optionally substituted with a C 3-6 cycloalkyl, heterocyclyl, C 3- ⁇ cycloalkyl, C 5-6 cycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R 3 ;
- A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R 4 ;
- R 3 is independently selected from halo, cyano, trifluoromethyl, methoxy and trifluoromethoxy;
- R 4 is independently selected from halo, cyano and methyl; provided that said bicyclic ring of A, is substituted with at least one R 4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
- R 5 is Ci- ⁇ alkyl optionally substituted with halogen, cyano, NH 2 , OH, CO 2 H, OCi ⁇ alkyl, SO 2 H, C(O)C 1-6 alkyl, C(O)OC 1-6 alkyl, C(O)NH 2 , C(O)NHC 1 .
- the present invention further provides potent BACE inhibitors of formula I:
- R 1 is selected from phenyl, heteroaryl, C ⁇ alkyl optionally substituted with a C 3- ⁇ cycloalkyl, C 2-6 alkenyl optionally substituted with a C 3 _ 6 cycloalkyl, heterocyclyl, C 3- ⁇ cycloalkyl, Cs- ⁇ cycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R 3 ;
- R 2 is independently selected from hydrogen, cyano or halo
- A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R 4 ;
- R 3 is independently selected from halo, cyano, C h alky!, trifluoromethyl, methoxy and trifluoromethoxy;
- R 4 is independently selected from halo, cyano and methyl; provided that said bicyclic ring of A, is substituted with at least one R 4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
- R 5 is Ci -6 alkyl optionally substituted with halogen, cyano, NH 2 , OH, CO 2 H, OCi- ⁇ alkyl, SO 2 H, C(O)C 1-6 alkyl, C(O)OC 1-6 alkyl, C(O)NH 2 , C(O)NHC 1-6 alkyl, C(O)N(C 1-6 alkyl) 2 ,
- NC(O)(aryl) 2 heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl) 2 , SO 2 heteroaryl, SO 2 NHheteroaryl,
- R 1 is phenyl, optionally substituted with one or more R 3 , or Ci -6 alkyl;
- R is independently selected from cyano and halo
- A is phenyl or heteroaryl, said phenyl or heteroaryl optionally fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R 4 ;
- R 3 is independently selected from halo, cyano, Ci- ⁇ alkyl, trifluoromethyl, methoxy, trifluoromethoxy and acetyl;
- R is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R 4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
- Ci- ⁇ alkyl optionally substituted with halogen, cyano, NH 2 , OH, CO 2 H, SO 2 H, C(O)Ci -6 alkyl, C(O)OC 1-6 alkyl, C(O)NH 2 , C(O)NHCi -6 alkyl, C(O)N(d. 6 alkyl) 2 , SO 2 C 1 - 6 alkyl ) SO 2 NHCi. 6 alkyl, SO 2 N(Ci -6 alkyl) 2 , NH(Ci -6 alkyl), N(Ci -6 alkyl) 2 ,
- NC(O)(aryl) 2 heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl) 2 , SO 2 heteroaryl, SO 2 NHheteroaryl,
- n 0, 1 or 2; provided that the following compounds are excluded:
- R 1 is phenyl. In another embodiment of this aspect, n is 1 and R 2 is fluoro.
- n 0.
- R 5 is Q- ⁇ alkyl
- R 5 is methyl
- A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system.
- A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is substituted with from one to four R 4 .
- said bicyclic ring system is substituted with two R 4 , said R 4 being methyl.
- said bicyclic ring system is substituted with one R 4 , said R 4 being cyano.
- A is selected from:
- A is:
- R 3 is independently selected from halo, cyano, methoxy, trifluoromethoxy and acetyl.
- R 1 is C ⁇ 2 C ⁇ 2 cyclopropyl
- R > 2 2 is independently selected from cyano and halo
- A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R 4 ;
- R 4 is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R 4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
- R 5 is Ci- ⁇ alkyl optionally substituted with halogen, cyano, NH 2 , OH, CO 2 H, OCi- ⁇ alkyl, SO 2 H, C(O)C I-6 alkyl, C(O)OCi -6 alkyl, C(O)NH 2 , C(O)NHC 1-6 alkyl, C(O)N(C 1-6 alkyl) 2 , SO 2 Ci -6 alkyl, SO 2 NHCi.
- n 1 and R 2 is fluoro.
- R 5 is C 1-6 alkyl.
- R 5 is methyl
- A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system, optionally substituted with from one to four R 4 .
- A is selected from:
- a compound selected from: 2-Amino-5-[3-(2-cyclopropylethyl)-4-fluorophenyl]-5-(3,4-dihydro-2H-chromen-6-yl)-3- methyI-3,5-dihydro-4H-imidazol-4-one; and 2-Amino-5-[3-(2-cyclopropylethyl)-4-fluorophenyl]-5-(2,3-dihydro-l-benzofuran-5-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
- composition comprising as active ingredient a therapeutically effective amount of a compound of formula I, in association with pharmaceutically acceptable excipients, carriers or diluents.
- a compound of formula I as a medicament for treating or preventing an A ⁇ -related pathology, wherein said A ⁇ -related pathology is Downs syndrome, a ⁇ -amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with
- Alzheimer Disease dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- a compound of formula I in the manufacture of a medicament for treating or preventing an A ⁇ -related pathology.
- use of a compound of formula I in the manufacture of a medicament for treating or preventing an A ⁇ -related pathology wherein said A ⁇ -related pathology is Downs syndrome, a ⁇ -amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- a method of treating or preventing an A ⁇ -related pathology in a mammal comprising administering to said patient a therapeutically effective amount of a compound of formula I.
- a method of treating or preventing an A ⁇ -related pathology in a mammal comprising administering to said patient a therapeutically effective amount of a compound of formula I, wherein said A ⁇ -related pathology is Downs syndrome, a ⁇ -amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- a method of treating or preventing Alzheimer's Disease comprising administering to said patient a therapeutically effective amount of a compound of formula I.
- a method of treating or preventing an A ⁇ -related pathology in a mammal comprising administering to said patient a therapeutically effective amount of a compound of formula I, wherein said A ⁇ -related pathology is Downs syndrome, a ⁇ -amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration, wherein said mammal is a human.
- MCI mimild cognitive impairment
- a method of treating or preventing an A ⁇ -related pathology in a mammal comprising administering to said patient a therapeutically effective amount of a compound of formula I and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor.
- a method of treating or preventing an A ⁇ -related pathology in a mammal comprising administering to said patient a therapeutically effective amount of a compound of formula I and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor, wherein said A ⁇ - related pathology is Downs syndrome, a ⁇ -amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- a method of treating or preventing an A ⁇ -related pathology in a mammal comprising administering to said patient a therapeutically effective amount of a compound of formula I and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor, wherein said A ⁇ - related pathology is Alzheimer Disease.
- a method of treating or preventing an A ⁇ -related pathology in a mammal comprising administering to said patient a therapeutically effective amount of a compound of formula I and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor, wherein said A ⁇ - related pathology is Downs syndrome, a ⁇ -amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration, wherein said mammal is a human.
- Some compounds of formula I may have stereogenic centres and/or geometric isomeric centres (E- and Z- isomers), and it is to be understood that the invention encompasses all such optical isomers, enantiomers, diastereoisomers, atropisomers and geometric isomers.
- the present invention relates to the use of compounds of formula I as hereinbefore defined as well as to the salts thereof. Salts for use in pharmaceutical compositions will be pharmaceutically acceptable salts, but other salts may be useful in the production of the compounds of formula I.
- the present invention provides compounds of formula I, or pharmaceutically acceptable salts, tautomers or in vzvo-hydroly sable precursors thereof, for use as medicaments.
- the present invention provides compounds described here in for use as medicaments for treating or preventing an A ⁇ -related pathology.
- the A ⁇ -related pathology is Downs syndrome, a ⁇ -amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- MCI mimild cognitive impairment
- the present invention provides use of compounds of formula I or pharmaceutically acceptable salts, tautomers or in vzvo-hydrolysable precursors thereof, in the manufacture of a medicament for the treatment or prophylaxis of A ⁇ -related pathologies.
- the A ⁇ -related pathologies include such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- MCI mimild cognitive impairment
- the present invention provides a method of inhibiting activity of BACE comprising contacting the BACE with a compound of the present invention.
- BACE is thought to represent the major ⁇ -secretase activity, and is considered to be the rate- limiting step in the production of amyloid- ⁇ -protein (A ⁇ ).
- a ⁇ amyloid- ⁇ -protein
- BACE is an important candidate for the development of drugs as a treatment and/or prophylaxis of A ⁇ -related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- a ⁇ -related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated
- the present invention provides a method for the treatment of A ⁇ - related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, presenile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration, comprising administering to a mammal (including human) a therapeutically effective amount of a compound of formula I, or a pharmaceutically acceptable salt, tautomer or in vz ' v ⁇ -hydrolysable precursor thereof.
- a ⁇ - related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angi
- the present invention provides a method for the prophylaxis of A ⁇ - related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, presenile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration comprising administering to a mammal (including human) a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt, tautomer or in v/v ⁇ -hydrolysable precursors.
- a ⁇ - related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy
- the present invention provides a method of treating or preventing A ⁇ -related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, presenile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration by administering to a mammal (including human) a compound of formula I or a pharmaceutically acceptable salt, tautomer or in vzv ⁇ -hydrolysable precursors and a cognitive and/or memory enhancing agent.
- a ⁇ -related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy
- the present invention provides a method of treating or preventing A ⁇ -related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, presenile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration by administering to a mammal (including human) a compound of formula I or a pharmaceutically acceptable salt, tautomer or in vzV ⁇ -hydrolysable precursors thereof wherein constituent members are provided herein, and a choline esterase inhibitor or anti-inflammatory agent.
- a ⁇ -related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such
- the present invention provides a method of treating or preventingA ⁇ -related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration, or any other disease, disorder, or condition described herein, by administering to a mammal (including human) a compound of the present inventionand an atypical antipsychotic agent.
- a ⁇ -related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders
- Atypical antipsychotic agents includes, but not limited to, Olanzapine (marketed as Zyprexa), Aripiprazole (marketed as Abilify), Risperidone (marketed as Risperdal), Quetiapine (marketed as Seroquel), Clozapine (marketed as Clozaril), Ziprasidone (marketed as Geodon) and Olanzapine/Fluoxetine (marketed as Symbyax).
- the mammal or human being treated with a compound of the invention has been diagnosed with a particular disease or disorder, such as those described herein. In these cases, the mammal or human being treated is in need of such treatment. Diagnosis, however, need not be previously performed.
- the present invention also includes pharmaceutical compositions, which contain, as the active ingredient, one or more of the compounds of the invention herein together with at least one pharmaceutically acceptable carrier, diluent or excipent.
- a variety of compounds in the present invention may exist in particular geometric or stereoisomeric forms.
- the present invention takes into account all such compounds, including cis- and trans isomers, R- and S- enantiomers, diastereomers, (D)-isomers, (L)- isomers, the racemic mixtures thereof, and other mixtures thereof, as being covered within the scope of this invention.
- Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention.
- the compounds herein described may have asymmetric centers. Compounds of the present invention containing an asymmetrically substituted atom may be isolated in optically active or racemic forms.
- optically active forms such as by resolution of racemic forms, by synthesis from optically active starting materials, or synthesis using optically active reagents.
- separation of the racemic material can be achieved by methods known in the art.
- Cis and trans geometric isomers of the compounds of the present invention are described and may be isolated as a mixture of isomers or as separated isomeric forms. All chiral, diastereomeric, racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated.
- substitution means that substitution is optional and therefore it is possible for the designated atom or moiety to be unsubstituted.
- substitution means that any number of hydrogens on the designated atom or moiety is replaced with a selection from the indicated group, provided that the normal valency of the designated atom or moiety is not exceeded, and that the substitution results in a stable compound.
- a substituent is methyl (i.e., CH 3 )
- 3 hydrogens on the carbon atom can be replaced.
- substituents include, but are not limited to: halogen, cyano, NH 2 , OH, CO 2 H, OCi -6 alkyl, CH 2 OH, SO 2 H, Ci -6 alkyl, OC ⁇ alkyl, C(O)C 1-6 alkyl, C(O)OC 1-6 alkyI, C(O)NH 2 , C(O)NHCi -6 alkyl, C(O)N(C 1-6 alkyl) 2 , SO 2 C 1-6 alkyl, SO 2 NHC 1-6 alkyl, SO 2 N(C 1-6 alkyl) 2 , NH(C 1-6 alkyl), N(C 1-6 alkyl) 2 , NHC(O)C 1-6 alkyl, NC(O)(C 1-6 alkyl) 2 , aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl) 2 , S0 2 aryl,
- alkyl used alone or as a suffix or prefix, is intended to include both branched and straight chain saturated aliphatic hydrocarbon groups having from 1 to 12 carbon atoms or if a specified number of carbon atoms is provided then that specific number would be intended.
- “Co -6 alkyl” denotes alkyl having O, 1, 2, 3, 4, 5 or 6 carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, t-butyl, pentyl, and hexyl.
- a subscript is the integer 0 (zero) the group to which the subscript refers to indicates that the group may be absent, i.e. there is a direct bond between the groups.
- alkenyl used alone or as a suffix or prefix is intended to include both branched and straight-chain alkene or olefin containing aliphatic hydrocarbon groups having from 2 to 12 carbon atoms or if a specified number of carbon atoms is provided then that specific number would be intended.
- C 2 - 6 alkenyl denotes alkenyl having 2, 3, 4, 5 or 6 carbon atoms.
- alkenyl examples include, but are not limited to, vinyl, allyl, 1-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 2-methylbut-2-enyl, 3-methylbut- 1-enyl, 1-pentenyl, 3-pentenyl and 4-hexenyl.
- aromatic refers to hydrocarbonyl groups having one or more unsaturated carbon ring(s) having aromatic characters, (e.g. 4n + 2 delocalized electrons) and comprising up to about 14 carbon atoms.
- heteroatoms such as nitrogen, oxygen or sulphur having aromatic character (e.g. 4n + 2 delocalized electrons).
- aryl refers to an aromatic ring structure made up of from 5 to 14 carbon atoms. Ring structures containing 5, 6, 7 and 8 carbon atoms would be single-ring aromatic groups, for example, phenyl. Ring structures containing 8, 9, 10, 11, 12, 13, or 14 would be polycyclic, for example naphthyl.
- the aromatic ring can be substituted at one or more ring positions with such substituents as described above.
- aryl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings (the rings are "fused rings") wherein at least one of the rings is aromatic, for example, the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls.
- ortho, meta and para apply to 1,2-, 1,3- and 1,4-disubstituted benzenes, respectively.
- the names 1,2-dimethylbenzene and ortho-dimethylbenzene are synonymous.
- cycloalkyl is intended to include saturated ring groups, having the specified number of carbon atoms. These may include fused or bridged polycyclic systems. Preferred cycloalkyls have from 3 to 10 carbon atoms in their ring structure, and more preferably have 3, 4, 5, and 6 carbons in the ring structure.
- C 3-6 cycloalkyl denotes such groups as cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl.
- cycloalkenyl is intended to include unsaturated ring groups, having the specified number of carbon atoms. These may include fused or bridged polycyclic systems. Preferred cycloalkenyls have from 3 to 10 carbon atoms in their ring structure, and more preferably have 3, 4, 5, and 6 carbons in the ring structure.
- C 3-6 cycloalkenyl denotes such groups as cyclopropenyl, cyclobutenyl, cyclopentenyl, or cyclohexenyl.
- halo or halogen refers to fluoro, chloro, bromo, and iodo.
- heteroaryl refers to a heteroaromatic heterocycle having at least one heteroatom ring member such as sulfur, oxygen, or nitrogen.
- Heteroaryl groups include monocyclic and polycyclic (e.g., having 2, 3 or 4 fused rings) systems. Examples of heteroaryl groups include without limitation, pyridyl (i.e., pyridinyl), pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl (i.e.
- the heteroaryl group has from 1 to about 20 carbon atoms, and in further embodiments from about 3 to about 20 carbon atoms.
- the heteroaryl group contains 3 to about 14, 4 to about 14, 3 to about 7, or 5 to 6 ring-forming atoms. In some embodiments, the heteroaryl group has 1 to about 4, 1 to about 3, or 1 to 2 heteroatoms. In some embodiments, the heteroaryl group has 1 heteroatom.
- heterocyclyl or “heterocyclic” or “heterocycle” refers to a saturated, unsaturated or partially saturated, monocyclic, bicyclic or tricyclic ring (unless otherwise stated) containing 3 to 20 atoms of which 1, 2, 3, 4 or 5 ring atoms are chosen from nitrogen, sulphur or oxygen, which may, unless otherwise specified, be carbon or nitrogen linked, wherein a -CH 2 - group is optionally be replaced by a -C(O)-; and where unless stated to the contrary a ring nitrogen or sulphur atom is optionally oxidised to form the N-oxide or S-oxide(s) or a ring nitrogen is optionally quarternized; wherein a ring -NH is optionally substituted by acetyl, formyl, methyl or mesyl; and a ring is optionally substituted by one or more halo.
- heterocyclyl group is bi- or tricyclic then at least one of the rings may optionally be a heteroaromatic or aromatic ring provided that at least one of the rings is non-heteroaromatic. If the said heterocyclyl group is monocyclic then it must not be aromatic.
- heterocyclyls include, but are not limited to, piperidinyl, N- acetylpiperidinyl, N-methylpiperidinyl, N-formylpiperazinyl, N-mesylpiperazinyl, homopiperazinyl, piperazinyl, azetidinyl, oxetanyl, morpholinyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, indolinyl, tetrahydropyranyl, dihydro-2H-pyranyl, tetrahydrofuranyl and 2,5-dioxoimidazolidinyl.
- protecting group means temporary substituents which protect a potentially reactive functional group from undesired chemical transformations.
- protecting groups include esters of carboxylic acids, silyl ethers of alcohols, and acetals and ketals of aldehydes and ketones respectively.
- the field of protecting group chemistry has been reviewed (Greene, T. W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3 rd ed.; Wiley: New York, 1999).
- pharmaceutically acceptable is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof.
- pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric acid.
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound that contains a basic or acidic moiety by conventional chemical methods.
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like diethyl ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are used.
- tautomer means other structural isomers that exist in equilibrium resulting from the migration of a hydrogen atom. For example, keto-enol tautomerism where the resulting compound has the porperties of both a ketone and an unsturated alcohol.
- stable compound and “stable structure” are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- Compounds of the invention further include hydrates and solvates.
- the present invention further includes isotopically-labelled compounds of the invention.
- An “isotopically” or “radio-labelled” compound is a compound of the invention where one or more atoms are replaced or substituted by an atom having an atomic mass or mass number different from the atomic mass or mass number typically found in nature (i.e., naturally occurring).
- Suitable radionuclides that may be incorporated in compounds of the present invention include but are not limited to 2 H (also written as D for deuterium), 3 H (also written as T for tritium), 11 C, 13 C, 14 C, 13 N, 15 N, 15 O, 17 O, 18 0, 18 F, 35 S, 36 Cl, 82 Br, 75 Br, 76 Br, 77 Br, 123 1, 124 1, 125 I and 131 I.
- the radionuclide that is incorporated in the instant radio-labelled compounds will depend on the specific application of that radio-labelled compound. For example, for in vitro receptor labelling and competition assays, compounds that incorporate 3 H, 14 C, 82 Br, 125 1 , 131 1, 35 S or will generally be most useful.
- radio-imaging applications 11 C, 18 F, 125 1, 123 1, 124 I, 131 I, 75 Br, 76 Br or 77 Br will generally be most useful.
- a "radio-labelled compound” is a compound that has incorporated at least one radionuclide.
- the radionuclide is selected from the group consisting of 3 H, 14 C, 125 1 , 35 S and 82 Br.
- the anti-dementia treatment defined herein may be applied as a sole therapy or may involve, in addition to the compound of the invention, conventional chemotherapy.
- chemotherapy may include one or more of the following categories of agents: acetyl cholinesterase inhibitors, anti-inflammatory agents, cognitive and/or memory enhancing agents or atypical antipsychotic agents.
- Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of the treatment.
- Such combination products employ the compounds of this invention.
- Compounds of the present invention may be administered orally, parenteral, buccal, vaginal, rectal, inhalation, insufflation, sublingually, intramuscularly, subcutaneously, topically, intranasally, intraperitoneally, intrathoracially, intravenously, epidurally, intrathecally, intracerebroventricularly and by injection into the joints.
- the dosage will depend on the route of administration, the severity of the disease, age and weight of the patient and other factors normally considered by the attending physician, when determining the individual regimen and dosage level as the most appropriate for a particular patient.
- An effective amount of a compound of the present invention for use in therapy of dementia is an amount sufficient to symptomatically relieve in a warm-blooded animal, particularly a human the symptoms of dementia, to slow the progression of dementia, or to reduce in patients with symptoms of dementia the risk of getting worse.
- inert, pharmaceutically acceptable carriers can be either solid or liquid.
- Solid form preparations include powders, tablets, dispersible granules, capsules, cachets, and suppositories.
- a solid carrier can be one or more substances, which may also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, or tablet disintegrating agents; it can also be an encapsulating material.
- the carrier is a finely divided solid, which is in a mixture with the finely divided active component.
- the active component is mixed with the carrier having the necessary binding properties in suitable proportions and compacted in the shape and size desired.
- a low-melting wax such as a mixture of fatty acid glycerides and cocoa butter is first melted and the active ingredient is dispersed therein by, for example, stirring. The molten homogeneous mixture is then poured into convenient sized molds and allowed to cool and solidify.
- Suitable carriers include magnesium carbonate, magnesium stearate, talc, lactose, sugar, pectin, dextrin, starch, tragacanth, methyl cellulose, sodium carboxymethyl cellulose, a low-melting wax, cocoa butter, and the like.
- the present invention provides a compound of formula I or a pharmaceutically acceptable salt thereof for the therapeutic treatment (including prophylactic treatment) of mammals including humans, it is normally formulated in accordance with standard pharmaceutical practice as a pharmaceutical composition.
- the pharmaceutical composition of this invention may also contain, or be co-administered (simultaneously or sequentially) with, one or more pharmacological agents of value in treating one or more disease conditions referred to herein.
- composition is intended to include the formulation of the active component or a pharmaceutically acceptable salt with a pharmaceutically acceptable carrier.
- this invention may be formulated by means known in the art into the form of, for example, tablets, capsules, aqueous or oily solutions, suspensions, emulsions, creams, ointments, gels, nasal sprays, suppositories, finely divided powders or aerosols or nebulisers for inhalation, and for parenteral use (including intravenous, intramuscular or infusion) sterile aqueous or oily solutions or suspensions or sterile emulsions.
- Liquid form compositions include solutions, suspensions, and emulsions.
- Sterile water or water-propylene glycol solutions of the active compounds may be mentioned as an example of liquid preparations suitable for parenteral administration.
- Liquid compositions can also be formulated in solution in aqueous polyethylene glycol solution.
- Aqueous solutions for oral administration can be prepared by dissolving the active component in water and adding suitable colorants, flavoring agents, stabilizers, and thickening agents as desired.
- Aqueous suspensions for oral use can be made by dispersing the finely divided active component in water together with a viscous material such as natural synthetic gums, resins, methyl cellulose, sodium carboxymethyl cellulose, and other suspending agents known to the pharmaceutical formulation art.
- the pharmaceutical compositions can be in unit dosage form.
- the composition is divided into unit doses containing appropriate quantities of the active component.
- the unit dosage form can be a packaged preparation, the package containing discrete quantities of the preparations, for example, packeted tablets, capsules, and powders in vials or ampoules.
- the unit dosage form can also be a capsule, cachet, or tablet itself, or it can be the appropriate number of any of these packaged forms.
- compositions may be formulated for any suitable route and means of administration.
- Pharmaceutically acceptable carriers or diluents include those used in formulations suitable for oral, rectal, nasal, topical (including buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous, intradermal, intrathecal and epidural) administration.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy.
- conventional non-toxic solid carriers include, for example, pharmaceutical grades of mannitol, lactose, cellulose, cellulose derivatives, starch, magnesium stearate, sodium saccharin, talcum, glucose, sucrose, magnesium carbonate, and the like may be used.
- Liquid pharmaceutically administrable compositions can, for example, be prepared by dissolving, dispersing, etc, an active compound as defined above and optional pharmaceutical adjuvants in a carrier, such as, for example, water, saline aqueous dextrose, glycerol, ethanol, and the like, to thereby form a solution or suspension.
- the pharmaceutical composition to be administered may also contain minor amounts of non-toxic auxiliary substances such as wetting or emulsifying agents, pH buffering agents and the like, for example, sodium acetate, sorbitan monolaurate, triethanolamine sodium acetate, sorbitan monolaurate, triethanolamine oleate, etc.
- auxiliary substances such as wetting or emulsifying agents, pH buffering agents and the like, for example, sodium acetate, sorbitan monolaurate, triethanolamine sodium acetate, sorbitan monolaurate, triethanolamine oleate, etc.
- the compounds of the invention may be derivatised in various ways.
- derivatives of the compounds includes salts (e.g. pharmaceutically acceptable salts), any complexes (e.g. inclusion complexes or clathrates with compounds such as cyclodextrins, or coordination complexes with metal ions such as Mn 2+ and Zn 2+ ), free acids or bases, polymorphic forms of the compounds, solvates (e.g. hydrates), prodrugs or lipids, coupling partners and protecting groups.
- prodrugs is meant for example any compound that is converted in vivo into a biologically active compound.
- Salts of the compounds of the invention are preferably physiologically well tolerated and non toxic. Many examples of salts are known to those skilled in the art. All such salts are within the scope of this invention, and references to compounds include the salt forms of the compounds.
- the compounds may form quaternary ammonium salts, for example by reaction with an alkylating agent according to methods well known to the skilled person. Such quaternary ammonium compounds are within the scope of the invention.
- Compounds containing an amine function may also form N-oxides.
- a reference herein to a compound that contains an amine function also includes the N-oxide.
- N-oxides are the N-oxides of a tertiary amine or a nitrogen atom of a nitrogen-containing heterocycle.
- N-Oxides can be formed by treatment of the corresponding amine with an oxidizing agent such as hydrogen peroxide or a per-acid (e.g. a peroxycarboxylic acid), see for example Advanced Organic Chemistry, by Jerry March, 4 th Edition, Wiley Interscience, pages. More particularly, N-oxides can be made by the procedure of L. W. Deady (Syn. Comm. 1977, 7, 509-514) in which the amine compound is reacted with w-chloroperoxybenzoic acid (MCPBA), for example, in an inert solvent such as dichloromethane.
- MCPBA w-chloroperoxybenzoic acid
- the quantity of the compound to be administered will vary for the patient being treated and will vary from about 100 ng/kg of body weight to 100 mg/kg of body weight per day and preferably will be from 10 pg/kg to 10 mg/kg per day.
- dosages can be readily ascertained by those skilled in the art from this disclosure and the knowledge in the art.
- the skilled artisan can readily determine the amount of compound and optional additives, vehicles, and/or carrier in compositions and to be administered in methods of the invention.
- Compounds of the present invention have been shown to inhibit beta secretase (including BACE) activity in vitro.
- Inhibitors of beta secretase have been shown to be useful in blocking formation or aggregation of A ⁇ peptide and therefore have beneficial effects in treatment of Alzheimer's Disease and other neurodegenerative diseases associated with elevated levels and/or deposition of A ⁇ peptide. Therefore, it is believed that the compounds of the present invention may be used for the treatment of Alzheimer disease and disease associated with dementia Hence, compounds of the present invention and their salts are expected to be active against age-related diseases such as Alzheimer, as well as other A ⁇ related pathologies such as Downs syndrome and ⁇ -amyloid angiopathy. It is expected that the compounds of the present invention would most likely be used as single agents but could also be used in combination with a broad range of cognition deficit enhancement agents.
- the present invention also relates to processes for preparing the compound of formula I as a free base or a pharmaceutically acceptable salt thereof.
- suitable protecting groups will be added to, and subsequently removed from the various reactants and intermediates in a manner that will be readily understood by one skilled in the art of organic synthesis.
- Conventional procedures for using such protecting groups as well as examples of suitable protecting groups are for example described in "Protective Groups in Organic Synthesis", T.W. Greene, P.G.M Wutz, Wiley-Interscience, New York, 1999. It is understood that microwaves can be used for the heating of reaction mixtures.
- reaction may be carried out by reaction with a suitable reagent such as 1,3-propanedithiol in the presence of an acid such as hydrochloric acid or/j-toluenesulfonic acid, or a Lewis acid such as boron trifluoride or titanium tetrachloride or ruthenium(III) chloride.
- a suitable reagent such as 1,3-propanedithiol in the presence of an acid such as hydrochloric acid or/j-toluenesulfonic acid, or a Lewis acid such as boron trifluoride or titanium tetrachloride or ruthenium(III) chloride.
- a suitable solvent such as dichloromethane, acetonitrile, chloroform, toluene or diethyl ether, at a temperature between -78 0 C and reflux.
- reaction III may be carried out by treating III with a suitable base such as an alkyllithium reagent e.g. «-butyl lithium or /-butyl lithium or lithium diisopropylamide, before addition of IV.
- a suitable base such as an alkyllithium reagent e.g. «-butyl lithium or /-butyl lithium or lithium diisopropylamide
- the reaction may be carried out in a solvent such as tetrahydrofuran or diethyl ether, or a mixture of tetrahydrofuran or diethyl ether with hexane, at a temperature between -100 0 C and 25 °C.
- the reaction may be aided by the presence of reagents such as hexamethylphosphoric triamide or N//,N ⁇ -tetramethyl-l,2-ethanediamine.
- reaction may be carried out by: a) reaction with a suitable reagent such as 1,1,1 -tris(acetyloxy)- 1 , 1 -dihydro- 1 ,2- benziodoxol-3(lH)-one, bis(trifluoroacetoxy)iodobenzene, N-bromosuccinimide, or a mixture of trifluoroacetic acid with either sodium nitrite or formaldehyde, in a suitable solvent such as dichloromethane, acetonitrile, chloroform, acetone or water or a mixture thereof, between -5 °C to 40 °C.
- a suitable reagent such as 1,1,1 -tris(acetyloxy)- 1 , 1 -dihydro- 1 ,2- benziodoxol-3(lH)-one, bis(trifluoroacetoxy)iodobenzene, N-bromosuccinimide, or a mixture of triflu
- a suitable reagent or reagent combination such as N- chlorosuccinimide and silver nitrate, N-iodosuccinimide, 3-chloroperoxybenzoic acid, ammonium cerium(TV) nitrate, thallium(III) nitrate, mercury(II) chloride and calcium carbonate, or mercery(II) acetate.
- the reaction may be preformed in a suitable solvent such as water, acetonitrile, methanol, acetone or diethyl ether or mixtures thereof, between -50 0 C and 50 0 C , followed or preceded by, oxidation with a reagent such as 1,1,1- tris(acetyloxy)- 1,1 -dihydro- l,2-benziodoxoI-3(lH)-one, manganese dioxide, hydrogen peroxide, potassium permanganate, pyridinium chlorochromate, copper sulfate or bromine, in a suitable solvent such as dichloromethane, water, acetonitrile, chloroform or dimethyl formamide, at between 0 °C and reflux.
- a suitable solvent such as water, acetonitrile, methanol, acetone or diethyl ether or mixtures thereof, between -50 0 C and 50 0 C , followed or preceded by, oxidation with a reagent such
- (VI) (VII) may be carried out by reaction with an appropriately N-substituted thiourea, such as N- methyl thiourea, in the presence of a suitable base such as potassium hydroxide or sodium hydroxide in a suitable solvent such as water, dimethyl sulfoxide, ethanol or methanol or mixtures thereof, between 20 0 C and reflux.
- a suitable base such as potassium hydroxide or sodium hydroxide
- a suitable solvent such as water, dimethyl sulfoxide, ethanol or methanol or mixtures thereof, between 20 0 C and reflux.
- alkylhydroperoxide such as 7-butylhydroperoxide in a solvent such as ethanol, methanol or water, or a mixture thereof, at 0 0 C to 50 °C.
- a suitable arylhalide such as a compound of formula II and an appropriate alkyne such as ethynyltrimethylsilane in the presence of copper(I) iodide and a suitable palladium catalyst such as dichlorobis(benzonitrile)palladium(II), bis(triphenylphosphine)palladium(ll) dichloride, palladium(II) chloride, palladium(O) tetrakistriphenylphosphine with or without a suitable ligand such as tri-fert-butylphosphine or triphenylphosphine, and a suitable base, such as trietylamine, diisopropylamine or piperidine may be used.
- the reaction may be performed in a solvent such as tetrahydrofuran or N,N-dimethylformamide, at temperatures between 20 °C and 100 0 C.
- reaction may be performed using silver(I) nitrate or a suitable base such as potassium hydroxide, sodium hydroxide, lithium hydroxide or potassium carbonate, or using a fluoride ion- mediated desilylation using a suitable compound such as tetrabutylammonium fluoride or potassium fluoride.
- a suitable base such as potassium hydroxide, sodium hydroxide, lithium hydroxide or potassium carbonate
- fluoride ion- mediated desilylation using a suitable compound such as tetrabutylammonium fluoride or potassium fluoride.
- the reaction may be performed in a solvent such as tetrahydrofuran, methanol, dichloromethane or water, or mixtures thereof, at temperatures between 0 °C and 100 0 C.
- reaction may be carried out by a reaction with: a) an alkyllithium such as butyllithium, or magnesium, and a suitable boron compound such as trimethyl borate or triisopropyl borate.
- the reaction may be performed in a suitable solvent such as tetrahydrofuran, hexane or dichloromethane in a temperature range
- a suitable boron species such as biscatecholatodiboron, bispinacolatodiboron or pinacolborane in the presence of a suitable palladium catalyst such as palladium(O) tetrakistriphenylphosphine, palladium diphenylphosphineferrocene dichloride or palladium
- I 0 acetate with or without a suitable ligand such as 2-(dicyclohexylphosphino)biphenyl, and a suitable base, such as a tertiary amine, such as trietylamine or diisopropylethylamine, or potassium acetate may be used.
- a suitable ligand such as 2-(dicyclohexylphosphino)biphenyl
- a suitable base such as a tertiary amine, such as trietylamine or diisopropylethylamine, or potassium acetate
- the reaction may be performed in a solvent such as dioxane, toluene, acetonitrile, water, ethanol or 1,2-dimethoxyethane, or mixtures thereof, at temperatures between 20 0 C and 160 0 C.
- reaction may be preformed using a suitable catalyst such as palladium-on-charcoal, Raney nickel or Wilkinson's catalyst, and hydrogen.
- a suitable catalyst such as palladium-on-charcoal, Raney nickel or Wilkinson's catalyst, and hydrogen.
- the reaction may be preformed in a suitable solvent such as ethyl acetate, methanol or ethanol, at temperatures between 20 0 C and reflux.
- 30 xv ⁇ xvi ⁇ may be preformed using a metal hydride reducing agent such as diisopropylaluminium hydride or lithium aluminum hydride.
- the reaction may be preformed in a suitable solvent such as dichlorometane, toluene or benzene, at a temperature between -78 0 C and 25 0 C.
- reaction may be carried out by a reaction with: a) an alkyllithium such as butyllithium, or magnesium, and a suitable boron compound such as trimethyl borate or triisopropyl borate.
- a) an alkyllithium such as butyllithium, or magnesium
- a suitable boron compound such as trimethyl borate or triisopropyl borate.
- the reaction may be performed in a suitable
- solvent such as tetrahydrofuran, hexane or dichloromethane in a temperature range between -78 0 C and 20 0 C; or, b) a suitable boron species such as biscatecholatodiboron, bispinacolatodiboron or pinacolborane in the presence of a suitable palladium catalyst such as palladium(O)
- the reaction may be performed in a solvent such as dioxane, toluene, acetonitrile, water, ethanol or 1,2-dimethoxyethane, or mixtures thereof, at temperatures between 20 °C and 160 0 C.
- a suitable reagent or mixture of reagents such as sodium periodate and ruthenium dioxide, iodine and dimethyl sulfoxide, palladium chloride and dimethyl sulfoxide, oxone, hydrogen peroxide, oxygen, potassium permanganate, ruthenium tetroxide, or selenium dioxide, in a suitable solvent such as dimethyl sulfoxide, dichloromethane, acetonitrile, water, acetone, chloroform or carbon tetrachloride at a temperature between -78 0 C and 150 0 C.
- the reaction may be aided by the presence of a catalyst such as ruthenium(III) chloride or iron(III) chloride.
- N-substituted guanidine such as N- methylguanidine, N-ethylguanidine, or N-propylguanidine
- a suitable base such as potassium hydroxide, sodium hydroxide or sodium carbonate
- a suitable solvent such as water, dimethyl sulfoxide, dioxane, ethanol or methanol or mixtures thereof, between 20 0 C and reflux.
- XXIII may be preformed by treating XXIII with: a) a formylation agent such as methylchloroformate, dichloromethoxymethane, dichlorobutoxymethane, formaldehyde, carbon monoxide, hydrogen cyanide or a combination of phosphorous oxychloride with N-methyl-N-phenyl formamide o ⁇ NJ ⁇ -o dimethylformamide.
- a formylation agent such as methylchloroformate, dichloromethoxymethane, dichlorobutoxymethane, formaldehyde, carbon monoxide, hydrogen cyanide or a combination of phosphorous oxychloride with N-methyl-N-phenyl formamide o ⁇ NJ ⁇ -o dimethylformamide.
- the reaction can be performed with or without a suitable reagent such as titanium tetrachloride, tin tetrachloride, aluminium trichloride and copper(I) chloride or combinations thereof.
- the reaction may be preformed with or without a suitable solvent such as dichloromethane, benzene, or 1,1,2,2-tetrachloroethane at a temperature between - 20 °C and reflux; 5 or, b) a bromination agent such as bromine or N-bromosuccinimide in a solvent such as chloroform, ⁇ N-dimethylformamide, tetrahydrofurane or hexane or mixtures thereof at a temperature between -78 0 C and reflux , followed by treatment with a base such as butyl lithium and subsequently a reagent such as N,N-dirnethylformamide or acetic acid in ao solvent such as tetrahydrofurane and/or hexane at a temperature between -78 0 C and 0 °C.
- a suitable solvent such as dichloromethane, benzene, or 1,1,2,2-tetrachloroethane
- a bromination agent such
- Another object of the invention are processes a or b or c for the preparation of compounds5 of general formula I, wherein R 1 , R 2 , R 3 , R 4 , R 5 and A unless otherwise specified, are defined as hereinbefore, and salts thereof.
- the free base may be treated with an acid such as a hydrogen halide such as hydrogen chloride, sulphuric acid, a sulphonic acid such as methane sulphonic acid or a carboxylic acid such as acetic or citric acid in a suitable solvent such as tetrahydrofuran, diethyl ether, methanol,o ethanol, chloroform or dichloromethane or mixtures thereof, the reaction may occur between -30 0 C to 50 0 C.
- a hydrogen halide such as hydrogen chloride, sulphuric acid, a sulphonic acid such as methane sulphonic acid or a carboxylic acid such as acetic or citric acid
- a suitable solvent such as tetrahydrofuran, diethyl ether, methanol,o ethanol, chloroform or dichloromethane or mixtures thereof
- reaction may be carried out by a de-halogen coupling with a suitable compound of formula XXVI.
- the reaction may be carried out by coupling of a compound of formula XXV with an appropriate aryl boronic acid or boronic ester of formula XXVI.
- the reaction may be carried out using a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium(0), palladium diphenylphosphineferrocene dichloride or palladium acetate, together with, or without, a suitable ligand such as t ⁇ -tert- butylphosphine or 2-(dicyclohexylphosphino)biphenyl, or using a nickel catalyst such as nickel on charcoal or l,2-bis(diphenylphosphino)ethanenickel dichloride together with zinc and sodium triphenylphosphinetrimetasulfonate.
- a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium(0), palladium diphenylphosphineferrocene dichloride or palladium acetate
- a suitable ligand such as t ⁇ -tert- butylphosphine or 2-(dic
- a suitable base such as cesium fluoride, an alkyl amine such as triethyl amine, or an alkali metal or alkaline earth metal carbonate or hydroxide such as potassium carbonate, sodium carbonate, cesium carbonate, or sodium hydroxide may be used in the reaction, which may be performed in a temperature range between 20 0 C and 160 0 C, in a suitable solvent such as toluene, tetrahydrofuran, dioxane, dimethoxyethane, water, ethanol or N,N-dimethylformamide, or mixtures thereof.
- a suitable solvent such as toluene, tetrahydrofuran, dioxane, dimethoxyethane, water, ethanol or N,N-dimethylformamide, or mixtures thereof.
- reaction may be preformed using a suitable catalyst such as palladium-on-charcoal, Raney nickel or Wilkinson's catalyst, and hydrogen.
- a suitable catalyst such as palladium-on-charcoal, Raney nickel or Wilkinson's catalyst, and hydrogen.
- the reaction may be preformed in a suitable solvent such as ethyl acetate, methanol or ethanol, at temperatures between 20 0 C and reflux.
- Microwave heating was performed in a Creator, Initiator or Smith Synthesizer Single- mode microwave cavity producing continuous irradiation at 2450 MHz.
- 1H NMR spectra were recorded in the indicated deuterated solvent at 400 MHz.
- the 400MHz spectra were obtained unless stated otherwise, using a Bruker av400 NMR spectrometer equipped with a 3 mm flow injection SEI 1 HTD- 13 C probe head with Z- gradients, using a BEST 215 liquid handler for sample injection, or using a Bruker DPX400 NMR spectrometer equipped with a 4-nucleus probehead with Z-gradients. Chemical shifts are given in ppm down- and upfield from TMS. Resonance multiplicities are denoted s, d, t, q, m and br for singlet, doublet, triplet, quartet, multiplet, and broad respectively.
- LC-MS analyses were recorded on a Waters LCMS equipped with a Waters X-Terra MS, C8-column, (3.5 ⁇ m, 100 mm x 3.0 mm i.d.).
- the mobile phase system consisted of A: 10 mM ammonium acetate in water/acetonitrile (95:5) and B: acetonitrile.
- a linear gradient was applied running from 0% to 100% B in 4-5 minutes with a flow rate of 1.0 mL/min.
- the mass spectrometer was equipped with an electrospray ion source (ESI) operated in a positive or negative ion mode.
- the capillary voltage was 3 kV and the mass spectrometer was typically scanned between m/z 100-700.
- LC-MS analyses were performed on a LC-MS consisting of a Waters sample manager 2111 C, a Waters 1525 ⁇ binary pump, a Waters 1500 column oven, a Waters ZQ single quadrupole mass spectrometer, a Waters PDA2996 diode array detector and a Sedex 85 ELS detector.
- the mass spectrometer was configured with an atmospheric pressure chemical ionisation
- APCI atmospheric pressure photo ionisation
- APPI atmospheric pressure photo ionisation
- the mass spectrometer scanned in the positive mode, switching between APCI and APPI mode.
- the mass range was set to m/z 120-800 using a scan time of 0.3 s.
- the APPI repeller and the APCI corona were set to 0.86 kV and 0.80 ⁇ A, respectively.
- the desolvation temperature (300°C), desolvation gas (400 L/Hr) and cone gas (5 L/Hr) were constant for both APCI and APPI mode.
- Mass spectra were run using an automated system with atmospheric pressure chemical (APCI or CI) or electrospray (+ESI) ionization. Generally, only spectra where parent masses are observed are reported. The lowest mass major ion is reported for molecules where isotope splitting results in multiple mass spectral peaks (for example when chlorine is present).
- HPLC assays were performed using an Agilent HPl 100 Series system equipped with a Waters X-Terra MS, C 8 column (3.0 x 100 mm, 3.5 ⁇ m). The column temperature was set to 40 °C and the flow rate to 1.0 rnL/min.
- the Diode Array Detector was scanned from 200-300 nm. A linear gradient was applied, run from 0% to 100% B in 4 min.
- Mobile phase A 10 mM ammonium acetate in water/acetonitrile (95:5)
- mobile phase B acetonitrile.
- Preparative HPLC was performed on a Waters Auto purification HPLC-UV system with a diode array detector using a Waters XTerra MS C 8 column (19x300 mm, 7 ⁇ m) and a linear gradient of mobile phase B was applied.
- Mobile phase A 0.1 M ammonium acetate in water/acetonitrile (95:5) and mobile phase B: acetonitrile.
- Flow rate 20 mL/min.
- TLC Thin layer chromatography
- 6-(l,3-Dithian-2-yl)-2,2-dimethylchromane (1.32 g, 4.71 mmol) was dissolved in anhydrous tetrahydrofuran (40 mL) under an atmosphere of argon, cooled to -78 °C and n- butyl lithium (2.1 mL, 2.5 M) was added dropwise. The solution was stirred at -78 °C for 20 min and then treated with 3-bromo-4-fluorobenzaldehyde (1.0 g, 4.9 mmol). Thes reaction was stirred for another 20 min at —78 °C and was then allowed to reach room temperature. Aqueous ammonium chloride was added and the mixture was extracted with dichloromethane.
- the reaction mixture was stirred at room temperature for 2 h.
- the crude mixture was diluted with ethyl acetate, washed with 1 M aqueous hydrochloric acid, water and saturated aqueous sodium hydrogen carbonate.
- the organic layer was dried over magnesium sulfate, filtered and concentrated.
- 6-[(3-Bromo-4-fluorophenyl)ethynyl]chromane (5.O g, 15.1 mmol) and s palladium(H)dichloride (0.27 g, 1.5 mmol) were heated in dimethyl sulfoxide (70 mL) at 150 °C under an atmosphere of argon for 3 h. After cooling to room temperature water was added and the mixture was extracted with dichloromethane. The combined organic layers were washed with 1 M hydrochloric acid, water and saturated aqueous sodium carbonate solution, dried over magnesium sulfate, filtered and concentrated.
- reaction mixture was diluted with water and the p ⁇ was adjusted to 4-5 by addition of aqueous hydrochloric acid (1 M).
- the mixture was extracted by dichloromethane. The combined organic layers were washed with water, dried over magnesium sulfate, filtrated and the solvent was evaporated.
- tert-Butyl hydroperoxide (24.0 mL, 154.5 mmol, 70 wt% aq) was added to a solution of 5- (3-bromo-4-fluorophenyl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-2- thioxoimidazolidin-4-one (4.5 g, 10.3 mmol) in a 1:3 mixture of methanol/ ammonium hydroxide (60:20 mL). The reaction was stirred at room temperature for 4 h, concentrated and the residue was dissolved in chloroform. The organic phase was washed with water, dried over magnesium sulfate, filtrated and the solvent was evaporated.
- Dess-Martin periodinane (14.0 g, 32.8 mmol) was added to a solution of (3-bromo-4- fluorophenyl)[2-(2,3-dihydro-l-benzofuran-5-yl)-l,3-dithian-2-yl]methanol (5.9 g, 13.1 mmol) and fer/-butanol (4.2 mL, 45.8 mmol) in dichloromethane (250 mL) under an atmosphere of argon and the reaction mixture was stirred over night. A solution of sodiumo thiosulfate (12.5 g) in saturated aqueous sodium hydrogencarbonate (200 mL) was added and the resulting mixture was stirred for 30 min.
- reaction mixture was diluted with water and the pH was adjusted to 4-5 by addition of aqueous hydrochloric acid (1 M).
- the mixture was extracted with dichloromethane.
- the combined organic layers were washed with water, dried over magnesium sulfate, filtrated and evaporated.
- Dess-Martin periodinane (1.3 g, 3.08 mmol) was added to a solution of 5- ⁇ 2-[(3-bromo-4- fluorophenyl)(hydroxy)methyl]-l ,3-dithian-2-yl ⁇ -2,3-dihydro- 1 -benzofuran-7-carbonitrile (0.64 g, 1.37 mmol) and tert-butanol (0.36 g, 4.80 mmol) in dichloromethane (10 mL) under an atmosphere of argon and the reaction mixture was stirred overnight. Aqueous sodium thiosulfate (2.5 g) was added and the resulting mixture was stirred for 30 min.
- Table 1 Representative examples synthesized as described for 5-[2-Amino-4-(4-fluoro-3- pyridin-3-ylphenyl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-4-yl]-2,3-dihydro-l- benzofuran-7-carbonitrile. AU the reactions were analyzed using LC-MS and those which showed a low level of conversion was irradiated in a microwave for another hour at 130 0C.
- the soluble part of the human ⁇ -Secretase (AA 1 - AA 460) was cloned into the ASP2- FclO-1-IRES-GFP-neoK mammalian expression vector.
- the gene was fused to the Fc domain of IgGl (affinity tag) and stably cloned into HEK 293 cells.
- Purified sBACE-Fc is stored in Tris buffer, pH 9.2 and has a purity of 95%.
- the enzyme was diluted to 43 ⁇ g/ml in 40 mM MES pH 5.0.
- the IGEN substrate was diluted to 12 ⁇ M in 40 mM MES pH 5.0.
- Compounds were diluted to the desired concentration in dimethyl sulfoxide (final dimethyl sulfoxide concentration in assay is
- the assay was performed in a 96 well PCR plate from Greiner (#650201). Compound in dimethyl sulfoxide (3 ⁇ L) and enzyme (27 ⁇ L) were added to the plate, and pre- incubated for 10 min. The reaction was started with substrate (30 ⁇ L). The final dilution of enzyme was 20 ⁇ g/ml and the final concentration of substrate was 6 ⁇ M. After 20 minutes reaction at room temperature (RT), the reaction was stopped by removing 10 ⁇ L of the reaction mix and diluting it 1 :25 in 0.2 M Trizma-HCl, pH 8.0.
- the product was quantified by adding 50 ⁇ L of a 1:5000 dilution of the neoepitope antibody to 50 ⁇ L of the 1:25 dilution of the reaction mix (all antibodies and the streptavidin coated beads were diluted in PBS containing 0.5% BSA and 0.5% Tween20). Then, 100 ⁇ L of 0.2 mg/mL streptavidin coated beads (Dynabeads M-280) and a 1:5000 dilution of ruthenylated goat anti-rabbit (Ru-GaR) antibody was added. The mixture was measured for electro- chemiluminescence in a BioVeris M8 Analyzer after 2 hours of incubation with shaking at RT. The dimethyl sulfoxide control defined 100% activity level and 0% activity was defined by exclusion of the enzyme (using 40 mM MES pH 5.0 buffer instead).
- the enzyme was diluted to 52 ⁇ g/ml in 40 mM MES pH 5.0.
- the substrate (Dabcyl-Edans) was diluted to 30 ⁇ M in 40 mM MES pH 5.0.
- Compounds were diluted to the desired concentration in dimethyl sulfoxide (final dimethyl sulfoxide concentration in assay is 5%).
- the assay is done in a Corning 384 well round bottom, low volume, non-binding surface plate (Corning #3676). Enzyme (9 ⁇ L) together with 1 ⁇ L of compound in dimethyl sulfoxide were added to the plate and pre-incubated for 10 min.
- Enzyme was diluted to 6 ⁇ g/mL and the substrate (Europium)CEVNLDAEFK(Qsy7) to 200 nM in reaction buffer (NaAcetate, chaps, triton x-100, EDTA pH 4.5). Compounds were diluted to the desired concentration in dimethyl sulfoxide (final dimethyl sulfoxide concentration in assay is 5%). The assay was done in a Costar 384 well round bottom, low volume, non-binding surface plate (Corning #3676). Enzyme (9 ⁇ L) and 1 ⁇ L of compound in dimethyl sulfoxide was added to the plate, mixed and pre-incubated for 10 min.
- Substrate (10 ⁇ L) was added and the reaction proceeded in the dark for 15 min at RT.
- the reaction was stopped with the addition of 7 ⁇ L NaAcetate, pH 9.
- the fluorescence of the product was measured on a Victor II plate reader with an excitation wavelength of 340 nm and an emission wavelength of 615 nm.
- the final concentration of the enzyme was 2.7 ⁇ g/ml and the final concentration of the substrate was 100 nM (Km of 290 nM).
- the dimethyl sulfoxide control defined the 100% activity level and 0% activity was defined by exclusion of the enzyme (using reaction buffer instead).
- BACE was assayed on a Biacore3000 instrument by attaching either a peptidic transition state isostere (TSI) or a scrambled version of the peptidic TSI to the surface of a Biacore CM5 sensor chip.
- TSI transition state isostere
- the surface of a CM5 sensor chip has 4 distinct channels that can be used to couple the peptides.
- the scrambled peptide KFES-statine-ETIAEVENV was coupled to channel 1 and the TSI inhibitor KTEEISEVN-statine-VAEF was coupled to channel 2 of the same chip.
- the two peptides were dissolved at 0.2 mg/mL in 20 mM sodium acetate pH 4.5, and then the solutions were centrifuged at 14K rpm to remove any particulates.
- Carboxyl groups on the dextran layer were activated by injecting a one to one mixture of 0.5 M N-ethyl-N' (3-dimethylarninopropyl)-carbodiimide and 0.5 M N- hydroxysuccinimide at 5 ⁇ L/min for 7 min. Then the stock solution of the control peptide was injected in channel 1 for 7 min at 5 ⁇ L/min., and then the remaining activated carboxyl groups were blocked by injecting 1 M ethanolamine for 7 min at 5 ⁇ L/min.
- the BACE Biacore assay was done by diluting BACE to 0.5 ⁇ M in sodium acetate buffer at pH 4.5 (running buffer minus dimethyl sulfoxide). The diluted BACE was mixed with dimethyl sulfoxide or compound diluted in dimethyl sulfoxide at a final concentration of 5% dimethyl sulfoxide. The BACE/inhibitor mixture was incubated for 30 minutes at RT before being injected over channel 1 and 2 of the CM5 Biacore chip at a rate of 20 ⁇ L/min. As BACE bound to the chip the signal was measured in response units (RU). BACE binding to the TSI inhibitor on channel 2 gave a certain signal.
- RU response units
- the presence of a BACE inhibitor reduced the signal by binding to BACE and inhibiting the interaction with the peptidic TSI on the chip. Any binding to channel 1 was non-specific and was subtracted from the channel 2 responses.
- the dimethyl sulfoxide control was defined as 100% and the effect of the compound was reported as percent inhibition of the dimethyl sulfoxide control.
- the pcDNA3.1 plasmid encoding the cDNA of human full-length APP695 was stably transfected into HEK-293 cells using the Lipofectamine transfection reagent according to manufacture's protocol (Invitrogen). Colonies were selected with 0.1-0.5 mg/mL of zeocin. Limited dilution cloning was performed to generate homogeneous cell lines. Clones were characterized by levels of APP expression and A ⁇ secreted in the conditioned media using an ELISA assay developed in-house.
- HEK293-APP695 HEK293 cells stably expressing human wild-type APP (HEK293-APP695) were grown at 37 0 C, 5% CO 2 in DMEM containing 4500 g/L glucose, GlutaMAX and sodium pyruvate supplemented with 10% FBS, 1% non-essential amino acids and 0.1 mg/mL of the selection antibiotic zeocin.
- HEK293-APP695 cells were harvested at 80-90% confluence and seeded at a concentration of 0.2xl0 6 cells/mL, 100 mL cell suspension/well, onto a black clear bottom 96-well poly-D-lysine coated plate. After over night incubation at 37 °C, 5% CO 2 , the cell medium was replaced with cell culture medium with penicillin and streptomycin (100 U/mL, 100 ⁇ g/mL, respectively) containing test compounds in a final dimethyl sulfoxide concentration of 1%. Cells were exposed to the test compounds for 24 h at 37 0 C, 5% CO 2 .
- test plate 100 ⁇ L cell medium was transferred to a round bottom polypropylene 96-well plate (assay plate). The cell plate was saved for the ATP assay, as described below.
- 50 ⁇ L of primary detection solution containing 0.5 ⁇ g/mL of the rabbit anti-A ⁇ 40 antibody and 0.5 ⁇ g/mL of the biotinylated monoclonal mouse 6E10 antibody in DPBS with 0.5 %BSA and 0.5% Tween-20 was added per well and incubated over night at 4 0 C.
- SH-SY5Y cells were grown 37 0 C with 5% CO 2 in DMEM/F-12 1:1 containing GlutaMAX supplemented with 1 mM HEPES, 10% FBS and 1% non-essential amino acids.
- sAPP ⁇ release assay
- SH-SY5Y cells were harvested at 80-90% confluence and seeded at a concentration of 1.5x10 6 cells/mL, 100 mL cell suspension/well, onto a black clear flat bottom 96-well tissue culture plate. After 7 hours of incubation at 37 0 C, 5% CO 2 , the cell medium was replaced with 90 ⁇ l cell culture medium with penicillin and streptomycin (100 U/mL, 100 ⁇ g/mL, respectively) containing test compounds in a final dimethyl sulfoxide concentration of 1%. Cells were exposed to the test compounds for 18 h at 37 0 C, 5% CO 2 .
- sAPP ⁇ microplates from Meso Scale Discovery were used and the assay was performed according to the manufacture's protocol. Briefly, 25 ⁇ L cell medium was transferred to a previously blocked MSD sAPP ⁇ microplate. The cell plate was saved for the ATP assay, as described below. The sAPP ⁇ was captured during shaking at RT for 1 hour, by antibodies spotted in the wells of the microplate. After multiple washes, SULFO-TAG labeled detection antibody was added (25 ⁇ L/well, final concentration InM) to the assay plate and the plate was incubated with shaking at RT for 1 hour. Following multiple washes, 150 ⁇ l/well of Read Buffer T was added to the plate. After 10 minutes at RT the plate was read in the SECTORTM Imager for electro-chemiluminescence .
- MSD Meso Scale Discovery
- ATP assay As indicated above, after transferring medium for analysis of A ⁇ 40 or sAPP ⁇ from the cell plate, the plate was used to analyze cytotoxicity using the ViaLightTM Plus cell proliferation/cytotoxicity kit from Cambrex BioScience that measures total cellular ATP. The assay was performed according to the manufacture's protocol. Briefly, 50 ⁇ L cell lysis reagent was added per well. The plates were incubated at RT for 10 min. Two min after addition of 100 ⁇ L reconstituted ViaLightTM Plus ATP reagent, the luminescence was measured in a Wallac Victor 2 1420 multilabel counter.
- hERG Assay Cell culture The hERG-expressing Chinese hamster ovary Kl (CHO) cells described by (Persson, Carlsson, Duker, & Jacobson, 2005) were grown to semi-confluence at 37 0 C in a humidified environment (5% CO 2 ) in F-12 Ham medium containing L-glutamine, 10% foetal calf serum (FCS) and 0.6 mg/ml hygromycin (all Sigma- Aldrich). Prior to use, the monolayer was washed using a pre-warmed (37°C) 3 ml aliquot of Versene 1 :5,000 (Invitrogen).
- CHO-KvI.5 cells which were used to adjust the voltage offset on IonWorksTM HT, were maintained and prepared for use in the same way.
- PatchPlate TM in which a recording is attempted in each well by using suction to position and hold a cell on a small hole separating two isolated fluid chambers. Once sealing has taken place, the solution on the underside of the PatchPlate is changed to one containing amphotericin B. This permeablises the patch of cell membrane covering the hole in each well and, in effect, allows a perforated, whole-cell patch clamp recording to be made.
- a ⁇ -test IonWorksTM HT from Essen Instrument was used. There is no capability to warm solutions in this device hence it was operated at room temperature ( ⁇ 21°C), as follows.
- the reservoir in the "Buffer” position was loaded with 4 ml of PBS and that in the "Cells” position with the CHO-hERG cell suspension described above.
- Each compound plate was laid-out in 12 columns to enable ten, 8- point concentration-effect curves to be constructed; the remaining two columns on the plate were taken up with vehicle (final concentration 0.33% DMSO), to define the assay baseline, and a supra-maximal blocking concentration of cisapride (final concentration 10 ⁇ M) to define the 100% inhibition level.
- the fluidics-head (F-Head) of IonWorksTM HT then added 3.5 ⁇ l of PBS to each well of the PatchPlateTM and its underside was perfused with "internal" solution that had the following composition (in mM): K-Gluconate 100, KCl 40, MgCl 2 3.2, EGTA 3 and HEPES 5 (all Sigma-Aldrich; pH 7.25-7.30 using 10 M KOH).
- the electronics-head (E-head) then moved round the PatchPlateTM performing a hole test (i.e. applying a voltage pulse to determine whether the hole in each well was open).
- the F-head then dispensed 3.5 ⁇ l of the cell suspension described above into each well of the PatchPlateTM and the cells were given 200 seconds to reach and seal to the hole in each well. Following this, the E-head moved round the PatchPlateTM to determine the seal resistance obtained in each well.
- the solution on the underside of the PatchPlateTM was changed to "access" solution that had the following composition (in mM): KCl 140, EGTA 1, MgCl 2 1 and HEPES 20 (pH 7.25-7.30 using 10 M KOH) plus 100 ⁇ g/ml of amphotericin B (Sigma-Aldrich).
- the E-head moved round the PatchPlateTM 48 wells at a time to obtain pre-compound hERG current measurements.
- the F-head then added 3.5 ⁇ l of solution from each well of the compound plate to 4 wells on the PatchPlateTM (the final DMSO concentration was 0.33% in every well). This was achieved by moving from the most dilute to the most concentrated well of the compound plate to minimise the impact of any compound carry-over.
- the E-head then moved around all 384- wells of the PatchPlateTM to obtain post-compound hERG current measurements.
- any offset was adjusted by determining the hERG tail current reversal potential in IonWorksTM HT, comparing it with that found in conventional electrophysiology (-82 mV) and then making the necessary offset adjustment in the IonWorksTM HT software.
- the current signal was sampled at 2.5 kHz.
- Pre- and post-scan hERG current magnitude was measured automatically from the leak subtracted traces by the IonWorksTM HT software by taking a 40 ms average of the current during the initial holding period at —70 mV (baseline current) and subtracting this from the peak of the tail current response.
- the acceptance criteria for the currents evoked in each well were: pre-scan seal resistance >60 M ⁇ , pre-scan hERG tail current amplitude >150 p A; post-scan seal resistance >60 M ⁇ .
- the degree of inhibition of the hERG current was assessed by dividing the post-scan hERG current by the respective pre-scan hERG current for each well.
- Typical IC50 values for the compounds of the present invention are in the range of about 1 to about 2,000 nM.
- Biological data on final compounds are given below in Table 2. TABLE 2.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Psychiatry (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hospice & Palliative Care (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
This invention relates to novel compounds having the structural formula I below: and to their pharmaceutically acceptable salt, compositions and methods of use. These novel compounds provide a treatment or prophylaxis of cognitive impairment, Alzheimer Disease, neurodegeneration and dementia.
Description
Novel 2-amino-5 , 5-diaryl-imidazol-4-ones
The present invention relates to novel compounds and their pharmaceutical compositions. In addition, the present invention relates to therapeutic methods for the treatment and/or
5 prevention of Aβ-related pathologies such as Downs syndrome, β-amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such aso Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
Background of the invention 5 Several groups have identified and isolated aspartate proteinases that have β-secretase activity (Hussain et al., 1999; Lin et. al, 2000; Yan et. al, 1999; Sϊnha et. al., 1999 and Vassar et. al., 1999). β-secretase is also known in the literature as Asp2 (Yan et. al, 1999), Beta site APP Cleaving Enzyme (BACE) (Vassar et. al., 1999) or memapsin-2 (Lin et al., 2000). BACE was identified using a number of experimental approaches such as ESTo database analysis (Hussain et al. 1999); expression cloning (Vassar et al. 1999); identification of human homologs from public databases of predicted C. elegans proteins (Yan et al. 1999) and finally utilizing an inhibitor to purify the protein from human brain (Sinha et al. 1999). Thus, five groups employing three different experimental approaches led to the identification of the same enzyme, making a strong case that BACE is a β-5 secretase. Mention is also made of the patent literature: WO96/40885, EP871720, U.S. Patents Nos. 5,942,400 and 5,744,346, EP855444, US 6,319,689, WO99/64587, WO99/31236, EP1037977, WO00/17369, WO01/23533, WO0047618, WO00/58479, WO00/69262, WO01/00663, WO01/00665, US 6,313,268. o BACE was found to be a pepsin-like aspartic proteinase, the mature enzyme consisting of the N-terminal catalytic domain, a transmembrane domain, and a small cytoplasmic
domain. BACE has an optimum activity at pH 4.0-5.0 (Vassar et al, 1999) and is inhibited weakly by standard pepsin inhibitors such as pepstatin. It has been shown that the catalytic domain minus the transmembrane and cytoplasmic domain has activity against substrate peptides (Lin et al, 2000). BACE is a membrane bound type 1 protein that is synthesized as a partially active proenzyme, and is abundantly expressed in brain tissue. It is thought to represent the major β-secretase activity, and is considered to be the rate-limiting step in the production of amyloid-β-protein (Aβ). It is thus of special interest in the pathology of Alzheimer's disease, and in the development of drugs as a treatment for Alzheimer's disease.
Aβ or amyloid-β-protein is the major constituent of the brain plaques which are characteristic of Alzheimer's disease (De Strooper et al, 1999). Aβ is a 39-42 residue peptide formed by the specific cleavage of a class 1 transmembrane protein called APP, or amyloid precursor protein. Cleavage of APP by BACE generates the extracellular soluble APPβ fragment and the membrane bound CTFβ (C99) fragment that is subsequently cleaved by γ-secretase to generate Aβ peptide.
Alzheimer's disease (AD) is estimated to afflict more than 20 million people worldwide and is believed to be the most common form of dementia. Alzheimer's disease is a progressive dementia in which massive deposits of aggregated protein breakdown products - amyloid plaques and neurofibrillary tangles accumulate in the brain. The amyloid plaques are thought to be responsible for the mental decline seen in Alzheimer's patients.
The likelihood of developing Alzheimer's disease increases with age, and as the aging population of the developed world increases, this disease becomes a greater and greater problem. In addition to this, there is a familial link to Alzheimer's disease and consequently any individuals possessing the double mutation of APP known as the Swedish mutation (in which the mutated APP forms a considerably improved substrate for BACE) have a much higher risk of developing AD, and also of developing the disease at an early age {see also US 6,245,964 and US 5,877,399 pertaining to transgenic rodents comprising APP-Swedish). Consequently, there is also a strong need for developing a
compound that can be used in a prophylactic fashion for these individuals.
The gene encoding APP is found on chromosome 21, which is also the chromosome found as an extra copy in Down's syndrome. Down's syndrome patients tend to develop Alzheimer's disease at an early age, with almost all those over 40 years of age showing Alzheimer's-type pathology (Oyama et al., 1994). This is thought to be due to the extra copy of the APP gene found in these patients, which leads to overexpression of APP and therefore to increased levels of Aβ causing the high prevalence of Alzheimer's disease seen in this population. Thus, inhibitors of BACE could be useful in reducing Alzheimer's-type pathology in Down's syndrome patients.
Drugs that reduce or block BACE activity should therefore reduce Aβ levels and levels of fragments of Aβ in the brain, or elsewhere where Aβ or fragments thereof deposit, and thus slow the formation of amyloid plaques and the progression of AD or other maladies involving deposition of Aβ or fragments thereof (Yankner, 1996; De Strooper and Konig, 1999). BACE is therefore an important candidate for the development of drugs as a treatment and/or prophylaxis of Aβ-related pathologies such as Downs syndrome, β- amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
It would therefore be useful to inhibit the deposition of Aβ and portions thereof by inhibiting BACE through inhibitors such as the compounds provided herein.
The therapeutic potential of inhibiting the deposition of Aβ has motivated many groups to isolate and characterize secretase enzymes and to identify their potential inhibitors (see, e.g., WO01/23533 A2, EP0855444, WO00/17369, WOOO/58479, WO00/47618,
WO00/77030, WO01/00665, WO01/00663, WO01/29563, WO02/25276, US5,942,400, US6,245,884, US6,221,667, US6,211,235, WO02/02505, WO02/02506, WO02/02512, WO02/02518, WO02/02520, WO02/14264, WO05/058311, WO05/097767, WO06/041404, WO06/041405, WO06/0065204, WO06/0065277, US2006287294, WO06/138265, US20050282826, US20050282825, US20060281729, WO06/138217, WO06/138230, WO06/138264, WO06/138265, WO06/138266, WO06/099379, WO06/076284, US20070004786, US20070004730, WO07/011833, WO07/011810, US20070099875, US20070099898, WO07/058601, WO07/058581, WO07/058580, WO07/058583, WO07/058582, WO07/058602, WO07/073284, WO07/049532, WO07/038271, WO07/016012, WO07/005366, WO07/005404, WO06/0009653).
The compounds of the present invention show beneficial properties compared to the potential inhibitors known in the art, e.g. improved hERG selectivity.
Disclosure of the invention
The present invention provides potent BACE inhibitors of formula I:

R1 is selected from phenyl, heteroaryl, d-όalkyl optionally substituted with a C3- ήcycloalkyl, C2-6alkenyl optionally substituted with a
 heterocyclyl, C3- ecycloalkyl, Cs^cycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R3;
heterocyclyl, C3- ecycloalkyl, Cs^cycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R3;
R2 is independently selected from hydrogen, cyano or halo;
A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R4;
R3 is independently selected from halo, cyano, d^alkyl, trifluoromethyl, methoxy, trifluoromethoxy and acetyl;
R4 is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
R5 is Ci-βalkyl optionally substituted with halogen, cyano, NH2, OH, CO2H, OC^alkyl, SO2H, C(O)C1-6alkyl,' C(O)OC1-6alkyl, C(O)NH2, C(O)NHC1-6alkyl, C(O)N(Ci-6alkyl)2, SO2C1-6alkyl, SO2NHC1-6alkyl, SO^d^U^, NH(d-6alkyl), N(Ci-6alkyl)2, NHC(O)C1-6alkyl, NC(O)(C1-6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl)2, S02aryI, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl, NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl)2, SO2heteroaryl, SO2NHheteroaryl, SO2N(heteroaryl)2, NH(heteroaryl), N(heteroaryl)2, NC(O)heteroaryl, NC(O)(heteroaryl)2, C5-6heterocyclyl, OCs-δheterocyclyl, C(O)C5-6heterocyclyl, C(O)OC5-6heterocyclyl, C(O)NHC5-6heterocyclyI, C(O)N(C5-6heterocyclyl)2, SO2C5-6heterocyclyl, SO2NHC5- όheterocyclyl, SO2N(C5-6heterocyclyl)2, NH(C5-6heterocyclyl), N(Cs,6heterocyclyl)2, NC(O)C5-6heterocyclyl or NC(O)(C5-6heterocyclyl)2; n is 0, 1 or 2; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
The present invention further provides potent BACE inhibitors of formula I:
 wherein
wherein
R1 is selected from phenyl, heteroaryl, Chalky! optionally substituted with a C3- βcycloalkyl, C2-6alkenyl optionally substituted with a C3-6cycloalkyl, heterocyclyl, C3. βcycloalkyl, Cs-όcycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R3;
R2 is independently selected from hydrogen, cyano or halo;
A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R4;
R3 is independently selected from halo, cyano, d-βalkyl, trifluoromethyl, methoxy, trifluoromethoxy and acetyl;
R is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
R5 is optionally substituted with halogen, cyano, NH2, OH, CO2H, OCi-βalkyl, SO2H, C(O)Ci-6alkyl, C(O)OC1-6alkyl, C(O)NH2, C(O)NHC 1-6alkyl, C(O)N(C1-6alkyl)2, SO2C1-6alkyl, SO2NHC1-6alkyl, SO2N(C1-6alkyl)2, NH(Ci-6alkyl), N(C1-6alkyl)2, NHC(O)C1-6alkyl, NC(O)(C1.6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl)2, S02aryl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl, NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl)2, SO2heteroaryl, SO2NHheteroaryl,
SO2N(heteroaryl)2, NH(heteroaryl), N(heteroaryl)2, NC(O)heteroaryl, NC(O)(heteroaryl)2,
C5-6heterocyclyl, OCs-eheterocyclyl, C(0)C5.6heterocyclyl, C(O)OC5-6heterocyclyl,
C(O)NHC5-6heterocyclyl, C(0)N(C5-6heterocyclyl)2, SO2C5-6heterocyclyl, SO2NHC5- όheterocyclyl, SO2N(C5-6heterocyclyl)2, NH(C5-6heterocycIyl), N(Cs-6lieterocyclyl)2, NC(O)C5-6heterocyclyl or NC(O)(C5-6heterocyclyl)2; n is 0, 1 or 2; provided that the following compounds are excluded:
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(3'-methoxybiphenyl-3-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one; 2-Anτino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2',5'-dimethoxybiphenyl-3-yl)-3-methyl-
3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2'-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[2-Amino-4-(2,3-dihydro-l-benzofuran-5-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-6-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-5-[3'-(trifluoromethoxy)biphenyl-3- yl]-3,5-dihydro-4H-imidazol-4-one; 2-Amino-5-(3'-chlorobiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one ;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(3'-methoxybiphenyl-3-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2',5'-dimethoxybiphenyl-3-yl)-3-methyl- 3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2'-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; 3'-[2-Amino-4-(3 ,4-dihydro-2H-chromen-6-yl)- 1 -methyl-5-oxo-4,5-dihydro- lH-imidazol-
4-yl]-4-fluorobiphenyl-3-carbonitriIe;
3 '-[2-Amino-4-(3 ,4-dihydro-2H-chromen-6-yl)- 1 -methyl-5-oxo-4,5-dihydro- lH-imidazol-
4-yl]-6-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-5-[3'-(trifluoromethoxy)biphenyl-3- yl]-3,5-dihydro-4H-imidazol-4-one; s 2-Amino-5-(3'-chlorobiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2',5'-dimethoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[4-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-l-methyl-5-oxo-4,5-dihydro-lH- I0 imidazol-4-yl]biphenyl-3-carbonitrile;
3'-[4-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-l-methyl-5-oxo-4,5-dihydro-lH- imidazol-4-yl] -4-fluorobipheny 1-3 -carbonitrile ;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2'-fluoro-5'-methoxybiphenyl-3- yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one; 15 5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2'-fluoro-3'-methoxybiphenyl-3- yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one;
S-Cl-Acetyl-l^^^^etrahydroquinolin-o-y^^-amino-S-P-CS-furyOphenylj-S-methyl-S^- dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-lH-isochromen-7-yl)-5-(2'-fluoro-5'-niethoxybiphenyl-3-yl)-3- 20 methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[2-Amino-4-(3,4-dihydro-lH-isochromen-7-yl)-l-methyl-5-oxo-4,5-dihydro-lH- imidazol-4-yl] -6-fluorobiphenyl-3 -carbonitrile; and
2-Ainino-5-(3,4-dihydro-lH-isochromen-7-yl)-5-[3-(3-furyl)phenyl]-3-methyl-3,5- dihydro-4H-imidazol-4-one; 25 as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
The present invention further provides potent BACE inhibitors of formula I:
 wherein
wherein
R1 is selected from phenyl, heteroaryl, C1-6alkyl optionally substituted with a C3- βcycloalkyl, C2-6alkenyl optionally substituted with a C3-6cycloalkyl, heterocyclyl, C3- δcycloalkyl, C5-6cycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R3;
R >2 i -s independently selected from hydrogen, cyano or halo;
A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R4;
R3 is independently selected from halo, cyano,
 trifluoromethyl, methoxy and trifluoromethoxy;
trifluoromethyl, methoxy and trifluoromethoxy;
R4 is independently selected from halo, cyano and methyl; provided that said bicyclic ring of A, is substituted with at least one R4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
R5 is Ci-βalkyl optionally substituted with halogen, cyano, NH2, OH, CO2H, OCi^alkyl, SO2H, C(O)C1-6alkyl, C(O)OC1-6alkyl, C(O)NH2, C(O)NHC1.6alkyl, C(O)N(C i-6alkyl)2, SO2Ci-6alkyl, SO2NHC1-6alkyl, SO2N(C1-6alkyI)2, NH(Ci-6alkyl), N(C1-6alkyl)2, NHC(O)Ci-6alkyl, NC(θχC1-6alkyl)2, aryl, Oaryl, C(O)aryI, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl)2, S02aryl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl, NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(0)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl)2, SO2heteroaryl, SO2NHheteroaryl,
SO2N(heteroaryl)2, NH(heteroaryl), N(heteroaryl)2, NC(O)heteroaryI, NC(O)(heteroaryl)2, C5_6heterocyclyl, OCs-βheterocyclyl, C(O)C5-6heterocyclyl, C(O)OC5-6heterocyclyl, C(O)NHC5-6heterocyclyl, C(O)N(C5-6heterocyclyl)2, SO2C5-6heterocyclyl, SO2NHC5- δheterocyclyl, SO2N(C5-6heterocyclyl)2, NH(C5.6heterocyclyl), N(C5-6lieterocyclyl)2, NC(O)C5.6heterocyclyl or NC(O)(C5-6heterocyclyl)2; n is 0, 1 or 2; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
The present invention further provides potent BACE inhibitors of formula I:

R1 is selected from phenyl, heteroaryl, C^alkyl optionally substituted with a C3- έcycloalkyl, C2-6alkenyl optionally substituted with a C3_6cycloalkyl, heterocyclyl, C3- βcycloalkyl, Cs-όcycloalkenyl, halo and cyano, said phenyl or heteroaryl optionally substituted with one or more R3;
R2 is independently selected from hydrogen, cyano or halo;
A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R4;
R3 is independently selected from halo, cyano, Chalky!, trifluoromethyl, methoxy and trifluoromethoxy;
R4 is independently selected from halo, cyano and methyl;
provided that said bicyclic ring of A, is substituted with at least one R4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
R5 is Ci-6alkyl optionally substituted with halogen, cyano, NH2, OH, CO2H, OCi-βalkyl, SO2H, C(O)C1-6alkyl, C(O)OC1-6alkyl, C(O)NH2, C(O)NHC1-6alkyl, C(O)N(C1-6alkyl)2,
SO2C1-6alkyl, SO2NHC1-6alkyl, SO2N(C1-6alkyl)2, NH(C1-6alkyl), N(C1-6alkyl)2,
NHC(O)Ci-6alkyl, NC(O)(C1-6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl,
C(O)N(aryl)2, S02aryl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl,
NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl)2, SO2heteroaryl, SO2NHheteroaryl,
SO2N(heteroaryl)2, NH(heteroaryl), N(heteroaryl)2, NC(O)heteroaryl, NC(O)(heteroaryl)2,
C5-6heterocyclyl, OCs-όheterocyclyl, C(O)C5-6heterocyclyl, C(O)OC5.6heterocyclyl,
C(O)NHC5-6heterocyclyl, C(O)N(C5-6heterocyclyl)2, SO2C5-6heterocyclyl, SO2NHC5- oheterocyclyl, SO2N(C5-6heterocyclyl)2, NH(C5-6heterocyclyl), N(C5.6heterocyclyl)2, NC(O)C5-6heterocyclyl or NC(O)(C5-6heterocyclyl)2; n is 0, 1 or 2; provided that the following compounds are excluded:
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(3'-methoxybiphenyl-3-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one; 2-Amino-5-(2,3-dihydro- 1 -benzofuran-5-yl)-5-(2I,5'-dimethoxybiphenyl-3-yl)-3-methyl-
3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2'-fluoro-3'-methoxybiphenyI-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
3 '-[2-Amino-4-(2,3-dihydro- 1 -benzofuran-5-yl)- 1 -methyl-S-oxo^S-dihydro- lH-imidazol-
4-yl]-6-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-5-[3'-(trifluoromethoxy)biphenyl-3- yl]-3,5-dihydro-4H-imidazol-4-one; 2-Amino-5-(3'-chlorobiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(3'-methoxybiphenyl-3-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one ;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2',5'-dimethoxybiphenyl-3-yl)-3-methyl-
3,5-dihydro-4H-imidazol-4-one; 5 2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3 , 5-dihydro-4H-imidazol-4-one;
2- Amino-5-(3 ,4-dihydro-2H-chromen-6-yl)-5 -(2'-fluoro-3 '-methoxybiphenyl-3 -yl)-3 - methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-o 4-yl]-4-fluorobiphenyl-3-carbonitrile;
3'-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-6-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-5-[3'-(trifluoromethoxy)biphenyl-3- yl]-3,5-dihydro-4H-imidazol-4-one; s 2-Amino-5-(3'-chlorobiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2',5'-dimethoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
3 '- [4-( 1 -Acetyl- 1,2,3 ,4-tetrahydroquinolin-6-yl)-2-amiαo- 1 -methyl-5 -oxo-4, 5 -dihydro- 1 H-o imidazol-4-yl]biphenyl-3-carbonitrile;
3'-[4-(l-Acetyl-l,2,3,4-tetτahydroquinolin-6-yl)-2-amino-l-methyl-5-oxo-4,5-dihydro-lH- imidazol-4-yl]-4-fluorobiphenyl-3-carbonitrile;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2'-fluoro-5'-πiethoxybiphenyl-3- yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one; 5 5-( 1 -Acetyl- 1 ,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2'-fluoro-3'-methoxybiphenyl-3- yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yI)-2-amino-5-[3-(3-fUryl)phenyl]-3-methyl-3,5- dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-lH-isochromen-7-yl)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3-o methyl-3 ,5-dihydro-4H-imidazol-4-one;
3 '-[2-Amino-4-(3 ,4-dihydro- lH-isochromen-7-yl)- 1 -methyl-S-oxo-^S-dihydro- IH- imidazol-4-yl]-6-fluorobiphenyl-3-carbonitrile; or
2-Amino-5-(3,4-dihydro-lH-isochroraen-7-yl)-5-[3-(3-furyl)phenyl]-3-methyl-3,5- dihydro-4H-imidazol-4-one; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
In one aspect of the invention there is provided compounds of formula I,

R1 is phenyl, optionally substituted with one or more R3, or Ci-6 alkyl;
R is independently selected from cyano and halo;
A is phenyl or heteroaryl, said phenyl or heteroaryl optionally fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R4;
R3 is independently selected from halo, cyano, Ci-βalkyl, trifluoromethyl, methoxy, trifluoromethoxy and acetyl;
R is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
R > 5 i ■s Ci-βalkyl optionally substituted with halogen, cyano, NH2, OH, CO2H,
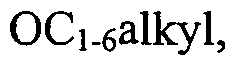 SO2H, C(O)Ci-6alkyl, C(O)OC 1-6alkyl, C(O)NH2, C(O)NHCi-6alkyl, C(O)N(d.6alkyl)2,
SO2C1-6alkyl) SO2NHCi.6alkyl, SO2N(Ci-6alkyl)2, NH(Ci-6alkyl), N(Ci-6alkyl)2,
SO2H, C(O)Ci-6alkyl, C(O)OC 1-6alkyl, C(O)NH2, C(O)NHCi-6alkyl, C(O)N(d.6alkyl)2,
SO2C1-6alkyl) SO2NHCi.6alkyl, SO2N(Ci-6alkyl)2, NH(Ci-6alkyl), N(Ci-6alkyl)2,
NHC(O)Ci-6alkyl, NC(O)(C1-6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl,
C(O)N(aryl)2, SO2aiyl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl,
NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl)2, SO2heteroaryl, SO2NHheteroaryl,
SO2N(heteroaryl)2, NH(heteroaryl), N(heteroaryl)2, NC(O)heteroaryl, NC(O)(heteroaryl)2,
C5-6heterocyclyl, OCs-δheterocyclyl, C(O)C5-6heterocyclyl, C(O)OC5.6heterocyclyl,
C(O)NHC5-6heterocyclyl, C(O)N(C5-6heterocyclyl)2, SO2C5-6heterocyclyl, SO2NHC5-
6heterocyclyl, SO2N(C5_6heterocyclyl)2j NH(C5-6heterocyclyl), N(C5.6heterocyclyl)2, NC(O)C5-6heterocyclyl or NC(O)(C5-6heterocyclyl)2; n is 0, 1 or 2; provided that the following compounds are excluded:
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(3'-methoxybiphenyl-3-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one; 2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2',5'-dirnethoxybiphenyl-3-yl)-3-methyl-
3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2'-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[2-Amino-4-(2,3-dihydro-l-benzofuran-5-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-6-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-5-[3'-(trifluorornethoxy)biphenyl-3- yl]-3,5-dihydro-4H-imidazol-4-one; 2-Amino-5-(3l-chlorobiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(3'-methoxybiphenyl-3-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one ;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2',5'-dimethoxybiphenyl-3-yl)-3-methyl- 3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yI)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2'-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-4-fluorobiphenyl-3-carbonitrile; 3 '-[2-Amino-4-(3 ,4-dihydro-2H-chromen-6-yl)- 1 -methyl-S-oxo^S-dihydro- lH-imidazol-
4-yl]-6-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-5-[3'-(txifluoromethoxy)biphenyl-3- yl]-3 ,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(3'-chlorobiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2',5'-dimethoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[4-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-l-methyl-5-oxo-4,5-dihydro-lH- imidazol-4-yl]biphenyl-3-carbonitrile; 3'-[4-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-l-methyl-5-oxo-4,5-dihydro-lH- imidazol-4-yl]-4-fluorobiphenyl-3-carbonitrile;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2'-fluoro-5'-methoxybiphenyl-3- yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2'-fluoro-3'-methoxybiphenyl-3- yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-[3-(3-furyl)phenyl]-3-methyl-3,5- dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-lH-isochromen-7-yl)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; 3'-[2-Amino-4-(3 ,4-dihydro- lH-isochromen-7-yl)- 1 -methyl-5-oxo-4,5-dihydro- IH- imidazol-4-yl]-6-fluorobiphenyl-3-carbonitrile; or
2-Ajnino-5-(3,4-dihydro-lH-isochromen-7-yl)-5-[3-(3-fiιryl)phenyl]-3-methyl-3,5- dihydro-4H-imidazol-4-one; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
In one embodiment of this aspect, R1 is phenyl.
In another embodiment of this aspect, n is 1 and R2 is fluoro.
In another embodiment of this aspect, n is 0.
In another embodiment of this aspect, R5 is Q-βalkyl.
In another embodiment of this aspect, R5 is methyl.
In another embodiment of this aspect, A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system.
In another embodiment of this aspect, A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is substituted with from one to four R4.
In another embodiment of this aspect, said bicyclic ring system is substituted with two R4, said R4 being methyl.
In another embodiment of this aspect, said bicyclic ring system is substituted with one R4, said R4 being cyano.
In another embodiment of this aspect, A is selected from:

In another embodiment of this aspect, A is:

In another embodiment of this aspect, R3 is independently selected from halo, cyano, methoxy, trifluoromethoxy and acetyl.
In another embodiment of this aspect, n is 0 or 1; R2 is fluoro; R5 is Ci-βalkyl; A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system, optionally substituted with one or two R4, said R4 being independently selected from methyl or cyano.
hi another embodiment of this aspect, there is provided a compound, selected from:
2-Amino-5-(2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl)-5-(6-fluoro-3'-methoxybiphenyl-
3-yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one hydrochloride;
5'-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol- 4-yl]-2'-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(6-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2',6-difluoro-3'-methoxybiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; 2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(6-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
5'-[2-Amino-4-(2,3-dihydro-l -benzofuran-5-yl)- 1 -methyl-5-oxo-4,5-dihydro- lH-imidazol-
4-yl]-2'-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(2',6-difluoro-3'-methoxybiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3- methyl-3 ,5-dihydro-4H-imidazol-4-one;
3'-[2-Amino-4-(2,3-dihydro-l-benzofuran-5-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-5-methoxybiphenyl-2-carbonitrile hydrochloride;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-[5'-methoxy-2'-(trifluoromethoxy)biphenyl-
3-yl]-3-methyl-3,5-dihydro-4H-imidazol-4-one hydrochloride;
2-Amino-5-(3',5'-dichlorobiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one hydrochloride;
3I-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-5-methoxybiphenyl-2-carbonitrile; 2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-[5'-methoxy-2'-(trifluoromethoxy)biphenyl-
3-yl]-3-methyl-3,5-dihydro-4H-imidazol-4-one hydrochloride;
2-Amino-5-(3',5'-dichlorobiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one hydrochloride;
5-(2'-Acetyl-5'-methoxybiphenyl-3-yl)-2-amino-5-(2,3-dihydro-l-benzofuran-5-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
5-(2'-Acetyl-5'-methoxybiphenyl-3-yl)-2-amino-5-(3,4-dihydro-2H-chromen-6-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
5-[2-Amino-4-(3',5'-dichloro-6-fluorobiphenyl-3-yl)-l-methyl-5-oxo-4,5-dihydro-lH- imidazol-4-yl]-2,3-dihydro-l-benzofuran-7-carbonitrile 0.25 acetate; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
In another aspect of the invention there is provided compounds of formula I, wherein
R1 is CΗ2CΗ2cyclopropyl;
R >22 is independently selected from cyano and halo;
A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R4;
R4 is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
R5 is Ci-βalkyl optionally substituted with halogen, cyano, NH2, OH, CO2H, OCi-βalkyl, SO2H, C(O)CI-6alkyl, C(O)OCi-6alkyl, C(O)NH2, C(O)NHC1-6alkyl, C(O)N(C1-6alkyl)2,
SO2Ci-6alkyl, SO2NHCi.6alkyl, SO2N(Ci-6alkyl)2, NH(C,-6alkyl), N(C1-6alkyl)2, NHC(O)Ci-6alkyl, NC(O)(C1-6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl)2, SO2aryl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl, NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl)2, SO2heteroaryl, SO2NHheteroaryl,
SO2N(heteroaryl)2, NH(heteroaryl), N(heteroaryl)2, NC(O)heteroaryl, NC(O)(heteroaryl)2, C5-6heterocyclyl, OCs-δheterocyclyl, C(O)C5-6heterocyclyl, C(O)OC5-6heterocyclyl, C(O)NHC5-6heterocyclyl, C(O)N(C5-6heterocyclyl)2, SChCs-eheterocyclyl, SO2NHC5- δheterocyclyl, SO2N(C5-6heterocyclyl)2, NH(Cs-6IIeIeTOCyCIyI), N(C5-6heterocyclyl)2, NC(O)C5-6heterocyclyl or NC(O)(C5-6heterocyclyl)2; n is 0, 1 or 2.
In one embodiment of this aspect, n is 1 and R2 is fluoro.
In another embodiment of this aspect, R5 is C1-6alkyl.
In another embodiment of this aspect, R5 is methyl.
In another embodiment of this aspect, A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system, optionally substituted with from one to four R4.
In another embodiment of this aspect, A is selected from:

In another embodiment of this aspect, there is provided a compound, selected from: 2-Amino-5-[3-(2-cyclopropylethyl)-4-fluorophenyl]-5-(3,4-dihydro-2H-chromen-6-yl)-3- methyI-3,5-dihydro-4H-imidazol-4-one; and
2-Amino-5-[3-(2-cyclopropylethyl)-4-fluorophenyl]-5-(2,3-dihydro-l-benzofuran-5-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
In another aspect of the invention there is provided a pharmaceutical composition comprising as active ingredient a therapeutically effective amount of a compound of formula I, in association with pharmaceutically acceptable excipients, carriers or diluents.
In another aspect of the invention there is provided a compound of formula I, or a pharmaceutically acceptable salt thereof, for use as a medicament.
In another aspect of the invention there is provided use of a compound of formula I as a medicament for treating or preventing an Aβ-related pathology.
In another aspect of the invention there is provided use of a compound of formula I as a medicament for treating or preventing an Aβ-related pathology, wherein said Aβ-related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with
Alzheimer Disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
In another aspect of the invention there is provided use of a compound of formula I as a medicament for treating or preventing Alzheimer Disease.
In another aspect of the invention there is provided use of a compound of formula I in the manufacture of a medicament for treating or preventing an Aβ-related pathology.
In another aspect of the invention there is provided use of a compound of formula I in the manufacture of a medicament for treating or preventing an Aβ-related pathology, wherein said Aβ-related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
In another aspect of the invention there is provided use of a compound of formula I in the manufacture of a medicament for treating or preventing Alzheimer's Disease.
In another aspect of the invention there is provided a method of inhibiting activity of BACE comprising contacting said BACE with a compound of formula I.
In another aspect of the invention there is provided a method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of formula I.
In another aspect of the invention there is provided a method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of formula I, wherein said Aβ-related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
In another aspect of the invention there is provided a method of treating or preventing Alzheimer's Disease, comprising administering to said patient a therapeutically effective amount of a compound of formula I.
In another aspect of the invention there is provided a method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of formula I, wherein said Aβ-related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration, wherein said mammal is a human.
In another aspect of the invention there is provided a method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of formula I and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor.
In another aspect of the invention there is provided a method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of formula I and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor, wherein said Aβ- related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
In another aspect of the invention there is provided a method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of formula I and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor, wherein said Aβ- related pathology is Alzheimer Disease.
In another aspect of the invention there is provided a method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of formula I and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor, wherein said Aβ- related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration, wherein said mammal is a human.
Some compounds of formula I may have stereogenic centres and/or geometric isomeric centres (E- and Z- isomers), and it is to be understood that the invention encompasses all such optical isomers, enantiomers, diastereoisomers, atropisomers and geometric isomers.
The present invention relates to the use of compounds of formula I as hereinbefore defined as well as to the salts thereof. Salts for use in pharmaceutical compositions will be pharmaceutically acceptable salts, but other salts may be useful in the production of the compounds of formula I.
It is to be understood that the present invention relates to any and all tautomeric forms of the compounds of formula I.
Compounds of the invention can be used as medicaments. In some embodiments, the present invention provides compounds of formula I, or pharmaceutically acceptable salts, tautomers or in vzvo-hydroly sable precursors thereof, for use as medicaments. In some embodiments, the present invention provides compounds described here in for use as medicaments for treating or preventing an Aβ-related pathology. In some further embodiments, the Aβ-related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
In some embodiments, the present invention provides use of compounds of formula I or pharmaceutically acceptable salts, tautomers or in vzvo-hydrolysable precursors thereof, in the manufacture of a medicament for the treatment or prophylaxis of Aβ-related pathologies. In some further embodiments, the Aβ-related pathologies include such as Downs syndrome and β-amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile
dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
In some embodiments, the present invention provides a method of inhibiting activity of BACE comprising contacting the BACE with a compound of the present invention. BACE is thought to represent the major β-secretase activity, and is considered to be the rate- limiting step in the production of amyloid-β-protein (Aβ). Thus, inhibiting BACE through inhibitors such as the compounds provided herein would be useful to inhibit the deposition of Aβ and portions thereof. Because the deposition of Aβ and portions thereof is linked to diseases such Alzheimer Disease, BACE is an important candidate for the development of drugs as a treatment and/or prophylaxis of Aβ-related pathologies such as Downs syndrome and β-amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
In some embodiments, the present invention provides a method for the treatment of Aβ- related pathologies such as Downs syndrome and β-amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, presenile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration, comprising administering to a mammal (including human) a therapeutically effective amount of a compound of formula
I, or a pharmaceutically acceptable salt, tautomer or in vz'vø-hydrolysable precursor thereof.
In some embodiments, the present invention provides a method for the prophylaxis of Aβ- related pathologies such as Downs syndrome and β-amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, presenile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration comprising administering to a mammal (including human) a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt, tautomer or in v/vø-hydrolysable precursors.
In some embodiments, the present invention provides a method of treating or preventing Aβ-related pathologies such as Downs syndrome and β-amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, presenile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration by administering to a mammal (including human) a compound of formula I or a pharmaceutically acceptable salt, tautomer or in vzvø-hydrolysable precursors and a cognitive and/or memory enhancing agent.
In some embodiments, the present invention provides a method of treating or preventing Aβ-related pathologies such as Downs syndrome and β-amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive
impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, presenile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration by administering to a mammal (including human) a compound of formula I or a pharmaceutically acceptable salt, tautomer or in vzVø-hydrolysable precursors thereof wherein constituent members are provided herein, and a choline esterase inhibitor or anti-inflammatory agent.
In some embodiments, the present invention provides a method of treating or preventingAβ-related pathologies such as Downs syndrome and β-amyloid angiopathy, such as but not limited to cerebral amyloid angiopathy, hereditary cerebral hemorrhage, disorders associated with cognitive impairment, such as but not limited to MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with diseases such as Alzheimer disease or dementia including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration, or any other disease, disorder, or condition described herein, by administering to a mammal (including human) a compound of the present inventionand an atypical antipsychotic agent. Atypical antipsychotic agents includes, but not limited to, Olanzapine (marketed as Zyprexa), Aripiprazole (marketed as Abilify), Risperidone (marketed as Risperdal), Quetiapine (marketed as Seroquel), Clozapine (marketed as Clozaril), Ziprasidone (marketed as Geodon) and Olanzapine/Fluoxetine (marketed as Symbyax).
In some embodiments, the mammal or human being treated with a compound of the invention has been diagnosed with a particular disease or disorder, such as those described herein. In these cases, the mammal or human being treated is in need of such treatment. Diagnosis, however, need not be previously performed.
The present invention also includes pharmaceutical compositions, which contain, as the active ingredient, one or more of the compounds of the invention herein together with at least one pharmaceutically acceptable carrier, diluent or excipent.
The definitions set forth in this application are intended to clarify terms used throughout this application. The term "herein" means the entire application.
A variety of compounds in the present invention may exist in particular geometric or stereoisomeric forms. The present invention takes into account all such compounds, including cis- and trans isomers, R- and S- enantiomers, diastereomers, (D)-isomers, (L)- isomers, the racemic mixtures thereof, and other mixtures thereof, as being covered within the scope of this invention. Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention. The compounds herein described may have asymmetric centers. Compounds of the present invention containing an asymmetrically substituted atom may be isolated in optically active or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of racemic forms, by synthesis from optically active starting materials, or synthesis using optically active reagents. When required, separation of the racemic material can be achieved by methods known in the art. Many geometric isomers of olefins, C=N double bonds, and the like can also be present in the compounds described herein, and all such stable isomers are contemplated in the present invention. Cis and trans geometric isomers of the compounds of the present invention are described and may be isolated as a mixture of isomers or as separated isomeric forms. All chiral, diastereomeric, racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated.
When a bond to a substituent is shown to cross a bond connecting two atoms in a ring, then such substituent may be bonded to any atom on the ring. When a substituent is listed without indicating the atom via which such substituent is bonded to the rest of the compound of a given formula, then such substituent may be bonded via any atom in such
substituent. Combinations of substituents, positions of substituents and/or variables are permissible only if such combinations result in stable compounds.
As used in this application, the term "optionally substituted," means that substitution is optional and therefore it is possible for the designated atom or moiety to be unsubstituted. In the event a substitution is desired then such substitution means that any number of hydrogens on the designated atom or moiety is replaced with a selection from the indicated group, provided that the normal valency of the designated atom or moiety is not exceeded, and that the substitution results in a stable compound. For example when a substituent is methyl (i.e., CH3), then 3 hydrogens on the carbon atom can be replaced. Examples of such substituents include, but are not limited to: halogen, cyano, NH2, OH, CO2H, OCi-6alkyl, CH2OH, SO2H, Ci-6alkyl, OC^alkyl, C(O)C1-6alkyl, C(O)OC1-6alkyI, C(O)NH2, C(O)NHCi-6alkyl, C(O)N(C 1-6alkyl)2, SO2C1-6alkyl, SO2NHC 1-6alkyl, SO2N(C1-6alkyl)2, NH(C1-6alkyl), N(C1-6alkyl)2, NHC(O)C1-6alkyl, NC(O)(C1-6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl)2, S02aryl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O) aryl, NC(O)(aryl)2, C^heterocyclyl, OCs-eheterocyclyl, C(O)C5- eheterocyclyl, C(O)OC5-6heterocyclyl, C(O)NHC5-6heterocyclyl, C(O)N(C5.6heterocyclyl)2, SO2C5-6heterocyclyl, SO2NHC5-6heterocyclyl, SO2N(C5-6heterocyclyl)2, NH(C5- eheterocyclyl), N(C5-6heterocyclyl)2, NC(O)C5.6heterocyclyl, NC(O)(C5-6heterocyclyl)2.
As used herein, "alkyl", used alone or as a suffix or prefix, is intended to include both branched and straight chain saturated aliphatic hydrocarbon groups having from 1 to 12 carbon atoms or if a specified number of carbon atoms is provided then that specific number would be intended. For example "Co-6 alkyl" denotes alkyl having O, 1, 2, 3, 4, 5 or 6 carbon atoms. Examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, t-butyl, pentyl, and hexyl. In the case where a subscript is the integer 0 (zero) the group to which the subscript refers to indicates that the group may be absent, i.e. there is a direct bond between the groups.
As used herein, "alkenyl" used alone or as a suffix or prefix is intended to include both branched and straight-chain alkene or olefin containing aliphatic hydrocarbon groups having from 2 to 12 carbon atoms or if a specified number of carbon atoms is provided
then that specific number would be intended. For example "C2-6alkenyl" denotes alkenyl having 2, 3, 4, 5 or 6 carbon atoms. Examples of alkenyl include, but are not limited to, vinyl, allyl, 1-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 2-methylbut-2-enyl, 3-methylbut- 1-enyl, 1-pentenyl, 3-pentenyl and 4-hexenyl. As used herein, "aromatic" refers to hydrocarbonyl groups having one or more unsaturated carbon ring(s) having aromatic characters, (e.g. 4n + 2 delocalized electrons) and comprising up to about 14 carbon atoms. In addition "heteroaromatic" refers to groups having one or more unsaturated rings containing carbon and one or more heteroatoms such as nitrogen, oxygen or sulphur having aromatic character (e.g. 4n + 2 delocalized electrons).
As used herein, the term "aryl" refers to an aromatic ring structure made up of from 5 to 14 carbon atoms. Ring structures containing 5, 6, 7 and 8 carbon atoms would be single-ring aromatic groups, for example, phenyl. Ring structures containing 8, 9, 10, 11, 12, 13, or 14 would be polycyclic, for example naphthyl. The aromatic ring can be substituted at one or more ring positions with such substituents as described above. The term "aryl" also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings (the rings are "fused rings") wherein at least one of the rings is aromatic, for example, the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls. The terms ortho, meta and para apply to 1,2-, 1,3- and 1,4-disubstituted benzenes, respectively. For example, the names 1,2-dimethylbenzene and ortho-dimethylbenzene are synonymous.
As used herein, the term "cycloalkyl" is intended to include saturated ring groups, having the specified number of carbon atoms. These may include fused or bridged polycyclic systems. Preferred cycloalkyls have from 3 to 10 carbon atoms in their ring structure, and more preferably have 3, 4, 5, and 6 carbons in the ring structure. For example, "C3-6 cycloalkyl" denotes such groups as cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl.
As used herein, the term "cycloalkenyl" is intended to include unsaturated ring groups, having the specified number of carbon atoms. These may include fused or bridged polycyclic systems. Preferred cycloalkenyls have from 3 to 10 carbon atoms in their ring
structure, and more preferably have 3, 4, 5, and 6 carbons in the ring structure. For example, "C3-6 cycloalkenyl" denotes such groups as cyclopropenyl, cyclobutenyl, cyclopentenyl, or cyclohexenyl.
As used herein, "halo" or "halogen" refers to fluoro, chloro, bromo, and iodo.
As used herein, "heteroaryl" refers to a heteroaromatic heterocycle having at least one heteroatom ring member such as sulfur, oxygen, or nitrogen. Heteroaryl groups include monocyclic and polycyclic (e.g., having 2, 3 or 4 fused rings) systems. Examples of heteroaryl groups include without limitation, pyridyl (i.e., pyridinyl), pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl (i.e. furanyl), quinolyl, isoquinolyl, thienyl, imidazolyl, thiazolyl, indolyl, pyrryl, oxazolyl, benzofuryl, benzothienyl, benzthiazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, indazolyl, 1,2,4-thiadiazolyl, isothiazolyl, benzothienyl, purinyl, carbazolyl, benzimidazolyl, indolinyl, and the like. In some embodiments, the heteroaryl group has from 1 to about 20 carbon atoms, and in further embodiments from about 3 to about 20 carbon atoms. In some embodiments, the heteroaryl group contains 3 to about 14, 4 to about 14, 3 to about 7, or 5 to 6 ring-forming atoms. In some embodiments, the heteroaryl group has 1 to about 4, 1 to about 3, or 1 to 2 heteroatoms. In some embodiments, the heteroaryl group has 1 heteroatom.
As used herein, the term "heterocyclyl" or "heterocyclic" or "heterocycle" refers to a saturated, unsaturated or partially saturated, monocyclic, bicyclic or tricyclic ring (unless otherwise stated) containing 3 to 20 atoms of which 1, 2, 3, 4 or 5 ring atoms are chosen from nitrogen, sulphur or oxygen, which may, unless otherwise specified, be carbon or nitrogen linked, wherein a -CH2- group is optionally be replaced by a -C(O)-; and where unless stated to the contrary a ring nitrogen or sulphur atom is optionally oxidised to form the N-oxide or S-oxide(s) or a ring nitrogen is optionally quarternized; wherein a ring -NH is optionally substituted by acetyl, formyl, methyl or mesyl; and a ring is optionally substituted by one or more halo. It is understood that when the total number of S and O atoms in the heterocyclyl exceeds 1, then these heteroatoms are not adjacent to one
another. If the said heterocyclyl group is bi- or tricyclic then at least one of the rings may optionally be a heteroaromatic or aromatic ring provided that at least one of the rings is non-heteroaromatic. If the said heterocyclyl group is monocyclic then it must not be aromatic. Examples of heterocyclyls include, but are not limited to, piperidinyl, N- acetylpiperidinyl, N-methylpiperidinyl, N-formylpiperazinyl, N-mesylpiperazinyl, homopiperazinyl, piperazinyl, azetidinyl, oxetanyl, morpholinyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, indolinyl, tetrahydropyranyl, dihydro-2H-pyranyl, tetrahydrofuranyl and 2,5-dioxoimidazolidinyl.
As used herein, the phrase "protecting group" means temporary substituents which protect a potentially reactive functional group from undesired chemical transformations. Examples of such protecting groups include esters of carboxylic acids, silyl ethers of alcohols, and acetals and ketals of aldehydes and ketones respectively. The field of protecting group chemistry has been reviewed (Greene, T. W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3rd ed.; Wiley: New York, 1999).
As used herein, "pharmaceutically acceptable" is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
As used herein, "pharmaceutically acceptable salts" refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like. The pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric acid.
The pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound that contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like diethyl ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are used.
As used herein, "tautomer" means other structural isomers that exist in equilibrium resulting from the migration of a hydrogen atom. For example, keto-enol tautomerism where the resulting compound has the porperties of both a ketone and an unsturated alcohol.
As used herein "stable compound" and "stable structure" are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
Compounds of the invention further include hydrates and solvates.
The present invention further includes isotopically-labelled compounds of the invention. An "isotopically" or "radio-labelled" compound is a compound of the invention where one or more atoms are replaced or substituted by an atom having an atomic mass or mass number different from the atomic mass or mass number typically found in nature (i.e., naturally occurring). Suitable radionuclides that may be incorporated in compounds of the present invention include but are not limited to 2H (also written as D for deuterium), 3H (also written as T for tritium), 11C, 13C, 14C, 13N, 15N, 15O, 17O, 180, 18F, 35S, 36Cl, 82Br, 75Br, 76Br, 77Br, 1231, 1241, 125I and 131I. The radionuclide that is incorporated in the instant radio-labelled compounds will depend on the specific application of that radio-labelled compound. For example, for in vitro receptor labelling and competition assays, compounds that incorporate 3H, 14C, 82Br, 1251 , 1311, 35S or will generally be most useful. For radio-imaging applications 11C, 18F, 1251, 1231, 124I, 131I, 75Br, 76Br or 77Br will generally be most useful.
It is understood that a "radio-labelled compound" is a compound that has incorporated at least one radionuclide. In some embodiments the radionuclide is selected from the group consisting of 3H, 14C, 1251 , 35S and 82Br.
The anti-dementia treatment defined herein may be applied as a sole therapy or may involve, in addition to the compound of the invention, conventional chemotherapy. Such chemotherapy may include one or more of the following categories of agents: acetyl cholinesterase inhibitors, anti-inflammatory agents, cognitive and/or memory enhancing agents or atypical antipsychotic agents.
Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of the treatment. Such combination products employ the compounds of this invention.
Compounds of the present invention may be administered orally, parenteral, buccal, vaginal, rectal, inhalation, insufflation, sublingually, intramuscularly, subcutaneously, topically, intranasally, intraperitoneally, intrathoracially, intravenously, epidurally, intrathecally, intracerebroventricularly and by injection into the joints.
The dosage will depend on the route of administration, the severity of the disease, age and weight of the patient and other factors normally considered by the attending physician, when determining the individual regimen and dosage level as the most appropriate for a particular patient.
An effective amount of a compound of the present invention for use in therapy of dementia is an amount sufficient to symptomatically relieve in a warm-blooded animal, particularly a human the symptoms of dementia, to slow the progression of dementia, or to reduce in patients with symptoms of dementia the risk of getting worse.
For preparing pharmaceutical compositions from the compounds of this invention, inert, pharmaceutically acceptable carriers can be either solid or liquid. Solid form preparations include powders, tablets, dispersible granules, capsules, cachets, and suppositories.
A solid carrier can be one or more substances, which may also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, or tablet disintegrating agents; it can also be an encapsulating material.
5
In powders, the carrier is a finely divided solid, which is in a mixture with the finely divided active component. In tablets, the active component is mixed with the carrier having the necessary binding properties in suitable proportions and compacted in the shape and size desired.
10
For preparing suppository compositions, a low-melting wax such as a mixture of fatty acid glycerides and cocoa butter is first melted and the active ingredient is dispersed therein by, for example, stirring. The molten homogeneous mixture is then poured into convenient sized molds and allowed to cool and solidify.
I5
Suitable carriers include magnesium carbonate, magnesium stearate, talc, lactose, sugar, pectin, dextrin, starch, tragacanth, methyl cellulose, sodium carboxymethyl cellulose, a low-melting wax, cocoa butter, and the like.
20 In some embodiments, the present invention provides a compound of formula I or a pharmaceutically acceptable salt thereof for the therapeutic treatment (including prophylactic treatment) of mammals including humans, it is normally formulated in accordance with standard pharmaceutical practice as a pharmaceutical composition.
25 In addition to the compounds of the present invention, the pharmaceutical composition of this invention may also contain, or be co-administered (simultaneously or sequentially) with, one or more pharmacological agents of value in treating one or more disease conditions referred to herein.
30 The term composition is intended to include the formulation of the active component or a pharmaceutically acceptable salt with a pharmaceutically acceptable carrier. For example this invention may be formulated by means known in the art into the form of, for example,
tablets, capsules, aqueous or oily solutions, suspensions, emulsions, creams, ointments, gels, nasal sprays, suppositories, finely divided powders or aerosols or nebulisers for inhalation, and for parenteral use (including intravenous, intramuscular or infusion) sterile aqueous or oily solutions or suspensions or sterile emulsions.
Liquid form compositions include solutions, suspensions, and emulsions. Sterile water or water-propylene glycol solutions of the active compounds may be mentioned as an example of liquid preparations suitable for parenteral administration. Liquid compositions can also be formulated in solution in aqueous polyethylene glycol solution. Aqueous solutions for oral administration can be prepared by dissolving the active component in water and adding suitable colorants, flavoring agents, stabilizers, and thickening agents as desired. Aqueous suspensions for oral use can be made by dispersing the finely divided active component in water together with a viscous material such as natural synthetic gums, resins, methyl cellulose, sodium carboxymethyl cellulose, and other suspending agents known to the pharmaceutical formulation art.
The pharmaceutical compositions can be in unit dosage form. In such form, the composition is divided into unit doses containing appropriate quantities of the active component. The unit dosage form can be a packaged preparation, the package containing discrete quantities of the preparations, for example, packeted tablets, capsules, and powders in vials or ampoules. The unit dosage form can also be a capsule, cachet, or tablet itself, or it can be the appropriate number of any of these packaged forms.
Compositions may be formulated for any suitable route and means of administration. Pharmaceutically acceptable carriers or diluents include those used in formulations suitable for oral, rectal, nasal, topical (including buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous, intradermal, intrathecal and epidural) administration. The formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy.
For solid compositions, conventional non-toxic solid carriers include, for example, pharmaceutical grades of mannitol, lactose, cellulose, cellulose derivatives, starch,
magnesium stearate, sodium saccharin, talcum, glucose, sucrose, magnesium carbonate, and the like may be used. Liquid pharmaceutically administrable compositions can, for example, be prepared by dissolving, dispersing, etc, an active compound as defined above and optional pharmaceutical adjuvants in a carrier, such as, for example, water, saline aqueous dextrose, glycerol, ethanol, and the like, to thereby form a solution or suspension. If desired, the pharmaceutical composition to be administered may also contain minor amounts of non-toxic auxiliary substances such as wetting or emulsifying agents, pH buffering agents and the like, for example, sodium acetate, sorbitan monolaurate, triethanolamine sodium acetate, sorbitan monolaurate, triethanolamine oleate, etc. Actual methods of preparing such dosage forms are known, or will be apparent, to those skilled in this art; for example, see Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pennsylvania, 15th Edition, 1975.
The compounds of the invention may be derivatised in various ways. As used herein "derivatives" of the compounds includes salts (e.g. pharmaceutically acceptable salts), any complexes (e.g. inclusion complexes or clathrates with compounds such as cyclodextrins, or coordination complexes with metal ions such as Mn2+ and Zn2+), free acids or bases, polymorphic forms of the compounds, solvates (e.g. hydrates), prodrugs or lipids, coupling partners and protecting groups. By "prodrugs" is meant for example any compound that is converted in vivo into a biologically active compound.
Salts of the compounds of the invention are preferably physiologically well tolerated and non toxic. Many examples of salts are known to those skilled in the art. All such salts are within the scope of this invention, and references to compounds include the salt forms of the compounds.
Where the compounds contain an amine function, these may form quaternary ammonium salts, for example by reaction with an alkylating agent according to methods well known to the skilled person. Such quaternary ammonium compounds are within the scope of the invention.
Compounds containing an amine function may also form N-oxides. A reference herein to a compound that contains an amine function also includes the N-oxide.
Where a compound contains several amine functions, one or more than one nitrogen atom may be oxidised to form an N-oxide. Particular examples of N-oxides are the N-oxides of a tertiary amine or a nitrogen atom of a nitrogen-containing heterocycle.
N-Oxides can be formed by treatment of the corresponding amine with an oxidizing agent such as hydrogen peroxide or a per-acid (e.g. a peroxycarboxylic acid), see for example Advanced Organic Chemistry, by Jerry March, 4th Edition, Wiley Interscience, pages. More particularly, N-oxides can be made by the procedure of L. W. Deady (Syn. Comm. 1977, 7, 509-514) in which the amine compound is reacted with w-chloroperoxybenzoic acid (MCPBA), for example, in an inert solvent such as dichloromethane.
Where the compounds contain chiral centres, all individual optical forms such as enantiomers, epimers and diastereoisomers, as well as racemic mixtures of the compounds are within the scope of the invention.
Compounds may exist in a number of different geometric isomeric, and tautomeric forms and references to compounds include all such forms. For the avoidance of doubt, where a compound can exist in one of several geometric isomeric or tautomeric forms and only one is specifically described or shown, all others are nevertheless embraced by the scope of this invention.
The quantity of the compound to be administered will vary for the patient being treated and will vary from about 100 ng/kg of body weight to 100 mg/kg of body weight per day and preferably will be from 10 pg/kg to 10 mg/kg per day. For instance, dosages can be readily ascertained by those skilled in the art from this disclosure and the knowledge in the art. Thus, the skilled artisan can readily determine the amount of compound and optional additives, vehicles, and/or carrier in compositions and to be administered in methods of the invention.
Compounds of the present invention have been shown to inhibit beta secretase (including BACE) activity in vitro. Inhibitors of beta secretase have been shown to be useful in blocking formation or aggregation of Aβ peptide and therefore have beneficial effects in treatment of Alzheimer's Disease and other neurodegenerative diseases associated with elevated levels and/or deposition of Aβ peptide. Therefore, it is believed that the compounds of the present invention may be used for the treatment of Alzheimer disease and disease associated with dementia Hence, compounds of the present invention and their salts are expected to be active against age-related diseases such as Alzheimer, as well as other Aβ related pathologies such as Downs syndrome and β-amyloid angiopathy. It is expected that the compounds of the present invention would most likely be used as single agents but could also be used in combination with a broad range of cognition deficit enhancement agents.
Methods of preparation The present invention also relates to processes for preparing the compound of formula I as a free base or a pharmaceutically acceptable salt thereof. Throughout the following description of such processes it is understood that, where appropriate, suitable protecting groups will be added to, and subsequently removed from the various reactants and intermediates in a manner that will be readily understood by one skilled in the art of organic synthesis. Conventional procedures for using such protecting groups as well as examples of suitable protecting groups are for example described in "Protective Groups in Organic Synthesis", T.W. Greene, P.G.M Wutz, Wiley-Interscience, New York, 1999. It is understood that microwaves can be used for the heating of reaction mixtures.
Preparation of Intermediates
The process, wherein R1, R2, R3, R4, R5 and A, unless otherwise specified, are as defined hereinbefore, comprises,
(i) conversion of a compound of formula II to obtain a compound of formula III, wherein R6 is a substituted phenyl,

(H) (πi)
may be carried out by reaction with a suitable reagent such as 1,3-propanedithiol in the presence of an acid such as hydrochloric acid or/j-toluenesulfonic acid, or a Lewis acid such as boron trifluoride or titanium tetrachloride or ruthenium(III) chloride. The reaction may be preformed in a suitable solvent such as dichloromethane, acetonitrile, chloroform, toluene or diethyl ether, at a temperature between -78 0C and reflux.
(ii) reaction between a compound of formula IV with a compound of formula III to obtain a compound of formula V, wherein R6 is a substituted phenyl and R7 is defined as in A above,

may be carried out by treating III with a suitable base such as an alkyllithium reagent e.g. «-butyl lithium or /-butyl lithium or lithium diisopropylamide, before addition of IV. The reaction may be carried out in a solvent such as tetrahydrofuran or diethyl ether, or a mixture of tetrahydrofuran or diethyl ether with hexane, at a temperature between -100 0C and 25 °C. The reaction may be aided by the presence of reagents such as hexamethylphosphoric triamide or N//,N^-tetramethyl-l,2-ethanediamine.
(iii) oxidative deprotection and oxidation of a compound of formula V to obtain a compound of formula VI, wherein R6 is a substituted phenyl and R7 is defined as in A above,

(V) (VI)
may be carried out by: a) reaction with a suitable reagent such as 1,1,1 -tris(acetyloxy)- 1 , 1 -dihydro- 1 ,2- benziodoxol-3(lH)-one, bis(trifluoroacetoxy)iodobenzene, N-bromosuccinimide, or a mixture of trifluoroacetic acid with either sodium nitrite or formaldehyde, in a suitable solvent such as dichloromethane, acetonitrile, chloroform, acetone or water or a mixture thereof, between -5 °C to 40 °C. The reaction may be aided by the presence of an alcohol such as J-butanol. or, b) hydrolysis by treatment with a suitable reagent or reagent combination such as N- chlorosuccinimide and silver nitrate, N-iodosuccinimide, 3-chloroperoxybenzoic acid, ammonium cerium(TV) nitrate, thallium(III) nitrate, mercury(II) chloride and calcium carbonate, or mercery(II) acetate. The reaction may be preformed in a suitable solvent such as water, acetonitrile, methanol, acetone or diethyl ether or mixtures thereof, between -50 0C and 50 0C , followed or preceded by, oxidation with a reagent such as 1,1,1- tris(acetyloxy)- 1,1 -dihydro- l,2-benziodoxoI-3(lH)-one, manganese dioxide, hydrogen peroxide, potassium permanganate, pyridinium chlorochromate, copper sulfate or bromine, in a suitable solvent such as dichloromethane, water, acetonitrile, chloroform or dimethyl formamide, at between 0 °C and reflux.
(iv) conversion of a compound of formula VI to obtain a compound of formula VII, wherein R6 is a substituted phenyl and R7 is defined as in A above,

(v) conversion of a compound of formula VII to obtain a compound of formula VHI, wherein R6 is a substituted phenyl and R7 is defined as in A above,

may be carried out by reaction with ammonia, or an ammonia equivalent, together with an alkylhydroperoxide such as 7-butylhydroperoxide in a solvent such as ethanol, methanol or water, or a mixture thereof, at 0 0C to 50 °C.
(vi) cross coupling of a compound of formula IX and a compound of formula X to obtain a compound of formula XI, wherein Halo is a halogen such as bromine, chlorine or iodine, R6 is a substituted phenyl, and R7 is defined as in A above or trimethylsilyl,
R-HaIo + ΞΞΞΞΞ≡— R6 - R7 Ξ^-R6
(IX) (X) (XI)
may be performed with a suitable arylhalide such as a compound of formula II and an appropriate alkyne such as ethynyltrimethylsilane in the presence of copper(I) iodide and a suitable palladium catalyst such as dichlorobis(benzonitrile)palladium(II), bis(triphenylphosphine)palladium(ll) dichloride, palladium(II) chloride, palladium(O) tetrakistriphenylphosphine with or without a suitable ligand such as tri-fert-butylphosphine or triphenylphosphine, and a suitable base, such as trietylamine, diisopropylamine or
piperidine may be used. The reaction may be performed in a solvent such as tetrahydrofuran or N,N-dimethylformamide, at temperatures between 20 °C and 1000C.
(viϊ) desilylation of a compound of formula XII to a compound of formula XIII, wherein R7 is defined as in A above,
R'- -Si(CH '3. '3 R'-
XII XIII
may be performed using silver(I) nitrate or a suitable base such as potassium hydroxide, sodium hydroxide, lithium hydroxide or potassium carbonate, or using a fluoride ion- mediated desilylation using a suitable compound such as tetrabutylammonium fluoride or potassium fluoride. The reaction may be performed in a solvent such as tetrahydrofuran, methanol, dichloromethane or water, or mixtures thereof, at temperatures between 0 °C and 1000C.
(viii) borylation of a compound of formula XIII to obtain a compound of formula XIV, wherein R8 is cyclopropyl and R9 may be a group outlined in Scheme I, wherein R10 and R11 are groups such as OH, Ci-βalkylO or C2-3alkyl0 fused together to form a 5 or 6 membered boron containing heterocycle and the alkyl, cycloalkyl or aryl moieties may be optionally substituted,
R'- Λ -R3
XIII XVI

5 between -78 0C and 20 0C; or, b) a suitable boron species such as biscatecholatodiboron, bispinacolatodiboron or pinacolborane in the presence of a suitable palladium catalyst such as palladium(O) tetrakistriphenylphosphine, palladium diphenylphosphineferrocene dichloride or palladium
I0 acetate, with or without a suitable ligand such as 2-(dicyclohexylphosphino)biphenyl, and a suitable base, such as a tertiary amine, such as trietylamine or diisopropylethylamine, or potassium acetate may be used. The reaction may be performed in a solvent such as dioxane, toluene, acetonitrile, water, ethanol or 1,2-dimethoxyethane, or mixtures thereof, at temperatures between 20 0C and 160 0C.
I5
(ix) reduction of a compound of formula XV to a compound of formula XVI, wherein R12 is cyclopropyl and R13 is a substituted aryl,
20 XV XVI
may be preformed using a suitable catalyst such as palladium-on-charcoal, Raney nickel or Wilkinson's catalyst, and hydrogen. The reaction may be preformed in a suitable solvent such as ethyl acetate, methanol or ethanol, at temperatures between 20 0C and reflux.
25
(x) reduction of a compound of formula XVII to a compound of formula XVIII, wherein R7 is defined as in A above,
O
R'-^N — 7X R7/ H
30 xvπ xviπ
may be preformed using a metal hydride reducing agent such as diisopropylaluminium hydride or lithium aluminum hydride. The reaction may be preformed in a suitable solvent such as dichlorometane, toluene or benzene, at a temperature between -78 0C and 25 0C.
(xi) borylation of a compound of formula XIX to obtain a compound of formula XX, wherein R14 is an optionally substituted aryl or heteroaryl and R15 may be a group outlined in Scheme II, wherein R16 and R17 are groups such as OH, Ci-βalkylO or C2-3alkylO fused together to form a 5 or 6 membered boron containing heterocycle and the alkyl, cycloalkyl
10 or aryl moieties may be optionally substituted,
R —halo R14-R15
XIX XX

may be carried out by a reaction with: a) an alkyllithium such as butyllithium, or magnesium, and a suitable boron compound such as trimethyl borate or triisopropyl borate. The reaction may be performed in a suitable
2o solvent such as tetrahydrofuran, hexane or dichloromethane in a temperature range between -78 0C and 200C; or, b) a suitable boron species such as biscatecholatodiboron, bispinacolatodiboron or pinacolborane in the presence of a suitable palladium catalyst such as palladium(O)
25 tetrakistriphenylphosphine, palladium diphenylphosphineferrocene dichloride or palladium acetate, with or without a suitable ligand such as 2-(dicyclohexylphosphino)biphenyl, and a suitable base, such as a tertiary amine, such as trietylamine or diisopropylethylamine, or potassium acetate may be used. The reaction may be performed in a solvent such as
dioxane, toluene, acetonitrile, water, ethanol or 1,2-dimethoxyethane, or mixtures thereof, at temperatures between 20 °C and 160 0C.
(xii) oxidation of a compound of formula XXI to obtain a compound of formula XXII, wherein R18 is defined as in A above and R19 is an optionally substituted phenyl,

(XXI) (XXII)
may be performed by reaction with a suitable reagent or mixture of reagents, such as sodium periodate and ruthenium dioxide, iodine and dimethyl sulfoxide, palladium chloride and dimethyl sulfoxide, oxone, hydrogen peroxide, oxygen, potassium permanganate, ruthenium tetroxide, or selenium dioxide, in a suitable solvent such as dimethyl sulfoxide, dichloromethane, acetonitrile, water, acetone, chloroform or carbon tetrachloride at a temperature between -78 0C and 150 0C. The reaction may be aided by the presence of a catalyst such as ruthenium(III) chloride or iron(III) chloride.
(xiii) conversion of a compound of formula VI to obtain a compound of formula VIII, wherein R6 is a substituted phenyl and R7 is defined as in A and C above,


may be carried out by reaction with an appropriately N-substituted guanidine such as N- methylguanidine, N-ethylguanidine, or N-propylguanidine, in the presence of a suitable base such as potassium hydroxide, sodium hydroxide or sodium carbonate in a suitable solvent such as water, dimethyl sulfoxide, dioxane, ethanol or methanol or mixtures thereof, between 20 0C and reflux.
(xiv) conversion of a compound of formula XXIII to obtain a compound of formula XXIV, wherein R20 is defined as in A above,

(XXIII) (XXIV)
5 may be preformed by treating XXIII with: a) a formylation agent such as methylchloroformate, dichloromethoxymethane, dichlorobutoxymethane, formaldehyde, carbon monoxide, hydrogen cyanide or a combination of phosphorous oxychloride with N-methyl-N-phenyl formamide oτNJϊ-o dimethylformamide. The reaction can be performed with or without a suitable reagent such as titanium tetrachloride, tin tetrachloride, aluminium trichloride and copper(I) chloride or combinations thereof. The reaction may be preformed with or without a suitable solvent such as dichloromethane, benzene, or 1,1,2,2-tetrachloroethane at a temperature between - 20 °C and reflux; 5 or, b) a bromination agent such as bromine or N-bromosuccinimide in a solvent such as chloroform, Λ^N-dimethylformamide, tetrahydrofurane or hexane or mixtures thereof at a temperature between -78 0C and reflux , followed by treatment with a base such as butyl lithium and subsequently a reagent such as N,N-dirnethylformamide or acetic acid in ao solvent such as tetrahydrofurane and/or hexane at a temperature between -78 0C and 0 °C.
Methods of Preparation of End products
Another object of the invention are processes a or b or c for the preparation of compounds5 of general formula I, wherein R1, R2, R3, R4, R5 and A unless otherwise specified, are defined as hereinbefore, and salts thereof. When it is desired to obtain the acid salt, the free base may be treated with an acid such as a hydrogen halide such as hydrogen chloride, sulphuric acid, a sulphonic acid such as methane sulphonic acid or a carboxylic acid such as acetic or citric acid in a suitable solvent such as tetrahydrofuran, diethyl ether, methanol,o ethanol, chloroform or dichloromethane or mixtures thereof, the reaction may occur between -30 0C to 50 0C.
These processes comprise;
(a) conversion of a compound of formula XXV to a compound of formula I, wherein halo represents halogen such as chlorine, bromine or iodine, R7 is as defined for A above, R21 is as defined for R5 above, R1, R2, R5 and A is as defined above, R14 is an optionally substituted aryl or heteroaryl and R15 may be a group outlined in Scheme II above,

CXXV) (XXVI) (I)
may be carried out by a de-halogen coupling with a suitable compound of formula XXVI. The reaction may be carried out by coupling of a compound of formula XXV with an appropriate aryl boronic acid or boronic ester of formula XXVI. The reaction may be carried out using a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium(0), palladium diphenylphosphineferrocene dichloride or palladium acetate, together with, or without, a suitable ligand such as tή-tert- butylphosphine or 2-(dicyclohexylphosphino)biphenyl, or using a nickel catalyst such as nickel on charcoal or l,2-bis(diphenylphosphino)ethanenickel dichloride together with zinc and sodium triphenylphosphinetrimetasulfonate. A suitable base such as cesium fluoride, an alkyl amine such as triethyl amine, or an alkali metal or alkaline earth metal carbonate or hydroxide such as potassium carbonate, sodium carbonate, cesium carbonate, or sodium hydroxide may be used in the reaction, which may be performed in a temperature range between 200C and 1600C, in a suitable solvent such as toluene, tetrahydrofuran, dioxane, dimethoxyethane, water, ethanol or N,N-dimethylformamide, or mixtures thereof.
(b) reduction of a compound of formula XXVII to a compound of formula I, wherein R7 is as defined for A above, R21 is as defined for R5 above, R2, R5 and A are as defined above, and R1 is CH2CH2cyclopropyl.

(XXVH) C)
may be preformed using a suitable catalyst such as palladium-on-charcoal, Raney nickel or Wilkinson's catalyst, and hydrogen. The reaction may be preformed in a suitable solvent such as ethyl acetate, methanol or ethanol, at temperatures between 20 0C and reflux.
General Methods
Starting materials used were available from commercial sources, or prepared according to literature procedures.
Microwave heating was performed in a Creator, Initiator or Smith Synthesizer Single- mode microwave cavity producing continuous irradiation at 2450 MHz. 1H NMR spectra were recorded in the indicated deuterated solvent at 400 MHz. The 400MHz spectra were obtained unless stated otherwise, using a Bruker av400 NMR spectrometer equipped with a 3 mm flow injection SEI 1HTD-13C probe head with Z- gradients, using a BEST 215 liquid handler for sample injection, or using a Bruker DPX400 NMR spectrometer equipped with a 4-nucleus probehead with Z-gradients. Chemical shifts are given in ppm down- and upfield from TMS. Resonance multiplicities are denoted s, d, t, q, m and br for singlet, doublet, triplet, quartet, multiplet, and broad respectively.
LC-MS analyses were recorded on a Waters LCMS equipped with a Waters X-Terra MS, C8-column, (3.5 μm, 100 mm x 3.0 mm i.d.). The mobile phase system consisted of A: 10 mM ammonium acetate in water/acetonitrile (95:5) and B: acetonitrile. A linear gradient was applied running from 0% to 100% B in 4-5 minutes with a flow rate of 1.0 mL/min. The mass spectrometer was equipped with an electrospray ion source (ESI) operated in a
positive or negative ion mode. The capillary voltage was 3 kV and the mass spectrometer was typically scanned between m/z 100-700. Alternative, LC-MS HPLC conditions were as follows: Column: Agilent Zorbax SB-C8 2mm ID X 50mm Flow: 1.4 mL/minGradient: 95% A to 90% B over 3 min. hold 1 minute ramp down to 95% A over 1 minute and hold 1 minute. Where A = 2% acetonitrile in water with 0.1% formic acid and B = 2% water in acetonitrile with 0.1% formic acid. UV-DAD 210-400 run. Or LC-MS analyses were performed on a LC-MS consisting of a Waters sample manager 2111 C, a Waters 1525 μ binary pump, a Waters 1500 column oven, a Waters ZQ single quadrupole mass spectrometer, a Waters PDA2996 diode array detector and a Sedex 85 ELS detector. The mass spectrometer was configured with an atmospheric pressure chemical ionisation
(APCI) ion source which was further equipped with atmospheric pressure photo ionisation (APPI) device. The mass spectrometer scanned in the positive mode, switching between APCI and APPI mode. The mass range was set to m/z 120-800 using a scan time of 0.3 s. The APPI repeller and the APCI corona were set to 0.86 kV and 0.80 μA, respectively. In addition, the desolvation temperature (300°C), desolvation gas (400 L/Hr) and cone gas (5 L/Hr) were constant for both APCI and APPI mode. Separation was performed using a Gemini column Cl 8, 3.0 mm x 50 mm, 3 μm, (Phenomenex) and run at a flow rate of 1 ml/min. A linear gradient was used starting at 100 % A (A: 10 mM ammonium acetate in 5% methanol) and ending at 100% B (methanol). The column oven temperature was set to 40 0C.
Mass spectra (MS) were run using an automated system with atmospheric pressure chemical (APCI or CI) or electrospray (+ESI) ionization. Generally, only spectra where parent masses are observed are reported. The lowest mass major ion is reported for molecules where isotope splitting results in multiple mass spectral peaks (for example when chlorine is present).
GC-MS analyses were performed on a Agilent 6890N GC equipped with a Chrompack CP- SiI 5CB column (25 m x 0.25 mm i.d. df = 0.25)), coupled to an Agilent 5973 Mass Selective Detector operating in a chemical ionization (CI) mode and the MS was scanned between m/z 50-500. HPLC assays were performed using an Agilent HPl 100 Series system equipped with a Waters X-Terra MS, C8 column (3.0 x 100 mm, 3.5 μm). The column temperature was set
to 40 °C and the flow rate to 1.0 rnL/min. The Diode Array Detector was scanned from 200-300 nm. A linear gradient was applied, run from 0% to 100% B in 4 min. Mobile phase A: 10 mM ammonium acetate in water/acetonitrile (95:5), mobile phase B: acetonitrile. Preparative HPLC was performed on a Waters Auto purification HPLC-UV system with a diode array detector using a Waters XTerra MS C8 column (19x300 mm, 7 μm) and a linear gradient of mobile phase B was applied. Mobile phase A: 0.1 M ammonium acetate in water/acetonitrile (95:5) and mobile phase B: acetonitrile. Flow rate: 20 mL/min. Thin layer chromatography (TLC) was performed on Merch TLC-plates (Silica gel 60 F254) and spots were UV visualized. Flash chromatography was performed using Merck Silica gel 60 (0.040-0.063 mm), or employing a Combi Flash® Companion™ system using RediSep™ normal-phase flash columns.
Compounds have been named using ACD/Name, version 9.0, software from Advanced Chemistry Development, Inc. (ACD/Labs), Toronto ON, Canada, www.acdlabs.com, 2004.
EXAMPLES
Below follows a number of non-limiting examples of compounds of the invention.
Example 1 6-(l,3-Dithian-2-yI)-2,2-dimethyIchromane

Propane-l,3-dithiol (541 mg, 5.0 mmol) was added to a solution of 2,2-dimethylchromane- 6-carbaldehyde (1.0 g, 5.26 mmol) in anhydrous dichloromethane (50 mL) and the mixture was stirred for 15 min and then cooled to -78 °C. Trifluoro(l,r-oxydiethane)boron (2.0 mL, 15.8 mmol) was added, the reaction was stirred at -78 0C for 15 min and the reaction was allowed to reach room temperature. Water was added and the mixture was diluted with dichloromethane. The layers were separated and the organic phase was washed with water
before the solvent was evaporated in vacuo. Purification by column chromatography, using a gradient of 0 to 40% ethyl acetate in n-heptane as the eluent, gave 1.32 g (89% yield) the title compound: 1H NMR (CDCl3) δ 7.23 - 7.10 (m, 2 H), 6.72 (d, J= 8.3 Hz, 1 H), 5.10 (s, 1 H), 3.12 - 3.10 (m, 2 H), 2.95 - 2.85 (m, 2 H), 2.76 (t, J= 6.7 Hz, 2 H), 2.22 - 2.11 (m, 1 5 H), 1.97 - 1.87 (m, 1 H), 1.79 (t, J= 6.7 Hz, 2 H), 1.33 (s, 6 H); MS (ES) m/z 281 [M+l]+.
Example 2
(S-Bromo^-fluorophenylJfl-Cl^-dimethyl-S^-dihydro-liϊ-chromen-ό-yO-l.S-dithian- 2-yl] methanol
o

6-(l,3-Dithian-2-yl)-2,2-dimethylchromane (1.32 g, 4.71 mmol) was dissolved in anhydrous tetrahydrofuran (40 mL) under an atmosphere of argon, cooled to -78 °C and n- butyl lithium (2.1 mL, 2.5 M) was added dropwise. The solution was stirred at -78 °C for 20 min and then treated with 3-bromo-4-fluorobenzaldehyde (1.0 g, 4.9 mmol). Thes reaction was stirred for another 20 min at —78 °C and was then allowed to reach room temperature. Aqueous ammonium chloride was added and the mixture was extracted with dichloromethane. The combined organic phases were evaporated and the residue was purified by column chromatography, using a gradient of 0 to 40% ethyl acetate in n- heptane as the eluent, to give 813 mg (36% yield) the title compound: 1H NMR (CDCl3) δo 7.45 (dd,J= 8.6, 2.5 Hz, 1 H), 7.17 (d, J= 2.3 Hz, 1 H), 7.14 - 7.064 (m, 1 H), 6.95 - 6.85 (m, 2 H), 6.76 (d, J= 8.6 Hz, 1 H), 3.06 (s, 1 H), 2.81 - 2.64 (m, 4 H), 1.97 - 1.90 (m, 2 H), 1.82 (t, J= 6.7 Hz, 2 H), 1.37 (s, 6 H), 0.94 - 0.86 (m, 2 H); MS (ES) m/z 465, 467 [M- water]+. 5 Example 3 l-(3-Bromo-4-fluorophenyl)-2-(2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl)ethane- 1,2-dione

(3-Bromo-4-fluorophenyl)[2-(2,2-dimethyl-3,4-dihydro-2H-chronien-6-yl)-l,3-ditliian-2- yljmethanol (1.32 g, 4.71 mmol) and fer/-butanol (436 mg, 5.89 mmol) was dissolved in anhydrous dichloromethane (40 mL) under an atmosphere of nitrogen. Dess-Martin Periodinane (1.6 g, 3.78 mmol) was added to the solution and the reaction was stirred over night. Sodium thiosulphate (30 mL, 1 M) was added and the layers were separated. The organic phase was washed with aqueous sodium hydrogen carbonate and concentrated in vacuo. The product was purified by column chromatography, using a gradient of 0 to 70% ethyl acetate in n-heptane as the eluent, to give 570 mg (87% yield) the title compound: 1H NMR (CDCl3) δ 8.26 - 8.17 (m, 1 H), 7.99 - 7.90 (m, 1 H), 7.77 - 7.70 (m, 2 H), 7.25 - 7.17 (m, 1 H), 6.87 (d, J= 8.3 Hz, 1 H), 2.82 (t, J= 6.7 Hz, 2 H), 1.85 (t, J= 6.8 Hz, 2 H), 1.37 (s, 6 H); MS (ES) m/z 391, 393 [M+l]+.
Example 4 5-(3-Bromo-4-fluorophenyI)-5-(2,2-dimethyl-3,4-dihydro-2H-chromen-6-yI)-3-methyl- 2-thioxoimidazolidin-4-one

Example 5 2-Amino-5-(3-bromo-4-fluorophenyl)-5-(2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl)- 3-methyl-3,5-dihydro-4H-imidazol-4-one

5-(3-Bromo-4-fluorophenyl)-5-(2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl)-3-methyl-2- thioxoimidazolidin-4-one (514 mg, 1.11 mmol) was dissolved in methanol (30 mL) and ammonium hydroxide (33%, 10 mL) and ferf-butyl hydroperoxide (1.5 g, 16.6 mmol) was added. The mixture was stirred at room temperature over night and then concentrated until approximately 50% of the volume remained. Water was added and the mixture was extracted with dichloromethane. The combined organic phases were washed with water and concentrated in vacuo. The product was purified by column chromatography, using a gradient of 0 to 10% 1:9 ammonium hydroxide/methanol in dichloromethane as the eluent, to give 248 mg (50% yield) of the title compound: MS (ES) m/z 446, 448 [M+l]+.
Example 6
2-Amino-5-(2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl)-5-(6-fluoro-3'- methoxybiphenyl-3-yl)-3-methyI-3,5-dihydro-4H-imidazol-4-one hydrochloride

A mixture of 2-amino-5-(3-bromo-4-fluorophenyl)-5-(2,2-dimethyl-3 ,4-dihydro-2H- chromen-6-yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one (83 mg, 0.186 mmol), (3- methoxyphenyl)boronic acid (30 mg, 0.242 mmol), [1,1'-
5 bis(diphenylphosphino)ferrocene]palladium(II) chloride dichloromethane adduct (15 mg, 0.018 mmol), potassium carbonate (154 mg, 1,12 mmol), 100 μL water and anhydrous tetrahydrofuran (3 mL) was irradiated in a microwave at 130 °C for 15 min. When cooled to room temperature the mixture was filtered and dimethyl sulfoxide (500 μL) was added. The solution was concentrated in vacuo to remove tetrahydrofuran and purified by
I0 preparative ΗPLC to give 49 mg of the base. The base was dissolved in 1 M hydrochloric acid in diethyl ether and the solvent was evaporated to give 64 mg (67% yield) of the title compound: 1U NMR (CDCl3) δ 7.58 (dd, J= 7.3 Hz, 1 H), 7.48 - 7.39 (m, 1 H), 7.34 (t, J = 8.0 Hz, 1 H), 7.18 - 7.03 (m, 5 H), 6.98 - 6.84 (m, 1 H), 6.73 (d, J= 8.3 Hz, 1 H), 3.84 (s, 3 H), 3.14 (s, 3 H), 2.73 (t, J= 6.8 Hz, 2 H), 1.77 (t,J= 6.7 Hz, 2 H), 1.31 (s, 6 H); MS (ES) is w/z 474 [M+l]+.
Example 7 (3,4-Dihydro-2H-chromen-6-ylethynyl)(trimethyI)silane
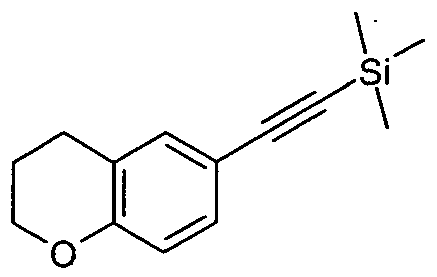
20 A mixture of 6-iodo-chroman (6.3 g, 24.2 mmol; described in: Togo H. et.al. J Chem. Soc. Perkin Trans. 1, 1997, 787-793)), triphenylphosphine (0.95 g, 3.6 mmol), cuprous iodide (0.23 g, 1.2 mmol) and triethylamine (12.1 mL, 87.2 mmol) in acetonitrile (100 mL) was
degassed with argon for 10 min. (Trimethylsilyl)acetylene (13.7 mL, 48.4 mmol) was added followed by palladium acetate (0.27 g, 1,2 mmol). The mixture was stirred under an atmosphere of argon for 2.5 h at room temperature, concentrated under reduced pressure and partitioned between water and dichloromethane. The organic layer was dried over magnesium sulfate, filtered and concentrated. The residue was slurried with hexane, filtered and concentrated to give 4.3 g of the crude title compound that was used in the next step without further purification.
Example 8 6-Ethynylchromane

(3,4-Dihydro-2H-chromen-6-ylethynyl)(trimethyl)silane (4.3 g, crude) was dissolved in methanol (150 mL) and stirred with potassium carbonate (2.0 g, 14.5 mL) for 2 h at room temperature. The mixture was filtered and concentrated under reduced pressure. The residue was purified by column chromatography, using heptane/ethyl acetate 10:1 to 5:1 as the eluent, to give 2.6 g (89% yield) of the title compound: 1H NMR (CDCl3) δ 7.18 - 7.25 (m, 2 H), 6.73 (d, J= 8.1 Hz, 1 H), 4.20 (t, 2 H), 2.76 (t, J= 6.4 Hz, 2 H), 1.97 - 2.05 (m, 2 H); MS (EI) m/z 158 [M+.].
Example 9
6-[(3-Bromo-4-fluorophenyl)ethynyl]chromane

To a mixture of bis(triphenylphospine)palladium(II)dichloride (0.12 g, 0.17 mmol) and cuprous iodide (0.032 g, 0.17 mmol) in anhydrous tetrahydrofuran (40 mL) was triethylamine (11.4 mL, 81.7 mmol) added dropwise under an atmosphere of argon. A solution of 6-ethynylchromane (2.6 g, 16,6 mmol) in anhydrous tetrahydrofuran (10 mL) was added dropwise followed by addition of 2-bromo-l-fluoro-4-iodobenzene (5.0 g, 16.6
mmol). The reaction mixture was stirred at room temperature for 2 h. The crude mixture was diluted with ethyl acetate, washed with 1 M aqueous hydrochloric acid, water and saturated aqueous sodium hydrogen carbonate. The organic layer was dried over magnesium sulfate, filtered and concentrated. The residue was purified by column 5 chromatography, using heptane/ethyl acetate 15:1 to 10:1 as the eluent, to give 5.0 g (91% yield) of the title compound: 1H NMR (CDCl3) δ 7.71 (dd, J= 6.6, 2.02 Hz, 1 H), 7.38 - 7.44 (m, 1 H), 7.21 - 7.26 (m, 2 H), 7.09 (t, J= 8.5 Hz, 1 H), 6.77 (d, J= 8.1 Hz, 1 H), 4.22 (t, 2 H), 2.79 (t, J= 6.6 Hz, 2 H), 1.99 - 2.07 (m, 2 H); MS (APPI+) m/z 331.0, 333.0 [M+H]+. 0
Example 10 l-(3-Bromo-4-fluorophenyl)-2-(3,4-dihydro-2H-chromen-6-yl)ethane-l,2-dione

6-[(3-Bromo-4-fluorophenyl)ethynyl]chromane (5.O g, 15.1 mmol) and s palladium(H)dichloride (0.27 g, 1.5 mmol) were heated in dimethyl sulfoxide (70 mL) at 150 °C under an atmosphere of argon for 3 h. After cooling to room temperature water was added and the mixture was extracted with dichloromethane. The combined organic layers were washed with 1 M hydrochloric acid, water and saturated aqueous sodium carbonate solution, dried over magnesium sulfate, filtered and concentrated. The residue was purifiedo by column chromatography, using heptane/ethyl acetate 8:1 to 7: 1 as the eluent, to give 4.1 g (75% yield) of the title compound: 1H NMR (CDCl3) δ 8.23 (dd, J= 6.6, 2.27 Hz, 1 H), 7.90 - 7.97 (m, 1 H), 7.73 (dd, J= 8.6, 2.27 Hz, 1 H), 7.66 - 7.70 (m, 1 H), 7.24 (t, 1 H), 6.88 (d, J= 8.6 Hz, 1 H), 4.28 (t, 2 H), 2.82 (t, J= 6.4 Hz, 2 H), 1.99 - 2.10 (m, 2 H); MS (ES) m/z 363.0, 365.0 [M+H]+. 5
Example 11
5-(3-Bromo-4-fluorophenyl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-2- thioxoimidazolidin-4-one

To a solution of l-(3-bromo-4-fluorophenyl)-2-(3,4-dihydro-2H-chromen-6-yl)ethane-l,2- dione (4.1 g, 11.3 mmol) in dimethyl sulfoxide (75 mL) was methyl-2-thiourea (2.0 g, 22.6 mmol) added. The solution was heated at 100 °C for 5 min and aqueous potassium hydroxide (18.8 mL, 22.6 mmol, 1.2 M) was added dropwise. The reaction mixture was heated at 100 °C for another 10 min and then cooled to room temperature. The reaction mixture was diluted with water and the pΗ was adjusted to 4-5 by addition of aqueous hydrochloric acid (1 M). The mixture was extracted by dichloromethane. The combined organic layers were washed with water, dried over magnesium sulfate, filtrated and the solvent was evaporated. The residue was purified by column chromatography, using heptane/ethyl acetate 10:1 to 6:1 as the eluent, to give 4.5 g (92% yield) of the title compound: 1H NMR (CDCl3) δ 8.76 (br s, 1 H), 7.59 (dd, J= 6.4, 2.4 Hz, 1 H), 7.29 - 7.35 (m, 1 H), 7.11 (t, J= 8.5 Hz, 1 H), 6.91 - 6.97 (m, 2 H), 6.76 (d, J= 8.3 Hz, 1 H), 4.04 - 4.34 (m, 2 H), 3.32 (s, 3 H), 2.74 (t, J= 6.4 Hz, 2 H), 1.94 - 2.04 (m, 2 H); MS (ES) m/z 435.0, 437.0 [M+H]+.
Example 12
2-Amino-5-(3-bromo-4-fluorophenyl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one

Example 13
5'-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-inethyl-5-oxo-4,5-dihydro-lH- imidazoI-4-yl]-2'-fluorobiphenyl-3-carbonitrile

2-Amino-5-(3-bromo-4-fluorophenyl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one (0.080 g, 0.191 mmol), (3-cyanophenyl)boronic acid (0.040 g, 0.027 mmol), [1,1 -bis (diphenylphosphino) ferrocene]palladium(II) chloride dichloromethane adduct (0.016 g, 0.019 mmol) and potassium carbonate (0.079 g, 0.57 mmol) in a mixture of 1,2-dimethoxyethane, water and ethanol (6:3:1, 3 mL) was irradiated under an atmosphere of argon in a microwave at 130 0C for 15 min. When cooled to room temperature the mixture was filtered, concentrated and purified by preparative ΗPLC to give 0.034 g (42% yield) of the title compound: 1H NMR (CDCl3) δ 7.86 - 7.97 (m, 2 H), 7.76 - 7.83 (m, 1 H), 7.70 (t,J= 7.8 Hz, 1 H), 7.60 (dd, J= 7.7, 2.1
Hz, 1 H), 7.49 - 7.57 (m, 1 H), 7.30 (dd, J= 10.4, 8.8 Hz, 1 H), 7.07 - 7.13 (m, 2 H), 6.64 (d, J= 9.1 Hz, 1 H), 4.07 (t, 2 H), 2.96 (s, 3 H), 2.67 (t, J= 6.4 Hz, 2 H), 1.81 - 1.90 (m, 2 H); MS (ES) m/z 439.24 [M-H]".
Example 14
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(6-fluoro-3'-methoxybiphenyl-3-yI)-3- methyl-3,5-dihydro-4H-imidazol-4-one

The title compound was synthesized as described for Example 13 in 56% yield, starting from (3-methoxyphenyl)boronic acid: 1H NMR (DMSO-^6) δ 7.55 (dd, J= 7.6, 2.3 Hz, 1 H), 7.43 - 7.48 (m, 1 H), 7.40 (t, J= 8.0 Hz, 1 H), 7.23 (dd, J= 10.5, 8.7 Hz, 1 H), 7.08 - 7.13 (m, 2 H), 6.94 - 7.03 (m, 3 H), 6.64 (d, J= 9.1 Hz, 1 H), 4.07 (t, 2 H), 2.96 (s, 3 H), 2.67 (t, J= 6.4 Hz, 1 H), 1.81 - 1.90 (m, 2 H); MS (ES) m/z 444.28 [M-H]\
Example 15
2-Amino-5-(2',6-difluoro-3'-methoxybiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)- 3-methyl-3,5-dihydro-4H-imidazoI-4-one

Example 16 l-K/rj-l-Cyclopropylvmyll-l^l-benzodioxaborole

I0 Ethynylcyclopropane (500 mg, 7.56 mmol) and 1,3,2-benzodioxaborole (1.361 g, 11.35 mmol) were dissolved in tetrahydrofuran (35 mL) and heated at reflux for 3 h. Upon cooling to room temperature the solvent was evaporated and the residue purified by column chromatography, using 0-60% ethyl acetate in heptane as the eluent. Residual 1,3,2-benzodioxaborole was precipitated from methanol by the addition of heptane and is removed by filtration. The residue was concentrated to give 640 mg (46% yield) of the title compound: 1H NMR (CDCl3) δ 7.22 - 7.19 (m, 2 H), 7.08 -7.05 (m, 2 H), 6.46 (dd, J= 17.7, 9.5 Hz, 1 H), 5.85 (d, J= 17.7 Hz, 1 H), 1.73 - 1.62 (m, 1 H), 0.97 - 0.91 (m, 2 H), 0.69 - 0.63 (m, 2 H). 0 Example 17
2-Amino-5-{3-[(JE)-2-cycIopropylvinyl]-4-fluorophenyI}-5-(3,4-dihydro-2H-chromen- 6-yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one

Example 18
2-Amino-5-[3-(2-cyclopropylethyl)-4-fluorophenyl]-5-(3,4-dihydro-2H-chromen-6-yl)-
3-methyl-3,5-dihydro-4H-imidazoI-4-one

A mixture of 2-amino-5- {3-[(E)-2-cyclopropylvinyl]-4-fluorophenyl} -5-(3,4-dihydro-2H- chromen-6-yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one (0.054 g, 0.13 mmol) and palladium on carbon (0.020 g, 10% wt) in ethyl acetate was shaken under an atmosphere of hydrogen for 13 h. The reaction mixture was filtered through celite, concentrated and purified by preparative ΗPLC to give 0.030 g (55% yield ) of the title compound: 1H NMR
(DMSO-J6) δ 7.34 (dd, J= 7.6, 2.3 Hz, 1 H), 7.21 - 7.30 (m, 1 H), 6.99 - 7.08 (m, 3 H), 6.62 (d, J= 8.8 Hz, 1 H), 6.56 (br s, 2 H), 4.01 - 4.13 (m, 2 H), 2.95 (s, 3 H), 2.59 - 2.69 (m, 4 H), 1.82 - 1.90 (m, 2 H), 1.33 - 1.43 (m, 2 H), 0.58 - 0.69 (m, 1 H), 0.30 - 0.38 (m, 2 H), 0.06 - 0.03 (m, 2 H); MS (ES) m/z 408.12 [M+H]+.
Example 19 5-(l,3-Dithian-2-yI)-2,3-dihydro-l-benzofuran
Boron trifluoride-diethyl etherate (9.0 mL, 70.1 mmol) was added dropwise to a cooled (0 °C) solution of 2,3-dihydrobenzofuran-5-carboxaldehyde (2,3 mL, 18.2 mmol) and 1, 3- propanedithiol (5.1 mL, 18.2 mmol) in dichloromethane (40 mL). The resulting mixture was stirred at 0 °C for 2 h. Saturated aqueous sodium bicarbonate was added and the aqueous phase was extracted with dichloromethane. The combined organic phases were washed with water, aqueous potassium hydroxide (10%) and water, dried over magnesium sulfate and the solvent was evaporated to give 4.4 g (100% yield) of the crude title compound: 1HNMR (CDCl3) δ 7.33 (s, 1 H), 7.20 (dd, J= 8.2, 1.9 Hz, 1 H), 6.73 (d, J= 8.3 Hz, 1 H), 4.57 (t, J= 8.7 Hz, 2 H), 3.20 (t, J= 8.7 Hz, 2 H), 3.00 - 3.11 (m, 2 H), 2.86 - 2.94 (m, 2 H), 2.11 - 2.22 (m, 1 H), 1.84 - 1.98 (m, 1 H); HR MS (ES) m/z 239.0559 [M+H]+.
Example 20
(3-Bromo-4-fluorophenyl)[2-(2,3-dihydro-l-benzofuran-5-yl)-l,3-dithian-2- yl] methanol

5 combined organic layers were dried over magnesium sulfate and the solvent was evaporated. The residue was purified by column chromatography, using heptane/ethyl acetate 5: 1 to 4: 1 as the eluent, to give 5.8 g (72% yield) of the title compound: 1H NMR (CDCl3) δ 7.46 (dd, J= 8.5, 2.1 Hz, 1 H), 7.40 (s, 1 H), 6.97 (dd, J= 6.8, 2.0 Hz, 1 H), 6.83 - 6.92 (m, 2 H), 6.74 (d, J= 8.6 Hz, 1 H), 4.88 (d, J= 3.0 Hz, 1 H), 4.62 (t, J= 8.6o Hz, 2 H), 3.17 (t, J= 8.7 Hz, 2 H), 2.65 - 2.82 (m, 4 H), 1.88 - 2.00 (m, 2 H); MS (ES) m/z 423.14, 425.14 [M-18]+.
Example 21 l-(3-Bromo-4-fluorophenyl)-2-(2,3-dihydro-l-benzofuran-5-yl)ethane-l,2-dione

Dess-Martin periodinane (14.0 g, 32.8 mmol) was added to a solution of (3-bromo-4- fluorophenyl)[2-(2,3-dihydro-l-benzofuran-5-yl)-l,3-dithian-2-yl]methanol (5.9 g, 13.1 mmol) and fer/-butanol (4.2 mL, 45.8 mmol) in dichloromethane (250 mL) under an atmosphere of argon and the reaction mixture was stirred over night. A solution of sodiumo thiosulfate (12.5 g) in saturated aqueous sodium hydrogencarbonate (200 mL) was added and the resulting mixture was stirred for 30 min. Dichloromethane was added and the organic phase was separated. The aqueous phase was extracted with dichloromethane and the combined organic phases were dried over magnesium sulfate and the solvent was evaporated. The residue was purified by column chromatography, using heptane/ethyl5 acetate 10:1 to 6:1 as the eluent, to give 3.5 g (76% yield) of the title compound: 1H NMR (CDCl3) δ 8.23 (dd, J= 6.6, 2.3 Hz, 1 H), 7.90 - 7.97 (m, 1 H), 7.85 (d, J= 1.3 Hz, 1 H), 7.79 (dd, J= 8.5, 1.9 Hz, 1 H), 7.21 - 7.27 (m, 1 H), 6.86 (d, J= 8.6 Hz, 1 H), 4.71 (t, J= 8.8 Hz, 2 H), 3.27 (t, J= 8.7 Hz, 2 H); MS (ES) m/z 349.0, 351.0 [M+H]+.
Example 22
5-(3-Bromo-4-fluorophenyl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyI-2- thioxoimidazolidin-4-one

Example 23 2-Amino-5-(3-bromo-4-fluorophenyI)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl- 3,5-dihydro-4H-imidazol-4-one
 tert-Bntyl hydroperoxide (23.2 niL, 150.0 mmol, 70 wt% aq) was added to a solution of 5- (3-bromo-4-fluorophenyl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-2- thioxoimidazolidin-4-one (4.2 g, 10.0 mmol) in a mixture of methanol (60 mL) and ammonium hydroxide (20 mL). The reaction was stirred for 4 h at room temperature, concentrated and the residue was dissolved in chloroform. The organic phase was washed with water and dried over magnesium sulfate, filtered and the solvent was evaporated. The residue was purified by column chromatography, using ethyl acetate/methanol 30:1 to 20:1 + 1% triethylamine as the eluent, to give 2.3 g (57% yield) of the title compound: 1H NMR (CDCl3) δ 7.70 (dd, J= 6.7, 2.4 Hz, 1 H), 7.37 - 7.43 (m, 1 H), 7.18 (s, 1 H), 7.13 (dd, J= 8.5, 2.1 Hz, 1 H), 7.03 (t, J= 8.5 Hz, 1 H), 6.70 (d, J= 8.3 Hz, 1 H), 4.98 (br s, 2 H), 4.55 (t, J= 8.7 Hz, 2 H), 3.14 (t, J= 8.5 Hz, 2 H), 3.10 (s, 3 H); MS (ES) m/z 402.19, 404.18 [M-H]".
tert-Bntyl hydroperoxide (23.2 niL, 150.0 mmol, 70 wt% aq) was added to a solution of 5- (3-bromo-4-fluorophenyl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-2- thioxoimidazolidin-4-one (4.2 g, 10.0 mmol) in a mixture of methanol (60 mL) and ammonium hydroxide (20 mL). The reaction was stirred for 4 h at room temperature, concentrated and the residue was dissolved in chloroform. The organic phase was washed with water and dried over magnesium sulfate, filtered and the solvent was evaporated. The residue was purified by column chromatography, using ethyl acetate/methanol 30:1 to 20:1 + 1% triethylamine as the eluent, to give 2.3 g (57% yield) of the title compound: 1H NMR (CDCl3) δ 7.70 (dd, J= 6.7, 2.4 Hz, 1 H), 7.37 - 7.43 (m, 1 H), 7.18 (s, 1 H), 7.13 (dd, J= 8.5, 2.1 Hz, 1 H), 7.03 (t, J= 8.5 Hz, 1 H), 6.70 (d, J= 8.3 Hz, 1 H), 4.98 (br s, 2 H), 4.55 (t, J= 8.7 Hz, 2 H), 3.14 (t, J= 8.5 Hz, 2 H), 3.10 (s, 3 H); MS (ES) m/z 402.19, 404.18 [M-H]".
Example 24
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(6-fluoro-3'-methoxybiphenyI-3-yl)-3- methyl-3,5-dihydro-42/-imidazoI-4-one

The title compound was synthesized as described for Example 13 in 56% yield, starting from (3-methoxyphenyl)boronic acid: 1H NMR (DMSO-cfe) δ 7.55 (dd, J = 7.6, 2.27 Hz, 1
H), 7.43 - 7.50 (m, 1 H), 7.40 (t, J= 8.0 Hz, 1 H), 7.20 - 7.30 (m, 2 H), 7.14 (d, J= 8.3 Hz, 1 H), 6.94 - 7.05 (m, 3 H), 6.68 (d, J= 8.3 Hz, 1 H), 4.47 (t, J= 8.7 Hz, 2 H), 3.78 (s, 3 H), 3.11 (t, J= 8.7 Hz, 2 H), 2.98 (s, 3 H); MS (ES) m/z 430.16 [M-H]".
Example 25
5'-[2-Amino-4-(2,3-dihydro-l-benzofuran-5-yl)-l-methyl-5-oxo-4,5-dihydro-lH- imidazoI-4-yl]-2'-fluorobiphenyl-3-carbonitrile

The title compound was synthesized as described for Example 13 in 33% yield starting from (3-cyanophenyl)boronic acid. 1H NMR (DMSO-^6) δ 7.93 (s, 1 H), 7.87 - 7.92 (m, 1 H), 7.76 - 7.84 (m, 1 H), 7.70 (t, J= 7.7 Hz, 1 H), 7.61 (dd, J= 7.6, 2.27 Hz, 1 H), 7.50 - 7.58 (m, 1 H), 7.24 - 7.36 (m, 2 H), 7.14 (d, J= 8.1 Hz, 1 H), 6.67 (d, J= 8.3 Hz, 1 H), 4.47 (t, J= 8.7 Hz, 2 H), 3.11 (t, J= 8.7 Hz, 2 H), 2.97 (s, 3 H); MS (ES) m/z 425.28 [M- H]-.
Example 26
2-Amino-5-(2',6-difluoro-3'-methoxybiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5- yO-S-methyl-SjS-dihydro^iϊ-imidazoM-one

Example 27 l-Amino-S-IS-KJE^-l-cyclopropylvinyll^-fluorophenyll-S-CljS-dihydro-l-benzofuran-
S-yty-S-methyl-SjS-dihydro^/Z-imidazoM-one

The title compound was synthesized as described for Example 17 in 73% yield, starting from 2-amino-5-(3-bromo-4-fluorophenyl)-5-(2,3-dihydro- 1 -benzofuran-5-yl)-3-methyl- 3,5-dihydro-4H-imidazol-4-one: 1H NMR (CDCl3) δ 7.51 (dd, J= 7.2, 2.4 Hz, 1 H), 7.20 7.26 (m, 1 H), 7.15 (s, 1 H), 7.11 (dd, J= 8.3, 2.0 Hz, 1 H), 6.93 (dd, J= 10.4, 8.6 Hz, 1 H), 6.69 (d, J= 8.3 Hz, 1 H), 6.54 (d, J= 15.9 Hz, 1 H), 5.79 (dd, J= 15.9, 9.1 Hz, 1 H), 5.00 (br s, 2 H), 4.54 (t, J= 8.7 Hz, 2 H), 3.02 - 3.22 (m, 5 H), 1.49 - 1.62 (m, 1 H), 0.77 - 0.86 (m, 2 H), 0.44 - 0.56 (m, 2 H); MS (ES) m/z 390.25 [M-H]".
Example 28 2-Amino-5-[3-(2-cyclopropylethyl)-4-fluorophenyl]-5-(2,3-dihydro-l-benzofuran-5- yO-S-methyl-SjS-dihydro^H-imidazoW-one
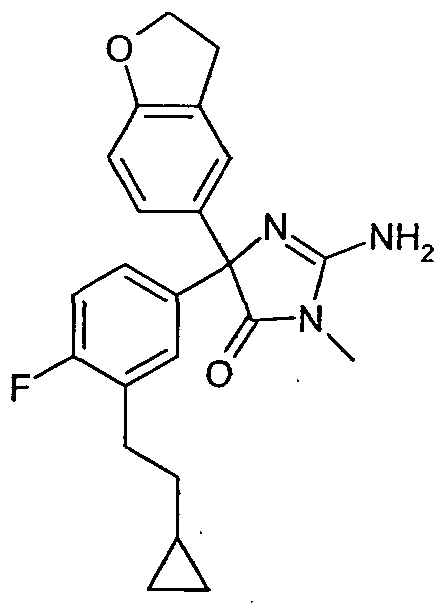
The title compound was synthesized as described for Example 18 in 43% yield, starting from 2-amino-5- {3-[(£)-2-cyclopropylvinyl]-4-fluorophenyl} -5-(2,3-dihydro-l - benzofuran-5-yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one: 1H NMR (DMSO-J6) δ 7.34 (dd, J = 7.6, 2.3 Hz, 1 H), 7.23 - 7.31 (m, 1 H), 7.19 (s, 1 H), 6.98 - 7.13 (m, 2 H), 6.64 (d, J= 8.3 Hz, 1 H), 6.58 (br s, 2 H), 4.46 (t, J= 8.7 Hz, 2 H), 3.09 (t, J= 8.7 Hz, 2 H), 2.95 (s, 3 H), 2.62 (t, J= 7.6 Hz, 2 H), 1.33 - 1.44 (m, 2 H), 0.55 - 0.70 (m, 1 H), 0.29 - 0.38 (m, 2 H), 0.07 - 0.04 (m, 2 H); MS (ES) m/z 392.30 [M-H]".
Example 29
4-Methoxy-2-(4,4,5,5-tetramethyl-l,3,2-dioxaboroIan-2-yl)benzonitriIe

A mixture of tris(dibenzylideneacetone)dipalladium (33 mg, 0.0357 mmol) and tricyclohexylphosphine (47 mg, 0167 mmol) in anhydrous dioxane (2.5 mL) was stirred for 30 min at room temperature under an argon atmosphere. 4,4,4',4',5,5,5',5'-Octamethyl-2,2'- bi-l,3,2-dioxaborolane (333 mg, 1.31 mmol) potassium acetate (175 mg, 1.785 mmol) and a solution of 2-chloro-4-methoxybenzonitrile (200 mg, 1.19 mmol) in anhydrous dioxane (2.5 mL) were sequentially added and the resulting mixture was irradiated in a microwave at 120 0C for 15 h. Concentration of the reaction mixture and purification by column chromatography, using a gradient of 0-30 % ethyl acetate in heptane as the eluent, gave 238 mg (77% yield) of the title compound: 1H NMR (CDCl3) δ 7.63 (d, J= 8.6 Hz, 1 H),
7.35 (d, J= 2.8 Hz, 1 H), 7.01 (dd,J= 8.6, 2.8 Hz, 1 H), 3.88 (s, 3 H), 1.39 (s, 12 H); MS (EI) m/z 259 [M+*].
Example 30 2-Chloro-4-methoxy-l-(trifluoromethoxy)benzene

A mixture of 3-chloro-4-(trifluoromethoxy)phenol (1.00 g, 4.71 mmol), dimethylsulfate (490 μL, 5.18 mmol), tetrabutylammonium sulfate, 2 M aqueous sodium hydroxide (2.59 mL, 5.18 mmol) and dichloromethane (25 mL) was vigorously stirred at room temperature for 18 h. The mixture was diluted with dichloromethane, washed with water, dried over sodium sulfate and concentrated in vacuo to give 780 mg (73% yield) of the title compound: 1H NMR (CDCl3) δ 7.22 - 7.26 (m, 1 H), 6.99 (d, J= 2.8 Hz, 1 H), 6.81 (dd, J = 9.1, 3.0 Hz, 1 H), 3.82 (s, 3 H).
Example 31
2-[5-Methoxy-2-(trifluoromethoxy)phenyl]-4,4,5,5-tetramethyI-l,3,2-dioxaborolane

The title compound was synthesized as described for Example 29 in 69% yield, starting from 2-chloro-4-methoxy-l-(trifluoromethoxy)benzene: 1H NMR (CDCl3) δ 7.25 (d, J= 3.3 Hz, 1 H), 7.16 (m, 1 H), 6.97 (dd, J= 9.0, 3.2 Hz, 1 H), 3.83 (s, 3 H), 1.36 (s, 12 H); MS (EI) m/z 318 [M+.].
Example 32 5-[(3-Bromophenyl)ethynyl]-2,3-dihydro-l-benzofuran

To a mixture of bis(triphenylphosphine)palladium(ll) dichloride (154 mg, 0.219 mmol) and copper iodide (42 mg, 0.219 mmol) in anhydrous tetrahydrofuran (50 mL) under an atmosphere of argon was sequentially added triethylamine (15 mL), a solution of 5-
5 ethynyl-2,3-dihydro-l-benzofuran (3.151 g, 21.86 mmol; described in: Walser A. et. al. J. Med Chem. 1991, 34, 1440-46) in anhydrous tetrahydrofuran (10 mL) and finally 3- bromo-1-iodobenzene (6.18 g, 21.86 mmol). The resulting solution was stirred at room temperature for 20 h. The crude mixture was diluted with ethyl acetate, washed sequentially with 1 M hydrochloric acid, water and a saturated sodium hydrogen carbonateo solution and dried over magnesium sulfate. The solvent was evaporated to give a solid material which was suspended in hexane, the solid was filtered, washed with hexane and dried in vacuo to give 5.30 g (81% yield) of the title compound: 1H NMR (CDCl3) δ 7.66 (m, 1 H), 7.43 (m, 2 H), 7.37 (m, 1 H), 7.31 (m, 1 H), 7.20 (t, J= 7.8 Hz, 1 H), 6.77 (d, J= 8.3, 1 H), 4.62 (t, J= 8.7 Hz, 2 H), 3.23 (t, J= 8.7 Hz, 2 H); MS (EI) m/z 298, 300 [M+.].5
Example 33 l-(3-Bromophenyl)-2-(2,3-dihydro-l-benzofuran-5-yl)ethane-l,2-dione

Example 34
5-(3-BromophenyI)-5-(2,3-dihydro-l-benzofuran-5-yI)-3-methyI-2-thioxoimidazolidin- 4-one

To a solution of l-(3-bromophenyl)-2-(2,3-dmydro-l-benzofuran-5-yl)ethane-l,2-dione (4.34 g, 13.11 mmol) in dimethyl sulfoxide (50 mL) was added N-methylthiourea (2.36 g, 26.21 mmol). The solution was heated at 100 °C for 5 min and then 1.2 M aqueous potassium hydroxide (22.4 mL, 26.82 mmol) was added dropwise over a period of 6-7 min. The mixture was heated for another 10 min and then cooled to room temperature. Water was added and the pH was adjusted to 5 by addition of 1 M hydrochloric acid. The mixture was extracted with dichloromethane. The combined extracts were washed twice with water, dried over sodium sulfate and evaporated to give 5.48 g (100% yield) of the title compound: 1H NMR (CDCl3) δ 7.72 (br s, 1 H), 7.49 - 7.53 (m, 2 H), 7.29 - 7.33 (m, 1 H), 7.24 - 7.28 (m, 1 H), 7.07 - 7.09 (m, 1 H), 6.97 - 7.01 (m, 1 H), 6.76 (d, J= 8.3 Hz, 1 H), 4.60 (t, J= 8.7 Hz, 2 H), 3.34 (s, 3 H), 3.20 (t, J= 8.7 Hz, 2 H); MS (ESI) m/z 401, 403 [M-H]-.
Example 35
2-Amino-5-(3-bromophenyI)-5-(2,3-dihydro-l-benzofuran-5-yI)-3-methyl-3,5- dihydro-4£T-imidazol-4-one

Example 36 3'-[2-Amino-4-(2,3-dihydro-l-benzofuran-5-yI)-l-methyl-5-oxo-4,5-dihydro-lH- imidazol-4-yl]-5-methoxybiphenyl-2-carbonitrile hydrochloride

A mixture of 2-amino-5-(3-bromophenyl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl- 3,5-dihydro-4H-imidazol-4-one (66 mg, 0.171 mmol), 4-methoxy-2-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)benzonitrile (58 mg, 0.222 mmol), [1,1'- bis(diphenylphosphino)ferrocene]palladium(II) dichloride dichloromethane adduct (14 mg, 0.017 mmol) and cesium carbonate (167 mg, 0.513 mmol) in a mixture of 1,2- dimethoxy ethane, water and ethanol (6:3:1, 5 mL) was irradiated in a microwave at 130 0C for 30 min, under an argon atmosphere. After cooling to room temperature the mixture was filtered through a plug of Silica, concentrated in vacuo and purified by preparative ΗPLC. The resulting residue was dissolved in hydrochloric acid (2 M in diethyl ether) and
dichloromethane. The solvents were evaporated to give 23 mg (31% yield) of the title compound: 1H NMR (DMSCM;) δ 7.86 (d, J= 8.6 Hz, 1 H), 7.69 (br s, 1 H), 7.51 - 7.56 (m, 1 H), 7.42 - 7.47 (m, 2 H), 7.32 (br s, 1 H), 7.18 - 7.22 (m, 1 H), 7.13 (dd, J= 8.6, 2.5 Hz, 1 H), 7.02 (d, J= 2.5 Hz, 1 H), 6.66 (d, J= 8.3 Hz, 1 H), 4.47 (t, J= 8.7 Hz, 2 H), 3.88 (S5 3 H), 3.11 (t, J= 8.7 Hz, 2 H), 2.97 (s, 3 H); MS (ESI) m/z 439.5
Example 37
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-[5'-methoxy-2'- (trifluoromethoxy)biphenyl-3-yl]-3-methyI-3,5-dihydro-4Hr-imidazol-4-one hydrochloride

The title compound was synthesized as described for Example 36 in 45% yield, starting from 2-[5-methoxy-2-(trifluoromethoxy)phenyl]-4,4,5,5-tetramethyl- 1,3 ,2-dioxaborolane: 1H NMR (free base) (CDCl3) δ 7.59 - 7.61 (m, 1 H), 7.45 - 7.50 (m, 1 H), 7.34 - 7.40 (m, 2 H), 7.23 - 7.25 (m, 1 H), 7.21 - 7.23 (m, 1 H), 7.16 - 7.20 (m, 1 H), 6.90 (d, J= 3.0 Hz, 1 H), 6.86 (dd, J= 9.0, 3.2 Hz, 1 H), 6.70 (d, J= 8.6 Hz, 1 H), 4.53 (t, J= 8.7 Hz, 2 H), 3.81 (s, 3 H), 3.10 (s, 3 H), 3.13 (d, J= 8.6 Hz, 3 H); MS (ESI) m/z 498.4 [M+H]+, 496.4 [M- H]--
Example 38
2-Amino-5-(3',5'-dichlorobiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl- 3,5-dihydro-4H-imidazol-4-one hydrochloride

The title compound was synthesized as described for Example 36 in 55% yield, starting from 3,5-dichlorophenylboronic acid: 1H NMR (free base) (CDCl3) δ 7.53 - 7.55 (m, 1 H), 7.50 - 7.53 (m, 1 H), 7.33 - 7.42 (m, 3 H), 7.32 (d, J= 2.5 Hz, 1 H), 7.26 - 7.28 (m, 1 H), 7.22 - 7.26 (m, 1 H), 7.18 - 7.22 (m, 1 H), 6.72 (d, J= 8.6 Hz, 1 H), 4.55 (t, J= 8.7 Hz, 2 H), 3.12 - 3.19 (m, 5 H); MS (ESI) m/z 452, 454, 456 [M+H]+, 450, 452, 454 [M-H]".
Example 39 6-[(3-Bromophenyl)ethynyl]chromane

The title compound was synthesized as described for Example 32 in 92% yield, starting from 6-iodochromane (described in: Togo H. et.al. J. Chem. Soc. Perkin Trans. 1, 1997, 787-793) and (3-bromophenyl)acetylene. Purification by column chromatography, using a gradient with increasing concentration of ethyl acetate in heptane (0 - 30 %), as the eluent: 1H NMR (CDCl3) δ 7.66 (t, J= 1.8 Hz, 1 H), 7.40 - 7.45 (m, 2 H), 7.23 - 7.27 (m, 2 H), 7.20 (t, J= 8.9 Hz, 1 H), 6.77 (d, J= 8.3 Hz, 1 H), 4.20 - 4.24 (m, 2 H), 2.79 (t, J= 6.6 Hz, 2 H), 1.99 - 2.06 (m, 2 H); MS (EI) m/z 312, 314 [M+.].
Example 40 l-(3-Bromophenyl)-2-(3,4-dihydro-2H-chromen-6-yl)ethane-l,2-dione

The title compound was synthesized as described for Example 33 in 73% yield, starting from 6-[(3-bromophenyi)ethynyl]chromane: 1H NMR (CDCl3) δ 8.12 (t, J= 1.6 Hz, 1 H), 7.89 (m, 1 H), 7.77 (m, 1 H), 7.73 (dd, J= 8.6, 2.3 Hz, 1 H), 7.69 (m, 1 H), 7.39 (t, J= 7.8 Hz, 1 H), 6.88 (d, J= 8.6 Hz, 1 H), 4.29 (m, 2 H) 2.83 (t, J= 6.4 Hz, 2 H), 2.05 (m, 2 H); MS (ESI) m/z 345.2, 347.3 [M+H]+.
Example 41
5-(3-BromophenyI)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-2-thioxoimidazoIidin-
4-one

The title compound was synthesized as described for Example 34 in 100% yield, starting from l-(3-bromophenyl)-2-(3,4-dihydro-2H-chromen-6-yl)ethane-l,2-dione: 1H NMR (CDCl3) δ 7.62 (br s, 1 H), 7.49 - 7.53 (m, 2 H), 7.28 - 7.32 (m, 1 H), 7.26 (t, J= 8.0 Hz, 1 H), 6.91 - 6.95 (m, 2 H), 6.76 - 6.80 (m, 1 H), 4.17 - 4.21 (m, 2 H), 3.33 (s, 3 H), 2.75 (t, J = 6.4 Hz, 2 H), 1.97 - 2.04 (m, 2 H); MS (ESI) m/z 415.3, 417.3 [M-H]".
Example 42 2-Amino-5-(3-bromophenyl)-5-(3,4-dihydro-2//-chromen-6-yI)-3-methyl-3,5-dihydro- 4H-imidazoI-4-one

Example 43
3^2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-inethyI-5-oxo-4,5-dihydro-lHr- imidazoI-4-yl]-5-methoxybiphenyl-2-carbonitriIe

The title compound was synthesized as described for Example 36 in 3% yield, starting from 2-arnino-5-(3-bromophenyl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one and 4-methoxy-2-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2- yl)benzonitrile without formation of the hydrochloric salt: 1H NMR (DMSO-J6) δ 7.86 (d, J= 8.8 Hz, 1 H), 7.68 - 7.70 (m, 1 H), 7.51 - 7.56 (m, 1 H), 7.41 - 7.47 (m, 2 H), 7.11 - 7.18 (m, 3 H), 7.02 (d, J= 2.5 Hz, 1 H), 6.61 - 6.65 (m, 1 H), 4.05 - 4.09 (m, 2 H), 3.88 (s, 3 H), 2.96 (s, 3 H), 2.66 (t, J= 6.4 Hz, 2 H), 1.83 - 1.90 (m, 2 H); MS (ESI) m/z 439.5 [M+H]+, 437.5 [M-H]".
Example 44
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-[5'-methoxy-2'- (trifluoromethoxy)biphenyl-3-yI]-3-methyl-3,5-dihydro-4H-imidazoI-4-one hydrochloride

The title compound was synthesized as described for Example 36 in 35% yield, starting from 2-amino-5-(3-bromophenyl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one and 2-[5-methoxy-2-(trifluoromethoxy)phenyl]-4 ,4,5,5- tetramethyl-l,3,2-dioxaborolane: 1R NMR (free base) (CDCl3) δ 7.59 - 7.61 (m, 1 Η), 7.47 - 7.50 (m, 1 Η), 7.35 - 7.41 (m, 2 Η), 7.21 - 7.25 (m, 1 Η), 7.12 (dd, J= 8.5, 2.4 Hz, 1 H), 7.07 - 7.09 (m, 1 H), 6.90 (d, J= 3.0 Hz, 1 H), 6.84 - 6.88 (m, 1 H), 6.72 (d, J= 8.6 Hz, 1 H), 4.13 - 4.17 (m, 2 H), 3.82 (s, 3 H), 3.11 (s, 3 H), 2.71 (t, J= 6.4 Hz, 2 H), 1.93 - 1.99 (m, 2 H); MS (ESI) m/z 512.4 [M+H]+, 510.4 [M-H]".
Example 45 2-Amino-5-(3',5'-dichlorobiphenyI-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-
3,5-dihydro-4iϊ-imidazol-4-one hydrochloride

The title compound was synthesized as described for Example 36 in 51% yield, stating from 2-amino-5-(3-bromophenyl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one and 3,5-dichlorophenylboronic acid: 1H NMR (CDCl3) δ 7.54 - 7.55 (m, 1 H), 7.50 - 7.53 (m, 1 H), 7.35 - 7.41 (m, 2 H), 7.32 - 7.35 (m, 1 H), 7.31 (d, J = 2.5 Hz, 1 H), 7.24 (dd, J= 8.6, 2.5 Hz, 1 H), 7.11 - 7.15 (m, 2 H), 6.73 (d, J= 8.6 Hz, 1 H),
4.14 - 4.17 (m, 2 H), 3.12 (s, 3 H), 2.71 - 2.75 (m, 2 H), 1.94 - 2.00 (m, 2 H); MS (ESI) m/z 466.3, 468.3, 470.3 [M+H]+, 464.5, 466.5, 468.5 [M-H]".
Example 46 5-(2'-AcetyI-5'-methoxybiphenyl-3-yl)-2-amino-5-(2,3-dihydro-l-benzofuran-5-yI)-3- methyl-3,5-dihydro-47Z-imidazol-4-one

A mixture of tris(dibenzylideneacetone)dipalladium (50 mg, 0.054 mmol), tricyclohexylphosphine (70 mg, 0.252 mmol), octamethyl-2,2'-bi-l,3,2-dioxaborolane (503 mg, 1.98 mmol), potassium acetate (265 mg, 2.70 mmol) and 2-chloro-4- methoxyacetophenone (330 mg, 1.79 mmol) in anhydrous dioxane (5 mL) was irradiated in a microwave at 130 °C for 5 h under an atmosphere of argon. The cooled mixture was filtered through a plug of silica and concentrated. A mixture of one third of the above material, 2-amino-5-(3-bromophenyl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one (95 mg, 0.246 mmol), [1,1'- bis(diphenylphosphino)ferrocene]palladium(II) chloride dichloromethane adduct (20 mg, 0.025 mmol), cesium carbonate (240 mg, 0.738 mmol), 1,2-dimethoxyethane (3 mL), water (1.5 mL) and ethanol (0.5 mL) was irradiated in a microwave at 130 0C for 30 min under an atmosphere of argon. The cooled mixture was filtered, concentrated and purified by preparative ΗPLC to give 17 mg ( 15% yield) of the title compound: 1H NMR (CDCl3) δ 7.58 (m, 2 H), 7.40 (t, J= 7.7 Hz, 1 H), 7.30 (m, 3 H), 7.22 (m, 1 H), 6.89 (dd, J= 8.6, 2.5 Hz, 1 H), 6.83 (d, J= 2.5 Hz, 1 H), 6.70 (d, J= 8.3 Hz, 1 H), 4.54 (t, J= 8.7 Hz, 2 H), 3.85 (s, 3 H), 3.16 (m, 5 H), 1.91 (s, 3 H); (ES) m/z 456.0 [M+H]+, 454.2 [M-H]".
Example 47
5-(2'-Acetyl-5t-methoxybiphenyl-3-yl)-2-amino-5-(3,4-dihydro-2H-chromen-6-yl)-3- methyl-3,5-dihydro-4H-imidazoI-4-one

The title compound was synthesized as described for 5-(2'-acetyl-5'-methoxybiphenyl-3- 5 yl)-2-amino-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one in 20% yield, starting from 2-amino-5-(3-bromophenyl)-5-(3,4-dihydro-2H-chromen-6-yl)- 3-methyl-3,5-dihydro-4H-imidazol-4-one: 1H NMR (CDCl3) δ 7.58 (m, 2 Η), 7.39 (t, J = 7.7 Hz, 1 H), 7.33 (m, 1 H), 7.28 (m, 1 H), 7.15 (m, 2 H), 6.89 (dd, J= 8.6, 2.8 Hz, 1 H), 6.83 (d, J= 2.5 Hz, 1 H), 6.71 (m, 1 H), 4.14 (m, 2 H), 3.85 (s, 3 H), 3.15 (s, 3 H), 2.73 (m,o 2 H), 1.96 (m, 2 H), 1.91 (s, 3 H); (ES) m/z 470.0 [M+H]+, 468.2 [M-H]".
Example 48 5-(l,3-Dithian-2-yI)-2,3-dihydro-l-benzofuran-7-carbonitrile

Example 49
5 5-{2-[(3-Bromo-4-fluorophenyI)(hydroxy)methyl]-l,3-dithian-2-yl}-2,3-dihydro-l- benzofuran-7-carbonitrile

5-(l,3-Dithian-2-yl)-2,3-dihydro-l-benzofuran-7-carbonitrile (0.2 g, 0.76 mmol) was dissolved in anhydrous tetrahydrofuran (4 mL). Lithium diisopropylamide (2 M in
I0 tetrahydrofuran, 0.57 mL, 0.83 mmol) was added over 15 min at 0 °C. 3-Bromo-4- fluorobenzaldehyde (0.16 g, 0.83 mmol) was added and the reaction mixture was stirred at 0 0C for 3 h. Saturated ammonium chloride was added and the mixture was extracted with dichloromethane. The combined organic phases were washed with brine, dried over magnesium sulfate and concentrated in vacuo. Purification by column chromatography, is using ethyl acetate/heptane (5-50%) as the eluent, gave 0.14 g (40% yield) of the title compound: 1H NMR (CDCl3) δ 7.76 (d, J= 2.0 Hz, 1 H), 7.53 - 7.56 (m, 1 H), 6.96 - 6.99 (m, 1 H), 6.94 (d, J= 8.3 Hz, 1 H), 6.83 - 6.89 (m, 1 H), 4.91 (s, 1 H), 4.80 (t, J= 8.7 Hz, 2 H), 3.24 (t, J= 8.7 Hz, 2 H), 2.94 (br s, 1 H), 2.73 - 2.82 (m, 2 H), 2.55 - 2.70 (m, 2 H), 1.87 - 2.02 (m, 2 H).
20
Example 50 5-[(3-Bromo-4-fluorophenyl)(oxo)acetyI]-2,3-dihydro-l-benzofuran-7-carbonitrile

Dess-Martin periodinane (1.3 g, 3.08 mmol) was added to a solution of 5-{2-[(3-bromo-4- fluorophenyl)(hydroxy)methyl]-l ,3-dithian-2-yl} -2,3-dihydro- 1 -benzofuran-7-carbonitrile (0.64 g, 1.37 mmol) and tert-butanol (0.36 g, 4.80 mmol) in dichloromethane (10 mL) under an atmosphere of argon and the reaction mixture was stirred overnight. Aqueous sodium thiosulfate (2.5 g) was added and the resulting mixture was stirred for 30 min. Dichloromethane was added and the aqueous phase was extracted with dichloromethane. The combined organic phases were dried over magnesium sulfate and the solvent was evaporated. Purification by column chromatography, using ethyl acetate/heptane (5-50%) as the eluent, gave 0.45 g (88% yield) of the title compound: 1H NMR (CDCl3) δ 8.23 (dd, J= 6.4, 2.1 Hz, 1 H), 8.00 - 8.09 (m, 2 H), 7.93 - 7.98 (m, 1 H), 7.44 - 7.49 (m, 1 H ), 4.91 (t, J= 8.8 Hz, 2 H), 3.39 (t, J= 8.7 Hz, 2 H).
Example 51 5-[2-Amino-4-(3-bromo-4-fluorophenyl)-l-methyI-5-oxo-4,5-dihydro-ljy-imidazol-4- yl]-2,3-dihydro-l-benzofuran-7-carbonitrile

5-[(3-Bromo-4-fluorophenyl)(oxo)acetyl]-2,3-dihydro-l-benzofuran-7-carbonitrile (0.45 g, 1.21 mmol) and 1-methylguanidine hydrochloride (0.61 g, 5.57 mmol) was dissolved in dioxane/ethanol (1:1, 20 mL) and stirred at room temperature for 15 min. Sodium carbonate (0.59 g, 5.57 mmol) and water (2.8 mL) was added and the reaction was heated at 85 °C for 45 min. The reaction mixture was concentrated in vacuo and the residue was
dissolved in dichloromethane and water. The phases were separated and the organic phase was washed with brine, dried over magnesium sulfate and concentrated in vacuo. Purification by column chromatography, using methanol/ethyl acetate (5-50% + 1% triethylamine) as the eluent, gave 0.34 g (65% yield) of the title compound: 1H NMR (CD3OD) δ 7.60 (dd, J= 6.6, 2.3 Hz5 1 H), 7.44 (d, J= 1.3 Hz, 1 H), 7.35 - 7.41 (m, 2 H), 7.18 (t, J= 8.6 Hz, 1 H), 4.73 (t, J= 8.7 Hz, 2 H), 3.25 (t, J= 8.7 Hz, 2 H), 3.11 (s, 3 H); MS (ES) m/z 429 [M-H]".
Example 52

Table 1 : Representative examples synthesized as described for 5-[2-Amino-4-(4-fluoro-3- pyridin-3-ylphenyl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-4-yl]-2,3-dihydro-l- benzofuran-7-carbonitrile. AU the reactions were analyzed using LC-MS and those which showed a low level of conversion was irradiated in a microwave for another hour at 130 0C.
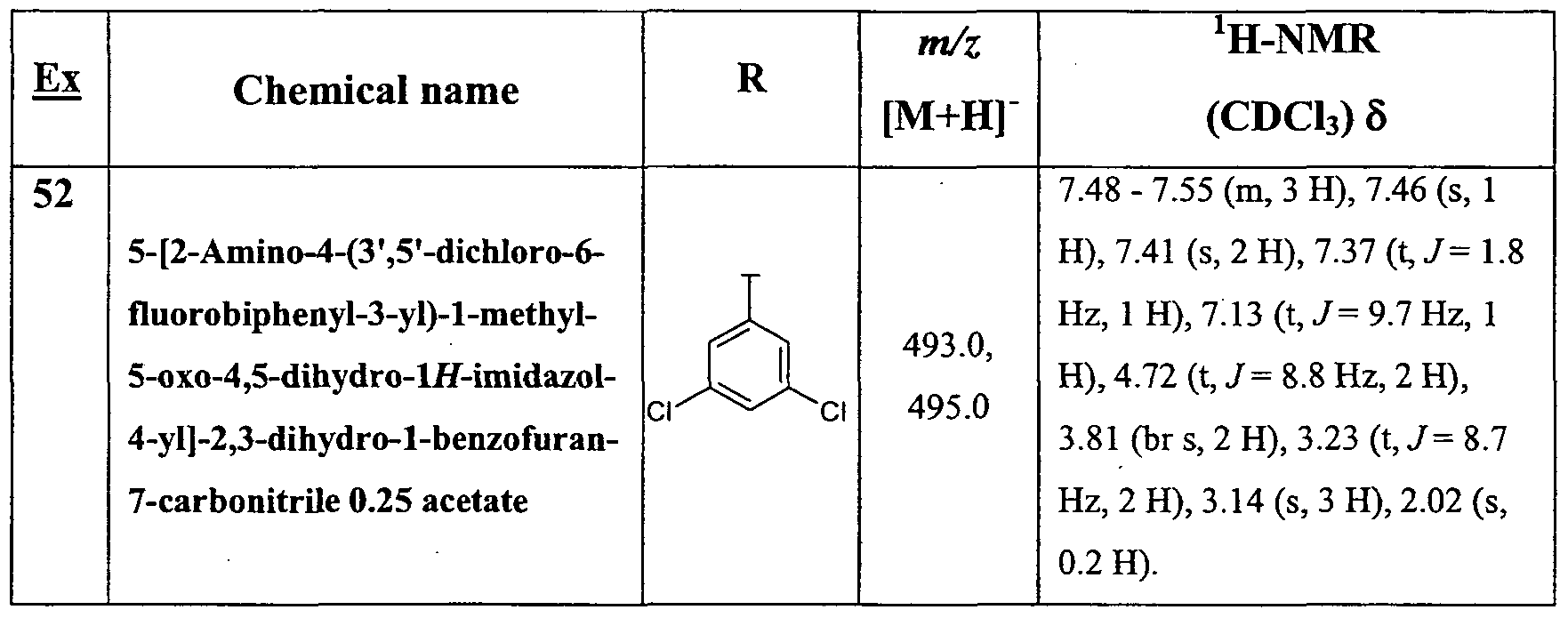
Compounds were tested in at least one of the following assays:
β-Secretase Enzyme The enzyme used in the IGEN Cleavage-, Fluorescent-, TR-FRET- and BiaCore assays is described as follows:
The soluble part of the human β-Secretase (AA 1 - AA 460) was cloned into the ASP2- FclO-1-IRES-GFP-neoK mammalian expression vector. The gene was fused to the Fc domain of IgGl (affinity tag) and stably cloned into HEK 293 cells. Purified sBACE-Fc is stored in Tris buffer, pH 9.2 and has a purity of 95%.
IGEN Cleavage Assay
The enzyme was diluted to 43 μg/ml in 40 mM MES pH 5.0. The IGEN substrate was diluted to 12 μM in 40 mM MES pH 5.0. Compounds were diluted to the desired concentration in dimethyl sulfoxide (final dimethyl sulfoxide concentration in assay is
5%). The assay was performed in a 96 well PCR plate from Greiner (#650201). Compound in dimethyl sulfoxide (3 μL) and enzyme (27 μL) were added to the plate, and pre- incubated for 10 min. The reaction was started with substrate (30 μL). The final dilution of enzyme was 20 μg/ml and the final concentration of substrate was 6 μM. After 20 minutes reaction at room temperature (RT), the reaction was stopped by removing 10 μL of the reaction mix and diluting it 1 :25 in 0.2 M Trizma-HCl, pH 8.0. The product was quantified by adding 50 μL of a 1:5000 dilution of the neoepitope antibody to 50 μL of the 1:25 dilution of the reaction mix (all antibodies and the streptavidin coated beads were diluted in PBS containing 0.5% BSA and 0.5% Tween20). Then, 100 μL of 0.2 mg/mL streptavidin coated beads (Dynabeads M-280) and a 1:5000 dilution of ruthenylated goat anti-rabbit (Ru-GaR) antibody was added. The mixture was measured for electro- chemiluminescence in a BioVeris M8 Analyzer after 2 hours of incubation with shaking at RT. The dimethyl sulfoxide control defined 100% activity level and 0% activity was defined by exclusion of the enzyme (using 40 mM MES pH 5.0 buffer instead).
Fluorescent Assay
The enzyme was diluted to 52 μg/ml in 40 mM MES pH 5.0. The substrate (Dabcyl-Edans) was diluted to 30 μM in 40 mM MES pH 5.0. Compounds were diluted to the desired concentration in dimethyl sulfoxide (final dimethyl sulfoxide concentration in assay is 5%). The assay is done in a Corning 384 well round bottom, low volume, non-binding surface plate (Corning #3676). Enzyme (9 μL) together with 1 μL of compound in dimethyl sulfoxide were added to the plate and pre-incubated for 10 min. Substrate (10 μL) was added and the reaction proceeded in the dark at RT for 25 min. The final dilution of enzyme was 23 μg/ml, and the final concentration of substrate was 15 μM (Km of 25 μM). The fluorescence of the product was measured on a Victor II plate reader with an excitation wavelength of 360 run and an emission wavelength of 485 nm using a protocol for labelled Edans peptide. The dimethyl sulfoxide control defined 100% activity level and 0% activity was defined by exclusion of the enzyme (using 40 mM MES pH 5.0 buffer instead).
TR-FRETAssay
Enzyme was diluted to 6 μg/mL and the substrate (Europium)CEVNLDAEFK(Qsy7) to 200 nM in reaction buffer (NaAcetate, chaps, triton x-100, EDTA pH 4.5). Compounds were diluted to the desired concentration in dimethyl sulfoxide (final dimethyl sulfoxide concentration in assay is 5%). The assay was done in a Costar 384 well round bottom, low volume, non-binding surface plate (Corning #3676). Enzyme (9 μL) and 1 μL of compound in dimethyl sulfoxide was added to the plate, mixed and pre-incubated for 10 min. Substrate (10 μL) was added and the reaction proceeded in the dark for 15 min at RT. The reaction was stopped with the addition of 7 μL NaAcetate, pH 9. The fluorescence of the product was measured on a Victor II plate reader with an excitation wavelength of 340 nm and an emission wavelength of 615 nm. The final concentration of the enzyme was 2.7 μg/ml and the final concentration of the substrate was 100 nM (Km of 290 nM). The dimethyl sulfoxide control defined the 100% activity level and 0% activity was defined by exclusion of the enzyme (using reaction buffer instead).
BA CE Biacore Sensor Ch ip Preparation
BACE was assayed on a Biacore3000 instrument by attaching either a peptidic transition state isostere (TSI) or a scrambled version of the peptidic TSI to the surface of a Biacore
CM5 sensor chip. The surface of a CM5 sensor chip has 4 distinct channels that can be used to couple the peptides. The scrambled peptide KFES-statine-ETIAEVENV was coupled to channel 1 and the TSI inhibitor KTEEISEVN-statine-VAEF was coupled to channel 2 of the same chip. The two peptides were dissolved at 0.2 mg/mL in 20 mM sodium acetate pH 4.5, and then the solutions were centrifuged at 14K rpm to remove any particulates. Carboxyl groups on the dextran layer were activated by injecting a one to one mixture of 0.5 M N-ethyl-N' (3-dimethylarninopropyl)-carbodiimide and 0.5 M N- hydroxysuccinimide at 5 μL/min for 7 min. Then the stock solution of the control peptide was injected in channel 1 for 7 min at 5 μL/min., and then the remaining activated carboxyl groups were blocked by injecting 1 M ethanolamine for 7 min at 5 μL/min.
BACE Biacore Assay Protocol
The BACE Biacore assay was done by diluting BACE to 0.5 μM in sodium acetate buffer at pH 4.5 (running buffer minus dimethyl sulfoxide). The diluted BACE was mixed with dimethyl sulfoxide or compound diluted in dimethyl sulfoxide at a final concentration of 5% dimethyl sulfoxide. The BACE/inhibitor mixture was incubated for 30 minutes at RT before being injected over channel 1 and 2 of the CM5 Biacore chip at a rate of 20 μL/min. As BACE bound to the chip the signal was measured in response units (RU). BACE binding to the TSI inhibitor on channel 2 gave a certain signal. The presence of a BACE inhibitor reduced the signal by binding to BACE and inhibiting the interaction with the peptidic TSI on the chip. Any binding to channel 1 was non-specific and was subtracted from the channel 2 responses. The dimethyl sulfoxide control was defined as 100% and the effect of the compound was reported as percent inhibition of the dimethyl sulfoxide control.
Beta-Secretase Whole Cell Assays Generation ofHEK293-APP695
The pcDNA3.1 plasmid encoding the cDNA of human full-length APP695 was stably transfected into HEK-293 cells using the Lipofectamine transfection reagent according to manufacture's protocol (Invitrogen). Colonies were selected with 0.1-0.5 mg/mL of zeocin. Limited dilution cloning was performed to generate homogeneous cell lines. Clones were
characterized by levels of APP expression and Aβ secreted in the conditioned media using an ELISA assay developed in-house.
Cell culture for HEK293-APP695 HEK293 cells stably expressing human wild-type APP (HEK293-APP695) were grown at 37 0C, 5% CO2 in DMEM containing 4500 g/L glucose, GlutaMAX and sodium pyruvate supplemented with 10% FBS, 1% non-essential amino acids and 0.1 mg/mL of the selection antibiotic zeocin.
Aβ40 release assay
HEK293-APP695 cells were harvested at 80-90% confluence and seeded at a concentration of 0.2xl06 cells/mL, 100 mL cell suspension/well, onto a black clear bottom 96-well poly-D-lysine coated plate. After over night incubation at 37 °C, 5% CO2, the cell medium was replaced with cell culture medium with penicillin and streptomycin (100 U/mL, 100 μg/mL, respectively) containing test compounds in a final dimethyl sulfoxide concentration of 1%. Cells were exposed to the test compounds for 24 h at 37 0C, 5% CO2. To quantify the amount of released Aβ, 100 μL cell medium was transferred to a round bottom polypropylene 96-well plate (assay plate). The cell plate was saved for the ATP assay, as described below. To the assay plate, 50 μL of primary detection solution containing 0.5 μg/mL of the rabbit anti-Aβ40 antibody and 0.5 μg/mL of the biotinylated monoclonal mouse 6E10 antibody in DPBS with 0.5 %BSA and 0.5% Tween-20 was added per well and incubated over night at 40C. Then, 50 μL of secondary detection solution containing 0.5 μg/mL of a ruthenylated goat anti-rabbit antibody and 0.2 mg/mL of streptavidin coated beads (Dynabeads M-280) was added per well. The plate was vigorously shaken at RT for 1-2 hours. The plate was then measured for electro- chemiluminescence in a BioVeris M8 Analyzer.
cell culture for SH-SY5Y
SH-SY5Y cells were grown 37 0C with 5% CO2 in DMEM/F-12 1:1 containing GlutaMAX supplemented with 1 mM HEPES, 10% FBS and 1% non-essential amino acids.
sAPPβ release assay
SH-SY5Y cells were harvested at 80-90% confluence and seeded at a concentration of 1.5x106 cells/mL, 100 mL cell suspension/well, onto a black clear flat bottom 96-well tissue culture plate. After 7 hours of incubation at 37 0C, 5% CO2, the cell medium was replaced with 90 μl cell culture medium with penicillin and streptomycin (100 U/mL, 100 μg/mL, respectively) containing test compounds in a final dimethyl sulfoxide concentration of 1%. Cells were exposed to the test compounds for 18 h at 37 0C, 5% CO2. To measure sAPPβ released into the cell medium, sAPPβ microplates from Meso Scale Discovery (MSD) were used and the assay was performed according to the manufacture's protocol. Briefly, 25 μL cell medium was transferred to a previously blocked MSD sAPPβ microplate. The cell plate was saved for the ATP assay, as described below. The sAPPβ was captured during shaking at RT for 1 hour, by antibodies spotted in the wells of the microplate. After multiple washes, SULFO-TAG labeled detection antibody was added (25μL/well, final concentration InM) to the assay plate and the plate was incubated with shaking at RT for 1 hour. Following multiple washes, 150 μl/well of Read Buffer T was added to the plate. After 10 minutes at RT the plate was read in the SECTOR™ Imager for electro-chemiluminescence .
ATP assay As indicated above, after transferring medium for analysis of Aβ40 or sAPPβ from the cell plate, the plate was used to analyze cytotoxicity using the ViaLight™ Plus cell proliferation/cytotoxicity kit from Cambrex BioScience that measures total cellular ATP. The assay was performed according to the manufacture's protocol. Briefly, 50 μL cell lysis reagent was added per well. The plates were incubated at RT for 10 min. Two min after addition of 100 μL reconstituted ViaLight™ Plus ATP reagent, the luminescence was measured in a Wallac Victor2 1420 multilabel counter.
hERG Assay Cell culture The hERG-expressing Chinese hamster ovary Kl (CHO) cells described by (Persson, Carlsson, Duker, & Jacobson, 2005) were grown to semi-confluence at 37 0C in a
humidified environment (5% CO2) in F-12 Ham medium containing L-glutamine, 10% foetal calf serum (FCS) and 0.6 mg/ml hygromycin (all Sigma- Aldrich). Prior to use, the monolayer was washed using a pre-warmed (37°C) 3 ml aliquot of Versene 1 :5,000 (Invitrogen). After aspiration of this solution the flask was incubated at 37 °C in an incubator with a further 2 ml of Versene 1 :5,000 for a period of 6 minutes. Cells were then detached from the bottom of the flask by gentle tapping and 10 ml of Dulbecco's Phosphate-Buffered Saline containing calcium (0.9 mM) and magnesium (0.5 mM) (PBS; Invitrogen) was then added to the flask and aspirated into a 15 ml centrifuge tube prior to centrifugation (50 g, for 4 mins). The resulting supernatant was discarded and the pellet gently re-suspended in 3 ml of PBS. A 0.5 ml aliquot of cell suspension was removed and the number of viable cells (based on trypan blue exclusion) was determined in an automated reader (Cedex; Innovatis) so that the cell re-suspension volume could be adjusted with PBS to give the desired final cell concentration. It is the cell concentration at this point in the assay that is quoted when referring to this parameter. CHO-KvI.5 cells, which were used to adjust the voltage offset on IonWorks™ HT, were maintained and prepared for use in the same way.
Electrophysiology
The principles and operation of this device have been described by (Schroeder, Neagle, Trezise, & Worley, 2003). Briefly, the technology is based on a 384-well plate
(PatchPlate™) in which a recording is attempted in each well by using suction to position and hold a cell on a small hole separating two isolated fluid chambers. Once sealing has taken place, the solution on the underside of the PatchPlate is changed to one containing amphotericin B. This permeablises the patch of cell membrane covering the hole in each well and, in effect, allows a perforated, whole-cell patch clamp recording to be made.
A β-test IonWorks™ HT from Essen Instrument was used. There is no capability to warm solutions in this device hence it was operated at room temperature (~21°C), as follows. The reservoir in the "Buffer" position was loaded with 4 ml of PBS and that in the "Cells" position with the CHO-hERG cell suspension described above. A 96-well plate (V-bottom, Greiner Bio-one) containing the compounds to be tested (at 3-fold above their final test
concentration) was placed in the "Plate 1" position and a PatchPlate™ was clamped into the PatchPlate™ station. Each compound plate was laid-out in 12 columns to enable ten, 8- point concentration-effect curves to be constructed; the remaining two columns on the plate were taken up with vehicle (final concentration 0.33% DMSO), to define the assay baseline, and a supra-maximal blocking concentration of cisapride (final concentration 10 μM) to define the 100% inhibition level. The fluidics-head (F-Head) of IonWorks™ HT then added 3.5 μl of PBS to each well of the PatchPlate™ and its underside was perfused with "internal" solution that had the following composition (in mM): K-Gluconate 100, KCl 40, MgCl2 3.2, EGTA 3 and HEPES 5 (all Sigma-Aldrich; pH 7.25-7.30 using 10 M KOH). After priming and de-bubbling, the electronics-head (E-head) then moved round the PatchPlate™ performing a hole test (i.e. applying a voltage pulse to determine whether the hole in each well was open). The F-head then dispensed 3.5 μl of the cell suspension described above into each well of the PatchPlate™ and the cells were given 200 seconds to reach and seal to the hole in each well. Following this, the E-head moved round the PatchPlate™ to determine the seal resistance obtained in each well. Next, the solution on the underside of the PatchPlate™ was changed to "access" solution that had the following composition (in mM): KCl 140, EGTA 1, MgCl2 1 and HEPES 20 (pH 7.25-7.30 using 10 M KOH) plus 100 μg/ml of amphotericin B (Sigma-Aldrich). After allowing 9 minutes for patch perforation to take place, the E-head moved round the PatchPlate™ 48 wells at a time to obtain pre-compound hERG current measurements. The F-head then added 3.5 μl of solution from each well of the compound plate to 4 wells on the PatchPlate™ (the final DMSO concentration was 0.33% in every well). This was achieved by moving from the most dilute to the most concentrated well of the compound plate to minimise the impact of any compound carry-over. After approximately 3.5 mins incubation, the E-head then moved around all 384- wells of the PatchPlate™ to obtain post-compound hERG current measurements. In this way, non-cumulative concentration-effect curves could be produced where, providing the acceptance criteria were achieved in a sufficient percentage of wells (see below), the effect of each concentration of test compound was based on recording from between 1 and 4 cells.
The pre- and post-compound hERG current was evoked by a single voltage pulse consisting of a 20 s period holding at -70 mV, a 160 ms step to -60 mV (to obtain an estimate of leak), a 100 ms step back to -70 mV, a 1 s step to + 40 mV, a 2 s step to -30 mV and finally a 500 ms step to -7OmV. In between the pre- and post-compound voltage pulses there was no clamping of the membrane potential. Currents were leak-subtracted based on the estimate of current evoked during the +1OmV step at the start of the voltage pulse protocol. Any voltage offsets in IonWorks™ HT were adjusted in one of two ways. When determining compound potency, a depolarising voltage ramp was applied to CHO- Kv 1.5 cells and the voltage noted at which there was an inflection point in the current trace (i.e. the point at which channel activation was seen with a ramp protocol). The voltage at which this occurred had previously been determined using the same voltage command in conventional electrophysiology and found to be -15 mV (data not shown); thus an offset potential could be entered into the IonWorks™ HT software using this value as a reference point. When determining the basic electrophysiological properties of hERG, any offset was adjusted by determining the hERG tail current reversal potential in IonWorks™ HT, comparing it with that found in conventional electrophysiology (-82 mV) and then making the necessary offset adjustment in the IonWorks™ HT software. The current signal was sampled at 2.5 kHz.
Pre- and post-scan hERG current magnitude was measured automatically from the leak subtracted traces by the IonWorks™ HT software by taking a 40 ms average of the current during the initial holding period at —70 mV (baseline current) and subtracting this from the peak of the tail current response. The acceptance criteria for the currents evoked in each well were: pre-scan seal resistance >60 MΩ, pre-scan hERG tail current amplitude >150 p A; post-scan seal resistance >60 MΩ. The degree of inhibition of the hERG current was assessed by dividing the post-scan hERG current by the respective pre-scan hERG current for each well.
Results Typical IC50 values for the compounds of the present invention are in the range of about 1 to about 2,000 nM. Biological data on final compounds are given below in Table 2.
TABLE 2.

Claims
1. A compound of formula I :

R . 1 : is phenyl, optionally substituted with one or more R , or C1-6 alkyl;
R2 is independently selected from cyano and halo;
A is phenyl or heteroaryl, said phenyl or heteroaryl optionally fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R4;
R3 is independently selected from halo, cyano, C1^aIlCyI, trifluoromethyl, methoxy, trifluoromethoxy and acetyl;
R4 is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R4 in the heterocyclyl part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
R5 is C^alkyl optionally substituted with halogen, cyano, NH2, OH, CO2H, OCi-βalkyl, SO2H, C(O)Ci-6alkyl, C(O)OC1-6alkyl, C(O)NH2, C(O)NHC1-6alkyl, C(O)N(C1-6alkyl)2, SO2C1-6alkyl, SO2NHC1-6alkyl, SO2N(Ci-6alkyl)2, NH(C1-6alkyl), N(C^aIlCyI)2,  NC(O)(C1-6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl)2, S02aryl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl, NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl,
NC(O)(C1-6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl)2, S02aryl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl, NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl,
C(O)NHheteroaryl, C(O)N(heteroaryl)2, SO2heteroaryl, SO2NHheteroaryl,
SO2N(heteroaryl)2, NH(heteroaryl), N(heteroaryl)2, NC(O)heteroaryl, NC(O)(heteroaryl)2,
C5-6heterocyclyl, OCs-βheterocyclyl, C(O)C5-6heterocyclyl, C(O)OC5-6heterocyclyl, C(O)NHC5-6heterocyclyl, C(O)N(C5-6heterocyclyl)2, SO2C5-6heterocyclyl, SO2NHC5- δheterocyclyl, SO2N(Cs^heterocyclyl)2, NH(C5-6heterocyclyl), N(C5-6heterocyclyl)2,
NC(O)C5-6heterocyclyl or NC(O)(C5.6heterocyclyl)2; n is 0, 1 or 2; provided that the following compounds are excluded: 2-Amino-5-(2,3-dihydro-l-benzoruran-5-yl)-5-(3'-methoxybiphenyl-3-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2',5'-dimethoxybiphenyl-3-yl)-3-methyl-
3,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2I-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
2-Arnino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(2'-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3 ,5-dihydro-4H-imidazol-4-one;
3'-[2-Arnino-4-(2,3-dihydro-l-benzofuran-5-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-6-fluorobiphenyl-3-carbonitrile; 2-Amino-5-(2,3-dihydro- 1 -benzofuran-5-yl)-3-methyl-5-[3'-(trifluoromethoxy)biphenyl-3- yl]-3 ,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(3'-chlorobiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(3'-metlioxybiphenyl-3-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2',5'-dirnethoxybiphenyl-3-yl)-3-methyl-
3 ,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2'-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; 2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(2'-fluoro-3l-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; 3'-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-4-fluorobiphenyl-3-carbonitrile;
3'-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-6-fluorobiphenyl-3-carbonitrile; 5 2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-5-[3'-(trifluoroπiethoxy)biphenyl-3- yl]-3 ,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(3'-chlorobiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2',5'-dimethoxybiphenyl-3-yl)-3-o methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[4-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-l-methyl-5-oxo-4,5-diliydro-lH- imidazol-4-yl]biphenyl-3-carbonitrile;
3'-[4-(I -Acetyl- 1 ,2,3 ,4-tetrahydroquinolin-6-yl)-2-amino- 1 -methyl-5-oxo-4,5-dihydro- IH- imidazol-4-yl]-4-fluorobiphenyl-3-carbonitrile; s 5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2'-fluoro-5'-methoxybiplienyl-3- yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one;
5-(l-Acetyl-l,2,3,4-tetrahydroquinolin-6-yl)-2-amino-5-(2'-fluoro-3'-methoxybiphenyl-3- yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one;
5-( 1 -Acetyl- 1 ,2,3 ,4-tetrahydroquinolin-6-yl)-2-amino-5-[3-(3 -furyl)phenyl]-3-methyl-3 ,5-o dihydro-4H-imidazol-4-one;
2-Amino-5-(3,4-dihydro-lH-isochromen-7-yl)-5-(2l-fluoro-5'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
3'-[2-Amino-4-(3,4-dihydro-lH-isochromen-7-yl)-l-methyl-5-oxo-4,5-dihydro-lH- imidazol-4-yl] -6-fluorobiphenyl-3 -carbonitrile; or 5 2-Amino-5-(3,4-dihydro-lH-isochromen-7-yl)-5-[3-(3-furyl)phenyl]-3-methyl-3,5- dihydro-4H-imidazol-4-one; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
2. A compound according to claim 1, wherein R1 is phenyl. 0
3. A compound according to claim 2, wherein n is 1 and R2 is fluoro.
4. A compound according to claim 2, wherein n is 0.
5. A compound according to claim 2, wherein R is methyl.
6. A compound according to claim 2, wherein A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system.
7. A compound according to claim 2, wherein A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system where the bicyclic ring system is substituted with from one to four R4.
8. A compound according to claim 7, wherein said bicyclic ring system is substituted with two R4, said R4 being methyl.
9. A compound according to claim 7, wherein said bicyclic ring system is substituted with one R4, said R4 being cyano.
10. A compound according to any one of claims 2 to 9, wherein A is selected from:

11. A compound according to any one of claims 2 to 9, wherein A is:

12. A compound according to any one of claims 2 to 9, wherein R3 is independently selected from halo, cyano, methoxy, trifluoromethoxy and acetyl.
13. A compound according to claim 2, wherein n is 0 or 1; R2 is fluoro; R5 is C1-6alkyl; A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system, optionally substituted with one or two R4, said R4 being independently selected from methyl or cyano.
14. A compound according to claim 1, selected from: 2-Amino-5-(2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl)-5-(6-fluoro-3'-methoxybiphenyl-
3-yl)-3-methyl-3,5-dihydro-4H-imidazol-4-one hydrochloride;
5'-[2-Amino-4-(3 ,4-dihydro-2H-chromen-6-yl)- 1 -methyl-5-oxo-4,5-dihydro- lH-imidazol-
4-yl]-2'-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-(6-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3 ,5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2l,6-difluoro-3'-methoxybiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3- methyl-3, 5-dihydro-4H-imidazol-4-one;
2-Amino-5-(2,3-dihydro-l-benzofuran-5-yl)-5-(6-fluoro-3'-methoxybiphenyl-3-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; 5'-[2-Amino-4-(2,3-dihydro-l-benzofuran-5-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-2'-fluorobiphenyl-3-carbonitrile;
2-Amino-5-(2',6-difluoro-3'-methoxybiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3- methyl-3 , 5 -dihydro-4H-imidazol-4-one;
3'-[2-Aniino-4-(2,3-dihydro-l-benzofuran-5-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol- 4-yl]-5-methoxybiphenyl-2-carbonitrile hydrochloride;
2-Ammo-5-(2,3-dmydro-l-benzofuran-5-yl)-5-[5'-methoxy-2'-(trifluoromethoxy)biphenyl-
3-yl]-3-methyl-3,5-dihydro-4H-imidazol-4-one hydrochloride;
2-Amino-5-(3',5'-dichlorobiphenyl-3-yl)-5-(2,3-dihydro-l-benzofuran-5-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one hydrochloride; 3'-[2-Amino-4-(3,4-dihydro-2H-chromen-6-yl)-l-methyl-5-oxo-4,5-dihydro-lH-imidazol-
4-yl]-5-methoxybiphenyl-2-carbonitrile; 2-Amino-5-(3,4-dihydro-2H-chromen-6-yl)-5-[5'-methoxy-2'-(trifluoromethoxy)biphenyl- 3-yl]-3-methyl-3,5-dihydro-4H-imidazol-4-one hydrochloride;
2-Amino-5-(3',5'-dichlorobiphenyl-3-yl)-5-(3,4-dihydro-2H-chromen-6-yl)-3-methyl-3,5- dihydro-4H-imidazol-4-one hydrochloride;
5 5-(2l-Acetyl-5'-methoxybiphenyl-3-yl)-2-amino-5-(2,3-dihydro-l-benzoruran-5-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
5-(2I-Acetyl-5'-methoxybiphenyl-3-yl)-2-amino-5-(3,4-dihydro-2H-chromen-6-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one;
S-P-Amino^-CS'^'-dichloro-ό-fluorobiphenyl-S-y^-l-methyl-S-oxo^jS-dihydro-lH- I0 imidazol-4-yl]-2,3-dihydro-l-benzofuran-7-carbonitτile 0.25 acetate; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
15. A compound according to claim 1, wherein
is R1 is CΗ2CΗ2cyclopropyl;
R2 is independently selected from cyano and halo;
A is phenyl or heteroaryl fused with a 5 or 6 membered heterocyclyl group to form a 20 bicyclic ring system where the bicyclic ring system is optionally substituted with one or more R4;
R4 is independently selected from halo, cyano, methyl and methoxy; provided that said bicyclic ring of A, is substituted with at least one R4 in the heterocyclyl 5 part, when it is a 1,3-benzodioxole or 2,3-dihydro-l,4-benzodioxine ring system;
R5 is C^alkyl optionally substituted with halogen, cyano, NH2, OH, CO2H, OQ-βalkyl, SO2H5 C(O)C1-6alkyl, C(O)OC1-6alkyl, C(O)NH2, C(O)NHC 1-6alkyl, C(O)N(C1-6alkyl)2, SO2C1-6alkyl, SO2NHC1-6alkyl, SO2N(C1-6alkyl)2, NH(d-6alkyl), N(d-6alkyl)2, 30 NHC(O)Ci-6alkyl, NC(O)(C 1-6alkyl)2, aryl, Oaryl, C(O)aryl, C(O)Oaryl, C(O)NHaryl, C(O)N(aryl)2, S02aryl, SO2NHaryl, SO2N(aryl)2, NH(aryl), N(aryl)2, NC(O)aryl, NC(O)(aryl)2, heteroaryl, Oheteroaryl, C(O)heteroaryl, C(O)Oheteroaryl, C(O)NHheteroaryl, C(O)N(heteroaryl)2, SO2heteroaryl, SO2NHheteroaryl, SO2N(heteroaryl)2, NH(heteroaryl), N(heteroaryl)2, NC(O)heteroaryl, NC(O)(heteroaryl)2, Cs-όheterocyclyl, OCs-δheterocyclyl, C(O)C5-6heterocyclyl, C(O)OC5-6heterocyclyl, C(O)NHC5-6heterocyclyl, C(O)N(C5-6heterocyclyl)2, SO2C5-6heterocyclyI, SO2NHC5- eheterocyclyl, SO2N(C5-6heterocyclyl)2, NH(C5-6heterocyclyl), N(C5-6heterocyclyl)2, NC(O)C5-6heterocyclyl or NC(O)(C5-6heterocyclyl)2; n is 0, 1 or 2.
16. A compound according to claim 15, wherein n is 1 and R2 is fluoro.
17. A compound according to claim 15, wherein R is methyl.
18. A compound according to claim 15, wherein A represents phenyl fused with a 5 or 6 membered heterocyclyl group to form a bicyclic ring system, optionally substituted with from one to four R4.
19. A compound according to claim 15, wherein A is selected from:

20. A compound according to claim 15, selected from:
2-Amino-5-[3-(2-cyclopropylethyl)-4-fluorophenyl]-5-(3,4-dihydro-2H-chromen-6-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; and 2-Amino-5-[3-(2-cyclopropylethyl)-4-fluorophenyl]-5-(2,3-dihydro- 1 -benzofuran-5-yl)-3- methyl-3,5-dihydro-4H-imidazol-4-one; as a free base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
21. A pharmaceutical composition comprising as active ingredient a therapeutically effective amount of a compound according to any one of claims 1 to 20 in association with pharmaceutically acceptable excipients, carriers or diluents.
22. A compound according to any one of claims 1 to 20, or a pharmaceutically acceptable salt thereof, for use as a medicament.
23. Use of a compound of any one of claims 1 to 20 as a medicament for treating or preventing an Aβ-related pathology.
24. Use of a compound of any one of claims 1 to 20 as a medicament for treating or preventing an Aβ-related pathology, wherein said Aβ-related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer Disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
25. Use of a compound of any one of claims 1 to 20 as a medicament for treating or preventing Alzheimer Disease.
26. Use of a compound of any one of claims 1 to 20 in the manufacture of a medicament for treating or preventing an Aβ-related pathology.
27. Use of a compound of any one of claims 1 to 20 in the manufacture of a medicament for treating or preventing an Aβ-related pathology, wherein said Aβ-related pathology is Downs syndrome, a β-amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
28. Use of a compound of any one of claims 1 to 20 in the manufacture of a medicament for treating or preventing Alzheimer's Disease.
29. A method of inhibiting activity of BACE comprising contacting said BACE with a compound of any one of claims 1 to 20.
30. A method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of any one of claims 1 to 20.
31. The method of claim 30, wherein said Aβ-related pathology is Downs syndrome, a β- amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
32. A method of treating or preventing Alzheimer's Disease, comprising administering to said patient a therapeutically effective amount of a compound of any one of claims 1 to 20.
33. The method of claim 31 or 32, wherein said mammal is a human.
34. A method of treating or preventing an Aβ-related pathology in a mammal, comprising administering to said patient a therapeutically effective amount of a compound of any one of claims 1 to 20 and at least one cognitive enhancing agent, memory enhancing agent, or choline esterase inhibitor.
35. The method of claim 34, wherein said Aβ-related pathology is Downs syndrome, a β- amyloid angiopathy, cerebral amyloid angiopathy, hereditary cerebral hemorrhage, a disorder associated with cognitive impairment, MCI ("mild cognitive impairment"), Alzheimer Disease, memory loss, attention deficit symptoms associated with Alzheimer disease, neurodegeneration associated with Alzheimer disease, dementia of mixed vascular origin, dementia of degenerative origin, pre-senile dementia, senile dementia, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
36. The method of claim 35, wherein said Aβ-related pathology is Alzheimer Disease.
37. The method of claim 35 or 36, wherein said mammal is a human.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US87093106P | 2006-12-20 | 2006-12-20 | |
| US60/870,931 | 2006-12-20 | ||
| US91799107P | 2007-05-15 | 2007-05-15 | |
| US60/917,991 | 2007-05-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008076043A1 true WO2008076043A1 (en) | 2008-06-26 |
Family
ID=39536553
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/SE2007/001116 WO2008076043A1 (en) | 2006-12-20 | 2007-12-18 | Novel 2-amino-5,5-diaryl-imidazol-4-ones |
| PCT/SE2007/001117 WO2008076044A1 (en) | 2006-12-20 | 2007-12-18 | Novel 2-amino-5, 5-diaryl-imidazol-4-ones |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/SE2007/001117 WO2008076044A1 (en) | 2006-12-20 | 2007-12-18 | Novel 2-amino-5, 5-diaryl-imidazol-4-ones |
Country Status (1)
| Country | Link |
|---|---|
| WO (2) | WO2008076043A1 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7855213B2 (en) | 2006-06-22 | 2010-12-21 | Astrazeneca Ab | Compounds |
| WO2011002408A1 (en) * | 2009-07-02 | 2011-01-06 | Astrazeneca Ab | Novel compounds for treatment of neurodegeneration associated with diseases, such as alzheimer's disease or dementia |
| WO2011002407A1 (en) * | 2009-07-02 | 2011-01-06 | Astrazeneca Ab | Novel compounds for treatment of neurodegeneration associated with diseases, such as alzheimer's disease or dementia |
| US7868000B2 (en) | 2005-06-14 | 2011-01-11 | Schering Corporation | Aspartyl protease inhibitors |
| EP2281824A1 (en) * | 2009-08-07 | 2011-02-09 | Noscira, S.A. | Furan-imidazolone derivatives, for the treatment of cognitive, neurodegenerative or neuronal diseases or disorders |
| US7973067B2 (en) | 2003-12-15 | 2011-07-05 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8030500B2 (en) | 2008-11-14 | 2011-10-04 | Astrazeneca Ab | Substituted isoindoles for the treatment and/or prevention of Aβ- related pathologies |
| WO2011134296A1 (en) | 2010-04-26 | 2011-11-03 | 上海阳帆医药科技有限公司 | (6,7-dihydro-2-nitro-5h-imidazol[2,1-b] [1,3]oxazin-6-yl) amide compounds, preparation methods and uses thereof |
| WO2012087237A1 (en) | 2010-12-22 | 2012-06-28 | Astrazeneca Ab | Compounds and their use as bace inhibitors |
| US8450308B2 (en) | 2008-08-19 | 2013-05-28 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| US8633212B2 (en) | 2009-03-13 | 2014-01-21 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| US8729071B2 (en) | 2009-10-08 | 2014-05-20 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| US8889703B2 (en) | 2010-02-24 | 2014-11-18 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| US8981112B2 (en) | 2012-03-05 | 2015-03-17 | Vitae Pharmaceuticals, Inc. | Inhibitors of β-secretase |
| US9000184B2 (en) | 2012-06-20 | 2015-04-07 | Astrazeneca Ab | Cyclohexane-1,2′-naphthalene-1′,2″-imidazol compounds and their use as BACE inhibitors |
| US9000183B2 (en) | 2012-06-20 | 2015-04-07 | Astrazeneca Ab | Cyclohexane-1,2′-indene-1′,2″-imidazol compounds and their use as BACE inhibitors |
| US9000185B2 (en) | 2012-06-20 | 2015-04-07 | Astrazeneca Ab | Cycloalkyl ether compounds and their use as BACE inhibitors |
| US9000182B2 (en) | 2012-06-20 | 2015-04-07 | Astrazeneca Ab | 2H-imidazol-4-amine compounds and their use as BACE inhibitors |
| US9018391B2 (en) | 2012-08-27 | 2015-04-28 | Boehringer Ingelheim International Gmbh | Inhibitors of beta-secretase |
| US9145426B2 (en) | 2011-04-07 | 2015-09-29 | Merck Sharp & Dohme Corp. | Pyrrolidine-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9181236B2 (en) | 2011-08-22 | 2015-11-10 | Merck Sharp & Dohme Corp. | 2-spiro-substituted iminothiazines and their mono-and dioxides as bace inhibitors, compositions and their use |
| US9221839B2 (en) | 2011-04-07 | 2015-12-29 | Merck Sharp & Dohme Corp. | C5-C6 oxacyclic-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9290477B2 (en) | 2012-09-28 | 2016-03-22 | Vitae Pharmaceuticals, Inc. | Inhibitors of β-secretase |
| US9650336B2 (en) | 2011-10-10 | 2017-05-16 | Astrazeneca Ab | Mono-fluoro beta-secretase inhibitors |
| WO2017093930A1 (en) | 2015-12-04 | 2017-06-08 | Viiv Healthcare Uk Limited | Isoindoline derivatives |
| US10548882B2 (en) | 2012-06-21 | 2020-02-04 | Astrazeneca Ab | Camsylate salt |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109096243B (en) * | 2018-07-26 | 2020-06-26 | 南京昊绿生物科技有限公司 | Synthesis process of deuterated ibrutinostat racemate |
| CN113030353B (en) * | 2019-12-25 | 2024-08-20 | 上海奥博生物医药股份有限公司 | Method for measuring chiral isomer content of (S) -1, 2-propanediol |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005058311A1 (en) * | 2003-12-15 | 2005-06-30 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| WO2006009653A1 (en) * | 2004-06-16 | 2006-01-26 | Wyeth | Amino-5,5-diphenylimidazolone derivatives for the inhibition of beta-secretase |
| WO2006065277A2 (en) * | 2004-12-13 | 2006-06-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| WO2007005366A1 (en) * | 2005-06-30 | 2007-01-11 | Wyeth | AMlNO-5-(5-MEMBERED)HETEROARYLIMIDAZOLONE COMPOUNDS AND THE USE THEREOF FOR β-SECRETASE MODULATION |
| WO2007005404A1 (en) * | 2005-06-30 | 2007-01-11 | Wyeth | AMINO-5-(6-MEMBERED)HETEROARYLIMIDAZOLONE COMPOUNDS AND THE USE THEREOF FOR ß-SECRETASE MODULATION |
| WO2007038271A1 (en) * | 2005-09-26 | 2007-04-05 | Wyeth | Amino-5- [4- (difluoromethoxy) phenyl] -5-phenylimidazolone compounds as inhibitors of the beta-secretase (bace) |
| WO2007058601A1 (en) * | 2005-11-21 | 2007-05-24 | Astrazeneca Ab | Novel 2-amino-imidazole-4-one compounds and their use in the manufacture of a medicament to be used in the treatment of cognitive impairment, alzheimer’s disease, neurodegeneration and dementia |
| WO2007058602A2 (en) * | 2005-11-21 | 2007-05-24 | Astrazeneca Ab | Novel 2-amino-imidazole-4-one compounds and their use in the manufacture of a medicament to be used in the treatment of cognitive impairment, alzheimer’s disease, neurodegeneration and dementia. |
| WO2007078813A2 (en) * | 2005-12-19 | 2007-07-12 | Wyeth | 2-AMINO-5-PIPERIDINYLIMIDAZOLONE COMPOUNDS AND USE THEREOF FOR β-SECRETASE MODULATION |
-
2007
- 2007-12-18 WO PCT/SE2007/001116 patent/WO2008076043A1/en active Application Filing
- 2007-12-18 WO PCT/SE2007/001117 patent/WO2008076044A1/en active Application Filing
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005058311A1 (en) * | 2003-12-15 | 2005-06-30 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| WO2006009653A1 (en) * | 2004-06-16 | 2006-01-26 | Wyeth | Amino-5,5-diphenylimidazolone derivatives for the inhibition of beta-secretase |
| WO2006065277A2 (en) * | 2004-12-13 | 2006-06-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| WO2007005366A1 (en) * | 2005-06-30 | 2007-01-11 | Wyeth | AMlNO-5-(5-MEMBERED)HETEROARYLIMIDAZOLONE COMPOUNDS AND THE USE THEREOF FOR β-SECRETASE MODULATION |
| WO2007005404A1 (en) * | 2005-06-30 | 2007-01-11 | Wyeth | AMINO-5-(6-MEMBERED)HETEROARYLIMIDAZOLONE COMPOUNDS AND THE USE THEREOF FOR ß-SECRETASE MODULATION |
| WO2007038271A1 (en) * | 2005-09-26 | 2007-04-05 | Wyeth | Amino-5- [4- (difluoromethoxy) phenyl] -5-phenylimidazolone compounds as inhibitors of the beta-secretase (bace) |
| WO2007058601A1 (en) * | 2005-11-21 | 2007-05-24 | Astrazeneca Ab | Novel 2-amino-imidazole-4-one compounds and their use in the manufacture of a medicament to be used in the treatment of cognitive impairment, alzheimer’s disease, neurodegeneration and dementia |
| WO2007058602A2 (en) * | 2005-11-21 | 2007-05-24 | Astrazeneca Ab | Novel 2-amino-imidazole-4-one compounds and their use in the manufacture of a medicament to be used in the treatment of cognitive impairment, alzheimer’s disease, neurodegeneration and dementia. |
| WO2007078813A2 (en) * | 2005-12-19 | 2007-07-12 | Wyeth | 2-AMINO-5-PIPERIDINYLIMIDAZOLONE COMPOUNDS AND USE THEREOF FOR β-SECRETASE MODULATION |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8242112B2 (en) | 2003-12-15 | 2012-08-14 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8937093B2 (en) | 2003-12-15 | 2015-01-20 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US8183252B2 (en) | 2003-12-15 | 2012-05-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US9416108B2 (en) | 2003-12-15 | 2016-08-16 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US7973067B2 (en) | 2003-12-15 | 2011-07-05 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8178513B2 (en) | 2003-12-15 | 2012-05-15 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7868000B2 (en) | 2005-06-14 | 2011-01-11 | Schering Corporation | Aspartyl protease inhibitors |
| US7855213B2 (en) | 2006-06-22 | 2010-12-21 | Astrazeneca Ab | Compounds |
| US8691831B2 (en) | 2007-02-23 | 2014-04-08 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US8829036B2 (en) | 2007-02-23 | 2014-09-09 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US8691833B2 (en) | 2007-02-23 | 2014-04-08 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US8450308B2 (en) | 2008-08-19 | 2013-05-28 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| US8030500B2 (en) | 2008-11-14 | 2011-10-04 | Astrazeneca Ab | Substituted isoindoles for the treatment and/or prevention of Aβ- related pathologies |
| US10336717B2 (en) | 2009-03-13 | 2019-07-02 | Vitae Pharmaceuticals, Llc | Inhibitors of beta-secretase |
| US9212153B2 (en) | 2009-03-13 | 2015-12-15 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| US8633212B2 (en) | 2009-03-13 | 2014-01-21 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| WO2011002407A1 (en) * | 2009-07-02 | 2011-01-06 | Astrazeneca Ab | Novel compounds for treatment of neurodegeneration associated with diseases, such as alzheimer's disease or dementia |
| WO2011002408A1 (en) * | 2009-07-02 | 2011-01-06 | Astrazeneca Ab | Novel compounds for treatment of neurodegeneration associated with diseases, such as alzheimer's disease or dementia |
| WO2011015646A3 (en) * | 2009-08-07 | 2011-03-31 | Noscira, S.A. | Furan-imidazolone derivatives, for the treatment of cognitive, neurodegenerative or neuronal diseases or disorders |
| EP2281824A1 (en) * | 2009-08-07 | 2011-02-09 | Noscira, S.A. | Furan-imidazolone derivatives, for the treatment of cognitive, neurodegenerative or neuronal diseases or disorders |
| US9428475B2 (en) | 2009-10-08 | 2016-08-30 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9029362B2 (en) | 2009-10-08 | 2015-05-12 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as brace inhibitors, compositions, and their use |
| US8729071B2 (en) | 2009-10-08 | 2014-05-20 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| US8940748B2 (en) | 2009-10-08 | 2015-01-27 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9475785B2 (en) | 2009-10-08 | 2016-10-25 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| US9687494B2 (en) | 2009-10-08 | 2017-06-27 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US8889703B2 (en) | 2010-02-24 | 2014-11-18 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| US9045500B2 (en) | 2010-02-24 | 2015-06-02 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| WO2011134296A1 (en) | 2010-04-26 | 2011-11-03 | 上海阳帆医药科技有限公司 | (6,7-dihydro-2-nitro-5h-imidazol[2,1-b] [1,3]oxazin-6-yl) amide compounds, preparation methods and uses thereof |
| US10231967B2 (en) | 2010-12-22 | 2019-03-19 | Astrazeneca Ab | Compounds and their use as BACE inhibitors |
| EP2655378A4 (en) * | 2010-12-22 | 2014-07-09 | Astrazeneca Ab | Compounds and their use as bace inhibitors |
| EP3176172A1 (en) * | 2010-12-22 | 2017-06-07 | Astrazeneca AB | Spiroimidazole compounds and their use as bace inhibitors |
| US9918985B2 (en) | 2010-12-22 | 2018-03-20 | Astrazeneca Ab | Compounds and their use as BACE inhibitors |
| EP2655378A1 (en) * | 2010-12-22 | 2013-10-30 | AstraZeneca AB | Compounds and their use as bace inhibitors |
| US9248129B2 (en) | 2010-12-22 | 2016-02-02 | Astrazeneca Ab | Compounds and their use as BACE inhibitors |
| JP2017008072A (en) * | 2010-12-22 | 2017-01-12 | アストラゼネカ・アクチエボラーグAstrazeneca Aktiebolag | Compounds and their use as BACE inhibitors |
| CN103380133B (en) * | 2010-12-22 | 2016-06-29 | 阿斯利康(瑞典)有限公司 | Compound and the purposes as BACE inhibitor thereof |
| CN103380133A (en) * | 2010-12-22 | 2013-10-30 | 阿斯利康(瑞典)有限公司 | Compounds and their use as BACE inhibitors |
| WO2012087237A1 (en) | 2010-12-22 | 2012-06-28 | Astrazeneca Ab | Compounds and their use as bace inhibitors |
| US9145426B2 (en) | 2011-04-07 | 2015-09-29 | Merck Sharp & Dohme Corp. | Pyrrolidine-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9221839B2 (en) | 2011-04-07 | 2015-12-29 | Merck Sharp & Dohme Corp. | C5-C6 oxacyclic-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9181236B2 (en) | 2011-08-22 | 2015-11-10 | Merck Sharp & Dohme Corp. | 2-spiro-substituted iminothiazines and their mono-and dioxides as bace inhibitors, compositions and their use |
| US9650336B2 (en) | 2011-10-10 | 2017-05-16 | Astrazeneca Ab | Mono-fluoro beta-secretase inhibitors |
| US9526727B2 (en) | 2012-03-05 | 2016-12-27 | Vitae Pharmaceutical, Inc. | Inhibitors of beta-secretase |
| US9949975B2 (en) | 2012-03-05 | 2018-04-24 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| US8981112B2 (en) | 2012-03-05 | 2015-03-17 | Vitae Pharmaceuticals, Inc. | Inhibitors of β-secretase |
| US9000182B2 (en) | 2012-06-20 | 2015-04-07 | Astrazeneca Ab | 2H-imidazol-4-amine compounds and their use as BACE inhibitors |
| US9000185B2 (en) | 2012-06-20 | 2015-04-07 | Astrazeneca Ab | Cycloalkyl ether compounds and their use as BACE inhibitors |
| US9000183B2 (en) | 2012-06-20 | 2015-04-07 | Astrazeneca Ab | Cyclohexane-1,2′-indene-1′,2″-imidazol compounds and their use as BACE inhibitors |
| US9000184B2 (en) | 2012-06-20 | 2015-04-07 | Astrazeneca Ab | Cyclohexane-1,2′-naphthalene-1′,2″-imidazol compounds and their use as BACE inhibitors |
| US10548882B2 (en) | 2012-06-21 | 2020-02-04 | Astrazeneca Ab | Camsylate salt |
| US9018391B2 (en) | 2012-08-27 | 2015-04-28 | Boehringer Ingelheim International Gmbh | Inhibitors of beta-secretase |
| US9290477B2 (en) | 2012-09-28 | 2016-03-22 | Vitae Pharmaceuticals, Inc. | Inhibitors of β-secretase |
| WO2017093930A1 (en) | 2015-12-04 | 2017-06-08 | Viiv Healthcare Uk Limited | Isoindoline derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008076044A1 (en) | 2008-06-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008076043A1 (en) | Novel 2-amino-5,5-diaryl-imidazol-4-ones | |
| US20080176862A1 (en) | New Compounds 617 | |
| US20080161269A1 (en) | Compounds 620 | |
| WO2007145568A9 (en) | Amino-imidazolones and their use as a medicament for treating cognitive impairment, alzheimer disease, neurodegeneration and dementia | |
| US20070299087A1 (en) | New Compounds 319 | |
| US20080293718A1 (en) | Novel 2-Amino-Imidazole-4-One Compounds and Their Use in the Manufacture of a Medicament to Be Used in the Treatment of Cognitive Impairment, Alzheimer's Disease, Neurodegeneration and Dementia | |
| US20080287460A1 (en) | New compounds 835 | |
| US20080214577A1 (en) | New Compounds 320 | |
| US20100324072A1 (en) | Pyrrolopyridine derivatives and their use as bace inhibitors | |
| EP2035378A1 (en) | Substituted isoindoles as bace inhibitors and their use | |
| US20080051420A1 (en) | New Compounds 317 | |
| JP2009539974A (en) | Aminoimidazolones and their use as pharmaceuticals to treat cognitive impairment, Alzheimer's disease, neurodegeneration and dementia | |
| KR20080070744A (en) | Novel 2-amino-heterocycles useful in the treatment of abeta-related pathologies | |
| EP2004630A1 (en) | 2-aminopyrimidin-4-ones and their use for treating or preventing a -related pathologies | |
| WO2008063114A1 (en) | Amino- imidazolones and their use as medicament for treating cognitive impairment alzheimer disease, neurodegeneration and dementia | |
| WO2009005470A1 (en) | Aryl and heteroaryl substituted isoindole derivatives as bace inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07852118 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07852118 Country of ref document: EP Kind code of ref document: A1 |